Embed Size (px)
Citation preview

This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
BAND 21 b ZEITSCHRIFT FÜR NATURFORSCHUNG IIEFT 11
Neue Ergebnisse der Säure-Basen-Katalyse 1
H e r m a n n S chm id u n d G ü n t h e r B a u er
Institut für Physikalische Chemie der Technischen Hochschule Wien
(Z. Naturforsdig. 21 b, 1009— 1014 [1966] ; eingegangen am 21. Juli 1966)
Von den Säure-Basen-Katalysen wurde zunächst die Mutarotation der Glucose einem eingehenden kinetischen Studium unter Anwendung der Ausgleichs- und Fehlerfortpflanzungs-Rechnung unterzogen. Die E y r i n g sehen Aktivierungsgrößen wurden für die Wasserkatalyse, die Ammoniakkatalyse, für eine Anzahl von Anionkatalysen und für die Wasserstoffionkatalyse ermittelt. Es wird gezeigt, daß die Wasserstoffionkatalyse und die Anionkatalysen auf der Primärreaktion der a-Glucose mit Wasser beruhen, wobei die richtende Wirkung der Kraftfelder der Ionen auf die polaren Wassermoleküle eine entscheidende Rolle spielt. Außerdem wird gezeigt, daß die heterogene Kupferkatalyse der Glucose-Mutarotation ebenfalls eine Wasserkatalyse ist, wobei die an Kupfer ausgerichteten Wasserdipole mit der a-Glucose in Reaktion treten.
Um einen tieferen Einblick in den Mechanismus der Säure — Basen — Katalyse zu gewinnen, muß man die freie Aktivierungsenthalpie, die Aktivierungsenthalpie und die Aktivierungsentropie möglichst genau kennen. Wir haben daher das Studium der Säure — Basen — Katalyse in der Weise in Angriff genommen, daß wir die Katalysekoeffizienten und die E y r i n g sehen Aktivierungsgrößen von Säure — Basen — Katalysen mit Hilfe der Ausgleidis- und Fehlerfortpflanzungs-Rechnung ermittelten. Die erste Reaktion, die wir einem derartigen kinetischen Studium unterzogen, ist die Mutarotation der Glucose, die seit B rön sted und L ow ry als klassisches Beispiel für die allgemeine Säure — Basen — Katalyse gilt.
OH OH OH H
H ch 2 o h h ch2 o h
Abb. 1. Mutarotation der Glucose.
Die landläufige Anschauung über den Medianismus der Glucose-Mutarotation2 ist bisher, daß sich das Proton der Säure an den Brückensauerstoff anlagert, während das Proton von der Hydroxylgruppe des dem Brückensauerstoff benachbarten Kohlenstoffatoms an die Base abgegeben wird, wobei der „offene“ Aldehyd gebildet wird. Neuerliche Ringschließung soll dann zur stereoisomeren Glucose führen.
1 Originalvortrag von H e r m a n n S c h m id am 29. Juni 1965 im Fritz-Haber-Institut der Max-Planck-Gesellschaft in Berlin- Dahlem.
Abb. 2. Bisherige Anschauung über den Mechanismus der Glucose-Mutarotation 3.
Die Versuche, auf die sich diese Anschauung gründet, sind aber zu ungenau. Es mußten daher die Messungen der Glucose Mutarotation neuerlich durchgeführt werden. Wir verfolgten den Fortschritt der Reaktion polarimetrisch.
Die Geschwindigkeitsgleidiung
- a) [ H Ga] — k z [H Gß] (1)
kann in die Form
- = ( i , + y ( [ « C . ] - [ H C . ] * ) (2)
gebracht werden, wenn [ H G a]* die Gleichgewichtskonzentration der a-Glucose ist. Integration von Gl. (2) und Einführung der Drehwinkel anstelle der Konzentrationen ergibt:
log (y - y * ) = - k l 2 ^ 2 t + log (y° - y * ) . (3)
7 ist der variable Drehwinkel der Glucose-Lösung, y* der Gleidigewichtsdrehwinkel und /° der Anfangsdrehwinkel .
2 Siehe H e r m a n n S c h m id , Handbuch der Katalyse (Herausgeber G. M. S c h w a b ) VII, S. 41, Springer-Verlag, Wien 1943.
3 Wobei hier Wasser als Säure reagiert.

1010 H. SCHMID UND G. BAUER
Wir berechnen nun die Summe der Geschwindigkeitskoeffizienten der Hin- und Riickreaktion (A + k2) aus einer Reihe von Versuchsdaten: log (ft — y*) und t\ mit Hilfe der Ausgleichsrechnung, der folgende Überlegung zugrunde liegt:
Der Absolutfehler bei der Messung eines Drehwinkels ist immer gleich groß — unabhängig von der Größe des Drehwinkels — und durch das verwendete Polarimeter bedingt. Da sich mit fortschreitender Versuchsdauer die Drehwinkeldifferenz ft — y* verringert, werden die Werte von ft — y* ungenauer. Es sind daher die Werte von log (ft — 7*) mit „Gewichten“ zu versehen, die mit abnehmendem log (ft — y*) und mit zunehmendem Fehler immer kleiner gemacht werden.
Die Gl. (3) stellt die Gleichung einer Geraden dar:
y = - a t + b . (4)
Für den Fehler der j-ten Messung erhält man:
fi = Yi + a t i - b . (5)
Multiplikation mit einem entsprechenden Gewicht gi ergibt:
figi = yigi + a t i g i - b gi . (6 )
Die Summe der Quadrate der mit den entsprechenden Gewichten multiplizierten Fehler soll ein Minimum sein. Es gilt daher:
3 ^ ( / i 8 i) 2
d b3^(/igi) 2 _ n
3a = 0 . (7)
Bezeichnet man den Absolutfehler der Drehwinkel- differenz ft — y * mit Ay, so findet man für den Absolutfehler von log (ft — y * ) näherungsweise A y l
(ft — 7*). Je kleiner dieser Fehler ist, um so größer soll das Gewicht von \ o g (y \ — y * ) sein. Es ist daher zweckmäßig, y-x — y * als Gewicht einzuführen:
■y\-y (8)
k1 ergibt sich aus (A:1 + ^2) zu:( k i + k 2) K
* 1 - K + 1 (9)
wenn K die Gleichgewichtskonstante der Glucose- Mutarotation ist.
Der Geschwindigkeitsterm für die Wasserkatalyse der Mutarotation der a-Glucose lautet:
d (H G a) dt = kyv[H20 ] [H Ga) (10)
wobei die Gin.
kyv[H20 ] —ki bzw. k \\ —(k t + k 2) K
[H 2 0 ] ( K + l ) [HoO] (11)
Der Geschwindigkeitskoeffizient der Hinreaktion
gelten. Der Katalysekoeffizient k \\ ergibt sich also unmittelbar aus dem experimentell gefundenen Wert (k1 + k2), der mit Hilfe der Ausgleichsrechnung ermittelt wurde. Die für eine Anzahl von Parallelversuchen gefundenen &w-Werte wurden gemittelt. Ist (JA:w)i die Abweichung des Einzelwertes (&w) i vom Mittelwert, so ist die Standardabweichung o
1 / 2 (A fcw) j2\ n {n — 1) ’ (12)
wenn n die Anzahl der Versuche ist.Enthält das Lösungsmittel Wasser einen weiteren
Katalysator, beispielsweise Wasserstoffion, so ist in verdünnter Lösung
kl - k w lH i O ] = k a °[H ® ]. (13)
(k1 — k \y[H 20 ] ) in Abhängigkeit von der Wasser- stoffionkonzentration stellt daher eine durch den Ursprung des Koordinatensystems gehende Gerade dar, deren Steigung der gesuchte Katalysekoeffizient — in diesem Falle kh® — ist. Die Ausgleichsrechnung ergibt:
/c h ® = (14)
Das Fehlerintervall für k^ : ± wird mit Hilfe der Fehlerfortpflanzungs-Rechnung erhalten. Der Fehler eines Ergebnisses E, das aus einer Reihe von Meßwerten X\ mit den dazugehörigen Fehlern (/x), durch beliebige Rechenoperationen gewonnen wurde, ergibt sich aus der Gleichung:
h - ] / ^ ( W i ( ! f f . (15)
In unserem Fall ist für das Ergebnis E der Katalysekoeffizient k n ® und für X[ = ( k t ) , — k w [ H 20 ] zu setzen.
Aus der E y r i n g sehen Gleichung für den Katalysekoeffizienten :
&H® =k T
expA Ga©
R T (16)
erechnet sich die freie Aktivierungsenthalpie AG* der Wasserstoffionkatalyse. Der Fehler der freien Aktivierungsenthalpie ist nach der eben besprochenen F ehlerfortpflanzungs-Rechnung
R. Tf j gh® = / kfl© • ( I 7)

NEUE ERGEBNISSE DER SÄURE-BASEN-KATALYSE 1011
Die Aktivierungsentropie ist nach der thermodynamischen Beziehung
/ 3 A G * \J S * = - ( 3T U <18)
und die Aktivierungsenthalpie nachAH* = AG* + T AS* (19)
berechenbar. Die Fehlerfortpflanzungs-Rechnung ergibt für den Fehler der Aktivierungsentropie:
h S* = T~— T \ V f^ G i+ fA G i (20)
und für den Fehler der Aktivierungsenthalpie:
V Y * Z t 21 2 2_ • (21)
In Tab. 1 sind die für verschiedene Katalysatoren nach der besprochenen Rechenmethode gefundenen Aktivierungsgrößen mit den Fehlerintervallen angeführt.
Die Aktivierungsenthalpie der Elementarreaktion zwischen a-Glucose und Wasser:
h 2oH Ga+ H.,0- akt. Kompl. (Ge. .. H20 .. . H30®)
(22)
ist viel höher als die zwischen a-Glucose und Ammoniak:
h 2oH G„ + NH3^= ^ akt. Kompl. (Ge ... H.,0 ... NH4®)
(23)(G° ist das Zeichen für das aktivierte Glucosation) entsprechend der schwächeren Basizität des Wassers.
Auch die Aktivierungsenthalpie der a-Glucose- Wasser-Reaktion reicht bei weitem nicht aus, den Ring der a-Glucose zu sprengen. Diese Schlußfolgerung läßt sich im Zusammenhalt mit den Ergebnissen von R i t t e n b e r g und G r a f f 5 über die Aius- tauschreaktion des Aldehydsauerstoffs 180 der Glucose mit dem Sauerstoff des Wassers ziehen. Sie finden für diese Reaktion eine Aktivierungsenergie
von 23,4 kcal Mol-1 und nehmen an, daß der Austausch zufolge der freien Aldehydform der Glucose erfolgt. Es ist daher die landläufige Anschauung über den Mechanismus der Glucose-Mutarotation, wonach zwischenzeitlich der „offene“ Aldehyd gebildet werden soll, unzutreffend. Vielmehr muß das aktivierte a-Glucosation ohne zwischenzeitliche Bildung des freien Aldehyds in das yS-Glucosation umklappen.
Die negative Aktivierungsentropie bei der a-Glu- cose-Wasser-Reaktion ist darauf zurückzuführen, daß der aktivierte Komplex in das aktivierte Glucosation und das protonierte Katalysatormolekül ionisiert und das elektrische Kraftfeld zwischen diesen Ionen den im aktivierten Komplex enthaltenen Lösungsmitteldipol in Richtung der Kraftlinien auszurichten trachtet.
Die Aktivierungsentropie der Elementarreaktion der a-Glucose mit Wasser ist negativer als die der aGlucose mit Ammoniak. Offenbar wird der im aktivierten Komplex enthaltene Lösungsmitteldipol von dem bei der Aktivierung der a-Glucose-Wasser- Reaktion entstandenen Hydroniumion, das eine stärkere Säure als Ammoniumion ist, stärker in Richtung der Kraftlinien ausgerichtet als von dem bei der Aktivierung der a-Glucose-Ammoniak-Reak- tion entstandenen Ammoniumion. Während sich die Aktivierungsenthalpie der Reaktion der a-Glucose mit der Base Ammoniak von der der a-Glucose-Was- ser-Reaktion um nahezu 4 kcal Mol-1 unterscheidet, entsprechen die Aktivierungsenthalpien der a-Glu- cose-Reaktion mit den Anionbasen, die zwischen 17 und 18 kcal Mol-1 liegen, nahezu der Aktivierungsenthalpie der a-Glucose-Wasser-Reaktion. Die Aktivierungsenthalpien für das Formiation und für das Glucosation stimmen mit der für Wasser besonders gut überein.
K atalysator5,0 °C
A G * (cal Mol“1)
15,0 °C 25,0 °C4A H *
(calMol-1)Zl£*
([Cl]Mol-i)
x h 3HoOh 3o ©HCOOeCHgCOO©C6H i i 0 6©OHe
21083 4- 8 17428 ± 47 15003 ± 16
18416 ± 10 24410 ± 3 20313 ± 5 21459 ± 10 21193 ± 10 17456 ± 27 14930 dt 20
18592 ± 10 24659 ± 2 20425 ± 17 21611 ± 10 21290 ± 12 17435 ± 35 14767 ± 14
13345 ± 410 17238 ± 117 17230 ± 499 17079 ± 410 18206 ± 202 17339 ± 836 18300 ± 317
- 1 7 ,6 ± 1,4- 24,9 ± 0,4 -1 0 ,7 ± 1,7 - 1 5 ,2 ± 1,4- 10,4 ± 0,7- 0,4 4- 2,9 + 11,8 ± 1,1
Tab. 1. Aktivierungsgrößen.
4 Mit dem Katalysator H30® wurden die Versuche bei 5 D. R i t t e n b e r g u . C h. G r a f f , J. Amer. chem. Soc. 8 0 , 3370 25,5 °C durchgeführt. [1958].

1012 H. SCHMID UND G. BAUER
Temperatur[°C] [CH3C 009] / hGoc /a c —
— —
25,0 0 bis 0.3 1.0015,0 0,3 bis 1,0 0,945,0 1,0 bis 3,0 0.89
Tab. 2. Konzentrationsabhängigkeit der B r ö n s t e d sehen Quotienten der Aktivitätskoeffizienten bei der Acetationkata-
lyse.
Der höhere Wert der Aktivierungsenthalpie für die Acetationkatalyse läßt sich auf folgende Weise erklären:
Für die bei tieferen Versuchstemperaturen angewendeten höheren Acetationkonzentrationen kann der Quotient der Aktivitätskoeffizienten in der Geschwindigkeitsgleichung im Sinne B r ö n s te d s
d (H Ga) } h g « /a c "- * a , - ed* [H G a] [A c Q]
(24)nicht mehr 1 gesetzt werden. / sind die entsprechenden Aktivitätskoeffizienten, /K© ist der .Aiktivitäts- koeffizient des aktivierten Komplexes, der nach der BRÖNSTEDschen Beziehung wie das Acetation einfach geladen ist. Unter der Annahme, daß für die bei 25 °C angewendeten Konzentrationen des Acetations von 0,1 bis 0,3 Mol 1_1 der Quotient der Aktivitätskoeffizienten noch 1 gesetzt werden kann, ergibt sich unter Zugrundelegung der Aktivierungsenthalpie von 17 200 cal Mol-1 für die bei 15 °C angewendeten Konzentrationen ein Quotient von 0,94 und für die bei 5 °C angewendeten Konzentrationen ein Quotient von 0,89; das sind also durchaus plausible Werte.
Die Anionen wurden durch Neutralisation der entsprechenden Säuren mit Natronlauge hergestellt. Die Glucosationkonzentration ergibt sich aus der analytischen Konzentration der Natronlauge und der wirklichen Konzentration der Hydroxidionen. Die Hy- droxidionen-Konzentration wurde dabei mit dem PH-Meßgerät bestimmt. Die Genauigkeit des pn* Meßgerätes ist mit i 0 ,0 2 pH angegeben. Ein ebenso großes Fehlerintervall für die Eichung mit Hilfe einer Pufferlösung würde eine Abweichung in der Aktivierungsenthalpie von + 1663 cal ergeben. Der in Tab. 1 angegebene Wert der Aktivierungsenthalpie für das Hydroxidion fällt daher nicht so sehr ins Gewicht wie die übrigen Werte der Aktivierungsenthalpie.
Man wird nicht fehlgehen, die Analogie der Aktivierungsenthalpie für die Anionen mit der Aktivie
rungsenthalpie für Wasser in der Weise zu erklären, daß die Hydrathülle der Anionen mit der a-Glucose in gleicher Weise in Reaktion tritt wie das Wasser. Der Unterschied liegt aber darin, daß bei der Anion- katalyse ein durch das Anion gerichtetes Wassermolekül der Hydrathülle mit der a-Glucose in Reaktion tritt. Die Aktivierungsentropie steigt um so mehr, je stärker das dabei verschwindende Wassermolekül gerichtet war. Dieser Richtungseffekt geht mit der Basizität des Anions symbat.
Die Aktivierungsentropie steigt von —24,9 [CI] Mol-1 bei reinem Wasser bis + 11,8 [CI] Mol-1 bei der Hydroxidionkatalyse. Bei Glucosation als Katalysator ist die Aktivierungsentropie entsprechend der Gleichung:
H Ga + Gae « akt. Kompl. (Ge. .. H20 .. . H Ga) (25)
Null. Der etwaige Entropieunterschied zwischen dem aktivierten und nicht aktivierten a-Glucoseation ist zu gering, um mit Sicherheit erfaßt werden zu können. Die Anionkatalyse ist nach dem Gesagten also entropisch.
Der Unterschied in den Katalysekoeffizienten (s. Tab. 3) ist entsprechend der E y r i n g sehen Gleichung:
k T ( AH* \ / ZJS* \ _ k r = h e x p ^ - R T R ) (26)
/|5*in erster Linie auf den Entropiefaktor exp ^ zu
rückzuführen. Das Verhältnis der Katalysekoeffizienten für Hydroxidion und für Wasser hat wegen des ausschlaggebenden Unterschiedes in den Aktivierungsentropien den hohen Wert von mehr als 7 Zehnerpotenzen.
Besonders bemerkenswert ist unser Befund, daß die Aktivierungsenthalpie der Wasserkatalyse der Mutarotation der a-Glucose identisch ist mit der Aktivierungsenthalpie der Wasserstoffionkatalyse der Mutarotation der a-Glucose:
AH h + = A H \\ (27)
Die Aiktivierungsentropie der Wasserstoffionkatalyse AS h+ ist hingegen weniger negativ als die der Wasserkatalyse ZlSw:
zISw AS h+ 0 . (28)
Da die Aktivierungsenthalpien der Wasserkatalyse und der Wasserstoffionkatalyse gleich sind, die Ak-
6 k r = Katalysekoeffizient, k = B o l t z m a n n sdie Konstante, li = P l a n c k sches Wirkungsquantum.

NEUE ERGEBNISSE DER SÄURE-BASEN-KATALYSE 1013
K atalysatorKatalysekoeffizienten [sec-1]
5,0 °C 15,0 °C 25,0 °C 4
N H 3 _ 0,0626 ± 0,0011 0,142 ± 0,003h 2o — (1.72 ± 0,01) IO“6 (4,90 ± 0 ,0 2 ) IO"6h 3o $ — (2,23 ± 0,02) IO“3 (6,67 ± 0,19) IO-3HCOOe — (3,06 ± 0,06) IO"4 (8,63 ± 0,15) IO- 4CHgCOO© (1,52 ± 0 ,0 2 ) 10~4 (4,88 ± 0,09) IO-4 (1,48 ± 0,03) 10-3C6H n 0 6e 0,114 ± 0,010 0,336 ± 0,016 1,00 ± 0,06OH0 9,18 ± 0,29 27,8 ± 1,0 90,7 ± 2.2
Tab. 3. Katalysekoeffizienten.
tivierungsentropie der Wasserstoffionkatalyse aber weniger negativ als die der Wasserkatalyse ist, wird die Schlußfolgerung 7 gezogen, daß die Wasserstoffionkatalyse eine Wasserkatalyse ist, bei der der in den aktivierten Komplex eintretende Lösungsmitteldipol durch das als Katalysator zugefügte Wasserstoffion bereits vorgerichtet ist. Die Wasserstoffionkatalyse kommt also nicht durch Anlagerung des Wasserstoff ions an den Brückensauerstoff der Glucose sondern durch die richtende Wirkung des elektrischen Kraftfeldes des Wasserstoff ions auf den Lösungsmitteldipol zustande.
Die Deutung der Wasserstoffionkatalyse der Glu- cose-Mutarotation als Wasserkatalyse muß zu dem Befund führen, daß die Geschwindigkeit der Wasserstoffionkatalyse proportional der Wasserstoff ionkon- zentration ist. Der Geschwindigkeitsterm der Wasserkatalyse der Mutarotation der a-Glucose [Gl. (1 0)] ist unter Verwendung der Gl. (26) :
d (H G a) \ _ k T d£ / W ~~ h eXP
[ H 20] [H Ga]
A Hy ~R T
exp (Ir)(29)
Durch Zugabe von Wasserstoffionen zu Wasser wird die Aktivierungsentropie des Wassers um AS h vergrößert, während die Aktivierungsenthalpie AHw unverändert bleibt:
AH'w R T
d ( H G a) \ k T df / H+ h CXP
exp R [ H20 ] [H Ga].(30)
Setzt man in Gl. (30) [H 20 ] = [ H Ga] =1, so erhält man für die Reaktionsgeschwindigkeit:
d (H G J 'd f /H < - Jew exp
[_as ; \ R
(31)
Madien wir für AS j* einen Ansatz in Analogie zur Entropiegleichung von B o l t z m a n n :
A S h = A S o + R \ n [ H ® ] , (32)
(33)
wobei für zlSo die Beziehung:
A S o = z l S h + — A S w
gilt, so erhalten wir:
_ _ i we x p (^ |!L)[H®] (34)
und mit
£w exp ( ) = k n ®: (35)
=Ah®[H®]- ( [HGJ-1) (36)
Die Wasserstoffionkatalyse und die Anionkatalyse der Glucose-Mutarotation in wäßrigen Lösungen beruhen also auf der Primärreaktion der a-Glucose mit Wasser, wobei die richtende Wirkung der Kraftfelder der Ionen auf die polaren Wassermoleküle eine entscheidende Rolle spielt.
Die richtende Wirkung von Katalysatoren auf polare Stoffe spielt auch in der heterogenen Katalyse eine große Rolle. So wird der Befund, daß die Geschwindigkeiten der Dehydrierung primärer Alkohole an Kupfer bei gegebener Temperatur alle gleich sind, in der Weise erklärt, daß die CH2OH-Gruppe der am Kupfer adsorbierten Alkoholmoleküle der Kupferoberfläche zugekehrt ist, die Kohlenwasserstoffketten hingegen von der Kupferoberfläche abgewendet sind 8. Es war daher zu erwarten, daß Kupfermetall auch die Wassermoleküle ausrichtet und sich als Katalysator für die Mutarotation der Glucose erweist.
Wir haben den Effekt des Kupfers auf die Mutarotation der a-Glucose vor kurzem untersucht. Die Versuche wurden in folgender Weise durchgeführt: Der Fortschritt der homogenen Mutarotation der a-Glucose bei der Versuchstemperatur wird in einem Teil der wäßrigen Lösung an der Änderung des Drehwinkels der Glucoselösung von der Zeit t = 0
7 H e r m a n n S c h m id , Mh. Chem. 9 5 , 454, 1009 [1964], 8 F. H. C o n s t a b l e , Proc. Roy. Soc. [London] Ser. A 108, 355.
1014 NEUE ERGEBNISSE DER SÄURE-BASEN-KATALYSE
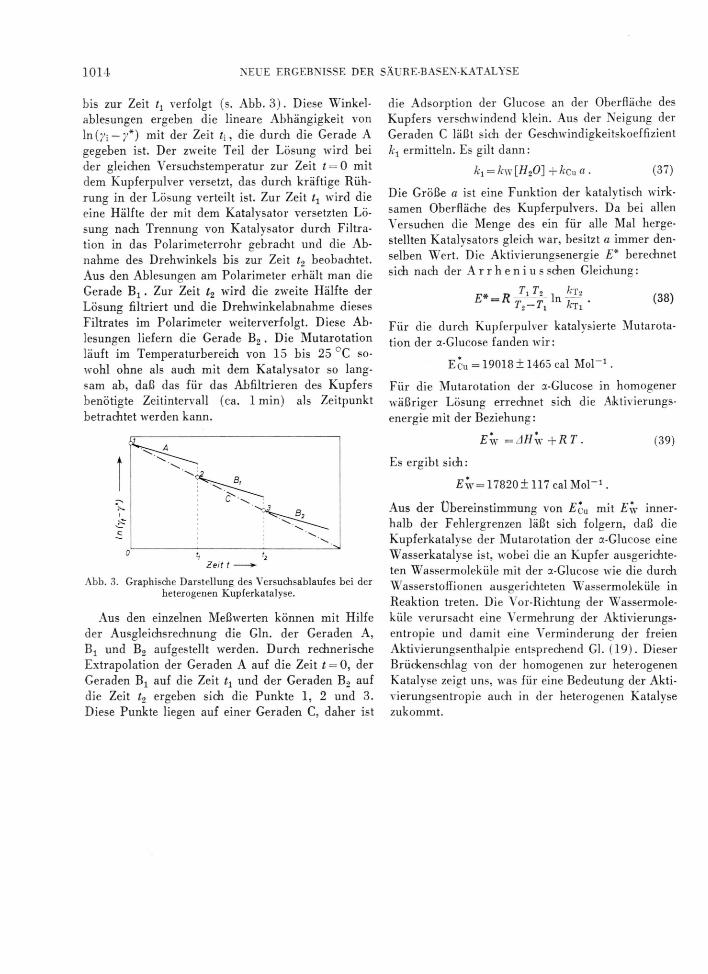
bis zur Zeit tx verfolgt (s. Abb. 3). Diese Winkelablesungen ergeben die lineare Abhängigkeit von ln (fr ~ 7*) mit der Zeit t[ , die durch die Gerade A gegeben ist. Der zweite Teil der Lösung wird bei der gleichen Versuchstemperatur zur Zeit / = 0 mit dem Kupferpulver versetzt, das durch kräftige Rührung in der Lösung verteilt ist. Zur Zeit tx wird die eine Hälfte der mit dem Katalysator versetzten Lösung nach Trennung von Katalysator durch Filtration in das Polarimeterrohr gebracht und die Abnahme des Drehwinkels bis zur Zeit U beobachtet. Aus den Ablesungen am Polarimeter erhält man die Gerade Bj. Zur Zeit t2 wird die zweite Hälfte der Lösung filtriert und die Drehwinkelabnahme dieses Filtrates im Polarimeter weiterverfolgt. Diese Ablesungen liefern die Gerade B2 . Die Mutarotation läuft im Temperaturbereich von 15 bis 25 °C sowohl ohne als auch mit dem Katalysator so langsam ab, daß das für das Abfiltrieren des Kupfers benötigte Zeitintervall (ca. 1 min) als Zeitpunkt betrachtet wTerden kann.
Abb. 3. Graphische Darstellung des Versuchsablaufes bei der heterogenen Kupferkatalyse.
Aus den einzelnen Meßwerten können mit Hilfe der Ausgleichsrechnung die Gin. der Geraden A, Bj und B2 aufgestellt werden. Durch rechnerische Extrapolation der Geraden A auf die Zeit t = 0, der Geraden Bj auf die Zeit tt und der Geraden B2 auf die Zeit Z2 ergeben sich die Punkte 1, 2 und 3. Diese Punkte liegen auf einer Geraden C, daher ist
die Adsorption der Glucose an der Oberfläche des Kupfers verschwindend klein. Aus der Neigung der Geraden C läßt sich der Geschwindigkeitskoeffizient k1 ermitteln. Es gilt dann:
kt = k \\ \H20] + kcu a . (37)
Die Größe a ist eine Funktion der katalytisch wirksamen Oberfläche des Kupferpulvers. Da bei allen Versuchen die Menge des ein für alle Mal hergestellten Katalysators gleich war, besitzt a immer denselben Wert. Die Aktivierungsenergie E* berechnet sich nach der Arrhenius sehen Gleichung:
o s )
Für die durch Kupferpulver katalysierte Mutarotation der a-Glucose fanden wir:
Ecu = 19018 ± 1465 cal Mol“1.
Für die Mutarotation der a-Glucose in homogener wäßriger Lösung errechnet sich die Aktivierungsenergie mit der Beziehung:
£w — + R T . (39)
Es ergibt sich:
Ew = 17820 ± 117 cal Mol'1.
Aus der Übereinstimmung von Ecu mit £w innerhalb der Fehlergrenzen läßt sich folgern, daß die Kupferkatalyse der Mutarotation der a-Glucose eine Wasserkatalyse ist, wobei die an Kupfer ausgerichteten Wassermoleküle mit der a-Glucose wie die durch Wasserstoffionen ausgerichteten Wassermoleküle in Reaktion treten. Die Vor-Richtung der Wassermoleküle verursacht eine Vermehrung der Aktivierungsentropie und damit eine Verminderung der freien Aktivierungsenthalpie entsprechend Gl. (19). Dieser Brückenschlag von der homogenen zur heterogenen Katalyse zeigt uns, was für eine Bedeutung der Aktivierungsentropie auch in der heterogenen Katalyse zukommt.









![Index [ ] · PDF file– Enol 1065 – Enolat 920 – Esterenolat 1009 – indirekte 1058 ... – 1H-NMR-Signale der Alkylgruppen 453 Außenelektron 8 Austrittvermögen 254](https://img.pdfslide.org/doc/110x75/5a9d80d57f8b9a21688bbf53/index-enol-1065-enolat-920-esterenolat-1009-indirekte-1058-.jpg)