Embed Size (px)
Citation preview

2 Symmetrie und Struktur
2.1 Ordnung in Festkörpern
2.1.1 Atomtheorie
Die griechischen Philosophen stellten als erste dieFrage, ob es möglich sei, einen bestimmten Körperbeliebig oft zu teilen. Demokrit von Abdera beant-wortete diese Frage als erster negativ, in dem er for-derte, dass alle Materie aus identischen Teilchen auf-gebaut sein sollte, den Atomen. Diese Ansicht wur-de dann von Aristoteles widersprochen, und erst im18 Jh. fanden die aufblühenden Naturwissenschaf-ten wieder Hinweise darauf, dass es doch solcheTeilchen geben sollte. Dafür sprachen insbesondereauch Beobachtungen der Kristallographen. Sie stell-ten fest, dass Kristalle, wenn sie wachsen oder wennsie gespalten werden, beinahe perfekte Oberflächenbilden, und dass zwischen verschiedenen solchenOberflächen nur ganz bestimmte Winkel auftreten.
100
110
Abbildung 2.1: NiO Kristall mit Wachstumsebenen.
Dieser Befund konnte relativ leicht erklärt werden,wenn man davon ausging, dass diese Kristalle auseiner Vielzahl von identischen Teilchen zusammen-gesetzt waren [2]. Abb. 2.1 zeigt als Beispiel einenNiO Kristall mit deutlichen Wachstumsebenen, so-wie ein Schema, wie man sich die Bildung solcherWachstumsebenen vorstellen kann.
Nicht nur beim Kristallwachstum erhält man Kri-stallflächen mit gleichen Winkeln, man findet auch,dass bestimmte Flächen beim Spalten von Kristal-
Abbildung 2.2: Spaltebenen.
len bevorzugt auftreten. Die Idee, dass Kristalle ausatomaren Einheiten bestehen, wurde später durchunterschiedliche Methoden betätigt, v.a. natürlichdurch Beugungsexperimente (Friedrich, Knippingund Laue, 1912).
Strom
Elektronik
Abbildung 2.3: Prinzip der Raster-Tunnelmikroskopie und damitgemessene Ni-Atome.
Seit einigen Jahren ist es auch möglich, die atoma-re Struktur von Festkörpern auch direkt zu beobach-ten, z.B. mit Hilfe der Tunnelmikroskopie (STM).Abb. 2.3 zeigt das Funktionsprinzip, sowie das Bildeiner Nickeloberfläche, die mit STM gemessen wur-de. Heute gehen wir deshalb selbstverständlich da-von aus, dass Festkörper aus Atomen oder Molekü-len aufgebaut sind.
17

2 Symmetrie und Struktur
2.1.2 Langreichweitige Ordnung
Die Atome oder Moleküle können auf unterschiedli-che Weise im Festkörper angeordnet sein. Man kannsie insbesondere auf Grund des Grades an Ordnungauf unterschiedlichen Längenskalen klassifizieren.
kristallin polykristallin
Abbildung 2.4: Kristalline vs. polykristalline Ord-nung.
• kristallin: periodische, langreichweitige Ord-nung. Dieses Idealbild ist Ausgangspunkt dermeisten Theorien im Bereich der Festkörper-physik.
• polykristallin: Auf kurzen Längenskalen sinddiese Systeme kristallin. Der makroskopischeKörper umfasst jedoch viele einzelne Kristalle.
Abbildung 2.5: Lokale 5-zählige Symmetrie.
• quasikristallin: Quasikristalle weisen lang-reichweitige Ordnung auf, sind aber nicht pe-riodisch. Sie besitzen 5- oder 10-zählige Sym-metrie.
Abbildung 2.6: Amorphe Materialien: Nahordnung,aber keine Fernordnung.
• amorph: In amorphen Materialien ist die direkteUmgebung eines Atoms oder Moleküls relativgut (aber nicht perfekt) definiert.
Auf einer Skala von typischerweise einigen Nano-metern nimmt der Grad der Ordnung ab und auf ei-ner Skala von mehr als 10 Nanometern sind amorpheMaterialien homogen und isotrop. Zu den amorphenMaterialien gehören v.a. Gläser und Polymere, dar-unter auch viele biologische Materialien. Viele Ei-genschaften von amorphen Materialien hängen starkvon ihrer Herstellung ab. So kann man Gläser als“unterkühlte Flüssigkeiten, welche zu kalt sind zumeinfrieren” betrachten: ihre Viskosität ist zu hoch alsdass sie in den energetisch tiefer liegenden kristal-linen Zustand übergehen könnten. Diese Abhängig-keit von der Herstellung ist ein wichtiger Grund da-für, dass z.B. die Herstellung von Gläsern lange Zeitmehr eine Kunst als eine Wissenschaft war.
2.1.3 Flüssigkristalle
Abbildung 2.7: Flüssigkristalle und Flüssigkristall-Polymere.
• flüssigkristallin: Flüssigkristalline Materialienzeigen langreichweitige Ordnung, wobei z.B.nur die Orientierung der Moleküle diese Ord-nung zeigen kann.
Auch bezüglich Fernordnung sind Flüssigkristallezwischen Flüssigkeiten und Kristallen angesiedelt.Viele besitzen keine Positions-Fernordnung, ande-re eine teilweise, wie in Abb.\,2.8 gezeigt. In ku-bischen Phasen kann auch in 3 Dimensionen eine
18

2 Symmetrie und Struktur
eindimensionale Positionsfernordnung:
zweidimensionale Positionsfernordnung:
smektisch lamellar
hexagonal Condis
Abbildung 2.8: Partielle Positionsfernordnung inFlüssigkristallen.
partielle Fernordnung auftreten. Sie besitzen jedochim Gegensatz zu Festkörpern keine Formbeständig-keit, d.h. ihr Schermodul verschwindet. Flüssigkri-stalle haben inzwischen in verschiedenen Bereicheneine wichtige Rolle erhalten, nicht nur in Anzeigen,sondern auch in Polymeren.
Abbildung 2.9: Flüssigkristalline Ordnung einerbiologischen Membran.
Flüssigkristalle spielen auch in der Biologie einewichtige Rolle: Membranen von Zellen sind flüs-sigkristallin, d.h. die Moleküle sind im Mittel allegleich ausgerichtet und befinden sich in einer Ebene.Diese Ebene ist jedoch leicht verformbar, da die Mo-leküle in der Ebene frei beweglich sind. Diese Mem-branen werden primär aus fettsäureähnlichen Mo-lekülen gebildet, ähnlich wie Seifenschaum. Darineingelagert “schwimmen” eingelagert Proteine.
Die Physik hat sich vor allem mit der Untersuchungperfekter Kristalle beschäftigt, wobei Defekte undVerunreinigungen als Störungen betrachtet wurden.Dieses Vorgehen hat enorme Erfolge gebracht undz.B. die Grundlagen für die Halbleiterindustrie ge-
legt. In den 80er und 90er Jahren des 20. Jahrhun-derts haben dann einige Physiker auch entdeckt, dassdie Physik auch zur Untersuchung von amorphenSystemen einiges beitragen kann.
Abbildung 2.10: Pierre Gilles de Gennes.
Ein wichtiger Schritt war hier die Verleihung desNobelpreises 1991 an Pierre Gilles de Gennes. DieUntersuchung von Materialien ohne langreichweiti-ge Ordnung dürfte in Zukunft eine zunehmend wich-tige Rolle spielen, da Polymere und Gläser (z.B. me-tallische Gläser, amorphes Silizium) auch industriellzunehmend wichtiger werden. In dieser Vorlesungwerden wir aber auf die detaillierte Diskussion sol-cher Systeme verzichten und uns auf Systeme mitTranslationssymmetrie beschränken. Der Grund da-für ist einerseits unser Curriculum, andererseits auchdie Tatsache, dass die Beschreibung von amorphenSystemen noch nicht so weit ist, dass sie sich füreinen Einführungskurs gut eignet.
2.1.4 Translationssymmetrie
Wie bereits erwähnt, betrachtet man in der Festkör-perphysik zunächst ideale Kristalle. Darunter stelltman sich einen unendlich ausgedehnten Körper mitperiodisch wiederholten Einheiten vor. Es soll hieraber klar gemacht werden, dass solche Körper in derNatur nicht existieren, und zwar aus 2 Gründen:
• Bei endlicher Temperatur ist ein System oh-ne Fehler, welches damit perfekt geordnet wäreund Entropie null hätte, thermodynamisch in-stabil.
• Ein idealer Kristall ist immer unendlich ausge-dehnt, da eine Oberfläche einen Bruch der Sym-
19

2 Symmetrie und Struktur
metrie bewirkt.
Diese Grundannahme bedeutet auch, dass Oberflä-cheneffekte (in dieser Näherung) nicht berücksich-tigt werden.
Abbildung 2.11: Kristallgitter.
Die Wiederholung der Grundeinheit erfolgt so, dassdie resultierende Anordnung Translationssymmetriezeigt. Das bedeutet, dass es möglich ist, diese Anord-nung um einen bestimmten Betrag zu verschieben,und dadurch das System in ein ununterscheidbaresSystem überzuführen. In Abb. 2.11 sind zwei sol-che Möglichkeiten dargestellt: Verschiebungen um~a1 und ~a2. Es gibt aber eine unendliche Zahl vonTranslationen ~T , welche diese Bedingung erfüllen.Es ist allerdings nicht nötig, diese Operationen ein-zeln aufzuzählen, man kann sie nach einer einfachenFormel zusammenfassen.
Abbildung 2.12: Basis-Translationsvektoren.
Man benötigt für jede Dimension einen Basis-Translationsvektor, welche wir als ~a1, ~a2 und ~a3 be-zeichnen. Eine allgemeine Translation ~T in drei Di-mensionen wird dann definiert als die Operation
~r0 =~r +u1~a1 +u2~a2 +u3~a3 =~r +~T ,
wobei die Indizes ui beliebige ganze Zahlen darstel-len. Diese Beziehung gilt für jeden Punkt des Kri-stalls, nicht nur für die Position der Atome. Die Ge-samtheit der Translationen ~T definiert das Raumgit-
ter oder Bravais1-Gitter. Ein idealer kristalliner Fest-körper ist dadurch definiert, dass diese TranslationenSymmetrieoperationen darstellen, dass sie also dieStruktur in sich selber überführen
quadratisch rechteckig hexagonal
Abbildung 2.13: Translationsgitter.
Je nach relativer Länge und Orientierung der erzeu-genden Translationsvektoren unterscheidet man ver-schiedene Arten von Translationsgittern. In zwei Di-mensionen kann man quadratisch (Vektoren senk-recht aufeinander, gleich lang), hexagonal (gleichlang, Winkel 60 oder 120 Grad), und rechteckig(senkrecht aufeinander unterscheiden.
Abbildung 2.14: Struktur von GaAs.
Die Tatsache, dass die meisten Festkörper, welcheaus wenigen Bauelementen zusammengesetzt sind,in periodischen Strukturen erstarren, lässt sich leichtals eine Konsequenz der Energieminimierung verste-hen: Wenn ein Atom, Ion oder Molekül in einer be-stimmten Umgebung die geringste Energie besitzt,so muss dies auch für alle anderen Atome, Ionenoder Moleküle der gleichen Art gelten. Die Nach-barschaft aller gleichartigen Atome sollte also diegleiche sein. Dies ist aber identisch mit der Aussa-ge, dass man die Nachbarschaft eines Atoms auf dieUmgebung eines anderen abbilden kann.
1Auguste Bravais (1811 - 1863)
20

2 Symmetrie und Struktur
2.1.5 Einheitszelle und Basis
Um eine Kristallstruktur zu definieren, braucht manoffensichtlich zusätzliche Information. Das Gittersagt, auf welche Art die Bausteine aneinander gefügtwerden müssen. Wir brauchen aber noch die Kennt-nis der Bausteine. Diese werden als Einheitszelle be-zeichnet, die darin enthaltenen Atome bilden die Ba-sis.
Die Position eines Atoms kann geschrieben werdenals
~r j = x j~a1 + y j~a2 + z j~a3,
mit j als Index des entsprechenden Atoms, ~ai einGittervektor und {x j,y j,z j} die Koordinaten desAtoms in der Einheitszelle. Üblicherweise wähltman diese im Bereich [0..1].
Abbildung 2.15: Gitter und Basis.
Wird die Basis jeweils um einen Translationsvektordes Gitters verschoben, so erhält man den gesamtenKristall. In Abb. 2.15 ist das für den zweidimensio-nalen Fall dargestellt. Jeder Translationsvektor desGitters schiebt alle Moleküle auf andere Moleküledes Kristalls, das Muster bleibt somit das gleiche.
Die Einheitszellen können auf beliebige Weise de-finiert werden, so lange sie unter den Translationendes Gitters den Kristall vollständig füllen. Eine naheliegende Möglichkeit zur Definition der Einheitszel-le ist deshalb die Menge aller Punkte, welche durch
~r = x1~a1 + x2~a2 + x3~a3 0 xi 1
bestimmt wird. Dies entspricht dem in Abb. 2.12gezeigten Parallel-Epiped. Das Volumen der Zelle
kann mit Hilfe der Vektoralgebra bestimmt werden:
V = |~a1 · (~a2 ⇥~a3)|.
2.1.6 Die Wigner-Seitz Konstruktion
WSZEZ
Abbildung 2.16: Wigner-Seitz Konstruktion (WSZ)der Einheitszelle.
Eine andere Methode zur Konstruktion einer Ein-heitszelle ist die von Wigner2 und Seitz3. Dazu ziehtman von einem Gitterpunkt Verbindungslinien zu al-len Nachbarn und fällt darauf die mittelhalbierendeEbene. Abb. 2.16 zeigt das zweidimensionale Ana-logon; in diesem Fall ist die Mittelhalbierende ei-ne Gerade. Die Kombination dieser Ebenen (Lini-en) begrenzt die Wigner-Seitz Zelle. Bei der Wigner-Seitz Zelle befindet sich der Gitterpunkt im Zentrumder Einheitszelle, im Gegensatz zur konventionel-len Wahl, wo die Punkte sich auf den Ecken befin-den. Die beiden Einheitszellen haben unterschiedli-che Form, aber das gleiche Volumen, resp. die glei-che Fläche.
Abbildung 2.17: Flächenfüllung mit der Wigner-Seitz Einheitszelle.
Auch mit der Wigner-Seitz Einheitszelle kann manjedoch den Raum füllen. Abb. 2.17 zeigt ein Beispielin zwei Dimensionen.
2Eugene Wigner (1902-1995)3Frederick Seitz (1911-2008)
21

2 Symmetrie und Struktur
Abbildung 2.18: Links: Wigner-Seitz Einheitszellein 3D; rechts: raumfüllende Anord-nung von WS-Zellen.
Ähnlich kann man das Wigner-Seitz Verfahren in 3Dimensionen anwenden. Man fällt hier jeweils diemittelhalbierende Ebene. In der linken Hälfte vonAbb. 2.18 wurde die Konstruktion auf ein raumzen-triertes Gitter angewendet. Das Zentrum der Ein-heitszelle ist im Zentrum eines Würfels, die nächstenNachbarn sitzen an den Ecken des Würfels. Auchdiese Einheitszelle füllt den gesamten Raum wennsie durch die Gitteroperationen verschoben wird. DieEinheitszelle enthält im allgemeinen mehrere Ato-me, auch bei primitiven Gittern. Einatomige Ein-heitszellen kommen nur bei Kristallen vor, welcheaus einer einzigen Atomsorte bestehen, und auchdann nur wenn sämtliche Atome durch Translatio-nen ineinander übergeführt werden können.
2.1.7 Punktsymmetrie-Operationen
Kristallgitter können nicht nur durch Translationenin sich selbst übergeführt werden, sondern auchdurch andere Symmetrieoperationen, insbesonde-re Drehungen und Spiegelungen. Ein wesentlicherUnterschied zwischen Punktsymmetrie-Operationenund Translationen ist, dass bei den Punktsymmetrienmindestens ein Punkt fest bleibt.
Wir betrachten zunächst den Effekt solcher Opera-tionen auf einzelne Elemente. Man unterscheidet diefolgenden Symmetrieelemente:
• Drehachsen Ci oder Ai.
• Inversion I oder i führt~r ! �~r über.
• Spiegelebene s : Invertiert die Komponentesenkrecht zur Ebene, z.B. (x,y,z) ! (x,y,�z)
• Drehinversionsachsen Si
C4
Abbildung 2.19: Transformation eines Objektsdurch eine 4-zählige Rotationsach-se.
Abb. 2.19 zeigt als Beispiel eine vierzählige Rotati-onsachse, welche die vier L-förmigen Objekte inein-ander überführt. Allgemein entspricht eine n-zähligeRotationsachse einer Symmetrieachse, welche Dre-hungen um ganzzahlige Vielfache von 2p/n bewirkt.Wie in den Übungen gezeigt wird, können als mög-liche Werte für n nur n = 1,2,3,4 und 6 auftreten.
linke Hand
Spiegelbild ~rechte Hand
Abbildung 2.20: Änderung der Händigkeit eines Ob-jekt bei Spiegelung.
Inversionszentrum und Spiegelebene ändern dieHändigkeit eines Objektes, sie führen z.B. eine lin-ke Hand in eine rechte Hand über. Kristalle mit in-trinsischer Händigkeit können somit keines dieserSymmetrieelemente enthalten. Ein Kristall, welcherMoleküle mit entgegengesetzter Händigkeit enthältkann hingegen Spiegelebenen enthalten, welche dieeine Form in die andere überführen.
Meistens treten die genannten Symmetrieelementenicht einzeln auf, sondern in Kombinationen. ImBeispiel von Abb. 2.19 existiert auch eine Spiege-lebene, welche senkrecht zur Rotationsachse liegtund durch die vier Elemente läuft. Wären die beidenSchenkel dieser Elemente gleich lang, so würden
22

2 Symmetrie und Struktur
ausserdem vier zweizählige Rotationsachsen existie-ren, welche in der Ebene liegen würden.
Es sind nicht beliebige Kombinationen von Symme-trieelementen möglich, da die Symmetrieelementeselber unter den Symmetrieoperationen der übrigenElemente auch erhalten bleiben müssen. So könneneinzelne Symmetrieachsen nur senkrecht zueinanderoder in einer Ebene liegen. Zwei Symmetrieebenenkönnen nur senkrecht zueinander stehen, aber dreiEbenen können einen Winkel von jeweils 60� unter-einander einschliessen. Ausserdem erzeugt die Kom-bination von zwei Elementen häufig ein drittes Ele-ment. So erzeugen zwei Symmetrieebenen, die senk-recht aufeinander stehen, eine zweizählige Drehach-se in ihrer Schnittgeraden.
2.1.8 Gruppen
Im mathematischen Sinn bildet die Menge der Sym-metrieoperationen, welche ein Objekt invariant lässt,eine Gruppe. Allgemein ist in der Mathematik ei-ne Gruppe G definiert als eine nicht leere MengeG = {Ai} von Objekten Ai und einer binären Ope-ration · zwischen den Objekten, welche folgende Ei-genschaften erfüllt:
• Das Resultat einer Operation Ai ·A j = Ak ist sel-ber ein Mitglied der Gruppe.
• Es existiert eine Einheit e mit der Eigenschafte ·Ai = Ai · e = Ai für alle Ai.
• Es existiert zu jedem Element ein inverses Ele-ment A�1
i mit Ai ·A�1i = A�1
i ·Ai = e.
Die verschiedenen Kombinationen von Symmetrie-elementen erfüllen diese Anforderungen. Sie werdenals Punktsymmetrie-Gruppen bezeichnet. Die ver-schiedenen Gruppen werden nach zwei verschiede-nen Systemen klassiert. Es existieren einerseits diesog. Schoenflies4-Symbole, andererseits die Klas-sifikation nach Hermann-Maugin, welche auch alsinternational bezeichnet wird. Für die Bezeichnun-gen nach Schoenflies verwendet man die folgendenSymbole:
4Arthur Moritz Schoenflies (1853 - 1928), deutscher Mathe-matiker
• Drehgruppen: Cn (n=2, 3, 4, 6) j-fache Rotati-onsachse. Die Drehgruppe Cn enthält die Ele-mente Cn = {e,Cn,C2
n , . . . ,Cn�1n }.
• Drehspiegelgruppen: Sn; wird durch eine Dreh-spiegelachse erzeugt. Diese entspricht einerDrehung um den Winkel 2p/n um die entspre-chende Achse, gefolgt von der Spiegelung aneiner Ebene, die senkrecht zur Drehachse steht.
• D j: Diedergruppen. Werden durch eine Ro-tationsachse Cn sowie n dazu senkrechte C2-Achsen erzeugt.
• T : Tetraedergruppen: vier 3-fache und drei 2-fache Rotationsachsen in einem Tetraeder.
• O: Ikosaedergruppen: 4 3-fache und 3 4-facheRotationsachsen in einem Oktaeder.
D3hC3
C2C2
C2
C4
C4
C4
C3C3C3
C3
+ 6 C2-Achsen
+ 9 Symmetrieebenen
Abbildung 2.21: Punktgruppe D3h (links) und Ok-taeder mit 3- und 4-zähligen Ro-tationsachsen (rechts). Nicht einge-zeichnet sind 6 C2-Achsen und 9Spiegelebenen.
Die Gruppen können neben den Rotationsachsenauch Spiegelebenen enthalten. Diese werden durchdie tiefgestellten Symbole h (für horizontal, d.h.senkrecht zu Cn), v (für vertikal, d.h. Cn liegt in derEbene) oder d (ebenfalls senkrecht, aber zwischenden horizontalen C2-Achsen) bezeichnet.
Daneben können auch Punktsymmetrien auftreten,also Inversion an einem Punkt. Befindet sich dieserPunkt im Ursprung, dann ist das Resultat der ent-sprechenden Operation ~r ! �~r. Inversionszentrenund Spiegelebenen invertieren die Händigkeit einesObjekts. Sie können also nur dann auftreten, wenndas Objekt keine Händigkeit aufweist.
23

2 Symmetrie und Struktur
2.1.9 Hermann-Maugin Notation
2 2m = mm
1 1m
Abbildung 2.22: Beispiele für Punktgruppen inHermann-Maugin Notation.
Abb. 2.22 zeigt für einige einfache Kombinationenvon Symmetrieelementen die entsprechenden Sym-bole nach Hermann-Maugin. Im letzten Beispiel sind2 Möglichkeiten gezeigt, wie die Symbole darge-stellt werden: Eine 2-zählige Drehachse senkrechtzu einer Spiegelebene erzeugt eine zweite Spiege-lebene. Umgekehrt ist die Schnittgerade von 2 senk-recht aufeinander stehenden Spiegelebenen auch ei-ne Drehachse.
aa
Abbildung 2.23: Schraubenachsen und Gleitspiege-lebenen.
Im Hermann-Maugin System verwendet man ger-ne weitere Symmetrieelemente, welche als Kom-bination von Translationen mit Punktsymmetrie-Operationen verstanden werden können. Abb. 2.23zeigt die Schraubenachse und die Gleitspiegelebene.Eine Schraubenachse erzeugt eine Translation umeinen Vektor ~a, der nicht zum Bravaisgitter gehört,gefolgt von einer Drehung um diesen Translations-vektor. Eine Gleitspiegelebene erzeugt ebenfalls ei-ne Translation um einen Vektor~a, der nicht zum Bra-vaisgitter gehört, gefolgt von einer Spiegelung an ei-
ner Ebene, die den Vektor enthält. Beide müssen so-mit durch Betrag und Richtung von~a, sowie um denDrehwinkel, respektive die Richtung der Ebene defi-niert werden.
2.2 Symmetrie und Gitter
2.2.1 Primitive und nichtprimitive Gitter
Die Menge der Translationsvektoren ergibt das Git-ter. Da sie die Symmetrieoperationen zusammenfas-sen, sind Kristallgitter ein wichtiges Hilfsmittel zurCharakterisierung von Kristallen. Das bedeutet abernicht, dass ein gegebener Kristall eindeutig zu einembestimmten Gitter zugeordnet werden kann. Häufiggibt es verschiedene Möglichkeiten, ein Gitter zuspezifizieren. Eine gegebene Anordnung von Ato-men oder Molekülen kann auf unterschiedliche Wei-se in eine Einheitszelle und ein Gitter zerlegt wer-den.
a1
a2
primitiv 2
a1
a2
nicht primitiv
a1a2
primitiv 1
Abbildung 2.24: Unterschiedliche Wahl der Ele-mentarzelle in einem hexagonalenGitter in 2 Dimensionen.
Abb. 2.24 zeigt eine zweidimensionale Anordnungvon Atomen, die in der Natur relativ häufig vor-kommt. Offensichtlich gibt es mehrere verschiedeneMöglichkeiten, die Gittervektoren~a1 und~a2 zu defi-nieren. Die ersten beiden Möglichkeiten sind hierbeigleichwertig; die Einheitszellen sind gleich groß undenthalten die gleiche Anzahl an Atomen. Die drit-te hingegen unterscheidet sich dadurch, dass es mitden hierdurch definierten Translationen nicht mög-lich ist, die dunklen Atome auf die Positionen der
24

2 Symmetrie und Struktur
hellen zu bringen. Dementsprechend enthält die drit-te Elementarzelle zwei Atome, während bei den er-sten beiden Varianten die Elementarzelle jeweils nurein Atom enthält. Man bezeichnet die ersten beidenGitter als primitiv, das dritte als nicht primitiv.
Bei der Ermittlung der Anzahl Atome pro Ele-mentarzelle muss berücksichtigt werden, dass dieAtome am Rand der Zelle zu mehreren Zellen beitra-gen, aber nur einmal gerechnet werden dürfen. Manhat die Wahl, entweder die Elementarzelle leicht zuverschieben, so dass alle Atome nur in einer Zelleliegen, oder man zählt bei einem Atom, welches zu nZellen beiträgt, jeweils nur 1/n. Offenbar entsprichtbei einem Atom in der Seitenfläche n = 2, auf einerKante n = 3 oder 4, und auf der Ecke eines Würfelsn = 8.
2.2.2 Punktsymmetrieklassen
Abbildung 2.25: Versuche, mit 5- oder 7-eckigenElementen die Ebene abzudecken.
Die Symmetrie eines Kristalls ergibt sich nun durchdie Kombination der Punktsymmetriegruppen, ange-wendet auf die Einheitszelle, mit der Translations-gruppe des Gitters. Nicht alle möglichen Punktsym-metriegruppen sind aber mit periodischen Gitternverträglich. Insbesondere sind 5- 7- oder 10-zähligeRotationsachsen nicht verträglich mit Translations-symmetrie. Abb. 2.25 illustrieret dies anhand einesVersuches, die Ebene mit 5- oder 7-eckigen Elemen-ten abzudecken.
Man kann einige Bedingungen definieren, welcheSymmetrielemente in einem Kristallgitter auftretenkönnen:
• Eine Einheitszelle als einfachste sich wiederho-lende Einheit in einem Kristall.
• Gegenüberliegende Flächen einer Einheitszellesind parallel.
• Der Rand der Einheitszelle verbindet jeweilsäquivalente Stellen.
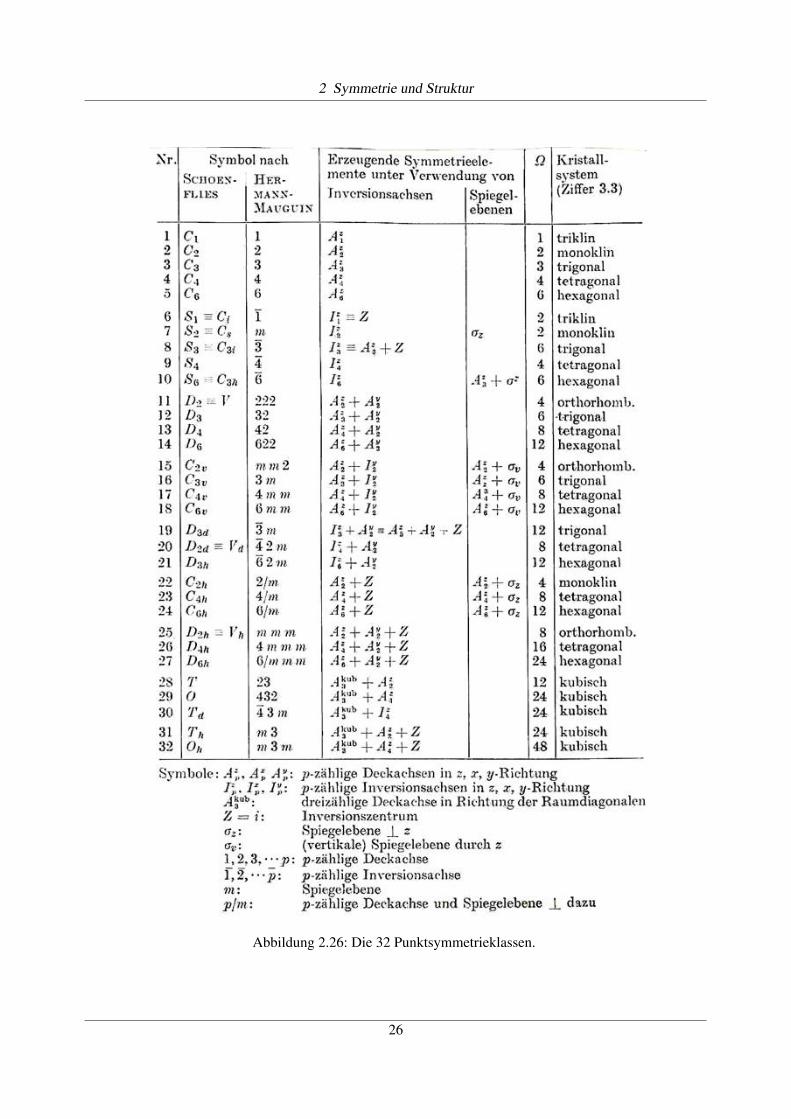
Insgesamt gibt es 32 Punktsymmetrieklassen, dieauch in periodischen Systemen vorkommen können.Diese enthalten Spiegelebenen, sowie Rotationsach-sen mit 2-, 3-, 4- und 6-zähliger Symmetrie.
Die Tabelle in Abb. 2.26 fasst alle 32 Punktsymme-triegruppen zusammen, welche mit Translationsgit-tern kompatibel sind. Die Bezeichnungen sind nachSchoenflies und nach Hermann-Maugin angegeben.Jede dieser Punktsymmetriegruppen kann durch ei-nes oder mehrere Symmetrieelemente erzeugt wer-den, wobei teilweise unterschiedliche Möglichkeitenbestehen, diese Elemente zu wählen. Die Zahl W be-zeichnet die Anzahl äquivalenter Positionen in allge-meiner Lage.
Bei allen Symmetrieoperationen bleibt eine Mengevon Gitterpunkten fest, nämlich die Punkte, welcheauf das Symmetrieelement fallen.
25
2 Symmetrie und Struktur
Abbildung 2.26: Die 32 Punktsymmetrieklassen.
26

2 Symmetrie und Struktur
2.2.3 Kristallsysteme
ab
c
Abbildung 2.27: Definition der Achsen und Winkel.
Die Kombination der Punktsymmetriegruppen mitdem Translationsgitter ergibt insgesamt 230 unter-schiedliche Raumgitter oder Raumgruppen. Diesewerden in mehreren hierarchischen Ebenen einge-teilt. Zunächst betrachtet man die Achsen a,b,c derEinheitszelle, sowie die Winkel a,b ,g zwischendiesen Achsen. Abb. 2.27 zeigt, zwischen welchenAchsen diese Winkel definiert sind: a zwischen bund c, b zwischen a und c und g zwischen a und b.Aufgrund der möglichen Werte dieser 6 Größen teiltman die Raumgruppen ein in sieben Kristallsysteme.
kubisch
� = � = � = 90�a = b = c
a 6= b 6= c� = � = 90� 6= �
monoklin
aa
triklin
a 6= b 6= c� = � = � = 90�
orthorombisch
rhomboedrisch (trigonal)a = b = c� = � = � 6= 90� < 120�
hexagonal
a = b 6= c
� = � = 90�, � = 120�
a = b 6= c
� = � = � = 90�
tetragonal
Abbildung 2.28: Übersicht über die Kristallsysteme.
Abb. 2.28 zeigt die 7 Kristallsysteme, zusammen mitden Bedingungen. Sie lauten
1. Triklin: a 6= b 6= c, a 6= b 6= g : keine Symmetrie
2. Monoklin: a 6= b 6= c, a = g = 90� 6= b : 1 C2;
3. Orthorombisch: a 6= b 6= c, a = b = g = 90� :3 C2
4. Hexagonal: a = b 6= c, a = b = 90�, g = 120�
: 1 C6
5. Rhomboedrisch (trigonal): a = b = c, a = b =g 6= 90� < 120�; : 1 C3
6. Tetragonal: a = b 6= c, a = b = g = 90�, : 1 C4
7. Kubisch: a = b = c, a = b = g = 90� : 4 C3
2.2.4 Bravais-Gitter
Diese sieben Kristallsysteme werden weiter diffe-renziert in 14 Bravais-Gitter. Ein primitives Bravais-Gitter ist definiert als die Menge aller Translations-vektoren
~T = u1~a1 +u2~a2 +u3~a3, (2.1)
welche die entsprechende, unendlich ausgedehnteKristallstruktur invariant lassen. Hier stellen ui gan-ze Zahlen dar. In einem nicht-primitiven Gitter wer-den zusätzliche Punkte eingefügt, so dass jede Ele-mentarzelle mehr als einen Punkt enthält, welchenicht durch die in (2.1) definierten Gittervektorenerreicht werden. Trotzdem ist die Umgebung dieserPunkte identisch zur Umgebung aller anderen Gitter-punkte.
Primitiv
Basiszentriert
a 6= b 6= c
� = � = 90� 6= �
ab
ca
b
c
Abbildung 2.29: Monoklines Kristallsystem mit un-terschiedlichen Möglichkeiten derBasis.
Zu jedem Kristallsystem gibt es ein primitives Gitter.Beim monoklinen gibt es ausserdem ein basiszen-triertes, d.h. die Einheitszelle besitzt nicht nur Gitter-punkte an den Ecken, sondern auch im Zentrum derdurch a und b aufgespannten Fläche (!Abb. 2.29).
27

2 Symmetrie und Struktur
Dieses Gitter ist also nicht primitiv. Beim orthorom-bischen gibt es ebenfalls ein basiszentriertes Gitter,sowie zusätzlich ein raumzentriertes (oder innenzen-triertes) und ein flächenzentriertes. Beim tetragona-len Gitter gibt es ein raumzentriertes und beim kubi-schen ein raumzentriertes und ein flächenzentriertes.
r2 = (1/2, 1/2, 1/2)
Abbildung 2.30: Kubisch primitives, innenzentrier-tes und flächenzentrierte Einheits-zellen.
Die vielleicht einfachste Kristallstruktur ist das pri-mitiv kubische Gitter (Abb. 2.30 links). Die Ato-me sind in diesem Fall auf den Ecken eines Wür-fels angeordnet, so dass jede Einheitszelle ein Atomenthält. In einem flächenzentrierten kubischen Gitter(Abb. 2.30 rechts) sind drei weitere Atome pro Ein-heitszelle vorhanden, zentriert in den Seitenflächendes Würfels.
~a2
~a3
~a1
Abbildung 2.31: fcc Gitter mit einer (alternativen)primitiven Einheitszelle.
Ein basiszentriertes oder raumzentriertes Gitter be-sitzen zwei Gitterpunkte pro Einheitszelle, ein flä-chenzentriertes Gitter vier. Natürlich wäre es bei al-len nichtprimitiven Gittern ebenfalls möglich, eineandere Einheitszelle zu wählen, sodass das Gitterprimitiv würde. Abb. 2.31 zeigt als Beispiel ein fccGitter mit einer alternativen Einheitszelle. Diese ent-spricht einem rhomboedrischen Gitter. Diese Ein-heitszelle enthält nur einen Gitterpunkt und ist damitvier mal kleiner. Eine der Möglichkeiten, eine pri-mitive Einheitszelle zu wählen, ist die Wigner-Seitz
Konstruktion. Häufig sind aber die Rechnungen ein-facher in einem nichtprimitiven Gitter durchzufüh-ren, z.B. wenn man dann ein orthonormiertes Koor-dinatensystem verwenden kann.
Insgesamt ergeben sich die folgenden 7 Kristallsy-steme und 14 Bravais-Gitter:
1. Triklin: a 6= b 6= c, a 6= b 6= g : keine Symmetrie
2. Monoklin: a 6= b 6= c, a = g = 90� 6= b : 1 C2;a) primitiv, b) basiszentriert
3. Orthorombisch: a 6= b 6= c, a = b = g = 90� :3 C2 a) primitiv, b) basiszentriert, c) raumzen-triert, d) flächenzentriert
4. Hexagonal: a = b 6= c, a = b = 90�, g = 120�
: 1 C6 primitiv
5. Rhomboedrisch (trigonal): a = b = c, a = b =g 6= 90� < 120�; : 1 C3 primitiv
6. Tetragonal: a = b 6= c, a = b = g = 90�, : 1 C4a) primitiv, b) raumzentriert
7. Kubisch: a = b = c, a = b = g = 90� : 4 C3 a)primitiv, b) raumzentriert, c) flächenzentriert
2.2.5 Raumgruppen
Bisher hatten wir angenommen, dass die Objekteselber sphärische Symmetrie aufweisen. Dies ist imAllgemeinen nicht der Fall.
AB
Spiegelebenen
A B
Abbildung 2.32: Links: zwei Atome in allgemeinerLage, mit Multiplizität 4. Rechts:3 Atome in spezieller Lage; A hatMultiplizität 2, B 2.
Werden die unterschiedlichen Symmetrien der Ba-sis mit berücksichtigt, dann ergibt sich eine weite-re Unterteilung der Bravais-Gitter in insgesamt 230
28

2 Symmetrie und Struktur
Raumgruppen. Abb. zeigt zwei Beispiele. In der lin-ken Hälfte sitzen die Atome auf allgemeinen Lagen,mit Multiplizität 4 aufgrund der beiden Spiegelebe-nen. In der rechten Hälfte sitzen sie auf symmetri-schen Positionen mit entsprechend niedrigerer Mul-tiplizität von 2 für A und 1 für B.
7 Kristallsysteme
Basis : primitiv / nicht primitiv
14 Bravaisgitter
z.B. triklin
z.B. kubisch innenzentriert
Symmetrie der Basis
230 Raumgruppen z.B. Amm2 /
Uran(V,VI)-oxid
C142v
Abbildung 2.33: Kristallsysteme, Bravais-Gitter undRaumgruppen.
Abb. 2.33 fasst diesen Übergang zusammen. AlsBeispiel ist die Raumgruppe von Uran(V,VI)-oxidgezeigt. Weitere Beispiele von kubischen Raum-gruppen werden in Kapitel 2.3.3 behandelt. Die voll-ständige Liste der Raumgruppen wurde von ArthurMoritz Schoenflies und Jewgraf Stepanowitsch Fjo-dorow erstellt.
2.3 Strukturen
2.3.1 Netzebenen und Miller Indizes
In der Kristallographie spielen die sog. Netzebeneneine große Rolle. Dabei handelt es sich um (gedach-te) Ebenen, die mit Atomen oder Gitterpunkten be-setzt sind. Wie man sich leicht überzeugen kann,sind die Atome in einer solchen Ebene ebenfallsperiodisch angeordnet, wobei die Periodizität grö-ßer sein kann als die Periodizität des Kristalls. Je-de Netzebene entspricht einer Netzebenenschar, d.h.einer unendlichen Schar von äquivalenten Ebenen,welche parallel zueinander in einem festen Abstand
liegen. Diese Netzebenen entsprechen auch mögli-chen Spaltflächen oder Wachstumsebenen von Kri-stallen. Außerdem reflektieren diese Ebenen Rönt-genstrahlung und spielen deshalb eine entscheidendeRolle bei der Strukturbestimmung (! Kap. 2.6).
Netzebenen können durch jeweils drei ganze Zah-len eindeutig charakterisiert werden. Diese werdenals Miller’sche Indizes5 bezeichnet und in der Form( jkl) geschrieben.
a
b
Abbildung 2.34: Zweidimensionale Netzebene mitAchsenabschnitten 3, 1.
Dafür bestimmt man die Abschnitte, an denen dieEbene die Achsen schneidet. Die Achsenabschnittewerden in Vielfachen der Einheitszelle (also nichtder primitiven Elementarzelle) bestimmt. Im Bei-spiel von Abb. 2.34 sind dies die Zahlen 3 und 1.Die Miller Indizes erhält man, indem man den Kehr-wert der Achsenabschnitte bildet (hier: 1/3, 1/1) unddas kleinste ganzzahlige Verhältnis bestimmt (hier:1, 3).
Abbildung 2.35: Netzebene in 3D; Achsenabschnit-te 3, 1, 2.
In drei Dimensionen geht man analog vor. Im Bei-spiel von Abb. 2.35 sind die Achsenabschnitte 3, 1,
5Nach dem Vorschlag von William Hallowes Miller(1801–1880).
29

2 Symmetrie und Struktur
2 und die Kehrwerte somit 1/3, 1, 1/2. GanzzahligeIndizes erhält man durch Erweitern mit 6 : (263). Ineinem zweiten Beispiel seien die Achsenabschnitte6, 2, 3. Daraus erhält man die Kehrwerte 1/6, 1/2 =3/6, 1/3 = 2/6 und damit Miller Indizes (132).
xy
z
(100)
z
xy
(110)
z
xy
(111)
xy
z
(200)x y
z
(100)-
Abbildung 2.36: Beispiele für Netzebenen.
Einige Beispiele von Miller Indizes für häufig ver-wendete Ebenen sind in Abb. 2.36 zusammenge-stellt. Liegt die Netzebene parallel zu einer Achse,so beträgt der entsprechende Achsenabschnitt un-endlich und der Index 0. Negative Achsenabschnittewerden mit einem Querstrich bezeichnet.
Meist sind aufgrund der Symmetrie des Gitters meh-rere Netzebenen äquivalent zueinander. Ein einfa-ches Beispiel sind die Ebenen (100), (010), und(001) des einfach kubischen Gitters. Solche Grup-pen von äquivalenten Netzebenen fasst man zusam-men, indem man die Indizes in geschweifte Klam-mern setzt, also z.B. {100}.
Für Richtungen im direkten Raum verwendet maneckige Klammern, also z.B. [hkl]. In einem kubi-schen Kristall stehen die Richtungen [hkl] senkrechtauf die Netzebenen (hkl).
2.3.2 Dichteste Kugelpackung
Festkörper bilden sich, weil die darin enthaltenenBausteine sich gegenseitig anziehen. Die Energie ei-nes Kristalls kann deshalb meist optimiert werden,wenn die Bestandteile möglichst dicht gepackt sind.Es stellt sich somit die Frage, welche Anordnung denRaum optimal füllt. Für die meisten Bestandteile ist
die Antwort nicht analytisch, aber für den wichtigenFall, dass die Bestandteile durch karte Kugeln ange-nähert werden könne, lässt sich die Frage beantwor-ten. Kugelförmige Bestandteile sind eine gute Nähe-rung für viele Ionenkristalle.
In einer Dimension wird die dichteste Kugelpackungdurch eine Reihe direkt aneinander gelegter Kugelnrealisiert.
Abbildung 2.37: Links: dichteste Kugelpackung ineiner Ebene; rechts: 2 hexagonaldichtest gepackte Ebenen gestapelt.
In zwei Dimensionen kann man Reihen von Ku-geln jeweils um eine halbe Gitterkonstante verscho-ben aneinander fügen und erhält eine dichteste Ku-gelpackung, welche einem hexagonalen Gitter ent-spricht. Fügt man zwei solcher Schichten aufeinan-der, so wird der Schichtabstand minimal, wenn sichdie Kugeln der oberen Lage über einer Lücke der un-teren Lage befinden (siehe Abb. 2.37 rechts).
Abbildung 2.38: 3 hexagonal dichtest gepackte Ebe-nen gestapelt.
Fügt man eine dritte Schicht auf diese beiden, sokann dies auf zwei Arten optimiert werden: Man legtdie dritte Schicht vertikal über die erste oder manverschiebt sie nochmals in die gleiche Richtung wiebeim ersten Schritt, so dass die dritte über die ge-meinsame Lücke der blauen und roten Schicht zu lie-gen kommt. Die erste Folge wird als ABAB charak-terisiert, die zweite als ABCABC. Beide Variantenkommen in der Natur vor, und es sind auch gemisch-te Fälle möglich, d.h. die Stapelfolge kann variie-
30

2 Symmetrie und Struktur
ren. In allen Fällen gilt für identische Kugeln, dassdas Volumen der Kugeln 74 % des Kristallvolumensausmacht. Das Verhältnis der Kugelvolumina zumgesamten Volumen wird als Raumfüllung bezeich-net. Da diese beiden Packungen die maximal mögli-che Raumfüllung aufweisen, werden sie als ‘dichte-ste Kugelpackung’ bezeichnet.
Schicht A
Schicht BSchicht A
A
B
CA
Abbildung 2.39: Anordnung der Schichten in der he-xagonal dichtesten Kugelpackung(links) und in der flächenzen-trierten dichtesten Kugelpackung(rechts).
Ist die Stapelfolge ABAB, so wählt man normaler-weise eine hexagonale Einheitszelle, wie in Abb.2.39 links dargestellt. Diese Struktur wird als hexa-gonal dichteste Kugelpackung bezeichnet oder kurzals hcp (=hexagonal close packed). Die Stapelrich-tung entspricht der c-Achse des hexagonalen Kri-stallsystems.
Abbildung 2.40: Strukturparameter der hexagonaldichtesten Kugelpackung.
Abb. 2.40 zeigt eine weitere Darstellung der hcpStruktur. Es gilt, wie immer im hexagonalen Kristall-system, a = b, g = 120� und a = b = 90�. Somit sinddie freien Parameter nur noch die beiden Kantenlän-gen c und a. Die Basis besteht hier aus zwei Atomen
mit den Koordinaten (0,0,0) und (2/3, 1/3, 1/2). JedesAtom hat 12 nächste Nachbarn; man bezeichnet die-se Zahl als Koordinationszahl. Für die ideale hcpStruktur gilt außerdem c =
p8/3a ⇡ 1,633a. Reale
Strukturen besitzen ein Verhältnis c/a, welches nahebei diesem Wert ist (siehe Abb. 2.40 rechts).
Für die Beschreibung des Gitters, das durch die Sta-pelfolge ABCABC erzeugt wird, verwendet mandas kubisch flächenzentrierte Gitter, welches in Abb.2.39 rechts dargestellt ist. Die Stapelrichtung ent-spricht der Raumdiagonale des Würfels. Dieser Fallwird kurz als fcc (=face centered cubic) bezeichnet.Die Raumfüllung beträgt in beiden Fällen (hcp undfcc) 74%. In einem kubisch innenzentrierten Gitter(bcc = (body centered cubic) ist die Raumfüllung68%, in einem einfachen kubischen Gitter 52%, undin einem Diamantgitter 34%.
2.3.3 Kubische Strukturen
0 0
0 0
01/2 1/2
1/2
1/2
1/4
1/4
3/4
3/4
Abbildung 2.41: Struktur von Diamant als 3D Dar-stellung und Projektion in die xy-Ebene mit den z-Koordinaten derAtome.
Eine relativ wichtige Struktur ist diejenige von Dia-mant. Zusätzlich zu einem flächenzentrierten kubi-schen Gitter enthält Diamant jeweils ein Atom an derStelle (1/4, 1/4, 1/4) und den entsprechenden äquiva-lenten Positionen.
Viele Halbleiter, wie z.B. Si oder GaAs kristallisie-ren in einer Struktur, welche von der Diamantstruk-tur abgeleitet werden kann. Bei den binären Halb-leitern werden die Gitterplätze abwechselnd mit Gaoder As belegt.
31

2 Symmetrie und Struktur
Abbildung 2.42: Struktur von GaAs.
Kristall Kristalla a
Tabelle 2.1: Größe der Eiheitszelle für unterschied-liche Materialien mit Zinkblende-Struktur.
Die Struktur eines Materials, welches in derZinkblende-Struktur kristallisiert, ist vollständig be-stimmt, wenn noch die Kantenlänge a der Einheits-zelle gegeben ist. Tabelle 2.1 zeigt diesen Parameterfür 12 unterschiedliche Verbindungen.
Auch SiC ist dadurch charakterisiert, dass ein SiAtom jeweils tetraedrisch durch kovalente Bindun-gen mit vier Kohlenstoff-Atomen verknüpft ist, undumgekehrt.
Cl–
Na+
a
Abbildung 2.43: Struktur von NaCl (links) und CsCl(rechts).
Kristalle, die aus mehr als einer Atomsorte beste-hen, enthalten dementsprechend mehrere Atome proEinheitszelle. Ein relativ einfaches Beispiel ist NaCl
(Kochsalz) (siehe Abb. 2.43 links). Da die Na+-Ionen kleiner sind als die Cl�-Ionen ist in diesemFall ein kubisch flächenzentriertes Gitter energetischam günstigsten. Dies bedeutet, dass in einem Unter-gitter, welches nur die Cl�, resp. Na+-Ionen enthält,jeweils Ecken und Flächenmittelpunkte eines Kubusbesetzt sind. Man kann das Gitter aber auch als pri-mitiv kubisches Gitter (mit der halben Gitterkonstan-te, d.h. 1/8 Volumen der Einheitszelle) beschreiben,bei dem die Gitterplätze alternierend mit Cl, resp. Nabesetzt sind.
Im CsCl-Kristall (Abb. 2.43 rechts) besetzen dieCs+-Ionen die Gitterpunkte eines einfach kubischenGitters. Das Raumgitter (Bravais-Gitter) ist damiteinfach kubisch (sc) mit einer zweiatomigen Basisaus einem Cs+-Ion bei (0,0,0) und einem Cl�-Ionbei (½,½,½). Jedes Atom befindet sich im Mittel-punkt eines Würfels der anderen Atomsorte. Die Ko-ordinationszahl ist somit 8.
dhcp-Struktur = double hexagonally close packed: Stapelfolge ABACABAC
No
Abbildung 2.44: Übersicht über die Struktur vonelementaren Kristallen.
Viele Elemente kristallisieren in kubischen Struktu-ren, Abb. 2.44 fasst sie zusammen. Die wichtigstensind bcc, fcc, hcp und dhcp (double hexagonally clo-se packed, mit der Stapelfolge ABACABAC), sowiedie Diamantstruktur.
2.3.4 Quasikristalle
Wie bereits erwähnt, sind fünfzählige Rotations-achsen in einem System mit Translationssymme-
32

2 Symmetrie und Struktur
trie nicht möglich. Auch in zwei Dimensionen istes nicht möglich, die Ebene mit Einheitszellen mitfünfzähliger Symmetrie abzudecken. Man hat des-halb lange Zeit geglaubt, dass solche Kristalle nichtexistieren würden. Erst 1984 wurden erstmals inBeugungsexperimenten 10-zählige Symmetrieach-sen gefunden, und etwas später konnte man dieseSymmetrie auch makroskopisch nachweisen.
Abbildung 2.45: Morphologie eines Quasikristalls(links) und zugehöriges Beugungs-muster (rechts).
Mit Hilfe der Elektronenmikroskopie findet man diefünfzählige Symmetrie sowohl in der Morphologieder Kristalle wie auch in der atomaren Struktur. Diegleiche Symmetrie findet man auch in hochauflö-senden Mikroskopie Bildern, welche direkt die ato-mare Struktur darstellen. Da diese Materialien zwareinen hohen Ordnungsgrad, aber keine Translations-symmetrie aufweisen, werden sie als Quasikristallebezeichnet. Die Details dieser Strukturen sind nochnicht in allen Fällen vollständig verstanden. Sie ba-sieren jedoch auf räumlich nichtperiodischen Struk-turen.
Abbildung 2.46: Zwei Beispiele, wie eine Ebenemit einem nichtperiodischen Mu-ster abgedeckt werden kann.
In zwei Dimensionen können Kombinationen von2 Elementen den Raum vollständig abdecken, ohnedass sie Translationssymmetrie aufweisen. Bekanntdafür sind vor allem die Elemente von Penrose6.
Quasikristalle wurden 1984 in bestimmten interme-tallischen Verbindungen entdeckt [4] und charakte-risiert. Für einige Jahre waren sie relativ umstritten,aber weitere Arbeiten und auch Messungen im di-rekten Raum haben ihre Existenz bestätigt. Die mei-sten Quasikristalle wurden in künstlich hergestelltenVerbindungen gefunden. Seit einigen Jahren gibt esjedoch auch Hinweise darauf, dass Quasikristalle innatürlich vorkommenden Mineralien vertreten sind[1].
2.3.5 Defekte
größeres Substitutionsatom
Leerstelle
Zwischengitteratom
Frenkel-Paar kleineres Substitutionsatom
Abbildung 2.47: Unterschiedliche Arten vonDefekten.
Ideale Kristalle stellen eine nützliche Fiktion dar. Sieexistieren jedoch nicht, sondern alle realen Kristalleenthalten Abweichungen vom idealen Gitter, welcheals Defekte bezeichnet werden. Du den wichtigstenDefekten gehören Leerstellen, Zwischengitteratome,Versetzungen und Substitution durch Fremdatome,sowie Kombinationen davon. Diese Defekte erhöhenim Allgemeinen die Energie des Systems, aber auchdie Entropie. Deshalb sind im thermodynamischenGleichgewicht immer Defekte vorhanden, und dieZahl der Defekte nimmt mit zunehmender Tempe-ratur zu. Eine Leerstelle in Kupfer besitzt z.B. eine
6Roger Penrose (* 1931)
33

2 Symmetrie und Struktur
Energie von 1,2 eV, ein Zwischengitteratom 3,4 eV.
Ag Cl
Abbildung 2.48: Frenkel Fehlstelle als Verschiebungeines Atoms auf einen Zwischen-gitterplatz, respektive als Kombi-nation einer Leerstelle mit einemZwischengitter-Atom.
Eine Kombination von 2 elementaren Defekten trittz.B. bei der Frenkel-Fehlstelle auf (! Abb. 2.48):hier wurde ein Atom von seinem eigentlichen Gitter-platz auf einen Zwischengitterplatz verschoben. Wiein der Abbildung rechts gezeigt ist, kann dieser De-fekt auch als Kombination einer Leerstelle mit einemZwischengitteratom gesehen werden.
Leerstelle
STM-Bild
NaCl
Schottky-Defekt in NaCl
Abbildung 2.49: Schottky Defekt in einem NaClKristall (links) und Leerstelle in ei-nem einatomigen Kristall (rechts).
Als Schottky-Defekt bezeichnet man eine Kombina-tion aus zwei Leerstellen von Atomen mit entgegen-gesetzter Ladung. Dieser Defekt ist damit, wie auchDer Frenkel-Defekt, ladungsneutral.
Eine weitere Kombination von elementaren Defek-ten ist das Stickstoff-Leerstellen Zentrum im Dia-mant (! Abb. 2.50). Es wird auch als NV-Zentrumbezeichnet (von Nitrogen-Vacancy). Es ist ein gutesBeispiel dafür, wie solche Defekte die Eigenschaften
N
V
Abbildung 2.50: Stickstoff-Leerstellen Defekt imDiamant.
eines Materials verändern können. Während reinerDiamant praktisch farblos ist, hat das NV-Zentrumeinen erlaubten Übergang im sichtbaren Bereich desSpektrums und führt deshalb zu einer Färbung desKristalls.
Abbildung 2.51: Struktur eines metallischen Glases(links) und eine Möglichkeit zu ih-rer Herstellung durch schnelles Ab-kühlen (rechts).
Defekte bilden sich nicht nur im thermodynami-schen Gleichgewicht, sie können auch z.B. auch beider Kristallisation entstehen, vor Allem wenn dieAbkühlung sehr schnell vor sich geht. Dies führt z.B.zu Gläsern oder amorphen Substanzen. Gläser kön-nen durchsichtig sein, aber es existieren auch soge-nannte metallische Gläser, welche interessante Ei-genschaften als Werkstoffe aufweisen.
Die genannten Defekte sind Punktdefekte. Dane-ben gibt es auch eindimensionale Defekte, wiez.B. Schraubenversetzungen oder Stufenversetzun-gen (siehe Abb. 2.52). Solche Defekte können durcheinen Burgersvektor charakterisiert werden (~b inAbb. 2.52 rechts). Er wird bestimmt, indem man ana-
34

2 Symmetrie und Struktur
b!
Abbildung 2.52: Stufenversetzung (links) und ihreCharakterisierung durch einen Bur-gersvektor~b.
loge Wege im perfekten und im defektbehafteten Teildes Kristalls vergleicht.
Zweidimensionale Defekte umfassen Korngrenzenund Stapelfehler. Auch dreidimensionale Defektekönnen charakterisiert werden.
2.4 Strukturbestimmung
Die atomare Struktur eines Körpers kann viele seinerEigenschaften erklären und ist deshalb immer vongroßem Interesse. Um diese Struktur zu bestimmenbenötigt man ein Werkzeug, welches in atomarenGrößen arbeiten kann. In erster Linie benutzt mandafür elektromagnetische Wellen mit kurzer Wel-lenlänge, d.h. Röntgenstrahlen. Auch Materiewellensind mit Erfolg eingesetzt worden, in erster LinieElektronen oder Neutronenstrahlen, aber neuerdingsauch Atomstrahlen.
2.4.1 Feld-Ionen Mikroskopie
Die erste Methode, welche Atome direkt sichtbarmachte, war die Feld-Ionen Mikroskopie. Es han-delt sich dabei um ein relativ einfaches Gerät: imWesentlichen benötigt man eine sehr scharfe Spitze,an die man eine positive elektrische Spannung an-legt. Dadurch erhält man an der Spitze ein sehr ho-hes elektrisches Feld. Ausserhalb der Spitze befin-det sich mit niedrigem Druck ein Gas, typischerwei-se Helium. Wenn ein Heliumatom in die Nähe der
+
V
He
Spitze
Schirm
Abbildung 2.53: Prinzip der Feldionenmikroskopie.
Spitze gelangt, wird es durch dieses enorme elek-trische Feld ionisiert, das heisst diese Metallspitzezieht eines der Elektronen des Heliumatoms weg.Dadurch wird das Heliumatom zu einem positiv ge-ladenen Heliumion und wird nun durch das starkeelektrische Feld sehr rasch von der Spitze weg be-schleunigt. Nach einer Distanz von etwa 10 cm trifftes auf einen Schirm, wo es sichtbar gemacht wird.Da sich die Atome auf dem direktesten Weg von derSpitze entfernen, entsteht dadurch auf dem Schirmein Bild der Spitze. Die Vergrößerung kommt durchdas Verhältnis des Radius der Spitze zur Distanz vomSchirm zustande und benötigt keine weiteren abbil-denden Elemente. Man erhält also auf diese Weiseauf dem Schirm ein Bild dieser Spitze mit sehr ho-her Auflösung. Allerdings ist das Bild ziemlich starkverzerrt.
Abbildung 2.54: Feldionenmikroskop-Aufnahmenvon Atomen, die sich auf einerMetallspitze bewegen. Das obereist ein Rhenium-, das untere einWolfram-Atom.
35

2 Symmetrie und Struktur
Diese Art von Mikroskopie ist inzwischen mehr als60 Jahre alt [3], sorgt aber immer noch für spek-takuläre Bilder, wie z.B. die Serie von Bildern inAbb. 2.54, welche zeigen, dass man damit nicht nuratomare Auflösung erhält, also einzelne Atome se-hen kann, sondern auch deren Bewegung über dieOberfläche beobachten kann. In Abb. 2.54 ist dieOberfläche einer Wolframspitze dargestellt, auf dersich zwei einzelne Atome bewegen, welche durchdie dreieckigen Pfeile markiert sind. Beim unterenhandelt es sich um ein Wolfram-Atom, beim obe-ren um ein Rhenium-Atom. (Aus T.T. Tsong, Atom-probe field ion microscopy, Cambridge UniversityPress, Cambridge (1990).)
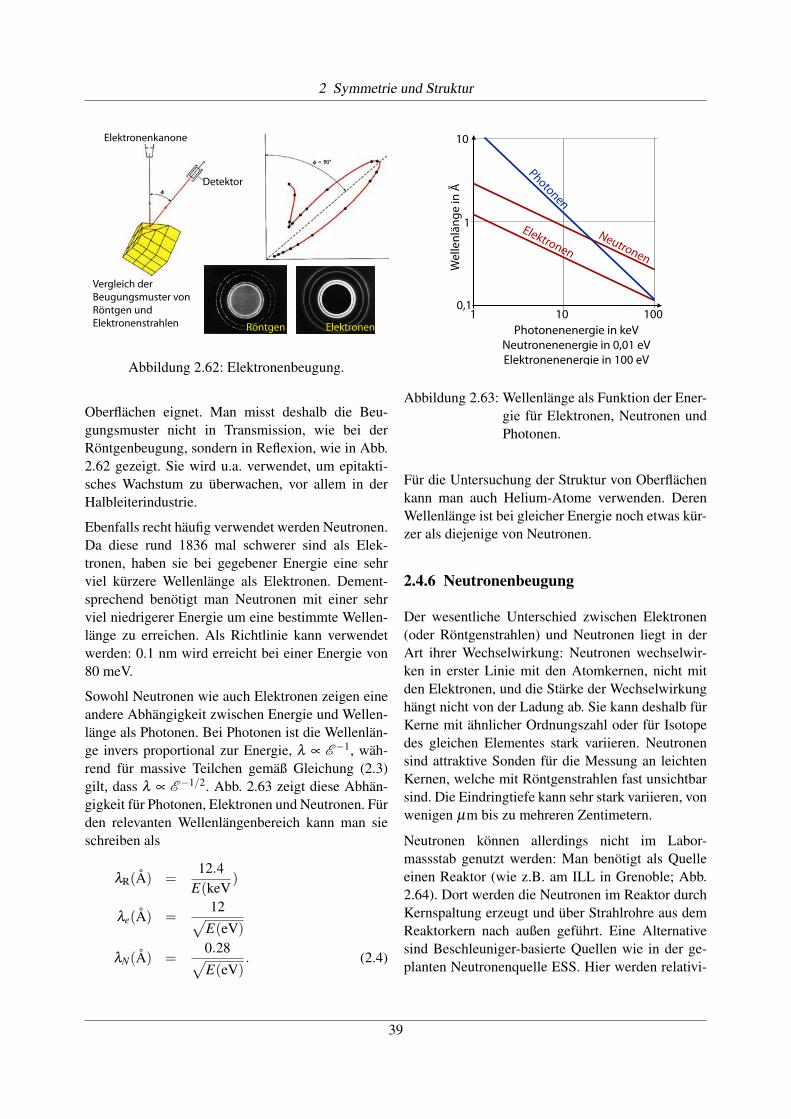
2.4.2 Elektronenmikroskopie
Um ein weniger verzerrtes Bild einer beliebigen ato-maren Struktur zu erhalten, benötigt man eine Ab-bildungsoptik, die unabhängig vom abzubildendenObjekt ist. Die Wellenlänge des abbildenden Feldesmuss dazu kleiner sein als die abzubildenden Struk-turen. Verwendet man elektromagnetische Wellen(d.h. Röntgenstrahlen), sind abbildende Linsen prak-tisch nicht herstellbar.
Abbildung 2.55: Funktionsprinzip eines Elektronen-mikroskops.
Verwendet man jedoch Elektronen für die Abbil-dung, so können Linsen mit elektromagnetischenFeldern erzeugt werden.
Abbildung 2.56: Elektronenmikroskopische Auf-nahme eines Molekülkristalls.
Hochgezüchtete Systeme sind in der Lage, Ato-me direkt abzubilden. Dafür muss allerdings eineVergrößerung um mindestens 7 Größenordnungenerreicht werden. Aufgrund der damit verbundenentechnischen Schwierigkeiten ist dies erst seit weni-gen Jahren möglich und stellt immer noch kein Rou-tineverfahren dar. Abb. 2.56 zeigt als Beispiel einenMolekülkristall mit atomarer Auflösung. Dies ist al-lerdings nur möglich, wenn die Orientierung geeig-net gewählt ist, so dass Stapel von übereinander lie-genden Molekülen aufeinander abgebildet werden.
2.4.3 Rastersonden Mikroskopie
Die Methode, mit der man die strukturelle Infor-mation erhält, hängt stark davon ab, welches dieserWerkzeuge man verwendet. Im Falle der Rasterson-den Mikroskopie ist die Methode sehr direkt: mantastet den Gegenstand mit der Probe ab und zeichnetdie Position der Probe auf, um so direkt ein Bild derOberfläche zu erhalten.
Diese Methode wurde 1982 von Binnig und Roh-rer am IBM Forschungslaboratorium in Rüschlikonentwickelt. Dabei wurde eine feine Spitze über eineOberfläche geführt, wobei der Abstand zwischen derSpitze und der Oberfläche konstant gehalten wur-de. Indem man die Position der Spitze aufzeichnete,konnte man ein Bild der Oberfläche erhalten. Mantastet also die Oberfläche mit einer Spitze ab, benutztalso eine Art verfeinerten Tastsinn, um die Oberflä-che sichtbar zu machen.
36

2 Symmetrie und Struktur
ElektronikStrom
Abbildung 2.57: Funktionsprinzip der Raster-Tunnelmikroskopie.
Abbildung 2.58: STM Bild eines Kreises aus 48 Ei-senatomen.
Insbesondere hat man auch gelernt, mit dem Mi-kroskop Atome zu verschieben, nicht nur zu beob-achten. Abb. 2.58 zeigt als Beispiel einen Ring aus48 Eisenatomen, welche mit einer Rastertunnelspit-ze auf der Oberfläche eingesammelt und an einen Ortgebracht wurden. Anschliessend wurde das gleicheMikroskop dafür verwendet, sie abzubilden.
Die Raster-Sonden Mikroskope verwendeten die ex-ponentielle Abhängigkeit des sog. Tunnelstroms, al-so eines elektrischen Stroms durch ein nichtleiten-des Medium wie das Vakuum, um ein Bild zu er-halten. Diese Technik wird deshalb als Tunnelmikro-skopie (STM = scanning tunneling microscopy) be-zeichnet. Die Notwendigkeit für einen elektrischenStrom beschränkt diese Technik auf leitende Ober-flächen. Später kamen andere Arten von Sonden da-zu, wie die Raster-Kraftmikroskopie (AFM = atomicforce microscopy) die magnetische Wechselwirkung(MFM = magnetic force microscopy) oder die opti-sche Nahfeld Mikroskopie (SNOM = scanning nearfield optical microscopy). Alle diese Techniken sind
hervorragend für die Untersuchung von bestimmtenOberflächen geeignet, jedoch nicht für die Untersu-chung von Volumenkristallen.
2.4.4 Röntgenbeugung
Vor der Entwicklung der direkten Methoden wardie einzige Möglichkeit, mit atomarer AuflösungInformationen über Kristallstrukturen zu erhalten,die Verwendung von Beugungsmethoden, also dieStreuung einer Welle an einer periodischen Struk-tur. Auch heute ist das für Volumenkristalle meistdie einzige Möglichkeit, da die direkten Methodennur für Oberflächen geeignet sind. Voraussetzung fürdie Verwendung von Beugungsmethoden ist, dassdie Wellenlänge der verwendeten Strahlung von dergleichen Größenordnung ist wie die Abstände zwi-schen den Atomen, also weniger als 1 nm.
Cl- Na+
a0
a0
Abbildung 2.59: Struktur von NaCl.
Die Beugung von Wellen an periodischen Strukturenwurde u.a. von den Braggs7 erklärt. Ihre Erklärungist sehr anschaulich und liefert das richtige Resultat.Man betrachtet dabei eine Reihe von parallelen Ebe-nen. Im Kristall sind dies natürlich keine wirklichenEbenen, sondern Netzebenen, also zweidimensiona-le Anordnungen von Atomen.
Jede dieser Ebenen reflektiert einen Teil der einfal-lenden Welle. Wie groß dieser Anteil ist, hängt vonder Welle selber ab, sowie von der Netzebene: wiedicht sind die Atome gepackt, was für eine Art vonAtomen sind es etc.
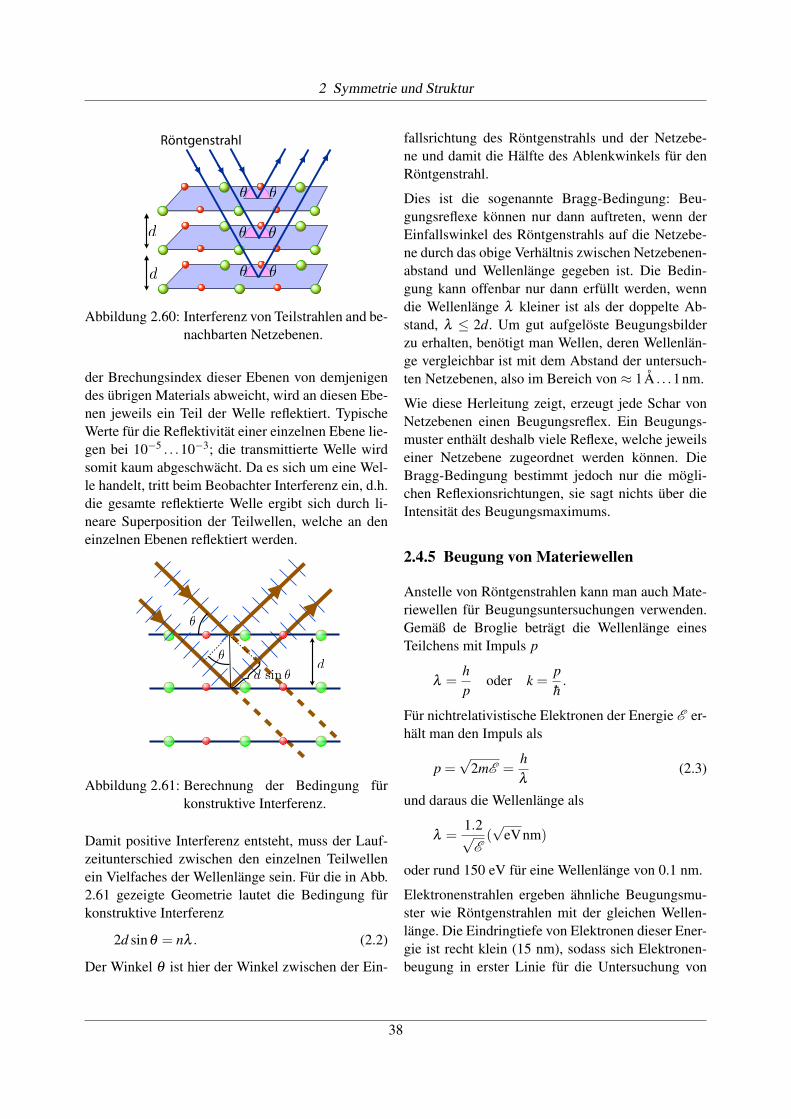
Für die Herleitung der Bragg-Bedingung bezeichnenwir den Abstand zwischen diesen Ebenen als d. Falls
7Sohn William Lawrence Bragg und Vater William HenryBragg, 1912
37
2 Symmetrie und Struktur
Röntgenstrahl
d
d
� �
� �
� �
Abbildung 2.60: Interferenz von Teilstrahlen and be-nachbarten Netzebenen.
der Brechungsindex dieser Ebenen von demjenigendes übrigen Materials abweicht, wird an diesen Ebe-nen jeweils ein Teil der Welle reflektiert. TypischeWerte für die Reflektivität einer einzelnen Ebene lie-gen bei 10�5 . . .10�3; die transmittierte Welle wirdsomit kaum abgeschwächt. Da es sich um eine Wel-le handelt, tritt beim Beobachter Interferenz ein, d.h.die gesamte reflektierte Welle ergibt sich durch li-neare Superposition der Teilwellen, welche an deneinzelnen Ebenen reflektiert werden.
�
�
d sin �d}
Abbildung 2.61: Berechnung der Bedingung fürkonstruktive Interferenz.
Damit positive Interferenz entsteht, muss der Lauf-zeitunterschied zwischen den einzelnen Teilwellenein Vielfaches der Wellenlänge sein. Für die in Abb.2.61 gezeigte Geometrie lautet die Bedingung fürkonstruktive Interferenz
2d sinq = nl . (2.2)
Der Winkel q ist hier der Winkel zwischen der Ein-
fallsrichtung des Röntgenstrahls und der Netzebe-ne und damit die Hälfte des Ablenkwinkels für denRöntgenstrahl.
Dies ist die sogenannte Bragg-Bedingung: Beu-gungsreflexe können nur dann auftreten, wenn derEinfallswinkel des Röntgenstrahls auf die Netzebe-ne durch das obige Verhältnis zwischen Netzebenen-abstand und Wellenlänge gegeben ist. Die Bedin-gung kann offenbar nur dann erfüllt werden, wenndie Wellenlänge l kleiner ist als der doppelte Ab-stand, l 2d. Um gut aufgelöste Beugungsbilderzu erhalten, benötigt man Wellen, deren Wellenlän-ge vergleichbar ist mit dem Abstand der untersuch-ten Netzebenen, also im Bereich von ⇡ 1A . . .1nm.
Wie diese Herleitung zeigt, erzeugt jede Schar vonNetzebenen einen Beugungsreflex. Ein Beugungs-muster enthält deshalb viele Reflexe, welche jeweilseiner Netzebene zugeordnet werden können. DieBragg-Bedingung bestimmt jedoch nur die mögli-chen Reflexionsrichtungen, sie sagt nichts über dieIntensität des Beugungsmaximums.
2.4.5 Beugung von Materiewellen
Anstelle von Röntgenstrahlen kann man auch Mate-riewellen für Beugungsuntersuchungen verwenden.Gemäß de Broglie beträgt die Wellenlänge einesTeilchens mit Impuls p
l =hp
oder k =ph.
Für nichtrelativistische Elektronen der Energie E er-hält man den Impuls als
p =p
2mE =hl
(2.3)
und daraus die Wellenlänge als
l =1.2p
E(p
eVnm)
oder rund 150 eV für eine Wellenlänge von 0.1 nm.
Elektronenstrahlen ergeben ähnliche Beugungsmu-ster wie Röntgenstrahlen mit der gleichen Wellen-länge. Die Eindringtiefe von Elektronen dieser Ener-gie ist recht klein (15 nm), sodass sich Elektronen-beugung in erster Linie für die Untersuchung von
38
2 Symmetrie und Struktur
Röntgen Elektronen
Vergleich der Beugungsmuster von Röntgen und Elektronenstrahlen
Elektronenkanone
Detektor
Abbildung 2.62: Elektronenbeugung.
Oberflächen eignet. Man misst deshalb die Beu-gungsmuster nicht in Transmission, wie bei derRöntgenbeugung, sondern in Reflexion, wie in Abb.2.62 gezeigt. Sie wird u.a. verwendet, um epitakti-sches Wachstum zu überwachen, vor allem in derHalbleiterindustrie.
Ebenfalls recht häufig verwendet werden Neutronen.Da diese rund 1836 mal schwerer sind als Elek-tronen, haben sie bei gegebener Energie eine sehrviel kürzere Wellenlänge als Elektronen. Dement-sprechend benötigt man Neutronen mit einer sehrviel niedrigerer Energie um eine bestimmte Wellen-länge zu erreichen. Als Richtlinie kann verwendetwerden: 0.1 nm wird erreicht bei einer Energie von80 meV.
Sowohl Neutronen wie auch Elektronen zeigen eineandere Abhängigkeit zwischen Energie und Wellen-länge als Photonen. Bei Photonen ist die Wellenlän-ge invers proportional zur Energie, l µ E �1, wäh-rend für massive Teilchen gemäß Gleichung (2.3)gilt, dass l µ E �1/2. Abb. 2.63 zeigt diese Abhän-gigkeit für Photonen, Elektronen und Neutronen. Fürden relevanten Wellenlängenbereich kann man sieschreiben als
lR(A) =12.4
E(keV)
le(A) =12p
E(eV)
lN(A) =0.28pE(eV)
. (2.4)
10
Photonenenergie in keVNeutronenenergie in 0,01 eVElektronenenergie in 100 eV
Wel
lenl
änge
in Å
Photonen
Neutronen
Elektronen
1 10 1000,1
1
Abbildung 2.63: Wellenlänge als Funktion der Ener-gie für Elektronen, Neutronen undPhotonen.
Für die Untersuchung der Struktur von Oberflächenkann man auch Helium-Atome verwenden. DerenWellenlänge ist bei gleicher Energie noch etwas kür-zer als diejenige von Neutronen.
2.4.6 Neutronenbeugung
Der wesentliche Unterschied zwischen Elektronen(oder Röntgenstrahlen) und Neutronen liegt in derArt ihrer Wechselwirkung: Neutronen wechselwir-ken in erster Linie mit den Atomkernen, nicht mitden Elektronen, und die Stärke der Wechselwirkunghängt nicht von der Ladung ab. Sie kann deshalb fürKerne mit ähnlicher Ordnungszahl oder für Isotopedes gleichen Elementes stark variieren. Neutronensind attraktive Sonden für die Messung an leichtenKernen, welche mit Röntgenstrahlen fast unsichtbarsind. Die Eindringtiefe kann sehr stark variieren, vonwenigen µm bis zu mehreren Zentimetern.
Neutronen können allerdings nicht im Labor-massstab genutzt werden: Man benötigt als Quelleeinen Reaktor (wie z.B. am ILL in Grenoble; Abb.2.64). Dort werden die Neutronen im Reaktor durchKernspaltung erzeugt und über Strahlrohre aus demReaktorkern nach außen geführt. Eine Alternativesind Beschleuniger-basierte Quellen wie in der ge-planten Neutronenquelle ESS. Hier werden relativi-
39

2 Symmetrie und Struktur
Abbildung 2.64: Erzeugung von Neutronen im For-schungsreaktor (ILL Grenoble).
stische Protonen auf ein Target geschossen, aus demdadurch Neutronen austreten.
Abbildung 2.65: Neutronen-FlugzeitspektrometerIN6 am ILL Grenoble.
Auch die eigentlichen Spektrometer sind sehr auf-wändige Großgeräte, welche nur an wenigen For-schungszentren zur Verfügung stehen, wie z.B. amInstitut Laue-Langevin (ILL) in Grenoble. Abb. 2.65zeigt schematisch ein solches Gerät.
Bezüglich der reinen Strukturaufklärung unterschei-den sich Neutronen von Röntgenstrahlung vor al-lem durch den Streuquerschnitt: sie bilden nicht dieElektronendichte ab, sondern die Position der Kerne.Deshalb sind sie z.B. nützlich für die Messung derPosition von Wasserstoffatomen, welche wegen ih-rer geringen Anzahl Elektronen in Röntgenmessun-gen schlecht sichtbar sind. Außerdem können sie zur
Neutronen Röntgen
Abbildung 2.66: Vergleich der Streuquerschnittevon unterschiedlichen Atomen fürNeutronen und Röntgenstrahlen.
Messung von Kernbewegungen, magnetischer Ord-nung und Isotopenverteilung eingesetzt werden. Wiein Abb. 2.66 gezeigt, unterscheiden sich die Streu-querschnitte auch für unterschiedliche Isotope desgleichen Elements.
2.5 Das reziproke Gitter
Die Bragg-Bedingung (2.2) liefert zwar eine Bedin-gung für das Auftreten von Röntgenreflexen, aber esist zum einen keine hinreichende Bedingung, zumzweiten liefert sie keine Intensitäten. Wie groß dieIntensität der gestreuten Welle ist, hängt davon ab,wie stark die einzelnen Ebenen reflektieren. Im Falleder Röntgenstrahlung ist die Beugungseffizienz imWesentlichen proportional zur Elektronendichte. Fürdie Berechnung der Streuintensität muss deshalb dieräumliche Abhängigkeit der Elektronendichte be-rücksichtig werden. Hier ist vor allem wichtig, diePeriodizität des Gitters zu berücksichtigen.
40

2 Symmetrie und Struktur
k0
b(k-a)
2ʌ/a
k0 2ʌa
4ʌa
6ʌa
x
Harmonische Funktionf(x) = cos(2πx/a)
x
Periodische Funktiong(x) Diskrete Funktion
F{f(x)} =
Z ��
��e�ikxf(x) dx
F{g(x)} =
Z ��
��e�ikxg(x) dx
Abbildung 2.67: Fourier-Zerlegung einer eindimen-sionalen Funktion.
2.5.1 Periodizität der Elektronendichte
Aufgrund der Periodizität des Kristalls muss dieElektronendichte n(~r) ebenfalls periodisch sein,
n(~r +~T ) = n(~r),
wobei ~T einen Translationsvektor
~T = u1~a1 +u2~a2 +u3~a3
darstellt.
Daraus folgt, dass man die Elektronendichte alsFourier-Reihe schreiben kann. In einer Dimensionwird sie dann
n(x) = n0 + Âp>0
Cp cos2p px
a+Sp sin
2p pxa
.
Hier stellt n0 den Mittelwert dar und p = 1,2, . . . einenatürliche Zahl. Alternativ kann die Reihe komplexgeschrieben werden:
n(x) =•
Âp=�•
npei2p px/a.
Damit die Elektronendichte reell wird, muss geltenn⇤
�p = np. Die Koeffizienten Cp, Sp oder np erhältman durch Fourier-Transformation der Elektronen-dichte (siehe Abb. 2.67).
Diese eindimensionale Betrachtung muss man fürKristalle auf drei Dimensionen erweitern. Die drei-dimensionale Elektronendichte ist periodisch in den
Richtungen, welche die Basisvektoren des Gittersangeben. Dies kann geschrieben werden als
n(~r) = Âpqs
npqsei2p px/aei2pqy/bei2psz/c
= Âpqs
npqsei2p( pxa + qy
b + szc ),
wobei p, q und s über alle (positiven und negativen)ganzen Zahlen laufen.
Der Exponent kann als Skalarprodukt geschriebenwerden:
2p(pxa
+qyb
+szc
) = ~G ·~r.
Damit wird die Elektronendichte
n(~r) = ÂG
n~Gei~G·~r,
Der Vektor
~G =
✓2p p
a,2pq
b,2ps
c
◆
wird definiert durch drei ganze Zahlen p,q,s. Erstellt also einen Punkt in einem Gitter dar, ähnlichwie die Translationsvektoren ~T . Dieses Gitter wirddurch drei Basisvektoren der Länge2p/a, 2p/b und2p/c aufgespannt. Es befindet sich allerdings nichtim gewöhnlichen dreidimensionalen Raum, sondernhat offenbar die Dimension einer inversen Länge. Eswird üblicherweise als reziprokes Gitter bezeichnet.
2.5.2 Definition des reziproken Gitters
Eine mögliche Definition des reziproken Gitters istdie folgende:
Das reziproke Gitter besteht aus denjenigen Wel-lenvektoren ~k, die eine Funktion ei~k·~r definieren,welche im direkten Raum die Periodizität des di-rekten Gitters aufweist.
Alternativ können wir das reziproke Gitter konstruk-tiv definieren, indem wir zunächst seine Basisvekto-ren~bi aus den Basisvektoren ~ai des direkten Gitters
41

2 Symmetrie und Struktur
konstruieren:
~b1 = 2p
~a2 ⇥~a3
~a1 · (~a2 ⇥~a3)= 2p
~a2 ⇥~a3
V
~b2 = 2p
~a3 ⇥~a1
V~b3 = 2p
~a1 ⇥~a2
V.
Aufgrund dieser Konstruktion steht~b1 senkrecht auf~a2 und ~a3 und entsprechendes gilt für die anderenVektoren. Sakalarprodukte zwischen Basisvektorendes direkten und reziproken Gitters werden somit
~bi ·~a j = 2pdi j.
Diese Konstruktion kann auch in Matrixform ge-schrieben werden. Wir definieren die Matrix
A = (~a1,~a2,~a3) =
0
@a1x a2x a3xa1y a2y a3ya1z a2z a3z
1
A
der primitiven Gittervektoren.
Entsprechend können wir eine Matrix B für die Ba-sisvektoren des reziproken Gitters definieren. Ausder Orthogonalitätsbeziehung folgt A†B = 2p1 oder
B = 2p
�A†��1
.
Damit ist es möglich, die Bestimmung des rezipro-ken Gitters auf eine Matrixinversion zurückzufüh-ren.
Aus der Konstruktion der Basisvektoren, resp. derOrthogonalitätsbeziehung~bi ·~a j = 2pdi j folgt für be-liebige Vektoren ~T des direkten Gitters und ~G desreziproken Gitters
~T · ~G = (u1v1 +u2v2 +u3v3)2p
= (ganzeZahl)2p
oder
ei~T ·~G = 1. (2.5)
Dies entspricht der ersten Definition des reziprokenGitters.
a reziproker Raum
2πa
Abbildung 2.68: Einheitszelle des kubisch primiti-ven Gitters und des zugehörigen re-ziproken Gitters.
2.5.3 Beispiele
Bei kubischen Strukturen mit Kantenlängen a derEinheitszelle ist das reziproke Gitter ebenfalls ei-ne kubische Struktur. Die Achsen haben die gleicheRichtung wie in der direkten Struktur, die Kanten-länge beträgt 2p/a. Die primitiven Gittervektorendes direkten und des reziproken Gitters sind dann
direktesGitter reziprokesGitter~a1 = a~ex ~b1 = 2p
a ~ex
~a2 = a~ey ~b2 = 2p
a ~ey
~a3 = a~ez ~b3 = 2p
a ~ez
V = a3 V =� 2p
a
�3
Die Situation wird etwas komplizierter für nicht-kubische Einheitszellen, insbesondere für schief-winklige.
~a2
~a3
~a1
Abbildung 2.69: Primitive EZ im fcc-Gitter mit denBasisvektoren des reziproken Git-ters.
Für die primitive Einheitszelle des kubisch flächen-zentrierten Gitters (siehe Abb. 2.69), z.B., erhalten
42

2 Symmetrie und Struktur
wir
A =a2
0
@1 0 11 1 00 1 1
1
A ,
wobei a wie üblich die Kantenlänge des Würfels dar-stellt. Damit wird die entsprechende Matrix für dasreziproke Gitter
B =2p
a
0
@1 �1 11 1 �1
�1 1 1
1
A .
Dies sind die primitiven Gittervektoren des bcc Git-ters (kubisch innenzentriert). Wie in Abb. 2.69 ge-zeigt, sind die Vektoren~bi in diesem Fall nicht par-allel zu ~ai, aber sie stehen senkrecht auf die übrigenVektoren ~ak für k 6= i.
Abbildung 2.70: Berechnung des reziproken Gittersfür das bcc Gitter.
Wie in Abb. 2.70 gezeigt, ergeben sich für die Ba-sisvektoren des reziproken Gitters einer bcc-Strukturdie primitiven Gittervektoren des fcc-Gitters.
2.5.4 Gitterelemente
Das gesamte Gitter erhält man wiederum durch Li-nearkombination der Basisvektoren
~G = v1~b1 + v2~b2 + v3~b3
mit ganzzahligen vi. ~G wird als Punkt oder Vektordes reziproken Gitters bezeichnet. Die Dimensiondieser Vektoren beträgt m�1, wie man leicht aus der
Definition der Basisvektoren ersieht. Falls die Vekto-ren ~ai die Basisvektoren des primitiven Gitters sind,so sind auch die Vektoren ~bi die Basisvektoren desprimitiven reziproken Gitters.
Die Punkte des reziproken Gitters sind Fourier-Komponenten des Kristalls und damit in erster Li-nie mathematische Hilfsmittel. Um sie doch etwaszu veranschaulichen, kann man sich aber vorstellen,dass sie ein Objekt des direkten Raumes beschrei-ben, welches eine bestimmte Periodizität besitzt. EinGitterpunkt, der im zweidimensionalen reziprokenRaum die Koordinaten (r,s) besitzt, entspricht derKomponente
sin2prx
asin
2psyb
.
(1,2)
Reziprokes Gitter
Direktes Gitter
a1
a2
a1
a2
~b1
~b2
(1,3)
~b1
~b2
Abbildung 2.71: Punkte im reziproken Gitter und ih-re Fouriertransformierten.
Abb. 2.71 zeigt zwei Beispiele. Ein Vektor des rezi-proken Gitters entspricht damit immer einer entspre-chenden Periodizität im direkten Raum. Damit ent-hält die Wellenfunktion des Kristalls eine Kompo-nente ei~k·~r. Aufgrund der Beziehung von de Brogliekann dies auch so interpretiert werden, dass ein Im-puls in Richtung~k vorhanden ist. Mit anderen Wor-ten: das reziproke Gitter ist eine Zerlegung des Fest-körperimpulses.
Zu den wichtigsten Eigenschaften des reziprokenGitters gehören
1. Das reziproke Gitter eines Bravais-Gitters istselbst ein Bravais-Gitter.
43

2 Symmetrie und Struktur
2. Das reziproke Gitter des reziproken Gitters istdas direkte Gitter.
3. Ist Vc das Volumen der von den primitiven Git-tervektoren aufgespannten Einheitszelle im di-rekten Gitter, so ist (2p)3/Vc das Volumen derZelle im reziproken Raum.
4. Die Länge der reziproken Gittervektoren istproportional zum Kehrwert der Länge der Git-tervektoren. Dies erklärt die Nomenklatur „re-ziprokes“ Gitter.
2.5.5 Reziproke Gittervektoren undEbenenscharen
Eine wichtige Beziehung besteht auch zu den Netze-benen des direkten Gitters: Ist eine Ebene durch dieMiller Indizes hkl gegeben, so steht der Vektor
~G =
0
@hkl
1
A = h~b1 + k~b2 + l~b3;
des reziproken Gitters senkrecht auf dieser Ebene.Beweis: wir zeigen, dass dieser Vektor senkrecht aufzwei linear unabhängigen Vektoren ~v1 und ~v2 steht,welche die Ebene (hkl) aufspannen. Wir wählen
~v1 =1h~a1 � 1
k~a2 , ~v2 =
1k~a2 � 1
l~a3.
~v1 =1
h~a1 � 1
k~a2
1
h~a1
1
k~a2
~a2
~a1
Abbildung 2.72: Definition von~v1.
Wie in Abb. 2.72 gezeigt, liegt der Vektor ~v1 in derSchnittgeraden von (hkl) und der Ebene, die von ~a1und ~a2 aufgespannt wird. Entsprechend liegt ~v2 inder Schnittgeraden von (hkl) und (~a2,~a3), und ge-meinsam spannen die beiden Vektoren die Netzebe-ne auf. Das Skalarprodukt mit dem reziproken Git-tervektor ~G ist
~G ·~v1 =⇣
h~b1 + k~b2 + l~b3
⌘·✓
1h~a1 � 1
k~a2
◆.
Die Orthogonalitätsrelation zwischen den Basisvek-toren des direkten und reziproken Raums ergibt
~G ·~v1 = ~G ·~v2 = 2p (1�1) = 0⌅.
Der kürzeste Vektor ~G des reziproken Gitters, dersenkrecht auf den Netzebenen steht, hat die Länge
|~G| = 2p
d,
wobei d den Abstand zwischen benachbarten Netze-benen darstellt. Diese Beziehung folgt aus der Tatsa-che, dass die Funktion ei~G·~r im direkten Raum einePeriode von 2p/|~G| hat, welche dem Abstand zwi-schen Netzebenen entsprechen muss.
~a2
~a1{a2
ka1
h{d2
Abbildung 2.73: Abstand der Netzebenen.
Für den Spezialfall eines rechteckigen Gitters in 2Dimensionen berechnen wir den Abstand d2 zwi-schen aufeinander folgenden Netzebenen gemäßAbb. 2.73
d2a1h
=a2kq
a21
h2 +a2
2k2
.
Daraus erhalten wir den Abstand
d2 =a1a2q
a21k2 +a2
2h2=
1qk2
a22+ h2
a21
=2p
|G|
in zwei Dimensionen.
2.5.6 Brillouin-Zonen
Im reziproken Gitter kann man genau so wie im di-rekten Gitter Einheitszellen definieren. Einige Bei-spiele wurden in Kapitel 2.5.3 diskutiert. Im rezipro-ken Raum spielt, im Gegensatz zum direkten Raum,
44
2 Symmetrie und Struktur
~a1
~a2
Mittel-senkrechte
Abbildung 2.74: 1. Brillouin-Zone als Wigner-Seitz Einheitszelle des reziprokenRaums.
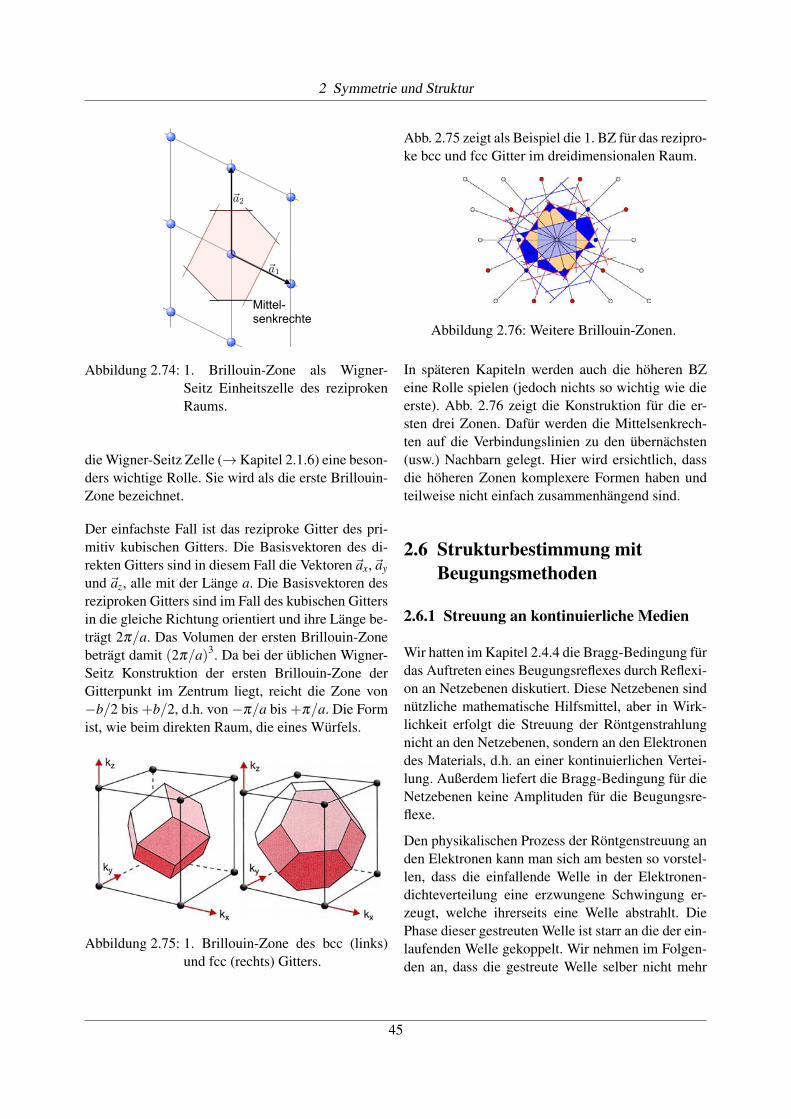
die Wigner-Seitz Zelle (! Kapitel 2.1.6) eine beson-ders wichtige Rolle. Sie wird als die erste Brillouin-Zone bezeichnet.
Der einfachste Fall ist das reziproke Gitter des pri-mitiv kubischen Gitters. Die Basisvektoren des di-rekten Gitters sind in diesem Fall die Vektoren~ax,~ayund ~az, alle mit der Länge a. Die Basisvektoren desreziproken Gitters sind im Fall des kubischen Gittersin die gleiche Richtung orientiert und ihre Länge be-trägt 2p/a. Das Volumen der ersten Brillouin-Zonebeträgt damit (2p/a)3. Da bei der üblichen Wigner-Seitz Konstruktion der ersten Brillouin-Zone derGitterpunkt im Zentrum liegt, reicht die Zone von�b/2 bis +b/2, d.h. von �p/a bis +p/a. Die Formist, wie beim direkten Raum, die eines Würfels.
Abbildung 2.75: 1. Brillouin-Zone des bcc (links)und fcc (rechts) Gitters.
Abb. 2.75 zeigt als Beispiel die 1. BZ für das rezipro-ke bcc und fcc Gitter im dreidimensionalen Raum.
Abbildung 2.76: Weitere Brillouin-Zonen.
In späteren Kapiteln werden auch die höheren BZeine Rolle spielen (jedoch nichts so wichtig wie dieerste). Abb. 2.76 zeigt die Konstruktion für die er-sten drei Zonen. Dafür werden die Mittelsenkrech-ten auf die Verbindungslinien zu den übernächsten(usw.) Nachbarn gelegt. Hier wird ersichtlich, dassdie höheren Zonen komplexere Formen haben undteilweise nicht einfach zusammenhängend sind.
2.6 Strukturbestimmung mitBeugungsmethoden
2.6.1 Streuung an kontinuierliche Medien
Wir hatten im Kapitel 2.4.4 die Bragg-Bedingung fürdas Auftreten eines Beugungsreflexes durch Reflexi-on an Netzebenen diskutiert. Diese Netzebenen sindnützliche mathematische Hilfsmittel, aber in Wirk-lichkeit erfolgt die Streuung der Röntgenstrahlungnicht an den Netzebenen, sondern an den Elektronendes Materials, d.h. an einer kontinuierlichen Vertei-lung. Außerdem liefert die Bragg-Bedingung für dieNetzebenen keine Amplituden für die Beugungsre-flexe.
Den physikalischen Prozess der Röntgenstreuung anden Elektronen kann man sich am besten so vorstel-len, dass die einfallende Welle in der Elektronen-dichteverteilung eine erzwungene Schwingung er-zeugt, welche ihrerseits eine Welle abstrahlt. DiePhase dieser gestreuten Welle ist starr an die der ein-laufenden Welle gekoppelt. Wir nehmen im Folgen-den an, dass die gestreute Welle selber nicht mehr
45

2 Symmetrie und Struktur
gestreut wird. Dies wird als erste Born’sche Nähe-rung bezeichnet und ist für die Streuung von Rönt-genlicht in Kristallen fast immer eine gute Nähe-rung. Mehrfachstreuung kann nur in wenigen Fäl-len überhaupt beobachtet werden. Der Grund dafürist der geringe Streuquerschnitt für die Streuung vonPhotonen an Elektronen: er ist von der Größenord-nung r2
e ⇡ 10�29 m2, wobei
re =1
4pe0
e2
mec2 ⇡ 2,818 ·10�15 m
den klassischen Elektronenradius darstellt.
0
Quelle Detektor
~r
dV
~k~k�
Abbildung 2.77: Streuungsbeitrag des Volumenele-ments dV.
Wir gehen aus von einem einfallenden Röntgen-strahl, der durch den Wellenvektor ~k beschriebenwird, und bestimmen die Intensität eines Strahls, derin Richtung ~k0 gestreut wird. Dazu berechnen wirden Beitrag jedes Volumenelementes des Kristalls.Ein Element dV an der Stelle ~r erzeugt einen Bei-trag, der proportional ist zur Elektronendichte n(~r)an diesem Ort. Wir gehen davon aus, dass die ein-laufende Welle als ebene Welle beschrieben werdenkann und dass der Detektor so weit vom Kristall ent-fernt ist, dass die gestreute Welle (welche einer Ku-gelwelle um dV entspricht) in guter Näherung beimDetektor ebenfalls als ebene Welle beschrieben wer-den kann.
Gegenüber einer Referenz-Phasenfläche durch denUrsprung O des Koordinatensystems erhält die ein-fallende Welle bis zum Volumenelement dV einePhasenverzögerung um~k ·~r. Die gestreute Welle er-hält auf dem Weg zum Detektor ebenfalls eine Pha-senverzögerung, um �~k0 ·~r. Somit ergibt sich insge-samt für den Beitrag des Volumenelements bei~r eine
Phasenverschiebung um den Betrag
j(~r) =~k ·~r �~k0 ·~r =⇣~k �~k0
⌘·~r = �D~k ·~r,
mit D~k =~k0 �~k als Änderung des Impulses beimStreuprozess. Bei elastischer Streuung sind die Be-träge der beiden Vektoren gleich, |~k| = |~k0|.
2.6.2 Bragg-Bedingung
Die gesamte Amplitude am Detektor erhalten wirdurch Integration über das Volumen des Kristalls,wobei die einzelnen Beiträge mit der entsprechen-den Elektronendichte gewichtet werden:
F =Z
dV n(~r)e�iD~k·~r. (2.6)
Das Integral entspricht einer Fouriertransformati-on. Damit ist die Streuamplitude proportional zurFourier-Amplitude der Elektronendichte n(~r) bei derräumlichen Frequenz D~k. Dies ist die Basis allerBeugungsmethoden für die Strukturbestimmung.
Da die Elektronendichte periodisch ist, können wirsie als Fourier-Reihe schreiben:
n(~r) = ÂG
n~Gei~G·~r.
Damit erhalten wir für die gestreute Amplitude
F =Z
dV ÂG
n~Gei(~G�D~k)·~r. (2.7)
Das Integral kann nur dann von Null verschiedensein, wenn der Integrand nicht oszilliert, d.h. wenn
~G = D~k,
d.h. wenn D~k ein Vektor des reziproken Gitters ist.Somit findet man nur dann einen Beugungsreflex,wenn der Streuvektor einem Vektor des reziprokenGitters entspricht. Dies ist einer der wesentlichstenGründe dafür, dass die Festkörperphysik in erster Li-nie kristalline Materialien diskutiert.
Abb. 2.78 zeigt als Beispiel die Beugungsreflexe vonMuskovit (KAl2(AlSi3O10)(F,OH)2). Die einzelnenReflexe sind als Vektoren des reziproken Gitters in-diziert. Die Schärfe dieser Bedingung ist begrenzt
46

2 Symmetrie und Struktur
b1
b2
Text
Abbildung 2.78: Beugungsreflexe von Muskovit(KAl2(AlSi3O10)(F,OH)2).
durch die Größe des Kristalls; die Unschärfe nimmtab mit der Anzahl der Elementarzellen, welche zurStreuung beitragen.
Diese Bedingung kann quantenmechanisch auch alsImpulserhaltung verstanden werden: h~k ist der Im-puls der einfallenden Welle, h~k0 der Impuls der ge-beugten Welle. Aufgrund der Impulserhaltung kannBeugung nur auftreten, wenn der entsprechende Im-pulsunterschied hD~k vom Material, d.h. vom Gitterzur Verfügung gestellt wird. Diese Möglichkeit istgenau dann gegeben, wenn ein entsprechender Vek-tor ~G = hD~k im reziproken Gitter existiert.
2.6.3 Röntgenstrahlung
Anodenspannung ~10-100 kV
Röntgenstrahlung
Thermische Elektronenemission
Hochvakuumröhre
Heiz-Spannung
Kathode Anode
Abbildung 2.79: Erzeugung von Röntgenstrahlungin einer Röntgenröhre.
Die verwendete Röntgenstrahlung wird meist miteiner Röntgenröhre erzeugt. Darin werden Elek-tronen aus einer Glühkathode ins Vakuum emit-tiert und zur Anode beschleunigt. Beim Auftreffen
auf die Anode erzeugen sie hochenergetische Strah-lung, welche 2 Komponenten enthält: breitbandi-ge Bremsstrahlung und schmalbandige charakteristi-sche Strahlung, welche für das Material der Anodecharakteristische Übergangsfrequenzen aufweist.
Einfallende Strahlung (breitbandig)
Monochromator-Kristall
Nicht abgelenkte Komponenten des Primärstrahls
Strahlung vom Monochromator
2θ Zur Probe
Photonenenergie
Log(
Inte
nsitä
t) KαKβ
Abbildung 2.80: Filterung der Röntgenstrahlung ineinem Monochromator.
Für viele Anwendungen ist es notwendig, mono-chromatische Strahlung zu verwenden. Dies stelltman her, indem man die unerwünschten Teile unter-drückt. Dies kann durch Bragg-Streuung an einemMonochromator-Kristall erfolgen, wie in Abb. 2.80gezeigt.
2.6.4 Ewald-Konstruktion
Mit dieser Bedingung allein könnte für jeden einfal-lenden Röntgenstrahl eine unendliche Zahl von Beu-gungsmaxima auftreten. Für die Strukturaufklärungist jedoch vor allem ein Spezialfall wichtig, nämlichder Fall der elastischen Streuung, d.h. dass die Wel-lenlänge der gebeugten Welle gleich derjenigen dereinfallenden Welle ist, |~k| = |~k0|. Mit dieser zusätzli-chen Bedingung ist die Bedingung für das Auftretenvon Beugung nicht mehr automatisch erfüllt.
Die Bedingung dafür, dass ein Röntgenreflex auftritt,kann mit Hilfe der Ewald-Konstruktion dargestelltwerden (siehe Abb. 2.81). Ausgangspunkt sind dieBedingungen
|~k| = |~k0| , ~k0 �~k = ~G
für das Auftreten eines Reflexes. Man stellt dabeiden einfallenden Röntgenstrahl durch einen Vektor
47

2 Symmetrie und Struktur
reziprokes Gitter
keinfallender Strahl
elastische Streuung:|k| = |k’|
k’
2e
6k
gestreuter Strahl
Abbildung 2.81: Ewald-Konstruktion.
~k dar, wobei seine Spitze auf einem Gitterpunkt desreziproken Raumes liegt. Der reflektierte Strahl wirddurch einen Vektor~k0 dargestellt, dessen Spitze wie-derum auf einem Gitterpunkt liegen muss und dessenUrsprung mit demjenigen des einfallenden Strahlszusammenfällt. Der Streuvektor D~k = ~G ist dann einVektor des reziproken Gitters. Der Winkel 2q zwi-schen den beiden Vektoren entspricht der Bragg-Bedingung.
Die Ewald-Konstruktion zeigt, dass das Auftretenvon Beugung nur für wenige spezielle Wellenvek-toren auftritt. Man findet diese Vektoren, wennman einen Kreis mit Radius k verschiebt, bis erdurch zwei Gitterpunkte läuft. Die Konstruktionzeigt auch, dass |~k| � 1
2 |~G|min sein muss, d.h. derBetrag des einfallenden Wellenvektors muss minde-stens gleich der Hälfte des Betrags des kleinsten Git-tervektors sein.
Abbildung 2.82 zeigt die Ewald-Konstruktion in dreiDimensionen.
2.6.5 Beugung an Pulvern
Da ein einfallender Röntgenstrahl i.A. keinen Re-flex erzeugt, sind verschiedene Methoden entwickeltworden, um Röntgenbeugung zu beobachten. Dieeinfachste Methode ist die Pulver- oder Debye-Scherrer Methode: man bestrahlt ein Pulver.
Ein Pulver besteht aus vielen Kristallen mit zufäl-liger Orientierung. Da alle möglichen Orientierun-
Abbildung 2.82: Die Ewald-Kugel.
nach Debye-Scherrer
Abbildung 2.83: Beugung an Pulvern (Debye-Scherrer).
gen vorkommen, sind immer einige Kristallite rich-tig orientiert, so dass Reflexe auftreten. Aus Sym-metriegründen ist die gebeugte Röntgenstrahlung indiesem Fall konisch, d.h. die Beugung hängt nurvom Winkel gegenüber der Strahlrichtung ab. Wiein Abb. 2.83 gezeigt, wird die Probe in das Zentrumeines Zylinders gelegt, und die Innenseite des Zylin-ders mit einem Film belegt. Auf dem Detektor findetman deshalb konzentrische Ringe. Da nicht bekanntist, wie der Kristallit, welcher den Reflex erzeugt,orientiert ist, eignet sich dieses Verfahren nicht füreine vollständige Strukturbestimmung. Es kann aberverwendet werden, um Gitterkonstanten zu bestim-men.
Abb. 2.84 zeigt das Beugungsmuster, welches vonSilizium-Pulver gemessen wurde. Die einzelnenBeugungsmaxima sind mit den zugehörigen Miller-Indizes bezeichnet. Im Bereich 0� < 2q < 180� fin-det man Reflexe zu allen Gittervektoren, welche kür-
48

2 Symmetrie und Struktur
Beugungswinkel (Grad)
Inte
nsitä
t
50 100
Abbildung 2.84: Beugungsmaxima für Si-Pulver.
zer sind als 2 |k|. Während ihre Richtung sich ausdem Pulvermuster nicht bestimmen lässt, erhält manihre Länge aus der Bedingung
|G| = 2k sinq .
2.6.6 Einkristall-Verfahren
Kollimatoren
Röntgenstrahl
transmittierter Strahl(nicht verwendeteWellenlängen)
Probenkristall
Detektor
Monochromator
�
Abbildung 2.85: Drehkristall-Verfahren.
Ein Verfahren, welches vollständige Strukturanaly-sen von Einkristallen erlaubt, ist das Bragg- oderDrehkristall-Verfahren. Dabei wird der Kristall ge-dreht. Da das reziproke Gitter starr an das direkteGitter gekoppelt ist, wird es dabei mit gedreht. Ineinem Koordinatensystem, welches an das rezipro-ke Gitter gekoppelt ist, wird damit die Ewald-Kugelgedreht und es treten bei bestimmten OrientierungenReflexe auf. Dabei werden alle Reflexe gemessen,welche im Lauf der Drehung auftreten.
Für diese Art von Messungen benötigt man mono-chromatische Röntgenstrahlung. Ist die verwende-te Quelle breitbandig, so wird deshalb ein Mono-
chromator benötigt, um die gewünschte Wellenlängeherauszufiltern. Dafür verwendet man normalerwei-se ebenfalls Bragg-Beugung an einem Kristall (sieheAbb. 2.85).
reziprokes Gitter
~k
~k�
Abbildung 2.86: Prinzip des Laue-Verfahrens(links)und Ewald-Konstruktion für dasLaue-Verfahren.
Eine weitere Möglichkeit für Messungen an Ein-kristallen ist das sogenannte Laue-Verfahren. Dabeibenutzt man kontinuierliche Röntgenstrahlung ausdem Bremsstrahlungsbereich. Wenn ein breiter Be-reich von k-Vektoren (und damit Radien der Ewald-Kugel) vorkommen, gibt es immer die Möglichkeit,die Bragg-Bedingung zu erfüllen. Dieses Verfahreneignet sich wiederum nicht für die Strukturbestim-mung, da man nicht weiss, welche Wellenlänge wel-chen Reflex erzeugt hat. Man kann das Verfahrenaber benutzen, um Änderungen von Zellkonstanten(z.B. mit der Temperatur) zu beobachten, oder umKristalle mit bekannter Struktur zu orientieren.
Insgesamt gibt es also 3 unterschiedliche Verfahren,um die Beugungsbedingung D~k = ~G zu erfüllen:
1. Der Einfallswinkel q wird konstant gehaltenund die Gleichung wird für unterschiedlicheWellenlängen l erfüllt: Laue-Verfahren.
2. Die Wellenlänge l wird konstant gehalten unddie Bragg-Gleichung wird für unterschiedlicheq erfüllt: Drehkristall-Verfahren.
3. Die Wellenlänge l wird konstant gehalten unddie Bragg-Gleichung wird für unterschiedlicheq dadurch erfüllt, dass in einer pulverförmigenProbe irgendein Kristallit immer „richtig“ liegt:Debye-Scherrer-Verfahren.
49

2 Symmetrie und Struktur
2.6.7 Laue-Bedingung
Unterschiedliche Formen der Bedingung für dasAuftreten eines Röntgenreflexes können bei derAnalyse von bestimmten Situationen nützlich sein.Allgemein gilt die Impulserhaltung, resp. die Bedin-gung, dass der einfallende und der gestreute Strahlsich um einen Vektor des reziproken Gitters unter-scheiden müssen,
~k0 =~k + ~G.
Für elastische Streuung können wir daraus eine Be-dingung für die Längen ableiten:
���~k + ~G���2= k2 oder 2~k · ~G+G2 = 0
oder, da dies auch für �~G gelten muss, welcherebenfalls ein Gittervektor ist,
2~k · ~G = G2.
Wenn wir beide Seiten dieser Gleichung durch 4 di-vidieren, erhalten wir
~k · 12~G =
✓12
G◆2
. (2.8)
Diese Bedingung eignet sich wiederum für eine geo-metrische Konstruktion.
D
G
O
12
k
Abbildung 2.87: Laue-Konstruktion der Beugungs-bedingung.
Ausgangspunkt ist diesmal der Streuvektor D~k = ~G,welcher die Punkte O und D im reziproken Gitterverbinden soll. Um diejenigen einfallenden Wellen-vektoren ~k zu finden, welche die Beugungsbedin-gung erfüllen, fällen wir die Mittelsenkrechte auf
den Vektor ~G. Jeder Vektor, dessen Ursprung in Oliegt und auf dieser Mittelsenkrechten endet, erfülltoffenbar die Bedingung (2.8).
Diese Konstruktion entspricht offenbar gerade derWigner-Seitz Konstruktion für die Einheitszelle, d.h.der ersten Brillouin-Zone. Streuung findet somit im-mer dann statt, wenn der Wellenvektor des einfallen-den Strahls auf der Grenze der Brillouin-Zone liegt.
2.7 Berechnung der gestreutenIntensität
Bisher wurden nur Auswahlregeln betrachtet, alsoob in die entsprechende Richtung überhaupt etwasgestreut wird. Als nächstes soll die Stärke eines Re-flexes berechnet werden.
2.7.1 Streuamplitude und Strukturfaktor
Wenn ein Reflex auftritt, d.h. wenn der Streuvek-tor D~k ein Vektor des reziproken Gitters ist, wirdexp(i(~G � D~k) ·~r) = 1. Damit vereinfacht sich derAusdruck (2.7) für die Streuamplitude zu F µ n~G.
qq=0
qq=2�
66k = G
qq=4�
Abbildung 2.88: Reflexion an der 100 Ebene.
Offenbar ist die räumliche Abhängigkeit im Inte-granden verschwunden. Dies bedeutet, dass alle Ein-heitszellen identische Beiträge zur Streuamplitudeliefern, welche durch die entsprechende Fourier-Komponente der Elektronendichte im reziprokenGitter gegeben sind. Diese ist definiert als
n~G =Z
dV n(~r)e�i~G·~r.
Damit wird die Streuamplitude
F = n~G =Z
dV n(~r)e�i~G·~r. (2.9)
50

2 Symmetrie und Struktur
Aufgrund der Periodizität der Elektronendichte kanndas Integral über den Kristall auf ein Integral über ei-ne Einheitszelle und eine Multiplikation mit der Zahlder Einheitszellen reduziert werden: Für ~G einenVektor des reziproken Gitters und ~T einen beliebi-gen Vektor des direkten Gitters gilt gemäß (2.5):
e�i~G·(~r+~T ) = e�i~G·~r.
Damit können wir das Integral in (2.9) auf eine Ein-heitszelle reduzieren,
F = NZ
EZdV n(~r)e�i~G·~r = N S~G,
wobei N die Anzahl Zellen im Kristall darstellt und
S~G =Z
EZdV n(~r)e�i~G·~r
als Strukturfaktor bezeichnet wird. Der Strukturfak-tor ist also die Fouriertransformierte der Elektronen-dichte n(~r) über eine Einheitszelle.
2.7.2 Atomare Beiträge
In vielen Fällen ist es nützlich, die Elektronendich-te n(~r) in Beiträge der einzelnen Atome aufzuteilen.Die Zuordnung einzelner Elektronen zu bestimmtenAtomen ist natürlich eine Näherung. Für Elektronenin der K-Schale ist diese Näherung sehr gut, für Va-lenzelektronen in kovalent gebundenen Atomen oderMetallen eher schlecht. Die Mehrheit der Elektronenist jedoch relativ gut lokalisiert, und die Näherunghilft sehr gut beim Verständnis für die Berechnungder Beugungsintensitäten.
Wir bezeichnen mit ~r j die Position eines Atoms.Dann stellt die Funktion n j(~r�~r j) den Beitrag diesesAtoms zur Elektronendichte dar. Die gesamte Elek-tronendichte am Ort~r ist gegeben durch die Summeüber die s Atome der Basis:
n(~r) =s
Âj=1
n j(~r �~r j).
Dies erlaubt uns, auch den Strukturfaktor in Beiträgeder einzelnen Atome aufzuteilen:
S~G =Z
EZdV
s
Âj=1
n j(~r �~r j)e�i~G·~r
dVrj
r
ll
Elektronenhülle des Atoms j
Streuzentrum
Abbildung 2.89: Relativkoordinaten zur Berech-nung der Streuamplitude.
Wir definieren die Koordinate ~r =~r �~r j des Elek-
trons bezogen auf die Position~r j des Kerns j. Damitwird~r = ~r j +~
r und der Strukturfaktor
S~G =Z
ZelledV
s
Âj=1
n j(~r)e�i~G·~r e�i~G·~r j .
Damit ist
f j =Z
dV n j(~r)e�i~G·~r (2.10)
der Beitrag des j-ten Atoms. Er wird als Atom-formfaktor bezeichnet. Die Integration erstreckt sichhier über den gesamten Raum. Der Atomformfaktorentspricht also im Wesentlichen der Fouriertransfor-mierten der Elektronendichte eines Atoms und kannin erster Näherung als eine atomare Eigenschaft be-trachtet werden. Diese Näherung impliziert, dass dieElektronendichte des Kristalls als Summe der ato-maren Elektronendichten geschrieben werden kann.
Mit dieser Definition können wir den Strukturfaktorschreiben als
S~G =s
Âj=1
f j e�i~G·~r j . (2.11)
d.h. der Strukturfaktor setzt sich additiv aus den Bei-trägen der einzelnen Atome zusammen, wobei jederBeitrag mit einem Phasenfaktor multipliziert wird,der seine Position codiert. Die Phase entspricht der-jenigen, welche eine Welle mit Wellenvektor ~G aufdem Weg vom Ursprung des Koordinatensystemszur Position~r j des Atoms akkumulieren würde.
51
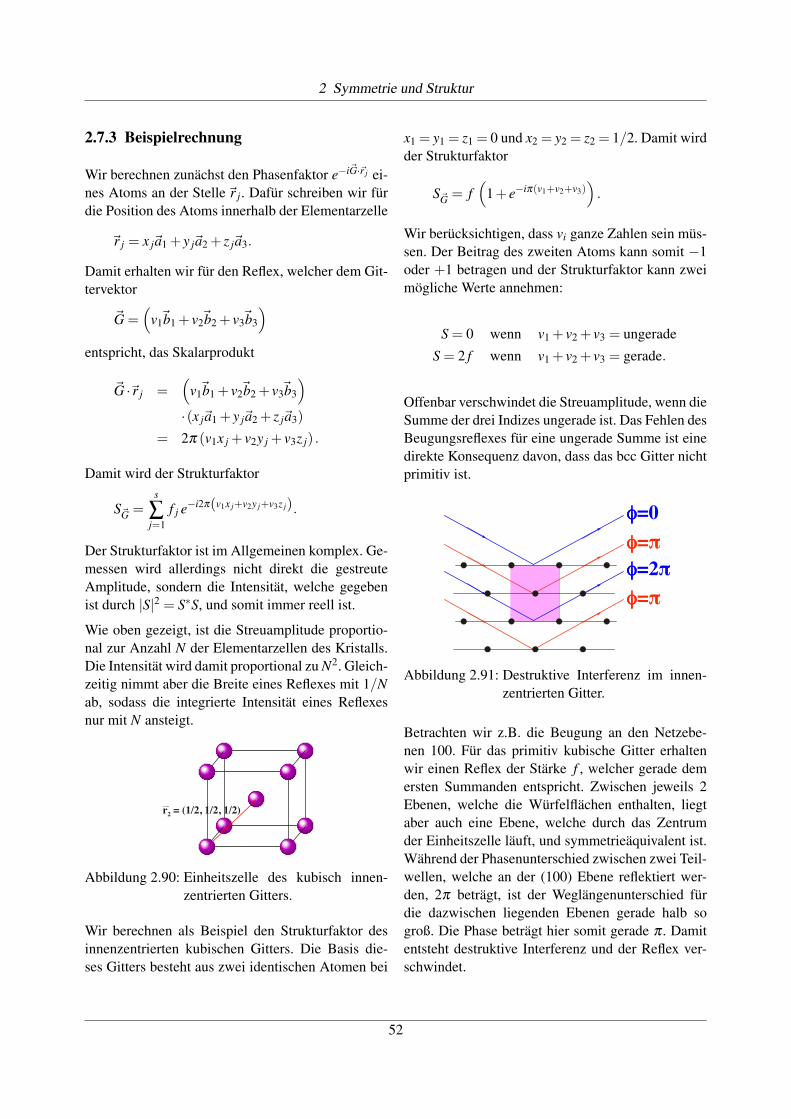
2 Symmetrie und Struktur
2.7.3 Beispielrechnung
Wir berechnen zunächst den Phasenfaktor e�i~G·~r j ei-nes Atoms an der Stelle~r j. Dafür schreiben wir fürdie Position des Atoms innerhalb der Elementarzelle
~r j = x j~a1 + y j~a2 + z j~a3.
Damit erhalten wir für den Reflex, welcher dem Git-tervektor
~G =⇣
v1~b1 + v2~b2 + v3~b3
⌘
entspricht, das Skalarprodukt
~G ·~r j =⇣
v1~b1 + v2~b2 + v3~b3
⌘
·(x j~a1 + y j~a2 + z j~a3)
= 2p (v1x j + v2y j + v3z j) .
Damit wird der Strukturfaktor
S~G =s
Âj=1
f j e�i2p(v1x j+v2y j+v3z j).
Der Strukturfaktor ist im Allgemeinen komplex. Ge-messen wird allerdings nicht direkt die gestreuteAmplitude, sondern die Intensität, welche gegebenist durch |S|2 = S⇤S, und somit immer reell ist.
Wie oben gezeigt, ist die Streuamplitude proportio-nal zur Anzahl N der Elementarzellen des Kristalls.Die Intensität wird damit proportional zu N2. Gleich-zeitig nimmt aber die Breite eines Reflexes mit 1/Nab, sodass die integrierte Intensität eines Reflexesnur mit N ansteigt.
r2 = (1/2, 1/2, 1/2)
Abbildung 2.90: Einheitszelle des kubisch innen-zentrierten Gitters.
Wir berechnen als Beispiel den Strukturfaktor desinnenzentrierten kubischen Gitters. Die Basis die-ses Gitters besteht aus zwei identischen Atomen bei
x1 = y1 = z1 = 0 und x2 = y2 = z2 = 1/2. Damit wirdder Strukturfaktor
S~G = f⇣
1+ e�ip(v1+v2+v3)⌘
.
Wir berücksichtigen, dass vi ganze Zahlen sein müs-sen. Der Beitrag des zweiten Atoms kann somit �1oder +1 betragen und der Strukturfaktor kann zweimögliche Werte annehmen:
S = 0 wenn v1 + v2 + v3 = ungeradeS = 2 f wenn v1 + v2 + v3 = gerade.
Offenbar verschwindet die Streuamplitude, wenn dieSumme der drei Indizes ungerade ist. Das Fehlen desBeugungsreflexes für eine ungerade Summe ist einedirekte Konsequenz davon, dass das bcc Gitter nichtprimitiv ist.
qq=0qq=�qq=2�qq=�
Abbildung 2.91: Destruktive Interferenz im innen-zentrierten Gitter.
Betrachten wir z.B. die Beugung an den Netzebe-nen 100. Für das primitiv kubische Gitter erhaltenwir einen Reflex der Stärke f , welcher gerade demersten Summanden entspricht. Zwischen jeweils 2Ebenen, welche die Würfelflächen enthalten, liegtaber auch eine Ebene, welche durch das Zentrumder Einheitszelle läuft, und symmetrieäquivalent ist.Während der Phasenunterschied zwischen zwei Teil-wellen, welche an der (100) Ebene reflektiert wer-den, 2p beträgt, ist der Weglängenunterschied fürdie dazwischen liegenden Ebenen gerade halb sogroß. Die Phase beträgt hier somit gerade p . Damitentsteht destruktive Interferenz und der Reflex ver-schwindet.
52
2 Symmetrie und Struktur
Atom A oder B
Atom B
Atom A
ungeordnet geordnet
Abbildung 2.92: Struktur von FeCo; links : ungeord-net; rechts : geordnet.
2.7.4 Symmetriebedingte Auslöschung
Man kann diesen Effekt z.B. in der Verbindung FeCodirekt beobachten: Die Intensität des 100 Reflexesist proportional zu ( fA � fB)2, wobei fA und fB dieAtomformfaktoren der Atome auf den Gitterplätzen(000) und ( 1
212
12 ) beschrieben. Im reinen Eisen oder
Kobalt verschwindet er deshalb (A = B). In der Ver-bindung FeCo sind die Ecken der Einheitszelle durchFe, das Zentrum durch Co besetzt (resp. umgekehrt,je nach Wahl der Einheitszelle). Dann sind die bei-den Formfaktoren leicht unterschiedlich und der Re-flex tritt auf. Die Zahl der Elektronen ist allerdingsrelativ ähnlich für die beiden Atome (Z(Fe) = 26,Z(Co) = 27), so dass diese Reflexe relativ schwachsind.
FeCo
Abbildung 2.93: Beugungsreflexe an FeCo.
Die Verbindung tritt jedoch auch in einer ungeordne-ten Struktur auf, in der jeder Gitterplatz im Schnittgleich häufig von Fe und Co besetzt ist. In diesemFall gilt im Schnitt wiederum fA = fB und der Re-flex verschwindet wieder, wie im unteren Teil vonAbb. 2.93 gezeigt. In diesem Fall wurde die Beugung
von Neutronen gemessen. Die Intensität der Reflexeist deshalb nicht proportional zur Elektronendichte,sondern zum Streuquerschnitt der Kerne.
Abbildung 2.94: Struktur von NaCl, KCl und KBr.
Man kann den Effekt auch an den beiden Substan-zen KBr und KCl beobachten. In beiden Substan-zen bilden die Kationen und die Anionen jeweils einkubisch flächenzentriertes Gitter, welche gegenein-ander um eine halbe Kantenlänge verschoben sind.Unterscheidet man nicht zwischen den Atomen er-hält man somit ein kubisch primitives Gitter mit derhalben Kantenlänge.
Streuwinkel 2θ80o60o40o20o
KCl
KBr
Streuwinkel 2θ80o60o40o20o
(200)
(220)
(222) (400) (420)
(200)
(220)
(222) (400) (420)(111)(311)
(331)
Abbildung 2.95: Vergleich der Beugungsreflexe vonKCl und KBr.
Im Fall von KCl besitzen K+ und Cl� jeweils 18Elektronen. Dadurch sind die Elektronendichten derbeiden Ionen fast gleich, so dass auch die Atom-
53

2 Symmetrie und Struktur
formfaktoren praktisch gleich sind und Auslöschungstattfindet. Man findet deshalb praktisch nur Reflexemit einer geraden Summe der Indizes. Brom hat ei-ne doppelt so große Zahl von Elektronen (Br : 36), sodass hier die beiden Atomarten deutlich unterschied-lich zum gestreuten Signal beitragen. Die (genäher-te) Symmetrie entfällt und man beobachtet auch un-geradzahlige Reflexe.
Abbildung 2.96: Einheitszelle von Kochsalz (NaCl).
Ähnliche Auslöschungen gibt es auch bei der Struk-tur von Kochsalz (NaCl). Abb. 2.96 zeigt die Ein-heitszelle, welche eine fcc-Struktur aufweist und da-mit 4 Atome von jeder Sorte enthält. Die Positionensind (0,0,0), (1/2, 1/2,0), (1/2,0, 1/2) und (0, 1/2, 1/2),die vier Cl-Atome sind jeweils um eine halbe Gitter-konstante versetzt.
Für die Na-Atome ergeben sich somit die Phasen
e0 + e�ip(v1+v2) + e�ip(v1+v3) + e�ip(v2+v3).
Die einzelnen Summanden sind jeweils ±1 für ge-rade / ungerade Summen. Sind alle drei Indizes ge-rade oder alle 3 ungerade, so sind alle Summanden= +1, die Summe also +4. Sind die Indizes ge-mischt (sowohl gerade wie ungerade), so verschwin-det die Summe.
Für die Beiträge der Cl-Atome gilt das gleiche, dochenthalten dort die Summanden wegen der Verschie-bung um a/2 einen zusätzlichen Faktor eip = �1.Die Summe aus den Beiträgen von Na und Cl ist so-mit
04 fNa +4 fCl4 fNa �4 fCl
9=
; furhkl
8<
:
gemischtallegerade
alleungerade.
2.7.5 Atomformfaktor
Röntgenstrahl
Phasendifferenz
2θ
Abbildung 2.97: Phasenverschiebung zwischen denTeilstrahlen.
Da die Wellenlänge der Röntgenstrahlung vergleich-bar ist mit der Ausdehnung eines Atoms, erhaltenBeiträge zur Streuamplitude aus unterschiedlichenBereichen der Elektronenhülle unterschiedliche Pha-senverschiebungen, wie in Abb. 2.97 gezeigt. Diesüberlagern sich im Atomformfaktor.
Die Berechnung des Atomformfaktors für ein Atommit kugelsymmetrischer Elektronendichteverteilungkann vereinfacht werden, wenn man Kugelkoordina-ten ~
r = (r,q ,j) einführt. Wir wählen ~G entlang derz-Achse. Damit wird (2.10) zu
f j =Z
dr r2 sinq dq dj n j(r)e�iGr cosq
= 2p
Zdr r2d(cosq)n j(r)e�iGr cosq .
Integration über cosq gibt
f j = 2p
Zdr r2 n j(r)
eiGr � e�iGr
iGr
= 4p
Zdr n j(r)r2 sin(Gr)
Gr.
Vorwärtsstreuung Rückwärtsstreuung
~k
~k�~G = �~k
~k~k�
~G = �~k
Abbildung 2.98: Streuvektoren bei Vorwärts- undRückwärtsstreuung.
Für kleine Streuvektoren, G ! 0, kann sin(Gr)/(Gr)über den Bereich des Atoms (r < 10�10m) nähe-rungsweise durch eins ersetzt werden. Damit redu-ziert sich das Integral auf die Anzahl der Elektronen.
54
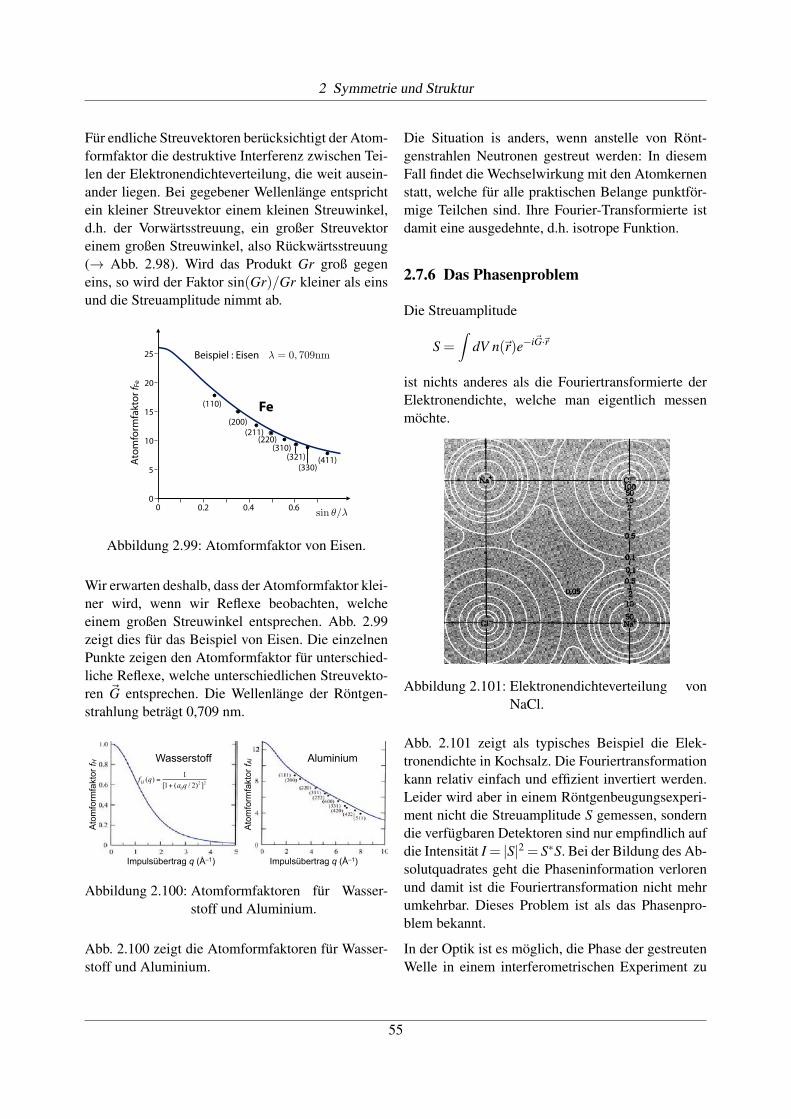
2 Symmetrie und Struktur
Für endliche Streuvektoren berücksichtigt der Atom-formfaktor die destruktive Interferenz zwischen Tei-len der Elektronendichteverteilung, die weit ausein-ander liegen. Bei gegebener Wellenlänge entsprichtein kleiner Streuvektor einem kleinen Streuwinkel,d.h. der Vorwärtsstreuung, ein großer Streuvektoreinem großen Streuwinkel, also Rückwärtsstreuung(! Abb. 2.98). Wird das Produkt Gr groß gegeneins, so wird der Faktor sin(Gr)/Gr kleiner als einsund die Streuamplitude nimmt ab.
Beispiel : Eisen
Fe(110)
(200)(211)
(220)(310)
(321)(330)
(411)
sin �/�0 0.2 0.4 0.6
0
5
10
15
20
25
Atom
form
fakt
or f F
e
Abbildung 2.99: Atomformfaktor von Eisen.
Wir erwarten deshalb, dass der Atomformfaktor klei-ner wird, wenn wir Reflexe beobachten, welcheeinem großen Streuwinkel entsprechen. Abb. 2.99zeigt dies für das Beispiel von Eisen. Die einzelnenPunkte zeigen den Atomformfaktor für unterschied-liche Reflexe, welche unterschiedlichen Streuvekto-ren ~G entsprechen. Die Wellenlänge der Röntgen-strahlung beträgt 0,709 nm.
2 20
1( )[1 ( / 2) ]
=+Hf qa q
Wasserstoff
Impulsübertrag q (Å–1) Impulsübertrag q (Å–1)
Ato
mfo
rmfa
ktor
f H
Ato
mfo
rmfa
ktor
f Al Aluminium
Abbildung 2.100: Atomformfaktoren für Wasser-stoff und Aluminium.
Abb. 2.100 zeigt die Atomformfaktoren für Wasser-stoff und Aluminium.
Die Situation is anders, wenn anstelle von Rönt-genstrahlen Neutronen gestreut werden: In diesemFall findet die Wechselwirkung mit den Atomkernenstatt, welche für alle praktischen Belange punktför-mige Teilchen sind. Ihre Fourier-Transformierte istdamit eine ausgedehnte, d.h. isotrope Funktion.
2.7.6 Das Phasenproblem
Die Streuamplitude
S =Z
dV n(~r)e�i~G·~r
ist nichts anderes als die Fouriertransformierte derElektronendichte, welche man eigentlich messenmöchte.
Abbildung 2.101: Elektronendichteverteilung vonNaCl.
Abb. 2.101 zeigt als typisches Beispiel die Elek-tronendichte in Kochsalz. Die Fouriertransformationkann relativ einfach und effizient invertiert werden.Leider wird aber in einem Röntgenbeugungsexperi-ment nicht die Streuamplitude S gemessen, sonderndie verfügbaren Detektoren sind nur empfindlich aufdie Intensität I = |S|2 = S⇤S. Bei der Bildung des Ab-solutquadrates geht die Phaseninformation verlorenund damit ist die Fouriertransformation nicht mehrumkehrbar. Dieses Problem ist als das Phasenpro-blem bekannt.
In der Optik ist es möglich, die Phase der gestreutenWelle in einem interferometrischen Experiment zu
55
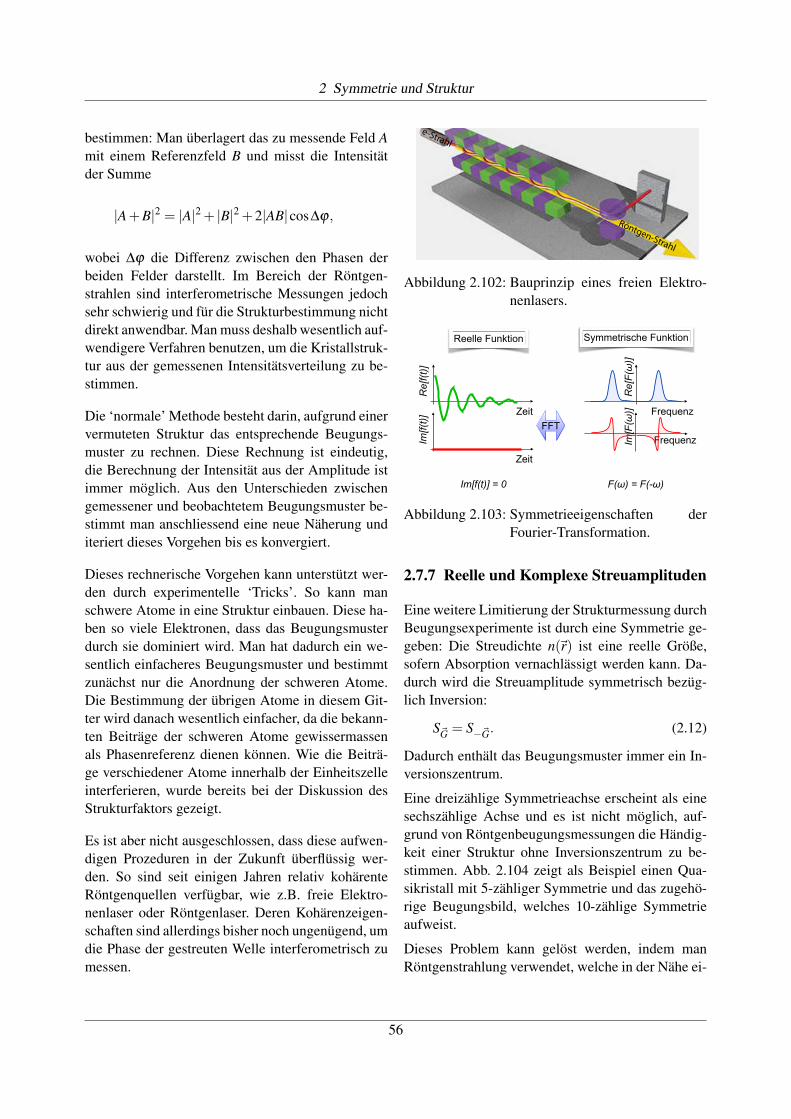
2 Symmetrie und Struktur
bestimmen: Man überlagert das zu messende Feld Amit einem Referenzfeld B und misst die Intensitätder Summe
|A+B|2 = |A|2 + |B|2 +2|AB|cosDj,
wobei Dj die Differenz zwischen den Phasen derbeiden Felder darstellt. Im Bereich der Röntgen-strahlen sind interferometrische Messungen jedochsehr schwierig und für die Strukturbestimmung nichtdirekt anwendbar. Man muss deshalb wesentlich auf-wendigere Verfahren benutzen, um die Kristallstruk-tur aus der gemessenen Intensitätsverteilung zu be-stimmen.
Die ‘normale’ Methode besteht darin, aufgrund einervermuteten Struktur das entsprechende Beugungs-muster zu rechnen. Diese Rechnung ist eindeutig,die Berechnung der Intensität aus der Amplitude istimmer möglich. Aus den Unterschieden zwischengemessener und beobachtetem Beugungsmuster be-stimmt man anschliessend eine neue Näherung unditeriert dieses Vorgehen bis es konvergiert.
Dieses rechnerische Vorgehen kann unterstützt wer-den durch experimentelle ‘Tricks’. So kann manschwere Atome in eine Struktur einbauen. Diese ha-ben so viele Elektronen, dass das Beugungsmusterdurch sie dominiert wird. Man hat dadurch ein we-sentlich einfacheres Beugungsmuster und bestimmtzunächst nur die Anordnung der schweren Atome.Die Bestimmung der übrigen Atome in diesem Git-ter wird danach wesentlich einfacher, da die bekann-ten Beiträge der schweren Atome gewissermassenals Phasenreferenz dienen können. Wie die Beiträ-ge verschiedener Atome innerhalb der Einheitszelleinterferieren, wurde bereits bei der Diskussion desStrukturfaktors gezeigt.
Es ist aber nicht ausgeschlossen, dass diese aufwen-digen Prozeduren in der Zukunft überflüssig wer-den. So sind seit einigen Jahren relativ kohärenteRöntgenquellen verfügbar, wie z.B. freie Elektro-nenlaser oder Röntgenlaser. Deren Kohärenzeigen-schaften sind allerdings bisher noch ungenügend, umdie Phase der gestreuten Welle interferometrisch zumessen.
e-Strahl
Röntgen-Strahl
Abbildung 2.102: Bauprinzip eines freien Elektro-nenlasers.
Reelle Funktion
ZeitR
e[f(t
)]
Zeit
Im[f(
t)] FFT
Im[f(t)] = 0
Frequenz
Re[
F(ω
)]
FrequenzIm[F
(ω)]
Symmetrische Funktion
F(ω) = F(-ω)
Abbildung 2.103: Symmetrieeigenschaften derFourier-Transformation.
2.7.7 Reelle und Komplexe Streuamplituden
Eine weitere Limitierung der Strukturmessung durchBeugungsexperimente ist durch eine Symmetrie ge-geben: Die Streudichte n(~r) ist eine reelle Größe,sofern Absorption vernachlässigt werden kann. Da-durch wird die Streuamplitude symmetrisch bezüg-lich Inversion:
S~G = S�~G. (2.12)
Dadurch enthält das Beugungsmuster immer ein In-versionszentrum.
Eine dreizählige Symmetrieachse erscheint als einesechszählige Achse und es ist nicht möglich, auf-grund von Röntgenbeugungsmessungen die Händig-keit einer Struktur ohne Inversionszentrum zu be-stimmen. Abb. 2.104 zeigt als Beispiel einen Qua-sikristall mit 5-zähliger Symmetrie und das zugehö-rige Beugungsbild, welches 10-zählige Symmetrieaufweist.
Dieses Problem kann gelöst werden, indem manRöntgenstrahlung verwendet, welche in der Nähe ei-
56

2 Symmetrie und Struktur
Abbildung 2.104: Quasikristall mit 5-zähliger Sym-metrie (links) und das zugehöri-ge Beugungsbild mit 10-zähligerSymmetrie (rechts).
ner Absorptionskante liegt. In diesem Fall wird einTeil der Röntgenstrahlung absorbiert und die Streu-amplitude wird dadurch komplex. Damit wird dieSymmetrie (2.12) gebrochen und das Vorzeichenkann bestimmt werden. Allerdings wird dadurch dieAnalyse des Beugungsmusters deutlich aufwändi-ger.
2.7.8 Thermische Bewegung
Bisher sind wir davon ausgegangen, dass die Ato-me perfekt auf bestimmten Gitterplätzen liegen. InWirklichkeit führen sie aber thermische Bewegun-gen um diese Gitterplätze aus, und sogar am abso-luten Nullpunkt besteht eine gewisse Ortsunschärfe.Interessanterweise führt diese Bewegung nicht zu ei-ner Verbreiterung der Reflexe. Sie führt aber zu einerReduktion der Intensität der Beugungsreflexe, da einTeil der einfallenden Strahlung inelastisch gestreutwird. Diese erscheint als diffuser Untergrund zwi-schen den Reflexen.
Um die Reduktion der Intensität zu berechnen,schreibt man die Position eines Atoms als
~r(t) =~r j +~u(t),
wobei~r j die Ruhelage darstellt und~u(t) eine Zufalls-bewegung um die Ruhelage (d.h. h~u(t)i = 0. Wennwir dies in die Definition (2.11) des Strukturfaktorseinsetzen und über die Zufallsbewegung mitteln, er-
halten wir
S~G = Âj
f je�i~G·~r jhe�i~G·~u(t)i.
Wir entwickeln die Exponentialfunktion in eineTaylor-Reihe und erhalten
he�i~G·~u(t)i = 1� ih~G ·~u(t)i
�12h⇣~G ·~u(t)
⌘2i+ . . . .
Da ~G und ~u statistisch nicht korreliert sind, könnenwir die Mittelwerte einzeln ausrechnen. Damit folgtfür den linearen Term h~G ·~u(t)i = ~Gh~u(t)i. Die Aus-lenkung ~u ist so definiert, dass ihr Mittelwert ver-schwindet, h~u(t)i = 0. Der lineare Term in der Tay-lorreihe verschwindet deshalb.
Für die Mittelung des quadratischen Terms setzenwir
h⇣~G ·~u(t)
⌘2i = G2hu2 cos2
b i = G2hu2ihcos2b i,
wobei u = |~u| und b den Winkel zwischen ~G und ~udarstellt und somit ebenfalls eine Zufallsgröße ist.
Bei der Mittelung über den Winkelanteil muss be-rücksichtig werden, dass nicht alle Werte gleichwahrscheinlich sind, sondern mit sinb gewichtetwerden müssen. Die Mittelung des Winkelanteilsüber alle möglichen Orientierungen ergibt deshalb
hcos2b i =
12
Zp
0db cos2
b sinb
=12
✓�1
3
◆cos3
b |p0 =13.
Damit erhalten wir
he�i~G·~u(t)i = 1� 16
G2h~u2i.
Wir betrachten dies als die ersten beiden Terme einerTaylor-Reihe, so dass wir den Strukturfaktor schrei-ben können als
S = S0e� G2hu2i6 .
57

2 Symmetrie und Struktur
Hier stellt S0 den Strukturfaktor für statische Ato-me dar. Gemessen wird allerdings die Streuintensität(d.h. das Quadrat der Amplitude)
I = I0e� G2hu2i3 . (2.13)
hu2i stellt hier die mittlere quadratische Verschie-bung des Atoms dar. Diese kann in erster Liniedurch thermische Anregung zustande kommen, aberauch durch die quantenmechanische Unschärfe imSchwingungs-Grundzustand.
2.7.9 Debye-Waller Faktor
Wir betrachten zunächst den Fall der thermischenAnregung. Dafür beschreiben wir die Bewegung desAtoms als harmonischen Oszillator mit der Frequenzw . Dafür können wir die mittlere quadratische Ver-schiebung aus der mittleren Energie berechnen, wel-che in drei Dimensionen 3kBT beträgt. Die mittle-re kinetische Energie Mhv2i/2 = Mhu2iw2/2 unddie mittlere potenzielle Energie Chu2i/2 betragen imMittel jeweils die Hälfte der thermischen Energie,
12
Chu2i =12
Mw
2hu2i =32
kBT
oder
hu2ith =3kBTMw
2 .
Dabei ist M die Masse des Atoms und C eine Kraft-konstante. Damit wird die Streuintensität (2.13)
I = I0e� G2kBTMw
2 .
Diese Reduktion der Intensität mit steigender Tem-peratur und Streuvektor wird als Debye-Waller Fak-tor bezeichnet. Es handelt sich hier um eine klas-sische Näherung, welche bei hohen Temperaturenrecht gut ist. Offenbar ist die Abnahme dann amkleinsten, wenn die Masse der Atome groß ist (d.h.für schwere Kerne) und wenn die Frequenz hoch ist(d.h. das Gitter starr ist). Der Effekt nimmt außer-dem mit dem Betrag des Streuvektors G zu, wie inAbb. 2.105 gezeigt.
Temperatur / K
Inte
nsitä
t / w
illk.
Ein
heite
n
(200)
(400)
(600)
(800)
(10,00)
100 200 3001
2
4
3
5
678
Abbildung 2.105: Temperaturabhängigkeit des De-bye-Waller Faktors von Alumini-um.
Bei niedrigen Temperaturen muss auch die Ortsun-schärfe aufgrund der Unschärfenrelation berücksich-tigt werden. Wir bestimmen sie über die Nullpunkts-Energie des harmonischen Oszillators. In drei Di-mensionen beträgt diese 3hw/2. Wir teilen sie wie-der zwischen kinetischer und potenzieller Energieauf, so dass
12
Mw
2hu2i =34
hw ! hu2iQM =3h
2Mw
und damit für die Intensität
I = I0e�hG2/2Mw .
Typische Zahlenwerte sind G = 1011m�1, M =10�25kg (entspricht etwa Nickel), w = 1014s�1. Un-ter diesen Bedingungen werden am absoluten Null-punkt rund 90% der maximalen Streuintensität er-reicht.
58





























