Embed Size (px)
Citation preview

This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
Thermolyse und Photolyse von cyclischen Diazoverbindungen [1]T herm olysis and Photolysis o f Cyclic D iazo C om pounds [1]
H an s-D ie trich S tachel*, H erm an n P oschenrieder, Ju tta R edlinInstitut für Pharmazie und Lebensmittelchemie der Universität München, Sophienstraße 10, D-80333 MünchenZ. Naturforsch. 51b, 1325-1333 (1996); eingegangen am 9. Mai 1996Diazoketones, Reductones, 2-Oxetanones, 2-Thietanones, 2-Azetidinones
The rhodium-catalyzed decomposition of the diazoketones 4 in teAY-butyl alcohol at 130 °C furnishes the monoenolethers 5 and, after deprotection, the ac/-reductones 6. In absence of intercepting agents the intermediate carbenes preferentially undergo Wolff rearrangement with ring contraction. In this case the /J-thiolactone 10b or the /J-lactone 13 or the /Mactam 14 are thermolysis products of the corresponding diazoketones. During photolysis of the diazoketones 4d/4g in presence of alcohols the 2-azetidinones lOc/d are formed.
Einleitung
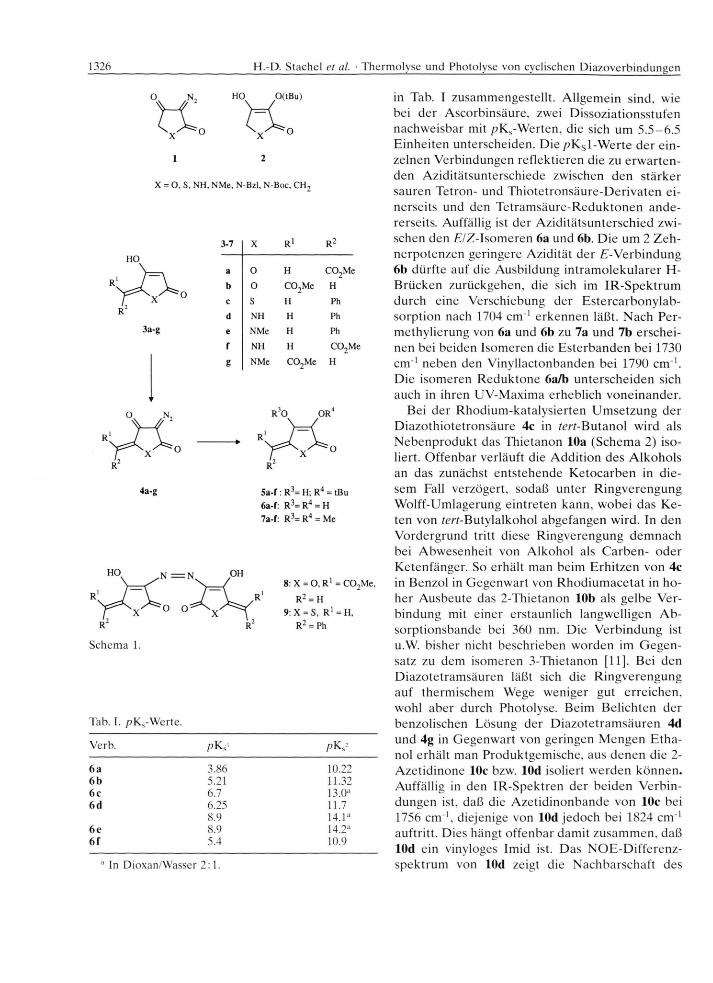
H eterocyclische A n alo g a d er R ed u k tin säu re [2] s te llen e ine in te re ssa n te Substanzklasse dar, un te r an d e rem als T e ils tru k tu ren von N atu rsto ffen wie A sco rb in säu re o d e r C h lo ro th ric in [3]. W ir haben kürzlich ü b e r d ie H erste llung von R ed u k to n en [4] aus cyclischen D iaz o k e to n en 1 (Schem a 1) berich te t [1, 5]. D er V orteil d ieser M ethode liegt darin , d aß m an leicht M o n o e th e r d er R ed u k to n e wie z.B. d ie te rf-B u ty le th e r 2 gew innen kann. D era rt geschü tzte R e d u k to n e haben w ir als A usgangsverb ind u n g en bei d e r Synthese von C arba- und H e te ro an a lo g en d e r A sco rb in säu re [6 -8 ] verw endet. Im Z u sam m en h a n g m it d iesen U n tersuchungen w aren w ir d a ran in te ressie rt, verg le ichend aus den D iaz o te tro n -, D iazo te tram - und D iazo th io te tron - säu ren 4 die en tsp re ch e n d en R ed u k to n e m it e iner A lk y lid e n se iten k e tte 6 herzustellen .
Ergebnisse und Diskussion
D ie D iaz o te tra m sä u re n 4d/e sind bere its früher von uns b esch rieb en w orden . A usgangsverb indungen w aren d ie T e tram säu ren 3d/e [5], D ie D iazo- g ru p p e n ü b e rtra g u n g au f die C H -aziden V erb in dungen erfo lg te in d e r üb lichen W eise m it Tosyl- o d e r M esylazid in G eg en w art von Basen. In d e rse lben W eise e rh ä lt m an die D iazo k eto n e 4f/g aus den T e tra m säu re n 3f7g. D ie M ethode versag t aber
* Sonderdruckanforderungen an Prof. Dr. H.-D. Stachel.
infolge von N e b e n re a k tio n e n bei den s tä rk e r azi- den T etron- und T h io te tro n säu ren 3a-c. E ine d ie ser N eb e n re ak tio n en ist die rasche U m setzung des sich b ildenden D iazo k e to n s m it restlichem Tetro- n a t bzw. T h io te tro n a t zu den s ta rk g efä rb ten Salzen von A zoverb in d u n g en . Bei d e r U m setzung von 3b bzw. 3c m it Tosylazid w urden in e rh e b licher M enge d ie V erb indungen 8 und 9 iso liert. Als D iazo tran sfer-R eag en z zu r H erste llu n g d e r D iazo- te tro n - und D iaz o th io te tro n sä u re n 4a-c eignet sich jed o ch 2 -A zido -3 -e thy lbenzth iazo lium -te tra fluo - ro b o ra t, da h ie rb e i keine B ase b en ö tig t w ird [9]. D ie ch a rak te ris tisch en IR -B an d e n d e r n euen D ia zo k e to n e 4 liegen zw ischen 2200 und 2130 c m '1, die V iny llac tonbanden von 4a/b bei 1810 c m 1.
Z u r R h o d iu m -k a ta ly sie rten Z e rse tzu n g d e r D ia zo k e to n e 4a-f w urden die D iazo v erb in d u n g en in rm -B u ta n o l im D ruckgefäß au f 130 °C erh itzt. In allen F ällen erfo lg te g la tte U m se tzu n g u n te r N 2- A b sp altu n g zu den teils ö ligen, te ils gut k rista llis ie ren d en te rf-B u ty le thern 5a-f. D iese w urden , z.T. oh n e vorherige R ein igung , m it T rifluoressigsäure zu den R e d u k to n e n 6a-f hydro lysiert. D ie V erb in dungen 6c, d w urden von uns b e re its au f anderem W ege e rh a lten und durch M eth y lie ru n g derivati- sie rt [10]. A uch die n eu en R e d u k to n e reag ieren m it D iazo m eth an u n te r B ildung d e r m eist k rista llinen D im e th y le th e r 7.
D ie R e d u k to n e liegen nach A ussage d er 'H - N M R -S p ek tren vollständig als E n d io le vor und en tfä rb en e rw artu n g sg em äß T illm ans R eagenz. M it L auge b ilden sich in tensiv gelb bzw. ro t gefärb te Salze. D ie p K s-W erte d e r R e d u k to n e 6 sind
0 9 3 2 -0 7 7 6 /9 6 /0 9 0 0 -1 3 2 5 $06.00 © 1996 Verlag der Zeitschrift für Naturforschung. A ll rights reserved. D
1326 H .-D. Stachel et al. • Therm olyse und Photolyse von cyclischen D iazoverbindungen
HO O(tBu)
X = O, S, NH, NMe, N-Bzl, N-Boc, CH2
HO
3a-g
w 2
V s > * <
4a-g
3-7 X R1 R2
a 0 H C02Meb o C 02Me Hc s H Phd NH H Phe NMe H Phf NH H c o 2m<
g NMe C02Me H
R30 OR4
r ‘ J r \'XR x = ^ 0
5a-f6a-f:7a-f:
r3= H; R4 = tBu r3= R4 = H r3= r 4 = Me
8: X = O, R1 = C 02Me,
R2 = H 9: X = S, R1 = H,
R2 = Ph
Schema 1.
Tab. I. pK s-Werte.
Verb. pK s> pKs2
6a 3.86 10.226b 5.21 11.326c 6.7 13.0a6d 6.25 11.7
8.9 14.l a6e 8.9 14.2a6f 5.4 10.9
ln Dioxan/Wasser 2:1.
in Tab. I zusam m engestellt. A llgem ein sind, wie bei d e r A scorb insäure , zwei D issozia tionsstu fen nachw eisbar m it /?KS-W erten , die sich um 5 .5 -6 .5 E in h e iten un terscheiden . D ie /?KS1-W erte d e r e in ze lnen V erb indungen re flek tie ren die zu e rw a rte n den A zid itä tsun tersch iede zw ischen d en s tä rk e r sau ren T etron- und T h io te tro n säu re -D eriv a ten e inerse its und den T e tram säu re -R ed u k to n e n a n d e rerseits. A uffällig ist d er A zid itä tsu n te rsch ied zw ischen den £7Z -Isom eren 6a und 6b. D ie um 2 Z e h n erp o te n ze n geringere A zid itä t d e r E -V erb indung 6b dü rfte auf die A usbildung in tra m o le k u la re r H- B rücken zurückgehen , die sich im IR -S p e k tru m durch eine V ersch iebung d er E ste rca rb o n y lab - so rp tio n nach 1704 c m '1 e rk e n n en läß t. N ach P e rm ethy lie rung von 6a und 6b zu 7a und 7b ersc h e in en bei beiden Isom eren die E s te rb a n d e n bei 1730 c m '1 neben den V iny llactonbanden bei 1790 c m '1. D ie isom eren R e d u k to n e 6a/b u n te rsc h e id en sich auch in ih ren U V -M axim a erheb lich v o n ein an d er.
Bei d e r R h o d ium -ka ta ly sie rten U m se tzu n g d er D iaz o th io te tro n sä u re 4c in te rt-B u tano l w ird als N eb e n p ro d u k t das T h ie tan o n 10a (S chem a 2) isoliert. O ffen b ar verläu ft die A d d itio n des A lk o h o ls an das zunächst en ts te h en d e K e to c a rb en in d ie sem Fall verzögert, sodaß u n te r R ingverengung W olff-U m lagerung e in tre te n kann , w obei das Ke- ten von terf-B utylalkohol abgefangen w ird. In den V o rderg rund tritt d iese R ingverengung dem nach bei A bw esenheit von A lkoho l als C a rb e n - o d e r K etenfänger. So erh ä lt m an beim E rh itz e n von 4c in B enzol in G egenw art von R h o d iu m ac e ta t in h o h e r A usbeu te das 2 -T h ietanon 10b als gelbe V erb indung m it e iner e rstaun lich langw elligen A b so rp tio n sb an d e bei 360 nm. D ie V erb in d u n g istu.W. b isher nicht besch rieben w orden im G e g e n satz zu dem isom eren 3 -T h ietanon [11]. B ei den D iazo te tram säu ren läßt sich die R ingverengung au f therm ischem W ege w eniger gu t e rre ich en , w ohl ab e r durch Photolyse. B eim B elich ten d er benzo lischen L ösung d er D iaz o te tra m sä u re n 4d und 4g in G egenw art von geringen M engen E th a nol e rh ä lt m an P roduk tgem ische, aus d en e n die 2- A ze tid in o n e 10c bzw. lOd iso liert w erd en k ö n n en . A uffällig in den IR -S p ek tren d e r b e id en V erb in dungen ist, daß die A ze tid in o n b an d e von 10c bei 1756 c m '1, d iejenige von lOd jedoch bei 1824 c m '1 au ftritt. D ies hängt o ffen b ar dam it zu sam m en , daß lOd ein vinyloges Im id ist. D as N O E -D iffe ren z- sp ek tru m von lOd zeigt die N ach b arsch a ft des

H .-D . Stachel et al. • Therm olyse und Photolyse von cyclischen D iazoverbindungen 1327
exocyclischen V iny lp ro tons und d e r N -M ethyl- gruppe. D ie Zs-Konfiguration ist also w äh ren d d e r U m lagerung e rh a lten geb lieben. E rfo lg t das B e lich ten von 4d in überschüssigem E th a n o l bzw. terr-B utanol, so w ird das zunächst en ts te h en d e A ze tid in o n 10c um g eestert und zu den M alone- s te rn 11 bzw. 12 aufgespalten . U n te r B each tung d ieser E rfah ru n g en gelang auch d ie H erste llu n g des 2 -O xe tanon -3 -ca rboneste rs 13 sow ie des 2- A ze tid inon -3 -ca rbonesters 14 d urch P ho to lyse von D iazo te tro n säu re (1, X = O ) bzw. N -M ethy ld iazo- te tram sä u re (1, X = N M e). Insgesam t w urden bei den b isherigen P ho to lyseversuchen n u r m äßige A usb eu ten erreich t. D ies geht z.T. d a ra u f zu rück , d aß eine T auchlam pe m it einem k o n tin u ie rlich en S p ek trum von 2 5 0 -6 0 0 nm v erw en d e t w urde.
D ie zur Synthese d e r D iazo k e to n e 4f/g b en ö tig ten T etram säu ren 3f/g w urden noch n ich t beschrie-
R
R2 X
lOa-d
PhCH.
CO,R
10 X R1 R2 R3
a S H Ph C 02(tBib s H Ph Hc NH H Ph C02Etd NMe CÜ2Me H C 02Et
CO Et
CO,R
NH,
MeO,C
II: R = Et 12: R = tBu
MeO
15
13: X = 0 14: X = NMe
MeO
MeO,C-CH,
(E)-18: R = Me (Z)-18: R = Me (Z)-19: R = H
HO |R
16:R = Me 17: R = H
MeO
v 0 -MeO,C
Schem a 2.
ben. Sie k ö n n en au f fo lgendem W eg hergeste llt w erden . D urch U m setzung des A lk y lid en te tro n - sä u re e th e rs 15 [12] m it M ethylam in en ts te h t un te r L ac to n sp a ltu n g das H ydroxylactam 16. D ie a n sch ließende D ehydra tis ie rung mit T hionylch lorid lie fert den T e tra m säu re e th e r 18 als säu len ch ro m atografisch tren n b a re s £V Z-Isom erengem isch [13]. H a u p tp ro d u k t ist das Z -Isom er. D as ’H -N M R - S p ek trum des E -Iso m eren zeigt durch einen positiven O v erh au se r-E ffek t die N achbarschaft des exocyclischen V iny lp ro tons und d er N -M ethyl- g ru p p e an. Im N M R -S pek trum von (E )-{ \S ) e r k en n t m an au ß e rd em eine A ufspaltung d e r beiden V iny lp ro tonensignale durch F ernkopp lung ü ber 5 B indungen , die m an auch zur U n tersch eid u n g der £ /Z -Iso m e re n an d e re r A lk y lid en te tram säu re - und A lk y lid e n te tro n säu red e riv a te m it e in er B u tad ien P a rtia ls tru k tu r b en u tz t ha t [12, 14].
D er T e tra m säu re e th e r (E )-18 reag iert m it Trifluo ressig säu re reg ioselek tiv u n te r E therspa ltung . M an e rh ä lt die T etram säu re 3g. D aß die K onfigura tio n w äh ren d d er E th e rsp a ltu n g u n v e rä n d e rt geb lieben ist, w ird dadu rch bew iesen, daß bei der M eth y lie ru n g von 3g m it D iazom ethan w ieder die A usgangsverb indung (E )-18 en ts teh t. Ü b e rra schenderw eise führt die E th e rsp a ltu n g des Te- tra m sä u re e th e rs (Z )-18 m it T rifluoressigsäure eben falls zu r T etram säu re 3g. D ie tre ib en d e K raft für d ie Isom erisierung ist o ffenbar die M öglichkeit zu r C h e la tis ie ru n g d er E ste rcarb o n y lg ru p p e bei dem E -Isom er. E in H inw eis h ie rau f ist dem 'H - N M R -S p ek tru m von 3g zu en tn eh m en , das volls tänd ige E no lisierung in C h lo ro fo rm anzeigt. G e w öhnlich liegen T etram säu ren in d iesem L ösungsm itte l überw iegend als K etone vor [15].
Bei d e r U m setzung von 15 mit A m m oniak wird das H ydroxy lac tam 17 gebildet. D arau s en ts teh t m it T h iony lch lo rid d e r T e tra m säu re e th e r 19 als sterisch einheitliche V erbindung. M an kann davon ausgehen , daß es sich um das Z -Iso m er handelt, das d ie M öglichkeit zu r C hela tisie rung der E ste rca rb o n y lg ru p p e hat. B ew iesen w ird die Z- K onfigura tion von 19 dadu rch , daß bei d e r N-M e- thy lierung u n te r B edingungen , u n te r d en en eine K onfig u ra tio n sän d eru n g nicht zu e rw arten ist [14], (Z )-18 en ts te h t. D as 'H -N M R -S p ek tru m von 19 zeigt d ie zu e rw arten d e F ernkopp lung zw ischen dem ringständ igem Vinyl- und dem N H -P ro to n , die nach H /D -A ustausch verschw indet [16] Die E th e rsp a ltu n g von (Z )-19 m it T rifluoressigsäure

1328 H.-D. Stachel et al. ■ Therm olyse und Photolyse von cyclischen D iazoverbindungen
verläu ft ohne Isom erisierung u n te r B ildung von 3f. D e r K onfigu ra tionserha lt kann durch O -M ethylie- rung von 3f zur A usgangsverb indung (Z )-19 m it D iazo m eth an bew iesen w erden .
Experimenteller Teil
S chm elzpunk te (unko rrig ie rt): S chm elzpunk ta p p a ra t nach D r. Tottoli (Fa. B üchi). - IR (in K B r): IR -S p e k tro m e te r In fraco rd 257, 710 B und 881 (Fa. P erk in -E lm er), B eckm an A cculab 6.- U V / V IS (in M ethano l, w enn nichts an d e res an g eg eben): U vikon 810 A nacom p 220 (Fa. K ontron A naly tik ). - 'H -N M R (60 M H z bzw. 400 M H z- S p ek tren , in n e re r S tan d a rd T etram ethylsilan): K ern re so n a n zsp e k tro m ete r A 60 A , EM 360 A (Fa. V arian), Jeol G SX 400 (Fa. Jeol). - E le m e n ta ranalysen: C H N -A nalyzer, M odell 185 (Fa. H ew le tt-P ack ard ). - D ie p K s-W erte w urden durch p o ten tio m e trisch e H albäqu iva lenzpunk tbes tim - m ung m it 0.1 N K O H in W asser, z.T. in D ioxan / W asser-M ischung (2:1), m it e in e r S tandard pH - E in s ta b m e ß k e tte (405 -60 -S 7 , Fa. Ingold) d u rc h geführt.
(Z)-Methyl-2-(2,5-dihydro-3-hy droxy -5-oxo- pyrrolyliden)-acetat (3f): M an läß t 0.36 g (2 m m ol) (Z)-19 in 3 ml T rifluoressigsäure 7 d bei R.T. s te hen. D as F lüchtige w ird en tfe rn t und d e r R ü c k stan d rek rista llis iert; b laßgelbe K ristalle, Schm p. 163 °C (aus D iiso p ro p y le th er/E th an o l), Ausb. 0.22 g (65% ). - UV: Amax (lg e) = 292 nm (4.13). - IR: v = 3235 c m 1, 2920, 1765, 1730, 1680, 1630. - 'H - N M R (CDC13): <5 = 9.30 (s, 1H ), 5.73 (s, 1H ), 3.83 (s, 3H ), 3.15 (s, 2H ). - ‘H -N M R ([D 6]D M SO ):9.30 (d, J = 1.2 H z, 1H), 5.38 (s, 1H ), 5.10 (d, J =1.2 Hz, 1H), 3.72 (s, 3H ).
C 7H 7N 0 4 (169.14)Ber. C 49.71 H 4 .1 7 N 8 .2 8 % ,Gef. C 50.08 H 4.49 N 8.22% .
(E)-Methyl-2-(2,5 -dihydro-3-hydroxy-l-methyl- 5-oxo-pyrrolyliden)-acetat (3g): D ie H erste llu n g erfo lg t analog 3f aus 0.40 g (2 m m ol) (Z)-18 m it T rifluoressigsäure; gelbliche K ristalle, Schmp. 114 °C (aus D iiso p ro p v le th er/E th an o l), Ausb. 0.22 g (61% ). - UV: Amax (lg e) = 207 nm (3.86), 299 (3.92). - IR: v = 2953 c m 1, 1738, 1687, 1621. - 'H -N M R (CDC13): d = 13.17 (s, 1H ), 5.52 (d, J =1.2 Hz, 1H), 5.34 (d, J = 1.2 H z, 1H ), 3.94, (s, 3H ),3.09 (s, 3H ).
C „ H yN 0 4 (183.17)Ber. C 52.46 H 4.95 N 7.64% ,Gef. C 52.53 H 4.91 N 7 .7 2 % .
(Z)-Methyl-2-(4-diazo-3,5-dioxo-2-oxo- lanyliden)-acetat (4a): 0.34 g (2 m m ol) 3a [17] und 0.73 g (2.5 m m ol) 2 -A zido-3-ethy lbenzth iazo lium - te tra f lu o ro b o ra t [9] w erd en in 30 ml M ethanol gerü h rt. N ach 24 h w ird das L ösungsm itte l en tfe rn t und d e r R ück stan d e rschöp fend m it D iethy le ther ex tra h ie rt. D anach rü h rt m an die L ösung 30 min m it etw as A k tivkoh le , filtriert und kristallisiert den V erd am p fu n g srü ck stan d um. Farblose K ristalle, Schm p. 109 °C (aus D iiso p ro p y le th e r/E th anol), A usb. 0.26 g (6 6 % ). - UV: X max (lg e) = 239 nm (4.29). - IR : v = 3075, 2172, 1810, 1701 c m 1. - 'H -N M R (C D C I3): ö = 6.03 (s, 1H), 3.83 (s,3H).
C 7H 4N 70 5 (196.12)Ber. C 42.87 H 2.06 N 14.28% ,Gef. C 42.98 H 2.31 N 14.18%.
(E)-Methyl-2-(4-dicizo-3,5-dioxo-2-oxo- lanyliden)-acetat (4b): D ie H erste llung erfo lg t an a log 4a aus 0.34 g (2 m m ol) 3b [12] und 0.73 g (2.5 m m ol) 2 -A zido -3-e thy lbenzth iazo lium -te tra fluo- ro b o ra t [9]. F arblose K ristalle, Schm p. 70 °C (aus D ie th y le th e r/H ex a n ), A usb. 0.23 g (58% ). - UV: l max (lg e) = 233 nm (4.23). - IR : v = 2926, 2195, 1808, 1735 c m 1. - 'H -N M R (CDC13): 6.00 (s, 1H), 3.81 (s, 3H ).
C 7H 4N 2 O s (196.12)B er. C 42.87 H 2.06 N 14.28% ,G ef. C 42.93 H 2.13 N 14.23% .
5-Benzyliden-3-diazo-2,4-thiolandion (4c): 0.20 g (2 m m ol) 3c [18] und 0.73 g (2.5 m m ol) 2-A zido-3 -e th y lb en z th iazo liu m -te tra flu o ro b o ra t [9] w erden in 50 ml M ethano l un d 0.5 ml E isessig gerührt. N ach 15 m in w ird m it W asser v e rd ü n n t und mit D ie th y le th e r ausgeschü tte lt. N ach T rocknen und E in d am p fen d er E th e rp h a se w ird säu lench rom atog raph isch gere in ig t. E lu tionsm itte l: D iethy lether. F arb lo se K ristalle , Schmp. 127 °C, Ausb. 0.3 g (65% ). - UV: Amax (lg e) = 239 nm (4.09), 276 (4.01), 302 (4.02), 335 (3.99). - IR : v = 2134, 1669, 1595 c m 1. - 'H -N M R (CDC13): 6 = 7.83 (s, 1H). 7 .5 5 -7 .4 3 (m , 5H ).
C , ,H 6N 70 , S (230.25)B er. C 57.38 H 2.63 N 12.18 S 13.93%, G ef. C 57.41 H 2.74 N 12.25 S 13.88%.
Methyl-2-(4-diazo-3,5-dioxo-2-pyrrolyliden)- acetat (4f): Z u e in e r e isgeküh lten Suspension von 0.34 g (2 m m ol) 3 f in 30 ml A ce ton itril gibt m an 0.48 g (4 m m ol) M esylazid und 0.2 g (2 m m ol) T riethy lam in in 10 ml A ce ton itril gelöst. N ach 1 h rein ig t m an den V erdam pfungsrückstand über eine K ieselgelsäule. E lu tionsm itte l: D iethy le ther/ P e tro le th e r (1:1). H ellgelbe K ristalle, Schmp. 183

H.-D. Stachel et al. • Therm olyse und Photolyse von cyclischen D iazoverbindungen 1329
°C (aus D iiso p ro p y le th e r/E th an o l). A usb. 0.1 g (51% ). - UV: Amax (lg e) = 247 nm (3.69), 285 (3.75). - IR: v - 3290 c m 1, 2145, 1747, 1707, 1673. - 'H -N M R (C D C l,): (3 = 9.18 (s, 1H), 5.84 (s, 1H), 3.80 (s, 3H ).
C 7H , N , 0 4 (195.13)Ber. C 43.09 H 2.58 N 21.53% ,Gef. C 43.05 H 2.61 N 21.52% .
Methyl-2-(4-diazo-3,5-dioxo-l -methyl-2-pyrro- lyliden)-acetat (4g): 0.18 g (1 m m ol) 3g w erden m it 0.20 g (2 m m ol) T riethy lam in und 0.20 g (1 m m ol) Tosylazid in 10 ml M ethano l 1 h bei -8 °C gerü h rt. Im E isfach scheidet sich ü b er N ach t 4g ab. F arb lose K ristalle, Schm p. 97 °C (aus M eth an o l). Ausb. 50 mg (24% ). - UV: Amax (lg e ) = 246 nm (4.62). - IR: v = 2155 c m 1, 1738, 1709, 1636. - 'H -N M R (CDC13): ö = 5.51 (s, 1H ), 3.85 (s, 3H ), 3.15 (s, 3H ).
C8H 7N , 0 4 (209.16)Ber. C 45.94 H 3.37 N 20.09% ,Gef. C 45.88 H 3.41 N 20.00% .
(E)-Methyl-2-(4-tert-butoxy-2,5-dihydro-3- hydroxy-5-oxo-2-furyliden)-acetat (5b): 0.30 g (1.5 m m ol) 4b w erden in 75 ml tro ck en em rm -B u ty la l- kohol und e iner S pate lsp itze R h 2(O A c )4 90 min im A utoklaven bei 130° erh itz t. N ach E n tfe rn u n g des L ösungsm ittels w ird d e r v e rb le ib en d e R ü c k stand ü b er eine K ieselgelsäule gerein ig t. E lu tio n sm ittel: D ie th y le th e r/P e tro le th e r (1:1; R t = 0.64). N ach E n tfe rn en des L ösungsm itte ls k rista llis iert d e r ölige R ückstand im E isbad. F arb lose K ristalle, Schmp. 60 °C, Ausb. 0.22 g (60% ). - UV: Amax (lg e) = 266 nm (4.11), 326 (3.83). - IR: v = 2928 cm ', 1803, 1699, 1650. - 'H -N M R (C D C l,): = 12.22 (s, 1H), 5.91 (s, 1H), 3.88 (s, 3H ), 1.43 (s, 9H ).
Cu H 140 6 (242.23)Ber. C 54.54 H 5.83% ,Gef. C 54.39 H 5 .9 2 % .
5-Benzyliden-3-tert-butoxy-4-hydroxy-2(5H)thio- phenon (5c): D ie H erste llung erfo lg t ana log 5b aus 0.23 g (1 m m ol) 4c. E lu tionsm itte l: C yclohexan/ D iisop ropy le ther (1:1; Rt = 0.23). Farb lose K ristalle, Schmp. 175 °C (aus A ce to n itril) , Ausb. 45 mg (16% ). - UV: Amax (lg e) = 235 nm (4.09), 330 (4.35). - IR: v = 2979 cm ' , 1663,1572. - 'H -N M R (C D C I3): ö = 7 .8 4 -7 .5 6 (m , 6 H ), 1.72 (s, 9H ).
C , ,H , öO ,S (276.36)Ber. C 65.19 H 5.84 S 11.60% ,Gef. C 65.27 H 5.87 S 11.59% .
5-Benzyliden-3-tert-butoxy-l,5-dihydro-4- hydroxy-2-pyrrolon (5d): 0.43 g (2 m m ol) 4d [5]
w erd en in 75 ml trockenem terr-B utylalkohol und e in e r S pate lsp itze R h2(O A c)4 90 m in im A u to k la ven bei 130 °C erh itz t. N ach E n tfe rn u n g des L ö sungsm itte ls w ird d e r R ückstand in D ioxan u n te r Z u sa tz von A ktivkoh le 15 min u n te r R ückfluß e r hitzt. D ie heiß filtrie rte Lösung w ird e ingedam pft und d e r R ückstand um krista llisiert. H ellgelbe K ristalle , Schmp. 175 °C (Z ers.), (aus A ce ton itril) , A usb. 0.32 g (62% ). - UV: Amax (lg e ) = 222 nm (3.98), 317 (4.44). - IR : v = 3431 c m 1, 2983, 1684, 1617. - 'H -N M R ([D 6]D M SO ): = 9.34 (s, 1H), 7 .6 4 -7 .2 3 (m, 5H ), 6.18 (s, 1H), 1,23 (s, 9H ).
C , sH , 7N O , (259.30)Ber. C 69.48 H 6.61 N 5.40% ,Gef. C 69.29 H 6.49 N 5.53% .
5-Benzyliden-3-tert-butoxy-l,5-dihydro-4- hydroxy-l-methyl-2-pyrrolon (5e): D ie H ers te llung erfo lg t ana log 5d aus 0.45 g 4e [5]. H ellgelbe K ristalle , Schmp. 153 °C (aus A ce to n itril) , Ausb. 0.35 g (65% ). - UV: Amax (lg e ) = 225 nm (3.99), 297 (4.35). - IR: v = 2985 c m 1, 1682, 1622. - 'H - N M R (C D C l,): ö = 7 .5 -7 .2 (m, 5H ), 6.60 (s, 1H), 2.80 (s, 3H ), 1.40 (s, 9H ).
C , 6H I9N 0 3 (273.33)Ber. C 70.31 H 7.00 N 5 .1 2 % ,Gef. C 70.20 H 7.05 N 5 .0 8 % .
Methyl-2-(4-tert-butoxy-2,5-dihydro-3-hydroxy- 5-oxo-2-pyrrolyliden)-acetat (5f): 0.19 g (1 m m ol) 4f w erd en in 75 ml trockenem rm -B u ty la lk o h o l und e in e r S pate lsp itze R h 2(O A c)4 2 h im A u to k la ven bei 130 °C erh itzt. N ach E n tfe rn u n g des L ösungsm itte ls w ird d e r verb le ibende R ückstand u m krista llis iert. H ellgelbe K ristalle. Schm p. 157 °C (aus A ce to n itril) , Ausb. 0.15 g (62% ). - UV: 2max (lg e ) = 289 nm (3.82). - IR: v = 3370 cm ', 2980, 2925, 1695, 1636. - 'H -N M R ([D 6]D M SO ): d =11.00 (s, 1H), 9.26 (s, 1H), 5.45 (s, 1H), 3.67 (s, 3H ), 1.28 (s, 9H ).
C 1i H I5N O s (241.24)Ber. C 54.77 H 6.27 N 5.80% ,Gef. C 54.78 H 6.30 N 5.75% .
(Z)-Methyl-2-(2,5-dihydro-3,4-dihydroxy-5-oxo- 2-furyliden)-acetat (6a): 0.3 g (1.5 m m ol) 4a w erden in 75 ml trockenem terr-B utylalkohol und e in e r S pate lsp itze R h 2(O A c)4 90 min im A utok laven bei 130 °C erh itzt. N ach E n tfern u n g des L ösungsm itte ls w ird d er ölige R ückstand m it 2 ml eisgek ü h lte r T rifluoressigsäure versetzt. Beim R ühren b ild e t sich bere its nach ku rzer Z eit ein N ied e rschlag, d e r nach V erdünnen m it D ie th y le th e r a b gesaug t wird. G elbe K ristalle, Schmp. 174 °C, Ausb. 0.15 g (53% ). - UV: Amax (lg e) = 218 nm

1330 H.-D. Stachel er ol. - T herm olyse und Photolyse von cyclischen D iazoverbindungen
(3.89), 260 (4.03), 322 (3.87). - IR: v = 3310 c m 1, 1822, 1798, 1677, 1620. - ’H -N M R ([D 6]D M SO ): d = 11.08 (s, 1H), 10.43 (s, 1H), 5.52 (s, 1H), 3.67 (s, 3H ).
C 7H 6N 0 6 (186.12)Ber. C 45.17 H 3 .2 5 % ,Gef. C 45.15 H 3 .2 7 % .
(E)-Methyl-2-(2,5-d ihy dro-3,4-d i hydroxy -5-oxo- 2-furyliden)-acetat (6b): Die H erste llung erfo lg t ana log 6a aus 0.3 g (1.5 m m ol) 4b. H ellgelbe K ristalle, Schm p. 200 °C (Zers.), Ausb. 0.17 g (60% ). - UV: Amax (lg e ) = 266 nm (3.79), 346 (3.91). - IR: v = 3347 cm ’1, 1772, 1704, 1667, 1636. - 'H -N M R ([D 6]D M SO ): ö = 11.56 (s, 1H ),11.06 (s, 1H), 6.12 (s, 1H), 3.82 (s, 3H ).
C 7H 6N 0 6 (186.12)Ber. C 45.17 H 3 .2 5 % ,Gef. C 45.11 H 3.31% .
5-Benzyliden-3,4-dihydroxy-2(5H)thiophenon (6c): 0.14 g (0.5 m m ol) 5c w erden m it 2 ml eisgek ü h lte r T rifluoressigsäure gerührt. N ach v o rü b e rg eh e n d e r L ösung b ilde t sich nach ku rze r Z eit ein N iedersch lag . M an v e rd ü n n t m it D ie th y le th e r und saugt ab. G elbe K ristalle, Schmp. und S p ek tra ld a ten identisch m it Lit. [10]. Ausb. 75 m g (68% ).
5-Benzyliden-l ̂ -d ih y d ro ^ A -d ih yd ro x y ^ -p y r - rolon (6d): D ie H erste llung erfo lg t ana log 6c aus 0.13 g (0.5 m m ol) 5d. H ellgelbe K ristalle, Schmp. und S p ek tra ld a te n iden tisch m it Lit. [10]. Ausb. 70 m g (63% ).
5-Benzy liden-l ,5-dihydro-3,4-dihydroxy-l - methyl-2-pyrrolon (6e): D ie H erste llung erfo lg t ana log 6c aus 0.27 g (1 m m ol) 5e. G elbe K ristalle, Schm p. 162 °C (Z ers.), Ausb. 0.16 g (74% ). - UV: l mas (lg e) = 222 nm (3.95), 296 (3.85). - IR: v = 3252 c m 1, 1663, 1618. - 'H -N M R ([D 6]D M SO ): (3 = 7 .5 -7 .2 (m , 5H ), 6.29 (s, 1H), 5.27 (b re it, 2H ), 2,68 (s, 3H ).
C n H , , N O ? (217.22)Ber. C 66.35 H 5.10 N 6 .4 5 % ,Gef. C 66.40 H 5.17 N 6 .3 2 % .
Methyl-2-(2,5-dihydro-3,4-dihydroxy-5-oxo-2- pyrrolyliden)-acetat (6f): D ie H erste llung erfo lgt ana log 6c aus 0.12 g (0.5 m m ol) 5f. G elbe K ristalle, Schm p. 205 °C (Z ers.), Ausb. 70 mg (75% ). - UV: Imax (lg e) = 270 nm (4.04). - IR: v = 3382 c m 1, 3222, 1704, 1651. - 'H -N M R ([D 6]D M SO ): (3 = 10.28 (s, 1H), 9.65 (s, 1H), 9.16 (s, 1H), 5.33 (s, 1H), 3.35 (s, 3H ).
C 7H 7N O , (185.13)Ber. C 45.41 H 3.81 N 7.57% ,Gef. C 45.48 H 3.95 N 7 .2 5 % .
(Z)-Methyl-2-(2,5-dihydro-3,4-dimethoxy-5-oxo- 2-furyliden)-acetat (7a): 93 m g (0.5 m m ol) 6a w erd en in M e th an o l gelöst und m it überschüssiger e th e risc h e r D iazo m eth an lö su n g versetzt. N ach A bk lin g en d e r G asen tw ick lung w ird das L ösungsm itte l en tfe rn t und d e r V erdam pfungsrückstand um krista llis iert. Farb lose K ristalle, Schmp. 93 °C (aus M e th an o l), Ausb. 50 m g (47% ). - UV: Amax (lg f ) = 262 nm (4.39). - IR : v = 1811 cm "', 1797, 1728, 1673, 1661. - 'H -N M R (CDC13): ö = 5.54 (s, 1H ), 4.15 (s, 3H ), 3.99 (s, 3H ), 3.78 (s, 3H ).
C 7H , 0O 6 (214.17)Ber. C 50.47 H 4 .7 1 % ,Gef. C 50.35 H 4 .8 1 % .
(E)-Methyl-2-(2,5-dihydro-3,4-dimethoxy-5-oxo- 2-furyliden)-acetat (7b): D ie H erste llu n g erfo lg t an a lo g 7a aus 93 m g (0.5 m m ol) 6b und D iazom e- th an . F arb lose K ristalle, Schm p. 60 °C, Ausb. 60 m g (56% ). - UV: Amax (lg e) = 209 nm (3.78), 260 (4.13) - IR : v = 1784 cm ', 1734, 1655. - 'H -N M R (CD C13): (3 = 5.87 (s, 1H ), 4.14 (s, 3H ), 3.99 (s, 3H ),3.76 (s, 3H ).
C 7H io0 6 (214.17)Ber. C 50.47 H 4 .7 1 % ,Gef. C 50.47 H 4.70% .
5-B enzy liden-l ,5-dihydro-3 A-dimethoxy-1 - methyl-2-pyrrolon (7 e ): D ie H erste llu n g erfolgt ana log 7a aus 0.11 g (0.5 m m ol) 6e und D iazom e- than . N ach säu len ch ro m ato g rap h isch e r R einigung (E lu tio n sm itte l: P e tro le th e r) e rh ä lt m an ein fa rb loses Öl. A usb. 50 m g (41% ). - UV: Amax (lg e) = 226 nm (3.87), 297 (4.33). - IR : v = 2948 c m '1, 1703, 1648. - 'H -N M R (CDC13): (3 = 7.30 (m, 5H ),6.40 (s, 1H ), 4.12 (s, 3 H ), 3.94 (s, 3H ), 2.78 (s, 3H ).
C I4H , ,N O , (245.28)Ber. C 68.56 H 6 .1 6 N 5 .7 1 % ,Gef. C 68.41 H 6.30 N 5 .6 1 % .
Methyl-2-(2,5-dihydro-3,4-dimethoxy-5-oxo-2- pyrrolyliden)-acetat (7f): D ie H erste llung erfolgt ana log 7a aus 92 m g (0.5 m m ol) 6f und D iazom e- than . F arb lose N adeln , Schm p. 125 °C (aus D iiso- p ro p y le th e r), A usb. 50 m g (47% ). - UV: Amax (lg e) = 286 nm (4.30). - IR : v = 3263 c m 1, 2992, 2953, 2857, 1736, 1702, 1678, 1645. - 'H -N M R (CDCI3): (3 = 8.44 (s, 1H), 5.46 (s, 1H), 4.06 (s, 3H ),3.98 (s, 3H ), 3.75 (s, 3H ).
C gH n N 0 5 (213.19)Ber. C 50.71 H 5.20 N 6.57% ,Gef. C 50.66 H 5.24 N 6.57% .
Dimethyl-4,4'-azodi[2-(2,5-dihydro-3-hydroxy- 5-oxo-2-furyliden) acetat] (8): 0.17 g (1 m m ol) 3b

H.-D. Stachel et al. • Therm olyse und Photolyse von cyclischen D iazoverbindungen 1331
[12] und 0.20 g (2 m m ol) T riethylam in w erden in10 ml M ethano l gelöst und au f -8 °C gekühlt. H ierzu tro p ft m an eine Lösung von 0.2 g (1 m m ol) Tosylazid in 3 ml M ethanol. N ach 1 h w ird das L ösungsm itte l en tfe rn t, der R ück stan d in w enig W asser au fgenom m en und m it 6 N H C l an g esäuert. D e r N iederschlag w ird abgesaug t und mit w enig W asser gew aschen. R o tes Pulver, Schmp. 224 °C (aus A ce ton itril) , Ausb. 0.15 g (82% ). - UV: Amax (lg e) = 225 nm (3.68), 250 (3.63), 460 (3.52). - IR : v = 2900-2400 c m 1, 1795, 1630. - 'H -N M R ([D 6]D M SO ): ö = 6.12 (s, 1H), 6.00 (s, 2H ), 3.91 (s, 6H ).
C 14Hk)N2Oio (366.24)Ber. C 45.91 H 2.75 N 7 .6 5 % ,Gef. C 45.79 H 2.83 N 7 .5 6 % .
5 ,5 ’-Dibenzyliden-4,4’-azodi[4-hydroxy-2(5H)- thiophenon] (9): D ie H erste llung erfo lg t ana log 8 aus 0.20 g (1 m m ol) 3c [18], 0.20 g (2 m m ol) T riethy lam in und 0.20 g (1 m m ol) Tosylazid. Schw arzro tes Pulver, Schmp. 239 °C (aus A c e to n itril), Ausb. 0.12 g (56% ). - UV: Amax (lg e) = 235 nm (3.95), 332 (3.86), 465 (3.79). - IR: v = 2 8 0 0 - 2400 c m 1, 1672, 1591. - 'H -N M R ([D 6]D M SO ): (3 = 7 .6 5 -7 .3 9 (m, 12H).
C , M l4N 20 4S2 (434.49)Ber. C 60.82 H 3.25 N 6.45% ,Gef. C 60.89 H 3.32 N 6 .4 1 % .
tert-Butyl-2-benzyliden-4-oxo-3-thiethan- carboxylat (10a): 10a en ts teh t bei d e r H erste llung von 5c. R einigung über eine K ieselgelsäule. E lu tionsm ittel: C yclohexan /D iisop ropy le the r (1/1; Rj = 0.69), gelbe K ristalle, Schmp. 98 °C, Ausb. 60 mg (22% ). - UV: Amax (lg e) = 241 nm (4.07), 267 (3.72), 360 (4.19). - IR: v - 2974 c m '1, 1757, 1725, 1615. - ‘H -N M R (C D C l,): ö = 7 .3 8 -7 .2 9 (m . 5H ),7.19 (s, 1H), 5.24 (s, 1H), 1.44 (s, 9H ).
C , ,H ,öO ,S (276.36)Ber. C 65.19 H 5.84 S 11.60% ,Gef. C 65.14 H 5.91 S 11.66%.
4-Benzyliden-2-thietanon (10b): 0.23 g (1 m m ol) 4c w erden in 80 ml B enzol gelöst und u n te r Z usa tz von 50 mg R h 2(O A c)4 4 h u n te r R ückfluß erh itzt. N ach E n tfern u n g des L ösungsm itte ls rein ig t m an den R ückstand säu lench rom atog raph isch , E lu tionsm ittel: C yclohexan /D iisop ropy le the r (1:1; Rf = 0.64), beiges Pulver, Schm p. 110 °C, Ausb. 0.16 g (90% ). - UV: Amax (lg e) = 241 nm (3.93), 360 (4.00). - IR : v = 2925 c m 1, 1748, 1609. - 'H - N M R (C D C l,): (3 = 7 .4 4 -7 .3 4 (m , 5H ), 7.27 (s, 1H), 4.46 (s, 2H ).
C ,()H hO S (176.24)Ber. C 68.15 H 4.58 S 18.19%,Gef. C 68.10 H 4.59 S 18.05%.
Ethyl-2-benzyliden-4-oxo-3-azetidin-carboxylat (10c): 0.21 g (1 m m ol) 4d [5] w erden in 110 ml abso lu tem B enzol gelöst und m it e iner äquim ola- ren M enge E th a n o l versetzt. M an belich te t 45 min bei R a u m te m p era tu r, destilliert das B enzol ab und rein ig t du rch F lash -C hrom atog raph ie , E lu tio n sm ittel: C yclohexan / E th y lace ta t (1/1; Rf - 0.12), farb lose K ristalle , Schmp. 166 °C, Ausb. 30 mg (13% ). - UV: Amax (lg e) = 231 nm (4.26), 250 (4.21). - IR : v = 3416 c n r 1, 2985, 1761, 1694, 1652. - 'H -N M R ([D 6]D M SO ): (3 = 8.73 (s, 1H), 7 .4 0 -7 .2 4 (m , 6H ), 4.50 (s, 1H ), 4.24 (q, J = 7 Hz, 2 H ), 1.32 (t, J = 7 Hz, 3H ).
C n H n N O , (231.25)B er. C 67.52 H 5.67 N 6.06% ,Gef. C 67.48 H 5.65 N 6 .1 0 % .
Ethyl-2-methoxy carbony Imethylen-1 -methy 1-4- oxo-3-azetidin-carboxylat (lOd): D ie H erste llung erfo lg t ana log 10c aus 0.21 g (1 m m ol) 4g ( Rf = 0.2), gelbe K ristalle, Schmp. 63 °C, A usb. 40 mg (18% ). - UV: Amax (lg e ) = 250 nm (4.19). - IR: v = 2986 c n r ' , 1824, 1715, 1662. - 'H -N M R (C D C l,): (3 = 5.30 (s, 1H), 4.70 (s, 1H), 4.24 (q, J =7 Hz, 2H ), 3.69 (s, 3H ), 3.03 (s, 3H ), 1.30 (t. .1 = 1 H z, 3H ). - N O E -D iffe ren zsp ek tru m : N ach E in strah lu n g bei (3 = 3.03 S ignalverstärkung bei ö = 5.30.
C ,0H n N O 5 (227.22)Ber. C 52.86 H 5.77 N 6 .1 6 % ,Gef. C 52.81 H 5.70 N 6 .1 0 % .
Diethyl-2-(l-amino-2-phenylethyliden)-malonat(11): 0.42 g (2 m m ol) 4d w erden in 110 ml tro ck e nem B enzol su sp en d iert und auf -8 °C gekühlt. M an fügt 2 ml E th an o l dazu und b elich te t 1 h. A n sch ließ en d w ird das L ösungsm itte l en tfe rn t und d e r R ü ck stan d ü b er eine K ieselgelsäule gereinigt. E lu tionsm itte l: D iiso p ro p y le th er (Rf= 0.85). F arb loses Ö l, A usb. 0.19 mg (34% ). - UV: Amax (lg e) =278 nm (4.31). - IR: v = 3419 c m '1, 3316, 2980, 1693, 1666. - 'H -N M R (C D C l,): (3 = 8.95 (s, 1H, H /D -A ust.), 7 .3 6 -7 .2 4 (m , 5H ), 4.85 (s, 1H. H /D - A ust.), 4.29 (q, J = 7 Hz, 2H ), 4.24 (q, J = 7 Hz, 2 H ), 3.83 (s, 2H ), 1.30 (t, J = 7 Hz, 3H ), 1.27 (t, .7 = 7 H z, 3H ).
C , , H , 9N 0 4 (277.32)B er. C 64.97 H 6.91 N 5.05% ,Gef. C 64.95 H 6.96 N 4.98% .
Di-tert-butyl-2-(l -amino-2-phenylethyliden)- malonat (12): D ie H erste llung erfo lg t analog 11

1332 H.-D. Stachel et al. • Therm olyse und Photolyse von cyclischen D iazoverbindungen
aus 0.42 g (2 mmol) 4d mit 2 ml terr-Butanol (R f =0.78). Farbloses Öl, Ausb. 0.17 g (25% ). - UV: Arnax (lg e) = 277 nm (4.36). - IR: v = 3471 cm '1, 3327, 2979, 1703, 1666. - 'H -N M R (CDC13): (3 = 7.27-7.24 (m, 5H), 3.72 (s, 2H), 1.55 (s, 9H), 1.49 (s, 9H).
C,yH77N 0 4 (333.43)Ber. C 68.44 H 8.16 N 4.20% ,Gef. C 68.37 H 8 . l l N 4.28% .
Ethyl-2-oxo-3-oxetan-carboxylat (13): Die H erstellung erfolgt analog 10c aus 0.13 g (1 mm ol) 3- Diazo-2,4 oxolandion [1] (Rf= 0.78). Farbloses Öl, Ausb. 30 mg (21)% . - UV: Amax (lg e) = 209 nm (3.86). - IR: v = 2986 c m 1, 1838,' 1737. - ‘H-N M R (CDC1,): r3 = 4.61 -4 .58 (m, 2H), 4.41 (t, J = 7 Hz, 1H), 4.29 (q. J = 1 Hz, 2H), 1.33 (t, J = 7 Hz, 3H).
C6H80 4 (144.13)Ber. C 50.00 H 5.60%,Gef. C 49.91 H 5.54%.
Ethyl-1 -methyl-2-oxo-3-azetidin-carboxylat (14): Die H erstellung erfolgt analog 10c aus 0.14 g (1 mmol) 3-Diazo-l-m ethyl-2,4-pyrrolidindion [19]. Elutionsm ittel: E thylacetat (R f = 0.8). Schwach gelbliches Öl. Ausb. 45 mg (29% ).- UV: Amax (lg e) = 206 nm (4.03). - IR: v = 2919 cm '1, 1765, 1731. - 'H -N M R (CDC13): (3 = 4.24 (q, J = 7 Hz, 2H ), 4.03 (dd, 7, = 5.1 Hz, J2 = 2.6 Hz, 1H), 3.55 (dd, 7, = 5.6 Hz, 7, = 2.6 Hz, 1H), 3.36 (t, J = 5.3 Hz, 1H), 2.88 (s, 3H), 1.30 (t, J = 7 Hz, 3H).
C7H ,,N O ? (157.17)Ber. C 53.49 H 7.05 N 8.91% ,Gef. C 53.42 H 7.10 N 8.84%.
M ethyl-2-(2,5-dihydro-2-hydroxy-3-methoxy-l- methyl-5-oxo-pyrrolyl)-acetat (16): M an läßt die Lösung von 0.74 g (4 mmol) 15 [12] in M ethanol mit 2 ml einer 33-proz. wäßrigen M ethylam inlösung 30 min bei R.T. stehen. Danach wird das Flüchtige i. Vak. entfernt und der getrocknete Rückstand umkristallisiert; farblose Kristalle, Schmp 122 °C (aus D iisopropylether/M ethanol), Ausb. 0.71 g (82% ). - UV: Amax (lg e) = 209 nm (4.15), 247 (3.44). - IR: v = 3223 c m 1, 2960, 1736, 1672, 1638. - 'H -N M R (CDCL): <3 = 5.20 (s, 1H),5.00 (s, 1H), 3.93 (s, 3H), 3.75 (s, 3H), 2.97 (s, 2H), 2.90 (s, 3H).
CgH n N O 5 (215.21)Ber. C 50.23 H 6.09 N 6.51% ,Gef. C 50.27 H 6.01 N 6.55%.
M ethyl-2 -(2,5-d ihyd ro-2-hyd roxy-3-m eth oxy-5- oxo-pyrrolyl)-acetat (17): Die H erstellung erfolgt analog 16 aus 0.74 g (4 mmol) 15 [12] mit 3 ml
konz. Am m oniaklösung. Farblose Kristalle, Schmp. 146 °C (aus E thylacetat), Ausb. 0.72 g (89% ). - UV: Amax (lg e ) = 254 nm (3.14). - IR: v = 3375 c m 1, 3250, 3100, 1710, 1690. - 'H -N M R ([D 6]DM SO): d = 7.70 (breit, 1H), 6.30 (s, 1H),4.97 (d, J = 2 Hz, 1H, nach H/D-Austausch s), 3.80 (s, 3H ), 3.60 (s, 3H), 2.76 (s, 2H).
C8H ,,N O 5 (201.18)Ber. C 47.76 H5.51 N 6.96%,Gef. C 47.69 H 5.35 N 6.96%.
(Z)-Methyl-2-(2,5-dihydro-3-meth oxy -1-methyl-5-oxo-pyrrolyliden)-acetat (Z-18): 1. Eine Lösung von 0.43 g (2 mmol) 16 in 10 ml Chloroform wird mit 2 ml Thionylchlorid 30 min unter Rückfluß e rhitzt. D er Verdam pfungsrückstand wird um kristallisiert. Die M utterlauge enthält (Z)-18 neben (E)- 18. Die K om ponenten w erden säulenchrom atographisch getrennt; (R t = 0.78, Elutionsm ittel: D iethylether), farblose Kristalle, Schmp. 132 °C (aus E thylacetat), Ausb. 0.25 g (63%). - 2. Eine Lösung von 0.55 g (3 mmol) (Z)-19 in 20 ml D im ethylform am id wird unter Kühlung portionsweise mit 0.18 g (4 mmol) Natrium hydrid (als 50-proz. ölige Suspension) versetzt. Nach 30 min werden zur Suspension 0.42 g (3 mmol) M ethyliodid gegeben. M an läßt 30 min rühren, verdünnt mit E iswasser und saugt das R ohprodukt ab; Ausb. 0.40 g (66% ). - UV: Amax (lg e) = 277 nm (4.19). - IR: v = 3107 cm '1, 1733, 1720, 1650, 1623. - 'H -N M R (CDCU): (3 = 5.62 (s, 1H), 5.18 (s, 1H), 3.86 (s, 3H), 3.75 (s, 3H), 3.32 (s, 3H).
C9H ,,N 0 4 (197.19)Ber. C 54.81 H 5.62 N 7.10%,Gef. C 54.90 H 5.59 N 7.08%.
(E)-Methyl-2-(2,5-dihydro-3-methoxy-l-methyl-5-oxo-pyrrolyliden)-acetat (E-18): 1. (E)-18 en tsteht als N ebenprodukt bei der Herstellung von (Z)-18, M ethode 1 und wird säulenchrom atographisch gewonnen (R f = 0.50; Elutionsmittel: D ie thylether), farblose Kristalle, Schmp. 90 °C (aus D iethylether), Ausb. 60 mg (15%). - 2. 90 mg (0.5 mmol) 3g werden in M ethanol gelöst und mit überschüssiger etherischer Diazom ethanlösung versetzt. Nach Abklingen der Gasentwicklung wird das Lösungsm ittel entfernt und der Verdam pfungsrückstand umkristallisiert. Ausb. 65 mg (66% ). - UV: Amax (lg e) = 271 nm (4.10). - IR: v = 2925 c m 1, 1727, 1700, 1646, 1600. - 'H -N M R (C D C l,): <3 = 5.47 (d, V = 1.2 Hz, 1H),5.25 (d ,5/ =1.2 Hz, 1H), 3.86 (s, 3H ), 3.77 (s, 3H), 3.02 (s, 3H). - N O E-D ifferenzspektrum : Nach E instrahlung bei ö = 5.47 bzw. 3.02 Signalverstärkung bei (3 = 3.02 bzw. 5.47.

H.-D. Stachel et al. • Thermolyse und Photolyse von cyclischen D iazoverbindungen 1333
C9H //N 0 4 (197.19)Ber. C 54.81 H 5.62 N7.1()% ,Gef. C 54.71 H 5.68 N 7.04%.
(Z)-Methyl-2-(2,5-dihy dro-3-methoxy-5-oxo- pyrrolyliden)-acetat (Z-19): Die Herstellung e rfolgt analog (Z)-18, M ethode 1, aus 0.40 g (2 mmol) 17 und 2 ml Thionylchlorid; farblose Kristalle, Schmp. 137 °C (aus E thylacetat), Ausb. 0.30
g (81% ). - UV: Amax (lg e) = 279 nm (4.28). - IR: v = 3340 c m 1, 3120, 2950, 1720. 1690. - 'H -N M R (CDC10: ö = 8.54 (breit. 1H), 5.53 (s, 1H), 5.21 (d. J = 1.5 Hz, 1H, nach H/D-Austausch s), 3.89 (s, 3H), 3.77 (s, 3H).
CsH gN 0 4 (183.17)Ber. C 52.46 H 4.95 N 7.64%,Gef. C 52.46 H 4.91 N 7.68%.
[1] Reduktone von Tetron-, Thiotetron- und Tetramsäuren. III; Teil II: H.-D. Stachel. H. Poschenrieder, J. Redlin. J. Schachtner, K. Zeitler, Liebigs Ann. Chem. 1994, 129-132.
[2] T. Reichstein. R. Oppenauer, Helv. Chim. Acta 16, 988-998 (1933); G. Hesse, E Bayer, P. Thieme, Chem. Ber. 99, 1810-1814 (1966).
[3] R. E. Ireland, W. J. Thompson, J. Org. Chem. 44, 3041-3052 (1979).
[4] K. Schank, Synthesis 1972, 176-190.[5] H. Poschenrieder, H.-D. Stachel, Arch. Pharm.
(Weinheim. Ger) 322, 301-302 (1989).[6] J. Schachtner, H.-D. Stachel, Tetrahedron 51, 9005-
9014 (1995).[7] H.-D. Stachel, J. Schachtner, Tetrahedron 49, 4871 -
4880 (1993).[8] H.-D. Stachel, K. Zeitler, H. Lotter, Liebigs Ann.
Chem. 1994, 1129-1134.H.-D. Stachel, K. Zeitler, Liebigs Ann. Chem. 1996,
103-107.[9] H. Balli, F. Kersing, Liebigs Ann. Chem. 647, 1-10
(1961).[10] H.-D. Stachel, H. Poschenrieder , H. Burghard, Z.
Naturforsch. 35b, 724-726 (1980).
11] A. J. Krubsack, T. Higa, W. E. Slack. J. Am. Chem. Soc. 92, 5258-5259 (1970).
12] H.-D. Stachel. M. Jungkenn, C. Koser-Gnoss, H. Poschenrieder, J. Redlin, Liebigs Ann. Chem. 1994, 129-132.
13] B. Gill, G.D. James, K.V. Oates, G. Pattenden, J. Chem. Soc. Perkin Trans. I, 2567-2579 (1993), haben Verbindung 18 (E/Z) als nicht getrenntes Isomerengemisch beschrieben.
14] H.-D. Stachel, H. Poschenrieder, Z. Naturforsch. 36b, 721-725 (1981).
15] H.-D. Stachel, K.K. Harigel, H. Poschenrieder, H. Burghard, J. Heterocycl. Chem. 17, 1195-1199(1980).
16] H.-D. Stachel, H. Poschenrieder , B. Wiesend, Z. Naturforsch. 38b, 988-991 (1983).
17] H.-D. Stachel, Arch. Pharm. Ber. Dtsch. Pharm. Ges. 296, 479-487 (1963).
18] D. Breternitz, Dissertation Universität München (1972).
19] J. Redlin, Dissertation Universität München (1991).





























