Embed Size (px)
Citation preview

Activation of Boron–Boron, Tin–Silicon, and Tin–Tin Bonds:
Application in Carbon–Element (E = B and Sn) Bond-
Forming Reactions and Site-Selective SUZUKI–MIYAURA
Cross-Coupling Reactions
vorgelegt von
Master of Science
Manish Pareek
geb. in Raila Road, India
Von der Fakultät II - Mathematik und Naturwissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
Dr. rer. nat.
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. Andreas Grohmann
Gutachter: Prof. Dr. Martin Oestreich
Gutachter: Prof. Dr. Philipp Heretsch
Tag der wissenschaftlichen Aussprache: 20. Juni 2017
Berlin 2017


This thesis was prepared at the Institut für Chemie, Technische Universität Berlin
between October 2013 and April 2017 under the supervision of Professor Dr. MARTIN
OESTREICH.
I thank Professor Dr. MARTIN OESTREICH for giving me an opportunity to work on
challenging and interesting projects. His endless support, constant encouragement,
patient guidance, and valuable suggestions allowed me to stay focused on my chemical
puzzles. In addition, he was always accessible and willing to help his students
with their research and beyond.
I would like to thank Professor Dr. PHILIPP HERETSCH for accepting the invitation to be
the external evaluator for this thesis.
I am grateful to Professor Dr. ANDREAS GROHMANN for acting as the chairman of the
doctoral commitee.
My gratitudes to Professor Dr. JOHANNES TEICHERT for organizing the synthesis
seminars, and for being my second supervisor for my DAAD fellowship. I would like to
thank Dr. HENDRIK KLARE for his valuable suggestions throughout my stay in the group.
I gratefully acknowledge the Deutscher Akademischer Austauschdienst (DAAD) for the
financial support. I would also like to thank all the members of the Berlin International
Graduate School of Natural Sciences and Engineering (BIG-NSE). Special thanks go to
Dr. JEAN-PHILIPPE LONJARET for his continuous help and perfect organization.
The analytic centers of the Institut für Chemie, Technische Universität Berlin are
acknowledged for their expert advice and help. I would like to especially thank
Dr. SEBASTIAN KEMPER and SAMANTHA VOGES for recording the NMR spectra, Dr.
ELISABETH IRRAN and PAULA NIXDORF from the X-ray crystallography service, as well as
Dr. MARIA SCHLANGEN-AHL and MARC GRIFFEL from the laboratory of mass-spectrometry
for outstanding service and advice. From the Bunker Team, I would like to thank ERIK
NEUMANN for friendly and efficient service.
I would like to acknowledge my correction team XICHANG DONG, JULIEN FUCHS, DIGVIJAY
PORWAL, and WEICHAO XUE for making writing of this thesis easy. I thank FRANCIS
FORSTER for translating the abstract.
I thank the present members of OESTREICH group, especially my Big Lab mates LUKAS
OMANN, PATRIZIO ORECCHIA, POLINA SHAYKHUTDINOVA, LARS SÜßE, MARIA VOLGER, and
DR. WEIMING YUAN, I would like to express my gratitudes to the fellow members of T-Lab
CAROLIN FOPP, DR. HAMIDEH HAZRATI, SOFIYA MARINOVA, DR. ENRIQUE E. MAROTO, SVEN
RICHTER, and JONAS SCHARFBIER, comrades from the French Lab SEBASTIAN KEEß,

WEIQIANG CHEN, and DR. QING-AN CHEN, die Bären from the Bär lab SUSANNE BÄHR and
PHILLIP “POMM BÄR” POMMERENING, members from the lab above SIMON WÜBBOLT, and
Dr. QIN YIN, the ones from Teichert Labs DR. MANAS DAS, THI NGOC THANH NGUYEN,
FELIX PAPE, DR. POLINA SMIRNOV, NIKLAS O. THIEL, and BIRTE ZIMMERMANN. I also thank
the past members from the OESTREICH group, Dr. THOMAS FALLON is gratefully
acknowledged for the initial help and introduction into NMR spectroscopy. I am grateful
to Prof. Dr. INDRANIL CHATTERJEE for the chemical and non-chemical discussions. I
would like to thank, Dr. TONI T. METSÄNEN and Dr. ANTOINE SIMONNEAU for useful
scientific discussions. STEPHANIE KROMBACH is thanked for helping me through all the
bureaucracy and paper work during the last three and a half years, I thank other non-
academic staff members MONIKA ULRICH and CORNELIA FISCHER for their help.
I thank Professor Dr. R. VIJAYA ANAND for his training during my master’s project. I would
like to thank all my friends who have helped me in the past, ABHILASHA, ANSHU, ANKIT,
ASIF, GAGAN, KESHAV, NITISH, SUMIT, and VIKESH.
Finally, I take this opportunity to express my profound gratitude to my family, my
parents, my in-laws, BAUJI, DADI, my sisters, my brother and sister in-laws, my nephews
and nieces, my cousins, my aunties, my uncles, and others for their constant support
and encouragement. My deepest appreciation has to go to my beloved wife TANU for her
love and care, whose support has always been my source of strength.

“You have control only over your actions but not over their results”
Bhagavad Gita
Chapter 2, Verse 47


PUBLICATIONS
Parts of this work have been published:
[1] “Platinum(0)-Catalyzed Indolyne Insertion into Bis(pinacolato)diboron
Followed by Site-Selective Suzuki–Miyaura Cross-Coupling”,
M. Pareek, T. Fallon, M. Oestreich, Org. Lett. 2015, 17, 2082–2085.
ORAL PRESENTATIONS
[1] M. Pareek, “Asymmetric Stannylation of α,β-Unsaturated Esters by
Interelement Bond Activation”,
BIG-NSE retreat, Schorfheide (Germany), May 19–20, 2016.
[2] M. Pareek, “Platinum(0)-Catalyzed Indolyne Insertion into
Bis(pinacolato)diboron Followed by Site-Selective Suzuki-Miyaura Cross-
Coupling”,
BIG-NSE retreat, Schorfheide (Germany), May 28–29, 2015.
[3] M. Pareek, “Transition Metal-Catalyzed Insertion of Indolynes into
Interelement Bonds”,
BIG-NSE retreat, Berlin (Germany), January 10, 2014.
POSTER PRESENTATIONS
[1] M. Pareek, T. Fallon, M. Oestreich, “Platinum(0)-Catalyzed Indolyne Insertion
into Bis(pinacolato)diboron Followed by Site-Selective Suzuki-Miyaura Cross-
Coupling”,
Tag der Chemie, Freie Universität. Berlin (Germany), June 18, 2015.


ZUSAMMENFASSUNG
Die vorliegende Arbeit fokussiert sich auf die Aktivierung von Interelementbindungen der
Elemente Bor, Silicium und Zinn. Der erste Teil dieser Arbeit beschreibt die Aktivierung
von Diborbindungen durch oxidative Addition von Bor–Bor-Bindungen an niedervalente
Platin(0)-Katalysatoren. Die Übertragung dieser aktivierten Diborspezies auf
ungesättigte Kohlenstoff–Kohlenstoff-Bindungen ist in der Literatur gut beschrieben.
YOSHIDA und Mitarbeiter untersuchten die Insertion von Arinen in die Diborbindung von
B2pin2 mittels Platin(0)-Katalysatoren. Die Erweiterung von YOSHIDAs Arbeit hin zu
Didehydroindolen erzeugte diborylierte Indole. Alle drei regioisomeren 4,5-, 5,6- und 6,7-
Didehydroindole wurden als Substrate verwendet. Anschließende SUZUKI–MIYAURA-
Kreuzkupplungsreaktionen der 4,5-diborylierten Indole verliefen nicht chemoselektiv und
lieferten einfach und zweifach gekuppelte Produkte. Erfreulicherweise zeigten 6,7-
diborylierte Indole Ortsselektivität in SUZUKI–MIYAURA-Kreuzkupplungsreaktionen. Die
Kupplungen erfolgten ausschließlich an der C7-Position der diborylierten Indole. Durch
nachfolgende Kupplungsreaktionen wurden bisher nicht beschriebene 6,7-diarylierte
Indole erhalten.
Der zweite Teil dieser Arbeit behandelt die Aktivierung von Zinn–Silicium- und Zinn–
Zinn-Bindungen, wodurch Stannylnukleophile generiert werden. Die anschließenden
asymmetrischen konjugierten Additionen solcher Nukleophile an α,β-ungesättigte Ester
wird beschrieben, wobei zur Erzeugung der Stannylnukleophile zwei unterschiedliche
Methoden genutzt wurden. Wie durch OESTREICH und Mitarbeiter gezeigt wurde, setzt
die chemoselektive Aktivierung von Zinn–Silicium-Bindungen durch Basen in Gegenwart
von Wasser Stannylnukleophile frei, welche für konjugierte Additionen verwendet
werden können. In dieser Arbeit wurden Phasentransfer-Katalysatoren genutzt, um
einerseits eine chirale Umgebung zu erzeugen und andererseits die Verwendung von
Wasser als Lösungsmittel zu ermöglichen. Aufgrund der Gegenwart von Wasser im
System wird ein Phasentransfer-Katalysator benötigt. Chinidin- und
cinchinidinabgeleitete, quartäre Ammoniumsalze lieferten die gewünschten Ausbeuten,
allerdings mit niedrigen Enantiomerenüberschüssen. Kontrollexperimente bestätigten
die Abwesenheit von Hintergrundreaktionen. Die Substratbreite für diese Reaktion
wurde untersucht, wobei elektronenreiche Systeme die Reaktion problemlos durchliefen
und β-stannylierte Ester in guten Ausbeuten und niedrigen Enantiomerenüberschüssen
lieferten. Die Aktivierung von Zinn–Zinn-Bindungen wurde durch eine basenvermittelte
Kupfer(I)-Katalyse erreicht. Ein chirales N-heterozyklisches Carben wurde als Ligand
verwendet, um moderate Enantiomerenüberschüsse in der konjugierten Stannylierung
von α,β-ungesättigten Estern zu erreichen, wenn auch in niedrigen Ausbeuten.


ABSTRACT
This thesis focuses on the activation of interelement bonds of boron, silicon and tin. The
first part of this thesis describes the activation of the diboron bonds via oxidative addition
of boron−boron bonds to low valent platinum(0) catalysts. The transfer of this activated
diboron species to unsaturated carbon−carbon bonds is well established in the literature.
YOSHIDA and co-workers studied the insertion of the arynes into the diboron bond of
B2Pin2 employing platinum(0) catalyst. The extension of YOSHIDA’s work to indolynes
generated diborylated indoles. All the three regioisomers of the (4,5-, 5,6-, and 6,7-)
indolynes were shown to undergo the diborylation. Subsequent SUZUKI−MIYAURA cross-
coupling reactions of the 4,5-diborylayed indole were not chemoselective and yielded
both mono- and bis-coupled products. Gratifyingly, the 6,7-diborylated indole showed
site-selectivity in the SUZUKI−MIYAURA cross-coupling reaction. The coupling was
performed exclusively at the C7 position of the indole. The second coupling reaction
afforded previously unprecedented 6,7-arylated indoles.
The second part of this thesis illustrates the activation of tin−silicon and tin−tin bonds.
The activation of these bonds generates stannyl nucleophiles, and subsequent
asymmetric conjugate addition of such nucleophiles to α,β-unsaturated esters is
reported. Two different approaches were used to access the stannyl nucleophiles. As
shown by OESTREICH and co-workers, the chemoselective activation of tin−silicon bonds
by base in presence of water liberates the stannyl nucleophile, and this nucleophile can
be used for the conjugate addition. In this thesis, a phase-transfer catalyst is used to
introduce the chiral environment in the reaction. The use of phase-transfer catalyst is
advantageous in the reaction because of the presence of the water in the system.
Quinidine- and cinchonine-derived quaternary ammonium salts furnished the desired
yields albeit with low enantiomeric excesses. Control experiments confirmed the
absence of background reactions. The substrate scope for this reaction was explored,
and electron-rich systems underwent the smooth reaction and afforded β-stannylated
esters with good yields and low enantiomeric excesses. Activation of tin−tin bond was
achieved by a base mediated copper(I) catalysis. A chiral N-heterocyclic carbene was
used as the ligand to afford moderate enantiomeric excesses in the conjugate
stannylation of α,β-unsaturated esters, however, with low yields.


TABLE OF CONTENTS


TABLE OF CONTENTS
THEORETICAL PART
1 INTRODUCTION 1 1.1 Activation of Interelement Bonds 2 1.2 Addition of Diboron Bonds to Unsaturated Carbon–Carbon
Bonds via Oxidative Addition 4
1.2.1 Reactivity of Diboron Bonds 4 1.2.2 Insertion of Alkynes into Diboron Bonds 6 1.2.3 Insertion of Aliphatic Alkenes into Diboron Bonds 9 1.2.4 Insertion of Styrenes into Diboron Bonds 11 1.2.5 Insertion of 1,3-Dienes into Diboron Bonds 13 1.2.6 Insertion of Allenes into Diboron Bonds 15 1.2.7 Insertion of Arynes into Diboron Bonds 16 1.3 Application of Carbon–Boron Bonds in Carbon–Carbon Bond Forming
Reactions 18
1.3.1 SUZUKI–MIYAURA Cross-Coupling Reaction 18 1.3.2 Orthogonal SUZUKI–MIYAURA Cross-Coupling Reaction 19 1.3.2.1 Orthogonal SUZUKI–MIYAURA Cross-Coupling Reaction Controlled by
Masked Boronates 20
1.3.2.2 Orthogonal SUZUKI–MIYAURA Cross-Coupling Reaction Controlled by Electronic and Steric Properties of the Substrate
22
1.4 Stannylation of Electrophilic Acceptors involving Activation of Tin–Silicon and Tin–Tin Bonds
24
1.4.1 Activation of Tin–Silicon Bonds by Nucleophiles 24 1.4.2 Stannylation of Electrophilic Acceptors involving Activation of Tin–Silicon
Bond by Nucleophiles 25
1.4.3 Activation of Tin–Tin Bonds by Transmetalation 28 1.4.4 Formation of Tin–Metal Bonds by using tin–tin Bonds 28 1.4.5 Conjugate Addition of Tin nucleophiles using Silyl-Cuprates to
Electrophilic Organic Compounds 29
1.5 Objective 31 2 PLATINUM(0)-CATALYZED INDOLYNE INSERTION INTO
BIS(PINACOLATO)DIBORON FOLLOWED BY SITE-SELECTIVE SUZUKI–MIYAURA CROSS-COUPLING
35
2.1 Introduction to Indolynes 35 2.2 Synthesis of Indolyne Precursors and Generation of Indolynes 36 2.3 Insertion of Indolynes into Bis(pinacolato)diboron 39 2.3.1 Insertion of 4,5-Indolyne 135 into Bis(pinacolato)diboron (1) 39 2.3.2 Insertion of 5,6-Indolyne 136 into Bis(pinacolato)diboron (1) 40 2.3.3 Insertion of 6,7-Indolyne 137 into Bis(pinacolato)diboron (1) 40 2.4 Site-Selective SUZUKI–MIYAURA Cross-Coupling 41 2.4.1 SUZUKI–MIYAURA Cross-Coupling of 4,5-Diborylated Indole 138 41 2.4.2 SUZUKI–MIYAURA Cross-Coupling of 6,7-Diboronated Indole 139 43 2.4.3 One-Pot SUZUKI–MIYAURA Cross-Coupling of 6,7-Diborylated Indole 140 47 2.5 Conclusion 48 3 ASYMMETRIC STANNYLATION OF Α,Β-UNSATURATED ESTERS INVOLVING 49

TIN–SILICON AND TIN–TIN BOND ACTIVATION 3.1 Asymmetric 1,4-Stannylation of α,β-Unsaturated Esters Using Tin−Silicon (Sn−Si)
Bond Activation 49
3.1.1 Introduction 49 3.1.2 Optimization Studies for the PTC-Catalyzed Asymmetric 1,4-Stannylation
of α,β-Unsaturated Esters 50
3.1.3 Control Experiments 57 3.1.4 Substrate Scope for the PTC-catalyzed Asymmetric 1,4-Stannylation of
α,β-Unsaturated Esters 58
3.1.5 Proposed mechanism for the PTC-catalyzed Asymmetric 1,4-Stannylation of α,β-Unsaturated Esters
60
3.2 Asymmetric 1,4-Stannylation of α,β-Unsaturated Esters Using Tin−Tin Bond Activation
61
3.2.1 Introduction 61 3.2.2 Optimization Studies for the Asymmetric 1,4-Stannylation of
α,β-Unsaturated Esters 62
3.3 Conclusion 69 4 SUMMARY 70
EXPERIMENTAL PART
1 GENERAL INFORMATION 75 2 GENERAL PROCEDURES 85 2.1 General Procedure for Cross-Coupling Experiments 85 2.1.1 General Procedures for the Synthesis of the 7-Aryl-6-boryl-Substituted
Indoles 141a−e and 172 (GP 1) 85
2.1.2 General Procedure for the Synthesis of the 6,7-Bisaryl-Substituted Indoles 142aa−ae (GP 2)
85
2.2 General Procedure for the Preparation of (Z)-α,β-Unsaturated Esters (GP 3)
86
2.3 General Procedure for the Asymmetric 1,4-Stannylation of the (Z)-α,β-Unsaturated Esters (GP 4)
86
3 DESCRIPTION OF EXPERIMENTS 89 3.1 Synthesis of the Indolyne Precursors 89 3.1.1 Synthesis of 4,5-Indolyne Precursor 132 and 5,6-Indolyne Precursor
133 89
3.1.2 Synthesis of 6,7-Indolyne Precursor 134 94 3.2 Synthesis of Diborylated Indoles 100 3.3 Synthesis of the 7-Aryl-6-boryl-Substituted Indoles 104 3.4 Synthesis of the 6,7-Bisaryl-Substituted Indoles 112 3.5 Synthesis of (Z)-α,β-Unsaturated Esters 118 3.6 Synthesis of Silylstannane 98 130 3.7 Asymmetric 1,4-Stannylation of the (Z)-α,β-Unsaturated Ester 134 3.8 Synthesis of Chiral Cu–NHC Catalyst ((S,S)-217) 142
APPENDIX
A1 X-RAY CRYSTAL STRUCTURE DATA 149 A1.1 Molecular Structure of cu-879 149 A2 ABBREVIATIONS 151 A3 BIBLIOGRAPHY 155 A4 CURRICULUM VITAE 162

THEORETICAL PART


1 Introduction 1
1 INTRODUCTION
Chemical bonds within the main-group element, and bonds between the main-group
elements and the transition metals are called ‘interelement bonds’.[1] The interelement bonds
(E–E) of boron, silicon, and tin have wide synthetic utility as they are known to undergo
carbon–element (C–E) bond formation via activation of E–E bonds.[2] The resulting C–E bond
can act as a linchpin to other transformations through various reactions, e.g., cross-coupling
reactions.[3] There can be six possible permutations (B–B, Si–Si, Sn–Sn, Si–B, Sn–Si, Sn–B)
of these three main group elements (Figure 1.1).
Figure 1.1: All possible permutations of interelement bonds of boron, silicon, and tin.
[1]
K. Tamao, S. Yamaguchi, J. Organomet. Chem. 2000, 611, 3–4. [2]
For general reviews on interelement bond activation and carbon–element bond formation
reaction, see: a) M. B. Ansell, O. Navarro, J. Spencer, Coord. Chem. Rev. 2017, doi:
http://dx.doi.org/10.1016/j.ccr.2017.01.003; b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, A.
Westcott, T. B. Marder, Chem. Rev. 2016, 116, 9091–9161; c) M. Iwasaki, Y. Nishihara, Chem.
Rec. 2016, 16, 2031–2045; d) M. Suginome, T. Matsuda, T. Ohmura, A. Seki, M. Murakami,
in Comprehensive Organometallic Chemistry III; Mingos, D. M. P.; Crabtree, R. H.; Ojima, I.,
Eds.; Elsevier: Oxford, 2007; Vol. 10, pp 725–787; e) H. E. Burks, J. P. Morken, Chem.
Commun. 2007, 4717–4725; f) I. Beletskaya, C. Moberg, Chem. Rev. 2006, 106, 2320–2354; g)
T. Ishiyama, N. Miyaura, Chem. Rec. 2004, 3, 271–280; h) I. Beletskaya, C. Moberg, Chem.
Rev. 1999, 99, 3435–3462; i) Y. J. Ito, Organomet. Chem. 1999, 576, 300–304; j) T. B. Marder,
N. C. Norman, Top. Catal. 1998, 5, 63–73; k) K. A. Horn, Chem. Rev. 1995, 95, 1371–1350. [3]
For carbon–carbon bond forming reactions, see: a) Metal Catalyzed Cross-Coupling Reactions and More, (Eds.: A. de Meijere, S. Brase, M, Oestreich), Wiley-VCH, Weinheim, 2014; b) Handbook of Organopalladium Chemistry for Organic Synthesis (Eds.: E. Negishi, A. de Meijere), Wiley, New York, 2002; c) N. Miyaura, A. Suzuki, Chem. Rev. 1995, 95, 2457–2483; d) Y. Hatanaka, T. Hiyama, J. Org. Chem. 1988, 53, 918–920; e) J. K. Stille, Angew. Chem. Int. Ed. 1986, 25, 508–524; Angew. Chem. 1986, 98, 504–519.

1.1 Activation of Interelement Bonds
The modes of activation of these interelement bonds have been described in detail by
Oestreich and co-workers and can be classified in four categories (Scheme1.1).[4]
Scheme 1.1: Classification of E–E’ bond activation modes.
The widely used method of activation is oxidative insertion of low-valent transition metals
(TM) II [e.g., Pt(0), Pd(0), Ni(0)] into the interelement bonds I to give the activated species III
(I+II→III). III then can be added across unsaturated carbon–carbon bonds. Activation by
transmetalation with anionic nucleophiles IV/V opened numerous ways for chemoselective
cleavage of unsymmetrical interelement bonds as the nucleophile tends to attack at the more
LEWIS-acidic atom (I+IV/V→VI/VII). However, a more pragmatic approach to metal-based
main group nucleophiles was through a σ-bond metathesis reaction between metal alkoxide
VIII and interelement bonds of I to give transition state IX‡, which then gives X in concerted
fashion (I+VIII→IX‡→X). This approach facilitated the use of transition metals such as Rh(I),
Cu(I), and Ni(II).The resulting main-group nucleophile is transferred to an electrophilic
carbon center. Another competently investigated mode of activation of E–E’ bonds is
[4]
M. Oestreich, E. Hartmann, M. Mewald, Chem. Rev. 2013, 113, 402–441.

1 Introduction 3
performed by the substrate itself. Several carbenoids XI by nucleophilic attack at the E–E’ I
generate the intermediate XII. This intermediate then migrates to the same carbon atom to
give expected XIII (I+XI→XII→XIII). Recently, a new approach has developed as catalytic
amounts of a LEWIS base or the LEWIS basic substrate XIV facilitates the release of main-
group nucleophiles through activation of E–E’ I bonds (I+XIV→XV). The following chapters
give an overview of the various modes of the E–E’ (B–B, Sn–Si, Sn–Sn) bond activation and
the use of such activated bonds to form new C–E (E = B, Sn) bonds. Chapter 1.2 presents
the well-established methods for the addition of diboron (B–B) bonds to unsaturated C–C
bonds. Chapter 1.3 gives an overview of the orthogonal SUZUKI–MIYAURA cross-coupling
reaction, where orthogonality is obtained at the transmetalation stage. Chapter 1.4 talks
about the activation of Sn–Si bonds to generate tin nucleophiles and the use of these
nucleophiles in making a C–Sn linkage. Chapter 1.5 discusses the activation of Sn–Sn
bonds by transmetalation with anionic nucleophiles and their use in the stannylation of
electrophilic compounds.

1.2 Addition of Diboron Bonds to Unsaturated Carbon–Carbon Bonds
via Oxidative Addition
1.2.1 Reactivity of Diboron Bonds
The advent of diboron compounds dates back to 1925 when STOCK, BRANDT and FISCHER
reported the first synthesis of B2Cl4.[5] The addition of such diboranes to unsaturated
substrates proceeded in the absence of a catalyst and often at low temperature. However,
the diboron starting material as well as the products are air and moisture sensitive.[6]
Therefore, recent developments have aimed at the synthesis of more stable, electron-rich
heteroatom-substituted diboron sources.[7] However, this leads to a decrease in LEWIS acidity
of these novel diboron sources and, hence, a catalyst is required for the activation of the
diboron bond. The facile synthesis, moderate reactivity, and stability of B2(OR)4 species have
made them the reagents of choice in synthetic chemistry. Particularly, bis(pinacolato)diboron
(B2pin2) (1), bis(catecholato)diboron (B2cat2) (2), and (pinacolato)(diaminonaphthaleno)-
diboron (BpinBdan) (3) have been used extensively (Figure: 1.2).
Figure 1.2: Most used diboron reagents, B2pin2 (1), B2cat2 (2), and BpinBdan (3).
Various low-valent transition-metal complexes have been developed for the activation of
diboron bonds. Platinum(0) complexes are proven to be the most common and effective
transition-metal catalysts for this reaction.[8] The broad substrate scope of this catalytic
system makes it one of the most used method to perform diborylation to alkynes, alkenes,
[5]
A. Stock, A. Brandt, H. Fischer, Ber. Dtsch. Chem. Ges. B 1925, 58, 643–657. [6]
R. Barbeyron, E. Benedetti, J. Cossy, J. J. Vasseur, S. Arseniyadis, M. Smietana, Tetrahedron
2014, 70, 8431–8452. [7]
S. A. Westcott, E. Fernandez, in Advances in Organometallic Chemistry; Academic Press:
Cambridge, 2015; pp 39–89. [8]
a) J. Takaya, N. Iwasawa, ACS Catal. 2012, 2, 1993–2006; b) M. V. Dembitsky, A. H. Ali, M.
Srebnik, in Advances in Organometallic Chemistry; Academic Press: Cambridge, 2004;
pp 193–250.
1 Introduction 5
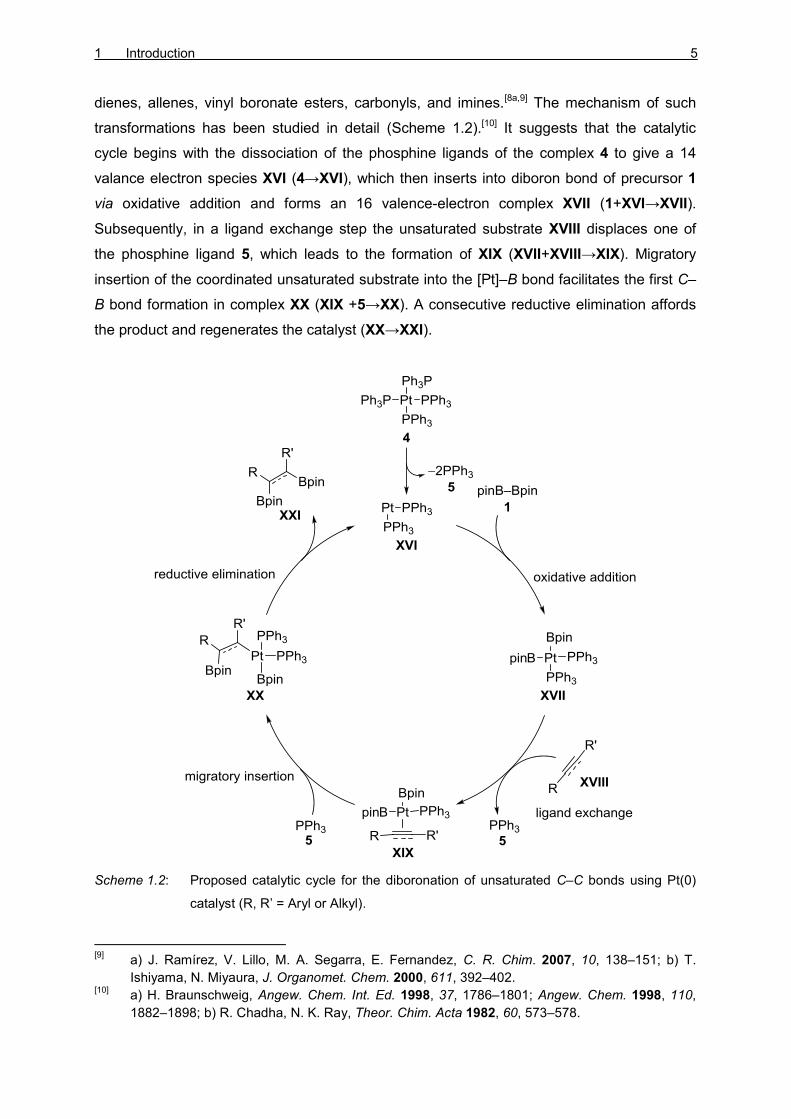
dienes, allenes, vinyl boronate esters, carbonyls, and imines.[8a,9] The mechanism of such
transformations has been studied in detail (Scheme 1.2).[10] It suggests that the catalytic
cycle begins with the dissociation of the phosphine ligands of the complex 4 to give a 14
valance electron species XVI (4→XVI), which then inserts into diboron bond of precursor 1
via oxidative addition and forms an 16 valence-electron complex XVII (1+XVI→XVII).
Subsequently, in a ligand exchange step the unsaturated substrate XVIII displaces one of
the phosphine ligand 5, which leads to the formation of XIX (XVII+XVIII→XIX). Migratory
insertion of the coordinated unsaturated substrate into the [Pt]–B bond facilitates the first C–
B bond formation in complex XX (XIX +5→XX). A consecutive reductive elimination affords
the product and regenerates the catalyst (XX→XXI).
Scheme 1.2: Proposed catalytic cycle for the diboronation of unsaturated C–C bonds using Pt(0)
catalyst (R, R’ = Aryl or Alkyl).
[9]
a) J. Ramírez, V. Lillo, M. A. Segarra, E. Fernandez, C. R. Chim. 2007, 10, 138–151; b) T.
Ishiyama, N. Miyaura, J. Organomet. Chem. 2000, 611, 392–402. [10]
a) H. Braunschweig, Angew. Chem. Int. Ed. 1998, 37, 1786–1801; Angew. Chem. 1998, 110,
1882–1898; b) R. Chadha, N. K. Ray, Theor. Chim. Acta 1982, 60, 573–578.

MARDER and co-workers isolated and characterized the complex [(Ph3P)2Pt (Bcat)2] (6).[11]
This complex shows a catalytic activity similar to [(Ph3P)4Pt] (4), suggesting that the catalytic
cycle probably goes through intermediate XVII. In the same study, two monodentate
phosphine ligands were replaced with one bidentate (1,2-bis(diphenylphosphino)ethane)
(dppe, 7) in the isolated complex 8, which is inactive in the catalysis (Scheme 1.3). This
indicated that the phosphine dissociation is a reasonable step in the diboron bond activation
reaction. The effects of ligands and metal-to-ligand ratio was further investigated by MARDER
and co-workers.[12] Screening of various phosphine ligands with Pt(0) catalysts revealed that
Cy3P and (o-Tol)PPh2 in 1:1 ratio with Pt(0) catalysts are the optimal ligands for the
diborylation reactions. MIYAURA, SUZUKI, and co-workers confirmed a syn addition of the
diboron species by stoichiometric reactions between cis-[(Ph3P)2Pt(Bpin)2] (XVII) and
alkynes.[13] MARDER, BAKER, and co-workers showed that other metals such as rhodium(I)
and iridium(I) also follow similar mechanism.[14] Later, PERUTZ and MARDER used osmium(0)
and cobalt(0) catalysts for this transformation.[15] MOROKUMA and co-workers performed a
density-functional study, showing that palladium(0) is catalytically inactive because of the
unstable palladium(II) species formed after oxidative addition of diboron bond of 1.[16]
Scheme 1.3: Effect of ligand replacement.
1.2.2 Insertion of Alkynes into Diboron Bonds
MIYAURA, SUZUKI, and co-workers reported the first addition of diboron compound 1 to a
terminal 9a and internal alkyne 9b using the [(Ph3P)4Pt] (4) complex to give cis-1,2-
[11]
G. Lesley, P. Nguyen, N. J. Taylor, T. B. Marder, A. J.Scott, W. Clegg, N. C. Norman,
Organometallics 1996, 15, 5137–5154. [12]
R. L. Thomas, F. E. S. Souza, T. B. Marder, J. Chem. Soc., Dalton Trans. 2001, 1650–1656. [13]
T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki, N. Miyaura, Organometallics 1996,
15, 713–720. [14]
a) W. Clegg, F. J. Lawlor, T. B. Marder, P. Nguyen, N. C. Norman, A. G. Orpen, M. J. Quayle,
C. R. Rice, E. G. Robins, A. J. Scott, F. E. S. Souza, G. Stringer, G. R. Whittell, J. Chem. Soc.,
Dalton Trans. 1998, 301–310; b) R. T. Baker, J. C. Calabrese, S. A. Westcott, P. Nguyen, T. B.
Marder, J. Am. Chem. Soc. 1993, 115, 4367–4368. [15]
a) M. V. Campian, J. L. Harris, N.Jasim, R. N. Perutz, T. B. Marder, A. C.Whitwood,
Organometallics 2006, 25, 5093–5104; b) C. Dai, G. Stringer, J. F. Corrigan, N. J. Taylor, T.B.
Marder, N. C. Norman, J. Organomet. Chem. 1996, 513, 273–275. [16]
Q. Cui, D. G. Musaev, K. Morokuma, Organometallics 1998, 17, 742–751.

1 Introduction 7
diboronated alkenes (10a−b) in good yields (Scheme 1.4, top), whereas diborylation with
Pd(0) and Rh(I) complexes was proven to be futile.[17] Later, the authors employed
sequential cross coupling reactions of 1,2 diboronated alkene (11) to give mono-coupled 12.
Trisubstituted alkene 13 was obtained by subsequent cross-coupling (Scheme 1.4,
bottom).[18]
Scheme 1.4: Diborylation of alkynes 9a−b using [(Ph3P)4Pt] (4) (top) and subsequent cross-
coupling reactions (bottom).
SREBNIK and co-workers reported the highly efficient Pt(0)-catalyzed addition of
bis(pinacolato)diboron (1) to 1-alkynylphosphonates 14a–b and 1-alkynylboronates
14c–d.[19] This delivered sterically bulky cis-1,2-diboronated vinylphosphonates 15a–b and
trisboronated alkenes 15c–d (Scheme 1.5).
Scheme 1.5: Diborylation of alkynylboronates 14a–b and 1-alkynylphosphonates 14c–d using a
Pt(0) catalyst.
SIEBERT and co-workers achieved highly substituted ethane and ethene frameworks through
reactions between catechol-substituted diborylacetylenes 16 and bis(catecholato)-
[17]
T. Ishiyama, N. Matsuda, N. Miyaura, A. Suzuki, J. Am. Chem. Soc. 1993, 115, 11018–11019. [18]
T. Ishiyama, M. Yamamoto, N. Miyaura, Chem. Lett. 1996, 1117–1118. [19]
H. Abu Ali, A. A. Al Quntar, I. Goldberg, M. Srebnik, Organometallics 2002, 21, 4533.

diboron (2) (Scheme 1.6). [20] The authors established that Pt(cod)2 gives tetraborylethene
17c, whereas [(Ph3P)2Pt(CH2═CH2)] yielded hexaborylethane 17a and 17b, depending upon
the reaction conditions.
Scheme 1.6: Diborylation of catechol-substituted diborylacetylenes 16 to give highly substituted
ethane and ethene frameworks 17a−c.
SUGINOME and co-workers were able to perform a regioselective insertion of phenyl
acetylene (18) into the unsymmetrical diboron bond of 3 employing Pt(dba)2 and [IrCl(COD)]2
catalysts.[21] This afforded 1-alkenyl-1,2-diboronic esters 19 with reactive internal boronyl
groups (Scheme 1.7). The authors also managed to do a chemoselsctive SUZUKI cross-
coupling reaction at the more reactive pinacolato-protected center to yield highly substituted
alkene 20.
Scheme 1.7: Regioselective diborylation of alkyne 18 and selective cross-coupling reactions.
[20]
M. Bluhm, A. Maderna, H. Pritzkow, S. Bethke, R. Gleiter, W. Siebert, Eur. J. Inorg. Chem.
1999, 1693–1700. [21] N. Iwadate, M. Suginome, J. Am. Chem. Soc. 2010, 132, 2548–2549.

1 Introduction 9
1.2.3 Insertion of Aliphatic Alkenes into Diboron Bonds
SCHLESINGER and co-workers reported the first diborylation of aliphatic alkenes in 1954 using
B2Cl4 as a diboron source.[22] However, the insertion into stable diboronic esters B2(OR)4
was challenging because of the weaker binding ability of alkenes to metal compared to
phosphine, making it resilient for alkene to replace phosphine ligands (XVII+XVIII→XIX).[17]
MIYAURA and SMITH independently reported the insertion of aliphatic alkene 21 into diboron
bond of 2, where each used Pt(0)-catalyzed insertion into diboron bond employing B2pin2 (1)
and B2cat2 (2) as diboron precursors (Scheme 1.8, top). The use of labile ligands such as
norbornene (23), 1,5-cyclooctadiene (24), and dibenzylideneacetone (25) promoted the
reaction (Scheme 1.8, middle). Interestingly, the authors also report the insertion of
norbornene (23) to afford 26, showing that the diborylation approach was extendable to
selected internal alkenes as well (Scheme 1.8, bottom).[23]
Scheme 1.8: Diborylation of α-olefin 21 employing Pt(0) catalyst (top), used ligands (middle), and
diborylation of internal aliphatic alkene 23 employing Pt(0) catalyst (bottom)
[22]
G. Urry, J. Kerrigan, T. D. Parsons, H. Schlesinger, J. Am. Chem. Soc. 1954, 76, 5299−5301. [23]
a) T. Ishiyama, M. Yamamoto, N. Miyaura, Chem. Commun. 1997, 689−690; b) C. N. Iverson,
M. R. Smith, Organometallics 1997, 16, 2757−2759; b) T. Oshiyama, M. Yamamoto, N.
Miyaura, Chem. Commun. 1997, 689−690.

The asymmetric diborylation of internal alkene 27 was reported by MORKEN and
co-workers.[24] High enantiomeric excesses were achieved using Rh(acac)(NBD) as catalyst
and (S)-quinap derivatives 28 as chiral ligands (Scheme 1.9, top). Later, the asymmetric
diborylation of the the terminal alkene 31 was investigated using Pt2(dba)3 and readily
available chiral phosphonite ligands 32 (Scheme 1.9, middle). In both cases, the products
were isolated as diols 30 and 34 after an oxidative degradation of the diborylated products
29 and 33. Recently, MORKEN and co-workers have performed an experimental and
computational study of enantioselective diborylation of alkene by Pt(dba)3.[25] In this study the
authors found that TADDOL-derived ligands 32 are optimal ligand. The reaction is facilitated
when 1 mol % of Pt(dba)3, and 1.2 mol % of ligand are in the system. The reaction was
proven to be in zero order with respect to B2pin2 (1) and alkene, establishing no role of the
substrate in the rate-determining step.
Scheme 1.9 Asymmetric diborylation of 27 using (S)-quinap derivative 28 as ligand (top) and
asymmetric diborylation of 31 using TADDOL-derived ligand 32 (middle).
[24] a) J. B. Morgan, S. P. Miller, J. P. Morken, J. Am. Chem. Soc. 2003, 125, 8702−8703; b) L. T.
Kliman, S. N. Mlynarski, J. P. Morken, J. Am. Chem. Soc. 2009, 131, 13210−13211. [25]
J. R. Coombs, F. Haeffner, L. T. Kliman, J. P Morken, J. Am. Chem. Soc. 2013, 135,
11222−11231.

1 Introduction 11
FERNÁNDEZ and co-workers reported Ag(I)-NHC 34-catalyzed insertion of terminal aliphatic
alkene 33 using B2cat2 (2) as the boron source Scheme 1.10).[26] The resulting diborylated
product 35 was converted to the diol 36 by oxidative migration of the C−B bonds.
Scheme 1.10 Ag(I)-catalyzed diborylation of aliphatic alkene 33.
1.2.4 Insertion of Styrenes into Diboron Bonds
BAKER, MARDER, and co-workers achieved the insertion of styrene 37 into the diboron bond
of 2 using RhCl(PPh3)3 as the catalyst (Scheme 1.11, top). However, the diborylated product
38a was obtained only as minor product in 10% yield; major products 38b and 38c were the
outcome of the β-hydride elimination. In the same report, the authors also rectified the
problem by using Au(I) catalyst albeit without any mechanistic explanation (Scheme 1.11,
bottom).[27]
[26]
a) J. Ramírez, R. Corberan, M. Sanau, E. Peris, E. Fernández, Chem. Commun. 2005,
3056−3058; b) V. Lillo, E. Mas-Marza, A. M. Segarra, J. J. Carbo, C. Bo, E. Peris, E.
Fernández, Chem. Commun. 2007, 3380−3382. [27]
R. T. Baker, P. Nguyen, T. B. Marder, S. A. Westcott, Angew. Chem. Int. Ed. 1995, 34,
1336−1338; Angew. Chem. 1995, 107, 1451–1452.
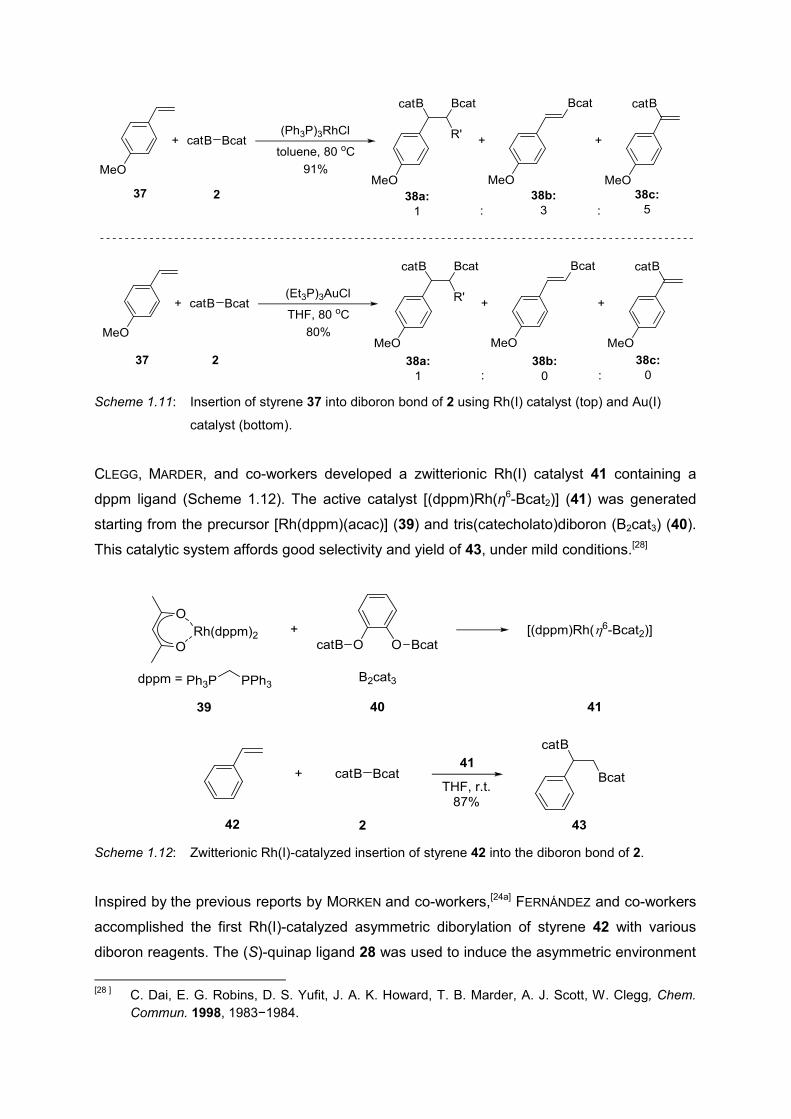
Scheme 1.11: Insertion of styrene 37 into diboron bond of 2 using Rh(I) catalyst (top) and Au(I)
catalyst (bottom).
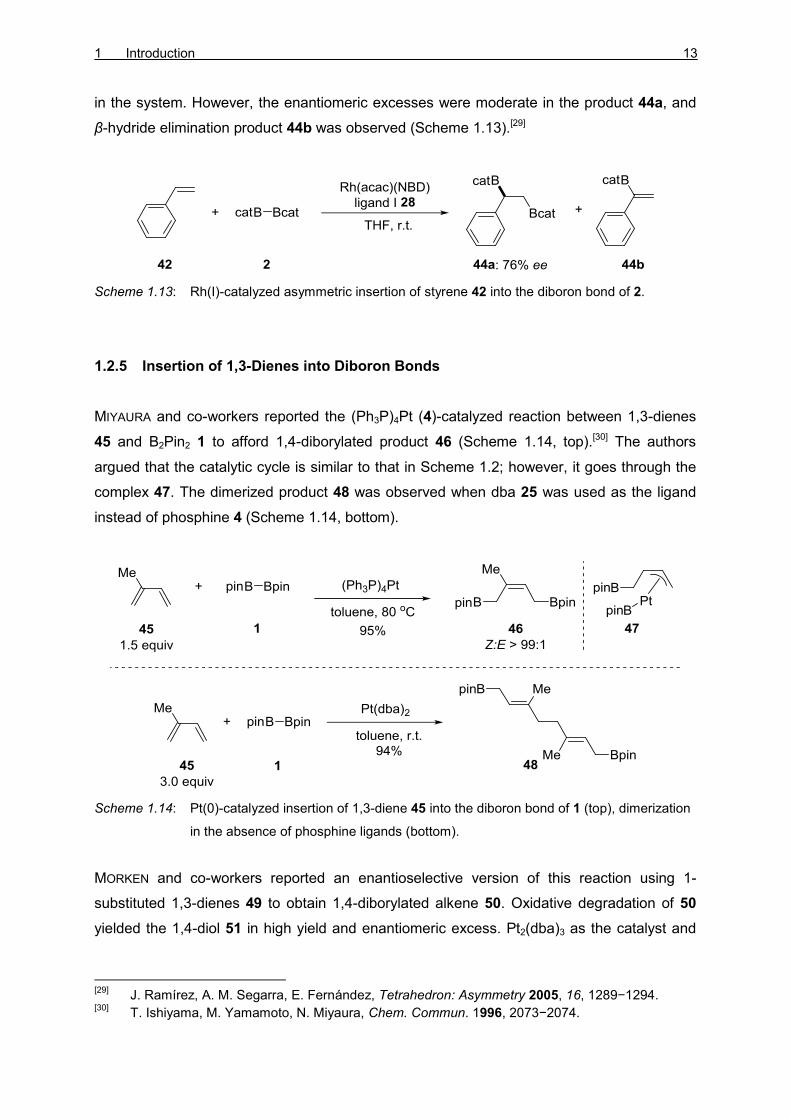
CLEGG, MARDER, and co-workers developed a zwitterionic Rh(I) catalyst 41 containing a
dppm ligand (Scheme 1.12). The active catalyst [(dppm)Rh(η6-Bcat2)] (41) was generated
starting from the precursor [Rh(dppm)(acac)] (39) and tris(catecholato)diboron (B2cat3) (40).
This catalytic system affords good selectivity and yield of 43, under mild conditions.[28]
Scheme 1.12: Zwitterionic Rh(I)-catalyzed insertion of styrene 42 into the diboron bond of 2.
Inspired by the previous reports by MORKEN and co-workers,[24a] FERNÁNDEZ and co-workers
accomplished the first Rh(I)-catalyzed asymmetric diborylation of styrene 42 with various
diboron reagents. The (S)-quinap ligand 28 was used to induce the asymmetric environment
[28 ]
C. Dai, E. G. Robins, D. S. Yufit, J. A. K. Howard, T. B. Marder, A. J. Scott, W. Clegg, Chem.
Commun. 1998, 1983−1984.
1 Introduction 13
in the system. However, the enantiomeric excesses were moderate in the product 44a, and
β-hydride elimination product 44b was observed (Scheme 1.13).[29]
Scheme 1.13: Rh(I)-catalyzed asymmetric insertion of styrene 42 into the diboron bond of 2.
1.2.5 Insertion of 1,3-Dienes into Diboron Bonds
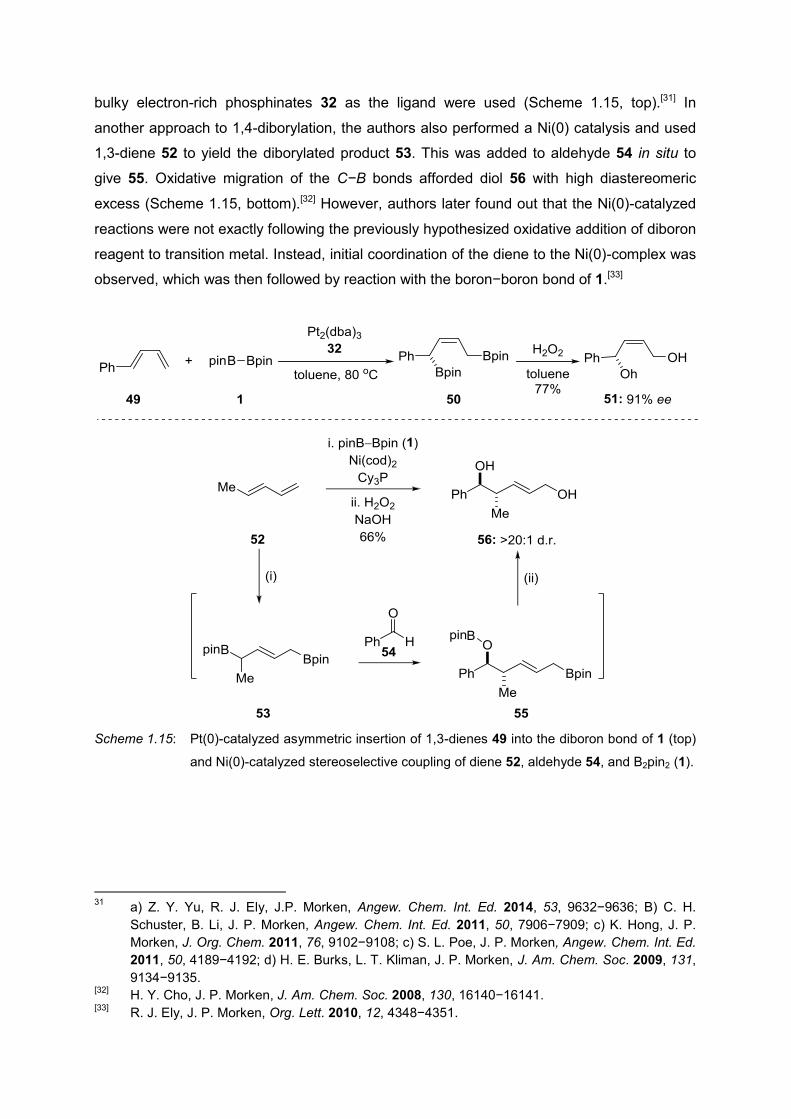
MIYAURA and co-workers reported the (Ph3P)4Pt (4)-catalyzed reaction between 1,3-dienes
45 and B2Pin2 1 to afford 1,4-diborylated product 46 (Scheme 1.14, top).[30] The authors
argued that the catalytic cycle is similar to that in Scheme 1.2; however, it goes through the
complex 47. The dimerized product 48 was observed when dba 25 was used as the ligand
instead of phosphine 4 (Scheme 1.14, bottom).
Scheme 1.14: Pt(0)-catalyzed insertion of 1,3-diene 45 into the diboron bond of 1 (top), dimerization
in the absence of phosphine ligands (bottom).
MORKEN and co-workers reported an enantioselective version of this reaction using 1-
substituted 1,3-dienes 49 to obtain 1,4-diborylated alkene 50. Oxidative degradation of 50
yielded the 1,4-diol 51 in high yield and enantiomeric excess. Pt2(dba)3 as the catalyst and
[29]
J. Ramírez, A. M. Segarra, E. Fernández, Tetrahedron: Asymmetry 2005, 16, 1289−1294. [30]
T. Ishiyama, M. Yamamoto, N. Miyaura, Chem. Commun. 1996, 2073−2074.
bulky electron-rich phosphinates 32 as the ligand were used (Scheme 1.15, top).[31] In
another approach to 1,4-diborylation, the authors also performed a Ni(0) catalysis and used
1,3-diene 52 to yield the diborylated product 53. This was added to aldehyde 54 in situ to
give 55. Oxidative migration of the C−B bonds afforded diol 56 with high diastereomeric
excess (Scheme 1.15, bottom).[32] However, authors later found out that the Ni(0)-catalyzed
reactions were not exactly following the previously hypothesized oxidative addition of diboron
reagent to transition metal. Instead, initial coordination of the diene to the Ni(0)-complex was
observed, which was then followed by reaction with the boron−boron bond of 1.[33]
Scheme 1.15: Pt(0)-catalyzed asymmetric insertion of 1,3-dienes 49 into the diboron bond of 1 (top)
and Ni(0)-catalyzed stereoselective coupling of diene 52, aldehyde 54, and B2pin2 (1).
31
a) Z. Y. Yu, R. J. Ely, J.P. Morken, Angew. Chem. Int. Ed. 2014, 53, 9632−9636; B) C. H.
Schuster, B. Li, J. P. Morken, Angew. Chem. Int. Ed. 2011, 50, 7906−7909; c) K. Hong, J. P.
Morken, J. Org. Chem. 2011, 76, 9102−9108; c) S. L. Poe, J. P. Morken, Angew. Chem. Int. Ed.
2011, 50, 4189−4192; d) H. E. Burks, L. T. Kliman, J. P. Morken, J. Am. Chem. Soc. 2009, 131,
9134−9135. [32]
H. Y. Cho, J. P. Morken, J. Am. Chem. Soc. 2008, 130, 16140−16141. [33]
R. J. Ely, J. P. Morken, Org. Lett. 2010, 12, 4348−4351.

1 Introduction 15
1.2.6 Insertion of Allenes into Diboron Bonds
The study of the advent of allene insertion into diboron bonds was studied by MIYAURA and
co-workers in 1998. 1,2-Diborylation of allenes was achieved by using Pt(0) catalysis.[34]
Monosubstituted allene 57 was diborylated at the internal double bond to afford 58b as the
major product and 58a as the minor product (Scheme 1.16, top), whereas disubstituted
allenes 59 was functionalized at terminal double bond to give 60a as the major product and
60b as the minor product (Scheme 1.16, middle). The authors also found that for substrate
such as para-nitrophenyl allene 61, (Ph3P)4Pt (4) yielded diborylation at the internal double
bond as in 62b and Pt(dba)2/PCy3 preferred diborylation at terminal double bond as in 62a
(Scheme 1.16, bottom).
[34]
T. Ishiyama, T. Kitano, N. Miyaura, Tetrahedron Lett. 1998, 39, 2357−2360.

Scheme 1.16: The insertion of monosubstituted allene 57 into the diboron bond of B2pin2 (1) (top),
the insertion of disubstituted allene 59 into the diboron bond of B2pin2 (1) (middle), and
effect of catalyst on the insertion of monosubstituted allene 61 into the diboron bond
of B2pin2 (1) (bottom).
Later, WU and co-workers found through computational studies that electron-withdrawing
substrates generally direct the diborylation at terminal double bonds and electron donating
substrates generally give the diborylation at internal double bonds.[35] Recently, SANTOS and
co-workers reported chemoselective diborylation of allene 63 using unsymmetrical diboron
reagent BpinBdan (3) employing Pt(dba)3 as the catalyst (Scheme 1.17).[36] The authors
found that the disubstituted allene 63 preferred the diborylation at the terminal double bonds,
and the Bdan group was transferred to the C3 position to give 64a as the major product and
64b as minor product. Alternatively, Pd(0)-and Pd(II)-catalyzed transformations have been
also studied by CHENG and co-workers where they have employed aryliodides as co-
catalysts.[37]
Scheme 1.17: The insertion of disubstituted allene 63 into unsymmetrical diboron reagent BpinBdan
(3)
1.2.7 Insertion of Arynes into Diboron Bonds
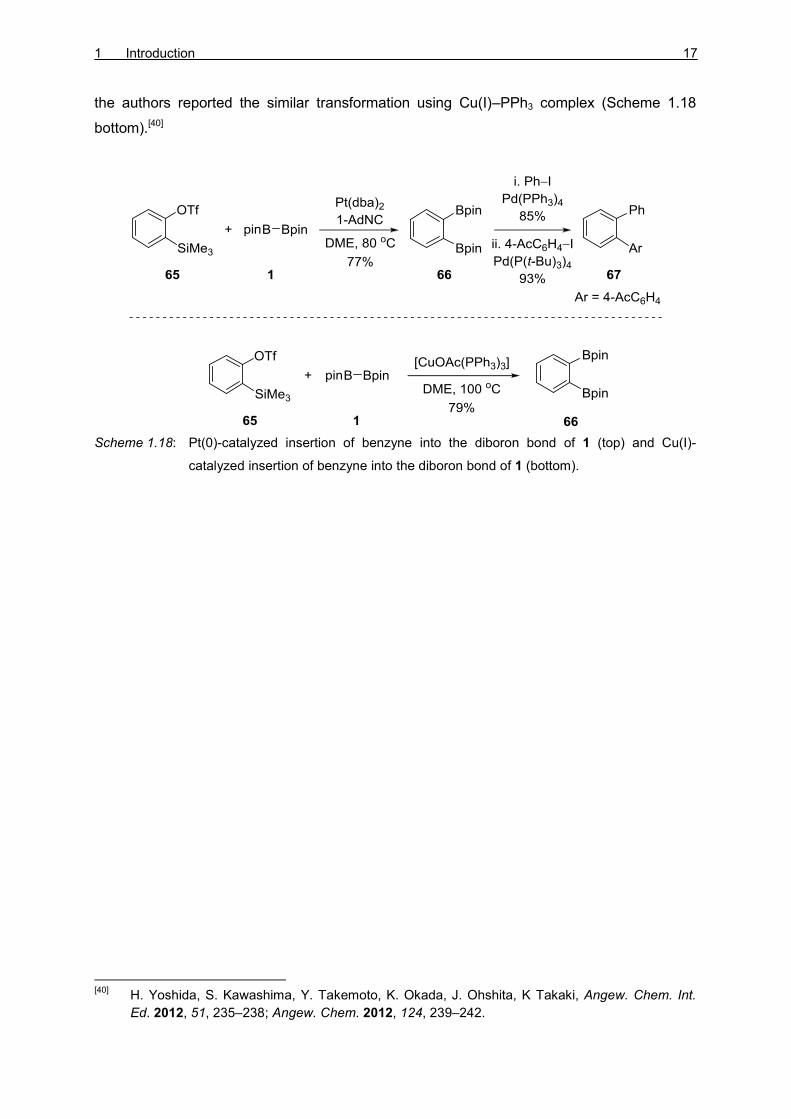
YOSHIDA and co-workers reported the first Pt(0)-catalyzed insertion of benzyne precursors 65
into the diboron bond of 1 in 2012 (Scheme 1.18 top).[38] Contrary to previously reported
procedures, this transformation was facilitated by isocyanide ligands instead of the
phosphine ligands. KOBAYASHI’s benzyne precursor 65 yielded diborylated product 66 in
presence of KF/18-Crown-6.[39] This diborylated product 66 was further subjected to a
stepwise SUZUKI–MIYAURA cross-coupling reaction to give a 1,2-diarylated product 67. Later,
[35]
M. Wang, L. Cheng, Z. Wu, Organometallics 2008, 27, 6464−6471. [36]
X. Guo, A. K. Nelson, C. Slebodnick, W. L. Santos, ACS Catal. 2015, 5, 2172−2176. [37]
F.-Y. Yang, C.-H. Cheng, J. Am. Chem. Soc. 2001, 123, 761−762. [38]
H. Yoshida, K. Okada, S. Kawashima, K. Tanino, J. Ohshita, Chem. Commun. 2010, 46, 1763–
1765. [39]
Y. Himeshima, T. Sonoda, H. Kobayashi, Chem. Lett. 1983, 1211−1214.
1 Introduction 17
the authors reported the similar transformation using Cu(I)–PPh3 complex (Scheme 1.18
bottom).[40]
Scheme 1.18: Pt(0)-catalyzed insertion of benzyne into the diboron bond of 1 (top) and Cu(I)-
catalyzed insertion of benzyne into the diboron bond of 1 (bottom).
[40]
H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita, K Takaki, Angew. Chem. Int.
Ed. 2012, 51, 235–238; Angew. Chem. 2012, 124, 239–242.

1.3 Application of Carbon–Boron Bonds in Carbon–Carbon Bond
Forming Reactions
1.3.1 SUZUKI–MIYAURA Cross-Coupling Reaction
SUZUKI and MIYAURA reported the catalytic use of palladium to couple 1-alkenylboronic
esters 68 with vinyl halides 69 to afford coupled product 70a and 70b in equal ratio (Scheme
1.19).[41] In the past, applications of the SUZUKI–MIYAURA cross-coupling in C–C bond-
forming reactions were studied in detail.[42]
Scheme 1.19: The SUZUKI–MIYAURA cross-coupling reaction between boronic ester 68 and vinyl
bromide 69.
The mechanism of the SUZUKI–MIYAURA cross-coupling has been explored in details
(Scheme 1.20).[43] The proposed catalytic cycle begins with the oxidative addition of halide
XXIII to activated 16-electron palladium species XXII to give the 18-electron complex XXIV
(XXII+XXIII→XXIV). There are two paths hypothesized for the transmetalation process.
According to the first pathway, the base XXVI first activates the boronic ester XXV to
generate boronate XXVII (XXV+XXVI→XXVII), which reacts with complex XXIV to afford the
[41]
a) N. Miyaura, K. Yamada, A. Suzuki, Tetrahedron Lett. 1979, 20, 3437–3440; b) N. Mivaura. K.
Yamada. H. Suginome, A. Suzuki. J. Am. Chem Soc. 1985, 107, 972–980. [42]
For the reviews and application of SUZUKI–MIYAURA reaction, see a) C. C. C. J. Seechurn, M. O.
Kitching, T. J. Colacot, V. Snieckus, Angew. Chem. Int. Ed. 2012, 51, 5062–5085; Angew.
Chem. 2012, 124, 5150–5174; b) A. Suzuki, Angew. Chem. Int. Ed. 2011, 50, 6722–6737;
Angew. Chem. 2011, 123, 6855–6869; c) J. S. Carey, D. Laffan, C. Thomson, M. T. Williams,
Org. Biomol. Chem. 2006, 4, 2337–2347. [43]
For a summary of studies of transmetalation in the SUZUKI–MIYAURA reaction, see: a) A. A.
Thomas, S. E. Denmark, Science 2016, 352, 329–332; b) B. P. Carrow, J. F. Hartwig, J. Am.
Chem. Soc. 2011, 133, 2116–2119; c) Y. Suzaki, K. Osakada, Organometallics 2006, 25, 3251–
3258; d) Y. Suzaki, K. Osakada, Organometallics 2006, 25, 3251–3258; e) A. A. C. Braga, N. H.
Morgen, G. Ujaque, F. Maseras, J. Am. Chem. Soc. 2005, 127, 9298–9307; f) L. J. Goossen, D.
Koley, H. L. Hermann, W. Thiel, J. Am. Chem. Soc. 2005, 127, 11102–11114; g) N. J. Miyaura,
J. Organomet. Chem. 2002, 653, 54–57.

1 Introduction 19
coupled product XXVIII (XXVII+XXIV→XXVIII). The second pathway starts with the
activation of complex XXIV with base XXVI to yield activated complex XXIX
(XXIV+XXVI→XXIX), which reacts with boronic acid XXV to produce product XXVIII and
activated 16 electron complex XXII (XXIX+XXV→XXVIII).
Scheme 1.20: Catalytic cycles for the SUZUKI–MIYAURA cross-coupling.
1.3.2 Orthogonal SUZUKI–MIYAURA Cross-Coupling Reaction
The SUZUKI–MIYAURA cross-coupling reaction provides an important approach to access
biaryl or polyene units in pharmaceutical and materials industries because of a facile
boronation.[44] The orthogonal SUZUKI–MIYAURA reaction facilitates the multiple
functionalization and gives a better control in target-oriented synthesis. The orthogonality is
generally achieved at the oxidative addition stage with respect to the halide (or
pseudohalide) components. The control over orthogonality is being either the nature of the
substrate or the catalyst system.[45] The trend of reactivity is X = N2+>I>Br>OTf>Cl.[46]
However, the focus of this thesis would be to look into the possible ways to achieve the
orthogonality at the transmetalation stage.
[44]
c) J. S. Carey, D. Laffan, C. Thomson, M. T. Williams, Org. Biomol. Chem. 2006, 4, 2337–2347;
b) A. D. Schlüter, J. Polym. Sci. 2001, 39, 1533–1556. [45]
a) A. R. Kapdi, D. Prajapati, RSC Adv. 2014, 4, 41245–41259; b) J.-R. Wang, K. Manabe,
Synthesis 2009, 1405–1427. [46]
P. Dobrounig, M. Trobe, R. Breinbauer, Monatsh. Chem. 2017, 148, 3–35.

1.3.2.1 Orthogonal SUZUKI–MIYAURA Cross-Coupling Reaction Controlled by Masked
Boronates
1.3.2.1.1 BF3K-Salts as the Masking Group
SANDROCK and MOLANDER synthesized the 1,2-diboryl species 73 by reacting alkenyl
trifluoroborate 71 with the 9-borabicyclo(3.3.1)nonane (9-BBN, 72) (Scheme 1.21).[47] When
this was subjected under water-free SUZUKI–MIYAURA conditions the trifluoroborate did not
undergo transmetalation as there is no possibility of fluoride-hydroxide exchange. This leads
to the reaction exclusively at the 9-BBN center to afford 74. The trifluoroborate 74 underwent
the reaction in presence of water, and the outcome of the sequence was 1,2-biarylated
ethane 75.
Scheme 1.21: Orthogonal SUZUKI–MIYAURA cross-coupling using trifluoroborate as the masking
group.
1.3.2.1.2 Boronic Amides as the Masking Group
Lone-pair donation from the nitrogen atom makes the boronic amides (Bdan) very stable,
yielding less reactive boron centers. This reactivity characteristic can be used to induce the
selectivity in the reaction, by having more reactive B(OR)2 groups in the system. SUGINOME
and co-workers used this strategy to obtain the orthogonal functionalization
[47]
a) G. A. Molander, D. L. Sandrock Org. Lett. 2009, 11, 2369–2372; b) G. A. Molander, D. L.
Sandrock, J. Am. Chem. Soc. 2008, 130,15792–15793.

1 Introduction 21
(Scheme 1.22).[48] In the earlier reports, biphenyl 78 was obtained from simple building
blocks 76 and 77. The authors have employed boronic amide 77 and boronic acid 76 in the
same system and selectively coupled the boronic acid center, while the Bdan center was left
as it is in the cross coupling reaction.
Scheme 1.22: Orthogonality using Bdan group and synthesis of 78.
1.3.2.1.3 N-Methyliminodiacetic Acid (MIDA) Boronates as the Masking Group
BURKE and co-workers reported an alternative masking group for boron by using the MIDA
group.[49] The LEWIS acidity of the boron atom in such moieties is reduced by the electron-
pair donation of the nitrogen. The use of the MIDA has been reported in various synthesis of
natural products.[50] WATSON reported exclusive coupling at the Bpin center of 79 in presence
of a B(MIDA) center in 80, followed by the subsequent conversion of MIDA to hydroxy group
in 81 (Scheme 1.23).[51]
Scheme 1.23: Orthogonal SUZUKI–MIYAURA cross-coupling using the MIDA group as the masking
group.
[48]
N. Iwadate, M. Suginome, Org. Lett. 2009, 11, 1899–1903; b) H. Noguchi, T. Shioda, C-M.
Chou, M. Suginome, Org. Lett. 2008, 10, 377–380; c) H. Noguchi, K. Hojo, M. Suginome, J. Am.
Chem. Soc. 2007, 129, 758–759. [49]
S. G. Ballmer, E. P. Gillis, M. D. Burke, Org. Synth. 2009, 86, 344–359. [50]
a) E. M. Woerly, J. Roy, M. D. Burke, Nat. Chem. 2014, 6, 484–491; b) S. Fujii, S. Y. Chang, M.
D. Burke, Angew. Chem. Int. Ed. 2011, 50, 7862–7864; Angew. Chem. 2011, 123, 8008–8010;
c) S. J. Lee, T. M. Anderson, M. D. Burke, Angew. Chem. Int. Ed. 2010, 49, 8860–8863; Angew.
Chem. 2010, 122, 9044–9047; d) E. M. Woerly, A. H. Cherney, E. K. Davis, M. D. Burke, J. Am.
Chem. Soc. 2010, 132, 6941–6943; e) E. P. Gillis, M. D. Burke, J. Am. Chem. Soc. 2007, 129,
6716–6718; [51]
J. W. B. Fyfe, E. Valverde, C. P. Seath, A. R. Kennedy, J. M. Redmond, N. A. Anderson, A. J.
B. Watson, Chem. Eur. J. 2015, 21, 8951–8964.

1.3.2.2 Orthogonal SUZUKI–MIYAURA Cross-Coupling Reaction Controlled by
Electronic and Steric Properties of the Substrate
Recently, the orthogonality in identically protected boronic esters has been explored. The
initial reports focused on 1,1-diborylalkanes, where one boryl group selectively underwent
SUZUKI–MIYAURA cross-coupling.[52] Structurally complex 1,2-diborylalkanes were studied by
MORKEN and co-workers, the authors were able to perform the asymmetric diborylation of
terminal alkene 33 to obtain 82 and subsequently coupling at the terminal boryl group
(Scheme 1.24).[53] The internal boryl group was converted into a hydroxy group, and the
overall sequence furnished the enantioselective synthesis of 2-arylated secondary alcohol
83.
Scheme 1.24: Asymmetric insertion of alkene 33 into the diboron bond of 1 and subsequent
orthogonal SUZUKI–MIYAURA cross-coupling.
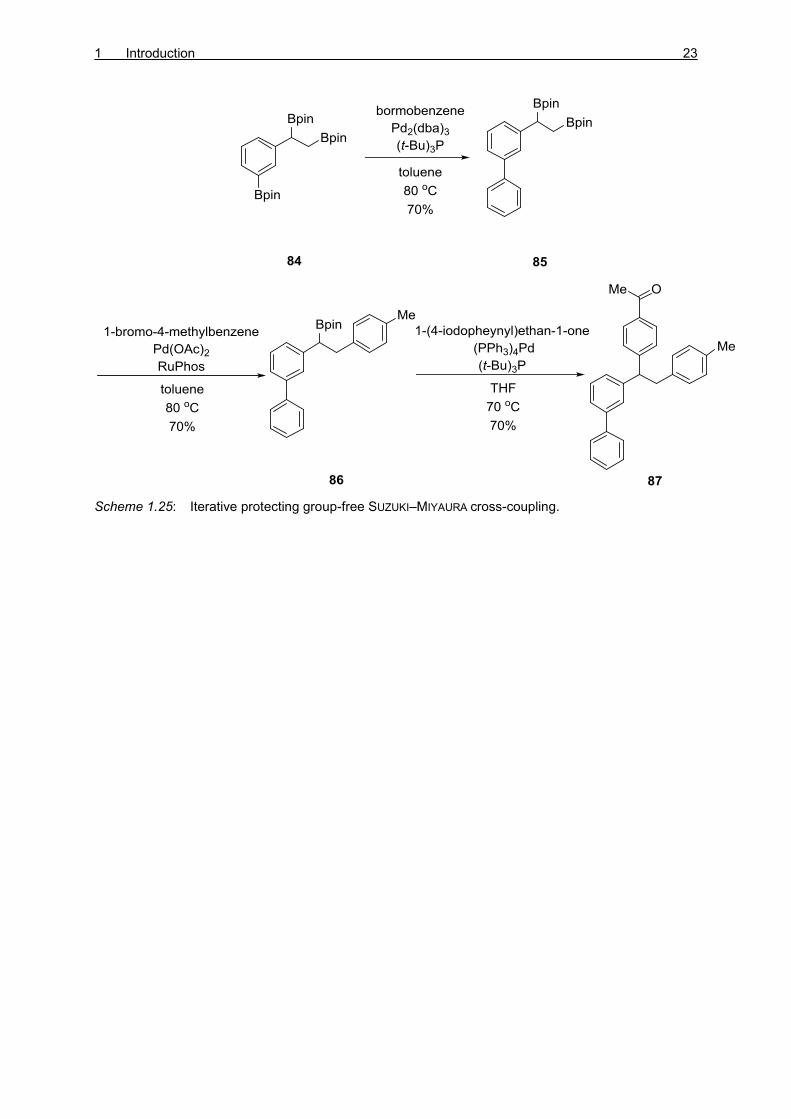
CRUDDEN and co-workers focused their work on the inherent differences in the
transmetalation efficiency of different C–B bonds to establish an iterative cross-coupling of
multiply borylated organic compounds having identical protecting group (Scheme 1.25).[54]
The authors chose the triborylated 84 as the starting material. The first coupling occurs at the
sp2 center when Pd2(dba)3 was used to give the di-borylated 85 which in presence of
Pd(OAc)2, afforded coupling at the terminal position to produce secondary borylated 86. The
last boryl group reacts to give branched 87.
[52]
K. Endo, T. Ohkubo, M. Hirokami, T. Shibata, J. Am. Chem. Soc. 2010 ,132, 11033–11035. [53]
S. N. Mlynarski, C. H. Schuster, J. P. Morken, Nature 2014, 505, 386–390. [54]
C. M Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo, S. Voth,
M. Hutchinson, V. S. Laberge, Y. Maekawa, D. Imao, Nat. Commun. 2016, 7, 11065–11071.
1 Introduction 23
Scheme 1.25: Iterative protecting group-free SUZUKI–MIYAURA cross-coupling.

1.4 Stannylation of Electrophilic Acceptors involving Activation of Tin–
Silicon and Tin–Tin Bonds
1.4.1 Activation of Tin–Silicon Bonds by Nucleophiles
The nucleophilic activation of tin–silicon bonds to generate stannyl anions was first reported
by CHENARD and co-workers (Scheme 1.26, top).[55] The authors utilized naked cyanide
anions 89 to generate the stannyl anion 90 from silylstannane 88, which was then
transferred to α,β-unsaturated ketones and acetylenes. It was believed that the mechanism
of the stannyl anion generation is ionic. Later, in the same year MORI and co-workers
produced the stannyl anion by the reaction between silylstannane 88 and quaternary
ammonium chloride 92 (Scheme 1.26, middle).[56] The authors hypothesized that the
generation of the stannyl anion was accomplished through a penta-coordinate silicon
species 93. In the same report the authors have also looked into the 1,4-stannylation of the
α,β-unsaturated ketones 95 to afford 96. Other silylstannanes 97 and 98 were also explored
in the past (Scheme 1.26, bottom).[57]
Scheme 1.26: The first activation of silylstannane 88 using cynide anions 89 (top), quaternary
ammonium chloride mediated stannylation of α,β-unsaturated ketones (middle), and
some common silylstannanes (bottom).
[55]
B. L. Chenard, E. D. Laganis, F. Davidson, T. V. R. Babu, J. Org. Chem. 1985, 50, 3666–3667. [56]
M. Mori, N. Kaneta, N. Isono, M. Shibasaki, J. Organomet. Chem. 1993, 455, 255–260. [57]
N. Isono, M. Mori, J. Org. Chem. 1998, 63, 1773–1779.

1 Introduction 25
1.4.2 Stannylation of Electrophilic Acceptors involving Activation of Tin–Silicon
Bond by Nucleophiles
CHENARD and co-workers transferred the stannyl anion to α,β-unsaturated ketones and
acetylenes.[55] Similar reactivity was observed by MORI and co-workers in their preliminary
studies.[56] Later, MORI and co-workers reported the 1,4-addition of stannyl anions to 99 to
give enolate 100, which then gave cyclized product 101 (Scheme 1.27, top).[58] FALCK and
co-worker accomplished the addition of Sn–Si bond over carbonyl group of 102, which after
hydrolysis, gave the α-hydroxy stannane 103 (Scheme 1.27, bottom). In the same report, the
authors accomplished chiral induction using chiral ammonium cyanide salt 104, however, the
enantiomeric excess was low.[59]
Scheme 1.27: 1,4-Stannylation of α,β-unsaturated ester 99 and subsequent cyclization (top) and
1,2-stannylation of aldehyde 102 (bottom).
MORI and co-workers observed an interesting reactivity of methyl propiolate 105 to obtain the
bis-stannylated compound 108 (Scheme 1.28, top). The proposed mechanism includes the
generation of allene 106, which yields the α,β-unsaturated ester 107 in situ, and a second
molecule of silylstannane 88 then reacts in a 1,4-fashion to give the bis-stannylated product
108.[60] OESTREICH and co-workers developed the first water tolerant base mediated
1,4-stannylation of α,β-unsaturated acceptors 109 and 95 to afford ester 110 and ketone 111
[58]
T. Honda, M. Mori, Chem. Lett. 1994, 1013–1016. [59]
R. K. Bhatt, J. Ye, J.R. Falck, Tetrahedron. Lett. 1994, 35. 4081–4084. [60]
a) N. Isono, M. Mori, Main Group Metal Chem. 1996, 19, 277–279; b) N. Isono, M. Mori,
Tetrahedron Lett. 1995, 36, 9345–9347.
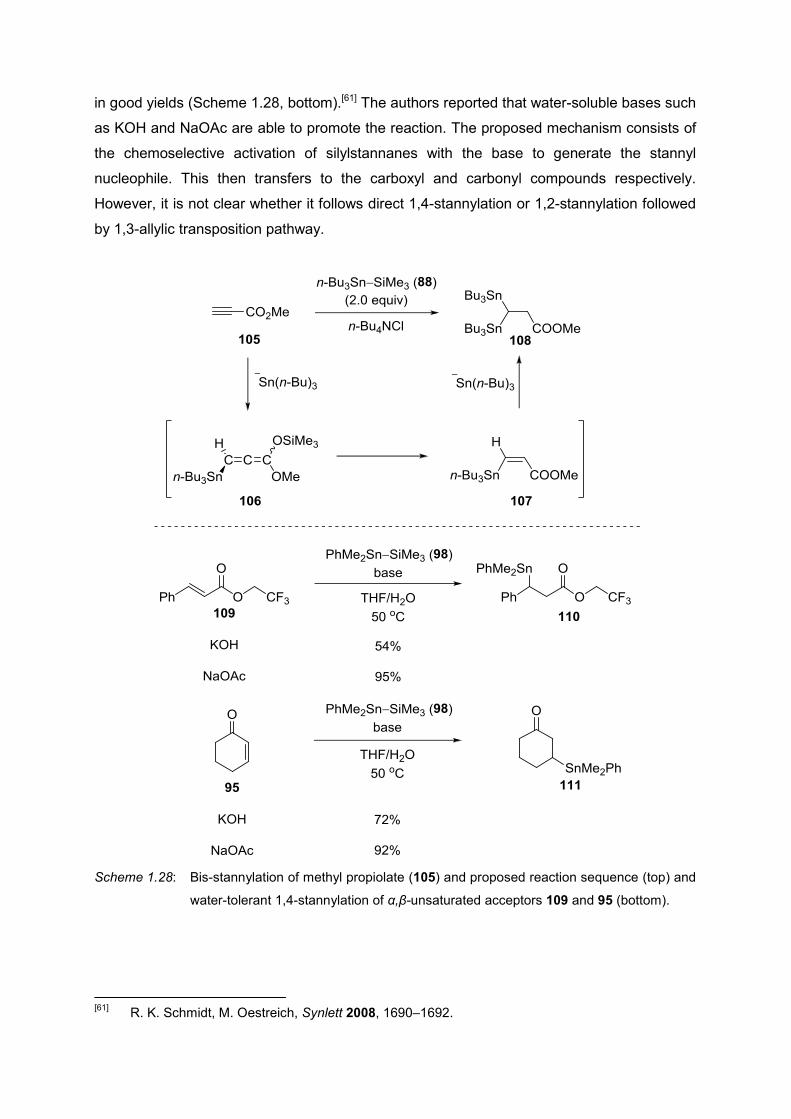
in good yields (Scheme 1.28, bottom).[61] The authors reported that water-soluble bases such
as KOH and NaOAc are able to promote the reaction. The proposed mechanism consists of
the chemoselective activation of silylstannanes with the base to generate the stannyl
nucleophile. This then transfers to the carboxyl and carbonyl compounds respectively.
However, it is not clear whether it follows direct 1,4-stannylation or 1,2-stannylation followed
by 1,3-allylic transposition pathway.
Scheme 1.28: Bis-stannylation of methyl propiolate (105) and proposed reaction sequence (top) and
water-tolerant 1,4-stannylation of α,β-unsaturated acceptors 109 and 95 (bottom).
[61]
R. K. Schmidt, M. Oestreich, Synlett 2008, 1690–1692.
1 Introduction 27
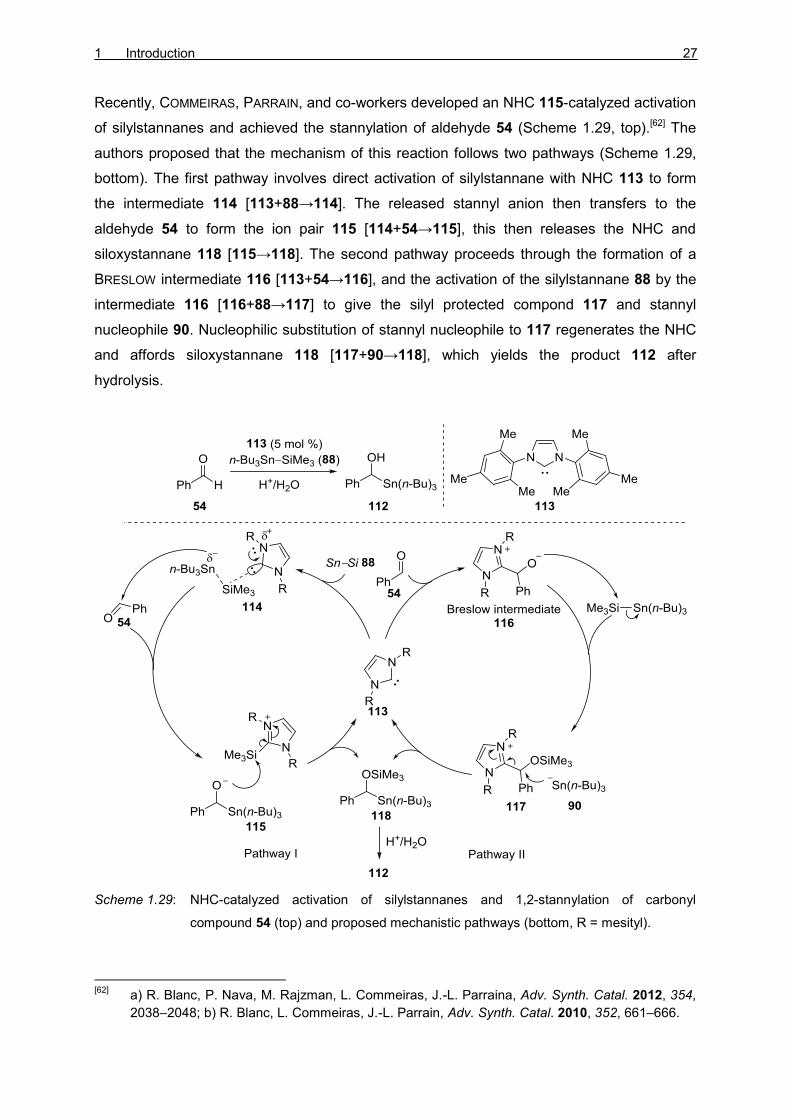
Recently, COMMEIRAS, PARRAIN, and co-workers developed an NHC 115-catalyzed activation
of silylstannanes and achieved the stannylation of aldehyde 54 (Scheme 1.29, top).[62] The
authors proposed that the mechanism of this reaction follows two pathways (Scheme 1.29,
bottom). The first pathway involves direct activation of silylstannane with NHC 113 to form
the intermediate 114 [113+88→114]. The released stannyl anion then transfers to the
aldehyde 54 to form the ion pair 115 [114+54→115], this then releases the NHC and
siloxystannane 118 [115→118]. The second pathway proceeds through the formation of a
BRESLOW intermediate 116 [113+54→116], and the activation of the silylstannane 88 by the
intermediate 116 [116+88→117] to give the silyl protected compond 117 and stannyl
nucleophile 90. Nucleophilic substitution of stannyl nucleophile to 117 regenerates the NHC
and affords siloxystannane 118 [117+90→118], which yields the product 112 after
hydrolysis.
Scheme 1.29: NHC-catalyzed activation of silylstannanes and 1,2-stannylation of carbonyl
compound 54 (top) and proposed mechanistic pathways (bottom, R = mesityl).
[62]
a) R. Blanc, P. Nava, M. Rajzman, L. Commeiras, J.-L. Parraina, Adv. Synth. Catal. 2012, 354,
2038–2048; b) R. Blanc, L. Commeiras, J.-L. Parrain, Adv. Synth. Catal. 2010, 352, 661–666.

1.4.3 Activation of Tin–Tin Bonds by Transmetalation
The activation of the Sn–Sn bond of 119 via transmetalation follows two pathways. First,
activation by a strong nucleophile or base which then transmetalates with a counter cation to
form an activated stannyl nucleophile 120 (Scheme 1.30, top). Second, activation pathway
involves the σ-bond metathesis between Sn–Sn bond of 119 and 121 to give transition state
122, which then collapses to give activated stannyl nucleophile equivalent 123 (Scheme
1.30, bottom). The transfer of stannyl nucleophiles generated by activation of Sn−Sn bond to
the electrophilic compounds is well established.[63] However, the most common method to
access the stannyl nucleophile via transmetalation of Sn−Sn bond with anionic nucleophiles.
Stannyl-nucleophile generation using transmetalation employing σ-bond metathesis between
Sn−Sn bond of the 119 and 121 has not been studied.
Scheme 1.30: Modes of activation of Sn−Sn bond using transmetalation approaches.
1.4.4 Formation of Tin–Metal Bonds by using Tin–Tin Bonds
Organotin compounds having Sn–M (M = Li 125, Na 126, K 127) bonds are important
reagents in synthesis. These reagents are generally synthesized by using transmetalation of
compound 124 with organo-lithium reagents, sodium naphthalenide, and potassium hydride
(Scheme 1.31).[63,64] The reactions are generally carried out in liquid ammonia or in an ether
solvent to facilitate the single electron-transfer process.[65] Sn–M bonds of alkali earth metals
and transition metals are not obtained by the direct transmetalation using anionic
nucleophiles.[63]
[63]
a) A. G. Davies, Organotin Chemistry, Wiley-VCH, Weinheim, 2004; b) M. S. Holt, W. L. Wilson,
J. H. Nelson, Chem. Rev. 1989, 89, 11–48. [64]
R. J. P. Corriu, C. Guerin, J. Organomet. Chem. 1980, 197, C19–C20. [65]
K. R. Wursthorn, H. G. Kuivila, G. F. Smith, J. Am. Chem. Soc. 1978, 100, 2779–2789.

1 Introduction 29
Scheme 1.31: Generation of Sn–M bond (M = Li, Na, K).
1.4.5 Conjugate Addition of Tin nucleophiles using Silyl-Cuprates to Electrophilic
Organic Compounds
The initial study from HUDAC established the first formation of stannnyl cuprates
(Me3Sn)2CuLi from trimethylstannyl lithium, which subsequently afforded 1,4-stannylation of
α,β-unsaturated carbonyl compounds.[66] However, the use of such cuprates was limited
because of the high reactivity of Sn–Li reagents. The first use of distannane 128 to
synthesize stannnyl cuprate 129 was reported by PIERS and co-workers (Scheme 1.32, top).
The authors achieved the transmetalation using methyllithium (MeLi) to obtain stannyl lithium
reagent, which was then reacted to form a thienyl stabilized stannnyl-cuprate complex 129.
The stannnylcuprate was convenient to use and underwent reactions with α,β-unsaturated
carbonyl compounds at higher temperatures (Scheme 1.32, bottom).[67]
Scheme 1.32: Thiophene stabilized tin-cuprate 129 formation (top), formation of mixed cuprate 130,
and 1,4 stannylation using 129 and 130.
[66]
J. Hudac, J. Chem. Soc.,Perkin Trans. 1 1975, 1020–1021. [67]
E. Piers, R. D. Tillyer, J. Org. Chem. 1988, 53, 5366–5369.

Later, SINGH and co-workers employed distannanes 128 to develop the mixed stannnyl
cuprate 130 with equimolar concentrations of MeLi and copper(I) source (Scheme 1.32,
middle). The mixed cuprate was able to selectively transfer stannyl group to α,β-unsaturated
carbonyl compound 95 to give 131 (Scheme 1.32, bottom).[68]
[68]
A. C. Cehlschlager, M. W. Hutzinger, R. Aksela, S. Sharma, S. M. Singh, Tetrahedron Lett. 1990,
31, 165–168.

1 Introduction 31
1.5 Objective
As showcased, the activation of various interelement bonds (E–E = B–B, Sn–Si, Sn–Sn) was
achieved by numerous methods in the past. One of the applications of such activation is the
formation of carbon–element bond, which acts as the linchpin for the formation of new
carbon–carbon bonds.
Previously, the activation of boron−boron bond afforded the borylation of different
unsaturated carbon–carbon bonds, and further SUZUKI–MIYAURA cross-coupling of these
borylated species formed new carbon–carbon bonds. The orthogonality at the
transmetalation stage of SUZUKI–MIYAURA cross-coupling has not been studied extensively.
YOSHIDA’s Pt(0) and Cu(I)-catalyzed insertions of benzyne into diboron bond generates two
boryl centers for the cross-coupling reaction.[38,40] However, these boron centers are
identical, and the issue of regioselectivity in SUZUKI–MIYAURA cross-coupling does not arise.
Indole moieties are ubiquitous in nature and many natural products have the indole core as
the main structural motif. Extension of YOSHIDA’s work to indolynes 135–137 will open the
access to rather unreactive 6-membered ring of indole. The unsymmetrical nature of indole,
however, raises the issue of regioselectivity in subsequent SUZUKI–MIYAURA cross-coupling
of the diborylated indole 138–140. The objective of this thesis is to synthesize precursors for
all the regioisomers of indolynes (132–134, Scheme 1.33, top), followed by insertion of
indolynes (135–137) into the diboron bond of 1 [135–137→138–140]. Two different
pathways are assumed for the iterative SUZUKI–MIYAURA cross-coupling reaction of 140
(Scheme 1.33, bottom), first, C7 position of indole undergoes reaction to give 141 which after
second coupling gives bis-coupled 142. Second, Bpin group at C6 reacts first to yield 143’,
and bis-coupled 144 in subsequent SUZUKI–MIYAURA cross-coupling. It is envigased that the
iterative coupling reaction will follow one of the pathways.
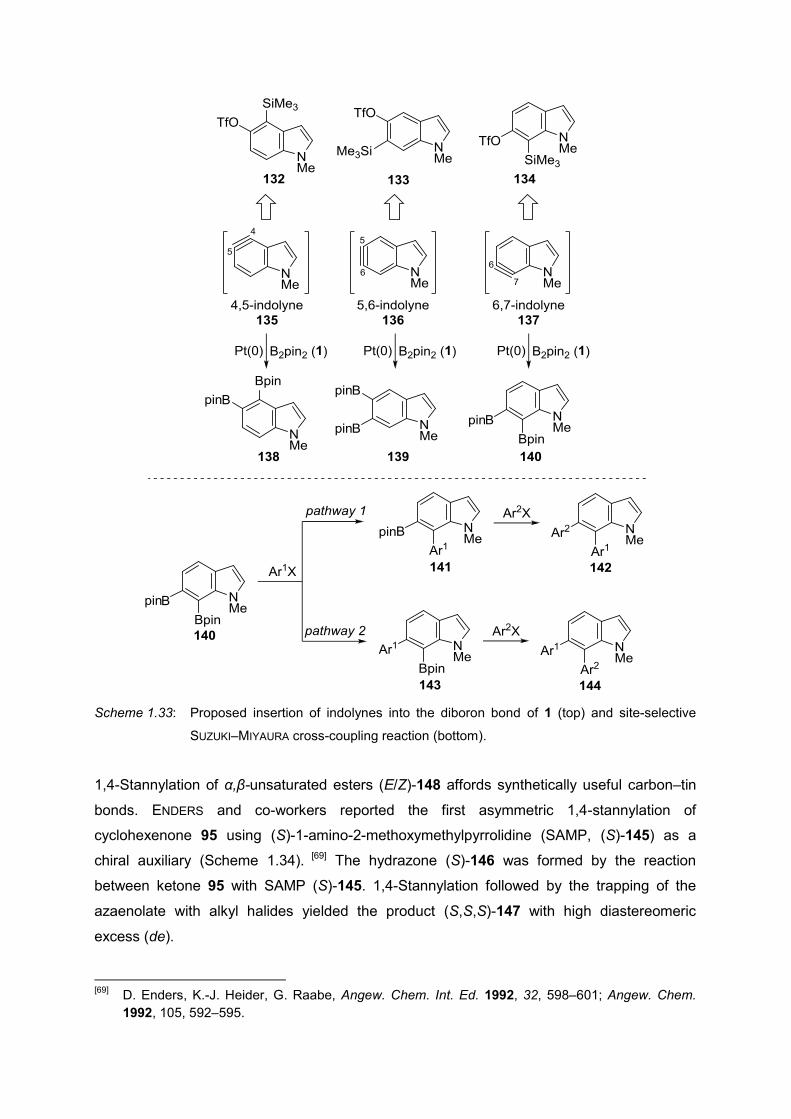
Scheme 1.33: Proposed insertion of indolynes into the diboron bond of 1 (top) and site-selective
SUZUKI–MIYAURA cross-coupling reaction (bottom).
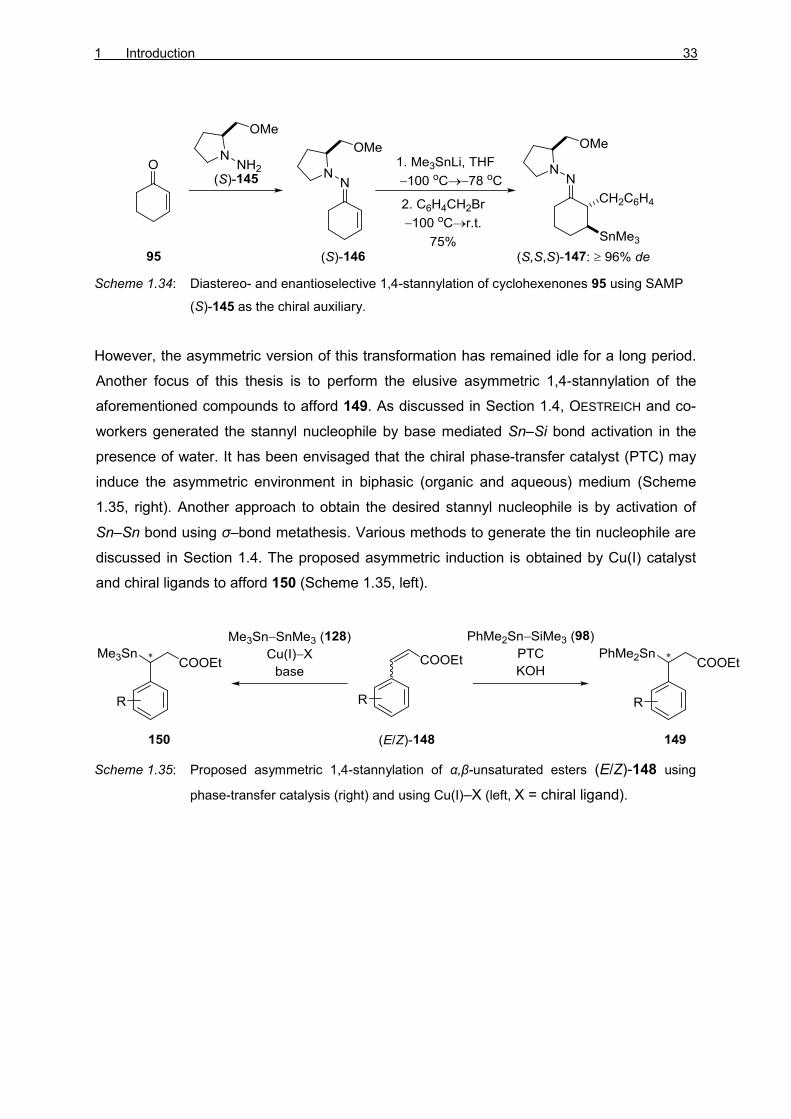
1,4-Stannylation of α,β-unsaturated esters (E/Z)-148 affords synthetically useful carbon–tin
bonds. ENDERS and co-workers reported the first asymmetric 1,4-stannylation of
cyclohexenone 95 using (S)-1-amino-2-methoxymethylpyrrolidine (SAMP, (S)-145) as a
chiral auxiliary (Scheme 1.34). [69] The hydrazone (S)-146 was formed by the reaction
between ketone 95 with SAMP (S)-145. 1,4-Stannylation followed by the trapping of the
azaenolate with alkyl halides yielded the product (S,S,S)-147 with high diastereomeric
excess (de).
[69]
D. Enders, K.-J. Heider, G. Raabe, Angew. Chem. Int. Ed. 1992, 32, 598–601; Angew. Chem.
1992, 105, 592–595.
1 Introduction 33
Scheme 1.34: Diastereo- and enantioselective 1,4-stannylation of cyclohexenones 95 using SAMP
(S)-145 as the chiral auxiliary.
However, the asymmetric version of this transformation has remained idle for a long period.
Another focus of this thesis is to perform the elusive asymmetric 1,4-stannylation of the
aforementioned compounds to afford 149. As discussed in Section 1.4, OESTREICH and co-
workers generated the stannyl nucleophile by base mediated Sn–Si bond activation in the
presence of water. It has been envisaged that the chiral phase-transfer catalyst (PTC) may
induce the asymmetric environment in biphasic (organic and aqueous) medium (Scheme
1.35, right). Another approach to obtain the desired stannyl nucleophile is by activation of
Sn–Sn bond using σ–bond metathesis. Various methods to generate the tin nucleophile are
discussed in Section 1.4. The proposed asymmetric induction is obtained by Cu(I) catalyst
and chiral ligands to afford 150 (Scheme 1.35, left).
Scheme 1.35: Proposed asymmetric 1,4-stannylation of α,β-unsaturated esters (E/Z)-148 using
phase-transfer catalysis (right) and using Cu(I)–X (left, X = chiral ligand).


2 Insertion of Indolynes into the B−B Bond and Site-Selective SUZUKI−MIYAURA Reaction 35
2 PLATINUM(0)-CATALYZED INDOLYNE INSERTION INTO
BIS(PINACOLATO)DIBORON FOLLOWED BY SITE-SELECTIVE
SUZUKI–MIYAURA CROSS-COUPLING
2.1 Introduction to Indolynes
Indolynes are a subclass of arynes GARG and co-workers studied the synthesis of various
regioisomers of indolyne precursors.[70] It is established that the indolynes show similar
reactivity as arynes because the indolynes also undergo representative reactions of arynes.
[4+2]-and [2+2]-Cycloaddition reactions were performed to afford indole 151, 152, and 153
(Scheme 2.1).
Scheme 2.1: [4+2]-and [2+2]-Cycloaddition reactions of indolyne 135.
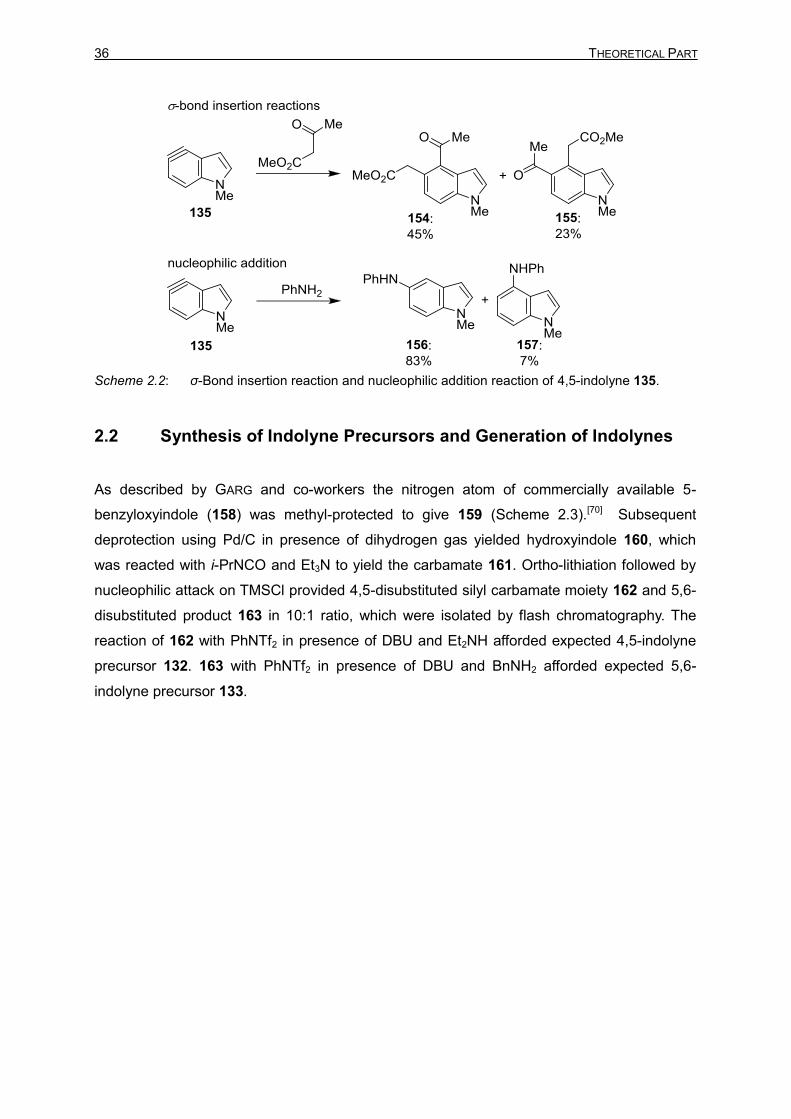
Insertion of indolyne 135 into a carbon−carbon σ-bond gave a mixture of 154 and 155.
Nucleophilic addition of aniline yielded 156 as the major product and 157 as the minor
product (Scheme 2.2). Later, the authors reported the computational proof for reactivity and
regioselectivity of various isomers of indolynes.[71]
[70]
a) G.-Y. J. Im, S. M. Bronner, A. E. Goetz, R. S. Paton, P. H.-Y. Cheong, K. N. Houk, N. K.
Garg, J. Am. Chem. Soc. 2010, 132, 17933–17944; b) S. M. Bronner, K. B. Bahnck, N. K. Garg,
Org. Lett. 2009, 11, 1007–1011. [71]
P. H.-Y. Cheong, R. S. Paton, S. M. Bronner, G.-Y. J. Im, N. K. Garg, K. N. Houk, J. Am. Chem.
Soc. 2010, 132, 1267–1269.
36 THEORETICAL PART
Scheme 2.2: σ-Bond insertion reaction and nucleophilic addition reaction of 4,5-indolyne 135.
2.2 Synthesis of Indolyne Precursors and Generation of Indolynes
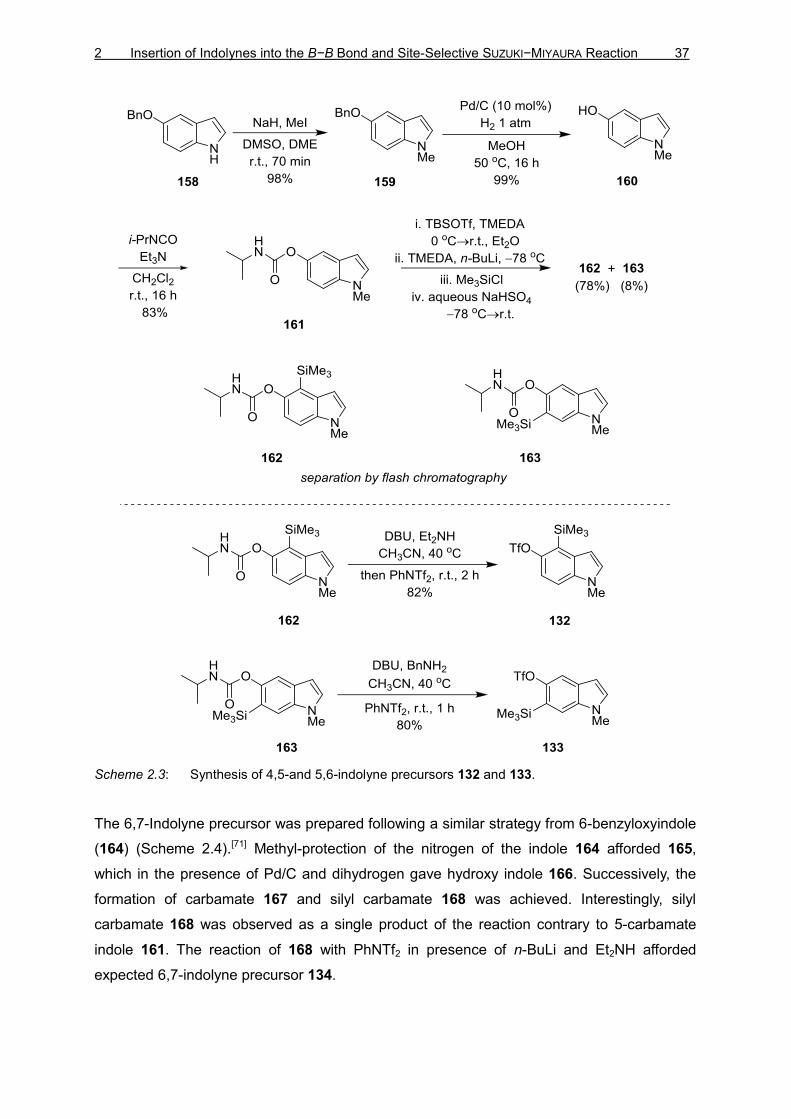
As described by GARG and co-workers the nitrogen atom of commercially available 5-
benzyloxyindole (158) was methyl-protected to give 159 (Scheme 2.3).[70] Subsequent
deprotection using Pd/C in presence of dihydrogen gas yielded hydroxyindole 160, which
was reacted with i-PrNCO and Et3N to yield the carbamate 161. Ortho-lithiation followed by
nucleophilic attack on TMSCl provided 4,5-disubstituted silyl carbamate moiety 162 and 5,6-
disubstituted product 163 in 10:1 ratio, which were isolated by flash chromatography. The
reaction of 162 with PhNTf2 in presence of DBU and Et2NH afforded expected 4,5-indolyne
precursor 132. 163 with PhNTf2 in presence of DBU and BnNH2 afforded expected 5,6-
indolyne precursor 133.
2 Insertion of Indolynes into the B−B Bond and Site-Selective SUZUKI−MIYAURA Reaction 37
Scheme 2.3: Synthesis of 4,5-and 5,6-indolyne precursors 132 and 133.
The 6,7-Indolyne precursor was prepared following a similar strategy from 6-benzyloxyindole
(164) (Scheme 2.4).[71] Methyl-protection of the nitrogen of the indole 164 afforded 165,
which in the presence of Pd/C and dihydrogen gave hydroxy indole 166. Successively, the
formation of carbamate 167 and silyl carbamate 168 was achieved. Interestingly, silyl
carbamate 168 was observed as a single product of the reaction contrary to 5-carbamate
indole 161. The reaction of 168 with PhNTf2 in presence of n-BuLi and Et2NH afforded
expected 6,7-indolyne precursor 134.

38 THEORETICAL PART
Scheme 2.4: Synthesis of 6,7-indolyne precursor 134.
The indolynes were generated using the protocol reported by GARG and co-workers,[70,71] the
authors established that the silyltriflates 132–134 afforded the indolynes 135–137 in situ,
when treated with a fluoride source (Scheme 2.5, top). To verify the formation of indolynes
using a fluride source, [4+2]-cycloaddition reactions were performed with 4,5-indolyne
precursor XX and CsF as the fluride source (Scheme 2.5, bottom). The results were in
accordance with those reported,[70a] proving the reproducibility of the transfomation.
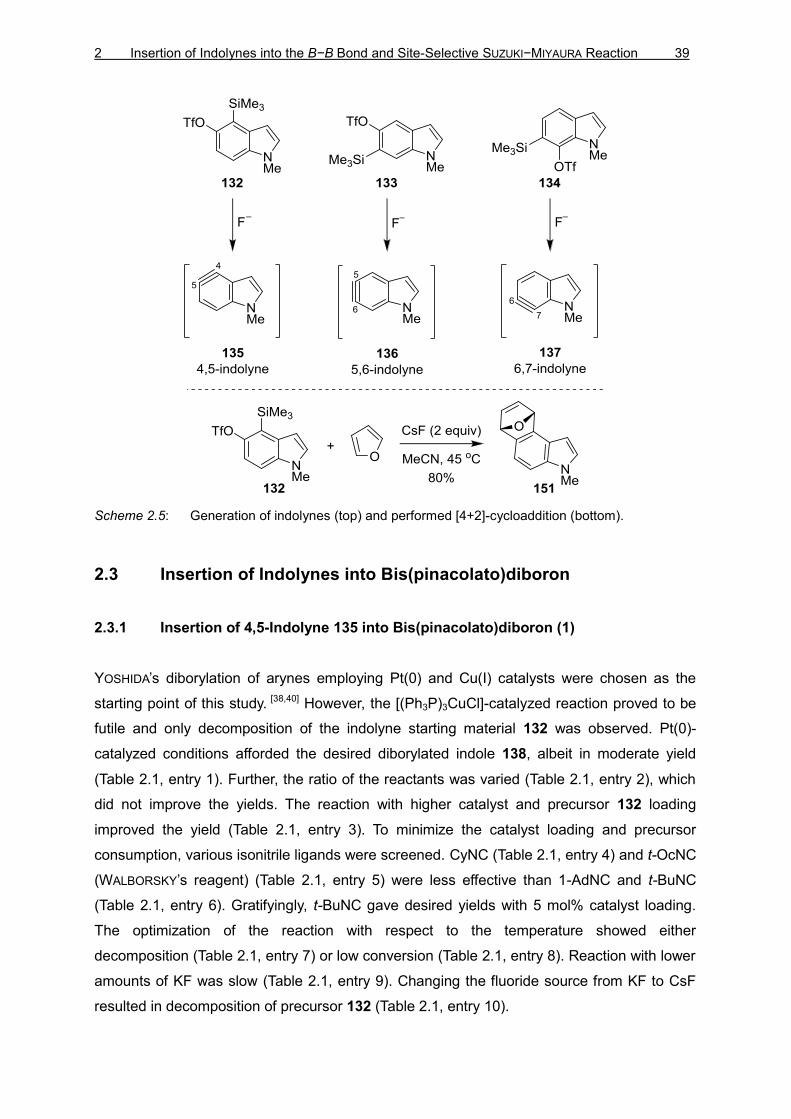
2 Insertion of Indolynes into the B−B Bond and Site-Selective SUZUKI−MIYAURA Reaction 39
Scheme 2.5: Generation of indolynes (top) and performed [4+2]-cycloaddition (bottom).
2.3 Insertion of Indolynes into Bis(pinacolato)diboron
2.3.1 Insertion of 4,5-Indolyne 135 into Bis(pinacolato)diboron (1)
YOSHIDA’s diborylation of arynes employing Pt(0) and Cu(I) catalysts were chosen as the
starting point of this study. [38,40] However, the [(Ph3P)3CuCl]-catalyzed reaction proved to be
futile and only decomposition of the indolyne starting material 132 was observed. Pt(0)-
catalyzed conditions afforded the desired diborylated indole 138, albeit in moderate yield
(Table 2.1, entry 1). Further, the ratio of the reactants was varied (Table 2.1, entry 2), which
did not improve the yields. The reaction with higher catalyst and precursor 132 loading
improved the yield (Table 2.1, entry 3). To minimize the catalyst loading and precursor
consumption, various isonitrile ligands were screened. CyNC (Table 2.1, entry 4) and t-OcNC
(WALBORSKY’s reagent) (Table 2.1, entry 5) were less effective than 1-AdNC and t-BuNC
(Table 2.1, entry 6). Gratifyingly, t-BuNC gave desired yields with 5 mol% catalyst loading.
The optimization of the reaction with respect to the temperature showed either
decomposition (Table 2.1, entry 7) or low conversion (Table 2.1, entry 8). Reaction with lower
amounts of KF was slow (Table 2.1, entry 9). Changing the fluoride source from KF to CsF
resulted in decomposition of precursor 132 (Table 2.1, entry 10).
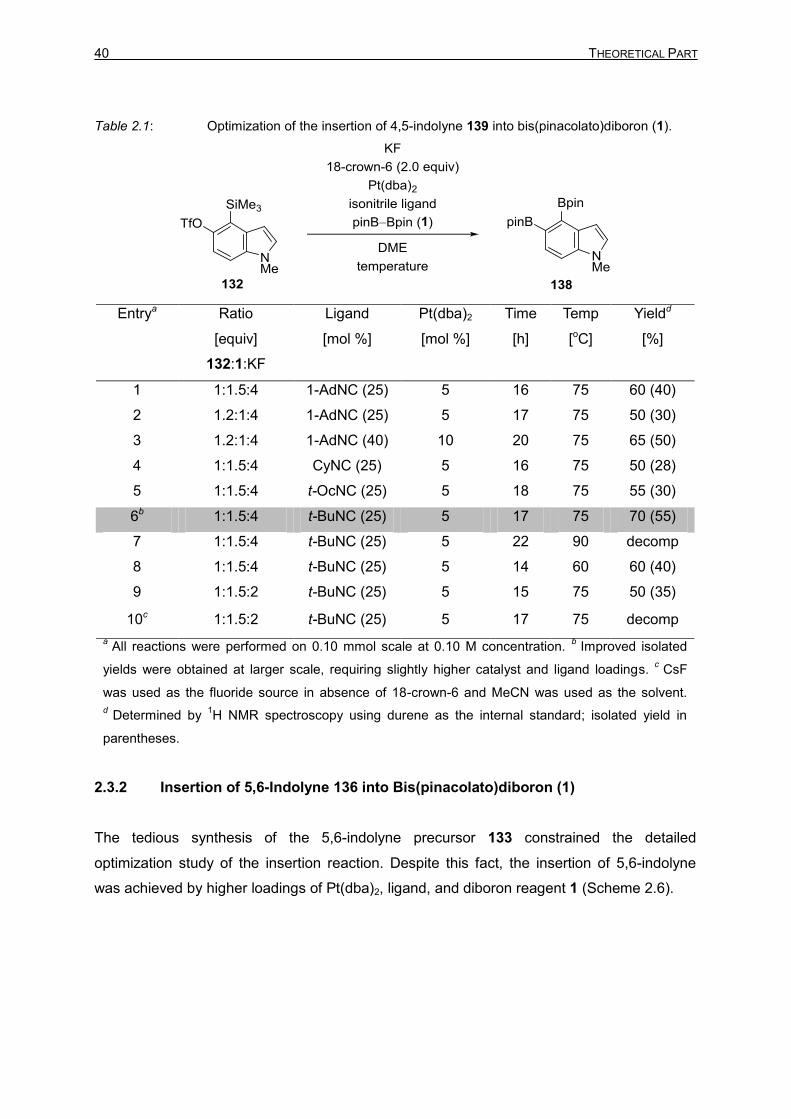
40 THEORETICAL PART
Table 2.1: Optimization of the insertion of 4,5-indolyne 139 into bis(pinacolato)diboron (1).
Entrya Ratio
[equiv]
132:1:KF
Ligand
[mol %]
Pt(dba)2
[mol %]
Time
[h]
Temp
[oC]
Yieldd
[%]
1 1:1.5:4 1-AdNC (25) 5 16 75 60 (40)
2 1.2:1:4 1-AdNC (25) 5 17 75 50 (30)
3 1.2:1:4 1-AdNC (40) 10 20 75 65 (50)
4 1:1.5:4 CyNC (25) 5 16 75 50 (28)
5 1:1.5:4 t-OcNC (25) 5 18 75 55 (30)
6b 1:1.5:4 t-BuNC (25) 5 17 75 70 (55)
7 1:1.5:4 t-BuNC (25) 5 22 90 decomp
8 1:1.5:4 t-BuNC (25) 5 14 60 60 (40)
9 1:1.5:2 t-BuNC (25) 5 15 75 50 (35)
10c 1:1.5:2 t-BuNC (25) 5 17 75 decomp
a All reactions were performed on 0.10 mmol scale at 0.10 M concentration.
b Improved isolated
yields were obtained at larger scale, requiring slightly higher catalyst and ligand loadings. c
CsF
was used as the fluoride source in absence of 18-crown-6 and MeCN was used as the solvent.
d Determined by
1H NMR spectroscopy using durene as the internal standard; isolated yield in
parentheses.
2.3.2 Insertion of 5,6-Indolyne 136 into Bis(pinacolato)diboron (1)
The tedious synthesis of the 5,6-indolyne precursor 133 constrained the detailed
optimization study of the insertion reaction. Despite this fact, the insertion of 5,6-indolyne
was achieved by higher loadings of Pt(dba)2, ligand, and diboron reagent 1 (Scheme 2.6).

2 Insertion of Indolynes into the B−B Bond and Site-Selective SUZUKI−MIYAURA Reaction 41
Scheme 2.6: Insertion of 5,6-Indolyne 136 into bis(pinacolato)diboron (1).
2.3.3 Insertion of 6,7-Indolyne 137 into Bis(pinacolato)diboron (1)
The optimization study for the insertion of 6,7-indolyne 137 into diboron reagent 1 was
commenced with the optimized conditions used for the insertion of 4,5-indolyne precursor
134. However, it afforded the expected product 140 in rather low yields (Table 2.2, entry 3).
Optimization with respect to the temperature proved to be essential in this case. Lowering
the temperature to 45 oC (Table 2.2, entry 1) and room temperature (Table 2.2, entry 2)
resulted in lower conversion. Increasing the temperature to 85 oC improved the yield
(Table 2.2, entry 4), Optimized conditions were obtained by decreasing the loading of
18-crown-6 at 85 oC (Table 2.2, entry 5). To improve the isolated yields the reaction was
performed at larger scale with 7.5 mol % of Pt(dba)2.
Table 2.2: Optimization of insertion of 6,7-indolyne 137 into bis(pinacolato)diboron (1).
Entrya Ratio
[equiv]
134:1:KF:18-crown-6
Temp
[oC]
Yieldc
[%]
1 1:1.5:4:2 r.t. 20 (─)
2 1:1.5:4:2 45 55 (30)
3 1:1.5:4:2 75 55 (28)
4 1:1.5:4:2 85 60 (40)
5b 1:1.5:2:2 85 70 (57)

42 THEORETICAL PART
a All reactions were performed on 0.06 mmol scale at 0.06 M concentration.
b Improved isolated yields
were obtained at larger scale, requiring slightly higher catalyst and ligand loadings. c
Determined by
1H NMR spectroscopy using durene as an internal standard; isolated yield in parentheses.
2.4 Site-Selective SUZUKI–MIYAURA Cross-Coupling
2.4.1 SUZUKI–MIYAURA Cross-Coupling of 4,5-Diborylated Indole 162
The study of the site-selective SUZUKI–MIYAURA cross-coupling of 4,5-diborylated indole 138
was initiated using the reaction conditions reported by YOSHIDA and co-workers.[38]
Employing (Ph3P)4Pd as the catalyst and KOH as the base resulted the decomposition of the
starting material 138 (Table 2.3, entry 1 and 2). Changing the catalyst to (dppf)PdCl2;
however, showed no selectivity and yielded mono-coupled 169, 170, and bis-coupled 171
products in 4:2:1 ratio (Table 2.3, entry 3). To reduce the ratio of the bis-coupled product
171, the ratio of 4,5-diborylated indole 138 to PhI was changed to 1.5:1, which gave 2:2:1
ratio of the products (Table 2.3, entry 4).
Table 2.3: Optimization of SUZUKI–MIYAURA cross-coupling of 4,5-diborylated indole 162
Entrya,b Ratio
[equiv]
138:PhI
Catalyst Base Solvent Temp.
[oC]
Product
ratioc
169:170:171
1 1:1 (Ph3P)4Pd KOH DME/H2O 45 decomp
2 1.2:1 (Ph3P)4Pd KOH DME/H2O 45 decomp
3 1:1 (dppf)PdCl2 KOH DME/H2O 45 4:2:1
4 1.5:1 (dppf)PdCl2 KOH DME/H2O 45 2:2:1
a All reactions were performed on 0.086 mmol scale (iodobenzene) at 0.072 M concentration. b
H2O
(2.16 mmol, 25 equiv) used, corresponding to a 30:1 ratio of DME and water. c
The structures of
regioisomers mono-169 and mono-170 were not assigned.
Considering the high reactivity of PhI, 2-bromomesitylene was employed in the reaction.
Comparatively less reactive (Ph3P)4Pd as the catalyst and carbonate bases did not improve
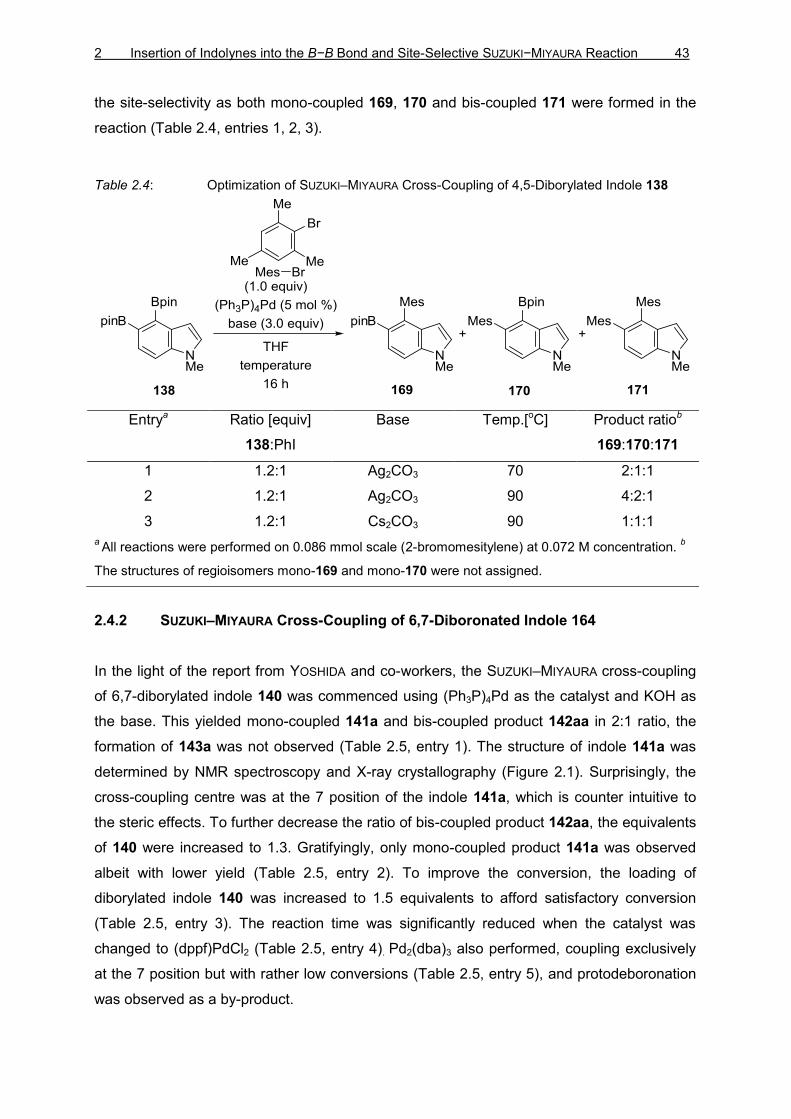
2 Insertion of Indolynes into the B−B Bond and Site-Selective SUZUKI−MIYAURA Reaction 43
the site-selectivity as both mono-coupled 169, 170 and bis-coupled 171 were formed in the
reaction (Table 2.4, entries 1, 2, 3).
Table 2.4: Optimization of SUZUKI–MIYAURA Cross-Coupling of 4,5-Diborylated Indole 138
Entrya Ratio [equiv]
138:PhI
Base Temp.[oC] Product ratiob
169:170:171
1 1.2:1 Ag2CO3 70 2:1:1
2 1.2:1 Ag2CO3 90 4:2:1
3 1.2:1 Cs2CO3 90 1:1:1
a All reactions were performed on 0.086 mmol scale (2-bromomesitylene) at 0.072 M concentration. b
The structures of regioisomers mono-169 and mono-170 were not assigned.
2.4.2 SUZUKI–MIYAURA Cross-Coupling of 6,7-Diboronated Indole 164
In the light of the report from YOSHIDA and co-workers, the SUZUKI–MIYAURA cross-coupling
of 6,7-diborylated indole 140 was commenced using (Ph3P)4Pd as the catalyst and KOH as
the base. This yielded mono-coupled 141a and bis-coupled product 142aa in 2:1 ratio, the
formation of 143a was not observed (Table 2.5, entry 1). The structure of indole 141a was
determined by NMR spectroscopy and X-ray crystallography (Figure 2.1). Surprisingly, the
cross-coupling centre was at the 7 position of the indole 141a, which is counter intuitive to
the steric effects. To further decrease the ratio of bis-coupled product 142aa, the equivalents
of 140 were increased to 1.3. Gratifyingly, only mono-coupled product 141a was observed
albeit with lower yield (Table 2.5, entry 2). To improve the conversion, the loading of
diborylated indole 140 was increased to 1.5 equivalents to afford satisfactory conversion
(Table 2.5, entry 3). The reaction time was significantly reduced when the catalyst was
changed to (dppf)PdCl2 (Table 2.5, entry 4). Pd2(dba)3 also performed, coupling exclusively
at the 7 position but with rather low conversions (Table 2.5, entry 5), and protodeboronation
was observed as a by-product.
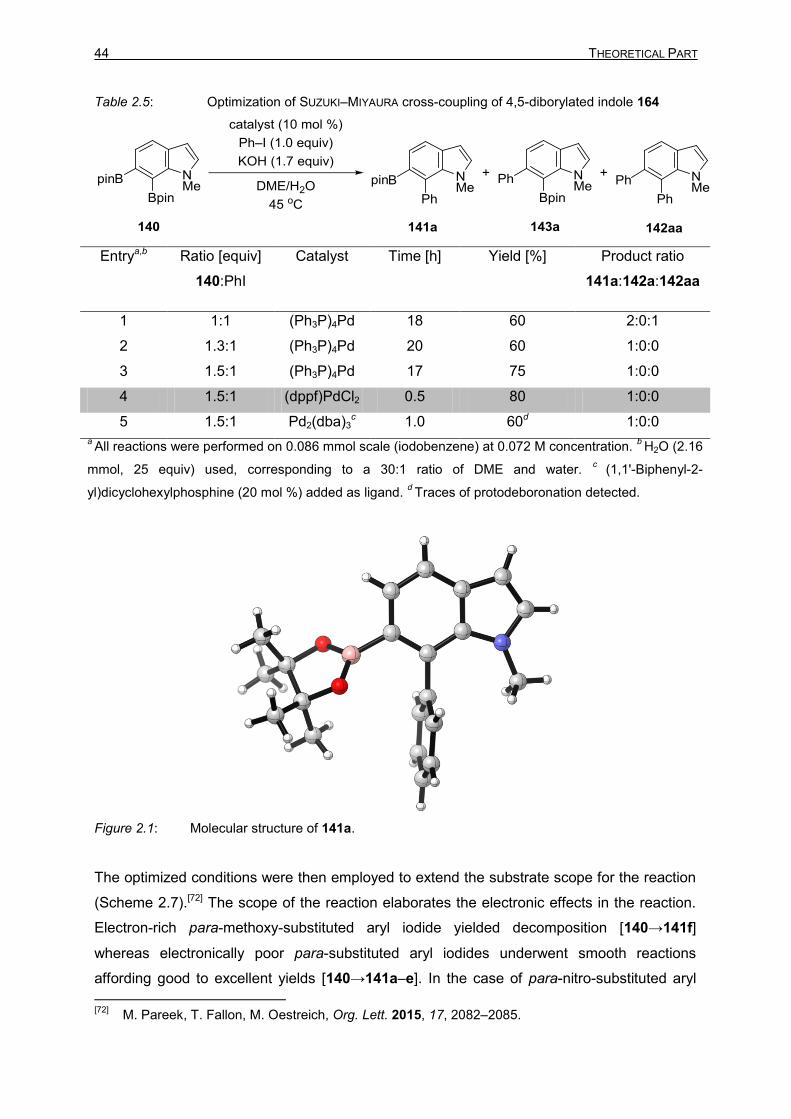
44 THEORETICAL PART
Table 2.5: Optimization of SUZUKI–MIYAURA cross-coupling of 4,5-diborylated indole 164
Entrya,b Ratio [equiv]
140:PhI
Catalyst Time [h] Yield [%]
Product ratio
141a:142a:142aa
1 1:1 (Ph3P)4Pd 18 60 2:0:1
2 1.3:1 (Ph3P)4Pd 20 60 1:0:0
3 1.5:1 (Ph3P)4Pd 17 75 1:0:0
4 1.5:1 (dppf)PdCl2 0.5 80 1:0:0
5 1.5:1 Pd2(dba)3c 1.0 60d 1:0:0
a All reactions were performed on 0.086 mmol scale (iodobenzene) at 0.072 M concentration.
b H2O (2.16
mmol, 25 equiv) used, corresponding to a 30:1 ratio of DME and water. c
(1,1'-Biphenyl-2-
yl)dicyclohexylphosphine (20 mol %) added as ligand. d Traces of protodeboronation detected.
Figure 2.1: Molecular structure of 141a.
The optimized conditions were then employed to extend the substrate scope for the reaction
(Scheme 2.7).[72] The scope of the reaction elaborates the electronic effects in the reaction.
Electron-rich para-methoxy-substituted aryl iodide yielded decomposition [140→141f]
whereas electronically poor para-substituted aryl iodides underwent smooth reactions
affording good to excellent yields [140→141a–e]. In the case of para-nitro-substituted aryl
[72]
M. Pareek, T. Fallon, M. Oestreich, Org. Lett. 2015, 17, 2082–2085.
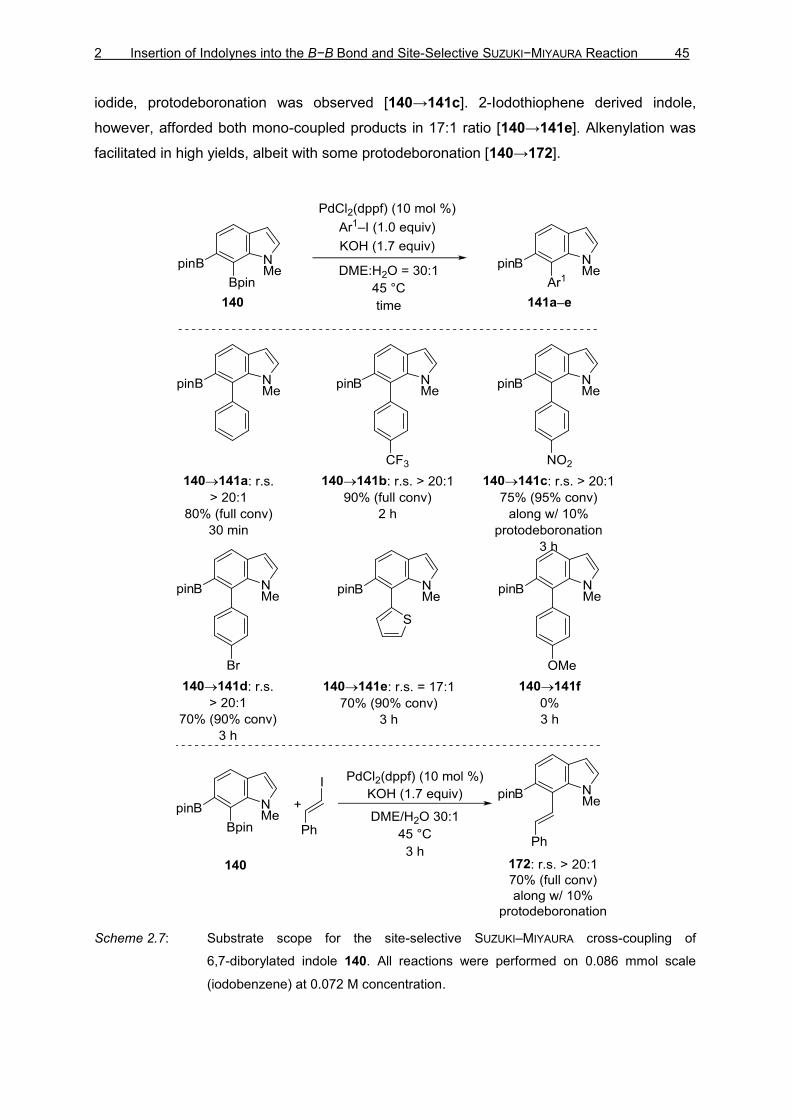
2 Insertion of Indolynes into the B−B Bond and Site-Selective SUZUKI−MIYAURA Reaction 45
iodide, protodeboronation was observed [140→141c]. 2-Iodothiophene derived indole,
however, afforded both mono-coupled products in 17:1 ratio [140→141e]. Alkenylation was
facilitated in high yields, albeit with some protodeboronation [140→172].
Scheme 2.7: Substrate scope for the site-selective SUZUKI–MIYAURA cross-coupling of
6,7-diborylated indole 140. All reactions were performed on 0.086 mmol scale
(iodobenzene) at 0.072 M concentration.
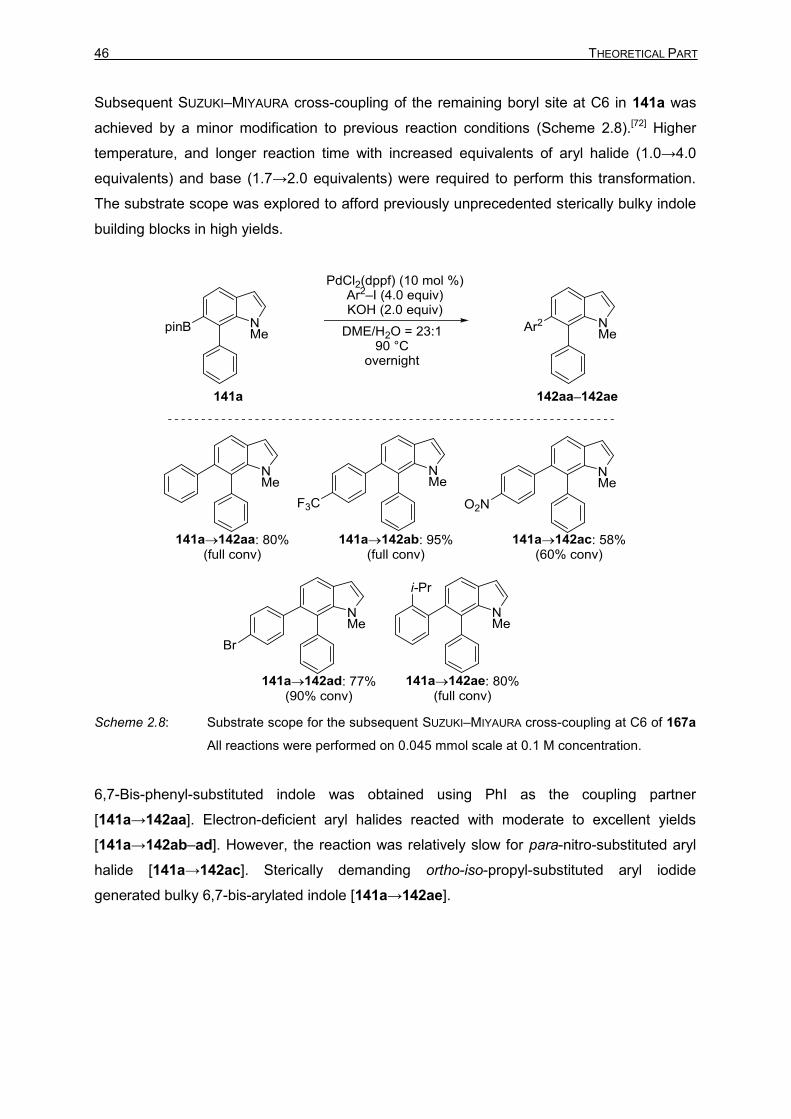
46 THEORETICAL PART
Subsequent SUZUKI–MIYAURA cross-coupling of the remaining boryl site at C6 in 141a was
achieved by a minor modification to previous reaction conditions (Scheme 2.8).[72] Higher
temperature, and longer reaction time with increased equivalents of aryl halide (1.0→4.0
equivalents) and base (1.7→2.0 equivalents) were required to perform this transformation.
The substrate scope was explored to afford previously unprecedented sterically bulky indole
building blocks in high yields.
Scheme 2.8: Substrate scope for the subsequent SUZUKI–MIYAURA cross-coupling at C6 of 167a
All reactions were performed on 0.045 mmol scale at 0.1 M concentration.
6,7-Bis-phenyl-substituted indole was obtained using PhI as the coupling partner
[141a→142aa]. Electron-deficient aryl halides reacted with moderate to excellent yields
[141a→142ab–ad]. However, the reaction was relatively slow for para-nitro-substituted aryl
halide [141a→142ac]. Sterically demanding ortho-iso-propyl-substituted aryl iodide
generated bulky 6,7-bis-arylated indole [141a→142ae].
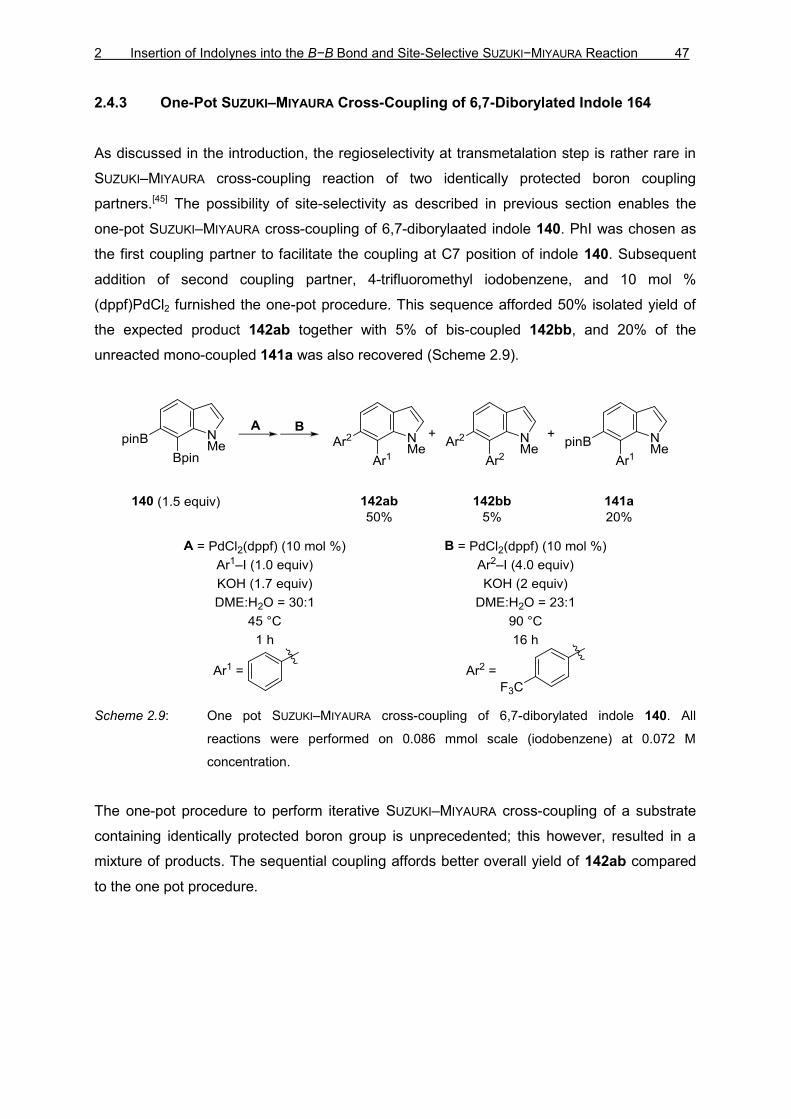
2 Insertion of Indolynes into the B−B Bond and Site-Selective SUZUKI−MIYAURA Reaction 47
2.4.3 One-Pot SUZUKI–MIYAURA Cross-Coupling of 6,7-Diborylated Indole 164
As discussed in the introduction, the regioselectivity at transmetalation step is rather rare in
SUZUKI–MIYAURA cross-coupling reaction of two identically protected boron coupling
partners.[45] The possibility of site-selectivity as described in previous section enables the
one-pot SUZUKI–MIYAURA cross-coupling of 6,7-diborylaated indole 140. PhI was chosen as
the first coupling partner to facilitate the coupling at C7 position of indole 140. Subsequent
addition of second coupling partner, 4-trifluoromethyl iodobenzene, and 10 mol %
(dppf)PdCl2 furnished the one-pot procedure. This sequence afforded 50% isolated yield of
the expected product 142ab together with 5% of bis-coupled 142bb, and 20% of the
unreacted mono-coupled 141a was also recovered (Scheme 2.9).
Scheme 2.9: One pot SUZUKI–MIYAURA cross-coupling of 6,7-diborylated indole 140. All
reactions were performed on 0.086 mmol scale (iodobenzene) at 0.072 M
concentration.
The one-pot procedure to perform iterative SUZUKI–MIYAURA cross-coupling of a substrate
containing identically protected boron group is unprecedented; this however, resulted in a
mixture of products. The sequential coupling affords better overall yield of 142ab compared
to the one pot procedure.

48 THEORETICAL PART
2.5 Conclusion
Precursors for all the three regioisomers of indolynes were synthesized and Pt(0)-catalyzed
insertion into diboron bond was accomplished. Contrary to YOSHIDA’s system,[38,40] the
unsymmetrical nature of diborylated indoles (138−140) poses the challenge of selectivity in
subsequent SUZUKI–MIYAURA cross-coupling reactions. The attempts to obtain the site-
selectivity with 4,5-diborylated indole 138 proved to be futile; however, excellent site-
selectivity was observed in the case of 6,7-diborylated indole 140. Sterically hindered C7
position of indole exclusively underwent the coupling reaction. Successively, C6 position was
also coupled giving previously unprecedented indole building blocks. The one pot reaction
sequence showed promising results. However, the excess of indole starting material proved
to be disadvantageous in this case.

3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 49
3 ASYMMETRIC STANNYLATION OF α,β-UNSATURATED
ESTERS INVOLVING TIN–SILICON AND TIN–TIN BOND
ACTIVATION
3.1 Asymmetric 1,4-Stannylation of α,β-Unsaturated Esters Using
Tin−Silicon (Sn−Si) Bond Activation
3.1.1 Introduction
The asymmetric conjugate stannylation reported by ENDERS was limited to only
cyclohexenone SAMP hydrazone (S)-146.[69] The 1,4-stannylation of linear α,β-unsaturated
acceptors was not investigated in detail. The seminal work from OESTREICH and co-workers
described the first base mediated 1,4-stannylation in biphasic media.[61] The requirement of
base in aqueous medium makes it an ideal setup for the use of asymmetric phase-transfer
catalyst (PTC) in order to induce the chiral environment. Asymmetric 1,4-addition of
nucleophilic carbon to α,β-unsaturated acceptors incorporating PTC is well-established.[73]
However, asymmetric 1,4-addition of nucleophilic main-group elements (E = B, Si, Sn) using
PTC is unprecedented. This report aims at base-mediated asymmetric 1,4-stannylation of
α,β-unsaturated esters employing PTC.
[73]
For general reviews and reports, see: a) S. Shirakawa, K. Maruoka, Angew. Chem. Int. Ed.
2013, 52, 4312–4348; Angew. Chem. 2013, 125, 4408–4445; b) Asymmetric Phase Transfer
Catalysis, (Ed.: K. Maruoka), Wiley-VCH, Weinheim, 2008; c) T. Ooi, K. Maruoka, Angew.
Chem. Int. Ed. 2007, 46, 4222–4266; Angew. Chem. 2007, 119, 4300–4345; d) T. Ooi, D.
Ohara, K. Fukumoto, K. Maruoka, Org. Lett. 2005, 7, 3195–3197; e) B. Lygo, B. Allbutt, E. H. M.
Kirton, Tetrahedron Lett. 2005, 46, 4461–4464; f) T. Ooi, S. Fujioka, K. Maruoka, J. Am. Chem.
Soc. 2004, 126, 11790–11793; g) M. Oku, S. Arai, K. Katayama, T. Shioiri, Synlett 2000, 493–
496; h) M. J. O’Donnell, F. Delgado, C. Hostettler, R. Schwesinger, Tetrahedron Lett. 1998, 39,
8775–8778; i) E. J. Corey, F. Xu, M. C. Noe, J. Am. Chem. Soc. 1998, 119, 12414–12416; j) E.
J. Corey, Y. Bo, J. Busch-Petersen, J. Am. Chem. Soc. 1998, 120, 13000–13002; k) B. B. Lygo,
P. G. Wainwright, Tetrahedron Lett. 1998, 39, 1599–1602; l) B. B. Lygo, P. G. Wainwright,
Tetrahedron Lett. 1997, 38, 8595–8598; m) K. Shishido, K. Goto, S. Miyoshi, Y. Takaishi, M.
Shibuya, J. Org. Chem. 1994, 59, 406–414; n) R. S. E. Conn, A. V. Lovell, S. Karady, L. M.
Weinstock, J. Org. Chem.1986, 51, 4710–4711; o) D. J. Cram, G. D. Y. Sogah, J. Chem. Soc.
1981, 625–628.

50 THEORETICAL PART
3.1.2 Optimization Studies for the PTC-Catalyzed Asymmetric 1,4-Stannylation of
α,β-Unsaturated Esters
The optimization study was initiated with the quinidine-derived PTC 188 and silylstannane 98
as stannyl anion precursor. The reaction conditions were similar to commonly used
1,4-addition of carbon nucleophile to α,β-unsaturated carbonyl compounds.[73n] The solvent
system was chosen to be a mixture of toluene and aqueous base solution (50% w/v) in 5:1
ratio. Initial study with cyclohex-2-ene-1-one (95) gave moderate yield with low ee (Table
3.1, entry 1). The scope was then extended to the acyclic acceptors; chalcone 173 showed
good reactivity but with a mixture of expected product 181 and reduced product in 1:1 ratio
with rather low ee (Table 3.1, entry 2). (E)-174 resulted in low yields with racemic product
182 (Table 3.1, entry 3). Other acceptors with oxazolidinone (E)-175, nitro (E)-176, and
cyano (E)-177 electron-withdrawing groups did not show any conversion to 183−185 (Table
3.1, entry 4−6). To form a better ion pair in the reaction mixture, acid (E)-178 was subjected
to the reaction conditions. However, the product 186 was not observed (Table 3.1, entry 7).
Ethyl cinnamate (E)-179 afforded low yields and ee (Table 3.1, entry 8), whereas the ester
(Z)-179 afforded product 187 in low yield but with relatively higher 36% ee (Table 3.1, entry
9). The tedious synthesis of (Z)-acceptors limited the further scope for improvement with
respect to the acceptors, and optimization was proceeded further with (Z)-179.
Table 3.1: Optimization with respect to the acceptor for 1,4-stannylation using PTC 188.
Entrya Acceptor Yield [%]
Enantiomeric excessc
[%]
1
95
60% 11%
2b
(E)-173
20% 21%
3
(E)-174
10% 17%

3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 51
4
(E)-175
0 ─
5
(E)-176
0 ─
6
(E)-177
0 ─
7
(E)-178
0 ─
8
(E)-179
23% 3%
9
(Z)-179
8% 36%
a All reactions were performed on 0.1 mmol scale at 0.1 M concentration in the mixture of toluene
and water in 5:1 ratio. b
1:1 mixture of expected product and 2,3-reduced product was observed.
c The absolute configuration of the product was not assigned.
The presence of high amounts of base resulted in the formation of distannane 190 from
98.[61,74] The distannae formation is presumed to involve the transition state 189‡. It is
envisaged that LEWIS base-activated hypevalent silicon intermediate is formed, which
releases the stannyl nucleophile. This tin nucleophile then forms the distannane 190 by
reacting with another molecule of silylstannae 98.
Scheme 3.1: The formation of distannane 190 from silylstannae 98.
To improve the isolated yields the loading of silylstannane 98 was increased (Table 3.2,
entries 1−3). Silylstannane in 3.0 equivalents afforded good yields; however the ee was still
low. Other hydroxide bases were explored, and NaOH showed similar reactivity as KOH
(Table 3.2, entry 4). CsOH and Ba(OH)2 failed to give any conversion to expected product
(Table 3.2, entry 5 and 6). Contrary to the report by OESTREICH,[61] sodium and potassium
acetates gave no conversion (Table 3.2, entries 7 and 8). To examine the effect of base
concentration, the solvent ratio was increased to 10:1 (Table 3.2, entry 9). This resulted in a
[74]
K Mochida, T Yamanishi, Bull. Chem. Soc. Jpn. 1987, 60, 3429−3430.
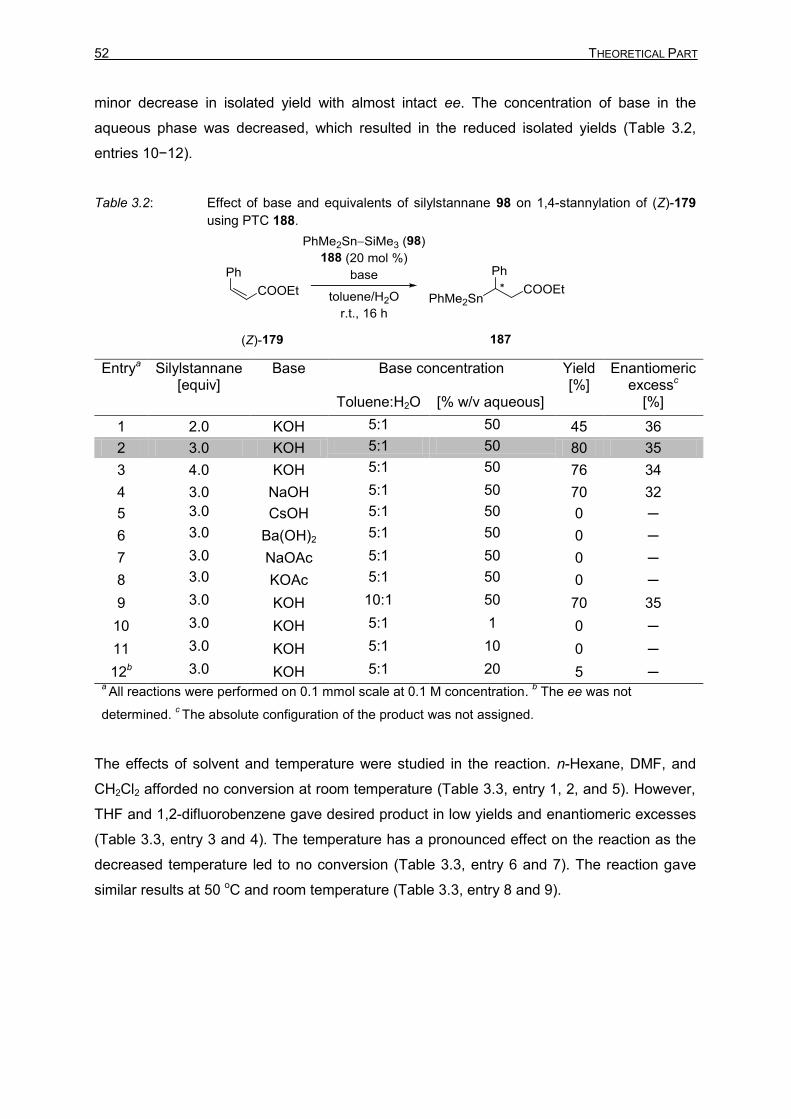
52 THEORETICAL PART
minor decrease in isolated yield with almost intact ee. The concentration of base in the
aqueous phase was decreased, which resulted in the reduced isolated yields (Table 3.2,
entries 10−12).
Table 3.2: Effect of base and equivalents of silylstannane 98 on 1,4-stannylation of (Z)-179
using PTC 188.
Entrya Silylstannane [equiv]
Base Base concentration
Yield [%]
Enantiomeric excessc
[%] Toluene:H2O [% w/v aqueous]
1 2.0 KOH 5:1 50 45 36
2 3.0 KOH 5:1 50 80 35
3 4.0 KOH 5:1 50 76 34
4 3.0 NaOH 5:1 50 70 32
5 3.0 CsOH 5:1 50 0 ─
6 3.0 Ba(OH)2 5:1 50 0 ─
7 3.0 NaOAc 5:1 50 0 ─
8 3.0 KOAc 5:1 50 0 ─
9 3.0 KOH 10:1 50 70 35
10 3.0 KOH 5:1 1 0 ─
11 3.0 KOH 5:1 10 0 ─
12b 3.0 KOH 5:1 20 5 ─ a All reactions were performed on 0.1 mmol scale at 0.1 M concentration.
b The ee was not
determined. c The absolute configuration of the product was not assigned.
The effects of solvent and temperature were studied in the reaction. n-Hexane, DMF, and
CH2Cl2 afforded no conversion at room temperature (Table 3.3, entry 1, 2, and 5). However,
THF and 1,2-difluorobenzene gave desired product in low yields and enantiomeric excesses
(Table 3.3, entry 3 and 4). The temperature has a pronounced effect on the reaction as the
decreased temperature led to no conversion (Table 3.3, entry 6 and 7). The reaction gave
similar results at 50 oC and room temperature (Table 3.3, entry 8 and 9).
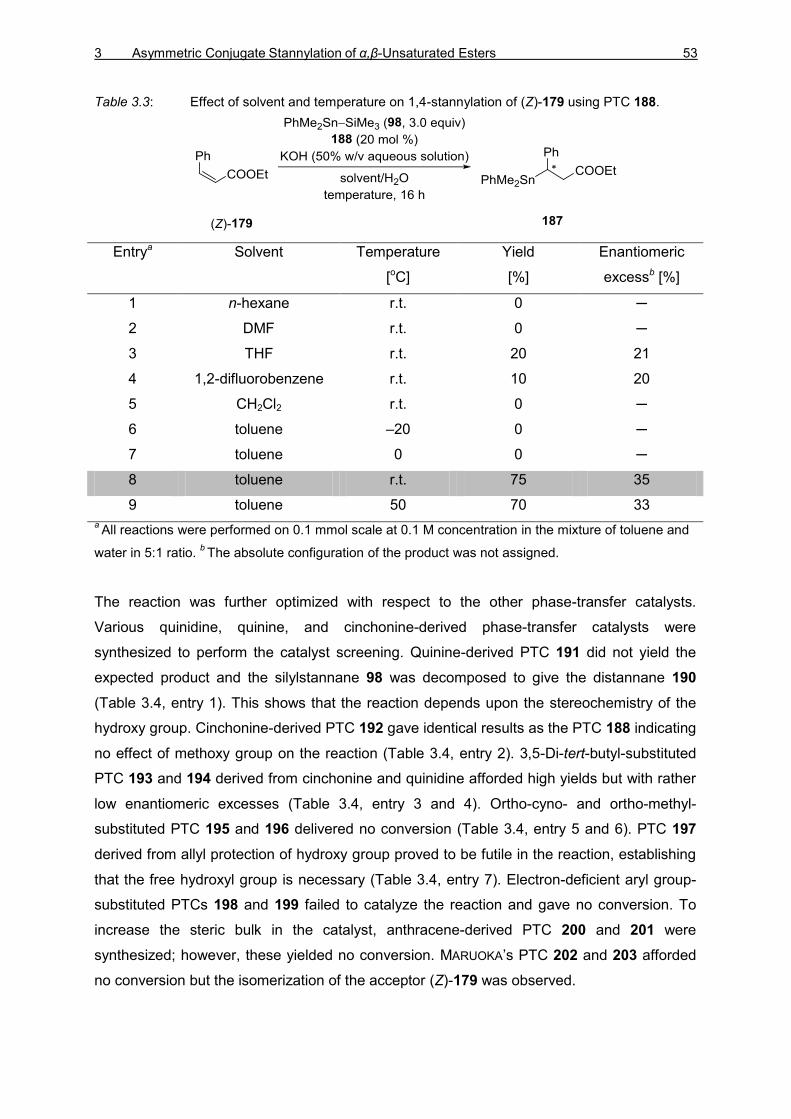
3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 53
Table 3.3: Effect of solvent and temperature on 1,4-stannylation of (Z)-179 using PTC 188.
Entrya Solvent Temperature
[oC]
Yield
[%]
Enantiomeric
excessb [%]
1 n-hexane r.t. 0 ─
2 DMF r.t. 0 ─
3 THF r.t. 20 21
4 1,2-difluorobenzene r.t. 10 20
5 CH2Cl2 r.t. 0 ─
6 toluene –20 0 ─
7 toluene 0 0 ─
8 toluene r.t. 75 35
9 toluene 50 70 33
a All reactions were performed on 0.1 mmol scale at 0.1 M concentration in the mixture of toluene and
water in 5:1 ratio. b The absolute configuration of the product was not assigned.
The reaction was further optimized with respect to the other phase-transfer catalysts.
Various quinidine, quinine, and cinchonine-derived phase-transfer catalysts were
synthesized to perform the catalyst screening. Quinine-derived PTC 191 did not yield the
expected product and the silylstannane 98 was decomposed to give the distannane 190
(Table 3.4, entry 1). This shows that the reaction depends upon the stereochemistry of the
hydroxy group. Cinchonine-derived PTC 192 gave identical results as the PTC 188 indicating
no effect of methoxy group on the reaction (Table 3.4, entry 2). 3,5-Di-tert-butyl-substituted
PTC 193 and 194 derived from cinchonine and quinidine afforded high yields but with rather
low enantiomeric excesses (Table 3.4, entry 3 and 4). Ortho-cyno- and ortho-methyl-
substituted PTC 195 and 196 delivered no conversion (Table 3.4, entry 5 and 6). PTC 197
derived from allyl protection of hydroxy group proved to be futile in the reaction, establishing
that the free hydroxyl group is necessary (Table 3.4, entry 7). Electron-deficient aryl group-
substituted PTCs 198 and 199 failed to catalyze the reaction and gave no conversion. To
increase the steric bulk in the catalyst, anthracene-derived PTC 200 and 201 were
synthesized; however, these yielded no conversion. MARUOKA’s PTC 202 and 203 afforded
no conversion but the isomerization of the acceptor (Z)-179 was observed.
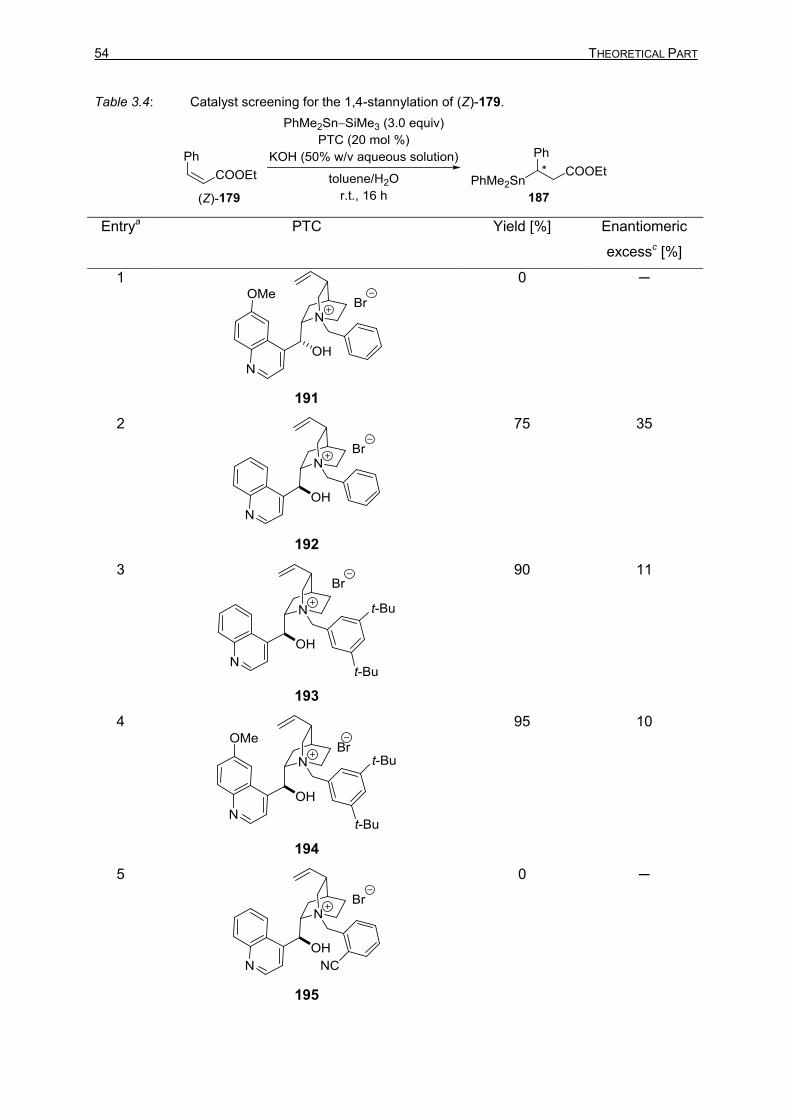
54 THEORETICAL PART
Table 3.4: Catalyst screening for the 1,4-stannylation of (Z)-179.
Entrya PTC Yield [%] Enantiomeric
excessc [%]
1
191
0 ─
2
192
75 35
3
193
90 11
4
194
95 10
5
195
0 ─
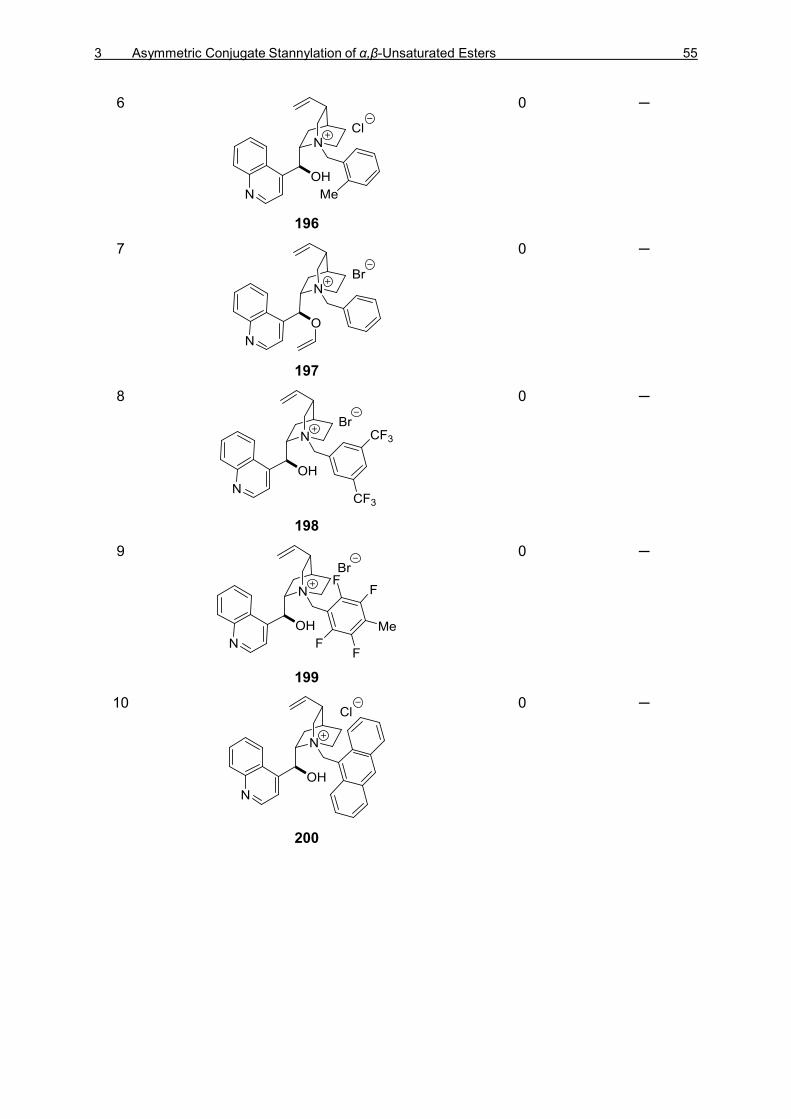
3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 55
6
196
0 ─
7
197
0 ─
8
198
0 ─
9
199
0 ─
10
200
0 ─
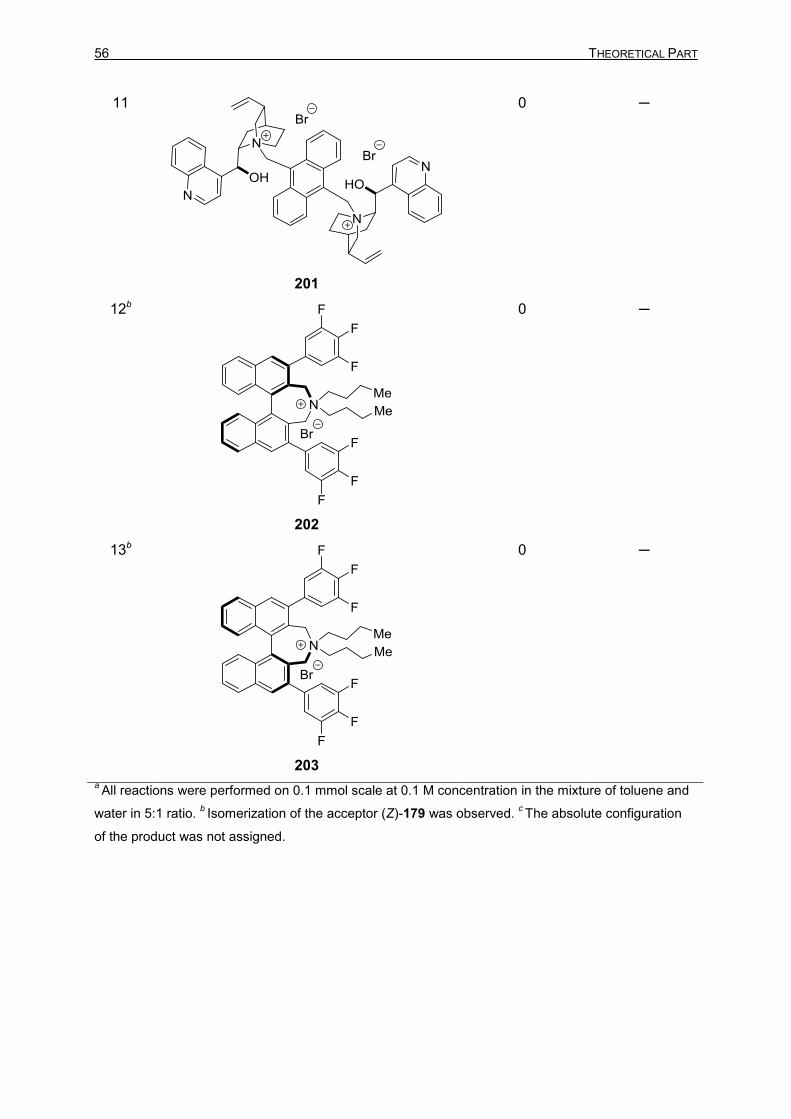
56 THEORETICAL PART
11
201
0 ─
12b
202
0 ─
13b
203
0 ─
a All reactions were performed on 0.1 mmol scale at 0.1 M concentration in the mixture of toluene and
water in 5:1 ratio. b Isomerization of the acceptor (Z)-179 was observed.
c The absolute configuration
of the product was not assigned.

3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 57
3.1.3 Control Experiments
To investigate the reasons behind the low ee in the reaction, various control experiments
were performed. First, no racemic background reaction was observed without the PTC (not
shown). Second, to figure out if the prolonged reaction time was causing racemization of the
product, the reaction was stopped after 2 h (Scheme 3.2, top). Conversion (30%) and ee
(42%) was determined, showing that there was only some racemization over the period of
time. Third, to probe if the base was playing any role in the racemization, the stannylated
product 187 was subjected to basic conditions in the absence of the silylstannane 98 and the
PTC 188 (Scheme 3.2, middle). HPLC analysis showed that there was no change in the ee,
ruling out the possibility of racemization caused by the base. Fourth, the reaction was carried
out with catalyst 188 and base without silylstannane 98. Again, no significant change of the
ee was observed (Scheme 3.2, bottom).
Scheme 3.2: Performed control experiments.
To verify the role of the alcohol functionality of the catalyst, the reaction was performed using
(n-Bu)4NBr as PTC (Scheme 3.3, top). This resulted in no conversion at all, demonstrating
the vital role of the alcohol group for turnover. To further confirm this, the alkoxide 204 was
generated using KH as the base (Scheme 3.3, middle). This species was directly used as the
catalyst in the model reaction, affording only racemic product rac-187 (Scheme 3.3, bottom).
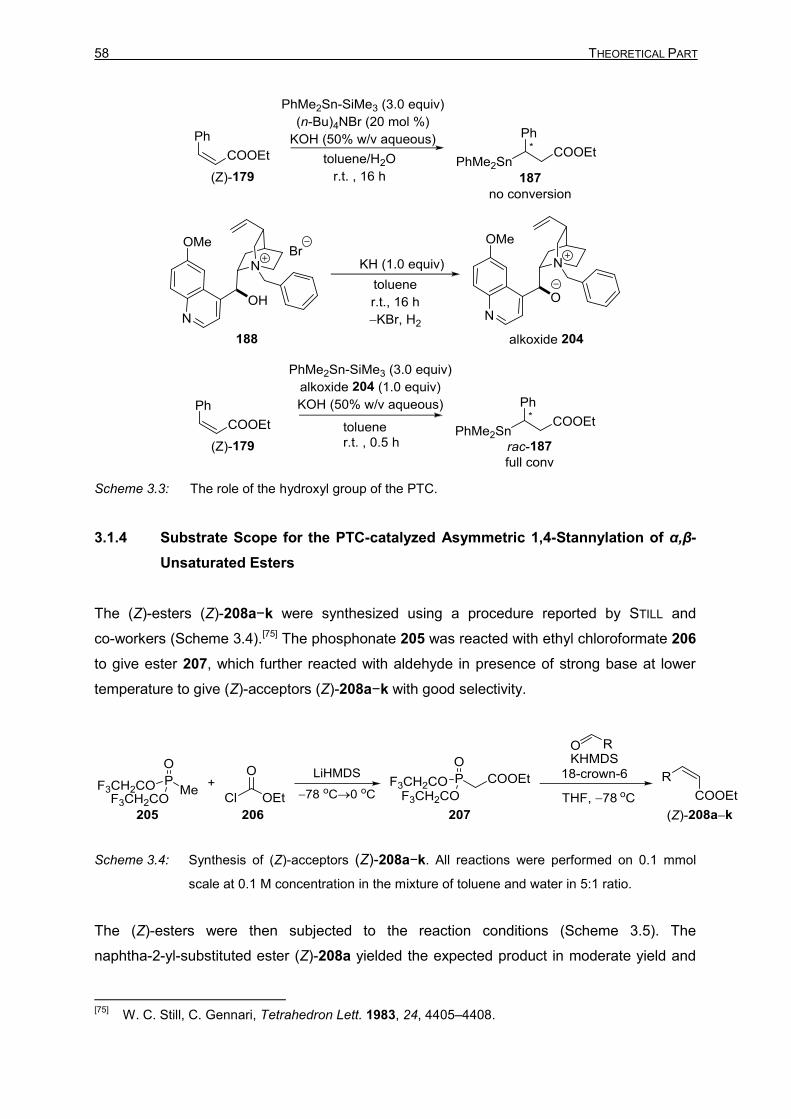
58 THEORETICAL PART
Scheme 3.3: The role of the hydroxyl group of the PTC.
3.1.4 Substrate Scope for the PTC-catalyzed Asymmetric 1,4-Stannylation of α,β-
Unsaturated Esters
The (Z)-esters (Z)-208a−k were synthesized using a procedure reported by STILL and
co-workers (Scheme 3.4).[75] The phosphonate 205 was reacted with ethyl chloroformate 206
to give ester 207, which further reacted with aldehyde in presence of strong base at lower
temperature to give (Z)-acceptors (Z)-208a−k with good selectivity.
Scheme 3.4: Synthesis of (Z)-acceptors (Z)-208a−k. All reactions were performed on 0.1 mmol
scale at 0.1 M concentration in the mixture of toluene and water in 5:1 ratio.
The (Z)-esters were then subjected to the reaction conditions (Scheme 3.5). The
naphtha-2-yl-substituted ester (Z)-208a yielded the expected product in moderate yield and
[75]
W. C. Still, C. Gennari, Tetrahedron Lett. 1983, 24, 4405–4408.

3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 59
ee [(Z)-208a→209a]. Electron-rich esters (Z)-208b and (Z)-208c delivered good yields with
low enantiomeric excesses [(Z)-208b−c→209b−c]. meta-Anisyl-substituted ester (Z)-208d
produced moderate conversion and yields [(Z)-208d→209d]. ortho-Fluorophenyl-substituted
acceptor (Z)-208e gave good conversion albeit with low yields [(Z)-208e→209e]. Electron-
deficient phenyl-substituted acceptors ((Z)-208f−h) gave no conversion [(Z)-
208f−h→209f−h]. Electron-rich esters ((Z)-208i−j) also failed to afford any conversion [(Z)-
208i−j→209i−j]. Alkyl-substituted acceptor (Z)-208k showed no conversion in the reaction
[(Z)-208k→209k].
Scheme 3.5: Substrate scope for the 1,4-stannylation of α,β-unsaturated esters. All reactions were
performed on 0.67 mmol scale at 0.1 M concentration in the mixture of toluene and
water in 5:1 ratio. The absolute configuration of the product was not assigned.

60 THEORETICAL PART
3.1.5 Proposed mechanism for the PTC-catalyzed Asymmetric 1,4-Stannylation of
α,β-Unsaturated Esters
STARK proposed the extraction mechanism for the phase-transfer catalysis.[76] In this
mechanism, the phase transfer catalyst moves back and forth across the organic and
aqueous phase (Scheme 3.6). The onium salt XXX equilibrates with the KOH in the aqueous
phase, and extracts hydroxide XXXI into the organic phase. The hydroxide then facilitates
the release of the stannyl nucleophile and the reactive intermediate XXXII is hypothesized to
form in the reaction. The intermediate XXXII transfers the nucleophilic stannyl group to the
acceptor (Z)-179, and the countercation in the XXXII is envisaged to induce the chirality in
the final product 187.
Scheme 3.6: Proposed mechanism for PTC-catalyzed asymmetric 1,4-stannylation of α,β-
unsaturated ester (Z)-179.
[76]
C. M. Starks, J. Am. Chem. Soc. 1971, 93, 195–199.

3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 61
3.2 Asymmetric 1,4-Stannylation of α,β-Unsaturated Esters Using
Tin−Tin (Sn−Sn) Bond Activation
3.2.1 Introduction
The main group nucleophile generated by activation of homonuclear interelement bonds
have been studied in details.[77,78] The generation of stannyl nucleophile transmetalation with
organometalic bases has been studied in detail. However, the nucleophilic stannyl addition to
electrophiles through transmetalation involving σ-bond metathesis is unprecedented.
OESTREICH and co-workers recently developed a rich chemistry associated with interelement
bond activation using transition metal catalysts.[79] This thesis envisaged to apply the same
strategy for the activation of Sn–Sn bonds using copper(I)-catalysis.[79e] Thus-generated tin
nucleophile could then participate in asymmetric carbon–tin bond formations.
[77]
a) G. A. Molander, S. R. Wisniewski, M. H. Sarvari, Adv. Synth. Catal. 2013, 355, 3037–3057; b)
S. B. Thorpe, J. A. Calderone, W. L. Santos, Org. Lett. 2012, 14, 1918–1921; c) G. A. Molander,
S. A. McKee, Org. Lett. 2011, 13, 4684–4687; d) W. J. Fleming, H. Miller-Bunz, V. Lillo, E.
Fernandez, P. J. Guiry, Org. Biomol. Chem. 2009, 7, 2520−2524; e) V. Lillo, A. Prieto, A. Bonet,
M. M. Diaz Requejo, J. Ramirez, P. J. Prez, E. Fernandez, Organometallics 2009, 28, 659–662;
f) H.-S. Sim, X. Feng, J. Yun, Chem. Eur. J. 2009, 15, 1939–1943; g) S. Mun, J.-E. Lee, J. Yun,
Org. Lett. 2006, 8, 4887–4889; h) H. Ito, H. Yamanaka, J. Tateiwa, A. Hosomi, Tetrahedron Lett.
2000, 41, 682–686; i) K. Takahashi, T. Isiyama, N. Miyaura, J. Organomet. Chem. 2001,
625−629; j) K. Takahashi, T. Isiyama, N. Miyaura, Chem. Lett. 2000, 982−983; k) Y. G. Lawson,
M. J. G. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, Chem. Commun. 1997, 2051−2052. [78]
a) K. Okamoto, T. Hayashi, Org. Lett. 2007, 9, 5067–5069; b) S. Ogoshi, S. Tomiyasu, M. Morita,
H Kurosawa, J. Am. Chem. Soc. 2002, 124, 11598–11599; c) H. Ito, T. Ishizuka, J.-I. Tateiwa, M.
Sonoda, A. Hosomi, J. Am. Chem. Soc. 1998, 120, 11196–11197; d) K. H. Pannell, H. K.
Sharma, Chem. Rev. 1995, 95, 1351–1374; e) K. A. Horn, Chem. Rev. 1995, 95, 1371–1350. [79]
a) A. Hensel, M. Oestreich, Chem. Eur. J. 2015, 21, 9062–9065; b) L. B. Delvos, A. Hensel, M.
Oestreich, Synthesis 2014, 46, 2957−2964; c) L. B. Delvos, M. Oestreich, Synthesis 2015, 47,
924−933; d) A. Hensel, K. Nagura, L. B. Delvos, M. Oestreich, Angew. Chem. Int. Ed. 2014, 53,
4964−4967; e) L. B. Delvos, D. J. Vyas, M. Oestreich, Angew. Chem. Int. Ed. 2013, 52,
4650−4653; Angew. Chem. 2013, 125, 4748–4751; f) E. Hartmann, D. J. Vyas, M. Oestreich,
Chem. Commun. 2011, 47, 7917−7932; g) D. J. Vyas, C. K. Hazra, M. Oestreich, Org. Lett. 2011,
13, 4462−4465; h) C. K. Hazra, E. Irran, M. Oestreich, Eur. J. Org. Chem. 2013, 4903–4908; i) D.
J. Vyas, R. Fröhlich, M. Oestreich, Org. Lett. 2011, 13, 2094–2097; j) D. J. Vyas, M. Oestreich,
Angew. Chem. Int. Ed. 2010, 49, 8513–8515; Angew. Chem. 2010, 122, 8692–8694; k) C.
Walter, R. Fröhlich, M. Oestreich, Tetrahedron 2009, 65, 5513−5520; l) C. Walter M. Oestreich,
Angew. Chem. Int. Ed. 2008, 47, 3818–3820; Angew. Chem. 2008, 120, 3878–3880; m) C.
Walter, G. Auer, M. Oestreich, Angew. Chem. Int. Ed. 2006, 45, 5675−5677; Angew. Chem.
2006,118, 5803–5805.

62 THEORETICAL PART
3.2.2 Optimization Studies for the Asymmetric 1,4-Stannylation of α,β-Unsaturated
Esters
Using the reaction conditions previously reported by OESTREICH and co-workers,[61] initial
optimization studies for the racemic reaction were performed. Hexamethyldistannane (128)
and (E)-179 with various copper salts and bases were employed. While metal salts such as
CuCl, CuCN as well as CuSCN performed poorly (Table 3.6, entries 1–3), copper(I) complex
of the N-heterocyclic carbene N,N-dimesityl imidazolium (IMes) proved to be a potent
catalyst for the conjugate stannylation (Table 3.6, entry 4). However, using ethyl ester (E)-15,
transesterfication was observed as a side reaction. Other bases than NaOMe and NaOt-Bu
led to lower conversions (Table 3.6, entries 5–7). The background reaction was observed
when the reaction was run without catalyst (Table 3.6, entry 8).
Table 3.5: Screening of various copper catalysts and bases for Sn–Sn activation.
Entrya Catalyst Base Conversion [%]
1 CuCl NaOMe <5
2 CuCN NaOMe 30
3b CuSCN NaOMe <5
4c IMesCuBr NaOMe 80
5d IMesCuBr NaOt-Bu 70
6 IMesCuBr KOt-Bu 40
7 IMesCuBr LiOt-Bu 20
8 ─ NaOt-Bu 40
a All reactions were performed on 0.05 mmol scale at 0.1 M concentration.
b 4,4'-Di-tert-butyl-2,2'-
bipyridine was used as ligand. c Transesterification was observed.
d 30% isolated yield.
To perform an asymmetric version, various chiral NHC ligands (S)-211−(S,S)-214 and
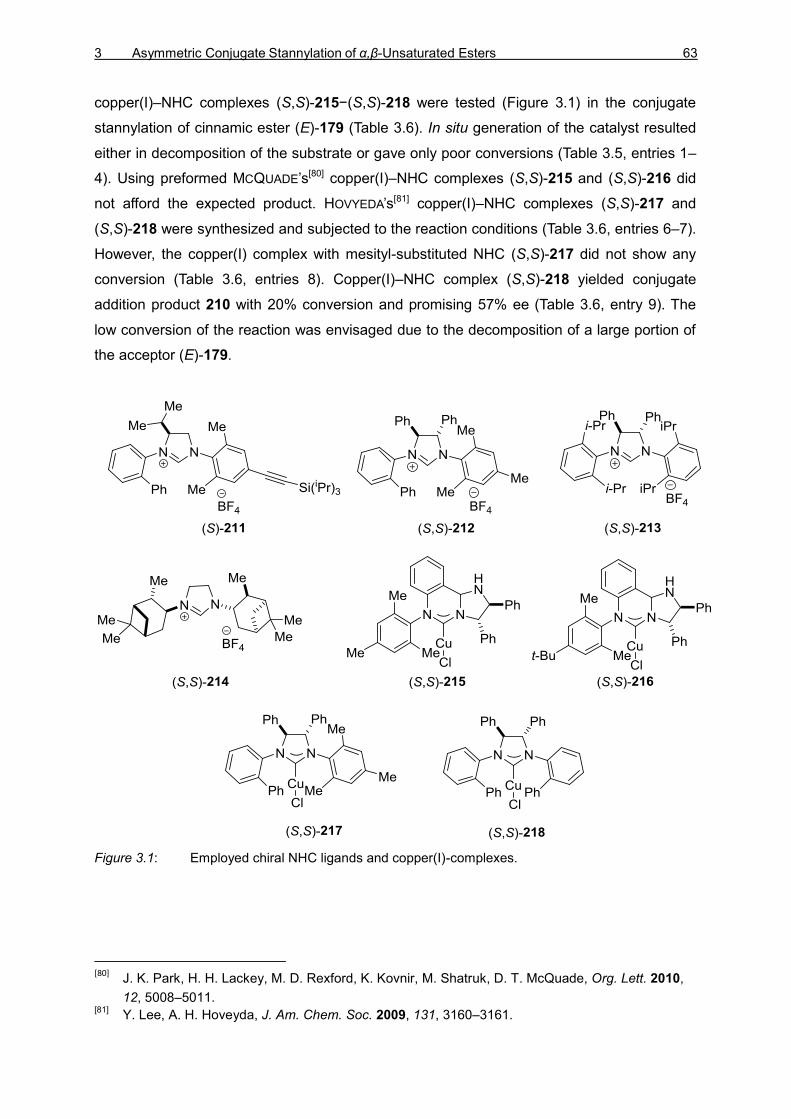
3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 63
copper(I)–NHC complexes (S,S)-215−(S,S)-218 were tested (Figure 3.1) in the conjugate
stannylation of cinnamic ester (E)-179 (Table 3.6). In situ generation of the catalyst resulted
either in decomposition of the substrate or gave only poor conversions (Table 3.5, entries 1–
4). Using preformed MCQUADE’s[80] copper(I)–NHC complexes (S,S)-215 and (S,S)-216 did
not afford the expected product. HOVYEDA’s[81] copper(I)–NHC complexes (S,S)-217 and
(S,S)-218 were synthesized and subjected to the reaction conditions (Table 3.6, entries 6–7).
However, the copper(I) complex with mesityl-substituted NHC (S,S)-217 did not show any
conversion (Table 3.6, entries 8). Copper(I)–NHC complex (S,S)-218 yielded conjugate
addition product 210 with 20% conversion and promising 57% ee (Table 3.6, entry 9). The
low conversion of the reaction was envisaged due to the decomposition of a large portion of
the acceptor (E)-179.
Figure 3.1: Employed chiral NHC ligands and copper(I)-complexes.
[80] J. K. Park, H. H. Lackey, M. D. Rexford, K. Kovnir, M. Shatruk, D. T. McQuade, Org. Lett. 2010,
12, 5008–5011. [81]
Y. Lee, A. H. Hoveyda, J. Am. Chem. Soc. 2009, 131, 3160–3161.
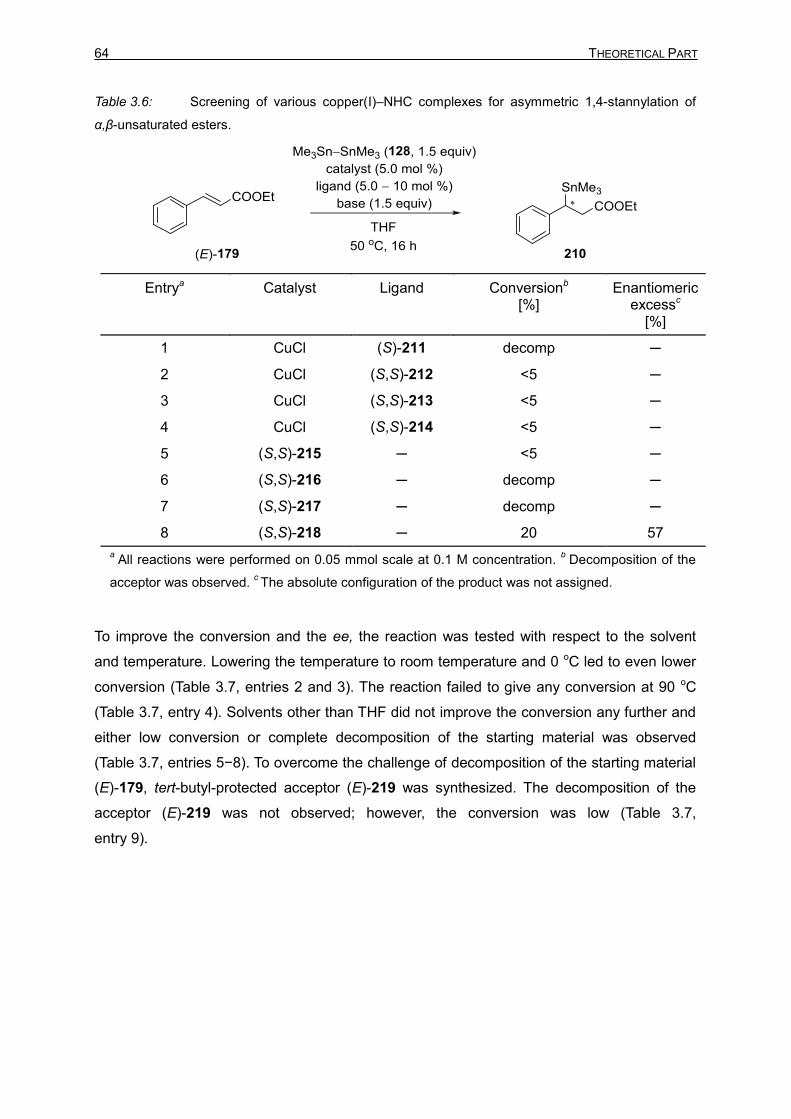
64 THEORETICAL PART
Table 3.6: Screening of various copper(I)–NHC complexes for asymmetric 1,4-stannylation of
α,β-unsaturated esters.
Entrya Catalyst Ligand Conversionb [%]
Enantiomeric excessc
[%]
1 CuCl (S)-211 decomp ─
2 CuCl (S,S)-212 <5 ─
3 CuCl (S,S)-213 <5 ─
4 CuCl (S,S)-214 <5 ─
5 (S,S)-215 ─ <5 ─
6 (S,S)-216 ─ decomp ─
7 (S,S)-217 ─ decomp ─
8 (S,S)-218 ─ 20 57
a All reactions were performed on 0.05 mmol scale at 0.1 M concentration.
b Decomposition of the
acceptor was observed. c The absolute configuration of the product was not assigned.
To improve the conversion and the ee, the reaction was tested with respect to the solvent
and temperature. Lowering the temperature to room temperature and 0 oC led to even lower
conversion (Table 3.7, entries 2 and 3). The reaction failed to give any conversion at 90 oC
(Table 3.7, entry 4). Solvents other than THF did not improve the conversion any further and
either low conversion or complete decomposition of the starting material was observed
(Table 3.7, entries 5−8). To overcome the challenge of decomposition of the starting material
(E)-179, tert-butyl-protected acceptor (E)-219 was synthesized. The decomposition of the
acceptor (E)-219 was not observed; however, the conversion was low (Table 3.7,
entry 9).
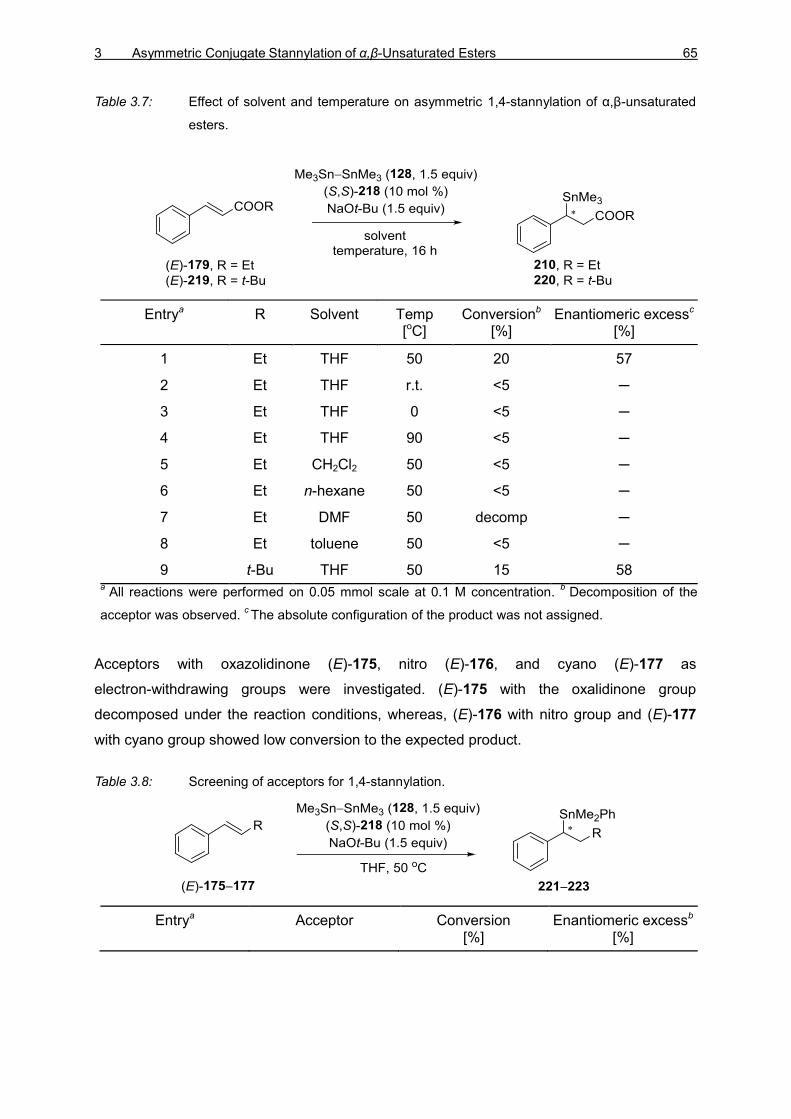
3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 65
Table 3.7: Effect of solvent and temperature on asymmetric 1,4-stannylation of α,β-unsaturated
esters.
Entrya R Solvent Temp [oC]
Conversionb [%]
Enantiomeric excessc [%]
1 Et THF 50 20 57
2 Et THF r.t. <5 ─
3 Et THF 0 <5 ─
4 Et THF 90 <5 ─
5 Et CH2Cl2 50 <5 ─
6 Et n-hexane 50 <5 ─
7 Et DMF 50 decomp ─
8 Et toluene 50 <5 ─
9 t-Bu THF 50 15 58 a
All reactions were performed on 0.05 mmol scale at 0.1 M concentration. b
Decomposition of the
acceptor was observed. c The absolute configuration of the product was not assigned.
Acceptors with oxazolidinone (E)-175, nitro (E)-176, and cyano (E)-177 as
electron-withdrawing groups were investigated. (E)-175 with the oxalidinone group
decomposed under the reaction conditions, whereas, (E)-176 with nitro group and (E)-177
with cyano group showed low conversion to the expected product.
Table 3.8: Screening of acceptors for 1,4-stannylation.
Entrya Acceptor Conversion [%]
Enantiomeric excessb [%]
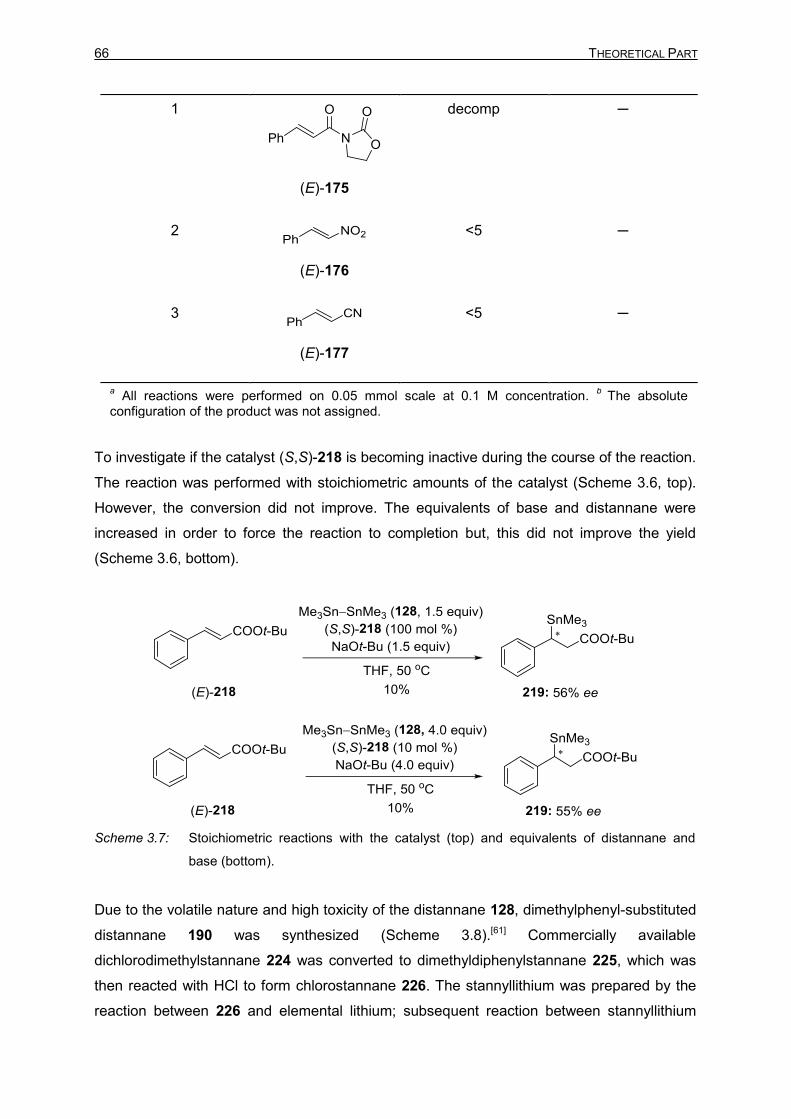
66 THEORETICAL PART
1
(E)-175
decomp ─
2
(E)-176
<5 ─
3
(E)-177
<5 ─
a All reactions were performed on 0.05 mmol scale at 0.1 M concentration.
b The absolute
configuration of the product was not assigned.
To investigate if the catalyst (S,S)-218 is becoming inactive during the course of the reaction.
The reaction was performed with stoichiometric amounts of the catalyst (Scheme 3.6, top).
However, the conversion did not improve. The equivalents of base and distannane were
increased in order to force the reaction to completion but, this did not improve the yield
(Scheme 3.6, bottom).
Scheme 3.7: Stoichiometric reactions with the catalyst (top) and equivalents of distannane and
base (bottom).
Due to the volatile nature and high toxicity of the distannane 128, dimethylphenyl-substituted
distannane 190 was synthesized (Scheme 3.8).[61] Commercially available
dichlorodimethylstannane 224 was converted to dimethyldiphenylstannane 225, which was
then reacted with HCl to form chlorostannane 226. The stannyllithium was prepared by the
reaction between 226 and elemental lithium; subsequent reaction between stannyllithium
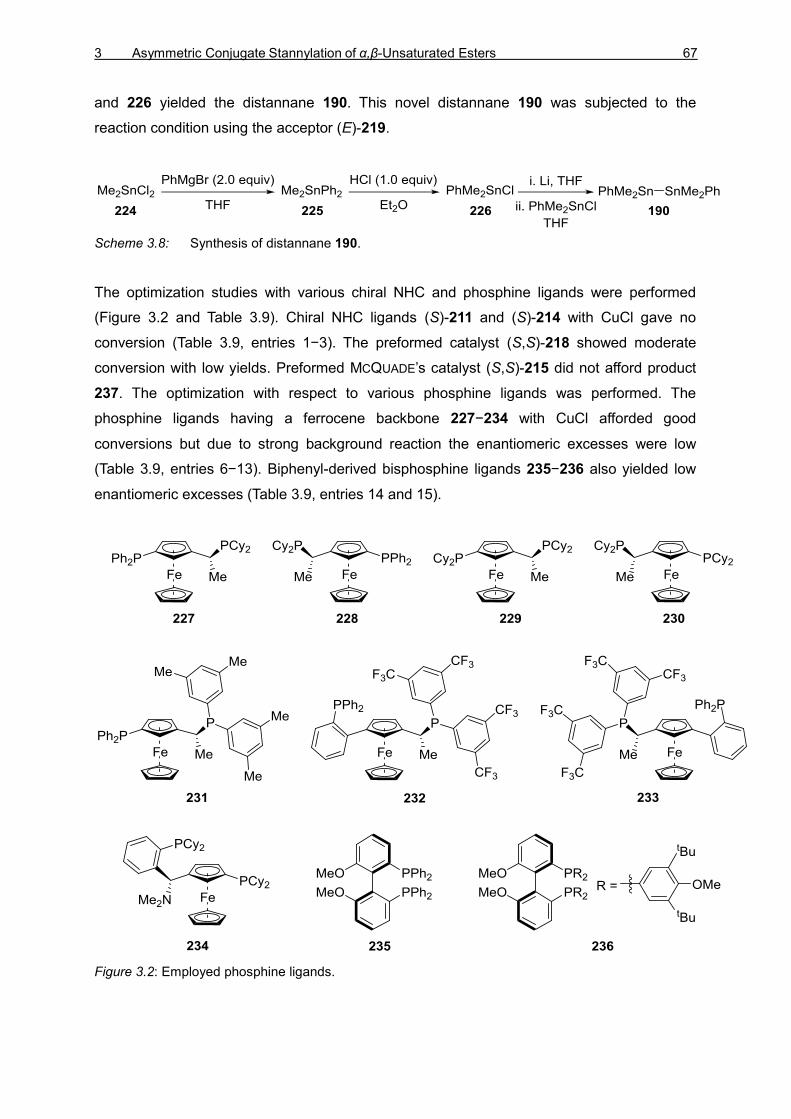
3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 67
and 226 yielded the distannane 190. This novel distannane 190 was subjected to the
reaction condition using the acceptor (E)-219.
Scheme 3.8: Synthesis of distannane 190.
The optimization studies with various chiral NHC and phosphine ligands were performed
(Figure 3.2 and Table 3.9). Chiral NHC ligands (S)-211 and (S)-214 with CuCl gave no
conversion (Table 3.9, entries 1−3). The preformed catalyst (S,S)-218 showed moderate
conversion with low yields. Preformed MCQUADE’s catalyst (S,S)-215 did not afford product
237. The optimization with respect to various phosphine ligands was performed. The
phosphine ligands having a ferrocene backbone 227−234 with CuCl afforded good
conversions but due to strong background reaction the enantiomeric excesses were low
(Table 3.9, entries 6−13). Biphenyl-derived bisphosphine ligands 235−236 also yielded low
enantiomeric excesses (Table 3.9, entries 14 and 15).
Figure 3.2: Employed phosphine ligands.
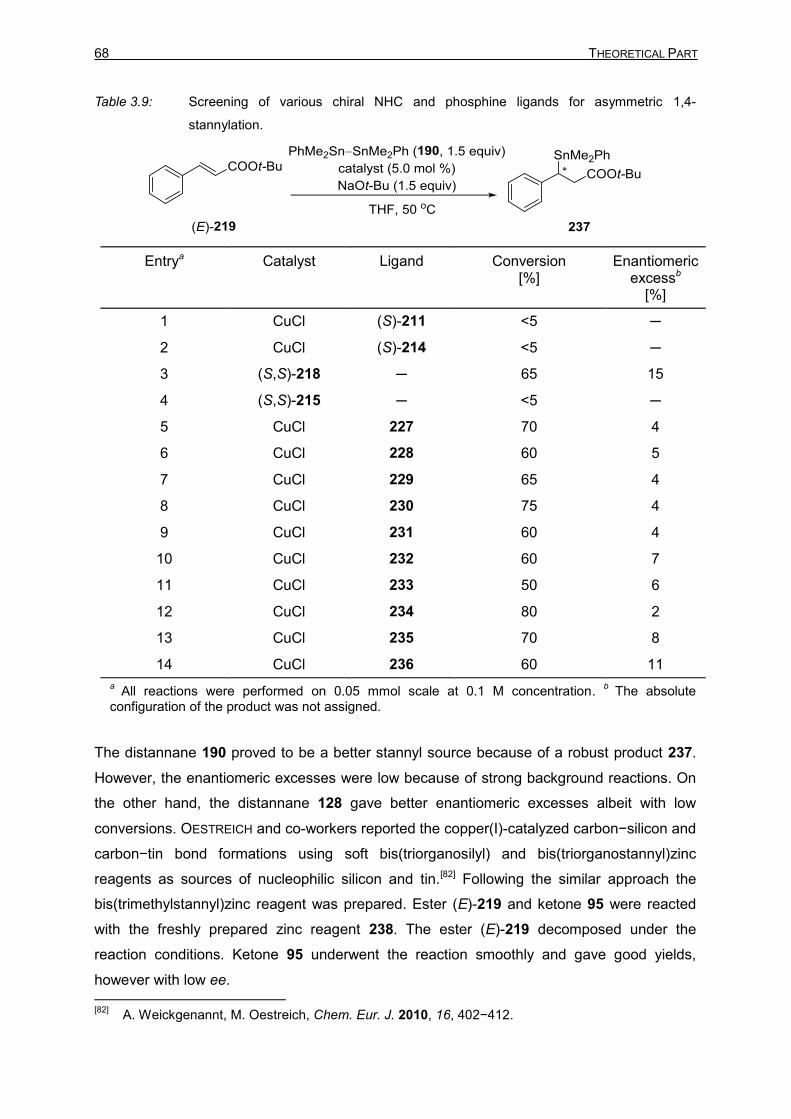
68 THEORETICAL PART
Table 3.9: Screening of various chiral NHC and phosphine ligands for asymmetric 1,4-
stannylation.
Entrya Catalyst Ligand Conversion [%]
Enantiomeric excessb
[%]
1 CuCl (S)-211 <5 ─
2 CuCl (S)-214 <5 ─
3 (S,S)-218 ─ 65 15
4 (S,S)-215 ─ <5 ─
5 CuCl 227 70 4
6 CuCl 228 60 5
7 CuCl 229 65 4
8 CuCl 230 75 4
9 CuCl 231 60 4
10 CuCl 232 60 7
11 CuCl 233 50 6
12 CuCl 234 80 2
13 CuCl 235 70 8
14 CuCl 236 60 11
a All reactions were performed on 0.05 mmol scale at 0.1 M concentration.
b The absolute
configuration of the product was not assigned.
The distannane 190 proved to be a better stannyl source because of a robust product 237.
However, the enantiomeric excesses were low because of strong background reactions. On
the other hand, the distannane 128 gave better enantiomeric excesses albeit with low
conversions. OESTREICH and co-workers reported the copper(I)-catalyzed carbon−silicon and
carbon−tin bond formations using soft bis(triorganosilyl) and bis(triorganostannyl)zinc
reagents as sources of nucleophilic silicon and tin.[82] Following the similar approach the
bis(trimethylstannyl)zinc reagent was prepared. Ester (E)-219 and ketone 95 were reacted
with the freshly prepared zinc reagent 238. The ester (E)-219 decomposed under the
reaction conditions. Ketone 95 underwent the reaction smoothly and gave good yields,
however with low ee.
[82]
A. Weickgenannt, M. Oestreich, Chem. Eur. J. 2010, 16, 402−412.

3 Asymmetric Conjugate Stannylation of α,β-Unsaturated Esters 69
Table 3.9: Use of bis(trimehylstannyl)zinc reagent 237 in copper(I)-catalyzed 1,4-stannylation.
Entry a Acceptor R3Sn
Yield [%] (R3Sn)2Zn+217
Enantiomeric excessb
[%]
1
(E)-219
Me3Sn decomp ─
2
95
Me3Sn 95 4
a All reactions were performed on 0.05 mmol scale at 0.1 M concentration.
b The absolute
configuration of the product was not assigned.
3.3 Conclusion
First, a PTC-catalyzed 1,4-stannylation of α,β-unsaturated esters was developed. The
nucleophilic tin was generated by base-mediated activation of Sn−Si bond. Although the
enantiomeric excess of 36% is still low, this transformation represents the first PTC-
catalyzed asymmetric conjugate stannylation. The substrate scope for this reaction was
explored and 1,4-stannylated products in good yields and moderate enantiomeric excesses
were obtained. In another approach an asymmetric 1,4-stannylation of an (E)-α,β-
unsaturated ester with distannanes using a chiral copper(I)–NHC catalyst for tin–tin bond
activation. An enantiomeric excess of 57% was obtained, and this result is a promising
starting point for further optimization.
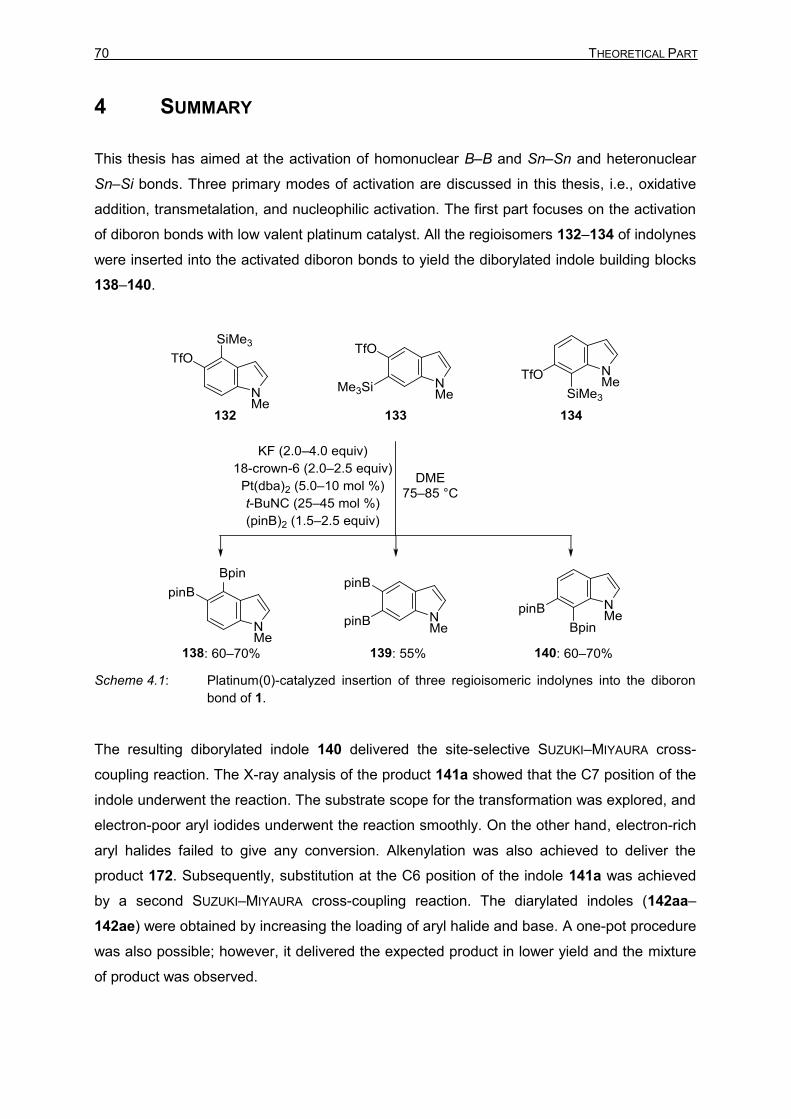
70 THEORETICAL PART
4 SUMMARY
This thesis has aimed at the activation of homonuclear B–B and Sn–Sn and heteronuclear
Sn–Si bonds. Three primary modes of activation are discussed in this thesis, i.e., oxidative
addition, transmetalation, and nucleophilic activation. The first part focuses on the activation
of diboron bonds with low valent platinum catalyst. All the regioisomers 132–134 of indolynes
were inserted into the activated diboron bonds to yield the diborylated indole building blocks
138–140.
Scheme 4.1: Platinum(0)-catalyzed insertion of three regioisomeric indolynes into the diboron
bond of 1.
The resulting diborylated indole 140 delivered the site-selective SUZUKI–MIYAURA cross-
coupling reaction. The X-ray analysis of the product 141a showed that the C7 position of the
indole underwent the reaction. The substrate scope for the transformation was explored, and
electron-poor aryl iodides underwent the reaction smoothly. On the other hand, electron-rich
aryl halides failed to give any conversion. Alkenylation was also achieved to deliver the
product 172. Subsequently, substitution at the C6 position of the indole 141a was achieved
by a second SUZUKI–MIYAURA cross-coupling reaction. The diarylated indoles (142aa–
142ae) were obtained by increasing the loading of aryl halide and base. A one-pot procedure
was also possible; however, it delivered the expected product in lower yield and the mixture
of product was observed.

4 Summary 71
Scheme 4.2: Subsequent site-selective SUZUKI–MIYAURA cross-coupling.
The second part of this thesis focuses on the unprecedented conjugate stannylation of the
α,β-unsaturated esters (E/Z)-179. Two methods were used to generate the nucleophilic tin.
First, as described by OESTREICH and co-workers, activation of Sn−Si bond with base
afforded the required tin nucleophile. The water tolerance of the reaction promoted the use
of chiral phase-transfer catalyst. Various phase-transfer catalysts were employed in order to
get the optimum ee. The quinidine-and cinchonine-derived PTCs 188 and 192 afforded the
desired product 187 in good yield albeit with low ee. The substrate scope for this reaction
was explored and electron-rich esters (Z)-208a-e underwent smooth reaction to give 1,4-
stannylated product 209a-e in good yields and low enantiomeric excesses. Second, the
activation of Sn−Sn bond was achieved using copper(I)-NHC catalyst in presence of NaOt-
Bu. Various chiral NHC ligands derived copper(I)-NHC complexes were synthesized and
subjected to the reaction condition. However, only HOVEYDA’s copper(I)-NHC catalyst
(S,S)-218 facilitated the reaction when hexamethylditin 128 was used as the tin source. Due
to the toxicity and volatile nature of 128 other tin source 190 was explored, which led to good
yields but with low enantiomeric excesses.
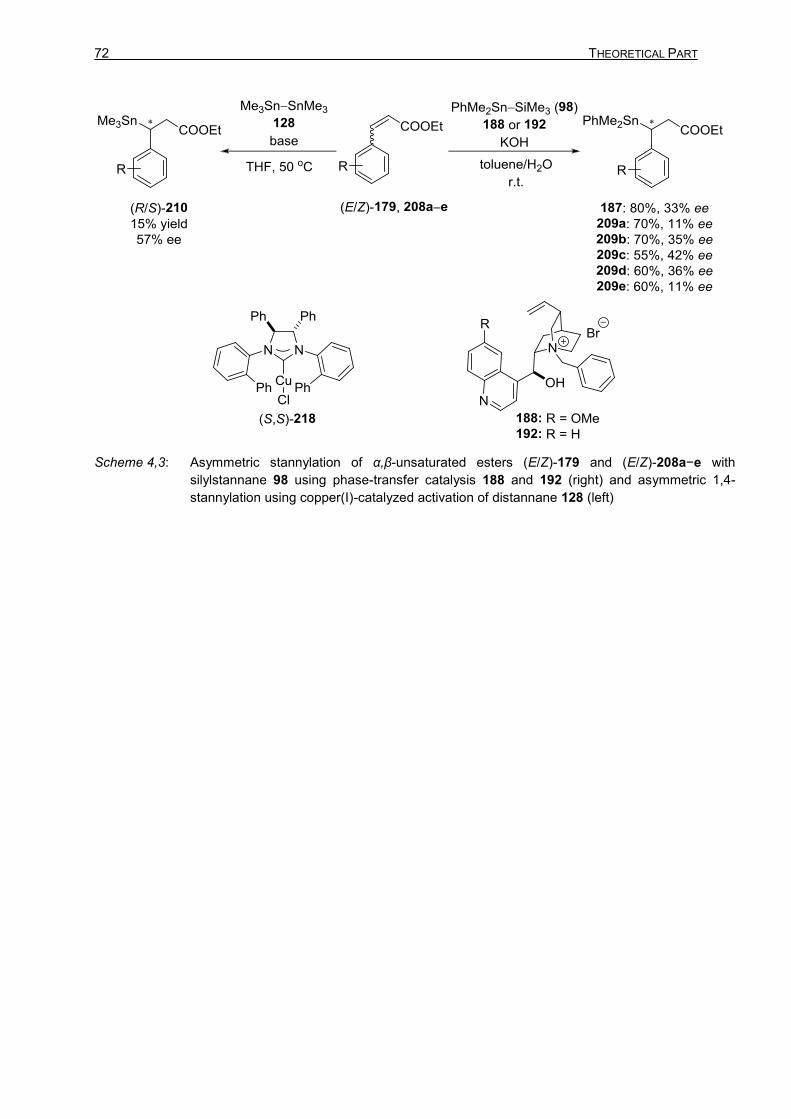
72 THEORETICAL PART
Scheme 4,3: Asymmetric stannylation of α,β-unsaturated esters (E/Z)-179 and (E/Z)-208a−e with
silylstannane 98 using phase-transfer catalysis 188 and 192 (right) and asymmetric 1,4-
stannylation using copper(I)-catalyzed activation of distannane 128 (left)

EXPERIMENTAL PART


1 General Information 75
1 GENERAL INFORMATION
All reactions were performed under argon or nitrogen atmosphere in flame-dried glassware.
For general cleaning, all laboratory glassware was kept overnight in an i-PrOH/KOH bath,
rinsed with distilled water, neutralized with saturated citric acid bath, rinsed again with
distilled water, and dried overnight at 120 °C. The glassware contaminated with transition
metals was initially rinsed with aqua regia (conc. HCl and conc. HNO3 in a ratio of 3:1) prior
to further cleaning. For the addition of reagents and solvents through silicon/rubber septa,
argon- or nitrogen-flushed disposable syringes and needles were used. All glass syringes
and stainless steel needles were used several times and stored at 120 °C. Solids were
added in a countercurrent of inert atmosphere or in solution. Low-temperature reactions
were either cooled by an ice bath, acetone/dry ice bath, or by using cryostats EK90 from
Haake or TC100E-F from Huber.
Physical Data
Melting Points (m.p.) were determined using a melting-point-determination apparatus from
Thompson Scientific and Stuart. The values are not corrected.
Chromatography
Qualitative thin-layer chromatography (TLC) was performed on glass plates with silica gel
60 F254 from Merck KGaA.
Preparative TLC was performed on glass plates with silica gel GF, UV 254, 1.5 mm
thickness from Analtech.
Following methods were used for indication of the analyte:
Exposure of the TLC plate to UV light (λ = 254 nm), UV absorption by the analyte.
Dipping the TLC plate into a solution of KMnO4 (3.0 g), K2CO3 (20 g), and
KOH (0.30 g) in distilled H2O (300 mL) and then heating with a heat gun.
Dipping the TLC plate into a solution of H3PMo12O40 (25 g), Ce(SO4)2 (10 g), and
conc. H2SO4 (60 mL) in distilled H2O (940 mL) and then heating with a heat gun.
Flash Chromatography was performed with silica gel from Merck of the grain size
40-63 µm, 230-400 mesh, ASTM.
Analytical gas-liquid chromatography (GLC) of the reaction mixtures and pure substances
were performed using gas chromatograph of the type 7890A from Agilent Technologies

76 EXPERIMENTAL PART
[equipped with a fused silica capillary column of the type HP-5 capillary column (Length:
30 m; inner diameter: 0.32 mm; film thickness of the covalently bonded stationary phase:
0.25 µm)].
All GLC analyses were performed using the following program:
Carrier gas N2; injector temperature 250 °C; detector temperature 300 °C; flow rate
1.7 mL/min; temperature program: starting temperature 40 °C, heating rate
10 °C/min, final temperature 280 °C for 10 min.
Qualitative analysis by high-performance liquid chromatography (HPLC) were performed
on an analytical HPLC system Series 1200 from Agilent Techonologies. The following
columns were used as a chiral stationary phase:
Daicel Chiralcel OD-H, OJ-H (normal phase)
NMR Spectroscopy
1H, 2H, 11B, 13C, 19F, 27Al 29Si, and 31P NMR spectra were recorded in CDCl3 (Eurisotop),
C6D6 (Eurisotop), CD2Cl2 (Sigma-Aldrich), or toluene-d8. (Eurisotop) on AV 400, AV 500, and
AV 700 instruments from Bruker at Institut für Chemie, Technische Universität Berlin. The 1H
and 13C chemical shifts are reported in parts per million (ppm) referenced to the residual
solvent resonance as the internal standard (CHCl3: δ = 7.26 ppm for 1H and CDCl3: δ =
77.16 ppm for 13C; C6D5H: δ = 7.16 ppm for 1H and C6D6: δ = 128.1 ppm for 13C; CDHCl2: δ
= 5.32 ppm for 1H and CD2Cl2: δ = 53.84 ppm for 13C; and toluene-d8: δ = 20.43, 125.13,
127.96, 128.87, 137.48 ppm).[83] For all other nuclei, the NMR resonance signals were
internally calibrated using the standardized scale for chemical shifts (unified chemical shift
scale).[84] Data are reported as follows: chemical shift, multiplicity (br s = broad singlet, s =
singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet, and mc =
centrosymmetric multiplet), coupling constant, integration, and assignment. The assignment
of signals refers to the numbering of the structures in the figures and is in accordance with
careful interpretations made from 2D NMR spectroscopy. In the case where the individual
assignment of the signal was not possible, corresponding atoms were marked with “*” , “**”
or “***” and are interchangable. The term ”Ar” refers to unspecified protons or carbon atoms
of an aromatic system and the term ”Alkyl” refers to unspecified protons or carbon atoms of
[83]
a) H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512–7515; b) G. R.
Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stolz, J. E. Bercaw,
K. I. Goldberg, Organometallics 2010, 29, 2176–2179. [84]
R. K. Harris, E. D. Becker, S. M. C. de Menezes, R. Goodfellow, P, Granger, Pure Appl.
Chem. 2001, 73, 1795–1818.

1 General Information 77
an aliphatic system. All samples were measured in reusable oven-dried standard NMR
tubes.
Mass Spectrometry
High Resolution Mass Spectrometry (HRMS) measurements were performed at the
analytical facilities of the Institut für Chemie, Technische Universität Berlin with an LTQ
Orbitrap XL [electrospray ionization (ESI)] or with a Finnigan MAT 95S (electron ionization,
70 eV) from Thermo Scientific. The in-detail fragmentation was omitted and only the
molecular ion peak or characteristic molecular fragments are considered.
Low Resolution Mass Spectrometry (LRMS) data were measured with the GC-MS-system
5975C from Agilent Technologies by electron ionization (EI). The GLC is equipped with a
fused silica capillary column of the type HP-5MS capillary column (Length: 30 m; inner
diameter: 0.25 mm; film thickness of the covalently bonded stationary phase: 0.25 µm)].
Analyses were typically performed using the following program:
Carrier gas He; injector temperature 300 °C; detector temperature 300 °C; flow rate
0.8 mL/min; temperature program: starting temperature 40 °C, heating rate 10
°C/min, final temperature 280 °C for 10 min.
Infrared Spectroscopy
Infrared (IR) spectra were recorded on a Cary 630 FT-IR from Agilent Techologies equipped
with an ATR unit and are reported (br = broad, w = weak, m, medium, s = strong) in
wavenumbers (cm−1).
Optical Rotation
The optical rotations were determined with a Polatronic H532 polarimeter from
Schmidt+Haensch. The analytes were measured as a solution in the reported solvent in
1 dm cuvettes, and the specific rotation was calculated using the following formula:
α λ
α
Where ”λ” is the wavelength (nm), ”T” is the measurement temperature (°C), [α] is the
polarimeter-determined rotation, ”c” is the concentration (g/100 mL) and ”d” is the length of
the cuvette (dm). The sodium D-line (λ = 589 nm) is used as the light source.

78 EXPERIMENTAL PART
X-Ray Crystal Structural Analysis
Data sets for X-ray crystal structure analyses were collected by PAULA NIXDORF on a Nonius
KappaCCD circle diffractometer equipped with Cu-Kα-radiation (λ = 154.178 pm) graphite
monochromator in the analytical facility at the Institut für Chemie, Technische Universität
Berlin and analyzed by Dr. ELISABETH IRRAN. Thermal ellipsoids are shown at the 50%
probability level; R-values are given for the observed reflections, wR2-values are given for all
reflections.
Software
GC-data were recorded and analyzed using EZChrom Elite Compact by Agilent. NMR data
was recorded and analyzed using Topspin 3.2 by Bruker. The stacked NMR spectra were
generated using GIMP 2.8.4 image manipulation program. GC-MS data was measured and
analyzed using Enhanced ChemStation 02.02.1431 by Agilent Technologies. The HRMS
data was analyzed using Mass++ 2.4.0 by Shimadzu and Eisai Co., Ltd. IR data was
recorded and analyzed using Microlab and Agilent Resolutions Pro 5.2.0 by Agilent
Technologies. X-ray structures were analyzed using Mercury 3.1.1 by CCDC. 3D graphics
were generated using CYLview 1.0b.[85] All schemes in this thesis were drawn in ChemDraw
Professional 15.1.0.144 by PerkinElmer. The references were retrieved using Chemistry
Reference Resolver.[86] The thesis is written using Microsoft Office 2010 by Microsoft.
Solvents and Reagents
Dichloromethane (CH2Cl2) and n-pentane were heated at reflux over CaH2 and distilled under
nitrogen atmosphere. Tetrahydrofuran (THF) and diethyl ether (Et2O) were heated at reflux
over potassium with benzophenone as indicator and distilled under nitrogen atmosphere.
Toluene was heated at reflux over sodium with benzophenone as indicator and distilled
under nitrogen atmosphere. Technical grade ethanol and acetone were used without further
purification unless stated otherwise. For extraction and flash chromatography, technical
grade solvents (tert-butyl methyl ether, cyclohexane, n-pentane, dichloromethane, diethyl
ether, and ethyl acetate) were distilled prior to use. Solvents with high purity standard were
used for the high-performance liquid chromatography (HPLC): n-heptane (Roth, Merck-
Schuchardt and Aldrich), i-PrOH (Roth and Aldrich), acetonitrile (Roth and Aldrich) and water
(Aldrich). All solvents and liquid reagents used in a glovebox were distilled and degassed by
the freeze-pump-thaw method.
[85]
CYLview, 1.0b; Legault, C. Y., Université de Sherbrooke, 2009. [86]
http://chemsearch.kovsky.net/

1 General Information 79
The following reagents were used in this thesis:
Reagent Supplier
Acetophenone Fluka
Acetophenone-d3 Sigma-Aldrich
Adamentyl isonitrile Sigma-Aldrich
Allyl bromide AlfaAesar
9,10-Bis(chloromethyl)anthracene ABCR
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) Sigma-Aldrich
Bis(pinacolato)diboron ABCR
Bis(2,2,2-trifluoroethyl) methylphosphonate Sigma-Aldrich
3,5-Bis(trifluoromethyl)benzyl bromide ABCR
Benzylbromide ABCR
5-(Benzyloxy)-1H-indole ABCR
6-(Benzyloxy)-1H-indole ABCR
4-Bromobanzaldehyde Sigma-Aldrich
1-Bromo-4-iodobenzene Sigma-Aldrich
2-(Bromomethyl)benzonitrile ABCR
tert-Butyl lithium (t-BuLi) ABCR
tert-Butyl isocyanide Sigma-Aldrich
Cesium fluoride Sigma-Aldrich
Chlorotrimethylsilane Sigma-Aldrich
Cinnamoyl chloride TCI
Cinchonine Merck
Copper(I) bromide AlfaAesar
Copper(I) chloride AlfaAesar
Copper(I) cyanide Sigma-Aldrich
Copper(I) thiocyanate Sigma-Aldrich
18-Crown-6 TCI
Cyclohex-2-en-1-one Sigma-Aldrich
Cyclohexyl isocyanide Sigma-Aldrich
1,8-Diazabicyclo(5.4.0)undec-7-ene (DBU) Sigma-Aldrich
Dichlorodimethylstannane TCI

80 EXPERIMENTAL PART
9-(Chloromethyl)anthracene ABCR
Diethylamine In-house stock
N,N-Diisopropylethylamine ABCR
3,5-Di-tert-butylbenzyl bromide Sigma-Aldrich
Di-tert-butylchlorophosphine Alfa Aesar
Di-n-butyl ether ABCR
Dimethylaminopyridine, DMAP Sigma-Aldrich
2,6-Dimethylpyridine Acros
(1S,2S)-(−)-1,2-Diphenylethylenediamine Alfa Aesar
Ethyl chloroformate Merck
Ethyl cinnamate Sigma-Aldrich
Ethyl phenylpropiolate Sigma-Aldrich
Hexabutylditin Acros
Hexamethylditin ABCR
Hexaphenylditin TCI
Hydrogen Air Liquide
Iodobenzene Sigma-Aldrich
1-iodo-2-isopropylbenzene Sigma-Aldrich
1-iodo-4-nitrobenzene ABCR
N-Iodosuccinimide ABCR
2-iodothiophene Acros
1-iodo-4-trifluoromethylbenzene Sigma-Aldrich
Isopropylisocynate Sigma-Aldrich
Lithium bis(trimethylsilyl)amide, LiHMDS Sigma-Aldrich
Magnesium In-house stock
meta-Anisaldehyde Alfa Aesar
4-Methoxyacetophenone Sigma-Aldrich
2-Methylbenzyl chloride ABCR
Methyl iodide Sigma-Aldrich
4-Methyl-2,3,5,6-tetrafluorobenzyl bromide ABCR
NHC 210 AK Teichert

1 General Information 81
NHC 211 AK Teichert
NHC 212 Dr. Lukas Delvos
NHC 213 Dr. Lukas Delvos
4-Nitro banzaldehyde ABCR
4-Nitro iodobenzene Sigma-Aldrich
Oxalyl chloride Sigma-Aldrich
2-Oxazolidinone ABCR
Palladium on charcoal 10%(w/w) ABCR
para-Anisaldehyde Acros
5-Phenoxy-1H-indole ABCR
N-Phenyl-bis(trifluoromethanesulfonimide) Sigma-Aldrich
Potassium bis(trimethylsilyl)amide, KHMDS Sigma-Aldrich
Potassium fluoride Sigma-Aldrich
Potassium hydride In-house stock
Potassium hydroxide In-house stock
Potassium-tert-butoxide, KOt-Bu Sigma-Aldrich
Quinidine In-house stock
Quinine Merck
Resorcinol Acros
Sodium hydride Sigma-Aldrich
Sodium methoxide,NaOMe Dr. ALEXANDER HENSEL
Sodium-tert-butoxide, NaOt-Bu In-house stock
Tetrakis(dimethylamino)diboron TCI
Tetrakis(triphenylphosphine)palladium(0) Sigma-Aldrich
Triethylamine, Et3N In-house stock
4-(Trifluoromethyl)acetophenone Sigma-Aldrich
4-(Trifluoromethyl)biphenyl Dr. A. SIMONNEAU
2-(Trimethylsilyl)phenyl trifluoromethanesulfonate TCI
Triphenylphosphite Sigma-Aldrich

82 EXPERIMENTAL PART
Literature Known Compounds :
The following compounds were prepared and characterized according to literature-known
procedures:
(E)-Chalcone ((E)-173),[87] Ethyl (Z)-3-phenylacrylate ((Z)-175),[88] 3-Cinnamoyloxazolidin-2-
one ((Z)-179),[89] (1S,2S,4S,5R)-1-Benzyl-2-((S)-hydroxy(6-methoxyquinolin-4-yl)methyl)-5-
vinylquinuclidin-1-ium bromide (188),[90] (1S,2S,4S,5R)-1-Benzyl-2-((R)-hydroxy(6-
methoxyquinolin-4-yl)methyl)-5-vinylquinuclidin-1-ium bromide (191),[90] (1S,2S,4S,5R)-1-
Benzyl-2-((S)-hydroxy(quinolin-4-yl)methyl)-5-vinylquinuclidin-1-ium bromide (192), [90]
(1S,2S,4S,5R)-1-(3,5-Di-tert-butylbenzyl)-2-((S)-hydroxy(quinolin-4-yl)methyl)-5-
vinylquinuclidin-1-ium bromide (193), [90] (1S,2S,4S,5R)-1-(3,5-Di-tert-butylbenzyl)-2-((S)-
hydroxy(6-methoxyquinolin-4-yl)methyl)-5-vinylquinuclidin-1-ium bromide (194), [90]
(1S,2S,4S,5R)-1-(2-Cyanobenzyl)-2-((S)-hydroxy(quinolin-4-yl)methyl)-5-vinylquinuclidin-1-
ium bromide (195), [90] (1S,2S,4S,5R)-2-((S)-Hydroxy(quinolin-4-yl)methyl)-1-(2-
methylbenzyl)-5-vinylquinuclidin-1-ium chloride (196), [90] (1S,2S,4S,5R)-1-Benzyl-2-((S)-
quinolin-4-yl(vinyloxy)methyl)-5-vinylquinuclidin-1-ium bromide (197), [90] (1S,2S,4S,5R)-1-
(3,5-Bis(trifluoromethyl)benzyl)-2-((S)-hydroxy(quinolin-4-yl)methyl)-5-vinylquinuclidin-1-ium
bromide (198),[91] (1S,2S,4S,5R)-2-((S)-Hydroxy(quinolin-4-yl)methyl)-1-(2,3,5,6-tetrafluoro-
4-methylbenzyl)-5-vinylquinuclidin-1-ium bromide (199), [90] (1S,2S,4S,5R)-1-(Anthracen-9-
ylmethyl)-2-((S)-hydroxy(quinolin-4-yl)methyl)-5-vinylquinuclidin-1-ium chloride (200),[92] 2-
((S)-Hydroxy(quinolin-4-yl)methyl)-1-((10-(((1S,2S,4S,5R)-2-((S)-hydroxy(quinolin-4-
yl)methyl)-5-vinylquinuclidin-1-ium-1-yl)methyl)anthracen-9-yl)methyl)-5-vinylquinuclidin-1-
ium (201),[93] 1,3-bis((1S,2S,3S,5R)-2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)-4,5-dihydro-1H-
imidazol-3-ium ((S,S)-214),[94] ((2S,3S)-6-Mesityl-2,3-diphenyl-2,3,6,10b-
tetrahydroimidazo[1,2-c]quinazolin-5(1H)-ylidene)copper(I) chloride ((S,S)-215),[80] ((2S,3S)-
6-(4-(tert-butyl)-2,6-dimethylphenyl)-2,3-diphenyl-2,3,6,10b-tetrahydroimidazo[1,2-
c]quinazolin-5(1H)-ylidene)copper(I) chloride ((S,S)-216), [80] ((4S,5S)-1-([1,1'-biphenyl]-2-yl)-
[87]
M.-Y. Chang, H.-Y. Tai, Y.-L. Chen, R. -T. Hsu, Tetrahedron 2012, 68, 7941−7948. [88]
S. Ueda, T. Okada, H. Nagasawa, Chem. Commun. 2010, 46, 2462−2464. [89]
A. C. Breman, S. E. M. Telderman, R. P. M. V. Santen, J. I. Scott, J. H. V. Maarseveen, S.
Ingemann, H. Hiemstra, J. Org. Chem. 2015, 80, 10561–10574. [90]
M. J. O’Donnell, S. Wu, J.C. Huffman, Tetrahedron 1994, 50, 4507–4509. [91]
P. Nun, V. Pérez, M. Calmès, J. Martinez, F. Lamaty, Chem. Eur. J. 2012, 18, 3773–3779. [92]
T. Kanemitsu, S. Koga, D. Nagano, M. Miyazaki, K. Nagata, T. Itoh, ACS Catal. 2011, 1, 1331–
1335. [93]
M. M. Parvez, N. Haraguchi, S. Itsuno, Macromolecules 2014, 47, 1922–1928. [94]
N. Ledoux, A. Linden, B. Allaert, H. V. Mierde, F. Verpoort, Adv. Synth. Catal. 2007, 349, 1692–1700.

1 General Information 83
3-mesityl-4,5-diphenylimidazolidin-2-ylidene)copper(I) chloride ((S,S)-217),[81] (E)-tert-Butyl
cinnamate ((E)-219).[95]
Nomenclature and Numbering
The numbering of compounds was done analogous to their representative structural drawing
and does not correspond to the IUPAC recommendations.
[95]
A. Armstrong, A. Ferguson, Beilstein J. Org. Chem. 2012, 8, 1747–1752.

84 EXPERIMENTAL PART

2 General Procedures 85
2 GENERAL PROCEDURES
2.1 General Procedure for Cross-Coupling Experiments
2.1.1 General Procedures for the Synthesis of the 7-Aryl-6-boryl-Substituted
Indoles 141a−e and 172 (GP 1)
A flame-dried SCHLENK tube was charged with [1,1′-bis-(diphenylphosphino)-
ferrocene]palladium(II) dichloride (6.3 mg, 9.0 µmol, 10 mol %), 1-methyl-6,7-bis(4,4,5,5-
tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140, 50 mg, 0.13 mmol, 1.5 equiv), KOH (8.3
mg, 0.15 mmol, 1.7 equiv), and the appropriate iodide (90 µmol, 1.0 equiv). DME (1.2 mL)
was subsequently added followed by water (40 µL). The reaction mixture was stirred in a
preheated oil bath at 45 oC. After a specified time, the reaction mixture was cooled to room
temperature and passed through a plug of silica gel using ethyl acetate as the eluting
solvent. The reaction mixture was evaporated in vacuo, and the residue was purified by flash
column chromatography on silica gel (n-pentane:ethyl acetate = 99:1).
2.1.2 General Procedure for the Synthesis of the 6,7-Bisaryl-Substituted Indoles
142aa−ae (GP 2)
A flame-dried SCHLENK tube was charged with [1,1′-bis-(diphenylphosphino)-
ferrocene]palladium(II) dichloride (3.3 mg, 4.5 µmol, 10 mol %), 1-methyl-7-phenyl-6-
(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (141a, 15 mg, 45 µmol, 1.0 equiv),
potassium hydroxide (5.1 mg, 90 µmol, 2.0 equiv), and the appropriate aryl iodide (0.18
mmol, 4.0 equiv). DME (0.4 mL) was subsequently added followed by water (17 µL). The
SCHLENK tube was sealed and the reaction mixture was stirred in a preheated oil bath at
90 oC. After the specified time, the reaction mixture was cooled to room temperature and
passed through a plug of silica gel using ethyl acetate as the eluting solvent. The reaction
mixture was evaporated in vacuo, and the residue was purified by flash column
chromatography on silica gel (n-pentane:ethyl acetate = 99:1).

86 EXPERIMENTAL PART
2.2 General Procedure for the Preparation of (Z)-α,β-Unsaturated
Esters (GP 3)
The esters were prepared according the procedure by STILL and GENNARI.[75] A flame-dried
SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5 mmol, 2.2 equiv). THF (8.0 mL)
was added, followed by the addition of phosphonate 207 (0.25 g, 0.75 mmol, 1.1 equiv). The
reaction mixture was cooled to –78 oC and a solution of KHMDS (0.15 g, 0.75 mmol, 1.1
equiv) in THF (1.2 mL) was subsequently added dropwise over 2 min. The resulting reaction
mixture was stirred for 30 min at –78 oC and corresponding aldehyde (0.67 mmol, 1.0 equiv)
was added dropwise over 5 min. The reaction mixture was then allowed to warm to 0 oC.
The reaction was monitored with TLC analysis and quenched with saturated aqueous NH4Cl
solution (10 mL) upon full conversion. The biphasic mixture was diluted with diethyl ether (10
mL) and washed with water (10 mL). The phases were separated, and the aqueous phase
was extracted with diethyl ether (3 x 10 mL). The organic phase was collected, and the
solvent was removed in vacuo. The residue was purified by flash column chromatography on
silica gel (cyclohexane:ethyl acetate = 99:1).
2.3 General Procedure for the Asymmetric 1,4-Stannylation of the
(Z)-α,β-Unsaturated Esters (GP 4)
A flame-dried SCHLENK tube was charged with (Z)-α,β-unsaturated ester (0.10 mmol, 1.0
equiv), followed by the phase-transfer catalyst (PTC) (188, 9.2 mg, 20 µmol, 20 mol %).
Toluene (1.0 mL) and 50% (w/v) aqueous solution of KOH in water (0.20 mL) were added,
and the resulting solution was stirred for 5 min. Silylstannane 98 (90mg, 0.30 mmol, 3.0
equiv) was added dropwise to the stirred solution. The reaction mixture was vigorously
stirred for 16 h at room temperature. The biphasic mixture was diluted with CH2Cl2 (2 mL)
and washed with water (2 mL). The phases were separated, and the aqueous phase was
extracted with diethyl ether (3 x 5 mL). The organic phases were combined, and the solvent
was removed in vacuo. The residue was purified by flash column chromatography on silica
gel (cyclohexane:ethyl acetate = 97:3).

2 Description of Experiments 87
3 DESCRIPTION OF EXPERIMENTS
3.1 Synthesis of the Indolyne Precursors
3.1.1 Synthesis of 4,5-Indolyne Precursor 132 and 5,6-Indolyne Precursor 133
3.1.1.1 5-Benzyloxy-1-methyl-1H-indole (159)
5-Benzyloxy-1-methyl-1H-indole (159) was prepared according to the procedure by GARG
and co-workers.[70] A flame-dried SCHLENK flask charged with a solution of 5-benzyloxyindole
(158) (6.0 g, 27 mmol, 1.0 equiv) in DME (75 mL), and DMSO (9 mL).
NaH (3.6 g, 90 mmol, 3.4 equiv) was added to this solution.[96] The reaction mixture was
stirred at room temperature for 40 min, followed by the dropwise addition of methyliodide
(5.7 g, 2.5 mL, 40 mmol, 1.5 equiv) over 10 min. The resulting mixture was stirred for 70 min,
and subsequently quenched with water (25 mL). The biphasic mixture was further diluted
with brine (120 mL) and ethyl acetate (120 mL). The phases were separated, and the
aqueous phase was extracted with ethyl acetate (3 x 50 mL). The organic phases were
combined, washed with brine, and dried over MgSO4. Evaporation of the solvent under
reduced pressure afforded 159 (6.2 g, 98% yield) as an orange solid, which was used in the
subsequent step without further purification.
Rf = 0.6 (cyclohexane:ethyl acetate = 75:25).
m.p.: 128 oC (CDCl3)
[96]
Commercially available NaH suspention 60% (w/w) in mineral oil was washed with n-pentane (3
x 20 mL). The resulting wet NaH was dried under vacuum prior to use.

88 EXPERIMENTAL PART
1H NMR (500 MHz, CDCl3): δ/ppm = 3.77 (s, 3H, N-CH3), 5.12 (s, 2H, H-1’), 6.40 (s, 1H, H-
4), 6.97 (d, 3J3,2 = 2.5 Hz, 1H, H-3), 7.02 (m, 1H, H-6)*, 7.18 (d, 3J2,3 = 2.5 Hz, 1H, H-2), 7.22
(m, 1H, Ar-H), 7.32–7.34 (m, 1 H, H-7)*, 7.39 (m, 2H, H-4’), 7.48 (d, 2H, H-3’).
13C NMR (125 MHz, CDCl3): δ/ppm = 33.0 (N-CH3), 71.0 (C-1’), 100.5 (C-3), 104.3 (C-7)*,
109.9 (C-6)*, 112.6 (C-4), 127.6 (C-3’)**, 127.7 (C-5’), 128.5 (C-2’)**, 128.8 (C-7a), 129.4 (C-
2), 132.2 (C-3a), 137.8 (C-2’), 153.2 (C-5).
HRMS (ESI) for C16H16NO+ [(M+H)+]: calculated 238.1226
found 238.1220
The analytical data are in accordance with those reported. [70]
3.1.1.2 1-Methyl-1H-indol-5-ol (160)
1-Methyl-1H-indol-5-ol (160) was prepared according to the procedure by GARG and co-
workers.[70] A flame-dried SCHLENK flask was charged with a solution of 5-benzyloxyindole
159 (6.0 g, 25 mmol, 1.0 equiv) in MeOH (60 mL). 10% (w/w) Pd on charcoal (5.4 g, 5 mmol,
20 mol % Pd) was added to the reaction mixture. The reaction mixture was placed under
hydrogen atmosphere (1 atm). The reaction was stirred overnight at 45 oC, the reaction
mixture was then allowed to cool to room temperature and passed through Celite® using
MeOH (100 mL) as the eluting solvent. The solvent was evaporated under reduced pressure
to afford hydroxyindole 160 (3.2 g, 88% yield) as a pink solid, which was used without further
purification.
Rf = 0.3 (cyclohexane:ethyl acetate = 75:25).
m.p.: 137 oC (CDCl3)

2 Description of Experiments 89
1H NMR (500 MHz, CDCl3): δ/ppm = 3.76 (s, 3H, N-CH3), 4.85 (brs, 1H, O-H), 6.30 (s, 1H,
H-4), 6.78 (d, 3J7,6 = 8.5 Hz, 1H, H-7), 7.10–7.12 (m, 2H, H-2, H-3), 7.22 (d, 3J6,7 = 8.5 Hz,
1H, H-6).
13C NMR (126 MHz, CDCl3): δ/ppm = 33.7 (N-CH3), 100.6 (C-3), 104.8 (C-7)*, 110.0 (C-6)*,
111.5 (C-4), 128.7 (C-7a), 129.5 (C-2), 134.3 (C-3a), 148.5 (C-5).
HRMS (ESI) for C9H10NO+ [(M+H)+]: calculated 148.0757
found 148.0754
The analytical data are in accordance with those reported.[70]
3.1.1.3 1-Methyl-1H-indol-5-yl isopropylcarbamate (161)
1-Methyl-1H-indol-5-yl isopropylcarbamate (161) was prepared according to the procedure
by GARG and co-workers.[70] A flame dried SCHLENK flask was charged with
hydroxyindole 160 (3.1 g, 21 mmol, 1.0 equiv) and CH2Cl2 (110 mL). i-PrNCO (2.7 g, 3.1 mL,
32 mmol, 1.5 equiv) was added to the reaction mixture, followed by the addition of Et3N
(0.36 g, 0.50 mL, 3.6 mmol, 0.3 equiv). The solution was stirred overnight at room
temperature and concentrated in vacuo. The residue was further purified by flash
chromatography (5 x 20 cm, cyclohexane:ethyl acetate = 70:30) to afford carbamate 161
(4.1 g, 83% yield) as a white solid.
Rf = 0.6 (cyclohexane:ethyl acetate = 50:50).
m.p.: 106 oC (CDCl3)
1H NMR (500 MHz, CDCl3): δ/ppm = 1.23 (d, 3J3’,2’ = 6.2 Hz, 6H, H-3’, H-4’), 3.77 (s, 3H, N-
CH3), 3.91-3.97 (m, 1H, H-2’), 4.85 (s, 1H, N-H), 6.44 (d, 3J3,2 = 2.3 Hz, 1H, H-3), 7.00 (d,

90 EXPERIMENTAL PART
3J7,6 = 8.6 Hz, 1H, H-7), 7.05 (d, 3J2,3 = 2.3 Hz, 1H, H-2), 7.25 (d, 3J6,7 = 8.6 Hz, 1H, H-6),
7.34 (s, 1H, C-4).
13C NMR (125 MHz, CDCl3): δ/ppm = 23.0 (C-3’)*, 27.0 (C-4’)*, 33.0 (N-CH3), 43.4 (C-2’),
101.1 (C-3), 109.4 (C-7)*, 113.0 (C-6)*, 116.0 (C-4), 128.6 (C-7a), 129.9 (C-2), 134.5 (C-3a),
144.5 (C-5), 154.8 (C-1’).
IR (ATR): /cm–1 = 3380 (m), 2969 (w), 1730 (m) 1445 (w), 1210 (w), 1118 (w), 976 (w),
843 (w), 743 (s).
HRMS (ESI) for C13H16N2O2+ [(M+H)+]: calculated 233.1285
found 233.1281
The analytical data are in accordance with those reported.[70]
3.1.1.4 1-Methyl-4-trimethylsilyl-1H-indol-5-yl isopropylcarbamate (162) and 1-
methyl-6-trimethylsilyl-1H-indol-5-yl isopropylcarbamate (163)
Silylcarbamates 162 and 163 were prepared according to the procedure by GARG and co-
workers.[70] A flame-dried SCHLENK flask was charged with carbamate 161 (3.5 g, 15 mmol,
1.0 equiv) and TMEDA (2.4 g, 3.1 mL, 21 mmol, 1.4 equiv). Diethylether (110 mL) and THF
(40 mL) were added, and the reaction mixture was cooled to 0 °C. A solution of TBSOTf
(4.1 mL, 17 mmol, 1.2 equiv) in n-pentane (13 mL) was added dropwise over 10 min. After
stirring for 5 min, the white suspension was allowed to warm to room temperature, TMEDA
(6.6 mL, 44 mmol, 3.0 equiv) was added, and the mixture was cooled to –78 oC. THF (100
mL) was added to dissolve the presipitated solid. A solution of n-BuLi (1.5 M in hexanes, 30
mL, 44 mmol, 3.0 equiv) was added dropwise over 30 min. The mixture was stirred at –78 oC
for 3 h, then TMSCl (13 mL, 103 mmol, 7.0 equiv) was added dropwise over 1 hour. The
resulting mixture was stirred at –78 oC for 1 hour, and quenched with saturated aqueous
NaHSO4 (150 mL). The reaction mixture was allowed to warm to room temperature with
~

2 Description of Experiments 91
vigorous stirring. The biphasic mixture was separated, and the organic phase was washed
with saturated aqueous NaHSO4 (100 mL) and brine (100 mL) then dried over Na2SO4. The
solvent was evaporated under reduced pressure to afford the crude products, which purified
by flash chromatography on silica gel (7.5 x 25 cm, cyclohexane:ethyl acetate = 91:9) to
afford silylcarbamates 162 (3.6 g, 80% yield) as white solid and 163 (0.29 g, 8% yield) as
viscous oil.
Analytical data for 162
Rf: 0.2 (cyclohexane:ethyl acetate = 70:30).
m.p.: 125 oC (CDCl3)
1H NMR (500 MHz, CDCl3): δ/ppm = 0.42 (s, 9H, Si-CH3), 1.24 (d, 3J3’,2’ = 6.4 Hz, 6H, H-3’),
3.76 (s, 3H, N-CH3), 3.90–3.96 (m, 1H, H-2’), 4.85 (s, 1H, N-H), 6.62 (m, 1H, H-3), 6.96 (d,
3J7,6 = 8.7 Hz, 1H, H-7), 7.08 (m, 1H, H-2), 7.30 (d, 3J6,7 = 8.7 Hz, 1H, H-6).
13C NMR (125 MHz, CDCl3): δ/ppm = 0.0 (Si-CH3), 22.1 (C-3’), 25.9 (C-3’), 32.0 (N-CH3),
42.3 (C-2’), 101.6 (C-3), 109.9 (C-6), 115.8 (C-7), 121.2 (C-4), 128.3 (C-2), 131.5 (C-7a),
132.7 (C-3a), 148.5 (C-5), 153.8 (C-1’).
HRMS (ESI) for C16H25N2O2Si+ [(M+H)+]: calculated 305.1680
found 305.1684
Analytical data for 163
Rf: 0.3 (cyclohexane:ethyl acetate = 70:30).
1H NMR (500 MHz, CDCl3): δ/ppm = 0.34 (s, 9H, Si-CH3), 1.24 (d, 3J3’,2’ = 6.4 Hz, 6H, H-3’),
3.79 (s, 3H, N-CH3), 3.91–3.98 (m, 1H, H-2’), 4.85 (s, 1H, N-H), 6.42 (d, 3J3,2 = 3.0 Hz, 1H,
H-3), 7.04 (d, 3J2,3 = 3.0 Hz, 1H, H-2), 7.34 (s, 1H, H-4), 7.35 (s. 1H, H-6).
13C NMR (125 MHz, CDCl3): δ/ppm = 0.0 (Si-CH3), 23.6 (C-3’), 33.4 (N-CH3), 43.9 (C-2’),
101.4 (C-3), 114.4 (C-4), 115.3 (C-7), 125.2 (C-7a), 130.5 (C-2), 135.2 (C-3a), 149.4 (C-5),
155.2 (C-1’).

92 EXPERIMENTAL PART
HRMS (ESI) for C16H25N2O2Si+ [(M+H)+]: calculated 305.1680
found 305.1674
The analytical data are in accordance with those reported.[70]
3.1.1.5 1-Methyl-4-trimethylsilyl-1H-indol-5-yl trifluoromethanesulfonate (132)
Silyltriflate 132 was prepared according to the procedure by GARG and co-workers.[70] A
flame-dried SCHLENK flask was charged with silyl carbamate 162 (3.3 g, 11 mmol, 1.0 equiv)
and MeCN (100 mL). Subsequently, DBU (4.2 g, 4.1 mL, 27 mmol, 2.5 equiv) and Et2NH
(1.6 g, 1.7 mL, 16 mmol, 1.5 equiv) were added. A solution of PhNTf2 (5.8 g, 16 mmol, 1.5
equiv) in MeCN (31 mL) was added dropwise over 5 min and the reaction mixture was
stirred for 2 h. The reaction mixture was passed through a plug of silica gel using ethyl
acetate (100 mL) as the eluing solvent. The solvent was evaporated in vacuo and the
residue purified by flash chromatography on silica gel (5 x 20 cm, cyclohexane:ethyl acetate
= 99:1) to afford silyltriflate 132 (3.0 g, 74% yield) as colorless oil.
Rf = 0.5 (cyclohexane:ethyl acetate = 70:30).
1H NMR (500 MHz, CDCl3): δ/ppm = 0.44 (s, 9H), 3.83 (s, 3H, N-CH3), 6.69 (d, 3J3,2 = 3.0
Hz, 1H, H-3), 7.17–7.19 (m, 2H, H-2, H-7), 7.33 (d, 3J6,7 = 8.8 Hz, 1H, H-6).
13C NMR (125 MHz, CDCl3): δ/ppm = 0.0 (Si-CH3), 32.1 (N-CH3), 102.4 (C-3), 110.3 (C-6),
113.9 (C-7), 117.7 (q, 1JC,F = 329 Hz, CF3), 122.9 (C-4), 129.5 (C-2), 131.9 (C-7a), 133.4 (C-
3a), 147.6 (C-5).
HRMS (ESI) for C13H17F3NO3SSi+ [(M+H)+]: calculated 352.0647
found 352.0643
The analytical data are in accordance with those reported.[70]

2 Description of Experiments 93
3.1.1.6 1-Methyl-6-trimethylsilyl-1H-indol-5-yl trifluoromethanesulfonate (133)
Silyltriflate 133 was prepared according to the procedure by GARG and co-workers.[70] A
flame-dried SCHLENK flask was charged with silyl carbamate 163 (0.20 g, 0.65 mmol,
1.0 equiv) and MeCN (7.0 mL). Subsequently, DBU (0.52 mL, 3.6 mmol, 2.5 equiv) and
BnNH2 (0.23 mL, 2.2 mmol, 1.5 equiv) were added to the reaction mixture. The resulting
mixture was stirred at 40 °C for 3 h, and allowed to cool to room temperature. A solution of
PhNTf2 (0.75 g, 2.2 mmol, 1.5 equiv) in MeCN (3.0 mL) was added dropwise over 5 min, and
stirred the resulting reaction mixture for 60 min. The reaction mixture was passed through a
plug of silica gel using ethyl acetate (100 mL) as the eluting solvent. The solvent was
evaporated in vacuo and the residue was purified by flash chromatography (3.5 x 20 cm,
cyclohexane:ethyl acetate = 99.5:0.5) to afford silyltriflate 133 (0.18 g, 80% yield) as yellow
oil.
Rf = 0.5 (cyclohexane:ethyl acetate = 70:30).
1H NMR (500 MHz, CDCl3): δ/ppm = 0.52 (s, 9H, Si-CH3), 3.80 (s, 3H, N-CH3), 6.69 (d,
3J3,2 = 3.2 Hz, 1H, H-3), 7.14 (d, 3J2,3 = 3.2 Hz, 1H, H-2), 7.41(s, 1H, H-4), 7.6 (s, 1H, H-6).
13C NMR (125 MHz, CDCl3): δ/ppm = 0.0 (Si-CH3), 33.4 (N-CH3), 101.9 (C-3), 111.9 (C-4),
115.4 (C-7), 119 (q, 1JC,F = 320 Hz, CF3), 124.8 (C-6), 130.1 (C-7a), 131.9 (C-2), 135.7 (C-
3a), 149.6 (C-5).
HRMS (ESI) for C13H17F3NO3SSi++ [(M+H)+]: calculated 352.0647
found 352.0645
The analytical data are in accordance with those reported.[70]

94 EXPERIMENTAL PART
3.1.2 Synthesis of 6,7-Indolyne Precursor 134
3.1.2.1 6-Benzyloxy-1-methyl-1H-indole (165)
6-Benzyloxy-1-methyl-1H-indole (165) was prepared according to the procedure by GARG
and co-workers.[71] A flame-dried SCHLENK flask charged with a solution of 6-benzyloxyindole
(164) (6.0 g, 27 mmol, 1.0 equiv) in DME (75 mL) and DMSO (9 mL).
NaH (3.6 g, 90 mmol, 3.4 equiv) was added to this solution.[96] The reaction mixture was
stirred at room temperature for 40 min, followed by the dropwise addition of methyliodide
(5.6 g, 2.5 mL, 39 mmol, 1.5 equiv) over 10 min. The resulting mixture was stirred for 70 min,
and subsequently quenched with water (25 mL). The biphasic mixture was further diluted
with brine (120 mL) and ethyl acetate (120 mL). The phases were separated, and the
aqueous phase was extracted with ethyl acetate (3 x 50 mL). The organic phases were
combined, washed with brine, and dried over MgSO4. Evaporation of the solvent under
reduced pressure afforded 165 (6.0 g, 94% yield) as an off white solid, which was used in
the subsequent step without further purification.
Rf = 0.5 (cyclohexane:ethyl acetate = 75:25).
m.p.: 139 oC (CDCl3)
1H NMR (500 MHz, CDCl3): δ/ppm = 3.72 (s, 3H, N-CH3), 5.15 (s, 2H, H-1’), 6.42 (d, 3J3,2 =
3.0, 1H, H-3), 6.87-6.89 (m, 2H, Ar-H), 6.96 (d, 3J2,3 = 3.0, 1H, H-2), 7.32–7.35 (m, 1H, Ar-
H), 7.41–7.47 (m, 2H, H-3’)*, 7.52–7.49 (m, 3H, H-4’, H-5’)*.
13C NMR (125 MHz, CDCl3): δ/ppm = 32.8 (N-CH3), 70.8 (C-1’), 94.4 (Ar-C), 100.8 (C-3),
109.9 (C-4), 121.4 (C-3’), 123.0 (Ar-C), 127.6 (Ar-C), 127.8 (Ar-C), 127.9 (Ar-C), 128.6 (C-2),
137.4 (C-3a), 137.5 (C-2‘), 155.4 (C-5).

2 Description of Experiments 95
HRMS (ESI) for C16H16NO+ [(M+H)+]: calculated 238.1226
found 238.1220
The analytical data are in accordance with those reported.[71]
3.1.2.2 1-Methyl-1H-indol-6-ol (166)
1-Methyl-1H-indol-6-ol (166) was prepared according to the procedure by GARG and co-
workers.[71] A flame-dried SCHLENK flask was charged with a solution of 6-benzyloxyindole
165 (5.5 g, 24 mmol, 1.0 equiv) in MeOH (60 mL). 10% (w/w) Pd on charcoal (1.4 g,
1.4 mmol, 5 mol % Pd) was added to the reaction mixture. The reaction mixture was placed
under hydrogen atmosphere (1 atm). The reaction was stirred overnight at 45 oC, the
reaction mixture was then allowed to cool down to room temperature and passed through
Celite® using MeOH (100 mL) as the eluting solvent. The solvent was evaporated under
reduced pressure to afford hydroxyindole 166 (3.4 g, 95% yield) as viscous oil, which was
used without further purification.
Rf = 0.4 (cyclohexane:ethyl acetate = 75:25).
1H NMR (400 MHz, CDCl3): δ/ppm = 3.68 (s, 3H, N-CH3), 5.51 (s, 1H, O-H), 6.39 (d, 3J3,2 =
3.1 Hz, 1H, H-3), 6.69 (dd, 3J5,4 = 8.3 Hz, 4J5,7 = 1.9 Hz 1H, H-5), 6.77 (d, 3J7,5 = 1.9 Hz, 1H,
H-7), 6.92 (d, 3J2,3 = 3.1, 1H, H-2), 7.44 (d, 3J4,5 = 8.3 Hz, 1H, H-4).
13C NMR (100 MHz, CDCl3): δ/ppm = 32.9 (N-CH3), 103.8 (C-3), 104.5 (C-7), 112.4 (C-5),
122.3 (C-4), 125.7 (C-7a), 130.2 (C-2), 138.4 (C-3a), 148.7 (C-6).
HRMS (ESI) for C9H10NO+ [(M+H)+]: calculated 148.0757
found 148.0759

96 EXPERIMENTAL PART
The analytical data are in accordance with those reported.[71]
3.1.2.3 1-Methyl-1H-indol-6-yl isopropylcarbamate (167)
1-Methyl-1H-indol-6-yl isopropylcarbamate (167) was prepared according to the procedure
by GARG and co-workers.[71] A flame-dried SCHLENK flask was charged with
hydroxyindole 166 (4.7 g, 29 mmol, 1.0 equiv) and DCM (120 mL). i-PrNCO (3.7 g, 4.3 mL,
44 mmol, 1.5 equiv) was added to the reaction mixture, followed by the addition of Et3N
(0.87 g, 1.2 mL, 8.7 mmol, 0.3 equiv). The solution was stirred overnight at room
temperature, and concentrated in vacuo. The residue was further purified by flash
chromatography on silica gel (5 x 20 cm, cyclohexane:ethyl acetate = 70:30) to afford
carbamate 167 (5.0 g, 74% yield) as a white solid.
Rf = 0.2 (cyclohexane:ethyl acetate = 75:25).
m.p.: 109 oC (CDCl3)
1H NMR (500 MHz, CDCl3): δ/ppm = 1.25 (d, 3J3’,2’ = 6.4 Hz, 6H, H-3’), 3.74 (s, 3H, N-CH3),
3.88–3.95 (m, 1H, H-2’), 4.86 (s, 1H, N-H), 6.45 (d, 3J3,2 = 3.0 Hz, 1H, H-3), 6.87 (d, 3J5,4 =
8.3 Hz, 1H, H-5), 7.02 (d, 3J2,3 = 3.0, 1H, H-2), 7.12 (s, 1H, H-7), 7.54 (d, 3J4,5 = 8.3 Hz, 1H,
H-4).
13C NMR (125 MHz, CDCl3): δ/ppm = 23.0 (C-3’), 32.9 (N-CH3), 43.4 (C-2’), 101.0 (C-3),
102.4 (C-7), 113.9 (C-5), 121.0 (C-4), 126.1 (C-7a), 129.3 (C-2), 136.7 (C-3a), 146.6 (C-6),
154.5 (C-1’).
HRMS (ESI) for C13H17N2O2+ [(M+H)+]: calculated 233.1285
found 233.1285
The analytical data are in accordance with those reported.[71]

2 Description of Experiments 97
3.1.2.4 1-Methyl-7-trimethylsilyl-1H-indol-6-yl isopropylcarbamate (168)
Silylcarbamates 168 was prepared according to the procedure by GARG and co-workers.[71]
A flame-dried SCHLENK flask was charged with carbamate 167 (4.8 g, 21 mmol, 1.0 equiv)
and TMEDA (3.3 g, 4.3 mL, 29 mmol, 1.4 equiv). Diethylether (150 mL) and THF (50 mL)
were added and the reaction mixture was cooled to 0 °C. A solution of TBSOTf
(5.5 g, 4.4 mL, 26 mmol, 1.2 equiv) in n-pentane (19 mL) was added dropwise over 15 min.
After stirring for 5 min, the white suspension was allowed to warm up to room temperature,
TMEDA (7.3 g, 11 mL, 63 mmol, 3.0 equiv) was added, and the mixture was cooled to –78
oC. THF (100 mL) was added to dissolve the crashed solid. A solution of n-BuLi (1.5 M in
hexanes, 50 mL, 63 mmol, 3.5 equiv) was added dropwise over 40 min. The mixture was
stirred at –78 oC for 3 h, then TMSCl (16 g, 19 mL, 147 mmol, 7.0 equiv) was added
dropwise over 1 hour. The resulting mixture was stirred at –78 oC for 1 hour, and quenched
with saturated aqueous NaHSO4 (200 mL). The reaction mixture was allowed to warm up to
room temperature with vigorous stirring. The biphasic mixture was separated, and the
organic phase was washed with saturated aqueous NaHSO4 (100 mL) and brine (100 mL),
and dried over Na2SO4. The solvent was evaporated under reduced pressure to afford the
crude product, which was purified by flash chromatography (7.5 x 10 cm, cyclohexane:ethyl
acetate = 85:15) to afford silylcarbamates 168 (5.1 g, 81% yield) as a white solid.
Rf: = 0.3 (75:25 cyclohexane/ethyl acetate).
m.p.: 148 oC (CDCl3)
1H NMR (500 MHz, CDCl3): δ/ppm = 0.50 (s, 9H, Si-CH3), 1.24 (d, 3J3’,2’ = 6.5 Hz, 6H, H-3’),
3.85 (s, 3H, N-CH3), 3.92-3.98 (m, 1H, H-2’), 4.84 (s, 1H, N-H), 6.52 (d, 3J3,2 = 3.0 Hz, 1H,
H-3), 6.83 (d, 3J5,4 = 8.1 Hz, 1H, H-5), 7.00 (d, 3J2,3 = 3.0, 1H, H-2), 7.59 (d, 3J4,5 = 8.1 Hz,
1H, H-4)

98 EXPERIMENTAL PART
13C NMR (126 MHz, CDCl3): δ/ppm = 1.2 (Si-CH3), 21.0 (C-3’), 35.8 (N-CH3), 41.4 (C-2’),
100.8 (C-3), 111.5 (C-7), 113.1 (C-5), 121.1 (C-4), 125.0 (C-7a), 129.9 (C-2), 140.5 (C-3a),
150.6 (C-6), 152.6 (C-1’).
HRMS (ESI) for C16H25N2O2Si+ [(M+H)+]: calculated 305.1680
found 305.1685
The analytical data are in accordance with those reported.[71]
3.1.2.5 1-Methyl-7-trimethylsilyl-1H-indol-6-yl trifluoromethanesulfonate (134)
Silyltriflate 134 was prepared following the procedure used by GARG and co-workers.[71] A
flame-dried SCHLENK flask containing a stirring bar was charged with silyl carbamate 168
(4.9 g, 14 mmol, 1.0 equiv) and THF (120 mL). The solution was cooled to –78 oC and n-
BuLi (1.5 M in hexanes, 13 mL, 19 mmol, 1.4 equiv) and Et2NH (2.8 g, 2.0 mL,
38 mmol, 2.7 equiv) were added. A solution of PhNTf2 (6.9 g, 21 mmol, 1.5 equiv) in THF (50
mL) was added dropwise and the reaction mixture was stirred for 1 hour at –78 oC. The
reaction was allowed to warm up to room temperature. The reaction mixture was
subsequently passed through a plug of silica gel using ethyl acetate (100 mL) as the eluting
solvent. The solvent was evaporated in vacuo, and the residue was purified by flash
chromatography on silica gel (7.5 x 10 cm, cyclohexane/ethyl acetate = 95:5) to afford
silyltriflate 134 (3.8 gm, 77% yield) as a white solid.
Rf = 0.5 (cyclohexane:ethyl acetate = 70:30).
m.p.: 95 oC (CDCl3)
1H NMR (500 MHz, CDCl3): δ/ppm = 0.53 (s, 9H, Si-CH3), 3.86 (s, 3H, N-CH3), 6.54 (d, 3J3,2
= 3.0 Hz, 1H, H-3), 6.98 (d, 3J5,4 = 8.3 Hz, 1H, H-5), 7.09 (d, 3J2,3 = 3.0, 1H, H-2), 7.60 (d,
3J4,5 = 8.3 Hz, 1H, H-4).

2 Description of Experiments 99
13C NMR (125 MHz, CDCl3): δ/ppm = 2.7 (Si-CH3), 37.7 (N-CH3), 103.0 (C-3), 113.3 (C-5),
115.6 (C-7), 118.8(q, 1JC,F = 320 Hz, CF3), 123.5 (C-4), 128.6 (C-7a), 133.58 (C-2), 142.8 (C-
3a), 151.1 (C-6).
HRMS (ESI) for C13H17F3NO3SSi+ [(M+H)+]: calculated 352.0645
found 352.0649
The analytical data are in accordance with those reported.[71]

100 EXPERIMENTAL PART
3.2 Synthesis of Diborylated Indoles
3.2.1 1-Methyl-4,5-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (138)
In a glove box, a flame-dried SCHLENK tube was charged with bis(dibenzylideneacetone)-
platinum(0) (36.0 mg, 540 µmol, 7.50 mol %), bis(pinacolato)diboron (1, 270 mg, 1.10 mmol,
1.50 equiv), 18-crown-6 (374 mg, 1.42 mmol, 2.00 equiv), and potassium fluoride (165 mg,
2.84 mmol, 4.00 equiv). The SCHLENK tube was removed from the glove box and connected
to a nitrogen SCHLENK line. tert-Butyl isocyanide (22.0 mg, 30.0 µL,
0.260 mmol, 0.370 equiv), and a solution of 1-methyl-4-trimethylsilyl-1H-indol-5-yl
trifluoromethanesulfonate (132, 250 mg, 0.710 mmol, 1.00 equiv) in DME (5.00 mL) were
added. The SCHLENK tube was sealed and placed in a preheated oil bath at 75 oC. After 16
h, the reaction mixture was cooled to room temperature and passed through a plug of silica
using ethyl acetate (50 mL) as the eluting solvent. The solvent was evaporated in vacuo and
the residue was purified by flash column chromatography on silica gel (3.5 x 20 cm,
cyclohexane:ethyl acetate = 97:3→95:5) to afford diborylated indole 138 (190 mg, 70%
yield) as yellow viscous oil.
Rf = 0.6 (cyclohexane:ethyl acetate = 70:30).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.37 (s, 12H, pin-CH3)*, 1.46 (s, 12H, pin-CH3)*, 3.75
(s, 3H, N-CH3), 6.66 (d, 3J3,2 = 2.95 Hz, 1H, H-3), 7.02 (d, 3J2,3 = 2.95 Hz, 1H, H-2) 7.30 (d,
3J7,6 = 8.3 Hz, 1H, H-7), 7.63 (d, 3J6,7 = 8.3 Hz, 1H, H-6).
13C NMR (126 MHz, CDCl3): δ/ppm = 24.9 (pin-CH3)*, 25.2 (pin-CH3)*, 32.7 (N-CH3), 83.5
(pin-Ctert)**, 83.7 (pin-Ctert)**, 102.5 (C-3), 109.3 (C-7), 127.7 (C-6), 128.7 (C-2), 132.2 (C-
7a), 137.2 (C-3a).

2 Description of Experiments 101
The signals corresponding to the carbon–boron bonds were not observed in the 13C NMR
spectrum.
11B NMR (160 MHz, CDCl3): δ/ppm = 32.7 (brs).
IR (ATR): /cm–1 = 2975 (m), 1527 (w), 1490 (w) 1354 (m), 1234 (w), 1198 (w) 1143 (s),
1115 (s), 852 (s).
HRMS (ESI) for C21H32B2NO4+ [(M+H)+]: calculated 384.2512
found 384.2510
3.2.2 1-Methyl-5,6-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (139)
In a glove box, a flame-dried sealed SCHLENK tube was charged with
bis(dibenzylideneacetone)platinum(0) (9.4 mg, 14 µmol, 10 mol %), 1-methyl-6-trimethylsilyl-
1H-indol-5-yl trifluoromethanesulfonate (133, 50 mg, 0.14 mmol, 1.0 equiv),
bis(pinacolato)diboron (1, 81 mg, 0.32 mmol, 2.5 equiv), 18-crown-6 (94 mg, 0.36 mmol, 2.0
equiv), and potassium fluoride (21 mg, 0.36 mmol, 2.5 equiv). The SCHLENK tube was
removed from the glove box and connected to a nitrogen SCHLENK line. tert-Butyl isocyanide
(5.4 mg, 7.5 µL, 64 µmol, 0.45 equiv) and DME (1.0 mL) were added. The SCHLENK tube
was sealed and placed in a preheated oil bath at 85 oC. After 16 h, the reaction mixture was
cooled to room temperature and passed through a plug of silica using ethyl acetate as the
eluting solvent. The solvent was evaporated in vacuo and the residue was purified by flash
column chromatography on silica gel (2.5 x 15 cm, cyclohexane:ethyl acetate = 97:3→95:5)
to afford diboronated indole 139 (29 mg, 55% yield) as pink viscous oil.
Rf = 0.6 (cyclohexane:ethyl acetate = 70:30).
~

102 EXPERIMENTAL PART
1H NMR (500 MHz, CDCl3): δ/ppm = 1.25 (s, 12H, pin-CH3)*, 1.26 (s, 12H, pin-CH3)*, 3.67
(s, 3H, N-CH3), 6.32 (d, 3J3,2 = 3.1 Hz, 1H, H-3), 7.90 (d, 3J2,3 = 3.1 Hz, 1H, H-2) 7.55 (s, 1H,
H-4) 7.86 (s, 1H, H-7).
13C NMR (126 MHz, CDCl3): δ/ppm = 23.9 (pin-CH3), 31.8 (N-CH3), 82.4 (pin-Ctert)*, 82.6
(pin-Ctert)*, 100.3 (C-3), 114.3 (C-7), 126.7 (C-4), 128.3 (C-7a), 128.4 (C-2), 136.3 (C-3a).
The signals corresponding to the carbon–boron bonds were not observed in the 13C NMR
spectrum.
11B NMR (160 MHz, CDCl3): δ/ppm = 32.6 (brs).
IR (ATR): /cm–1 = 2970 (m), 1624 (w), 1576 (w), 1472 (w), 1384 (m), 1293 (w), 1202 (w)
1139 (s), 856 (s).
HRMS (ESI) for C21H32B2NO4+ [(M+H)+]: calculated 384.2512
found 384.2510
3.2.3 1-Methyl-6,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140)
In a glove box, a flame-dried screw-capped SCHLENK tube was charged with
bis(dibenzylideneacetone)platinum(0) (36.0 mg, 54.0 µmol, 7.50 mol %), 1-methyl-7-
trimethylsilyl-1H-indol-6-yl trifluoromethanesulfonate (134, 250 mg, 0.710 mmol, 1.00 equiv),
bis(pinacolato)diboron (1, 274 mg, 1.08 mmol, 1.50 equiv), 18-crown-6 (372 mg, 1.42 mmol,
2.00 equiv), and potassium fluoride (80.0 mg, 1.40 mmol, 2.00 equiv). The SCHLENK tube
was removed from the glove box and connected to a nitrogen Schlenk line. tert-Butyl
isocyanide (21 mg, 29 µL, 0.25 mmol, 0.35 equiv) and DME (5.00 mL) were added. The
SCHLENK tube was sealed and placed in a preheated oil bath at 85 oC. After 16 h, the
~

2 Description of Experiments 103
reaction mixture was cooled to room temperature and passed through a plug of silica eluting
with ethyl acetate. The solvent was evaporated in vacuo and the residue was purified using
flash column chromatography on silica gel (3.5 x 20 cm, cyclohexane:ethyl acetate =
97:3→95:5) to afford the title compound 140 (185 mg, 68% yield)as a yellow solid.
Rf = 0.6 (70:30 cyclohexane/ethyl acetate).
m.p.: 145 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.37 (s, 12H, pin-CH3)*, 1.50 (s, 12H, pin-CH3)*, 3.96
(s, 3H, N-CH3), 6.42 (d, 3J3,2 = 3.1 Hz, 1H, H-3), 7.02 (d, 3J2,3 = 3.1 Hz, 1H, H-2) 7.60-7.63
(m, 2H, H-4, H-5).
13C NMR (126 MHz, CDCl3): δ/ppm = 24.8 (pin-CH3)*, 26.0 (pin-CH3)*, 35.3 (N-CH3), 83.6
(pin-Ctert)**, 84.3 (pin-Ctert)**, 100.6 (C-3), 120.9 (C-7), 126.3 (C-6), 130.8 (C-7a), 131.0 (C-
2), 139.0 (C-3a).
The signals corresponding to the carbon–boron bonds were not observed in the 13C NMR
spectrum.
11B NMR (160 MHz, CDCl3): δ/ppm = 32.2 (brs).
IR (ATR): /cm–1 = 2972 (m), 1527 (w), 1478 (w), 1311 (m), 1280 (w), 1210 (w) 1136 (s),
1060 (w), 818 (s).
HRMS (ESI) for C21H32B2NO4+ [(M+H)+]: calculated 384.2512
found 384.2509
~

104 EXPERIMENTAL PART
3.3 Synthesis of the 7-Aryl-6-boryl-Substituted Indoles
3.3.1 1-Methyl-7-phenyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole
(141a)
According to GP 1, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (6.3 mg, 9.0 µmol, 10 mol %), 1-
methyl-6,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140, 50 mg, 0.13
mmol, 1.5 equiv), KOH (8.3 mg, 0.15 mmol, 1.7 equiv), and iodobenzene (18 mg, 9.7 µL,
0.087 mmol, 1.0 equiv). DME (1.2 mL) was subsequently added followed by water (40 µL).
The reaction mixture was stirred in a preheated oil bath at 45 oC. After 30 min, the reaction
mixture was cooled to room temperature and passed through a plug of silica gel using ethyl
acetate as the eluting solvent. The reaction mixture was evaporated in vacuo, and the
residue was purified by flash column chromatography on silica gel (n-pentane:ethyl acetate
= 99:1) to afford 141a (23 mg, 80% yield) as a white solid.
Rf: 0.7 (cyclohexane:ethyl acetate = 90:10).
m.p.: 120 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.08 (s, 12H, pin-CH3), 3.17 (s, 3H, N-CH3), 6.50 (d,
3J3,2 = 3.1 Hz, 1H, H-3), 6.95 (d, 3J2.3 = 3.1 Hz, 1H, H-2) 7.32–7.39 (m, 5H, Ar-H), 7.43 (d,
3J5,4 = 8.0 Hz, 1H, H-5), 7.60 (d, 3J4,5 = 8.0 Hz, 1H, H-4).
13C NMR (126 MHz, CDCl3): δ/ppm = 24.5 (pin-CH3), 36.7 (N-CH3), 82.9 (pin-Ctert), 100.7 (C-
3), 119.5 (C-4), 124.6 (C-5), 126.7 (C-3’), 126.8 (C-4’), 130.9 (C-2’), 131.2 (C-7a), 131.9 (C-
2), 132.3 (C-7), 134.0 (C-3a), 140.6 (C-1’).

2 Description of Experiments 105
The signal corresponding to the carbon–boron bond was not observed in the 13C NMR
spectrum.
11B NMR (160 MHz, CDCl3): δ/ppm = 32.1 (brs).
IR (ATR): /cm–1 = 2972 (m), 1580 (w), 1510 (w), 1467 (w), 1390 (w), 1356 (s), 1142 (s),
1074 (w), 850 (s).
HRMS (ESI) for C21H25BNO2+ [(M+H)+]: calculated 334.1973
found 334.1971
3.3.2 1-Methyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-7-(4-
trifluoromethylphenyl)-1H-indole (141b)
According to GP 1, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (6.3 mg, 9.0 µmol, 10 mol %), 1-
methyl-6,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140, 50 mg, 0.13
mmol, 1.5 equiv), KOH (8.3 mg, 0.15 mmol, 1.7 equiv), and 1-iodo-4-trifluoromethylbenzene
(24 mg, 13 µL, 0.087 mmol, 1.0 equiv). DME (1.2 mL) was subsequently added followed by
water (40 µL). The reaction mixture was stirred in a preheated oil bath at 45 oC. After 2 h, the
reaction mixture was cooled to room temperature and passed through a plug of silica gel
using ethyl acetate as the eluting solvent. The reaction mixture was evaporated in vacuo,
and the residue was purified by flash column chromatography on silica gel (n-pentane:ethyl
acetate = 99:1) to afford 141b (31 mg, 90% yield) as a white solid.
Rf = 0.7 (ethyl cyclohexane:ethyl acetate = 90:10).
~

106 EXPERIMENTAL PART
m.p.: 152 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.06 (s, 12H, pin-CH3), 3.19 (s, 3H, N-CH3), 6.52 (d,
3J3,2 = 3.1 Hz, 1H, H-3), 6.98 (d, 3J2.3 = 3.1 Hz, 1H, H-2), 7.48 (d, 3J5,4 = 7.9 Hz, 1H, H-5),
7.50–7.52 (m, 2H, H-2’), 7.61–7.64 (m, 3H, H-4, H-3’),.
13C NMR (126 MHz, CDCl3): δ/ppm = 24.5 (pin-CH3), 37.0 (N-CH3), 83.1 (pin-Ctert), 100.9 (C-
3), 120.0 (C-4), 123.6 (q, 3JC-3’,F = 3.8 Hz, C-3’), 124.5 (q, 1JC-5’,F = 272 Hz, C-5’), 125.1 (C-5),
129.2 (q, 2JC-4’,F = 32 Hz, C-4’), 130.9 (C-7), 131.2 (C-2’), 131.6 (C-2), 132.2 (C-7a), 133.6
(C-3a), 144.8 (C-1’)
The signal corresponding to the carbon–boron bond was not observed in the 13C NMR
spectrum.
19F NMR (471 MHz, CDCl3): δ/ppm = –62.3.
11B NMR (160 MHz, CDCl3): δ/ppm = 32.2 (brs).
IR (ATR): /cm–1 = 2976 (m), 1623 (w), 1542 (w), 1422 (w), 1319 (m), 1106 (s), 857 (s).
HRMS (ESI) for C22H24BF3NO2+ [(M+H)+]: calculated 402.1847
found 402.1843
3.3.3 1-Methyl-7-(4-nitrophenyl)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-
indole (141c)
~

2 Description of Experiments 107
According to GP 1, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (6.3 mg, 9.0 µmol, 10 mol %), 1-
methyl-6,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140, 50 mg, 0.13
mmol, 1.5 equiv), KOH (8.3 mg, 0.15 mmol, 1.7 equiv), and 1-iodo-4-nitrobenzene (22 mg,
0.087 mmol, 1.0 equiv). DME (1.2 mL) was subsequently added followed by water (40 µL).
The reaction mixture was stirred in a preheated oil bath at 45 oC. After 3 h, the reaction
mixture was cooled to room temperature and passed through a plug of silica gel using ethyl
acetate as the eluting solvent. The reaction mixture was evaporated in vacuo, and the
residue was purified by flash column chromatography on silica gel (n-pentane:ethyl acetate
= 99:1) to afford 141c (25 mg, 75% yield) as a yellow solid.
Rf = 0.5 (cyclohexane:ethyl acetate = 90:10).
m.p.: 184 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.08 (s, 12H, pin-CH3), 3.17 (s, 3H, N-CH3), 6.53 (d,
3J3,2 = 3.1 Hz, 1H, H-3), 6.99 (d, 3J2,3 = 3.1 Hz, 1H, H-2), 7.53 (d, 3J5,4 = 7.9 Hz, 1H, H-5),
7.57 (d, 3J2’,3’ = 8.6 Hz, 2H, H-2’), 7.66 (d, 3J4,5 = 7.9 Hz, 1H, H-4), 8.23 (d, 3J3’,2’ = 8.6 Hz,
2H, H-3’).
13C NMR (126 MHz, CDCl3): δ/ppm = 24.5 (pin-CH3), 37.0 (N-CH3), 83.2 (pin-Ctert), 101.2 (C-
3), 120.4 (C-4), 121.8 (2C, C-3’), 125.4 (C-5), 130.1 (C-7), 131.7 (2C, C-2’), 131.8 (C-2),
132.5 (C-7a), 133.5 (C-3a), 147.1 (C-4’), 148.4 (C-1’).
The signal corresponding to the carbon–boron bond was not observed in the 13C NMR
spectrum.
11B NMR (160 MHz, CDCl3): δ/ppm = 32.2 (brs).
IR (ATR): /cm–1 = 2972 (s), 1628 (w), 1518 (s), 1429 (w) 1344 (m), 1142 (s), 1027 (w), 970
(w), 854 (s).
HRMS (ESI) for C21H24BN2O4+ [(M+H)+]: calculated 379.1824
found 379.1820
~

108 EXPERIMENTAL PART
3.3.4 7-(4-Bromophenyl)-1-methyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-
1H-indole (141d)
According to GP 1, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (6.3 mg, 9.0 µmol, 10 mol %), 1-
methyl-6,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140, 50 mg, 0.13
mmol, 1.5 equiv), KOH (8.3 mg, 0.15 mmol, 1.7 equiv), and 1-bromo-4-iodobenzene (25 mg,
0.087 mmol, 1.0 equiv). DME (1.2 mL) was subsequently added followed by water (40 µL).
The reaction mixture was stirred in a preheated oil bath at 45 oC. After 3 h, the reaction
mixture was cooled to room temperature and passed through a plug of silica gel using ethyl
acetate as the eluting solvent. The reaction mixture was evaporated in vacuo, and the
residue was purified by flash column chromatography on silica gel (n-pentane:ethyl acetate
= 99:1) to afford 141d (26 mg, 71% yield) as a white solid.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
m.p.: 184 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.10 (s, 12H, pin-CH3), 3.20 (s, 3H, N-CH3), 6.49 (d,
3J3,2 = 3.1 Hz, 1H, H-3), 6.95 (d, 3J2,3 = 3.1 Hz, 1H, H-2), 7.24–7.27 (m, 2H, H-2’), 7.44 (d,
3J5,4 = 7.9 Hz, 1H, H-5), 7.43–7.49 (m, 2H, H-3’), 7.60 (d, 3J4,5 = 7.9 Hz, 1H, H-4).
13C NMR (126 MHz, CDCl3): δ/ppm = 24.5 (pin-CH3), 37.0 (N-CH3), 83.1 (pin-Ctert), 100.8 (C-
3), 119.8 (C-4), 121.0 (C-7), 124.9 (C-4’), 129.8 (C-5), 130.9 (2C, C-3’), 131.4 (C-7), 132.1
(C-7a), 132.6 (C-2), 133.8 (C-3a), 139.8 (C-1’).
The signal corresponding to the carbon–boron bond was not observed in the 13C NMR
spectrum.

2 Description of Experiments 109
11B NMR (160 MHz, CDCl3): δ/ppm = 32.2 (brs).
IR (ATR): /cm–1 = 2979 (m), 1518 (s), 1440 (w), 1360 (m), 1145 (s), 1114 (s) 1027 (w),
990 (w), 825 (s).
HRMS (ESI) for C21H24BBrNO2+ [(M+H)+]: calculated 412.1078
found 412.1074
3.3.5 7-(4-Bromophenyl)-1-methyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-
1H-indole (141e)
According to GP 1, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (6.3 mg, 9.0 µmol, 10 mol %), 1-
methyl-6,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140, 50 mg, 0.13
mmol, 1.5 equiv), KOH (8.3 mg, 0.15 mmol, 1.7 equiv), and 2-iodothiophene (18 mg, 9.6 µL,
0.087 mmol, 1.0 equiv). DME (1.2 mL) was subsequently added followed by water (40 µL).
The reaction mixture was stirred in a preheated oil bath at 45 oC. After 3 h, the reaction
mixture was cooled to room temperature and passed through a plug of silica gel using ethyl
acetate as the eluting solvent. The reaction mixture was evaporated in vacuo, and the
residue was purified by flash column chromatography on silica gel (n-pentane:ethyl acetate
= 99:1) to afford 141e (15 mg, 58% yield) as a viscous oil.
Rf = 0.8 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.07 (s, 12H, pin-CH3), 3.27 (s, 3H, N-CH3), 6.41 (d,
3J3,2 = 3.1 Hz, 1H, H-3), 6.90 (d, 3J2,3 = 3.1 Hz, 1H, H-2) 6.95–6.97 (m, 2H, H-3’, H-5’), 7.26–
7.28 (m, 1H, H-4’), 7.33 (d, 3J5,4 = 7.9 Hz, 1H, H-5), 7.55 (d, 3J4,5 = 7.9 Hz, 1H, H-4).
~

110 EXPERIMENTAL PART
13C NMR (126 MHz, CDCl3): δ/ppm = 24.5 (pin-CH3), 35.8 (N-CH3), 83.1 (pin-Ctert), 100.8 (C-
3), 120.7 (C-4), 123.7 (C-7), 124.5 (C-5), 125.1 (C-4’), 125.8 (C-3’), 129.2 (C-5’), 131.2 (C-
7a), 132.0 (C-2), 135.0 (C-3a), 140.9 (C-1’).
The signal corresponding to the carbon–boron bond was not observed in the 13C NMR
spectrum.
11B NMR (160 MHz, CDCl3): δ/ppm = 34.3 (brs).
IR (ATR): /cm–1 = 2990 (m), 1529 (w), 1421 (w), 1392 (m), 1167 (s), 1120 (s) 1070 (w),
995 (w), 835 (s), 755 (s).
HRMS (ESI) for C19H23BNO2S+ [(M+H)+]: calculated 340.1537
found 340.1532
3.3.6 (E)-1-Methyl-7-styryl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole
(172)
According to GP 1, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (6.3 mg, 9.0 µmol, 10 mol %), 1-
methyl-6,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (140, 50 mg, 0.13
mmol, 1.5 equiv), KOH (8.3 mg, 0.15 mmol, 1.7 equiv), and E-(2-iodo-vinyl)benzene (20 mg,
0.087 mmol, 1.0 equiv). DME (1.2 mL) was subsequently added followed by water (40 µL).
The reaction mixture was stirred in a preheated oil bath at 45 oC. After 3 h, the reaction
mixture was cooled to room temperature and passed through a plug of silica gel using ethyl
acetate as the eluting solvent. The reaction mixture was evaporated in vacuo, and the
~

2 Description of Experiments 111
residue was purified by flash column chromatography on silica gel (n-pentane:ethyl acetate
= 99:1) to afford 172 (22 mg, 70% yield) as a yellow viscous oil.
Rf: 0.6 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.27 (s, 12H, pin-CH3), 3.91 (s, 3H, N-CH3), 6.47 (d,
3J3,2 = 3.1 Hz, 1H, H-3), 6.56 (d, 3J1’,2’ = 16 Hz, 1H, H-1’), 7.01 (d, 3J2,3 = 3.1 Hz, 1H, H-2),
7.29 (d, 3J5,4 = 8.3 Hz, 1H, H-5), 7.36–7.39 (m, 2H, H-3’’)*, 7.45 (d, 3J4,5 = 8.3 Hz, 1H, H-4),
7.52–7.55 (m, 3H, H-2’’, H-4’’)*, 7.96 (d, 3J2’,1’ = 16 Hz, 1H, H-2’).
13C NMR (126 MHz, CDCl3): δ/ppm = 24.8 (pin-CH3), 37.7 (N-CH3), 83.3 (pin-Ctert), 101.1 (C-
3), 119.6 (C-4), 125.7 (C-5), 126.5 (2C, C-2’’), 127.4 (C-4’’), 128.2 (C-2’), 128.6 (C-3’’), 129.8
(C-7), 131.7 (C-7a), 132.0 (C-2), 134.5 (C-3a), 134.6 (C-1’), 137.7 (C-1’’).
The signal corresponding to the carbon–boron bond was not observed in the 13C NMR
spectrum.
11B NMR (160 MHz, CDCl3): δ/ppm = 32.5 (brs).
IR (ATR): /cm–1 = 2915 (m), 1590 (s), 1353 (s), 1259 (m), 1104 (m), 1018 (s), 800 (s).
HRMS (ESI) for C23H27BNO2+ [(M+H)+]: calculated 360.2129
found 360.2128
~

112 EXPERIMENTAL PART
3.4 Synthesis of the 6,7-Bisaryl-Substituted Indoles
3.4.1 1-Methyl-6,7-diphenyl-1H-indole (142aa)
According to GP 2, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (3.3 mg, 4.5 µmol, 10 mol %), 1-
methyl-7-phenyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (141a, 15 mg, 45
µmol, 1.0 equiv), KOH (5.1 mg, 90 µmol, 2.0 equiv), and iodobenzene (37 mg, 20 µL,
0.18 mmol, 4.0 equiv). DME (0.4 mL) was subsequently added followed by water (17 µL).
The SCHLENK tube was sealed and the reaction mixture was stirred in a preheated oil bath at
90 oC. After 16 h, the reaction mixture was cooled to room temperature and passed through
a plug of silica using ethyl acetate as the eluting solvent. The reaction mixture was
evaporated in vacuo, and the residue was purified by flash column chromatography on silica
gel (n-pentane:ethyl acetate = 99:1) to afford 142aa (10 mg, 80% yield) as a white solid.
Rf = 0.8 (cyclohexane:ethyl acetate = 90:10).
m.p.: 143 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 3.16 (s, 3H, N-CH3), 6.55 (d, 3J3,2 = 3.1 Hz, 1H, H-3),
6.97 (d, 3J2,3 = 3.1 Hz, 1H, H-2), 7.09–7.13 (m, 5H, Ar-H), 7.15 (d, 3J5,4 = 8.1 Hz, 1H, H-5),
7.21–7.25 (m, 5H, Ar-H), 7.66 (d, 3J4,5 = 8.1 Hz, 1H, H-4).
13C NMR (126 MHz, CDCl3): δ/ppm = 36.8 (N-CH3), 100.7 (C-3), 119.9 (C-4), 122.2 (C-5),
124.8 (C-7), 125.6 (Ar-C), 126.9 (Ar-C), 127.16 (Ar-C), 127.20 (Ar-C), 128.9 (C-7a), 130.4
(Ar-C), 131.7 (Ar-C), 131.8 (C-2), 134.4 (C-3a), 135.7 (Ar-C), 138.3 (C-1’’)*, 142.4 (C-1’)*.

2 Description of Experiments 113
IR (ATR): /cm–1 = 1597 (w), 1561 (w), 1512 (w), 1438 (m), 1408 (m), 1362 (w), 809 (s),
752 (s), 697 (s).
HRMS (ESI) for C21H18N+ [(M+H)+]: calculated 284.1434
found 284.1429
3.4.2 1-Methyl-7-phenyl-6-(4-trifluoromethylphenyl)-1H-indole (142ab)
According to GP 2, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (3.3 mg, 4.5 µmol, 10 mol %), 1-
methyl-7-phenyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (141a, 15 mg, 45
µmol, 1.0 equiv), KOH (5.1 mg, 90 µmol, 2.0 equiv), and 1-iodo-4-trifluoromethylbenzene (49
mg, 26 µL, 0.18 mmol, 4.0 equiv). DME (0.4 mL) was subsequently added followed by water
(17 µL). The SCHLENK tube was sealed and the reaction mixture was stirred in a preheated
oil bath at 90 oC. After 16 h, the reaction mixture was cooled to room temperature and
passed through a plug of silica using ethyl acetate as the eluting solvent. The reaction
mixture was evaporated in vacuo, and the residue was purified by flash column
chromatography on silica gel (n-pentane:ethyl acetate = 99:1) to afford 142ab (15 mg, 95%
yield) as a white solid.
Rf = 0.8 (cyclohexane:ethyl acetate = 90:10).
m.p.: 118 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 3.17 (s, 3H, N-CH3), 6.57 (d, 3J3,2 = 3.1 Hz, 1H, H-3),
7.00 (d, 3J2,3 = 3.1 Hz, 1H, H-2), 7.11 (d, 3J5,4 = 8.1 Hz, 1H, H-5), 7.22–7.27 (m, 7H, Ar-H),
7.37–7.38 (m, 2H, H-3’’), 7.68 (d, 3J4,5 = 8.0 Hz, 1H, H-4).
~

114 EXPERIMENTAL PART
13C NMR (126 MHz, CDCl3): δ/ppm = 36.8 (N-CH3), 100.8 (C-3), 120.2 (C-4), 121.7 (C-5),
124.2 (q, 3JC-3’’,F = 3.8 Hz, C-3’’), 124.4 (q, 1JC-5’’,F = 272 Hz, C-5’’), 124.8 (C-7), 127.3 (C-4’),
127.4 (C-3’)*, 127.7 (q, 2JC-4’’,F = 32 Hz, H-4’’), 129.4 (C-7a), 130.6 (C-2’)*, 131.7 (C-2’’)*,
132.2 (C-2), 134.2 (C-6), 134.3 (C-3a), 137.8 (C-1’), 146.3 (C-1’’).
19F NMR (471 MHz, CDCl3): δ/ppm = –62.3.
IR (ATR): /cm–1 = 1612 (s), 1572 (m), 1439 (m), 1406 (m), 1319 (s), 1159 (s), 1100 (s),
811 (s), 700 (s), 663 (s).
HRMS (ESI) for C22H17F3N+ [(M+H)+]: calculated 352.1308
found 352.1304
3.4.3 1-Methyl-6-(4-nitrophenyl)-7-phenyl-1H-indole (142ac)
According to GP 2, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (3.3 mg, 4.5 µmol, 10 mol %), 1-
methyl-7-phenyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (141a, 15 mg, 45
µmol, 1.0 equiv), KOH (5.1 mg, 90 µmol, 2.0 equiv), and 1-iodo-4-nitrobenzene (45 mg, 0.18
mmol, 4.0 equiv). DME (0.4 mL) was subsequently added followed by water (17 µL). The
SCHLENK tube was sealed and the reaction mixture was stirred in a preheated oil bath at 90
oC. After 16 h, the reaction mixture was cooled to room temperature and passed through a
plug of silica using ethyl acetate as the eluting solvent. The reaction mixture was evaporated
in vacuo, and the residue was purified by flash column chromatography on silica gel (n-
pentane:ethyl acetate = 99:1) to afford 142ac (8.6 mg, 58% yield) as a yellow solid.
~

2 Description of Experiments 115
Rf = 0.6 (cyclohexane:ethyl acetate = 90:10).
m.p.: 189 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 3.17 (s, 3H, N-CH3), 6.56 (d, 3J3,2 = 3.1 Hz, 1H, H-3),
7.00 (d, 3J2,3 = 3.1 Hz, 1H, H-2), 7.14 (d, 3J5,4 = 8.0 Hz, 1H, H-5), 7.23–7.26 (m, 7H, Ar-H),
7.69 (d, 3J4,5 = 8.0 Hz, 1H, H-4), 7.96–7.97 (m, 2H, Ar-H).
13C NMR (126 MHz, CDCl3): δ/ppm = 36.9 (N-CH3), 100.9 (C-3), 120.4 (C-4), 121.4 (C-5),
122.6 (C-3’’), 124.8 (C-7), 127.5 (C-4’), 127.6 (2C, C-3’), 129.8 (C-7a), 131.1 (C-2’’), 131.7
(C-2’), 132.5 (Ar-C), 133.3 (C-6), 134.2 (C-3a), 137.4 (C-1’), 145.8 (C-1’’), 149.8 (C-4’’).
IR (ATR): /cm–1 = 3049 (m), 2917 (m), 1672 (w), 1597 (w), 1458 (m), 1405 (m), 1305 (m),
1090 (m), 1022 (m), 772 (s), 749 (s), 697 (s), 660 (s).
HRMS (ESI) for C21H17N2O2+ [(M+H)+]: calculated 329.1285
found 329.1280
3.4.4 6-(4-Bromophenyl)-1-methyl-7-phenyl-1H-indole (142ad)
According to GP 2, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (3.3 mg, 4.5 µmol, 10 mol %), 1-
methyl-7-phenyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (141a, 15 mg, 45
µmol, 1.0 equiv), KOH (5.1 mg, 90 µmol, 2.0 equiv), and 1-bromo-4-iodobenzene (51 mg,
0.18 mmol, 4.0 equiv). DME (0.4 mL) was subsequently added followed by water (17 µL).
The SCHLENK tube was sealed and the reaction mixture was stirred in a preheated oil bath at
90 oC. After 16 h, the reaction mixture was cooled to room temperature and passed through
~

116 EXPERIMENTAL PART
a plug of silica using ethyl acetate as the eluting solvent. The reaction mixture was
evaporated in vacuo, and the residue was purified by flash column chromatography on silica
gel (n-pentane:ethyl acetate = 99:1) to afford 142ad (13 mg, 77% yield) as a white solid.
Rf = 0.6 (cyclohexane:ethyl acetate = 90:10).
m.p.: 135 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 3.15 (s, 3H, N-CH3), 6.54 (d, 3J3,2 = 3.1 Hz, 1H, H-3),
6.97 (d, 3J2,3 = 3.1 Hz, 1H, H-2), 6.97-6.99 (m, 2H, Ar-H), 7.09 (d, 3J5,4 = 8.0 Hz, 1H, H-5),
7.22–7.26 (m, 7H, Ar-H), 7.65 (d, 3J4,5 = 8.0 Hz, 1H, H-4).
13C NMR (126 MHz, CDCl3): δ/ppm = 36.8 (N-CH3), 100.7 (C-3), 119.9 (C-7), 121.0 (C-4),
121.8 (C-5), 124.7 (C-6), 125.7 (C-4’’), 127.1 (C-4’), , 127.4 (2C, C-3’), 129.1 (C-7a), 130.4
(2C, C-2’), 131.7 (2C, C-3’’), 131.9 (C-2), 132.0 (2C, C-2’’), 134.3 (C-3a), 138.0 (C-1’’), 141.4
(C-1’).
IR (ATR): /cm–1 = 1559 (w), 1441 (w), 1406 (w), 1308 (m), 1258 (m), 1070 (m), 1008 (s),
933 (w), 805 (s), 702 (s).
HRMS (ESI) for C21H17BrN+ [(M+H)+]: calculated 362.0539
found 362.0534
3.4.5 6-(2-Isopropylphenyl)-1-methyl-7-phenyl-1H-indole (142ae)
According to GP 2, a flame-dried SCHLENK tube was charged with [1,1′-bis-
(diphenylphosphino)-ferrocene]palladium(II) dichloride (3.3 mg, 4.5 µmol, 10 mol %), 1-
~

2 Description of Experiments 117
methyl-7-phenyl-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole (141a, 15 mg, 45
µmol, 1.0 equiv), KOH (5.1 mg, 90 µmol, 2.0 equiv), and 1-iodo-2-isopropylbenzene (44 mg,
28 µL, 0.18 mmol, 4.0 equiv). DME (0.4 mL) was subsequently added followed by water (17
µL). The SCHLENK tube was sealed and the reaction mixture was stirred in a preheated oil
bath at 90 oC. After 16 h, the reaction mixture was cooled to room temperature and passed
through a plug of silica using ethyl acetate as the eluting solvent. The reaction mixture was
evaporated in vacuo, and the residue was purified by flash column chromatography on silica
gel (n-pentane:ethyl acetate = 99:1) to afford 142ae (12 mg, 80% yield) as a white solid.
Rf = 0.9 (90:10 cyclohexane/ethyl acetate).
m.p.: 101 oC (CDCl3).
1H NMR (500 MHz, CDCl3): δ/ppm = 0.97 (d, 3J H-2’’’,1’’’ = 7.0, 3H, H-2’’’)*, 1.06 (d, 3J H-3’’’,1’’’ =
7.0, 3H, H-3’’’)*, 2.82 (sept, 3J1’’’, iPr-CH3 = 7.0, 1H, H-1’’’), 3.15 (s, 3H, N-CH3), 6.56 (d, 3J3,2 =
3.1 Hz, 1H, H-3), 6.94–6.96 (m, 1H, H-4’’), 6.97 (d, 3J2,3 = 3.1 Hz, 1H, H-2), 6.99 (d, 3J5,4 =
8.0 Hz, 1H, H-5), 7.02–7.04 (m, 1H, Ar-H), 7.11–7.13 (m, 2H, Ar-H), 7.15–7.18 (m, 4H, Ar-
H), 7.23–7.25 (m, 1H) 7.62 (d, 3J4,5 = 8.0 Hz, 1H, H-4).
13C NMR (126 MHz, CDCl3): δ/ppm = 22.7 (C-2’’’)*, 25.4 (C-3’’’)*, 29.9 (C-1’’’), 36.6 (N- CH3),
100.7 (C-3), 119.6 (C-4), 122.1 (C-5), 124.0 (C-4’’), 124.7 (C-3’’), 125.01 (C-7), 126.8 (Ar-C),
126.87 (Ar-C), 126.90 (Ar-C), 127.0 (Ar-C), 128.6 (C-7a), 130.8 (Ar-C), 131.3 (C-2), 131.7
(Ar-C), 134.4 (C-3a), 135.3 (C-6), 138.1 (C-1’), 140.4 (C-1’’), 146.9 (C-2’’).
IR (ATR): /cm–1 = 1595 (m), 1513 (s), 1462 (w), 1344 (s), 1107 (w), 847 (m), 814 (w), 753
(w), 705 (m).
HRMS (ESI) for C24H24N+ [(M+H)+]: calculated 326.1903
found 326.1899
~

118 EXPERIMENTAL PART
3.5 Synthesis of (Z)-α,β-Unsaturated Esters
3.5.1 Ethyl (Z)-3-(naphthalen-2-yl)acrylate ((Z)-208a)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and 2-
naphthaldehyde (0.10 g, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min. The
reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208a (90 mg, 60% yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.24 (t, 3J1,2 = 7.1 Hz, 3H, H-1), 4.19 (q, 3J2,1 = 7.1 Hz,
2H, H-2), 6.02 (d, 3J4,5 = 12.6 Hz, 1H, H-4), 7.10 (d, 3J5,4 = 12.6 Hz, 1H, H-5), 7.45–7.51 (m,
2H, Ar-H), 7.71–7.74 (m, 1H, Ar-H), 7.79–7.85 (m, 3H, Ar-H), 8.03−8.05 (m, 1H, Ar-H)
13C NMR (126 MHz, CDCl3): δ/ppm = 14.2 (C-1), 60. 5 (C-2), 120.1 (C-4), 126.3 (C-14)*,
126.8 (C-15)*, 127.1 (Ar-C), 127.5 (C-7), 127.7 (Ar-C), 128.6 (Ar-C), 129.9 (Ar-C), 132.5 (C-
6)**, 133.0 (C-8)**, 133.5 (C-13), 143.1 (C-5), 166.5 (C-3).

2 Description of Experiments 119
IR (ATR): /cm–1 = 3053 (w), 2978 (w), 1709 (s), 1619 (m), 1440 (w), 1172 (s), 1027 (m),
860 (m), 746 (m).
HRMS (ESI) for C15H15O2+ [(M+H)+]: calculated 227.1067
found 227.1061
3.5.2 Ethyl (Z)-3-(4-methoxyphenyl)acrylate ((Z)-208b)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and p-
anisaldehyde (90 mg, 80 µL, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min. The
reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208b (60 mg, 50% yield) as colorless oil.
Rf = 0.6 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.27 (t, 3J1,2 = 7.2 Hz, 3H, H-1), 3.82 (s, 3H, O-CH3),
4.18 (q, 3J2,1 = 7.2 Hz, 2H, H-2), 5.82 (d, 3J4,5 = 12.7 Hz, 1H, H-4), 6.82 (d, 3J4,5 = 12.7 Hz,
1H, H-5), 6.83–6.86 (m, 2H, H-8), 7.67–7.69 (m, 2H, H-7).
~

120 EXPERIMENTAL PART
13C NMR (126 MHz, CDCl3): δ/ppm = 14.4 (C-1), 31.1 (O-CH3), 60.3 (C-2), 113.6 (C-8),
117.5 (C-4), 127.5 (C-6), 132.3 (C-7), 143.3 (C-5), 160.5 (C-9), 166.6 (C-3).
IR (ATR): /cm–1 = 2977 (w), 1711 (s), 1599 (s), 1509 (s), 1458 (m), 1254 (s), 1152 (s),
1026 (m), 843 (m).
HRMS (ESI) for C12H15O3+ [(M+H)+]: calculated 207.1016
found 207.1011
3.5.3 Ethyl (Z)-3-(4-allyloxyphenyl)acrylate ((Z)-208c)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and 24-
allyloxybenzaldehyde (0.11 g, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min. The
reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvent was removed in vacuo. The residue was purified by flash column
chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to afford
(Z)-208c (80 mg, 51% yield) as colorless oil.
Rf = 0.5 (cyclohexaneethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.27 (t, 3J1,2 = 7.3 Hz, 3H, H-1), 4.19 (q, 3J2,1 = 7.3 Hz,
2H, H-2), 4.19 (dt, 3J10,11 = 5.3 Hz, 4J10,12 = 1.5 Hz, 2H, H-10), 5.30 (dq, 3J12b,11 = 10.6 Hz,
~

2 Description of Experiments 121
4J12b,10 = 1.4 Hz, 1H, H-12b), 5.41 (dq, 3J12a,11 = 17.3 Hz, 4J12a,10 = 1.4 Hz, 1H, H-12a), 5.82
(d, 3J4,5 = 12.9 Hz, 1H, H-4), 6.00–6.09 (m, 1H, H-11), 6.84 (d, 3J5,4 = 12.9 Hz, 1H, H-5),
6.87–6.90 (m, 2H, H-8), 7.66–7.70 (m, 2H, H-7)
13C NMR (126 MHz, CDCl3): δ/ppm = 14.3 (C-1), 60.3 (C-2), 68.9 (C-10), 114.3 (C-8), 117.4
(C-4), 118.1 (C-12), 127.6 (C-6), 132.3 (C-7), 133.1 (C-11), 143.3 (C-5), 159.5 (C-9), 166.6
(C-3).
IR (ATR): /cm–1 = 2979 (w), 1711 (s), 1598 (s), 1507 (s), 1457 (w), 1250 (m), 1151 (s),
1019 (s), 925 (m), 840 (m).
HRMS (ESI) for C14H15O3+ [(M+H)+]: calculated 231.1016
found 231.1005
3.5.4 Ethyl (Z)-3-(3-methoxyphenyl)acrylate ((Z)-208d)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and m-
anisaldehyde (90 mg, 80 µL, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min. The
reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208d (90 mg, 65% yield) as colorless oil.
~

122 EXPERIMENTAL PART
Rf = 0.6 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.25 (t, 3J1,2 = 7.2 Hz, 3H, H-1), 3.82 (s, 3H, OCH3),
4.17 (q, 3J2,1 = 7.2 Hz, 2H, H-2), 5.94 (d, 3J4,5 = 12.7 Hz, 1H, H-4), 6.87–6.91 (m, 2H, H-5, H-
7), 7.11–7.12 (m, 1H, H-9), 7.24–7.27 (m, 2H, Ar-H).
13C NMR (126 MHz, CDCl3): δ/ppm = 14.3 (C-1), 55.4 (OCH3), 60.5 (C-2), 114.9 (C-10),
115.1 (C-7), 120.3 (C-4), 122.5 (C-9), 129.1 (C-11), 136.4 (C-6), 142.7 (C-5), 159.4 (C-8),
166.3 (C-3).
IR (ATR): /cm–1 = 2979 (w), 1714 (s), 1626 (m), 1577 (s), 1458 (m), 1258 (m), 1157 (s),
1027 (s), 955 (w), 783 (m).
HRMS (ESI) for C12H15O3+ [(M+H)+]: calculated 207.1016
found 207.1010
3.5.5 Ethyl (Z)-3-(2-fluorophenyl)acrylate ((Z)-208e)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and p-
anisaldehyde (90 mg, 80 µL, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min. The
reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
~

2 Description of Experiments 123
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208e (0.12 g, 93% yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = = 1.21 (t, 3J1,2 = 7.2 Hz, 3H, H-1), 4.16 (q, 3J2,1 = 7.2 Hz,
2H, H-2), 6.07 (d, 3J4,5 = 12.4 Hz, 1H, H-4), 7.02–7.07 (m, 2H, H-5, H-11), 7.10–7.13 (mc,
1H, H-9), 7.28–7.33 (m, 1H, H-8), 7-57–7.60 (mc, 1H, H-10).
13C NMR (126 MHz, CDCl3): δ/ppm = 14.2 (C-1), 60.5 (C-2), 115.3 (d, 2J8,F = 22.1 Hz, C-8),
122.5 (C-4), 123.3 (d, 2J6,F = 13.3 Hz, C-6), 123.6 (d, 3J9,F = 3.5 Hz, C-9), 130.7 (d, 3J11,F =
8.5 Hz, C-11), 130.0 (d, 4J10,F = 2.5 Hz, C-10), 135.5 (d, 3J5,F = 3.5 Hz, C-5), 160.3 (d, 1J7,F =
250 Hz, C-7), 165.9 (C-3).
IR (ATR): /cm–1 = 2981 (w), 1717 (s), 1636 (m), 1575 (w), 1453 (m), 1408 (w), 1160 (s),
1096 (m), 1027 (m), 754 (s).
HRMS (ESI) forC11H12FO2+ [(M+H)+]: calculated 195.0816
found 195.0807
3.5.6 Ethyl (Z)-3-(2-bromophenyl)acrylate ((Z)-208f)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
~

124 EXPERIMENTAL PART
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and 2-
bromobenzaldehyde (0.10 g, 70 µL, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min.
The reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208f (0.14 g, 81% yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = = 1.17 (t, 3J1,2 = 7.1 Hz, 3H, H-1), 4.10 (q, 3J2,1 = 7.1 Hz,
2H, H-2), 6.05 (d, 3J4,5 = 12.2 Hz, 1H, H-4), 7.08 (d, 3J5,4 = 12.2 Hz, 1H, H-5), 7.16–7.19 (m,
1H, H-9), 7.25–7.29 (m, 1H, H-10), 7.46–7.47 (m, 1H, H-8), 7.56–7.58 (m, 1H, H-11).
13C NMR (126 MHz, CDCl3): δ/ppm = 14.1 (C-1), 60.5 (C-2), 121.9 (C-4), 123.2 (C-7), 126.8
(C-8), 129.9 (C-10), 130.9 (C-9), 132.4 (C-11), 136.1 (C-6), 142.6 (C-5), 165.7 (C-3).
IR (ATR): /cm–1 = 2979 (w), 1717 (s), 1635 (w), 1465 (w), 1268 (w), 1157 (s), 1023 (s),
949 (w), 755 (m).
HRMS (ESI) for C11H12BrO2+ [(M+H)+]: calculated 256.9995
found 256.9927
3.5.10 Ethyl (Z)-3-(4-bromophenyl)acrylate ((Z)-208g)
~

2 Description of Experiments 125
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and 4-
bromobanzaldehyde (0.10 g, 70 µL, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min.
The reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208g (0.12 g, 70% yield) as colorless oil.
Rf = 0.7 (90:10 cyclohexane/ethyl acetate).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.26 (t, 3J1,2 = 7.2 Hz, 3H, H-1), 4.17 (q, 3J2,1 = 7.2 Hz,
2H, H-2), 5.97 (d, 3J4,5 = 12.6 Hz, 1H, H-4), 6.85 (d, 3J5,4 = 12.9 Hz, 1H, H-5), 7.46–7.49 (m,
4H, Ar-H).
13C NMR (126 MHz, CDCl3): δ/ppm = 14.3 (C-1), 60.6 (C-2), 120.7 (C-4), 123.4 (C-9), 131.3
(C-8), 131.5 (C-7), 133.9 (C-6), 142.0 (C-5), 166.1 (C-3).
IR (ATR): /cm–1 = 2978 (w), 1714 (s), 1628 (m), 1584 (w), 1388 (w), 1157 (s), 1070 (m),
947 (w), 845 (m).
HRMS (ESI) for C11H12BrO2+ [(M+H)+]: calculated 256.0015
found 256.0006
3.5.7 Ethyl (Z)-3-(4-nitrophenyl)acrylate ((Z)-208h)
~

126 EXPERIMENTAL PART
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and subsequently
a solution of KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and 4-
nitrobenzaldehyde (0.10 g, 65 µL, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min.
The reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208h (90 mg, 60% yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CDCl3): δ/ppm = 1.24 (t, 3J1,2 = 7.3 Hz, 3H, H-1), 4.17 (q, 3J2,1 = 7.3 Hz,
2H, H-2), 6.12 (d, 3J4,5 = 12.6 Hz, 1H, H-4), 7.00 (d, 3J5,4 = 12.5 Hz, 1H, H-5), 7.65–7.68 (m,
2H, H-7), 8.19–8.21 (m, 2H, H-8).
13C NMR (126 MHz, CDCl3): δ/ppm = 14.2 (C-1), 60.9 (C-2), 123.3 (C-8), 123.4 (C-4), 130.3
(C-7), 140.7 (C-5), 141.6 (C-6), 147.7 (C-9), 165.4 (C-3).
IR (ATR): /cm–1 = 3038 (w), 2981 (w) 1702 (s), 1514 (s), 1342 (s), 1271 (s), 1213 (m),
1025 (m), 872 (m), 700 (m).
HRMS (ESI) for C11H12NO4+ [(M+H)+]: calculated 222.0761
found 222.0759
~

2 Description of Experiments 127

128 EXPERIMENTAL PART
3.5.9 Ethyl (Z)-3-(4-diethylaminophenyl)acrylate ((Z)-208i)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and 4-
diethylaminobenzaldehyde (0.12 g, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min.
The reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208i (0.11 g, 66% yield) as yellow oil.
Rf = 0.6 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 1.16 (t, 3J10,11 = 7.1 Hz, 6H, H-10), 1.27 (t, 3J1,2 = 7.2
Hz, 3H, H-1), 3.39 (q, 3J11,10 = 7.1 Hz, 2H, H-11), 4.15 (q, 3J2,1 = 7.2 Hz, 2H, H-2), 5.59 (d,
3J4,5 = 12.8 Hz, 1H, H-4), 6.59–6.61 (m, 2H, H-8), 6.70 (d, 3J4,5 = 12.8 Hz, 1H, H-5), 7.71–
7.73 (m, 2H, H-7).
13C NMR (126 MHz, CD2Cl2): δ/ppm = 12.7 (C-10), 14.2 (C-1), 44.7 (C-11), 60.1 (C-2), 110.6
(C-8), 113.2 (C-4), 121.9 (C-6), 133.4 (C-7), 144.4 (C-5), 149.0 (C-9), 167.1 (C-3).
IR (ATR): /cm–1 = 2970 (w), 1705 (m), 1581 (s), 1517 (s), 1443 (m), 1351 (m), 1269 (m),
1127 (s), 1030 (m), 827 (m).
~

2 Description of Experiments 129
HRMS (ESI) for C15H22NO2+ [(M+H)+]: calculated 248.1645
found 248.1629
3.5.8 Ethyl (Z)-3-(4-(dimethylamino)phenyl)acrylate ((Z)-208j)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate 207
(0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a solution of
KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added dropwise
over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and 4-
dimethylaminobenzaldehyde (99 mg, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min.
The reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208j (0.10 g, 68% yield) as yellow oil.
Rf = 0.6 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 1.27 (t, 3J1,2 = 7.2 Hz, 3H, H-1), 2.99 (s, 6H, N-CH3),
4.15 (q, 3J2,1 = 7.3 Hz, 2H, H-2), 5.64 (d, 3J4,5 = 12.8 Hz, 1H, H-4), 6.64–6.66 (m, 2H, H-8),
6.74 (d, 3J4,5 = 12.6 Hz, 1H, H-5), 7.72–7.76 (m, 2H, H-7).
13C NMR (126 MHz, CD2Cl2): δ/ppm = 14.2 (C-1), 40.3 (N-CH3), 60.9 (C-2), 111.3 (2C, C-8),
114.1 (C-4), 122.9 (C-6), 132.9 (2C, C-7), 144.2 (C-5), 151.6 (C-9), 164.1 (C-3).

130 EXPERIMENTAL PART
IR (ATR): /cm–1 = 2977 (w), 2895 (w), 1704 (s), 1583 (s), 1520 (s), 1360 (m), 1126 (s),
1029 (m), 940 (m), 827 (m).
HRMS (ESI) for C13H18NO2+ [(M+H)+]: calculated 220.1332
found 220.1326
3.5.11 Ethyl (Z)-non-2-enoate ((Z)-208k)
According to GP 3, a flame-dried SCHLENK flask was charged with 18-crown-6 (0.40 g, 1.5
mmol, 2.2 equiv). THF (8.0 mL) was added, followed by the addition of phosphonate
207 (0.25 g, 0.75 mmol, 1.1 equiv). The reaction mixture was cooled to –78 oC and a
solution of KHMDS (0.15 g, 0.75 mmol, 1.1 equiv) in THF (1.2 mL) was subsequently added
dropwise over 2 min. The resulting reaction mixture was stirred for 30 min at –78 oC and n-
heptaldehyde (77 mg, 91 µL, 0.67 mmol, 1.0 equiv) was added dropwise over 5 min. The
reaction mixture was then allowed to warm to 0 oC. After 16 h, saturated aqueous NH4Cl
solution (10 mL) was added into the reaction mixture. The biphasic mixture was diluted with
diethyl ether (10 mL) and washed with water (10 mL). The phases were separated and the
aqueous phase was extracted with diethyl ether (3 x 10 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (3.5 x 10 cm, cyclohexane:ethyl acetate = 99:1), to
afford (Z)-208k (50 mg, 40% yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.88–0.91 (m, 3H, C-11), 1.25–1.36 (m, 9H, Alkyl-H),
1.42–1.47 (m, 2H, Alkyl-H), 2.60–2.66 (m, 2H, H-6), 4.11–4.17 (m, 2H, H-2), 5.73– 5.76 (m,
1H, H-4), 6.20–6.26 (m, 1H, H-5).
13C NMR (126 MHz, CD2Cl2): δ/ppm = 14.2 (C-11), 14.7 (C-1), 23.0 (C-6)*, 29.3 (C-7)*,
29.36 (C-8)**, 29.37 (C-9)**, 32.1 (C-10), 60.1 (C-2), 119.9 (C-4), 150.8 (C-5), 160.6 (C-3).
~

2 Description of Experiments 131
IR (ATR): /cm–1 = 2924 (m), 2855 (m), 1719 (s), 1642 (w), 1164 (s), 1034 (m), 820 (m),
724 (w).
HRMS (ESI) for C11H21O2+ [(M+H)+]: calculated 185.1536
found 185.1524
3.6 Synthesis of Silylstannane 98
3.6.1 Dimethyldiphenylstannane (225)
Dimethyldiphenylstannane (225) was prepared according to the procedure by OESTREICH
and co-workers.[61] A flame-dried SCHLENK flask was charged with dichlorodimethylstannane
(224, 5.0 g, 23 mmol, 1.0 equiv) and THF (20 mL). The solution was cooled to 0 oC and
phenylmagnesium bromide (1M in THF, 50 mmol, 2.2 equiv) was added dropwise over 10
min at 0 oC. After the addition, the reaction mixture was allowed to warm to room
temperature and stirred overnight. The solvent was concentrated in vacuum. Cyclohexane
(100 mL) was added, and the mixture was filtered over a pad of silica gel. The filtrate was
collected, and the solvents were evaporated in vacuo. The residue was purified by flash
column chromatography on silica gel (7.5 x 10 cm, cyclohexane). Dimethyldiphenylstannane
(225, 6.2 g, 90% yield) was obtained as colorless oil
Rf = 0.9 (cyclohexane).
1H NMR (500 MHz, CDCl3): δ/ppm = 0.50 (s, 6H, Sn-CH3), 7.33–7.36 (m, 6H, Ar-H), 7.50–
7.52 (m, 4H, Ar-H).
13C NMR (126 MHz, CDCl3): δ/ppm = –9.9 (Sn-CH3), 128.4 (C-3’)*, 128.7 (C-4’), 136.4 (C-
2’)*, 140.8 (C-1’).
~

132 EXPERIMENTAL PART
119Sn NMR (186 MHz, CD2Cl2): δ/ppm = –58.7.
IR (ATR): /cm–1 = 1479 (w), 1426 (m), 1372 (w), 1330 (w), 1239 (w), 1189 (w), 1074 (m),
694 (s).
HRMS (ESI) for C14H17Sn+ [(M+H)+]: calculated 305.0347
found 305.0344
The analytical data are in accordance with those reported.[61]
3.6.2 Chlorodimethylphenylstannane (226)
Chlorodimethyl(phenyl)stannane (226) was prepared according to the procedure by
OESTREICH and co-workers.[61] A flame-dried SCHLENK flask was charged with
dimethyldiphenylstannane (225, 5.5 g, 18 mmol, 1.0 equiv) solution in THF (10 mL). The
reaction mixture was cooled to 0 oC and HCl solution (2M in HCl, 9 mL, 18 mmol, 1.0 equiv)
was added dropwise over 10 min at this temperature. The reaction was allowed to warm to
room temperature and stirred for 16 h. The solvent was evaporated in vacuum to afford the
analytically pure chlorodimethyl(phenyl)stannane (226, 4.5 g, 95% yield) as colorless oil.
Rf = 0.4 (cyclohexane).
1H NMR (500 MHz, CDCl3): δ/ppm = 0.85 (s, 6H, Sn-CH3), 7.41–7.46 (m, 3H, Ar-H), 7.51–
7.65 (m, 2H, Ar-H).
13C NMR (126 MHz, CDCl3): δ/ppm = –2.1 (Sn-CH3), 128.9 (C-3’)*, 130.2 (C-4’), 135.2 (C-
2’)*, 140.6 (C-1’).
119Sn NMR (186 MHz, CD2Cl2): δ/ppm = 99.1.
~

2 Description of Experiments 133
IR (ATR): /cm–1 = 1494 (w), 1450 (m), 1330 (m), 1309 (w), 1250 (m), 1014 (m), 854(m),
687 (s).
HRMS (ESI) for C8H12ClSn+ [(M+H)+]: calculated 262.9644
found 262.9640
The analytical data are in accordance with those reported.[61]
3.6.3 (Dimethylphenylstannyl)trimethylsilane (98)
Chlorodimethyl(phenyl)stannane (98) was prepared according to the procedure by
OESTREICH and co-workers.[61] A flame-dried SCHLENK flask was charged with Li chunks
(0.53 g, 76 mmol, 10 equiv) in THF (10 mL). Chlorotrimethylsilane (0.17 g, 0.19 mL,
1.5 mmol, 20 mol %) was added to the solution, and the reaction mixture was stirred
vigorously at room temperature. After Li surface turned metallic from black, the solvent was
removed with syringe. The Li chunks were washed with THF (3 x 10 mL). THF (20 mL) was
added into the reaction mixture and cooled to 0 oC. Chlorodimethylphenylstannane (226,
2 g, 7.6 mmol, 1.0 equiv) was added dropwise, and the reaction mixture was stirred
vigorously. The reaction mixture started to turn yellow after 10 min which became dark green
over the period of 3 h. The supernatant was transferred using a cannula to a different flame-
dried SCHLENK flask and the reaction mixture was cooled to 0 oC. Chlorotrimethylsilane
(1.0 g, 1.2 mL, 9.1 mmol, 1.2 equiv) was added dropwise and the reaction was stirred at 0
oC for 10 min and subsequently warmed to room temperature. The reaction mixture was
stirred for 16 h and the solvent was removed in vacuum. The residue was purified using
flash column chromatography on silica gel (5 x 10 cm, cyclohexane). The final product
(dimethyl(phenyl)stannyl)trimethylsilane (98, 0.91 g, 40% yield) was obtained as colorless
oil.
Rf = 0.8 (cyclohexane).
~

134 EXPERIMENTAL PART
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.26 (s, 6H, Sn-CH3), 0.29 (s, 9H, Si-CH3), 7.23–7.29
(m, 3H, Ar-H), 7.40–7.50 (m, 2H, Ar-H).
13C NMR (126 MHz, CD2Cl2): δ/ppm = –11.9 (Sn-CH3), 0.8 (Si-CH3), 127.9 (C-4), 128.3 (C-
3)*, 136.9 (C-2)*, 142.6 (C-1).
29Si NMR (100 MHz, CD2Cl2): δ/ppm = –9.9.
119Sn NMR (186 MHz, CD2Cl2): δ/ppm = –138.6.
IR (ATR): /cm–1 = 1426 (w), 1296 (w), 1244 (m), 1072 (w), 833 (s), 720 (s), 694 (s).
HRMS (ESI) for C11H21SiSn+ [(M+H)+]: calculated 301.0429
found 301.0547
The analytical data are in accordance with those reported.[61]
~

2 Description of Experiments 135
3.7 Asymmetric 1,4-Stannylation of the (Z)-α,β-Unsaturated Ester
3.7.1 Ethyl-3-(dimethylphenylstannyl)-3-phenylpropanoate (187)
According to GP 4, a flame-dried SCHLENK tube was charged with ethyl (Z)-3-phenylacrylate
((Z)-179, 18 mg, 0.10 mmol, 1.0 equiv), followed by the phase-transfer catalyst 188 (9.2 mg,
20 µmol, 20 mol %). Toluene (1.0 mL) and 50% (w/v) aqueous solution of KOH in water
(0.20 mL) were added, and the resulting solution was stirred for 5 min. Silylstannane 98 (90
mg, 0.30 mmol, 3.0 equiv) was added dropwise to the stirred solution. The reaction mixture
was stirred vigorously for 16 h at room temperature. The biphasic mixture was diluted with
CH2Cl2 (2 mL) and washed with water (2 mL). The phases were separated, and the aqueous
phase was extracted with diethyl ether (3 x 5 mL). The organic phases were combined, and
the solvents were removed in vacuo. The residue was purified by flash column
chromatography on silica gel (2.5 x 20 cm, cyclohexane:ethyl acetate = 97:3) to afford
187 (30 mg, 75 % yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.19 (s, 3H, Sn-CH3)*, 0.20 (s, 3H, Sn-CH3)*, 1.15 (t,
3J1,2 = 7.2 Hz, 3H, H-1), 2.86–2.91 (m, 1H, H-4a), 2.94–2.99 (m, 1H, H-4b), 3.11 (t, 3J5,4 = 7.8
Hz, H-5), 4.02 (q, 3J2,1 = 7.2, 2H, H-2), 6.98–6.99 (m, 2H, H-2‘), 7.02–7.05 (m, 1H, H-4‘),
7.18–7.21 (mC, 2H, H-3‘), 7.29–7.30 (m, 3H, Ar-H), 7.35–7.37 (m, 2H, Ar-H).
13C NMR (126 MHz, CD2Cl2): δ/ppm = –10.5 (Sn-CH3)*, –10.2 (Sn-CH3)*, 14.4 (C-1), 30.7
(C-4), 37.1 (C-5), 60.9 (C-2), 124.5 (C-3‘), 126.4 (C-4‘), 128.4 (C-3’’), 128.7 (C-4‘‘), 128.8 (C-
2‘), 136.6 (C-2’’), 141.6 (C-1‘‘), 145.6 (C-1‘), 173.9 (C-3).
19Sn NMR (186 MHz, CD2Cl2): δ/ppm = –22.6.

136 EXPERIMENTAL PART
IR (ATR): /cm–1 = 2979 (w), 1723 (s), 1597 (w), 1491 (m), 1372 (m), 1176 (m), 1031 (w),
758 (s), 725 (s), 696 (s).
Opitical rotation: = – 6.0 (c = 1.8, CHCl3, 33% ee). The enantiomeric excess was
determined by HPLC analysis on a chiral stationary phase (Daicel Chiralcel OJ-H column,
column temperature 20 °C, solvent n-heptane:iso-propanol = 99.5:0.5, flow rate 0.70
mL/min, λ = 254 nm): tR/S = 17.1 min for (R/S)-187, tS/R = 28.4 min for (S/R)-187.
HRMS (ESI) for C19H25O2Sn+ [(M+H)+]: calculated 405.0871
found 405.0893
3.7.2 Ethyl-3-(dimethyl(phenyl)stannyl)-3-(3-methoxyphenyl)propanoate (209a)
According to GP 4, a flame-dried SCHLENK tube was charged with ethyl (Z)-3-(naphthalen-2-
yl)acrylate ((Z)-208a, 23 mg, 0.10 mmol, 1.0 equiv), followed by the phase-transfer catalyst
188 (9.2 mg, 20 µmol, 20 mol %). Toluene (1.0 mL) and 50% (w/v) aqueous solution of KOH
in water (0.20 mL) were added, and the resulting solution was stirred for 5 min. Silylstannane
98 (90 mg, 0.30 mmol, 3.0 equiv) was added dropwise to the stirring solution. The reaction
mixture was stirred vigorously for 16 h at room temperature. The biphasic mixture was
diluted with CH2Cl2 (2 mL) and washed with water (2 mL). The phases were separated, and
the aqueous phase was extracted with diethyl ether (3 x 5 mL). The organic phases were
combined, and the solvents were removed in vacuo. The residue was purified by flash
column chromatography on silica gel (2.5 x 20 cm, cyclohexane:ethyl acetate = 97:3) to
afford 209a (30 mg, 68% yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
~

2 Description of Experiments 137
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.22 (s, 3H, Sn-CH3)*, 0.24 (s, 3H, Sn-CH3)*, 1.18 (t,
3J1,2 = 7.2 Hz, 3H, H-1), 3.00–3.05 (m, 1H, H-4a), 3.09–3.14 (m, 1H, H-4b), 3.33 (t, 3J5,4 = 7.9
Hz, H-5), 4.04 (q, 3J2,1 = 7.1 Hz, 2H, H-2), 7.17–7.21 (m, 1H, H-2’), 7.31–7.35 (m, 3H, H-2’’,
H-4’’), 7.37–7.50 (m, 5H, Ar-H, H-2’’), 7.71–7.73 (m, 2H, Ar-H), 7.78–7.79 (m, 1H, H-10’).
13C NMR (126 MHz, CD2Cl2): δ/ppm = –10.5 (Sn-CH3)*, –10.2 (Sn-CH3)*, 13.9 (C-1), 30.6
(C-4), 36.6 (C-5), 60.4 (C-2), 122.6 (C-9’)**, 124.4 (C-10’)**, 125.8 (C-2’), 126.2 (C-2’’),
127.0 (C-4’’), 127.4 (C-4’)***, 127.7 (C-5’)***, 127.9 (C-6’)***, 128.3 (C-7’)***, 131.1 (C-3’’),
133.8 (C-3’), 136.2 (C-8’), 141.0 (C-1’’), 142.9 (C-1’), 173.4 (C-3).
19Sn NMR (186 MHz, CD2Cl2): δ/ppm = –21.6
IR (ATR): /cm–1 = 2976 (w), 1790 (s), 1580 (m), 1547 (w), 1468 (w), 1421 (w), 1393 (w),
1272(m), 1174 (m), 678 (s).
Opitical rotation: = – 0.8.0 (c = 1.1, CHCl3, 11% ee). The enantiomeric excess was
determined by HPLC analysis on a chiral stationary phase (Daicel Chiralcel OD-H column,
column temperature 20 °C, solvent n-heptane:iso-propanol = 98:2, flow rate 0.70 mL/min, λ
= 254 nm): tR/S = 17.1 min for (R/S)-209a, tS/R = 26.7 min for (S/R)-209a.
HRMS (ESI) for C23H27O2Sn+ [(M+H)+]: calculated 455.1028
Found 455.1037
3.7.3 Ethyl-3-(dimethyl(phenyl)stannyl)-3-(4-methoxyphenyl)propanoate
(209b)
According to GP 4, a flame-dried SCHLENK tube was charged with ethyl (Z)-3-(4-
methoxyphenyl)acrylate ((Z)-208b, 21 mg, 0.10 mmol, 1.0 equiv), followed by the phase-
~

138 EXPERIMENTAL PART
transfer catalyst 188 (9.2 mg, 20 µmol, 20 mol %). Toluene (1.0 mL) and 50% (w/v) aqueous
solution of KOH in water (0.20 mL) were added, and the resulting solution was stirred for 5
min. Silylstannane 98 (90 mg, 0.30 mmol, 3.0 equiv) was added dropwise to the stirring
solution. The reaction mixture was stirred vigorously for 16 h at room temperature. The
biphasic mixture was diluted with CH2Cl2 (2 mL) and washed with water (2 mL). The phases
were separated, and the aqueous phase was extracted with diethyl ether (3 x 5 mL). The
organic phases were combined, and the solvents were removed in vacuo. the residue was
purified by flash column chromatography on silica gel (2.5 x 20 cm, cyclohexane:ethyl
acetate = 97:3) to afford 209b (31 mg, 70% yield) as colorless oil.
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.18 (s, 3H, Sn-CH3)*, 0.19 (s, 3H, Sn-CH3)*, 1.14 (t,
3J1,2 = 7.3 Hz, 3H, H-1), 2.82–2.87 (m, 1H, H-4a), 2.89–2.94 (m, 1H, H-4b), 3.05 (t, 3J5,4 = 8.0
Hz, H-5), 3.74 (s, 3H, OCH3), 4.00 (q, 3J2,1 = 7.2 Hz, 2H, H-2), 6.74–6.77 (m, 2H, H-2‘), 6.89–
6.92 (m, 2H, H-3‘), 7.30–7.31 (m, 3H, Ar-H), 7.35–7.37 (m, 2H, Ar-H).
13C NMR (126 MHz, CD2Cl2): δ/ppm = –10.6 (Sn-CH3)*, –10.2 (Sn-CH3)*, 14.3 (C-1), 29.7
(C-4), 37.6 (C-5), 55.5 (O-CH3), 60.8 (C-2), 114.2 (C-3’), 127.4 (C-2’), 128.3 (C-3’’), 128.7
(C-4‘‘), 136.6 (C-2’’), 137.4 (C-1‘), 141.7(C-1‘‘), 157.2 (C-4‘), 173.9 (C-3).
19Sn NMR (186 MHz, CD2Cl2): δ/ppm = –24.6
IR (ATR): /cm–1 = 2980 (w), 1722 (s), 1604 (w), 1506 (s), 1461 (w), 1243 (s), 1176 (s),
1034 (s), 750 (m), 724 (s).
Opitical rotation: = – 7.8 (c = 1.4, CHCl3, 37% ee). The enantiomeric excess was
determined by HPLC analysis on a chiral stationary phase (Daicel Chiralcel OD-H column,
column temperature 20 °C, solvent n-heptane:iso-propanol = 99:1, flow rate 0.70 mL/min, λ
= 254 nm): tR/S = 13.5 min for (R/S)-209b, tS/R = 14.9 min for (S/R)-209b.
HRMS (ESI) for C20H25O3Sn+ [(M+H)+]: calculated 435.0977
found 435.0972
~

2 Description of Experiments 139
3.7.4 Ethyl-3-(dimethylphenylstannyl)-3-(3-methoxyphenyl)propanoate
(209c)
According to GP 4, a flame-dried SCHLENK tube was charged with ethyl (Z)-3-(4-
allyloxyphenyl)acrylate ((Z)-208c, 23 mg, 0.10 mmol, 1.0 equiv), followed by the phase
transfer catalyst 188 (9.2 mg, 20 µmol, 20 mol %). Toluene (1.0 mL) and 50% (w/v) aqueous
solution of KOH in water (0.20 mL) were added, and the resulting solution was stirred for 5
min. Silylstannane 98 (90 mg, 0.30 mmol, 3.0 equiv) was added dropwise to the stirring
solution. The reaction mixture was stirred vigorously for 16 h at room temperature. The
biphasic mixture was diluted with CH2Cl2 (2 mL) and washed with water (2 mL). The phases
were separated, and the aqueous phase was extracted with diethyl ether (3 x 5 mL). The
organic phases were combined, and the solvents were removed in vacuo. The residue was
purified by flash column chromatography on silica gel (2.5 x 20 cm, cyclohexane:ethyl
acetate = 97:3) to afford 209c (24 mg, 55% yield) as colorless oil.
Rf = 0.6 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.21 (s, 3H, Sn-CH3)*, 0.22 (s, 3H, Sn-CH3)*, 1.18 (t,
3J1,2 = 7.0 Hz, 3H, H-1), 2.85–2.90 (m, 1H, H-4a), 2.92–2.97 (m, 1H, H-4b), 3.08 (t, 3J5,4 = 8.0
Hz, H-5), 4.04 (q, 3J2,1 = 7.1 Hz, 2H, H-2), 4.51 (dt, 3J5’,6’ = 5.3 Hz, 4J5’,7’ = 1.5 Hz, 2H, H-5’),
5.28 (dq, 3J7’b,6’ = 10.5 Hz, 4J7’b,5’ = 1.5 Hz, 1H, H-7’b), 5.42 (dq, 3J7’a,6’ = 17.3 Hz, 4J7’a,5’ = 1.5
Hz, 1H, H-7’a), 6.04–6.12 (m, 1H, H-6’), 6.79–6.80 (m, 2H, H-3’)**, 6.91–6.95 (m, 2H, H-
2’)**, 7.31–7.34 (m, 3H, H-2’’, H-4’’)***, 7.37–7.40 (m, 2H, H-2’’)***.
13C NMR (126 MHz, CD2Cl2): δ/ppm = –10.5 (Sn-CH3)*, –10.2 (Sn-CH3)*, 13.9 (C-1), 29.2
(C-4), 37.2 (C-5), 60.4 (C-2), 68.8 (C-5’), 114.7 (C-3’), 116.9 (C-4’’), 126.9 (C-3’’), 127.9 (C-
7’), 128.2 (C-2’’) 133.8 (C-2’), 133.1 (C-6’), 136.2 (C-1’’), 148.3 (C-1’), 155.5 (C-4’), 155.7 (C-
5’), 173.5 (C-3).

140 EXPERIMENTAL PART
19Sn NMR (186 MHz, CD2Cl2): δ/ppm = –24.4
IR (ATR): /cm–1 = 2962 (w), 1750 (s), 1582 (m), 1490 (w), 1428 (w), 1257 (m), 1203 (w),
1154 (m), 667 (s).
Opitical rotation: = – 7.2 (c = 1.5, CHCl3, 42% ee). The enantiomeric excess was
determined by HPLC analysis on a chiral stationary phase (Daicel Chiralcel OD-H column,
column temperature 20 °C, solvent n-heptane:iso-propanol = 99.5:0.5, flow rate 0.70
mL/min, λ = 254 nm): tR/S = 19.0 min for (R/S)-209c, tS/R = 24.7 min for (S/R)-209c.
HRMS (ESI) for C22H29O3Sn+ [(M+H)+]: calculated 461.1133
found 461.1150
3.7.5 Ethyl-3-(dimethylphenylstannyl)-3-(3-methoxyphenyl)propanoate
(209d)
According to GP 4, a flame-dried SCHLENK tube was charged with ethyl (Z)-3-(3-
methoxyphenyl)acrylate ((Z)-208d, 20 mg, 0.10 mmol, 1.0 equiv), followed by the phase
transfer catalyst 188 (9.2 mg, 20 µmol, 20 mol %). Toluene (1.0 mL) and 50% (w/v) aqueous
solution of KOH in water (0.20 mL) were added, and the resulting solution was stirred for 5
min. Silylstannane 98 (90 mg, 0.30 mmol, 3.0 equiv) was added dropwise to the stirring
solution. The reaction mixture was stirred vigorously for 16 h at room temperature. The
biphasic mixture was diluted with CH2Cl2 (2 mL) and washed with water (2 mL). The phases
were separated, and the aqueous phase was extracted with diethyl ether (3 x 5 mL). The
organic phases were combined, and the solvents were removed in vacuo. The residue was
purified by flash column chromatography on silica gel (2.5 x 20 cm, cyclohexane:ethyl
acetate = 97:3) to afford 209d (26 mg, 60% yield) as colorless oil.
~

2 Description of Experiments 141
Rf = 0.7 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.20 (s, 3H, Sn-CH3)*, 0.21 (s, 3H, Sn-CH3)*, 1.16 (t,
3J1,2 = 7.2 Hz, 3H, H-1), 2.84–2.89 (m, 1H, H-4a), 2.93–2.98 (m, 1H, H-4b), 3.09 (t, 3J5,4 = 7.9,
H-5), 3.69 (s, 3H, OCH3), 4.02 (q, 3J2,1 = 7.1, 2H, H-2), 6.48–6.49 (m, 1H, H-2‘), 6.56–6.58
(m, 2H, H-4‘, H-6‘), 7.09–7.11 (m, 1H, H-5‘), 7.29–7.30 (m, 3H, Ar-H), 7.35–7.37 (m, 2H, Ar-
H).
13C NMR (126 MHz, CD2Cl2): δ/ppm = –10.5 (Sn-CH3)*, –10.2 (Sn-CH3)*, 14.4 (C-1), 30.8
(C-4), 36.9 (C-5), 55.3 (O-CH3), 60.9 (C-2), 110.1 (C-4‘), 111.9 (C-2‘), 118.8 (C-6‘), 128.4 (C-
4‘‘), 128.7 (C-3’’), 129.7(C-5‘), 136.6 (C-2’’), 141.6 (C-1‘‘), 147.3 (C-1‘), 160.2 (C-3‘), 173.9
(C-3).
19Sn NMR (186 MHz, CD2Cl2): δ/ppm = –22.0
IR (ATR): /cm–1 = 2980 (w), 1725 (s), 1597 (m), 1484 (w), 1428 (w), 1161 (m), 698 (s).
Opitical rotation: = – 5.0 (c = 1.3, CHCl3, 36% ee). The enantiomeric excess was
determined by HPLC analysis on a chiral stationary phase (Daicel Chiralcel OD-H column,
column temperature 20 °C, solvent n-heptane:iso-propanol = 98:2, flow rate 0.70 mL/min, λ
= 254 nm): tR/S = 21.0 min for (R/S)-209d, tS/R = 22.0 min for (S/R)-209d.
HRMS (ESI) for C20H27O3Sn+ [(M+H)+]: calculated 435.0977
found 435.0991
3.7.6 Ethyl-3-(dimethyl(phenyl)stannyl)-3-(2-fluorophenyl)propanoate (209e)
According to GP 4, a flame-dried SCHLENK tube was charged with ethyl (Z)-3-(2-
fluorophenyl)acrylate ((Z)-208e, 19 mg, 0.10 mmol, 1.0 equiv), followed by the phase
~

142 EXPERIMENTAL PART
transfer catalyst 188 (9.2 mg, 20 µmol, 20 mol %). Toluene (1.0 mL) and 50% (w/v) aqueous
solution of KOH in water (0.20 mL) were added, and the resulting solution was stirred for 5
min. Silylstannane 98 (90 mg, 0.30 mmol, 3.0 equiv) was added dropwise to the stirring
solution. The reaction mixture was stirred vigorously for 16 h at room temperature. The
biphasic mixture was diluted with CH2Cl2 (2 mL) and washed with water (2 mL). The phases
were separated, and the aqueous phase was extracted with diethyl ether (3 x 5 mL). The
organic phases were combined, and the solvents were removed in vacuo. The residue was
purified by flash column chromatography on silica gel (2.5 x 20 cm, cyclohexane:ethyl
acetate = 97:3) to afford 209e (25 mg, 60% yield) as colorless oil.
Rf = 0.8 (cyclohexane:ethyl acetate = 90:10).
1H NMR (500 MHz, CD2Cl2): δ/ppm = 0.14–0.20 (m, 3H, Sn-CH3)*, 0.23–0.28 (m, 3H,
Sn-CH3)*, 1.13 (t, 3J1,2 = 7.1 Hz, 3H, H-1), 2.91–2.92 (m, 1H, H-4a), 2.93–2.94 (m, 1H, H-4b),
3.21 (t, 3J5,4 = 8.6 Hz, H-5), 3.74 (s, 3H, OCH3), 3.99–4.03 (mc, 2H, H-2), 6.92–6.96 (m, 1H,
H-3’), 7.01–7.04 (m, 3H, Ar-H), 7.27–7.33 (m, 3H, Ar-H), 7.36–7.42 (m, 2H, Ar-H).
13C NMR (126 MHz, CD2Cl2): δ/ppm = –10.4 (Sn-CH3)*, –10.0 (Sn-CH3)*, 14.3 (C-1), 23.7
(C-4), 36.2 (C-5), 60.9 (C-2), 115.2 (2JC-3’,F = 23 Hz, C-3’), 124.5 (4JC-5’,F Hz, C-5’), 125.8 (3JC-
6’,F = 8.0 Hz, C-6’), 127.6 (3JC-4’,F = 4.5 Hz, C-4’), 128.3 (C-3’’), 128.7 (C-4‘‘), 132.9 (2JC-1’,F =
15 Hz, C-1’), 136.5 (C-2’’), 141.4 (C-1‘‘), 160.2 (1JC-2’,F = 241 Hz, C-2’), 173.7 (C-3).
19Sn NMR (186 MHz, CD2Cl2): δ/ppm = –16.1.
IR (ATR): /cm–1 = 2979 (w), 1724 (s), 1576 (w), 1487 (m), 1426 (m), 1426 (m), 1225 (m),
1179 (s), 1105 (m), 750 (s).
Opitical rotation: = – 1.1 (c = 1.5, CHCl3, 11% ee). The enantiomeric excess was
determined by HPLC analysis on a chiral stationary phase (Daicel Chiralcel OD-H column,
column temperature 20 °C, solvent n-heptane:iso-propanol = 98:2, flow rate 0.70 mL/min, λ
= 360 nm): tR/S = 9.8 min for (R/S)-209e, tS/R = 11.2 min for (S/R)-209e.
HRMS (ESI) for C19H24FO2Sn+ [(M+H)+]: Calculated 423.0777
Found 423.0793
~

2 Description of Experiments 143
3.8 Synthesis of Chiral Cu–NHC Catalyst ((S,S)-217)
3.8.1 (1S,2S)-N1,N2-di([1,1'-biphenyl]-2-yl)-1,2-diphenylethane-1,2-diamine
((S,S)-239)
Diamine (S,S)-239 was prepared according to procedure reported by HOVEYDA and co-
workers.[81] A flame-dried SCHLENK flask was charged with Pd(OAc)2 (87 mg, 0.78 mmol, 10
mol %), (rac)-BINAP (0.48 g, 1.6 mmol, 20 mol %), (–)-(S,S)-1,2-diphenylethylenediamine
(0.83 g, 7.8 mmol, 1.0 equiv), and NaOt-Bu (2.2 g, 23 mmol, 3.0 equiv). Toluene (100 mL)
was added and the reaction mixture was stirred at 110 oC for 24 h. The reaction was cooled
down to room temperature and quenched with water (20 mL). The biphasic mixture was
separated, and the aqueous phase was extracted with CH2Cl2 (3 x 50 mL). The solvents
were evaporated in vacuo, and the residue was purified by flash column chromatography on
silica gel (5.0 x 10 cm, cyclohexane: ethyl ether = 80:20) afforded diamine (S,S)-239 (1.2 g,
30% yield) as yellow solid.
1H NMR (500 MHz, CD2Cl2): δ/ppm = 4.41–4.42 (m, 2H, H-1), 4.48–4.49 (m, 2H, N-H), 5.99–
6.01 (m, 2H, H-6’), 6.51–6.54 (m, 2H, H-4’), 6.77–6.80 (m, 2H, H-5’), 6.87–6.89 (m, 2H, H-
3’), 6.92–6.94 (m, 4H, H-3), 7.06–7.15 (m, 10H, H-4, H-4’’, H-2’’)*, 7.32–7.34 (m, 2H, H-5)*,
7.39–7.42 (m, 4H, H-3’’).
13C NMR (126 MHz, CD2Cl2): δ/ppm = 64.1 (C-1), 112.1 (C-6‘), 117.8 (C-4‘), 127.1 (C-3),
127.9 (C-4), 128.4 (C-5‘), 128.6 (C-5)*, 128.9 (C-3‘‘), 129.3 (C-2‘‘), 129.8 (C-3‘), 130.0 (C-
4‘‘)*, 133.5 (C-1‘)**, 139.4 (C-2‘)**, 140.1 (C-2), 144.1 (C-1’’)**.

144 EXPERIMENTAL PART
IR (ATR): /cm–1 = 3023 (w), 1578 (w), 1504 (m), 1434 (m), 1351 (w), 1308 (m), 1279 (w),
1069 (w), 746 (s), 698 (s).
Opitical rotation: reported = + 1.0 (c = 0.40, CHCl3)
[81], measured = – 149 (c =
1.4, CHCl3).
HRMS (ESI) for C38H33N2+ [(M+H)+]: calculated 517.2638
found 517.2646
The analytical data are in accordance with those reported. [81]
3.8.2 Chiral NHC Precursor ((S,S)-240)
NHC precursor (S,S)-240 prepared according to procedure reported by HOVEYDA and co-
workers.[81] A flame-dried SCHLENK flask was charged with diamine (S,S)-239 (1.03 g, 1.99
mmol, 1.00 equiv), ammonium tetrafluoroborate (252 mg, 2.38 mmol, 1.20 equiv), and
triethyl orthoformate (5.00 mL). The reaction mixture was heated at 120 oC for 16 h, cooled
the reaction mixture to room temperature, and filtered the solid residue. The residue was
purified using flash column chromatography on silica gel (5.0 x 10 cm, CH2Cl2:MeOH = 20:1)
to afford NHC precursor (S,S)-240 (787 mg, 64% yield) as off white solid.
1H NMR (500 MHz, CD2Cl2): δ/ppm = 4.77 (s, 2H, H-2), 6.70–6.72 (m, 4H, H-2’), 7.23–7.26
(mc, 4H, H-3’’’)*, 7.29–7.34 (m, 6H, H-3’, H-4’)**, 7.37–7.46 (m, 8H, H-3’’, H-4’’, H-5’’, H-6’’),
7.57–7.60 (m, 2H, H-4‘‘‘)**, 7.63–7.67 (m, 4H, H-2‘‘‘)*, 8.98 (s, 1H, H-1).
13C NMR (126 MHz, CD2Cl2): δ/ppm = 75.5 (C-2), 128.4 (C-4‘)*, 128.5 (C-2‘), 129.2 (C-4‘‘‘)*,
129.5 (C-3‘‘)**, 129.6 (C-2‘‘, C-3‘)***, 129.8 (C-3‘‘)***, 130.4 (C-4‘‘)**, 130.5 (C-5‘‘)**, 131.8
(C-2‘‘), 131.9 (C-6‘), 133.9 (C-1‘‘‘), 138.3 (C-1‘), 138.5 (C-1‘‘), 157.5 (C-1).
~

2 Description of Experiments 145
IR (ATR): /cm–1 = 1669 (w), 1614 (m), 1453 (w), 1262 (w), 1223 (w), 1051 (m), 1011 (m),
940 (m), 810 (w), 747 (m), 696 (s).
Opitical rotation: reported = – 339 (c = 1.0, CHCl3)
[78], measured = – 378 (c = 1.5,
CHCl3).
HRMS (ESI) for C39H31N2+ [(M–BF4)
+]: calculated 527.2482
found 527.2483
The analytical data are in accordance with those reported.[81]
~

146 EXPERIMENTAL PART
3.8.3 Chiral Cu–NHC catalyst ((S,S)-217)
The chiral Cu–NHC catalyst (S,S)-217 was synthesized as reported by FERNÁNDEZ and co-
workers.[97] The NHC precursor (S,S)-240 (200 mg, 0.320 mmol, 1.00 equiv) was suspended
in THF (5 mL). KOtBu solution (1M in THF, 384 µL, 0.384 mmol, 1.20 equiv) was added
dropwise at room temperature. The reaction mixture was stirred for 1 hour. CuCl (32.2 mg,
0.320 mmol, 1.00 equiv) was added to the solution, and the reaction mixture was stirred for 4
h at room temperature. The reaction mixture was filtered over Celite® and washed with THF
(10 mL). The filtrate was evaporated and recrystallized from DCM and n-pentane at –20 oC
to afford the Cu–NHC catalyst (S,S)-217 (177 mg, 88% yield) as green solid.
1H NMR (500 MHz, CD2Cl2): δ/ppm = 4.42 (s, 2H, H-2), 6.89–6.90 (m, 4H, H-2’), 7.38–7.41
(m, 4H, H-3’’’)*, 7.46–7.49 (m, 3H, Ar-H), 7.61–7.68 (m, 6H, Ar-H), 7.82–7.95 (m, 11H, Ar-
H).
13C NMR (126 MHz, CD2Cl2): δ/ppm = 69.4 (C-2), 127.9 (C-4‘), 128.0 (C-2‘), 128.4 (C-4‘‘‘)*,
128.5 (Ar-C), 128.6 (C-3‘‘‘)*, 129.1 (Ar-C), 129.4 (C-6‘), 129.6 (Ar-C), 131.3 (Ar-C), 136.0 (C-
1‘‘‘), 138.0 (C-1‘), 140.4 (C-1‘‘).
IR (ATR): /cm–1 = 1701 (w), 1477 (w), 1435 (w), 1281 (w), 1026 (m), 916 (w), 876 (w), 741
(m), 697 (s).
Opitical rotation: reported = – 296 (c = 0.5, CHCl3)
[81], measured = – 160 (c = 1.5,
CHCl3).
[97]
V. Lillo, A. Prieto, A. Bonet, M. M. Díaz-Requejo, J. Ramírez, P. J. Pérez, E. Fernández, Organometallics 2009, 28, 659−662.
~

2 Description of Experiments 147
HRMS (ESI) for C39H31CuN2+ [(M–Cl)+]: calculated 589.1778
found 589.1701
The analytical data are in accordance with those reported.[97]

APPENDIX

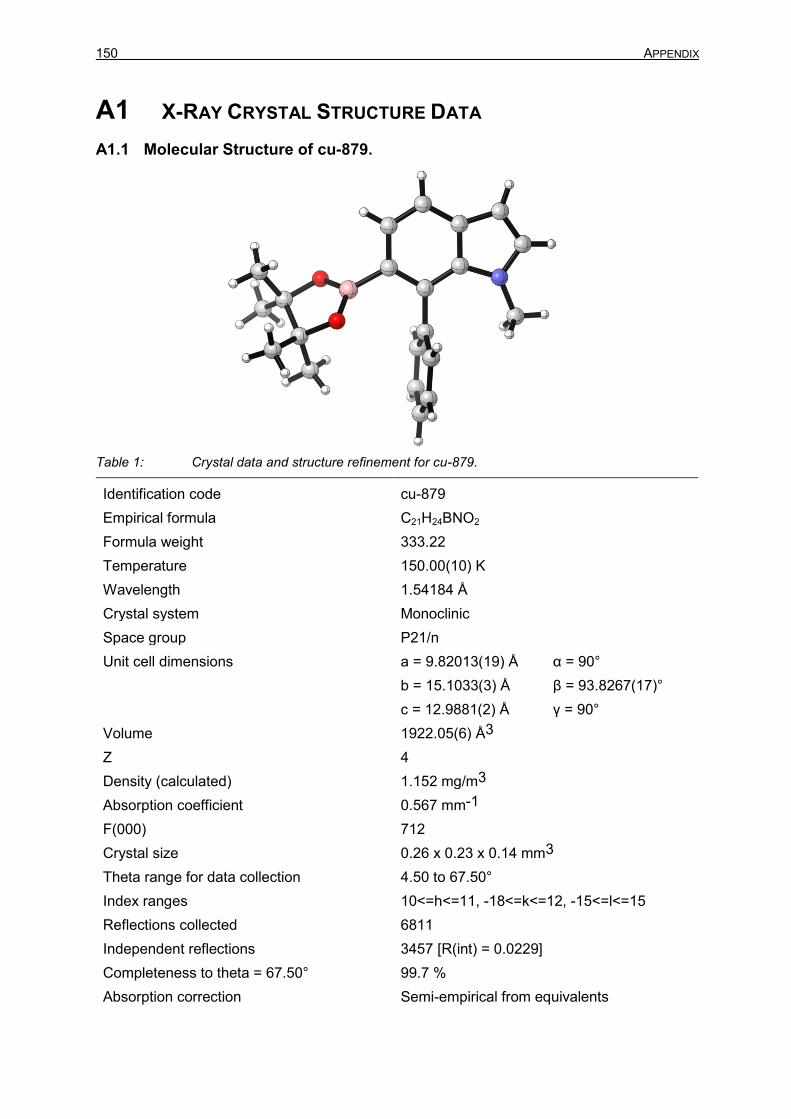
150 APPENDIX
A1 X-RAY CRYSTAL STRUCTURE DATA
A1.1 Molecular Structure of cu-879.
Table 1: Crystal data and structure refinement for cu-879.
Identification code cu-879
Empirical formula C21H24BNO2
Formula weight 333.22
Temperature 150.00(10) K
Wavelength 1.54184 Å
Crystal system Monoclinic
Space group P21/n
Unit cell dimensions a = 9.82013(19) Å α = 90°
b = 15.1033(3) Å β = 93.8267(17)°
c = 12.9881(2) Å γ = 90°
Volume 1922.05(6) Å3
Z 4
Density (calculated) 1.152 mg/m3
Absorption coefficient 0.567 mm-1
F(000) 712
Crystal size 0.26 x 0.23 x 0.14 mm3
Theta range for data collection 4.50 to 67.50°
Index ranges 10<=h<=11, -18<=k<=12, -15<=l<=15
Reflections collected 6811
Independent reflections 3457 [R(int) = 0.0229]
Completeness to theta = 67.50° 99.7 %
Absorption correction Semi-empirical from equivalents

APPENDIX 151
Max. and min. transmission 0.9248 and 0.8665
Refinement method Full-matrix least-squares on F2
Data / restraints / parameters 3457 / 0 / 231
Goodness-of-fit on F2 1.030
Final R indices [I>2sigma(I)] R1 = 0.0402, wR2 = 0.0988
R indices (all data) R1 = 0.0464, wR2 = 0.1040
Largest diff. peak and hole 0.351 and -0.200 e.Å-3


A2 Abbreviations 151
A2 ABBREVIATIONS
[α] specific rotation
δ chemical shift
λ wavelenght
~ wavenumber
Å Ångström
Ac acetyl
APCI atmospheric pressure chemical ionization
Ar aryl
ATR attenuated total reflection
b.p. boiling point
br broad
n-Bu n-butyl
i-Bu iso-butyl
t-Bu tert-butyl
°C degree Celsius
c concentration
cat. catalytic
cat catechol
cm centimeter
cod 1,5-cyclooctadiene
conv conversion
COSY correlation spectroscopy
Cy cyclohexyl
d doublet
dan diaminonaphthalene
decomp Decomposition
DEPT distortionless enhancement by polarization transfer
DIBAL diisobutylaluminum
dm decimeter
DMAP 4-dimethylaminopyridine

152 APPENDIX
d.r. diastereomeric ratio
E main-group element
ee enantiomeric excess
EI electronic Ionization
e.r. enantiomeric ratio
ESI electron spray ionization
Et ethyl
equiv equivalent
g gram
GLC gas-liquid chromatography
GP General Procedure
h hour
n-Hept n-heptyl
n-Hex n-hexyl
HMBC heteronuclear multiple bond coherence
HMQC heteronuclear multiple quantum coherence
HPLC high-performance liquid chromatography
HSQC heteronuclear single quantum coherence
IMes 1,3-dimesityl-1H-imidazol-3-ium-2-ide
IR infrared spectroscopy
J coupling constant
k rate constant
KHMDS potassium bistrimethylsilylamide
L ligand or liter
LA LEWIS acid
LB LEWIS base
LIHMDS lithium bistrimethylsilylamide
M molecular mass or metal or mega

A2 Abbreviations 153
M molar
m multiplet or medium or milli or meter
m meta
mc centrosymmetric multiplet
Me methyl
Mes mesityl
min minute
mol % mole percent
m.p. melting point
MS mass spectrometry
n number of units
NHC N-heterocyclic carbine
NMR nuclear magnetic resonance
o ortho
n-Oct n-octyl
OTf triflate
p para
Ph phenyl
pin pinacolato
ppm parts per million
i-Pr isopropyl
q quartet
R organic rest or as defined in the text
Rf retention factor
rac racemic
r.s. regio selectivity
r.t. room temperature
s singlet or strong
t triplet
tR retention time

154 APPENDIX
Temp temperature
THF tetrahydrofuran
TLC thin-layer chromatography
TOF turnover frequency
Tol tolyl
TON turnover number
UV ultraviolet
w weak
X heteroatom
(9-BBN) 9-borabicyclo(3.3.1)nonane

A3 Bibliography 155
A3 BIBLIOGRAPHY
[1] K. Tamao, S. Yamaguchi, J. Organomet. Chem. 2000, 611, 3–4.
[2] For general reviews on interelement bond activation and carbon–element bond
formation reaction, see: a) M. B. Ansell, O. Navarro, J. Spencer, Coord. Chem. Rev.
2017, doi: http://dx.doi.org/10.1016/j.ccr.2017.01.003; b) E. C. Neeve, S. J. Geier, I. A.
I. Mkhalid, A. Westcott, T. B. Marder, Chem. Rev. 2016, 116, 9091–9161; c) M.
Iwasaki, Y. Nishihara, Chem. Rec. 2016, 16, 2031–2045; d) M. Suginome, T. Matsuda,
T. Ohmura, A. Seki, M. Murakami, in Comprehensive Organometallic Chemistry III;
Mingos, D. M. P.; Crabtree, R. H.; Ojima, I., Eds.; Elsevier: Oxford, 2007; Vol. 10, pp
725–787; e) H. E. Burks, J. P. Morken, Chem. Commun. 2007, 4717–4725; f) I.
Beletskaya, C. Moberg, Chem. Rev. 2006, 106, 2320–2354; g) T. Ishiyama, N.
Miyaura, Chem. Rec. 2004, 3, 271–280; h) I. Beletskaya, C. Moberg, Chem. Rev.
1999, 99, 3435–3462; i) Y. J. Ito, Organomet. Chem. 1999, 576, 300–304; j) T. B.
Marder, N. C. Norman, Top. Catal. 1998, 5, 63–73; k) K. A. Horn, Chem. Rev. 1995,
95, 1371–1350.
[3] For carbon–carbon bond forming reactions, see: a) Metal Catalyzed Cross-Coupling
Reactions and More, (Eds.: A. de Meijere, S. Brase, M, Oestreich), Wiley-VCH,
Weinheim, 2014; b) Handbook of Organopalladium Chemistry for Organic Synthesis
(Eds.: E. Negishi, A. de Meijere), Wiley, New York, 2002; c) N. Miyaura, A. Suzuki,
Chem. Rev. 1995, 95, 2457–2483; d) Y. Hatanaka, T. Hiyama, J. Org. Chem. 1988, 53,
918–920; e) J. K. Stille, Angew. Chem. Int. Ed. 1986, 25, 508–524; Angew. Chem.
1986, 98, 504–519.
[4] M. Oestreich, E. Hartmann, M. Mewald, Chem. Rev. 2013, 113, 402–441.
[5] A. Stock, A. Brandt, H. Fischer, Ber. Dtsch. Chem. Ges. B 1925, 58, 643–657.
[6] R. Barbeyron, E. Benedetti, J. Cossy, J. J. Vasseur, S. Arseniyadis, M. Smietana,
Tetrahedron 2014, 70, 8431–8452.
[7] S. A. Westcott, E. Fernandez, in Advances in Organometallic Chemistry; Academic
Press: Cambridge, 2015; pp 39–89.
[8] a) J. Takaya, N. Iwasawa, ACS Catal. 2012, 2, 1993–2006; b) M. V. Dembitsky, A. H.
Ali, M. Srebnik, in Advances in Organometallic Chemistry; Academic Press:
Cambridge, 2004; pp 193–250.
[9] a) J. Ramírez, V. Lillo, M. A. Segarra, E. Fernandez, C. R. Chim. 2007, 10, 138–151;
b) T. Ishiyama, N. Miyaura, J. Organomet. Chem. 2000, 611, 392–402.

156 APPENDIX
[10] a) H. Braunschweig, Angew. Chem. Int. Ed. 1998, 37, 1786–1801; Angew. Chem.
1998, 110, 1882–1898; b) R. Chadha, N. K. Ray, Theor. Chim. Acta 1982, 60,
573–578.
[11] G. Lesley, P. Nguyen, N. J. Taylor, T. B. Marder, A. J.Scott, W. Clegg, N. C. Norman,
Organometallics 1996, 15, 5137–5154.
[12] R. L. Thomas, F. E. S. Souza, T. B. Marder, J. Chem. Soc., Dalton Trans. 2001, 1650–
1656.
[13] T. Ishiyama, N. Matsuda, M. Murata, F. Ozawa, A. Suzuki, N. Miyaura,
Organometallics 1996, 15, 713–720.
[14] a) W. Clegg, F. J. Lawlor, T. B. Marder, P. Nguyen, N. C. Norman, A. G. Orpen, M. J.
Quayle, C. R. Rice, E. G. Robins, A. J. Scott, F. E. S. Souza, G. Stringer, G. R.
Whittell, J. Chem. Soc., Dalton Trans. 1998, 301–310; b) R. T. Baker, J. C. Calabrese,
S. A. Westcott, P. Nguyen, T. B. Marder, J. Am. Chem. Soc. 1993, 115, 4367–4368.
[15] a) M. V. Campian, J. L. Harris, N.Jasim, R. N. Perutz, T. B. Marder, A. C.Whitwood,
Organometallics 2006, 25, 5093–5104; b) C. Dai, G. Stringer, J. F. Corrigan, N. J.
Taylor, T.B. Marder, N. C. Norman, J. Organomet. Chem. 1996, 513, 273–275.
[16] Q. Cui, D. G. Musaev, K. Morokuma, Organometallics 1998, 17, 742–751.
[17] T. Ishiyama, N. Matsuda, N. Miyaura, A. Suzuki, J. Am. Chem. Soc. 1993, 115, 11018–
11019.
[18] T. Ishiyama, M. Yamamoto, N. Miyaura, Chem. Lett. 1996, 1117–1118.
[19] H. Abu Ali, A. A. Al Quntar, I. Goldberg, M. Srebnik, Organometallics 2002, 21, 4533.
[20] M. Bluhm, A. Maderna, H. Pritzkow, S. Bethke, R. Gleiter, W. Siebert, Eur. J. Inorg.
Chem. 1999, 1693–1700.
[21] N. Iwadate, M. Suginome, J. Am. Chem. Soc. 2010, 132, 2548–2549.
[22] G. Urry, J. Kerrigan, T. D. Parsons, H. Schlesinger, J. Am. Chem. Soc. 1954, 76,
5299−5301.
[23] a) T. Ishiyama, M. Yamamoto, N. Miyaura, Chem. Commun. 1997, 689−690; b) C. N.
Iverson, M. R. Smith, Organometallics 1997, 16, 2757−2759; b) T. Oshiyama, M.
Yamamoto, N. Miyaura, Chem. Commun. 1997, 689−690.
[24] a) J. B. Morgan, S. P. Miller, J. P. Morken, J. Am. Chem. Soc. 2003, 125, 8702−8703;
b) L. T. Kliman, S. N. Mlynarski, J. P. Morken, J. Am. Chem. Soc. 2009, 131,
13210−13211.
[25] J. R. Coombs, F. Haeffner, L. T. Kliman, J. P Morken, J. Am. Chem. Soc. 2013, 135,
11222−11231.
[26] a) J. Ramírez, R. Corberan, M. Sanau, E. Peris, E. Fernández, Chem. Commun. 2005,
3056−3058; b) V. Lillo, E. Mas-Marza, A. M. Segarra, J. J. Carbo, C. Bo, E. Peris, E.
Fernández, Chem. Commun. 2007, 3380−3382.

A3 Bibliography 157
[27] R. T. Baker, P. Nguyen, T. B. Marder, S. A. Westcott, Angew. Chem. Int. Ed. 1995, 34,
1336−1338; Angew. Chem. 1995, 107, 1451–1452.
[28] C. Dai, E. G. Robins, D. S. Yufit, J. A. K. Howard, T. B. Marder, A. J. Scott, W. Clegg,
Chem. Commun. 1998, 1983−1984.
[29] J. Ramírez, A. M. Segarra, E. Fernández, Tetrahedron: Asymmetry 2005, 16,
1289−1294.
[30] T. Ishiyama, M. Yamamoto, N. Miyaura, Chem. Commun. 1996, 2073−2074.
[31] a) Z. Y. Yu, R. J. Ely, J.P. Morken, Angew. Chem. Int. Ed. 2014, 53, 9632−9636; B) C.
H. Schuster, B. Li, J. P. Morken, Angew. Chem. Int. Ed. 2011, 50, 7906−7909; c) K.
Hong, J. P. Morken, J. Org. Chem. 2011, 76, 9102−9108; c) S. L. Poe, J. P. Morken,
Angew. Chem. Int. Ed. 2011, 50, 4189−4192; d) H. E. Burks, L. T. Kliman, J. P.
Morken, J. Am. Chem. Soc. 2009, 131, 9134−9135.
[32] H. Y. Cho, J. P. Morken, J. Am. Chem. Soc. 2008, 130, 16140−16141.
[33] R. J. Ely, J. P. Morken, Org. Lett. 2010, 12, 4348−4351.
[34] T. Ishiyama, T. Kitano, N. Miyaura, Tetrahedron Lett. 1998, 39, 2357−2360.
[35] M. Wang, L. Cheng, Z. Wu, Organometallics 2008, 27, 6464−6471.
[36] X. Guo, A. K. Nelson, C. Slebodnick, W. L. Santos, ACS Catal. 2015, 5, 2172−2176.
[37] F.-Y. Yang, C.-H. Cheng, J. Am. Chem. Soc. 2001, 123, 761−762.
[38] H. Yoshida, K. Okada, S. Kawashima, K. Tanino, J. Ohshita, Chem. Commun. 2010,
46, 1763–1765.
[39] Y. Himeshima, T. Sonoda, H. Kobayashi, Chem. Lett. 1983, 1211−1214.
[40] H. Yoshida, S. Kawashima, Y. Takemoto, K. Okada, J. Ohshita, K Takaki, Angew.
Chem. Int. Ed. 2012, 51, 235–238; Angew. Chem. 2012, 124, 239–242.
[41] a) N. Miyaura, K. Yamada, A. Suzuki, Tetrahedron Lett. 1979, 20, 3437–3440; b) N.
Mivaura. K. Yamada. H. Suginome, A. Suzuki. J. Am. Chem Soc. 1985, 107, 972–980.
[42] For the reviews and application of SUZUKI–MIYAURA reaction, see a) C. C. C. J.
Seechurn, M. O. Kitching, T. J. Colacot, V. Snieckus, Angew. Chem. Int. Ed. 2012, 51,
5062–5085; Angew. Chem. 2012, 124, 5150–5174; b) A. Suzuki, Angew. Chem. Int.
Ed. 2011, 50, 6722–6737; Angew. Chem. 2011, 123, 6855–6869; c) J. S. Carey, D.
Laffan, C. Thomson, M. T. Williams, Org. Biomol. Chem. 2006, 4, 2337–2347.
[43] For a summary of studies of transmetalation in the SUZUKI–MIYAURA reaction, see: a)
A. A. Thomas, S. E. Denmark, Science 2016, 352, 329–332; b) B. P. Carrow, J. F.
Hartwig, J. Am. Chem. Soc. 2011, 133, 2116–2119; c) Y. Suzaki, K. Osakada,
Organometallics 2006, 25, 3251–3258; d) Y. Suzaki, K. Osakada, Organometallics
2006, 25, 3251–3258; e) A. A. C. Braga, N. H. Morgen, G. Ujaque, F. Maseras, J. Am.
Chem. Soc. 2005, 127, 9298–9307; f) L. J. Goossen, D. Koley, H. L. Hermann, W.

158 APPENDIX
Thiel, J. Am. Chem. Soc. 2005, 127, 11102–11114; g) N. J. Miyaura, J. Organomet.
Chem. 2002, 653, 54–57.
[44] c) J. S. Carey, D. Laffan, C. Thomson, M. T. Williams, Org. Biomol. Chem. 2006, 4,
2337–2347; b) A. D. Schlüter, J. Polym. Sci. 2001, 39, 1533–1556.
[45] a) A. R. Kapdi, D. Prajapati, RSC Adv. 2014, 4, 41245–41259; b) J.-R. Wang, K.
Manabe, Synthesis 2009, 1405–1427.
[46] P. Dobrounig, M. Trobe, R. Breinbauer, Monatsh. Chem. 2017, 148, 3–35.
[47] a) G. A. Molander, D. L. Sandrock Org. Lett. 2009, 11, 2369–2372; b) G. A. Molander,
D. L. Sandrock, J. Am. Chem. Soc. 2008, 130,15792–15793.
[48] N. Iwadate, M. Suginome, Org. Lett. 2009, 11, 1899–1903; b) H. Noguchi, T. Shioda,
C-M. Chou, M. Suginome, Org. Lett. 2008, 10, 377–380; c) H. Noguchi, K. Hojo, M.
Suginome, J. Am. Chem. Soc. 2007, 129, 758–759.
[49] S. G. Ballmer, E. P. Gillis, M. D. Burke, Org. Synth. 2009, 86, 344–359.
[50] a) E. M. Woerly, J. Roy, M. D. Burke, Nat. Chem. 2014, 6, 484–491; b) S. Fujii, S. Y.
Chang, M. D. Burke, Angew. Chem. Int. Ed. 2011, 50, 7862–7864; Angew. Chem.
2011, 123, 8008–8010; c) S. J. Lee, T. M. Anderson, M. D. Burke, Angew. Chem. Int.
Ed. 2010, 49, 8860–8863; Angew. Chem. 2010, 122, 9044–9047; d) E. M. Woerly, A.
H. Cherney, E. K. Davis, M. D. Burke, J. Am. Chem. Soc. 2010, 132, 6941–6943; e) E.
P. Gillis, M. D. Burke, J. Am. Chem. Soc. 2007, 129,
6716–6718;
[51] J. W. B. Fyfe, E. Valverde, C. P. Seath, A. R. Kennedy, J. M. Redmond, N. A.
Anderson, A. J. B. Watson, Chem. Eur. J. 2015, 21, 8951–8964.
[52] K. Endo, T. Ohkubo, M. Hirokami, T. Shibata, J. Am. Chem. Soc. 2010 ,132, 11033–
11035.
[53] S. N. Mlynarski, C. H. Schuster, J. P. Morken, Nature 2014, 505, 386–390.
[54] C. M Crudden, C. Ziebenhaus, J. P. G. Rygus, K. Ghozati, P. J. Unsworth, M. Nambo,
S. Voth, M. Hutchinson, V. S. Laberge, Y. Maekawa, D. Imao, Nat. Commun. 2016, 7,
11065–11071.
[55] B. L. Chenard, E. D. Laganis, F. Davidson, T. V. R. Babu, J. Org. Chem. 1985, 50,
3666–3667.
[56] M. Mori, N. Kaneta, N. Isono, M. Shibasaki, J. Organomet. Chem. 1993, 455, 255–
260.
[57] N. Isono, M. Mori, J. Org. Chem. 1998, 63, 1773–1779.
[58] T. Honda, M. Mori, Chem. Lett. 1994, 1013–1016.
[59] R. K. Bhatt, J. Ye, J.R. Falck, Tetrahedron. Lett. 1994, 35. 4081–4084.
[60] a) N. Isono, M. Mori, Main Group Metal Chem. 1996, 19, 277–279; b) N. Isono, M.
Mori, Tetrahedron Lett. 1995, 36, 9345–9347.

A3 Bibliography 159
[61] R. K. Schmidt, M. Oestreich, Synlett 2008, 1690–1692.
[62] a) R. Blanc, P. Nava, M. Rajzman, L. Commeiras, J.-L. Parraina, Adv. Synth. Catal.
2012, 354, 2038–2048; b) R. Blanc, L. Commeiras, J.-L. Parrain, Adv. Synth. Catal.
2010, 352, 661–666.
[63] a) A. G. Davies, Organotin Chemistry, Wiley-VCH, Weinheim, 2004; b) M. S. Holt, W.
L. Wilson, J. H. Nelson, Chem. Rev. 1989, 89, 11–48.
[64] R. J. P. Corriu, C. Guerin, J. Organomet. Chem. 1980, 197, C19–C20.
[65] K. R. Wursthorn, H. G. Kuivila, G. F. Smith, J. Am. Chem. Soc. 1978, 100, 2779–2789.
[66] J. Hudac, J. Chem. Soc.,Perkin Trans. 1 1975, 1020–1021.
[67] E. Piers, R. D. Tillyer, J. Org. Chem. 1988, 53, 5366–5369.
[68] A. C. Cehlschlager, M. W. Hutzinger, R. Aksela, S. Sharma, S. M. Singh, Tetrahedron
Lett. 1990, 31, 165–168.
[69] D. Enders, K.-J. Heider, G. Raabe, Angew. Chem. Int. Ed. 1992, 32, 598–601; Angew.
Chem. 1992, 105, 592–595.
[70] a) G.-Y. J. Im, S. M. Bronner, A. E. Goetz, R. S. Paton, P. H.-Y. Cheong, K. N. Houk,
N. K. Garg, J. Am. Chem. Soc. 2010, 132, 17933–17944; b) S. M. Bronner, K. B.
Bahnck, N. K. Garg, Org. Lett. 2009, 11, 1007–1011.
[71] P. H.-Y. Cheong, R. S. Paton, S. M. Bronner, G.-Y. J. Im, N. K. Garg, K. N. Houk, J.
Am. Chem. Soc. 2010, 132, 1267–1269.
[72] M. Pareek, T. Fallon, M. Oestreich, Org. Lett. 2015, 17, 2082–2085.
[73] For general reviews and reports, see: a) S. Shirakawa, K. Maruoka, Angew. Chem. Int.
Ed. 2013, 52, 4312–4348; Angew. Chem. 2013, 125, 4408–4445; b) Asymmetric
Phase Transfer Catalysis, (Ed.: K. Maruoka), Wiley-VCH, Weinheim, 2008; c) T. Ooi,
K. Maruoka, Angew. Chem. Int. Ed. 2007, 46, 4222–4266; Angew. Chem. 2007, 119,
4300–4345; d) T. Ooi, D. Ohara, K. Fukumoto, K. Maruoka, Org. Lett. 2005, 7, 3195–
3197; e) B. Lygo, B. Allbutt, E. H. M. Kirton, Tetrahedron Lett. 2005, 46, 4461–4464; f)
T. Ooi, S. Fujioka, K. Maruoka, J. Am. Chem. Soc. 2004, 126, 11790–11793; g) M.
Oku, S. Arai, K. Katayama, T. Shioiri, Synlett 2000, 493–496; h) M. J. O’Donnell, F.
Delgado, C. Hostettler, R. Schwesinger, Tetrahedron Lett. 1998, 39, 8775–8778; i) E.
J. Corey, F. Xu, M. C. Noe, J. Am. Chem. Soc. 1998, 119, 12414–12416; j) E. J.
Corey, Y. Bo, J. Busch-Petersen, J. Am. Chem. Soc. 1998, 120, 13000–13002; k) B. B.
Lygo, P. G. Wainwright, Tetrahedron Lett. 1998, 39, 1599–1602; l) B. B. Lygo, P. G.
Wainwright, Tetrahedron Lett. 1997, 38, 8595–8598; m) K. Shishido, K. Goto, S.
Miyoshi, Y. Takaishi, M. Shibuya, J. Org. Chem. 1994, 59, 406–414; n) D. J. Cram, G.
D. Y. Sogah, J. Chem. Soc. 1981, 625–628.
[74] K Mochida, T Yamanishi, Bull. Chem. Soc. Jpn. 1987, 60, 3429−3430.
[75] W. C. Still, C. Gennari, Tetrahedron Lett. 1983, 24, 4405–4408.

160 APPENDIX
[76] C. M. Starks, J. Am. Chem. Soc. 1971, 93, 195–199.
[77] a) G. A. Molander, S. R. Wisniewski, M. H. Sarvari, Adv. Synth. Catal. 2013, 355,
3037–3057; b) S. B. Thorpe, J. A. Calderone, W. L. Santos, Org. Lett. 2012, 14, 1918–
1921; c) G. A. Molander, S. A. McKee, Org. Lett. 2011, 13, 4684–4687; d) W. J.
Fleming, H. Miller-Bunz, V. Lillo, E. Fernandez, P. J. Guiry, Org. Biomol. Chem. 2009,
7, 2520−2524; e) V. Lillo, A. Prieto, A. Bonet, M. M. Diaz Requejo, J. Ramirez, P. J.
Prez, E. Fernandez, Organometallics 2009, 28, 659–662; f) H.-S. Sim, X. Feng, J.
Yun, Chem. Eur. J. 2009, 15, 1939–1943; g) S. Mun, J.-E. Lee, J. Yun, Org. Lett.
2006, 8, 4887–4889; h) H. Ito, H. Yamanaka, J. Tateiwa, A. Hosomi, Tetrahedron Lett.
2000, 41, 682–686; i) K. Takahashi, T. Isiyama, N. Miyaura, J. Organomet. Chem.
2001, 625−629; j) K. Takahashi, T. Isiyama, N. Miyaura, Chem. Lett. 2000, 982−983;
k) Y. G. Lawson, M. J. G. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, Chem.
Commun. 1997, 2051−2052.
[78] a) K. Okamoto, T. Hayashi, Org. Lett. 2007, 9, 5067–5069; b) S. Ogoshi, S. Tomiyasu,
M. Morita, H Kurosawa, J. Am. Chem. Soc. 2002, 124, 11598–11599; c) H. Ito, T.
Ishizuka, J.-I. Tateiwa, M. Sonoda, A. Hosomi, J. Am. Chem. Soc. 1998, 120, 11196–
11197; d) K. H. Pannell, H. K. Sharma, Chem. Rev. 1995, 95, 1351–1374; e) K. A.
Horn, Chem. Rev. 1995, 95, 1371–1350.
[79] a) A. Hensel, M. Oestreich, Chem. Eur. J. 2015, 21, 9062–9065; b) L. B. Delvos, A.
Hensel, M. Oestreich, Synthesis 2014, 46, 2957−2964; c) L. B. Delvos, M. Oestreich,
Synthesis 2015, 47, 924−933; d) A. Hensel, K. Nagura, L. B. Delvos, M. Oestreich,
Angew. Chem. Int. Ed. 2014, 53, 4964−4967; e) L. B. Delvos, D. J. Vyas, M.
Oestreich, Angew. Chem. Int. Ed. 2013, 52, 4650−4653; Angew. Chem. 2013, 125,
4748–4751; f) E. Hartmann, D. J. Vyas, M. Oestreich, Chem. Commun. 2011, 47,
7917−7932; g) D. J. Vyas, C. K. Hazra, M. Oestreich, Org. Lett. 2011, 13, 4462−4465;
h) C. K. Hazra, E. Irran, M. Oestreich, Eur. J. Org. Chem. 2013, 4903–4908; i) D. J.
Vyas, R. Fröhlich, M. Oestreich, Org. Lett. 2011, 13, 2094–2097; j) D. J. Vyas, M.
Oestreich, Angew. Chem. Int. Ed. 2010, 49, 8513–8515; Angew. Chem. 2010, 122,
8692–8694; k) C. Walter, R. Fröhlich, M. Oestreich, Tetrahedron 2009, 65,
5513−5520; l) C. Walter M. Oestreich, Angew. Chem. Int. Ed. 2008, 47, 3818–3820;
Angew. Chem. 2008, 120, 3878–3880; m) C. Walter, G. Auer, M. Oestreich, Angew.
Chem. Int. Ed. 2006, 45, 5675−5677; Angew. Chem. 2006,118, 5803–5805.
[80] J. K. Park, H. H. Lackey, M. D. Rexford, K. Kovnir, M. Shatruk, D. T. McQuade, Org.
Lett. 2010, 12, 5008–5011.
[81] Y. Lee, A. H. Hoveyda, J. Am. Chem. Soc. 2009, 131, 3160–3161.
[82] A. Weickgenannt, M. Oestreich, Chem. Eur. J. 2010, 16, 402−412.

A3 Bibliography 161
[83] a) H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512–7515; b) G.
R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stolz, J.
E. Bercaw, K. I. Goldberg, Organometallics 2010, 29, 2176–2179.
[84] R. K. Harris, E. D. Becker, S. M. C. de Menezes, R. Goodfellow, P, Granger, Pure
Appl. Chem. 2001, 73, 1795–1818.
[85] CYLview, 1.0b; Legault, C. Y., Université de Sherbrooke, 2009.
[86] http://chemsearch.kovsky.net/
[87] M. -Y. Chang, H. -Y. Tai, Y. -L. Chen, R. -T. Hsu, Tetrahedron 2012, 68, 7941−7948.
[88] S. Ueda, T. Okada, H. Nagasawa, Chem. Commun. 2010, 46, 2462−2464.
[89] A. C. Breman, S. E. M. Telderman, R. P. M. V. Santen, J. I. Scott, J. H. V.
Maarseveen, S. Ingemann, H. Hiemstra, J. Org. Chem. 2015, 80, 10561–10574.
[90] M. J. O’Donnell, S. Wu, J.C. Huffman, Tetrahedron 1994, 50, 4507–4509.
[91] P. Nun, V. Pérez, M. Calmès, J. Martinez, F. Lamaty, Chem. Eur. J. 2012, 18, 3773–
3779.
[92] T. Kanemitsu, S. Koga, D. Nagano, M. Miyazaki, K. Nagata, T. Itoh, ACS Catal. 2011,
1, 1331–1335.
[93] M. M. Parvez, N. Haraguchi, S. Itsuno, Macromolecules 2014, 47, 1922–1928.
[94] N. Ledoux, A. Linden, B. Allaert, H. V. Mierde, F. Verpoort, Adv. Synth. Catal. 2007,
349, 1692–1700.
[94] A. Armstrong, A. Ferguson, Beilstein J. Org. Chem. 2012, 8, 1747–1752.
[96] Commercially available NaH suspention 60% (w/w) in mineral oil was washed with n-
pentane (3 x 20 mL). The resulting wet NaH was dried under vacuum prior to use.
[97] V. Lillo, A. Prieto, A. Bonet, M. M. Díaz-Requejo, J. Ramírez, P. J. Pérez, E.
Fernández, Organometallics 2009, 28, 659−662.

A3 Curriculum Vitae 162
CURRICULUM VITAE
Personal Details
Name Manish Pareek
Date and place of birth July 1st 1990 in Raila Road, India
Maritial Status Married
Education
October 2013 – April 2017 Technische Universität Berlin, Berlin
Dissertation under the supervision of Prof. Dr.
MARTIN OESTREICH,
Thesis: “Activation of Boron–Boron, Tin–Silicon,
and Tin–Tin Bonds: Application in Carbon–Element
(E = B and Sn) Bond-Forming Reactions and Site-
Selective SUZUKI–MIYAURA Cross-Coupling
Reactions”
August 2012 – April 2013 Indian Institute of Science Education and Research,
Mohali, India
Master thesis under the supervision of Prof. Dr. R.
VIJAYA ANAND,
Thesis: “Base Mediated 5-endo-dig Cyclization of
N-propargyl Proline Derivatives: A Facile Entry to
Pyrrolizidine Scaffolds “
August 2008 – May 2013 Indian Institute of Science Education and Research,
Mohali, India
Integrated BS – MS dual degree
Matriculation Examination
May 2007 Rajasthan Board of Secondary Education, Ajmer





























