Embed Size (px)
Citation preview

Dennis Bensinger
Den Ursachen von Krebs auf den Grund gehen
Schon vor über hundert Jahren postulierte der deutsche Biologe Theodor Boveri (1862-1915), dass die Tumorbildung mit einer fehlerhaften Verteilung der Chromosomen einer einzigen Zelle beginnt, wodurch diese aus dem Gleichgewicht gerät und „eine abnorme, dem Plan des Ganzen zuwiderlaufende Wucherung eintritt“.
Die direkte Zuordnung eines regelmäßigen Gendefektes mit einer Tumorerkrankung gelang Peter Nowell und David Hungerford 1960. Sie fanden das verkürzte Philadelphia-Chromosom (Ph+) bei Patienten mit chronischer myeloischer Leukämie (CML) und benannten es nach dem Ort seiner Entdeckung. Von besonderer Bedeutung für die Tumorenstehung ist dabei, dass durch diese chromosomale Umlagerung ein vorher in dieser Form in der Zelle nicht vorhandenes Enzym BCR-Abl entsteht, das der Leukämiezelle ein dauerhaftes Überleben ermöglicht. Die Entwicklung des BCR-Abl hemmenden Wirkstoffes Imatinib, die vor allem von dem Onkologen Brian Druker mit der Pharmafirma Ciba-Geigy (heute Novartis) vorangetrieben wurde, war mit dessen Zulassung 2001 ein spektakulärer Erfolg.
Die Geschichte begründete einen Paradigmenwechsel in der Krebstherapie: weg von der ungerichteten Hemmung der DNA-Vermehrung schnell wachsender, aber auch gesunder Zellen, hin zur Untersuchung des geschädigten Genoms und der daraus resultierenden Proteine und deren molekularer Beeinflussung.
Doch die nächste Herausforderung wartet bereits: Auch bei Einsatz maßgeschneiderter Tumortherapeutika werden Resistenzbildungen beobachtet, die deren Wirkung verpuffen lassen und die wiederum neue Therapiekonzepte erfordern. Wie können die Erkenntnisse der molekularen Onkologie beitragen, Krebs zu stoppen?
Krebs: Eine Krankheit der Gene
Krebs ist heute in den Industrieländern nach Herz-Kreislauf-Erkrankungen die häufigste Todesursache. Im Jahr 2004 erkrankten in Deutschland 436 000 Menschen an Krebs, 206 000 starben daran. Das Durchschnittsalter aller Patienten liegt bei etwa 70 Jahren. Die Mortalität unterscheidet sich für verschiedene Krebsarten erheblich, so liegt die Überlebensrate für Brustkrebs bei 80-90 %, während sie bei Lungen- und Pankreaskarzinomen immer noch sehr gering ist.
Leukämien sind Krebserkrankungen des blutbildenden Systems und führen zu einer starken Vermehrung von weißen Blutkörperchen (Leukozyten). Dieser Artikel befasst sich vor allem mit der Therapie der chronischen myeloische Leukämie (CML), die 15 bis 20 % aller Leukämien ausmacht. In den USA treten ca. 5000 Fälle dieser Tumorerkrankung pro Jahr auf.
Die Aktuelle-Wochenschau© der GDCh – Fachgruppe Biochemie 47/2013
„Molekulares Teamplay in der Tumortherapie - Krebstherapeutika der nächsten Generation“

Ursachen der CML
Die chronische myeloische Leukämie wurde klinisch erstmals 1845 von Virchow und Benett als „Weißes Blut“ beschrieben, die molekulare Pathogenese wurde jedoch erst 1984 mit der Verknüpfung des Philadelphia-Chromosoms mit der Bildung des Proteins BCR-Abl verstanden. Es zeigte sich, dass durch chromosomale Umlagerung das Gen der Abelson-Tyrosinkinase (Abl) auf Chromosom 9 mit der Breakpoint Cluster Region (BCR) auf Chromsom 22 fusioniert wird. Das nach der Proteinsynthese entstehende Fusionsprotein BCR-Abl ist in 95 % aller Patienten mit CML nachweisbar. Offenbar wirkt dieses als molekularer Schalter und bewirkt eine unkontrollierte Zellteilung. Das Einschleusen dieser Translokation in transgene Mäuse, die daraufhin eine Leukämie entwickeln, beweist, dass es sich hierbei um ein onkogenes Ereignis handelt.
Was macht eine Protein-Kinase? Wie kann ein einzelnes Enzym eine so verheerende Krankheit auslösen? Und wie kann dieses Wissen zur Therapie der Erkrankung eingesetzt werden?
Signalwege verstehen und blockieren
Protein-Kinasen bilden eine große Gruppe von eng miteinander verwandten Enzymen, die Adenosin-Triphosphat (ATP) binden und von diesem eine Phosphatgruppe auf einen bestimmten Aminosäure-Rest eines Substratproteins übertragen, wodurch eine Signalkaskade angestoßen wird. Tyrosinkinasen (Abbildung 1) haben deshalb eine Bindetasche für ATP und eine Interaktionsdomäne für die Erkennung des Proteins, auf das eine Phosphatgruppe übertragen wird. In der ATP Bindungstasche ist insbesondere der sogenannte „Gatekeeper-Residue“ für die Funktion einer Kinase wichtig, da er ein ATP-Molekül so positioniert, dass eine Phosphatgruppenübertragung ermöglicht wird. Bei der Abelson-Kinase (Abl) spielen mehrere Domänen bei der Regulation ihrer Aktivität eine Rolle: die Src-Homologiedomänen SH2 und SH3, die Aktivierungsschleife, die das aktive Zentrum für das Substrat zugänglich macht oder versperrt, und die regulatorische N-Schleife (siehe Abbildung 1). Diese Selbsthemmung geht durch die Bildung des chimären Fusionsproteins BCR-Abl bei der CML verloren, da Tyr412 durch die fehlende N-Schleife dauerhaft reaktiv ist.
Es resultiert eine zu hohe Aktivität und eine geringere Substratspezifität, wodurch eine Phosphatgruppe auch auf andere Proteine in der Zelle übertragen wird und diese dadurch aktiviert werden und letztendlich eine unkontrollierte Zellteilung bewirken. BCR-Abl ist zudem autokatalytisch aktiv,
es phosphoryliert also bestimmte Tyrosin-Reste in seiner eigenen Peptidstruktur und aktiviert sich dadurch selbst.
Die Aktuelle-Wochenschau© der GDCh – Fachgruppe Biochemie 47/2013
Abbildung 1: Die Phosphorylierungsreaktion der Tyrosinkinase und der Aufbau der Kinase c-Abl. Die Aktivierungsschleife steuert die Funktion der Kinase, Tyr412 ist katalytisch aktiv. Myristinsäure (blau) dient der Membranverankerung, SH2 und SH3 interagieren mit dem Substratprotein. Die N-Schleife reguliert die Kinase-Aktivität (Abbildung auf Basis des PDB-Eintrags 1IEP erstellt).
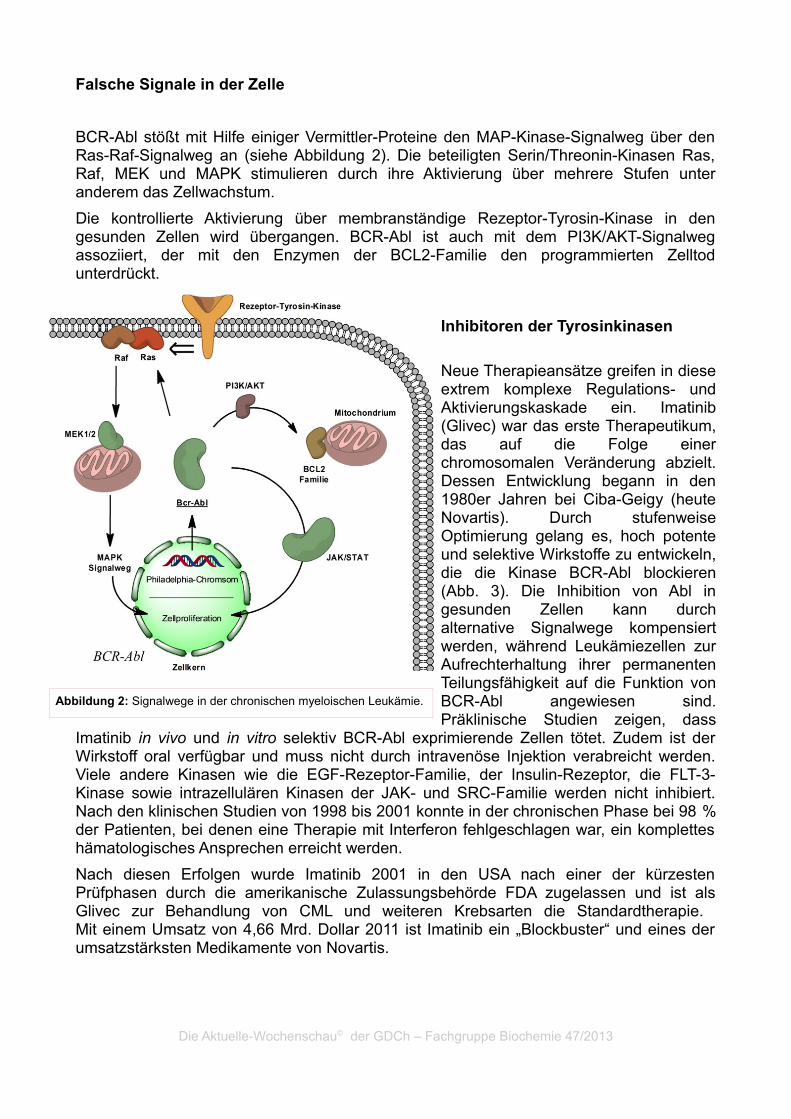
Falsche Signale in der Zelle
BCR-Abl stößt mit Hilfe einiger Vermittler-Proteine den MAP-Kinase-Signalweg über den Ras-Raf-Signalweg an (siehe Abbildung 2). Die beteiligten Serin/Threonin-Kinasen Ras, Raf, MEK und MAPK stimulieren durch ihre Aktivierung über mehrere Stufen unter anderem das Zellwachstum.
Die kontrollierte Aktivierung über membranständige Rezeptor-Tyrosin-Kinase in den gesunden Zellen wird übergangen. BCR-Abl ist auch mit dem PI3K/AKT-Signalweg assoziiert, der mit den Enzymen der BCL2-Familie den programmierten Zelltod unterdrückt.
Inhibitoren der Tyrosinkinasen
Neue Therapieansätze greifen in diese extrem komplexe Regulations- und Aktivierungskaskade ein. Imatinib (Glivec) war das erste Therapeutikum, das auf die Folge einer chromosomalen Veränderung abzielt. Dessen Entwicklung begann in den 1980er Jahren bei Ciba-Geigy (heute Novartis). Durch stufenweise Optimierung gelang es, hoch potente und selektive Wirkstoffe zu entwickeln, die die Kinase BCR-Abl blockieren (Abb. 3). Die Inhibition von Abl in gesunden Zellen kann durch alternative Signalwege kompensiert werden, während Leukämiezellen zur Aufrechterhaltung ihrer permanenten Teilungsfähigkeit auf die Funktion von BCR-Abl angewiesen sind. Präklinische Studien zeigen, dass
Imatinib in vivo und in vitro selektiv BCR-Abl exprimierende Zellen tötet. Zudem ist der Wirkstoff oral verfügbar und muss nicht durch intravenöse Injektion verabreicht werden. Viele andere Kinasen wie die EGF-Rezeptor-Familie, der Insulin-Rezeptor, die FLT-3-Kinase sowie intrazellulären Kinasen der JAK- und SRC-Familie werden nicht inhibiert. Nach den klinischen Studien von 1998 bis 2001 konnte in der chronischen Phase bei 98 % der Patienten, bei denen eine Therapie mit Interferon fehlgeschlagen war, ein komplettes hämatologisches Ansprechen erreicht werden.
Nach diesen Erfolgen wurde Imatinib 2001 in den USA nach einer der kürzesten Prüfphasen durch die amerikanische Zulassungsbehörde FDA zugelassen und ist als Glivec zur Behandlung von CML und weiteren Krebsarten die Standardtherapie. Mit einem Umsatz von 4,66 Mrd. Dollar 2011 ist Imatinib ein „Blockbuster“ und eines der umsatzstärksten Medikamente von Novartis.
Die Aktuelle-Wochenschau© der GDCh – Fachgruppe Biochemie 47/2013
Abbildung 2: Signalwege in der chronischen myeloischen Leukämie.

Resistenzen überwinden
Bei 25 % der Patienten in der fortgeschrittenen Phase treten nach Behandlung mit Imatinib Resistenzen auf, so dass Imatinib BCR-Abl nicht mehr blockiert. Die Ursache sind zufällig entstandene Mutationen im BCR-ABl Gen, die dazu führen, dass das aktive Zentrum etwas anders aufgebaut ist und Imatinib dort nicht mehr gut bindet. Die daraufhin entwickelten Imatinib-Derivate Nilotinib und Dasatinib (Abbildung 3) binden mit 300facher Affinität an BCR-Abl und können Resistenzen somit überwinden. Die Ausnahme ist die Mutation des Türsteher-Restes 315 der Kinase-Domäne, wobei Threonin durch Isoleucin ausgetauscht wird. Die Röntgenkristallstruktur der c-Abl Kinase im Komplex mit Imatinib zeigt wieso: Threonin bildet eine Wasserstoffbrücke mit Imatinib
aus. Da Isoleucin größer als Threonin ist und in die Bindungstasche ragt, kann sich Imatinib nicht mehr positionieren und die Affinität sinkt. Kinase-Aktivität und ATP-Bindungskapazität bleiben jedoch erhalten (Abbildung 4).
Gegen die CML mit Mutation des Türsteher-Restes ist noch keine Therapie verfügbar. Ein 2010 veröffentlichter neuer Ansatz der Kombination von Inhibitoren der ATP-Bindetasche mit sogenannten allosterischen Modulatoren in der Bindungstasche der Myristinsäure (GNF-2) ermöglichten jedoch erstmals eine wirksame Inhibition der Enzymaktivität (siehe Abbildung 4). Studien in vivo zeigten einen signifikanten Tumorrückgang bei T315I positiven Mäusen. Die Kokristallisation von Imatinib mit GNF-2 zeigten die hydrophobe Wechselwirkung von GNF-2 in der Myristoyl-Bindetasche. Die Anlagerung des Modulators induziert eine Drehung der benachbarten α-Helix um 90 ° und zwingt dadurch die SH3-Domäne in eine Position, die zur Schließung der ATP-Bindungstasche führt und die Bindung von Imatinib ermöglicht.
Ausgehend von dieser Entdeckung wird intensiv an den Struktur-Aktivität-Beziehungen und dem Aufbau von Screening-Methoden zur Durchmusterung großer Bibliotheken von potenziellen allosterischen Modulatoren geforscht.
Zusammenfassung und Ausblick
Imatinib stellt mit dem Konzept der zielgerichteten Krebstherapie einen Paradigmenwechsel dar. Die genetische Analyse zur Aufklärung der molekularen Pathogenese erlaubt die zielgerichtete und nebenwirkungsarme Therapie einer Krankheit. Der Erfolg kann mit dem Wissen um das Krebsgenom auf weitere maligne Erkrankungen übertragen werden. Dennoch zeigt Imatinib bei vielen Krebsarten keinen therapeutischen Effekt, obwohl diese auch Kinasen exprimieren, an die Imatinib binden kann. Die Krebszellen sind jedoch nicht von diesen Kinasen abhängig und ihre Funktionen können von anderen Kinasen übernommen werden.
Die Aktuelle-Wochenschau© der GDCh – Fachgruppe Biochemie 47/2013
Abbildung 3: a) Inhibition des BCR-Abl Fusionsproteins. In der geschlossenen Konformation (rot) ist das Enzym inaktiv, die Bindung von Glivec stabilisiert diese Konformation. Die offene Konformation (grün) führt zur Bindung von ATP und die folgende Signalkaskade zur Zellproliferation.b) Inhibitoren in der ATP-Bindungstasche.c) Allosterische Modulatoren der Myristoyl-Bindetasche.

Das Zusammenspiel von Imatinib, Dasatinib oder Nilotinib mit anderen Wirkstoffen, die am selben Enzym aber an einer anderen Stelle angreifen und dessen Raumstruktur dadurch verändern, konnte Resistenzen gegen einen Wirkstoff alleine überwinden und stellt somit ein neues Therapiekonzept dar.
Wenn es möglich ist, solche Konzepte auf weitere Tumorarten zu übertragen, besteht bei manchen Tumorerkrankungen die berechtigte Hoffnung, dass es durch neue Wirkstoffe und Wirkstoffkombinationen gelingt, das Fortschreiten der Erkrankung soweit zu verlangsamen, dass Krebs zu einer Krankheit wird, mit der es sich lange leben lässt.
In Kürze
• Imatinib ist das erste Medikament das zielgerichtet auf ein abnormes Protein wirkt und begründet mit seinen Heilungserfolgen einen Paradigmenwechsel in der Krebstherapie.
• Mutationen in der Krebszelle führen jedoch zu Resistenzen, die nur Wirkstoffe der nächsten Generation überwinden können.
• Neue Forschungsarbeiten ermöglichen die Überwindung dieser Resistenz durch die Kombination von Wirkstoffen, die an verschiedenen Stellen im Zielprotein angreifen.
Die Aktuelle-Wochenschau© der GDCh – Fachgruppe Biochemie 47/2013
Abbildung 4: Kombination von allosterischen Modulatoren und ATP-kompetitiven Inhibitoren stabilisiert die inaktive Konformation der Kinase. Imatinib (grün) bildet Wasserstoffbrückenbindungen mit Thr315, Met318, Asp381 und Glu286 aus während GNF-2 in der Bindetasche der Myristinsäure liegt (Abbildung auf Basis des PDB-Eintrag 3K5V erstellt).

Kontakt: Schlauer Fuchs
Der Artikel wurde im Rahmen des Studienprojektes HighChem des Fachbereichs Chemie der Technischen Universität Darmstadt verfasst (s. Woche 2).
Der Autor, Dennis Bensinger, ist Studierender im Masterstudiengang Chemie
(E-Mail: [email protected]).
Wissenschaftliche Betreuung: Prof. Dr. Harald Kolmar (E-Mail: [email protected]).
Unsere Schlaue-Fuchs-Frage zu diesem Beitrag lautete:
Was ist BCR-Abl? Wie erklärt sich die Abkürzung?
Literatur:
[1] T. Boveri, Über mehrpolige Mitosen als Mittel zur Analyse des Zellkerns. Verhandlungen der physicalisch-medizinischen Gesselschaft zu Würzburg. Neu Folge 35, 67-90 (1902).
[2] Science 1960, 132, 1497.
[3] B. J. Druker, S. Tamura, E. Buchdunger, S. Ohno, G. M. Segal, S. Fanning, J. Zimmermann, N. B. Lydon, Nature Medicine 1996, 2, 6.
[4] C. Wagener, O. Müller, Molekulare Onkologie, 3 ed., Georg Thieme Verlag KG, Stuttgart, Germany, 2010.
[5] H. Pluk, K. Dorey, G. Superti-Furga, Cell 2002, 108, 13.
[6] M. E. Gorre, M. Mohammed, K. Ellwood, N. Hsu, R. Paquette, P. N. Rao, C. L. Sawyers, Science 2001, 293, 5.
[7] B. Nagar, W. G. Bornmann, P. Pellicena, T. Schindler, D. R. Veach, W. T. Miller, B. Clarkson, J. Kuriyan, Cancer Research 2002, 62, 8.
[8] J. Zhang, F. J. Adrian, W. Jahnke, S. W. Cowan-Jacob, A. G. Li, R. E. Iacob, T. Sim, J. Powers, C. Dierks, F. Sun, G. R. Guo, Q. Ding, B. Okram, Y. Choi, A. Wojciechowski, X. Deng, G. Liu, G. Fendrich, A. Strauss, N. Vajpai, S. Grzesiek, T. Tuntland, Y. Liu, B. Bursulaya, M. Azam, P. W. Manley, J. R. Engen, G. Q. Daley, M. Warmuth, N. S. Gray, Nature 2010, 463, 501-506.
Die Aktuelle-Wochenschau© der GDCh – Fachgruppe Biochemie 47/2013




