Embed Size (px)
Citation preview

Anionenanalytik mit der On-line-Kopplung
von Ionenchromatographie und
induktiv gekoppelter Plasma-Massenspektrometrie
Dissertation
zur
Erlangung des Doktorgrades
der Naturwissenschaften
(Dr. rer. nat.)
dem
Fachbereich Chemie
der Philipps-Universität Marburg
vorgelegt von
Thomas Eickhorst
aus
Osterode am Harz
Marburg/Lahn 2005

Vom Fachbereich Chemie der Philipps-Universität Marburg als Dissertation am 31.10.2005
angenommen.
Erstgutachter: Prof. Dr. Andreas Seubert
Zweitgutachter: Prof. Dr. Wolfgang Ensinger
Tag der mündlichen Prüfung am: 02.11.2005

Die vorliegende Dissertation wurde in der Zeit von März 2002 bis September 2005 am Institut
für Anorganische und Analytische Chemie der Philipps-Universität Marburg unter Leitung
von Herrn Prof. Dr. Andreas Seubert angefertigt.
Mein besonderer Dank gilt meinem Doktorvater Prof. Dr. Andreas Seubert, der mir nicht nur
die Möglichkeit gegeben hat, ein hochinteressantes Forschungsthema zu bearbeiten, sondern
der mir auch durch stete Gesprächsbereitschaft und konstruktive Ratschläge zur Seite stand.
Für die Übernahme des Korreferats danke ich Prof. Dr. Wolfgang Ensinger.
Allen Mitgliedern des Arbeitskreises danke ich für die gute Zusammenarbeit und das ange-
nehme Arbeitsklima. Hervorheben möchte ich die lange gemeinsame Zeit in der Analytischen
Chemie mit Herrn Sven Holland. Die Zeit hat mich auf meinem Weg in dieser Fachrichtung
sehr bestärkt. Außerdem bin ich ihm, genauso wie Frau Sandra Schütze und Herrn Michael
Raskop, zu Dank verpflichtet für die Synthese von stationären Phasen für die Ionenchromato-
graphie, ohne die meine Forschung nicht möglich gewesen wäre. Ferner danke ich für ihre
Insider-Tipps während meiner praktischen Arbeit bzw. für das Korrekturlesen meiner Disser-
tation. Für Korrekturen bin ich darüber hinaus Herrn Oliver Happel, Herrn Andreas Grimm
und nicht zuletzt Frau Birgit Barthelmes dankbar.
Für die liebevolle Unterstützung auf vielerlei Art danke ich meiner Freundin Astrid.
Herrn Roland Fleischer möchte ich meine Anerkennung aussprechen, da er mir mit seinem
überragenden technischen Wissen über ICP-MS-Geräte oftmals entweder tatkräftig vor Ort
oder telefonisch bei Geräteproblemen geholfen hat.
Bei den Mitarbeitern der Haushandwerker- der Feinmechanik- und der Elektronikwerkstatt
des Fachbereichs Chemie der Universität Marburg bedanke ich mich für die jederzeit freund-
liche Zusammenarbeit bei Umbau- oder Reparaturmaßnahmen.

Meinen Eltern

Kurzzusammenfassung
Thomas Eickhorst
Anionenanalytik mit der On-line-Kopplung von Ionenchromatographie und induktiv
gekoppelter Plasma-Massenspektrometrie
Stichworte: Ionenchromatographie, ICP-MS, Anionen, stationäre Phasen,
Germaniumdioxid, interner Standard
Die On-line-Kopplung von Ionenchromatographie und ICP-MS wurde zur Spurenanalyse
anorganischer Anionen in wässriger Lösung verwendet. Es gelang eine Methode zu
entwickeln, mit der sich bis zu 15 Anionen gleichzeitig innerhalb von 35 min bestimmen
ließen. Verschiedene stationäre Phasen wurden untersucht und ihre Trenneigenschaften
miteinander verglichen. Es kamen Latex-Anionenaustauscher, gepfropfte und oberflächen-
funktionalisierte Trennmaterialien zum Einsatz. Die Praxistauglichkeit der IC-ICP-MS wurde
durch die Analyse von Mineralwasserproben auf die Anionen Bromid, Bromat, Iodid und
Iodat demonstriert, für die Nachweisgrenzen von 0.1 µg L−1, 0.07 µg L−1, 0.15 µg L−1 und
0.05 µg L−1 erreicht wurden.
Des Weiteren wurde festgestellt, dass den verwendeten Ammoniumnitrat-Eluenten
Germaniumdioxid als interner Standard zugegeben werden kann, und dass dieses sich
ausgezeichnet zur Signalkorrektur von Brom- und Iodspezies eignet. Das kontinuierliche
Germaniumsignal ist sowohl zur Echtzeitkorrektur nicht-spezifischer Interferenzen, als auch
zur schnellen Systemdiagnose nutzbar. Es konnte gezeigt werden, dass das Signalverhältnis
von Analyt-zu-GeO2 für brom- und iodhaltige Analyten weitgehend unabhängig von ver-
änderten Messbedingungen ist, so dass damit eine über Wochen stabile Kalibration erhalten
werden kann. Dadurch wird eine Quantifizierung ohne zeitintensive tägliche Messung von
Kalibrationsreihen ermöglicht.

Abstract
Thomas Eickhorst
Analysis of Anions by On-line-Coupling Ion Chromatography and Inductively Coupled
Plasma Mass Spectrometry
Keywords: Ion Chromatography, ICP-MS, Anion Analysis, Stationary Phases,
Germanium Dioxide, Internal Standard
The on-line coupling of ion chromatography and ICP-MS was used for trace analysis of
inorganic anions in aqueous solution. A method for the simultaneous determination of up to
15 anions within 35 min was developed. Different stationary phases, – latex anion
exchangers, grafted and surface-functionalized materials, were investigated and their
separation characteristics were compared. The IC-ICP-MS system was successfully applied to
analyze bromide, bromate, iodide and iodate in mineral water samples achieving detection
limits of 0.1 µg L−1, 0.07 µg L−1, 0.15 µg L−1 and 0.05 µg L−1 respectively.
Germanium dioxide added directly to the aqueous ammonium nitrate eluent was found to be
well-suited as an internal standard for bromine and iodine species. The measured continuous
germanium signal serves for real-time correction of non-specific interferences and also as a
tool for system diagnostics. It was shown that the signal ratio of analyte-to-GeO2 for bromine-
and iodine-containing analytes remains unaltered to a large extent despite changed system
parameters. Thus a calibration with high long-term-stability over several weeks is made
possible allowing quantification without time-consuming daily measurement of calibration
series.

Inhaltsverzeichnis
- I -
Inhaltsverzeichnis
1 Einleitung und Zielsetzung ................................................................................................. 1
2 Theorie.................................................................................................................................. 7
2.1 Induktiv gekoppelte Plasma-Massenspektrometrie.................................................. 7 2.1.1 Einleitung und historische Entwicklung ............................................................. 7 2.1.2 Funktionsweise und instrumentelle Grundlagen der ICP-MS ............................ 8 2.1.3 Interferenzen ..................................................................................................... 21
2.2 Ionenchromatographie .............................................................................................. 23 2.2.1 Einleitung .......................................................................................................... 23 2.2.2 Trennmechanismen ........................................................................................... 23 2.2.3 Aufbau eines Ionenchromatographen ............................................................... 26 2.2.4 Grundlegende chromatographische Kenngrößen.............................................. 27 2.2.5 Stationäre Phasen .............................................................................................. 32
2.3 Kopplungstechniken .................................................................................................. 33 2.3.1 Übersicht ........................................................................................................... 33 2.3.2 Die On-Line-Kopplung IC-ICP-MS ................................................................. 35
2.4 Analyten für die Anionen-IC-ICP-MS..................................................................... 36 2.4.1 Übersicht ........................................................................................................... 36 2.4.2 Wichtige analytspezifische Parameter .............................................................. 37 2.4.3 Polarisierbare Anionen...................................................................................... 41
3 Literaturübersicht: Anionenanalytik mit der IC-ICP-MS............................................ 48
4 Ergebnisse .......................................................................................................................... 54
4.1 Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS.................................................................................................. 54 4.1.1 Einleitung .......................................................................................................... 54 4.1.2 Latex-Anionenaustauscher................................................................................ 57 4.1.3 VBC-Latex-Anionenaustauscher ...................................................................... 59 4.1.4 GMA-Latex-Anionenaustauscher ..................................................................... 74 4.1.5 Oberflächenfunktionalisierte Anionenaustauscher ........................................... 80 4.1.6 Gepfropfte Anionenaustauscher........................................................................ 85 4.1.7 Zusammenfassender Vergleich der untersuchten stationären Phasen anhand der
relativen Retention ............................................................................................ 91

Inhaltsverzeichnis
- II -
4.2 Vergleich von Nitrat- und Carbonat/Bicarbonat-Eluenten ................................... 96
4.3 Nachweisgrenzen........................................................................................................ 99
4.4 Anwendungsbeispiele der IC-ICP-MS-Kopplung ................................................ 101 4.4.1 Analyse von Speisesalzen und Salz für Sole-Bäder auf Bromat..................... 101 4.4.2 Analyse von Mineralwässern auf Bromid, Bromat, Iodid und Iodat .............. 106
4.5 Germaniumdioxid als interner Standard .............................................................. 109 4.5.1 Methode des internen Standards ..................................................................... 109 4.5.2 Auswahlkriterien für interne Standards bei der IC-ICP-MS........................... 109 4.5.3 Transiente Messungen und zeitliche Aspekte der IS-Korrektur ..................... 112 4.5.4 Vergleich dreier potentieller ISs und Evaluierung der Ladung ...................... 115 4.5.5 Zuführungsmethoden des internen Standards bei transienten Messungen ..... 117 4.5.6 Germaniumdioxid zur Überwachung instrumenteller Probleme oder
nichtspezifischer Interferenzen ....................................................................... 117
4.6 Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse ..... 119 4.6.1 Berechnung des Rn-Werts ............................................................................... 119 4.6.2 Langzeitstabilität der Rn-Werte ....................................................................... 120 4.6.3 Weitere nützliche Kenngrößen zur Messwertüberwachung und Auswertung 124 4.6.4 Anwendungsbeispiele zur Quantifizierung mit Hilfe des Rn-Werts ............... 126
4.7 Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern ............................................................................................................... 128 4.7.1 Vergleich der IC-ICP-MS und der FI-ICP-MS............................................... 128 4.7.2 Rn-Abhängigkeit von den Tuningbedingungen: 1. IE des Tune-Elements ..... 131 4.7.3 Rn-Abhängigkeit von den Tuningbedingungen: Masse des Tune-Elements... 134 4.7.4 Ursachen zu hoher Rn-Werte der Iodspezies................................................... 134 4.7.5 Variation der Samplingtiefe ............................................................................ 137 4.7.6 Variation der Generatorleistung...................................................................... 139 4.7.7 Variation der Eluentkonzentration .................................................................. 143 4.7.8 Variation der Flussrate .................................................................................... 144 4.7.9 Variation der Analytkonzentration – Ermittlung des linearen Bereichs ......... 145 4.7.10 Variation von Quadrupoleinstellungen ........................................................... 147 4.7.11 Variation der Zerstäuber ................................................................................. 154 4.7.12 Zusammenfassung: Abhängigkeiten der Rn-Werte ......................................... 155
5 Zusammenfassung und Ausblick ................................................................................... 156

Inhaltsverzeichnis
- III -
6 Anhang.............................................................................................................................. 162
6.1 Instrumentelles ......................................................................................................... 162 6.1.1 Die IC-ICP-MS-Kopplung .............................................................................. 162 6.1.2 Das FI-ICP-MS-System .................................................................................. 164 6.1.3 Manuelle Massenkalibration ........................................................................... 167 6.1.4 Verwendete Geräte.......................................................................................... 169
6.2 Verwendete Chemikalien ........................................................................................ 171
6.3 Arbeitsvorschriften .................................................................................................. 172 6.3.1 Ansetzen von Bromid-, Bromat-, Iodid-, und Iodatstandards......................... 172 6.3.2 Probenlösungen/Probenvorbereitung .............................................................. 173 6.3.3 Ansetzen eines Ammoniumnitrat-Eluents mit Germaniumdioxid als IS........ 174 6.3.4 Formular zur Protokollierung der Betriebsbedingungen des ICP-MS............ 175
7 Literaturverzeichnis ........................................................................................................ 176
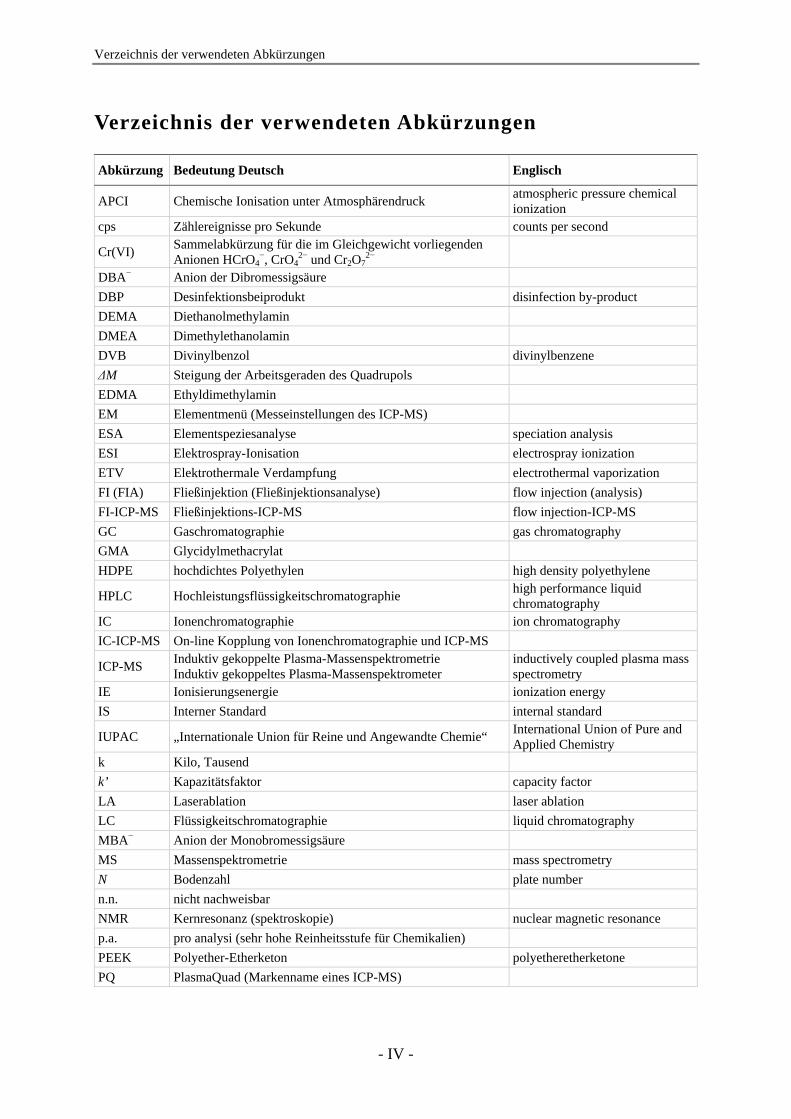
Verzeichnis der verwendeten Abkürzungen
- IV -
Verzeichnis der verwendeten Abkürzungen
Abkürzung Bedeutung Deutsch Englisch
APCI Chemische Ionisation unter Atmosphärendruck atmospheric pressure chemical ionization
cps Zählereignisse pro Sekunde counts per second
Cr(VI) Sammelabkürzung für die im Gleichgewicht vorliegenden Anionen HCrO4
−, CrO42− und Cr2O7
2−
DBA− Anion der Dibromessigsäure DBP Desinfektionsbeiprodukt disinfection by-product DEMA Diethanolmethylamin DMEA Dimethylethanolamin DVB Divinylbenzol divinylbenzene ∆M Steigung der Arbeitsgeraden des Quadrupols EDMA Ethyldimethylamin EM Elementmenü (Messeinstellungen des ICP-MS) ESA Elementspeziesanalyse speciation analysis ESI Elektrospray-Ionisation electrospray ionization ETV Elektrothermale Verdampfung electrothermal vaporization FI (FIA) Fließinjektion (Fließinjektionsanalyse) flow injection (analysis) FI-ICP-MS Fließinjektions-ICP-MS flow injection-ICP-MS GC Gaschromatographie gas chromatography GMA Glycidylmethacrylat HDPE hochdichtes Polyethylen high density polyethylene
HPLC Hochleistungsflüssigkeitschromatographie high performance liquid chromatography
IC Ionenchromatographie ion chromatography IC-ICP-MS On-line Kopplung von Ionenchromatographie und ICP-MS
ICP-MS Induktiv gekoppelte Plasma-Massenspektrometrie Induktiv gekoppeltes Plasma-Massenspektrometer
inductively coupled plasma mass spectrometry
IE Ionisierungsenergie ionization energy IS Interner Standard internal standard
IUPAC „Internationale Union für Reine und Angewandte Chemie“ International Union of Pure and Applied Chemistry
k Kilo, Tausend k’ Kapazitätsfaktor capacity factor LA Laserablation laser ablation LC Flüssigkeitschromatographie liquid chromatography MBA− Anion der Monobromessigsäure MS Massenspektrometrie mass spectrometry N Bodenzahl plate number n.n. nicht nachweisbar NMR Kernresonanz (spektroskopie) nuclear magnetic resonance p.a. pro analysi (sehr hohe Reinheitsstufe für Chemikalien) PEEK Polyether-Etherketon polyetheretherketone PQ PlasmaQuad (Markenname eines ICP-MS)
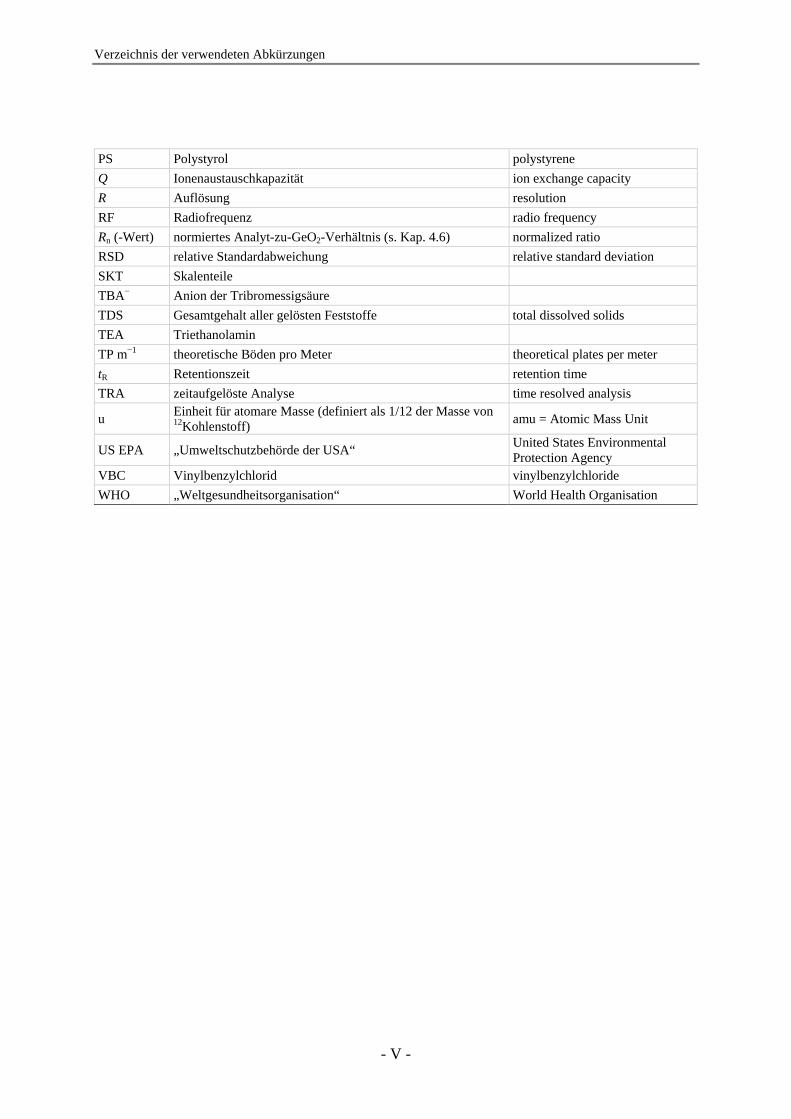
Verzeichnis der verwendeten Abkürzungen
- V -
PS Polystyrol polystyrene Q Ionenaustauschkapazität ion exchange capacity R Auflösung resolution RF Radiofrequenz radio frequency Rn (-Wert) normiertes Analyt-zu-GeO2-Verhältnis (s. Kap. 4.6) normalized ratio RSD relative Standardabweichung relative standard deviation SKT Skalenteile TBA− Anion der Tribromessigsäure TDS Gesamtgehalt aller gelösten Feststoffe total dissolved solids TEA Triethanolamin TP m−1 theoretische Böden pro Meter theoretical plates per meter tR Retentionszeit retention time TRA zeitaufgelöste Analyse time resolved analysis
u Einheit für atomare Masse (definiert als 1/12 der Masse von 12Kohlenstoff) amu = Atomic Mass Unit
US EPA „Umweltschutzbehörde der USA“ United States Environmental Protection Agency
VBC Vinylbenzylchlorid vinylbenzylchloride WHO „Weltgesundheitsorganisation“ World Health Organisation

Einleitung und Zielsetzung
- 1 -
1 Einleitung und Zielsetzung
Die Belastung der Umwelt durch verschiedene organische und anorganische Schadstoffe
anthropogenen Ursprungs stellt in der modernen Industriegesellschaft ein drängendes Problem
dar. Um wirkungsvolle Maßnahmen zum Schutz der Umwelt ergreifen zu können, ist das
Verständnis biochemischer und geochemischer Stoffkreisläufe von großer Wichtigkeit.
Hierbei spielt die anorganische Spurenanalytik eine Schlüsselrolle, und es werden immer
effizientere Verfahren zur Detektion und Quantifizierung benötigt.
Erst in den letzten zwei Jahrzehnten wurde in der Forschung verstärkt erkannt, dass die
Bestimmung der Gesamtkonzentration eines chemischen Elements allein nicht ausreicht, um
sinnvolle Aussagen über die Auswirkungen auf ökologische Systeme und biologische
Organismen treffen zu können. Sowohl die Toxizität, als auch die Nützlichkeit eines Elements
hängen neben der Konzentration von dessen Mobilität und Bioverfügbarkeit ab. Die letzt-
genannten Faktoren werden maßgeblich durch die Vorkommensform eines Elements, die so
genannte Elementspezies bestimmt. Verschiedene Spezies eines Elements unterscheiden sich
durch ihre Molekül- oder Komplexstruktur oder durch ihre Oxidationsstufe. Die Identifi-
zierung und Quantifizierung einzelner Elementspezies in einer Probe wird als Element-
speziesanalyse (ESA) bezeichnet. Erst durch diese wird eine sinnvolle Risiko/Nutzen-
Abschätzung der Elemente in der Umwelt oder eine sichere Qualitätskontrolle von Nahrungs-
und Pharmaprodukten möglich. Aus diesem Grund hat sich die ESA zu einem der wichtigsten
Forschungsgebiete der anorganischen Analytik entwickelt und in den letzten Jahren wurde
eine Vielzahl an Reviewartikeln [1-7] und Monographien [8, 9] veröffentlicht.
Die am häufigsten angewendete Strategie zur ESA besteht in der Kombination von Trenn-
methoden mit elementselektiven oder molekülselektiven Detektionssystemen und bildet die
Basis dieses Forschungszweigs [1]. Eine erfolgreiche Methode stellt die Kopplung der Ionen-
chromatographie (IC) mit der induktiv gekoppelten Plasma-Massenspektrometrie (ICP-MS)
dar. Die moderne IC wurde 1975 von Small, Stevens und Baumann eingeführt [10] und hat
sich für anorganische Anionen in Wässern zur Hauptanalysenmethode entwickelt [11-13].
Obwohl die ersten ICP-MS-Geräte erst Mitte der 1980er Jahre kommerziell erhältlich waren,
ist die ICP-MS mittlerweile zur leistungsfähigsten Methode der Elementanalytik avanciert.

Einleitung und Zielsetzung
- 2 -
Die On-line-Kopplung IC-ICP-MS vereint die Vorteile der beiden Einzelmethoden: Mit Hilfe
der IC werden ionische Spezies nach Ladung und Größe getrennt, – somit ist sie spezies-
selektiv. Die Vorteile der ICP-MS im Einzelnen sind die Fähigkeit zur Detektion fast aller
Elemente, die hohe Empfindlichkeit und sehr niedrigen Nachweisgrenzen, die Isotopenunter-
scheidung, der große lineare Arbeitsbereich und die relativ einfachen Massenspektren mit
wenigen spektralen Interferenzen. Auf Grund der Elementselektivität der Detektion ist es bei
der IC-ICP-MS unproblematisch, wenn sich chromatographische Peaks von Spezies
verschiedener Elemente überlagern. Eine Auflösung aller Peaks ist nicht zwingend notwendig
und viele Messungen können daher schneller durchgeführt werden. Zudem ist es möglich,
mehr Analyten gleichzeitig zu bestimmen. Darüber hinaus sind unbekannte Spezies durch
Retentionszeitvergleich zusammen mit der zusätzlichen Elementinformation leichter zu
identifizieren.
Die Unterscheidung der Elementspezies ist von großer Bedeutung, was anhand der folgenden,
für diese Dissertation wichtigen Beispiele verdeutlicht werden soll:
• Chrom (III) ist essentiell und z.B. Bestandteil des Glucosemetabolismus. Chrom (VI)
ist dagegen hochtoxisch und kanzerogen [14].
• Sowohl anorganisches Arsen (III) als auch Arsen (V) sind toxisch. Methylierte
Arsenspezies zeigen bereits geringere Toxizität, und Arsenobetain, das in hoher
Konzentration in Meereslebewesen vorkommt, ist überhaupt nicht toxisch, da es nicht
metabolisiert wird [15, 16].
• Bromid ist für den menschlichen Organismus unschädlich, Bromat hingegen wird
schon in kleinsten Konzentrationen als kanzerogen angesehen. Auch kleine organische
bromhaltige Moleküle, die z.B. bei der Desinfektion von Trinkwasser entstehen, sind
aus toxikologischer Sicht bedenklich, da neueste Forschungsergebnisse darauf
hinweisen, dass sie noch krebserregender sind als ihre chlorierten Analoga [17].
• Iod wird im menschlichen Körper zum Aufbau von Schilddrüsenhormonen benötigt,
so dass ein Iodmangel eine Vielzahl von Krankheiten zur Folge hat. Liegt Iod aber in
Trinkwasser als Iodid vor, so kann dies zu einer Verschlechterung des Geruchs oder
Geschmacks des Wassers führen. Die Spezies Iodat zeigt hingegen nicht diese
negativen Eigenschaften [18].
• Die Chlorspezies Perchlorat kann bereits in niedriger Konzentration die Hormon-
produktion der Schilddrüse beeinträchtigen. Eine Kontamination durch Ammonium-

Einleitung und Zielsetzung
- 3 -
perchlorat ist ein großes Problem in vielen Gegenden, in denen es im Zusammenhang
mit seiner Verwendung als Sprengstoff oder Raketentreibstoff ins Grundwasser
gelangt ist. Die US Environmental Protection Agency (US EPA) hat im Februar 2005
die offiziell als unschädlich angesehene Referenzdosis für Perchlorat auf
0.0007 mg kg−1 Tag−1 festgelegt [19]. Daraus ergibt sich ein empfohlener Trink-
wassergrenzwert von 24.5 µg L−1. Zur Kontrolle des Grenzwerts sind geeignete
Analysenverfahren erforderlich, die Perchlorat neben einer Fülle von anderen
Chlorspezies bestimmen können.
In den letzten Jahren ist insbesondere die ESA in Trinkwasser in das Forschungsinteresse
gerückt. Um Trinkwasser für den Menschen aufzubereiten, ist es unerlässlich, Bakterien,
Viren und andere pathogene Mikroorganismen mit Hilfe von Desinfektionsmitteln unschäd-
lich zu machen. Hierzu wird schon seit Anfang des 20. Jahrhunderts meistens das effektive
und einfach anzuwendende Desinfektionsmittel Chlor benutzt [20]. In den 1970er Jahren
wurde allerdings erkannt, dass durch Chlorung neben einem schlechten Geschmack und üblen
Geruch auch halogenierte Kohlenwasserstoffe erzeugt werden [21, 22], von denen sich die
Trihalogenmethane als besonders kanzerogen herausstellten.
Daraufhin traten alternative Desinfektionsmittel wie Chlordioxid, Hypochlorige Säure,
Chloramin und Ozon in den Mittelpunkt wissenschaftlicher Untersuchungen, da sie alle
weniger Trihalogenmethane bilden als Chlor [23]. Am wenigsten Halogenkohlenwasserstoffe
als Nebenprodukte entstehen bei der Ozonierung, die deswegen zunächst als „perfekte“
Desinfektionstechnologie betrachtet wurde. Dies änderte sich, nachdem man herausfand, dass
bei ungünstiger Reaktionsführung das in Wasser enthaltene Bromid durch Ozon zu Bromat
oxidiert wird. Auf Grund toxikologischer Studien [24-26] ist Bromat sowohl von der World
Health Organisation (WHO) [27], als auch von der US EPA [28] als potentiell krebserregend
eingestuft worden. Schon eine Bromatkonzentration von 0.05 µg L−1 steht im Verdacht,
Nierenkrebs auslösen zu können. Schätzungen der US EPA zu Folge liegt das Krebsrisiko für
Menschen, die ihr Leben lang einer Konzentration von 5 µg L−1 Bromat im Trinkwasser
ausgesetzt sind, bei 1:10000 [24, 28].
Es hat sich gezeigt, dass mit den in großen Trinkwasseraufbereitungsanlagen gebräuchlichen
Desinfektionsmitteln, – Chlor, Chlordioxid, Ozon und Chloramin –, teilweise unterschiedliche
Desinfektionsbeiprodukte (DBPs) in veränderlichen Anteilen entstehen. Allein für die
Chlorung von Trinkwasser wurden mittlerweile mehr als 500 DBPs veröffentlicht, von denen

Einleitung und Zielsetzung
- 4 -
aber bisher nur etwa 40 % identifiziert und die wenigsten quantifiziert, geschweige denn
toxikologisch beurteilt werden konnten [29]. In Trinkwasser können DBPs aber selbst in
niedrigen µg L−1-Konzentrationen ein signifikantes Gesundheitsrisiko darstellen. Angesichts
der noch bevorstehenden sehr umfangreichen analytischen Aufgabe werden weltweit
Informationen über DBPs zusammengetragen und Prioritätenlisten der Analyten aufge-
stellt [30]. Es wird angestrebt, bei der Wasserdesinfektion die Entstehung gefährlicher
Substanzen, die sich als toxisch und/oder krebserregend herausgestellt haben, durch geeignete
Maßnahmen zu minimieren. Zur Identifizierung, Quantifizierung und laufenden Kontrolle
werden neue, leistungsfähige analytische Methoden benötigt. Eine solche stellt die Kopplung
von Ionenchromatographie und ICP-MS dar.
Zielsetzung
Ziel dieser Arbeit ist es, auf verschiedene Weise die Anionenchromatographie mit ICP-MS als
Detektor zu optimieren. Mit Hilfe der IC-ICP-MS-Kopplung soll eine Methode entwickelt
werden, mit der diverse DBPs sowie eine Reihe weiterer Anionen gleichzeitig gemessen
werden können. Bei den DBPs handelt es sich um Bromat, Chlorit, Chlorat und die drei
Bromessigsäuren. Da diese Spezies anionisch vorliegen, können sie mit Hilfe der Ionen-
chromatographie separiert und anschließend mittels ICP-MS elementselektiv detektiert
werden. Weitere Analyten sind Chlorid, Bromid, Iodid, Iodat, Phosphat, Arsenat, Chromat,
Thiocyanat und Perchlorat. Für alle genannten 15 Spezies müssen die geeigneten Proben-
vorbereitungsschritte sowie Messbedingungen gefunden werden.
Die Untersuchung von stationären Phasen für die IC unter Einsatz eines Ammoniumnitrat-
Elutionssystem ist ein wichtiger Bestandteil dieser Dissertation. Bisher werden in den meisten
veröffentlichten Arbeiten zur Elementspeziesanalyse nur kommerzielle Trennsäulen ver-
wendet, ohne genauer auf die Eigenschaften der verschiedenen Trennmaterialien einzugehen.
Dies lässt sich damit begründen, dass sich die Herstellerangaben zum chemischen Aufbau der
stationären Phasen oft auf wenig aussagekräftige Beschreibungen wie „pellikularer Anionen-
austauscher auf Basis von Polystyrol/Divinylbenzol mit quartären Ammoniumgruppen“
beschränken. Eine Aussage dieser Art trifft aber im Prinzip auf die Mehrheit aller modernen
Anionenaustauscher zu. Selbst wenn eine hohe Hydrophilie des Anionenaustauschers als
besonderes Attribut angegeben ist, wird der dafür verantwortliche chemische und physika-
lische Aufbau des Materials nicht genau beschrieben. Spezielle verwendete Monomere oder

Einleitung und Zielsetzung
- 5 -
die Substituenten an den Austauscherfunktionen werden zumeist geheim gehalten. Dies hat
zur Folge, dass besondere Vor- oder Nachteile bei der Trennung bestimmter Spezies nicht
erklärt und deswegen nicht die optimalen Ergebnisse erzielt werden können.
Für die ionenchromatographischen Trennungen im Rahmen dieser Dissertation wurden daher
ausschließlich stationäre Phasen verwendet, die im Arbeitskreis von Prof. Dr. A. Seubert
hergestellt wurden. Die Materialien sollen möglichst genau charakterisiert werden, so dass
ihre Trenneigenschaften, wie z.B. eine spezielle Selektivität oder die Effizienz, sowohl auf
morphologische, als auch auf chemische Aufbauprinzipien zurückgeführt werden können. Mit
dem darauf basierenden Wissen sollen in Zukunft für spezielle Trennprobleme die besten
bereits vorhandenen Chromatographiesäulen eingesetzt oder neue, ideal angepasste stationäre
Phasen hergestellt werden können.
Da die ICP-MS eine kostenintensive Detektionsmethode ist, sollen möglichst viele Anionen-
spezies verschiedener Elemente simultan erfasst werden. Eine besondere Herausforderung
besteht darin, solche stationäre Phasen zu finden, die frühzeitig eluierende Anionen
ausreichend separieren können, aber unter den gleichen Bedingungen ein geringes Retentions-
vermögen für polarisierbare Anionen aufweisen. Dies sind die Voraussetzungen, um
möglichst kurze Analysenzeiten für die gleichzeitige Trennung aller Analyten bei iso-
kratischer Elution zu gewährleisten.
Bei quantitativen Analysen mit einem ICP-MS ist es üblich, Empfindlichkeitsschwankungen
über die Methode des internen Standards zu korrigieren. Indem Matrixeffekte und nicht-
spezifische Interferenzen, die z.B. eine Signaldrift hervorrufen, korrigiert werden, lässt sich
die Präzision der Messergebnisse erhöhen. Da die ESA mit vorgeschalteter chromato-
graphischer Trennung zeitintensiver ist als eine einfache Elementbestimmung, kommt dem
Einsatz eines internen Standards für die IC-ICP-MS-Kopplung eine besonders große Bedeu-
tung zu. Ein gut geeigneter interner Standard muss viele Voraussetzungen erfüllen, von denen
an dieser Stelle nur die wichtigsten genannt werden sollen: Aus ionenchromatographischen
Gesichtspunkten ist es vorteilhaft, wenn der interne Standard neutral aber trotzdem gut
wasserlöslich ist. Im ICP-MS sollte sich das zu detektierende Element ähnlich wie die Analyt-
elemente verhalten. Es gilt eine Verbindung mit entsprechenden Eigenschaften ausfindig zu
machen und ein nutzbringendes Potential unter Beweis zu stellen.

Einleitung und Zielsetzung
- 6 -
Ein weiteres Ziel im Rahmen dieser Dissertation ist die Entwicklung neuer Strategien für die
Datenauswertung. Es soll untersucht werden, wie stabil das Signalverhältnis bestimmter
Analyten zum verwendeten internen Standard ist. Solch ein unter veränderten Betriebs-
bedingungen stabiles Signalverhältnis lässt sich für eine einfache und schnelle Quanti-
fizierung nutzen.

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 7 -
2 Theorie
2.1 Induktiv gekoppelte Plasma-Massenspektrometrie
2.1.1 Einleitung und historische Entwicklung
Die Plasma-Massenspektrometrie unter Verwendung eines induktiv gekoppelten Plasmas
(ICP-MS von engl.: Inductively Coupled Plasma Mass Spectrometry) ist heute die leistungs-
fähigste Methode für die Spurenelementanalytik. Sie findet in geologischen, biologischen,
umweltanalytischen und industriellen Bereichen breite Anwendung. Besonders die Halbleiter-
industrie hat sich schon frühzeitig für die Analyse hochreiner Ausgangsstoffe der ICP-MS
bedient, und damit die Entwicklung der Analysenmethode vorangetrieben. Der Erfolg der
ICP-MS beruht auf den folgenden Vorzügen:
• Die exzellenten Nachweisgrenzen, die in wässriger Lösung für die meisten bestimm-
baren Elemente im ng L−1-Bereich oder darunter liegen, sind eine wichtige Voraus-
setzung für Spurenanalysen.
• Durch die Multielementfähigkeit sind bis auf wenige Ausnahmen alle Elemente des
Periodensystems nahezu gleichzeitig bestimmbar.
• Der geringe Probenverbrauch ermöglicht eine vollständige Elementanalyse mit
weniger als 1 mL Probenvolumen.
• Auf Grund kurzer Analysenzeiten ist ein hoher Probendurchsatz erreichbar.
• Der dynamische Messbereich erstreckt sich über mehr als sechs Größenordnungen.
• Es ergeben sich übersichtliche Massenspektren und relativ wenige Interferenzen im
Vergleich z.B. mit ICP-AES-Messungen.
• Die massenspektrometrische Unterscheidung von Isotopen lässt sich zur Bestimmung
von Isotopenverhältnissen und zur Quantifizierung mittels Isotopenverdünnung
nutzen.
• Mit der ICP-MS können schnelle halbquantitative Übersichtsanalysen durchführt
werden, die trotzdem sehr aussagekräftig sind.
Nur die hohen Anschaffungs- und Betriebskosten der ICP-MS-Geräte beschränken ihren
Einsatz meist noch auf größere Einrichtungen.

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 8 -
Die ICP-MS-Technik ist mittlerweile über 20 Jahre alt. Die maßgebliche Entwicklung wurde
von drei Arbeitsgruppen geleistet: Erste Kopplungen eines Plasmas mit einem Massen-
spektrometer wurden von Gray und Date [31, 32] an der britischen Universität Surrey in den
1970er Jahren mit dem Gleichstromplasma durchgeführt. Sie wechselten Anfang der
Achtziger auf das ICP [33]. Etwa zeitgleich startete ein Programm unter Houk und Fassel [34]
an der amerikanischen Iowa State University zur ICP-MS-Kopplung. Die Konstruktion einer
Verbindung aus dem inneren Plasmakanal in das Vakuum eines Massenspektrometers stellte
wohl die größte Hürde dar, wurde aber mit einem von Douglas und French [35] in Toronto
zuerst verwendeten zweistufigem Interface gelöst. Die kanadische Gruppe wechselte 1983
vom mikrowelleninduzierten Plasma zum ICP. Die ersten ICP-MS-Spektren wurden 1981 von
Gray [36] aufgenommen. Kommerzielle Geräte der Unternehmen VG Isotopes (UK) und
Sciex (Kanada) waren erstmals ab 1983 auf dem Markt.
2.1.2 Funktionsweise und instrumentelle Grundlagen der ICP-MS
Übersicht
Die Basis aller massenspektrometrischen Methoden ist die Erzeugung von Ionen, eine
nachfolgende Trennung nach Masse-zu-Ladungs-Verhältnis und die Detektion. Bei der
ICP-MS dient das ICP als Ionenquelle. Über ein spezielles Interface werden die Ionen in das
Massenspektrometer eingebracht, wo sie zuerst in der Ionenoptik fokussiert und von nicht
geladenen Teilchen separiert werden. Danach gelangen die Ionen in den Massenanalysator.
Als gebräuchlichster Massenanalysator wird der Quadrupolmassenfilter verwendet, der ein
ausreichendes Auflösungsvermögen besitzt, um Elementionen mit einer Massendifferenz von
1 u zu unterscheiden. Die Detektion der Ionen und die Bestimmung ihrer Zählrate für die
Konzentrationsbestimmung erfolgt mit einem Sekundärelektronenvervielfacher.
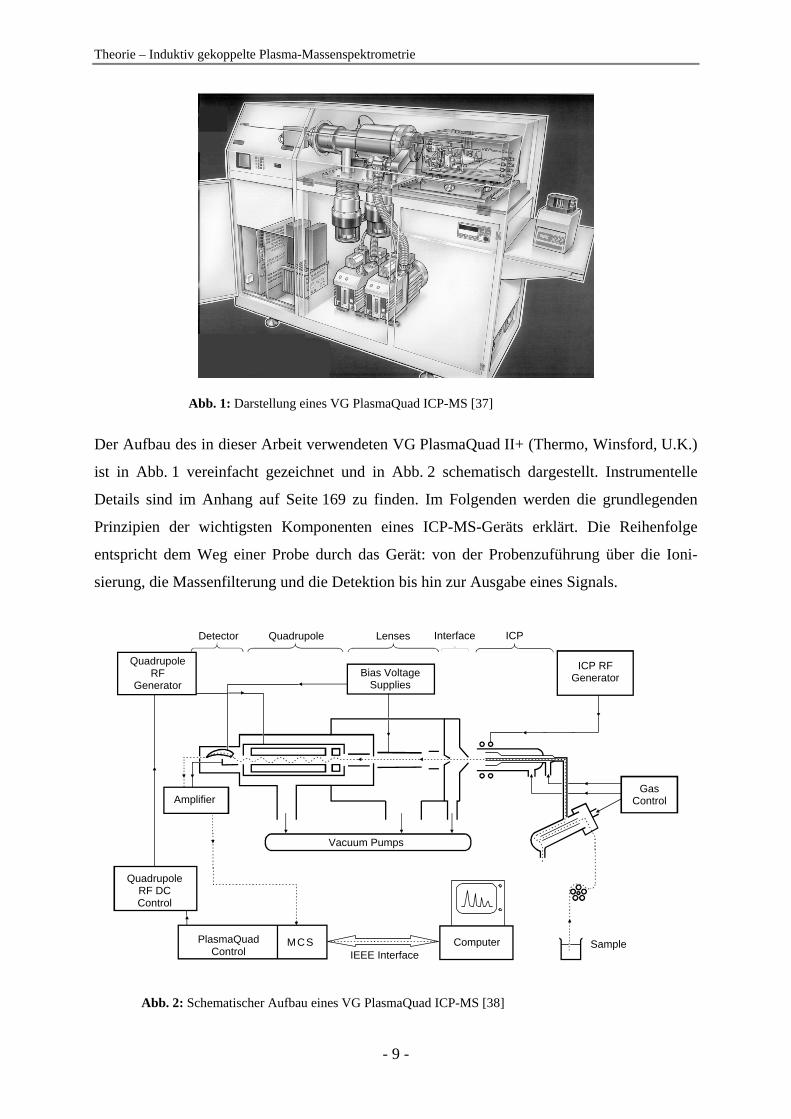
Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
Abb. 1: Darstellung eines VG PlasmaQuad ICP-MS [37]
Der Aufbau des in dieser Arbeit verwendeten VG PlasmaQuad II+ (Thermo, Winsford, U.K.)
ist in Abb. 1 vereinfacht gezeichnet und in Abb. 2 schematisch dargestellt. Instrumentelle
Details sind im Anhang auf Seite 169 zu finden. Im Folgenden werden die grundlegenden
Prinzipien der wichtigsten Komponenten eines ICP-MS-Geräts erklärt. Die Reihenfolge
entspricht dem Weg einer Probe durch das Gerät: von der Probenzuführung über die Ioni-
sierung, die Massenfilterung und die Detektion bis hin zur Ausgabe eines Signals.
Quadrupole RF
Generator
Detector Quadrupole Lenses Interface ICP
Bias VoltageSupplies
ICP RF Generator
Amplifier
Vacuum Pumps
Gas Control
QuadrupoleRF DC Control
Abb. 2: Sc
P MCS Computer Sample
lasmaQuadControlhematischer Aufbau eines VG PlasmaQuad ICP-MS [38]
IEEE Interface
- 9 -

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
Probenzuführung
Die Probe muss als Gas, Aerosol oder in Form von feinen Partikeln in das ICP gelangen. Am
häufigsten werden wässrige Proben analysiert, die mit einer peristaltischen Pumpe angesaugt
und mit einem pneumatischen Zerstäuber in ein feines Aerosol überführt werden. Die pneu-
matischen Zerstäubungssysteme basieren darauf, dass ein Flüssigkeitsstrom durch einen
Gasstrom von hoher Geschwindigkeit in feine Tröpfchen zerrissen wird.
Kz
Za
P
Z O
IkR
Z
Abb. 3: Verschiedene Zerstäubertypen [39]: oben: Meinhanach Scott [40], unten links: V-Spalt-Typ, unten rechts: Bu
Drei pneumatische Zerstäubertypen sind in Abb
die konzentrische nach Meinhard [41], und die n
keits- und der Gasstrom in einem Winkel aufein
Vergleich zu den Babington-Zerstäubern höher
Bauformen nach dem Babington-Prinzip besitz
hohe Salzfrachten sind. Vertreter dieser Zerstäu
[42] und der Burgener-Zerstäuber.
Alle bisher vorgestellten Zerstäubertypen haben
halb von 10 % liegt. Zur besseren Aerosolausbe
- 10
ugelschliffverbindung ur Plasmafackel
erstäuberkammer-dapter
konventioneller konzentrischer Zerstäuber
robenlösung
l
erstäubergas-Ring-Dichtung
nnere onzentrische öhre
Abf
P
rd-Zerstärgener-T
. 3 geze
ach dem
ander tr
e Aeros
en den
bertype
gemeins
ute wu
-
al
ProbenlösungZa
O
Zerstäubergasrobenlösung
erstäubergas
uber in einer Doppelpayp.
igt. Die wichtigste
Babington-Prinz
effen. Meinhard-Z
olausbeuten und s
Vorteil, dass sie
n sind der V-Spalt
am, dass ihre Aero
rden so genannte
erstäuberkörper us Teflon
-Ringe
ss-Zerstäuberkammer
n Bauformen sind
ip, wo der Flüssig-
erstäuber haben im
ind pulsationsarm.
unanfälliger gegen
-, der Cross-Flow-
solausbeute unter-
hocheffektive Zer-

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 11 -
stäuber wie der Ultraschallzerstäuber (USN) oder der Mikrokonzentrische Zerstäuber (MCN)
entwickelt. Außerdem gibt es noch Direktzerstäuber (DIN oder DIHEN), mit denen ein
Aerosol komplett ins Plasma gelangt. Hier soll nur erwähnt werden, dass die hocheffektiven
und die Direktzerstäuber für Anwendungen mit minimalem Probenverbrauch nützlich sind,
aber im Allgemeinen nicht so robust arbeiten wie die zuerst beschriebenen Zerstäuber vom
Meinhard oder Babington-Typ. Detaillierte Abhandlungen über den Aufbau, die Leistungs-
daten sowie die Vor- und Nachteile verschiedener Zerstäuber sind in der Literatur zu
finden [39, 43].
Die Zerstäuber- oder Sprühkammer (s. Abb. 3) dient dazu, große Flüssigkeitströpfchen abzu-
trennen, denn nur Tröpfchen bis zu einer Größe von 10 µm können im Plasma effektiv
desolvatisiert, atomisiert und ionisiert werden. Zu schwere Tröpfchen prallen an die Wände
der Kammer und werden in den Abfall befördert. Die Zerstäuberkammer wird auf eine
Temperatur von etwa 4 °C gekühlt, so dass die Dichte des Wassers am höchsten und der
Selektionsprozess am effektivsten ist. Durch die Sprühkammer werden Zerstäubungs-
fluktuationen gedämpft und somit das Messsignal stabilisiert. Zerstäuberkammern bestehen
meist aus Quarzglas, aber z.B. für die Analyse von fluoridhaltigen Lösungen sind auch
Anfertigungen aus Teflon erhältlich. Wichtig ist ein gutes Ablaufverhalten der eingesetzten
Kammer, da größere an den Wandungen hängen bleibende Tröpfchen das Messsignal beein-
flussen und zu Memoryeffekten führen können. Ein Vergleich moderner Zerstäuberkammern
sowie eine Übersicht über flüssige Probenzuführung in der Plasma-Spektrometrie ist in
Reviewartikeln von Maestre et al. zu finden [44, 45].
Weitere Arten der Probenzuführung sind die Laserablation (LA) [46, 47], die elektrothermale
Verdampfung (ETV) [48] oder die gasförmige Probenzuführung z.B. durch Hydrid-
generation [49].
Die ICP-Einheit
Als Plasma bezeichnet man in der Physik ein gasförmiges System, welches zu einem
nennenswerten Anteil freie Ladungsträger (Elektronen, Atom- und Molekülionen) enthält. In
einem solchen System liegen zum Teil in angeregten Zuständen Atome, Ionen, Moleküle,
Radikale und freie Elektronen nebeneinander vor. Wegen der Rekombination von Teilchen
und durch Übergänge von höheren in niedrigere energetische Zustände sendet ein Plasma

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
elektromagnetische Strahlung aus. Es ist elektrisch leitfähig aber nach außen hin neutral. Der
Plasmazustand wird auch als vierter Aggregatzustand bezeichnet.
Im Falle des ICPs erfolgt die Energiezufuhr in ein Gas (meist Argon) durch induktive
Kopplung in einem Hochfrequenzschwingkreis. Die wichtigsten Bestandteile der ICP-Einheit
stellen der Hochfrequenzgenerator, die Induktionsspule und die Plasmafackel dar. Des
Weiteren wird ein System zur Stabilisierung des Plasmas (impedance matching network)
sowie eine Gasversorgungseinheit mit präziser Regelungsmöglichkeit der Gasflüsse benötigt.
Atomisierungs-zone (Interferenzen)
Atom-linien I
l
Rekombinations-zone instabiles Plasma
Luft-Plasma- Wechsel-wirkungszone
Plasmagas Hilfsgas
Zerstäubergas
Abb. 4: Schematischer Aufbau der Torch vom Fassel-Typ [50] und verschiedene Zonen in e
Die Plasmafackel (Torch) besteht aus drei konzentrischen Quarzglasröhren (
eine typische Länge von 10–12 cm und einen Durchmesser von etwa 1.8 cm
Röhren strömt im Betrieb Argon, wobei der innerste Gasstrom mit 0.5–1 L
zuführt, der mittlere mit 0–1.5 L min−1 zur Formgebung und Positionierung
und der dritte mit 10–18 L min−1 den Hauptteil des Plasmagases liefert un
Torchwandungen kühlt. Die Spitze der Torch befindet sich innerhalb einer
Induktionsspule aus Kupfer, die an einen Radiofrequenz-Wechselspannun
schlossen ist. Der Generator liefert eine Leistung von 800–2000 W be
Frequenzen von 27.12 MHz oder 40 MHz.
Mit Hilfe eines Zündfunkens einer Teslaspule werden erste Elektronen
gebracht. Über das durch den Wechselstrom in der Spule erzeugte magn
trische Wechselfeld wird Energie auf die Ladungsträger übertragen. D
Kreisbahnen innerhalb der Spule stark beschleunigt. Wenn die Elektrone
kollidieren, werden angeregte Atome und Ionen erzeugt und das induktiv g
entsteht.
- 12 -
onen-inien
inem ICP [39].
s. Abb. 4). Sie hat
. Durch alle drei
min−1 die Probe
des Plasmas dient
d gleichzeitig die
wassergekühlten
gsgenerator ange-
i gebräuchlichen
in den Gasstrom
etische und elek-
iese werden auf
n mit Gasatomen
ekoppelte Plasma

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
Im ICP werden Temperaturen von über 7000 K erreicht. Aus diesem Grund ist es eine ideale
Ionenquelle für die Elementanalytik, da bei diesen Temperaturen praktisch sämtliche chemi-
schen Bindungen eingebrachter Proben aufgebrochen und die entstandenen Atome effektiv
ionisiert werden. Die Ionisierung der Analytatome erfolgt hauptsächlich durch Stoßreaktionen
sowohl mit hochenergetischen Argonionen, als auch mit Elektronen. Der Ionisierungsgrad
einzelner Elemente ist von ihrer Ionisierungsenergie abhängig (s.a. Tab. 3 und Tab. 26).
Das Interface
Der Extraktionsschritt, der die Ionen unbeeinflusst aus dem heißen Plasma in das Hoch-
vakuum eines Massenspektrometers transportiert, verlangte jahrelange Entwicklung und
wurde mit einem zweistufigen Interface (s. Abb. 5) gelöst.
Abb. 5: Typisches ICP-MS Interfa
Das Interface besteht aus zwei konusförmigen Blenden („
kleine Öffnung an der Spitze haben und mit Wasser gekü
Lochblende, des „Samplers“, hat einen Durchmesser von
die Expansionskammer, die die erste Stufe zur Druck
schieberpumpe senkt hier den Druck von Atmosphärend
breitet sich mit Überschallgeschwindigkeit in die Expansio
auf ca. 580 K ab. Die zweite Lochblende, der „Skimme
etwa 0.6 mm und stellt den Eingang ins Hochvakuum dar
richtige Abstand von Sampler und Skimmer sind esse
Hinter dem Sampler befindet sich im Plasmabetrieb eine E
teile, deren Wellenfront als Machscheibe bezeichnet wird
S
Extraktionslinse
Skimmer- Cone
SC
zur Expansionskammerpumpe
- 13 -
pule
ampler- one
ce [50].
Cones“) aus Nickel, die jeweils eine
hlt werden. Die Öffnung der ersten
etwa 1 mm, und ist der Einlass in
reduzierung darstellt. Eine Dreh-
ruck auf unter 5 mbar. Das Plasma
nskammer aus und kühlt sich dabei
r“, hat einen Lochdurchmesser von
. Die geometrische Form sowie der
ntiell für eine gute Ionenausbeute.
xpansionszone der Plasmabestand-
. Die Position der Machscheibe ist

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 14 -
abhängig vom Druckgefälle und vom Öffnungsdurchmesser des Samplers. Um eine maximale
Ionenausbeute zu erhalten, sollte die Skimmer-Spitze bei zwei Drittel der Länge der
Expansionszone positioniert sein [51, 52].
Viele spezifische und nichtspezifische Interferenzen (s. Kap. 2.1.3) haben ihren Ursprung im
Interface, da es dort durch die verminderte Temperatur und die hohe Ionendichte zur Bildung
von Molekülionen kommen kann. Sowohl der Sampler als auch der Skimmer bedürfen daher
einer permanenten Beobachtung ihres Zustands. Ablagerungen an den Lochblenden
verstärken Interferenzen und machen eine Reinigung notwendig. Bei zu starker Erosion und
Vergrößerung der Cone-Öffnungen ist ein Austausch durch neue Lochblenden notwendig.
Das Vakuumsystem
Für die Leistungsfähigkeit eines ICP-MS ist ein gutes Vakuum essentiell. Die Flugbahn der
Ionen im Massenspektrometer sollte so wenig wie möglich gestört werden, um die höchsten
Zählraten für Analyten zu erhalten. Gleichzeitig verringert ein gutes Vakuum die Untergrund-
zählrate. Für diese kann in modernen Geräten ein Wert kleiner als 1 cps erreicht werden.
Somit ist das Vakuumsystem ein wichtiger Einflussfaktor auf die erreichbaren instrumentellen
Nachweisgrenzen eines ICP-MS. Bei schlechtem Vakuum besteht außerdem das Risiko
elektrischer Überschläge durch die hohen Spannungen, die z.B. an einen Sekundärelektronen-
vervielfacher, an einen Quadrupol und an die Ionenlinsen angelegt werden (Bauteile s.u.).
Das Vakuum in ICP-MS-Geräten wird meist in drei Bereiche unterteilt: In der Expansions-
oder Interface-Region wird der Druck mittels einer Drehschieberpumpe von Normaldruck auf
wenige Millibar reduziert. Eine oder mehrere Turbomolekularpumpen oder Öldiffusions-
pumpen erzeugen danach das Hochvakuum mit weniger als 1·10−4 mbar in der so genannten
Zwischenregion der Ionenlinsen und weniger als 1·10−5 mbar in der Analysatorregion
(Quadrupol, Detektor).

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
Die Ionenoptik
Hinter dem Skimmer liegt die Ionenoptik, in der ein Vakuum von mindestens 1·10−4 mbar
herrscht und in das die Kationen mit Hilfe der Extraktionslinse (typisches Potential: −150 V
bis −400 V) extrahiert werden. So genannte Ionenlinsen sind stromlose Elektroden in Form
von Zylindern und Blenden, an die jeweils ein verstellbares positives oder negatives Potential
angelegt wird, um die Ionen zu bündeln oder in eine gewünschte Richtung zu lenken.
Abb. 6: Anordnung der Ionenlinsen im PQ II [38]
Das in Abb. 6 schematisch dargestellte System d
On-Axis-Bauweise gebaut. Bei dieser Bauweise
Extraktionsrichtung der Ionen liegendes Plättche
Kollektorlinse liegt, aufgefangen. Die Kollekto
Extraktionslinse weniger negatives Potential den
meisten Ionen, den Photonenstopp zu umgehen. D
Refokussieren des Ionenstrahls. Den gleichen Zw
eine Bündelung nach Eintritt der Ionen durch d
bessere Vakuum der Analysatorkammer nochmals
ICP-MS-Systeme verschiedener Hersteller untersc
Anordnung der Ionenlinsen. Die verschiedenen
Transmissionseffizienz verschiedener Ionen,
(s. Kap. 2.1.3), und sind verantwortlich für die
L4 L3 DA L2
Photonen-stopp
Kl
El
Sv
L1
- 15 -
ollektor-inse
er Ionenlins
werden st
n (Photone
rlinse weite
Ionenstrahl
ie weiteren
eck haben
ie DA-Lins
nötig ist.
heiden sich
Bauweisen
insbesonder
so genannte
xtraktions-inse
chiebe-entil
en des VG PlasmaQuads ist in
örende Photonen durch ein in
nstopp), das gleich hinter der
t durch ihr im Vergleich zur
auf und ermöglicht es so den
Linsen L1 und L2 dienen zum
auch die Linsen L3 und L4, da
e (differential aperture) in das
stark durch die Anzahl und die
haben Auswirkungen auf die
e durch Raumladungseffekte
Massenempfindlichkeitskurve

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 16 -
(s. Seite 19). In der Off-Axis Bauweise werden die Ionen nicht um einen Photonenstopp
herum geleitet, sondern auf eine leicht verschobene parallele Flugbahn umgelenkt und auf
diese Weise eine Abtrennung von Licht und neutralen Teilchen erreicht. Zu den neuesten
Entwicklungen gehört der Einsatz eines Ionenspiegels, der Ionen komplett reflektieren und
gleichzeitig fokussieren kann [53]. Durch einen Ionenspiegel soll eine hohe Ionenausbeute
erreicht werden und gleichzeitig das Kontaminationspotential innerhalb eines Massenspektro-
meters deutlich reduziert werden.
Einen revolutionären Einfluss auf die Leistungsfähigkeit der ICP-MS hatte die Entwicklung
von Reaktionszellen und Kollisionszellen, die seit Ende der 1990er Jahre in ICP-MS-Geräten
eingesetzt werden und sich dort zwischen der klassischen Ionenoptik und dem Quadrupol
befinden. Kollisions- oder Reaktionszellen dienen zur Eliminierung von Interferenzen durch
Molekülionen. Zur Fokussierung und Führung der Ionen in den Zellen, nutzt man einen
Hexapol oder Oktopol. Eine Einführung in die Grundlagen der Reaktions- bzw. Kollisions-
zellentechnologie erhält man bei Thomas [54] und eine ausführliche Beschreibung in einem
Review von Tanner [55].
Der Massenanalysator
Nach der Ionenoptik gelangen die Ionen in einen Massenanalysator, z.B. in einen Quadrupol,
der den am häufigsten verwendeten Massenanalysator darstellt. Ein Quadrupol besteht aus
vier Stäben (meist aus Molybdän), die parallel und im gleichen Abstand zueinander montiert
sind (s. Abb. 7). An alle Stäbe wird eine Gleichspannung angelegt, die von einer Wechsel-
spannung überlagert wird. Jeweils zwei gegenüber liegende Stäbe haben das gleiche
Vorzeichen für die Gleich- und die Wechselspannung. Somit ist an nebeneinander liegende
Stäbe eine um 180° phasenverschobene Wechselspannung umgekehrter Polarität angelegt.
Nur Ionen mit einem Masse-zu-Ladungs-Verhältnis, das zur Resonanzfrequenz führt, können
das Feld auf spiralförmigen Bahnen passieren. Auf Ionen mit geringerer Masse als die
Resonanzmasse wird Energie übertragen, ihre Oszillationsamplitude nimmt zu, und sie treffen
entweder auf die Stäbe oder verlassen das Feld. Flugbahnen von Ionen, die eine größere
Masse als die Resonanzmasse haben, werden hauptsächlich durch die Gleichspannung
abgelenkt. Weitere Informationen zu speziellen Quadrupolparametern befinden sich in
Kap. 4.7.10.

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
++-
-
instabile Flugbahn
stabile Flugbahn
-U - V cos ( t)ω
+U + V cos ( t)ω Abb. 7: Prinzipieller Aufbau eines Quadrupols
Ein Quadrupol ist ein sequentiell arbeitender Massenanalysator, d.h. Ionen werden nachein-
ander getrennt und zum Detektor geleitet. Es können nicht alle Massen gleichzeitig wie z.B.
mit einem Flugzeitanalysator (s.u.) analysiert werden. Mit einem Quadrupol ist es aber
möglich, sehr schnell (in etwa 0.1 s) den gesamten analytisch sinnvollen Massenbereich von
5–255 u zu scannen, weswegen man von quasisimultaner Detektion spricht.
Alternativ zum Quadrupol werden in ICP-MS-Geräten auch Sektorfeld- oder Flugzeitanalysa-
toren eingesetzt. Die höchste Auflösung von bis zu 100000 lässt sich mit einem Sektorfeld
erreichen, bei dem die Ionen sowohl durch Ablenkung in einem magnetischen als auch einem
elektrischen Feld getrennt werden. Durch so genannte Doppelfokussierung werden auch
Unterschiede der Anfangsgeschwindigkeit und -flugrichtung ausgeglichen. Die hochauf-
lösende Sektorfeldmethode ist aber sehr teuer, insbesondere wenn viele Isotope simultan
gemessen werden sollen und deshalb viele Detektoren notwendig werden (Multikollektor-
ICP-MS) [56, 57].
Flugzeitanalysatoren (engl.: Time-of-Flight, TOF) erlangen mit bis zu 10000 ebenfalls eine
höhere Auflösung als Quadrupolmassenfilter und sind nicht viel teurer als diese. In einem
TOF-Analysator werden Ionen zu einem genau definierten Startpunkt auf eine Driftstrecke
beschleunigt. Die Fluggeschwindigkeit ist abhängig vom Masse-zu-Ladungs-Verhältnis der
Ionen, so dass sie nach unterschiedlicher Flugdauer auf den Detektor treffen. Da alle Ionen
gleichzeitig in die Driftstrecke extrahiert werden ist der TOF-Analysator ein simultaner
Massenanalysator und kein Filter wie der Quadrupol. Die Möglichkeit der simultanen Detek-
tion ist äußerst vorteilhaft für die Kopplung mit Analysentechniken, bei denen nur kurze
Signale erhalten werden, wie zum Beispiel LA-ICP-MS. Durch die Extraktion aller Ionen
gleichzeitig ergibt sich aber auch die Schwierigkeit, dass störende Ionen in hoher Konzen-
tration aus der Driftröhre entfernt werden müssen, um den Detektor nicht zu überlasten.
- 17 -

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
Diesbezüglich sowie bei der schnellen Datenaquisition sind noch Weiterentwicklungen nötig.
Bisher ist der Untergrund von TOF-ICP-MS-Geräten zu hoch und die Transmission der
Analytionen zu gering. Daher sind die erreichbaren Nachweisgrenzen noch mehrere Größen-
ordnungen schlechter als bei der Quadrupol-ICP-MS.
Der Detektor und die Auswertungseinheit
Die mit Hilfe des Massenanalysators selektierten Ionen werden üblicherweise mit einem
Sekundärelektronenvervielfacher (Multiplier) registriert.
Ein kontinuierlicher Mul
Horns. Ein einzelnes Io
Elektronen herausschlage
liegende Seite beschleun
sich so oft bis die Elektr
dann als Strompuls de
Diskriminators, d.h. eine
definierten Schwellenwe
zu hohen Konzentratione
einen unempfindlicheren
über den im Detektor ent
von bis zu acht Größenor
Die Auswerteeinheit, im
Analyzer), steuert den Qu
einem bestimmten Masse
aI
Hv
uftreffendes on
Abb. 8: Der Sekundärele
tiplier ist etwa 7 cm l
n mit ausreichender
n, welche durch die
igt werden und dort
onenlawine ein Maxim
r Auswerteeinheit r
s elektronischen Filter
rts ausblenden. Mode
n automatisch aus de
Modus um. Höhere Z
standenen Strom gem
dnungen ermöglicht.
PlasmaQuad die
adrupol an, registrier
-zu-Ladungs-Verhältn
aE
- 1
ochspannungs-ersorgung
Kollektor
usgelöste lektronenlawine
ktronenvervielfacher [50]
ang und hat die Form eines krumm gebogenen
kinetischer Energie kann aus der Oberfläche
angelegte Hochspannung auf die gegenüber-
weitere Elektronen auslösen. Dies wiederholt
um von etwa 108 Elektronen erreicht hat, die
egistriert werden können. Mit Hilfe eines
s, lassen sich Untergrundpulse unterhalb eines
rne Multiplier stellen sich beim Auftreten von
m besonders empfindlichen Pulszählmodus in
ählraten werden in diesem Modus analog, d.h.
essen. So wird ein großer linearer Messbereich
so genannte MCA-Einheit (Multi Channel
t über eine festgelegte Zeit (dwell time) alle zu
is gehörenden Zählereignisse und speichert sie
8 -
Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
in für die jeweilige Masse zugewiesenen Kanälen. Deren Informationen werden am Ende
einer Messung ausgelesen und zur Darstellung eines Massenspektrums und zur weiteren
Datenverarbeitung in einen Computer weitergegeben.
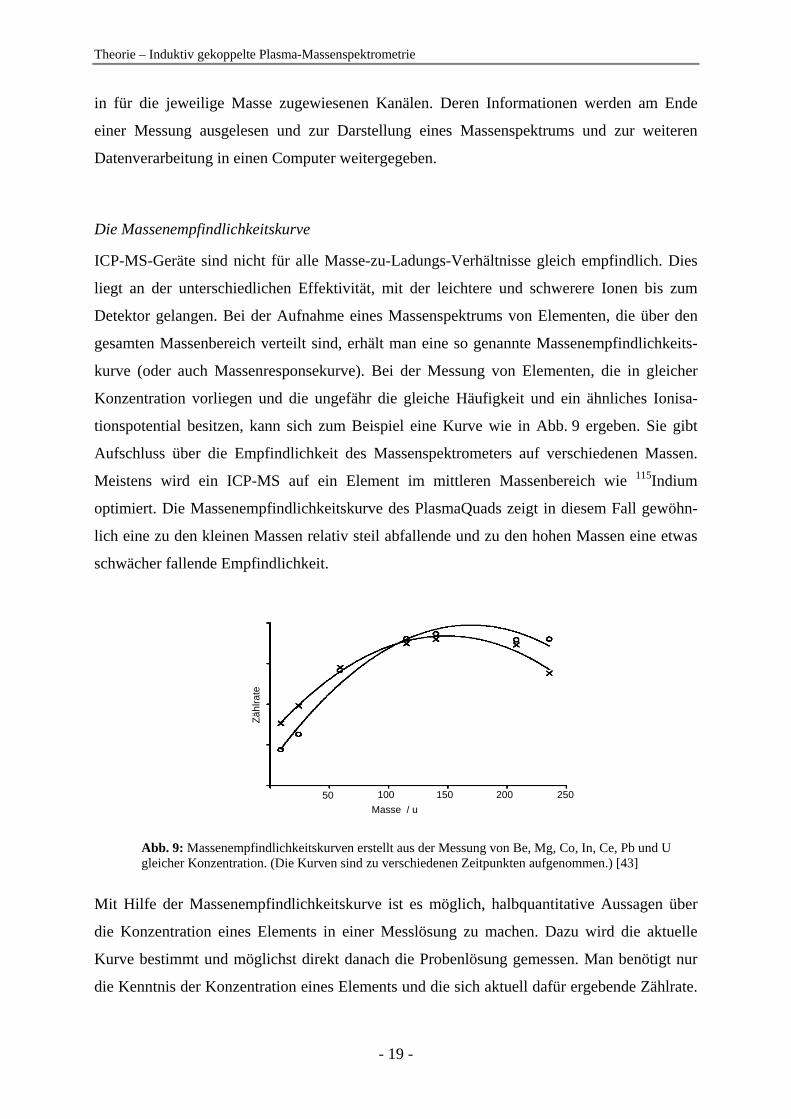
Die Massenempfindlichkeitskurve
ICP-MS-Geräte sind nicht für alle Masse-zu-Ladungs-Verhältnisse gleich empfindlich. Dies
liegt an der unterschiedlichen Effektivität, mit der leichtere und schwerere Ionen bis zum
Detektor gelangen. Bei der Aufnahme eines Massenspektrums von Elementen, die über den
gesamten Massenbereich verteilt sind, erhält man eine so genannte Massenempfindlichkeits-
kurve (oder auch Massenresponsekurve). Bei der Messung von Elementen, die in gleicher
Konzentration vorliegen und die ungefähr die gleiche Häufigkeit und ein ähnliches Ionisa-
tionspotential besitzen, kann sich zum Beispiel eine Kurve wie in Abb. 9 ergeben. Sie gibt
Aufschluss über die Empfindlichkeit des Massenspektrometers auf verschiedenen Massen.
Meistens wird ein ICP-MS auf ein Element im mittleren Massenbereich wie 115Indium
optimiert. Die Massenempfindlichkeitskurve des PlasmaQuads zeigt in diesem Fall gewöhn-
lich eine zu den kleinen Massen relativ steil abfallende und zu den hohen Massen eine etwas
schwächer fallende Empfindlichkeit.
Abb. 9: Massenempfindlichkeitskurven erstellt aus der Messung von Be, Mg, Co, In, Ce, Pb und U gleicher Konzentration. (Die Kurven sind zu verschiedenen Zeitpunkten aufgenommen.) [43]
Mit Hilfe der Massenempfindlichkeitskurve ist es möglich, halbquantitative Aussagen über
die Konzentration eines Elements in einer Messlösung zu machen. Dazu wird die aktuelle
Kurve bestimmt und möglichst direkt danach die Probenlösung gemessen. Man benötigt nur
die Kenntnis der Konzentration eines Elements und die sich aktuell dafür ergebende Zählrate.
Zähl
rate
Masse / u 100 150 200 250 50
- 19 -

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 20 -
Am aktuellsten erhält man diese Information, wenn allen Messlösungen ein interner Standard
zugegeben wird. Über die bekannte Massenempfindlichkeit lassen sich den Intensitäten
anderer Elemente in den Proben auch ohne Kalibration Konzentrationen zuordnen. Allerdings
sind die Ergebnisse mit hoher Unsicherheit behaftet, wenn z.B. Interferenzen durch die
Probenmatrix und unterschiedliche Ionisierungsausbeuten nicht berücksichtigt werden. Man
versucht deshalb die Ergebnisse durch weitere Korrekturalgorithmen zu verbessern. Eine auf
diese Weise erhältliche halbquantitative Übersicht der Zusammensetzung einer Probe ist auf
jeden Fall hilfreich zur Planung weiterer Quantifizierungsschritte.
ICP-MS-Messmodi
Als die beiden wichtigsten Messmodi bei ICP-MS-Geräten werden der Scan- und der Peak-
Jump-Modus (auch Peak-Hop) unterschieden. Beim Scan-Modus wird ein Spektrum des
gesamten bzw. eines gewünschten Massenbereichs aufgenommen. Dabei selektiert der
Quadrupol in sehr kleinen Schritten (z.B. 20 Messpositionen u−1) die zu messenden Massen.
Für das Überstreichen (sweep) des gesamten Elementmassenbereichs wird je nach dwell-Zeit
und Anzahl der Messpositionen nur etwa 0.1–1 s benötigt. Wegen dieser sehr kurzen Zeit
wird die Messung als „quasisimultan“ bezeichnet.
Beim Peak-Jump-Modus springt der Quadrupol nur zu ausgewählten Peaks. Dies geschieht
zwar auf Kosten des Gesamtüberblicks, jedoch wird hierdurch die Messzeit effektiver
ausgenutzt. Es wird eine bessere Zählstatistik erreicht, aus der niedrigere Nachweisgrenzen
resultieren.
Der Time-Resolved-Analysis-Modus (TRA) wird angewendet, um die Informationen Masse
und Intensität zeitaufgelöst (transient) zu erhalten.

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 21 -
2.1.3 Interferenzen
Die Spektren von ICP-MS-Messungen sind im Vergleich zu ICP-AES-Spektren einfacher, da
sie viel weniger Signale enthalten und spektrale Überlappungen seltener sind. Die leichtere
Interpretation der Spektren ist ein großer Vorteil der ICP-MS. Dennoch gibt es einige Störun-
gen, die die Leistungsfähigkeit zum Teil stark beeinträchtigen. In Abb. 10 soll ein Überblick
über die wichtigsten verschiedenen Interferenzen und ihre Einteilung in Gruppen gegebenen
werden. In der ICP-MS ist es sinnvoll (element)spezifische von nicht(element)spezifischen
Interferenzen zu unterscheiden. In der Literatur wird zudem oft der Begriff „Matrixeffekte“
verwendet, um die Störungen zu beschreiben, die durch die Probenmatrix hervorgerufen
werden. Genau genommen werden aber nahezu alle Interferenzen direkt oder indirekt durch
die Matrix hervorgerufen. Mit dem Begriff „Matrixeffekte“ werden aber ausschließlich die
nichtspezifischen Interferenzen gemeint.
Spezifische Interferenzen • Isobare Überlagerungen (griech. iso: gleich, baros: Schwere)
(Spektrale Interferenzen) – Interelementüberlappungen wie z.B. 58Ni (68 %) und 58Fe (0.3 %)
Führen zu fälschlicher – Molekülionen (= polyatomare Störungen), dazu gehören z.B.:
Signalerhöhung typische Addukte aus Argon, Luft, Wasser u. Säuren wie:
[ArO]+, [Ar2]+, [ArH]+, [ArN]+, [ArOH]+, [ArH2]+...,
gebildete Molekülionen der Analyt- und Matrixelemente sowie
nicht vollständig atomisierte Oxide von Refraktärmetallen
(Ti, V, Cr, Zr, Nb, Mo, Hf, Ta, W, Re, Th, U)
• Doppelt geladene Ionen (vor allem bei Atomen, deren zweite Ionisie-
rungsenergie (IE) kleiner ist als die erste IE des Argons von 15.76 eV)
Nichtspezifische Interferenzen • Unterschiede bei der Ionenextraktion und dem Ionentransport durch das
(Nichtspektrale Interferenzen) Massenspektrometer (besonders bei der Fokussierung in der Ionenoptik)
Oft als Matrixeffekte bezeichnet – Eine Hauptursache sind Raumladungseffekte
Führen zu Signalerniedrigung • Transportinterferenzen
oder -erhöhung – Andere Zerstäubungseffektivität z.B. wegen veränderter Viskosität
– Physikalische Verstopfung (Zerstäuber, Injektorrohr oder Cones)
• Veränderte Ionisierungsausbeute
– Wechselnde Plasmaparameter wie die Temperatur z.B. durch
Abkühlen bei Einbringung einer hohen Salzfracht
• Elektrische oder elektromagnetische Interferenzen
Abb. 10: Spezifische und nichtspezifische Interferenzen in der ICP-MS

Theorie – Induktiv gekoppelte Plasma-Massenspektrometrie
- 22 -
Massendiskriminierung und Raumladungseffekte
Die Hauptursache für Massendiskriminierung (engl.: mass bias) im ICP-MS wird heute in den
so genannten Raumladungseffekten (engl.: space charge) vermutet. Das Plasma ist nach außen
hin neutral, da gleich viele positive wie negative Ionen vorhanden sind. Bei einem
entnommenen Plasmaanteil kommt es aber im Interface beim Übergang aus dem Skimmer in
die Ionenoptik und besonders beim Durchgang durch die Ionenlinsen zu einer räumlichen
Ladungstrennung. Drei verschiedene Mechanismen zur Entstehung von Raumladungseffekten
werden in der Literatur beschrieben [39]:
1. Liegt im Skimmer eine hohe Ladungsdichte vor, überlappen die Debye-Sphären der
Ladungsträger, wodurch deren elektrostatische Wechselwirkung abgeschirmt wird.
Die auf Grund ihrer geringeren Größe mobileren Elektronen diffundieren im neuen
Feld an die Wände des Skimmers und in der Mitte verbleibt eine positive Raum-
ladung.
2. Beim Übergang vom Skimmer in die Ionenoptik kommt es zu einem starken Druck-
abfall. Die leichten Elektronen diffundieren schneller aus dem „Skimmerstrahl“ heraus
als schwerere Teilchen. Auch wenn die Kationen aus Neutralitätsgründen dann
schneller mit diffundieren, entsteht trotzdem auf der Achse des Strahls eine positive
Nettoladung.
3. Durch die negativ geladenen Ionenlinsen werden Elektronen abgestoßen und kaum
durchgelassen, so dass dort ein positiver Ionenstrahl auf engem Raum erzeugt wird.
Durch die verschiedenen Mechanismen zur Ladungstrennung entsteht eine positive Raum-
ladung, die sich selber defokussiert. Von der Defokussierung sind leichtere Ionen stärker
betroffen als schwere, da sich schwere Ionen nicht so stark von ihrer Flugbahn zum Detektor
ablenken lassen. Dies ist die Hauptursache der Diskriminierung leichterer Isotope bei
Isotopenverhältnismessungen. Auch die häufig zu beobachtende Signaldepression leichter
Elemente durch ein Vorhandensein vieler schwerer Elemente in der Matrix lässt sich durch
die Raumladungseffekte erklären.

Theorie – Ionenchromatographie
- 23 -
2.2 Ionenchromatographie
2.2.1 Einleitung
Unter dem Begriff Ionenchromatographie werden moderne, effiziente Methoden zur
Trennung und Bestimmung von Ionen zusammengefasst [13, 58]. Die heute angewendete IC
ist ein Variante der Hochleistungsflüssigkeitschromatographie (engl.: High Performance
Liquid Chromatography, HPLC) und wurde 1975 von Small, Stevens und Baumann
eingeführt [10].
Das grundlegende Prinzip der Chromatographie kann wie folgt erklärt werden: Die in einer
mobilen Phase gelösten Analyten werden durch eine stationäre Phase bewegt. Bei der wieder-
holten Verteilung zwischen den beiden Phasen werden verschiedene Probenkomponenten
unterschiedlich stark von der stationären Phase zurückgehalten und können getrennt von-
einander detektiert werden.
2.2.2 Trennmechanismen
Die Trennmechanismen, auf denen die Ionenchromatographie basiert, sind Ionenaustausch,
Ionenausschluss und Ionenpaarbildung. In der Literatur werden teilweise uneinheitliche
Abkürzungen für diese Trennmechanismen verwendet. In Tab. 1 sind die verschiedenen
gebräuchlichen Abkürzungen veranschaulicht.
Tab. 1: Abkürzungen verschiedener Trennmechanismen in der Ionenchromatographie
Trennmechanismus Abkürzung Englische Bezeichnung
unterschiedlich (drei mögliche)
IC HPIC
Ion Chromatography High Performance Ion Chromatography
Ionenaustausch
IC HPIC IEX IEC
Ion Chromatography High Performance Ion Chromatography Ion Exchange (Chromatography) Ion Exchange Chromatography
Ionenausschluss IEC ICE HPICE
Ion Exclusion Chromatography Ion Chromatography Exclusion High Performance Ion Chromatography Exclusion
Ionenpaarbildung
IPC IIC MPIC PIC
Ion Pairing Chromatography Ion Interaction Chromatography Mobile Phase Ion Chromatography Paired-Ion Chromatography

Theorie – Ionenchromatographie
- 24 -
Die Methoden, die auf Ionenausschluss oder Ionenpaarbildung als Trennprinzip beruhen, sind
wesentlich seltener anzutreffen als die Ionenaustauschchromatographie und sollen an dieser
Stelle nur kurz vorgestellt werden: Die zur Ionenausschlusschromatographie eingesetzten
stationären Phasen sind in der Regel hochkapazitive, sulfonierte Kationenaustauscher. Als
Eluent werden wässrige Mineralsäurelösungen eingesetzt. Bei Kontakt mit dem Eluenten
bildet sich eine Hydrathülle um die polaren Sulfonsäuregruppen, die dann teilweise dissoziiert
vorliegen. Es resultiert eine negative Partialladung im Bereich der Hydrathülle, eine gedachte
Schicht, die für Anionen undurchlässig ist und die Donnan-Membran genannt wird. Die
Donnan-Membran ist permeabel für undissoziierte Moleküle, die deshalb längere Zeit auf der
stationären Phase verbringen und von geladenen Substanzen getrennt werden können. Die
Ionenausschlusschromatographie dient vor allem der Analyse schwacher organischer oder
anorganischer Säuren, die in Reihenfolge ihre Säurestärke separiert werden.
Als stationäre Phasen für die Ionenpaarchromatographie werden die aus der Verteilungs-
chromatographie bekannten unpolaren Reversed-Phase-Materialien verwendet. Um Wechsel-
wirkungen von Ionen mit diesen Phasen zu ermöglichen, wird dem Eluenten ein so genanntes
Ionenpaarreagenz zugegeben. Dabei handelt es sich meist um kationische bzw. anionische
Tenside, die mit den Analyten ein nach außen ungeladenes Ionenpaar bilden können. Die
Retention erfolgt durch hydrophobe Wechselwirkungen des unpolaren Rests des Ionenpaar-
reagenzes mit der stationären Phase. Dabei gibt es zwei Modellvorstellungen: Bei der einen
bildet sich erst das Ionenpaar, das sich danach an der stationären Phase retardieren lässt. Die
andere Vorstellung geht davon aus, dass das Ionenpaarreagenz sich zuerst allein an die
stationäre Phase anlagert und somit temporäre Ionenaustauscherplätze zu Verfügung stellt.
In der vorliegenden Dissertation wird ausschließlich die Ionenaustauschchromatographie
verwendet und mit IC abgekürzt. Die IC beruht auf Austauschvorgängen zwischen Ionen in
Lösung und Ionen gleichen Vorzeichens an der Oberfläche der stationären Phase. Kationen
werden durch elektrostatische Wechselwirkungen (Coulombkräfte) an negativ geladene
Austauscherfunktionen wie z. B. Sulfonat- oder Carbonatgruppen gebunden, während sich
Anionen an positiv geladene Funktionen, typischerweise quartäre Ammoniumgruppen,
anlagern. Der Name „Austauscher“ rührt daher, dass Ionen sich wiederholt gegenseitig von
den Wechselwirkungsplätzen verdrängen.

Theorie – Ionenchromatographie
Das sich in wässriger Lösung einstellende Austauschgleichgewicht für einen Anionen-
austauscher kann wie folgt beschrieben werden:
Harz-NR3+ E− + Harz-NR3
+ A− + E−A−Gl. 1
Gl. 2 ( ) ( )
( ) ( )
( ) ( )
( ) ( )m,As,E
m,Es,A
AENRHarz
EANRHarzEA,
3
3
−−
−−
−−+
−−+
⋅
⋅=
⋅
⋅=
−
−
cc
cc
cc
ccK
E− = Eluention A− = Analytion c = Konzentration s = stationäre Phase m = mobile Phase
Die Lage des Gleichgewichts ist abhängig von der Affinität des Analytions zur stationären
Phase und wird durch die Konstante KA,E (Selektivitätskoeffizient) beschrieben. Zwei
Analyten können nur voneinander getrennt werden, wenn sich ihre Selektivitätskoeffizienten
unterscheiden (s.a. Seite 28, Gl. 6). Die Affinität verschiedener Ionen zu den Austausch-
plätzen ist vor allem von dem Verhältnis ihrer Ladung zu ihrem hydrodynamischen Radius
abhängig. Die Retention steigt mit höherer Ladung und mit kleiner werdendem Radius des
Ions inklusive seiner Solvathülle. Die Güte der Trennung wird entscheidend durch das ver-
wendete Säulenmaterial bestimmt und kann weiterhin über die Wahl des Eluenten und der
anderen chromatographischen Bedingungen gelenkt werden.
- 25 -

Theorie – Ionenchromatographie
2.2.3 Aufbau eines Ionenchromatographen
Der Aufbau eines typischen Ionenchromatographen (s. Abb. 11) ähnelt dem klassischer
HPLC-Geräte. Benötigt wird eine möglichst pulsationsfrei arbeitende Pumpe zur Förderung
des Elutionsmittels, ein Dreiwege-Injektionsventil für die Zugabe der Probe, die Trennsäule
sowie ein Detektor, der zur Datenverarbeitung an einen PC angeschlossen wird. Die in der IC
am häufigsten angewendete Detektionsart ist die suppressierte Leitfähigkeitsdetektion. Mit
Hilfe eines Suppressors wird die Eigenleitfähigkeit des Eluenten herab- und, wenn möglich,
die Leitfähigkeit des Analyten heraufgesetzt. Dadurch wird das Nettosignal der Analytionen
erhöht und die Empfindlichkeit der Leitfähigkeitsdetektion verbessert.
06.7
A
Abb. 11: Schematischer Aufbau eines typischen Ionenchromatographs mit Säulensuppressor und Leitfähigkeits-detektion. Ventil in Befüllungsposition (abgeändert nach [59])
Die Säulen und Verbindungsschläuche müssen aus Inertmaterialien, wie z.B. Stahl oder
PEEK (Polyether-Etherketon) bestehen und auch hohen Druck von bis zu 300 bar verkraften.
Die Proben werden typischerweise mit Injektionsvolumina im Bereich von 10–50 µL in den
mit einer Flussrate von ca. 0.3–3 mL strömenden Eluenten injiziert. Werden spezielle
Anreicherungsverfahren oder alternative Detektionsmethoden wie die ICP-MS genutzt sind
auch höhere Injektionsvolumina bis in den mL-Bereich üblich.
Eluent
HPLC-Pumpe
Probe
Probenschleife
Injektionsventil
Abfall
Abfall
Detektor
Auswertung
IC-S
äule
Supp
ress
orsy
stem
- 26 -
Theorie – Ionenchromatographie
2.2.4 Grundlegende chromatographische Kenngrößen
Eine chromatographische Trennung lässt sich mit Hilfe eines Chromatogramms darstellen,
welches durch Auftragung des Detektorsignals als Funktion der Zeit registriert wird. Unter
Retention versteht man die messbare Verzögerung einer Substanz während des Trenn-
prozesses, die durch Wechselwirkungen mit der stationären Phase zu Stande kommt. Die Zeit,
die ein Analyt zum Passieren einer Trennsäule bis hin zu Detektor benötigt, wird als
(Brutto-) Retentionszeit tR bezeichnet. Sie setzt sich additiv aus der Totzeit t0* (s. Gl. 4) und
der Nettoretentionszeit t’R zusammen:
Gl. 3 R*0R t' tt +=
Als Nettoretentionszeit t’R ist die Zeit definiert, in der eine Substanz nicht wandert. Unter der
Totzeit t0 wird die Zeit verstanden, die eine Substanz zum Durchfließen einer Trennsäule
benötigt, ohne mit der stationären Phase in Wechselwirkung zu treten. Die gesamte Totzeit t0*
beinhaltet zusätzlich eine mögliche weitere Flusszeit außerhalb der Trennsäule. Die verwen-
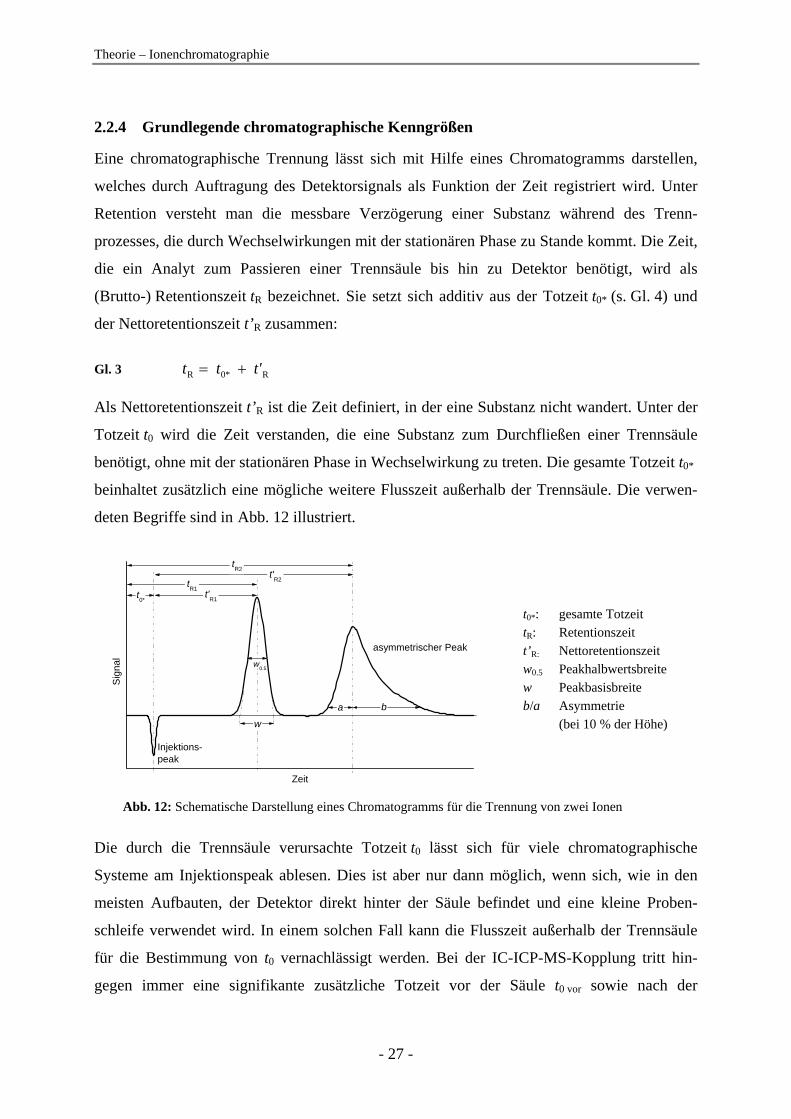
deten Begriffe sind in Abb. 12 illustriert.
t0*: gesamte Totzeit : Reten
it
he)
. 12: Schematische Darstellung eines Chromatogramms für die Trennung von zwei Ionen
Die durch die Trennsäule verursachte Totzeit t0 lässt sich für viele chromatographische
Sig
nal
Zeit
t0*
tR1
tR2
a b
w0.5
Injektions-peak
asymmetrischer Peak
t'R1
w
t'R2
tR
’tionszeit
ze t R: Nettoretentions w0.5 Peakhalbwertsbreite
w b/a
Peakbasisbreite Asymmetrie
(bei 10 % der Hö
Abb
Systeme am Injektionspeak ablesen. Dies ist aber nur dann möglich, wenn sich, wie in den
meisten Aufbauten, der Detektor direkt hinter der Säule befindet und eine kleine Proben-
schleife verwendet wird. In einem solchen Fall kann die Flusszeit außerhalb der Trennsäule
für die Bestimmung von t0 vernachlässigt werden. Bei der IC-ICP-MS-Kopplung tritt hin-
gegen immer eine signifikante zusätzliche Totzeit vor der Säule t0 vor sowie nach der
- 27 -

Theorie – Ionenchromatographie
Säule t0 nach auf. Die gesamte Totzeit t0* am Injektionspeak setzt sich im Fall der IC-ICP-MS-
Kopplung wie folgt zusammen:
- 28 -
Gl. 4 Säulenach 0Säulevor 00*0 t t tt ++=
Die Totzeit nach der Säule wird durch die notwendige Verwendung einer längeren Verbin-
al zu injizierende Probenmenge ist abhängig von der Säulenkapazität Q. Diese ist
Für die Charakterisierung der Affinität eines Ions zur stationären Phase gibt man weniger die
Gl. 5
dungskapillare vom Säulenausgang bis zum ICP-MS und durch den Weg der Probe durch die
Zerstäuberkammer bis ins Plasma bedingt. Da die IC-ICP-MS nicht auf den Einsatz kleiner
Probenschleifen limitiert ist, kann zudem eine beträchtliche Totzeit vor der Säule zu Stande
kommen.
Die maxim
im Gegensatz zur IC-ICP-MS bei der IC mit suppressierter Leitfähigkeitsdetektion auf
Q < 100 µeq Säule−1 limitiert, da nur eine begrenzte Auswahl an suppressierbaren Elutions-
mitteln zu Verfügung steht (z.B. Carbonat/Bicarbonat oder Hydroxid). Diese Eluentanionen
sind nur bis zu einem maximalen Konzentrationslimit suppressierbar und besitzen nur relativ
schwache Elutionskraft z.B. im Vergleich zu Nitrationen.
die Retentionszeit tR, sondern vielmehr den von apparativen Bedingungen unabhängigen
Kapazitätsfaktor k’ an. Dieser ermöglicht z.B. auch bei Trennungen mit unterschiedlicher
Flussrate oder bei unterschiedlichen Säulendimensionen eine vergleichende Aussage über die
Retention einer Substanz. Der dimensionslose Kapazitätsfaktor wird aus dem Quotient der
Nettoretentionszeit t’R und der Totzeit t0 gebildet.
0
0R
0
R
t-tt
tt' k' *==
Bei kleinem Wert von k’ eluiert ein Ion nahe am Injektionspeak, was bedeutet, dass es nur
Die Selektivität α, auch relative Retention genannt, ist ein Maß für die relative Signallage
Gl. 6
wenig von nichtionischen Substanzen abgetrennt werden kann.
zweier Ionen und hängt folgendermaßen mit dem Kapazitätsfaktor zusammen (Gl. 6):
12R1
R2
1
2 mit k' k' t't'
k'k'α ≥==

Theorie – Ionenchromatographie
Die Selektivität ist eine thermodynamische Größe, die bei konstanter Temperatur nur von den
stoffspezifischen Eigenschaften der zu trennenden Probenkomponenten und von den Eigen-
schaften der stationären und der mobilen Phase abhängt.
Je höher der Wert von α ist, desto besser ist die Trennung. Allerdings vergrößert sich bei zu
großer relativer Retention der Zeitaufwand für eine Trennung. Für Ionen, die zu sehr von
einander getrennt werden, versucht man daher die relative Retention zu verringern.
Definitionsgemäß ist die Selektivität α immer größer als eins. Im Rahmen dieser Dissertation
wird zwischen der Selektivität und der relativen Retention ein Unterschied gemacht: Die
relative Retention verschiedener Ionen wird in Bezug auf ein ausgewähltes Anion ermittelt,
dessen Kapazitätsfaktor bzw. Nettoretentionszeit immer im Nenner steht, auch wenn der Wert
größer als der Zähler sein sollte. Deshalb ergeben sich im Fall früh eluierender Analyten für
die relative Retention Werte, die kleiner als eins sind.
Da die Selektivität α nur die Lage von Peaks zu einander, nicht aber die Peakbreite berück-
sichtigt, beinhaltet sie keine Information über die Effizienz einer Trennung. Die Güte der
Trennung zweier benachbarter Peaks lässt sich mit Hilfe der Auflösung R beurteilen. Die
Berechnung der Auflösung ist über folgende Beziehungen möglich:
Gl. 7 (2)0.5,(1)0.5,
R1R2
(2)0.5,(1)0.5,
R1R2
21
R1R2 181
2)(
35424
2)(
wwtt.ww
.
ttwwttR
+−
=+
−=
+−
=
Die Umrechnungsfaktoren für die Peakbreite beruhen auf der Annahme, dass die Form eines
chromatographischen Peaks in erster Näherung als Gaußkurve dargestellt werden kann.
Chromatographische Signale sind nicht unendlich schmal, da sie auf Grund statistischer
Diffusions- und Strömungsprozesse mit steigender Retentionszeit verbreitert werden. Unter
der Annahme, dass die Peaks die Form einer Gaußverteilung annehmen, lassen sich mit Hilfe
der Peakbasisbreite w oder Peakhalbwertsbreite w0.5 charakteristische Kenngrößen zur
Beurteilung der Güte einer Trennung berechnen.
Die Peakbasisbreite ist durch die Schnittpunkte der Wendetangenten mit der Abszisse
definiert und beträgt das Vierfache der Standardabweichung σ der Gaußverteilung. Die Peak-
halbwertsbreite ist die Peakbreite bei halber Höhe eines Signals und entspricht dem
2.354-fachen der Standardabweichung (s. Abb. 13).
- 29 -

Theorie – Ionenchromatographie
1.000
0.500
0.882
0.134
Höh
e
Zeit
0.607
σ
2σw0.5=2.354σ
w=4σ
Abb. 13: Die Gaußverteilung mit ihren wichtigsten Kenngrößen
Ist die Differenz der Retentionszeiten zweier Peaks groß im Verhältnis zu ihren Basis- oder
Halbwertsbreiten, so liegt eine hohe Auflösung vor. Unter Voraussetzung einer idealen
Peaksymmetrie können zwei Komponenten bei R = 0.5 noch unterschieden und identifiziert
werden. Etwa ab R = 1.2 spricht man von einer Basislinientrennung. Für eine Quantifizierung
strebt man eine Auflösung von R = 1.5–2 an. Auflösungen von R > 2 sind nicht erwünscht, da
sich die Analysenzeiten dadurch zu sehr verlängern.
Eine exakt gaußförmige Peakform wird in der Realität bei chromatographischen Trennungen
nur selten erreicht. Mit Hilfe der Asymmetrie As wird die Abweichung von der idealen Peak-
form quantifiziert. Die Asymmetrie ist direkt aus einem Chromatogramm bestimmbar. Dazu
wird am Peakmaximum eine Senkrechte auf die Abszisse gefällt. Bei 10 % der Peakhöhe
werden die Strecken a und b bestimmt (s. Abb. 12) und zueinander ins Verhältnis gesetzt:
Gl. 8 abA =s
Für perfekt symmetrische Signale gilt As = 1. Ein Peak, mit einer auf der rechten Seite länger
auslaufenden Flanke, wie der zweite Peak in Abb. 12, besitzt eine Asymmetrie As > 1. Man
spricht in diesem Fall von einem „Tailing“ des Peaks. Beim umgekehrten Fall mit As < 1
bezeichnet man die Asymmetrie als „Fronting“.
Die Ursachen für nicht ideale Peakformen sind vielfältig: Zum Beispiel führen Überladungs-
effekte zu einem Fronting. Für das Tailing ionenchromatographischer Peaks werden meist
sekundäre Wechselwirkungen der Analyten mit der stationären Phase (s. Kap. 2.4.3)
verantwortlich gemacht. Qualitätsmängel der Säulenpackung können beide Asymmetrie-
formen und sogar Doppelpeaks erzeugen.
- 30 -

Theorie – Ionenchromatographie
Ein Maß für die Effizienz einer Säule ist die so genannte Bodenzahl N, auch Trennstufenzahl
genannt. Je größer die Bodenzahl für einen Peak bei einer bestimmten Retentionszeit, desto
schmaler ist der Peak. Hohe Bodenzahlen wirken sich nicht nur auf das Auflösungsvermögen
positiv aus, sondern auch auf die Nachweisgrenzen, denn bei geringer Peakverbreiterung
werden die Signale für eine bestimmte Analytkonzentration höher. Die Bodenzahl kann über
die folgenden Beziehungen direkt aus einem Chromatogramm berechnet werden:
Gl. 9 2
0.5
R2
R2
R 54516 ⎟⎟⎠
⎞⎜⎜⎝
⎛=⎟
⎠⎞
⎜⎝⎛=⎟
⎠⎞
⎜⎝⎛=
wt' .
wt'
σt' N
Um Bodenzahlen von Säulen verschiedener Länge besser miteinander vergleichen zu können
werden Bodenzahlen oft in theoretischen Böden pro Meter angegeben (TP m−1, von engl.:
theoretical plates per meter).
Die Bodenzahl N, die Selektivität α und der Kapazitätsfaktor k’ können zusammen in einer
Gleichung mit der Auflösung in Beziehung gebracht werden:
Gl. 10 114 k'α +
Gemäß Gl. 10 gibt es drei Mö
21 k'αNR ⋅−
⋅=
glichkeiten, die Auflösung zweier benachbarter Peaks zu
ver
1. N durch Erhöhung der Säulenlänge oder durch die Wahl
2. ndung anderer stationärer Phasen oder durch
3. s k’ durch Änderung der Konzentration und der
Ionenstärke des Elutionsmittels.
bessern:
Erhöhung der Bodenzahl
einer effizienteren Säule.
Erhöhung der Selektivität α durch Verwe
Änderung der Eluentzusammensetzung.
Änderung des Kapazitätsfaktor
- 31 -

Theorie – Ionenchromatographie
- 32 -
2.2.5 Stationäre Phasen
In der IC werden vorwiegend stationäre Phasen aus organischen Polymerharzen eingesetzt, in
der Kationen-IC auch vermehrt Silicagel. Für die Anionen-IC werden meist folgende Arten
stationärer Phasen auf der Basis von organischen Polymerharzen verwendet:
• Latex-Anionenaustauscher
• oberflächenfunktionalisierte Anionenaustauscher
• gepfropfte Anionenaustauscher
Der Aufbau und die resultierenden Trenneigenschaften werden in Kap. 4.1 beschrieben. Des
Weiteren werden dort ausführlich die Vor- und Nachteile verschiedener stationärer Phasen
insbesondere für die Multianionentrennung diskutiert.
Als gebräuchlichste Harze für stationäre Phasen in der IC finden Polystyrol/Divinylbenzol-
Copolymere (PS/DVB) Anwendung, daneben existieren auch Materialien auf der Basis von
Methacrylat- oder Polyvinyl-(co-)polymeren. Die Durchmesser der Polymerpartikel liegen
typischerweise bei 3–25 µm. Die Polymere lassen sich auch über ihre Porosität unterscheiden,
wobei man bei mittleren Porengrößen von 2–50 nm von mesoporösen und bei 50–400 nm-
Poren von makroporösen Materialien spricht. Die Materialien besitzen eine große Oberfläche
von 200–500 m2 g−1, die nach der Funktionalisierung für den Ionenaustausch genutzt werden
kann. Weitere wichtige physikalische Eigenschaften der Packungsmaterialien sind Druck-
stabilität bis ca. 300 bar sowie pH- und Lösungsmittelstabilität, damit die stationäre Phase
unter dem Einfluss verschiedener Elutionsmittel möglichst wenig schrumpft oder aufquillt.
Eine wichtige Kenngröße einer stationären Phase ist ihre (Austausch-) Kapazität Q, die durch
die Anzahl der Austauscherfunktionen bestimmt wird. Die Kapazität wird meist in mmol g−1
oder in mmol Säule−1 angegeben. Statt der Austauscherplätze ist es leichter, die entsprechende
Stoffmenge an gebundenen Gegenionen zu messen, die mit meq (von engl.: milliequivalents)
abgekürzt wird. Die Angabe „pro Gramm“ bezieht sich, wenn nicht anders angegeben, auf das
getrocknete Material in der H-Form bei Kationen- und in der Cl-Form bei Anionen-
austauschern. Wegen Ungenauigkeiten bei der Massenbestimmung durch den schwankenden
Wassergehalt der Polymere, ist es als dritte Alternative auch üblich, sich auf das Volumen der
Materialien zu beziehen (meq mL−1). Für einen Vergleich der Kapazität von Ionen-
austauschern sind die verschiedenen Arten der Angabe unbedingt zu beachten. Aus

Theorie – Ionenchromatographie
- 33 -
praktischen Gesichtspunkten ist die Angabe der Kapazität pro Säule sinnvoll, da damit am
schnellsten die notwendige Eluentkonzentration abgeschätzt werden kann.
2.3 Kopplungstechniken
2.3.1 Übersicht
Die Kopplung von Analysentechniken kann zu einem beträchtlichen Synergieeffekt führen.
Das Sprichwort „Das Ganze ist mehr als die Summe seiner Teile“ trifft auch auf den
Informationsgehalt und weitere Vorteile von Kopplungstechniken zu.
Als Kopplungstechnik wird die Kombination von zwei in der Regel eigenständigen Analysen-
techniken bezeichnet. Im Englischen hat sich besonders der Begriff „hyphenation“ zu einem
Schlagwort der analytischen Chemie entwickelt. Die Bezeichnung „hyphenated techniques“
wurde zuerst von Hirschfeld [60] vorgeschlagen und bezieht sich auf die Namen der
Kopplungsmethoden, die häufig durch einen Bindestrich zwischen den Abkürzungen der
Einzeltechniken gekennzeichnet werden.
Bei Kopplungsmethoden wird in den meisten Fällen eine Trenntechnik mit einer spektro-
metrischen Detektion verbunden [61]. Die wichtigsten Trenntechniken in diesem Zusammen-
hang sind die Gaschromatographie (GC), die verschiedenen Arten der Hochleistungsflüssig-
keitschromatographie (HPLC), und elektrophoretische Trenntechniken. Anhand der verwen-
deten Spektrometrie lässt sich im Allgemeinen erkennen, ob eine Kopplungstechnik im
Bereich der organischen oder der anorganischen Analytik eingesetzt wird:
− Kopplungen für die organische Analytik bedienen sich meist molekülselektiver Detek-
tionsmethoden [61, 62]. Als nützlichste Detektionsarten für die Identifizierung von
komplizierten oder unbekannten Molekülen sind die Kernresonanzspektroskopie
(NMR) und die Molekülmassenspektrometrie zu nennen [63].
− In der anorganischen Analytik wird hauptsächlich elementselektive Detektion z.B.
durch Atomspektrometrie genutzt. Die Kopplungsmethoden finden dort die häufigste
Anwendung im Bereich der Elementspeziesanalyse [1, 64].

Theorie – Kopplungstechniken
- 34 -
Die On-line-Kopplung HPLC-Atomspektrometrie lässt sich nach Seubert folgendermaßen
definieren [65]:
„Als On-line-Kopplung Flüssigchromatographie-Atomspektrometrie werden Analysenver-
fahren bezeichnet, welche die Analyten im Eluat einer auf flüssigchromatographischen Wege
ablaufenden Trennung direkt mit Hilfe atomspektrometrischer Methoden detektieren.“
Neben den klassischen Methoden der Atomabsorptions- und Atomemissionsspektroskopie
wird auch die im Rahmen dieser Dissertation verwendete ICP-Massenspektrometrie in das
Gebiet der Atomspektrometrie eingeordnet, da die analytischen Aussagen der Methoden
ähnlich sind.
Die erste weit verbreitete Massenspektrometriekopplung stellte die Gaschromatographie-
Massenspektrometrie (GC-MS) dar, die heute längst zu einer Routinemethode der organi-
schen Analytik zur Identifizierung von verdampfbaren Substanzen in komplexen Gemischen
geworden ist.
Die Kopplung von Flüssigchromatographie und Massenspektrometrie (LC-MS) gestaltete sich
wesentlich schwieriger, da es lange nicht möglich war, das für die MS benötigte Hochvakuum
auch bei hoher Flüssigkeitszufuhr durch den Eluenten aufrecht zu erhalten. Erst die Ein-
führung von Ionisierungstechniken, die auch bei Normaldruck Ionen erzeugen können,
verhalf der LC-MS zum Durchbruch: Als Ionenquellen für die molekülselektive MS stehen
heute z.B. APCI (Atmospheric Pressure Chemical Ionization) und ESI (Electrospray
Ionization) zu Verfügung. Für Kopplungsmethoden mit elementselektiver MS spielt die
ICP-MS eine dominierende Rolle.
Es werden Off-line- und On-line-Kopplungen unterschieden. Bei Off-line-Methoden werden
zwei Techniken mit einer zeitlichen Unterbrechung nacheinander und/oder an verschiedenen
Orten durchgeführt. Der Begriff On-line-Kopplung weist auf den direkten Zusammenbau von
zwei Techniken und ihre kontinuierliche Durchführung hin. Neben Analysentechniken
können auch viele physikalisch-chemische Probenvorbereitungen, die früher in einem sepa-
raten Arbeitsschritt durchgeführt wurden, wie zum Beispiel Extraktionen von Proben oder
Anreicherung durch Adsorption, in eine On-line-Form gebracht werden.
Off-Line-Analysen haben den Vorteil gegenüber On-line-Methoden, dass kein spezielles
Interface zwischen Trenntechnik und Spektrometrie benötigt wird und dass die Qualitäts-
kontrolle von Analysen einfacher zu gestalten ist [66]. Der letztere Punkt ist darin begründet,

Theorie – Kopplungstechniken
dass bei off-line Arbeitsweise Probenfraktionen aufbewahrt und wiederholt gemessen werden
können. Die allgemeinen Vorteile der On-line-Kopplungen sind die leichtere Automa-
tisierbarkeit und der höhere Probendurchsatz. Des Weiteren werden sowohl die Kontami-
nationsgefahr, als auch Probleme mit der Stabilität von Analyten durch eine direkte
Verbindung verringert.
2.3.2 Die On-Line-Kopplung IC-ICP-MS
Im Fall der On-line-Kopplung von Ionenchromatographie und ICP-MS (IC-ICP-MS) wird die
Speziesselektivität der IC mit der Elementselektivität der ICP-MS kombiniert. Dies
ermöglicht die Durchführung von Elementspeziesanalysen. Ein weiterer Vorteil ist, dass
chromatographische Peaks nicht mehr in jedem Fall von einander aufgelöst werden müssen,
da Komponenten auch durch ihre Elementzusammensetzung unterscheidbar sind. Dadurch
lässt sich die Analysenzeit verringern und es können mehr Substanzen gleichzeitig analysiert
werden. Auch konventionelle Elementanalysen können verbessert werden, da mit der IC eine
Analytanreicherung bei gleichzeitiger Abtrennung von der störenden Probenmatrix erreicht
werden kann. Das grundlegende Konzept zur IC-ICP-MS-Kopplung ist in Abb. 14 illustriert.
Ionenchromatographie ICP-Massenspektrometrie
elementspezifische Detektionmit sehr niedrigen Nachweisgrenzen
On-line-KopplungIC-ICP-MS
speziesselektive Wechselwirkungen(Ladung, Größe)
Elementspeziesanalyse Anreicherung/Matrixabtrennung=> UltraspurenanalyseMultianionenbestimmung
Abb. 14: Grundlegendes Konzept der IC-ICP-MS und einige sich daraus ergebende Anwendungsgebiete (verändert nach [65])
- 35 -

Theorie – Analyten für die Anionen-IC-ICP-MS
- 36 -
2.4 Analyten für die Anionen-IC-ICP-MS
2.4.1 Übersicht
Mit der Kopplung von Ionenchromatographie und ICP-MS lässt sich eine Vielzahl von
Anionen bis in den Spurenbereich bestimmen. Eine wichtige Vorraussetzung für die Bestim-
mung ist, dass die Anionen ein mit der ICP-MS detektierbares Element enthalten. Da mit der
ICP-MS mehr als 75 Elemente in Spurenkonzentrationen analysierbar sind, ist diese Detek-
tionsmethode auch fast universell für alle Anionen einsetzbar. Ungeeignet ist sie aber z.B. für
Spezies, die ausschließlich aus Sauerstoff, Stickstoff, Kohlenstoff, Wasserstoff und Fluor
bestehen, da diese Elemente in zu hohen Konzentrationen im Plasma vorliegen und/oder nicht
ausreichend ionisiert werden können. In der folgenden Übersicht werden ausgesuchte
Analyten für die Anionen-IC-ICP-MS aufgeführt.
Halogenverbindungen
− Cl−, ClO−, ClO2−, ClO3
−, ClO4−, Chloressigsäuren
− Br−, BrO−, BrO3−, Bromessigsäuren
− I−, IO3−, IO4
−, I3−, (IX2
−, I2X−), Iodessigsäuren
− kleine organische halogenhaltige Moleküle mit saurem Charakter Verbindungen von Elementen der sechsten Hauptgruppe
− S2−, SO32−, SO4
2−, S2O32−, SCN−
− SeO32−, SeO4
2−, CH3SeCH2CH2CH(NH2)COOH
− TeO32−, H4TeO6
2− Verbindungen von Elementen der fünften Hauptgruppe
− PO23−, PO3
3−, PO43−, P2O7
4−, P3O105−, P4O13
6−, PO3F22−
− AsO33−, AsO4
3−, CH3AsO(OH)2, (CH3)2AsOOH, C6H5AsO(OH)2, (CH3)3As+CH2COO−, (CH3)3As+CH2CH2OH − SbO3
3− Verbindungen von Elementen der vierten und dritten Hauptgruppe
− SiO3−, SiF6
2− − GeO32−
− B4O72−, BF4
− Verbindungen von Nebengruppenelementen
− VO43−, CrO4
2−, MoO42−, WO4
2−, MnO4−, TcO4
−, ReO4−, Fe(CN)6
3−, RuO42−, OsO4
2− Metalle in anionischen Komplexen
− Metall-Komplexe mit: Ethylendiamintetraacetat (EDTA), Nitrilotriacetat (NTA), Citrat, Tartrat u.v.m
− Chlorokomplexe der Platingruppenelemente IC-ICP-MS ungeeignet
− F−, OH−, CN−, OCN−, NO2−, NO3
−, N3−, CO3
2−, HCO3−
Abb. 15: Potentielle Analyten für die Anionen-IC-ICP-MS (u. Angabe einiger schlecht detektierbarer Anionen)

Theorie – Analyten für die Anionen-IC-ICP-MS
- 37 -
2.4.2 Wichtige analytspezifische Parameter
Für die Multianionenanalyse mit der IC-ICP-MS sind verschiedene Parameter zu beachten,
durch welche sich entscheidet, wie viele und welche Anionen gleichzeitig bestimmt werden
können, und wie niedrig die erreichbaren Nachweisgrenzen sind:
• Die Valenz, Größe und Polarisierbarkeit der Anionen bestimmt hauptsächlich ihre
Retention und Trennbarkeit in einem vorgegebenen Trennsystem (stationäre Phase,
Eluent, Temperatur u.a.). Des Weiteren kann die Molekülgeometrie Einfluss auf die
Selektivität haben. Chromatographisch nicht getrennte Spezies verschiedener Ele-
mente können mit Hilfe der Elementinformation des ICP-MS unterschieden werden.
• Die Redoxreaktivität nebeneinander zu bestimmender Spezies darf nicht zu hoch sein,
damit diese nicht abreagieren.
• Es dürfen keine unlöslichen Salze entstehen.
• Die Nachweisgrenzen sind zum einen abhängig vom Ausmaß der chromatographi-
schen Bandenverbreiterung, die mit der Retentionszeit steigt, aber zum anderen vor
allem von der Nachweisstärke der ICP-MS. Die Nachweisstärke der ICP-MS für das
jeweilig zu detektierende Element wird bestimmt von
o der ersten Ionisierungsenergie
o der Isotopenhäufigkeit
o Interferenzen auf dem Masse-zu-Ladungs-Verhältnis
o und der Massenempfindlichkeit des ICP-MS.
Auf Seiten der Ionenchromatographie wird die Trennung von Ionen hauptsächlich durch ihre
Valenz und Größe bzw. der damit verbundenen Polarisierbarkeit beeinflusst. Ob eine Analyt-
spezies bei einem bestimmten pH-Wert des Eluenten mehrfach geladen oder überhaupt an-
ionisch vorliegt, hängt von dem pKs-Wert ihrer korrespondierenden Säure ab (s. Tab. 2). Im
Rahmen dieser Dissertation wird als Eluent Ammoniumnitrat im leicht sauren Milieu
(pH = 4–6) verwendet. Dies hat den Vorteil, dass die meisten Anionen einfach geladen
vorliegen. Mehrfach geladene Anionen hätten teilweise wesentlich längere Retentionszeiten,
wodurch sich die Trennzeit unnötig erhöhen würde. Schwache Säuren, wie z.B. Arsenige
Säure, Borsäure und Germaniumsäure werden in einem leicht sauren Eluenten nicht deproto-
niert und somit nicht durch ionische Wechselwirkungen retardiert. Sie sind als Analyten
weniger geeignet, aber als kontinuierliche interne Standards einsetzbar (s. Kap. 4.5.4).

Theorie – Analyten für die Anionen-IC-ICP-MS
- 38 -
Tab. 2: Dissoziationskonstanten einiger Säuren (aus [67, 68])
Name Formel T / °C pKs1 pKs2 pKs3
Arsenige Säure H3AsO3 25 9.29 Arsensäure H3AsO4 25 2.26 6.76 11.29 Borsäure H3BO3 20 9.27 >14 Bromwasserstoffsäure HBr 25 −9 Hypobromige Säure HBrO 25 8.55 Bromsäure HBrO3 25 ~0 Bromessigsäure BrCH2COOH 25 2.90 Brompropansäure C3H5BrO2 25 4.0 Chlorwasserstoffsäure HCl 25 −7 Hypochlorige Säure HClO 25 7.40 Chlorige Säure HClO2 25 1.94 Chlorsäure HClO3 25 −2.7 Perchlorsäure HClO4 20 −10 Chromsäure H2CrO4 25 0.74 6.49 Dichromsäure H2Cr2O7 25 <<0 0.07 Germaniumsäure H2GeO3 25 9.01 12.3 Hypoiodige Säure HIO 25 10.5 Iodsäure HIO3 25 0.78 Iodessigsäure IH2CCOOH 25 3.18 Phosphorige Säure H3PO3 20 1.3 6.70 Phosphorsäure H3PO4 25 2.16 7.21 12.32 Schwefelwasserstoff H2S 25 7.05 19 Schwefelige Säure H2SO3 25 1.85 7.2 Schwefelsäure H2SO4 25 −3 1.99 Thiocyansäure HSCN 25 −1.8 Selenige Säure H2SeO3 25 2.62 8.32 Selensäure H2SeO4 25 <0 1.7 Tellurige Säure H2TeO3 25 2.48 7.70 Ortho-Tellursäure H6TeO6 18 7.68 11.0
Ein leicht saurer Eluent hat außerdem den Vorteil, dass viele Salze, insbesondere von Schwer-
metallen, besser löslich sind als im Alkalischen. Der pH-Wert sollte allerdings nicht niedriger
als etwa drei liegen, da sonst die Redoxaktivität einiger Ionen zu sehr ansteigt. Die Redox-
reaktionen der Oxyhalogenide werden zum Beispiel durch Protonen katalysiert.
Im Spurenbereich liegen auch Redoxspezies nebeneinander vor, die gemäß ihrer Standard-
potentiale miteinander reagieren könnten. Durch die geringe Konzentration der Ionen ist die
Wahrscheinlichkeit aber gering, dass sie auf einander treffen und es zu einer erfolgreichen
Reaktion kommt. So ist zum Beispiel der geschwindigkeitsbestimmende Schritt der Kompro-
portionierungsreaktion von Bromid und Bromat zu Brom im Spurenbereich so langsam, dass
keine Konzentrationsänderung messbar ist. Andere Oxidationsmittel, wie z.B. Chlorit,
reagieren dagegen schneller und können somit nur kurz (~30 Minuten, je nach Temperatur)
neben starken Reduktionsmitteln, wie z.B. Hydrogensulfid oder Iodid, vorliegen.

Theorie – Analyten für die Anionen-IC-ICP-MS
Bei Trennung an einigen stationären Phasen wird vermutet, dass sich auch die Molekül-
geometrie bzw. die dreidimensionale Ladungsverteilung von Anionen merklich auf die
Selektivität auswirkt (s. Kap. 4.1.6). Mögliche Geometrien von Anionen und typische
Beispiele sind in Abb. 16 und Abb. 17 zusammengestellt. Außer den sphärischen Anionen
haben auch die mit den besonders symmetrischen tetraedrischen oder oktaedrischen
Strukturen eine nahezu kugelsymmetrische Ladungsverteilung. Die Ladungsverteilung bei
linearen, planaren und trigonal-pyramidalen Anionen ist dagegen oft nicht in alle
Raumrichtungen gleich.
- 39 -
Abb. 16: Molekülgeometrie von Anionen (nach [69, 70])
trigonal-pyramidal ClO
Abb. 17: Molekülgeometrie von Chlorat, Bromat und Iodat
Die Empfindlichkeit und die Nachweisgrenzen der IC-ICP-MS werden vor allem auf Seiten
der ICP-MS bestimmt. Die Nachweisstärke der ICP-MS ist für Nichtmetalle auf Grund der
hohen Ionisierungsenergie schlechter als für Metalle. Der Ionisierungsgrad für Chlor liegt
zum Beispiel nur bei 0.9 % und für Brom bei 5 % (s. Tab. 3). Trotzdem lassen sich für
bromhaltige Anionen im Vergleich zu anderen Methoden immer noch sehr gute Nachweis-
grenzen erreichen, die im unteren ng L−1-Bereich liegen (s. Kap. 4.3).
sphärisch F−, Cl−, Br−, I−, S2−
linear CN−, N3
−, OH−, SCN−, [ AuCN2] −
trigonal- bzw. quadratisch-planar CO3
2−, NO3−, CH3COO−
bzw. PdCl42−
tetraedrisch SO4
2−, PO43−, ClO4
−, CrO4
−, TcO4−, ReO4
−
VO4−, SeO4
−, MnO4−
oktaedrisch [Fe(CN)6]4−, [Co(CN)6]3− ,
PF6−, SiF6
2−
3−, BrO3
−, IO3−
2

Theorie – Analyten für die Anionen-IC-ICP-MS
- 40 -
Tab. 3: Berechneter Ionisierungsgrad (M+ und M2+) verschiedener Elemente bei einer Temperatur von 7500 K und einer Elektronendichte im Plasma von 1015 cm−3 (aus [50]).
Ionisierungs-grad
Ionisierungs-energie / eV Elemente
> 95 % < 7.5 Li, Na, Mg, Al, K, Ca, Sc, Ti, V, Cr, Fe, Ga, Rb, Sr, Y, Zr, Nb, Mo, Ru, In, Sn, Cs, Hf, Ta, Tl, Pb, Ce
90–95 % 7–8.5 Mn, Co, Ni, Cu, Ge, Rh, Pd, Ag, Ba, La, W, Re, Bi
70–90 % 8–10 Be, Si, Zn, Os 50–70 % 8–10 B, As, Cd, Sb, Te, Pt, Au 20–50 % 9.7–10.5 P, Se, I ,Hg 10–20 % 10–11 S 1–10 % 11–12 C, Br
0–1 % > 13 N, O, F, Cl
Tab. 4: Einige mögliche Interferenzen für Phosphor, Schwefel, Chlor, Brom und Iod bei der ICP-MS-Detektion.
Isotop (Häufigkeit) Mögliche Interferenzen
31P (100%) [16O14NH]+, [15N16O]+, [15N2H]+ 32S (95.02%) [16O2]+, [16O15NH]+, [14N18O]+
35Cl (75.77%) [16O18OH]+, [34SH]+, [70Ge]2+
79Br (50.69%) [40Ar39K]+, [40Ar38ArH]+, [78KrH]+, [38Ar41K]+, [44Ca35Cl]+, [42Ca37Cl]+, [63Cu16O]+, [65Cu14N]+, [67Zn12C]+, [62Ni16OH]+, [60Ni19F]+, [48Ca31P]+, [39K2H]+, [78SeH]+, [31P16O3]+
81Br (49.31%) [40Ar2H]+, [40Ar41K]+, [80KrH]+, [32S16O3H]+, [33S16O3]+
127I (100%) [95Mo16O2]+, [238U16O]2+, [115Sn12C]+
In Tab. 4 werden die wichtigsten spezifischen Interferenzen für einige Elemente aufgeführt,
die im Rahmen dieser Dissertation bedeutsam sind. Da viele Messungen von Bromspezies
durchgeführt wurden, werden für 79Brom die möglichen Interferenzen zwar am ausführ-
lichsten angegeben, die meisten davon treten aber so gut wie nie auf. Die Detektion des
Isotops 79Brom ist nachweisstärker als die des Isotops 81Brom, da das Untergrundsignal auf
dem Masse-zu-Ladungs-Verhältnis 81 durch das immer im Plasma vorliegende [40Ar2H]+
höher ist. Die relevanten Bromstörungen für 79Brom sind vor allem [40Ar39K]+ sowie der
permanente Plasmauntergrund durch [40Ar38ArH]+.
Für Schwefel, Phosphor, und Chlor liegen wesentlich intensivere permanente Störungen
durch Molekülionen vor, die die Nachweisgrenzen für diese Elemente beeinträchtigen. Die
stärkste Interferenz tritt für das Isotop 32Schwefel durch das Molekülion [16O2]+ auf, so dass
auf diesem Masse-zu-Ladungs-Verhältnis sogar der Sekundärelektronenvervielfacher

Theorie – Analyten für die Anionen-IC-ICP-MS
- 41 -
überlastet wird und zur Detektion auf das Isotop 34Schwefel oder das Schwefeloxidion
[32S16O]+ ausgewichen werden muss. Nach Goessler et al. [71] sind für Schwefelspezies
Nachweisgrenzen zwischen 100 und 300 µg L−1 (bezogen auf S) erreichbar. Eine positive
Ausnahme stellt das Sulfid dar (Nachweisgrenze = 35 µg L−1), das auf Grund der Bildung von
gasförmigen Schwefelwasserstoffs effektiver ins Plasma befördert und somit empfindlicher
nachgewiesen werden kann.
Die Bildungsrate des Molekülions [SO]+ ist abhängig von der Bindungsstärke zwischen
Schwefel und Sauerstoff, die nur 518 kJ mol−1 beträgt (zum Vergleich Ce-O = 795 kJ mol−1).
Bessere Nachweisgrenzen für Schwefelspezies lassen sich z.B. durch Einsatz der Reaktions-
zellentechnologie erreichen, mit deren Hilfe man entweder die Bildungsrate des auf 32Schwefel störenden [O2]+ erniedrigt [72] oder die Bildungsrate von [SO]+ durch
Sauerstoffzugabe erhöht. Durch diese Technologie wird allerdings die Multielementfähigkeit
des ICP-MS verringert.
2.4.3 Polarisierbare Anionen
Auf Grund verschiedener Effekte, auf die in diesem Kapitel noch näher eingegangen wird,
werden polarisierbare Anionen in der Anionenchromatographie meist deutlich länger
retardiert als nicht polarisierbare Anionen. Deswegen ist es schwierig diese verschieden-
artigen Anionen zusammen in einem chromatographischen Lauf zu bestimmen. Das Problem
besteht darin, dass für die Elution der polarisierbaren Anionen starke Eluenten nötig sind.
Durch die Verwendung solch starker Eluenten können aber früh eluierende Ionen manchmal
nicht mehr ausreichend separiert werden. Für die Multianionenanalyse stellt die gleichzeitige
Analyse polarisierbarer und nicht polarisierbarer Anionen eine wichtige Voraussetzung dar.
Deswegen werden die Polarisierbarkeit von Anionen und die sich daraus ergebenden
Eigenschaften im Folgenden detailliert beschrieben.
Definition der elektronischen Polarisierbarkeit
Die Ladungen aus denen Atome und Moleküle bestehen (Kerne, Elektronen, Ionenrümpfe)
sind nicht starr verbunden. Sie sind elastisch, d.h. durch Kräfte, die in erster Näherung
proportional zur Auslenkung sind, an ihre Ruhelage gebunden.

Theorie – Analyten für die Anionen-IC-ICP-MS
Durch ein äußeres elektrisches Feld können solche Ladungen verschoben werden, wodurch
ein Dipolmoment p entsteht:
Gl. 11: αEp =
p = Dipolmoment α = Polarisierbarkeit E = Feldstärke
Die elektronische Polarisierbarkeit α ist charakteristisch für ein Teilchen (Atom, Molekül,
Anion…). In der Literatur wird für die Polarisierbarkeit meistens der griechische Buchstabe α
verwendet, welcher in der Chromatographie der Selektivität zugeordnet ist (s. Kap. 2.2.4).
Verwechslungen in dieser Dissertation sind aber ausgeschlossen, da das Symbol α für die
Polarisierbarkeit nur für die Definition in diesem Kapitel angegeben wird.
Unter der Annahme, dass die Rückstellkraft für die verschobene Ladung dem Coulomb-
Gesetz folgt, lässt sich die Polarisierbarkeit α näherungsweise auch mit folgender Formel be-
schreiben [73]:
Gl. 12: 302 rπεα ≈
ε0 = stoffspezifische Konstante r ≈ Teilchenradius
Man erkennt, dass die Polarisierbarkeit eines Teilchens neben einer stoffspezifischen
Konstante besonders von seiner Größe (Radius) abhängt.
Die spezielle Struktur von Wasser und seine speziellen Lösungseigenschaften
Obwohl Wasser einer der wichtigsten und am meisten erforschten Stoffe ist, ist die genaue
Struktur von flüssigem Wasser immer noch weitgehend ungeklärt [74]. Das Wassermolekül
ist ein Dipol und ist außerordentlich gut für die Bildung von Wasserstoffbrücken geeignet.
Die Wechselwirkungen durch Wasserstoffbrücken sind der Grund, dass Wassermoleküle im
flüssigen Zustand nicht chaotisch verteilt sind, sondern kurzzeitig geordnete Strukturen
bilden. Es werden Ketten, Ringe, Käfige und ganze Netzwerke in flüssigem Wasser vermutet,
die meisten solcher Angaben beruhen aber auf Berechnungen [74]. Mit Untersuchungen durch
Synchrotronstrahlung lassen sich auch „Momentaufnahmen“ von Wasser auf molekularer
Ebene machen, die nahe legen, dass Wasser zumindest aus Ketten und Ringen besteht [75].
- 42 -
Um ein Ion in Wasser zu lösen wird die Wasserstruktur unter Aufbrechen von Wasserstoff-
brücken zerstört (Lochbildungs- oder Cavity-Effekt). Die erforderliche Lochbildungsenergie
ist umso höher, je größer das Ion ist. Die Anordnung von Wassermolekülen um ein Ion

Theorie – Analyten für die Anionen-IC-ICP-MS
(Hydratation) führt gleichzeitig zu einem Energiegewinn auf Grund von Ion-Dipol-Wechsel-
wirkungen. Die Ion-Dipol-Wechselwirkungen sind umso stärker, je kleiner der Ionenradius
und je höher die Ionenladung ist. Die Hydratation steigt daher mit abnehmendem Ionenradius.
Polarisierbare Ionen sind groß und deswegen nur schwach hydratisiert. Aus diesem Grund
ergibt sich wiederum ein kleiner Radius entsprechender Ionen zusammen mit ihrer Hydrat-
hülle (hydrodynamischer Radius, s. Ionenbeweglichkeit oder Grenzleitfähigkeit z.B. in [76]).
Eigenschaften polarisierbarer Anionen
In der Ionenchromatographie hat die Polarisierbarkeit eines Anions einen entscheidenden
Einfluss auf seine Retention an Anionenaustauschern. Bei polarisierbaren Ionen ist die
Ladung auf ein großes Volumen verteilt und die Elektronenwolke ist leicht deformierbar. Die
Ionen besitzen einen lipophileren Charakter und daher eine höhere Affinität zu lipophilen
Phasen. Die polarisierbaren Anionen gehen verstärkt nicht-ionische, so genannte sekundäre
Wechselwirkungen mit Ionenaustauschern ein. Bei Polymerharzen auf PS/DVB-Basis lassen
sich Adsorptionseffekte hauptsächlich auf π-π-Wechselwirkungen mit dem aromatischen
Grundgerüst zurückführen [13]. Polarisierbare Ionen können zudem auf Grund ihrer
Lipophilie leichter in die Poren der Polymerteilchen eindringen und verweilen länger auf der
stationären Phase. Unterschiedlich lange Diffusionszeiten in Verbindung mit sekundären
Wechselwirkungen führen zu Peakverbreiterung sowie Peaktailing.
Die Polarisierbarkeit von Anionen ist eng mit ihrem Radius und ihrer Hydratationsenthalpie
verknüpft. Die Definition des so genannten thermochemischen Radius rth nach Roobottom
und Jenkins lautet [77]:
„Der thermochemische Radius rth wird zur Berechnung von Gitterenthalpien anorganischer
Salze mit Hilfe der Kapustinskii-Gleichung verwendet und beschreibt in diesem Zusammen-
hang den effektiven Ionenradius eines Ions.“
Die Standardenthalpieänderung für den Vorgang
- 43 -
M −+ +→+ , ∆HHydrGl. 13: (g)A (g) −+ (aq)A (aq)M
wird als Solvatationsenthalpie ∆HSolv bzw. Hydratationsenthalpie ∆HHydr im Falle von Wasser
bezeichnet. Die Hydratationsenthalpien ∆HHydr einzelner Ionen sind zwar nicht messbar,
lassen sich aber mit Hilfe von tabellierten Werten [67] für die Gitterenthalpien ∆HL (z.B. der

Theorie – Analyten für die Anionen-IC-ICP-MS
- 44 -
∆HSol (Lösung in Wasser) nach Gl. 16 berechnen:
+=
in Wasser Lösungfür ∆∆∆ HHH +=
Natriumsalze) und ihren Lösungsenthalpien
Gl. 14: H LSolvSol ∆∆∆ HH
Gl. 15: LHydrSol
Gl. 16: +−+−+− −−=MHydr,AML,AMSol,AHydr,
∆∆∆∆ HHHH
∆HSol = Lösungsenthalpie ∆HSolv = Solvatationsenthalpie, für Wasser = ∆HHydr
+ −
Die Werte von Gitterenthalpien sind positiv, während Hydratationsenthalpien für Ionen
ber noch aussagekräftiger als die Hydratationsenthalpie ∆HHydr ist
e ydratationsenthalpie ∆GHydr (Gibbs-Energie), bei der auch noch entropische Effekte
Gl. 17:
Wasser) zu bewegen; dabei wird das Lösungsmittel als kontinuierliches Dielektrikum mit der
n ielektrizitätskonstante εrel behandelt [76]. Die Bornsche Gleichung lautet wie folgt:
∆HL = Gitterenthalpie M = Kation A = Anion
Wert für ∆HHydr, Na+ = 405 kJ mol−1 nach [76]
negative Werte besitzen, da die Hydratation von Ionen immer zu einem Energiegewinn führt.
Schwieriger zu bestimmen a
die frei H
mit berücksichtigt werden:
HydrHydrHydr
T = Temperatur ∆S
∆∆∆ STHG ⋅−=
Hydr = Hydratationsentropie
Die Entropieänderung beim Hydratisieren eines Ions hängt davon ab, in welchem Maß die
Hydrathülle um ein Ion herum geordnet ist, bzw. wie stark der Ordnungszustand von Wasser
erhöht oder erniedrigt wird. Kleine hochgeladene Ionen bewirken eine stärker geordnete
Struktur der Hydrathülle als große, einfach geladene Ionen. Experimentell bestimmbar sind
freie Hydratationsenthalpien oder Entropiedaten durch Extraktionsexperimente mit quartären
Ammoniumbasen [78-80]. Mit Hilfe einer Beziehung, die von Max Born abgeleitet wurde,
kann man ∆GHydr auch näherungsweise berechnen. Er ordnete ∆GHydr der elektrischen Arbeit
zu, die aufgewendet werden muss, um ein Ion aus dem Vakuum in das Lösungsmittel (hier
relative D
)11(∆ A22 NezG −⋅−=Gl. 18:
8 rel0Hydr εrπε
z = Ionenladung r = Radius des Ions NA = Avogadro-Konstante
ε0 = Dielektrizitätskonstante des Mediums bei 25°C
εrel = relative Dielektrizitätskonstante (für Wasser bei 25°C = 78.54)
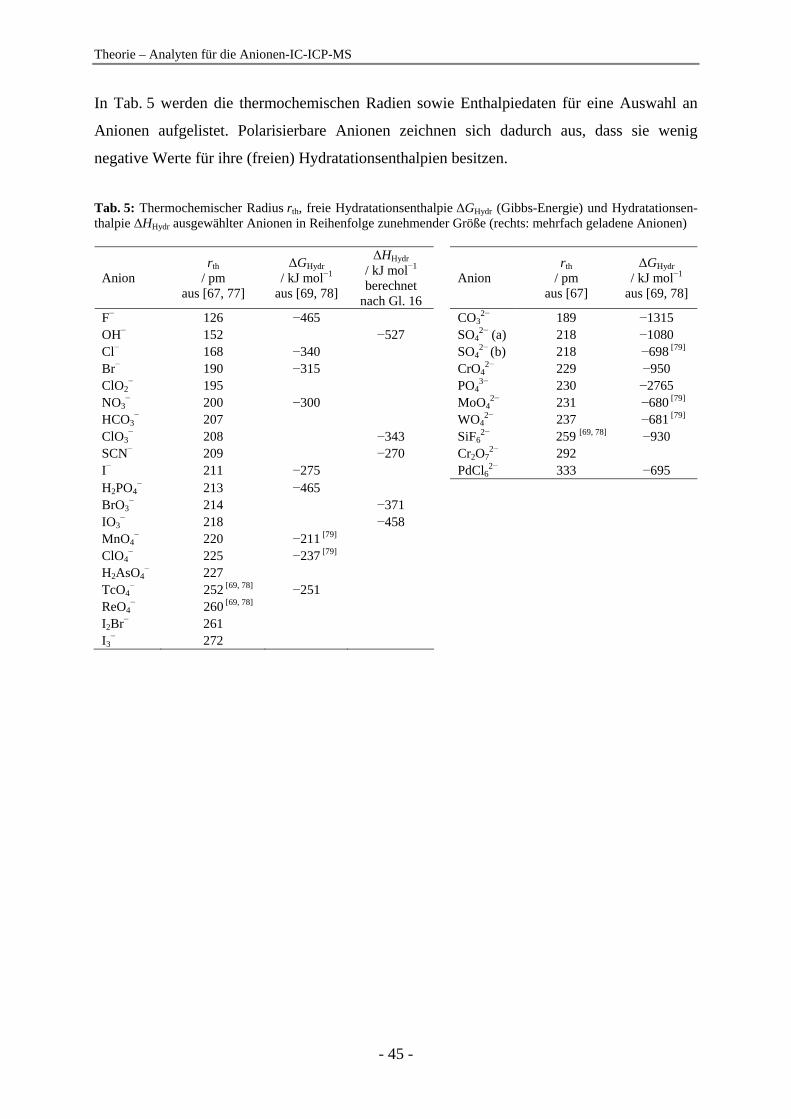
Theorie – Analyten für die Anionen-IC-ICP-MS
- 45 -
In Tab. 5 werden die thermochemischen Radien sowie Enthalpiedaten für eine Auswahl an
Anionen aufgelistet. Polarisierbare Anionen zeichnen sich dadurch aus, dass sie wenig
negative Werte für ihre (freien) Hydratationsenthalpien besitzen.
Tab. 5: Thermochemischer Radius rth, freie Hydratationsenthalpie ∆GHydr (Gibbs-Energie) und Hydratationsen-thalpie ∆HHydr ausgewählter Anionen in Reihenfolge zunehmender Größe (rechts: mehrfach geladene Anionen)
Anion rth
/ pm aus [67, 77]
∆GHydr / kJ mol−1
aus [69, 78]
∆HHydr / kJ mol−1
berechnet nach Gl. 16
Anion rth
/ pm aus [67]
∆GHydr / kJ mol−1
aus [69, 78]
F− 126 −465 CO32− 189 −1315
OH− 152 −527 SO42− (a) 218 −1080
Cl− 168 −340 SO42− (b) 218 −698 [79]
Br− 190 −315 CrO42− 229 −950
ClO2− 195 PO4
3− 230 −2765 NO3
− 200 −300 MoO42− 231 −680 [79]
HCO3− 207 WO4
2− 237 −681 [79]
ClO3− 208 −343 SiF6
2− 259 [69, 78] −930 SCN− 209 −270 Cr2O7
2− 292 I− 211 −275 PdCl6
2− 333 −695 H2PO4
− 213 −465 BrO3
− 214 −371 IO3
− 218 −458 MnO4
− 220 −211 [79] ClO4
− 225 −237 [79] H2AsO4
− 227 TcO4
− 252 [69, 78] −251 ReO4
− 260 [69, 78] I2Br− 261 I3− 272

Theorie – Analyten für die Anionen-IC-ICP-MS
- 46 -
Im Folgenden sind die Eigenschaften von polarisierbaren Anionen zusammenfassend
aufgelistet:
• wenig negative Werte für die freie Hydratationsenthalpie
• relativ hydrophob
• großer Radius ohne Hydrathülle (thermochemischer Radius)
• kleiner Radius des Ions zusammen mit der Hydrathülle
• leicht deformierbare Elektronenwolke
• zerstören die Struktur von Wasser (chaotrop)
• starke Wechselwirkungen mit π-Elektronen
• hoher Brechungsindex
• relativ leicht oxidierbar
• korrespondierende Säuren haben niedrige pKs-Werte
Andere Konzepte zur Beschreibung polarisierbarer Anionen
Verschiedene Konzepte aus der Biochemie, der anorganischen sowie der physikalischen
Chemie hängen mit der Polarisierbarkeit zusammen. Da sich mit diesen Konzepten ebenfalls
diverse Eigenschaften von Anionen erklären lassen, sollen sie näher erläutert werden.
Schon im Jahr 1888 wurden von Hofmeister unterschiedliche, für die Biochemie wichtige
Eigenschaften von Anionen bei der Lösung in Wasser festgestellt [81]. Er bemerkte, dass
durch Zugabe von bestimmten Salzen die Löslichkeit von Proteinen in wässriger Lösung
verbessert wurde, während andere Salze die Ausfällung von Proteinen verursachten. Die so
genannte „Hofmeister-Reihe“ ordnet Anionen nach ihrer Hydrophobie (und damit nach dem
Ausmaß ihrer Hydratation):
hydrophob - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - hydrophil geringe Hydratation - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - starke Hydratation
ClO4− > SCN− > NO3
− > Br− > NO2− > Cl− >> CH3COO− > CO3
2− > SO42−
verbessern Löslichkeit von Proteinen - - - - - - - - - - - - - - - - - - - - - verschlechtern Löslichkeit von Proteinen
Abb. 18: Hofmeister-Reihe [69, 81]

Theorie – Analyten für die Anionen-IC-ICP-MS
- 47 -
In der anorganischen Chemie wird das von R.G. Pearson 1963 eingeführte „Prinzip der harten
und weichen Säuren“ (HSAB-Prinzip) genutzt, mit dem sich die Stabilität von Lewis-Säure-
Base-Komplexen erklären lässt [82]. Dabei wird zwischen „harten“ (schwerer polarisierbaren)
und „weichen“ (leichter polarisierbaren) Lewis-Säuren und -Basen unterschieden. Das
HSAB-Prinzip besagt, dass für eine Komplexbildung die Kombination von harten Säuren mit
harten Basen bzw. von weichen Säuren mit weichen Basen bevorzugt ist. In der theoretischen
Betrachtung mit Hilfe des Orbitalmodells werden die energetischen Lagen des höchsten be-
setzten Molekülorbitals (HOMO) und des niedrigsten unbesetzten Molekülorbitals (LUMO)
zur Erklärung des HSAB-Prinzips herangezogen. Weiche Basen sind gekennzeichnet durch
energetisch hoch liegende HOMOs, die energetisch ähnlich sind wie die LUMOs von weichen
Säuren. Dadurch können die Orbitale gut überlappen und eine Bindung ermöglichen. Die
Bindung zwischen weichen Säuren und Basen ist orbitalkontrolliert und besitzt mehr
kovalenten Charakter. Dagegen ist die Wechselwirkung zwischen harten Säuren und Basen
ladungskontrolliert und die Bindung hat mehr ionischen Charakter. Die Klassifizierung
einiger Anionen nach Pearson wird in Tab. 6 gezeigt.
Tab. 6: Klassifizierung einiger Basen als hart oder weich nach dem HSAB-Prinzip von Pearson [82]
hart Grenzbereich weich
OH−, F−, CH3CO2−,
PO43−, SO4
2−, Cl−, CO3
2−, ClO4−, NO3
−
N3−, Br−, NO2
−, SO3
2−, RS−, I−, SCN−, S2O3
2−, CN−
Die Einteilung als hart oder weich nach dem HSAB-Prinzip ist nur bedingt nützlich für die
Vorhersage der Retention von Anionen an Anionenaustauschern. Zwar sind die als weich
eingestuften Anionen alle auch polarisierbar und weisen somit eine hohe Retention in der
Anionenchromatographie auf. Auf der Seite der als hart eingestuften Ionen ist allerdings die
Übereinstimmung von Härte und geringer Polarisierbarkeit nicht streng gültig, denn Ionen
wie Nitrat und insbesondere Perchlorat sind polarisierbar und werden deswegen an stationären
Phasen auf Basis organischer Polymere meist stark retardiert.

Literaturübersicht: Anionenanalytik mit der IC-ICP-MS
- 48 -
3 Literaturübersicht:
Anionenanalytik mit der IC-ICP-MS
In diesem Kapitel wird ein Überblick über die aktuelle Forschungsliteratur auf dem Gebiet der
Anionenanalytik mit der Kopplung von Ionenchromatographie und ICP-MS gegeben. Auf
Grund der Vielzahl von Veröffentlichungen, die in diesen Themenbereich fallen, ist es
schwerlich möglich, auf alle Arbeiten umfassend einzugehen. Vielmehr werden einige
wichtige Veröffentlichungen ausgewählt, von denen wiederum nur ein paar detaillierter
beschrieben werden. Die Selektion ergibt sich aus einer subjektiven Bewertung der Wichtig-
keit verschiedener Artikel. Als Bewertungskriterien liegen unter anderem die Qualität, die
Aktualität aber auch die Würdigung von Pionierleistungen zu Grunde. Entscheidend ist
allerdings die inhaltliche Relevanz für die vorliegende Dissertation: Priorität genießen die
Veröffentlichungen, die sich mit der Spurenanalyse von Halogenspezies und/oder anorga-
nischen Anionen befassen. Besonderer Wert wird zudem auf Arbeiten gelegt, die die
gleichzeitige Bestimmung möglichst vieler dieser Analyten beschreiben.
Hervorzuheben sind die Anionenanalytik in Wässern und die Elementspeziesanalyse, wobei
sich diese Arbeitsgebiete oft überschneiden. An dieser Stelle wird noch einmal darauf
hingewiesen, dass in jüngerer Zeit die ESA ein besonders stark wachsendes Forschungsgebiet
darstellt, zu dem auch die IC-ICP-MS einen Beitrag leistet. Das stetig steigende
wissenschaftliche Interesse an der ESA zeigt sich z.B. an der Anzahl von Publikationen zu
diesem Thema, die von etwa 250 im Jahr 1990 auf über 600 im Jahr 2004 angestiegen ist [83].
Ein Überblick über den Stand der Technik zur Analyse von Anionen mit der IC-ICP-MS lässt
sich z.B. durch Reviewartikel im Bereich der ESA gewinnen [1-7, 84, 85]. Insbesondere bei
Sutton und Caruso wird ausführlich auf IC- und andere LC-Kopplungen mit der ICP-MS
eingegangen [5]. Ein Übersichtsartikel über die Bestimmung von Anionen mit der Ionen-
chromatographie, in dem auch Methoden mit der ICP-MS-Detektion aufgegriffen werden,
wurde von Lopez-Ruiz verfasst [12]. Ein Review von Brede und Pedersen-Bjergaard aus dem
Jahr 2004 beschreibt vor allem den Stand der Technik für die halogenselektive Detek-
tion [86]. Der Hauptfokus dieses Übersichtsartikels liegt auf der Gaschromatographie mit
ICP-MS- oder AES-Detektion für organische Substanzen. Des Weiteren ist auch ein Kapitel
über die Kopplung von LC mit der ICP-MS ist zu finden.

Literaturübersicht: Anionenanalytik mit der IC-ICP-MS
- 49 -
Auf dem Gebiet der Anionenanalytik in Wässern sind viele Informationen, insbesondere in
Bezug auf Halogenspezies, in diversen Reviews über die Analyse von Desinfektions-
beiprodukten erhältlich [17, 20, 87-89].
Im Folgenden werden einzelne, veröffentlichte Forschungsarbeiten vorgestellt:
Die Ionenchromatographie mit Leitfähigkeits- und ICP-MS-Detektion nacheinander wurde
von Dudoit und Pergantis zur Multielement-Speziesanalyse verwendet [90]. Mit der
vorgestellten Methode ist es möglich, in Trinkwasser ohne Probenvorbereitung Spuren von
Bromat, Bromid, Arsenat, Arsenit, Iodat und Iodid in niedrigen µg L−1-Konzentrationen, und
gleichzeitig die höher konzentrierten Anionen Chlorid, Nitrat und Sulfat im mg L−1-Bereich
zu bestimmen. Für die Trennung wurde eine AS14®-Säule (Dionex, Sunnyvale) im Carbo-
nat/Bicarbonat-Elutionssystem eingesetzt. Diese Säule enthält einen gepfropften Anionenaus-
tauscher. Dudoit und Pergantis stellten fest, dass eine stabile Detektion mittels ICP-MS
dadurch ermöglicht wurde, dass Natrium- und Kaliumionen mit Hilfe eines Membran-
suppressors aus dem Eluenten entfernt wurden. Es ließen sich auch Eluenten mit hoher
Ionenstärke einsetzen, ohne dass es zu Salzablagerungen oder zur Bildung von Molekülionen
von Kalium oder Natrium im ICP-MS kam. Die erreichten Nachweisgrenzen für die neun
untersuchten Analyten sind in Tab. 7 aufgelistet. Die Methode wurde getestet, indem
Leitungswasser sowie in Flaschen abgefülltes Wasser analysiert wurde. Die ermittelten
Konzentrationen lagen unter den von der US EPA und einer europäischen Kommission für
Trinkwasser empfohlenen Grenzwerten. Im weiteren Verlauf der Untersuchung wurden
Wiederfindungsexperimente durchgeführt und dabei für die meisten Analyten gute Wieder-
findungsraten in Höhe von 96 % bis 101 % ermittelt. Für Chlorid und Iodid belief sich der
Wert nur auf etwa 90 %. Die Analyse von Iodid bereitete wegen des starken Peaktailings und
der zu langen Retentionszeit Probleme. Für Arsenit und Arsenat konnte nur die Gesamt-
wiederfindung angegeben werden, da eine Speziesumwandlung zwischen diesen beiden
Anionen beobachtet wurde.
In einer ähnlichen Arbeit nutzten Fernández et al. die Kopplung von Ionenchromatographie
und ICP-MS zur Analyse von sieben anorganischen Anionen [91]. Sie verwendeten einen
Carbonat/Bicarbonat-Eluenten, der mittels Leitfähigkeitssuppression von Natriumionen be-
freit wurde, wodurch sich Ablagerungsprobleme im ICP-MS verhindern ließen. Eine Leit-
fähigkeitsmesszelle und ein ICP-MS wurden in Serie geschaltet, was einen direkten Vergleich
der beiden Detektionsmethoden ermöglichte (Nachweisgrenzen s. Tab. 7). Die Gruppe be-
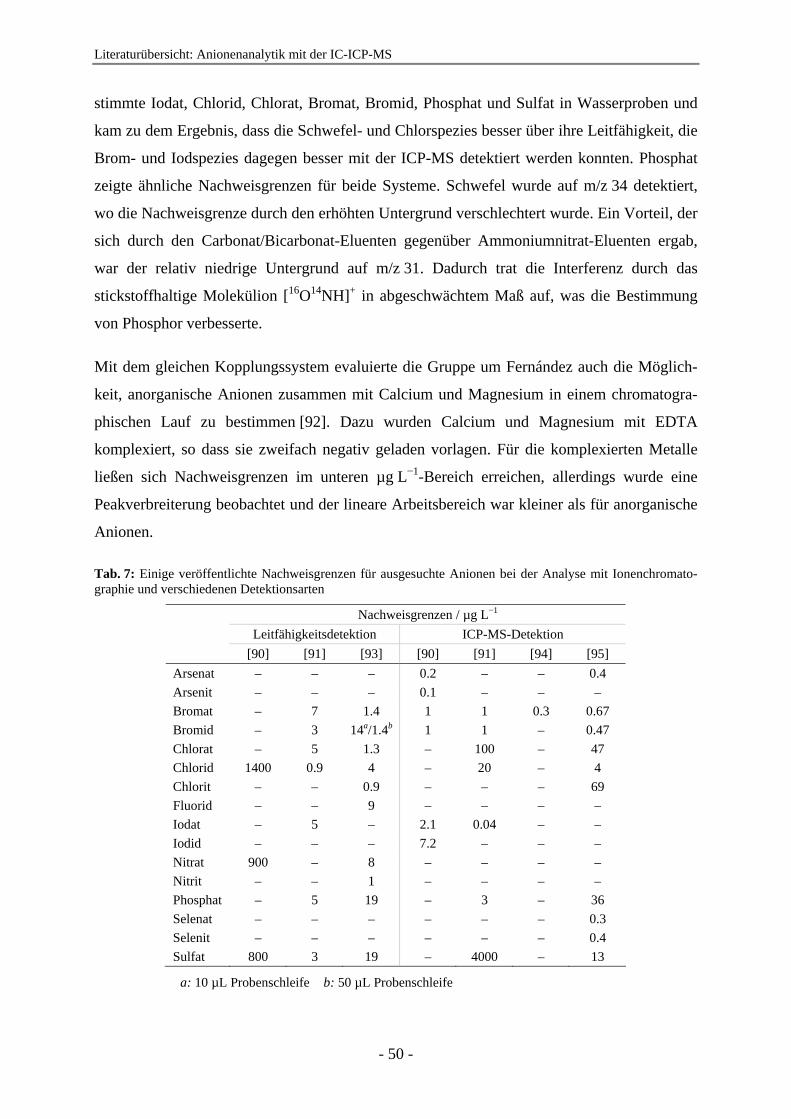
Literaturübersicht: Anionenanalytik mit der IC-ICP-MS
- 50 -
stimmte Iodat, Chlorid, Chlorat, Bromat, Bromid, Phosphat und Sulfat in Wasserproben und
kam zu dem Ergebnis, dass die Schwefel- und Chlorspezies besser über ihre Leitfähigkeit, die
Brom- und Iodspezies dagegen besser mit der ICP-MS detektiert werden konnten. Phosphat
zeigte ähnliche Nachweisgrenzen für beide Systeme. Schwefel wurde auf m/z 34 detektiert,
wo die Nachweisgrenze durch den erhöhten Untergrund verschlechtert wurde. Ein Vorteil, der
sich durch den Carbonat/Bicarbonat-Eluenten gegenüber Ammoniumnitrat-Eluenten ergab,
war der relativ niedrige Untergrund auf m/z 31. Dadurch trat die Interferenz durch das
stickstoffhaltige Molekülion [16O14NH]+ in abgeschwächtem Maß auf, was die Bestimmung
von Phosphor verbesserte.
Mit dem gleichen Kopplungssystem evaluierte die Gruppe um Fernández auch die Möglich-
keit, anorganische Anionen zusammen mit Calcium und Magnesium in einem chromatogra-
phischen Lauf zu bestimmen [92]. Dazu wurden Calcium und Magnesium mit EDTA
komplexiert, so dass sie zweifach negativ geladen vorlagen. Für die komplexierten Metalle
ließen sich Nachweisgrenzen im unteren µg L−1-Bereich erreichen, allerdings wurde eine
Peakverbreiterung beobachtet und der lineare Arbeitsbereich war kleiner als für anorganische
Anionen.
Tab. 7: Einige veröffentlichte Nachweisgrenzen für ausgesuchte Anionen bei der Analyse mit Ionenchromato-graphie und verschiedenen Detektionsarten
Nachweisgrenzen / µg L−1
Leitfähigkeitsdetektion ICP-MS-Detektion [90] [91] [93] [90] [91] [94] [95]
Arsenat – – – 0.2 – – 0.4 Arsenit – – – 0.1 – – – Bromat – 7 1.4 1 1 0.3 0.67 Bromid – 3 14a/1.4b 1 1 – 0.47 Chlorat – 5 1.3 – 100 – 47 Chlorid 1400 0.9 4 – 20 – 4 Chlorit – – 0.9 – – – 69 Fluorid – – 9 – – – – Iodat – 5 – 2.1 0.04 – – Iodid – – – 7.2 – – – Nitrat 900 – 8 – – – – Nitrit – – 1 – – – – Phosphat – 5 19 – 3 – 36 Selenat – – – – – – 0.3 Selenit – – – – – – 0.4 Sulfat 800 3 19 – 4000 – 13
a: 10 µL Probenschleife b: 50 µL Probenschleife

Literaturübersicht: Anionenanalytik mit der IC-ICP-MS
- 51 -
Eine detaillierte Studie über die Bestimmung anionischer Metallkomplexe von verschiedenen
Chelatkomplexbildnern mit der IC-ICP-MS ist bei Amman zu finden [96, 97]. In dieser wur-
den Komplexe von Kobalt, Nickel, Kupfer, Zink, Cadmium und Blei an einer stationären Pha-
se mit hydrophilen Alkanolsubstituenten an den Austauschergruppen (Säule AS11®, Dionex,
Sunnyvale) getrennt. Als Eluent wurde Ammoniumnitrat bei pH 7 mit einem Konzentrations-
gradienten von 20−170 mmol L−1 eingesetzt. Die Gradientenelution ermöglichte ein breites
analytisches Trennfenster, d.h. viele Komplexe, auch solche mit sehr unterschiedlicher
Retention, konnten in einem Lauf bestimmt werden. Experimente zur Beurteilung des Trenn-
mechanismus für die anionischen Metallkomplexe zeigten, dass die Trennung hauptsächlich
auf Anionenaustausch und nur zu geringem Maß auf hydrophoben Wechselwirkungen beruht.
Divjak, Novic und Goessler beschreiben die Optimierung der IC-ICP-MS für die
Bromid/Bromat-Analyse und gehen zusätzlich auf die gleichzeitige Bestimmung von zehn
Anionen ein [95]. Dabei handelt es sich um Bromid, Bromat, Chlorit, Chlorid, Chlorat, Sulfat,
Phosphat, Selenit, Selenat und Arsenat. Die Trennung der Analyten an einer Dionex IonPac®
AS12A®-Säule wurde in weniger als 4 min mit einem 12 mmol L−1 Ammoniumcarbonat-
Eluenten erreicht. Durch die Verwendung eines ammoniumhaltigen statt eines natrium-
haltigen Eluenten wurden Ablagerungsprobleme im ICP-MS verhindert, weil sich aus
Ammonium nur gasförmige Substanzen bilden. Auf den Einsatz eines Suppressors konnte
daher verzichtet werden. Das Probenzuführungssystem des ICP-MS beinhaltete einen Ultra-
schallzerstäuber mit Membrandesolvator, mit dem sich hohe Aerosolausbeuten und gute
Nachweisgrenzen erzielen lassen (s. Tab. 7).
Für die Analyse von Schwefelspezies empfehlen Divjak und Goessler spezielle Betriebs-
bedingungen [71]. Hierfür setzten sie Natriumhydroxidlösung als Eluenten ein und verwen-
deten dann einen Membransuppressor zur Natriumentfernung vor dem Einlass in das ICP-MS.
Die Nutzung dieses Eluents und das Tuning auf möglichst starke Oxidbildung führte dazu,
dass Schwefel empfindlich auf der Masse des Molekülions [32S16O]+ detektiert werden konnte
und dass sich bessere Nachweisgrenzen ergaben als bei der Detektion des Isotops 34Schwefel.
Eine der ersten Arbeiten, in der gleichzeitig acht Halogenide und Oxyhalogenide mit der
IC-ICP-MS bestimmt wurden, stammt von Pantsar-Kallio und Manninem [98]. Sie führten in
Wasser aus finnischen Trinkwasseraufbereitungsanlagen vor und nach der Desinfektion Ele-
mentspeziesanalysen durch und bestimmten Chlorid, Chlorit, Chlorat, Perchlorat, Bromid,
Bromat, Iodid und Iodat. Aus der Differenz der Totalhalogenkonzentration und der Summe

Literaturübersicht: Anionenanalytik mit der IC-ICP-MS
- 52 -
der einzelnen Spezieskonzentrationen wurde geschlossen, dass außer den anorganischen
Halogenspezies auch noch ein erheblicher Anteil an organischen Halogenspezies im desinfi-
zierten Trinkwasser vorliegen musste. Die ermittelten Nachweisgrenzen für alle Chlorspezies
betrugen 500 µg L−1, für die Bromspezies 10 µg L−1 und für Iodid und Iodat 0.1 µg L−1 bzw.
0.2 µg L−1.
Auch Salov et al. versuchten schon 1992 sechs Halogenanionen ionenchromatographisch zu
trennen und mit der ICP-MS zu detektieren [99]. Mit der verwendeten IC-Methode konnte
aber bei niedrigen Konzentrationen keine lineare Kalibrationsfunktion für Iodid erhalten
werden. Des Weiteren erwies sich die Retentionszeit als zu lang. Die Gruppe wechselte daher
zur Größenausschlusschromatographie, mit der eine Trennung von Iodat, Bromat, Chlorid,
Chlorat, Bromid und Iodid in ca. 7 min gelang.
Die US EPA Methode 300.1 stellt eine sehr weit verbreitete Methode zur Bestimmung
anorganischer Anionen in Trinkwasser mit Ionenchromatographie dar [93]. Da die Methode
häufig zum Vergleich mit IC-ICP-MS-Messungen herangezogen wird, wird an dieser Stelle
auf sie eingegangen, obwohl bei ihr ausschließlich die suppressierte Leitfähigkeitsdetektion
zum Einsatz kommt. In der Methode 300.1 wird die Analyse der so genannten sieben
„Standardanionen“ Bromid, Chlorid, Fluorid, Nitrat, Nitrit, Phosphat und Sulfat zusammen
mit einigen anorganischen Desinfektionsbeiprodukten beschrieben. Unter die DBPs fallen
Bromat, Bromid, Chlorit und Chlorat, wobei Bromid eigentlich nur als „precursor“ von
Bromat angesehen wird. Für die Trennung ist die Säule AS9-HC® (Dionex, Sunnyvale) bzw.
eine äquivalente Säule vorgeschrieben und als Eluent ist 9 mmol L−1 Natriumcarbonat zu
verwenden. Die erreichbaren Nachweisgrenzen sind zum Vergleich mit den ICP-MS-
Methoden in Tab. 7 aufgeführt.
Eine weitere Veröffentlichung der US EPA, die Methode 321.8, geht auf die Bestimmung von
Bromat in Trinkwasser mit Hilfe der IC-ICP-MS-Kopplung ein [94]. Die Trennung erfolgt
hier an der Säule PA-100 (Dionex, Sunnyvale) bzw. einer äquivalenten. Als Eluent wird eine
Lösung aus 5 mmol L−1 Salpetersäure und 25 mmol L−1 Ammoniumnitrat bei einer Flussrate
von 1 mL L−1 angegeben. Bei der Aufgabe eines Probenvolumens von 580 µL und der
Detektion des Isotops 79Brom wird mit der Instrumentation in Methode 321.8 eine Nachweis-
grenze von 0.3 µg L−1 für Bromat erreicht. Mögliche Störungen durch Halogenessigsäuren
werden vermieden, indem diese vor der Trennung mit Hilfe einer Adsorptionskartusche
entfernt werden. Die Autoren halten es für nötig, die Proben und Kalibrationsstandards auf
pH 10 einzustellen, um zu verhindern, dass Bromat auf Grund extrem niedriger Ionenstärke

Literaturübersicht: Anionenanalytik mit der IC-ICP-MS
- 53 -
der Lösungen an den Wänden der verwendeten Kunststoffprobenschleifen adsorbiert wird
(s.a. [100]). Ein solches Problem wurde für Bromat im Rahmen der vorliegenden Dissertation
nicht beobachtet. Ein Übersichtsartikel über die verschiedenen US EPA Methoden zur
Bestimmung von Bromat wurde von Hautman et al. verfasst [101].
Die meisten Veröffentlichungen über die Analyse von Halogenspezies mit der IC-ICP-MS
konzentrieren sich auf die ausschließliche Bestimmung von Bromat. Diese wurde meistens in
Wässern vorgenommen [102-108], aber es wurde auch Brot 1 auf Bromat untersucht [109,
110]. Drei Gruppen zeigten die Möglichkeiten und Vorteile der Isotopenverdünnungsanalyse
bei der Analyse von Bromat mit der IC-ICP-MS auf [111-114]. In weiteren Artikeln wird
auch darauf eingegangen wie Störungen durch Bromessigsäuren bei der Bromatanalyse ver-
mieden werden können, oder wie diese Bromspezies nebeneinander bestimmt werden
können [107, 115-117]. Cai et al. nutzen die On-line-Kopplung von Ionenchromatographie
und ICP-MS, um Bromat zusammen mit drei nur Brom enthaltenden Essigsäuren sowie mit
drei weiteren gemischten Brom-/Chloressigsäuren in Wasser zu analysieren [116]. Die
Trennung wurde an einer AS11-HC®-Säule (Dionex, Sunnyvale) mit einem 100 mmol L−1
Ammoniumnitrat-Eluenten vollzogen. Verschiedene Parameter, wie die Eluentflussrate, das
Probenvolumen, ein erhöhter Gehalt an organischen Lösungsmitteln oder Bromid, wurden auf
ihren Einfluss auf die Trennung und ICP-MS-Detektion hin untersucht. Die ermittelten Nach-
weisgrenzen auf Basis einer 500 µL-Probenschleife lagen zwischen 0.13 und 0.36 µg L−1.
Iod kann mit der ICP-MS auf Grund der höheren Ionisierungsausbeute und Interferenzfreiheit
besonders präzise und mit noch besseren Nachweisgrenzen detektiert werden als Brom.
Deswegen stellen Iodspezies ideale Targetanalyten für die ICP-MS dar. Ein Vergleich der
ICP-MS- mit der ESI-MS-Detektion für die Spurenanalyse von Iodid nach IC-Trennung ist
bei Buchberger et al. zu finden [118]. Heumann und Mitarbeiter bestimmten anorganische
sowie organische Iodspezies in Wasserproben [119]. Caruso et al. entwickelten eine
IC-ICP-MS Methode um das Flammschutzmittel 2,4,6-Triiodophenol und seine Metabolite in
Urin zu messen [120]. Die gleiche Gruppe studierte die organische und anorganische
Speziesverteilung von Iod in Algen mit mehreren LC-ICP-MS-Methoden [121]. Von Sacher,
Raue und Brauch wurde gezeigt, dass die IC-ICP-MS ausgezeichnet für die Analyse iodierter
Röntgenkontrastmittel in wässriger Lösung geeignet ist [122].
1 Bromat wird zur Verbesserung der Brotproduktion oft dem Mehl zugegeben, wird aber beim Backen zu Bromid reduziert.

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 54 -
4 Ergebnisse
4.1 Chromatographische Charakterisierung stationärer Phasen für die
Anionen-IC-ICP-MS
4.1.1 Einleitung
In diesem Kapitel sollen verschiedene Säulen für die Anionenchromatographie beschrieben
und miteinander verglichen werden. Dazu werden jeweils kurz der Aufbau der verwendeten
Ionenaustauschermaterialien dargestellt und die ermittelten Leistungskenngrößen für Säulen
angegeben 2. Anhand von Beispielchromatogrammen wird diskutiert, welche chromatogra-
phischen Eigenschaften, wie Trennleistung, Peakformen, Retentionszeiten und Elutions-
reihenfolge aus den Materialien resultieren. Als Elutionsmittel für die IC-ICP-MS-Messungen
dient Ammoniumnitrat mit einem pH-Wert zwischen drei und sieben. Die Eluentkonzen-
tration muss auf die jeweilige Säulenkapazität angepasst werden und liegt typischerweise
zwischen 10 und 100 mmol L−1 Ammoniumnitrat. (s.a. Kap. 4.2)
Durch die Kopplung von Ionenchromatographie und ICP-MS werden die Anionenselektivität
der Säulen und die elementselektive Detektion der ICP-MS kombiniert. Diese kombinierte
Selektivität lässt sich nutzen, um möglichst viele Analyten schnell zu analysieren
(„Multianionenanalyse“). So ist in Abb. 30 die simultane Analyse von 15 Anionen in weniger
als 40 min gezeigt.
Die in dieser Arbeit wichtigsten und am häufigsten gemessenen Analyten sind Bromid,
Bromat, Iodid und Iodat, die mit der ICP-MS mit sehr guten Nachweisgrenzen von etwa
0.1 µg L−1 detektierbar sind. Für den Vergleich verschiedener Säulenmaterialien wurden des
weiteren Multianionenstandards mit 10–15 Anionen analysiert. Die Nachweisstärke der
IC-ICP-MS-Kopplung für die verschiedenen Ionen ist äußerst unterschiedlich. Die Ionen
Dihydrogenarsenat, Mono-, Di-, und Tribromacetat (MBA−, DBA−, TBA−) sind gut bis in den
unteren µg L−1-Bereich messbar. Dagegen lassen sich die Analyten Chlorid, Chlorit, Chlorat,
Perchlorat, Thiocyanat und Dihydrogenphosphat nur in höheren Konzentrationen detektieren.
Die Nachweisstärke für diese Ionen ist im Vergleich mit anderen Detektionsarten wie z.B. der
2 Auf weitere wichtige physikalische Kenngrößen der Polymerträgermaterialien, wie die durchschnittliche Partikelgröße, die Partikelgrößenverteilung, der Quervernetzungsgrad, die Porengröße oder das Gesamtporen-volumen, wird in Arbeiten von S. Schütze, S. Holland, und M. Raskop eingegangen. ([123-125])

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 55 -
Leitfähigkeitsmessung nur gleich gut oder schlechter. Der Vorteil der IC-ICP-MS liegt aber in
diesem Fall darin, dass sehr viele Ionen in einem einzigen chromatographischen Lauf
gemessen werden können, da die Peaks verschiedener Elemente durch das Massenspektro-
meter differenziert werden können und somit nicht zwingend chromatographisch aufgelöst
werden müssen. Dies führt zu einem enormen Zeitgewinn. Führte man ähnliche Messungen
mit Leitfähigkeitsdetektion durch, würden die Analysenzeiten wesentlich länger werden, da
alle Analyten chromatographisch von einander separiert werden müssten. Der Einsatz einer
Gradiententechnik wäre unabdingbar, während man bei der IC-ICP-MS meist unter
isokratischen Bedingungen messen kann. Durch die Kopplung von Ionenchromatographie und
ICP-MS erreicht man sowohl eine Zeitersparnis, als auch eine Steigerung des Informations-
gehalts.
Die gleichzeitige chromatographische Trennung einer großen Anzahl von Anionen lässt auf
schnellem Wege eine umfassende Charakterisierung der verwendeten Säulenmaterialien zu
und das unter „möglichst realen Bedingungen“. Außerdem können Interferenzen oder eine
eventuelle gegenseitige Beeinflussung der Ionen erkannt werden. Der Aussage „möglichst
reale Bedingungen“ liegt die Erwartung zu Grunde, dass die Messung von Standards mit
besonders vielen Analyten, auch zukünftigen realen Anwendungen nahe kommt. Bei
Routineanwendungen wird wegen der relativ hohen Betriebskosten eines ICP-MS ein
wirtschaftlicher Vorteil nur dann erreicht, wenn möglichst viele Analyten gleichzeitig
bestimmt werden und wenn ein hoher Probendurchsatz vorliegt. Deswegen ist davon auszu-
gehen, dass sich langfristig im Routinebereich vermehrt Multispeziesbestimmungsmethoden
durchsetzen werden.
Die in einem Multianionenstandard enthaltenen Analyten müssen für eine sichere Charakteri-
sierung des chromatographischen Systems aber auch einzeln gemessen werden. Dabei ist zu
überprüfen, ob die Retentionszeit und die Peakfläche der Einzelstandards mit den Werten aus
der Messung des Multistandards identisch sind. Abweichungen der Retentionszeit lassen auf
Überladungseffekte schließen, und Änderungen der Peakfläche zeigen an, dass eine Spezies-
umwandlung stattgefunden hat. So lässt sich z.B. die Haltbarkeit der Multistandards beurtei-
len. Wenn chemische Reaktionen stattfinden, können auch deren Geschwindigkeitskonstanten
berechnet werden, indem eine Messreihe mit festgelegten zeitlichen Abständen aufgenommen
wird.

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 56 -
Über das Retentionsverhalten und die Peakform jedes einzelnen Ions eines Multistandards
sowie über die relative Retention zueinander ergeben sich Rückschlüsse auf das verwendete
Säulenmaterial. Auf folgende Parameter wird in der Diskussion insbesondere eingegangen:
• die relative Retention von Bromid und Iodid
• die Auflösung des Totvolumenpeaks vom Iodatsignal
• die Auflösung der in den ersten fünf bis acht Minuten eluierenden Anionen
• die Elutionsreihenfolge
• die Peakform aller Analyten, insbesondere die der polarisierbaren Anionen
• die Trennleistung für alle Analyten
Die Halogenide und Oxyhalogenide zur Beurteilung der Trenneigenschaften
Am Beispiel der Halogenide und Oxyhalogenide von Brom und Iod sollen einige spezielle
Anforderungen aufgezeigt werden, die an die Trennsäulen in dieser Arbeit gestellt werden.
Das Iodat eluiert bei der Trennung an Latex-Anionenaustauschern meist direkt nach dem
Injektionspeak, wenn man die Nitratkonzentration des Eluenten so einstellt, dass das Iodid
noch innerhalb von 20 min von der Säule eluiert wird. Unter diesen Vorraussetzungen ist das
Iodat nicht immer vollständig vom darauf folgenden Bromat aufgelöst (z.B. Abb. 22). Die
Trennung dieser beiden Ionen ist dank der Elementselektivität des ICP-MS nicht unbedingt
notwendig. Eine gute Auflösung von Iodat und Bromat bei gleichzeitig kurzer Retentionszeit
des Iodids ist aber ein nützliches Qualitätskriterium zur Erkennung von Anionenaustauschern,
die sich für die Multianionentrennung eignen.
In der Praxis ist die Auflösung des Iodats vom Wasserdip (negativer Injektionspeak) von
Bedeutung. Bei zu früher Elution des Iodats ist eine interne-Standard-Korrektur mit Hilfe
eines kontinuierlichen IS im Eluenten kaum möglich, denn in einem solchen Fall tritt das
folgende Problem auf: Für ein Signal zu nahe am Wasserdip führt die Echtzeitkorrektur mit
Hilfe des IS zu einer Verschlechterung der Präzision, weil das IS-Signal zu diesem Zeitpunkt
noch nicht wieder auf ein stabiles Niveau angestiegen ist. Wie schon erwähnt, kann die
Elutionskraft des Eluenten nicht beliebig abgeschwächt werden, um dieses Problem zu
beheben, da dann die stark retardierten Ionen nicht mehr schnell genug eluiert werden. Eine
zu lange Trennzeit wäre erstens unwirtschaftlich, und zweitens würden sich auf Grund der
Peakverbreiterung die Nachweisgrenzen verschlechtern. Deswegen sollte eine ideale Säule

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 57 -
eine relativ hohe Retention der früh eluierenden Ionen wie Iodat und Bromat, aber gleich-
zeitig eine möglichst geringe Retention der polarisierbaren Ionen wie Iodid bewirken.
Im Allgemeinen dürfen bei der IC-ICP-MS-Kopplung Peaks von Anionen, die über verschie-
dene Elemente detektiert werden, koeluieren. Durch die ICP-MS-Detektion steht als
zusätzliches Unterscheidungskriterium die Masse zu Verfügung. Verschiedene Spezies eines
Elements müssen dagegen chromatographisch getrennt werden. Für die Auflösung gelten
Werte von R = 1.5–2 als ideal, da dann die Peaks ausreichend getrennt sind, und die Gesamt-
messzeit nicht unnötig verlängert wird.
4.1.2 Latex-Anionenaustauscher
Allgemeine Definition sowie Aufbau und Eigenschaften von Latex-Anionenaustauschern
Für die heutige Hochleistungsanionenchromatographie werden häufig pellikulare Anionen-
austauscher verwendet (lat.: pellicula = dünne Haut). In der Chromatographie versteht man
unter einer pellikularen Packung laut IUPAC ein Säulenmaterial, bei dem die eigentliche
stationäre Phase eine poröse Außenhaut auf einem nichtpermeablen Trägerteilchen
bildet [126]. Die so genannten „Latex-Ionenaustauscher“ oder auch „agglomerierten Ionen-
austauscher“ sind eine spezielle Art pellikularer stationärer Phasen, im allgemeinen Sprach-
gebrauch der IC werden alle drei Bezeichnungen allerdings oft synonym verwendet. Latex-
Ionenaustauscher wurden 1975 von Small et al. erstmals vorgestellt [10]. Dieses Datum gilt
als Beginn der modernen Hochleistungsionenchromatographie.
Der schematische Aufbau typischer Latex-Ionenaustauscher für die Anionenchromatographie
ist in Abb. 19 gezeigt. Sie bestehen zum einen aus einem oberflächensulfonierten, sphärischen
Substrat meist auf der Basis von Polystyrol/Divinylbenzol (PS/DVB) mit einem Teilchen-
durchmesser von 3–25 µm. Zum anderen befinden sich auf der Substratoberfläche total
aminierte Teilchen aus Polyvinylbenzylchlorid/Divinylbenzol (VBC/DVB) oder aus Poly-
methacrylatderivaten, die als Latexteilchen bezeichnet werden [13]. Die Latexteilchen
zeichnen sich durch eine hohe Kapazität und einen wesentlich kleineren Durchmesser von
20–500 nm aus. Sie sind hauptsächlich durch elektrostatische und Van der Waals- Wechsel-
wirkungen an der Oberfläche des Substrats agglomeriert. Als Latex (lat.: Latex = Flüssigkeit)
wurde ursprünglich nur der milchige Saft, der durch Anritzen von Kautschukbäumen

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
gewonnen wurde, benannt. Heute werden als Latex ganz allgemein kolloidale Lösungen von
Polymeren in wässrigen Medien bezeichnet [127].
SO3- +R N3
NR3+
NR3+
NR3+
+R N3
+R N3
SO3-
SO3-
SO3-
SO3-
SO3-
SO3-
R = Methyl, Ethyl, Ethanol (= Hydroxyethyl) EDMA = Ethyldimethylamin DMEA = Dimethylethanolamin DEMA = Diethanolmethylamin TEA = Triethanolamin
Abb. 19: Schematischer Aufbau von Latex-Anionenaustauschern und Auflistung einiger zur Funktionalisierung eingesetzter Amine in Reihenfolge ansteigender Polarität.
Die im Rahmen dieser Dissertation verwendeten Latex-Anionenaustauscher überzeugen durch
ihre sehr hohen Trennleistungen, die mit Carbonat/Bicarbonat-Eluenten typischerweise 30000
bis 80000 TP m−1 betragen. Die gute Effizienz der Säulen ist bedingt durch schnelle Aus-
tauschprozesse (Massentransfer), und wird durch die geringe Größe der Latexpartikel gewähr-
leistet. Die Peaks, die man bei Trennungen erhält, sind sehr symmetrisch und für viele
Analyten nahezu gaußförmig. Der Trennprozess spielt sich hauptsächlich an der Oberfläche
dieser Anionenaustauscher ab, und es kommt wenig zu sekundären Wechselwirkungen mit
dem Trägermaterial. Auch die polarisierbaren Anionen (s. Kap. 2.4.3) ergeben mit Latex-
säulen gute Peakformen, allerdings nur, wenn geeignete funktionelle Gruppen verwendet
werden (s. Seite 61). Das Substrat neigt auf Grund der hohen Quervernetzung und der
Abschirmung durch die Oberflächenfunktionalisierung kaum zu Schwell- oder Schrumpf-
prozessen, wodurch eine hohe Reproduzierbarkeit der Trennungen gewährleistet werden
kann. Eine gute mechanische Stabilität käuflicher Materialien ist laut Herstellerangaben bis zu
einem Arbeitsdruck von 270 bar gegeben.
Latex-Ionenaustauscher sind über einen weiten pH-Bereich nutzbar, was auch die Anwendung
von stark alkalischen Eluenten ermöglicht, die mit stationären Phasen auf Silicabasis nicht
kompatibel wären.
- 58 -

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 59 -
4.1.3 VBC-Latex-Anionenaustauscher
4.1.3.1 Überladungseffekte bei Latex-Anionenaustauschern
Die Kapazitäten der im Rahmen dieser Dissertation verwendeten Anionenaustauscher mit den
Säulendimensionen 100 × 4 mm erreichen typischerweise Werte von 20–80 µeq Säule−1. Sie
liegen damit fünf- bis zehnmal niedriger als die Kapazitäten oberflächenfunktionalisierter
Materialien, wie sie in Kap. 4.1.5 beschrieben werden. Die Gesamtkapazität einer Säule
bestimmt die maximale Beladungsmenge mit Ionen, die auf der Säule getrennt werden kann,
ohne dass Überladungseffekte auftreten.
Laut theoretischen Betrachtungen hat streng genommen jede noch so kleine Veränderung der
aufgegebenen Probenmenge einen Effekt auf die Peakform und die Retentionszeit [128]. Die
kleinen Änderungen, die bei niedrigen Konzentrationen auftreten, sind aber meist nicht
messbar, weil die Präzision der Messungen nicht ausreicht. Erst bei hohen Probenmengen, die
störenden Effekte wie die Verkürzung der Retentionszeit und eine unsymmetrische Peakform
verursachen, spricht man von Überladung. Dies kommt bei der Spurenanalytik in wässrigen
Lösungen mit relativ geringer ionischer Matrix selten vor. Aus diesem Grund spielt dabei die
Kapazität der Säulen als Qualitätsmerkmal meist nur eine untergeordnete Rolle. Zum Beispiel
handelt es sich bei Trinkwasserproben oder auch Multianionenstandards im niedrigen
mg L−1-Bereich und darunter um Lösungen mit so geringer Ionenkonzentration, dass auch bei
niedrigkapazitiven Säulen keine Überladungsprobleme auftreten.
Es soll an dieser Stelle aber darauf hingewiesen werden, dass pellikulare Anionenaustauscher
wegen ihrer geringeren Kapazität auch nur eine fünf- bis zehnmal niedrigere Gesamt-
ionenkonzentration tolerieren als oberflächenfunktionalisierte Säulenmaterialien. Dies ist bei
Proben mit sehr konzentrierter Matrix zu beachten, z.B. bei der Messung von 0.1–1%igen
Salzlösungen in Kap. 4.4.1, aber auch bei der Aufgabe von Multistandards mit außerordent-
lich hoher Gesamtionenkonzentration, besonders wenn Anionen mit starker Elutionskraft
darin enthalten sind. Es kommt zu störenden Überladungseffekten, wenn sich die molare
Gesamtanionenkonzentration in Proben zu sehr der Eluentkonzentration annähert. Folgende
Zahlenwerte sollen einen ungefähren Überblick für Latex-Anionenaustauscher und Nitrat als
Eluentanion vermitteln: Bei einem Aufgabevolumen von 153 µL ist bei einer Kapazität von
20−80 µeq Säule−1 erfahrungsgemäß ab Anionenkonzentrationen im Bereich von
50−500 mg L−1 (oder >0.01 % TDS) mit störenden Überladungseffekten zu rechnen. Größere
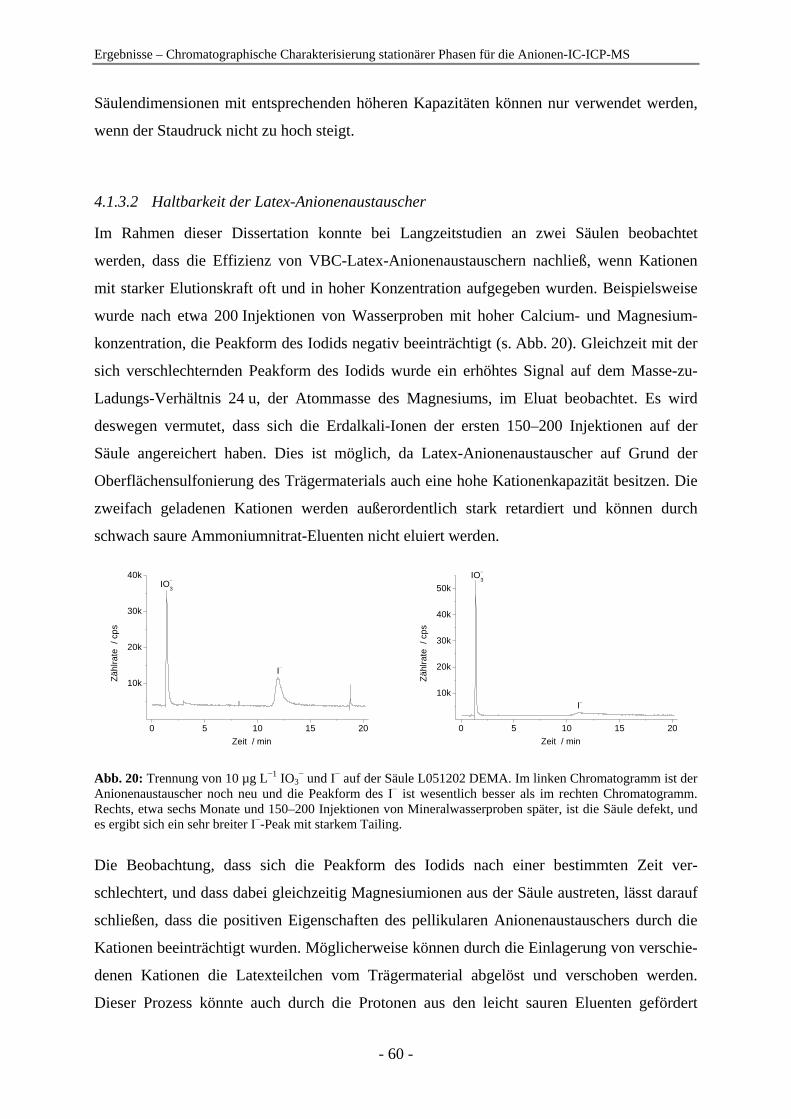
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
Säulendimensionen mit entsprechenden höheren Kapazitäten können nur verwendet werden,
wenn der Staudruck nicht zu hoch steigt.
4.1.3.2 Haltbarkeit der Latex-Anionenaustauscher
Im Rahmen dieser Dissertation konnte bei Langzeitstudien an zwei Säulen beobachtet
werden, dass die Effizienz von VBC-Latex-Anionenaustauschern nachließ, wenn Kationen
mit starker Elutionskraft oft und in hoher Konzentration aufgegeben wurden. Beispielsweise
wurde nach etwa 200 Injektionen von Wasserproben mit hoher Calcium- und Magnesium-
konzentration, die Peakform des Iodids negativ beeinträchtigt (s. Abb. 20). Gleichzeit mit der
sich verschlechternden Peakform des Iodids wurde ein erhöhtes Signal auf dem Masse-zu-
Ladungs-Verhältnis 24 u, der Atommasse des Magnesiums, im Eluat beobachtet. Es wird
deswegen vermutet, dass sich die Erdalkali-Ionen der ersten 150–200 Injektionen auf der
Säule angereichert haben. Dies ist möglich, da Latex-Anionenaustauscher auf Grund der
Oberflächensulfonierung des Trägermaterials auch eine hohe Kationenkapazität besitzen. Die
zweifach geladenen Kationen werden außerordentlich stark retardiert und können durch
schwach saure Ammoniumnitrat-Eluenten nicht eluiert werden.
0 5 10 15 20
10k
20k
30k
40k
Zähl
rate
/ c
ps
Zeit / min
IO−
3
I−
00 5 10 15 2
10k
20k
30k
40k
50k
Zähl
rate
/ c
ps
Zeit / min
IO−
3
I−
Abb. 20: Trennung von 10 µg L−1 IO3
− und I− auf der Säule L051202 DEMA. Im linken Chromatogramm ist der Anionenaustauscher noch neu und die Peakform des I− ist wesentlich besser als im rechten Chromatogramm. Rechts, etwa sechs Monate und 150–200 Injektionen von Mineralwasserproben später, ist die Säule defekt, und es ergibt sich ein sehr breiter I−-Peak mit starkem Tailing.
Die Beobachtung, dass sich die Peakform des Iodids nach einer bestimmten Zeit ver-
schlechtert, und dass dabei gleichzeitig Magnesiumionen aus der Säule austreten, lässt darauf
schließen, dass die positiven Eigenschaften des pellikularen Anionenaustauschers durch die
Kationen beeinträchtigt wurden. Möglicherweise können durch die Einlagerung von verschie-
denen Kationen die Latexteilchen vom Trägermaterial abgelöst und verschoben werden.
Dieser Prozess könnte auch durch die Protonen aus den leicht sauren Eluenten gefördert
- 60 -

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 61 -
werden. Der Latex wird aber nicht vollständig von der Säule befördert, da die beobachteten
Retentionszeiten und damit auch die Kapazität gleich blieben.
Eine zweite Möglichkeit besteht darin, dass die Sulfonatgruppen des Trägermaterials durch
die starke Wechselwirkung mit Erdalkali-Ionen neutralisiert werden. Dadurch könnte die
Abschirmung des Trägermaterials durch Donnan-Ausschlusskräfte verloren gehen. Bei beiden
Möglichkeiten, sowohl durch die Verschiebung der Latexteilchen, als auch durch eine
vergrößerte Anionendurchlässigkeit der Donnan-Membran, entstehen mehr sekundäre
Wechselwirkungen mit dem Substrat, die für das beobachtete Peaktailing des Iodids verant-
wortlich sein können.
Ein weiterer möglicher Grund für die verkürzte Lebensdauer der Säulen sind Packungsfehler,
die durch Schwellprozesse bei der Anlagerung von Calcium, Magnesium und anderen
Kationen im sulfonierten Trägermaterial entstehen könnten. Die Entstehung von Packungs-
ungleichmäßigkeiten lässt sich am besten durch einen höheren Druck von 300–500 bar beim
Packen der stationären Phasen vermeiden [129].
4.1.3.3 Einfluss verschiedener Alkylreste an der funktionellen Gruppe
Der schematische Aufbau von Latex-Anionenaustauschern sowie die vier verwendeten
Ammoniumaustauschergruppen der Latices sind in Abb. 19 gezeigt. Bei den untersuchten
VBC-Latex-Anionenaustauschern bestehen sowohl die Trägerteilchen, als auch die Latex-
partikel aus polymerisierten Styrolderivaten. Neben dem grundsätzlichen Aufbau der
pellikularen Austauscher üben die Substituenten der quartären Ammoniumfunktionen einen
wichtigen Einfluss auf das Retentionsverhalten und die Peakform der getrennten Ionen
aus [123, 130, 131].
Im Allgemeinen gelten Latex-Ionenaustauscher wegen ihres pellikularen Aufbaus als gut
geeignet für die Trennung polarisierbarer Ionen [13]. Dafür werden zwei Gründe angeführt:
Zum einen hemmt die Oberflächensulfonierung der Trägerpartikel auf Grund von Donnan-
Ausschlusskräften das Eindringen von Ionen. Zum anderen sind nur auf den Latexteilchen
Anionenaustauscherfunktionen angebracht, und die Latexschicht befindet sich ausschließlich
auf der Oberfläche der Trägerpartikel. Die Ionenaustauschprozesse finden daher bei Latex-
Ionenaustauschern im Vergleich zu oberflächenfunktionalisierten Ionenaustauschern in
größerer Entfernung vom Trägermaterial statt. Dies reduziert die Wahrscheinlichkeit, dass
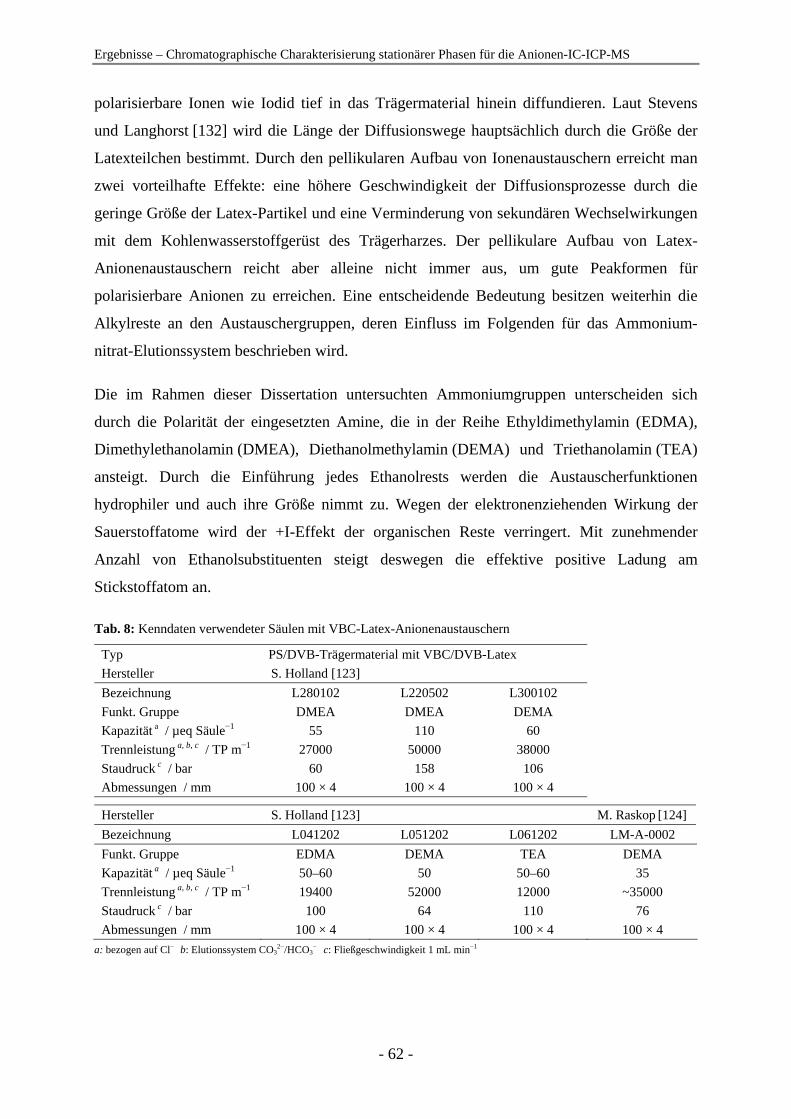
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 62 -
polarisierbare Ionen wie Iodid tief in das Trägermaterial hinein diffundieren. Laut Stevens
und Langhorst [132] wird die Länge der Diffusionswege hauptsächlich durch die Größe der
Latexteilchen bestimmt. Durch den pellikularen Aufbau von Ionenaustauschern erreicht man
zwei vorteilhafte Effekte: eine höhere Geschwindigkeit der Diffusionsprozesse durch die
geringe Größe der Latex-Partikel und eine Verminderung von sekundären Wechselwirkungen
mit dem Kohlenwasserstoffgerüst des Trägerharzes. Der pellikulare Aufbau von Latex-
Anionenaustauschern reicht aber alleine nicht immer aus, um gute Peakformen für
polarisierbare Anionen zu erreichen. Eine entscheidende Bedeutung besitzen weiterhin die
Alkylreste an den Austauschergruppen, deren Einfluss im Folgenden für das Ammonium-
nitrat-Elutionssystem beschrieben wird.
Die im Rahmen dieser Dissertation untersuchten Ammoniumgruppen unterscheiden sich
durch die Polarität der eingesetzten Amine, die in der Reihe Ethyldimethylamin (EDMA),
Dimethylethanolamin (DMEA), Diethanolmethylamin (DEMA) und Triethanolamin (TEA)
ansteigt. Durch die Einführung jedes Ethanolrests werden die Austauscherfunktionen
hydrophiler und auch ihre Größe nimmt zu. Wegen der elektronenziehenden Wirkung der
Sauerstoffatome wird der +I-Effekt der organischen Reste verringert. Mit zunehmender
Anzahl von Ethanolsubstituenten steigt deswegen die effektive positive Ladung am
Stickstoffatom an.
Tab. 8: Kenndaten verwendeter Säulen mit VBC-Latex-Anionenaustauschern
Typ PS/DVB-Trägermaterial mit VBC/DVB-Latex Hersteller S. Holland [123] Bezeichnung L280102 L220502 L300102 Funkt. Gruppe DMEA DMEA DEMA Kapazität a / µeq Säule−1 55 110 60 Trennleistung a, b, c / TP m−1 27000 50000 38000 Staudruck c / bar 60 158 106 Abmessungen / mm 100 × 4 100 × 4 100 × 4
Hersteller S. Holland [123] M. Raskop [124] Bezeichnung L041202 L051202 L061202 LM-A-0002 Funkt. Gruppe EDMA DEMA TEA DEMA Kapazität a / µeq Säule−1 50–60 50 50–60 35 Trennleistung a, b, c / TP m−1 19400 52000 12000 ~35000 Staudruck c / bar 100 64 110 76 Abmessungen / mm 100 × 4 100 × 4 100 × 4 100 × 4
a: bezogen auf Cl− b: Elutionssystem CO32−/HCO3
− c: Fließgeschwindigkeit 1 mL min−1
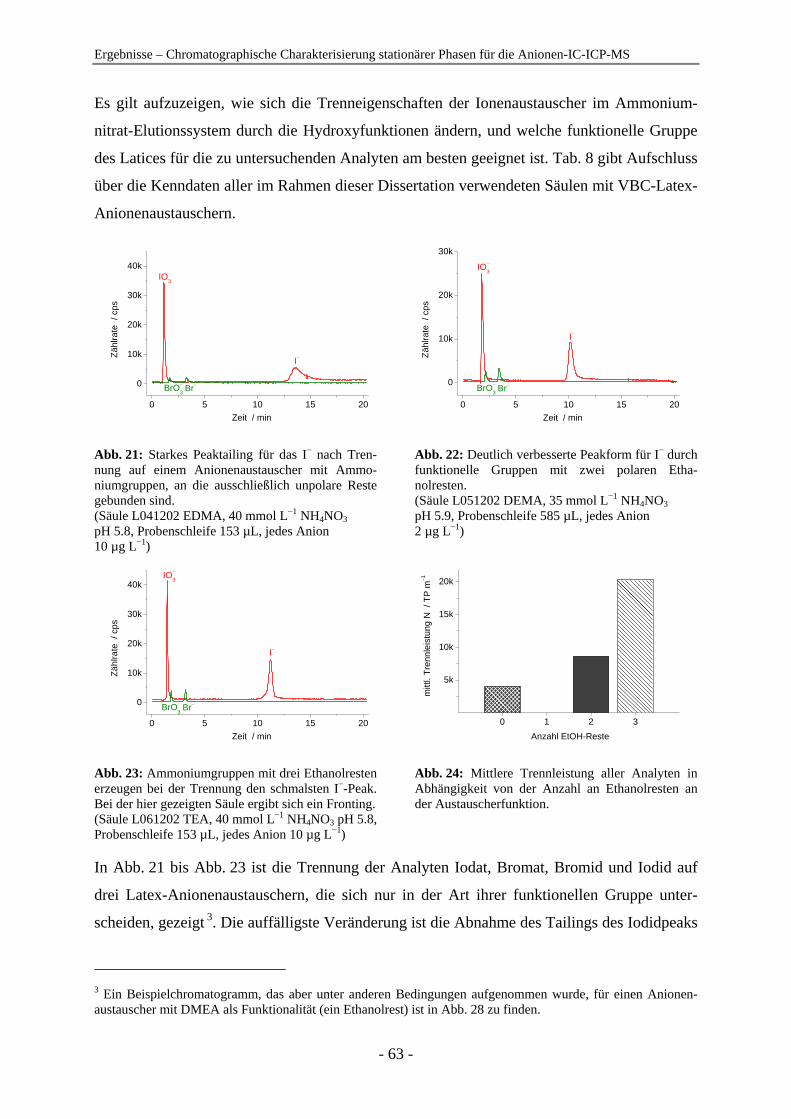
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
Es gilt aufzuzeigen, wie sich die Trenneigenschaften der Ionenaustauscher im Ammonium-
nitrat-Elutionssystem durch die Hydroxyfunktionen ändern, und welche funktionelle Gruppe
des Latices für die zu untersuchenden Analyten am besten geeignet ist. Tab. 8 gibt Aufschluss
über die Kenndaten aller im Rahmen dieser Dissertation verwendeten Säulen mit VBC-Latex-
Anionenaustauschern.
0 5 10 15 20
0
10k
20k
30k
40k
BrO−
3
Zähl
rate
/ c
ps
Zeit / min
IO−
3
Br−
I−
0 5 10 15 20
0
10k
20k
30k
- 63 -
BrO−
3 Br−
Zähl
rate
/ c
psZeit / min
IO−
3
I−
Abb. 21: Starkes Peaktailing für das I− nach Tren-nung auf einem Anionenaustauscher mit Ammo-niumgruppen, an die ausschließlich unpolare Reste gebunden sind. (Säule L041202 EDMA, 40 mmol L−1 NH4NO3 pH 5.8, Probenschleife 153 µL, jedes Anion 10 µg L−1)
Abb. 22: Deutlich verbesserte Peakform für I− durch funktionelle Gruppen mit zwei polaren Etha-nolresten. (Säule L051202 DEMA, 35 mmol L−1 NH4NO3 pH 5.9, Probenschleife 585 µL, jedes Anion 2 µg L−1)
0 5 10 15 20
0
10k
20k
30k
40k
BrO−
3
Zähl
rate
/ c
ps
Zeit / min
IO−
3
Br−
I−
0 1 2 3
5k
10k
15k
20k
Anzahl EtOH-Reste
mitt
l. Tr
ennl
eist
ung
N /
TP
m−1
Abb. 23: Ammoniumgruppen mit drei Ethanolrestenerzeugen bei der Trennung den schmalsten I
−-Peak.
Bei der hier gezeigten Säule ergibt sich ein Fronting.−1(Säule L061202 TEA, 40 mmol L NH4NO3 pH 5.8,
Probenschleife 153 µL, jedes Anion 10 µg L−1)
Abb. 24: Mittlere Trennleistung aller Analyten in Abhängigkeit von der Anzahl an Ethanolresten an der Austauscherfunktion.
In Abb. 21 bis Abb. 23 ist die Trennung der Analyten Iodat, Bromat, Bromid und Iodid auf
drei Latex-Anionenaustauschern, die sich nur in der Art ihrer funktionellen Gruppe unter-
scheiden, gezeigt 3. Die auffälligste Veränderung ist die Abnahme des Tailings des Iodidpeaks
3 Ein Beispielchromatogramm, das aber unter anderen Bedingungen aufgenommen wurde, für einen Anionen-austauscher mit DMEA als Funktionalität (ein Ethanolrest) ist in Abb zu finden. . 28

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 64 -
bei der Verwendung stärker polarer Austauschergruppen. Die in Abb. 23 dargestellte Tren-
nung durch die Säule L061202 mit TEA-Gruppen führt sogar zu einem Peak mit Fronting.
Das Iodidsignal wird in Korrelation mit den funktionellen Gruppen vom EDMA mit null,
DEMA mit zwei und TEA mit drei Ethanolresten deutlich schmaler und höher. Dieser Effekt
macht das Iodidsignal zu einem geeigneten Indikator für die Hydrophilie der verwendeten
Ammoniumgruppe.
In der Reihe vom EDMA-, zum DEMA- bis zum TEA-Austauscher wird die durchschnittliche
Trennleistung der vier Analyten von 4000 TP m−1 über 8600 TP m−1 bis hin zu 20300 TP m−1
jeweils mehr als verdoppelt (Abb. 24). Dies wirkt sich positiv auf die Auflösung benachbarter
Peaks aus. Allerdings wird trotzdem mit keiner dieser Säulen eine ausreichende Auflösung
von Totvolumenpeak und Iodat oder von Iodat und Bromat erreicht, wenn das Iodid innerhalb
von 15 min eluiert (s. Seite 56).
Die relative Retention des Iodids in Bezug auf Bromid wird verkürzt, wenn sich Hydroxy-
gruppen an den Ammoniumfunktionen befinden. Die Verringerung der Selektivität für polari-
sierbare Anionen ist von Vorteil, da dadurch die Gesamtmesszeit von Multianionenanalysen
verkürzt werden kann. Wie sich in der Untersuchung des Einflusses der Alkylreste an den
funktionellen Gruppen gezeigt hat, sind Latex-Anionenaustauscher mit DEMA oder TEA als
funktioneller Gruppe am besten für die Multianionenanalyse und insbesondere für die
Analyse des Iodids geeignet.
Eine Ursache für die veränderte Iodidselektivität und für die verbesserte Peakform durch
Etablierung von Ethanolresten an den Austauscherfunktionen liegt in der Verringerung von
sekundären Wechselwirkungen. Nichtionische Wechselwirkungen mit Kohlenwasserstoff-
einheiten können bei genauer theoretischer Betrachtung an drei verschiedenen Stellen von
Latex-Anionenaustauschern auftreten:
• am Polymergerüst der Trägerteilchen (auf der äußeren und inneren Oberfläche)
• am Polymergerüst der Latexpartikel
• und an den Alkylsubstituenten der Austauscherfunktionen des Latex.
Die Veränderung der Substituenten der Ammoniumgruppen kann sich auf alle drei Positionen
auswirken, wenn man die folgende Modellvorstellung zu Grunde legt. Erstens gehen die
polarisierbaren Ionen weniger sekundäre Wechselwirkungen mit den Substituenten am

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 65 -
quartären Amin ein, wenn diese polarer sind. Zweitens steigt durch die Hydroxygruppen der
Wassergehalt in dem aromatischen Kohlenstoffgerüst der Latexteilchen an, was die Retention
der wenig hydratisierten polarisierbaren Ionen dort verringert. Drittens ist ein hydrophilerer
Latex auf der Oberfläche des Trägermaterials besonders undurchlässig für lipophile Analyten,
so dass das Iodid oder andere polarisierbare Ionen daran gehindert werden, bis in das
Trägermaterial einzudringen.
4.1.3.4 Hindernisse bei der Analyse von Iodid:
Instabilität des Analyten und spezielle Anforderungen an die stationäre Phase
Im Gegensatz zu den Ionen Iodat, Bromat oder Bromid, bereitet die Messung von Iodid
verschiedene, wesentlich größere Schwierigkeiten, die erkannt und beseitigt werden müssen.
Das Iodid erscheint in manchen Chromatogrammen nur als intensitätsschwacher, breiter Peak,
der zudem oft ein Tailing aufweist. Bei Trennung auf besonders ungeeigneten Säulen kann
das Iodid im unteren µg L−1-Bereich kaum oder gar nicht mehr detektiert werden. Ein solches
Beispiel ist in Abb. 25 dargestellt. Man erkennt, dass der Peak des Iodids, trotz einer relativ
kurzen Retentionszeit von nur zehn Minuten, breit ist und sich kaum vom Untergrund abhebt.
Nach etwa vier Minuten ist außerdem der Peak einer unbekannten Iodspezies zu sehen.
Für die Schwierigkeiten bei der Iodidwiederfindung lassen sich zwei Hauptursachen identi-
fizieren: Zum einen sind Iodidstandards nur begrenzt haltbar und zum anderen ist Iodid in
niedrigen Konzentrationen nicht mehr detektierbar, wenn es mit den Ionenaustauschern zu
starke sekundäre Wechselwirkungen eingehen kann. Im Falle, dass diesbezüglich ungeeignete
Ionenaustauscher verwendet werden, kann es zu einer vollständigen Entfernung des Iodids
durch irreversible 4 Adsorption an den Harzen bis zur Sättigung aller Adsorptionsstellen
kommen, oder das Iodid ergibt Peaks mit so starkem Tailing, dass sie sich nicht mehr genug
vom Untergrund abheben. Weitere Betrachtungen zu sekundären Wechselwirkungen und
polarisierbaren Anionen sind in Kap. 2.4.3 und in Kap. 4.1.3.3 zu finden.
4 Der Gebrauch des Adjektivs „irreversibel“ soll hier eine wesentlich längere Dauer der Adsorption im Vergleich mit der Dauer von ionenchromatographischen Verdrängungsprozessen andeuten.
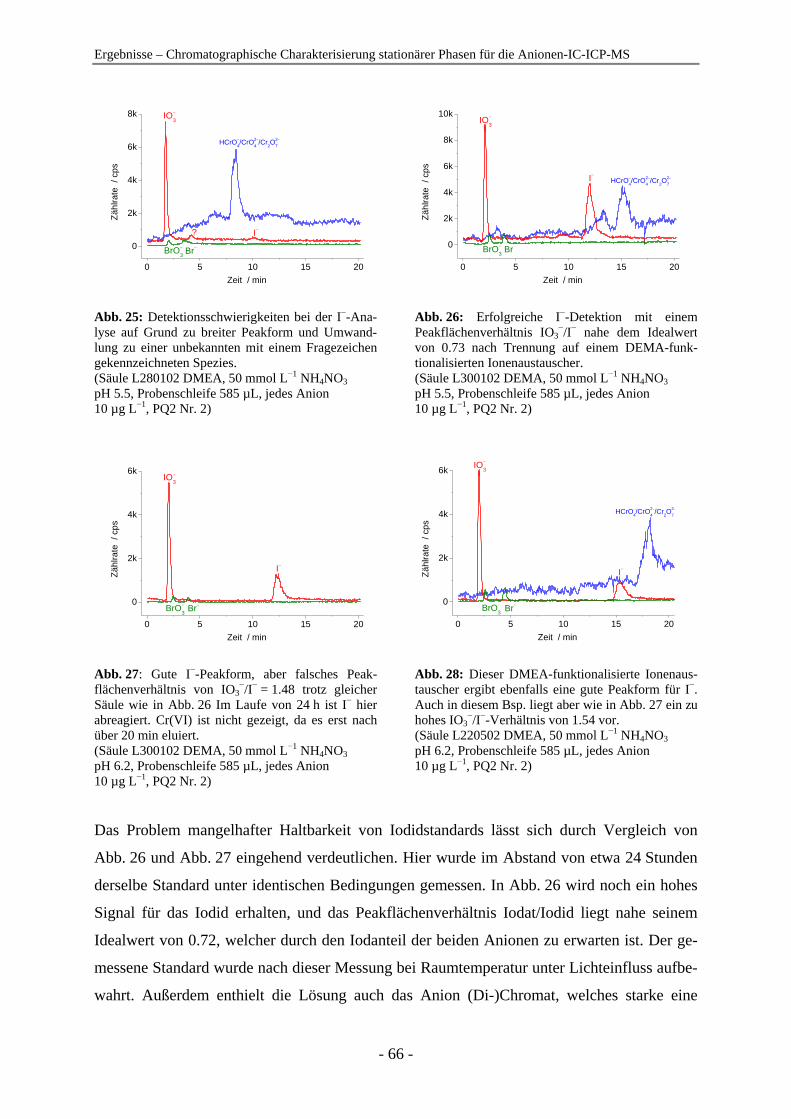
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
0 5 10 15 20
0
2k
4k
6k
8k
HCrO−
4/CrO2−4 /Cr2O
2−7
?
BrO−
3
Zähl
rate
/ c
ps
Zeit / min
IO−
3
Br−I−
0 5 10 15 20
0
2k
4k
6k
8k
10k
HCrO−
4/CrO2−4 /Cr2O
2−7
- 66 -
BrO−
3 Br−
Zähl
rate
/ c
ps
Zeit / min
IO−
3
I−
Abb. 25: Detektionsschwierigkeiten bei der I−-Ana-lyse auf Grund zu breiter Peakform und Umwand-lung zu einer unbekannten mit einem Fragezeichengekennzeichneten Spezies. (Säule L280102 DMEA, 50 mmol L−1 NH4NO3 pH 5.5, Probenschleife 585 µL, jedes Anion 10 µg L−1, PQ2 Nr. 2)
Abb. 26: Erfolgreiche I−-Detektion mit einem Peakflächenverhältnis IO3
−/I− nahe dem Idealwert von 0.73 nach Trennung auf einem DEMA-funk-tionalisierten Ionenaustauscher. (Säule L300102 DEMA, 50 mmol L−1 NH4NO3 pH 5.5, Probenschleife 585 µL, jedes Anion 10 µg L−1, PQ2 Nr. 2)
0 5 10 15 20
0
2k
4k
6k
BrO−
3
Zähl
rate
/ c
ps
Zeit / min
IO−
3
Br−
I−
0 5 10 15 20
0
2k
4k
6k
HCrO−
4/CrO2−4 /Cr2O
2−7
BrO−
3 Br−
Zähl
rate
/ c
ps
Zeit / min
IO−
3
I−
Abb. 27: Gute I−-Peakform, aber falsches Peak-flächenverhältnis von IO3
−/I− = 1.48 trotz gleicherSäule wie in Abb. 26 Im Laufe von 24 h ist I− hier abreagiert. Cr(VI) ist nicht gezeigt, da es erst nachüber 20 min eluiert. (Säule L300102 DEMA, 50 mmol L−1 NH4NO3 pH 6.2, Probenschleife 585 µL, jedes Anion 10 µg L−1, PQ2 Nr. 2)
Abb. 28: Dieser DMEA-funktionalisierte Ionenaus-tauscher ergibt ebenfalls eine gute Peakform für I−. Auch in diesem Bsp. liegt aber wie in Abb. 27 ein zu hohes IO3
−/I−-Verhältnis von 1.54 vor. −1(Säule L220502 DMEA, 50 mmol L NH4NO3
pH 6.2, Probenschleife 585 µL, jedes Anion 10 µg L−1, PQ2 Nr. 2)
Das Problem mangelhafter Haltbarkeit von Iodidstandards lässt sich durch Vergleich von
Abb. 26 und Abb. 27 eingehend verdeutlichen. Hier wurde im Abstand von etwa 24 Stunden
derselbe Standard unter identischen Bedingungen gemessen. In Abb. 26 wird noch ein hohes
Signal für das Iodid erhalten, und das Peakflächenverhältnis Iodat/Iodid liegt nahe seinem
Idealwert von 0.72, welcher durch den Iodanteil der beiden Anionen zu erwarten ist. Der ge-
messene Standard wurde nach dieser Messung bei Raumtemperatur unter Lichteinfluss aufbe-
wahrt. Außerdem enthielt die Lösung auch das Anion (Di-)Chromat, welches starke eine

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 67 -
Oxidationskraft besitzt. Abb. 27 zeigt, wie das Iodid nur 24 Stunden später durch Luft-
sauerstoff und/oder das (Di-)Chromat teilweise zu Iodat oxidiert worden ist. Das Iodat/Iodid-
Verhältnis liegt bei der erneuten Messung bei 1.48 und ist damit viel zu hoch. Außer zu Iodat
kann das Iodid aber auch zu elementarem Iod (I2) und weiter zu Spuren von noch leichter
flüchtigen Organoiodverbindungen umgesetzt werden. Ein sichtbarer Peak einer unbekannten
Iodspezies wie in Abb. 25 entsteht jedoch noch keineswegs innerhalb von 24 h, sondern erst
bei mehrere Wochen alten, nicht gekühlt gelagerten Lösungen. An den Beispielen wird
deutlich, dass Iodid enthaltende Proben unbedingt kalt, luft- und lichtgeschützt aufbewahrt
und die Standards regelmäßig neu angesetzt werden müssen. Eine detaillierte allgemeine
Arbeitsanweisung zum Ansetzen und Aufbewahren von verschiedenen Standards ist im
Anhang 6.3.1 zu finden.
Da bei den Messungen in Abb. 25–Abb. 28 auch das Anion (Di-)Chromat mit gemessen
wurde, soll an dieser Stelle kurz darauf eingegangen werden. Das (Di-)Chromat weist sehr
unterschiedliche Retentionszeiten auf, da es in einem pH-Wert-abhängigen Gleichgewicht
dreier Chromspezies vorliegt (s. Kap. 4.1.3.5). Außerdem wird bei diesem Analyt des Öfteren
eine schlechte Peakform beobachtet. Diese kommt allerdings im Gegensatz zum Iodid meist
nicht durch das Säulenmaterial zu Stande, sondern durch den schwankenden Untergrund des
ICP-MS auf dem Masse-zu-Ladungs-Verhältnis 52, wo eine Interferenz mit [40Ar12C]+
vorliegt (s. Kap. 2.4.2).
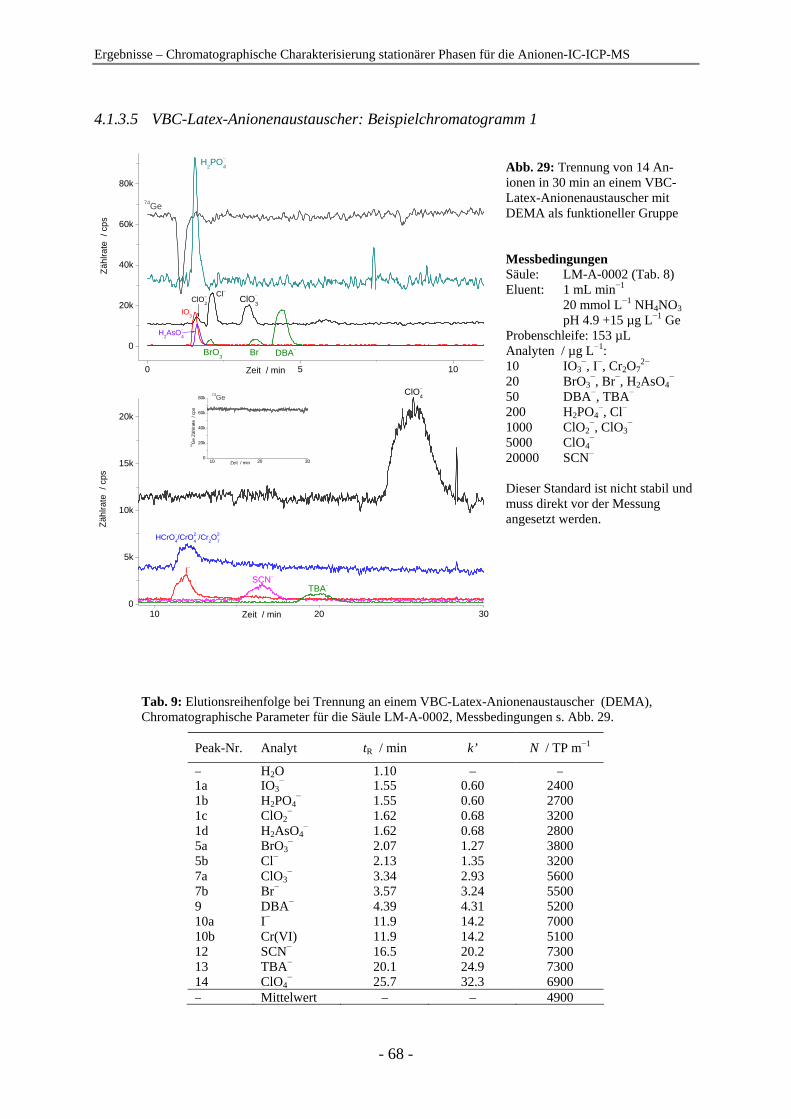
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
4.1.3.5 VBC-Latex-Anionenaustauscher: Beispielchromatogramm 1
- 68 -
Abb. 29: Trennung von 14 An-ionen in 30 min an einem VBC-Latex-Anionenaustauscher mit DEMA als funktioneller Gruppe
10 20 300
5k
10k
15k
20k
10 20 300
20k
40k
60k
80k
0 5
0
20k
40k
60k
80k
Messbedingungen Säule: LM-A-0002 (Tab. 8) Eluent: 1 mL min−1
20 mmol L−1 NH4NO3 pH 4.9 +15 µg L−1 Ge Probenschleife: 153 µL Analyten / µg L−1: 10 IO3
−, I−, Cr2O72−
20 BrO3−, Br−, H2AsO4
−
50 DBA−, TBA−
200 H2PO4−, Cl−
1000 ClO2−, ClO3
−
5000 ClO4−
20000 SCN−
Dieser Standard ist nicht stabil und muss direkt vor der Messung angesetzt werden.
Tab. 9: Elutionsreihenfolge bei Trennung an einem VBC-Latex-Anionenaustauscher (DEMA), Chromatographische Parameter für die Säule LM-A-0002, Messbedingungen s. Abb. 29.
Peak-Nr. Analyt tR / min k’ N / TP m−1
– H2O 1.10 – – 1a IO3
− 1.55 0.60 2400 1b H2PO4
− 1.55 0.60 2700 1c ClO2
− 1.62 0.68 3200 1d H2AsO4
− 1.62 0.68 2800 5a BrO3
− 2.07 1.27 3800 5b Cl− 2.13 1.35 3200 7a ClO3
− 3.34 2.93 5600 7b Br− 3.57 3.24 5500 9 DBA− 4.39 4.31 5200 10a I− 11.9 14.2 7000 10b Cr(VI) 11.9 14.2 5100 12 SCN− 16.5 20.2 7300 13 TBA− 20.1 24.9 7300 14 ClO4
− 25.7 32.3 6900 – Mittelwert – – 4900
10
I−
TBA−
ClO−
4
Zähl
rate
/ c
ps
Zeit / min
SCN−
HCrO−
4/CrO2−4 /Cr2O
2−7
74Ge
74G
e Zä
hlra
te /
cps
Zeit / min
Cl−
DBA−BrO−
3 Br−
Zähl
rate
/ c
ps
Zeit / min
ClO−
3ClO−
2
74Ge
H2PO−
4
H2AsO−
4
IO−
3

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 69 -
Die Eigenschaften von VBC-Latex-Anionenaustauschern bei der Multianionentrennung wer-
den anhand von zwei Beispielchromatogrammen beschrieben. Die jeweils verwendeten
stationären Phasen unterscheiden sich nur durch die Ammoniumgruppen. Es wurden die laut
Kap. 4.1.3.3 am besten geeigneten funktionellen Gruppen eingesetzt: In der Säule
LM-A-0002 die Ammoniumgruppe DEMA (Abb. 29) und in der Säule L061202 die Funktio-
nalität TEA (Abb. 30).
Das erste Beispiel in Abb. 29 zeigt, dass mit der Säule LM-A-0002 (Tab. 8) in einem
30-minütigen Chromatogramm 14 Anionen gemessen werden können. Die ersten 8 der
14 Analyten eluieren schon innerhalb von fünf Minuten (Tab. 9). Iodat, Phosphat, Arsenat
und Chlorit verlassen die Säule nahezu gleichzeitig nach 1 min 35 s und damit direkt nach
dem Totvolumen. Die Basispeakbreiten 5 betragen in diesem Anfangsbereich des Chromato-
gramms ca. 30 s. Die Effizienz dieser Säule ist nicht mehr optimal, was sich in den niedrigen
Bodenzahlen unter 8000 TP m−1 für alle Analyten ausdrückt. Der Grund dafür liegt in der
außerordentlich langen und häufigen Verwendung dieser Säule (s. Kap. 4.1.3.2).
Phosphor- und Arsensäure haben pKs2-Werte von ca. sieben. Sie liegen deswegen bei einem
pH-Wert des Eluenten von vier bis sechs größtenteils einfach deprotoniert vor und eluieren
frühzeitig. Erst ungefähr ab einem pH-Wert von sieben macht sich ihre pH-Wert-
Abhängigkeit bemerkbar. Es entsteht eine höhere Gleichgewichtskonzentration der zweifach
deprotonierten Spezies und die Retention nimmt zu.
Der Bromatpeak nach 2 min 4 s ist mit einer Auflösung von 1.4 gerade von der ersten
Peakgruppe getrennt. Bromat eluiert zusammen mit Chlorid, welches nur minimal später
detektiert wird. Chlorat und Bromid haben in diesem System schon deutlich längere
Retentionszeiten von 3 min 20 s bzw. 3 min 34 s, wobei ihre Peaks ebenfalls überlappen.
MBA− lässt sich unter den gewählten Bedingungen über den DEMA-funktionalisierten Latex-
Ionenaustauscher nicht gleichzeitig messen, da es mit Bromat koeluiert. DBA− wird mit einer
Retentionszeit von 4 min 23 s knapp vom Bromid abgetrennt (R = 1.3). Erst sieben Minuten
später erscheinen die ersten Peaks der stark polarisierbaren oder teilweise zweifach geladenen
Anionen.
5 Die Peakbreiten sind ungefähr gleich, da die Anionenkonzentrationen im Multistandard so gewählt wurden, dass sie gut erkennbare Peaks ergeben. Die Konzentrationen sind der jeweiligen Nachweisstärke angepasst, so dass sich die Peakhöhen bei gleicher Skalierung nicht mehr als etwa das 25-fache unterscheiden, wie in z.B. der Phosphatpeak und die Signale der Bromspezies.
Abb. 29

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 70 -
Iodid eluiert unter den Bedingungen von Abb. 29 nach 12 min und hat das stärkste Peak-
tailing. Dennoch ist die Peakform des Iodids als vergleichsweise gut anzusehen
(vergl. Abb. 20). Die Integrationsgrenzen lassen sich sicher festlegen, so dass die Peakfläche
präzise ermittelt werden kann. Chromat eluiert gleichzeitig mit Iodid, aber die Position des
Chromatpeaks kann sich verschieben. Dies ist darin begründet, dass Chrom (VI) in Form von
drei anionischen Spezies vorliegt. Die Geschwindigkeit der Gleichgewichtsreaktionen ist so
schnell, dass nur ein Peak für HCrO4−/CrO4
2−/Cr2O72− erscheint. Die Retentionszeit kann sich
durch Verschiebung des Gleichgewichts mit dem pH-Wert und der Temperatur verändern.
Das Anion Thiocyanat gehört zu den so genannten Pseudohalogeniden. Als Pseudohalogenide
werden Molekülanionen bezeichnet, die den Halogeniden hinsichtlich ihrer physikalischen
und chemischen Eigenschaften ähnlich sind [68]. Für die beiden Ionen Thiocyanat und Iodid
sind insbesondere auch der thermochemische Radius und die Hydratationsenthalpie fast
gleich (s. Tab. 5), weswegen sie ähnliche ionenchromatographische Eigenschaften aufweisen.
Wie in Abb. 29 ersichtlich eluiert das Thiocyanat später als das Iodid. Außerdem ist
erkennbar, dass Schwefelspezies mit dem verwendeten ICP-MS-Gerät nur in 20000-fach
höheren Konzentrationen detektiert werden können. Das Elemention 32S+ kann nicht direkt
gemessen werden, da der Untergrund des ICP-MS auf dieser Masse durch die Interferenz mit
[16O2]+ zu hoch ist. Man ist gezwungen das Molekülion [32S16O]+ zu messen, obwohl dessen
Bildungsrate bei normalen ICP-MS-Bedingungen unter 1 % liegt, und weswegen sich keine
optimalen Nachweisgrenzen ergeben (s.a. Kap. 2.4.2).
Trotz starker Polarisierbarkeit der Ionen Thiocyanat und insbesondere Perchlorat sind ihre
Peakformen im Vergleich zu Iodid erstaunlich symmetrisch. Dies lässt sich durch die hohen
Konzentrationen von Thiocyanat und Perchlorat erklären: Wenn die Positionen für sekundäre
Wechselwirkungen auf dem Säulenmaterial durch Thiocyanat und Perchlorat abgesättigt
werden, können vorwiegend nur noch ionische Wechselwirkungen zu Retention führen, was
sich positiv auf die Peakform auswirkt.
Die beiden zuletzt eluierenden Anionen TBA− und Perchlorat haben Retentionszeiten von
20 min und 26 min. Im Gegensatz zu einigen anderen Säulenmaterialien (s. Abb. 30 oder
Kap. 4.1.6) eluiert TBA− bei Trennung an dem VBC-Latex-Anionenaustauscher noch vor
Perchlorat.
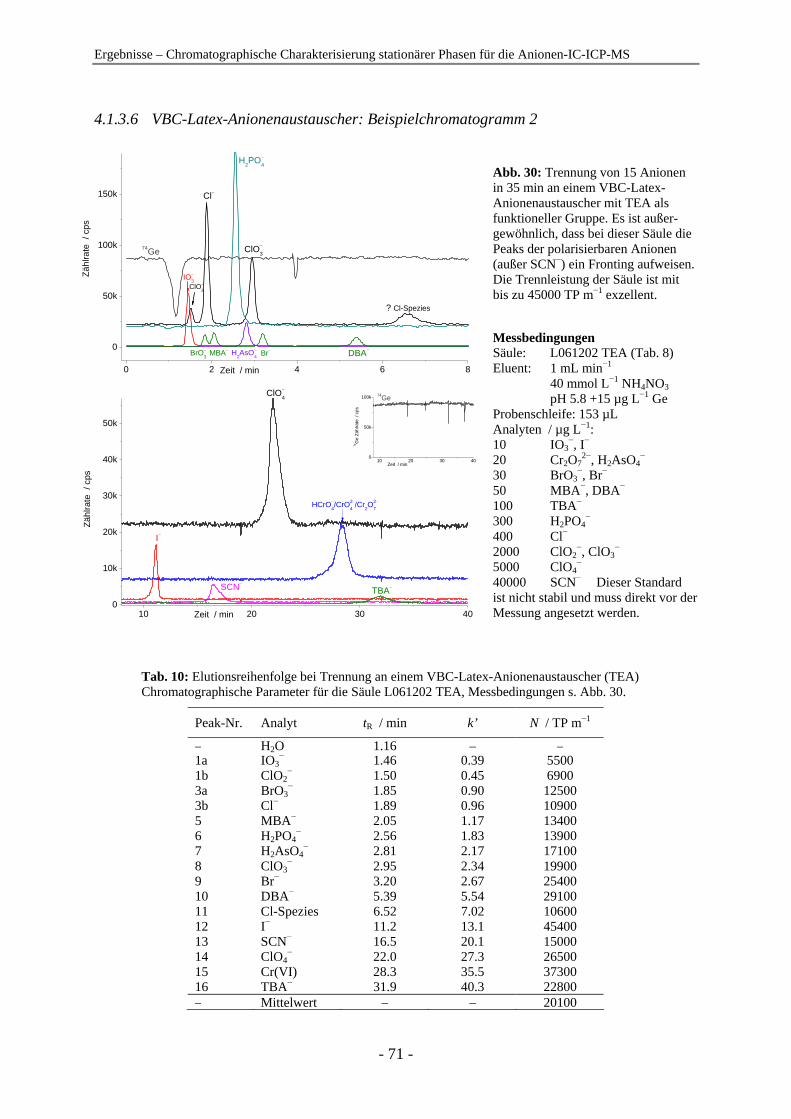
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
4.1.3.6 VBC-Latex-Anionenaustauscher: Beispielchromatogramm 2
- 71 -
ung angesetzt werden.
Tab. 10: Elutionsreihenfolge bei Trennung an einem VBC-Latex-Anionenaustauscher (TEA) Chromatographische Parameter für die Säule L061202 TEA, Messbedingungen s. Abb. 30.
Peak-Nr. Analyt tR / min k’ N / TP m−1
Abb. 30: Trennung von 15 Anionen in 35 min an einem VBC-Latex-Anionenaustauscher mit TEA als funktioneller Gruppe. Es ist außer-gewöhnlich, dass bei dieser Säule die Peaks der polarisierbaren Anionen (außer SCN−) ein Fronting aufweisen. Die Trennleistung der Säule ist mit bis zu 45000 TP m−1 exzellent. Messbedingungen Säule: L061202 TEA (Tab. 8) Eluent: 1 mL min−1 40 mmol L−1 NH4NO3 pH 5.8 +15 µg L−1 Ge Probenschleife: 153 µL Analyten / µg L−1: 10 IO3
−, I−20 Cr2O7
2−, H2AsO4−
30 BrO3−, Br−
50 MBA−, DBA−
100 TBA−
300 H2PO4−
400 Cl−2000 ClO2
−, ClO3−
5000 ClO4−
40000 SCN− Dieser Standard ist nicht stabil und muss direkt vor der Mess
– H2O 1.16 – – 1a IO3
− 1.46 0.39 5500 1b ClO2
− 1.50 0.45 6900 3a BrO3
− 1.85 0.90 12500 3b Cl− 1.89 0.96 10900 5 MBA− 2.05 1.17 13400 6 H2PO4
− 2.56 1.83 13900 7 H2AsO4
− 2.81 2.17 17100 8 ClO3
− 2.95 2.34 19900 9 Br− 3.20 2.67 25400 10 DBA− 5.39 5.54 29100 11 Cl-Spezies 6.52 7.02 10600 12 I− 11.2 13.1 45400 13 SCN− 16.5 20.1 15000 14 ClO4
− 22.0 27.3 26500 15 Cr(VI) 28.3 35.5 37300 16 TBA− 31.9 40.3 22800 – Mittelwert – – 20100
80 2 4 6
0
50k
100k
150k
10 20 30 400
10k
20k
30k
40k
50k
10 20 30 400
50k
100k
ClIO−
3O−
2
? Cl-Spezies
74Ge
HCrO−
4/CrO2−4 /Cr2O
2−7
Cl−
Zähl
rate
/ c
ps
Zeit / min
ClO−
3
H2PO−
4
H2AsO−
4 DBA−BrO−
3 MBA− Br−
I−
SCN−?
ClO−
4
Zähl
rate
/ c
ps
Zeit / min
74Ge
74G
e Zä
hlra
te /
cps
Zeit / min
TBA−

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 72 -
Als zw rennung
eines 15-Anionenstandards an einer stationären Phase mit der funktionellen Gruppe TEA
dargestellt. Das w ste Me al des m funktionalisiert ials ist die hohe
Trennleistung, die es im at-Elutionssystem aufweist (s. 10). Mittelwert für die
Effizienz aller Ionen beträgt 20100 TP m ußerdem t der A austauscher eine
besonders hohe elektivi die Anion er Brom säuren für Phosphat und
Arsenat. So wird immerhi uflösung 1.2 für B at und M rreicht, was sonst
mit keinem anderen untersuchten Latex-Ionenaustauscher gelang. Dies ist von Vorteil z.B. für
die Unterscheidung von Nebenprodukten bei der Desinfektion von Wa
DBA− eluiert bei diesem Säulenmaterial deutlich später als Bromid, und TBA− ist sogar der
am spätesten el ende A Die Reten der Anio der Bro säuren sowie von
Phosphat und enat w sichtlich rch die rophilie EA-Austauscher-
s Chlorit in einem
rend der Trennung anzeigt. Eine Quantifizierung in einer so reaktiven Lösung ist nicht mehr
allgemein bei der Kombination von Ammoniumnitrat-Eluenten und TEA-funktionalisierten
Latex-Austauschern auftritt, da nur diese eine Säule zu Verfügung stand. Auf Grund der
eites Beispiel für einen VBC-Latex-Anionenaustauscher ist in Abb. 30 die T
ichtig rkm it TEA en Mater
Nitr Tab. Der −1. A zeig nionen
S tät für en d essig sowie
n eine A von rom BA e−
sser.
uier nalyt. tion nen messig
Ars ird offen du H dy der T
gruppen verstärkt. Der umgekehrte Effekt tritt für die polarisierbaren Ionen wie Iodid,
Thiocyanat und Perchlorat auf, die relativ früh eluiert werden.
Auch bei TEA-Funktionalisierung lässt sich eine fast gemeinsame Elution der Ionen
Bromat/Chlorid und Chlorat/Bromid beobachten. Diese beiden Peakpaare und die
Elutionsreihenfolge Bromat < Chlorid < Chlorat < Bromid sind bei allen Messungen im
Rahmen dieser Dissertation ein typisches Merkmal pellikularer Anionenaustauscher.
Die notwendige Auflösung der beiden Chlorspezies Chlorit und Chlorid wird mit R = 2.2
erreicht. Es ist bemerkenswert, dass sich ein starkes Oxidationsmittel wie da
Multistandard zusammen mit leicht oxidierbaren Spezies, wie insbesondere dem Iodid, nach-
weisen lässt. Dafür muss die Lösung allerdings mit kaltem Wasser und direkt vor der
Messung angesetzt werden. In Abb. 30 ist zu erkennen, dass solch ein Standard nicht lange
stabil ist, da eine unidentifizierte Chlorspezies entstanden ist. Außerdem befindet sich
zwischen dem Chlorit- und dem Chloridpeak ein Plateau, was eine Speziesumwandlung wäh-
möglich bzw. stellt immer nur eine Momentaufnahme der Konzentrationsverhältnisse dar.
Die Peaks von Chlorat, Iodid, Perchlorat und Chromat zeigen ein Fronting. Leider kann keine
Aussage darüber getroffen werden, ob dieser Effekt nur bei der Säule L061202 oder

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 73 -
t
dies die Messung dar, in der die meisten Analyten gleichzeitig in einem chromatographischen
geringen Reaktivität des Triethanolamins ist es schwierig, damit funktionalisierte Ionen-
austauscher mit ausreichend hoher Kapazität zu erhalten.
Wenn ausgesprochen polare Ammoniumgruppen mit vielen Hydroxygruppen als Aus-
tauscherfunktionen eingesetzt werden, weisen VBC-Latex-Anionenaustauscher eine hohe
Retention der früh eluierenden Ionen bei gleichzeitig verringerter Retention der polarisierbare
Ionen auf. Durch diese spezielle Selektivität und durch die hohe Trennleistung ist es möglich,
besonders viele Anionen innerhalb kurzer Zeit zu trennen. Bei der Messung in Abb. 30 gelang
die Trennung von 15 Anionen in weniger als 35 min. Im Rahmen dieser Dissertation stell
Lauf getrennt wurden. Dies macht VBC-Latex-Anionenaustauscher mit TEA als funktioneller
Gruppe sehr interessant für die Multianionenanalyse.

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
4.1.4 GMA-Latex-Anionenaustauscher
Der grundsätzliche Aufbau dieses Säulentyps entspricht dem in Abb. 19. Als Latexteilchen
werden hier keine VBC/DVB-Copolymere, sondern Polymethacrylatderivate verwendet. Der
wichtigste Ausschnitt6 der Strukturformel des in den untersuchten Säulen enthaltenen
Methacrylatlatices ist in Abb. 31 gezeigt, und die Kenndaten der Säulen sind in Tab. 11
aufgeführt. Das zur Herstellung eingesetzte präfunktionelle Monomer ist Glycidylmetha-
crylat (GMA). Die zum Ionenaustausch dienende quartäre Ammoniumgruppe wird erst nach
der Polymerisation durch eine Epoxid-Ringöffnungsreaktion eingeführt, wodurch man den
funktionalisierten Methacrylatlatex erhält. Eine wichtige Eigenschaft eines Methacrylatlatex
ist, dass er im Vergleich mit VBC/DVB-Copolymeren wesentlich polarer ist und keine
aromatischen Strukturkomponenten mehr enthält. Diese Eigenschaften führen zu einer
Änderung der Selektivität. Durch die Hydrophilie der stationären Phase soll die Retention
stark hydratisierter Anionen erhöht werden. Die Retentionszeit der lipophilen, polarisierbaren
Ionen hingegen soll verkürzt und deren Peakform verbessert werden, indem π-π-Wechsel-
wirkungen mit dem Latexmaterial ausgeschlossen werden [123].
- 74 -
NR3-funktionalisierterPolymethacrylat-Latex
O
OO
O
OOH
Hn
M er GMA= ethacrylat
H
N
onom Glycidyl-M
R3R = Me, Et, EtOH
A her Aufbau des verw GMA-Late
Ta mit GM ex-Anionena hern
PS/D ermaterial + Methacrylatlatex
bb. 31: Chemisc endeten x
b. 11: Kennda n der Säulente A-Lat ustausc
Typ VB-TrägHersteller S. Holland [1 3] 2Bezeichnung M191103 M201103 M211103 Funkt. Gruppe EDMA DMEA DEMA Kapazität a / µeq Säule−1 56 56 43 Trennleistung a, b / TP m−1 61000 d 58000 d 4670 c
Staudruck / bar 190 d 208 d 203 c
Abmessungen / mm 100 × 4 100 × 4 100 × 4 a: bezogen auf Cl− b: Elutionssystem CO3
2−/HCO3− c: Flussrate 1 mL min−1 d: Flussrate 0.7 mL min−1
6 Aus Gründen der Übersichtlichkeit ist nur die Struktur ohne Quervernetzung abgebildet.
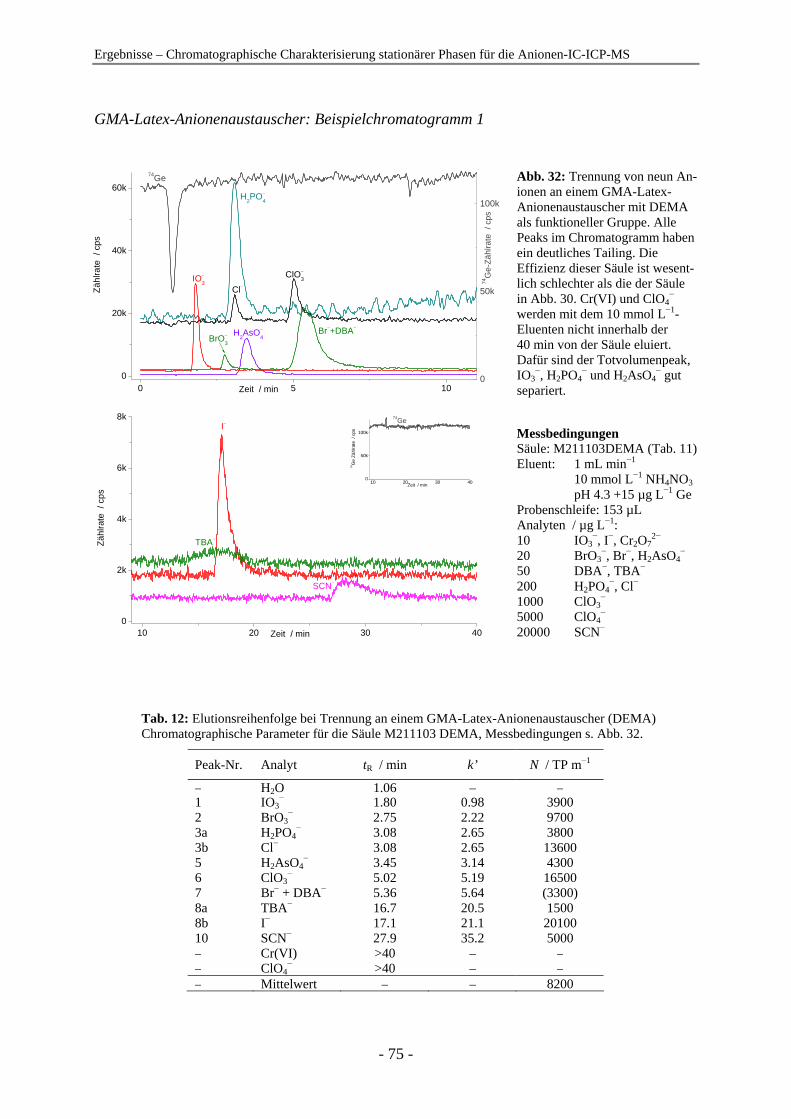
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
GMA-Latex-Anionenaustauscher: Beispielchromatogramm 1
- 75 -
n-
Tab. 11) 1 mL min
NH NO
rob schl
r2O 2−
O −
Tab. 12: Elutionsreihenfolge bei Trennung an einem GMA-Latex-Anionenaustauscher (DEMA) Chromatographische Parameter für die Säule M211103 DEMA, Messbedingungen s. Abb. 32.
Peak-Nr. Analyt tR / min k’ N / TP m−1
Abb. 32: Trennung von neun Aionen an einem GMA-Latex-Anionenaustauscher mit DEMA als funktioneller Gruppe. Alle Peaks im Chromatogramm haben ein deutliches Tailing. Die Effizienz dieser Säule ist wesent-lich schlechter als die der Säule in Abb. 30. Cr(VI) und ClO4
− werden mit dem 10 mmol L−1-Eluenten nicht innerhalb der 40 min von der Säule eluiert. Dafür sind der Totvolumenpeak, IO3
−, H2PO4− und H2AsO4
− gut separiert.
10 20 30 400
2k
4k
6k
8k
10 20 30 400
50k
100k
0 50
20k
40k
60k
Messbedingungen Säule: M211103DEMA (Eluent: −1
10 mmol L−1 4 3 pH 4.3 +15 µg L Ge P en eife: 153 µL
−1
Analyten / µg L−1: 10 IO3
−, I−, C 720 BrO3
−, Br−, H2As 450 DBA−, TBA−
200 H2PO4−, Cl−
1000 ClO3−
5000 ClO4−
20000 SCN−
– H2O 1.06 – – 1 IO3
− 1.80 0.98 3900 2 BrO3
− 2.75 2.22 9700 3a H2PO4
− 3.08 2.65 3800 3b Cl− 3.08 2.65 13600 5 H2AsO4
− 3.45 3.14 4300 6 ClO3
− 5.02 5.19 16500 7 Br− + DBA− 5.36 5.64 (3300) 8a TBA− 16.7 20.5 1500 8b I− 17.1 21.1 20100 10 SCN− 27.9 35.2 5000 – Cr(VI) >40 – – – ClO4
− >40 – – – Mittelwert – – 8200
100
50k
100k
74GeI−
TBA−
Zähl
rate
/ c
ps
Zeit / min
SCN−
74G
e Zä
hlra
te /
cps
Zeit / min
74Ge
Cl−Zähl
rate
/ c
ps
Zeit / min
ClO−
3
H2PO−
4
H2AsO−
4
74 G
e-Zä
hlra
te /
cps
IO−
3
Br−+DBA−
BrO−
3

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 76 -
Im Be t die
starke Asymmetrie aller Peaks auf. Auch früh eluierende Analyten zeigen ein Tailing. Die
Trennleistung m TP −1 (Cl t (Tab. 11) und
ist damit schlechter als fü texsäulen üblich. Die negativen Eigenschaften sind aber nicht
auf den Materialtyp zurückzuführen, denn sie wurden nur speziell bei dieser Säule beobachtet.
So könnten beispielsweise Aufreinigungs- oder Packp e für die schlechte Qualität
verantwortlich in. Mit ule la sich ke ptimale bnisse bei einer
quantitativen Analyse erhalten, aber es werden einige ssante E chaften bezüglich
Selektivität und Retentionsreihenfolge deutlich.
Bei Verwendun eines 1 −1 Ammoniumnitrat-Eluenten gelingt es zwar, Iodat vom
jektionspeak zu trennen (R = 2.1). Dies liegt aber vor allem an der niedrigen Eluentkonzen-
Retentionszeiten von über drei Minuten.
zusammen mit dem Iodid und erst nach
28 min das Thiocyanat im Chromatogramm zu sehen. Chromat und Perchlorat können inner-
it
über 2000 cps ungewöhnlich hoch ist. Das Untergrundsignal auf dem Masse-zu-Ladungs-
Verhältnis 79 lag bei Messungen mit anderen Säulen zu dieser Zeit bei 300–800 cps. Es
ispielchromatogramm in Abb. 32 für die Säule M211103 DEMA fällt zunächs
it Carbonat-Eluent wurde zu nur 4670 m −) bestimm
r La
roblem
se dieser Sä ssen ine o n Erge
intere igens
g 0 mmol L
In
tration, mit der einige polarisierbare Anionen schon nicht mehr innerhalb von 40 min eluiert
werden können. Mit stärkerem Eluenten reicht die Iodatselektivität des Methacrylatlatices
nicht aus, um es vom Injektionspeak aufzulösen.
Durch die Verwendung von Latexpartikeln auf Methacrylatbasis wird eine deutlich stärkere
Retention der Ionen Phosphat und Arsenat erlangt. Im Gegensatz zu dem Beispiel mit
VBC-Latex und DEMA-Funktionalisierung (Abb. 29), bei dem Phosphat und Arsenat direkt
nach dem Injektionspeak eluieren, verlassen diese Ionen bei Trennung auf dem hydrophileren
Methacrylatlatex die Säule erst nach Bromat mit
Ein Nachteil in diesem Trennsystem ist, dass sowohl Bromat und MBA− als auch Bromid und
DBA− exakt gleichzeitig eluieren. Die Koelution des Bromids und des Dibromacetats ist an
der großen Fläche des entsprechenden Peaks in Abb. 32 zu erkennen.
Nach einer Retentionszeit von 17 min ist das TBA−
halb von 40 min gar nicht von der Säule eluiert werden. Die Retentionszeiten sind zu lang für
eine schnelle Multianionenanalyse und die Peakformen, insbesondere die vom Thiocyanat,
sind zu schlecht. Der Thiocyanatpeak in Abb. 32 zeigt starkes Tailing.
Bei der Skalierung von 0–8000 cps in Abb. 32 ist erkennbar, dass der Bromuntergrund m

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 77 -
g gemacht werden, aber
es ist wichtig, solche Unterschiede des Bromuntergrunds durch bestimmte verwendete Säulen
scheinen bromhaltige Substanzen von der Säule gespült zu werden, obwohl der GMA-Latex-
Ionenaustauscher ohne bromhaltige Reagenzien hergestellt wurde. Die Identität und Ursache
der möglichen Bromkontamination können zwar bisher nicht ausfindi
zu kontrollieren und auszuschalten, um optimale Nachweisgrenzen für die Bromspezies zu
gewährleisten.
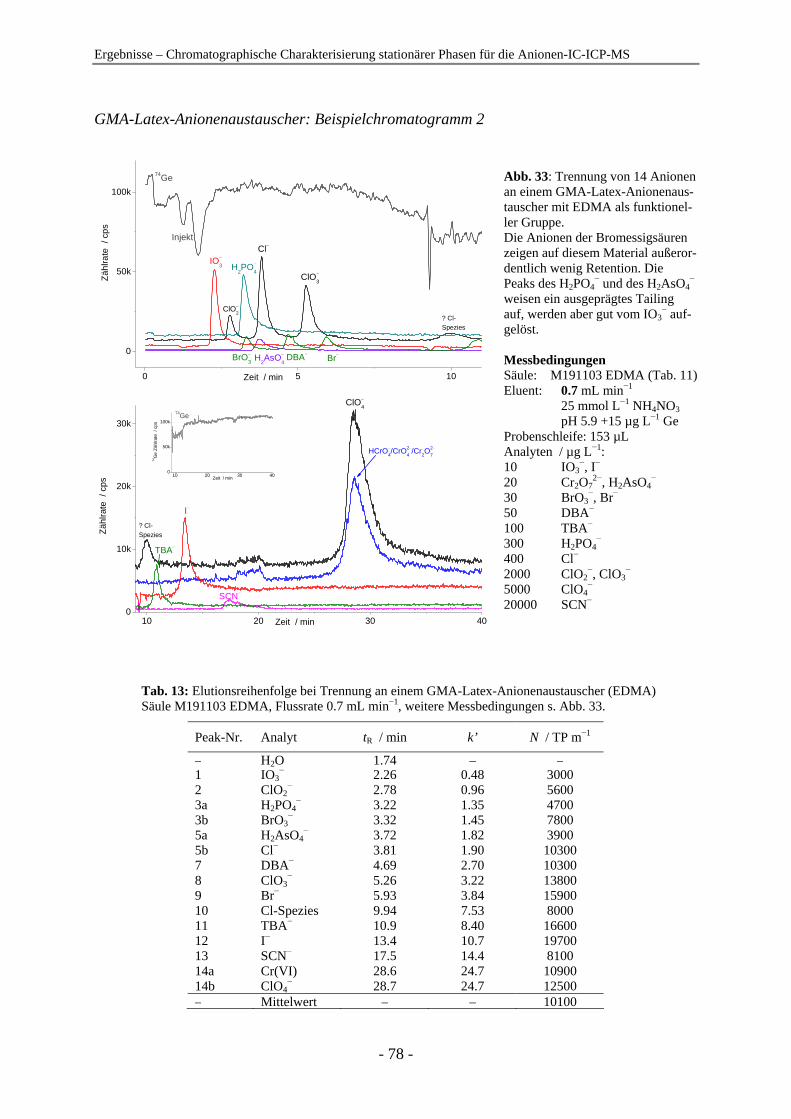
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
GMA-Latex-Anionenaustauscher: Beispielchromatogramm 2
- 78 -
-
Tab. 11) 0.7 mL min
O
robe schl
AsO −
−
, ClO3−
Tab. 13: Elutionsreihenfolge bei Trennung an einem GMA-Latex-Anionenaustauscher (EDMA) Säule M191103 EDMA, Flussrate 0.7 mL min−1, weitere Messbedingungen s. Abb. 33.
Peak-Nr. Analyt tR / min k’ N / TP m−1
Abb. 33: Trennung von 14 Anionen an einem GMA-Latex-Anionenaus-tauscher mit EDMA als funktionel-ler Gruppe.
10 20 30 400
10k
20k
30k
10 20 30 400
50k
100k
0 5
0
50k
100k
Die Anionen der Bromessigsäuren zeigen auf diesem Material außerordentlich wenig Retention. Die Peaks des H2PO4
− und des H2AsO4−
weisen ein ausgeprägtes Tailing auf, werden aber gut vom IO3
− auf-gelöst. Messbedingungen Säule: M191103 EDMA (Eluent: −1
25 mmol L−1 NH4N 3 pH 5.9 +15 µg L−1 Ge P n eife: 153 µL Analyten / µg L−1: 10 IO3
−, I−20 Cr2O7
2−, H2 430 BrO3
−, Br−50 DBA−
100 TBA−
300 H2PO4400 Cl−2000 ClO25000 ClO
−
4−
20000 SCN−
– H2O 1.74 – – 1 IO3
− 2.26 0.48 3000 2 ClO2
− 2.78 0.96 5600 3a H2PO4
− 3.22 1.35 4700 3b BrO3
− 3.32 1.45 7800 5a H2AsO4
− 3.72 1.82 3900 5b Cl− 3.81 1.90 10300 7 DBA− 4.69 2.70 10300 8 ClO3
− 5.26 3.22 13800 9 Br− 5.93 3.84 15900 10 Cl-Spezies 9.94 7.53 8000 11 TBA− 10.9 8.40 16600 12 I− 13.4 10.7 19700 13 SCN− 17.5 14.4 8100 14a Cr(VI) 28.6 24.7 10900 14b ClO4
− 28.7 24.7 12500 – Mittelwert – – 10100
10
? Cl-Spezies
HCrO−
4/CrO2−4 /Cr2O
2−7
74Ge
I−
TBA−
Zähl
rate
/ c
ps
Zeit / min
SCN−
74G
e Zä
hlra
te /
cps
Zeit / min
? Cl-Spezies
ClO−
2
Cl−
74Ge
Injekt
ClO−
3Zähl
rate
/ c
ps
Zeit / min
ClO−
4
H2PO−
4
H2AsO−
4
IO−
3
DBA− Br−BrO−
3

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 79 -
Auch d zeigt
eine hohe Selektivität für Phosphat und Arsenat. Für die Trennung in Abb. 33 wurde die
Eluentkonzentration so hoch gewählt, dass das Perchlorat und das (Di-)Chromat innerhalb
von 35 min eluiert werden. Trotz des starken Eluenten weisen die Peaks des Phosphats und
des Arsenats ei deutlic nung vom ektionspeak auf. Dies trifft aber nicht für das
Iodat zu, welch auch h e bei stat Phase –, kurz nach
dem Injektion k elu ird. Ein Merkmal der pellikularen naustauscher mit
Methacrylatlatex, sowohl 32, als auch in Abb.
lösung des Brom vom I
Die Retention der Anionen der Bromessigsäuren ist rig im V ich zu Anionen-
austauschern m atexp s VBC/DVB, bei denen z.B. das das DBA− nach dem
Bromid eluiert (s. Kap. 4.1.3.5 u. Kap. 4.1.3.6). Noch viel ausgeprägter ist der Unterschied zu
oberflächenfunktionalisierten und gepfropften Materialien (Kap. 4.1.5 bzw. Kap. 4.1.6.2), auf
denen die Anionen der Bromessigsäuren eine extrem starke Retention erfahren. Im ersten
Beispiel (DEMA-funktionalisierter Methacrylatlatex) eluieren DBA− und Bromid gleichzeitig
(Abb. 32). Durch die unpolare EDMA-Austauschergruppe am Methacrylatlatex im zweiten
Beispiel wird die Retention von DBA− weiter erniedrigt, so dass es zwischen Bromat und
Bromid eluiert (Abb. 33).
Alle getesteten pellikularen Anionenaustauscher mit Latex auf Methacrylatbasis erzeugten
einen starken Staudruck. Trotz der verringerten Flussrate von 0.7 mL min−1 für die Säule
M191103 EDMA, betrug der Betriebsdruck immer noch 193 bar. Wenn man das Monitor-
signal des internen Standards 74Germanium in Abb. 33 betrachtet, fallen ein Doppelpeak
durch die Injektion und starke Intensitätsveränderungen auf. Ursache für die Artefakte im
Monitorsignal können Packungsprobleme des Säulenmaterials oder Pumpenschwankungen
durch den hohen Staudruck sein.
Ein weiterer Nachteil dieser Säule ist, dass viele Peaks, insbesondere der des Thiocyanats aber
auch die nicht polarisierbarer Ionen wie Phosphat und Arsenat, ein Tailing aufweisen. Die
mittlere Trennleistung aller Anionen für die Säule M191103 EDMA mit Nitrat als Eluention
beträgt 10100 TP m−1.
er Methacrylatlatex-Anionenaustauscher mit EDMA als funktioneller Gruppe
ne he Tren Inj
es ier, – wi ionären n mit VBC/DVB-Latex
spea iert w Ione
in Abb. 33 ist die außerordentlich hohe Auf-
ats odat (R = 3).
nied ergle
it L artikeln au

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
4.1.5 Oberflächenfunktionalisierte Anionenaustauscher
Die untersuchte Säule P130198 MN IV enthält das im Rahmen der Dissertation von
Köhler [133] hergestellte PS/DVB-Copolymer P130198, welches durch Nowak [130] mit
Dimethylethanolamin oberflächenfunktionalisiert wurde. Die Funktionalisierung erfolgte
durch Alkylierung am Aromaten mit 6-Brom-1-Hexen, woraus sich ein C-5-Spacer ergibt,
und anschließenden Austausch des Broms gegen eine quartäre Ammoniumgruppe. Abb. 34
illustriert den schematischen Aufbau dieser Art von oberflächenfunktionalisierten Anionen-
austauschern.
- 80 -
NR3+
NR3+
NR3+
NR3+
NR3+
NR3+
NR3+
NPS/DVB
SpacerSubstrat Austauscher-Funktion
OH
Abb. 34: Schematischer Aufbau des verwendeten oberflächenfunktionalisierten Anionenaustauschers
t wie die in dieser Arbeit verwendeten Latexsäulen. Das
ößere Probenvolumina bzw. Proben mit hohem
robenvolumina bei geringer Verdünnung der Proben können gute
Nachweisgrenzen erzielt werden.
(funktionalisiert mit DMEA)
Komplett oberflächenaminierte Materialien zeichnen sich im Allgemeinen durch eine
wesentlich höhere Kapazität als die der Latex-Ionenaustauscher aus. Sie gehören zur Gruppe
der hochkapazitiven Ionenaustauscher, in die man Materialien mit einer Kapazität von mehr
als 200 µeq Säule−1 einordnet [130]. Die Säule P130198 MN IV hat mit 475 µeq Säule−1 eine
etwa zehnmal so hohe Kapazitä
bedeutet, dass auf diese Trennsäule gr
Matrixgehalt aufgegeben werden können, ohne dass Überladungseffekte auftreten. Aus
diesem Grund wurde die Säule unter anderem für die Spurenanalyse von Bromat in
hochkonzentrierten, bis zu 1%igen Salzlösungen verwendet (s. Kap. 4.4.1). Durch die
Injektion von hohen P

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 81 -
Die Trennleistung der direkt oberflächenfunktionalisierten Materialien ist aber deutlich
en Phasen. Durch die breiteren
nur einem Drittel der Höhe erhalten werden, weswegen sie bei
iedrigen Konzentrationen schlechter vom Untergrund unterscheidbar werden.
Tab. 14: Kenndaten der oberflächenfunktionalisierten Säule P130198 MN IV
Typ Oberflächenfunktionalisiert PS/DVB-Trägermaterial
niedriger als die der pellikularen oder gepfropften stationär
Peaks, die so genannte „chromatographische Verdünnung“ kommt es wieder zu Einbußen bei
den Nachweisgrenzen. Die ermittelte Anzahl theoretischer Böden bei der Säule
P130198 MN IV beträgt für Bromat und Bromid im Ammoniumnitrat-Elutionssystem nur
ca. 6000 TP m−1 (Tab. 15). Bei Verwendung guter Latexsäulen (s. Tab. 10) können mehr als
dreimal so hohe Bodenzahlen erreicht werden. Das bedeutet, dass durch die Trennung auf
oberflächenfunktionalisierten Säulen im Vergleich zur Trennung auf Latexsäulen, Peaks mit
dreifacher Breite aber
n
Hersteller M. Nowak [130] Bezeichnung P130198 MN IV Funkt. Gruppe DMEA Kapazität a / µeq Säule−1 475 Trennleistung a, b, c / TP m−1 ~15000 Staudruck c / bar 40 Abm gen / mm 125 × 4 essun
a: bezogen auf Cl− b: Elutionssystem CO32−/HCO3
− c: Fließgeschwindigkeit 1 mL min−1
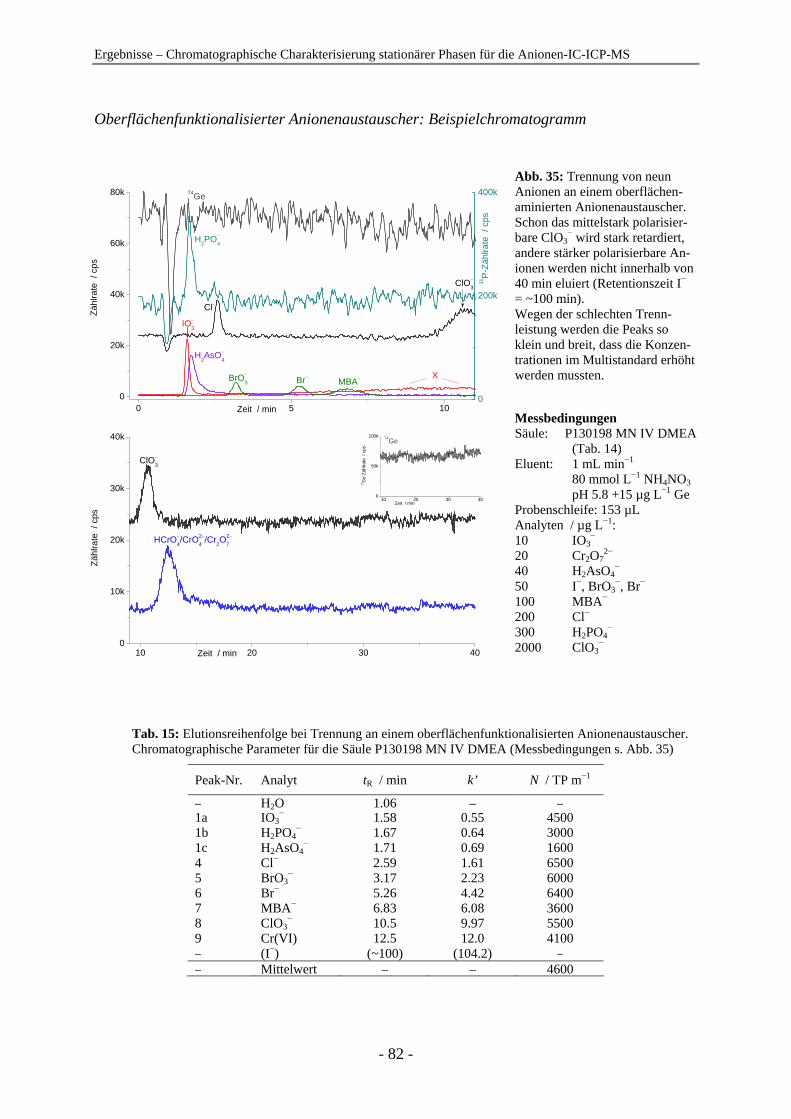
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 82 -
die Konzen-dard erhöht
werden mussten.
ingungen P130198 MN IV DMEA
(Tab. 14) : 1 mL min−1
80 mmol L−1 NH4NO3 pH 5.8 +15 µg L−1 Ge Probenschleife: 153 µL Analyten / µg L−1: 10 IO3
−
20 Cr2O72−
40 H AsO4−
O3−, Br−
100 MBA−
200 Cl−
300 H2PO4−
2000 ClO3−
Tab. 15: Elutionsreihenfolge bei Trennung an einem oberflächenfunktionalisierten Anionenaustauscher. Chromatographische Parameter für die Säule P130198 MN IV DMEA (Messbedingungen s. Abb. 35)
Peak-Nr. Analyt tR / min k’ N / TP m−1
Oberflächenfunktionalisierter Anionenaustauscher: Beispielchromatogramm
Abb. 35: Trennung von neun Anionen an einem oberflächen-aminierten Anionenaustauscher. Schon das mittelstark polarisier-bare ClO3
− wird stark retardiert, andere stärker polarisierbare An-ionen werden nicht innerhalb von 40 min eluiert (Retentionszeit I− = ~100 min). Wegen der schlechten Trenn-leistung werden die Peaks so klein und breit, dasstrationen im Multistan
MessbedSäule: Eluent
250 I−, Br
– H2O 1.06 – – 1a IO3
− 1.58 0.55 4500 1b H2PO4
− 1.67 0.64 3000 1c H2AsO4
− 1.71 0.69 1600 4 Cl− 2.59 1.61 6500 5 BrO3
− 3.17 2.23 6000 6 Br− 5.26 4.42 6400 7 MBA− 6.83 6.08 3600 8 ClO3
− 10.5 9.97 5500 9 Cr(VI) 12.5 12.0 4100 – (I−) (~100) (104.2) – – Mittelwert – – 4600
0 5 100
20k
40k
60k
80k
10 20 30 400
20k
30k
40k
10k
10 20 300
50k
100k
40
0
200k
400k
XBrO−
HCrO−
4/CrO
Zähl
rate
/ c
ps
Zeit / min
−−
ClO−
3
74Ge
Cl−
IO−
3
H2PO−
4
H2AsO−
4
3 MBABr
2−4 /Cr2O
2−7
ClO−
3
?
Zähl
rate
/ c
ps
Zeit / min
74Ge
74G
e Zä
hlra
te /
cps
Zeit / min
31P
-Zäh
lrate
/ c
ps

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 83 -
Die hohe Kapazität der oberflächenfunktionalisierten Materialien bringt nicht nur Vorteile.
Beispiel von
bb. 35 schon bei 80 mmol L−1 Ammoniumnitrat. Die maximal dauerhaft einsetzbare Eluent-
, denn darüber treten Löslich-
führen können. Typischerweise erscheinen in
chen Aussalzungen an der heißen Seite im
a
gen. Außerdem kommt es bei zu hohen Ammoniumnitratkonzentrationen ebenfalls leichter zu
Verstopfungen im Zerstäuber.
Eine weitere Auffälligkeit bei der Verwendung eines stärker konz
Eluenten ist der hohe Untergrund auf dem Masse-zu-Ladungs-Verhältnis 31. In Abb. 35
musste für die Darstellung des Phosphatsignals eine zusätzliche Ordinate verwendet werden,
da das Blindwertsignal mit etwa 200k cps fünf- bis zehnmal höher ist als bei niedriger
konzentrierten Eluenten. Durch die hohe Nitratkonzentration wird die Bi
ions [14N16OH]+, welches die Hauptinterferenz auf der Mass s .
Hochkapazitive Säulenmaterialien sind demnach auf Grund der zur Elution nötigen hohen
Nitratkonzentrationen für die Detektion von Phosphoranalyten von Nachteil. Eine alternative
Verstärkung von Nitrat-Eluenten ist auch durch Zugabe von 0.1–1 rchlorat
möglich, was aber zu weiteren Interferenzen durch chlorhaltige Molekülionen führt.
Die oberflächenfunktionalisierten Anionenaustauscher zeigen eine besonders ausgeprägte
ffinität zu polarisierbaren Anionen. Dies ist in Abb. 35 daran zu erkennen, dass das leicht
polarisierbare Anion Chlorat wesentlich später eluiert wird als das Bromid. Die Retentions-
reihen
eluiert das Chlorat noch vor dem Bromid. Die stark polarisierbaren Anionen wie Iodid,
Thiocyanat un erchlor nnen unter Bedingun en von Ab 35 gar nicht mehr
innerhalb von in vo onalisierten Anionenaustauscher verdrängt
werden 7.
Die starke Retention der polarisierbaren Anionen lässt sich damit begründen, dass diese Ionen
nicht nur an r Oberf des Ionen auschers trostatische Wechselwirkungen
eingehen, sond n vielm ief in d germa eindring s. Kap. 2.4.3 und
Die Eluentkonzentration muss der Kapazität angepasst werden und liegt im
A
konzentration für Ammoniumnitrat beträgt etwa 120 mmol L−1
keitsprobleme auf, die zu Störungen im ICP-MS
einem solchen Fall als erste sichtbare Anzei
Probeninjektionsrohr der Torch. Diese führen zu Plasma- und d mit auch Signalveränderun-
entrierten Ammoniumnitrat-
ldung des Molekül-
ün tigte 31 u darstellt, beg
mmol L−1 Pe
A
folge ist bei einer entsprechenden Trennung auf Latex-Austauschern umgekehrt. Dort
d P at kö den g b.
40 m n dem oberflächenfunkti
de läche aust elek
er ehr t as Trä terial e (n
7 Die mit X gekennzeichnete Untergrundanhebung in der Iodspur, ist das Iodidsignal einer vorherigen Messung, woraus sich für Iodid eine Retentionszeit von mehr als 100 min ableiten lässt.

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 84 -
enfalls nicht vom Untergrund zu unterscheiden sind. Bei ober-
flächenfunktionalisierten Anionenaustauschern kommt es zudem oft zu Memoryproblemen,
ist es
unmöglich, durch eine isokratische Trennung eine Auflösung der nicht polarisierbaren Ionen
Kap. 4.1.3.3). Bei oberflächenaminierten Ionenaustauschern gibt es keine negativ geladene
Sperrschicht aus Sulfonatgruppen, die durch Donnan-Ausschlusskräfte das Eindringen der
Anionen in das Trägermaterial verringert, wie es bei Latex-Ionenaustauschern der Fall ist.
Polarisierbare Anionen sind nur schwach hydratisiert, haben deswegen einen gewissen
organischen Charakter und können tief in die Poren des lipophilen Trägerpolymers hinein und
nur langsam wieder heraus diffundieren. Als sekundäre Wechselwirkungen sind bei den
polarisierbaren Ionen insbesondere π-π-Wechselwirkungen mit den aromatischen Systemen
des PS/DVB-Materials von Bedeutung [13, 134]. Aus dem schlechten Massentransfer resul-
tieren breite Peaks, die gegeben
wenn polarisierbare Anionen aufgegeben werden. Es dauert lange, Ionen mit starker
Retention, z.B. das Perchlorat, wieder zu eluieren.
Iodat, Phosphat und Arsenat werden auf dem oberflächenfunktionalisierten Anionen-
austauscher unter den Bedingungen wie in Abb. 35 kaum retardiert und sind nur schlecht vom
Injektionspeak aufgelöst. Die mittlere Trennleistung der Säule P130198 MN IV DMEA liegt
bei nur 4600 TP m−1, was nur einem Viertel des Werts für den besten pellikularen Anionen-
austauscher entspricht.
Mit dem oberflächenfunktionalisierten Anionenaustauscher P130198 MN IV DMEA
zu erreichen und unter den gleichen Bedingungen die polarisierbaren innerhalb von 45 min zu
eluieren. Diese Art der stationären Phasen ist daher nicht für eine schnelle Multianionen-
analyse geeignet. Der einzige Vorteil ist die hohe Kapazität, die aber nur benötigt wird, wenn
konzentrierte Lösungen oder hohe Probenvolumina gemessen werden sollen.

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 85 -
ypus von stationären Phasen, bei denen
tung wird beispielsweise über adsorptive Wechselwirkungen fixiert
oder indem Monomere um ein fertiges Teilchen herum polymerisiert werden.
PS/DVB-Trägermaterial
4.1.6 Gepfropfte Anionenaustauscher
4.1.6.1 Allgemeine Definition und Beschreibung
Man spricht von Pfropf(co)polymeren, wenn an vorgefertigte Polymere zu einem späteren
Zeitpunkt weitere Monomere oder Polymere kovalent gebunden werden. Der Begriff
Aufpfropfen (engl.: grafting) ist der Terminologie des Gartenbaus entlehnt, wo er eine häufig
verwendete Praxis zur Veredelung von Obstbäumen bezeichnet. Für Ionenaustauscher werden
meist sphärische Partikel im 3–10 µm-Bereich als Pfropf-Substrat verwendet. Die ange-
koppelten Teilchen werden als Pfropf-Äste oder -Zweige bezeichnet und an sie sind die
positiv geladenen funktionellen Gruppen für den Anionenaustausch gebunden.
In der Chromatographie gibt es einen weiteren T
ebenfalls ein vorgefertigtes Polymer mit einem anderen kombiniert wird, die so genannten
beschichteten Trägermaterialien (engl.: coated polymers). Im Unterschied zu Pfropfcopoly-
meren sind dies Polymermischungen, weil die einzelnen Polymere nicht über kovalente
Bindungen verfügen, sondern über andere Mechanismen zusammen gehalten werden. Eine
solche Polymerbeschich
Die im Folgenden beschriebenen gepfropften Anionenaustauscher basieren auf sphärischen
PS/DVB Trägermaterialien. An diese wurde ein bereits aminiertes Monomer aufgepfropft. In
Tab. 16 sind die Kenndaten der verwendeten Säulen aufgeführt.
Tab. 16: Kenndaten der Säulen, die über Pfropfpolymerisation hergestellt worden
Typ Aufgepfropft
Hersteller M. Raskop [124]
Bezeichnung DR48VBC DEMA 4 EVO3
P3162120 EVO3
Funkt. Gruppe DEMA DEMA Kapazität a / µeq Säule−1 ~50 ~80 Trennleistung a, b, c / TP m−1 ~30000 46000 Staudruck c / bar 156 61 Abmessungen / mm 150 × 3 (Stahl) 100 × 4 (PEEK)
a: bezogen auf Cl− b: Elutionssystem CO32−/HCO3
− c: Fließgeschwindigkeit 1 mL min−1
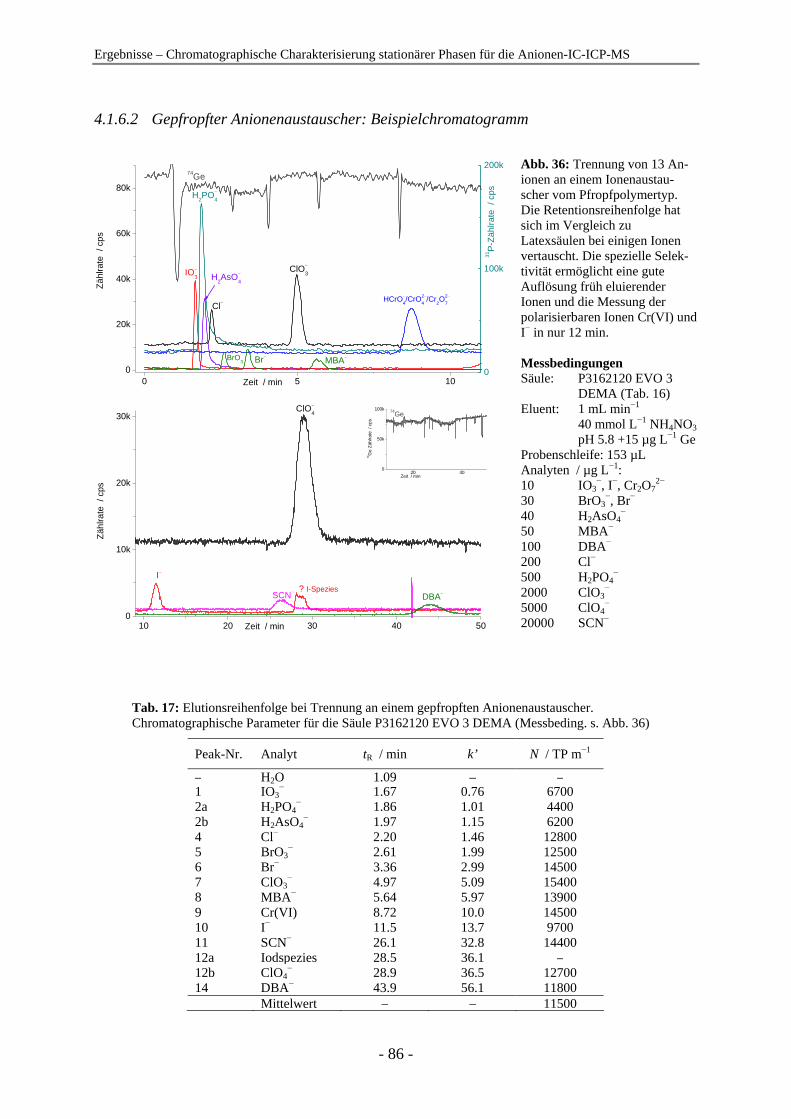
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 86 -
Beispielchromatogramm
Abb. 36: Trennung von 13 An-ionen an einem Ionenaustau-
gen 20 EVO 3
DEMA (Tab. 16)
4−
2000 ClO3−
−
. 36)
tR / min TP m−1
4.1.6.2 Gepfropfter Anionenaustauscher:
scher vom Pfropfpolymertyp. Die Retentionsreihenfolge hat sich im Vergleich zu Latexsäulen bei einigen Ionen vertauscht. Die spezielle Selek-tivität ermöglicht eine gute Auflösung früh eluierender Ionen und die Messung der polarisierbaren Ionen Cr(VI) und I− in nur 12 min. MessbedingunSäule: P31621
Eluent: 1 mL min−1 40 mmol L−1 NH4NO3 pH 5.8 +15 µg L−1 Ge Probenschleife: 153 µL Analyten / µg L−1: 10 IO3
−, I−, Cr2O72−
30 BrO3−, Br−
40 H AsO2 4−
50 MBA−
100 DBA−
200 Cl−
500 H2PO
5000 ClO420000 SCN−
Tab. 17: Elutionsreihenfolge bei Trennung an einem gepfropften Anionenaustauscher. Chromatographische Parameter für die Säule P3162120 EVO 3 DEMA (Messbeding. s. Abb
Peak-Nr. Analyt k’ N /
– H2O 1.09 – – 1 IO3
−
2PO4− 1.86 1.01
sO4− 1.97 1.15
2.20 1.46 00 2.61 1.99
1.67 0.76 6700 2a H 44002b H2A 62004 Cl− 128
1255 Br −O3 00 6 Br− 3.36 2.99 0
4.97 5.09 00 5. 97
I
11 SCN− 26.1 32.8 14400 12a Iods
14507 ClO3
− 1548 MBA− 64 5. 13900 9 Cr(V ) 8.72 10.0 14500 10 I− 11.5 13.7 9700
pezies 28.5 36.1 – 12b ClO4
− 28.9 36.5 12700 14 DBA− 43.9 56.1 11800 Mittelwert – – 11500
0 5 10
80k
0
20k
40k
60k
10 20 30 40 500
10k
20k
30k
20 400
50k
100k
0
100k
200k
BrO−
3
HCrO−
4/CrO2−4 /Cr2O
2−7Cl−
Zähl
rate
/ c
ps
MBA−Br−
ClO−
3IO−
3
H2PO−
4
H2AsO−
4
74Ge
Zeit / min
I−
ClO−
4
Zähl
rate
/ c
ps
? I-Spezies
74Ge
74G
e Zä
hlra
te /
cps
Zeit / min
SCN−?
DBA−
Zeit / min
31P
-Zäh
lrate
/ c
ps

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 87 -
Die Auswertung des Chromatogramms für ein gepfropftes Anionenaustauschmaterial in
atogrammen für Latex-
u ,
wodurch sich die Retentionsreihenfolge einiger Ionen verändert. tz
zur Trennung an Latex-Ionenaustauschern Chlorid vor Bromat t
eluiert. Die organischen Anionen MBA− und DBA− erfahren so starke Retention, dass in
Abb. 36 das MBA− erst nach Bromid eluiert und dass das DB e
Retentionszeit aller injizierten Ionen besitzt.
Der größte Vorteil der verwendeten Säule P3162120 EVO3 liegt darin, dass einerseits die
früh eluierenden Ionen gut voneinander aufgelöst werden, und andererseits die polarisierbaren
Anionen unter den gleichen Bedingungen kurze Retentionszeiten
schließlich mit diesem gepfropften Anionenaustauschmaterial, ein frühe Elution des Iodids zu
erreichen und unter Verwendung des gleichen Eluenten, das oda ak
aufzulösen (R = 2.3). Auch die Auflösung von Iodat und Brom eutlich
größer als bei Latex-Anionenaustauschern. Die Peaks von Broma r urch
die verstärke Retention des Bromats näher zusammen als bei pellikularen Ionenaustauschern,
sind aber mit R = 2.6 immer noch mehr als ausreichend aufgel es ren ist eine
außergewöhnlich gute Abtrenn den anderen aufgegebene sp egeben. So
tritt keine Koelution von Anionen der Bromessigsäuren mit Brom
bei vielen Latex-Ionenaustauschern der Fall ist. Diese sichere U des Bromats
on anderen Bromspezies ist vorteilhaft für eine der wichtigsten Anwendungen: die Kontrolle
bromhaltiger Nebenprodukte bei der Desinfektion von Wasser.
Die Trenneffizienz der Säule P3162120 mit Nitrat-Eluent ist mit einem über alle Ionen
gemittelten Wert von 11500 TP m−1 sehr hoch und vergleichbar mit der Effizienz von Latex-
Anionenaustauschern in Kap. 4.1.3. Eine Besonderheit ist, dass die Trennleistung des
gepfropften Materials für alle Analyten, mit der Ausnahme von Iodat, Phosphat und Arsenat,
etwa gleich gut ist (nur 13 % relative Standardabweichung). Iodat, Phosphat und Arsenat sind
die einzigen Anionen mit unsymmetrischen Peakformen. Die Peaks dieser drei früh
eluierenden Analyten zeigen ein Tailing, wodurch sich auch die geringere Bodenzahl ergibt.
Das für früh eluierende Ionen ungewöhnliche Peaktailing wurde auch bei einer weiteren Säule
mit dem gleichen gepfropften Trennmaterial beobachtet (Säule DR48VBC DEMA 4,
s. Tab. 16).
Abb. 36 ergibt gravierende Unterschiede im Vergleich mit den Chrom
tlich andere Selektivitäten
So wird z.B. im Gegensa
und Bromid vor Chlora
A− mit 44 min die längst
Ionenaustauscher. Der gepfropfte Anionenaustauscher zeigt de
besitzen. So gelingt es aus-
I t vom Injektionspe
at ist mit R = 4.0 d
t und B omid liegen d
öst. D Weite
ung von n Brom ezies g
at oder Bromid auf, wie es
nterscheidung
v

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 88 -
4.1.6.
Die Ursachen der unterschiedlichen Selektivität gepfropfter Anionenaustauscher im Vergleich
zu Latex-Anion austaus sollen aufg
dem gepfropftem Ionenaustauscher P3162120 EVO3 bb. 36) werden die folgenden
Beobachtungen gemacht:
1. Die Retention der Anionen Chlorid, Bromid, (D romat, Iodid und Perchlorat ist
relativ g ng.
2. Die Re ion de Iodat, B t, Chlorat, Thiocyanat und der Anionen der
Bromessigsäuren ist relativ hoch.
3. Insbesondere die Anionen, die Kohlenstoffatome enthalten, d.h. Thiocyanat sowie die
Anionen der Brom n, werde besonders s rk retardi
4. Die Peaks einiger früh eluierender Ionen zeigen ein Tailing. Dies ist für Iodat und in
stärkerem Maße für Dihydrogenphosphat und -arsenat zu beobachten.
Während einige polarisierbare Anionen auf dem gepfropften Anionenaustauscher eine relativ
kurze Retentionszeit aufweisen, erfahren andere, wie das Thiocyanat und das Chlorat und
besonders die organischen Anionen, eine längere Retention als auf pellikularen Materialien.
Deswegen kann nicht alleinig damit argumentiert werden, dass polarisierbare Ionen oder
allgemein Ionen mit organischem Charakter auf dem gepfropften Ionenaustauscher weniger
sekundäre Wechselwirkungen eingehen. Es muss noch andere Wechselwirkungen geben, die
die besondere Selektivität des Materials verursachen. Es ist anzunehmen, dass diese Wechsel-
wirkungen von der Struktur der Anionen bzw. von der Ladungsverteilung abhängig sind, denn
die in Beobachtung 1 aufgeführten Ionen Chlorid, Bromid, Chromat, Iodid und Perchlorat
besitzen alle eine nahezu kugelsymmetrische Ladungsverteilung, während die Ladung der
unter Beobachtung 2 genannten Ionen Iodat, Bromat, Chlorat, Thiocyanat, MBA− und DBA−
nicht kugelsymmetrisch verteilt ist.
Im Folgenden wird ein Modell zur Erklärung der Selektivität aufgestellt, das die drei-
dimensionale Ladungsverteilung der Analyten mit einbezieht. Dazu werden die nötigen
Konzepte der „effektiven Ladung“ und der „lokalen Kapazität“ vorgestellt. Des Weiteren
wird die Auswirkung von Doppelbindungen mit Kohlenstoff bei den Analytionen berück-
sichtigt.
3 Ursachen spezieller Selektivität
en chern eklärt werden. Bei einer Multianionentrennung an
(s. A
i-)Ch
eri
tent r Ionen roma
essigsäure n ta ert.

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
Die Ladungsverteilung wirkt sich zusammen mit weiteren Faktoren wie z.B. der Hydrati-
sierung auf die effektive Ladung der Ionen aus. Als effektive Ladung wird bei der Ver-
wendung von monoanionischen Eluenten das Verhältnis der Ladung von Analytanion und
Eluentanion bezeichnet. Der Wert lässt sich mit Hilfe von Retentionsmodellen für die
Anionenchromatographie bestimmen [58, 130, 135]. Einen quantitativen Zusammenhang zwi-
schen dem Retentionsfaktor k’ und diversen experimentell zugänglichen Parametern, wie der
Konzentration des Eluenten und der Austauschkapazität stellt die folgende Gleichung her:
- 89 -
Gl. 19 ( )−−++= y c yyyy MEEA,A
x Q x K k' loglogloglog1log Φ
M
SmitVV
Φ =
k’: Retentionsfaktor A: Analytanion x: Ladung des Analytanions
E: Eluentanion y: Ladung des Eluentanions Q: Kapazität c: Konzentration
KA,E: Gleichgewichtskonstante des Ionenaustauschs zwischen stationärer und mobiler Phase
Φ: Phasenvolumenverhältnis V: Volumen Index S: stationäre Phase Index M: mobile Phase
Wenn alle Bedingungen außer der Kapazität konstant gehalten und zu C zusammengefasst
werden, vereinfacht sich die Formel zu:
Gl. 20 C yQ
yx k' += loglog A
Die graphische Auftragung von log k’A gegen log Q/y ergibt eine Gerade, deren Steigung der
effektiven Ladung entspricht (s.a. Abb. 37).
Zum Verständnis der Unterschiede der stationären Phasen muss die Kapazität differenziert
betrachtet werden. Die Säulenkapazität ist ein Maß für die Anzahl an Austauschergruppen in
einer Säule (s. Kap. 2.2.5). Aus makroskopischer Sicht sind die Austauscherfunktionen durch
die gleichmäßige Packung monodisperser Trägerpartikel statistisch über das Säulenvolumen
verteilt. Betrachtet man aber ein einzelnes sphärisches Trägerpartikel auf molekularer Ebene,
so ist die Verteilung der für Analyten zugänglichen Austauschergruppen nicht gleichmäßig,
da sich die Austauscherplätze ausschließlich auf der „Kugeloberfläche“ bzw. auf der
Oberfläche von Makroporen befinden. Der Verteilungsgrad der Austauscherplätze lässt sich
mit den Begriffen „Funktionalisierungsdichte“ oder „lokale Kapazität“ beschreiben. Auch
wenn diese Parameter auf molekularer Ebene für einzelne Polymerpartikel bisher nicht

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 90 -
e n mit diesem Konzept aber durch theoretische
für die Funktionalisierung mittels Pfropfpolymerisation,
Mo
artige Eigenschaften, da sie sowohl eine positive Ladung als auch einen hydrophoben Rest
bes
die Oberfläche der Harzpartikel anzulagern, wobei sich die Monomere auf Grund ihrer
pos
Abstan satz einer geringen
Me
sierung on verwendeten
gepfropften Austauscher zeichnen sich durch eine niedrige lokale Kapazität aus.
experim ntell bestimmbar sind, könne
Überlegungen Selektivitätsunterschiede zwischen Latex-Anionenaustauschern und
gepfropften Anionenaustauschern gedeutet werden.
Die lokale Kapazität gepfropfter Austauscher ist meist wesentlich geringer als die von
Latexteilchen. Das liegt daran, dass
nomere mit einer Ammoniumgruppe eingesetzt werden. Diese Moleküle zeigen tensid-
itzen. Eine Eigenart der tensidartigen Monomere ist es, sich mit ihrer lipophilen Seite an
itiven Ladung gegenseitig abstoßen, so dass sich ein gleichmäßiger, möglichst großer
d zwischen den funktionellen Gruppen ergibt. Durch den Ein
nge an Pfropfmonomeren lassen sich gezielt stationäre Phasen mit niedriger Funktionali-
sdichte herstellen [124]. Die im Rahmen der vorliegenden Dissertati
Im Gegensatz dazu sind Latex-Anionenaustauscher dafür bekannt, eine außerordentlich hohe
lokale Kapazität aufzuweisen. Latexteilchen müssen nicht so druckstabil wie Trägerpartikel
sein, und werden deswegen gewöhnlich mit einem niedrigen Quervernetzungsgrad hergestellt.
Dadurch lässt sich eine große Oberfläche mit sehr hoher Funktionalisierungsdichte erreichen.
Latex-Anionen-austauscher
BrO−
3
log
k'
Cl−
log Qlokal
gepfropfterAnionen-austauscher
ie eine niedrige lokale Kapazität besitzen, eine höhere Retention auf als Chlorid.
Wie in Abb. 37 bei der Auftragung von log k’ gegen log Qlokal erkennbar, muss die Geraden-
Abb. 37: Graphische Darstellung von Gl. 20 für Chlorid und Bromat
Die Auswirkung der lokalen Kapazität auf den Retentionsfaktor soll am Beispiel der Analyten
Chlorid und Bromat verdeutlicht werden, deren Elutionsreihenfolge sich bei Trennung an
pellikularen und gepfropften Anionenaustauschern umdreht. Bromat weist an stationären
Phasen, d
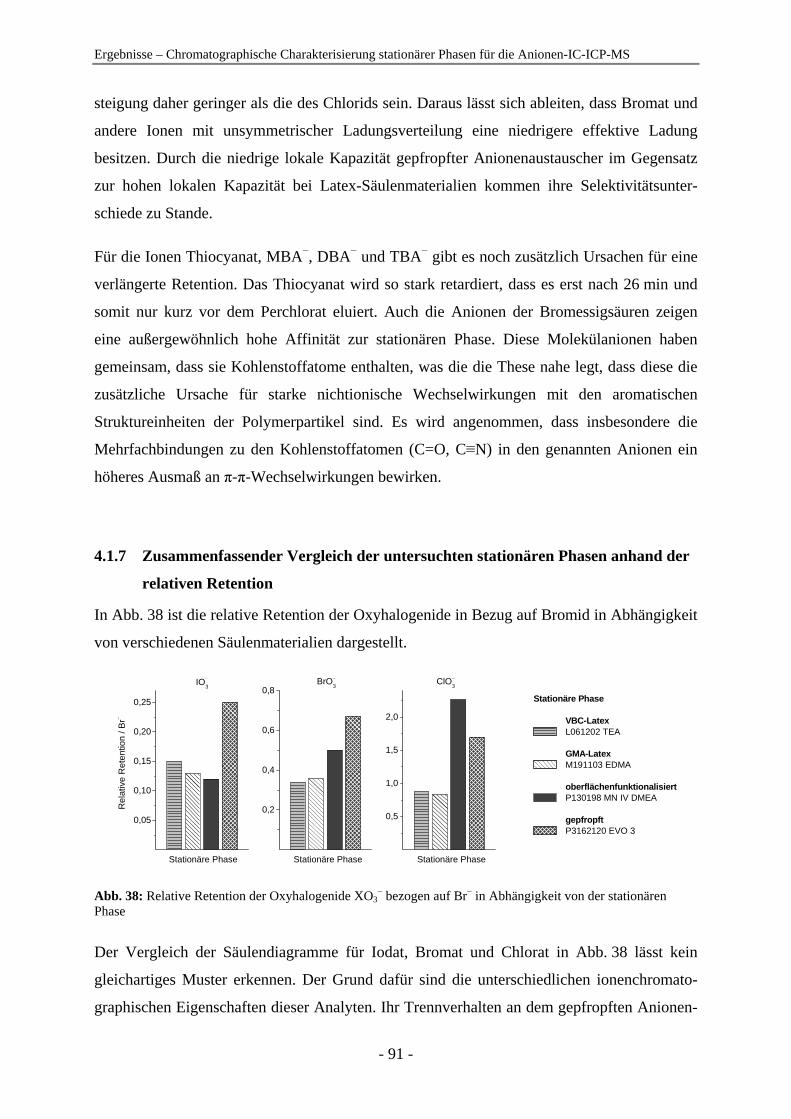
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 91 -
ine
verlängerte Retention. Das Thiocyanat wird so stark retardiert, dass es erst nach 26 min und
nu kurz vor dem Perchlorat eluiert. Auch die Anionen der Bromessigsäuren zeigen
eine außergewöhnlich hohe Affinität zur stationären Phase. Diese Molekülanionen haben
ie Kohlen fatome enthalten, was die die These nahe legt, dass diese die
it den aromatischen
an ere die
ein
4.1.7 Zusammenfassender Vergleich der untersuchten stationären Phasen anhand der
relativen Retention
ellt.
steigung daher geringer als die des Chlorids sein. Daraus lässt sich ableiten, dass Bromat und
andere Ionen mit unsymmetrischer Ladungsverteilung eine niedrigere effektive Ladung
besitzen. Durch die niedrige lokale Kapazität gepfropfter Anionenaustauscher im Gegensatz
zur hohen lokalen Kapazität bei Latex-Säulenmaterialien kommen ihre Selektivitätsunter-
schiede zu Stande.
Für die Ionen Thiocyanat, MBA−, DBA− und TBA− gibt es noch zusätzlich Ursachen für e
somit r
gemeinsam, dass s stof
zusätzliche Ursache für starke nichtionische Wechselwirkungen m
Struktureinheiten der Polymerpartikel sind. Es wird genommen, dass insbesond
Mehrfachbindungen zu den Kohlenstoffatomen (C=O, C≡N) in den genannten Anionen
höheres Ausmaß an π-π-Wechselwirkungen bewirken.
In Abb. 38 ist die relative Retention der Oxyhalogenide in Bezug auf Bromid in Abhängigkeit
von verschiedenen Säulenmaterialien dargest
0,05
0,10
0,15
0,20
0,25
Stationäre Phase
ClO−
3IO−
3
Rel
ativ
e R
eten
tion
/ Br−
BrO−
3
0,5
1,0
1,5
2,0
Stationäre Phase
0,2
0,4
0,6
0,8
Stationäre Phase
Stationäre Phase
VBC-Latex L061202 TEA
GMA-LatexM191103 EDMA
oberflächenfunktionalisiert P130198 MN IV DMEA
gepfropft P3162120 EVO 3
Abb. 38: Relative Retention der Oxyhalogenide XO3− bezogen auf Br− in Abhängigkeit von der stationären
Phase
Der Vergleich der Säulendiagramme für Iodat, Bromat und Chlorat in Abb. 38 lässt kein
gleichartiges Muster erkennen. Der Grund dafür sind die unterschiedlichen ionenchromato-
graphischen Eigenschaften dieser Analyten. Ihr Trennverhalten an dem gepfropften Anionen-
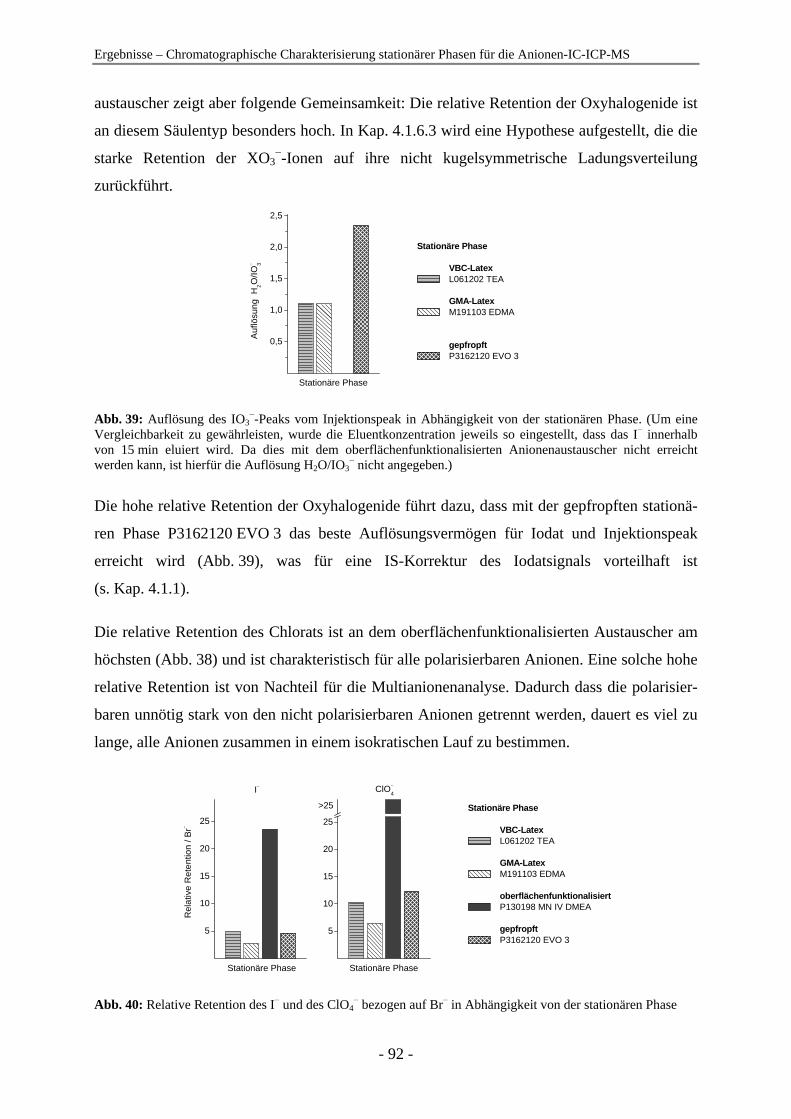
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 92 -
wird eine Hypothese aufgestellt, die die
starke Retention der XO −-Ionen auf ihre nicht kugelsymmetrische Ladungsverteilung
austauscher zeigt aber folgende Gemeinsamkeit: Die relative Retention der Oxyhalogenide ist
an diesem Säulentyp besonders hoch. In Kap. 4.1.6.3
3
zurückführt.
0,5
1,0
1,5
2,0
2,5
Stationäre Phase
Stationäre Phase
VBC-Latex L061202 TEA
GMA-LatexM191103 EDMA
gepfropft P3162120 EVO 3
Auf
lösu
ng H
2O/IO
− 3
Abb. 39: Auflösung des IO3−-Peaks vom Injektionspeak in Abhängigkeit von der stationären Phase. (Um eine
Vergleichbarkeit zu gewährleisten, wurde die Eluentkonzentration jeweils so eingestellt, dass das I− innerhalb von 15 min eluiert wird. Da dies mit dem oberflächenfunktionalisierten Anionenaustauscher
− nicht erreicht
werden kann, ist hierfür die Auflösung H2O/IO3 nicht angegeben.)
Die relative Retention des Chlorats ist an dem oberflächenfunktionalisierten Austauscher am
höchsten (Abb. 38) und ist charakteristisch für alle polarisierbaren Anionen. Eine solche hohe
relative Retention ist von Nachteil für die Multianionenanalyse. Dadurch dass die polarisier-
baren unnötig stark von den nicht polarisierbaren Anionen getrennt werden, dauert es viel zu
lange, alle Anionen zusammen in einem isokratischen Lauf zu bestimmen.
Die hohe relative Retention der Oxyhalogenide führt dazu, dass mit der gepfropften stationä-
ren Phase P3162120 EVO 3 das beste Auflösungsvermögen für Iodat und Injektionspeak
erreicht wird (Abb. 39), was für eine IS-Korrektur des Iodatsignals vorteilhaft ist
(s. Kap. 4.1.1).
5
10
15
20
25
Stationäre Phase
Rel
ativ
e R
eten
tion
/ Br
5
10
15
20
I−
−
ClO−
25
4
Stationäre Phase
VBC-Latex L061202 TEA
GMA-LatexM191103 EDMA
oberflächenfunktionalisiert P130198 MN IV DMEA
gepfropft P3162120 EVO 3
Stationäre Phase
>25
Abb. 40: Relative Retention des I− und des ClO4− bezogen auf Br− in Abhängigkeit von der stationären Phase
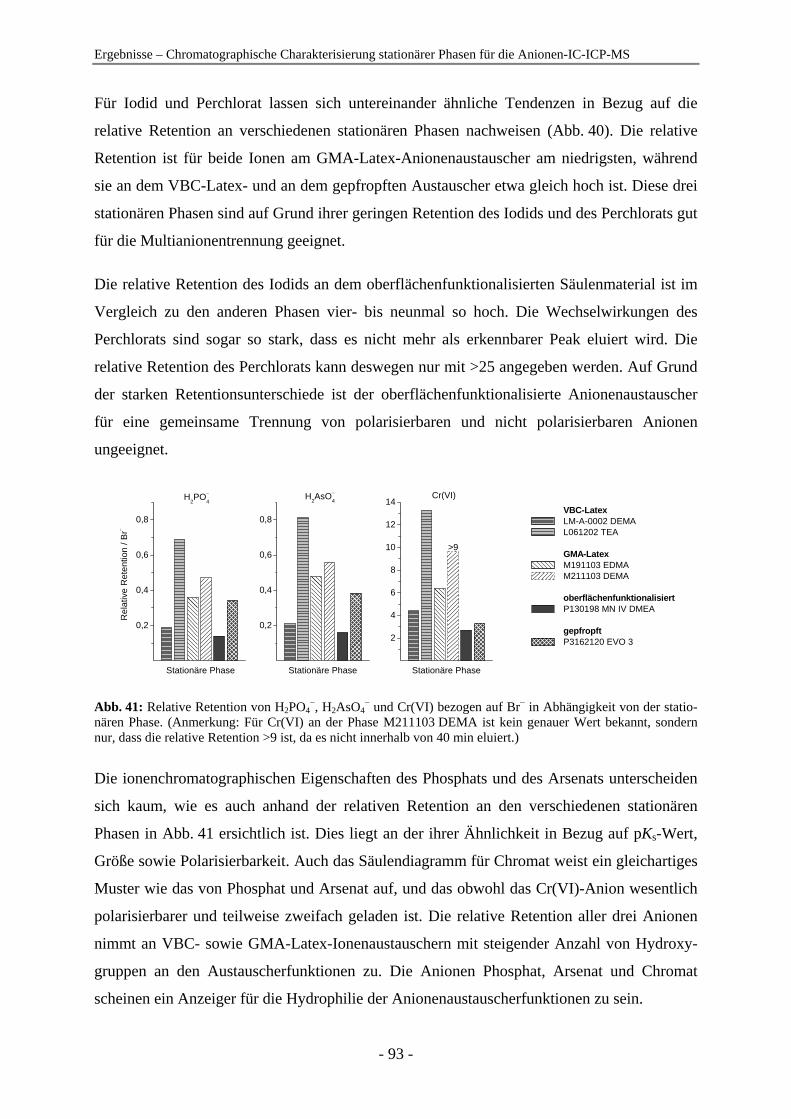
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 93 -
tex- und an dem gepfropften Austauscher etwa gleich hoch ist. Diese drei
stationären Phasen sind auf Grund ihrer geringen Retention des Iodids und des Perchlorats gut
Für Iodid und Perchlorat lassen sich untereinander ähnliche Tendenzen in Bezug auf die
relative Retention an verschiedenen stationären Phasen nachweisen (Abb. 40). Die relative
Retention ist für beide Ionen am GMA-Latex-Anionenaustauscher am niedrigsten, während
sie an dem VBC-La
für die Multianionentrennung geeignet.
Die relative Retention des Iodids an dem oberflächenfunktionalisierten Säulenmaterial ist im
Vergleich zu den anderen Phasen vier- bis neunmal so hoch. Die Wechselwirkungen des
Perchlorats sind sogar so stark, dass es nicht mehr als erkennbarer Peak eluiert wird. Die
relative Retention des Perchlorats kann deswegen nur mit >25 angegeben werden. Auf Grund
der starken Retentionsunterschiede ist der oberflächenfunktionalisierte Anionenaustauscher
für eine gemeinsame Trennung von polarisierbaren und nicht polarisierbaren Anionen
ungeeignet.
0,2
0,4
0,
0,
6
8
Stationäre Phase
Cr(VI)H2PO−
4
ntio
n /
H2AsO−
4
Stationäre Phase
2
8
10
14
Stationäre Phase
VBC-Latex
Br−
0,6
0,8 LM-A-0002 DEMA L061202 TEA
12
GMA-Latex M191103 EDMA
Rel
ativ
e R
ete
0,2
0,4 M211103 DEMA
oberflächenfunktionalisiert
4
6
P130198 MN IV DMEA
gepfropft P3162120 EVO 3
>9
1: Relative Retention von H2PO4−, H2AsO4
− und Cr(VI) bezogen auf Br− in Abhängigkeit von der statio-nären Phase. (Anmerkung: Für Cr(VI) an der Phase M211103 DEMA ist kein genauer Wert bekannt, sondern
r und teilweise zweifach geladen ist. Die relative Retention aller drei Anionen
nimmt an VBC- sowie GMA-Latex-Ionenaustauschern mit steigender Anzahl von Hydroxy-
gruppen an den Austauscherfunktionen zu. Die Anionen Phosphat, Arsenat und Chromat
scheinen ein Anzeiger für die Hydrophilie der Anionenaustauscherfunktionen zu sein.
Abb. 4
nur, dass die relative Retention >9 ist, da es nicht innerhalb von 40 min eluiert.)
Die ionenchromatographischen Eigenschaften des Phosphats und des Arsenats unterscheiden
sich kaum, wie es auch anhand der relativen Retention an den verschiedenen stationären
Phasen in Abb. 41 ersichtlich ist. Dies liegt an der ihrer Ähnlichkeit in Bezug auf pKs-Wert,
Größe sowie Polarisierbarkeit. Auch das Säulendiagramm für Chromat weist ein gleichartiges
Muster wie das von Phosphat und Arsenat auf, und das obwohl das Cr(VI)-Anion wesentlich
polarisierbare
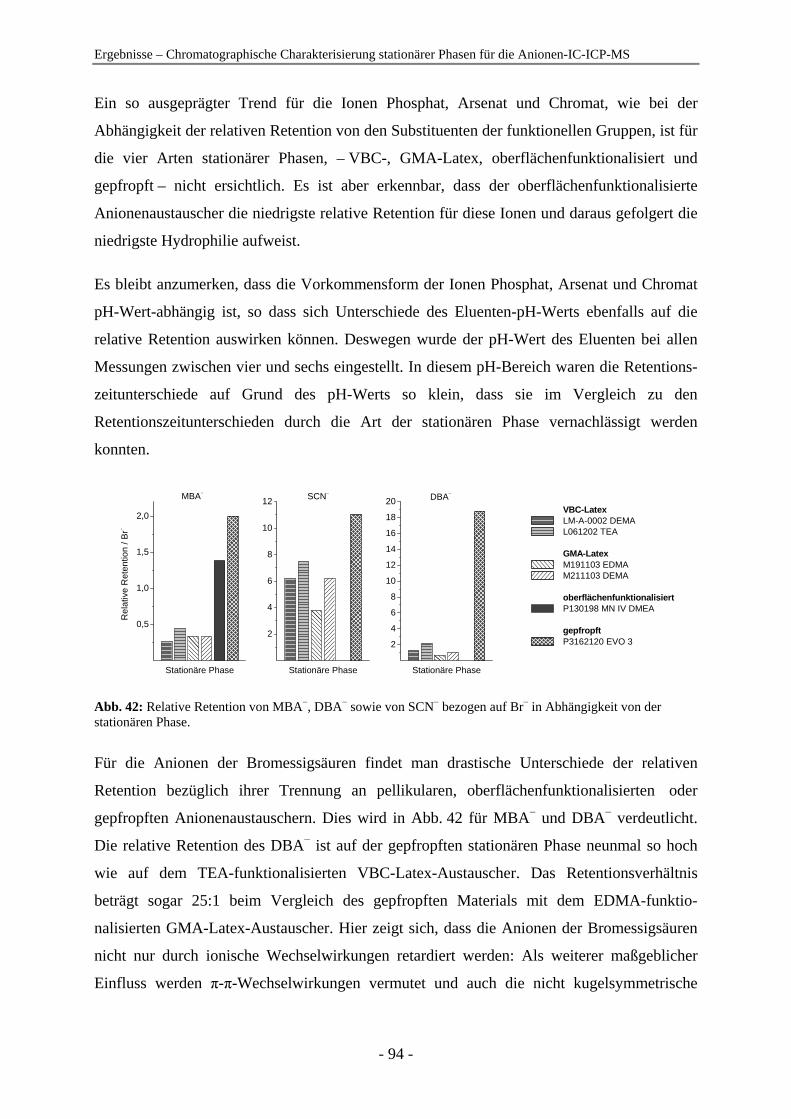
Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 94 -
Ein so ausgeprägter Trend für die Ionen Phosphat, Arsenat und Chromat, wie bei der
Abhängigkeit der relativen Retention von den Substituenten der funktionellen Gruppen, ist für
die vier Arten stationärer Phasen, – VBC-, GMA-Latex, oberflächenfunktionalisiert und
gepfropft – nicht ersichtlich. Es ist aber erkennbar, dass der oberflächenfunktionalisierte
Anionenaustauscher die niedrigste relative Retention für diese Ionen und daraus gefolgert die
niedrigste Hydrophilie aufweist.
Es bleibt anzumerken, dass die Vorkommensform der Ionen Phosphat, Arsenat und Chromat
pH-Bereich waren die Retentions-
pH-Wert-abhängig ist, so dass sich Unterschiede des Eluenten-pH-Werts ebenfalls auf die
relative Retention auswirken können. Deswegen wurde der pH-Wert des Eluenten bei allen
Messungen zwischen vier und sechs eingestellt. In diesem
zeitunterschiede auf Grund des pH-Werts so klein, dass sie im Vergleich zu den
Retentionszeitunterschieden durch die Art der stationären Phase vernachlässigt werden
konnten.
Stationäre Phase
MBA− DBA−SCN−
12
Stationäre Phase
VBC-Latex LM-A-0002 DEMA L061202 TEA
GMA-Latex M191103 EDMA M211103 DEMA
oberflächenfunktionalisiert P130198 MN IV DMEA
gepfropft P312
4
6
8
10
12
14
16
18
0,5
1,0
1,5
2,0
Rel
ativ
e R
eten
tion
/ Br−
2
4
6
8
10
62120 EVO 3
20
Stationäre Phase
stationären Phase.
pfropften Materials mit dem EDMA-funktio-
Abb. 42: Relative Retention von MBA−, DBA− sowie von SCN− bezogen auf Br− in Abhängigkeit von der
Für die Anionen der Bromessigsäuren findet man drastische Unterschiede der relativen
Retention bezüglich ihrer Trennung an pellikularen, oberflächenfunktionalisierten oder
gepfropften Anionenaustauschern. Dies wird in Abb. 42 für MBA− und DBA− verdeutlicht.
Die relative Retention des DBA− ist auf der gepfropften stationären Phase neunmal so hoch
wie auf dem TEA-funktionalisierten VBC-Latex-Austauscher. Das Retentionsverhältnis
beträgt sogar 25:1 beim Vergleich des ge
nalisierten GMA-Latex-Austauscher. Hier zeigt sich, dass die Anionen der Bromessigsäuren
nicht nur durch ionische Wechselwirkungen retardiert werden: Als weiterer maßgeblicher
Einfluss werden π-π-Wechselwirkungen vermutet und auch die nicht kugelsymmetrische

Ergebnisse – Chromatographische Charakterisierung stationärer Phasen für die Anionen-IC-ICP-MS
- 95 -
etention des Thiocyanats an der gepfropften stationären Phase ist zwar bei
weitem nicht so stark wie die des DBA−, aber die Säulendiagramme für DBA−, MBA− und
Thiocyanat weisen ein vergleichbares Muster auf (Abb. 42). Dies lässt auf ähnliche ionen-
chromatographische Eigenschaften dieser Ionen schließen.
In der Literatur wird berichtet, dass es häufig zu Problemen bei der Auflösung der Peaks von
Halogenessigsäuren und anorganischen Anionen kommt [20, 100, 136]. Als Lösung wird ein
Temperaturprogramm während der Trennung vorgeschlagen. Der Einsatz der im Rahmen
dieser Dissertation untersuchten stationären Phasen stellt eine neue Alternative dar. Die
gepfropften stationären Phasen zeigen eine völlig andere Selektivität für die Anionen der
gegenseitig ergänzen: Bei Trennung an dem gepfropften Anionenaustauscher kommt es zu
Ladungsverteilung der Bromacetate scheint die Retention an gepfropften Anionenaustau-
schern zu verstärken (s. Kap. 4.1.6.3).
Die relative R
Bromessigsäuren verglichen mit den Latex-Anionenaustauschern. Dies lässt sich für die
sichere Identifizierung aller Bromspezies ausnutzen, da sich die beiden Säulentypen
keiner Koelution aufgegebener Bromspezies, allerdings ist die Retention des TBA− zu stark.
Das TBA− kann aber dafür an pellikularen Austauschern getrennt und schnell eluiert werden.
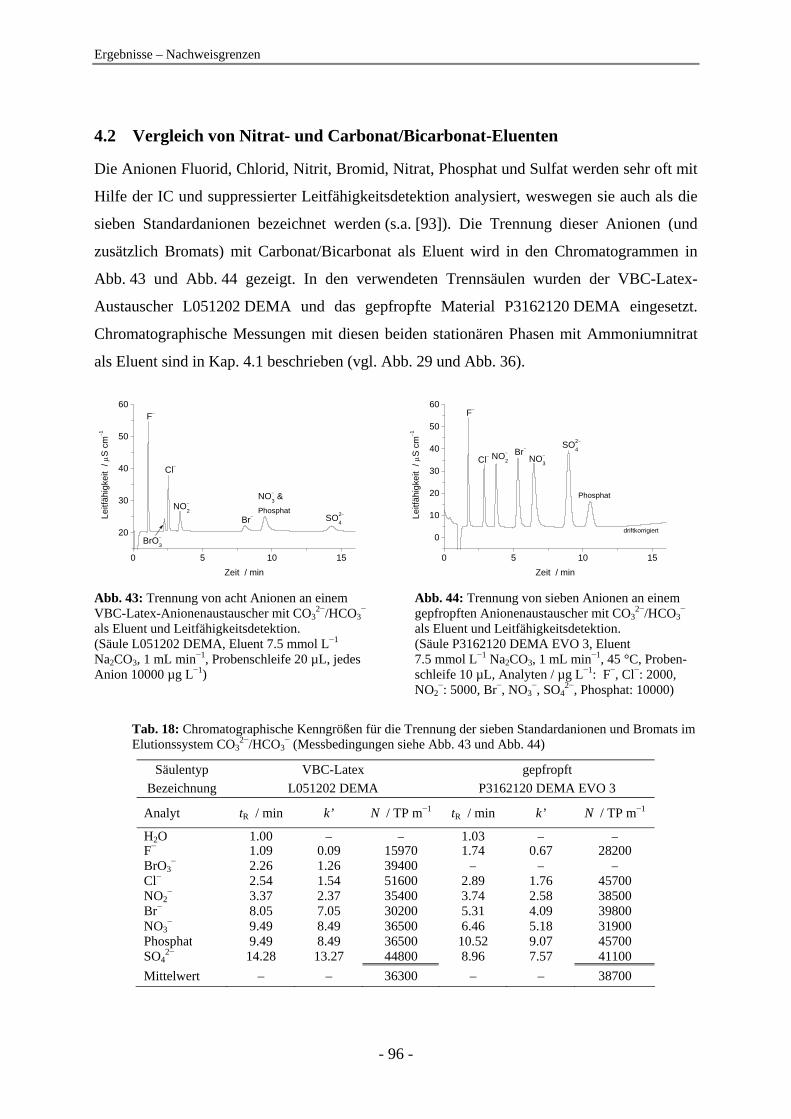
Ergebnisse – Nachweisgrenzen
- 96 -
andardanionen bezeichnet werden (s.a. [93]). Die Trennung dieser Anionen (und
zusätzlich Bromats) mit Carbonat/Bicarbonat als Eluent wird in den Chromatogrammen in
44 gezeigt. In den verwendeten Trennsäulen wurden der VBC-Latex-
51202 DEMA und das gepfropfte Material P3162120 DEMA eingesetzt.
4.2 Vergleich von Nitrat- und Carbonat/Bicarbonat-Eluenten
Die Anionen Fluorid, Chlorid, Nitrit, Bromid, Nitrat, Phosphat und Sulfat werden sehr oft mit
Hilfe der IC und suppressierter Leitfähigkeitsdetektion analysiert, weswegen sie auch als die
sieben St
Abb. 43 und Abb.
Austauscher L0
Chromatographische Messungen mit diesen beiden stationären Phasen mit Ammoniumnitrat
als Eluent sind in Kap. 4.1 beschrieben (vgl. Abb. 29 und Abb. 36).
0 5 10 15
20
30
40
50
60
Leitf
ähig
keit
/ µS
cm
−1
Zeit / min
F−
BrO−
3
Cl−
NO−
2
Br−
NO−
3 &Phosphat
SO2−4
0 5 10 15
0
10
20
30
40
50
60Le
itfäh
igke
it / µS
cm
−1
Zeit / min
F−
Cl− NO−
2Br−
NO−
3
SO2−4
Phosphat
driftkorrigiert
Abb. 43: Trennung von acht Anionen an einem VBC-Latex-Anionenaustauscher mit CO3
2−/HCals Eluent und Leitfähigkeitsdetektion.
Abb. 44: Trennung von sieben Anionen an einem O3
−
(Säule L051202 DEMA, Eluent 7.5 mmol L−1
gepfropften Anionenaustauscher mit CO32−/HCO3
− als Eluent und Leitfähigkeitsdetektion. (Säule P3162120 DEMA EVO 3, Eluent
d Abb. 44)
Säulentyp VBC-Latex gepfropft
Na2CO3, 1 mL min−1, Probenschleife 20 µL, jedes Anion 10000 µg L−1)
7.5 mmol L−1 Na2CO3, 1 mL min−1, 45 °C, Proben-schleife 10 µL, Analyten / µg L−1: F−, Cl−: 2000, NO2
−: 5000, Br−, NO3−, SO4
2−, Phosphat: 10000)
Tab. 18: Chromatographische Kenngrößen für die Trennung der sieben Standardanionen und Bromats im Elutionssystem CO3
2−/HCO3− (Messbedingungen siehe Abb. 43 un
Bezeichnung L051202 DEMA P3162120 DEMA EVO 3
Analyt tR / min k’ N / TP m−1 tR / min k’ N / TP m−1
H2O 1.00 – – 1.03 – – F− 1.09 0.09 15970 1.74 0.67 28200 BrO3
− 2.26 1.26 39400 – – – Cl− 2.54 1.54 51600 2.89 1.76 45700 NO2
− 3.37 2.37 35400 3.74 2.58 38500 Br− 8.05 7.05 30200 5.31 4.09 39800 NO3
− 9.49 8.49 36500 6.46 5.18 31900 Phosphat 9.49 8.49 36500 10.52 9.07 45700 SO4
2− 14.28 13.27 44800 8.96 7.57 41100 Mittelwert – – 36300 – – 38700

Ergebnisse – Nachweisgrenzen
- 97 -
luorid, Nitrit sowie Nitrat nicht und Sulfat nur schlecht mit der ICP-MS detektierbar. Fluor
ird im Plasma nicht ausreichend ionisiert. Die Stickstoffspezies sind nicht messbar, da
neben der geringen Ionisierungsausbeute sowohl der Stickstoffuntergrund im ICP-MS durch
die Umgebungsluft zu hoch ist, als auch der Eluent aus Nitrat besteht. Die Nachweisgrenzen
für Schwefelspezies sind mit einem ICP-MS ohne Kollisionszelle relativ hoch verglichen mit
Iod- oder Bromspezies, da die Messung des häufigsten Schwefelisotops 32Schwefel durch das
Molekülion [16O2]+ gestört wird, so dass die Detektion über das Schwefeloxid [32S16O]+ oder
das Isotop 34Schwefel (4.21 % Häufigkeit) erfolgen muss. Für die meisten anderen Anionen
(Kap. 2.4), insbesondere wenn die Anionen Metallatome enthalten, ist aber die ICP-MS-
Detektion der Leitfähigkeitsdetektion in Bezug auf die Nachweisstärke und durch die
Elementselektivität überlegen.
Die vier Ionen Bromat, Chlorid, Bromid und Phosphat wurden sowohl mit Carbonat/Bicar-
bonat-Eluenten, als auch mit dem Nitrat-Elutionssystem an den gleichen stationären Phasen
getrennt. Für diese Ionen liegen daher chromatographische Parameter vor, die zu einem
Vergleich des Eluenteinflusses herangezogen werden können.
Die mittlere Trennleistung aller gemessenen Ionen liegt trotz Verwendung der gleichen
Trennsäulen mit Carbonat/Bicarbonat-Eluenten immer höher als mit Nitrat-Eluenten. Für
Chlorid beispielsweise beträgt die Trennstufenzahl an dem gepfropften Anionenaustauscher
P3162120 DEMA mit Carbonat/Bicarbonat-Eluent 46000 TP m−1 (Tab. 16) und mit Nitrat-
Eluent 12800 TP m−1 (Tab. 17). Auch bei Latex-Anionenaustauschern wird die Trennleistung
für Chlorid durch Verwendung des Nitrats als Verdrängerion um den Faktor drei bis vier
verringert. Die Ursache ist die stärkere Affinität des Nitrats zu den Austauscherfunktionen der
stationären Phase im Vergleich zum Carbonat. Ein Austausch der Nitrationen an der
stationären Phase durch die Analytionen findet deswegen seltener statt. Dies erniedrigt die
„dynamische Kapazität“ einer Säule sowie die Anzahl an Ionenaustauschprozessen und führt
zur Verringerung der Trennstufenzahl.
Im Gegensatz zum Nitrat besitzen Carbonat und Bicarbonat nur eine schwache Elutionskraft.
Deswegen ist es mit Carbonat/Bicarbonat-Eluenten schwierig, polarisierbare Ionen wie Iodid
oder Perchlorat von Latex-Anionenaustauschern zu eluieren, insbesondere wenn die
Austauscher mit relativ hydrophoben funktionellen Gruppen ausgestattet sind. Für die Elution
Die typische Analytpalette der IC mit Carbonat/Bicarbonat-Eluenten und der IC-ICP-MS mit
Ammoniumnitrat-Eluenten unterscheidet sich deutlich. Von den sieben Standardanionen sind
F
w

Ergebnisse – Nachweisgrenzen
- 98 -
werden Eluentzusätze, wie z.B. p-Cyanophenol, und hohe Carbonatkonzentrationen benötigt.
ngt durch die hohe
43 verwendeten, bei der
Elution mit Carbonat-Eluenten häufig ein Tailing des Bromidpeaks auf. Dagegen gibt es im
Nitrat-Elutionssystem keine Symmetrieprobleme für Bromid, und auch die Peakformen der
noch stärker polarisierbaren Ionen werden verbessert.
Carbonat/Bicarbonat-Eluenten können ausschließlich im alkalischen Bereich verwendet
werden. Im Gegensatz dazu wird mit Ammoniumnitrat im sauren Milieu, typischerweise bei
pH-Werten zwischen drei und sieben, gearbeitet. Dies ist ein Pluspunkt für pH-Wert-
abhängige Ionen, da diese bei niedrigem pH-W rt in einer weniger deprotonierten Form
luie t we in
her geladen un it Nitrat-
alyten r
d Elementselektivität kein Pro ze
Retentionszeiten die Nachweisgrenzen der Analyten verbessert.
Der g r IC-ICP-MS ist, dass
dieser im sschließli zerfällt, die au r gasförmig
vorliege leichtf Verb ung S , Wa rsto erstoff,
Wasser, Stickstoffoxide usw. Des tstehen auch bei hohen Eluentkonzentrationen
keine A rungen an küh Zon Inter oder Massenspektrometer.
Anders ält es sich Elue , die 3/Na 3 ent n: De atz einer
Suppressortechnik ist notwendig, die Natriumeinbringung ini-
mieren e Natr atrix afür nt, so durc lagerungen, als auch
durch Signalunterdrückungs- oder Signalverstärkungseffekte die Stabilität von ICP-MS-
essungen zu beeinträchtigen [137-139]. Nach der Suppression enthält der Eluent aber
Dadurch kann es zu Problemen bei der Suppression kommen. Bedi
Grundleitfähigkeit können Hohlfasermembransuppressoren nicht eingesetzt werden und
herkömmliche Suppressorsäulen müssen sehr oft regeneriert werden [13].
Diese Probleme treten mit ICP-MS-Detektion nicht auf. In der relativ hohen Polarisierbarkeit
des Nitrats (s. Tab. 5) liegt ein großer Vorteil des Nitrat-Eluenten: Da das Nitrat ein
polarisierbares Anion ist, lassen sich damit andere polarisierbare Anionen leichter eluieren.
Der Einfluss nichtionischer Wechselwirkungen zwischen Analyten und stationärer Phasen
wird mit Nitrat-Eluenten zurückgedrängt. Dies wirkt sich positiv auf die Peakformen aus.
Beispielsweise tritt an Latex-Ionenaustauschern, wie dem in Abb.
e
vorliegen und entsprechend schneller e
Carbonat/Bicarbonat-Eluenten hö
r rden. So ist beispielsweise das Phosphat
d eluiert wesentlich später als m
Eluenten. Die frühe Elution mehrerer An
ICP-MS dank er
gleichzeitig stellt bei der Detektion mit de
blem dar. Vielmehr werden durch kur
rößte Vorteil des Ammoniumnitrat-Eluenten für den Einsatz in de
Plasma au ch in Elemente ch bei Raumtemperatu
n oder lüchtige ind en bilden: tickstoff sse ff und Sau
wegen en
blage den leren en im f eac im
verh bei nten Na2CO HCO halte r Eins
um in das ICP-MS zu m
[90]. Ein iumm ist d bekan wohl h Ab
M

Ergebnisse – Nachweisgrenzen
- 99 -
stimmung der Nachweisgrenze erfolgt nach verschiedenen
lten in der unmittelbaren Nähe
e Blindlösung siebenmal gemessen und die folgende
immer noch Kohlenstoffdioxid. Dies stellt ebenfalls ein Problem dar, da durch den vermehrt
ins Plasma eingebrachten Kohlenstoff isobare Interferenzen entstehen können, und es zu
Kohlenstoffablagerungen am Sampler- und am Skimmer-Cone kommen kann [98].
4.3 Nachweisgrenzen
Die Nachweisgrenze ist als Maß für die Nachweisstärke ein wichtiges Kriterium zur
Bewertung der Leistungsfähigkeit eines analytischen Verfahrens. Die Nachweisgrenze stellt
den kleinsten Messwert dar, der mit einer vorgegebenen Sicherheit vom Blindwert
unterschieden werden kann. Sie ist eine Entscheidungsgrenze für das Vorhandensein eines
Bestandteils, aber dessen Konzentration kann nicht notwendigerweise als genauer Wert
quantifiziert werden. Die Be
Verfahren. Eine zuverlässige und vergleichbare Angabe muss immer auch das Berechnungs-
modell sowie die wichtigsten Messparameter enthalten.
Gemäß DIN 32645 (Kalibriergeradenmethode) kann die Nachweisgrenze mit Hilfe der
Regressionsdaten einer Kalibriergeraden bei sehr kleinen Geha
der Nachweisgrenze ermittelt werden [140]. Das Verhältnis von der errechneten Nachweis-
grenze zu dem höchsten Kalibrierwert soll dabei den Faktor zehn nicht überschreiten. Bei
dieser Berechnungsart der Nachweisgrenze werden die Einflussgrößen des gesamten
Kalibrierverfahrens mit einbezogen.
Eine weitere weniger aufwendige Möglichkeit zur Nachweisgrenzenberechnung wird in der
Methode 321.8 der US EPA angewendet [94]. Diese Bestimmung ist ähnlich zu der Leerwert-
methode nach DIN 32645. Da bei der IC-ICP-MS für Reinstwasser durch eine Leerwertprobe
kein integrierbarer Peak erzeugt wird, wird das Reinstwasser mit einer kleinen Analytmenge
verstärkt (engl.: fortified blank = verstärkte Blindlösung), so dass die Analytkonzentration
dem zwei- bis fünffachen Wert der erwarteten Nachweisgrenze entspricht. Zur Bestimmung
der Nachweisgrenze wird die verstärkt
Berechnung durchgeführt:
Ergebnisse – Nachweisgrenzen
- 100 -
s: Standardabweichung
Nachweisgrenzen / µg L−1
Gl. 21: s t α- ⋅= = % 99 1,nNWG
tn−1, α=99 %: t-Wert nach Student für ein Signifikanzniveau α von 99 % und n−1 Freiheitsgrade (t = 3.14 für sieben Wiederholungsmessungen) n: Anzahl der Wiederholungsmessungen α: Signifikanzniveau
In Tab. 19 sind die mit den zwei vorgestellten Methoden ermittelten Nachweisgrenzen für
Bromid, Bromat, Iodid und Iodat mit und ohne Korrektur durch den internen Standard
Germaniumdioxid aufgeführt. Typische bisher von anderen Arbeitsgruppen für IC-ICP-MS
veröffentlichte Nachweisgrenzen liegen in einem Bereich von 0.05–3.5 µg L−1 [107]. Wie in
Tab. 19 ersichtlich befinden sich die im Rahmen dieser Dissertation ermittelten Nachweis-
grenzen somit im Bereich der niedrigsten publizierten Werte.
Tab. 19: Nachweisgrenzen der IC-ICP-MS für Iodat, Bromat, Bromid und Iodid.
niveau mit IS GeO2 Iodat Bromat Bromid Iodid
Signifikanz- Korrektur
nein 0.064 0.088 0.129 – Kalibriergeraden-methode a 95 %
ja 0.049 0.066 0.074 – nein 0.084 0.069 0.124 0.182 US EPA-
Methode b 99 % ja 0.045 0.071 0.104 0.151
a DIN 32645 [140], Zehn äquidistante Kalibrationspunkte von 0.1–1.0 µg L−1 in Reinstwasser, Probenschleife 585 µL, Säule VBC-Latex-Anionenaustauscher L051202 DEMA, Ausreißer eliminiert b US EPA-Methode [94], 3.14-fache Standardabweichung von sieben Wiederholmessungen, Konzentration 0.5 µg L−1 in Reinstwasser, Probenschleife 585 µL, Säule VBC-Latex-Anionenaustauscher L300102 DEMA

Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
4.4 Anwendungsbeispiele der IC-ICP-MS-Kopplung
- 101 -
.4.1 Analyse von Speisesalzen und Salz für Sole-Bäder auf Bromat
erschiedene Salze wurden mit der IC-ICP-MS auf Bromatspuren untersucht. Zum einen
handelte es sich um Badesalze, wie bei der Probe „Sodener Salz“ für ein Sole-Heilbad oder
er Salz“, welches als Badewannenzusatz verkauft wird 8. Zum
eitfähigkeit ausgeschlossen. Für die
Chlorid/Bromat-Unterscheidung wird zusätzlich die Elementselektivität durch das ICP-MS
4
V
bei der Probe „Totes Me
anderen wurden auch im Handel erhältliche Speisesalze verschiedener Hersteller auf Bromat
analysiert. Die Analysen sind anspruchsvoll, da Bromat in den Messlösungen, wenn
überhaupt, nahe der Nachweisgrenze (s. Kap. 4.3) vorliegt, und es neben einer um sieben
Größenordnungen konzentrierteren Anionenmatrix bestimmt werden muss. Zur Lösung des
Analysenproblems wird die ionenchromatographische Matrixabtrennung und gleichzeitige
Aufkonzentrierung des Analyten ausgenutzt. Wegen der sehr hohen Chloridkonzentration ist
es allerdings nicht möglich, das Bromat chromatographisch gut genug vom Chlorid
aufzulösen. Dadurch wird eine Detektion über die L
benötigt.
50k
100k
150k
200k
250k
0 5 10 15 20
0
Cl−?HCO−
3?
Pumpe
74Ge
Zähl
rate
/ c
ps IO−
3
Br−
BrO−
3
Zeit / min
Abb. 45: Aufgabe von 585 µL 0.5%iger Lösung des „Sodener Salzes“, der 20 µg L−1 BrO3
− zugesetzt wurden. Bei Latex-Anionenaustauschern treten zu starke Überladungseffekte auf. (Säule L051202 DEMA, Q = 50−60 µeq Säule−1, Eluent: 20 mmol L−1 NH4NO3 pH 5.6).
Wie in Abb. 45 ersichtlich, führt die Trennung von Proben mit hohem Matrixgehalt an Latex-
Anionenaustauschern zu Überladungsstörungen, so dass Bromat keinen schmalen, definierten 8 Sole (aus spätmittelhochdt.: sul, sol = Salzbrühe) ist ursprünglich eine Bezeichnung für Natriumchlorid- oder Steinsalzlösungen, die durch Einleiten von Wasser in Steinsalzlager erhalten wurden [127]. In der Medizin werden auch Kochsalzlösungen mit einem Salzgehalt von 1.5–6 % (teilweise auch bis 30 %) als Sole bezeichnet. Die medizinische Wirksamkeit von Sole-Anwendungen bei einigen Hautkrankheiten ist unbestritten. Bei anderen Anwendungsgebieten wird die Wirksamkeit in Frage gestellt. Tatsache ist, dass es Sole-Trinkkuren, Sole-Spülungen, Sole-Bäder, Sole-Einreibungen, Sole-Umschläge und Sole-Inhalationen gibt [141].
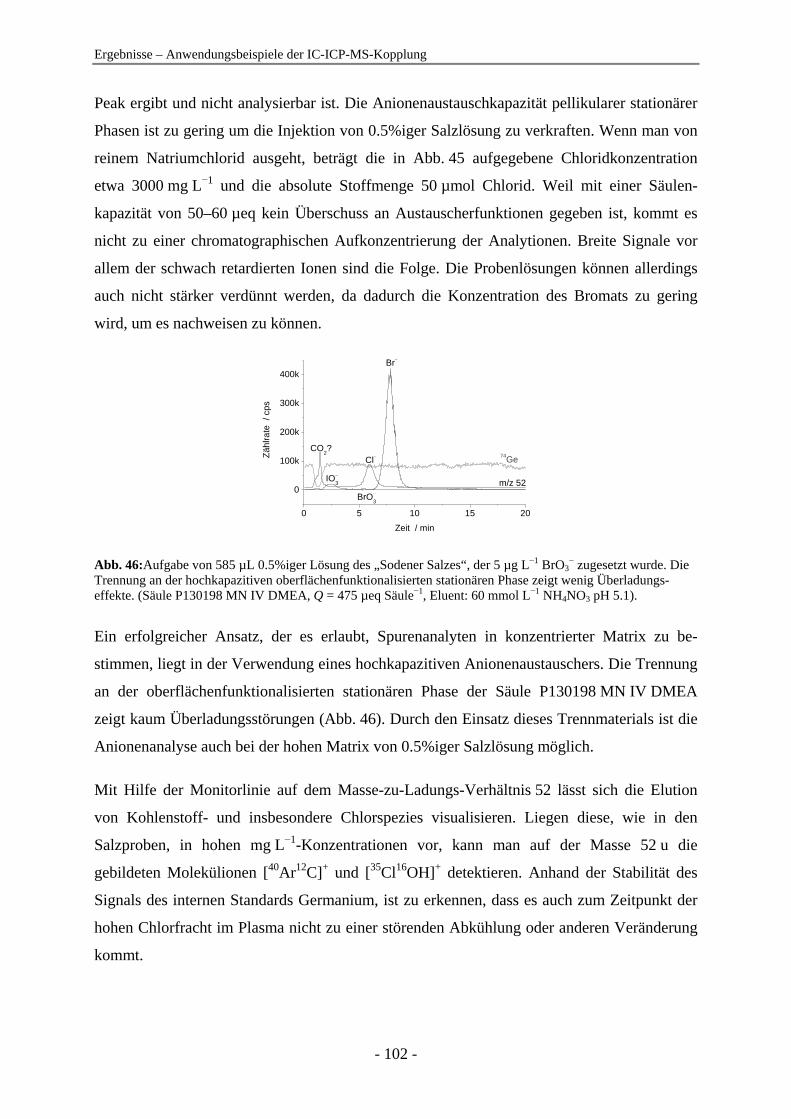
Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
- 102 -
g alysierbar i enaustauschkapazität pellikularer stationärer
egebene Chloridkonzentration
Stoffmenge 50 µmol Chlorid. Weil mit einer Säulen-
stauscherfunktionen gegeben ist, kommt es
300k
Peak er ibt und nicht an st. Die Anion
Phasen ist zu gering um die Injektion von 0.5%iger Salzlösung zu verkraften. Wenn man von
reinem Natriumchlorid ausgeht, beträgt die in Abb. 45 aufg
etwa 3000 mg L−1 und die absolute
kapazität von 50–60 µeq kein Überschuss an Au
nicht zu einer chromatographischen Aufkonzentrierung der Analytionen. Breite Signale vor
allem der schwach retardierten Ionen sind die Folge. Die Probenlösungen können allerdings
auch nicht stärker verdünnt werden, da dadurch die Konzentration des Bromats zu gering
wird, um es nachweisen zu können.
400k
0 5 10 15 20
0
Br−
100k
200k
CO2?Zäh
BrO−
3
lps
Zeit /
rate
/ c
min
Cl−
IO3
− −
Ein erfolgreicher Ansatz, der es erlaubt, Spurenanalyten in konzentrierter Matrix zu be-
timmen, liegt in der Verwendung eines hochkapazitiven Anionenaustauschers. Die Trennung
an der oberflächenfunktionalisierten stationären Phase der Säule P130198 MN IV DMEA
zeigt kaum Überladungsstörungen (Abb. 46). Durch den Einsatz dieses Trennmaterials ist die
Anionenanalyse auch bei der hohen Matrix von 0.5%iger Salzlösung möglich.
Mit Hilfe der Monitorlinie auf dem Masse-zu-Ladungs-Verhältnis 52 lässt sich die Elution
von Kohlenstoff- und insbesondere Chlorspezies visualisieren. Liegen diese, wie in den
Salzproben, in hohen mg L−1-Konzentrationen vor, kann man auf der Masse 52 u die
gebildeten Molekülionen [40Ar12C]+ und [35Cl16OH]+ detektieren. Anhand der Stabilität des
Signals des internen Standards Germanium, ist zu erkennen, dass es auch zum Zeitpunkt der
hohen Chlorfracht im Plasma nicht zu einer störenden Abkühlung oder anderen Veränderung
kommt.
−
m/z 52
74Ge
1Abb. 46:Aufgabe von 585 µL 0.5%iger Lösung des „Sodener Salzes“, der 5 µg L BrO3 zugesetzt wurde. Die
Trennung an der hochkapazitiven oberflächenfunktionalisierten stationären Phase zeigt wenig Überladungs-fekte. (Säule P130198 MN IV DMEA, Q = 475 µeq Säule−1, Eluent: 60 mmol L−1 NH NOef 4 3 pH 5.1).
s
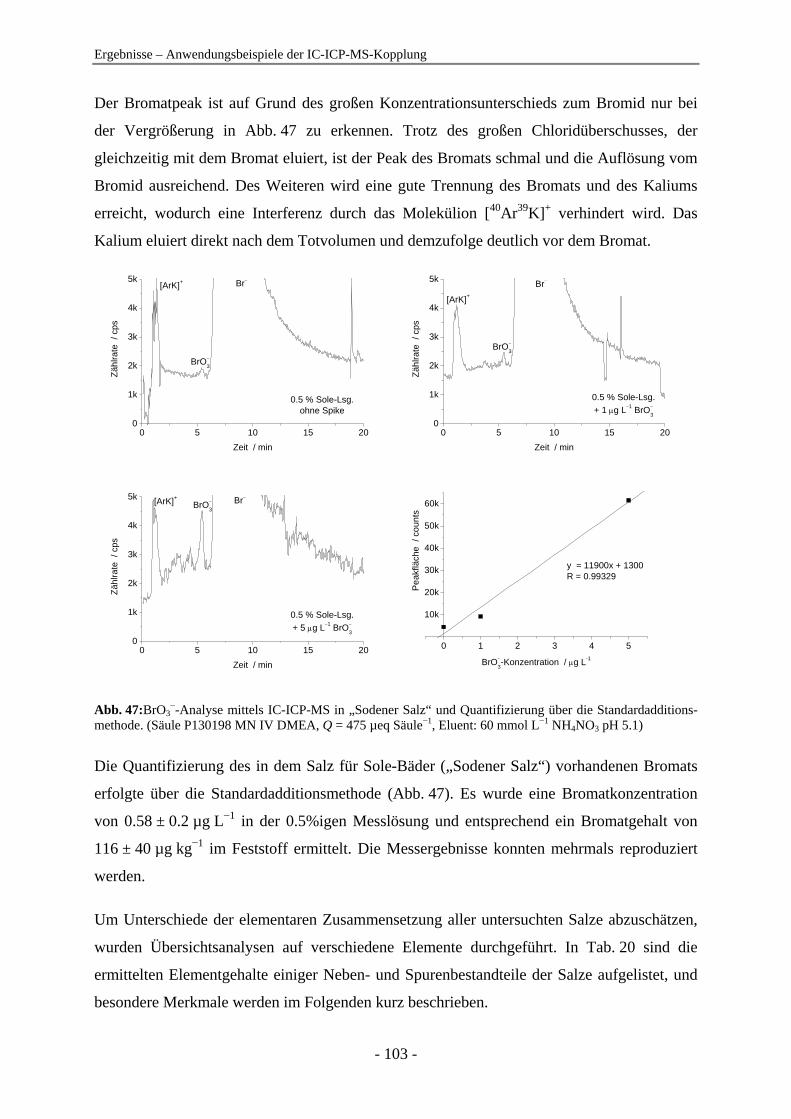
Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
- 103 -
Der Bromatpeak ist auf Grund des großen Konzentrationsunterschieds zum Bromid nur bei
ßen Chloridüberschusses, der
gleichzeitig mit dem Bromat eluiert, ist der Peak des Bromats schmal und die Auflösung vom
d ts und des Kaliums
der Vergrößerung in Abb. 47 zu erkennen. Trotz des gro
Bromi ausreichend. Des Weiteren wird eine gute Trennung des Broma
erreicht, wodurch eine Interferenz durch das Molekülion [40Ar39K]+ verhindert wird. Das
Kalium eluiert direkt nach dem Totvolumen und demzufolge deutlich vor dem Bromat.
0 5 10 15 200
1k
2k
3k
4k
5k[ArK]+
Zähl
rate
/ c
ps
Zeit / min
0.5 % Sole-Lsg.ohne Spike
BrO−
3
Br−
0 5 10 15 200
1k
2k
3k
4k
5k
[ArK]+
Zähl
rate
/ c
ps
Zeit / min
0.5 % Sole-Lsg.+ 1 µg L−1 BrO−
3
BrO−
3
Br−
3k
4k
5k
0 5 10 15 200
[ArK]+
cps
BrO−
3Br−
50k
60k
1k
2k
Zä
0.5 % Sole-Lsg.+ 5 µg L−1 BrO−
3
10k
20k
hlra
te /
30k
40k
Zeit / min
0 1 2 3 4 5
unts
Pea
kflä
che
/ co
y = 11900x + 1300R = 0.99329
Um Unterschiede der elementaren Zusammensetzung aller untersuchten Salze abzuschätzen,
BrO-3-Konzentration / µg L-1
Abb. 47:BrO3−-Analyse mittels IC-ICP-MS in „Sodener Salz“ und Quantifizierung über die Standardadditions-
methode. (Säule P130198 MN IV DMEA, Q = 475 µeq Säule−1, Eluent: 60 mmol L−1 NH4NO3 pH 5.1)
Die Quantifizierung des in dem Salz für Sole-Bäder („Sodener Salz“) vorhandenen Bromats
erfolgte über die Standardadditionsmethode (Abb. 47). Es wurde eine Bromatkonzentration
von 0.58 ± 0.2 µg L−1 in der 0.5%igen Messlösung und entsprechend ein Bromatgehalt von
116 ± 40 µg kg−1 im Feststoff ermittelt. Die Messergebnisse konnten mehrmals reproduziert
werden.
wurden Übersichtsanalysen auf verschiedene Elemente durchgeführt. In Tab. 20 sind die
ermittelten Elementgehalte einiger Neben- und Spurenbestandteile der Salze aufgelistet, und
besondere Merkmale werden im Folgenden kurz beschrieben.
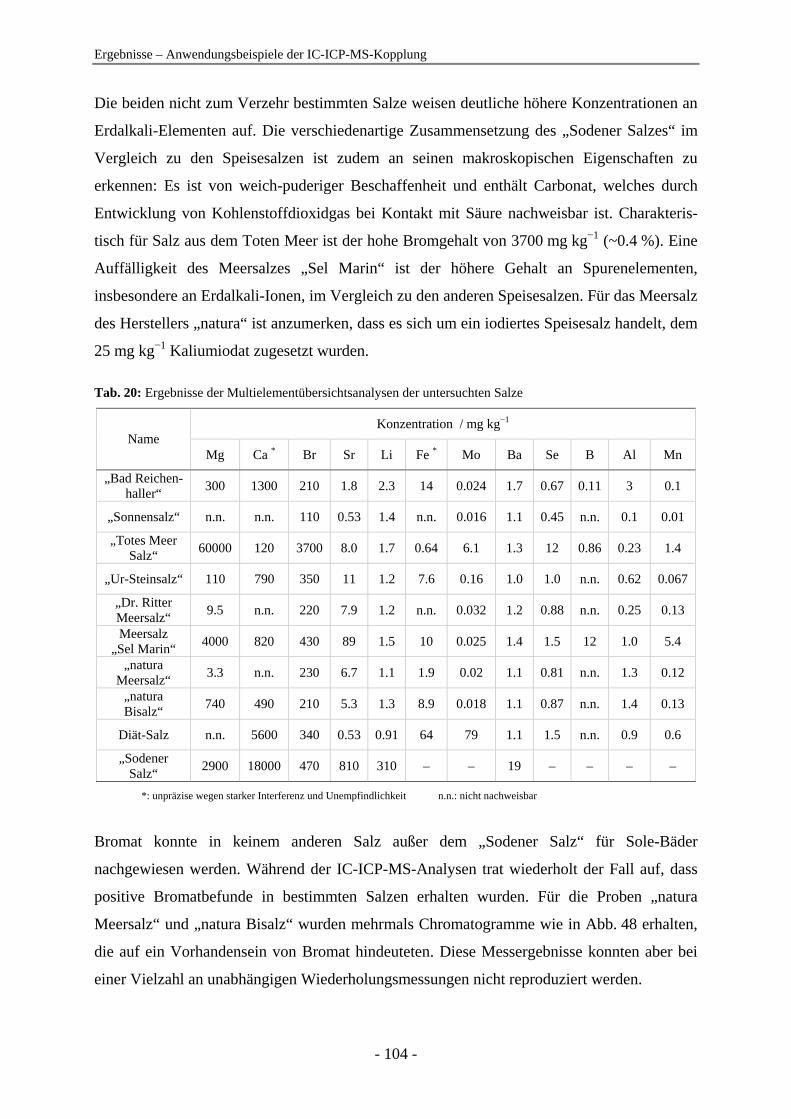
Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
- 104 -
iedenartige Zusammensetzung des „Sodener Salzes“ im
Vergleich zu den Speisesalzen ist zudem an seinen makroskopischen Eigenschaften zu
erkennen: Es ist von weich-puderiger Beschaffenheit und enthält Carbonat, welches durch
Entwicklung von Kohlenstoffdioxidgas bei Kontakt mit Säure nachweisbar ist. Charakteris-
tisch für Salz aus dem Toten Meer ist der hohe Bromgehalt von 3700 mg kg−1 (~0.4 %). Eine
Auffälligkeit des Meersalzes „Sel Marin“ ist der höhere Gehalt an Spurenelementen,
insbesondere an Erdalkali-Ionen, im Vergleich zu den anderen Speisesalzen. Für das Meersalz
des Herstellers „natura“ ist anzumerken, dass es sich um ein iodiertes Speisesalz handelt, dem
25 mg kg−1 Kaliumiodat zugesetzt wurden.
Konzentration / mg kg−1
Die beiden nicht zum Verzehr bestimmten Salze weisen deutliche höhere Konzentrationen an
Erdalkali-Elementen auf. Die versch
Tab. 20: Ergebnisse der Multielementübersichtsanalysen der untersuchten Salze
Name Mg Ca * Br Sr Li Fe * Mo Ba Se B Al Mn
„Bad Reichen-haller“ 300 1300 210 1.8 2.3 14 0.024 1.7 0.67 0.11 3 0.1
„Sonnensalz“ n.n. n.n. 110 0.53 1.4 n.n. 0.016 1.1 0.45 n.n. 0.1 0.01
„Totes Meer Salz“ 60000 120 3700 8.0 1.7 0.64 6.1 1.3 12 0.86 0.23 1.4
„Ur-Steinsalz“ 110 790 350 11 1.2 7.6 0.16 1.0 1.0 n.n. 0.62 0.067
„Dr. Ritter Meersalz“ 9.5 n.n. 220 7.9 1.2 n.n. 0.032 1.2 0.88 n.n. 0.25 0.13
Meersalz „Sel Marin“ 4000 820 430 89 1.5 10 0.025 1.4 1.5 12 1.0 5.4
„natura Meersalz“ 3.3 n.n. 230 6.7 1.1 1.9 0.02 1.1 0.81 n.n. 1.3 0.12
„natura Bisalz“ 740 490 210 5.3 1.3 8.9 0.018 1.1 0.87 n.n. 1.4 0.13
Diät-Salz n.n. 5600 340 0.53 0.91 64 79 1.1 1.5 n.n. 0.9 0.6
„SodeSalz
ner “ 2900 18000 470 810 310 – – 19 – – – –
*: unpräzise wegen starker Interferenz und Unempfindlichkeit n.n.: nicht nachweisbar
er bei
einer Vielzahl an unabhängigen Wiederholungsmessungen nicht reproduziert werden.
Bromat konnte in keinem anderen Salz außer dem „Sodener Salz“ für Sole-Bäder
nachgewiesen werden. Während der IC-ICP-MS-Analysen trat wiederholt der Fall auf, dass
positive Bromatbefunde in bestimmten Salzen erhalten wurden. Für die Proben „natura
Meersalz“ und „natura Bisalz“ wurden mehrmals Chromatogramme wie in Abb. 48 erhalten,
die auf ein Vorhandensein von Bromat hindeuteten. Diese Messergebnisse konnten ab

Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
0 5 10 15 200
200
400
600
800
1000
[ArK]+
Zähl
rate
/ c
ps
Zeit / min
0.5 % "naturaMeersalz"
BrO−
3?
Br−
0 5 10 15 200
200
400
600
800
1000
[ArK]+
Zähl
rate
/ c
ps
Zeit / min
0.5 % "natura Meer-salz" + 1 µg L−1 BrO−
3
BrO−
3Br−
Abb. 48: Beispiel einer BrO3
−-Analyse in Speisesalz. Ein BrO3−-Gehalt in „natura Meersalz“ ist nicht sicher
nachweisbar. (Säule P130198 MN IV, Q = 475 µeq Säule−1, Eluent: 30 mmol L−1 NH4NO3 pH 6.2)
Es stellt sich die Frage, wodurch falsch positive Bromatbefunde entstehen, bzw. wodurch
unregelmäßig Bromatpeaks vorgetäuscht werden können?
Um möglichst sicher zu gehen, dass eine Kontamination mit Bromat ausgeschlossen ist,
wurden viele Maßnahmen getroffen. Dazu gehörte die wiederholte gründliche Reinigung aller
Gefäße und Hilfsmittel beim Ansetzen von Lösungen genauso wie Spülschritte zwischen zwei
aufeinander folgenden Messungen, um mögliche Bromatreste aus der Probenschleife und den
f, Nitrat oder einem anderen Oxidationsmittel unter bestimmten Temperatur- oder
Lichtbedingungen (z.B. Sonneneinstrahlung) geschehen.
nnten z.B. durch [40Ar39K]+,
[31P16O3]+, [40Ar38Ar1H]+, [44Ca35Cl]+, [42Ca37Cl]+ verursacht werden (s.a. Tab. 4). Diese
Verbindungskapillaren zu entfernen.
Weitere Möglichkeiten, die nicht ausgeschlossen werden können, sind die Entstehung von
Bromat in Probengefäßen oder eine Entstehung erst während der Trennung durch Reaktion
mit dem Eluenten. Möglicherweise bildet sich bei hohen Bromidgehalten eine kleine Gleich-
gewichtskonzentration an Bromat. Dies könnte durch Oxidation des Bromids mit Luft-
sauerstof
Eine weitere mögliche Ursache sind durch Molekülionen verursachte Interferenzen, die sich
nur unregelmäßig unter bestimmten plasmaphysikalischen Vorraussetzungen bemerkbar
machen. Isobare Störungen auf der Masse des Broms kö
Fehlerquelle kann allerdings umgangen werden, indem das Bromisotop mit der Masse 81 u
gemessen wird.
Es wurden viele Optimierungsversuche unternommen, um die Nachweisstärke und Repro-
duzierbarkeit der Bromatmessung mit IC-ICP-MS noch weiter zu erhöhen. Dazu gehörten ein
- 105 -

Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
- 106 -
der Eluentkonzentration sowie des Eluent-pH-
Werts, Addition von Perchlorat zum Eluenten sowie Bestimmung der optimalen Proben-
aufgabemenge (Volumen und Verdünnung).
4 e v i w n B id ro o u
D tio r A en m r t, d d r ist t
nur abhängig von der La n lo h e fe t d agerstätten, sondern auch
von zur Herstellung benötigten qualitätssichernd e l e fe t
Mineralwasser- und Tafelwasserverordnung zwar ka
z.B. die Abtrennung von Eisen-, Mangan- und Schwefelverbindungen sowie Entzug oder
Zugabe von Kohlenstoffdioxid [127]. Zur Enteisenung und Entmanganung werden die
lös bon lze t m u e ff nl e is I ro bz
Brau oxid Ab uc ar Oxidationsmit ie o n z
kom ch A oc e i n ang yd d filtriert [142].
Für die Analysenauftraggeber lag der Kernpunkt der analytischen Fragestellung in folgenden
Aspekten: Inwiefern wirken sich bestimmte Methoden der Mineralwasseraufbereitung positiv
ität des
Bromats ausgeschlossen werden.
spezielles Tuning des ICP-MS auf die Masse 79 u von Brom, eine Verlängerung der dwell-
Zeit (s.a. Kap. 4.5.3) und die Erhöhung der Plasmaleistung, um die Ionisierungsausbeute zu
steigern. Diese Optimierungsversuche führten ebenso wenig zum Erfolg wie folgende Verän-
derungen der chromatographischen Bedingungen: Erprobung verschiedener oberflächenfunk-
tionalisierter Anionenaustauscher, Variation
.4.2 Analys on M neral ässer auf rom , B mat, I did nd Iodat
ie Konzentra n de nion Bro id, B oma Iodi und Io at in Mine alwässern nich
ge u d geo gisc en B schaf nhei er L
en Maßnahm n. Minera wäss r dür n lau
um behandelt werden, erlaubt sind aber
lichen Car atsa meis it L ftsau rsto zu u öslich m E en(II )hyd xid w. zu
nstein iert. er a h st ke tel w Oz n kö nen um Einsatz
men. Na usfl kung w rden die E sen- u d M anh roxi e ab
oder negativ auf die Speziesverteilung der Halogenide und Oxyhalogenide des Broms und des
Iods aus? Eine Oxidation des in Wässern vorhandenen Iodids zu Iodat gilt als vorteilhaft, da
schon geringe Mengen an Iodid den Geschmack und den Geruch beeinträchtigen können [18].
Der Grund dafür ist, dass Iodid in Mineralwasser bei Luftkontakt Spuren von leichtflüchtigem
elementarem Iod freisetzt. Im Gegensatz zu den Iodspezies muss eine Oxidation des in
Wässern vorhandenen Bromids zu Bromat auf Grund der potentiellen Kanzerogen
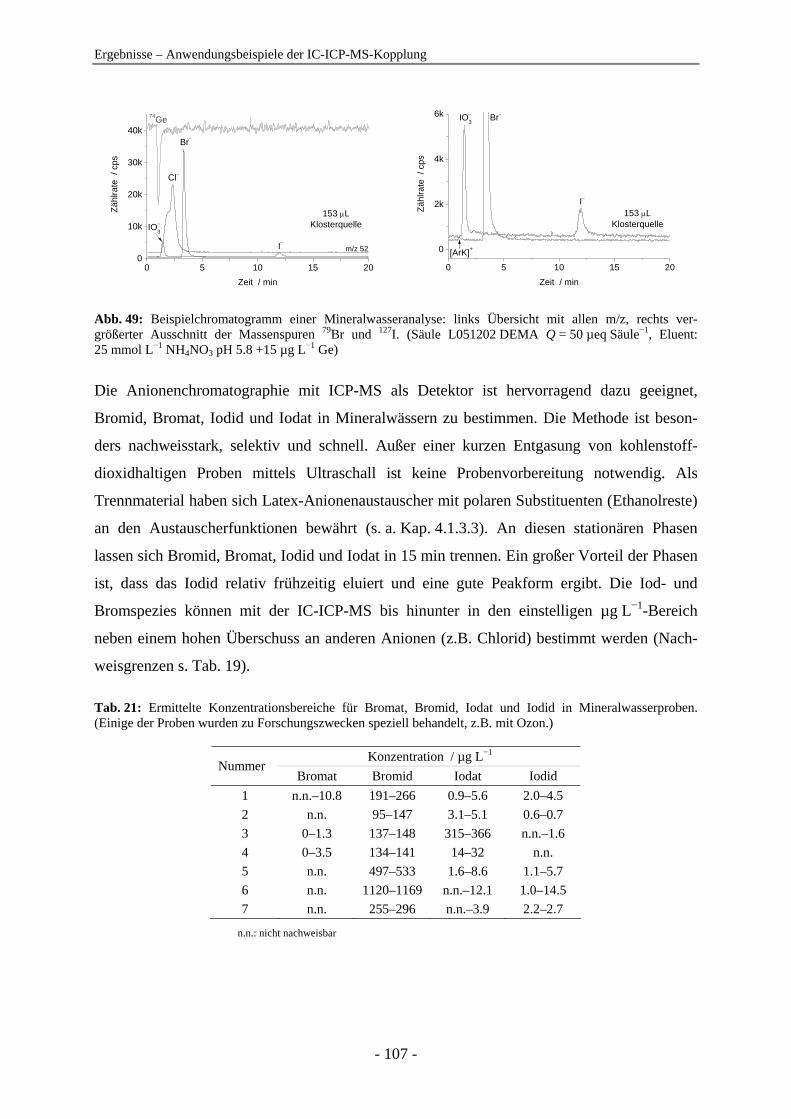
Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
0 5 10 15 200
10k
20k
30k
40k
153 µLKlosterquelle
Zähl
rate
/ c
ps
Zeit / min
74Ge
Cl−
Br−
I−IO−
3
m/z 52
0 5 10 15 20
0
2k
4k
6k
[ArK]+
Zähl
rate
/ c
ps
Zeit / min
153 µLKlosterquelle
Br−
I−
IO−
3
Abb. 49: Beispielchromatogramm einer Mineralwasseranalyse: links Übersicht mit allen m/z, rechts ver-größerter Ausschnitt der Massenspuren 79Br und 127I. (Säule L051202 DEMA Q = 50 µeq Säule−1, Eluent:
−1 −125 mmol L NH4NO3 pH 5.8 +15 µg L Ge)
en. Die Methode ist beson-
d Iodat in 15 min trennen. Ein großer Vorteil der Phasen
ist, dass das Iodid relativ frühzeitig eluiert und eine gute Peakform ergibt. Die Iod- und
Konzentration / µg L−1
Die Anionenchromatographie mit ICP-MS als Detektor ist hervorragend dazu geeignet,
Bromid, Bromat, Iodid und Iodat in Mineralwässern zu bestimm
ders nachweisstark, selektiv und schnell. Außer einer kurzen Entgasung von kohlenstoff-
dioxidhaltigen Proben mittels Ultraschall ist keine Probenvorbereitung notwendig. Als
Trennmaterial haben sich Latex-Anionenaustauscher mit polaren Substituenten (Ethanolreste)
an den Austauscherfunktionen bewährt (s. a. Kap. 4.1.3.3). An diesen stationären Phasen
lassen sich Bromid, Bromat, Iodid un
Bromspezies können mit der IC-ICP-MS bis hinunter in den einstelligen µg L−1-Bereich
neben einem hohen Überschuss an anderen Anionen (z.B. Chlorid) bestimmt werden (Nach-
weisgrenzen s. Tab. 19).
Tab. 21: Ermittelte Konzentrationsbereiche für Bromat, Bromid, Iodat und Iodid in Mineralwasserproben. (Einige der Proben wurden zu Forschungszwecken speziell behandelt, z.B. mit Ozon.)
Nummer Bromat Bromid Iodat Iodid
1 n.n.–10.8 191–266 0.9–5.6 2.0–4.5 2 n.n. 95–147 3.1–5.1 0.6–0.7 3 0–1.3 137–148 315–366 n.n.–1.6 4 0–3.5 134–141 14–32 n.n. 5 n.n. 497–533 1.6–8.6 1.1–5.7 6 n.n. 1120–1169 n.n.–12.1 1.0–14.5 7 n.n. 255–296 n.n.–3.9 2.2–2.7
n.n.: nicht nachweisbar
- 107 -

Ergebnisse – Anwendungsbeispiele der IC-ICP-MS-Kopplung
- 108 -
stark (Tab. 21). Die durchgeführten Messungen
dingungen auch Bromat bis zu einer Konzen-
tration von 10.8 µg L−1 in den Mineralwässern entstand. Des Weiteren konnte aus den Mess-
senungsproze-
In Tab. 21 sind die ermittelten Konzentrationsbereiche für Bromid, Bromat, Iodid und Iodat
aus einer Vielzahl von Mineralwasserproben angegeben. Die verschiedenen Sorten, Quellorte
oder Hersteller wurden mit Zahlen anonymisiert. Die Proben wurden teilweise an unterschied-
lichen Punkten im Herstellungsprozess genommen oder speziell behandelt, z.B. mit Ozon.
Die festgestellten Bromidkonzentrationen in den untersuchten Mineralwässern unterschieden
sich mit Werten zwischen 90–1200 µg L−1
zeigten, dass unter bestimmten Behandlungsbe
ergebnissen geschlossen werden, dass Iodid bei Entmanganungs- und Entei
duren größtenteils zu Iodat oxidiert wurde.

Ergebnisse – Germaniumdioxid als interner Standard
- 109 -
4.5 Germaniumdioxid als interner Standard
4.5.1 Methode des internen Standards
Ein interner Standard (IS) ist eine Substanz, die in genau bekannter Menge jeder Probe und
jedem Kalibrationsstandard zugesetzt wird. Im Falle von Fließinjektions- oder chromatogra-
phischen Messungen kann der IS auch dem Eluenten zugegeben werden. Die Methode des
in ndards dient zur Korrektur nichtspez ischer Interferenzen 9 (s. Kap. 2.1.3).
gemessen. Zur Auswertung wird das Verhältnis der beiden Messsignale gebildet:
ternen Sta if
Die Signale des IS und des Analyten werden möglichst gleichzeitig oder kurz nacheinander
Gl. 22 IS
Analytkorrigiert Signal
SignalSignal =
Durch die Verhältnisbildung werden alle Störungen korrigiert, die einen Analyten und den IS
in gleichem Maße betreffen. Dies hat eine Verbesserung der Präzision der Messungen zur
Folge. Um eine erfolgreiche IS-Korrektur zu erreichen, muss ein IS diverse Vorraussetzungen
erfüllen, auf die in den nächsten Kapiteln ausführlich eingegangen wird.
4.5.2 Auswahlkriterien für interne Standards bei der IC-ICP-MS
Für die Ionenchromatographie ist es wichtig einen IS zu finden, der einerseits gut genug
wasserlöslich ist, aber andererseits nicht mit der stationären Phase in Wechselwirkung tritt.
zusetzen. Es resultiert ein kontinuierliches Messsignal, an m zu jeder Zeit die aktuelle
Leistungsfähigkeit des System lese n ka as fü Korrektur, Echtzeit-
quantifizierung und Fehlerübe id
Als allgemeine Voraus tzung g ss ei misch stabil sei Bei Nutzung in der
Ionenchromatographie llte de auße ht nau prozess teilnehmen,
damit er nicht von be nders h ode n ze n beeinflusst wird,
und da tze blockiert werden. Heutzutage gibt es eine sehr
Hat man eine entsprechende Verbindung gefunden, kann man den IS einfach dem Eluenten
de
s abge n werde nn, w r IS-
rwachung eal ist.
se ilt, da n IS che n s. mus
so r IS rdem nic am Ione stausch
so ohen r niedrige Ionenkon ntratione
mit durch ihn keine Austauscherplä
Wird der IS schon ganz am Anfang des analytischen Prozesses zugegeben, können damit auch Analytverluste
bei der Probenvorbereitung oder Ungleichmäßigkeiten bei der Probenaufgabe korrigiert werden. 9

Ergebnisse – Germaniumdioxid als interner Standard
- 110 -
auf
Grund besserer Trennleistung häufig verwendet werden, sind fast immer zwitterionisch
lwirkungen mit dem Material eingehen.
Weitere Aspekte werden in Kap. 4.5.4 erwähnt und zeigen, dass das Germanium (IV) in den
benutzten Eluenten neutral vorliegt und damit gut für alle Ionenchromatographiematerialien
geeignet ist.
Für die Detektion mit der ICP-MS muss ein geeigneter IS ebenfalls einige spezielle
Bedingungen erfüllen [143-146]. Eine wichtige Voraussetzung ist eine Ähnlichkeit der zu
detektierenden Elemente von den Analytspezies und vom IS, damit sich die Elementionen im
ICP-MS gleichartig verhalten. Bei einer IS-Korrektur geht man von der Annahme aus, dass
sich nichtspezifische Störungen auf die Signale der Analyten und des IS gleich auswirken.
Die Ähnlichkeit der Atommassen hat in diesem Zusammenhang den größten Einfluss, weil sie
die Flugbahn der Ionen im Massenspektrometer bestimmt. Die Masse unterscheidet sich
zwischen 74Germanium und 79Brom nur um fünf atomare Masseneinheiten, aber auch 127Iod
liegt noch in derselben Massenregion.
Eine andere wichtige Elementeigenschaft ist die erste Ionisierungsenergie (IE). Sie bestimmt
den Ionisierungsgrad der verschiedenen Elemente im Plasma. Bei sehr großen Differenzen der
Ionisierungsenergien des IS und der Analyten wäre z.B. ein unterschiedliches Verhalten bei
Temperaturänderungen zu erwarten. Brom und Iod haben mit 11.85 bzw. 10.46 eV fast die
höchsten ersten IEs aller mit der ICP-MS messbaren Elemente. Germanium liegt mit 7.89 eV
im oberen Drittel und lässt damit noch genug Ähnlichkeit erwarten.
Des Weiteren muss ein IS mindestens ein interferenzfreies Isotop besitzen. Germanium hat
fünf natürlich vorkommende Isotope. Die in Tab. 22 angegebenen möglichen isobaren Inter-
große Anzahl an unterschiedlich aufgebauten Säulen für die Anionenchromatographie. Einige
enthalten nur die für den Anionenaustausch benötigten kationischen funktionellen Gruppen,
weshalb in einem solchen Fall neutrale aber auch positiv geladene ISs verwendet werden
können, da sie keine Wechselwirkungen eingehen. Agglomerierte Ionenaustauscher, die
aufgebaut. Sie bestehen meist aus kugelförmigen PS/DVB-Trägermaterialien, die sulfoniert
sind, um über elektrostatische Wechselwirkungen kleinere Latexpartikel auf der Oberfläche
zu fixieren (s. Kap. 4.1.2). Die Latexpartikel tragen die eigentlichen, positiv geladenen
Anionenaustauschergruppen. Für solche Säulen, bei denen Ammonium- und Sulfonatgruppen
nebeneinander vorliegen, werden zwingend neutrale ISs benötigt, damit sie vor der Säule
zugegeben werden können und keine Wechse

Ergebnisse – Germaniumdioxid als interner Standard
- 111 -
ferenzen sind zwar zahlreich, wurden aber bei der Analyse typischer Wasserproben fast nie
uftrennung werden einige mögliche
Interferenzen eliminiert. Meistens kann auf dem Isotop der Masse 74 u, welches die größte
werden. Die möglichen Interferenzen sollten aber
Verhältnis 76 ist zwar durch [40Ar36Ar]+ erhöht, aber es gibt keine Eisen- und nur eine seltene
Tab. 22: Mögliche Interferenzen für Germanium. In runden Klammern ist die Häufigkeit bzw. das Produkt der n ange ben. Bildungsraten für Molekülionen sind dabei nicht berücksichtigt.
beobachtet. Durch die ionenchromatographische A
Häufigkeit aufweist, problemlos gemessen
stets im Auge behalten werden, denn z.B. bei sehr chlorid- oder eisenhaltigen Proben sind
störende Molekülionen zu erwarten. Diese bleiben durch die vorgeschaltete ionenchromato-
graphische Trennung zeitlich begrenzt. Wenn trotzdem eine Interferenz vorliegt, kann z.B. auf
das Isotop 76Germanium ausgewichen werden. Der Untergrund auf dem Masse-zu-Ladungs-
Chlorinterferenz. Durch die vielen Germaniumisotope ist eine große Flexibilität gegeben.
Häufigkeite ge
Isotop Mögliche Interferenzen
70Ge (20.5 %) [40ArNO]+ (99.0 %), [35Cl2]+ (57.4 %), 70Zn+ (0.6 %), [54CrO]+ (2.36 %), [54FeO]+ (5.79 %), [56FeN]+ (91.4 %), (La, Ce, Pr)2+
72Ge (27.4 %) [36Ar2]+ (0.0011 %), [35Cl37Cl]+ (18.4 %), [40Ar32S]+ (94.6 %) [56FeO[
]+ (91.5 %), 2 )2+
73Ge (7.8 %) [36Ar2H]+ (1.1·10−3 %), [40Ar32SH]+ (94.6 %), [57FeO]+ (2.19 %), [56FeOH]+ (91.5 %), [36Ar37Cl]+ (0.08 %), [38Ar35Cl]+ (0.05 %), [43CaO2]+ (0.13 %), (Nd, Sm)2+
3 (68.1 %), [ FeO] (0.28 %), [ NiN] (26.0 %), [ K Cl] (70.7 %), [ CaO ]+ (0.64 %), (Sm, Nd)2+
58NiN]+ (68.0 %) [55MnOH]+ (99.7 %), [40CaO ]+ (96.5 %), (Nd, Sm
74Ge ( 6.5 %) [36Ar38Ar]+ (2.1·10−4 %), 74Se+ (0.9 %), [37Cl2]+ (5.87 %), [58NiO]+
58 + 60 + 39 35 + 422
76Ge (7.8 %) [40Ar36Ar]+ (0.34 %), [38Ar2]+ (4.0·10−5 %), 76Se+ (9.0 %), [75AsH]+ (99.99 %), [60NiO]+ (26.0 %), [39K37Cl]+ (22.6 %), [44CaO2]+ (2.1 %), (Eu, Sm, Gd)2+
Das Element, das als IS zugegeben werden soll, darf in den zu analysierenden Proben auf
keinen Fall schon vorher vorhanden sein, da dies die Korrektur verschlechtern oder
verhindern könnte. Deswegen sollte das IS-Element möglichst selten sein. Dies ist bei
Germanium der Fall. Es ist zwar weltweit verbreitet, aber nur in sehr geringen Konzen-
trationen. Der Gesamtanteil in der äußeren 16 km dicken Erdkruste wird auf etwa 0.00056 %
geschätzt [127]. Germanium kommt in höheren Konzentrationen in der Natur gewöhnlich nur
als Begleiter von Kupfer und Zinkerzen oder in einigen Kohlen beziehungsweise angereichert
in deren Flugaschen vor.

Ergebnisse – Germaniumdioxid als interner Standard
- 112 -
echseln
der Massen braucht, die Dauer eines Durchlaufs (sweep o. run). Bei normalen nicht zeit-
lösten (transienten) Messmodus wird dagegen die Zählrate jedes einzelnen
chend schnelles Umstellen des Quadrupols von einer
Masse zur nächsten sind vorteilhaft, weil durch die zeitnahe Verhältnisbildung auch
.
4.5.3 Transiente Messungen und zeitliche Aspekte der IS-Korrektur
Mit einem Quadrupol können der IS und die Analyten nicht gleichzeitig, wie z.B. mit einem
Time-of-Flight- oder Multikollektoranalysator gemessen werden. Es ist aber möglich, Ionen
verschiedener Masse im Abstand von Millisekunden zu detektieren, oder den gesamten sinn-
vollen analytischen Massenbereich von 5–255 u in nur 0.1 s zu scannen. Deswegen spricht
man bei der Quadrupol-MS von einer quasisimultanen Detektion.
In so genannten Elementmenüs werden die zu detektierenden Elemente, die Anzahl der Mess-
positionen pro Massenpeak und die Messzeit für jede Messposition (dwell time) festgelegt.
Daraus ergibt sich nach Addition der Beruhigungszeit, die der Quadrupol für das W
aufgelösten Messungen werden die Zählereignisse aller einzelnen Durchläufe während einer
gewählten Gesamtmesszeit (acquisition time) addiert, um so die Zählstatistik zu verbessern.
Im zeitaufge
Durchlaufs gegen die Zeit aufgetragen. Ein Durchlauf wird in dieser Betriebsweise als
Zeitscheibe bezeichnet.
Bei der Wahl der Messzeiten pro Element ergeben sich gegenläufige Trends, was die
Präzision der Messungen angeht [147-149]. Sowohl zu lange als auch zu kurze Messzeiten
können die Qualität von Verhältnismessungen verschlechtern. Dies betrifft insbesondere die
IS-Korrektur und die Bestimmung von Isotopenverhältnissen. Die folgenden Aspekte müssen
beachtet und eine Kompromisseinstellung gefunden werden:
• Kurze Zeitscheiben und entspre
die Korrektur von kurzfristigen Signalschwankungen ermöglicht und somit die
Präzision erhöht wird.
• Bei häufigem Wechsel des Masse-zu-Ladungs-Verhältnisses geht allerdings Messzeit
verloren, da ein Quadrupol 3–10 ms Stabilisierungszeit braucht, um ein neues Ver-
hältnis einzustellen. Zu viele schnelle Sprünge mit dem Quadrupol verringern daher
die effektive Messzeit, wodurch die Präzision schlechter wird
• Die Anzahl der Elemente soll möglichst hoch sein, um die Multielementfähigkeit,
einen der großen Vorteile des ICP-MS, auszunutzen. Dies verkürzt aber die effektive

Ergebnisse – Germaniumdioxid als interner Standard
- 113 -
nals
verantwortlich sind, unterschieden. Ein Rauschanteil wird als Flacker- oder Flimmerrauschen
Zerstäubungsunterschiede oder Pumpenpulsationen, und es ist unabhängig von der
Konzentration. Bei simultaner Messung mehrerer Elemente lässt sich dieser Rauschanteil
d eei
D aus lstatistik“
(Poisson-Statistik) bezeichnet.
gem Sig ufälliges Auf-
t lsc Stande. Der
Einfluss der Zählstatistik lässt
A n b en durch die
Zählstatistik 1000½ = 31.6 counts und somit einer relativen Standardabweichung von 3.16 %.
verwendet wurde. Mit diesem Menü wird immer nur auf einer
osition pro Masse gemessen. Die Messzeiten für die Masse-zu-Ladungs-Verhältnisse sind im
Vergleich zu den Quadrupolwartezeiten lang. Dadurch wird die Gesamtmesszeit möglichst
effektiv ausgenutzt, was sich vorteilhaft auf die Präzision durch die Zählstatistik auswirkt. Die
gewählte Verweildauer auf jeder Masse hängt von der Anzahl der zu detektierenden
Elemente, der Empfindlichkeit mit der sie detektiert werden können und von ihrer erwarteten
Messzeit pro Element und vergrößert die Zeit zwischen der Messung des IS und den
Analyten.
• Die Dauer einer Zeitscheibe darf nicht zu lange sein, damit genügend zeitliche
Auflösung der transienten Messungen gewährleistet ist. Bei typischen ionenchromato-
graphischen Bedingungen in dieser Arbeit ist eine Zeitscheibenlänge bis maximal 1.5 s
geeignet, da dann auch noch der schmalste transiente Peak aus ca. 15 Datenpunkten
besteht.
Es werden vor allem zwei Ursachen, die für das kurzfristige Rauschen eines ICP-Sig
bezeichnet. Es kommt hauptsächlich durch die Probenzuführung zu Stande, z.B. durch kleine
urch einen g gneten IS korrigieren.
er zweite R chanteil ist von fundamentaler Art und wird als „Einfluss der Zäh
Dieses Rauschen ist direkt proportional zur Quadratwurzel des
nals und damit konzentrationsabhängig. Es kommt durch zessenen
reffen von fa hen Teilchen (Photonen, Elektronen, Ionen) auf den Detektor zu
sich an einem einfachen Rechenbeispiel verdeutlichen:
ei einem Signal von 1000 counts entspricht das Rauschngenomme
Die relative Standardabweichung bei gleicher Rechnung für ein Signal von 10000 counts
beträgt dann nur noch 1 %. Der Rauschanteil durch die Zählstatistik ist nicht korrigierbar und
er wird umso höher, je niedriger die Analytkonzentration oder die Messdauer ist.
Für die ionenchromatographischen Messungen in dieser Arbeit wurden gewöhnlich zwischen
40 und 400 ms Messzeit pro Element eingestellt. Als Beispiel ist in Abb. 50 die Aufteilung
der Messzeit auf die vier Massenpositionen in einer Zeitscheibe des Elementmenüs Nr. 11
gezeigt, welches häufig
P

Ergebnisse – Germaniumdioxid als interner Standard
- 114 -
oder LA-ICP-MS
ür einige
ber 0.5–5 min ermittelt, und zwar an einer Position, an der das Signal
möglichst stabil war. Die Analytpeakflächen wurden mit der so ermittelten IS-Signalhöhe ins
Konzentration ab. Bei anderen transienten Messmethoden wie z.B. GC-
werden wesentlich kürzere Signale erzeugt. Deshalb müssen bei diesen Methoden kleinere
Zeitscheiben programmiert werden.
Die beiden bisher vorgestellten Einflüsse auf das Signalrauschen sind die Hauptursache der
typischen Kurzzeitmessunsicherheit von 1–2 %, die bei modernen ungekoppelten ICP-MS-
Geräten im µg L−1-Bereich erreicht wird. Diese Unsicherheit ist zwar zu groß f
Anwendungen bei Isotopenverhältnismessungen, aber bei Chromatographiekopplungs-
methoden fällt sie nicht so stark ins Gewicht, denn dort ist die zusätzliche Langzeit-
messunsicherheit durch die chromatographische Trennung meist höher. Die gesamte relative
Standardabweichung liegt bei der IC-ICP-MS etwa bei 7–15 % und mit Verwendung eines
internen Standards bei 5–10 %. Eine Korrektur von Kurzzeitschwankungen ist zwar
anzustreben, die Berichtigung von längerfristigen Effekten, die zu Empfindlichkeits-
schwankungen oder zu einer Drift im Laufe eines Messtages führen, hat aber höhere Priorität.
Dazu wurde in dieser Arbeit bei jeder transienten Messung ein bis zweimal der Mittelwert der
Germaniumsignalhöhe ü
Verhältnis gesetzt, was dazu führte, dass alle längerfristigen Signaländerungen innerhalb von
Messreihen oder im Laufe von verschiedenen Messtagen ausgeglichen wurden. Durch diese
Langzeit-IS-Korrektur mit sicherer visueller Ermittlung der Germaniumsignalhöhe wurden
Analyt-zu-GeO2-Verhältnisse erhalten, die trotz verschiedensten Messbedingungen über
Wochen und Monate stabil blieben.
0 100 200 300 400 500 600 700 800 900
200 ms 127I400 ms 79Br00 ms m/z 52
Zeit / ms
40 ms74Ge
: Messzeiten für die vier vers
2
Abb. 50 chiedenen m/z in einer Zeitscheibe von Elementmenü 11. (Die dunkel-grau Zo
Für ein
leichter
Zeitsch hen im Millisekunden-
ber h
der Qu
zwischen der Messung des IS 74Germanium und 79Brom nur etwa 50 ms, bis zum 127Iod sind
en nen sind die Stabilisierungszeiten des Quadrupols.)
e IS-Korrektur von Schwankungen im Sekunden- bis Minutenbereich und für eine
e Automatisierung müsste das Signalverhältnis von Analyt-zu-GeO2 in jeder einzelnen
eibe eines Chromatogramms gebildet werden. Das Flackerrausc
eic kann aber nicht oder nur schlecht korrigiert werden, da der IS und die Analyten mit
adrupoltechnik nicht simultan gemessen werden. Wie in Abb. 50 erkennbar, liegen

Ergebnisse – Germaniumdioxid als interner Standard
- 115 -
es imm
erwarten als für Iod. Diese mit dem zeitlichen Abstand größer werdende Messungenauigkeit,
die e
„precis
gern, w
IS zu m
Zeitbereich wären besser auszugleichen.
gt noch an. Die starke Signalveränderung zu diesem
Zeitpunkt macht eine Echtzeitkorrektur mit Hilfe des IS im Eluenten unmöglich. Stattdessen
erhin schon 420 ms. Deswegen ist für Brom eine etwas bessere IS-Korrektur zu
b i allen scannenden Massenspektrometern auftritt, wird als „spectral skew“ oder
ion skew“ (skew: engl. = Schieflage) bezeichnet [150]. Den Effekt könnte man verrin-
enn es möglich wäre, mit einem Quadrupol innerhalb einer Zeitscheibe wiederholt den
essen. Dann wäre die Korrektur zeitnaher und auch Flackerschwankungen in diesem
Auch die Verhältnisbildung des Analyt- und des IS-Signals aller einzelnen Zeitscheiben
wurde in dieser Arbeit getestet, aber es kam dabei zu Komplikationen bei der Korrektur der
Iodatsignale. Iodat eluiert bei der Anionenchromatographie meist direkt mit dem Totzeitpeak.
Es erreicht mit dem injizierten Wasser und damit teilweise noch vor dem Germanium aus dem
Eluenten den Detektor. Während der Iodatelution hat sich das Germaniumsignal noch nicht
wieder vollständig stabilisiert und stei
muss man den Wert des Germaniumsignals an einer etwas späteren Position im Chromato-
gramm nutzen. Für die Korrektur von Iodat hätte eine Nachsäulenzugabe des IS Vorteile, da
dort kein Injektionspeak in der Signalspur des IS aufträte (s. Kap. 4.1.1, Seite 56). Als weitere
Möglichkeit ließe sich durch Verringerung der Eluentstärke die Auflösung von Injektions-
und Iodatpeak vergrößern, aber dies hat meist zur Folge das stark retardierte Analyten nicht
mehr innerhalb von 40 min von der Säule eluiert werden. Eine bessere Lösung sind Säulen-
materialien, die selektiv die Retention von Iodat erhöhen (s. Kap. 4.1.6).
4.5.4 Vergleich dreier potentieller ISs und Evaluierung der Ladung
Drei als IS in Frage kommende Verbindungen wurden auf ihre Wechselwirkung mit einem
typischen agglomerierten Anionenaustauscher hin untersucht: Borsäure (pKs1 9.25), Germa-
niumdioxid und Tellur(IV)-Lösung 10. Die Anhydride Germaniumdioxid und Tellurdioxid
können in Lösung in die korrespondierenden Säuren Germaniumsäure (pKs1 9.01) bzw.
Tellurige Säure (pKs1 2.48) überführt werden. In den verwendeten Ammoniumnitrat-Eluenten
mit einem pH-Wert von vier bis sechs würden die beiden sehr schwachen Säuren gemäß ihren 10 Die verwendete Tellurlösung war leider nicht gut definiert. Es ist nicht auszuschließen, dass auch Te (VI) vorlag. Ortho-Tellursäure H6TeO6 ist ebenfalls gut wasserlöslich und mit einem pKs1 von 7.7 eine schwache Säure. Sie würde im Eluenten in protonierter Form vorliegen. Im Sauren ist sie aber ein sehr starkes Oxidations-mittel und würde schnell zu Tellur(IV) abreagieren.
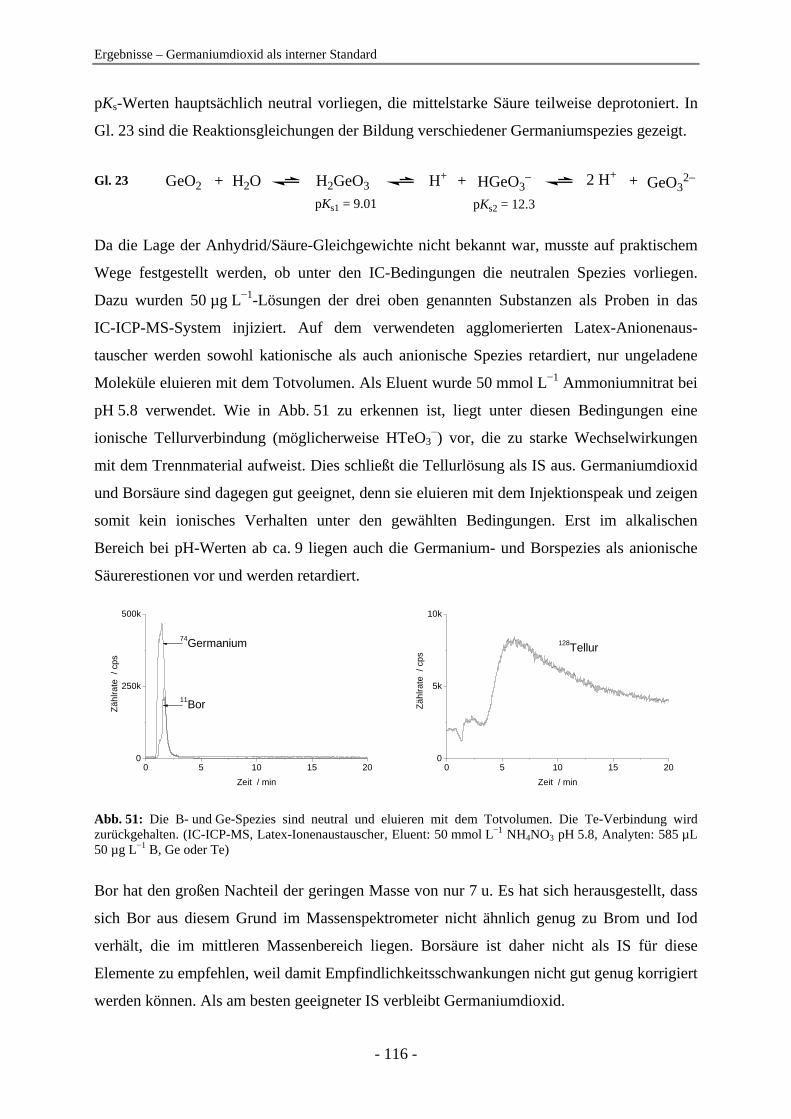
Ergebnisse – Germaniumdioxid als interner Standard
pKs-Werten hauptsächlich neutral vorliegen, die mittelstarke Säure teilweise deprotoniert. In
Gl. 23 sind die Reaktionsgleichungen der Bildung verschiedener Germaniumspezies gezeigt.
- 116 -
Gl. 23
Da die Lage der Anhydrid/Säure-Gleichgewichte nicht bekannt war, musste auf praktischem
Wege festgestellt werden, ob unter den IC-Bedingungen die neutralen Spezies vorliegen.
Dazu wurden 50 µg L−1-Lösungen der drei oben genannten Substanzen als Proben in das
IC-ICP-MS-System injiziert. Auf dem verwendeten agglomerierten Latex-Anionenaus-
tauscher werden sowohl kationische als auch anionische Spezies retardiert, nur ungeladene
Moleküle eluieren mit dem Totvolumen. Als Eluent wurde 50 mmol L−1 Ammoniumnitrat bei
pH 5.8 verwendet. Wie in Abb. 51 zu erkennen ist, liegt unter diesen Bedingungen eine
ionische Tellurverbindung (möglicherweise HTeO3−) vor, die zu starke Wechselwirkungen
mit dem Trennmaterial aufweist. Dies schließt die Tellurlösung als IS aus. Germaniumdioxid
und Borsäure sind dagegen gut geeignet, denn sie eluieren mit dem Injektionspeak und zeigen
somit kein ionisches Verhalten unter den gewählten Bedingungen. Erst im alkalischen
Bereich bei pH-Werten ab ca. 9 liegen auch die Germanium- und Borspezies als anionische
Säurerestionen vor und werden retardiert.
500k
0 5 10 15 200
250k11BorZä
hlra
te /
cps
Zeit / min
74Germanium
10k
0 5 10 15 200
5k
Zähl
rate
/ c
p
Abb. 51: Die B- und Ge-Spezies sind neutral und eluieren mit dem Totvolumen. Die Te-Verbindung wird
−1
werden können. Als am besten geeigneter IS verbleibt Germaniumdioxid.
2−2 H+HGeO3−H+H2GeO3H2OGeO2 ++ +
s
128Tellur
3pKs1 = 9.01 pKs2 = 12.3
GeO
Zeit / min
zurückgehalten. (IC-ICP-MS, Latex-Ionenaustauscher, Eluent: 50 mmol L NH4NO3 pH 5.8, Analyten: 585 µL 50 µg L−1 B, Ge oder Te)
Bor hat den großen Nachteil der geringen Masse von nur 7 u. Es hat sich herausgestellt, dass
sich Bor aus diesem Grund im Massenspektrometer nicht ähnlich genug zu Brom und Iod
verhält, die im mittleren Massenbereich liegen. Borsäure ist daher nicht als IS für diese
Elemente zu empfehlen, weil damit Empfindlichkeitsschwankungen nicht gut genug korrigiert

Ergebnisse – Germaniumdioxid als interner Standard
- 117 -
Am Beispiel der IC-ICP-MS-Kopplung erklärt, ist ein gepulster IS ein Ion, das in die Proben
uss, sind in
ap. 4.5.2 beschrieben. Die Vorsäulenzugabe in den Eluenten macht kaum Arbeitsaufwand
und es wird kein weiteres technisches Zubehör benötigt. Nur auf diese Art unterliegt der IS
indlichkeitsschwan-
nützliche Monitorfunktion. Wenn eine Messung normal verläuft, kann ein konstantes Signal
4.5.5 Zuführungsmethoden des internen Standards bei transienten Messungen
Bei transienten Messungen sind verschiedene Zugabemethoden für ISs möglich. Transiente
Signale erhält man z.B. bei Laserablation, elektrothermaler Verdampfung, Fließinjektions-
analyse, Chromatographie u.a. Man unterscheidet zwischen gepulster (diskreter) oder
kontinuierlicher IS-Zugabe [151] sowie bei Verwendung von Säulen zwischen Vor- und
Nachsäulenaddition.
und Standards zugegeben wird und dann als einzelner Peak im Chromatogramm erscheint.
Dagegen wird ein kontinuierlicher IS entweder durch Vorsäulenaddition dem Eluenten zu-
gesetzt oder erst durch Nachsäulenzugabe über eine weitere Pumpe und ein T-Stück in den
Eluentenstrom eingemischt. Kontinuierliche ISs haben den Vorteil, dass sie während der
ganzen Messzeit ein Signal liefern, so dass eine Systembeobachtung und die IS-Korrektur
zeitnah durchgeführt werden können. Ein positiver Aspekt der Nachsäulenzugabe ist, dass
eine große Anzahl an möglichen ISs zu Verfügung steht, da hierfür keine chromato-
graphischen Einschränkungen beachtet werden müssen. Nachteilig zu bewerten ist die
Notwendigkeit eines Mischungsmoduls und einer zusätzlichen Pumpe sowie dass sehr auf
konstante Pumpgeschwindigkeiten und Mischungsverhältnisse geachtet werden muss, um ein
stabiles Signal zu erhalten. Die Abnutzung von Peristaltikpumpenschläuchen ist z.B. eine
typische Fehlerquelle. Generell erhöht sich auch bei gut funktionierenden Systemen durch den
Mischungsvorgang das Signalrauschen. Dies kann durch Vorsäulenzugabe des IS verhindert
werden. Die Voraussetzungen, die ein IS für die Ionenchromatographie erfüllen m
K
genau den gleichen Bedingungen wie die Analyten und kann auch Empf
kungen, die z.B. durch die HPLC-Pumpe zu Stande kommen, anzeigen und korrigieren.
4.5.6 Germaniumdioxid zur Überwachung instrumenteller Probleme oder nicht-
spezifischer Interferenzen
Das kontinuierliche Signal des IS Germaniumdioxid bietet schon während der Messung eine
beobachtet werden, das nur durch den negativen Wasserpeak der Probeninjektion unter-
brochen wird (s. z.B. Abb. 55). Die Höhe und das Rauschen des Germaniumsignals (und des
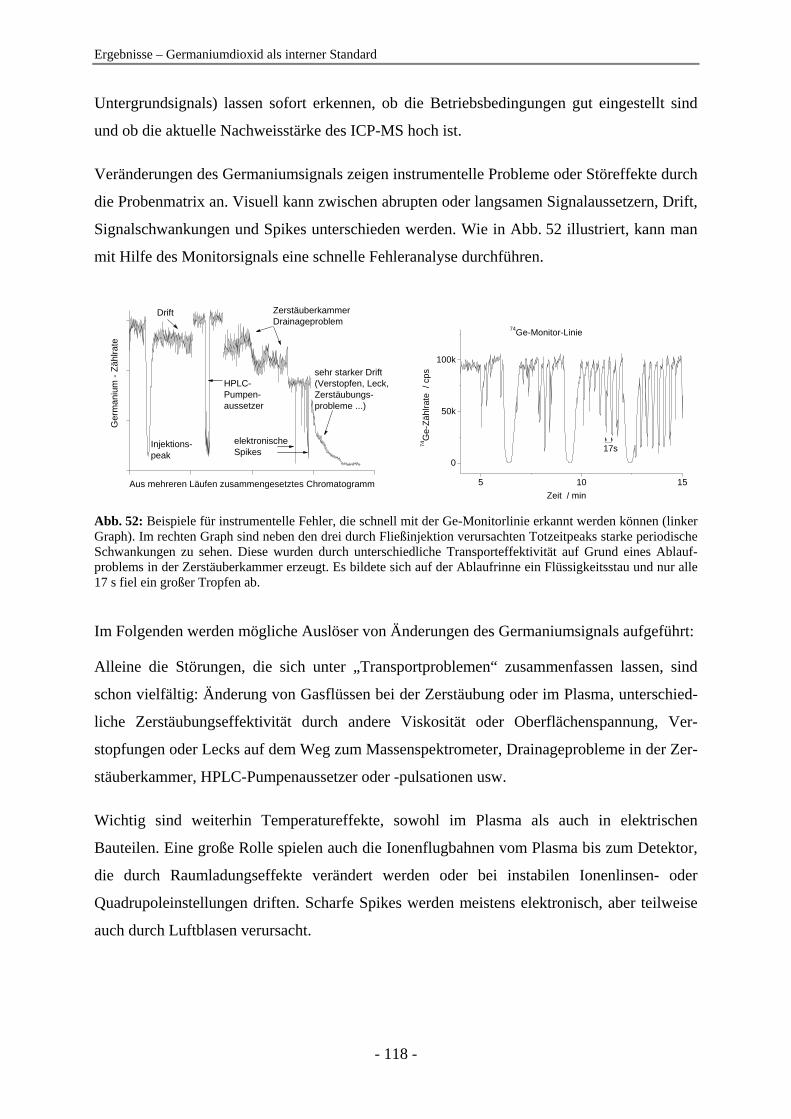
Ergebnisse – Germaniumdioxid als interner Standard
Untergrundsignals) lassen sofort erkennen, ob die Betriebsbedingungen gut eingestellt sind
und ob die aktuelle Nachweisstärke des ICP-MS hoch ist.
- 118 -
erungen des Germaniumsignals zeigen instrumentelle Probleme oder Störeffekte durch
die Probenmatrix an. Visuell kann zwischen abrupten oder langsamen Signalaussetzern, Drift,
Signalschwankungen und Spikes unterschieden werden. Wie in Abb. 52 illustriert, kann man
Veränd
mit Hilfe des Monitorsignals eine schnelle Fehleranalyse durchführen.
Ger
man
ium
- Zä
hlra
te
Aus mehreren Läufen zusammengesetztes Chromatogramm
Injektions-peak
Drift
HPLC-Pumpen-aussetzer
ZerstäuberkammerDrainageproblem
elektronischeSpikes
sehr starker Drift(Verstopfen, Leck,Zerstäubungs-probleme ...)
5 10 15
0
50k
100k
74G
e-Zä
hlra
te /
cps
Zeit / min
74Ge-Monitor-Linie
17s
Abb. 52: Beispiele für instrumentelle Fehler, die schnell mit der Ge-Monitorlinie erkannt werden können (linker Graph). Im rechten Graph sind neben den drei durch Fließinjektion verursachten Totzeitpeaks starke periodische Schwankungen zu sehen. Diese wurden durch unterschiedliche Transporteffektivität auf Grund eines Ablauf-problems in der Zerstäuberkammer erzeugt. Es bild17 s fiel ein großer Tropfen ab.
ete sich auf der Ablaufrinne ein Flüssigkeitsstau und nur alle
Im Folgenden werden mögliche Auslöser von Änderungen des Germaniumsignals aufgeführt:
Alleine die Störungen, die sich unter „Transportproblemen“ zusammenfassen lassen, sind
schon vielfältig: Änderung von Gasflüssen bei der Zerstäubung oder im Plasma, unterschied-
liche Zerstäubungseffektivität durch andere Viskosität oder Oberflächenspannung, Ver-
stopfungen oder Lecks auf dem Weg zum Massenspektrometer, Drainageprobleme in der Zer-
stäuberkammer, HPLC-Pumpenaussetzer oder -pulsationen usw.
die durch Raumladungseffekte verändert werden oder bei instabilen Ionenlinsen- oder
Wichtig sind weiterhin Temperatureffekte, sowohl im Plasma als auch in elektrischen
Bauteilen. Eine große Rolle spielen auch die Ionenflugbahnen vom Plasma bis zum Detektor,
Quadrupoleinstellungen driften. Scharfe Spikes werden meistens elektronisch, aber teilweise
auch durch Luftblasen verursacht.

Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 119 -
: ratio). Wird R auf ein festes molares Verhältnis normiert, erhält es die
Bezeichnung Rn. Dazu bezieht man sich auf eine bestimmte Konzentration des
4.6 Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
4.6.1 Berechnung des Rn-Werts
Das Verhältnis von Analytpeakfläche und Germaniumsignalhöhe gemäß Gl. 24 wird als R
bezeichnet (von engl.
Analyten (10 µg L−1), des Germaniums (15 µg L−1) sowie auf ein Probenschleifenvolumen
von 100 µL (Gl. 25). Werden die Konzentration des IS und das Probenschleifenvolumen für
alle Messungen konstant gehalten, sind die Normierungsterme nicht nötig. Messungen, bei
denen die aufgegebenen Stoffmengen des Analyten und des Germaniums unterschiedlich
waren, können durch die Normierung gut verglichen werden. Die Rn-Werte sind dann nur
noch von instrumentellen Parametern abhängig. Eine Abhängigkeit des Verhältniswerts tritt
aber nur auf, wenn sich die Empfindlichkeiten für Analyt und Germanium unterschiedlich
ändern, ansonsten bleibt das Verhältnis konstant.
Gl. 24 Ge
A
SignalhöhePeakfläche
=R
Gl. 25 c
c
V R R
-
-k A,
1
1Ge
sn
L µg10 L µg15
µL 100⋅⋅⋅=
Gl. 26 11
Ge
sn xA, L µg 10
L µg15µL 100 -
- c
V
RRc ⋅⋅⋅=
R: Analyt-zu-GeO2-Verhältnis
Rn: R normiert auf ein festes molares Verhältnis von Analyt zu Germanium und bei einer
Eluentflussrate von 1 mL min−1. Der Wert ist bei konstanter Flussrate langzeitstabil und
bekannt (oder wird durch einmalige Messung ermittelt).
A: Analyt = BrO3−, Br−, IO3
− o. I−
ion (Probe)
Vs: Volumen der Probenschleife
cA, k: bekannte Konzentration (Standard)
cA, x: unbekannte Konzentrat
Durch die in Kap. 4.6.2 beschriebene Langzeitstabilität, können die Rn-Werte als Bezugspunkt
für die Konzentrationsbestimmung nach Gl. 26 genutzt werden. Rn-Werte stellen weiterhin ein
Gütemaß für die Korrektur durch den internen Standard dar: Bleibt Rn bei Empfindlichkeits-

Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 120 -
Parametervariationen, wie in sie in
für Kap. 4.7 durchgeführt wurden.
geführt. Dabei ließ sich beobachten, dass das gemessene Verhältnis Rn von Analytpeakfläche
zur Signalhöhe Germaniums bei normierten Molmengen und konstanter Eluentflussrate sehr
stabil bleibt. Die Analyten wurden in verschiedenen wässrigen Matrices und über einen sehr
großen Konzentrationsbereich von 0.5–1000 µg L−1 bestimmt. Standards in Reinstwasser lie-
ferten die gleichen Rn-Werte wie Mineralwasserproben mit sehr unterschiedlichen Salzgehal-
ten. Es wurden mehrere Arten von Säulenmaterialien benutzt oder im Falle der Fließinjek-
tionsanalyse auf eine Trennsäule verzichtet. Die Rn-Werte blieben trotz Veränderung von
effektivität oder der Massenempfindlichkeit, ist eine gute Korrelation der Signale gegeben.
n
te darf als gering angesehen
auch besonders Schwankungen durch die Peak-
ration relevant.
änderungen stabil, ist eine gute IS-Korrektur gegeben. Signaländerungen können durch echte
Interferenzen zu Stande kommen oder durch beabsichtigte
4.6.2 Langzeitstabilität der Rn-Werte
Es wurde eine große Anzahl an Messungen von Bromid, Bromat, Iodid und Iodat durch-
Massenspektrometereinstellungen oder Wechseln von Cones, Zerstäubern, Torches und
anderen Glasteilen stabil. Dies bedeutet, dass sich Brom, Iod und Germanium im ICP-MS
ähnlich verhalten. Auch bei Änderung von elementspezifischen Größen, wie der Ionisierungs-
In Abb. 53 ist der zeitliche Verlauf der R -Werte über einen Zeitraum von 36 Monaten
dargestellt. Gut lässt sich die Langzeitstabilität und die Streuung der Ergebnisse um einen
Mittelwert erkennen. Größere Änderungen und Ausnahmen durch außergewöhnliche Vor-
kommnisse sind mit eingekreisten Ziffern gekennzeichnet und werden im Folgenden
detaillierter beschrieben. Untersuchungen über die Abhängigkeiten von Rn von bestimmten
Parametern sind in Kap. 4.7 zu finden. Die Streuung der Rn-Wer
werden, wenn man bedenkt, dass es sich um einen sehr langen Aquisitionszeitraum und um
Messungen sehr verschiedener Proben unter variierenden Bedingungen handelt. Außerdem
sind die Daten in Abb. 53 nicht ausreißerkorrigiert. Einzelne ausreißende Punkte resultieren
meistens aus sehr geringen Konzentrationen unter 1 µg L−1, bei denen eine große Messun-
sicherheit zu erwarten ist. Hier werden
integ
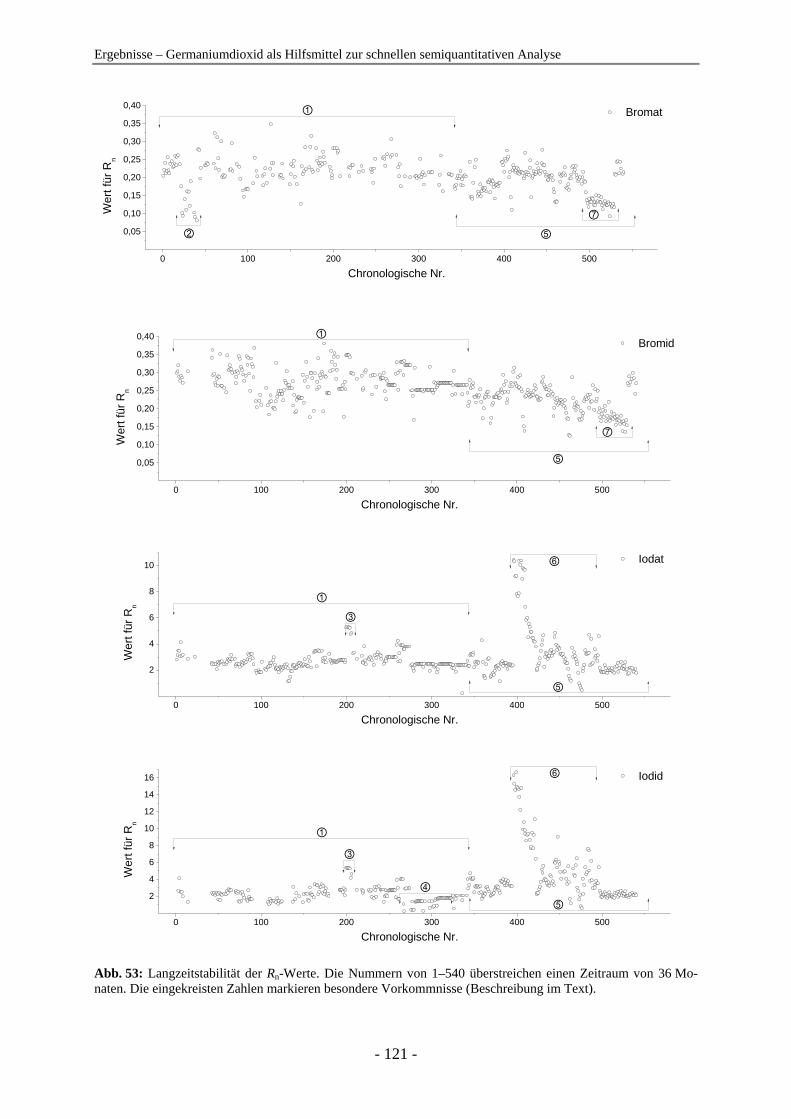
Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 121 -
0,25
0,
0,
0,40
0 100 200 300 400 500
0,05
0,10
1
30
35 Bromat
nW
ert f
ür R
0,20
0,15
5
Chronologische Nr.
2
7
0 100 200
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
300 400 500
Bromid
Wer
t für
Rn
Chronologische Nr.
5
7
1
8
10
0 100 200 300 400 500
2
4
6
6 Iodat
Wer
tn
Chronologische Nr.
5
3
1
für R
0 100 200 300 400 500
2
4
36
8
10
12
14
16 Iodid
Wer
t
4
für R
n
Chronologische Nr.
5
6
1
Abb. 53: Langzeitstabilität der Rn-Werte. Die Nummern von 1–540 überstreichen einen Zeitraum von 36 Mo-naten. Die eingekreisten Zahlen markieren besondere Vorkommnisse (Beschreibung im Text).

Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 122 -
1 Im Zeitraum von März 2003 bis Juni 2004 wurden keine Fließinjektionsanalysen
sondern nur IC-ICP-MS-Messungen und durchgeführt. Die Rn-Werte betragen für
0.28 ± 0.040, für Iodat 2.7 ± 0.66 und für Iodid
a
um ein Viertel größer sein als für Iodat, da Iodat nur einen zu detektierenden Iodanteil
speziellen Tuningbedingungen oder Komponenten gelegen haben, denn
nach leichter Modifikation der Bauteile und Neutuning wurden auch an diesem
ICP-MS Rn-Werte im Normalbereich gemessen.
Beschreibung spezieller Zeiträume und Ausnahmen von der Langzeitstabilität in Abb. 53:
Bromat 0.22 ± 0.043, für Bromid
2.3 ± 1.1. Die relativen Standardabweichungen sind mit 19, 14 und 24 % bei den
ersten drei Anionen viel besser als für das Iodid mit 48 %. (Die unterschiedliche
Skalierung kann in den Graphen für Iod eine scheinbar bessere Stabilität vortäuschen.)
Die hohe relative Standardabweichung macht die typischen Probleme bei der Messung
des stark polarisierbaren, weichen Anions Iodid deutlich. Bei Verwendung von nicht
optimalen Ionenaustauschern wird die Peakform des Iodids durch sekundäre Wechsel-
wirkungen verschlechtert, und die Peakflächenbestimmung wird dadurch sehr
ungenau. Außerdem kann bei sehr kleinen Iodidkonzentrationen ein erheblicher Anteil
nicht mehr detektiert werden, woraus sich zu geringe Rn-Werte ergeben. Dies ist z.B.
im Bereich 4 der Fall, und die Werte sind als zu niedrige Ausreißer zu betrachten.
Auch nach dem Eliminieren von Ausreißern bleibt der Iodid-Rn-Wert kleiner als der
für das Iodat. Nach der theoretischen Berechnung müsste Rn für Iodid eigentlich etw
von 73 % enthält. Dies zeigt, dass die Wiederfindung bei der IC-ICP-MS für Iodid
geringer ist als für Iodat.
2 Bei diesen Messungen handelt es sich um Bromatbestimmungen in Salzen. Die
Rn-Werte sind auffällig niedrig (Ausreißer). Hier wurden sehr niedrige Konzen-
trationen von 0.1–5 µg L−1 in schwieriger Matrix gemessen. Die Proben wiesen einen
Anteil von bis zu 1 % an gelösten Feststoffen auf (TDS).
3 Diese Messungen wurden an einem anderen ICP-MS-Gerät durchgeführt: einem
X7-Modell der Firma Thermo (Winsford, U.K.) mit Kollisionszelle, Meinhard-
Zerstäuber, Xi-Cone und bei einer Plasmaleistung von 1500 W. Der genaue Grund für
die ungewöhnlich hohen Rn-Werte der Iodspezies konnte nicht ermittelt werden. Es
muss aber an

Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 123 -
4 Hier wurde eine Säule verwendet, die defekt war bzw. die das Ende ihrer Lebensdauer
erreicht hatte. Die Peaks des Iodids waren extrem flach und wiesen ein starkes Tailing
auf. Die ermittelten Rn-Werte für Iodid sind zu niedrig und als Ausreißer zu werten,
weil nicht das gesamte aufgegebene Iod erfasst wurde.
5 Im Zeitraum von Juli 2004 bis März 2005 wurden größtenteils FI-ICP-MS-Messungen
durchgeführt. Im Bereich 6 kam es zu einer Kontamination des ICP-MS Geräts mit
Iod. Die Rn-Werte (ohne Ausreißer) bei der FI-ICP-MS liegen mit 0.21 ± 0.028 für
Bromat, 0.24 ± 0.031 für Bromid, 2.3 ± 0.57 für Iodat tendenziell etwas niedriger als
bei der IC-ICP-MS-Kopplung. Im Gegensatz zu den ionenchromatographischen
Messungen ist der Rn für Iodid aber bei der Fließinjektion mit 2.7 ± 0.73 größer als für
Iodat, wie es auf Grund der höheren molaren Konzentration an Iod zu erwarten ist. Die
Streuung während der FI-ICP-MS-Messungen 5 bleibt, außer im Zeitraum 6, trotz
der absichtlichen drastischen Parametervariationen gering.
6 Das ICP-MS war in diesem Zeitraum mit Iod kontaminiert. Die Analyse von sehr
iodhaltigen Proben hatte dazu geführt, dass der Ioduntergrund danach auf einige
hunderttausend cps angestiegen war. Auch nach dem Austauschen aller Glasteile,
Schläuche und der Cones verlieb eine zu hohe Zählrate auf dem Masse-zu-Ladungs-
Verhältnis 127 von ca. 50000 cps, die sich nur langsam im Laufe der Messungen und
gesamten Interfacebereichs (Expansionskammer) konnte die Kontamination wieder
entfernt werden.
Die Störung durch die Kontamination hatte großen Einfluss auf die Iod-Rn-Werte. Da
der Signalanteil des Ioduntergrunds bei der Berechnung von Nettopeakflächen
subtrahiert wird, war dies zunächst nicht zu erwarten. Es muss durch die Konta-
mination noch weitere Effekte gegeben haben, wodurch sich nur die Elutionspeaks
von Iodat und Iodid vergrößerten. Diesem Phänomen wird in Kap. 4.7.4 genauer
nachgegangen.
7 Bei diesen FI-ICP-MS Messungen wurden unterdurchschnittlich niedrige Rn-Werte für
Bromat und Bromid ermittelt. Die gemessenen Standardlösungen in diesem Zeitraum
stammen alle aus den gleichen Stammlösungen. Bei diesen scheint es einen Ansetz-
fehler gegeben zu haben, denn nachdem wieder frische Stammlösungen hergestellt
worden waren, erreichten die Analyt-zu-GeO2-Verhältnisse wieder höhere Werte im
durch Spülprozeduren verringern ließ. Erst durch eine gründliche Reinigung des
Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 124 -
on
Tendenzen, die durch Variation von Parametern hervorgerufen werden, aber bei der
Die ge
Elimin
Tab.
Durchschnittsbereich. Die zu niedrigen Rn störten nicht bei der Untersuchung v
Ermittlung der Langzeitstabilität sind sie als Ausreißer nicht mit einzubeziehen.
mittelten Langzeit-Rn-Werte für Bromat, Bromid, Iodat und Iodid, die sich nach
ierung von Ausreißern ergeben, sind in Tab. 23 zusammengefasst.
23: Langzeitmittelwerte von Rn bei der IC- und der FI-ICP-MS (Ausreißer aus Datensatz eliminiert)
Rn-Wert Bromat Bromid Iodat Iodid IC-ICP-MS Mittelwert 0.23 ± 0.03 0.27 ± 0.04 2.7 ± 0.5 2.5 ± 0.6 RSD / % 14 14 18 24 Anzahl n 158 261 251 137 FI-ICP-MS Mittelwert 0.21 ± 0.03 0.24 ± 0.03 2.3 ± 0.6 2.7 ± 0.7 RSD / % 13 13 25 27 Anzahl n 142 147 91 92
4.6.3
Neben
Iodat/Io
gezoge
Volum . Mit dem ICP-MS wird nur der Halogenanteil der Spezies
detektiert. Bei gleicher Masse/Volumen-Konzentration der Anionen ist die Peakfläche von
Bromat wegen des Brom
Gleiches gilt für Iodat m
man auch das Verhältnis
falls die ein essenen Verhältnisse sollten
sich
möglic
viel de
konnte
Verhäl
Weitere nützliche Kenngrößen zur Messwertüberwachung und Auswertung
den Rn-Werten kann das Verhältnis der Peakflächen von Bromat/Bromid und
did als ein weiterer wichtiger Anhaltspunkt zur Kontrolle der Messungen heran-
n werden. Die Konzentration der Standards wird üblicherweise in Masse (Anion) pro
en z.B. µg L−1 angegeben
anteils von nur 62 % daher kleiner als die Fläche für Bromid.
it 73 % Iodanteil und Iodid. Statt des Peakflächenverhältnisses kann
der Rn-Werte von Oxohalogenid/Halogenid bilden. Es spiegelt eben-
gesetzten molaren Konzentrationen wider. Die gem
den theoretisch berechneten Werten von 0.62 (Bromat/Bromid) und 0.73 (Iodat/Iodid)
hst annähern. Typischerweise werden aber oft bis zu 30 % höhere Werte, also etwas zu
r oxidierten Form bzw. zu wenig der reduzierten Form erhalten. Der Grund hierfür
nicht ermittelt werden, spielt aber bei einer Kalibration keine Rolle, solange die
tnisse stabil bleiben.

Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 125 -
Bei
folgend
•
• Mangelnde Stabilität der Spezies (Reaktion von Halogenid zu Oxohalogenid oder
•
•
•
Die M
um in
Konzen olekülions [35ClOH]+ auf
dem Masse-zu-Ladungs-Verhältnis 52. Heilwässer haben z.B. oft eine wesentlich höhere
Chl
koeluie
großen
Ionen,
könnte
benutze
zu emp
Das Iso
Masse-
Wenn
z.B. [40
Masse
[40Ar41 einem Kaliumisotop geringerer Häufigkeit.
groben Abweichungen der Verhältniswerte von Bromat/Bromid und Iodat/Iodid können
e Ursachen zugrunde liegen:
Fehler beim Ansetzen der Standards
umgekehrt, oder Umsetzung zu einer anderen Verbindung)
Fehler bei der Datenauswertung
Chromatographische Probleme (z.B. führt schlechte Peakform zu fehlerhafter Integra-
tion oder verringerter Wiederfindung)
Nicht korrigierte Empfindlichkeitsschwankung
asse 52 u wird nicht nur gemessen, um 52Chromspezies nachzuweisen, sondern auch,
Mineralwasser- oder Salzproben hohe Chloridkonzentrationen anzuzeigen. Bei hoher
tration von Chlorid bildet sich ein deutlicher Peak des M
oridkonzentration als normale Mineralwässer. Der Peak wird dann sehr breit und Chlorid
rt sowohl mit Bromid, als auch mit Bromat. Das Vorhandensein eines besonders
Chloridpeaks kann die Ursache dafür sein, wenn kürzere Retentionszeiten der anderen
ungewöhnlich schlechte Peakformen oder Signalschwankungen auftreten. Alternativ
man auch das Masse-zu-Ladungs-Verhältnis 51 für [35ClO]+ oder 53 für [37ClO]+
n. Die direkte Messung auf 35Cl oder 37Cl ist bei hohen Matrixkonzentrationen nicht
fehlen, da dies schnell zu Detektorüberlauf führen kann.
top 79Brom lässt sich mit der ICP-MS empfindlicher messen als 81Brom, denn auf dem
zu-Ladungs-Verhältnis 81 ist der Untergrund durch das Molekülion [40Ar2H]+ höher.
man sich allerdings nicht sicher ist, ob auf der Masse 79 u eine Interferenz (s. Tab. 4),
Ar39K]+ oder [31P16O ]3+ vorliegt, kann dies durch eine gleichzeitige Messung der
81 u kontrolliert werden. Dort liegt zwar ebenfalls eine Kaliuminterferenz durch
K]+ vor, aber mit
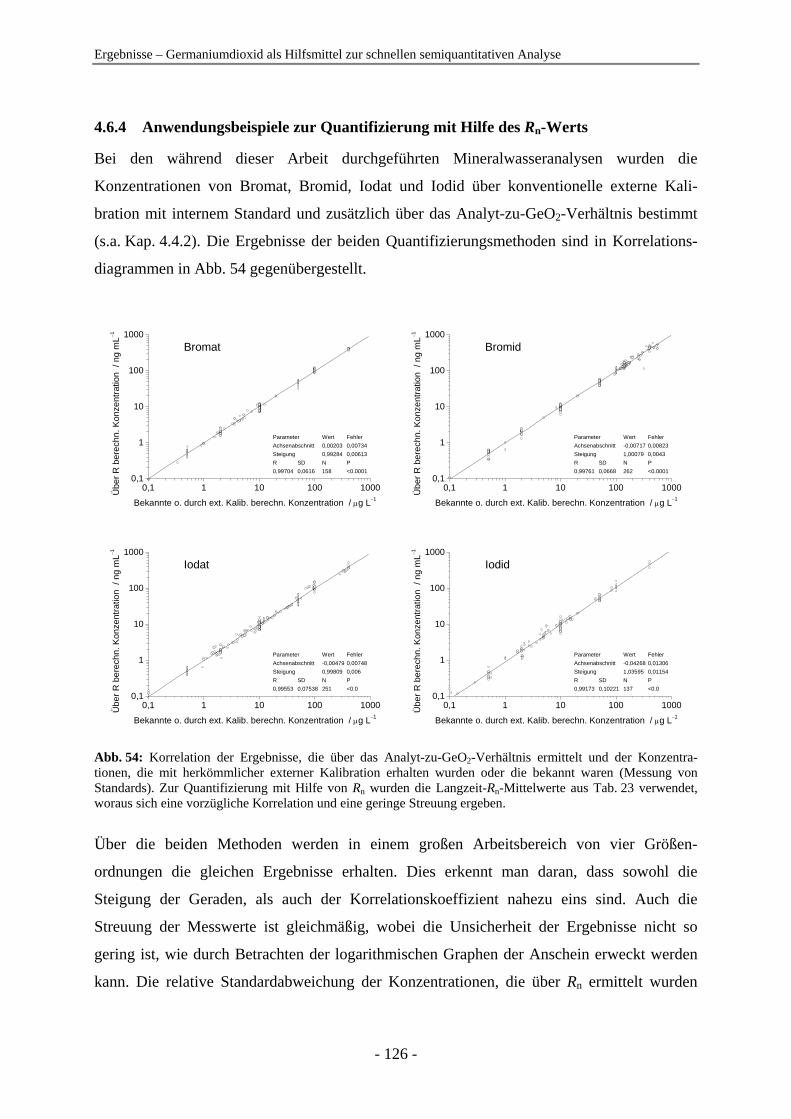
Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 126 -
4.6.4
Bei d die
Konzentrationen von Bromat, Bromid, Iodat und Iodid über konventionelle externe Kali-
Anwendungsbeispiele zur Quantifizierung mit Hilfe des Rn-Werts
en während dieser Arbeit durchgeführten Mineralwasseranalysen wurden
bration mit internem Standard und zusätzlich über das Analyt-zu-GeO2-Verhältnis bestimmt
(s.a. Kap. 4.4.2). Die Ergebnisse der beiden Quantifizierungsmethoden sind in Korrelations-
diagrammen in Abb. 54 gegenübergestellt.
0,1 1 10 100 10000,1
1
10
100
1000
Übe
r R b
erec
hn. K
onze
ntra
tion
/ ng
mL−1
B
10
100
1000
ekannte o. durch ext. Kalib. berechn. Konzentration / µg L−1
0,1 1 10 100 1
Bromat
Parameter WertAchsenabschnitt 0,00203 0,00734Steigung 0,99284 0,00613R SD N P0,99704 0,0616 158 <0.0001
Fehler
0000,1
1
Übe
r R b
erec
hn. K
onze
/ n
g
L−1
Br
Pa Wert FehlerAchsenabschnitt -0,00717 0,00823Steigung 1,00079 0,0043R SD N P0,99761 0,0668 262 <0.0001
ntra
tion
mL−1
omid
rameter
Bekannte o. durch ext. Kalib. berechn. Konzentration / µg
0,1 1 10 100 10000,1
1
10
100
1000
Übe
r R b
erec
hn. K
onze
ntra
tion
/ ng
mL−1
Bekannte o. durch ext. Kalib. berechn. Konzentration / µg L−1
Iodat
Parameter Wert FehlerAchsenabschnitt -0,00479 0,00748Steigung 0,99809 0,006R SD N P0,99553 0,07538 251 <0.0
0,1 1 10 100 10000,1
1
10
100
1000
Übe
r R b
erec
hn. K
onze
ntra
tion
/ ng
mL−1
Bekannte o. durch ext. Kalib. berechn. Konzentration / µg L−1
Iodid
Parameter Wert FehlerAchsenabschnitt -0,04268 0,01306Steigung 1,03595 0,01154R SD N P0,99173 0,10221 137 <0.0
Abb. 54: Korrelation der Ergebnisse, die über das Analyt-zu-GeO2-Verhältnis ermittelt und der Konzentra-tionen, die mit herkömmlicher externer Kalibration erhalten wurden oder die bekannt waren (Messung von Standards). Zur Quantifizierung mit Hilfe von Rn wurden die Langzeit-Rn-Mittelwerte aus Tab. 23 verwendet, woraus sich eine vorzügliche Korrelation und eine geringe Streuung ergeben.
Über die beiden Methoden werden in einem großen Arbeitsbereich von vier Größen-
ordnungen die gleichen Ergebnisse erhalten. Dies erkennt man daran, dass sowohl die
Steigung der Geraden, als auch der Korrelationskoeffizient nahezu eins sind. Auch die
Streuung der Messwerte ist gleichmäßig, wobei die Unsicherheit der Ergebnisse nicht so
gering ist, wie durch Betrachten der logarithmischen Graphen der Anschein erweckt werden
kann. Die relative Standardabweichung der Konzentrationen, die über Rn ermittelt wurden
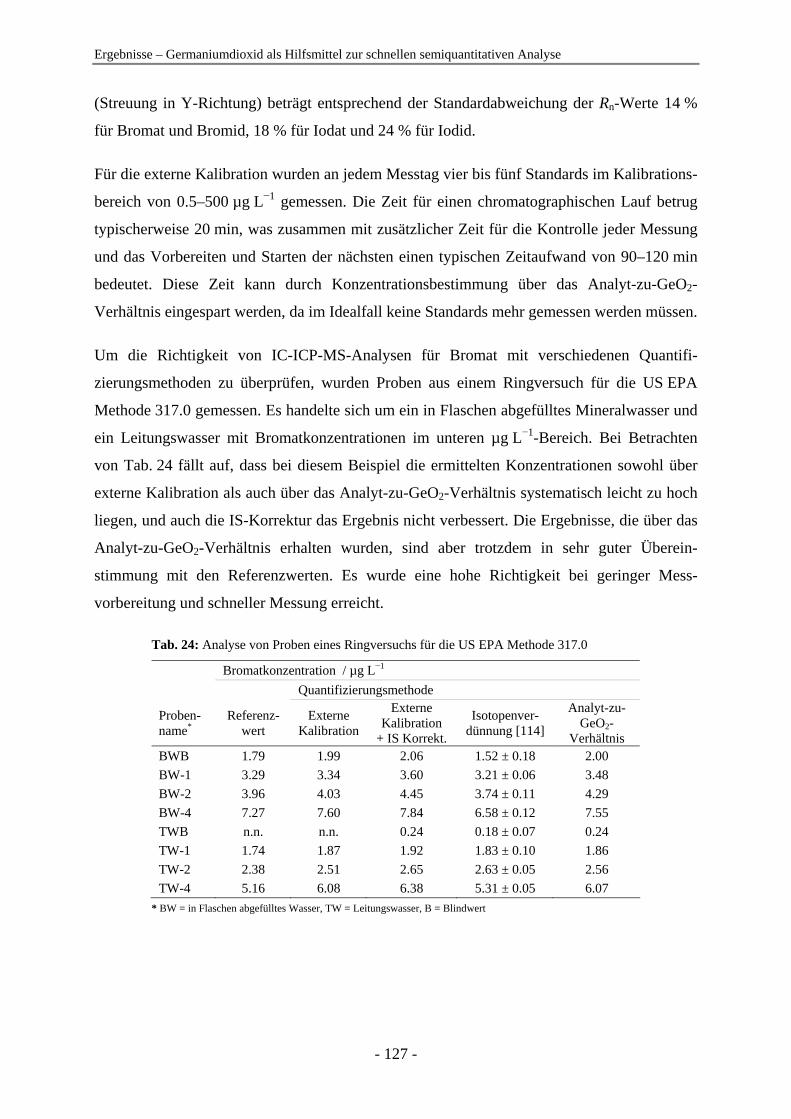
Ergebnisse – Germaniumdioxid als Hilfsmittel zur schnellen semiquantitativen Analyse
- 127 -
at und 24 % für Iodid.
Für e Messtag vier bis fünf Standards im Kalibrations-
ber h
typisch e Kontrolle jeder Messung
und das Vorbereiten und Starten der nächsten einen typischen Zeitaufwand von 90–120 min
bed te
Verhäl eine Standards mehr gemessen werden müssen.
Um i Bromat mit verschiedenen Quantifi-
c
(Streuung in Y-Richtung) beträgt entsprechend der Standardabweichung der Rn-Werte 14 %
für Bromat und Bromid, 18 % für Iod
di externe Kalibration wurden an jedem
eic von 0.5–500 µg L−1 gemessen. Die Zeit für einen chromatographischen Lauf betrug
erweise 20 min, was zusammen mit zusätzlicher Zeit für di
eu t. Diese Zeit kann durch Konzentrationsbestimmung über das Analyt-zu-GeO2-
tnis eingespart werden, da im Idealfall k
d e Richtigkeit von IC-ICP-MS-Analysen für
zierungsmethoden zu überprüfen, wurden Proben aus einem Ringversuch für die US EPA
Methode 317.0 gemessen. Es handelte sich um ein in Flaschen abgefülltes Mineralwasser und
ein Leitungswasser mit Bromatkonzentrationen im unteren µg L−1-Bereich. Bei Betrachten
von Tab. 24 fällt auf, dass bei diesem Beispiel die ermittelten Konzentrationen sowohl über
externe Kalibration als auch über das Analyt-zu-GeO2-Verhältnis systematisch leicht zu hoch
liegen, und auch die IS-Korrektur das Ergebnis nicht verbessert. Die Ergebnisse, die über das
Analyt-zu-GeO2-Verhältnis erhalten wurden, sind aber trotzdem in sehr guter Überein-
stimmung mit den Referenzwerten. Es wurde eine hohe Richtigkeit bei geringer Mess-
vorbereitung und schneller Messung erreicht.
Tab. 24: Analyse von Proben eines Ringversu hs für die US EPA Methode 317.0
Bromatkonzentration / µg L−1
Quantifizierungsmethode
Proben-name*
Referenz-wert
Externe Kalibration
Externe Kalibration
+ IS Korrekt.
Isotopenver-dünnung [114]
Analyt-zu-GeO2-
Verhältnis BWB 1.79 1.99 2.06 1.52 ± 0.18 2.00 BW-1 3.29 3.34 3.60 3.21 ± 0.06 3.48 BW-2 3.96 4.03 4.45 3.74 ± 0.11 4.29 BW-4 7.27 7.60 7.84 6.58 ± 0.12 7.55 TWB n.n. n.n. 0.24 0.18 ± 0.07TW-1 1.74 1.87 1.92 1.83 ± 0.10
0.24 1.86
TW-2 2.38 2.51 2.65 2.63 ± 0.05 2.56 TW-4 5.16 6.08 6.38 5.31 ± 0.05 6.07
* BW = in Flaschen abgefülltes Wasser, TW = Leitungswasser, B = Blindwert

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 128 -
tellen
erbaren Parametern auf die Rn-Werte zeitsparend
essen zu können. Für die Berechnung von Rn werden die Analytpeakflächen und die
Germaniumsignalhöhe einer transienten Messung benötigt, welche bei der Fließinjektions-
analyse – genauso wie bei der IC-Kopplung – geliefert werden. Bei Verwendung von
Bromat/Iodat- und Bromid/Iodid-Doppelstandards ist eine Trennung der Spezies nicht nötig,
da das ICP-MS elementselektiv detektiert.
Ein ionenchromatographischer Lauf würde mit dem in dieser Arbeit verwendeten System für
die vier Analyten etwa 15 min dauern (s. z.B. Kap. 4.1.3.3). Diese Zeit wird gebraucht, um
eine Auflösung von Iodat und Bromat zu erreichen, und um das Iodid trotz seiner starken
Retention zu eluieren. Für eine systematische Überprüfung von vielen Parametern mit
Wiederholungsmessungen ist diese Messzeit zu lang. Deswegen wurde die FI-ICP-MS
verwendet, mit der eine Doppelbestimmung in nur 5 min 40 s möglich ist. Die Messzeit
reduzierte sich somit auf etwa ein Fünftel.
Alle Verbindungskapillaren wurden möglichst kurz gehalten, um schnelle Messungen zu
erreichen und um Adsorptions- und Diffusionseinflüsse zu minimieren. Bei einer Standard-
flussrate von 1 mL min−1 brauchte ein Analyt bei Fließinjektion von der Aufgabe bis zur
Detektion nur etwa 9 s und die vollständige Elution war nach weiteren 30 s erreicht.
chromatographie übertragen
4.7 Untersuchungen zur Abhängigkeit des Rn-Werts von instrumen
Parametern
4.7.1 Vergleich der IC-ICP-MS und der FI-ICP-MS
Der Fließinjektions-ICP-MS-Aufbau (s. Anhang Kap. 6.1.2) wurde verwendet, um die Aus-
wirkungen einer großen Anzahl an veränd
m
Als erstes musste sichergestellt werden, dass sich die mit Fließinjektion gewonnenen
Erkenntnisse über die Analyt-zu-GeO2-Verhältnisse auf die Ionen
lassen. Für diesen Vorversuch wurden Messungen mit der IC-ICP-MS und der FI-ICP-MS
unter gleichen Bedingungen durchgeführt, um einen direkten Vergleich der Rn-Werte zu
ermöglichen. Die Messparameter sind Abb. 55 und Abb. 56 zu entnehmen, in denen ein
Beispiel eines erhaltenen Chromatogramms und Fließinjektionsprofile abgebildet sind.
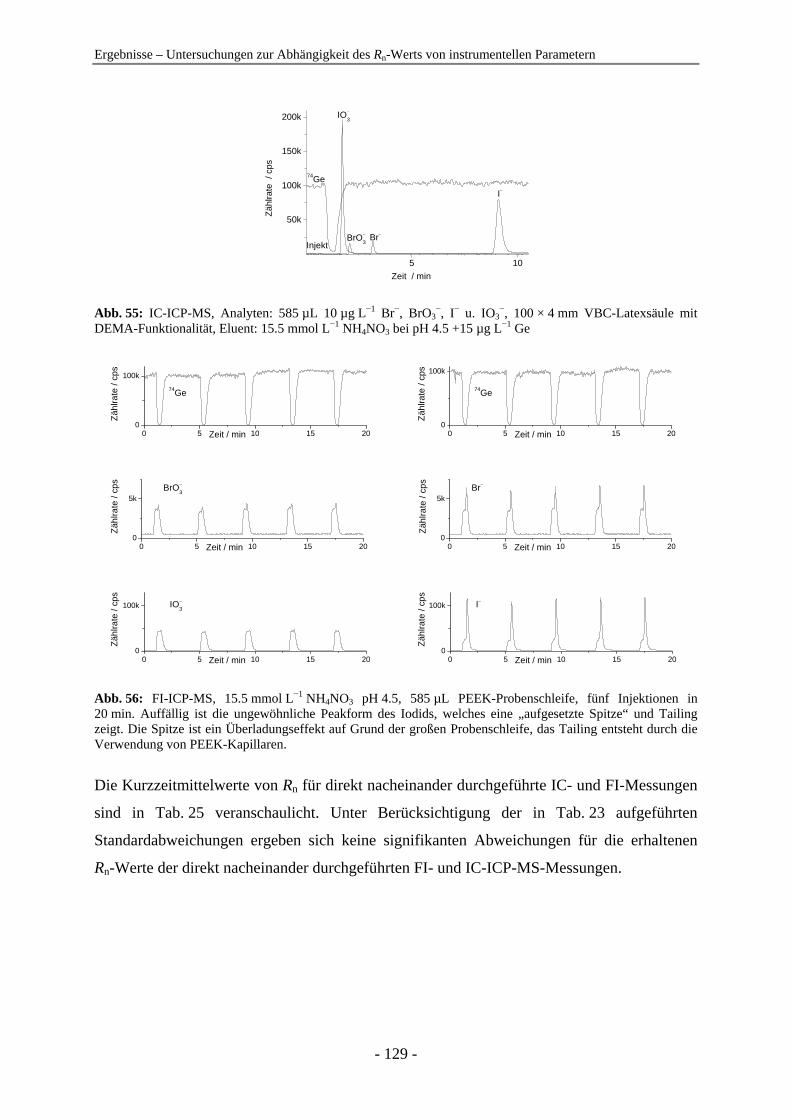
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 129 -
5 10
50k
100k
150k
200k IO−
3
74Ge
InjektBrO−
3
Zähl
rate
/ c
psZeit / min
Br−
I−
Abb. 55: IC-ICP-MS, Analyten: 585 µL 10 µg L−1 Br−, BrO3
−, I− u. IO3−, 100 × 4 mm VBC-Latexsäule mit
−1 −1DEMA-Funktionalität, Eluent: 15.5 mmol L NH4NO3 bei pH 4.5 +15 µg L Ge
0 5 10 15 200
100k
Zähl
rate
/ cp
s
Zeit / min
74Ge
0 5 10 15 20
0
100k
Zähl
rate
/ cp
sZeit / min
74Ge
0 5 10 15 200
5kBrO−
3
Zähl
rate
/ cp
s
Zeit / min
0 5 10 15 20
0
5kBr−
Zähl
rate
/ cp
s
Zeit / min
100k
0 5 10 15 20
IO−
3
te /
cps
0
Zäh
0
lra
Zeit /
100k
min 0 5 10 15 20
I−
lrate
/ cp
s
Zeit / min
Abb. 56: F -ICP-MS, 15.5 mol L−
4 3 L PEEK-Probenschlei jektionen in 20 min. Au di nlich rm de elc esetzte Spitze“ und Tailing zeigt. Die S t ein Üb ngse r s T ht durch die Verwendung von PEEK-Kapillaren.
Die Kurzz telwer n Rn für direkt nacheinander durchgeführte IC- und FI-Messungen
sind in vera aulicht ter Be Tab aufgeführten
Standarda hungen eben s eine sig anten gen f e erhaltenen
Rn-Werte ekt na ander d geführte und IC Messu .
Zäh
I m 1 NH NO pH 4.5, 585 µs Iodids, w
fe, fünf In
ailing entsteffällig istpitze is
e ungewöherladu
e Peakfoffekt auf Grun
hes eine „aufgobenschleife, dad der großen P
eitmit te vo
Tab. 25 nsch . Un rücksichtigung der in . 23
bweic erg ich k nifik Abweichun ür di
der dir chein urch n FI- -ICP-MS- ngen

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 130 -
Tab. 25: Kurzzeitmittelwerte von Rn von IC- bzw. FI-ICP-MS im Vergleich
Rn ert
(Messungen unter gleichen Bedingungen und zeitnah)
-WMessmethode Bromat Bromid Iodat Iodid IC-ICP-MS 0.18 0.23 2.4 3.2 FI-ICP-MS 0.20 0.25 2.8 3.6
Betrachtet man die Langzeit-Rn-Werte aller FI- und IC-ICP-MS Messungen (Tab. 23), so sind
auch dort die Abweichungen beider Methoden so gering, dass bei FI beobachtete Tendenzen
auf IC-ICP-MS-Messungen übertragen werden können. Es zeigen sich folgende geringfügige
Unterschiede für die Kopplungsmethoden: Der Langzeitmittelwert von Rn ist für die Ionen
Iodat, Bromat und Bromid bei IC-Messungen höher, und für das Iodid niedriger als bei FI-
Messungen.
Für die Beobachtung, dass Iodid bei der IC-ICP-MS niedrigere Rn-Werte ergibt als im
führt. Entgegen der theoretischen Berechnung
werden in einem solchen Fall für Iodid meist kleinere R -Werte als für Iodat ermittelt. Eine
P-MS
festgestellt. Daher können die durch Parametervariation mit der FI-ICP-MS erhaltenen Ergeb-
nisse auf das IC-ICP-MS-System übertragen werden.
Vergleich mit Iodat zu erwarten wäre, finden sich plausible Erklärungen. Fließinjektions-
profile haben für alle Analyten ungefähr die gleiche Form. Dagegen sind chromatographische
Peaks früh eluierender Analyten wegen des Aufkonzentrierungseffekts schmaler als Fließ-
injektionsprofile. Analyten mit hoher chromatographischer Retention wie Iodid ergeben auf
Grund der längeren Diffusionszeit und nicht-ionischen Wechselwirkungen wesentlich breitere
Peaks. Einige stationäre Phasen erzeugen so breite Iodidpeaks mit Tailing, dass dies zu
Schwankungen der Rn-Werte des Iodids
n
Erklärung dafür ist, dass aufgegebene Iodidionen durch Adsorption auf Ionenaustauschern
oder wegen zu breiter Peaks nicht mehr detektiert werden. Diese Effekte werden bei
ungeeigneten Säulentypen oder bei Latex-Ionenaustauschern am Ende ihrer Lebensdauer
beobachtet und darauf wird in Kap. 4.1.3.2 näher eingegangen.
Die Signalhöhe des Germaniums bleibt beim Wechsel zwischen FI und IC konstant. Ein
unterschiedlicher Einfluss auf die Rn-Werte durch die Germaniumsignalhöhe ist nicht zu
beobachten und auch nicht zu erwarten, da das Germaniumdioxid keine Wechselwirkungen
mit dem Ionenaustauscher eingeht.
Es wurden nur leichte Unterschiede zwischen den Rn-Werten von IC- und FI-IC

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 131 -
4.7.2 Rn-Abhängigkeit von den Tuningbedingungen: 1. IE des Tune-Elements
Bei dieser Messreihe sollte untersucht werden, wie sich die Rn-Werte verhalten, wenn man
das Massenspektrometer auf Elemente mit unterschiedlicher erster Ionisierungsenergie (IE)
optimiert. Neben der Isotopenhäufigkeit und den Untergrundstörungen hat die IE einen
großen Einfluss auf die Nachweisstärke der ICP-MS für verschiedene Elemente (s. Kap. 2.4).
Durch die IE wird die Ionenausbeute bestimmt, die bei einer bestimmten Temperatur aus dem
Plasma erhalten werden kann. Etwa drei Viertel aller mit ICP-MS messbaren Elemente haben
Ionisierungsenergien von unter 8 eV. Sie liegen im Plasma zu mi destens 90 % ionisiert
n Halogene, haben hohe
IEs und entsprechend niedrige Ionisierungsgrade (Iod = 29 %, Brom = 5 %). Veränderungen
der Plasmabedingungen oder der Position im Plasma, an der die Ionen extrahiert werden,
können die Ionenausbeute verändern. Der Rn-Wert würde nicht stabil bleiben, wenn sich der
Ionisierungsgrad eines Analyten und des German s bei speziellem Tuning unterschiedlich
stark verändert.
Für das Experiment wurde nicht nur ein einzelner, sondern es wurden alle relevanten
Parameter so variiert, dass eine möglichst hohe Zählrate für das jeweilige Tuningelement
erreicht wurde. Nach jeder Änderung wurde ein Massenscan aufgenommen, um sicher zu
gehen, dass sich die Messposition auf den Massenpeaks nicht verändert hatte. Wenn sich die
Peakmaxima nicht mehr exakt auf den richtigen Massenpositionen befanden, wurde die
Massenkalibration in dem Programm PQ Vision manuell korrigiert (s.a. 6.1.3).
T 6: Ionisierungsenergien u. -grade der Tune-Elemente u. Germaniums sowie Signal-zu-Untergrund-Werte
n
vor [152]. Nichtmetalle, insbesondere die in dieser Arbeit untersuchte
ium
ab. 2
Tune- Element
1. IE/ eV
Ionisierungsgrad e/ % nach [152]
Konzentration / µg L−1 Isotop
Zählrate a/ cps
Signal-zu-Untergrund bBr I
133Cs 3.89 100 10 420000 60 17 115In 5.79 99 9.6 250000 69 38 74Ge 7.90 90 5.5 – – – 127I c 10.46 29 7.3 110000 72 46
79Br d 11.85 5 31.7 90000 70 40 a: Massenpeakhöhe, b: Peakfläche Halogenspezies/Signalhöhe Untergrund, c: 10 µg L−1 IO3
−, d: 100 µg L−1 BrO3−
e: Plasmatemperatur: 7500 K, Elektronendichte: 1.3 × 1015 cm−3
Tab. 26 zeigt die Ionisierungsparameter und erhaltene Signal-zu-Untergrund-Verhältnisse der
vier Elemente, auf die optimiert wurde: 133Cäsium, 115Indium, 127Iod und 79Brom und
zusätzlich 74Germanium zum Vergleich. An den Daten kann auch abgelesen werden, mit
welchem Tune-Element sich das ICP-MS so einstellen lässt, dass man die besten Nachweis-

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 132 -
grenzen für die asse zu Iod
besitzt, ist das Signal-zu-Untergrund-Verhältnis aller Halogenspezies beim Tuning auf
Cäsium deutlich schle O u In Br oder Iodat. Cäsium ist
demnach wegen seiner sehr geringen IE nicht so gut für die Optimierung geeignet. Die
Unterschiede zwischen den anderen drei Tune-Spezies sind dagegen gering.
Halogenspezies erhält. Obwohl Cäsium eine sehr ähnliche M
chter als bei ptimier ng auf dium, omat
Beim Tuning mit den schwer ionisierbaren Halogenen wurde die Plasmaleistung von 1350 W
auf 1600 W erhöht. Durch die höhere Temperatur ergaben sich zwar Rekordzählraten für alle
Analyten, da aber der Untergrund ebenso anstieg, konnte das Signal-zu-Untergrund-
Verhältnis kaum verbessert werden.
0 5 100
100k
200k
Zähl
ra
Zeit / min
0 5 100
5k
10k
te /
cps 74Ge
Br−
Zähl
rate
/ c
ps
Zeit / min
BrO−
3
0 5 100
100k
200kI−
Zähl
rate
/ c
ps
Zeit / min
IO−
3
Abb. 57: Beispiel einer Messung mit der FI-ICP-MS-Kopplung. (Messbedingungen: Tuning auf 133Cs, Eluent: 15 mmol L−1 NH4NO3 pH 4.6, Injektion 1–3 = 10 µg L−1 BrO3
−/IO3−, Injektion 4–6 = 10 µg L−1 Br−/I−, 153 µL-Probenschleife aus PEEK)
57 sind repräsentative Signale der
verschiedenen Massenspuren bei der Fließinjektion gezeigt. Für die nachfolgenden Messun-
ührt. Wie sich herausstellte, lag die
schlechte Peakform des Iodids an den verwendeten PEEK-Kapillaren (s.a. Kap. 6.1.2).
Um die Auswirkungen verschiedener Tuningbedingungen auf Rn zu beobachten, wurde
jeweils direkt nach dem Tuning eine zehnminütige FI-ICP-MS-Messung gestartet. Nach der
Optimierung auf Cäsium und Indium wurden Dreifachinjektionen von 10 µg L−1
Iodat/Bromat bzw. Iodid/Bromid durchgeführt. In Abb.
gen (Iodat- und Bromat-Tuning) mussten die Wiederholungen auf Zweifachinjektionen redu-
ziert werden, um die Spülzeit erhöhen zu können. Das in Abb. 57 erkennbare Peaktailing des
Iodids hätte sonst zu starken Memoryeffekten gef
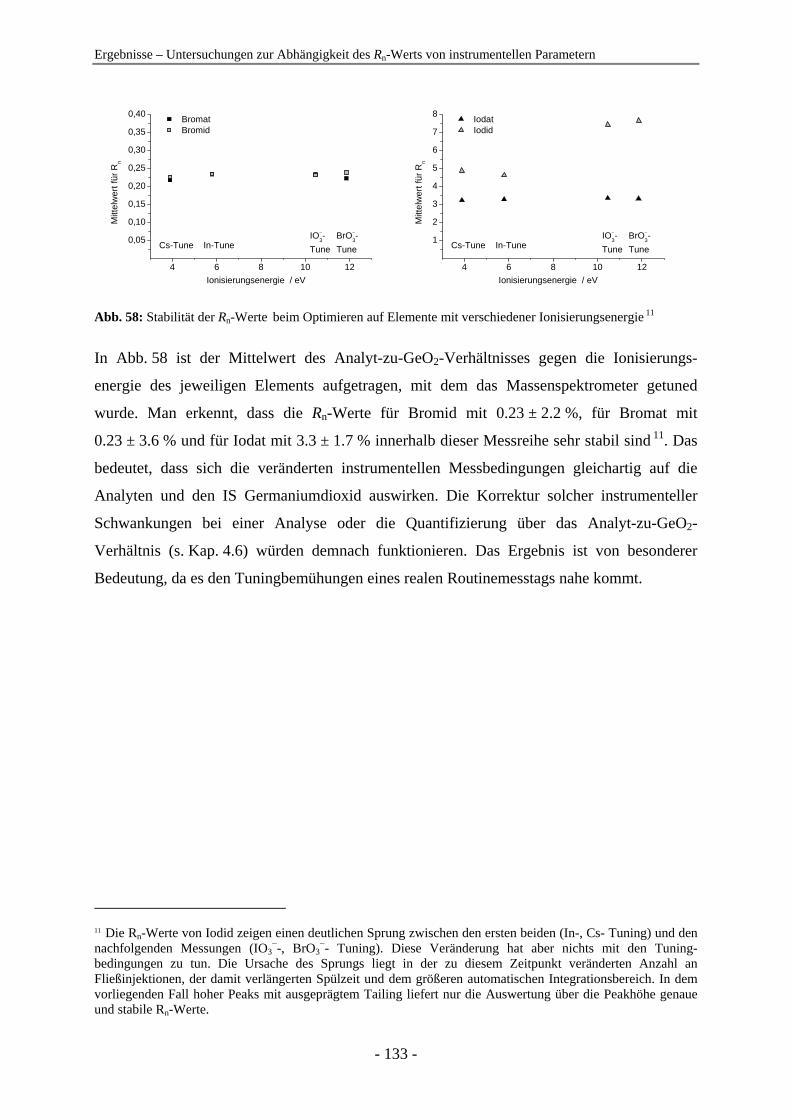
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
0,
- 133 -
4 6 8 10 12
0,05
0,10
0,15
0,20
0,25
0,30
0,35
40 8 Bromat
BrO−
3-Tune
IO−
3-TuneIn-TuneCs-Tune
Bromid
Mitt
elw
ert f
ür R
n
Ionisierungsenergie / eV
4 6 8 10 12
1
2
3
4
5
6
7 Iodat Iodid
Mitt
elw
ert f
ür R
n
Ionisierungsenergie / eV
Cs-Tune In-TuneIO−
3-Tune
BrO−
3-Tune
Abb. 58: Stabilität der Rn-Werte beim Optimieren auf Elemente mit verschiedener Ionisierungsenergie 11
In Abb. 58 ist der Mittelwert des Analyt-zu-GeO2-Verhältnisses gegen die Ionisierungs-
energie des jeweiligen Elements aufgetragen, mit dem das Massenspektrometer getuned
wurde. Man erkennt, dass die Rn-Werte für Bromid mit 0.23 ± 2.2 %, für Bromat mit
0.23 ± 3.6 % und für Iodat mit 3.3 ± 1.7 % innerhalb dieser Messreihe sehr stabil sind 11. Das
bedeutet, dass sich die veränderten instrumentellen Messbedingungen gleichartig auf die
Analyten und den IS Germaniumdioxid auswirken. Die Korrektur solcher instrumenteller
Schwankungen bei einer Analyse oder die Quantifizierung über das Analyt-zu-GeO2-
Verhältnis (s. Kap. 4.6) würden demnach funktionieren. Das Ergebnis ist von besonderer
Bedeutung, da es den Tuningbemühungen eines realen Routinemesstags nahe kommt.
11 Die Rn-Werte von Iodid zeigen einen deutlichen Sprung zwischen den ersten beiden (In-, Cs- Tuning) und den nachfolgenden Messungen (IO3
−-, BrO3−- Tuning). iese Verän rung hat a r nichts mit den Tuning-
bedingungen zu tun. Die Ursache des Sprungs liegt in der zu diesem Zeitpunkt veränderten Anzahl an Fließinjektio n, der damit verlängerten Spülzeit und dem größeren automatischen Integrationsbereich. In dem vorliegenden Fall hoher Peaks mit ausgeprägtem Tailing liefert nur die Auswertung über die Peakhöhe genaue
D de be
ne
und stabile Rn-Werte.

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 134 -
Tuning auf ein
besonders leichtes Element wie 9Beryllium, ein mittelschweres wie 115Indium bzw. ein
2
4.7.3 Rn-Abhängigkeit von den Tuningbedingungen: Masse des Tune-Elements
Das Massenspektrometer wird bei Messungen von Brom- und Iodspezies normalerweise mit
Lösungen von Elementen optimiert, die im gleichen Massenbereich liegen, z.B. 115Indium, 74Germanium, 79Brom oder 89Yttrium. Es galt herauszufinden, ob ein
schweres wie 238Uran den Rn-Wert signifikant verändert. Es wurden nach erfolgtem Tuning
jeweils FI-ICP-MS-Doppelbestimmungen von 10 µg L−1 Iodat/Bromat bzw. Iodid/Bromid
durchgeführt und die Rn-Werte berechnet. Wie in Abb. 59 ersichtlich, bleibt das Verhältnis
von Analyt-zu-GeO trotz der verschiedenen Tuningbedingungen stabil.
0 50 100 150 200 250
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
238U-Tune
115In-Tune
9Be-Tune
Bromat Bromid
Mitt
elw
ert f
ür R
n
0 50 100 150 200 250
1
2
3
4
5
6
7
8 Iodat Iodid
Mitt
elw
ert f
ür R
n
Masse / u
9Be-Tune
115In
Masse / u
-Tune
238U-Tune
ttlerer und hoher Masse 12
n des
Germaniumsignals oder durch überproportional ansteigende Flächen der Analytpeaks zu
Stande kommen. Bei den beobachteten Ausreißern blieb die Germaniumzählrate konstant und
nur überproportional gestiegene Iodpeakflächen waren für die hohen Rn-Werte verantwortlich.
Abb. 59: Stabilität der Rn-Werte beim Optimieren auf Elemente mit niedriger, mi
4.7.4 Ursachen zu hoher Rn-Werte der Iodspezies
Bei einigen Experimenten wurden deutlich nach oben ausreißende Rn-Werte der Iodspezies
beobachtet. Diese können nicht mit bestimmten in Kap. 4.7 variierten instrumentellen
Parametern in Verbindung gebracht werden. Es scheint mehrere Gründe für die Ausreißer zu
geben, da sie in Messsituationen ohne sichtbare Gemeinsamkeiten oder an verschiedenen
ICP-MS-Geräten auftraten. Prinzipiell kann eine Veränderung des normierten
Analyt-zu-GeO2-Verhältnisses durch überproportional kleiner werdende Zählrate
12 Problembehaftet ist bei dieser Messreihe die Integration der FI-Elutionspeaks des Iodids wegen des starken Tailings und der Memoryeffekte (zur Lösung des Problems s. Kap. 6.1.2). Deswegen musste ein ausreißender Datenpunkt vom 238U-Tuning weggelassen werden.

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 135 -
Der Mittelwert der Rn-Werte aller Fließinjektionsmessungen ohne Ausreißer liegt für Iodat
bei 2.3 ± 0.6 und für Iodid bei etwa 2.7 ± 0.7 (Tab. 23). Die höchsten je erhaltenen Rn-Werte
betragen 10.5 für Iodat bzw. 17.2 für Iodid. Diese sehr hohen Rn-Werte ergaben sich einen
Tag, nach dem aufgeschlossene Blutproben gemessen wurden, die große Mengen an Iod
enthielten. Deswegen war das Messsystem mit Iod kontaminiert. Der Geräteblindwert für das
Masse-zu-Ladungs-Verhältnis 127 lag bei ca. 60000 cps und fiel an den folgenden Messtagen
durch Ausspülen ab. Bei den in dieser Zeit durchgeführten Experimenten wurden ent-
sprechend fallende Iod-Rn-Werte gemessen (s. 53). Erst nach mehreren Wochen und
rund-
in
ile des
FI-ICP-MS-Systems bis zum Vakuumbereich wurden gewechselt. Reste des Iods können nur
noch im (gereinigten) Interface, den Ionenlinsen und im Quadrupol vorhanden gewesen sein.
Im Folgenden werden zwei Hypothesen zur Erklärung erhöhter Iod-zu-GeO2-Verhältnisse bei
FI-ICP-MS-Messungen aufgestellt:
Hypothese A
Das Iodsignal wird erhöht, wenn es zu einer durch die Probeninjektion induzierten
Iodauswaschung (im Interface) kommt. Der entscheidende Faktor ist die Temperaturdifferenz.
Wenn ein aufgegebener Standard bei der FIA ins Plasma gelangt, kommen die Analyten
hauptsächlich zusammen mit Wasser an. Im Vergleich mit dem davor und danach
gemessenen konzentrierteren Eluenten müssen weniger schwer dissoziierbare Moleküle
atomisiert und ionisiert werden. Deswegen kommt es zu einem Temperaturanstieg im Plasma,
Abb.
nach gründlicher Reinigung des Interfacebereichs konnten wieder ein normaler Unterg
wert von wenigen Tausend cps und Iod-Rn-Werte im durchschnittlichen Bereich erreicht
werden. Das lässt darauf schließen, dass die Kontamination des Geräts der Grund für die sehr
hohen Rn-Werte war.
Diese Beobachtung ist überraschend, da der Systemblindwert eigentlich keinen E fluss
haben dürfte, wenn mit den Nettopeakflächen der Analyten gerechnet wird. In jedem
Chromatogramm wird von dem additiven Signal aus Untergrund und Analyt (Bruttosignal)
der Untergrund subtrahiert. Dieser wird immer aktuell ermittelt, indem von einem Punkt links
und einem rechts neben dem Peak eine Linie gezogen und die Fläche darunter berechnet wird.
Wieso kann der Iodblindwert dann noch eine Rolle spielen? Alle austauschbaren Te
der zu einem verstärkten Ablösen eines Iodniederschlags im Interface führt. Die Hypothese A
wird durch die folgenden Beobachtungen gestützt:

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 136 -
A.1) Solange das Gerät noch mit Iod kontaminiert war, stiegen die Rn-Werte für Iod-
n Ein-
l erhalten. Dieser
Blindwert könnte von Iodverunreinigungen (z.B. flüchtigen Iodkohlenwasserstoffen
im Plasma-Argon) herrühren. Auch ein solches von einer Iodauswaschung zu unter-
scheidendes Untergrundsignal kann während des Wasserpeaks durch eine Erhöhung
der Plasmatemperatur und des Ionisierungsgrads ansteigen.
Hypothese B
Die zweite Hypothese verhält sich invers zu der ersten, weil hier eine Temperaturabsenkung
durch die Probe und nicht durch den Eluenten angenommen wird. Bei einer Plasmatemperatur
von 7500 K, liegt der nach der Saha-Gleichung berechnete Ionisierungsgrad von Iod bei nur
chen iodhaltigen Fraktion ins Plasma angelangt (Peakmaximum), kühlt sich das Plasma
durch die hohe aufzubringende Ionisierungsenergie für das Halogen lokal ab, weshalb der
gelangt dagegen eine gleiche Stoff-
n
spezies bei geringerer Samplingtiefe und bei höherer Leistung. Die beide
stellungen haben ähnliche Auswirkungen. Eine geringere Samplingtiefe bedeutet, dass
die Ionen an einer Plasmaposition mit höherer Temperatur extrahiert werden. Ebenso
wird mit der Plasmaleistung die Temperatur erhöht. Zudem wird das Plasma aber auch
vergrößert, was indirekt zu einer Verringerung der Samplingtiefe führt.
A.2) Auch wenn sichergestellt ist, dass keine Iodkontamination durch Ablagerungen
im ICP-MS vorhanden ist, wird bei der Injektion von Reinstwasser in das FI-ICP-MS-
Kopplungssystem vereinzelt immer noch ein kleines Iodsigna
29 % (s. Tab. 3 u. Tab. 26). In dem Augenblick, in dem der Hauptteil der chromatographi-
s
Ionisierungsgrad sinkt. Bei breiten Peaks mit Tailing
menge an Iodatomen über einen etwas längeren Zeitabschnitt und damit „verdünnter“ ins
Plasma. Die Temperatur und die Ionisierungsausbeute bleiben höher, und es ergibt sich damit
ein höheres Signal für eine gleiche Anzahl an Iodatomen. Die Hypothese B wird durch zwei
Beobachtungen genährt:
B.1) Sehr kleine Iodkonzentrationen ergeben bei der FIA etwas größere Rn-Werte als
konzentrierte Standards (s. Kap. 4.7.9).
B.2) Je stärker das Tailing der Iodidpeaks bei der FIA ausgeprägt ist, desto größere
Iodsignale bzw. R -Werte werden berechnet.

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 137 -
Samplingtiefe
Die Torch lässt sich zusammen mit der Zerstäuberkammer und allen Verbindungen während
r die Ionen extrahiert werden längs des inneren
. Dabei werden ein Temperaturgradient und eine leicht veränderliche lokale
n aus Tab. 27 entnommen werden.
Die beiden Hypothesen zum Einfluss des Ioduntergrunds sind nicht auf die IC-ICP-MS-
Kopplung übertragbar. Im Unterschied zur FI kommen die Analyten bei der IC nicht alle
zusammen mit Reinstwasser im Plasma an, sondern von einander getrennt und gleichzeitig
mit dem Ammoniumnitrat-Eluenten. Bei den ionenchromatographischen Messungen haben
die hohe Retention Iodids und Tailingprobleme einen größeren Einfluss auf den Iodid-Rn-
Wert. Wenn der Iodidpeak zu breit ist wird die Peakintegration schwierig und unpräzise. Auf
manchen Säulen kann sogar bei kleinen Konzentrationen von Iodid ein Anteil auf der Säule
adsorbiert werden oder das Signal verschwindet auf Grund starken Tailings im Untergrund-
rauschen. Dies ist die Ursache für zu niedrige Iodid-Rn-Werte bei der IC-ICP-MS.
4.7.5 Variation der
des Betriebs mit Hilfe von Mikrometerschrauben in den drei Raumrichtungen positionieren.
Der hellste und damit heißeste Teil des Aerosolkanals im innern des Plasmas liefert die
höchste Analytionenausbeute. Als Samplingtiefe wird der Abstand von der Spitze des
Sampler-Cones bis zur Mitte der ersten Spulenwindung verstanden. Der Ausdruck Sampling-
tiefe ist zuweilen etwas irreführend, da damit nicht die Eindringtiefe des Sampler-Cones
gemeint ist, sondern die „Tiefe“ oder Länge des Plasmas von der Spule aus gemessen. Die
Ionenzählraten steigen im Allgemeinen, je näher man das Plasma an die Lochblende
heranbewegt, also mit kleiner werdender Samplingtiefe. So erhöht sich die Signalintensität
beim Übergang von größter zu kleinster Samplingtiefe etwa um den Faktor vier. Beim
Verstellen verschiebt sich die Position, an de
Aerosolkanals
Plasmazusammensetzung abgefahren, so dass die Bildung von Element- und Molekülionen
unterschiedlich ist. Durch die Ermittlung von Rn-Werten lässt sich verdeutlichen, ob sich
unter diesen Bedingungen die Signalstärke der Analytionen und die des internen Standards
proportional verhalten.
Da an der Mikrometerschraube keine Skala für die Samplingtiefe angebracht ist, wird am
besten mit einem Messschieber der Abstand zwischen den beiden Klötzen, zwischen denen
die Schraube steckt, gemessen. Beim Verkleinern der Samplingtiefe werden die Klötze weiter
auseinander geschoben. Die Zusammenhänge könne
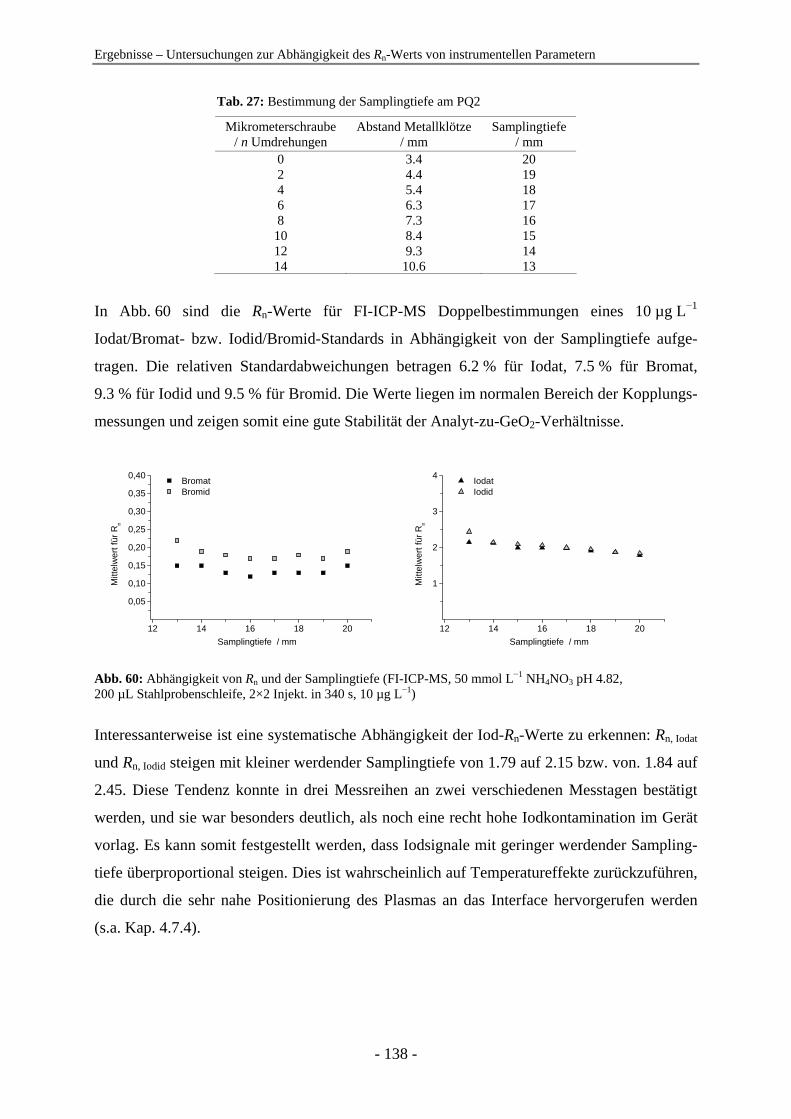
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 138 -
Tab. 27: Bestimmung der Samplingtiefe am PQ2
Mikrometerschraube / n Umdrehungen
Abstand Metallklötze / mm
Samplingtiefe / mm
0 3.4 20 2 4.4 19 4 5.4 18 6 6.3 17 8 7.3 16
10 8.4 15 12 9.3 14 14 10.6 13
In Ab
Iodat/B
tragen.
9.3 % f
messun
b. 60 sind die Rn-Werte für FI-ICP-MS Doppelbestimmungen eines 10 µg L−1
romat- bzw. Iodid/Bromid-Standards in Abhängigkeit von der Samplingtiefe aufge-
Die relativen Standardabweichungen betragen 6.2 % für Iodat, 7.5 % für Bromat,
ür Iodid und 9.5 % für Bromid. Die Werte liegen im normalen Bereich der Kopplungs-
gen und zeigen somit eine gute Stabilität der Analyt-zu-GeO2-Verhältnisse.
12 14 16 18 20
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40 Bromat Bromid
Mitt
elw
ert f
ür R
n
Samplingtiefe / mm
12 14 16 18 20
1
2
3
4 Iodat Iodid
Mitt
elw
ert f
ür R
n
Samplingtiefe / mm
Abb. 60: Abhängigkeit von Rn und der Samplingtiefe (FI-ICP-MS, 50 mmol L−1 NH4NO3 pH 4.82, 200 µL Stahlprobenschleife, 2×2 Injekt. in 340 s, 10 µg L−1)
Interessanterweise ist eine systematische Abhängigkeit der Iod-Rn-Werte zu erkennen: Rn, Iodat
und Rn, Iodid steigen mit kleiner werdender Samplingtiefe von 1.79 auf 2.15 bzw. von. 1.84 auf
2.45. Diese Tendenz konnte in drei Messreihen an zwei verschiedenen Messtagen bestätigt
werden, und sie war besonders deutlich, als noch eine recht hohe Iodkontamination im Gerät
vorlag.
tiefe überproportional steigen. Dies ist wahrscheinlich auf Temperatureffekte zurückzuführen,
die durch die sehr nahe Positionierung des Plasmas an das Interface hervorgerufen werden
(s.a. Kap. 4.7.4).
Es kann somit festgestellt werden, dass Iodsignale mit geringer werdender Sampling-
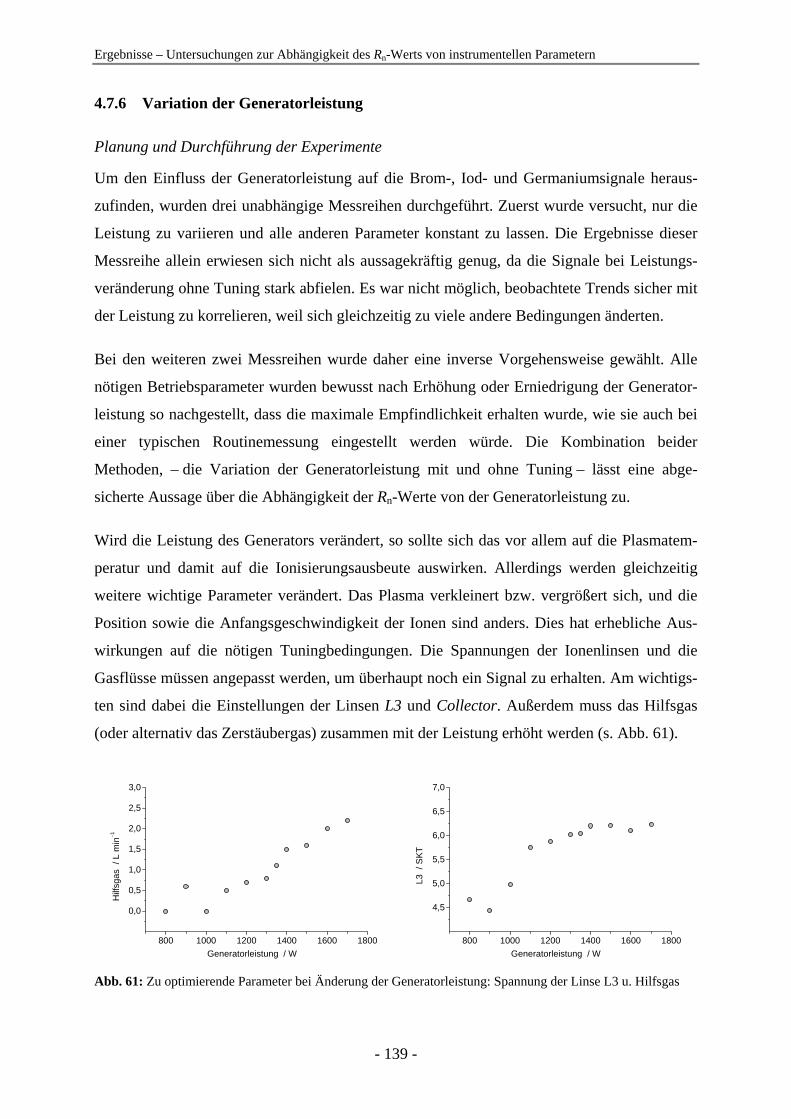
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 139 -
erten.
ei den weiteren zwei Messreihen wurde daher eine inverse Vorgehensweise gewählt. Alle
nötigen Betriebsparameter wurden bewusst nach Erhöhung oder Erniedrigung der Generator-
ale Empfindlichkeit erhalten wurde, wie sie auch bei
4.7.6 Variation der Generatorleistung
Planung und Durchführung der Experimente
Um den Einfluss der Generatorleistung auf die Brom-, Iod- und Germaniumsignale heraus-
zufinden, wurden drei unabhängige Messreihen durchgeführt. Zuerst wurde versucht, nur die
Leistung zu variieren und alle anderen Parameter konstant zu lassen. Die Ergebnisse dieser
Messreihe allein erwiesen sich nicht als aussagekräftig genug, da die Signale bei Leistungs-
veränderung ohne Tuning stark abfielen. Es war nicht möglich, beobachtete Trends sicher mit
der Leistung zu korrelieren, weil sich gleichzeitig zu viele andere Bedingungen änd
B
leistung so nachgestellt, dass die maxim
einer typischen Routinemessung eingestellt werden würde. Die Kombination beider
Methoden, – die Variation der Generatorleistung mit und ohne Tuning – lässt eine abge-
sicherte Aussage über die Abhängigkeit der Rn-Werte von der Generatorleistung zu.
Wird die Leistung des Generators verändert, so sollte sich das vor allem auf die Plasmatem-
peratur und damit auf die Ionisierungsausbeute auswirken. Allerdings werden gleichzeitig
weitere wichtige Parameter verändert. Das Plasma verkleinert bzw. vergrößert sich, und die
Position sowie die Anfangsgeschwindigkeit der Ionen sind anders. Dies hat erhebliche Aus-
wirkungen auf die nötigen Tuningbedingungen. Die Spannungen der Ionenlinsen und die
Gasflüsse müssen angepasst werden, um überhaupt noch ein Signal zu erhalten. Am wichtigs-
ten sind dabei die Einstellungen der Linsen L3 und Collector. Außerdem muss das Hilfsgas
(oder alternativ das Zerstäubergas) zusammen mit der Leistung erhöht werden (s. Abb. 61).
1,5
2,0
2,5
3,0
800 1000 1200 1400 1600 1800
0,0
0,5
min
−1
6,5
7,0
1,0
Hilf
s
Generatorleistung / W
800 1000 1200 1400 1600 1800
4,5
5,0
5,5
6,0
gas
/ L
L3
Generatorleistung / W
Abb. 61: Zu optimierende Parameter bei Änderung der Generatorleistung: Spannung der Linse L3 u. Hilf
/ S
KT
sgas
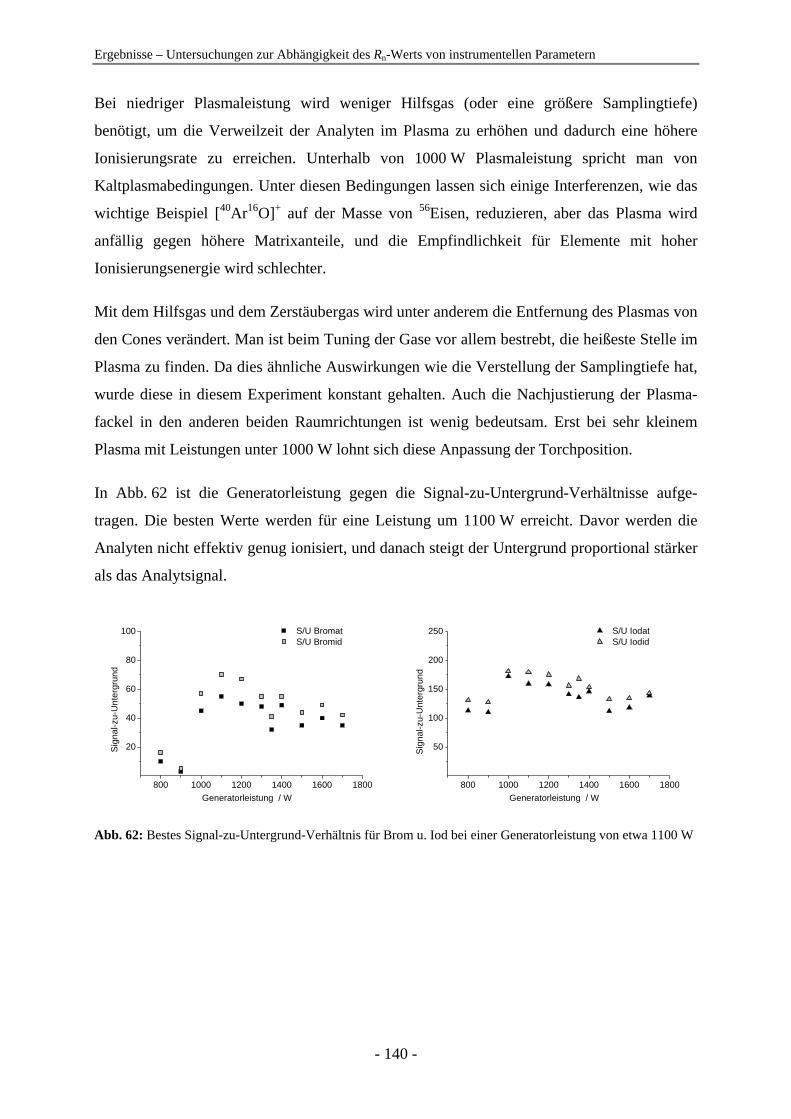
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 140 -
r eine größere Samplingtiefe)
benötigt, um die Ve ly er durch eine höhere
Ionisierungsrate zu erreichen. Unterhalb von 1000 W Plasma tung spricht man von
Kaltplasmabedingungen. Unter diesen Bedingungen lassen sich einige Interferenzen, wie das
wichtige Beispiel [40Ar16O] uf der Masse vo Eisen, reduzieren, aber das Plasma wird
anfällig gegen höhere Matrixanteile, und di pfindlichkeit für Elemente mit hoher
Ionisierungsenergie wird schlechter.
r kleinem
Plasma mit Leistungen unter 1000 W lohnt sich diese Anpassung der Torchposition.
In Abb. 62 ist die Generatorleistung gegen die Signal-zu-Untergrund-Verhältnisse aufge-
tragen. Die besten Werte werden für eine Leistung um 1100 W erreicht. Davor werden die
Analyten nicht effektiv genug ionisiert, und danach steigt der Untergrund proportional stärker
als das Analytsignal.
80
100
Bei niedriger Plasmaleistung wird weniger Hilfsgas (ode
rweilzeit der A an ten im Plasma zu höhen und da
leis
+ a n 56
e Em
Mit dem Hilfsgas und dem Zerstäubergas wird unter anderem die Entfernung des Plasmas von
den Cones verändert. Man ist beim Tuning der Gase vor allem bestrebt, die heißeste Stelle im
Plasma zu finden. Da dies ähnliche Auswirkungen wie die Verstellung der Samplingtiefe hat,
wurde diese in diesem Experiment konstant gehalten. Auch die Nachjustierung der Plasma-
fackel in den anderen beiden Raumrichtungen ist wenig bedeutsam. Erst bei seh
800 1000 1200 1400 1600 1800
20
40
60
S/U Bromat S/U Bromid
zu-
d
200
250
Sig
nal-
Generatorleistung / W
800 1000 1200 1400 1600 1800
50
S/U Iodat S/U Iodid
Unt
ergr
un
150
Unt
ergr
und
100u-S
igna
l-z
Generatorleistung / W
Abb. 62: Bestes Signal-zu-Untergrund-Verhältnis für Brom u. Iod bei einer Generatorleistung von etwa 1100 W

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 141 -
Optimale Durchführung des Experiments
en, die sich nach mehreren Messreihen als am
mit guten Peakformen für Iodid erreicht. Als Eluent wurde 30 mmol L Ammoniumnitrat bei
msignale
die Massenkalibration überprüft, die aber bei diesem Experiment stabil blieb.
kt
lässt sich aber nicht, oder zumindest nicht alleine auf die Leistung zurückführen, denn die
Ionen waren durch ihre veränderte Position und andere Anfangsgeschwindigkeit ohne Tuning
besonders schlechten Extraktions- und Transmissionsbedingungen im Massenspektrometer
ausgesetzt.
Im Folgenden wird die Messmethode angegeb
besten herausgestellt hat. Die damit erhaltenen Ergebnisse sind in Abb. 64 gezeigt.
Für jede eingestellte Generatorleistung, wurde eine transiente Messung mit einer Gesamt-
dauer von 5 min 40 s durchgeführt. In dieser Zeit wurden je zweimal ein 10 µg L−1
Bromat/Iodat- bzw. ein 10 µg L−1 Bromid/Iodid-Standard mit der FI-ICP-MS gemessen.
Durch eine 200 µL Probenschleife aus Stahl wurden Signale ohne Überladungseffekte und −1
pH 4.8 verwendet. Die Generatorleistung wurde in willkürlicher Reihenfolge variiert. Nach
einer Änderung, wurden wiederholt einige wichtige ICP-MS-Parameter in folgender
Reihenfolge optimiert: Hilfsgas, XYZ-Position der Torch, Collector, L3, andere Ionenlinsen.
Andere Parameter wurden konstant gehalten: Extraction, Pole Bias, Resolution, ∆M und das
Zerstäubergas. Das Tuning erfolgte über den internen Standard im Eluenten 74Germanium.
Der Eluent wurde außerdem vor jeder Messung gescannt und anhand der Germaniu
Diskussion der Ergebnisse
In Abb. 63 sind die Rn-Werte in Abhängigkeit von der Generatorleistung ohne Durchführung
eines Tunings und in Abb. 64 mit Tuning dargestellt. Trotz noch nicht optimaler Messdurch-
führung sind schon in Abb. 63 stabile normierte Analyt-zu-GeO2-Verhältnisse für Bromat und
Bromid erkennbar. Für die Iodspezies zeigt sich bei diesem Experiment ein Maximum der
Rn-Werte genau bei der Leistung, auf die das Gerät am Anfang optimiert wurde. Dieser Effe
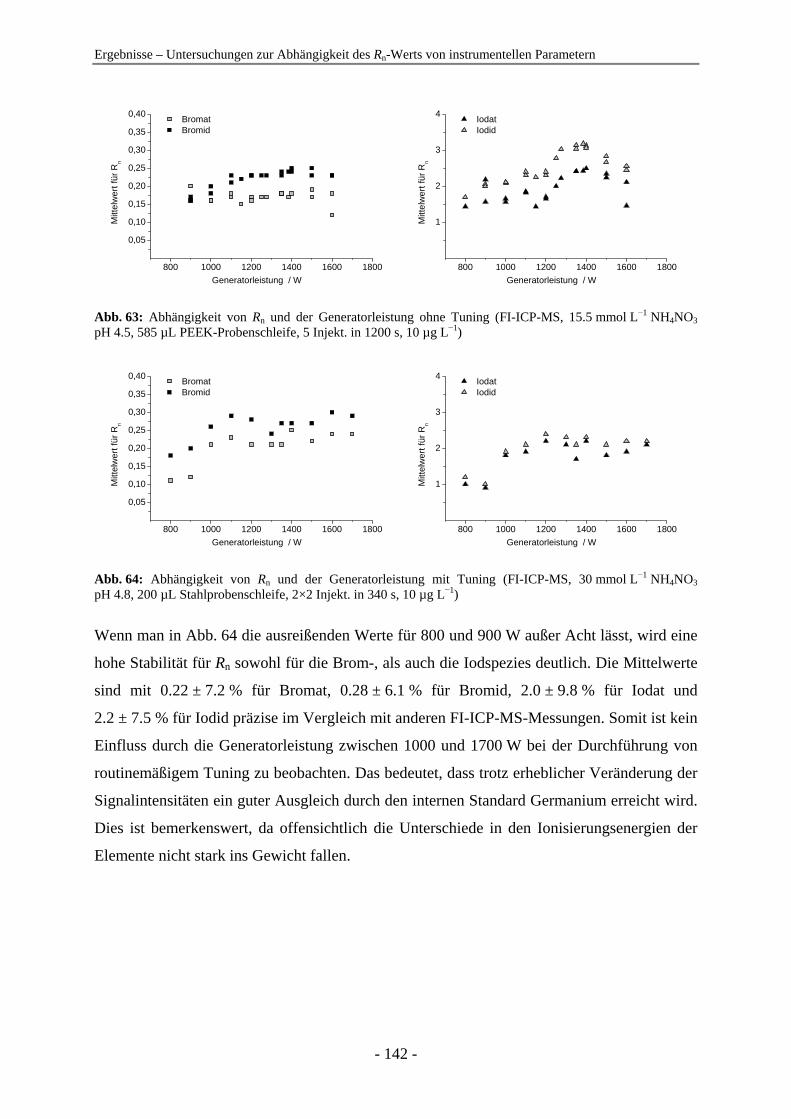
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
800 1000 1200 1400 1600 1800
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40 Bromat Bromid
Mitt
elw
ert f
ür R
n
Generatorleistung / W800 1000 1200 1400 1600 1800
1
2
3
4 Iodat Iodid
Mitt
elw
ert f
ür R
n
Generatorleistung / W
Abb. 63: Abhängigkeit von Rn und der Generatorleistung ohne Tuning (FI-ICP-MS, 15.5 mmol L−1 NH4NO3
H 4.5, 585 µL PEEK-Probenschleife, 5 Injekt. in 1200 s, 10 µg L−1) p
800 1000 1200 1400 160
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
- 142 -
0 1800
Bromat Bromid
Mitt
elw
ert f
ür R
n
2
3
4
800 1000 1200 1400 1600 1800
1
Iodat Iodid
Mitt
elw
ert f
ür R
n
Generatorleistung / W Generatorleistung / W
rt, da offensichtlich die Unterschiede in den Ionisierungsenergien der
Elemente nicht stark ins Gewicht fallen.
Abb. 64: Abhängigkeit von Rn und der Generatorleistung mit Tuning (FI-ICP-MS, 30 mmol L−1 NH4NO3 pH 4.8, 200 µL Stahlprobenschleife, 2×2 Injekt. in 340 s, 10 µg L−1)
Wenn man in Abb. 64 die ausreißenden Werte für 800 und 900 W außer Acht lässt, wird eine
hohe Stabilität für Rn sowohl für die Brom-, als auch die Iodspezies deutlich. Die Mittelwerte
sind mit 0.22 ± 7.2 % für Bromat, 0.28 ± 6.1 % für Bromid, 2.0 ± 9.8 % für Iodat und
2.2 ± 7.5 % für Iodid präzise im Vergleich mit anderen FI-ICP-MS-Messungen. Somit ist kein
Einfluss durch die Generatorleistung zwischen 1000 und 1700 W bei der Durchführung von
routinemäßigem Tuning zu beobachten. Das bedeutet, dass trotz erheblicher Veränderung der
Signalintensitäten ein guter Ausgleich durch den internen Standard Germanium erreicht wird.
Dies ist bemerkenswe
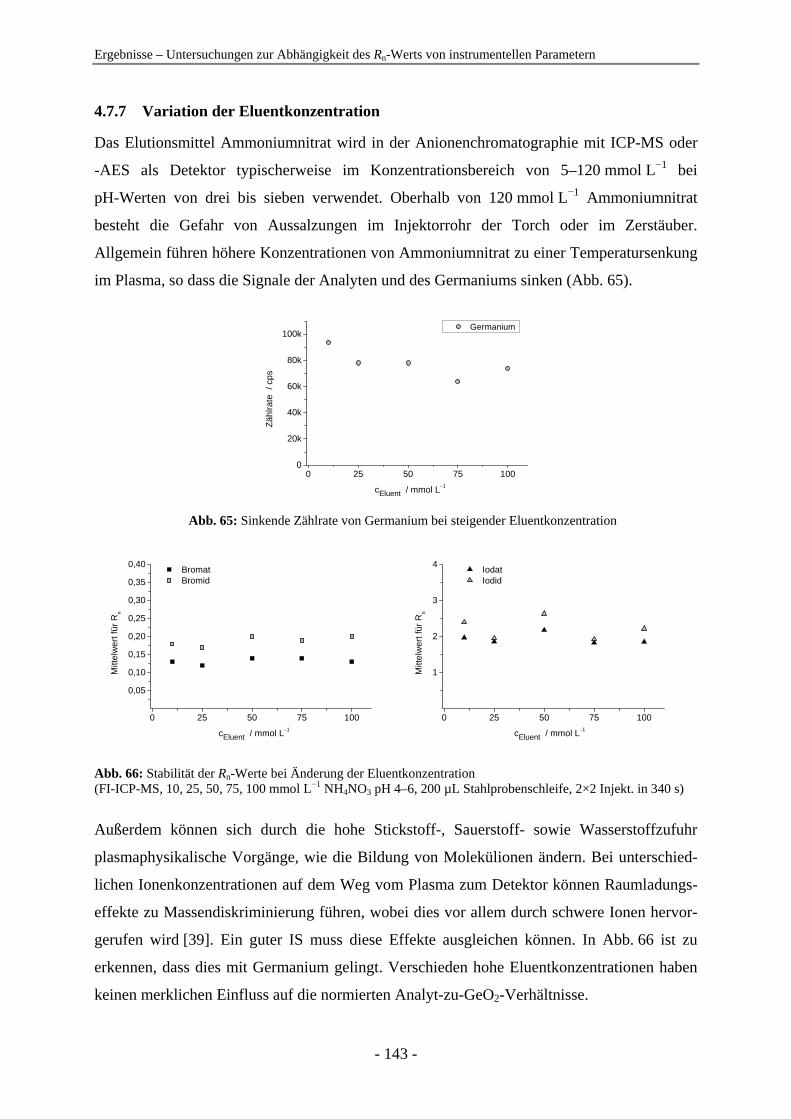
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 143 -
4.7.7 Variation der Eluentkonzentration
Das Elutionsmittel Ammoniumnitrat wird in der Anionenchromatographie mit ICP-MS oder
-AES als Detektor typischerweise im Konzentrationsbereich von 5–120 mmol L−1 bei
pH-Werten von drei bis sieben verwendet. Oberhalb von 120 mmol L−1 Ammoniumnitrat
besteht die Gefahr von Aussalzungen im Injektorrohr der Torch oder im Zerstäuber.
Allgemein führen höhere Konzentrationen von Ammoniumnitrat zu einer Temperatursenkung
im Plasma, so dass die Signale der Analyten und des Germaniums sinken (Abb. 65).
0 25 50 75 1000
20k
40k
60k
80k
100k
Zähl
rate
/ c
ps
cEluent / mmol L−1
Germanium
Abb. 65: Sinkende Zählrate von Germanium bei steigender Eluentkonzentration
0 25 50 75 100
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40 Bromat Bromid
Mitt
elw
ert f
ür R
n
cEluent / mmol L−1
0 25 50 75 100
1
2
3
4 Iodat Iodid
Mitt
elw
ert f
ür R
n
cEluent / mmol L−1
Abb. 66: Stabilität der Rn-Werte bei Änderung der Eluentkonzentration (FI-ICP-MS, 10, 25, 50, 75, 100 mmol L−1 NH4NO3 pH 4–6, 200 µL Stahlprobenschleife, 2×2 Injekt. in 340 s)
Außerdem können sich durch die hohe Stickstoff-, Sauerstoff- sowie Wasserstoffzufuhr
plasmaphysikalische Vorgänge, wie die Bildung von Molekülionen ändern. Bei unterschied-
lichen Ionenkonzentrationen auf dem Weg vom Plasma zum Detektor können Raumladungs-
effekte zu Massendiskriminierung führen, wobei dies vor allem durch schwere Ionen hervor-
gerufen wird [39]. Ein guter IS muss diese Effekte ausgleichen können. In Abb. 66 ist zu
erkennen, dass dies mit Germanium gelingt. Verschieden hohe Eluentkonzentrationen haben
keinen merklichen Einfluss auf die normierten Analyt-zu-GeO2-Verhältnisse.
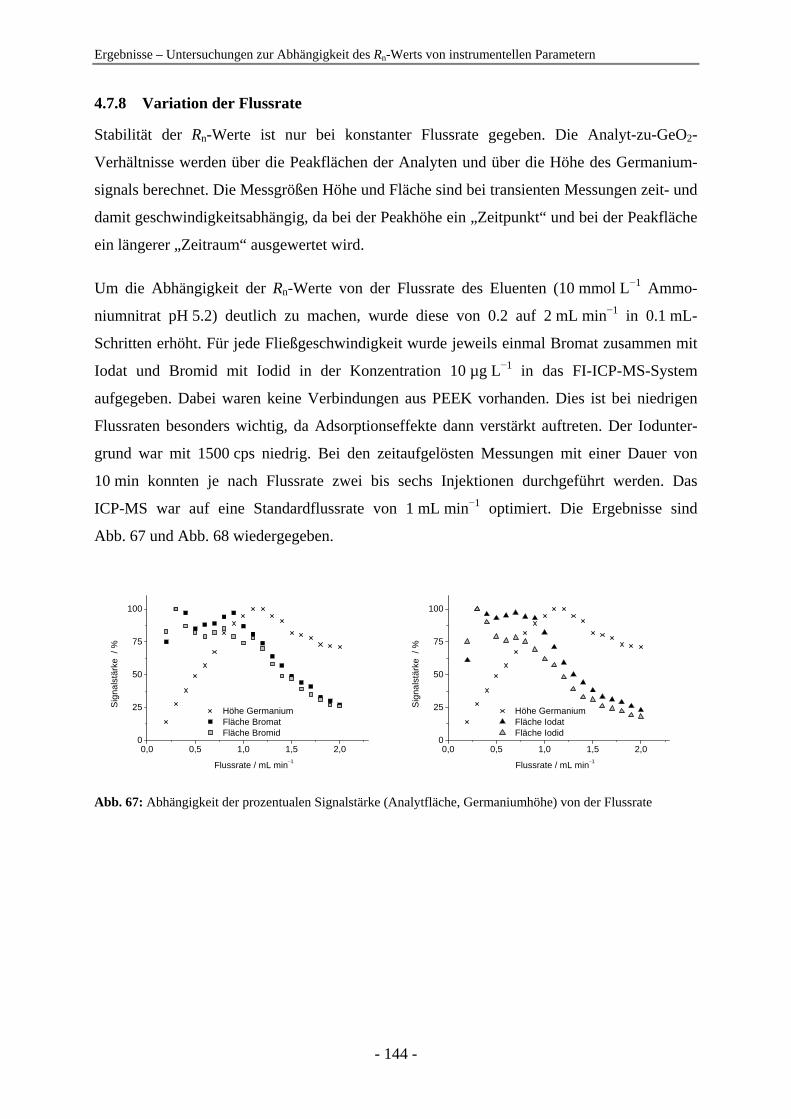
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 144 -
4.7.8 Variation der Flussrate
Stabilität der R -Werte ist nur bei konstanter Flussrate gegeben. Die Analyt-zu-GeO -
zeit- und
n 2
Verhältnisse werden über die Peakflächen der Analyten und über die Höhe des Germanium-
signals berechnet. Die Messgrößen Höhe und Fläche sind bei transienten Messungen
damit geschwindigkeitsabhängig, da bei der Peakhöhe ein „Zeitpunkt“ und bei der Peakfläche
ein längerer „Zeitraum“ ausgewertet wird.
Um die Abhängigkeit der Rn-Werte von der Flussrate des Eluenten (10 mmol L−1 Ammo-
niumnitrat pH 5.2) deutlich zu machen, wurde diese von 0.2 auf 2 mL min−1 in 0.1 mL-
Schritten erhöht. Für jede Fließgeschwindigkeit wurde jeweils einmal Bromat zusammen mit
Iodat und Bromid mit Iodid in der Konzentration 10 µg L−1 in das FI-ICP-MS-System
aufgegeben. Dabei waren keine Verbindungen aus PEEK vorhanden. Dies ist bei niedrigen
Flussraten besonders wichtig, da Adsorptionseffekte dann verstärkt auftreten. Der Iodunter-
grund war mit 1500 cps niedrig. Bei den zeitaufgelösten Messungen mit einer Dauer von
10 min konnten je nach Flussrate zwei bis sechs Injektionen durchgeführt werden. Das
ICP-MS war auf eine Standardflussrate von 1 mL min−1 optimiert. Die Ergebnisse sind
Abb. 67 und Abb. 68 wiedergegeben.
0,0 0,5 1,0 1,5 2,00
25
50
75
100
Sig
nals
tärk
e /
%
Flussrate / mL min−1
Höhe Germanium Fläche Bromat Fläche Bromid
0,0 0,5 1,0 1,5 2,00
25
50
75
100
Höhe Germanium Fläche Iodat Fläche Iodid
Sig
nals
tärk
e /
%
Flussrate / mL min−1
Abb. 67: Abhängigkeit der prozentualen Signalstärke (Analytfläche, Germaniumhöhe) von der Flussrate
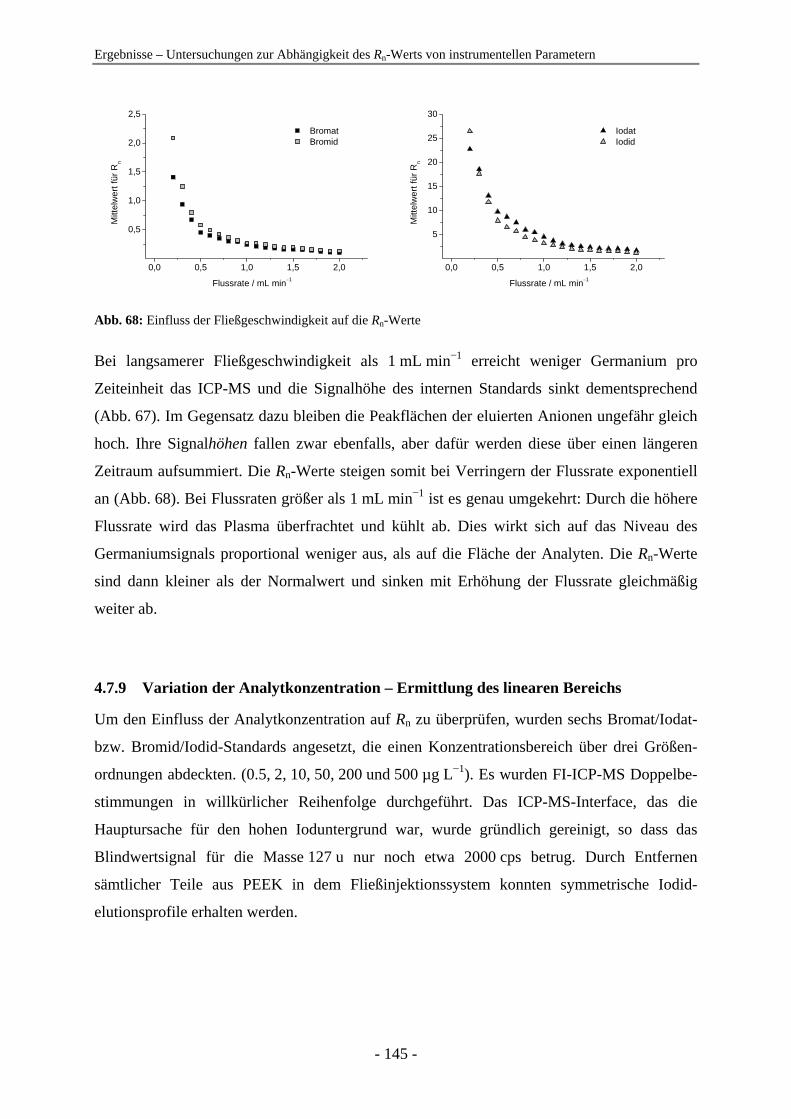
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
0,0 0,5 1,0 1,5 2,0
0,5
1,0
1,5
2,0
2,5
Bromat Bromid
Mitt
elw
ert f
ür R
n
Flussrate / mL min−1
0,0 0,5 1,0 1,5 2,0
5
10
15
20
25
30
Iodat Iodid
Mitt
elw
ert f
ür R
n
Flussrate / mL min−1
Abb. 68: Einfluss der Fließgeschwindigkeit auf die Rn-Werte
Bei langsamerer Fließgeschwindigkeit als 1 mL min−1 erreicht weniger Germanium pro
Zeiteinheit das ICP-MS und die Signalhöhe des internen Standards sinkt dementsprechend
(Abb. 67). Im Gegensatz dazu bleiben die Peakflächen der eluierten Anionen ungefähr gleich
hoch. Ihre Signalhöhen fallen zwar ebenfalls, aber dafür werden diese über einen längeren
Zeitraum aufsummiert. Die Rn-Werte steigen somit bei Verringern der Flussrate exponentiell
an (Abb. 68). Bei Flussraten größer als 1 mL min−1 ist es genau umgekehrt: Durch die höhere
Flussrate wird das Plasma überfrachtet und kühlt ab. Dies wirkt sich auf das Niveau des
Germaniumsignals proportional weniger aus, als auf die Fläche der Analyten. Die Rn-Werte
ur noch etwa 2000 cps betrug. Durch Entfernen
sämtlicher Teile aus PEEK in dem Fließinjektionssystem konnten symmetrische Iodid-
elutionsprofile erhalten werden.
sind dann kleiner als der Normalwert und sinken mit Erhöhung der Flussrate gleichmäßig
weiter ab.
4.7.9 Variation der Analytkonzentration – Ermittlung des linearen Bereichs
Um den Einfluss der Analytkonzentration auf Rn zu überprüfen, wurden sechs Bromat/Iodat-
bzw. Bromid/Iodid-Standards angesetzt, die einen Konzentrationsbereich über drei Größen-
ordnungen abdeckten. (0.5, 2, 10, 50, 200 und 500 µg L−1). Es wurden FI-ICP-MS Doppelbe-
stimmungen in willkürlicher Reihenfolge durchgeführt. Das ICP-MS-Interface, das die
Hauptursache für den hohen Ioduntergrund war, wurde gründlich gereinigt, so dass das
Blindwertsignal für die Masse 127 u n
- 145 -
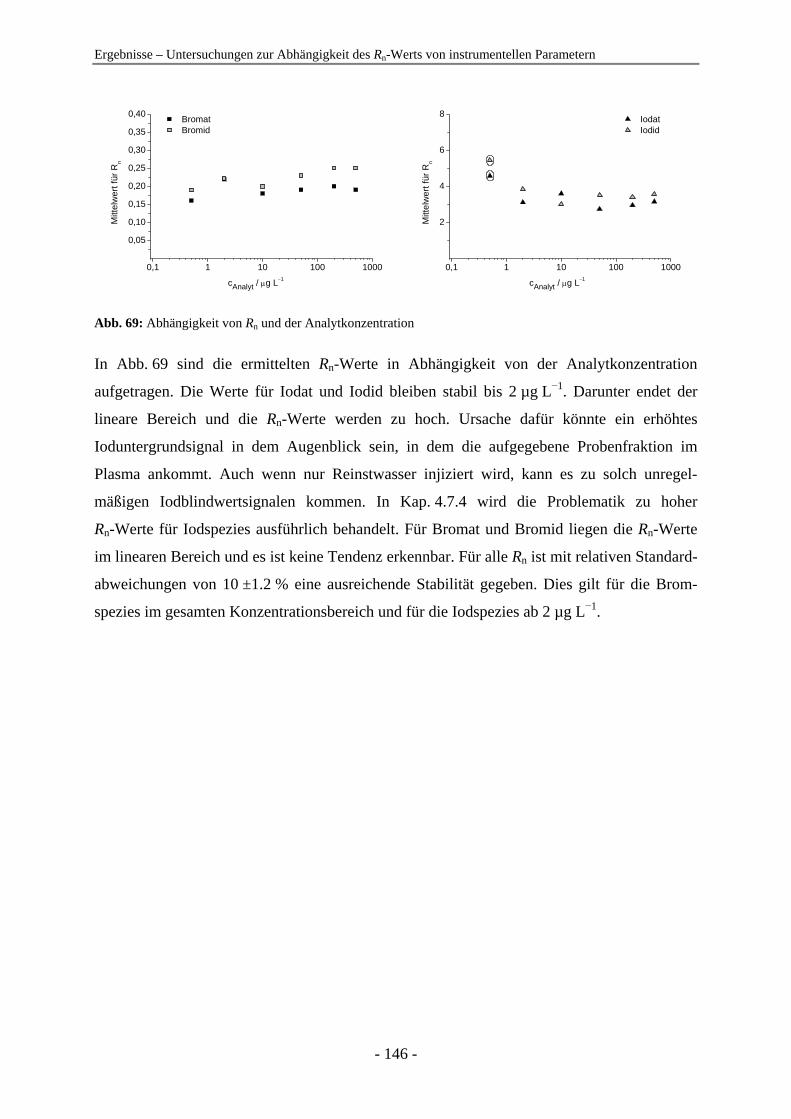
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
0, 04
- 146 -
0,1 1 10 100 1000
0,05
0,10
0,15
0,20
0,25
0,30
0,35 Bromat Bromid
Mitt
elw
ert f
ür R
n
cAnalyt / µg L−1
0,1 1 10 100
2
4
6
8
1000
Iodat Iodid
Mitt
elw
ert f
ür R
n
cAnalyt / µg L−1
Abb. 69: Abhängigkeit von Rn und der Analytkonzentration
In Abb. 69 sind die ermittelten Rn-Werte in Abhängigkeit von der Analytkonzentration
aufgetragen. Die Werte für Iodat und Iodid bleiben stabil bis 2 µg L−1. Darunter endet der
lineare Bereich und die Rn-Werte werden zu hoch. Ursache dafür könnte ein erhöhtes
Ioduntergrundsignal in dem Augenblick sein, in dem die aufgegebene Probenfraktion im
Plasma ankommt. Auch wenn nur Reinstwasser injiziert wird, kann es zu solch unregel-
mäßigen Iodblindwertsignalen kommen. In Kap. 4.7.4 wird die Problematik zu hoher
Rn-Werte für I e Rn-Werte
im linearen Bereich und es ist keine Tendenz erkennbar. Für alle Rn ist mit relativen Standard-
abweichungen von 10 ±1.2 % eine ausreichende Stabilität gegeben. Dies gilt für die Brom-
spezies im gesamten Konzentrationsbereich und für die Iodspezies ab 2 µg L−1.
odspezies ausführlich behandelt. Für Bromat und Bromid liegen di

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 147 -
rupolsteuerung vorgestellt. Es wird zudem auf
4.7.10 Variation von Quadrupoleinstellungen
Theoretische Grundlagen des Quadrupolmassenfilters
Die prinzipielle Funktionsweise des Quadrupolmassenfilters wird in Kap. 2.1.2 (Seite 16)
beschrieben. Um die Auswirkungen von Quadrupoleinstellungen einschätzen zu können,
werden im Folgenden die Parameter zur Quad
die zugrunde liegenden Gleichungen und auf die Auswirkungen der Steuerparameter
eingegangen.
Die stabilen Trajektorien der Ionen im Quadrupol werden durch die zwei Parameter q und a
bestimmt (Gl. 27 und Gl. 28), wobei q von der Amplitude V der Wechselspannung abhängt
und a eine Funktion der angelegten Gleichspannung U ist.
Gl. 27 20
2
8 rm ω
z Ua =
Gl. 28 20
2
4 rm ω
z V q =
U = Gleichspannung z = Ladung des Ions m = Masse des Ions
r0 = Radius des RF-Felds
V = Amplitude der Wechselspannung ω = 2 π f, f = Frequenz der Wechselspannung
In den Stabilitätsdiagrammen in Abb. 70 ist eine Region mit stabilen Flugbahnen erkennbar.
Bei einem Massenscan wird die so genannte Arbeitsgerade abgefahren, d.h. U und V werden
gleichmäßig erhöht, so dass ihr Verhältnis immer gleich bleibt. Jeder Dreiecksplot umschließt
den Bereich, in dem für eine bestimmte Masse die Flugbahn gleichzeitig in X- und in
Y-Richtung stabil ist. Dort wo die Arbeitsgerade die Fläche schneidet, wird der Quadrupol für
kte in
t das Auflösungsvermögen sowie die Intensität und damit die Empfind-
lichkeit des Massenspektrometers.
das zugehörige Masse-zu-Ladungs-Verhältnis durchlässig. Der Abstand der Schnittpun
X-Richtung bestimm
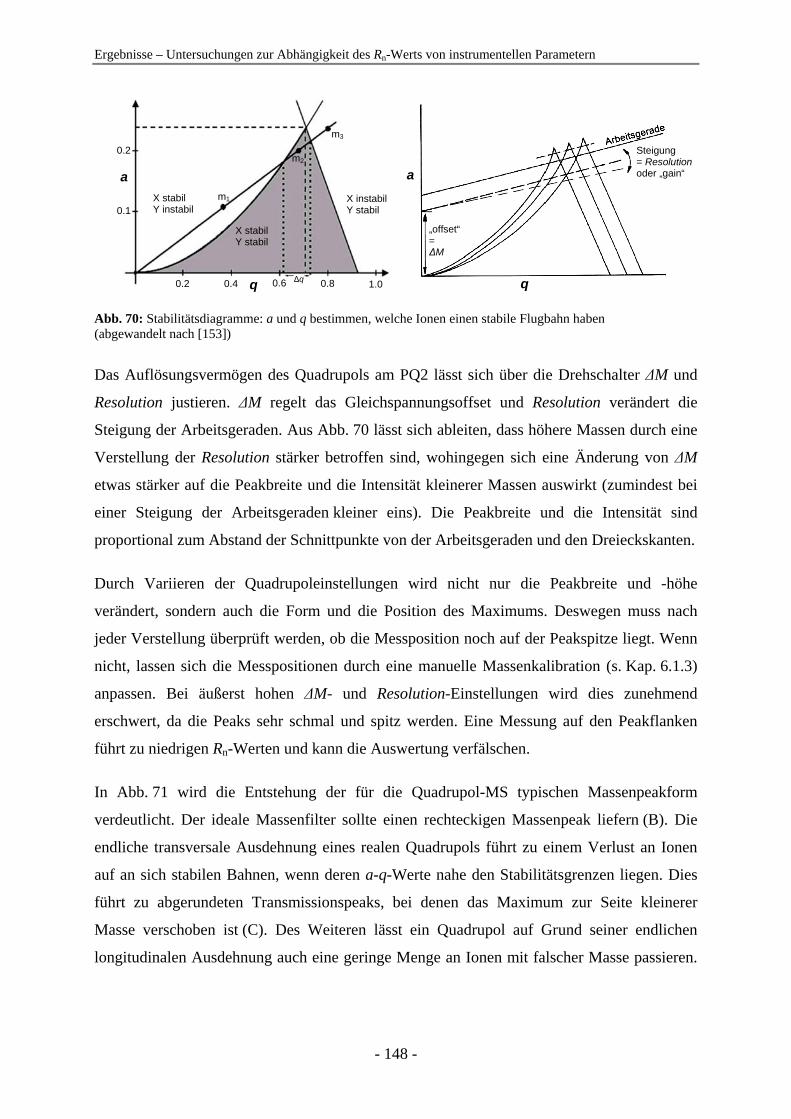
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
Abb. 70: Stabilitätsdiagramme: a und(abgewandelt nach [153])
sondern auch die F
der Verstellung überprüft wer
nicht, lassen sich die Messposi
Masse verschoben ist (C). Des
longitudinalen Ausdehnung auc
a a
q ∆
0.2
0.1
0.2 0.4 0.6
m1
m2
m3
X stabil Y instabil
X stabil Y stabil
X instabil Y stabil
„offset“= ∆M
Steigung = Resolution oder „gain“
Das Auflösungsvermögen des
Resolution justieren. ∆M rege
Steigung der Arbeitsgeraden. A
Verstellung der Resolution stär
etwas stärker auf die Peakbreit
einer Steigung der Arbeitsger
proportional zum Abstand der S
Durch Variieren der Quadrup
verändert,
je
anpassen. Bei äußerst hohen
erschwert, da die Peaks sehr s
führt zu niedrigen Rn-Werten un
In Abb. 71 wird die Entstehu
verdeutlicht. Der ideale Masse
endliche transversale Ausdehnu
auf an sich stabilen Bahnen, w
führt zu abgerundeten Transm
q
- 148 -
q bestimmen, welche Ionen einen stabile Flugbahn haben
.
orm und die Position des Maximums. Deswegen muss nach
den, ob die Messposition noch auf der Peakspitze liegt. Wenn
tionen durch eine manuelle Massenkalibration (s. Kap. 6.1.3)
unehmend
bei denen das Maximum zur Seite kleinerer
Weiteren lässt ein Quadrupol auf Grund seiner endlichen
h eine geringe Menge an Ionen mit falscher Masse passieren.
q 0.8 1.0
Quadrupols am PQ2 lässt sich über die Drehschalter ∆M und
lt das Gleichspannungsoffset und Resolution verändert die
us Abb. 70 lässt sich ableiten, dass höhere Massen durch eine
ker betroffen sind, wohingegen sich eine Änderung von ∆M
e und die Intensität kleinerer Massen auswirkt (zumindest bei
aden kleiner eins) Die Peakbreite und die Intensität sind
chnittpunkte von der Arbeitsgeraden und den Dreieckskanten.
oleinstellungen wird nicht nur die Peakbreite und -höhe
∆M- und Resolution-Einstellungen wird dies z
chmal und spitz werden. Eine Messung auf den Peakflanken
d kann die Auswertung verfälschen.
ng der für die Quadrupol-MS typischen Massenpeakform
nfilter sollte einen rechteckigen Massenpeak liefern (B). Die
ng eines realen Quadrupols führt zu einem Verlust an Ionen
enn deren a-q-Werte nahe den Stabilitätsgrenzen liegen. Dies
issionspeaks,

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 149 -
Daraus resultieren gemäß (D) Ausläufer an den Massenpeaks, die zur kleineren Massenseite
hin stärker ausgeprägt sind.
Abb. 71: Entwicklung der realen Massenpeakform beim Quadrupol [153]: (A) Spitze des Stabilitätsbereichs, (B) erwartete Peakform für ein ideales unendliches Feld, (C) für ein transversal limitiertes Feld (D) für ein transversal und longitudinal limitiertes Feld
Variation der Steigung der Arbeitsgeraden Resolution
Planung und Durchführung der Experimente
Die Abhängigkeit der Rn-Werte von der Resolution wurde in drei unabhängigen Messreihen
bestimmt. Bei der Reihe 040712/16 wurde mit einem Messpunkt pro Massenpeak gearbeitet,
wie es der Standardmethode entspricht. Allerdings gibt es einige Probleme, die die zu diesem
Zeitpunkt durchgeführten Messungen betreffen. Zum einen war der Ioduntergrund im
ICP-MS noch übermäßig hoch, was einen Drift der Rn-Werte für Iod verursachte und die
Aussagekraft der Messungen verringerte (s. Kap. 4.6.2 und Kap. 4.7.4). Zum anderen wurde
die Messposition auf den Massenpeaks nach Ändern der Resolution nicht überprüft und keine
manuelle Massenkalibration durchgeführt. Deswegen ist es nicht auszuschließen, dass die
Messposition bei extremen Werten der Resolution auf den Peakflanken gelegen hat.
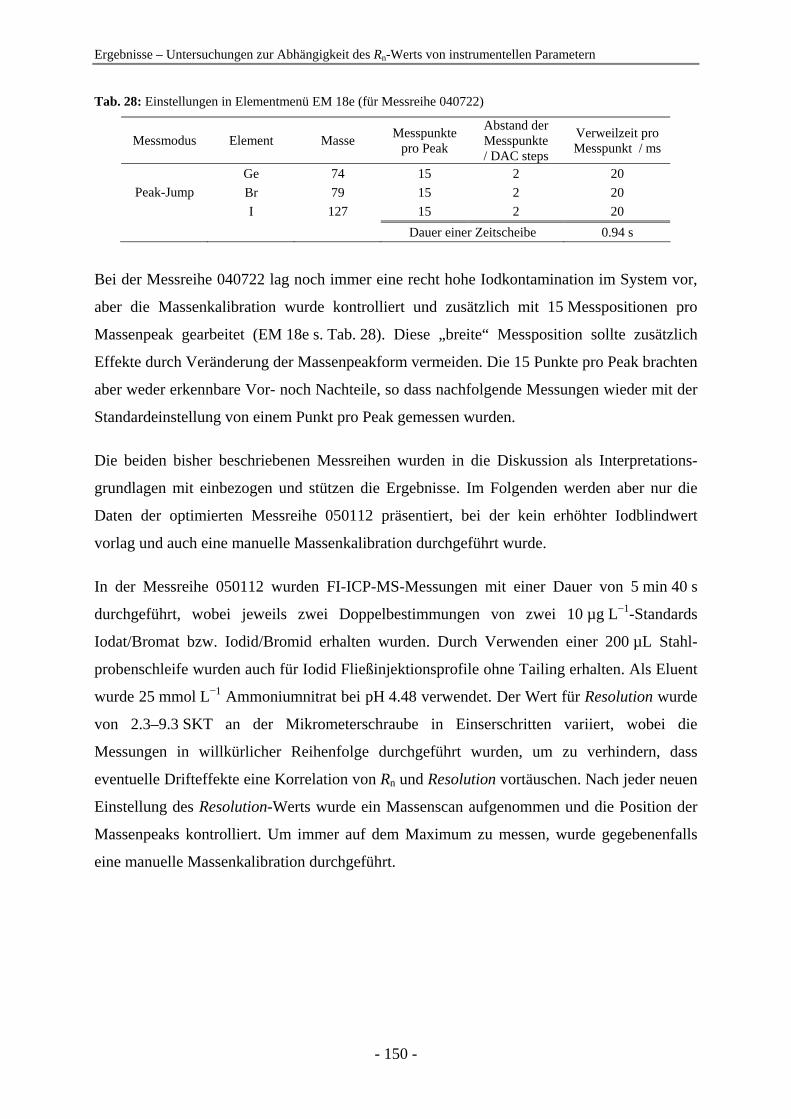
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 150 -
Tab. 28: Einstellungen in Elementmenü EM 18e (für Messreihe 040722)
spunkte pro Peak
Abstand der Messpunkte / DAC steps
Verweilzeit pro Messpunkt / ms Messmodus Element Masse Mes
Ge 74 15 2 20 Br 79 15 2 20 Peak-Jump I 127 15 2 20
Dauer einer Zeitscheibe 0.94 s
Bei der Messreihe 040722 lag noch immer eine recht hohe Iodkontamination im System vor,
aber die Massenkalibration wurde kontrolliert und zusätzlich mit 15 Messpositionen pro
Massenpeak gearbeitet (EM 18e s. Tab. 28). Diese „breite“ Messposition sollte zusätzlich
urden.
id n bis er beschriebenen Messreihen wurden in die Diskussion als Interpretations-
ag n mit einbezogen und stützen die Ergebnisse. Im Folgenden werden aber nur die
Daten der optimierten Messreihe 050112 präsentiert, bei der kein erhöhter Iodblindwert
40 s
durchgeführt, wobei jeweils zwei Doppelbestimmungen von zwei 10 µg L−1-Standards
er auf dem Maximum zu messen, wurde gegebenenfalls
eine manuelle Massenkalibration durchgeführt.
Effekte durch Veränderung der Massenpeakform vermeiden. Die 15 Punkte pro Peak brachten
aber weder erkennbare Vor- noch Nachteile, so dass nachfolgende Messungen wieder mit der
Standardeinstellung von einem Punkt pro Peak gemessen w
Die be e h
grundl e
vorlag und auch eine manuelle Massenkalibration durchgeführt wurde.
In der Messreihe 050112 wurden FI-ICP-MS-Messungen mit einer Dauer von 5 min
Iodat/Bromat bzw. Iodid/Bromid erhalten wurden. Durch Verwenden einer 200 µL Stahl-
probenschleife wurden auch für Iodid Fließinjektionsprofile ohne Tailing erhalten. Als Eluent
wurde 25 mmol L−1 Ammoniumnitrat bei pH 4.48 verwendet. Der Wert für Resolution wurde
von 2.3–9.3 SKT an der Mikrometerschraube in Einserschritten variiert, wobei die
Messungen in willkürlicher Reihenfolge durchgeführt wurden, um zu verhindern, dass
eventuelle Drifteffekte eine Korrelation von Rn und Resolution vortäuschen. Nach jeder neuen
Einstellung des Resolution-Werts wurde ein Massenscan aufgenommen und die Position der
Massenpeaks kontrolliert. Um imm
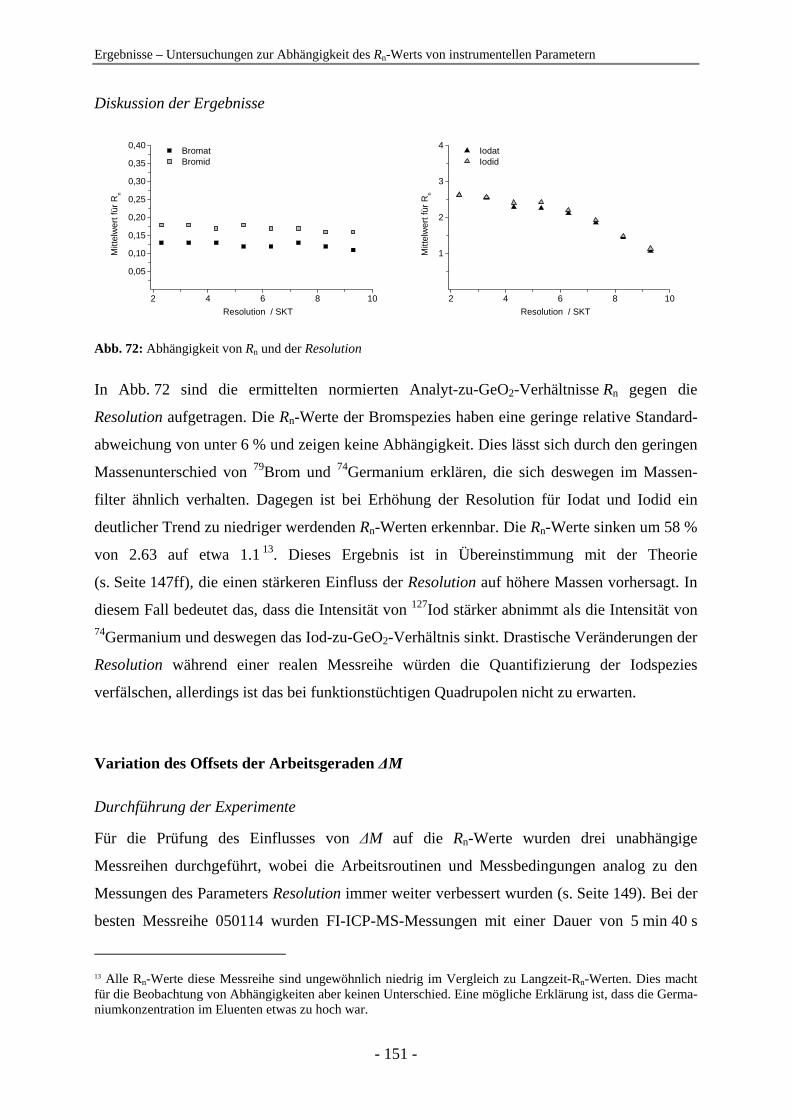
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 151 -
Diskussion der Ergebnisse
02 4 6 8 1
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40 Bromat Bromid
Mitt
elw
ert f
ür R
n
Resolution / SKT2 4 6 8 1
1
2
3
4
0
Iodat Iodid
Mitt
elw
ert f
ür R
n
Resolution / SKT
Abb. 72: Abhängigkeit von Rn und der Resolution
In Abb. 72 sind die ermittelten normierten Analyt-zu-GeO2-Verhältnisse Rn gegen die
Resolution aufgetragen. Die Rn-Werte der Bromspezies haben eine geringe relative Standard-
abweichung von unter 6 % und zeigen keine Abhängigkeit. Dies lässt sich durch den geringen
Massenunterschied von 79Brom und 74Germanium erklären, die sich deswegen im Massen-
filter ähnlich verhalten. Dagegen ist bei Erhöhung der Resolution für Iodat und Iodid ein
deutlicher Trend zu niedriger werdenden Rn-Werten erkennbar. Die Rn-Werte sinken um 58 %
von 2.63 auf etwa 1.1 13. Dieses Ergebnis ist in Übereinstimmung mit der Theorie
(s. Seite 147ff), die einen stärkeren Einfluss der Resolution auf höhere Massen vorhersagt. In
diesem Fall bedeutet das, dass die Intensität von 127Iod stärker abnimmt als die Intensität von 74Germanium und deswegen das Iod-zu-GeO2-Verhältnis sinkt. Drastische Veränderungen der
Resolution während einer realen Messreihe würden die Quantifizierung der Iodspezies
verfälschen, allerdings ist das bei funktionstüchtigen Quadrupolen nicht zu erwarten.
Variation des Offsets der Arbeitsgeraden ∆M
Durchführung der Experimente
Für die Prüfung des Einflusses von ∆M auf die Rn-Werte wurden drei unabhängige
Messreihen durchgeführt, wobei die Arbeitsroutinen und Messbedingungen analog zu den
Messungen des Parameters Resolution immer weiter verbessert wurden (s. Seite 149). Bei der
besten Messreihe 050114 wurden FI-ICP-MS-Messungen mit einer Dauer von 5 min 40 s
13 Alle Rn-Werte diese Messreihe sind ungewöhnlich niedrig im Vergleich zu Langzeit-Rn-Werten. Dies macht für die Beobachtung von Abhängigkeiten aber keinen Unterschied. Eine mögliche Erklärung ist, dass die Germa-niumkonzentration im Eluenten etwas zu hoch war.
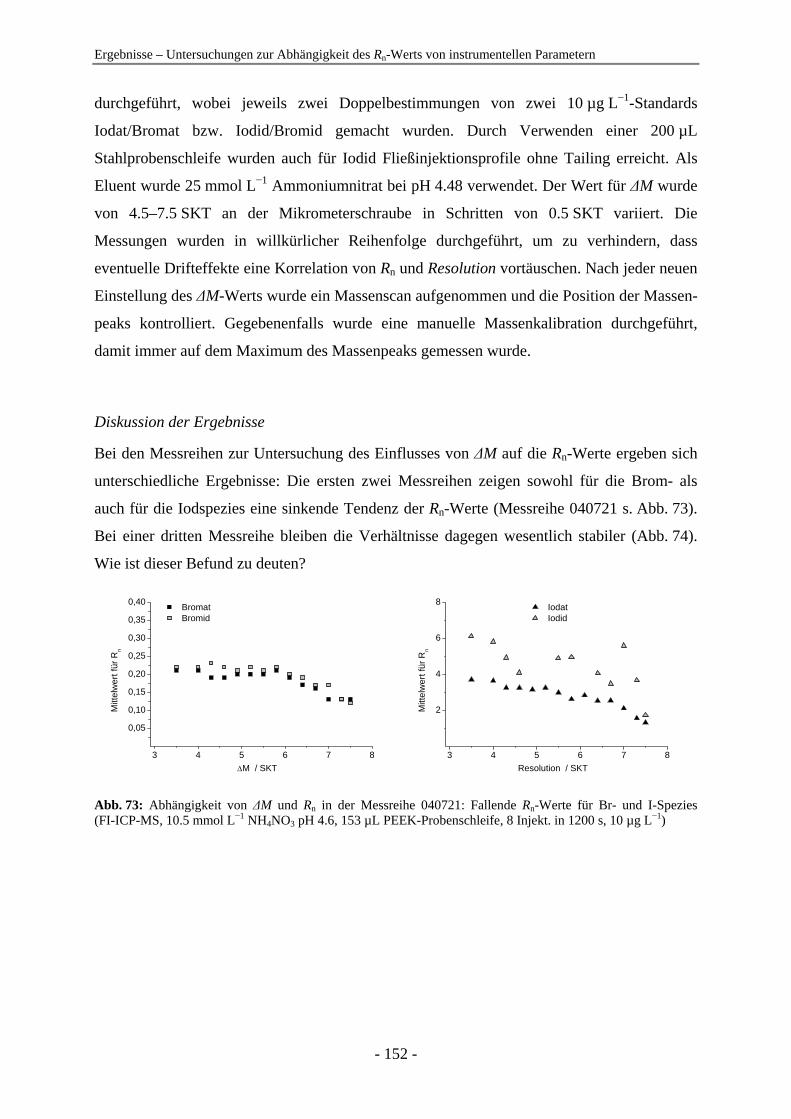
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 152 -
romid gemacht wurden. Durch Verwenden einer 200 µL
Stahlprobenschleife wurden auch für Iodid Fli tionsprofile ohne Tailing erreicht. Als
in Schritten von 0.5 SKT variiert. Die
essungen wurden in willkürlicher Reihenfolge durchgeführt, um zu verhindern, dass
tion vortäuschen. Nach jeder neuen
Einstellung des ∆M-Werts wurde ein Massenscan aufgenommen und die Position der Massen-
ine manuelle Massenkalibration durchgeführt,
0,15
0,
0,30
0,40
durchgeführt, wobei jeweils zwei Doppelbestimmungen von zwei 10 µg L−1-Standards
Iodat/Bromat bzw. Iodid/B
eßinjek
Eluent wurde 25 mmol L−1 Ammoniumnitrat bei pH 4.48 verwendet. Der Wert für ∆M wurde
von 4.5–7.5 SKT an der Mikrometerschraube
M
eventuelle Drifteffekte eine Korrelation von Rn und Resolu
peaks kontrolliert. Gegebenenfalls wurde e
damit immer auf dem Maximum des Massenpeaks gemessen wurde.
Diskussion der Ergebnisse
Bei den Messreihen zur Untersuchung des Einflusses von ∆M auf die Rn-Werte ergeben sich
unterschiedliche Ergebnisse: Die ersten zwei Messreihen zeigen sowohl für die Brom- als
auch für die Iodspezies eine sinkende Tendenz der Rn-Werte (Messreihe 040721 s. Abb. 73).
Bei einer dritten Messreihe bleiben die Verhältnisse dagegen wesentlich stabiler (Abb. 74).
Wie ist dieser Befund zu deuten?
3 4 5 6 7 8
0,05
0,
Bromat
10
20
0,25
0,35 Bromid
Mitt
elw
ert f
ür R
n
∆M / SKT
8
6
8
3 4 5 6 7
2
4
Iodat Iodid
Resolution / SKT
Mitt
elw
ert f
ür R
n
Abb. 73: Abhängigkeit von ∆M und Rn in der Messreihe 040721: Fallende Rn-Werte für Br- und I-Spezies (FI-ICP-MS, 10.5 mmol L−1 NH4NO3 pH 4.6, 153 µL PEEK-Probenschleife, 8 Injekt. in 1200 s, 10 µg L−1)
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 153 -
4 5 6 7 8
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40 Bromat Bromid
lwer
t für
Rn
2
3
4M
itte
∆M / SKT
4 5 6 7 8
1
Iodat Iodid
lwer
t für
Rn
Mitt
e
sehr spitz und das Maximum kann sich
erschieben. Die Intensität ist nur noch gering, woraus eine schlechte Zählstatistik und eine
ungleichmäßige Peakform resultieren. Dies macht es besonders schwer, eine Massenkali-
bei der die Messpositionen genau auf den Maxima liegen. Wenn
einer oder mehrere dieser Effekte bei den gemessenen Massen 74, 79 und 127 u unterschied-
lich auftreten, kommt es zu einer Veränderung der Rn-Werte wie in den Abb. Abb. 73.
Da die Steigung der Arbeitsgeraden beim Quadrupol normalerweise kleiner eins ist, hat eine
Verschiebung des Achsenabschnitts eine größere Auswirkung auf kleine Massen. Wenn
folglich leichtere Elemente stärker an Intensität verlieren, so müsste das Verhältnis von 79Brom zu 74Germanium der Theorie nach kaum merklich fallen und das von 127Iod zu 74Germanium steigen. Diese Theorie kann durch Abb. 74 bestätigt werden. Wie erwähnt
icht überlagern. In Abb. 73 fallen die Iod-Rn-Werte
z.B. mit steigendem ∆M deutlich ab. Dies kann durch Auswaschen des derzeitig hohen
∆M / SKT
Abb. 74: Abhängigkeit von ∆M und Rn in der Messreihe 050114: Alle Rn sehr stabil, wobei Rn für Br-Spezies leicht fallend, Rn für I-Spezies leicht steigend (FI-ICP-MS, 50 mmol L−1 NH4NO3 pH 4.82, 200 µL Stahlproben-schleife, 2×2 Injekt. in 340 s, 10 µg L−1)
Durch Erhöhen des Gleichspannungoffsets des Quadrupols ∆M wird die Arbeitsgerade
angehoben und die Filterfunktion erhöht. Man gewinnt, wie auch über den Parameter
Resolution, an Auflösungsvermögen wohingegen man an Intensität verliert. Die Veränderung
von ∆M wirkt sich allerdings im untersuchten SKT- bzw. Spannungsbereich drastischer aus:
Bei hohen Werten werden die Massenpeaks
v
brierung zu gewährleisten,
können andere Effekte diese Tendenzen le
Ioduntergrunds erklärt werden (s. Kap. 4.6.2).
Abschließend bleibt festzuhalten, dass sich zwar leichte Tendenzen abzeichnen, diese aber bei
guter Kontrolle der Messposition nur relative Standardabweichen für Rn von ca. 9.5 % ± 1 %
ergeben. Die Korrektur der veränderten Quadrupolbedingungen durch den internen Standard
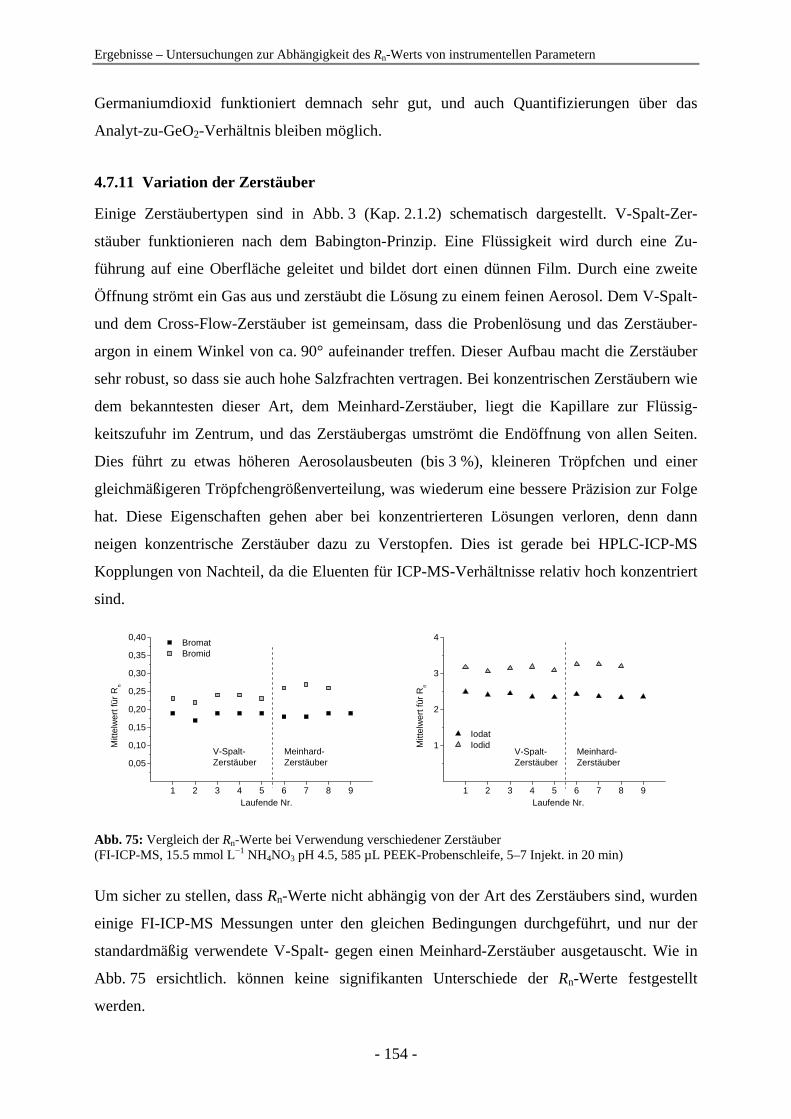
Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 154 -
ine Zu-
hrung auf eine Oberfläche geleitet und bildet dort einen dünnen Film. Durch eine zweite
Öffnung strömt ein Gas aus und zerstäubt die Lösung zu einem feinen Aerosol. Dem V-Spalt-
dass die Probenlösung und das Zerstäuber-
ffen. Dieser Aufbau macht die Zerstäuber
Germaniumdioxid funktioniert demnach sehr gut, und auch Quantifizierungen über das
Analyt-zu-GeO2-Verhältnis bleiben möglich.
4.7.11 Variation der Zerstäuber
Einige Zerstäubertypen sind in Abb. 3 (Kap. 2.1.2) schematisch dargestellt. V-Spalt-Zer-
stäuber funktionieren nach dem Babington-Prinzip. Eine Flüssigkeit wird durch e
fü
und dem Cross-Flow-Zerstäuber ist gemeinsam,
argon in einem Winkel von ca. 90° aufeinander tre
sehr robust, so dass sie auch hohe Salzfrachten vertragen. Bei konzentrischen Zerstäubern wie
dem bekanntesten dieser Art, dem Meinhard-Zerstäuber, liegt die Kapillare zur Flüssig-
keitszufuhr im Zentrum, und das Zerstäubergas umströmt die Endöffnung von allen Seiten.
Dies führt zu etwas höheren Aerosolausbeuten (bis 3 %), kleineren Tröpfchen und einer
gleichmäßigeren Tröpfchengrößenverteilung, was wiederum eine bessere Präzision zur Folge
hat. Diese Eigenschaften gehen aber bei konzentrierteren Lösungen verloren, denn dann
neigen konzentrische Zerstäuber dazu zu Verstopfen. Dies ist gerade bei HPLC-ICP-MS
Kopplungen von Nachteil, da die Eluenten für ICP-MS-Verhältnisse relativ hoch konzentriert
sind.
0,10
0,15
0,20
0,25
0,30
0,35
0,40
1 2 3 4 5 6 7 8 9
0,05
Laufende Nr.
ZerstäuberZerstäuberMeinhard-V-Spalt-
Bromat Bromid
Mitt
elw
ert f
ür R
n
1 2 3 4 5 6 7 8 9
1
2
3
4
Iodat IodidM
ittel
wer
t für
Rn
V-Spa
Laufende Nr.
Meinhard-Zerstäuber
lt-Zerstäuber
einige FI-ICP-MS Messungen unter den gleichen Bedingungen durchgeführt, und nur der
standardmäßig verwendete V-Spalt- gegen einen Meinhard-Zerstäuber ausgetauscht. Wie in
Abb. 75: Vergleich der Rn-Werte bei Verwendung verschiedener Zerstäuber (FI-ICP-MS, 15.5 mmol L−1 NH4NO3 pH 4.5, 585 µL PEEK-Probenschleife, 5–7 Injekt. in 20 min)
Um sicher zu stellen, dass Rn-Werte nicht abhängig von der Art des Zerstäubers sind, wurden
Abb. 75 ersichtlich. können keine signifikanten Unterschiede der Rn-Werte festgestellt
werden.

Ergebnisse – Untersuchungen zur Abhängigkeit des Rn-Werts von instrumentellen Parametern
- 155 -
ter aufgelistet bei deren Änderung die Rn-Werte weitgehend
stabil blieben:
− Ionisierungsenergie des Tune-Elements
− Masse des Tune-Elements
− Generatorleistung (1000-1700 W)
− Samplingtiefe (für Iodspezies leichter Anstieg der Rn-Werte mit geringerer Samplingtiefe)
− Zerstäuber
− Eluentkonzentration
Konzentration)
− Achsenabschnitt der Arbeitsgerade des Quadrupols ∆M
(für Bromspezies minimal geringere, für Iodspezies minimal höhere Rn-Werte bei
Erhöhung von ∆M)
− Steigung der Arbeitsgerade des Quadrupols Resolution:
Die Rn-Werte bleiben nur im Falle von Bromspezies stabil!
Die Rn-Werte bleiben nicht stabil bei V a eter und Mess-
− Resolution
4.7.12 Zusammenfassung: Abhängigkeiten der Rn-Werte
Im Folgenden werden die Parame
− Analytkonzentration (für Iodspezies leichter Anstieg der Rn-Werte mit geringerer
ari tion nachstehender Param
bedingungen:
Eine Erhöhung dieser Quadrupoleinstellung führt zu fallenden Rn-Werten für die
Iodspezies.
− Messposition auf den Massenpeaks
Zur sicheren Festlegung der Messposition auf den Massenpeaks ist eine manuelle
Massenkalibration erforderlich (s. Kap. 6.1.3)
− Ioduntergrund
Ein erhöhter Ioduntergrund im ICP-MS wirkt sich bei FI-ICP-MS-Messungen signifikant
auf die Iod-Rn-Werte aus, so dass zu hohe Rn-Werte resultieren (s. Kap. 4.7.4)

Zusammenfassung und Ausblick
- 156 -
r d
lorid, Bromid und Phosphat, zusammen mit in
ischer Spezies von sieben Elementen (Chlor,
Brom, Iod, Schwefel, Phosphor, Arsen und Chrom). Einen großen Vorteil stellte hierbei die
hkeit durch den elementselektiven Detektor dar, weil es
und kaum spezifische Interferenzen auf ihren Masse-zu-Ladungs-Verhältnissen
auftreten. Die ermittelten Nachweisgrenzen liegen sehr niedrig und betragen für Iodat
0.05 µg L−1, für Bromat 0.07 µg L−1, für Bromid 0.1 µg L−1 und für Iodid 0.15 µg L−1.
5 Zusammenfassung und Ausblick
In den meisten bisher veröffentlichten Arbeiten auf dem Gebiet der IC-ICP-MS konzen-
t ierten sich ie Forscher entweder auf wenige Analyten oder auf die Speziesanalyse von nur
einem oder zwei Elementen. Um die Multielementfähigkeit des ICP-MS voll auszuschöpfen,
wurde im Rahmen dieser Dissertation das Ziel verfolgt, möglichst viele Analyten bzw. die
Spezies zahlreicher verschiedener Elemente gleichzeitig zu bestimmen. Die Analyse gängiger
Anionen für die Ionenchromatographie, wie Ch
Wasser seltenen Spezies, wie z.B. Arsenat oder den Anionen der Bromessigsäuren, sollte
diesem Gedanken Rechnung tragen.
Durch den Einsatz der On-line-Kopplung IC-ICP-MS gelang es, bis zu 15 Anionen
gleichzeitig in einem chromatographischen Lauf innerhalb von 35 min zu analysieren. Dies
entsprach einer parallelen Bestimmung anion
zusätzliche Unterscheidungsmöglic
dadurch nicht zwingend notwendig war, alle Anionen chromatographisch voneinander zu
trennen. Es konnten mehr Analyten zusammen gemessen und die Analysenzeiten verkürzt
werden, was eine erhebliche Kostenersparnis in der Spurenanalytik bewirkt. Ein Haupt-
augenmerk lag auf der simultanen Analyse toxischer oder krebserregender Spezies, wie z.B.
Bromat, Chromat, Arsenat, Perchlorat und den Bromessigsäuren, da hier routinemäßige
Analysen zur Grenzwertüberwachung schon gesetzlich vorgeschrieben oder in nächster Zeit
zu erwarten sind. Die gesamte Liste der Analyten bestand aus Iodat, Iodid, Bromat, Bromid,
MBA−, DBA−, TBA−, Chromat, Phosphat, Arsenat, Thiocyanat, Chlorid, Chlorat, Chlorit und
Perchlorat. Der in diesem Zusammenhang eingeführte Begriff „Multianionenanalyse“ weist
auf die Vielzahl der gleichzeitig analysierbaren Anionenspezies hin.
Wenn man die Nachweisstärke der IC-ICP-MS für halogen-, phosphor- und schwefelhaltige
Verbindungen vergleicht, so fällt sie für Elementspezies von Brom und insbesondere von Iod
sehr gut aus, da eine ausreichende Ionisierung dieser Elemente im Plasma erreicht werden
kann

Zusammenfassung und Ausblick
- 157 -
Für die ionenchromatographischen Trennungen sind im Arbeitskreis von Prof. Dr. Seubert
hergestellte Anionenaustauscher eingesetzt worden. Vier Arten von stationären Phasen
wurden anhand ihrer Retentionsparameter charakterisiert: VBC- und GMA-Latex-Anionen-
austauscher sowie oberflächenfunktionalisierte und gepfropfte Anionenaustauscher. Die
stationären Phasen unterschieden sich darüber hinaus in ihren Austauschergruppen, die mit
den Aminen EDMA, DMEA, DEMA oder TEA funktionalisiert waren. Im Folgenden werden
die wichtigsten Trenneigenschaften, die für die untersuchten stationären Phasen im Ammo-
niumnitrat-Elutionssystem festgestellt wurden, zu ammengefasst:
gerung der Retention des polarisierbaren Anions Iodid in Relation zu Bromid und
eine Verbesserung der Trennleistung. Die über alle Anionen gemittelte Trennleistung für
mmoniumnitrat-Elutionssystem
n Gruppen erwiesen sich als gut geeignet für die Multianionenanalyse. Ein
auffälliger Unterschied im Vergleich zu gepfropften Anionenaustauschern bestand in der
anionen. Dies ermöglichte zwar einerseits deren
immung, hatte aber andererseits teilweise eine Koelution von
id und DBA− zur Folge.
Durch die hohe Hydrophilie der GMA-Latexpartikel zeigte sich auch diese Art stationärer
Phasen prinzipiell für die Trennung polarisierbarer Anionen geeignet. Diese und ebenso
Da die bisher verfügbaren Säulen allerdings
öglichkeiten eingeschränkt.
− alisierte Anionenaustauscher
n auf Grund ihrer hohen Kapazität
n z.B. sogar in 0.1%igen Salz-
lösungen Spuren von Bromat im unteren µg L−1-Bereich bestimmt werden. Diese Art von
s
− VBC-Latex-Anionenaustauscher
Mit VBC-Latex-Anionenaustauschern ließ sich sowohl eine symmetrische Peakform, als
auch eine relativ kurze Retentionszeit für polarisierbare Anionen erreichen. Dies setzte
voraus, dass Austauschergruppen mit hydrophilen Substituenten vorhanden waren. Die
Erhöhung der Anzahl von Ethanolgruppen an den Ammoniumfunktionen bewirkte eine
Verrin
die Säule L061202 TEA betrug 20000 TP m−1, was im A
einer hohen Trenneffizienz entspricht. VBC-Latex-Anionenaustauscher mit hydrophilen
funktionelle
geringen Retention von Bromessigsäure
schnelle gleichzeitige Best
Bromat und MBA− bzw. von Brom
− GMA-Latex-Anionenaustauscher
die Anionen der Bromessigsäuren wiesen an GMA-Latexanionenaustauschern eine
besonders niedrige relative Retention auf.
einen sehr hohen Staudruck besaßen, waren ihre Einsatzm
Oberflächenfunktion
Oberflächenfunktionalisierte stationäre Phasen tolerierte
die Aufgabe von Proben mit hohem Matrixanteil. So konnte

Zusammenfassung und Ausblick
- 158 -
Anionenaustauschern eignete sich allerdings nicht für die Multianionenanalyse, da sich die
Retentionszeiten für viele Anionen zu stark unterschieden. Für polarisierbare Anionen wie
funktionalisierten stationären Phasen zu eluieren. Die Retention der Anionen
DBA− und TBA− erwies sich dafür ebenfalls als zu hoch. Die durchschnittliche Trenn-
ler Anionen im Ammoniumnitrat-Elutionssystem betrug für die Säule
ersuchten
hasen relativ niedrig.
−
er ließ sich eine Auflösung des Iodatpeaks
peak erreichen, ohne dass zugleich die Retentionszeiten für polarisierbare
en internen
Standard deutlich. Zudem gelang innerhalb von 40 min und unter isokratischen Bedin-
gungen die Elution des Perchlorats bei immer noch guter Trennung früh eluierender
Anionen. Auf Grund dieser Eigenschaften ist der Einsatz gepfropfter Anionenaustauscher
für die Multianionenanalyse zu empfehlen. Mit der Säule P3162120 EVO 3 DEMA wurde
eine durchschnittliche Trennleistung für alle Anionen von 11500 TP m−1 erzielt.
Die Retention der Anionen der Bromessigsäuren erwies sich an dieser Art von Anionen-
austauschern als außergewöhnlich hoch. So eluierte MBA− nach Bromat und DBA−
deutlich später als Bromid, wodurch eine sichere Unterscheidung dieser typischen
bromierten Desinfektionsbeiprodukte vorgenommen werden kann.
Der Einsatz einer wässrigen Lösung von Ammoniumnitrat als mobile Phase beinhaltete eine
Reihe von Vorteilen. Nach der Atomisierung dieses Eluents im Plasma können sich auch im
kühleren Interfacebereich ausschließlich gasförmige Stoffe bilden. Dies erhöht die Stabilität
der Messungen, da es nicht zu Ablagerungen von Partikeln auf den Cones des ICP-MS-Geräts
kommt. Mit Nitrat als Eluentanion lässt sich darüber hinaus im Vergleich zum Carbo-
nat/Bicarbonat-Elutionssystem die Peakform polarisierbarer Anionen verbessern. Auch der
eingestellte pH-Wert zwischen vier und sechs im Ammoniumnitrat-Elutionssystem hat einen
positiven Einfluss, weil hierdurch mehrfach deprotonierbare Säuren, wie z.B. Phosphor-,
Arsen- oder Chromsäure, größtenteils einfach geladen vorliegen. Dies verringert ihre
Perchlorat und Iodid war es zum Beispiel nicht möglich, sie innerhalb vertretbarer Zeit von
oberflächen
leistung al
P130198 DMEA 4500 TP m−1 und war damit im Vergleich zu den anderen unt
stationären P
Gepfropfte Anionenaustauscher
Nur mit einem gepfropften Anionenaustausch
vom Injektions
Anionen zu lang wurden. Eine solche gute Auflösung des Iodat- vom Injektionspeak
verbessert die Echtzeitkorrektur des Iodatsignals durch einen kontinuierlich

Zusammenfassung und Ausblick
- 159 -
Retention, verbessert somit ihre Nachweisgrenzen und hält die Gesamttrennzeiten für
xid als IS direkt dem Eluenten zugegeben werden kann
(Vorsäulenzugabe). Mit dem daraus resultierenden kontinuierlichen Signal kann zu jeder Zeit
tifizie-
rung mit nachfolgender Isotopenverdünnungsanalyse in ausgesuchten Proben zu sehen. Für
Multianionenanalysen niedrig.
Mit Germaniumdioxid ist ein bestens geeigneter interner Standard für die IC-ICP-MS mit
Ammoniumnitrat-Eluenten gefunden worden [154]. Alle Anforderungen, die an ISs für diese
Methode gestellt werden, werden erfüllt: Erstens ist das Molekül neutral und trotzdem aus-
reichend wasserlöslich. Zweitens hat das Element Germanium große Ähnlichkeit zu den
Analytelementen Brom und Iod in Bezug auf Masse und Ionisierungsenergie. Zu guter letzt
kommt es in Wasserproben selten vor und ist mit der ICP-MS interferenzfrei messbar.
Von Vorteil ist, dass Germaniumdio
der Messung eine IS-Korrektur durchgeführt werden. In diesem Zusammenhang wurde
erörtert, dass die Vorsäulenzugabe des IS die robustere Methodik im Vergleich zur Nach-
säulenzugabe über ein Mischungsmodul ist. Des Weiteren wurde anhand von Beispielen
gezeigt, dass instrumentelle Probleme und nicht-spezifische Interferenzen mit Hilfe des
kontinuierlichen Germaniumsignals in Echtzeit erkannt werden können. Dadurch lassen sich
schnell Maßnahmen zur Fehlerbehebung ergreifen und viele Fehlmessungen vermeiden.
Das auf definierte Stoffmengen normierte Signalverhältnis von Analyt-zu-GeO2 wurde als
Rn-Wert definiert. Die Stabilität der Rn-Werte lässt sich über ihre Standardabweichung
angeben und kann als Gütekriterium für die IS-Korrektur verwendet werden. Für die Analyten
Bromid, Bromat, Iodid und Iodat konnten hervorragende Korrekturqualitäten des IS Germa-
niumdioxid aufgezeigt werden. Die Rn-Werte erwiesen sich über einen Zeitraum von
36 Monaten und unter verschiedenen instrumentellen Bedingungen als außergewöhnlich
stabil. Die Langzeitstandardabweichungen der Rn-Werte beliefen sich auf 14 % für Bromid
und Bromat, 18 % für Iodat und 24 % für Iodid.
Basierend auf der Stabilität der Rn-Werte ist eine dauerhafte Ein-Punkt-Kalibration mit
(quasi-) Echtzeitkorrektur durch einen IS gegeben. Dadurch wird eine unaufwendige und
schnelle semiquantitative Analyse ermöglicht, da langwierige, regelmäßige Kalibrations-
reihen nicht mehr nötig sind. Zwar resultiert daraus eine verringerte Richtigkeit und Präzision,
die jedoch ohne weiteres für viele Problemstellungen in Kauf genommen werden kann. Eine
effiziente Kombination ist z.B. im schnellen Screening über die Analyt-zu-GeO2-Quan

Zusammenfassung und Ausblick
- 160 -
t. Die erhal-
tenen Ergebnisse zeugten von einer guten Korrelation mit den über konventionelle
onen. Ferner wurden Wasserproben aus einem Ring-
ve
erh
Um
wurden Fließinjektions-ICP-MS-Messungen unter
Bedingunge
Massenpeaks (m
ICP-MS oder im
stabil. Eine Ausnahm
Quadrupolparam
Quadrupols festgelegt wird. Unter realen
Änderung dieser Einstellung zu erwart
problem
n dieser Dissertation erzielt wurden, können weitere
Forschungen auf de
die Trenncharakteristika versch
system
oder neu z
schiedenen Trennm
Analysefragestellungen heranzie
nn der neue interne Standard für die
IC
ein
sch
eine erfolgreiche Durchführung der Isotopenverdünnung, mit der sehr genaue Ergebnisse
erzielt werden, ist schon vorher eine ungefähre Kenntnis der Analytkonzentration notwendig.
Mit Hilfe der Analyt-zu-GeO2-Quantifizierung kann diese besonders schnell ermittelt werden.
Aus den Screening-Daten können außerdem die Proben selektiert werden, für die sich eine
zusätzliche Isotopenverdünnungsanalyse lohnt.
Als Anwendungsbeispiel für die Quantifizierung mit Hilfe der Rn-Werte wurde in einer
Vielzahl von Mineralwasseranalysen Bromid, Bromat, Iodid und Iodat bestimm
Kalibration ermittelten Konzentrati
rsuch auf Bromat analysiert. Auch die hierbei über das Analyt-zu-GeO2-Verhältnis
altenen Ergebnisse stimmten mit den vorhandenen Referenzwerten überein.
die Stabilität der Rn-Werte zu überprüfen und mögliche Abhängigkeiten aufzudecken,
systematisch variierten instrumentellen
n durchgeführt. Unter Vorraussetzung konstanter Messpositionen auf den
anuelle Massenkalibration) und eines nicht erhöhten Ioduntergrunds im
Plasma-Argon, blieben die Rn-Werte bei Änderung der meisten Parameter
e stellten fallende Rn-Werte für Iodspezies bei der Erhöhung des
eters Resolution dar, mit dem die Steigung der Arbeitsgeraden des
Messbedingungen ist aber keine drastische
en, so dass dann das Analyt-zu-GeO2-Verhältnis
los zur Quantifizierung genutzt werden kann.
An die Ergebnisse, die im Rahme
m Gebiet der IC-ICP-MS anknüpfen: Die gewonnenen Kenntnisse über
iedener stationärer Phasen im Ammoniumnitrat-Elutions-
helfen, geeignete stationäre Phasen für ein bestimmtes Trennproblem auszusuchen
u entwickeln. Die vorgestellten Beispiele für Multianionenanalysen mit ver-
aterialien kann man als Ausgangspunkt zur Bearbeitung neuartiger
hen. Die Zugabe von Germaniumdioxid in Ammoniumnitrat-
Eluenten ist hierbei ein nützliches Hilfsmittel, de
-ICP-MS kann zur Systemüberwachung, Signalkorrektur und schnellen Quantifizierung
gesetzt werden. Bisher sind die IS-Eigenschaften Germaniums in der IC-ICP-MS aus-
ließlich für Brom- und Iodspezies untersucht worden, weshalb als nächstes überprüft

Zusammenfassung und Ausblick
- 161 -
we
we
äh
Die Forschung m
beschränkte sich m
Ins
sauren funktionellen Gruppen hi en. Solche Moleküle liegen in neutralen Ammo-
niumnitrat-Eluenten anionisch vor, was sie für die Ionenchromatographie zugänglich macht.
Für die Anionenanalytik m
steigerungen denkbar. Um die Ge
könnte während der Trennung die Elutionskraft de
werden. In Hinblick auf die Soft
nachfolgenden Chrom
wert. Des Weiteren kann dank moderner Gerä
Nachweisgrenzen für Phosphor-,
Elem
Masse-zu-L gebildete
Molekülionen komm
elim
Nachweisstärke und Präzision m
insbesondere diese E en.
techniken ausgleichen.
rden könnte, inwieweit damit eine Signalkorrektur und die einfache Quantifizierung für
itere Elemente möglich ist. Es ist z.B. davon auszugehen, dass Germanium aufgrund der
nlichen Elementmassen ebenfalls gut als IS für Arsen- und Selenspezies geeignet ist.
it dem im Rahmen dieser Dissertation verwendeten Kopplungssystem
it Ausnahme der Anionen der Bromessigsäuren auf anorganische Anionen.
ofern könnten als neue Analytspezies weitere organische, halogenhaltige Moleküle mit
nzukomm
it der IC-ICP-MS sind noch diverse instrumentelle Leistungs-
samttrennzeiten für die Multianionenanalysen zu verkürzen,
s Eluenten durch Gradiententechnik erhöht
ware ist eine stärkere Automatisierung der Messungen, der
atogrammdarstellung sowie der benötigten Berechnungen erstrebens-
tetechnik daran gearbeitet werden, die
Schwefel und Chlorspezies zu senken. Die Detektion dieser
ente mit der ICP-MS besitzt noch ein großes Verbesserungspotential, da es auf ihren
adungs-Verhältnissen zu spezifischen Interferenzen durch im Plasma
t. Solche Interferenzen können mit Hilfe der Reaktionszellentechnologie
iniert werden. Die Möglichkeit auch Chlor, Phosphor und Schwefel mit höherer
essen zu können, würde viele neue Perspektiven eröffnen, da
lemente in biologischen Systemen häufig vorkomm
Zukünftig werden mit der ICP-MS gekoppelte Trennmethoden vermehrt Einzug in die Life
Sciences halten. In diesem Zusammenhang gewinnt die Analyse komplexer Proben, wie
biologische Flüssigkeiten oder Gewebeextrakte, an Bedeutung. Für solche Proben mit noch
vielen unidentifizierten Elementspezies zeigt sich die Anwendung verschiedener HPLC-MS-
Methoden angebracht. Beispielsweise liegt die Stärke der ESI-MS in der Fähigkeit zur
sicheren Identifizierung von Elementspezies auf Grund der Strukturinformationen. Der
Hauptvorteil der ICP-MS hingegen ist die robuste und leistungsfähige Quantifizierung. Durch
die komplementären Informationen molekülselektiver MS-Methoden und elementselektiver
ICP-MS-Detektion lassen sich die Stärken und Schwächen einzelner Massenspektrometrie-

Anhang
6 Anhang
- 162 -
det, ist
6.1 Instrumentelles
6.1.1 Die IC-ICP-MS-Kopplung
Die IC-ICP-MS-Kopplung (s. Abb. 76) besteht aus einem Eluentenvorratsbehälter, einer
HPLC-Pumpe, einem 6-Port/3-Wege-Ventil, der Trennsäule und dem ICP-MS als Detektor.
Da HPLC-Pumpen erst funktionieren, wenn sich Flüssigkeit in den Pumpkolben befin
es ist empfehlenswert, das Eluentenvorratsgefäß höher als die HPLC-Pumpe aufzustellen,
damit die Pumpkolben mit Hilfe des hydrostatischen Drucks leichter befüllt werden können.
Durch Hochstellen des Eluenten wird des Weiteren verhindert, dass sich während eines
Dauerbetriebs Luftblasen in den Kolben sammeln, die zu Pumpaussetzern führen können.
06.7
Säule3-Wege-Ventil
Spritze
Probe
ProbenschleifeEluent
HPLC-Pumpe
ICP-MS
Abb. 76: Schematische Darstellung der IC-ICP-MS-Kopplung (Ventil in Injektionsposition)
Das Dreiwegeventil hat zwei unterschiedliche Stellungen: die Befüllungs- und die Injektions-
position. In ersterer Position lässt sich eine Probenschleife (typisches Volumen 50–1000 µL)
befüllen. Bei vielen HPLC-Aufbauten ist es üblich, die Probe mit einer Spritze in die Proben-
schleife zu injizieren. Dabei besteht die Gefahr, eine Luftblase aus der Spritze mit aufzu-
geben. Durch die umgekehrte Durchführung, bei der eine Probe mit einer Spritze angesaugt
wird, lässt sich dieses Problem vermeiden. Man kann zudem bei durchsichtigen Kapillaren
visuell sicherstellen, dass keine undichten Verbindungen bestehen, durch die Luft mit
angesaugt wird.
In der zweiten Stellung des Ventils, der Injektionsposition, lässt sich der Flussweg durch die
Probenschleife zur Säule schalten und somit die Trennung einer aufgegebenen Probe starten.

Anhang
- 163 -
der IC-Säule mit dem
Ansaugschlauch zum Zerstäuber des ICP-MS verbunden wird. Eine typische chromato-
Allgemeine Messbedingungen
Zwischen Injektionsventil und Trennsäule sollte ein 2 µm-In-Line-Filter eingebaut werden,
der Partikel, Bakterien oder Algen aus Probenlösungen oder Eluenten entfernt.
Die Kopplung des Ionenchromatographiesystems mit einem ICP-MS ist denkbar einfach. Sie
wird normalerweise dadurch erreicht, dass die Ausgangskapillare
graphische Flussrate von 1 mL min−1 ist mit den meisten pneumatischen Zerstäubern
kompatibel. Als Kopplungsadapter eignet sich ein weiches Schlauchstück, es sind aber auch
kommerzielle Schraubverbinder erhältlich. Der Flussweg zwischen der Trennsäule und dem
Detektor ist möglichst kurz zu halten, um die Bandenverbreiterung zu minimieren.
Die IC-ICP-MS-Messungen wurden im so genannten TRA-Modus der ICP-MS-Software
PQ Vision 4.3 durchgeführt. Dazu wurde ein Elementmenü (EM) eingestellt, in dem die zu
messenden Isotope und die Anzahl und Dauer der Messpunkte für jedes Isotop festgelegt sind.
Aus diesen Parametern ergibt sich die Länge einer Einzelmessung (Zeitscheibe). Eine
komplette transiente Messung setzt sich aus einer bestimmten Anzahl an Zeitscheiben
zusammen, die wiederum von der gewählten Gesamtdauer der Messung abhängt. Bei den
ionenchromatographischen Messungen zur Bestimmung von Bromid, Bromat, Iodid und
Iodat, wurde das EM 11 nach Tab. 29 verwendet. Für alle Multianionenanalysen kam das
EM 20a (Tab. 30) zum Einsatz.
Tab. 29: Einstellungen im Elementmenü EM 11
Messmodus Element Masse Messpunkte pro Peak
Verweilzeit pro Messpunkt / ms
Cr, [35ClOH]+ 52 1 200 Ge 74 1 40 Br 79 1 400
Peak-Jump
I 127 1 200 Dauer einer Zeitscheibe 0.89 s

Anhang
- 164 -
Tab. 30: Einstellungen im Elementmenü EM 20a
Messmodus Element Masse Messpunkte pro Peak
Verweilzeit pro Messpunkt / ms
P 31 1 100 Cl 35 1 100
S bzw. [SO]+ 48 1 100 Cr 52 1 100 Ge 74 1 100 As 75 1 100 Br 79 1 100
Peak-Jump
I 127 1 100 Dauer einer Zeitscheibe 0.88 s
Datenauswertung
Die transienten Daten wurden im Programm PQ-Vision® (Thermo, Winsford) als ASCII-
Textdateien im csv-Format (comma separated variable) abgespeichert. Zur weiteren Ver-
arbeitung wurden die Daten in Tabellenkalkulationsprogramme importiert. Vor der Peak-
integration wurden die Chromatogramme über den Savitzky-Golay-Algorithmus geglättet
(zweite Ordnung, je vier Punkte nach links und rechts). Die Berechnung chromatographischer
Kenngrößen erfolgte gemäß den Gleichungen in Kap. 2.2.4.
Messungen, bei denen nur eine oder zwei Massenspuren (z.B. nur Brom- und Iodspezies)
ausgewertet werden mussten, wurden auch in dem Programm IC-Net® (Metrohm, Herisau)
bearbeitet. Die Konvertierung in das von IC-Net® importierbare Textformat erfolgte mit
Tabellenkalkulationsprogrammen.
6.1.2 Das FI-ICP-MS-System
Der allgemeine Aufbau einer FI-ICP-MS-Kopplung ist in Abb. 77 dargestellt. Der Aufbau
unterscheidet sich von der IC-ICP-MS hauptsächlich darin, dass die Probe nicht über eine
Säule geführt wird. Da HPLC-Pumpen für den Betrieb unter Hochdruck konzipiert sind, sollte
aber eine Lastsäule vor der Probenschleife eingebaut werden. Der dadurch erzeugte
Gegendruck führt zu einer gleichmäßigeren Förderleistung der HPLC-Pumpe. Das Pack-
material in der Lastsäule ist beliebig, muss nur rein und einige Zeit konditioniert worden sein,
um den Eluenten nicht zu kontaminieren.

Anhang
06.7
LastsäuleSpritze
Probe
Probenschleife
3-Wege-Ventil
Eluent
HPLC-Pumpe
ICP-MS
sen
der Fall ist.
Das in der Chromatographie häufig verwendete Polymer PEEK ist für die FIA nicht zu
empfehlen, wenn polarisierbare Anionen wie Iodid analysiert werden sollen. Iodid wird auch
von kurzen PEEK-Kapillaren schon merklich zurückgehalten, was zu einem ausgeprägten
Tailing der Signale und zu Memoryeffekten führt. Die Retention von Iodid auf PEEK-
Oberflächen lässt sich hauptsächlich durch π-π-Wechselwirkungen an den aromatischen
d Kapillaren aus
Stahl oder PTFE (Polytetrafluorethylen, Teflon) wird die Form des Fließinjektionspeaks für
Abb. 77: Schematische Darstellung des FI-ICP-MS-Aufbaus (Ventil in Injektionsposition)
Bei der Fließinjektionsanalyse haben die Länge und das Material der Kapillaren große
Bedeutung. Alle Verbindungen sollten so kurz wie möglich gehalten werden, um die Peak-
verbreiterung durch Diffusion zu minimieren. Die Elutionsprofile der Fließinjektionsanalyse
sind breiter als chromatographische Peaks, da es nicht zu einer Anreicherung der Analyten
kommen kann, wie es durch Wechselwirkungen mit chromatographischen stationären Pha
Systemen des Polymers erklären. Durch Verwendung von Probenschleifen un
Iodid wesentlich verbessert (s. Abb. 78). Ein kleiner Nachteil entsteht durch Stahlproben-
schleifen: Beim Germaniumsignal des Eluenten wird ein breiterer Injektionspeak beobachtet,
der sich nur langsam wieder der Ausgangssignalhöhe annähert. Offensichtlich wird das
Germaniumdioxid in einer Probenschleife aus Stahl retardiert. Durch den breiten Injektions-
peak wird die Präzision der an dieser Stelle ermittelten Germaniumsignalhöhe vermindert.
- 165 -
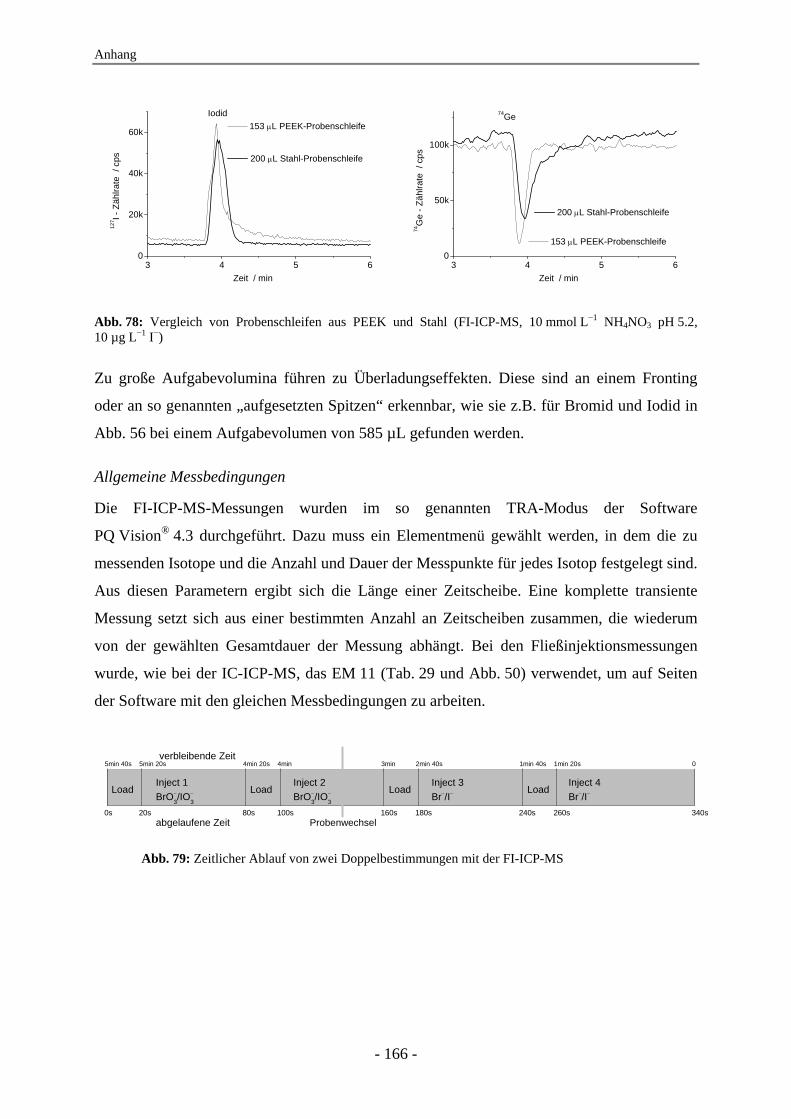
Anhang
- 166 -
3 4 5 60
20k
40k
60k 153 µL PEEK-ProbenschleifeIodid
200 µL Stahl-Probenschleifeps12
7 I - Z
ählra
te /
c
Zeit / min
3 4 5 60
50k
100k
200 µL Stahl-Probenschleife
153 µL PEEK-Probenschleife
74Ge
74G
e - Z
ählra
te /
cp
Zeit / min
Abb. 78: Vergleich von Probenschleifen aus PEEK und Stahl (FI-ICP-MS, 10 mmol L
s
3
ina führen zu Überladungseffekten. Diese sind an einem Fronting
tzten Spitzen“ erkennbar, wie sie z.B. für Bromid und Iodid in
11 (Tab. 29 und Abb. 50) verwendet, um auf Seiten
it den gleichen Messbedingungen zu arbeiten.
−1 NH4NO pH 5.2, 10 µg L−1 I−)
Zu große Aufgabevolum
oder an so genannten „aufgese
Abb. 56 bei einem Aufgabevolumen von 585 µL gefunden werden.
Allgemeine Messbedingungen
Die FI-ICP-MS-Messungen wurden im so genannten TRA-Modus der Software
PQ Vision® 4.3 durchgeführt. Dazu muss ein Elementmenü gewählt werden, in dem die zu
messenden Isotope und die Anzahl und Dauer der Messpunkte für jedes Isotop festgelegt sind.
Aus diesen Parametern ergibt sich die Länge einer Zeitscheibe. Eine komplette transiente
Messung setzt sich aus einer bestimmten Anzahl an Zeitscheiben zusammen, die wiederum
von der gewählten Gesamtdauer der Messung abhängt. Bei den Fließinjektionsmessungen
wurde, wie bei der IC-ICP-MS, das EM
der Software m
0s 20s 80s 100s 160s 180s 240s 260s 340s
5min 40s 5min 20 4min 2 2min 40s 1min 0
4Inject 3Br−/I−
Loadad
v
Inject 1BrO−
3/IO−
3
Load
ab
3−
3
serbleibende Zeit
0s 4min 3min 40s 1min 20s
Inject Br− −/I
Lo
gelaufene Zeit
LoadInject 2
−/IOBrO
Probenwechse
Abb. 79: itlicher Ablauf wei Dop FI-ICP-MS
l
Ze von z pelbestimmungen mit der

Anhang
- 167 -
estimmungen betrug 5 min 40 s. 14 Das
zugehörige mm enwec d Inje Ab llt. Es sind
jeweils 20 s vorgesehen, um ittels ein Spritze in die Probenschleife zu
ziehen. Dabei wird mindestens das Doppelte des Probenschleifenvolum ufgesogen, um
gleichzeitig Reste vorh
Proben ist zusätzlich eine Spülung der Beladungskapillare mit Reinstwasser empfehlenswert.
Die festgelegten Umschaltzeiten des Ventils auf „Load“ sind einzuhalten. Das Ventil darf erst
kurz vor der jeweiligen nächsten Injektion die Positio ur Befüllung der Probenschleife
umgestellt werd n, da es beim Schalten z ignalschwankung kommt. Eine
ersten Mal nach 20 s und danach alle 80 s injiziert.
ssung folgende Datenverarbeitung wurde analog zu den IC-ICP-MS-
Massen zugeordnet. Dazu wird
ein Standard gescannt, der fünf bis acht über den gesamten Massenbereich verteilte Elemente
ion werden die Positionen der restlichen Massen kalibriert.
ie über diese Funktion festgelegten DAC-Werte bestimmen die Peak-Jump-Messpositionen
bei transienten Messungen. Wenn nicht stärkere Drifteffekte auftreten, ist die Massen-
es oder auch darüber hinaus stabil.
Die optimale Gesamtmessdauer für zwei Doppelb
Zeitprogra für Prob hsel un ktionen ist b. 79 dargeste
die Probenlösung m er
ens a
eriger Messungen auszuspülen. Nach Aufgabe sehr konzentrierter
auf n z
e u einer Druck- und S
neue Probe wird beim
Die nach der Me
Messungen (Seite 164) durchgeführt.
6.1.3 Manuelle Massenkalibration
Die im Quadrupol selektierten und am Detektor gezählten Ionen werden mit Hilfe der
Massenkalibration in der Betriebssoftware ihren zugehörigen
enthält. Der gesamte Messbereich von 1–300 u ist im Quadrupol in etwa 64000 Einzelschritte,
so genannte DAC-steps unterteilt (DAC = direct to analog converter). Nach dem Scan weist
eine automatische Peakerkennung diesen DAC-Werten die Masse der jeweiligen Elemente zu.
Über eine lineare Regressionsfunkt
D
kalibration während eines Messtag
14 Erste FI-Messungen hatten noch längere Messzetenden Dateien reduzierte, aber aus zwei Gründen
iten von bis zu 30 min, was zwar die Anzahl der zu bearbei- nicht zu empfehlen ist: Im transienten Modus kann es in der
Software PQ-Vision zu Abstürzen kommen, weswegen möglichst häufig gespeichert werden sollte. Die ist nur bei kurzen Messungen möglich. Des Weiteren bleibt die Auswertung übersichtlicher, wenn pro Datei immer nur ein Parameter variiert wurde.
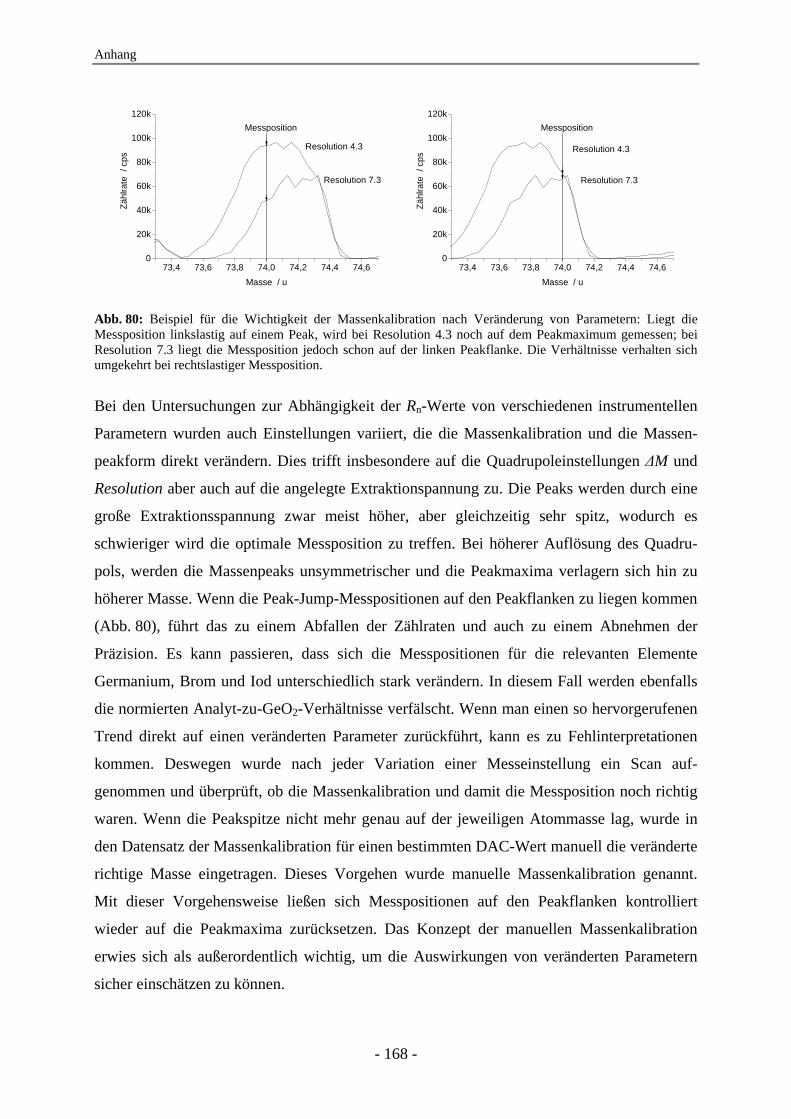
Anhang
- 168 -
73,4 73,6 73,8 74,0 74,2 74,4 74,60
20k
40k
60k
80k
100k
120k
Resolution 7.3
Zähl
rate
Masse / u
/ c
psMessposition
Resolution 4.3
73,4 73,6 73,8 74,0 74,2 74,4 74,60
20k
40k
60k
80k
100k
120k
Resolution 7.3
Zähl
rate
Masse / u
Messposition
solution 4.3Re
/ c
ps
Abb. 80: BeisMessposition l
piel für die Wichtigkeit der Massenkalibration nach Veränderung von Parametern: Liegt die inkslastig auf einem Peak, wird bei Resolution 4.3 noch auf dem Peakmaximum gemessen; bei
Resolution 7.3 liegt die Messposition jedoch schon auf der linken Peakflanke. Die Verhältnisse verhalten sich
lls
die normierten Analyt-zu-GeO2-Verhältnisse verfälscht. Wenn man einen so hervorgerufenen
Trend direkt auf einen veränderten Parameter zurückführt, kann es zu Fehlinterpretationen
kommen. Deswegen wurde nach jeder Variation einer Messeinstellung ein Scan auf-
genommen und überprüft, ob die Massenkalibration und damit die Messposition noch richtig
waren. Wenn die Peakspitze nicht mehr genau auf der jeweiligen Atommasse lag, wurde in
den Datensatz der Massenkalibration für einen bestimmten DAC-Wert manuell die veränderte
richtige Masse eingetragen. Dieses Vorgehen wurde manuelle Massenkalibration genannt.
Mit dieser Vorgehensweise ließen sich Messpositionen auf den Peakflanken kontrolliert
wieder auf die Peakmaxima zurücksetzen. Das Konzept der manuellen Massenkalibration
erwies sich als außerordentlich wichtig, um die Auswirkungen von veränderten Parametern
sicher einschätzen zu können.
umgekehrt bei rechtslastiger Messposition.
Bei den Untersuchungen zur Abhängigkeit der Rn-Werte von verschiedenen instrumentellen
Parametern wurden auch Einstellungen variiert, die die Massenkalibration und die Massen-
peakform direkt verändern. Dies trifft insbesondere auf die Quadrupoleinstellungen ∆M und
Resolution aber auch auf die angelegte Extraktionspannung zu. Die Peaks werden durch eine
große Extraktionsspannung zwar meist höher, aber gleichzeitig sehr spitz, wodurch es
schwieriger wird die optimale Messposition zu treffen. Bei höherer Auflösung des Quadru-
pols, werden die Massenpeaks unsymmetrischer und die Peakmaxima verlagern sich hin zu
höherer Masse. Wenn die Peak-Jump-Messpositionen auf den Peakflanken zu liegen kommen
(Abb. 80), führt das zu einem Abfallen der Zählraten und auch zu einem Abnehmen der
Präzision. Es kann passieren, dass sich die Messpositionen für die relevanten Elemente
Germanium, Brom und Iod unterschiedlich stark verändern. In diesem Fall werden ebenfa

Anhang
- 169 -
6.1.4 Verwendete Geräte
ICP-MS Nr. 1
Bezeichnung VG PlasmaQuad 2+ E
Abkürzung: PQ2+ E, (E für “Enhanced Interface”)
neuer Firmenname von „VG Elemental“ ist „Thermo“ (Winsford, U.K.)
Baujahr ca. 1991 (spätere Aufrüstungen)
Probenzuführung − peristaltische Pumpe
− Scott-Zerstäuberkammer (auf −4 °C oder +1 °C gekühlt)
ICP-Einheit − RF-Generator ICP20P (Solid State): 27.13 MHz, Standardleistung: 1350 W,
− µ-Sampler und µ-Skimmer aus Nickel
durchmesser: ~1 mm, ~0.7 mm
Vision 4.30)
Tab. 31: Standardbetriebsbedingungen für die ICP-MS
Parameter Betriebsbedingungen
− V-Spalt-Zerstäuber (oder Meinhard-Zerstäuber)
maximale Leistung: 2000 W
− Torch vom Fassel-Typ, Länge: 13 cm
Interface − High Performance Interface
Öffnungs
Massenanalysator − Quadrupol SX300 (Arbeitsbereich: 0–300 u, Auflösung: 0.5–1 u)
Detektor − Sekundärelektronenvervielfacher BURLE Channeltron 4710, max. 4000 V
Vakuumsystem − Drehschieberpumpen:
Leybold Trivac S25B (Endvakuum: 2.5·10−2 mbar, Saugvermögen: 25 m3 h−1, für
die Expansionskammer) und Edwards E2M18 (Endvakuum: 10−3 mbar, 17 m3 h−1,
liefert das Vorvakuum für die Turbomolekularpumpen)
− Zwei Turbomolekularpumpen Leybold Turbovac 340M (magnetgelagert,
51600 Umdreh. min−1, Endvakuum <10−10 mbar, Saugvermögen 400 L s−1)
Elektronik − STE-Controller (angesteuert über Software PQ
Plasmaleistung 1350 W Kühlgas/Plasmagas 13 L min−1
Hilfsgas 0.5–1.5 L minZerstäubergas 0.83–0.93 L min−1 (V-Spalt-Zerstäuber)Detektormodus Pulszählmodus Betriebsvakuum 9·10−7–4·10−6 mbar Software OS/2 PQ-Vision 4.30 TRA-Modus
−1

Anhang
- 170 -
die Expansions-
e Diffusionspumpen)
− Öldiffusionspumpen (Endvakuum 3·10−8 mbar):
Edwards Diffstak MK2 Serie Modell 63 (135 L s−1) und Modell 160 (700 L s−1)
n der ersten Generation
ICP-MS Nr. 2
Bis auf wenige Ausnahmen, auf die im Text hingewiesen wird, wurden alle Messungen am
ICP-MS Nr. 1 durchgeführt. Das ICP-MS Nr. 2 war ebenfalls ein PQ2+-Gerät, aber von
älterer Bauweise. Nennenswert unterschied es sich nur in folgenden Punkten:
Bezeichnung: VG PlasmaQuad 2+ (PQ2+)
Baujahr: 1989
ICP-Einheit: − RF-Generator von Henry Radio, USA (27.12 MHz, max. 2000 W)
Interface: − kein High Performance Interface
Vakuumsystem: − Drehschieberpumpen:
Edwards E2M-30 (Endvakuum: 1·10−4 mbar, 32.2 m3 h−1, für
kammer) und Edwards E1M18 (Endvakuum: 3·10−2 mbar, 20.5 m3 h−1, für das
Vorvakuum für di
Elektro ik − Controller-Einheit
Chromatographiezubehör
Gerät/Zubehör Typ/Ausführung Hersteller
HPLC-Pumpe Modell 709 Metrohm, Herisau Injektionsventil, elektrisch 6-Port/3-Wege VICI AG, Schenkon Kapillaren und Fittings PEEK, PTFE* verschiedene Probenschleifen PEEK, Stahl verschiedene Vorratsgefäße für Eluenten 2 L, Glas Schott Duran, Mainz Säulenkörper PEEK, 100×4 mm Metrohm, Herisau
* PTFE-Kapillaren sind nur für Verbindungen anwendbar, bei denen sich kein Hochdruck aufbaut (nach der Säule und bei Fließinjektionsmessungen).
Sonstige Geräte
Gerät/Zubehör Typ/Ausführung Hersteller
Kunststoffgefäße 30–120 mL, HDPE, FEP verschiedene pH-Glasselektrode 6.0202.000 Metrohm, Herisau pH-Meter Modell 744 Metrohm, Herisau Pipetten, elektrisch Research Pro Eppendorf, Hamburg Ultraschallbad Sonorex Super RK514BH Bandelin, Berlin Vakuumaufsatz für Glasflaschen Falcon 7111 Becton Dickinson Labware, New Jersey Waage Modell 770 Kern, Balingen

Anhang
- 171 -
6.2 Verwendete Chemikalien
Bezeichnung Qualität Bezugsquelle
Ammoniumhydroxid in H2O >25 % Traceselect Fluka, Buchs Argon 4.6 Air Liquide, Krefeld (früher Messer Griesheim)Arsenlösung 1000 mg L−1 Titrisol Merck, Darmstadt Borlösung 1000 mg L−1 für Atomspektrometrie Fluka, Buchs Cäsiumlösung 1000 mg L−1 für Atomspektrometrie Fluka, Buchs Dibromessigsäure puriss. p.a. Fluka, Buchs Dinatriumhydrogenphosphat · H2O purum p.a. Fluka, Buchs Germaniumdioxid 99.99 % Heraeus, Karlsruhe
Monobromessigsäure zur Synthese Gruppe Prof. Koert, Uni Marburg
12
Indiumlösung 1000 mg L−1 für Atomspektrometrie Fluka, Buchs Kaliumdichromat p.a. Merck, Darmstadt Kaliumiodat puriss. p.a. Fluka, Buchs
Natriumbromat p.a. Fluka, Buchs Natriumbromid p.a. Riedel-de-Haën, Seelze Natriumchlorat p.a. Fluka, Buchs Natriumchlorid p.a. Riedel-de-Haën, Seelze Natriumchlorit 80 % puriss. p.a. Fluka, Buchs Natriumiodid p.a. Merck, Darmstadt Natriumthiocyanat rein zentrales Chemikalienlager, Uni Marburg Perchlorsäure 70 % p.a. Riedel-de-Haën, Seelze Salpetersäure p.a. * Riedel-de-Haën, Seelze Salpetersäure 69 % selectipur Merck, Darmstadt Tellurlösung p.a. Uni Marburg, Abteilung Routineanalytik Tribromessigsäure purum Fluka, Buchs Wasser
reinst, 18.2 MΩ
Millipore, Schwalbach (Reinstwasseranlage Milli-Q Gradient + Vorreinigungsanlage Elix)
* gereinigt durch doppelte Oberflächendestillation

Anhang
- 172 -
6.3 Arbeitsvorschriften
on Bromid-, Bromat-, Iodid-, und Iodatstandards
zelstammlösungen 1000 mg L−1
Für die 1000 mg L−1 r jeweiligen
uf Wägeschi lze müssen dazu vorher getrocknet und im
ewah
Bromat: 0.1180 N
0.1218 K
Bromid: 0.1288 g N
Iodid: 0.1181 N
Nach Einwiegen de ge mit einem 100 mL HDPE-Gefäß
(HDPE = high density polyethylene) tariert. Da ägeschiffchen in das Gefäß
d dieses m
ard k L suprapurer Salpetersäure stabilisiert werden.
Die Standards müssen st
mindestens monatlich f
6.3.1 Ansetzen v
Anionen-Ein
Anionen-Einzelstammlösungen werden folgende Massen de
Salze a ffchen eingewogen. Die Sa
Exsikkator aufb rt worden sein:
g aBrO3 p.a.
Iodat: g IO3 pur. p.a. 15
aBr p.a.
g aI p.a.
s jeweiligen Salzes wird die Waa
s Salz wird vom W
überführt un it Reinstwasser auf 100 g aufgefüllt. Sowohl der Bromat- als auch 17
16
der Iodatstand ann zusätzlich mit 1 m
dunkel im Kühlschrank aufbewahrt werden. Der Iodidstandard i
risch anzusetzen. Die anderen Standards sind länger haltbar.
15 Natriumsalze sind Kal m ]+ auf dem Masse-zu-
adungs-Verhältnis 79 des Broms zu vermeiden (s. Interferenzen in Kap. 2.4.2). 16 Beim Einwiegen wird die Dichte der Standards bei allen anzusetzenden Lösungen mit 1 g mL−1 angenommen.
n zwar etwas genauer, da man Ansetzfehler auf Grund der
geringeren Dichte von Reinstwasser (~0.997 g mL−1) beträgt ca. 0.3 %. 17 Die Bromat- und Iodat- Einzelstandards können mit 1 mL Salpetersäure pro 100 mL Standardlösung stabilisiert werden (supr ure, boiled = oberflächendestilliert). Es wird angenommen, dass durch eine höhere Io n Nitratüberschuss Wandeffekte (Adsorptionseffekte) verringert werden könn werd n pH-Werten Mikroorganismen abgetötet, die möglicherweise durch M pezie u Bromid und Iodid darf keinesfalls Salpetersäure en, d zw. Iod reagieren. Auch unbekannte Proben oder ards Anionen dürfen niemals angesäuert werden, da durch Protonen Redoxrea og n.
roben und Standards könn essen mit [155]. Eventuell vorhandene puren von Metallen werden damit abgefangen und somit eine katalytische Beschleunigung der Oxidation oder
Reduktion von Spezies verhindert. Außerdem fängt Ethylendiamin überschüssiges Ozon in ozonierten Proben ab und stoppt eine weitere Oxidation von z.B. Bromid zu Bromat.
iu salzen vorzuziehen, um die Bildung des Molekülions [39K40ArL
Durch volumetrisches Ansetzen in Messkolben aus Glas wären die Konzentrationekeinen „Dichtefehler“ macht, dafür gibt es aber einen Umfüllschritt mehr. Der
apure Salpetersä engl.: subnenstärke [100] bzw. durch eine
en. Außerdem en bei niedrigeetabolisierung S zugesetzt werd
skonzentrationen verändern könnten. Za diese Spezies dadurch zu Bromat b
gemischte Stand mit mehreren ktionen der Oxyhal enide katalysiert werde
PS
en stattd Ethylendiamin stabilisiert werden

Anhang
- 173 -
t/Iodid/Iodat-Viererstandard 1000 µg L−1 (Zwischenverdünnung)
Einzelstandards
a. 95 mL Reinstwasser in das Gefäß füllen. Diese Reihenfolge verhindert, dass Iodid von der
alpeter d Iodatstandards aufoxidiert wird. Der Vierer-
ist für quan e Analysen wöchentlich neu anzusetzen und im Kühlschrank aufzu-
Verdünnungsreihe 0.1–
Iodid enthaltende Mess ich müssen für quantitative Bestim-
ungen täglich neu u asser angesetzt werden. Für
Lösungen mit einem G alt
sind FEP- andeffekte zu minimieren. (FEP =
etrafluorethylen-Perfluorpropylen, PFA = Perfluor-Alkoxyalkan). Standards mit besonders
<1 µg L−1 sollten möglichst kurz vor der Messung eingewogen
werden.
n/Probe g
ngen sollten ein G lt an gelösten Feststoffen (TDS) von 0.1 % nicht
werden. Trinkwas it der
müssen entgast werden. Dazu werden sie
gegebenenfalls in geeignete HDPE-Gefäße umgefüllt. Die Entgasung erfolgt im Ultraschall-
keine Bläschen mehr entstehen (ca. 15 min). Die Entgasungszeiten sollten
en die Proben auch erwärmt und es werden
reaktive Hydroxylradikale und Wasserstoffperoxid gebildet. Bei zu langer Ultraschallbehand-
wegen zu Spezi lungen komm
Bromid/Broma
Je 100 µL 1000 mg L−1 Bromid, Bromat, Iodid und Iodat werden mit Reinstwasser in einer
braunen HDPE-Flasche (Lichtschutz) auf 100 g aufgefüllt. Falls Bromat und Iodat in
verdünnter Salpetersäure angesetzt wurden, sollte man schon vor den
c
verdünnten S säure in den Bromat- un
standard titativ
bewahren.
500 µg L−1
lösungen im unteren µg L−1-Bere
m a s der Zwischenverdünnung mit Reinstw
ehalt >1 µg L−1 sind Gefäße aus HDPE ausreichend, bei einem Geh
<1 µg L−1 oder PFA-Gefäße vorzuziehen, um W
T
niedrigen Konzentrationen
6.3.2 Probenlösunge nvorbereitun
In Probenlösu esamtgeha
überschritten ser (Leitungswasser, Mineralwasser) kann unverdünnt m
IC-ICP-MS auf Anionen gemessen werden. Mit Kohlenstoffdioxid versetzte Mineralwässer
der andere kohlenstoffdioxidhaltige Proben o
bad, solange bis
nicht länger sein, denn durch Ultraschall werd
lung kann es des esumwand en.

Anhang
- 174 -
nitrat-Eluents mit Germaniumdioxid als IS
Für die Eluenten werden Reinstwasser und folgende möglichst suprapure Chemikalien
benötigt: Ammoniak (25 % in Wasser), konzentrierte oberflächendestillierte Salpetersäure,
Germaniumlösung un ür starke Eluen e
Einstellun Ammoniak fäßen auf ungefähr 5 %,
dünnt. A r säure ist eine Konzentration
eeignet (69.0 m Salpetersä .7 mL 70%ige Salpetersäure
am best h Schott Duran, Mainz) an-
man einen Vakuum Falcon ickinson Labware, New
ntgasung au . Es wird hineingelegt
rkierung bei der -Füllhöhe angezeichnet. Die gewünschte Menge 18 an
etersäure und üblicherweise 5 mL 6 m lösung 19 werden in die
geben und fast bi it Reinstwas ird unter kräftigem
s Magnetrührers e H-Wert 20 zwischen vier und sechs durch Zugabe von
estellt. Bei die rozedur lösen s . Um
n zu erhalten, h entgast werden. Dazu ist eine Ultraschall-
ne nicht ausrei ne Entgasung durch Anlegen eines Vakuums lässt
ilfe eines Vak fsatzes und e bran-
durchführen und hat sich effek
6.3.3 Ansetzen eines Ammonium
6 mg L−1 d f ten zusätzlich Perchlorsäure. Für ein
stufenweise pH-Wert- g wird in PE-Vorratsge
0.5 % und 0.025 % ver ls Stammlösung fü die Salpeter
von 1 mol L−1 g L 65%ige ure bzw. 63
auf 1 L).
Die Eluenten werden en in 2 L-Glasflasc en (z.B. von
gesetzt, auf die aufsatz 7111 (Becton D
Jersey, USA) zur E fsetzen kann ein Rührfisch in das Gefäß
und eine Ma 2 L
1 mol L−1 Salp g L−1 Germanium
Glasflasche ge s 2 L m ser aufgefüllt. Dann w
Rühren eine in p
Ammoniak eing ser P ich relativ viele Gase im Eluenten
stabile Messunge muss dieser noc
behandlung allei chend. Ei
sich leicht mit H uumau iner Wasserstrahlpumpe oder Mem
pumpe als wesentlich tiver herausgestellt.
18 Die Konzentration von Ammoniumnitrat-Eluenten liegt typischerweise zwischen 10 und 120 mmol L−1. Über 120 mmol L−1 kann man nicht beliebig lang mit dem ICP-MS messen, da es mit der Zeit zu Salzablagerungen im Injektorrohr der Plasmafackel kommen kann. Will man den Eluenten weiter verstärken, sollte man 0.1−10 mmol L−1 Perchlorsäure zusetzen. Perchlorat ist ein sehr starkes Verdrängeranion. Gleichzeitig wirkt es als „Peaksymmetriefix“ für andere stark polarisierbare Anionen. 19 Die Konzentration von 15 µg L−1 Germanium im Eluenten ergibt auf der Isotopenmasse 74 u am PQ2 typischerweise eine Zählrate von 50k−120k cps. Die konzentrierteste Germaniumstammlösung wurde durch Einwiegen von 0.0897 g GeO2 (99.99 %, Heraeus, Karlsruhe) auf 100 mL Reinstwasser und Lösen mit Hilfe von Ultraschall hergestellt. Die Konzentration nach 1:100-Verdünnung beträgt 8.97 mg L−1 bezogen auf Germaniumdioxid bzw. 6.23 mg L−1 bezogen auf Germa-nium, müsste aber zur genauen Bestimmung titriert werden. Die Verwendung einer käuflichen 1000 mg L−1 Germaniumstandardlösung (Bernd Kraft GmbH, Duisburg) ist nicht empfehlenswert. Es wurde festgestellt, dass diese aus Germaniumtetrachlorid hergestellte Lösung zu viele Brom- und Iodverunreinigungen enthält. 20 Unterhalb des pH-Werts von drei besteht die Gefahr, dass einige Anionen in ihre nicht retardierbare korrespondierende Säure überführt werden. Außerdem wird die Oxidationskraft der Salpetersäure zu stark und Oxyhalogenide und Halogenide können komproportionieren. Bei einem pH-Wert größer als sieben liegt Germanium zudem teilweise als (Hydrogen-)Germanat vor und kann somit retardiert werden, was für einen internen Standard unerwünscht ist.

Anhang
- 175 -
ollierung der Betriebsbedingungen des ICP-MS 6.3.4 Formular zur Protok

Literaturverzeichnis
- 176 -
Andrade F, Gamez G, McClenathan D, Rogers D, Schilling G, Wetzel W, Hieftje GM, J. Chromatogr. A 2004, 1050:3-34 "Plasma-source mass spectrometry for speciation analysis: state-of-the-art".
gr. A 1999, 856:243-58 ely coupled plasma mass spectrometry".
J
y, New York.
] Ure AM, Davidson CM, Chemical Speciation in the Environment 1995, Blackie, Glasgow.
[10] Small H, Stevens TS, Baumann WC, Anal. Chem. 1975, 47:1801.
29
SD, Trac-Trends Anal. Chem. 2003, 22:666-84 "Disinfection by-products and other emerging contaminants in drinking water".
[18] Bichsel Y, von Gunten U, Anal. Chem. 1999, 71:34-38 "Determination of iodide and iodate by ion chromatography with postcolumn reaction and UV/visible
detection".
7 Literaturverzeichnis
[1] Caruso JA, Klaue B, Michalke B, Rocke DM, Ecotox. Environ. Safe. 2003, 56:32-44 "Group assessment: elemental speciation".
[2] Caruso JA, Montes-Bayon M, Ecotox. Environ. Safe. 2003, 56:148-63 "Elemental speciation studies - new directions for trace metal analysis".
[3] Michalke B, Ecotox. Environ. Safe. 2003, 56:122-39 "Element speciation definitions, analytical methodology, and some examples".
[4] Ray SJ,
[5] Sutton KL, Caruso JA, J. Chromato "Liquid chromatography-inductiv
[6] Rodriguez-Gonzalez P, Marchante-Gayon JM, Alonso IG, Sanz-Medel A, Spectrochim. Acta, Part B 2005, 60:151-207
"Isotope dilution analysis for elemental speciation: A tutorial review".
[7] Waddell R, Lewis C, Hang W, Hassell C, Majidi V, Appl. Spectrosc. Rev. 2005, 40:33-69 "Inductively coupled plasma mass spectrometry for elemental speciation: Applications in the new
millennium".
[8] Cornelis R, Caruso JA, Crews H, Heumann KG, Handbook of Elemental Speciation, Vol.1: Techniques and Methodology, Vol. 2: Species in the Environment, Food, Medicine and Occupational Health 2005, Wile
[9
[11] Jackson PE, Trac-Trends Anal. Chem. 2001, 20:320- "Determination of inorganic ions in drinking water by ion chromatography".
[12] Lopez-Ruiz B, J. Chromatogr. A 2000, 881:607-27 "Advances in the determination of inorganic anions by ion chromatography".
[13] Weiß J, Ionenchromatographie 2001, 3. Aufl., Wiley-VCH, Weinheim.
[14] Marques MJ, Salvador A, Morales-Rubio A, de la Guardia M, Fresenius J. Anal. Chem. 2000, 367:601-13 "Chromium speciation in liquid matrices: a survey of the literature".
[15] B'Hymer C, Caruso JA, J. Chromatogr. A 2004, 1045:1-13 "Arsenic and its speciation analysis using high-performance liquid chromatography and inductively
coupled plasma mass spectrometry".
[16] Le XC, Lu X, Li X-F, Anal. Chem. 2004, 76:26A-33A "Arsenic speciation".
[17] Richardson

Literaturverzeichnis
- 177 -
tand: .02.05
ation of haloforms during chlorination of natural waters".
"Disinfection byproducts in drinking water: The analytical challenge".
[30] US EPA, Weinberg HS, Krasner SW, Richardson SD 2002, EPA/600/R-02/068,
"A plasma source for mass analysis".
"Mass spectrometric analysis of solutions using an atmospheric pressure ion source".
[33] Date AR, Gray AL, Analyst 1981, 106:1255-67 "Plasma source mass spectrometry using an inductively coupled plasma and a high resolution quadrupole
mass filter".
[34] Houk RS, Fassel VA, Flesch GD, Svec HJ, Gray AL, Taylor CE, Anal. Chem. 1980, 52:2283-89 "Inductively Coupled Argon Plasma as an Ion Source for Mass Spectrometric Determination of Trace
Elements".
[35] Douglas DJ, French JB, Anal. Chem. 1981, 53:37-41 "Elemental analysis with a microwave induced plasma quadrupole mass spectrometer system".
[36] Gray AL, PhD thesis 1982, University of Surrey "The use of an inductively coupled plasma as an ion source for atomic mass spectrometry".
[19] US EPA 2005, Integrated Risk Information System, http://www.epa.gov/iris/subst/1007.htm S18
"Perchlorate and Perchlorate Salts".
[20] Paull B, Barron L, J. Chromatogr. A 2004, 1046:1-9 "Using ion chromatography to monitor haloacetic acids in drinking water: a review of current
technologies".
[21] Bellar TA, Lichtenberg JJ, J. Am. Water Works Assoc. 1974, 66:739-44 "Determining volatile organics at microgram-per-liter levels by gas chromatography".
[22] Rook JJ, Water Treatment Examination 1974, 23:234 "Form
[23] US EPA, Clark RM, Boutin BK 2001, EPA/600/R-01/110 "Controlling Disinfection By-Products and Microbial Contaminants in Drinking Water".
[24] US EPA 2001, Integrated Risk Information System, EPA/635/R-01/002, http://www.epa.gov/iris/subst/1002.htm, Abruf am: 20.07.2005
"Toxicological Review of Bromate".
[25] DeAngelo AB, George MH, Kilburn SR, Moore TM, Wolf DC, Toxicol. Pathol. 1998, 26:587-94 "Carcinogenicity of potassium bromate administered in the drinking water to male B6C3F(1) mice and
F344/N rats".
[26] Kurokawa Y, Maekawa A, Takahashi M, Hayashi Y, Environ. Health Perspect. 1990, 87:309 "Toxicity and carcinogenicity of potassium bromate - a new renal carcinogen".
[27] WHO, Guidelines for Drinking-Water Quality 2004, vol. 1, 3. ed., WHO, Genf, http://www.who.int/water_sanitation_health/dwq/gdwq3/en/index.html, Abruf am: 13.07.2005.
[28] US EPA, Fed. Reg. 1998, 63, (241):69389-476 "Disinfectants and Disinfection Byproducts; Final Rule".
[29] Weinberg H, Anal. Chem. 1999, 71:801A-08A
www.epa.gov/athens/publications/DBP.html, Abruf am: 22.09.2005 "The Occurrence of Disinfection By-Products (DBPs) of Health Concern in Drinking Water: Results of a
Nationwide DBP Occurrence Study".
[31] Gray AL, Proc. Soc. Anal. Chem. 1974, 11:182-83
[32] Gray AL, Analyst 1975, 100:289-99

Literaturverzeichnis
- 178 -
[37] Fisons Plc, Poster 1991, VGE/SM/PM/016 Issue 1
38] PlasmaQuad System Manual 1988, ed. 2b, VG Elemental.
[39] Montaser A (Hrsg.), Inductively Coupled Plasma Mass Spectrometry 1998, Wiley, New York.
0] Scott RH, Fassel VA, Kniseley RW, Nixon DE, Anal. Chem. 1974, 46:75-80 "Inductively coupled plasma optical emission analytical spectrometry. A compact facility for trace
analysis of solutions".
[41] Meinhard JE, ICP Inform. Newslett. 1978, 3:489 "The concentric glass nebulizer".
[42] Ivaldi JC, Slavin W, J. Anal. At. Spectrom. 1990:359-64 "Cross-flow Nebulisers and Testing Procedures for Inductively Coupled Plasma Nebulisers".
[43] Wollenweber D, Dissertation 2000, Universität Hannover "Instrumentelle Leistungssteigerungen der ICP-MS Ultraspurenanalytik durch neue Zerstäuber oder
Kaltplasma und Einsatz der Isotopenverdünnung".
[44] Maestre S, Mora J, Todoli JL, Canals A, J. Anal. At. Spectrom. 1999, 14:61-67 "Evaluation of several commercially available spray chambers for use in inductively coupled plasma
atomic emission spectrometry".
[45] Mora J, Maestre S, Hernandis V, Todoli JL, Trac-Trends Anal. Chem. 2003, 22:123-32 "Liquid-sample introduction in plasma spectrometry".
[46] Gunther D, Hattendorf B, Trac-Trends Anal. Chem. 2005, 24:255-65 "Solid sample analysis using laser ablation inductively coupled plasma mass spectrometry".
[47] Russo RE, Mao XL, Liu HC, Gonzalez J, Mao SS, Talanta 2002, 57:425-51 "Laser ablation in analytical chemistry - a review".
[48] Vanhaecke F, Resano M, Moens L, Anal. Bioanal. Chem. 2002, 374:188-95 "Electrothermal vaporisation ICP-mass spectrometry (ETV-ICP-MS) for the determination and speciation
of trace elements in solid samples - A review of real-life applications from the author's lab".
[49] Nakahara T, Anal. Sci. 2005, 21:477-84 "Development of gas-phase sample-introduction techniques for analytical atomic spectrometry".
[50] Jarvis KE, Gray AL, Houk RS, Handbook of Inductively Coupled Plasma Mass Spectrometry 1992, Blackie, Glasgow.
[51] Douglas DJ, Kerr LA, J. Anal. At. Spectrom. 1988, 3:749-52 "Study of solids deposition on inductively coupled plasma mass spectrometry samplers and skimmers".
[52] Ross BS, Hieftje GM, Spectrochim. Acta, Part B 1991, 46:1263-73 "Alteration of ion-optic lens configuration to eliminate mass-dependent matrix interference effects in
inductively coupled plasma mass spectrometry".
[53] Elliott S, Knowles M, Kalinitchenko I, Spectroscopy 2004, 19:30 "A new direction in ICP-MS".
[54] Thomas R, Spectroscopy 2002, 17:42 "A beginner's guide to ICP-MS - Part IX - Mass analyzers: Collision/reaction cell technology".
[55] Tanner SD, Baranov VI, Bandura DR, Spectrochim. Acta, Part B 2002, 57:1361-452 "Reaction cells and collision cells for ICP-MS: a tutorial review".
"VG PlasmaQuad Variations on a Theme".
[
[4

Literaturverzeichnis
- 179 -
[56] Moldovan M, Krupp EM, Holliday AE, Donard OFX, J. Anal. At. Spectrom. 2004, 19:815-22 multicollector ICP-MS as tools for trace metal speciation in
instrumentation, measurement techniques and analytical
capabilities".
8] ns 1990, Journal of Chromatography Library, vol. 46, Elsevier.
9] "Funktionalisierung und Charakterisierung neuer Ionenaustauscher auf Methacrylat-Basis für die
0] "The hyphenated methods".
1] "Hyphenation and hypernation - The practice and prospects of multiple hyphenation".
[62] -Oates J, Analyst 2005, 130:13-17 y: on-line or off-line".
2:25-26
[64] Heumann KG, Anal. Bioanal. Chem. 2002, 373:323-24
[65] ionsschrift 1995, Universität Hannover "Neue Einsatzgebiete der On-line-Kopplung Hochleistungsflüssigkeitschromatographie-
"The coupling of LC to ICP-MS in element speciation: I. General aspects".
7] .9 2002, CRC Press, Boca Raton.
8] ufl., Walter de Gruyter, Berlin, New York.
[69] Gloe K, Stephan H, Grotjahn M, Chemie Ingenieur Technik 2002, 74:767-77
[70] Beer PD, Gale PA, Angew. Chem. 2001, 113:502-32 d und Perspektiven".
[71] . A 1999, 844:161-69 "Ion chromatographic separation of sulfur-containing inorganic anions with an ICP-MS as element-
[72] Profrock D, Leonhard P, Prange A, Anal. Bioanal. Chem. 2003, 377:132-39 ke proteins using capillary
h an octopole reaction cell".
"High resolution sector field ICP-MS andenvironmental studies: a review".
[57] Rehkamper M, Schonbachler M, Stirling CH, Geostand. Newsl. 2001, 25:23-40 "Multiple collector ICP-MS: Introduction to
[5 Haddad PR, Jackson PE, Ion Chromatography Principles and Applicatio
[5 Holland S, Diplomarbeit 2001, Philipps-Universität Marburg
Chromatographie von Anionen".
[6 Hirschfeld T, Anal. Chem. 1980:279A
[6 Wilson ID, Brinkman UAT, J. Chromatogr. A 2003, 1000:325-56
Edwards E, Thomas "Hyphenating liquid phase separation techniques with mass spectrometr
[63] Albert K, Krucker M, Glaser T, Schefer A, Lienau A, Zeeb D, Anal. Bioanal. Chem. 2002, 37 "Hyphenated techniques".
"Hyphenated techniques - the most commonly used method for trace elemental speciation analysis".
Seubert A, Habilitat
Atomspektrometrie in der Elementanalytik".
[66] Michalke B, Trac-Trends Anal. Chem. 2002, 21:142-53
[6 Lide DR (Hrsg.), Handbook of Chemistry and Physics on CD-ROM, Version 0
[6 Hollemann AF, Wiberg E, Lehrbuch der Anorganischen Chemie 1995, 101. A
"Quo Vadis Anionenextraktion?"
"Erkennung und Nachweis von Anionen: gegenwärtiger Stan
Divjak B, Goessler W, J. Chromatogr
specific detector".
"Determination of sulfur and selected trace elements in metallothionein-lielectrophoresis hyphenated to inductively coupled plasma mass spectrometry wit
[73] Gerthsen C, Vogel H, Physik 1997, 19. Aufl., Springer, Berlin, Heidelberg, New York.

Literaturverzeichnis
- 180 -
on Clustern in die Flüssigkeit".
[75] Wernet P, Nordlund D, Bergmann U, Cavalleri M, Odelius M, Ogasawara H, Naslund LA, Hirsch TK, nce 2004, 304:995-99
[76] Atkins PW, Physikalische Chemie 1996, 2. Aufl., VCH, Weinheim.
7] http://www.csv.warwick.ac.uk/fac/sci/Chemistry/thermochemistry/index.htm, Abruf am: 15.08.2005.
ecular Chemistry of Anions 1997, Wiley-VCH, New York.
9] -73 "Hydration and Extraction of Oxoanions".
[80] h Guanidinium-Rezeptoren ist entropiebestimmt".
[82] m. Chem. Soc. 1963, 85:3533.
4-87 nd ICP-MS".
A 2004, 1050:45-62 and F-containing compounds in gas
chromatography and liquid chromatography".
[87]
[88] Zwiener C, Richardson SD, Trac-Trends Anal. Chem. 2005, 24:613-21 ter by LC-MS and related MS techniques".
[89] Richardson SD, Ternes TA, Anal. Chem. 2005, 77:3807-38 ants and Current Issues".
[90] Dudoit A, Pergantis SA, J. Anal. At. Spectrom. 2001, 16:575-80 detection and inductively coupled plasma-mass
[91] Fernandez RG, Alonso JIG, Sanz-Medel A, J. Anal. At. Spectrom. 2001, 16:1035-39 of
[92] Garcia-Fernandez R, Garcia-Alonso JI, Sanz-Medel A, J. Chromatogr. A 2004, 1033:127-33 um and magnesium by suppressed ion
[74] Ludwig R, Angew. Chem. 2001, 113:1856-76 "Wasser: v
Ojamae L, Glatzel P, Pettersson LGM, Nilsson A, Scie "The structure of the first coordination shell in liquid water".
[7 Roobottom HK, Jenkins HDB, Thermochemistry of inorganic salts,
[78] Bianchi A, Bowman-James K, Garcia-Espana E (Hrsg.), Supramol
[7 Abramov AA, Dzhigirkhanov MS-A, Iofa BZ, Volkova SV, Radiochemistry 2002, 44:270
Berger M, Schmidtchen FP, Angew. Chem. 1998, 110:2840-42 "Die Erkennung von Sulfationen durc
[81] Hofmeister F, Arch. Exp. Pathol. Pharmakol. 1888, 24:247.
Pearson R, J. A
[83] Francesconi KA, Sperling M, Analyst 2005, 130:998-1001 "Speciation analysis with HPLC-mass spectrometry: time to take stock".
[84] Das AK, de la Guardia M, Cervera ML, Talanta 2001, 55:1-28 "Literature survey of on-line elemental speciation in aqueous solutions".
[85] Seubert A, Trac-Trends Anal. Chem. 2001, 20:27 "On-line coupling of ion chromatography with ICP-AES a
[86] Brede C, Pedersen-Bjergaard S, J. Chromatogr. "State-of-the art of selective detection and identification of I-, Br-, Cl-,
Michalski R, Pol. J. Environ. Stud. 2005, 14:257-68 "Inorganic oxyhalide by-products in drinking water and ion chromatographic determination methods".
"Analysis of disinfection by-products in drinking wa
"Water Analysis: Emerging Contamin
"Ion chromatography in series with conductivity spectrometry for the determination of nine halogen, metalloid and non-metal species in drinking water".
"Coupling of ICP-MS with ion chromatography after conductivity suppression for the determination anions in natural and waste waters".
"Simultaneous determination of inorganic anions, calcichromatography".

Literaturverzeichnis
- 181 -
DP, Munch DJ 1997, EPA/600/R-98/118, f, Abruf am: 05.09.2005
"Determination of Inorganic Anions in Drinking Water by Ion Chromatography".
[94] US EPA Method 321.8, Creed JT, Brockhoff CA, Martin TD 1998,
"Determination of bromate in drinking waters by ion chromatography inductively coupled plasma / mass
[95] essler W, J. Chromatogr. A 1999, 862:39-47 "Determination of bromide, bromate and other anions with ion chromatography and an inductively
ific detector".
[96] Ammann AA, J. Chromatogr. A 2002, 947:205-16 l complexes by anion-exchange chromatography
[98] Pantsar-Kallio M, Manninen PKG, Anal. Chim. Acta 1998, 360:161-66 pled plasma mass
[99] Salov VV, Yoshinaga J, Shibata Y, Morita M, Anal. Chem. 1992, 64:2425-8 oupled argon
31:2059-63 aphy with
inductively coupled plasma mass spectrometric detection".
01] ich BV, J. Chromatogr. A 2001, 920:221-29 "Review of the methods of the US Environmental Protection Agency for bromate determination and
romate".
[102] "Determination of bromate in drinking waters by ion chromatography with inductively coupled plasma
[103] Yamanaka M, Sakai T, Kumagai H, Inoue Y, Journal of Chromatography, A 1997, 789:259-65 mn
ductively-coupled plasma mass spectrometry".
-80
ely coupled plasma mass spectrometry".
25-30 ination of bromate in drinking waters using flow injection ICP mass
spectrometry".
07] "Bromate assay in water by inductively coupled plasma mass spectrometry combined with solid-phase
[93] US EPA Method 300.1 Rev. 1.0, Pfaff JD, Hautman www.epa.gov/safewater/methods/met300.pd
www.epa.gov/nerlcwww/m_321_8.pdf, Abruf am: 22.09.2005
spectrometry".
Divjak B, Novic M, Go
coupled plasma mass spectrometer as element-spec
"Determination of strong binding chelators and their metaand inductively coupled plasma mass spectrometry".
[97] Ammann AA, Anal. Bioanal. Chem. 2002, 372:448-52 "Speciation of heavy metals in environmental water by ion chromatography coupled to ICP-MS".
"Speciation of halogenides and oxyhalogens by ion chromatography inductively couspectrometry".
"Determination of inorganic halogen species by liquid chromatography with inductively cplasma mass spectrometry".
[100] Creed JT, Magnuson ML, Brockhoff CA, Environ. Sci. Technol. 1997, "Determination of bromate in the presence of brominated haloacetic acids by ion chromatogr
[1 Hautman DP, Munch DJ, Frebis C, Wagner HP, Pep
validation of Method 317.0 for disinfection by-product anions and low-level b
Creed JT, Magnuson ML, Pfaff JD, Brockhoff C, J. Chromatogr. A 1996, 753:261-67
mass spectrometric detection".
"Specific determination of bromate and iodate in ozonized water by ion chromatography with postcoluderivatization and in
[104] Seubert A, Nowak M, Fresenius J. Anal. Chem. 1998, 360:777 "Trace analysis of bromate in drinking waters by means of on- line coupling IC-ICP-MS".
[105] Nowak M, Seubert A, Anal. Chim. Acta 1998, 359:193-204 "Ultra-trace determination of bromate in drinking waters by means of microbore column ion
chromatography and on-line coupling with inductiv
[106] Elwaer AR, McLeod CW, Thompson KC, Anal. Chem. 2000, 72:57 "On-line separation and determ
[1 Cai Q, Guo Z-X, Yu C, Zhang W, Yang Z, Anal. Bioanal. Chem. 2003, 377:740-48
extraction cartridges".

Literaturverzeichnis
- 182 -
waer AR,
ng water: development of laboratory and field methods".
aki T, Yomota C,
rmination of bromate in bread by ion chromatography with ICP-MS".
tection by inductively coupled plasma atomic emission spectroscopy and inductively coupled plasma mass spectrometry to ion chromatography of foods".
] ate in drinking water matrixes by ion chromatography with inductively
coupled plasma mass spectrometric detection".
[112] senius J. Anal. Chem. 1997, 357:74-79 "Bromide/bromate speciation by NTI-IDMS and ICP-MS coupled with ion exchange chromatography".
] "Rapid, calibration-free determination of bromate in water using IC-ICP-MS coupling".
] "Isotope dilution analysis of bromate in tap water using on-line coupling IC-ICP-MS utilising collision cell
[115] Creed JT, Magnuson ML, Brockhoff CA, Environ. Sci. Technol. 1997, 31:2059-63 haloacetic acids by ion chromatography with
18:1396-99 tively coupled
95 atography-inductively
coupled plasma mass spectrometry".
[118] rger W, Czizsek B, Hann S, Stingeder G, J. Anal. At. Spectrom. 2003, 18:512-14 "Preliminary comparison of inductively coupled plasma mass spectrometry and electrospray mass
[119] Heumann KG, Gallus SM, Radlinger G, Vogl J, Spectrochim. Acta, Part B 1998, 53:273-87 aphic systems with ICP-MS
J. Anal. At. Spectrom. 2004, 19:1442-47 exchange
chromatography with ICP-MS detection".
21] ies in commercially available seaweed by coupling different chromatographic
techniques with UV and ICP-MS detection".
]
d plasma mass spectrometry".
"Untersuchungen zur Herstellung von agglomerierten Anionenaustauschern auf Styrol- und Methacrylat-Basis".
[108] Ingrand V, Guinamant JL, Bruchet A, Brosse C, Noij THM, Brandt A, Sacher F, McLeod C, ElCroue JP, Quevauviller P, Trac-Trends Anal. Chem. 2002, 21:1-12 "Determination of bromate in drinki
[109] Akiyama T, Yamanaka M, Date Y, Kubota H, Nagaoka MH, Kawasaki Y, YamazMaitani T, Shokuhin Eiseigaku Zasshi 2002, 43:348-51 "Specific dete
[110] Heitkemper DT, Kaine LA, Jackson DS, Wolnik KA, J. Chromatogr. A 1994, 671:101-8 "Practical applications of element-specific de
[111 Creed JT, Brockhoff CA, Anal. Chem. 1999, 71:722-26 "Isotope dilution analysis of brom
Diemer J, Heumann KG, Fre
[113 Seubert A, G.I.T. Lab. J., Europe 2003, 7:97-99
[114 Seubert A, Godfrey J, Int. Lab. 2001, 31:18-19
technology".
"Determination of bromate in the presence of brominatedinductively coupled plasma mass spectrometric detection".
[116] Guo Z-X, Cai Q, Yu C, Yang Z, J. Anal. At. Spectrom. 2003, "Determination of bromate and bromoacetic acids in water by ion chromatography-induc
plasma mass spectrometry".
[117] Liu Y, Mou S, Chen D, J. Chromatogr. A 2004, 1039:89- "Determination of trace-level haloacetic acids in drinking water by ion chrom
Buchbe
spectrometry hyphenated with ion chromatography for trace analysis of iodide".
"Accurate determination of element species by on-line coupling of chromatogrusing isotope dilution technique".
[120] Wuilloud RG, Selar N, Kannamkumarath SS, Caruso JA, "Determination of 2,4,6-triiodophenol and its metabolites in human urine by anion-
[1 Shah M, Wuilloud RG, Kannamkumarath SS, Caruso JA, J. Anal. At. Spectrom. 2005, 20:176-82 "Iodine speciation stud
[122 Sacher F, Raue B, Brauch HH, J. Chromatogr. A 2005, 1085:117-23 "Analysis of iodinated X-ray contrast agents in water samples by ion chromatography and inductively-couple
[123] Holland S, Dissertation 2005, Universität Marburg

Literaturverzeichnis
- 183 -
arburg.
[125] Schütze S, Dissertation 2004, Universität Marburg
[126] McNaught AD, Wilkinson A, Compendium of Chemical Terminology - The Gold Book 1997, 2. ed., ndex.html, Abruf am: 13.07.2005.
emie Lexikon auf CD-ROM Vers. 1.0
[129] Schäfer H, Metrohm Ltd., Herisau, Schweiz, Persönl. Mitteilung 16.06.2005
30] annover "Stationäre Phasen für die Anionenchromatographie – Neue Strategien zur Herstellung und
[131] Pohl CA, Stillian JR, Jackson PE, J. Chromatogr. A 1997, 789:29-41 n chromatography".
, 54:950.
[134] Slingsby RW, Pohl CA, J. Chromatogr. A 1988, 458:241-53 matography".
[135] Janos P, J. Chromatogr. A 1997, 789:3-19 de equilibria in ion-exchange chromatography of
07-15
chromatography".
l. Chim. Acta 1988, 211:91-8 nal
standard".
38] "Matrix-effect observations in inductively coupled plasma mass spectrometry".
39] "Suppression of analyte signal by various concomitant salts in inductively coupled plasma mass
[140] "Nachweis-, Erfassungs- und Bestimmungsgrenze".
]
[142] Mädl W, FAQ zur Enteisenung und Entmanganung, Firma GBT Grund-, Brauch-,
[143] De Ridder F, Pintelon R, Schoukens J, Navez J, Andre L, Dehairs F, J. Anal. At. Spectrom. 2002, 17:1461-70
"An improved multiple internal standard normalization for drift in LA-ICP-MS measurements".
[124] Raskop M, laufende Dissertation, Universität M
"Stationäre Phasen für die Anionenchromatographie - Neue Strategien zur Synthese und Modifizierung makroporöser Methacrylat-Copolymere".
Blackwell Science, http://www.iupac.org/publications/compendium/i
[127] Autorenkollektiv, Falbe J, Regitz M (Hrsg.), CD Römpp - Römpp Ch1995, 9. Aufl., Thieme Verlag, Stuttgart, New York.
[128] Autorenkollektiv, Ullmann's Encyclopedia of Industrial Chemistry 2002, 6th ed., Wiley-VCH.
[1 Nowak M, Dissertation 1999, Universität H
Charakterisierung".
"Factors controlling ion-exchange selectivity in suppressed io
[132] Stevens TS, Langhorst MA, Anal. Chem. 1982
[133] Köhler K, Dissertation 1998, Universität Hannover.
"Anion-exchange selectivity in latex-based columns for ion chro
"Retention models in ion chromatography: the role of siinorganic cations and anions".
[136] Barron L, Nesterenko Pavel N, Paull B, J. Chromatogr. A 2005, 1072:2 "Use of temperature programming to improve resolution of inorganic anions, haloacetic acids and
oxyhalides in drinking water by suppressed ion
[137] Vandecasteele C, Nagels M, Vanhoe H, Dams R, Ana "Suppression of analyte signal in inductively-coupled plasma/mass spectrometry and the use of an inter
[1 Tan SH, Horlick G, J. Anal. At. Spectrom. 1987, 2:745-63
[1 Olivares JA, Houk RS, Anal. Chem. 1986, 58:20-5
spectrometry".
DIN 32645 1994, Beuth Verlag, Berlin
[141 Wikipedia - Die freie Enzyklopädie, http://de.wikipedia.org/wiki/Sole, Abruf am: 30.06.2005.
Trinkwasseraufbereitung, Industrie- und Umwelttechnik, http://www.wasseraufbereitungsseiten.de/enteisenung_faq.htm, Abruf am: 30.06.2005.

Literaturverzeichnis
- 184 -
ndards for inductively coupled
a-mass spectrometry".
[146] anhoe H, Dams R, Vandecasteele C, Talanta 1992, 39:737-42 "The use of internal standards in ICP-MS".
]
2000, 407:301-09
[149] Laborda F, Medrano JS, Castillo JR, Spectrochim. Acta, Part B 2004, 59:857-70 njection and liquid chromatography transient
:158A-66A
[151] Stefanova V, Kmetov V, Canals A, J. Anal. At. Spectrom. 2003, 18:1171-74 introduction
[152] Houk RS, Anal. Chem. 1986, 58:97A-105A
[153] oderne Experimente mit gekühlten und gespeicherten Ionen 2004, Universität Mainz,
[154] Eickhorst T, Seubert A, J. Chromatogr. A 2004, 1050:103-09 bromide, iodate
[155] US EPA Method 326.0 Rev. 1.0, Wagner HP, Pepich BV, Hautman DP, Munch DJ, Salhi E,
postcolumn reagent for trace bromate analysis".
[144] Tangen A, Lund W, Spectrochim. Acta, Part B 1999, 54B:1831-38 "A multivariate study of the acid effect and the selection of internal staplasma mass spectrometry".
[145] Thompson JJ, Houk RS, Appl. Spectros. 1987, 41:801-6 "A study of internal standardization in inductively coupled plasm
Vanhaecke F, V
[147 Denoyer ER, Atom. Spectrosc. 1994, 15:7-16 "Optimization Of Transient Signal Measurements In ICP-MS".
[148] Laborda F, Medrano J, Castillo JR, Anal. Chim. Acta "Data acquisition of transient signals in inductively coupled plasma mass spectrometry".
"Estimation of the quantification uncertainty from flow isignals in inductively coupled plasma mass spectrometry".
[150] McClenathan DM, Ray SJ, Wetzel WC, Hieftje GM, Anal. Chem. 2004, 76 "Plasma source TOFMS".
"Application of internal standardization in ICP-QMS through discrete sample methodologies".
"Mass Spectrometry of Inductively Coupled Plasmas".
Blaum K, Spektroskopie in Ionenfallen – Grundlagen und m
http://www.physik.uni-mainz.de/quantum/lectures/ws0405_ionenfallen/content_de.html, Abruf am: 18.07.2005.
"Germanium dioxide as internal standard for simplified trace determination of bromate,and iodide by on-line coupling ion chromatography-inductively coupled plasma mass spectrometry".
von Gunten U 2002 "Determination of inorganic oxyhalide disinfection by-product in drinking water using ion
chromatography incorporating the addition of a suppressor acidified





















![Ioduhr Oxidation von Iodid mit Peroxodisulfat - Guido Petri · Die in der Tabelle 1[2] angegebenen Volumina der Kaliumiodid- und Kaliumchlorid-Lösung werden ins Reagenzglas A pipettiert](https://img.pdfslide.org/doc/110x75/5e02ff8ed9e2ea2f204147fb/ioduhr-oxidation-von-iodid-mit-peroxodisulfat-guido-die-in-der-tabelle-12-angegebenen.jpg)







![chemistry.mdma...Die Oxvdation $ Jahrg. 74 ist 3/194 Il N -LP- (2-Methoxy-phenyl) -äthyl]-pyridiniunj bromid (VI). Atis 2 g Bromid und 0.75 g Pyridin wie oben dargestellt. Das Produkt](https://img.pdfslide.org/doc/110x75/60dd9527f6bf256fb62c2936/-die-oxvdation-jahrg-74-ist-3194-il-n-lp-2-methoxy-phenyl-thyl-pyridiniunj.jpg)