Embed Size (px)
Citation preview

Fakultät für Biologie und Biotechnologie Lehrstuhl für Biologie der Mikroorganismen
Bakterielle Phospholipid N-Methyltransferasen: Charakterisierung enzymatischer
Eigenschaften und Substratspezifitäten
Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften
der Fakultät für Biologie und Biotechnologie der Ruhr-Universität Bochum
angefertigt am
Lehrstuhl für Biologie der Mikroorganismen
vorgelegt von
Jan Gleichenhagen
aus
Essen
Referent: Prof. Dr. Franz Narberhaus Korreferent: Prof. Dr. Eckhard Hofmann
Bochum, 2012

Department for Biology and Biotechnology Microbial biology
Bacterial phospholipid N-methyltransferases: Characterisation of enzymatic properties and
substrate specificity
Dissertation to obtain the degree Doctor Rerum Naturalium (Dr. rer. nat.) at the Faculty of Biology and Biotechnology
International Graduate School of Biosciences Ruhr-University Bochum
Department of Microbial Biology
submitted by
Jan Gleichenhagen
from
Essen
Referent: Prof. Dr. Franz Narberhaus Korreferent: Prof. Dr. Eckhard Hofmann
Bochum, 2012


Meinen Dank…
meinem Doktorvater Herrn Prof. Dr. Franz Narberhaus für seine fortwährende
Unterstützung, die zahlreichen Anregungen und vielen Ideen, die zur Durchführung
und zum stetigem Fortgang dieser Arbeit beigetragen haben.
Herrn Prof. Dr. Eckhard Hofmann für seine Unterstützung, die stete Diskussions-
bereitschaft und die Übernahme des Zweitgutachtens.
Frau Prof. Dr. Nicole Frankenberg-Dinkel für die vielen Anregungen und Vorschläge
bei biochemischen Fragestellungen.
Frau Dr. Meriyem Aktas für die vielen konstruktiven Diskussionen, die Unterstützung
und Hilfe nicht nur in wissenschaftlichen Fragen: Benim sana ihtiyacim oldugunda
benim hep yanimdaydin bunun icin sana tesekkür etmek istiyorum!
Herrn Dr. Bernd Masepohl für seine stete Hilfsbereitschaft und hilfreichen
Ratschläge.
Frau Dr. Holländer-Czytko für die freundliche Unterstützung bei den radioakiven
Bindestudien und ihre Diskussionsbereitschaft.
Michael Schäkermann, Kai Westphal und Roman Moser für die stete Hilfs- und
Diskussionsbereitschaft und für die unterhaltsame Abwechslung neben dem
Laboralltag.
Christiane Fritz für die tatkräftige Unterstützung im Labor in der Endphase meiner
Arbeit.
„meinen“ Bachelorstudenten Linna M. Danne und Christian Harak für die gute
Zusammenarbeit und ihren Anteil an dieser Arbeit.
allen übrigen Mitarbeitern des Lehrstuhls für ein angenehmes Arbeitsklima und gute
Zusammenarbeit.
all meinen Freunden für ihr Verständnis in den letzen Jahren und die moralische
Unterstützung.
meinen Eltern und meiner Schwester, die mich immer unterstützt haben und zu mir
standen. Ohne Euch wäre ich nicht da, wo ich jetzt bin!

Inhaltsverzeichnis
I
Inhaltsverzeichnis
1. Einleitung 1
1.1 Lipide und Membranen 1
1.1.1 Lipide 1
1.2 Die Bedeutung von Phosphatidylcholin 5
1.2.1 Vorkommen und Bedeutung von Phosphatidylcholin 5
1.2.2 PC-Biosynthese in Eukaryoten 5
1.2.3 Bedeutung von PC für Bakterien-Wirts-Interaktionen 9
1.2.4 PC-Biosynthese in Prokaryoten 10
1.2.5 Die Phospholipid N-Methyltransferase PmtA aus Agrobacterium
tumefaciens 14
1.3 S-Adenosylmethionin-abhängige Methyltransferasen 16
1.3.1 S-Adenosylmethionin 16
1.3.2 Strukturelle Merkmale SAM-abhängiger Methyltransferasen 16
1.3.3 SAM-Bindemotive 18
1.4 Zielsetzung 21
2. Material und Methoden 22
2.1 Verwendete Stämme 22
2.2 Plasmide 22
2.3 Oligonukleotide 24
2.4 Anzucht von Bakterien 26
2.4.1 Nährmedien 26
2.4.2 Anzucht von Escherichia coli 27
2.4.3 Messung der optischen Dichte 27
2.5 Molekularbiologische Methoden 28
2.5.1 Isolierung von Plasmid-DNA 28
2.5.2 Hydrolytische Spaltung von DNA durch Restriktions-
endonukleasen 29
2.5.3 Agarose-Gelelektrophorese 29

Inhaltsverzeichnis
II
2.5.4 Amplifikation von DNA mittels Polymerasekettenreaktion und
ortsspezifische Mutagenese 30
2.5.5 Herstellen transformationskompetenter Zellen 32
2.5.6 Transformation 32
2.5.7 DNA-Konzentrationsbestimmung 33
2.6 Herstellung von Liposomen und Micellen 34
2.6.1 Herstellung von Liposomen 34
2.6.2 Herstellung von Mizellen 34
2.7 Proteinbiochemische Methoden 35
2.7.1 Überexpression rekombinanter Proteine in E. coli BL21(DE3) 35
2.7.2 Zellaufschluss und Gewinnung von Rohextrakt 35
2.7.3 Reinigung rekombinanter Proteine mittels Ni-IDA-Affinitäts-
chromatographie 36
2.7.4 Denaturierende SDS-Polyacrylamid-Gelelektrophorese 36
2.7.5 Proteinkonzentrationsbestimmung 39
2.7.6 Umpufferung und aufkonzentrieren von Proteinen 39
2.7.7 Größenausschlusschromatographie 39
2.7.8 In vitro-Aktivitätsassay 40
2.7.9 S-Adenosylmethionin-Bindestudien 40
2.7.10 Circular Dichroismus-Spektroskopie (CD-Spektroskopie) 41
2.7.11 Tryptophanfluoreszenzmessung 42
2.7.12 Fluoreszenzmessung mit Hilfe von Fluorescein-
Phosphatidylethanolamin 43
2.7.13 Protein-Lipid-Overlay-Assay 44
2.7.14 Limitierte Proteolyse 45
2.7.15 Tryptischer Verdau im Gel und Massenspektrometrie 45
2.8 Lipidanalytik 47
2.8.1 Lipidextraktion 47
2.8.2 Dünnschichtchromatographie (DC) 47
2.9 Chemikalienliste 49
2.10 Geräteliste 50
2.11 Software - / Linkliste 51

Inhaltsverzeichnis
III
3. Ergebnisse 52
3.1 SAM-Bindeeigenschaften der Phospholipid N-
Methyltransferase aus Agrobacterium tumefaciens 52
3.1.1 Bioinformatische Identifizierung putativer SAM-Bindemotive in
PmtA 52
3.1.2 Einfluss von Punktmutationen in putativen SAM-Bindemotiven
auf die PmtA-Aktivität 54
3.1.3 Beeinflussen die Punktmutationen die Faltung von PmtA-
Derivaten? 55
3.1.4 SAM-Bindefähigkeit von PmtA und PmtA-Derivaten 57
3.2 Einfluss der Membranlipidzusammensetzung auf die
Aktivität und Membranbindung von PmtA 60
3.2.1 Negativ geladene Lipide stimulieren die PmtA-Aktivität 60
3.2.2 Haben anionische Lipide einen Einfluss auf die SAM-Bindung
von PmtA? 62
3.2.3 Substratspezifität von PmtA 64
3.2.4 Beeinflussen PC-Analoga/Derivate die PmtA-Aktivität? 66
3.2.5 Rekrutierung von PmtA zur Membran via elektrostatischer
Interaktionen 68
3.2.6 Etablierung von verschiedenen Methoden zur quantitativen und
qualitativen Analyse der Lipid-Bindeeigenschaften von PmtA 73
3.2.7 Limitierte Proteolyse von PmtA in Gegenwart von
Modellmembranen 73
3.2.8 Massenspektrometrische Analyse der PmtA-Fragmente 76
3.2.9 CD-spektroskopische Analyse von PmtA in Gegenwart von
verschiedenen Lipiden 78
3.2.10 Analyse der Lipidbindeeigenschaften von PmtA mittels
Fluorescein-Phosphatidylethanolamin 82
3.2.11 Tryptophanfluoreszenzspektroskopie zur Analyse der
Lipidinteraktion von PmtA 86
3.2.12 Bioinformatische Analyse putativer Lipidbindestellen in PmtA 89
3.2.13 Einfluss verschiedener Punktmutationen auf die PmtA-
Aktivität 90
3.2.14 Die Bedeutung der Aminosäure Threonin 36 für die Lipidbindung
von PmtA 93

Inhaltsverzeichnis
IV
4. Diskussion 96
4.1 SAM-Bindeeigenschaften der agrobakteriellen
Phospholipid N-Methyltransferase PmtA 96
4.2 PmtA-Lipid Interaktion 102
4.2.1 Anionische Lipide stimulieren die PmtA-Aktivität 102
4.2.2 Elektrostatische Interaktion von PmtA mit Membranen 103
4.2.3 Substratspezifität von PmtA 106
4.2.4 Inhibierung von PmtA durch PC-Analoga 108
4.2.5 Die Bedeutung des N-Terminus für eine PmtA-Membran-
Interaktion 109
4.2.6 Die PmtA-Lipid-Interaktion ist hoch-affin und führt zu einer
Konformationsänderung 113
4.2.7 Gibt es in PmtA essentielle Aminosäuren für eine
Lipidinteraktion? 118
4.2.8 Postuliertes Modell für die PmtA-Membran-Interaktion 121
5. Zusammenfassung 123
6. Summary 124
7. Literaturverzeichnis 125
8. Anhang 138
8.1 Vorveröffentlichungen der Dissertation 138
8.2 Lebenslauf 139
8.3 Erklärung 140
Abkürzungsverzeichnis
V
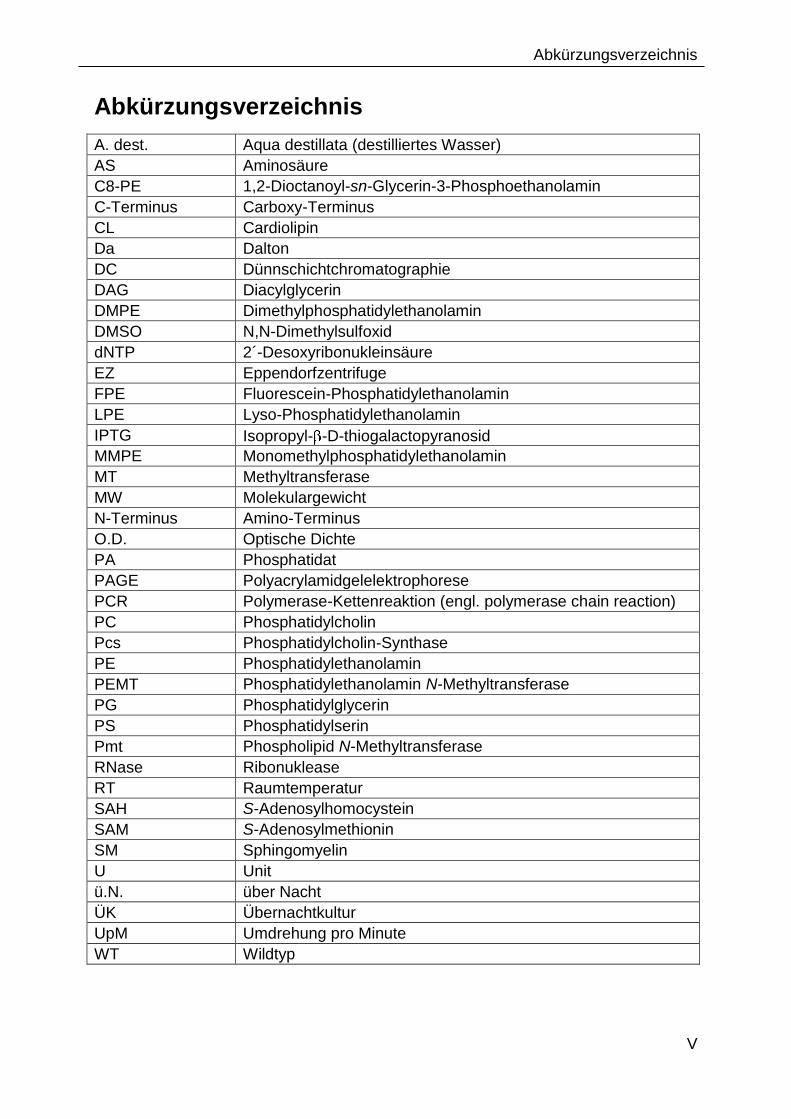
Abkürzungsverzeichnis
A. dest. Aqua destillata (destilliertes Wasser)
AS Aminosäure
C8-PE 1,2-Dioctanoyl-sn-Glycerin-3-Phosphoethanolamin
C-Terminus Carboxy-Terminus
CL Cardiolipin
Da Dalton
DC Dünnschichtchromatographie
DAG Diacylglycerin
DMPE Dimethylphosphatidylethanolamin
DMSO N,N-Dimethylsulfoxid
dNTP 2´-Desoxyribonukleinsäure
EZ Eppendorfzentrifuge
FPE Fluorescein-Phosphatidylethanolamin
LPE Lyso-Phosphatidylethanolamin
IPTG Isopropyl- -D-thiogalactopyranosid
MMPE Monomethylphosphatidylethanolamin
MT Methyltransferase
MW Molekulargewicht
N-Terminus Amino-Terminus
O.D. Optische Dichte
PA Phosphatidat
PAGE Polyacrylamidgelelektrophorese
PCR Polymerase-Kettenreaktion (engl. polymerase chain reaction)
PC Phosphatidylcholin
Pcs Phosphatidylcholin-Synthase
PE Phosphatidylethanolamin
PEMT Phosphatidylethanolamin N-Methyltransferase
PG Phosphatidylglycerin
PS Phosphatidylserin
Pmt Phospholipid N-Methyltransferase
RNase Ribonuklease
RT Raumtemperatur
SAH S-Adenosylhomocystein
SAM S-Adenosylmethionin
SM Sphingomyelin
U Unit
ü.N. über Nacht
ÜK Übernachtkultur
UpM Umdrehung pro Minute
WT Wildtyp

Einleitung
- 1 -
1. Einleitung
1.1 Lipide und Membranen
1.1.1 Lipide
Lipide sind wasserunlösliche, organische Moleküle, die eine polare Kopfgruppe und
einen hydrophoben Kohlenwasserstoffrest besitzen (Abbildung 1). Dabei können die
hydrophoben Kohlenwasserstoffreste sowohl in ihrer Länge, als auch im Vorkommen
von Doppelbindungen variieren. Auch die Kopfgruppe ist variabel und kann zum
Beispiel positiv oder negativ geladen sein. Der Aufbau aus sowohl hydrophoben, als
auch hydrophilen Komponenten verleiht Lipiden einen amphiphilen Charakter.
Abbildung 1: Allgemeiner Aufbau eines Phospholipides.
Allgemein werden Lipide aufgrund ihrer strukturellen und chemischen Eigenschaften
in acht Klassen eingeteilt (Tabelle 1). Da für diese Arbeit die Klasse der
Glycerophospholipide eine besondere Bedeutung hat, werden im Folgenden der
Aufbau und die Funktion dieser Lipide näher betrachtet. Phospholipide sind ein
Hauptbestandteil biologischer Membranen und stellen eine der wichtigsten
Lipidklassen dar. Sie bestehen aus einer polaren Kopfgruppe, die über eine
Phosphorsäurediesterbrücke an ein Glycerin gekoppelt ist, sowie zwei Fettsäuren,

Einleitung
- 2 -
die jeweils mit dem Glycerin verestert sind (Abbildung 1). Ein Glycerophospholipid
kann aufgrund verschiedener Fettsäurekombinationen mehrere Molekülspezies
umfassen. Die Phosphatgruppe ist bei neutralem pH negativ geladen. Die
Kopfgruppe hingegen kann sowohl negativ, als auch positiv geladen sein.
Tabelle 1: Übersicht der verschiedenen Lipidklassen.
Klasse Beispiele
Fettsäuren Oleat, Stearoyl-Coa, Palmitoylcarnitin
Glycerolipide Di- und Triacylglycerine
Glycerophospholipide Phosphatidylcholin, Phosphatidylserin
Sphingolipide Sphingomyelin, Gangliosid GM2
Sterinlipide Cholesterin, Progesteron, Gallensäuren
Prenyllipide Farnesol, Geraniol, Retinol, Ubichinon
Polyketide Tetracyclin, Aflatoxin B1
Saccharolipide Lipopolysaccharid
Beispiele für Lipide mit einer positiv geladenen Kopfgruppe sind
Phosphatidylethanolamin (PE) oder Phosphatidylcholin (PC). Bei einem
physiologischen pH sind diese Lipide neutral, da die positive Ladung der Kopfgruppe
(+1) die negative Ladung der Phosphatgruppe (-1) ausgleicht. Ferner werden daher
PE und PC als zwitterionische Lipide bezeichnet. Phosphatidylglycerin (PG) oder
Phosphatidylserin (PS) besitzen hingegen eine negativ geladene Kopfgruppe und
zählen deshalb zu den anionischen Lipiden.
Weiterhin ist für diese Arbeit das Sphingolipid Sphingomyelin wichtig. Der Aufbau von
Sphingolipiden ist Phospholipiden generell sehr ähnlich. Sie besitzen ebenfalls zwei
Fettsäuren und eine polare Kopfgruppe. Die Grundstruktur eines Sphingolipides
bezeichnet man als Ceramid. Die Besonderheit bei einem Ceramid ist, dass eine
Fettsäure über eine Amidbrücke an das C2-Atom geknüpft ist. Sphingomyelin besitzt
als Kopfgruppe Cholin, das auch über eine Phosphorsäurediesterbindung verbunden
ist. Die Struktur und die Ladung von Sphingomyelin ist PC sehr ähnlich.
Sphingomyeline sind insbesondere in Plasmamembranen von Nervenzellen zu
finden. Eine Übersicht ausgewählter Lipide ist in Abbildung 2 dargestellt.

Einleitung
- 3 -
Abbildung 2: Strukturformeln ausgewählter Phospholipide und Sphingomyelin. CL: Cardiolipin; DMPE: Dimethylphosphatidylethanolamin; DPPG: Lysyl-PG; LPE: Lyso-PE; MMPE: Monomethylphosphatidylethanolamin; PA: Phosphatidat; PC: Phosphatidylcholin; PE: Phosphatidyl-ethanolamin; PG: Phosphatidylglycerin; PI: Phosphatidylinositol; PS: Phosphatidylserin, SM: Sphingomyelin.

Einleitung
- 4 -
Aufgrund ihres amphiphilen Charakters können Lipide in wässrigen Lösungen
Membranen ausbilden, die zwei Kompartimente voneinander trennen. Neben der
strukturgebenden Funktion, dienen Lipide auch als Energiespeicher und haben eine
enorme Bedeutung für die Faltung, Stabilität und Regulation von integralen oder
assoziierten Membranproteinen (Dowhan, 1997; van Meer et al., 2008). So benötigt
beispielsweise der Multidrugtransporter LmpR aus Lactococcus lactis für seine
Transporterfunktion die Anwesenheit von PE. In Abwesenheit von PE ist LmpR
inaktiv (Hakizimana et al., 2008). Ein Beispiel für die regulatorische Funktion von
Lipiden ist die cytosolische Protease Lon aus E. coli. Lon wird durch Cardiolipin an
die Membran gebunden und dadurch inaktiviert (Minami et al., 2011). Neben der
Bedeutung für Membranproteine, können Lipide auch als Signalmoleküle dienen.
Exponiertes PS in der äußeren Membranschicht von Eukaryoten ist beispielsweise
ein Zeichen für den programmierten Zelltod (Bevers et al., 1983; Rosing et al., 1985;
Tait & Gibson 1994). In Abbildung 3 sind die wichtigsten Funktionen von Lipiden kurz
dargestellt.
Abbildung 3: Übersicht über die verschiedenen Funktionen von Lipiden. Lipide haben neben der Funktion als strukturgebendes Element biologischer Membranen noch weitere Bedeutung für Membranproteine, als Signalstoffe oder Energiespeicher.

Einleitung
- 5 -
1.2 Die Bedeutung von Phosphatidylcholin
1.2.1 Vorkommen und Bedeutung von Phosphatidylcholin
Phosphatidylcholin (PC) besitzt als Kopfgruppe Cholin, das eine positiv geladene,
quartäre Ammoniumverbindung ist. PC ist das häufigste membranbildende Lipid in
Eukaryoten (Kent, 1990; Vance, 2002). Neben der strukturgebenden Funktion für die
Membran reguliert PC auch zahlreiche zelluläre Prozesse. PC ist unter anderem bei
der lipidabhängigen Signalübertragung (engl. lipid signaling) und der Aktivierung von
membranassoziierten Proteinen beteiligt (Kent, 1990; Exton, 1994). PC dient zudem
als Reservoir für Cholin, das unter anderem für die Synthese des Neurotransmitters
Acteylcholin benötigt wird. Desweiteren ist PC ein Vorläufermolekül für sekundäre
Lipidbotenstoffe. Dazu zählen beispielsweise Lyso-PC, Phosphatidat, Diacylglycerin,
Lyso-Phosphatidat, Thrombozyten-aktivierender Faktor (PAF) oder Arachidonsäure
(Exton, 1994; Zeisel & Blusztajn, 1994). Ebenfalls ist PC das Hauptlipid von
Lipoproteinen im Blut (Kent, 1990; Leblanc et al., 1998). Im Gegensatz zu
Eukaryoten ist das Vorkommen sowie die Bedeutung von PC in prokaryotischen
Organismen bisher nur geringfügig untersucht. Ein Grund hierfür ist, dass die
Modellorganismen Bacillus substilis und Escherichia coli kein PC besitzen.
Bioinformatische Genomstudien haben allerdings gezeigt, dass wahrscheinlich 10 %
aller Bakterien PC synthetisieren können (Sohlenkamp et al., 2003). Häufig ist PC in
Bakterien präsent, die mit einem eukaryotischen Wirt interagieren, sodass PC
wahrscheinlich eine zentrale Rolle bei einer Bakterien-Wirts-Interaktion spielt
(López-Lara et al., 2003; Sohlenkamp et al., 2003; Aktas et al., 2010).
1.2.2 PC-Biosynthese in Eukaryoten
In Eukaryoten gibt es zwei PC-Biosynthesewege. Zum einen kann PC über einen
Methylierungsweg synthetisiert werden, zum anderen über einen Cytidindiphosphat-
Cholin (CDP-Cholin) Weg. Der Methylierungsweg ist eine de novo Synthese und
beruht auf einer dreifachen Methylierung von Phosphatidylethanolamin (PE) zu PC.
Dabei entstehen die Intermediate Monomethylphosphatidylethanolamin (MMPE) und
Dimethylphosphatidylethanolamin (DMPE). Der Methyldonor für diese Reaktion ist

Einleitung
- 6 -
S-Adenosylmethionin (SAM), der zu S-Adenosylhomocystein (SAH) umgewandelt
wird (Bremer & Greenberg, 1959; Vance & Ridgway, 1988; Ridgway et al., 1989).
In der Hefe Saccharomyces cerevisiae katalysieren zwei Phosphatidylethanolamin N-
Methyltransferasen (PEMT) den Methylierungsweg. Diese sind eingeteilt in Typ I und
Typ II PEMT (Kodaki & Yamashita, 1987; Kanipes & Henry, 1997;
Kanipes et al., 1998). Das Typ II Enzym PEMT1/CHO2 kann nur PE zu MMPE
methylieren. Die PEMT2/OPI3 katalysiert hingegen vorwiegend die nachfolgenden
Methylierungsschritte von MMPE zu DMPE und PC. Eine Methylierung von PE zu
MMPE ist auch durch PEMT2/OPI3 möglich, jedoch sehr ineffizient (Kodaki &
Yamashita, 1987; Kanipes & Henry, 1997). CHO2 und OPI3 sind Proteine, die im
endoplasmatischen Reticulum lokalisiert sind (Gaynor & Carman, 1990). Das
PEMT1-Enzym der Hefe ist homolog zu den Phospholipid N-Methyltransferasen
(Pmt) aus der Ratte und Maus (Vance, 2002). Weiterhin haben PEMT1 und 2
zueinander einen sehr hohen Verwandschaftsgrad (Kodaki & Yamashita, 1987).
Daher ist es wahrscheinlich, dass beide Proteine ursprünglich von einem
gemeinsamen Vorläufergen abstammen (Kanipes & Henry, 1997). Die Pmt-Enzyme
der Hefe konnten bisher nicht isoliert werden. Zur Analyse der enzymatischen und
kinetischen Eigenschaften von OPI3 und CHO2 wurden S. cerevisiae-
Deletionsmutanten verwendet. Die Enzyme OPI3 und CHO2 unterscheiden sich im
pH-Optimum, in den benötigten Kofaktoren und der Thermostabilität (Gaynor &
Carman, 1990).
Im Gegensatz zur Hefe besitzen höhere Säugetiere nur ein pemt-Gen, das für ein
ca. 20 kDa großes PEMT-Enzym kodiert. Dieses Enzym katalysiert alle drei
Methylierungsschritte von PE zu PC. Signifikante Methylierungsaktivität wurde
allerdings nur im Lebergewebe gefunden. Die PEMT der Rattenleber ist ein 18 kDa
großes, monomeres Protein, das in zwei Isoformen vorkommt. Die PEMT1 ist ein
integrales Membranprotein, das in der Membran des endoplasmatischen Reticulums
lokalisiert ist. Die PEMT2 ist hingegen in der mitochondrialen Membran lokalisiert
(Ridgway & Vance, 1987; Cui et al., 1993; Vance et al., 1997). Es wird vermutet,
dass posttranslationale Modifikationen für die unterschiedliche Lokalisation
verantwortlich sind (Vance et al., 1997). Die kinetischen Eigenschaften und der
Mechanismus dieser PEMT-Enzyme sind gut untersucht (Ridgway & Vance, 1987;
Ridgway & Vance, 1988; Ridgway & Vance, 1992; Vance et al., 1997). Die Katalyse

Einleitung
- 7 -
erfolgt nach einem Bi-Bi Mechanismus: d.h. das Lipid bindet als erster Ligand und
dissoziiert als letzter Ligand vom Enzym. Die Substrate PE, MMPE, DMPE und PC
konkurrieren dabei jeweils um die gleiche Bindestelle. Erst nach einer erfolgreichen
Substratbindung wird der Methyldonor SAM gebunden und die
Transmethylierungsreaktion kann durchgeführt werden (Ridgway & Vance, 1987;
Ridgway & Vance, 1988; Ridgway & Vance, 1992; Vance et al., 1997). Eine
Regulation der PEMT-Enzyme erfolgt sehr wahrscheinlich durch die inhibierenden
Endprodukte PC und SAH (Ridgway & Vance, 1988).
Im Gegensatz zum Methylierungsweg wird durch den alternativen PC-
Biosyntheseweg, dem CDP-Cholin-Weg, PC nicht de novo synthetisiert. Beim CDP-
Cholin-Weg wird CDP-Cholin mit 1,2-Diacylglycerin (DAG) direkt verknüpft. Freies
Cholin kann dafür aus exogenen Quellen stammen oder intern durch den Abbau von
PC durch die Phospholipasen C und D freigesetzt werden. Die Aufnahme von Cholin
erfolgt über Cholin-Transporter (Kent, 1990). Es sind drei Transportsysteme bekannt:
1. Hoch-affiner, Na+-abhängiger Transport; 2. Intermediäraffiner, Na+-unabhängiger
Transport und 3. Niedrig-affiner Transport (Michel et al., 2006). Sobald das Cholin in
die Zelle gelangt, wird es durch die Cholinkinase (CKI) phosphoryliert.
Cholinphosphat wird durch die CTP-Phosphocholin Cytidylyltransferase (CCT) unter
Verbrauch eines Moleküls Cytidintriphosphat (CTP) zu CDP-Cholin umgewandelt.
Die Cholingruppe wird dann durch die 1, 2-DAG Cholinphosphotransferase (CPT) mit
DAG zu PC verknüpft, wobei ein CMP-Molekül freigesetzt wird
(Kennedy & Weiss, 1956; Weiss et al., 1958; Kennedy, 1989). Die CKI ist im Cytosol
lokalisiert und kann neben Cholin auch Ethanolamin phosphorylieren (Brophy et al.,
1977; Yamashita & Hosaka, 1997). Die generelle Regulation des CDP-Cholin-Weges
erfolgt jedoch über die CCT (Vance, 2002). Die CCT ist ein amphitrophes Protein,
das in löslicher Form inaktiv im Nucleolus lokalisiert ist. Erst eine Rekrutierung und
Assoziation an die Membran des endoplasmatischen Reticulums aktiviert die CCT
(Cornell & Northwood, 2000).
Generell ist die präferierte Nutzung eines PC-Biosyntheseweges abhängig vom
Organismus oder vom Gewebetyp bei höheren Eukaryoten. Hefen und Pilze nutzen
hauptsächlich den Methylierungsweg (Kanipes & Henry, 1997). Säugetiere hingegen
nutzen bevorzugt den CDP-Cholin-Weg (Vance & Schneider, 1981;
Vance & Ridgway, 1988; Vance et al., 2007). Dieser ist essentiell in tierischen Zellen

Einleitung
- 8 -
(Walkey et al., 1998). Die PC-Synthese in Pflanzen ist bisher relativ unerforscht,
wobei PC hier vermutlich auch hauptsächlich über den CDP-Cholin-Weg synthetisiert
wird. In Abbildung 4 sind die hier vorgestellten PC-Biosynthesewege in Eukaryoten
schematisch dargestellt.
Abbildung 4: Gegenüberstellung der verschiedenen eukaryotischen PC-Biosynthesewege (nach Sohlenkamp et al., 2003) . In Hefen wird PC hauptsächlich über den Methylierungsweg synthetisiert. Die Enzyme CHO2 und OPI3 methylieren dafür PE über die Intermediate MMPE und DMPE zu PC. Säugetiere besitzen auch einen Methylierungsweg, diese gesamte Methylierungsreaktion wird jedoch nur von einem PEMT-Enzym katalysiert. Vorwiegend wird in höheren Säugetieren der CDP-Cholin-Weg zur PC-Biosynthese genutzt, bei dem CDP-DAG direkt mit Cholin verknüpft wird. ADP: Adenosindiphosphat; ATP: Adenosintriphosphat; CKI: Cholinkinase; CCT: CTP-Phosphocholin Cytidylyltransferase; CMP: Cytidinmonophosphat; CPT: Cholinphosphotransferase; CTP: Cytidintriphosphat; DAG: Diacylglycerin; DMPE: Dimethyl-PE; MMPE: Monomethyl-PE; PA: Phosphatidat; PC: Phosphatidylcholin; PE: Phosphatidylethanolamin; PEMT: PE N-Methyltransferase; PLC: Phospholipase C; PLD: Phospholipase D; PPi: Pyrophosphat; R: Kohlenwasserstoffrest; SAM: S-Adenosylmethionin; SAH: S-Adenosylhomocystein.

Einleitung
- 9 -
1.2.3 Bedeutung von PC für Bakterien-Wirts-Interaktionen
Eine bakterielle Membran setzt sich in der Regel aus den Phospholipiden PE
(70 - 80 mol%), PG (20 – 25 mol%) und CL (<5 mol%) zusammen (Dowhan, 1976;
Linde et al., 2004). Phototrophe Bakterien mit vielen internen Membranen und auch
Bakterien, die mit Eukaryoten assoziiert leben, besitzen meist zusätzlich PC
(Goldfine, 1984). Genomstudien haben gezeigt, dass wahrscheinlich 10 % aller
Bakterien PC synthetisieren können (López-Lara et al., 2001; Sohlenkamp et al.,
2003). Der Anteil von PC in der Membran ist dabei von Organismus zu Organismus
sehr variabel. Bei Pseudomonas aeruginosa beträgt der Anteil von PC nur einige
wenige Prozent der Gesamtlipidmenge (Goldfine, 1984), bei Acetobacter aceti
hingegen beträgt der PC-Gehalt in der Membran bis zu 73 % (Hanada et al., 2001).
Da die prokaryotischen Modellorganismen E. coli und B. subtilis kein PC besitzen,
wurde PC in Bakterien lange Zeit keine Aufmerksamkeit geschenkt.
Für das Überleben von Bakterien ist PC in der Membran nicht essentiell. Eine PC-
defiziente Mutante von Rhodobacter sphaeroides oder Zymomonas mobilis weist ein
normales Wachstum auf. Dementgegen gibt es Bakterien, die zwar ohne PC
überleben, aber einen deutlichen Phänotyp zeigen. Die Wachstumsrate und
maximale Zelldichte ist bei einer A. aceti PC-defizienten Mutante deutlich reduziert.
Eine Bradyrhizobium japonicum-Mutante, die nur sehr geringe Mengen an PC
besitzt, weist eine reduzierte Symbioseeffizienz mit seiner Wirtspflanze auf. Hierbei
werden weniger Bakteroide gebildet, was zu einer geringeren Stickstofffixierung
(18 % des Wildtyps) führt (Minder et al., 2001). Eine PC-Defizienz führt bei
Sinorhizobium meliloti zu einem noch drastischeren Symbiosedefekt. Ohne PC ist
S. meliloti nicht mehr in der Lage Knöllchen zu bilden, die für eine Symbiose
essentiell sind (Sohlenkamp et al., 2000). Neben symbiontischen Bakterien zeigen
auch pathogene Bakterien einen deutlichen Phänotyp in Abwesenheit von PC.
Brucella abortus hat zum Beispiel ohne PC einen deutlichen Virulenzdefekt (Comerci
et al., 2006). Legionella pneumophila produziert ohne PC weniger Flagellin, wodurch
die Motilität eingeschränkt ist und das Bakterium daher einfacher von Makrophagen
gebunden werden kann. Desweiteren ist die Funktion des Typ IV-
Sekretionssystemes Dot/Icm in L. pneumophila stark beeinträchtigt (Conover et al.,
2008). Eine PC-defiziente Mutante des pflanzenpathogenen Bakteriums
Agrobacterium tumefaciens zeigt ebenfalls einen gravierenden Virulenzdefekt. Das

Einleitung
- 10 -
Fehlen von PC führt zum Verlust des Typ IV-Sekretionssystems in der Membran.
Diese Beispiele zeigen, dass PC in bakteriellen Membranen eine wichtige Rolle für
eine effiziente Symbiose oder Infektion spielt. Bakterielle PC-Biosyntheseenzyme
sind daher potentielle Angriffspunkte für Antibiotika.
1.2.4 PC-Biosynthese in Prokaryoten
Bakterien können PC wie Eukaryoten über den Methylierungsweg synthetisieren,
wobei die bakteriellen PC-Biosyntheseenzyme des Methylierungsweges meist
cytosolisch lokalisiert sind. Bisher sind nur sehr wenige Bakterien bekannt, die einen
CDP-Cholin-Weg zur PC-Synthese nutzen. Ein Beispiel dafür ist Treponema
denticola (Kent, 2005). Ein Großteil der PC-synthetisierenden Bakterien nutzt
hingegen einen dem CDP-Cholin-Weg ähnlichen PC-Syntheseweg. Hier wird aus
Cholin und CDP-DAG PC produziert. Dieser Syntheseweg wird durch die
Phosphatidylcholinsynthase (Pcs), die bisher nur in Bakterien identifiziert wurde,
katalysiert. Viele Bakterien besitzen sowohl den Methylierungsweg, als auch den
Pcs-Weg (Sohlenkamp et al., 2000; Wessel et al., 2006) (Abbildung 5). Einige
Bakterien dagegen haben nur einen der beiden Wege (Arondel et al., 1993).
Beispielsweise synthetisieren Pseudomonas aeruginosa, B. abortus und Borrelia
burgdorferi PC nur über den Pcs-Weg (Wilderman et al., 2002; Martínez-Morales et
al., 2003; Comerci et al., 2006). Im Gegensatz dazu nutzen S. meliloti,
A. tumefaciens, Mesorhizobium loti und L. pneumophila sowohl den Pcs-Weg, als
auch den Methylierungsweg (Sohlenkamp et al., 2003; Wessel et al., 2006).

Einleitung
- 11 -
Abbildung 5: Bakterielle PC-Biosynthese (nach Wessel et al., 2006). Bakterien besitzen einen zu Eukaryoten homologen Methylierungsweg. Die PC-Biosynthese via Pcs ist wahrscheinlich exklusiv für Bakterien. CDP: Cytidindiphosphat; CMP: Cytidinmonophosphat; DAG: Diacylglycerin; Pcs: Phosphatidylcholinsynthase; Pmt: Phospholipid N-Methyltransferase; SAH: S-Adenosylhomocystein; SAM: S-Adenosylmethionin.
Phosphatidylcholinsynthase-Weg
Der Pcs-Weg unterscheidet sich vom Kennedy-Weg insofern, dass die Pcs kein
CDP-aktiviertes Cholin verwendet, sondern ein CDP-aktiviertes Phospholipid
(de Rudder et al., 1997; de Rudder et al., 1999; Sohlenkamp et al., 2000;
Martínez-Morales et al., 2003). Durch eine Kondensierung von Cholin mit CDP-
Diacylglycerin entsteht PC und das Nebenprodukt CMP. Pcs-Enzyme besitzen
6 - 8 Transmembranhelices und sind integrale Membranproteine. Üblicherweise
besitzen Pcs-Enzyme ein modifiziertes Motiv (DGx2ARx12Gx3Dx3D), das
charakteristisch für CDP-Alkoholphosphatidyltransferasen (CDP-OH-P)
(DGx2ARx8Gx3Dx3D) ist (Sohlenkamp et al., 2000; Sohlenkamp et al., 2003). Das
Pcs-Enzym aus S. meliloti wurde als erstes Enzym dieser Klasse charakterisiert. Es
weist eine Länge von 291 Aminosäuren auf, ist stark hydrophob und hat ein pH-
Optimum von 8. Desweiteren ist die Enzymaktivität abhängig von divalenten

Einleitung
- 12 -
Kationen wie Mg2+ oder Mn2+. Kinetische Analysen zeigen für sinorhizobielles Pcs
eine sigmoidale Bindungskurve für Cholin bei Konzentrationen über 100 mM.
Unterhalb einer Konzentration von 100 mM Cholin beobachtet man jedoch eine
Michaelis-Menten-Kinetik für Pcs. Daher ist zum einen möglich, dass die Pcs-Aktivität
einem positiven kooperativen Effekt unterliegt, d.h. die Bindung eines Moleküls
Cholin erleichtert die Bindung eines weiteren Moleküls Cholin, oder aber Pcs wird
durch Cholin inhibiert (de Rudder et al., 1999). Die Topologie der sinorhizobiellen
Pcs, sowie einige wichtige Aminosäuren für die Enzymaktivität wurden kürzlich
identifiziert. Konservierte Aminosäuren in den CDP-OH-P sind in einem
cytoplasmatischen Loop und teilweise in einer Transmembranhelix lokalisiert
(Solís-Oviedo et al., 2012). Pcs-Enzyme sind weiterhin homolog zu
Phosphatidylserinsynthasen (Pss). Die Pss katalysiert die Synthese von
Phosphatidylserin aus CDP-DAG und Serin.
Für die PC-Synthese benötigt die bakterielle Pcs freies Cholin. Dies ist
beispielsweise in tierischem und menschlichem Blut vorhanden (Sheard et al., 1986).
Die pathogenen Bakterien P. aeruginosa, B. abortus und L. pneumophila könnten
dieses Cholin als Quelle für die PC-Biosynthese nutzen. Darüber hinaus können
pathogene Organismen auch Phospholipasen sekretieren. Diese greifen die
eukaryotische Wirtsmembran an und können PC an spezifischen Stellen spalten,
sodass Cholin freigesetzt wird. Dieses freigesetzte Cholin kann wiederum für die PC-
Biosynthese genutzt werden. In P. aeruginosa wird beispielsweise die Expression der
Phospholipase C und einer Phosphatase durch Cholin induziert (Lisa et al., 1984;
Lucchesi et al., 1989).
Eine S. meliloti- oder A. tumefaciens-Mutante, deren Methylierungsweg
ausgeschaltet ist, kann PC in wildtypischen Mengen über den Pcs-Weg
synthetisieren (de Rudder et al., 1997; Wessel et al., 2006). S. meliloti kann hierbei
Cholin von der Wirtspflanze mit Hilfe eines hoch-affinen ABC-Transporters
aufnehmen (de Rudder et al., 1997; Dupont et al., 2004). In A. tumefaciens wurde
kürzlich ein ähnliches hoch-affines Cholin-Transportersystem (ChoXWV) identifiziert
und charakterisiert. Allerdings wird dieses System für eine PC-Synthese durch Pcs
nicht benötigt. Es ist daher sehr wahrscheinlich, dass Pcs extrazelluläres Cholin im
Periplasma binden und umsetzen kann (Aktas et al., 2011b).

Einleitung
- 13 -
Das Bereitstellen oder Vorkommen von freiem Cholin durch den Wirtsorganismus
scheint somit eine wichtige Rolle für die Nutzung des Pcs-Weges in Bakterien zu
spielen.
Methylierungsweg
Neben dem Pcs-Weg können Bakterien PC auch über einen Methylierungsweg
synthetisieren. Dafür katalysiert eine Phospholipid N-Methyltransferase (Pmt) die
schrittweise Methylierung von PE zu PC. Als Intermediate der PC-Biosynthese
entstehen MMPE und DMPE. Der Methyldonor für diese Reaktion ist SAM und nach
Abspalten der Methylgruppe entsteht SAH. Bakterielle Pmt-Enzyme unterscheiden
sich untereinander stark in ihrer Aminosäuresequenz. Die meisten bakteriellen Pmt-
Enzyme sind cytosolische Proteine, die an der Membran peripher assoziiert sind.
Bisher ist jedoch keine dreidimensionale Struktur eines bakteriellen Pmt-Enzyms
bekannt. Man unterscheidet bei bakteriellen Pmt-Enzymen zwischen zwei Familien.
Die sinorhizobiellen Pmt-Enzyme besitzen Sequenzhomologien zur Pmt aus
S. meliloti. Desweiteren haben diese Pmt-Enzyme Ähnlichkeit mit rRNA-
Methyltransferasen. Zum anderen gibt es die rhodobakteriellen Pmt-Enzyme, welche
auf Sequenzebene homolog zur Pmt aus R. sphaeroides sind (Sohlenkamp et al.,
2003). Rhodobakterielle Pmts zeigen zudem Homologien zu Ubiquinon/Menaquinon
Biosynthesemethyltransferasen. Die einzige Gemeinsamkeit zwischen
sinorhizobiellen, rhodobakteriellen und eukaryotischen Pmts ist das SAM-
Bindemotiv I (Abschnitt 1.3.3) (Haydock et al., 1991).
Als erstes bakterielles Pmt-Enzym wurde das Pmt aus R. sphaeroides (RPmt)
untersucht. RPmt ist ein monomeres 23 kDa großes Protein. In E. coli, das
natürlicherweise kein PC synthetisieren kann, wurde nach heterologer Expression
von RPmt PC-Synthese nachgewiesen. Allerdings wurden keine Intermediate
detektiert (Arondel et al., 1993). Weitere Vertreter dieses Pmt-Typs sind Pmt-Enzyme
aus A. aceti und L. pneumophila (Hanada et al., 2001; Martínez-Morales et al., 2003).
Später wurde das sinorhizobielle Pmt (SPmt) isoliert und charakterisiert. SPmt ist
22 kDa groß und synthetisiert deutliche Mengen an MMPE und PC nach
Überexpression in E. coli (Arondel et al., 1993; de Rudder et al., 2000). Weitere
Beispiele für SPmt-ähnliche Enzyme findet man in A. tumefaciens und B. japonicum.
Einen dritten, bisher relativ unbeschriebenen bakteriellen Pmt-Typ, besitzt

Einleitung
- 14 -
Zymomonas mobilis. Dieses Protein ist membrangebunden und als Homotrimer aktiv,
wobei eine Untereinheit dieses Trimers 42 kDa groß ist (Tahara et al., 1986; Tahara
et al., 1987a; Tahara et al., 1987b; Tahara et al., 1994).
Genomanalysen lassen vermuten, dass einige Bakterien zwei oder mehrere Pmt-
Enzyme für die PC-Biosynthese besitzen (López-Lara et al., 2003). Verschiedene
Pmts wurde in B. japonicum bereits nachgewiesen. Hierbei katalyisert PmtA
hauptsächlich den ersten Methylierungsschritt von PE zu MMPE
(Hacker et al., 2008). Die Methylierung von MMPE zu DMPE und PC erfolgt dann
über ein zweites Pmt-Enzym (PmtX1) (Minder et al., 2001; Hacker et al., 2008).
Wahrscheinlich synthetisieren auch Mesorhizobium loti, Rhizobium leguminosarum
und Rhodopseudomonas palustris PC durch mehrere Pmt-Enzyme
(López-Lara et al., 2003).
1.2.5 Die Phospholipid N-Methyltransferase PmtA aus
Agrobacterium tumefaciens
Im Vorfeld dieser Arbeit wurden grundlegende enzymatische Eigenschaften der
agrobakteriellen Phospholipid N-Methyltransferase (PmtA) untersucht. PmtA konnte
in E. coli heterolog überexprimiert und mittels Affinitätschromatographie isoliert
werden. Der Großteil des Proteins war jedoch unlöslich (Aktas & Narberhaus, 2009).
Die isolierte PmtA ist ein 22 kDa großes monomeres Protein, das sowohl heterolog in
vivo als auch in vitro aktiv ist. Es kann sowohl isolierte E. coli-Membranen, als auch
reines PE, MMPE und DMPE als Phospholipidsubstrat nutzen. Die
Methylierungsaktivität von PmtA wird durch PG stimuliert. Die Substratlipide PE,
MMPE, DMPE und das Produkt PC werden von PmtA in vitro gebunden. Darüber
hinaus bindet PmtA auch die anionischen Lipide PG und PI. Der Methyldonor für die
PC-Biosynthese ist SAM. Ähnlich wie dem eukaryotischen PEMT muss zunächst
eines der Phospholipidsubstrate gebunden werden, bevor der Kofaktor SAM
gebunden wird. Die katalysiert Mehrsubstratreaktion von PmtA unterliegt sehr
wahrscheinlich einem Bi-Bi Mechanismus: d.h. das Lipidsubstrat bindet als erster
Ligand an PmtA und dissoziiert als letzter Ligand vom Enzym. Die enzymatische
Aktivität von PmtA wird durch die Endprodukte PC und SAH inhibiert (Abbildung 6).

Einleitung
- 15 -
Abbildung 6: Reaktionsmechanismus und Regulation (-/+) des agrobakteriellen PmtA-Enzyms.

Einleitung
- 16 -
1.3 S-Adenosylmethionin-abhängige Methyltransferasen
Da bakterielle und eukaryotische Phospholipid N-Methyltransferasen zur Klasse der
SAM-abhängigen Methyltransferasen (SAM-MT) gehören, sollen in diesem Kapitel
die wichtigsten Eigenschaften dieser Enzymklasse kurz erläutert werden.
1.3.1 S-Adenosylmethionin
SAM ist ein Konjugat aus Adenosin und Methionin (Kresge et al., 2005). In der Zelle
ist SAM ein essentieller Metabolit (Loenen, 2006) und fungiert als Methyldonor für
verschiedenste Substrate. Es werden zum einen monovalente Ionen wie
beispielsweise Arsen, Chlorid oder Brom, zum anderen auch höher molekulare
Verbindungen wie tRNA, rRNA, DNA, Lipide und sogar Proteine methyliert (Kozbial &
Mushegian, 2005). Auf atomarer Ebene wird die Methylgruppe an ein Kohlenstoff-,
Sauerstoff-, Stickstoff- oder Schwefelatom gekoppelt. Durch diesen Methyltransfer
wird SAM zu SAH umgewandelt. Nach ATP ist SAM das häufigst genutzte
Enzymsubstrat und wird meist bevorzugt gegenüber anderen energetisch
ungünstigeren Methyldonoren wie beispielsweise Folat genutzt (Schubert et al.,
2003). Die physiologische Funktion einer Methylierung kann je nach Substrat
variieren. So ist beispielsweise die Methylierung der DNA ein Schutz gegen eigene
Restriktionsendonukleasen. Die Methylierung von Proteinen hat hingegen meist
regulatorische Wirkung.
1.3.2 Strukturelle Merkmale SAM-abhängiger Methyltransferasen
Der Großteil der SAM-abhängigen Methyltransferasen (SAM-MT) besitzt eine
Rossmann-Faltung, die charakteristisch für Nukleotid-bindende Enzyme ist. Nur ein
kleiner Teil zählt zur TIM-Barrel-Klasse, die sich durch acht alternierende α-Helices
und β-Faltblätter auszeichnet (Kozbial & Mushegian, 2005). Die Sequenzhomologie
von SAM-MTs beträgt teilweise nur 10 %. Es liegt jedoch eine hohe strukturelle
Konservierung vor. SAM-MTs sind in fünf verschiedene „Faltungsklassen“ eingeteilt
(Schubert et al., 2003). Die meisten der SAM-MTs gehören zur Faltungsklasse 1.
Diese bildet meist ein großes, zentrales β-Faltblatt aus. Es wird flankiert von jeweils

Einleitung
- 17 -
drei α-Helices. Eine allgemeine Anordnung der β-Faltblätter entspricht dem Muster
3 2 1 4 5 (7) 6 (Abbildung 7), wobei das siebte β-Faltblatt antiparallel zu den
restlichen β-Faltblättern angeordnet ist (Kozbial & Mushegian, 2005). Das N-
terminalste β-Faltblatt ist zentral lokalisiert. Einige wenige SAM-MTs besitzen eine
zusätzliche α-Helix (Martin & McMillan, 2002) oder bei anderen fehlen die β-
Faltblätter 6 und/oder 7 (Bujnicki, 1999).
Abbildung 7: Schematischer Aufbau von SAM-abhängigen Methyltransferasen. (A.) Strukturformel von S-Adenosylmethionin. (B.) Schematische Darstellung der Sekundärstruktur von SAM-MTs der Faltungsklasse 1. Im N-terminalen Bereich einer SAM-MT wird der Kofaktor SAM gebunden. Die Substratbinderegion ist C-terminal lokalisiert. N: Aminoterminus; C: Carboxyterminus. Gelbe Markierungen zeigen die Lokalisation der SAM-Bindemotive an. Rote Pfeile: β-Faltblatt; Blaue Zylinder: α-Helix
Weitergehend werden die SAM-MTs nach der Beschaffenheit ihres
Methylierungssubstrates eingeteilt (bspw. DNA- oder RNA- SAM-MT) (Martin &
McMillan, 2002). Vereinfacht betrachtet besteht eine SAM-MT aus einer SAM-
Bindedomäne und einer Substratbindedomäne. Die Bereiche und Aminosäuren der

Einleitung
- 18 -
Substratbindedomäne erkennen und binden das Methylierungssubstrat, sind jedoch
aufgrund der hohen Substratvielfalt sehr variabel. Die SAM-Bindedomäne wird aus
Aminosäuren gebildet, die den Kofaktor SAM binden und die Methylgruppe auf das
Substrat übertragen (Kozbial & Mushegian, 2005; Faumann et al., 1999). Eine der
ersten beschriebenen und charakterisierten SAM-MTs war die Catechol
O-Methyltransferase (COMT) (Vidgren et al., 1994). Es folgte die strukturelle
Aufklärung weiterer SAM-MTs, deren Substrate Nukleinsäuren, Proteine oder
kleinere Moleküle sind (Reinisch et al., 1995; Pattanayek et al., 1998; O’Gara et al.,
1999; Zhang et al., 2000). Die Aminosäurereste, die SAM binden, sind generell kaum
konserviert. Die SAM-Bindung ist jedoch immer an strukturell äquivalenten
Positionen lokalisiert. Diese Bereiche innerhalb der Protein-Struktur bezeichnet man
als SAM-Bindemotive, welche im nächsten Abschnitt näher beschrieben werden.
1.3.3 SAM-Bindemotive
Die SAM-Binderegion befindet sich im N-terminalen Bereich, der durch die Loops
und β-Faltblätter 1, 2 und 3 gebildet wird (Martin & McMillan, 2002). Bisher wurden
zehn SAM-Bindemotive (SAM-I – X-Motiv) beschrieben, wobei die ersten sechs für
alle SAM-MTs gelten (Cheng et al., 1993; Kumar et al., 1994; Malone et al., 1995;
Kozbial & Mushegian, 2005). Die Motive sieben bis zehn sind spezielle Bindemotive
für die Klasse der SAM-abhängigen DNA-Methyltransferasen (Kozbial & Mushegian,
2005). Die sechs universellen SAM-Bindemotive treten immer in der gleichen
linearen Abfolge auf. Dabei ist nicht die Aminosäure selbst, sondern die Eigenschaft
des Aminosäurerestes (sauer, hydrophob, basisch) immer an der strukturell
äquivalenten Position innerhalb der Sekundärstruktur konserviert. Das SAM-
Bindemotiv I (SAM-I-Motiv) ist ein glycinreicher Loop zwischen der α-Helix αA und β-
Faltblatt 1 (GxGxG). Die Glycine sind nicht universell konserviert und können durch
ähnlich kleine, unpolare Aminosäuren ersetzt sein (Kozbial & Mushegian, 2005).
Desweiteren gehört eine saure Aminosäure (D/E) im mittleren Bereich des β-
Faltblattes 1 und eine oder mehrere basische Aminosäuren am Ende des β-
Faltblattes 1 zum SAM-I-Motiv. Formell wird das SAM-I-Motiv durch D/ExGxGxG
beschrieben (Kozbial & Mushegian, 2005). Eine saure Aminosäure (D/E) am C-
Terminus des β-Faltblattes 2 oder im benachbarten Loop zeichnet das SAM-II-Motiv

Einleitung
- 19 -
aus. Das SAM-III-Motiv weist ebenfalls eine saure Aminosäure (D/E) im C-terminalen
Bereich des β-Faltblattes 3 auf.
Im N-terminalen Bereich des β-Faltblattes 4 oder den flankierenden Loops ist eine
saure Aminosäure (D/E) oder ein Asparagin (N) für die SAM-Bindung verantwortlich
(SAM-IV-Motiv). Auf das β-Faltblatt 5 folgt eine α-Helix (αD) mit außergewöhnlich
hohem Anteil an hydrophoben Aminosäuren. Diese stellt das SAM-V-Motiv dar. Ein
einzelnes oder auch doppeltes Glycin nach dem β-Faltblatt 4 ist das SAM-VI-Motiv
(Kozbial & Mushegian, 2005). Die Motive SAM-VII und SAM-VIII sind hinsichtlich der
Aminosäurereste kaum konserviert. Für die DNA-Methyltransferase M.HhaI besteht
das SAM-VII-Motiv aus einem Aspartat und einem Tyrosin zwischen der α-Helix αE
und dem β-Faltblatt 6. Das SAM-VIII-Motiv ist als ein konserviertes Phenylalanin
zwischen dem β-Faltblatt 6 und 7 oder als eine Aminosäuresequenz QxRxR
beschrieben (Malone et al., 1995).
Das aktive Zentrum bei SAM-MTs bilden die Motive SAM-I bis SAM-III. Das SAM-I-
Motiv interagiert dabei mit dem Carboxylterminus des Methionins von SAM. Neben
direkter ionischer Interaktion wird auch die Koordination über ein Wassermolekül
durch die saure Aminosäure des SAM-I-Motives diskutiert (Kozbial & Mushegian,
2005). Das SAM-II-Motiv interagiert über Wasserstoffbrückenbindungen mit den
Hydroxylgruppen der Ribose. Das SAM-III-Motiv interagiert mit dem freien Aminrest
des Adenins von SAM (Abbildung 8) (Kozbial & Mushegian, 2005). Das SAM-V-Motiv
interagiert mit dem Adeninringsystem (Sankpal & Rao, 2002). Die Motive SAM-IV
und SAM-VI sind direkt im aktiven Zentrum lokalisiert und sind wahrscheinlich für die
Katalyse verantwortlich. Es wird postuliert, dass außer dem SAM-IV-Motiv noch
weitere Aminosäuren C-terminal des β-Faltblattes 4 mit der Ammonium- und
Sulfoniumgruppe von SAM interagieren (Kozbial & Mushegian, 2005). Die
Aminosäuren der Motive SAM-V und SAM-VI sind hauptsächlich für die strukturelle
Stabilität der SAM-MT verantwortlich (Kumar et al., 1994; Malone et al., 1995;
Faumann et al., 1999).
Der durch SAM-MTs katalysierte Methyltransfer gleicht dem einer SN2-Reaktion. Die
Methylgruppe wird direkt auf das Substrat übertragen, während das Substrat
synchron deprotoniert wird (Kozbial & Mushegian, 2005). Eine besondere Funktion
könnte hierbei die saure Aminosäure des SAM-I-Motivs übernehmen. Es wäre
möglich, dass diese Aminosäure zu Beginn der Reaktion die positive Ladung des

Einleitung
- 20 -
Sulfoniumions stabilisiert. Gleichzeitig werden Wasserstoffbrückenbindungen zum
Substrat aufgebaut, um dieses zu binden. Wird das Substrat durch die saure
Aminosäure deprotoniert, kann die Methylgruppe auf das Substrat übertragen
werden (Kozbial & Mushegian, 2005).
Abbildung 8: Postulierte Interaktionsstellen der SAM-Bindemotive I-V mit S-Adenosylmethionin. Das SAM-I-Motiv interagiert mit der Carboxylgruppe von SAM. Das SAM-II-Motiv baut Wasserstoffbrückenbindungen zu den Hydroxylgruppen der Ribose auf. Das SAM-III-Motiv wechselwirkt mit der Amingruppe des Adeninrings. Das SAM-V-Motiv interagiert mit dem Adeninringsystem, wohingegen das SAM-IV-Motiv direkt an der Übertragung der Methylgruppe beteiligt ist.

Zielsetzung
- 21 -
1.4 Zielsetzung
Zu Beginn dieser Arbeit war bekannt, dass die Phospholipid N-Methyltransferase
PmtA aus A. tumefaciens PE zu PC methyliert. Der Methyldonor für diese Reaktion
ist SAM. PmtA gehört daher zu den SAM-abhängigen Methyltransferasen (SAM-MT)
(Aktas & Narberhaus, 2009).
In dieser Arbeit sollen die Lipid- und SAM-Bindeeigenschaften von PmtA näher
charakterisiert werden. Durch gezielte Mutagenese sollen PmtA-Derivate generiert
werden, um wichtige Aminosäuren für die Lipid- und SAM-Bindung zu identifizieren.
Dazu werden die PmtA-Derivate hinsichtlich ihrer enzymatischen Aktivität, Lipid- und
SAM-Bindefähigkeit untersucht, um die Bedeutung der jeweiligen mutierten
Aminosäure für die Lipid- bzw. SAM-Bindung zu klären.
Zudem soll geklärt werden, warum PmtA die Anwesenheit eines
Phospholipidsubstrates benötigt, um SAM binden zu können. Dazu sollen u.a.
spektroskopische Methoden zur Analyse der Sekundärstruktur von PmtA etabliert
und angewendet werden, da eine Substrat-abhängige Konformationsänderung
vermutet wird. Diese spektroskopischen Methoden sollen weiterhin zur
Quantifizierung und Bestimmung der Bindeaffinität von PmtA zu verschiedenen
Modellmembranen dienen.
Weiterhin ist bisher ungeklärt, weshalb PmtA neben den Substratlipiden auch PG
und PI bindet und die PmtA-Aktivität durch PG stimuliert wird (Aktas & Narberhaus,
2009). Mit Hilfe eines bereits etablierten in vitro-Aktivitätsassays soll geklärt werden,
ob PmtA durch PG spezifisch stimuliert wird oder ob dies auch durch andere Lipide
möglich ist. Mit den zuvor erwähnten spektroskopischen Methoden und einem
immunochemischen Detektionsverfahren soll die Bindung von PmtA an verschiedene
Lipide nachgewiesen werden.
Zudem sollen zur Analyse der Substratspezifität von PmtA verschiedene Substrat-
Derivate eingesetzt werden.

Material und Methoden
- 22 -
2. Material und Methoden
2.1 Verwendete Stämme
Tabelle 2: Genotyp der verwendeten Bakterienstämme.
Stamm Genotyp Referenz
Escherichia coli
DH5
supE44 Δ(lacZYA-argF)U196
(Φ80ΔlacZM15) hsdR17 recA1 endA1 gyrA96 thi-1
relA1
Woodcock et al., 1989
Escherichia coli
BL21(DE3)
F-
ompT hsdSB(r
B
-
mB
-)
gal dem (λIts857 indI Sam7 nin5 lacUV5-
T7gene1)
Studier & Moffatt, 1986
Agrobacterium tumefaciens C58
Wildtyp Baron C., Montreal,
Canada
2.2 Plasmide
Tabelle 3: Übersicht der in dieser Arbeit verwendeten Vektoren.
Vektoren für E. coli Charakteristika Referenz/Quelle
pET28b(+)
KanR, Expressionsvektor mit einem T7 lac-Promotor und die Möglichkeit, ein Fusions-Protein
mit sechs C- und/oder N-terminalen Histidin-Resten (His-
Tag) zu konstruieren
Novagen, Madison, USA

Material und Methoden
- 23 -
Tabelle 4: Übersicht der in dieser Arbeit verwendeten rekombinanten Plasmide. Die angegebene
Position des Aminosäureaustausch bezieht sich auf die primäre Aminosäuresequenz von PmtA.
Rekombinante Plasmide Bemerkung Referenze
pET28b_pmtA (pBO832)
Wildtyp PmtA Aktas & Narberhaus,
2009
pET28b_pmtA Q26A (pBO2717)
Austausch von Glutamin an Position 26 zu Alanin
Diese Arbeit
pET28b_pmtA K28A (pBO2718)
Austausch von Lysin an Position 28 zu Alanin
Diese Arbeit
pET28b_pmtA K29A (pBO2719)
Austausch von Lysin an Position 29 zu Alanin
Diese Arbeit
pET28b_pmtA K28A,K29A (pBO2732)
Austausch von Lysinen an Position 28 und 29 je zu Alanin
Diese Arbeit
pET28b_pmtA I33A (pBO1247)
Austausch von Isoleucin an Position 33 zu Alanin
Diese Arbeit
pET28b_pmtA T36A (pBO2720)
Austausch von Threonin an Position 36 zu Alanin
Diese Arbeit
pET28b_pmtA K42A (pBO2728)
Austausch von Lysin an Position 42 zu Alanin
Diese Arbeit
pET28b_pmtA K43A (pBO2721)
Austausch von Lysin an Position 43 zu Alanin
Diese Arbeit
pET28b_pmtA K42A,K43A (pBO2729)
Austausch von Lysinen an Position 42 und 43 je zu Alanin
Diese Arbeit
pET28b_pmtA E58A (pBO1222)
Austausch von Glutamat an Position 58 zu Alanin
Diese Arbeit
pET28b_pmtA G60A (pBO1228)
Austausch von Glycin an Position 60 zu Alanin
Diese Arbeit
pET28b_pmtA P61A (pBO1223)
Austausch von Prolin an Position 61 zu Alanin
Diese Arbeit
pET28b_pmtA G62A (pBO1229)
Austausch von Glycin an Position 62 zu Alanin
Diese Arbeit
pET28b_pmtA G64A (pBO1230)
Austausch von Glycin an Position 64 zu Alanin
Diese Arbeit
pET28b_pmtA E84A (pBO866)
Austausch von Glutamat an Position 84 zu Alanin
Aktas et al., 2011a
pET28b_pmtA F89A (pBO876)
Austausch von Phenylalanin an Position 89 zu Alanin
Aktas, unveröffentlicht
pET28b_pmtA D106A (pBO871)
Austausch von Aspartat an Position 106 zu Alanin
Aktas et al., 2011a

Material und Methoden
- 24 -
pET28b_pmtA D106E (pBO1244)
Austausch von Aspartat an Position 106 zu Glutamat
Diese Arbeit
pET28b_pmtA I126A (pBO1248)
Austausch von Isoleucin an Position 126 zu Alanin
Diese Arbeit
pET28b_pmtA V129T (pBO1249)
Austausch von Valin an Position 129 zu Threonin
Diese Arbeit
pET28b_pmtA Q158E (pBO1250)
Austausch von Glutamin an Position 158 zu Glutamat
Diese Arbeit
pET28b_pmtA Q158R (pBO2700)
Austausch von Glutamin an Position 158 zu Arginin
Diese Arbeit
pET28b_pmtA S160A (pBO2701)
Austausch von Serin an Position 160 zu Alanin
Diese Arbeit
pET28b_pmtA G162A (pBO873)
Austausch von Glycin an Position 162 zu Alanin
Aktas et al., 2011a
pET28b_pmtA I186A (pBO2702)
Austausch von Isoleucin an Position 186 zu Alanin
Diese Arbeit
pET28b_pmtA P187T (pBO2703)
Austausch von Prolin an Position 187 zu Threonin
Diese Arbeit
pET28b_pmtA W192A (pBO867)
Austausch von Tryptophan an Position 192 zu Alanin
Aktas, unveröffentlicht
pET28b_pmtA Y194A (pBO874)
Austausch von Tyrosin an Position 194 zu Alanin
Aktas, unveröffentlicht
pET28b_pmtA T195A (pBO2730)
Austausch von Threonin an Position 195 zu Alanin
Diese Arbeit
pET28b_pmtA R196A (pBO2731)
Austausch von Arginin an Position 196 zu Alanin
Diese Arbeit
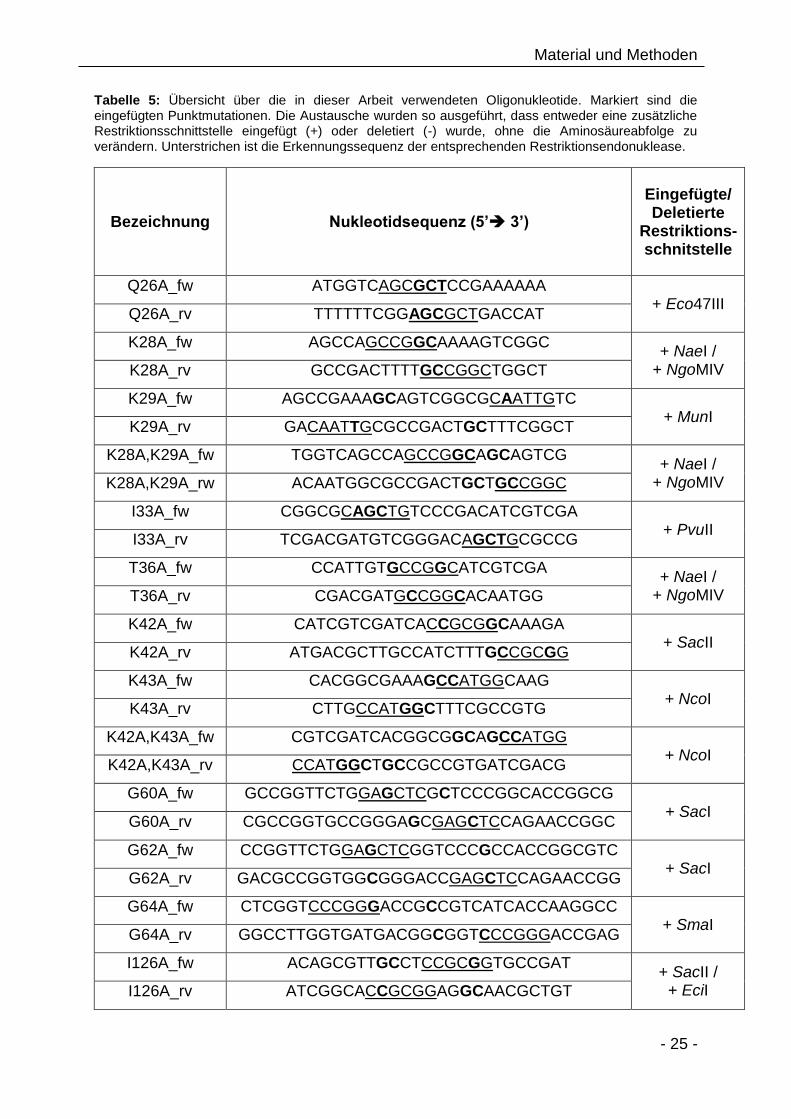
2.3 Oligonukleotide
Die synthetischen Oligonukleotide wurden von der Firma MWG Biotech AG in HPSF-
gereinigter und lyophilisierter Form bezogen. Nach Herstellerangaben wurden diese
im angegebenen Volumen A. dest. gelöst, um eine Konzentration von 100 pmol/µl zu
erhalten.
Material und Methoden
- 25 -
Tabelle 5: Übersicht über die in dieser Arbeit verwendeten Oligonukleotide. Markiert sind die eingefügten Punktmutationen. Die Austausche wurden so ausgeführt, dass entweder eine zusätzliche Restriktionsschnittstelle eingefügt (+) oder deletiert (-) wurde, ohne die Aminosäureabfolge zu verändern. Unterstrichen ist die Erkennungssequenz der entsprechenden Restriktionsendonuklease.
Bezeichnung Nukleotidsequenz (5’ 3’)
Eingefügte/ Deletierte
Restriktions-schnitstelle
Q26A_fw ATGGTCAGCGCTCCGAAAAAA + Eco47III
Q26A_rv TTTTTTCGGAGCGCTGACCAT
K28A_fw AGCCAGCCGGCAAAAGTCGGC + NaeI /
+ NgoMIV K28A_rv GCCGACTTTTGCCGGCTGGCT
K29A_fw AGCCGAAAGCAGTCGGCGCAATTGTC + MunI
K29A_rv GACAATTGCGCCGACTGCTTTCGGCT
K28A,K29A_fw TGGTCAGCCAGCCGGCAGCAGTCG + NaeI /
+ NgoMIV K28A,K29A_rw ACAATGGCGCCGACTGCTGCCGGC
I33A_fw CGGCGCAGCTGTCCCGACATCGTCGA + PvuII
I33A_rv TCGACGATGTCGGGACAGCTGCGCCG
T36A_fw CCATTGTGCCGGCATCGTCGA + NaeI /
+ NgoMIV T36A_rv CGACGATGCCGGCACAATGG
K42A_fw CATCGTCGATCACCGCGGCAAAGA + SacII
K42A_rv ATGACGCTTGCCATCTTTGCCGCGG
K43A_fw CACGGCGAAAGCCATGGCAAG + NcoI
K43A_rv CTTGCCATGGCTTTCGCCGTG
K42A,K43A_fw CGTCGATCACGGCGGCAGCCATGG + NcoI
K42A,K43A_rv CCATGGCTGCCGCCGTGATCGACG
G60A_fw GCCGGTTCTGGAGCTCGCTCCCGGCACCGGCG + SacI
G60A_rv CGCCGGTGCCGGGAGCGAGCTCCAGAACCGGC
G62A_fw CCGGTTCTGGAGCTCGGTCCCGCCACCGGCGTC
+ SacI G62A_rv GACGCCGGTGGCGGGACCGAGCTCCAGAACCGG
G64A_fw CTCGGTCCCGGGACCGCCGTCATCACCAAGGCC + SmaI
G64A_rv GGCCTTGGTGATGACGGCGGTCCCGGGACCGAG
I126A_fw ACAGCGTTGCCTCCGCGGTGCCGAT + SacII /
+ EciI I126A_rv ATCGGCACCGCGGAGGCAACGCTGT

Material und Methoden
- 26 -
V129T_fw CGCCACGCCGATGCTGAATTTTCCCATGG + NcoI
V129T_rv CCATGGGAAAATTCAGCATCGGCGTGGCG
Q158E_fw GACCCGTAGTAGAAATATCCTACGG - EcoRV
Q158E_rv CCGTAGGATATTTCTACTACGGGTC
Q158R_fw GACCCGTAGTCCGGATATCC + AccIII
Q158R_rv GGATATCCGGACTACGGGTC
S160A_fw AGATAGCCTACGGTCCGATTTCCCCAATCG - PvuI
/ - EcoRV S160A_rv CGATTGGGGAAATCGGACCGTAGGCTATCT
I186A_fw ATCGTGCGCAATGCTCCGCCGGCG - SspI / + BsrDI I186A_rv CGCCGGCGGAGCATTGCGCACGAT
P187T_fw TCGTGCGCAATATTACACCGGCGCA - EciI / - NaeI / - NgoMIV P187T_rv TGCGCCGGTGTAATATTGCGCACGA
T195A_fw CTATGCCCGGGCCCTCGAGTGAGT + SmaI
T195A_rv ACTCACTCGAGGGCCCGGGCATAG
R196A_fw CTATACGGCGGCCCTCGAGTGAGT - ApaI
R196A_rv ACTCACTCGAGGGCCGCCGTATAG
2.4 Anzucht von Bakterien
Die verwendeten Nährmedien wurden vor dem Gebrauch bei 121 °C und 200 kPa für
20 min autoklaviert. Komponenten, die hitzelabil sind, wurden sterilfiltriert (Millipore
Membranfilter; Porendurchmesser von 0.2 µm) und den jeweiligen Medien nach dem
Autoklavieren hinzugegeben. Um auf die plasmidkodierten Resistenzen zu
selektieren, wurde dem Medium Kanamycin (Km) (Endkonzentration 50 µg/ml)
zugegeben.
2.4.1 Nährmedien
LB-Flüssigmedium (Sambrook et al., 1989):
Trypton 10 g
NaCl 10 g
Hefeextrakt 5 g
A. dest ad 1000 ml

Material und Methoden
- 27 -
LB-Agar:
Trypton 10 g
NaCl 10 g
Hefeextrakt 5 g
Agar 15 g
A. dest ad 1000 ml
2.4.2 Anzucht von Escherichia coli
Die Anzucht von E. coli erfolgte bei 37 °C auf LB-Agar oder in LB-Flüssigmedium auf
einem Rundschüttler bei 200 UpM. Kulturen mit einem Volumen bis zu 5 ml wurden
in einem Reagenzglas auf einem Brutroller bebrütet. Größere Kulturen wurden in
einem Erlenmeyerkolben (Kulturvolumen max. 1/5 des Gefäßvolumens) angezogen.
Übernachtkulturen wurden in LB-Medium mit einer Einzelkolonie inokuliert und für
mindestens 16 Stunden inkubiert.
Für Gefrierkulturen wurden Zellen einer 20 ml Übernachtkultur zunächst bei 4 °C und
4000 UpM (Heraeus Megafuge 1.0R) geerntet. Anschließend wurden die Zellen in
autoklaviertem Glycerin und LB-Medium in einem Verhältnis 1:1 resuspendiert und
bei -80 °C gelagert.
2.4.3 Messung der optischen Dichte
Das Zellwachstum wurde durch photometrische Messung der optischen Dichte (O.D.)
bei 580 nm verfolgt. Eine O.D.580 von 1 einer E. coli Kultur entspricht dabei ca.
2 x 109 Zellen/ml. Als Referenz diente zellfreies Medium.

Material und Methoden
- 28 -
2.5 Molekularbiologische Methoden
2.5.1 Isolierung von Plasmid-DNA
Die Plasmid-DNA-Isolierung aus E. coli erfolgte nach einer modifizierten Methode
von Birnboim (Birnboim & Doly, 1979), beruhend auf dem Prinzip der alkalischen
Lyse.
Aus einer 5 ml Übernachtkultur wurden 4 ml Zellen per Zentrifugation bei 13000 UpM
in einer Eppendorfzentrifuge (EZ) geerntet (1 min, RT). Der Überstand wurde restlos
abgenommen und die Zellen in 300 µl Mix I mit 3 µl RNase (20 mg/ml) resuspendiert.
Nach Inkubation von einer Minute bei Raumtemperatur (RT) wurden 300 µl Mix II
hinzugegeben und das Reaktionsgefäß 6 – 8 mal invertiert, bevor es für weitere
5 min bei RT inkubiert wurde. Anschließend wurden 300 µl Mix III hinzugegeben und
die Probe weitere 6 – 8 mal invertiert. Die entstandenen Protein-SDS-Präzipitate, die
gefällte chromosomale DNA, sowie Zelltrümmer wurden dann durch Zentrifugation
(15 min, 13000 UpM, EZ, RT) sedimentiert. Der Überstand wurde abgenommen und
ein zweites Mal zentrifugiert, um letzte Verunreinigungen der Plasmid-DNA zu
sedimentieren. Durch Zugabe von 630 µl Isopropanol und Inkubation von mindestens
30 min bei Raumtemperatur wurde die Plasmid-DNA gefällt. Nach Zentrifugation (10
min, 13000 UpM, EZ, RT) wurde der Überstand verworfen und das Pellet mit 400 –
500 µl kaltem 70 %igem Ethanol gewaschen (5 min, 13000 UpM, EZ, RT). Nach
vollständigem Abnehmen des Überstandes wurde das Pellet bei ca. 50 °C
getrocknet, bis sämtliches Ethanol verdunstet war.
Je nach Kopienzahl des Plasmides wurde das DNA-Pellet in 30 – 50 µl TE-Puffer
oder H2O bei ca. 50 °C für 10 min gelöst.
Mix I: pH 8.0 Mix II:
Tris-HCl 50 mM NaOH 200 mM
EDTA 10 mM SDS 1 % (w/v)
Mix III: pH 4.8 TE-Puffer: pH 8.0
Kaliumacetat 3 M Tris-HCl 10 mM
Formiat 1,8 M EDTA 1 mM

Material und Methoden
- 29 -
2.5.2 Hydrolytische Spaltung von DNA durch Restriktions-
endonukleasen
Zur gezielten hydrolytischen Spaltung von DNA wurden Restriktionsendonukleasen
des Typ II (Bindungsstelle identisch mit Schnittstelle) eingesetzt
(Sambrook et al., 1989). Um eine vollständige Hydrolyse zu gewährleisten, wurden
pro μg DNA 1 U der entsprechenden Endonuklease im Restriktionsansatz eingesetzt
und für 1 bis 2 Stunden bei der vom Hersteller angegebenen Temperatur inkubiert.
Die vom Hersteller mitgelieferten Restriktionspuffer sorgten für optimale
Reaktionsbedingungen. Doppelrestriktionen konnten in einem Schritt durchgeführt
werden, wenn die entsprechenden Enzyme genügend hohe Aktivitäten im
verwendeten Restriktionspuffer aufwiesen. Die Reaktion wurde durch Inkubation der
Ansätze für 20 min bei der vom den Hersteller angegebenen Temperatur gestoppt
und die hydrolytische Spaltung der DNA mittels Agarose-Gelelektrophorese
überprüft.
2.5.3 Agarose-Gelelektrophorese
Die Agarose-Gelelektrophorese diente der Analyse von DNA-Restriktionen, der
Mengen- und Größenabschätzung linearisierter DNA, sowie der präparativen
Isolierung von DNA-Fragmenten. Bei diesem Trennverfahren wird die
unterschiedliche Wanderungsgeschwindigkeit verschieden großer Moleküle im
elektrischen Feld ausgenutzt. Die Proben wurden vor dem Auftragen mit
1/5 Volumen 5-fach konzentriertem DNA-Probenpuffer versetzt. Zur Auftrennung von
DNA-Fragmentgrößen bis 1 kb wurden Gele mit einer 2-%igen Agarosekonzentration
in 1-fachem TAE-Puffer verwendet, bei größeren Fragmenten ein 1-%iges
Agarosegel. Die Auftrennung erfolgte in einer horizontalen Gelkammer mit einer
maximalen Stromstärke von 500 mA und konstanten Spannung von 100 – 120 V.
Um die DNA-Fragmente sichtbar zu machen, wurde das Agarosegel nach der
gelelektrophoretischen Auftrennung in einem 0.05 % (w/v) Ethidiumbromidbad für
mindestens 15 min inkubiert und anschließend bei UV-Bestrahlung (λ=254 nm) unter
einer Videodokumentationsanlage fotografiert. Als Molekulargewichtsstandard diente
der “1-kb-ladder“ der Firma Invitrogen.

Material und Methoden
- 30 -
5-fach DNA-Probenpuffer: pH 8.0
Bromphenolblau 0.05 % (w/v)
EDTA 250 mM
Glycerol 43 % (v/v)
TAE-Puffer: pH 7.8
Tris-HCl 40 mM
Natriumacetat 10 mM
EDTA 1 mM
2.5.4 Amplifikation von DNA mittels Polymerasekettenreaktion und
ortsspezifische Mutagenese
Die Polymerase-Kettenreaktion (PCR) dient generell der Amplifikation von DNA. In
dieser Arbeit wurde die PCR-Reaktion für die gerichtete Punktmutagenese des pmtA-
Gens aus A. tumefaciens verwendet. Bei der PCR-Reaktion vervollständigen DNA-
Polymerasen einzelsträngige Matrizen-DNA zu einem Doppelstrang, wenn ein kurzer
doppelsträngiger Bereich als Startpunkt für die DNA-Polymerase zur Verfügung
steht. Dieser doppelsträngige Bereich setzt sich zum einen aus der Matrizen-DNA
und zum anderen aus einem Primer zusammen. Primer sind chemisch synthetisierte
Oligonukleotide, die komplementär zu den Randbereichen der zu amplifizierenden
Sequenz sind und unter geeigneten Bedingungen spezifisch an den Matrizenstrang
binden.
Sequenzspezifische Mutationen des agrobakteriellen pmtA-Gens wurden nach der
QuikChange® Methode eingeführt. Dafür wurden die Primer so konstruiert, dass die
Mutation durch einen Basenaustausch mit dem Einfügen oder Deletieren einer
Restriktionsschnittstelle einhergeht. Als Template-DNA diente das Plasmid pBO832,
welches das wildtyp pmtA-Gen besitzt. Ein Standard-PCR Ansatz setzte sich wie
folgt zusammen:

Material und Methoden
- 31 -
Template DNA 15 ng
Primer 1 75 pmol
Primer 2 75 pmol
dNTPs 0.2 mM
10 x Pfu-Polymerase- Puffer 5 µl
Pfu – Polymerase 2 U
A. dest. ad 50 µl
Die PCR-Reaktion wurde in einem „Eppendorf Mastercycler personal“ mit
nachstehendem Programm durchgeführt:
Denaturierung der DNA 95 °C 2 min
Denaturierung der DNA 95 °C 0.5 min
Annealing 55 °C 1 min
Elongation 68 °C 2 min / kb Produktgröße
Die Elongationszeit wurde dabei so gewählt, dass die Template-DNA vollständig
repliziert werden konnte. Um die Transformationseffizienz für mutationstragende
Plasmide zu erhöhen, wurden 10 µl des PCR-Ansatzes entnommen und mit DpnI
verdaut. Dies ist eine Restriktionsendonuklease, die spezifisch nur methylierte DNA
an der Erkennungssequenz schneidet. Ein Standard-Ansatz setzte sich wie folgt
zusammen:
PCR-Ansatz 10 µl
DpnI (10U/µl) 1 µl
10 x Reaktionspuffer 1 µl
Der Ansatz wurde für eine Stunde bei 37 °C inkubiert. Anschließend wurden weitere
1 µl DpnI zugegeben und eine weitere Stunde restringiert.
Schließlich wurde der gesamte Ansatz für die Transformation in E. coli-Zellen
verwendet (Abschnitt 2.5.6).
x 16

Material und Methoden
- 32 -
2.5.5 Herstellen transformationskompetenter Zellen
Für die Herstellung kompetenter E. coli Zellen wurden 70 ml LB-Medium mit je 0.7 ml
einer 500 mM Lösung MgCl2, sowie MgSO4 versetzt und anschließend mit 0.7 ml
einer Übernachtkultur angeimpft. Die Bakterienzellen wurden in einem
Erlenmeyerkolben bei 37 °C bis zu einer optischen Dichte von ca. 0.5 bei 580 nm
inkubiert. Nun wurden je 25 ml der Kultur in einem sterilen Zentrifugenröhrchen bei
4 °C und 4000 UpM (Heraeus Megafuge 1.0R) für 10 min zentrifugiert. Der
Überstand wurde verworfen und die sedimentierten Zellen in je 12.5 ml kaltem TMF-
Puffer resuspendiert und eine Stunde auf Eis inkubiert. Anschließend wurden die
Zellen 10 min bei 4 °C und 4000 UpM (Heraeus Megafuge 1.0R) zentrifugiert. Die
Zellpellets wurden in je 2.5 ml TMF-Puffer auf Eis resuspendiert und jeder Ansatz mit
750 µl autoklaviertem Glycerol gemischt. Die kompetenten Zellen wurden dann in
200 µl Portionen aliquotiert und bei -80 °C gelagert.
TMF-Puffer:
CaCl2 100 mM
MnCl 40 mM
RbCl 50 mM
2.5.6 Transformation
Für die Transformation eines Plasmides mit kompetenten E. coli Zellen, wurden
diese mit Plasmid-DNA oder 20 µl Ligationsansatz bei 4 °C für mindestens 30 min
inkubiert. Anschließend wurden die Zellen einem 2 minütigen Hitzeschock bei 42 °C
unterzogen. Nach weiteren 5 min Inkubation bei 4 °C wurde den Zellen 700 µl LB-
Flüssigmedium hinzugefügt und für mindestens zwei Stunden bei 37 °C auf einem
Roller inkubiert. Die transformierten Zellen wurden dann auf LB-Agar-Platten mit
entsprechendem Antibiotikum ausplattiert.

Material und Methoden
- 33 -
2.5.7 DNA-Konzentrationsbestimmung
Die DNA-Konzentration wurde mit einem Nano-Drop Spectrophotometer von der
Firma Peqlab bei 260 nm bestimmt, wobei eine Absorptionseinheit einer
Konzentration von 50 µg doppelsträngiger DNA/ml entspricht (Sambrook &
Russell, 2001). Die Reinheit der Nukleinsäure wurde durch Berechnung des
Quotienten 260/280 nm bestimmt. Für DNA sollte dieser bei ca. 1.8 liegen.

Material und Methoden
- 34 -
2.6 Herstellung von Liposomen und Micellen
2.6.1 Herstellung von Liposomen
Zur Herstellung von Liposomen wurde eine Lipidmenge eingesetzt, um eine finale
Konzentration von 1 mM Lipid im Ansatz zu erhalten. Im Falle von A. tumefaciens
oder E. coli Gesamtlipidextrakt wurden jeweils 4 ml einer Übernachtkultur geerntet
und die Lipide extrahiert. Die Gesamtlipidmenge der extrahierten Lipide wurde
anhand einer Dünnschichtchromatographie und im Vergleich zu einem definierten
Lipidstandard (3 µl 10 mg/ml PE) geschätzt. Zuerst wurden die in Chloroform
gelösten Lipide in einem Reagenzglas mittels Argon- oder Stickstoffbegasung
getrocknet, bis das Lösungsmittel vollständig verdampfte. Anschließend wurde 1 ml
Puffer (50 mM KH2PO4 oder 100 mM Tris-HCl, jeweils pH 8) hinzugegeben und die
Lösung für mindestens eine Stunde bei RT inkubiert, um eine Rehydrierung zu
ermöglichen. Für die Ausbildung von Liposomen wurde die Lösung mittels Ultraschall
für 30 min bei 30 °C beschallt. Um die Liposomen auf eine einheitliche Maximalgröße
einzustellen, wurde ein Extruder und eine Membran mit 100 nm Porengröße
verwendet. Die Liposomen passierten dabei den Extruder 10-mal. Die Liposomen
wurden maximal für 2 Wochen bei 4 °C gelagert.
2.6.2 Herstellung von Mizellen
Zur Herstellung von Mizellen wurde eine Lipidmenge eingesetzt, um eine finale
Konzentration von 400 µM Lipid zu erhalten. Zu den in Chloroform:Methanol gelösten
Lipiden wurde Triton X-100 (Endkonzentration 0.02% (v/v)) gegeben. Das Gemisch
wurden in einer SpeedVac (UNIV PO 100 H) getrocknet, bis das Lösungsmittel
vollständig verdampfte. Anschließend wurden die Lipide in 100 µl des jeweils
verwendeten Puffers gelöst.

Material und Methoden
- 35 -
2.7 Proteinbiochemische Methoden
2.7.1 Überexpression rekombinanter Proteine in E. coli BL21(DE3)
Für die Überexpression von PmtA oder PmtA-Derivaten wurde das jeweilige
Expressionsplasmid in den Expressionsstamm E. coli BL21(DE3) transformiert. 20 ml
einer Übernachtkultur der resultierenden Stämme wurden in 1 l selektivem LB-
Flüssigmedium verdünnt und bei 37 °C bis zu einer O.D.580 von 0.5 - 0.7 angezogen.
Die Expression des pmtA-Genes im verwendeten pET-System wird durch einen lac-
Promotor kontrolliert, der mit IPTG (Isopropyl- -D-thiogalactopyranosid) induzierbar
ist. Die Überexpression wurde durch Zugabe von 0.4 mM IPTG induziert und die
Kultur für 20 Stunden bei 18 °C inkubiert. Die Zellen wurden durch Zentrifugation (10
min, 6000 UpM, SLA-3000, 4 °C) geerntet und mit LEW-Puffer gewaschen.
Anschließend wurde das Pellet bei -20 °C gelagert oder direkt weiter verarbeitet.
LEW-Puffer pH 8.0
NaH2PO4 50 mM
NaCl 300 mM
2.7.2 Zellaufschluss und Gewinnung von Rohextrakt
Das Zellpellet (2.7.1) wurde in 30 ml LEW-Puffer mit 200 µg/ml Lysozym sowie
5 µg/ml DNAse resuspendiert und mindestens 30 min auf Eis inkubiert. Anschließend
erfolgte der Zellaufschluss mittels Constant Cell Disruption System (Constant
Systems Ltd), wobei die Probe zwei Aufschlussdurchgänge durchlief. Nach dem
Zellaufschluss wurde dem Ansatz Triton X-100 (Endkonzentration 0.05 % (v/v))
beigefügt und der Ansatz 30 min auf Eis inkubiert. Anschließend wurden die
Zelltrümmer bei 20000 UpM (SS-34) bei 4 °C für 30 min sedimentiert.

Material und Methoden
- 36 -
2.7.3 Reinigung rekombinanter Proteine mittels Ni-IDA-Affinitäts-
chromatographie
Für die Überexpression und Reinigung mittels Metall-Ionen-Affinitätschromatographie
(IMAC) von Proteinen wurde das „Purification of Polyhistidine-Tagged Proteins“ -
System von Macherey & Nagel verwendet. Durch Klonierung geeigneter DNA-
Fragmente in den Expressionsvektor pET28b(+) werden „in-frame“-Fusionen
zwischen dem zu exprimierenden Zielgen und dem Vektor-kodierten His-Tag
erzeugt. Der His-Tag besteht aus sechs aufeinander folgenden Histidinen, die mit
einem immobilisierten Ni2+ - Ion interagieren und dadurch das Protein an die
Säulenmatrix binden.
Für die His-Tag-Affinitätschromatographie wurde der Rohextrakt auf eine mit LEW-
Puffer equilibrierte Protino® Ni-IDA Säule (2 ml Säulenvolumen) der Firma Macherey
& Nagel gegeben. Nach Beladen der Säule mit dem Proteinrohextrakt, wurde die
Säule mit 16 Säulenvolumen (SV) LEW-Puffer mit 10 mM Imidazol gewaschen.
Anschließend folgten weitere Waschschritte mit 16 SV LEW-Puffer mit 30 mM
Imidazol und 16 SV LEW-Puffer mit 50 mM Imidazol.
Die Elution der Proteine erfolgte mit Elutionspuffer (LEW mit 250 mM Imidazol).
Imidazol konkuriert mit dem His-Tag des überexprimierten Proteins um die Bindung
zum Ni2+ - Ion. Deshalb kann Imidazol im Überschuss eine gezielte Freisetzung des
Zielproteins bewirken und in niedrigeren Konzentrationen unspezifisch gebundene
Proteine verdrängen.
2.7.4 Denaturierende SDS-Polyacrylamid-Gelelektrophorese
Mit Hilfe der SDS-PAGE (Sodiumdodecylsulfat-Polyacrylamidgel-Elektrophorese)
können Proteine entsprechend ihrem Molekulargewicht aufgetrennt werden. Das
anionische Detergenz Natriumdodecylsulfat (SDS) bindet an die durch
Hitzebehandlung denaturierten und in ihre Untereinheiten zerfallenen Proteine und
überdeckt ihre Eigenladung so effektiv, dass Mizellen mit konstanter negativer
Ladung pro Masseeinheit entstehen. Der Prozess der Denaturierung wird durch die
zusätzliche Zugabe von 2-Mercaptoethanol noch verstärkt, da dieses Reagenz
Cystein-Schwefelbrücken reduziert.

Material und Methoden
- 37 -
Durch diesen Effekt ist die Wanderungsrichtung und -geschwindigkeit im Gel von der
Eigenladung des Proteins unabhängig, so dass eine elektrophoretische Auftrennung
nach dem Molekulargewicht erfolgen kann.
Zur elektrophoretischen Auftrennung der Proteine wurden 12.5-%ige SDS-
Polyacrylamidgele nach Laemmli (Laemmli, 1970) eingesetzt. Die Elektrophorese
erfolgte in einer Mini-Protean II Zelle (Biorad) bei einer konstanten Spannung von
120 V in Elektrophorese-Puffer. Die Proteinproben (15 μl) wurden vor dem Auftragen
auf das Gel mit 5 µl SDS-Probenpuffer versetzt und 10 min aufgekocht.
Trenngel (Acrylamid-Konz. 12.5 %)
A. dest. 1.800 ml
4 x Trenngelpuffer 1.250 ml
40 % Acrylamid/Bisacrylamid 1.565 ml
87 % Glycerin 0.050 ml
10 % (w/v) SDS 0.050 ml
10 % (w/v) APS 0.038 ml
TEMED 1.7 μl
Sammelgel (Acrylamid-Konz. 6%)
A. dest. 1.570 ml
4 x Sammelgelpuffer 0.625 ml
40 % Acrylamid/Bisacrylamid 0.350 ml
10 % (w/v) SDS 0.025 ml
10 % (w/v) APS 0.025 ml
TEMED 3.8 μl
4-fach Sammelgelpuffer pH 6.8
Tris/HCl 0.5 M
4-fach Trenngelpuffer pH 8.8
Tris/HCl 1.5 M

Material und Methoden
- 38 -
10-fach Elektrophoresepuffer pH 8.3
Tris/HCl 0.25 M
Glycin 1.92 M
SDS 1 % (w/v)
5-fach SDS-Probenpuffer pH 6.8
4-fach Sammelgelpuffer 1.25 ml
Glycerin 10 % (v/v)
β-Mercaptoethanol 5 % (v/v)
SDS 3 % (w/v)
Bromphenolblau 1 % (w/v)
A.dest. ad 10 ml
Nach elektrophoretischer Auftrennung wurden die Proteine im Polyacrylamidgel mit
Coomassie angefärbt. Das in der Färbelösung enthaltene Methanol dient dazu, den
Coomassiefarbstoff zu lösen. Zusätzlich ist in der Färbelösung Essigsäure enthalten,
welche die Proteine im Polyacrylamidgel fixiert. Der Farbstoff Coomassie Brilliant
Blue R-250 besitzt die Eigenschaft, sich an basischen und aromatischen
Seitenketten von Aminosäuren anzulagern und färbt dadurch alle Proteine
unspezifisch an.
Die Färbung erfolgte 10 min bei Raumtemperatur. Anschließend wurde der
Gelhintergrund durch Inkubation in Entfärberlösung entfernt. Als
Molekulargewichtsstandard wurde “Benchmark Ladder“ der Firma Invitrogen
verwendet.
Coomassie-Färbelösung Entfärber
Methanol 50 % (v/v) Methanol 20 % (v/v)
Essigsäure 10 % (v/v) Essigsäure 7 % (v/v)
Coomassie Brilliant blue R 250 0.1 % (w/v)

Material und Methoden
- 39 -
2.7.5 Proteinkonzentrationsbestimmung
Die Konzentration von gereinigten Proteinen wurde spektrophotometrisch durch
Absorptionsmessung bei 280 nm bestimmt. Bei dieser Wellenlänge weisen die
aromatischen Aminosäurereste (vor allem Tryptophan) ein Absorptionsmaximum auf
(Nelson & Cox, 2009). Mit Hilfe des Lambert-Beerschen Gesetzes (E280 = c . d . )
wurde die Proteinkonzentration errechnet. Der molare theoretische Extinktions-
koeffizient des jeweiligen Proteins wurde mit dem Programm ProtParam bestimmt.
2.7.6 Umpufferung und aufkonzentrieren von Proteinen
Proteinlösungen wurden mit Hilfe von Konzentratoren aufkonzentriert oder
umgepuffert. Das aufzukonzentrierende Protein wurde in ein Amicon Ultra 10-
Röhrchen (MWCO 10000) überführt und bei 2500 UpM (Heraeus Megafuge 1.0R) bei
4 °C zentrifugiert, bis die gewünschte Proteinkonzentration erreicht wurde.
2.7.7 Größenausschlusschromatographie
Die Größenausschlusschromatographie (oder auch Gelfiltration genannt) ist ein
säulenbasiertes Trennverfahren, das Moleküle aufgrund ihrer Größe und Form
voneinander separiert. Die Säulenmatrix besteht aus porösen Kugeln, deren Poren
eine definierte Größe besitzen. Diese limitieren die Permeation großer Moleküle in
die Kugeln, so dass sie einen kürzeren Weg durch die Matrix haben, als Moleküle,
die kleiner als die Poren sind. Daher eluieren große Moleküle früher als kleinere. Bei
einem Gemisch aus Molekülen unterschiedlicher Größe eluieren Moleküle, die die
Porengröße der Matrix überschreiten, im Ausschlussvolumen. Diese Moleküle
passieren die Säule mit der gleichen Geschwindigkeit, wie der Fluss des Puffers. Für
die Gelfiltration wurde eine Superdex 75 HR 10/30-Säule (Pharmacia) verwendet.
Die Säule hat ein Gesamtvolumen von 24 ml und ein Ausschlussvolumen von 8 ml.
Für die Gelfiltration von rekombinanten Proteinen, die mittels IMAC isoliert wurden,
wurden 2 ml Elutionsfraktionen aufgetragen und in 50 mM Kaliumdihydrogen-
phosphatpuffer (pH 8) oder 100 mM Tris-Puffer (pH 8) mit einer Fließgeschwindigkeit
von 0.4 ml/min bei 4 °C aufgetrennt.

Material und Methoden
- 40 -
2.7.8 In vitro-Aktivitätsassay
Die in vitro-Aktivitätsassays wurden nach Aktas (Aktas & Narberhaus, 2009)
durchgeführt. Dazu wurden 10 µM rekombinantes Protein in einem Volumen von
100 µl (100 mM Tris-HCl pH 8.0) mit 400 µM Liposomen und 1.84 mM SAM bei
30 °C für 1 Stunde inkubiert. Im Anschluss wurde eine Lipidextraktion (2.8.1) und
eine dünnschichtchromatographische Analyse der Reaktionsprodukte durchgeführt.
2.7.9 S-Adenosylmethionin-Bindestudien
Um die SAM-Bindefähigkeit von PmtA und PmtA-Derivaten zu untersuchen, wurde
ein Filterbindeassay mit radioaktiv markiertem SAM (S-[Methyl-14C] Adenosyl-L-
Methionin; Hartmann Analytics) verwendet (Aktas & Narberhaus, 2009). Das Prinzip
dieser Methode beruht auf einer Bindung eines Enzym-Radioliganden-Komplexes an
eine Filtermembran. Der ungebundene Radioligand wird weggewaschen und der
gebundene Anteil mittels Flüssig-Szintillationszähler quantifiziert. Dabei steht die
Bindung des Radioliganden in einem proportionalen Verhältnis zur detektierten
Radioaktivität. Ein Standard-Ansatz setzte sich wie folgt zusammen:
Protein 50 µM
Radioligand 2.5 µCi
Phospholipid 400 µM
Bindepuffer ad. 50 µl
Die spezifische Aktivität des Radioliganden betrug 48.8 mCi/mmol und die
radioaktive Konzentration 0.05 mCi/2.5 ml. Phospholipide wurden als Mizellen dem
Ansatz beigefügt. Der Ansatz wurde für 10 min bei 30 °C inkubiert und anschließend
auf eine zuvor mit Puffer equilibrierte Nitrozellulosemembran der Firma Millipore
(HAWP02500; 0.45 µm) gebunden. Nach viermaligem Waschen mit 0.3 ml
Bindepuffer (Tris-HCl 100 mM, pH 8.0) mittels einer Vakuumfiltrationsanlage, wurde
die Nitrozellulosemembran in 4 ml Szintillationsflüssigkeit Hydroluma (J.T. Baker)
aufgelöst (ca. 3 Stunden). Anschließend wurde die Probe bei einer Zählzeit von
10 min im Szintillationszähler LS-6000 TA (Beckman) gemessen.

Material und Methoden
- 41 -
2.7.10 Circular Dichroismus-Spektroskopie (CD-Spektroskopie)
Die Circular Dichroismus-Spektroskopie (CD-Spektroskopie) ist eine Methode zur
Aufklärung der Sekundärstruktur von optisch aktiven Molekülen. Optisch aktive
Moleküle wechselwirken mit polarisiertem Licht und absorbieren rechts- bzw.
linksgerichtetes zirkular polarisiertes Licht unterschiedlich stark. Die unterschiedlich
starke Absorption von links bzw. rechts gerichtetem Licht führt zu einer Drehung der
Polarisationsebene. Diese Drehung bzw. Differenz im unterschiedlichen
Absorptionsverhalten von rechts oder links gerichtetem Licht bezeichnet man als
Elliptizität. Die Elliptizität wird in Abhängigkeit zur Wellenlänge gemessen und als
CD-Spektrum aufgezeichnet (Nelson & Cox, 2009).
Proteine sind aufgrund des chiralen Cα-Atoms ihrer Aminosäuren optisch aktive
Moleküle. Daher kann die CD-Spektroskopie bei Proteinen und Peptiden zur Analyse
der Sekundärstruktur verwendet werden. Die einzelnen Sekundärstrukturelemente,
wie α-Helix, β-Faltblatt und Loops, haben ein unterschiedliches Absorptionsverhalten
gegenüber linear polarisiertem Licht. Ein CD-Spektrum eines Peptides/Proteins stellt
daher die Gesamtheit aller Sekundärstrukturelemente, deren Signale sich
überlagern, dar (Kelly & Price, 2000). Weiterhin können mittels CD-Spektroskopie die
Auswirkungen von beispielsweise pH, Temperatur oder Ligandeninteraktion auf die
Sekundärstruktur von Proteinen untersucht werden (Kelly & Price, 2000).
Für die CD-spektroskopischen Analysen von PmtA wurden 7.5 µM Protein in einem
50 mM Kaliumdihydrogenphosphatpuffer (pH 8.0) bei 20 °C mit einem Jasco J-715
CD-Spektrometer vermessen. Es wurde ein CD-Spektrum zwischen 190 – 260 nm in
0.5 nm Abständen aufgezeichnet, wobei das finale CD-Spektrum eine Akkumulation
aus 10 Einzelspektren darstellt. In Tabelle 6 sind die verwendeten Parameter
aufgelistet.
Für die Thermostabilitätsmessungen wurde das CD-Signal bei 222 nm in
Abhängigkeit zur Temperatur aufgezeichnet. Die Temperatur wurde in einem Bereich
von 5 - 80 °C in 2 °C-Schritten erhöht.
Für den Einfluss von Liposomen auf die PmtA-Sekundärstruktur wurden steigende
Konzentrationen an Liposomen in einem Bereich zwischen 0.5 und 150 µM hinzu
titriert.

Material und Methoden
- 42 -
Tabelle 6: Paramter für CD-spektroskopische Analysen.
Parameter Einstellungen
Empfindlichkeit 100 mdeg
Wellenlängenbereich 190 – 260 nm
Messpunktabstand 0.5 nm
Abtastgeschwindigkeit 50 nm/min
Antwortverhalten 1 s
Bandbreite 1 nm
Akkumulationen 10
2.7.11 Tryptophanfluoreszenzmessung
Ein photochemisch angeregtes Molekül kann entweder durch Relaxation
(Schwingungsenergie/Wärmeenergie) oder durch die Emmision eines Photons
wieder in seinen energetischen Grundzustand gelangen (Nelson & Cox, 2009).
Letzteres wird als Fluoreszenz bezeichnet und kann in einem Fluorimeter gemessen
und quantifiziert werden.
In dieser Arbeit wurde die intrinsische Fluoreszenz der Aminosäure Tryptophan
genutzt. Tryptophan wird spezifisch durch eine Extinktion von 295 nm angeregt,
sodass eine Fluoreszenzemission zwischen 300 – 350 nm beobachtet werden kann.
Die Tryptophanfluoreszenz bietet die Möglichkeit auf die Polarität der Umgebung in
Abhängigkeit des Emissionsmaximums zu schließen, da das Fluoreszenzmaximum
von Tryptophan in polarer bzw. unpolarer Umgebung unterschiedlich ist (Nelson &
Cox, 2009; Kraft et al., 2009). Die Tryptophanfluoreszenz ist weiterhin ein häufig
verwendetes Verfahren, um die Interaktion von Proteinen und Liganden zu
quantifizieren. Dazu zählt auch die Interaktion von Proteinen mit Lipiden
(Kraft et al., 2009). Um Bindungskonstanten zu bestimmen, kann sowohl eine
veränderte Fluoreszenzintensität, als auch die Verschiebung des
Fluoreszenzmaximums genutzt werden (Kraft et al., 2009).
Um den Einfluss von Lipiden auf die PmtA-Tryptophanfluoreszenz zu messen,
wurden 10 µM Protein in 100 mM Tris-HCl Puffer in einem Thermo Amicon Bowman
II Luminescence Spectrometer vermessen. Die Tryptophanfluoreszenz wurde mit
295 nm angeregt und das Emissionspektrum zwischen 300 – 400 nm aufgezeichnet.

Material und Methoden
- 43 -
Die Spaltbreite wurde auf 4 nm eingestellt. Die Abtastgeschwindigkeit betrug 5 nm/s
wobei Messpunkte im Abstand von 5 nm genommen wurden. Zum Protein wurden
steigende Konzentrationen von Liposomen in einem Bereich zwischen 0 – 100 µM
hinzutitriert, und anschließend die Änderung der relativen Fluoreszenzintensität
gegen die Liposomenkonzentration aufgetragen. Zur Korrektur des
Verdünnungseffektes wurden ein Ansatz mit Protein und Puffer als Titrand
verwendet. Dieser Ansatz und ein Ansatz ohne Protein dienten als Negativkontrollen,
wobei anhand von letzterem eine Eigenfluoreszenz der verwendeten Liposomen
ausgeschlossen werden konnte.
2.7.12 Fluoreszenzmessung mit Hilfe von Fluorescein-
Phosphatidylethanolamin
Das Lipid Fluorescein-Phosphatidylethanolamin (FPE) besteht aus einer
Fluoresceingruppe und Phosphatidylethanolamin (PE). Das Fluorescein ist dabei
über die Ethanolaminkopfgruppe an das PE gekoppelt. Dies ermöglicht die
Integration von FPE in eine Membran und somit die Quantifizierung des
elektrostatischen Potentials einer Membran (Wall et al., 1995). Wenn eine Membran,
die FPE beinhaltet, mit einem geladenen Molekül interagiert, führt dies zu einer
Änderung des Membranpotentials, was wiederum den Protonierungszustand des
Xanthenringes beeinflusst. Dies bewirkt eine Änderung der Fluoreszenzintensität und
kann zur Quantifizierung einer Membraninteraktion genutzt werden. Da diese
Methode jedoch auf einer Änderung des Membranpotentials basiert, können nur
geladene Modellmembranen verwendet werden. Membranen aus zwitterionischen
Lipiden können nicht verwendet werden, da diese kein Membranpotential aufweisen.
Proteine besitzen auf ihrer Oberfläche größtenteils geladene Aminosäuren und sind
daher auch geladene Moleküle. Eine Protein-Membran Interaktion führt daher auch
zu einer Änderung des Membranpotentials und kann mittels FPE gemessen werden.
Für eine Messung wurden 10 µM FPE-markierter Liposomen (0.5 mol-% FPE)
verwendet, zu denen PmtA (0 - 45 µM) hinzu titriert wurde. Die Fluoreszenz von FPE
wurde mit 490 nm angeregt und das Emissionsspektrum zwischen 500 – 600 nm
aufgezeichnet. Das Spektrum wurde mit einer Abtastgeschwindigkeit von 5 nm/s und
im Abstand von 2 nm aufgezeichnet. Die Spaltbreite wurde auf 4 nm eingestellt.

Material und Methoden
- 44 -
2.7.13 Protein-Lipid-Overlay-Assay
Der Protein-Lipid-Overlay-Assay wurde nach Dowler (Dowler et al., 2002)
durchgeführt. Dafür wurden zuvor getrocknete Lipide in einem
Methanol:Chloroform:Wasser Gemisch (2 : 1 : 0.8) aufgenommen und 2 oder
14 nmol auf eine C-Hybond Membran (Amersham) pipettiert. Als Kontrolle, um
Membranschäden auszuschließen, wurde auch das Lösungsmittel in äquivalentem
Volumen aufgetragen. Anschließend wurden die Membranen mind. 1 Stunde bei RT
getrocknet. Die Blockierung der Membran erfolgte mit TBST mit 2 mg/ml BSA für
1 Stunde. Nach erfolgreicher Blockierung, wurde in frische Blockierungslösung
25 µM PmtA hinzugegeben. Um eine Protein-Lipid-Interaktion zu ermöglichen
wurden die Ansätze für 2 Stunden auf einer Kippwaage inkubiert. Ungebundenes
Protein wurde mit zweimaligem Waschen mit TBST und dreimaligem Waschen in
TBS für jeweils 8 min weggewaschen. Zur Detektion der His-getaggeten PmtA wurde
ein Penta-His-Hrp-Konjugat nach Herstellerangaben der Firma Qiagen verwendet.
Der Nachweis erfolgte unter Verwendung des ECL-Systems (GE-Healthcare) nach
Angaben des Herstellers.
TBS-Puffer pH 8.0
Tris-HCl 150 mM
NaCl 50 mM
TBST-Puffer pH 8.0
Tris-HCl 150 mM
NaCl 50 mM
Tween-20 0.5 % (v/v)

Material und Methoden
- 45 -
2.7.14 Limitierte Proteolyse
Die limitierte Proteolyse ist eine Methode zur qualitativen Analyse einer Protein-
Liganden-Interaktion. Das Prinzip beruht auf dem unterschiedlichen Abbau eines
Proteins durch eine Protease in Ab- und Anwesenheit eines Liganden. Da ein Abbau
von Proteinen durch Proteasen sehr schnell erfolgt, wurde dieses Experiment unter
limitierenden Bedingungen für die Protease durchgeführt. Limitierende Bedingungen
sind beispielsweise die Temperatur oder die Dauer der Inkubation mit der Protease
(Fontana et al., 2004).
In dieser Arbeit wurde die limitierte Proteolyse zur Untersuchung der
Lipidbindeeigenschaften von PmtA verwendet. Dafür wurde der Abbau von PmtA in
An- und Abwesenheit von unterschiedlichen Lipiden untersucht. PmtA wurde mit
verschiedenen Liposomen für eine Stunde bei 4 °C vorinkubiert, um eine Anlagerung
von PmtA an die Liposomen zu gewährleisten. Die Konzentration an eingesetztem
Protein betrug 30 μM, die Konzentration der Phospholipide 150 μM. Als
Vergleichsansatz wurde ein Ansatz ohne Liposomen verwendet. Die Degradation
wurde durch Zugabe der Protease Chymotrypsin oder Trypsin initiiert. Die
Inkubationszeit mit der Protease erfolgte für 0, 2, 5, 7, 10, 15, und 30 min. Zur
Beendigung des Proteinverdaus wurden jeweils 20 μl aus den entsprechenden
Ansätzen entnommen, 5 µl SDS-Probenpuffer hinzugegeben und aufgekocht. Im
Anschluß wurden die Proben über eine SDS-PAGE aufgetrennt und die Proteine
mittels Coomassiefärbung visualisiert.
2.7.15 Tryptischer Verdau im Gel und Massenspektrometrie
Aus den Banden des SDS-Gels wurden mit einem Skalpell 1.5 – 3 mm große Stücke
ausgeschnitten. Zur Entfärbung und Dehydradation der Gel-Stücke wurde 150 μl
50 mM Ammoniumbicarbonat (pH 8.5) und 50 % Acetonitril hinzu gegeben. Die
Proben wurden dann für 30 min bei 37 °C inkubiert. Die Lösung wurde nach der
Inkubation abgenommen. Danach wurden 5 μl 5 % Iodacetamid in 50 mM
Ammoniumbicarbonat (pH 8.5) zugegeben und für 20 min bei 37 °C inkubiert.
Anschließend wurden die Gel-Stücke 2-mal für 10 min mit 100 μl Wasser
gewaschen. Der Peptidverdau erfolgte durch Trypsin für 16 Stunden bei 37 °C. Um

Material und Methoden
- 46 -
daraufhin die Peptide aus dem Gel zu extrahieren wurden zu jedem Gel-Stück 15 μl
60 % Acetonitril / 1% Tetraflur-Essigsäure (TFA) zugegeben und für eine Stunde
inkubiert. Danach wurde die Lösung in neue Eppendorfgefäße überführt und bis zu
einem Volumen von 2 μl eingedampft. Anschließend wurde 1 μl der Probe auf ein
MALDI-ToF-Target gespottet und in Kooperation mit der Arbeitsgruppe Redox
Proteomics (Ruhr-Uni Bochum, Jun. Prof. Lars Leichert) in einem
Massenspektrometer (Ultraflex II MALDI TOF/TOF) vermessen und ausgewertet.

Material und Methoden
- 47 -
2.8 Lipidanalytik
2.8.1 Lipidextraktion
Um die Membranlipidkomposition einer Überexpressionskultur oder die
Reaktionsprodukte eines in vitro-Aktivitätsassays zu analysieren, wurde eine
Lipidextraktion nach Bligh und Dyer durchgeführt (Bligh & Dyer, 1959). Dafür wurden
2 ml Zellen durch Zentrifugation (13000 UpM für 1 min, EZ, RT) geerntet. Der
Überstand wurde verworfen und das Pellet mit 500 µl H2O gewaschen. Anschließend
wurden die Zellen in 100 µl H2O resuspendiert. Die resuspendierten Zellen oder der
Ansatz eines in vitro-Aktivitätsassays wurde mit 375 µl Methanol:Chloroform 2:1
versetzt. Nach 15 Sekunden Vortexen wurde die Probe 3 Minuten bei 13 000 UpM
zentrifugiert (EZ, RT). Der flüssige Überstand wurde vorsichtig abgenommen und in
ein neues Eppendorfgefäß überführt. Dieser Überstand wurde mit 100 µl Chloroform
und 100 µl H2O durch 15 sekündiges Vortexen gemischt. Nach einem weiteren
Zentrifugationsschritt (13000 UpM, 3 min, EZ, RT) wurde die Chloroformphase
(~200 µl untere Phase) abgenommen und in einer SpeedVac (UNIV PO 100 H)
getrocknet. Die getrockneten Lipide wurden in 20 µl Methanol:Chloroform 1:1 gelöst
und auf eine Dünnschichtchromatographieplatte aufgetragen.
2.8.2 Dünnschichtchromatographie (DC)
Die DC-Platten (Merck; HPTLC-Aluminiumfolien, 20 x 20 cm, Kieselgel 60;
Schichtdicke 0.2 mm) wurden in einem Laufmittel (3 : 2 : 2 : 1
n-Propanol : Propionsäure : Chloroform : Wasser) in einer DC-Kammer entwickelt.
Zur Visualisierung der Lipide wurde entweder das „Molybdenum Blue Spray“ der
Firma Sigma verwendet oder eine Kupfersulfat-Färbung durchgeführt. Die
Molybdänblau-Färbung ist ein spezifisches Nachweisverfahren für Phospholipide, bei
dem das Molybdänblau mit der Phosphatgruppe der Phospholipide reagiert.
Die Kupfersulfat-Färbung ist hingegen ein unspezifisches Nachweisverfahren, das
auf der Verkohlung von Kohlenstoffatomen basiert. Für die Kupfersulfat-Färbung
wurde die getrocknete DC-Platte für ca. 3 Sekunden in ein Kupfer-Sulfat-Reagenz
getaucht, getrocknet und anschließend bei 170 °C in einem Wärmeschrank erhitzt.

Material und Methoden
- 48 -
Phospholipide und eventuell noch andere organische Verbindungen wurden nach ca.
6 – 8 min sichtbar.
Kupfersulfat-Färbereagenz
Kupfersulfatpentahydrat 75 g
Phosphorsäure (85 %) 100 ml
H2O ad 1000 ml
Material und Methoden
- 49 -
2.9 Chemikalienliste
Chemikalien Hersteller
-Mercaptoethanol Merck
40 % Acrylamid/Bisacrylamid Biorad
Agar Roth
Ammoniumpersulfat Pharmacia Biotech
Ampicillin Serva
Borsäure J.T.Baker
Bromphenolblau Riedel-de Haën
C8-PE Avanti Polar Lipids
Calciumsulfat J.T.Baker
Chloroform Prolabo
Cholin Sigma Aldrich
Coomassie Brilliant Blue R 250 Serva
Dinatriumhydrogenphosphat J.T.Baker
DMSO J.T.Baker
EDTA Merck
Eisen(III)Chlorid Fulka
Essigsäure J.T.Baker
Formiat VWR Prolabo
Fluorescein-Phosphatidyltehanolamin Avanti Polar Lipids
Glycerin J.T.Baker
Glycin Applichem
Hefeextrakt Becton Dickinson
IPTG Applichem
Kaliumacetat J.T.Baker
Kaliumnitrat Riedel-de Haën
Kanamycin Serva
Kobaldchlorid Riedel-de Haën
Kupfersulfat Riedel-de Haën
Lysozym Serva
Lyso-Phosphatidylethanolamin Sigma Aldrich
Magnesiumchlorid J.T.Baker
Magnesiumsulfat Applichem
Manganchlorid J.T.Baker
Mangansulfat Riedel-de Haën
Methanol J.T.Baker
NaCl J.T.Baker
Natriumacetat J.T.Baker
Natriummolybdat Riedel-de Haën
Natriumnitrat Riedel-de Haën

Material und Methoden
- 50 -
Natronlauge Waldeck
Nitrilessigsäure Sigma
n-Propanol J.T.Baker
Dimethylphosphatidylethanolamin Sigma Aldrich
Monomethylphosphatidylethanolamin Sigma Aldrich
Phosphatidat Sigma Aldrich
Phosphatidylcholin Sigma Aldrich
Phosphatidylethanolamin Sigma Aldrich
Phosphorylethanolamin Sigma Aldrich
Phosphocholin Sigma Aldrich
Phosphorsäure J.T.Baker
Propionsäure J.T.Baker
Rubidiumchlorid Sigma
Schwefelsäure J.T.Baker
SDS Applichem
TEMED Pharmacia Biotech
Tris-HCl AppliChem
Triton X-100 Serva
Trypton Becton Dickinson
Zinksulfat Riedel-de Haën
2.10 Geräteliste
Gerät Hersteller
CD-Spektrometer Spectropolarimeter Jasco J-715
Elektrophoresekammern BioRad Mini-Protean Gelkammer II Dual Slab Cell
Ruhr-Universität Bochum Eigenbauten
Feinwaage Sartorius 2004 MP
Fluorimeter Thermo Amicon Bowman II Luminescence
Spectrometer
Constant Cell Disruption System
Constant Systems Ltd
Gelfiltrationssäule Pharmacia Superdex 200 HR 10/30
Gelfiltrationssystem Amersham Äkta Explorer
Inkubationsschüttler New Brunswick Scientific Innova 4230 Ruhr-Universität-Bochum Eigenbauten
Microplatereader Biotek µQuant

Material und Methoden
- 51 -
pH-Meter Mettler Toledo pH-Meter SevenEasy
Spannungsgeber Biometra Power Pack P25 T
BioRad PowerSupply 200V/2.0A
Spektralphotometer Amersham Biochrom Ultrospec II
Peqlab Nano-drop Pharmacia Biotech Novaspec II
Szintillationszähler Beckman Counter (LS-6000 TA)
Videodokumentationsanlage Alpha Innotech Multi Image Light Carbinet
Wärmeschrank Heraeus Instruments Typ UT6420
Zentrifugen
Eppendorf Tischzentrifuge 5415D Heraeus Biofuge Megafuge 1.0R
Sorvall Kühlzentrifuge RC-5B (Rotoren SS-34, SLA 3000)
Alle nicht aufgeführten Kleingeräte entsprechen dem allgemein üblichen
Laborstandard.
2.11 Software - / Linkliste
Programm Hersteller / Link
BLAST http://blast.ncbi.nlm.nih.gov/Blast.cgi
Clonemanager Sci-Ed
ClustalW http://www.ebi.ac.uk/Tools/clustalw2/index.html
GeneDoc http://www.nrbsc.org/gfx/genedoc/index.html
I-TASSER http://zhanglab.ccmb.med.umich.edu/I-TASSER/
K2D2 http://www.ogic.ca/projects/k2d2/
Peptidecutter http://web.expasy.org/peptide_cutter/
Protein Calculator http://www.scripps.edu/~cdputnam/protcalc.html
ProtParam http://www.expasy.org/tools/protparam.html
PSI Pred http://bioinf.cs.ucl.ac.uk/psipred/
Pymol DeLano Scientific LLC; Version 0.99rc6b
Seqman DNASTAR
SigmaPlot 9.0 Systat
Stride http://webclu.bio.wzw.tum.de/cgi-bin/stride/stridecgi.py

Ergebnisse
- 52 -
3. Ergebnisse
Kapitel 1
3.1 SAM-Bindeeigenschaften der Phospholipid N-
Methyltransferase aus Agrobacterium tumefaciens
Die agrobakterielle Phospholipid N-Methyltransferase PmtA katalysiert die
Methylierung von PE zu PC. Als Methyldonor für diese Reaktion dient SAM, das zu
SAH umgesetzt wird. Putative SAM-Bindemotive wurden in PmtA bioinformatisch
identifiziert und die konservierten Aminosäuren mittels QuikChange® Mutagenese
ausgetauscht. PmtA und PmtA-Derivate wurden in E. coli heterolog überexprimiert
und mittels Affinitätschromatographie (Hexahistidin-Tag) isoliert, um die SAM-
Bindeeigenschaften zu analysieren.
3.1.1 Bioinformatische Identifizierung putativer SAM-Bindemotive
in PmtA
PmtA zählt aufgrund seiner Aminosäuresequenz zu den sinorhizobiellen Pmt-
Enzymen. Durch einen Aminosäuresequenzvergleich mit verschiedenen Enzymen
des sinorhizobiellen-Typs wurden putative SAM-Bindemotive für PmtA identifiziert.
Das SAM-Bindemotiv I (SAM-I-Motiv) ist in allen SAM-MTs hoch konserviert. Es
besteht aus einer sauren Aminosäure mit einem anschließenden glycinreichen Loop
(D/ExGxGxG). Dieses Motiv ist auch in allen Pmt-Enzymen hoch konserviert und im
N-terminalen Bereich lokalisiert (Abbildung 9). Die Aminosäuren E58, G60, G62 und
G64 bilden das putative SAM-I-Motiv in PmtA. Die Aminosäuren der weiteren SAM-
Bindemotive sind in verschiedenen SAM-MTs zum Teil sehr variabel. Einige
konservierte Aminosäuren dieser SAM-Bindemotive befinden sich in verschiedenen
Proteinen an strukturell äquivalenten Positionen. Daher wurden diese konservierten
Aminosäuren in PmtA mit Hilfe einer Sekundärstrukturvorhersage identifiziert
(I-TASSER; Zhang, 2008). Für das SAM-II-Motiv ist eine saure Aminosäure (D/E) am
Ende des β-Faltblattes 2 oder im daran angrenzenden Loop charakteristisch. An
Position 84 in PmtA befindet sich ein Glutamat (E), das diese Kriterien erfüllt. Für das

Ergebnisse
- 53 -
SAM-III-Motiv wird eine weitere saure Aminosäure im C-terminalen Bereich des β-
Faltblattes 3 als typisches Merkmal beschrieben. Das Aspartat (D) an Position 106 in
PmtA stellt die konservierte Aminosäure des putativen SAM-III-Motives für PmtA dar.
Abbildung 9: Aminosäuresequenzvergleich bakterieller Phospholipid N-Methyltransferasen (Pmt). Hoch konservierte Aminosäuren sind schwarz unterlegt. Die putativen SAM-Bindemotive sind eingezeichnet (SAM-I – III). Über der Aminosäuresequenz ist die vorhergesagte Sekundärstruktur (I-TASSER, Zhang, 2008) für PmtA eingezeichnet. *: unlösliche PmtA-Derivate; Atum: Agrobacterium tumefaciens; Smel: Sinorhizobium meliloti; Bjap: Bradyrhizobium japonicum; Mloti: Mesorhizobium loti;
Ml:mlr5374: putatives Pmt-Enzym aus Mloti; SAM-I / II / III: SAM-Bindemotiv I / II / III.
Um die Bedeutung der identifizierten Aminosäuren für die SAM-Bindung zu klären
(SAM-I-Motiv: E58, G60, G62, G64; SAM-II-Motiv: E84; SAM-III-Motiv: D106),
wurden diese mittels QuikChange® Mutagenese gegen ein Alanin ausgetauscht. Im
Folgenden wurde die enzymatische Aktivität, Faltung und SAM-Bindefähigkeit der
entstandenen PmtA-Derivate analysiert. Als Kontrolle diente ein PmtA-Derivat
(G162A) mit einem Aminosäureaustausch außerhalb der beschriebenen putativen
SAM-Bindemotive an Position 162.

Ergebnisse
- 54 -
3.1.2 Einfluss von Punktmutationen in putativen SAM-Bindemotiven
auf die PmtA-Aktivität
Die PmtA-Derivate E58A, G60A, P61A, G62A, G64A, E84A, D106A und G162A
wurden in E. coli BL21 (DE3) mittels des T7-Expressionssystems heterolog
überexprimiert. Alle PmtA-Derivate wurden erfolgreich überexprimiert (Abbildung
10A). Die Derivate G64A und D106A waren allerdings vollständig unlöslich. Im Falle
von D106A verbesserte auch ein Austausch von Glutamat zu Aspartat die Löslichkeit
des Proteins nicht. Daher konnten keine weiterführenden Analysen für diese PmtA-
Derivate durchgeführt werden. Alle anderen PmtA-Derivate konnten mittels
Affinitätschromatographie isoliert und näher charakterisiert werden.
Abbildung 10: Heterologe Überexpression und Enzymaktivität verschiedener PmtA-Varianten. (A.) Analyse der Synthese von verschiedenen PmtA-Derivate in E. coli BL21 (DE3) mittels SDS-PAGE. (B.) Dünnschichtchromatographische Auftrennung von Gesamtlipiden aus E. coli BL21 (DE3) Zellen, die verschiedene PmtA-Derivate exprimieren. Die Lipide wurden mit einem Kupersulfatreagenz visualisiert.
Die Aktivität der PmtA-Derivate wurde durch Nachweis der Biosyntheseprodukte
analysiert. Dafür wurden die Gesamtlipide der PmtA synthetisierenden E. coli
Stämme isoliert und mittels Dünnschichtchromatograhpie aufgetrennt. Mit einem
Kufpersulfatreagenz wurden die aufgetrennten Lipide visualisiert (Abbildung 10B).

Ergebnisse
- 55 -
E. coli ist natürlicherweise nicht in der Lage PC zu synthetisieren. Eine
Überexpression von PmtA in E. coli führt zur PC-Biosynthese, was für diesen
Versuch als Basis zur Aktivitätsanalyse genutzt wurde (Aktas & Narberhaus, 2009).
Es zeigte sich, dass das Kontroll-Derivat G162A wildtyp-ähnliche PmtA-Aktivität
besitzt (Abbildung 10B). P61 ist eine nur in Pmt-Enzymen konservierte Aminosäure
innerhalb des SAM-I-Motivs, jedoch hat diese Aminosäure vermutlich keine
Bedeutung für die SAM-Bindung (Abbildung 9 und Abschnitt 1.3.3). Eine
Punktmutation an dieser Position hat daher keinen Einfluss auf die enzymatische
Aktivität. Im Gegensatz dazu führt eine Mutation der konservierten Glycine an den
Postionen 60 und 62 zu einer drastischen Reduktion der PmtA-Aktivität (Abbildung
10B). Die PmtA-Derivate E58A und E84A waren vollständig inaktiv, da weder MMPE,
DMPE noch PC nachgewiesen werden konnten. Somit sind die Aminosäuren E58,
G60, G62 und E84 für die PmtA-Aktivität essentiell.
3.1.3 Beeinflussen die Punktmutationen die Faltung von PmtA-
Derivaten?
Im vorherigen Abschnitt wurde gezeigt, dass Punktmutationen einen erheblichen
Einfluss auf die PmtA-Aktivität haben. Um auszuschließen, dass der Aktivitätsverlust
auf einem strukturellen Defekt beruht, wurde die Faltung dieser Proteine überprüft.
Die Circular Dichroismus-Spektroskopie (CD-Spektroskopie) ist eine Methode, mit
der die Sekundärstruktur und Faltung eines Proteins untersucht werden kann (Kelly &
Price, 2000). Ein CD-Spektrum stellt dabei die Gesamtheit aller einzelnen
Sekundärstrukturanteile eines Proteins (α-Helices, β-Faltblätter und Loops) dar und
kann mithilfe eines passenden Algorithmus dekonvoliert werden. Dadurch erhält man
Informationen über den prozentualen Anteil an α-Helices, β-Faltblättern und
ungefalteten Regionen eines Proteins (Kelly & Price, 2000). Durch diese Methode ist
es daher möglich die PmtA-Derivate auf eventuelle strukturelle Unterschiede zu
untersuchen, die die Aktivität beeinflussen könnten.
Das CD-Spektrum für PmtA hat ein Minimum an Elliptizität bei ca. 208 - 210 nm
(Abbildung 11A). Das Spektrum weist auf einen hohen Anteil an α-Helices in der
Sekundärstruktur hin. Eine Dekonvolierung des CD-Spektrums mit CDSSTR und
K2D2 (Sreerama & Woody, 2000; Perez-Iratxeta & Andrade-Navarro, 2008) lässt auf

Ergebnisse
- 56 -
einen α-helikalen Anteil von ca. 42 % bzw. 39 % schließen. Der Anteil an β-
Faltblättern beträgt ca. 11 %. Die experimentell ermittelten Daten für die
Sekundärstruktur werden durch in silico-Vorhersagen gestützt (Abbildung 11B). Die
Programme GOR IV und PHD (Rost & Sander, 1993; Garnier et al., 1996) nutzen die
Primärsequenz von Proteinen, um den Sekundärstrukturanteil zu berechnen. Stride
hingegen nutzt eine Struktur oder ein Homologiemodell, um das α/β-Verhältnis zu
bestimmen (Heinig & Frishman, 2004) (Abbildung 11B).
Abbildung 11: CD-spektroskopische Analyse von PmtA. (A.) CD-Spektrum von PmtA. Die minimale Elliptizität liegt bei ca. 210 nm. (B.) Übersicht der prozentualen Anteile an Sekundärstrukturelementen in PmtA. GOR IV und PHD berechnen den Sekundärstrukturanteil anhand der Primärsequenz eines Proteins. Für eine Sekundärstrukturanalyse nutzt Stride bekannte Proteinstrukturen oder Homologiemodelle. CDSSTR und K2D2 sind Algorithmen zur Dekonvolierung von experimentellen CD-Spektren. Mdeg: millidegree
Die strukturelle Integrität der PmtA-Derivate E58A, G60A, G62A und E84A wurde
mittels CD-Spektroskopie untersucht und mit wildtyp PmtA verglichen. Alle PmtA-
Derivate wiesen ein wildtyp-ähnliches CD-Spektrum auf, was für eine native Faltung
der PmtA-Derivate spricht (Abbildung 12). Die eingefügten Mutationen hatten somit
keinen Einfluss auf die Sekundärstruktur von PmtA. Einen weiteren Hinweis für eine
native Faltung liefert das Aufschmelzverhalten von PmtA und der PmtA-Derivate. Der
Übergang von einer geordneten zu einer ungeordneten Struktur stellt den
Wendepunkt einer Schmelzkurve dar (Nelson & Cox, 2009). Der Schmelzpunkt (Tm)
von PmtA liegt bei ca. 35.9 °C. Alle PmtA-Derivate hatten einen vergleichbaren
Schmelzpunkt von ca. 35.5 °C (Abbildung 12B und C). Eine Missfaltung der Proteine

Ergebnisse
- 57 -
kann somit als Grund für den enzymatischen Aktivitätsverlust ausgeschlossen
werden. Sehr wahrscheinlich ist, dass durch den Aminosäureaustausch die SAM-
Bindung negativ beeinflusst wird. Daher wurde im Folgenden die SAM-Bindefähigkeit
der PmtA-Derivate untersucht.
Abbildung 12: Sekundärstrukturvergleich und Thermostabilität von PmtA und PmtA-Derivaten. (A.) CD-Spektrum von PmtA und den PmtA-Derivaten. Die Proteine weisen eine native Faltung auf. (B.) Thermostabilität von PmtA. Die Tm für PmtA beträgt ca. 35.9 °C. (C.) Vergleich der
Schmelzpunkte von PmtA und den PmtA-Derivaten.
3.1.4 SAM-Bindefähigkeit von PmtA und PmtA-Derivaten
Die PmtA-Derivate E58A, G60A, G62A und E84A zeigten einen deutlichen
enzymatischen Defekt (Abbildung 10), jedoch wiesen alle PmtA-Derivate eine native
Faltung auf (Abbildung 12). Um die SAM-Bindefähigkeit der PmtA-Derivate zu
untersuchen, wurde ein Filterbindeassay verwendet (Aktas & Narberhaus, 2009).
Dafür wurde PmtA bzw. die PmtA-Derivate mit radioaktiv markiertem SAM
(S-[methyl-14C]adenosyl-L-methionin) inkubiert und die gebundene Menge 14C-SAM
mittels Szintillationszähler detektiert. Die SAM-Bindefähigkeit des wildtyp PmtA (WT)
wurde als 100 % gesetzt. Für die Bestimmung der Dissoziationskonstanten (KD) von
SAM zu PmtA bzw. zu den PmtA-Derivaten wurden Ansätze mit steigenden SAM-
Konzentrationen verwendet.
Abbildung 13 zeigt einen relativen Vergleich der SAM-Bindefähigkeit von PmtA (WT)
und den PmtA-Derivaten. Die enzymatisch aktiven PmtA-Derivate P61A und G162A
waren in der Lage SAM vergleichbar effizient wie wildtyp PmtA (WT) zu binden (74 –
78 %). E58A, G60A und G62A konnten hingegen nur sehr geringe Mengen an SAM

Ergebnisse
- 58 -
binden. Einen drastischeren Effekt zeigte E84A, da dieses PmtA-Derivat nicht mehr
fähig war detektierbare Mengen an SAM zu binden.
Abbildung 13: Relative SAM-Bindung der PmtA-Derivate E58A, G60A, P61A, G62A, E84A und G162A. Es wurden 10 µM rekombinantes Protein mit 100 µM S-[methyl-
14C]adenosyl-L-methionin
(48.8 mCi/mmol) in Gegenwart von 100 µM PE inkubiert. 100 % an S-[methyl-14
C]adenosyl-L-
methionin entspricht 143 757 dpm.
Um die SAM-Bindung von PmtA zu charakterisieren, wurden Titrationsexperimente
mit radioaktiv markiertem SAM durchgeführt. Für PmtA (WT) und die PmtA-Derivate
P61A sowie G162A wurde eine Dissoziationskonstante im mikromolaren Bereich
ermittelt (P61A: 22 µM; G162A: 30 µM; PmtA: 25 µM). Da die PmtA-Derivate E58A,
G60A und G62A kaum SAM binden, konnten keine Dissoziationskonstanten
bestimmt werden.
Abbildung 14: SAM-Titrationskurven von PmtA (WT) und PmtA-Derivaten. Das Reaktionsvolumen betrug 50 µl, wobei 10 µM PmtA und 100 µM PE-Lipsosomen eingesetzt wurden. Aufgetragen wurde die Änderung von gebundenem
14C-SAM [dpm] gegen die Konzentration des eingesetzten
14C-SAM [µM]. Titrationskurve mit PmtA (A.), den PmtA-Derivate P61A und G162A (B.), sowie E58A,
G60A, G62A und E84A (C.).

Ergebnisse
- 59 -
In vorangehenden Experimenten wurde gezeigt, dass die SAM-Bindung durch PmtA
nur in Gegenwart von Phospholipidsubstraten erfolgt (Aktas & Narberhaus, 2009).
Möglicherweise wird erst durch die Lipidsubstratbindung die SAM-Bindetasche
zugänglich, sodass SAM gebunden werden kann. Um auszuschließen, dass der
Verlust der SAM-Bindung der PmtA-Derivate auf einem Verlust der Lipidsubstrat-
bindung beruht, wurde die Lipidbindung von PmtA und den PmtA-Derivaten mit
einem „Protein-Lipid-Overlay-Assay“ getestet. Dabei konnte gezeigt werden, dass
alle PmtA-Derivate in der Lage sind sowohl Gesamtlipidextrakt aus A. tumefaciens,
als auch kommerziell erhältliches PE zu binden (Abbildung 15).
Abbildung 15: Protein-Lipid-Overlay-Assay mit PmtA und den PmtA-Derivaten E58A, G60A, P61A, G62A, E84A und G162A. Als Lipidsubstrat wurden gleiche Mengen an Gesamtlipidextrakt aus A. tumefaciens oder 14 nmol Phosphatidylethanolamin verwendet. Gebundenes Protein wurde mit
einem Anti-Penta-His HRP-Konjugat der Firma Qiagen detektiert.
Die in diesem Kapitel präsentierten Ergebnisse zeigen eindeutig, dass die
Aminosäuren E58, G60, G62 und E84 in PmtA für die SAM-Bindung essentiell sind
und höchstwahrscheinlich die SAM-Bindetasche formen. Eine solche Funktion wird
später anhand eines Strukturmodells diskutiert (Abschnitt 4.1).
Weiterhin konnten diese Aminosäuren den für SAM-MTs bekannten SAM-
Bindemotiven zugeordnet werden. Das SAM-I-Motiv bilden dabei die Aminosäuren
E58, G60 und G62. Die Aminosäure E84 konnte als konservierte Aminosäure des
SAM-II-Motivs für PmtA identifiziert und verifiziert werden.

Ergebnisse
- 60 -
Kapitel 2
3.2 Einfluss der Membranlipidzusammensetzung auf die
Aktivität und Membranbindung von PmtA
Das Membranlipid PE ist das natürliche Phospholipid-Substrat von PmtA. Das Enzym
PmtA gehört daher zu den peripheren Membranproteinen, deren Substrat ein
Bestandteil der bakteriellen Membran ist. Die detaillierte Funktionsweise bakterieller
Pmt-Enzyme ist jedoch bis heute relativ unklar. Bisher konnte gezeigt werden, dass
die Bindung eines Phospholipidsubstrates der erste Schritt der Transmethlyierungs-
reaktion ist (Aktas & Narberhaus, 2009). Unklar ist jedoch, warum eine
Phospholipidsubstratbindung als initialer Schritt erfolgen muss, damit der Kofaktor
SAM binden kann. Weiterhin stellt sich die Frage, wie Phosphatidylglycerin (PG) die
Aktivität von PmtA stimuliert, obwohl es die SAM-Bindung nicht begünstigt (Aktas &
Narberhaus, 2009). Daher soll im Folgenden die Protein-Lipid-Interaktion von PmtA
und der Einfluss der Membranlipidkomposition auf die Aktivität näher untersucht
werden.
3.2.1 Negativ geladene Lipide stimulieren die PmtA-Aktivität
Eine wichtige Fragestellung dieser Arbeit war, ob die PmtA-Aktivität spezifisch durch
PG stimuliert wird oder ob die PmtA-Aktivität auch durch andere Lipide beeinflusst
wird. PG ist ein anionisches Phospholipid, das in bakteriellen Membranen vorkommt.
Geladene Lipide wie PG können die Aktivität oder Lokalisation von membran-
assoziierten Proteinen beeinflussen (Cornell, 1991a; Johnson & Cornell, 1999;
Linde et al., 2004; Mulgrew-Nesbitt et al., 2006; Minami et al., 2011). Um zu klären
ob nur PG oder auch andere Lipide die enzymatische Aktivität von PmtA
beeinflussen, wurde die PmtA-Aktivität in Gegenwart verschiedener Lipide
untersucht. Eine Übersicht der Kopfgruppen der verwendeten geladenen Lipide ist in
Abbildung 16 dargestellt. Um die PmtA-Aktivität in An- und Abwesenheit dieser Lipide
zu untersuchen, wurde ein bereits etabliertes in vitro-Aktivitätsassay (Aktas &
Narberhaus, 2009) eingesetzt.

Ergebnisse
- 61 -
Abbildung 16: Übersicht der Kopfgruppen ausgewählter geladener Lipide. Rot (-) kennzeichnet
negativ geladene Lipidkopfgruppen, blau (+) hingegen positiv geladene Lipidkopfgruppen.
PmtA wurde mit PE als Substratlipid in Liposomenform und SAM als Methyldonor
inkubiert. Um den Effekt verschiedener geladener Lipide auf die PmtA-Aktivität zu
untersuchen, wurden diese mit dem Substratlipid vermischt und anschließend dem
Ansatz als „Mischmembranen“ hinzugegeben. Nach Ablauf der Reaktionszeit von
einer Stunde, wurden die Reaktionsprodukte isoliert, mittels Dünnschicht-
chromatographie (DC) aufgetrennt und mit spezifischen Färbemethoden visualisiert.
Abbildung 17A zeigt, dass PmtA PE-Liposomen als Substrat nutzen kann. Über die
Zwischenstufen MMPE und DMPE wird PE zum Endprodukt PC methyliert.
Rekonstituiert man in diese künstliche PE-Membran zusätzlich PG, so wird die PmtA-
Aktivität stimuliert und die Produkte MMPE, DMPE sowie PC werden vermehrt
gebildet. Dies wurde bereits in vorherigen Arbeiten (Aktas & Narberhaus, 2009)
gezeigt und konnte in dieser Arbeit bestätigt werden.
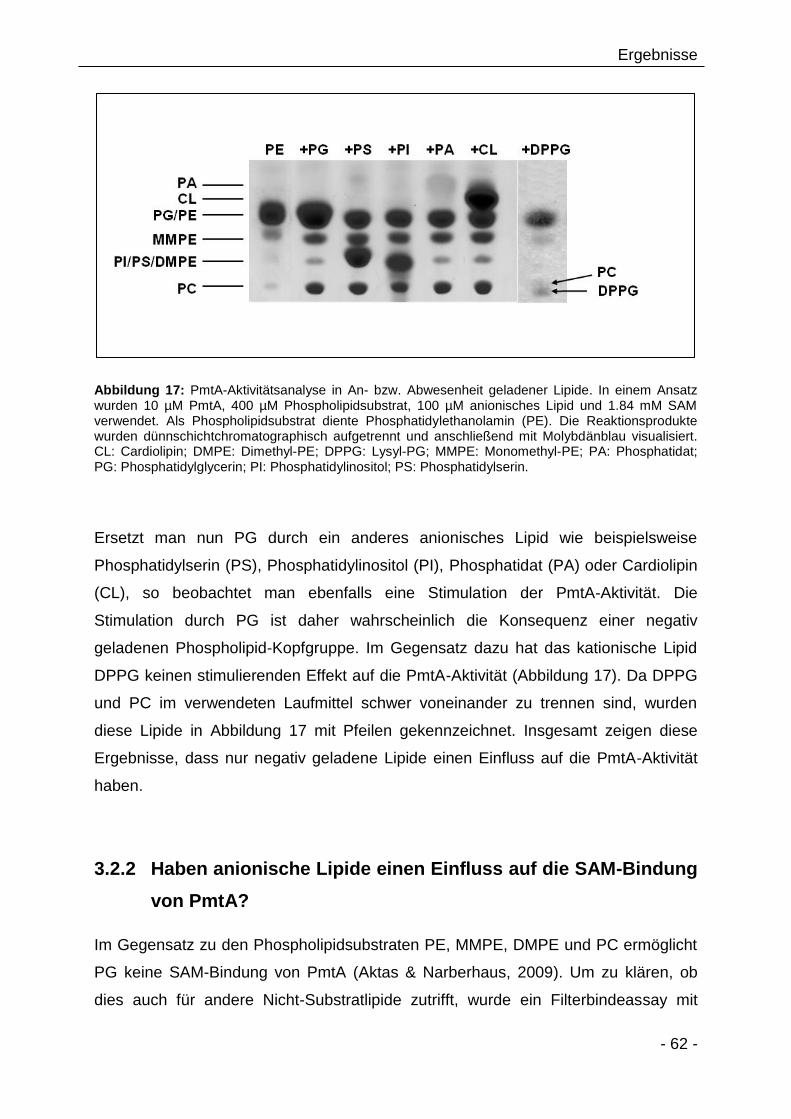
Ergebnisse
- 62 -
Abbildung 17: PmtA-Aktivitätsanalyse in An- bzw. Abwesenheit geladener Lipide. In einem Ansatz wurden 10 µM PmtA, 400 µM Phospholipidsubstrat, 100 µM anionisches Lipid und 1.84 mM SAM verwendet. Als Phospholipidsubstrat diente Phosphatidylethanolamin (PE). Die Reaktionsprodukte wurden dünnschichtchromatographisch aufgetrennt und anschließend mit Molybdänblau visualisiert. CL: Cardiolipin; DMPE: Dimethyl-PE; DPPG: Lysyl-PG; MMPE: Monomethyl-PE; PA: Phosphatidat; PG: Phosphatidylglycerin; PI: Phosphatidylinositol; PS: Phosphatidylserin.
Ersetzt man nun PG durch ein anderes anionisches Lipid wie beispielsweise
Phosphatidylserin (PS), Phosphatidylinositol (PI), Phosphatidat (PA) oder Cardiolipin
(CL), so beobachtet man ebenfalls eine Stimulation der PmtA-Aktivität. Die
Stimulation durch PG ist daher wahrscheinlich die Konsequenz einer negativ
geladenen Phospholipid-Kopfgruppe. Im Gegensatz dazu hat das kationische Lipid
DPPG keinen stimulierenden Effekt auf die PmtA-Aktivität (Abbildung 17). Da DPPG
und PC im verwendeten Laufmittel schwer voneinander zu trennen sind, wurden
diese Lipide in Abbildung 17 mit Pfeilen gekennzeichnet. Insgesamt zeigen diese
Ergebnisse, dass nur negativ geladene Lipide einen Einfluss auf die PmtA-Aktivität
haben.
3.2.2 Haben anionische Lipide einen Einfluss auf die SAM-Bindung
von PmtA?
Im Gegensatz zu den Phospholipidsubstraten PE, MMPE, DMPE und PC ermöglicht
PG keine SAM-Bindung von PmtA (Aktas & Narberhaus, 2009). Um zu klären, ob
dies auch für andere Nicht-Substratlipide zutrifft, wurde ein Filterbindeassay mit

Ergebnisse
- 63 -
radioaktiv markiertem SAM eingesetzt. Dabei wurde die durch PmtA gebundene
Menge an radioaktivem SAM (S-[methyl-14C]adenosyl-L-methionin) mittels
Szintillationszähler detektiert. Es wurde bereits gezeigt, dass die Anwesenheit eines
Phospholipidsubstrates für PmtA notwendig ist, um SAM binden zu können (Aktas &
Narberhaus, 2009). In dieser Arbeit wurde sowohl die Notwendigkeit der
Anwesenheit eines Phospholipidsubstrates für die SAM-Bindung durch PmtA, als
auch die Tatsache, dass PG keine SAM-Bindung ermöglicht, bestätigt. Weiterhin
wurde gezeigt, dass die anionischen Lipide CL, PS, PA und PI ebenfalls keinen
Einfluss auf die SAM-Bindung von PmtA haben. Auch die Anwesenheit des
kationische DPPG ermöglicht keine SAM-Bindung durch PmtA.
Abbildung 18: SAM-Bindefähigkeit von PmtA in An- und Abwesenheit verschiedener Phospholipide. Für ein Filterbindeassay wurden 10 µM PmtA und 100 µM
14C-SAM (S-[methyl-
14C]adenosyl-L-
methionin) (48.8 mCi/mmol) in Gegenwart von 100 µM Phospholipid inkubiert. 100 % S-[methyl-14
C]adenosyl-L-methionin entsprechen 188 583 dpm. CL: Cardiolipin; DMPE: Dimethyl-PE; DPPG: Lysyl-PG; MMPE: Monomethyl-PE; PA: Phosphatidat; PE: Phosphatidylethanolamin; PG: Phosphatidylglycerin; PI: Phosphatidylinositol; PS: Phosphatidylserin.
Die Stimulierung der PmtA-Aktivität durch anionische Lipide beruht demnach nicht
auf der Stimulierung der SAM-Bindung. Der Bedeutung anionische Lipide für PmtA
wird in Abschnitt 3.2.5 nachgegangen. Im Folgenden soll nun zunächst die
Substratspezifität von PmtA betrachtet werden.
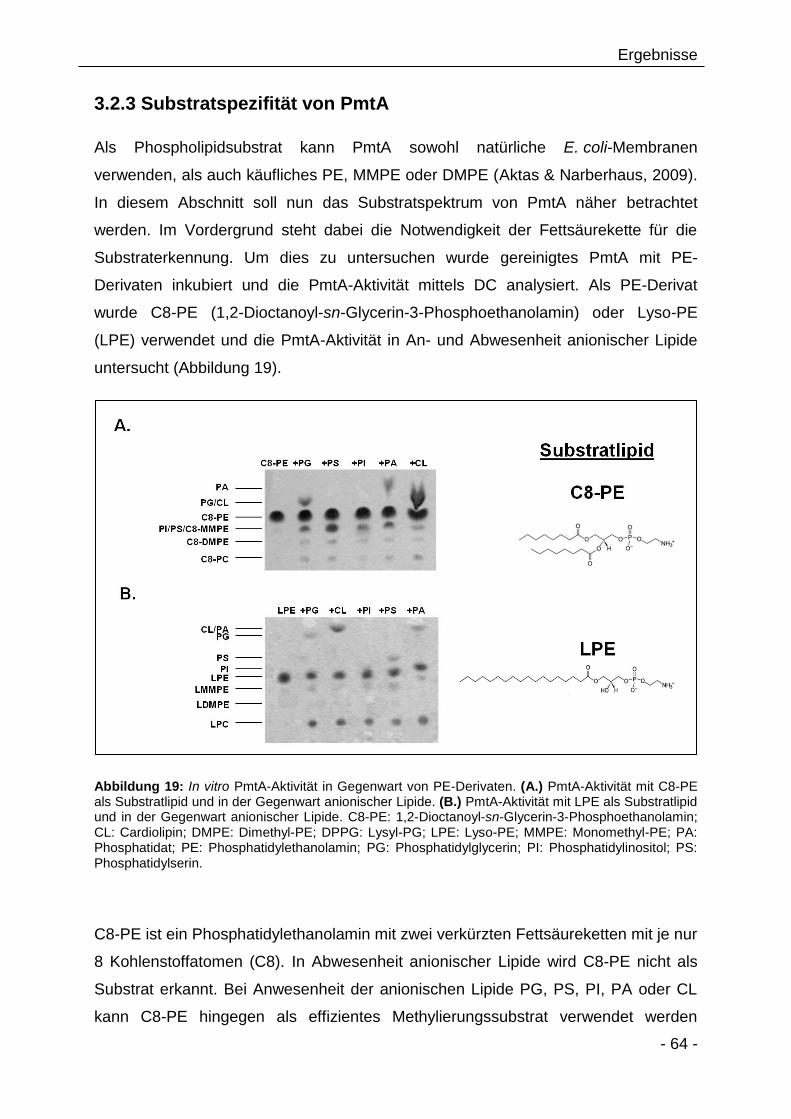
Ergebnisse
- 64 -
3.2.3 Substratspezifität von PmtA
Als Phospholipidsubstrat kann PmtA sowohl natürliche E. coli-Membranen
verwenden, als auch käufliches PE, MMPE oder DMPE (Aktas & Narberhaus, 2009).
In diesem Abschnitt soll nun das Substratspektrum von PmtA näher betrachtet
werden. Im Vordergrund steht dabei die Notwendigkeit der Fettsäurekette für die
Substraterkennung. Um dies zu untersuchen wurde gereinigtes PmtA mit PE-
Derivaten inkubiert und die PmtA-Aktivität mittels DC analysiert. Als PE-Derivat
wurde C8-PE (1,2-Dioctanoyl-sn-Glycerin-3-Phosphoethanolamin) oder Lyso-PE
(LPE) verwendet und die PmtA-Aktivität in An- und Abwesenheit anionischer Lipide
untersucht (Abbildung 19).
Abbildung 19: In vitro PmtA-Aktivität in Gegenwart von PE-Derivaten. (A.) PmtA-Aktivität mit C8-PE als Substratlipid und in der Gegenwart anionischer Lipide. (B.) PmtA-Aktivität mit LPE als Substratlipid und in der Gegenwart anionischer Lipide. C8-PE: 1,2-Dioctanoyl-sn-Glycerin-3-Phosphoethanolamin; CL: Cardiolipin; DMPE: Dimethyl-PE; DPPG: Lysyl-PG; LPE: Lyso-PE; MMPE: Monomethyl-PE; PA: Phosphatidat; PE: Phosphatidylethanolamin; PG: Phosphatidylglycerin; PI: Phosphatidylinositol; PS: Phosphatidylserin.
C8-PE ist ein Phosphatidylethanolamin mit zwei verkürzten Fettsäureketten mit je nur
8 Kohlenstoffatomen (C8). In Abwesenheit anionischer Lipide wird C8-PE nicht als
Substrat erkannt. Bei Anwesenheit der anionischen Lipide PG, PS, PI, PA oder CL
kann C8-PE hingegen als effizientes Methylierungssubstrat verwendet werden

Ergebnisse
- 65 -
(Abbildung 19A). Man erkennt deutlich die Zwischenstufen C8-MMPE und C8-DMPE,
sowie das Endprodukt C8-PC. Ähnlich wie C8-PE kann auch LPE als Substrat
dienen. LPE ist ein Signallipid, das nur in sehr geringen Mengen in einer
Zellmembran vorhanden ist. Weiterhin ist LPE ein Abbauprodukt der
Phospholipase A. LPE besitzt im Vergleich zu PE oder C8-PE nur eine
Fettsäurekette. Dieses PE-Derivat kann in Abwesenheit anionischer Lipide nicht als
Phospholipidsubstrat genutzt werden. Die Gegenwart von PG, CL, PS, PI oder PA
führt hingegen zur Synthese von Lyso-MMPE, Lyso-DMPE und Lyso-PC (Abbildung
19B). Im Gegensatz zu diesen beiden PE-Derivaten wurde wasserlösliches
Phosphorylethanolamin (P-Etn) von PmtA nicht als Substrat erkannt.
Bei einem Filterbindeassay mit 14C-SAM wurde durch die Gegenwart von P-Etn wie
zu erwarten keine SAM-Bindung durch PmtA beobachtet. Die Anwesenheit von C8-
PE führte hingegen zur Bindung geringer SAM-Mengen durch PmtA. Dementgegen
wurde durch die Anwesenheit von LPE keine signifikante SAM-Bindung durch PmtA
begünstigt (Abbildung 20). Dies liegt wahrscheinlich daran, dass PmtA eine geringere
Spezifität zu LPE aufweist, als zu C8-PE.
Abbildung 20: SAM-Bindung von PmtA in Gegenwart von verschiedenen PE-Derivaten. Für ein Filterbindeassay wurden 10 µM PmtA und 100 µM
14C-SAM (S-[methyl-
14C]adenosyl-L-methionin)
(48.8 mCi/mmol) in Gegenwart von 100 µM Phospholipid inkubiert. 100 % S-[methyl-14
C]adenosyl-L-
methionin entsprechen 188 583 dpm.
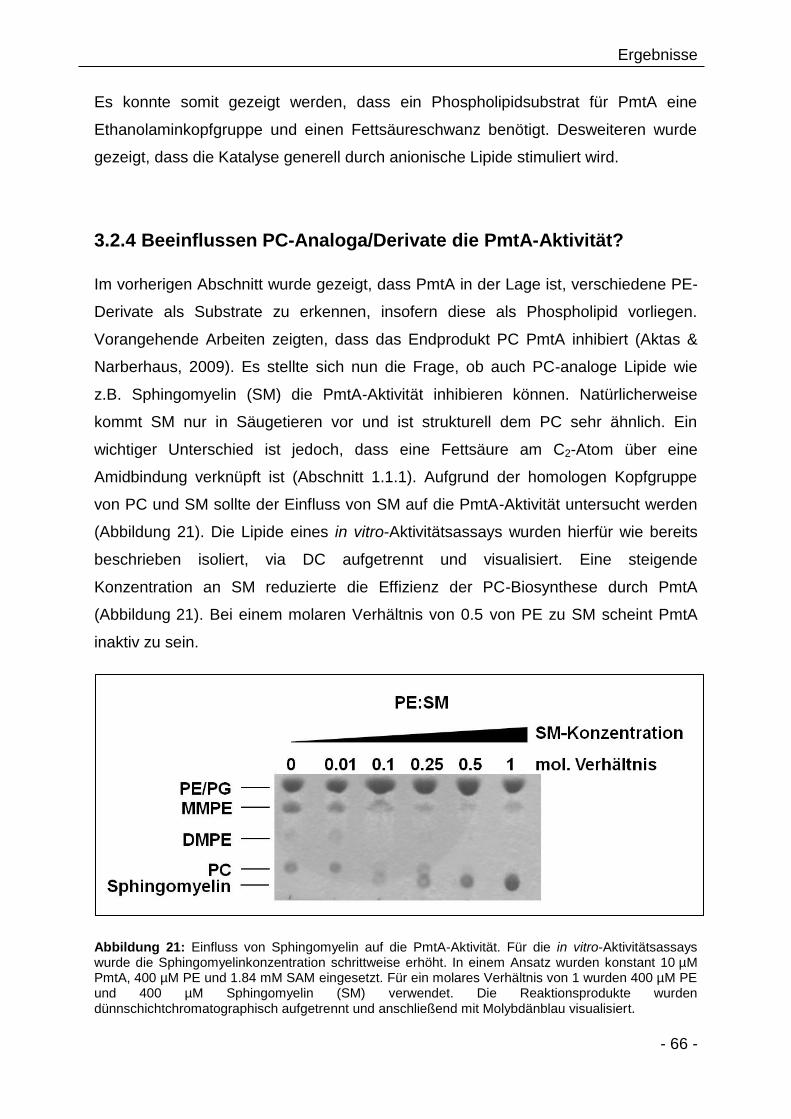
Ergebnisse
- 66 -
Es konnte somit gezeigt werden, dass ein Phospholipidsubstrat für PmtA eine
Ethanolaminkopfgruppe und einen Fettsäureschwanz benötigt. Desweiteren wurde
gezeigt, dass die Katalyse generell durch anionische Lipide stimuliert wird.
3.2.4 Beeinflussen PC-Analoga/Derivate die PmtA-Aktivität?
Im vorherigen Abschnitt wurde gezeigt, dass PmtA in der Lage ist, verschiedene PE-
Derivate als Substrate zu erkennen, insofern diese als Phospholipid vorliegen.
Vorangehende Arbeiten zeigten, dass das Endprodukt PC PmtA inhibiert (Aktas &
Narberhaus, 2009). Es stellte sich nun die Frage, ob auch PC-analoge Lipide wie
z.B. Sphingomyelin (SM) die PmtA-Aktivität inhibieren können. Natürlicherweise
kommt SM nur in Säugetieren vor und ist strukturell dem PC sehr ähnlich. Ein
wichtiger Unterschied ist jedoch, dass eine Fettsäure am C2-Atom über eine
Amidbindung verknüpft ist (Abschnitt 1.1.1). Aufgrund der homologen Kopfgruppe
von PC und SM sollte der Einfluss von SM auf die PmtA-Aktivität untersucht werden
(Abbildung 21). Die Lipide eines in vitro-Aktivitätsassays wurden hierfür wie bereits
beschrieben isoliert, via DC aufgetrennt und visualisiert. Eine steigende
Konzentration an SM reduzierte die Effizienz der PC-Biosynthese durch PmtA
(Abbildung 21). Bei einem molaren Verhältnis von 0.5 von PE zu SM scheint PmtA
inaktiv zu sein.
Abbildung 21: Einfluss von Sphingomyelin auf die PmtA-Aktivität. Für die in vitro-Aktivitätsassays wurde die Sphingomyelinkonzentration schrittweise erhöht. In einem Ansatz wurden konstant 10 µM PmtA, 400 µM PE und 1.84 mM SAM eingesetzt. Für ein molares Verhältnis von 1 wurden 400 µM PE und 400 µM Sphingomyelin (SM) verwendet. Die Reaktionsprodukte wurden dünnschichtchromatographisch aufgetrennt und anschließend mit Molybdänblau visualisiert.
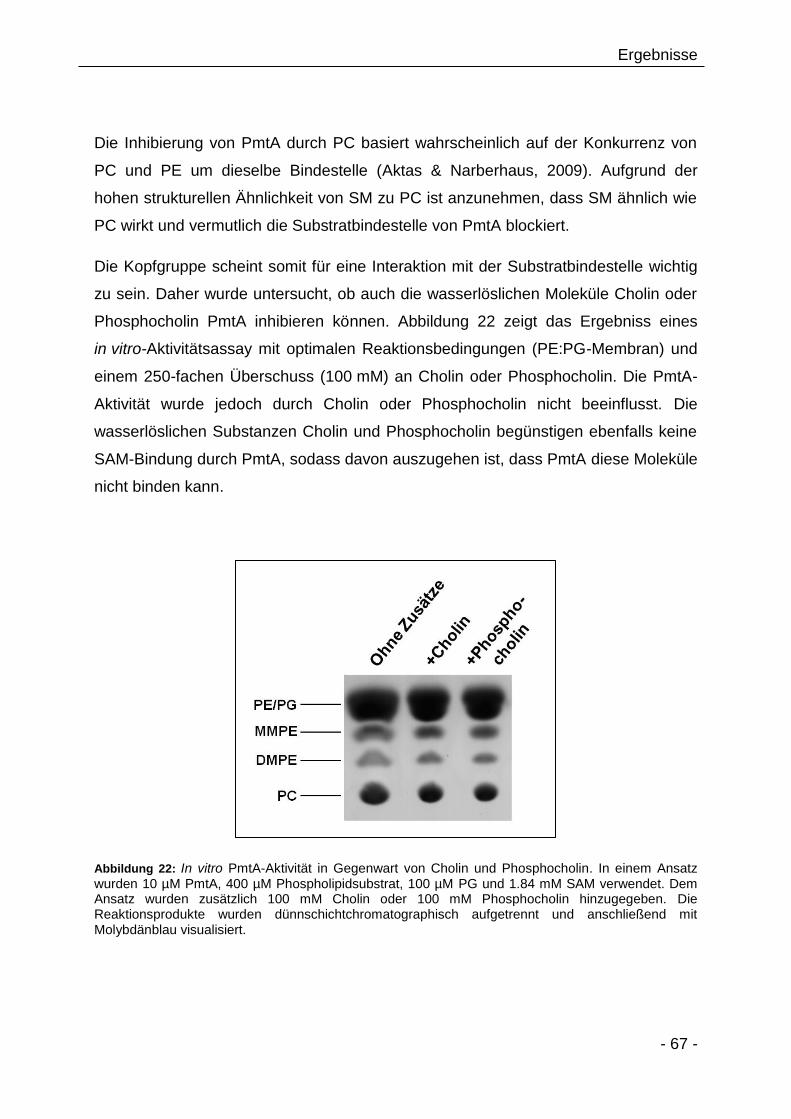
Ergebnisse
- 67 -
Die Inhibierung von PmtA durch PC basiert wahrscheinlich auf der Konkurrenz von
PC und PE um dieselbe Bindestelle (Aktas & Narberhaus, 2009). Aufgrund der
hohen strukturellen Ähnlichkeit von SM zu PC ist anzunehmen, dass SM ähnlich wie
PC wirkt und vermutlich die Substratbindestelle von PmtA blockiert.
Die Kopfgruppe scheint somit für eine Interaktion mit der Substratbindestelle wichtig
zu sein. Daher wurde untersucht, ob auch die wasserlöslichen Moleküle Cholin oder
Phosphocholin PmtA inhibieren können. Abbildung 22 zeigt das Ergebniss eines
in vitro-Aktivitätsassay mit optimalen Reaktionsbedingungen (PE:PG-Membran) und
einem 250-fachen Überschuss (100 mM) an Cholin oder Phosphocholin. Die PmtA-
Aktivität wurde jedoch durch Cholin oder Phosphocholin nicht beeinflusst. Die
wasserlöslichen Substanzen Cholin und Phosphocholin begünstigen ebenfalls keine
SAM-Bindung durch PmtA, sodass davon auszugehen ist, dass PmtA diese Moleküle
nicht binden kann.
Abbildung 22: In vitro PmtA-Aktivität in Gegenwart von Cholin und Phosphocholin. In einem Ansatz wurden 10 µM PmtA, 400 µM Phospholipidsubstrat, 100 µM PG und 1.84 mM SAM verwendet. Dem Ansatz wurden zusätzlich 100 mM Cholin oder 100 mM Phosphocholin hinzugegeben. Die Reaktionsprodukte wurden dünnschichtchromatographisch aufgetrennt und anschließend mit Molybdänblau visualisiert.

Ergebnisse
- 68 -
3.2.5 Rekrutierung von PmtA zur Membran via elektrostatischer
Interaktionen
In Abschnitt 3.2.2 wurde gezeigt, dass anionische Lipide die PmtA-Aktivität
stimulieren, aber keine SAM-Bindung durch PmtA begünstigen. Die Experimente des
folgenden Abschnittes sollen die Bedeutung von anionischen Lipiden für die PmtA-
Aktivität näher aufklären.
Für eine Protein-Membran-Interaktion gibt es verschiedene Interaktions-
möglichkeiten. Zum einen können Proteine fest in eine Membran integriert sein
(integrale Membranproteine) oder zum anderen peripher an eine Membran assoziiert
(periphere Membranproteine) vorliegen. Bei peripheren Membranproteinen
unterscheidet man weiterhin zwischen Proteinen, die einen Lipidanker besitzen und
Proteinen die mittels Lipidbindemotiven oder elektrostatischen Interaktionen an eine
Membran binden (Abbildung 23).
Abbildung 23: Protein-Membran-Interaktion. Man unterscheidet zwischen peripher-assoziierten Proteinen und integralen Membranproteinen. Periphere Membranproteine binden die Membran über spezifische Lipidbindemotive oder mit Hilfe eines Lipidankers, der eine sehr starke Membran-Protein Interaktion ermöglicht.
Die Interaktion via elektrostatischer Interaktionen ist ein weit verbreiteter
Mechanismus für die Membranassoziation von peripheren Membranproteinen

Ergebnisse
- 69 -
(Mulgrew-Nesbitt et al., 2006). PmtA ist ein lösliches Protein, das keinen Lipidanker
besitzt und dessen Substrat Bestandteil einer Membran ist. Möglicherweise wird
PmtA durch elektrostatische Interaktionen an die Membran gebunden. In silico-
Vorhersagen für PmtA mit dem Programm Proteincalculator zeigen, dass PmtA eine
Nettoladung von +7.4 bei einem physiologischen pH besitzt. Betrachtet man die
theoretische Oberflächenladung eines PmtA-Homologiemodells (erstellt mit I-
TASSER, Zhang, 2008), so fällt auf, dass es zwei stark positiv geladene Bereiche
gibt (Abbildung 24). Daher kann vermutet werden, dass PmtA mittels
elektrostatischer Interaktionen mit negativ geladenen Membranlipiden interagieren
könnte.
Abbildung 24: Die theoretische Oberflächenladung von PtmA wurde auf der Basis eines Homologiemodells von PmtA (I-TASSER, Zhang, 2008) mit Pymol (www.pymol.org) kalkuliert und visualisiert. Es fallen zwei stark positiv geladene Bereiche (blau) auf der PmtA-Oberfläche auf. Rot: negativ geladene Oberfläche; Weiß: ungeladene Oberfläche.
Um diese Hypothese zu prüfen, wurde zunächst die enzymatische Aktivität von PmtA
bei steigender ionischer Stärke in vitro untersucht. Dafür wurde die NaCl-
Konzentration in den in vitro-Ansätzen schrittweise erhöht. Als Modellmembran
diente eine künstliche Membran aus PE mit anionischen Lipiden, um eine
elektrostatische Interaktion zu ermöglichen. Beispielhaft ist in Abbildung 25 das
Ergebnis für PE:PG gezeigt. Anhand des Endproduktes PC ist deutlich zu erkennen,
dass die enzymatische Aktivität von PmtA bei steigender NaCl-Konzentration
abnimmt. PmtA ist bis zu einer NaCl-Konzentration von 0.1 M aktiv. Erhöht man die
Konzentration von NaCl auf 0.25 M, so sind nur noch Spuren an PC zu beobachten.
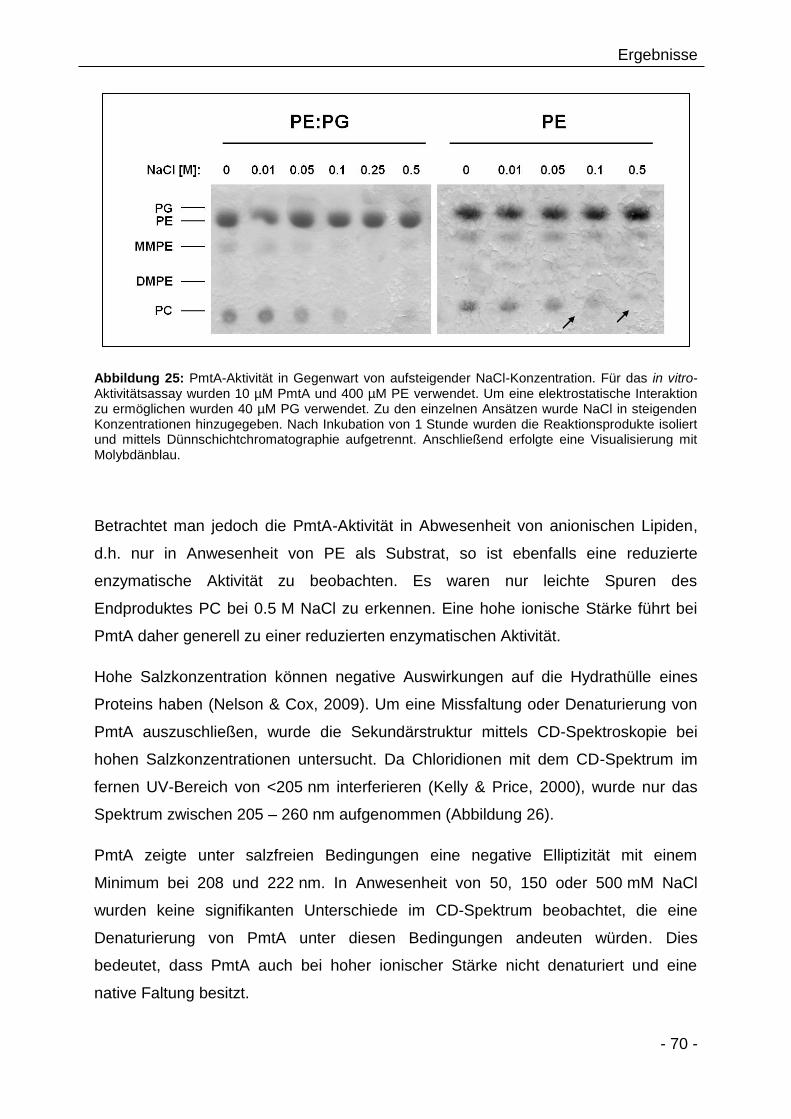
Ergebnisse
- 70 -
Abbildung 25: PmtA-Aktivität in Gegenwart von aufsteigender NaCl-Konzentration. Für das in vitro-Aktivitätsassay wurden 10 µM PmtA und 400 µM PE verwendet. Um eine elektrostatische Interaktion zu ermöglichen wurden 40 µM PG verwendet. Zu den einzelnen Ansätzen wurde NaCl in steigenden Konzentrationen hinzugegeben. Nach Inkubation von 1 Stunde wurden die Reaktionsprodukte isoliert und mittels Dünnschichtchromatographie aufgetrennt. Anschließend erfolgte eine Visualisierung mit Molybdänblau.
Betrachtet man jedoch die PmtA-Aktivität in Abwesenheit von anionischen Lipiden,
d.h. nur in Anwesenheit von PE als Substrat, so ist ebenfalls eine reduzierte
enzymatische Aktivität zu beobachten. Es waren nur leichte Spuren des
Endproduktes PC bei 0.5 M NaCl zu erkennen. Eine hohe ionische Stärke führt bei
PmtA daher generell zu einer reduzierten enzymatischen Aktivität.
Hohe Salzkonzentration können negative Auswirkungen auf die Hydrathülle eines
Proteins haben (Nelson & Cox, 2009). Um eine Missfaltung oder Denaturierung von
PmtA auszuschließen, wurde die Sekundärstruktur mittels CD-Spektroskopie bei
hohen Salzkonzentrationen untersucht. Da Chloridionen mit dem CD-Spektrum im
fernen UV-Bereich von <205 nm interferieren (Kelly & Price, 2000), wurde nur das
Spektrum zwischen 205 – 260 nm aufgenommen (Abbildung 26).
PmtA zeigte unter salzfreien Bedingungen eine negative Elliptizität mit einem
Minimum bei 208 und 222 nm. In Anwesenheit von 50, 150 oder 500 mM NaCl
wurden keine signifikanten Unterschiede im CD-Spektrum beobachtet, die eine
Denaturierung von PmtA unter diesen Bedingungen andeuten würden. Dies
bedeutet, dass PmtA auch bei hoher ionischer Stärke nicht denaturiert und eine
native Faltung besitzt.

Ergebnisse
- 71 -
Abbildung 26: CD-Spektrum von PmtA in An- und Abwesenheit verschiedener NaCl-Konzentrationen. Für ein CD-Spektrum wurden 7.5 µM PmtA verwendet. Die Spektren wurden bei RT aufgezeichnet und 10-fach akkumuliert. RT: Raumtemperatur.
Die vorangegangenen Experimente zeigen somit, dass hohe ionische Stärke die
PmtA-Aktivität reduziert, jedoch hat die hohe ionische Stärke keinen Einfluss auf die
PmtA-Faltung. Um zu überprüfen, ob die Lipidbindung von PmtA durch hohe ionische
Stärke beeinflusst wird, wurde ein „Protein-Lipid-Overlay-Assay“ bei hoher ionischer
Stärke durchgeführt (Dowler et al., 2002). Dafür wurden Lipide auf einer Membran
fixiert und diese mit PmtA inkubiert. PmtA, das an Lipide gebunden vorlag, wurde
immunochemisch detektiert. Da Lipide nicht wasserlöslich sind, wurden diese in
einem Chloroform-Methanol-Wasser – Gemisch gelöst. Als Kontrolle wurde auch das
verwendete Lösungsmittelgemisch auf die Membran aufgetragen, um chloroform-
bzw. methanolbedingte Membranbeschädigungen auszuschließen (Abbildung 27).
Weiterhin wurde PmtA direkt auf die Membran aufgetragen, um eine Beeinflussung
der immunochemischen Detektion durch hohe Salzkonzentrationen auszuschließen.
Eine Bindung von PmtA an PE oder an isolierte Lipide aus A. tumefaciens wurde bis
zu einer Konzentration von 150 mM NaCl deutlich nachgewiesen. Im Gegensatz
dazu unterschied sich die Bindefähigkeit von PmtA an die anionischen Lipide PG, CL
und PS. Bei einer NaCl Konzentration von 50 mM wurden noch signifikante Mengen

Ergebnisse
- 72 -
an PmtA detektiert. Erhöhte sich die ionische Stärke jedoch auf 150 mM NaCl, so
waren nur noch sehr geringe PmtA-Mengen detektierbar. Eine Konzentration von
500 mM NaCl verdrängte PmtA vollständig. Dies war auch für PE bzw.
A. tumefaciens-Lipide zu beobachten. Der enzymatische Aktivitätsverlust von PmtA
bei hoher ionischer Stärke ist daher vermutlich auf einen Verlust der Lipidbindung
gegenüber anionischen Lipiden zurückzuführen. Es ist daher wahrscheinlich, dass
die Bindung von PmtA an anionische Lipide auf einer elektrostatischen Interaktion
beruht.
Abbildung 27: Lipid-Bindung von PmtA in Gegenwart aufsteigender NaCl-Konzentrationen. Protein-Lipid-Overlay-Assay mit steigenden NaCl-Konzentrationen. Auf die Membran wurden gleiche Mengen an Lipiden fixiert. Die Membranen wurden mit 10 µM PmtA inkubiert und das gebundene Protein mit einem Anti-His Antikörper (Qiagen) detektiert.

Ergebnisse
- 73 -
3.2.6 Etablierung von verschiedenen Methoden zur quantitativen
und qualitativen Analyse der Lipid-Bindeeigenschaften von
PmtA
Um ein besseres Verständnis für die PmtA-Lipid-Interaktion zu bekommen, mussten
zunächst geeignete Methoden für eine sowohl qualitative, als auch quantitative
Analyse etabliert werden. Es wurde die PmtA-Lipid-Interaktion zunächst qualitativ
durch limitierte Proteolyse untersucht, um eine erste Vorstellung von dieser
Interaktion und der zugrunde liegenden Mechanismen zu bekommen. Um ein
weitaus detaillierteres Verständnis der Mechanismen der PmtA-Lipid-Interaktion zu
bekommen, wurden spektroskopische Verfahren zur Quantifizierung und Analyse
dieser Interaktion etabliert und angewendet.
3.2.7 Limitierte Proteolyse von PmtA in Gegenwart von
Modellmembranen
Die limitierte Proteolyse ist eine Methode, um den Einfluss einer Protein-
Substratbindung auf die Proteinstruktur zu untersuchen (Fontana et al., 2004). Dabei
wird der proteolytische Abbau eines Proteins in An- und Abwesenheit eines
Substrates analysiert. Der Proteaseverdau führt zu einer Fragmentierung des
Proteins, die gelelektrophoretisch aufgetrennt und anschließend visualisiert werden
kann. Durch eine Substratbindung des zu untersuchenden Proteins ist es zum einen
möglich, dass eine Proteaseschnittstelle blockiert wird. Zum anderen kann eine
Substratbindung eine Konformationsänderung bewirken, durch die eine
Proteaseschnittstelle entweder nicht mehr zugänglich ist oder dadurch zugänglich
wird (Abbildung 28). Da eine Proteolyse durch Proteasen sehr schnell erfolgt, wird
ein solches Experiment unter limitierten Bedingungen, wie z.B. eine Inkubation auf
Eis oder eine geringe Inkubationszeit, durchgeführt.
Um den Einfluss einer Phospholipidbindung auf die Proteolyse von PmtA zu
untersuchen wurde PmtA mit und ohne Lipid auf Eis mit Chymotrypsin oder Trypsin
verdaut. Zu definierten Zeitpunkten wurden Proben entnommen. Anschließend
wurden die Proben gelelektrophoretisch aufgetrennt und das Fragmentierungs-
muster analysiert. Da keine Unterschiede in der Fragmentierung durch Chymotrypsin

Ergebnisse
- 74 -
oder Trypsin erkennbar waren, werden nur die Ergebnisse für Chymotrypsin
präsentiert.
Abbildung 28: Das Prinzip der limitierten Proteolyse. Rote Punkte kennzeichnen mögliche Proteaseschnittstellen in PmtA. Das frei vorliegende Enzym bietet eventuell mehr zugängliche Proteaseschnittstellen (A.) als PmtA, das an eine Membran/Liposom gebunden ist (B.).
In Abwesenheit von Lipiden zeigten sich schon nach 2 min erste Abbauprodukte von
PmtA (~24 kDA). Nach 7 min sind nur noch sehr geringe Mengen an
Volllängenprotein vorhanden. Der Abbau erfolgt über eine Zwischenstufe
(ca. 20 kDa) zu einem Fragment mit einer ungefähren Größe von 18 kDa. Dieses
Fragment scheint stabil zu sein, da auch nach 30 min kein weiterer Abbau zu
kleineren Fragmenten erfolgte (Abbildung 29). Eine proteaseunabhängige
Degradation von PmtA in einem parallelen Ansatz wurde nicht beobachtet. In
Anwesenheit von PE schien PmtA stabiler gegenüber dem proteolytischen Abbau zu
sein. Unter diesen Bedingungen wurden erste deutliche Abbauprodukte nach 5 min
beobachtet. Nach 15 min waren im Gegensatz zum Ansatz ohne Lipid noch deutliche
Mengen an Volllängenprotein (~24 kDa) vorhanden. Erst nach 30 min lag PmtA
größtenteils als 18 kDa großes Abbaufragment vor. Ein Vergleich der Stabilität von
PmtA unter beiden Bedingungen zeigt, dass PmtA stabiler in Gegenwart von PE als
in Abwesenheit von Lipid ist. Eine noch höhere Stabilität zeigte sich mit PG oder PI.
Bei Anwesenheit dieser Lipide war kaum ein Abbau von PmtA zu erkennen. Auch
nach 30 min waren deutliche Mengen an Volllängenprotein vorhanden. Dies
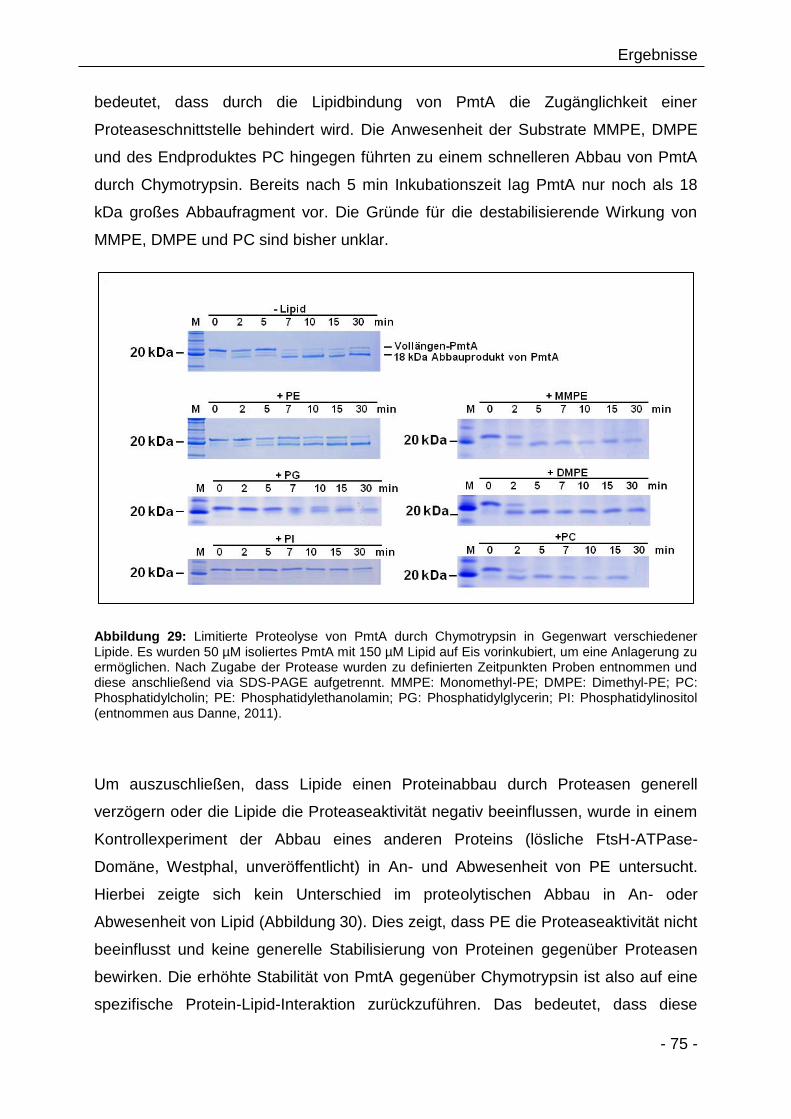
Ergebnisse
- 75 -
bedeutet, dass durch die Lipidbindung von PmtA die Zugänglichkeit einer
Proteaseschnittstelle behindert wird. Die Anwesenheit der Substrate MMPE, DMPE
und des Endproduktes PC hingegen führten zu einem schnelleren Abbau von PmtA
durch Chymotrypsin. Bereits nach 5 min Inkubationszeit lag PmtA nur noch als 18
kDa großes Abbaufragment vor. Die Gründe für die destabilisierende Wirkung von
MMPE, DMPE und PC sind bisher unklar.
Abbildung 29: Limitierte Proteolyse von PmtA durch Chymotrypsin in Gegenwart verschiedener Lipide. Es wurden 50 µM isoliertes PmtA mit 150 µM Lipid auf Eis vorinkubiert, um eine Anlagerung zu ermöglichen. Nach Zugabe der Protease wurden zu definierten Zeitpunkten Proben entnommen und diese anschließend via SDS-PAGE aufgetrennt. MMPE: Monomethyl-PE; DMPE: Dimethyl-PE; PC: Phosphatidylcholin; PE: Phosphatidylethanolamin; PG: Phosphatidylglycerin; PI: Phosphatidylinositol (entnommen aus Danne, 2011).
Um auszuschließen, dass Lipide einen Proteinabbau durch Proteasen generell
verzögern oder die Lipide die Proteaseaktivität negativ beeinflussen, wurde in einem
Kontrollexperiment der Abbau eines anderen Proteins (lösliche FtsH-ATPase-
Domäne, Westphal, unveröffentlicht) in An- und Abwesenheit von PE untersucht.
Hierbei zeigte sich kein Unterschied im proteolytischen Abbau in An- oder
Abwesenheit von Lipid (Abbildung 30). Dies zeigt, dass PE die Proteaseaktivität nicht
beeinflusst und keine generelle Stabilisierung von Proteinen gegenüber Proteasen
bewirken. Die erhöhte Stabilität von PmtA gegenüber Chymotrypsin ist also auf eine
spezifische Protein-Lipid-Interaktion zurückzuführen. Das bedeutet, dass diese

Ergebnisse
- 76 -
Interaktion entweder zu einer Blockierung der Schnittstellen in PmtA führt oder eine
Lipidbindung eine Konformationsänderung der PmtA-Struktur induziert. Durch diese
Konformationsänderung wird eine Proteaseschnittstelle nicht mehr zugänglich, was
wiederum die PmtA-Stabilität erhöht.
Abbildung 30: Negativkontrolle mit der löslichen ATPase-Domäne von Thermotoga maritimaFtsH. Es wurden 50 µM isoliertes FtsH mit 150 µM Lipid auf Eis vorinkubiert. Nach Zugabe der Protease wurden zu definierten Zeitpunkten Proben entnommen und die Proteine anschließend via SDS-PAGE aufgetrennt (entnommen aus Danne, 2011).
3.2.8 Massenspektrometrische Analyse der PmtA-Fragmente
Im vorherigen Abschnitt wurde gezeigt, dass der proteolytische Abbau von PmtA bis
zu einem stabilem 18 kDa großen Fragment erfolgt. Da eine Spaltung von PmtA an
einer zentralen Position in der Peptidkette zu weitaus kleineren Fragmenten geführt
hätte, wird PmtA vermutlich am N- oder C-Terminus geschnitten. Um dies zu klären,
wurden Proben von dem Volllängenprotein und dem 18 kDa-Fragment aus dem
SDS-Gel ausgestochen, tryptisch verdaut und mittels Massenspektrometrie
analysiert (Abbildung 31). Für das Volllängenprotein wurde ein Peptidfragment
identifiziert, das im N-terminalen Bereich von PmtA liegt. Dieses Peptidfragment
konnte für das 18 kDa-Fragment nicht nachgewiesen werden. Jedoch wurde für das
18 kDa-Fragment ein Peptid identifiziert, das Bestandteil des C-Terminus von PmtA
ist. PmtA besitzt 15 vorhergesagte Schnittstellen für Chymotrypsin. Eine putative
Proteaseschnittstelle befindet sich an Position 38 (RFFKG). Ein Schnitt an dieser
Stelle würde ein 18.3 kDa und ein 6 kDa großes Fragment erzeugen. Diese

Ergebnisse
- 77 -
theoretischen Peptidgrößen stimmen mit den experimentell beobachteten
Fragmentgrößen überein.
Abbildung 31: Massenspektrometrische Analyse der PmtA-Fragmente. (A.) SDS-PAGE nach limitierter Proteolyse von PmtA in Gegenwart von PE. Es wurden aus dem Gel Proben ausgestochen und mittels MALDI-TOF analysiert. (B.) MS/MS-Spektren der ausgewählten Proben. Markiert sind die Peaks der identifizierten Peptide. (C.) Aminosäuresequenz von PmtA. Das identifizierte Peptid für das Vollängenprotein ist in Pink markiert. Es umfasst den N-terminalen His-Tag (Aminosäuren 2-17). Türkis markiert ist das identifizierte Peptid für das 18 kDa Abbauprodukt (Aminosäure 205-216) (entnommen aus Danne, 2011).
Die Degradation von PmtA erfolgt somit sehr wahrscheinlich N-terminal. Daher ist es
wahrscheinlich, dass der N-Terminus von PmtA durch eine Protein-Lipid-Interaktion
direkt oder indirekt beeinflusst wird. Eine mögliche Erklärung für die erhöhte Stabilität
gegenüber proteolytischem Abbau wäre, dass PmtA in Folge einer Protein-Lipid-
Interaktion eine Konformationsänderung vollzieht und dadurch die

Ergebnisse
- 78 -
Proteaseschnittstelle verdeckt wird. Um dies näher zu untersuchen, wurde die
Konformation von PmtA durch spektroskopische Methoden in An- und Abwesenheit
verschiedener Lipide analysiert.
3.2.9 CD-spektroskopische Analyse von PmtA in Gegenwart von
verschiedenen Lipiden
Die CD-Spektroskopie ist eine Methode zur Analyse der Sekundärstruktur von
Proteinen (Abschnitt 3.1.3). Mit Hilfe der CD-Spektroskopie können auch Einflüsse
von beispielsweise Substraten oder Kofaktoren auf die Sekundärstruktur von
Proteinen untersucht werden (Kelly & Price, 2000). Eine Bedingung dafür ist jedoch,
dass die Additive kein eigenes CD-Signal besitzen, welches das CD-Signal des
Proteins verfälscht. Phospholipide in Liposomenform weisen kein signifikantes CD-
Signal auf. Der Einfluss von Lipiden auf Protein-Sekundärstrukturelemente oder
ganze Proteine wurde bereits in vielen Arbeiten mittels CD-Spektroskopie untersucht
(Jo et al., 2000, Vallée et al., 2001; Constantinescu & Lafleur, 2004; Thomas et al.,
2006; Shih et al., 2011). Für PmtA ist bereits bekannt, dass für die SAM-Bindung
eine Substratlipidbindung essentiell ist (Aktas & Narberhaus, 2009). Möglicherweise
führt eine Substratlipidbindung zu einer Konformationsänderung von PmtA, sodass
eine SAM-Bindung begünstigt wird. Eine Möglichkeit, um solch eine Änderung der
PmtA-Konformation durch Lipide nachzuweisen, ist die CD-Spektroskopie.
Als Kontrolle diente ChoX (Aktas et al., 2011b) und HO3 (Gisk et al., 2010). ChoX ist
ein periplasmatisches Cholin-Bindeprotein und HO3 eine cytosolische
Hämoxygenase. Beide Proteine interagieren nicht mit der Membran oder mit
Phospholipiden. Die Zugabe von verschiedenen Phospholipiden hatte daher keinen
Einfluss auf das CD-Spektrum und somit auch nicht auf die Sekundärstruktur dieser
Proteine (Abbildung 32).
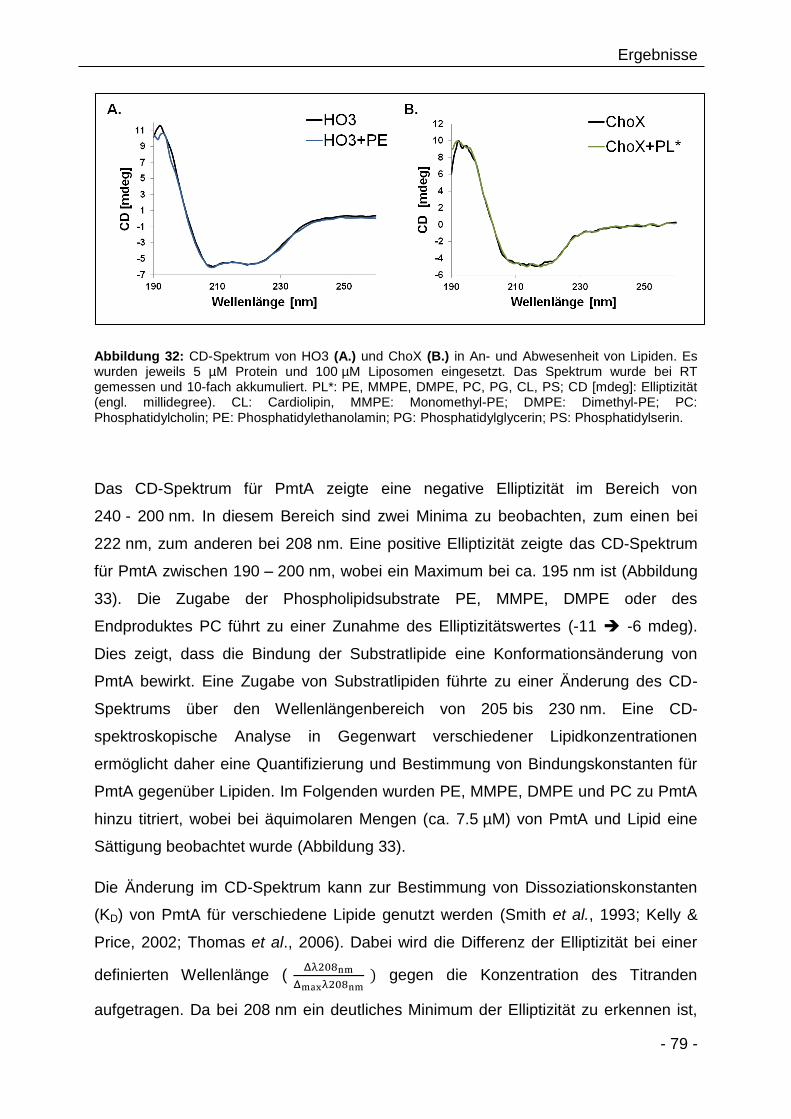
Ergebnisse
- 79 -
Abbildung 32: CD-Spektrum von HO3 (A.) und ChoX (B.) in An- und Abwesenheit von Lipiden. Es wurden jeweils 5 µM Protein und 100 µM Liposomen eingesetzt. Das Spektrum wurde bei RT gemessen und 10-fach akkumuliert. PL*: PE, MMPE, DMPE, PC, PG, CL, PS; CD [mdeg]: Elliptizität (engl. millidegree). CL: Cardiolipin, MMPE: Monomethyl-PE; DMPE: Dimethyl-PE; PC: Phosphatidylcholin; PE: Phosphatidylethanolamin; PG: Phosphatidylglycerin; PS: Phosphatidylserin.
Das CD-Spektrum für PmtA zeigte eine negative Elliptizität im Bereich von
240 - 200 nm. In diesem Bereich sind zwei Minima zu beobachten, zum einen bei
222 nm, zum anderen bei 208 nm. Eine positive Elliptizität zeigte das CD-Spektrum
für PmtA zwischen 190 – 200 nm, wobei ein Maximum bei ca. 195 nm ist (Abbildung
33). Die Zugabe der Phospholipidsubstrate PE, MMPE, DMPE oder des
Endproduktes PC führt zu einer Zunahme des Elliptizitätswertes (-11 -6 mdeg).
Dies zeigt, dass die Bindung der Substratlipide eine Konformationsänderung von
PmtA bewirkt. Eine Zugabe von Substratlipiden führte zu einer Änderung des CD-
Spektrums über den Wellenlängenbereich von 205 bis 230 nm. Eine CD-
spektroskopische Analyse in Gegenwart verschiedener Lipidkonzentrationen
ermöglicht daher eine Quantifizierung und Bestimmung von Bindungskonstanten für
PmtA gegenüber Lipiden. Im Folgenden wurden PE, MMPE, DMPE und PC zu PmtA
hinzu titriert, wobei bei äquimolaren Mengen (ca. 7.5 µM) von PmtA und Lipid eine
Sättigung beobachtet wurde (Abbildung 33).
Die Änderung im CD-Spektrum kann zur Bestimmung von Dissoziationskonstanten
(KD) von PmtA für verschiedene Lipide genutzt werden (Smith et al., 1993; Kelly &
Price, 2002; Thomas et al., 2006). Dabei wird die Differenz der Elliptizität bei einer
definierten Wellenlänge ( gegen die Konzentration des Titranden
aufgetragen. Da bei 208 nm ein deutliches Minimum der Elliptizität zu erkennen ist,
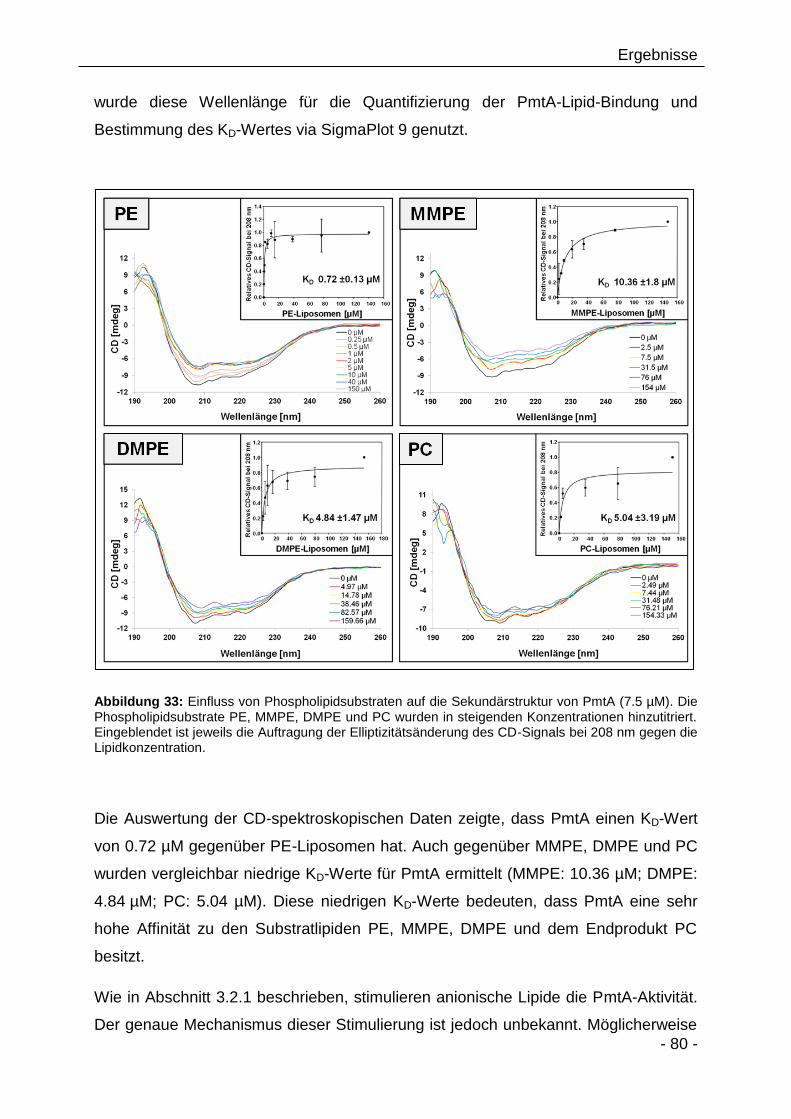
Ergebnisse
- 80 -
wurde diese Wellenlänge für die Quantifizierung der PmtA-Lipid-Bindung und
Bestimmung des KD-Wertes via SigmaPlot 9 genutzt.
Abbildung 33: Einfluss von Phospholipidsubstraten auf die Sekundärstruktur von PmtA (7.5 µM). Die Phospholipidsubstrate PE, MMPE, DMPE und PC wurden in steigenden Konzentrationen hinzutitriert. Eingeblendet ist jeweils die Auftragung der Elliptizitätsänderung des CD-Signals bei 208 nm gegen die Lipidkonzentration.
Die Auswertung der CD-spektroskopischen Daten zeigte, dass PmtA einen KD-Wert
von 0.72 µM gegenüber PE-Liposomen hat. Auch gegenüber MMPE, DMPE und PC
wurden vergleichbar niedrige KD-Werte für PmtA ermittelt (MMPE: 10.36 µM; DMPE:
4.84 µM; PC: 5.04 µM). Diese niedrigen KD-Werte bedeuten, dass PmtA eine sehr
hohe Affinität zu den Substratlipiden PE, MMPE, DMPE und dem Endprodukt PC
besitzt.
Wie in Abschnitt 3.2.1 beschrieben, stimulieren anionische Lipide die PmtA-Aktivität.
Der genaue Mechanismus dieser Stimulierung ist jedoch unbekannt. Möglicherweise
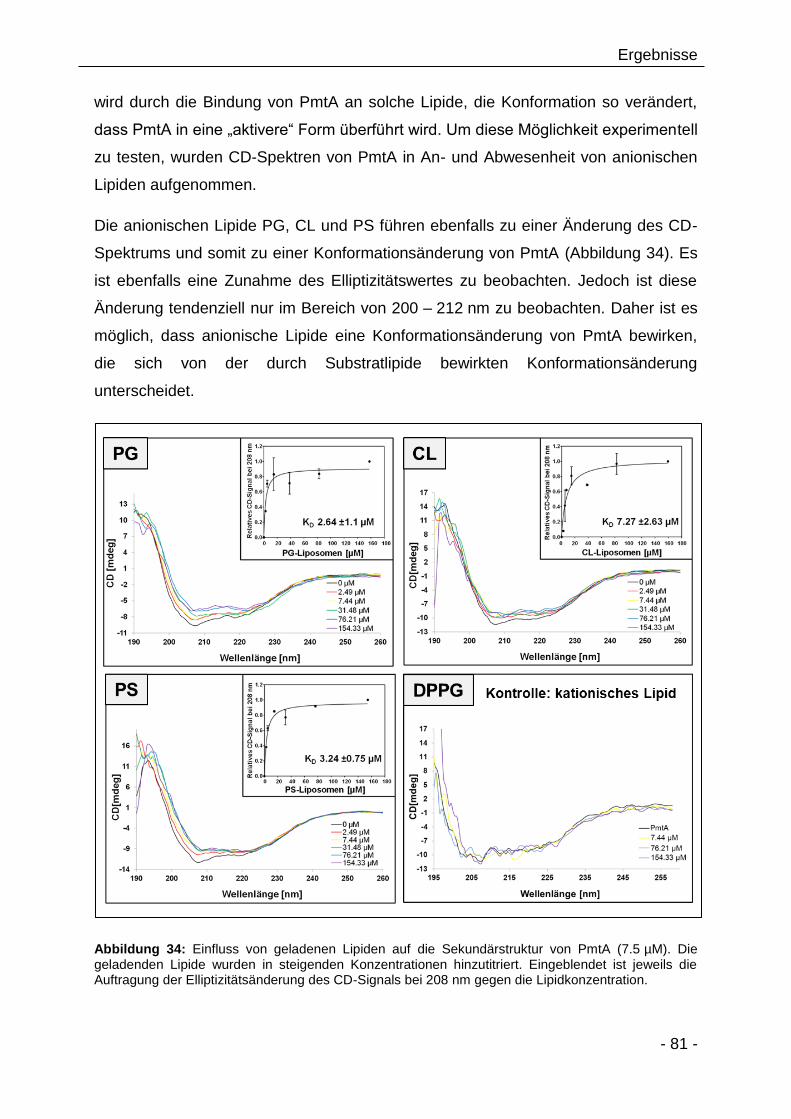
Ergebnisse
- 81 -
wird durch die Bindung von PmtA an solche Lipide, die Konformation so verändert,
dass PmtA in eine „aktivere“ Form überführt wird. Um diese Möglichkeit experimentell
zu testen, wurden CD-Spektren von PmtA in An- und Abwesenheit von anionischen
Lipiden aufgenommen.
Die anionischen Lipide PG, CL und PS führen ebenfalls zu einer Änderung des CD-
Spektrums und somit zu einer Konformationsänderung von PmtA (Abbildung 34). Es
ist ebenfalls eine Zunahme des Elliptizitätswertes zu beobachten. Jedoch ist diese
Änderung tendenziell nur im Bereich von 200 – 212 nm zu beobachten. Daher ist es
möglich, dass anionische Lipide eine Konformationsänderung von PmtA bewirken,
die sich von der durch Substratlipide bewirkten Konformationsänderung
unterscheidet.
Abbildung 34: Einfluss von geladenen Lipiden auf die Sekundärstruktur von PmtA (7.5 µM). Die geladenden Lipide wurden in steigenden Konzentrationen hinzutitriert. Eingeblendet ist jeweils die Auftragung der Elliptizitätsänderung des CD-Signals bei 208 nm gegen die Lipidkonzentration.

Ergebnisse
- 82 -
Weiterhin wurde die Bindungsaffinität von PmtA zu anionischen Lipiden mit Hilfe der
Elliptizitätsdifferenz untersucht. Die KD-Werte für PG-, PS- und CL-Liposomen
wurden analog wie für die Substratlipide bestimmt. Es zeigte sich, dass die Affinität
von PmtA zu anionischen Lipide vergleichbar mit der Affinität zu Substratlipiden ist.
Die ermittelten KD-Werte liegen in einem mikromolaren Bereich (PG: 2.64 µM;
CL: 7.27 µM; PS: 3.24 µM). Die Zugabe des kationischen Lipides DPPG hatte
hingegen keinen Einfluss auf das CD-Spektrum und somit auch nicht auf
Konformation von PmtA (Abbildung 34).
Durch CD-spektroskopische Analysen konnte somit gezeigt werden, dass sowohl
Phospholipidsubstrate (PE, MMPE, DMPE oder PC), als auch anionische Lipide (PG,
PS oder CL) eine Konformationsänderung von PmtA bewirken. Diese Ergebnisse
bestätigen somit die Vermutung, dass die SAM-Bindung von PmtA durch eine
Substrat-abhängige Konformationsänderung ermöglicht wird. Die Tatsache, dass
auch anionische Lipide eine Konformationsänderung von PmtA bewirken, obwohl
diese keine SAM-Bindung ermöglichen, wirft neue Fragen hinsichtlich der Bedeutung
anionischer Lipide für die PmtA-Aktivität auf.
3.2.10 Analyse der Lipidbindeeigenschaften von PmtA mittels
Fluorescein-Phosphatidylethanolamin
Als eine weitere Methode zur Bestimmung der PmtA-Lipid-Bindeeigenschaften wurde
ein auf Fluorescein-Phosphatidylethanolamin (FPE) basierende Bindestudie etabliert
und Bindeaffinitäten von PmtA gegenüber Modellmembranen bestimmt. Bei dieser
Methode wurde, im Gegensatz zur CD-Spektroskopie, PmtA zu einer definierten
Menge Liposomen hinzu titriert. Die verschiedenen Liposomen bestanden zu einem
0.5 % molaren Anteil aus FPE.
FPE ist ein PE-Derivat, an dessen Kopfgruppe eine Fluoresceingruppe geknüpft ist
(Abbildung 35). Diese Fluoresceingruppe fluoresziert nach Anregung mit einer
Wellenlänge bei 490 – 495 nm. Die Fluoreszenzeigenschaften ändern sich, abhängig
von einer Protonierung des Xanthenrings. Da FPE ein Lipid ist, kann es als eine Art
Sonde in eine Membran integriert werden, um mit Hilfe der Fluorescein-Kopfgruppe

Ergebnisse
- 83 -
Änderungen des elektrostatischen Potentials von geladenen Membranoberflächen zu
messen (Wall et al., 1995).
Abbildung 35: Strukturformel des Fluorescein-Phosphatidylethanolamin.
Interagiert ein geladenes Molekül/Protein mit einer Membran, so ändert sich das
elektrostatische Potential und dadurch die Fluoreszenzintensität von FPE. Die
Fluoreszenz von FPE wurde bei 490 nm angeregt und das Fluoreszenzspektrum im
spektralen Bereich von 500 – 600 nm aufgezeichnet. Ein Nachteil dieser Methode ist,
dass nur Membranen untersucht werden können, die ein Membranpotential besitzen.
PE, MMPE, DMPE oder PC sind zwitterionische Lipide und daher neutral. Eine
Titration von PmtA hatte daher wie erwartet keinen Einfluss auf die Fluoreszenz. Im
Folgenden wurden deshalb nur geladene Membranen verwendet. Dazu zählen
rekonstituierte Membranen aus isolierten A. tumefaciens-Lipiden (At) und E. coli-
Lipiden (Ec), sowie künstliche Membranen aus den anionischen Lipiden PG, CL und
PS.
Als Negativkontrolle diente das periplasmatische Cholin-Bindeprotein ChoX
(Aktas et al., 2011b). Eine Zugabe von ChoX in 10-fachem Überschuss hatte keinen
signifikanten Einfluss auf die Fluoreszenzintensität (Abbildung 36). Desweiteren
konnte ausgeschlossen werden, dass PmtA im Wellenlängenbereich von
500 - 600 nm eine Eigenfluoreszenz besitzt.

Ergebnisse
- 84 -
Abbildung 36: FPE-basiertes Membranbindeassay mit ChoX. FPE wurde mit 490 nm angeregt und das Emissionsspektrum zwischen 500 und 600 nm aufgezeichnet. Es wurden 10 µM Liposomen und ein 10-facher Überschuss an ChoX verwendet. Für die PmtA-Kontrolle wurde PmtA in steigenden Konzentrationen in Tris-Puffer titriert. At: Gesamtlipidextrakt aus A. tumefaciens, CL: Cardiolipin; Ec: Gesamtlipidextrakt aus E. coli; PG: Phosphatidylglycerin; PS: Phosphatidylserin
Eine Titration von PmtA zu einer FPE-markierten rekonstituierten A. tumefaciens
Membran führte zu einer deutlichen Erhöhung der Fluoreszenzintensität (Abbildung
37). Ein Maximum war dabei bei 525 nm zu erkennen. Die Fluoreszenzintensität
nahm um ca. 87 % zu. Im Vergleich dazu änderte sich die Fluoreszenzintensität bei
einer E. coli Membran um ca. 62 %. Für die Modellmembranen aus PG, CL und PS
wurde bei 525 nm eine Zunahme in der Fluoreszenzintensität um jeweils 32 %,
137 % und 95 % beobachtet.

Ergebnisse
- 85 -
Abbildung 37: FPE-basiertes Membranbindeassay mit PmtA. Es wurden 10 µM Liposomen verwendet. FPE wurde mit 490 nm angeregt und das Emissionsspektrum zwischen 500 - 600 nm aufgezeichnet. Zu den Liposomen wurde PmtA in steigenden Konzentrationen hinzutitriert. At: Gesamtlipidextrakt aus A. tumefaciens, CL: Cardiolipin; Ec: Gesamtlipidextrakt aus E. coli; PG:
Phosphatidylglycerin; PS: Phosphatidylserin.
Zur Bestimmung des KD-Wertes von PmtA zu der jeweiligen Membran wurde die
Änderung in der Fluoreszenzintensität bei 525 nm genutzt. Es wurde für PmtA
gegenüber einer rekonstituierten A. tumefaciens Membran ein KD-Wert von 3.20 µM
ermittelt. Gegenüber einer E. coli Membran beträgt der KD-Wert 1.6 µM. Auch
gegenüber PG (3.5 µM), CL (4.73 µM) und PS (5.72 µM) besitzt PmtA einen KD-Wert

Ergebnisse
- 86 -
in einem vergleichbaren mikromolaren Bereich. Die Ergebnisse dieses Abschnittes
bestätigen somit die zuvor bestimmte hohe Affinität von PmtA gegenüber
anionischen Lipiden.
3.2.11 Tryptophanfluoreszenzspektroskopie zur Analyse der
Lipidinteraktion von PmtA
Eine weitere Methode zur Analyse der PmtA-Lipid-Bindung ist die
Tryptophanfluoreszenzspektroskopie. Bei dieser Methode wird die Fluoreszenz der
Aminosäure Tryptophan zur Bestimmung von Protein-Liganden Affinitäten genutzt.
Da Tryptophan sehr sensibel gegenüber der Polarität seiner Umgebung ist, kann es
auch für die Bestimmung von Affinitäten von Proteinen zu Phospholipiden genutzt
werden (Hammel et al., 2001; Constantinescu & Lafleur, 2004; Grobenko et al., 2007;
Kraft et al., 2009). Ein weiterer Vorteil dieser Methode ist, dass eine Änderung der
intrinsischen Tryptophanfluoreszenz von Proteinen oftmals eine Konformations-
änderung als Folge einer Substratbindung bedeutet. Desweiteren korreliert bei einer
Protein-Membran-Interaktion die Verschiebung des Fluoreszenzmaximums mit der
Tiefe der Insertion des Tryptophans in die Membran. Je größer die Verschiebung ist,
desto tiefer inseriert das Tryptophan in die Membran (Ren et al., 1997).
PmtA besitzt nur ein Tryptophan, das am C-Terminus an Position 192 lokalisiert ist.
Die Tryptophanfluoreszenz wurde mit 295 nm angeregt und das Fluoreszenz-
spektrum im Bereich von 300 – 400 nm aufgezeichnet. Für diese Methode wurde
zuvor ausgeschlossen, dass die verwendeten Lipide fluoreszieren. Die Lipidzugabe
störte die Messung somit nicht. Das Fluoreszenzspektrum von PmtA wies ein
Maximum bei ca. 340 nm auf (Abbildung 38). Titriert man PE-Liposomen hinzu, so
wurde die Fluoreszenzintensität bei einer Sättigung von PmtA mit Lipiden um 66 %
verringert (Abbildung 38). Auch die weiteren Phospholipidsubstrate führten zu einer
vergleichbaren Änderung der Fluoreszenzintensität (MMPE: 70 %; DMPE: 61 %; PC:
71 %). Die Änderung der intrinsischen Tryptophanfluoreszenz von PmtA nach
Lipidzugabe bestätigt somit erneut, dass eine Interaktion von PmtA mit den Lipiden
PE, MMPE, DMPE und PC zu einer Konformationsänderung führt.
Anhand der Änderung der Fluoreszenzintensität wurden Dissoziationskonstanten
(KD) für die PmtA-Lipid-Interaktion ermittelt. Mittels der Fluoreszenzintensität wurde

Ergebnisse
- 87 -
für PmtA zu PE-Liposomen ein KD-Wert von 1.71 µM ermittelt. Zu MMPE, DMPE und
PC besitzt PmtA vergleichbare KD-Werte mit 1.19 µM, 0.84 µM und 1.57 µM. Alle
ermittelten KD-Werte liegen in einem vergleichbaren mikromolaren Bereich, ähnlich
zu den KD-Werten, die anhand der zuvor beschriebenen Methoden bestimmt wurden.
Abbildung 38: Tryptophanfluoreszenz von PmtA in Gegenwart von Substratlipiden und dem Endprodukt PC. Für die Tryptophanfluoreszenz wurden 10 µM PmtA mit steigender Konzentration von Liposomen inkubiert. Die Fluoreszenz wurde bei 290 nm angeregt und das Spektrum zwischen 300 - 400 nm aufgezeichnet. Für die Bestimmung der KD-Werte wurde die Abnahme der Fluoreszenz verwendet (F/F0).
Neben der Fluoreszenintensitätsänderung wurde auch eine geringfügige
Blauverschiebung des Fluoreszenzmaximums nach Lipidzugabe beobachtet
(ca. 4 nm). Dies könnte für eine nur geringfügige Insertion des Tryptophanrestes in
die Membran sprechen. Diese Verschiebung war jedoch nicht signifikant genug, um
diese für weitere Analysen oder Bestimmung von Bindeaffinität von PmtA zu Lipiden
zu nutzen.

Ergebnisse
- 88 -
Die Tryptophanfluoreszenz von PmtA wurde weiterhin dazu genutzt, um die
Interaktion mit PG, CL und PS zu untersuchen (Abbildung 39). Es zeigte sich hierbei,
dass diese Lipide ebenfalls eine Verringerung der Fluoreszenzintensität von PmtA
bewirken. PG führte dabei zu einer Verringerung der Fluoreszenzintensität um 66 %,
CL um 72 % und PS um 50 %, wenn eine Sättigung von PmtA mit Lipiden vorlag.
Abbildung 39: Tryptophanfluoreszenz von PmtA in Gegenwart anionischer Lipide. Für die Tryptophanfluoreszenz wurden 10 µM PmtA mit steigenden Konzentration PG-, CL- und PS-Liposomen inkubiert. Die Fluoreszenz wurde bei 290 nm angeregt und das Spektrum zwischen 300 - 400 nm aufgezeichnet. Für die Bestimmung der KD-Werte wurde die Abnahme der Fluoreszenz verwendet (F/F0).
Auch eine leichte Verschiebung der Fluoreszenzmaxima war zu beobachten, diese
war jedoch vergleichbar gering wie bei der Zugabe von Substratlipiden. Zur
Bestimmung der KD-Werte wurde daher die Fluoreszenzintensität verwendet. Für
PG, CL und PS wurden KD-Werte im mikromolaren Bereich (PG: 1.1 µM,
CL: 0.53 µM, PS: 2.25 µM) ermittelt.

Ergebnisse
- 89 -
3.2.12 Bioinformatische Analyse putativer Lipidbindestellen in PmtA
Die vorherigen Abschnitte haben gezeigt, dass PmtA eine hohe Affinität zu
Phospholipiden besitzt. Die Bindung von Substratlipiden durch PmtA beruht dabei
wahrscheinlich auf einer spezifischen Enzym-Substrat-Interaktion, wohingegen die
Bindung anionischer Lipide auf einer elektrostatischen Interaktion basiert. Die
Struktur und der detaillierte Ablauf der Transmethylierungsreaktion bakterieller
Phospholipid N-Methyltransferasen ist weitgehend unklar. Obwohl PmtA löslich ist
und für biochemische Analysen in ausreichenden Mengen isoliert werden kann, war
eine Strukturaufklärung mittels Röntgenstrukturanalyse oder NMR bisher nicht
möglich. Putative Aminosäuren, die für eine PmtA-Lipid-Interaktion essentiell sind,
konnten daher nicht auf struktureller Basis identifiziert werden. Weiterhin besitzten
Pmt-Enzyme kein bekanntes Phospholipidbindemotiv (bspw.: Ca2+-Domäne, FYVE
oder PH-Domäne). Daher wurden auf Basis eines Aminosäuresequenzvergleiches
konservierte Aminosäuren und strukturell benachbarte Aminosäuren, die
möglicherweise an der PmtA-Lipid-Interaktion beteiligt sind, mutiert (Abbildung 40).
Weiterhin wurden nur solche Aminosäuren ausgewählt, die vermutlich keine Rolle für
die SAM-Bindung spielen (Abschnitt 3.1), aber womöglich für eine Lipid-Interaktion
von Bedeutung sind. Die ausgewählten Aminosäuren wurden gegen ein Alanin oder
eine Aminosäure mit konträrer Ladung ausgetauscht.
Da PmtA wahrscheinlich auch über elektrostatische Interaktionen mit der Membran
interagiert, wurden zusätzlich basische Aminosäuren gegen ein Alanin ausgetauscht.
Diese Aminosäuren sind in den stark positiv geladenen Bereichen auf der PmtA-
Oberfläche des Homologiemodells lokalisiert (Abbildung 24). Die Mutation einer
positiv geladenen Aminosäure in diesem Bereich könnte die Membranbindung
beeinflussen.

Ergebnisse
- 90 -
Abbildung 40: Aminosäuresequenzvergleich mehrerer Pmt-Enzyme. Mit Pfeil gekennzeichnet sind die eingefügten Punktmutationen (schwarze Pfeile: putative Aminosäuren für eine PmtA-Substratlipid Interkation; blaue Pfeile: basische Aminosäuren innerhalb der positiv geladenen Bereiche der PmtA-Oberfläche des Homologiemodells). Pmt: Phospholipid N-Methyltransferase. Atum: Agrobacterium tumefaciens; Smel: Sinorhizobium meliloti; Bjap: Bradyrhizobium japonicum; Mloti: Mesorhizobium loti;
Ml:mlr5374: putatives Pmt-Enzym aus Mloti.
3.2.13 Einfluss verschiedener Punktmutationen auf die PmtA-
Aktivität
Die durch Quik-Change® Mutagenese generierten PmtA-Derivate wurden mittels T7-
Expressionssystem in E. coli BL21 (DE) überexprimiert. Durch eine SDS-PAGE
wurde bestätigt, dass alle PmtA-Derivate bis auf K42A, K43A und K42A,K43A
erfolgreich überexprimiert wurden (Abbildung 41A und C). Die Gründe für die
fehlende Expression der PmtA-Derivate K42A, K43A und K42A,K43A sind ungeklärt.
Eine mögliche Erklärung ist, dass durch eine Mutation in diesem Bereich die PmtA-
Struktur zu stark beeinflusst wurde. Dies würde dann wahrscheinlich zu einer
Missfaltung und sofortigen Degradation des Proteins führen.
Um den Einfluss der verschiedenen Punktmutationen auf PC-Bioysnthese von PmtA
zu analysieren, wurden die Lipide der Überexpressionskulturen isoliert,
dünnschichtchromatographisch aufgetrennt und die Reaktionsprodukte durch
Kupfersulfat visualisiert.

Ergebnisse
- 91 -
Ein Alaninaustausch der Aminosäuren Q26, K28, K29 und K28A,K29A beeinflusste
die PC-Synthese nicht, wohingegen die Punktmutation des T36 zu einer deutlichen
Akkumulation von MMPE im Vergleich zu PmtA (WT) führte (Abbildung 41B). Für die
PmtA-Derivate K42A, K43A und K42A,K43A konnte keine PC-Biosynthese
nachgewiesen werden, was aufgrund der fehlenden Expression zu erwarten war.
Abbildung 41: Überexpression und Einfluss von Punktmutationen konservierter oder basischer Aminosäuren in PmtA. Die Überexpression von PmtA (WT) oder der PmtA-Derivate wurde mittels SDS-PAGE analysiert (A. und C.). Die Lipide der Überexpressionskulturen wurden isoliert und mittels Dünnschichtchromatographie aufgetrennt. Anschließend erfolgte eine Visualisierung der Lipide mittels Kupfersulfat (B. und D.). LV: Überexpressionskultur mit Leervektor.
Die PmtA-Derivate I33A, I126A und S160A zeigten eine marginal reduzierte PC-
Biosynthese (Abbildung 41D). Jedoch wurden die Zwischenstufen MMPE und DMPE,
sowie das Endprodukt PC in deutlichen Mengen synthetisiert. Ein drastischer
enzymatischer Defekt war für die PmtA-Derivate F89A, V129T, Q158E/R, P187T und

Ergebnisse
- 92 -
W192A zu beobachten. Eine Mutation dieser Aminosäuren reduzierte die PC-
Biosynthese deutlich (F89A, V129T, P187A) oder bewirkte einen vollständigen
Verlust der PC-Biosynthesefähigkeit (Q158E, Q158R und W192A). Die PmtA-
Derivate V129T, Q158E/R, P187T und W192A waren jedoch bis auf F89A unlöslich.
Es ist daher sehr wahrscheinlich, dass die Aminosäuren V129, Q158, P187 und
W192 eine wesentliche Bedeutung für die Faltung und strukturelle Stabilität von
PmtA haben. Ein Alaninaustausch der Aminosäuren I186, Y194, T195 und R196
hatte hingegen keinen Einfluss auf die PC-Biosyntheseaktivität von PmtA.
Der Einfluss der verschiedenen Punktmutationen auf die PmtA-Aktivität ist in Tabelle
7 zusammengefasst. Auffällig ist, dass eine Mutation im N-terminalen Bereich
(Aminosäure 1-33) kaum einen Einfluss auf die Aktivität und Löslichkeit von PmtA
hat. Eine Mutation im Bereich von Aminosäure 42 – 159 führt meist zu einer
reduzierten PmtA-Aktivität. Punktmutationen in diesem Bereich haben jedoch oftmals
zur Folge, dass das generierte PmtA-Derivat unlöslich ist. Eine Punktmutation am C-
Terminus hat hingegen keinen Einfluss auf die PmtA-Aktivität.
Tabelle 7: Übersicht der Punktmutanten von PmtA. +: PmtA(-Derivat) ist aktiv/ wird überexprimiert/ ist löslich. -: PmtA(-Derivat) ist inaktiv/ wird nicht überexprimiert/ ist unlöslich. Klammern deuten eine geringe Aktivität/Löslichkeit an. Pfeil: Akkumulation.
PmA-Variante In vivo Aktivität Heterologe Expression In vitro Löslichkeit
WT + + +
Q26A + + +
K28A + + +
K29A + + +
KK28/29AA + + +
I33A + + +
T36A + (MMPE↑) + +
K42A - - -
K43A - - -
KK42/43AA - - -
F89A (+) + +
I126A + + (+)
V129T (+) + -
Q158E - + -
Q158R - + -
S160A + + (+)
I186A + + (+)
P187T ((+)) + -
W192A - + -
Y194A + + (+)
T195A + + (+)
R196A + + (+)

Ergebnisse
- 93 -
3.2.14 Die Bedeutung der Aminosäure Threonin 36 für die
Lipidbindung von PmtA
Ein Austausch der Aminosäure Threonin gegen ein Alanin an Position 36 in PmtA
führte in vivo zu einer Akkumulation von MMPE verglichen mit PmtA (WT) (Abbildung
41). Solch ein Einfluss einer Punktmutation wurde zuvor für PmtA noch nicht
beobachtet. Eine weiterführende Analyse dieser Mutante bietet die Möglichkeit zum
besseren Verständnis der Funktionsweise von PmtA. Das PmtA-Derivat T36A ist im
Gegensatz zu anderen PmtA-Derivaten löslich und wurde in ausreichenden Mengen
isoliert, um die enzymatische Aktivität in vitro zu untersuchen. Dafür wurde die
enzymatische Aktivität von PmtA und T36A in Gegenwart von PE und von PE+PG
verglichen. PmtA kann bei Anwesenheit von PE signifikante Mengen an PC
synthetisieren. Eine Zugabe von PG stimuliert wie erwartet die PmtA-Aktivität und es
wird mehr PC synthetisiert. T36A kann wie PmtA (WT) in vitro PC synthetisieren,
jedoch sind die PC-Mengen deutlich geringer. Um zu überprüfen, ob T36 eine
Bedeutung für die Bindung von PmtA an anionische Lipide hat, wurde die Aktivität in
Anwesenheit von PG untersucht. Eine Zugabe von PG stimulierte zwar die Aktivität
von T36A, jedoch wurden verglichen mit PmtA geringere Mengen an PC
synthetisiert. Weiterhin unterscheiden sich die Mengen an Reaktionsprodukten die
in vivo und in vitro beobachtet wurden. In vivo wurde eine deutlich höhere
Akkumulation von MMPE bei T36A im Vergleich zu PmtA detektiert. Dies konnte bei
einem in vitro Ansatz nicht beobachtet werden. Die Zwischenprodukte MMPE und
DMPE sind in vergleichbaren Mengen vorhanden (Abbildung 42).
Abbildung 42: In vitro-Aktitivät von PmtA (WT) und dem PmtA-Derivat T36A. Es wurden jeweils 10 µM Protein, 400 µM PE und 100 µM PG eingesetzt. Die Reaktion wurde durch Zugabe von 1.8 mM SAM gestartet. Nach Reaktionszeit von 1 Stunde wurden die Lipide extrahiert und mittels Dünnschichtchromatographie aufgetrennt und mittels Kupfersulfat visualisiert.

Ergebnisse
- 94 -
Daher ist anzunehmen, dass eine Mutation des Threonins an Position 36 die
Reaktionsgeschwindigkeit der PC-Biosynthese beeinflusst. Denkbar wäre eine
Beeinträchtigung des katalytischen Prozesses, der Konformationsänderung oder
eine reduzierte Lipidbindeaffinität. Um die Konformation und die Lipidbindeaffinität zu
überprüfen, wurden die Lipidbindeeigenschaften von T36A mittels CD-Spektroskopie
untersucht (Abbildung 43).
Abbildung 43: Einfluss von geladenen Lipiden auf die Sekundärstruktur des PmtA-Derivates T36A. Zu 7.5 µM T36A wurden Liposomen in steigenden Konzentrationen hinzu titriert. Eingeblendet ist jeweils die Auftragung der Elliptizitätsänderung des CD-Signals bei 208 nm gegen die Lipidkonzentration und der ermittelte KD-Wert.

Ergebnisse
- 95 -
Das CD-Spektrum von T36A zeigte, dass dieses PmtA-Derivat eine native Faltung
wie PmtA besitzt. Weiterhin wies das CD-Spektrum eine negative Elliptizität und
Minima bei ca. 208 nm sowie bei 222 nm auf (Abbildung 43). Ein Maximum der
Elliptizität war bei ca. 195 nm zu beobachten. Anhand des CD-Spektrums kann somit
eine Missfaltung ausgeschlossen werden. Das Spektrum ist vergleichbar mit dem
CD-Spektrum von PmtA. Die Zugabe von PE, MMPE, DMPE, PC oder PG führte zu
einer Zunahme des Elliptizitätwertes. Die Änderung des CD-Spektrums ist
vergleichbar mit der für PmtA beschriebenen Änderung nach Lipidzugabe
(Abschnitt 3.2.9). Eine quantitative Analyse dieser Änderung zeigte weiterhin eine
hohe Affinität von T36A zu den getesteten Lipiden. Dafür wurde die
Elliptizitätsänderung bei 208 nm zur Bestimmung der Dissoziationskonstante (KD)
genutzt. T36A besitzt gegenüber PE-Liposomen einen KD-Wert von 5.25 µM. Dieser
liegt im mikromolaren Bereich, ist jedoch geringfügig höher, als der von PmtA
(0.72 µM). Die ermittelten KD-Werte für MMPE (9.82 µM), DMPE (3.39 µM),
PC (3.30 µM) und PG (1.59 µM) sind fast äquivalent zu denen von wildtyp PmtA
(MMPE: 10.36 µM; DMPE: 4.84 µM; PC 5.04 µM; PG: 2.64 µM).
Die CD-spektroskopischen Daten und die anhand dieser ermittelten KD-Werte legen
nahe, dass die Phospholipidbindung von T36A nicht beeinflusst ist. Weiterhin hat der
Austausch von Threonin zu Alanin keinen Einfluss auf die Faltung oder
Konformationsänderung. Es ist also eher wahrscheinlich, dass T36 ein Teil des
katalytischen Zentrums ist und die Mutation die Reaktionsgeschwindigkeit negativ
beeinflusst.

Diskussion
- 96 -
4. Diskussion
4.1 SAM-Bindeeigenschaften der agrobakteriellen
Phospholipid N-Methyltransferase PmtA
Das pflanzenpathogene Bakterium A. tumefaciens ist in der Lage, PC über den
Methylierungsweg oder den PC-Synthaseweg zu synthetisieren. PC ist für die
Virulenz von A. tumefaciens essentiell (Wessel et al., 2006), jedoch ist über die
Funktionsweise bakterieller PC-Biosyntheseenzyme bisher sehr wenig bekannt
(Wessel et al., 2006; Klüsener et al., 2009; Aktas & Narberhaus, 2009). Die
agrobakterielle Phospholipid N-Methyltransferase PmtA ist ein PC-
Biosyntheseenzym, das PE zu PC methyliert. Für diese Reaktion nutzt PmtA SAM
als Methyldonor und gehört daher zur Klasse der SAM-MT (Kaneshiro & Law, 1964;
Aktas & Narberhaus, 2009). Im Rahmen dieser Arbeit wurden die SAM-
Bindeeigenschaften von PmtA analysiert und essentielle Aminosäuren für die SAM-
Bindung identifiziert.
Mit Hilfe von radioaktiv markiertem SAM (14C-SAM) wurde die SAM-Bindeaffinität für
PmtA und für PmtA-Derivate bestimmt. PmtA besitzt einen KD-Wert von 25 µM
gegenüber SAM und somit eine hohe Affinität für den Kofaktor. Für die eukaryotische
Phosphatidylethanolamin N-Methyltransferase (PEMT) aus der Rattenleber wurde
ein vergleichbarer KD-Wert mit 30 µM ermittelt (Schneider & Vance, 1979; Vance &
Ridgway, 1988). Die KD-Werte der PmtA und eukaryotischen PEMT gegenüber SAM
liegen in einem für SAM-MT charakteristischen Bereich von 0.1 – 30 µM (Mashhoon
et al., 2004; Basturea & Deutscher, 2007; Savic et al., 2008). Eine alternative
Methode zur Bestimmung der Bindeaffinität für SAM via NMR-Spektroskopie zeigte
hingegen einen 3 – 4 fach höheren KD-Wert (91 µM) für PmtA gegenüber SAM
(Aktas et al., 2011a). Da eine Messung mittels NMR-Spektroskopie sehr zeitintensiv
ist, wurde dem Ansatz eine S-Adenosylhomocysteinnucleosidase hinzugefügt. Diese
wurde eingesetzt, um entstehendes SAH enzymatisch abzubauen, sodass SAH nicht
in Konkurrenz zu SAM steht und die Bindung von SAM an PmtA beeinflusst. Eine
mögliche Erklärung ist, dass zu geringe Mengen an SAH-Nucleosidase verwendet
wurden. Somit kann nicht ausgeschlossen werden, dass Reste von SAH vorhanden
waren und diese die Messung beeinflusst haben. Weitere Titrationsexperimente via

Diskussion
- 97 -
NMR-Spektroskopie mit SAH zeigten für PmtA einen KD-Wert von 25 µM. Dieser
Wert entspricht der KD für 14C-SAM. PmtA besitzt somit eine vergleichbare Affinität
gegenüber SAH und SAM. Der für SAM ermittelte Wert mittels NMR-Spektroskopie,
wurden sehr wahrscheinlich durch SAH-Reste verfälscht (Aktas et al., 2011a).
Durch Aminosäuresequenzvergleich und Topologieanalysen von PmtA konnten
putative SAM-Bindemotive (SAM-I-, SAM-II- und SAM-III-Motiv) identifiziert werden.
Das SAM-I-Motiv (D/ExGxGxG) bilden die Aminosäuren E58, G60, G62 und G64.
Die Aminosäure E84 ist die charakteristische Aminosäure des putativen SAM-II-
Motivs für PmtA. Das SAM-III-Motiv besteht wahrscheinlich aus der Aminosäure
D106 (Abbildung 44). Punktmutationen der identifizierten Aminosäuren führten zu
einem vollständigen Verlust oder zu einer drastisch reduzierten Enzymaktivität. Die
PmtA-Derivate G64A und D106A bzw. D106E konnten nicht gereinigt werden,
sodass eine weitere biochemische Charakterisierung nicht möglich war. Eine
mögliche Erklärung für die mangelnde Löslichkeit dieser PmtA-Derivate ist, dass
diese Aminosäuren eventuell eine bedeutende Rolle für die Faltung und Stabilität des
Proteins besitzen. Die Faltung und die Lipidbindung der übrigen PmtA-Derivate
wurden durch die Punktmutation nicht beeinflusst. Der enzymatische Defekt beruhte
allein auf dem Verlust der SAM-Bindefähigkeit. Die Phosphoethanolamin N-
Methyltransferase (PfPMT) aus Plasmodium falciparum ist ebenfalls eine SAM-MT,
die eine Methylgruppe auf Phosphoethanolamin transferiert. Das synthetisierte
Cholin kann unter anderem in Form von PC in die Membran eingebaut werden
(Witola et al., 2008). Die Motive SAM-I, SAM-II und SAM-III von PfPMT wurden in
einer ähnlichen Studie bioinformatisch identifiziert und anschließend die Bedeutung
konservierter Aminosäuren durch Punktmutationen analysiert. Die Strukturintegrität
wurde durch die Punktmutation nicht oder nur sehr geringfügig beeinflusst. Dies
zeigten CD-spektroskopische Analysen, sowie das Schmelzverhalten der PfPMT-
Varianten (Reynolds et al., 2008). Mutationen innerhalb des SAM-I-Motivs oder des
SAM-II-Motivs führten bei PfPMT ähnlich wie bei PmtA zu einer drastisch reduzierten
Enzymaktivität (Reynolds et al., 2008). Die Motive SAM I-III sind in eukaryotischen
Pmt-Enzymen nicht vorhanden oder nur schwach konserviert (Shields et al., 2003).
Punktmutationen haben jedoch eine ähnliche Auswirkung bei der humanen PEMT.
Mutationen im SAM-I-Motiv (G98 und G100) führten im Falle von G98 zu einer
Verringerung der SAM-Bindefähigkeit um ca. 25 %. Ein Alaninaustausch von G100
führte hingegen zu einem vollständigen Verlust der SAM-Bindefähigkeit (Shields et

Diskussion
- 98 -
al., 2003). Eine Abfolge saurer Aminosäuren (E180 und E181) ist ebenfalls wichtig
für die SAM-Bindung der PEMT. Eine Mutation von E180 zu Alanin bewirkte einen
vollständigen Verlust der SAM-Bindefähigkeit. Dies zeigt, dass die Aminosäuren
G100 und E180 essentiell für die SAM-Bindung der PEMT sind (Shields et al., 2003).
Diese Aminosäuren sind in räumlicher Nachbarschaft und bilden womöglich die
SAM-Bindestelle für PEMT (Shields et al., 2003).
Für bakterielle Pmt-Enzyme konnten bisher keine Proteinstrukturen gelöst werden.
Auf Basis der Aminosäuresequenz konnte jedoch ein Homologiemodell für PmtA
erstellt werden (I-TASSER; Zhang, 2008). Dieses Modell zeigt, dass die Reste der
Aminosäuren E58, G60, G62 und E84 in räumlicher Nähe zueinander orientiert sind
(Abbildung 44). Diese Aminosäuren könnten eine putative SAM-Bindetasche formen.
Um diese Vermutung zu bestätigen, wäre es nötig eine Strukturaufklärung von PmtA
inklusive gebundenem Kofaktor durchzuführen. Das Homologiemodell postuliert für
PmtA eine α-β-α Sandwichstruktur, was auch bereits bei anderen SAM-MTs
beobachtet wurde. Das zentrale, planare β-Faltblatt besteht aus 5 parallelen (β1-4)
und einem anti-parallelen β-Faltblatt (β5). Die α-Helices αE, αF, αG zum einen und
αB, αC, αD zum anderen flankieren das zentrale β-Faltblatt. Eine N-terminale α-Helix
(αA) ist über dem zentralen β-Faltblatt exponiert.
Die tertiäre Struktur und der Reaktionsmechanismus der PfPMT wurden kürzlich
aufgeklärt (Lee et al., 2012). Die Struktur der PfPMT ist homolog zu anderen SAM-
MTs. PfPMT besitzt ein zentrales, planares β-Faltblatt, das von mehreren α-Helices
flankiert wird. Zusätzlich besitzt dieses Enzym eine α-helicale Deckeldomäne, die für
die Substratbindung und Abgrenzung vom wässrigen Milieu der Umgebung von
enormer Bedeutung ist (Lee et al., 2012). Eine Strukturanalyse von PfPMT im
Komplex mit SAH ermöglichte die Identifizierung wichtiger Aminosäuren für die
Kofaktorbindung. Auf Basis dieses Strukturmodells sind die Aminosäuren D61, G63,
D85 und D110 essentielle Interaktionspartner für die SAM-Bindung der PfPMT.

Diskussion
- 99 -
Abbildung 44: Putative SAM-Bindetasche im PmtA-Homologiemodell. (A.): Cartoon-Darstellung des PmtA-Homologiemodells. Hervorgehoben sind essentielle Aminosäuren für die SAM-Bindung. (B.): Detailansicht der putativen SAM-Bindetasche mit gebundem SAM. (C.): Modell zur Interaktion wichtiger Aminosäuren mit SAM. SAM: S-Adenosylmethionin.
Dabei interagiert G63 mit dem Aminoanteil des Kofaktors über die Carboxylgruppe
des Glycins. Über D61 werden zwei Wassermoleküle koordiniert, die den Kofaktor in
die richtige Konformation orientieren. Diese Aminosäuren sind Teil des glycinreichen
Loops des SAM-I-Motivs.
Für andere SAM-MTs wurde ein ähnliches Interaktionsmodell des SAM-I-Motivs und
dessen Bedeutung für die Koordination von SAM postuliert (Cheng et al., 1993;
Kozbial & Mushegian, 2005). Für das SAM-II-Motiv wird eine Interaktion mit den
Ribosylhydroxylgruppen angenommen und das SAM-III-Motiv bindet den Adeninring
von SAM (Cheng et al., 1993; Kozbial & Mushegian, 2005). Dies wurde auch für die

Diskussion
- 100 -
PfPMT gezeigt, wobei D85 die konservierte Aminosäure des SAM-II-Motivs ist und
D110 die konservierte Aminosäure des SAM-III-Motivs (Lee et al., 2012). Die
Aminosäuren D61, G63, D85 und D110 formen so eine SAM-Bindetasche.
Untersuchungen von PfPMT-Derivaten mit Punktmutationen von Aminosäuren, die
für die SAM-Bindung wichtig sind, unterstützen das postulierte Interaktionsmodell.
(Lee et al., 2012). Ein Vergleich der PfPMT und des Homologiemodells von PmtA
zeigt, dass beide Proteine strukturell sehr ähnlich sind. Die kritischen Aminosäuren
für die SAM-Bindung sind teilweise an identischen Positionen innerhalb der Struktur
lokalisiert. Somit kann spekuliert werden, dass die Aminosäuren E58, G60, E84 und
D106 in PmtA eine analoge Funktion zu den Aminosäuren D61, G63, D85 und D110
in PfPMT einnehmen (Abbildung 45). Einen deutlichen Unterschied in der Struktur
zeigt sich am C-Terminus. PfPMT besitzt dort eine α-helicale Deckelstruktur, die für
die Substratbindung wichtig ist. Insgesamt stimmen die experimentell ermittelten
Daten zur SAM-Bindung von PmtA sehr gut mit den bisherigen bioinformatischen
Erkenntnissen und bekannten Daten anderer SAM-MTs überein. Somit konnte im
Rahmen dieser Arbeit Aminosäuren, die für die SAM-Bindung von PmtA essentiell
sind, identifiziert werden.
Abbildung 45: Strukturvergleich des Homologiemodells für PmtA (grau) und PfPMT (gelb). (A.): Vergleich der Tertiärstruktur auf Basis einer Cartoon-Darstellung. Bis auf den C-Terminus zeigen die beiden SAM-MTs hohe strukturelle Ähnlichkeiten. (B.): Detailansicht der SAM-Binderegion. Die essentiellen Aminosäuren der PfPMT sind in Gelb dargestellt, wobei schwarze Striche die Interaktionen skizzieren. In Grau sind die essentiellen Aminosäuren für die SAM-Bindung in PmtA dargestellt.

Diskussion
- 101 -
Neben der Bindung von SAM, spielt auch die Übertragung der Methylgruppe eine
wichtige Rolle für den enzymatischen Mechanismus. Die Transmethylierungsreaktion
durch SAM-MTs ähnelt einer SN2-Reaktion. Die saure Aminosäure des SAM-I-Motivs
koordiniert dafür ein Wassermolekül, um einen nukleophilen Angriff zu ermöglichen
(Bügl et al., 2000; Kozbial & Mushegian, 2005). Das Wassermolekül kann dabei als
Nukleophil dienen oder die Abspaltung der Methylgruppe vom Schwefelatom
begünstigen. Ein SN2-ähnlicher Reaktionsmechanismus wird auch für PfPMT
angenommen (Lee et al., 2012).
Es konnte gezeigt werden, dass das Tyrosin an Position 19 und das Histidin an
Position 132 essentiell für die Katalyse sind. Die Deprotonierung des Y19 durch das
H132 ermöglicht wiederum eine Deprotonierung der Amingruppe des Substrates
Phosphoethanolamin. Infolge der Substratdeprotonierung erfolgt eine Bindung der
Methylgruppe von SAM an das Stickstoffatom des Phosphoethanolamins
(Lee et al., 2012). Punktmutationen der Aminosäuren Y19 und H132 haben gezeigt,
dass die Substrat- und Kofaktorbindung durch diese Aminosäuren nicht beeinflusst
wird. Dies unterstützt die spezifische Bedeutung der Aminosäuren Y19 und H132 für
die Katalyse (Lee et al., 2012). Ein derartiger Reaktionsmechanismus lässt sich
zurzeit nicht direkt auf PmtA übertragen, da PmtA keine dazu homologen
Aminosäuren in ähnlicher räumlicher Anordnung aufweist.
Die Untersuchung der konservierten Aminosäuren des SAM-I- und SAM-II-Motivs in
PmtA haben gezeigt, dass die SAM-Bindemotive in PmtA ähnlich wie in anderen
SAM-MTs lokalisiert sind. Die SAM-Binderegion von PmtA konnte auf den N-
Terminus eingegrenzt werden und weiterhin wurden Aminosäuren identifiziert, die für
die SAM-Bindung essentiell sind. Die Phospholipidsubstratbindung könnte analog zu
anderen SAM-MTs am C-Terminus lokalisiert sein (Martin & McMillan, 2002; Kozbial
& Mushegian, 2005).

Diskussion
- 102 -
4.2 PmtA-Lipid Interaktion
PmtA synthetisiert PC durch dreifache Methylierung von PE. Der Mechanismus der
PmtA-Lipid Interaktion und die Bedeutung verschiedener Membranlipide für die
enzymatische Aktivität von PmtA ist bisher unbekannt. In Rahmen dieser Arbeit
wurden verschiedene Methoden angewendet, um die Interaktion von PmtA mit
Lipiden und die Bedeutung einer PmtA-Lipid Bindung näher zu charakterisieren.
4.2.1 Anionische Lipide stimulieren die PmtA-Aktivität
Vorangegangene Arbeiten zeigten, dass die PmtA-Aktivität durch das anionische
Lipid PG stimuliert wird (Aktas & Narberhaus, 2009). PG ist ein Bestandteil vieler
bakterieller Membranen und ist auch in der agrobakteriellen Membran präsent
(Sherr & Law, 1965; Karnezis et al., 2002). PG ist jedoch kein Substratlipid für PmtA.
In dieser Arbeit wurde gezeigt, dass auch andere anionische Lipide wie CL, PA, PI
oder PS einen stimulierenden Effekt auf die PmtA-Aktivität haben. Im Gegensatz zu
den Substratlipiden vermitteln anionische Lipide jedoch keine SAM-Bindung. Das
kationische Lipid DPPG hat keinen Einfluss auf die PmtA-Aktivität. Daher ist davon
auszugehen, dass nur anionische Lipide einen Einfluss auf die PmtA-Aktivität haben.
Zukünftig könnte untersucht werden, ob auch pflanzliche Sulfolipide, die zu den
anionischen Lipiden gehören und nicht in Bakterien vorkommen, die PmtA-Aktivität in
vitro stimulieren.
Die Bedeutung und Wirkung von anionischen Lipiden auf andere Proteine ist bereits
gut untersucht. Oftmals üben anionische Lipide eine regulatorische Funktion auf
periphere Membranproteine aus, wie zum Beispiel auf die ATP-abhängige Protease
Lon, den Replikationsinitiator DnaA, die Phosphatidylserinsynthase Pss aus E. coli,
die eukaryotische CTP:Phosphocholin Cytidylyltransferase (CTP) oder die
Phosphatidylinositol-spezifische Phospholipase C aus Bacillus thuringiensis
(Cornell, 1991a; Sekimizu & Kornberg, 1987; Salamon et al., 2000;
Linde et al., 2004; Minami et al., 2011; Shih et al., 2011). Die Regulation dieser
Proteine erfolgt oftmals über die Lokalisation bzw. Bindung an die Membran. Die
cytosolische Protease Lon kann beispielsweise CL binden und wird durch die
Membranbindung inhibiert (Minami et al., 2011). Im Gegensatz dazu werden die

Diskussion
- 103 -
peripheren Membranproteine DnaA, Pss und CTP durch anionische Lipide aktiviert
bzw. die Aktivität stimuliert (Cornell, 1991a; Sekimizu & Kornberg, 1987;
Salamon et al., 2000; Linde et al., 2004). In E. coli spielt MinD bei der Zellteilung eine
wichtige Rolle und ist an den Zellpolen lokalisiert. Dabei assoziiert MinD vorwiegend
an das anionische CL. Weiterhin ist die Kurvatur der Membran, die durch Lipide wie
CL beeinflusst wird, für die Lokalisation von MinD von Bedeutung. (Szeto et al., 2002;
Renner et al., 2011). Das „high potential iron-sulfor protein“ (HiPIP) aus
Allochromatium vinosum ist ein periplasmatisches Protein, das aus dem Cytosol über
die Membran ins Periplasma transportiert werden muss. Es wurde gezeigt, dass das
cytoplasmatische Vorläuferprotein durch eine elektrostatische Interaktion der
Signalsequenz mit anionische Lipiden zuerst an die Membran rekrutiert wird, bevor
es über die Membran transportiert wird (Brehmer et al., 2012). Die PmtA-Aktivität und
Lokalisation an die Membran könnte auf ähnliche Weise durch anionische Lipide
beeinflusst werden. Potentielle Interaktionsstellen für anionische Lipide sind die stark
positiv geladenen Bereiche auf der Oberfläche des PmtA-Homologiemodells. Eine
derartige Wechselwirkung zwischen Lipiden und Proteinen bezeichnet man als
elektrostatische Interaktion und soll im Folgenden für PmtA diskutiert werden.
4.2.2 Elektrostatische Interaktion von PmtA mit Membranen
In dieser Arbeit wurde gezeigt, dass nicht nur PG, sondern anionische Lipide generell
die PmtA-Aktivität stimulieren. Weiterhin wurde mittels CD-Spektroskopie und
Fluoreszenzstudien gezeigt, dass die Bindung von PmtA an anionische Lipide hoch-
affin ist. Die Bindung an anionische Lipide bewirkt zwar eine Konformations-
änderung, jedoch ist dadurch keine SAM-Bindung möglich. Anionische Lipide sind
zudem kein potentielles Substratlipid für PmtA, weshalb sich die Frage stellte: warum
bindet PmtA diese Lipide und weshalb wird die Aktivität stimuliert? Das für die
Zellform wichtige MARCKS-Protein, das Phosphatidylethanolaminbindeprotein
PEBP, die CTP: Phosphocholincytidylyltransferase, das Tumorsupressorprotein
PTEN oder des Saposin-ähnliche Protein Na-SLP-1 sind Beispiele für Proteine, die
ebenfalls mit anionischen Lipiden interagieren, obwohl diese kein Substrat für das
jeweilige Protein darstellen (Kim et al., 1994; Cornell & Northwood, 2000;
Das et al., 2002; Vallée et al., 2001; Willis et al., 2011). Die Bindung beruht dabei auf

Diskussion
- 104 -
einer elektrostatischen Interaktion zwischen basischen Aminosäuren und negativ
geladenen Lipiden. PmtA besitzt ein großes Cluster an basischen Aminosäuren auf
der Protein-Oberfläche. Das elektrostatische Potential der PmtA-Oberfläche,
basierend auf der Poisson-Boltzmann Gleichung, wurde für das Homologiemodell
mittels ABPS (Baker et al., 2001) bestimmt und mit Pymol visualisiert (Abbildung 46).
Abbildung 46: Elektrostatisches Potential der Oberfläche des PmtA-Homologiemodells basierend auf der Poisson-Boltzmann Gleichung. Die elektrostatische Potential wurde auf +5 kT/e (blau; positive Ladung), bzw. -5 kT/e (rot; negative Ladung) gesetzt.
Es ist ein stark positiv geladenes Potential (blau) auf der Oberfläche des PmtA-
Homologiemodells zu erkennen. Möglicherweise interagiert PmtA ähnlich wie die
oben aufgeführten Beispielproteine auch via elektrostatischer Interaktionen mit
anionischen Lipiden in einer Membran.
Um eine solche Interaktion nachzuweisen, wurde die Aktivität, Faltung und
Lipidbindung von PmtA bei hoher ionischer Stärke untersucht. Eine hohe ionische
Stärke führt dabei zu einer Minderung des negativen Membranpotentials, das durch
anionische Lipide ausgebildet wird. Die hohe ionische Stärke stört daher die
elektrostatischen Interaktionen zwischen Proteinen und Membranen
(McLaughlin, 1977; McLaughlin, 1989). Es wurde gezeigt, dass die PmtA-Aktivität bei
steigender ionischer Stärke abnimmt. Ähnliche Effekte sind für andere
membranassoziierte Proteine beschrieben. Die hohe ionische Stärke führte dabei zu
einer Dissoziation bzw. Beeinträchtigung der Membranbindung dieser Proteine.

Diskussion
- 105 -
Beispiele dafür sind die Phosphatidylserinsynthase aus E. coli, das MARCKS-
Protein, α-Synuclein, PEBP und das Gag-Protein des HIV-1 Virus (Louie et al., 1985;
Kim et al., 1994; Jo et al., 2000; Vallée et al., 2001; Dalton et al., 2007).
Die strukturelle Integrität von PmtA blieb durch hohe ionische Stärke unbeeinflusst.
Auch bei 500 mM NaCl war PmtA noch nativ gefaltet. Die Bindung von PmtA an die
Membran wurde jedoch durch hohe ionische Stärke gestört. PmtA konnte PE und
A. tumefaciens Lipidextrakt nur bis zu einer ionischen Stärke von 150 mM NaCl
binden. Eine Bindung an die anionischen Lipide CL, PG oder PS war jedoch nur bis
zu einer ionische Stärke von 50 mM NaCl effizient möglich. PmtA wird daher
vermutlich durch elektrostatische Interaktion basischer Aminosäuren und anionischer
Lipide an die Membran rekrutiert und gebunden. Durch die gestörte
Membraninteraktion bei hoher ionischer Stärke hat PmtA keinen Zugang zum
Substratlipid PE. Dies führte zu einer reduzierten Enzymaktivität. Jedoch war die
PmtA-Aktivität auch reduziert, wenn die Membran nur aus PE besteht. Hohe
Salzkonzentrationen können ebenfalls die spezifische Enzym-Substrat Bindung
beeinträchtigen, wenn diese auf ionischen Interaktionen oder
Wasserstoffbrückenbindungen basiert. Diese werden ebenfalls durch eine hohe
ionische Stärke beeinflusst. Eine reine PE-Membran ist jedoch eine nicht
physiologische Modellmembran. Eine physiologische Membran für E. coli besteht zu
ca. 75 % aus PE, 20 % aus PG und bis zu 5 % aus CL. Die agrobakterielle Membran
besitzt ebenfalls PG und sehr wahrscheinlich auch CL (Sherr & Law, 1965; Karnezis
et al., 2002). Eine Rekrutierung von PmtA via elektrostatischer Interaktionen ist daher
möglich.
Für periphere Membranproteine, die über elektrostatische Wechselwirkungen mit
einer Membran interagieren, ist bereits bekannt, dass diese oftmals durch
Lokalisation bzw. Delokalisation reguliert werden. Eine Delokalisation kann durch
Protein-Protein-Interaktion erfolgen, wenn das regulatorische Protein eine
ausgeprägte negative Ladung besitzt. Ein solcher Mechanismus ist für das
MARCKS-Protein bekannt, das aufgrund einer Interaktion mit dem stark negativ
geladenen Calmodulin von der Membran dissoziiert (Kim et al., 1994;
André et al., 2004). Für PmtA sind bisher keine Protein-Protein-Interaktionspartner
bekannt. Eine Alternative für die Regulation peripherer Membranproteine bietet eine
Phosphorylierung. Die stark negative Phosphatgruppe ändert dabei die

Diskussion
- 106 -
Oberflächenladung des Proteins, sodass die positive Ladung der kationischen
Aminosäurereste neutralisiert wird. Ein Beispiel dafür ist auch das MARCKS-Protein
oder das Tumorsuppressorprotein PETN (Kim et al., 1994; Das et al., 2002). Das
Programm NetPhosBac (Miller et al., 2009) postuliert putative Phosphorylierungs-
stellen in bakteriellen Proteinen. Für PmtA wurden sechs putative
Phosphorylierungsstellen vorhergesagt (S38, S52, S96, S124, S127, S160). Eine
putative Phosphorylierungsstelle für eine Neutralisierung des positiven
elektrostatischen Potentials bietet das S52. Dieses befindet sich in räumlicher Nähe
zu den basischen Aminosäuren K42 und K43, die eine positive Ladung auf der
Oberfläche des PmtA-Homologiemodells ausbilden. Eine Phosphorylierung an dieser
Position könnte das positive Ladungspotential der PmtA-Oberfläche mindern und
dadurch die Membranassoziation durch elektrostatische Interaktionen
beeinträchtigen.
4.2.3 Substratspezifität von PmtA
Neben der Bedeutung anionischer Lipide wurde auch die Substratspezifität von PmtA
anhand von PE-Derivaten untersucht. PmtA ist in der Lage sowohl Lyso-PE (LPE),
als auch 1,2-Dioctanoyl-sn-Glycerin-3-Phosphoethanolamin (C8-PE) als Substrate zu
nutzen. Das wasserlösliche Phosphorylethanolamin diente hingegen nicht als
Substrat. Modellmembranen, die nur aus LPE oder C8-PE bestehen, bieten jedoch
keine optimalen Methylierungsbedingungen. LPE und C8-PE sind in Abwesenheit
anionischer Lipide für PmtA kaum als Substrat nutzbar. Auch bindet PmtA 14C-SAM
in Gegenwart dieser Lipide nur sehr gering (C8-PE) oder nur in unspezifischen
Mengen (LPE). In Anwesenheit von anionischen Lipiden konnte jedoch gezeigt
werden, dass PmtA C8-PE und LPE als Substrate nutzen kann. Eine Abhängigkeit
der Enzymaktivität von der Fettsäurekettenlänge eines Lipides wurde bereits für
andere periphere Membranproteine gezeigt, wie beispielsweise für die eukaryotische
CTP:Phosphocholin Cytidylyltransferase (CTP) (Cornell, 1991b). Die CTP benötigt
für ihre enzymatische Aktivität eine Membran aus Lipiden, deren Fettsäureketten aus
mindestens 12 Kohlenstoffatome bestehen. Für optimale Aktivität sollte diese Kette
jedoch länger als 14 Kohlenstoffatome sein (Cornell, 1991b). Im Gegensatz dazu
benötigt die Proteinkinase C nur eine Fettsäurekettenlänge von 6 Kohlenstoffatomen
für optimale enzymatische Aktivität (Ebeling et al., 1985). Die Bedeutung der

Diskussion
- 107 -
Fettsäurenkettenlänge für die Enzymaktivität liegt womöglich in einer Stabilisierung
der Membranbindung. Über eine hydrophobe Interaktion von aromatischen
Aminosäuren mit den Fettsäureketten innerhalb der Membran wird das Protein
besser assoziiert (Lomize et al., 2007). Eine andere Bedeutung der
Fettsäurenkettenlänge ist für die PEMT der Hefe bekannt. Diese besitzt eine
Substratspezifität in Abhängigkeit zur Fettsäureketteneigenschaften und methyliert
hauptsächlich PE mit einer ungesättigten Fettsäurekette zu PC (Boumann et al.,
2004). Diese PEMT ist jedoch ein integrales Membranprotein. Als peripheres
Membranprotein, das an der Oberfläche einer Membran lokalisiert ist, sind die
Fettsäureketteneigenschaften der Lipide, wie beispielsweise Doppelbindungen, für
PmtA sehr wahrscheinlich von geringer Bedeutung.
Abbildung 47: Strukturformeln für das Etherlipid Plasmalogen (A.) oder N-(Acyl)-Sphing-4-Enin-1-Phosphoethanolamin (B.).
Es zeigt sich also, dass ein PmtA-Substrat sowohl eine Ethanolaminkopfgruppe, als
auch eine Fettsäurekette benötigt. Weiterhin muss dieses Substrat ein Bestandteil
einer Membran sein. Das Etherlipid Plasmalogen oder das Spingolipid N-(Acyl)-
Sphing-4-Enin-1-Phosphoethanolamin könnten weitere potentielle Substrate für

Diskussion
- 108 -
PmtA sein, die in zukünftigen Studien getestet werden können (Abbildung 47). Auch
sollte zukünftig untersucht werden, ob durch die Anwesenheit von anionischen
Lipiden und C8-PE bzw. LPE nicht nur die Aktivität von PmtA, sondern auch die
SAM-Bindefähigkeit erhöht wird.
4.2.4 Inhibierung von PmtA durch PC-Analoga
PmtA bindet neben seinen Substratlipiden PE, MMPE und DMPE auch das
Endprodukt PC. Weiterhin ist bekannt, dass PmtA neben SAH auch durch das
Endprodukt PC inhibiert wird. Dabei scheint PC mit PE um dieselbe
Substratbindestelle zu konkurrieren (Aktas & Narberhaus, 2009). Es stellte sich die
Frage, ob diese Inhibierung nur auf der Cholinkopfgruppe basiert. Daher wurde die
Wirkung des PC-homologen Sphingomyelins (SM) getestet. Die polare Kopfgruppe
von SM ist wie bei PC Cholin. Der einzige Unterschied zwischen PC und SM besteht
in der Verknüpfung der Fettsäuren. Bei SM ist eine Fettsäure über eine Amidbrücke
verknüpft. In Abschnitt 3.2.2 wurde gezeigt, dass steigende SM-Konzentrationen die
PmtA-Aktivität reduzieren. Da SM und PC strukturell sehr ähnlich sind, interagiert SM
möglicherweise ähnlich wie PC mit PmtA. Dies würde bedeuten, dass SM die
Substratbindestelle von PmtA besetzt und so in Konkurrenz zu PE steht. Im
Gegensatz zu SM hatte das wasserlösliche Cholin und auch Phosphocholin keinen
Einfluss auf die PmtA-Aktivität. Ähnliche Beobachtungen wurden für die pflanzliche
Phosphatidylethanolamin N-Methyltransferase gemacht. Sowohl Cholin als auch
Glycinbetain (ein Cholin-Analogon) reduzieren die enzymatischen Aktivität nicht (Jost
et al., 2009). Daher scheint es ähnlich wie bei den PE-Derivaten von entscheidender
Bedeutung zu sein, dass die polare Kopfgruppe an ein Lipid gekoppelt ist und in der
Membran integriert ist. Eine Inhibierung von PmtA ist daher auch durch andere
cholintragende Membrankomponenten denkbar. Ein putativer Kandidat wäre hierfür
das Isoprenoid Cholesterylphosphorylcholin. Dies könnte durch ein in vitro-
Aktivitätsassay in Gegenwart von PE und Cholesterylphosphorylcholin gezeigt
werden.

Diskussion
- 109 -
4.2.5 Die Bedeutung des N-Terminus für eine PmtA-Membran-
Interaktion
Um die Interaktion von PmtA mit Membranlipiden und die Konsequenzen einer
Protein-Lipid-Interaktion für PmtA näher zu charakterisieren, wurden sowohl
qualitative, als auch quantitative Methoden angewendet. Eine qualitative Methode
zum Nachweis der PmtA-Lipid-Interaktion ist die limitierte Proteolyse. Die Ergebnisse
dieser Methode lieferten erste Hinweise auf eine Konformationsänderung von PmtA
nach Lipidbindung. Weiterhin lassen sich aufgrund dieser Ergebnisse erste
Hypothesen aufstellen, welche Bereiche der PmtA-Struktur von der
Konformationsänderung betroffen sind. Es zeigte sich, dass PmtA durch
Membraninteraktion vor proteolytischer Degradation geschützt wird. PmtA wurde in
Gegenwart von Lipiden, mit Ausnahme von MMPE, DMPE und PC, nicht oder nur bis
zu einem 18 kDa großen Fragment abgebaut. Die Gründe für den schnelleren Abbau
von PmtA in Gegenwart von MMPE, DMPE und PC sind bisher unbekannt.
Eine massenspektrometrische Analyse der entstandenen Fragmente hat gezeigt,
dass der N-Terminus von PmtA in Gegenwart von Lipiden vor einem proteolytischen
Abbau geschützt wird. PmtA besitzt mit der Aminosäuresequenz 19FFK21 eine
putative Schnittstelle für Chymotrypsin im N-terminalen Bereich. Eine Spaltung an
dieser Stelle würde ein ca. 18 kDa großes PmtA-Fragment erzeugen. Durch die
Membranbindung könnte diese Schnittstelle blockiert werden und PmtA somit vor
einem proteolytischen Abbau geschützt werden. Entsprechend einer Sekundär-
strukturvorhersage für PmtA bilden die ersten 20 Aminosäuren eine α-Helix αA aus
(I-TASSER; Zhang, 2008). Das Homologiemodel für PmtA postuliert, dass diese
α-Helix über der globulären Kernstruktur exponiert ist (Abbildung 48). Einen
ähnlichen strukturellen Aufbau zeigt die eukaryotische CTP (Dunne et al., 1996). Die
N-terminale Helix der CTP ist ein Teil der Membranbindedomäne (Domäne M). Diese
Helix ist amphiphil und besitzt eine Nettoladung von +1. Basische Aminosäuren in
dieser Helix ermöglichen eine selektive Bindung an anionische Lipide (Taneva et al.,
2008). Die Selektivität für anionische Lipide basiert auf elektrostatischer Interaktion,
wobei die positiv geladenen Aminosäurereste mit den negativ geladenen
Lipidkopfgruppen interagieren (Johnson & Cornell, 1999). In der N-terminalen Helix
von PmtA sind ebenfalls basische Aminosäuren vorhanden (K6, R8, K12, R18 und
K21). Die Nettoladung dieser postulierten Helix αA beträgt +1. Möglicherweise

Diskussion
- 110 -
interagiert PmtA mit Hilfe dieser Helix auch über elektrostatische Interaktion mit der
Membran.
Abbildung 48: Putative Proteaseschnitstellen für PmtA. (A.): In magenta sind putative Schnittstellen für die Protease Chymotrypsin im PmtA-Homologiemodell markiert. (B.): Aminosäuresequenz von PmtA mit den putativen Schnittstellen für Chymotrypsin (via PeptidCutter; Gasteiger et al., 2003).
Das MinE Protein ist Teil des Min-Systems in E. coli, dass für die Lokalisierung des
Zellteilungsapperates wichtig ist. MinE und die eukaryotische Tyrosinhydroxylase
sind weitere Beispiele für Proteine, die mittels α-helikaler Strukturelemente mit
anionischen Lipiden interagieren (Halskau et al., 2009; Shih et al., 2011).
Desweiteren hat die N-terminale Helix von PmtA auch einen amphiphilen Charakter.
Sie besitzt mehrere Phenylalanine mit aromatischen Seitenketten. Diese könnten
nach Lipidbindung zur Membran hin orientiert werden und/oder teilweise in
hydrophobe Bereiche der Membran eintauchen. Eine hydrophobe Interaktion der
aromatischen Seitenketten und der Fettsäureketten könnte so die PmtA-
Membranassoziation stabilisieren.

Diskussion
- 111 -
Abbildung 49: PmtA-Homologiemodell mit Detailansicht der N-terminalen Helix αA von oben. Hervorgehoben sind die Phenylalanine der Helix αA. Hydrophobe Abschnitte der Helix sind in grün dargestellt, hydrophile Elemente sind blau dargestellt.
Eine solche Funktion konnte für die aromatische Aminosäure Tryptophan 142 im
humanen Glycolipidtransferprotein gezeigt werden (Kamlekar et al., 2010). Um in
Zukunft eine ähnliche Bedeutung für die Phenylalanine in PmtA nachzuweisen, bietet
sich ein Aminosäureaustausch der Phenylalanine zu Tryptophan an.
Tryptophanfluoreszenzmessungen der putativen N-terminalen α-Helix αA könnten
dann Aufschluss über eine derartige Membraninteraktion geben (Kraft et al., 2009).
Das Homologiemodell für PmtA zeigt, dass der hydrophobe Teil der Helix zum
Proteininneren hin orientiert ist (Abbildung 49). Durch eine Rotation der Helix über
den flexiblen Loop zwischen Aminosäure 24 bis 27 wäre es möglich, dass der
hydrophobe Teil der Helix zur Membran hin orientiert wird. Dadurch würde eine
Interaktion zwischen den aromatischen Seitenketten mit den Acylketten der Membran
ermöglicht. Ein solcher Mechanismus wird für das MinE-Protein aus Neisseria
gonorrhoeae postuliert (Shih et al., 2011).
Desweiteren könnte PmtA ähnlich wie andere Proteine durch eine Interaktion einer
amphiphilen Helix mit der Membran in seine korrekte Orientierung gebracht werden.
Ein Beispiel dafür ist das HIV-1 Gagprotein, das über elektrostatische Interaktionen

Diskussion
- 112 -
der N-terminalen Matrixdomäne mit der Membran interagiert. Diese elektrostatischen
Interaktionen sind notwendig, um das Protein auf der Membran funktional zu
orientieren (Lomize et al., 2007).
Die Bedeutung des N-Terminus für die PmtA-Aktivität zeigte sich bereits in
vorangegangenen Studien. N-terminale Deletionen von PmtA wirkten sich
unterschiedlich auf die PmtA-Aktivität aus, wobei generell eine erhöhte Löslichkeit
des Proteins zu beobachten war (Aktas, unveröffentlicht). Eine am N-Terminus um
10 Aminosäuren verkürzte PmtA-Variante (ΔN10) ist im Gegensatz zu einer Variante,
die um 20 Aminosäuren verkürzt ist (ΔN20), noch katalytisch aktiv. Beide Varianten
sind in der Lage Lipid zu binden (Aktas, unveröffentlicht). Die N-terminale Helix ist
daher wahrscheinlich nur partiell an der Membranbindung beteiligt. Möglicherweise
stabilisieren insbesondere die hydrophoben Aminosäurereste im N-Terminus von
PmtA die Membranassoziation und verhindern so eine rasche Dissoziation.
Eine alternative Funktion der N-terminalen Helix wäre die Blockierung des
katalytischen Zentrums. Eine solche Funktion ist für die Membranbindedomäne der
Phospholipase cPLA2 bekannt. Die N-terminale Domäne der cPLA2 gleicht einer
beweglichen Deckelstruktur und verhindert zunächst die Substratbindung. Durch
Membranassoziation erfolgt eine Konformationsänderung dieser Domäne, wodurch
eine Substratbindung im aktiven Zentrum möglich ist (Dessen et al., 1999). Die
Konformation von PmtA wird durch Membranbindung ebenfalls beeinflusst.
Möglicherweise ist von dieser Konformationsänderung auch der N-Terminus
betroffen. Die N-terminale Helix hätte dadurch eine entscheidende Bedeutung für die
Konformationsänderung. Die ΔN20-Variante ist zwar nativ gefaltet, aber es ist
möglich, dass durch die fehlenden N-terminalen Aminosäuren die Lipid-abhängige
Konformationsänderung nicht funktional ist. Auch eine negative Beeinflussung der
Substratspezifität kann durch die Deletion des N-Terminus nicht ausgeschlossen
werden.

Diskussion
- 113 -
4.2.6 Die PmtA-Lipid-Interaktion ist hoch-affin und führt zu einer
Konformationsänderung
Anhand von CD-spektroskopischen Stuiden wurde die Bedeutung von verschiedenen
Lipiden für die PmtA Struktur und Funktion näher analysiert. Die CD-Spektroskopie
ermöglicht die Analyse von Sekundärstrukturelementen ganzer Proteine oder
löslicher Proteindomänen (Kelly & Price, 2000). Auch kann die Interaktion von
Proteinen/Peptiden mit künstlichen Membranen in Form von Liposomen untersucht
werden (Vallée et al., 2001; Shih et al., 2011). Es konnte bereits gezeigt werden,
dass PmtA nur in Anwesenheit von Substratlipiden SAM bindet (Aktas &
Narberhaus, 2009). Eine mögliche Erklärung dafür ist, dass eine Substratlipidbindung
eine Konformationsänderung bewirkt, die die SAM-Bindung durch PmtA begünstigt
(Aktas & Narberhaus, 2009). Um dies zu klären, wurde mittels CD-Spektroskopie der
Einfluss von Lipiden auf die PmtA-Struktur untersucht. In Gegenwart von Liposomen
wurde die Elliptizität von PmtA beeinflusst, sodass sich der Elliptizitätswert erhöhte.
Dies war für die Substratlipide PE, MMPE, DMPE und PC über den gesamten
Bereich von 208 – 230 nm zu beobachten. Diese Veränderung des CD-Spektrums
könnte eine Erhöhung des β-Faltblattanteils und eine gleichzeitige Verringerung des
α-helikalen Anteils bedeuten. Auch könnten durch eine Lipidbindung bestimmte
Bereiche im Protein in flexible Loopregionen übergehen. Diese flexiblen
Loopregionen könnten eine Bindung von cytosolischem SAM ermöglichen. Für
eukaryotische PEMT ist beispielsweise bekannt, dass die SAM-Binderegion in
cytosolischen Loopregionen lokalisiert ist (Shields et al., 2003). PEMTs sind jedoch
integrale Membranproteine. Die Lipide PG, PS und CL führen auch zu einer
Änderung des CD-Spektrums. Die Änderung wurde jedoch tendenziell eher im
Bereich zwischen 208 – 210 nm beobachtet. Dies legt nahe, dass diese Lipide eine
geringfügig andere Konformationsänderung von PmtA im Vergleich zu den
Substratlipiden bewirken. Anhand der relativen Elliptizitätsänderung konnte die
Bindeaffinität von PmtA zu Modellmembranen bestimmt werden. Die
Dissoziationskonstanten (KD) lagen in einem mikromolaren Bereich zwischen
1 - 10 µM. Dies bedeutet, dass PmtA eine hohe Membranbindeaffinität besitzt. Eine
konformelle Änderung der Struktur durch Membraninteraktion ist eine bekannte
Konsequenz für periphere Membranproteine. Diese sind in cytosolischer oder
ungebundener Form oftmals inaktiv. Durch die Membranbindung und die daraus

Diskussion
- 114 -
resultierende Konformationsänderung gehen diese Proteine in eine aktive Form über.
Die Konformation der Pyruvatoxidase aus E. coli ändert sich beispielsweise erst nach
Membranbindung. Die Konformationsänderung führt zu einer Exponierung des
autoinhibierenden C-Terminus, wodurch eine Substratbindung möglich ist
(Neumann et al., 2008). Für die Phosphoethanolamin N-Methyltransferasen aus
Haemonchus contortus oder Plasmodium falciparum sind ähnliche konformelle
Änderungen bekannt (Lee et al., 2011; Lee et al., 2012). Durch Ligandenbindung
findet in der PfPMT aus P. falciparum eine Neuorientierung der N-terminalen Region
statt (Lee et al., 2012). Es konnte gezeigt werden, dass sich die Position und
Orientierung der Aminosäuren Y28, F31 und R179 der PfPMT ändert (Lee et al.,
2012). Eine homologe Aminosäure zu F31 befindet sich innerhalb des PmtA-
Homologiemodells nur an Position 20 (F20). Diese Aminosäure ist an einer ähnlichen
Position innerhalb der Struktur wie F31 der PfPMT lokalisiert. Möglicherweise übt
F20 in PmtA eine ähnliche Funktion wie in PfPMT aus und eine Umlagerung dieser
Aminosäure ist notwendig, um eine Substratbindung und Katalyse zu ermöglichen.
Die Glycin N-Methyltransferase, die Histamin Methyltransferase, die humane
Phenylethanolamin Methyltransferase oder die Guanidinoacetat Methyltransferase
sind weitere Beispiele für Proteine, deren Konformation sich nach Substratbindung
ändert (Horton et al., 2001; Takata et al., 2003; Komoto et al., 2004; Wu et al., 2005).
Speziell für periphere Membranproteine gibt es auch Beispiele für
Konformationsänderungen, die durch eine Interaktion mit Lipiden hervorgerufen wird.
So ändern beispielsweise Lysozyme, die cytosolische Domäne von Fis1, α-Synuclein
oder das Phosphatidylethanolaminbindeprotein (PEBP) ihre Struktur durch eine
Protein-Lipid Interaktion (Jo et al., 2000; Vallée et al., 2001; Gorbenko et al., 2007;
Wells & Hill, 2011). Im Gegensatz zu PmtA wird bei diesen Proteinen/Peptiden nach
einer Interaktion mit Lipiden meist eine Zunahme des α-helikalen Anteils beobachtet.
Die Sekundärstruktur von Peptiden ist größtenteils ungeordnet, wenn diese im
Cytosol lokalisiert sind oder in vitro in Lösung vorliegen. Erst durch eine
Membranbindung erfolgt die Ausbildung einer α-Helix. Dies gilt auch für das HiPIP-
Protein aus A. vinosum, dessen ungeordneter N-Terminus durch Membranbindung
α-helikale Strukturen ausbildet (Brehmer et al., 2012).
Die CD-spektroskopischen Daten für eine Lipid-abhängige Konformationsänderung
werden durch die Ergebnisse der PmtA-Tryptophanfluoreszenz gestützt. Eine
Lipidzugabe führt zur Abnahme der Fluoreszenzintensität und zu einer minimalen

Diskussion
- 115 -
Blauverschiebung des Fluoreszenzmaximums. Die Änderung der intrinsischen
Tryptophanfluoreszenz eines Proteins ist oftmals die Konsequenz einer
Konformationsänderung. (Kraft et al., 2009). Beispielsweise wurden für das humane
β2-Glycoprotein I (β2GPI), das wichtig für die Blutgerinnung ist (Brigthon et al., 1996),
eine deutliche Abnahme der Fluoreszenzintensität und eine Verschiebung des
Fluoreszenzmaximums nach Interaktion mit CL beobachtet (Hammel et al., 2001). Im
Gegensatz zu β2GPI besitzt PmtA nur ein Tryptophan (W192). Daher kann der
Ursprungsort der beobachteten Fluoreszenzänderung auf eine bestimmte Region
innerhalb der PmtA-Struktur eingegrenzt werden. Eine mögliche Erklärung für die
Fluoreszenzabnahme ist, dass eine Amidgruppe nach Lipidbindung in räumliche
Nähe zum Tryptophan hin orientiert wird. Arginin, Asparagin, Glutamin und Lysin sind
Aminosäuren mit einer Amidgruppe und können die Emmision der
Tryptophanfluoreszenz beeinflussen (Bushueva et al., 1975; Clark et al., 1996;
Henneke et al., 1997). Die Reste der Aminosäuren Q158, Q170, R184, N185 und
Q190 besitzen Amidgruppen, die in räumlicher Nähe von W192 lokalisiert sind
(Abbildung 50).
Abbildung 50: Detailansicht des Tryptophans in PmtA. Hervorgehoben ist das Tryptophan (rot) und amidgruppentragende Aminosäuren (blau) in der räumlichen Nachbarschaft. Grüne Striche zeigen den Abstand der Amidgruppen zum Tryptophan an. Ǻ: Angström.
Diese sind zwischen 3 und 11 Å von W192 entfernt und könnten durch eine
Konformationsänderung näher zum Tryptophan hin orientiert werden.

Diskussion
- 116 -
Neben der Fluoreszenzabnahme wurde eine Blauverschiebung des Fluoreszenz-
maximums nach Lipidzugabe beobachtet. Allerdings ist die Verschiebung um
maximal 4 nm sehr gering. Eine Blauverschiebung des Maximums bei einer Protein-
Lipid-Interaktion bedeutet, dass das Tryptophan in einer lipophilen Umgebung
exponiert ist. Dies könnte durch eine geringfügige Einbettung in die Membran
geschehen, ähnlich wie beim β-Glycoprotein I (Hammel et al., 2001;
Ren et al., 1997). Auch ist es möglich, dass das Tryptophan zur Acylkette des
gebunden Phospholipides hin orientiert wird. Ähnliche Beobachtungen wurden
mittels Tryptophanmessungen für das Phosphatidylethanolaminbindeprotein (PEBP)
oder der CTP gemacht (Vallée et al., 2001; Lavor et al., 2007).
Eine alternative Methode zur Quantifizierung der Protein-Lipid-Interaktion basiert auf
Fluorescein-PE (FPE). Dabei wird zu einer definierten Konzentration an FPE-
markierten Membranen das Zielprotein hinzu titriert. Anhand der Änderung der
Fluoreszenzintensität kann eine Protein-Lipid Interaktion quantifiziert werden. Die
Fluoreszenzintensität ist dabei abhängig vom Membranpotential, das wiederum
direkte Auswirkung auf den Protonierungsgrad des Xanthenringes der
Fluoresceingruppe hat. Eine Deprotonierung des Xanthenringes führt letztendlich zur
Änderung der Fluoreszenzintensität (O´Toole et al., 1999). Mit Hilfe von FPE wurde
bereits erfolgreich die Bindungsaffinität von Spectrin zu Modellmembranen bestimmt
(O´Toole et al., 1999). Die Zugabe von PmtA zu FPE-markierten Membranen
bewirkte eine Zunahme der Fluoreszenzintensität. Die Erhöhung der
Fluoreszenzintensität war jedoch für die verschiedenen Modellmembranen
unterschiedlich stark ausgeprägt. Dies ist wahrscheinlich auf ein unterschiedlich
großes Membranpotential der Modellmembranen zurückzuführen. Das
Ladungspotential einer Membran ist abhängig von der Beschaffenheit des
verwendeten Lipides. Beispielsweise ist CL doppelt so negativ geladen wie PG
(Abbildung 2). Eine Modellmembran aus CL ist daher wahrscheinlich insgesamt
negativer geladen, als eine Membran, die aus PG besteht. Möglicherweise ist daher
die Fluoreszenzzunahme bei CL-Membranen stärker, als bei PG-Membranen.
Anhand der relativen Fluoreszenzerhöhung wurden für PmtA Bindungskonstanten
gegenüber den jeweiligen Modellmembranen bestimmt. Die ermittelten KD-Werte (3 –
5 µM) sind in derselben Größenordnung wie die durch CD-Spektroskopie ermittelten
Werte (1 – 10 µM).

Diskussion
- 117 -
Die Ergebnisse der CD- und Fluoreszenzspektroskopie sind in Tabelle 8
zusammengestellt. Insgesamt zeigen die ermittelten Daten Bindungskonstanten im
mikromolaren Bereich für die PmtA-Lipid Interaktion. Dies bedeutet, dass die
Bindung von PmtA an Lipide hoch-affin ist.
Tabelle 8: Vergleich der Bindungsaffinitäten von PmtA zu verschiedenen Lipiden. At: A. tumefaciens-Lipide; CD: CD-Spektroskopie; CL: Cardiolipin; DMPE: Dimethyl-PE; Ec: E. coli-Lipide; FPE: Fluorescein-PE; KD: Dissoziationskonstante; MMPE: Monomethyl-PE; PC: Phosphatidylcholin; PE: Phosphatidylethanolamin; PG; Phosphatidylglycerin; PS: Phosphatidylserin.
Für Enzym-Substrat-Komplexe sind KD –Werte zwischen 10-7 bis 10-3 M üblich
(Nelson & Cox, 2009). Für periphere Membranproteine wurden KD-Werte von 500 µM
bis hin zu 33 nM gegenüber Membranen beschrieben (Johnson & Cornell, 1999). Die
ermittelten KD-Werte für PmtA liegen daher in einer bekannten Größenordnung für
die Interaktion eines peripheren Proteins mit einer Membran. PmtA besitzt also eine
hohe Affinität gegenüber Membranen und weiterhin führt die Interaktion mit Lipiden
zu einer Konformationsänderung von PmtA.

Diskussion
- 118 -
4.2.7 Gibt es in PmtA essentielle Aminosäuren für eine
Lipidinteraktion?
Für bakterielle Pmt-Enzyme konnten bisher keine Proteinstrukturen aufgeklärt
werden. Weiterhin besitzen bakterielle Pmt-Enzyme keine bekannten
Lipidbindemotive wie beispielsweise PH-Homologie oder das Ca2+-Motiv
(DiNitto el al., 2003). Daher konnten Aminosäuren, die für eine Lipidinteraktion
essentiell sind nur aufgrund von Aminosäuresequenzvergleichen identifiziert werden.
In silico-Analysen des PmtA-Homologiemodells postulieren darüber hinaus zwei
Bereiche auf der PmtA-Oberfläche, die stark positiv geladen sind. Basische
Aminosäuren, die diese positive Ladung ausbilden, sind potentielle
Interaktionspartner für anionische Lipide. Daher wurden, neben konservierten
Aminosäuren, basischer Aminosäuren dieser PmtA-Oberfläche mutiert und der
Einfluss der Punktmutation auf die PmtA-Aktivität untersucht.
Eine Mutation basischer und konservierter Aminosäuren im N-terminalen Bereich von
PmtA beeinflusste die PmtA-Aktivität nicht oder nur sehr geringfügig. Dies legt nahe,
dass diese Aminosäuren für die PmtA-Aktivität und Membranassoziation
wahrscheinlich nicht von Bedeutung sind. Nur eine Doppelmutante K28A,K29A
zeigte eine geringfügig reduzierte PmtA-Aktivität. Dies könnte strukturell bedingt sein,
da ein doppelter Aminosäureaustausch wahrscheinlich einen größeren Einfluss auf
die Faltung und Stabilität eines Proteins hat, als eine einzelne Punktmutation. Eine
Mutation der Aminosäuren K42 oder K43 führten hingegen zu einem drastischen
Aktivitätsverlust. Es konnte jedoch keine Überexpression dieser Proteine
nachgewiesen werden. Es ist daher möglich, dass diese Mutation die PmtA-Struktur
und Stabilität stark beeinflusst und wahrscheinlich zu einer Missfaltung des Proteins
führt. Das überexprimierte PmtA wird vermutlich durch Proteasen im Rahmen der
Qualitätskontrolle schnell degradiert. Eine Mutation von Lysin zu Alanin ist dabei ein
drastischer Austausch bezüglich der molekularen Größe der Aminosäure. Ein
Aminosäureaustausch von Lysin zu Glutamat oder Glutamin würde die Faltung
möglicherweise geringfügiger beeinflussen. Die konträre Ladung könnte
elektrostatische Interaktion mit anionischen Lipiden jedoch mindern. Die Bedeutung
von kationischen Aminosäuren für die Membranbindung konnte bereits für die
Phospholipase cPLA2 und die CTP: Phosphocholin Cytidylyltransferase gezeigt
werden (Snitko et al., 1997; Johnson et al., 2003). Eine Mutation von Lysin zu

Diskussion
- 119 -
Glutamin in der Domäne M der CTP reduziert die Membranassoziation. Durch diese
Mutation wurde die elektrostatische Bindung eliminiert (Johnson et al., 2003).
Weiterhin konnte gezeigt werden, dass auch drei aufeinanderfolgende Glutamate in
der Domäne M teilweise an der Membranassoziation beteiligt sind. Eine Mutation
dieser Aminosäuren führte zu einer stärkeren Membranbindung bei hoher ionischer
Stärke (Johnson et al., 2003). Eine Anordnung von drei aufeinanderfolgenden
Glutamaten ist auch in der N-terminalen Helix von PmtA lokalisiert. Möglicherweise
besitzen diese Aminosäuren eine ähnliche Bedeutung für die PmtA-Membran
Interaktion. In zukünftigen Arbeiten sollte daher auch die Bedeutung dieser
Glutamate näher aufgeklärt werden.
Einen interessanten und bisher unbekannten Effekt zeigte eine Mutation des
Threonins an Position 36 in PmtA. Eine Mutation der Aminosäure T36 zu Alanin
führte in vivo zu einer vermehrten Akkumulation von MMPE im Vergleich zu wildtyp
PmtA. Die Aminosäure T36 ist am N-Terminus und an der Oberfläche des PmtA-
Homologiemodells lokalisiert (Abbildung 51). T36A konnte im Gegensatz zu anderen
PmtA-Derivaten in ausreichenden Mengen isoliert werden. In vitro konnte jedoch
keine erhöhte Akkumulation von MMPE im Vergleich zu wildtyp PmtA nachgewiesen
werden. Verglichen mit PmtA synthetisierte T36A nur geringere Mengen des
Endproduktes PC. CD-spektroskopische Analysen zeigten, dass T36A nativ gefaltet
ist. Auch die Affinität zu Phospholipidsubstraten oder PG wurde durch die Mutation
nicht beeinflusst, sodass T36A möglicherweise im effizienten Transfer der
Methylgruppe von SAM auf das Phospholipidsubstrat oder generell in der
Reaktionsgeschwindigkeit beeinträchtig ist. Für das humane RAS-Proteine wurde
ebenfalls ein Threonin an Position 50 identifiziert, dass mit PC interagiert
(Cristea et al., 2009). Die Interaktion beruht dabei auf Wasserstoffbrückenbindungen
zwischen der Hydroxylgruppe des Threonins und der Phosphatgruppe des PCs. Eine
ähnliche Interaktion könnte auch T36 in PmtA ausführen. Eine Bedeutung von T36
für die Orientierung, Substratbindung oder auch Konformationsänderung von PmtA,
könnte nur durch eine Strukturaufklärung mit gebundenem Phospholipidsubstrat
eindeutig geklärt werden. Ein Strukturvergleich von PmtA mit PfPMT zeigt, dass T36
in PmtA an einer äquivalenten Position wie S37 in PfPMT lokalisiert ist. Die
Aminosäuren Threonin und Serin besitzen beide eine Hydroxylgruppe in ihrer
funktionellen Seitenkette.

Diskussion
- 120 -
Abbildung 51: Struktureller Vergleich von PmtA (grau) und PfPMT (gelb). Hervorgehoben sind die
Aminosäuren Threonin 36 in PmtA und Serin 37 in PfPMT.
Für PfPMT wird postuliert, dass S37 mit der Carboxylgruppe von SAM interagiert.
Dadurch wird SAM für die Transmethylierungsreaktion korrekt orientiert
(Lee et al., 2012). Eine derartige Funktion könnte T36 auch in PmtA übernehmen.
Daher ist es wahrscheinlich, dass T36 eher eine Bedeutung für die SAM-Bindung, als
für die Lipidbindung von PmtA hat.
Zudem wurden Punktmutationen konservierter Aminosäuren im zentralen und C-
terminalen Bereich der PmtA-Struktur eingefügt. Mutationen im zentralen Bereich der
(Aminosäure 44 – 160 und 187 – 192) bewirkte größtenteils eine stark reduzierte
Enzymaktivität von PmtA. Jedoch waren die generierten PmtA-Derivate unlöslich,
weshalb die Ursachen des enzymatischen Defektes vorerst ungeklärt bleiben.
Möglich ist es, dass die eingefügte Mutation die Stabilität der PmtA-Struktur zu sehr
beeinflusst. Mutationen am C-Terminus hatten bis auf das unlösliche PmtA-Derivat
W192A keinen Einfluss auf die PmtA-Aktivität. Zur Identifizierung essentieller
Aminosäuren von PmtA für die Interaktion mit Lipiden, scheint eine zukünftige
Strukturaufklärung von PmtA die vielversprechendste Basis zu bieten.

Diskussion
- 121 -
4.2.8 Postuliertes Modell für die PmtA-Membran-Interaktion
In dieser Arbeit wurde gezeigt, dass PmtA an anionische Lipide durch
elektrostatische Interaktionen bindet. Die Bindung an anionische Lipide, sowie an
Substratlipide bewirkt eine Konformationsänderung von PmtA. Diese
Konformationsänderung ist wiederum notwendig, um den Methyldonor SAM binden
zu können. Anhand dieser Resultate, kann folgendes Modell zur Membran-
rekrutierung und Substratbindung von PmtA postuliert werden (Abbildung 52):
Abbildung 52: Das postulierte PmtA-Membran-Interaktionsmodell: PmtA wird durch elektrostatische Interaktionen zunächst an die Membran rekrutiert. Die Membran- und Substratbindung bewirken eine Konformationsänderung. Diese ermöglicht die Bindung des Kofaktors SAM und die Methylierungsreaktion, bei der PC synthetisiert wird. Als Nebenprodukt entsteht SAH. PC und SAH inhibieren PmtA. SAM: S-Adenosylmethionin. SAH: S-Adenosylhomocystein.
PmtA liegt zunächst als inaktives, cytosolisch lokalisiertes Protein vor. Durch
elektrostatische Interaktion basischer Aminosäuren mit anionischen Lipiden wird
PmtA an die Membran rekrutiert (Abbildung 52; 1.). Die elektrostatische Interaktion
beschleunigt jedoch nur die Membranassoziation und ist nicht essentiell für eine
Membranbindung, da PmtA die Substratlipide auch in Abwesenheit von anionischen
Lipiden binden kann. Nach der Membranbindung erfolgt eine konformelle

Diskussion
- 122 -
Umlagerung bzw. Neuordnung der PmtA-Struktur. Dadurch können die Reste
aromatischer Aminosäuren zur Membran hin exponiert werden, um die Bindung an
die Membran zu verstärken (Abbildung 52; 2.). Im nächsten Schritt bindet PmtA sein
Substratlipid, das ein Bestandteil der Membran ist. Die Bindung eines Substratlipides
ermöglicht dann die Bindung des Kofaktors SAM (Abbildung 52; 3.). Als letzter Schritt
erfolgt die Transmethylierungsreaktion, wobei die Methylgruppe von SAM auf das
Substratlipid übertragen wird. Dadurch entsteht das Nebenprodukt SAH und das
Endprodukt PC. Diese beiden Reaktionsprodukte inhibieren die PmtA-Aktivität.
Dieses Modell liefert einen ersten Einblick in die Funktionsweise bakterieller Pmt-
Enzyme. Auf Basis der etablierten Methoden und erzielten Resultate dieser Arbeit
können zukünftig die Auswirkungen von Mutationen auf die PmtA-Lipid-Interaktion
analysiert werden. Eine initiale Rekrutierung von PmtA an die Membran scheint
durch die Interaktion basischer Aminosäuren mit anionischen Lipiden von besonderer
Bedeutung zu sein. Es wäre daher lohnenswert die Rolle der positiv geladenen
PmtA-Oberfläche durch gezielte Mutagenese näher zu charakterisieren. Ebenfalls
bedarf die Bedeutung der N-terminalen Helix für die Lipidbindung weiterer Analysen.
Einen detaillierten Einblick in die Funktionsweise von PmtA und die Interaktion mit
Lipiden würde eine Strukturaufklärung in Anwesenheit von Lipiden liefern. Dies
würde weiterhin die Identifizierung wichtiger Aminosäuren für die spezifische PmtA-
Lipid-Interaktion erleichtern.

Zusammenfassung
- 123 -
5. Zusammenfassung
Für alle drei Domänen des Lebens sind biologische Membranen und deren
spezifische Lipidzusammensetzung essentiell. Eine bakterielle Membran besteht
meist aus den Lipiden Phosphatidylethanolamin (PE), Phosphatidylglycerin (PG) und
Cardiolipin (CL). Phosphatidylcholin (PC) ist hingegen eher selten und meist nur in
Membranen von Bakterien zu finden, die mit einem eukaryotischen Wirt interagieren.
Agrobacterium tumefaciens ist ein pflanzenpathogenes Bakterium, das in der Lage
ist, PC mittels einer Phospholipid N-Methyltransferase (PmtA) zu synthetisieren.
PmtA methyliert PE zu PC und nutzt als Methyldonor S-Adenosylmethionin (SAM).
Diese Reaktion wird durch das anionische Membranlipid PG stimuliert. Im Rahmen
dieser Arbeit sollten die SAM- und die Lipid-Bindeeigenschaften von PmtA näher
charakterisiert werden. Weiterhin sollte geklärt werden, auf welchem Mechanismus
der stimulierende Effekt von PG auf die PmtA-Aktivität beruht.
Mit Hilfe von Mutagenesestudien und bioinformatischen Analysen konnten
Aminosäuren in PmtA identifiziert werden, die für die SAM-Bindung spezifisch und
essentiell sind. Dabei konnte ausgeschlossen werden, dass die generierten
Punktmutationen die Proteinfaltung oder Lipidbindefähigkeit beeinflussten. Mit Hilfe
eines Homologiemodells von PmtA wurde die SAM-Bindetasche identifiziert, sowie
ein Bindungsmechanismus postuliert, der wichtige Grundlagen für die SAM-Bindung
durch bakterielle Pmt-Enzyme aufzeigt.
Zudem wurde durch verschiedene spektroskopische Methoden die Lipidbindung von
PmtA quantifiziert. Dissoziationskonstanten für die PmtA-Lipid-Interaktion liegen im
mikromolaren Bereich und deuten somit auf eine hoch-affine Bindung hin.
Basierend auf der in dieser Arbeit erzielten Resultate wurde ein Modell für die
Membran-Rekrutierung und Substratbindung von PmtA aufgestellt: Durch Bindung an
anionische Lipide wird der erste Kontakt zur Membran hergestellt. Die Bindung an
anionische Lipide führt zu einer Konformationsänderung, welche die Lipid-
Substratbindung erleichtert. Die Substratbindung wiederum geht mit einer weiteren
Konformationsänderung einher, die erst die Bindung des Methyldonors SAM
ermöglicht.
Die Resultate dieser Arbeit tragen somit zum Verständnis der Funktionsweise von
bakteriellen Phospholipid N-Methyltransferasen bei.

Summary
- 124 -
6. Summary
Biological membranes and their specific lipid composition are essential for all
domains of life. A typical bacterial membrane is composed of phosphatidyl-
ethanolamine (PE), phosphatidylglycerol (PG) and cardiolipin (CL).
Phosphatidylcholine (PC), a typical eukaryotic membrane lipid, is also present in a
restricted number of bacteria, especially in those interacting with eukaryotes. PC
plays a crucial role in bacteria-host interactions and is essential for the virulence of
the plant pathogen Agrobacterium tumefaciens. Its phospholipid N-methyltransferase
PmtA catalyzes the formation of PC by a three-step methylation of PE. The methyl
group is provided by S-adenosylmethionine (SAM), which is converted to S-
adenosylhomocysteine. This reaction is stimulated by the anionic lipid PG.
The aim of this work was to characterise the SAM- and lipid-binding properties of
PmtA and to elucidate the mechanism for the stimulation of PmtA activity by PG.
Using bioinformatic and mutagenesis analysis residues essential for SAM binding by
PmtA were identified. The point mutations did not influence PmtA folding and lipid
binding as shown by CD spectroscopy. A PmtA homology model supported the
mutagenesis data and provided a useful basis for SAM-binding mechanism and
structure. The postulated SAM-binding mechanism for PmtA illustrates basic
principles for SAM binding by SAM-methyltransferases.
Furthermore, lipid binding of PmtA was analysed and quantified using different
independent spectroscopic approaches. These studies revealed binding constants
for the PmtA-lipid interaction in a low micromolar range indicating a high-affinity lipid
binding.
Based on these results, a model for membrane recruitment and substrate binding of
PmtA can be postulated: The initial contact between PmtA and the membrane
depends on anionic lipids. This interaction induces a conformational change in PmtA
facilitating lipid-substrate binding, which in turn causes a second conformational
change finally allowing SAM binding.
This study provides valuable insights into mechanism and structural features of
bacterial Pmt enzymes.

Literaturverzeichnis
- 125 -
7. Literaturverzeichnis
Aktas, M. and F. Narberhaus. 2009. In vitro characterization of the enzyme properties of the phospholipid N-methyltransferase PmtA from Agrobacterium tumefaciens. J Bacteriol. 191: 2033-2041.
Aktas, M., Gleichenhagen, J., Stoll, R., and F. Narberhaus. 2011a. S-Adenosylmethionine-binding properties of a bacterial phospholipid N-methyltransferase. J Bacteriol. 193: 3473-3481.
Aktas, M., Jost, K.A., Fritz, C., and F. Narberhaus. 2011b. Choline uptake in Agrobacterium tumefaciens by the high-affinity ChoXWV transporter. J Bacteriol. 193: 5119-5129.
Aktas, M., Wessel, M., Hacker, S., Klüsener, S., Gleichenhagen, J. and F. Narberhaus. 2010. Phosphatidylcholine biosynthesis and its significance in bacteria interacting with eukaryotic cells. Eur J Cell Biol. 89: 888-894.
André, I., Kesvatera, T., Jönsson, B., Akerfeldt, K.S. and S. Linse. 2004. The role of electrostatic interactions in calmodulin-peptide complex formation. Biophys J. 87: 1929-1938.
Arondel, V., Benning, C. and C.R. Somerville. 1993. Isolation and functional expression in Escherichia coli of a gene encoding phosphatidylethanolamine methyltransferase (EC 2.1.1.17) from Rhodobacter sphaeroides. J Biol Chem. 268: 16002-16008.
Baker, N.A., Sept, D., Joseph, S., Holst, M.J. and J.A. McCammon. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 98: 10037-10041.
Basturea, G.N. and M.P. Deutscher. 2007. Substrate specificity and properties of the Escherichia coli 16S rRNA methyltransferase, RsmE. RNA. 13: 1969-1976.
Bevers, E.M., Comfurius, P., and R.F.A. Zwaal. 1983. Changes in membrane phospholipid distribution during platelet activation. Biochim Biophy Acta. 736: 57-66.
Birnboim, H.C. and J. Doly. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7: 1513-1523.
Bligh, E.G. and W.J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 37: 911-917.
Boumann, H.A., Chin, P.T., Heck, A.J., De Kruijff, B. and Al. De Kroon. 2004. The yeast phospholipid N-methyltransferases catalyzing the synthesis of phosphatidylcholine preferentially convert di-C16:1 substrates both in vivo and in vitro. J Biol Chem. 279: 40314-40319.
Brehmer, T., Kerth, A., Graubner, W., Malesevic, M., Hou, B., Brüser, T. and A. Blume. 2012. Negatively charged phospholipids trigger the interaction of a bacterial tat substrate precursor protein with lipid monolayers. Langmuir. 28: 3534-3541.

Literaturverzeichnis
- 126 -
Bremer, J. and D. M. Greenberg. 1959. Mono- and dimethylethanolamine isolated from ratliver phospholipids. Biochim Biophys Acta. 35: 287-288.
Brighton, T.A., Hogg, P.J., Dai, Y.P., Murray, B.H., Chong, B.H. and C.N. Chesterman. 1996. β2-glycoprotein I in thrombosis: evidence for a role as a natural anticoagulant. Br J Haematol. 93: 185-194.
Brophy, P.J., Choy, P.C., Toone, J.R. and D.E. Vance. 1977. Choline kinase and ethanolamine kinase are separate, soluble enzymes in rat liver. Eur J Biochem. 78: 491-495.
Bruscella, P., Cassagnaud, L., Ratouchniak, J., Brasseur, G., Lojou, E., Amilis, R. and V. Bonnefoy. 2005. The HiPIP from the acidophilic Acidithiobacillus ferrooxidans is correctly processed and translocated in Escherichia coli, in spite of the periplasm pH difference between these two micro-organisms. Microbiology. 151: 1421-1431.
Bujnicki, J. M. 1999. Comparison of protein structures reveals monophyletic origin of the AdoMet-dependent methyltransferase family and mechanistic convergence rather than recent differentiation of N4-cytosine and N6-adenine DNA methylation. In Silico Biol. 1: 175-182.
Bushueva, T.L., Busel, E.P. and E.A. Burstein. 1975. The interaction of protein functional groups with indole chromophore. III. Amine, amide and thiol groups. Studia Biophys 52: 41-52
Bügl, H., Fauman, E.B., Staker, B.L., Zheng, F., Kushner, S.R., Saper, M.A., Bardwell, J.C and U. Jakob. 2000. RNA methylation under heat shock control. Mol Cell. 6: 349-360.
Cheng, X., Kumar, S., Posfai, J., Pflugrath, J.W. and R.J. Roberts. 1993. Crystal structure of the HhaI DNA methyltransferase complexed with S-Adenosyl-L-methionine. Cell 74: 299-307.
Cirstea, I.C., Kutsche, K., Dvorsky, R., Gremer, L., Carta, C., Horn, D., Roberts, A.E., Lepri, F., Merbitz-Zahradnik, T., König, R., Kratz, C.P., Pantaleoni, F., Dentici, M.L., Joshi, V.A., Kucherlapati, R.S., Mazzanti, L., Mundlos, S., Patton, M.A., Silengo, M.C., Rossi, C., Zampino, G., Digilio, C., Stuppia, L., Seemanova, E., Pennacchio, L.A., Gelb, B.D., Dallapiccola, B., Wittinghofer, A., Ahmadian, M.R., Tartaglia, M. and M. Zenker. 2010. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 42: 27-29.
Clark, P.L., Liu, Z.P., Zhang, J. and L.M. Gierasch. 1996. Intrinsic tryptophans of CRABPI as probes of structure and folding. Protein Sci. 5: 1108-1117.
Comerci, D.J., Altabe, S., de Mendoza, D. and R.A. Ugalde. 2006. Brucella abortus synthesizes phosphatidylcholine from choline provided by the host. J Bacteriol. 188: 1929-1934.

Literaturverzeichnis
- 127 -
Conover, G.M., Martínez-Morales, F., Heidtman, M.I., Luo Z.Q., Tang, M., Chen, C., Geiger, O. and R.R. Isberg. 2008. Phosphatidylcholine synthesis is required for optimal function of Legionella pneumophila virulence determinants. Cell Microbiol. 10: 514-528.
Constantinescu, I. and M. Lafleur. 2004. Influence of the lipid composition on the kinetics of concerted insertion and folding of melittin in bilayers. Biochim Biophys Acta. 1667: 26-37.
Cornell, R.C. 1991a. Regulation of CTP:phosphocholine cytidylyltransferase by lipids 1. Negative surface charge dependence for activation. Biochemistry. 30: 5873-5880.
Cornell, R.C. 1991b. Regulation of CTP:phosphocholine cytidylyltransferase by lipids 2. Surface curvature, acyl chain length and lipid-phase dependence for activation. Biochemistry. 30: 5881-5888.
Cornell, R.B. and I.C. Northwood. 2000. Regulation of CTP:phosphocholine cytidyltransferase by amphitropism and relocclization. Trends Biochem Sci. 25: 441-447.
Cui, Z., Vance, J.E., Chen, M.H., Voelker, D.R. and D.E. Vance. 1993. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J Biol Chem. 268: 16655-16663.
Dalton, A.K., Ako-Adjei, D., Murray, P.S., Murray, D. and V.M. Vogt. 2007. Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J Virol. 81: 6435-6445.
Danne, L.M. 2010. In vitro characterization of the phospholipid-binding properties of the phospholipid N-methyltransferase PmtA from Agrobacterium tumefaciens. Bachelorarbeit, Ruhr-Universität Bochum.
Das, S. Dixon, J.E. and W. Cho. 2002. Membrane-binding and activation mechanism of PTEN. PNAS. 100: 7491-7496
de Rudder, K.E.E., Thomas-Oates, J.E. and O. Geiger. 1997. Rhizobium meliloti mutants deficient in phospholipid N-methyltransferase still contain phosphatidylcholine. J Bacteriol. 179: 6921-6928.
de Rudder, K.E.E., Sohlenkamp, C. and O. Geiger. 1999. Plant-exuded choline is used for rhizobial membrane lipid biosynthesis by phosphatidylcholine synthase. J Biol Chem. 274: 20011-20016.
de Rudder, K.E.E., López-Lara, I.M. and O. Geiger. 2000. Inactivation of the gene for phospholipid N-methyltransferase in Sinorhizobium meliloti: phosphatidylcholine is required for normal growth. Mol Microbiol. 37: 763-772.
Dessen, A., Tang, J., Schmidt, H., Stahl, M., Clark, J.D., Seehra, J. and W.S. Somers. 1999. Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell. 97: 349-360.

Literaturverzeichnis
- 128 -
DiNitto, J.P., Cronin, T.C. and D.G. Lambright. 2003. Membrane recognition and targeting by lipid-binding domains. Sci STKE. 2003:re16.
Dowhan, W. 1997. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu Rev Biochem. 66: 199-232.
Dowler, S., Kular, G. and D.R. Alessi. 2002. Protein lipid overlay assay. Sci STKE. 2002: pl6.
Dunne, S.J., Cornell, R.B., Johnson, J.E., Glover, N.R. and A.S. Tracey. 1996. Structure of the membrane binding domain of CTP:phosphocholine cytidylyltransferase. Biochemistry. 35: 11975-11984.
Dupont, L., Garcia, I., Poggi, M.C., Alloing, G., Mandon, K. and D. Le Rudulier. 2004. The Sinorhizobium meliloti ABC transporter Cho is highly specific for choline and expressed in bacteroids from Medicago sativa nodules. J Bacteriol. 186: 5988-5996.
Ebeling, J.G., Vandenbark, G.R., Kuhn, L.J., Ganong, B.R., Bell, R.M and J.E. Niedel. 1985. Diacylglycerols mimic phorbol diester induction of leukemic cell differentiation. Prot Natl Acad Sci USA. 82: 815-819.
Exton, J.H. 1994. Phosphatidylcholine breakdown and signal transduction. Biochim Biophys Acta. 1212: 26-42.
Fauman E.B., Blumenthal R.M. and X. Cheng. 1999. Structure and evolution of AdoMetdependent MTases. In: Cheng X., Blumenthal, R. M. (ed.): S-Adenosylmethionine-dependent methyltransferases: structures and functions, vol. I. World Scientific Inc., Singapore: 38.
Fontana, A., de Lauretto, P.P., Spolaore, B., Frare, E. Picotti, P. and M. Zambonin. 2004. Probing protein structure by limited proteolysis. Acta Biochim Pol. 51: 299-321.
Garnier, J., Gibrat, J.F. and B. Robson. 1996. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 266: 540-553.
Gastieger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R.D. and A. Bairoch. 2003. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31: 3784-3788.
Gaynor, P.M. and G.M. Carman. 1990. Phosphatidylethanolamine methyltransferase and phospholipid methyltransferase activities from Saccharomyces cerevisiae. Enzymological and kinetic properties. Biochim Biophys Acta. 1045: 156-163.
Gisk, B., Yasui, Y., Kohchi, T. and N. Frankenberg-Dinkel. 2010. Characterization of the haem oxygenase protein family in Arabidopsis thaliana reveals a diversity of functions. Biochem J. 425: 425-434.
Goldfine, H. 1984. Bacterial membranes and lipid packing theory. J Lipid Res. 25: 1501-1507.

Literaturverzeichnis
- 129 -
Grobenko, G.P., Ioffe, V.M. and P.K. Kinnunen. 2007. Binding of lysozyme to phospholipid bilayers: evidence for protein aggregation upon membrane association. Biophys J. 93: 140-153.
Hacker S., Sohlenkamp C., Aktas M., Geiger O. and F. Narberhaus. 2008. Multiple phospholipid N-methyltransferases with distinct substrate specificities are encoded in Bradyrhizobium japonicum. J Bacteriol 190: 571-580.
Hakizimana, P., Masureel, M., Gbaquidi, B., Ruysschaert, J.M. and C. Govaerts. 2008. Interactions between phosphatidylethanolamine headgroup and LmrP, a multidrug transporter: a conserved mechanism for proton gradient sensing? J Biol Chem. 283: 9359-9376.
Halskau, Ø Jr., Ying, M. Baumann, A., Kleppe, R., Rodriquez-Larrea, D., Almås, B., Haavik, J. and A. Martinez. 2009. Three-way interaction between 14-3-3 proteins, the N-terminal region of tyrosine hydroxylase, and negatively charged membranes. J Biol Chem. 284: 32758-32769.
Hammel, M., Schwarzenbacher, R., Gries, A., Kostner, G.M., Laggner, P. and R. Prassl. 2001. Mechanism of the interaction of β2-glycoprotein I with negatively charged phospholipid membranes. Biochemistry. 40: 14173-14181.
Hanada, T., Kashima, Y., Kosugi, A., Koizumi, Y., Yanagida, F. and S. Udaka. 2001. A gene encoding phosphatidylethanolamine N-methyltransferase from Acetobacter aceti and some properties of its disruptant. Biosci Biotechnol Biochem. 65: 2741-2748.
Haydock, S.F., Dowson, J.A., Dhillon, N., Roberts, G.A., Cortes, J. and P.F. Leadlay. 1991. Cloning and sequence analysis of genes involved in erythromycin biosynthesis in Saccharopolyspora erythraea: sequence similarities between EryG and a family of S-Adenosylmethionine-dependent methyltransferases. Mol Gen Genet. 230: 120-128.
Heinig, M. and D. Frishman. 2004. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res. 32: W500-W502.
Hennecke, J., Sillen, A., Huber-Wunderlich, M., Engelborghs, Y. and R. Glockshuber. 1997. Quenching of tryptophan fluorescence by the active-site disulfide bridge in the DsbA protein from Escherichia coli. Biochemistry. 36: 6391-6400.
Horton, J.R., Sawada, K., Nishibori, M., Zhang, X., and X. Cheng. 2001. Two polymorphic forms of human histamine methyltransferase: structural, thermal, and kinetic comparisons. Structure. 9: 837–849
Jo, E., McLaurin, J., Yip, C.M., St. George-Hyslop, P and P.E. Fraser. 2000. α-Synuclein membrane interactions and lipid specificity. J Biol Chem. 275: 34328-34334.

Literaturverzeichnis
- 130 -
Johnson, J.E. and R.B. Cornell. 1999. Amphitropic proteins: regulation by reversible membrane interactions (review). Mol Membr Biol. 16: 217-235.
Johnson, J.E., Xie, M., Singh, L.M., Edge, R. and R.B. Cornell. 2003. Both acidic and basic amino acids in an amphitropic enzyme, CTP:phosphocholine cytidylyltransferase, dictate its selectivity for anionic membranes. J Biol Chem. 278: 514-522.
Jost, R., Berkowitz, O., Shaw, J. and J. Masle. 2009. Biochemical characterization of two wheat phosphoethanolamine N-methyltransferase isoforms with different sensitivities to inhibition by phosphatidic acid. J Biol Chem. 284: 31962-31971.
Kamlekar, R.K., Gao, Y., Kenoth, R., Molotkovsky, J.G., Prendergast, F.G., Malinina, L., Patel, D.J., Wessels, W.S., Venyaminov, S.Y. and R.E. Brown. 2010. Human GLTP: Three distinct functions for the three tryptophans in a novel peripheral amphitropic fold. Biophys J. 99: 2626-2635.
Kaneshiro, T. and J.H. Law. 1964. Phosphatidylcholine synthesis in Agrobacterium tumefaciens. I. Purification and properties of a phosphatidylethanolamine N-methyltransferase. J Biol Chem. 239: 1705-1713.
Kanipes, M.I. and S.A. Henry. 1997. The phospholipid methyltransferases in yeast. Biochim Biophys Acta. 1348: 134-141.
Kanipes, M.I., Hill J.E., and S. A. Henry. 1998. The Schizosaccharomyces pombe cho1+ gene encodes a phospholipid methyltransferase. Genetics. 150: 553-562.
Karnezis, T., Fisher, H.C., Neumann, G.M., Stone, B.A. and V.A. Stanisisch. 2002. Cloning and characterization of the phosphatidylserine synthase gene of Agrobacterium sp. strain ATCC 31749 and effect of its inactivation on production of high-molecular-mass (1-->3)-β-D-glucan (curdlan). J Bacteriol. 184: 4114-4123.
Kelly, S.M. and N.C. Price. 2000. The use of circular dichroism in the investigation of protein structure and function. Curr Protein Pept Sci. 1: 349-384.
Kennedy, E.P. and S.B. Weiss. 1956. The function of cytidine coenzymes in the biosynthesis of phospholipids. J Biol Chem. 222: 193-214.
Kennedy, E.P. 1989. Discovery of the pathways for the biosynthesis of phosphatidylcholine. In: Vance D.E. (ed.): Phosphatidylcholine metabolism. CRC Press, Boca Raton, Florida, USA: 1-8.
Kent, C. 1990. Regulation of phosphatidylcholine biosynthesis. Prog Lipid Res. 29: 87-105.
Kent, C. 2005. Regulatory enzymes of phosphatidylcholine biosynthesis: a personal perspective. Biochim Biophys Acta. 1733: 53-66.
Kim, J., Shishido, T., Jiang, X., Aderem, A. and S. McLaughlin. 1994. Phosphorylation, high ionic strength, and calmodulin reverse the binding of MARCKS to phospholipid vesicles. J Biol Chem. 269: 28214-28219.

Literaturverzeichnis
- 131 -
Klüsener, S., Aktas, M., Thormann, K.M., Wessel, M. and F. Narberhaus. 2009. Expression and physiological relevance of Agrobacterium tumefaciens phosphatidylcholine biosynthesis genes. J Bacteriol. 191: 365-374.
Kodaki, T. and S. Yamashita. 1987. Yeast phosphatidylethanolamine methylation pathway. Cloning and characterization of two distinct methyltransferase genes. J Biol Chem. 262: 15428-15435.
Komoto, J., Yamada, T., Takata, Y., Konishi, K., Ogawa, H., Gomi, T., Fujioka, M., and F. Takusagawa. 2004. Catalytic mechanism of guanidinoacetate methyltransferase: crystal structures of guanidinoacetate methyltransferase ternary complexes. Biochemistry. 43: 14385-14394
Kozbial, P.Z. and A.R. Mushegian. 2005. Natural history of S-Adenosylmethionine-binding proteins. BMC Struct Biol. 5: 19.
Kraft, C.A., Garrido, J.L., Leiva-Vega, L. and G. Romero. 2009. Quantitative analysis of protein-lipid interactions using tryptophan fluorescence. Sci Signal. 2: pl4.
Kresge, N., Tabor, H., Simoni, R.D. and R.L. Hill. 2005. An escape from Italy, the discovery of S-Adenosylmethionine, and the biosynthesis of creatine by Giulio L. Cantoni. J Biol Chem. 280: e35.
Kumar, S., Cheng, X., Klimasauskas, S., Mi, S., Posfai, J., Roberts R. J. and G.G. Wilson. 1994. The DNA (cytosine-5) methyltransferases. Nucleic Acids Res.
22: 1-10.
Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227: 680-685.
LeBlanc M. J., Gavino V., Perea A., Yousef I. M., Levy E. and B. Tuchweber. 1998. The role of dietary choline in the beneficial effects of lecithin on the secretion of biliary lipids in rats. Biochim Biophys Acta. 1393: 223-234.
Lee, S.G., Haakenson, W., McCarter, J.P., Williams, D.J., Hresko, M.C. and J.M. Jez. 2011. Thermodynamic evaluation of ligand binding in the plant-like phosphoethanolamine methyltransferases of the parasitic nematode Haemonchus contortus. J Biol Chem. 286: 38060-38068.
Lee, S.G., Kim, Y., Alpert, T.D., Nagata, A. and J.M. Jez. 2012. Structure and reaction mechanism of phosphoethanolamine methyltransferase from the malaria parasite Plasmodium falciparum: an antiparasitic drug target. J Biol Chem. 287: 1426-1435.
Linde, K. Gröbner, G. and L. Rilfors. 2004. Lipid dependence and activity control of phosphatidylserine synthase from Escherichia coli. FEBS Lett. 575: 77-80.
Lisa, T.A., Garrido M.N. and C.E. Domenech. 1984. Pseudomonas aeruginosa acid phosphatase and cholinesterase induced by choline and its metabolic derivatives may contain a similar anionic peripheral site. Mol Cell Biochem. 63: 113-118.
Loenen, W.A. 2006. S-Adenosylmethionine: jack of all trades and master of everything? Biochem Soc Trans. 34: 330-333.

Literaturverzeichnis
- 132 -
Lomize, A.L., Pogozheva, I.D., Lomize, M.A. and H.I. Mosberg. 2007. The role of hydrophobic interactions in positioning of peripheral proteins in membranes. BMC Struct Biol. 7: 44.
López-Lara, I.M., Sohlenkamp, C. and O. Geiger. 2003. Membrane lipids in plant-associated bacteria: their biosyntheses and possible functions. Mol Plant Microbe Interact. 16: 567-579.
Louie, K., Chen, Y.C. and W. Dowhan. 1986. Substrate-induced membrane association of phosphatidylserine synthase from Escherichia coli. J Bacteriol. 165: 805-812.
Lucchesi, G.I., Lisa, T.A. and C.E. Domenech. 1989. Choline and betaine as inducer agents of Pseudomonas aeruginosa phospholipase C activity in high phosphate medium. FEMS Microbiol Lett. 48: 335-338.
Malone, T., Blumenthal R.M. and X. Cheng. 1995. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol. 253: 618-632.
Martin, J.L. and F.M. McMillan. 2002. SAM (dependent) I AM: the S-adenosylmethionine dependent methyltransferase fold. Curr Opin Struct Biol. 12: 783-793.
Martínez-Morales, F., Schobert M., López-Lara, I.M. and O. Geiger. 2003. Pathways for phosphatidylcholine biosynthesis in bacteria. Microbiology. 149: 3461-3471.
Mashhoon, N., Carroll, M., Pruss, C., Eberhard, J., Ishikawa, S., Estabrook, R.A. and N. Reich. 2004. Functional characterization of Escherichia coli DNA adenine methyltransferase, a novel target for antibiotics. J Biol Chem. 279: 52075-52081.
McLaughlin, S. 1977. Electrostatic potentials at membrane-solution interfaces. Curr. Top. Membr. Transp. 9:71-144.
McLaughlin, S. 1989. The electrostatic properties of membranes. Annu Rev Biophys Biophys Chem. 18: 113-136.
Michel, V., Yuan, Z., Ramsubir, S. and M. Bakovic. 2006. Choline transport for phospholipid synthesis. Exp. Biol. Med. (Maywood) 231:490–504.
Miller, M.L., Soufi, B., Jers, C., Blom, N., Macek, B. and I. Mijakovic. 2009. NetPhosBac – a predictor for Ser/Thr phosphorlyation sites in bacterial proteins. Proteomics. 9: 116-125.
Minami, N., Yasuda, T., Ishii, Y., Fujimori, K. and F. Amano. 2011. Regulatory role of cardiolipin in the activity of an ATP-dependent protease, Lon, from Escherichia coli. J Biochem. 149: 519-527.
Minder, A.C., de Rudder, K.E., Narberhaus, F., Fischer, H.M., Hennecke, H. and O. Geiger. 2001. Phosphatidylcholine levels in Bradyrhizobium japonicum membranes are critical for an efficient symbiosis with the soybean host plant. Mol Microbiol. 39: 1186-1198.

Literaturverzeichnis
- 133 -
Mulgrew-Nessbitt, A., Diraviyam, K., Wang, J., Singh, S., Murray, P., Li, Z., Rogers, L., Mirkovic, N. and D. Murray. 2006. The role of electrostatics in protein-membrane interactions. Biochim Biophys Acta. 1761: 812-826.
Nelson, D. and M. Cox. 2009. Lehninger Principles of Biochemistry (4th Ed). Springer-Verlag, Berlin Heidelberg.
Neumann, P., Weidner, A., Pech, A., Stubbs, M.T. and K. Tittmann. 2008. Structural basis for membrane binding and catalytic activation of the peripheral membrane enzyme pyruvate oxidase from Escherichia coli. Proc Natl Acad Sci USA. 105: 17390-17395.
O`Toole, P.J., Morrison, I.E. and R.J. Cherry. 2000. Investigations of spectrin-lipid interactions using fluoresceinphosphatidylethanolamine as a membrane probe. Biochim Biophys Acta. 1466: 39-46.
O`Gara, M., Zhang, X., Roberts, R.J. and X. Cheng. 1999. Structure of a binary complex of HhaI methyltransferase with S-Adenosylmethionine formed in the presence of a short nonspecific DNA oligonucleotide. J Mol Biol. 287: 201-209.
Pattanayek, R., Newcomer, M.E. and C. Wagner. 1998. Crystal structure of apo-glycine N-methyltransferase (GNMT). Protein Sci. 7: 1326-1331.
Perez-Iratxeta, C. and M.A. Andrade-Navarro. 2008. K2D2: estimation of protein secondary structure from circular dichroism spectra. BMC Struct Biol. 8: 25.
Reinisch, K.M., Chen, L., Verdine, G.L. and W.N. Lipscomb. 1995. The crystal structure of HaeIII methyltransferase convalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Cell. 82: 143-153.
Ren, J., Lew, S., Wang, Z. and E. London. 1997. Transmembrane orientation of hydrophobic alpha-helices is regulated both by the relationship of helix length to bilayer thickness and by the cholesterol concentration. Biochemistry.
36: 10213-10220.
Reynolds, J.M., Takebe, S., Choi, J.Y., El Bissati, K., Witola, W.H., Bobenchik, A.M., Hoch, J.C., Voelker, D.R. and C.B. Mamoun. 2008. Biochemical and genetic analysis of the phosphoethanolamine methyltransferase of the human malaria parasite Plasmodium falciparum. J Biol Chem. 283: 7894-7900.
Ridgway, N.D. and D.E. Vance. 1987. Purification of phosphatidylethanolamine N-methyltransferasefrom rat liver. J Biol Chem. 262: 17231-17239.
Ridgway N.D., Yao, Z. and D.E. Vance. 1989. Phosphatidylethanolamine levels and regulation of phosphatidylethanolamine N-methyltransferase. J Biol Chem. 264: 1203-1207.
Ridgway, N.D. and D.E. Vance. 1992. Phosphatidylethanolamine N-methyl-transferase fromrat liver. Methods Enzymol. 209: 366-374.
Rosing, J., van Rijn, J.L., Bevers, E.M., van Dieijen, G., Comfurius, P. and R.F. Zwaal. 1985. The role of activated human platelets in prothrombin and factor X activation. Blood. 65: 319-332.

Literaturverzeichnis
- 134 -
Rost, B. and C. Sander. 1993. Prediction of protein secondary structure at better than 70% accuracy. J Mol Biol. 232:584-599.
Salamon, Z., Lindblom, G., Rilfors, L., Linde, K. and G. Tollin. 2000. Interaction of phosphatidylserine synthase from E. coli with lipid bilayers: coupled plasmon-waveguide resonance spectroscopy studies. Biophys J. 78: 1400-1412.
Sambrook, J., Fritsch, E.F. and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. 2nd ed. Plainview, N.Y.: Cold Spring Harbor Laboratory Press.
Sambrook, J. and D.W. Russell. 2001. Molecular cloning: A laboratory manual, 3rd edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
Sankpal, U.T. and D.N. Rao. 2002. Mutational analysis of conserved residues in HhaI DNA methyltransferase. Nucleic Acids Res. 30: 2628-2638.
Savic, M., Ilic-Tomic, T., Macmaster, R., Vasilijevic, B. and G.L. Coon. 2008. Critical residues for cofactor binding and catalytic activity in the aminoglycoside resistance methyltransferase Sgm. J Bacteriol. 190: 5855-5861.
Schneider, W.J. and D.E. Vance. 1979. Conversion of phosphatidylethanolamine to phosphatidylcholine in rat liver. Partial purification and characterization of the enzymatic activities. J Biol Chem. 254: 3886-3891.
Schubert H.L., Blumenthal R.M. and X. Cheng. 2003. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 28: 329-335.
Sekimizu, K. and A. Kornberg. 1988. Cardiolipin activation of dnaA protein, the initiation protein of replication in Escherichia coli. J Biol Chem. 263: 7131-7135.
Sheard, N.F., Tayek, J.A., Bistrian, B.R., Blackburn, G.L. and S.H. Zeisel. 1986. Plasma choline concentration in humans fed parenterally. Am J Clin Nutr. 43: 219-224.
Sherr, S.I. and J.H. Law. 1965. Phosphatidylcholine synthesis in Agrobacterium tumefaciens. II. Uptake and utilization of choline. J Biol Chem. 240: 3760-3765.
Shields, D.J., Altarejos, J.Y., Wang, X., Agellon, L.B. and D.E. Vance. 2003. Molecular dissection of the S-Adenosylmethionine-binding site of phosphatidylethanolamine N-methyltransferase. J Biol Chem. 278: 35826-35836.
Shih, Y.L., Huang, K.F., Lai, H.M., Liao, J.H., Lee, C.S., Chang, C.M., Mak, H.M., Hsieh, C.W. and C.C. Lin. 2011. The N-terminal amphipathic helix of the topological specificity factor MinE is associated with shaping membrane curvature. PLoS One. 6: e21425.
Smith, L., Greenfield, N.J. and S.E. Hitchcock-DeGregori. 1994. The effects of deletion of the amino-terminal helix on troponin C function and stability. J Biol Chem. 269: 9857-9863.

Literaturverzeichnis
- 135 -
Snitko, Y., Koduri, R.S., Han, S.K., Othman, R., Baker, S.F., Molini, B.J., Wilton, D.C., Gelb, M.H. and W. Cho. 1997. Mapping the interfacial binding surface of human secretory group IIa phospholipase A2. Biochemistry. 36: 14325-14333.
Sohlenkamp, C., de Rudder, K.E., Rohrs, V., López-Lara, I.M. and O. Geiger. 2000. Cloning and characterization of the gene for phosphatidylcholine synthase. J Biol Chem. 275: 18919-18925.
Sohlenkamp, C., López-Lara, I.M. and O. Geiger. 2003. Biosynthesis of phosphatidylcholine in bacteria. Prog lipid Res. 42: 115-162.
Solís-Oviedo, R.L., Martínez-Morales, F., Geiger, O. and C. Sohlenkamp. 2012. Functional and topological analysis of phosphatidylcholine synthase from Sinorhizobium meliloti. Biochim Biophys Acta. 1821: 573-581.
Sreerama, N. and R.W. Woody. 2000. Estimation of protein secondary structure from CD spectra: Comparison of CONTIN, SELCON and CDSSTR methods with an expanded reference set. Anal Biochem. 287: 252-260.
Studier, F.W. and B.A. Moffatt. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 189: 113-130.
Szeto, T.H., Rowland, S.L., Rothfield, L.I. and G.F. King. 2002. Membrane localization of MinD is mediated by a C-terminal motif that is conserved across eubacteria, archaea and chloroplasts. Proc Natl Acad Sci USA. 99: 15693-15698.
Tahara, Y., Ogawa, Y., Sakakibara, T. and Y. Yamada. 1986. Phosphatidyl-ethanolamine N-methyltransferase from Zymomonas mobilis - purification and characterization. Agric Biol Chem. 50: 257-259.
Tahara, Y., Ogawa, Y., Sakakibara, T., and Y. Yamada. 1987a. Purification and characterizationof phosphatidylethanolamine N-methyltransferase from Zymomonas mobilis. Agric Biol Chem. 51: 1425-1430.
Tahara, Y., Yamashita, T., Sogabe, A., Ogawa, Y. and Y. Yamada. 1987b. Zymomonas mobilis mutant defective in phosphatidylethanolamine N-methyltransferase. Agric Biol Chem. 51: 3179-3181.
Tahara Y., Yamashita T., Sogabe A. and Y. Ogawa. 1994. Isolation and characterization of Zymomonas mobilis mutant defective in phosphatidylethanolamine N-methyltransferase. J Gen Appl Microbiol. 40: 389-396.
Tait, J.F. and D. Gibson. 1994. Measurement of membrane phospholipid asymmetry in normal and sickle-cell erythrocytes by means of annexin V binding. J Lab Clin Med. 123: 741-748.
Takata, Y., Huang, Y., Komoto, J., Yamada, T., Konishi, K., Ogawa, H., Gomi, T., Fujioka, M., and F. Takusagawa. 2003. Catalytic mechanism of glycine N- methyltransferase. Biochemistry. 42: 8394–8402

Literaturverzeichnis
- 136 -
Taneva, S., Dennis, M.K., Ding, Z., Smith, J.L. and R.B. Cornell. 2008. Contribution of each membrane binding domain of the CTP:phosphocholine cytidylyltransferase-alpha dimer to its activation, membrane binding, and membrane cross-bridging. J Biol Chem. 283: 28137-28148.
Thomas, G.H., Southworth, T., León-Kempis, M.R., Leech, A. and D.J. Kelly. 2006. Novel ligands for the extracellular solute receptors of two bacterial TRAP transporters. Microbiology. 152: 187-198.
Vallée, B.S., Tauc, P., Brochon, J.C., Maget-Dana, R., Lelièvre, D., Metz-Boutique, M.H., Bureaud, N. and F. Schoentgen. 2001. Behaviour of bovine phosphatidylethanolamine-binding protein with model membranes. Evidence of affinity for negatively charged membranes. Eur J Biochem. 268: 5831-5841.
van Meer, G., Voelker, D.R. and G.W. Feigenson. 2008. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 9: 112-124.
Vance, D.E. and W.J. Schneider. 1981. Conversion of phosphatidylethanolamine to phosphatidylcholine. Methods Enzymol. 71: 581-588.
Vance D.E. and N.D. Ridgway. 1988. The methylation of phosphatidylethanol-amine. Prog Lipid Res. 27: 61-79.
Vance, D.E., Walkey, C.J. and Z. Cui. 1997. Phosphatidylethanolamine N-methyltransferase from liver. Biochim Biophys Acta. 1348: 142-150.
Vance, D.E. 2002. Phospholipid biosynthesis in eukaryotes. In: Vance D. E. & Vance J. E. (ed.): Biochemistry of lipids, lipoproteins and membranes (4th Edn.). Elsevier B V, Amsterdam, The Netherlands: 205-232.
Vance, D.E., Li, Z. and R.L. Jacobs. 2007. Hepatic phosphatidylethanolamine N-methyltransferase, unexpected roles in animal biochemistry and physiology. J Biol Chem. 282: 33237-33241.
Vidgren, J., Svensson, L.A. and A. Liljas. 1994. Crystal structure of catechol O-methyltransferase. Nature. 368: 354-358.
Walkey, C.J., Yu, L., Agellon, L.B. and D.E. Vance. 1998. Biochemical and evolutionary significance of phospholipid methylation. J Biol Chem. 273: 27043-27046.
Wall, J., Golding, C.A., Van Veen, M. and P. O´Shea. 1995. The use of fluoresceinphosphatidylethanolamine (FPE) as a real-time probe for peptide-membrane interactions. Mol Membr Biol. 12: 183-192.
Weiss, S.B., Smith, S.W. and E.P. Kennedy. 1958. The enzymatic formation of lecithin from cytidine diphosphate choline and D-1,2-diglyceride. J Biol Chem. 231: 53-64.
Wells, R.C. and R.B. Hill. 2011. The cytosolic domain of Fis1 binds and reversibly clusters lipid vesicles. PLoS One. 6: e21384.

Literaturverzeichnis
- 137 -
Wessel, M., Klüsener, S., Gödeke, J., Fritz, C., Hacker, S. and F. Narberhaus. 2006. Virulence of Agrobacterium tumefaciens requires phosphatidylcholine in the bacterial membrane. Mol Microbiol. 62: 906-915.
Wilderman, P.J., Vasil, A.I., Martin, W.E., Murphy, R.C. and M.L. Vasil. 2002. Pseudomonas aeruginosa synthesizes phosphatidylcholine by use of the phosphatidylcholinesynthase pathway. J Bacteriol. 184: 4792-4799.
Willis, C., Wang, C.K., Osman, A., Simon, A., Pickering, D., Mulvenna, J., Riboldi-Tunicliffe, A., Jones, M.K., Loukas, A and A. Hofmann. 2011. Insights into the membrane interactions of the saposin-like proteins Na-SLP-1 and Ac-SLP-1 from human and dog hookworm. PLoS One. 6: e25369.
Witola, W.H., El Bissati, K., Pessi, G., Xie, C., Roepe, P.D. and C.B. Mamoun. 2008. Disruption of the Plasmodium falciparum PfPMT gene results in a complete loss of phosphatidylcholine biosynthesis via the serine-decarboxylase-phosphoethanolamine-methyltransferase pathway and severe growth and survival defects. J Biol Chem. 283: 27636-27643.
Woodcock, D.M., Crother, P.J., Doherty, J., Jefferson, S., DeCruz, E., Noyer-Weidner, M., Smith, S.S., Michael, M.Z. and M.W. Graham. 1989. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 17: 3469-3478.
Wu, Q., Gee, C. L., Lin, F., Tyndall, J.D., Martin, J.L., Grunewald, G.L., and M.J. McLeish. 2005. Structural, mutagenic, and kinetic analysis of the binding of substrates and inhibitors of human phenylethanolamine N-methyltransferase. J Med Chem. 48: 7243–7252
Yamashita, S. and K. Hosaka. 1997. Choline kinase from yeast. Biochim Biophys Acta. 1348: 63-69.
Zeisel, S.H. and J.K. Blusztajn. 1994. Choline and human nutrition. Annu Rev Nutr. 14: 269-296.
Zhang, X., Zhou, L. and X. Cheng. 2000. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. Embo J. 19: 3509-3519.
Zhang, Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioionformatics. 9: 40.

Anhang
- 138 -
8. Anhang
8.1 Vorveröffentlichungen der Dissertation
Teilergebnisse aus dieser Arbeit wurden mit Genehmigung des Fachbereiches für
Biologie der Mikroorganismen, vertreten durch den Mentor dieser Arbeit, in folgenden
Beiträgen vorab veröffentlicht:
Publikationen
Aktas, M., Wessel, M., Hacker, S., Klüsener, S., Gleichenhagen, J. and F. Narberhaus. 2010. Phosphatidylcholine biosynthesis and its significance in bacteria interacting with eukaryotic cells. Eur J Cell Biol. 89: 888-894
10 % Anfertigung des Manuskriptes.
Aktas, M., Gleichenhagen, J., Stoll, R., and F. Narberhaus. 2011. S-Adenosylmethionine-binding properties of a bacterial phospholipid N-methyltransferase. J Bacteriol. 193: 3473-3481.
50 % Planung und Durchführung der Experimente, sowie
20 % Anfertigung des Manuskriptes.
Tagungsbeiträge
Gleichenhagen, J., Aktas, M., Hacker, S., Wessel, M. and F. Narberhaus. 2010. Poster: Pathogenic and symbiotic plant-microbe interaction require bacterial phosphatidylcholine. VAAM-Jahrestagung, Hannover, Book of Abstracts: 147
Gleichenhagen, J., Aktas, M., Köster, S., Stoll, R. and F. Narberhaus. 2011. Poster: Exploring Agrobacterium tumefaciens phosphatidylcholine biosynthesis enzymes. Annual Symposium - Recent advances in membrane biochemistry, Cambridge (UK), Book of Abstracts: 42
Gleichenhagen, J., Wessel, M., Aktas, M., Klüsener, S., Hacker, S., Fritz, C. and F. Narberhaus. 2011. Talk: A typical eukaryotic lipid in prokaryotic membranes: Synthesis and necessity of phosphatidylcholine in Agrobacterium tumefaciens. VAAM-Jahrestagung, Karlsruhe, Book of Abstracts: 178

Anhang
- 139 -
8.2 Lebenslauf
Name: Jan Gleichenhagen
Anschrift: Goebenstraße 32, 44866, Bochum
Geburtsdatum: 18.09.1982
Geburtsort: Essen
Familienstand: ledig
Staatsangehörigkeit: Deutsch
Schulbildung
1989 – 1993 Tuttmannschule in Essen-Stoppenberg
1993 – 2002 Gymnasium Essen Nord Ost
06/2002 Abitur
Wehrdienst
10/2002 – 12/2002 Marineoperationsschule Bremerhaven
01/2003 – 06/2003 Marinefernmeldegruppe 21 (Führungsunterstützungszentrum B)
Studium
10/2003 Beginn Biologiestudium an der Ruhr-Universität Bochum
09/2005 Vordiplom Biologie
10/2005 – 05/2007 Hauptstudium Biologie an der Ruhr-Universität Bochum
07/2007 – 05/2008 Diplomarbeit am Lehrstuhl Biologie der Mikroorganismen an der
Ruhr-Universität Bochum
Promotion
Seit 07/2008 Anfertigung der Doktorarbeit
Lehrstuhl Biologie der Mikroorganismen
Ruhr-Universität Bochum
Berufstätigkeit
11/2007-03/2008 Studentische Hilfskraft
Lehrstuhl für Biologie der Mikroorganismen
Ruhr-Universität Bochum
Seit 07/2008 Wissenschaftlicher Mitarbeiter
Lehrstuhl für Biologie der Mikroorganismen
Ruhr-Universität Bochum

Anhang
- 140 -
8.3 Erklärung
Hiermit erkläre ich, dass ich die Arbeit selbständig verfasst und bei keiner anderen
Fakultät eingereicht und dass ich keine anderen als die angegebenen Hilfsmittel
verwendet habe. Es handelt sich bei der heute von mir eingereichten Dissertation um
sechs in Wort und Bild völlig übereinstimmende Exemplare.
Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten
und in keinem Fall inhaltsverändernde Bildbearbeitung vorgenommen wurde.
Bochum, den
___________________________
(Jan Gleichenhagen)

![İŞ GELİŞTİRME MERKEZLERİ (İŞGEM) · Mancuso’nun iş stratejisi ve bunun iş yaratmadaki potansiyeli hakkında farkındalığı arttıkça bu fikir daha çok tutuldu [5]](https://img.pdfslide.org/doc/110x75/5f03995f7e708231d409d8d3/-geltrme-merkezler-gem-mancusoanun-i-stratejisi-ve-bunun-i.jpg)







