Embed Size (px)
Citation preview

Biochemie
Cre-Rekombinase-exprimierende Mäuse zur örtlichen Mutagenese in der Niere.
Entwicklung und Analyse eines Tiermodells zur Untersuchung der
Autosomal Dominanten Polycystischen Nierenerkrankung (ADPKD) beim Menschen
Inaugural-Dissertation
zur Erlangung des Doktorgrades
der Naturwissenschaften im Fachbereich Chemie und Pharmazie
der Mathematisch-Naturwissenschaftlichen Fakultät
der Westfälischen Wilhelms-Universität Münster
vorgelegt von
Lambrini Tontsidou
aus Wuppertal
- 2008 -

Dekan: Univ.-Prof. Dr. F. Ekkehardt Hahn
Erster Gutachter: Univ.-Prof. Dr. Hans-Joachim Galla
Zweiter Gutachter: Univ.-Prof. Dr. Jürgen Horst
Tag der mündlichen Prüfung: 28.05.2008
Tag der Promotion: 28.05.2008

„Das Ziel weicht ständig vor uns zurück. Genugtuung liegt im Einsatz, nicht im Erreichen.
Ganzer Einsatz ist ganzer Erfolg“.
Mahatma Gandhi (1869-1948)

Für meinen Sohn

Teile dieser Arbeit wurden publiziert:
• Tontsidou L, Bilbilis K, Pennekamp P, Skryabin B , Galla H-J, Horst J , Dworniczak B
(2002), Specific expression of Cre recombinase in kidney tubular cells of mice, 52nd
Annual Meeting of the American Society of Human Genetics“ in Baltimore Maryland
USA 10/2002, Program Nr: 891 from 2002 ASHG Annual Meeting
(http://www.ashg.org/genetics/ashg/ashgmenu.htm)
Specific expression of Cre recombinase in kidney tubular cells of mice Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common
monogenic diseases in humans with an incidence of 1:600. The cardinal finding is the
formation of fluid filled cysts in both kidneys - predominantly in the proximal tubules -
frequently resulting in end stage renal failure. In the vast majority of cases, the disease is
caused by mutations in PKD1 and/or PKD2. However, despite extensive research only little
is known concerning the pathogenesis of ADPKD. Cystogenesis in ADPKD kidneys in
humans is a focal process: patients inherit one germline mutation and acquire somatic
mutations affecting the normal allele in a subset of cells (second hit). In the last couple of
years conventional knockout mouse models for Pkd1 and Pkd2 have been generated. All of
them are prenatal lethal making it impossible to analyze the pathogenesis of ADPKD after
birth/over a longer period in adult mice. These limitations caused by embryonic lethality can
be circumvented by conditional mutagenesis in which the loss of gene function is placed
under temporal and/or spatial control. In order to establish this system we have generated a
transgenic mouse line expressing the Cre-recombinase under the control of a truncated
mouse type II GGT promoter. We expected that this promoter allows the spatial excision of
loxP flanked genes in proximal tubular cells. The recombinase activity was demonstrated by
crossing these mice with Cre- reporter lines (Z/AP and ROSA26). Both lines carry a marker
gene whose expression requires excision of loxP flanked stop sequences. Using alkaline
phosphatase histochemistry in Z/AP mice as well as b-Galactosidase histochemistry in
ROSA26 mice we observed a strong and specific expression of Cre-recombinase in proximal
tubular renal cells. This Cre transgenic mouse line, which induces efficient Cre-mediated
excision of DNA in proximal tubules, should provide a useful genetic resource to elucidate
the role of loxP manipulated genetic targets in cyst formation of ADPKD.

• Lambrini Tontsidou, Konstantinos Bilbilis, Petra Pennekamp, Boris Skryabin, Hans-
Joachim Galla, Jürgen Horst, Bernd Dworniczak, GGT-Cre mice for tissue-specific
gene ablation in proximal kidney tubules, Proceedings (poster or lecture-abstract) of
the annual fall meeting, German Society for Biochemistry and Molecular Biology
(GBM), Münster (Westfalen), Germany, September 19-22, 2004 (www.gbm-
online.de).
GGT-Cre mice for tissue-specific gene ablation in proximal kidney tubules Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common
monogenic diseases in humans with an incidence of 1:1000. The main characteristic of the
disease is the formation of fluid filled cysts in both kidneys (originating from the proximal
tubules), frequently resulting in end stage renal failure. In the vast majority of cases the
disease is caused by a mutation in the PKD1 or PKD2 gene. In the last couple of years
numerous conventional Pkd1 and Pkd2 knockout mouse models have been generated. All of
them are prenatal lethal, thus making it impossible to analyze the pathogenesis of ADPKD
after birth. These limitations can be circumvented by conditional mutagenesis in which the
loss of gene function is placed under temporal and/or spatial control. In order to establish this
system we generated a transgenic mouse line expressing the Cre-recombinase under the
control of a truncated mouse type II GGT promoter. We expected that this promoter allows
the spatial excision of loxP flanked genes in proximal tubular cells.
The recombinase activity was demonstrated by crossing these mice with Cre-reporter lines
(Z/AP and ROSA26). Both lines carry a marker gene whose expression requires excision of
loxP flanked stop sequences. Using alkaline phosphatase histochemistry in Z/AP mice as
well as Beta-Galactosidase histochemistry in ROSA26 mice we observed a strong and
specific expression of Cre-recombinase in proximal tubular renal cells.
This GGT-Cre transgenic mouse line, which induces efficient Cre-mediated excision of DNA
in proximal tubules, should provide a useful genetic resource to elucidate the role of loxP
manipulated genetic targets in cyst formation of ADPKD.

INHALTSVERZEICHNIS I
I Inhaltsverzeichnis
II Abkürzungsverzeichnis ........................................................................................ V 1 Einleitung ................................................................................................................1 1.1 Autosomal Dominante Polyzystische Nierenerkrankung ...............................................1
1.1.1 Genetik der ADPKD ...............................................................................................1
1.1.2 Genprodukte von PKD1/PKD2...............................................................................2
1.1.2.1 Expression von Polycystin 1 und 2.................................................................2
1.1.3 Zystenentwicklung .................................................................................................3
1.2 Herstellung transgener Tiermodelle...............................................................................4
1.2.1 Tiermodelle der ADPKD.........................................................................................5
1.2.1.1 Pkd1 ...............................................................................................................5
1.2.1.2 Pkd2 ...............................................................................................................6
1.3 Cre/loxP-Rekombinationsmechanismus........................................................................6
1.3.1 Gewebespezifische Cre-Expression ......................................................................9
1.3.2 Zeitlich regulierbare Cre-exprimierende Tiermodelle ...........................................12
1.3.2.1 Tet-Repressor/Aktivator-System ..................................................................12
1.3.2.2 Interferon-Rezeptor-Rekonstitution ..............................................................14
1.3.2.3 Murines Östrogen-induzierbares System .....................................................14
1.3.2.4 Humanes Östrogen-induzierbares System ..................................................14
1.4 Ziele.............................................................................................................................16
2 Material und Methoden.........................................................................................17 2.1 Datenbanken ...............................................................................................................17
2.2 Molekularbiologische Methoden ..................................................................................18
2.2.1 Kultivierung von Escherichia coli (E. coli) ............................................................18
2.2.2 Anlegen einer Gefrierkultur ..................................................................................18
2.2.3 Plasmid-Isolierung aus E. coli ..............................................................................18
2.2.3.1 Plasmid-Minipräparation aus E. coli .............................................................18
2.2.3.2 Qiaprep-Plasmid-Mini-/Maxi-Präparation aus E. coli....................................19
2.2.4 DNA-Isolierung aus Gewebe................................................................................19
2.2.5 Reinigung isolierter DNA......................................................................................19
2.2.5.1 Phenol/Chloroform-Extraktion ......................................................................19
2.2.5.2 Aussalzung...................................................................................................20
2.2.5.3 Isopropanol-Fällung......................................................................................20
2.2.5.4 Ethanol-Fällung ............................................................................................20
2.2.5.5 Dialyse..........................................................................................................20
2.2.6 Konzentrationsbestimmung von Nukleinsäuren...................................................20

INHALTSVERZEICHNIS II
2.2.7 Gelelektrophorese von Nukleinsäuren.................................................................21
2.2.8 Extraktion und Rückgewinnung von DNA aus Agarosegelen ..............................22
2.2.9 Modifikation und Klonierung von Nukleinsäuren..................................................22
2.2.9.1 Restriktion ....................................................................................................22
2.2.9.2 Dephosphorylierung der 5´-Enden ...............................................................22
2.2.9.3 Auffüllen von 5´-Überhängen mit Klenow-Polymerase.................................23
2.2.9.4 Ligation.........................................................................................................23
2.2.9.5 Klonierung von PCR-Produkten ...................................................................24
2.2.9.6 Klonierung von Restriktionsfragmenten........................................................24
2.2.10 Transformation von E. coli .................................................................................24
2.2.11 Polymerasekettenreaktion (PCR).......................................................................24
2.2.12 DNA-Sequenzierung ..........................................................................................25
2.2.12.1 Proben-Aufreinigung ..................................................................................26
2.2.12.2 Vorbereitungen der PCR-Reaktionsansätze ..............................................26
2.2.12.3 Vorbereitung der Sequenzierungsansätze für den ABI PRISM® 310 ........27
2.2.12.4 Vorbereitung der Sequenzierungsansätze für den ABI PRISM® 3700 ......27
2.2.13 Hybridisierungstechnik.......................................................................................27
2.2.13.1 Southern-Transfer ......................................................................................27
2.2.13.2 Radioaktive DNA-Markierung.....................................................................28
2.2.13.3 Radioaktive Hybridisierung.........................................................................28
2.3 Etablierung der Mauslinien ..........................................................................................29
2.3.1 Mikroinjektion .......................................................................................................29
2.3.2 Genotypisierung der Mauslinien...........................................................................30
2.3.3 Dokumentierung der Mauslinien ..........................................................................30
2.3.4 Kreuzungen mit Reportermäusen ........................................................................31
2.3.4.1 Kreuzungen mit der Z/AP-Reportermauslinie...............................................32
2.3.4.2 Kreuzungen mit der ROSA26-Reportermauslinie.........................................33
2.4 Histologische Techniken..............................................................................................34
2.4.1 Vorbereitung der Gefrierschnitte ..........................................................................34
2.4.1.1 Entnahme des Gewebes ..............................................................................34
2.4.1.2 Entnahme der Embryonen ...........................................................................34
2.4.1.3 Anfertigungen der Gefrierschnitte ................................................................34
2.4.2 Enzymhistochemische Färbungen.......................................................................34
2.4.2.1 Detektion der β-Galaktosidase .....................................................................34
2.4.2.2 Detektion der alkalischen Phosphatase .......................................................35
2.4.3 Histologische Übersichtsfärbung..........................................................................35
2.4.4 Immunfluoreszenz-Untersuchungen ....................................................................36

INHALTSVERZEICHNIS III
2.4.4.1 Lektin-Markierung.........................................................................................36
2.4.4.2 Anti-Cre-Rekombinase-Markierung..............................................................36
2.4.5 Auswertung der histochemischen Präparate .......................................................37
2.5 Sicherheitsbestimmungen ...........................................................................................39
2.6 Verwendete Materialien ...............................................................................................39
2.6.1 Plasmide ..............................................................................................................39
2.6.2 Sonde...................................................................................................................39
2.6.3 Oligonukleotide ....................................................................................................40
2.6.4 Organismen .........................................................................................................40
2.6.5 Antikörper.............................................................................................................41
2.6.6 Puffer ...................................................................................................................42
2.7 Geräte..........................................................................................................................46
2.8 Bezugsfirmen...............................................................................................................47
3 Ergebnisse ............................................................................................................48 3.1 Promotor-Recherche ...................................................................................................48
3.2 Klonierung der Zielvektoren.........................................................................................51
3.2.1 Klonierung des Vektors pGGT-CRE ....................................................................51
3.2.1.1 Klonierung des Vektors pGGT-PFU-1 - Genomische PCR -........................52
3.2.1.2 Umklonierung des Vektors pGGT-PFU-1 .....................................................55
3.2.1.3 Isolierung des GGT-CRE-Transgens aus dem Zielvektor ............................56
3.2.2 Klonierung des Vektors pGGT-RTTA...................................................................58
3.2.2.1 Amplifikation des GGT-Promotors................................................................59
3.2.2.2 Isolierung des GGT-RTTA-Transgens aus dem Zielvektor ..........................61
3.2.3 Klonierung des Vektors pBI-CRE-lacZ.................................................................63
3.2.3.1 Einführung einer HindIII-Schnitttstelle in pBI-3 an Position nt 5188.............64
3.2.3.2 Klonierung des Vektors pBI-3-H3.................................................................64
3.2.3.3 Isolierung des Transgens BI-CRE-lacZ aus dem Zielvektor.........................65
3.3 Generierung transgener Tiere .....................................................................................69
3.4 Identifizierung möglicher transgener Mauslinien .........................................................70
3.4.1 PCR-Genotypisierung ..........................................................................................70
3.4.2 Southern-Analysen ..............................................................................................72
3.5 Generierung stabiler Linien..........................................................................................74
3.6 Das Expressionsprofil des murinen GGT-Promotors...................................................75
3.6.1 Untersuchungen der Cre-Expression mittels Z/AP-Reportermäuse ....................75
3.6.1.1 Verpaarung der GGT-CRE-Mäuse mit Z/AP-Reportermäuse ......................75
3.6.1.2 Charakterisierung der doppeltransgenen GGT-CRE+/Z/AP+-Mäuse............77
3.6.1.3 Cre-Expression in adulten GGT-CRE-Mäusen.............................................77

INHALTSVERZEICHNIS IV
3.6.2 Untersuchungen der Cre-Expression mittels ROSA26-Reportermäuse ..............79
3.6.2.1 Verpaarung der GGT-CRE-Mäuse mit ROSA26-Reportermäuse ................79
3.6.2.2 Charakterisierung der GGT-CRE+/ROSA26+-Mauslinien .............................79
3.6.2.3 Cre-Rekombinase-Expression zum Zeitpunkt E14,5....................................81
3.6.2.4 Cre-Rekombinase-Expression zum Zeitpunkt E15,5....................................82
3.6.2.5 Cre-Rekombinase-Expression zum Zeitpunkt E17,5....................................84
3.6.2.6 Cre-Rekombinase-Expression bei neugeborenen Mäusen..........................86
3.6.2.7 Cre-Rekombinase-Expression bei adulten GGT-CRE-Mäusen ...................89
3.6.3 Expression des GGT-CRE-Transgens in der adulten Niere ................................91
4 Diskussion ............................................................................................................93 4.1 Nierenspezifische Cre-exprimierende Mausmodelle ...................................................94
4.2 Generierung der transgenen GGT-CRE-Maus............................................................95
4.2.1 Relevanz des proximalen Tubulus für die Zystenbildung.....................................95
4.2.2 Relevanz der Promotorgröße...............................................................................98
4.3 Muriner 346-bp-GGT-Minimalpromotor ......................................................................99
4.3.1 GGT-pII-346/β-Galaktosidase-Maus ..................................................................100
4.4 Klonierung des GGT-CRE-Transgens ......................................................................100
4.5 Das Expressionsprofil des murinen GGT-Promotors ................................................100
4.5.1 Cre/loxP-Rekombination in GGT-CRE+/Z/AP+-Mäuse .......................................100
4.5.2 Früheste Cre/loxP-Rekombination in GGT-CRE+/ROSA26+-Mäuse..................101
4.5.3 Cre/loxP-Rekombination in neonatalen/adulten GGT-CRE+/ROSA26+-Mäusen102
4.6 Expression des GGT-Transgens in der Niere............................................................103
4.7 Ausblick: Zeitlich induzierbare nierenspezifische Cre-Mäuse....................................104
4.7.1 Generierung der Konstrukte GGT-RTTA und BI-CRE-lacZ ...............................104
5 Zusammenfassung.............................................................................................107 6 Literatur ...............................................................................................................108 7 Anhang ................................................................................................................121 7.1 Restriktionsendonukleasen .......................................................................................121
7.2 Plasmidkarten ...........................................................................................................122
7.3 DNA-Sequenzen .......................................................................................................127
7.3.1 Sequenzname: pGGT-CRE ...............................................................................127
7.3.2 DNA-Sequenz der Sonde S1 .............................................................................130
Danksagung......................................................................................................................131
Lebenslauf.........................................................................................................................132

ABKÜRZUNGSVERZEICHNIS V
II Abkürzungsverzeichnis A Adenin
Abb. Abbildung
AmpR Ampicillin-Resistenz
Aqua dest. (lat.: Aqua destillata) destilliertes Wasser
AS Aminosäuren
bp Basenpaar
BSA (engl.: bovine serum albumin) Rinderserumalbumin
C Cytosin
CAG (engl.: chicken β-actin gene) Hühnchen-β-Actin-Gen
CAT Chloramphenicol-Acetyltransferase
cDNA komplementäre DNA
CMV Cytomegalievirus
Cre (engl.: cyclization recombination) Cre-Rekombinase
CreERT Tamoxifen-aktivierbare Cre-Rekombinase mit ERT-LBD
CreERT2 Tamoxifen-aktivierbare Cre-Rekombinase mit ERT2-LBD
Cy3 Indocarbocyanin
ddNTP 2’,3’-Didesoxynukleosid-5’-triphosphat
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
DNase Desoxyribonuklease
dNTP 2’-Desoxynukleosid-5’-triphosphat
ds doppelsträngig
DTT Dithiothreitol
EDTA Ethylendiamin-N,N,N´,N´-Tetraessigsäure-Dinatriumsalz
E. coli Escherichia coli
et al. (lat.: et allii) und andere
EtBr Ethidiumbromid
EtOH Ethanol
ER Östrogenrezeptor
ES-Zellen embryonale Stammzellen
F (engl.: forward) vorwärts
FITC Fluorescein-Isothiocyanat
G Guanin
g Erdbeschleunigung (9,81 m s-2)
gefloxt loxP-flankiert
HEPES N-2-Hydroxyethylpiperazin-N-2-ethansulfonsäure

ABKÜRZUNGSVERZEICHNIS VI
HSV Herpes-Simplex-Virus
IPTG Isopropyl-β-D-thiogalactopyranosid
IRES (engl.: Internal Ribosomal Entry Site) interne ribosomale Eintrittsstelle
kb Kilobase
kDa Kilodalton
lacZ β-Galaktosidasegen
LB Luria-Bertani
LBD Ligandenbindungsdomäne
loxP (engl.: locus of crossing over of P1) DNA-Substrat von Cre
MCS (engl.: multiple cloning site) Multiple Klonierungsstelle
mRNA messenger RNA
neo Neomycinresistenz-Gen
nt Nukleotide
OD optische Dichte
ORF (engl.: open reading frame) offener Leserahmen
pA Polyadenylierungssignal
PBS (engl.: phosphate buffered saline) Phosphat-gepufferte Salzlösung
PCR (engl.: polymerase chain reaction) Polymerasekettenreaktion
PGK Phosphoglycerat Kinase
pT-Zellen proximale Tubuluszellen
R (engl.: reverse) rückwärts
RNA Ribonukleinsäure
rpm (engl.: rotations per minute) Umdrehungen pro Minute
RNase Ribonuklease
RT Raumtemperatur
SDS Natriumdodecylsulfat
SV40 simian virus 40
T Thymin
TBE Tris/Borat-EDTA
Tris Tris[hydroxymethyl]-aminomethan
U (engl.: unit) Einheit
ü. N. über Nacht
V Volt
Wt Wildtyp
X-Gal 5-Brom-4-chloro-3-indolyl-β-D-Galactosid
Allgemein gebräuchliche Maßeinheiten und umgangssprachliche Abkürzungen sind nicht
angegeben. Die Restriktionsendonukleasen werden in der üblichen Schreibweise aufgeführt.

EINLEITUNG 1
1 Einleitung
1.1 Autosomal Dominante Polyzystische Nierenerkrankung Die Autosomal Dominante Polyzystische Nierenerkrankung (ADPKD) ist eine der häufigsten
erblichen monogenen Erkrankungen. Weltweit liegt die Inzidenz zwischen 1:500 bis 1:1000
(Gabow & Grantham, 1997). Ein Charakteristikum ist die progressive Entwicklung und
Vergrößerung flüssigkeitsgefüllter renaler Zysten, was bei nahezu der Hälfte aller ADPKD-
Patienten zum vollständigen Nierenversagen führt (Torra et al., 2000). Dabei entstammen
die Nierenzysten aus renalen Tubulusstrukturen. Es handelt sich hierbei um die proximalen
Tubuli den distalen Tubuli und dem Sammelrohr (Nadasdy et al., 1995). Obwohl
Nierenfunktionsstörungen klinisch im Vordergrund stehen, handelt es sich bei der ADPKD
um eine systemische Erkrankung (Bogdanova et al., 2002). Die auffälligste
Begleiterscheinung ist der arterielle Bluthochdruck, der bereits lange vor dem
Nierenversagen auftritt (Torres, 1999). Bei der häufigsten extrarenalen Manifestation handelt
es sich um die Zystenbildung in den epithelialen Organen (Perrone, 1997). Bis zum heutigen
Zeitpunkt existiert keine effiziente Therapie zur Behandlung von ADPKD-Patienten.
1.1.1 Genetik der ADPKD ADPKD kann durch Mutationen in zwei - inzwischen im Detail charakterisierten - Genen
verursacht werden: PKD1 und PKD2 (Kimberling et al., 1988, Peters & Sandkuijl, 1992). Die
Existenz eines dritten verantwortlichen Gens PKD3 ist von De Almeida (1995) postuliert
worden, wobei seine Genomlokalisation bislang noch nicht bestimmt werden konnte. PKD1-
Mutationen sind verantwortlich für 85% der Erkrankungen und etwa 14% können auf Defekte
im PKD2-Gen zurückgeführt werden. Für die übrigen ADPKD-Fälle sind Mutationen in bisher
nicht identifizierten PKD-Genen verantwortlich.
PKD1 ist auf dem Chromosom 16p13.3 lokalisiert. Dabei handelt sich um eines der größeren
humanen Gene, bestehend aus 46 Exons und verteilt auf über 53 kb der genomischen DNA
(The International Polycystic Kidney Disease Consortium, 1994, Hughes et al., 1995). Das
Gen PKD1 kodiert für ein 14,1 kb mRNA-Transkript, welches in das so genannte Polycystin
translatiert wird. Das zweite ADPKD-Gen -PKD2- wurde 1996, aufgrund seiner partiellen
Sequenzhomologie zum PKD1-Gen, nach entsprechenden Datenbankrecherchen
identifiziert. PKD2 ist auf dem Chromosom 4q21–23 lokalisiert und kodiert für ein 5,3 kb
großes mRNA-Transkript. Es ist in 15 Exons aufgeteilt und weist eine ca. 25%ige Homologie
zum PKD1 Gen auf. Obwohl ADPKD einem autosomal dominanten Erbgang folgt wirkt sich
auf zellulärer Ebene ein rezessiver Mutationsmechanismus aus (Torra et al., 1999; Qian &
Watnick, 1999). ADPKD setzt eine Keimbahnmutation in einem Allel voraus, jedoch wird die
Zystenbildung durch eine zweite somatische Mutation im anderen Allel initiiert. Solch ein

EINLEITUNG 2
Mutationsmechanismus wurde häufig in Tumorsuppressorgenen nachgewiesen und bereits
1971 als die so genannte "two hit hypothesis" von Knudson am Beispiel von Retinoblastoma
postuliert (Knudson, 1971).
1.1.2 Genprodukte von PKD1/PKD2 Die Genprodukte von PKD1 und PKD2 gehören zu den so genannten Polycystinen. Sie
spielen eine entscheidende Rolle in einer Vielzahl von biologischen Prozessen, wie z.B. der
Fertilisation und der mechanischen Reizübermittlung (Igarashi & Somlo, 2002; Nauli et al.,
2003). PKD1 kodiert für Polycystin 1, einem großen transmembranen Glykoprotein mit
verschiedenen extrazellulären Motiven (The American PKD1 Consortium, 1995; Hughes et
al., 1995; The International Polycystic Kidney Disease Consortium, 1995). Das Genprodukt
von PKD2 - Polycystin 2 - ist ein Kalziumkanal-bildendes Protein, das vorwiegend im
endoplasmatischen Retikulum, aber auch in der Zellmembran lokalisiert ist (Mochizuki et al.,
1996; Koulen et al. 2002). Durch „patch clamp“ -Analysen konnte gezeigt werden, dass
Polycystin 2 einen nicht-selektiven Kationenkanal mit einer Leitungsfähigkeit für Ca2+-Ionen
bildet (Hanaoka et al., 2000; Gonzalez-Perrett et al., 2001; 2002; Vassilev et al., 2001;
Koulen et al., 2002; Igarashi & Somlo, 2002).
Es wird allgemein akzeptiert, dass Polycystin 1 mit Polycystin 2 interagieren kann. In vitro-
Experimente zeigten Wechselwirkungen zwischen den carboxyterminalen Domänen auf.
Darüber hinaus konnte ein nativer Polycystinkomplex in transgene, nicht transgene und
humane renale Tubulusstrukturen nachgewiesen werden (Qian et al., 1997; Tsiokas et al.,
1997; Newby et al., 2002). Ferner aktiviert und stabilisiert Polycystin 1 den Kalziumkanal (Xu
et al., 2003). Da Mutationen von Polycystin 1 bzw. Polycystin 2 zur Ausbildung von
Nierenzysten führen, wird vermutet, dass dieser Proteinkomplex einen Mechanosensor
bildet, welcher die Geometrie der Tubuli in der Niere reguliert. Es ist jedoch gegenwärtig
unklar, welche zellulären Programme durch die flussbedingte Zilienbewegung reguliert
werden (Nauli et al., 2003).
1.1.2.1 Expression von Polycystin 1 und 2 Durch in situ-Hybridisierungen und immunhistochemische Methoden wurde die Expression
der ADPKD-Proteine analysiert. Aufgrund der Schwierigkeit, adäquate Antikörper gegen die
Polycystine zu gewinnen, sowie der Verwendung unterschiedlicher Gewebepräparationen
führten diese Analysen zu widersprüchlichen Ergebnissen. Sowohl Polycystin 1 als auch
Polycystin 2 weisen ein überraschend komplexes Expressionsmuster auf. Zu den Geweben
mit der höchsten Expressionsrate gehören Nieren, Pankreas, Leber, Lunge, Darm, Gehirn,
reproduktive Organe und Thymus (Griffin et al., 1996, 1997a, 1997b; Peters et al., 1996,
1999; Ward et al., 1996; Weston et al., 1997; Geng et al., 1996; 1997; van Adelsberg, 1999a;

EINLEITUNG 3
1999b; Obermüller et al., 1999; Ong et al., 1999a, 1999b, 1999c; Guillaume & Trudel, 2000;
Foggensteiner et al., 2000; Wilson, 2001). Expressionsorte sind vor allem Epithel- und
Endothelzellen und aufzweigende Strukturen wie Nierentubuli, Gallengänge und
Pankreasgänge (Ibraghimov-Beskrovnaya et al., 1997).
1.1.3 Zystenentwicklung Verschiedene zelluläre Prozesse können zur Entwicklung von Nierenzysten führen. Zu
nennen sind: Zellproliferation, Apoptose, Veränderungen der Zellpolarität/Sekretion und
Anomalien der extrazellulären Matrix (Lanoix et al., 1996).
Eine hohe Zellteilungsrate konnte im frühen Stadium der Zystenentwicklung nachgewiesen
werden (Ebihara et al., 1988; Montesano et al., 1991; Trudel et al., 1991; Wilson, 1991,
Wilson et al., 1992, Wilson, 1997). Durch Induktion mit endogenen Wachstumsfaktoren wie
z.B. dem epidermalen Wachstumsfaktor EGF (Ogborn & Sareen, 1996), zeigte sich bei
Tiermodellen zunächst eine Nierenausdehnung mit anschließender progressiver
Zystenentwicklung. Ferner entwickelt das ADPKD-Epithelium eine erhöhte Sensitivität
gegenüber dem EGF, dem Glukokortikoid Hydrocortison und dem Peptidhormon Insulin
(Nauta et al., 1995). Histologisch betrachtet geht die Zystenbildung einher mit einer
Abflachung des tubulären Epithels, einer Verdickung der Basalmembran und einer
Veränderung der epithelialen Polarität. Ergänzend verlieren die Epithelzellen ihre
charakteristischen Eigenschaften wie z.B. den Bürstensaum der proximalen Tubuli und die
basolateralen Einfaltungen. Ferner liegen in humanen ADPKD Epithelien die Proteine Na/K-
ATPase, Fodrin, Ankyrin und EGF-Rezeptoren unregelmäßig lokalisiert vor (Du & Wilson
1995; Brill et al., 1996).
Um nun die Frage aufzugreifen, wie die Zysten entstehen, muss zunächst auf die Genetik
verwiesen werden. Genetisch spricht viel dafür, dass beide Allele von PKD1 und PKD2
inaktiviert sein müssen, damit sich Zysten bilden (Pei, 2001). In Analogie zu den
Tumorsuppressorgenen wird zur Zeit davon ausgegangen, dass das in der Keimbahn
gesunde Allel im Laufe des Lebens eine Mutation erleidet, die betroffenen Zellen erhalten
möglicherweise einen Wachstumsvorteil und die Zystenbildung beginnt, weil ihr
entsprechendes “Stoppsignal” ausgefallen ist. In diesem Zusammenhang sei erwähnt, dass
die renalen Tubulusstrukturen aus einem bindegewebigen Vorläufergewebe differenzieren.
Das bedeutet das Lumen der Tubuli entwickelt sich erst im Laufe der Zeit. Zu einem genau
definierten Zeitpunkt muss die Lumenbildung allerdings zu einem Ende kommen, denn die
Nierentubuli sind sehr ausgedehnt. Wenn das Stoppsignal folglich nicht mehr existiert, wird
mehr Lumen gebildet und es entstehen Zysten (Gallagher et al., 2000). Über die Natur des
Sensors gibt es bestimmte Vorstellungen. Möglicherweise kommt primären Zilien, eine
maßgebliche chemo- und/oder mechanosensorische Rolle zu. Die dafür sprechenden

EINLEITUNG 4
Befunde sind der Nachweis verschiedener Zystenproteine in primären Zilien (Yoder et al.,
2002) und der Verlust ihrer mechanosensorischen Eigenschaften bei Ausfall von Polycystin 1
und 2 (Nauli et al., 2003).
Abschließend stellt sich die Frage, wo die Zystebildung initiiert wird. Bei Betrachtung von
transgenen Tiermodellen mit einer polycystischen Nierenerkrankung oder der humanen
ADPKD zeigte sich, dass sowohl im renalen Sammeltubulus als auch in den distalen und
hauptsächlich in den proximalen Tubulussegmenten sich Zysten ausbilden (Witzgall, 1999).
1.2 Herstellung transgener Tiermodelle Zur Erforschung menschlicher Erkrankungen wird häufig ein Tiermodell herangezogen,
wobei sich Mäuse bei genetisch bedingten Erkrankungen bewährt haben. Um eine definierte
Veränderung im Genom zu erreichen gibt es verschiedene methodische Ansätze:
1. Mikroinjektion von DNA-Konstrukten in befruchtete Eizellen
2. Genetische Veränderung von embryonalen Stammzellen (ES-Zellen) und
Erzeugung chimärer Transgene
3. Intramuskuläre DNA-Injektion
4. Klonierung von Tieren aus genetisch veränderten Zellen
5. Gentransfer mittels retroviraler Vektoren
Bei der ersten Methode mittels Mikroinjektion erfolgt die Einführung gentechnisch
veränderter DNA in den Pronukleus kurz vor der Karyogamie (Pronukleusinjektion), wobei
sich das Transgen zufällig in das murine Genom integrieren kann (Meisler, 1992; Hogan et
al., 1994; Simoni, 1994; Wight & Wagner, 1994). Darüber hinaus sind zu dieser Methode
noch folgende Eigenschaften erwähnenswert: Es liegt eine stabile Integration des DNA-
Konstrukts in das Genom vor, es können häufig viele Kopien (ca. 10-100) tandemartig
vorliegen und die transgenen Tiere besitzen genetisch einheitliche Zellen. Mittels
Pronukleusinjektion generierte transgene Tiere werden z.B. für folgende Studien
angewendet:
• Expressions- und Promotorstudien durch Einsatz von Reportergenen
• Über- oder Fehlexpressionsstudien durch Einsatz genfremder Promotoren
• Korrektur eines vorhandenen Gendefektes durch Einbau eines Wildtyp-Gens
Hingegen wird bei der zweiten Methode die genetisch modifizierte DNA gezielt durch
homologe Rekombination in murine embryonale Stammzellen (ES-Zellen) integriert
(Capecchi, 1989; Shastry, 1994, 1995, 1998; Nomura, 1997; Müller, 1999). Es resultiert eine

EINLEITUNG 5
vollständige oder partielle Defizienz eines Gens und dessen Genprodukts. Mitunter wird
diese Methodik auch als konventionelle Gen-„knockout“-Technologie bezeichnet. Bei der
Methode der intramuskulären Injektion ist die Durchführung zwar relativ einfach, das
Transgen wird aber nicht an die Nachkommen weitergegeben, wobei die Klonierung von
Tieren aus differenzierten Zellen prinzipiell die gleichen Möglichkeiten zum Gentransfer wie
die Verwendung von ES-Zellen bietet.
1.2.1 Tiermodelle der ADPKD Um die ADPKD in vivo besser analysieren zu können und neuere Therapieansätze zu
entwickeln, wurden mehrere konventionelle Gen-„knockout“-Tiermodelle generiert. Tabelle
1.1 gibt einen Überblick über die generierten transgenen Mäuse.
Tabelle 1.1: ADPKD-Mausmodelle
Name verändertes Gen Literatur
Pkd1del34/del34 Pkd1 (Exon 34) Lu et al.,1997
Pkd1L/L Pkd1 (Exon 43-45) Kim et al., 2000
Pkd1S-lacZ/S-lacZ Pkd1 (Exon 17-21) [IRES-lacZ]
Boulter et al., 2001
Pkd1β-gal/β-gal Pkd1 (Exon 2-3) ["gene trap", lacZ]
Piontek, 5th Int. Workshop on PKD (11.05.-12.05.2000)
Pkd2-/- Pkd2 (Exon 1) Wu et al., 1998 Wu et al., 2000
Pkd2-/-lacZ+/+ Pkd2 (Exon 1) [lacZ] Pennekamp, 2000
1.2.1.1 Pkd1 1997 ist die erste Pkd1-„knockout- Maus konstruiert worden (Lu et al., 1997). Eine Deletion
im Exon 34 konnte durch homologe Rekombination in murine ES-Zellen verursacht werden.
In dieser Arbeit zeigte sich zunächst bei Pkd1-heterozygoten Tieren kein erkennbarer
Phänotyp. Aufgrund einer massiven Entwicklung von Nierenzysten, Pankreaszysten und
einer pulmonären Hypoplasie verstarben jedoch homozygote Pkd1-Mäuse perinatal. Die
renale Zystenentwicklung beginnt am embryonalen Tag 15,5 (E 15,5) in den proximalen
Tubulusstrukturen. 1999 berichtete die Arbeitsgruppe Lu et al., dass auch heterozygote
Pkd1-Mäuse im Alter von 9-14 Monaten Nierenzysten ausbilden. Es zeigte sich, dass
homozygot mutante Embryonen darüber hinaus Ödeme, Gefäßstörungen sowie Brüche in
den Blutgefäßen entwickelten. Die Autoren erklärten diese Beobachtung mit der Beteiligung
von Polycystin 1 an endothelialen Zell-Zell-Verbindungen und infolgedessen an der Integrität
von Blutgefäßen (Kim et al., 2000).

EINLEITUNG 6
1.2.1.2 Pkd2 1998 ist eine Pkd2-„knockout“-Maus mit einem unstabilen Pkd2-Allel beschrieben worden
(Wu et al., 1998). Das Allel durchläuft eine somatische Inaktivierung durch intragenische
homologe Rekombination und produziert dadurch ein reales Nullallel. Sowohl heterozygote
als auch homozygote Mausmutanten entwickeln polycystische Nieren mit Leberschäden. In
einer späteren Arbeit wurde ergänzend über Herzdefekte bei homozygoten Pkd2-
Mausmutanten berichtet (Wu et al., 2000).
Bei der Pkd2-„knockout“-Mauslinie Pkd2-/-lacZ+/+ ist einzigartig, dass das Reportergen lacZ zum
einen unter der Promotorkontrolle des Pkd2-Gens steht und zum anderen Pkd2 zusätzlich
inaktiviert (Pennekamp, 2000). Mit diesem Tiermodell war es möglich, sowohl die Expression
von Pkd2 während der Entwicklung als auch den Phänotyp der ADPKD2 in der Maus zu
analysieren. Zusätzlich zu der progressiven Zystenbildung in beiden Nieren und im Pankreas
was auch die anderen ADPKD-Mausmodelle aufzeigten sind hier zum ersten Mal skelettale
Defekte, Lebernekrosen und eine Fehllage der Organe, verursacht durch eine fehlerhafte
Determinierung der embryonalen Links-Rechts-Achse, beobachtet worden (Pennekamp et
al., 2002).
1.3 Cre/loxP-Rekombinationsmechanismus Die Erforschung von genetischen Erkrankungen wurde durch die Inaktivierung von
spezifischen Genen (Gen-„knockout“-Technologie) durch homologe Rekombination in der
Maus revolutioniert (Thomas & Capecchi, 1987). Allerdings verfügen die bisher vorgestellten
konventionellen ADPKD-Tiermodelle über erhebliche Nachteile, da eine permanente
genetische Modifikation in der Keimbahn der Maus erzeugt wurde. Das konventionelle
Ausschalten eines Gens führt in allen Zellen des Tieres während der gesamten prä- und
postnatalen Entwicklung zum Verlust der Genfunktion. Alle Pkd1- und -2 homozygot
mutanten Mäuse versterben aufgrund erheblicher Defekte bereits vor der Geburt. Folgerichtig werden durch die embryonale Letalität und den komplex pleiotropen Phänotyp
postnatale Untersuchungen am Tier, besonders präventive und therapeutische
Langzeitstudien, unmöglich gemacht. Insbesondere kann die zeit- und zelltypspezifische
Funktion eines Gens an konventionellen Nullmutanten nicht analysiert werden. Ferner eignet
sich die Technik des konventionellen Ausschaltens eines Gens auch nicht zur Erzeugung
komplexer chromosomaler Veränderungen, wie großer Deletionen, Duplikationen,
Inversionen und Translokationen. Es kann auch nicht ausgeschlossen werden, dass die
beobachteten Phänotypen nicht nur ein Ergebnis gezielter Mutationen sind, sondern auch
aus ihren unterschiedlichen genetischen Hintergrund resultieren. Darüber hinaus üben
diverse Gene in verschiedenen Zelltypen während der Ontogenese aber auch postnatal
einen Einfluss aus, wie es in der ADPKD in den extrarenalen Manifestationen zu beobachten

EINLEITUNG 7
ist. Um diesen pleiotropen Phänotyp zu umgehen und einen präziseren Einblick in die
Zystenentstehung zu gewinnen, wird ein konditionales Mausmodell benötigt (Nagy, 2000).
Dieses Modell würde ermöglichen gezielt zeit- und/oder gewebespezifisch Gene
auszuschalten. Dafür bietet sich in idealer Weise das Cre/loxP-Rekombinationssystem an.
Bei diesem System wurden klassische Methoden zur Modifikation von Genen mit
Rekombinationssystemen kombiniert, wobei Rekombinasen einen Austausch zwischen zwei
kurzen DNA Erkennungssequenzen katalysieren. Der Bakteriophage P1 aus Escherichia coli
(E. coli) kodiert für dieses ortspezifische Rekombinationssystem, dass sich aus zwei
Komponenten zusammensetzt: eine loxP-(locus of recombination)-DNA-Sequenz an der das
Rekombinationsereignis erfolgt und ein Cre-Rekombinase-Protein, welches die
Rekombination ausführt (Sternberg & Hamilton, 1981; Sternberg et al., 1981). Die Cre-
Rekombinase ist ein einzelnes exogenes Polypeptid und ein Mitglied der Integrase-Familie.
Es handelt sich um ein 38,5 kDa großes Protein (343 AS), welches in der Lage ist, zwischen
34 bp großen loxP-DNA-Sequenzen anzubinden und sowohl in vivo als auch in vitro zu
rekombinieren (Sternberg & Hamilton, 1981; Abremski & Hoess, 1984). Die natürliche
katalytische Funktion des Cre-Proteins als endogenes Produkt des Bakteriophagen P1 ist
das Verpacken von DNA zwischen zwei loxP-Stellen (Sternberg & Hamilton, 1981; Sternberg
et al., 1981). Jede loxP-Stelle weist zwei invertierte 13 bp große gegenläufig angeordnete,
flankierende Sequenzen, die als Bindestellen für jeweils ein Cre-Monomer fungieren und
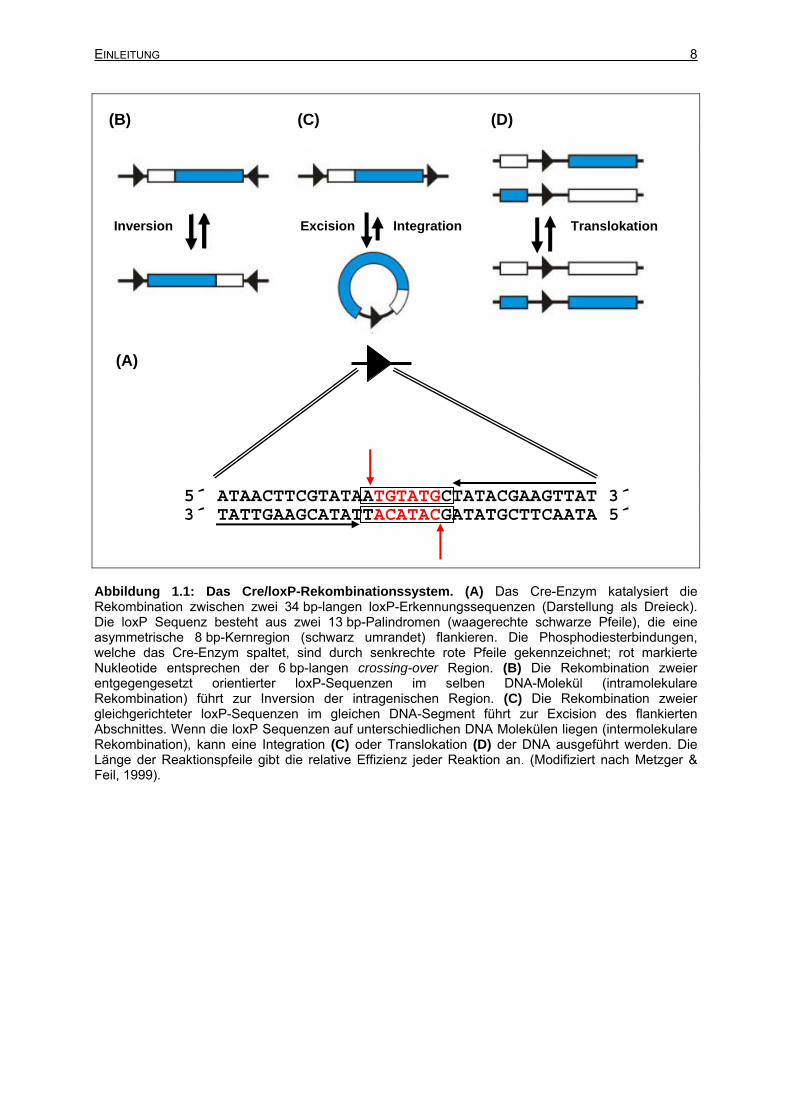
durch eine 8 bp große Region getrennt ist (Hoess et al., 1982). Abbildung 1.1.A stellt die
loxP-Sequenz dar. Unter Bildung eines Cre-Protein Tetramers entsteht der
Rekombinationskomplex. Das Cre-Protein enthält DNA-bindende amino- und
carboxyterminale Domänen, wobei nur der Carboxyterminus die Rekombination zwischen
den loxP-Sequenzen vermittelt (Wierzbicki et al., 1987; Hoess et al., 1990; Guo et al., 1997).
Innerhalb der 8 bp langen, zentralen Region führt der nukleophile Angriff eines in der C-
terminalen, katalytischen Domäne befindlichen Tyrosins zu Einzelstrangbrüchen (Hoess &
Abremski, 1985). Das Rekombinationsprodukt entsteht durch den koordinierten Austausch
der Stränge unter Bildung eines Rekombinationsintermediates mit einer Holliday-Struktur
(Holliday, 1964). Nach Studien von Mack et al. (1992) konnte ermittelt werden, dass jeweils
ein Cre-Rekombinase Molekül an eine loxP-Motivwiederholung bindet. Abhängig von der
Orientierung der loxP-Sequenzen zueinander und dem Integrationsort kann eine Inversion,
Excision/Integration oder Translokation von DNA erzeugt werden (Abbildung 1.1). Die
zentrale Region ist asymmetrisch, welche dadurch eine Richtung erhält. Bei gleicher
Orientierung der loxP-Sequenzen führt die Rekombination zur Excision des
eingeschlossenen Fragmentes (Abbildung 1.1C), bei entgegen gesetzter Orientierung zur
Inversion (Abbildung 1.1B). Intermolekulare Rekombinationsereignisse können zu
Integrationen oder Translokationen führen (Abbildung 1.1D).
EINLEITUNG 8
5´ ATAACTTCGTATAATGTATGCTATACGAAGTTAT 3´ 3´ TATTGAAGCATATTACATACGATATGCTTCAATA 5´ Abbildung 1.1: Das Cre/loxP-Rekombinationssystem. (A) Das Cre-Enzym katalysiert die Rekombination zwischen zwei 34 bp-langen loxP-Erkennungssequenzen (Darstellung als Dreieck). Die loxP Sequenz besteht aus zwei 13 bp-Palindromen (waagerechte schwarze Pfeile), die eine asymmetrische 8 bp-Kernregion (schwarz umrandet) flankieren. Die Phosphodiesterbindungen, welche das Cre-Enzym spaltet, sind durch senkrechte rote Pfeile gekennzeichnet; rot markierte Nukleotide entsprechen der 6 bp-langen crossing-over Region. (B) Die Rekombination zweier entgegengesetzt orientierter loxP-Sequenzen im selben DNA-Molekül (intramolekulare Rekombination) führt zur Inversion der intragenischen Region. (C) Die Rekombination zweier gleichgerichteter loxP-Sequenzen im gleichen DNA-Segment führt zur Excision des flankierten Abschnittes. Wenn die loxP Sequenzen auf unterschiedlichen DNA Molekülen liegen (intermolekulare Rekombination), kann eine Integration (C) oder Translokation (D) der DNA ausgeführt werden. Die Länge der Reaktionspfeile gibt die relative Effizienz jeder Reaktion an. (Modifiziert nach Metzger & Feil, 1999).
Integration Excision Inversion Translokation
(B) (C) (D)
(A)

EINLEITUNG 9
1.3.1 Gewebespezifische Cre-Expression Das Cre/loxP-Rekombinationssystem hat sich in Säugerzellen als sehr effizient erwiesen und
wird verwendet, um gezielt genetische Veränderungen in ES- und in Somazellen der Maus
zu induzieren (Sauer, 1994). Es bietet ein großes Spektrum an Variationen, wie das Genom
modifiziert werden kann, wobei es in vivo am häufigsten zur Inaktivierung von Genen durch
Cre-vermittelte Excision loxP-flankierter („gefloxter“) DNA-Abschnitte eingesetzt wird (Nagy,
2000). Eine gewebespezifische Modifikation des Genoms ist in vielen Fällen informativer als
bei der konventionellen Gen-„knockout“-Maus, wo zu jedem Zeitpunkt der Entwicklung und in
jeder Zelle der Genverlust vorliegt. Dabei basiert die konditionale somatische Mutagenese in
der Maus auf der gewebespezifischen oder induzierbaren Expression von Cre über
geeignete Promotoren. Unter der Kontrolle des Promotors ergibt sich somit die Möglichkeit
einer sehr selektiven Cre-Expression und infolgedessen einer gewebespezifischen
Gendeletion. In der Abbildung 1.2 wird das Wirkprinzip der konditionalen Mutagenese
dargestellt.
Abbildung 1.2 Konditionale Mutagenese in der Maus. Durch Expression der Cre-Rekombinase wird das „gefloxte“ Zielgen herausgeschnitten. Rote Dreiecke stellen loxP-Sequenzen dar.
Bei der Erzeugung konditionaler Mausmutanten wird zunächst mittels pronukleärer Injektion
bei einer Mauslinie ein DNA-Konstrukt injiziert, welches die Cre-Rekombinase unter der
Kontrolle eines gewebespezifischen Promotors stellt (Abbildung 1.3 A). Die loxP-
Erkennungssequenzen werden bei einer weiteren Mauslinie durch homologe Rekombination
gezielt in das Genom integriert. Dabei werden die loxP-Sequenzen in der Weise eingeführt,
dass sie ein bestimmtes Zielgen flankieren. Daraus resultieren so genannte „gefloxte“
Mäuse, welche sich phänotypisch vom Wildtyp nicht unterscheiden lassen (Abbildung 1.3 B).
Die „gefloxten“ Mäuse werden mit Cre-exprimierenden Mäusen verkreuzt, um eine Excision
Cre-Rekombinase
CRE
Zielgen
Promotor Cre-Rekombinase
CRE
Zielgen
Promotor

EINLEITUNG 10
der loxP-flankierten DNA-Sequenz zu bewirken (Abbildung 1.3 C). In den Zellen, wo der
gewebespezifische Promotor aktiv ist, kann nach Expression der Cre-Rekombinase eine
Rekombination zwischen den eingefügten loxP-Sequenzen bewirkt werden, was letztendlich
zu einem Herausschneiden des „gefloxten“ DNA-Segments führt (Abbildung 1.3 D) (Sauer,
1998; Metzger & Feil, 1999; Nagy, 2000; Metzger & Chambon, 2001). Alle anderen Zellen
verbleiben in ihrem ursprünglichen Genotyp (Abbildung 1.3 E).
.P Cre . .ZG .
.P Cre .
ZG
X
ZGCre
spezifische Zellen alle andere Zellen
(a) (b)
(c)
(d) (f)
..P Cre . .ZG ..ZG .
.P Cre .
ZG .P Cre .
ZG
X
ZGZGCreCre
spezifische Zellen alle andere Zellen
(a) (b)
(c)
(d) (f)
Abbildung 1.3: Gewebespezifische Geninaktivierung. (A) Cre-exprimierende Mauslinie; (B) „gefloxte“ Maus. Nach Kreuzung von (A) und (B) resultiert eine konditionale Mauslinie (C). In den speziellen Zellen mit katalytisch aktiver Cre-Rekombinase wird das „gefloxte“ Zielgen (ZG) herausgeschnitten (D). Bei alle anderen Zellen ohne Aktivität des gewebespezifischen Promotors, bleibt die Cre-Expression aus und infolgedessen ZG unverändert.
Mit Hilfe gewebespezifischer konditionaler Mäuse kann auch unterschieden werden, ob sich
der beobachtete Phänotyp aus der primären Funktion des Gens ergibt, oder ob die
Gendeletion sekundär auch andere Gewebe beeinflusst. Transgene Mauslinien mit
gewebespezifischer Cre-Expression haben bereits mehrfach zur Aufklärung biologischer
Fragestellungen beigetragen; wie z.B. bei Insulin-Rezeptoren in murine Skelettmuskelzellen
oder bei Zytokin-Rezeptoren in Herzmuskelzellen (Brüning et al., 1998; Chen et al., 1998;
Hirota et al., 1999; Kulkarni et al., 1999). Die Gewebespezifität hängt stark von dem
verwendeten Promotor ab. In der Regel werden endogene verkürzte Promotoren
(Minimalpromotoren) eingesetzt, deren Aktivitätsmuster hinreichend charakterisiert ist. Für
eine Untersuchung der Zystenentwicklung bei der ADPKD mit Hilfe des konditionalen
Mausmodels richtet sich das Augenmerk auf die Niere. Aus diesem Grunde wäre hier ein
(A)
(C)
(D) spezifische Zellen (E) alle anderen Zellen
(B)
EINLEITUNG 11
Promotor von Interesse, der zellspezifisch in der Niere und präferenziell im Nephron aktiv ist,
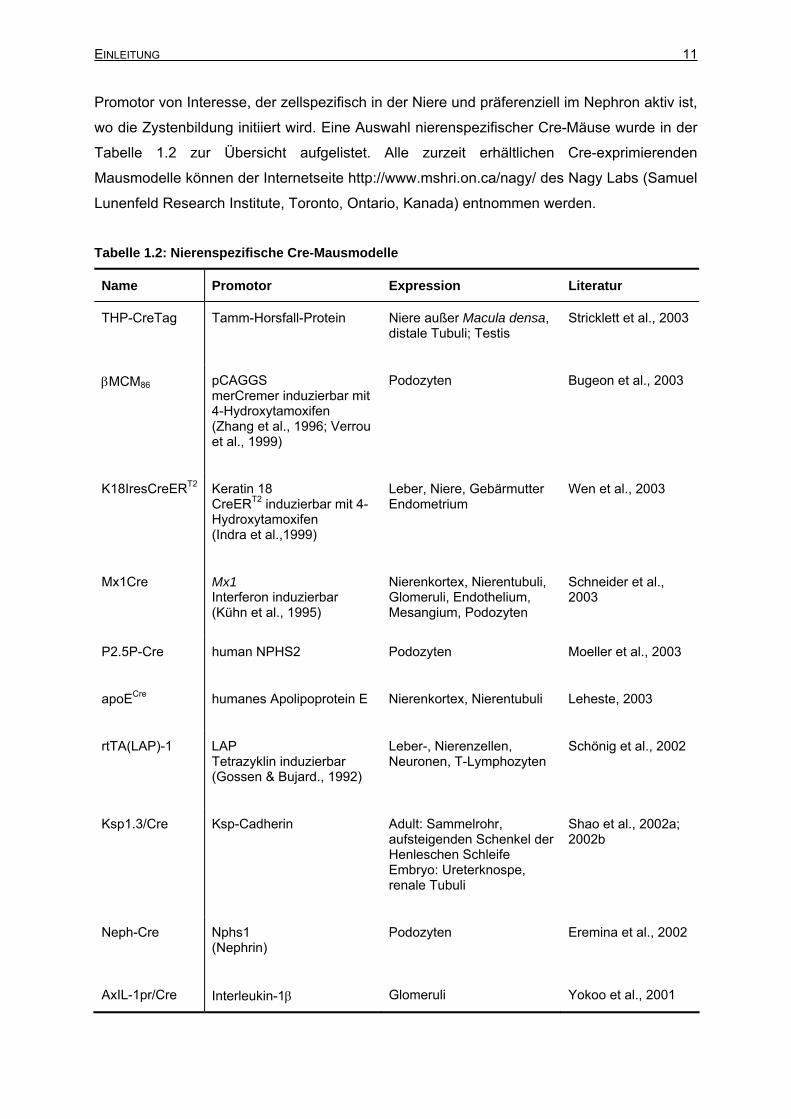
wo die Zystenbildung initiiert wird. Eine Auswahl nierenspezifischer Cre-Mäuse wurde in der
Tabelle 1.2 zur Übersicht aufgelistet. Alle zurzeit erhältlichen Cre-exprimierenden
Mausmodelle können der Internetseite http://www.mshri.on.ca/nagy/ des Nagy Labs (Samuel
Lunenfeld Research Institute, Toronto, Ontario, Kanada) entnommen werden.
Tabelle 1.2: Nierenspezifische Cre-Mausmodelle
Name Promotor Expression Literatur
THP-CreTag Tamm-Horsfall-Protein Niere außer Macula densa, distale Tubuli; Testis
Stricklett et al., 2003
βMCM86 pCAGGS merCremer induzierbar mit 4-Hydroxytamoxifen (Zhang et al., 1996; Verrou et al., 1999)
Podozyten Bugeon et al., 2003
K18IresCreERT2 Keratin 18 CreERT2 induzierbar mit 4-Hydroxytamoxifen (Indra et al.,1999)
Leber, Niere, Gebärmutter Endometrium
Wen et al., 2003
Mx1Cre Mx1 Interferon induzierbar (Kühn et al., 1995)
Nierenkortex, Nierentubuli, Glomeruli, Endothelium, Mesangium, Podozyten
Schneider et al., 2003
P2.5P-Cre human NPHS2 Podozyten Moeller et al., 2003
apoECre humanes Apolipoprotein E Nierenkortex, Nierentubuli Leheste, 2003
rtTA(LAP)-1 LAP Tetrazyklin induzierbar (Gossen & Bujard., 1992)
Leber-, Nierenzellen, Neuronen, T-Lymphozyten
Schönig et al., 2002
Ksp1.3/Cre Ksp-Cadherin Adult: Sammelrohr, aufsteigenden Schenkel der Henleschen Schleife Embryo: Ureterknospe, renale Tubuli
Shao et al., 2002a; 2002b
Neph-Cre Nphs1 (Nephrin)
Podozyten Eremina et al., 2002
AxIL-1pr/Cre Interleukin-1β Glomeruli Yokoo et al., 2001

EINLEITUNG 12
1.3.2 Zeitlich regulierbare Cre-exprimierende Tiermodelle Eine enge zeitliche Kontrolle der Genexpression kann jedoch grundsätzlich nicht alleine über
den Promotor erzielt werden, da verkürzte Minimalpromotoren häufig konstitutiv aktiv sind.
Diese zeitliche Kontrolle wird über eine exogene Beeinflussung der Cre-Expression erreicht.
Die besten Erfahrungen wurden hierbei mit der hormonellen Induktion gemacht. Für eine
zeitlich induzierbare Expression der Cre-Rekombinase in transgenen Tieren sind bislang
mehrere Regulationsmodelle veröffentlich worden. Im folgenden Abschnitt werden die
induzierbaren Systeme vorgestellt.
1.3.2.1 Tet-Repressor/Aktivator-System Als Pioniere entwickelten Gossen und Bujard 1992 ein Kontrollsystem, welches die
Expression individueller Gene durch exogene Zugabe von Tetrazyklin-Analoga reguliert
(Gossen & Bujard, 1992). Dieses System zur induzierbaren Genexpression basiert auf zwei
regulatorischen Elementen: dem Transkriptionsaktivator tTA und einem Operator. Der
Transkriptionsaktivator tTA ist ein Fusionsprotein bestehend aus dem Tet-Repressor vom
tn10 des Bakteriums E. coli und der Aktivierungsdomäne vom viralen Protein VP16 des
Herpes simplex Virus (HSV). Beim Operator handelt es sich um einen humanen
Cytomegalovirus (CMV) Promotor kombiniert mit multiplen tet-Operatorsequenzen (tetO)
(Gossen & Bujard, 1992; 1993; Kuhn et al., 1995; Gossen et al., 1995; Kistner et al., 1996).
Der Transkriptionsaktivator tTA kann nur in seiner aktiven Form an den Operator anbinden
und die Transkription initiieren. Diese aktive Form wird tTA nur in Abwesenheit von
Tetrazyklin beibehalten. Durch die Zugabe von Tetrazyklin kommt es zu einer
Konformationsänderung vom tTA und wird dadurch inaktiviert. Infolgedessen kann der
inaktive tTA nicht an den Operator anbinden und die Transkription unterbleibt.
Der reverse Transaktivator rtTA ist mit dem tTA identisch mit Ausnahme von vier
Aminosäure-Substitutionen im tTA-Lokus, die den reversen Phänotyp vermitteln (Gossen et
al., 1995; Lindeberg & Ebendal, 1999). Hierbei wird rtTA im Gegensatz zu tTA, der als
Repressor fungiert, erst durch eine Tetrazyklin-Zugabe (bzw. seines Analogons Doxycyclin
(Dox)) aktiviert und kann am Operator anbinden. Erst nach Anbindung des aktiven rtTA
erfolgt die Transkription. Zum besseren Verständnis ist die induzierbare Genexpression mit
rtTA in Abbildung 1.4 bildlich dargestellt. Eine konditionale Expression wird ermöglicht, wenn
die Cre-Rekombinase unter der Kontrolle vom Tetrazyklin-Regulationssystem gestellt wird.
Eine herausragende Arbeit wurde 2000 von Yamamoto et al., (2000) veröffentlicht. Die
Arbeitsgruppe etablierte mittels des tTA-Regulationssystem und des Cre-lox/P-
Rekombinationsmechanismus ein konditionales transgenes Mausmodell um die
neurodegenerative Erkrankung Huntington Disease (HD) zeitlich zu regulieren. HD ist
charakterisiert durch progressive und putativ irreversible neuropathologische Symptome

EINLEITUNG 13
durch Einlagerung von Proteinaggregaten, so genannten Huntingtinen, in Neuronen. Die
entwickelten HD-Mausmodelle exprimieren ein mutiertes Hungtingtin-Fragment, wodurch
sich der charakteristische HD-Phänotyp einstellt. Durch Zugabe von Tetrazyklin konnte tTA
inaktiviert werden, somit ist die Huntingtin Expression inhibiert. Bemerkenswerterweise
stellten sich eine Regeneration der Neuronen und ein Rückgang der HD-Symptomatik ein.
Somit zeigte sich, dass diese Erkrankung, was lange Zeit als nicht möglich erachtet wurde,
reversibel ist. Diesbezüglich sind weitere zeitlich induzierbare Tiermodelle entwickelt worden
(Hennighausen et al., 1995; Utomo at al., 1999; Lindeberg & Ebendal, 1999; Saam &
Gordon, 1999; Schönig et al., 2002; Radomska et al., 2002).
Abbildung 1.4: Induzierbare Genexpression vermittelt durch rtTA. Nach Zugabe von Doxycyclin (Dox) wird rtTA aktiviert und bindet an die tet-Operatorsequenzen (tetO). Die Transkription des gewählten Transgens wird somit aktiviert. Abbildung modifiziert nach Gossen et al., 1995.
-Dox +Dox
rtTA
x (A)n
tetO
inaktives rtTA
x (A)n
tetO
aktives rtTA
rtTA
Promoter (A)n
-Dox +Dox
rtTA
x (A)nx (A)n
tetO
inaktives rtTA
x (A)nx (A)n
tetO
aktives rtTA
rtTA
Promoter (A)n(A)n

EINLEITUNG 14
1.3.2.2 Interferon-Rezeptor-Rekonstitution Kühn etablierte 1995 eine Mx1-Cre-Maus, bei welcher die Cre-Rekombinase unter der
Kontrolle des Interferons induzierbaren Mx1-Promotors steht (Kühn et al., 1995). Die
Interferon induzierbare Mx1-Cre-Maus ist von mehreren Arbeitsgruppen verwendet worden
(Schneider et al., 2003; Radtke et al., 1999; Tomita et al., 2000; Horcher et al., 2001;
Ioannidis et al., 2001; Yang et al., 2001; Akiyama et al., 2002; Han et al., 2002; Raffai et al.,
2002; Lieu et al., 2003).
1.3.2.3 Murines Östrogen-induzierbares System Ein weiteres System zur Regulation der Cre-Rekombinase ist 1996 von Zhang et al. (1996)
entwickelt und 1999 von Verrou et al. weitergehend modifiziert worden. Es wurden
Expressionsvektoren konstruiert für ein Fusionsprotein, welches aus der Cre-Rekombinase
und einer mutierten Hormonbindungsdomäne des murinen Östrogenrezeptors besteht. Die
Bindungsdomäne wurde sowohl am C- als auch am N-Terminus der Cre-Rekombinase
kloniert. An der Hormonbindungsdomäne kann noch das synthetische Anti-Östrogen
Tamoxifen anbinden, aber nicht mehr das endogen vorkommende 17-β-Östradiol. Durch 4-
Hydroxytamoxifen wird folglich die chimäre Cre-Rekombinase merCremer (mer: murine
oestrogen receptor) aktiviert. Dieses Modell wurde von Bugeon et al. (2003) angewendet um
eine zeitlich induzierbare Cre-Expression in Podozyten zu erzielen. 2003 fand diese Methode
auch zur konditionalen Cre-Expression in Herzmuskelzellen Verwendung (Petrich et al.,
2003).
1.3.2.4 Humanes Östrogen-induzierbares System Ein weiteres System für die zeitlich und örtlich regulierte Cre-Expression stellt das
gemeinsam von Metzger et al. (1995) entwickelte Cre-ERT-System dar. Die Fusion der
Ligandenbindungsdomänen (LBD: ligand-binding domain) aus steroid Hormonrezeptoren
(ER: estrogen receptor) mit einer Vielzahl von heterologen Proteinen zeigten eine liganden-
abhängige Aktivitätskontrolle. Es wurde eine ligandeninduzierbare Cre-Rekombinase durch
Fusionierung des Cre-Strukturgens mit der Ligandenbindungsdomäne des humanen
Östrogen-Rezeptors (hER) generiert. Das entstandene chimäre Protein Cre-ER führte zu
einer östradiolabhängigen Cre/loxP-Rekombination. Sowohl das endogene 17β-Östradiol als
auch das synthetische Anti-Östrogen 4-Hydroxytamoxifen aktiviert das Fusionsprotein Cre-
ER (Metzger et al., 1995). Die Spezifität dieses Rezeptors wurde nachfolgend durch
Mutagenese so variiert, dass das Fusionsprotein nicht mehr durch das endogene Östrogen,
sondern nur noch durch Tamoxifen aktiviert werden kann. Durch die mutierte
Ligandenbindungsdomäne resultierte das Protein Cre-ERT (Feil et al., 1996, 1997). Auf
molekularer Ebene ist Tamoxifen für die Translokation des cytoplasmatisch lokalisierten Cre-

EINLEITUNG 15
ERT in Richtung Nukleus verantwortlich (Brocard et al., 1997). Erst im Nukleus kann das Cre
als DNA-Bindeprotein enzymatisch aktiv werden. Eine Tamoxifen-induzierbare retinoid-X-
Rezeptoren-Expression wurde in Keratinozyten (Li et al., 2000) und in Adipozyten
(Fettzellen) ausgeführt (Imai et al., 2001). 2003 erfolgte eine spezifische Cre-ERT2 -
Expression in myelinbildenden Schwann-Zellen (Leone et al., 2003).

EINLEITUNG 16
1.4 Ziele Die Herstellung transgener Tiermodelle zur Funktionsanalyse neuer Gene oder
Genvarianten und die Gen-„knockout/knockin“-Technologie, durch die Gene oder Gen-
Abschnitte inaktiviert, addiert oder modifiziert werden können, haben sich mittlerweile zu
einer vielfach genutzten Technik entwickelt. Diese komplexen experimentellen Ansätze sind
insbesondere dann von Bedeutung, wenn die Keimbahnmutationen letal oder die
Auswirkungen einer Mutation nur in bestimmten Geweben oder Organen von Wichtigkeit
sind. Diese Limitierungen zeigten sich bei den ADPKD-„knockout“-Mäusen. Konventionell
generierte Mausmutanten zeigen nur den ersten relevanten Expressionszeitpunkt auf. In
anderen Fällen wird ein komplexer pleiotroper Phänotyp verursacht, der mit dem
menschlichen ADPKD-Phänotyp nicht vergleichbar ist. Die konditionale Mutagenese in der
Maus ist ein neuer Ansatz zur gewebespezifischen, funktionellen Analyse von Genen. Ein
Meilenstein zur Generierung konditionaler Mausmutanten ist das mittlerweile vielfach
eingesetzte Cre/loxP-Rekombinationssystem. Ziel dieser Arbeit war es eine transgene
Mauslinie zu entwickeln, bei welcher nephronspezifisch die Cre-Rekombinase exprimiert
wird. Diese Maus wäre in Zukunft nicht nur für die Analyse der ADPKD, sondern auch für
Funktionsstudien vieler weiterer nierenspezifischer Gene von großer Bedeutung.

MATERIAL UND METHODEN
17
2 Material und Methoden
Alle in den folgenden Kapiteln verwendeten Materialien und Geräte sind in Kapitel 2.6-2.7
aufgeführt. Die Bezugsfirmen und deren Adressen sind in Kapitel 2.8 aufgelistet.
2.1 Datenbanken Zwecks Identifizierung geeigneter Oligonukleotide zur PCR und zur Sequenzierung wurden
folgende Programme verwendet:
• OLIGO 4.0, Primer Analysis Software, National BioSciences
• http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi (Whitehead Institute
Center for Genome Research, Cambridge, MA, USA)
Für die Standard-Sequenzanalyse (Transkription, Translation, Restriktion, Editierung der
Sequenzinformationen) sind folgende Programme verwendet worden:
• DNASISTM Version 6.0, Pharmacia Biotech (Freiburg)
• http://genius.embnet.dkfz-heidelberg.de/menu/w2h/w2hdkfz/index.shtml,DKFZ
Heidelberg, HUSAR
Zur Sequenzauswertung des ABI PRISM® 310 und des ABI PRISM® 3700 wurde folgendes
Programm verwendet:
• Chromas, Version 1.43 (32-bit) Conor Mc Carthy School of Biomolecular and
Biomedical Science Faculty of Science Griffith University Brisbane, Queensland
Australien
Zur Darstellung von Plasmidkarten wurde folgendes Programm verwendet:
• pDRAW32 1.0 Revision 1.1.61, Kjeld Olesen Acaclone software,
http://www.acaclone.com
Datenbankrecherchen erfolgten über folgende Internetseite:
• http://www.ncbi.nlm.nih.gov/PubMed/ National Center for Biotechnology Information
U.S National Library of Medicine 8600 Rochville Pike, Bethesda, MD 20894, USA
Zur Dokumentierung der in dieser Arbeit verwendeten Mauslinien ist folgendes Programm
verwendet worden:
• Microsoft® Access 2002, Microsoft Corporation 1992-2001

MATERIAL UND METHODEN
18
2.2 Molekularbiologische Methoden
2.2.1 Kultivierung von Escherichia coli (E. coli) Die Anzucht der E. coli-Bakterien erfolgte in LB-Medium bei 37°C. Flüssigkulturen wurden
mit 200 rpm geschüttelt. Um plasmidtragende Zellen zu selektionieren, wurde Ampicillin in
einer Endkonzentration von 100 µg/ml zugegeben. Die Herstellung der Nährmedien erfolgte
nach Sambrook et al. (1989). Für Festmedien wurde 1% - 2,5% (w/v) Agar-Agar zugesetzt.
2.2.2 Anlegen einer Gefrierkultur Unter Selektionsdruck wurde zunächst eine Einzelkolonie in Flüssigmedium über Nacht
angezogen. 700 µl von der Flüssigkultur sind anschließend in ein Kryoröhrchen, in dem
150 µl Glyzerin (87%) vorgelegt waren, überführt worden. Die Gefrierkultur wurde bei -70°C
gelagert und vor Gebrauch im Schüttelwasserbad aufgetaut.
2.2.3 Plasmid-Isolierung aus E. coli In Abhängigkeit von der gewünschten Qualität und Quantität der Plasmid-DNA wurden drei
unterschiedliche Isolierungsmethoden durchgeführt. Für geringe DNA-Mengen ohne hohen
Reinheitsgrad, erfolgte die konventionelle Plasmid-Isolation aus E. coli (2.2.3.1). Um einen
höheren Reinheitsgrad und/oder größere DNA-Mengen zu erzielen, ist ein Kit für Plasmid-
Mini-/Maxi-Präparation (2.2.3.2) der Firma Qiagen (Hilden) verwendet worden. Alle
Methoden der Plasmid-Isolation beruhen auf der alkalischen Denaturierung mit
anschließender DNA-Neutralisierung (Birnboim & Doly, 1979).
2.2.3.1 Plasmid-Mini-Präparation aus E. coli
Nach Animpfung einer E. coli-Einzelkolonie erfolgte die Anzucht unter entsprechendem
Selektionsdruck über Nacht (37°C) in 7 ml LB-Medium. Nach Zentrifugation von 1,5 ml der
Flüssigkultur (15000 g, 3 min) wurden die Zellen in 50 µl TE-Puffer resuspendiert. Durch
Zugabe von 300 µl des alkalischen Lysispuffers (TENS-Puffer 1) fanden die Zell-Lyse und
die DNA-Denaturierung statt. Die Plasmid-DNA renaturierte durch die anschließende Zugabe
von 150 µl Neutralisierungspuffer (TENS-Puffer 2). Nach Durchmischen der Lösung wurden
die chromosomale DNA, die Proteine, sowie die Zelltrümmer abzentrifugiert (15000 g,
15 min). Um die DNA zu präzipitieren, wurde der Überstand in einen Volumen Isopropanol
(-20°C) überführt. Nach erneuter Zentrifugation (15000 g, 30 min, 4°C) wurde das Pellet mit
70% Ethanol (-20°C) gewaschen. Der Ansatz ist abschließend zentrifugiert (s. o.) und das
entstandene Pellet in 20 µl TE-Puffer gelöst und mit 1 µl RNase (1 U/µg) vermengt worden.

MATERIAL UND METHODEN
19
2.2.3.2 QIAprep-Plasmid-Mini-/Maxi-Präparation aus E. coli
Durch die QIAprep-Plasmid-Mini-/Maxi-Präparation (Qiagen, Hilden) wurde Plasmid-DNA
mittels einer Anionenaustauschersäule aus E. coli Bakterien-Flüssigkultur gewonnen. Die
Präparationen bakterieller Plasmid-DNA erfolgten mit dem QIAprep-Mini-/Maxi-Kit (Qiagen,
Hilden) gemäß dem mitgelieferten Protokoll aus 50 ml- bzw. 500 ml-Kulturen.
2.2.4 DNA-Isolierung aus Gewebe Genomische DNA aus Gewebe wurde in dieser Arbeit mittels zwei unterschiedlichen
Methoden gewonnen:
• Phenol-Chloroform-Extraktion (2.2.5.1)
• Aussalzung (2.2.5.2)
Beide Methoden beruhen auf den Zellaufschluss durch SDS und Proteinase K. Bei der
anschließender Beseitigung der Proteine unterscheiden sie sich durch die Zugabe von
entweder einer Phenol/Chloroform- oder einer gesättigten NaCl-Lösung. Das Gewebe wurde
zunächst mit einem Skalpell zerkleinert und in 0,5-5 ml Lysispuffer überführt
(Mäuseschwänze: 750 µl, andere Gewebe: 5 ml). Nach Zugabe von 50 µl Proteinase K
(10 mg/ml) / ml Lysispuffer wurde die Lösung über Nacht in einem auf 55°C temperiertes
Schüttelwasserbad inkubiert. Die DNA wurde am folgenden Tag mittels Phenol/Chloroform-
Lösung extrahiert (2.2.5.1) oder ausgesalzen (2.2.5.2). Abschließend erfolgte die DNA-
Fällung mit Isopropanol (2.2.5.3) oder mit Ethanol (2.2.5.4).
2.2.5 Reinigung isolierter DNA 2.2.5.1 Phenol/Chloroform-Extraktion Zur Abtrennung von Nukleinsäuren aus proteinhaltigen Lösungen wurden
Phenol/Chloroform-Extraktionen durchgeführt. Die wässrige Lösung ist mit einem Volumen
TE- gesättigtem Phenol versetzt und gut gemischt worden. Dabei bleiben die Nukleinsäuren
aufgrund ihrer hydrophilen Gruppen in der oberen wässrigen Phase gelöst, während sich die
Proteine zwischen beiden Phasen, in der so genannten Interphase, anordnen. Nach
anschließender 5-minütiger Zentrifugation bei 13.000 rpm (RT) wurde die obere wässrige
Phase in ein neues Reaktionsgefäß überführt. Nach Zugabe von einem Volumen
Chloroform:Isoamylalkohol (24:1) auf die wässrigen Phase wurde das Gefäß geschüttelt und
erneut zentrifugiert (13000 rpm, RT, 5´), so dass die vorhandenen Phenolreste extrahiert
wurden. Anschließend ist die obere DNA-haltige wässrige Phase vorsichtig abgenommen
und in ein neues Reaktionsgefäß überführt worden. Mit 800 µl Isopropanol (2.2.5.3) oder
Ethanol erfolgte abschließend die DNA-Fällung (2.2.5.4).

MATERIAL UND METHODEN
20
2.2.5.2 Aussalzung Bei der Methode der Aussalzung wurden 550 µl DNA-Lösung mit 154 µl 6 M NaCl vermengt
und 15 Sekunden gemischt. Es folgte eine Zentrifugation für 5 Minuten bei 13.000 rpm (RT).
Der DNA-haltige Überstand ist vorsichtig abgenommen und mit 600 µl Isopropanol gefällt
worden (2.2.5.3).
2.2.5.3 Isopropanol-Fällung Nach Zugabe von 0,7 fachen Volumens Isopropanol (4°C) zu der DNA-haltigen Lösung
wurde das Reaktionsgefäß leicht geschwenkt und anschließend abzentrifugiert (15000 g,
30 min, 4°C) worden. Die Reinigung des Pellets erfolgte durch Zugabe von 70% Ethanol und
einer erneuten Zentrifugation (15000 g, 5 min, 4°C). Das entstandene Pellet wurde
abschließend luftgetrocknet und in einer gewünschten Menge Aqua dest. oder TE-Puffer
gelöst (über Nacht, RT).
2.2.5.4 Ethanol-Fällung 1/10 Volumen Na-Acetat (3M, pH 5,2) und das 2,5-fache Volumen Ethanol abs. (-20°C)
wurden in die DNA-Lösung überführt. Das Reaktionsgefäß wurde leicht geschwenkt und
nach Zentrifugation (13000 rpm, 30 min, 4°C) konnte der Überstand abgegossen und das
entstandene Präzipitat mit 70%igem Ethanol (4°C) gewaschen werden. Abschließend wurde
das DNA-Pellet an der Luft getrocknet und in einer gewünschten Menge Aqua dest. oder TE-
Puffer aufgenommen (über Nacht, RT).
2.2.5.5 Dialyse Niedermolekulare Verunreinigungen wie z.B. Zucker oder Salze können enzymatische
Reaktionen inhibieren. Um sie zu entfernen, wurde bis zu 50 µl DNA-Lösung auf ein
Mikrofilterblättchen (Millipore, Schwalbach) mit einem Porendurchmesser von 0,025 µm
übertragen. Das Filterblatt lag 15 min auf der Oberfläche einer mit Aqua dest. gefüllter
Petrischale. Die semipermeable Membran des Mikrofilterblättchens ist für Nukleinsäuren
undurchlässig, wodurch die niedermolekularen Verbindungen aus der Probe entlang des
osmotischen Gradienten in die umgebende Flüssigkeit diffundieren.
2.2.6 Konzentrationsbestimmung von Nukleinsäuren Die Bestimmung der Reinheit und die Quantifizierung von Nukleinsäuren erfolgte durch
Messung der optischen Dichte (OD) mit dem Photometer ”Genequant” (Pharmacia, Freiburg)
bei 360 nm, 280 nm und 260 nm.

MATERIAL UND METHODEN
21
2.2.7 Gelelektrophorese von Nukleinsäuren Die Nukleinsäuren wandern beim Anlegen eines elektrischen Feldes aufgrund der negativen
Ladung ihres Zucker-Phosphat-Rückrats in Richtung Anode. Als Folge des
Molekularsiebeffektes der Gelmatrix vollzieht sich eine Auftrennung der Makromoleküle nach
ihrer Größe (Aaij & Borst, 1972). Tabelle 2.1 stellt den Auftrennungsbereich linearer DNA-
Moleküle in Gelen bei unterschiedlichen Agarosekonzentrationen dar.
Tabelle 2.1: Effizienter Trennbereich linearer DNA-Moleküle in Gelen unterschiedlicher Agarosekonzentration (Maniatis et al., 1989). Agarosekonzentration [%] 0,3 0,6 0,7 0,9 1,2 1,5 2,0
Auftrennungsbereich [kb] 60-5 20-1 10-0,8 7-0,5 6-0,4 4-0,2 3-0,2
Die Agarose wurde in Elektrophoresepuffer (1x-TBE-Puffer/Größenbestimmung; 1×-TEP-
Puffer/DNA-Transfer-Gele) suspendiert, und mit Ethidiumbromid versetzt. Nach Erstarren
des Agarosegels wurde mit Elektrophoresepuffer überschichtet. Anschließend sind die
Proben mit 1/10 Volumen Gelladepuffer versetzt und in die Probetaschen pipettiert worden.
Als Längenstandard dienten 500 ng der 100 bp-Leiter (Invitrogen, Karlsruhe) und/oder 500
ng des Markers GeneRuler™ 1 kb DNA Leiter (Fermentas, St. Leon-Rot; Abbildung 2.1). Die
Gelelektrophorese erfolgte je nach Agarosekonzentration, Größe des Gels und
Zusammensetzung der Fragmente bei einer Feldstärke von 0,5 bis 5 V/cm in einem Zeitraum
von 1-12 Stunden. Abschließend konnte die DNA unter UV-Licht betrachtet werden.
Abbildung 2.1: DNA-Längenstandards (A): 100 bp-DNA-Leiter (Invitrogen, Karlsruhe); (B): GeneRuler™ 1 kb DNA-Leiter (Fermentas, St. Leon-Rot).
(A)
-500
-600
-300
-100
-200
-400
-1.500
-2.072
bp bp (B)

MATERIAL UND METHODEN
22
2.2.8 Extraktion und Rückgewinnung von DNA aus Agarosegelen Mit Hilfe des “QIAquick Gel Extraction Kit” (Qiagen, Hilden) erfolgte die Extraktion von DNA-
Fragmenten (<10 kb) aus Agarosegelen, entsprechend dem mitgelieferten Protokoll. Die
Gelmatrix wird durch Natriumperchlorat zersetzt und die DNA adsorbiert an die
Säulenmatrix. Nach Reinigung durch mehrere Waschschritte kann die DNA aus den
Säulchen mit wahlweise 30 µl TE-Puffer (65°C) oder Aqua dest. eluiert werden.
2.2.9 Modifikation und Klonierung von Nukleinsäuren Die Klonierung von Nukleinsäuren ist definiert als Einbau von DNA-Fragmenten in Vektoren
mit anschließender Transformation in E. coli-Bakterien. Ziel dieser Transformation ist die in
vivo Vermehrung der exogenen Nukleinsäuren. Dabei setzt jede Klonierungsstrategie eine
Modifikation der DNA voraus. Alle einklonierten DNA-Fragmente wurden nach
abgeschlossener Klonierung zur Kontrolle sequenziert und anschließend mit der
Ausgangssequenz verglichen. Somit konnte gewährleistet werden, dass nur einklonierte
DNA-Fragmente ohne Sequenzveränderungen verwendet worden sind.
2.2.9.1 Restriktion Die Restriktion von DNA erfolgte durch Restriktionsendonukleasen der Klasse II, welche dem
Anhang zu entnehmen sind. Es wurden Enzyme der Firmen Roche (Mannheim), Invitrogen
(Karlsruhe) oder New England Biolabs (Frankfurt/Main) bezogen und unter Verwendung der
mitgelieferten Puffersysteme nach Angaben des Herstellers eingesetzt. Bei Mehrfach-
Restriktionen mit Enzymen, die unterschiedliche Puffer benötigten, wurde der Puffer gewählt,
bei dem die geringsten Aktivitätsverluste zu erwarten war. Falls erforderlich, erfolgte nach
der Restriktion eine Hitzeinaktivierung. Die Restriktionsreaktionen wurden abschließend über
eine Gelelektrophorese (2.2.7) kontrolliert und analysiert.
2.2.9.2 Dephosphorylierung der 5´-Enden Um bei Klonierungen eine Vektor-Religation zu vermeiden, können die 5´-Enden der DNA-
Vektoren mit Hilfe der alkalischen Phosphatase dephosphoryliert werden. Hierfür wurde die
SAP-alkalische-Phosphatase der Firma Roche (Mannheim) eingesetzt. Nach Restriktion
(2.2.9.1) mit anschließender Hitzeinaktivierung der Restriktionsendonukleasen erfolgte die
Zugabe folgender Lösungen: pro 50 ng Vektor-DNA: 0,9 µl SAP-Puffer (10x) und 1 U
alkalischer Phosphatase. Bei überstehenden DNA-Enden schloss sich eine
Inkubationsphase von 10 min bei 37°C an und bei glatten DNA-Enden 60 min bei 37°C.
Nach der Dephosphorylierungsphase folgte die Hitzeinaktivierung der alkalischen
Phosphatase bei 65°C für 15 min, in der sie irreversibel denaturiert. Die dephosphorylierte
DNA konnte dann für weitere Ligationen eingesetzt werden. Alternativ kann CIP („calf
MATERIAL UND METHODEN
23
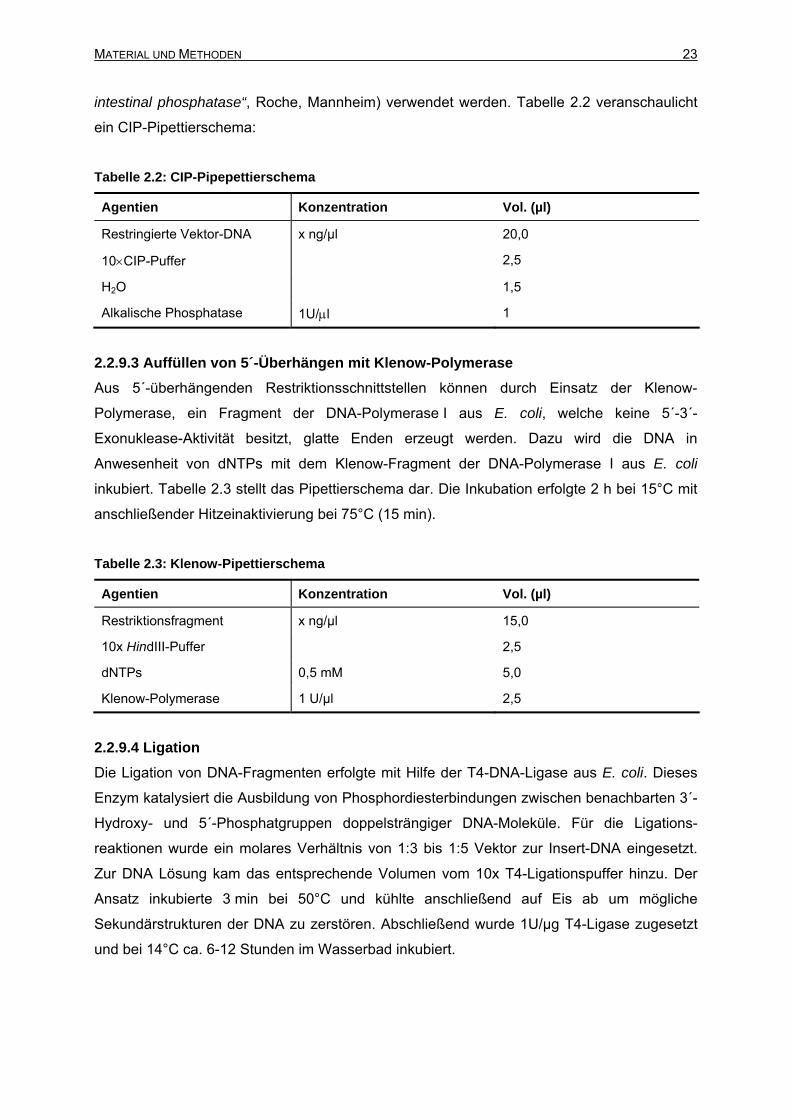
intestinal phosphatase“, Roche, Mannheim) verwendet werden. Tabelle 2.2 veranschaulicht
ein CIP-Pipettierschema:
Tabelle 2.2: CIP-Pipepettierschema
Agentien Konzentration Vol. (µl)
Restringierte Vektor-DNA x ng/µl 20,0
10×CIP-Puffer 2,5
H2O 1,5
Alkalische Phosphatase 1U/μl 1
2.2.9.3 Auffüllen von 5´-Überhängen mit Klenow-Polymerase Aus 5´-überhängenden Restriktionsschnittstellen können durch Einsatz der Klenow-
Polymerase, ein Fragment der DNA-Polymerase I aus E. coli, welche keine 5´-3´-
Exonuklease-Aktivität besitzt, glatte Enden erzeugt werden. Dazu wird die DNA in
Anwesenheit von dNTPs mit dem Klenow-Fragment der DNA-Polymerase I aus E. coli
inkubiert. Tabelle 2.3 stellt das Pipettierschema dar. Die Inkubation erfolgte 2 h bei 15°C mit
anschließender Hitzeinaktivierung bei 75°C (15 min).
Tabelle 2.3: Klenow-Pipettierschema
Agentien Konzentration Vol. (µl)
Restriktionsfragment x ng/µl 15,0
10x HindIII-Puffer 2,5
dNTPs 0,5 mM 5,0
Klenow-Polymerase 1 U/µl 2,5
2.2.9.4 Ligation Die Ligation von DNA-Fragmenten erfolgte mit Hilfe der T4-DNA-Ligase aus E. coli. Dieses
Enzym katalysiert die Ausbildung von Phosphordiesterbindungen zwischen benachbarten 3´-
Hydroxy- und 5´-Phosphatgruppen doppelsträngiger DNA-Moleküle. Für die Ligations-
reaktionen wurde ein molares Verhältnis von 1:3 bis 1:5 Vektor zur Insert-DNA eingesetzt.
Zur DNA Lösung kam das entsprechende Volumen vom 10x T4-Ligationspuffer hinzu. Der
Ansatz inkubierte 3 min bei 50°C und kühlte anschließend auf Eis ab um mögliche
Sekundärstrukturen der DNA zu zerstören. Abschließend wurde 1U/µg T4-Ligase zugesetzt
und bei 14°C ca. 6-12 Stunden im Wasserbad inkubiert.

MATERIAL UND METHODEN
24
2.2.9.5 Klonierung von PCR-Produkten Um PCR-Produkte zu klonieren, sind in der vorliegenden Arbeit zwei Techniken angewandt
worden. Erstens die Topoisomerase-Methode (Shuman, 1994) mit Hilfe des TOPOTM TA
Cloning Version E-Kits (Invitrogen, Karlsruhe) und zweitens konventionell mittels Einführung
von Restriktionsschnittstellen in die Leitsequenzen (Primer). Die Taq-Polymerase hat eine
terminale Transferase-Aktivität, durch die einzelne Desoxyadenosine (A) an den 3´-Enden
der resultierenden PCR-Produkte kovalent angebunden werden. Ein im Kit mitgelieferter
linearisierter Vektor (TOPOTM -Klonierungsvektor) verfügt über einzelne überhängende 3´-
Desoxythymidine (T). Das Enzym Topoisomerase I katalysiert die Ligation des TOPOTM -
Klonierungsvektors mit den PCR-Amplifikaten. Gemäß den Angaben des Herstellers wurde
die Klonierung durchgeführt. Mit der konventionellen Methode sind Oligonukleotide durch
Restriktionsschnittstellen generiert worden (Anhang). Abschließend wurden die
Ligationsansätze in E. coli-Zellen transformiert (2.2.10).
2.2.9.6 Klonierung von Restriktionsfragmenten Um DNA-Fragmente zu klonieren, wurde die DNA zunächst restringiert (2.2.9.1) und nach
gelelektrophoretischer Auftrennung (2.2.7) aus dem Agarosegel extrahiert (2.2.8). Zwecks
Vermeidung einer Religation des Vektors bei Verwendung eines Restriktionsenzyms erfolgte
vor der Ligation eine Dephosphorylierung mit der alkalischen Phosphatase (2.2.9.2). Bei der
Klonierung von DNA mit unterschiedlichen Restriktionsfragmenten wurden mittels der
Klenow-Polymerase (2.2.9.3) glatte Enden produziert und anschließend ligiert (2.2.9.4). Zur
Vervielfältigung der klonierten Fragmente, sind die Ligationsansätze abschließend in E. coli-
Zellen transformiert worden (2.2.10).
2.2.10 Transformation von E. coli Als Transformation wird die Veränderung des Genotyps einer Zelle durch die exogene
Aufnahme von DNA-Molekülen bezeichnet (Singer & Berg, 1992). Um DNA aufnehmen zu
können, müssen die Zellen chemisch vorbehandelt werden. Zur Transformation wurden
E. coli-Top10-Zellen (Invitrogen, Karlsruhe) gemäß den Angaben des Herstellers verwendet.
2.2.11 Polymerasekettenreaktion (PCR) Bei der PCR-Methode handelt es sich um ein effizientes Verfahren zur in vitro-Vermehrung
von DNA (Mullis, 1990). Einzelne DNA-Fragmente liegen nach 20 bis 40 Vermehrungszyklen
in milliardenfacher Kopie vor. Folgende Reaktionskomponenten sind für die PCR-
Durchführung erforderlich: doppelsträngige Matrizen-DNA; Oligonukleotide (PCR-Primer),
welche zur Matrizen-DNA komplementär sind; freie Desoxynukleotide (dNTPs); Mg2+-Ionen
und thermostabile DNA-Polymerasen. In dieser Arbeit wurden die Taq-Polymerase aus dem
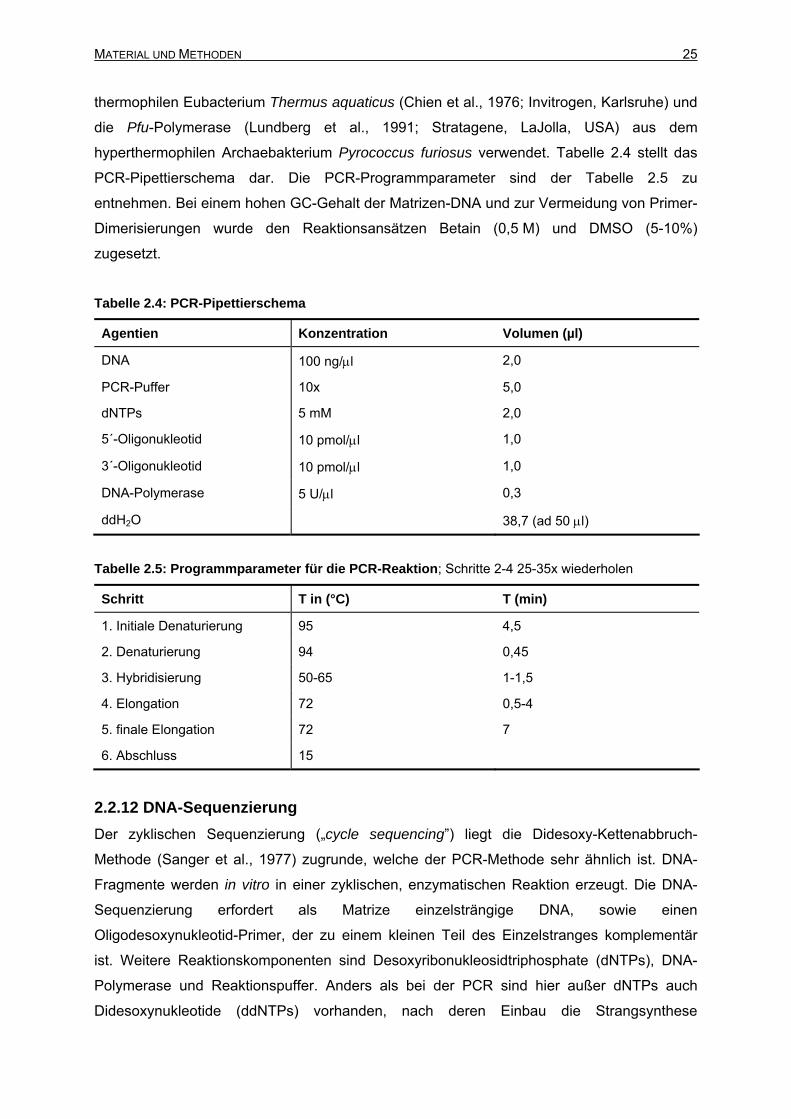
MATERIAL UND METHODEN
25
thermophilen Eubacterium Thermus aquaticus (Chien et al., 1976; Invitrogen, Karlsruhe) und
die Pfu-Polymerase (Lundberg et al., 1991; Stratagene, LaJolla, USA) aus dem
hyperthermophilen Archaebakterium Pyrococcus furiosus verwendet. Tabelle 2.4 stellt das
PCR-Pipettierschema dar. Die PCR-Programmparameter sind der Tabelle 2.5 zu
entnehmen. Bei einem hohen GC-Gehalt der Matrizen-DNA und zur Vermeidung von Primer-
Dimerisierungen wurde den Reaktionsansätzen Betain (0,5 M) und DMSO (5-10%)
zugesetzt.
Tabelle 2.4: PCR-Pipettierschema
Agentien Konzentration Volumen (µl)
DNA 100 ng/μl 2,0
PCR-Puffer 10x 5,0
dNTPs 5 mM 2,0
5´-Oligonukleotid 10 pmol/μl 1,0
3´-Oligonukleotid 10 pmol/μl 1,0
DNA-Polymerase 5 U/μl 0,3
ddH2O 38,7 (ad 50 μl)
Tabelle 2.5: Programmparameter für die PCR-Reaktion; Schritte 2-4 25-35x wiederholen
Schritt T in (°C) T (min)
1. Initiale Denaturierung 95 4,5
2. Denaturierung 94 0,45
3. Hybridisierung 50-65 1-1,5
4. Elongation 72 0,5-4
5. finale Elongation 72 7
6. Abschluss 15
2.2.12 DNA-Sequenzierung Der zyklischen Sequenzierung („cycle sequencing”) liegt die Didesoxy-Kettenabbruch-
Methode (Sanger et al., 1977) zugrunde, welche der PCR-Methode sehr ähnlich ist. DNA-
Fragmente werden in vitro in einer zyklischen, enzymatischen Reaktion erzeugt. Die DNA-
Sequenzierung erfordert als Matrize einzelsträngige DNA, sowie einen
Oligodesoxynukleotid-Primer, der zu einem kleinen Teil des Einzelstranges komplementär
ist. Weitere Reaktionskomponenten sind Desoxyribonukleosidtriphosphate (dNTPs), DNA-
Polymerase und Reaktionspuffer. Anders als bei der PCR sind hier außer dNTPs auch
Didesoxynukleotide (ddNTPs) vorhanden, nach deren Einbau die Strangsynthese

MATERIAL UND METHODEN
26
abgebrochen wird. Dabei wird die Reaktion durch das Fehlen der 3´-Hydroxylgruppe des
Didesoxynukleotids abgebrochen. Da zu jedem Zeitpunkt der Strangsynthese der Zufall
entscheidet, ob ein dNTP oder das modifizierte Nukleotid ddNTP eingebaut wird, entstehen
insgesamt DNA-Fragmente jeder Länge, die jeweils mit der Primersequenz beginnen und mit
einem ddNTP enden. Zur Sequenzierung wurde das ABI PRISM BigDyeTM, Terminator Cycle
Sequencing Kit (Applied Biosystems, Darmstadt) gemäß den Angaben des Herstellers
verwendet. Die gekoppelten Fluoreszenzfarbstoffe der eingesetzten ddNTPs wurden auf den
ABI PRISM® 310 oder ABI PRISM® 3700 DNA Analyzer (Applied Biosystems, Darmstadt)
nach Anregung mit Laserlicht detektiert. Es erfolgte die Sequenzierung des kodogenen
Stranges und des Gegenstranges.
2.2.12.1 Proben-Aufreinigung Die PCR-Produkte wurden mittels eines kommerziellen Pre-Sequencing-Kits (USB
Corporation, Cleveland Ohio, USA) aufgereinigt. Nach Beendigung der PCR verbleiben
unverbrauchte dNTPs und Primer im PCR-Reaktionsgemisch, welche sich störend auf die
nachfolgende Sequenzierung auswirken würden. Die im Kit vorliegenden hydrolytischen
Enzyme alkalische Garnellen-Phosphatase und Exonuklease I entfernen unerwünschte
dNTPs und Primer. Dabei beseitigt die Exonuklease I die übrigen einzelsträngigen Primer
und überflüssige einzelsträngige DNA, und die alkalische Garnellen-Phosphatase die
verbliebenen dNTPs. Beide Enzyme lassen sich bei 80°C inaktivieren. Folgender Ansatz
wurde vorbereitet:
3 µl PCR-Produkt
0,6 µl Exonuklease I
0,6 µl Alkalische Phosphatase
Es folgte eine Inkubation von 15 min bei 37°C mit abschließender Hitzeinaktivierung (15 min;
80°C). Die Vektor-DNA kann nach einer Plasmid-Präparation (2.2.3) ohne weitere
Reinigungsschritte sofort eingesetzt werden. Optional konnten mittels Dialyse (2.2.5.5)
niedermolekulare Verunreinigungen entfernt werden.
2.2.12.2 Vorbereitungen der PCR-Reaktionsansätze Pro Reaktion wurden zwei Ansätze gemäß den Angaben des Herstellers (Applied
Biosystems, Darmstadt) vorbereitet. Jeder Ansatz enthält die Matrizen-DNA, den Strang-
bzw. Gegenstrang-Sequenzierungs-Primer, das Reaktionsgemisch (ABI PRISM BigDye
Terminator Cycle Sequencing Kit, Applied Biosystems, Darmstadt) und Aqua dest. Der
Reaktionsansatz sollte zum einen auf Eis stehen, um Aktivitätsverluste der DNA-Polymerase

MATERIAL UND METHODEN
27
vorzubeugen und zum anderen, wegen der Lichtempfindlichkeit des ddNTP-Chromophors,
Licht vermieden werden. Anschließend erfolgte die Sequenzreaktion (2.2.12) im T3-
Thermocycler (Biometra, Göttingen). Tabelle 2.6 stellt die Programmparameter der
Sequenzreaktion dar.
Tabelle 2.6: Programmparameter für die Sequenzreaktion; Schritte 2-4 25x wiederholen
Schritt T (°C) t
1. Denaturierung 96 2 min
2. Denaturierung 94 10 sec
3. Hybridisierung 50 5 sec
4. Elongation 60 2 min
2.2.12.3 Vorbereitung der Sequenzierungsansätze für den ABI PRISM® 310 Der Reaktionsansatz (10 µl) wurde nach der Sequenzreaktion in 90 µl Aqua dest. überführt
und nach Zugabe von 3M NaAc (pH 5,2) und 275 µl abs. Ethanol für 5 min bei RT inkubiert.
Nach einer Zentrifugation (10 min; 13000 rpm) wurde der Überstand mittels einer
Wasserstrahlpumpe abgezogen. Das Pellet wurde zunächst in 30 µl HPLC-Wasser gelöst
dann in Spezial-Behälter für den ABI PRISM® 310 überführt und abschließend mit Gummi-
Septen verschlossen.
2.2.12.4 Vorbereitung der Sequenzierungsansätze für den ABI PRISM® 3700 Der ABI PRISM® 3700 DNA Analysator ist ein vollautomatisiertes Multikapillar-
Elektrophorese-Gerät, das die parallele Sequenzanalyse von 96 Proben in einigen Stunden
ermöglicht. Um die Sequenzreaktionen einzusetzen, wurden sie zunächst mittels einer
Sephadex-Multiscreen Filterplatte (Multiscreen: Millipore Billerica, USA; SephadexTM G-50
Fine: Amersham Biosciences, Braunschweig) aufgereinigt und mittels Zentrifugation in „96-
well optical Reaktionsplatten“ (Applied Biosystems, Darmstadt) überführt.
2.2.13 Hybridisierungstechnik 2.2.13.1 Southern-Transfer Um den Southern-Transfer (Southern, 1975) durchzuführen wurden die zu identifizierenden
DNA-Fragmente nach Restriktion (2.2.9.1) in einem Agarosegel neben einem
Längenstandard (2.2.7) gelelektrophoretisch aufgetrennt (2.2.7). Anschließend wurde das
Agarosegel mit einem Lineal als Orientierungshilfe photographiert, um später die
radioaktiven Signale distinkter DNA-Banden zuordnen zu können. Um den Transfer großer
Fragmente (>10 kb) zu erleichtern, erfolgte eine Depurinierung in 0,25 M HCl für 5 min. Das
Agarosegel inkubierte dann in die Denaturierungslösung für 30 min. Nachdem das Gel kurz

MATERIAL UND METHODEN
28
mit Aqua dest. gewaschen wurde, folgte eine weitere 30-minütige Inkubationsphase in der
Neutralisierungslösung. Abschließend äquilibrierte das Gel in dem SSC-Puffer (2x).
Für den Transfervorgang wurde das äquilibrierte Gel luftblasenfrei auf einen Stapel
Whatmann 3MM Filterpapier gelegt, welches sich in einer Schale mit SSC-Puffer (20x)
befand. Um den Kapillarstrom vom Gel auf die Nylonmembran zu sichern, sind die Gelränder
mit Folie abgedeckt worden. Anschließend wurde eine - passend zum Gel geschnittene -
positiv geladene Nylon-Membran (Roche, Mannheim) auf das Gel gelegt. Um den
essentiellen Kapillarsog zu erhöhen, sind auf der Nylon-Membran drei Lagen mit SSC-
Puffer (2x) angefeuchtetes 3MM-Papier, mehrere Lagen Zellstoff und ein Gewicht von ca.
1 kg gestellt worden. Der DNA-Transfer erfolgte über Nacht.
Nachfolgend wurde die Nylonmembran mit SSC-Puffer (2x) gewaschen, an der Luft
getrocknet und für 2 Stunden bei 80°C erhitzt. Aufgrund der hohen Bindungskapazität der
positiv geladenen Nylon Membran (Roche, Mannheim), wurde alternativ ein modifiziertes
alkalisches Verfahren für den Southern-Transfer verwendet (Chomczynski & Qasha, 1984;
Reed & Mann, 1985). Hierbei erfolgte der Transfer von Agarosegelen nach der
Depurinierung in 0,4 M NaOH. Zur Bindung der DNA an die Membran war eine UV-
Vernetzung für 3 min im Transilluminator erforderlich. Bei alkalisch behandelten Membranen
musste die Lagerung bei 4°C erfolgen.
2.2.13.2 Radioaktive DNA-Markierung Als Hybridisierungssonden wurden entweder PCR- oder genomische DNA-Fragmente
verwendet. Dabei erfolgte die Markierung der Sonden enzymatisch durch Einbau des
radioaktiven Isotops α-P32-dCTP mit dem “High Prime Kit” (Boehringer Ingelheim, Ingelheim
am Rhein). Hierzu wurden 50 bis 100 ng Sonde in 11 µl H2O für 10 min im Wasserbad
gekocht, 5 min auf Eis abgekühlt und mit 5 µl α-P32-dCTP und 4 µl Reaktionsmix gemischt.
Es folgte eine Inkubationsphase von 1 h bei 37°C. Abschließend wurde die radioaktiv
markierte DNA-Sonde über QIAquick-Säulchen (Qiagen, Hilden) nach Angaben des
Herstellers aufgereinigt.
2.2.13.3 Radioaktive Hybridisierung Die Southern-Transfer-Membranen (2.2.13.1) wurden zuerst mit SSC-Puffer (2x) benetzt und
anschließend für 1 h in Prähybridisierungspuffer (1ml/cm2) bei 68°C vorinkubiert. Zur
Hybridisierung wurde zunächst der Hybridisierungspuffer (1ml/cm2) mit 50-100 ng der frisch
denaturierten 32P-markierten DNA-Sonde gemischt und auf die Membran gegeben (Der
Prähybridisierungspuffer wurde entfernt). Die Inkubation erfolgte in einem
Hybridisierungsofen (Biometra, Göttingen) bei 68°C über Nacht. Nach Beendigung der
Hybridisierungsphase wurde der Hybridisierungspuffer abgegossen und die Filter zweimal je

MATERIAL UND METHODEN
29
15 min bei RT und anschließend dreimal je 15 min bei 68°C mit der Waschlösung 1
gewaschen. Die Membranen wurden anschließend in Folien eingeschweißt. Unter
Verwendung eines BioMAX-MR-Röntgenfilms (Kodak, Stuttgart) erfolgte die
Autoradiographie für 2-24 h (-70°C). Alternativ wurde als Hybridisierungslösung
ExpressHypTM (Clontech, Heidelberg) verwendet und nach Angaben des Herstellers
eingesetzt.
2.3 Etablierung der Mauslinien Die Mäuse wurden von der Firma Charles River (Sulzfeld) und der Firma Harlan (Horst,
Niederlande) bezogen und in der zentralen tierexperimentellen Einrichtung (ZTE) der
medizinischen Fakultät des Universitätsklinikums Münster in Standardkäfigen zu
Vierergruppen mit Wasser ad libitum und Standarddiät (Altromin 1324, Altromin, Lage)
gehalten. Sie waren einem 12 Stunden Tag-Nacht-Rhythmus (07.00 - 19.00 Uhr) und einer
Temperatur von 22 ± 2°C ausgesetzt. Alle Versuche sind gemäß dem deutschen
Tierschutzgesetz durchgeführt und mit der Nummer G10/98 genehmigt worden.
2.3.1 Mikroinjektion
Transgene Mäuse wurden mittels der Mikroinjektions-Methode nach Hogan et al. (1994)
erzeugt. Um zunächst eine ausreichende Menge an Plasmid-DNA zu erhalten, die für die
Mikroinjektion in Oocyten vorgesehen war, erfolgte eine Plasmid-Präparation (2.2.3.2). Die
FVB/N-Oocyten sind von FVB/N-Männchen befruchtet worden. Anschließend wurden in den
fertilisierten Oocyten mittels einer feinen Kanüle die Konstrukte eingeführt. Dabei kann eine
zufällige Integration des Transgens ins murine Genom erfolgen. Die Oocyten wurden
nachfolgend in pseudoschwangere CD1-Weibchen überführt. Durch die vorherige
Verpaarung der Weibchen mit vasektomierten Männchen wird die Ovulation durch den
mechanischen Reiz ausgelöst, worauf die Weibchen zur Aufnahme anderer fertilisierter
Oocyten wie auch zum Austragen der Embryonen bereit sind. Die Injektion der in dieser
Arbeit klonierten DNA-Konstrukte in fertilisierte FVB/N-Oocyten und die anschließende
Implantation der Oocyten in scheinschwangere CD1-Mäuse wurde freundlicherweise von
Herrn Dr. Dr. Boris Skryabin (Institut für Experimentelle Pathologie; ZMBE Münster)
durchgeführt.

MATERIAL UND METHODEN
30
2.3.2 Genotypisierung der Mauslinien Um zu überprüfen, ob die injizierten DNA-Konstrukte ins Genom der Embryonen erfolgreich
integriert und auch an deren Nachkommen weitergegeben wurden, erfolgte die
Genotypisierung über PCR- (2.2.11) und Southern-Transfer-Analysen (2.2.13.1). Es wurden
ebenfalls auch diejenigen Mäuse genotypisiert, die aus Kreuzungen zwischen transgenen
und Reportermauslinien resultierten. Der Genotyp wurde durch spezifische PCR-Formate mit
den zugehörigen Primern festgestellt, welche der Tabelle 2.7 zu entnehmen sind. Southern-
Transfer Analysen erfolgten bei der transgenen Mauslinie GGT-CRE mit der Sonde S1
(Abbildung 2.6).
Tabelle 2.7: Primer zur Genotypisierung transgener Mäuse
Transgene Mauslinie Primer Sequenz 5´- 3´
GGT-CRE Cre-3-F Cre-3-R
GGT TCG TTC ACT CAT GGA AAA GCA TGA TCT CCG GTA TTG AAA
5470-GGT-IN-F 5523-Cre-R2
GTG ACG TCA GCG TTG ACC ATC CTC C CGA TGA GTT GCT TCA AAA ATC CCT TCC
RTTA-GGT-CRE 5525-RTTA-F1 5528-RTTA-R2
CGG CGC TCT GGA ATT ACT CAA TGG CGC ACT TTA GCT GTT TCT CCA GGC CAC
ROSA26 5588-rosa26-F 5589-rosa26-R1 5590-rosa26-R2
AAA GTC GCT CTG AGT TGT TAT GCG AAG AGT TTG TCC TCA ACC GGA GCG GGA GAA ATG GAT ATG
Z/AP 5573-lacZ-F 5574-lacZ-R
CTG CGT TGG AGT GAC GGC AGT TAT C GTA GTG TGA CGC GAT CGG CAT AAC C
2.3.3 Dokumentierung der Mauslinien Um alle Mäuse zu registrieren, wurden erst die Männchen und dann die Weibchen eines
Wurfs durchnummeriert und ihre Identitätsnummer, Stamm/Hybridstamm, Geburtsdatum,
Geschlecht, Fellfarbe, Nummer der Eltern, Generation, Geschwisterzugehörigkeit und
genetische Sicherheitsstufe sowohl im Zuchtbuch als auch auf der Käfigkarte dokumentiert.
Alle erfassten Daten sind in Microsoft-Access Datenbanken gespeichert und katalogisiert
worden. Zur eindeutigen Identifizierung der Tiere wurden Ohrmarkierungen angebracht.
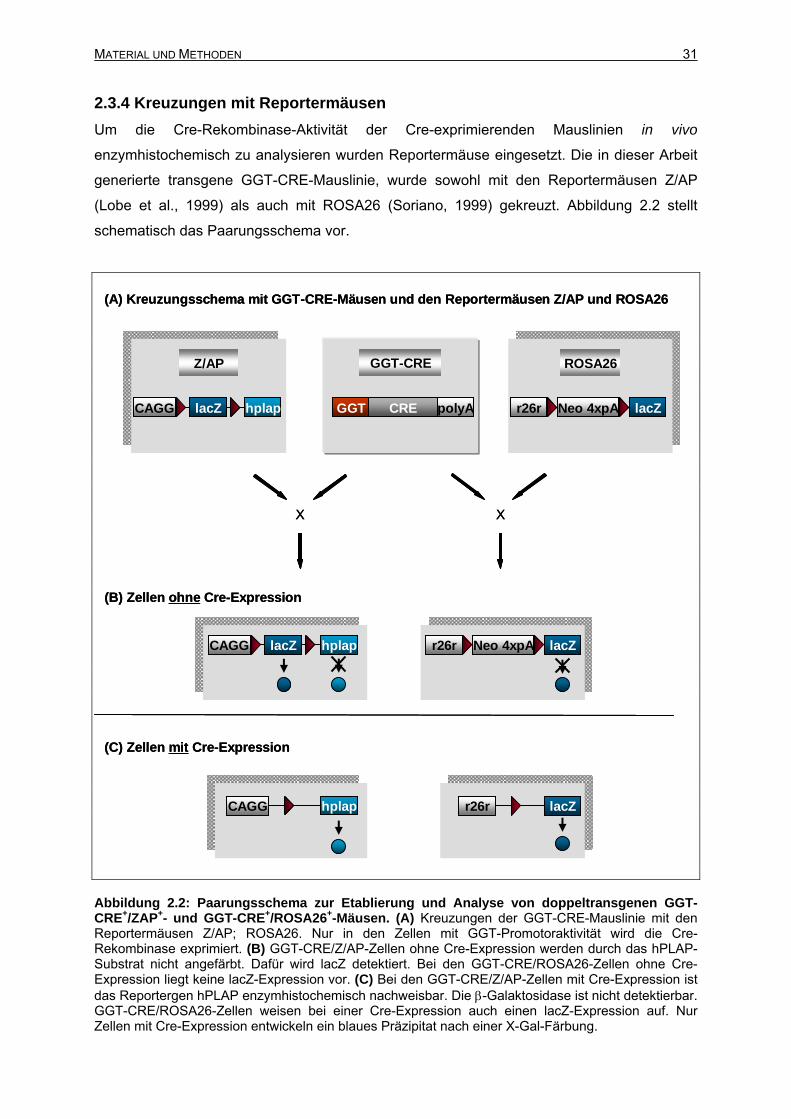
MATERIAL UND METHODEN
31
2.3.4 Kreuzungen mit Reportermäusen Um die Cre-Rekombinase-Aktivität der Cre-exprimierenden Mauslinien in vivo
enzymhistochemisch zu analysieren wurden Reportermäuse eingesetzt. Die in dieser Arbeit
generierte transgene GGT-CRE-Mauslinie, wurde sowohl mit den Reportermäusen Z/AP
(Lobe et al., 1999) als auch mit ROSA26 (Soriano, 1999) gekreuzt. Abbildung 2.2 stellt
schematisch das Paarungsschema vor.
Abbildung 2.2: Paarungsschema zur Etablierung und Analyse von doppeltransgenen GGT-CRE+/ZAP+- und GGT-CRE+/ROSA26+-Mäusen. (A) Kreuzungen der GGT-CRE-Mauslinie mit den Reportermäusen Z/AP; ROSA26. Nur in den Zellen mit GGT-Promotoraktivität wird die Cre-Rekombinase exprimiert. (B) GGT-CRE/Z/AP-Zellen ohne Cre-Expression werden durch das hPLAP-Substrat nicht angefärbt. Dafür wird lacZ detektiert. Bei den GGT-CRE/ROSA26-Zellen ohne Cre-Expression liegt keine lacZ-Expression vor. (C) Bei den GGT-CRE/Z/AP-Zellen mit Cre-Expression ist das Reportergen hPLAP enzymhistochemisch nachweisbar. Die β-Galaktosidase ist nicht detektierbar. GGT-CRE/ROSA26-Zellen weisen bei einer Cre-Expression auch einen lacZ-Expression auf. Nur Zellen mit Cre-Expression entwickeln ein blaues Präzipitat nach einer X-Gal-Färbung.
CAGG hplap GGT CRE polyA
GGT-CRE
r26r Neo 4xpA lacZ
(B) Zellen ohne Cre-Expression
lacZ hplap
X
(C) Zellen mit Cre-Expression
X
lacZ
hplap r26r lacZ
r26r Neo 4xpA lacZ
Z/AP ROSA26
CAGG
CAGG
(A) Kreuzungsschema mit GGT-CRE-Mäusen und den Reportermäusen Z/AP und ROSA26
CAGG hplap GGT CRE polyA
GGT-CRE
r26r Neo 4xpA lacZr26r Neo 4xpA lacZ
(B) Zellen ohne Cre-Expression
lacZ hplap
X
(C) Zellen mit Cre-Expression
X
lacZ
hplap r26r lacZ
r26r Neo 4xpA lacZ
Z/AP ROSA26
CAGG
CAGG
(A) Kreuzungsschema mit GGT-CRE-Mäusen und den Reportermäusen Z/AP und ROSA26

MATERIAL UND METHODEN
32
2.3.4.1 Kreuzungen mit der Z/AP-Reportermauslinie Die Z/AP-Reportermaus Tg(ACTB-Bgeo/ALPP)1Lbe (Lobe et al., 1999) exprimiert konstitutiv
das Reportergen lacZ unter der Kontrolle des CAGG-Promotors (Niwa et al., 1991). Sowohl
embryonal als auch adult liegt eine ubiquitäre lacZ-Expression vor. Ausnahmen sind
Erythrozyten, Chondrocyten und Adipozyten. Der CAGG-Promotor besteht aus dem CMV-
Enhancer und dem Hühnchen-β-Aktin-Promotor. Dem Promotorbereich schließen sich zwei
Reportergene an: lacZ und humane plazentale alkalische Phosphatase (hPLAP, DePrimo et
al., 1996; Millan, 1986). Die β-Galaktosidaseaktivität wird durch eine βgeo-Kassette vermittelt
(Friedrich & Soriano, 1991). Es handelt sich hierbei um die Fusion einer lacZ-Sequenz mit
einer Neomycinkassette. Das Reportergen lacZ wird von loxP-Stellen flankiert. Stromabwärts
befinden sich drei Kopien des SV40-polyA Signals (Zinyk et al., 1998), sowie das zweite
Reportergen hPLAP (Abbildung 2.3 A). Diese besondere Konstellation hat zur Folge, dass
die β-Galaktosidase ubiquitär exprimiert; dafür kann hPLAP aufgrund der vorgeschaltenen
SV40-polyA Signale nicht translatiert werden. Erst nach Kreuzung der Z/AP-Mauslinie mit
einer Cre-exprimierenden Maus kann die gefloxte lacZ-Kasette durch das Cre-Enzym
entfernt und folgerichtig hPLAP ungehindert transkribiert werden (Abbildung 2.3 B). Somit
wird nur in den Cre-aktiven Zellen die lacZ Expression durch die alkalische Phosphatase
Expression ersetzt. Durch die Detektion der alkalischen Phosphatase (2.4.2.2) konnten bei
den doppeltransgenen GGT-CRE/Z/AP-Mäusen, Rückschlüsse auf den Expressionsort der
Cre-Rekombinase und ihrer katalytische Aktivität gezogen werden.
Abbildung 2.3: Schematische Darstellung und Cre/loxP-Rekombination des Z/AP-Konstrukts. (A) Der Promotor besteht aus dem CMV-Verstärker und dem Huhn-Aktin-Promotor. Die „gefloxte“ (dargestellt durch schwarze Dreiecke) β−geo-Sequenz kodiert für ein Fusionsprotein aus β-Galaktosidase (lacZ) und Neomycin-Phosphotransferase (Friedrich & Soriano, 1991). Nachgeschaltet sind drei Kopien des SV40-polyA Signals (3xpA). Beim zweiten Reportergen handelt es sich um das humane plazentale alkalische Phosphatase Gen (hPLAP). (B) Die Cre-Rekombinase hat den gefloxten DNA-Bereich deletiert, was die Transkription von hPLAP zur Folge hat. Zeichnung modifiziert nach Lobe et al., 1999.
CMV enhancer
pA´ βgeo hPLAP 3xpAloxP loxP
Huhn-βActin Promotor
Zielbereich der Cre-Rekombinase
+ CRE
hPLAP (B)
(A)
pA´

MATERIAL UND METHODEN
33
2.3.4.2 Kreuzungen mit ROSA26-Reportermäusen Bei der in dieser Arbeit eingesetzten ROSA26-Maus handelt es sich um die lacZ-
Reportermauslinie B6, 129S-Gtrosa26tm1Sor (Friedrich & Soriano, 1991; Zambrowicz, et al.,
1997; Soriano, 1999). Ihr Transgen besteht aus einer Spleiß-Akzeptor-Sequenz (SA) gefolgt
von einer gefloxten Neomycinkassette mit einer vierfachen polyA-Sequenz und dem
Reportergen lacZ am 3´-Ende (Maxwell et al., 1989). Die vierfache polyA-Sequenz führt zu
einem Transkriptiosstillstand der RNA-Polymerase. Demzufolge kann das stromabwärts
gelegene lacZ-Reportergen nicht transkribiert werden. (Soriano, 1999). Zur besseren
Übersicht ist das ROSA26-Konstrukt in Abbildung 2.4 A schematisch wiedergegeben. Erst
durch die enzymatische Aktivität der Cre-Rekombinase wird der gefloxte DNA-Bereich
entfernt. Demzufolge kann die stromabwärts liegende lacZ-Kassette transkribiert und die β-
Galaktosidase exprimiert werden (Abbildung 2.4 B). Dieser Arbeit standen sowohl
heterozygote als auch homozygote ROSA26-Mauslinien zur Verfügung. Nach Verkreuzung
von GGT-CRE-Mäusen mit ROSA26-Reportermäusen erfolgte zunächst eine
Genotypisierung um den transgenen Status zu ermitteln (2.3.2). Eine Charakterisierung der
in vivo Cre-Expression erfolgte mittels einer X-Gal-Färbung (2.4.2.1) von Gefrierschnitten
doppeltransgener GGT-CRE+/ROSA26+-Mäusen. Als Kontrolle dienten GGT-CRE-
/ROSA26+-Mäuse. Nur in diesem Gewebe wo das Cre-Protein exprimiert wurde und
katalytisch aktiv war, konnte die β-Galaktosidase nachgewiesen werden.
Abbildung 2.4: Schematische Darstellung und Cre/loxP-Rekombination des ROSA26-Konstrukts. (A) Der Spleiß-Akzeptor-Sequenz (SA) folgt eine gefloxte Neomycin Kassette mit einer vierfachen polyA-Sequenz (PGK neo 4xpA). Dem schließt sich eine lacZ-Kasette und eine dreifache polyA-Sequenz (bPA) an. (modifiziert nach Soriano, 1999). (B) Nach Anbindung einer enzymatisch aktiven Cre-Rekombinase entfernt Cre die „gefloxte“ PGK neo 4xpA Sequenz. Demzufolge kann lacZ transkribiert werden. Die loxP-Stellen sind als schwarze Pfeile dargestellt.
SA PGK neo 4xpA lacZ bPA
SA lacZ bPA
+ CRE
(B)
(A)

MATERIAL UND METHODEN
34
2.4 Histologische Techniken
2.4.1 Vorbereitung der Gefrierschnitte 2.4.1.1 Entnahme des Gewebes Den Versuchstieren entnommenes Gewebe wurde in PBS-Puffer pH 7,4 (1mM MgCl2)
gewaschen und anschließend in -20°C flüssiges Isopentan überführt. Durch diese
Gefrierungstechnik wird eine spätere Herstellung von Gefrierschnitten am Mikrotom
ermöglicht. Die Lagerung erfolgte bei -20°C. Bei adulten Tieren wurden folgende Organe
entnommen: Gehirn, Leber, Nieren, Milz, Lunge, Herz, Pankreas, Haut, Muskelgewebe,
Ovar/Testis.
2.4.1.2 Entnahme der Embryonen Um den frühesten Expressionszeitpunkt und den Expressionsprofil der Cre-Rekombinase
festzustellen, wurden transgene murine Embryonen in verschiedenen embryonalen
Entwicklungsstufen angefärbt und analysiert. Dafür wurden Mäuse an einen definierten
Zeitpunkt verpaart. Der Kopulationsplug zeigt den Anfang der Schwangerschaft an und wird
als Tag 0,5 der Embryogenese festgelegt. Die Embryonen wurden nach Entnahme
narkotisiert und in -20°C flüssiges Isopentan überführt. Die Lagerung erfolgte bei -20°C
2.4.1.3 Anfertigungen der Gefrierschnitte Die Präparate sind mit dem Gefriermedium GSV1 (Slee Technik, Mainz) als
Haftungsvermittler auf Objektgläser mit entsprechenden Haftrillen aufgefroren worden.
Anschließend wurde das vorbereitete Gewebe bzw. die Embryonen mit dem Mikrotom-
Kryostat HM 500 OM geschnitten. Die Schnitte (6-15 µm) wurden auf Superfrost-
Objektträger (R. Langenbrinck, Emmendingen) übertragen.
2.4.2 Enzymhistochemische Färbungen 2.4.2.1 Detektion der β-Galaktosidase
Das E. coli lacZ-Gen - ein klassisches Reportergen - kodiert für die β-Galaktosidase
(Beckwith, 1980). Bei der β-Galaktosidase handelt es sich um das bakterielle Äquivalent zur
Lactase. Dieses Enzym spaltet die β-1,4-glykosidische Bindung der Laktose. Das meist
verwendete Substrat zur Detektion der β-Galaktosidase ist ein synthetisches
Substratanalogon, ein Indol-Derivat 5-bromo-4-chloro-3-indolyl-b-D-galactosid (X-Gal, Holt &
Sadler, 1958). Dabei wird X-Gal in β-Galaktose und ein blaues Chromophor (Indigofarbstoff)
hydrolysiert. Die β-Galaktosidase wurde auf Gefrierschnitten nach Matsusaka et al. (1999)
detektiert. In einer 2%igen Glutaraldehyd-Fixierungslösung wurden die Gefrierschnitte
(2.4.1.3) für 5 min auf Eis inkubiert. Nach dreimaligem Waschen mit PBS-Puffer für je 10 min

MATERIAL UND METHODEN
35
(1 mM MgCl2) wurden die Gefrierschnitte mit der X-Gal-Färbelösung für maximal 10 Stunden
bei 30°C inkubiert. Abschießend wurde mit PBS-Puffer gewaschen. Die endogene β-
Galaktosidase lässt sich, anders als bei der endogenen alkalischen Phosphatase, wo eine
Hitzeeinwirkung (20 min.) erforderlich ist, durch eine entsprechend alkalischen pH-Wert (8)
und reduzierter Inkubations-Temperatur (30°C) inaktivieren.
2.4.2.2 Detektion der alkalischen Phosphatase Das Gen hPLAP (DePrimo et al., 1996; Millan, 1986) kodiert für ein hitzestabiles Protein,
was die Unterscheidung zu der thermolabilen endogenen murinen Phosphatase ermöglicht.
Die Gefrierschnitte wurden eingangs in 0,2% Glutaraldehyd (in PBS-Puffer pH 7,4 + 1mM
MgCl2) fixiert. Es folgten drei Waschschritte (je 5 min in PBS-Puffer pH 7,4 + 1mM MgCl2)
mit anschließender Hitzeinaktivierung der endogenen alkalischen Phosphatase (20 min;
70°C). Anschließend inkubierten die Präparate 10 min in AP-Puffer. Die Darstellung der
alkalischen Phosphatase (AP) erfolgte mittels einem chromogenen Substrat für alkalische
Phosphatase (BM Purple AP Substrat; Roche, Mannheim). Ein blaues Präzipitat zeigt AP-
Aktivität an.
2.4.3 Histologische Übersichtsfärbung Eine Übersichtsfärbung der Schnitte erfolgte mit Eosin Y. Dabei handelt es sich um einen
schwach sauren Farbstoff der Fluorescein-Gruppe, welcher acidophile Cytoplasmaproteine,
Kollagenfasern und Bindegewebe rot anfärbt. Die Präparate wurden zunächst in einer
0,1%igen Eosin-Lösung für 15 min inkubiert und anschließend mit Aqua dest. für 2 min
gespült. Die Differenzierung erfolgte für 2 min in 80%iges Ethanol, wo sich eine Alkoholreihe
anschloss: 2 min 90% Ethanol, 2 min 100% Ethanol. Abschließend wurden die Schnitte
zweimal je 2 min in Xylol inkubiert und mit Eukitt eingedeckelt.
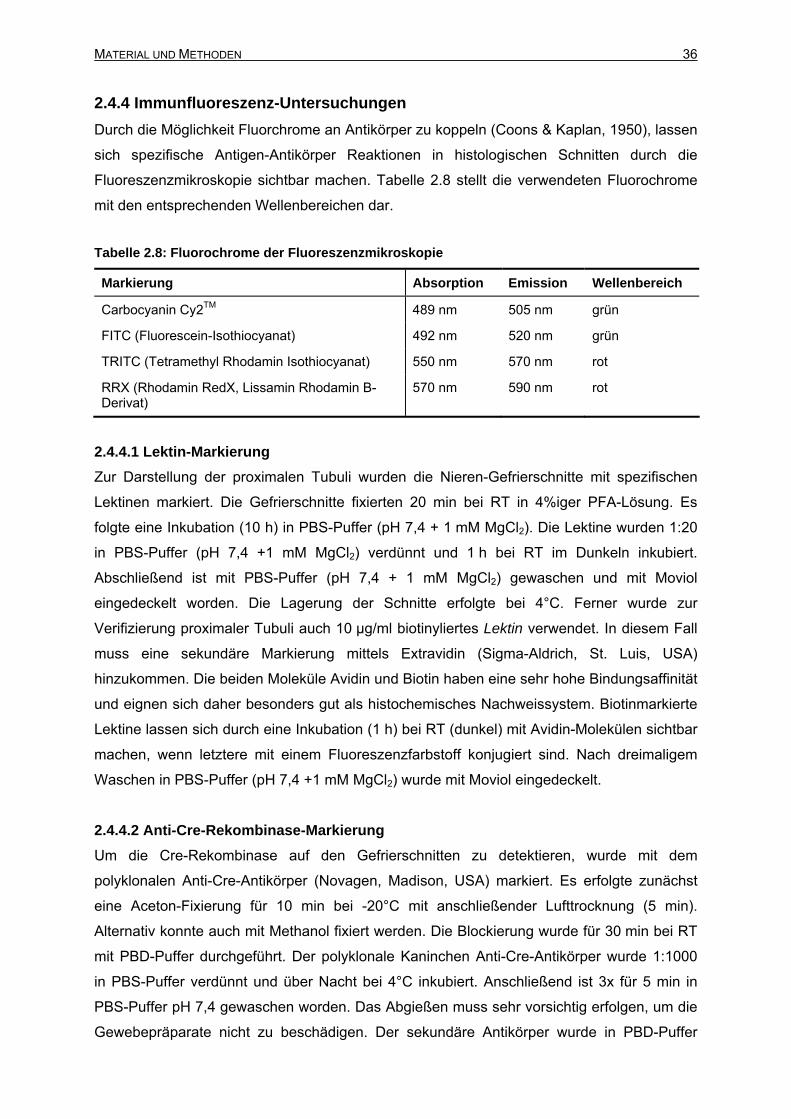
MATERIAL UND METHODEN
36
2.4.4 Immunfluoreszenz-Untersuchungen Durch die Möglichkeit Fluorchrome an Antikörper zu koppeln (Coons & Kaplan, 1950), lassen
sich spezifische Antigen-Antikörper Reaktionen in histologischen Schnitten durch die
Fluoreszenzmikroskopie sichtbar machen. Tabelle 2.8 stellt die verwendeten Fluorochrome
mit den entsprechenden Wellenbereichen dar.
Tabelle 2.8: Fluorochrome der Fluoreszenzmikroskopie
Markierung Absorption Emission Wellenbereich
Carbocyanin Cy2TM 489 nm 505 nm grün
FITC (Fluorescein-Isothiocyanat) 492 nm 520 nm grün
TRITC (Tetramethyl Rhodamin Isothiocyanat) 550 nm 570 nm rot
RRX (Rhodamin RedX, Lissamin Rhodamin B-Derivat)
570 nm 590 nm rot
2.4.4.1 Lektin-Markierung Zur Darstellung der proximalen Tubuli wurden die Nieren-Gefrierschnitte mit spezifischen
Lektinen markiert. Die Gefrierschnitte fixierten 20 min bei RT in 4%iger PFA-Lösung. Es
folgte eine Inkubation (10 h) in PBS-Puffer (pH 7,4 + 1 mM MgCl2). Die Lektine wurden 1:20
in PBS-Puffer (pH 7,4 +1 mM MgCl2) verdünnt und 1 h bei RT im Dunkeln inkubiert.
Abschließend ist mit PBS-Puffer (pH 7,4 + 1 mM MgCl2) gewaschen und mit Moviol
eingedeckelt worden. Die Lagerung der Schnitte erfolgte bei 4°C. Ferner wurde zur
Verifizierung proximaler Tubuli auch 10 µg/ml biotinyliertes Lektin verwendet. In diesem Fall
muss eine sekundäre Markierung mittels Extravidin (Sigma-Aldrich, St. Luis, USA)
hinzukommen. Die beiden Moleküle Avidin und Biotin haben eine sehr hohe Bindungsaffinität
und eignen sich daher besonders gut als histochemisches Nachweissystem. Biotinmarkierte
Lektine lassen sich durch eine Inkubation (1 h) bei RT (dunkel) mit Avidin-Molekülen sichtbar
machen, wenn letztere mit einem Fluoreszenzfarbstoff konjugiert sind. Nach dreimaligem
Waschen in PBS-Puffer (pH 7,4 +1 mM MgCl2) wurde mit Moviol eingedeckelt.
2.4.4.2 Anti-Cre-Rekombinase-Markierung Um die Cre-Rekombinase auf den Gefrierschnitten zu detektieren, wurde mit dem
polyklonalen Anti-Cre-Antikörper (Novagen, Madison, USA) markiert. Es erfolgte zunächst
eine Aceton-Fixierung für 10 min bei -20°C mit anschließender Lufttrocknung (5 min).
Alternativ konnte auch mit Methanol fixiert werden. Die Blockierung wurde für 30 min bei RT
mit PBD-Puffer durchgeführt. Der polyklonale Kaninchen Anti-Cre-Antikörper wurde 1:1000
in PBS-Puffer verdünnt und über Nacht bei 4°C inkubiert. Anschließend ist 3x für 5 min in
PBS-Puffer pH 7,4 gewaschen worden. Das Abgießen muss sehr vorsichtig erfolgen, um die
Gewebepräparate nicht zu beschädigen. Der sekundäre Antikörper wurde in PBD-Puffer

MATERIAL UND METHODEN
37
verdünnt (1:100) und für 1 h im Dunkeln auf das Gefrierpräparat aufgetragen. Es folgten 3
Waschschritte mit PBS-Puffer pH 7,4 je 5 min. Abschließend wurden die markierten
Gefrierschnitte mit Moviol eingedeckelt und bei -20°C gelagert.
2.4.5 Auswertung der histochemischen Präparate Für die Auswertung der Präparate wurde ein „ZEISS Axioskop 2 plus“-Mikroskop (Carl Zeiss,
Jena) mit dem Filtersatz 23 (DBP485/20 und DBP 546/12) für kombinierte Visualisierung der
Fluoreszenz-Farbstoffe verwendet. Zum besseren Verständnis der Nierenanatomie wird in
Abbildung 2.5 A ein Sagittalschnitt der Niere dargestellt. Die Niere gliedert sich in drei
Abschnitte auf: Nierenbecken (Pelvis renalis), Nierenmark (Medulla renalis) und Nierenrinde
(Cortex renalis). Von der Nierenrinde reichen kegelförmige Markpyramiden (Pyramides
renales) durch die Nierenmarksubstanz bis hin zum Nierenbecken. Die Nierenrinde liegt
unter der gesamten Oberfläche, dringt aber in den Bertin´schen Säulen (Columnae renales)
zwischen den Markpyramiden (Pyramides renales) bis an die Nierenbucht (Sinus renalis)
vor. Die Nieren bestehen aus etwa einer Million Nephronen. Nephrone sind strukturelle und
funktionelle Einheiten der Niere und bestehen aus dem Nierenkörperchen (Glomerulus) und
den Nierenkanälchen (renale Tubuli), welche im Sammelrohr münden. Die renalen Tubuli
sind dünne Kanälchen mit einem einschichtigen Epithel; hier erfolgt die Rückresorption aber
auch die Sekretion von bestimmten Stoffen. Der epitheliale Tubulusapparat besteht aus
einem gewundenem Teil, dem proximalen Tubulus, einem geraden Abschnitt der in das
Nierenmark absteigt (Pars recta), einer haarnadelförmigen Schleife (Henlesche Schleife),
einem gerade aufsteigendem Abschnitt und einem zweiten gewundenen Teil - dem distalen
Tubulus - der in der Nähe des zugehörigen Nierenkörperchens liegt (Abbildung 2.5 B).

MATERIAL UND METHODEN
38
(A)
(B)
Abbildung 2.5: Niere/Nephron (A): Schematische Darstellung einer Niere (Sagittalschnitt). (B): Illustrierung eines Nephrons. Die Pfeile zeigen die Richtung des Flüssigkeitsstroms an.
Nierenpyramiden (Pyramides renales)
Nierenbecken (Pelvis renalis)
Harnleiter (Ureter)
Nierenkapsel (Capsula fibrosa)
Nierenrinde (Cortex renalis)
Nierenkelch (Calix renalis)
Nierenpapillen (Papillae renales)
Bertin´schen Säulen (Columnae renales)
Unterer Nierenpol (Extremitas inferior)
Oberer Nierenpol (Extremitas superior)
Nierenmark (Medulla renalis)
pT dT Hs Sr tW Bo Gl zA aA
proximaler Tubulus distaler Tubulus Henlesche Schleife Sammelrohr Tubuluswand Bowmansche Kapsel Glomerulum zuführende Arterie abführende Arterie
pT dT
Hs
Sr
Bo
Gl
zA aA
tW

MATERIAL UND METHODEN
39
2.5 Sicherheitsbestimmungen Alle gentechnischen Arbeiten wurden gemäß dem Gentechnikgesetz vom 22. März 2004
durchgeführt. Die Anmeldung der Arbeiten erfolgte gemäß § 8 Abs. 1 und § 11 des
Gentechnikgesetzes. Die Arbeiten wurden gemäß der Sicherheitsstufen und
Sicherheitsmaßnahmen (§ 7 Abs. 1 Gentechnikgesetz) durchgeführt.
2.6 Verwendete Materialien Alle benutzten Chemikalien wurden im analytischen Reinheitsgrad von folgenden Firmen
bezogen: Amersham Buchler (Freiburg, D), Biozym (Hess. Oldendorf, D), Fluka AG (Buchs,
CH), Hartmann Analytics (Braunschweig, D), Invitrogen (Karlsruhe), MERCK (Darmstadt, D),
Pharmacia LKB (Freiburg, D), SERVA Feinbiochemica (Heidelberg, D) und Sigma-Aldrich
(St. Louis, USA).
2.6.1 Plasmide Tabelle 2.9: Plasmide. Die zugehörigen Plasmidkarten sind im Anhang dargestellt.
Name Große (kb) Herkunft
pSL300/PgkNeoL 4,9 Dr. Dr. B. Skryabin, ZMBE, Münster
pHD2-Cre1 4,5 Dr. C. Kellendonk, DKFZ, Heidelberg
pGGT-PFU-1 4,5 diese Arbeit
pGGT-CRE 7,2 diese Arbeit
pUHDrtTA2S-M2 4,2 Prof. Dr. W. Paulus, Institut für Neuropathologie, Münster
pGGT-RTTA 3,8 diese Arbeit
pBI-3 7,7 BD Biosciences Clontech, Heidelberg
pBI-3-H3 7,7 diese Arbeit
pBI-CRE-lacZ 9,7 diese Arbeit
2.6.2 Sonde
Abbildung 2.6: Sonde S1. BglII/EcoRI-DNA-Fragment (1129 bp) aus dem Vektor pHD2. Die schwarze Box stellt die NLS-Sequenz dar; der graue Pfeil den zweiten offenen Leserahmen der P1-Cre-Rekombinase. Die DNA-Sequenz ist dem Anhang zu entnehmen.
NLS-Cre-Rekombinase
BglII-1 EcoRI-1129 bp
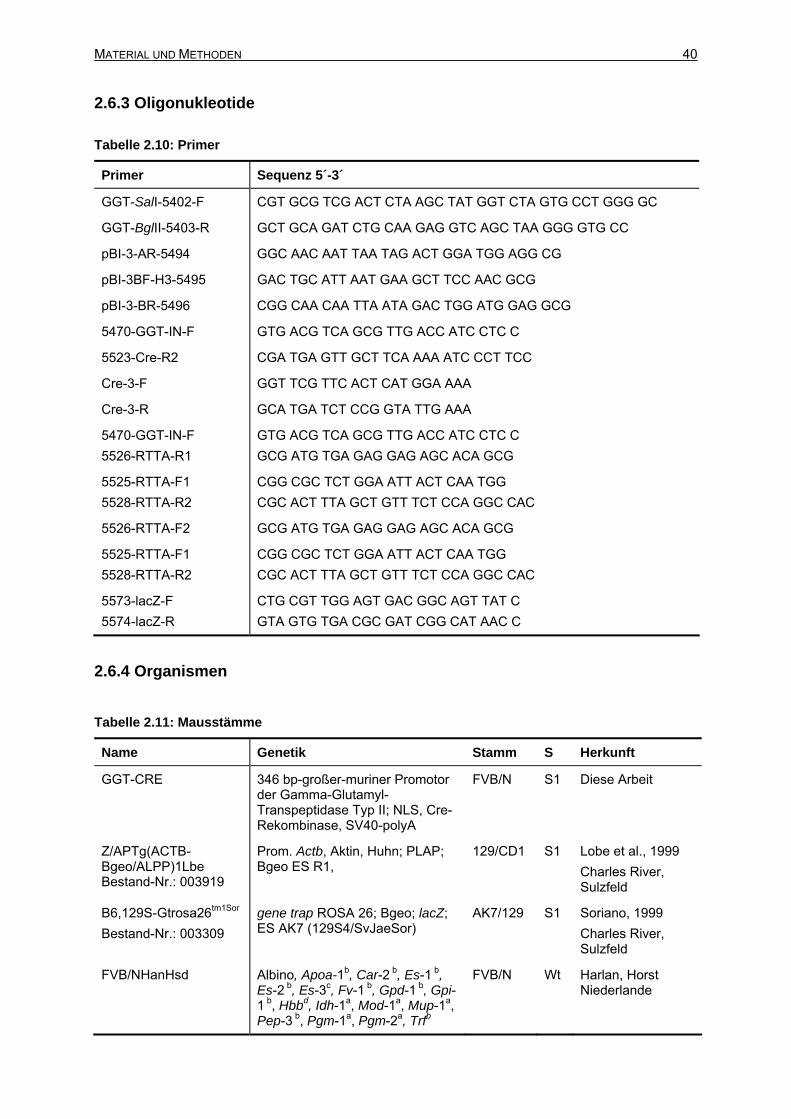
MATERIAL UND METHODEN
40
2.6.3 Oligonukleotide Tabelle 2.10: Primer
Primer Sequenz 5´-3´
GGT-SalI-5402-F CGT GCG TCG ACT CTA AGC TAT GGT CTA GTG CCT GGG GC
GGT-BglII-5403-R GCT GCA GAT CTG CAA GAG GTC AGC TAA GGG GTG CC
pBI-3-AR-5494 GGC AAC AAT TAA TAG ACT GGA TGG AGG CG
pBI-3BF-H3-5495 GAC TGC ATT AAT GAA GCT TCC AAC GCG
pBI-3-BR-5496 CGG CAA CAA TTA ATA GAC TGG ATG GAG GCG
5470-GGT-IN-F GTG ACG TCA GCG TTG ACC ATC CTC C
5523-Cre-R2 CGA TGA GTT GCT TCA AAA ATC CCT TCC
Cre-3-F GGT TCG TTC ACT CAT GGA AAA
Cre-3-R GCA TGA TCT CCG GTA TTG AAA
5470-GGT-IN-F 5526-RTTA-R1
GTG ACG TCA GCG TTG ACC ATC CTC C GCG ATG TGA GAG GAG AGC ACA GCG
5525-RTTA-F1 5528-RTTA-R2
CGG CGC TCT GGA ATT ACT CAA TGG CGC ACT TTA GCT GTT TCT CCA GGC CAC
5526-RTTA-F2 GCG ATG TGA GAG GAG AGC ACA GCG
5525-RTTA-F1 5528-RTTA-R2
CGG CGC TCT GGA ATT ACT CAA TGG CGC ACT TTA GCT GTT TCT CCA GGC CAC
5573-lacZ-F 5574-lacZ-R
CTG CGT TGG AGT GAC GGC AGT TAT C GTA GTG TGA CGC GAT CGG CAT AAC C
2.6.4 Organismen Tabelle 2.11: Mausstämme
Name Genetik Stamm S Herkunft
GGT-CRE 346 bp-großer-muriner Promotor der Gamma-Glutamyl-Transpeptidase Typ II; NLS, Cre-Rekombinase, SV40-polyA
FVB/N S1 Diese Arbeit
Z/APTg(ACTB-Bgeo/ALPP)1Lbe Bestand-Nr.: 003919
Prom. Actb, Aktin, Huhn; PLAP; Bgeo ES R1,
129/CD1 S1 Lobe et al., 1999 Charles River, Sulzfeld
B6,129S-Gtrosa26tm1Sor Bestand-Nr.: 003309
gene trap ROSA 26; Bgeo; lacZ; ES AK7 (129S4/SvJaeSor)
AK7/129 S1 Soriano, 1999 Charles River, Sulzfeld
FVB/NHanHsd Albino, Apoa-1b, Car-2 b, Es-1 b, Es-2 b, Es-3c, Fv-1 b, Gpd-1 b, Gpi-1 b, Hbbd, Idh-1a, Mod-1a, Mup-1a, Pep-3 b, Pgm-1a, Pgm-2a, Trfb
FVB/N Wt Harlan, Horst Niederlande

MATERIAL UND METHODEN
41
Tabelle 2.12: E. coli-Bakterienstamm
Stamm Genotyp Referenz
TOP10 F-, mcrA, Δ(mrr-hsdRMS-mcrBC), Δ80 lacZ Δ M15, ΔlacX74, deoR, recA1, araD139, Δ(ara-leu)7697, galU, galK, rpsL, endA1, nupG
Invitrogen, Karlsruhe
2.6.5 Antikörper Tabelle 2.13: Antikörper
Antikörper Markierung Firma
Polyklonaler Anti-Cre-Antikörper
- Novagen, Madison, USA
Donkey Anti-Rabbit IgG (H+L) sekundärer Antikörper
RRX Jackson ImmunoResearch, West Grove, USA
Goat Anti-Rabbit IgH (H+L) sekundärer Antikörper
CyTM2 Jackson ImmunoResearch West Grove, USA
Lektin-PHA-E (Phaseolus vulgaris-Erythroagglutinin)
TRITC Sigma-Aldrich, St. Luis, USA
Lektin-PHA-E (Phaseolus vulgaris-Erythroagglutinin)
Biotin Vector Laboratories, Burlingame, USA
Extravidin FITC Sigma-Aldrich, St. Luis, USA

MATERIAL UND METHODEN
42
2.6.6 Puffer
Ampicillin Stammlösung 100 mg/ml in Aqua dest. AP-Puffer 10 mM MgCl2 100 mM Tris-HCl pH 9,5 100 mM NaCl 0,01% Natrium-Deoxycholat 0,02% NP-40 In PBS-Puffer
Betain Stammlösung 5 M Betain in Aqua dest. Lagern bei -20°C
Denaturierungslösung, 0,5 M NaOH 1,5 M NaCl
dNTPs 5 mM dATP 100 mM 5 mM dCTP 100 mM 5 mM dGTP 100 mM 5 mM dTTP 100 mM In Aqua dest. Eosin-Lösung 0,1% Aqua dest.
Ethidiumbromid-Lösung
1 µl 1 ml
Ethidiumbromid-Stammlösung Aqua dest
Ethidiumbromid-Stammlösung 5 mg Ethidiumbromid/ml Aqua dest
Gelladepuffer 6 g Saccharose (60% Saccharose (w/v)) 0,3% Bromphenol-Blau 0,3% Xylen-Cyanol ad 10 ml Aqua dest. Glutaraldehyd-Fixierungslösung 2% Glutaraldehyde (50%) 0,01% Natrium-Deoxycholat 0,02% IGAEPAL-CA630 1 mM MgCl2 in PBS-Puffer pH 8.0

MATERIAL UND METHODEN
43
Hybridisierungspuffer 2-6x SSC (20x) 0,1-0,5% SDS (20%) 200 ng/µl Herings-Sperma-DNA 5x Denhardts (50x) IPTG-Stammlösung 100 mM IPTG in Aqua dest.
LB -Medium (LB: Low salt) 1,0% Trypton 0,5% Hefe-Extrakt
0,5% NaCl pH 7,0 Lysispuffer 10 mM Tris, pH 8 10 mM EDTA 10 mM NaCl 0.6% SDS Mowiol 2,4 g Mowiol 4-88 (Hoechst, Frankfurt/Main) 6 g Glycerol, bei 50°C ca. 1 h lösen 12 ml 200 mM Tris-HCl Bei 50°C ca. 3 h lösen 5 min/2500 rpm zentrifugieren Überstand abnehmen
Neutralisierungslösung 0.5 M Tris Southern-Transfer 1.5 M NaCl pH 7 mit HCl konz. Paraformaldehyd (PFA)-Lösung 4% (w/v) PFA 1x PBS-Puffer 1 mM MgCl2 pH 7,4
PBD-Puffer 1x PBS-Puffer 10% Eselserum pH 7,4 PBS-Puffer (10x) 1,37 M NaCl 0,027M KCl 0,080M Na2PO4 0,020M Na2PO4

MATERIAL UND METHODEN
44
PCR-Puffer (10x) 500 mM KCl 100 mM Tris 15,0 mM MgCl2 0,1% Gelatine 1,0% Triton X
Prähybridisierungspuffer 2-6x SSC (20x) 0,1-0,5% SDS (20%) 200 ng/µl Herings-Sperma-DNA 5x Denhardts (50x) Aqua dest. SDS-Puffer 20% 20 g Natriumdodecylsulfat (SDS) ad 100 ml Aqua dest.
SOC-Medium 2,0% Trypton 0,5% Hefe-Extrakt 10 mM NaCl 2,5 mM KCl 10 mM MgCl2 x 6H2O 20 mM Glucose
SSC (20x) 3 M NaCl 0,3 M Natriumcitrat pH 7 mit HCl
TBE-Puffer (10x) 1 M Tris-Borat 20 mM EDTA pH 8,3 TEP-Puffer (10x) 218 g Tris 20,07 g NaH2PO4-H2O 18,6 g EDTA
TE-Puffer 10 mM Tris 1 mM EDTA pH 7,6 TENS-Puffer 1 10 ml TE-Puffer (1x) 0,1 M NaOH 0,5% w/v SDS

MATERIAL UND METHODEN
45
TENS-Puffer 2 3 M Kaliumacetat pH 4,8 Waschlösung 1 2x SSC 0,1% SDS; RT
Waschlösung 2 0,1x SSC 0,5% SDS; 60-68°C
X-Gal-Stammlösung 40 mg/ml X-Gal in DMF lösen lagern bei -20°C X-Gal-Färbelösung 0,01% Natrium-Deoxycholat 0,02% 0,02% IGAEPAL-CA630 1mM MgCl2 2mM K3Fe(CN)6 2mM K4Fe(CN) 6 1mg/ml X-Gal-Stammlösung in PBS-Puffer pH 8.0

MATERIAL UND METHODEN
46
2.7 Geräte
DNA Thermal Cycler Biometra T3-Thermocycler Biometra Filmentwickler Kodak, RPX-OMat Prozessor Gelelektrophorese-Kammer Pharmacia LKB GNA 100
Pharmacia LKB GNA 200 Gelphotographie Polaroid MP-4 Land Camera Heizblöcke Eppendorf Thermostat 5320
Stuart Test Tube Heater Inkubator, Schüttler Edmund Buhler, Laborgerätebau, Glastechnik,
Umwelttechnik, SM 5 Kamera RICO XR/X 3000D, SLR Kamera Kaltlicht-Leuchte Leica Kühlzentrifugen Sorvall® RC-5B Refrigerated Superspeed Centrifuge
Rotoren: Sorvall SS-34, Sorvall SH-MT Mikroskop Axioskop, Zeiss Mikrotom Mikrotom-Kryostat HM 500 OM, MICROM Netzgeräte Pharmacia GPS 200/400
Pharmacia EPS 3500 pH-Meter Knick, ph-mV-Meter Photometer Genequant, Pharmacia Schüttler New Brunswick Scientific, Edison, N.J., U.S.A.
Biodancer Stereolupe Leica MZ 12 Sterilbank Heraeus Herasafe Tischzentrifugen
Heraeus Sepatech Biofuge A Heraeus Sepatech Biofuge 13 Heraeus Labofuge I;
UV-Lampe BACHOFER 302 nm; Vilbert Lourenat Waagen Sartorius Type 1801
Sartorius L2200S Zentrifugen BECKMAN CS-15R Centrifuge, Rotoren: F2402,
F0630, S1480

MATERIAL UND METHODEN
47
2.8 Bezugsfirmen
Altromin Lage, Deutschland Amersham Biosciences Braunschweig, Deutschland Applied Biosystems Darmstadt, Deutschland BD Biosciences Clontech Heidelberg, Deutschland Bio-Rad Laboratories München, Deutschland Biometra Göttingen, Deutschland Biozym Diagnostik Hessisch Oldendorf, Deutschland Boehringer Ingelheim Ingelheim am Rhein, Deutschland Carl Zeiss Jena, Deutschland Charles River Sulzfeld, Deutschland Chroma Köngen, Deutschland Dianova Hamburg, Deutschland Epicentre Technologies Madison, USA Eppendorf Hamburg, Deutschland Fermentas St. Leon-Rot, Deutschland Fluka Chemie Buchs Schweiz Greiner Solingen, Deutschland Heraeus Düsseldorf, Deutschland Hoechst Frankfurt am Main, Deutschland Interactiva Ulm, Deutschland Invitrogen Karlsruhe, Deutschland Jackson ImmunoResearch West Grove, USA Kodak Stuttgart, Deutschland Nalgene-Nunc Wiesbaden, Deutschland New England Biolabs Frankfurt am Main, Deutschland Merck Darmstadt, Deutschland Microm International Walldorf, Deutschland MWG Biotech Ebersberg, Deutschland Novagen Madison, USA Pharmacia Amersham Freiburg, Deutschland Promega GmbH Mannheim, Deutschland Qiagen Hilden, Deutschland Research Genetics, Inc. Huntsville, AL, USA Roche Diagnostics Mannheim, Deutschland Roche Applied Science Mannheim, Deutschland R.Langenbrinck Emmendingen, Deutschland Schleicher& Schuell Dassel, Deutschland SERVA Electrophoresis Heidelberg, Deutschland SIGMA-ALDRICH St. Louis, USA Slee Technik GmbH Mainz, Deutschland Stratagene La Jolla CA, USA USB Corporation Cleveland Ohio,USA Vector Laboratories Burlinghame, USA Whatman Biometra Göttingen, Deutschland

ERGEBNISSE
48
3 Ergebnisse
3.1 Promotor-Recherche Um den geeigneten Promotor für die Generierung einer gewebespezifisch Cre-Rekombinase
exprimierenden Maus zu finden, erfolgte eine intensive Literatur-Recherche. Dabei richtete
sich das Augenmerk auf Promotoren mit einer spezifischen Aktivität in der Niere,
insbesondere in den renalen Tubuluszellen. Das Expressionsprofil des idealen Promotors
sollte - außer in murinen Zelllinien - auch in transgenen Tieren mittels stromabwärts
klonierter Reportergene getestet sein. Unter den Reportergenen waren sowohl lacZ aus E.
coli als auch GFP aus der Qualle Aequorea victoria (Prasher, 1995) beschrieben. Darüber
hinaus konnte außer einem Reportergen auch c-DNA unter der Kontrolle des zu
untersuchenden Promotors vorhanden sein. Anschließende Untersuchungen sollten mittels
Nothern-blots, in situ Hybridisierungen oder immunhistochemischen Untersuchungen durch
geeignete Antikörper erfolgt sein. Im Rahmen dieser Recherche ist eine entsprechende
Auswahl von geeigneten Promotoren in Tabelle 3.1 aufgelistet worden. Dieser Liste können
die zugehörigen Gene der Promotoren und die Literaturquellen entnommen werden. Ferner
wird die Promotor-Länge und das stromabwärts klonierte Reportergen bzw. die c-DNA,
sowie das Expressionsprofil des Transgens aufgeführt. Nach genauer Abwägung von Größe,
Spezifität und Voruntersuchungen der ausgewählten Promotoren ist die Entscheidung auf
den 346 bp-großen-Minimalpromotor der murinen Gamma-Glutamyl-Transpeptidase Typ II
(GGT-Promotor) gefallen. Wichtige Kriterien sind seine geringe Größe und die eingehenden
Promotoranalysen in transgenen lacZ-exprimierenden Mäusen (Sepulveda et al., 1997). Eine
detaillierte Erläuterung dieser Entscheidung wird im Diskussionsteil aufgeführt.
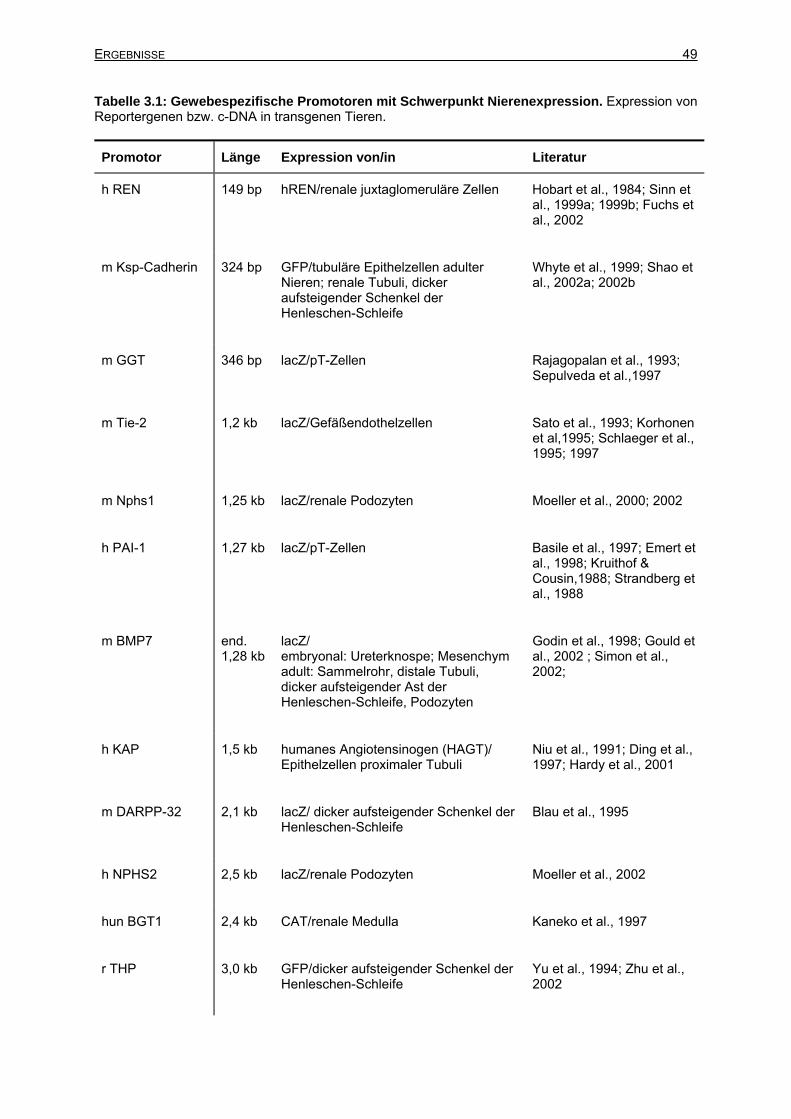
ERGEBNISSE
49
Tabelle 3.1: Gewebespezifische Promotoren mit Schwerpunkt Nierenexpression. Expression von Reportergenen bzw. c-DNA in transgenen Tieren.
Promotor Länge Expression von/in Literatur
h REN 149 bp hREN/renale juxtaglomeruläre Zellen Hobart et al., 1984; Sinn et al., 1999a; 1999b; Fuchs et al., 2002
m Ksp-Cadherin 324 bp GFP/tubuläre Epithelzellen adulter Nieren; renale Tubuli, dicker aufsteigender Schenkel der Henleschen-Schleife
Whyte et al., 1999; Shao et al., 2002a; 2002b
m GGT 346 bp lacZ/pT-Zellen Rajagopalan et al., 1993; Sepulveda et al.,1997
m Tie-2 1,2 kb lacZ/Gefäßendothelzellen Sato et al., 1993; Korhonen et al,1995; Schlaeger et al., 1995; 1997
m Nphs1 1,25 kb lacZ/renale Podozyten Moeller et al., 2000; 2002
h PAI-1 1,27 kb lacZ/pT-Zellen Basile et al., 1997; Emert et al., 1998; Kruithof & Cousin,1988; Strandberg et al., 1988
m BMP7 end. 1,28 kb
lacZ/ embryonal: Ureterknospe; Mesenchym adult: Sammelrohr, distale Tubuli, dicker aufsteigender Ast der Henleschen-Schleife, Podozyten
Godin et al., 1998; Gould et al., 2002 ; Simon et al., 2002;
h KAP 1,5 kb humanes Angiotensinogen (HAGT)/ Epithelzellen proximaler Tubuli
Niu et al., 1991; Ding et al., 1997; Hardy et al., 2001
m DARPP-32 2,1 kb lacZ/ dicker aufsteigender Schenkel der Henleschen-Schleife
Blau et al., 1995
h NPHS2 2,5 kb lacZ/renale Podozyten Moeller et al., 2002
hun BGT1 2,4 kb CAT/renale Medulla Kaneko et al., 1997
r THP 3,0 kb GFP/dicker aufsteigender Schenkel der Henleschen-Schleife
Yu et al., 1994; Zhu et al., 2002
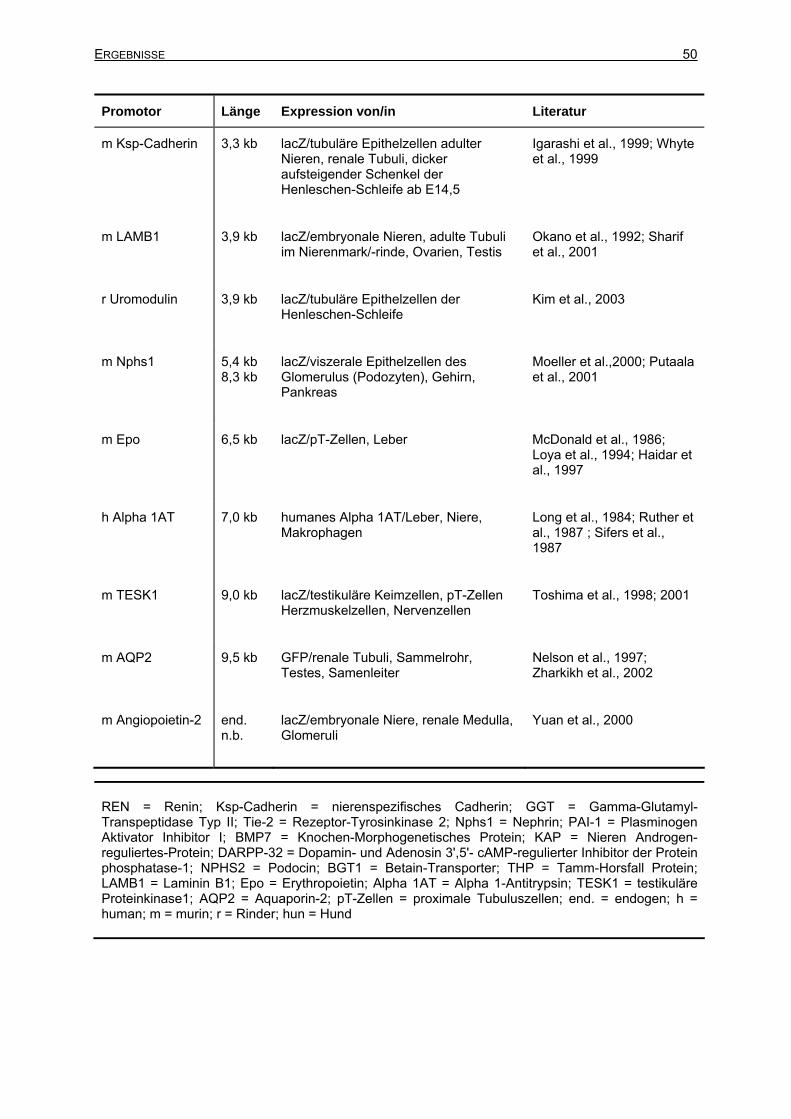
ERGEBNISSE
50
Promotor Länge Expression von/in Literatur
m Ksp-Cadherin 3,3 kb lacZ/tubuläre Epithelzellen adulter Nieren, renale Tubuli, dicker aufsteigender Schenkel der Henleschen-Schleife ab E14,5
Igarashi et al., 1999; Whyte et al., 1999
m LAMB1 3,9 kb lacZ/embryonale Nieren, adulte Tubuli im Nierenmark/-rinde, Ovarien, Testis
Okano et al., 1992; Sharif et al., 2001
r Uromodulin 3,9 kb lacZ/tubuläre Epithelzellen der Henleschen-Schleife
Kim et al., 2003
m Nphs1 5,4 kb 8,3 kb
lacZ/viszerale Epithelzellen des Glomerulus (Podozyten), Gehirn, Pankreas
Moeller et al.,2000; Putaala et al., 2001
m Epo 6,5 kb lacZ/pT-Zellen, Leber McDonald et al., 1986; Loya et al., 1994; Haidar et al., 1997
h Alpha 1AT 7,0 kb humanes Alpha 1AT/Leber, Niere, Makrophagen
Long et al., 1984; Ruther et al., 1987 ; Sifers et al., 1987
m TESK1 9,0 kb lacZ/testikuläre Keimzellen, pT-Zellen Herzmuskelzellen, Nervenzellen
Toshima et al., 1998; 2001
m AQP2 9,5 kb GFP/renale Tubuli, Sammelrohr, Testes, Samenleiter
Nelson et al., 1997; Zharkikh et al., 2002
m Angiopoietin-2 end. n.b.
lacZ/embryonale Niere, renale Medulla, Glomeruli
Yuan et al., 2000
REN = Renin; Ksp-Cadherin = nierenspezifisches Cadherin; GGT = Gamma-Glutamyl-Transpeptidase Typ II; Tie-2 = Rezeptor-Tyrosinkinase 2; Nphs1 = Nephrin; PAI-1 = Plasminogen Aktivator Inhibitor I; BMP7 = Knochen-Morphogenetisches Protein; KAP = Nieren Androgen-reguliertes-Protein; DARPP-32 = Dopamin- und Adenosin 3',5'- cAMP-regulierter Inhibitor der Protein phosphatase-1; NPHS2 = Podocin; BGT1 = Betain-Transporter; THP = Tamm-Horsfall Protein; LAMB1 = Laminin B1; Epo = Erythropoietin; Alpha 1AT = Alpha 1-Antitrypsin; TESK1 = testikuläre Proteinkinase1; AQP2 = Aquaporin-2; pT-Zellen = proximale Tubuluszellen; end. = endogen; h = human; m = murin; r = Rinder; hun = Hund

ERGEBNISSE
51
3.2 Klonierung der Zielvektoren In dieser Arbeit wurden die Zielvektoren pGGT-CRE, pGGT-RTTA und pBI-CRE-lacZ
kloniert. Zum besseren Verständnis werden in den folgenden Abschnitten 3.2.1 bis 3.2.3 die
jeweiligen Klonierungsschritte anschaulich dargestellt.
3.2.1 Klonierung des Vektors pGGT-CRE Zur Erstellung des Zielvektors pGGT-CRE wurden folgende funktionelle Elemente benötigt:
1. 346 bp-Minimalpromotor Typ II der murinen Gamma-Glutamyl-Transpeptidase (GGT)
(Sepulveda et al., 1997)
2. Kernlokalisationssignal (NLS, Kalderon et al., 1984) vom SV40 large T Antigen
3. kodierender Bereich der P1-Cre-Rekombinase (Sternberg et al., 1986)
4. SV40-polyA Region (Fiers et al., 1978)
5. Ampicillin-Resistenz (ampR)
Beim Zielvektor pGGT-CRE steht der kodierende Bereich der P1-Cre-Rekombinase unter
der Kontrolle des gewebespezifischen GGT-Miniminalpromotors. Wobei die vorgeschaltene
NLS-Sequenz zur Translokation der Cre-Rekombinase in den Nukleus dienen soll. Im 3´-
Bereich wurde die SV40-polyA Region einkloniert, um einen Transkriptionsstillstand der
RNA-Polymerase sicherzustellen. Im folgenden Verlauf werden die einzelnen
Klonierungsschritte für den Zielvektor pGGT-CRE bis zur Isolierung des Transgens GGT-
CRE dargestellt. In der Tabelle 3.2 sind alle verwendeten Plasmide bzw. genomischen DNA-
Fragmente für die in vitro-Konstruktion des Vektors pGGT-CRE mit den zugehörigen
funktionellen Kassetten aufgelistet.
Tabelle 3.2: Verwendete Plasmide bzw. genomische DNA-Fragmente zur Klonierung des Zielvektors pGGT-CRE. Plasmid/DNA-Bezeichnung funktionelle Kassette
amplifizierte murine genomische DNA GGT-Promotor
pHD2-Cre1 NLS
Cre-Rekombinase
SV40-polyA
pSL300/PgkNeoL ampR

ERGEBNISSE
52
3.2.1.1 Klonierung des Vektors pGGT-PFU-1 - Genomische PCR - Ein 424 bp großes DNA-Fragment mit anteilig 346 bp des GGT-Promotors Typ II (Abbildung
3.1) wurde aus der genomischen murinen DNA amplifiziert. Die PCR-Reaktion fand unter
Verwendung der Oligonukleotide 5402/5403 (unterstrichen) und der pfu-DNA-Polymerase
(Stratagene) statt.
SalI -346
1 GTCGACTCTA AGCTATGGTC TAGTGCCTGG GGCTACCCCT ACTTTATTGC 51 CTCAGGAACA TTTTTGAGGA CATCATGTCT GTCTCTAAAT TCACAGAGGG 101 ACTTAGCAGG GGCTGTCTGG AAGGGTTCAC CGGTGGCCTC TGCCCCTTAG 151 AGGGAACCAA ATCTGGAAAG TGGGGGAGCC CCTTTCCCAG GCTCTTAGTA 201 CCTCCCTCCT CCCCCGCCCA GAGCCCTCCT GGGGCTGCAT GAACTTTAGT 251 CACCTGGTCA CAAGCCTGAC GCTGCGCCTC CTTGCGGCTG TGACGTCAGC 301 GTTGACCATC CTCCCAGCCT ATCTGAACCT CTTACAGAGG ACTCCCTACC +1 351 TCACACCCCA CTCAGAAGCC TTCAGATGCC CTTCTGGGAT GCTGGGCACC BglII 401 CCTTAGCTGA CCTCTTGCAG ATCT
Abbildung 3.1: 424 bp-DNA-Sequenz mit dem GGT-Minimalpromotor. Die Sequenz stellt die 346-bp-Promotorregion des murinen GGT Typ II dar (eingerahmt). Die Restriktionsschnittstellen sind grau hervorgehoben und die Primersequenzen (5402/5403) sind unterstrichen. +1 zeigt den Transkriptionsstartpunkt an (Sepulveda et al., 1994). Die vorliegende genomische Sequenz von 412 bp (gelb hervorgehoben) ist kongruent mit der Genbank Accession Nr.: NT_039496.4 nt 1354210 -1354621 (NCBI) von Mus musculus Chromosom 10.
Mit den Oligonukleotiden GGT-SalI-5402-F und GGT-BglII-5403-R (Tabelle 3.3) wurde am
5´-Ende des Amplifikats eine SalI-Schnittstelle und am 3´-Ende eine BglII-Schnittstelle
eingeführt.
Tabelle 3.3: Oligonukleotide mit eingeführten Restriktionsschnittstellen. Die Primer 5402/5403 wurden in der genomischen PCR zur Amplifikation des murinen GGT-Promotors eingesetzt. Die Restriktionsenzyms-Erkennungssequenzen sind grau unterlegt (R: reverse; F: forward).
Primer-Name Oligonukleotid-Sequenz 5´- 3´
GGT-SalI-5402-F 5´- CGT GCG TCG ACT CTA AGC TAT GGT CTA GTG CCT GGG GC - 3´
GGT-BglII- 5403-R 5´- GCT GCA GAT CTG CAA GAG GTC AGC TAA GGG GTG CC -3´
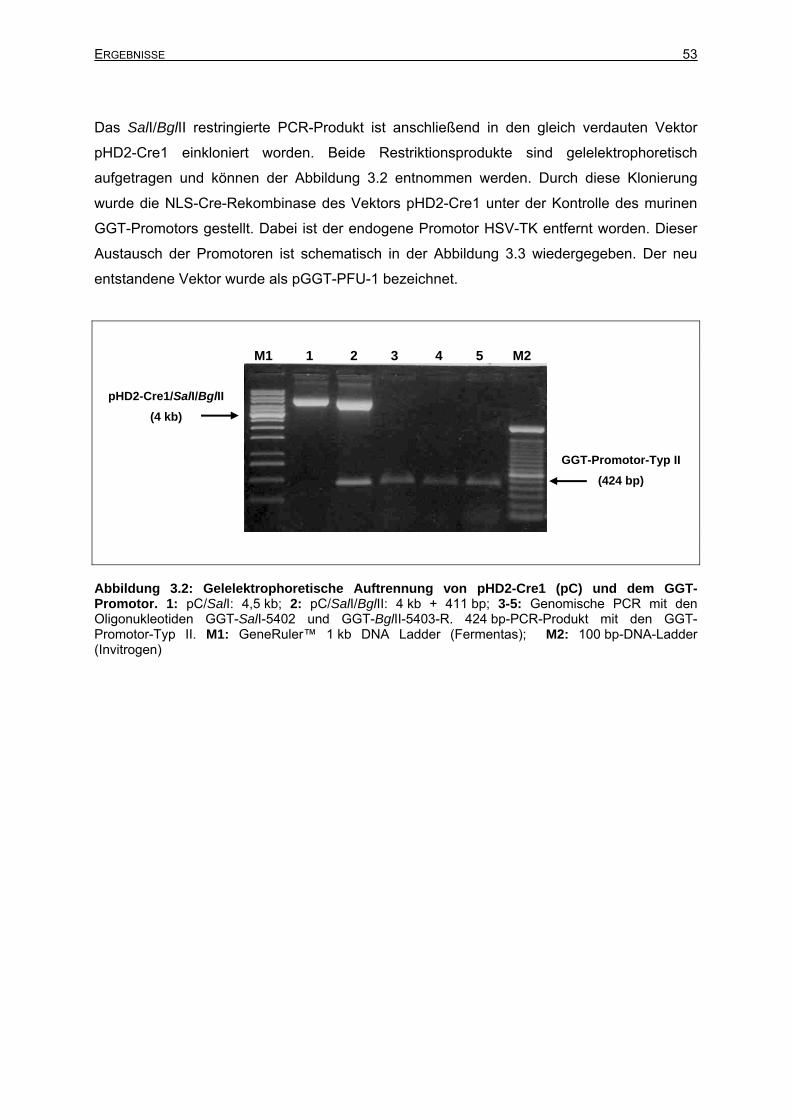
ERGEBNISSE
53
Das SalI/BglII restringierte PCR-Produkt ist anschließend in den gleich verdauten Vektor
pHD2-Cre1 einkloniert worden. Beide Restriktionsprodukte sind gelelektrophoretisch
aufgetragen und können der Abbildung 3.2 entnommen werden. Durch diese Klonierung
wurde die NLS-Cre-Rekombinase des Vektors pHD2-Cre1 unter der Kontrolle des murinen
GGT-Promotors gestellt. Dabei ist der endogene Promotor HSV-TK entfernt worden. Dieser
Austausch der Promotoren ist schematisch in der Abbildung 3.3 wiedergegeben. Der neu
entstandene Vektor wurde als pGGT-PFU-1 bezeichnet.
M1 1 2 3 4 5 M2
Abbildung 3.2: Gelelektrophoretische Auftrennung von pHD2-Cre1 (pC) und dem GGT-Promotor. 1: pC/SalI: 4,5 kb; 2: pC/SalI/BglII: 4 kb + 411 bp; 3-5: Genomische PCR mit den Oligonukleotiden GGT-SalI-5402 und GGT-BglII-5403-R. 424 bp-PCR-Produkt mit den GGT-Promotor-Typ II. M1: GeneRuler™ 1 kb DNA Ladder (Fermentas); M2: 100 bp-DNA-Ladder (Invitrogen)
pHD2-Cre1/SalI/BglII (4 kb)
GGT-Promotor-Typ II(424 bp)

ERGEBNISSE
54
Abbildung 3.3: Schematische Darstellung der Klonierung des Vektors pGGT-PFU-1. Der Vektor pHD2-Cre1 wurde mit SalI/BglII restringiert und das entstandene ca. 4 kb große DNA-Fragment mit dem 418 bp-SalI/BglII-PCR-Produkt (GGT-Promotor Typ II) ligiert. Die roten Pfeile stellen die Primer 5402/5403 dar und der Transkriptionsstartpunkt ist mit einem schwarzen Pfeil verdeutlicht. pA: SV40-polyA. Zur Verdeutlichung werden nur die Inserts mit den funktionellen Kassetten (SalI/NotI-Fragmente) der Vektoren pHD2-Cre1 und pGGT-PFU-1 dargestellt.
Entfernen der HSV-TK (411 bp)
pA
pA
SalI BglII
GGT X SalI/BglII Ligation mit PCR-Produkt
pGGT-PFU-1
pHD2-Cre1
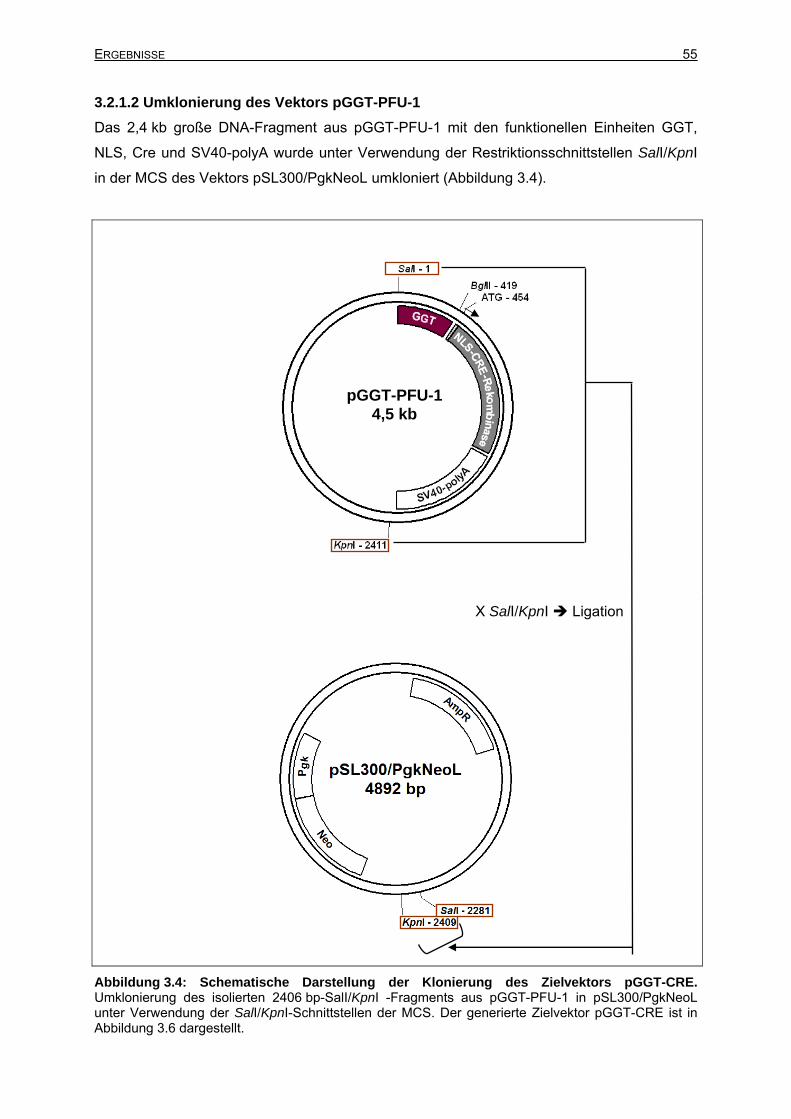
ERGEBNISSE
55
3.2.1.2 Umklonierung des Vektors pGGT-PFU-1 Das 2,4 kb große DNA-Fragment aus pGGT-PFU-1 mit den funktionellen Einheiten GGT,
NLS, Cre und SV40-polyA wurde unter Verwendung der Restriktionsschnittstellen SalI/KpnI
in der MCS des Vektors pSL300/PgkNeoL umkloniert (Abbildung 3.4).
Abbildung 3.4: Schematische Darstellung der Klonierung des Zielvektors pGGT-CRE. Umklonierung des isolierten 2406 bp-SalI/KpnI -Fragments aus pGGT-PFU-1 in pSL300/PgkNeoL unter Verwendung der SalI/KpnI-Schnittstellen der MCS. Der generierte Zielvektor pGGT-CRE ist in Abbildung 3.6 dargestellt.
X SalI/KpnI Ligation
pGGT-PFU-1 4,5 kb

ERGEBNISSE
56
3.2.1.3 Isolierung des GGT-CRE-Transgens aus dem Zielvektor Für die nachfolgende Mikroinjektion wurde aus dem Zielvektor pGGT-CRE, unter
Verwendung der SalI/KpnI-Schnittstellen, das 2,4 kb-große Transgen GGT-CRE isoliert. Zur
Charakterisierung des Zielvektors wurde pGGT-CRE vollständig sequenziert und die DNA-
Sequenz kann dem Anhang entnommen werden. Des Weiteren sind Restriktionskontrollen
durchgeführt worden (Abbildung 3.5). Die Abbildung 3.6 stellt anschaulich die Isolierung des
GGT-CRE dar.
M1 1 2 3 4 5 6 7 8 9 10 11 12 M2
Abbildung 3.5 (A): Restriktionskontrolle des Konstruktes pGGT-CRE (pG). 1: pG/SalI/KpnI 2430 bp + 4744 bp; 2: pG/SalI 7174 bp; 3: pG/KpnI 7174 bp; 4: pG/EcoRV 1460 bp + 5714 bp; 5: pG/HincII 300 bp + 1964 bp + 4910 bp; 6: pG/HpaI 7174 bp; 7: pG/EcoNI 7174 bp; 8: pG/SspI 3597 bp + 551 bp + 153 bp + 1942 bp + 800 bp + 131 bp; 9: pG/CvnI 7174 bp; 10: pGGT-PFU-1/CvnI 4,7 kb; 11: pSL300/pGKNeoL/CvnI 4901 bp; 12: pGGT-CRE unverdaut; M1: GeneRuler™ 1 kb DNA Ladder (Fermentas); M2: 100 bp-DNA-Ladder (Invitrogen).
A
Transgen GGT-CRE (2,4 kb)
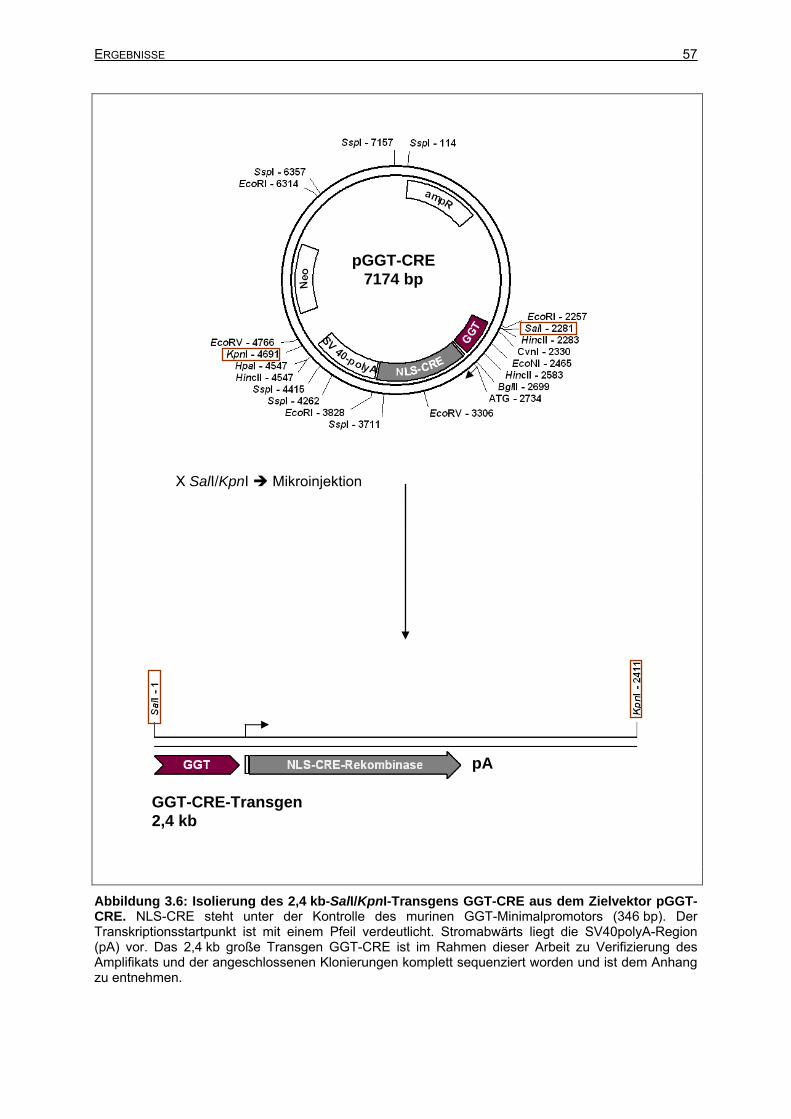
ERGEBNISSE
57
X SalI/KpnI Mikroinjektion
Abbildung 3.6: Isolierung des 2,4 kb-SalI/KpnI-Transgens GGT-CRE aus dem Zielvektor pGGT-CRE. NLS-CRE steht unter der Kontrolle des murinen GGT-Minimalpromotors (346 bp). Der Transkriptionsstartpunkt ist mit einem Pfeil verdeutlicht. Stromabwärts liegt die SV40polyA-Region (pA) vor. Das 2,4 kb große Transgen GGT-CRE ist im Rahmen dieser Arbeit zu Verifizierung des Amplifikats und der angeschlossenen Klonierungen komplett sequenziert worden und ist dem Anhang zu entnehmen.
pA
2,4 kb
pGGT-CRE 7174 bp
GGT-CRE-Transgen 2,4 kb

ERGEBNISSE
58
3.2.2 Klonierung des Vektors pGGT-RTTA Für eine zeitliche und gewebespezifische kontrollierte Genexpression beim tTA-System
(Gossen et al., 1995) sind wie eingangs schon erwähnt zwei genetische Elemente
erforderlich: Ein Doxycyclin-abhängiger Transkriptionsaktivator (rtTA), welcher unter
Kontrolle des gewebespezifischen Promotors steht und ein Tet Operator (tetO). Aus diesem
Grund sind in dieser Arbeit zwei weitere Vektoren konstruiert worden: pGGT-RTTA und pBI-
CRE-lacZ. Durch die Kombination beider genetischer Elemente wird eine, durch Doxycyclin
beeinflussbare, konstitutive Expression der Cre-Rekombinase ermöglicht.
Für die Konstruktion des Vektors pGGT-RTTA sind folgende funktionelle Einheiten
verwendet worden:
1. 346 bp-GGT-Minimalpromotor
2. kodierende Region des Transkriptionsaktivators rtTA2S-M2
3. SV40-polyA Region
4. Ampicillin-Resistenz (ampR)
Der Vektor pGGT-RTTA enthält als funktionelle Einheit die kodierende Region des
Transkriptionsaktivators rtTA2S-M2. Es handelt sich hierbei um einer modifizierten Version
vom rtTA mit höherer Stabilität in eukaryotischen Zellen, größerer Sensitivität gegenüber
Doxycyclin und reduzierter Hintergrundaktivität bei dessen Abwesenheit (Urlinger et al.,
2000). Der modifizierte Transkriptionsaktivator rtTA2S-M2 wurde unter die Kontrolle des
GGT-Minimalpromotors gestellt. Im 3´- Bereich schließt das Transgen mit einer SV40- polyA
Region ab. Tabelle 3.4 listet die benötigten Plasmide bzw. DNA-Fragmente und deren
funktionellen Einheiten zur Erstellung des Vektors pGGT-RTTA auf.
Tabelle 3.4: Verwendete Plasmide und genomische DNA-Fragmente zur Klonierung des Zielvektors pGGT-CRE
Plasmid/DNA-Bezeichnung funktionelle Kassette
amplifizierte murine genomische DNA GGT-Promotor
pUHDrtTA2S-M2 rtTA2S-M2
SV40-polyA
ampR

ERGEBNISSE
59
3.2.2.1 Amplifikation des GGT-Promotors Aus muriner genomischer DNA wurde ein 424 bp-großes-PCR-Produkt mit anteilig 346 bp
des GGT-Promotors Typ II amplifiziert und gelelektrophoretisch aufgetragen (Abbildung
3.8 B). Die eingesetzten Primer GGT-XhoI-5442-F und GGT-EcoRI-5443-R werden in der
Tabelle 3.5 dargestellt. Daraus ergeben sich die Restriktionsschnittstellen XhoI/EcoRI,
welche den GGT Promotor Typ II flankieren.
Tabelle 3.5: Oligonukleotide GGT-XhoI-5442-F und GGT-EcoRI-5443-R mit internen Restriktionsschnittstellen zur Amplifikation des murinen GGT-Promotors. Die Restriktionsenzym-Erkennungssequenzen sind grau hervorgehoben; (R: reverse; F: forward).
Primer-Name Oligonukleotid-Sequenz 5´- 3´
GGT-XhoI-5442-F 5´- CGT GCC TCG AGT CTA AGC TAT GGT CTA GTG CCT GGG GC - 3´
GGT-EcoRI-5443-R 5´- GCT GCG AAT TCG CAA GAG GTC AGC TAA GGG GTG CC -3´
Nach Isolation aus dem Agarosegel wurde das PCR-Produkt unter Verwendung der neu
generierten Restriktionsschnittstellen XhoI/EcoRI geschnitten. Anschließend erfolgte eine
Ligation mit dem gleich restringierten pUHDrtTA2S-M2-Fragment (3433 bp). Aus dieser
Klonierung resultierte der Zielvektors pGGT-RTTA (Abbildung 3.7), wo der endogene
Promotor hCMV durch den GGT-Promotor Typ II ausgetauscht wurde. Somit stand die Cre-
Rekombinase beim Zielvektor pGGT-RTTA unter der Kontrolle vom Doxycyclin-abhängigen
Transkriptionsaktivator rtTA2S-M2.
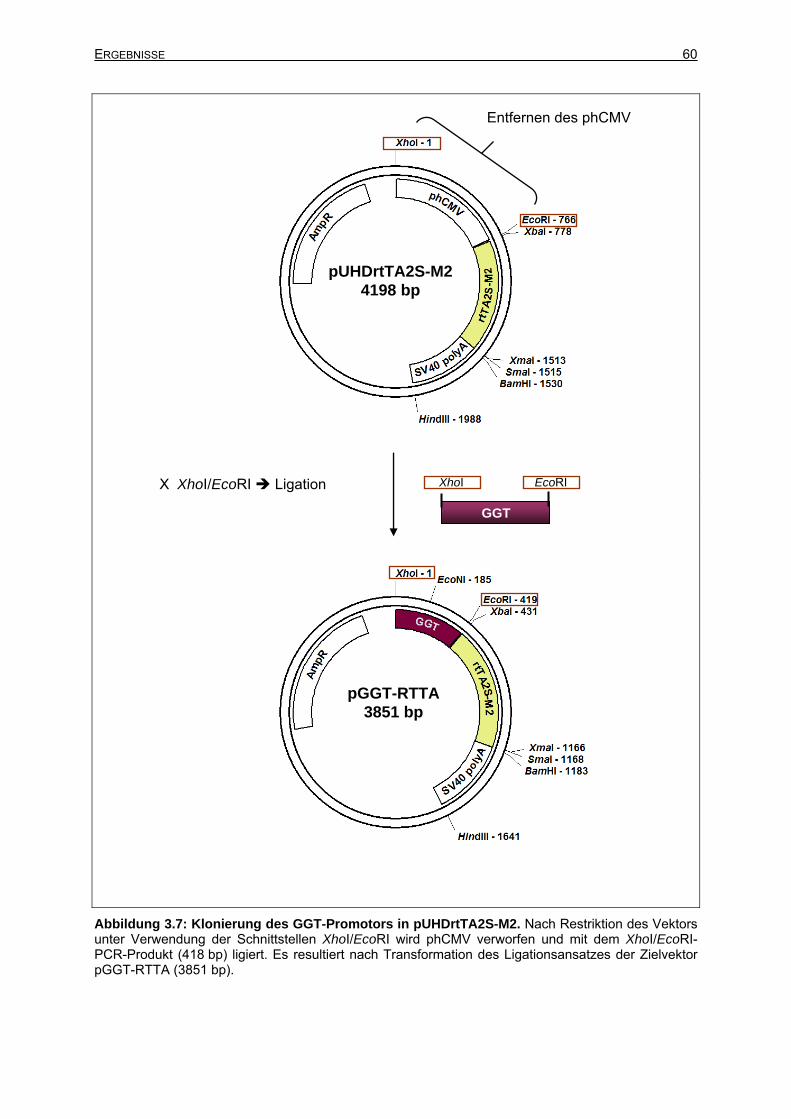
ERGEBNISSE
60
X XhoI/EcoRI Ligation
Abbildung 3.7: Klonierung des GGT-Promotors in pUHDrtTA2S-M2. Nach Restriktion des Vektors unter Verwendung der Schnittstellen XhoI/EcoRI wird phCMV verworfen und mit dem XhoI/EcoRI-PCR-Produkt (418 bp) ligiert. Es resultiert nach Transformation des Ligationsansatzes der Zielvektor pGGT-RTTA (3851 bp).
Entfernen des phCMV
XhoI EcoRI
GGT
pGGT-RTTA 3851 bp
pUHDrtTA2S-M2 4198 bp

ERGEBNISSE
61
Vor der Ligation wurde der Vektor pUHDrtTA2S-M2 mittels Restriktionsverdau (siehe
Abbildung 3.8 A) und Resequenzierung kontrolliert.
M1 1 2 3 4 5 6 M2
Abbildung 3.8: (A): Restriktionskontrolle des Vektors pUHDrtTA2S-M2 (pUH). 1: pUH/XhoI/HindIII 2,21 kb + 1,98 kb; 2: pUH/XhoI/EcoRI 3,43 kb + 0,76 kb; 3: pUH/EcoRI/HindIII 2,97 kb + 1,22 kb; 4: pUH/SmaI/EcoRI 3,44 kb + 0,74 kb; (B): Murine genomische PCR. Es wurden die Oligonukleotide GGT-XhoI-5442-F und GGT-EcoRI-5443-R eingesetzt. Daraus resultiert ein 424 bp-PCR-Produkt mit den GGT-Promotor-Typ II (Spur 5). 6: Wasserkontrolle mit den Primern GGT-XhoI-5442-F und GGT-EcoRI-5443-R. M1: GeneRuler™ 1 kb DNA Ladder (Fermentas); M2: 100 bp-DNA-Ladder (Invitrogen)
3.2.2.2 Isolierung des GGT-RTTA-Transgens aus dem Zielvektor Unter Verwendung der Restriktionsendonukleasen XhoI und HindIII konnte das 1646 bp
große Transgen GGT-RTTA aus dem Vektorrückrat des pGGT-RTTA isoliert werden
(Abbildung 3.9). In der Abbildung 3.10 ist die Isolierung des Transgens schematisch
dargestellt.
M1 1 M2
Abbildung 3.9: Restriktionskontrolle des GGT-RTTA (GT). 1: GT/XhoI/HindIII 2,2 kb + 1,6 kb; M1: GeneRuler™ 1 kb DNA Ladder (Fermentas); M2: 100 bp-DNA-Ladder (Invitrogen)
Transgen
GGT-RTTA (1,6 kb)
A B
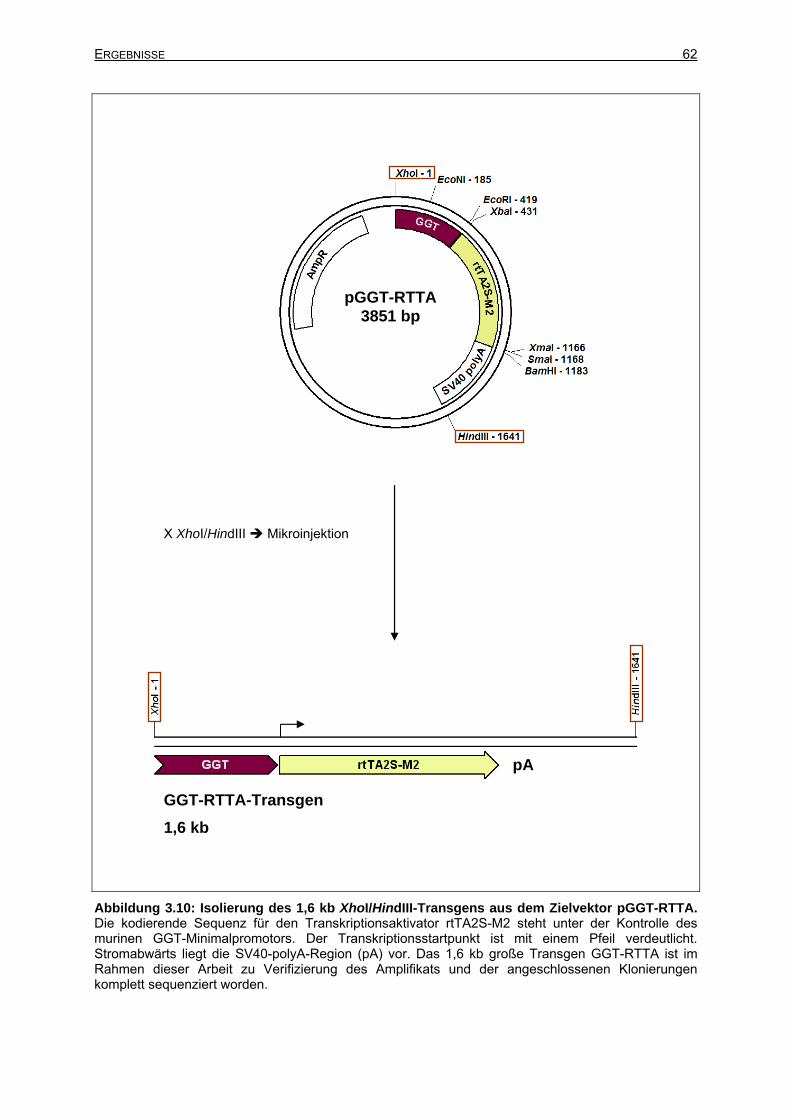
ERGEBNISSE
62
Abbildung 3.10: Isolierung des 1,6 kb XhoI/HindIII-Transgens aus dem Zielvektor pGGT-RTTA. Die kodierende Sequenz für den Transkriptionsaktivator rtTA2S-M2 steht unter der Kontrolle des murinen GGT-Minimalpromotors. Der Transkriptionsstartpunkt ist mit einem Pfeil verdeutlicht. Stromabwärts liegt die SV40-polyA-Region (pA) vor. Das 1,6 kb große Transgen GGT-RTTA ist im Rahmen dieser Arbeit zu Verifizierung des Amplifikats und der angeschlossenen Klonierungen komplett sequenziert worden.
pA
X XhoI/HindIII Mikroinjektion
1,6 kb
pGGT-RTTA 3851 bp
GGT-RTTA-Transgen 1,6 kb

ERGEBNISSE
63
3.2.3 Klonierung des Vektors pBI-CRE-lacZ Zur Klonierung des Vektors pBI-CRE-lacZ wurden folgende funktionelle Einheiten verwendet:
1. bidirektioneller Promotor Pbi-1 (Baron et al., 1995)
2. sieben tet-Operatorsequenzen (tetO)
3. NLS-lacZ
4. NLS-P1-Cre-Rekombinase
5. SV40-polyA
6. Kaninchen β-Globin-Intron/polyA (Lanford et al., 1988)
7. ampR
Nach entsprechender Modifizierung des kommerziellen Vektors pBI-3 (BD Biosciences
Clontech, Heidelberg) wurde die kodierende Region der Cre-Rekombinase mit einer
vorgeschaltenen NLS-Sequenz einkloniert. Der Vektor pBI-3 weist als wichtiges funktionelles
Element den bidirektionellen Promotor Pbi-1 auf (Baron et al., 1995). Diese Konstellation
ermöglicht die gleichzeitige äquimolare Expression eines Reportergens (z.B. lacZ) und eines
zweiten Gens, hier in dieser Arbeit die Cre-Rekombinase. Die bidirektionelle Aktivierung des
Reportergens und der Cre-Rekombinase erlaubt sehr einfach die Kontrolle der
vorgenommenen Transfektion. Dadurch ist es möglich, dass z.B. In einem transgenen Tier
neben der leicht nachweisbaren β-Galaktosidase auch die Cre-Rekombinase exprimiert wird.
Die eigentliche Induzierbarkeit der Genexpression resultiert aber aus den vorliegenden tet
Operator-Sequenzen (tetO). Erst durch die Anbindung eines Doxycyclin-aktiviertes rtTA-
Proteins erfolgt die gleichzeitige Transkription beider klonierten Gene. Die
Klonierungsstrategie des Vektors pBI-CRE-lacZ und die anschließende Transgen-Isolierung
(BI-CRE-lacZ) werden in den folgenden Abschnitten (3.2.3.1-3.2.3.3) erörtert. In der Tabelle
3.6 sind die Plasmide für die in vitro-Konstruktion des Zielvektors pBI-CRE-lacZ aufgelistet
worden.
Tabelle 3.6: Verwendete Plasmide zur Klonierung des Zielvektors pBI-CRE-lacZ
Plasmid-Bezeichnung funktionelle Kassette
pBI-3 Pbi-1 tetO NLS-lacZ
β-Globin-polyA ampR
pHD2-Cre1 NLS-Cre-Rekombinase
SV40-polyA

ERGEBNISSE
64
3.2.3.1 Einführung einer HindIII-Schnitttstelle in pBI-3 an Position nt 5188 Die Generierung einer singulären HindIII-Restriktionsschnittstelle im Vektor pBI-3 (Clontech)
an Position nt 5188 war erforderlich, um das Transgen am 3´-Ende des b-Globin-polyA-
Bereichs isolieren zu können. Mittels PCR und der Primer 5494 und 5495 (Tabelle 3.7)
wurde eine HindIII-Schnittstelle eingeführt. Als DNA-Matrize diente der Ausgangsvektor pBI-
3, wobei ein 1235 bp AseI-Fragment amplifiziert wurde.
Tabelle 3.7: Oligonukleotide zur Einführung einer HindIII-Schnittstelle in pBI-3 an Position nt 5188. Die HindIII-Erkennungssequenz ist eingerahmt. Grau hervorgehoben sind die endogenen AseI-Restriktionsschnittstellen (R: reverse; F: forward).
Primer-Name Oligonukleotid-Sequenz 5´- 3´
pBI-3-AR-5494 5´- GGC AAC AAT TAA TAG ACT GGA TGG AGG CG-3´
pBI-3BF-H3-5495 5´- GAC TGC ATT AAT GAA GCT TCC AAC GCG-3´
Die Einführung der zusätzlichen HindIII-Erkennungssequenz im AseI-Fragment des Vektors
pBI-3, erfolgte durch den Primer pBI-3BF-H3-5495 (Tabelle 3.7). Dieses Fragment wurde
anschließend in korrekter Orientierung mit dem AseI-pBI-3-Fragment (6,4 kb) kloniert
(Abbildung 3.13). Daraus resultierte der so benannte Vektor pBI-3-H3 (7,7 kb). Das
Klonierungsergebnis wurde mittels Restriktionskontrolle (Abbildung 3.11) und
Resequenzierung verifiziert.
M 1 2 3 4
Abbildung 3.11: Restriktionskontrolle von pBI-3-H3 (pBI-H). 1: pBI-H /HindIII/PstI 5,2 kb + 2,5 kb 2: pBI-H /HindIII/PstI 5,2 kb + 2,5 kb 3: pBI-H/HindIII linearisiert 4: pBI-H/HindIII linearisiert. M: GeneRuler™ 1 kb DNA Ladder (Fermentas)
3.2.3.2 Klonierung des Vektors pBI-3-H3 Um das NLS-Cre-Rekombinase-Gen mit der SV40-polyA-Region unter der Kontrolle des
tetO abhängigen Minimalpromotors vom Pbi-1 zu setzen, wurde über die endogenen
Schnittstellen PstI und NotI aus pHD2-Cre-1 geschnitten und im Vektor pBI-3-H3 über
7,7 kb-pBI-3-H3

ERGEBNISSE
65
PstI/NotI kloniert. Aus dieser Klonierungsstrategie entstand der Zielvektor pBI-CRE-lacZ
(9,6 kb). Abbildung 3.14 stellt die Klonierung schematisch dar.
3.2.3.3 Isolierung des Transgens BI-CRE-lacZ aus dem Zielvektor Die Entfernung des Vektorrückrats zur Erlangung des 7,16 kb große Transgens BI-CRE-lacZ
erfolgte unter Verwendung der endogenen NotI- und der neu generierten HindIII-
Schnittstelle. In Abbildung 3.12 ist die gelelektrophoretische Auftrennung des Transgens und
des Vektorrückrats dargestellt und die Isolierung in Abbildung 3.15 schematisch
wiedergegeben. Das Transgen BI-CRE-lacZ ist im Rahmen dieser Arbeit komplett
resequenziert worden.
M 1
Abbildung 3.12: Restriktionsverdau des Vektors pBI-CRE-lacZ. 1: pBI-CRE-lacZ/NotI/HindIII 7,16 kb (Transgen BI-CRE-lacZ) + 2,5 kb (Vektorrückrat) M: GeneRuler™ 1 kb DNA Ladder (Fermentas)
7,16 kb BI-CRE-lacZ
2,5 kb Vektorrückrat
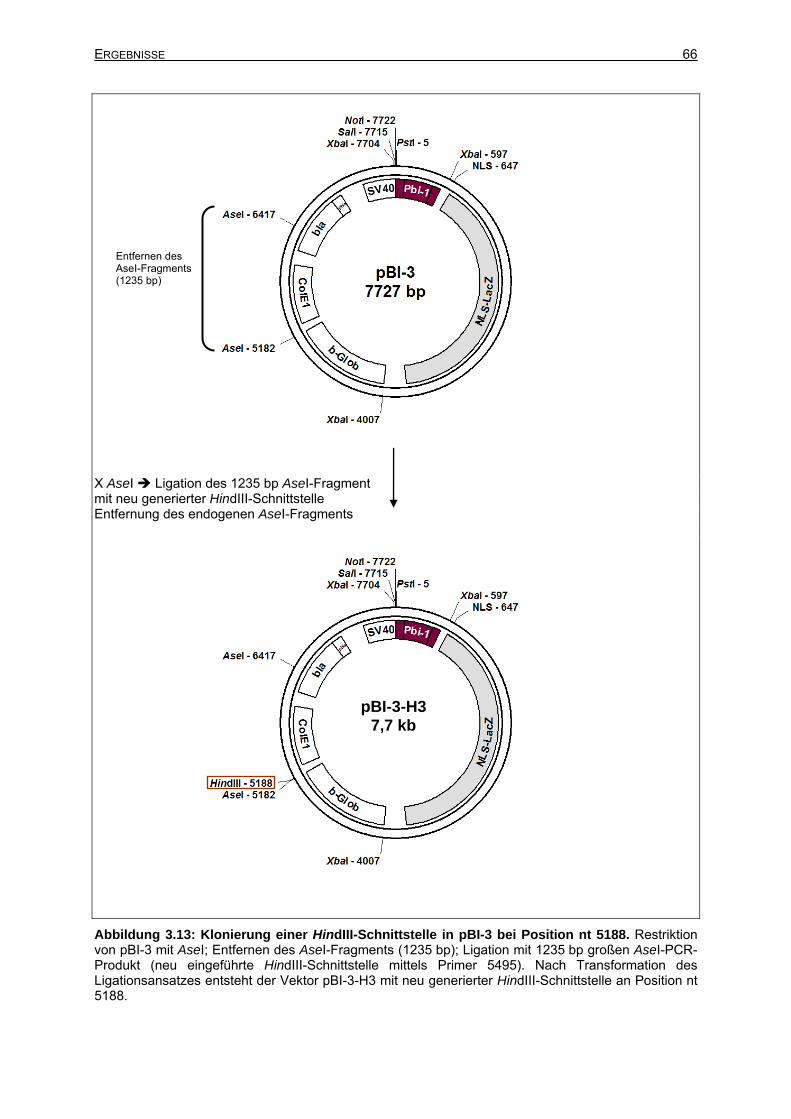
ERGEBNISSE
66
X AseI Ligation des 1235 bp AseI-Fragment mit neu generierter HindIII-Schnittstelle Entfernung des endogenen AseI-Fragments
Abbildung 3.13: Klonierung einer HindIII-Schnittstelle in pBI-3 bei Position nt 5188. Restriktion von pBI-3 mit AseI; Entfernen des AseI-Fragments (1235 bp); Ligation mit 1235 bp großen AseI-PCR-Produkt (neu eingeführte HindIII-Schnittstelle mittels Primer 5495). Nach Transformation des Ligationsansatzes entsteht der Vektor pBI-3-H3 mit neu generierter HindIII-Schnittstelle an Position nt 5188.
Entfernen des AseI-Fragments (1235 bp)
pBI-3-H3 7,7 kb
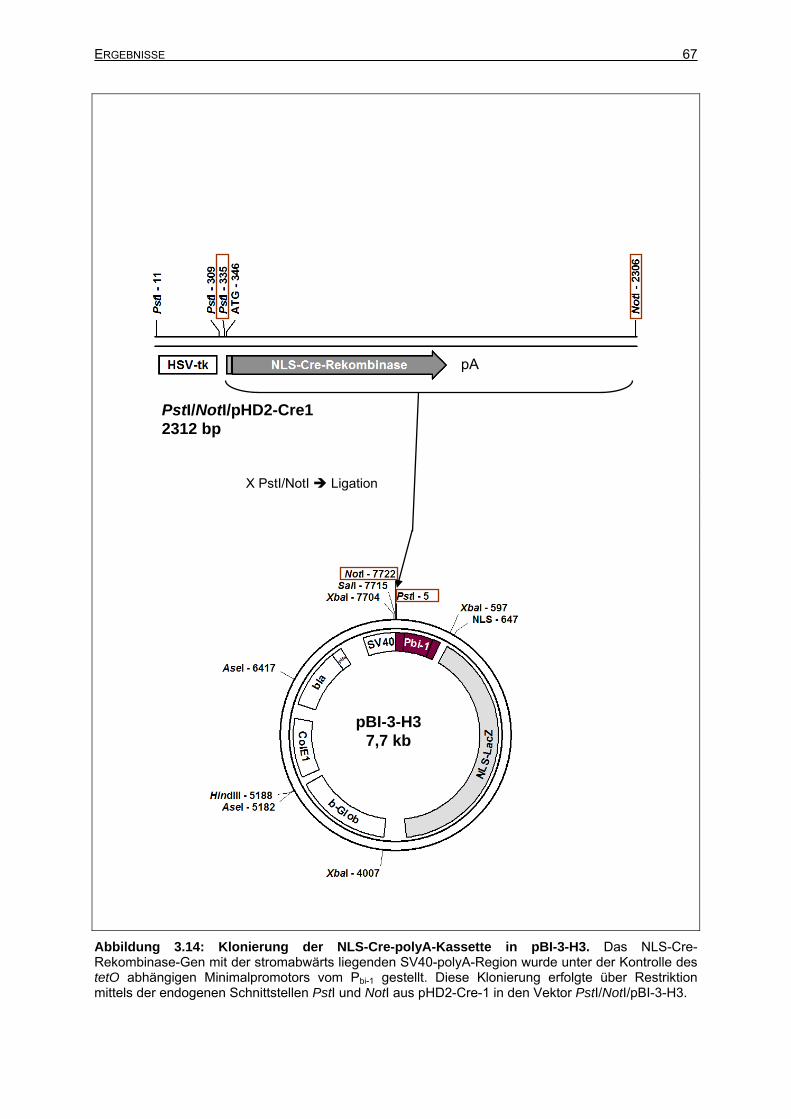
ERGEBNISSE
67
X PstI/NotI Ligation
Abbildung 3.14: Klonierung der NLS-Cre-polyA-Kassette in pBI-3-H3. Das NLS-Cre-Rekombinase-Gen mit der stromabwärts liegenden SV40-polyA-Region wurde unter der Kontrolle des tetO abhängigen Minimalpromotors vom Pbi-1 gestellt. Diese Klonierung erfolgte über Restriktion mittels der endogenen Schnittstellen PstI und NotI aus pHD2-Cre-1 in den Vektor PstI/NotI/pBI-3-H3.
pA
pBI-3-H3 7,7 kb
PstI/NotI/pHD2-Cre1 2312 bp
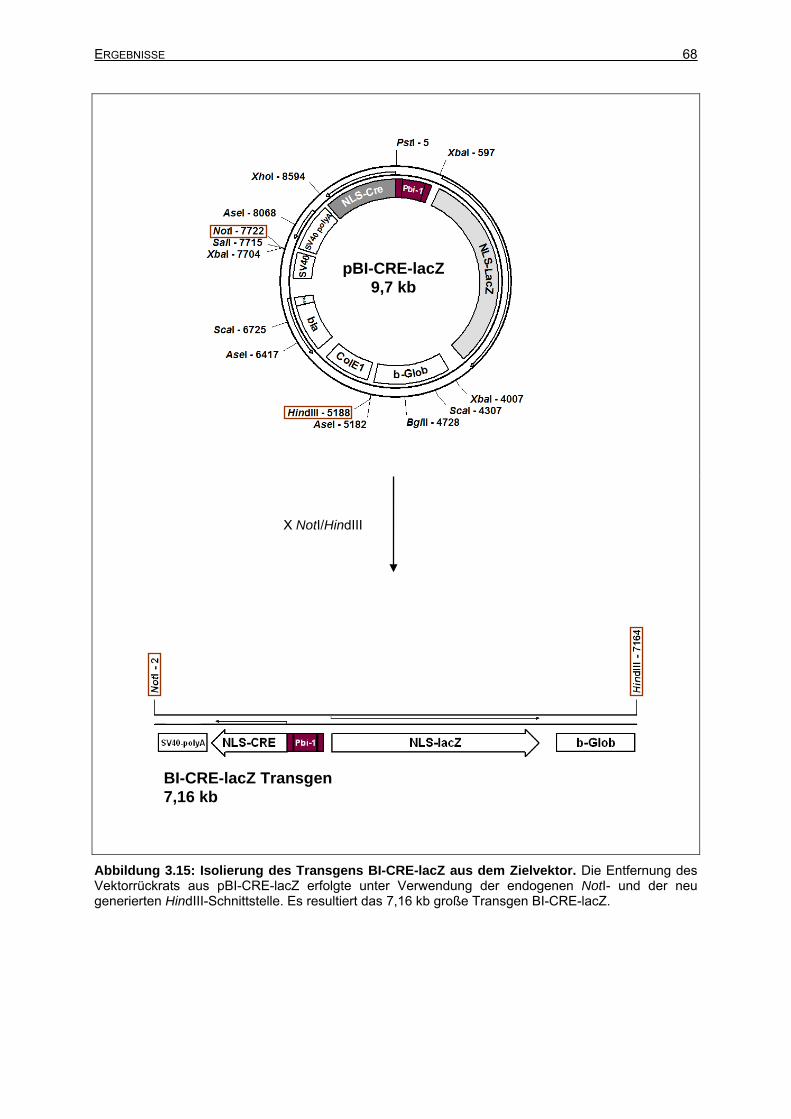
ERGEBNISSE
68
Abbildung 3.15: Isolierung des Transgens BI-CRE-lacZ aus dem Zielvektor. Die Entfernung des Vektorrückrats aus pBI-CRE-lacZ erfolgte unter Verwendung der endogenen NotI- und der neu generierten HindIII-Schnittstelle. Es resultiert das 7,16 kb große Transgen BI-CRE-lacZ.
X NotI/HindIII
BI-CRE-lacZ Transgen 7,16 kb
pBI-CRE-lacZ 9,7 kb
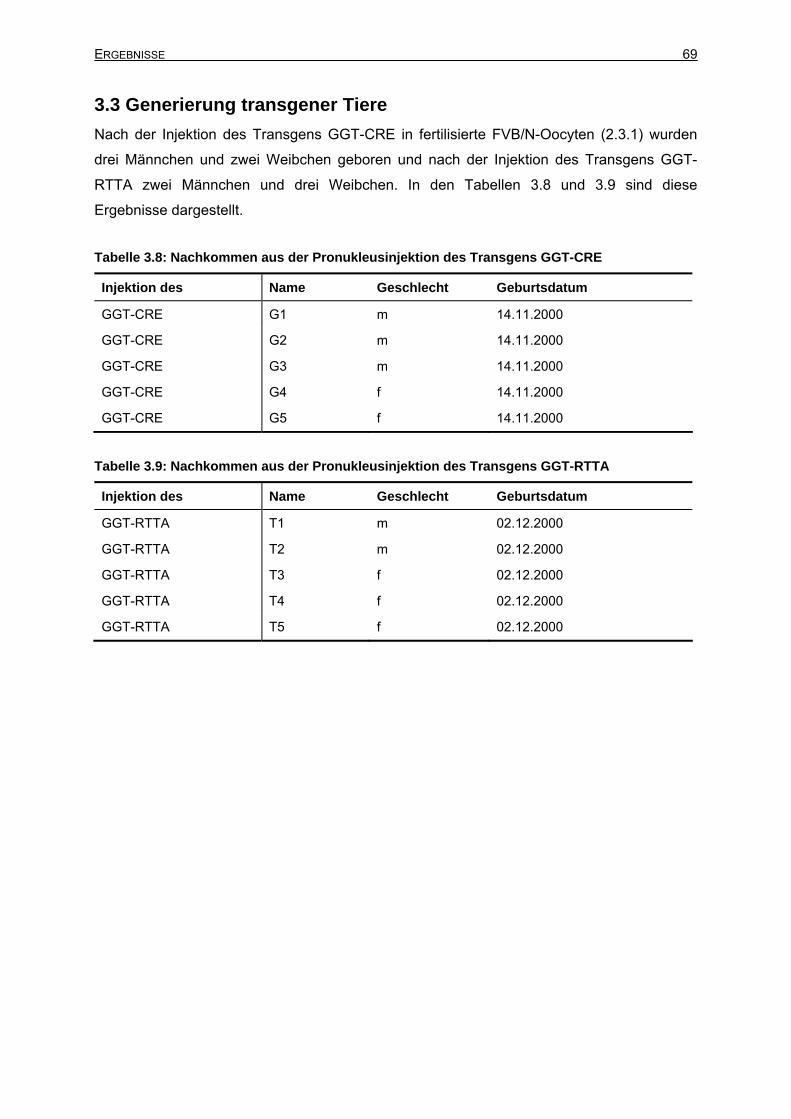
ERGEBNISSE
69
3.3 Generierung transgener Tiere Nach der Injektion des Transgens GGT-CRE in fertilisierte FVB/N-Oocyten (2.3.1) wurden
drei Männchen und zwei Weibchen geboren und nach der Injektion des Transgens GGT-
RTTA zwei Männchen und drei Weibchen. In den Tabellen 3.8 und 3.9 sind diese
Ergebnisse dargestellt.
Tabelle 3.8: Nachkommen aus der Pronukleusinjektion des Transgens GGT-CRE
Injektion des Name Geschlecht Geburtsdatum
GGT-CRE G1 m 14.11.2000 GGT-CRE G2 m 14.11.2000 GGT-CRE G3 m 14.11.2000 GGT-CRE G4 f 14.11.2000 GGT-CRE G5 f 14.11.2000
Tabelle 3.9: Nachkommen aus der Pronukleusinjektion des Transgens GGT-RTTA
Injektion des Name Geschlecht Geburtsdatum
GGT-RTTA T1 m 02.12.2000
GGT-RTTA T2 m 02.12.2000
GGT-RTTA T3 f 02.12.2000
GGT-RTTA T4 f 02.12.2000
GGT-RTTA T5 f 02.12.2000
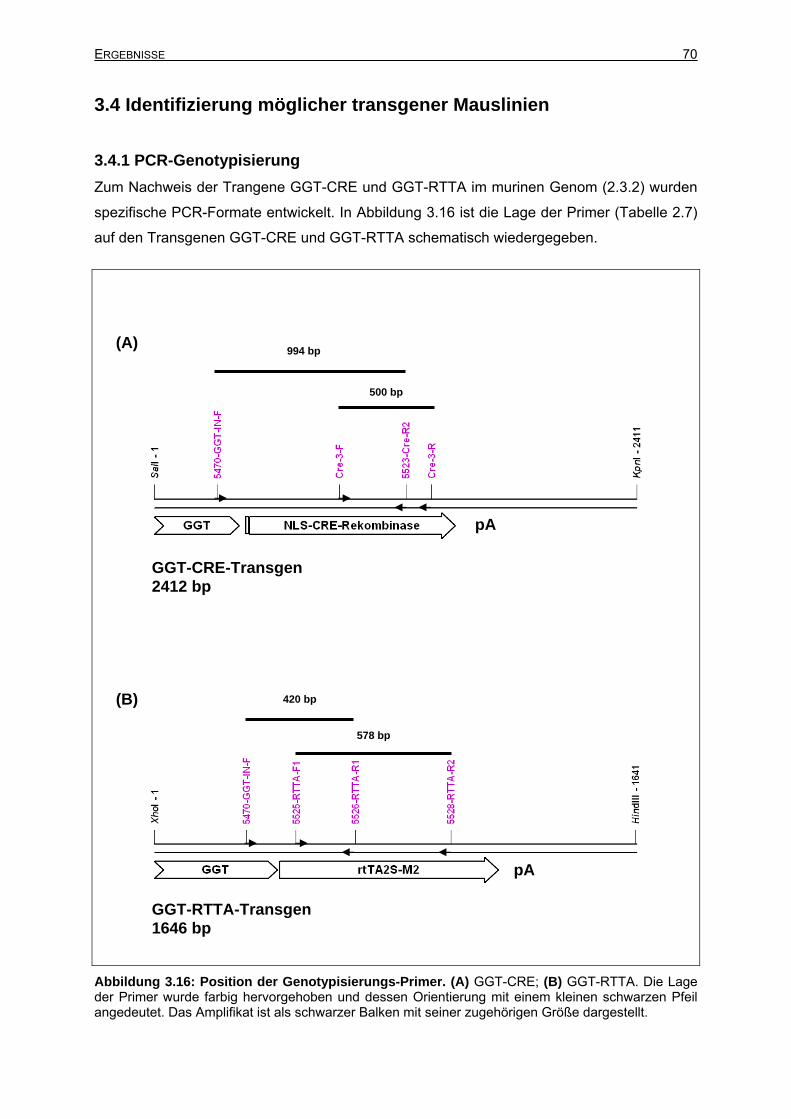
ERGEBNISSE
70
3.4 Identifizierung möglicher transgener Mauslinien
3.4.1 PCR-Genotypisierung Zum Nachweis der Trangene GGT-CRE und GGT-RTTA im murinen Genom (2.3.2) wurden
spezifische PCR-Formate entwickelt. In Abbildung 3.16 ist die Lage der Primer (Tabelle 2.7)
auf den Transgenen GGT-CRE und GGT-RTTA schematisch wiedergegeben.
Abbildung 3.16: Position der Genotypisierungs-Primer. (A) GGT-CRE; (B) GGT-RTTA. Die Lage der Primer wurde farbig hervorgehoben und dessen Orientierung mit einem kleinen schwarzen Pfeil angedeutet. Das Amplifikat ist als schwarzer Balken mit seiner zugehörigen Größe dargestellt.
994 bp
500 bp
pA
420 bp
578 bp
pA
GGT-CRE-Transgen 2412 bp
GGT-RTTA-Transgen 1646 bp
(A)
(B)

ERGEBNISSE
71
Nach der PCR (2.2.11) zur Genotypisierung potentieller GGT-CRE-transgener Tiere wurden
die resultierenden Amplifikate gelektrophoretisch aufgetrennt (2.2.7). Die Ergebnisse können
der Abbildung 3.17 entnommen werden. Als Kontrolle zur Spezifität der verwendeten PCR-
Primer wurde auch genomische DNA aus nicht transgenen FVB/N Mäusen amplifiziert (WT).
Die Zielvektoren pGGT-CRE bzw. pGGT-RTTA dienten als Positivkontrollen. Eine distinkte
PCR-Bande in Höhe von 994 bp ist nur beim Weibchen G5 zu erkennen. Bei den
Geschwistern G1, G2, G3 und G4 konnte kein Transgen nachgewiesen werden. Wie
erwartet liegt nach Amplifikation der genomischen DNA vom Wildtyp (WT) keine Bande vor.
Anders bei der vektoriellen DNA, wo eine sehr starke Bande zu erkennen ist. Das Transgen
GGT-RTTA konnte bei den Nachkommen T1-T5 trotz mehrmaliger PCR-Ansätze nicht
nachgewiesen werden.
Genomische PCR; Primer 5470-GGT-IN-F/5523-Cre-R2
M1 G1 G2 G3 G4 G5 Wt V M2
Abbildung 3.17: Genomische PCR unter Verwendung der Primer 5470-GGT-IN-F/5523-Cre-R2. G1-G5: Genomische DNA aus G1-G5; Wt: Genomische DNA aus FVB/N-Mäusen; V: pGGT-CRE; M1: GeneRuler™ 1 kb DNA Ladder (Fermentas); M2: 100 bp-DNA-Ladder (Invitrogen); Der Pfeil zeigt auf das ca. 0,9 kb große PCR-Produkt.
ca. 0.9 kb

ERGEBNISSE
72
3.4.2 Southern-Analysen
Um die PCR-Resultate der Genotypisierung von GGT-CRE zu verifizieren erfolgte ein
genomischer Southern-Blot mit der Sonde S1 (2.6.2). Dabei handelt es sich um ein 1,1 kb
großes BglII/EcoRI-Fragment isoliert aus dem Zielvektor pGGT-CRE (Abbildung 3.18).
Innerhalb dieses Fragmentes liegt der gesamte kodierende Bereich der Cre-Rekombinase.
Nach radioaktiver Markierung mit dem Isotop 32P wurde die Sonde S1 für die Hybridisierung
der mit dem Restriktionsenzym HincII verdauten genomischen DNA eingesetzt. Die genaue
Position der Sonde S1 im Transgen GGT-CRE zeigt die Abbildung 3.19 auf.
M 1
Abbildung 3.18: Restriktion des Vektors pGGT-CRE (pGC) zur Isolation der Sonde S1. 1: pGC/BglII/EcoRI: 3117 bp + 2486 bp + 1129 bp (S1) + 442 bp M: GeneRuler™ 1 kb DNA Ladder (Fermentas)
Abbildung 3.19: Schematische Darstellung der Sonden-Position (S1) im GGT-CRE.
1129 bp Sonde S1
Sonde S1 (1129 bp)
1964 bp-HincII-Fragment
GGT-CRE-Transgen 2,4 kb

ERGEBNISSE
73
Für den Southern-Transfer wurde außer HincII-restringierter genomische DNA aus G1 bis
G5 auch HincII-restringierte DNA einer FVB/N-Maus (WT) zur Kontrolle aufgetragen. Nach
der Hybridisierung mit der S1-Sonde ist ein Hybridisierungssignal bei G5 auf der Höhe von
ca.1,9 kb deutlich zu erkennen. Dies bestätigt somit das PCR-Resultat für die G5-Maus. Das
Radiogramm ist in Abbildung 3.20 dargestellt.
Genomische Southern-Analyse; Sonde S1 WT G1 G2 G3 G4 G5
Abbildung 3.20: Southern-Analyse unter Verwendung der Sonde S1. WT: HincII-restringierte genomische DNA aus FVB/N-Mäusen G1-G5: HincII-restringierte genomische DNA aus G1-G5. Das Hybridisierungssignal wird mit einem Pfeil markiert.
ca. 1,9 kb

ERGEBNISSE
74
3.5 Generierung stabiler Linien Das Weibchen G5 wurde sowohl mit FVB/N Männchen als auch mit den Reportermäusen
ROSA26- bzw. Z/AP-Mäusen (2.3.4) verpaart. Für die Genotypisierung der F1-Generation
wurden sowohl PCR- als auch genomische Southern-Analysen durchgeführt. In der
Abbildung 3.21 sind die Ergebnisse der Genotypisierung für folgende Nachkommen
dargestellt: G16, G17, G22, G25, G26, G27 und GR13. Bei den Mäusen G16-G27 handelt es
sich um Nachkommen aus Kreuzungen mit FVB/N-Mäusen. GR13 resultierte aus der
Verpaarung von G5 mit einer ROSA26-Reportermaus. Es konnte sowohl durch PCR- als
auch durch Southern-Blot-Analysen das Transgen GGT-CRE bei den Nachkommen
nachgewiesen werden. Somit zeigte sich, dass das Weibchen G5 sein Transgen GGT-CRE
an die Nachkommen weitergeben kann. Abbildung 3.21 stellt parallel zum PCR-Resultat das
Hybridisierungsergebnis dar.
G5 G16 G17 G25 G27 M
G5 GR13 G17 G22 G26
Abbildung 3.21: Ergebnisse der genomischen PCR und der Southern-Analysen A: Genomische PCR unter Verwendung der Primer Cre-3-F/Cre-3-R. Der Pfeil zeigt auf das 0,5 kb große PCR-Produkt B: Southern-Transfer-Analyse der GGT-CRE-Mäuse zur Verifizierung der PCR Resultate. Der Nachweis erfolgte nach Restriktion der DNA mit HincII und Hybridisierung mit der Sonde S1. Das Hybridisierungssignal wird mit einem Pfeil markiert. M: GeneRuler™ 1 kb DNA Ladder (Fermentas)
ca. 0,5 kb
ca. 1,9 kb
(A) Genomische PCR; Primer Cre-3-F/Cre-3-R
(B) Genomische Southern-Analyse; Sonde S1

ERGEBNISSE
75
3.6 Das Expressionsprofil des murinen GGT-Promotors Die transgene Maus G5 trägt das DNA-Konstrukt GGT-CRE (Abbildung 3.6), welches eine
346 bp-große Region des murinen GGT-Promotors (Sepulveda et al., 1997) enthält.
Stromabwärts liegt unter dessen Kontrolle der kodierende Bereich der P1-Cre-Rekombinase
mit vorgeschaltenen Kern-Lokalisations-Signal (NLS). Dieses Signal führt zur Translokation
des Cre-Proteins vom Cytoplasma in den Nukleus. Es ist insofern von signifikanter
Bedeutung, da es sich bei der Cre-Rekombinase um ein DNA-Bindeprotein handelt. Die
gewebespezifische Expression als auch die enzymatische Rekombinationsaktivität des Cre-
Proteins wurde experimentell nachgewiesen. Zu diesem Zweck erfolgte die
Charakterisierung der Cre-vermittelten Rekombination in Reportermauslinien. Das zeitliche
und örtliche Expressionsmuster der Cre-Rekombinase, sowie ihre katalytische Aktivität
wurde sowohl mittels Z/AP- (Lobe et al., 1999) als auch mittels ROSA26 -Reportermäusen
(Soriano, 1999) aufgezeigt. Zur besseren Übersicht ist das Paarungsschema der Etablierung
und der Analyse doppeltransgener GGT-CRE+/ZAP+- und GGT-CRE+/ROSA26+-Mäuse
sehr anschaulich in der Abbildung 2.2 dargestellt.
3.6.1 Untersuchungen der Cre-Expression mittels Z/AP-Reportermäuse Im folgenden Abschnitt werden die Ergebnisse der Cre-Rekombinase-Expression bei adulten
GGT-CRE-Mäusen durch Verpaarung mit der Z/AP-Reportermauslinie vorgestellt.
3.6.1.1 Verpaarung der GGT-CRE-Mäuse mit Z/AP-Reportermäuse Die Untersuchung des Expressionsprofils der Cre-Rekombinase, wie auch ihre katalytischen
Aktivität in vivo erfolgte zunächst mit der Z/AP-Reportermaus. Diese Z/AP-Mauslinie (Lobe et
al., 1999) exprimiert lacZ konstitutiv und annähernd ubiquitär unter Kontrolle des CAGG
Promotors (Niwa et al.; 1991). Dabei liegt die Expression sowohl embryonal als auch adult
weit verbreitet vor. Das lacZ-Reportergen wird von loxP-Erkennungssequenzen flankiert und
kann somit durch die katalytische Aktivität der Cre-Rekombinase deletiert werden. Ferner
liegen im 3´-Bereich drei polyA-Signale vor (Zinyk et al., 1998), damit eine unbeabsichtigte
Transkription des weiter stromabwärts gelegenen Gens der humanen plazentalen
alkalischen Phosphatase (hPLAP; DePrimo et al., 1996; Millan, 1986) unterbleibt. Eine von
der Cre-Rekombinase erfolgreich vermittelte Rekombination kann somit histochemisch
nachgewiesen werden. Unrekombinierte Zellen und Gewebe sind lacZ-positiv.
Rekombinierte Bereiche sind hingegen β-Galactosidase-negativ. Sie exprimieren hPLAP,
welches ebenfalls durch histochemische Methoden detektiert werden kann. Zum Nachweis
der Cre-Rekombinase-Expression sind GGT-CRE- mit Z/AP-Reportermäusen verpaart
worden. In der Abbildung 3.22 wird zum besseren Verständnis der Cre/loxP-
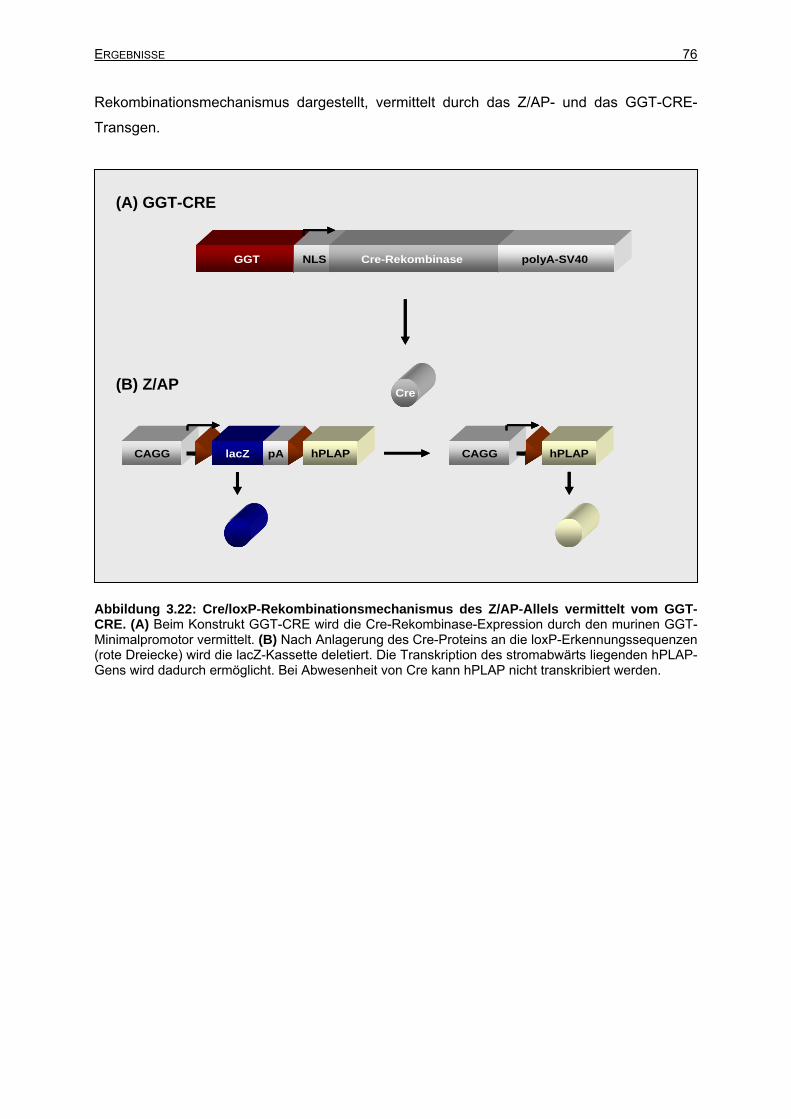
ERGEBNISSE
76
Rekombinationsmechanismus dargestellt, vermittelt durch das Z/AP- und das GGT-CRE-
Transgen.
Cre
GGT NLS Cre-Rekombinase polyA-SV40
lacZ pA hPLAPCAGG CAGG hPLAP
Cre
GGT NLS Cre-Rekombinase polyA-SV40
lacZ pA hPLAPCAGG CAGG hPLAP
Cre
GGT NLS Cre-Rekombinase polyA-SV40
lacZ pA hPLAPCAGG CAGG hPLAP
Abbildung 3.22: Cre/loxP-Rekombinationsmechanismus des Z/AP-Allels vermittelt vom GGT-CRE. (A) Beim Konstrukt GGT-CRE wird die Cre-Rekombinase-Expression durch den murinen GGT-Minimalpromotor vermittelt. (B) Nach Anlagerung des Cre-Proteins an die loxP-Erkennungssequenzen (rote Dreiecke) wird die lacZ-Kassette deletiert. Die Transkription des stromabwärts liegenden hPLAP-Gens wird dadurch ermöglicht. Bei Abwesenheit von Cre kann hPLAP nicht transkribiert werden.
(A) GGT-CRE
(B) Z/AP

ERGEBNISSE
77
3.6.1.2 Charakterisierung der doppeltransgenen GGT-CRE+/Z/AP+-Mäuse Nach Verpaarung von heterozygoten Z/AP- mit heterozygoten GGT-CRE-Mäusen wurde
zunächst der Genotyp bestimmt. Die Genotypisierung für das Z/AP-Allel erfolgte mittels einer
spezifischen lacZ-PCR unter Verwendung der Primer 5573-lacZ-F/5574-lacZ-R (Tabelle 2.7).
Dabei wurde ein PCR-Produkt der Größe 310 bp amplifiziert. Das Transgen GGT-CRE
wurde - wie bereits im Abschnitt 3.4.1 beschrieben - sowohl durch PCR unter Verwendung
der Primer Cre-3-F/Cre-3-R bzw. 5470-GGT-IN-F/5523-Cre-R2, als auch mittels Southern-
Blot-Analysen (Sonde S1) nachgewiesen. Nach der Genotypisierung sind doppeltransgene
GGT-CRE+/Z/AP+-Mäuse und zur Kontrolle GGT-CRE-/Z/AP+-Mäuse experimentell
charakterisiert worden. Um das Expressionsprofil des GGT-Promotors und die katalytische
Aktivität des Cre-Enzyms bei adulten GGT-CRE+/Z/AP+-Mauslinien zu analysieren, wurden
zunächst Gefrierschnitte von GGT-CRE+/Z/AP+ - und als Kontrolle von GGT-CRE-/Z/AP+-
Mäusen erstellt. Zum Nachweis des hitzestabilen hPLAP-Proteins erfolgten
enzymhistochemische Färbungen, durch Zugabe eines chromogenen Substrats der
alkalischen Phosphatase (AP; 2.4.2.2).
3.6.1.3 Cre-Expression in adulten GGT-CRE-Mäusen Die hPLAP-Detektion bei den Doppeltransgenen ist nur in den Zellen möglich, in denen eine
Cre/loxP-Rekombination stattgefunden hat. Bei einer vorliegenden hPLAP-Expression
kommt es nach Zugabe des chromogenen AP-Substrates zu einer Blaufärbung. Das Z/AP-
Allel vermittelt im unrekombinierten Fall keine ungewünschte Expression der hitzestabilen
hPLAP, weshalb eine Blaufärbung unterbleibt. Nach Zugabe des AP-Substrates konnte,
außer bei den Nieren, bei keinem weiteren entnommenen Organ (Gehirn, Leber, Milz,
Lunge, Herz, Pankreas, Haut, Muskelgewebe, Ovar/Testis) eine Blaufärbung festgestellt
werden (Abbildungen nicht gezeigt). Abbildung 3.23 zeigt die AP-Färbung von
Gefrierschnitten einer Niere, welche aus einem 6 Wochen alten GGT-CRE+/Z/AP+-Weibchen
stammen. Bei der mikroskopischen Darstellung des Gefrierschnittes (Abbildung 3.23 A: 2,5x)
ist eine Blaufärbung an der äußeren Nierenregion deutlich zu erkennen. Die AP-Färbung
erstreckt sich von der Nierenrinde bis hin zum Außenstreifen der Nierenmarkregion. In
diesem Abschnitt, vor allem im Rindengewebe der Niere liegt das Hauptstück des
proximalen Tubulus als proximales Konvolut (Pars convoluta) vor. Bei 40-facher
Vergrößerung des Nieren-Kortex sind die blau gefärbten proximalen Tubuluszellen
lichtmikroskopisch zu erkennen (Abbildung 3.23 B: 40x). Die Abbildungen 3.2.3 D und E
stellen Gefrierpräparate aus GGT-CRE-/Z/AP+ Kontrollnieren dar. Beim Fehlen der Cre-
Rekombinase Aktivität kann keine alkalische Phosphatase Färbung beobachtet werden.
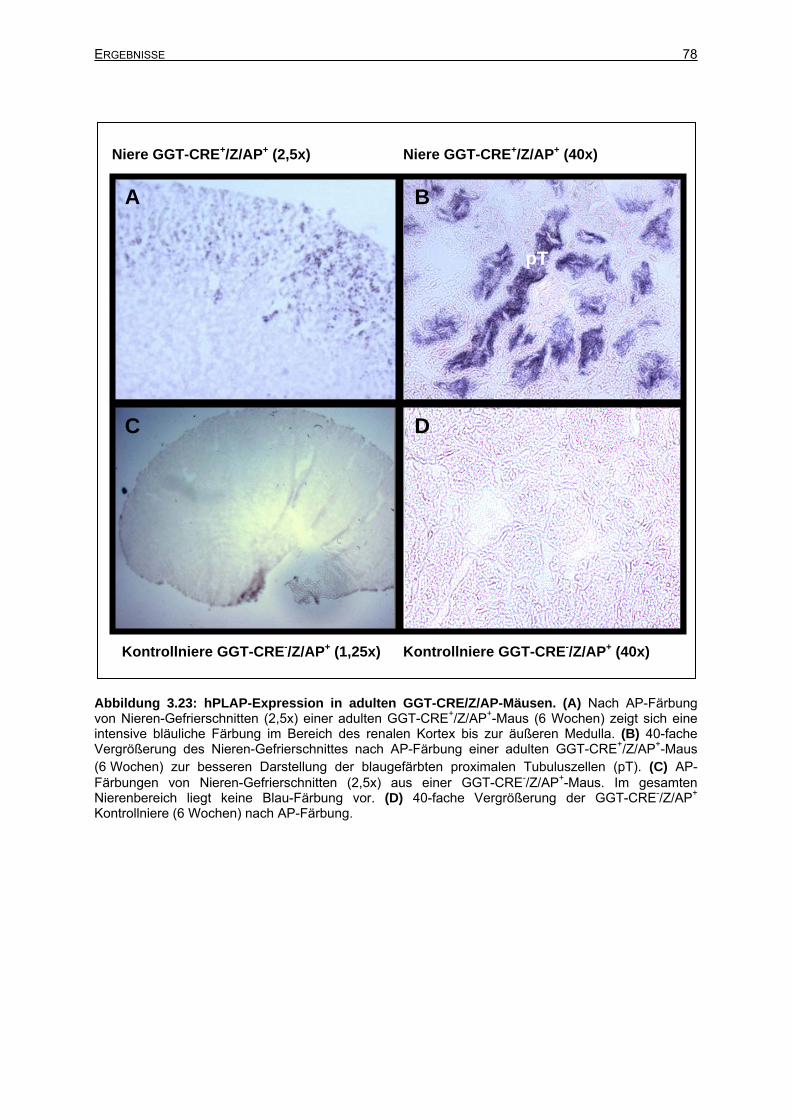
ERGEBNISSE
78
Abbildung 3.23: hPLAP-Expression in adulten GGT-CRE/Z/AP-Mäusen. (A) Nach AP-Färbung von Nieren-Gefrierschnitten (2,5x) einer adulten GGT-CRE+/Z/AP+-Maus (6 Wochen) zeigt sich eine intensive bläuliche Färbung im Bereich des renalen Kortex bis zur äußeren Medulla. (B) 40-fache Vergrößerung des Nieren-Gefrierschnittes nach AP-Färbung einer adulten GGT-CRE+/Z/AP+-Maus (6 Wochen) zur besseren Darstellung der blaugefärbten proximalen Tubuluszellen (pT). (C) AP-Färbungen von Nieren-Gefrierschnitten (2,5x) aus einer GGT-CRE-/Z/AP+-Maus. Im gesamten Nierenbereich liegt keine Blau-Färbung vor. (D) 40-fache Vergrößerung der GGT-CRE-/Z/AP+ Kontrollniere (6 Wochen) nach AP-Färbung.
Niere GGT-CRE+/Z/AP+ (2,5x) Niere GGT-CRE+/Z/AP+ (40x)
pT
A B
D
Kontrollniere GGT-CRE-/Z/AP+ (1,25x) Kontrollniere GGT-CRE-/Z/AP+ (40x)
DC D

ERGEBNISSE
79
3.6.2 Untersuchungen der Cre-Expression mittels ROSA26-Reportermäuse Im folgenden Abschnitt werden die Ergebnisse der Cre-Rekombinase-Expression vorgestellt,
nach Verpaarung der GGT-CRE-Mäuse mit der ROSA26-Reportermauslinie. Folgende
Stadien werden erläutert: E12,5; E14,5; E15,5; E17,5; neugeboren und adult.
3.6.2.1 Verpaarung der GGT-CRE-Mäuse mit ROSA26-Reportermäuse ROSA26-Reportermäuse wurden mit den in dieser Arbeit generierten transgenen GGT-CRE-
Mäusen verpaart (2.3.4.2). Beim Reporterstamm ROSA26 (Soriano, 1999) liegt eine gefloxte
Neomycinkassette mit einer vierfachen polyA-Sequenz und das stromabwärts gelegene
Reportergen lacZ vor. Erst durch die Anbindung des Cre-Proteins an die loxP-
Erkennungssequenzen kann diese Terminationskassette entfernt und folgerichtig eine lacZ-
Expression initiiert werden. Demzufolge liegt bei diesen ROSA26-Reportermäusen eine lacZ-
Expression nur in den Zellen vor, in denen die Cre-Rekombinase katalytisch aktiv ist. Bei
Abwesenheit des Cre-Enzyms unterbleibt die Transkription des lacZ-Gens, wodurch nach
enzymhistochemischen Färbereaktionen eine Unterscheidung zwischen Cre-exprimierenden
und nicht-exprimierenden Zellen ermöglicht wird. Mittels dieser Unterscheidung kann das
Expressionsprofil des murinen GGT-Minimalpromotors durch Kreuzung der GGT-CRE- mit
ROSA26-Mauslinien charakterisiert werden.
3.6.2.2 Charakterisierung der GGT-CRE+/ROSA26+-Mauslinien Homozygote und heterozygote ROSA26-Mäuse können, anders als bei den Z/AP-
Reportermäusen, durch PCR-Analysen unterschieden werden. Bei der PCR-Genotypisierung
des ROSA26-Allels wurde für die Amplifikation des mutanten Allels die Primer 5588/5589
(250 bp Amplifikat) und für das Wildtyp-Allel das Oligonukleotidpaar 5588 und 5590 (500 bp
Amplifikat) verwendet (Tabelle 2.7).
Zur Generierung doppeltransgener GGT-CRE+/ROSA26+-Mäuse sind demzufolge
homozygote ROSA26+- mit heterozygoten GGT-CRE+/--Mäusen verpaart worden. Ziel dieser
Analysen war es das Expressionsprofil des GGT-Promotors und die Rekombinationsfähigkeit
der stromabwärts gelegenen Cre-Rekombinase, sowohl in den embryonalen als auch in den
adulten murinen Stadien zu analysieren. Bei den GGT-CRE+/ROSA26+-Tieren kann die
Aktivität der Cre-Rekombinase durch das Vorliegen des lacZ-Reportergens
enzymhistochemisch visualisiert werden. In den speziellen Zellen, wo das Cre-Enzym
exprimiert wird, findet eine enzymatische Rekombination an den loxP-Stellen statt und die
gefloxte Terminationskassette wird folglich deletiert. Es erfolgt die Transkription des
stromabwärts gelegenen lacZ-Gens. Falls die Rekombination durch die Abwesenheit des
Cre-Enzyms unterbleibt behindert die Terminationskassette das Ablesen des Reportergens
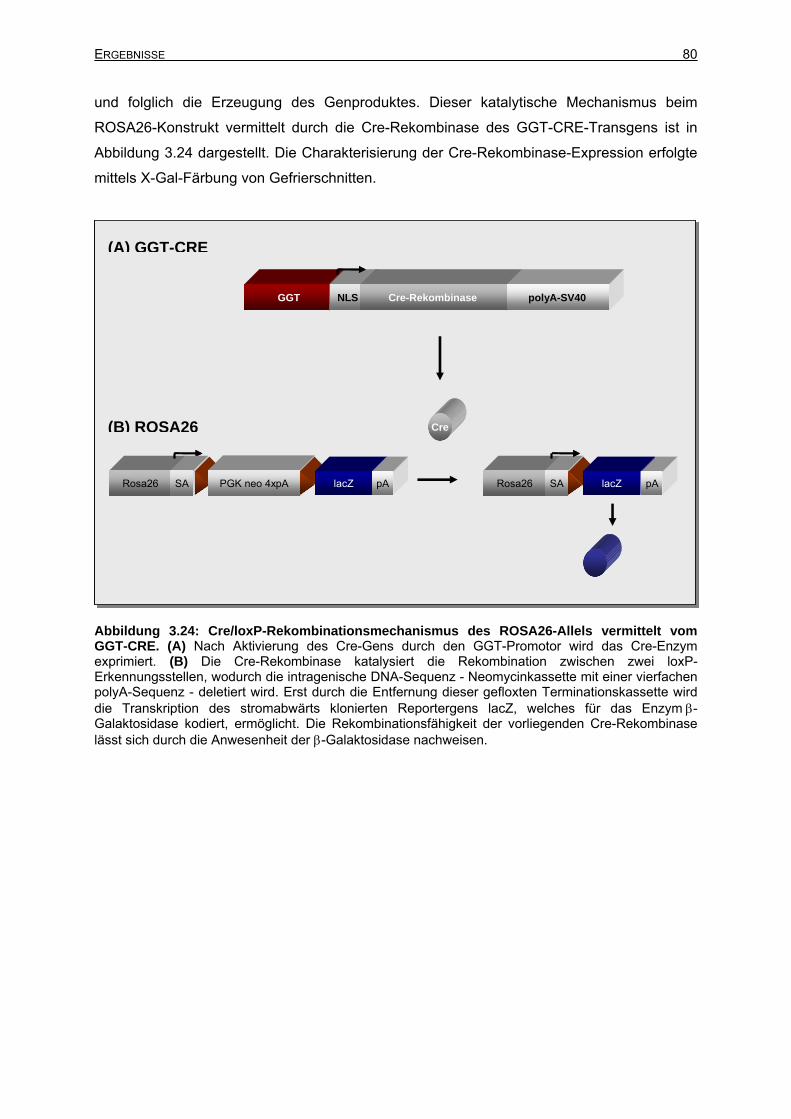
ERGEBNISSE
80
und folglich die Erzeugung des Genproduktes. Dieser katalytische Mechanismus beim
ROSA26-Konstrukt vermittelt durch die Cre-Rekombinase des GGT-CRE-Transgens ist in
Abbildung 3.24 dargestellt. Die Charakterisierung der Cre-Rekombinase-Expression erfolgte
mittels X-Gal-Färbung von Gefrierschnitten.
Rosa26Rosa26
Cre
GGT NLS Cre-Rekombinase polyA-SV40
SA PGK neo 4xpA lacZ pA SA lacZ pARosa26Rosa26
Cre
GGT NLS Cre-Rekombinase polyA-SV40
SA PGK neo 4xpA lacZ pA SA lacZ pARosa26Rosa26
Cre
GGT NLS Cre-Rekombinase polyA-SV40
SA PGK neo 4xpA lacZ pA SA lacZ pARosa26Rosa26
Cre
GGT NLS Cre-Rekombinase polyA-SV40
SA PGK neo 4xpA lacZ pA SA lacZ pA
Abbildung 3.24: Cre/loxP-Rekombinationsmechanismus des ROSA26-Allels vermittelt vom GGT-CRE. (A) Nach Aktivierung des Cre-Gens durch den GGT-Promotor wird das Cre-Enzym exprimiert. (B) Die Cre-Rekombinase katalysiert die Rekombination zwischen zwei loxP-Erkennungsstellen, wodurch die intragenische DNA-Sequenz - Neomycinkassette mit einer vierfachen polyA-Sequenz - deletiert wird. Erst durch die Entfernung dieser gefloxten Terminationskassette wird die Transkription des stromabwärts klonierten Reportergens lacZ, welches für das Enzym β-Galaktosidase kodiert, ermöglicht. Die Rekombinationsfähigkeit der vorliegenden Cre-Rekombinase lässt sich durch die Anwesenheit der β-Galaktosidase nachweisen.
(A) GGT-CRE
(B) ROSA26
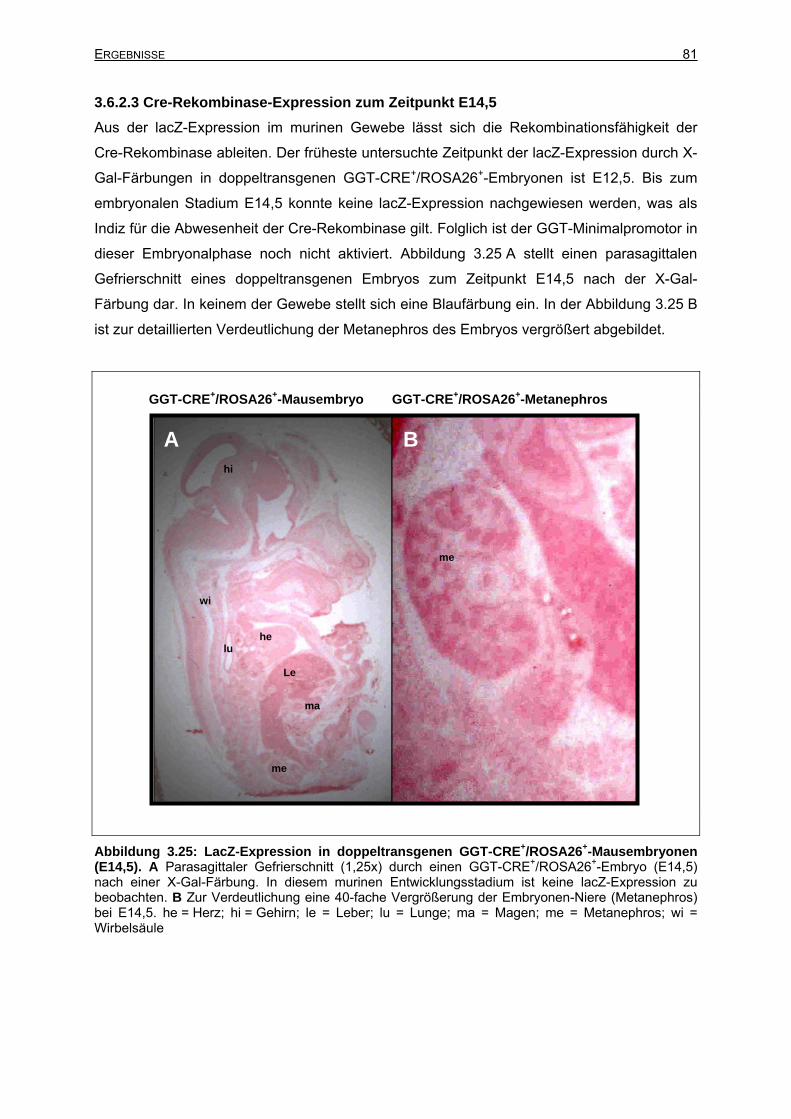
ERGEBNISSE
81
3.6.2.3 Cre-Rekombinase-Expression zum Zeitpunkt E14,5 Aus der lacZ-Expression im murinen Gewebe lässt sich die Rekombinationsfähigkeit der
Cre-Rekombinase ableiten. Der früheste untersuchte Zeitpunkt der lacZ-Expression durch X-
Gal-Färbungen in doppeltransgenen GGT-CRE+/ROSA26+-Embryonen ist E12,5. Bis zum
embryonalen Stadium E14,5 konnte keine lacZ-Expression nachgewiesen werden, was als
Indiz für die Abwesenheit der Cre-Rekombinase gilt. Folglich ist der GGT-Minimalpromotor in
dieser Embryonalphase noch nicht aktiviert. Abbildung 3.25 A stellt einen parasagittalen
Gefrierschnitt eines doppeltransgenen Embryos zum Zeitpunkt E14,5 nach der X-Gal-
Färbung dar. In keinem der Gewebe stellt sich eine Blaufärbung ein. In der Abbildung 3.25 B
ist zur detaillierten Verdeutlichung der Metanephros des Embryos vergrößert abgebildet.
GGT-CRE+/ROSA26+-Mausembryo GGT-CRE+/ROSA26+-Metanephros
Abbildung 3.25: LacZ-Expression in doppeltransgenen GGT-CRE+/ROSA26+-Mausembryonen (E14,5). A Parasagittaler Gefrierschnitt (1,25x) durch einen GGT-CRE+/ROSA26+-Embryo (E14,5) nach einer X-Gal-Färbung. In diesem murinen Entwicklungsstadium ist keine lacZ-Expression zu beobachten. B Zur Verdeutlichung eine 40-fache Vergrößerung der Embryonen-Niere (Metanephros) bei E14,5. he = Herz; hi = Gehirn; le = Leber; lu = Lunge; ma = Magen; me = Metanephros; wi = Wirbelsäule
A B
Le
me
hi
ma
wi
lu he
me

ERGEBNISSE
82
3.6.2.4 Cre-Rekombinase-Expression zum Zeitpunkt E15,5 Nach der X-Gal-Färbung von Gefrierschnitten eines GGT-CRE+/ROSA26+-Mausembryos
konnte erstmalig zum Zeitpunkt E15,5 eine leichte Blaufärbung in der embryonalen Niere
(Metanephros) und dem Darm beobachtet werden. Im Darm liegende Bakterien weisen
endogene β-Galaktosidasen auf, welche das X-Gal mit hydrolisieren. Kein weiteres
embryonales Gewebe weist eine lacZ-Expression auf. In der Abbildung 3.26 sind
Gefrierschnitte sowohl aus Cre-/ROSA26+-Kontrollembryonen (Abbildung A-D), als auch aus
doppeltransgenen GGT-CRE+/ROSA26+-Mausembryonen (Abbildung E-F) in
unterschiedlichen mikroskopischen Vergrößerungen dokumentiert worden. Bei den
Kontrollembryonen lässt sich eine Blaufärbung nach der X-Gal-Färbung nur im Darm
visualisieren. Kein weiteres Gewebe konnte angefärbt werden. In den Abbildungen 3.26 B, C
und D sind Gefrierschnitte embryonaler Nieren ohne nachweisbare lacZ-Expression zu
sehen. Zur besseren mikroskopischen Visualisierung ist in Abbildung 3.26 D eine
Kontrollniere vergrößert (10x) dargestellt. Verglichen mit den Kontrollembryonen weisen
doppeltransgene GGT-CRE+/ROSA26+-Embryonen vereinzelte leichte Blaufärbungen in der
äußeren renalen Medulla-Region auf (Abbildungen 3.26 E-F). Schwarze Pfeile zeigen auf die
β-Galaktosidase-exprimierenden blau gefärbten renale Regionen. Dieses Ergebnis steht für
eine anfängliche gewebespezifische Cre/loxP-Rekombination eines enzymatisch aktiven
Cre-Rekombinase-Proteins. Ab dem embryonalen Zeitpunkt E15,5 wird nach diesen
vorliegenden Ergebnissen der gewebespezifische Minimalpromotor des Transgens GGT-
CRE aktiviert. Folgerichtig setzt die Transkription der stromabwärts klonierten Cre-
Rekombinase setzt ein. Durch das Vorliegen des ROSA26-Allels kann die früheste
katalytische Cre-Rekombinase Aktivität zum Zeitpunkt E15,5 in der embryonalen Niere
erfasst werden.
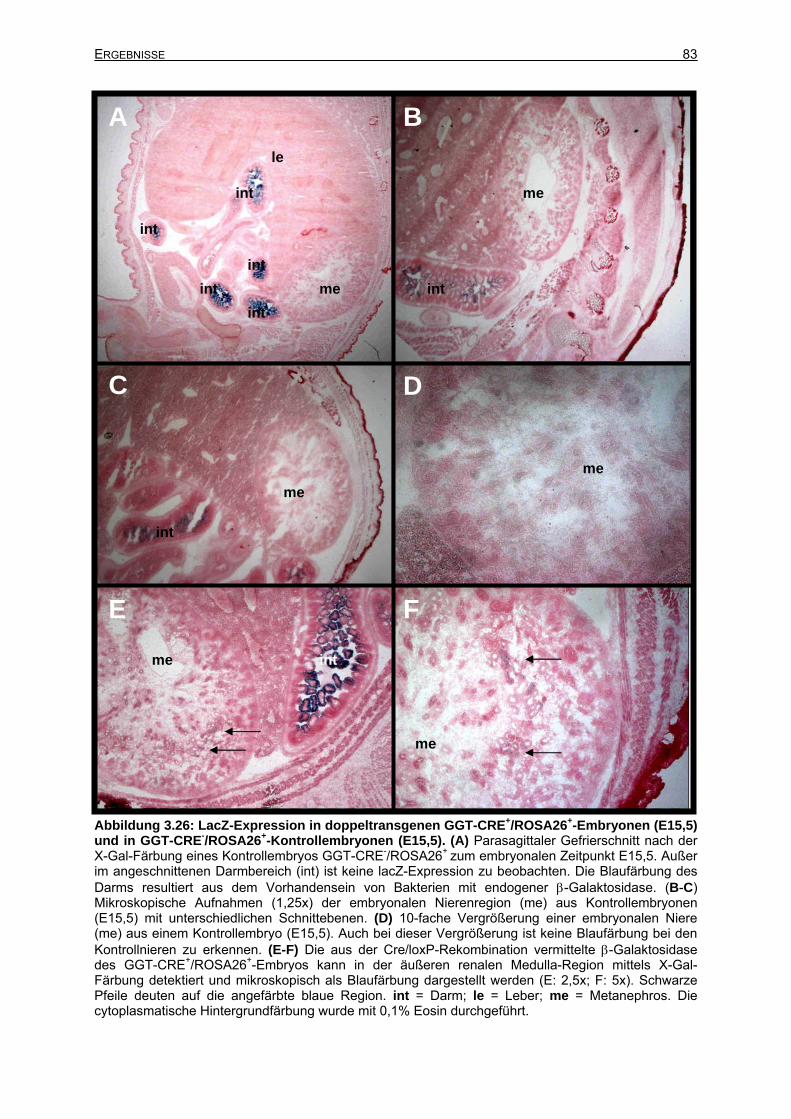
ERGEBNISSE
83
Abbildung 3.26: LacZ-Expression in doppeltransgenen GGT-CRE+/ROSA26+-Embryonen (E15,5) und in GGT-CRE-/ROSA26+-Kontrollembryonen (E15,5). (A) Parasagittaler Gefrierschnitt nach der X-Gal-Färbung eines Kontrollembryos GGT-CRE-/ROSA26+ zum embryonalen Zeitpunkt E15,5. Außer im angeschnittenen Darmbereich (int) ist keine lacZ-Expression zu beobachten. Die Blaufärbung des Darms resultiert aus dem Vorhandensein von Bakterien mit endogener β-Galaktosidase. (B-C) Mikroskopische Aufnahmen (1,25x) der embryonalen Nierenregion (me) aus Kontrollembryonen (E15,5) mit unterschiedlichen Schnittebenen. (D) 10-fache Vergrößerung einer embryonalen Niere (me) aus einem Kontrollembryo (E15,5). Auch bei dieser Vergrößerung ist keine Blaufärbung bei den Kontrollnieren zu erkennen. (E-F) Die aus der Cre/loxP-Rekombination vermittelte β-Galaktosidase des GGT-CRE+/ROSA26+-Embryos kann in der äußeren renalen Medulla-Region mittels X-Gal-Färbung detektiert und mikroskopisch als Blaufärbung dargestellt werden (E: 2,5x; F: 5x). Schwarze Pfeile deuten auf die angefärbte blaue Region. int = Darm; le = Leber; me = Metanephros. Die cytoplasmatische Hintergrundfärbung wurde mit 0,1% Eosin durchgeführt.
A B
C D
E F
me
le
int
int int
int
int
me
int
int
me me
me
int
me

ERGEBNISSE
84
3.6.2.5 Cre-Rekombinase-Expression zum Zeitpunkt E17,5 Im embryonalen Zeitpunkt E17,5 zeigt sich bei den doppeltransgenen GGT-CRE+/ROSA26+-
Mäusen nach der X-Gal-Färbung eine vergleichsweise stärkere lacZ-Expression als zum
embryonalen Zeitpunkt E15,5. Auch während der Embryonalphase E17,5 ist bei den
Doppeltransgenen eine charakteristische Blau-Färbung in der äußeren Medulla der
Nierenregion und im Darmbereich zu erkennen. Dies bestätigt das Vorhandensein eines
katalytisch aktiven Cre-Enzyms im Metanephros, welches unter transkriptioneller Kontrolle
des murinen GGT-Minimalpromotors steht. Bei den GGT-CRE-/ROSA26+- Kontrollmäusen ist
die β-Galaktosidase außer in der angeschnittenen Darmregion nicht nachzuweisen. Diese
Ergebnisse lassen den Schluss zu, dass die Cre-Rekombinase bei den GGT-CRE-Mäusen
zum embryonalen Zeitpunkt E17,5 an katalytischer Aktivität gewonnen hat. In der Abbildung
3.27 werden mikroskopische Aufnahmen von X-Gal-gefärbten Gefrierschnitten aus
Kontrollembryonen GGT-CRE-/ROSA26+ (Abbildung A-C) und doppeltransgenen GGT-
CRE+/ROSA26+ (Abbildung D-H) im embryonalen Zeitpunkt E17,5 dargestellt.
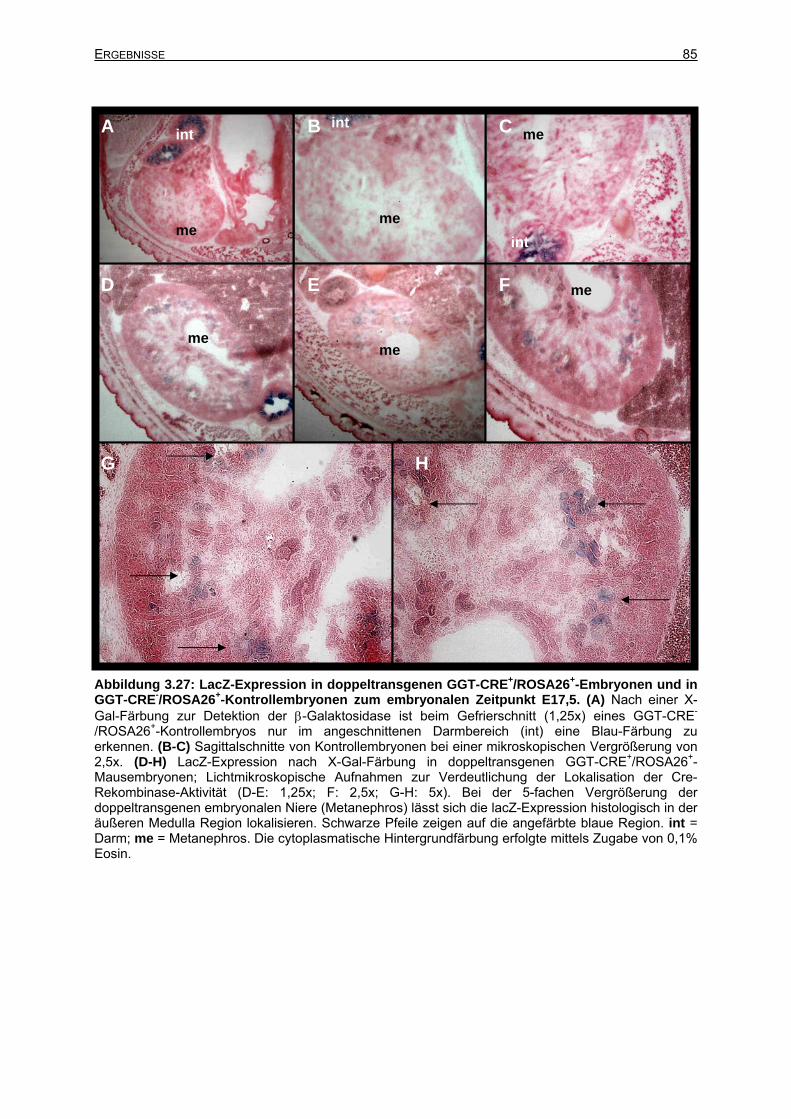
ERGEBNISSE
85
Abbildung 3.27: LacZ-Expression in doppeltransgenen GGT-CRE+/ROSA26+-Embryonen und in GGT-CRE-/ROSA26+-Kontrollembryonen zum embryonalen Zeitpunkt E17,5. (A) Nach einer X-Gal-Färbung zur Detektion der β-Galaktosidase ist beim Gefrierschnitt (1,25x) eines GGT-CRE-
/ROSA26+-Kontrollembryos nur im angeschnittenen Darmbereich (int) eine Blau-Färbung zu erkennen. (B-C) Sagittalschnitte von Kontrollembryonen bei einer mikroskopischen Vergrößerung von 2,5x. (D-H) LacZ-Expression nach X-Gal-Färbung in doppeltransgenen GGT-CRE+/ROSA26+-Mausembryonen; Lichtmikroskopische Aufnahmen zur Verdeutlichung der Lokalisation der Cre-Rekombinase-Aktivität (D-E: 1,25x; F: 2,5x; G-H: 5x). Bei der 5-fachen Vergrößerung der doppeltransgenen embryonalen Niere (Metanephros) lässt sich die lacZ-Expression histologisch in der äußeren Medulla Region lokalisieren. Schwarze Pfeile zeigen auf die angefärbte blaue Region. int = Darm; me = Metanephros. Die cytoplasmatische Hintergrundfärbung erfolgte mittels Zugabe von 0,1% Eosin.
E
A B C
D F
G H
int
int
int
me me
me
me me
me

ERGEBNISSE
86
3.6.2.6 Cre-Rekombinase-Expression bei neugeborenen Mäusen Bei den neugeborenen doppeltransgenen GGT-CRE+/ROSA26+-Mäusen zeichnet sich nach
X-Gal-Färbungen von Gesamt-Gefrierschnitten (Abbildung 3.28 D-F; H) eine signifikant
intensivere lacZ-Expression in der Niere ab, als zum embryonalen Entwicklungsstadium
E17,5. Ferner konnte erst zu diesem Entwicklungsstadium durch Lektin-markierte
Gefrierpräparate eine präzisere Aussage zur Lokalisation der Cre-Rekombinase-Expression
getroffen werden (Abbildung 3.29).
Um den lacZ-Expressionsort genau einzuordnen, welcher mit den Expressionsort der Cre-
Rekombinase bei doppeltransgenen GGT-CRE+/ROSA26+-Mäusen kongruent ist, wurden
Gefrierschnitte mit Lektin PHAE-TRITC aus Phaseolus vulgaris immunhistochemisch
markiert. Das Lektin PHAE-TRITC bindet spezifisch an Oberflächenproteine der proximalen
Tubuli (rot) und ermöglicht dadurch eine Differenzierung aller renalen Tubulusstrukturen.
Aufgrund der Tatsache, dass X-Gal-gefärbte Präparate mit Glutaraldehyd fixiert wurden,
konnten keine nachfolgenden immunhistochemischen Markierungen auf dem gleichen
Gefrierschnitt durchgeführt werden. Bei diesem Fixierungsmittel liegt bei
fluoreszenzmikroskopischer Untersuchung eine starke Eigen-Emission vor, was eine
Darstellung des Chromophors TRITC vom Lektin undurchführbar machte. Die Fixierung für
Präparate, die immunhistochemisch markiert wurden, erfolgte mittels Aceton. Aus diesem
Grund wurden parallele Gefrierpräparate zur Verdeutlichung der Expressionsorte verwendet.
Es zeigt sich sehr deutlich, dass es sich bei den blau gefärbten Segmenten, die aus der Cre-
vermittelten lacZ-Expression resultieren, um proximale Tubulusstrukturen handelt. Abbildung
3.29 stellt zwei parallele Nieren-Gefrierschnitte einer neonatalen GGT-CRE+/ROSA26+-Maus
dar: (A) immunfluorimetrische Aufnahme nach der Lektin-Markierung und (B) parallel dazu
nach der X-Gal-Färbung.
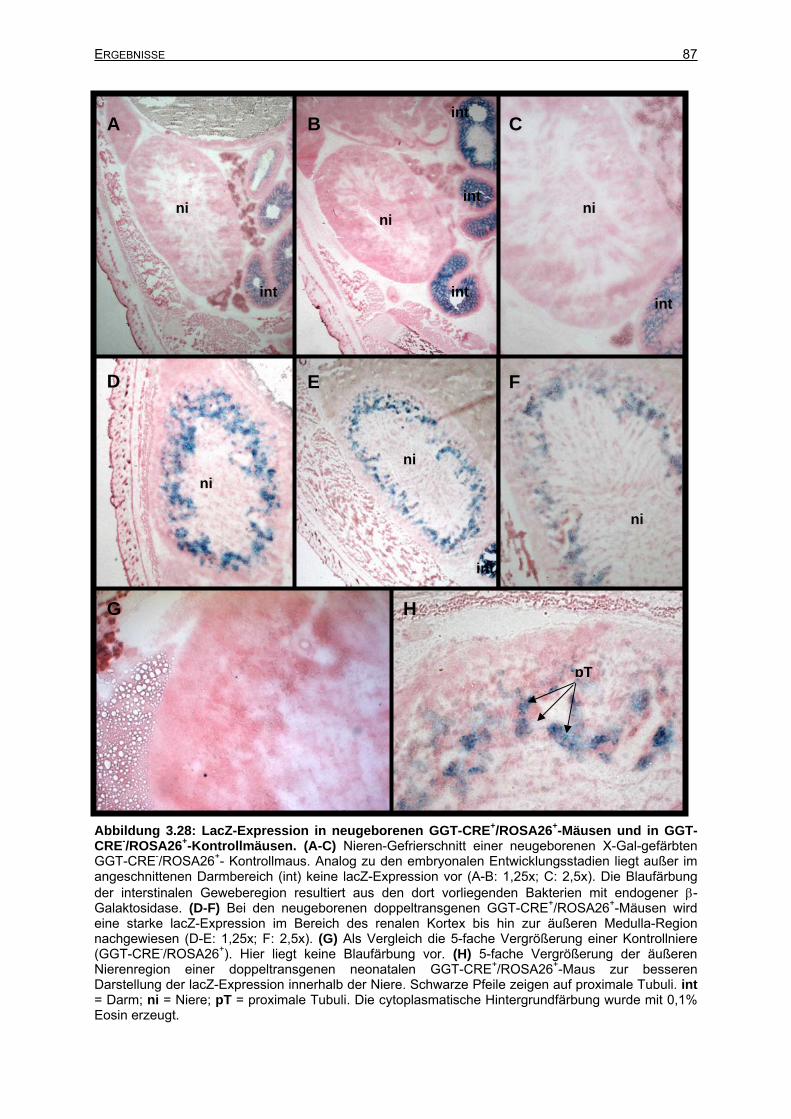
ERGEBNISSE
87
Abbildung 3.28: LacZ-Expression in neugeborenen GGT-CRE+/ROSA26+-Mäusen und in GGT-CRE-/ROSA26+-Kontrollmäusen. (A-C) Nieren-Gefrierschnitt einer neugeborenen X-Gal-gefärbten GGT-CRE-/ROSA26+- Kontrollmaus. Analog zu den embryonalen Entwicklungsstadien liegt außer im angeschnittenen Darmbereich (int) keine lacZ-Expression vor (A-B: 1,25x; C: 2,5x). Die Blaufärbung der interstinalen Geweberegion resultiert aus den dort vorliegenden Bakterien mit endogener β-Galaktosidase. (D-F) Bei den neugeborenen doppeltransgenen GGT-CRE+/ROSA26+-Mäusen wird eine starke lacZ-Expression im Bereich des renalen Kortex bis hin zur äußeren Medulla-Region nachgewiesen (D-E: 1,25x; F: 2,5x). (G) Als Vergleich die 5-fache Vergrößerung einer Kontrollniere (GGT-CRE-/ROSA26+). Hier liegt keine Blaufärbung vor. (H) 5-fache Vergrößerung der äußeren Nierenregion einer doppeltransgenen neonatalen GGT-CRE+/ROSA26+-Maus zur besseren Darstellung der lacZ-Expression innerhalb der Niere. Schwarze Pfeile zeigen auf proximale Tubuli. int = Darm; ni = Niere; pT = proximale Tubuli. Die cytoplasmatische Hintergrundfärbung wurde mit 0,1% Eosin erzeugt.
A B C
D E F
G H
int int
int
int
int
ni ni
ni
ni ni
ni
int
pT
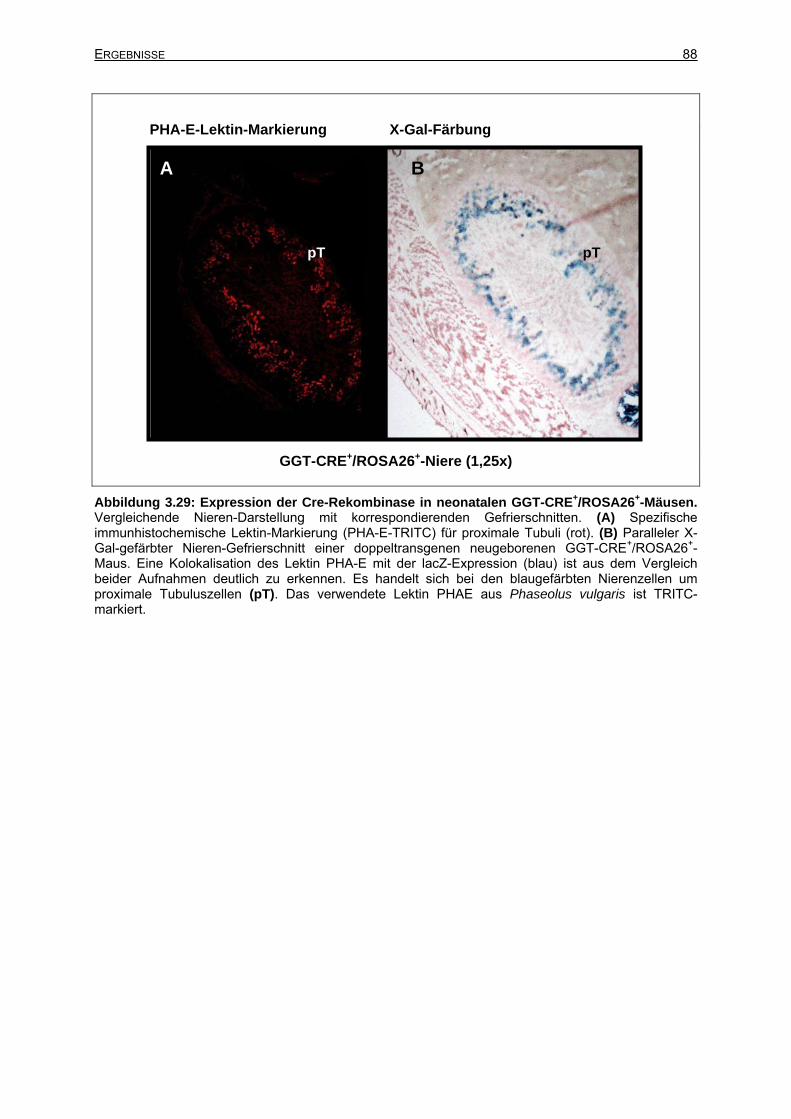
ERGEBNISSE
88
PHA-E-Lektin-Markierung X-Gal-Färbung
GGT-CRE+/ROSA26+-Niere (1,25x)
Abbildung 3.29: Expression der Cre-Rekombinase in neonatalen GGT-CRE+/ROSA26+-Mäusen. Vergleichende Nieren-Darstellung mit korrespondierenden Gefrierschnitten. (A) Spezifische immunhistochemische Lektin-Markierung (PHA-E-TRITC) für proximale Tubuli (rot). (B) Paralleler X-Gal-gefärbter Nieren-Gefrierschnitt einer doppeltransgenen neugeborenen GGT-CRE+/ROSA26+-Maus. Eine Kolokalisation des Lektin PHA-E mit der lacZ-Expression (blau) ist aus dem Vergleich beider Aufnahmen deutlich zu erkennen. Es handelt sich bei den blaugefärbten Nierenzellen um proximale Tubuluszellen (pT). Das verwendete Lektin PHAE aus Phaseolus vulgaris ist TRITC-markiert.
A B
pT pT

ERGEBNISSE
89
3.6.2.7 Cre-Rekombinase-Expression bei adulten GGT-CRE-Mäusen Aus adulten doppeltransgenen GGT-CRE+/ROSA26+-Mäusen, sowie aus GGT-CRE-
/ROSA26+-Kontrollmäusen erfolgte eine Organentnahme (siehe Abschnitt 2.4.1.1) um
weiterführende histochemische Untersuchungen durchzuführen. Ziel war es eine genaue
Aussage zu treffen, ob die Cre-Rekombinase-Expression auch im adulten
Entwicklungsstadium der Maus zellspezifisch vorliegt. Nach erfolgter Detektion der β-
Galaktosidase mittels X-Gal-Färbung konnte außer in den Nieren und dem Darmtrakt keine
weitere Blaufärbung beobachtet werden. Um experimentell nachzuweisen, ob die Expression
der Cre-Rekombinase in adulten Nieren aus 4-Wochen alten GGT-CRE+/ROSA26+-Mäusen
in den proximalen Tubulusstrukturen beschränkt ist, folgten immunhistochemische
Markierungen. Dabei wurden Nieren-Gefrierschnitte von CRE+/ROSA26+-Mäusen (4
Wochen) und CRE-/ROSA26+-Kontrollmäusen (4 Wochen) mit TRITC-gekoppeltem PHA-E-
Lektin angefärbt. Korrespondierende Gefrierschnitte wurden zum Nachweis der β-
Galaktosidase-Expression mit X-Gal angefärbt. In Abbildung 3.30 A wird der histochemische
Nachweis der lacZ-Expression einer CRE+/ROSA26+-Niere neben einer GGT-CRE-
/ROSA26+-Kontrollniere (Abbildung 3.30 B) präsentiert. Bestimmte histologische Areale des
doppeltransgenen Nierenpräparates wurden eingegrenzt (K1-K3). Diese Regionen werden
neben den Lektin-PHA-E TRITC-markierten korrespondierenden Gefrierschnitten in
Abbildung 3.31 vergrößert vergleichend dargestellt. Es zeigt sich eine Kolokalisation der
Lektin-markierten (rot) mit den blaugefärbten Nierenregionen. Dies lässt zum einem den
Schluss zu, dass eine Cre-Rekombinase-Expression zellspezifisch in den proximalen
Tubuluszellen vorliegt und zum anderen dass bis zum adulten Stadium die katalytische
Aktivität des Cre-Proteins beibehalten wird.
Abbildung 3.30: LacZ-Expression in adulten Nieren aus doppeltransgenen GGT-CRE+/ROSA26+-Mäusen und GGT-CRE-/ROSA26+-Kontrollmäusen (4 Wochen). (A) Sagittaler Gefrierschnitt nach der X-Gal-Färbung einer doppeltransgenen 4-Wochen alten GGT-CRE+/ROSA+-Maus (1,25x). Die lacZ-Expression dehnt sich kranzformig von der äußeren renalen Medulla bis hin zum Nierenkortex aus. (B) Lichtmikroskopische Aufnahme (2,5x) einer adulten Kontrollniere (GGT-CRE-/ROSA+). Eine Blaufärbung liegt hier nicht vor. Durch Zugabe von 0,1% Eosin wurde das Cytoplasma rot angefärbt.
A B K1
K2
K3
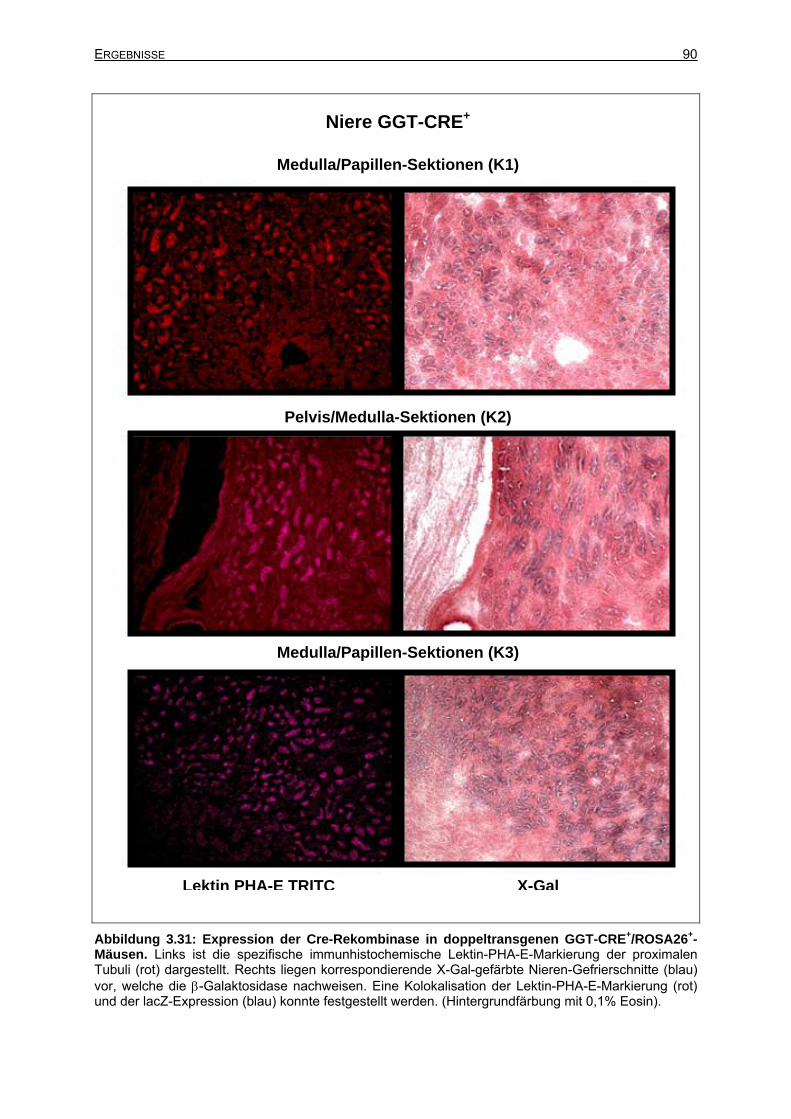
ERGEBNISSE
90
Abbildung 3.31: Expression der Cre-Rekombinase in doppeltransgenen GGT-CRE+/ROSA26+-Mäusen. Links ist die spezifische immunhistochemische Lektin-PHA-E-Markierung der proximalen Tubuli (rot) dargestellt. Rechts liegen korrespondierende X-Gal-gefärbte Nieren-Gefrierschnitte (blau) vor, welche die β-Galaktosidase nachweisen. Eine Kolokalisation der Lektin-PHA-E-Markierung (rot) und der lacZ-Expression (blau) konnte festgestellt werden. (Hintergrundfärbung mit 0,1% Eosin).
Niere GGT-CRE+
Medulla/Papillen-Sektionen (K1)
Pelvis/Medulla-Sektionen (K2)
Medulla/Papillen-Sektionen (K3)
Lektin PHA-E TRITC X-Gal

ERGEBNISSE
91
3.6.3 Expression des GGT-CRE-Transgens in der adulten Niere Nieren aus adulten GGT-CRE+-Transgenen (4-84 Wochen) wurden entnommen, um nach
Anfertigung von Gefrierschnitten das Expressionsprofil der Cre-Rekombinase durch
Markierung mit einem spezifischen primären Cre-Antikörper (Novagen) zu untersuchen. Die
Abbildungen 3.32 A-C zeigen eine immunfluormetrische Darstellung der äußeren renalen
Medulla von einer adulten transgenen GGT-CRE+-Maus auf. Bei dieser
immunhistochemischen Markierung ist der primäre polyklonale Antikörper (Novagen) gegen
die Cre-Rekombinase, sowie ein sekundärer Antikörper verwendet worden, der mit dem
roten Fluorophor Rhodamin (Jackson ImmunoResearch) gekoppelt ist. Das Fluorophor
Rhodamin lässt sich mittels der Fluoreszenzmikroskopie im roten Wellenbereich gut
visualisieren. Eine deutliche rote Markierung ist in den Nuklei renaler Tubuli aus transgenen
Mäusen zu erkennen, was mit der Präsenz eines Kernlokalisationssignals (NLS) im
Transgen GGT-CRE übereinstimmt (Abbildung 3.32 A-C). Das Kernlokalisationssignal ist
von großer Bedeutung, weil das Enzym Cre-Rekombinase als DNA-Bindeprotein nur im Kern
katalytisch aktiv sein kann. Nieren-Gefrierschnitte aus nicht transgenen Tieren (FVB/N)
zeigen nach gleicher immunhistochemischer Analyse keine rote Markierung auf, was für die
Spezifität des verwendeten Cre-Antikörpers spricht (Abbildung 3.34 D). Außer den
Rhodamin-markierten sekundären Antikörper wurde auch ein Cy2-gekoppelter Antikörper
(Jackson ImmunoResearch) zur Darstellung der Cre-Rekombinase auf Gefrierschnitten
transgener GGT-CRE+ und nicht transgener Mäuse eingesetzt. In diesem Fall lassen sich die
Kerne grün darstellen (Abbildung 3.32 E-G). Als Kontrolle wurden Nieren-Gefrierschnitte
nicht transgener Tiere unter gleichen immunhistochemischen Konditionen markiert. Bei
Nieren-Gefrierschnitten nicht transgener FVB/N-Kontrollmäuse, ist keine signifikante grüne
Markierung zu erkennen (Abbildung 3.32 H). Des Weiteren sind Gefrierschnitte folgender
Organe auf das Vorliegen der Cre-Rekombinase mittels dem primären Cre-Antikörper und
dem sekundären Rhodamin-gekoppelten Antikörper immunhistochemisch analysiert worden:
Gehirn, Leber, Milz, Lunge, Herz, Pankreas, Haut, Muskelgewebe und Ovar/Testis
(Abbildung nicht gezeigt). Bei keinen der genannten Organe konnte die Cre-Rekombinase
unter Verwendung der Fluoreszenzmikroskopie detektiert werden. Um eine histologische
Identifizierung der Cre-Rekombinase-exprimierenden Zellen zu erzielen, wurden Nieren-
Gefrierschnitte transgener und nicht transgener Tiere doppelt markiert. Für die
immunhistochemische Doppellmarkierung wurde zum einem der Cre-Antikörper mit dem
sekundären Rhodamin-markierten Antikörper eingesetzt und zum anderen ein FITC-
gekoppeltes biotinyliertes Lektin PHA-E, welches unverändert spezifisch an
Oberflächenproteine proximaler Tubuluszellen anbindet und im grünen Wellenbereich
emittiert. In den Abbildungen 3.32 I-K sind fluoreszenzmikroskopische Aufnahmen dieser
immunhistochemischen Doppelmarkierung dargestellt. Bei den Nieren-Gefrierschnitten
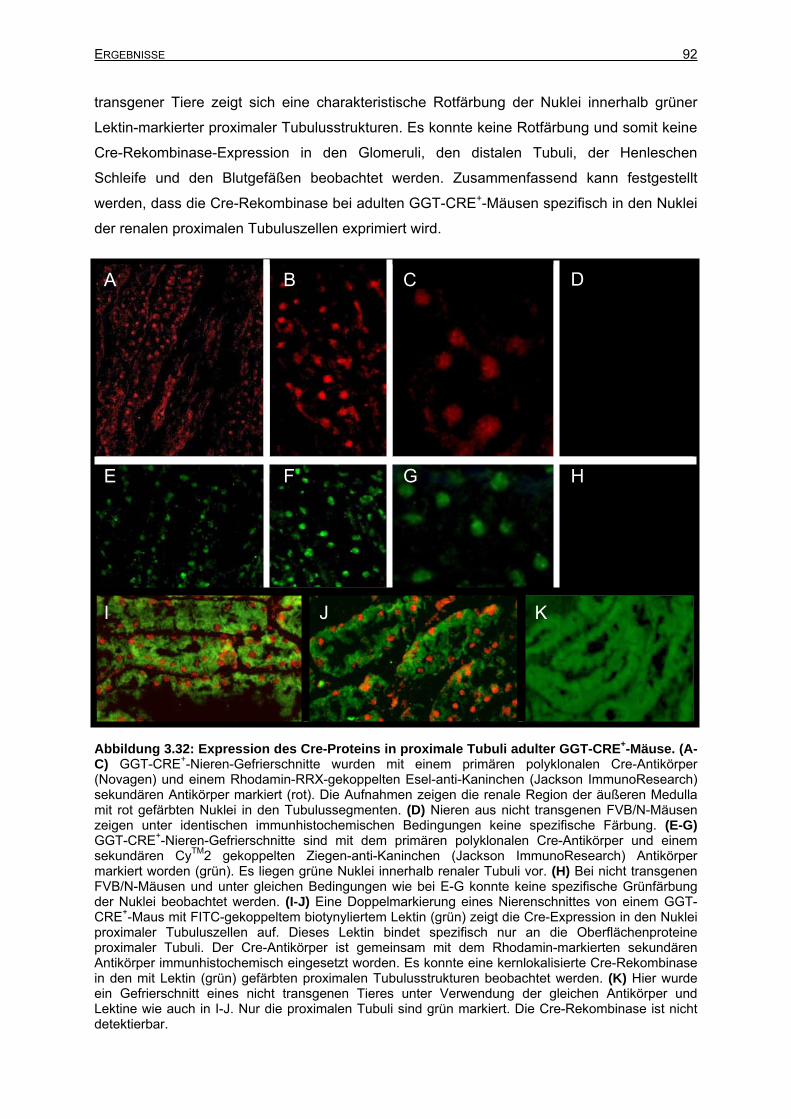
ERGEBNISSE
92
transgener Tiere zeigt sich eine charakteristische Rotfärbung der Nuklei innerhalb grüner
Lektin-markierter proximaler Tubulusstrukturen. Es konnte keine Rotfärbung und somit keine
Cre-Rekombinase-Expression in den Glomeruli, den distalen Tubuli, der Henleschen
Schleife und den Blutgefäßen beobachtet werden. Zusammenfassend kann festgestellt
werden, dass die Cre-Rekombinase bei adulten GGT-CRE+-Mäusen spezifisch in den Nuklei
der renalen proximalen Tubuluszellen exprimiert wird.
Abbildung 3.32: Expression des Cre-Proteins in proximale Tubuli adulter GGT-CRE+-Mäuse. (A-C) GGT-CRE+-Nieren-Gefrierschnitte wurden mit einem primären polyklonalen Cre-Antikörper (Novagen) und einem Rhodamin-RRX-gekoppelten Esel-anti-Kaninchen (Jackson ImmunoResearch) sekundären Antikörper markiert (rot). Die Aufnahmen zeigen die renale Region der äußeren Medulla mit rot gefärbten Nuklei in den Tubulussegmenten. (D) Nieren aus nicht transgenen FVB/N-Mäusen zeigen unter identischen immunhistochemischen Bedingungen keine spezifische Färbung. (E-G) GGT-CRE+-Nieren-Gefrierschnitte sind mit dem primären polyklonalen Cre-Antikörper und einem sekundären CyTM2 gekoppelten Ziegen-anti-Kaninchen (Jackson ImmunoResearch) Antikörper markiert worden (grün). Es liegen grüne Nuklei innerhalb renaler Tubuli vor. (H) Bei nicht transgenen FVB/N-Mäusen und unter gleichen Bedingungen wie bei E-G konnte keine spezifische Grünfärbung der Nuklei beobachtet werden. (I-J) Eine Doppelmarkierung eines Nierenschnittes von einem GGT-CRE+-Maus mit FITC-gekoppeltem biotynyliertem Lektin (grün) zeigt die Cre-Expression in den Nuklei proximaler Tubuluszellen auf. Dieses Lektin bindet spezifisch nur an die Oberflächenproteine proximaler Tubuli. Der Cre-Antikörper ist gemeinsam mit dem Rhodamin-markierten sekundären Antikörper immunhistochemisch eingesetzt worden. Es konnte eine kernlokalisierte Cre-Rekombinase in den mit Lektin (grün) gefärbten proximalen Tubulusstrukturen beobachtet werden. (K) Hier wurde ein Gefrierschnitt eines nicht transgenen Tieres unter Verwendung der gleichen Antikörper und Lektine wie auch in I-J. Nur die proximalen Tubuli sind grün markiert. Die Cre-Rekombinase ist nicht detektierbar.
A D C B
E F G H
I J K
F G H

DISKUSSION
93
4 Diskussion
Charakteristisch für die ADPKD ist die langsam fortschreitende Bildung und Vergrößerung
von Zysten in beiden Nieren, welche aus Keimbahnmutationen in einem Allel des PKD1 oder
des PKD2 Gens resultieren (Torra et al., 1999; Qian & Watnick, 1999) und im weiteren
Verlauf des Lebens sich nach der Zweitreffer-Theorie (Knudson, 1971) eine zweite
somatische Mutation im anderen Allel ereignet. Durch eine kontinuierliche Proliferation der
klonalen Zellen, welche aus unterschiedlichen Nephronsegmenten entstammen, kommt es
dann zur Zystenbildung.
Sowohl spontan entstandene genetische Modelle für die polycystische Nierenerkrankung,
wie cpk (Preminger et al., 1982; Davisson et al., 1991; Simon et al., 1994; Hou et al., 2002),
orpk (Yoder et al., 1995; 1996; 2000b; Murcia et al., 2000; Taulman et al., 2001) und inv
(Mochizuki et al., 1998) als auch die konventionell entwickelten Pkd1- (Lu et al., 1997; Kim et
al., 2000; Boulter et al., 2001; Lu et al., 2001) und Pkd2-defizienten Mausmutanten (Wu et
al., 1998; Pennekamp et al., 2002) wiesen u. a. Nierenzysten auf. Diese Tiermodelle lieferten
wichtige Daten über die Pathogenese der Zystenentstehung, welche unabhängig vom
betroffenen Gen, auf einen gemeinsamen Mechanismus der renalen Zystogenese schließen
lassen. Ferner konnte gezeigt werden, dass Polycystine eine Schlüsselrolle bei der renalen-,
kardiovaskulären-, pankreatischen- und der Knochenentwicklung spielen. Polycystin 2 ist
zudem essentiell für die Links-Rechts-Polarität (Pennekamp et al., 2002). Bei allen bislang
generierten ADPKD-Tiermodellen sind die endogenen Pkd-Gene in allen Organen und
während der gesamten Lebenszeit des Organismus ausgeschaltet. Daraus ergeben sich
erhebliche Nachteile wie die embryonale Letalität im homozygoten Zustand und ein
komplexer pleiotroper Phänotyp. Beides macht eine Untersuchung im adulten Tier, also
präventive und therapeutische Langzeitstudien, undurchführbar. Um diese Limitierungen zu
umgehen, ist vor allem eine örtliche somatische (konditionale) Geninaktivierung notwendig.
Hierdurch wäre es möglich, im adulten Tier die Genfunktion nur in einem bestimmtem
Gewebe, Organ oder Zelltyp auszuschalten. Mit einer konditionalen Mausmutante könnte die
Zystenentwicklung gezielt im Ursprungsgewebe untersucht werden.
Für eine konditionale Genaktivierung wurde in den letzten Jahren das so genannte Cre/loxP-
Rekombinationssystem entwickelt und fortlaufend modifiziert (Lakso et al., 1992; Gu et al.,
1993; Sauer 1998). Zur Realisierung einer konditionalen Mausmutante sind eine „gefloxte“
Maus und eine gewebespezifisch Cre-Rekombinase-exprimierende Maus erforderlich. Beim
gefloxten Mausmodell werden essentielle Bereiche eines ausgewählten Zielgens mit
Erkennungssequenzen (loxP-Stellen) flankiert, die die Transkription nicht beeinflussen. Die
Cre-Rekombinase bindet spezifisch an die loxP-Sequenzen und katalysiert eine somatische
Rekombination zwischen den loxP-Motiven (Sternberg & Hamilton 1981). Nach der

DISKUSSION
94
Verpaarung wird der ansonsten phänotypisch unauffälligen gefloxten Maus in den Cre-
Rekombinase exprimierenden Zellen der gefloxte DNA-Bereich herausgeschnitten. Ziel
dieser Arbeit war es eine transgene Maus zu generieren, welche nierenspezifisch die Cre-
Rekombinase exprimiert, wobei die geeignete Auswahl des Promotors mit der erforderlichen
Gewebespezifität von großer Bedeutung war. Dabei sollte die Promotoraktivität spezifisch in
den renalen Tubuli vorliegen, da Nierenzysten in diesem Gewebe initiiert werden.
4.1 Nierenspezifische Cre-exprimierende Mausmodelle In der Tabelle 4.1 sind transgene Mauslinien mit nierenspezifischer Expression der Cre-
Rekombinase aufgelistet, die in der Literatur schon beschrieben sind.
Tabelle 4.1: Nierenspezifische Cre-exprimierende Mäuse. h: human; m: murin
Transgen Promotor Expression Literatur
AQP2-CreTag h Aquaporin-2 Renales Sammelrohr, Epithelzellen der Testes
Nelson et al., 1998
Neph-Cre m Nephrin Podozyten Eremina et al., 2002
Ksp1.3/Cre m Ksp-Cadherin Embryos: abzweigende Ureterknospe, renale Tubuli, Wolffscher/ Müllerscher Gang Adult: renale Tubuli, Sammelrohr, Pars ascendens
Shao et al., 2002a; 2002b
THP-CreTag m Tamm-Horsfall-Protein
Niere außer Macula densa, distale Tubuli; leichte Expression Testis, Gehirn
Stricklett et al., 2003
P2.5P-Cre h NPHS2 Podozyten Moeller et al., 2003
Die AQP2-CreTag-Maus exprimiert die Cre-Rekombinase, welche unter der Kontrolle des
humanen 13 kb Aquaporin-2 Promotors steht, im renalen Sammelrohr und den Epithelzellen
der Testes (Nelson et al., 1998). Hier ist zu erwähnen, dass die embryonale Expression
unbekannt ist. 4125 bp des murinen Nephrin-Promotors wurden bei der Neph-Cre Maus der
Cre-Rekombinase vorgeschaltet, was zu einer spezifischen Expression in den renalen
Podozyten führt (Eremina et al., 2002). Die Ksp1.3/Cre Maus wurde sowohl embryonal als
auch adult auf ihre Cre-Rekombinase-Expression hin untersucht (Shao et al., 2002a; 2002b).
Bei diesem Transgen steht das Cre-Gen unter der Kontrolle des murinen Ksp-Cadherin
Promotors (1329 bp). Hierbei liegt das Cre-Protein embryonal in den sich entwickelnden
renalen Tubuli, der abzweigenden Ureterknospe und den Gonodukten (Wolffscher/
Müllerscher Gang) vor. Im adulten Stadium ist eine Expression immunhistochemisch im
renalen Sammelrohr, dem dicken aufsteigenden Ast (Pars ascendens) der Henleschen
Schleife und der renalen Tubuli -vorwiegend distaler Tubulus- zu erkennen. Stricklett et al.

DISKUSSION
95
(2003) entwickelten eine transgene THP-CreTag Maus, wobei der murine 3,7 kb-große-THP-
Promotor dem Cre-Gen vorgeschaltet wurde. In diesem Fall konnte bei adulten Tieren eine
Cre-Expression hauptsächlich im dicken aufsteigenden Ast der Henleschen Schleife
beobachtet werden. Des Weiteren konnte CreTag-mRNA in den Testes und im Gehirn
detektiert werden. Spezifisch in den Podozyten und nur postnatal wird die Cre-Rekombinase
bei der transgenen P2.5P-Cre Maus mit dem humanen 2,5 kb-NPHS2-Promotor exprimiert
(Moeller et al., 2003). Außer der Neph-Cre-Maus, die spezifisch in den Podozyten Cre
exprimiert, weisen alle genannten putativ nierenspezifischen Mäuse in unterschiedlichen
Nierenregionen eine Cre-Expression auf. Zum Teil liegt sogar eine extrarenale Expression
vor. Bei keiner der bislang generierten Cre-exprimierenden Mäuse war ausschließlich in den
renalen Tubulussegmenten eine spezifische Cre-Expression vorliegend.
Die in dieser Arbeit entwickelte transgene GGT-CRE-Maus wäre demnach die Erste, die
sowohl embryonal als auch adult spezifisch in den proximalen Tubuluszellen der Niere eine
katalytisch aktive Cre-Rekombinase exprimiert.
4.2 Generierung der transgenen GGT-CRE-Maus Bei der Generierung der GGT-CRE-Maus war zunächst die Auswahl eines geeigneten
gewebespezifischen Promotors von hoher Relevanz, was eine intensive Literaturrecherche
erforderte. Im Ergebnissteil (Tabelle 3.1) sind alle potentiellen nierenspezifischen
Promotoren der Größe nach tabellarisch aufgeführt. Letztendlich fiel die Entscheidung auf
den murinen Minimalpromotor Gamma-Glutamyl-Transpeptidase Typ II (GGT).
Ausschlaggebend für die Auswahl des GGT-Promotors bei der Generierung der Cre-
exprimierenden transgenen Maus waren folgende zwei Hauptkriterien, welche in den
Abschnitten 4.2.1-4.2.2 diskutiert werden:
• Ort der Promotoraktivität • Promotorlänge
4.2.1 Relevanz des proximalen Tubulus für die Zystenbildung Der proximale Tubulus der Niere wird bei der Zystenentstehung im Vergleich zum distalen
Tubulus und dem Sammelrohr unterschiedlich beeinträchtigt und wird im folgenden Absatz
erörtert.

DISKUSSION
96
Die cpk/cpk Maus war das erste beschriebene Maus-Modell mit der Symptomatik der
polycystischen Nierenerkrankung. Es stellte sich ein autosomal rezessives Krankheitsbild
ein. Die Zystenbildung wird bei der cpk/cpk Maus im proximalen Tubulus initiiert und betrifft
im späteren Verlauf auch das Sammelrohr (Nidess et al., 1984; Avner et al., 1987). Ein
analoges Muster dieser pathologischen Nierenveränderung konnte gleichfalls bei der
homozygoten Pkd1-defizienten-Maus (Lu et al., 1997) und auch bei zwei weiteren ARPKD-
Mausmodellen beobachtet werden: der bpk/bpk Maus (Nauta et al., 1995) und der orpk/orpk
Maus (Moyer et al., 1994). Nierenanalysen bei der transgenen Han:SPRD cy/+ Ratte - ein
ADPKD Tiermodell - zeigten ebenfalls auf, dass die Nierenzysten dem proximalen Tubulus
entstammen (Cowley et al., 1993; Schäfer et al., 1994; Obermüller et al., 1997). Dieses
Muster der pathologischen Nierenveränderung konnte auch beim ADPKD-Mausmodell
Pkd2-/-lacZ+/+ beobachtet werden (Pennekamp, 2000). Es lag eine Zystenbildung in den
Grenzbereichen von Cortex und Medulla in den Glomeruli und proximalen Abschnitte der
Tubuli vor. Im Gegensatz dazu entstammen die renalen Zysten bei der Pkd2-Knockout-Maus
Pkd2-/- von Wu et al. (1998), beim pcy/pcy Mausmodell (Takahashi et al., 1991) und dem
chi/chi Rattenmodell (Inage et al., 1991) überwiegend aus den distalen Tubulusstrukturen
und dem Sammelrohr. Ein Grund für diese widersprüchlichen Ergebnisse kann die
verwendete Untersuchungsmethodik sein. Wenn ausschließlich Licht- und
Elektronenmikroskopische Methoden durchgeführt wurden, um den Ursprung der Zysten
nachzuweisen, ist eine Differenzierung zwischen den einzelnen Tubuli und dem Sammelrohr
erheblich erschwert. Die charakteristischen histologischen Unterscheidungsmerkmale, wie
z.B. der Bürstensaum beim proximalen Tubulus gehen im Laufe der Zystenentwicklung
verloren. Ferner unterbleibt bei zystisch veränderten Nephronsegmenten die Expression
deren spezifischer Marker, so dass ein spezieller Markernachweis nicht mehr zur Verfügung
steht. Folglich führt dessen Anwendung nicht zu einer sicheren Charakterisierung einzelner
Nephronabschnitte (Obermüller et al., 1997). Manche transgene Tiermodelle der ADPKD
ähneln der humanen Form der polycystischen Nierenerkrankung so sehr, dass die
Vermutung nahe liegt, dass die Zysten bei der humanen Krankheitsform ebenfalls aus den
proximalen Tubuli und dem Sammelrohr entstammen (Bachinsky et al., 1995; Devuyst et al.,
1996). Nach einer Erhebung aller in diesem Abschnitt genannten Ergebnisse wird dem
proximalen Tubulus bei der Zystenbildung eine besondere Bedeutung zugesprochen. Bei
dieser Nephroneinheit liegt eine erhöhte Affinität zur Zystenbildung vor. Diese Anfälligkeit ist
möglicherweise darin begründet, dass die vorliegenden Mutationen bei den ADPKD-
Tiermodellen Strukturen betreffen, wie z.B. Zell-Zell-Kontakte, Zell-Matrix-Kontakte, welche
für eine reguläre Funktion der proximalen Tubuli wichtiger sind als für die übrigen
Nephronsegmente. Die mutierten Proteine würden zwar nicht das eigentliche
Differenzierungsprogramm der proximalen Tubuli behindern, könnten aber eventuell ein

DISKUSSION
97
essentielles Stoppsignal verhindern, so dass fortdauernd Zystenzellen proliferieren und sich
pathologisch Lumen ausbilden.
Aufgrund der erhöhten Anfälligkeit des proximalen Tubulus zur Zystenbildung bietet sich
dieser Expressionsort für die Cre-Rekombinase in idealer Weise an. In der Literatur sind fünf
Promotoren beschrieben worden, die nach in vivo-Untersuchungen, eine Aktivität
vorwiegend in renalen proximalen Tubuli aufwiesen (Tabelle 4.2).
Tabelle 4.2: Auflistung gewebespezifischer Promotoren mit Hauptaktivität in pT-Zellen. Expression von Reportergenen bzw. c-DNA in transgenen Tieren.
Promotor Länge Expression von/in Literatur
m GGT 346 bp lacZ/pT-Zellen Rajagopalan et al., 1993; Sepulveda et al.,1997
h PAI-1 1,27 kb lacZ/pT-Zellen Kruithof & Cousin, 1988; Strandberg et al., 1988; Emert et al., 1998
h KAP 1,5 kb HAGT/pT-Epithelzellen Niu et al., 1991 Ding et al., 1997; Hardy et al., 2001
m Epo 6,5 kb lacZ/pT-Zellen, Leber McDonald et al., 1986; Loya et al., 1994; Haidar et al., 1997
m TESK1
9,0 kb lacZ/testikuläre Keimzellen, pT-Zellen, Herzmuskelzellen, Nervenzellen
Toshima et al., 1998; 2001
GGT = Gamma-Glutamyl-Transpeptidase Typ II; PAI-1 = Plasminogen Aktivator Inhibitor I; KAP = Nieren Androgen-reguliertes-Protein; Epo = Erythropoietin; TESK1 = testikuläre Proteinkinase1; humanes Angiotensinogen; HAGT = humanes Angiotensinogen; pT-Zellen = proximale Tubuluszellen; h = human; m = murin
9,0 kb des murinen testikulären Proteinkinase-1-Promotors (Toshima et al., 1998) wurden
dem Reportergen lacZ vorgeschaltet um dessen Expressionsprofil zu analysieren.
Tatsächlich konnte in den renalen proximalen Tubuluszellen der adulten TESK1/lacZ-Maus
die β-Galaktosidase nachgewiesen werden (Toshima et al., 2001). Ferner lag extrarenal in
den testikulären Keimzellen, den Herzmuskelzellen und den Nervenzellen ebenfalls eine
erhöhte lacZ-Expression vor. Da keine nierenspezifische Promotoraktivität vorlag, wurde der
murine TESK1-Promotor ausgeschlossen.
Analog zu den Ergebnissen des murinen TESK1 Promotors zeigte sich beim murinen
Erythropoietin-Promotor (McDonald et al., 1986) außer im proximalen Tubulus auch eine
extrarenale Aktivität in der Leber (Loya et al., 1994; Haidar et al., 1997).

DISKUSSION
98
Der KAP-, PAI-1- und GGT-Promotor kamen in die engere Auswahl. Grund dafür waren die
in vivo Expressionsuntersuchungen, welche eine Promotoraktivität primär im proximalen
Tubulus nachwiesen (Ding et al., 1997; Emert et al., 1998; Sepulveda et al., 1997). Bei der
so genannten KAP-HAGT-Maus (Ding et al., 1997) wurden 1,5 kb vom Promotor des
humanen Nieren Androgen-regulierten Proteins mit der kodierenden Region des humanen
Angiotensinogen (hAGT) Gens fusioniert. Durch in situ Hybridisierungen, unter Verwendung
von anti-sense-Proben aus hAGT-mRNA, konnte eine sehr spezifische KAP-hAGT-
Expression in den Epithelzellen des proximalen Tubulus detektiert werden (Ding et al.,
1997). Es lag adult keine extrarenale Expression vor. Sowohl der PAI-1-Promotor als auch
der GGT-Minimalpromotor wurden stromaufwärts vom lacZ kloniert, um dessen
Expressionsprofil in transgenen Tieren aufzuzeigen. Bei beiden transgenen Tieren zeigte
sich eine äußerst spezifische β-Galaktosidase-Expression ausschließlich im proximalen
Tubulus der Niere.
4.2.2 Relevanz der Promotorgröße Hinsichtlich ihres örtlichen Aktivitätsprofils unterscheiden sich der KAP-, PAI-1- und GGT-
Promotor kaum voneinander. Promotoraktivitäten wurden mittels histochemischer
Expressionsuntersuchungen ausschließlich in den renalen proximalen Tubuluszellen
nachgewiesen. Ein wichtiger Unterschied ist jedoch die Promotorlänge. Beim KAP-Promotor
sind es 1,5 kb, beim PAI-1-Promotor 1,27 kb, wohingegen beim GGT-Promotor nur 346 bp
für eine Gewebespezifität ausreichen.
Eine geeignete und flexible Klonierungsstrategie zum Einbringen eines Promotors vor ein
Strukturgen ist mittels der PCR-Technologie zu realisieren. Der Promotorbereich wird mit
den entsprechenden homologen Oligonukleotidsequenzen aus genomischer DNA heraus
amplifiziert. Am 5´- und 3´-Ende der Primersequenz werden Restriktionsschnittstellen
eingefügt, um eine weiterführende Klonierung in einen Vektor mit analogen Schnittstellen zu
ermöglichen. Bei dieser Klonierungsmethodik sind kürzere Promotorbereiche von Vorteil. Je
größer die zu amplifizierende DNA-Sequenz ist, desto höher ist die Wahrscheinlichkeit eines
Synthesefehlers. Aus diesem Grund fiel letztendlich die Wahl auf den 346 bp großen GGT-
Promotors. Zur Amplifikation der murinen genomischen 346 bp-DNA-Sequenz wurde die pfu-
DNA-Polymerase eingesetzt, die aufgrund einer 3´-5´- Exonuklease-Aktivität im Vergleich zu
anderen thermostabilen DNA-Polymerasen die niedrigste Fehlerrate besitzt (Cline et al.,
1996; Shafikhani, 2002).

DISKUSSION
99
4.3 Muriner 346-bp-GGT-Minimalpromotor Bei der Gamma-Glutamyl-Transpeptidase - eine membrangebundene Ektopeptidase -
handelt es sich um ein Schlüsselenzym im Glutathion-Metabolismus (Vina, 1990; Meister,
1995; Lieberman et al., 1995; Meister & Larsson, 1995; Shi et al., 1995). Das Protein wird in
unterschiedliche epitheliale Zellen exprimiert (Whitfield, 2001; Hanigan & Frierson, 1996) mit
der höchsten Expressionsrate präferenziell in der Niere, Dünndarm, Pankreas, fetaler Leber
und weiteren Organen mit sekretorischen und absorptiven Eigenschaften (Vina, 1990;
Lieberman et al., 1995). 1993 konnte erstmals die Struktur von unterschiedlichen GGT-
mRNAs in murinen Nieren charakterisiert werden (Rajagopalan et al., 1993). Mittlerweile
wurde auch die kodierende Region des murinen GGT-Gens entschlüsselt (Shi et al., 1995).
Es ist auf dem Chromosom 10 lokalisiert (LocusID: 14598, NCBI), umfasst 12589 bp (NT
039496, NCBI) und wird in 12 Exons unterteilt. GGT wird von sieben unterschiedlichen
Promotoren in sieben mRNAs transkribiert, welche zwar den gleichen offenen Leserahmen
einnehmen, sich aber in der 5´-nicht translatierten Region unterscheiden (Rajagopalan et al.,
1993; Sepulveda et al., 1994; Carter et al., 1994; Habib et al., 1995; Lieberman, 1995; Shi et
al., 1996; Chikhi et al., 1999). Die sieben Promotoren mit den zugehörigen mRNAs werden
als Typ I - Typ VII bezeichnet. Obwohl GGT dank der unterschiedlichen Promotoren in einer
relativ ubiquitären Weise exprimiert wird, ergibt sich dennoch eine Gewebespezifität durch
die mRNAs (Lieberman et al., 1995; Chikhi et al., 1999). Zellbiologische
Transfektionsexperimente mit dem CAT-Gen, welches als Nachweis für die
Promotoraktivierung fungiert zeigten auf, dass der GGT-Promotor-Typ II den murinen
proximalen Tubuluszellen eine signifikant höhere CAT-Aktivität verleiht als den Fibroblasten.
Für eine zellspezifische Expression eines Reportergens in murine Zellen reichen die cis-
aktiven Elemente des GGT-Promotors Typ II aus (Sepulveda et al., 1994). Zusätzlich zu den
Expressionsstudien wurden Promotoruntersuchungen durchgeführt, um die exakte
Lokalisation der betreffenden gewebespezifischen DNA-Elemente nachzuweisen. Durch
Mutagenese ergaben sich unterschiedliche 5´-Deletionen, welche durch weiterführende
Transfektionsexperimente in murinen proximalen Tubuluszellen (MPT-Zellen) analysiert
wurden. Es zeigte sich, dass die 346 bp-großen Promotorelemente des murinen GGT-
Promotor-Typ II die höchste Aktivität aufwiesen (Sepulveda et al., 1997). Um einen Einblick
zu gewinnen, ob der murine GGT-Promotor-Typ II ausreichend cis-aktive Elemente für eine
eingeschränkte in vivo-Expression besitzt, folgte die Generierung eines transgenes
Tiermodells (Sepulveda et al., 1997).

DISKUSSION
100
4.3.1 GGT-pII-346/β-Galaktosidase-Maus Nach Ermittlung der geeigneten Promotorlänge wurde ein Transgen mit stromabwärts
geschalteter lacZ-Kassette unter Kontrolle des 346 bp-großen Minimalpromotors GGT
gesetzt (Sepulveda et al., 1997). Das Konstrukt pII-346/β-Galaktosidase wurde zunächst in
MPT-Zellen transfiziert, um die β-Galaktosidase Aktivität zu testen. Nach Generierung der
GGT-pII-346/β-Galaktosidase-Maus sind verschiedene Organe (Niere, Nebenniere, Leber,
Dünndarm, Milz, Lunge, Herz, Gehirn und Uterus) zur Detektion der β-Galaktosidase
histochemisch analysiert worden. Nur die Nieren-Sektionen und hier nur die proximalen
Tubulusstrukturen ließen sich blau anfärben, was auf eine spezifische Promotoraktivität
hinwies.
4.4 Klonierung des GGT-CRE-Transgens Um eine zellspezifische Expression der Cre-Rekombinase im proximalen Tubulus zu
erreichen, wurde in dieser Arbeit das Strukturgen der Cre-Rekombinase (ORF2; Sternberg et
al., 1981) unter die Kontrolle des oben beschriebenen 346 bp großen GGT-Promotors
gestellt. Stromabwärts des ATG-Startkodons des Cre-Gens sind im Leseraster 21 bp des
Kernlokalisationssignals (NLS; Kalderon et al., 1984) vom SV40 large T Antigen inseriert
worden. NLS führt zur Translokation der Cre-Rekombinase in den Nukleus. Das DNA-
Bindeprotein Cre-Rekombinase muss im Kern vorliegen, um die gewünschte Rekombination
zu katalysieren. Stromabwärts wurde abschließend eine SV40-polyA-Region einkloniert. Das
so entstandene Transgen GGT-CRE wurde zur Erzeugung transgener Tiere in den
Pronukleus fertilisierter FVB/N-Oocyten injiziert und anschließend in scheinschwangere
CD1-Mäuse nach Hogan et al., (1994) implantiert. Aus der Implantierung resultierte eine
transgene weibliche Maus (G5). Mittels Southern-Transfer-Analysen und PCR-
Genotypisierungen konnte nachgewiesen werden, dass das Transgen GGT-CRE auch bei
den Nachkommen vorlag.
4.5. Das Expressionsprofil des murinen GGT-Promotors Zuerst konnte gezeigt werden, dass die Cre-Rekombinase des GGT-CRE-Konstruktes
katalytisch aktiv war. Dafür wurden GGT-CRE-Mäuse sowohl mit Z/AP-Reportermäusen
(Lobe et al., 1999) als auch mit ROSA26-Reportermäusen (Soriano, 1999) verpaart.
4.5.1 Cre/loxP-Rekombination in GGT-CRE+/Z/AP+-Mäuse Mittels enzymhistochemischer Detektionsmethoden für die hitzestabile humane plazentale
alkalischen Phosphatase (hPLAP) des Z/AP-Allels konnte bei den doppeltransgenen GGT-
CRE+/Z/AP+-Mäusen ein Rekombinationsereignis der Cre-Rekombinase nachgewiesen

DISKUSSION
101
werden. In unrekombinierten Zellen verhindert eine vorgeschaltene gefloxte βgeo-Kassette
(Friedrich & Soriano, 1991) die Transkription des hPLAP-Gens. Eine hPLAP-Expression
stellt sich nur ein, wenn eine katalytisch aktive Cre-Rekombinase exprimiert und dadurch die
gefloxte Terminationseinheit deletiert wird. Infolgedessen kann hPLAP mittels dessen
Substratzugabe detektiert werden. Nieren-Gefrierschnitte von adulten GGT-CRE+/Z/AP+-
Mäusen wiesen eine intensive bläuliche Färbung der hPLAP im Bereich des renalen Kortex
bis zur äußeren Medulla auf. Es handelte sich nach einer lichtmikroskopischen
Charakterisierung um proximale Tubulusstrukturen. GGT-CRE-/Z/AP+ Kontroll-Tiere wiesen
unter gleichen histochemischen Bedingungen keine hPLAP-Expression auf.
Schlussfolgernd lässt sich durch diese Experimente eine katalytische in vivo-Aktivität der
Cre-Rekombinase in adulten transgenen GGT-CRE-Mäusen nachweisen.
4.5.2 Früheste Cre/loxP-Rekombination in GGT-CRE+/ROSA26+-Mäusen ROSA26-Reportermäuse können eine Cre/loxP-Rekombination mittels einer β-
Galaktosidase-Expression aufzeigen. Auf dem ROSA26-Allel liegt als Terminationseinheit
eine gefloxte Neomycinkassette mit einer vierfachen polyA-Sequenz vor, wodurch der
Transkriptionsstillstand der RNA Polymerase gewährleistet wird. Das stromabwärts gelegene
Reportergen lacZ kann folglich nicht translatiert werden. Erst durch die Anbindung des Cre-
Enzyms an die loxP-Erkennungssequenzen erfolgt die Entfernung der Terminationskassette
und demzufolge die Initiierung der lacZ-Transkription. In dieser Arbeit wurden sowohl
embryonale als auch adulte GGT-CRE+/ROSA26+-Mäuse überprüft. Als Kontrolle dienten
GGT-CRE-/ROSA26+-Mäuse. Zur embryonalen Untersuchung der Cre/loxP-Rekombination
wurden Gefrierschnitte ganzer Embryonen eingesetzt. Der erste Analysezeitpunkt war E12,5.
Erst zum embryonalen Zeitpunkt E15,5 bei doppeltransgenen GGT-CRE+/ROSA26+-
Embryonen konnte eine leichte lacZ-Expression in der embryonalen Niere (Metanephros)
und dem Darm beobachtet werden. In der Kontrollgruppe lag nur im Darmbereich eine lacZ-
Expression vor. Im Darm lebende Bakterien enthalten die endogene β-Galaktosidase, die
das vorgelegte X-Gal mitverwertet. Aus diesem Grunde wurde eine Blaufärbung auch bei
Kontroll-Mäusen interstinal beobachtet. Bei früheren embryonalen Stadien konnte durch
lichtmikroskopische Betrachtungen keine Blaufärbung im gesamten Embryo erfasst werden.
In der Literatur finden sich zum Vergleich Informationen über den ersten
Expressionszeitpunkt der murinen GGT. 1988 wurde an murinen Zellen quantitativ durch
Fluorimetrie-Messverfahren die spezifische Aktivität der Transpeptidase mit parallelen
immunhistochemischen GGT-Nachweismethoden (Curto et al., 1988) festgestellt. Beim
Embryonalstadium E13 wurde in der fetalen murinen Niere 1 mU/mg Protein gemessen.
Zwischen den Gestationstagen 13-21 ergab sich eine 50-fach höhere spezifische
Transpeptidase-Aktivität. Am 13. Tag der Gestation wurde GGT immunhistochemisch an der

DISKUSSION
102
apikalen Oberfläche der sich entwickelnden murinen renalen Vesikel und dem proximalen
Segment der S-förmigen Tubuli dargestellt. Am 15. Tag der Gestation findet sich GGT
vorwiegend an der apikalen Oberfläche der sich entwickelten proximalen Segmente. Der 17.
Gestationstag geht einher mit der Nierenorganisation in diskreten Zonen. Hier findet sich ein
ziemlich hoher Aktivitätsanstieg des Enzyms, was mit einem starken immunhistochemischen
Signal am proximalen Tubulus der inneren Kortex korrelierte. Im Vergleich dazu konnte
humane GGT mittels immunhistochemischer Analysen in menschlichen proximalen
Tubuluszellen eines 12 Wochen alten Fötus lokalisiert werden (Hanigan & Frierson, 1996).
Die Diskrepanz des in doppeltransgenen GGT-CRE+/ROSA26+-Mäusen festgestellten ersten
lacZ-Expressionszeitpunkts bei E15,5 und der in der Literatur beschriebenen ersten
Expression des GGT-Proteins kann zum einem an der verkürzten Promotorregion des
Transgens liegen. Zum anderen könnte eine Cre/loxP-Rekombination erst durch einen
gewissen Dosis-Effekt gewährleistet werden. Wie bei Curto et al., (1988) beschrieben erhöht
sich die Transpeptidase Aktivität um das 50-fache zwischen den Embryonaltagen 13-21, was
auf eine stetig ansteigende Aktivität des Promotors hinweist.
4.5.3 Cre/loxP-Rekombination in neonatalen und adulten GGT-CRE+/ROSA26+- Mäusen Bei den neugeborenen GGT-CRE+/ROSA26+-Mäusen wurde eine genaue Charakterisierung
der lacZ-exprimierenden Nierenzellen mittels einer Lektin-Markierung, welche spezifisch für
proximale Tubuluszellen ist, durchgeführt. Eine Kolokalisation des Lektins PHA-E aus
Phaseolus vulgaris und der lacZ-Expression ist bei den Gefrierschnitten von ganzen
doppeltransgenen Neugeborenen Tieren deutlich zu erkennen. Es handelte sich beim
blaugefärbten Gewebe um proximale Tubuluszellen. Ganzkörperschnitte von neugeborenen
GGT-CRE+/ROSA26+- und GGT-CRE-/ROSA26+-Mäusen wurden für weiterführende X-Gal-
Färbungen und Lektin-Markierungen eingesetzt. Eine lacZ-Expression konnte bei neonatalen
Mäusen ausschließlich im renalen proximalen Tubulus beobachtet werden. Diese
Ergebnisse demonstrieren, dass eine gewebespezifische Cre/loxP-Rekombination auch an
diesem Entwicklungszeitpunkt durch den GGT-Minimalpromotor vermittelt wird. Wichtig
hierbei ist auch die Tatsache, dass Ganzkörperschnitte von Neugeborenen histochemisch
analysiert wurden. Somit wurden praktisch alle Geweberegionen berücksichtigt. Das Fehlen
einer lacZ-Expression im extrarenalen Bereich ergibt die Schlussfolgerung, dass die Cre-
Rekombinase zumindest bei der neugeborenen transgenen GGT-CRE+-Maus nur im
proximalen Tubulus katalytisch aktiv ist. Analog zu den GGT-CRE+/Z/AP+-Mäusen konnte
auch bei den adulten GGT-CRE+/ROSA26+-Mäusen eine spezifische Anfärbung der
proximalen Tubuluszellen beobachtet werden. Mit dem Unterschied, dass bei den adulten
GGT-CRE+/ROSA26+-Mäusen die lacZ-Expression außer durch lichtmikroskopischer

DISKUSSION
103
Beobachtung auch durch nephronspezifische Lektine die Lokalisation der Cre-Rekombinase
genauer untersucht werden konnte. Zusammenfassend kann postuliert werden, dass bei der
transgenen GGT-CRE-Maus die katalytische Aktivität der Cre-Rekombinase im proximalen
Tubulus ab den Embryonalstadium E15,5 bis hin zum adulten Stadium durchgängig vorliegt.
4.6 Expression des GGT-CRE-Transgens in der Niere Die genaue Lokalisation des Cre-Proteins innerhalb der Zelle bei der GGT-CRE-Maus
konnte durch die Verwendung von Cre-Rekombinase-Antikörpern dargestellt werden.
Ausschließlich in den Zellkernen renaler proximaler Tubuli wurde die Cre-Rekombinase auf
Nieren-Gefrierschnitten GGT-CRE+-Mäusen immunhistochemisch detektiert. Eine genaue
Charakterisierung des Nephronsegments erfolgte mit Hilfe eines biotinylierten Lektins,
welches spezifisch an Oberflächenproteine proximaler Tubuluszellen bindet. Für die
immunhistochemische Doppelmarkierung wurde zum einem der Cre-Antikörper mit dem
sekundären Rhodamin-markierten Antikörper und zum anderen ein FITC-gekoppeltes
biotinyliertes Lektin eingesetzt. Nieren-Gefrierschnitte proximaler Tubuluszellen mit
immunhistochemischer Doppelmarkierung zeigten ein eindeutiges Resultat. Die Cre-
Rekombinase liegt im Zellkern des proximalen Tubulus vor. Eine Kernlokalisation war zu
erwarten, da das angebundene Kernlokalisationssignal die Translokation der Cre-mRNA in
den Nukleus aktiv vermittelt.
Das Muster der Cre-Expression ist vergleichbar mit den Ergebnissen der Arbeitsgruppe
Sepulveda et al., (1997). Ihre Resultate ergaben sich durch Einbringen des bakteriellen
Reportergens lacZ unter die transkriptionelle Kontrolle des verkürzten Minimalpromotors.
Dabei konnte die β-Galaktosidase durch X-Gal Färbungen ausschließlich im proximalen
Tubulus detektiert werden. Unklar blieb bei deren Untersuchungen, ob extrarenale lacZ-
Expression vorlag. Durch Markierung von GGT-CRE-Gefrierschnitten des Gehirns, Leber,
Milz, Lunge, Herz, Pankreas, Haut, Muskelgewebe und des Ovars/Testis mit dem primären
polyklonalen Antikörper gegen die Cre-Rekombinase und dem sekundärer Rhodamin-
gekoppelten Antikörper konnte bei diesem Gewebe keine Cre-Rekombinase detektiert
werden.
Zusammenfassend kann gesagt werden, dass die in dieser Arbeit entwickelte transgene
GGT-CRE-Maus die Erste beschriebene ist, die sowohl embryonal als auch adult spezifisch
in den proximalen Tubuluszellen der Niere eine katalytisch aktive Cre-Rekombinase
exprimiert. Dieses Ergebnis konnte sowohl durch den Einsatz von zwei unterschiedlichen
Reportermäusen als auch durch Cre-Antikörper eindeutig gezeigt werden.

DISKUSSION
104
Die GGT-CRE-Maus repräsentiert ein neues experimentelles Reagenz für
gewebespezifische Knockouts in der Niere, und genauer im proximalen Tubulus. Da die Cre-
Rekombinase unter der Kontrolle des GGT-Promotors steht und ihre Expression sich auf den
proximalen Tubulus beschränkt, wird sich die Geninaktivierung nach Verpaarung mit
gefloxten Mäusen nur in diesem Gewebe ereignen. Zudem wird das Transgen der GGT-
CRE-Maus nicht vor dem embryonalen Zeitpunkt E15,5 exprimiert. Genau diese Eigenschaft
lässt sich zur Untersuchung von Genen mit essentiellen Funktionen bei der
Embryonalentwicklung aneignen, die sonst durch konventionelle Inaktivierung zur
embryonalen Letalität führen. Sie kann letztendlich einen großen Beitrag zur Erforschung der
Zystenentwicklung bei der ADPKD beitragen, welche unter anderem in den proximalen
Tubulussegmenten initiiert werden.
4.7 Ausblick: Zeitlich induzierbare nierenspezifische Cre-Mäuse
4.7.1 Generierung der Konstrukte GGT-RTTA und BI-CRE-lacZ Um sowohl eine örtliche als auch eine zeitlich definierte Expression der Cre-Rekombinase zu
ermöglichen wurden in dieser Arbeit zwei weitere Konstrukte zur Generierung transgener
Tiere entwickelt: GGT-RTTA und BI-CRE-lacZ. Für eine zeitliche Kontrolle der Cre-
Rekombinase wurde das binäre Transkriptions-Kontrollsystem von Gossen und Bujard
(1992) verwendet, welches die Expression individueller Gene durch exogene Zugabe von
Doxycyclin reguliert. Bei diesem regulatorischen System sind zwei regulatorische Elemente
erforderlich: Der Transkriptionsaktivator tTA und der Operator. Methodisch werden diese
genetischen Elemente in zwei unterschiedlichen Mauslinien pronuklear injiziert, um erst
durch späteres Verkreuzen in Wechselwirkung zu treten. Beim Transgen GGT-RTTA wurde
der reverse Doxycyclin-abhängige Transkriptionsaktivator (rtTA) unter Kontrolle des
Minimalpromotors GGT gesetzt. Der Transkriptionsaktivator rtTA könnte gewebespezifisch
durch den GGT-Promotor exprimiert und zeitlich durch exogene Doxycyclin-Zugabe reguliert
werden. Erst durch die Zugabe des Doxycyclins wäre rtTA aktiviert und könnte an spezielle
Operatoren (tetO) anbinden, welche auf dem zweiten Konstrukt BI-CRE-lacZ vorliegen. Das
Konstrukt BI-CRE-lacZ weist außer den tetO-Sequenzen auch den bidirektionellen Promotor
Pbi-1 auf (Baron et al., 1995). Diese Konstellation erlaubt die gleichzeitige äquimolare
Expression zweier Gene. Der aktivierte Transkriptionsaktivator rtTA aus dem Konstrukt GGT-
RTTA würde spezifisch an den tetO-Sequenzen anbinden und infolgedessen die
Transkription der zwei divergent geschalteten Zielgene Cre und lacZ induzieren. Abbildung
4.1 zeigt schematisch das Zusammenwirken der Transgene pGGT-RTTA mit pBI-CRE-lacZ
auf.
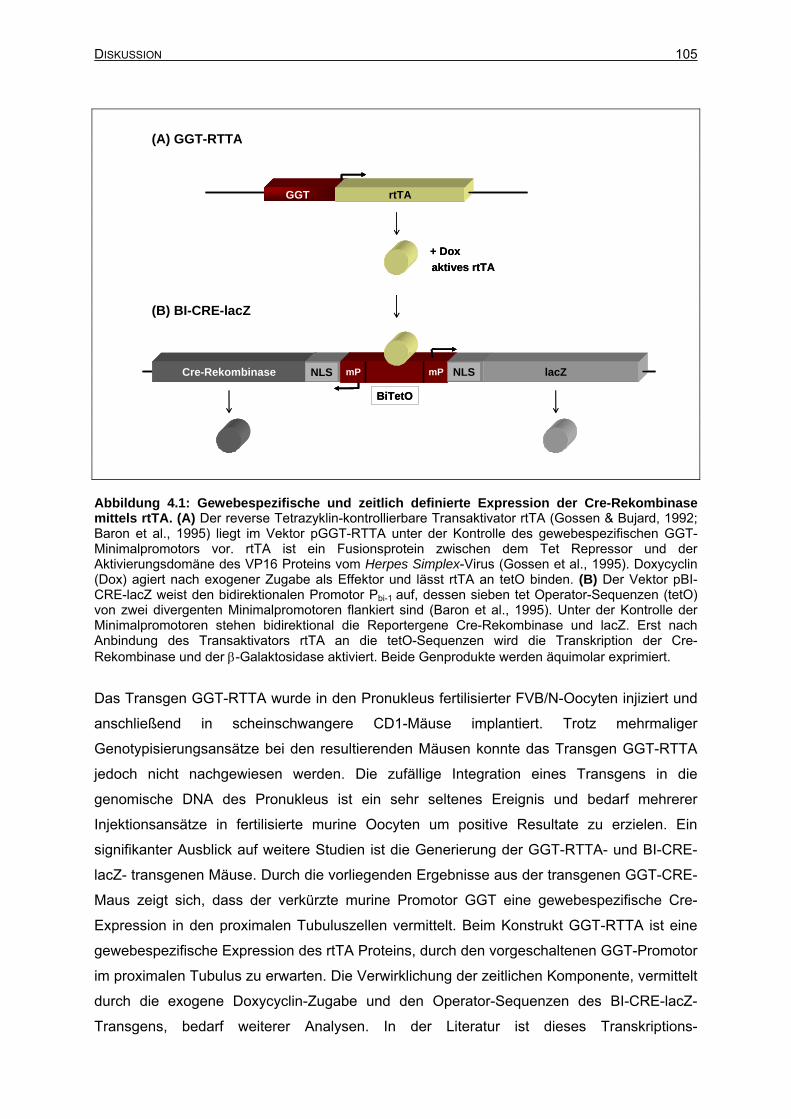
DISKUSSION
105
Abbildung 4.1: Gewebespezifische und zeitlich definierte Expression der Cre-Rekombinase mittels rtTA. (A) Der reverse Tetrazyklin-kontrollierbare Transaktivator rtTA (Gossen & Bujard, 1992; Baron et al., 1995) liegt im Vektor pGGT-RTTA unter der Kontrolle des gewebespezifischen GGT-Minimalpromotors vor. rtTA ist ein Fusionsprotein zwischen dem Tet Repressor und der Aktivierungsdomäne des VP16 Proteins vom Herpes Simplex-Virus (Gossen et al., 1995). Doxycyclin (Dox) agiert nach exogener Zugabe als Effektor und lässt rtTA an tetO binden. (B) Der Vektor pBI-CRE-lacZ weist den bidirektionalen Promotor Pbi-1 auf, dessen sieben tet Operator-Sequenzen (tetO) von zwei divergenten Minimalpromotoren flankiert sind (Baron et al., 1995). Unter der Kontrolle der Minimalpromotoren stehen bidirektional die Reportergene Cre-Rekombinase und lacZ. Erst nach Anbindung des Transaktivators rtTA an die tetO-Sequenzen wird die Transkription der Cre-Rekombinase und der β-Galaktosidase aktiviert. Beide Genprodukte werden äquimolar exprimiert.
Das Transgen GGT-RTTA wurde in den Pronukleus fertilisierter FVB/N-Oocyten injiziert und
anschließend in scheinschwangere CD1-Mäuse implantiert. Trotz mehrmaliger
Genotypisierungsansätze bei den resultierenden Mäusen konnte das Transgen GGT-RTTA
jedoch nicht nachgewiesen werden. Die zufällige Integration eines Transgens in die
genomische DNA des Pronukleus ist ein sehr seltenes Ereignis und bedarf mehrerer
Injektionsansätze in fertilisierte murine Oocyten um positive Resultate zu erzielen. Ein
signifikanter Ausblick auf weitere Studien ist die Generierung der GGT-RTTA- und BI-CRE-
lacZ- transgenen Mäuse. Durch die vorliegenden Ergebnisse aus der transgenen GGT-CRE-
Maus zeigt sich, dass der verkürzte murine Promotor GGT eine gewebespezifische Cre-
Expression in den proximalen Tubuluszellen vermittelt. Beim Konstrukt GGT-RTTA ist eine
gewebespezifische Expression des rtTA Proteins, durch den vorgeschaltenen GGT-Promotor
im proximalen Tubulus zu erwarten. Die Verwirklichung der zeitlichen Komponente, vermittelt
durch die exogene Doxycyclin-Zugabe und den Operator-Sequenzen des BI-CRE-lacZ-
Transgens, bedarf weiterer Analysen. In der Literatur ist dieses Transkriptions-
(A) GGT-RTTA
GGT
Cre-Rekombinase NLS
+ Dox
BiTetO
mP mP
rtTA
NLS lacZ
aktives rtTA
GGTGGT
Cre-Rekombinase NLSNLS
+ Dox
BiTetO
mP mP
rtTArtTA
NLS lacZ
aktives rtTA
(B) BI-CRE-lacZ

DISKUSSION
106
Kontrollsystem von Gossen und Bujard (1992) schon erfolgreich zum Einsatz gekommen
und der Tabelle 4.3 zu entnehmen (Hennighausen et al., 1995; Utomo at al., 1999;
Lindeberg & Ebendal, 1999; Saam & Gordon, 1999; Schönig et al., 2002; Radomska et al.,
2002). Schönig et al., entwickelte 2002 ein effizientes rtTA/Cre/loxP-Regulationssystem. Es
zeigte sich durch Kreuzungen der generierten βMCM86-Mäusen mit ROSA26-
Reportermäusen, dass durch die exogene Zugabe von Tetrazyklinen eine effiziente
Cre/loxP-Rekombination außer in Nierenzellen auch in Hepatozyten, Neuronen und in T-
Lymphozyten stattfindet.
Tabelle 4.3: Zeitlich regulierbare Cre-exprimierende transgene Mäuse in der Niere.
Transgen Promotor Expression Literatur
βMCM86 pCAGGS merCremer induzierbar mit 4-Hydroxytamoxifen (Zhang et al., 1996; Verrou et al., 1999)
Podozyten Bugeon et al., 2003
K18IresCreERT2 Keratin 18 CreERT2 induzierbar mit 4-Hydroxytamoxifen (Indra et al.,1999)
Leber, Niere, Gebärmutter
Wen et al., 2003
Mx1Cre Mx1 Interferon induzierbar (Kühn et al., 1995)
Nierenkortex/-tubuli, Glomeruli, Endothelium, Mesangium, Podozyten
Schneider et al., 2003
rtTA(LAP)-1 LAP Tetrazyklin induzierbar (Gossen & Bujard., 1992)
Hepatozyten, Nierenzellen, Neuronen und T-Lymphozyten
Schönig et al., 2002
Die Bedeutung einer zeitlich und örtlich gerichteten Expression der Cre-Rekombinase im
proximalen Tubulus ergibt sich aus der Tatsache, dass es sich bei der Zystenentstehung
beim ADPKD um einen fokalen zweistufigen Prozess handelt. ADPKD setzt eine
Keimbahnmutation in einem Allel voraus, jedoch wird die Zystenbildung durch eine zweite
somatische Mutation zu einem späteren Lebenszeitpunkt im anderen Allel renaler tubulärer
Zellen initiiert. Eine Anregung zur Zystenbildung kann durch eine gerichtete Inaktivierung des
Polycystin1 oder 2 in den proximalen Tubulussegmenten ermöglicht werden und somit zur
Verwirklichung der Entwicklung therapeutischer Ansätze zur Beeinflussung von ADPKD,
verholfen werden.

ZUSAMMENFASSUNG
107
5. Zusammenfassung Die Zystogenese bei Nieren von ADPKD-Patienten ist ein fokaler Prozess: Obwohl diese
Erkrankung einem autosomal dominanten Erbgang folgt, wirkt sich auf zellulärer Ebene ein
rezessiver Mutationsmechanismus aus. Es liegt zunächst eine Keimbahnmutation vor und
erst im Laufe des Lebens ereignen sich somatische Mutationen im normalen Allel (second
hit). In den letzten Jahren sind mehrere konventionelle Mausmutanten für die Gene Pkd1
und Pkd2 generiert worden. Durch die pränatale Letalität ergaben sich Limitierungen bei der
Analyse der ADPKD-Pathogenese. Ferner kann auch trotz Vorliegen lebensfähiger
Nullmutanten die spezifische Funktion des Gens in einem bestimmten Gewebe nicht
untersucht werden, da das Zielgen in diesen Mäusen in jeder Körperzelle, bereits von Beginn
der Embryonalentwicklung an, inaktiviert ist. Diese Limitierungen der permanenten
Gendeletion können durch die Methode der konditionalen somatischen Mutagenese unter
Einsatz des Cre/loxP-Rekombinationssystems umgangen werden. Mittels transgener
Mauslinien, die die Cre-Rekombinase gewebespezifisch exprimieren, können loxP-flankierte
(„gefloxte“) Zielgene selektiv nur in einem bestimmten Zelltyp aus dem Chromosom
herausgeschnitten und damit inaktiviert werden. Somit kann mittels konditionaler
Mutagenese die embryonale Letalität umgangen werden. Ziel dieser Arbeit war es transgene
Mäuse zu entwickeln, die gewebespezifisch im proximalen Tubulus der Niere die Cre-
Rekombinase exprimieren. Es wurde die GGT-CRE-Mauslinie generiert, welche die Cre-
Rekombinase unter der Kontrolle des murinen Minimalpromotors Gamma-Glutamyl-
Transpeptidase Typ II stellt. Dieser Promotor erlaubt eine nierenspezifische Expression des
Cre-Proteins in den proximalen Tubuluszellen. Die erzeugte GGT-CRE-Mauslinie wurde mit
Cre-Reportermäusen (ROSA26 und Z/AP) verkreuzt und auf ihre Expressions- und
Rekombinationseigenschaften hin untersucht. Beide Reporterlinien tragen ein Markergen,
welches erst nach einer Cre-vermittelten Excision gefloxter Terminationseinheiten exprimiert
werden kann. Unter Verwendung von histochemischen Methoden für die alkalische
Phosphatase bei der Z/AP-Mauslinie und für die β-Galaktosidase bei der ROSA26-Mauslinie
konnte sehr deutlich die Expression einer katalytisch aktiven Cre-Rekombinase in den
proximalen Tubuluszellen der Niere doppeltransgener Tiere beobachtet werden. Ferner
konnte die genaue zelluläre Lokalisation der Cre-Rekombinase, welche sich im Zellkern der
proximalen Tubuluszellen befand, durch immunhistochemischer Markierung mittels Cre-
Antikörpern und gewebespezifischen Lektinen demonstriert werden. Die GGT-CRE-
Mauslinie kann durch die Einführung genomischer Veränderungen (Cre/loxP-Rekombination)
spezifisch in den proximalen Tubuluszellen der Niere einen wichtigen Beitrag bei der
Erklärung der Mechanismen für die Zystenformierung leisten. Ferner erlaubt diese transgene
Mauslinie durch ihre embryonale gewebespezifische Cre-Expression eine Untersuchung
diverser Gene welche speziell in der Nierenentwicklung involviert sind.

LITERATUR
108
6 Literatur
Aaij C, Borst P (1972), The gel electrophoresis of DNA. Biochim Biophys Acta 269: 192-200
Abremski K, Hoess R (1984), Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. J Biol Chem 259: 1509-14
Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K, Pimprale S, Lee YH, Ricote M, Glass CK, Brewer HB Jr, Gonzalez FJ (2002), Conditional disruption of the peroxisome proliferator-activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Mol Cell Biol 22: 2607-19
Avner ED, Studnicki FE, Young MC, Sweeney WE Jr, Piesco NP, Ellis D, Fettermann GH (1987), Congenital murine polycystic kidney disease. I. The ontogeny of tubular cyst formation. Pediatr Nephrol 1: 587-96
Bachinsky DR, Sabolic I, Emmanouel DS, Jefferson DM, Carone FA, Brown D, Perrone RD (1995), Water channel expression in human ADPKD kidneys. Am J Physiol 268: F398-F403
Baron U, Freundlieb S, Gossen M, Bujard H (1995), Co-regulation of two gene activities by tetracycline via a bidirectional promoter. Nucleic Acids Res. 23: 3605-6
Basile DP, Emut M, Sorenson C, Rogers J, Hammerman MR, Billadella J (1997), The human PAI-1 promoter directs transgene expression in proximal tubules (abstract). J Am Soc Nephrol1997;8: 357A
Beckwith, JR (1980), Lac: The genetic system. In The Operon (ed. J.H. Miller and W.S. Reznikoff). Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
Birnboim W, Doly J (1979), A rapid alkaline procedure for screening recombinant plasmid. DNA. Nucleic Acids Res 7: 1513-23
Blau S, Daly L, Fienberg A, Teitelman G, Ehrlich ME (1995), DARPP-32 promoter directs transgene expression to renal thick ascending limb of loop of Henle. Am J Physiol. 269: F564-70
Bogdanova N, Markoff A, Horst J (2002), Autosomal dominant polycystic kidney disease - clinical and genetic aspects. Kidney Blood Press Res 25: 265-83
Boulter C, Mulroy S, Webb S, Fleming S, Brindle K, Sandford R (2001), Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci U S A. 98: 12174-9
Brill SR, Ross KE, Davidow CJ, Ye M, Grantham JJ, Caplan MJ (1996), Immunolovalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc Natl Acad Sci U S A 93: 10206-11
Brocard J, Warot X, Wendling O, Messaddeq N, Vonesch JL, Chambon P, Metzger D (1997), Spatio-temporally controlled site-specific somatic mutagenesis in the mouse. Proc Natl Acad Sci U S A 94: 14559-63
Brüning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR (1998), A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2: 559-69
Bugeon L, Danou A, Carpentier D, Langridge P, Syed N, Dallman MJ (2003), Inducible gene silencing in podocytes: a new tool for studying glomerular function. J Am Soc Nephrol 14: 786-91
Capecchi MR (1989), The new mouse genetics: altering the genome by gene targeting. Trends Genet. 5: 70-6
Carter BZ, Habib GM, Sepulveda AR, Barrios R, Wan DF, Lebovitz RM, Lieberman MW (1994), Type VI RNA is the major gamma-glutamyl transpeptidase RNA in the mouse small intestine. J Biol Chem. 269: 24581-5
Chen J, Kubalak SW, Chien KR (1998), Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 125: 1943-9
Chien A, Edgar DB, Trela JM (1976), Deoxyribonucleic acid polymerase from the extreme thermophile Thermus aquaticus. J Bacteriol. 127: 1550-7

LITERATUR
109
Chikhi N, Holic N, Guellaen G, Laperche Y (1999), Gamma-glutamyl transpeptidase gene organization and expression: a comparative analysis in rat, mouse, pig and human species. Comp Biochem Physiol B Biochem Mol Biol. 122: 367-80.
Chomczynski P, Qasha PK (1984), Alkaline transfer of DNA to plastic membrane. Biochem Biophys Res Commun 122 : 340-4
Cline J, Braman JC, Hogrefe HH (1996), PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 24: 3546-51
Coons AH, Kaplan MH (1950), Localization of antigen in tissue cells; improvements in a method for the detection of antigen by means of fluorescent antibody. J Exp Med. 91: 1-13
Cowley BD, Gudapaty S, Kraybill AL, Barash BD, Harding MA, Calvet JP, Gattone VH (1993), Autosomal-dominant polycystic kidney disease in the rat. Kidney Int 43: 522-34.
Curto KA, Sweeney WE, Avner ED, Piesco NP, Curthoys NP (1988), Immunocytochemical localization of gamma-glutamyltranspeptidase during fetal development of mouse kidney. J Histochem Cytochem. 36: 159-66
Davisson MT, Guay-Woodford LM, Harris HW, D'Eustachio P (1991), The mouse polycystic kidney disease mutation (cpk) is located on proximal chromosome 12, Genomics 9: 778-781
De Almeida S, de Almeida E, Peters D, Pinto JR, Tavora I, Lavinha J, Breuning M, Prata MM (1995), Autosomal dominant polycystic kidney disease: evidence for the existence of a third locus in a Portuguese family. Hum. Genet. 96: 83-8
DePrimo SE, Stambrook PJ, Stringer JR (1996), Human placental alkaline phosphatase as a histochemical marker of gene expression in transgenic mice. Transgenic Res. 5: 459-66
Devuyst O, Burrow CR, Smith BL, Agre P, Knepper MA, Wilson PD (1996), Expression of aquaporins-1 and 2 during nephrogenesis and in autosomal dominant polycystic kidney disease. Am J Physiol 271: F169-183
Ding Y, Davisson RL, Hardy DO, Zhu LJ, Merrill DC, Catterall JF, Sigmund CD (1997), The kidney androgen-regulated protein promoter confers renal proximal tubule cell-specific and highly androgen-responsive expression on the human angiotensinogen gene in transgenic mice. J Biol Chem. 272: 28142-8.
Du J, Wilson PD (1995), Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am. J. Physiol. 269: C487-95
Ebihara I, Killen PD, Laurie GW, Huang T, Yamada Y, Martin GR, Brown KS (1988), Altered mRNA expression of basement membrane components in a murine model of polycystic kidney disease. Lab Invest 58: 262-9
Emert MP, Sorenson CM, Basile DP, Rogers JG, Hammerman MR, Billadello JJ (1998), The human plasminogen activator inhibitor type I gene promoter targets to kidney. Am J Physiol. 274: F405-12
Eremina V, Wong MA, Cui S, Schwartz L, Quaggin SE (2002), Glomerular-specific gene excision in vivo. J Am Soc Nephrol 13: 788-93
Feil R, Brocard J, Mascrez B, LeMeur M, Metzger D, Chambon P (1996), Ligand-activated site-specific recombination in mice. Proc Natl Acad Sci U S A 93: 10887-90
Feil R, Wagner J, Metzger D, Chambon P (1997), Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun 237: 752-7
Fiers W, Contreras R, Haegemann G, Rogiers R, Van de Voorde A, Van Heuverswyn H, Van Herreweghe J, Volckaert G, Ysebaert M (1978), Complete nucleotide sequence of SV40 DNA. Nature 273: 113-20
Foggensteiner L, Bevan AP, Thomas R, Coleman N, Boulter C, Bradley J, Ibraghimov-Beskrovnaya O, Klinger K, Sandford R (2000), Cellular and subcellular distribution of polycystin-2, the protein product of the PKD2 gene [In Process Citation]. J. Am. Soc. Nephrol. 11: 814-27
Friedrich G, Soriano P (1991), Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev 5: 1513-23

LITERATUR
110
Fuchs S, Germain S, Philippe J, Corvol P, Pinet F (2002), Expression of renin in large arteries outside the kidney revealed by human renin promoter/LacZ transgenic mouse. Am J Pathol 161: 717-25
Gabow PA, Grantham JJ (1997), Polycystic kidney disease. In: Diseases of the Kidney. edited by Schrier RW, Gottschalk CW, Boston, Little, Brown: 521-60
Gallagher AR, Obermuller N, Cedzich A, Gretz N, Witzgall R (2000), An ever-expanding story of cyst formation. Cell Tissue Res. 300: 361-71
Geng L, Segal Y, Peissel B, Deng N, Pei Y, Carone F, Rennke HG, Glucksmann-Kuis AM, Schneider MC, Ericsson M, Reeders ST, Zhou J (1996), Identification and localization of polycystin, the PKD1 gene product, J.Clin.Invest 98: 2674-82
Geng L, Segal Y, Pavlova A, Barros EJ, Lohning C, Lu W, Nigam SK, Frischauf AM, Reeders ST, Zhou J (1997), Distribution and developmentally regulated expression of murine polycystin. Am J Physiol. 272: F451-9
Godin RE, Takaesu NT, Robertson EJ, Dudley AT (1998), Regulation of BMP7 expression during kidney development. Development. 125: 3473-82
Gonzalez-Perrett S, Kim K, Ibarra C, Damiano AE, Zotta E, Batelli M, Harris PC, Reisin IL, Arnaout MA, Cantiello HF (2001), Polycystin-2, the protein mutated in autosomal dominant polycystic kidney disease (ADPKD), is a Ca2+-permeable nonselective cation channel. Proc Natl Acad Sci U S A 98: 1182-7
Gonzalez-Perrett S, Batelli M, Kim K, Essafi M, Timpanaro G, Moltabetti N, Reisin IL, Arnaout MA, Cantiello HF (2002), Voltage dependence and pH regulation of human polycystin-2-mediated cation channel activity. J Biol Chem 277: 24959-66
Gossen M, Bujard H (1992), Tight control of gene expression in mammalian cells by tetracycline responsive promoters. Proc. Natl Acad. Sci. U S A, 89: 5547-51
Gossen M, Bujard H (1993), Anhydrotetracycline, a novel effector for tetracycline controlled gene expression systems in eukaryotic cells. Nucleic Acids Res 21: 4411-2
Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H (1995), Transcriptional activation by tetracyclines in mammalian cells. Science 268: 1766-9
Gould SE, Day M, Jones SS, Dorai H (2002), BMP-7 regulates chemokine, cytokine, and hemodynamic gene expression in proximal tubule cells. Kidney Int. 61: 51-60
Griffin MD, Torres VE, Grande JP, Kumar R (1996), Immunolocalization of polycystin in human tissues and cultured cells. Proc.Assoc.Am.Physicians 108: 185-97
Griffin MD, O' Sullivan DA, Torres VE, Grande JP, Kanwar YS, Kumar R (1997a), Expression of polycystin in mouse metanephros and extra-metanephric tissues. Kidney Int. 52: 1196-205
Griffin MD, Torres VE, Grande JP, Kumar R (1997b), Vascular expression of polycystin, J.Am.Soc.Nephrol. 8: 616-26
Gu H, Zou YR, Rajewsky K (1993), Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 73: 1155-64
Guillaume R, Trudel M (2000), Distinct and common developmental expression patterns of the murine Pkd2 and Pkd1 genes. Mech.Dev. 93: 179-83
Guo F, Gopaul D, Van Duyne G (1997), Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389: 40-6
Habib GM, Carter BZ, Sepulveda AR, Shi ZZ, Wan DF, Lebovitz RM, Lieberman MW (1995). Identification of a sixth promoter that directs the transcription of gamma-glutamyl transpeptidase type III RNA in mouse. J Biol Chem. 270: 13711-5
Haidar MA, Loya F, Yang Y, Lin H, Glassman A, Goldwasser E, Albitar M (1997), Electron microscopic localization of lacZ expression in the proximal convoluted tubular cells of the kidney in transgenic mice carrying chimeric erythropoietin/lacZ gene constructs. J Struct Biol. 1997 118: 220-5

LITERATUR
111
Han H, Tanigaki K, Yamamoto N, Kuroda K, Yoshimoto M, Nakahata T, Ikuta K, Honjo T (2002), Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol 14: 637-45
Hanaoka K, Qian F, Boletta A, Bhunia AK, Piontek K, Tsiokas L, Sukhatme VP, Guggino WB, Germino GG (2000), Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 408: 990-4
Hanigan MH, Frierson HF Jr (1996), Immunohistochemical detection of gamma-glutamyl transpeptidase in normal human tissue. J Histochem Cytochem 44: 1101-8
Hardy DO, Niu EM, Catterall JF (2001), promoter analysis in vivo: a regulatory role for a truncated L1 repeat. Mol Cell Endocrinol. 181: 57-67
Hennighausen L, Wall RJ, Tillmann U, Li M, Furth PA (1995), Conditional gene expression in secretory tissues and skin of transgenic mice using the MMTV-LTR and the tetracycline responsive system. J Cell Biochem 59: 463-72
Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J Jr, Muller W, Chien KR.(1999), Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 97: 189-98
Hobart PM, Fogliano M, O'Connor BA, Schaefer IM, Chirgwin JM (1984), Human renin gene: structure and sequence analysis. Proc Natl Acad Sci U S A. 81: 5026-30
Hoess R, Ziese M, Sternberg N (1982), P1 site-specific recombination: Nucleotide sequence of the recombination sites. Proc Natl Acad Sci USA 79: 3398-402
Hoess RH, Abremski K (1985), Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J Mol Biol. 181: 351-62
Hoess R, Abremski K, Irwin S, Kendall M, Mack A (1990), DNA specificity of the Cre recombinase resides in the 25 kDa carboxyl domain of the protein. J Mol Biol 216: 873-82.
Hogan B, Beddington R, Costantini F, Laca E (1994), Manipulating the Mouse Embryo – A laboratory manual. Cold Spring Harbor Laboratory Press, New York.
Holt SJ, Sadler PW (1958), Studies in enzyme cytochemistry III. Relationships between solubility, molecular association and structure in indigoid dyes. Proc R Soc Lond B Biol Sci 148: 495-505
Holliday R (1964), A mechanism for gene conversion in fungi. Genet. Res. 5: 282-304
Horcher M, Souabni A, Busslinger M (2001), Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity 14: 779-90
Hou X, Mrug M, Yoder BK, Lefkowitz EJ, Kremmidiotis G, D' Eustachio P, Beier DR, Guay-Woodford LM (2002), Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J Clin Invest 109: 533-40
Hughes J, Ward CJ, Peral B, Aspinwall R, Clark K, San Millan JL, Gamble V, Harris PC (1995), The polycystic kidney disease1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat Genet 10: 151-9
Ibraghimov-Beskrovnaya O, Dackovski WR, Foggensteiner L, Coleman N, Thiru S, Petry LR, Burn TC, Connors TD, van Raay T, Bradley J, Qian F, Onuchic LF, Watnick TJ, Piontek K, Hakim R, Landes GM, Germino GG, Sanford R, Klinger KW (1997), Polycystin: In vitro synthesis, in vivo tissue expression, and subcellular localization identifies a large membrane-associated protein. Proc Natl Acad Sci USA 94: 6397-402
Igarashi P, Shashikant CS, Thomson RB, Whyte DA, Liu-Chen S, Ruddle FH, Aronson PS (1999), Ksp-cadherin gene promoter. II. Kidney-specific activity in transgenic mice. Am J Physiol. 277: F599-610
Igarashi P, Somlo S (2002), Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol 13: 2384-98
Imai T, Jiang M, Chambon P, Metzger D (2001), Impaired adipogenesis and lipolysis in the mouse upon selective ablation of the retinoid X receptor alpha mediated by a tamoxifen-inducible chimeric Cre recombinase (Cre-ERT2) in adipocytes. Proc Natl Acad Sci USA 98: 224-8

LITERATUR
112
Inage Z, Kikkawa Y, Minato M, Owada M, Kitagawa T, Ohno K, Kondo K, Ueda Y, Iidaka K (1991), Autosomal recessive polycystic kidney in rats. Nephron 59: 637-40
Indra AK, Warot X, Brocard J, Bornert JM, Xiao JH, Chambon P, Metzger D (1999), Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res 27: 4324-7
Ioannidis V, Zimmer J, Beermann F, Held W (2001), Cre recombinase-mediated inactivation of H-2Dd transgene expression: evidence for partial missing self-recognition by Ly49A NK cells. J Immunol 167: 6256-62
Kalderon D, Roberts BL, Richardson WD, Smith AE (1984) A short amino acid sequence able to specify nuclear location. Cell. 39: 499-509
Kaneko T, Takenaka M, Okabe M, Yoshimura Y, Yamauchi A, Horio M, Kwon HM, Handler JS, Imai E (1997), Osmolarity in renal medulla of transgenic mice regulates transcription via 5)- flanking region of canine BGT1 gene. Am J Physiol 272: F610–6
Kellendonk C, Tronche F, Monaghan AP, Angrand PO, Stewart F, Schutz G (1996), Regulation of Cre recombinase activity by the synthetic steroid RU 486. Nucleic Acids Res. 24: 1404-11
Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K, Arnaout MA (2000), Polycystin 1 is required for the structural integrity of blood vessels. Proc Natl Acad Sci U S A 97: 1731-6
Kim HT, Song IY, Piedrahita J (2003), Kidney-specific activity of the bovine uromodulin promoter. Transgenic Res. 12: 191-201
Kimberling WJ, Fain PR, Kenyon JB, Goldgar D, Sujansky E, Gabow PA (1988), Linkage heterogeneity of autosomal dominant polycystic kidney disease. N Engl J Med 319: 913-8
Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H (1996), Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci U S A 93: 10933-8
Knott A, Garke K, Urlinger S, Guthmann J, Muller Y, Thellmann M, Hillen W (2002), Tetracycline-dependent gene regulation: combinations of transregulators yield a variety of expression windows. Biotechniques. 32: 796, 798, 800
Knudson AG Jr (1971), Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A 68: 820-3
Koponen JK, Kankkonen H, Kannasto J, Wirth T, Hillen W, Bujard H, Yla-Herttuala S (2003), Doxycycline-regulated lentiviral vector system with a novel reverse transactivator rtTA2S-M2 shows a tight control of gene expression in vitro and in vivo. Gene Ther. 10: 459-66
Korhonen J, Lahtinen I, Halmekyto M, Alhonen L, Janne J, Dumont D, Alitalo K (1995) Endothelial-specific gene expression directed by the tie gene promoter in vivo. Blood 86: 1828-35
Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S (2002), Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 4: 191-7
Kruithof EK, Cousin E (1988), Plasminogen activator inhibitor 2. Isolation and characterization of the promoter region of the gene. Biochem Biophys Res Commun. 156: 383-8
Kühn R, Schwenk F, Aguet M, Rajewsky K (1995), Inducible gene targeting in mice. Science 269: 1427-9
Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR (1999), Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96: 329-39
Lakso M, Sauer B, Mosinger B Jr, Lee EJ, Manning RW, Yu SH, Mulder KL, Westphal H (1992), Targeted oncogene activation by site-specific recombination in transgenic mice. Proc. Natl. Acad. Sci. USA 89: 6232-6
Lanford RE, White RG, Dunham RG, Kanda P. (1988), Effect of basic and nonbasic amino acid substitutions on transport induced by simian virus 40 T-antigen synthetic peptide nuclear transport signals. Mol Cell Biol. 8: 2722-9

LITERATUR
113
Lanoix J, D'Agati V, Szabolcs M Trudel M (1996), Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 13: 1153-60
Leheste JR, Melsen F, Wellner M, Jansen P, Schlichting U, Renner-Muller I, Andreassen TT, Wolf E, Bachmann S, Nykjaer A, Willnow TE (2003), Hypocalcemia and osteopathy in mice with kidney-specific megalin gene defect. FASEB J 17: 247-9
Leone DP, Genoud S, Atanasoski S, Grausenburger R, Berger P, Metzger D, Macklin WB, Chambon P, Suter U (2003), Tamoxifen-inducible glia-specific Cre mice for somatic mutagenesis in oligodendrocytes and Schwann cells. Mol Cell Neurosci 22: 430-40
Li M, Indra AK, Warot X, Brocard J, Messaddeq N, Kato S, Metzger D, Chambon P (2000), Skin abnormalities generated by temporally controlled RXRalpha mutations in mouse epidermis. Nature 407: 633-6
Lieberman MW, Barrios R, Carter BZ, Habib GM, Lebovitz RM, Rajagopalan S, Sepulveda AR, Shi ZZ, Wan DF (1995), Gamma-Glutamyl transpeptidase. What does the organization and expression of a multipromoter gene tell us about its functions? Am J Pathol. 147: 1175-85
Lieu HD, Withycombe SK, Walker Q, Rong JX, Walzem RL, Wong JS, Hamilton RL, Fisher EA, Young SG (2003), Eliminating atherogenesis in mice by switching off hepatic lipoprotein secretion. Circulation 107: 1315-21
Lindeberg J, Ebendal T (1999), Use of an internal ribosome entry site for bicistronic expression of Cre recombinase or rtTA transactivator. Nucleic Acids Res 27: 1552-4
Lobe CG, Koop KE, Kreppner W, Lomeli H, Gertsenstein M, Nagy A (1999), Z/AP, a double reporter for cre-mediated recombination. Dev Biol. 208: 281-92
Long GL, Chandra T, Woo SL, Davie EW, Kurachi K (1984), Complete sequence of the cDNA for human alpha 1-antitrypsin and the gene for the S variant. Biochemistry. 23: 4828-37
Loya F, Yang Y, Lin H, Goldwasser E, Albitar M (1994), Transgenic mice carrying the erythropoietin gene promoter linked to lacZ express the reporter in proximal convoluted tubule cells after hypoxia. Blood. 84: 1831-6
Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, Larson C, Brent G, Zhou J (1997), Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet 17: 179-81
Lu W, Fan X, Basora N, Babakhanlou H, Law T, Rifai N, Harris PC, Perez-Atayde AR, Rennke HG, Zhou J (1999), Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat Genet 21: 160-1
Lu W, Shen X, Pavlova A, Lakkis M, Ward CJ, Pritchard L, Harris PC, Genest DR, Perez-Atayde AR, Zhou J (2001), Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum Mol Genet. 10: 2385-96.
Lundberg KS, Shoemaker DD, Adams MW, Short JM, Sorge JA, Mathur EJ (1991), High-fidelity amplification using a thermostable DNA polymerase isolated from Pyrococcus furiosus. Gene 108: 1-6
Mack A, Sauer B, Abremski K, Hoess R (1992), Stoichiometry of the Cre recombinase bound to the lox recombining site. Nucleic Acids Res 20: 4451-5
Maniatis T, Sambrook J, Fritsch EF (1989), Molecular cloning: A laboratory manual, second edition. Cold Spring Harbor Laboratory, New York
Matsusaka T, Katori H, Inagami T, Fogo A, Ichikawa I (1999), Communication between myocytes and fibroblasts in cardiac remodeling in angiotensin chimeric mice. J Clin Invest 103: 1451-8
Maxwell IH, Harrison GS, Wood WM, Maxwell F (1989), A DNA cassette containing a trimerized SV40 polyadenylation signal which efficiently blocks spurious plasmid-initiated transcription. Biotechniques. 7: 276-80
McDonald JD, Lin FK, Goldwasser E (1986), Cloning, sequencing, and evolutionary analysis of the mouse erythropoietin gene. Mol Cell Biol. 6: 842-8
Meisler MH (1992), Insertional mutation of 'classical' and novel genes in transgenic mice. Trends Genet 8: 341-4

LITERATUR
114
Meister A (1995), Glutathione metabolism. Methods Enzymol. 251: 3-7
Meister A, Larsson A (1995), Glutathione synthetase deficiency and other disorders of the g-glutamyl cycle. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. 7th ed. New York: McGraw-Hill: 1461-77
Metzger D, Clifford J, Chiba H, Chambon P (1995), Conditional site-specific recombination in mammalian cells using a ligand-dependent chimeric Cre recombinase. Proc Natl Acad Sci U S A 92: 6991-5
Metzger D, Feil R. (1999), Engineering the mouse genome by site-specific recombination. Curr Opin Biotechnol. 10: 470-6
Metzger D, Chambon P (2001), Site- and time-specific gene targeting in the mouse. Methods 24: 71-80
Millan JL (1986), Molecular cloning and sequence analysis of human placental alkaline Phosphatase. J Biol Chem. 261: 3112-5
Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, Kimberling WJ, Breuning MH, Deltas CC, Peters DJ, Somlo S (1996), PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 272: 1339-42
Mochizuki T, Saijoh Y, Tsuchiya K, Shirayoshi Y, Takai S, Taya C, Yonekawa H, Yamada K, Nihei H, Nakatsuji N, Overbeek PA, Hamada H, Yokoyama T (1998), Cloning of inv, a gene that controls left/right asymmetry and kidney development. Nature 395: 177-81
Moeller MJ, Kovari IA, Holzman LB (2000), Evaluation of a new tool for exploring podocyte biology: mouse Nphs1 5' flanking region drives LacZ expression in podocytes. J. Am. Soc. Nephrol. 11: 2306-14
Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB (2002), Two gene fragments that direct podocyte-specific expression in transgenic mice. J Am Soc Nephrol. 13: 1561-7
Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB (2003), Podocyte-specific expression of cre recombinase in transgenic mice. Genesis 35: 39-42
Montesano R, Schaller G, Orci L (1991), Induction of epithelial tubular morphogenesis in vitro by fibroblast- derived soluble factors. Cell 66: 697-711
Moyer JH, Lee-Tischler MJ, Kwon HY, Schrick JJ, Avner ED, Sweeney WE, Godfrey VL, Cacheiro NL, Wilkinson JE, Woychik RP (1994), Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 264: 1329-33
Müller U (1999), Ten years of gene targeting: targeted mouse mutants, from vector design to phenotype analysis, Mech.Dev. 82: 3-21
Mullis KB (1990), The unusual origin of the polymerase chain reaction. Sci Am 262: 56-61, 64-5
Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP (2000), The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development 127: 2347-55
Nadasdy T, Laszik Z, Lajoie G, Blick KE, Wheeler DE, Silva FG (1995), Proliferative activity of cyst epithelium in human renal cystic diseases. J Am Soc Nephrol 5: 1462-8
Nagy A (2000), Cre recombinase: The universal reagent for genome tailoring Special Issue: Tissue Specific Expression of Cre Recombinase in Mice. Genesis 26: 99-109
Nelson RD, Stricklett PK, Ausiello DA, Brown D, Kohan DE (1997), Expression of the aquaporin-2 promoter in transgenic mice: Tissue localization and potential use in cell-specific knockouts (abstract). J Am Soc Nephrol 8: 23A
Nelson RD, Stricklett P, Gustafson C, Stevens A, Ausiello D, Brown D, Kohan DE (1998), Expression of an AQP2 Cre recombinase transgene in kidney and male reproductive system of transgenic mice. Am J Physiol. 275: C216-26
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J (2003), Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129-37

LITERATUR
115
Nauta J, Sweeney WE, Rutledge JC, Avner ED (1995), Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr. Res 37: 755-63
Newby LJ, Streets AJ, Zhao Y, Harris PC, Ward CJ, Ong AC (2002), Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J. Biol. Chem. 277: 20763-73
Nidess R, Koch WE, Fried FA, McFarland E, Mandell J (1984), Development of the embryonic murine kidney in normal and congenital polycystic kidney disease: Characterization of approximatic tubular degenerative process as the first observable light microscopic defect. J Urol 131: 156-62
Niu EM, Meseguer A, Catterall J (1991), Genomic organization and DNA sequence of the mouse kidney androgen-regulated protein (KAP) gene. DNA Cell Biol. 10: 41-8
Niwa H, Yamamura K, Miyazaki J (1991), Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108: 193-9
Nomura T (1997), Practical development of genetically engineered animals as human disease models. Lab Anim. Sci 47: 113-7
Obermüller N, Gretz N, Kriz W, van der Woude FJ, Reilly RF, Murer H, Biber J, Witzgall R (1997), Differentiation and cell polarity during renal cyst formation in the Han:SPRD (cy/+) rat, a model for ADPKD. Am J Physiol 273: F357-71
Obermüller N, Gallagher AR, Cai Y, Gassler N, Gretz N, Somlo S, Witzgall R (1999), The rat pkd2 protein assumes distinct subcellular distributions in different organs. Am J Physiol. 277: F914-25
Ogborn MR, Sareen S (1996), Transforming growth factor alpha and epidermal growth factor expression in experimental murine polycystic kidney disease. Pediatr Nephrol. 10: 181-4
Okano R, Mita T, Matsui T (1992), Characterization of a novel promoter structure and its transcriptional regulation of the murine laminin B1 gene. Biochim Biophys Acta. 1132: 49-57
Ong AC, Harris PC, Biddolph S, Bowker C, Ward CJ (1999a), Characterisation and expression of the PKD-1 protein, polycystin, in renal and extrarenal tissues. Kidney Int. 55: 2091-116
Ong AC, Harris PC, Davies DR, Pritchard L, Rossetti S, Biddolph S, Vaux DJ, Migone N, Ward CJ (1999b), Polycystin-1 expression in PKD1, early-onset PKD1, and TSC2/PKD1 cystic tissue. Kidney Int. 56: 1324-33
Ong AC, Ward CJ, Butler RJ, Biddolph S, Bowker C, Torra R, Pei Y, Harris PC (1999c), Coordinate expression of the autosomal dominant polycystic kidney disease proteins, polycystin-2 and polycystin-1, in normal and cystic tissue, Am. J. Pathol. 154: 1721-9
Pei, Y. (2001), A 'two-hit' model of cystogenesis in autosomal dominant polycystic kidney disease. Trends Mol. Med. 7: 151-6
Pennekamp P (2000), Tiermodelle zur Analyse humangenetischer Erkrankungen am Beispiel der autosomal dominanten polyzystischen Nierenerkrankung Typ 2 (ADPKD2). Dissertation Westfälische Wilhelms-Universität, Münster.
Pennekamp P, Karcher C, Fischer A, Schweickert A, Skryabin B, Horst J, Blum M, Dworniczak B (2002), The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol 12: 938-43
Perrone RD (1997), Extrarenal manifestations of ADPKD. Kidney Int 51: 2022-36
Peters DJ, Sandkuijl LA (1992), Genetic heterogeneity of polycystic kidney disease in Europe. Contrib Nephrol 97: 128-39
Peters DJ, Spruit L, Klingel R, Prins F, Baelde HJ, Giordano PC, Bernini LF, de Heer E, Breuning MH, Bruijn JA (1996). Adult, fetal, and polycystic kidney expression of polycystin, the polycystic kidney disease-1 gene product. Lab Invest 75: 221-30
Peters DJ, van de WA, Spruit L, Saris JJ, Breuning MH, Bruijn JA, de Heer E (1999), Cellular localization and tissue distribution of polycystin-1. J.Pathol. 188: 439-46

LITERATUR
116
Petrich BG, Molkentin JD, Wang Y (2003), Temporal activation of c-Jun N-terminal kinase in adult transgenic heart via cre-loxP-mediated DNA recombination. FASEB J 17: 749-51
Piontek KB (2000) 5th Int. Workshop on PKD (11.05.-12.05.2000)
Prasher DC (1995), Using GFP to see the light. Trends Genet.11: 320-3
Preminger GM, Koch WE, Fried FA, McFarland E, Murphy ED, Mandell J (1982), Murine congenital polycystic kidney disease: a model for studying development of cystic disease. J Urol 127: 556-60
Putaala H, Soininen R, Kilpelainen P, Wartiovaara J, Tryggvason K (2001), The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet. 10: 1-8
Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG (1997), PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 16: 179-83
Qian F, Watnick TJ (1999), Somatic mutation as mechanism for cyst formation in autosomal dominant polycystic kidney disease. Mol Genet Metab 68: 237-42
Radomska HS, Gonzalez DA, Okuno Y, Iwasaki H, Nagy A, Akashi K, Tenen DG, Huettner CS (2002), Transgenic targeting with regulatory elements of the human CD34 gene. Blood 100: 4410-9
Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M. Deficient T (1999), cell fate specification in mice with an induced inactivation of Notch1. Immunity 10: 547-58
Raffai RL, Weisgraber KH (2002), Hypomorphic apolipoprotein E mice: a new model of conditional gene repair to examine apolipoprotein E-mediated metabolism. Biol Chem 277: 11064-8
Rajagopalan S, Wan DF, Habib GM, Sepulveda AR, McLeod MR, Lebovitz RM, Lieberman MW (1993), Six mRNAs with different 5' ends are encoded by a single gamma-glutamyltransferase gene in mouse. Proc Natl Acad Sci U S A 90: 6179-83
Reed KC, Mann DA (1985), Rapid transfer of DNA from agarose gels to nylon membranes. Nucleic Acids Res. 13: 7207-21
Ruther U, Tripodi M, Cortese R, Wagner EF (1987), The human alpha-1-antitrypsin gene is efficiently expressed from two tissue-specific promotors in transgenic mice. Nucleic Acids Res. 15: 7519-29
Saam JR, Gordon JI (1999), Inducible gene knockouts in the small intestinal and colonic epithelium. J Biol Chem 274: 38071-82
Sambrook J, Fritsch EF, Maniatis T (1989), Molecular cloning: A laboratory manual, second edition. Cold Spring Harbor Laboratory, New York
Sanger F, Nicklen S, Coulson AR (1977), DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 74: 5463-7
Sato TN, Qin Y, Kozak CA, Audus KL (1993), Tie-1 and tie-2 define another class of putative receptor tyrosine kinase genes expressed in early embryonic vascular system. Proc Natl Acad Sci U S A. 90: 9355-8
Sauer B (1994), Site-specific recombination: developments and applications. Curr Opin Biotechnol. 5: 521-7
Sauer B (1998), Inducible gene targeting in mice using the Cre/lox system. Methods 14: 381-92
Schäfer K, Gretz N, Bader M, Oberbäumer I, Eckardt K-U, Kriz W, Bachmann S (1994), Characterization of the Han-SPRD rat model for hereditary polycystic kidney disease. Kidney Int 46: 134-52
Schlaeger TM, Qin Y, Fujiwara Y, Magram J, Sato TN (1995), Vascular endothelial cell lineage-specific promoter in transgenic mice. Development. 121: 1089-98
Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN (1997), Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci U S A 94: 3058-63

LITERATUR
117
Schneider A, Zhang Y, Guan Y, Davis LS, Breyer MD (2003), Differential, inducible gene targeting in renal epithelia, vascular endothelium, and viscera of Mx1Cre mice. Am J Physiol Renal Physiol 284: F411-7
Schönig K, Schwenk F, Rajewsky K, Bujard H (2002), Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res 30: e134
Sepulveda AR, Carter BZ, Habib GM, Lebovitz RM, Lieberman MW (1994), The mouse gamma-glutamyl transpeptidase gene is transcribed from at least five separate promoters. J Biol Chem. 269: 10699-705
Sepulveda AR, Huang SL, Lebovitz RM, Lieberman MW (1997), A 346-base pair region of the mouse gamma-glutamyl transpeptidase type II promoter contains sufficient cis-acting elements for kidney-restricted expression in transgenic mice. J Biol Chem. 272: 11959-67
Shafikhani S (2002), Factors affecting PCR-mediated recombination. Environ Microbiol. 4: 482-6
Shao X, Johnson JE, Richardson JA, Hiesberger T, Igarashi P (2002a), A minimal Ksp-cadherin promoter linked to a green fluorescent protein reporter gene exhibits tissue-specific expression in the developing kidney and genitourinary tract. J Am Soc Nephrol 13: 1824-36
Shao X, Somlo S, Igarashi P (2002b), Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol 13: 1837-46
Sharif KA, Li C, Gudas LJ (2001), cis-acting DNA regulatory elements, including the retinoic acid response element, are required for tissue specific laminin B1 promoter/lacZ expression in transgenic mice. Mech Dev. 103: 13-25
Shastry BS (1994), More to learn from gene knockouts. Mol Cell Biochem 136:171-82
Shastry BS (1995), Genetic knockouts in mice: an update. Experimentia 51: 1028-39
Shastry BS (1998), Gene disruption in mice: models of development and disease. Mol Cell Biochem 181: 163-79
Shi ZZ, Habib GM, Lebovitz RM, Lieberman MW (1995), Cloning of cDNA and genomic structure of the mouse gamma-glutamyl transpeptidase-encoding gene. Gene 167: 233-7
Shi ZZ, Carter BZ, Habib GM, He X, Sazer S, Lebovitz RM, Lieberman MW (1996), A single mouse glutathione synthetase gene encodes six mRNAs with different 5' ends. Arch Biochem Biophys. 331: 215-24
Shuman S (1994), Novel approach to molecular cloning and polynucleotide synthesis using vaccinia DNA topoisomerase. J Biol Chem. 269: 32678-84
Sifers RN, Carlson JA, Clift SM, DeMayo FJ, Bullock DW, Woo SL (1987), Tissue specific expression of the human alpha-1-antitrypsin gene in transgenic mice. Nucleic Acids Res. 15: 1459-75
Simon EA, Cook S, Davisson MT, D'Eustachio P, Guay-Woodford LM (1994), The mouse congenital polycystic kidney (cpk) locus maps within 1.3 cM of the chromosome 12 marker D12Nyu2. Genomics. 21: 415-8
Simon M, Feliers D, Arar M, Bhandari B, Abboud HE (2002), Cloning of the 5'-flanking region of the murine bone morphogenetic protein-7 gene. Mol Cell Biochem. 233: 31-7
Simon, P (1995) Prognosis of autosomal dominant polycystic kidney disease. Nephron 71: 247-8
Simoni M (1994), Transgenic animals in male reproduction research. Exp.Clin.Endocrinol. 102: 419-33
Singer M, Berg P (1992), Gene und Genome. Spektrum Akademischer Verlag. Heidelberg, Berlin, New York
Sinn PL, Davis DR, Sigmund CD (1999a), Highly regulated cell type-restricted expression of human renin in mice containing 140- or 160-kilobase pair P1 phage artificial chromosome transgenes. J Biol Chem. 274: 35785-93
Sinn PL, Zhang X, Sigmund CD (1999b), JG cell expression and partial regulation of a human renin genomic transgene driven by a minimal renin promoter. Am J Physiol. 277: F634-42
Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21: 70-1

LITERATUR
118
Southern EM (1975), Detection of specific sequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98: 503-17
Sternberg N, Hamilton D (1981), Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J Mol Biol 150: 467-86
Sternberg N, Hamilton D, Hoess R (1981), Bacteriophage P1 site-specific recombination. II. Recombination between loxP and the bacterial chromosome. J Mol Biol 150: 487-507
Sternberg N, Sauer B, Hoess R, Abremski K (1986), Bacteriophage P1 cre gene and its regulatory region. Evidence for multiple promoters and for regulation by DNA methylation. J Mol Biol. 187: 197-212
Strandberg L, Lawrence D, Ny T (1988), The organization of the human-plasminogen-activator-inhibitor-1 gene. Implications on the evolution of the serine-protease inhibitor family. Eur. J. Biochem. 176: 609-16
Stricklett PK, Taylor D, Nelson RD, Kohan DE (2003), Thick ascending limb-specific expression of Cre recombinase. Am J Physiol Renal Physiol 285: F33-9
Takahashi H, Calvet JP, Dittemore-Hoover D, Yoshida K, Grantham JJ, Gattone VH (1991), A hereditary model of slowly progressive polycystic kidney disease in the mouse. J Am Soc Nephrol 1: 980-9
Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK (2001), Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell 12: 589-99
The American PKD1 Consortium (1995), Analysis of the genomic sequence for the autosomal dominant polycystic kidney disease (PKD1) gene predicts the presence of a leucine-rich repeat. Hum. Mol. Genet. 4: 575-82
The International Polycystic Kidney Disease Consortium (1994), The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 77: 881–94
The International Polycystic Kidney Disease Consortium (1995), Polycystic kidney disease: The complete structure of the PKD1 gene and its protein. Cell 81: 289-98
Thomas KR, Capecchi MR (1987), Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51: 503-12
Tomita S, Sinal CJ, Yim SH, Gonzalez FJ (2000), Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1alpha. Mol Endocrinol 14: 1674-81
Torra R, Badenas C, San Millan JL, Perez-Oller L, Estivill X, Darnell A (1999), A loss-of-function model for cystogenesis in human autosomal dominant polycystic kidney disease type 2, Am.J.Hum.Genet. 65: 345-52
Torra R, Badenas C, Perez-Oller L, San Millan JL, Nicolau C, Oppenheimer F, Mila M, Darnell A (2000), Increased prevalence of polycystic kidney disease type 2 among elderly polycystic patients. Am J Kidney Dis 36: 728-34
Torres VE (1999), Extrarenal manifestations of autosomal dominant polycystic kidney disease. Am J Kidney Dis 34: xlv-xlviii
Toshima J, Nakagawara K, Mori M, Noda T, Mizuno K. Structural organization and chromosomal localization of the mouse tesk1 (testis-specific protein kinase 1) gene. Gene. 1998 Jan 12; 206(2): 237-45
Toshima J, Toshima JY, Suzuki M, Noda T, Mizuno K (2001), Cell-type-specific expression of a TESK1 promoter-linked lacZ gene in transgenic mice. Biochem Biophys Res Commun. 286: 566-73
Trudel M, D'Agati V, Costantini F (1991), C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int. 39: 665-71
Tsiokas L, Kim E, Arnould T, Sukhatme VP, Walz G (1997), Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2. Proc Natl Acad Sci U S A. 94: 6965-70

LITERATUR
119
Urlinger S, Baron U, Thellmann M, Hasan MT, Bujard H, Hillen W (2000), Exploring the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield expanded range and sensitivity. Proc Natl Acad Sci U S A. 97: 7963-8
Utomo AR, Nikitin AY, Lee WH (1999), Temporal, spatial, and cell type-specific control of Cre-mediated DNA recombination in transgenic mice. Nat Biotechnol 17: 1091-6
van Adelsberg J (1999a), Peptides from the PKD repeats of polycystin, the PKD1 gene product, modulate pattern formation in the developing kidney. Dev.Genet. 24: 299-308
van Adelsberg J (1999b), The role of the polycystins in kidney development. Pediatr.Nephrol. 13: 454-9
Vassilev PM, Guo L, Chen XZ, Segal Y, Peng JB, Basora N, Babakhanlou H, Cruger G, Kanazirska M, Ye Cp, Brown EM, Hediger MA, Zhou J (2001), Polycystin-2 is a novel cation channel implicated in defective intracellular Ca2+ homeostasis in polycystic kidney disease. Biochem Biophys Res Commun 282: 341-50
Verrou C, Zhang Y, Zurn C, Schamel WW, Reth M (1999), Comparison of the tamoxifen regulated chimeric Cre recombinases MerCreMer and CreMer. Biol Chem 380: 1435-8
Vina J (1990), In Glutathione: Metabolism and Physiologic Functions, CRC Press, Boca Raton, FL: 79-89
Ward CJ, Turley H, Ong AC, Comley M, Biddolph S, Chetty R, Ratcliffe PJ, Gattner K, Harris PC (1996), Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney, Proc.Natl.Acad.Sci.U.S.A 93: 1524-8
Wen F, Cecena G, Munoz-Ritchie V, Fuchs E, Chambon P, Oshima RG (2003), Expression of conditional cre recombinase in epithelial tissues of transgenic mice. Genesis 35: 100-6
Weston BS, Jeffery S, Jeffrey I, Sharaf SF, Carter N, Saggar-Malik A, Price RG (1997), Polycystin expression during embryonic development of human kidney in adult tissues and ADPKD tissue, Histochem.J. 29: 847-56
Whitfield JB (2001), Gamma glutamyl transferase. Crit Rev Clin Lab Sci. 38: 263-355
Whyte DA, Li C, Thomson RB, Nix SL, Zanjani R, Karp SL, Aronson PS, Igarashi P (1999), Ksp-cadherin gene promoter. I. Characterization and renal epithelial cell-specific activity. Am J Physiol. 277: F587-98
Wierzbicki A, Kendall M, Abremski K, Hoess R (1987), A mutational analysis of the bacteriophage P1 recombinase Cre. J Mol Biol 195: 785-94
Wight DC, Wagner TE (1994), Transgenic mice: a decade of progress in technology and research. Mutat Res. 307: 429-40
Wilson PD (1991), Aberrant epithelial cell growth in autosomal dominant polycystic kidney disease, Am.J.Kidney Dis. 17: 634-7
Wilson PD, Hreniuk D, Gabow PA (1992), Abnormal extracellular matrix and excessive growth of human adult polycystic kidney disease epithelia. J Cell Physiol 150: 360-9
Wilson PD (1997), Epithelial cell polarity and disease. Am J Physiol. 272: F434-42
Wilson PD (2001), Polycystin: new aspects of structure, function, and regulation. J Am Soc Nephrol 12: 834-45
Witzgall R (1999), The proximal tubule phenotype and its disruption in acute renal failure and polycystic kidney disease. Exp Nephrol 7: 15-9
Wu G, D'Agati V, Cai Y, Markowitz G, Park JH, Reynolds DM, Maeda Y, Le TC, Hou H Jr, Kucherlapati R, Edelmann W, Somlo S (1998), Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93: 177-188
Wu G, Markowitz GS, Li L, D' Agati VD, Factor SM, Geng L, Tibara S, Tuchman J, Cai Y, Park JH, van Adelsberg J, Hou H Jr, Kucherlapati R, Edelmann W, Somlo S (2000), Cardiac defects and renal failure in mice with targeted mutations in Pkd2. Nat Genet 24: 75-8
Xu GM, Gonzalez-Perrett S, Essafi M, Timpanaro GA, Montalbetti N, Arnaout MA, Cantiello HF (2003), Polycystin-1 activates and stabilizes the polycystin-2 channel. J Biol Chem 278: 1457-62

LITERATUR
120
Yamamoto A, Lucas JJ, Hen R (2000), Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57-66
Yang J, Goldstein JL, Hammer RE, Moon YA, Brown MS, Horton JD (2001), Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc Natl Acad Sci U S A 98: 13607-12
Yoder BK, Richards WG, Sweeney WE, Wilkinson JE, Avener ED, Woychik RP (1995), Insertional mutagenesis and molecular analysis of a new gene associated with polycystic kidney disease. Proc Assoc Am Physicians 107: 314-23
Yoder BK, Richards WG, Sommardahl C, Sweeney WE, Michaud EJ, Wilkinson JE, Avner ED, Woychik RP (1996), Functional correction of renal defects in a mouse model for ARPKD through expression of the cloned wild-type Tg737 cDNA, Kidney Int. 50: 1240-8
Yoder BK, Hou X, Guay-Woodford LM (2002a), The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol 13: 2508-16
Yoder BK, Tousson A, Millican L, Wu JH, Bugg CE Jr, Schafer JA, Balkovetz DF (2002b), Polaris, a protein disrupted in orpk mutant mice, is required for assembly of renal cilium. Am J Physiol Renal Physiol 282: F541-52
Yokoo T, Ohashi T, Utsunomiya Y, Shiba H, Shen JS, Hisada Y, Eto Y, Kawamura T, Hosoya T (2001), Inflamed glomeruli-specific gene activation that uses recombinant adenovirus with the Cre/loxP system. J Am Soc Nephrol 12: 2330-7
Yu H, Papa F, Sukhatme VP (1994), Bovine and rodent tamm-horsfall protein (THP) genes: cloning, structural analysis, and promoter identification. Gene Expr. 4: 63-75
Yuan HT, Suri C, Landon DN, Yancopoulos GD, Woolf AS (2000), Angiopoietin-2 is a site-specific factor in differentiation of mouse renal vasculature. J Am Soc Nephrol. 11: 1055-66
Zambrowicz BP, Imamoto A, Fiering S, Herzenberg LA, Kerr WG, Soriano P (1997), Disruption of overlapping transcripts in the ROSA beta geo 26 gene trap strain leads to widespread expression of beta-galactosidase in mouse embryos and hematopoietic cells. Proc Natl Acad Sci U S A 94: 3789-94
Zhang Y, Riesterer C, Ayrall AM, Sablitzky F, Littlewood TD, Reth M (1996), Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res 24: 543-8
Zharkikh L, Zhu X, Stricklett PK, Kohan DE, Chipman G, Breton S, Brown D, Nelson RD (2002), Renal principal cell-specific expression of green fluorescent protein in transgenic mice. Am J Physiol Renal Physiol. 283: F1351-64
Zhu X, Cheng J, Gao J, Lepor H, Zhang ZT, Pak J, Wu XR (2002), Isolation of mouse THP gene promoter and demonstration of its kidney-specific activity in transgenic mice. Am J Physiol Renal Physiol. 282: F608-17
Zinyk DL, Mercer EH, Harris E, Anderson DJ, Joyner AL (1998), Fate mapping of the mouse midbrain-hindbrain constriction using a site-specific recombination system. Curr Biol. 8: 665-8

ANHANG
121
7 Anhang
7.1 Restriktionsendonukleasen
Restriktionsenzym Erkennungsstelle
BamH1 G!GATCC
BglII A!GATCT
Eco47III AGC!GCT
EcoRI G!AATTC
EcoRV GAT!ATC
HincII GTY!RAC
HindIII A!AGCTT
HpaI GTT!AAC
KpnI GGTAC!C
NcoI C!CATGG
NheI G!CTAGC
NotI GC!GGCCGC
PstI CTGCA!G
PvuII CAG!CTG
SalI G!TCGAC
SphI GCATG!C
XbaI T!CTAGA
XhoI C!TCGAG

ANHANG
122
7.2 Plasmidkarten
pSL300/PgkNeoL (B. Skryabin, ZMBE, Münster); Resistenz: Ampicillin, Kanamycin/Geneticin (G418); Wirt: E. coli; eukaryotische Zellen
pHD2-Cre1 (C. Kellendonk, German Cancer Research Center, Heidelberg, Kellendonk et al., 1996); NLS-Cre-Rekombinase (pMC-Cre: Gu et al., 1993); (NLS : Kalderon et al., 1984; Cre: Sternberg et al., 1986) ; HSV-TK, SV40-polyA; Resistenz: Ampicillin; Wirt: E. coli
pHD2-Cre1 4,5 kb
pSL300/pgkNeoL 4892 bp
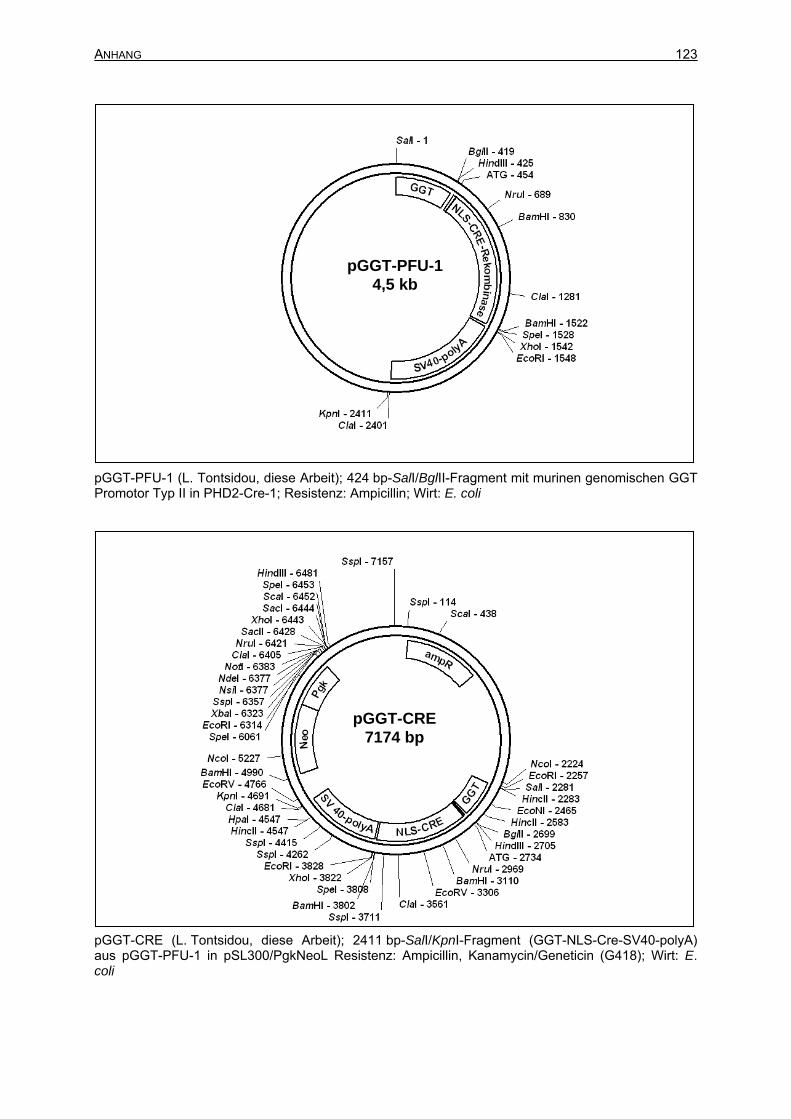
ANHANG
123
pGGT-PFU-1 (L. Tontsidou, diese Arbeit); 424 bp-SalI/BglII-Fragment mit murinen genomischen GGT Promotor Typ II in PHD2-Cre-1; Resistenz: Ampicillin; Wirt: E. coli
pGGT-CRE (L. Tontsidou, diese Arbeit); 2411 bp-SalI/KpnI-Fragment (GGT-NLS-Cre-SV40-polyA) aus pGGT-PFU-1 in pSL300/PgkNeoL Resistenz: Ampicillin, Kanamycin/Geneticin (G418); Wirt: E. coli
pGGT-PFU-1 4,5 kb
pGGT-CRE 7174 bp
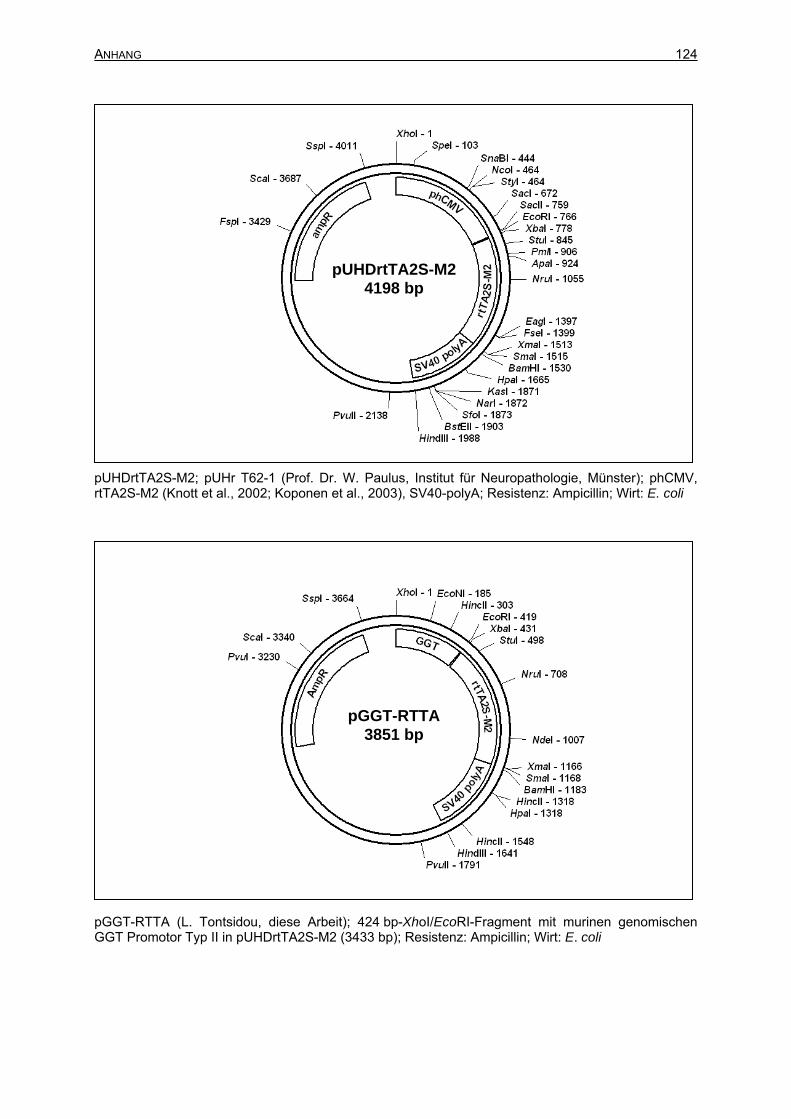
ANHANG
124
pUHDrtTA2S-M2; pUHr T62-1 (Prof. Dr. W. Paulus, Institut für Neuropathologie, Münster); phCMV, rtTA2S-M2 (Knott et al., 2002; Koponen et al., 2003), SV40-polyA; Resistenz: Ampicillin; Wirt: E. coli
pGGT-RTTA (L. Tontsidou, diese Arbeit); 424 bp-XhoI/EcoRI-Fragment mit murinen genomischen GGT Promotor Typ II in pUHDrtTA2S-M2 (3433 bp); Resistenz: Ampicillin; Wirt: E. coli
pGGT-RTTA 3851 bp
pUHDrtTA2S-M2 4198 bp
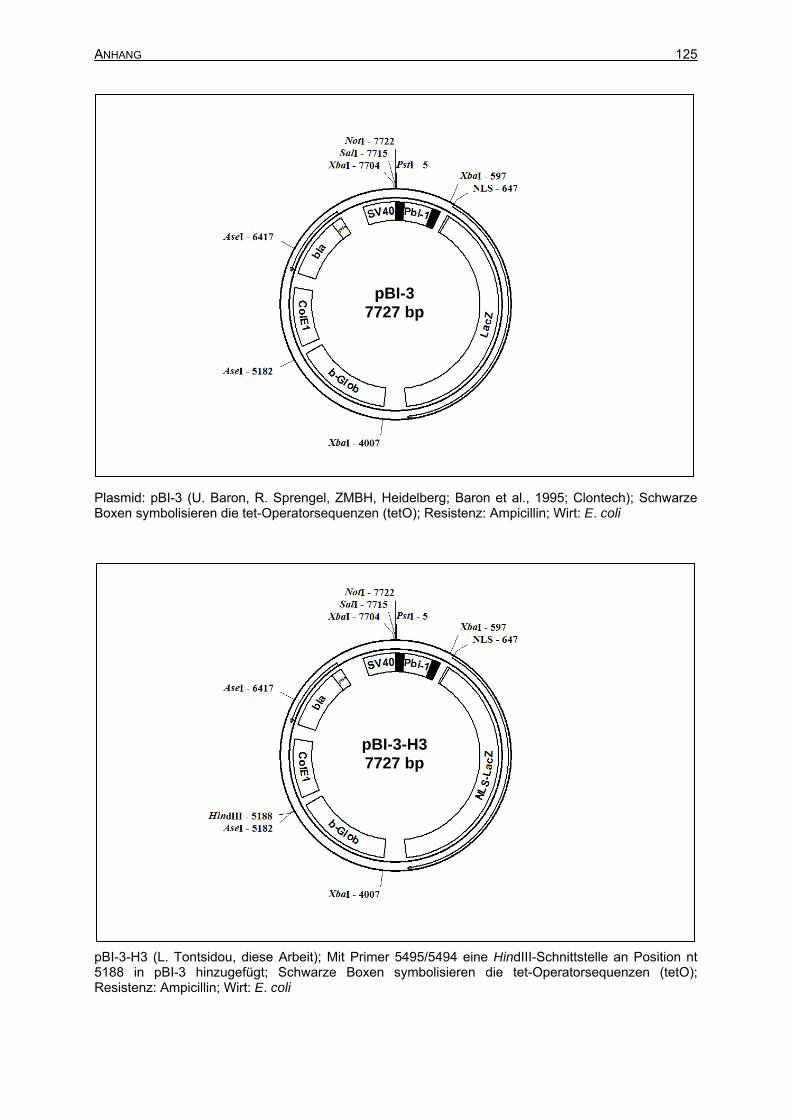
ANHANG
125
Plasmid: pBI-3 (U. Baron, R. Sprengel, ZMBH, Heidelberg; Baron et al., 1995; Clontech); Schwarze Boxen symbolisieren die tet-Operatorsequenzen (tetO); Resistenz: Ampicillin; Wirt: E. coli
pBI-3-H3 (L. Tontsidou, diese Arbeit); Mit Primer 5495/5494 eine HindIII-Schnittstelle an Position nt 5188 in pBI-3 hinzugefügt; Schwarze Boxen symbolisieren die tet-Operatorsequenzen (tetO); Resistenz: Ampicillin; Wirt: E. coli
pBI-3 7727 bp
pBI-3-H3 7727 bp

ANHANG
126
pBI-CRE-lacZ (L. Tontsidou, diese Arbeit); ein PstI/NotI-Fragment aus PHD2-Cre kloniert in pBI-3-H3; Schwarze Boxen symbolisieren die tet-Operatorsequenzen (tetO); Resistenz: Ampicillin; Wirt: E. coli.
pBI-CRE-lacZ 9696 bp

ANHANG
127
7.3 DNA-Sequenzen 7.3.1 Sequenzname: pGGT-CRE Länge: 7174 bp Vektorrückgrat : nt 1-2286 GGT Promotor II: nt 2287-2704 ATG: nt 2734-2736 NLS: nt 2737-2757 Cre-ORF2: nt 2758-3786 SV40-polyA: nt 3834-4682 Vektorrückgrat: nt 4683-7174 1 CAGGTGGCAC TTTTCGGGGA AATGTGCGCG GAACCCCTAT TTGTTTATTT 51 TTCTAAATAC ATTCAAATAT GTATCCGCTC ATGAGACAAT AACCCTGATA 101 AATGCTTCAA TAATATTGAA AAAGGAAGAG TATGAGTATT CAACATTTCC 151 GTGTCGCCCT TATTCCCTTT TTTGCGGCAT TTTGCCTTCC TGTTTTTGCT 201 CACCCAGAAA CGCTGGTGAA AGTAAAAGAT GCTGAAGATC AGTTGGGTGC 251 ACGAGTGGGT TACATCGAAC TGGATCTCAA CAGCGGTAAG ATCCTTGAGA 301 GTTTTCGCCC CGAAGAACGT TTTCCAATGA TGAGCACTTT TAAAGTTCTG 351 CTATGTGGCG CGGTATTATC CCGTATTGAC GCCGGGCAAG AGCAACTCGG 401 TCGCCGCATA CACTATTCTC AGAATGACTT GGTTGAGTAC TCACCAGTCA 451 CAGAAAAGCA TCTTACGGAT GGCATGACAG TAAGAGAATT ATGCAGTGCT 501 GCCATAACCA TGAGTGATAA CACTGCGGCC AACTTACTTC TGACAACGAT 551 CGGAGGACCG AAGGAGCTAA CCGCTTTTTT GCACAACATG GGGGATCATG 601 TAACTCGCCT TGATCGTTGG GAACCGGAGC TGAATGAAGC CATACCAAAC 651 GACGAGCGTG ACACCACGAT GCCTGTAGCA ATGGCAACAA CGTTGCGCAA 701 ACTATTAACT GGCGAACTAC TTACTCTAGC TTCCCGGCAA CAATTAATAG 751 ACTGGATGGA GGCGGATAAA GTTGCAGGAC CACTTCTGCG CTCGGCCCTT 801 CCGGCTGGCT GGTTTATTGC TGATAAATCT GGAGCCGGTG AGCGTGGGTC 851 TCGCGGTATC ATTGCAGCAC TGGGGCCAGA TGGTAAGCCC TCCCGTATCG 901 TAGTTATCTA CACGACGGGG AGTCAGGCAA CTATGGATGA ACGAAATAGA 951 CAGATCGCTG AGATAGGTGC CTCACTGATT AAGCATTGGT AACTGTCAGA 1001 CCAAGTTTAC TCATATATAC TTTAGATTGA TTTAAAACTT CATTTTTAAT 1051 TTAAAAGGAT CTAGGTGAAG ATCCTTTTTG ATAATCTCAT GACCAAAATC 1101 CCTTAACGTG AGTTTTCGTT CCACTGAGCG TCAGACCCCG TAGAAAAGAT 1151 CAAAGGATCT TCTTGAGATC CTTTTTTTCT GCGCGTAATC TGCTGCTTGC 1201 AAACAAAAAA ACCACCGCTA CCAGCGGTGG TTTGTTTGCC GGATCAAGAG 1251 CTACCAACTC TTTTTCCGAA GGTAACTGGC TTCAGCAGAG CGCAGATACC 1301 AAATACTGTT CTTCTAGTGT AGCCGTAGTT AGGCCACCAC TTCAAGAACT 1351 CTGTAGCACC GCCTACATAC CTCGCTCTGC TAATCCTGTT ACCAGTGGCT 1401 GCTGCCAGTG GCGATAAGTC GTGTCTTACC GGGTTGGACT CAAGACGATA 1451 GTTACCGGAT AAGGCGCAGC GGTCGGGCTG AACGGGGGGT TCGTGCACAC 1501 AGCCCAGCTT GGAGCGAACG ACCTACACCG AACTGAGATA CCTACAGCGT 1551 GAGCATTGAG AAAGCGCCAC GCTTCCCGAA GGGAGAAAGG CGGACAGGTA 1601 TCCGGTAAGC GGCAGGGTCG GAACAGGAGA GCGCACGAGG GAGCTTCCAG 1651 GGGGAAACGC CTGGTATCTT TATAGTCCTG TCGGGTTTCG CCACCTCTGA 1701 CTTGAGCGTC GATTTTTGTG ATGCTCGTCA GGGGGGCGGA GCCTATGGAA 1751 AAACGCCAGC AACGCGGCCT TTTTACGGTT CCTGGCCTTT TGCTGGCCTT 1801 TTGCTCACAT GTTCTTTCCT GCGTTATCCC CTGATTCTGT GGATAACCGT 1851 ATTACCGCCT TTGAGTGAGC TGATACCGCT CGCCGCAGCC GAACGACCGA 1901 GCGCAGCGAG TCAGTGAGCG AGGAAGCGGA AGAGCGCCCA ATACGCAAAC 1951 CGCCTCTCCC CGCGCGTTGG CCGATTCATT AATGCAGCTG GCACGACAGG 2001 TTTCCCGACT GGAAAGCGGG CAGTGAGCGC AACGCAATTA ATGTGAGTTA 2051 GCTCACTCAT TAGGCACCCC AGGCTTTACA CTTTATGCTT CCGGCTCGTA 2101 TGTTGTGTGG AATTGTGAGC GGATAACAAT TTCACACAGG AAACAGCTAT

ANHANG
128
2151 GACCATGATT ACGCCAAGCT CGAAATTAAC CCTCACTAAA GGGAACAAAA 2201 GCTGGTACGA GGACAGGCTG GAGCCATGGC TGGTGACCAC GTCGTGGAAT 2251 GCCTTCGAAT TCAGCACCTG CACATGGGAC GTCGACTCTA AGCTATGGTC 2301 TAGTGCCTGG GGCTACCCCT ACTTTATTGC CTCAGGAACA TTTTTGAGGA 2351 CATCATGTCT GTCTCTAAAT TCACAGAGGG ACTTAGCAGG GGCTGTCTGG 2401 AAGGGTTCAC CGGTGGCCTC TGCCCCTTAG AGGGAACCAA ATCTGGAAAG 2451 TGGGGGAGCC CCTTTCCCAG GCTCTTAGTA CCTCCCTCCT CCCCCGCCCA 2501 GAGCCCTCCT GGGGCTGCAT GAACTTTAGT CACCTGGTCA CAAGCCTGAC 2551 GCTGCGCCTC CTTGCGGCTG TGACGTCAGC GTTGACCATC CTCCCAGCCT 2601 ATCTGAACCT CTTACAGAGG ACTCCCTACC TCACACCCCA CTCAGAAGCC 2651 TTCAGATGCC CTTCTGGGAT GCTGGGCACC CCTTAGCTGA CCTCTTGCAG 2701 ATCTAAGCTT CCCAATTCCT GCAGCCCTCG ACCATGCCCA AGAAGAAGAG 2751 GAAGGTGTCC AATTTACTGA CCGTACACCA AAATTTGCCT GCATTACCGG 2801 TCGATGCAAC GAGTGATGAG GTTCGCAAGA ACCTGATGGA CATGTTCAGG 2851 GATCGCCAGG CGTTTTCTGA GCATACCTGG AAAATGCTTC TGTCCGTTTG 2901 CCGGTCGTGG GCGGCATGGT GCAAGTTGAA TAACCGGAAA TGGTTTCCCG 2951 CAGAACCTGA AGATGTTCGC GATTATCTTC TATATCTTCA GGCGCGCGGT 3001 CTGGCAGTAA AAACTATCCA GCAACATTTG GGCCAGCTAA ACATGCTTCA 3051 TCGTCGGTCC GGGCTGCCAC GACCAAGTGA CAGCAATGCT GTTTCACTGG 3101 TTATGCGGCG GATCCGAAAA GAAAACGTTG ATGCCGGTGA ACGTGCAAAA 3151 CAGGCTCTAG CGTTCGAACG CACTGATTTC GACCAGGTTC GTTCACTCAT 3201 GGAAAATAGC GATCGCTGCC AGGATATACG TAATCTGGCA TTTCTGGGGA 3251 TTGCTTATAA CACCCTGTTA CGTATAGCCG AAATTGCCAG GATCAGGGTT 3301 AAAGATATCT CACGTACTGA CGGTGGGAGA ATGTTAATCC ATATTGGCAG 3351 AACGAAAACG CTGGTTAGCA CCGCAGGTGT AGAGAAGGCA CTTAGCCTGG 3401 GGGTAACTAA ACTGGTCGAG CGATGGATTT CCGTCTCTGG TGTAGCTGAT 3451 GATCCGAATA ACTACCTGTT TTGCCGGGTC AGAAAAAATG GTGTTGCCGC 3501 GCCATCTGCC ACCAGCCAGC TATCAACTCG CGCCCTGGAA GGGATTTTTG 3551 AAGCAACTCA TCGATTGATT TACGGCGCTA AGGATGACTC TGGTCAGAGA 3601 TACCTGGCCT GGTCTGGACA CAGTGCCCGT GTCGGAGCCG CGCGAGATAT 3651 GGCCCGCGCT GGAGTTTCAA TACCGGAGAT CATGCAAGCT GGTGGCTGGA 3701 CCAATGTAAA TATTGTCATG AACTATATCC GTAACCTGGA TAGTGAAACA 3751 GGGGCAATGG TGCGCCTGCT GGAAGATGGC GATTAGCCAT TAACGCGTGG 3801 GGGATCCACT AGTTCTAGGG GCTCGAGGAA TTCTCTTTGT GAAGGAACCT 3851 TACTTCTGTG GTGTGACATA ATTGGACAAA CTACCTACAG AGATTTAAAG 3901 CTCTAAGGTA AATATAAAAT TTTTAAGTGT ATAATGTGTT AAACTACTGA 3951 TTCTAATTGT TTGTGTATTT TAGATTCCAA CCTATGGAAC TGATGAATGG 4001 GAGCAGTGGT GGAATGCCTT TAATGAGGAA AACCTGTTTT GCTCAGAAGA 4051 AATGCCATCT AGTGATGATG AGGCTACTGC TGACTCTCAA CATTCTACTC 4101 CTCCAAAAAA GAAGAGAAAG GTAGAAGACC CCAAGGACTT TCCTTCAGAA 4151 TTGCTAAGTT TTTTGAGTCA TGCTGTGTTT AGTAATAGAA CTCTTGCTTG 4201 CTTTGCTATT TACACCACAA AGGAAAAAGC TGCACTGCTA TACAAGAAAA 4251 TTATGGAAAA ATATTCTGTA ACCTTTATAA GTAGGCATAA CAGTTATAAT 4301 CATAACATAC TGTTTTTTCT TACTCCACAC AGGCATAGAG TGTCTGCTAT 4351 TAATAACTAT GCTCAAAAAT TGTGTACCTT TAGCTTTTTA ATTTGTAAAG 4401 GGGTTAATAA GGAATATTTG ATGTATAGTG CCTTGACTAG AGATCATAAT 4451 CAGCCATACC ACATTTGTAG AGGTTTTACT TGCTTTAAAA AACCTCCCAC 4501 ACCTCCCCCT GAACCTGAAA CATAAAATGA ATGCAATTGT TGTTGTTAAC 4551 TTGTTTATTG CAGCTTATAA TGGTTACAAA TAAAGCAATA GCATCACAAA 4601 TTTCACAAAT AAAGCATTTT TTTCACTGCA TTCTAGTTGT GGTTTGTCCA 4651 AACTCATCAA TGTATCTTAT CATGTCTGCA TCGATGGGTA CCGGTTGTTG 4701 ATCTATAGAT CATGAGTGGA GGAATGAGCT GGCCCTTAAT TTGGTTTTGC 4751 TTGTTTAAAT TATGATATCC AACTATGAAA CATTATCATA AAGCAATAGT 4801 AAAGAGCCTT CAGTAAAGAG CAGGCATTTA TCTAATCCCA CCCCACCCCC 4851 ACCCCCGTAG CTCCAATCCT TCCATTCAAA ATGTAGGTAC TCTGTTCTCA 4901 CCCTTCTTAA CAAAGTATGA CAGTAAAAAA CTTCCATTTT AGTGGACATC 4951 TTTATTGTTT AATAGATCAT CAATTTCTGC AGACTTACAG GATCCCCTCA

ANHANG
129
5001 GAAGAACTCG TCAAGAAGGC GATAGAAGGC GATGCGCTGC GAATCGGGAG 5051 CGGCGATACC GTAAAGCACG AGGAAGCGGT CAGCCCATTC GCCGCCAAGC 5101 TCTTCAGCAA TATCACGGGT AGCCAACGCT ATGTAATGAT AGCGGTCCGC 5151 CACACCCAGC CGGCCACAGT CGATGAATCC AGAAAAGCGG CCATTTTCCA 5201 CCATGATATT CGGCAAGCAG GCATCGCCAT GGGTCACGAC GAGATCCTCG 5251 CCGTCGGGCA TGCGCGCCTT GAGCCTGGCG AACAGTTCGG CTGGCGCGAG 5301 CCCCTGATGC TCTTCGTCCA GATCATCCTG ATCGACAAGA CCGGCTTCCA 5351 TCCGAGTACG TGCTCGCTCG ATGCGATGTT TCGCTTGGTG GTCGAATGGG 5401 CAGGTAGCCG GATCAAGCGT ATGCAGCCGC CGCATTGCAT CAGCCATGAT 5451 GGATACTTTC TCGGCAGGAG CAAGGTGAGA TGACAGGAGA TCCTGCCCCG 5501 GCACTTCGCC CAATAGCAGC CAGTCCCTTC CCGCTTCAGT GACAACGTCG 5551 AGCACAGCTG CGCAAGGAAC GCCCGTCGTG GCCAGCCACG ATAGCCGCGC 5601 TGCCTCGTCC TGCAGTTCAT TCAGGGCACC GGACAGGTCG GTCTTGACAA 5651 AAAGAACCGG GCGCCCCTGC GCTGACAGCC GGAACACGGC GGCATCAGAG 5701 CAGCCGATTG TCTGTTGTGC CCAGTCATAG CCGAATAGCC TCTCCACCCA 5751 AGCGGCCGGA GAACCTGCGT GCAATCCATC TTGTTCAATG GCCGATCCCA 5801 TATTGGCTGC AGGTCGAAAG GCCCGGAGAT GAGGAAGAGG AGAACAGCGC 5851 GGCAGACGTG CGCTTTTGAA GCGTGCAGAA TGCCGGGCTC CGGAGGACCT 5901 TCGCGCCCGC CCCGCCCCTG AGCCCGCCCC TGAGCCCGCC CCCGGACCCA 5951 CCCCTTCCCA GCCTCTGAGC CCAGAAAGCG AAGGAGCAAA GCTGCTATTG 6001 GCCGCTGCCC CAAAGGCCTA CCCGCTTCCA TTGCTCAGCG GTGCTGTCCA 6051 TCTGCACGAG ACTAGTGAGA CGTGCTACTT CCATTTGTCA CGTCCTGCAC 6101 GACGCGAGCT GCGGGGCGGG GGGGAACTTC CTGACTAGGG GAGGAGTAGA 6151 AGGTGGCGCG AAGGGGCCAC CAAAGAACGG AGCCGGTTGG CGCTACCGGT 6201 GGATGTGGAA TGTGTGCGAG GCCAGAGGCC ACTTGTGTAG CGCCAAGTGC 6251 CAGCGGGGCT GCTAAAGCGC ATGCTCCAGA CTGCCTTGGG AAAAGCGCCT 6301 CCCCTACCCG GTAGAATTCC AGTCTAGAAC GTTAGCCGGC TACGTATACT 6351 CCGGAATATT AATAGGCCTA GGATGCATAT GGCGGCCGCC TGCAGCTGGC 6401 GCCATCGATA CGCGTACGTC GCGACCGCGG ACATGTACAG AGCTCGAGAA 6451 GTACTAGTGG CCACGTGGGC CGTGCACCTT AAGCTTGCCT AATCGGACGA 6501 AAAAATGACC ATGATTACGC CAAGCTCCAA TTCGCCCTAT AGTGAGTCGT 6551 ATTACAATTC ACTGGCCGTC GTTTTACAAC GTCGTGACTG GGAAAACCCT 6601 GGCGTTACCC AACTTAATCG CCTTGCAGCA CATCCCCCTT TCGCCAGCTG 6651 GCGTAATAGC GAAGAGGCCC GCACCGATCG CCCTTCCCAA CAGTTGCGCA 6701 GCCTGAATGG CGAATGGGAC GCGCCCTGTA GCGGCGCATT AAGCGCGGCG 6751 GGTGTGGTGG TTACGCGCAG CGTGACCGCT ACACTTGCCA GCGCCCTAGC 6801 GCCCGCTCCT TTCGCTTTCT TCCCTTCCTT TCTCGCCACG TTCGCCGGCT 6851 TTCCCCGTCA AGCTCTAAAT CGGGGGCTCC CTTTAGGGTT CCGATTTAGT 6901 GCTTTACGGC ACCTCGACCC CAAAAAACTT GATTAGGGTG ATGGTTCACG 6951 TAGTGGGCCA TCGCCCTGAT AGACGGTTTT TCGCCCTTTG ACGTTGGAGT 7001 CCACGTTCTT TAATAGTGGA CTCTTGTTCC AAACTGGAAC AACACTCAAC 7051 CCTATCTCGG TCTATTCTTT TGATTTATAA GGGATTTTGC CGATTTCGGC 7101 CTATTGGTTA AAAAATGAGC TGATTTAACA AAAATTTAAC GCGAATTTTA 7151 ACAAAATATT AACGTTTACA ATTT

ANHANG
130
7.3.2 DNA-Sequenz der Sonde S1
1 AGATCTAAGC TTCCCAATTC CTGCAGCCCT CGACCATGCC CAAGAAGAAG 51 AGGAAGGTGT CCAATTTACT GACCGTACAC CAAAATTTGC CTGCATTACC 101 GGTCGATGCA ACGAGTGATG AGGTTCGCAA GAACCTGATG GACATGTTCA 151 GGGATCGCCA GGCGTTTTCT GAGCATACCT GGAAAATGCT TCTGTCCGTT 201 TGCCGGTCGT GGGCGGCATG GTGCAAGTTG AATAACCGGA AATGGTTTCC 251 CGCAGAACCT GAAGATGTTC GCGATTATCT TCTATATCTT CAGGCGCGCG 301 GTCTGGCAGT AAAAACTATC CAGCAACATT TGGGCCAGCT AAACATGCTT 351 CATCGTCGGT CCGGGCTGCC ACGACCAAGT GACAGCAATG CTGTTTCACT 401 GGTTATGCGG CGGATCCGAA AAGAAAACGT TGATGCCGGT GAACGTGCAA 451 AACAGGCTCT AGCGTTCGAA CGCACTGATT TCGACCAGGT TCGTTCACTC 501 ATGGAAAATA GCGATCGCTG CCAGGATATA CGTAATCTGG CATTTCTGGG 551 GATTGCTTAT AACACCCTGT TACGTATAGC CGAAATTGCC AGGATCAGGG 601 TTAAAGATAT CTCACGTACT GACGGTGGGA GAATGTTAAT CCATATTGGC 651 AGAACGAAAA CGCTGGTTAG CACCGCAGGT GTAGAGAAGG CACTTAGCCT 701 GGGGGTAACT AAACTGGTCG AGCGATGGAT TTCCGTCTCT GGTGTAGCTG 751 ATGATCCGAA TAACTACCTG TTTTGCCGGG TCAGAAAAAA TGGTGTTGCC 801 GCGCCATCTG CCACCAGCCA GCTATCAACT CGCGCCCTGG AAGGGATTTT 851 TGAAGCAACT CATCGATTGA TTTACGGCGC TAAGGATGAC TCTGGTCAGA 901 GATACCTGGC CTGGTCTGGA CACAGTGCCC GTGTCGGAGC CGCGCGAGAT 951 ATGGCCCGCG CTGGAGTTTC AATACCGGAG ATCATGCAAG CTGGTGGCTG 1001 GACCAATGTA AATATTGTCA TGAACTATAT CCGTAACCTG GATAGTGAAA 1051 CAGGGGCAAT GGTGCGCCTG CTGGAAGATG GCGATTAGCC ATTAACGCGT 1101 GGGGGATCCA CTAGTTCTAG GGGCTCGAGG AATTC

DANKSAGUNG
131
Danksagung
Bei Herrn Univ.-Prof. Dr. Hans-Joachim Galla möchte ich mich an dieser Stelle ganz herzlich
für die Betreuung meiner Promotionsarbeit bedanken.
Herrn Univ.-Prof. Dr. Jürgen Horst danke ich ganz herzlich für die Möglichkeit, im Institut für
Humangenetik meine Promotionsarbeit anfertigen zu dürfen. Seine stetige hilfreiche
Unterstützung und seine Diskussionsbereitschaft trugen sehr zum Erfolg dieser Arbeit bei.
Frau Univ.-Prof. Dr. Angelika Barnekow danke ich ganz herzlich für ihre Bereitschaft als
Prüferin tätig zu werden.
Bei Herrn Dr. Dr. Boris Skryabin möchte ich mich sehr herzlich für die Mikroinjektionen zur
Generierung transgener Tiere bedanken.
Ein großes Dankeschön gilt allen alten und neuen Mitarbeitern des Instituts für
Humangenetik, die durch ihre Zusammenarbeit zum Gelingen dieser Arbeit beigetragen
haben.
Allen, die mich im Laufe der Zeit, in der diese Arbeit entstanden ist, begleitet haben, möchte
ich meinen Dank aussprechen.
Meiner Famille, die immer für mich da war, gebührt mein ganz besonderer Dank. Vor allem
danke ich meinem Mann, der mir stets hilfreich zur Seite stand und mich immerwährend
motiviert und begeistert hat. Schließlich möchte ich mich ganz besonders liebevoll bei
meinem Sohn Alex bedanken, dessen erstes Lebensjahr von der Fertigstellung dieser Arbeit
beeinflusst war und dem diese Arbeit gewidmet ist.

132























![[German translation] Global CRE Trends Infographic 2013](https://img.pdfslide.org/doc/110x75/55cc546ebb61ebbb2c8b46a5/german-translation-global-cre-trends-infographic-2013.jpg)