Embed Size (px)
Citation preview

Z. anorg. allg. Chem. ,585 (1990) 177-158 J. A. Bartli, Leipzig
Darstellung und Reaktivitat von Tris(dialkylthiophosphiny1)- phosphanen
M. SCHEER, F. UHLIG, T. T. NAN, M. DARGATZ, H. D. SCHADLER und E. HERRMA"*
H a lle /S aa Ie , Sektion Cliemie der Rlartiii-Luther-Universitat Halle-Wittenberg
Professor Karl-Heinz Thiele zum 60. Geburtstage gewidmet
Inha l t subers ich t . Die Untersucliung verschiedener Synthesemethoden zur Darstellung der Tit elverbindungen ergab, dalj am besten die Reaktion von LiP(SiMe,), mit Thiophosphinslure- chloriden geeignet ist. Es werden Tris(dialkylthiophosphiny1)pliosphane des Typs P[P(S)R,], (R = Me, Et, n-Pr) in guten Ausbeuten erhalten und mittels lH-, 31P-XMR- und Massenspektrometrie charakterisiert. Durch HMO-Berechnungen werden Stabilitaten verschiedener iso-Tetraphosphane verglichen und Voraussagen zur Ladungsverteilung in den Molekulen getroffen. Diese Ergebnisse werden durch Reaktivitatsuntersuchungen der Titelverbindungen gegenuber OH-, HCl und Br, bestatigt.
Synthesis and Reactivity of Tris (dialkylthiophosphiny1)phosphanes Abst rac t . Investigation of different methods for the synthesis of the title compounds shows
that the reaction of LiP(SiMe,), with chlorides of thiophosphinic acid is the most effective method. Thereby tris(dialkylthiophosp1iinyl)pliosphanes of the type P[P(S)R,], (R = Me, Et, n-Pr) are ob- tained in good yields. They were characterized by lH, 31P NMR, and mass spectrometry. The sta- bility of different iso-tetraphosphanes and the intramolecular charge distribution is compared on t.he basis of HMO calculations. These results are confirmed by investigations of the reactivity of the title compounds with OH-, HCI, and Br,.
Einleitung
Iso-Tetraphosphane der allgemeinen Formel P(PZ,), (Z = dlkyl, Aryl) sind seit langerer Zeit Gegeiistand intensiver Untersuchungen [ 1 - 41. Dagegen sind Verbindungen des Typs Y[P(X)Z,], bisher lediglich fur X = 0 und Z = Alkoxy beltannt [5]. Sie wurden von FLUCK u. Mitarb. entsprechend G1. (1) durch Reak- tion von PC1, mit Dialkylphosphiten in Gegenmart von Pyridin als Base darge- stellt :
(1) 3 P g , - 6 0 ° C
PCI, + 3 HP(O)(OR), --3PyHr+ P[P(O)(OR),l, R = Alkyl
Unser Interesse richtete sich auf die Darstellung der bisher unbekannten Tris- (thiophosphiny1)phosphane der allgemeinen Formel P[P(S)R,], (R = Alkyl) .
178 Z. anorg. allg. Chem. 585 (1990)
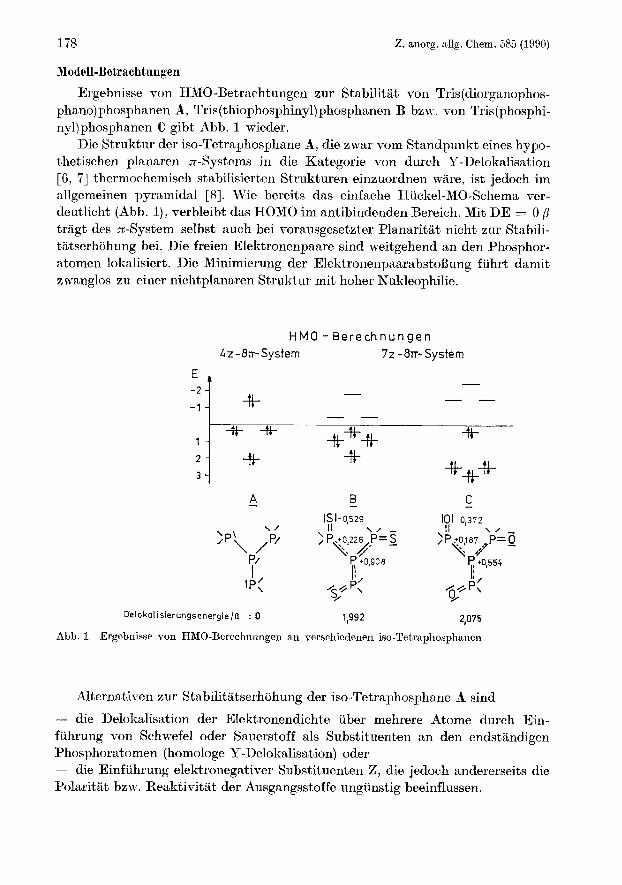
~~Odcll-Bctrachtnngen Ergebnisse von HMO-Betrachtungen zur Stabilitat von Tris(diorgan0phos-
phano)phosphanen A, Tris(thiophosphiny1)phosphanen B bzw. von Tris(phosphi- ny1)phosphanen C gibt Abb. 1 wieder.
Die Struktur der iso-Tetraphosphane A, die zwar vom Standpunkt eines hypo- thetischen planaren n-Systems in die Kategorie von durch P-Delokalis a t' ion [ 6, 71 thermochemisch stabilisierten Strukturen einzuordnen ware, ist jedoch im allgemeinen pyramidal [ 81. Wie bereits das einfache Hiickel-MO-Schema ver- deutlicht (Abb. l), verbleibt das HOMO im antibindenden Bereich. Mit DE = O P tragt des 7c-System selbst auch bei vorausgesetzter Planaritat nicht zur Stabili- tiitserh6hung bei. Die freien Elektronenpaare sind weitgehend an den Phosphor- atomen lokalisiert. Die Minimierung der ElektronenpaarabstoBung fiihrt damit zwanglos zu einer nichtplanaren Struktur mit hoher Nukleophilie.
HMO - B e r e c h n u n g e n 4z -8rr-System
E 4
-1 -*I x
2 3 1-7 A -
Delokalisierungsenergie/fi : 0
72 -8rr-System
-
xstx x. x
1,992 2,075
Abb. 1 Ergcbnisse yon HMO-Berechnungen an verscliicdenen iso-Tetraphosphanen
Alternativen zur Stnbilitatserhohung der iso-Tetraphosphane A sind
- die Delokalisation der Elektronendichte iiber mehrere Atome durch Ein- f uhrung von Schwefel oder Sauerstoff als Substituenten an den endstandigen Phosphoratomen (homologe Y-Delokalisation) oder - die Einfiihrung elektronegativer Substituenten Z, die jedoch andererseits die Polaritat bzw. Reaktivitat der Ausgangsstoffe ungiinstig beeinflussen.

M. SCHEER u. a., Tris(dialkylthiophosphiny1)pliosphane 179
Bereits das qualitative HMO-Mode11 191, (Parameter aus [lo]), laBt folgende SchluBfolgerungen zu (vgl. Abb. 1) :
1. Durch Einfuhrung von Sauerstoff oder Schwefel wird das HOMO zu einem bindenden Orbital, d. h., das gesamte Molekul wird im n-System stabilisiert. Die Nukleophilie wird dadurch herabgesetzt.
2. Das Sauerstoffsystem C ist etwas stabiler als das Schwefelsystem B. Der Beitrag des z-Systems zur thermodynamischen Stabilisierung liegt in beiden Fallen in der GroBenordnung von 2 p (vgl. Benzen !) .
3. Die Ladungsverteilung zeigt, daB der Schwefel Lrotz geringerer Elektro- negativitiit die negative Ladung besser ubernehmen kann als der Sauerstoff (vgl. auch Carbanionenstabilisierung durch S !) .
4. Aus der berechneten Ladungsverteilung laBt sich ebenfalls der EinfluB von am endstandigen Phosphoratom befindlichen Substituenten abschatzen. Da- nach tragen +I- bzw. +M-Substituenten zusatzlich zur Stabilitat des betrach- teten Systems P[P(X)Z,], mit X = 0, S bei, wahrend -I- und -M-Substituenten einen entsprechenden umgekehrten Effekt hervorrufen.
5. Fur die Reaktion mit Nukleophilen lassen die Ladungsverhaltnisse den SchluB zu, daB der Angriff am zentralen Phosphoratom entsprechend G1. (2) erfolgt .
(2) Untersuchungen von PLUCK und WEBElt uber die Reaktivitat von Tris(phos- phory1)phosphanen scheinen dies zu bestatigen [Ill.
P[P(X)Z,], + Nu- --f RuP[P(X)Z,], + P(X)Z,-.
Ergebnissc und Diskussion Versuche zur Synthese von Tris(thiophosphiny1)phosphanen analog G1. (1)
durch Umsetzung von PC1, mit den Thiophosphinigsauren Me,P(S)H bzw. Ph,P(S)H entsprechend G1. (3) fuhrten nur bei der Methylverbindung zum ge- wiinschten Zielprodukt l.
(3) 3Et,N, -fiO"C
PCI, + 3 HP(S)R, -3[Et,Nlf]C1 * P[P(f3R,),
1 la : R = Me
Durch 31P-NMR-Spektroskopie konnte nachgewiesen werden, daB im Reak- tionsgemisch aber nur bis zu 36% des eingesetzten Phosphors als Tris(dimethy1- thiophosphiny1)phosphan 1 a vorliegen. Als weitere Reaktionsprodukte wurden die Diphosphanderivate R,PP(S)R, (2a, b ; R = Me, Ph) und R,P(S)P(S)R, (3 b ; R = Ph) , sowie das entsprechende Thiophosphinsaurechlorid R,P(S)Cl (4a, b ; R = Me, Yh) identifiziert (vgl. Tab. 4).
Zu noch schlechteren Ergebnissen fuhrte die Umsetzung von PCl, mit dem Lithiumdimethylthiophosphinit (Gl. (4)).
(4) PCl, + 3 LiP(S)Me, zi:&+ P[P(S)Me,l,.
l a

180 Z. anorg. allg. Chem. 586 (1990)
Der Anteil an l a war geringer und neben den entsprechenden Diphosphanmono- und -disulfiden 2a und 3a trat eine noch grol3ere Anzahl nicht identifizierter Ne- benprodukte auf (vgl. Tab. 4).
Eine Isolierung von l a gelang aus keinem der Ansatze nach den Gln. (3) und (4). Deshalb wurden andere Synthesewege untersucht.
Durch Umsetzung einer PNMe,- und einer PH-Verbindung in siedendem Toluen gelang MAIER [ l2] die Kniipfung von (P-P) -Bindungen unter Abspaltung von Dimethylamin. Die Reaktion von Tris(dimethy1amino)phosphan mit Di- methyl- bzw. Diphenylthiophosphinigsauren sollte dementsprechend durch drei- malige Wiederholung von G1. (5)
--P--NMe, + HP(S)R, --f >P-P(S)R, +- HNMe, (6)
zu den Tris(thiophiny1)phosphanen fiihren. Bei der Umsetzung in siedendem Toluen konnte jedoch nicht die Bildung von iso-Tetraphosphan-Derivaten beob- achtet werden. Als Reaktionsprodukt trat ein in organischen Losungsmitteln unloslicber Feststoff mit hohem Phosphorgehalt auf. In der Losung lieBen sich 31P-NMR-spektroskopisch neben einem betrachtlichen Anteil an der unum- gesetzten Thiophosphinigsaure die Diphosphanderivate 2a bzw. 2 b und 3 b und die Signale einer Reihe nicht identifizierter Produkte nachweisen. Bei der Um- setzung der Diphenylthiophosphinigsaure konnte auf3erdem das Dimethylammo- niumdiphenyldithiophosphinat [Me,NH,][Ph,PS,] isoliert werden.
Eine andere Mijglichkeit der Kniipfung von PP-Bindungen besteht in der Reaktion einer PSiMe,- mit einer PC1-Verbindung unter Eliminierung von Tri- methylchlorsilan, die vor allem von der Arbeitsgruppe von FRITZ fur die Darstel- lung von iso-Tetraphosphanen genutzt wurde [ 81.
Tris(trimethylsily1)phosphan reagiert tatsachlich gemail3 G1. (6) mit Dimethyl- thiophosphinsaurechlorid in siedendem Benzen unter kontinuierlichem Entfernen von Me,SiCl zu la .
Das aus der Losung nach dem Einengen susfallende l a kann nach mehrmaligem Umkristallisieren aus heifiem Benzen analytjsch rein, jedoch nur in Ausbeuten von 20% gewonnen werden. Das 31P-NMR-Spektrum des Rohprodukts weist hin- gegen neben 3a und Spuren von 2a Anteile von gebildetem l a in etwa 40 Mo1.-% des Gessmtphosphors auf.
Das Resultat kann auf die thermische Empfindlichkeit von 1 a zuruckgefiihrt werden, da es sich (s. u. G1. (15)) wahrend der Reaktion bzw. des Umkristalli- sierens in Me,Y(S)P(S)&le, und eine polymere, unlosliche Phosphorverbindung zersetzt,
Wir suchten deshalb nach einer schonenderen Synthesevariante und fanden, dalj sich 1 bei der Umsetzung von LiP(SiMe,) , mit Thiophosphinsaurechloriden

M. SCHEER u. a., Tris(dialkylthiophosphiny1)phosphane 181
in THF unter bestimmten Bedingungen bildet. Dabei ist die Reaktionsfuhrung entscheidend. Wird bei - 40 O C zum Saurechlorid das Lithiumphosphid zugetropft, erhalt man das Diphosphandisulfid 3 als Hauptprodukt (Gl. (7) und (8)). Wahr- scheinlich ist das intermediar gebildete Dialkylthiophosphinyl-bis(trimethy1- sily1)phosphan ZIX instabil und reagiert mit iiberschussigem Thiophosphinsaure- chlorid gemaB G1. (8) zu 3 und polymeren P-Verbindungen.
( 7 )
R,P(S)P(SiMe,), + CIP(S)R, R,P(S)P(S)R, + . . . (8)
Gibt man hingegen das entsprechende Thiophosphinsaurechlorid unterhalb von - 2OoC zu einer Losung des Phosphids im Molverhaltnis 1 : 1, lassen sich ent- sprechend G1. (9) die Tris(thiophosphiny1)phosphane l a , C, d (R = Me, Et, n-Pr) in Ausbeuten his zu 45% isolieren. Als weiteres Reaktionsprodukt lal3t sich Tris- (trimethylsilyl) phosphan erhalten.
THF R,P(S)CI + LiP(SiMe,), -Lia+ R,P(S)P(SiMe,),
3 R = Me, Et
-20°C 3 LiP(SiMe,), + 3 CIP(S)R,
l I a c d
P[P(S)R,], + 2 P(SiMe3),. (9)
R I Me Et n-Pr
Fur den Fall des Dipropylthiophosphinyl-Derivates konnten mittels 31P-NMR- Spektroskopie einige Zwischenprodukte des stufenweisen Aufbaues des iso-Tetra- phosphans 1 d heobachtet werden. Nach einer 90minutigen Reaktionsdauer (-30°C) sind im Reaktionsgemisch neben dem Produkt I d und P(SiMe,), das Diphosphan Pr,P(S)P(SiMe,)Li, 5 d’, und das Triphosphan [Pr,P(S)],PLi, 6 d’, nachweisbar (Tab. 1). An Hand dieser Zwischenstufen kann der in den Gln. (10) his (14) gezeigte Reaktionsmechanismus angenommen werden.
LiP(SiMe,), + R,P(S)CI (Me,Si),PP(S)R, (10)
6d
LiP(SiMe,), + Sd ---+ -P(SiMe& Li(Me,Si)PP(S)R, (11)
5 d’
R,P(S)CI 4 541’ (Me,Si)P[P(S)R,], (12)
(13)
6 d
LiP(Siiw, + 641 --P(SiMe,t, + LiP[P(S)R,],
R,P(S)CI + xl+ P[P(S)R,)I, (14)
6d‘
I d R = n-Pr
Im ersten Reaktionsschritt (Gl. (10)) wird unter Abspaltung von LiCl das Diphosphan 5 d gebildet. Der Uberschul3 an LiP(SiMe,), im ReaktionsgefaB

182 Z. anorg. allg. Chem. 585 (1990)
bewirkt gemail3 G1. (11) eine Metallierung des Silylphosphors unter Austritt von P( SiMe,) 3, worauf das metallierte Diphosphan 5 (2' mit dem Thiophosphinsaure- chlorid zum Triphosphan 6 d weiterreagiert (Gl. (12)). Weiter erfolgt entsprechend G1. (13) die Metallierung des zentralen Phosphoratoms. Die anschlieBende Reak- tion mit dem Thiophosphinsaurechlorid (Gl. (14)) fuhrt zur Bildung des iso-Tetra- phosphans 1 d. Wahrend die Methyl- und Ethylvertreter l a , c analytisch rein iso- liert wurden, gelang dies im Falle des Propylderivates I d nicht.
Tabelle 1 31P-NMR-spektroskopische Daten der Verbindungen 5d', Gd', I d und P(SiMe,),
Verbindung chem. Verschiebung 4PPm P A PB
~ ~
V(PP) /Hz
Die Tris(dialkylthiophosphiny1)phosphane 1 a, c sind gelbe, kristalline Sub- stanzen, die unter Zersetzung schmelzen. Im festen Zustand konnen sie kurzzeitig an der Luft gehandhabt werden, zerfallen jedoch uber langere Zeit unter Bildung polymerer, unloslicher Derivate und Verbindungen, die eine 31P-NMR-chemische Verschiebung bei 81,2 ppm (R = Me) und 85,2 ppm (R = Et) haben. Unter Inertgasatmosphare sind 1 a, c unbegrenzt lagerbar. Sie sind in organischen Losungsmitteln wie Toluen, Benzen, CS,, THF und CH2C12 gut loslich, in Et,O sind sie wenig und in gesattigten Kohlenwasserstoffen unloslich. Ihre Losungen sind luftempfindlich. Thermolyseuntersuchungen in Toluen (Gl. (15)) zeigen, da13 sie sich in die entsprechenden Tetraalkyldiphosphandisulfide und unlosliche Produkte zersetzen.
-+ Me,P(S)P(S)Me, + . . . T oluen, A P[P(S)Mezl, ___
l a
Wahrend bei Raumtemperatur innerhalb von 1 2 h keine Reaktion zwischen l a und H,O beobachtet wird, erfolgt in alkalischer Losung die vollstgndige Zer- setzung von l a gemail3 G1. (16).
(16)
Ein ganz analoges Reaktionsverhalten zeigt Verbindung 1 a gegenuber HCl. Ent- sprechend G1. (17) wird PC13 und Dimethylphosphansulfid gebildet, so daB unter Beriicksichtigung von G1. (3) das System PCl,, HP(S)Me,/P[P(S)Me,], ungeachtet von Nebenreaktionen einem reversiblen Gleichgewicht unterliegt .
P[P(S)Me,], + 3 H,O -OH- + H,PO, + 3 HP(S)Me,.
l a
P[P(S)Me,], + 3 HCI -Etao+ PCI, + 3 HP(S)Me,.
la

31. SCHEER u. a., Tris(dialkylthiop1iosphinyl)pliosphnne 18.3
Die Natur der Reaktionsprodukte gemaB Gln. (16) und (17) belegen die Richtigkeit der nach den HMO-Berechnungen getroffenen Ladungsverteilung in den Tris- (thiophosphiny1)phosphanen. Danach erfolgt der Angriff des Nukleophils am Zentralphosphoratom von 1.
Aus der Umsetzung von l a mit Brom ergeben sich erwartungsgemaB PBr, und Dimethylthiophosphinsiiurebromid (Gl. (18)).
P[P(S)Me,], + 3 Br, --f PBr, + 3 BrP(S)Me,.
l a
I m Gegensatz zu Tris(phosphory1)phosphanen [ 111 reagiert 1 a mit sekundaren Aminen nicht (Gl. (19)).
(19) P[P(S)Me,], + 2 HNEt, ___ I- + [NH,Et,][hle,(S)PPP(S)lle,]. -EtJP(S)JIe,
l a
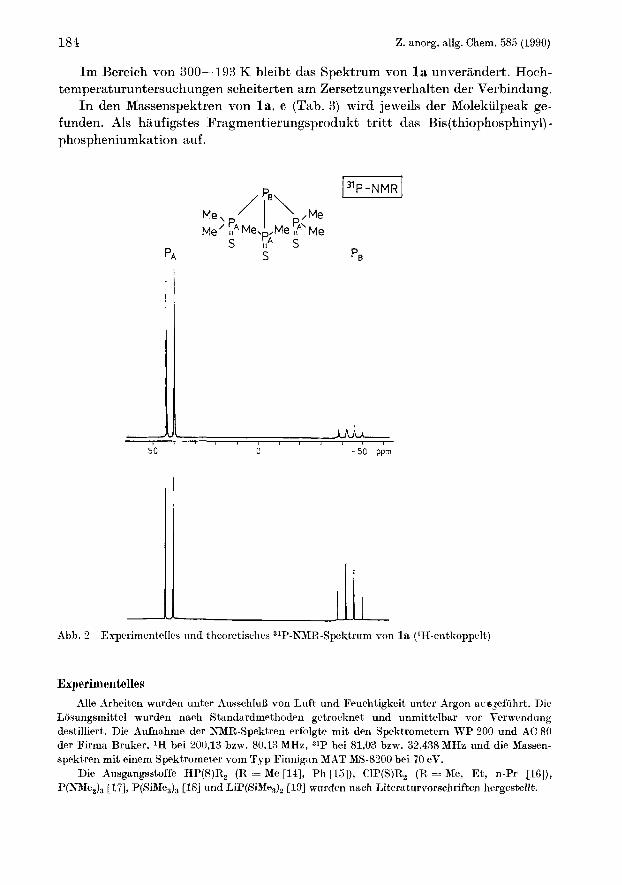
Die Ergebnisse der NMR-Untersuchungen an den Tris(thiophosphiny1)phosphanen sind in Tab. 2 zusammengestellt. In Abb. 2 ist dns IH-entkoppelte ,IP-NMR- Spektrum von l a abgebildet. Die Simulation zeigt, daB es vom AM,-Typ ist, wobei nur die IJ(PP) fur das Aufspaltungsbild verantwortlich ist. Fur iso-Tetra- phosphane sind solche 31P-NMX-Spektren typisch [3, 4, 131.
Tabelle 2 hWR-Daten der Tris(thiophosphiny1)phosphane P,[Pa(S)R,], (R = hIe, Et) la, c in C,D,
Kern Verb. chem. Verschiebung G/ppm Kopplungskonst. J/Hz 'A '€I -CH, -CH, 1J(PP) "(PCH) 3J(PPCH)
1H l a "0 12,3 3,7 l o 2,65 1,25 23,s
3 1 p l a 41,8 -44,2 303,9 l c 63,s -76,l 320,6
Tabelle 3
l a (R = Me) I C (R = Et) Zuordnung
Molekiilpeak und die wiclitigsten Fragmente in den Mnssenspektren von P[P(S)R,],
Masse/u &el.% Masse/u &el.%
310 5 394 3 Nf "7 50 331 5 x+ - PS
306 50 M+ - PR, 217 100 273 100 M'-P(S)R, 18.5 20 "1 60 M+ - P(S),R,
184 Z. anorg. allg. Chem. 585 (1990)
Im Bereich von 300-193 K bleibt das Spektrum von l a unverandert. Hoch- temperaturuntersuchungen scheiterten am Zersetzungsverhalten der Verbindung.
I n den Massenspektren von la., B (Tab. 3) wird jeweils der Molekulpeak ge- funden. Als haufigstes Fragmentierungsprodukt tritt das Bis(thiophosphiny1)- phospheniumkation auf.
50 0 - 5 0 ppm
Abb. 2 Experimentelles und theoretisches 31P-P\’MR-Spektrum von l a (lH-entkoppelt)
Experimentelles Alle Arbeiten wurden unter AusschluO von Luft und Feuehtigkeit unter Argon at szefiihrt. Die
Losungsmittel wurden nach Standardmethoden getrocknet und unmittelbar vor Verwendung destilliert. Die Aufnahme drr NMR-Spektren erfolgte mit den Spektrometern FVP 200 und AC 80 der Firma Bruker, ‘H bei 200,13 bzw. 80,lR MHz, slP bri 81,03 bzw. 32,438 MHz und die Massen- spektren mit einem Spektrometer vom Typ Finnigan MAT MS-8200 bei T O eV.
Die Ausgangsstoffe HP(S)R, (R = Me [14], P h [15]), CIP(S)R, (K = Me, Et, n-Pr [IC]), P(NMe,), [U], P(SiMe,), [lSj und LiP(Si,l.le,), [19] wiirden nacli Literaturvorschriften hergestellt.

M. SCHEER u. a., Tris(dialkylthiophosphiny1)phosphane 185
U m s e t z u n g e n v o n PCI, m i t T hi o pho s p hi nigs 1 u r e n i n Ge g e n war t vo n KEt,. Zu 18 mmol der entsprechenden Thiophosphinigsaure HP(S)R, (R = Me, Ph) und 1,s g (18 mmol) NEt, in 40 ml CH,C1, werden bei -60°C unter starkem Riihren 0,s g (6 mmol) PCI, in 20 ml CH,CI, wlthrend 30 min zugetropft. AnschlieBend wird eine Stunde boi --GO"C geruhrt und nach Entfernen der Kuhlung auf Raumtemperatur erwarmt. Es werden 20 ml Petrolether hinzugefugt und der ausfdlende Nieder- schlag von NEt3HC1 abfiltriert. Das Filtrat wird im Vakuum eingeengt und 31P-RT~R-spektroskopisch unt>ersacht. Die Ergebnisse sind in Tab. 4 zusammengestellt.
U m s e t z u n g v a n PCI, m i t LiP(S)Me,. Zn 1,9 g (20 mmol) HP(S)Me, in 100 ml Toluen wird bei - i8"C die Lquimolare Menge an Butyllithium in Pentan zugetropft. Es wird eine Stunde bei -40°C und eine neitere Stunde bei Raumtemperatur bis zum Ausfallen eines weiBen Peststoffes geruhrt. Zu dieser Suspension werden bei -440°C bis -50°C 0,91g (G,Gmmol) PCI,, gelost in 30ml Toluen, getropft. Es wird 2 Stunden nachgeruhrt, auf Raumperatur erwiirmt und vom ausgefallenen LiCl abfiltriert. Das Filtrat wird im Vakuum aufkonzentriert und mittels 31P-NMR-Spektroskopie cha- rakterisiert (Tab. 4).
Tabelle 4 von Umsetzungen gemLU Gln. (3)-(6) und (16)-(18)
Durch 31P-NMR-Spektroskopie bestimmte Zusammensetzung der Reaktionslosungen
Gleichung WPPm Losungsmittel
*J(PP)/Hz Zuordnung Mol.-% P
(3) R = Me CH,CI,
(3)
CH,CI, R == P h
(4) R = Me Toluen
303,1
220,l
30,8
2%5,7
70
303,1
214,2
Ph$BPA(S)Ph,
Ph,P(S)P(S)Ph, Ph,P(S)Cl
a)
3G
23
13
8
49
13 8
19
20
40
20 5
i 15

186 Z. anorg. allg. Chem. 585 (1990)
Tabelle 4 (Fortsetzung)
Gleichung SlPPm IJ(PP)/Hz Zuordnung Dlol.-~o P Losungsmit'tel
( t i )
R = M e Toluen
(6) R = P h Toluen
(6) R = Me Benzen
(16) R = Me EtOH
(17) R = M e Benzen
(18) R = Me Benzen
4,s
32,9 53,5 64,3
4-8
2,G 4-9
81,2 3 3 3 43,5
63,6 237,s
-s05,5 -113,l
-47,4
214,2 Me,P,P,(S)Me,
")
30 ")
Me,P(S)H
Ph,P(S)H
225,s Ph,PBPA(s)Ph,
Ph,P(S)P(S)Ph, ") ")
214,2 Me,PBPA(S)Me,
303,l PB[PA(S)Me213
Me2P(S)P(S)Me,
Me,P(S)H H3P0, Me,P(S)P(S)Me, ") ")
Me,P(S)Br PBr, OPBr, SPBr, "1
1s 29
12
41
34
28
8 10
7
40
40
20
60 20 5
15
60 20 15
2 3
45 15 5
10 25
") Signale nicht zugeordnct.
U m s e t z u n g y o n P(NMe,), m i t Thiophosphinigsi iuren. 1 g (6,l mmol) P(NMe,), und 18,3 mmol der entspreehenden Thiophosphinigsaure HP(S)R, (R = Me, Ph) werden in 30 ml Tolnen fur 8 Stunden am RiickfluB erhitzt. Der ausfallende, gelbe Nicderschlag wird abfiltriert nnd mit Benzen ; von den iJoslichen Brstandteilen befreit. Die nach dem Einengen der benzenischen Losung auf etwa SO ml auskristallisierendcn Verbindnngen (Mr,P(S)P(S)Me,: 0,7 g, 41%; - _

M. SCHEER u. a., Tris(dialky1thiophosphinyl)phosphane 187
[H,NMe,][Ph,P(S)S]: 1,5 g, 27%) werden abfiltriert und aus Benzen umkristallisiert. Die Restlosung (Toluen) wird 31P-NMR-spektroskopisch untersucht. Die Ergebnisse sind in Tab. 4 ZusammengefaBt. Zusammensetzung der unloslichen Reaktionsprodukte: R = Me: 0,7 g; C 14,70; H (i,29; N 8J9; S 0,53; P 47,20%. R = Ph: 0,s g; C 16,33; H 3,26; N 3,89; S 8,09; P 60,5yo.
Umsetzung P(SiMe,), rnit Dimetyhlthiophosphinsaurechlorid. Zu 3,04 g (24 mmol) CIP(S)Me, in 200 ml Benzen werden unter starkem Riihren innerhalb von 2 Stunden 2,O g (8 mmol) P(SiMe,),, gelost in 100 ml Benzen, zugetropft. Wahrenddessen wird kontinuierlich Benzen abdestil- liert bis etwa 80 ml des Losungsmittels verbleiben. Das restliche Benzen wird im Vakuum soweit entfernt, bis eine Kristallisation einsetzt. Es wird dekantiert, der verbleibende Feststoff mit Hexan gewaschen und aus Benzen umkristallisiert. Man erhalt 0,5 g (20%) von la.
Umsetzung von LiP(SiMe,), mi t Thiophosphinsaurechloriden. Zu einer Losung von 5 g (16,l mmol) LiP(SiMe,), in 50 ml THF wird unter starkem Riihren bei -7O"C, innerhalb von 2 Stunden, die aquimolare Menge des entsprechenden Dialkylthiophosphinsiiurechlorides, gelost in 20 ml THF, zugetropft. AnschlieBend wird bis auf -20°C erwarmt und 3 Stunden geruhrt. Wahrend weiterer 3 Stunden wird auf Raumtemperatur erwarmt. Das Losungsmittelvolumen wird im Vakuum reduziert, der ausfallende Feststoff abfiltriert und rnit Hexan gewaschen. Der verbleibende, feste Riickstand wird in Benzen aufgenommen und vom LiCl abfiltriert. Im AnschluB wird im Vakuum bis zum Einsetzen der Kristallisation eingeengt. Das Produkt wird abfiltriert, rnit je 10 ml H,O, MeOH und Hexan gewaschen und im Vakuum getrocknet. Man erhlilt 0,75 g (45% der Theorie) von l a vom Schmp. 172°C bzw. 0,75 g (35% der Theorie) von l c vom Schmp. 160°C.
l a , C,H,,S,P,, MG 309,9536 (MS) (ber. 309,9621); C 23,21 (ber. 23,63); H 5,92 (5,92); S 30,96 (31J6); P 39,73 (40,00)%.
l o , C,,H,,S,P,, MG 394,0468 (MS) (ber. 394,0460); C 35,30 (ber. 36,5); H 7,04 (7,60); S 24,24
Thermolyse von l a . 0,2 g (0,64 mmol) l a werden in 10 ml Toluen fur 8 Stunden am Ruck- fluB erhitzt. Der gebildete gelbe Niederschlsg wird abfiltriert und die Losung 31P-NMR-spektrosko- pisoh vermessen (Singulett bei 34,2 ppm).
Umsetzung von l a rnit NaOH/H,O. 0,l g (0,32 mmol) l a werden rnit 0,06 g (1,6 mmol) NaOH in 5 ml H,O zusammengegeben und 10 Stunden bei Raumtemperatur geruhrt. Die Losnng wird 31P-NMR-spektroskopisch untersucht (Tab. 4).
Umsetzung von l a mit HCI. Zu einer Losung von 0,175 g (0,56 mmol) l a in 5 ml Toluen werden 1,7 mmol HCl in 4 ml Et,O gegeben. Es wird 20 Stunden bei Raumtemperatur geruhrt und im An- schluB der Ether abdestilliert. Die Ergebnisse der NMR-Untersuchung der Losung sind in Tab. 4 zusammengestellt.
Umsetzung von l a rnit Br,. Zu einer Losung von 0,17 mmol l a in 10ml Benzen werden 0,26 ml (0,51 mmol) Brom, gelost in 5 ml Benzen, getropft und 8 Stunden bei Raumtemperatur geriihrt. Die Losung wird im Vakuum auf 3 ml aufkonzentriert und mittels der 31P-NMR-Spektrosko- pie untersucht (Tab. 4).
Umsetzung von l a rnit HNEt,. Zu einer Losung von 0,16 g (0,48 mmol) l a in 40 ml Et,O/ Benzen (1 : l ) werden 0,11 ml(O,9 mmol) HNEt,, gelost in 10 ml Et,O, getropft. Es wird 72 Stunden bei Raumtemperatur geriihrt, das Losungsmittel im Vakuum entfernt, der verbleibende Feststoff in 5 ml THF aufgenommen und 31P-P\TMR-spektroskopisch analysiert. Es handelt sich dabei aus- schlicBlich um la.
(24,37)%.
Literatur [I] BAUDLER, M.: Angew. Chem. '34 (1982) 620. [2] BAUDLER, M.; HELLNAXN, J.: Z. Naturforsch. 3810 (1983) 637. [8] STOLL, K.: Dissertation, Univ. Kitrlsruhe 1986.

188 Z. anorg. allg. Chem. 585 (1990)
[4] JARMER, M.: Dissertation, Univ. Karlsruhe 1987. [ri] FLUCK, E.: Topics in Phosphorus Chemistry 10 (1980) 193. [GI GRUND, P.: J . Chem. Educ. 4!1(1972) 100. [7] SCHADLER, H. D.: Z. Chem. 28 (1988) 278. [S] FTITZ, G.; STOLL, K.; HONLE, W.; TON SCHNERING, H. G.: Z. anorg. allg. Chem. 544 (1986) 127. [9] HEILBRONNER, E. ; BOCK, H.: Das HMO-Mode11 und seine Anwendung; Weinheim: Verlag
Chemie 1968. [lo] BAIRD, N. C.; WHITEHEAD, M. A.: Can. J. Chem. 44 (1966) 1933. [ll] FLUCK, E.; WEBER, D.: Z. Naturforsch. 30b (1976) 60; 31b (1976) 81. [12] MAIER, L.: Helv. Chim. Acta 4!) (1966) 1119. [13] HAHN, J.: 31P-NMR-Spectroscopy in Stereochemical Analysis, VCH Publisher, inc. 1987, 331. [14] SASSE, K.: Methoden der org. Chcmie; Stuttgart: G. Thieme, Verlag 1963, Bd. XII / l , S. 212. [16] PETERS, G.: J. Org. Chem. 27 (1962) 2198. [ l G ] PERSHALL, G. W.; STOCKS, R. C.; QWINN, L. D.: Inorg. Synthesis XV (1974) 191. [17] SASSE, K.: Methoden der org. Chemie; Stuttgart: G. Thieme Verlag 1964, Bd. XI112 S. 108. [lS] BECKER, G.; HOLDERICH, W.: Chem. Ber. 108 (1979) 2484. [19] FRITZ, G.; HOLDERICH, W.: Z. anorg. allg. Chcm. 422 (1976) 104.
Bei der Redaktion eingegangen am 9. November 1989.
Anschr. d. Vcrf.: Dr. M. SCHEER, DC F. UHLIG, DC T. T. N-~M, DC M. DARGATZ, Doz. Dr. H. D. SCHADLER und Prof. Dr. E. HERRNIANN, Sektion Chemie der Martin-Luther-Univ., Postfach, Halle/Snale, DDR-4010





![Tris[4,4'-(ethene-1,2-diyl)dipyridinium] decavanadate ...journals.iucr.org/e/issues/2010/03/00/bg2319/bg2319.pdf · Tris[4,4000-(ethene-1,2-diyl)dipyridinium] decavanadate dihydrate](https://img.pdfslide.org/doc/110x75/5fa3b6c93bca6f76e64e095f/tris44-ethene-12-diyldipyridinium-decavanadate-tris44000-ethene-12-diyldipyridinium.jpg)


![Synthese, Struktur und Reaktivität von [1 ...darwin.bth.rwth-aachen.de/opus/volltexte/2000/58/pdf/Koblinski_Carsten.pdf · Synthese, Struktur und Reaktivität von [1]Borametallocenophanen](https://img.pdfslide.org/doc/110x75/5d55a09588c993f8298b4abc/synthese-struktur-und-reaktivitaet-von-1-synthese-struktur-und-reaktivitaet.jpg)











![Tris[tris(ethane-1,2-diamine)cobalt(II)] bis[octacyanidomolybdate(V)] dihydratemypage.just.edu.cn/_upload/article/files/30/fc/684d... · 2017. 6. 8. · Tris[tris(ethane-1,2-diamine)cobalt(II)]](https://img.pdfslide.org/doc/110x75/60d9aab9684bc31d7d4fd5e5/tristrisethane-12-diaminecobaltii-bisoctacyanidomolybdatev-2017-6-8.jpg)








