Embed Size (px)
Citation preview

Chemisehes Institut der Universit~t KSln und Max-Planck-Institut ffir Eisenforschnng Diisseldorf
Die analytische Charakteristik des k~iuflichen und des gereinigten Sehwefels ~
Beitriige zur Chemie des Schwefels, 54
Von F. FEH]~R~ S. ECKHARD und K. H. SAUER**
~i~ 8 Textabbildungen
(Eingegangen am 16. Januar 1959)
Der flfissige Schwefel besteht im wesentlichen aus den beiden Form- arten S~ (achtgliedrige Schwefelringe) und S, (Ketten verschiedener Ls Die Ss-Ringe brechen mit steigender Temperatur auf und bilden durch Assoziation die polymere Erscheinungsform. Es besteht die Auf- fassung, dab die Entstehung hochgliedriger Ket ten durch Anlagerung yon Fremdsubstanzen verhindert wird. Verunreinigungen haben dem- nach einen Einflul3 auf die Struktur und damit auf die physikalisch- ehemischen Eigenschaften des flfissigen Schwefels. Zur Messung physi- kalischer Konstanten wird deshalb in neuerer Zeit grSl3ter Wert auf die Reinigung des Schwefels gelegt. Besonders ein yon BAco~ u. FA- NELLI I entwickeltes Verfahren scheint den Reinheitsforderungen weit- gehend zu entsprechen. Eine Entscheidung fiber die Leistungsfi~higkeit dieser Methode kann erst getroffen werden, wenn die Schwefelproben analytisch eharakterisiert sind*** ; erst dann kann der EinfluB der Fremd- stoffe auf die Struktur des fliissigen Schwefels systematisch untersueht werden.
Es erschien uns deshalb angebracht, geeignete Analysenverfahren zu ermitteln bzw. bekannte Bestimmungsmethoden ffir die einzelnen Elemente so abzuwandeln, da• eine schnelle quanti tat ive Untersuehung aller Verunreinigungen im Schwefel mSglieh ist. Es wurden zwei k/~uf- hehe Sehwefelsorten, die beide aus Gasreinigungsmasse gewonnen sind, vor und naeh der Reinigung untersucht. Die l~einigung erfolgte nach der yon Bxco~ u. FA~ELLI besehriebenen Methode.
Vortrag, gehalten auf der Tagung der Gesellsehaft Deutscher Chemiker -- Fachgruppe ,,Analytische Chemie" -- am 25. Oktober 1958 in 1Vfarburg/Lahn.
~r Dissertation, Chemisches Institut der Universit~tt KSln. *** Mit der Untersuchung eines naeh v. W~T~,NBEI~O [Z. anorg, allg. Chem.
286, 243 (1956)] gereinigten Sehwefeis sind wit gegenw/~rtig beschMtigt und werden demn~ehst dariiber berichten.

Analytik des k~ufliehen und gereinigten Schwefels 89
I)iese Autoren reinigen den S chwefel durch 120 stiindiges Koehen mit Magnesium- oxyd und durch mehrmaliges Filtrieren. Durch das li~ngere Kochen werden Kohlen- stoff-Sehwefelverbindungen und Sulfane zersetzt. Schwefelwasserstoff entweicht g~s- fSrmig, der Kohlenstoff wird elementar ausgeschieden. Metallisehe Verunreinigungen bilden Sulfide, die ebenfalls ausfallen und mit dem ausgesehiedenen Kohlenstoff und dem zugesetzten Magnesiumoxyd abfiltriert werden. Der Zusatz yon ?r dient neben der besseren Abseheidung des Kohlenstoffs zur Neutralisation anwesender Siuren.
Die Kohlenstoffbestimmung
Fi i r die Bes t immung des Kohlenstoffs erschien ein yon Scm~IDTs u. BAASCE li entwickel tes Lei tf /~higkei tsverfahren geeignet , dessen An- wendung an f die Ana lyse yon Kohlens tof f in S tah l Koc t I u. MALISSA s beschr ieben haben.
Nach diesem Verfahren wird die Stahll0robe in einem Verbrennungsofen im Sauerstoffstrom verbrannt und die Leitfihigkeitsinderung gemessen, die eine ver- diinnte Natronlauge beim Durchleiten dos entstehenden Kohlendioxyds erf~hrt. Die Analysenmegzelle und eine VergleichsmeBstreeke sind zu Beginn einer Be- stimmung mit Natronlauge gleicher Konzentration gefiillt. ])as kohlendioxydhaltige Verbrennungsgas strOmt nut dutch die MeBzelle. W/~hrend der Absorption wird die Leiff/~higkeitsdifferenz beider Megstreeken yon einem Kompensationsliniensehreiber aufgezeiehnet. Dutch die Anwendung der Differenzmessung wird das Verfahren weitgehend yon systematischen MeBfehlern befreit. Die Verbrennungsgase diirfer~ kein Schwefeldioxyd enthalten. ] ) a im Stahl auch Spuren Schwefel enthalten sind, wird vet die Megzelle tin Gef/iB mit Lewatit zur Absorption yon S0~ gesehaltet.
__,f" I
02
P~ms )~
z D ) -3 -3 ) ) D )
Zu- and AUuus dep A~sorp//aosld~un 9
Abb. 1. Appara~ur ftir (lie Kohlenstoffbestimmung. J/0 l~fel~zelle; M1 Vergleichsmel]strecke
Dieses Ana lysenve r fah ren i s t im w e s e n t l i c h e n / i b e r n o m m e n worden. Ledigl ieh die bei der Verbrennung des Sehwefels en t s t ehenden grogen Mengen s t6render Sehwefeloxyde lassen sieh du tch den Lew~ti t - aus tauseher n ieh t entfernen. Die Ana lysensubs tanz wird yon uns deshalb

90 F. FEtt~P~, S. ECKttARD und K. 1-I. SAV~R:
mit der 10faehen lVfenge Bleidioxyd vermiseht und dann verbrannt. Bus Bleidioxyd binder je naeh den Verbrennungsbedingungen 40--60o/o des Sehwefels als Bleisulfat. Die Verbrennungsgase str6men dann durch eine Reaktionsflfissigkeit, die aus Schwefels~ure und Wasserstoffperoxyd besteht und das restliche 802 zu SO a oxydiert. I)iese Vorlage wird auf - - 5 0 ~ abgekiihlt, um den Dampfdruok der Schwefels~ure so niedrig wie m6glich zu halten. Bei - -50 ~ C ist das Gemiseh noch d/innfl/issig. Zur Sicherung wird attch die Lewatitvorlage beibehalten. Abb. 1 (S. 89) zeigt sehematiseh die Apparatur ffir die Kohlenstoffbestimmung.
Die yon K o c ~ u. MALISSA ffir das Verfahren angegebene Empfindlieh- keit betrug 2 #g Kohlenstoff je Millimeter Sohreiberaussehlag. Sie ver- wandten als Absorptionsfl/issigkeit eine 0,005 n Natronlauge. Wit be- nutzten 0,001 n Natronlauge, woduroh die Empfindliehkeit auf etwa 0,5 #g C/ram Ausschlag gesteigert werden konnte. Die Kohlenstoffanalyse ben6tigte eine Einwaage yon 100 mg Sehwefel. Die Dauer einer Analyse betrug 2- -3 rain.
Die Anreicherung der anorganischen Yerunreinigungen Zur Anreicherung der anorganischen Verunreinigungen im Schwefel
werden gr6Bere Mengen Schwefel verbrannt und die l~fickst~nde unter- sucht. Um Verluste, die dutch Verdampfen leicht fliichtiger Fremdstoffe auftreten k6nnen, mit Sicherheit zu vermeiden, erfolgt die Verbrennung in der in Abb. 2 dargesteIlten Verbrennungsapparatur aus Quarz.
02
Decke/und 6'~ f~] zur #chwel~lauFnahrne "~--- 02
~WoschYlaschsn /~nungsraum
Abb. 2. Verbrennungsapparatur
Der Verbrennungsraum ist durch ein mit einem Schliff versehenes, sich nach unten zu einer Capill~re verjiingendes Gefi~l~ versch]iel~b~r. Dieses client gleich- zeitig zur Auihahme des flfissigen Schwefels, der durch das Cupill~rrohr in einen Mikrop]atintiegel (1 ml FassungsvermSgen) fiiel~t, wo er verbrennt. Die t~eaktions- geschwindigkeit wird vermittels eines gereinigten S~uerstoffstroms so geregelt, dab

Analytik des k~uflichen und gereinigten Schwefels 91
der verbrannte Sehwefel dureh neu zufliel3enden gerade erggnzt wird. Die l~eak- tionsw~rme reicht aus, den Sehwefel im Vorratsgef/iB flfissig zu halten. H~ufig erfolgt im Verbrennungsraum die Umsetzung zu SO 2 nieht vollst~ndig. Man ffihrt noehmals Sauerstoff zu und erhitzt zur Nachverbrennung die kugelfSrmige Er- weiterung im Ableitungsrohr. Die leichtflfichtigen Bestandteile werden dureh 3 Waschflasehen zuriickgehalten, die zusammen mit etwa 15 ml dreifach dest. Wasser beschickt sind. Nach der Verbrennung wird die Apparatur sorgf/~ltig mit ebenfalls dreifach dest. Wasser ausgewaschen, wozu 25 ml gentigen. Die Spfilfliissigkeit iiber- fiihrt man in den Platintiegel und dampft ein. Aus je 500 g der beiden gereinigten Schwefelsorten erhielten wir auf diese Weise 0,8 bzw. 1,4 mg Riickstand.
Die Analyse der Verbrennungsriickst~inde 1. Qualitative Analyse
Die Durchff ihrung einer quan t i t a t i ven Mikroanalyse wird wesentlich erleiehtert, wenn die qual i ta t ive Zusammense tzung der Analysensubs tanz b e k a n n t ist. Eine qual i ta t ive Ubersiehtsanalyse erh~lt m a n sehnell und
sieher spektralanalyt isch.
In die Bohrung einer Graphitelektrode werden etwa 0,3 mg Verbrennungs- riickst/~nde gebraeht und im Wechselstromhochspannungsbogen angeregt. Als Gegenelektrode dient ebenfalls eine Kohleelektrode. Die Aufnahme der Spektren erfolgt mit einem leistungsfiihigen Spektrographen zwischen 2100 und 4600 ~_. In diesem Gebiet liegen nahezu alle empfindlichen Linien der mit der Emissions- spektralanalyse erfagbaren Elemente. Ffir den qualitativen Nachweis empfiehlt sieh die Benutzung des grSl3ten vorhandenen Spektrographen, um die besten Bedingungen ffir die Spurenanalyse zu erreichen. Wit verwandten einen 1,5 m- Konkavgitterspektrographen (ARL). Die Kohleelektroden wurden vorher spektral- analytiseh unter den gleichen Bedingungen auf ihre Reinheit gepriift. StSrende Verunreinigungen liegen sieh nicht nachweisen. Bei der Auswertung der Spektren wurden in den Sehwefelriicksg~nden mit Sicherheit Aluminium, Silber, Barium, Wismut, Calcium, Chrom, Kupfer, Eisen, Kalium, Magnesium, Natrium, l~{angan, Nickel, Phosphor, Blei, Silicium, Strontium, Titan nnd Zink identifiziert. Fiir den Naehweis der Alkalien lieferte die Anregung in einer Flamme die beste Naehweis- empfindlichkeit. Die flammenphotometrisehe Untersuehung der l~/ickst~nde er- braehte jecloch keine weiteren Alkalien. Naeh der beschriebenen Methode konnten praktiseh die Elemente mit einer h6heren Ordnungszahl als Wismut, feruer Gase, I-Ialogene, die Seltenen Erden, Kohlenstoff, Selen und Tellur nieht erfaBt werden.
Die im Schwefel ebenfalls als Verunreinigung vermuteten Elemente Arsen, Antimon, Selen und Tellur konnten weder spektroehemiseh noeh mikrochemiseh ermittelt werden. Zur Kontrolle der angewendeten Verfahren wurde Sehwefel mit kleinen bekannten Mengen dieser Elemente verunreinigt und analysiert. Es ergab sieh, dal~ der vorliegende Schwefel weniger als 10 -G ~ an diesen Elementen ent- halten mug. Das Fehlen des st/~ndigen Sehwefelbegleiters Selen erseheint zun~chst bemerkenswert, erkl/irt sich aber dutch die Vorgesehiehte dieser Sehwefelsorten, die aus Gasreinigungsmasse gewonnen wurden.
Spektroehenisehe Methoden vereinigen liohe Naehweisempfindlieh- keit mi t geringem Substanzverbrauch. Die dutch die Ubersichtsanalyse gefundenen Verunre in igungen wurden daher mi t Ausnahme des Phosphors quantitativ spektralanalytiseh bestimmt.

92 F. FEH]~I~, S. ECKIIARD und K. H. SAUEI~:
2. Quantitative Bestimmungen durch LSsungsspektralanalyse Zur quanti tat iven Untersuehung der Verbrennungsrtickstgnde ist es
vorteilhaft, das Probegut aufzusehlieBen und eine L6sungsspektral- analyse durehzuffihren. ECKttAI~D, KooIt n. MAHI~ a haben ein Verfahren zur spektroehemisehen Analyse isolierter Gefiigebestandteile des Stahls angegeben, das mit Substanzmengen yon weniger als 1 nag durehfiihrbar ist und uns fiir die Analyse der Schwefelriieksts brauchbar erschien.
Es werden etwa 0,5 mg Substanz mit der 40fachen Nenge spektralreinem Borax aufgeschlossen. Die Schme]ze 15st man in einer kobaltchloridhaltigen 10~ Citronenss welche alle Metalle nebeneinander komplex in LSsung h~lt. Kobalt wird im festen VerhMtnis zur Einwaage zugesetzt und dient als Vergleiehselement (innerer Standard). Die L6sung wird auf spektralreine Koh]e aufgebraeht und im Hochspannungsfunken angeregt. Diese Methode ist ffir die Elemente Eisen, Chrom, Aluminium, Mangan, Silicium, Titan, Calcium und Magne- sium besehrieben. Als Spektrogmph wird der Einprismenquarzspektrograph Q24 (Zeiss) benutzt, die GehMte der untersuchten Substanzen an den einzelnen Elementen betragen 1~ und mehr.
Abb. 3. StOrung dureh Cyanbanden; oben: Kohlebogen in Lnft ; unten: Kohlebogen in Argon
Da die qualitative Untersuehung die Abwesenheit yon Kobal t er- braeht hatte, konnte es als innerer Standard beibehalten werden. Es durfte jedoeh nieht erwartet werden, dab im iibrigen die Methode un- ver/~ndert fibernommen und lediglieh dutch Aufstellung neuer Eieh- kurven auf die qualitativ gefundenen Elemente ausgedehnt werden konnte. In den linienreiehen Spektren war beim Auftreten neuer Elemente mit Koinzidenzen zu reehnen, zumindest bei Benutzung des Quarzspektrographen Q 24 im langwelligen Ultraviolett. Weiter lag die Konzentrat ion der Elemente in den R/iekst~nden der gereinigten Sehwefelsorten mit Ausnahme yon Eisen und Silieium unter 1~ so dan die Naehweisgrenze des besehriebenen Verfahrens untersehritten wurde. Nur die Eisen- und Silieiumbestimmung konnten unver/~ndert durch- gefiihrt werden.
Znr Analyse der/ibrigen Elemente muBten wit neue Wege besehreiten. F/ir die Bestimmung yon AI, Ca, Cu, Mg, Mn, Zn und Ti setzten wit den 1,5 m-Git terspektrographen ein. Die Anregung erfolgte im Weehsel- stromhoehspannungsbogen (4400 V, 2 Amp.). Damit wurde die Naeb- weisempfindliehkeit sowohl dutch den leistungsf~higeren Spektrographen als aueh dureh die empfindliehere Bogenanregung verbessert. Die Analyse
Ana.lytik des k~uflichen und gereinigten Sohwefels 93
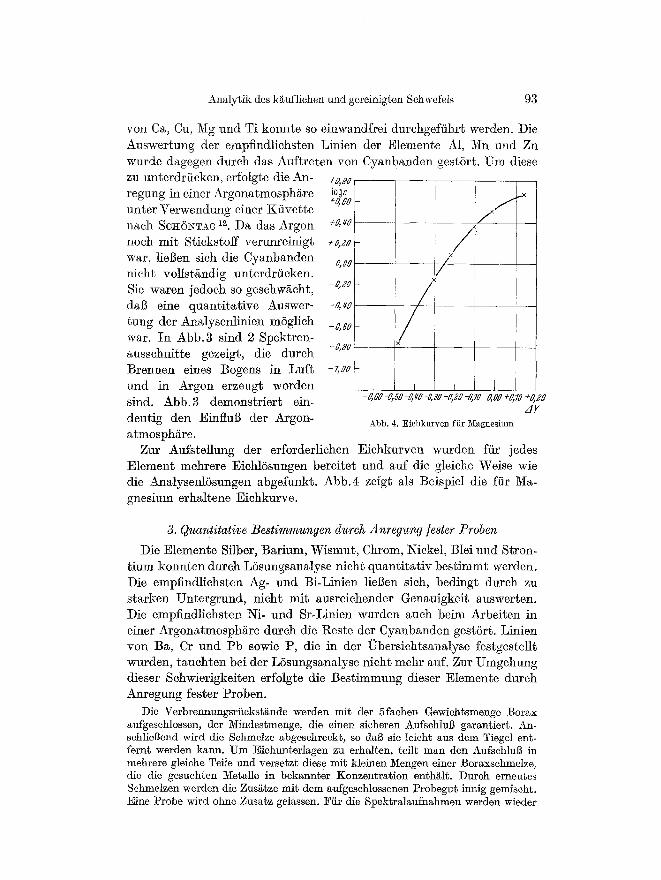
yon Ca, Cu, Mg und Ti konnte so einwandfrei darchgeffihrt werden. Die Auswertung der empfindliehsten Linien der Elemente A1, ~ n und Zn wurde dagegen dutch das Auftreten yon Cyanbanden gest6rt. Um diese zu unterdrficken, erfolgte die An- regung in einer Argonatmosph/tre unter Verwendung einer Kiivette naeh SCit6~TAG i~. Da das Argon noch mit Stickstoff verunreinigt war, liegen sich die Cyanbanden nieht vollst/indig unterdr/icken. Sic waren jedoeh so geschw/icht, dab eine quantitative Answer- tung der Analysenlinien m6glich war. In Abb.3 sind 2 Spektren- ausschnitte gezeigt, die dutch Brennen sines Bogens in Luft und in Argon erzengt worden sind. Abb.3 demonstriert ein- deutig den Einflug der Argon- atmosphgre.
+g<~o r iogc /
+0,~o I- +0,20 I
o, ug
-o, zg
-ggO
-geo
-0,80
-7,00
/ /
i / !
I I i r i I i -o,60-o,5o-o,~-o, so-o,3o-u/o 4oo +o/o +o,3n
Lily
Abb. 4. Eichkurven fiir N~agnesium
Zar Aufstellung der erforderliehen Eichkurven wurden ffir jedes Element mehrere Eichl6snngen bereitet und auf die gleiche Weise wie die Analysenl6sungen abgefunkt. Abb.4 zeigt als Beispiel die f~r Ma- gnesium erhaltene Eiehkurve.
3. Quantitative Best immungen dutch Anregung [ester Proben
Dis Elements Silber, Barium, Wismut, Chrom, Nickel, Blei und Stron- t ium konnten dutch LSsungsanalyse nicht quantitativ bestimmt werden. Die empfindliehsten Ag- und Bi-Linien lieBen sich, bedingt durch zu starken Untergrund, nicht mit ausreiehender Genauigkeit auswerten. Die empfindliehsten Ni- und Sr-Linien warden auch beim Arbeiten in einer Argonatmosph~re dutch die Reste der Cyanbanden gestSrt. Linien yon Ba, Cr und Pb sowie P, die in der iJbersiehtsanalyse festgestellt warden, tauchten bei der L6sungsanalyse nieht mehr auf. Zur Umgehung dieser Sehwierigkeiten erfolgte die Bestimmung dieser Elements durch Anregung fester Proben.
Die Verbrennungsr/ickst~nde werden mit der 5fachea Gewichtsmenge Borax aufgeschlossen, der IMindestmenge, dig einen sicheren AufsehluB garantier& An- schlieBend wird die Schmelze abgeschreekt, so da• sic leicht aus dem Tiegel ent- fernt werden karm. Um Eiehunterlagen zu erhalten, teilt man den Aufsehlul~ in mehrere gleiche Teile und versegzt diese mit kleinen Mengen einer Boraxsehmelze, die die gesuchten Metalle in bekannger Konzentra~ion en~h/~lt. Dutch erneutes Sehmelzen werden die Zus/~tze mit dem aufgeschlossenen Probegut innig gemiseht. Eine Probe wird ohne Zusatz gelassen. Fiir die Spekgralaufnahmen werden wieder

94 F. FEIK~R, S. EGK~_ARD und K. i . S•
spektralreine Kohlen benutzt. Als Kathode dient ein Stiff, dessen Ende in einem stumpfen Kegel auslauft. Die Anode ist mi~ einer Bohrung yon 1 mm Tiefe und 1,5 mm Durchmesser versehen. In diese Bohrung gib~ man 0,4--0,7 mg der be- reiteten Schmelze. Damit zu Beginn der Anregung keine Verluste durch Spritzen entstehen, wird das beschiekte Ende der Anode mit einem Gleiehs~rombogen, der dutch eine in halber H6he der Elektrode seitlieh angesetzte Hilfselektrode erzeugt wird, kurz bis zur l~otglut erhitzt (Abb. 5). Dabei fliel]en die aufgebraehten Teile der Boraxsehmelze zu einer Perle zusammen, die naeh dem Erkalten fest in der Bohrnng der Elektrode haftet. Es kann jetzt ohne Spritzverluste angeregt werden. Die Aufn~hme der mit dem hochempfindlichen Gleichstromdauerbogen (160V, 3,3Amp.) erzengten Spektren erfolgte mit dem 1,5m-Gitterspektro-
k i
i i
Ui ' . . . . i
Abb. 5. Spek~ralanalyse der Rfiekst~tnde: Ein~ehraelzen mit Hilfselektrode
graphen. Die Belichtungs- zeit betrug 30 see. In dieser Zeit war die Analysensub- stanz vollst~ndig verdampft, wie naehfo]gende Auf- nahmen ohne neue Besehik- kung zeigten. Damit ist die Voraussetzung fiir eine gute quantitative Analyse aueh im Gleichstromdauerbogen erfiillt.
Aus den In t ens i t~ t en der Analysen l in ien u n d den bekann ten Zuss werden nach e inem yon GATTERER 5 zuers~ be- schr iebenen Verfahren
die K a n z e n t r a t i o n e n der gesuchten E lemente berechnet . Das Verfahren se tz t die ffir geringe Konzen t r a t i onen beobach te t e s t renge Propor t io - na l i t s zwisehen K o n z e n t r a t i o n und Lin ienin tens i t / i t voraus , so dab aus mehreren, mi t Zusi~tzen versehenen P roben die K o n z e n t r a t i o n in der urspr i ingl iehen Probe berechnet werden kann. Als Vergle ichselement client Eisen, das in ausre ichenden Mengen in den Verbrennungsr i iek- sti~nden en tha l t en ist.
4. Bestimmung der Alkalien Die quantitative Bestimmung yon Natrium und Kalium erfolgt flammenphoto-
metrisch. 0,5--5 mg Verbrennungsrfickstand werden mit FluBss und Schwefel- s~ure abgeraucht, um die Alkalien mit Sicherheit in Sulfate zu iiberfiihren. Man nimmt nun mit ch-eifach dest. Wasser auf und verspriiht die LSsung in eine Acetylen- Luft-Flamme. Um den st6renden Einflul~ der Anionen zu vermeiden, wird der AlkaHgehalt mit ttflfe yon Konzentrationsreihen bestimmt.
5. Milcrochemische Analyse yon Phosphor und Chlor
Die Bes t immung des Phosphors erfolgt durch In tens i t i i t smessungen der Orangegelbf~rbung, die eine phospha tha l t ige sa lpe te rsaure LSsung mi t A m m o n i u m m o l y b d a t und A m m o n i u m v a n a d a t ergibt l~ la.

Ana134ik des kaufliehen und gereinigten Schwefels 95
Es werden 0,5--5 mg Verbrennungsrfiekstand mit Salpeters~ure und FluBs~ure aufgesehlossen und eingedampft. Den Riickstand versetzt man mit salpetersaurer Ammoniummolybdat- und VanadatlSsung und verdfinnt mit Wasser his auf 1 ml. Nach 30 min wird die Intensit~t der Gelbf~rbung in einem Photometer mit dem Filter Hg 436 gegen eine ]31indprobe gemessen. Die Empfindliehkeit des Verfahrens betragt 0,1 #g/mi.
Zur Halogenbestimmung wird der Sehwefel in der bereits besehriebenen Appara- tur (vgl. Abb.2, S. 90) verbrannt. Naeh der Verbrennung versetzt man die Spfil- fltissigkeit, die aueh die gesamten Riiekstgnde enth/~lt, mit etwas Natronlauge und dampft ein. Die Lauge client zur Neutralisation der bei der Verbren- nung des Schwefe]s in geringem MaSe entstehenden Sohwefelsgure, die beim Einengen Halogen unter
Halogenwasserstoffentwietdung zersetzen wfirde.
Das so erhaltene Salzgemiseh wird zuerst qualitativ auf seinen Halogengehalt untersueht. Im t~eaktionsgefgB erfolgt die Oxy- dation der Substanz in Anlehnung an J~rDA~ 6 dureh Sehwefels&ure und Silberdiehromat. Das Reak- tionsgef&g (Abb. 6) ist fiber einen Absorber mit einer Kfihlfalle ver- bunden. Der Absorber entfernt Sehwefeldioxyd, das sieh bei der nassen Oxydation sehwefelhaltiger Substanzen bilden kann, dutch ein Gemiseh yon Kaliumpermanganat- 16sung und Sehwefelsgure. Ein sehwaeher Stiekstoffstrom fiber- ftihrt das entwiekelte Halogen in die dureh flfissige Luf~ gek/ihlte Falle. Diese wird dann yon der fibrigen Apparatur getrennt und
/rUq/~//e "+ / I 1 Z'i:tl~disi:]~i'ok:" I I
-A&o:~e:
Abb. 6. knordnung zur qual%ativen Halogenbes$immung
die Gase werden spektrographisch untersucht. Die Kfihlfalle dient hierbei als Entladungsrohr. Ein RShrengenerator erzeugt das anregende Hochfrequenzfeld. Die Aufnahme des Spektrums erfolgt dureh efi3en Spektrographen mit guter Dispersion im sic.htbaren Bereich. Sie geschieht hier aus iiugeren Grfinden mit einem Dreiprismenglasspektrographen, f = 1600 mm (SteinheiI).
Die Auswer tung der Spek t r en ergab, dab in den Verbrennungsrf ick- s t~nden auBer Chlor iden keine nachweisbaren Halogenide en tha l t en waren. Der gefundene Chlorgehal t yon 2 . 1 0 -8 ~ reoht fer t ig te die An- nahme, dab die Gehal te an Brom und J o d hOehstens bei 10 -v ~ lagen, da Chlor wesent l ich schwerer an regbar i s t als Br und J .
Das yon JUDAH zur q u a n t i t a t i v e n Chlorbes t immung vorgeschlageneVer- fahren b r ing t S chwier igkei ten mi t s ich.Es wird daher wie folgt abgewandel t :
Die Kfihlfalle wird durch einen mit KaliumjodidlOsung geffillten Absorber ersetzt. Im Reaktionsgefiil3 erfolgt auf die besehriebene Art die Oxydation der

96 F. FEI~gIt, S. ECKItARD und K. H. SAVE~:
An~lysensubstanz. Durch das Chlor wird in dem Absorber Jodid zu Jod oxydiert. ~ach Beendigung der l~eaktion schtittelt man die jodhaltige K~liumjodidlSsung mit insgesamt 1 ml Chloroform aus and bestimmt die Violettfgrbung des organischen LSsungsmittels in einer 1 ral-Kfivette unter Verwendung des Filters S 53 photo- metrisch. Naeh dem Verfahren kSnnen noeh 0,5 #g Chlor n~chgewiesen werden.
Bestimmung der geliisten Gase Wie schon seit l~ngem bekannt ist, gibt Sehwefel beim Erhitzen im
Vakuum Gase ab. Nach TtnC]~L~ALL U. BI~I~AI~L~ZY 14 wird bei ver- mindertem Druek - - 50 Torr - - bis zum Siedepunkt Gasentwicklung beobaehtet. Wird dieser Sehwefel nach dem Erstarren erneut auf 240~ erhitzt, so soll es zu keiner Gasbildung mehr kommen. Dagegen finder MALUS 9 selbst nach 80m~tigem aufeinanderfolgendem Schmelzen unter einem Druck yon 20-- 30 Torr ernente Gasentwicklung. BAco~ n. FANELLI ~ haben ebenfalls beim mehrmaligen Schmelzen im Vakuum (1 mm Hg) erneute Gasentwicklung beobaehtet. Nach Ansicht dieser Autoren mul~ der Vorgang des Krist~llisierens und Sehmelzens 12m~l wiederholt werden, wenn der Sehwefel ohne Gasentwicklung schmelzen soll.
1. Ex t rak t ion der Gase
Angesiehts dieser widersprechendcn Angaben gait es zun~chst festzu- stellen, ob und unter welchen Bedingungen sich der Schwefel im Vukuum
vollst~ndig entgasen lieB und welche Mengen an Gas dabei in Freiheit
cLcod gesetzt wurden. - - Dazu
wird die in Abb. 7 sche-
Versuehsanordnung be- nutzt. Sie besteht aus einem Extraktionsgef~g,
und eine Kfihlfalle mit /herln~el~trl'sekes ttunlialle ~ dem Sammelgef~B ver-
f/gnome~st fxtrakt/omsgefizfl bnnden ist. Znm Eva- Abb. 7. hnordnung zxar Extr~ktion der Gase kuieren dient eine 01-
rotat ionspumpe; die Druckmessung erfolgt durch ein McLeod- and ein thermoelektrisches Manometer.
Um Schwefelproben mit ldeiner und gleichm~l~iger Oberfls zu erhalten, giel~t man fitissigen, gereinigten Schwefel in l~eagensgli~ser und schmelzt diese zu. Eine eingeschmolzene Probe ~_rd erst kurz vor einem Versuch Bus dem Glasrohr entfernt und sofort in das Extraktions- gefitB eingebr~eht. Man evakuiert bis auf 0,10 Torr und erw~rmt das

Analytik des k~uflichen und gereinigten Sehwefels 97
Extraktionsgef~tg auf 125 ~ C. W/~hrend des Aufsehmelzens wird eine kr/iftige Gasentwieklung beobaehtet. Aus dem vSllig gesehmolzenen Sehwefel entweichen keine Gasblasen mehr. Dann liBt man auf Zimmer- temperatur abkiihlen und mil]t den Druek. Es wird nun wieder bis auf 0,10 Torr evakuiert und der Schwefel erneut gesehmolzen. Naeh dem Ab- kiihlen der Sehmelze haben wir keine Druckinderung festgestellt. Nun wird der Sehwefel aus dem Extraktionsgef/iB in die Vorlage destilliert. Die auf - - 2 0 ~ gekfihlte Falle verhinderg ein Eindringen yon Schwefel- dampfen in das Sammelgef/~B. Erneute Druekmessung ergibt, dag aueh beim Destillieren kein meBbares Gasvolumen mehr in Freiheit gesetzt wird. Die verwendeten Manometer gestat ten eine genaue Druck- ablesung auf 0,01 Torr. Aus diesen Beobaehtungen folgern wir, dab der Schwefel dureh einmaliges Schmelzen im Vakuum miter einem Druck yon 0,1 Torr vSllig entgas~ wird.
2. Die qualitative Untersuchung der extrahierten Gaze
KocH, ECx~A~D u. STRZCK~ 7 berichten fiber eine quantitat ive spektrographische Methode zttr Bestimmung der Gase im Stahl. Die beschriebene Versuchsanorduung wurde yon uns zur qualitativen Analyse der aus dem Sehwefel extrahierten Gase benutzt. In einem Entladungs- rohr, das mi t dem Extraktionsgef~B verbunden ist, erfolgt die Anregung der Gase. Die Spektren werden mit einem Steinheil-Universalspek~ro- graphen aufgenommen, da die Nachweislinien bzw. -banden der Gase hauptsiehlich im Bereich um 4000 ~ liegen und die Spektren nur sehwer aufl6sbar sind.
Zur Durchfiihrung der qualitativen Analyse gibt man eine Schwefelstange in das Extraktionsgefiig, kfiM~ die Kiihffalle auf --20 ~ C und evakuier~ his auf 10-4Tort. Nun wird die Apparagur mit Argon gefiilR, erneut evakuiert und nochmals mit Argon geffillt. Man vermindert jetzt den Druck bis auf 0,1 Tort und erw/irmt das Extraktionsgefal] auf 125 ~ C. Das Spiilen mit Argon bewirkt, dab der Sehwefel in einer luftfreien Atmosphire gesehmolzen werden kann. Bei der qualitativen Auswertung des Spektrums liBt sieh sonst nicht entseheiden, ob gefundene Sauer- sgoff- und S~ickstoffbanden dutch die extrahierten Gase oder die schon vorher vorhandene Luft hervorgerufen werden.
Die im Sehwefel gel6sten Gase bestehen aus Sauerstoff, Stickstoff und Sehwefeldioxyd. Wasserstoff konnte nieht nachgewiesen werden. Die Analysenempfmdliehkeit gestatteg es, noeh 0,05#g I t in 100g Schwefel gut zu erkennen. ])er gereinigte Sehwefel engMlt also weniger als 5 �9 10 -s ~ Wasserstoff.
Die quantitative Bestimmung dieser Gase naeh der Methode yon KOCH u. Mitarb. hi~tte umfangreiche Eieharbeiten erfordert, die dutch Be- nutzung der gasvolumetrischen Mikromethode nach F E I C H T I N G E R 4 v e r -
mieden werden konnten. Z. analyt. Chem., Bd. 168 7

98 F. FEH]~R, ~. ECEHARD und K. H. SAUE~:
3. Quantitative Untersuchung der Gase
Dureh sine Quecksilbermembranpumpe werden die Gase aus der Extraktionsvorrichtung in den Mikrogasanalys~tor fibergefiihrt (Abb. 8). Der Analysator gestattet es, 0,1--2,0 ml Gas zu untersuehen. Die Gas- probe wird in dem Ger~t yon einem Absorptionsraum quantitativ in den anderen gebracht. Zwischen den einzelnen Reaktionsrs be- finder sieh Queeksilber. Durch dieses neutrale Verdr/~ngungsmittel wird die Gasblase welter gedrfiekt und beim Weiterwandern mit der zur Verdr~ngung notwendigen Hflfsfl/issigkeit gemessen. Diese indirekte
#xlrokl/on~eibpi~hlung
..PwnTpe
Abb. 8. Anordnung zur quantitativen Analyse der Gase
Volumenermittlung erfolgt mit einem rnikromefrischen Mel~kolbensystem auf 4 Stellen genau; die n~chste Dezimale kann noch gesch~tzt werden.
Im Analysator bestimmt man zuerst das Gesamtvolumen. Anschliel~end drtiekt man die Gasblase zur Sauerstoff- und Schwefeldioxydabsorption durch die mit alkaHscher PyrogallollSsung geffillten Reaktionsr~ume und mil~t erneut das Volumen. Das Restgas besteht aus Stickstoff. Eine weitere Gasprobe wird durch Kalilauge geleitet und dadureh das Schwefeldioxydvolumen ermittelt. Dieses zieht man yon der dureh die Pyrogalloll6sung absorbierten Gasmenge ab und erhalt so das Sauer- stoffvolumen.
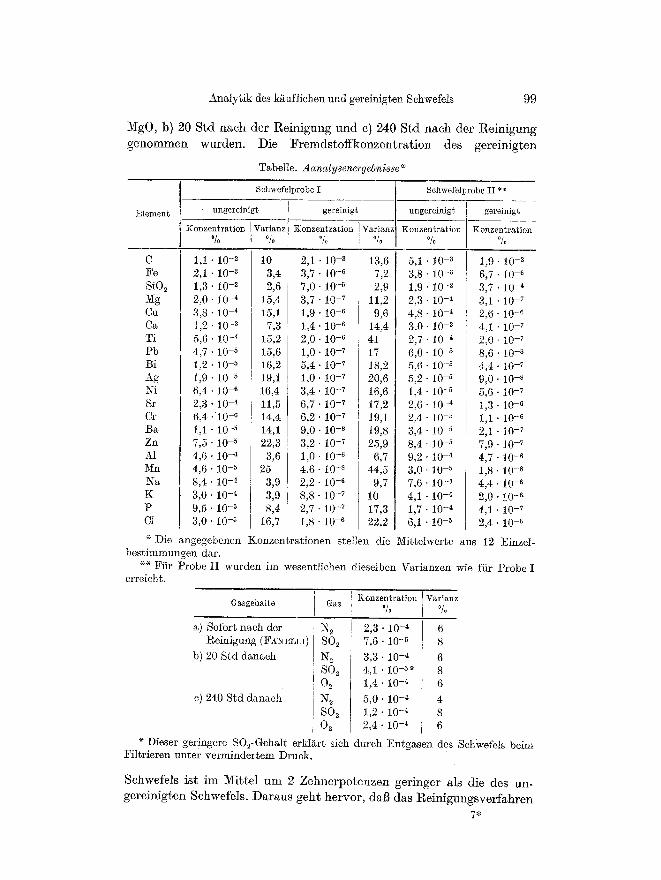
Analysenergebnisse In der Tabelle sind die Analysenergebrdsse mif den Varianzen f/Jr
gereinigten und ungereinigten Schwefel zusammengestellt. Zur Gas- analyse dienten Schwefelproben, die a) sofort nach dem Koehen mit
Analyt ik des k/~uflichen und gereinigten Schwefels 99
M g O , b ) 20 S t d n a c h d e r g e i n i g u n g u n d c) 240 S t d n a c h d e r R e i n i g u n g
g e n o m m e n w u r d e n . D i e F r e m d s t o f f k o n z e n t r a t i o n d e s g e r e i n i g t e n
Tabelle. Aanalysenergebnisse ~
Schwefelprobe I Schwefell)robe II **
Element~ ungereinigt gereinigt ungerein~gt gereinigt
Konzentration Varianz :Konzen?.ration ] Varianz Konzentration Konzen~ratJon % O/o % I ~ % ~/o
C 1,1 �9 10 -2 10 2,1 �9 10 -a 13,6 5,i 10 -3 1,9" 10 -3 Fe 2,1 - 1O -~ 3,4 3,7" 10 -6 7,2 3,8 10 -3 6 ,7 ' 10 -s Si02 1,3" 10 -2 2,6 7,0- 10 -5 2,9 1,9 10 -2 3,7 ' 10 -4 5{g 2,0 �9 10 -4 15,4 3,7 " 10 -7 11,2 2,3 10 -4 3,1 �9 10 -7 Ca 3,8 - 10 -4 15,1 1,9" 10 -6 9,6 4,8 10 4 2,6 �9 t0 -6 Ca 1,2 �9 10 -~ 7,3 1 , 4 . 1 0 -6 14,4 3,0 10 ~ 4,1 �9 10 -7 Ti 5,6- 10 -4 15,2 2 ) 0 . 1 0 -6 41 2,7 10 -4 2,0- 10 -7 Pb 4 , 7 . 1 0 -5 15,6 1 , 0 . 1 0 -7 17 6,0 10 5 8 , 6 . 1 0 -s Bi 1 , 2 . 1 0 -5 16,2 5 , 4 . 1 0 -7 18,2 5,6 10 -5 4,4- 10 -7 Ag 1,9 �9 10 -5 19,1 1 , 0 . 1 0 -7 20,6 5,2 10 -~ 9 , 0 . 1 0 -s ~Ni 6 , 4 . 1 0 -~ 16,4 3 ,4 ' 10 -7 16,6 1,4 10 -5 5,6 10 -7 Sr 2,3 . 10 -4 11,5 6,7 �9 10 -7 17,2 2,6 10 -4 1,3 10 -6 Cr 6 , 4 . 1 0 -6 14,4 6 , 2 . 1 0 -7 19,1 2,4 10 -5 1,1 10 -s Ba 1,1 �9 10 -5 14,1 9,0 - l0 - s t9,8 3,4 10 -5 2,1 10 -7 Zn 7,5 �9 10 5 22,3 3,2 �9 10 -7 25,9 8,4 10 -5 7,9 10 -7 A1 4 , 6 . 1 0 -4 3,6 1 , 0 . 1 0 -~ 6,7 9,2 10 -4 4,7 10 -6 Mn 4,6 - 10 -5 25 4,6 - 10 -s 44,5 3,0- 10 -5 1,8 10 -s Na 8 , 4 . 1 0 -4 3,9 2 , 2 . 1 0 -s 9,7 7,6" 10 -4 4,4 10 -6 K 3,0 �9 10 -4 3,9 8,8 - 10 -7 10 4,1 - 10 -4 2,0 10 -6 P 9,5" 10 -5 8,4 2,7 �9 10 -7 17,3 1,7 - 10 -~ 4,1 10 -3 C1 3,0 �9 10 -5 16,7 1,8 �9 10 -6 22,2 6,1 ' 10 ~ 2,4 10 -~
Die angegebenen Konzen t ra t ionen stellen die Mittelwerte aus 12 Einzel- bes t immungen dar.
�9 ~ Ffir Probe I I wurden im wesentl ichen dieselben Varianzen wie fiir Probe I erreicht.
Konzentration Varianz Gasgehalte Gas % %
a) Sofort naeh der N 2 2,3 �9 10 -4 6 l~einigung (F,~ELLI) SO 2 7,6 " 10 -5 8
b) 20 Std danach N e 3,3 �9 10 -4 6 S02 4,1 �9 10 - ~ 8 0 2 1,4" 10 -4 6
c) 240 Std danach N~ 5,0 �9 10 -4 4 SO 2 1 , 2 . 1 0 -4 8 O~ 2,4- 10 -4 6
Dieser geringere SO2-Gehalt erklart sich durch Entgasen des Schwefels beim Fil tr ieren un te r ve rminder tem Druck.
S c h w e f e l s i s t i m M i t t e l u m 2 Z e h n e r p o t e n z e n g e r i n g e r a l s d ie d e s u n -
g e r e i n i g t e n S c h w e f e l s . D a r a u s g e h t h e r v o r , dal3 d a s R e i n i g u n g s v e r f a h r e n
7*

lO0 F ~ , E c ~ I ) u. SAUER: Analytik d. k~uflichen und gereinigten Schwefels
nach FANELLI sehr wirksam ist. Die Unterschiede im Reinheitsgr~d der beiden gereinigten Sehwefelproben, die unterschiedlieh verunreinigt waren, weisen jedoch dar~uf hill, d~l~ des Reinigungsverfahren keinen Schwefel mit bestimmtem Reinheitsgrad liefert. Art und Menge der noch im gereinigten Sehwefel verbleibenden Fremdsubstanzen h~ngen yon den im Rohsehwefel enthaltenen Verunreinigungen ~b. Eine Steige- rung des Reinheitsgrades l~l]t sich erwarten, wenn der naeh FA~ELLI gereinigte Sehwefel ansehliel]end noeh im Vakuum sublimiert wird.
Zusammenfassung Der ehemisehe Reinheitsgrad des elementaren Sehwefels hut Einflul]
auf seine physikalisehen Eigenschaften. Da aber Art und Menge der Verunreinigungen im Sehwefel bisher nur unsieher bekannt waren, fehlte ~uch eine absolut zuverl~ssige Methode, einen fiir physikaliseh-ehemisehe Messungen geniigend reinen Schwefel herzustellen.
Die Verff. haben den Sehwefel qualitativ und quantitativ auf alle Beimengungen systemafiseh untersucht und dazu geeignete Analysen- verfahren herangezogen und fiir den speziellen Zweek abgewandelt. Die wichtige Bestimmung des Kohlenstoffs ist dureh Verbrennung des Sehwefels im Sauerstoffstrom und ansehliel~ende Leitf~higkeitsdiiferenz- messung der aufgefangenen Verbrennungsg~se durchgeffihrt worden. Die metallisehen Verunreinigungen konnten mit hoher Empfindliehkeit d~dureh erfaBt werden, dab gr58ere Sehwefelmengen unter Vorsiehtsmal~- nahmen verbrannt und so die niehtgasfSrmigen Verbrennungsprodukte angereiohert wurden. Die Analyse der angereieherten Substanzen erfolgte im wesentliehen spektralanalytiseh. Es warden im Mittel Nachweis- empfindliehkeiten yon 10 -6 ~ erreieht. Der Phosphor und die Halogene warden mikroehemiseh untersucht. Der Gasgehalt im Schwefel wurde dureh Aufschmelzen unter vermindertem Druek und Anregung in einem Gasentladungsrohr qualitativ spektralanalytisch naehgewiesen. Die quantitative Bestimmung erfolgte dann gasvolumetriseh.
Untersueht vatrden k~ufliehe Schwefelsorten und Schwefelproben, die naeh FA~Er,LI gereinigt wurden. Die besehriebenen Verfahren er- mSgliehten eine einwandfreie Beurteilung des chemischen Reinheitsgrades yon Schwefelproben. Des Reinigungsverfahren nach FANEL~ scheint den erwtinsehten l~einheitsforderungen weitgehend zu entspreehen.
Literatur 1 BAcon, 1~. F., u. l~. FA~ELLI: Ind. Engng. Chem. 34, 1043 (i942). --
B).co~, 1%. F., u. t~. FA~ELLI: J. Amer. chem. Soc. 65, 639 (1943). -- aECK~AI~O, S., W. Koc~ u. C. h / I ~ : Angew. Chem. 68, 296 (1956); vgl. diese Z. 156, 51 (1957). -- Vgl. auch Koch, W., u. S. EcK~m): Arch. Eisenhfit~enwes. 27, 165 (1956); vgl. diese Z. 151, 3i3 (1956). -- ~ Fnle~I~G~, H.: Arch. Eisenhiittenwes. 26, 127 (1955); vgl. diese Z. 150, 359 (1956). -- ~ G~-TTERER, A.: Spectrochim. Acta

A. J. LE]~B und i~. ttv, CHT: Cu-Bestimmung in Metallen und Legierungen 101
(London) 1, 513 (1941). -- ~ JvI)A~, J. D. : Biochemie. J. 45, 60 (1949); vgl. diese Z. 132, 301 (1951). -- 7 Koch, W., S. E e ~ D u. F. STriCKEn: Die ]ichtelektrische Spektral~nalyse der Gase im Stahl. KS]n/Ololaden 1958. (Forsehungsber. Wirt- schafts- u. Verkehrsmin. Nordrhein~Westfalen, Nr. 600). -- s Koch, W., u. H. MA~ssA: Arch. Eisenhiittenwes. 27, 695 (1956); vgl. diese Z. 158, 61 (1957). -- 9 MALVS, C.: Ann. Chim. Physique 24, 491 (1901). - - ~o MIsso~, G.: Chemiker-Ztg. 32, 633 (1908); vgl. diese Z. 56, 402 (1917). -- 1~ Scm~I])TS, W., u. D. BAASCH: Entwicklung einer chemisch-physikalischen Apparatur zur Bestimmung kleinster Kohlenoxyd-Konzentr~tionen. KSln/Opladen 1954. (Forschungsber. Wh'tschafts- u. Verkehrsmin. ~ordrhein-Westfalen, Nr. 67). -- 1~ Sc~5~T~, A.: Mikrechim. Acta (Wien) 1955, 376. -- TM Scm~SDV,~, R. : Stuh] u. Eisen 29, 1158 (1909); vgl. diese Z. 56, 403 (1917). -- ~ T~LF~LL, ~., U. J. I-~. D. BREAICL]~Y: Prec. Roy. See. (London) 56, 32 (1894).
Prof. Dr. F. FEH~R, Chem. Institut der Universit~t, KOln, Zfilpicher Strat~e 47
Aus dem chemisehen Laboratorinm der Schoeller-Bleckmann Stahlwerke-A.G., Ternitz, und der Lehrkanzel ffir analytisehe Chemie im II. Chemischen Universitats-
Institut Wien
Die Bestimmung yon Kupfer in Stahl, Ferrolegierungen, Aluminium, Chrom, Kobalt, Mangan und Nickel
Extraktionsverfahren mit 2,2' -Diehinolyl
Von A. J. LEEB und F. HECHT
Mit 1 Textabbildung
(Eingegangen am 26. Januar 1959)
Sei t den Un te r suchungen yon B~]~CXENRI])GE, LwW/S u. QVICK 1, welche ergaben, dab sich 2 ,2 ' -Dich ino ly l (ffir das auch der N a m e ,,Cu- p ro in" vorgeschlagen wurde) zum empfindl ichen :Nachweis und zur color imetr ischen Bes t immung yon K u p f e r eignet, erschien eine Anzah l yon Arbe i t en fiber die Bes t immung des Kupfe r s in verschiedenen Mater ia l ien mi t t e l s dieses Reagenses 3. Vor e inigen J a h r e n un te r such te ELW~LL 2 die color imetr ische Bes t immung yon K u p f e r in St~hlen m i t Cuproin nochmals genau und te i l te eine Arbe i t svorschr f f t zur E r m i t t l u n g yon 0,001 bis 1~ Cu in unlegier ten , legier ten und hochlegier ten St~hlen mi t . E r wies ferner d a r a u f hin, dal3 m a n K u p f e r au f diese Weise auch in einigen Fer ro leg ie rungen (Fer rowolf ram, -mangan, -molybd~n, -vana- d in und - t i tan) be s t immen kSnne.
U n t e r Zugrundelegung der obigen Arbe i ten , besonders der VerSffent- l ichung yon ELWELL, a rbe i t e t en wir in unseren L a b o r a t o r i e n ein Ver- fahren aus, naeh dem wir nun das K u p f e r in hochlegier ten Sti~hlen, v e t











![T O N I K U M 19 - APVzierung, besitzen bereits die spagyri-schen Urtinkturen über ein hohes energetisches Potenzial [2,4]. den die drei gereinigten Prinzipien Ausserdem sind die](https://img.pdfslide.org/doc/110x75/60fe776cd2dc1910f43e6057/t-o-n-i-k-u-m-19-apv-zierung-besitzen-bereits-die-spagyri-schen-urtinkturen-ber.jpg)

















