Embed Size (px)
Citation preview

Die extrazelluläre RNA-Welt von Listeria monocytogenes
und deren Potential zur Typ-I-IFN Induktion
Proteomanalyse der von Listeria monocytogenes
produzierten Membrane Vesicles
Inauguraldissertation
zur Erlangung des Grades eines Doktors der Humanbiologie
des Fachbereichs Medizin
der Justus-Liebig-Universität Gießen
Vorgelegt von Frantz, Renate
aus Siebenbürgen (Rumänien)
Gießen 2018

Aus dem Zentrum für Medizinische Mikrobiologie und Virologie
unter der Leitung von Prof. Dr. Trinad Chakraborty
des Fachbereichs Medizin der Justus-Liebig-Universität Gießen
1. Gutachter: Prof. Dr. Trinad Chakraborty
2. Gutachter: Prof. Dr. Günther Lochnit
Tag der Disputation: 19.11.2018

Inhaltsverzeichnis
I
Inhaltsverzeichnis
1. Einleitung ................................................................................................................................................ 1
1.1. Listeria monocytogenes .................................................................................................................... 1
1.2. Immunantwort gegen L. monocytogenes .......................................................................................... 3
1.3. Induktion von Typ-I-Interferon (Typ-I-IFN) .................................................................................... 5
1.3.1. Induktion von Typ-I-IFN während einer Infektion mit L. monocytogenes ............................... 8
1.4. Produktion von Vesikeln (OMVs bzw. MVs) durch Bakterien ...................................................... 11
1.5. Sekretion von RNA durch Bakterien .............................................................................................. 14
1.6. Zielsetzung dieser Arbeit ................................................................................................................ 17
2. Material und Methoden ....................................................................................................................... 18
2.1. Bakterienstämme und Virusstamm ................................................................................................. 18
2.2. Zelllinien ........................................................................................................................................ 18
2.3. Antibiotika-Stammlösungen ........................................................................................................... 18
2.4. Antikörper ...................................................................................................................................... 19
2.5. Molekulargewichtsmarker .............................................................................................................. 19
2.6. Vektoren ......................................................................................................................................... 20
2.6.1. Plasmidkarte des pCR2.1-TOPO-Vektors .............................................................................. 20
2.6.2. Multiple Cloning Site (MCS) des pCR2.1-TOPO-Vektors .................................................... 21
2.6.3. Plasmidkarte des pERL3-Vektors ........................................................................................... 22
2.7. Oligonukleotide (Primer) ............................................................................................................... 22
2.7.1. Primer für die Real-Time PCR (RT-PCR) .............................................................................. 22
2.7.2. Primer für die Herstellung der 25 RNA-Kandidaten .............................................................. 23
2.7.3. Primer für die Herstellung der rli32-Motive ........................................................................... 25
2.7.4. Primer für die Herstellung von Lm-pERL3-rli32 .................................................................... 25
2.7.5. Primer für die Herstellung von Lm-pERL3-ssrS .................................................................... 26
2.8. Chemikalien ................................................................................................................................... 26
2.9. Kits, Enzyme, gebrauchsfertige Lösungen und Chemikalien ......................................................... 27
2.10. Medien, Puffer und Enzyme für die Zellkultur ............................................................................ 28
2.11. Medien für die Anzucht von Bakterien ........................................................................................ 28
2.12. Gele und Puffer ............................................................................................................................ 31
2.13. Verbrauchsmaterialien .................................................................................................................. 37
2.14. Geräte ........................................................................................................................................... 37
2.15. Arbeiten mit DNA ........................................................................................................................ 39
2.15.1. Polymerase-Ketten-Reaktion (PCR) ..................................................................................... 39
2.15.2. Agarosegel-Elektrophorese von DNA-Fragmenten .............................................................. 40
2.15.3. Aufreinigung von DNA-Fragmenten .................................................................................... 41
2.15.4. Extraktion von DNA-Fragmenten aus dem Agarosegel ....................................................... 41
2.15.5. Isolierung von Plasmid-DNA ............................................................................................... 42
2.15.6. Bestimmung der Nukleinsäure-Konzentration ..................................................................... 42
2.15.7. Klonierung ............................................................................................................................ 43
2.15.7.1. Konierung durch TA Cloning ....................................................................................... 43

Inhaltsverzeichnis
II
2.15.7.2. Klonierung durch Restriktionsverdau und Ligation ...................................................... 44
2.16. Bakteriologische Arbeiten ............................................................................................................ 46
2.16.1. Kultivierung von Bakterien .................................................................................................. 46
2.16.2. Messung des Bakterienwachstums ....................................................................................... 46
2.16.3. Kompetenz und Transformation ........................................................................................... 46
2.16.3.1. Herstellung von elektro-kompetenten E. coli-Zellen .................................................... 47
2.16.3.2. Herstellung von chemisch-kompetenten E. coli-Zellen ................................................ 47
2.16.3.3. Herstellung von elektro-kompetenten L. monocytogenes-Zellen .................................. 48
2.16.3.4. Transformation von elektro-kompetenten E. coli-Zellen .............................................. 48
2.16.3.5. Transformation von chemisch-kompetenten E. coli-Zellen .......................................... 49
2.16.3.6. Transformation von elektro-kompetenten L. monocytogenes-Zellen ............................ 49
2.17. Arbeiten mit RNA ........................................................................................................................ 49
2.17.1. Transfektion von RNA in eukaryotischen Zellen ................................................................. 49
2.17.2. In vivo Transfektion von RNA ............................................................................................. 50
2.17.3. Isolierung der zellulären RNA aus eukaryotischen Zellen ................................................... 51
2.17.4. Isolierung der zellulären RNA aus Bakterien ....................................................................... 52
2.17.5. Isolierung von RNA aus Mausorganen ................................................................................. 53
2.17.6. Qualitätskontrolle der RNA und RNA-Profilanalyse ........................................................... 54
2.17.7. Reverse Transkription (cDNA-Synthese) ............................................................................. 54
2.17.8. Real-Time PCR (RT-PCR) ................................................................................................... 55
2.17.9. Isolierung von MVs-RNA .................................................................................................... 56
2.17.10. Isolierung von sec-RNA ..................................................................................................... 57
2.17.11. Sequenzierung .................................................................................................................... 58
2.17.12. Herstellung von RNA durch in vitro Transkription (IVT) .................................................. 58
2.17.13. Verdau des DNA-templates ................................................................................................ 59
2.17.14. Dephosphorylierung von RNA ........................................................................................... 59
2.17.15. Denaturierende Polyacrylamidgel-Elektrophorese ............................................................. 60
2.17.16. Extraktion von RNA aus dem Polyacrylamidgel ................................................................ 60
2.17.17. Phenol/Chloroform-Fällung von RNA ............................................................................... 60
2.18. Proteinanalytische Methoden ....................................................................................................... 61
2.18.1. Isolierung von Membrane Vesicles (MVs), die von L. monocytogenes produziert werden .. 61
2.18.2. Bestimmung der Proteinkonzentration mit der Bradford-Methode ...................................... 61
2.18.3. Bestimmung der Proteinkonzentration mit der BCA-Methode ............................................ 62
2.18.4. Probenvorbereitung für die Elektronenmikroskopie (EM) ................................................... 63
2.18.5. SDS-Polyacrylamidgel-Elektrophorese (SDS-PAGE) ......................................................... 63
2.18.6. Western blot-Analyse von MVs ........................................................................................... 64
2.18.7. Untersuchung der Internalisierung von MVs in PtK-Zellen ................................................. 65
2.18.8. Hämolyse-Assay ................................................................................................................... 65
2.19. Zellkultur-Methoden .................................................................................................................... 66
2.19.1. Kultivierung von Zelllinien .................................................................................................. 66
2.19.2. Gewinnung von primären Knochenmark-Makrophagen aus der Maus ................................ 67
2.19.2.1. Gewinnung von M-CSF ................................................................................................ 68
2.19.3. Infektion von HEK293-Zellen mit A/PR/8/34 (H1N1) ........................................................ 68
2.19.3.1. Virusquantifizierung durch Focus Forming Assay (FFA) ............................................ 68
2.20. Statistische Analyse ...................................................................................................................... 69
3. Ergebnisse ............................................................................................................................................. 70
3.1. Isolierung und Identifizierung der von L. monocytogenes sekretierten RNA................................. 70
3.1.1. Co-Isolierung von sec-RNA und MVs-RNA .......................................................................... 70

Inhaltsverzeichnis
III
3.1.2. Profilanalyse von sec-RNA und MVs-RNA ........................................................................... 71
3.1.3. Untersuchung der Fähigkeit von sec-RNA und MVs-RNA IFN-β zu induzieren .................. 72
3.1.4. Analyse der Zusammensetzung von sec-RNA und MVs-RNA .............................................. 73
3.1.5. Herstellung einzelner sRNAs durch in vitro Transkription .................................................... 74
3.1.6. Untersuchung der Fähigkeit einzelner sRNAs IFN-β zu induzieren ...................................... 78
3.1.6.1. Untersuchung der Abhängigkeit der IFN-β Induktion von dem Rezeptor RIG-I............ 80
3.1.6.2. Untersuchung der Abhängigkeit der IFN-β Induktion von dem Rezeptor MDA5 .......... 81
3.1.6.3. Untersuchung der rli32-Motive hinsichtlich der Fähigkeit IFN-β zu induzieren ............ 82
3.1.7. Untersuchung der Inhibition der viralen Replikation durch rli32 und rli47 ........................... 83
3.1.7.1. Untersuchung der Abhängigkeit der viralen Inhibition von den Rezeptoren RIG-I und
MDA5 .......................................................................................................................................... 85
3.1.7.2. Untersuchung der Abhängigkeit der viralen Inhibition von der RNA-Konzentration .... 86
3.1.7.3. Untersuchung der Inhibition der viralen Replikation durch weitere sRNAs................... 87
3.1.8. Untersuchung der Fähigkeit von rli32 Typ-I-IFN in der Maus zu induzieren ........................ 88
3.1.9. Überexpression von rli32 und ssrS in L. monocytogenes ....................................................... 90
3.1.9.1. Untersuchung der IFN-β Induktion durch sec-RNA und MVs-RNA in BMDMs .......... 91
3.1.9.2. Untersuchung der IFN-β Induktion durch sec-RNA und MVs-RNA in HEK293-Zellen92
3.1.9.3. Untersuchung der Abhängigkeit der IFN-β Induktion durch sec-RNA und MVs-RNA
von dem Rezeptor RIG-I.............................................................................................................. 93
3.1.9.4. Untersuchung der Fähigkeit von MVs IFN-β zu induzieren ........................................... 94
3.2. Charakterisierung der von L. monocytogenes produzierten MVs ................................................... 95
3.2.1. Elektronenmikroskopische Visualisierung von L. monocytogenes und isolierten MVs ......... 95
3.2.2. Bestimmung der Größe, Anzahl und Proteinkonzentration von isolierten MVs .................... 96
3.2.3. Proteomanalyse (Massenspektrometrie) von MVs ................................................................. 98
3.2.3.1. Klassifizierung der MVs-Proteine nach Funktion ........................................................... 98
3.2.3.2. Klassifizierung der MVs-Proteine nach Lokalisation ................................................... 105
3.2.3.3. Die 20 häufigsten Proteine in den MVs ........................................................................ 107
3.2.3.4. Virulenzfaktoren in den MVs ....................................................................................... 109
3.2.4. SDS-PAGE- und Western blot-Analyse von MVs ............................................................... 111
3.2.5. Untersuchung der hämolytischen Aktivität von MVs........................................................... 112
3.2.6. Untersuchung der Fähigkeit von MVs zur Internalisierung in PtK-Zellen ........................... 113
4. Diskussion ........................................................................................................................................... 115
4.1. Die von L. monocytogenes sekretierte RNA ist in den Fraktionen sec-RNA und MVs-RNA
gegenwärtig ......................................................................................................................................... 115
4.2. Unterschiedliche Zusammensetzung von sec-RNA, MVs-RNA und zellulärer RNA ................. 116
4.3. Anreicherung von kleinen nicht-kodierenden RNAs in MVs-RNA ............................................. 117
4.4. Unterschiedliche IFN-β Induktion durch sRNAs, die in sec-RNA und MVs-RNA identifiziert
worden sind ......................................................................................................................................... 118
4.4.1. RIG-I-Abhängigkeit der IFN-β Induktion durch die sRNAs ................................................ 119
4.4.2. Protektion von HEK293-Zellen gegen das Influenza A Virus durch die sRNAs ................. 120
4.5. Das Motiv 2 ist für die hohe IFN-β Induktion durch rli32 verantwortlich ................................... 121
4.6. Hohe Typ-I-IFN Induktion durch rli32 in der Maus .................................................................... 122
4.7. Erhöhte IFN-β Induktion durch die sekretierten RNA-Fraktionen und MVs nach Überexpression
von rli32 in L. monocytogenes ............................................................................................................ 122
4.8. Produktion von MVs durch L. monocytogenes ............................................................................ 124
4.9. MVs enthalten Enzyme, die möglicherweise an deren Biogenese beteiligt sind .......................... 125

Inhaltsverzeichnis
IV
4.10. MVs enthalten Proteine aus allen subzellulären Fraktionen: Cytoplasma, Cytoplasmamembran,
Zellwand und Extrazellulär ................................................................................................................. 126
4.11. MVs enthalten zum Teil vollständige Stoffwechselwege zur Energiegewinnung ..................... 127
4.12. MVs enthalten Virulenzfaktoren, die während der Infektion eine Rolle spielen ........................ 129
5. Zusammenfassung .............................................................................................................................. 131
5.1. Summary ...................................................................................................................................... 132
6. Abkürzungsverzeichnis...................................................................................................................... 134
7. Literaturverzeichnis ........................................................................................................................... 135
8. Anhang ................................................................................................................................................ 152
8.1. Veröffentlichungen ....................................................................................................................... 152
8.2. Kongressteilnahme ....................................................................................................................... 153
8.3. Danksagung .................................................................................................................................. 154
8.4. Eidesstattliche Erklärung .............................................................................................................. 155
8.5. Tab. 1: Einteilung der COGs in 25 funktionelle Kategorien ........................................................ 156
8.6. Tab. 2: Berechnung des Anteils an MVs-Proteine in den funktionellen Kategorien und Gruppen
anhand des NSAF-Wertes in % ........................................................................................................... 157
8.7. Tab. 3: Funktionelle Klassifizierung der 516 MVs-Proteine (EggNOG 4.5.1) ............................ 158

1. Einleitung
1
1. Einleitung
1.1. Listeria monocytogenes
L. monocytogenes (Lm) ist ein stäbchenförmiges, gram-positives und fakultativ
intrazelluläres Bakterium, das sowohl für den Menschen als auch für das Tier pathogen
ist. Es gehört zur Gattung Listeria mit insgesamt 17 Arten, darunter ein weiteres
Pathogen, L. ivanovii, das vorwiegend Tiere infiziert (Orsi und Wiedmann, 2016).
L. monocytogenes ist weit verbreitet und kommt in zahlreichen Tierarten und in der
Umwelt vor, wie beispielsweise im Boden, Wasser und Abwasser (Weis und Seeliger,
1975; Nightingale et al, 2005). Dieses Bakterium ist äußerst resistent gegenüber
extremen Umweltbedingungen. Es toleriert einen weiten Temperatur- (0-45°C) und pH-
Bereich (4,5-9,0) sowie hohe Salzkonzentrationen (10%) (Koutsoumanis et al, 2003;
Johnson et al, 1988). Aufgrund dieser Eigenschaften und wegen der ubiquitären
Verbreitung erfolgt die Infektion mit L. monocytogenes in der Regel durch den Verzehr
von verunreinigten Lebensmitteln in Form von rohen Milch- und Fleischprodukten
sowie ungewaschenem Obst und Gemüse (Bille et al, 2006; McLauchlin et al, 1990).
Nach der oralen Aufnahme gelangt L. monocytogenes zunächst in den Magen-Darm-
Trakt. Der weitere Verlauf der Infektion hängt von der immunologischen Fitness des
Wirtes ab. Das Krankheitsbild der Listeriose reicht dabei von Durchfall und grippe-
ähnlichen Symptomen bis hin zu Entzündungen der Hirnhäute (Meningitis) und des
Gehirns (Enzephalitis) (Roberts et al, 2003; Todd und Notermans, 2011). Bei immun-
kompetenten Menschen tritt eine nicht-invasive Listeriose auf, die sich in einer
fiebrigen Gastroenteritis äußert. Immunsupprimierte Menschen, darunter ältere
Menschen, Schwangere und Neugeborene, sind besonders für eine invasive Listeriose
anfällig. Die Mortalität bei einer Erkrankung an invasive Listeriose beträgt 20-30%
(Farber und Peterkin, 1991; Mead et al, 1999). Bei der invasiven Listeriose durchbricht
L. monocytogenes die intestinale Barriere und breitet sich über Blut und Lymphe in eine
Vielzahl von Organen aus. L. monocytogenes ist in der Lage weitere Barrieren im
menschlichen Körper zu durchbrechen. Zum einen die Blut-Hirn-Schranke, woraus die
Infektion der Hirnhäute und des Gehirns resultiert, und zum anderen die Placenta-
Barriere, die zu einer Infektion des Fötus mit Fehl- und Totgeburt führen kann (Lecuit
et al, 2004; Lecuit, 2005).
1. Einleitung
2
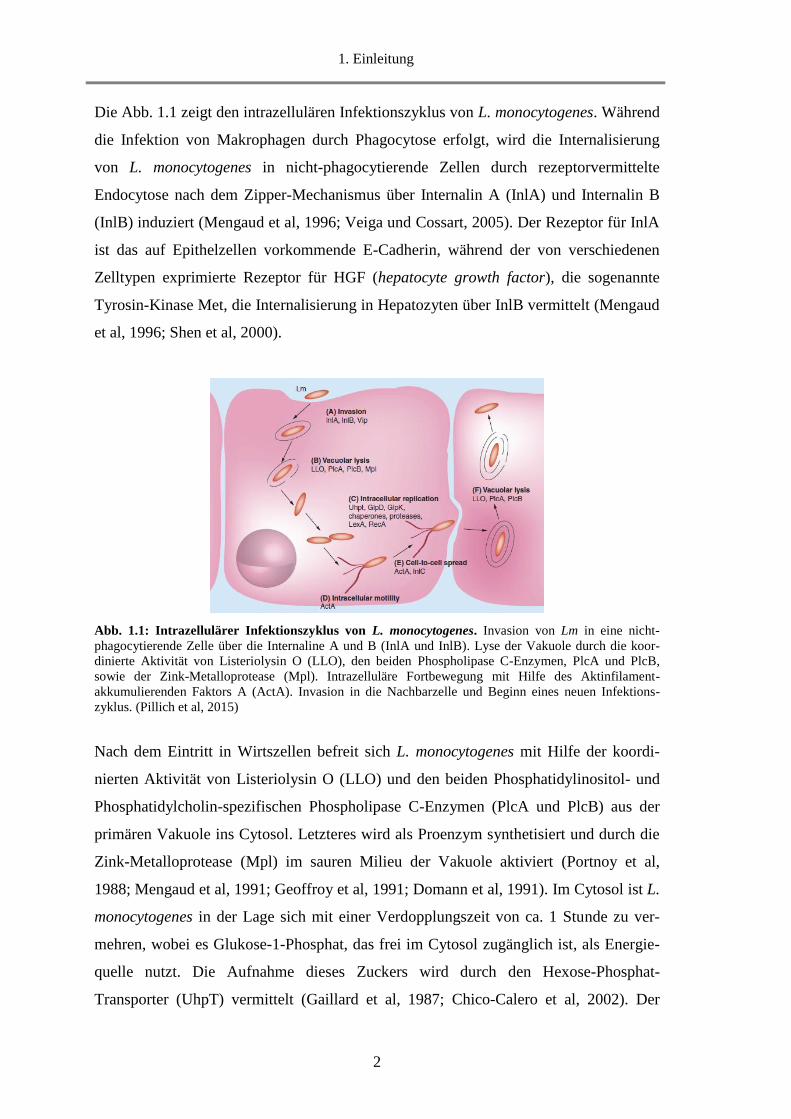
Die Abb. 1.1 zeigt den intrazellulären Infektionszyklus von L. monocytogenes. Während
die Infektion von Makrophagen durch Phagocytose erfolgt, wird die Internalisierung
von L. monocytogenes in nicht-phagocytierende Zellen durch rezeptorvermittelte
Endocytose nach dem Zipper-Mechanismus über Internalin A (InlA) und Internalin B
(InlB) induziert (Mengaud et al, 1996; Veiga und Cossart, 2005). Der Rezeptor für InlA
ist das auf Epithelzellen vorkommende E-Cadherin, während der von verschiedenen
Zelltypen exprimierte Rezeptor für HGF (hepatocyte growth factor), die sogenannte
Tyrosin-Kinase Met, die Internalisierung in Hepatozyten über InlB vermittelt (Mengaud
et al, 1996; Shen et al, 2000).
Abb. 1.1: Intrazellulärer Infektionszyklus von L. monocytogenes. Invasion von Lm in eine nicht-
phagocytierende Zelle über die Internaline A und B (InlA und InlB). Lyse der Vakuole durch die koor-
dinierte Aktivität von Listeriolysin O (LLO), den beiden Phospholipase C-Enzymen, PlcA und PlcB,
sowie der Zink-Metalloprotease (Mpl). Intrazelluläre Fortbewegung mit Hilfe des Aktinfilament-
akkumulierenden Faktors A (ActA). Invasion in die Nachbarzelle und Beginn eines neuen Infektions-
zyklus. (Pillich et al, 2015)
Nach dem Eintritt in Wirtszellen befreit sich L. monocytogenes mit Hilfe der koordi-
nierten Aktivität von Listeriolysin O (LLO) und den beiden Phosphatidylinositol- und
Phosphatidylcholin-spezifischen Phospholipase C-Enzymen (PlcA und PlcB) aus der
primären Vakuole ins Cytosol. Letzteres wird als Proenzym synthetisiert und durch die
Zink-Metalloprotease (Mpl) im sauren Milieu der Vakuole aktiviert (Portnoy et al,
1988; Mengaud et al, 1991; Geoffroy et al, 1991; Domann et al, 1991). Im Cytosol ist L.
monocytogenes in der Lage sich mit einer Verdopplungszeit von ca. 1 Stunde zu ver-
mehren, wobei es Glukose-1-Phosphat, das frei im Cytosol zugänglich ist, als Energie-
quelle nutzt. Die Aufnahme dieses Zuckers wird durch den Hexose-Phosphat-
Transporter (UhpT) vermittelt (Gaillard et al, 1987; Chico-Calero et al, 2002). Der

1. Einleitung
3
Aktinfilament-akkumulierende Faktor A (ActA), der die polarisierte Polymerisation von
Aktin der Wirtszelle induziert, vermittelt sowohl die intra- als auch die interzelluläre
Bewegung von L. monocytogenes, wobei es zur Infektion von Nachbarzellen kommt
(Kocks et al, 1992; Domann et al, 1992). Nach der Invasion in Nachbarzellen befreit
sich das Pathogen erneut durch Lyse der sekundären Vakuole und kann einen neuen
Infektionszyklus beginnen. Mit dieser Strategie kann sich L. monocytogenes von Zelle
zu Zelle ausbreiten, wodurch es der humoralen Immunantwort des Wirtes entkommt.
Das Genom von L. monocytogenes EGD-e (Serotyp 1/2a), der Stamm der in dieser
Arbeit verwendet wurde, ist im Jahr 2001 vollständig sequenziert worden. Dieser besitzt
ein zirkuläres Chromosom, das ein GC-Gehalt von 39% aufweist und aus 2944528 Bp
besteht. Die Anzahl der proteinkodierenden Gene beträgt 2853 (Glaser et al, 2001).
Die für den Infektionszyklus essentiellen Virulenzgene prfA, plcA, hly, mpl, actA und
plcB sind in dem 9 Kb langen Virulenzgencluster LIPI-1 organisiert (Listeria pathoge-
nicity island 1) (Chakraborty et al, 2000). Dieser wird von dem positiven Regulations-
faktor A (PrfA), der selbst Teil des Clusters ist, positiv reguliert. Die PrfA-abhängige
Transkription unterliegt einer Thermoregulation und wird erst bei 37°C induziert
(Chakraborty et al, 1992; Renzoni et al, 1999; Johansson et al, 2002). Das inlAB-Operon
ist an einer anderen Stelle des Genoms lokalisiert und wird nur teilweise von PrfA
reguliert (Lingnau et al, 1995). Dies gilt auch für Internalin C (InlC), ein Homolog der
beiden Internaline A und B, dem aufgrund der stark induzierten Transkription während
der Vermehrung von L. monocytogenes im Cytosol eine Rolle in der späten Phase des
Infektionszyklus zugeschrieben wird (Engelbrecht et al, 1996).
1.2. Immunantwort gegen L. monocytogenes
In der frühen Phase einer Infektion mit L. monocytogenes wirkt das angeborene
Immunsystem unspezifisch und wirksam zur raschen Eindämmung der Infektion.
Hierbei spielen die Aktivierung und Rekrutierung einer Reihe von Immunzellen eine
entscheidende Rolle. In Milz und Leber, die primären Zielorgane von L. monocytogenes
nach einer systemischen Infektion, wird die Mehrheit der Bakterien von gewebs-
ständigen Makrophagen phagocytiert (Mackaness, 1962; Conlan und North, 1992).
Diese produzieren daraufhin die pro-inflammatorischen Cytokine TNF-α und IL-12.
Dadurch werden Natürliche Killer-Zellen (NK-Zellen) zur Produktion von IFN-γ
stimuliert (Havell, 1989; Wherry et al, 1991; Tripp et al, 1993). IFN-γ ist eines der

1. Einleitung
4
wichtigsten Cytokine in der frühen Phase einer Infektion mit L. monocytogenes. Es
führt zur Aktivierung von Makrophagen, indem es deren Effektorfunktionen, wie
beispielsweise die Produktion reaktiver Sauerstoff- (ROS) und Stickstoffspezies (RNS),
sowie die Sekretion von pro-inflammatorischen Cytokinen steigert (Buchmeier und
Schreiber, 1985; Huang et al, 1993; Beckerman et al, 1993). In der frühen Phase einer
Infektion mit L. monocytogenes sind NK-Zellen die Hauptproduzenten von IFN-γ.
Jedoch stellen T-Zellen des adaptiven Immunsystems eine alternative Quelle an
anfänglichen IFN-γ dar, wodurch diese die angeborene Immunabwehr unterstützen.
CD4+- und CD8+-T-Zellen werden in Anwesenheit von IL-12 und IFN-γ selbst zur
Produktion von IFN-γ angeregt. Dieser Prozess findet in der frühen Phase der Infektion
ohne Antigenstimulation über MHC-Moleküle (major histocompatibility complex) statt
und wird ausschließlich von Cytokinen ausgelöst (Andersson et al, 1998; Berg et al,
2002; Lertmemongkolchai et al, 2001).
Bei der Bekämpfung von L. monocytogenes in der frühen Phase der Infektion sind
neben Makrophagen weitere unspezifische Immunzellen beteiligt. Neutrophile Granulo-
zyten werden durch Chemokine, die beispielsweise von infizierten Hepatozyten
sekretiert werden, an die Infektionsorte gelockt (Rogers et al, 1996). Ähnlich wie
Makrophagen verfügen diese über antimikrobielle Mechanismen, indem sie u.a.
phagocytierte Bakterien durch die Bildung von ROS und RNS töten (Bortolussi et al,
1987; Conlan und North, 1991; Rogers und Unanue, 1993). Ly6C+ Monocyten sind
weitere Effektorzellen, die durch das Chemokin CCL2 (CC-chemokine ligand 2), das
von infizierten Makrophagen sekretiert wird, rekrutiert werden. Diese wandern aus dem
Knochenmark in die infizierten Organe ein und differenzieren zu TipDCs (TNF and
inducible nitric oxide synthase (iNOS) producing dendritic cells). Deren hohe
Produktion an TNF und iNOS ist für die Begrenzung der Infektion unentbehrlich
(Serbina et al, 2003; Serbina und Pamer, 2006).
Aufgrund der intrazellulären Lebensweise dieses Pathogens spielt die humorale Immun-
antwort keine wesentliche Rolle. Für die Überwindung einer Infektion mit L.
monocytogenes ist die Aktivität von T-Zellen des adaptiven Immunsystems notwendig.
Sowohl CD4+- als auch CD8+-T-Zellen sind darin involviert, jedoch ist die Aktivität
von antigenspezifischen cytotoxischen CD8+-T-Zellen für die vollständige Elimi-
nierung des Pathogens essentiell (Ladel et al, 1994). Während CD4+-T-Zellen als TH1-
Helferzellen die Funktion von Makrophagen antigenspezifisch über den T-Zell-

1. Einleitung
5
Rezeptor regulieren, töten cytotoxische CD8+-T-Zellen infizierte Zellen. Diese werden
durch die Aktivität von DCs, den wichtigsten antigenpräsentierenden Zellen des
Immunsystems (APCs), aktiviert. Im Gegensatz zu Makrophagen sind DCs in der Lage
Antigene über MHC-Klasse-I-Moleküle an naive CD8+-T-Zellen zu präsentieren (Jung
et al, 2002). Unreife DCs, die im peripheren Gewebe patrouillieren, nehmen Antigene
auf und reifen daraufhin zu APCs. Diese wandern aus den infizierten Organen in die
Lymphknoten ein und präsentieren Antigene an naive CD8+-T-Zellen. Diese
differenzieren daraufhin zu antigenspezifischen cytotoxischen CD8+T-Zellen und
lysieren infizierte Körperzellen. Dies führt zur Freisetzung der intrazellulären Listerien
und zur deren Tötung durch aktivierte Makrophagen. Weil DCs kein bedeutendes
Reservoir für Listerien darstellen, findet das Priming von CD8+-T-Zellen über
Kreuzpräsentation exogener Antigene statt (Albert et al, 1998).
Voraussetzung für die Bildung listerienspezifischer CD8+T-Zellen ist die Invasion von
L. monocytogenes ins Cytosol der infizierten Zellen. LLO-defiziente Listerien, die nicht
ins Cytosol der Wirtszellen eindringen können, führen nicht zur Bildung von listerien-
spezifischen CD8+T-Zellen (Berche et al, 1987; Brunt et al, 1990). Ausschließlich im
Cytosol erhalten Listerien-Antigene, die während der Vermehrung des Pathogens
sekretiert werden, Zugang zu dem MHC-Klasse-I-Antigen-Präsentationsweg. Dabei
agiert das Hämolysin LLO als eines der wirksamsten Listerien-Antigene (Berche et al,
1987; Safley et al, 1991). Ein weiteres Antigen, das eine ebenso effektive CD8+-T-Zell-
vermittelte Immunantwort gegen L. monocytogenes auslöst, ist das Autolysin P60 (Sijts
et al, 1997). Die antigenspezifischen cytotoxischen CD8+T-Zellen sind sowohl bei einer
primären als auch bei einer sekundären Infektion mit L. monocytogenes essentiell. Die
Ausbildung einer protektiven Immunität gegen L. monocytogenes erfolgt nur bei einer
Immunisierung mit lebenden Bakterien, nicht mit HKLM (heat-killed Lm) oder LLO-
defizienten Stämmen (von Koenig und Finger, 1982).
1.3. Induktion von Typ-I-Interferon (Typ-I-IFN)
Typ-I-IFN umfasst eine Vielzahl von Interferonen, darunter IFN-β und verschiedene
IFN-α Subtypen. Die vielfältigen antiviralen Mechanismen, die nach deren Bindung an
den interferon alpha and beta receptor (IFNAR) ausgelöst werden, zählen zu der am
besten charakterisierten Immunantwort gegen virale Infektionen (Isaacs und
Lindemann, 1957; Müller et al, 1994; Sadler und Williams, 2008). Neben der

1. Einleitung
6
antiviralen Aktivität wirken Interferone auch antiproliferativ auf Tumorzellen,
immunmodulatorisch und als Signalüberträger zwischen Immunzellen (Trinchieri,
2010).
Typ-I-IFN wird von verschiedenen Zelltypen als Antwort auf virale und bakterielle
Infektionen produziert. Die Produktion von Typ-I-IFN wird nach Detektion von
sogenannten PAMPs (pathogen-associated molecular patterns) durch PRRs (pattern
recognition receptors) des angeborenen Immunsystems über intrazelluläre Signal-
kaskaden ausgelöst (Abb. 1.2). Hierbei sind sowohl membrangebundene TLRs (Toll-
like receptors) als auch cytosolische PRRs involviert (Meylan und Tschopp, 2006; Kato
et al, 2011).
Abb.1.2: Induktion von Typ-I-IFN nach Detektion bakterieller und viraler Strukturen durch
verschiedene Immunrezeptoren. Die Erkennung von LPS erfolgt durch TLR4 an der Zelloberfläche.
Die Detektion von dsRNA, ssRNA und DNA erfolgt durch TLR3, TLR7/8 und TLR9 im Endosom.
TLR4 und TLR3 nutzen TRIF als Adapterprotein für die Signalübermittlung, TLR7/8 und TLR9 hin-
gegen MyD88. RIG-I und MDA5 erkennen RNA im Cytosol und nutzen IPS-1 als Adapterprotein.
STING ist der Rezeptor für cyklische Dinukleotide und dient gleichzeitig als Adapterprotein. Die Signal-
kaskaden münden in die Aktivierung der Transkriptionsfaktoren IRF3 und IRF7, die zur Produktion von
Typ-I-IFN führen. (Abbildung angepasst nach Kawai und Akira, 2008)
TLR4, der Lipopolysaccharide (LPS) der äußeren Membran von gram-negativen
Bakterien erkennt, ist in der Cytoplasmamembran lokalisiert. Die in der Membran von
Endosomen verankerten TLR3, TLR7/8 und TLR9 detektieren die von außen
aufgenommenen Nukleinsäuren. Während TLR3 beispielsweise vom Reovirus
abgeleitete dsRNA erkennt, erkennt TLR7/8 ssRNA des Human Immunodeficiency
Virus (HIV). TLR9 hingegen detektiert unmethylierte CpG-DNA-Motive von Bakterien

1. Einleitung
7
und Viren (Perry et al, 2005; Takeuchi, 2009). Für die Signalübermittlung nutzen TLR4
und TLR3 das Adapterprotein TRIF (TIR-domain-containing adaptor protein inducing
IFN-β). Die von plasmazytoiden DCs (pDCs) besonders stark exprimierten TLR7/8 und
TLR9 hingegen nutzen das Adapterprotein MyD88 (myeloid differentiation primary
response protein 88) (Kawai und Akira, 2010).
Zu den cytosolischen Immunrezeptoren, die nach Detektion viraler RNA die Produktion
von Typ-I-IFN auslösen, zählen die Rezeptoren der RLR-Familie (RIG-I-like recep-
tors), RIG-I (retinoic acid-inducible gene-I) und MDA5 (melanoma differentiation-
associated gene 5) (Kato et al, 2006; Kato et al, 2011). Im Gegensatz zu den TLRs, die
nur von bestimmten Zellen des Immunsystems, wie beispielsweise DCs und
Makrophagen, exprimiert werden, kommen diese Rezeptoren im Cytosol der meisten
kernhaltigen Zellen vor (Kato et al, 2006). Diese unterscheiden sich in ihrem RNA-
Erkennungsmuster und detektieren infolgedessen verschiedene RNA-Viren. RIG-I
erkennt 5`-Triphosphat-ssRNA mit einer sogenannten panhandle structure, wie sie
beispielsweise von dem Influenza A Virus (IAV), dem Newcastle Disease Virus (NDV)
und dem Sendaivirus (SeV) generiert wird. MDA5 hingegen erkennt lange dsRNA, die
von Picornaviren, wie beispielsweise dem Encephalomyocarditis Virus (EMCV),
erzeugt wird (Kato et al, 2011; Yoneyama et al, 2015).
RIG-I und MDA5 bestehen aus zwei N-terminalen CARD-Domänen (caspase
activation and recruitment domain), einer zentralen DExD/H box Helikase-Domäne
und einer C-terminalen Domäne (CTD) (Yoneyama et al, 2015). Zwei Studien an RIG-I
haben durch Kristallstrukturanalyse und NMR-Spektroskopie gezeigt, dass nach
Bindung des Liganden durch die CTD die ATPase-Aktivität der Helikase aktiviert wird.
Durch die daraufhin ausgelöste Konformationsänderung werden die ansonsten
„maskierten“ CARD-Domänen, die für die Dimerisierung und Signalübermittlung
verantwortlich sind, exponiert. Nach Dimerisierung von RIG-I Molekülen erfolgt die
Interaktion mit dem an der Mitochondrienmembran lokalisierten Adapterprotein IPS-1
(IFN-β-promoter stimulator 1) (Cui et al, 2008; Takahasi et al, 2008).
Sowohl der TRIF- als auch der IPS-1-abhängige Signalweg führt zur Aktivierung der
Serin/Threonin-Proteinkinase TBK1 (TANK-binding kinase 1). STING (stimulator of
inteferon genes), ein weiterer cytosolischer Immunrezeptor, der nach Bindung von
cyklischen Dinukleotiden die Produktion von Typ-I-IFN auslöst, nutzt ebenfalls TBK1
für die Signalweitergabe. Die Aktivierung von TBK1 führt zur Phosphorylierung und

1. Einleitung
8
Dimerisierung des Transkriptionsfaktors IRF3 (interferon regulatory factor 3), der nach
Translokation in den Zellkern die Initiation der Transkription von Typ-I-IFN induziert.
Der MyD88-abhängige Signalweg in pDCs führt über IRF7 zur Produktion von Typ-I-
IFN. Darüber hinaus erfolgt über Aktivierung des MAPK- (mitogen-activated protein
kinase) und NF-ҡB-Signalwegs (nuclear factor-kappa B) die Produktion von pro-
inflammatorischen Cytokinen (Honda und Taniguchi, 2006; Kawai und Akira, 2010;
Burdette et al, 2011).
1.3.1. Induktion von Typ-I-IFN während einer Infektion mit L. monocytogenes
L. monocytogenes befällt nach einer systemischen Infektion zunächst Milz und Leber
des Wirtes. Hier werden die Bakterien von gewebsständigen Phagocyten aufgenommen.
Es kommt zu einer effizienten Eliminierung des Pathogens durch IFN-γ-aktivierte
Makrophagen. Einige Bakterien sind dennoch in der Lage aus dem Phagosom zu
entkommen und sich im Cytosol der infizierten Zellen zu vermehren. Die Invasion von
L. monocytogenes ins Cytosol führt, neben der Produktion von pro-inflammatorischen
Cytokinen, zur Produktion von IFN-β (Abb. 1.3). Nach einer Infektion der Maus mit L.
monocytogenes sind Makrophagen die wichtigsten Produzenten von IFN-β (Stockinger
et al, 2009). Dabei ist die von L. monocytogenes ausgelöste IFN-β Antwort von
cytosolischen Immunrezeptoren abhängig. LLO-defiziente Listerien, die nicht ins
Cytosol der Wirtszellen eindringen können, führen nicht zur Produktion von IFN-β
(O`Riordan et al, 2002; McCaffrey et al, 2004; Leber et al, 2008). Infolgedessen spielen
membrangebundene TLRs in der IFN-β Induktion im Kontext einer Infektion mit L.
monocytogenes keine wesentliche Rolle. In peritoneale Makrophagen allerdings trägt
TLR2, der durch Bestandteile der Zellwand im Phagosom aktiviert wird, maßgeblich
zur Produktion von IFN-β bei (Aubry et al, 2012).
Die sogenannte späte Antwort (late response), die von cytosolischen Listerien ausgelöst
wird, wird hauptsächlich von IFN-β und die daraufhin aktivierten IRGs (IFN response
genes) dominiert (McCaffrey et al, 2004). Die cytosolische Immunüberwachung dient
der Detektion von virulenten Bakterien, die aus dem Phagosom ins Cytosol der Wirts-
zelle entkommen sind. Hierfür ist eine Reihe von verschiedenen Immunrezeptoren von
Bedeutung. Diese erkennen diverse bakterielle Produkte, die von L. monocytogenes
während der Vermehrung im Cytosol sekretiert werden.
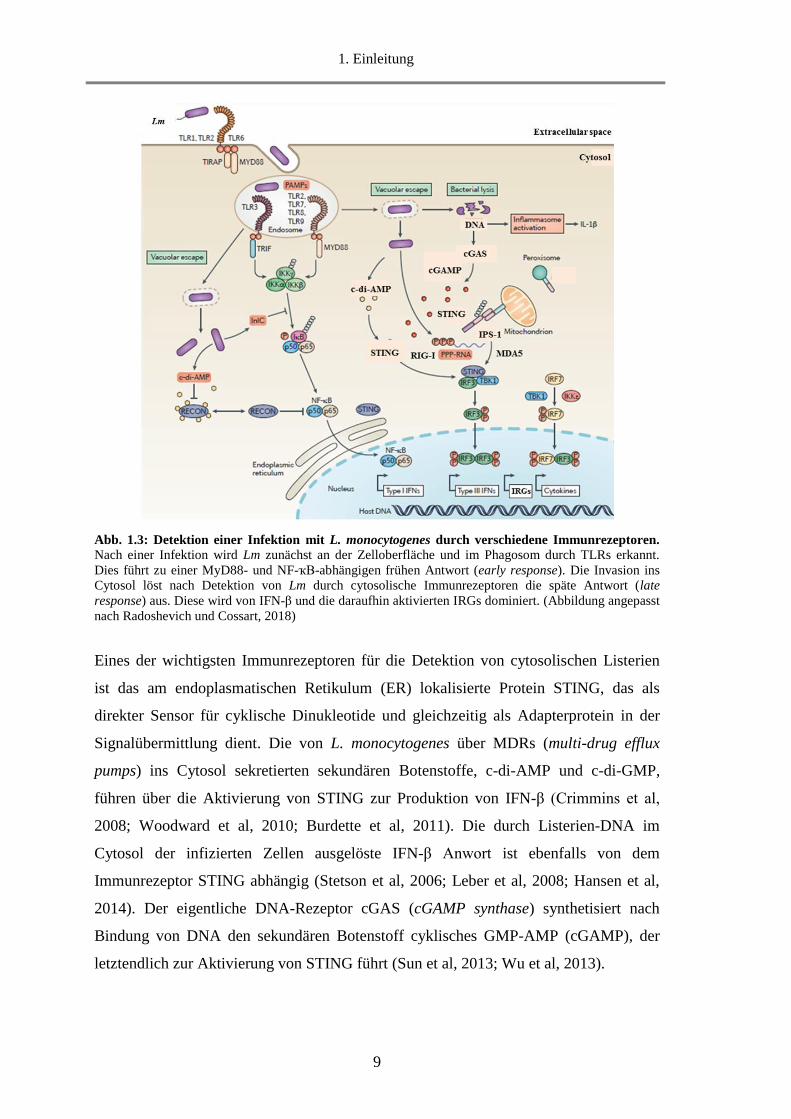
1. Einleitung
9
Abb. 1.3: Detektion einer Infektion mit L. monocytogenes durch verschiedene Immunrezeptoren.
Nach einer Infektion wird Lm zunächst an der Zelloberfläche und im Phagosom durch TLRs erkannt.
Dies führt zu einer MyD88- und NF-ҡB-abhängigen frühen Antwort (early response). Die Invasion ins
Cytosol löst nach Detektion von Lm durch cytosolische Immunrezeptoren die späte Antwort (late
response) aus. Diese wird von IFN-β und die daraufhin aktivierten IRGs dominiert. (Abbildung angepasst
nach Radoshevich und Cossart, 2018)
Eines der wichtigsten Immunrezeptoren für die Detektion von cytosolischen Listerien
ist das am endoplasmatischen Retikulum (ER) lokalisierte Protein STING, das als
direkter Sensor für cyklische Dinukleotide und gleichzeitig als Adapterprotein in der
Signalübermittlung dient. Die von L. monocytogenes über MDRs (multi-drug efflux
pumps) ins Cytosol sekretierten sekundären Botenstoffe, c-di-AMP und c-di-GMP,
führen über die Aktivierung von STING zur Produktion von IFN-β (Crimmins et al,
2008; Woodward et al, 2010; Burdette et al, 2011). Die durch Listerien-DNA im
Cytosol der infizierten Zellen ausgelöste IFN-β Anwort ist ebenfalls von dem
Immunrezeptor STING abhängig (Stetson et al, 2006; Leber et al, 2008; Hansen et al,
2014). Der eigentliche DNA-Rezeptor cGAS (cGAMP synthase) synthetisiert nach
Bindung von DNA den sekundären Botenstoff cyklisches GMP-AMP (cGAMP), der
letztendlich zur Aktivierung von STING führt (Sun et al, 2013; Wu et al, 2013).

1. Einleitung
10
Weitere Immunrezeptoren, die eine essentielle Rolle in der Detektion von virulenten
Listerien im Cytosol spielen, sind RIG-I und MDA5. Diese detektieren die von L.
monocytogenes sekretierte RNA und führen daraufhin zur Produktion von IFN-β
(Abdullah et al, 2012). Vor allem in Nicht-Immunzellen, wie beispielsweise Epithel-
zellen, die keine funktionsfähige STING-Signalübermittlung besitzen, spielt RIG-I eine
wesentliche Rolle in der IFN-β Produktion während einer Infektion mit L. monocyto-
genes (Hagmann et al, 2013). Die Einbeziehung verschiedener Liganden und deren
individuelle Erkennung durch die entsprechenden Immunrezeptoren stellt ein
Mechanismus dar, der die Identifizierung von virulenten Listerien im Cytosol
sicherstellt.
In der Signalübermittlung, die nach Detektion von cytosolischen Listerien zur
Produktion von IFN-β führt, sind die Serin/Threonin-Proteinkinase TBK1 und der
Transkriptionsfaktor IRF3 involviert. Die Aktivierung von TBK1 durch die Adapter-
proteine STING und IPS-1 führt über IRF3 zur Produktion von IFN-β (O`Connell et al,
2004; O`Connell et al, 2005; Leber et al, 2008; McWhirter et al, 2009).
Anders als bei viralen Infektionen hat die IFN-β Antwort, die durch bakterielle
Infektionen ausgelöst wird, unterschiedliche Auswirkungen auf den Wirt. Diese erhöht
die Anfälligkeit des Wirtes bei einer Infektion mit L. monocytogenes und fördert dessen
Vermehrung, wobei verschiedene Mechanismen eine Rolle spielen. IFN-β induziert
nach Bindung an den entsprechenden Rezeptor (IFNAR) die Expression einer Vielzahl
von Genen mit antiviraler und antiproliferativer Wirkung, darunter auch die
Proteinkinase R (PKR). Diese spielt bei der IFN-β-vermittelten Hemmung der
Proteinsynthese eine entscheidende Rolle (Williams, 2001). Im Zuge der Translations-
hemmung kommt es zu einer Cap-unabhängigen Translation des pro-apoptotischen
Transkriptionsfaktors ATF4 (activating transcription factor 4). Die daraufhin
ausgelöste integrierte Stressantwort, ISR (integrated stress resonse), führt zur Apoptose
der betroffenen Zellen (Valderrama et al, 2017). So haben beispielsweise ältere Studien
eine massive Apoptose von Makrophagen und Lymphozyten in den mit Listerien
infizierten Mäusen gezeigt (Carrero et al, 2004; Stockinger et al, 2002). Infolgedessen
sind IFNAR-defiziente Mäuse deutlich resistenter gegenüber einer Listerien-Infektion
als Wildtyptiere. Diese weisen in den primären Zielorganen, Milz und Leber, eine
deutlich reduzierte Bakterienlast auf (Carrero et al, 2004; O`Connell et al, 2004;
Auerbuch et al, 2004). Darüber hinaus wird die Immunantwort des Wirtes durch das

1. Einleitung
11
anti-inflammatorische Cytokin IL-10, das von Makrophagen nach Phagocytose der
apoptotischen Immunzellen produziert wird, gedrosselt (Carrero et al, 2006). Zudem
unterdrückt die von L. monocytogenes ausgelöste IFN-β Antwort die Aktivierung von
Phagocyten durch eine reduzierte Expression von IFN-γ, eines der wichtigsten Cytokine
in der frühen Phase einer Infektion mit diesem Pathogen (Rayamajhi et al, 2010).
1.4. Produktion von Vesikeln (OMVs bzw. MVs) durch Bakterien
Die Produktion von Vesikeln ist in allen bislang auf diesen Prozess hin untersuchten
Organismen nachgewiesen worden. Dies ist daher ein universeller Prozess, der in
Prokaryoten, Eukaryoten und Archaeen während des Wachstums stattfindet. Die
Produktion von Vesikeln durch Bakterien ist, bezüglich deren Anzahl und Inhalt, von
äußeren Faktoren, wie beispielsweise Temperatur, Antibiotikaeinwirkung und
Wachstumsmedium, abhängig (McBroom und Kuehn, 2007; Kadurugamuwa und
Beveridge, 1995; Sidhu et al, 2008).
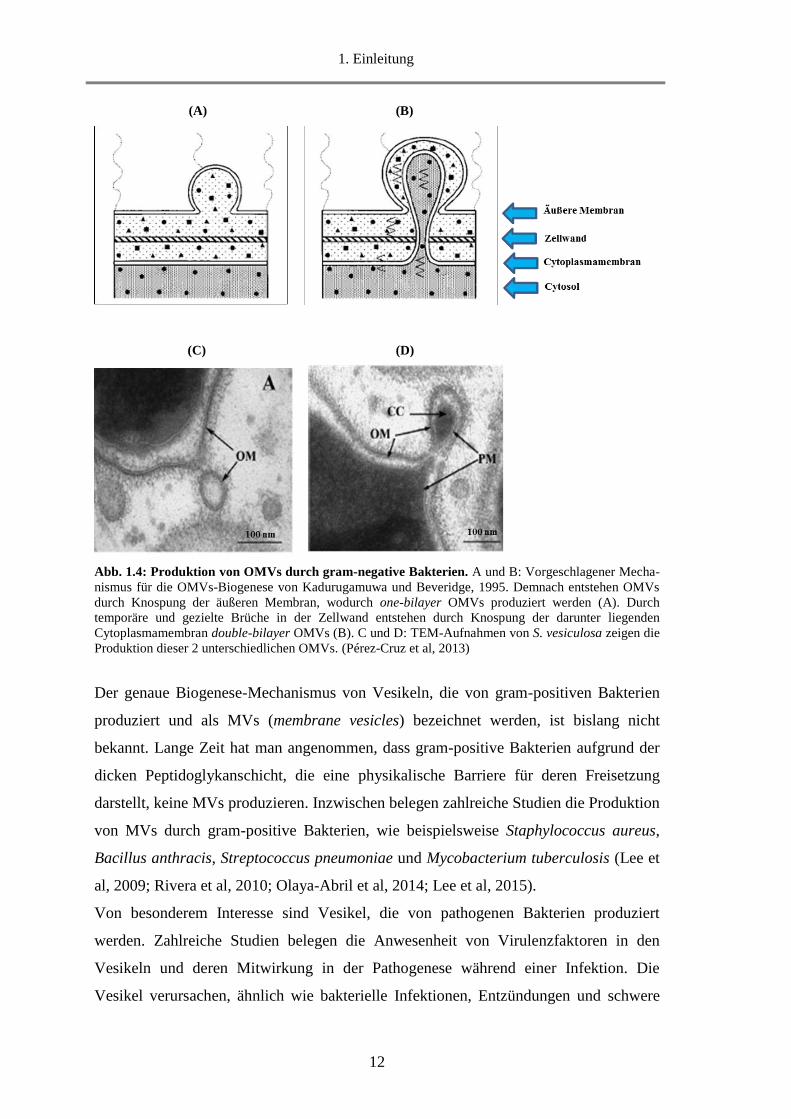
In gram-negative Bakterien entstehen Vesikel durch Knospung der äußeren Membran
und werden daher als OMVs (outer membrane vesicles) bezeichnet (Abb. 1.4A und
1.4C). Diese enthalten verschiedene Proteine und Lipide der äußeren Membran und des
Periplasmas (Kadurugamuwa und Beveridge, 1995; Kulp und Kuehn, 2010;
Schwechheimer und Kuehn, 2015; Lappann et al, 2013; Roier et al, 2016). Darüber
hinaus wird für eine OMVs-Subpopulation ein weiterer Biogenese-Mechanismus
vermutet, in dem auch die Cytoplasmamembran involviert ist (Abb. 1.4B).
Entscheidend für deren Freisetzung sind kontrollierte Brüche in der Peptido-
glykanschicht durch die gezielte Einwirkung von Autolysinen (Kadurugamuwa und
Beveridge, 1995). Dieses vorgeschlagene Modell wurde Jahre später durch
Transmissionselektronenmikroskopie (TEM) von Shewanella vesiculosa bestätigt (Abb.
1.4D). Infolgedessen sind diese OMVs von einer doppelten Membran umgeben und
enthalten darüber hinaus cytosolische Proteine, RNA und DNA (Pérez-Cruz et al,
2013).
1. Einleitung
12
(A) (B)
(C) (D)
Abb. 1.4: Produktion von OMVs durch gram-negative Bakterien. A und B: Vorgeschlagener Mecha-
nismus für die OMVs-Biogenese von Kadurugamuwa und Beveridge, 1995. Demnach entstehen OMVs
durch Knospung der äußeren Membran, wodurch one-bilayer OMVs produziert werden (A). Durch
temporäre und gezielte Brüche in der Zellwand entstehen durch Knospung der darunter liegenden
Cytoplasmamembran double-bilayer OMVs (B). C und D: TEM-Aufnahmen von S. vesiculosa zeigen die
Produktion dieser 2 unterschiedlichen OMVs. (Pérez-Cruz et al, 2013)
Der genaue Biogenese-Mechanismus von Vesikeln, die von gram-positiven Bakterien
produziert und als MVs (membrane vesicles) bezeichnet werden, ist bislang nicht
bekannt. Lange Zeit hat man angenommen, dass gram-positive Bakterien aufgrund der
dicken Peptidoglykanschicht, die eine physikalische Barriere für deren Freisetzung
darstellt, keine MVs produzieren. Inzwischen belegen zahlreiche Studien die Produktion
von MVs durch gram-positive Bakterien, wie beispielsweise Staphylococcus aureus,
Bacillus anthracis, Streptococcus pneumoniae und Mycobacterium tuberculosis (Lee et
al, 2009; Rivera et al, 2010; Olaya-Abril et al, 2014; Lee et al, 2015).
Von besonderem Interesse sind Vesikel, die von pathogenen Bakterien produziert
werden. Zahlreiche Studien belegen die Anwesenheit von Virulenzfaktoren in den
Vesikeln und deren Mitwirkung in der Pathogenese während einer Infektion. Die
Vesikel verursachen, ähnlich wie bakterielle Infektionen, Entzündungen und schwere

1. Einleitung
13
pathologische Veränderungen von Gewebe, wie beispielsweise Lungenschäden bis hin
zum Zelltod (Fiocca et al, 1999; Lee et al, 2012; Gurung et al, 2011; Thay et al, 2013).
Die Sekretion von Virulenzfaktoren über die Produktion von Vesikeln stellt einen
zusätzlichen Sekretionsmechanismus dar, der von pathogenen Bakterien genutzt wird.
Helicobacter pylori beispielsweise sekretiert ein Teil (25%) des Exotoxins VacA über
OMVs (Ricci et al, 2005). Die Aminopeptidase PaAP, eines der Hauptproteine in den
OMVs, die von Pseudomonas aeruginosa (cystische Fibrose-Isolate) produziert werden,
fördert zudem deren Assoziation und Aufnahme durch Lungenepithelzellen (Bauman
und Kuehn, 2006; Bauman und Kuehn, 2009). Ähnlich vermittelt das hitzelabile Entero-
toxin LT, das von ETEC (Enterotoxigenic Escherichia coli) hauptsächlich über OMVs
sekretiert wird (mehr als 95%), die Anheftung der OMVs an Wirtszellen (Kesty et al,
2004). Vesikel fungieren daher als Transportvehikel, die speziell gerichtet die
Aufnahme von Virulenzfaktoren durch Wirtszellen vermitteln. Darüber hinaus bietet die
Sekretion von Virulenzfaktoren über Vesikel weitere Vorteile. Hierdurch können
Virulenzfaktoren gleichzeitig und über lange Strecken hinweg transportiert werden. Des
Weiteren sind diese vor Abbau durch Proteasen im extrazellulären Milieu geschützt.
Vesikel nutzen verschiedene Mechanismen für deren Internalisierung in Wirtszellen.
Hierzu zählt die Fusion von OMVs, die von P. aeruginosa produziert werden, mit der
Plasmamembran von Wirtszellen (Bomberger et al, 2009). Für Shigella flexneri-OMVs
wurde gezeigt, dass diese durch Phagocytose in Wirtszellen gelangen (Kadurugamuwa
und Beveridge, 1998). OMVs, die von Porphyromonas gingivalis und H. pylori
produziert werden, gelangen über Endocytose in Wirtszellen (Furuta et al, 2009; Parker
et al, 2010).
Aufgrund der komplexen Zusammensetzung aus verschiedenen biologisch aktiven
Biomolekülen stellen Vesikel eine Quelle für Antigene dar. Eines der Hauptbestandteile
von OMVs, die von gram-negativen Bakterien produziert werden, ist LPS der äußeren
Membran. Durch die Internalisierung von OMVs gelangt LPS in die Wirtszellen und
löst die Produktion von IL-1β und IL-18 durch Aktivierung von Caspase-11 aus (Vanaja
et al, 2016). Durch die Produktion von Vesikeln können selbst nicht-intrazelluläre
Pathogene die Aktivierung des Inflammasoms und weiterer cytosolischer Immun-
rezeptoren auslösen. Peptidoglykan beispielsweise wird über Vesikel in Wirtszellen
transportiert und löst NOD-abhängig (nucleotide-binding and oligomerization domain)
die Produktion von IL-8 aus (Kaparakis et al, 2010). Weitere pro-inflammatorische

1. Einleitung
14
Cytokine, wie beispielsweise TNF-α and IL-12, werden von Makrophagen und DCs
nach deren Aktivierung durch OMVs, die von Salmonella typhimurium produziert
werden, sekretiert. Darüber hinaus werden diese Immunzellen zu einer gesteigerten
Expression von MHC-Klasse-II-Molekülen stimuliert (Alaniz et al, 2007). Als APCs
sind diese mit den adaptiven T- und B-Lymphozyten verlinkt, woraus protektive
Immunantworten resultieren. Zahlreiche Studien haben im Mausmodell gezeigt, dass
eine Immunisierung mit Vesikeln, die von verschiedenen pathogenen Bakterien
produziert werden, vor der anschließenden Infektion schützt (Schild et al, 2008; Avila-
Calderón et al, 2012; Olaya-Abril et al, 2014; Alaniz et al, 2007). Aufgrund der
immunogenen Eigenschaften werden Vesikel zur Formulierung von Vakzinen
eingesetzt. Ein auf OMVs-basierendes Vakzin gegen Neisseria meningitidis der Gruppe
B wird beispielsweise derzeit erfolgreich eingesetzt (Uli et al, 2006; Holst et al, 2009;
Masforrol et al, 2017). Die Nutzung von Vesikeln für die Antigen-Präsentation bietet
den Vorteil, dass oberflächenassoziierte Antigene, wie beispielsweise das immun-
dominante Antigen Porin PorA in den von N. meningitidis produzierten OMVs, deren
native Struktur behalten. Daraufhin wird eine robuste Antikörperproduktion ausgelöst,
die vor invasiven Meningokokken-Erkrankungen schützt.
Ein weiterer bedeutsamer Aspekt der Produktion von Vesikeln durch Bakterien ist der
Transfer von Antibiotikaresistenzen. Beta-Laktamase positive Moraxella catarrhalis-
Stämme beispielsweise vermitteln Amoxicillin-Resistenz durch den Transfer von
OMVs auf andere Bakterienspezies mit denen diese häufig in Mischinfektionen
auftreten (Schaar et al, 2011). Der Transfer von Resistenzgenen über OMVs wurde
anhand eines Carbapenemase-Resistenzgens innerhalb verschiedener Acinetobacter
baumannii-Stämmen gezeigt (Rumbo et al, 2011). Weitere Studien belegen den
horizontalen Transfer von DNA über Vesikel zwischen Bakterien (Yaron et al, 2000;
Renelli et al, 2004; Klieve et al, 2005; Fulsundar et al, 2014).
1.5. Sekretion von RNA durch Bakterien
Die Sekretion von RNA durch Bakterien wurde erstmal 1989 im Zusammenhang mit
OMVs, die von Neisseria gonorrhoeae produziert werden, gezeigt (Dorward et al,
1989). Erst Jahre nach dieser Entdeckung rückte die von Bakterien sekretierte RNA in
den Fokus wissenschaftlichen Interesses. In jüngster Zeit beschreiben zahlreiche
Studien die Anwesenheit von RNA in den Vesikeln, die von verschiedenen Bakterien-

1. Einleitung
15
spezies produziert werden (Biller et al, 2014; Sjöström et al, 2015; Ghosal et al, 2015;
Resch et al, 2016; Koeppen et al, 2016; Blenkiron et al, 2016). Deren umfassende
Untersuchung über RNA-Sequenzierung hat gezeigt, dass es sich hierbei um eine
komplexe Zusammensetzung aus RNAs verschiedener Größe und zellulärer Funktion
handelt. Die von UPEC (UroPathogenic E. coli) über OMVs sekretierte RNA beispiels-
weise beinhaltet sämtliche zelluläre RNA-Spezies, rRNA, tRNA, mRNA und sRNA.
Diese Reihenfolge entspricht der Häufigkeit der identifizierten RNA-Spezies in den
OMVs (Blenkiron et al, 2016). Eine ähnliche Zusammensetzung ergab die Unter-
suchung der RNA in den MVs, die von Streptococcus pyogenes produziert werden
(Resch et al, 2016).
Von entscheidender Bedeutung ist die Feststellung, dass sich die über Vesikel
sekretierte RNA im Auftreten und in der Häufigkeit bestimmter RNA-Spezies von dem
intrazellulären RNA-Profil unterscheidet. Daher wird ein selektiver Export von RNA
durch Bakterien vermutet. Ähnliche Beobachtungen wurden über die extrazelluläre
RNA, die von eukaryotischen Zellen zum Teil über Exosomen sekretiert wird, berichtet
(Valadi et al, 2007; Nolte-'t Hoen et al, 2012; Li et al, 2013). Der Mechanismus hinter
der Verpackung von RNA in den Vesikeln ist bislang nicht bekannt. Deren Funktion
wird in der interzellulären Kommunikation vermutet, in der die RNA Spezies- und
kingdom-übergreifend eine regulatorische Rolle einnimmt. Dies wird durch Studien
begründet, die den Transport und die Lieferung von RNA über Vesikel in Wirtszellen
gezeigt haben (Blenkiron et al, 2016). Darüber hinaus hat eine weitere Studie gezeigt,
dass P. aeruginosa die Immunantwort des Wirtes herunterreguliert, indem es kleine
nicht-kodierende RNAs über OMVs in Wirtszellen transportiert (Koeppen et al, 2016).
Hierfür verantwortlich wurde ein tRNA-Fragment identifiziert, das durch Inhibition der
Expression von MAPK-Kinasen zu einer Reduktion der IL-8 Produktion führt.
Zahlreiche Hinweise deuten auf eine Einflussnahme pathogener Bakterien auf ihre
Wirtszellen über sekretierte RNA hin. So wurden beispielsweise zahlreiche RNA-
Moleküle mit Charakteristika von miRNA-Vorläufermolekülen in den Genomen von
Krankheitserregern durch computerbasierende Vorhersagen identifiziert. Als mögliche
targets der daraus resultierenden miRNAs wurden Genprodukte, die in verschiedenen
Erkrankungen involviert sind, ausgemacht (Shmaryahu et al, 2014). Ein weiterer
Hinweis auf einen wahrscheinlichen RNAi-Mechanismus (RNA interference) durch
bakterielle RNAs kam von einer Studie aus dem gleichen Jahr. Nach Infektion von

1. Einleitung
16
humanen Zelllinien mit verschiedenen pathogenen Bakterien wurden zahlreiche
bakterielle RNAs, die mit dem RISC-Komplex (RNA-induced silencing complex)
assoziiert waren, identifiziert (Furuse et al, 2014). Eine Manipulation der Genexpression
durch bakterielle RNA während einer Infektion setzt voraus, dass diese unbeschadet in
die Wirtszellen gelangt. Vesikel als Transportvehikel für RNA zu nutzen, bietet Schutz
vor Abbau durch allgegenwärtige RNasen und ermöglicht zudem deren Transport über
lange Distanzen (Bomberger et al, 2009).
Zahlreiche Studien aus der Eukaryoten-Forschung haben bereits Jahre zuvor gezeigt,
dass der Großteil der extrazellulären RNA (bis zu 90%) in einer freien, nicht
Exosomen-assoziierten Form sekretiert wird (Wang et al, 2010; Turchinovich et al,
2011; Arroyo et al, 2011). Analog zu den Eukaryoten wird RNA auch von den
Bakterien in einer freien, nicht Vesikel-assoziierten Form sekretiert. Im Jahr 2015
wurde diese RNA-Fraktion, die von E. coli sekretiert wird, erstmals umfassend
untersucht (Ghosal et al, 2015). Diese Untersuchung hat gezeigt, dass die freie, nicht
Vesikel-assoziierte RNA eine vergleichbare, komplexe Mischung verschiedener RNA-
Spezies besitzt wie die Vesikel-assoziierte RNA. Der funktionelle Unterschied
zwischen den beiden sekretierten RNA-Fraktionen ist nicht bekannt. Dies gilt auch für
den Sekretionsmechanismus der freien, nicht Vesikel-assoziierten RNA. Für L. mono-
cytogenes wurde gezeigt, dass die SecA2-ATPase der Sec-Translokase, die einen
generellen Sekretionsweg für Proteine darstellt, teilweise an der RNA-Sekretion
beteiligt ist. Die Deletion dieses Proteins resultiert in einer reduzierten Sekretion von
RNA ins extrazelluläre Milieu (Desvaux und Hébraud, 2006; Abdullah et al, 2012).
Die Sekretion von RNA durch Bakterien hat einen essentiellen immunologischen
Aspekt während einer Infektion. Die Detektion von bakterieller RNA signalisiert dem
Immunsystem die Lebendigkeit und damit das Ausmaß der infektiösen Gefahr. Die
RNA gehört zur Klasse der vita-PAMPs (viability-associated PAMPs) und dient als
Kriterium für das Immunsystem, um lebende von toten Bakterien unterscheiden zu
können (Sander et al, 2011). Die sekretierte RNA spielt beispielsweise eine essentielle
Rolle in der Detektion virulenter Listerien, die ins Cytosol der infizierten Zellen
gelangen (Abdullah et al, 2012). Die Translokation von RNA ins Cytosol der
Wirtszellen während einer Infektion mit L. monocytogenes wurde anhand einer
Fluoreszenzmarkierung gezeigt. Daraufhin führt die Detektion der RNA durch den
Immunrezeptor RIG-I zur Produktion von Typ-I-IFN (Hagmann et al, 2013). Die

1. Einleitung
17
während einer Infektionen mit S. pyogenes durch RNA ausgelöste Typ-I-IFN Antwort
trägt maßgeblich zur Ausbildung einer protektiven Immunantwort gegen dieses
Pathogen bei (Gratz et al, 2011). Darüber hinaus löst die bakterielle RNA die
Produktion von pro-inflammatorischen Cytokinen, wie beispielsweise IL-12, aus (Koski
et al, 2004). Hierzu zählen des Weiteren IL-1β und IL-18, die nach Aktivierung des
Inflammasoms durch RNA produziert werden (Kanneganti et al, 2006; Sha et al, 2014).
Die bakterielle RNA spielt eine essentielle Rolle in der Bekämpfung von Pathogenen
durch Aktivierung des Immunsystems über Typ-I-IFN und pro-inflammatorische
Cytokine.
1.6. Zielsetzung dieser Arbeit
Ziel dieser Arbeit war es die von L. monocytogenes sekretierte RNA aus den Über-
ständen von Bakterienkulturen zu isolieren und deren Zusammensetzung über
Sequenzierung zu identifizieren. Anschließend sollte das Potential einzelner sekretierter
RNAs eine IFN-β Antwort in Wirtszellen auszulösen, untersucht werden. Dies sollte
durch Transfektion der hergestellten RNAs in Epithelzellen und BMDMs (bone
marrow-derived macrophages) durchgeführt werden. Hierbei war es von besonderem
Interesse herauszufinden, ob Unterschiede in der IFN-β Induktion durch die einzelnen
RNAs festzustellen sind. Darüber hinaus sollte untersucht werden welcher cytosolischer
Rezeptor der RLR-Familie für die Detektion der einzelnen RNAs und der daraufhin
ausgelösten IFN-β Antwort verantwortlich ist. Dies sollte in Zelllinien, die eine
Defizienz des jeweiligen Rezeptors aufweisen, untersucht werden.
Ein weiteres Ziel dieser Arbeit war es die von L. monocytogenes produzierten MVs aus
den Überständen von Bakterienkulturen zu isolieren und zu charakterisieren, bezüglich
Größe, Proteinkonzentration und Anzahl. Darüber hinaus sollte über Massen-
spektrometrie der Proteininhalt der MVs identifiziert werden. Es galt herauszufinden
welche zelluläre Funktion und Lokalisation die Proteine in den MVs, die von L.
monocytogenes produziert werden, haben. Des Weiteren sollten die MVs auf die Anwe-
senheit von RNA untersucht werden. Es galt herauszufinden welche RNA-Spezies über
die Produktion von MVs exportiert werden. Anschließend sollte deren IFN-β Induk-
tionspotential durch Transfektion in verschiedenen Zelllinien untersucht werden.
2. Material und Methoden
18
2. Material und Methoden
2.1. Bakterienstämme und Virusstamm
Bakterienstämme Herkunft
Escherichia coli DH10β Invitrogen
Escherichia coli TOP10 Invitrogen
Listeria monocytogens EGD-e (Lm) Glaser et al, 2001
Lm-pERL3-rli32 Diese Arbeit
Lm-pERL3-ssrS Diese Arbeit
2.2. Zelllinien
2.3. Antibiotika-Stammlösungen
Ampicillin: Stock-Konzentration: 100 mg/ml. Gelöst in 70% EtOH.
Erythromycin: Stock-Konzentration: 30 mg/ml. Gelöst in 50% EtOH.
Penicillin: Stock-Konzentration: 10 mg/ml. Gelöst in H2O. Anschließend steril-filtriert.
Die Stocklösungen wurden bei -20°C gelagert.
Virusstamm Ursprung Wirt Herkunft
Influenza A Virus
(H1N1)
Puerto Rico
(1934)
Mensch Medizinische Virologie
(JLU Gießen)
Zelllinie Name Spezies Herkunft
HEK293 human embryonic kidney cell Mensch Niere
MDCK-II Madin Darby canine kidney cells Hund Niere
PtK rat-kangaroo epithelial kidney cells Rattenkänguru Niere
BMDMs bone marrow-derived macrophages Maus Knochenmark
L929 mouse fibroblast L929 cells Maus Fibroblasten

2. Material und Methoden
19
2.4. Antikörper
Primäre Antikörper
Sekundäre Antikörper
2.5. Molekulargewichtsmarker
A B C D
Abb. 2.1: DNA-, RNA- und Protein-Größenstandards. A: GeneRuler Ultra Low Range DNA Ladder
(Thermo Scientific). B: 1 Kb Plus DNA Ladder (Invitrogen). C: RNA Century Marker-Plus
(ThermoFisher Scientific). D: Precision Plus Protein Standard (Bio-Rad).
Name Spezies Hersteller
anti-LLO Maus Eigene Herstellung
anti-InlB Maus Eigene Herstellung
anti-P60 Maus Eigene Herstellung
anti-β-Aktin Hase Cell signaling
anti-IAV-Nukleoprotein Maus S. Ludwig, Münster
anti-mouse IgG-HRP Ziege Santa Cruz B
anti-rabbit IgG-HRP Ziege Santa Cruz B
2. Material und Methoden
20
2.6. Vektoren
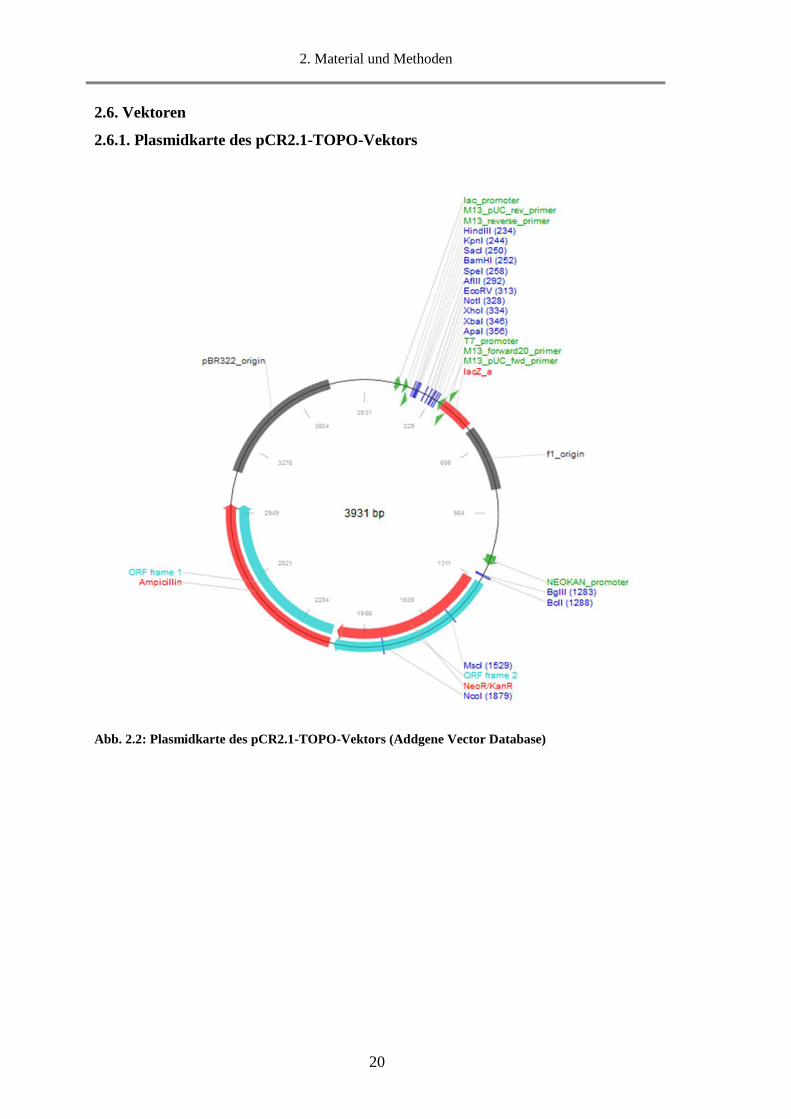
2.6.1. Plasmidkarte des pCR2.1-TOPO-Vektors
Abb. 2.2: Plasmidkarte des pCR2.1-TOPO-Vektors (Addgene Vector Database)
2. Material und Methoden
21
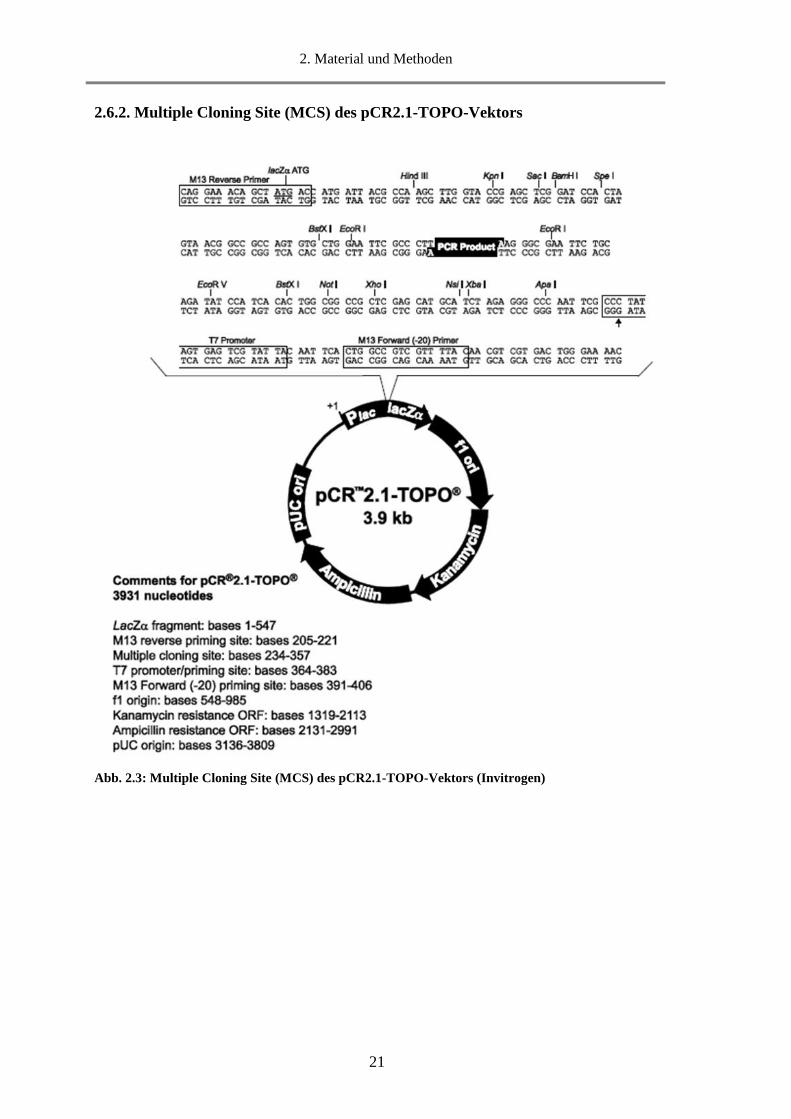
2.6.2. Multiple Cloning Site (MCS) des pCR2.1-TOPO-Vektors
Abb. 2.3: Multiple Cloning Site (MCS) des pCR2.1-TOPO-Vektors (Invitrogen)

2. Material und Methoden
22
2.6.3. Plasmidkarte des pERL3-Vektors
Abb. 2.4: Plasmidkarte des pERL3-Vektors
2.7. Oligonukleotide (Primer)
Die Primer für die RT-PCR wurden bei Qiagen bestellt. Die Primer für die PCR wurden
selbst entworfen und bei Eurofins Genomics bestellt.
2.7.1. Primer für die Real-Time PCR (RT-PCR)
Gen Name Spezies
IFN-β Ifnb1_1_SG QuantiTect Primer Assay (QT00249662) Maus
IFN-α4 Ifna4_2_SG QuantiTect Primer Assay (QT01774353) Maus
Rplp Rplp0_1_SG QuantiTect Primer Assay (QT00249375) Maus
IFN-β IFNB1_1_SG QuantiTect Primer Assay (QT00203763) Mensch
PPIA PPIA_1_SG QuantiTect Primer Assay (QT00052311) Mensch
2. Material und Methoden
23
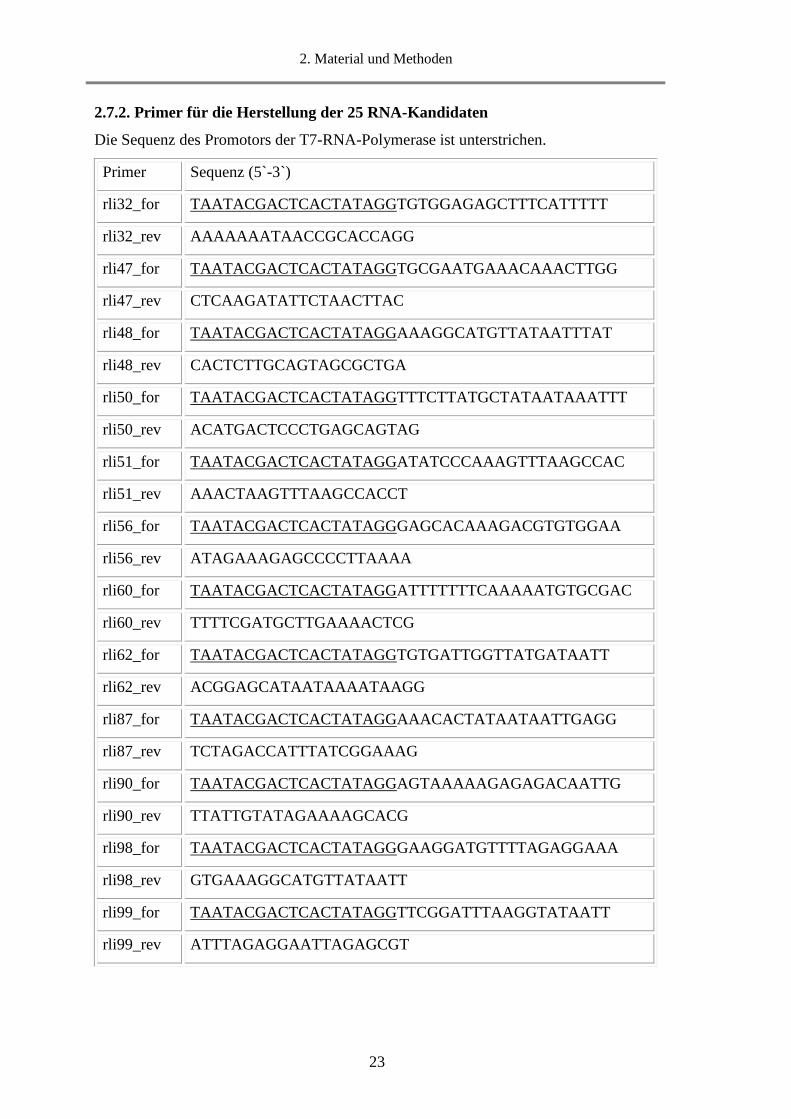
2.7.2. Primer für die Herstellung der 25 RNA-Kandidaten
Die Sequenz des Promotors der T7-RNA-Polymerase ist unterstrichen.
Primer Sequenz (5`-3`)
rli32_for TAATACGACTCACTATAGGTGTGGAGAGCTTTCATTTTT
rli32_rev AAAAAAATAACCGCACCAGG
rli47_for TAATACGACTCACTATAGGTGCGAATGAAACAAACTTGG
rli47_rev CTCAAGATATTCTAACTTAC
rli48_for TAATACGACTCACTATAGGAAAGGCATGTTATAATTTAT
rli48_rev CACTCTTGCAGTAGCGCTGA
rli50_for TAATACGACTCACTATAGGTTTCTTATGCTATAATAAATTT
rli50_rev ACATGACTCCCTGAGCAGTAG
rli51_for TAATACGACTCACTATAGGATATCCCAAAGTTTAAGCCAC
rli51_rev AAACTAAGTTTAAGCCACCT
rli56_for TAATACGACTCACTATAGGGAGCACAAAGACGTGTGGAA
rli56_rev ATAGAAAGAGCCCCTTAAAA
rli60_for TAATACGACTCACTATAGGATTTTTTTCAAAAATGTGCGAC
rli60_rev TTTTCGATGCTTGAAAACTCG
rli62_for TAATACGACTCACTATAGGTGTGATTGGTTATGATAATT
rli62_rev ACGGAGCATAATAAAATAAGG
rli87_for TAATACGACTCACTATAGGAAACACTATAATAATTGAGG
rli87_rev TCTAGACCATTTATCGGAAAG
rli90_for TAATACGACTCACTATAGGAGTAAAAAGAGAGACAATTG
rli90_rev TTATTGTATAGAAAAGCACG
rli98_for TAATACGACTCACTATAGGGAAGGATGTTTTAGAGGAAA
rli98_rev GTGAAAGGCATGTTATAATT
rli99_for TAATACGACTCACTATAGGTTCGGATTTAAGGTATAATT
rli99_rev ATTTAGAGGAATTAGAGCGT
2. Material und Methoden
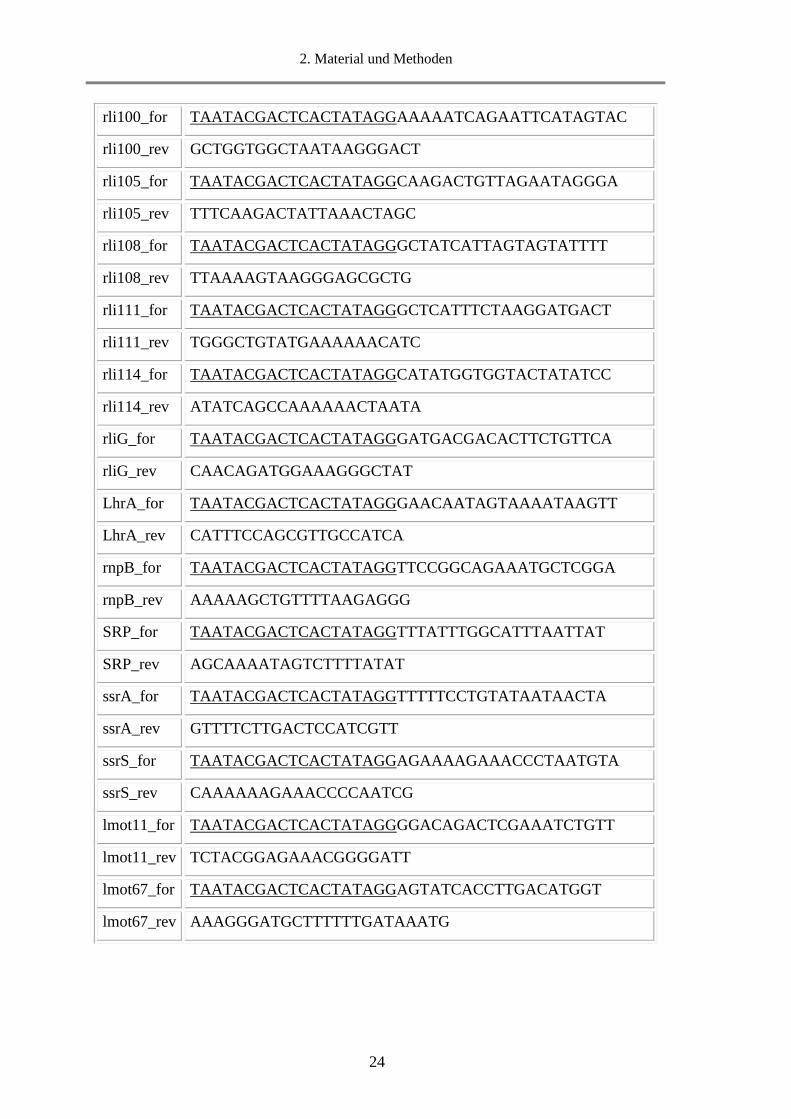
24
rli100_for TAATACGACTCACTATAGGAAAAATCAGAATTCATAGTAC
rli100_rev GCTGGTGGCTAATAAGGGACT
rli105_for TAATACGACTCACTATAGGCAAGACTGTTAGAATAGGGA
rli105_rev TTTCAAGACTATTAAACTAGC
rli108_for TAATACGACTCACTATAGGGCTATCATTAGTAGTATTTT
rli108_rev TTAAAAGTAAGGGAGCGCTG
rli111_for TAATACGACTCACTATAGGGCTCATTTCTAAGGATGACT
rli111_rev TGGGCTGTATGAAAAAACATC
rli114_for TAATACGACTCACTATAGGCATATGGTGGTACTATATCC
rli114_rev ATATCAGCCAAAAAACTAATA
rliG_for TAATACGACTCACTATAGGGATGACGACACTTCTGTTCA
rliG_rev CAACAGATGGAAAGGGCTAT
LhrA_for TAATACGACTCACTATAGGGAACAATAGTAAAATAAGTT
LhrA_rev CATTTCCAGCGTTGCCATCA
rnpB_for TAATACGACTCACTATAGGTTCCGGCAGAAATGCTCGGA
rnpB_rev AAAAAGCTGTTTTAAGAGGG
SRP_for TAATACGACTCACTATAGGTTTATTTGGCATTTAATTAT
SRP_rev AGCAAAATAGTCTTTTATAT
ssrA_for TAATACGACTCACTATAGGTTTTTCCTGTATAATAACTA
ssrA_rev GTTTTCTTGACTCCATCGTT
ssrS_for TAATACGACTCACTATAGGAGAAAAGAAACCCTAATGTA
ssrS_rev CAAAAAAGAAACCCCAATCG
lmot11_for TAATACGACTCACTATAGGGGACAGACTCGAAATCTGTT
lmot11_rev TCTACGGAGAAACGGGGATT
lmot67_for TAATACGACTCACTATAGGAGTATCACCTTGACATGGT
lmot67_rev AAAGGGATGCTTTTTTGATAAATG

2. Material und Methoden
25
2.7.3. Primer für die Herstellung der rli32-Motive
Die Sequenz des Promotors der T7-RNA-Polymerase ist unterstrichen.
Die Einzelstränge der DNA-templates für die Motive 1 und 3 wurden bei Eurofins
Genomics bestellt. In einem weiteren Schritt erfolgte das annealing der ssDNA zur
dsDNA. Die Sequenz des Promotors der T7-RNA-Polymerase ist unterstrichen.
2.7.4. Primer für die Herstellung von Lm-pERL3-rli32
Die Schnittstellen für die Restriktionsenzyme und die überstehenden Sequenzen für die
Fusion von hly-Promotor und hly-Terminator an rli32 sind unterstrichen.
Primer Sequenz (5`-3`)
Motiv 2_for TAATACGACTCACTATAGGAATAATGTTAGAGCAATAA
Motiv 2_rev AATAATATTAAAGCGAGCCA
ssDNA Sequenz (5`-3`)
Motiv 1_for TAATACGACTCACTATAGGGCTTTCATTTTTCCCAAGAGAA
AGC
Motiv 1_rev GCTTTCTCTTGGGAAAAATGAAAGCCCTATAGTGAGTCGT
ATTA
Motiv 3_for TAATACGACTCACTATAGGCCTAATACTCCCTTAACCGCAT
CCCCCTGGTGCGGTTATTTTTTT
Motiv 3_rev AAAAAAATAACCGCACCAGGGGGATGCGGTTAAGGGAGT
ATTAGGCCTATAGTGAGTCGTATTA
Primer Sequenz (5`-3`) Restriktionsenzym
hly-p_for GCGCGTCGACGTGACTTTTATGTTGAGGCA Sal I
hly-p_rev GCTCTCCACAGCTTGCTTTATAGCTTTAT
rli32_for TAAAGCAAGCTGTGGAGAGCTTTCATTTTT
rli32_rev ACTTTTACAAAAAAAAATAACCGCACCAGG
hly-t_for TTATTTTTTTTTGTAAAAGTAATAAAAAATT
hly-t_rev GCGCCCCGGGGCTTATATTATATGGATAAA Xma I

2. Material und Methoden
26
2.7.5. Primer für die Herstellung von Lm-pERL3-ssrS
Die Schnittstellen für die Restriktionsenzyme und die überstehenden Sequenzen für die
Fusion von hly-Promotor und hly-Terminator an ssrS sind unterstrichen.
2.8. Chemikalien
Roth: NaCl, Agarose, Borsäure, Harnstoff, Tris, Glycin, SDS (Sodium Dodecyl Sulfate),
Roti-Phenol/Chloroform-Isoamylalkohol (25:24:1, pH = 4,5-5,0), Roti-Aqua-Phenol,
Roti-Chloroform, Ficoll 400, Ampicillin, KH2PO4, CaCl2 × 2 H2O, Xylencyanol,
Deionisiertes Formamid, Saccharose, Ethidiumbromid, DTT (Dithiothreitol)
SIGMA ALDRICH: Ethanol, 2-Propanol, β-Mercaptoethanol, Penicillin, Erythromycin,
MgSO4 × 7 H2O, Riboflavin, Thiamin, L-Arginin, L-Leucin, L-Isoleucin, L-Methionin,
L-Valin, L-Cystein, L-Glutamin, BSA (Bovine Serum Albumin), PMSF (Phenylmethyl-
sulfonyl Fluoride), Mutanolysin, Na-Deoxycholat
Merck: DMSO (Dimethylsulfoxid), Na2HPO4, NaH2PO4 × H2O, NaOAc, Glukose,
Essigsäure, MgCl2 × 6 H2O, MnCl2 × 4 H2O, KAc, Glycerin, APS (Ammonium-
persulfat), Eisencitrat
SERVA: Tween 20, Triton X-100, HEPES (Hydroxyethylpiperazin-Ethansulfonsäure),
EDTA (Ethylendiamintetraacetat), Bromphenolblau
GERBU: Coomassie Brilliant Blue R
VWR: Lysozym, Methanol
BD: BHI (Brain Heart Infusion), Trypton, Hefeextrakt, Agar
Primer Sequenz (5`-3`) Restriktionsenzym
hly-p_for GCGCGTCGACGTGACTTTTATGTTGAGGCA Sal I
hly-p_rev TTTCTTTTCTGCTTGCTTTATAGCTTTAT
ssrS_for TAAAGCAAGCAGAAAAGAAACCCTAATGTA
ssrS_rev ACTTTTACAACAAAAAAGAAACCCCAATCG
hly-t_for TTCTTTTTTGTTGTAAAAGTAATAAAAAATT
hly-t_rev GCGCGGATCCGCTTATATTATATGGATAAA BamH I
2. Material und Methoden
27
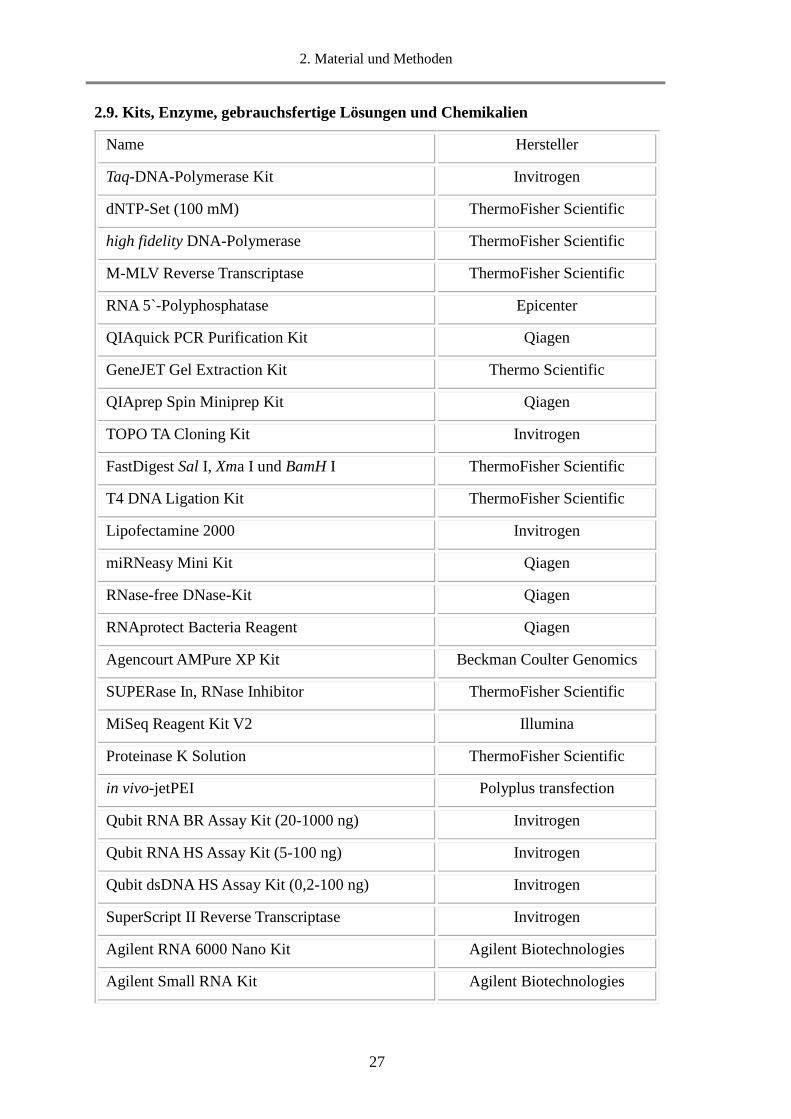
2.9. Kits, Enzyme, gebrauchsfertige Lösungen und Chemikalien
Name Hersteller
Taq-DNA-Polymerase Kit Invitrogen
dNTP-Set (100 mM) ThermoFisher Scientific
high fidelity DNA-Polymerase ThermoFisher Scientific
M-MLV Reverse Transcriptase ThermoFisher Scientific
RNA 5`-Polyphosphatase Epicenter
QIAquick PCR Purification Kit Qiagen
GeneJET Gel Extraction Kit Thermo Scientific
QIAprep Spin Miniprep Kit Qiagen
TOPO TA Cloning Kit Invitrogen
FastDigest Sal I, Xma I und BamH I ThermoFisher Scientific
T4 DNA Ligation Kit ThermoFisher Scientific
Lipofectamine 2000 Invitrogen
miRNeasy Mini Kit Qiagen
RNase-free DNase-Kit Qiagen
RNAprotect Bacteria Reagent Qiagen
Agencourt AMPure XP Kit Beckman Coulter Genomics
SUPERase In, RNase Inhibitor ThermoFisher Scientific
MiSeq Reagent Kit V2 Illumina
Proteinase K Solution ThermoFisher Scientific
in vivo-jetPEI Polyplus transfection
Qubit RNA BR Assay Kit (20-1000 ng) Invitrogen
Qubit RNA HS Assay Kit (5-100 ng) Invitrogen
Qubit dsDNA HS Assay Kit (0,2-100 ng) Invitrogen
SuperScript II Reverse Transcriptase Invitrogen
Agilent RNA 6000 Nano Kit Agilent Biotechnologies
Agilent Small RNA Kit Agilent Biotechnologies

2. Material und Methoden
28
2.10. Medien, Puffer und Enzyme für die Zellkultur
ThermoFisher Scientific: Opti-MEM Reduced Serum Medium, RPMI 1640 Medium
(ATCC modification), DMEM (Dulbecco's Modified Eagle Medium), MEM (Minimum
Essential Medium)
Merck: FKS (Fötales Kälberserum), PBS (Dulbecco’s Phosphate Buffered Saline), NEA
(Nicht-essenzielle Aminosäuren), Trypsin
2.11. Medien für die Anzucht von Bakterien
Herstellung von Brain Heart Infusion-Medium (BHI-Medium)
Für die Herstellung von BHI-Medium wurden 37 g BHI in 1 L H2O gelöst. Anschlie-
ßend wurde das Medium autoklaviert. Für die Herstellung von BHI-Agarplatten wurden
dem Medium 15 g Agar/L zugesetzt.
High sensitivity DNA Kit Agilent Biotechnologies
RNaseOut Invitrogen
QuantiTect SYBR Green PCR Kit Qiagen
RNase, DNase-free Roche
T7-RNA-Polymerase NEB
rNTP-Set (100 mM) GE Healthcare
DNase I, RNase-free ThermoFisher Scientific
Alkaline Phosphatase, Calf intestinal (CIAP) NEB
ECL Plus Western Blotting Detection Reagent GE Healthcare
AEC Staining Kit SIGMA ALDRICH
Protein Assay Dye Reagent Concentrate Bio-Rad
Bicinchoninic Acid Protein Assay Kit SIGMA ALDRICH
Rotiphorese Gel 30 Roth
Tetramethylethylendiamin (TEMED) SERVA
RNase-Away Reagent ThermoFisher Scientific
0,5 M EDTA, pH = 8,0 ThermoFisher Scientific
Penicillin-Streptomycin (10,000 U/mL) ThermoFisher Scientific

2. Material und Methoden
29
Herstellung von Luria Bertani-Medium (LB-Medium)
Zusammensetzung LB-Medium:
Die Reagenzien wurden zunächst in H2O gelöst und anschließend autoklaviert. Für die
Herstellung von LB-Agarplatten wurden dem Medium 15 g Agar/L zugesetzt.
Herstellung von SOC-Medium
Das SOC-Medium in dem die E. coli-Zellen nach der Transformation zur Expression
der Antibiotika-Resistenzgene aufgenommen wurden, bestand aus SOB-Medium, das
20 mM Glukose enthielt.
Zusammensetzung SOB-Medium (Super Optimal Broth):
Die Reagenzien wurden zunächst in H2O gelöst und anschließend autoklaviert.
Reagenz Konzentration
Trypton 1% (w/v)
Hefeextrakt 0,5% (w/v)
NaCl 0,5% (w/v)
Reagenz Konzentration
Trypton 2% (w/v)
Hefeextrakt 0,5% (w/v)
NaCl 10 mM
KCl 2,5 mM
MgCl2 × 6 H2O 10 mM
MgSO4 × 7 H2O 10 mM
2. Material und Methoden
30
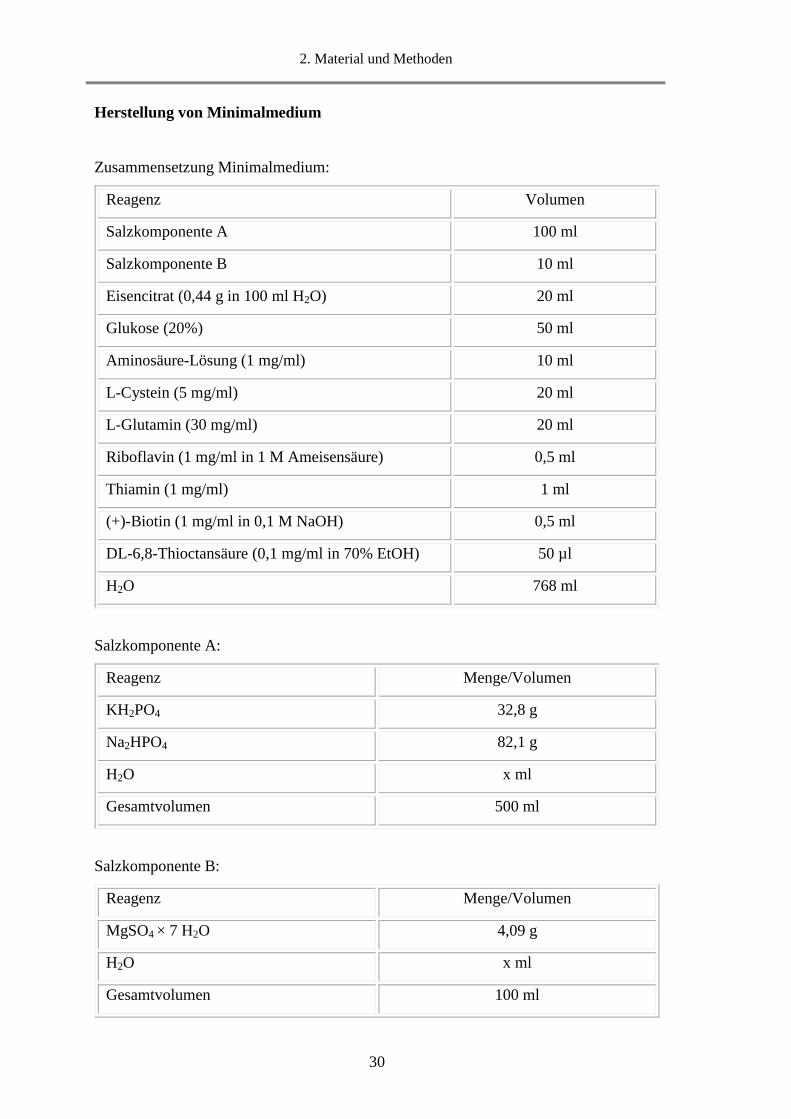
Herstellung von Minimalmedium
Zusammensetzung Minimalmedium:
Reagenz Volumen
Salzkomponente A 100 ml
Salzkomponente B 10 ml
Eisencitrat (0,44 g in 100 ml H2O) 20 ml
Glukose (20%) 50 ml
Aminosäure-Lösung (1 mg/ml) 10 ml
L-Cystein (5 mg/ml) 20 ml
L-Glutamin (30 mg/ml) 20 ml
Riboflavin (1 mg/ml in 1 M Ameisensäure) 0,5 ml
Thiamin (1 mg/ml) 1 ml
(+)-Biotin (1 mg/ml in 0,1 M NaOH) 0,5 ml
DL-6,8-Thioctansäure (0,1 mg/ml in 70% EtOH) 50 µl
H2O 768 ml
Salzkomponente A:
Salzkomponente B:
Reagenz Menge/Volumen
MgSO4 × 7 H2O 4,09 g
H2O x ml
Gesamtvolumen 100 ml
Reagenz Menge/Volumen
KH2PO4 32,8 g
Na2HPO4 82,1 g
H2O x ml
Gesamtvolumen 500 ml
2. Material und Methoden
31
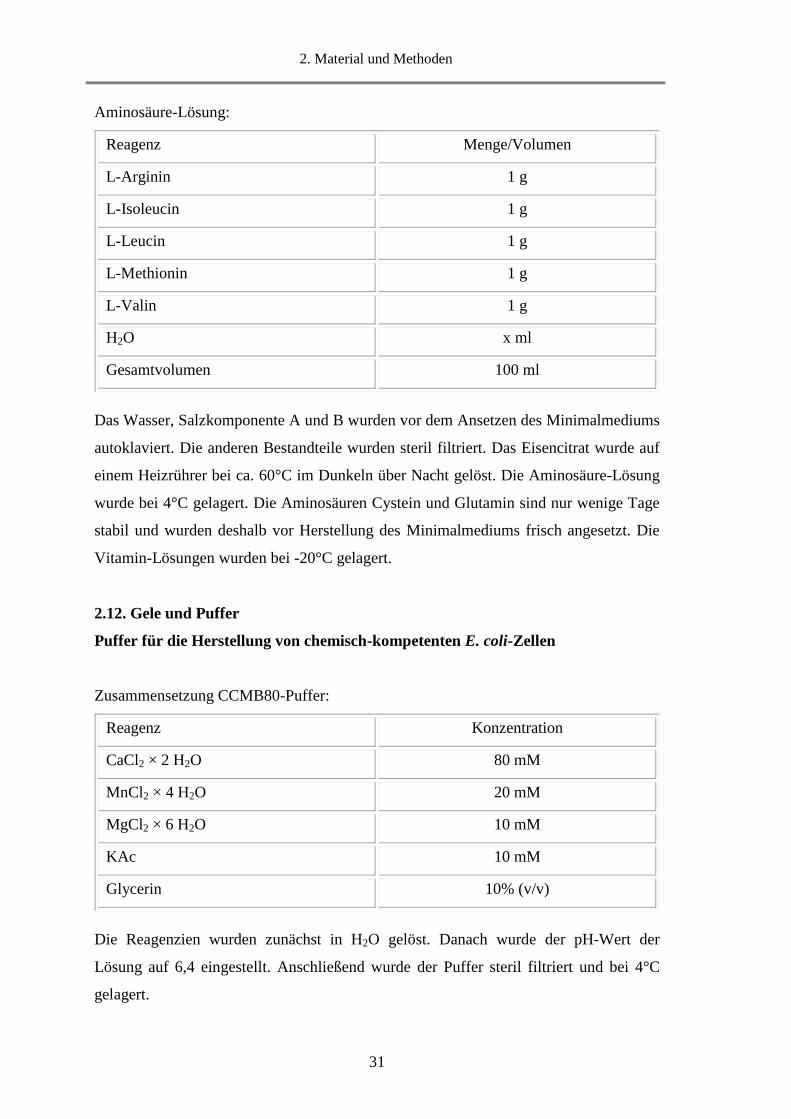
Aminosäure-Lösung:
Das Wasser, Salzkomponente A und B wurden vor dem Ansetzen des Minimalmediums
autoklaviert. Die anderen Bestandteile wurden steril filtriert. Das Eisencitrat wurde auf
einem Heizrührer bei ca. 60°C im Dunkeln über Nacht gelöst. Die Aminosäure-Lösung
wurde bei 4°C gelagert. Die Aminosäuren Cystein und Glutamin sind nur wenige Tage
stabil und wurden deshalb vor Herstellung des Minimalmediums frisch angesetzt. Die
Vitamin-Lösungen wurden bei -20°C gelagert.
2.12. Gele und Puffer
Puffer für die Herstellung von chemisch-kompetenten E. coli-Zellen
Zusammensetzung CCMB80-Puffer:
Die Reagenzien wurden zunächst in H2O gelöst. Danach wurde der pH-Wert der
Lösung auf 6,4 eingestellt. Anschließend wurde der Puffer steril filtriert und bei 4°C
gelagert.
Reagenz Menge/Volumen
L-Arginin 1 g
L-Isoleucin 1 g
L-Leucin 1 g
L-Methionin 1 g
L-Valin 1 g
H2O x ml
Gesamtvolumen 100 ml
Reagenz Konzentration
CaCl2 × 2 H2O 80 mM
MnCl2 × 4 H2O 20 mM
MgCl2 × 6 H2O 10 mM
KAc 10 mM
Glycerin 10% (v/v)
2. Material und Methoden
32
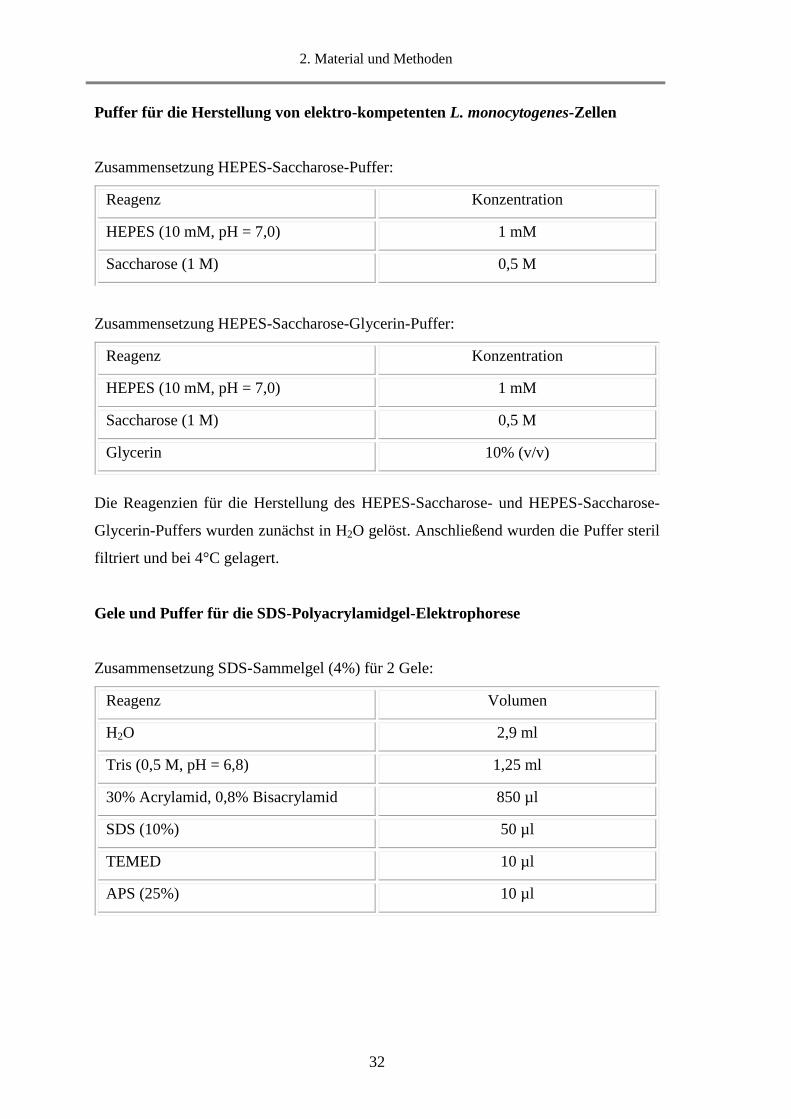
Puffer für die Herstellung von elektro-kompetenten L. monocytogenes-Zellen
Zusammensetzung HEPES-Saccharose-Puffer:
Zusammensetzung HEPES-Saccharose-Glycerin-Puffer:
Die Reagenzien für die Herstellung des HEPES-Saccharose- und HEPES-Saccharose-
Glycerin-Puffers wurden zunächst in H2O gelöst. Anschließend wurden die Puffer steril
filtriert und bei 4°C gelagert.
Gele und Puffer für die SDS-Polyacrylamidgel-Elektrophorese
Zusammensetzung SDS-Sammelgel (4%) für 2 Gele:
Reagenz Konzentration
HEPES (10 mM, pH = 7,0) 1 mM
Saccharose (1 M) 0,5 M
Reagenz Konzentration
HEPES (10 mM, pH = 7,0) 1 mM
Saccharose (1 M) 0,5 M
Glycerin 10% (v/v)
Reagenz Volumen
H2O 2,9 ml
Tris (0,5 M, pH = 6,8) 1,25 ml
30% Acrylamid, 0,8% Bisacrylamid 850 µl
SDS (10%) 50 µl
TEMED 10 µl
APS (25%) 10 µl
2. Material und Methoden
33
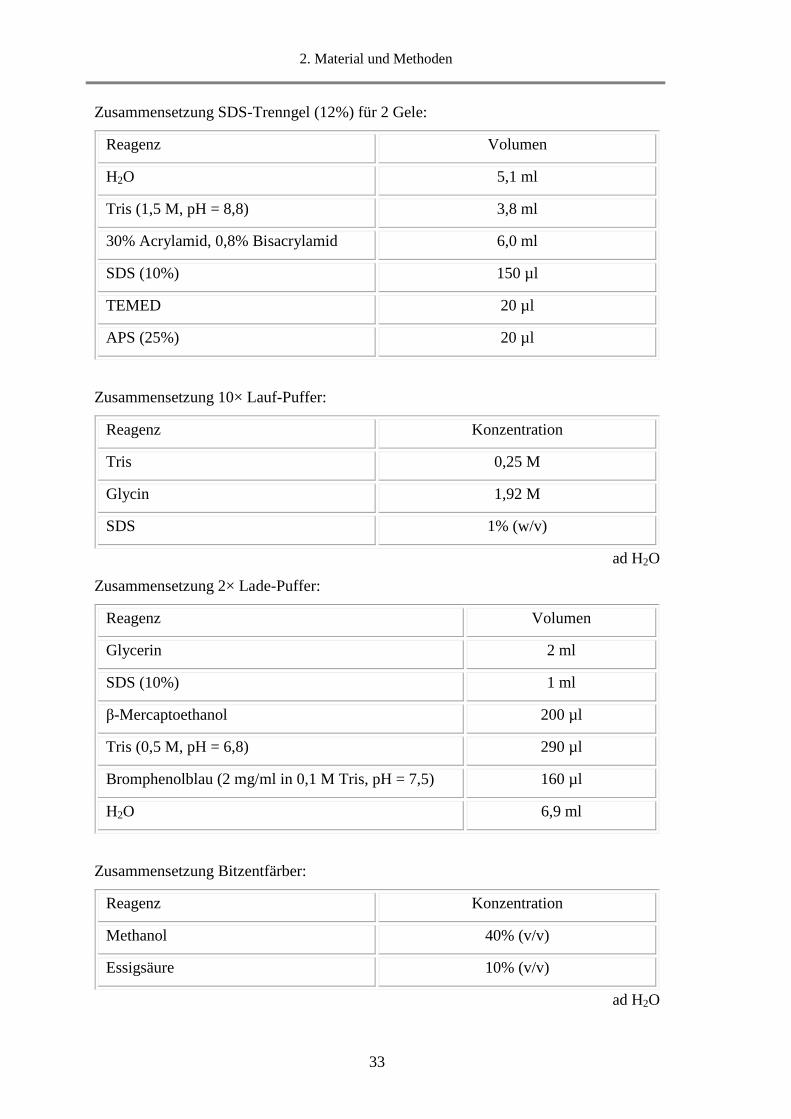
Zusammensetzung SDS-Trenngel (12%) für 2 Gele:
Zusammensetzung 10× Lauf-Puffer:
ad H2O
Zusammensetzung 2× Lade-Puffer:
Zusammensetzung Bitzentfärber:
ad H2O
Reagenz Volumen
H2O 5,1 ml
Tris (1,5 M, pH = 8,8) 3,8 ml
30% Acrylamid, 0,8% Bisacrylamid 6,0 ml
SDS (10%) 150 µl
TEMED 20 µl
APS (25%) 20 µl
Reagenz Konzentration
Tris 0,25 M
Glycin 1,92 M
SDS 1% (w/v)
Reagenz Volumen
Glycerin 2 ml
SDS (10%) 1 ml
β-Mercaptoethanol 200 µl
Tris (0,5 M, pH = 6,8) 290 µl
Bromphenolblau (2 mg/ml in 0,1 M Tris, pH = 7,5) 160 µl
H2O 6,9 ml
Reagenz Konzentration
Methanol 40% (v/v)
Essigsäure 10% (v/v)
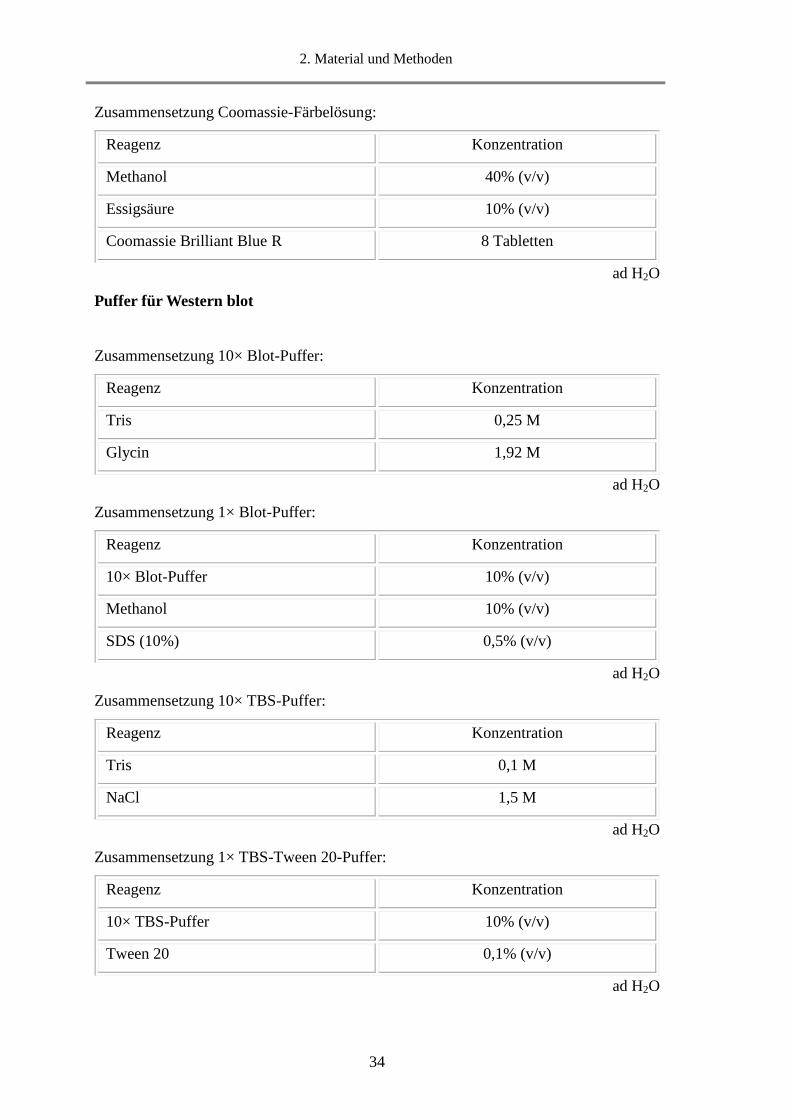
2. Material und Methoden
34
Zusammensetzung Coomassie-Färbelösung:
ad H2O
Puffer für Western blot
Zusammensetzung 10× Blot-Puffer:
ad H2O
Zusammensetzung 1× Blot-Puffer:
ad H2O
Zusammensetzung 10× TBS-Puffer:
ad H2O
Zusammensetzung 1× TBS-Tween 20-Puffer:
ad H2O
Reagenz Konzentration
Methanol 40% (v/v)
Essigsäure 10% (v/v)
Coomassie Brilliant Blue R 8 Tabletten
Reagenz Konzentration
Tris 0,25 M
Glycin 1,92 M
Reagenz Konzentration
10× Blot-Puffer 10% (v/v)
Methanol 10% (v/v)
SDS (10%) 0,5% (v/v)
Reagenz Konzentration
Tris 0,1 M
NaCl 1,5 M
Reagenz Konzentration
10× TBS-Puffer 10% (v/v)
Tween 20 0,1% (v/v)
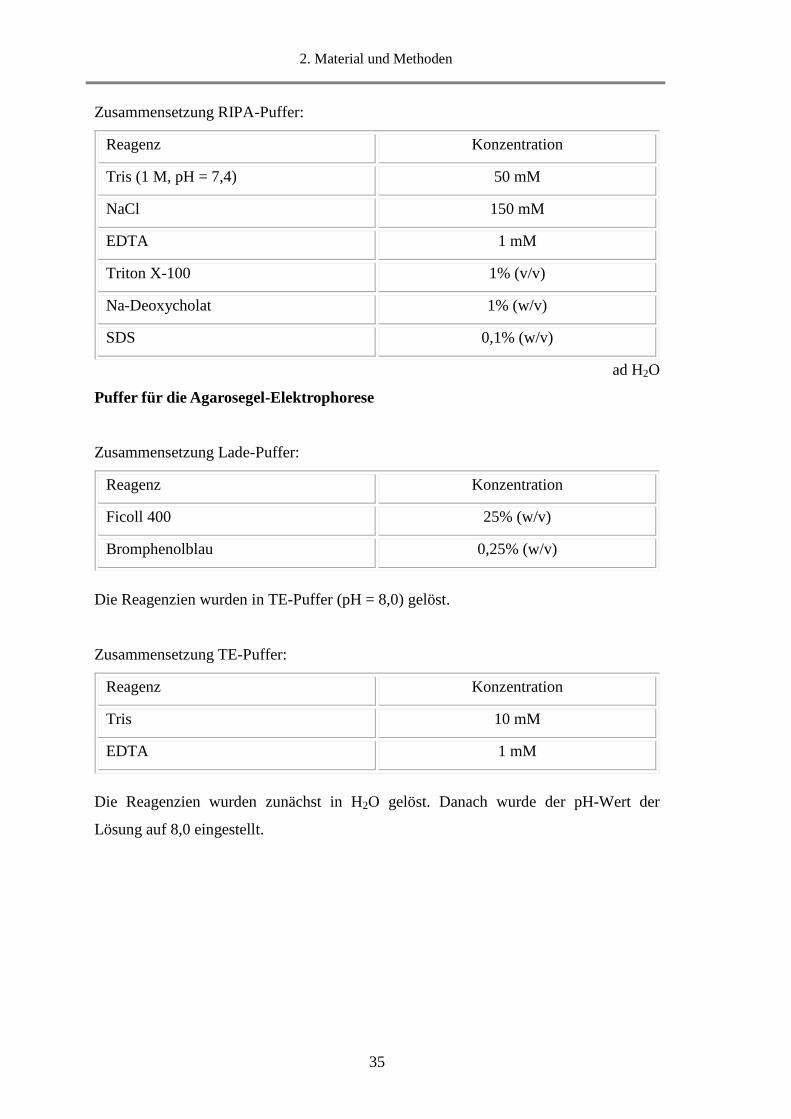
2. Material und Methoden
35
Zusammensetzung RIPA-Puffer:
ad H2O
Puffer für die Agarosegel-Elektrophorese
Zusammensetzung Lade-Puffer:
Die Reagenzien wurden in TE-Puffer (pH = 8,0) gelöst.
Zusammensetzung TE-Puffer:
Reagenz Konzentration
Tris 10 mM
EDTA 1 mM
Die Reagenzien wurden zunächst in H2O gelöst. Danach wurde der pH-Wert der
Lösung auf 8,0 eingestellt.
Reagenz Konzentration
Tris (1 M, pH = 7,4) 50 mM
NaCl 150 mM
EDTA 1 mM
Triton X-100 1% (v/v)
Na-Deoxycholat 1% (w/v)
SDS 0,1% (w/v)
Reagenz Konzentration
Ficoll 400 25% (w/v)
Bromphenolblau 0,25% (w/v)
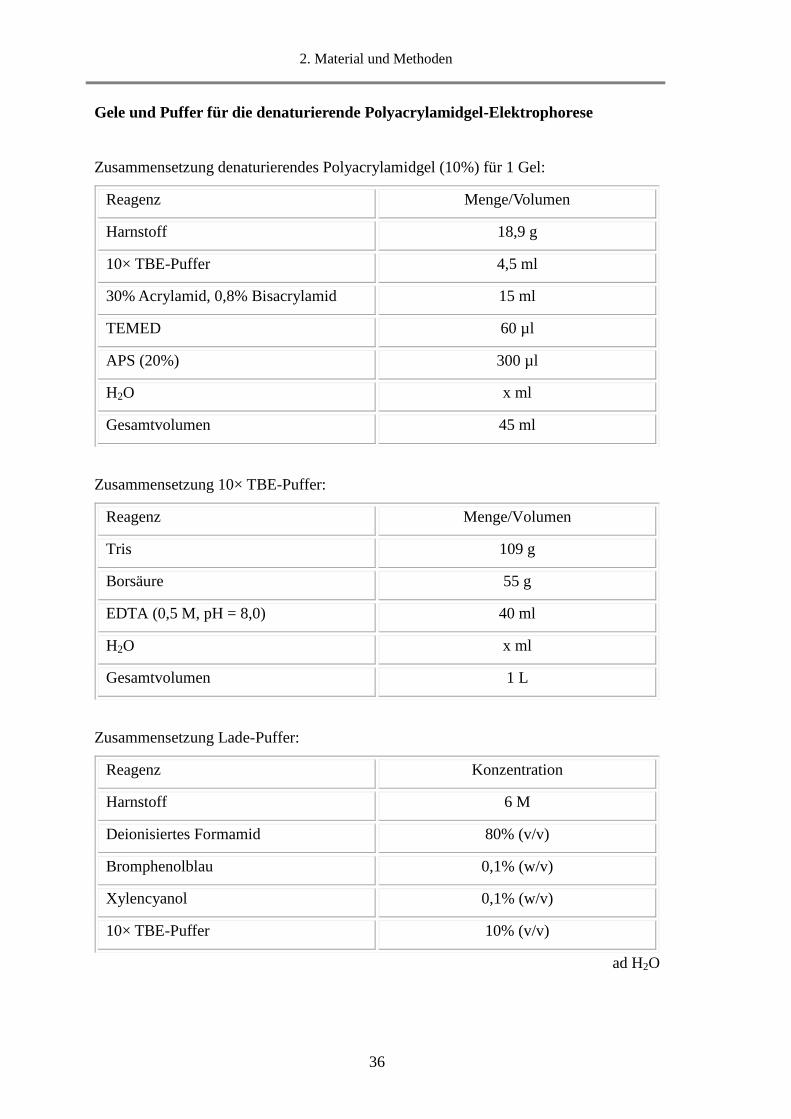
2. Material und Methoden
36
Gele und Puffer für die denaturierende Polyacrylamidgel-Elektrophorese
Zusammensetzung denaturierendes Polyacrylamidgel (10%) für 1 Gel:
Zusammensetzung 10× TBE-Puffer:
Zusammensetzung Lade-Puffer:
ad H2O
Reagenz Menge/Volumen
Harnstoff 18,9 g
10× TBE-Puffer 4,5 ml
30% Acrylamid, 0,8% Bisacrylamid 15 ml
TEMED 60 µl
APS (20%) 300 µl
H2O x ml
Gesamtvolumen 45 ml
Reagenz Menge/Volumen
Tris 109 g
Borsäure 55 g
EDTA (0,5 M, pH = 8,0) 40 ml
H2O x ml
Gesamtvolumen 1 L
Reagenz Konzentration
Harnstoff 6 M
Deionisiertes Formamid 80% (v/v)
Bromphenolblau 0,1% (w/v)
Xylencyanol 0,1% (w/v)
10× TBE-Puffer 10% (v/v)

2. Material und Methoden
37
2.13. Verbrauchsmaterialien
ThermoFisher Scientific: MicroAmp Optical Adhesive Film, MicroAmp Fast Optical
96-Well Reaction Plate (0,1 ml), RNase-freie Eppendorf-Reaktionsgefäße (1,5 ml und 2
ml), 12er-Well Platten für die Zellkultur, große Zellkulturflaschen, 10 cm Zellkultur-
schalen, Petrischalen, Kryoröhrchen
Merck: Express PLUS 0,22 µm Filtereinheit, Millex-GV 0,22 µm Filtereinheit
SARSTEDT: Eppendorf-Reaktionsgefäße (1,5 ml und 2 ml)
VWR: Poly-D-Lysin 12er-Well Platten für die Zellkultur, Zellsieb, Zellschaber
Roche: PVDF-Membran
Beranek: 500 ml Becher, Ultrazentrifugenröhrchen
Braun: Feindosierungsspritzen
TERUMO NEOLUS: Einmalkanülen
Spectrum Laboratories: Hollow Filter Module (>30 kDa-Membran)
Bio-Rad: Elektroporationsküvetten
Bertin Technologies: Precellys Lysing Kit (Soft tissue homogenizing CK14-Röhrchen)
Greiner Bio-One: Greiner-Röhrchen (15 ml und 50 ml), 96er-Well Platten mit F- und
U-Boden
2.14. Geräte
Agilent 2100 Bioanalyzer (Agilent Biotechnologies)
MiSeq Next Generation Sequencer (Illumina)
Ultrafiltrationsgerät: KrosFlo Research II TFF Systems (Spectrum Laboratories)
Thermomixer: Eppendorf Thermomixer comfort
Thermocycler: Applied Biosystems 2720 Thermal Cycle
Heizblock (Eigenbau der Institutswerkstatt)
Tischzentrifuge: Eppendorf Centrifuge 5424
Kühlzentrifuge: Eppendorf Centrifuge 5424 R
Zentrifuge für Greiner-Röhrchen: Heraeus Megafuge 1.0R
Zentrifuge für 500 ml Becher: Sorvall Superspeed RC2-B
Ultrazentrifuge: L8 60M ultracentrifuge (Beckman Coulter)
SW41 Ti-Rotor: Sorvall TH-641
F10 Rotor: Piramoon Technologies F10-6x500y Fiberlite
pH-Messgerät: SevenEasy (Mettler Toledo)

2. Material und Methoden
38
Magnetrührer: IKAMAG RED
Elektrophorese-Kammer für Protein-, DNA-, und RNA-Gele (Eigenbau der Instituts-
werkstatt)
Spannungsgerät: Electrophoresis Power Supply EPS 601 (Amersham Biosciences)
Gel-Dokumentationssystem (Bio-Rad)
Elektroporationsgerät (Bio-Rad electroporation system)
Taumelnder Plattformschüttler: Heidolph Polymax 1040 (Heraeus)
Schüttler für Bakterienkulturen: INFORS HAT (Ecotron)
Brutschrank für Bakterienkulturen (Heraeus)
Brutschrank für Zellkulturen (Thermo Scientific Forma Steri-Cycle CO2 Incubators)
Wasserbad für Bakterienkulturen (Eigenbau der Institutswerkstatt)
Wasserbad für Zellkulturen (Grant Instruments)
-20°C Gefrierschrank (Bosch) und -80°C Gefrierschrank (Thermo Scientific)
Kühlschrank (Liebherr)
365 nm UV-Tisch (Vilber)
Plattenphotometer: Infinite M200 PRO NonoQuant (Tecan)
Photometer für Messung des Bakterienwachstums: Ultrospec 10 (Amersham Bio-
sciences)
Vortexer: Vortex Genie 2 (Scientific Industries)
Bachofer Speed Vacuum Concentrator
Mikrowelle (Moulinex)
Pipetten (Eppendorf)
Sterilbank: HERAsafe Sicherheitswerkbänke (Thermo Scientific)
Sterilbank: NuAire Biological Safety Cabinets (Thermo Scientific)
StepOnePlus Real-Time PCR System (Applied Biosystems)
Mikroskop: Wilovert S (Hund Wetzlar)
Digitalkamera (Canon)
NanoDrop Technologies (ThermoFisher Scientific)
Qubit 2.0 Fluorometer (Life Technologies)
Waage und Feinwaage (KERN)
Precellys 24-Homogenisator (Bertin Technologies)
NanoSight Gerät (Malvern Instruments)
Kritische-Punkt-Trocknungseinrichtung: CPD 030 (Balzers, Liechtenstein)
2. Material und Methoden
39
Sputtercoater: SCD 500 (Balzers, Liechtenstein)
Elektronenmikroskop (Carl Zeiss)
2.15. Arbeiten mit DNA
2.15.1. Polymerase-Ketten-Reaktion (PCR)
Die PCR ist ein Verfahren zur Amplifikation von DNA-Sequenzen. Die Reaktion findet
in einem Thermocycler statt und benötigt: DNA-template, Vorwärts- und Rückwärts-
primer, dNTPs und eine hitzestabile DNA-Polymerase, wie beispielsweise die Taq-
DNA-Polymerase aus dem thermophilen Bakterium Thermus aquaticus. Die PCR läuft
in 3 sich wiederholenden Reaktionsschritten ab. Im ersten Reaktionsschritt wird das
DNA-template durch Erhitzen auf 95°C denaturiert. Im zweiten Reaktionsschritt wird
die Temperatur soweit abgesenkt, dass sich die komplementären Primer an die DNA-
Einzelstränge anlagern. Im dritten Reaktionsschritt findet die DNA-Synthese durch die
DNA-Polymerase statt, wobei die Primer als Startersequenzen dienen. Dadurch
entstehen neue DNA-Doppelstränge, die im nächsten PCR-Zyklus ebenfalls als
template dienen. Infolgedessen steigt die synthetisierte DNA-Menge pro Zyklus
exponentiell an.
Die PCR-Reaktionen wurden mit dem Taq-DNA-Polymerase Kit (Invitrogen)
durchgeführt.
Pipettierschema pro Reaktionsansatz:
Reagenz Menge/Volumen
DNA-template 50-100 ng
Taq-DNA-Polymerase (5 U/µl) 0,15 µl
10× Taq-DNA-Polymerase-Puffer ohne MgCl2 5 µl
MgCl2 (50 mM) 1,5 µl
dNTP-Mix (1,25 mM) 3 µl
Vorwärtsprimer (10 µM) 1 µl
Rückwärtsprimer (10 µM) 1 µl
H2O x µl
Gesamtvolumen 50 µl
2. Material und Methoden
40
PCR-Programm:
Die Schritte 2 bis 4 wurden 30mal wiederholt.
2.15.2. Agarosegel-Elektrophorese von DNA-Fragmenten
In der Agarosegel-Elektrophorese findet die Auftrennung von DNA nach Größe statt,
wobei eine Agarosegel-Matrix als Trägermaterial dient. Aufgrund des Zucker-Phosphat-
Rückgrats sind DNA-Moleküle negativ geladen und wandern infolgedessen in einem
elektrischen Feld zum positiven Pol.
Agarose löst sich beim Erhitzen in 1× TBE-Puffer und bildet beim Abkühlen ein
dreidimensionales Netzwerk, das je nach Agarose-Konzentration unterschiedlich große
Poren aufweist. Mit steigender Agarose-Konzentration steigt der Vernetzungsgrad der
Agarose. Die Konzentration der eingesetzten Agarose hängt von der Größe der aufzu-
trennenden DNA-Fragmente ab. Die in dieser Arbeit verwendeten Gele bestanden aus
1% und 2% Agarose. Hierfür wurde die Agarose zunächst abgewogen und danach in 1×
TBE-Puffer in einem Erlenmeyer-Kolben durch Aufkochen in einer Mikrowelle gelöst.
Nach kurzem Abkühlen der Agarose wurde Ethidiumbromid zur Färbung der DNA
hinzugefügt. Anschließend wurde die gelöste Agarose in den dafür vorbereiteten Gel-
träger gegossen. Nach Auspolymerisierung der Agarose wurde das Gel in eine
horizontale Kammer reingelegt. Die Kammer wurde zuvor mit 1× TBE-Puffer befüllt.
Vor dem Auftragen auf das Gel wurden die DNA-Proben mit Ladepuffer versetzt. Die
Elektrophorese wurde für 1 Stunden bei 150 V, 150 mA und 100 W durchgeführt. Um
die Größe der DNA-Fragmente bestimmen zu können, wurde ein DNA-Größenstandard
verwendet (Abb. 2.1). Nach der Elektrophorese wurden die DNA-Fragmente visualisiert
und fotografiert.
Schritte Temperatur [°C] Zeit
1 95 5 Minuten
2 95 20 Sekunden
3 55 30 Sekunden
4 72 30 Sekunden
5 4 ∞

2. Material und Methoden
41
2.15.3. Aufreinigung von DNA-Fragmenten
Die Aufreinigung von PCR- und Verdau-Produkten sowie cDNA wurde mit dem
QIAquick PCR Purification Kit (Qiagen) nach Angaben des Herstellers durchgeführt.
Dieser Kit eignet sich für die Aufreinigung von DNA-Fragmenten mit einer Größe von
100 Bp bis 10 Kb.
Zunächst wurden 5 V PB-Puffer zu den DNA-Proben pipettiert. Danach wurden die
Proben gevortext und auf dafür vorgesehenen Säulen gegeben. Die Säulen wurden für 1
Minute bei 13200 rpm und RT zentrifugiert. An die Säulen gebundene DNA wurde mit
750 µl PE-Puffer gewaschen. Dabei wurden die Säulen erneut für 1 Minute bei 13200
rpm und RT zentrifugiert. Es folgte eine Zentrifugation von 3 Minuten bei 13200 rpm
und RT. Nach dem die Säulen auf neue Eppis draufgesetzt wurden, wurde die DNA mit
50 µl H2O von den Säulen eluiert. Dabei wurden die Säulen für 1 Minute bei 13200 rpm
und RT zentrifugiert. Die Konzentration der DNA wurde mit dem NanoDrop gemessen.
2.15.4. Extraktion von DNA-Fragmenten aus dem Agarosegel
Durch unspezifische Bindung der Primer an das DNA-template können Nebenprodukte
während der PCR entstehen. In diesem Fall wird das entsprechende DNA-Fragment aus
dem Agarosegel extrahiert. Die Extraktion der DNA-Fragmente aus dem Agarosegel
wurde mit dem GeneJET Gel Extraction Kit (Thermo Scientific) nach Angaben des
Herstellers durchgeführt. Dieser Kit eignet sich für die Extraktion von DNA-
Fragmenten mit einer Größe von 25 Bp bis 20 Kb.
Nach Auftrennung der PCR-Produkte durch Agarosegel-Elektrophorese wurden die
entsprechenden DNA-Banden mit einem Skalpell aus dem Gel herausgeschnitten und in
Eppis überführt. Zur Bestimmung der DNA-Größe wurde ein DNA-Größenstandard
verwendet (Abb. 2.1). Dabei wurde die DNA auf einem UV-Tisch (365 nm) visualisiert.
Um die DNA aus dem Agarosegel zu extrahieren, wurden die Gelstücke zunächst mit
einem Skalpell in kleine Stücke geschnitten und danach mit 100 µl Binding-Puffer/100
mg Gel versetzt. Anschließend wurden die Proben solange auf dem Heizblock bei 50-
60°C inkubiert bis sich das Gel aufgelöst hatte. Zur schnelleren Auflösung des Gels
wurden die Proben in dieser Zeit öfters kräftig gevortext. Anschließend wurden die
Proben auf dafür vorgesehenen Säulen gegeben. Diese wurden für 1 Minute bei 6000
rpm und RT zentrifugiert. An die Säulen gebundene DNA wurde mit 700 µl Wash-

2. Material und Methoden
42
Puffer gewaschen. Dabei wurden die Säulen erneut für 1 Minute bei 6000 rpm und RT
zentrifugiert. Es folgte eine Zentrifugation von 1 Minute bei 13200 rpm und RT.
Nach dem die Säulen auf neue Eppis draufgesetzt wurden, wurde die DNA mit 50 µl
H2O von den Säulen eluiert. Hierfür wurden die Säulen für 1 Minute bei 13200 rpm und
RT zentrifugiert. Die DNA-Konzentration wurde mit dem NanoDrop gemessen.
2.15.5. Isolierung von Plasmid-DNA
Die Isolierung von Plasmid-DNA aus E. coli-Zellen wurde mit dem QIAprep Spin
Miniprep Kit (Qiagen) nach Angaben des Herstellers durchgeführt. Hierfür wurden
Übernachtkulturen von plasmidhaltigen E. coli-Zellen verwendet. Diese wurden
zunächst in Eppis überführt und für 3 Minuten bei 13200 rpm abzentrifugiert. Der
Überstand wurde verworfen. Anschließend wurde das Bakterienpellet in 250 µl P1-
Puffer resuspendiert. Es folgte die Zugabe von 250 µl P2-Puffer. Die Eppis wurden
mehrmals invertiert und für 5 Minuten bei RT inkubiert. Hierdurch erfolgte die Lyse der
Bakterienzellen unter alkalischen Bedingungen. Zur Neutralisation und Renaturierung
der Plasmid-DNA wurden 350 µl N3-Puffer hinzugefügt. Die Eppis wurden erneut
mehrmals invertiert. Anschließend wurden die Eppis für 10 Minuten bei 13200 rpm und
RT zentrifugiert. Der Überstand mit der darin gelösten Plasmid-DNA wurde auf dafür
vorgesehenen Säulen gegeben. Die Säulen wurden für 1 Minute bei 6000 rpm und RT
zentrifugiert. An die Säulen gebundene Plasmid-DNA wurde mit 750 µl PE-Puffer
gewaschen. Dabei wurden die Säulen erneut für 1 Minute bei 6000 rpm und RT
zentrifugiert. Es folgte eine Zentrifugation von 3 Minuten bei 13200 rpm und RT. Nach
dem die Säulen auf neue Eppis draufgesetzt wurden, wurde die DNA mit 50 µl H2O von
den Säulen eluiert. Dabei wurden die Säulen für 1 Minute bei 13200 rpm und RT
zentrifugiert. Die Konzentration der isolierten Plasmid-DNA wurde mit dem NanoDrop
gemessen.
2.15.6. Bestimmung der Nukleinsäure-Konzentration
Nach Isolierung von Plasmid-DNA aus E. coli-Zellen und Isolierung von RNA aus
eukaryotischen Zellen und Bakterienzellen sowie nach Aufreinigung von cDNA, PCR-
und Verdau-Produkten wurde die Nukleinsäure-Konzentration mit dem NanoDrop
gemessen. Nukleinsäuren haben, aufgrund der Absorption durch die quasi-aromatischen
Purine und Pyrimidine, ein Absorptionsmaximum bei 260 nm. Eine Absorption von 1

2. Material und Methoden
43
entspricht einer Nukleinsäure-Konzentration von 50 µg/ml dsDNA, 33 µg/ml ssDNA
und 40 µg/ml ssRNA.
Der Quotient aus A260/A280 gibt die Reinheit einer DNA- bzw. RNA-Lösung an.
Proteine haben, aufgrund der Absorption durch die aromatischen Aminosäuren, ein
Absorptionsmaximum bei 280 nm. Ein A260/A280-Quotient von >1,8 spricht für eine
reine DNA-Isolierung, ein Verhältnis von >2,0 für eine reine RNA-Isolierung. Ist die
Nukleinsäure-Lösung mit Proteinen oder Phenol kontaminiert, so beträgt der A260/A280-
Quotient <1,5.
2.15.7. Klonierung
Bei der Klonierung wird ein bestimmtes DNA-Fragment in einen Vektor, der als
Transportvehikel zur Übertragung der gewünschten DNA-Sequenz in eine Wirtszelle
dient, integriert. Durch Selektionsdruck mittels Antibiotikum im Wachstumsmedium
wird erreicht, dass sich nur die vektorhaltigen Zellen vermehren. Zu diesem Zweck
enthalten Vektoren Antibiotika-Resistenzgene und weitere Elemente, die beispielweise
für deren Vervielfältigung in den Wirtszellen essentiell sind.
2.15.7.1. Konierung durch TA Cloning
Durch TA Cloning können PCR-Produkte mit glatten Enden (blunt ends) in den
pCR2.1-TOPO-Vektor kloniert werden (Abb. 2.2). Hierfür macht man sich zunutze,
dass die Taq-DNA-Polymerase am 3`-Ende der synthetisierten DNA-Produkte nicht
template-abhängig ein A-Nukleotid hinzufügt. Infolgedessen besitzen diese PCR-
Produkte an den 3`-Enden jeweils ein überstehendes A-Nukleotid. Der linearisierte
pCR2.1-TOPO-Vektor besitzt komplementär dazu an den 3`-Enden jeweils ein
überstehendes T-Nukleotid. Daraufhin integrieren die PCR-Produkte über TA-
Basenpaarung in den pCR2.1-TOPO-Vektor (Abb. 2.3).
Für die Herstellung von Lm-pERL3-rli32 und Lm-pERL3-ssrS wurden die entspre-
chenden PCR-Produkte zunächst über TA-Basenpaarung in den pCR2.1-TOPO-Vektor
kloniert. Dabei wurde der TOPO TA Cloning Kit (Invitrogen) nach Angaben des
Herstellers verwendet.
2. Material und Methoden
44
Pipettierschema pro Reaktionsansatz:
Anschließend wurden die Proben für 10 Minuten bei RT inkubiert. Es folgte die
Transformation von kompetenten E. coli-Zellen. Für die Transformation von elektro-
kompetenten E. coli-Zellen wurde die Salt Solution 1:4 in H2O verdünnt.
2.15.7.2. Klonierung durch Restriktionsverdau und Ligation
Eine weitere Methode, um gewünschte DNA-Fragmente in einen Vektor zu klonieren,
wird mittels Restriktionsenzymen durchgeführt. Hierfür werden im ersten Schritt
Vektor und DNA-Fragment, das in den Vektor kloniert werden soll, mit den gleichen
Restriktionsenzymen geschnitten. Vorzugsweise verwendet man Restriktionsenzyme
die versetzt schneiden, so dass überstehende Enden (sticky ends) entstehen. Durch
Hybridisierung der komplementären Enden wird das gewünschte DNA-Fragment in den
Vektor kloniert. Im zweiten Schritt wird durch Ligation mittels DNA-Ligase das
Phosphodiester-Band kovalent geschlossen.
Für die Herstellung von Lm-pERL3-rli32 und Lm-pERL3-ssrS wurden die entspre-
chenden DNA-Fragmente zunächst durch Restriktionsverdau aus dem pCR2.1-TOPO-
Vektor herausgeschnitten. Diese wurden anschließend durch Ligation in den pERL3-
Vektor kloniert (Abb. 2.4).
Restriktionsverdau
Zunächst wurde ein Restriktionsverdau beider Vektoren, pCR2.1-TOPO und pERL3,
mit FastDigest Restriktionsenzymen (ThermoFisher Scientific) durchgeführt. Hierfür
wurden die entsprechenden DNA-Fragmente aus dem pCR2.1-TOPO-Vektor mit Sal I
und Xma I (für rli32) bzw. Sal I und BamH I (für ssrS) herausgeschnitten. In einem
parallelen Ansatz wurde der pERL3-Vektor mit Sal I und Xma I (für rli32) bzw. Sal I
und BamH I (für ssrS) linearisiert.
Reagenz Menge/Volumen
pCR2.1-TOPO-Vektor 1 μl
Salt Solution 1 μl
PCR-Produkt 4 μl (150-200 ng)
Gesamtvolumen 6 µl
2. Material und Methoden
45
Pipettierschema pro Reaktionsansatz:
Anschließend wurden die Proben für 2 Stunden bei 37°C auf dem Heizblock inkubiert.
In einem weiteren Schritt wurden die Verdau-Produkte elektrophoretisch in einem
1%igen Agarosegel aufgetrennt, visualisiert und fotografiert.
Ligation
Die Ligation wurde mit dem T4 DNA Ligation Kit (ThermoFisher Scientific) nach
Angaben des Herstellers durchgeführt.
Pipettierschema pro Reaktionsansatz:
Die Ligationsansätze wurden über Nacht bei 16°C im Wasserbad inkubiert. Am
nächsten Tag wurde die T4-DNA-Ligase zunächst durch Hitze inaktiviert. Hierfür
wurden die Proben für 20 Minuten bei 65°C auf dem Heizblock inkubiert. Es folgte die
Transformation von kompetenten E. coli-Zellen.
Reagenz Menge/Volumen
Sal I 1 μl
Xma I bzw. BamH I 1 μl
10× Restriktionsenzym-Puffer 2 μl
pCR2.1-TOPO- bzw. pERL3-Vektor 1 µg
H2O x µl
Gesamtvolumen 20 µl
Reagenz Menge/Volumen
T4-DNA-Ligase (1 U/µl) 1,5 μl
10× T4-DNA-Ligase-Puffer 2 μl
Verdautes pCR2.1-TOPO-Vektor 4 μl (150-300 ng)
Verdautes pERL3-Vektor 1,5 μl (50-100 ng)
H2O x µl
Gesamtvolumen 20 µl

2. Material und Methoden
46
2.16. Bakteriologische Arbeiten
2.16.1. Kultivierung von Bakterien
Die Kultivierung von E. coli-Zellen erfolgte in LB-Medium oder auf LB-Agarplatten.
Für die Isolierung von Plasmid-DNA wurden am Vortag Übernachtkulturen von
plasmidhaltigen E. coli-Zellen in 10 ml LB-Medium mit entsprechendem Antibiotikum
in 100 ml Erlenmeyer-Kolben angesetzt. Hierfür wurden die Bakterien mit einer Öse
von der Platte eingeimpft. Zuvor wurde das Antibiotikum ins Medium pipettiert, 100
µg/ml Ampicillin für die Anzucht von pCR2.1-TOPO-haltigen E. coli-Zellen und 300
µg/ml Erythromycin für die Anzucht von pERL3-haltigen E. coli-Zellen. Die Über-
nachtkulturen für die Herstellung von kompetenten E. coli-Zellen wurden in LB-
Medium ohne Antibiotikum angesetzt.
L. monocytogenes wurde in BHI-Medium oder auf BHI-Agarplatten sowie in Minimal-
medium kultiviert. Die Übernachtkulturen für die Isolierung von MVs (MVs-RNA),
sec-RNA und zellulärer RNA der Stämme Lm-pERL3-rli32 und Lm-pERL3-ssrS
wurden in BHI-Medium mit 5 µg/ml Erythromycin angesetzt. Diese wurden 1:50 in
Minimalmedium eingeimpft. Zuvor wurden 5 µg/ml Erythromycin ins Medium
pipettiert. Lm wurde ohne Zugabe von Antibiotikum im Wachstumsmedium angezogen.
Die Bakterienkulturen wurden bei 37°C und 180 rpm auf einen Schüttler angezogen.
2.16.2. Messung des Bakterienwachstums
Das Wachstum der Bakterienkulturen wurde durch Messung der optischen Dichte
überprüft. Diese wurde mit einem Spektralphotometer in Einmal-Mikroküvetten bei
einer Wellenlänge von 600 nm gemessen. Bei OD600nm-Werten über 1 wurden die
Bakterienkulturen verdünnt. Infolgedessen wurde bei der Bestimmung der optischen
Dichte der Verdünnungsfaktor berücksichtigt.
2.16.3. Kompetenz und Transformation
Die Transformation ist ein Prozess, bei dem Bakterien frei in der Umgebung
vorliegende Fremd-DNA aufnehmen. Voraussetzung für die Transformation ist die
Kompetenz, die Fähigkeit von Bakterienzellen DNA aufzunehmen. Nur wenige
Bakterienarten, wie beispielsweise Bacillus subtilis, zeigen eine natürliche Kompetenz.
Deshalb müssen Bakterienzellen zunächst für die Aufnahme von DNA kompetent
gemacht werden. Danach erfolgt die Aufnahme von DNA durch Poren, die

2. Material und Methoden
47
vorübergehend durch Elektroporation oder Hitzeschock in der Zellmembran erzeugt
werden.
2.16.3.1. Herstellung von elektro-kompetenten E. coli-Zellen
Eine E. coli-Übernachtkultur wurde 1:50 in 50 ml LB-Medium in einem 300 ml
Erlenmeyer-Kolben eingeimpft und bis zum Erreichen einer OD600nm von 0,5 bis 0,6 bei
37°C und 180 rpm auf dem Schüttler inkubiert. Anschließend wurde die Bakterienkultur
in ein 50 ml Greiner-Röhrchen überführt und für 8 Minuten bei 5000 rpm und 4°C
abzentrifugiert. Das Röhrchen wurde auf Eis gestellt und der Überstand wurde mit der
Pipette abgesaugt. Das Bakterienpellet wurde insgesamt 3mal mit jeweils 30 ml kaltem
10%igem Glycerin gewaschen. Dabei wurden die Bakterienzellen durch Zentrifuga-
tionen von jeweils 8 Minuten bei 5000 rpm und 4°C pelletiert. Nach jeder Zentri-
fugation wurde das Röhrchen auf Eis gestellt und der Überstand wurde mit der Pipette
abgesaugt. Im letzten Schritt wurde das Bakterienpellet in 500 µl kaltem 10%igem
Glycerin resuspendiert und als 50 µl Aliquot in Kryoröhrchen überführt. Die Bakterien-
zellen wurden in flüssigem Stickstoff schockgefroren und bis zur deren Verwendung bei
-80°C gelagert.
2.16.3.2. Herstellung von chemisch-kompetenten E. coli-Zellen
Eine E. coli-Übernachtkultur wurde 1:50 in 50 ml LB-Medium in einem 300 ml
Erlenmeyer-Kolben eingeimpft und bis zum Erreichen einer OD600nm von 0,3 bei 37°C
und 180 rpm auf dem Schüttler inkubiert. Anschließend wurde die Bakterienkultur in
ein 50 ml Greiner-Röhrchen überführt und für 10 Minuten auf Eis gestellt. Die
Bakterienzellen wurden durch eine Zentrifugation von 10 Minuten bei 4000 rpm und
4°C pelletiert. Das Röhrchen wurde auf Eis gestellt und der Überstand wurde mit der
Pipette abgesaugt. Das Bakterienpellet wurde zunächst in 1/3 V (17 ml) CCMB80-
Puffer resuspendiert und danach für weitere 10 Minuten auf Eis gestellt. Anschließend
wurden die Bakterienzellen erneut durch eine Zentrifugation von 10 Minuten bei 4000
rpm und 4°C pelletiert. Das Röhrchen wurde auf Eis gestellt und der Überstand wurde
mit der Pipette abgesaugt. Im letzten Schritt wurde das Bakterienpellet in 1/12 V (4,2
ml) CCMB80-Puffer resuspendiert und als 200 µl Aliquot in Kryoröhrchen überführt.
Die Bakterienzellen wurden in flüssigem Stickstoff schockgefroren und bis zur deren

2. Material und Methoden
48
Verwendung bei -80°C gelagert. Der CCMB80-Puffer wurde vor dessen Verwendung in
den Kühlschrank gestellt.
2.16.3.3. Herstellung von elektro-kompetenten L. monocytogenes-Zellen
Eine L. monocytogenes-Übernachtkultur wurde zunächst 1:100 in 200 ml 1× BHI-
Medium + 0,5 M Saccharose in einem 1 L Erlenmeyer-Kolben eingeimpft und danach
bei 37°C und 180 rpm auf dem Schüttler inkubiert. Nach Erreichen einer OD600nm von
0,2 wurden 200 µg Penicillin hinzugefügt. Danach wurde die Bakterienkultur für
weitere 2 Stunden auf dem Schüttler inkubiert. Anschließend wurde die Bakterienkultur
in 4× 50 ml Greiner-Röhrchen überführt und für 10 Minuten bei 5000 rpm und 4°C
abzentrifugiert. Die Röhrchen wurden auf Eis gestellt und der Überstand wurde mit der
Pipette abgesaugt. Die Bakterienpellets wurden in jeweils 10 ml kaltem HEPES-
Saccharose-Puffer resuspendiert und in ein 50 ml Greiner-Röhrchen zusammengeführt.
Danach wurden die Bakterienzellen erneut für 10 Minuten bei 5000 rpm und 4°C
pelletiert. Das Bakterienpellet wurde insgesamt 3mal mit jeweils 10 ml kaltem HEPES-
Saccharose-Puffer gewaschen. Dabei wurden die Bakterienzellen durch Zentrifu-
gationen von jeweils 15 Minuten bei 6000 rpm und 4°C pelletiert. Nach jeder
Zentrifugation wurde das Röhrchen auf Eis gestellt und der Überstand wurde mit der
Pipette abgesaugt. Im letzten Schritt wurde das Bakterienpellet in 500 µl kaltem
HEPES-Saccharose-Glycerin-Puffer resuspendiert und als 50 µl Aliquot in
Kryoröhrchen überführt. Die Bakterienzellen wurden in flüssigem Stickstoff
schockgefroren und bei -80°C gelagert.
2.16.3.4. Transformation von elektro-kompetenten E. coli-Zellen
Die bei -80°C gelagerten elektro-kompetenten E. coli-Zellen wurden zunächst auf Eis
aufgetaut. Danach wurden 2 µl aus den Klonierungsansätzen (TA-Cloning bzw.
Ligation) hinzugefügt. Anschließend wurden die Zellen in dafür vorgesehenen Küvetten
überführt und auf Eis gestellt. Es folgte die Elektroporation durch einen kurzen
Stromstoß (200 Ω, 1,8 V und 125 µF) mittels Elektroporationsgerät. Nach der
Elektroporation wurden die Zellen in 500 µl SOC-Medium aufgenommen und für 1
Stunde (pCR2.1-TOPO-Vektor) bzw. für 3 Stunden (pERL3-Vektor) bei 37°C und 180
rpm auf dem Schüttler inkubiert. Anschließend wurden die Zellen auf LB-Agarplatten

2. Material und Methoden
49
mit entsprechendem Antibiotikum ausplattiert und über Nacht bei 37°C im Brutschrank
inkubiert.
2.16.3.5. Transformation von chemisch-kompetenten E. coli-Zellen
Die bei -80°C gelagerten chemisch-kompetenten E. coli-Zellen wurden zunächst auf Eis
aufgetaut und danach mit 5 µl aus den Klonierungsansätzen (TA-Cloning bzw.
Ligation) versetzt. Die Zellen wurden für weitere 10 Minuten auf Eis gestellt. Danach
wurden die Zellen einem kurzen Hitzeschock von 90 Sekunden bei 42°C im Wasserbad
ausgesetzt. Es folgte eine erneute Inkubation von 10 Minuten auf Eis. Anschließend
wurden die Zellen in 500 µl SOC-Medium überführt und für 1 Stunde (pCR2.1-TOPO-
Vektor) bzw. für 3 Stunden (pERL3-Vektor) bei 37°C und 180 rpm auf dem Schüttler
inkubiert. Nach dieser Inkubation wurden die Zellen auf LB-Agarplatten mit entspre-
chendem Antibiotikum ausplattiert. Die Platten wurden über Nacht bei 37°C im
Brutschrank inkubiert.
2.16.3.6. Transformation von elektro-kompetenten L. monocytogenes-Zellen
Für die Herstellung von Lm-pERL3-rli32 und Lm-pERL3-ssrS wurden elektro-
kompetente Lm-Zellen mit den entsprechenden pERL3-Vektoren transformiert. Hierfür
wurden die bei -80°C gelagerten Zellen zunächst auf Eis aufgetaut. Danach wurden 50-
100 ng entsprechender pERL3-Vektor hinzugefügt. Für die anschließende Elektro-
poration wurden die Zellen in dafür vorgesehenen Küvetten überführt und auf Eis
gestellt. Die Zellen wurden zunächst einem kurzen Stromstoß (400 Ω, 1 V und 125 µF)
mittels Elektroporationsgerät ausgesetzt. Danach wurden die Zellen in 500 µl 1× BHI-
Medium + 0,5 M Saccharose aufgenommen. Anschließend wurden die Zellen für 3
Stunden bei 37°C und 180 rpm auf dem Schüttler inkubiert. Die Zellen wurden auf
BHI-Platten mit 5 µg/ml Erythromycin ausplattiert und für 2 Tage bei 37°C im
Brutschrank inkubiert.
2.17. Arbeiten mit RNA
2.17.1. Transfektion von RNA in eukaryotischen Zellen
Unter Transfektion versteht man das Einschleusen von Nukleinsäuren (DNA bzw.
RNA) in eukaryotische Zellen. Für die Transfektion von RNA in HEK293-Zellen und
BMDMs wurde Lipofectamine 2000 (Invitrogen) nach Angaben des Herstellers

2. Material und Methoden
50
verwendet. Lipofectamine 2000 besteht aus kationischen Lipide, die an das negativ
geladene Zucker-Phosphat-Rückgrat von Nukleinsäuren binden und diese in Liposomen
einschließen. Die positive Ladung der Liposomen vermittelt die Wechselwirkung mit
der negativ geladenen Zellmembran. Über Endocytose gelangt die Nukleinsäure in die
Zellen.
HEK293-Zellen wurden am Vortag in DMEM + 10% FKS in 12er-Well Platten
ausgesät. Zum Zeitpunkt der Transfektion lag die Konfluenz der Zellen bei 60-70%,
deren Zahl betrug ca. 7 × 105 Zellen/Well.
BMDMs wurden aus flüssigem Stickstoff aufgetaut, in BMDM-Medium + 30% M-CSF
aufgenommen, gezählt und in 12er-Well Platten ausgesät. Am nächsten Tag (24
Stunden später) wurde das Medium der Zellen gewechselt. Zum Zeitpunkt der
Transfektion (48 Stunden später) lag die Konfluenz der Zellen bei 80-90%, deren Zahl
betrug ca. 1 × 106 Zellen/Well. Das BMDM-Medium setzte sich wie folgt zusammen:
RPMI-Medium + 10% FKS + 50 µM β-Mercaptoethanol.
Vor der Transfektion der RNA in die Zellen wurden die RNA-Lipofectamine 2000-
Komplexe hergestellt. Hierfür wurden 50 bzw. 100 ng RNA in jeweils 50 µl Opti-MEM
Reduced Serum Medium pipettiert. In einem parallelen Ansatz wurden 0,25 µg Trans-
fektionsreagenz ebenfalls in jeweils 50 µl Opti-MEM Reduced Serum Medium
pipettiert. Anschließend wurde die RNA zu dem Transfektionsreagenz pipettiert, mit
der Pipette vorsichtig vermischt und für 20 Minuten bei RT inkubiert. In dieser Zeit
wurde das Medium der Zellen gewechselt. Zuvor wurden 100 U/ml Penicillin und 100
µg/ml Streptomycin zu dem Medium hinzugefügt. Im letzten Schritt wurden die RNA-
Lipofectamine 2000-Komplexe ins Medium zu den Zellen pipettiert. Die Zellen wurden
im Brutschrank bei 37°C und 5% CO2 inkubiert. Dabei wurden die HEK293-Zellen für
24 Stunden inkubiert. Die BMDMs wurden für 6 Stunden inkubiert. In beiden Fällen
folgte die Untersuchung der Typ-I-IFN Antwort über RNA-Isolierung, cDNA-Synthese
und RT-PCR.
2.17.2. In vivo Transfektion von RNA
Die von rli32 und ssrS ausgelöste Typ-I-IFN Antwort in vivo wurde in Milz und Leber
der Maus untersucht. Als Transfektionsreagenz wurde in vivo-jetPEI (Polyplus
transfection) nach Angaben des Herstellers verwendet. Dieses Reagenz enthält eine von
dem kationischen Polymer Polyethylenimin (PEI) abgeleitete Substanz als Transport-

2. Material und Methoden
51
molekül für Nukleinsäuren. Nach einer intravenösen Injektion wird die Nukleinsäure
von in vivo-jetPEI u.a. in Leber und Milz der Maus geliefert.
Für diesen Versuch wurden insgesamt 20 Mäuse, die in 4 Gruppen eingeteilt wurden,
verwendet. Eine Gruppe aus 5 Mäusen diente als Kontrolle. Diese Tiere wurden nur mit
dem Transfektionsreagenz intravenös injiziert. Die Tiere in den restlichen 3 Gruppen (5
Tiere/Gruppe) wurden mit jeweils 40 µg RNA (rli32, ssrS bzw. eukaryotische RNA)
intravenös injiziert. Vor der in vivo Transfektion der RNA wurden die RNA-in vivo-
jetPEI-Komplexe hergestellt. Hierfür wurde die RNA (jeweils 40 µg) in jeweils 50 µl
einer 10%igen Glukose-Lösung aufgenommen und auf 100 µl mit H2O aufgefüllt. In
einem parallelen Ansatz wurde das Transfektionsreagenz (jeweils 6,4 µl) ebenfalls in
jeweils 50 µl einer 10%igen Glukose-Lösung aufgenommen und auf 100 µl mit H2O
aufgefüllt. Anschließend wurden die Ansätze miteinander vermischt, gevortext und für
15 Minuten bei RT inkubiert. Es folgte die intravenöse Injektion der Mäuse mit den
RNA-in vivo-jetPEI-Komplexen.
2.17.3. Isolierung der zellulären RNA aus eukaryotischen Zellen
Die Isolierung der zellulären RNA aus eukaryotischen Zellen wurde mit dem miRNeasy
Mini Kit (Qiagen) nach Angaben des Herstellers durchgeführt. Zunächst wurde das
Medium der Zellen in den 12er-Well Platten abgesaugt. Anschließend wurden die
Zellen mit 700 µl QIAzol/Well lysiert. Das Zelllysat wurde in Eppis überführt. Nach
Zugabe von 0,2 V (140 µl) Chloroform wurden die Eppis kräftig geschüttelt und für 3
Minuten bei RT inkubiert. Zur Phasentrennung wurden die Eppis für 15 Minuten bei
13200 rpm und 4°C zentrifugiert. Die obere wässrige Phase wurde in neue Eppis
überführt und mit 1,5 V 100% EtOH versetzt. Nach kräftigem Mischen mit der Pipette
wurden die Proben auf miRNeasy Mini Säulen gegeben. Danach wurden die Säulen für
30 Sekunden bei 10000 rpm und RT zentrifugiert. An die Säulen gebundene RNA
wurde mit 350 µl RWT-Puffer gewaschen, wobei die Säulen erneut für 30 Sekunden bei
10000 rpm und RT zentrifugiert wurden.
Es folgte ein DNase-Verdau mit dem RNase-free DNase-Kit (Qiagen) nach Angaben
des Herstellers. Hierfür wurden 80 µl (10 µl DNase I + 70 µl RDD-Puffer)/Säule
eingesetzt. Der DNase-Verdau wurde für 30 Minuten bei 30°C auf dem Thermomixer
durchgeführt. An die Säulen gebundene RNA wurde anschließend erneut mit 350 µl
RWT-Puffer gewaschen. Hierfür wurden die Säulen für 30 Sekunden bei 10000 rpm

2. Material und Methoden
52
und RT zentrifugiert. Es folgten 2 weitere Waschschritte mit jeweils 500 µl RPE-Puffer,
wobei im letzten Waschschritt die Säulen für 2 Minuten bei 10000 rpm und RT
zentrifugiert wurden. Anschließend wurden die Säulen für 1 Minute bei 13200 rpm und
RT zentrifugiert. Nach dem die Säulen auf neue Eppis draufgesetzt wurden, wurde die
RNA mit 40 µl H2O von den Säulen eluiert. Dabei wurden die Säulen für 1 Minute bei
13200 rpm und RT zentrifugiert.
Die RNA-Konzentration wurde mit dem NanoDrop gemessen. Die Qualität der
isolierten RNA wurde mit dem Agilent 2100 Bioanalyzer untersucht.
2.17.4. Isolierung der zellulären RNA aus Bakterien
Für die Isolierung der zellulären RNA wurden Lm-, Lm-pERL3-rli32- und Lm-pERL3-
ssrS-Übernachtkulturen zunächst 1:50 in 10 ml Minimalmedium eingeimpft und danach
bei 37°C und 180 rpm auf dem Schüttler inkubiert. Zuvor wurden 5 µg/ml Erythro-
mycin ins Medium der beiden überexprimierenden Stämme gegeben. Nach Erreichen
einer OD600nm von 1 wurden 0,5 ml aus den Bakterienkulturen entnommen, mit 1 ml
RNAprotect Bacteria Reagent (Qiagen) versetzt, gevortext und für 5 Minuten bei RT
inkubiert. Anschließend wurden die Bakterienzellen für 5 Minuten bei 13200 rpm und
RT pelletiert. Der Überstand wurde verworfen. Die Bakterienpellets wurden bis zur
RNA-Isolierung bei -80°C gelagert.
Für die Isolierung der zellulären RNA wurden die Bakterienzellen zunächst lysiert.
Hierfür wurde das Bakterienpellet in 200 µl SET-Puffer [50 mM NaCl, 5 mM EDTA
und 30 mM Tris-HCl (pH = 7,0)] + 10% SDS (v/v) resuspendiert. Danach wurden die
Bakterienzellen durch eine Zentrifugation von 3 Minuten bei 13200 rpm und RT
abzentrifugiert. Der Überstand wurde vorsichtig mit der Pipette abgezogen. Anschlie-
ßend wurde das Bakterienpellet in 83 µl 50 mM Tris-HCl (pH = 6,5) + 50 mg/ml
Lysozym resuspendiert. Es folgte die Zugabe von 2 µl SUPERase In, RNase Inhibitor
(20 U/µl), 5 µl Mutanolysin (5 U/µl) und 10 µl Proteinase K (20 mg/ml). Die Proben
wurden gevortext und für 45 Minuten bei 37°C und 350 rpm auf dem Thermomixer
inkubiert.
Nach Lyse der Bakterienzellen wurden zunächst 600 µl QIAzol dazugegeben. Danach
wurden die Eppis gevortext und für 5 Minuten bei RT inkubiert. Nach Zugabe von 0,2
V (140 µl) Chloroform wurden die Eppis kräftig geschüttelt und für weitere 3 Minuten
bei RT inkubiert. Zur Phasentrennung wurden die Eppis für 15 Minuten bei 13200 rpm

2. Material und Methoden
53
und 4°C zentrifugiert. Die obere wässrige Phase wurde in Eppis überführt und mit 1,5 V
100% EtOH versetzt. Nach kräftigem Mischen mit der Pipette wurden die Proben auf
miRNeasy Mini Säulen (Qiagen) gegeben. Die weiteren Schritte wurden, wie bereits im
Abschnitt 2.17.3 beschrieben, durchgeführt.
Im letzten Schritt wurde die RNA mit 40 µl H2O von den Säulen eluiert, wobei die
Säulen für 1 Minute bei 13200 rpm und RT zentrifugiert wurden. Die Konzentration der
isolierten RNA wurde mit dem NanoDrop gemessen. Die RNA-Qualität wurde mit dem
Agilent 2100 Bioanalyzer untersucht.
2.17.5. Isolierung von RNA aus Mausorganen
5 Stunden nach der intravenösen Injektion von Mäusen mit RNA wurden Milz und
Leber entnommen, mit einer Präzisionswaage gewogen, in Eppis überführt und in
flüssigem Stickstoff schockgefroren.
Für die RNA-Isolierung wurden Teile von Milz und Leber (in tiefgefrorenem Zustand)
in 700 µl QIAzol in 2 ml Soft tissue homogenizing CK14-Röhrchen (Bertin
Technologies) überführt und auf Eis gestellt. Die Röhrchen enthalten 1,4 mm ceramic
beads und eignen sich für die Homogenisierung von 20-200 mg Gewebe. Die
Homogenisierung der Organe wurde mit einem Precellys 24-Homogenisator durch-
geführt, wobei die Proben zwischendurch auf Eis gestellt wurden. Anschließend wurden
die Proben für 5 Minuten bei RT inkubiert. Nach Zugabe von 0,2 V (140 µl)
Chloroform wurden die Proben kräftig geschüttelt und für weitere 3 Minuten bei RT
inkubiert. Zur Phasentrennung wurden die Proben für 15 Minuten bei 13200 rpm und
4°C zentrifugiert. Die obere wässrige Phase wurde in Eppis überführt und mit 1,5 V
100% EtOH versetzt. Nach kräftigem Mischen mit der Pipette wurden die Proben auf
miRNeasy Mini Säulen (Qiagen) gegeben. Die weiteren Schritte wurden, wie bereits im
Abschnitt 2.17.3 beschrieben, durchgeführt.
Im letzten Schritt wurde die RNA mit 40 µl H2O und eine Zentrifugation von 1 Minute
bei 13200 rpm und RT von den Säulen eluiert. Die Konzentration der isolierten RNA
wurde mit dem NanoDrop gemessen. Die Qualität der RNA wurde mit dem Agilent
2100 Bioanalyzer untersucht.

2. Material und Methoden
54
2.17.6. Qualitätskontrolle der RNA und RNA-Profilanalyse
Der Agilent 2100 Bioanalyzer wird zur Qualitätsüberprüfung von isolierter RNA
verwendet. Ähnlich herkömmlicher Standardverfahren zur RNA-Analyse wird die RNA
in eine Gelmatrix, die als Trägermaterial dient, elektrophoretisch nach Größe aufge-
trennt. Dabei wird die RNA durch eingelagerten Fluoreszenzfarbstoff detektiert. Die
Analyse von RNA-Proben mit dem Agilent 2100 Bioanalyzer liefert sogenannte
Elektropherogramme (Peaks), wobei die y-Achse die RNA-Menge in FU (fluorescence
units) und die x-Achse die RNA-Länge in Nukleotiden angibt. Diese liefert darüber
hinaus gelartige Bilder (Banden) von RNA-Proben. Diese bildlichen Darstellungen
decken selbst kleine Degradierungseffekte in den Proben auf. Zusätzlich wird die RNA-
Qualität als RIN-Wert (RNA-Integritätsnummer) angegeben.
Diese Technologie wurde zur Qualitätsüberprüfung von RNA, die aus eukaryotischen
Zellen, Bakterienzellen und Organe der Maus isoliert wurde, verwendet. Diese wurde
des Weiteren zur Profilanalyse der von L. monocytogenes sekretierten RNA-Fraktionen,
sec-RNA und MVs-RNA, verwendet.
2.17.7. Reverse Transkription (cDNA-Synthese)
Die Untersuchung der Typ-I-IFN Antwort nach der in vitro und in vivo Transfektion
von RNA erfolgte über RT-PCR. Hierfür wurde die isolierte RNA zunächst in cDNA
durch Reverse Transkription mit der SuperScript II Reverse Transcriptase (Invitrogen)
umgeschrieben. Zum Umschreiben der RNA in cDNA wurden ein Oligo (dT) 21-
Primer, der an den Poly(A)-Schwanz von mRNAs bindet, und ein Random-Primer (N9)
verwendet.
Pipettierschema pro Reaktionsansatz:
Reagenz Menge/Volumen
RNA 500 ng
Oligo (dT) 21-Primer (20 µM) 1 µl
N9 (100 µM) 2 µl
H2O x µl
Gesamtvolumen 11 µl
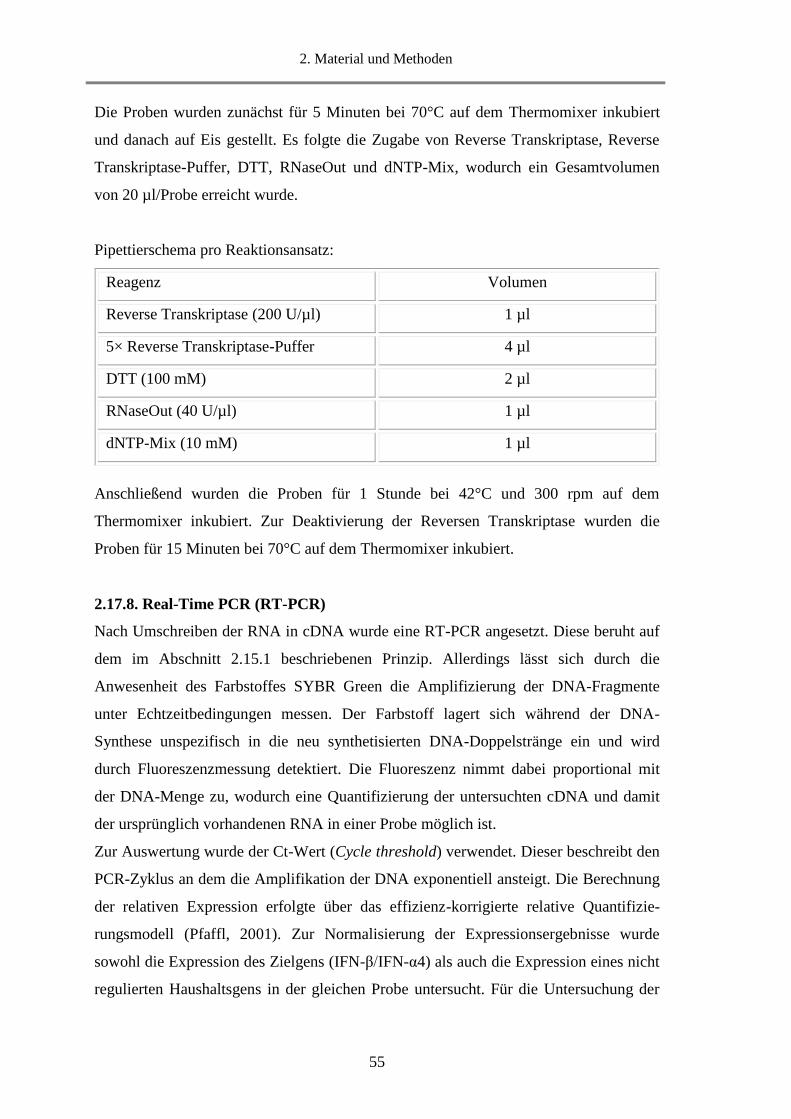
2. Material und Methoden
55
Die Proben wurden zunächst für 5 Minuten bei 70°C auf dem Thermomixer inkubiert
und danach auf Eis gestellt. Es folgte die Zugabe von Reverse Transkriptase, Reverse
Transkriptase-Puffer, DTT, RNaseOut und dNTP-Mix, wodurch ein Gesamtvolumen
von 20 µl/Probe erreicht wurde.
Pipettierschema pro Reaktionsansatz:
Anschließend wurden die Proben für 1 Stunde bei 42°C und 300 rpm auf dem
Thermomixer inkubiert. Zur Deaktivierung der Reversen Transkriptase wurden die
Proben für 15 Minuten bei 70°C auf dem Thermomixer inkubiert.
2.17.8. Real-Time PCR (RT-PCR)
Nach Umschreiben der RNA in cDNA wurde eine RT-PCR angesetzt. Diese beruht auf
dem im Abschnitt 2.15.1 beschriebenen Prinzip. Allerdings lässt sich durch die
Anwesenheit des Farbstoffes SYBR Green die Amplifizierung der DNA-Fragmente
unter Echtzeitbedingungen messen. Der Farbstoff lagert sich während der DNA-
Synthese unspezifisch in die neu synthetisierten DNA-Doppelstränge ein und wird
durch Fluoreszenzmessung detektiert. Die Fluoreszenz nimmt dabei proportional mit
der DNA-Menge zu, wodurch eine Quantifizierung der untersuchten cDNA und damit
der ursprünglich vorhandenen RNA in einer Probe möglich ist.
Zur Auswertung wurde der Ct-Wert (Cycle threshold) verwendet. Dieser beschreibt den
PCR-Zyklus an dem die Amplifikation der DNA exponentiell ansteigt. Die Berechnung
der relativen Expression erfolgte über das effizienz-korrigierte relative Quantifizie-
rungsmodell (Pfaffl, 2001). Zur Normalisierung der Expressionsergebnisse wurde
sowohl die Expression des Zielgens (IFN-β/IFN-α4) als auch die Expression eines nicht
regulierten Haushaltsgens in der gleichen Probe untersucht. Für die Untersuchung der
Reagenz Volumen
Reverse Transkriptase (200 U/µl) 1 µl
5× Reverse Transkriptase-Puffer 4 µl
DTT (100 mM) 2 µl
RNaseOut (40 U/µl) 1 µl
dNTP-Mix (10 mM) 1 µl

2. Material und Methoden
56
Typ-I-IFN Antwort in HEK293-Zellen wurde das Haushaltsgen PPIA (peptidylprolyl
isomerase A) verwendet. In BMDMs und in den Mausorganen wurde Rplp (ribosomal
protein, large, P0) verwendet.
Die RT-PCR wurde mit dem QuantiTect Primer Assay (Qiagen) und dem QuantiTect
SYBR Green PCR Kit (Qiagen) durchgeführt.
Pipettierschema pro Reaktionsansatz:
Anschließend wurden 12,5 µl QuantiTect SYBR Green PCR Master Mix dazugegeben.
Die Proben wurden in 96er-Well Platten pipettiert. Für jede Probe wurden
Dreifachbestimmungen durchgeführt. Die RT-PCR wurde mit dem StepOnePlus Real-
Time PCR System (Applied Biosystems) durchgeführt.
PCR-Programm:
Die Schritte 2 bis 4 wurden 40mal wiederholt.
2.17.9. Isolierung von MVs-RNA
Vor der Isolierung von MVs-RNA wurde ein RNase-Verdau durchgeführt, um die an
der äußeren Oberfläche von MVs assoziierte RNA zu verdauen. Zu 50 µg MVs-Probe
wurden 2,5 µg RNase, DNase-free (Roche) pipettiert. Die Proben wurden für 30
Reagenz Menge/Volumen
cDNA 100 ng
QuantiTect Primer Assay 7,5 µl
H2O x µl
Gesamtvolumen 12,5 µl
Schritte Temperatur [°C] Zeit
1 95 15 Minuten
2 95 20 Sekunden
3 55 30 Sekunden
4 72 30 Sekunden
5 4 ∞

2. Material und Methoden
57
Minuten bei 37°C auf dem Thermomixer inkubiert. Anschließend wurden die MVs-
Proben auf 500 µl mit PBS aufgefüllt. Nach Zugabe von 1,5 V (750 µl)
Phenol/Chloroform-Isoamylalkohol (25:24:1, pH = 4,5-5,0) wurden die Proben gevor-
text und für 5 Minuten bei RT inkubiert. Zur Phasentrennung wurden die Proben für 15
Minuten bei 13200 rpm und 4°C zentrifugiert. Anschließend wurde die obere wässrige
Phase in Eppis überführt und mit 0,1 V 3 M NaOAc (pH = 4,8-5,2) und 2 V 100%
EtOH versetzt. Die Proben wurden zunächst für mindestens 1 Stunde bei -80°C
inkubiert. Danach wurden die Proben für 15 Minuten bei 13200 rpm und 4°C
zentrifugiert. Der Überstand wurde verworfen und das RNA-Pellet wurde über Nacht
getrocknet.
Im letzten Schritt wurde das RNA-Pellet in 50 µl H2O resuspendiert. Die Konzentration
der isolierten MVs-RNA wurde mit dem Qubit gemessen. Die RNA-Profilanalyse
wurde mit dem Agilent 2100 Bioanalyzer durchgeführt.
2.17.10. Isolierung von sec-RNA
Nach dem die MVs in dem eingeengten Überstand (ca. 30 ml) von L. monocytogenes-
Kulturen durch Ultrazentrifugation pelletiert worden sind, wurde der MVs-freie
Überstand für die Isolierung von sec-RNA verwendet. Dieser wurde in ein 50 ml
Greiner-Röhrchen überführt, in einem Verhältnis von 1:1 mit 100% EtOH versetzt und
über Nacht bei -20°C inkubiert.
Am nächsten Tag wurde das Röhrchen für 30 Minuten bei 6000 rpm und 4°C
zentrifugiert. Der Überstand wurde verworfen und das Pellet über Nacht getrocknet.
Anschließend wurde das Pellet in 3 ml H2O resuspendiert und in Eppis (500 µl/Eppi)
überführt. Nach Zugabe von 1,5 V (750 µl) QIAzol wurden die Proben gevortex und für
5 Minuten bei RT inkubiert. Anschließend wurden 0,2 V (250 µl) Chloroform
dazugegeben und die Proben kräftig geschüttelt. Nach einer Inkubation von 3 Minuten
bei RT wurden die Proben zur Phasentrennung für 15 Minuten bei 13200 rpm und 4°C
zentrifugiert. Die obere wässrige Phase wurde in Eppis überführt und mit 1,5 V 100%
EtOH versetzt. Nach kräftigem Mischen mit der Pipette wurden die Proben auf
miRNeasy Mini Säulen (Qiagen) gegeben. Die weiteren Schritte wurden, wie bereits im
Abschnitt 2.17.3 beschrieben, durchgeführt.
Im letzten Schritt wurde die RNA mit 50 µl H2O von den Säulen eluiert. Dabei wurden
die Säulen für 1 Minute bei 13200 rpm und RT zentrifugiert. Die Konzentration der

2. Material und Methoden
58
isolierten sec-RNA wurde mit dem Qubit gemessen. Die RNA-Profilanalyse wurde mit
dem Agilent 2100 Bioanalyzer durchgeführt.
2.17.11. Sequenzierung
Zunächst wurde die RNA mit einer RNA 5'-Polyphosphatase (Epicenter) behandelt.
Anschließend wurden Oligonukleotid-Adapter an den 5`- und 3`- Enden der RNA
ligiert. Die first-strand cDNA-Synthese wurde mit der M-MLV Reverse Transcriptase
(ThermoFisher Scientific) durchgeführt, wobei der 3`-Adapter als Primer diente. Die
resultierende cDNA wurde mit einer high fidelity DNA-Polymerase (ThermoFisher
Scientific) über PCR amplifiziert. Anschließend wurde die cDNA mit dem Agencourt
AMPure XP Kit (Beckman Coulter Genomics) aufgereinigt. Die Menge und
Größenverteilung der cDNA wurde mit dem Agilent 2100 Bioanalyzer analysiert.
Anschließend wurden die cDNA libraries verdünnt und mit dem MiSeq Next
Generation Sequencer mittels MiSeq Reagent Kit V2 (Illumina) sequenziert und
analysiert. Die Adapter-Sequenzen wurden mittels Sequence Quality Trimming mit
cutadapt 1 entfernt (Martin, 2011). Die reads wurden gegen das Referenzgenom von L.
monocytogenes mittels CLC Genomics Workbench (Qiagen) gemappt (Glaser et al,
2001).
2.17.12. Herstellung von RNA durch in vitro Transkription (IVT)
Die 25 ausgesuchten RNA-Kandidaten, die in den sekretierten RNA-Fraktionen von L.
monocytogenes identifiziert worden sind, wurden enzymatisch durch in vitro Trans-
kription mittels T7-RNA-Polymerase (NEB) hergestellt. Zunächst wurde das DNA-
template durch PCR hergestellt. Dabei enthielt der Vorwärtsprimer am 5`-Ende die
Sequenz des Promotors der T7-RNA-Polymerase (5`-taatacgactcactatagg-3`). Anschlie-
ßend wurden die PCR-Produkte in 1%ige und 2%ige Agarosegele aufgetrennt,
visualisiert und fotografiert. Nach Aufreinigung der PCR-Produkte wurde die DNA-
Konzentration mit dem NanoDrop gemessen. Es folgte die in vitro Transkription der
DNA in RNA.

2. Material und Methoden
59
Pipettierschema pro Reaktionsansatz:
Anschließend wurden die Proben für 3 Stunden bei 37°C auf dem Thermomixer
inkubiert.
2.17.13. Verdau des DNA-templates
Nach der in vitro Transkription wurde das DNA-template in den Proben durch DNase-
Verdau mit der DNase I, RNase-free (ThermoFisher Scientific) entfernt. Hierfür wurden
5 µl DNase I (2 U/µl) und 5 µl 10× DNase I-Puffer zu den Proben hinzugefügt. Das
Gesamtvolumen der Proben betrug 50 µl. Danach wurden die Proben für 1 Stunde bei
37°C auf dem Thermomixer inkubiert. Anschließend wurde die DNase I durch Zugabe
von 5 µl EDTA (50 mM) und eine Inkubation von 10 Minuten bei 75°C auf dem
Thermomixer deaktiviert.
2.17.14. Dephosphorylierung von RNA
Um die 5`-Triphosphat-Gruppe der RNAs, die durch in vitro Transkription hergestellt
worden sind, sowie der RNAs in sec-RNA und MVs-RNA zu entfernen, wurde eine
Dephosphorylierung mit der Alkaline Phosphatase, Calf intestinal (NEB) durchgeführt.
Hierfür wurden 1 µl Alkalische Phosphatase (10 U/µl) und 5 µl 10× Alkalische
Phosphatase-Puffer zu 1 µg RNA hinzugefügt. Das Gesamtvolumen der Proben betrug
50 µl. Die Proben wurden für 1 Stunde bei 37°C auf dem Thermomixer inkubiert.
Anschließend wurde die RNA durch Phenol/Chloroform-Fällung aufgereinigt.
Reagenz Menge/Volumen
DNA 1 µg
T7-RNA-Polymerase (50 U/µl) 2 µl
10× T7-RNA-Polymerase-Puffer 5 µl
DTT (100 mM) 5 µl
rNTP-Mix (25 mM) 4 µl
RNaseOut (40 U/µl) 2 µl
H2O x µl
Gesamtvolumen 50 µl

2. Material und Methoden
60
2.17.15. Denaturierende Polyacrylamidgel-Elektrophorese
Nach Herstellung der 25 ausgesuchten RNA-Kandidaten durch in vitro Transkription
wurden diese unter denaturierenden Bedingungen (7 M Harnstoff) in 10%ige und
12%ige Polyacrylamidgele elektrophoretisch aufgetrennt. Die Zugabe von Harnstoff
verhindert die Bildung von Sekundärstrukturen, wodurch eine Auftrennung der RNA
nach Länge erfolgt. Hierfür wurden jeweils 10 µl aus den in vitro Transkriptions-
ansätzen auf das Gel aufgetragen. Vor dem Auftragen wurden die RNA-Proben mit
Ladepuffer in einem Verhältnis von 1:1 vermischt und für 10 Minuten bei 65°C auf dem
Thermomixer inkubiert. Danach wurden die Proben auf Eis gestellt.
Die Auftrennung der RNA erfolgte in 20 cm × 20 cm große Glasplatten, die in vertikale
Kammern reingelegt wurden. Die Kammern wurden zuvor mit 1× TBE-Puffer befüllt.
Die Elektrophorese wurde für 2 Stunden bei 400 V, 150 mA und 150 W durchgeführt.
Zur Färbung der RNA wurden die Gele in ethidiumbromidhaltiges 1× TBE-Puffer für
10 Minuten reingelegt. Anschließend wurde die RNA auf einem UV-Tisch (365 nm)
visualisiert und fotografiert. Um die Größe der RNA bestimmen zu können, wurde ein
RNA-Größenstandard verwendet (Abb. 2.1). Die entsprechenden RNA-Banden wurden
mit einem Skalpell aus dem Gel herausgeschnitten, in Eppis überführt und bei -80°C
gelagert.
2.17.16. Extraktion von RNA aus dem Polyacrylamidgel
Um die RNA aus dem Polyacrylamidgel zu extrahieren, wurden die Gelstücke zunächst
mit einem Skalpell in kleine Stücke geschnitten und danach mit 500 µl Elutionspuffer
[0,5 M NaOAc (pH = 5,0), 1 mM EDTA (pH = 8,0) und 2,5% Phenol (v/v)] versetzt.
Anschließend wurde die RNA durch wiederholtes Einfrieren in flüssigem Stickstoff,
Auftauen und Vortexen sowie einer Inkubation über Nacht bei 1500 rpm auf dem
Thermomixer aus dem Gel extrahiert. Es folgte die Aufreinigung der RNA durch
Phenol/Chloroform-Fällung.
2.17.17. Phenol/Chloroform-Fällung von RNA
Nach Dephosphorylierung der RNA und Extraktion der RNA aus dem Polyacrylamidgel
wurde die RNA durch Phenol/Chloroform-Fällung aufgereinigt. Hierfür wurden die
RNA-Proben mit 1,5 V (750 µl) Phenol/Chloroform-Isoamylalkohol (25:24:1, pH = 4,5-
5,0) versetzt und gevortext. Das Gesamtvolumen der Proben betrug 500 µl. Nach einer

2. Material und Methoden
61
Inkubation von 5 Minuten bei RT folgte die Phasentrennung durch eine Zentrifugation
von 15 Minuten bei 13200 rpm und 4°C. Die obere wässrige Phase wurde in Eppis
überführt und mit 0,1 V 3 M NaOAc (pH = 4,8-5,2) und 2 V 100% EtOH versetzt.
Anschließend wurden die Proben für mindestens 1 Stunde bei -80°C inkubiert. Nach
dieser Inkubation wurden die Proben für 15 Minuten bei 13200 rpm und 4°C zentri-
fugiert. Der Überstand wurde verworfen und das RNA-Pellet wurde über Nacht
getrocknet. Im letzten Schritt wurde das RNA-Pellet in 50 µl H2O resuspendiert. Die
RNA-Konzentration wurde mit dem Qubit gemessen.
2.18. Proteinanalytische Methoden
2.18.1. Isolierung von Membrane Vesicles (MVs), die von L. monocytogenes
produziert werden
Für die Isolierung von MVs, die von Lm, Lm-pERL3-rli32 und Lm-pERL3-ssrS
produziert werden, wurden Übernachtkulturen in BHI-Medium angezogen. Das Mini-
malmedium (2 L) wurde nach Fertigstellung auf 6× 1 L Erlenmeyer-Kolben aufgeteilt
(ca. 300 ml Medium/Kolben). Anschließend wurden die Übernachtkulturen 1:50 in
Minimalmedium eingeimpft und die Kolben bei 37°C und 180 rpm auf dem Schüttler
inkubiert. Zuvor wurden 5 µg/ml Erythromycin ins Medium der beiden überexpri-
mierenden Stämme pipettiert. Nach Erreichen einer OD600nm von 1 wurden die
Bakterienkulturen in 6× 500 ml Zentrifugenbecher überführt und für 15 Minuten bei
9000 rpm und 4°C abzentrifugiert. Der Überstand wurde mit einer Express PLUS 0,22
µm Filtereinheit steril filtriert. Anschließend wurde der Überstand durch Ultrafiltration
mit einer Membran mit einem Cut Off von 30 kDa auf ca. 30 ml eingeengt. Der
eingeengte Überstand (das Ultrafiltrat) wurde zunächst mit einer Millex-GV 0,22 µm
Filtereinheit steril filtriert. Danach wurde das Ultrafiltrat in 3 Röhrchen für den SW41
Ti-Rotor der Ultrazentrifuge überführt und für 3 Stunden bei 30000 rpm und 4°C
abzentrifugiert. Hierdurch wurden die während des Wachstums produzierten und ins
Medium abgegebenen MVs pelletiert. Das MVs-Pellet wurde zunächst getrocknet,
danach in 300 µl PBS resuspendiert und bei -80°C gelagert.
2.18.2. Bestimmung der Proteinkonzentration mit der Bradford-Methode
Die Messung der Proteinkonzentration von isolierten MVs wurde mit der Bradford-
Methode durchgeführt. Diese Methode beruht auf der Anlagerung des Farbstoffs

2. Material und Methoden
62
Coomassie an kationische und nichtpolare, hydrophobe Seitenketten von Proteinen.
Dadurch kommt es zu einer Verschiebung des Absorptionsmaximums des Farbstoffs
von 450 zu 595 nm. Da die Zunahme der Absorption bei 595 nm direkt proportional zur
Proteinkonzentration in der Lösung ist, lässt sich unter Verwendung einer Eichkurve die
Proteinkonzentration bestimmen.
Für die Messung wurden jeweils 10 µl MVs-Probe (1:3 in PBS verdünnt) zu 200 µl
Bradford-Reagenz (1:5 in H2O verdünnt) in einer 96er-Well Platte pipettiert und
vermischt. In einem parallelen Ansatz wurde zur Herstellung einer Eichkurve eine
BSA-Verdünnungsreihe in PBS angesetzt. Anschließend wurden jeweils 10 µl aus der
BSA-Verdünnungsreihe zu 200 µl Bradford-Reagenz pipettiert und vermischt. Sowohl
für die MVs-Proben als auch für die Eichkurve wurden Doppelbestimmungen durch-
geführt. Die Platte wurde für 5 Minuten bei RT inkubiert. Anschließend wurde die
optische Dichte der Proben bei 595 nm mit einem Photometer gemessen. Zur Aus-
wertung wurde eine Eichkurve hergestellt, wobei die Absorption (x-Achse) gegen die
Proteinkonzentration (y-Achse) des BSA-Standards aufgetragen wurde. Für die
Berechnung der Proteinkonzentration wurde die Gleichung verwendet, die man nach
dem Erstellen der Regressionsgerade erhält.
2.18.3. Bestimmung der Proteinkonzentration mit der BCA-Methode
Die Fähigkeit von MVs in PtK-Zellen zu internalisieren, wurde durch Detektion des
Autolysins P60 im Zelllysat über Western blot untersucht. Hierfür wurde zunächst die
Proteinkonzentration des Zelllysats der PtK-Zellen mit der BCA-Methode bestimmt.
Grundlage für die BCA-Methode ist die Reduktion von Cu2+
zu Cu+ durch
Peptidbindungen unter alkalischen Bedingungen. Das entstandene Cu+ bildet mit
Bicinchoninsäure (BCA) einen Farbkomplex mit einem Absorptionsmaximum bei 562
nm.
Für die Messung der Proteinkonzentration des Zelllysats wurden jeweils 6,25 µl Probe
(1:5 in RIPA-Puffer verdünnt) zu 50 µl BCA-Reagenz in einer 96er-Well Platte
pipettiert und vermischt. In einem parallelen Ansatz wurde zur Herstellung einer
Eichkurve eine BSA-Verdünnungsreihe in RIPA-Puffer angesetzt. Anschließend
wurden jeweils 6,25 µl aus der BSA-Verdünnungsreihe zu 50 µl BCA-Reagenz
pipettiert und vermischt. Sowohl für die Proben als auch für die Eichkurve wurden
Doppelbestimmungen durchgeführt. Die Platte wurde für 30 Minuten bei 37°C

2. Material und Methoden
63
inkubiert. Anschließend wurde die optische Dichte der Proben bei 562 nm mit einem
Photometer gemessen. Die Auswertung erfolgte ähnlich wie bei der Bradford-Methode.
2.18.4. Probenvorbereitung für die Elektronenmikroskopie (EM)
Für die Transmissionselektronenmikroskopie wurde zunächst ein Tropfen aus den MVs-
Proben auf ein Trägermaterial (Grid) pipettiert. Anschließend wurde zur Kontrastierung
ein Tropfen aus einer 3%igen Uranylacetat-Lösung auf die Proben pipettiert und
vermischt. Nach einer Inkubation von wenigen Minuten bei RT wurde die Flüssigkeit
mit einem Filterpapier abgezogen. Anschließend wurden die Proben analysiert.
Für die Rasterelektronenmikroskopie wurden die Lm-Proben zunächst mit 5%
Formaldehyd und 1% Glutaraldehyd chemisch fixiert. Anschließend wurden die Proben
durch eine aufsteigende Aceton-Reihe (10-100%) dehydriert und durch Kritische-
Punkt-Trocknung (KPT) mit CO2 in der KPT-Anlage getrocknet. Um eine Aufladung
der Proben zu vermeiden, wurden elektrisch leitende Schichten aus Gold-Palladium auf
die Oberfläche der Proben durch Sputtern aufgebracht. Anschließend wurden die
Proben analysiert.
2.18.5. SDS-Polyacrylamidgel-Elektrophorese (SDS-PAGE)
Durch SDS-Polyacrylamidgel-Elektrophorese werden Proteine in einem Netzwerk aus
Acrylamid/Bisacrylamid elektrophoretisch nach Größe aufgetrennt. Durch Zugabe des
Detergenz SDS (Sodium Dodecyl Sulfate) werden nicht-kovalente Bindungen in den
Proteinen gelöst. Zur Reduktion von Disulfidbrücken wird β-Mercaptoethanol im
Ladepuffer hinzugefügt. Darüber hinaus wurden die Proben vor dem Auftragen auf das
Gel für 5 Minuten bei 95°C aufgekocht. Zuvor wurden die Proben mit 2× Ladepuffer
vermischt. Durch die Einwirkung von SDS, Reduktionsmittel und Hitze werden die
Proteine vollständig denaturiert. Durch die Bindung von SDS-Anionen an die Haupt-
kette erhalten die Proteine eine konstant negative Ladungsverteilung. Daraufhin
wandern die Proteine in einem elektrischen Feld zum positiven Pol.
Die in dieser Arbeit verwendeten Gele bestanden aus einem Sammelgel (4%) und einem
Trenngel (12%). Das Sammelgel dient zum Bündeln der aufgetragenen Proben und
ermöglicht für alle Proteine den gleichen Startpunkt für die Trennung im Trenngel.
Die Auftrennung der Proteine erfolgte in 10 cm × 10 cm große Glasplatten, die in
vertikale Kammern reingelegt wurden. Die Kammern wurden zuvor mit 1×

2. Material und Methoden
64
Elektrophorese-Puffer befüllt. Die Elektrophorese wurde für 90 Minuten bei 125 V und
25 mA/Gel durchgeführt. Die Gele wurden über Nacht in Coomassie-Färbelösung
gefärbt und anschließend mittels Bitzentfärber teilentfärbt.
2.18.6. Western blot-Analyse von MVs
Die Anwesenheit des Autolysins P60 und der beiden Virulenzfaktoren, InlB und LLO,
in den von L. monocytogenes produzierten MVs wurde über Western blot untersucht.
Dies ist ein Verfahren zur Detektion von bestimmten Proteinen in einem Protein-
gemisch über spezifische Antikörperbindung.
Die durch SDS-PAGE aufgetrennten Proteine in den MVs-Proben wurden zunächst
elektrophoretisch aus dem Gel auf eine PVDF-Membran übertragen. Diese wurde zuvor
für 1 Minute in 100% Methanol inkubiert, anschließend in H2O geschwenkt und für
eine weitere Minute in 1× Blot-Puffer inkubiert. Anschließend wurde das Gel in einem
Stapel aus Whatman-Filterpapieren, die zuvor für 1 Minute in 1× Blot-Puffer inkubiert
wurden, auf die PVDF-Membran in der Blotting-Apparatur gelegt. Der Transfer der
Proteine aus dem Gel auf die Membran erfolgte für 90 Minuten bei 50 V und 54
mA/Gel.
Um freie Bindungsstellen auf der PVDF-Membran zu blockieren, wurde diese für 1
Stunde bei RT in 1× TBS-Tween 20-Puffer* geschwenkt. Anschließend wurde die
Membran mit dem primären Antikörper (mouse anti-P60, mouse anti-InlB bzw. mouse
anti-LLO) über Nacht bei 4°C geschwenkt. Die Antikörper wurden wie folgt in 1×
TBS-Tween 20-Puffer* verdünnt: mouse anti-P60 (1:50), mouse anti-InlB (1:10) und
mouse anti-LLO (1:10). Am nächsten Tag wurde die PVDF-Membran 3mal für jeweils
5 Minuten in 1× TBS-Tween 20-Puffer bei RT geschwenkt. Es folgte die Inkubation der
Membran mit dem sekundären Antikörper für 1 Stunde bei RT. Hierfür wurde ein goat
anti-mouse IgG-HRP, der 1:2000 in 1× TBS-Tween 20-Puffer* verdünnt wurde,
verwendet. Anschließend wurde die Membran erneut 3mal für jeweils 5 Minuten in 1×
TBS-Tween 20-Puffer bei RT geschwenkt. Der 1× TBS-Tween 20-Puffer* enthielt 5%
Milch (w/v).
Die Detektion der Proteine erfolgte durch Chemilumineszenz mit dem ECL-System
(GE Healthcare). Hierfür wurde die Membran zunächst für 1 Minute in ECL-Lösung 1
und 2 (1:1) inkubiert. Dadurch wurde die von der Meerrettich-Peroxidase (HRP) kataly-

2. Material und Methoden
65
sierte Reaktion gestartet. Durch anschließendes Auflegen eines Röntgenfilms wurden
die Proteinbanden sichtbar gemacht.
2.18.7. Untersuchung der Internalisierung von MVs in PtK-Zellen
Am Vortag wurden PtK-Zellen in MEM + 10% FKS + 1% NEA in einer 24er-Well
Platte ausgesät. Vor der Zugabe von MVs wurde das Medium der Zellen gewechselt.
Anschließend wurden 5 bzw. 10 µg MVs ins Medium zu den Zellen pipettiert. Als
Negativkontrolle dienten Zellen, die nicht mit MVs inkubiert wurden. Nach einer
Inkubation von 24 Stunden wurde das Medium abgesaugt und die Zellen wurden 3mal
mit jeweils 500 µl PBS/Well gewaschen. Anschließend wurden die Zellen mit 300 µl
PBS/Well abgedeckt, mit einem Zellschaber abgeschabt und in Eppis überführt. Die
Zellen wurden für 5 Minuten bei 13200 rpm und 4°C abzentrifugiert. Der Überstand
wurde verworfen und die Zellen wurden mit 10 µl RIPA-Puffer + Protease-Inhibitoren
lysiert. Hierfür wurden die Proben für 30 Minuten bei 4°C und 300 rpm auf dem
Thermomixer inkubiert. Nach einer Zentrifugation von 10 Minuten bei 13200 rpm und
4°C wurde der Überstand (das Zelllysat) in Eppis überführt.
Zunächst wurde die Proteinkonzentration des Zelllysat mit der BCA-Methode bestimmt.
Danach wurden die Proteine im Zelllysat durch SDS-PAGE nach Größe aufgetrennt,
wobei 15 µg Protein/Ansatz aufgetragen wurden. Anschließend wurden die Proteine
durch Western blot auf eine PVDF-Membran übertragen. Es folgte die Detektion des
Autolysins P60 im Zelllysat über spezifische Antikörperbindung. Hierfür wurden mouse
anti-P60 (1:50) und goat anti-mouse IgG-HRP (1:2000) verwendet. Beide Antikörper
wurden in 1× TBS-Tween 20-Puffer* verdünnt. Um sicherzustellen, dass von jedem
Ansatz die gleiche Proteinmenge aufgetragen wurde, wurde das Haushaltsgen β-Aktin
als Ladekontrolle verwendet. Für die Detektion von β-Aktin wurden rabbit anti-β-Aktin
(1:2000 in 1× TBS-Tween 20-Puffer** verdünnt) und goat anti-rabbit IgG-HRP (1:2000
in 1× TBS-Tween 20-Puffer* verdünnt) verwendet. Der 1× TBS-Tween 20-Puffer*
enthielt 5% Milch (w/v). Der 1× TBS-Tween 20-Puffer** enthielt 5% BSA (w/v).
2.18.8. Hämolyse-Assay
Die hämolytische Aktivität von MVs wurde anhand der Lyse von Schafserythrozyten
untersucht. Zunächst wurde das Schafsblut in ein 50 ml Greiner-Röhrchen überführt und
solange mit PBS gewaschen bis der Überstand klar war. Hierfür wurde das Röhrchen

2. Material und Methoden
66
mehrmals invertiert und für 5 Minuten bei 4000 rpm und 4°C zentrifugiert. Anschlie-
ßend wurde eine 1%ige Erythrozyten-Lösung mit Hämolyse-Puffer hergestellt. Für die
Herstellung des Hämolyse Puffers wurden 50 mM NaH2PO4 × H2O und 150 mM NaCl
abgewogen und in H2O gelöst. Nach dem der pH-Wert auf 6,6 eingestellt wurde,
wurden 0,1% BSA (w/v) und 5 mM DTT hinzugefügt.
In einer 96er-Well Platte wurden zunächst 50 µl/Well Hämolyse-Puffer vorgelegt.
Anschließend wurden 10 µg MVs in einem Volumen von 50 µl im ersten Well pipettiert,
vermischt und in das nächste Well der Reihe überführt. Hierdurch wurde die MVs-Probe
1:2 verdünnt. Diese Verdünnungsreihe wurde mit allen nachfolgenden Wells der Reihe
durchgeführt. Aus dem letzten Well der Reihe wurden 50 µl verworfen. In der 2ten
Reihe wurde eine weitere Verdünnungsreihe mit PBS, das als Negativkontrolle diente,
angesetzt. In einem weiteren Well wurden 50 µl H2O zu den 50 µl Hämolyse-Puffer
pipettiert. Der durch H2O erzielte hämolytische Effekt entspricht der maximalen
Hämolyse (100%-Wert). Nach der Zugabe von 50 µl 1%ige Erythrozyten-Lösung/Well
und Mischen wurde die Platte für 1 Stunde bei 37°C inkubiert. Anschließend wurde die
Platte zentrifugiert und die Überstände in eine 96er-Well Platte überführt. Die optische
Dichte der Überstände wurde bei 405 nm mit einem Photometer gemessen.
2.19. Zellkultur-Methoden
2.19.1. Kultivierung von Zelllinien
Die Epithelzelllinien HEK293 und MDCK-II wurden in 10 ml DMEM + 10% FKS in
10 cm Zellkulturschalen kultiviert. Für die Epithelzelllinie PtK wurde MEM + 10%
FKS + 1% NEA verwendet. Nach Erreichen einer Konfluenz von 80-90% wurde das
Medium abgesaugt, die Zellen 1mal mit PBS gewaschen und mit 3 ml Trypsin/Schale
abgelöst. Die Zellen wurden für die weitere Kultivierung 1:3 in frischem Medium
verdünnt und in Schalen überführt. Die Zellen wurden bei 37°C und 5% CO2 im Brut-
schrank inkubiert. Vor jedem Versuch wurde die Zahl der Epithelzellen mikroskopisch
durch Auszählen der Zellen mittels Neubauer-Zählkammer bestimmt. Hierfür wurden
die Epithelzellen 1mal mit PBS gewaschen, mit Trypsin abgelöst, abzentrifugiert und in
ein bestimmtes Volumen an Medium aufgenommen. Anschließend wurde die Zellzahl
in einem 10 μl Aliquot gezählt und diese pro Milliliter berechnet.
Die BMDMs wurden 48 Stunden vor der Transfektion aus flüssigem Stickstoff auf-
getaut, in BMDM-Medium + 30% M-CSF aufgenommen, gezählt und in 12er-Well

2. Material und Methoden
67
Platten ausgesät. Am nächsten Tag wurde das Medium der Zellen gewechselt. Die
Zellen wurden bei 37°C und 5% CO2 im Brutschrank inkubiert. Das BMDM-Medium
setzte sich wie folgt zusammen: RPMI-Medium + 10% FKS + 50 µM β-Mercapto-
ethanol.
2.19.2. Gewinnung von primären Knochenmark-Makrophagen aus der Maus
Die von den einzelnen sRNAs, den sekretierten RNA-Fraktionen und MVs ausgelöste
Typ-I-IFN Antwort wurde in Knochenmark-Makrophagen (BMDMs = bone marrow-
derived macrophages) untersucht. Hierfür wurden BMDMs aus C57BL/6-Mäusen
isoliert.
Zunächst wurden Femur (Oberschenkel-Knochen) und Tibia (Schienbein) vom Fleisch
befreit und in 70% EtOH reingelegt. Anschließend wurden die Knochen in PBS rein-
gelegt. Unter sterilen Bedingungen wurden die Röhrenknochen an den Enden geöffnet
und die Markhöhle mit kaltem BMDM*-Medium mit Hilfe einer Kanüle durchgespült.
Die Knochenmarkzellen wurden zunächst durch ein Zellsieb passiert und danach durch
eine Zentrifugation von 5 Minuten bei 1500 rpm pelletiert. Anschließend wurden die
Zellen in BMDM*-Medium + 30% M-CSF resuspendiert und in 10 cm unbeschichteten
Petrischalen ausgesät. Die Schalen wurden bei 37°C und 5% CO2 inkubiert.
Die Ausdifferenzierung der Zellen zu BMDMs erfolgte in einem Zeitraum von 6 Tagen,
wobei am 4ten Tag ein Mediumwechsel durchgeführt wurde. Am 6ten Tag wurden die
Zellen durch Kryokonservierung eingefroren. Hierfür wurde zunächst das Medium
abgesaugt. Danach wurden die Zellen mit 5 ml BMDM*-Medium abgedeckt, mit einem
Zellschaber abgeschabt und in ein 50 ml Greiner-Röhrchen überführt. Die Zellen
wurden durch eine Zentrifugation von 5 Minuten bei 1500 rpm pelletiert. Anschließend
wurden die Zellen in FKS + 10% DMSO (Dimethylsulfoxid) resuspendiert. Die Zellen
wurden in Kryoröhrchen überführt und in einem speziellen Einfrierbehälter, der mit 2-
Propanol gefüllt war, zunächst bei -80°C gelagert. Diese Vorgehensweise gewährleistet
ein schrittweises Abkühlen von 1°C/Minute. Danach wurden die Zellen in flüssigem
Stickstoff überführt.
Das BMDM*-Medium setzte sich wie folgt zusammen: RPMI-Medium + 10% FKS +
50 µM β-Mercaptoethanol + 100 U/ml Penicillin + 100 µg/ml Streptomycin.

2. Material und Methoden
68
2.19.2.1. Gewinnung von M-CSF
Der Makrophagen-Differenzierungsfaktor M-CSF (macrophage colony stimulating
factor) wird von mouse fibroblast L929 cells produziert. Dieser unterstützt die Reifung
und das Wachstum von Makrophagen, die aus dem Knochenmark von Mäusen isoliert
werden (Ladner et al, 1988).
Für die Gewinnung von L929-Zellüberstand wurden die Zellen zunächst aus flüssigem
Stickstoff aufgetaut und danach bis zur Konfluenz in DMEM + 10% FKS in 10 cm
Zellkulturschalen kultiviert. Anschließend wurde das Medium abgesaugt, die Zellen
1mal mit PBS gewaschen und mit 3 ml Trypsin/Schale abgelöst. Die Zellen wurden in
DMEM + 5% FKS aufgenommen und auf große Zellkulturflaschen umgesetzt. Die
Zellen wurden 5 bis 6 Tage bei 37°C und 5% CO2 im Brutschrank inkubiert. Anschlie-
ßend wurde der Zellüberstand in 50 ml Greiner-Röhrchen überführt und für 5 Minuten
bei 3000 rpm abzentrifugiert. Der Überstand wurde abgenommen, in 50 ml Greiner-
Röhrchen überführt und bis zur Verwendung bei -20°C gelagert.
2.19.3. Infektion von HEK293-Zellen mit A/PR/8/34 (H1N1)
24 Stunden nach der Transfektion von HEK293-Zellen mit den einzelnen sRNAs
wurden die Zellen zunächst mit PBS++
gewaschen und anschließend mit A/PR/8/34
(H1N1) für 1 Stunde bei RT infiziert. Dabei wurde eine MOI von 0,1 in PBS++
/BSA/P/S
eingesetzt. Nach dem das Virusinokulum abgesaugt wurde, wurden die Zellen mit
DMEM/FKS/P/S + 2 µg/ml Trypsin für 24 Stunden bei 37°C und 5% CO2 inkubiert.
2.19.3.1. Virusquantifizierung durch Focus Forming Assay (FFA)
Der FFA verwendet zur Detektion von infektiösen Viruspartikeln die Anfärbung von
virusspezifischen Antigenen mittels Antikörpern (Ma et al, 2010).
In einer 96er-Well Platte ausgesäte MDCK-II-Zellen wurden zunächst mit PBS++
gewaschen und anschließend mit 50 µl/Well einer 1:3 in PBS++
/BSA/P/S angesetzten
Verdünnungsreihe des virushaltigen Überstandes für 1 Stunde bei RT infiziert. Nach
dem das Virusinokulum abgesaugt wurde, wurden die Zellen mit Avicel-Medium
(DMEM/FKS + 1,25% Avicel + 2 µg/ml Trypsin) für 24 Stunden bei 37°C und 5% CO2
inkubiert. Die Überschichtung der Zellen mit einem halbfesten Overlay-Medium
verhindert die Ausbreitung von infektiösen Viruspartikeln, wodurch lokalisierte
Infektionsherde entstehen. Um diese zu detektieren wurden die Zellen zunächst mit 4%

2. Material und Methoden
69
Paraformaldehyd + 1% Triton X-100 in PBS++
für 1 Stunde bei 4°C fixiert und
permeabilisiert. Danach wurden die Zellen 3mal mit PBS++
+ 0,05% Tween 20
gewaschen. Die Zellen wurden mit 50 µl/Well des primären Antikörpers (mouse anti-
IAV-Nukleoprotein) für 1 Stunde bei RT inkubiert. Dieser wurde zuvor 1:100 in PBS++
+ 3% BSA verdünnt. Anschließend wurden die Zellen erneut 3mal mit PBS++
+ 0,05%
Tween 20 gewaschen. Es folgte die Zugabe von 50 µl/Well des sekundären Antikörpers
(goat anti-mouse IgG-HRP). Dieser wurde zuvor 1:1000 in PBS++
+ 3% BSA verdünnt.
Nach einer Inkubation von 1 Stunde bei RT wurden die Zellen 3mal mit PBS++
+ 0,05%
Tween 20 gewaschen und mit 40 µl/Well AEC staining solution inkubiert. Anschlie-
ßend wurden die Platten gescannt und mit dem Photoshop software package analysiert.
Das PBS++
setzte sich wie folgt zusammen: PBS + 1 mM MgCl2 + 0,9 mM CaCl2.
Das PBS++
/BSA/P/S setzte sich wie folgt zusammen: PBS++
+ 0,2% BSA + 100 U/ml
Penicillin + 100 µg/ml Streptomycin.
2.20. Statistische Analyse
Die statistische Analyse wurde mit dem zweiseitigen ungepaarten t-Test durchgeführt.
Alle Daten sind mit Mittelwert ± Standardabweichung (MW ± SA) dargestellt.

3. Ergebnisse
70
3. Ergebnisse
3.1. Isolierung und Identifizierung der von L. monocytogenes sekretierten RNA
3.1.1. Co-Isolierung von sec-RNA und MVs-RNA
Um die von L. monocytogenes sekretierte RNA zu isolieren, wurde ein Protokoll
entwickelt, das zunächst die Trennung beider RNA-Fraktionen und anschließend die
Isolierung der RNA beinhaltet (Abb. 3.1). Um eine Freisetzung von zellulärer RNA
durch Lyse der Bakterienzellen zu vermeiden, wurden die sekretierten RNA-Fraktionen
aus dem Überstand von Bakterienkulturen isoliert, die sich in der exponentiellen
Wachstumsphase befanden. Des Weiteren wurde ein chemisch definiertes Medium
verwendet, um eine Kontamination durch RNA, die in einem komplexen Medium wie
BHI-Medium vorhanden ist, auszuschließen (Premaratne et al, 1991).
Abb. 3.1: Co-Isolierung von sec-RNA und MVs-RNA. Lm wurde in 2 L Minimalmedium bis OD600nm
von 1 kultiviert und anschließend abzentrifugiert. Der steril filtrierte Überstand wurde durch Ultra-
filtration auf ca. 30 ml eingeengt. Anschließend wurden die MVs durch Ultrazentrifugation pelletiert. Das
MVs-Pellet diente für die Isolierung von MVs-RNA. Der MVs-freie Überstand wurde für die Isolierung
von sec-RNA verwendet.
Nach Abzentrifugation der Bakterienzellen wurde der Überstand steril filtriert und
durch Ultrafiltration mit einer Membran mit einem Cut Off von 30 kDa von 2 L auf ca.
30 ml eingeengt. Dies ist ein Verfahren um bestimmte Moleküle, abhängig von der
Porengröße der dabei verwendeten Membran, zu konzentrieren. Dabei wird die
Flüssigkeit aus der Lösung entfernt. Durch anschließende Ultrazentrifugation wurden
die MVs in dem eingeengten Überstand von L. monocytogenes-Kulturen pelletiert. Das
MVs-Pellet wurde in PBS aufgenommen. Nach Bestimmung der Proteinkonzentration
mit der Bradford-Methode erfolgte die Isolierung der MVs-RNA. Zuvor wurde ein
RNase-Verdau durchgeführt, um die an der äußeren Oberfläche von MVs assoziierte
RNA zu entfernen. Aus dem eingeengten MVs-freien Überstand von L. monocytogenes-
Kulturen wurde die sec-RNA isoliert.

3. Ergebnisse
71
Aus 2 L L. monocytogenes-Kulturen wurden anhand dieses Protokolls 150-200 µg MVs
isoliert. Daraus wurden in einem weiteren Schritt ca. 12 µg MVs-RNA isoliert. Die
Konzentration an sec-RNA, die aus 2 L L. monocytogenes-Kulturen isoliert wurde,
beträgt ca. 120 µg.
3.1.2. Profilanalyse von sec-RNA und MVs-RNA
Nach Isolierung und Quantifizierung von sec-RNA und MVs-RNA wurde eine
Profilanalyse der beiden sekretierten RNA-Fraktionen mittels Agilent 2100 Bioanalyzer
durchgeführt (Abb. 3.2). Diese Technologie liefert eine visuelle Darstellung der Grö-
ßenverteilung von RNAs, die in einer RNA-Population vorhandenen sind. Die RNA
wird, ähnlich herkömmlicher Standardverfahren zur RNA-Analyse, elektrophoretisch
nach Größe aufgetrennt, wobei eine Gelmatrix als Trägermaterial dient. Dabei wird auf
der y-Achse die RNA-Menge in FU (fluorescence units) angegeben, während die x-
Achse die RNA-Länge in Nukleotiden (Nt) angibt.
(A) (B) (C)
Abb. 3.2: Profilanalyse von sec-RNA, MVs-RNA und zelluläre RNA. Die Profilanalyse der 3 RNA-
Fraktionen von Lm wurde mit dem Agilent 2100 Bioanalyzer durchgeführt. Hierdurch wurde die Menge
(FU) und die Länge (Nt) der RNAs in den beiden sekretierten RNA-Fraktionen von Lm erstmals
visualisiert. Der Peak zwischen 25-200 Nt zeigt die Anwesenheit von kleinen nicht-kodierenden RNAs in
sec-RNA (A) und MVs-RNA (B). Die beiden Peaks in der zellulären RNA zeigen die ribosomalen RNAs,
16S- und 23S-rRNA (C).
Das typische Bild einer zellulären RNA-Präparation zeigt die beiden ribosomalen
RNAs, die 16S- und die 23S-rRNA (Abb. 3.2C). Die beiden sekretierten RNA-
Fraktionen, sec-RNA und MVs-RNA, hingegen weisen ein völlig anderes RNA-Profil
auf (Abb. 3.2A und 3.2B). In beide RNA-Fraktionen sind kleine nicht-kodierende
RNAs mit einer Größe, die weniger als 200 Nt beträgt, zu sehen. Diese überwiegen in
MVs-RNA, während in sec-RNA auch längere RNAs, darunter ribosomale RNAs, zu
sehen sind.
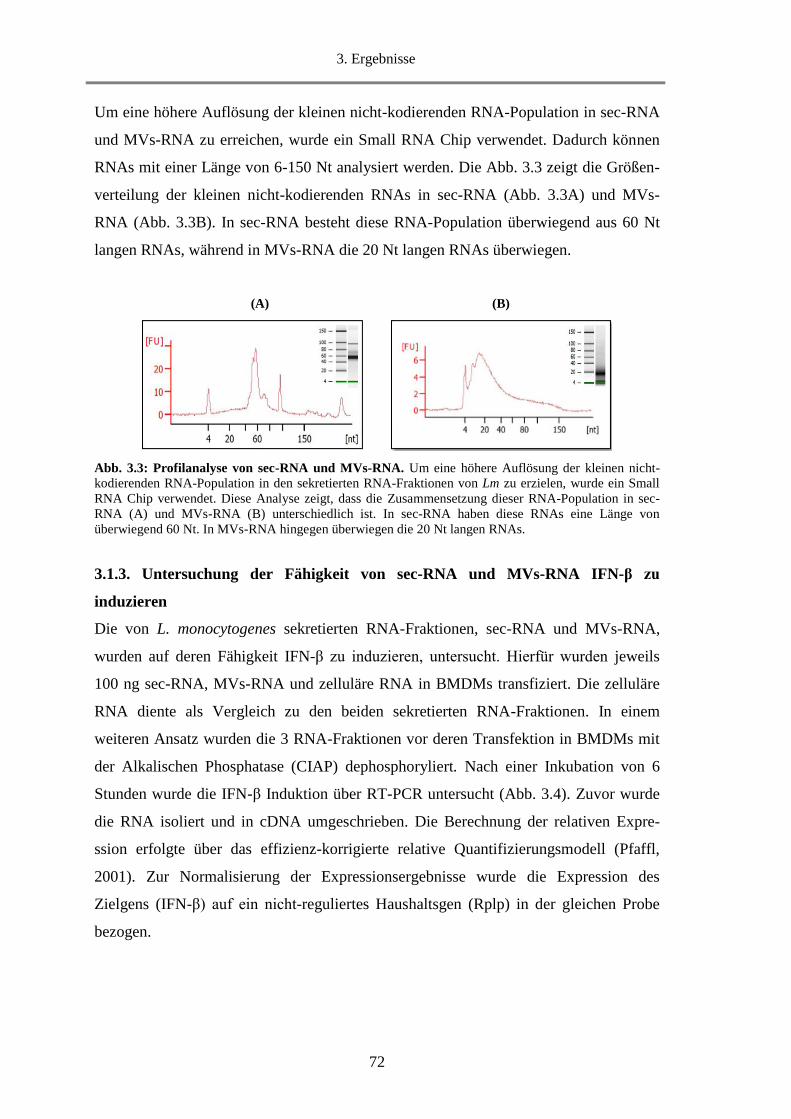
3. Ergebnisse
72
Um eine höhere Auflösung der kleinen nicht-kodierenden RNA-Population in sec-RNA
und MVs-RNA zu erreichen, wurde ein Small RNA Chip verwendet. Dadurch können
RNAs mit einer Länge von 6-150 Nt analysiert werden. Die Abb. 3.3 zeigt die Größen-
verteilung der kleinen nicht-kodierenden RNAs in sec-RNA (Abb. 3.3A) und MVs-
RNA (Abb. 3.3B). In sec-RNA besteht diese RNA-Population überwiegend aus 60 Nt
langen RNAs, während in MVs-RNA die 20 Nt langen RNAs überwiegen.
(A) (B)
Abb. 3.3: Profilanalyse von sec-RNA und MVs-RNA. Um eine höhere Auflösung der kleinen nicht-
kodierenden RNA-Population in den sekretierten RNA-Fraktionen von Lm zu erzielen, wurde ein Small
RNA Chip verwendet. Diese Analyse zeigt, dass die Zusammensetzung dieser RNA-Population in sec-
RNA (A) und MVs-RNA (B) unterschiedlich ist. In sec-RNA haben diese RNAs eine Länge von
überwiegend 60 Nt. In MVs-RNA hingegen überwiegen die 20 Nt langen RNAs.
3.1.3. Untersuchung der Fähigkeit von sec-RNA und MVs-RNA IFN-β zu
induzieren
Die von L. monocytogenes sekretierten RNA-Fraktionen, sec-RNA und MVs-RNA,
wurden auf deren Fähigkeit IFN-β zu induzieren, untersucht. Hierfür wurden jeweils
100 ng sec-RNA, MVs-RNA und zelluläre RNA in BMDMs transfiziert. Die zelluläre
RNA diente als Vergleich zu den beiden sekretierten RNA-Fraktionen. In einem
weiteren Ansatz wurden die 3 RNA-Fraktionen vor deren Transfektion in BMDMs mit
der Alkalischen Phosphatase (CIAP) dephosphoryliert. Nach einer Inkubation von 6
Stunden wurde die IFN-β Induktion über RT-PCR untersucht (Abb. 3.4). Zuvor wurde
die RNA isoliert und in cDNA umgeschrieben. Die Berechnung der relativen Expre-
ssion erfolgte über das effizienz-korrigierte relative Quantifizierungsmodell (Pfaffl,
2001). Zur Normalisierung der Expressionsergebnisse wurde die Expression des
Zielgens (IFN-β) auf ein nicht-reguliertes Haushaltsgen (Rplp) in der gleichen Probe
bezogen.
3. Ergebnisse
73
Abb. 3.4: IFN-β Induktion in BMDMs durch sec-RNA und MVs-RNA. Jeweils 100 ng aus den 3
RNA-Fraktionen, sec-RNA, MVs-RNA und zelluläre RNA, von Lm wurden in BMDMs transfiziert. 6
Stunden später wurde die IFN-β Induktion über RT-PCR untersucht. Sec-RNA und MVs-RNA haben zu
einem signifikanten Anstieg der IFN-β Induktion im Vergleich zu der zellulären RNA geführt. Die
Dephosphorylierung der sekretierten RNA-Fraktionen vor deren Transfektion in BMDMs hat zu einer
signifikanten Reduktion der IFN-β Induktion geführt. (*P ˂0,05; **P ˂0,01; ns = nicht signifikant)
Die Abb. 3.4 zeigt, dass nach Transfektion der 3 RNA-Fraktionen in BMDMs sec-RNA
die höchste IFN-β Antwort ausgelöst hat, gefolgt von MVs-RNA. Beide sekretierte
RNA-Fraktionen haben zu einem signifikanten Anstieg der IFN-β Induktion im
Vergleich zu der zellulären RNA geführt. Diese zeigen ein Vielfach höheres IFN-β
Induktionspotential als die zelluläre RNA. Die in BMDMs ausgelöste IFN-β Antwort ist
durch sec-RNA ca. 70fach und durch MVs-RNA ca. 20fach höher im Vergleich zu der
zellulären RNA. Die Dephosphorylierung von sec-RNA und MVs-RNA mit der
Alkalischen Phosphatase (CIAP) vor deren Transfektion in BMDMs hat zu einer
signifikanten Reduktion der IFN-β Induktion geführt.
3.1.4. Analyse der Zusammensetzung von sec-RNA und MVs-RNA
Um die Zusammensetzung der sekretierten RNA-Fraktionen von L. monocytogenes zu
identifizieren, wurde eine Sequenzierung durchgeführt. Die zelluläre RNA, die als
Vergleich diente, wurde ebenfalls sequenziert. Für die Sequenzierung wurden von jeder
RNA-Fraktion 3 biologische Replikate verwendet. Die Abb. 3.5 zeigt die Anzahl der
total reads, die an das Referenzgenom von L. monocytogenes gemappt haben (Glaser et
al, 2001). Diese weisen eine hohe Reproduzierbarkeit auf.
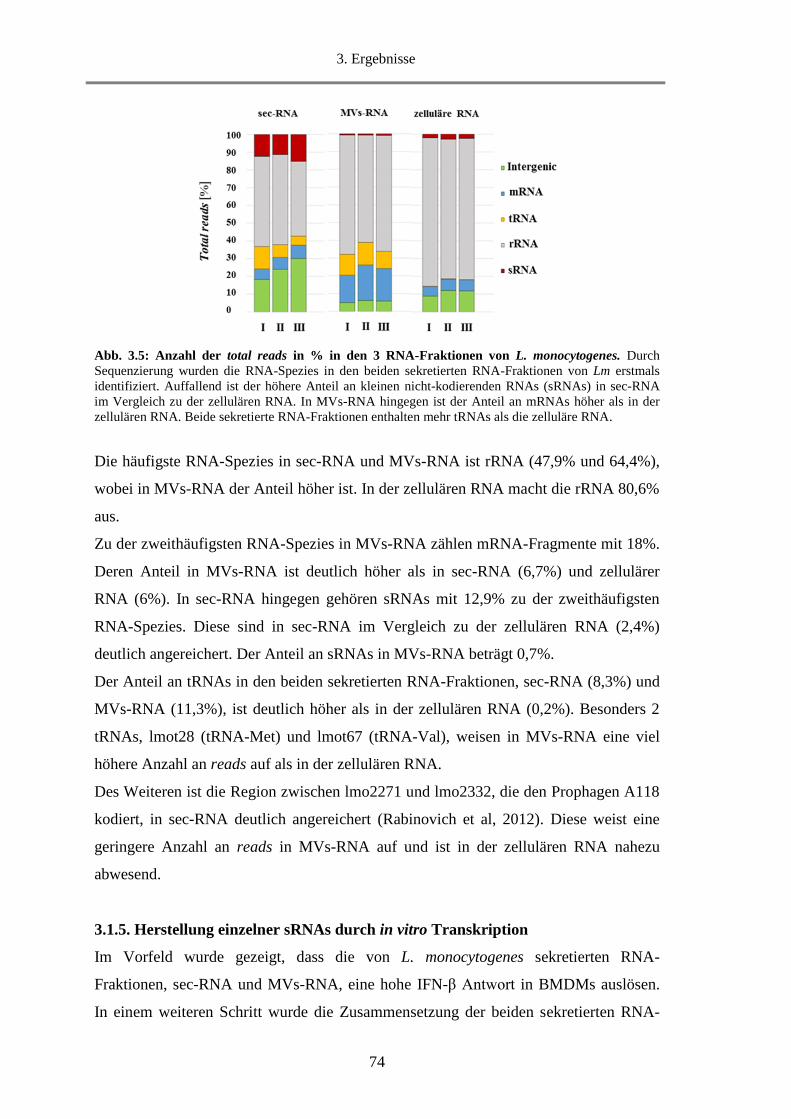
3. Ergebnisse
74
Abb. 3.5: Anzahl der total reads in % in den 3 RNA-Fraktionen von L. monocytogenes. Durch
Sequenzierung wurden die RNA-Spezies in den beiden sekretierten RNA-Fraktionen von Lm erstmals
identifiziert. Auffallend ist der höhere Anteil an kleinen nicht-kodierenden RNAs (sRNAs) in sec-RNA
im Vergleich zu der zellulären RNA. In MVs-RNA hingegen ist der Anteil an mRNAs höher als in der
zellulären RNA. Beide sekretierte RNA-Fraktionen enthalten mehr tRNAs als die zelluläre RNA.
Die häufigste RNA-Spezies in sec-RNA und MVs-RNA ist rRNA (47,9% und 64,4%),
wobei in MVs-RNA der Anteil höher ist. In der zellulären RNA macht die rRNA 80,6%
aus.
Zu der zweithäufigsten RNA-Spezies in MVs-RNA zählen mRNA-Fragmente mit 18%.
Deren Anteil in MVs-RNA ist deutlich höher als in sec-RNA (6,7%) und zellulärer
RNA (6%). In sec-RNA hingegen gehören sRNAs mit 12,9% zu der zweithäufigsten
RNA-Spezies. Diese sind in sec-RNA im Vergleich zu der zellulären RNA (2,4%)
deutlich angereichert. Der Anteil an sRNAs in MVs-RNA beträgt 0,7%.
Der Anteil an tRNAs in den beiden sekretierten RNA-Fraktionen, sec-RNA (8,3%) und
MVs-RNA (11,3%), ist deutlich höher als in der zellulären RNA (0,2%). Besonders 2
tRNAs, lmot28 (tRNA-Met) und lmot67 (tRNA-Val), weisen in MVs-RNA eine viel
höhere Anzahl an reads auf als in der zellulären RNA.
Des Weiteren ist die Region zwischen lmo2271 und lmo2332, die den Prophagen A118
kodiert, in sec-RNA deutlich angereichert (Rabinovich et al, 2012). Diese weist eine
geringere Anzahl an reads in MVs-RNA auf und ist in der zellulären RNA nahezu
abwesend.
3.1.5. Herstellung einzelner sRNAs durch in vitro Transkription
Im Vorfeld wurde gezeigt, dass die von L. monocytogenes sekretierten RNA-
Fraktionen, sec-RNA und MVs-RNA, eine hohe IFN-β Antwort in BMDMs auslösen.
In einem weiteren Schritt wurde die Zusammensetzung der beiden sekretierten RNA-

3. Ergebnisse
75
Fraktionen identifiziert. Als nächstes wurde untersucht welchen Beitrag einzelne in sec-
RNA und MVs-RNA identifizierte RNAs an der IFN-β Antwort haben. Hierfür wurden
insgesamt 25 RNA-Kandidaten, 23 sRNAs und 2 tRNAs, ausgesucht und auf deren
Fähigkeit IFN-β zu induzieren, untersucht. Hierbei handelt es sich um sRNAs, die in
sec-RNA eine höhere Anzahl an reads aufweisen als in der zellulären RNA (rli48, rli62,
rliG, SRP, ssrA und ssrS). Diese zählen in MVs-RNA zu den sRNAs mit der höchsten
Anzahl an reads. Darüber hinaus wurden auch solche sRNAs ausgesucht, die in beiden
sekretierten RNA-Fraktionen eine geringe Anzahl an reads aufweisen (rli100, rli105
und rli114). Ein weiteres Kriterium für die Auswahl der sRNAs war deren Trans-
kriptionslevel während des intra- bzw. extrazellulären Wachstums von L. monocyto-
genes. So zählen rli32, rli47, rli48, rli51, rli98, rli105 und rliG zu den sRNAs deren
Transkriptionslevel während des intrazellulären Wachstums höher ist. Andere hingegen,
wie rli62, rli87, rli90, rli99, rli108, rli114, rnpB, ssrA und ssrS, weisen während des
extrazellulären Wachstums einen höheren Transkriptionslevel auf (Mraheil et al, 2011).
Des Weiteren wurden 2 tRNAs ausgesucht, die in den sekretierten RNA-Fraktionen
häufiger vertreten sind als in der zellulären RNA, zum einen lmot11 (tRNA-Serin) in
sec-RNA und zum anderen lmot67 (tRNA-Val) in MVs-RNA.
Der erste Schritt bestand darin die 25 ausgesuchten RNA-Kandidaten herzustellen. Die
Abb. 3.6 zeigt die einzelnen Schritte, die für die Herstellung der RNAs durchgeführt
worden sind. Die RNAs wurden enzymatisch durch in vitro Transkription mit der T7-
RNA-Polymerase hergestellt. Diese katalysiert die Synthese von ssRNA in 5`-3`-
Richtung von einem DNA-template aus, das stromabwärts vom T7-spezifischen
Promotor (5`-taatacgactcactataGg-3`) liegt. Die T7-RNA-Polymerase bevorzugt an der
Position +1 ein G (in der Sequenz groß geschrieben), daher beginnen die RNA-
Transkripte mit 5´-GG. Darüber hinaus besitzen die so hergestellten RNAs am 5`-Ende
eine Triphosphat-Gruppe.

3. Ergebnisse
76
Abb. 3.6: Schematische Darstellung der einzelnen Schritte für die Herstellung der 25 RNA-
Kandidaten. Im ersten Schritt wurde das DNA-template durch PCR hergestellt. Nach Aufreinigung der
PCR-Produkte wurde die in vitro Transkription (IVT) mit der T7-RNA-Polymerase durchgeführt.
Anschließend wurden die RNAs in denaturierende Polyacrylamidgele aufgetrennt. Im letzten Schritt
wurden die RNAs aus dem Gel herausgeschnitten und aufgereinigt.
Die DNA-templates für die Herstellung der einzelnen RNAs durch in vitro
Transkription wurden durch PCR hergestellt. Dabei enthielt der Vorwärtsprimer am 5`-
Ende die Sequenz des Promotors der T7-RNA-Polymerase. Die PCR-Produkte wurden
zunächst in 1%ige und 2%ige Agarosegele aufgetrennt und danach visualisiert und
fotografiert (Abb. 3.7). Nach Aufreinigung der PCR-Produkte folgte die in vitro
Transkription der DNA in RNA. Die hergestellten RNAs wurden unter denaturierenden
Bedingungen (7 M Harnstoff) in 10%ige und 12%ige Polyacrylamidgele aufgetrennt
(Abb. 3.8). Durch die Zugabe von Harnstoff wurde die Bildung von Sekundärstrukturen
verhindert, so dass die RNAs nach Länge aufgetrennt wurden.
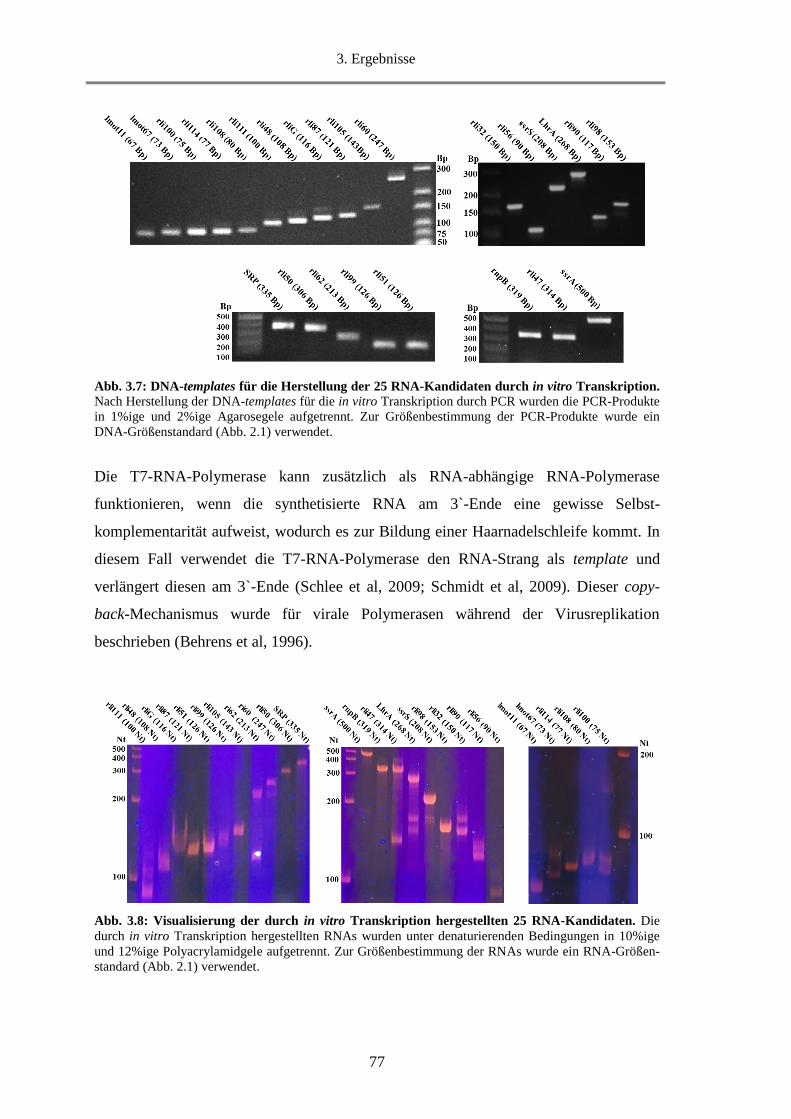
3. Ergebnisse
77
Abb. 3.7: DNA-templates für die Herstellung der 25 RNA-Kandidaten durch in vitro Transkription.
Nach Herstellung der DNA-templates für die in vitro Transkription durch PCR wurden die PCR-Produkte
in 1%ige und 2%ige Agarosegele aufgetrennt. Zur Größenbestimmung der PCR-Produkte wurde ein
DNA-Größenstandard (Abb. 2.1) verwendet.
Die T7-RNA-Polymerase kann zusätzlich als RNA-abhängige RNA-Polymerase
funktionieren, wenn die synthetisierte RNA am 3`-Ende eine gewisse Selbst-
komplementarität aufweist, wodurch es zur Bildung einer Haarnadelschleife kommt. In
diesem Fall verwendet die T7-RNA-Polymerase den RNA-Strang als template und
verlängert diesen am 3`-Ende (Schlee et al, 2009; Schmidt et al, 2009). Dieser copy-
back-Mechanismus wurde für virale Polymerasen während der Virusreplikation
beschrieben (Behrens et al, 1996).
Abb. 3.8: Visualisierung der durch in vitro Transkription hergestellten 25 RNA-Kandidaten. Die
durch in vitro Transkription hergestellten RNAs wurden unter denaturierenden Bedingungen in 10%ige
und 12%ige Polyacrylamidgele aufgetrennt. Zur Größenbestimmung der RNAs wurde ein RNA-Größen-
standard (Abb. 2.1) verwendet.
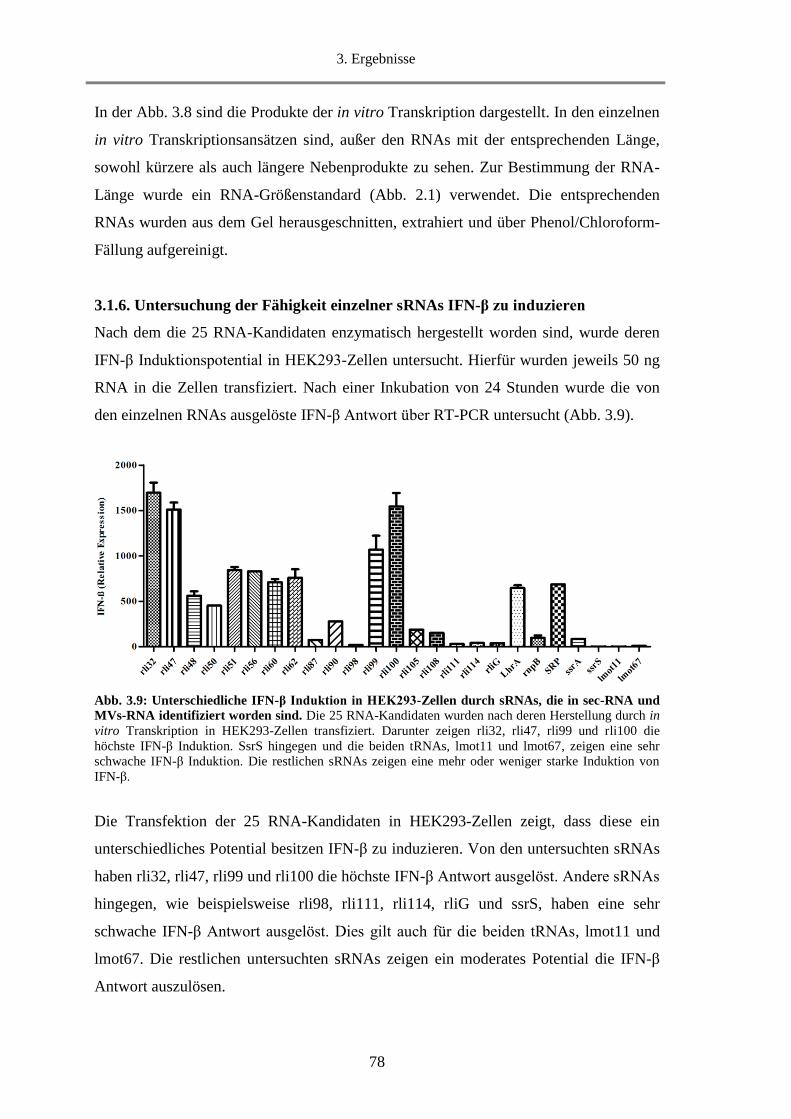
3. Ergebnisse
78
In der Abb. 3.8 sind die Produkte der in vitro Transkription dargestellt. In den einzelnen
in vitro Transkriptionsansätzen sind, außer den RNAs mit der entsprechenden Länge,
sowohl kürzere als auch längere Nebenprodukte zu sehen. Zur Bestimmung der RNA-
Länge wurde ein RNA-Größenstandard (Abb. 2.1) verwendet. Die entsprechenden
RNAs wurden aus dem Gel herausgeschnitten, extrahiert und über Phenol/Chloroform-
Fällung aufgereinigt.
3.1.6. Untersuchung der Fähigkeit einzelner sRNAs IFN-β zu induzieren
Nach dem die 25 RNA-Kandidaten enzymatisch hergestellt worden sind, wurde deren
IFN-β Induktionspotential in HEK293-Zellen untersucht. Hierfür wurden jeweils 50 ng
RNA in die Zellen transfiziert. Nach einer Inkubation von 24 Stunden wurde die von
den einzelnen RNAs ausgelöste IFN-β Antwort über RT-PCR untersucht (Abb. 3.9).
Abb. 3.9: Unterschiedliche IFN-β Induktion in HEK293-Zellen durch sRNAs, die in sec-RNA und
MVs-RNA identifiziert worden sind. Die 25 RNA-Kandidaten wurden nach deren Herstellung durch in
vitro Transkription in HEK293-Zellen transfiziert. Darunter zeigen rli32, rli47, rli99 und rli100 die
höchste IFN-β Induktion. SsrS hingegen und die beiden tRNAs, lmot11 und lmot67, zeigen eine sehr
schwache IFN-β Induktion. Die restlichen sRNAs zeigen eine mehr oder weniger starke Induktion von
IFN-β.
Die Transfektion der 25 RNA-Kandidaten in HEK293-Zellen zeigt, dass diese ein
unterschiedliches Potential besitzen IFN-β zu induzieren. Von den untersuchten sRNAs
haben rli32, rli47, rli99 und rli100 die höchste IFN-β Antwort ausgelöst. Andere sRNAs
hingegen, wie beispielsweise rli98, rli111, rli114, rliG und ssrS, haben eine sehr
schwache IFN-β Antwort ausgelöst. Dies gilt auch für die beiden tRNAs, lmot11 und
lmot67. Die restlichen untersuchten sRNAs zeigen ein moderates Potential die IFN-β
Antwort auszulösen.
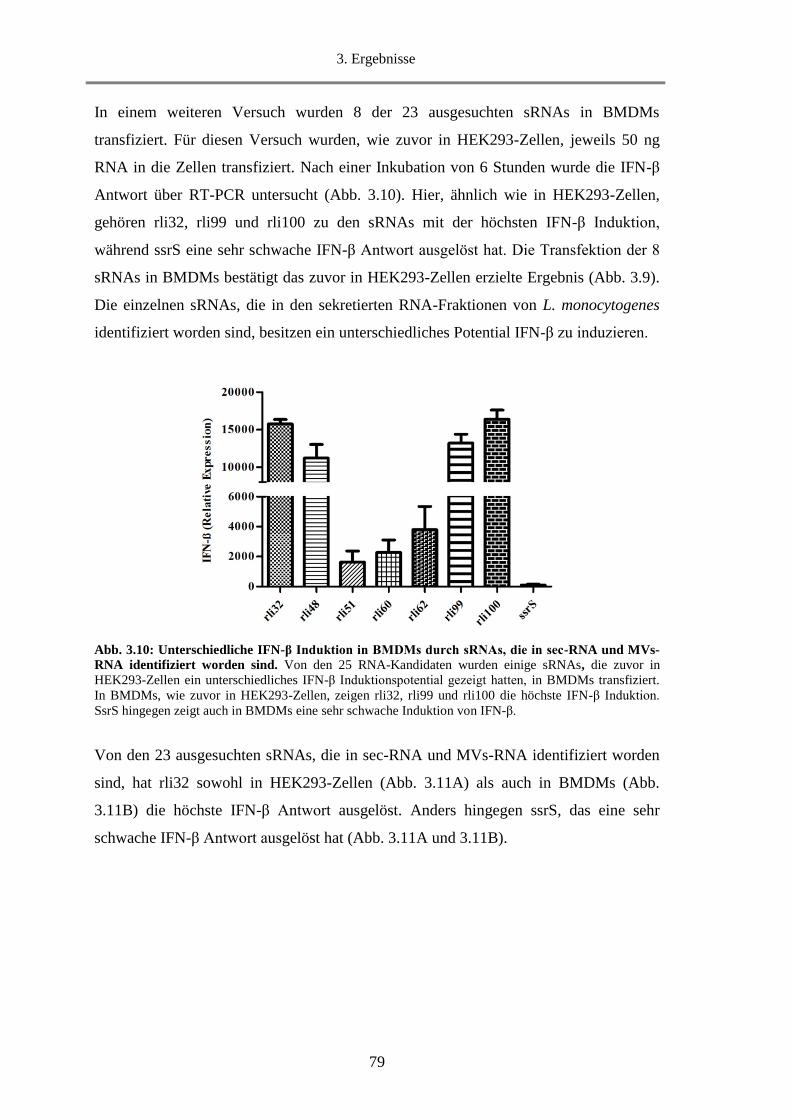
3. Ergebnisse
79
In einem weiteren Versuch wurden 8 der 23 ausgesuchten sRNAs in BMDMs
transfiziert. Für diesen Versuch wurden, wie zuvor in HEK293-Zellen, jeweils 50 ng
RNA in die Zellen transfiziert. Nach einer Inkubation von 6 Stunden wurde die IFN-β
Antwort über RT-PCR untersucht (Abb. 3.10). Hier, ähnlich wie in HEK293-Zellen,
gehören rli32, rli99 und rli100 zu den sRNAs mit der höchsten IFN-β Induktion,
während ssrS eine sehr schwache IFN-β Antwort ausgelöst hat. Die Transfektion der 8
sRNAs in BMDMs bestätigt das zuvor in HEK293-Zellen erzielte Ergebnis (Abb. 3.9).
Die einzelnen sRNAs, die in den sekretierten RNA-Fraktionen von L. monocytogenes
identifiziert worden sind, besitzen ein unterschiedliches Potential IFN-β zu induzieren.
Abb. 3.10: Unterschiedliche IFN-β Induktion in BMDMs durch sRNAs, die in sec-RNA und MVs-
RNA identifiziert worden sind. Von den 25 RNA-Kandidaten wurden einige sRNAs, die zuvor in
HEK293-Zellen ein unterschiedliches IFN-β Induktionspotential gezeigt hatten, in BMDMs transfiziert.
In BMDMs, wie zuvor in HEK293-Zellen, zeigen rli32, rli99 und rli100 die höchste IFN-β Induktion.
SsrS hingegen zeigt auch in BMDMs eine sehr schwache Induktion von IFN-β.
Von den 23 ausgesuchten sRNAs, die in sec-RNA und MVs-RNA identifiziert worden
sind, hat rli32 sowohl in HEK293-Zellen (Abb. 3.11A) als auch in BMDMs (Abb.
3.11B) die höchste IFN-β Antwort ausgelöst. Anders hingegen ssrS, das eine sehr
schwache IFN-β Antwort ausgelöst hat (Abb. 3.11A und 3.11B).
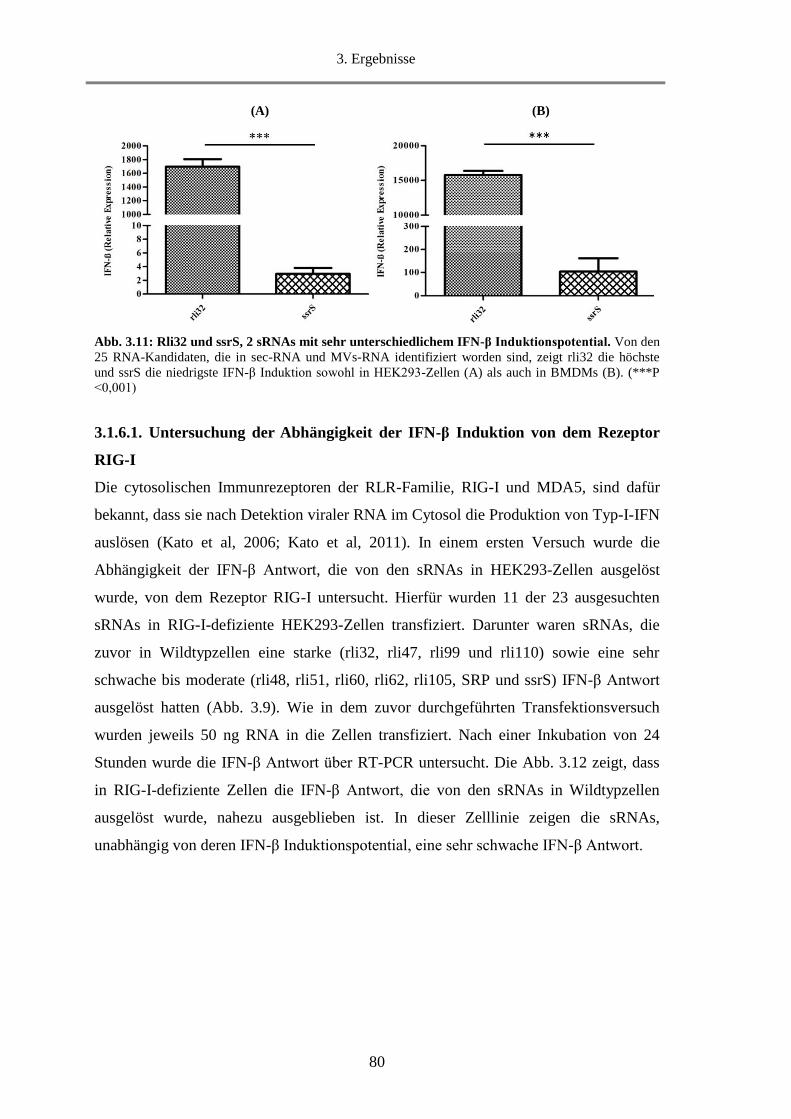
3. Ergebnisse
80
(A) (B)
Abb. 3.11: Rli32 und ssrS, 2 sRNAs mit sehr unterschiedlichem IFN-β Induktionspotential. Von den
25 RNA-Kandidaten, die in sec-RNA und MVs-RNA identifiziert worden sind, zeigt rli32 die höchste
und ssrS die niedrigste IFN-β Induktion sowohl in HEK293-Zellen (A) als auch in BMDMs (B). (***P
˂0,001)
3.1.6.1. Untersuchung der Abhängigkeit der IFN-β Induktion von dem Rezeptor
RIG-I
Die cytosolischen Immunrezeptoren der RLR-Familie, RIG-I und MDA5, sind dafür
bekannt, dass sie nach Detektion viraler RNA im Cytosol die Produktion von Typ-I-IFN
auslösen (Kato et al, 2006; Kato et al, 2011). In einem ersten Versuch wurde die
Abhängigkeit der IFN-β Antwort, die von den sRNAs in HEK293-Zellen ausgelöst
wurde, von dem Rezeptor RIG-I untersucht. Hierfür wurden 11 der 23 ausgesuchten
sRNAs in RIG-I-defiziente HEK293-Zellen transfiziert. Darunter waren sRNAs, die
zuvor in Wildtypzellen eine starke (rli32, rli47, rli99 und rli110) sowie eine sehr
schwache bis moderate (rli48, rli51, rli60, rli62, rli105, SRP und ssrS) IFN-β Antwort
ausgelöst hatten (Abb. 3.9). Wie in dem zuvor durchgeführten Transfektionsversuch
wurden jeweils 50 ng RNA in die Zellen transfiziert. Nach einer Inkubation von 24
Stunden wurde die IFN-β Antwort über RT-PCR untersucht. Die Abb. 3.12 zeigt, dass
in RIG-I-defiziente Zellen die IFN-β Antwort, die von den sRNAs in Wildtypzellen
ausgelöst wurde, nahezu ausgeblieben ist. In dieser Zelllinie zeigen die sRNAs,
unabhängig von deren IFN-β Induktionspotential, eine sehr schwache IFN-β Antwort.
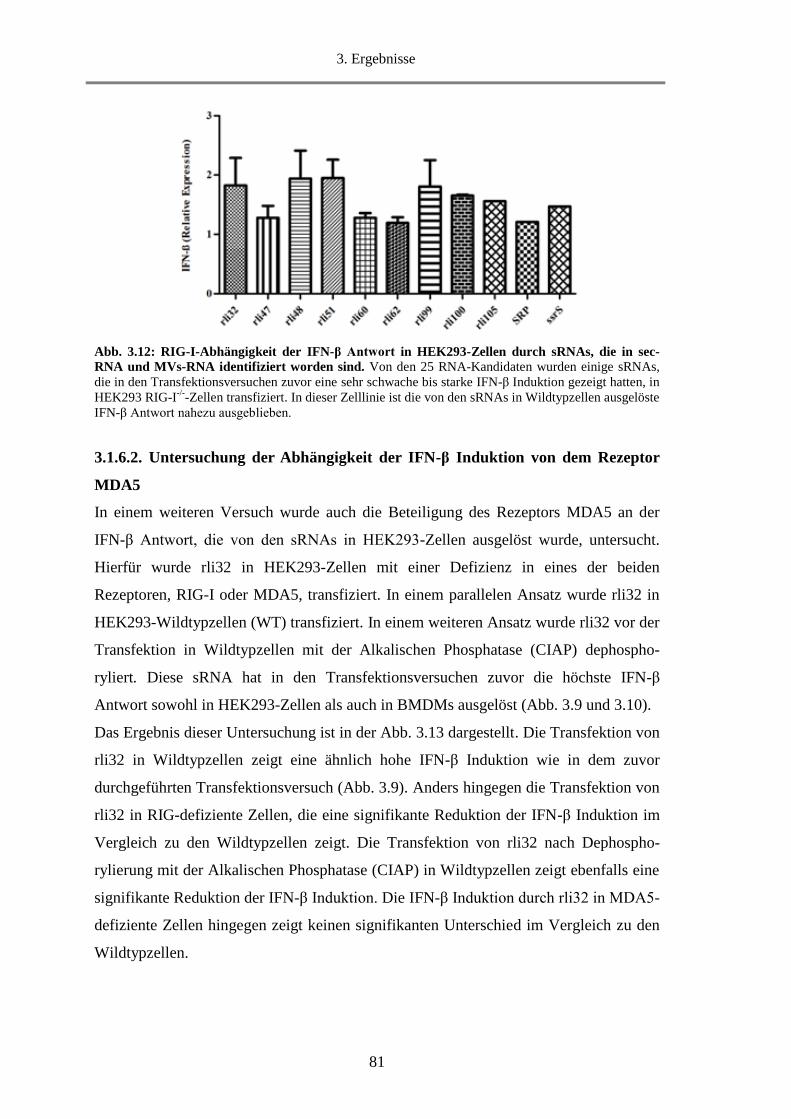
3. Ergebnisse
81
Abb. 3.12: RIG-I-Abhängigkeit der IFN-β Antwort in HEK293-Zellen durch sRNAs, die in sec-
RNA und MVs-RNA identifiziert worden sind. Von den 25 RNA-Kandidaten wurden einige sRNAs,
die in den Transfektionsversuchen zuvor eine sehr schwache bis starke IFN-β Induktion gezeigt hatten, in
HEK293 RIG-I-/-
-Zellen transfiziert. In dieser Zelllinie ist die von den sRNAs in Wildtypzellen ausgelöste
IFN-β Antwort nahezu ausgeblieben.
3.1.6.2. Untersuchung der Abhängigkeit der IFN-β Induktion von dem Rezeptor
MDA5
In einem weiteren Versuch wurde auch die Beteiligung des Rezeptors MDA5 an der
IFN-β Antwort, die von den sRNAs in HEK293-Zellen ausgelöst wurde, untersucht.
Hierfür wurde rli32 in HEK293-Zellen mit einer Defizienz in eines der beiden
Rezeptoren, RIG-I oder MDA5, transfiziert. In einem parallelen Ansatz wurde rli32 in
HEK293-Wildtypzellen (WT) transfiziert. In einem weiteren Ansatz wurde rli32 vor der
Transfektion in Wildtypzellen mit der Alkalischen Phosphatase (CIAP) dephospho-
ryliert. Diese sRNA hat in den Transfektionsversuchen zuvor die höchste IFN-β
Antwort sowohl in HEK293-Zellen als auch in BMDMs ausgelöst (Abb. 3.9 und 3.10).
Das Ergebnis dieser Untersuchung ist in der Abb. 3.13 dargestellt. Die Transfektion von
rli32 in Wildtypzellen zeigt eine ähnlich hohe IFN-β Induktion wie in dem zuvor
durchgeführten Transfektionsversuch (Abb. 3.9). Anders hingegen die Transfektion von
rli32 in RIG-defiziente Zellen, die eine signifikante Reduktion der IFN-β Induktion im
Vergleich zu den Wildtypzellen zeigt. Die Transfektion von rli32 nach Dephospho-
rylierung mit der Alkalischen Phosphatase (CIAP) in Wildtypzellen zeigt ebenfalls eine
signifikante Reduktion der IFN-β Induktion. Die IFN-β Induktion durch rli32 in MDA5-
defiziente Zellen hingegen zeigt keinen signifikanten Unterschied im Vergleich zu den
Wildtypzellen.
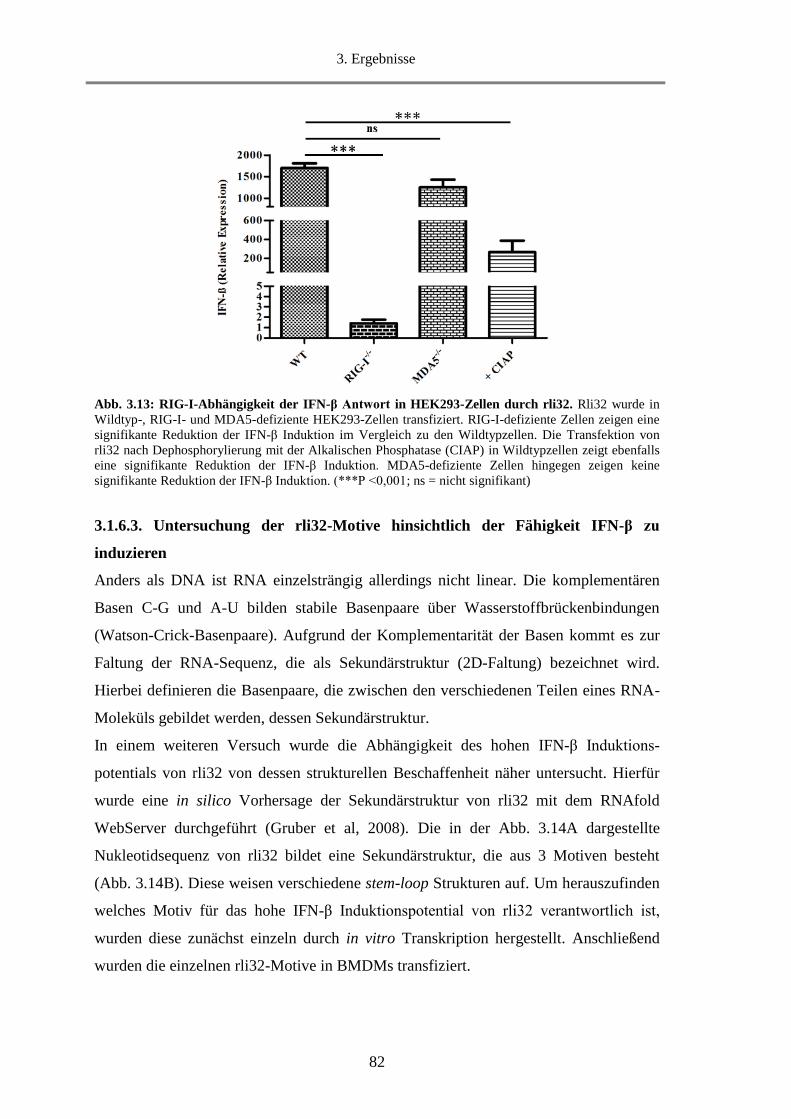
3. Ergebnisse
82
Abb. 3.13: RIG-I-Abhängigkeit der IFN-β Antwort in HEK293-Zellen durch rli32. Rli32 wurde in
Wildtyp-, RIG-I- und MDA5-defiziente HEK293-Zellen transfiziert. RIG-I-defiziente Zellen zeigen eine
signifikante Reduktion der IFN-β Induktion im Vergleich zu den Wildtypzellen. Die Transfektion von
rli32 nach Dephosphorylierung mit der Alkalischen Phosphatase (CIAP) in Wildtypzellen zeigt ebenfalls
eine signifikante Reduktion der IFN-β Induktion. MDA5-defiziente Zellen hingegen zeigen keine
signifikante Reduktion der IFN-β Induktion. (***P ˂0,001; ns = nicht signifikant)
3.1.6.3. Untersuchung der rli32-Motive hinsichtlich der Fähigkeit IFN-β zu
induzieren
Anders als DNA ist RNA einzelsträngig allerdings nicht linear. Die komplementären
Basen C-G und A-U bilden stabile Basenpaare über Wasserstoffbrückenbindungen
(Watson-Crick-Basenpaare). Aufgrund der Komplementarität der Basen kommt es zur
Faltung der RNA-Sequenz, die als Sekundärstruktur (2D-Faltung) bezeichnet wird.
Hierbei definieren die Basenpaare, die zwischen den verschiedenen Teilen eines RNA-
Moleküls gebildet werden, dessen Sekundärstruktur.
In einem weiteren Versuch wurde die Abhängigkeit des hohen IFN-β Induktions-
potentials von rli32 von dessen strukturellen Beschaffenheit näher untersucht. Hierfür
wurde eine in silico Vorhersage der Sekundärstruktur von rli32 mit dem RNAfold
WebServer durchgeführt (Gruber et al, 2008). Die in der Abb. 3.14A dargestellte
Nukleotidsequenz von rli32 bildet eine Sekundärstruktur, die aus 3 Motiven besteht
(Abb. 3.14B). Diese weisen verschiedene stem-loop Strukturen auf. Um herauszufinden
welches Motiv für das hohe IFN-β Induktionspotential von rli32 verantwortlich ist,
wurden diese zunächst einzeln durch in vitro Transkription hergestellt. Anschließend
wurden die einzelnen rli32-Motive in BMDMs transfiziert.
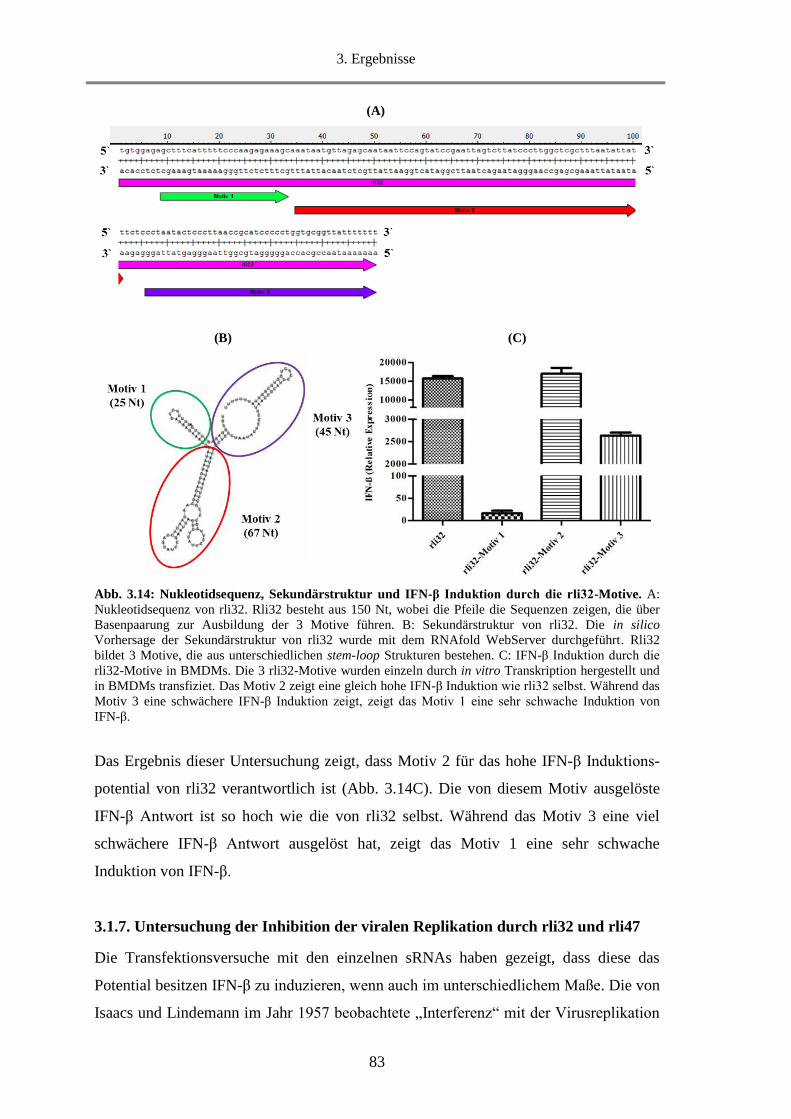
3. Ergebnisse
83
(A)
(B) (C)
Abb. 3.14: Nukleotidsequenz, Sekundärstruktur und IFN-β Induktion durch die rli32-Motive. A:
Nukleotidsequenz von rli32. Rli32 besteht aus 150 Nt, wobei die Pfeile die Sequenzen zeigen, die über
Basenpaarung zur Ausbildung der 3 Motive führen. B: Sekundärstruktur von rli32. Die in silico
Vorhersage der Sekundärstruktur von rli32 wurde mit dem RNAfold WebServer durchgeführt. Rli32
bildet 3 Motive, die aus unterschiedlichen stem-loop Strukturen bestehen. C: IFN-β Induktion durch die
rli32-Motive in BMDMs. Die 3 rli32-Motive wurden einzeln durch in vitro Transkription hergestellt und
in BMDMs transfiziet. Das Motiv 2 zeigt eine gleich hohe IFN-β Induktion wie rli32 selbst. Während das
Motiv 3 eine schwächere IFN-β Induktion zeigt, zeigt das Motiv 1 eine sehr schwache Induktion von
IFN-β.
Das Ergebnis dieser Untersuchung zeigt, dass Motiv 2 für das hohe IFN-β Induktions-
potential von rli32 verantwortlich ist (Abb. 3.14C). Die von diesem Motiv ausgelöste
IFN-β Antwort ist so hoch wie die von rli32 selbst. Während das Motiv 3 eine viel
schwächere IFN-β Antwort ausgelöst hat, zeigt das Motiv 1 eine sehr schwache
Induktion von IFN-β.
3.1.7. Untersuchung der Inhibition der viralen Replikation durch rli32 und rli47
Die Transfektionsversuche mit den einzelnen sRNAs haben gezeigt, dass diese das
Potential besitzen IFN-β zu induzieren, wenn auch im unterschiedlichem Maße. Die von
Isaacs und Lindemann im Jahr 1957 beobachtete „Interferenz“ mit der Virusreplikation
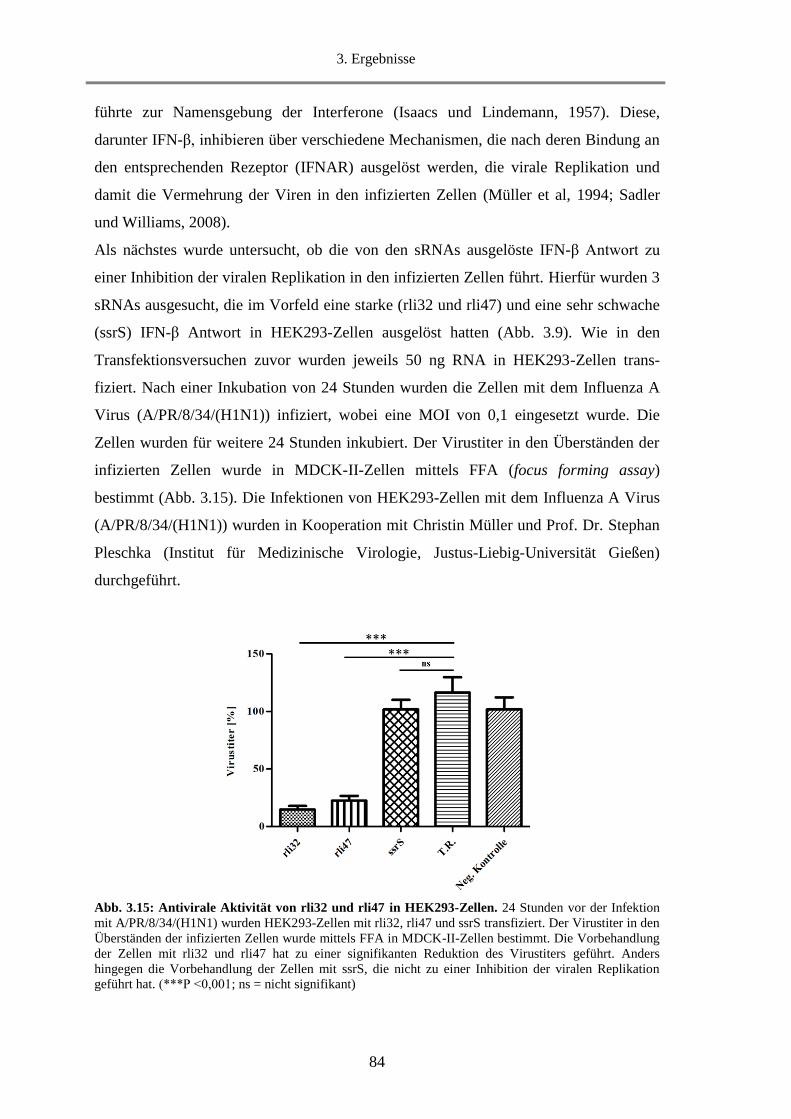
3. Ergebnisse
84
führte zur Namensgebung der Interferone (Isaacs und Lindemann, 1957). Diese,
darunter IFN-β, inhibieren über verschiedene Mechanismen, die nach deren Bindung an
den entsprechenden Rezeptor (IFNAR) ausgelöst werden, die virale Replikation und
damit die Vermehrung der Viren in den infizierten Zellen (Müller et al, 1994; Sadler
und Williams, 2008).
Als nächstes wurde untersucht, ob die von den sRNAs ausgelöste IFN-β Antwort zu
einer Inhibition der viralen Replikation in den infizierten Zellen führt. Hierfür wurden 3
sRNAs ausgesucht, die im Vorfeld eine starke (rli32 und rli47) und eine sehr schwache
(ssrS) IFN-β Antwort in HEK293-Zellen ausgelöst hatten (Abb. 3.9). Wie in den
Transfektionsversuchen zuvor wurden jeweils 50 ng RNA in HEK293-Zellen trans-
fiziert. Nach einer Inkubation von 24 Stunden wurden die Zellen mit dem Influenza A
Virus (A/PR/8/34/(H1N1)) infiziert, wobei eine MOI von 0,1 eingesetzt wurde. Die
Zellen wurden für weitere 24 Stunden inkubiert. Der Virustiter in den Überständen der
infizierten Zellen wurde in MDCK-II-Zellen mittels FFA (focus forming assay)
bestimmt (Abb. 3.15). Die Infektionen von HEK293-Zellen mit dem Influenza A Virus
(A/PR/8/34/(H1N1)) wurden in Kooperation mit Christin Müller und Prof. Dr. Stephan
Pleschka (Institut für Medizinische Virologie, Justus-Liebig-Universität Gießen)
durchgeführt.
Abb. 3.15: Antivirale Aktivität von rli32 und rli47 in HEK293-Zellen. 24 Stunden vor der Infektion
mit A/PR/8/34/(H1N1) wurden HEK293-Zellen mit rli32, rli47 und ssrS transfiziert. Der Virustiter in den
Überständen der infizierten Zellen wurde mittels FFA in MDCK-II-Zellen bestimmt. Die Vorbehandlung
der Zellen mit rli32 und rli47 hat zu einer signifikanten Reduktion des Virustiters geführt. Anders
hingegen die Vorbehandlung der Zellen mit ssrS, die nicht zu einer Inhibition der viralen Replikation
geführt hat. (***P ˂0,001; ns = nicht signifikant)
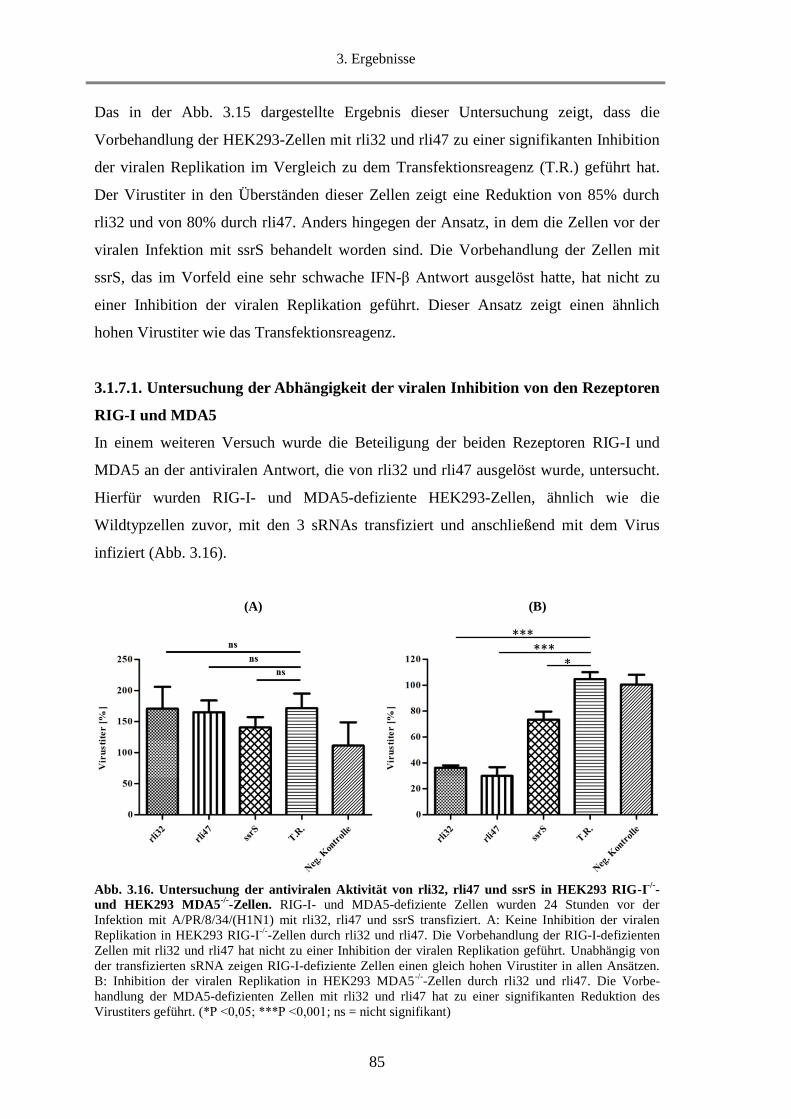
3. Ergebnisse
85
Das in der Abb. 3.15 dargestellte Ergebnis dieser Untersuchung zeigt, dass die
Vorbehandlung der HEK293-Zellen mit rli32 und rli47 zu einer signifikanten Inhibition
der viralen Replikation im Vergleich zu dem Transfektionsreagenz (T.R.) geführt hat.
Der Virustiter in den Überständen dieser Zellen zeigt eine Reduktion von 85% durch
rli32 und von 80% durch rli47. Anders hingegen der Ansatz, in dem die Zellen vor der
viralen Infektion mit ssrS behandelt worden sind. Die Vorbehandlung der Zellen mit
ssrS, das im Vorfeld eine sehr schwache IFN-β Antwort ausgelöst hatte, hat nicht zu
einer Inhibition der viralen Replikation geführt. Dieser Ansatz zeigt einen ähnlich
hohen Virustiter wie das Transfektionsreagenz.
3.1.7.1. Untersuchung der Abhängigkeit der viralen Inhibition von den Rezeptoren
RIG-I und MDA5
In einem weiteren Versuch wurde die Beteiligung der beiden Rezeptoren RIG-I und
MDA5 an der antiviralen Antwort, die von rli32 und rli47 ausgelöst wurde, untersucht.
Hierfür wurden RIG-I- und MDA5-defiziente HEK293-Zellen, ähnlich wie die
Wildtypzellen zuvor, mit den 3 sRNAs transfiziert und anschließend mit dem Virus
infiziert (Abb. 3.16).
(A) (B)
Abb. 3.16. Untersuchung der antiviralen Aktivität von rli32, rli47 und ssrS in HEK293 RIG-I
-/--
und HEK293 MDA5-/-
-Zellen. RIG-I- und MDA5-defiziente Zellen wurden 24 Stunden vor der
Infektion mit A/PR/8/34/(H1N1) mit rli32, rli47 und ssrS transfiziert. A: Keine Inhibition der viralen
Replikation in HEK293 RIG-I-/-
-Zellen durch rli32 und rli47. Die Vorbehandlung der RIG-I-defizienten
Zellen mit rli32 und rli47 hat nicht zu einer Inhibition der viralen Replikation geführt. Unabhängig von
der transfizierten sRNA zeigen RIG-I-defiziente Zellen einen gleich hohen Virustiter in allen Ansätzen.
B: Inhibition der viralen Replikation in HEK293 MDA5-/-
-Zellen durch rli32 und rli47. Die Vorbe-
handlung der MDA5-defizienten Zellen mit rli32 und rli47 hat zu einer signifikanten Reduktion des
Virustiters geführt. (*P ˂0,05; ***P ˂0,001; ns = nicht signifikant)
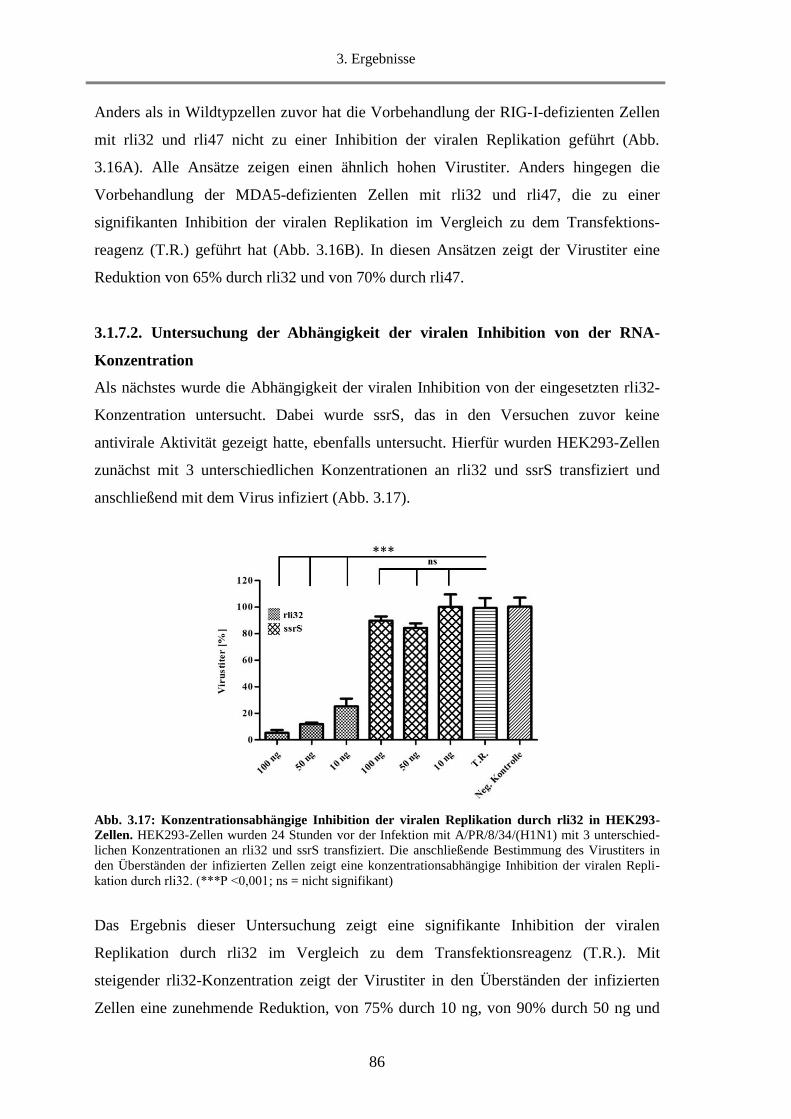
3. Ergebnisse
86
Anders als in Wildtypzellen zuvor hat die Vorbehandlung der RIG-I-defizienten Zellen
mit rli32 und rli47 nicht zu einer Inhibition der viralen Replikation geführt (Abb.
3.16A). Alle Ansätze zeigen einen ähnlich hohen Virustiter. Anders hingegen die
Vorbehandlung der MDA5-defizienten Zellen mit rli32 und rli47, die zu einer
signifikanten Inhibition der viralen Replikation im Vergleich zu dem Transfektions-
reagenz (T.R.) geführt hat (Abb. 3.16B). In diesen Ansätzen zeigt der Virustiter eine
Reduktion von 65% durch rli32 und von 70% durch rli47.
3.1.7.2. Untersuchung der Abhängigkeit der viralen Inhibition von der RNA-
Konzentration
Als nächstes wurde die Abhängigkeit der viralen Inhibition von der eingesetzten rli32-
Konzentration untersucht. Dabei wurde ssrS, das in den Versuchen zuvor keine
antivirale Aktivität gezeigt hatte, ebenfalls untersucht. Hierfür wurden HEK293-Zellen
zunächst mit 3 unterschiedlichen Konzentrationen an rli32 und ssrS transfiziert und
anschließend mit dem Virus infiziert (Abb. 3.17).
Abb. 3.17: Konzentrationsabhängige Inhibition der viralen Replikation durch rli32 in HEK293-
Zellen. HEK293-Zellen wurden 24 Stunden vor der Infektion mit A/PR/8/34/(H1N1) mit 3 unterschied-
lichen Konzentrationen an rli32 und ssrS transfiziert. Die anschließende Bestimmung des Virustiters in
den Überständen der infizierten Zellen zeigt eine konzentrationsabhängige Inhibition der viralen Repli-
kation durch rli32. (***P ˂0,001; ns = nicht signifikant)
Das Ergebnis dieser Untersuchung zeigt eine signifikante Inhibition der viralen
Replikation durch rli32 im Vergleich zu dem Transfektionsreagenz (T.R.). Mit
steigender rli32-Konzentration zeigt der Virustiter in den Überständen der infizierten
Zellen eine zunehmende Reduktion, von 75% durch 10 ng, von 90% durch 50 ng und
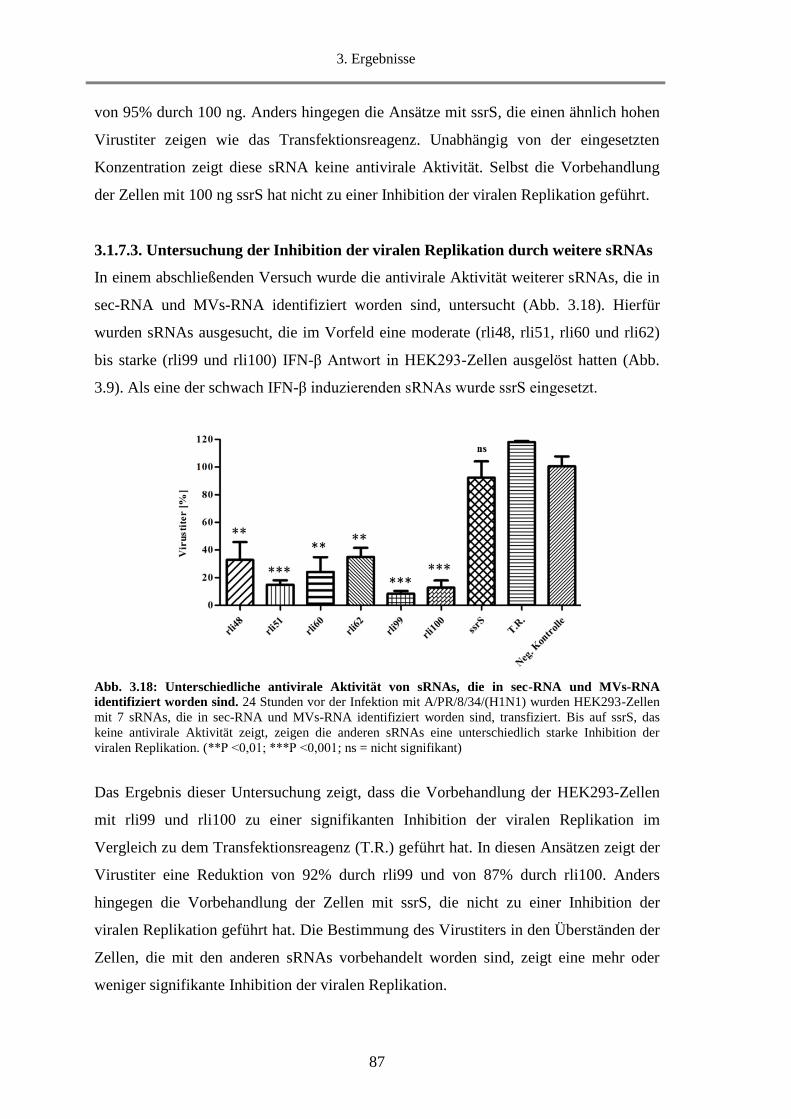
3. Ergebnisse
87
von 95% durch 100 ng. Anders hingegen die Ansätze mit ssrS, die einen ähnlich hohen
Virustiter zeigen wie das Transfektionsreagenz. Unabhängig von der eingesetzten
Konzentration zeigt diese sRNA keine antivirale Aktivität. Selbst die Vorbehandlung
der Zellen mit 100 ng ssrS hat nicht zu einer Inhibition der viralen Replikation geführt.
3.1.7.3. Untersuchung der Inhibition der viralen Replikation durch weitere sRNAs
In einem abschließenden Versuch wurde die antivirale Aktivität weiterer sRNAs, die in
sec-RNA und MVs-RNA identifiziert worden sind, untersucht (Abb. 3.18). Hierfür
wurden sRNAs ausgesucht, die im Vorfeld eine moderate (rli48, rli51, rli60 und rli62)
bis starke (rli99 und rli100) IFN-β Antwort in HEK293-Zellen ausgelöst hatten (Abb.
3.9). Als eine der schwach IFN-β induzierenden sRNAs wurde ssrS eingesetzt.
Abb. 3.18: Unterschiedliche antivirale Aktivität von sRNAs, die in sec-RNA und MVs-RNA
identifiziert worden sind. 24 Stunden vor der Infektion mit A/PR/8/34/(H1N1) wurden HEK293-Zellen
mit 7 sRNAs, die in sec-RNA und MVs-RNA identifiziert worden sind, transfiziert. Bis auf ssrS, das
keine antivirale Aktivität zeigt, zeigen die anderen sRNAs eine unterschiedlich starke Inhibition der
viralen Replikation. (**P ˂0,01; ***P ˂0,001; ns = nicht signifikant)
Das Ergebnis dieser Untersuchung zeigt, dass die Vorbehandlung der HEK293-Zellen
mit rli99 und rli100 zu einer signifikanten Inhibition der viralen Replikation im
Vergleich zu dem Transfektionsreagenz (T.R.) geführt hat. In diesen Ansätzen zeigt der
Virustiter eine Reduktion von 92% durch rli99 und von 87% durch rli100. Anders
hingegen die Vorbehandlung der Zellen mit ssrS, die nicht zu einer Inhibition der
viralen Replikation geführt hat. Die Bestimmung des Virustiters in den Überständen der
Zellen, die mit den anderen sRNAs vorbehandelt worden sind, zeigt eine mehr oder
weniger signifikante Inhibition der viralen Replikation.

3. Ergebnisse
88
3.1.8. Untersuchung der Fähigkeit von rli32 Typ-I-IFN in der Maus zu induzieren
Die im Vorfeld durchgeführten in vitro Transfektionsversuche haben gezeigt, dass von
den untersuchten sRNAs rli32 das höchste IFN-β Induktionspotential besitzt. Als
nächstes wurde die von rli32 ausgelöste Typ-I-IFN Antwort (IFN-β und IFN-α4) in vivo
im Mausversuch untersucht. Dabei wurde auch ssrS, das im Vorfeld ein sehr schwaches
Potential zur IFN-β Induktion gezeigt hatte, untersucht. Des Weiteren wurde zelluläre
RNA von HEK293-Zellen (eukaryotische RNA), die als Vergleich zu den bakteriellen
sRNAs diente, eingesetzt. Für diesen Versuch wurden insgesamt 20 Mäuse verwendet.
Davon wurden 15 Mäuse mit jeweils 40 µg rli32, ssrS und eukaryotische RNA (5
Tiere/Gruppe) und 5 Mäuse nur mit dem Transfektionsreagenz intravenös injiziert. Die
Mausversuche wurden in Kooperation mit Jessica Wingerath und Dr. Thomas Wirth
(Abteilung für Gastroenterologie, Hepatologie und Endokrinologie, Medizinische
Hochschule Hannover) durchgeführt.
Als Transfektionsreagenz wurde in vivo-jetPEI verwendet, das jede Art von
Nukleinsäure in eine Reihe von Organen effizient liefert. Hierbei bestimmt die
Injektionsart der Nukleinsäure-in vivo-jetPEI-Komplexe weitgehend die target-Organe.
Nach einer intravenösen Injektion wird die Nukleinsäure von in vivo-jetPEI u.a. in
Leber und Milz geliefert. Diese Organe wurden 5 Stunden nach Injektion der RNA und
des Transfektionsreagenz entnommen. Die dort ausgelöste Typ-I-IFN Antwort wurde
nach Isolierung der RNA und Umschreibung dieser in cDNA über RT-PCR untersucht
(Abb. 3.19 und 3.20). In der Gruppe ssrS ist eine Maus gestorben. Eine weitere Maus in
dieser Gruppe hat keine Typ-I-IFN Antwort gezeigt. Diese wurden von der Auswertung
ausgeschlossen.
Das Ergebnis dieser Untersuchung zeigt, dass rli32 in beide Organe der Maus eine hohe
IFN-β Antwort ausgelöst hat. In der Leber wurde eine ca. 10000fache (Abb. 3.19A) und
in der Milz eine ca. 14000fache (Abb. 3.19B) IFN-β Induktion festgestellt. Insgesamt ist
die IFN-β Antwort in der Milz höher ausgefallen als in der Leber. Anders als in den in
vitro Transfektionsversuchen hat ssrS in der Maus eine hohe IFN-β Antwort ausgelöst.
In der Milz hat ssrS eine höhere IFN-β Antwort ausgelöst (ca. 20000fache) als rli32
(Abb. 3.19B). Die eukaryotische RNA hat, anders als die beiden sRNAs, eine schwache
IFN-β Antwort ausgelöst. Im Vergleich zu der eukaryotischen RNA zeigen beide
sRNAs eine signifikant höhere IFN-β Induktion in beide Organe der Maus.
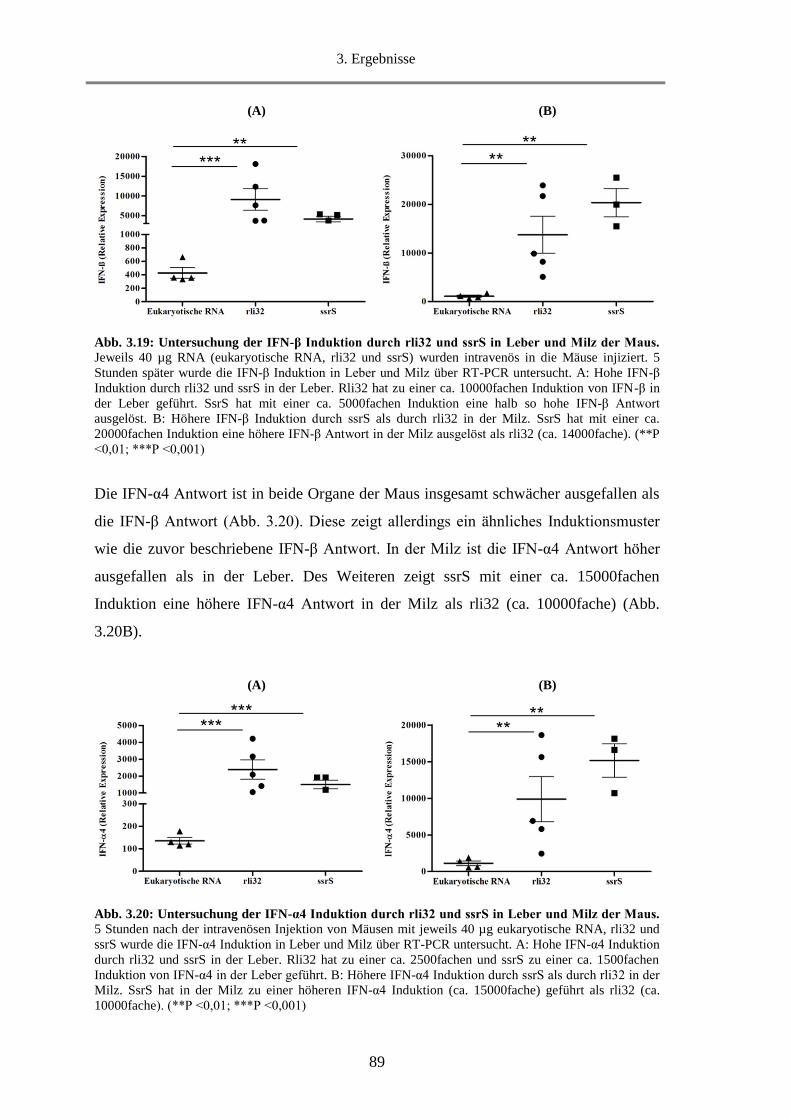
3. Ergebnisse
89
(A) (B)
Abb. 3.19: Untersuchung der IFN-β Induktion durch rli32 und ssrS in Leber und Milz der Maus.
Jeweils 40 µg RNA (eukaryotische RNA, rli32 und ssrS) wurden intravenös in die Mäuse injiziert. 5
Stunden später wurde die IFN-β Induktion in Leber und Milz über RT-PCR untersucht. A: Hohe IFN-β
Induktion durch rli32 und ssrS in der Leber. Rli32 hat zu einer ca. 10000fachen Induktion von IFN-β in
der Leber geführt. SsrS hat mit einer ca. 5000fachen Induktion eine halb so hohe IFN-β Antwort
ausgelöst. B: Höhere IFN-β Induktion durch ssrS als durch rli32 in der Milz. SsrS hat mit einer ca.
20000fachen Induktion eine höhere IFN-β Antwort in der Milz ausgelöst als rli32 (ca. 14000fache). (**P
˂0,01; ***P ˂0,001)
Die IFN-α4 Antwort ist in beide Organe der Maus insgesamt schwächer ausgefallen als
die IFN-β Antwort (Abb. 3.20). Diese zeigt allerdings ein ähnliches Induktionsmuster
wie die zuvor beschriebene IFN-β Antwort. In der Milz ist die IFN-α4 Antwort höher
ausgefallen als in der Leber. Des Weiteren zeigt ssrS mit einer ca. 15000fachen
Induktion eine höhere IFN-α4 Antwort in der Milz als rli32 (ca. 10000fache) (Abb.
3.20B).
(A) (B)
Abb. 3.20: Untersuchung der IFN-α4 Induktion durch rli32 und ssrS in Leber und Milz der Maus.
5 Stunden nach der intravenösen Injektion von Mäusen mit jeweils 40 µg eukaryotische RNA, rli32 und
ssrS wurde die IFN-α4 Induktion in Leber und Milz über RT-PCR untersucht. A: Hohe IFN-α4 Induktion
durch rli32 und ssrS in der Leber. Rli32 hat zu einer ca. 2500fachen und ssrS zu einer ca. 1500fachen
Induktion von IFN-α4 in der Leber geführt. B: Höhere IFN-α4 Induktion durch ssrS als durch rli32 in der
Milz. SsrS hat in der Milz zu einer höheren IFN-α4 Induktion (ca. 15000fache) geführt als rli32 (ca.
10000fache). (**P ˂0,01; ***P ˂0,001)

3. Ergebnisse
90
3.1.9. Überexpression von rli32 und ssrS in L. monocytogenes
Die in vitro Transfektionsversuche haben gezeigt, dass einzelne in sec-RNA und MVs-
RNA identifizierte sRNAs ein unterschiedliches IFN-β Induktionspotential besitzen.
Unter den 23 ausgesuchten sRNAs hat sich rli32 als die sRNA mit dem höchsten IFN-β
Induktionspotential herausgestellt. Im Laufe dieser Arbeit kam die Frage auf, ob eine
Überexpression von rli32 in L. monocytogenes eine höhere IFN-β Antwort durch die
sekretierten RNA-Fraktionen und MVs in Wirtszellen auslöst. Um dies zu überprüfen,
wurde rli32 in L. monocytogenes überexprimiert. In einem parallelen Ansatz wurde ssrS
ebenfalls in L. monocytogenes überexprimiert. Hierfür wurde die Nukleotidsequenz der
beiden sRNAs von chromosomaler DNA aus über PCR amplifiziert und mit dem hly-
Promotor stromaufwärts und dem hly-Terminator stromabwärts von der kodierenden
Sequenz ausgestattet. Anschließend wurden diese Transkriptionseinheiten in den
pERL3-Vektor über Restriktionsverdau und Ligation kloniert. Durch Transformation
von L. monocytogenes mit den pERL3-Vektoren mit entsprechender sRNA-Kassette
wurden 2 Stämme konstruiert, die als Lm-pERL3-rli32 und Lm-pERL3-ssrS bezeichnet
wurden. Es folgte die Isolierung von sec-RNA, MVs-RNA und zellulärer RNA der
Stämme Lm (WT), Lm-pERL3-rli32 und Lm-pERL3-ssrS. Die RNA, die vom Wildtyp
isoliert wurde, diente als Vergleich zu den beiden überexprimierenden Stämmen.
In einem weiteren Schritt wurde die Überexpression der beiden sRNAs in L. monocyto-
genes über RT-PCR überprüft. Zuvor wurde die isolierte RNA in cDNA umge-
schrieben. Das Ergebnis dieser Untersuchung ist in der Abb. 3.21 dargestellt. Der
sogenannte Ct-Wert (Cycle threshold) auf der y-Achse ist ein Maß für die Quantität der
untersuchten cDNA und damit der ursprünglich vorhandenen RNA in einer Probe. Je
niedriger der Ct-Wert desto mehr DNA und infolgedessen desto mehr RNA ist in der
untersuchten Probe vorhanden. Das Ergebnis dieser Untersuchung zeigt, dass die
Überexpression von rli32 und ssrS in L. monocytogenes zu einem Anstieg der
entsprechenden sRNA-Transkripte in allen 3 RNA-Fraktionen der beiden Stämme
geführt hat.
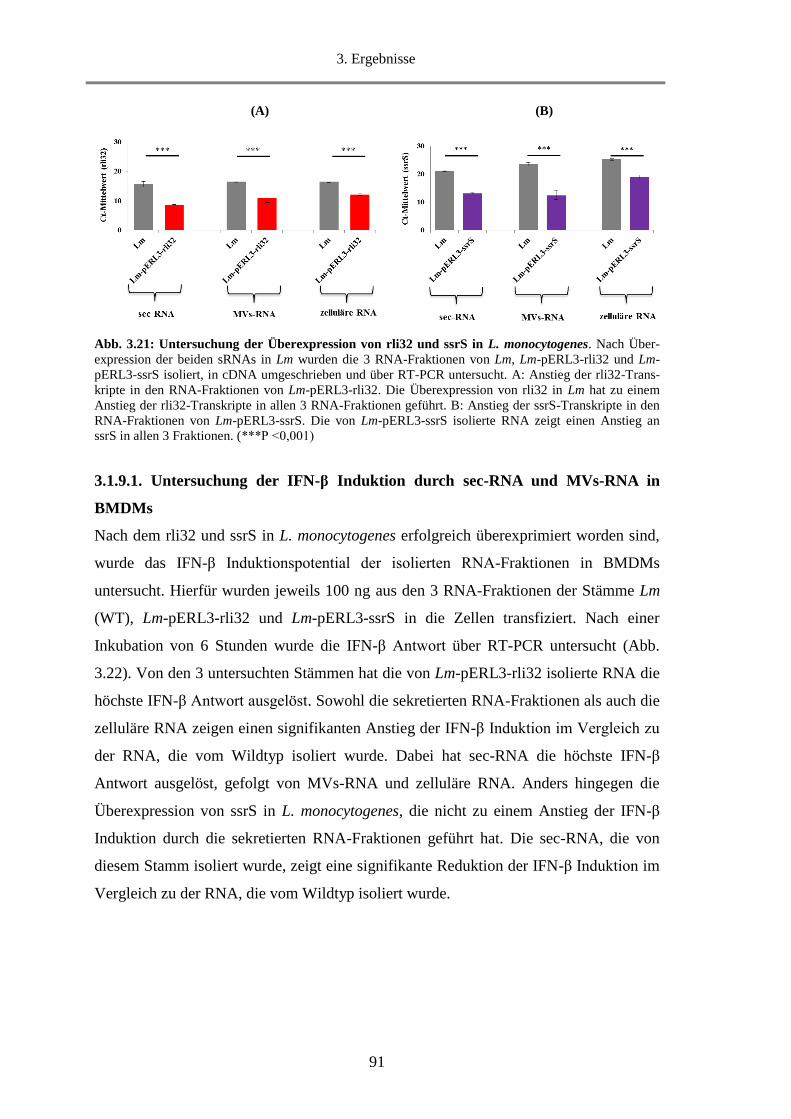
3. Ergebnisse
91
(A) (B)
Abb. 3.21: Untersuchung der Überexpression von rli32 und ssrS in L. monocytogenes. Nach Über-
expression der beiden sRNAs in Lm wurden die 3 RNA-Fraktionen von Lm, Lm-pERL3-rli32 und Lm-
pERL3-ssrS isoliert, in cDNA umgeschrieben und über RT-PCR untersucht. A: Anstieg der rli32-Trans-
kripte in den RNA-Fraktionen von Lm-pERL3-rli32. Die Überexpression von rli32 in Lm hat zu einem
Anstieg der rli32-Transkripte in allen 3 RNA-Fraktionen geführt. B: Anstieg der ssrS-Transkripte in den
RNA-Fraktionen von Lm-pERL3-ssrS. Die von Lm-pERL3-ssrS isolierte RNA zeigt einen Anstieg an
ssrS in allen 3 Fraktionen. (***P ˂0,001)
3.1.9.1. Untersuchung der IFN-β Induktion durch sec-RNA und MVs-RNA in
BMDMs
Nach dem rli32 und ssrS in L. monocytogenes erfolgreich überexprimiert worden sind,
wurde das IFN-β Induktionspotential der isolierten RNA-Fraktionen in BMDMs
untersucht. Hierfür wurden jeweils 100 ng aus den 3 RNA-Fraktionen der Stämme Lm
(WT), Lm-pERL3-rli32 und Lm-pERL3-ssrS in die Zellen transfiziert. Nach einer
Inkubation von 6 Stunden wurde die IFN-β Antwort über RT-PCR untersucht (Abb.
3.22). Von den 3 untersuchten Stämmen hat die von Lm-pERL3-rli32 isolierte RNA die
höchste IFN-β Antwort ausgelöst. Sowohl die sekretierten RNA-Fraktionen als auch die
zelluläre RNA zeigen einen signifikanten Anstieg der IFN-β Induktion im Vergleich zu
der RNA, die vom Wildtyp isoliert wurde. Dabei hat sec-RNA die höchste IFN-β
Antwort ausgelöst, gefolgt von MVs-RNA und zelluläre RNA. Anders hingegen die
Überexpression von ssrS in L. monocytogenes, die nicht zu einem Anstieg der IFN-β
Induktion durch die sekretierten RNA-Fraktionen geführt hat. Die sec-RNA, die von
diesem Stamm isoliert wurde, zeigt eine signifikante Reduktion der IFN-β Induktion im
Vergleich zu der RNA, die vom Wildtyp isoliert wurde.
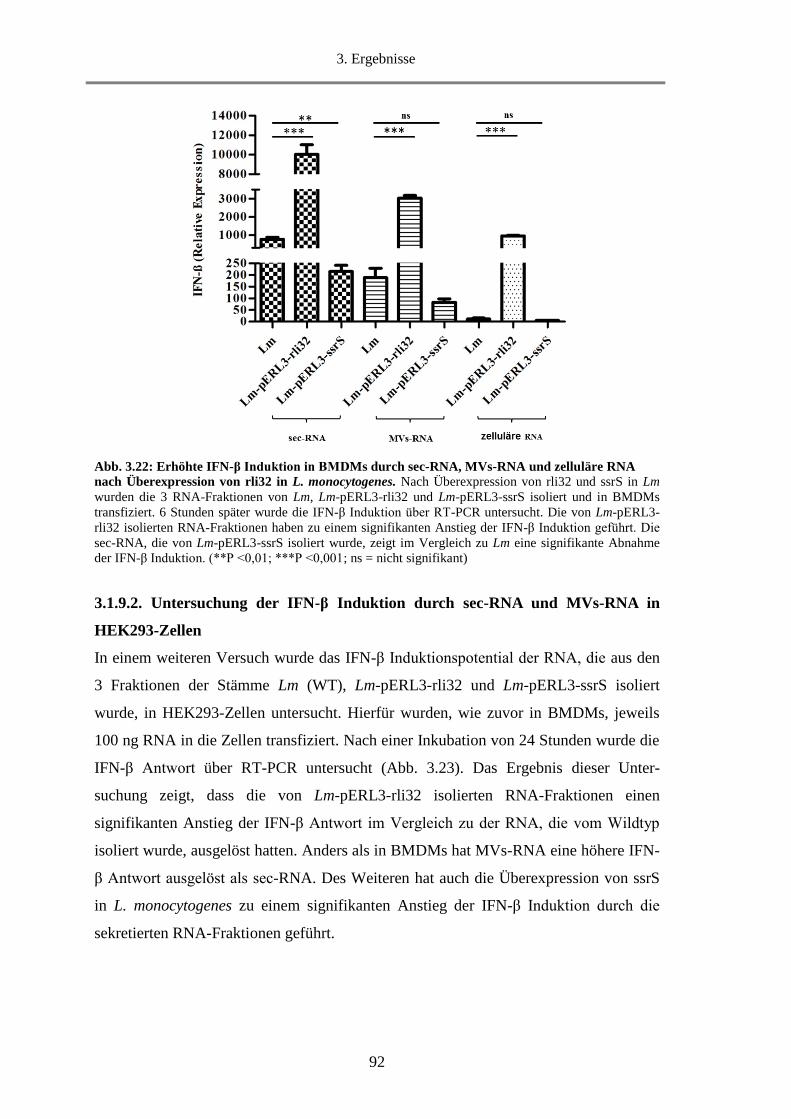
3. Ergebnisse
92
Abb. 3.22: Erhöhte IFN-β Induktion in BMDMs durch sec-RNA, MVs-RNA und zelluläre RNA
nach Überexpression von rli32 in L. monocytogenes. Nach Überexpression von rli32 und ssrS in Lm
wurden die 3 RNA-Fraktionen von Lm, Lm-pERL3-rli32 und Lm-pERL3-ssrS isoliert und in BMDMs
transfiziert. 6 Stunden später wurde die IFN-β Induktion über RT-PCR untersucht. Die von Lm-pERL3-
rli32 isolierten RNA-Fraktionen haben zu einem signifikanten Anstieg der IFN-β Induktion geführt. Die
sec-RNA, die von Lm-pERL3-ssrS isoliert wurde, zeigt im Vergleich zu Lm eine signifikante Abnahme
der IFN-β Induktion. (**P ˂0,01; ***P ˂0,001; ns = nicht signifikant)
3.1.9.2. Untersuchung der IFN-β Induktion durch sec-RNA und MVs-RNA in
HEK293-Zellen
In einem weiteren Versuch wurde das IFN-β Induktionspotential der RNA, die aus den
3 Fraktionen der Stämme Lm (WT), Lm-pERL3-rli32 und Lm-pERL3-ssrS isoliert
wurde, in HEK293-Zellen untersucht. Hierfür wurden, wie zuvor in BMDMs, jeweils
100 ng RNA in die Zellen transfiziert. Nach einer Inkubation von 24 Stunden wurde die
IFN-β Antwort über RT-PCR untersucht (Abb. 3.23). Das Ergebnis dieser Unter-
suchung zeigt, dass die von Lm-pERL3-rli32 isolierten RNA-Fraktionen einen
signifikanten Anstieg der IFN-β Antwort im Vergleich zu der RNA, die vom Wildtyp
isoliert wurde, ausgelöst hatten. Anders als in BMDMs hat MVs-RNA eine höhere IFN-
β Antwort ausgelöst als sec-RNA. Des Weiteren hat auch die Überexpression von ssrS
in L. monocytogenes zu einem signifikanten Anstieg der IFN-β Induktion durch die
sekretierten RNA-Fraktionen geführt.
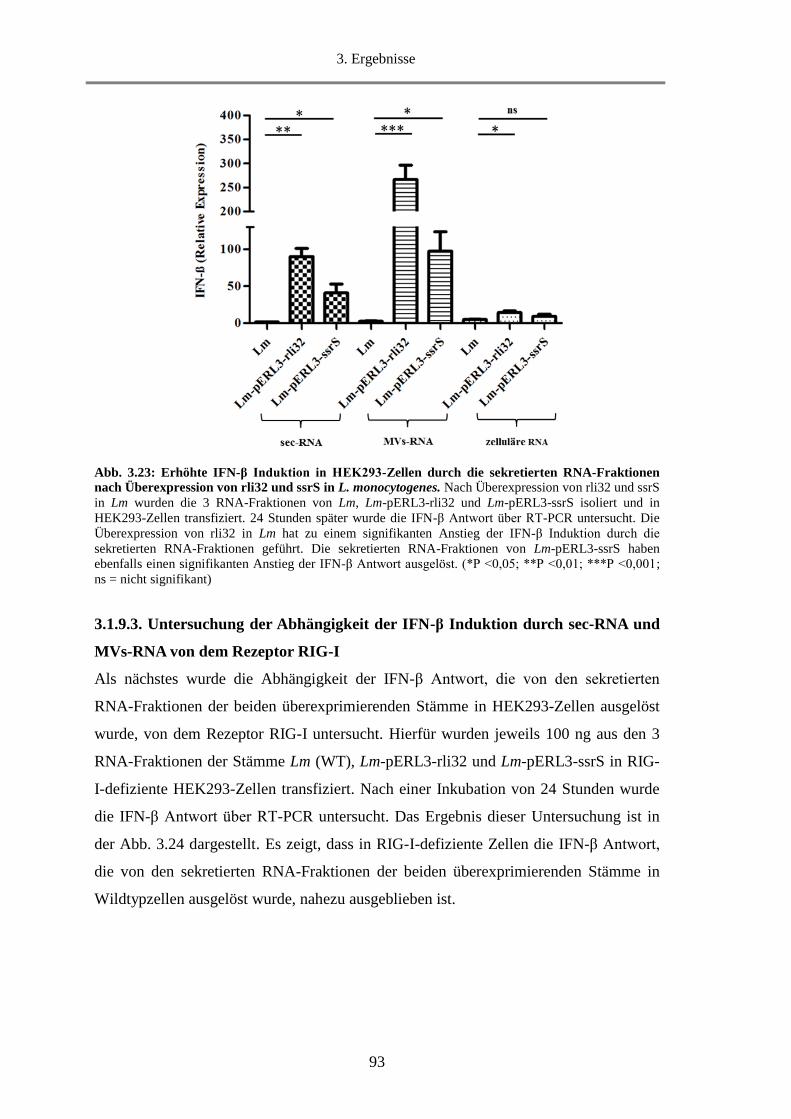
3. Ergebnisse
93
Abb. 3.23: Erhöhte IFN-β Induktion in HEK293-Zellen durch die sekretierten RNA-Fraktionen
nach Überexpression von rli32 und ssrS in L. monocytogenes. Nach Überexpression von rli32 und ssrS
in Lm wurden die 3 RNA-Fraktionen von Lm, Lm-pERL3-rli32 und Lm-pERL3-ssrS isoliert und in
HEK293-Zellen transfiziert. 24 Stunden später wurde die IFN-β Antwort über RT-PCR untersucht. Die
Überexpression von rli32 in Lm hat zu einem signifikanten Anstieg der IFN-β Induktion durch die
sekretierten RNA-Fraktionen geführt. Die sekretierten RNA-Fraktionen von Lm-pERL3-ssrS haben
ebenfalls einen signifikanten Anstieg der IFN-β Antwort ausgelöst. (*P ˂0,05; **P ˂0,01; ***P ˂0,001;
ns = nicht signifikant)
3.1.9.3. Untersuchung der Abhängigkeit der IFN-β Induktion durch sec-RNA und
MVs-RNA von dem Rezeptor RIG-I
Als nächstes wurde die Abhängigkeit der IFN-β Antwort, die von den sekretierten
RNA-Fraktionen der beiden überexprimierenden Stämme in HEK293-Zellen ausgelöst
wurde, von dem Rezeptor RIG-I untersucht. Hierfür wurden jeweils 100 ng aus den 3
RNA-Fraktionen der Stämme Lm (WT), Lm-pERL3-rli32 und Lm-pERL3-ssrS in RIG-
I-defiziente HEK293-Zellen transfiziert. Nach einer Inkubation von 24 Stunden wurde
die IFN-β Antwort über RT-PCR untersucht. Das Ergebnis dieser Untersuchung ist in
der Abb. 3.24 dargestellt. Es zeigt, dass in RIG-I-defiziente Zellen die IFN-β Antwort,
die von den sekretierten RNA-Fraktionen der beiden überexprimierenden Stämme in
Wildtypzellen ausgelöst wurde, nahezu ausgeblieben ist.
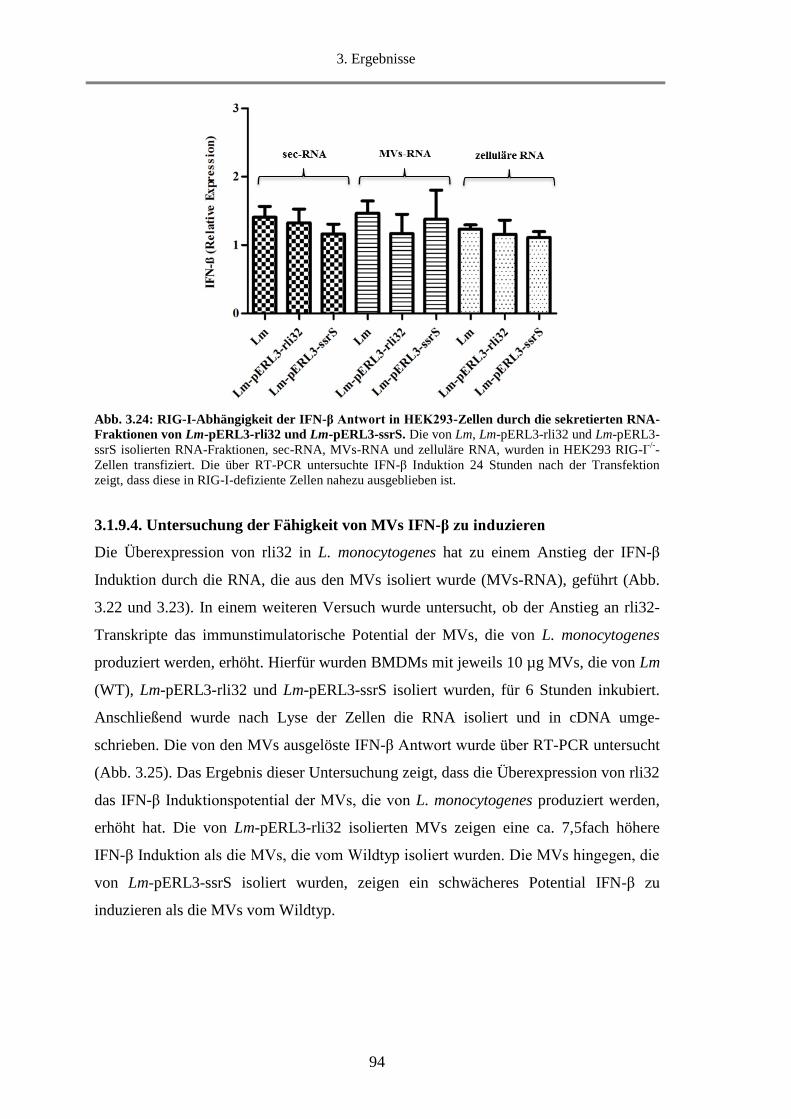
3. Ergebnisse
94
Abb. 3.24: RIG-I-Abhängigkeit der IFN-β Antwort in HEK293-Zellen durch die sekretierten RNA-
Fraktionen von Lm-pERL3-rli32 und Lm-pERL3-ssrS. Die von Lm, Lm-pERL3-rli32 und Lm-pERL3-
ssrS isolierten RNA-Fraktionen, sec-RNA, MVs-RNA und zelluläre RNA, wurden in HEK293 RIG-I-/-
-
Zellen transfiziert. Die über RT-PCR untersuchte IFN-β Induktion 24 Stunden nach der Transfektion
zeigt, dass diese in RIG-I-defiziente Zellen nahezu ausgeblieben ist.
3.1.9.4. Untersuchung der Fähigkeit von MVs IFN-β zu induzieren
Die Überexpression von rli32 in L. monocytogenes hat zu einem Anstieg der IFN-β
Induktion durch die RNA, die aus den MVs isoliert wurde (MVs-RNA), geführt (Abb.
3.22 und 3.23). In einem weiteren Versuch wurde untersucht, ob der Anstieg an rli32-
Transkripte das immunstimulatorische Potential der MVs, die von L. monocytogenes
produziert werden, erhöht. Hierfür wurden BMDMs mit jeweils 10 µg MVs, die von Lm
(WT), Lm-pERL3-rli32 und Lm-pERL3-ssrS isoliert wurden, für 6 Stunden inkubiert.
Anschließend wurde nach Lyse der Zellen die RNA isoliert und in cDNA umge-
schrieben. Die von den MVs ausgelöste IFN-β Antwort wurde über RT-PCR untersucht
(Abb. 3.25). Das Ergebnis dieser Untersuchung zeigt, dass die Überexpression von rli32
das IFN-β Induktionspotential der MVs, die von L. monocytogenes produziert werden,
erhöht hat. Die von Lm-pERL3-rli32 isolierten MVs zeigen eine ca. 7,5fach höhere
IFN-β Induktion als die MVs, die vom Wildtyp isoliert wurden. Die MVs hingegen, die
von Lm-pERL3-ssrS isoliert wurden, zeigen ein schwächeres Potential IFN-β zu
induzieren als die MVs vom Wildtyp.
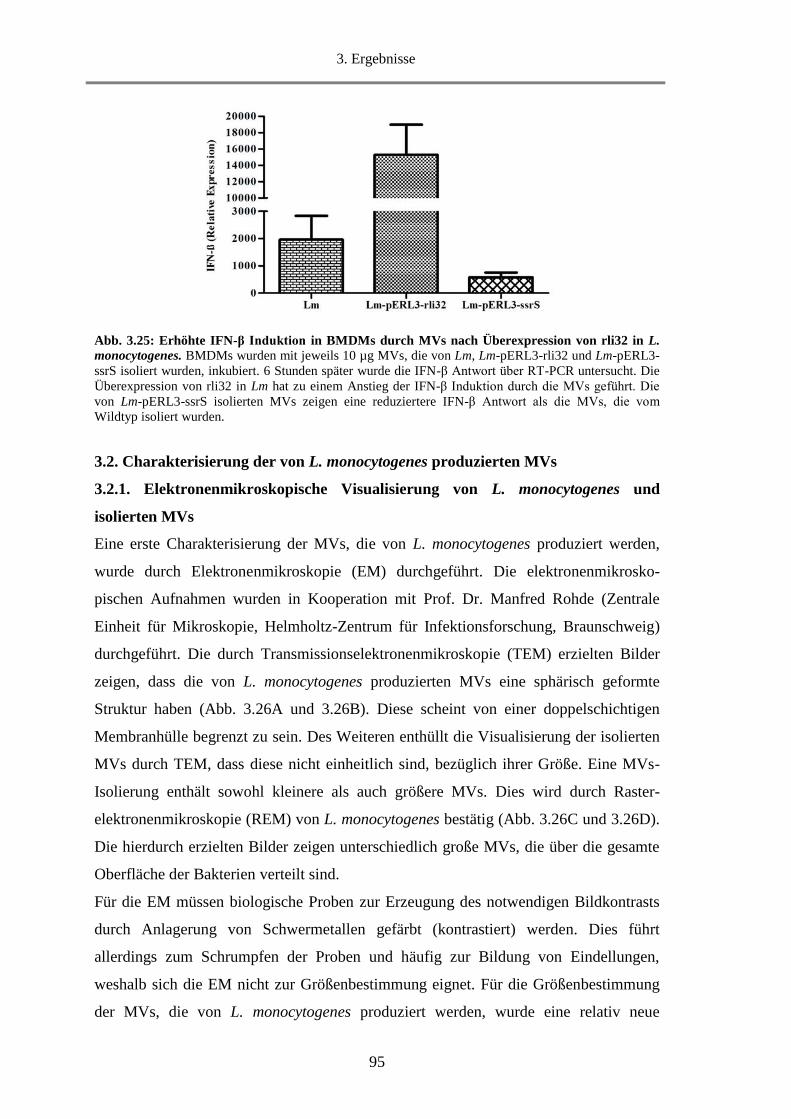
3. Ergebnisse
95
Abb. 3.25: Erhöhte IFN-β Induktion in BMDMs durch MVs nach Überexpression von rli32 in L.
monocytogenes. BMDMs wurden mit jeweils 10 µg MVs, die von Lm, Lm-pERL3-rli32 und Lm-pERL3-
ssrS isoliert wurden, inkubiert. 6 Stunden später wurde die IFN-β Antwort über RT-PCR untersucht. Die
Überexpression von rli32 in Lm hat zu einem Anstieg der IFN-β Induktion durch die MVs geführt. Die
von Lm-pERL3-ssrS isolierten MVs zeigen eine reduziertere IFN-β Antwort als die MVs, die vom
Wildtyp isoliert wurden.
3.2. Charakterisierung der von L. monocytogenes produzierten MVs
3.2.1. Elektronenmikroskopische Visualisierung von L. monocytogenes und
isolierten MVs
Eine erste Charakterisierung der MVs, die von L. monocytogenes produziert werden,
wurde durch Elektronenmikroskopie (EM) durchgeführt. Die elektronenmikrosko-
pischen Aufnahmen wurden in Kooperation mit Prof. Dr. Manfred Rohde (Zentrale
Einheit für Mikroskopie, Helmholtz-Zentrum für Infektionsforschung, Braunschweig)
durchgeführt. Die durch Transmissionselektronenmikroskopie (TEM) erzielten Bilder
zeigen, dass die von L. monocytogenes produzierten MVs eine sphärisch geformte
Struktur haben (Abb. 3.26A und 3.26B). Diese scheint von einer doppelschichtigen
Membranhülle begrenzt zu sein. Des Weiteren enthüllt die Visualisierung der isolierten
MVs durch TEM, dass diese nicht einheitlich sind, bezüglich ihrer Größe. Eine MVs-
Isolierung enthält sowohl kleinere als auch größere MVs. Dies wird durch Raster-
elektronenmikroskopie (REM) von L. monocytogenes bestätig (Abb. 3.26C und 3.26D).
Die hierdurch erzielten Bilder zeigen unterschiedlich große MVs, die über die gesamte
Oberfläche der Bakterien verteilt sind.
Für die EM müssen biologische Proben zur Erzeugung des notwendigen Bildkontrasts
durch Anlagerung von Schwermetallen gefärbt (kontrastiert) werden. Dies führt
allerdings zum Schrumpfen der Proben und häufig zur Bildung von Eindellungen,
weshalb sich die EM nicht zur Größenbestimmung eignet. Für die Größenbestimmung
der MVs, die von L. monocytogenes produziert werden, wurde eine relativ neue

3. Ergebnisse
96
Technologie, die NTA (Nanoparticle Tracking Analysis), verwendet. Die NTA wurde
erstmals im Jahr 2011 von unabhängigen Gruppen für die Charakterisierung von
eukaryotischen Exosomen genutzt und beschrieben. Seitdem wird diese Technologie für
die Größenbestimmung von Vesikeln allgemein verwendet (Sokolova et al, 2011;
Dragovic et al, 2011).
(A) (B)
(C) (D)
Abb. 3.26: Visualisierung von MVs und L. monocytogenes durch Elektronenmikroskopie. A und B:
TEM-Aufnahmen von Lm-MVs. Die MVs wurden von Lm-Kulturen isoliert, die in Minimalmedium bis
OD600nm von 1 gezüchtet wurden. Eine MVs-Isolierung enthält unterschiedlich große MVs mit einer
sphärisch geformten Struktur. C und D: SEM-Aufnahmen von Lm. Sphärisch geformte MVs sind über die
gesamte Oberfläche der Bakterien verteilt.
3.2.2. Bestimmung der Größe, Anzahl und Proteinkonzentration von isolierten
MVs
Die Größe der von L. monocytogenes produzierten MVs wurde durch NTA mittels
NanoSight-Gerät bestimmt. Diese Technologie ermöglicht die Detektion und Verfol-
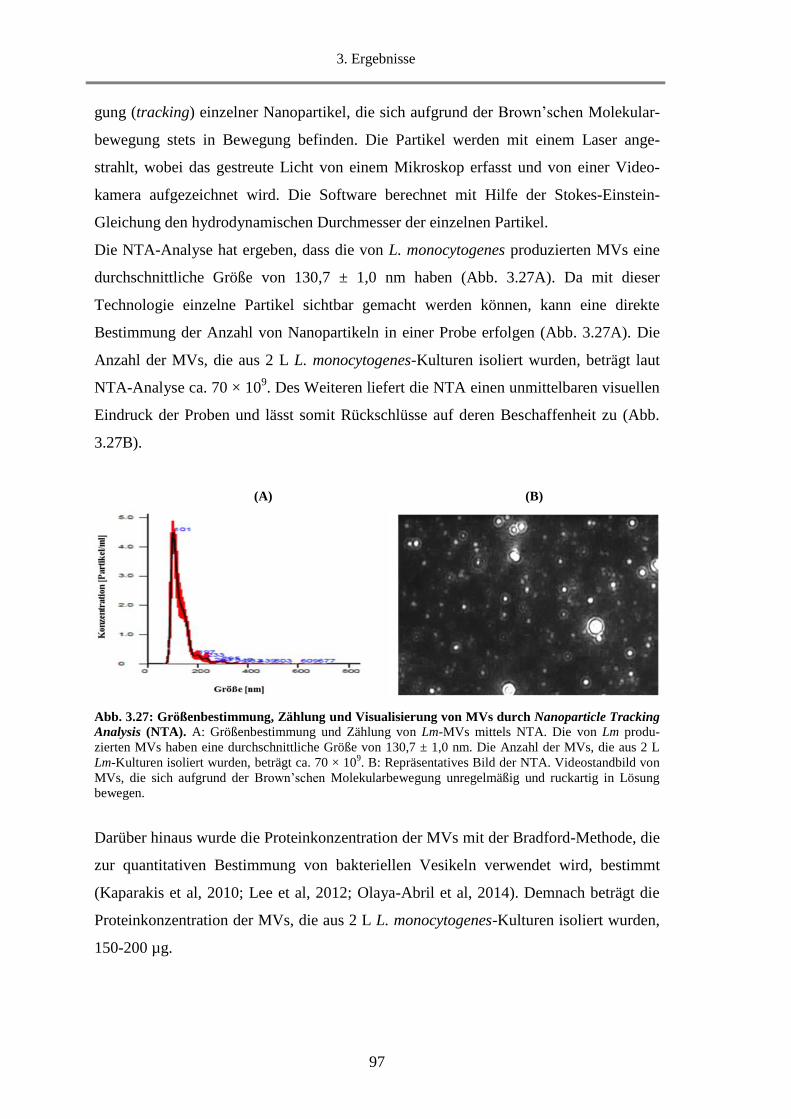
3. Ergebnisse
97
gung (tracking) einzelner Nanopartikel, die sich aufgrund der Brown’schen Molekular-
bewegung stets in Bewegung befinden. Die Partikel werden mit einem Laser ange-
strahlt, wobei das gestreute Licht von einem Mikroskop erfasst und von einer Video-
kamera aufgezeichnet wird. Die Software berechnet mit Hilfe der Stokes-Einstein-
Gleichung den hydrodynamischen Durchmesser der einzelnen Partikel.
Die NTA-Analyse hat ergeben, dass die von L. monocytogenes produzierten MVs eine
durchschnittliche Größe von 130,7 ± 1,0 nm haben (Abb. 3.27A). Da mit dieser
Technologie einzelne Partikel sichtbar gemacht werden können, kann eine direkte
Bestimmung der Anzahl von Nanopartikeln in einer Probe erfolgen (Abb. 3.27A). Die
Anzahl der MVs, die aus 2 L L. monocytogenes-Kulturen isoliert wurden, beträgt laut
NTA-Analyse ca. 70 × 109. Des Weiteren liefert die NTA einen unmittelbaren visuellen
Eindruck der Proben und lässt somit Rückschlüsse auf deren Beschaffenheit zu (Abb.
3.27B).
(A) (B)
Abb. 3.27: Größenbestimmung, Zählung und Visualisierung von MVs durch Nanoparticle Tracking
Analysis (NTA). A: Größenbestimmung und Zählung von Lm-MVs mittels NTA. Die von Lm produ-
zierten MVs haben eine durchschnittliche Größe von 130,7 ± 1,0 nm. Die Anzahl der MVs, die aus 2 L
Lm-Kulturen isoliert wurden, beträgt ca. 70 × 109. B: Repräsentatives Bild der NTA. Videostandbild von
MVs, die sich aufgrund der Brown’schen Molekularbewegung unregelmäßig und ruckartig in Lösung
bewegen.
Darüber hinaus wurde die Proteinkonzentration der MVs mit der Bradford-Methode, die
zur quantitativen Bestimmung von bakteriellen Vesikeln verwendet wird, bestimmt
(Kaparakis et al, 2010; Lee et al, 2012; Olaya-Abril et al, 2014). Demnach beträgt die
Proteinkonzentration der MVs, die aus 2 L L. monocytogenes-Kulturen isoliert wurden,
150-200 µg.

3. Ergebnisse
98
3.2.3. Proteomanalyse (Massenspektrometrie) von MVs
Die Identifizierung der Proteine in den MVs, die von L. monocytogenes produziert
werden, erfolgte über Massenspektrometrie (MS). Gleichzeitig wurde eine markierungs-
freie relative Proteinquantifizierung durchgeführt. Diese basierte auf den Vergleich der
identifizierten MS/MS-Spektren eines Proteins zwischen LC-MS/MS-Läufen (spectral
counting) und den zur Normalisierung herangezogenen NSAF-Wert (normalized
spectral abundance factor) (Zybailov et al, 2005; Zybailov et al, 2006; Zhu et al, 2010).
Die MS-Analyse wurde von Dr. Andreas Otto und Prof. Dr. Dörte Becher (Institut für
Mikrobiologie, Abteilung für Mikrobielle Proteomics und Massenspektrometrie, Ernst-
Moritz-Arndt-Universität Greifswald) durchgeführt.
Die MS-Analyse führte zur Identifizierung von insgesamt 612 Proteinen in den von L.
monocytogenes produzierten MVs. Für weitere Analysen wurden ausschließlich
Proteine verwendet, die in mindestens 2 von 3 biologischen Replikaten identifiziert
worden sind. Nach Abzug der Proteine, die nur in eine MVs-Isolierung identifiziert
worden sind, wurden die restlichen 516 Proteine näher analysiert (Tab. 3). Anhand des
NSAF-Wertes wurde der Anteil jedes Proteins an der Gesamtmasse der Proteine in den
MVs in % berechnet.
3.2.3.1. Klassifizierung der MVs-Proteine nach Funktion
Um herauszufinden welche zelluläre Funktion die in den MVs identifizierten Proteine
haben, wurde eine funktionelle Klassifizierung durchgeführt. Hierfür wurde die
EggNOG 4.5.1-Datenbank (evolutionary genealogy of genes: Non-supervised
Orthologous Groups), die auf der COG-Ontologie basiert, verwendet (Jensen et al,
2008; Huerta-Cepas et al, 2016). Jedes COG (Cluster of Orthologous Groups) enthält
orthologe Proteine aus mindestens 3 unterschiedlichen Spezies. Sämtliche COGs
werden dabei in 25 funktionelle Kategorien, die eine Vielzahl von biologischen
Prozessen in der Zelle beschreiben, eingeteilt (Tab. 1). Diese werden den folgenden
funktionellen Gruppen zugeordnet: Metabolismus, Zelluläre Prozesse und Signale sowie
Informationsspeicherung und -prozessierung. In der Gruppe Wenig charakterisiert
werden Proteine eingeteilt, die nur eine allgemeine Funktion beschrieben haben oder
deren Funktion unbekannt ist. Die Proteine für die es in der EggNOG 4.5.1-Datenbank
kein Treffer gibt, werden der Gruppe Kein Ortholog gefunden zugeordnet. Für jede
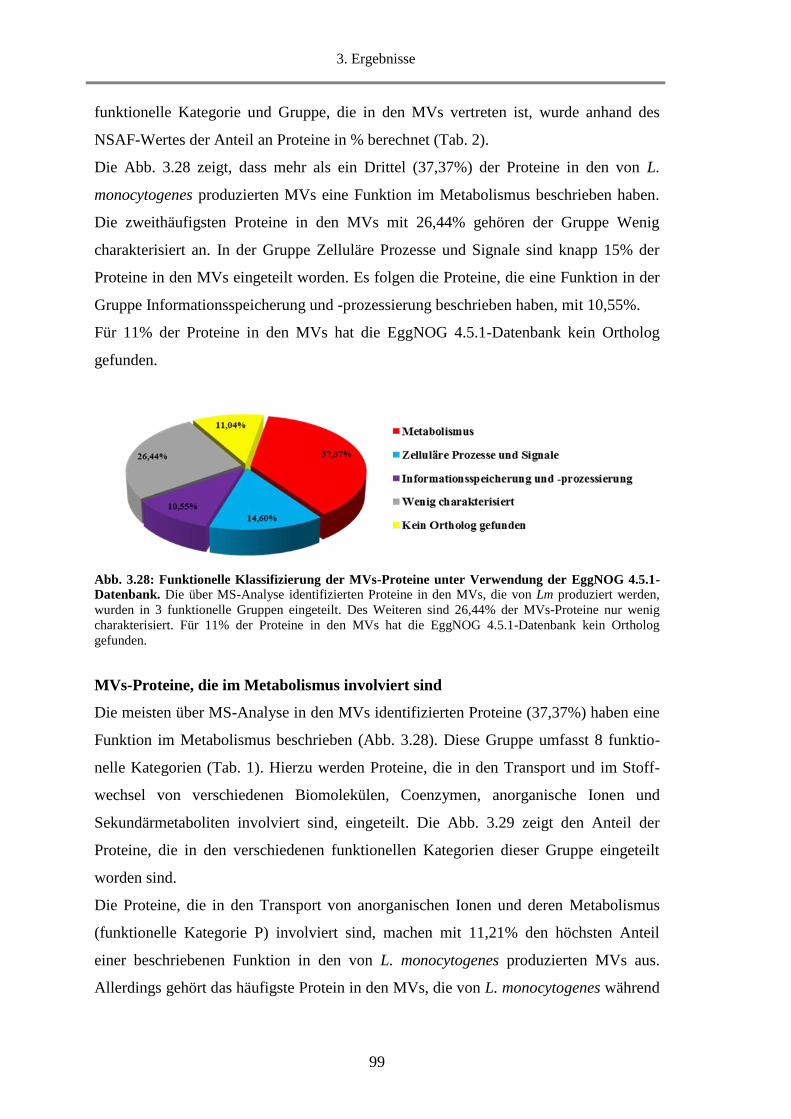
3. Ergebnisse
99
funktionelle Kategorie und Gruppe, die in den MVs vertreten ist, wurde anhand des
NSAF-Wertes der Anteil an Proteine in % berechnet (Tab. 2).
Die Abb. 3.28 zeigt, dass mehr als ein Drittel (37,37%) der Proteine in den von L.
monocytogenes produzierten MVs eine Funktion im Metabolismus beschrieben haben.
Die zweithäufigsten Proteine in den MVs mit 26,44% gehören der Gruppe Wenig
charakterisiert an. In der Gruppe Zelluläre Prozesse und Signale sind knapp 15% der
Proteine in den MVs eingeteilt worden. Es folgen die Proteine, die eine Funktion in der
Gruppe Informationsspeicherung und -prozessierung beschrieben haben, mit 10,55%.
Für 11% der Proteine in den MVs hat die EggNOG 4.5.1-Datenbank kein Ortholog
gefunden.
Abb. 3.28: Funktionelle Klassifizierung der MVs-Proteine unter Verwendung der EggNOG 4.5.1-
Datenbank. Die über MS-Analyse identifizierten Proteine in den MVs, die von Lm produziert werden,
wurden in 3 funktionelle Gruppen eingeteilt. Des Weiteren sind 26,44% der MVs-Proteine nur wenig
charakterisiert. Für 11% der Proteine in den MVs hat die EggNOG 4.5.1-Datenbank kein Ortholog
gefunden.
MVs-Proteine, die im Metabolismus involviert sind
Die meisten über MS-Analyse in den MVs identifizierten Proteine (37,37%) haben eine
Funktion im Metabolismus beschrieben (Abb. 3.28). Diese Gruppe umfasst 8 funktio-
nelle Kategorien (Tab. 1). Hierzu werden Proteine, die in den Transport und im Stoff-
wechsel von verschiedenen Biomolekülen, Coenzymen, anorganische Ionen und
Sekundärmetaboliten involviert sind, eingeteilt. Die Abb. 3.29 zeigt den Anteil der
Proteine, die in den verschiedenen funktionellen Kategorien dieser Gruppe eingeteilt
worden sind.
Die Proteine, die in den Transport von anorganischen Ionen und deren Metabolismus
(funktionelle Kategorie P) involviert sind, machen mit 11,21% den höchsten Anteil
einer beschriebenen Funktion in den von L. monocytogenes produzierten MVs aus.
Allerdings gehört das häufigste Protein in den MVs, die von L. monocytogenes während
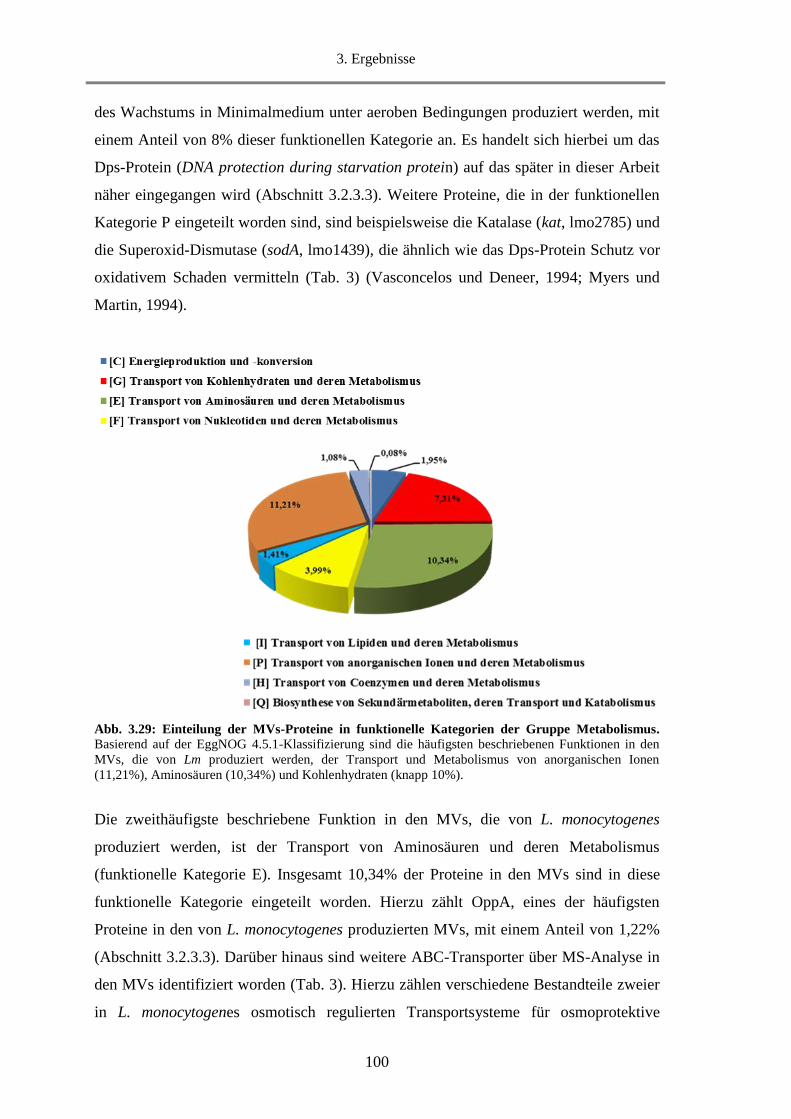
3. Ergebnisse
100
des Wachstums in Minimalmedium unter aeroben Bedingungen produziert werden, mit
einem Anteil von 8% dieser funktionellen Kategorie an. Es handelt sich hierbei um das
Dps-Protein (DNA protection during starvation protein) auf das später in dieser Arbeit
näher eingegangen wird (Abschnitt 3.2.3.3). Weitere Proteine, die in der funktionellen
Kategorie P eingeteilt worden sind, sind beispielsweise die Katalase (kat, lmo2785) und
die Superoxid-Dismutase (sodA, lmo1439), die ähnlich wie das Dps-Protein Schutz vor
oxidativem Schaden vermitteln (Tab. 3) (Vasconcelos und Deneer, 1994; Myers und
Martin, 1994).
Abb. 3.29: Einteilung der MVs-Proteine in funktionelle Kategorien der Gruppe Metabolismus.
Basierend auf der EggNOG 4.5.1-Klassifizierung sind die häufigsten beschriebenen Funktionen in den
MVs, die von Lm produziert werden, der Transport und Metabolismus von anorganischen Ionen
(11,21%), Aminosäuren (10,34%) und Kohlenhydraten (knapp 10%).
Die zweithäufigste beschriebene Funktion in den MVs, die von L. monocytogenes
produziert werden, ist der Transport von Aminosäuren und deren Metabolismus
(funktionelle Kategorie E). Insgesamt 10,34% der Proteine in den MVs sind in diese
funktionelle Kategorie eingeteilt worden. Hierzu zählt OppA, eines der häufigsten
Proteine in den von L. monocytogenes produzierten MVs, mit einem Anteil von 1,22%
(Abschnitt 3.2.3.3). Darüber hinaus sind weitere ABC-Transporter über MS-Analyse in
den MVs identifiziert worden (Tab. 3). Hierzu zählen verschiedene Bestandteile zweier
in L. monocytogenes osmotisch regulierten Transportsysteme für osmoprotektive

3. Ergebnisse
101
Substanzen, der Glycin-Betain Transporter Gbu und der Carnitin-Transporter OpuC (Ko
und Smith, 1999; Fraser et al, 2000). Zu dieser funktionellen Kategorie zählen des
Weiteren 2 Enzyme, die über α-Ketoglutarat den Kohlenhydratstoffwechsel mit der
Fixierung von Ammonium verbinden, Glutamat-Dehydrogenase (lmo0560) und
Glutamin-Synthetase (glnA, lmo1299) (Tab. 3).
Die Proteine in den funktionellen Kategorien C (Energieproduktion und -konversion)
und G (Transport von Kohlenhydraten und deren Metabolismus) ergeben gemeinsam
knapp 10% und stellen damit die dritthäufigste beschriebene Funktion in den von L.
monocytogenes produzierten MVs dar. In diese funktionellen Kategorien eingeteilte
Proteine sind in den Transport und im Stoffwechsel von Kohlenhydraten zur Energie-
gewinnung involviert. Diese Arbeit hat MVs untersucht, die von L. monocytogenes
während des Wachstums in Minimalmedium mit Glukose als Energiequelle unter
aeroben Bedingungen produziert werden. Unter diesen Bedingungen wird Glukose über
die Glykolyse und den Pentosephosphat-Weg zur Energiegewinnung und Bereitstellung
von Vorstufen für Biosynthesen abgebaut (Müller-Herbst et al, 2014). Sämtliche
Enzyme beider Stoffwechselwege sind über MS-Analyse in den MVs identifiziert
worden (Tab. 3). Das aus der Glykolyse resultierende Pyruvat wird unter aeroben
Bedingungen zum Teil durch den Pyruvat-Dehydrogenase-Komplex (pdhABCD), der
ebenfalls in den MVs identifiziert worden ist, zu Acetyl-CoA oxidiert und in den Citrat-
Zyklus eingeschleust (Kim et al, 2006). Verschiedene Enzyme, sowohl des oxidativen
als auch des reduktiven Zweigs des Citrat-Zyklus, sind in den MVs identifiziert worden
(Tab. 3). Darüber hinaus sind verschiedene Bestandteile der Atmungskette, die in L.
monocytogenes relativ einfach aufgebaut ist, über MS-Analyse in den MVs identifiziert
worden (Tab. 3) (Glaser et al, 2001; Patchett et al, 1991). Hierzu zählen 2 NADH-
Dehydrogenasen, lmo2389 und lmo2638, die durch Oxidation Reduktionsäquivalente
regenerieren und gleichzeitig Elektronen in die Atmungskette einschleussen.
Verschiedene Bestandteile der beiden terminalen Oxidasen, cytochrome aa3
(qoxABCD) und cytochrome bd (cydABCD), die diese Elektronen auf Sauerstoff
übertragen, zählen ebenfalls dazu (Müller-Herbst et al, 2014).
Des Weiteren sind Enzyme der Substratkettenphosphorylierung, die von L. monocyto-
genes auch unter aeroben Bedingungen genutzt wird, in den MVs identifiziert worden
(Tab. 3). Hierzu zählen Fumarat-Reduktase (lmo0355) und Laktat-Dehydrogenasen

3. Ergebnisse
102
(ldh1, lmo0210 und ldh2, lmo1534), deren Aktivität zur Oxidation von Reduktions-
äquivalenten und Bildung von Säuren führt (Romick et al, 1996).
Verschiedene Transporter für Kohlenhydrate, die über MS-Analyse in den MVs
identifiziert worden sind, sind ebenfalls in den funktionellen Kategorien C und G
eingeteilt worden (Tab. 3). Hierzu zählen beispielsweise verschiedene Bestandteile
zweier Phosphotransferase-Systeme (PTS), PTSMan
-2/Mpt (mptACD, lmo0096-0098)
und PTSMan
-3/Mpo (mpoABCD, lmo0781-0784). Diese Transporter befördern zusätz-
lich zur Mannose auch Glukose (Stoll und Goebel, 2010). Ein weiterer in den MVs
identifizierter Transporter für Glukose ist der PrfA-regulierte Hexose-Phosphat-
Transporter UhpT (uhpT, lmo0838), der während des Wachstums von L. monocyto-
genes im Cytosol der Wirtszellen eine essentielle Rolle spielt (Chico-Calero et al,
2002).
MVs-Proteine, die in Zelluläre Prozesse und Signale involviert sind
Knapp 15% der Proteine in den MVs, die von L. monocytogenes produziert werden,
haben eine Funktion in der Gruppe Zelluläre Prozesse und Signale beschrieben (Abb.
3.28). Diese Gruppe umfasst 10 funktionelle Kategorien (Tab. 1). Die funktionellen
Kategorien Nukleäre Struktur [Y], Zytoskelett [Z] und Extrazelluläre Strukturen [W]
sind in den MVs nicht vertreten. Die restlichen funktionellen Kategorien umfassen
Proteine, die in verschiedene zelluläre Prozesse, darunter Biogenese der Zellwand und
Cytoplasmamembran, Proteinsekretion, Signaltransduktion und Verteidigung, involviert
sind. In der Abb. 3.30 sind die verschiedenen funktionellen Kategorien und der
jeweilige Anteil an Proteine in den von L. monocytogenes produzierten MVs dargestellt.
Die Proteine, die in der funktionellen Kategorie M (Biogenese der
Zellwand/Membran/Hülle) eingeteilt worden sind, gehören mit 7,10% zu der
vierthäufigsten beschriebenen Funktion in den von L. monocytogenes produzierten
MVs. Hierbei handelt es sich um Proteine, die überwiegend in der Umgestaltung
(remodeling) der Zelloberfläche, ein für Zellwachstum und Zellteilung essentieller
Prozess, involviert sind (Tab. 3). Dieser Prozess beinhaltet u.a. ein koordiniertes
Zusammenspiel von Abbau und Biosynthese der Zellwand durch die entsprechenden
Enzyme. In den von L. monocytogenes produzierten MVs sind Enzyme, die an beide
Vorgänge beteiligt sind, über MS-Analyse identifiziert worden. So zählen
beispielsweise MurA (lmo2691), Ami (ami, lmo2558), P60 (iap, lmo0582) und P45
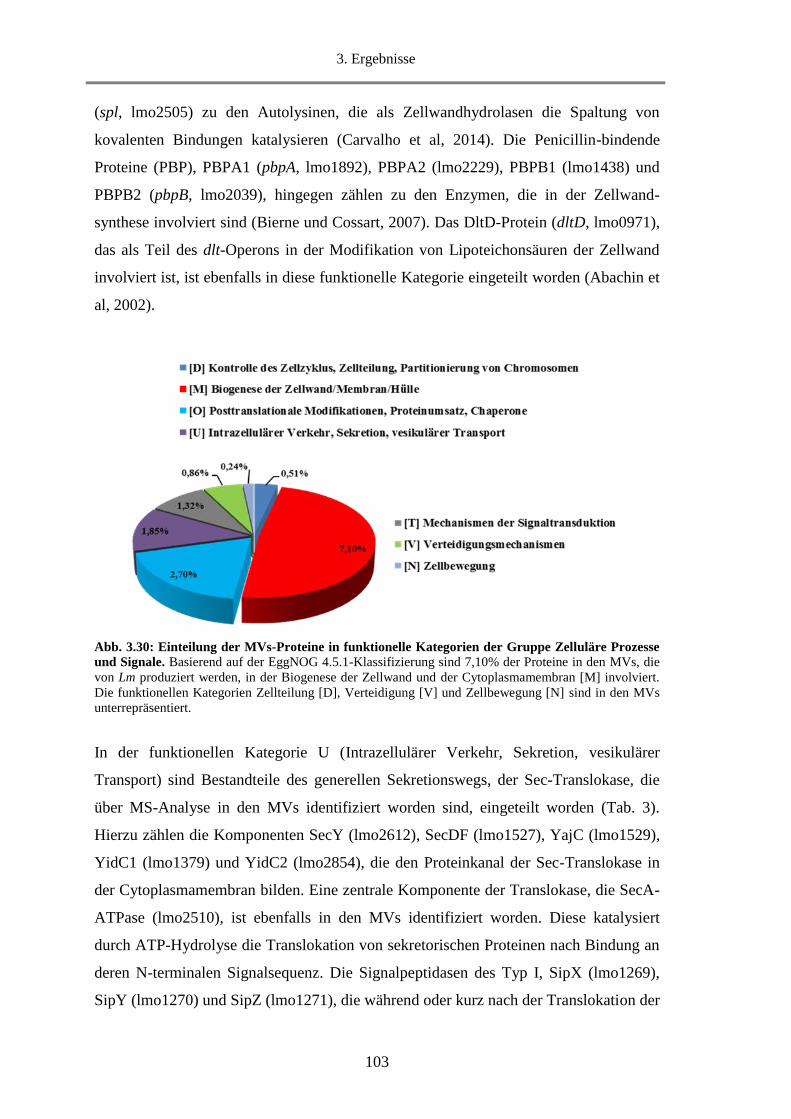
3. Ergebnisse
103
(spl, lmo2505) zu den Autolysinen, die als Zellwandhydrolasen die Spaltung von
kovalenten Bindungen katalysieren (Carvalho et al, 2014). Die Penicillin-bindende
Proteine (PBP), PBPA1 (pbpA, lmo1892), PBPA2 (lmo2229), PBPB1 (lmo1438) und
PBPB2 (pbpB, lmo2039), hingegen zählen zu den Enzymen, die in der Zellwand-
synthese involviert sind (Bierne und Cossart, 2007). Das DltD-Protein (dltD, lmo0971),
das als Teil des dlt-Operons in der Modifikation von Lipoteichonsäuren der Zellwand
involviert ist, ist ebenfalls in diese funktionelle Kategorie eingeteilt worden (Abachin et
al, 2002).
Abb. 3.30: Einteilung der MVs-Proteine in funktionelle Kategorien der Gruppe Zelluläre Prozesse
und Signale. Basierend auf der EggNOG 4.5.1-Klassifizierung sind 7,10% der Proteine in den MVs, die
von Lm produziert werden, in der Biogenese der Zellwand und der Cytoplasmamembran [M] involviert.
Die funktionellen Kategorien Zellteilung [D], Verteidigung [V] und Zellbewegung [N] sind in den MVs
unterrepräsentiert.
In der funktionellen Kategorie U (Intrazellulärer Verkehr, Sekretion, vesikulärer
Transport) sind Bestandteile des generellen Sekretionswegs, der Sec-Translokase, die
über MS-Analyse in den MVs identifiziert worden sind, eingeteilt worden (Tab. 3).
Hierzu zählen die Komponenten SecY (lmo2612), SecDF (lmo1527), YajC (lmo1529),
YidC1 (lmo1379) und YidC2 (lmo2854), die den Proteinkanal der Sec-Translokase in
der Cytoplasmamembran bilden. Eine zentrale Komponente der Translokase, die SecA-
ATPase (lmo2510), ist ebenfalls in den MVs identifiziert worden. Diese katalysiert
durch ATP-Hydrolyse die Translokation von sekretorischen Proteinen nach Bindung an
deren N-terminalen Signalsequenz. Die Signalpeptidasen des Typ I, SipX (lmo1269),
SipY (lmo1270) und SipZ (lmo1271), die während oder kurz nach der Translokation der

3. Ergebnisse
104
Proteine das Signalpeptid abspalten, sind ebenfalls in den MVs identifiziert worden
(Desvaux und Hébraud, 2006). Weitere in diesem Prozess involvierte Proteine, die über
MS-Analyse in den von L. monocytogenes produzierten MVs identifiziert worden sind,
sind Chaperone, die das targeting von sekretorischen Proteinen zur Sec-Translokase
vermitteln (Tab. 3). Hierzu zählen PrsA2 (prsA2, lmo2219), DnaK (dnaK, lmo1473),
GroEL (groEL, lmo2068) und GroES (groES, lmo2069) (Desvaux und Hébraud, 2006;
Beha et al, 2003). PrsA2 gehört mit einem Anteil von 1,34% zu den häufigsten
Proteinen in den MVs, die von L. monocytogenes produziert werden (Abschnitt 3.2.3.3).
Diese Chaperone sind in der funktionellen Kategorie O (Posttranslationale Modifika-
tionen, Proteinumsatz, Chaperone) eingeteilt worden.
Zu den Proteinen, die eine Funktion in der Signaltransduktion (funktionelle Kategorie
T) beschreiben haben, gehört beispielsweise das RNA-bindende Protein Hfq (lmo1295)
mit einem Anteil von 0,66% (Tab. 3). Dieses RNA-Chaperon hat eine Schlüsselfunktion
in der post-transkriptionellen Genregulation, indem es die Stabilität und Bindung von
sRNAs an deren mRNA-targets vermittelt (Møller et al, 2002; Geissmann und Touati,
2004).
Die funktionelle Kategorie V (Verteidigungsmechanismen) umfasst 17 Proteine, die mit
einem Anteil von 0,86% zu den unterrepräsentierten Proteinen in den MVs, die von L.
monocytogenes produziert werden, gehören (Tab. 3). Hierzu zählen überwiegend ABC-
Transporter, darunter die Strukturproteine BceA (lmo2114) und BceB (lmo2115) des
BceAB-Transporters, der Resistenz gegen das Antibiotikum Bacitracin vermittelt
(Bernard et al, 2007; Rietkötter et al, 2008).
MVs-Proteine, die in der Speicherung, Weitergabe und Umsetzung der genetischen
Information involviert sind
In der funktionellen Gruppe Informationsspeicherung und -prozessierung sind 10,55%
der MVs-Proteine, die über MS-Analyse identifiziert worden sind, eingeteilt worden
(Abb. 3.28). Diese Gruppe umfasst 5 funktionelle Kategorien (Tab. 1). Die funktio-
nellen Kategorien RNA-Prozessierung und -Modifikation [A] sowie Chromatinstruktur
und deren Dynamik [B] sind in den MVs nicht vertreten. Die restlichen funktionellen
Kategorien umfassen Proteine, die in der Replikation, Rekombination, Reparatur und
Transkription der DNA sowie Ribosomen-Aufbau und Proteinsynthese involviert sind
(Abb. 3.31).
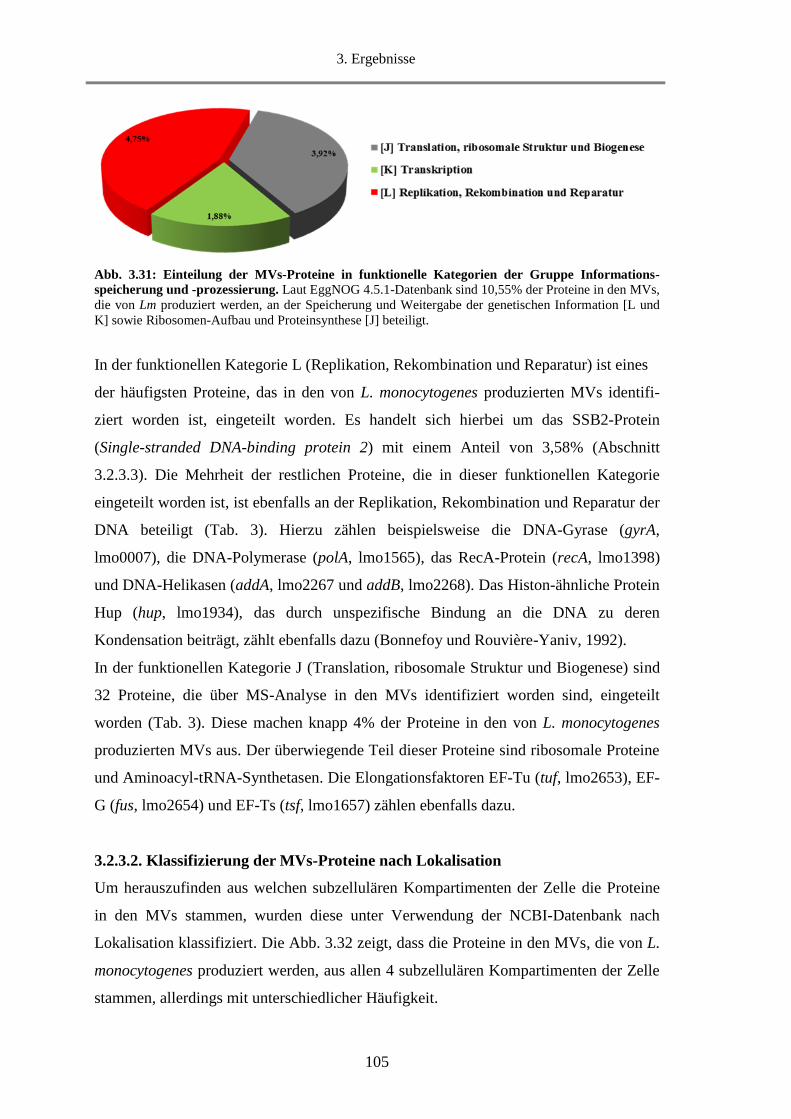
3. Ergebnisse
105
Abb. 3.31: Einteilung der MVs-Proteine in funktionelle Kategorien der Gruppe Informations-
speicherung und -prozessierung. Laut EggNOG 4.5.1-Datenbank sind 10,55% der Proteine in den MVs,
die von Lm produziert werden, an der Speicherung und Weitergabe der genetischen Information [L und
K] sowie Ribosomen-Aufbau und Proteinsynthese [J] beteiligt.
In der funktionellen Kategorie L (Replikation, Rekombination und Reparatur) ist eines
der häufigsten Proteine, das in den von L. monocytogenes produzierten MVs identifi-
ziert worden ist, eingeteilt worden. Es handelt sich hierbei um das SSB2-Protein
(Single-stranded DNA-binding protein 2) mit einem Anteil von 3,58% (Abschnitt
3.2.3.3). Die Mehrheit der restlichen Proteine, die in dieser funktionellen Kategorie
eingeteilt worden ist, ist ebenfalls an der Replikation, Rekombination und Reparatur der
DNA beteiligt (Tab. 3). Hierzu zählen beispielsweise die DNA-Gyrase (gyrA,
lmo0007), die DNA-Polymerase (polA, lmo1565), das RecA-Protein (recA, lmo1398)
und DNA-Helikasen (addA, lmo2267 und addB, lmo2268). Das Histon-ähnliche Protein
Hup (hup, lmo1934), das durch unspezifische Bindung an die DNA zu deren
Kondensation beiträgt, zählt ebenfalls dazu (Bonnefoy und Rouvière-Yaniv, 1992).
In der funktionellen Kategorie J (Translation, ribosomale Struktur und Biogenese) sind
32 Proteine, die über MS-Analyse in den MVs identifiziert worden sind, eingeteilt
worden (Tab. 3). Diese machen knapp 4% der Proteine in den von L. monocytogenes
produzierten MVs aus. Der überwiegende Teil dieser Proteine sind ribosomale Proteine
und Aminoacyl-tRNA-Synthetasen. Die Elongationsfaktoren EF-Tu (tuf, lmo2653), EF-
G (fus, lmo2654) und EF-Ts (tsf, lmo1657) zählen ebenfalls dazu.
3.2.3.2. Klassifizierung der MVs-Proteine nach Lokalisation
Um herauszufinden aus welchen subzellulären Kompartimenten der Zelle die Proteine
in den MVs stammen, wurden diese unter Verwendung der NCBI-Datenbank nach
Lokalisation klassifiziert. Die Abb. 3.32 zeigt, dass die Proteine in den MVs, die von L.
monocytogenes produziert werden, aus allen 4 subzellulären Kompartimenten der Zelle
stammen, allerdings mit unterschiedlicher Häufigkeit.
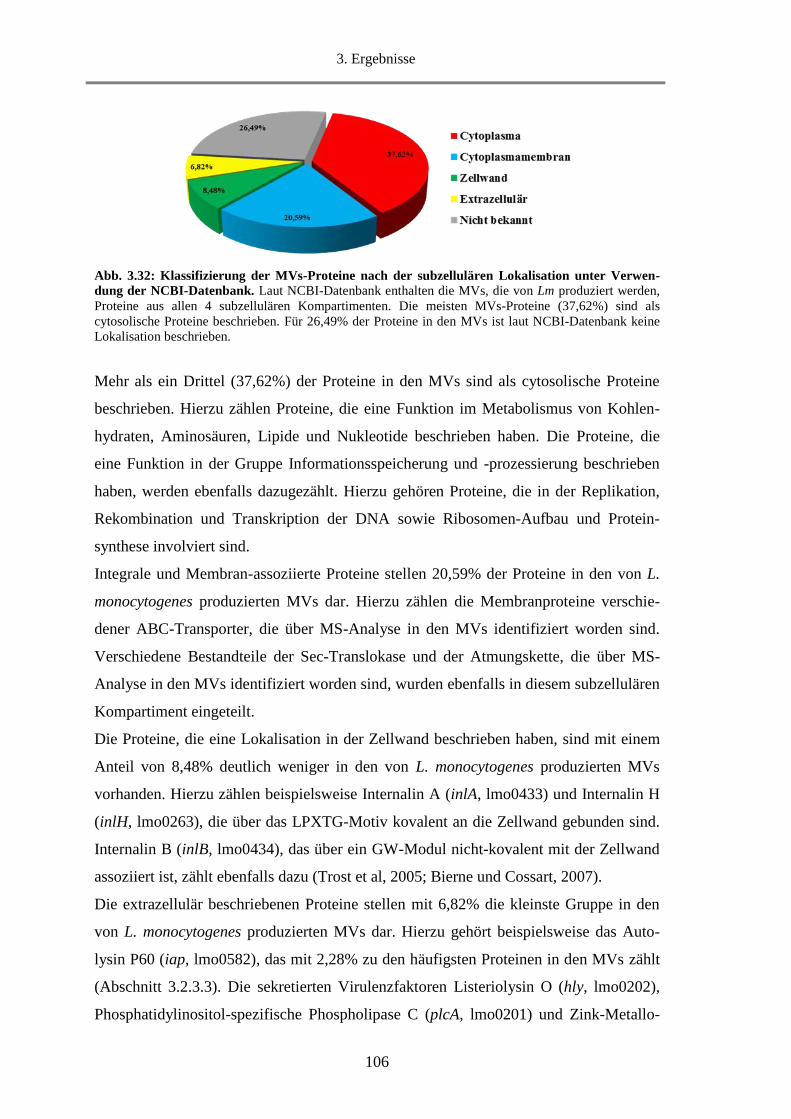
3. Ergebnisse
106
Abb. 3.32: Klassifizierung der MVs-Proteine nach der subzellulären Lokalisation unter Verwen-
dung der NCBI-Datenbank. Laut NCBI-Datenbank enthalten die MVs, die von Lm produziert werden,
Proteine aus allen 4 subzellulären Kompartimenten. Die meisten MVs-Proteine (37,62%) sind als
cytosolische Proteine beschrieben. Für 26,49% der Proteine in den MVs ist laut NCBI-Datenbank keine
Lokalisation beschrieben.
Mehr als ein Drittel (37,62%) der Proteine in den MVs sind als cytosolische Proteine
beschrieben. Hierzu zählen Proteine, die eine Funktion im Metabolismus von Kohlen-
hydraten, Aminosäuren, Lipide und Nukleotide beschrieben haben. Die Proteine, die
eine Funktion in der Gruppe Informationsspeicherung und -prozessierung beschrieben
haben, werden ebenfalls dazugezählt. Hierzu gehören Proteine, die in der Replikation,
Rekombination und Transkription der DNA sowie Ribosomen-Aufbau und Protein-
synthese involviert sind.
Integrale und Membran-assoziierte Proteine stellen 20,59% der Proteine in den von L.
monocytogenes produzierten MVs dar. Hierzu zählen die Membranproteine verschie-
dener ABC-Transporter, die über MS-Analyse in den MVs identifiziert worden sind.
Verschiedene Bestandteile der Sec-Translokase und der Atmungskette, die über MS-
Analyse in den MVs identifiziert worden sind, wurden ebenfalls in diesem subzellulären
Kompartiment eingeteilt.
Die Proteine, die eine Lokalisation in der Zellwand beschrieben haben, sind mit einem
Anteil von 8,48% deutlich weniger in den von L. monocytogenes produzierten MVs
vorhanden. Hierzu zählen beispielsweise Internalin A (inlA, lmo0433) und Internalin H
(inlH, lmo0263), die über das LPXTG-Motiv kovalent an die Zellwand gebunden sind.
Internalin B (inlB, lmo0434), das über ein GW-Modul nicht-kovalent mit der Zellwand
assoziiert ist, zählt ebenfalls dazu (Trost et al, 2005; Bierne und Cossart, 2007).
Die extrazellulär beschriebenen Proteine stellen mit 6,82% die kleinste Gruppe in den
von L. monocytogenes produzierten MVs dar. Hierzu gehört beispielsweise das Auto-
lysin P60 (iap, lmo0582), das mit 2,28% zu den häufigsten Proteinen in den MVs zählt
(Abschnitt 3.2.3.3). Die sekretierten Virulenzfaktoren Listeriolysin O (hly, lmo0202),
Phosphatidylinositol-spezifische Phospholipase C (plcA, lmo0201) und Zink-Metallo-

3. Ergebnisse
107
protease (mpl, lmo0203) werden ebenfalls dazu gezählt (Trost et al, 2005). Auf diese
Virulenzfaktoren wird im Abschnitt 3.2.3.4 näher eingegangen.
Für die restlichen Proteine (26,49%), die über MS-Analyse in den von L. monocyto-
genes produzierten MVs identifiziert worden sind, hat die NCBI-Datenbank keine
Lokalisation beschrieben. Die Mehrheit dieser Proteine ist weitgehend nicht charakte-
risiert.
3.2.3.3. Die 20 häufigsten Proteine in den MVs
In der Abb. 3.33 sind die 20 häufigsten Proteine in den MVs, die von L. monocytogenes
während des Wachstums in Minimalmedium unter aeroben Bedingungen produziert
werden, dargestellt. Deren Anteil an der Gesamtmasse der Proteine in den MVs liegt bei
jeweils über 1%. Diese 20 Proteine machen knapp die Hälfte der Masse (47,18%) aller
Proteine in den MVs aus. Die restlichen 496 Proteine, deren Anteil bei jeweils unter 1%
liegt, machen knapp 53% aus. Dieser Abschnitt beschreibt einige der häufigsten
Proteine in den von L. monocytogenes produzierten MVs.
Das häufigste Protein in den MVs ist das DNA protection during starvation protein
(dps, lmo0943) mit einem Anteil von 8,06%. Dieses Protein wurde ursprünglich in E.
coli identifiziert als eines der Hauptproteine, die in der stationären Wachstumsphase
induziert werden (Altuvia et al, 1994). Das Dps-Protein ist ein Ferritin-ähnliches
Protein, das die Bildung toxischer Hydroxyl-Radikale über die Fenton-Reaktion
verhindert, indem es frei in der Zelle vorliegendes Eisen bindet. Es reduziert darüber
hinaus vorhandenes H2O2 zu H2O, wobei das gebundene Eisen als Reduktionsmittel
dient (Zhao et al, 2002). Dadurch werden die beiden reaktiven Komponenten aus der
Fenton-Reaktion entfernt. Das Dps-Protein bietet zusätzlichen Schutz vor oxidativem
Schaden, indem es unspezifisch an DNA bindet (Martinez und Kolter, 1997). Aufgrund
dieser Eigenschaften spielt das Dps-Protein eine essentielle Rolle in der intrazellulären
Vermehrung und damit in der Virulenz von L. monocytogenes (Olsen et al, 2005).
Ein weiteres häufiges Protein in den von L. monocytogenes produzierten MVs ist das
Single-stranded DNA-binding protein 2 (ssb2, lmo2308) mit einem Anteil von 3,58%.
Durch die Bindung an DNA vermittelt das SSB2-Protein u.a. Schutz vor Abbau durch
Exo- und Endonukleasen unter Stressbedingungen (Meyer und Laine, 1990).
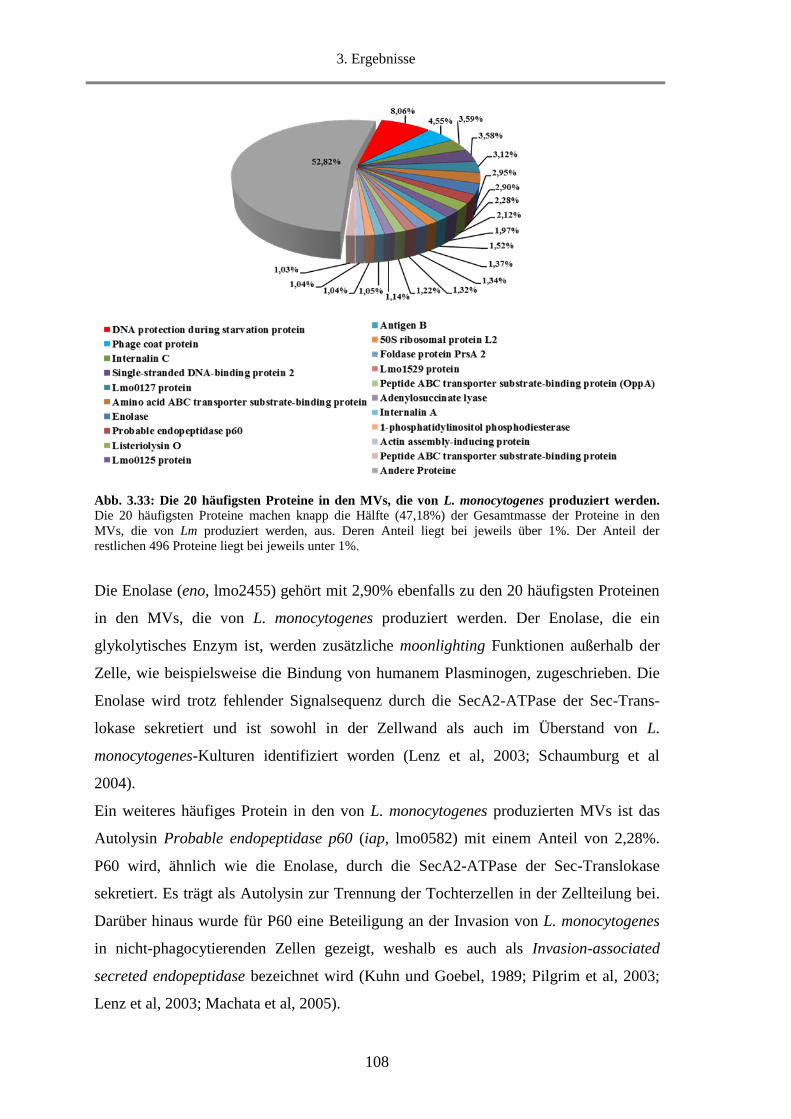
3. Ergebnisse
108
Abb. 3.33: Die 20 häufigsten Proteine in den MVs, die von L. monocytogenes produziert werden.
Die 20 häufigsten Proteine machen knapp die Hälfte (47,18%) der Gesamtmasse der Proteine in den
MVs, die von Lm produziert werden, aus. Deren Anteil liegt bei jeweils über 1%. Der Anteil der
restlichen 496 Proteine liegt bei jeweils unter 1%.
Die Enolase (eno, lmo2455) gehört mit 2,90% ebenfalls zu den 20 häufigsten Proteinen
in den MVs, die von L. monocytogenes produziert werden. Der Enolase, die ein
glykolytisches Enzym ist, werden zusätzliche moonlighting Funktionen außerhalb der
Zelle, wie beispielsweise die Bindung von humanem Plasminogen, zugeschrieben. Die
Enolase wird trotz fehlender Signalsequenz durch die SecA2-ATPase der Sec-Trans-
lokase sekretiert und ist sowohl in der Zellwand als auch im Überstand von L.
monocytogenes-Kulturen identifiziert worden (Lenz et al, 2003; Schaumburg et al
2004).
Ein weiteres häufiges Protein in den von L. monocytogenes produzierten MVs ist das
Autolysin Probable endopeptidase p60 (iap, lmo0582) mit einem Anteil von 2,28%.
P60 wird, ähnlich wie die Enolase, durch die SecA2-ATPase der Sec-Translokase
sekretiert. Es trägt als Autolysin zur Trennung der Tochterzellen in der Zellteilung bei.
Darüber hinaus wurde für P60 eine Beteiligung an der Invasion von L. monocytogenes
in nicht-phagocytierenden Zellen gezeigt, weshalb es auch als Invasion-associated
secreted endopeptidase bezeichnet wird (Kuhn und Goebel, 1989; Pilgrim et al, 2003;
Lenz et al, 2003; Machata et al, 2005).

3. Ergebnisse
109
Foldase protein PrsA2 (prsA2, lmo2219) zählt mit 1,34% ebenfalls zu den häufigsten
Proteinen in den MVs, die von L. monocytogenes produziert werden. Es unterstützt als
Chaperon die korrekte Faltung von sekretierten Proteinen im Anschluss an die Trans-
lokation (Alonzo und Freitag, 2010). PrsA2 trägt zur Virulenz von L. monocytogenes
bei, indem es die Faltung und Stabilität und dadurch die Aktivität der Virulenzfaktoren
LLO und PlcB kontrolliert. Die PrsA2-Deletionsmutante zeigt eine verringerte
Hämolyse- und Phospholipase-Aktivität und infolgedessen eine abgeschwächte
Virulenz im Mausinfektionsmodell (Alonzo et al, 2009). Ein weiterer Hinweis für die
Bedeutung von PrsA2 in der Virulenz von L. monocytogenes ist dessen Hochregulation
während des intrazellulären Wachstums in Makrophagen aufgrund der PrfA-abhängigen
Regulation (Chatterjee et al, 2006; Port und Freitag, 2007).
Ein weiteres häufiges Protein in den von L. monocytogenes produzierten MVs ist das
Oligopeptid-bindende Protein OppA (lmo2196) mit einem Anteil von 1,22%. Dieses
Protein ist Teil des Oligopeptidpermease-Operons (Opp), das den Transport von
Peptiden in die Zelle vermittelt (Borezee et al, 2000). Während des intrazellulären
Wachstums von L. monocytogenes vermittelt OppA die Aufnahme von Peptiden, die als
Quelle für Aminosäuren genutzt werden. Infolgedessen spielt OppA eine essentielle
Rolle in der Virulenz dieses Pathogens (Borezee et al, 2000; Marquis et al, 1993).
Darüber hinaus wird diesem Protein eine Rolle in der Kälteadaptation von L.
monocytogenes zugeschrieben (Borezee et al, 2000).
3.2.3.4. Virulenzfaktoren in den MVs
In den von L. monocytogenes produzierten MVs ist eine Vielzahl von Virulenzfaktoren,
die für den intrazellulären Infektionszyklus dieses Pathogens essentiell sind, über MS-
Analyse identifiziert worden. Diese machen 10% der Proteine in den MVs aus (Abb.
3.34).
Mit einem Anteil von 3,59% ist Internalin C (inlC, lmo1786) der häufigste Virulenz-
faktor in den MVs, die von L. monocytogenes produziert werden. Es handelt sich
hierbei um ein sekretiertes Protein, das homolog zu den beiden Internaline A und B ist
(Engelbrecht et al, 1996). Aufgrund der induzierten Expression während der
Vermehrung von L. monocytogenes im Cytosol von Wirtszellen wird diesem Internalin
eine Rolle in der späten Phase des Infektionszyklus zugeschriebe (Engelbrecht et al,
1996). Darüber hinaus wurde für Internalin C eine unterstützende Rolle in der InlA-

3. Ergebnisse
110
induzierten Internalisierung in nicht-phagocytierenden Zellen gezeigt (Bergmann et al,
2002). Die reduzierte Virulenz der InlC-Deletionsmutante im Mausinfektionsmodell
bestätigt dessen Rolle als Virulenzfaktor von L. monocytogenes (Engelbrecht et al,
1996).
Abb. 3.34: Virulenzfaktoren in den von L. monocytogenes produzierten MVs. Die MS-Analyse hat
zur Identifizierung von Virulenzfaktoren, die in den intrazellulären Infektionszyklus von Lm involviert
sind, in den MVs geführt. Diese machen 10% der Proteine in den MVs aus, wobei Internalin C mit einem
Anteil von knapp 4% der häufigste Virulenzfaktor ist.
In den von L. monocytogenes produzierten MVs sind weitere Internaline, Internalin A
(inlA, lmo0433) und Internalin B (inlB, lmo0434), identifiziert worden. Diese vermitteln
die Internalisierung von L. monocytogenes in nicht-phagocytierenden Zellen (Abb. 1.1)
(Mengaud et al, 1996; Shen et al, 2000).
Die für die Befreiung von L. monocytogenes aus dem Phagosom der Wirtszellen
essentiellen Virulenzfaktoren Listeriolysin O (hly, lmo0202), die beiden Phosphatidyl-
inositol- und Phosphatidylcholin-spezifischen Phospholipase C-Enzyme (plcA, lmo0201
und plcB, lmo0205) sowie die Zink-Metalloprotease (mpl, lmo0203) sind ebenfalls in
den MVs identifiziert worden (Gedde et al, 2000; Leimeister-Wächter et al, 1991;
Geoffroy et al, 1991; Domann et al, 1991). Dies gilt auch für den Aktinfilament-
akkumulierenden Faktor A (actA, lmo0204), der die intra- und interzelluläre Bewegung
von L. monocytogenes über die Akkumulierung von Aktin der Wirtszelle vermittelt
(Abb. 1.1) (Kocks et al, 1992; Domann et al, 1992).
Nach InlC ist LLO mit 2,12% der zweithäufigste Virulenzfaktor in den MVs, die von L.
monocytogenes produziert werden. Es folgen InlA, PlcA, ActA und PlcB mit jeweils
1%. Mpl und InlB machen jeweils 0,15% der Proteine in den MVs aus (Abb. 3.34).
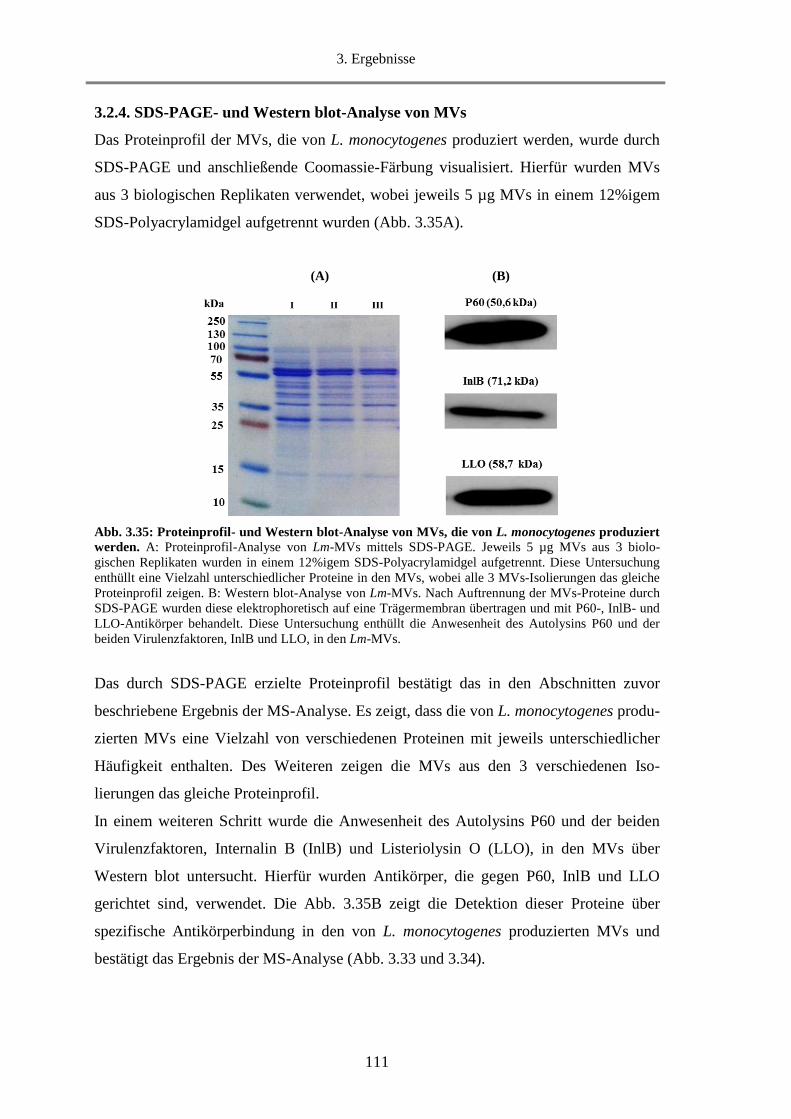
3. Ergebnisse
111
3.2.4. SDS-PAGE- und Western blot-Analyse von MVs
Das Proteinprofil der MVs, die von L. monocytogenes produziert werden, wurde durch
SDS-PAGE und anschließende Coomassie-Färbung visualisiert. Hierfür wurden MVs
aus 3 biologischen Replikaten verwendet, wobei jeweils 5 µg MVs in einem 12%igem
SDS-Polyacrylamidgel aufgetrennt wurden (Abb. 3.35A).
(A) (B)
Abb. 3.35: Proteinprofil- und Western blot-Analyse von MVs, die von L. monocytogenes produziert
werden. A: Proteinprofil-Analyse von Lm-MVs mittels SDS-PAGE. Jeweils 5 µg MVs aus 3 biolo-
gischen Replikaten wurden in einem 12%igem SDS-Polyacrylamidgel aufgetrennt. Diese Untersuchung
enthüllt eine Vielzahl unterschiedlicher Proteine in den MVs, wobei alle 3 MVs-Isolierungen das gleiche
Proteinprofil zeigen. B: Western blot-Analyse von Lm-MVs. Nach Auftrennung der MVs-Proteine durch
SDS-PAGE wurden diese elektrophoretisch auf eine Trägermembran übertragen und mit P60-, InlB- und
LLO-Antikörper behandelt. Diese Untersuchung enthüllt die Anwesenheit des Autolysins P60 und der
beiden Virulenzfaktoren, InlB und LLO, in den Lm-MVs.
Das durch SDS-PAGE erzielte Proteinprofil bestätigt das in den Abschnitten zuvor
beschriebene Ergebnis der MS-Analyse. Es zeigt, dass die von L. monocytogenes produ-
zierten MVs eine Vielzahl von verschiedenen Proteinen mit jeweils unterschiedlicher
Häufigkeit enthalten. Des Weiteren zeigen die MVs aus den 3 verschiedenen Iso-
lierungen das gleiche Proteinprofil.
In einem weiteren Schritt wurde die Anwesenheit des Autolysins P60 und der beiden
Virulenzfaktoren, Internalin B (InlB) und Listeriolysin O (LLO), in den MVs über
Western blot untersucht. Hierfür wurden Antikörper, die gegen P60, InlB und LLO
gerichtet sind, verwendet. Die Abb. 3.35B zeigt die Detektion dieser Proteine über
spezifische Antikörperbindung in den von L. monocytogenes produzierten MVs und
bestätigt das Ergebnis der MS-Analyse (Abb. 3.33 und 3.34).

3. Ergebnisse
112
3.2.5. Untersuchung der hämolytischen Aktivität von MVs
Die Anwesenheit von Listeriolysin O (LLO) in den MVs, die von L. monocytogenes
produziert werden, wurde über MS- und Western blot-Analyse identifiziert (Abb. 3.34
und 3.35B). Die hämolytische Aktivität dieses Virulenzfaktors vermittelt u.a. die
Befreiung von L. monocytogenes aus dem Phagosom der Wirtszellen (Abb. 1.1). In
einem weiteren Schritt wurde untersucht, ob das in den MVs identifizierte LLO
hämolytisch aktiv ist. Hierfür wurde ein Hämolyse-Assay durchgeführt. In einer 96er-
Well Platte wurden 5 µg MVs heruntertitriert und mit einer 1%igen Schafserythrozyten-
Lösung für 1 Stunde bei 37°C inkubiert. Anschließend wurde die Platte abzentrifugiert
und die Überstände wurden in eine neue Platte überführt (Abb. 3.36).
(A)
(B)
Abb. 3.36: Konzentrationsabhängige Lyse von Schafserythrozyten durch MVs, die von L.
monocytogenes produziert werden. 5 µg Lm-MVs wurden heruntertitriert und mit einer 1%igen
Schafserythrozyten-Lösung inkubiert. Anschließend wurde die Platte abzentrifugiert und die Überstände
wurden in eine neue Platte überführt. Die Verfärbung der Überstände in den einzelnen Wells zeigt den
Grad der Hämolyse an (B). Dabei ist eine konzentrationsabhängige Lyse von Schafserythrozyten durch
die MVs zu sehen.
Das Ausmaß der Lyse von Erythrozyten lässt sich anhand der Verfärbung, die durch
Freisetzung von Hämoglobin verursacht wird, in den einzelnen Wells optisch feststellen
und photometrisch messen. Der durch H2O erzielte hämolytische Effekt entspricht der
maximalen Hämolyse (100%-Wert). Als Negativkontrolle diente PBS. Die Abb. 3.36

3. Ergebnisse
113
zeigt, dass LLO in den von L. monocytogenes produzierten MVs hämolytisch aktiv ist.
Es zeigt des Weiteren eine konzentrationsabhängige Lyse von Schafserythrozyten durch
die MVs. Die Messung der optischen Dichte bei 405 nm ergab, dass 0,078 µg MVs
einer hämolytischen Einheit (HU) und damit 50% der durch H2O verursachten
Hämolyse entsprechen.
3.2.6. Untersuchung der Fähigkeit von MVs zur Internalisierung in PtK-Zellen
Um herauszufinden, ob die von L. monocytogenes produzierten MVs die Fähigkeit zur
Internalisierung in Wirtszellen haben, wurden PtK-Zellen mit 2 verschiedenen MVs-
Konzentrationen für 24 Stunden inkubiert. Anschließend wurden die Zellen gründlich
gewaschen und lysiert. Die Anwesenheit des Autolysins P60 im Zelllysat der PtK-
Zellen wurde über Western blot untersucht (Abb. 3.37). Hierfür wurde ein Antikörper,
der gegen P60 gerichtet ist, verwendet. P60 wurde zuvor über MS- und Western blot-
Analyse in den MVs identifiziert (Abb. 3.33 und 3.35B). Es gehört zu den häufigsten
Proteinen in den MVs, die von L. monocytogenes produziert werden.
Abb. 3.37: Internalisierung von MVs, die von L. monocytogenes produziert werden, in PtK-Zellen.
Nach Inkubation von PtK-Zellen mit 5 und 10 µg MVs wurde die Anwesenheit des Autolysins P60 im
Zelllysat über Western blot untersucht. Während die unbehandelten Zellen (Neg. Kontrolle) kein Signal
zeigen, ist eine zunehmende Signalintensität in den behandelten Ansätzen zu sehen. Die Ladekontrolle (β-
Aktin) zeigt in allen 3 Ansätzen die gleiche Intensitätsstärke.
Die Abb. 3.37 zeigt die Detektion des Autolysins P60 im Zelllysat der PtK-Zellen, die
mit den MVs inkubiert wurden. Die Signalintensität ist in dem Ansatz, der mit einer
höheren MVs-Konzentration (10 µg) inkubiert wurde, stärker. Dies spricht für eine
dosenabhängige Internalisierung von MVs in PtK-Zellen. Im Zelllysat der Zellen, die
nicht mit MVs inkubiert wurden und als Negativkontrolle dienten, ist kein Signal zu
sehen. Dies zeigt, dass der verwendete P60 Antikörper nicht unspezifisch an Zell-
strukturen von PtK-Zellen bindet. Des Weiteren zeigt die Ladekontrolle (β-Aktin) in
allen Ansätzen die gleiche Signalintensität. Das Ergebnis dieser Untersuchung zeigt,

3. Ergebnisse
114
dass die von L. monocytogenes produzierten MVs die Fähigkeit zur Internalisierung in
Wirtszellen haben.

4. Diskussion
115
4. Diskussion
4.1. Die von L. monocytogenes sekretierte RNA ist in den Fraktionen sec-RNA und
MVs-RNA gegenwärtig
Diese Arbeit hat gezeigt, dass die von L. monocytogenes sekretierte RNA in zwei
Fraktionen vorkommt, in den MVs und in einer freien, nicht MVs-assoziierten Fraktion.
Daraufhin wurden die Begriffe MVs-RNA und sec-RNA eingeführt, um eine
Unterscheidung dieser beiden sekretierten RNA-Fraktionen machen zu können. Diese
Unterscheidung wurde von früheren Studien, die sich mit der von L. monocytogenes
sekretierten RNA beschäftigt haben, nicht gemacht (Abdullah et al, 2012; Hagmann et
al, 2013). Die Erkenntnis, dass die von L. monocytogenes sekretierte RNA in zwei
Fraktionen vorkommt, wurde erst im Laufe dieser Arbeit gewonnen. Dabei wurde ein
Protokoll etabliert, das zunächst die Trennung beider RNA-Fraktionen und anschlie-
ßend die Isolierung der RNA beinhaltet (Abb. 3.1). Sowohl MVs als auch sec-RNA
werden von L. monocytogenes während des Wachstums ins Medium freigesetzt, daher
war der Rohstoff für die Isolierung beider RNA-Fraktionen der bakterienfreie Über-
stand. Nach dem dieser durch Ultrafiltration eingeengt wurde, wurden die MVs
pelletiert. Daraus erfolgte in einem weiteren Schritt die Isolierung von MVs-RNA.
Der resultierende MVs-freie Überstand diente als Grundlage für die Isolierung von sec-
RNA. Anhand dieses Protokolls wurden aus 2 L L. monocytogenes-Kulturen ca. 12 µg
MVs-RNA und ca. 120 µg sec-RNA isoliert. Infolgedessen sekretiert L. monocytogenes
10mal mehr RNA in einer freien, nicht MVs-assoziierten Form. Bislang gibt es keine
Studie, die eine Quantifizierung der RNA, die von den Bakterien sekretiert wird,
durchgeführt hat. Daher ist an dieser Stelle ein Vergleich mit anderen Bakterienspezies
nicht möglich. Dies ist allerdings analog zu eukaryotischen Zellen, die den Großteil der
extrazellulären RNA in einer freien, nicht Exosomen-assoziierten Form sekretieren
(Wang et al, 2010; Arroyo et al, 2011; Turchinovich et al, 2011).
Dass Bakterien die Produktion von Vesikeln u.a. für den Export von RNA nutzen,
wurde von weiteren Studien gezeigt, wenngleich der Mechanismus für die selektive
Sortierung von RNA in die Vesikel weitgehend unbekannt ist (Ho et al, 2015; Sjöström
et al, 2015; Resch et al, 2016). Aufgrund der Tatsache, dass L. monocytogenes 10mal
mehr RNA in einer freien, nicht MVs-assoziierten Form sekretiert, muss es für deren
Export weitere Sekretionswege geben. Eine Studie aus dem Jahr 2012 hat gezeigt, dass

4. Diskussion
116
die SecA2-ATPase der Sec-Translokase, die einen generellen Sekretionsweg für
Proteine darstellt, teilweise an der Sekretion von RNA durch L. monocytogenes beteiligt
ist. Die SecA2-Deletionsmutante zeigt eine reduzierte Sekretion von RNA ins extra-
zelluläre Milieu (Desvaux und Hébraud, 2006; Abdullah et al, 2012). Die SecA2-
ATPase vermittelt u.a. die Sekretion von sogenannten moonlighting Proteinen, wie
beispielsweise DnaK, GroEL, EF-Tu, Enolase, RNA-Polymerase und ribosomale
Proteine, die für deren RNA-Bindung bekannt sind (Lenz et al, 2003; Georgellis et al,
1995; Py et al, 1994; Miczak et al, 1996; Vanzo et al, 1998). Es wäre daher denkbar,
dass L. monocytogenes RNA über Proteinsekretionswege in Verbindung mit RNA-
bindenden Proteinen sekretiert. Eine Sekretion von RNA über RNA-bindende Proteine,
wie beispielsweise Argonaute 2 (AGO2), Nucleophosmin (NPM1) und High Density
Lipoprotein (HDL), wird auch bei Eukaryoten vermutet (Wang et al, 2010; Turchi-
novich et al, 2011; Vickers et al, 2011). Dies würde die Stabilität der freien, nicht MVs-
assoziierten RNA im extrazellulären Milieu erklären. Darüber hinaus könnte diese
Transportform deren Aufnahme durch target-Zellen vermitteln, wie das bereits für
HDL-assoziierte miRNAs gezeigt wurde (Vickers et al, 2011).
4.2. Unterschiedliche Zusammensetzung von sec-RNA, MVs-RNA und zellulärer
RNA
Diese Arbeit hat erstmals die Zusammensetzung der RNA, die von L. monocytogenes
sekretiert wird, identifiziert. Durch die Sequenzierung von sec-RNA und MVs-RNA
wurde gezeigt, dass die von L. monocytogenes sekretierte RNA alle bekannten RNA-
Spezies enthält, rRNA, tRNA, mRNA und sRNA (Abb. 3.5). Darüber hinaus wurde
gezeigt, dass die beiden sekretierten RNA-Fraktionen sich sowohl untereinander als
auch von der zellulären RNA unterscheiden, bezüglich Vorkommen und Häufigkeit
bestimmter RNA-Spezies. Diese Feststellung und die hohe Reproduzierbarkeit der
erzielten Sequenzierungsdaten aus drei biologischen Replikaten weisen auf einen
selektiven Export von RNA durch L. monocytogenes hin. Ähnliche Beobachtungen
wurden von weiteren Studien, die sich mit der von verschiedenen Bakterienspezies
sekretierten RNA beschäftigt haben, berichtet (Ghosal et al, 2015; Koeppen et al, 2016;
Blenkiron et al, 2016). Daher wird für die sekretierte RNA eine Funktion in der
interzellulären Kommunikation, in der pathogene Bakterien die Genexpression ihrer
Wirtszellen manipulieren, vermutet. Dies wird zum einen durch Studien begründet, die

4. Diskussion
117
den Transfer von RNA über Vesikel in Wirtszellen gezeigt haben (Koeppen et al, 2016;
Blenkiron et al, 2016). Zum anderen wurde eine veränderte Expression von bestimmten
Proteinen, die von Vesikel-assoziierten RNAs in Wirtszellen ausgelöst wurde, gezeigt
(Koeppen et al, 2016). Weitere Analysen wären notwendig, um eine genaue Aussage
darüber machen zu können, ob die von L. monocytogenes sekretierte RNA eine
Funktion in der interzellulären Kommunikation erfüllt.
Diese Arbeit hat allerdings gezeigt, dass die von L. monocytogenes sekretierten RNA-
Fraktionen, sec-RNA und MVs-RNA, eine hohe IFN-β Antwort nach Transfektion in
Wirtszellen auslösen (Abb. 3.4). Hierdurch wurde gezeigt, dass die in BMDMs ausge-
löste IFN-β Antwort durch sec-RNA ca. 70fach und durch MVs-RNA ca. 20fach höher
ist im Vergleich zu der zellulären RNA. Wie bereits eingangs erwähnt, erhöht die von L.
monocytogenes während einer Infektion ausgelöste IFN-β Antwort die Anfälligkeit des
Wirtes gegenüber diesem Pathogen und fördert dessen Vermehrung (Carrero et al,
2004; O`Connell et al, 2004). So wurde beispielsweise diesbezüglich gezeigt, dass die
von L. monocytogenes ausgelöste Typ-I-IFN Antwort die polarisierte Polymerisation
von Aktin der Wirtszelle durch den Virulenzfaktor ActA fördert, wodurch dessen
intrazelluläre Bewegung und Infektion von Nachbarzellen unterstützt wird (Osborne et
al, 2017). Die Induktion von IFN-β über die Sekretion von RNA ist daher als eine Form
von Einflussnahme durch L. monocytogenes auf die Wirtszellen anzusehen.
4.3. Anreicherung von kleinen nicht-kodierenden RNAs in MVs-RNA
Ein erster Eindruck über die Zusammensetzung von sec-RNA und MVs-RNA wurde
durch Profilanalyse mittels Agilent 2100 Bioanalyzer erzielt. Hierdurch wurde die
Anwesenheit von kleinen nicht-kodierenden RNAs in beide sekretierten Fraktionen
gezeigt (Abb. 3.2). Die höhere Auflösung dieser RNA-Population hat gezeigt, dass in
MVs-RNA der überwiegende Teil aus 20 Nt langen RNAs besteht (Abb. 3.3B). Diese
Länge entspricht der von eukaryotischen miRNAs, die als kleine nicht-kodierende
RNAs eine Schlüsselfunktion in der post-transkriptionellen Genregulation haben. RNAs
mit dieser Länge wurden in verschiedenen Bakterienspezies, darunter E. coli und
Streptococcus mutans, identifiziert und werden als msRNAs (microRNA-size, small
RNAs) bezeichnet (Kang et al, 2013; Lee und Hong, 2011). Deren Funktion ist
weitgehend unbekannt, allerdings wird aufgrund der Häufigkeit und der Diversität ihres
Vorkommens in den Bakterienzellen eine regulatorische Funktion angenommen.

4. Diskussion
118
Eine weitere Studie hat die Anwesenheit von msRNAs in den MVs, die von Strepto-
coccus sanguinis produziert werden, gezeigt (Choi et al, 2016). Eine mögliche Funktion
dieser RNA-Spezies in den von Bakterien produzierten MVs wird in der interzellulären
Kommunikation vermutet. Diese Vermutung wird von zahlreichen Studien aus der
Eukaryoten-Forschung, die den Export von miRNAs über Exosomen gezeigt haben,
begründet (Chevillet et al, 2014; Baldwin et al, 2017). Darüber hinaus wurde eine
Einflussnahme auf die Genregulation von Empfängerzellen nach Transfer von miRNAs
über Exosomen gezeigt (Kogure et al, 2011).
4.4. Unterschiedliche IFN-β Induktion durch sRNAs, die in sec-RNA und MVs-
RNA identifiziert worden sind
Aufgrund der höheren IFN-β Induktion durch die sekretierten RNA-Fraktionen im
Vergleich zu der zellulären RNA wurde untersucht welchen Beitrag einzelne RNAs, die
in sec-RNA und MVs-RNA identifiziert worden sind, an der IFN-β Antwort haben. Die
Transfektion der einzelnen sRNAs, die durch in vitro Transkription hergestellt worden
sind, in HEK293-Zellen hat gezeigt, dass diese ein unterschiedliches Potential besitzen
IFN-β zu induzieren (Abb. 3.9). So haben rli32, rli47, rli99 und rli100 die höchste IFN-
β Antwort ausgelöst. Andere hingegen, wie rli98, rli111, rli114, rliG, ssrA und ssrS,
haben eine sehr schwache IFN-β Antwort ausgelöst. Weitere sRNAs, wie rli50, rli51,
rli56, rli60, rli62, LhrA und SRP, haben ein moderates IFN-β Induktionspotential
gezeigt. In einem weiteren Versuch, in dem einige dieser sRNAs in BMDMs transfiziert
worden sind, wurde das unterschiedliche Potential einzelner sRNAs IFN-β zu indu-
zieren, bestätigt (Abb. 3.10).
Die Fähigkeit einer gegebenen sRNA IFN-β zu induzieren, korreliert nicht mit der
Häufigkeit ihres Vorkommens in den beiden sekretierten RNA-Fraktionen. So zählen
beispielsweise ssrA und ssrS zu den häufigsten sRNAs in sec-RNA und MVs-RNA,
dennoch haben diese sRNAs eine sehr schwache IFN-β Antwort ausgelöst. SRP, die
häufigste sRNA in den beiden sekretierten RNA-Fraktionen, hat eine moderate IFN-β
Antwort nach Transfektion in HEK293-Zellen ausgelöst. Anders hingegen rli100, das
im Vergleich dazu eine viel geringere Anzahl an reads in sec-RNA und MVs-RNA
aufweist und dennoch zu den sRNAs mit der höchsten IFN-β Induktion zählt.
Einige in den sekretierten RNA-Fraktionen von L. monocytogenes identifizierten
sRNAs sind von früheren Studien beschrieben worden. Für rli32 beispielsweise wird,

4. Diskussion
119
aufgrund der höheren Transkription während des intrazellulären Wachstums von L.
monocytogenes und dessen möglichen mRNA-targets, eine Rolle in der Virulenz
vermutet (Mraheil et al, 2011; Toledo-Arana et al, 2009). Ähnliches wird für rli47
angenommen, dessen Transkription von dem alternativen Sigma Faktor B, der die
generelle Stressantwort in der stationären Wachstumsphase und während der Infektion
kontrolliert, abhängt (Hain et al, 2008; Mraheil et al, 2011; Oliver et al, 2009). Des
Weiteren wurde über Transkriptom-Analyse gezeigt, dass LhrA in der stationären
Wachstumsphase die Expression von ca. 300 Genen reguliert. Experimentell wurde die
chiA-mRNA, die für eine der beiden Chitinasen von L. monocytogenes kodiert, als
target nachgewiesen (Christiansen et al, 2006; Nielsen et al, 2011). Für rli60 wurde eine
Rolle in der Anpassung von L. monocytogenes an verschiedenen Stressbedingungen und
in der Biofilmbildung gezeigt. Für rli50 hingegen wird, aufgrund der reduzierten Viru-
lenz der rli50-Deletionsmutante im Mausinfektionsmodell, eine Rolle in der intra-
zellulären Vermehrung von L. monocytogenes vermutet (Peng et al, 2016; Mraheil et al,
2011).
4.4.1. RIG-I-Abhängigkeit der IFN-β Induktion durch die sRNAs
Diese Arbeit hat gezeigt, dass die von L. monocytogenes sekretierten sRNAs ein
unterschiedliches Potential besitzen IFN-β zu induzieren. In einem weiteren Schritt
wurde die Abhängigkeit der IFN-β Antwort, die von den einzelnen sRNAs in HEK293-
Zellen ausgelöst wurde, von den Rezeptoren RIG-I und MDA5 untersucht. Hierfür
wurden einige der 23 ausgesuchten sRNAs in RIG-I-defiziente Zellen transfiziert.
Darunter waren sRNAs, die in Wildtypzellen eine starke (rli32, rli47, rli99 und rli110)
sowie eine sehr schwache bis moderate (rli48, rli51, rli60, rli62, rli105, SRP und ssrS)
IFN-β Antwort ausgelöst hatten (Abb. 3.9). In RIG-I-defiziente Zellen haben diese
sRNAs, unabhängig von deren in Wildtypzellen gezeigten IFN-β Induktionspotential,
eine sehr schwache IFN-β Antwort ausgelöst (Abb. 3.12). Hierdurch wurde gezeigt,
dass die von den sRNAs in HEK293-Zellen ausgelöste IFN-β Antwort von dem
Rezeptor RIG-I abhängig ist. Dies wurde durch Transfektion von rli32, das unter den
ausgesuchten sRNAs das höchste IFN-β Induktionspotential gezeigt hatte, in HEK293-
Zellen mit einer Defizienz in eines der beiden Rezeptoren, RIG-I oder MDA5, bestätigt
(Abb. 3.13). RIG-I-defiziente Zellen zeigen eine signifikante Reduktion der IFN-β
Antwort im Vergleich zu den Wildtypzellen. Anders hingegen MDA5-defiziente Zellen,

4. Diskussion
120
die keinen signifikanten Unterschied in der IFN-β Antwort im Vergleich zu den
Wildtypzellen zeigen. Die signifikante Reduktion der IFN-β Induktion durch rli32 nach
Entfernung der 5`-Triphosphat-Gruppe durch Dephosphorylierung weist ebenfalls auf
eine RIG-I-Abhängigkeit der IFN-β Antwort durch die sRNAs in HEK293-Zellen hin.
Die erforderliche molekulare Struktur von RNAs für die Bindung und Aktivierung des
Rezeptors RIG-I wurde von zwei Studien identifiziert. Diese haben gezeigt, dass RNAs,
die am 5`-Ende eine basengepaarte Struktur mit einer Triphosphat-Gruppe besitzen, der
optimale Ligand für diesen Rezeptor darstellen. Dabei ist die Anwesenheit einer
Triphosphat-Gruppe am 5`-Ende von RNAs eine Grundvoraussetzung für deren
Bindung durch RIG-I (Schlee et al, 2009; Schmidt et al, 2009). Nach Entfernung der 5`-
Triphosphat-Gruppe durch Dephosphorylierung verliert rli32 die Fähigkeit einer effek-
tiven Bindung durch RIG-I und infolgedessen das hohe immunstimulatorische Potential.
4.4.2. Protektion von HEK293-Zellen gegen das Influenza A Virus durch die
sRNAs
Zahlreiche Studien haben die Bedeutung des Rezeptors RIG-I in der antiviralen Immun-
antwort gezeigt. Nach Detektion viraler RNA im Cytosol der infizierten Zellen löst
RIG-I über die Produktion von Typ-I-IFN antivirale Mechanismen aus, die zur Inhibi-
tion der viralen Vermehrung führen (Pichlmair et al, 2006; Kato et al, 2006; Kato et al,
2011). In einem weiteren Schritt wurde untersucht, ob die von den sRNAs ausgelöste
IFN-β Antwort die virale Replikation nach Infektion von HEK293-Zellen mit dem
Influenza A Virus (A/PR/8/34/(H1N1)) inhibiert. Diese Untersuchung hat gezeigt, dass
die Vorbehandlung von HEK293-Zellen mit rli32 und rli47, die im Vorfeld eine starke
IFN-β Antwort ausgelöst hatten, zu einer signifikanten Reduktion des Virustiters in den
Überständen der infizierten Zellen geführt hat (Abb. 3.15). Infolgedessen hat die von
den sRNAs ausgelöste IFN-β Antwort zu einer Inhibition der viralen Replikation in den
infizierten Zellen geführt. Anders hingegen ssrS, das nicht zu einer Inhibition der
viralen Replikation geführt hat. Diese sRNA hat im Vorfeld eine sehr schwache IFN-β
Antwort in HEK293-Zellen ausgelöst. Aufgrund des sehr schwachen Potentials von ssrS
eine IFN-β Antwort auszulösen, blieb die Inhibition der viralen Replikation in den
infizierten Zellen aus. Hierdurch wurde gezeigt, dass das unterschiedliche IFN-β
Induktionspotential einzelner sRNAs zu einer entsprechend mehr oder weniger starken
Inhibition der viralen Replikation geführt hat.

4. Diskussion
121
Des Weiteren wurde eine konzentrationsabhängige Inhibition der viralen Replikation
durch rli32 gezeigt. Die Vorbehandlung von HEK293-Zellen mit zunehmenden rli32
Konzentrationen hat zu einer zunehmenden Reduktion des Virustiters in den Über-
ständen der infizierten Zellen geführt (Abb. 3.17). Infolgedessen hat der Einsatz einer
höheren rli32 Konzentration zu einer höheren IFN-β Antwort mit entsprechend höherer
antiviraler Aktivität in den Zellen geführt.
In einem weiteren Versuch wurde die direkte Beteiligung von RIG-I an der antiviralen
Antwort, die von den sRNAs ausgelöst wurde, gezeigt. Die Vorbehandlung von RIG-I-
defizienten Zellen mit rli32 und rli47 hat nicht zu einer Inhibition der viralen Repli-
kation geführt (Abb. 3.16A). In MDA5-defiziente Zellen hingegen wurde eine signifi-
kante Reduktion des Virustiters festgestellt (Abb. 3.16B). Hierdurch hat diese Arbeit
erstmals die RIG-I-abhängige antivirale Aktivität von individuellen bakteriellen sRNAs
gezeigt. Verschiedene Studien haben die antivirale Aktivität von RIG-I-aktivierenden
RNA-Oligonukleotiden gezeigt. So wurde beispielsweise gezeigt, dass diese synthe-
tischen RIG-I-Liganden humane Lungenepithelzelllinien vor der Infektion mit den von
1918 und 2009 pandemischen Influenza A Viren schützen (Ranjan et al, 2010). Eine
weitere Studie hat gezeigt, dass die intravenöse Injektion von Mäusen mit einem
solchen RNA-Oligonukleotid Schutz gegen eine letale Infektion mit dem Influenza A
Virus bietet (Coch et al, 2017).
4.5. Das Motiv 2 ist für die hohe IFN-β Induktion durch rli32 verantwortlich
Rli32 hat sich unter den 23 ausgesuchten sRNAs, die in sec-RNA und MVs-RNA
identifiziert worden sind, als die sRNA mit dem höchsten IFN-β Induktionspotential
herausgestellt. Eine in silico Vorhersage der Sekundärstruktur von rli32 mit dem
RNAfold WebServer hat gezeigt, dass diese sRNA aus 3 Motiven besteht, die
verschiedene stem-loop Strukturen aufweisen (Gruber et al, 2008). Um herauszufinden
welche strukturelle Beschaffenheit für die hohe immunstimulatorische Aktivität von
rli32 verantwortlich ist, wurden die einzelnen Motive nach deren Herstellung in
BMDMs transfiziert. Diese Untersuchung hat Unterschiede, bezüglich der Fähigkeit
einzelner Motive IFN-β zu induzieren, gezeigt (Abb. 3.14). Während die Motive 1 und
3 eine sehr schwache bis moderate IFN-β Antwort ausgelöst hatten, hat das Motiv 2
eine ähnlich hohe IFN-β Antwort ausgelöst wie rli32 selbst. Basierend auf dieses
Ergebnis ist Motiv 2 für das hohe IFN-β Induktionspotential von rli32 verantwortlich.

4. Diskussion
122
Darüber hinaus hat diese Untersuchung gezeigt, dass Unterschiede in der strukturellen
Beschaffenheit von RNAs für die unterschiedliche IFN-β Antwort verantwortlich sind.
Zusätzlich zu der 5`-Triphosphat-Gruppe bestimmt die Struktur einer gegebenen RNA
die Bindung und Aktivierung des Rezeptors RIG-I. Dies ist möglicherweise der Grund
für die unterschiedliche IFN-β Induktion durch die von L. monocytogenes sekretierten
sRNAs.
4.6. Hohe Typ-I-IFN Induktion durch rli32 in der Maus
Die Untersuchung des immunstimulatorischen Potentials von rli32 in vivo hat gezeigt,
dass diese sRNA in beide Organe der Maus, Leber und Milz, eine hohe Typ-I-IFN
Antwort ausgelöst hat (Abb. 3.19 und 3.20). SsrS, das sich in den in vitro Transfektions-
versuchen als eine sRNA mit einem sehr schwachen IFN-β Induktionspotential
herausgestellt hatte, hat ebenfalls eine hohe Typ-I-IFN Antwort ausgelöst. Darüber
hinaus hat ssrS in der Milz eine höhere Typ-I-IFN Antwort ausgelöst als rli32. Die Typ-
I-IFN Antwort ist insgesamt in der Milz höher ausgefallen als in der Leber. Es wurde
gezeigt, dass die DCs-Subpopulationen in der Leber schwache Aktivatoren des Immun-
systems sind. Diese sind weniger reif, erfassen weniger Antigene und induzieren eine
schwächere T-Zelle Stimulation als die DCs in de Milz (Pillarisetty et al, 2004). Dies
erklärt die schwächere Typ-I-IFN Antwort in der Leber gegenüber der Milz.
Die von ssrS ausgelöste hohe Typ-I-IFN Antwort in der Maus ist möglicherweise auf
dessen Detektion durch pDCs, die das TLR-System und das Adapterprotein MyD88 für
die Produktion von Typ-I-IFN nutzen, zurückzuführen (Kawai und Akira, 2010). Diese
DCs-Subpopulation ist für die hohe Typ-I-IFN Antwort nach Detektion viraler RNA
bekannt (Cella et al, 1999; Colonna et al, 2002; Diebold et al, 2003). So lösen beispiels-
weise pDCs einen antiviralen Status nach Detektion des Hepatitis C Virus (HCV) über
TLR7 aus (Takahashi et al, 2010). Die Erkennung von ssrS durch die aktiveren pDCs in
der Milz ist möglicherweise der Grund dafür, dass ssrS eine höhere Typ-I-IFN Antwort
in der Milz ausgelöst hat als rli32.
4.7. Erhöhte IFN-β Induktion durch die sekretierten RNA-Fraktionen und MVs
nach Überexpression von rli32 in L. monocytogenes
Die Überexpression von rli32 in L. monocytogenes hat zu einem signifikanten Anstieg
der IFN-β Induktion durch alle RNA-Fraktionen, die von diesem Stamm isoliert und in

4. Diskussion
123
BMDMs transfiziert wurden, geführt (Abb. 3.22). Dabei hat sec-RNA die höchste IFN-
β Antwort ausgelöst, gefolgt von MVs-RNA und zellulärer RNA. Dadurch wurde das
IFN-β Induktionspotential der sec-RNA von L. monocytogenes um ein 10faches erhöht,
von ca. 1000fach auf ca. 10000fach. Darüber hinaus hat diese Untersuchung gezeigt,
dass nicht nur durch in vitro Transkription hergestelltes rli32 die Fähigkeit besitzt eine
hohe IFN-β Antwort auszulösen. Das von den Bakterienzellen selbst transkribierte rli32
besitzt das Potential IFN-β zu induzieren. Die von dem ssrS-überexprimierenden Stamm
isolierte RNA hingegen hat nicht zu einem Anstieg der IFN-β Antwort nach Transfek-
tion in BMDMs geführt. Dieses Ergebnis bestätigt die im Vorfeld durchgeführten in
vitro Transfektionsversuche und verdeutlicht, dass einzelne in den sekretierten RNA-
Fraktionen vorhandene sRNAs ein unterschiedliches Potential besitzen IFN-β zu
induzieren. Infolgedessen bestimmt die Zusammensetzung der sekretierten RNA-
Fraktionen aus verschiedenen RNAs mit unterschiedlichem immunstimulatorischem
Potential letztendlich die Höhe der IFN-β Antwort.
In HEK293-Zellen zeigen die sekretierten RNA-Fraktionen von Lm-pERL3-rli32,
ähnlich wie in BMDMs, eine höhere IFN-β Antwort als die zelluläre RNA (Abb. 3.23).
Anders als in BMDMs hat MVs-RNA eine höhere IFN-β Antwort ausgelöst als sec-
RNA. Ein weiterer Unterschied ist die höhere IFN-β Induktion durch die sekretierten
RNA-Fraktionen von Lm-pERL3-ssrS im Vergleich zu den RNA-Fraktionen, die vom
Wildtyp isoliert wurden. In einem weiteren Versuch wurde allerdings gezeigt, dass die
von den sekretierten RNA-Fraktionen der Stämme Lm-pERL3-rli32 und Lm-pERL3-
ssrS ausgelöste IFN-β Antwort in HEK293-Zellen von dem Rezeptor RIG-I abhängig ist
(Abb. 3.24). In BMDMs wurde eine signifikante Reduktion der IFN-β Antwort nach
Dephosphorylierung und Transfektion von sec-RNA und MVs-RNA festgestellt (Abb.
3.4). Dies spricht dafür, dass auch in BMDMs der Rezeptor RIG-I in der Detektion der
RNA, die von L. monocytogenes sekretiert wird, involviert ist. Allerdings könnten hier
auch andere Rezeptoren, wie beispielsweise TLRs, eine Rolle spielen. Weitere Analy-
sen wären notwendig, um eine genaue Aussage darüber machen zu können.
Des Weiteren hat die Überexpression von rli32 in L. monocytogenes das immun-
stimulatorische Potential der MVs erhöht (Abb. 3.25). Die von Lm-pERL3-rli32
isolierten MVs haben eine höhere IFN-β Antwort in BMDMs ausgelöst als die MVs, die
vom Wildtyp isoliert wurden. Das IFN-β Induktionspotential der MVs hingegen, die
von Lm-pERL3-ssrS isoliert wurden, liegt unter dem Niveau der MVs vom Wildtyp.

4. Diskussion
124
Infolgedessen hat das unterschiedliche IFN-β Induktionspotential der beiden sRNAs zu
der unterschiedlichen IFN-β Antwort durch die MVs der beiden Stämme geführt.
4.8. Produktion von MVs durch L. monocytogenes
Diese Arbeit hat die Produktion von MVs durch L. monocytogenes in der exponentiellen
Phase während des Wachstums in Minimalmedium unter aeroben Bedingungen gezeigt.
Die Charakterisierung der von L. monocytogenes produzierten MVs unter diesen
Bedingungen beinhaltete u.a. die Visualisierung und Bestimmung der Größe, Anzahl
und Proteinkonzentration. Die optische Darstellung der MVs wurde durch
Transmissions- und Rasterelektronenmikroskopie durchgeführt (Abb. 3.26). Diese
Visualisierung zeigt, dass L. monocytogenes sphärisch geformte MVs, die mehr oder
weniger einheitlich sind in ihrer Größe, produziert. Die Größenbestimmung der MVs
durch NTA (Nanoparticle Tracking Analysis) ergab, dass die von L. monocytogenes
produzierten MVs eine durchschnittliche Größe von 130,7 ± 1,0 nm haben (Abb. 3.27).
Eine Studie, die im Jahr 2013 erstmals die Produktion von MVs durch L. monocyto-
genes gezeigt hatte, hat dabei eine Größe von 20-100 nm festgestellt (Lee et al, 2013).
Allerdings hat diese Studie MVs untersucht, die von L. monocytogenes in der
stationären Phase während des Wachstums in BHI-Medium unter aeroben Bedingungen
produziert werden. Die Anzahl der MVs, die aus 2 L L. monocytogenes-Kulturen
isoliert wurden, beträgt laut NTA-Analyse ca. 70 × 109. Die Bestimmung der
Proteinkonzentration mit der Bradford-Methode ergab 150-200 µg/2 L Bakterienkultur.
Die im Jahr 2013 durchgeführte Studie hat eine Proteinkonzentration von 121 ± 6,2
µg/1 L L. monocytogenes-Kultur angegeben. Diese Unterschiede zwischen der
vorliegenden Arbeit und der Studie von Lee et al (2013) zeigen, dass die Produktion
von MVs durch L. monocytogenes von äußeren Faktoren, wie beispielsweise das
Wachstumsmedium, beeinflusst wird. Dass die Produktion von Vesikeln durch
Bakterien von äußeren Bedingungen beeinflusst wird, wurde auch von anderen Studien
gezeigt. Eine davon beispielsweise beschreibt den Einfluss des Wachstumsmediums auf
die MVs-Produktion durch das Pflanzen-Pathogen Xanthomonas campestris (McBroom
und Kuehn, 2007; Kadurugamuwa und Beveridge, 1995; Sidhu et al, 2008).

4. Diskussion
125
4.9. MVs enthalten Enzyme, die möglicherweise an deren Biogenese beteiligt sind
Die durch Rasterelektronenmikroskopie (REM) erzielten Bilder zeigen die Produktion
von MVs durch L. monocytogenes (Abb. 3.26C und 3.26D). Darauf kann man erkennen,
dass die Produktion von MVs über die gesamte Oberfläche von L. monocytogenes
erfolgt. Der genaue Biogenese-Mechanismus für die Produktion von MVs durch gram-
positive Bakterien ist weitgehend unbekannt. Ein Modell, das für die Produktion von
OMVs durch gram-negative Bakterien von zwei Studien vorgeschlagen wurde,
beschreibt die MVs-Produktion durch L. monocytogenes am plausibelsten (Kaduru-
gamuwa und Beveridge, 1995; Pérez-Cruz et al, 2013). Basierend auf dieses Modell und
aufgrund der Bilder, die durch REM erzielt wurden, kann man davon ausgehen, dass die
von L. monocytogenes produzierten MVs von der Cytoplasmamembran abgeschnürt
und ins extrazelluläre Milieu freigesetzt werden. Vor dem Abschnüren werden die MVs
mit diversem zellulärem Material von der Mutterzelle befüllt. Dies würde die Anwesen-
heit von RNA und zahlreicher cytosolischer Proteine, die mit 37,62% die häufigsten
Proteine in den von L. monocytogenes produzierten MVs darstellen, erklären (Abb.
3.32). Der Unterschied in der Größe spiegelt wahrscheinlich den Beladungszustand der
MVs wieder.
Wegen der Zellwand, die eine physikalische Barriere für deren Freisetzung darstellt,
findet die Produktion von MVs sehr wahrscheinlich während des Wachstums und der
Zellteilung von L. monocytogenes statt. Diese Prozesse beinhalten die Umgestaltung
(remodeling) der Zelloberfläche durch eine Vielzahl von unterschiedlichen Proteinen,
darunter Enzyme, die den Abbau und die Biosynthese der Zellwand vermitteln. Die
funktionelle Klassifizierung der über MS-Analyse identifizierten Proteine unter Ver-
wendung der EggNOG 4.5.1-Datenbank zeigt, dass 7,10% der Proteine in den MVs in
der Biogenese der Zellwand/Membran/Hülle involviert sind (Abb. 3.30). Diese
funktionelle Kategorie ist die vierthäufigste beschriebene Funktion in den von L.
monocytogenes produzierten MVs. Hierzu gehören die Autolysine MurA, Ami, P60 und
P45, die als Zellwandhydrolasen die Spaltung von kovalenten Bindungen katalysieren
(Carvalho et al, 2014). Eine Vielzahl von verschiedenen Penicillin-bindenden Proteinen
(PBP), die in der Zellwandsynthese involviert sind, zählt ebenfalls dazu (Bierne und
Cossart, 2007). Die Anwesenheit weiterer Proteine, die in der Biogenese der Zellhülle
involviert sind, spricht ebenfalls dafür, dass die Produktion von MVs durch L. mono-
cytogenes an Stellen erfolgt wo ein aktives remodeling der Zelloberfläche stattfindet.

4. Diskussion
126
Der hohe Anteil an Proteine, die laut NCBI-Datenbank eine Lokalisation in der
Cytoplasmamembran (20,59%) und in der Zellwand (8,48%) beschrieben haben, ist ein
weiterer Hinweis dafür, dass die Zellhülle in der Biogenese der MVs involviert ist (Abb.
3.32).
4.10. MVs enthalten Proteine aus allen subzellulären Fraktionen: Cytoplasma,
Cytoplasmamembran, Zellwand und Extrazellulär
Die über MS-Analyse identifizierten Proteine in den MVs wurden unter Verwendung
der NCBI-Datenbank nach Lokalisation eingeteilt. Dies ergab, dass die MVs, die von L.
monocytogenes produziert werden, Proteine aus allen subzellulären Fraktionen enthalten
(Abb. 3.32). Allerdings sind die Proteine aus den subzellulären Fraktionen Cytoplasma,
Cytoplasmamembran, Zellwand und Extrazellulär mit jeweils unterschiedlichem Anteil
in den MVs vertreten. Die meisten Proteine in den MVs mit 37,62% sind als
cytosolische Proteine beschrieben. Der Grund hierfür ist der hohe Anteil an Proteine,
die eine Funktion im Metabolismus beschrieben haben (Abb. 3.28). Diese stellen mit
37,37% die häufigsten Proteine in den von L. monocytogenes produzierten MVs dar.
Ein ähnlich hoher Anteil an cytosolischen Proteinen wurde auch in den Vesikeln, die
beispielsweise von Acinetobacter radioresistens (gram-negativ) und Mycobacterium
tuberculosis (gram-positiv) produziert werden, identifiziert (Fulsundar et al, 2015; Lee
et al, 2015). Aufgrund des hohen Anteils an cytosolischen Proteinen kann man davon
ausgehen, dass L. monocytogenes die Produktion von MVs u.a. für die Sekretion von
cytosolischen Proteinen nutzt. Dies würde zum Teil die Sekretion von Proteinen, die
keine Signalsequenz besitzen und für die es keinen beschriebenen Sekretionsweg gibt,
erklären. Des Weiteren wäre eine Sekretion von bestimmten Proteinen sowohl über
Proteinsekretionswege als auch über MVs denkbar. So wurde beispielsweise für die
sogenannten moonlighting Proteinen, wie DnaK, GroEL, EF-Tu, Enolase, RNA-
Polymerase und ribosomale Proteine, eine Sekretion über die SecA2-ATPase der Sec-
Translokase gezeigt (Lenz et al, 2003). Diese Proteine sind auch in den MVs über MS-
Analyse identifiziert worden. Das glykolytische Enzym Enolase beispielsweise zählt
mit 2,90% zu den häufigsten Proteinen in den MVs, die von L. monocytogenes
produziert werden (Abb. 3.33). Ein weiteres Protein für das eine Sekretion über die
SecA2-ATPase gezeigt wurde, ist das Autolysin P60, das zur Trennung der Tochter-
zellen in der Zellteilung beiträgt (Pilgrim et al, 2003; Machata et al, 2005; Lenz et al,

4. Diskussion
127
2003). Ähnlich wie die Enolase zählt P60 mit 2,28% zu den häufigsten Proteinen in den
MVs. Dies zeigt, dass L. monocytogenes für die Sekretion von bestimmten Proteinen ins
extrazelluläre Milieu sowohl Proteinsekretionswege als auch die Produktion von MVs
nutzt. Dass Bakterien beide Formen der Sekretion für den Export von bestimmten
Proteinen nutzen, wurde auch von anderen Studien gezeigt. So wurde beispielsweise das
Shiga-Toxin (Stx), das von E. coli O104:H4 produziert wird, teils in den OMVs (30%)
und teils in den OMVs-freien Überstand (70%) identifiziert (Kunsmann et al, 2015).
Weitere Analysen wären notwendig, um eine genaue Aussage darüber machen zu
können welche Proteine von L. monocytogenes ausschließlich über MVs sekretiert
werden.
Die Proteine, die von der NCBI-Datenbank als extrazellulär beschrieben sind, stellen
mit 6,82% die kleinste Gruppe in den von L. monocytogenes produzierten MVs dar
(Abb. 3.32). Dies spricht dafür, dass L. monocytogenes die Produktion von MVs nicht
für die Sekretion von Proteinen, die für den Export bestimmt sind, nutzt.
Proteine aus zwei weiteren subzellulären Fraktionen sind ebenfalls in den MVs
identifiziert worden. Laut NCBI-Datenbank haben 20,59% und 8,48% der Proteine in
den MVs eine Lokalisation in der Cytoplasmamembran und in der Zellwand beschrie-
ben. Wie bereits erwähnt ist die Zellhülle möglicherweise in der Biogenese der MVs
involviert. Dies würde die Anwesenheit zahlreicher sowohl Integraler und Membran-
assoziierter Proteine als auch Zellwand-assoziierter Proteine erklären.
4.11. MVs enthalten zum Teil vollständige Stoffwechselwege zur Energie-
gewinnung
Die funktionelle Klassifizierung der Proteine unter Verwendung der EggNOG 4.5.1-
Datenbank zeigt, dass die meisten in den MVs identifizierten Proteine (37,37%) im
Metabolismus involviert sind (Abb. 3.28). Diese funktionelle Gruppe beinhaltet u.a.
Proteine, die den Transport und den Stoffwechsel von Kohlenhydraten vermitteln. Diese
machen knapp 10% der Proteine in den MVs aus und stellen damit die dritthäufigste
beschriebene Funktion dar (Abb. 3.29). Eine genaue Analyse dieser Proteine ergab, dass
die von L. monocytogenes produzierten MVs zum Teil vollständige Stoffwechselwege,
die zur Energiegewinnung genutzt werden, enthalten. Die Glukose, die als einzige
Energiequelle dem Minimalmedium zugefügt wurde, wird von L. monocytogenes unter
aeroben Bedingungen über die Glykolyse und den Pentosephosphat-Weg zur

4. Diskussion
128
Energiegewinnung und Bereitstellung von Vorstufen für Biosynthesen abgebaut
(Müller-Herbst et al, 2014; Kim et al, 2006). Sämtliche Enzyme beider Stoffwechsel-
wege sind über MS-Analyse in den MVs identifiziert worden. Es wäre daher denkbar,
dass diese Stoffwechselwege nach der Freisetzung der MVs ins extrazelluläre Milieu
weiterhin aktiv sind. Dies würde bedeuten, dass in den von L. monocytogenes produ-
zierten MVs Energie in Form von ATP über die Glykolyse erzeugt wird. Der
Pentosephosphat-Weg würde als alternativer Abbauweg für die Glukose u.a.
Zwischenprodukte für die Glykolyse bereitstellen.
Die für die aerobe Atmung notwendigen Enzyme des Citrat-Zyklus und der
Atmungskette sind nicht vollständig aber zum großen Teil ebenfalls in den MVs identi-
fiziert worden. Diese dienen dem weiteren Abbau von Glukose, wodurch Vorstufen für
Biosynthesen entstehen und die Regenerierung von Reduktionsäquivalenten erfolgt.
Denkbar wäre es, dass manche Reaktionen dieser Stoffwechselwege in den MVs statt-
finden, aufgrund der darin vorhandenen Enzyme.
Basierend auf der Anwesenheit sämtlicher Enzyme der Glykolyse und ganz entschei-
dend der Laktat-Dehydrogenase wäre eine Energiegewinnung durch Substratketten-
phosphorylierung in Form einer Milchsäuregärung in den MVs denkbar. Die Regene-
rierung von Reduktionsäquivalenten würde in diesem Fall über Oxidation durch die
Laktat-Dehydrogenase stattfinden und damit ein Weiterlaufen der Glykolyse sicher-
stellen.
Die Vermutung, dass in den von L. monocytogenes produzierten MVs energie-
erzeugende Prozesse stattfinden, wird durch die Anwesenheit von Transporter für
Kohlenhydrate unterstützt. So wurden über MS-Analyse Bestandteile verschiedener
Phosphotransferase-Systeme (PTS), darunter PTSMan
-2/Mpt und PTSMan
-3/Mpo, in den
MVs identifiziert (Stoll und Goebel, 2010). Es wäre daher denkbar, dass diese
Transporter die Aufnahme von Glukose in die MVs vermitteln, woraus letztendlich
ATP über die vorhin erwähnten Stoffwechselwege erzeugt wird. Die Erzeugung von
ATP würde bedeuten, dass in den von L. monocytogenes produzierten MVs biologische
Prozesse stattfinden, die Energie benötigen. Um eine genaue Aussage darüber machen
zu können, wären weitere Untersuchungen notwendig, wie beispielsweise die Messung
der Aktivität verschiedener Enzyme und des ATP-Levels. Eine aktuelle Studie hat
bereits gezeigt, dass in den von Bacteroides fragilis produzierten OMVs verschiedene

4. Diskussion
129
biochemische Reaktionen, die in dem Stoffwechsel von Kohlenhydraten, Aminosäuren
und Nukleotiden eine Rolle spielen, aktiv sind (Zakharzhevskaya et al, 2017).
4.12. MVs enthalten Virulenzfaktoren, die während der Infektion eine Rolle
spielen
Diese Arbeit hat die Anwesenheit von Virulenzfaktoren in den MVs, die von L.
monocytogenes produziert werden, gezeigt. Diese wurden durch MS- und teils durch
Western blot-Analyse identifiziert (Abb. 3.34 und 3.35). Hierbei handelt es sich um
Virulenzfaktoren, die für den intrazellulären Infektionszyklus von L. monocytogenes
essentiell sind (Abb. 1.1). Diese machen 10% der Proteine in den MVs aus. Die im Jahr
2013 durchgeführte Studie hat ebenfalls die Anwesenheit verschiedener Virulenz-
faktoren in den MVs, die von L. monocytogenes während des Wachstums in BHI-
Medium produziert werden, gezeigt (Lee et al, 2013). Demnach nutzt L. monocyto-
genes, selbst in unterschiedlichen Wachstumsmedien, die Produktion von MVs u.a. für
die Sekretion von Virulenzfaktoren. Eine ähnliche Beobachtung wurde beispielsweise
von einer Studie über das gram-negative Bakterium Xanthomonas campestris berichtet.
Die von diesem Pflanzen-Pathogen produzierten OMVs enthalten als konstanten
Bestandteil, trotz deren Produktion in unterschiedlichen Wachstumsmedien und einem
entsprechend unterschiedlichem Proteininhalt, Virulenzfaktoren (Sidhu et al, 2008). Die
Sekretion von Virulenzfaktoren über die Produktion von Vesikeln wurde für verschie-
dene pathogene Bakterienspezies gezeigt (Rivera et al, 2010; Thay et al, 2013;
Kunsmann et al, 2015). ETEC (Enterotoxigenic Escherichia coli) beispielsweise
sekretiert den überwiegenden Teil des hitzelabilen Enterotoxins LT (mehr als 95%) über
OMVs (Kesty et al, 2004). Daher stellen Vesikel, neben den bekannten Sekretionswege
für Proteine, einen zusätzlichen Sekretionsmechanismus für Virulenzfaktoren und
weitere Proteine dar.
Diese Arbeit hat des Weiteren die Internalisierung von MVs, die von L. monocytogenes
produziert werden, in die Epithelzelllinie PtK gezeigt. Dies wurde durch Detektion des
Autolysins P60 im Zelllysat der PtK-Zellen nach deren Inkubation mit MVs über
Western blot-Analyse gezeigt (Abb. 3.37). Eine aktuelle Studie hat die Internalisierung
von MVs, die von L. monocytogenes produziert werden, in die Epithelzelllinie HeLa
gezeigt. Hierfür wurde allerdings kein Mechanismus gezeigt (Vdovikova et al, 2017).

4. Diskussion
130
Eine rezeptorvermittelte Endocytose über die Internaline A und B, die durch MS- und
teils durch Western blot-Analyse in den MVs identifiziert worden sind, wäre für deren
Internalisierung denkbar (Abb. 3.34 und 3.35B). Diese Virulenzfaktoren, die für die
Internalisierung von L. monocytogenes in nicht-phagocytierenden Zellen essentiell sind,
könnten in ähnlicher Weise die Internalisierung der MVs vermitteln (Seveau et al,
2004). Um eine genaue Aussage über die Beteiligung der Internaline an dem Interna-
lisierungsprozess von MVs in eukaryotische Zellen machen zu können, wären weitere
Untersuchungen notwendig. Dass Virulenzfaktoren in den Vesikeln, die von verschie-
denen pathogenen Bakterienspezies produziert werden, deren Assoziation und
Aufnahme durch eukaryotische Zellen vermitteln, wurde von weiteren Studien gezeigt
(Bauman und Kuehn, 2006; Bauman und Kuehn, 2009; Kesty et al, 2004).
Die Funktionalität der Virulenzfaktoren in den von L. monocytogenes produzierten MVs
wurde durch Hämolyse-Assay gezeigt (Abb. 3.36). Hierdurch wurde eine konzen-
trationsabhängige Lyse von Schafserythrozyten durch die MVs gezeigt. Dieser Effekt
wurde durch Listeriolysin O (LLO), das mit 2,12% zu den häufigsten Proteinen in den
MVs zählt, verursacht (Abb. 3.33). Dies wurde von einer aktuellen Studie bestätig, die
gezeigt hat, dass die von der LLO-Deletionsmutante produzierten MVs keinen
hämolytischen Effekt haben (Vdovikova et al, 2017). Diese Studie hat zudem gezeigt,
dass LLO in einer nicht-aktiven Form in den MVs vorhanden ist. Listeriolysin O ist ein
Hämolysin, das seine maximale Aktivität bei einem leicht saurem pH-Wert, wie das
beispielsweise im Lysosom der Fall ist, entfaltet (Glomski et al, 2002). Allerdings
gelangen MVs nach der Internalisierung im Lysosom, woraus sie möglicherweise durch
die Aktivität des MVs-assoziierten LLO entkommen (Vdovikova et al, 2017).

5. Zusammenfassung
131
5. Zusammenfassung
Diese Arbeit hat erstmals die RNA, die von L. monocytogenes während des Wachstums
sekretiert wird, umfassend untersucht. Im Rahmen dieser Untersuchung wurde gezeigt,
dass L. monocytogenes die Produktion von Membrane vesicles (MVs) für die Sekretion
von RNA (MVs-RNA) nutzt. Darüber hinaus hat diese Arbeit gezeigt, dass der
überwiegende Teil der extrazellulären RNA in einer freien, nicht MVs-assoziierten
Form (sec-RNA) sekretiert wird. Die umfassende Untersuchung von sec-RNA und
MVs-RNA hat die einzigartige Zusammensetzung und Vielfalt der RNA, die von L.
monocytogenes sekretiert wird, enthüllt. Hierdurch wurde gezeigt, dass sec-RNA und
MVs-RNA sich sowohl untereinander als auch von der zellulären RNA unterscheiden,
bezüglich Vorkommen und Häufigkeit bestimmter RNA-Spezies. Darüber hinaus hat
diese Arbeit die besondere Eigenschaft von sec-RNA und MVs-RNA eine höhere IFN-β
Antwort in Wirtszellen auszulösen als die zelluläre RNA gezeigt. Hierdurch wurde
gezeigt, dass die Induktion von IFN-β ein Charakteristikum der RNA, die von L.
monocytogenes sekretiert wird, darstellt. Diese Erkenntnis weist auf eine funktionelle
Abgrenzung von sec-RNA und MVs-RNA von der zellulären RNA hin. Für die
unterschiedliche IFN-β Antwort wurde die diverse Zusammensetzung von sec-RNA,
MVs-RNA und zelluläre RNA aus RNAs mit unterschiedlichem immun-
stimulatorischem Potential ausgemacht. Dies beruht auf der Erkenntnis, dass
individuelle RNAs, die in sec-RNA und MVs-RNA identifiziert worden sind, ein
unterschiedliches Potential besitzen eine IFN-β Antwort in Wirtszellen auszulösen. Im
Rahmen dieser Untersuchung wurden zwei sRNAs, rli32 mit einem sehr hohen und ssrS
mit einem sehr schwachen IFN-β Induktionspotential, identifiziert. In diesem
Zusammenhang hat diese Arbeit erstmals die antivirale Aktivität von individuellen
bakteriellen sRNAs gezeigt. Hierdurch wurde die Wirksamkeit der RIG-I-abhängigen
IFN-β Antwort, die von den sRNAs ausgelöst wird, im Kontext einer viralen Infektion
gezeigt.
Im zweiten Teil dieser Arbeit wurde gezeigt, dass die von L. monocytogenes
produzierten MVs eine Vielzahl von unterschiedlichen Proteinen, die in verschiedenen
zellulären Prozessen involviert sind, enthalten. Im Rahmen dieser Untersuchung hat
diese Arbeit erstmals die Anwesenheit zum Teil vollständiger Stoffwechselwege und
Kohlenhydrattransporter, die zur Energiegewinnung genutzt werden, gezeigt. Diese
Erkenntnis weist auf aktive biologische Prozesse in den MVs, die von L.

5. Zusammenfassung
132
monocytogenes produziert werden, hin. Darüber hinaus hat diese Arbeit die Anwesen-
heit von Virulenzfaktoren in den MVs und damit deren potentielle Beteiligung an dem
Infektionsprozess von L. monocytogenes gezeigt.
5.1. Summary
This work has for the first time extensively studied the RNA secreted by L.
monocytogenes during growth. In the context of this research it was shown that L.
monocytogenes are using the production of membrane vesicles (MVs) for the secretion
of RNA (MVs-RNA). In addition, this work has shown that the majority of the extra-
cellular RNA is being secreted in a free, not MVs-associated form (sec-RNA). The
comprehensive study of sec-RNA and MVs-RNA has revealed the unique composition
and diversity of RNA, which is secreted by L. monocytogenes. It was shown that sec-
RNA and MVs-RNA differ in their occurrence and abundance of specific RNA species,
both among each other and from the cellular RNA. Moreover, this work has revealed
the special characteristic of sec-RNA and MVs-RNA to elicit a higher IFN-β response
in host cells than the cellular RNA. Thru this it was shown that the induction of IFN-β is
a characteristic of the RNA secreted by L. monocytogenes. This finding points to a
functional demarcation of sec-RNA and MVs-RNA from cellular RNA. It was shown
that the diverse composition of sec-RNA, MVs-RNA and cellular RNA consisting of
RNAs with different immune stimulatory potential is responsible for the different IFN-β
response. This is based on the finding that individual RNAs, which have been identified
in sec-RNA and MVs-RNA, have a different potential to trigger an IFN-β response in
host cells. Within this study, two sRNAs, rli32 with a very high and ssrS with a very
weak IFN-β induction potential, were identified. In this context, this work has for the
first time demonstrated the antiviral activity of individual bacterial sRNAs. This
demonstrated the effectiveness of the RIG-I-dependent IFN-β response triggered by the
sRNAs in the context of a viral infection.
In the second part of this work, it has been shown that the MVs produced by L.
monocytogenes contain a large number of different proteins which are involved in
different cellular processes. In the context of this study, this work has for the first time
shown the presence of partially complete metabolic pathways and carbohydrate
transporters used for energy production. This finding points to active biological
processes in the MVs produced by L. monocytogenes. In addition, this work has shown

5. Zusammenfassung
133
the presence of virulence factors in the MVs and with that their potential involvement in
the infection process of L. monocytogenes.

6. Abkürzungsverzeichnis
134
6. Abkürzungsverzeichnis
BMDMs = bone marrow-derived macrophages
Bp = Basenpaare
BSA = Bovine Serum Albumin
CIAP = Alkaline Phosphatase, Calf intestinal
DCs = Dendritic cells
dsDNA = double-stranded DNA
dsRNA = double-stranded RNA
FKS = Fötales Kälberserum
IVT-RNA = in vitro transkribierte RNA
Lm = Listeria monocytogenes
MDA5 = melanoma differentiation-associated gene 5
MS = Massenspektrometrie
MVs = Membrane Vesicles
Nt = Nukleotide
NTA = Nanoparticle Tracking Analysis
OMVs = Outer Membrane Vesicles
PBS = Dulbecco’s Phosphate Buffered Saline
PCR = Polymerase chain reaction
P/S = Penicillin/Streptomycin
REM = Rasterelektronenmikroskopie
RIG-I = retinoic acid-inducible gene I
Rli = RNA in Listeria
RLR = RIG-I-like receptors
RT = Raumtemperatur
RT-PCR = Real-Time PCR
ssRNA = single-stranded RNA
sRNA = small non-coding RNA
TEM = Transmissionselektronenmikroskopie
T.R. = Transfektionsreagenz
WT = Wildtyp

7. Literaturverzeichnis
135
7. Literaturverzeichnis
Abachin, Eric; Poyart, Claire; Pellegrini, Elisabeth; Milohanic, Eliane; Fiedler, Franz; Berche,
Patrick; Trieu-Cuot, Patrick (2002): Formation of D-alanyl-lipoteichoic acid is required for
adhesion and virulence of Listeria monocytogenes. In: Molecular microbiology 43 (1), S. 1–14.
Abdullah, Zeinab; Schlee, Martin; Roth, Susanne; Mraheil, Mobarak Abu; Barchet, Winfried;
Böttcher, Jan et al. (2012): RIG-I detects infection with live Listeria by sensing secreted
bacterial nucleic acids. In: The EMBO journal 31 (21), S. 4153–4164.
Alaniz, Robert C.; Deatherage, Brooke L.; Lara, Jimmie C.; Cookson, Brad T. (2007):
Membrane vesicles are immunogenic facsimiles of Salmonella typhimurium that potently
activate dendritic cells, prime B and T cell responses, and stimulate protective immunity in
vivo. In: Journal of immunology (Baltimore, Md.: 1950) 179 (11), S. 7692–7701.
Albert, M. L.; Sauter, B.; Bhardwaj, N. (1998): Dendritic cells acquire antigen from apoptotic
cells and induce class I-restricted CTLs. In: Nature 392 (6671), S. 86–89.
Alonzo, Francis; Freitag, Nancy E. (2010): Listeria monocytogenes PrsA2 is required for
virulence factor secretion and bacterial viability within the host cell cytosol. In: Infection and
immunity 78 (11), S. 4944–4957.
Alonzo, Francis; Port, Gary C.; Cao, Min; Freitag, Nancy E. (2009): The posttranslocation
chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. In:
Infection and immunity 77 (7), S. 2612–2623.
Altuvia, S.; Almirón, M.; Huisman, G.; Kolter, R.; Storz, G. (1994): The dps promoter is
activated by OxyR during growth and by IHF and sigma S in stationary phase. In: Molecular
microbiology 13 (2), S. 265–272.
Andersson, A.; Dai, W. J.; Di Santo, J. P.; Brombacher, F. (1998): Early IFN-gamma
production and innate immunity during Listeria monocytogenes infection in the absence of NK
cells. In: Journal of immunology (Baltimore, Md.: 1950) 161 (10), S. 5600–5606.
Arroyo, Jason D.; Chevillet, John R.; Kroh, Evan M.; Ruf, Ingrid K.; Pritchard, Colin C.;
Gibson, Donald F. et al. (2011): Argonaute2 complexes carry a population of circulating
microRNAs independent of vesicles in human plasma. In: Proceedings of the National Academy
of Sciences of the United States of America 108 (12), S. 5003–5008.
Aubry, Camille; Corr, Sinéad C.; Wienerroither, Sebastian; Goulard, Céline; Jones, Ruth;
Jamieson, Amanda M. et al. (2012): Both TLR2 and TRIF contribute to interferon-β production
during Listeria infection. In: PloS one 7 (3), e33299.
Auerbuch, Victoria; Brockstedt, Dirk G.; Meyer-Morse, Nicole; O'Riordan, Mary; Portnoy,
Daniel A. (2004): Mice lacking the type I interferon receptor are resistant to Listeria
monocytogenes. In: The Journal of experimental medicine 200 (4), S. 527–533.
Avila-Calderón, Eric Daniel; Lopez-Merino, Ahidé; Jain, Neeta; Peralta, Humberto; López-
Villegas, Edgar Oliver; Sriranganathan, Nammalwar et al. (2012): Characterization of outer
membrane vesicles from Brucella melitensis and protection induced in mice. In: Clinical &
developmental immunology 2012, S. 352493.
Baldwin, Scott; Deighan, Clayton; Bandeira, Elga; Kwak, Kwang J.; Rahman, Mohammad;
Nana-Sinkam, Patrick et al. (2017): Analyzing the miRNA content of extracellular vesicles by
fluorescence nanoparticle tracking. In: Nanomedicine : nanotechnology, biology, and medicine
13 (2), S. 765–770.
Bauman, Susanne J.; Kuehn, Meta J. (2006): Purification of outer membrane vesicles from
Pseudomonas aeruginosa and their activation of an IL-8 response. In: Microbes and infection 8
(9-10), S. 2400–2408.

7. Literaturverzeichnis
136
Bauman, Susanne J.; Kuehn, Meta J. (2009): Pseudomonas aeruginosa vesicles associate with
and are internalized by human lung epithelial cells. In: BMC microbiology 9, S. 26.
Beckerman, K. P.; Rogers, H. W.; Corbett, J. A.; Schreiber, R. D.; McDaniel, M. L.; Unanue, E.
R. (1993): Release of nitric oxide during the T cell-independent pathway of macrophage
activation. Its role in resistance to Listeria monocytogenes. In: Journal of immunology
(Baltimore, Md. : 1950) 150 (3), S. 888–895.
Beha, Daniel; Deitermann, Sandra; Müller, Matthias; Koch, Hans-Georg (2003): Export of beta-
lactamase is independent of the signal recognition particle. In: The Journal of biological
chemistry 278 (24), S. 22161–22167.
Behrens, S. E.; Tomei, L.; Francesco, R. de (1996): Identification and properties of the RNA-
dependent RNA polymerase of hepatitis C virus. In: The EMBO journal 15 (1), S. 12–22.
Berche, P.; Gaillard, J. L.; Sansonetti, P. J. (1987): Intracellular growth of Listeria
monocytogenes as a prerequisite for in vivo induction of T cell-mediated immunity. In: Journal
of immunology (Baltimore, Md.: 1950) 138 (7), S. 2266–2271.
Berche, P.; Gaillard, J. L.; Geoffroy, C.; Alouf, J. E. (1987): T cell recognition of listeriolysin O
is induced during infection with Listeria monocytogenes. In: Journal of immunology (Baltimore,
Md.: 1950) 139 (11), S. 3813–3821.
Berg, Rance E.; Cordes, Christoph J.; Forman, James (2002): Contribution of CD8+ T cells to
innate immunity. IFN-γ secretion induced by IL-12 and IL-18. In: Eur. J. Immunol. 32 (10), S.
2807–2816.
Bergmann, Birgit; Raffelsbauer, Diana; Kuhn, Michael; Goetz, Monika; Hom, Silke; Goebel,
Werner (2002): InlA- but not InlB-mediated internalization of Listeria monocytogenes by non-
phagocytic mammalian cells needs the support of other internalins. In: Molecular microbiology
43 (3), S. 557–570.
Bernard, Remi; Guiseppi, Annick; Chippaux, Marc; Foglino, Maryline; Denizot, François
(2007): Resistance to bacitracin in Bacillus subtilis. Unexpected requirement of the BceAB
ABC transporter in the control of expression of its own structural genes. In: Journal of
bacteriology 189 (23), S. 8636–8642.
Bierne, Hélène; Cossart, Pascale (2007): Listeria monocytogenes surface proteins. From
genome predictions to function. In: Microbiology and molecular biology reviews: MMBR 71
(2), S. 377–397.
Bille, J.; Blanc, D. S.; Schmid, H.; Boubaker, K.; Baumgartner, A.; Siegrist, H. H. et al. (2006):
Outbreak of human listeriosis associated with tomme cheese in northwest Switzerland, 2005. In:
Euro surveillance: bulletin Europeen sur les maladies transmissibles = European
communicable disease bulletin 11 (6), S. 91–93.
Biller, Steven J.; Schubotz, Florence; Roggensack, Sara E.; Thompson, Anne W.; Summons,
Roger E.; Chisholm, Sallie W. (2014): Bacterial vesicles in marine ecosystems. In: Science
(New York, N.Y.) 343 (6167), S. 183–186.
Blenkiron, Cherie; Simonov, Denis; Muthukaruppan, Anita; Tsai, Peter; Dauros, Priscila;
Green, Sasha et al. (2016): Uropathogenic Escherichia coli Releases Extracellular Vesicles That
Are Associated with RNA. In: PloS one 11 (8), e0160440.
Bomberger, Jennifer M.; Maceachran, Daniel P.; Coutermarsh, Bonita A.; Ye, Siying; O'Toole,
George A.; Stanton, Bruce A. (2009): Long-distance delivery of bacterial virulence factors by
Pseudomonas aeruginosa outer membrane vesicles. In: PLoS pathogens 5 (4), e1000382.
Bonnefoy, E.; Rouvière-Yaniv, J. (1992): HU, the major histone-like protein of E. coli,
modulates the binding of IHF to oriC. In: The EMBO journal 11 (12), S. 4489–4496.

7. Literaturverzeichnis
137
Borezee, E.; Pellegrini, E.; Berche, P. (2000): OppA of Listeria monocytogenes, an
oligopeptide-binding protein required for bacterial growth at low temperature and involved in
intracellular survival. In: Infection and immunity 68 (12), S. 7069–7077.
Bortolussi, R.; Vandenbroucke-Grauls, C. M.; van Asbeck, B. S.; Verhoef, J. (1987):
Relationship of bacterial growth phase to killing of Listeria monocytogenes by oxidative agents
generated by neutrophils and enzyme systems. In: Infection and immunity 55 (12), S. 3197–
3203.
Brunt, L. M.; Portnoy, D. A.; Unanue, E. R. (1990): Presentation of Listeria monocytogenes to
CD8+ T cells requires secretion of hemolysin and intracellular bacterial growth. In: Journal of
immunology (Baltimore, Md.: 1950) 145 (11), S. 3540–3546.
Buchmeier, N. A.; Schreiber, R. D. (1985): Requirement of endogenous interferon-gamma
production for resolution of Listeria monocytogenes infection. In: Proceedings of the National
Academy of Sciences of the United States of America 82 (21), S. 7404–7408.
Burdette, Dara L.; Monroe, Kathryn M.; Sotelo-Troha, Katia; Iwig, Jeff S.; Eckert, Barbara;
Hyodo, Mamoru et al. (2011): STING is a direct innate immune sensor of cyclic di-GMP. In:
Nature 478 (7370), S. 515–518.
Carrero, Javier A.; Calderon, Boris; Unanue, Emil R. (2004): Type I interferon sensitizes
lymphocytes to apoptosis and reduces resistance to Listeria infection. In: The Journal of
experimental medicine 200 (4), S. 535–540.
Carrero, Javier A.; Calderon, Boris; Unanue, Emil R. (2006): Lymphocytes are detrimental
during the early innate immune response against Listeria monocytogenes. In: The Journal of
experimental medicine 203 (4), S. 933–940.
Carvalho, Filipe; Sousa, Sandra; Cabanes, Didier (2014): How Listeria monocytogenes
organizes its surface for virulence. In: Frontiers in cellular and infection microbiology 4, S. 48.
Cella, M.; Jarrossay, D.; Facchetti, F.; Alebardi, O.; Nakajima, H.; Lanzavecchia, A.; Colonna,
M. (1999): Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large
amounts of type I interferon. In: Nature medicine 5 (8), S. 919–923.
Chakraborty, T.; Hain, T.; Domann, E. (2000): Genome organization and the evolution of the
virulence gene locus in Listeria species. In: International journal of medical microbiology :
IJMM 290 (2), S. 167–174.
Chakraborty, T.; Leimeister-Wächter, M.; Domann, E.; Hartl, M.; Goebel, W.; Nichterlein, T.;
Notermans, S. (1992): Coordinate regulation of virulence genes in Listeria monocytogenes
requires the product of the prfA gene. In: Journal of bacteriology 174 (2), S. 568–574.
Chatterjee, Som Subhra; Hossain, Hamid; Otten, Sonja; Kuenne, Carsten; Kuchmina, Katja;
Machata, Silke et al. (2006): Intracellular gene expression profile of Listeria monocytogenes. In:
Infection and immunity 74 (2), S. 1323–1338.
Chevillet, John R.; Kang, Qing; Ruf, Ingrid K.; Briggs, Hilary A.; Vojtech, Lucia N.; Hughes,
Sean M. et al. (2014): Quantitative and stoichiometric analysis of the microRNA content of
exosomes. In: Proceedings of the National Academy of Sciences of the United States of America
111 (41), S. 14888–14893.
Chico-Calero, Isabel; Suárez, Mónica; González-Zorn, Bruno; Scortti, Mariela; Slaghuis, Jörg;
Goebel, Werner; Vázquez-Boland, José A. (2002): Hpt, a bacterial homolog of the microsomal
glucose- 6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. In:
Proceedings of the National Academy of Sciences of the United States of America 99 (1), S.
431–436.
Choi, Ji-Woong; Kwon, Tae-Yub; Hong, Su-Hyung; Lee, Heon-Jin (2016): Isolation and
Characterization of a microRNA-size Secretable Small RNA in Streptococcus sanguinis. In:
Cell biochemistry and biophysics.

7. Literaturverzeichnis
138
Christiansen, Janne K.; Nielsen, Jesper S.; Ebersbach, Tine; Valentin-Hansen, Poul; Søgaard-
Andersen, Lotte; Kallipolitis, Birgitte H. (2006): Identification of small Hfq-binding RNAs in
Listeria monocytogenes. In: RNA (New York, N.Y.) 12 (7), S. 1383–1396.
Coch, Christoph; Stümpel, Jan Phillip; Lilien-Waldau, Vanessa; Wohlleber, Dirk; Kümmerer,
Beate M.; Bekeredjian-Ding, Isabelle et al. (2017): RIG-I Activation Protects and Rescues from
Lethal Influenza Virus Infection and Bacterial Superinfection. In: Molecular therapy: the
journal of the American Society of Gene Therapy 25 (9), S. 2093–2103.
Colonna, Marco; Krug, Anne; Cella, Marina (2002): Interferon-producing cells. On the front
line in immune responses against pathogens. In: Current opinion in immunology 14 (3), S. 373–
379.
Conlan, J. W.; North, R. J. (1991): Neutrophil-mediated dissolution of infected host cells as a
defense strategy against a facultative intracellular bacterium. In: The Journal of experimental
medicine 174 (3), S. 741–744.
Conlan, J. W.; North, R. J. (1992): Roles of Listeria monocytogenes virulence factors in
survival. Virulence factors distinct from listeriolysin are needed for the organism to survive an
early neutrophil-mediated host defense mechanism. In: Infection and immunity 60 (3), S. 951–
957.
Crimmins, Gregory T.; Herskovits, Anat A.; Rehder, Kai; Sivick, Kelsey E.; Lauer, Peter;
Dubensky, Thomas W.; Portnoy, Daniel A. (2008): Listeria monocytogenes multidrug
resistance transporters activate a cytosolic surveillance pathway of innate immunity. In:
Proceedings of the National Academy of Sciences of the United States of America 105 (29), S.
10191–10196.
Cui, Sheng; Eisenächer, Katharina; Kirchhofer, Axel; Brzózka, Krzysztof; Lammens, Alfred;
Lammens, Katja et al. (2008): The C-terminal regulatory domain is the RNA 5'-triphosphate
sensor of RIG-I. In: Molecular cell 29 (2), S. 169–179.
Desvaux, Mickaël; Hébraud, Michel (2006): The protein secretion systems in Listeria. Inside
out bacterial virulence. In: FEMS microbiology reviews 30 (5), S. 774–805.
Diebold, Sandra S.; Montoya, Maria; Unger, Hermann; Alexopoulou, Lena; Roy, Polly;
Haswell, Linsey E. et al. (2003): Viral infection switches non-plasmacytoid dendritic cells into
high interferon producers. In: Nature 424 (6946), S. 324–328.
Domann, E.; Leimeister-Wächter, M.; Goebel, W.; Chakraborty, T. (1991): Molecular cloning,
sequencing, and identification of a metalloprotease gene from Listeria monocytogenes that is
species specific and physically linked to the listeriolysin gene. In: Infection and immunity 59
(1), S. 65–72.
Domann, E.; Wehland, J.; Rohde, M.; Pistor, S.; Hartl, M.; Goebel, W. et al. (1992): A novel
bacterial virulence gene in Listeria monocytogenes required for host cell microfilament
interaction with homology to the proline-rich region of vinculin. In: The EMBO journal 11 (5),
S. 1981–1990.
Dorward, D. W.; Garon, C. F.; Judd, R. C. (1989): Export and intercellular transfer of DNA via
membrane blebs of Neisseria gonorrhoeae. In: Journal of bacteriology 171 (5), S. 2499–2505.
Dragovic, Rebecca A.; Gardiner, Christopher; Brooks, Alexandra S.; Tannetta, Dionne S.;
Ferguson, David J. P.; Hole, Patrick et al. (2011): Sizing and phenotyping of cellular vesicles
using Nanoparticle Tracking Analysis. In: Nanomedicine: nanotechnology, biology, and
medicine 7 (6), S. 780–788.
Engelbrecht, F.; Chun, S. K.; Ochs, C.; Hess, J.; Lottspeich, F.; Goebel, W.; Sokolovic, Z.
(1996): A new PrfA-regulated gene of Listeria monocytogenes encoding a small, secreted
protein which belongs to the family of internalins. In: Molecular microbiology 21 (4), S. 823–
837.

7. Literaturverzeichnis
139
Farber, J. M.; Peterkin, P. I. (1991): Listeria monocytogenes, a food-borne pathogen. In:
Microbiological reviews 55 (3), S. 476–511.
Fiocca, R.; Necchi, V.; Sommi, P.; Ricci, V.; Telford, J.; Cover, T. L.; Solcia, E. (1999):
Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and
budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric
epithelium. In: The Journal of pathology 188 (2), S. 220–226.
Fraser, K. R.; Harvie, D.; Coote, P. J.; O'Byrne, C. P. (2000): Identification and characterization
of an ATP binding cassette L-carnitine transporter in Listeria monocytogenes. In: Applied and
environmental microbiology 66 (11), S. 4696–4704.
Fulsundar, Shweta; Harms, Klaus; Flaten, Gøril E.; Johnsen, Pål J.; Chopade, Balu Ananda;
Nielsen, Kaare M. (2014): Gene transfer potential of outer membrane vesicles of Acinetobacter
baylyi and effects of stress on vesiculation. In: Applied and environmental microbiology 80
(11), S. 3469–3483.
Fulsundar, Shweta; Kulkarni, Heramb M.; Jagannadham, Medicharla V.; Nair, Rashmi; Keerthi,
Sravani; Sant, Pooja et al. (2015): Molecular characterization of outer membrane vesicles
released from Acinetobacter radioresistens and their potential roles in pathogenesis. In:
Microbial pathogenesis 83-84, S. 12–22.
Furuse, Yuki; Finethy, Ryan; Saka, Hector A.; Xet-Mull, Ana M.; Sisk, Dana M.; Smith,
Kristen L. Jurcic et al. (2014): Search for microRNAs expressed by intracellular bacterial
pathogens in infected mammalian cells. In: PloS one 9 (9), e106434.
Furuta, Nobumichi; Tsuda, Kayoko; Omori, Hiroko; Yoshimori, Tamotsu; Yoshimura,
Fuminobu; Amano, Atsuo (2009): Porphyromonas gingivalis outer membrane vesicles enter
human epithelial cells via an endocytic pathway and are sorted to lysosomal compartments. In:
Infection and immunity 77 (10), S. 4187–4196.
Gaillard, J. L.; Berche, P.; Mounier, J.; Richard, S.; Sansonetti, P. (1987): In vitro model of
penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like
cell line Caco-2. In: Infection and immunity 55 (11), S. 2822–2829.
Gedde, M. M.; Higgins, D. E.; Tilney, L. G.; Portnoy, D. A. (2000): Role of listeriolysin O in
cell-to-cell spread of Listeria monocytogenes. In: Infection and immunity 68 (2), S. 999–1003.
Geissmann, Thomas A.; Touati, Danièle (2004): Hfq, a new chaperoning role. Binding to
messenger RNA determines access for small RNA regulator. In: The EMBO journal 23 (2), S.
396–405.
Geoffroy, C.; Raveneau, J.; Beretti, J. L.; Lecroisey, A.; Vazquez-Boland, J. A.; Alouf, J. E.;
Berche, P. (1991): Purification and characterization of an extracellular 29-kilodalton
phospholipase C from Listeria monocytogenes. In: Infection and immunity 59 (7), S. 2382–
2388.
Georgellis, D.; Sohlberg, B.; Hartl, F. U.; Gabain, A. von (1995): Identification of GroEL as a
constituent of an mRNA-protection complex in Escherichia coli. In: Molecular microbiology 16
(6), S. 1259–1268.
Ghosal, Anubrata; Upadhyaya, Bimal Babu; Fritz, Joëlle V.; Heintz-Buschart, Anna; Desai,
Mahesh S.; Yusuf, Dilmurat et al. (2015): The extracellular RNA complement of Escherichia
coli. In: MicrobiologyOpen.
Glaser, P.; Frangeul, L.; Buchrieser, C.; Rusniok, C.; Amend, A.; Baquero, F. et al. (2001):
Comparative genomics of Listeria species. In: Science (New York, N.Y.) 294 (5543), S. 849–
852.
Glomski, Ian J.; Gedde, Margaret M.; Tsang, Albert W.; Swanson, Joel A.; Portnoy, Daniel A.
(2002): The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize

7. Literaturverzeichnis
140
activity and prevent damage to infected host cells. In: The Journal of cell biology 156 (6), S.
1029–1038.
Gratz, Nina; Hartweger, Harald; Matt, Ulrich; Kratochvill, Franz; Janos, Marton; Sigel, Stefanie
et al. (2011): Type I interferon production induced by Streptococcus pyogenes-derived nucleic
acids is required for host protection. In: PLoS pathogens 7 (5), e1001345.
Gruber, Andreas R.; Lorenz, Ronny; Bernhart, Stephan H.; Neuböck, Richard; Hofacker, Ivo L.
(2008): The Vienna RNA websuite. In: Nucleic acids research 36 (Web Server issue), W70-4.
Gurung, Mamata; Moon, Dong Chan; Choi, Chi Won; Lee, Jung Hwa; Bae, Yong Chul; Kim,
Jungmin et al. (2011): Staphylococcus aureus produces membrane-derived vesicles that induce
host cell death. In: PloS one 6 (11), e27958.
Hagmann, Cristina Amparo; Herzner, Anna Maria; Abdullah, Zeinab; Zillinger, Thomas;
Jakobs, Christopher; Schuberth, Christine et al. (2013): RIG-I detects triphosphorylated RNA of
Listeria monocytogenes during infection in non-immune cells. In: PloS one 8 (4), e62872.
Hain, Torsten; Hossain, Hamid; Chatterjee, Som S.; Machata, Silke; Volk, Ute; Wagner, Sandra
et al. (2008): Temporal transcriptomic analysis of the Listeria monocytogenes EGD-e sigmaB
regulon. In: BMC microbiology 8, S. 20.
Hansen, Kathrine; Prabakaran, Thaneas; Laustsen, Anders; Jørgensen, Sofie E.; Rahbæk, Stine
H.; Jensen, Søren B. et al. (2014): Listeria monocytogenes induces IFNβ expression through an
IFI16-, cGAS- and STING-dependent pathway. In: The EMBO journal 33 (15), S. 1654–1666.
Havell, E. A. (1989): Evidence that tumor necrosis factor has an important role in antibacterial
resistance. In: Journal of immunology (Baltimore, Md. : 1950) 143 (9), S. 2894–2899.
Ho, Meng-Hsuan; Chen, Chin-Ho; Goodwin, J. Shawn; Wang, Bing-Yan; Xie, Hua (2015):
Functional Advantages of Porphyromonas gingivalis Vesicles. In: PloS one 10 (4), e0123448.
Holst, Johan; Martin, Diana; Arnold, Richard; Huergo, Concepcion Campa; Oster, Philipp;
O'Hallahan, Jane; Rosenqvist, Einar (2009): Properties and clinical performance of vaccines
containing outer membrane vesicles from Neisseria meningitidis. In: Vaccine 27 Suppl 2, B3-
12.
Honda, Kenya; Taniguchi, Tadatsugu (2006): IRFs. Master regulators of signalling by Toll-like
receptors and cytosolic pattern-recognition receptors. In: Nature reviews. Immunology 6 (9), S.
644–658.
Huang, S.; Hendriks, W.; Althage, A.; Hemmi, S.; Bluethmann, H.; Kamijo, R. et al. (1993):
Immune response in mice that lack the interferon-gamma receptor. In: Science (New York, N.Y.)
259 (5102), S. 1742–1745.
Huerta-Cepas, Jaime; Szklarczyk, Damian; Forslund, Kristoffer; Cook, Helen; Heller, Davide;
Walter, Mathias C. et al. (2016): eggNOG 4.5. A hierarchical orthology framework with
improved functional annotations for eukaryotic, prokaryotic and viral sequences. In: Nucleic
acids research 44 (D1), D286-93.
Isaacs, A.; Lindemann, J. (1957): Virus interference. I. The interferon. In: Proceedings of the
Royal Society of London. Series B, Biological sciences 147 (927), S. 258–267.
Jensen, Lars Juhl; Julien, Philippe; Kuhn, Michael; Mering, Christian von; Muller, Jean;
Doerks, Tobias; Bork, Peer (2008): eggNOG. Automated construction and annotation of
orthologous groups of genes. In: Nucleic acids research 36 (Database issue), D250-4.
Johansson, Jörgen; Mandin, Pierre; Renzoni, Adriana; Chiaruttini, Claude; Springer, Mathias;
Cossart, Pascale (2002): An RNA thermosensor controls expression of virulence genes in
Listeria monocytogenes. In: Cell 110 (5), S. 551–561.

7. Literaturverzeichnis
141
Johnson, J. L.; Doyle, M. P.; Cassens, R. G.; Schoeni, J. L. (1988): Fate of Listeria
monocytogenes in tissues of experimentally infected cattle and in hard salami. In: Applied and
environmental microbiology 54 (2), S. 497–501.
Jung, Steffen; Unutmaz, Derya; Wong, Phillip; Sano, Gen-Ichiro; los Santos, Kenia de;
Sparwasser, Tim et al. (2002): In vivo depletion of CD11c+ dendritic cells abrogates priming of
CD8+ T cells by exogenous cell-associated antigens. In: Immunity 17 (2), S. 211–220.
Kadurugamuwa, J. L.; Beveridge, T. J. (1995): Virulence factors are released from
Pseudomonas aeruginosa in association with membrane vesicles during normal growth and
exposure to gentamicin. A novel mechanism of enzyme secretion. In: Journal of bacteriology
177 (14), S. 3998–4008.
Kadurugamuwa, J. L.; Beveridge, T. J. (1998): Delivery of the non-membrane-permeative
antibiotic gentamicin into mammalian cells by using Shigella flexneri membrane vesicles. In:
Antimicrobial agents and chemotherapy 42 (6), S. 1476–1483.
Kang, Sung-Min; Choi, Ji-Woong; Lee, Youngkyun; Hong, Su-Hyung; Lee, Heon-Jin (2013):
Identification of microRNA-size, small RNAs in Escherichia coli. In: Current microbiology 67
(5), S. 609–613.
Kanneganti, Thirumala-Devi; Ozören, Nesrin; Body-Malapel, Mathilde; Amer, Amal; Park,
Jong-Hwan; Franchi, Luigi et al. (2006): Bacterial RNA and small antiviral compounds activate
caspase-1 through cryopyrin/Nalp3. In: Nature 440 (7081), S. 233–236.
Kaparakis, Maria; Turnbull, Lynne; Carneiro, Leticia; Firth, Stephen; Coleman, Harold A.;
Parkington, Helena C. et al. (2010): Bacterial membrane vesicles deliver peptidoglycan to
NOD1 in epithelial cells. In: Cellular microbiology 12 (3), S. 372–385.
Kato, Hiroki; Takahasi, Kiyohiro; Fujita, Takashi (2011): RIG-I-like receptors. Cytoplasmic
sensors for non-self RNA. In: Immunological reviews 243 (1), S. 91–98.
Kato, Hiroki; Takeuchi, Osamu; Sato, Shintaro; Yoneyama, Mitsutoshi; Yamamoto, Masahiro;
Matsui, Kosuke et al. (2006): Differential roles of MDA5 and RIG-I helicases in the recognition
of RNA viruses. In: Nature 441 (7089), S. 101–105.
Kawai, Taro; Akira, Shizuo (2008): Toll-like receptor and RIG-I-like receptor signaling. In:
Annals of the New York Academy of Sciences 1143, S. 1–20.
Kawai, Taro; Akira, Shizuo (2010): The role of pattern-recognition receptors in innate
immunity. Update on Toll-like receptors. In: Nature immunology 11 (5), S. 373–384.
Kesty, Nicole C.; Mason, Kevin M.; Reedy, Mary; Miller, Sara E.; Kuehn, Meta J. (2004):
Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. In: The
EMBO journal 23 (23), S. 4538–4549.
Kim, Hyun-Jin; Mittal, Meghna; Sonenshein, Abraham L. (2006): CcpC-dependent regulation
of citB and lmo0847 in Listeria monocytogenes. In: Journal of bacteriology 188 (1), S. 179–
190.
Klieve, Athol V.; Yokoyama, Melvin T.; Forster, Robert J.; Ouwerkerk, Diane; Bain, Peter A.;
Mawhinney, Erin L. (2005): Naturally occurring DNA transfer system associated with
membrane vesicles in cellulolytic Ruminococcus spp. of ruminal origin. In: Applied and
environmental microbiology 71 (8), S. 4248–4253.
Ko, R.; Smith, L. T. (1999): Identification of an ATP-driven, osmoregulated glycine betaine
transport system in Listeria monocytogenes. In: Applied and environmental microbiology 65
(9), S. 4040–4048.
Kocks, C.; Gouin, E.; Tabouret, M.; Berche, P.; Ohayon, H.; Cossart, P. (1992): L.
monocytogenes-induced actin assembly requires the actA gene product, a surface protein. In:
Cell 68 (3), S. 521–531.

7. Literaturverzeichnis
142
Koenig, Carl Heinz Wirsing von; Finger, Horst; Hof, Herbert (1982): Failure of killed Listeria
monocytogenes vaccine to produce protective immunity. In: Nature 297 (5863), S. 233–234.
Koeppen, Katja; Hampton, Thomas H.; Jarek, Michael; Scharfe, Maren; Gerber, Scott A.;
Mielcarz, Daniel W. et al. (2016): A Novel Mechanism of Host-Pathogen Interaction through
sRNA in Bacterial Outer Membrane Vesicles. In: PLoS pathogens 12 (6), e1005672.
Kogure, Takayuki; Lin, Wen-Lang; Yan, Irene K.; Braconi, Chiara; Patel, Tushar (2011):
Intercellular nanovesicle-mediated microRNA transfer. A mechanism of environmental
modulation of hepatocellular cancer cell growth. In: Hepatology (Baltimore, Md.) 54 (4), S.
1237–1248.
Koski, G. K.; Kariko, K.; Xu, S.; Weissman, D.; Cohen, P. A.; Czerniecki, B. J. (2004): Cutting
Edge. Innate Immune System Discriminates between RNA Containing Bacterial versus
Eukaryotic Structural Features That Prime for High-Level IL-12 Secretion by Dendritic Cells.
In: Journal of immunology (Baltimore, Md.: 1950) 172 (7), S. 3989–3993.
Koutsoumanis, K. P.; Kendall, P. A.; Sofos, J. N. (2003): Effect of Food Processing-Related
Stresses on Acid Tolerance of Listeria monocytogenes. In: Applied and environmental
microbiology 69 (12), S. 7514–7516.
Kuhn, M.; Goebel, W. (1989): Identification of an extracellular protein of Listeria
monocytogenes possibly involved in intracellular uptake by mammalian cells. In: Infection and
immunity 57 (1), S. 55–61.
Kulp, Adam; Kuehn, Meta J. (2010): Biological functions and biogenesis of secreted bacterial
outer membrane vesicles. In: Annual review of microbiology 64, S. 163–184.
Kunsmann, Lisa; Rüter, Christian; Bauwens, Andreas; Greune, Lilo; Glüder, Malte; Kemper,
Björn et al. (2015): Virulence from vesicles. Novel mechanisms of host cell injury by
Escherichia coli O104:H4 outbreak strain. In: Scientific reports 5, S. 13252.
Ladel, C. H.; Flesch, I. E.; Arnoldi, J.; Kaufmann, S. H. (1994): Studies with MHC-deficient
knock-out mice reveal impact of both MHC I- and MHC II-dependent T cell responses on
Listeria monocytogenes infection. In: Journal of immunology (Baltimore, Md. : 1950) 153 (7),
S. 3116–3122.
Ladner, M. B.; Martin, G. A.; Noble, J. A.; Wittman, V. P.; Warren, M. K.; McGrogan, M.;
Stanley, E. R. (1988): cDNA cloning and expression of murine macrophage colony-stimulating
factor from L929 cells. In: Proceedings of the National Academy of Sciences of the United
States of America 85 (18), S. 6706–6710.
Lappann, Martin; Otto, Andreas; Becher, Dörte; Vogel, Ulrich (2013): Comparative proteome
analysis of spontaneous outer membrane vesicles and purified outer membranes of Neisseria
meningitidis. In: Journal of bacteriology 195 (19), S. 4425–4435.
Leber, Jess H.; Crimmins, Gregory T.; Raghavan, Sridharan; Meyer-Morse, Nicole P.; Cox,
Jeffery S.; Portnoy, Daniel A. (2008): Distinct TLR- and NLR-mediated transcriptional
responses to an intracellular pathogen. In: PLoS pathogens 4 (1), e6.
Lecuit, M. (2005): Understanding how Listeria monocytogenes targets and crosses host barriers.
In: Clinical microbiology and infection: the official publication of the European Society of
Clinical Microbiology and Infectious Diseases 11 (6), S. 430–436.
Lecuit, Marc; Nelson, D. Michael; Smith, Steve D.; Khun, Huot; Huerre, Michel; Vacher-
Lavenu, Marie-Cécile et al. (2004): Targeting and crossing of the human maternofetal barrier by
Listeria monocytogenes. Role of internalin interaction with trophoblast E-cadherin. In:
Proceedings of the National Academy of Sciences of the United States of America 101 (16), S.
6152–6157.
Lee, Eun-Young; Choi, Do-Young; Kim, Dae-Kyum; Kim, Jung-Wook; Park, Jung Ok; Kim,
Sungjee et al. (2009): Gram-positive bacteria produce membrane vesicles. Proteomics-based

7. Literaturverzeichnis
143
characterization of Staphylococcus aureus-derived membrane vesicles. In: Proteomics 9 (24), S.
5425–5436.
Lee, Heon-Jin; Hong, Su-Hyung (2012): Analysis of microRNA-size, small RNAs in
Streptococcus mutans by deep sequencing. In: FEMS microbiology letters 326 (2), S. 131–136.
Lee, Jaewook; Kim, Si-Hyun; Choi, Dong-Sic; Lee, Jong Seok; Kim, Dae-Kyum; Go,
Gyeongyun et al. (2015): Proteomic analysis of extracellular vesicles derived from
Mycobacterium tuberculosis. In: Proteomics 15 (19), S. 3331–3337.
Lee, Je Chul; Lee, Eun Jeoung; Lee, Jung Hwa; Jun, So Hyun; Choi, Chi Won; Kim, Seung Il et
al. (2012): Klebsiella pneumoniae secretes outer membrane vesicles that induce the innate
immune response. In: FEMS microbiology letters 331 (1), S. 17–24.
Lee, Jung Hwa; Choi, Chi-Won; Lee, Taewon; Kim, Seung Il; Lee, Je-Chul; Shin, Ji-Hyun
(2013): Transcription factor σB plays an important role in the production of extracellular
membrane-derived vesicles in Listeria monocytogenes. In: PloS one 8 (8), e73196.
Leimeister-Wächter, M.; Domann, E.; Chakraborty, T. (1991): Detection of a gene encoding a
phosphatidylinositol-specific phospholipase C that is co-ordinately expressed with listeriolysin
in Listeria monocytogenes. In: Molecular microbiology 5 (2), S. 361–366.
Lenz, Laurel L.; Mohammadi, Sina; Geissler, Aimee; Portnoy, Daniel A. (2003): SecA2-
dependent secretion of autolytic enzymes promotes Listeria monocytogenes pathogenesis. In:
Proceedings of the National Academy of Sciences of the United States of America 100 (21), S.
12432–12437.
Lertmemongkolchai, G.; Cai, G.; Hunter, C. A.; Bancroft, G. J. (2001): Bystander activation of
CD8+ T cells contributes to the rapid production of IFN-gamma in response to bacterial
pathogens. In: Journal of immunology (Baltimore, Md. : 1950) 166 (2), S. 1097–1105.
Li, Cheryl C. Y.; Eaton, Sally A.; Young, Paul E.; Lee, Maggie; Shuttleworth, Rupert;
Humphreys, David T. et al. (2013): Glioma microvesicles carry selectively packaged coding and
non-coding RNAs which alter gene expression in recipient cells. In: RNA biology 10 (8), S.
1333–1344.
Lingnau, A.; Domann, E.; Hudel, M.; Bock, M.; Nichterlein, T.; Wehland, J.; Chakraborty, T.
(1995): Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products
mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent
mechanisms. In: Infection and immunity 63 (10), S. 3896–3903.
Ma, Wenjun; Brenner, Dominique; Wang, Zhongfang; Dauber, Bianca; Ehrhardt, Christina;
Högner, Katrin et al. (2010): The NS segment of an H5N1 highly pathogenic avian influenza
virus (HPAIV) is sufficient to alter replication efficiency, cell tropism, and host range of an
H7N1 HPAIV. In: Journal of virology 84 (4), S. 2122–2133.
Machata, Silke; Hain, Torsten; Rohde, Manfred; Chakraborty, Trinad (2005): Simultaneous
deficiency of both MurA and p60 proteins generates a rough phenotype in Listeria
monocytogenes. In: Journal of bacteriology 187 (24), S. 8385–8394.
Mackaness, G. B. (1962): Cellular resistance to infection. In: The Journal of experimental
medicine 116, S. 381–406.
Marquis, H.; Bouwer, H. G.; Hinrichs, D. J.; Portnoy, D. A. (1993): Intracytoplasmic growth
and virulence of Listeria monocytogenes auxotrophic mutants. In: Infection and immunity 61
(9), S. 3756–3760.
Martin, Marcel (2011): Cutadapt removes adapter sequences from high-throughput sequencing
reads. In: EMBnet j. 17 (1), S. 10.
Martinez, A.; Kolter, R. (1997): Protection of DNA during oxidative stress by the nonspecific
DNA-binding protein Dps. In: Journal of bacteriology 179 (16), S. 5188–5194.

7. Literaturverzeichnis
144
Masforrol, Yordanka; Gil, Jeovanis; García, Darien; Noda, Jesús; Ramos, Yassel; Betancourt,
Lázaro et al. (2017): A deeper mining on the protein composition of VA-MENGOC-BC®. An
OMV-based vaccine against N. meningitidis serogroup B and C. In: Human vaccines &
immunotherapeutics 13 (11), S. 2548–2560.
McBroom, Amanda J.; Kuehn, Meta J. (2007): Release of outer membrane vesicles by Gram-
negative bacteria is a novel envelope stress response. In: Molecular microbiology 63 (2), S.
545–558.
McCaffrey, Ramona L.; Fawcett, Paul; O'Riordan, Mary; Lee, Kyung-Dall; Havell, Edward A.;
Brown, Patrick O.; Portnoy, Daniel A. (2004): A specific gene expression program triggered by
Gram-positive bacteria in the cytosol. In: Proceedings of the National Academy of Sciences of
the United States of America 101 (31), S. 11386–11391.
McLauchlin, J.; Greenwood, M. H.; Pini, P. N. (1990): The occurrence of Listeria
monocytogenes in cheese from a manufacturer associated with a case of listeriosis. In:
International journal of food microbiology 10 (3-4), S. 255–262.
McWhirter, Sarah M.; Barbalat, Roman; Monroe, Kathryn M.; Fontana, Mary F.; Hyodo,
Mamoru; Joncker, Nathalie T. et al. (2009): A host type I interferon response is induced by
cytosolic sensing of the bacterial second messenger cyclic-di-GMP. In: The Journal of
experimental medicine 206 (9), S. 1899–1911.
Mead, P. S.; Slutsker, L.; Dietz, V.; McCaig, L. F.; Bresee, J. S.; Shapiro, C. et al. (1999):
Food-related illness and death in the United States. In: Emerging infectious diseases 5 (5), S.
607–625.
Mengaud, J.; Braun-Breton, C.; Cossart, P. (1991): Identification of phosphatidylinositol-
specific phospholipase C activity in Listeria monocytogenes. A novel type of virulence factor?
In: Molecular microbiology 5 (2), S. 367–372.
Mengaud, J.; Ohayon, H.; Gounon, P.; Mege, R-M; Cossart, P. (1996): E-cadherin is the
receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial
cells. In: Cell 84 (6), S. 923–932.
Meyer, R. R.; Laine, P. S. (1990): The single-stranded DNA-binding protein of Escherichia coli.
In: Microbiological reviews 54 (4), S. 342–380.
Meylan, Etienne; Tschopp, Jürg (2006): Toll-like receptors and RNA helicases. Two parallel
ways to trigger antiviral responses. In: Molecular cell 22 (5), S. 561–569.
Miczak, A.; Kaberdin, V. R.; Wei, C. L.; Lin-Chao, S. (1996): Proteins associated with RNase E
in a multicomponent ribonucleolytic complex. In: Proceedings of the National Academy of
Sciences of the United States of America 93 (9), S. 3865–3869.
Møller, Thorleif; Franch, Thomas; Højrup, Peter; Keene, Douglas R.; Bächinger, Hans Peter;
Brennan, Richard G.; Valentin-Hansen, Poul (2002): Hfq. A bacterial Sm-like protein that
mediates RNA-RNA interaction. In: Molecular cell 9 (1), S. 23–30.
Mraheil, Mobarak A.; Billion, André; Mohamed, Walid; Mukherjee, Krishnendu; Kuenne,
Carsten; Pischimarov, Jordan et al. (2011): The intracellular sRNA transcriptome of Listeria
monocytogenes during growth in macrophages. In: Nucleic acids research 39 (10), S. 4235–
4248.
Müller, U.; Steinhoff, U.; Reis, L. F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R. M.; Aguet, M.
(1994): Functional role of type I and type II interferons in antiviral defense. In: Science (New
York, N.Y.) 264 (5167), S. 1918–1921.
Müller-Herbst, Stefanie; Wüstner, Stefanie; Mühlig, Anna; Eder, Daniela; M Fuchs, Thilo;
Held, Claudia et al. (2014): Identification of genes essential for anaerobic growth of Listeria
monocytogenes. In: Microbiology (Reading, England) 160 (Pt 4), S. 752–765.

7. Literaturverzeichnis
145
Myers, Eric R.; Martin, Scott E. (1994): Virulence of Listeria monocytogenes Propagated in
NaCl Containing Media at 4, 25 and 37°C. In: Journal of Food Protection 57 (6), S. 475–478.
Nielsen, Jesper S.; Larsen, Marianne Halberg; Lillebæk, Eva Maria Sternkopf; Bergholz, Teresa
M.; Christiansen, Mie H. G.; Boor, Kathryn J. et al. (2011): A small RNA controls expression of
the chitinase ChiA in Listeria monocytogenes. In: PloS one 6 (4), e19019.
Nightingale, Kendra K.; Fortes, Esther D.; Ho, Alphina J.; Schukken, Ynte H.; Grohn, Yrjo T.;
Wiedmann, Martin (2005): Evaluation of farm management practices as risk factors for clinical
listeriosis and fecal shedding of Listeria monocytogenes in ruminants. In: Journal of the
American Veterinary Medical Association 227 (11), S. 1808–1814.
Nolte-'t Hoen, Esther N. M.; Buermans, Henk P. J.; Waasdorp, Maaike; Stoorvogel, Willem;
Wauben, Marca H. M.; Hoen, Peter A. C. 't (2012): Deep sequencing of RNA from immune
cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes
with potential regulatory functions. In: Nucleic acids research 40 (18), S. 9272–9285.
O'Connell, R. M.; Vaidya, S. A.; Perry, A. K.; Saha, S. K.; Dempsey, P. W.; Cheng, G. (2005):
Immune Activation of Type I IFNs by Listeria monocytogenes Occurs Independently of TLR4,
TLR2, and Receptor Interacting Protein 2 but Involves TANK-Binding Kinase 1. In: Journal of
immunology (Baltimore, Md.: 1950) 174 (3), S. 1602–1607.
O'Connell, Ryan M.; Saha, Supriya K.; Vaidya, Sagar A.; Bruhn, Kevin W.; Miranda, Gustavo
A.; Zarnegar, Brian et al. (2004): Type I interferon production enhances susceptibility to
Listeria monocytogenes infection. In: The Journal of experimental medicine 200 (4), S. 437–
445.
Olaya-Abril, Alfonso; Prados-Rosales, Rafael; McConnell, Michael J.; Martín-Peña, Reyes;
González-Reyes, José Antonio; Jiménez-Munguía, Irene et al. (2014): Characterization of
protective extracellular membrane-derived vesicles produced by Streptococcus pneumoniae. In:
Journal of proteomics 106, S. 46–60.
Oliver, Haley F.; Orsi, Renato H.; Ponnala, Lalit; Keich, Uri; Wang, Wei; Sun, Qi et al. (2009):
Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase
and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs.
In: BMC genomics 10, S. 641.
Olsen, Katja N.; Larsen, Marianne H.; Gahan, Cormac G. M.; Kallipolitis, Birgitte; Wolf, Xenia
A.; Rea, Rosemary et al. (2005): The Dps-like protein Fri of Listeria monocytogenes promotes
stress tolerance and intracellular multiplication in macrophage-like cells. In: Microbiology
(Reading, England) 151 (Pt 3), S. 925–933.
O'Riordan, Mary; Yi, Caroline H.; Gonzales, Ramona; Lee, Kyung-Dall; Portnoy, Daniel A.
(2002): Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. In:
Proceedings of the National Academy of Sciences of the United States of America 99 (21), S.
13861–13866.
Orsi, Renato H.; Wiedmann, Martin (2016): Characteristics and distribution of Listeria spp.,
including Listeria species newly described since 2009. In: Applied microbiology and
biotechnology 100 (12), S. 5273–5287.
Osborne, Suzanne E.; Sit, Brandon; Shaker, Andrew; Currie, Elissa; Tan, Joël M. J.; van Rijn,
Jorik et al. (2017): Type I interferon promotes cell-to-cell spread of Listeria monocytogenes. In:
Cellular microbiology 19 (3).
Parker, Heather; Chitcholtan, Kenny; Hampton, Mark B.; Keenan, Jacqueline I. (2010): Uptake
of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. In: Infection and
immunity 78 (12), S. 5054–5061.
Patchett, R. A.; Kelly, A. F.; Kroll, R. G. (1991): Respiratory activity in Listeria
monocytogenes. In: FEMS microbiology letters 62 (1), S. 95–98.

7. Literaturverzeichnis
146
Peng, Ye-Long; Meng, Qing-Ling; Qiao, Jun; Xie, Kun; Chen, Cheng; Liu, Tian-Li et al.
(2016): The Regulatory Roles of ncRNA Rli60 in Adaptability of Listeria monocytogenes to
Environmental Stress and Biofilm Formation. In: Current microbiology 73 (1), S. 77–83.
Pérez-Cruz, Carla; Carrión, Ornella; Delgado, Lidia; Martinez, Gemma; López-Iglesias,
Carmen; Mercade, Elena (2013): New type of outer membrane vesicle produced by the Gram-
negative bacterium Shewanella vesiculosa M7T. Implications for DNA content. In: Applied and
environmental microbiology 79 (6), S. 1874–1881.
Perry, Andrea K.; Chen, Gang; Zheng, Dahai; Tang, Hong; Cheng, Genhong (2005): The host
type I interferon response to viral and bacterial infections. In: Cell research 15 (6), S. 407–422.
Pfaffl, M. W. (2001): A new mathematical model for relative quantification in real-time RT-
PCR. In: Nucleic acids research 29 (9), e45.
Pichlmair, Andreas; Schulz, Oliver; Tan, Choon Ping; Näslund, Tanja I.; Liljeström, Peter;
Weber, Friedemann; Reis e Sousa, Caetano (2006): RIG-I-mediated antiviral responses to
single-stranded RNA bearing 5'-phosphates. In: Science (New York, N.Y.) 314 (5801), S. 997–
1001.
Pilgrim, Sabine; Kolb-Mäurer, Annette; Gentschev, Ivaylo; Goebel, Werner; Kuhn, Michael
(2003): Deletion of the gene encoding p60 in Listeria monocytogenes leads to abnormal cell
division and loss of actin-based motility. In: Infection and immunity 71 (6), S. 3473–3484.
Pillarisetty, Venu G.; Shah, Alaap B.; Miller, George; Bleier, Joshua I.; DeMatteo, Ronald P.
(2004): Liver dendritic cells are less immunogenic than spleen dendritic cells because of
differences in subtype composition. In: Journal of immunology (Baltimore, Md. : 1950) 172 (2),
S. 1009–1017.
Pillich, Helena; Chakraborty, Trinad; Mraheil, Mobarak Abu (2015): Cell-autonomous
responses in Listeria monocytogenes infection. In: Future microbiology 10 (4), S. 583–597.
Port, Gary C.; Freitag, Nancy E. (2007): Identification of novel Listeria monocytogenes
secreted virulence factors following mutational activation of the central virulence regulator,
PrfA. In: Infection and immunity 75 (12), S. 5886–5897.
Portnoy, D. A.; Jacks, P. S.; Hinrichs, D. J. (1988): Role of hemolysin for the intracellular
growth of Listeria monocytogenes. In: The Journal of experimental medicine 167 (4), S. 1459–
1471.
Premaratne, R. J.; Lin, W. J.; Johnson, E. A. (1991): Development of an improved chemically
defined minimal medium for Listeria monocytogenes. In: Applied and environmental
microbiology 57 (10), S. 3046–3048.
Py, B.; Causton, H.; Mudd, E. A.; Higgins, C. F. (1994): A protein complex mediating mRNA
degradation in Escherichia coli. In: Molecular microbiology 14 (4), S. 717–729.
Rabinovich, Lev; Sigal, Nadejda; Borovok, Ilya; Nir-Paz, Ran; Herskovits, Anat A. (2012):
Prophage excision activates Listeria competence genes that promote phagosomal escape and
virulence. In: Cell 150 (4), S. 792–802.
Radoshevich, Lilliana; Cossart, Pascale (2018): Listeria monocytogenes. Towards a complete
picture of its physiology and pathogenesis. In: Nature reviews. Microbiology 16 (1), S. 32–46.
Ranjan, Priya; Jayashankar, Lakshmi; Deyde, Varough; Zeng, Hui; Davis, William G.; Pearce,
Melissa B. et al. (2010): 5'PPP-RNA induced RIG-I activation inhibits drug-resistant avian
H5N1 as well as 1918 and 2009 pandemic influenza virus replication. In: Virology journal 7, S.
102.
Rayamajhi, Manira; Humann, Jessica; Penheiter, Kristi; Andreasen, Karl; Lenz, Laurel L.
(2010): Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage
activation by IFN-gamma. In: The Journal of experimental medicine 207 (2), S. 327–337.

7. Literaturverzeichnis
147
Renelli, Marika; Matias, Valério; Lo, Reggie Y.; Beveridge, Terry J. (2004): DNA-containing
membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation
potential. In: Microbiology (Reading, England) 150 (Pt 7), S. 2161–2169.
Renzoni, A.; Cossart, P.; Dramsi, S. (1999): PrfA, the transcriptional activator of virulence
genes, is upregulated during interaction of Listeria monocytogenes with mammalian cells and in
eukaryotic cell extracts. In: Molecular microbiology 34 (3), S. 552–561.
Resch, Ulrike; Tsatsaronis, James Anthony; Le Rhun, Anaïs; Stübiger, Gerald; Rohde, Manfred;
Kasvandik, Sergo et al. (2016): A Two-Component Regulatory System Impacts Extracellular
Membrane-Derived Vesicle Production in Group A Streptococcus. In: mBio 7 (6).
Ricci, Vittorio; Chiozzi, Valentina; Necchi, Vittorio; Oldani, Amanda; Romano, Marco; Solcia,
Enrico; Ventura, Ulderico (2005): Free-soluble and outer membrane vesicle-associated VacA
from Helicobacter pylori. Two forms of release, a different activity. In: Biochemical and
biophysical research communications 337 (1), S. 173–178.
Rietkötter, Eva; Hoyer, Diana; Mascher, Thorsten (2008): Bacitracin sensing in Bacillus
subtilis. In: Molecular microbiology 68 (3), S. 768–785.
Rivera, Johanna; Cordero, Radames J. B.; Nakouzi, Antonio S.; Frases, Susana; Nicola, André;
Casadevall, Arturo (2010): Bacillus anthracis produces membrane-derived vesicles containing
biologically active toxins. In: Proceedings of the National Academy of Sciences of the United
States of America 107 (44), S. 19002–19007.
Roberts, A. J.; Wiedmann, M. (2003): Pathogen, host and environmental factors contributing to
the pathogenesis of listeriosis. In: Cellular and molecular life sciences: CMLS 60 (5), S. 904–
918.
Rogers, H. W.; Callery, M. P.; Deck, B.; Unanue, E. R. (1996): Listeria monocytogenes induces
apoptosis of infected hepatocytes. In: Journal of immunology (Baltimore, Md.: 1950) 156 (2), S.
679–684.
Rogers, H. W.; Unanue, E. R. (1993): Neutrophils are involved in acute, nonspecific resistance
to Listeria monocytogenes in mice. In: Infection and immunity 61 (12), S. 5090–5096.
Roier, Sandro; Zingl, Franz G.; Cakar, Fatih; Schild, Stefan (2016): Bacterial outer membrane
vesicle biogenesis. A new mechanism and its implications. In: Microbial cell (Graz, Austria) 3
(6), S. 257–259.
Romick, T. L.; Fleming, H. P.; McFeeters, R. F. (1996): Aerobic and anaerobic metabolism of
Listeria monocytogenes in defined glucose medium. In: Applied and environmental
microbiology 62 (1), S. 304–307.
Rumbo, Carlos; Fernández-Moreira, Esteban; Merino, María; Poza, Margarita; Mendez, Jose
Antonio; Soares, Nelson C. et al. (2011): Horizontal transfer of the OXA-24 carbapenemase
gene via outer membrane vesicles. A new mechanism of dissemination of carbapenem
resistance genes in Acinetobacter baumannii. In: Antimicrobial agents and chemotherapy 55
(7), S. 3084–3090.
Sadler, Anthony J.; Williams, Bryan R. G. (2008): Interferon-inducible antiviral effectors. In:
Nature reviews. Immunology 8 (7), S. 559–568.
Safley, S. A.; Cluff, C. W.; Marshall, N. E.; Ziegler, H. K. (1991): Role of listeriolysin-O
(LLO) in the T lymphocyte response to infection with Listeria monocytogenes. Identification of
T cell epitopes of LLO. In: Journal of immunology (Baltimore, Md.: 1950) 146 (10), S. 3604–
3616.
Sander, Leif E.; Davis, Michael J.; Boekschoten, Mark V.; Amsen, Derk; Dascher, Christopher
C.; Ryffel, Bernard et al. (2011): Detection of prokaryotic mRNA signifies microbial viability
and promotes immunity. In: Nature 474 (7351), S. 385–389.

7. Literaturverzeichnis
148
Schaar, Viveka; Nordström, Therése; Mörgelin, Matthias; Riesbeck, Kristian (2011): Moraxella
catarrhalis outer membrane vesicles carry β-lactamase and promote survival of Streptococcus
pneumoniae and Haemophilus influenzae by inactivating amoxicillin. In: Antimicrobial agents
and chemotherapy 55 (8), S. 3845–3853.
Schaumburg, Jessica; Diekmann, Oliver; Hagendorff, Petra; Bergmann, Simone; Rohde,
Manfred; Hammerschmidt, Sven et al. (2004): The cell wall subproteome of Listeria
monocytogenes. In: Proteomics 4 (10), S. 2991–3006.
Schild, Stefan; Nelson, Eric J.; Camilli, Andrew (2008): Immunization with Vibrio cholerae
outer membrane vesicles induces protective immunity in mice. In: Infection and immunity 76
(10), S. 4554–4563.
Schlee, Martin; Roth, Andreas; Hornung, Veit; Hagmann, Cristina Amparo; Wimmenauer,
Vera; Barchet, Winfried et al. (2009): Recognition of 5' triphosphate by RIG-I helicase requires
short blunt double-stranded RNA as contained in panhandle of negative-strand virus. In:
Immunity 31 (1), S. 25–34.
Schmidt, Andreas; Schwerd, Tobias; Hamm, Wolfgang; Hellmuth, Johannes C.; Cui, Sheng;
Wenzel, Michael et al. (2009): 5'-triphosphate RNA requires base-paired structures to activate
antiviral signaling via RIG-I. In: Proceedings of the National Academy of Sciences of the United
States of America 106 (29), S. 12067–12072.
Schwechheimer, Carmen; Kuehn, Meta J. (2015): Outer-membrane vesicles from Gram-
negative bacteria. Biogenesis and functions. In: Nature reviews. Microbiology 13 (10), S. 605–
619.
Serbina, Natalya V.; Pamer, Eric G. (2006): Monocyte emigration from bone marrow during
bacterial infection requires signals mediated by chemokine receptor CCR2. In: Nature
immunology 7 (3), S. 311–317.
Serbina, Natalya V.; Salazar-Mather, Thais P.; Biron, Christine A.; Kuziel, William A.; Pamer,
Eric G. (2003): TNF/iNOS-producing dendritic cells mediate innate immune defense against
bacterial infection. In: Immunity 19 (1), S. 59–70.
Seveau, Stéphanie; Bierne, Hélène; Giroux, Stéphanie; Prévost, Marie-Christine; Cossart,
Pascale (2004): Role of lipid rafts in E-cadherin-- and HGF-R/Met--mediated entry of Listeria
monocytogenes into host cells. In: The Journal of cell biology 166 (5), S. 743–753.
Sha, Wenwen; Mitoma, Hiroki; Hanabuchi, Shino; Bao, Musheng; Weng, Leiyun; Sugimoto,
Naoshi et al. (2014): Human NLRP3 inflammasome senses multiple types of bacterial RNAs.
In: Proceedings of the National Academy of Sciences of the United States of America 111 (45),
S. 16059–16064.
Shen, Y.; Naujokas, M.; Park, M.; Ireton, K. (2000): InIB-dependent internalization of Listeria
is mediated by the Met receptor tyrosine kinase. In: Cell 103 (3), S. 501–510.
Shmaryahu, Amir; Carrasco, Margarita; Valenzuela, Pablo D. T. (2014): Prediction of bacterial
microRNAs and possible targets in human cell transcriptome. In: Journal of microbiology
(Seoul, Korea) 52 (6), S. 482–489.
Sidhu, Vishaldeep K.; Vorhölter, Frank-Jörg; Niehaus, Karsten; Watt, Steven A. (2008):
Analysis of outer membrane vesicle associated proteins isolated from the plant pathogenic
bacterium Xanthomonas campestris pv. campestris. In: BMC microbiology 8, S. 87.
Sijts, A. J.; Pilip, I.; Pamer, E. G. (1997): The Listeria monocytogenes-secreted p60 protein is
an N-end rule substrate in the cytosol of infected cells. Implications for major histocompatibility
complex class I antigen processing of bacterial proteins. In: The Journal of biological chemistry
272 (31), S. 19261–19268.
Sjöström, Annika E.; Sandblad, Linda; Uhlin, Bernt Eric; Wai, Sun Nyunt (2015): Membrane
vesicle-mediated release of bacterial RNA. In: Scientific reports 5, S. 15329.

7. Literaturverzeichnis
149
Sokolova, Viktoriya; Ludwig, Anna-Kristin; Hornung, Sandra; Rotan, Olga; Horn, Peter A.;
Epple, Matthias; Giebel, Bernd (2011): Characterisation of exosomes derived from human cells
by nanoparticle tracking analysis and scanning electron microscopy. In: Colloids and surfaces.
B, Biointerfaces 87 (1), S. 146–150.
Stetson, Daniel B.; Medzhitov, Ruslan (2006): Recognition of cytosolic DNA activates an
IRF3-dependent innate immune response. In: Immunity 24 (1), S. 93–103.
Stockinger, Silvia; Kastner, Renate; Kernbauer, Elisabeth; Pilz, Andreas; Westermayer, Sandra;
Reutterer, Benjamin et al. (2009): Characterization of the interferon-producing cell in mice
infected with Listeria monocytogenes. In: PLoS pathogens 5 (3), e1000355.
Stockinger, Silvia; Materna, Tilo; Stoiber, Dagmar; Bayr, Lourdes; Steinborn, Ralf; Kolbe,
Thomas et al. (2002): Production of type I IFN sensitizes macrophages to cell death induced by
Listeria monocytogenes. In: Journal of immunology (Baltimore, Md.: 1950) 169 (11), S. 6522–
6529.
Stoll, Regina; Goebel, Werner (2010): The major PEP-phosphotransferase systems (PTSs) for
glucose, mannose and cellobiose of Listeria monocytogenes, and their significance for extra-
and intracellular growth. In: Microbiology (Reading, England) 156 (Pt 4), S. 1069–1083.
Sun, Lijun; Wu, Jiaxi; Du, Fenghe; Chen, Xiang; Chen, Zhijian J. (2013): Cyclic GMP-AMP
synthase is a cytosolic DNA sensor that activates the type I interferon pathway. In: Science
(New York, N.Y.) 339 (6121), S. 786–791.
Takahashi, Ken; Asabe, Shinichi; Wieland, Stefan; Garaigorta, Urtzi; Gastaminza, Pablo;
Isogawa, Masanori; Chisari, Francis V. (2010): Plasmacytoid dendritic cells sense hepatitis C
virus-infected cells, produce interferon, and inhibit infection. In: Proceedings of the National
Academy of Sciences of the United States of America 107 (16), S. 7431–7436.
Takahasi, Kiyohiro; Yoneyama, Mitsutoshi; Nishihori, Tatsuya; Hirai, Reiko; Kumeta,
Hiroyuki; Narita, Ryo et al. (2008): Nonself RNA-sensing mechanism of RIG-I helicase and
activation of antiviral immune responses. In: Molecular cell 29 (4), S. 428–440.
Takeuchi, Osamu; Akira, Shizuo (2009): Innate immunity to virus infection. In: Immunological
reviews 227 (1), S. 75–86.
Thay, Bernard; Wai, Sun Nyunt; Oscarsson, Jan (2013): Staphylococcus aureus α-toxin-
dependent induction of host cell death by membrane-derived vesicles. In: PloS one 8 (1),
e54661.
Todd, E.C.D.; Notermans, S. (2011): Surveillance of listeriosis and its causative pathogen,
Listeria monocytogenes. In: Food Control 22 (9), S. 1484–1490.
Toledo-Arana, Alejandro; Dussurget, Olivier; Nikitas, Georgios; Sesto, Nina; Guet-Revillet,
Hélène; Balestrino, Damien et al. (2009): The Listeria transcriptional landscape from
saprophytism to virulence. In: Nature 459 (7249), S. 950–956.
Trinchieri, Giorgio (2010): Type I interferon. Friend or foe? In: The Journal of experimental
medicine 207 (10), S. 2053–2063.
Tripp, C. S.; Wolf, S. F.; Unanue, E. R. (1993): Interleukin 12 and tumor necrosis factor alpha
are costimulators of interferon gamma production by natural killer cells in severe combined
immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. In:
Proceedings of the National Academy of Sciences of the United States of America 90 (8), S.
3725–3729.
Trost, Matthias; Wehmhöner, Dirk; Kärst, Uwe; Dieterich, Guido; Wehland, Jürgen; Jänsch,
Lothar (2005): Comparative proteome analysis of secretory proteins from pathogenic and
nonpathogenic Listeria species. In: Proteomics 5 (6), S. 1544–1557.

7. Literaturverzeichnis
150
Turchinovich, Andrey; Weiz, Ludmila; Langheinz, Anne; Burwinkel, Barbara (2011):
Characterization of extracellular circulating microRNA. In: Nucleic acids research 39 (16), S.
7223–7233.
Uli, Liliam; Castellanos-Serra, Lila; Betancourt, Lazaro; Domínguez, Francisco; Barberá,
Ramón; Sotolongo, Franklin et al. (2006): Outer membrane vesicles of the VA-MENGOC-BC
vaccine against serogroup B of Neisseria meningitidis. Analysis of protein components by two-
dimensional gel electrophoresis and mass spectrometry. In: Proteomics 6 (11), S. 3389–3399.
Valadi, Hadi; Ekström, Karin; Bossios, Apostolos; Sjöstrand, Margareta; Lee, James J.; Lötvall,
Jan O. (2007): Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of
genetic exchange between cells. In: Nature cell biology 9 (6), S. 654–659.
Valderrama, Carolina; Clark, Amy; Urano, Fumihiko; Unanue, Emil R.; Carrero, Javier A.
(2017): Listeria monocytogenes induces an interferon-enhanced activation of the integrated
stress response that is detrimental for resolution of infection in mice. In: European journal of
immunology 47 (5), S. 830–840.
Vanaja, Sivapriya Kailasan; Russo, Ashley J.; Behl, Bharat; Banerjee, Ishita; Yankova, Maya;
Deshmukh, Sachin D.; Rathinam, Vijay A. K. (2016): Bacterial Outer Membrane Vesicles
Mediate Cytosolic Localization of LPS and Caspase-11 Activation. In: Cell 165 (5), S. 1106–
1119.
Vanzo, N. F.; Li, Y. S.; Py, B.; Blum, E.; Higgins, C. F.; Raynal, L. C. et al. (1998):
Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. In:
Genes & development 12 (17), S. 2770–2781.
Vasconcelos, J. A.; Deneer, H. G. (1994): Expression of superoxide dismutase in Listeria
monocytogenes. In: Applied and environmental microbiology 60 (7), S. 2360–2366.
Vdovikova, Svitlana; Luhr, Morten; Szalai, Paula; Nygård Skalman, Lars; Francis, Monika K.;
Lundmark, Richard et al. (2017): A Novel Role of Listeria monocytogenes Membrane Vesicles
in Inhibition of Autophagy and Cell Death. In: Frontiers in cellular and infection microbiology
7, S. 154.
Veiga, Esteban; Cossart, Pascale (2005): Listeria hijacks the clathrin-dependent endocytic
machinery to invade mammalian cells. In: Nature cell biology 7 (9), S. 894–900.
Vickers, Kasey C.; Palmisano, Brian T.; Shoucri, Bassem M.; Shamburek, Robert D.; Remaley,
Alan T. (2011): MicroRNAs are transported in plasma and delivered to recipient cells by high-
density lipoproteins. In: Nature cell biology 13 (4), S. 423–433.
Wang, Kai; Zhang, Shile; Weber, Jessica; Baxter, David; Galas, David J. (2010): Export of
microRNAs and microRNA-protective protein by mammalian cells. In: Nucleic acids research
38 (20), S. 7248–7259.
Weis, J.; Seeliger, H. P. (1975): Incidence of Listeria monocytogenes in nature. In: Applied
microbiology 30 (1), S. 29–32.
Wherry, J. C.; Schreiber, R. D.; Unanue, E. R. (1991): Regulation of gamma interferon
production by natural killer cells in scid mice. Roles of tumor necrosis factor and bacterial
stimuli. In: Infection and immunity 59 (5), S. 1709–1715.
Williams, B. R. (2001): Signal integration via PKR. In: Science's STKE: signal transduction
knowledge environment 2001 (89), re2.
Woodward, Joshua J.; Iavarone, Anthony T.; Portnoy, Daniel A. (2010): c-di-AMP secreted by
intracellular Listeria monocytogenes activates a host type I interferon response. In: Science
(New York, N.Y.) 328 (5986), S. 1703–1705.

7. Literaturverzeichnis
151
Wu, Jiaxi; Sun, Lijun; Chen, Xiang; Du, Fenghe; Shi, Heping; Chen, Chuo; Chen, Zhijian J.
(2013): Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by
cytosolic DNA. In: Science (New York, N.Y.) 339 (6121), S. 826–830.
Yaron, S.; Kolling, G. L.; Simon, L.; Matthews, K. R. (2000): Vesicle-mediated transfer of
virulence genes from Escherichia coli O157:H7 to other enteric bacteria. In: Applied and
environmental microbiology 66 (10), S. 4414–4420.
Yoneyama, Mitsutoshi; Onomoto, Koji; Jogi, Michihiko; Akaboshi, Teppei; Fujita, Takashi
(2015): Viral RNA detection by RIG-I-like receptors. In: Current opinion in immunology 32, S.
48–53.
Zakharzhevskaya, Natalya B.; Vanyushkina, Anna A.; Altukhov, Ilya A.; Shavarda, Aleksey L.;
Butenko, Ivan O.; Rakitina, Daria V. et al. (2017): Outer membrane vesicles secreted by
pathogenic and nonpathogenic Bacteroides fragilis represent different metabolic activities. In:
Scientific reports 7 (1), S. 5008.
Zhao, Guanghua; Ceci, Pierpaolo; Ilari, Andrea; Giangiacomo, Laura; Laue, Thomas M.;
Chiancone, Emilia; Chasteen, N. Dennis (2002): Iron and hydrogen peroxide detoxification
properties of DNA-binding protein from starved cells. A ferritin-like DNA-binding protein of
Escherichia coli. In: The Journal of biological chemistry 277 (31), S. 27689–27696.
Zhu, Wenhong; Smith, Jeffrey W.; Huang, Chun-Ming (2010): Mass spectrometry-based label-
free quantitative proteomics. In: Journal of biomedicine & biotechnology 2010, S. 840518.
Zybailov, Boris; Coleman, Michael K.; Florens, Laurence; Washburn, Michael P. (2005):
Correlation of relative abundance ratios derived from peptide ion chromatograms and spectrum
counting for quantitative proteomic analysis using stable isotope labeling. In: Analytical
chemistry 77 (19), S. 6218–6224.
Zybailov, Boris; Mosley, Amber L.; Sardiu, Mihaela E.; Coleman, Michael K.; Florens,
Laurence; Washburn, Michael P. (2006): Statistical analysis of membrane proteome expression
changes in Saccharomyces cerevisiae. In: Journal of proteome research 5 (9), S. 2339–2347.

8. Anhang
152
8. Anhang
8.1. Veröffentlichungen
Fischer, Sabrina; Lamping, Matthias; Gold, Maike; Röttger, Yvonne; Brödje, Dörte;
Dodel, Richard; Frantz, Renate; Mraheil, Mobarak Abu; Chakraborty, Trinad (2017):
Synthesis of a biological active β-hairpin peptide by addition of two structural motifs.
In: Bioorganic & medicinal chemistry 25 (2), S. 603–608.
Mraheil, Mobarak Abu; Frantz, Renate; Teubner, Lisa; Wendt, Heiko; Linne, Uwe;
Wingerath, Jessica; Wirth, Thomas; Chakraborty, Trinad (2017): Requirement of the
RNA-binding protein SmpB during intracellular growth of Listeria monocytogenes. In:
International journal of medical microbiology: IJMM 307 (3), S. 166–173.
Die in dieser Arbeit gezeigten Daten über die von L. monocytogenes sekretierten sRNAs
und deren Potential zur Induktion von IFN-β wurden als Manuskript bei Nature
Communications unter folgendem Namen submittiert: Comprehensive analysis of the
secRNome of Listeria monocytogenes reveals small non-coding RNA as potent inducers
of IFN-β response.
Folgende Autoren: Frantz, Renate; Schultze, Tilman; Teubner, Lisa; Wingerath, Jessica;
Müller, Christin; Gwozdzinski, Konrad; Pillich, Helena; Hain, Torsten; Gerlach,
Michaela; Mostafa, Ahmed; Weber, Friedemann; Rohde, Manfred; Pleschka, Stephan;
Wirth, Thomas; Chakraborty, Trinad; Mraheil, Mobarak Abu

8. Anhang
153
8.2. Kongressteilnahme
GGL (2013)
Poster: Detection of nucleic acids secreted by L. monocytogenes
GGL (2014)
Poster: Characterization of membrane vesicles (MVs) produced by L. monocytogenes
GGL Retreat (2015)
Vortrag: Secreted nucleic acids and membrane vesicles (MVs) released by L. monocyto-
genes
Proantilis, Porto (2015)
Vortrag: Mechanisms affecting secretion and shedding of bacterial components.
Production of Membrane Vesicles and NAs secretion
Vorgetragen von Dr. Abu Mraheil
Isopol, Paris (2016)
Poster: Immunogenic capacity of membrane vesicles (MVs) produced by L. monocyto-
genes
Proantilis, Paris (2017)
Vortrag: Secretion mechanisms of nucleic acids by L. monocytogenes
Vorgetragen von Dr. Abu Mraheil
Proantilis, Gießen (2017)
Vortrag: Analysis of the secRNome of L. monocytogenes
Vorgetragen von Dr. Abu Mraheil

8. Anhang
154
8.3. Danksagung
Mein besonderer Dank gilt Prof. Dr. Chakraborty für die Bereitstellung des Projekts und
die Möglichkeit diese Arbeit im Institut für Medizinische Mikrobiologie anfertigen zu
können. Des Weiteren möchte ich mich bei Ihm für die Betreuung und Unterstützung
der Arbeit bedanken.
Weiterhin möchte ich mich bei Dr. Abu Mraheil für die kontinuierliche Betreuung der
Arbeit bedanken.
Des Weiteren möchte ich mich bei den Kooperationspartner Dr. Andreas Otto, Prof. Dr.
Dörte Becher, Christin Müller, Prof. Dr. Stephan Pleschka, Jessica Wingerath, Dr.
Thomas Wirth und Prof. Dr. Manfred Rohde für deren Unterstützung bedanken.
Mein herzlicher Dank gilt Lisa Teubner für ihre unermüdlich gute Laune, die mich in
dieser Zeit aufgebaut hat. Des Weiteren möchte ich mich bei Ihr für die tatkräftige
Unterstützung in allen Belangen des alltäglichen Laboraltags ganz lieb bedanken.
Des Weiteren möchte ich mich bei den anderen Kollegen im Labor bedanken, Tilman
Schultze, Nelli und Juri Schklarenko, Sylvia Krämer, Martina Hudel, Besim Berisha,
Luigi La Pietra, Konrad Gwozdzinski, Helena Pillich, Christina Gerstmann und
Alexandra Amend.
Ein herzlicher Dank gilt der Familie Keil, besonders Frau Keil und Jochen, für deren
aufmerksame Unterstützung und Hilfsbereitschaft in jeglichen Lebenssituationen.
Im Besonderen möchte ich mich bei meinen Eltern für deren Hilfe und Beistand in
dieser Zeit bedanken. Dies gilt auch für meine Zwillingsschwester und Bruder, die
immer für mich da waren. Abschließend danke ich meinen Eltern für das Verständnis
dafür, dass ich aufgrund dieser Arbeit nicht so viel Zeit mit Ihnen verbringen konnte.

8. Anhang
155
8.4. Eidesstattliche Erklärung
„Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig und ohne
unzulässige Hilfe oder Benutzung anderer als der angegebenen Hilfsmittel
angefertigt habe. Alle Textstellen, die wörtlich oder sinngemäß aus
veröffentlichten oder nichtveröffentlichten Schriften entnommen sind, und alle
Angaben, die auf mündlichen Auskünften beruhen, sind als solche kenntlich
gemacht. Bei den von mir durchgeführten und in der Dissertation erwähnten
Untersuchungen habe ich die Grundsätze guter wissenschaftlicher Praxis, wie sie
in der „Satzung der Justus-Liebig-Universität Gießen zur Sicherung guter
wissenschaftlicher Praxis“ niedergelegt sind, eingehalten sowie ethische,
datenschutzrechtliche und tierschutzrechtliche Grundsätze befolgt. Ich versichere,
dass Dritte von mir weder unmittelbar noch mittelbar geldwerte Leistungen für
Arbeiten erhalten haben, die im Zusammenhang mit dem Inhalt der vorgelegten
Dissertation stehen, oder habe diese nachstehend spezifiziert. Die vorgelegte
Arbeit wurde weder im Inland noch im Ausland in gleicher oder ähnlicher Form
einer anderen Prüfungsbehörde zum Zweck einer Promotion oder eines anderen
Prüfungsverfahrens vorgelegt. Alles aus anderen Quellen und von anderen
Personen übernommene Material, das in der Arbeit verwendet wurde oder auf das
direkt Bezug genommen wird, wurde als solches kenntlich gemacht. Insbesondere
wurden alle Personen genannt, die direkt und indirekt an der Entstehung der
vorliegenden Arbeit beteiligt waren. Mit der Überprüfung meiner Arbeit durch
eine Plagiatserkennungssoftware bzw. ein internetbasiertes Softwareprogramm
erkläre ich mich einverstanden.“
_____________________ ______________________________
Ort, Datum Unterschrift

8. Anhang
156
8.5. Tab. 1: Einteilung der COGs in 25 funktionelle Kategorien
Metabolismus
[C] Energieproduktion und -konversion
[G] Transport von Kohlenhydraten und deren Metabolismus
[E] Transport von Aminosäuren und deren Metabolismus
[F] Transport von Nukleotiden und deren Metabolismus
[I] Transport von Lipiden und deren Metabolismus
[P] Transport von anorganischen Ionen und deren Metabolismus
[H] Transport von Coenzymen und deren Metabolismus
[Q] Biosynthese von Sekundärmetaboliten, deren Transport und Katabolismus
Zelluläre Prozesse und Signale
[D] Kontrolle des Zellzyklus, Zellteilung, Partitionierung von Chromosomen
[M] Biogenese der Zellwand/Membran/Hülle
[O] Posttranslationale Modifikationen, Proteinumsatz, Chaperone
[U] Intrazellulärer Verkehr, Sekretion, vesikulärer Transport
[T] Mechanismen der Signaltransduktion
[V] Verteidigungsmechanismen
[N] Zellbewegung
[Y] Nukleäre Struktur
[Z] Zytoskelett
[W] Extrazelluläre Strukturen
Informationsspeicherung und -prozessierung
[A] RNA-Prozessierung und -Modifikation
[B] Chromatinstruktur und deren Dynamik
[J] Translation, ribosomale Struktur und Biogenese
[K] Transkription
[L] Replikation, Rekombination und Reparatur
Wenig charakterisiert
[R] Nur generelle Funktion vorhergesagt
[S] Funktion unbekannt
Kein Ortholog gefunden
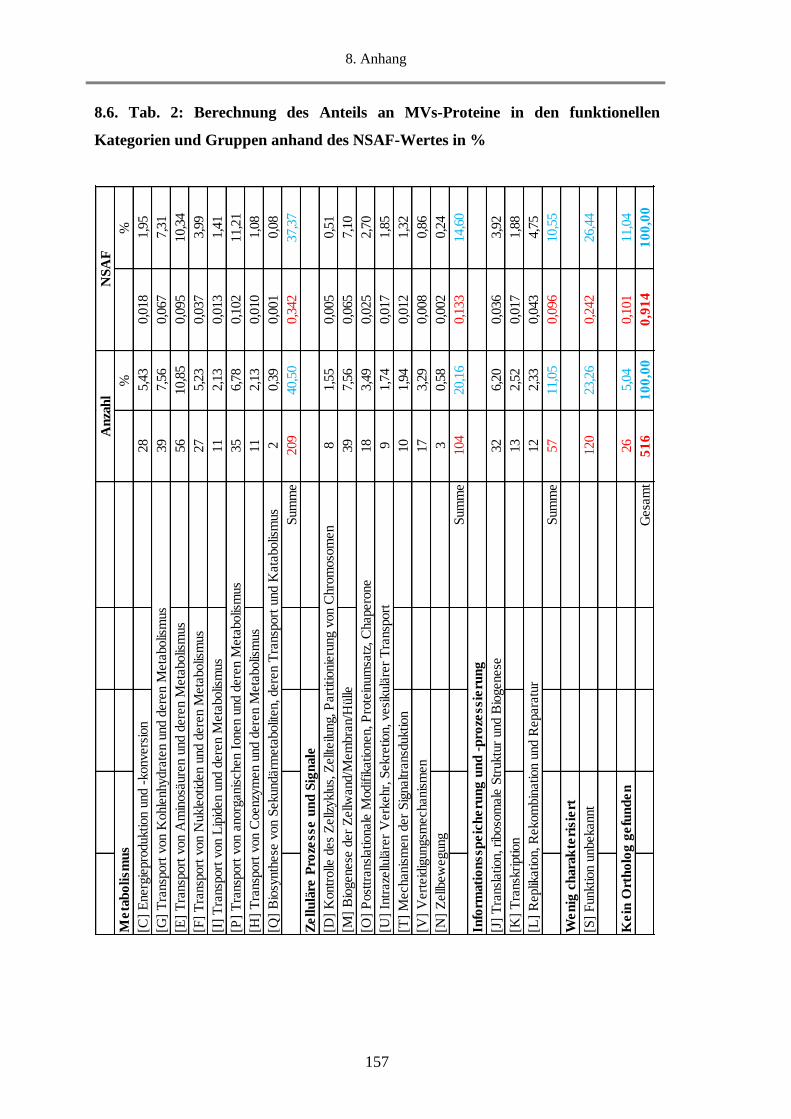
8. Anhang
157
8.6. Tab. 2: Berechnung des Anteils an MVs-Proteine in den funktionellen
Kategorien und Gruppen anhand des NSAF-Wertes in %
Me
tab
oli
sm
us
%%
[C]
Energ
iepro
duktion u
nd -
konvers
ion
28
5,4
30,0
18
1,9
5
[G]
Tra
nsp
ort
von K
ohle
nhydra
ten u
nd d
ere
n M
eta
bolis
mus
39
7,5
60,0
67
7,3
1
[E]
Tra
nsp
ort
von A
min
osä
ure
n u
nd d
ere
n M
eta
bolis
mus
56
10,8
50,0
95
10,3
4
[F]
Tra
nsp
ort
von N
ukle
otiden u
nd d
ere
n M
eta
bolis
mus
27
5,2
30,0
37
3,9
9
[I]
Tra
nsp
ort
von L
ipid
en u
nd d
ere
n M
eta
bolis
mus
11
2,1
30,0
13
1,4
1
[P]
Tra
nsp
ort
von a
norg
anis
chen I
onen u
nd d
ere
n M
eta
bolis
mus
35
6,7
80,1
02
11,2
1
[H]
Tra
nsp
ort
von C
oenzy
men u
nd d
ere
n M
eta
bolis
mus
11
2,1
30,0
10
1,0
8
[Q]
Bio
synth
ese
von S
ekundärm
eta
bolit
en, dere
n T
ransp
ort
und K
ata
bolis
mus
20,3
90,0
01
0,0
8
Sum
me
209
40,5
00,3
42
37,3
7
Ze
llu
läre
Pro
zesse
un
d S
ign
ale
[D]
Kontr
olle
des
Zellz
yklu
s, Z
ellt
eilu
ng, P
art
itio
nie
rung v
on C
hro
moso
men
81,5
50,0
05
0,5
1
[M]
Bio
genese
der
Zellw
and/M
em
bra
n/H
ülle
39
7,5
60,0
65
7,1
0
[O]
Post
transl
ationale
Modif
ikationen, P
rote
inum
satz
, C
hapero
ne
18
3,4
90,0
25
2,7
0
[U]
Intr
aze
llulä
rer
Verk
ehr,
Sekre
tion, vesi
kulä
rer
Tra
nsp
ort
91,7
40,0
17
1,8
5
[T]
Mechanis
men d
er
Sig
naltra
nsd
uktion
10
1,9
40,0
12
1,3
2
[V]
Vert
eid
igungsm
echanis
men
17
3,2
90,0
08
0,8
6
[N]
Zellb
ew
egung
30,5
80,0
02
0,2
4
Sum
me
104
20,1
60,1
33
14,6
0
Info
rmati
on
ssp
eic
he
run
g u
nd
-p
roze
ssie
run
g
[J]
Tra
nsl
ation, ri
boso
male
Str
uktu
r und B
iogenese
32
6,2
00,0
36
3,9
2
[K]
Tra
nsk
ription
13
2,5
20,0
17
1,8
8
[L]
Replik
ation, R
ekom
bin
ation u
nd R
epara
tur
12
2,3
30,0
43
4,7
5
Sum
me
57
11,0
50,0
96
10,5
5
We
nig
ch
ara
kte
risie
rt
[S]
Funktion u
nbekannt
120
23,2
60,2
42
26,4
4
Ke
in O
rth
olo
g g
efu
nd
en
26
5,0
40,1
01
11,0
4
Gesa
mt
51
61
00
,00
0,9
14
10
0,0
0
NS
AF
An
zah
l
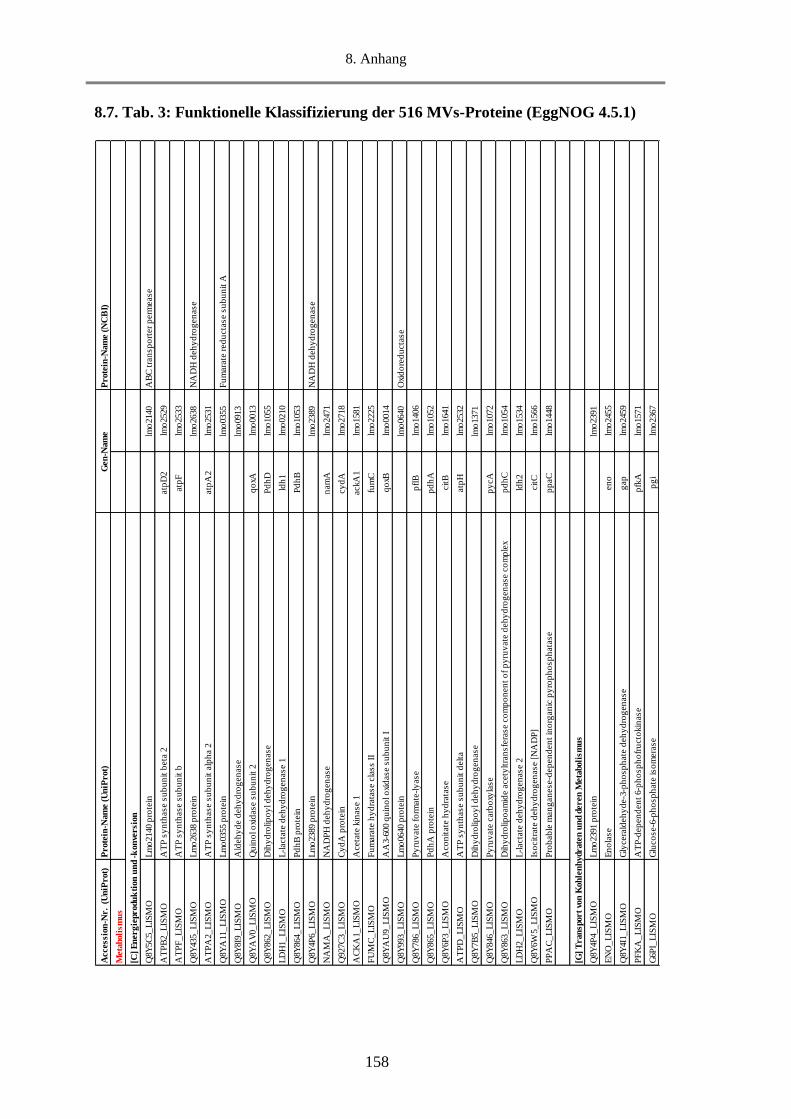
8. Anhang
158
8.7. Tab. 3: Funktionelle Klassifizierung der 516 MVs-Proteine (EggNOG 4.5.1)
Accessio
n-N
r. (
Un
iProt)
Prote
in-N
am
e (
Un
iProt)
Prote
in-N
am
e (
NC
BI)
Meta
boli
sm
us
[C]
En
erg
ieprodu
kti
on
un
d -
kon
versio
n
Q8Y
5C
5_
LIS
MO
Lm
o2140 p
rote
in
lmo
2140
AB
C t
ran
sp
ort
er
perm
ease
AT
PB
2_
LIS
MO
AT
P s
yn
thase s
ub
un
it b
eta
2
atp
D2
lmo
2529
AT
PF
_L
ISM
OA
TP
sy
nth
ase s
ub
un
it b
atp
F
lmo
2533
Q8Y
435_
LIS
MO
Lm
o2638 p
rote
in
lmo
2638
NA
DH
deh
yd
rog
en
ase
AT
PA
2_
LIS
MO
AT
P s
yn
thase s
ub
un
it a
lph
a 2
atp
A2
lmo
2531
Q8Y
A11_
LIS
MO
Lm
o0355 p
rote
in
lmo
0355
Fu
mara
te r
ed
ucta
se s
ub
un
it A
Q8Y
8I9
_L
ISM
OA
ldeh
yd
e d
eh
yd
rog
en
ase
lmo
0913
Q8Y
AV
0_
LIS
MO
Qu
ino
l o
xid
ase s
ub
un
it 2
q
oxA
lm
o0013
Q8Y
862_
LIS
MO
Dih
yd
rolip
oy
l d
eh
yd
rog
en
ase
Pd
hD
lm
o1055
LD
H1_
LIS
MO
L-l
acta
te d
eh
yd
rog
en
ase 1
ld
h1
lmo
0210
Q8Y
864_
LIS
MO
Pd
hB
pro
tein
P
dh
B
lmo
1053
Q8Y
4P
6_
LIS
MO
Lm
o2389 p
rote
in
lmo
2389
NA
DH
deh
yd
rog
en
ase
NA
MA
_L
ISM
ON
AD
PH
deh
yd
rog
en
ase
nam
A
lmo
2471
Q927C
3_
LIS
MO
Cy
dA
pro
tein
cy
dA
lm
o2718
AC
KA
1_
LIS
MO
Aceta
te k
inase 1
ackA
1
lmo
1581
FU
MC
_L
ISM
OF
um
ara
te h
yd
rata
se c
lass I
I fu
mC
lm
o2225
Q8Y
AU
9_
LIS
MO
AA
3-6
00 q
uin
ol o
xid
ase s
ub
un
it I
q
oxB
lm
o0014
Q8Y
993_
LIS
MO
Lm
o0640 p
rote
in
lmo
0640
Oxi
do
red
ucta
se
Q8Y
786_
LIS
MO
Py
ruv
ate
fo
rmate
-ly
ase
pfl
B
lmo
1406
Q8Y
865_
LIS
MO
Pd
hA
pro
tein
p
dh
A
lmo
1052
Q8Y
6P
3_
LIS
MO
Aco
nit
ate
hy
dra
tase
cit
B
lmo
1641
AT
PD
_L
ISM
OA
TP
sy
nth
ase s
ub
un
it d
elt
a
atp
H
lmo
2532
Q8Y
7B
5_
LIS
MO
Dih
yd
rolip
oy
l d
eh
yd
rog
en
ase
lmo
1371
Q8Y
846_
LIS
MO
Py
ruv
ate
carb
oxy
lase
py
cA
lm
o1072
Q8Y
863_
LIS
MO
Dih
yd
rolip
oam
ide a
cety
ltra
nsfe
rase c
om
po
nen
t o
f p
yru
vate
deh
yd
rog
en
ase c
om
ple
x p
dh
C
lmo
1054
LD
H2_
LIS
MO
L-l
acta
te d
eh
yd
rog
en
ase 2
ld
h2
lmo
1534
Q8Y
6W
5_
LIS
MO
Iso
cit
rate
deh
yd
rog
en
ase [
NA
DP
] cit
C
lmo
1566
PP
AC
_L
ISM
OP
rob
ab
le m
an
gan
ese-d
ep
en
den
t in
org
an
ic p
yro
ph
osp
hata
se
pp
aC
lm
o1448
[G]
Tran
sport
von
Koh
len
hydrate
n u
nd d
eren
Meta
boli
sm
us
Q8Y
4P
4_
LIS
MO
Lm
o2391 p
rote
in
lmo
2391
EN
O_
LIS
MO
En
ola
se
en
o
lmo
2455
Q8Y
4I1
_L
ISM
OG
lycera
ldeh
yd
e-3
-ph
osp
hate
deh
yd
rog
en
ase
gap
lm
o2459
PF
KA
_L
ISM
OA
TP
-dep
en
den
t 6-p
ho
sp
ho
fru
cto
kin
ase
pfk
A
lmo
1571
G6P
I_L
ISM
OG
luco
se-6
-ph
osp
hate
iso
mera
se
pg
i lm
o2367
Gen
-Nam
e

8. Anhang
159
Q8Y
AM
0_
LIS
MO
Lm
o0098 p
rote
in
lmo
0098
PT
S m
an
no
se t
ran
sp
ort
er
su
bu
nit
IID
Q8Y
6W
1_
LIS
MO
Py
ruv
ate
kin
ase
py
kA
lm
o1570
Q8Y
8W
1_
LIS
MO
Lm
o0781 p
rote
in
lmo
0781
PT
S m
an
no
se t
ran
sp
ort
er
su
bu
nit
IID
TA
L1_
LIS
MO
Pro
bab
le t
ran
sald
ola
se 1
ta
l1
lmo
2743
Q8Y
7B
0_
LIS
MO
6-p
ho
sp
ho
glu
co
nate
deh
yd
rog
en
ase, d
ecarb
oxy
lati
ng
lm
o1376
Q8Y
7H
4_
LIS
MO
Tra
nsketo
lase
tkt
lmo
1305
Q8Y
5S
7_
LIS
MO
Glu
co
se-6
-ph
osp
hate
1-d
eh
yd
rog
en
ase
zwf
lmo
1978
LA
CD
_L
ISM
OT
ag
ato
se 1
,6-d
iph
osp
hate
ald
ola
se
lacD
lm
o0539
TP
IS1_
LIS
MO
Tri
osep
ho
sp
hate
iso
mera
se 1
tp
iA1
lmo
2457
Q8Y
498_
LIS
MO
Fb
aA
pro
tein
fb
aA
lm
o2556
Q8Y
9F
4_
LIS
MO
Lm
o0574 p
rote
in
lmo
0574
Beta
-glu
co
sid
ase
Q8Y
AM
2_
LIS
MO
Lm
o0096 p
rote
in
lmo
0096
PT
S m
an
no
se t
ran
sp
ort
er
su
bu
nit
IIA
B
Q8Y
AE
9_
LIS
MO
Lm
o0181 p
rote
in
lmo
0181
Su
gar
AB
C t
ran
sp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
GP
MA
_L
ISM
O2,3
-bis
ph
osp
ho
gly
cera
te-d
ep
en
den
t p
ho
sp
ho
gly
cera
te m
uta
se
gp
mA
lm
o2205
PT
1_
LIS
MO
Ph
osp
ho
en
olp
yru
vate
-pro
tein
ph
osp
ho
tran
sfe
rase
pts
I lm
o1003
Q8Y
681_
LIS
MO
Rib
ulo
se-p
ho
sp
hate
3-e
pim
era
se
lmo
1818
Q8Y
4U
6_
LIS
MO
Fru
A p
rote
in
fru
A
lmo
2335
Q8Y
404_
LIS
MO
Lm
o2674 p
rote
in
lmo
2674
Rib
ose-5
-ph
osp
hate
iso
mera
se B
Q8Y
AM
1_
LIS
MO
Lm
o0097 p
rote
in
lmo
0097
PT
S m
an
no
se t
ran
sp
ort
er
su
bu
nit
IIC
Q8Y
9W
8_
LIS
MO
Lm
o0401 p
rote
in
lmo
0401
Alp
ha-m
an
no
sid
ase
Q8Y
7L
9_
LIS
MO
Lm
o1255 p
rote
in
lmo
1255
PT
S t
reh
alo
se t
ran
sp
ort
er
su
bu
nit
IIB
C
Q8Y
8Q
8_
LIS
MO
Uh
pT
pro
tein
u
hp
T
lmo
0838
Q8Y
9V
5_
LIS
MO
Lm
o0415 p
rote
in
lmo
0415
En
do
-1,4
-beta
-xy
lan
ase
PG
K_
LIS
MO
Ph
osp
ho
gly
cera
te k
inase
pg
k
lmo
2458
Q8Y
3Z
4_
LIS
MO
Perm
ease I
IC c
om
po
nen
t lm
o2684
Q8Y
8J4
_L
ISM
OL
mo
0907 p
rote
in
lmo
0907
Ph
osp
ho
gly
cera
te m
uta
se
Q8Y
7E
4_
LIS
MO
Lm
o1339 p
rote
in
lmo
1339
Glu
co
se k
inase
Q8Y
990_
LIS
MO
Tra
nsald
ola
se
fsa_
like
lmo
0643
Q8Y
A88_
LIS
MO
Lm
o0271 p
rote
in
lmo
0271
Ph
osp
ho
-beta
-glu
co
sid
ase
Q8Y
4G
8_
LIS
MO
Lm
o2475 p
rote
in
lmo
2475
Ph
osp
ho
glu
co
mu
tase
Q929P
4_
LIS
MO
Lm
o2125 p
rote
in
lmo
2125
Su
gar
AB
C t
ran
sp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
9K
6_
LIS
MO
Lm
o0521 p
rote
in
lmo
0521
6-p
ho
sp
ho
-beta
-glu
co
sid
ase
Q8Y
904_
LIS
MO
Lm
o0738 p
rote
in
lmo
0738
PT
S b
eta
-glu
co
sid
e t
ran
sp
ort
er
su
bu
nit
IIA
BC
[E]
Tran
sport
von
Am
inosäu
ren
un
d d
eren
Meta
boli
sm
us
Q7A
P53_
LIS
MO
Lm
o2193 p
rote
in
lmo
2193
Pep
tid
e A
BC
tra
nsp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
6E
9_
LIS
MO
Lm
o1738 p
rote
in
lmo
1738
Am
ino
acid
AB
C t
ran
sp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q7A
P52_
LIS
MO
Lm
o2196 p
rote
in
lmo
2196
Pep
tid
e A
BC
tra
nsp
ort
er
su
bstr
ate
-bin
din
g p
rote
in

8. Anhang
160
Q8Y
AJ0
_L
ISM
OL
mo
0135 p
rote
in
lmo
0135
Pep
tid
e A
BC
tra
nsp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
6E
7_
LIS
MO
Lm
o1740 p
rote
in
lmo
1740
Am
ino
acid
AB
C t
ran
sp
ort
er
perm
ease
Q8Y
7I0
_L
ISM
OG
luta
min
e s
yn
theta
se
gln
A
lmo
1299
Q8Y
6E
8_
LIS
MO
Lm
o1739 p
rote
in
lmo
1739
Am
ino
acid
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
9G
8_
LIS
MO
Glu
tam
ate
deh
yd
rog
en
ase
lmo
0560
Q8Y
6V
2_
LIS
MO
Ala
nin
e d
eh
yd
rog
en
ase
lmo
1579
HIS
7_
LIS
MO
Imid
azo
leg
lycero
l-p
ho
sp
hate
deh
yd
rata
se
his
B
lmo
0566
Q7A
P65_
LIS
MO
Op
uC
A p
rote
in
op
uC
A
lmo
1428
Q8Y
8C
6_
LIS
MO
Lm
o0982 p
rote
in
lmo
0982
Pep
tid
ase
Q8Y
775_
LIS
MO
Lm
o1422 p
rote
in
lmo
1422
Gly
cin
e/b
eta
ine A
BC
tra
nsp
ort
er
perm
ease
Q8Y
581_
LIS
MO
Lm
o2192 p
rote
in
lmo
2192
Pep
tid
e A
BC
tra
nsp
ort
er
AT
P-b
ind
ing
pro
tein
PR
TA
_L
ISM
OZ
inc m
eta
llo
pro
tein
ase
mp
l lm
o0203
Q8Y
AC
3_
LIS
MO
Cy
ste
ine s
yn
thase
cy
sK
lm
o0223
Q7A
P67_
LIS
MO
Op
uC
C p
rote
in
op
uC
C
lmo
1426
DA
PE
L_
LIS
MO
N-a
cety
ldia
min
op
imela
te d
eacety
lase
lmo
1012
GL
YA
_L
ISM
OS
eri
ne h
yd
roxy
meth
ylt
ran
sfe
rase
gly
A
lmo
2539
GC
SP
B_
LIS
MO
Pro
bab
le g
lycin
e d
eh
yd
rog
en
ase (
decarb
oxy
lati
ng
) su
bu
nit
2
gcv
PB
lm
o1350
Q8Y
638_
LIS
MO
Lm
o1862 p
rote
in
lmo
1862
Q7A
P68_
LIS
MO
Op
uC
D p
rote
in
op
uC
D
lmo
1425
Q8Y
AH
4_
LIS
MO
Lm
o0152 p
rote
in
lmo
0152
Pep
tid
e A
BC
tra
nsp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
486_
LIS
MO
Lm
o2569 p
rote
in
lmo
2569
Pep
tid
e A
BC
tra
nsp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
8P
9_
LIS
MO
Lm
o0847 p
rote
in
lmo
0847
Glu
tam
ine A
BC
tra
nsp
ort
er
GC
SP
A_
LIS
MO
Pro
bab
le g
lycin
e d
eh
yd
rog
en
ase (
decarb
oxy
lati
ng
) su
bu
nit
1
gcv
PA
lm
o1349
Q7A
P76_
LIS
MO
Gb
uA
pro
tein
g
bu
A
lmo
1014
Q7A
P69_
LIS
MO
Lm
o1421 p
rote
in
lmo
1421
Gly
cin
e/b
eta
ine A
BC
tra
nsp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
730_
LIS
MO
Lm
o1493 p
rote
in
lmo
1493
Olig
op
ep
tid
ase
Q8Y
583_
LIS
MO
Lm
o2188 p
rote
in
lmo
2188
Olig
oen
do
pep
tid
ase
Q8Y
8D
0_
LIS
MO
Bra
nch
ed
-ch
ain
-am
ino
-acid
am
ino
tran
sfe
rase
lmo
0978
DA
AA
_L
ISM
OD
-ala
nin
e a
min
otr
an
sfe
rase
dat
lmo
1619
DA
PB
_L
ISM
O4-h
yd
roxy
-tetr
ah
yd
rod
ipic
olin
ate
red
ucta
se
dap
B
lmo
1907
Q8Y
898_
LIS
MO
Gb
uC
pro
tein
g
bu
C
lmo
1016
Q8Y
AB
2_
LIS
MO
Seri
ne a
cety
ltra
nsfe
rase
cy
sE
lm
o0238
AR
OA
_L
ISM
O3-p
ho
sp
ho
sh
ikim
ate
1-c
arb
oxy
vin
ylt
ran
sfe
rase
aro
A
lmo
1923
Q8Y
527_
LIS
MO
Arp
J p
rote
in
arp
J lm
o2250
Q8Y
9X
2_
LIS
MO
Py
rro
lin
e-5
-carb
oxy
late
red
ucta
se
pro
C
lmo
0396
Q8Y
8Y
7_
LIS
MO
Lm
o0755 p
rote
in
lmo
0755
Q8Y
6T
0_
LIS
MO
Lm
o1603 p
rote
in
lmo
1603
Am
ino
pep
tid
ase
Q8Y
8P
8_
LIS
MO
Lm
o0848 p
rote
in
lmo
0848
Am
ino
acid
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
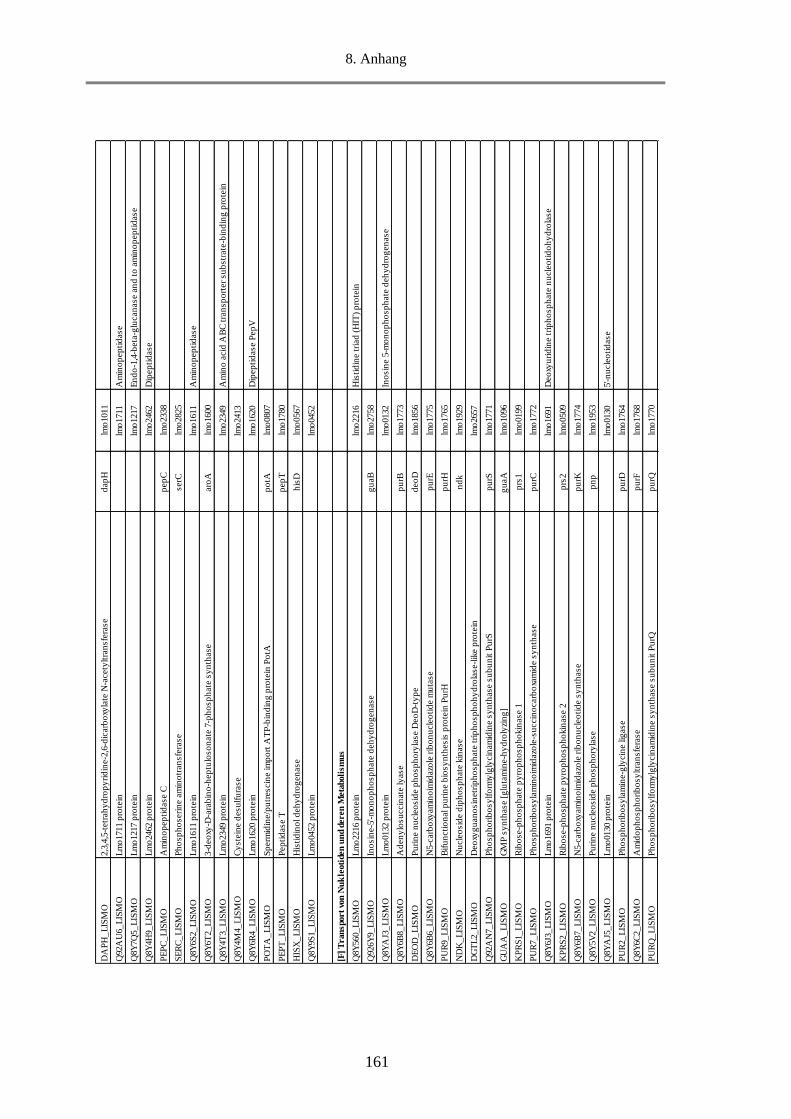
8. Anhang
161
DA
PH
_L
ISM
O2,3
,4,5
-tetr
ah
yd
rop
yri
din
e-2
,6-d
icarb
oxy
late
N-a
cety
ltra
nsfe
rase
dap
H
lmo
1011
Q92A
U6_
LIS
MO
Lm
o1711 p
rote
in
lmo
1711
Am
ino
pep
tid
ase
Q8Y
7Q
5_
LIS
MO
Lm
o1217 p
rote
in
lmo
1217
En
do
-1,4
-beta
-glu
can
ase a
nd
to
am
ino
pep
tid
ase
Q8Y
4H
9_
LIS
MO
Lm
o2462 p
rote
in
lmo
2462
Dip
ep
tid
ase
PE
PC
_L
ISM
OA
min
op
ep
tid
ase C
p
ep
C
lmo
2338
SE
RC
_L
ISM
OP
ho
sp
ho
seri
ne a
min
otr
an
sfe
rase
serC
lm
o2825
Q8Y
6S
2_
LIS
MO
Lm
o1611 p
rote
in
lmo
1611
Am
ino
pep
tid
ase
Q8Y
6T
2_
LIS
MO
3-d
eo
xy-D
-ara
bin
o-h
ep
tulo
so
nate
7-p
ho
sp
hate
sy
nth
ase
aro
A
lmo
1600
Q8Y
4T
3_
LIS
MO
Lm
o2349 p
rote
in
lmo
2349
Am
ino
acid
AB
C t
ran
sp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
4M
4_
LIS
MO
Cy
ste
ine d
esu
lfu
rase
lmo
2413
Q8Y
6R
4_
LIS
MO
Lm
o1620 p
rote
in
lmo
1620
Dip
ep
tid
ase P
ep
V
PO
TA
_L
ISM
OS
perm
idin
e/p
utr
escin
e im
po
rt A
TP
-bin
din
g p
rote
in P
otA
p
otA
lm
o0807
PE
PT
_L
ISM
OP
ep
tid
ase T
p
ep
T
lmo
1780
HIS
X_
LIS
MO
His
tid
ino
l d
eh
yd
rog
en
ase
his
D
lmo
0567
Q8Y
9S
1_
LIS
MO
Lm
o0452 p
rote
in
lmo
0452
[F]
Tran
sport
von
Nu
kle
oti
den
un
d d
eren
Meta
boli
sm
us
Q8Y
560_
LIS
MO
Lm
o2216 p
rote
in
lmo
2216
His
tid
ine t
riad
(H
IT)
pro
tein
Q926Y
9_
LIS
MO
Ino
sin
e-5
'-m
on
op
ho
sp
hate
deh
yd
rog
en
ase
gu
aB
lm
o2758
Q8Y
AJ3
_L
ISM
OL
mo
0132 p
rote
in
lmo
0132
Ino
sin
e 5
-mo
no
ph
osp
hate
deh
yd
rog
en
ase
Q8Y
6B
8_
LIS
MO
Ad
en
ylo
su
ccin
ate
ly
ase
pu
rB
lmo
1773
DE
OD
_L
ISM
OP
uri
ne n
ucle
osid
e p
ho
sp
ho
ryla
se D
eo
D-t
yp
e
deo
D
lmo
1856
Q8Y
6B
6_
LIS
MO
N5-c
arb
oxy
am
ino
imid
azo
le r
ibo
nu
cle
oti
de m
uta
se
pu
rE
lmo
1775
PU
R9_
LIS
MO
Bif
un
cti
on
al p
uri
ne b
iosy
nth
esis
pro
tein
Pu
rH
pu
rH
lmo
1765
ND
K_
LIS
MO
Nu
cle
osid
e d
iph
osp
hate
kin
ase
nd
k
lmo
1929
DG
TL
2_
LIS
MO
Deo
xyg
uan
osin
etr
iph
osp
hate
tri
ph
osp
ho
hy
dro
lase-l
ike p
rote
in
lmo
2657
Q92A
N7_
LIS
MO
Ph
osp
ho
rib
osy
lfo
rmy
lgly
cin
am
idin
e s
yn
thase s
ub
un
it P
urS
p
urS
lm
o1771
GU
AA
_L
ISM
OG
MP
sy
nth
ase [
glu
tam
ine-h
yd
roly
zin
g]
gu
aA
lm
o1096
KP
RS
1_
LIS
MO
Rib
ose-p
ho
sp
hate
py
rop
ho
sp
ho
kin
ase 1
p
rs1
lmo
0199
PU
R7_
LIS
MO
Ph
osp
ho
rib
osy
lam
ino
imid
azo
le-s
uccin
ocarb
oxa
mid
e s
yn
thase
pu
rC
lmo
1772
Q8Y
6J3
_L
ISM
OL
mo
1691 p
rote
in
lmo
1691
Deo
xyu
rid
ine t
rip
ho
sp
hate
nu
cle
oti
do
hy
dro
lase
KP
RS
2_
LIS
MO
Rib
ose-p
ho
sp
hate
py
rop
ho
sp
ho
kin
ase 2
p
rs2
lmo
0509
Q8Y
6B
7_
LIS
MO
N5-c
arb
oxy
am
ino
imid
azo
le r
ibo
nu
cle
oti
de s
yn
thase
pu
rK
lmo
1774
Q8Y
5V
2_
LIS
MO
Pu
rin
e n
ucle
osid
e p
ho
sp
ho
ryla
se
pn
p
lmo
1953
Q8Y
AJ5
_L
ISM
OL
mo
0130 p
rote
in
lmo
0130
5'-n
ucle
oti
dase
PU
R2_
LIS
MO
Ph
osp
ho
rib
osy
lam
ine-g
lycin
e lig
ase
pu
rD
lmo
1764
Q8Y
6C
2_
LIS
MO
Am
ido
ph
osp
ho
rib
osy
ltra
nsfe
rase
pu
rF
lmo
1768
PU
RQ
_L
ISM
OP
ho
sp
ho
rib
osy
lfo
rmy
lgly
cin
am
idin
e s
yn
thase s
ub
un
it P
urQ
p
urQ
lm
o1770
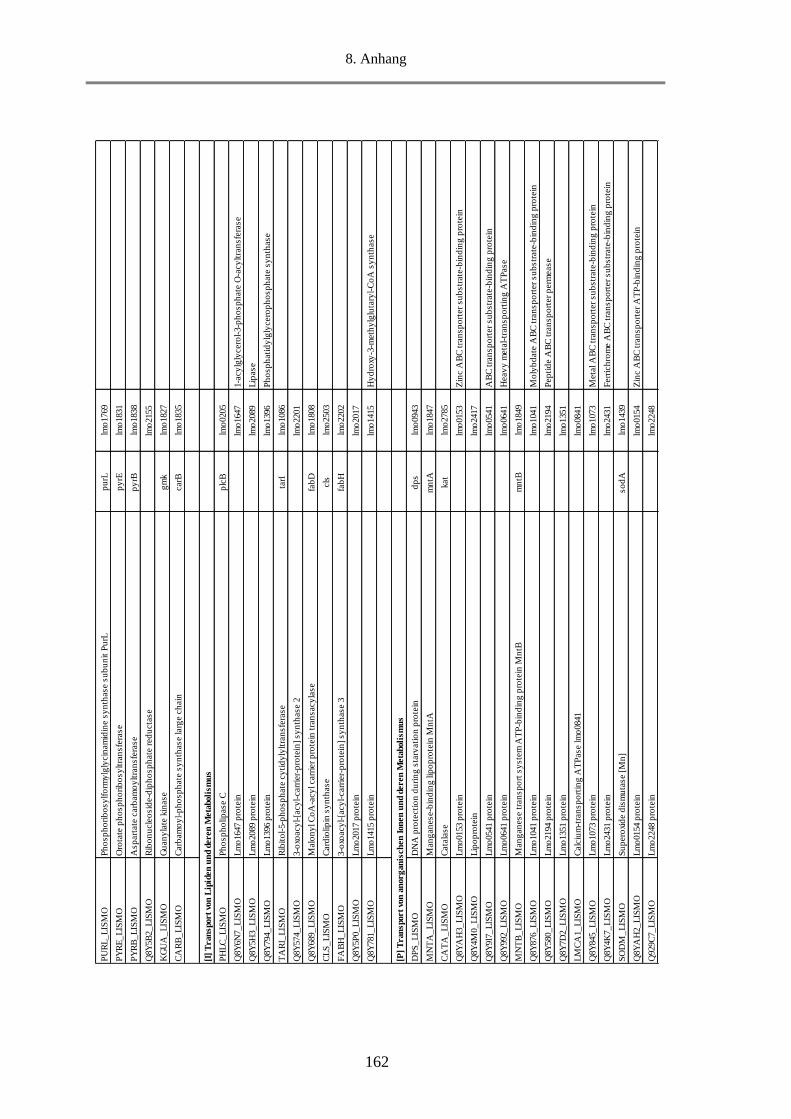
8. Anhang
162
PU
RL
_L
ISM
OP
ho
sp
ho
rib
osy
lfo
rmy
lgly
cin
am
idin
e s
yn
thase s
ub
un
it P
urL
p
urL
lm
o1769
PY
RE
_L
ISM
OO
rota
te p
ho
sp
ho
rib
osy
ltra
nsfe
rase
py
rE
lmo
1831
PY
RB
_L
ISM
OA
sp
art
ate
carb
am
oy
ltra
nsfe
rase
py
rB
lmo
1838
Q8Y
5B
2_
LIS
MO
Rib
on
ucle
osid
e-d
iph
osp
hate
red
ucta
se
lmo
2155
KG
UA
_L
ISM
OG
uan
yla
te k
inase
gm
k
lmo
1827
CA
RB
_L
ISM
OC
arb
am
oy
l-p
ho
sp
hate
sy
nth
ase larg
e c
hain
carB
lm
o1835
[I]
Tran
sport
von
Lip
iden
un
d d
eren
Meta
boli
sm
us
PH
LC
_L
ISM
OP
ho
sp
ho
lip
ase C
p
lcB
lm
o0205
Q8Y
6N
7_
LIS
MO
Lm
o1647 p
rote
in
lmo
1647
1-a
cy
lgly
cero
l-3-p
ho
sp
hate
O-a
cy
ltra
nsfe
rase
Q8Y
5H
3_
LIS
MO
Lm
o2089 p
rote
in
lmo
2089
Lip
ase
Q8Y
794_
LIS
MO
Lm
o1396 p
rote
in
lmo
1396
Ph
osp
hati
dy
lgly
cero
ph
osp
hate
sy
nth
ase
TA
RI_
LIS
MO
Rib
ito
l-5-p
ho
sp
hate
cy
tid
yly
ltra
nsfe
rase
tarI
lm
o1086
Q8Y
574_
LIS
MO
3-o
xoacy
l-[a
cy
l-carr
ier-
pro
tein
] sy
nth
ase 2
lm
o2201
Q8Y
689_
LIS
MO
Malo
ny
l C
oA
-acy
l carr
ier
pro
tein
tra
nsacy
lase
fab
D
lmo
1808
CL
S_
LIS
MO
Card
iolip
in s
yn
thase
cls
lm
o2503
FA
BH
_L
ISM
O3-o
xoacy
l-[a
cy
l-carr
ier-
pro
tein
] sy
nth
ase 3
fa
bH
lm
o2202
Q8Y
5P
0_
LIS
MO
Lm
o2017 p
rote
in
lmo
2017
Q8Y
781_
LIS
MO
Lm
o1415 p
rote
in
lmo
1415
Hy
dro
xy-3
-meth
ylg
luta
ryl-
Co
A s
yn
thase
[P]
Tran
sport
von
an
org
an
isch
en
Ion
en
un
d d
eren
Meta
boli
sm
us
DP
S_
LIS
MO
DN
A p
rote
cti
on
du
rin
g s
tarv
ati
on
pro
tein
d
ps
lmo
0943
MN
TA
_L
ISM
OM
an
gan
ese-b
ind
ing
lip
op
rote
in M
ntA
m
ntA
lm
o1847
CA
TA
_L
ISM
OC
ata
lase
kat
lmo
2785
Q8Y
AH
3_
LIS
MO
Lm
o0153 p
rote
in
lmo
0153
Zin
c A
BC
tra
nsp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
4M
0_
LIS
MO
Lip
op
rote
in
lmo
2417
Q8Y
9I7
_L
ISM
OL
mo
0541 p
rote
in
lmo
0541
AB
C t
ran
sp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
992_
LIS
MO
Lm
o0641 p
rote
in
lmo
0641
Heav
y m
eta
l-tr
an
sp
ort
ing
AT
Pase
MN
TB
_L
ISM
OM
an
gan
ese t
ran
sp
ort
sy
ste
m A
TP
-bin
din
g p
rote
in M
ntB
m
ntB
lm
o1849
Q8Y
876_
LIS
MO
Lm
o1041 p
rote
in
lmo
1041
Mo
lyb
date
AB
C t
ran
sp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
580_
LIS
MO
Lm
o2194 p
rote
in
lmo
2194
Pep
tid
e A
BC
tra
nsp
ort
er
perm
ease
Q8Y
7D
2_
LIS
MO
Lm
o1351 p
rote
in
lmo
1351
LM
CA
1_
LIS
MO
Calc
ium
-tra
nsp
ort
ing
AT
Pase lm
o0841
lmo
0841
Q8Y
845_
LIS
MO
Lm
o1073 p
rote
in
lmo
1073
Meta
l A
BC
tra
nsp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q8Y
4K
7_
LIS
MO
Lm
o2431 p
rote
in
lmo
2431
Ferr
ich
rom
e A
BC
tra
nsp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
SO
DM
_L
ISM
OS
up
ero
xid
e d
ism
uta
se [
Mn
] so
dA
lm
o1439
Q8Y
AH
2_
LIS
MO
Lm
o0154 p
rote
in
lmo
0154
Zin
c A
BC
tra
nsp
ort
er
AT
P-b
ind
ing
pro
tein
Q929C
7_
LIS
MO
Lm
o2248 p
rote
in
lmo
2248
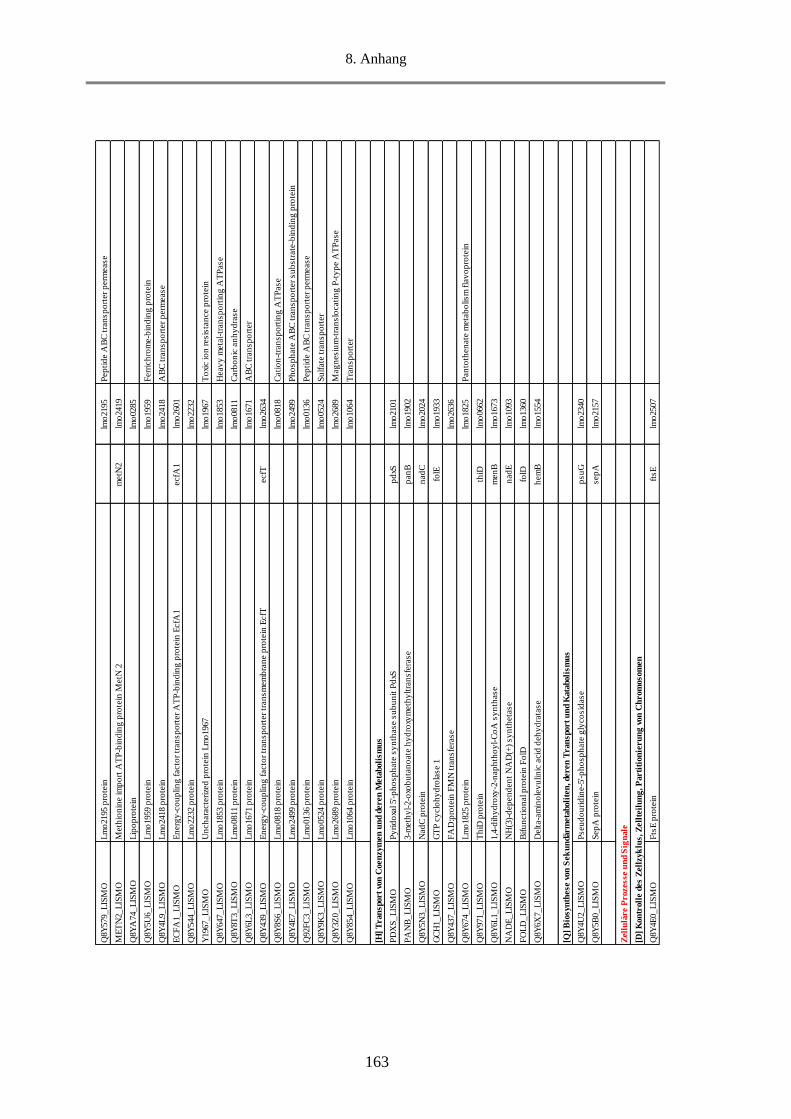
8. Anhang
163
Q8Y
579_
LIS
MO
Lm
o2195 p
rote
in
lmo
2195
Pep
tid
e A
BC
tra
nsp
ort
er
perm
ease
ME
TN
2_
LIS
MO
Meth
ion
ine im
po
rt A
TP
-bin
din
g p
rote
in M
etN
2
metN
2
lmo
2419
Q8Y
A74_
LIS
MO
Lip
op
rote
in
lmo
0285
Q8Y
5U
6_
LIS
MO
Lm
o1959 p
rote
in
lmo
1959
Ferr
ich
rom
e-b
ind
ing
pro
tein
Q8Y
4L
9_
LIS
MO
Lm
o2418 p
rote
in
lmo
2418
AB
C t
ran
sp
ort
er
perm
ease
EC
FA
1_
LIS
MO
En
erg
y-c
ou
plin
g f
acto
r tr
an
sp
ort
er
AT
P-b
ind
ing
pro
tein
EcfA
1
ecfA
1
lmo
2601
Q8Y
544_
LIS
MO
Lm
o2232 p
rote
in
lmo
2232
Y1967_
LIS
MO
Un
ch
ara
cte
rize
d p
rote
in L
mo
1967
lmo
1967
To
xic io
n r
esis
tan
ce p
rote
in
Q8Y
647_
LIS
MO
Lm
o1853 p
rote
in
lmo
1853
Heav
y m
eta
l-tr
an
sp
ort
ing
AT
Pase
Q8Y
8T
3_
LIS
MO
Lm
o0811 p
rote
in
lmo
0811
Carb
on
ic a
nh
yd
rase
Q8Y
6L
3_
LIS
MO
Lm
o1671 p
rote
in
lmo
1671
AB
C t
ran
sp
ort
er
Q8Y
439_
LIS
MO
En
erg
y-c
ou
plin
g f
acto
r tr
an
sp
ort
er
tran
sm
em
bra
ne p
rote
in E
cfT
ecfT
lm
o2634
Q8Y
8S
6_
LIS
MO
Lm
o0818 p
rote
in
lmo
0818
Cati
on
-tra
nsp
ort
ing
AT
Pase
Q8Y
4E
7_
LIS
MO
Lm
o2499 p
rote
in
lmo
2499
Ph
osp
hate
AB
C t
ran
sp
ort
er
su
bstr
ate
-bin
din
g p
rote
in
Q92F
C3_
LIS
MO
Lm
o0136 p
rote
in
lmo
0136
Pep
tid
e A
BC
tra
nsp
ort
er
perm
ease
Q8Y
9K
3_
LIS
MO
Lm
o0524 p
rote
in
lmo
0524
Su
lfate
tra
nsp
ort
er
Q8Y
3Z
0_
LIS
MO
Lm
o2689 p
rote
in
lmo
2689
Mag
nesiu
m-t
ran
slo
cati
ng
P-t
yp
e A
TP
ase
Q8Y
854_
LIS
MO
Lm
o1064 p
rote
in
lmo
1064
Tra
nsp
ort
er
[H]
Tran
sport
von
Coen
zym
en
un
d d
eren
Meta
boli
sm
us
PD
XS
_L
ISM
OP
yri
do
xal 5'-p
ho
sp
hate
sy
nth
ase s
ub
un
it P
dxS
p
dxS
lm
o2101
PA
NB
_L
ISM
O3-m
eth
yl-
2-o
xob
uta
no
ate
hy
dro
xym
eth
ylt
ran
sfe
rase
pan
B
lmo
1902
Q8Y
5N
3_
LIS
MO
Nad
C p
rote
in
nad
C
lmo
2024
GC
H1_
LIS
MO
GT
P c
yclo
hy
dro
lase 1
fo
lE
lmo
1933
Q8Y
437_
LIS
MO
FA
D:p
rote
in F
MN
tra
nsfe
rase
lmo
2636
Q8Y
674_
LIS
MO
Lm
o1825 p
rote
in
lmo
1825
Pan
toth
en
ate
meta
bo
lism
fla
vo
pro
tein
Q8Y
971_
LIS
MO
Th
iD p
rote
in
thiD
lm
o0662
Q8Y
6L
1_
LIS
MO
1,4
-dih
yd
roxy
-2-n
ap
hth
oy
l-C
oA
sy
nth
ase
men
B
lmo
1673
NA
DE
_L
ISM
ON
H(3
)-d
ep
en
den
t N
AD
(+)
sy
nth
eta
se
nad
E
lmo
1093
FO
LD
_L
ISM
OB
ifu
ncti
on
al p
rote
in F
olD
fo
lD
lmo
1360
Q8Y
6X
7_
LIS
MO
Delt
a-a
min
ole
vu
lin
ic a
cid
deh
yd
rata
se
hem
B
lmo
1554
[Q]
Bio
syn
these v
on
Sek
un
därm
eta
boli
ten
, deren
Tran
sport
un
d K
ata
boli
sm
us
Q8Y
4U
2_
LIS
MO
Pseu
do
uri
din
e-5
'-p
ho
sp
hate
gly
co
sid
ase
psu
G
lmo
2340
Q8Y
5B
0_
LIS
MO
Sep
A p
rote
in
sep
A
lmo
2157
Zell
ulä
re P
rozesse u
nd S
ign
ale
[D]
Kon
troll
e d
es Z
ell
zyk
lus, Z
ell
teil
un
g, P
arti
tion
ieru
ng
von
Ch
rom
osom
en
Q8Y
4E
0_
LIS
MO
Fts
E p
rote
in
ftsE
lm
o2507

8. Anhang
164
Q8Y
4E
1_
LIS
MO
Cell d
ivis
ion
pro
tein
Fts
X
ftsX
lm
o2506
Q8Y
5N
7_
LIS
MO
Div
IVA
pro
tein
d
ivIV
A
lmo
2020
EZ
RA
_L
ISM
OS
ep
tati
on
rin
g f
orm
ati
on
reg
ula
tor
Ezr
A
ezr
A
lmo
1594
Q8Y
5M
4_
LIS
MO
Cell d
ivis
ion
pro
tein
Fts
A
ftsA
lm
o2033
Q8Y
5M
5_
LIS
MO
Cell d
ivis
ion
pro
tein
Fts
Z
ftsZ
lm
o2032
Q8Y
6Y
7_
LIS
MO
Sit
e-d
ete
rmin
ing
pro
tein
m
inD
lm
o1544
FT
SK
_L
ISM
OD
NA
tra
nslo
case F
tsK
ft
sK
lm
o1386
[M]
Bio
gen
ese d
er Z
ell
wan
d/M
em
bran
/Hü
lle
TA
GH
_L
ISM
OT
eic
ho
ic a
cid
s e
xpo
rt A
TP
-bin
din
g p
rote
in T
ag
H
tag
H
lmo
1075
Q8Y
707_
LIS
MO
Lm
o1521 p
rote
in
lmo
1521
N-a
cety
lmu
ram
oy
l-L
-ala
nin
e a
mid
ase
Q8Y
4C
8_
LIS
MO
Lm
o2522 p
rote
in
lmo
2522
Cell w
all-b
ind
ing
pro
tein
Q8Y
3Y
8_
LIS
MO
Lm
o2691 p
rote
in
lmo
2691
Au
toly
sin
P60_
LIS
MO
Pro
bab
le e
nd
op
ep
tid
ase p
60
iap
lm
o0582
Q8Y
3S
7_
LIS
MO
Lm
o2754 p
rote
in
lmo
2754
D-a
lan
yl-
D-a
lan
ine c
arb
oxy
pep
tid
ase
Q7A
P49_
LIS
MO
Pep
tid
og
lycan
ly
tic p
rote
in P
45
sp
l lm
o2505
MS
CL
_L
ISM
OL
arg
e-c
on
du
cta
nce m
ech
an
osen
sit
ive c
han
nel
mscL
lm
o2064
Q8Y
8H
6_
LIS
MO
Lm
o0927 p
rote
in
lmo
0927
Q8Y
AJ6
_L
ISM
OL
mo
0129 p
rote
in
lmo
0129
N-a
cety
lmu
ram
oy
l-L
-ala
nin
e a
mid
ase
Q8Y
8H
5_
LIS
MO
Lm
o0929 p
rote
in
lmo
0929
So
rtase
Q8Y
4E
2_
LIS
MO
Lm
o2504 p
rote
in
lmo
2504
Cell w
all-b
ind
ing
pro
tein
Q8Y
496_
LIS
MO
Au
toly
sin
, am
idase
am
i lm
o2558
Q8Y
763_
LIS
MO
Lm
o1438 p
rote
in
lmo
1438
Pen
icillin
-bin
din
g p
rote
in
Q8Y
610_
LIS
MO
Pb
pA
pro
tein
p
bp
A
lmo
1892
MR
EC
_L
ISM
OC
ell s
hap
e-d
ete
rmin
ing
pro
tein
Mre
C
mre
C
lmo
1547
Q8Y
AD
8_
LIS
MO
Lm
o0193 p
rote
in
lmo
0193
Q8Y
8L
7_
LIS
MO
Lm
o0880 p
rote
in
lmo
0880
Wall a
sso
cia
ted
pro
tein
pre
cu
rso
r
SP
5G
2_
LIS
MO
Pu
tati
ve s
ep
tati
on
pro
tein
Sp
oV
G 2
sp
oV
G2
lmo
0197
Q7A
P54_
LIS
MO
Lm
o2185 p
rote
in
lmo
2185
Q7A
P78_
LIS
MO
Dlt
D p
rote
in f
or
D-a
lan
ine e
ste
rifi
cati
on
of
lip
ote
ich
oic
acid
an
d w
all t
eic
ho
ic a
cid
d
ltD
lm
o0971
Q8Y
5J3
_L
ISM
OL
mo
2067 p
rote
in
lmo
2067
Bile a
cid
hy
dro
lase
Q8Y
5W
4_
LIS
MO
Lm
o1941 p
rote
in
lmo
1941
Q8Y
989_
LIS
MO
Lm
o0644 p
rote
in
lmo
0644
Q7A
P48_
LIS
MO
Lm
o2550 p
rote
in
lmo
2550
Gly
co
sy
l tr
an
sfe
rase
Q8Y
836_
LIS
MO
Lm
o1082 p
rote
in
lmo
1082
dT
DP
-su
gar
ep
imera
se
Q8Y
645_
LIS
MO
Lm
o1855 p
rote
in
lmo
1855
D-a
lan
yl-
D-a
lan
ine c
arb
oxy
pep
tid
ase
Q8Y
547_
LIS
MO
Lm
o2229 p
rote
in
lmo
2229
Pen
icillin
-bin
din
g p
rote
in
Q8Y
899_
LIS
MO
Lm
o1013 p
rote
in
lmo
1013

8. Anhang
165
Q8Y
649_
LIS
MO
Lm
o1851 p
rote
in
lmo
1851
Carb
oxy
-term
inal p
rocessin
g p
rote
inase
Q8Y
5L
8_
LIS
MO
Pb
pB
pro
tein
p
bp
B
lmo
2039
DIV
IB_
LIS
MO
Cell d
ivis
ion
pro
tein
Div
IB
div
IB
lmo
2034
Q8Y
6E
3_
LIS
MO
Lm
o1744 p
rote
in
lmo
1744
Q8Y
591_
LIS
MO
Pu
tati
ve p
ep
tid
og
lycan
bo
un
d p
rote
in (
LP
XT
G m
oti
f)
lmo
2178
Pep
tid
og
lycan
bin
din
g p
rote
in
LG
T_
LIS
MO
Pro
lip
op
rote
in d
iacy
lgly
cery
l tr
an
sfe
rase
lgt
lmo
2482
Y1318_
LIS
MO
Pu
tati
ve z
inc m
eta
llo
pro
tease L
mo
1318
lmo
1318
Q8Y
4A
0_
LIS
MO
Lm
o2554 p
rote
in
lmo
2554
Gala
cto
sy
ltra
nsfe
rase
Q8Y
8Q
4_
LIS
MO
Pu
tati
ve p
ep
tid
og
lycan
bo
un
d p
rote
in (
LP
XT
G m
oti
f)
lmo
0842
Q8Y
841_
LIS
MO
Lm
o1077 p
rote
in
lmo
1077
Teic
ho
ic a
cid
bio
sy
nth
esis
pro
tein
B
GL
MU
_L
ISM
OB
ifu
ncti
on
al p
rote
in G
lmU
g
lmU
lm
o0198
[O]
Postt
ran
sla
tion
ale
Modif
ikati
on
en
, P
rote
inu
msatz
, C
haperon
e
PR
SA
2_
LIS
MO
Fo
ldase p
rote
in P
rsA
2
prs
A2
lmo
2219
Q8Y
A67_
LIS
MO
Lm
o0292 p
rote
in
lmo
0292
Heat-
sh
ock p
rote
in h
trA
seri
ne p
rote
ase
CH
60_
LIS
MO
60 k
Da c
hap
ero
nin
g
roL
lm
o2068
CH
10_
LIS
MO
10 k
Da c
hap
ero
nin
g
roS
lm
o2069
Q8Y
AC
6_
LIS
MO
AT
P-d
ep
en
den
t zi
nc m
eta
llo
pro
tease F
tsH
ft
sH
lm
o0220
CL
PP
_L
ISM
OA
TP
-dep
en
den
t C
lp p
rote
ase p
rote
oly
tic s
ub
un
it
clp
P
lmo
2468
HS
LV
_L
ISM
OA
TP
-dep
en
den
t p
rote
ase s
ub
un
it H
slV
h
slV
lm
o1278
Q8Y
4M
3_
LIS
MO
Lm
o2414 p
rote
in
lmo
2414
Am
ino
tran
sfe
rase
Q7A
P51_
LIS
MO
L-a
lan
oy
l-D
-glu
tam
ate
pep
tid
ase
lysA
lm
o2278
Q8Y
7Y
1_
LIS
MO
AT
P-d
ep
en
den
t C
lp p
rote
ase p
rote
oly
tic s
ub
un
it
clp
P
lmo
1138
HT
PX
_L
ISM
OP
rote
ase H
tpX
ho
mo
log
h
tpX
lm
o0963
CO
XX
_L
ISM
OP
roto
hem
e I
X f
arn
esy
ltra
nsfe
rase
cta
B
lmo
2057
CL
PB
_L
ISM
OC
hap
ero
ne p
rote
in C
lpB
clp
B
lmo
2206
TR
XB
_L
ISM
OT
hio
red
oxi
n r
ed
ucta
se
trxB
lm
o2478
DN
AK
_L
ISM
OC
hap
ero
ne p
rote
in D
naK
d
naK
lm
o1473
Q8Y
798_
LIS
MO
Lm
o1392 p
rote
in
lmo
1392
Pep
tid
ase
Q8Y
4E
4_
LIS
MO
Lm
o2502 p
rote
in
lmo
2502
Q8Y
4M
2_
LIS
MO
Lm
o2415 p
rote
in
lmo
2415
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
[U]
Intr
azell
ulä
rer V
erk
eh
r, S
ek
reti
on
, ve
sik
ulä
rer T
ran
sport
Q8Y
701_
LIS
MO
Lm
o1529 p
rote
in
lmo
1529
Q8Y
7K
8_
LIS
MO
Sig
nal p
ep
tid
ase I
lm
o1269
Q8Y
703_
LIS
MO
Pro
tein
tra
nslo
case s
ub
un
it S
ecD
secD
lm
o1527
YID
C2_
LIS
MO
Mem
bra
ne p
rote
in in
sert
ase Y
idC
2
yid
C2
lmo
2854
Q8Y
7K
6_
LIS
MO
Sig
nal p
ep
tid
ase I
lm
o1271

8. Anhang
166
Q8Y
7K
7_
LIS
MO
Sig
nal p
ep
tid
ase I
lm
o1270
YID
C1_
LIS
MO
Mem
bra
ne p
rote
in in
sert
ase Y
idC
1
yid
C1
lmo
1379
SE
CA
1_
LIS
MO
Pro
tein
tra
nslo
case s
ub
un
it S
ecA
1
secA
1
lmo
2510
Q8Y
448_
LIS
MO
Pro
tein
tra
nslo
case s
ub
un
it S
ecY
secY
lm
o2612
[T]
Mech
an
ism
en
der S
ign
alt
ran
sdu
kti
on
Q92C
58_
LIS
MO
RN
A-b
ind
ing
pro
tein
Hfq
h
fq
lmo
1295
Q8Y
6V
1_
LIS
MO
Un
ivers
al str
ess p
rote
in
lmo
1580
Q8Y
679_
LIS
MO
Lm
o1820 p
rote
in
lmo
1820
Seri
ne/t
hre
on
ine p
rote
in k
inase
Q8Y
9J6
_L
ISM
OL
mo
0531 p
rote
in
lmo
0531
Q8Y
8K
9_
LIS
MO
Rsb
R p
rote
in
Rsb
R
lmo
0889
Q92D
C5_
LIS
MO
Rsb
S p
rote
in
rsb
S
lmo
0890
Q8Y
6E
6_
LIS
MO
Lm
o1741 p
rote
in
lmo
1741
His
tid
ine k
inase
RS
BW
_L
ISM
OS
eri
ne-p
rote
in k
inase R
sb
W
rsb
W
lmo
0894
Q8Y
5K
8_
LIS
MO
Lm
o2051 p
rote
in
lmo
2051
Q7A
P63_
LIS
MO
Lm
o1467 p
rote
in
lmo
1467
Ph
osp
hate
sta
rvati
on
-in
du
ced
pro
tein
Ph
oH
[V]
Verte
idig
un
gsm
ech
an
ism
en
Q8Y
5F
0_
LIS
MO
Lm
o2114 p
rote
in
lmo
2114
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
4R
2_
LIS
MO
Lm
o2372 p
rote
in
lmo
2372
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
6P
8_
LIS
MO
Lm
o1636 p
rote
in
lmo
1636
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
4R
3_
LIS
MO
Lm
o2371 p
rote
in
lmo
2371
AB
C t
ran
sp
ort
er
perm
ease
Q8Y
475_
LIS
MO
Lm
o2580 p
rote
in
lmo
2580
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
474_
LIS
MO
Lm
o2581 p
rote
in
lmo
2581
Q8Y
9C
5_
LIS
MO
Lm
o0607 p
rote
in
lmo
0607
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
5U
1_
LIS
MO
Lm
o1964 p
rote
in
lmo
1964
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
9C
4_
LIS
MO
Lm
o0608 p
rote
in
lmo
0608
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
6E
0_
LIS
MO
Lm
o1747 p
rote
in
lmo
1747
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
3T
0_
LIS
MO
Lm
o2751 p
rote
in
lmo
2751
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
3W
4_
LIS
MO
Cy
dD
pro
tein
cy
dD
lm
o2715
Q8Y
8Y
6_
LIS
MO
Lm
o0756 p
rote
in
lmo
0756
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
3V
6_
LIS
MO
Lm
o2725 p
rote
in
lmo
2725
Q8Y
5E
9_
LIS
MO
Lm
o2115 p
rote
in
lmo
2115
AB
C t
ran
sp
ort
er
perm
ease
Q8Y
3W
3_
LIS
MO
Cy
dC
pro
tein
cy
dC
lm
o2716
Q8Y
3S
9_
LIS
MO
Lm
o2752 p
rote
in
lmo
2752
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
[N]
Zell
bew
eg
un
g
Q8Y
464_
LIS
MO
Su
rface p
rote
in (
GW
rep
eat)
sim
ilar
to N
-acety
lmu
ram
idase
lmo
2591
N-a
cety
lmu
ram
oy
l-L
-ala
nin
e a
mid
ase
Q8Y
7Q
7_
LIS
MO
Lm
o1215 p
rote
in
lmo
1215
N-a
cety
lmu
ram
oy
l-L
-ala
nin
e a
mid
ase
Q8Y
842_
LIS
MO
Lm
o1076 p
rote
in
lmo
1076
Au
toly
sin
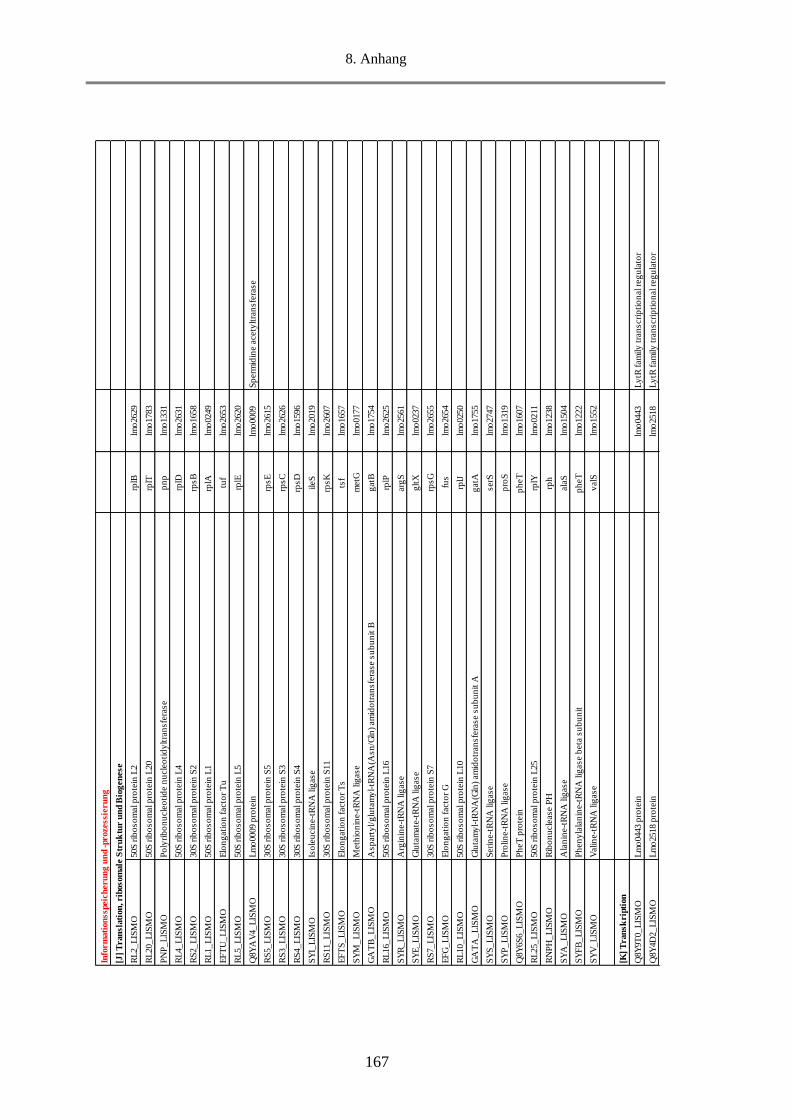
8. Anhang
167
Info
rm
ati
on
sspeic
heru
ng
un
d -
prozessie
ru
ng
[J]
Tran
sla
tion
, rib
osom
ale
Str
uk
tur u
nd B
iog
en
ese
RL
2_
LIS
MO
50S
rib
oso
mal p
rote
in L
2
rplB
lm
o2629
RL
20_
LIS
MO
50S
rib
oso
mal p
rote
in L
20
rplT
lm
o1783
PN
P_
LIS
MO
Po
lyri
bo
nu
cle
oti
de n
ucle
oti
dy
ltra
nsfe
rase
pn
p
lmo
1331
RL
4_
LIS
MO
50S
rib
oso
mal p
rote
in L
4
rplD
lm
o2631
RS
2_
LIS
MO
30S
rib
oso
mal p
rote
in S
2
rpsB
lm
o1658
RL
1_
LIS
MO
50S
rib
oso
mal p
rote
in L
1
rplA
lm
o0249
EF
TU
_L
ISM
OE
lon
gati
on
facto
r T
u
tuf
lmo
2653
RL
5_
LIS
MO
50S
rib
oso
mal p
rote
in L
5
rplE
lm
o2620
Q8Y
AV
4_
LIS
MO
Lm
o0009 p
rote
in
lmo
0009
Sp
erm
idin
e a
cety
ltra
nsfe
rase
RS
5_
LIS
MO
30S
rib
oso
mal p
rote
in S
5
rpsE
lm
o2615
RS
3_
LIS
MO
30S
rib
oso
mal p
rote
in S
3
rpsC
lm
o2626
RS
4_
LIS
MO
30S
rib
oso
mal p
rote
in S
4
rpsD
lm
o1596
SY
I_L
ISM
OIs
ole
ucin
e-t
RN
A lig
ase
ileS
lm
o2019
RS
11_
LIS
MO
30S
rib
oso
mal p
rote
in S
11
rpsK
lm
o2607
EF
TS
_L
ISM
OE
lon
gati
on
facto
r T
s
tsf
lmo
1657
SY
M_
LIS
MO
Meth
ion
ine-t
RN
A lig
ase
metG
lm
o0177
GA
TB
_L
ISM
OA
sp
art
yl/
glu
tam
yl-
tRN
A(A
sn
/Gln
) am
ido
tran
sfe
rase s
ub
un
it B
g
atB
lm
o1754
RL
16_
LIS
MO
50S
rib
oso
mal p
rote
in L
16
rplP
lm
o2625
SY
R_
LIS
MO
Arg
inin
e-t
RN
A lig
ase
arg
S
lmo
2561
SY
E_
LIS
MO
Glu
tam
ate
-tR
NA
lig
ase
glt
X
lmo
0237
RS
7_
LIS
MO
30S
rib
oso
mal p
rote
in S
7
rpsG
lm
o2655
EF
G_
LIS
MO
Elo
ng
ati
on
facto
r G
fu
slm
o2654
RL
10_
LIS
MO
50S
rib
oso
mal p
rote
in L
10
rplJ
lm
o0250
GA
TA
_L
ISM
OG
luta
my
l-tR
NA
(Gln
) am
ido
tran
sfe
rase s
ub
un
it A
g
atA
lm
o1755
SY
S_
LIS
MO
Seri
ne-t
RN
A lig
ase
serS
lm
o2747
SY
P_
LIS
MO
Pro
lin
e-t
RN
A lig
ase
pro
S
lmo
1319
Q8Y
6S
6_
LIS
MO
Ph
eT
pro
tein
p
heT
lm
o1607
RL
25_
LIS
MO
50S
rib
oso
mal p
rote
in L
25
rplY
lm
o0211
RN
PH
_L
ISM
OR
ibo
nu
cle
ase P
H
rph
lm
o1238
SY
A_
LIS
MO
Ala
nin
e-t
RN
A lig
ase
ala
S
lmo
1504
SY
FB
_L
ISM
OP
hen
yla
lan
ine-t
RN
A lig
ase b
eta
su
bu
nit
p
heT
lm
o1222
SY
V_
LIS
MO
Valin
e-t
RN
A lig
ase
valS
lm
o1552
[K]
Tran
sk
rip
tion
Q8Y
9T
0_
LIS
MO
Lm
o0443 p
rote
in
lmo
0443
Ly
tR f
am
ily
tra
nscri
pti
on
al re
gu
lato
r
Q8Y
4D
2_
LIS
MO
Lm
o2518 p
rote
in
lmo
2518
Ly
tR f
am
ily
tra
nscri
pti
on
al re
gu
lato
r
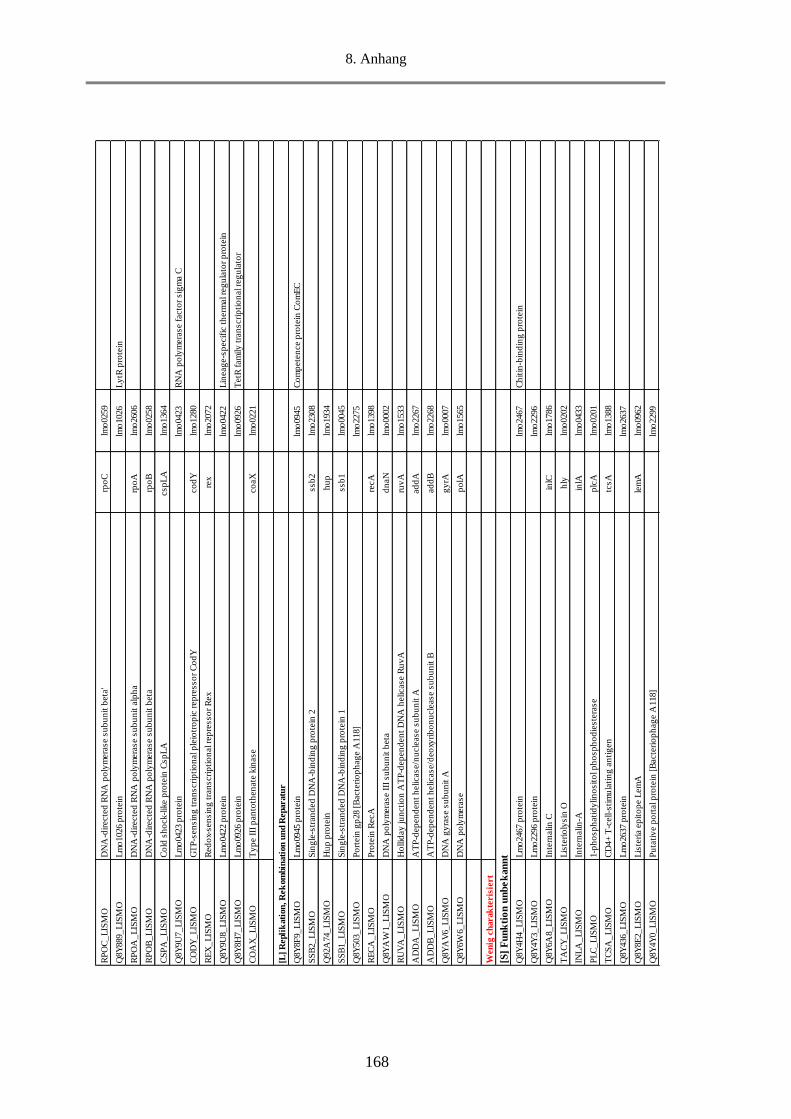
8. Anhang
168
RP
OC
_L
ISM
OD
NA
-dir
ecte
d R
NA
po
lym
era
se s
ub
un
it b
eta
' rp
oC
lm
o0259
Q8Y
889_
LIS
MO
Lm
o1026 p
rote
in
lmo
1026
Ly
tR p
rote
in
RP
OA
_L
ISM
OD
NA
-dir
ecte
d R
NA
po
lym
era
se s
ub
un
it a
lph
a
rpo
A
lmo
2606
RP
OB
_L
ISM
OD
NA
-dir
ecte
d R
NA
po
lym
era
se s
ub
un
it b
eta
rp
oB
lm
o0258
CS
PA
_L
ISM
OC
old
sh
ock-l
ike p
rote
in C
sp
LA
csp
LA
lm
o1364
Q8Y
9U
7_
LIS
MO
Lm
o0423 p
rote
in
lmo
0423
RN
A p
oly
mera
se f
acto
r sig
ma C
CO
DY
_L
ISM
OG
TP
-sen
sin
g t
ran
scri
pti
on
al p
leio
tro
pic
rep
resso
r C
od
Y
co
dY
lm
o1280
RE
X_
LIS
MO
Red
ox-
sen
sin
g t
ran
scri
pti
on
al re
pre
sso
r R
ex
rex
lmo
2072
Q8Y
9U
8_
LIS
MO
Lm
o0422 p
rote
in
lmo
0422
Lin
eag
e-s
pecif
ic t
herm
al re
gu
lato
r p
rote
in
Q8Y
8H
7_
LIS
MO
Lm
o0926 p
rote
in
lmo
0926
TetR
fam
ily
tra
nscri
pti
on
al re
gu
lato
r
CO
AX
_L
ISM
OT
yp
e I
II p
an
toth
en
ate
kin
ase
co
aX
lm
o0221
[L]
Repli
kati
on
, R
ek
om
bin
ati
on
un
d R
eparatu
r
Q8Y
8F
9_
LIS
MO
Lm
o0945 p
rote
in
lmo
0945
Co
mp
ete
nce p
rote
in C
om
EC
SS
B2_
LIS
MO
Sin
gle
-str
an
ded
DN
A-b
ind
ing
pro
tein
2
ssb
2
lmo
2308
Q92A
74_
LIS
MO
Hu
p p
rote
in
hu
p
lmo
1934
SS
B1_
LIS
MO
Sin
gle
-str
an
ded
DN
A-b
ind
ing
pro
tein
1
ssb
1
lmo
0045
Q8Y
503_
LIS
MO
Po
rtein
gp
28 [
Bacte
rio
ph
ag
e A
118]
lmo
2275
RE
CA
_L
ISM
OP
rote
in R
ecA
re
cA
lm
o1398
Q8Y
AW
1_
LIS
MO
DN
A p
oly
mera
se I
II s
ub
un
it b
eta
d
naN
lm
o0002
RU
VA
_L
ISM
OH
ollid
ay
ju
ncti
on
AT
P-d
ep
en
den
t D
NA
helicase R
uv
A
ruv
A
lmo
1533
AD
DA
_L
ISM
OA
TP
-dep
en
den
t h
elicase/n
ucle
ase s
ub
un
it A
ad
dA
lm
o2267
AD
DB
_L
ISM
OA
TP
-dep
en
den
t h
elicase/d
eo
xyri
bo
nu
cle
ase s
ub
un
it B
ad
dB
lm
o2268
Q8Y
AV
6_
LIS
MO
DN
A g
yra
se s
ub
un
it A
g
yrA
lm
o0007
Q8Y
6W
6_
LIS
MO
DN
A p
oly
mera
se
po
lA
lmo
1565
Wen
ig c
harak
teris
iert
[S]
Fu
nk
tio
n u
nb
ek
an
nt
Q8Y
4H
4_
LIS
MO
Lm
o2467 p
rote
in
lmo
2467
Ch
itin
-bin
din
g p
rote
in
Q8Y
4Y
3_
LIS
MO
Lm
o2296 p
rote
in
lmo
2296
Q8Y
6A
8_
LIS
MO
Inte
rnalin
C
inlC
lm
o1786
TA
CY
_L
ISM
OL
iste
rio
lysin
O
hly
lm
o0202
INL
A_
LIS
MO
Inte
rnalin
-A
inlA
lm
o0433
PL
C_
LIS
MO
1-p
ho
sp
hati
dy
lin
osit
ol p
ho
sp
ho
die
ste
rase
plc
A
lmo
0201
TC
SA
_L
ISM
OC
D4+
T-c
ell-s
tim
ula
tin
g a
nti
gen
tc
sA
lm
o1388
Q8Y
436_
LIS
MO
Lm
o2637 p
rote
in
lmo
2637
Q8Y
8E
2_
LIS
MO
Lis
teri
a e
pit
op
e L
em
A
lem
A
lmo
0962
Q8Y
4Y
0_
LIS
MO
Pu
tati
ve p
ort
al p
rote
in [
Bacte
rio
ph
ag
e A
118]
lmo
2299

8. Anhang
169
Q8Y
AK
2_
LIS
MO
Lm
o0122 p
rote
in
lmo
0122
Q8Y
4Y
4_
LIS
MO
Pro
tein
gp
8 [
Bacte
rio
ph
ag
e A
118]
lmo
2295
Q7A
P93_
LIS
MO
An
tig
en
A
lmaA
lm
o0118
Q7A
P59_
LIS
MO
Lm
o1601 p
rote
in
lmo
1601
Gen
era
l str
ess p
rote
in
Q8Y
AK
1_
LIS
MO
Lm
o0123 p
rote
in
lmo
0123
Q8Y
850_
LIS
MO
Lm
o1068 p
rote
in
lmo
1068
Q8Y
6T
1_
LIS
MO
Lm
o1602 p
rote
in
lmo
1602
Q8Y
AR
7_
LIS
MO
Lm
o0047 p
rote
in
lmo
0047
Q8Y
6N
5_
LIS
MO
Lm
o1649 p
rote
in
lmo
1649
Y392_
LIS
MO
UP
F0365 p
rote
in lm
o0392
lmo
0392
Q8Y
7E
8_
LIS
MO
Lm
o1334 p
rote
in
lmo
1334
Q8Y
639_
LIS
MO
Lm
o1861 p
rote
in
lmo
1861
Q8Y
795_
LIS
MO
Lm
o1395 p
rote
in
lmo
1395
Q8Y
980_
LIS
MO
Lm
o0653 p
rote
in
lmo
0653
Q8Y
3I3
_L
ISM
OL
mo
2853 p
rote
in
lmo
2853
Q8Y
8V
1_
LIS
MO
Lm
o0791 p
rote
in
lmo
0791
Q8Y
690_
LIS
MO
Fab
G p
rote
in
fab
G
lmo
1807
Q8Y
6L
8_
LIS
MO
Pep
tid
og
lycan
lin
ked
pro
tein
(L
PxT
G)
lmo
1666
Q8Y
5Y
4_
LIS
MO
Lm
o1919 p
rote
in
lmo
1919
Q8Y
7E
9_
LIS
MO
Lm
o1333 p
rote
in
lmo
1333
Q8Y
6D
1_
LIS
MO
Lm
o1757 p
rote
in
lmo
1757
INL
B_
LIS
MO
Inte
rnalin
B
inlB
lm
o0434
Q929B
9_
LIS
MO
Lm
o2256 p
rote
in
lmo
2256
Q8Y
5M
8_
LIS
MO
Lm
o2029 p
rote
in
lmo
2029
Q8Y
559_
LIS
MO
Lm
o2217 p
rote
in
lmo
2217
Q8Y
3W
9_
LIS
MO
Lm
o2710 p
rote
in
lmo
2710
Q8Y
AK
3_
LIS
MO
Lm
o0121 p
rote
in
lmo
0121
Ph
ag
e t
ail p
rote
in
Q8Y
4W
1_
LIS
MO
Lm
o2318 p
rote
in
lmo
2318
Y1776_
LIS
MO
UP
F0316 p
rote
in lm
o1776
lmo
1776
YH
AM
_L
ISM
O3'-5' e
xori
bo
nu
cle
ase Y
haM
y
haM
lm
o2220
Q8Y
9E
6_
LIS
MO
Lm
o0584 p
rote
in
lmo
0584
Y2113_
LIS
MO
Pu
tati
ve h
em
e-d
ep
en
den
t p
ero
xid
ase lm
o2113
lmo
2113
DA
CA
_L
ISM
OD
iad
en
yla
te c
ycla
se
dacA
lm
o2120
Q8Y
5T
9_
LIS
MO
Lm
o1966 p
rote
in
lmo
1966
Q8Y
605_
LIS
MO
Lm
o1898 p
rote
in
lmo
1898
Q8Y
725_
LIS
MO
Lm
o1499 p
rote
in
lmo
1499
Q8Y
8F
4_
LIS
MO
Lm
o0950 p
rote
in
lmo
0950
Q8Y
6P
7_
LIS
MO
Lm
o1637 p
rote
in
lmo
1637
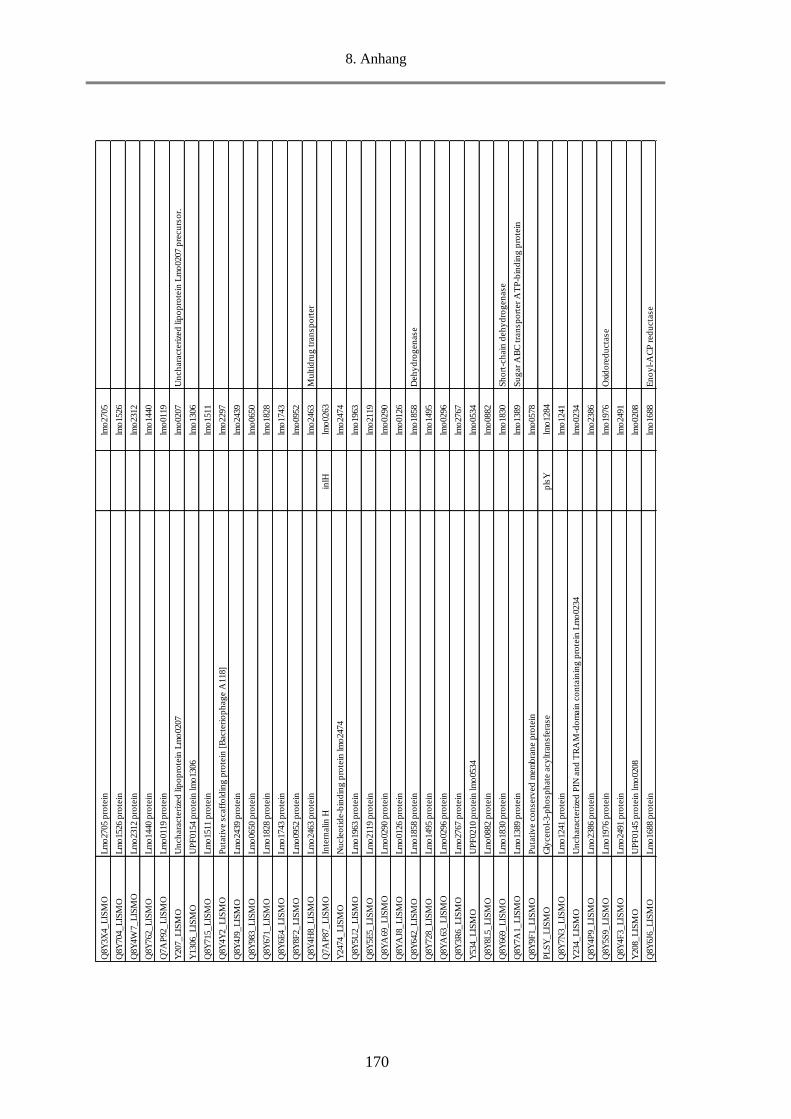
8. Anhang
170
Q8Y
3X
4_
LIS
MO
Lm
o2705 p
rote
in
lmo
2705
Q8Y
704_
LIS
MO
Lm
o1526 p
rote
in
lmo
1526
Q8Y
4W
7_
LIS
MO
Lm
o2312 p
rote
in
lmo
2312
Q8Y
762_
LIS
MO
Lm
o1440 p
rote
in
lmo
1440
Q7A
P92_
LIS
MO
Lm
o0119 p
rote
in
lmo
0119
Y207_
LIS
MO
Un
ch
ara
cte
rize
d lip
op
rote
in L
mo
0207
lmo
0207
Un
ch
ara
cte
rize
d lip
op
rote
in L
mo
0207 p
recu
rso
r.
Y1306_
LIS
MO
UP
F0154 p
rote
in lm
o1306
lmo
1306
Q8Y
715_
LIS
MO
Lm
o1511 p
rote
in
lmo
1511
Q8Y
4Y
2_
LIS
MO
Pu
tati
ve s
caff
old
ing
pro
tein
[B
acte
rio
ph
ag
e A
118]
lmo
2297
Q8Y
4J9
_L
ISM
OL
mo
2439 p
rote
in
lmo
2439
Q8Y
983_
LIS
MO
Lm
o0650 p
rote
in
lmo
0650
Q8Y
671_
LIS
MO
Lm
o1828 p
rote
in
lmo
1828
Q8Y
6E
4_
LIS
MO
Lm
o1743 p
rote
in
lmo
1743
Q8Y
8F
2_
LIS
MO
Lm
o0952 p
rote
in
lmo
0952
Q8Y
4H
8_
LIS
MO
Lm
o2463 p
rote
in
lmo
2463
Mu
ltid
rug
tra
nsp
ort
er
Q7A
P87_
LIS
MO
Inte
rnalin
H
inlH
lm
o0263
Y2474_
LIS
MO
Nu
cle
oti
de-b
ind
ing
pro
tein
lm
o2474
lmo
2474
Q8Y
5U
2_
LIS
MO
Lm
o1963 p
rote
in
lmo
1963
Q8Y
5E
5_
LIS
MO
Lm
o2119 p
rote
in
lmo
2119
Q8Y
A69_
LIS
MO
Lm
o0290 p
rote
in
lmo
0290
Q8Y
AJ8
_L
ISM
OL
mo
0126 p
rote
in
lmo
0126
Q8Y
642_
LIS
MO
Lm
o1858 p
rote
in
lmo
1858
Deh
yd
rog
en
ase
Q8Y
728_
LIS
MO
Lm
o1495 p
rote
in
lmo
1495
Q8Y
A63_
LIS
MO
Lm
o0296 p
rote
in
lmo
0296
Q8Y
3R
6_
LIS
MO
Lm
o2767 p
rote
in
lmo
2767
Y534_
LIS
MO
UP
F0210 p
rote
in lm
o0534
lmo
0534
Q8Y
8L
5_
LIS
MO
Lm
o0882 p
rote
in
lmo
0882
Q8Y
669_
LIS
MO
Lm
o1830 p
rote
in
lmo
1830
Sh
ort
-ch
ain
deh
yd
rog
en
ase
Q8Y
7A
1_
LIS
MO
Lm
o1389 p
rote
in
lmo
1389
Su
gar
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
9F
1_
LIS
MO
Pu
tati
ve c
on
serv
ed
mem
bra
ne p
rote
in
lmo
0578
PL
SY
_L
ISM
OG
lycero
l-3-p
ho
sp
hate
acy
ltra
nsfe
rase
pls
Y
lmo
1284
Q8Y
7N
3_
LIS
MO
Lm
o1241 p
rote
in
lmo
1241
Y234_
LIS
MO
Un
ch
ara
cte
rize
d P
IN a
nd
TR
AM
-do
main
co
nta
inin
g p
rote
in L
mo
0234
lmo
0234
Q8Y
4P
9_
LIS
MO
Lm
o2386 p
rote
in
lmo
2386
Q8Y
5S
9_
LIS
MO
Lm
o1976 p
rote
in
lmo
1976
Oxi
do
red
ucta
se
Q8Y
4F
3_
LIS
MO
Lm
o2491 p
rote
in
lmo
2491
Y208_
LIS
MO
UP
F0145 p
rote
in lm
o0208
lmo
0208
Q8Y
6J6
_L
ISM
OL
mo
1688 p
rote
in
lmo
1688
En
oy
l-A
CP
red
ucta
se
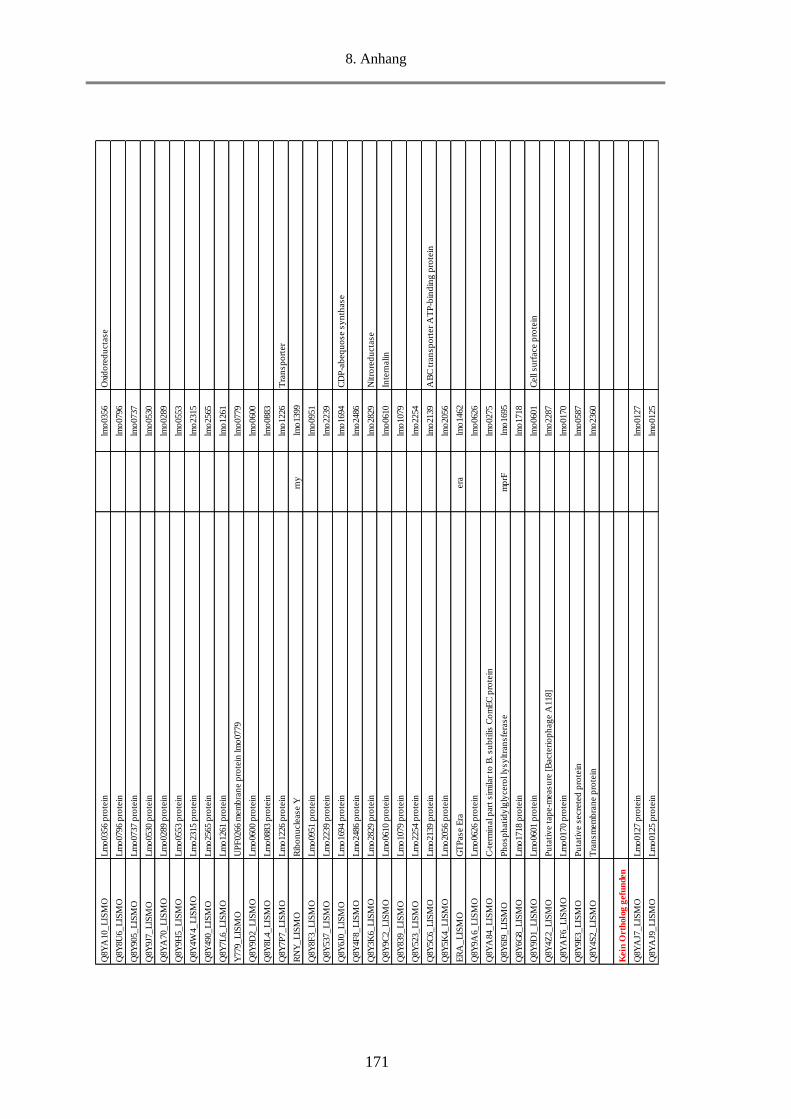
8. Anhang
171
Q8Y
A10_
LIS
MO
Lm
o0356 p
rote
in
lmo
0356
Oxi
do
red
ucta
se
Q8Y
8U
6_
LIS
MO
Lm
o0796 p
rote
in
lmo
0796
Q8Y
905_
LIS
MO
Lm
o0737 p
rote
in
lmo
0737
Q8Y
9J7
_L
ISM
OL
mo
0530 p
rote
in
lmo
0530
Q8Y
A70_
LIS
MO
Lm
o0289 p
rote
in
lmo
0289
Q8Y
9H
5_
LIS
MO
Lm
o0553 p
rote
in
lmo
0553
Q8Y
4W
4_
LIS
MO
Lm
o2315 p
rote
in
lmo
2315
Q8Y
490_
LIS
MO
Lm
o2565 p
rote
in
lmo
2565
Q8Y
7L
6_
LIS
MO
Lm
o1261 p
rote
in
lmo
1261
Y779_
LIS
MO
UP
F0266 m
em
bra
ne p
rote
in lm
o0779
lmo
0779
Q8Y
9D
2_
LIS
MO
Lm
o0600 p
rote
in
lmo
0600
Q8Y
8L
4_
LIS
MO
Lm
o0883 p
rote
in
lmo
0883
Q8Y
7P
7_
LIS
MO
Lm
o1226 p
rote
in
lmo
1226
Tra
nsp
ort
er
RN
Y_
LIS
MO
Rib
on
ucle
ase Y
rn
y
lmo
1399
Q8Y
8F
3_
LIS
MO
Lm
o0951 p
rote
in
lmo
0951
Q8Y
537_
LIS
MO
Lm
o2239 p
rote
in
lmo
2239
Q8Y
6J0
_L
ISM
OL
mo
1694 p
rote
in
lmo
1694
CD
P-a
beq
uo
se s
yn
thase
Q8Y
4F
8_
LIS
MO
Lm
o2486 p
rote
in
lmo
2486
Q8Y
3K
6_
LIS
MO
Lm
o2829 p
rote
in
lmo
2829
Nit
rore
du
cta
se
Q8Y
9C
2_
LIS
MO
Lm
o0610 p
rote
in
lmo
0610
Inte
rnalin
Q8Y
839_
LIS
MO
Lm
o1079 p
rote
in
lmo
1079
Q8Y
523_
LIS
MO
Lm
o2254 p
rote
in
lmo
2254
Q8Y
5C
6_
LIS
MO
Lm
o2139 p
rote
in
lmo
2139
AB
C t
ran
sp
ort
er
AT
P-b
ind
ing
pro
tein
Q8Y
5K
4_
LIS
MO
Lm
o2056 p
rote
in
lmo
2056
ER
A_
LIS
MO
GT
Pase E
ra
era
lm
o1462
Q8Y
9A
6_
LIS
MO
Lm
o0626 p
rote
in
lmo
0626
Q8Y
A84_
LIS
MO
C-t
erm
inal p
art
sim
ilar
to B
. su
bti
lis C
om
EC
pro
tein
lm
o0275
Q8Y
6I9
_L
ISM
OP
ho
sp
hati
dy
lgly
cero
l ly
sy
ltra
nsfe
rase
mp
rF
lmo
1695
Q8Y
6G
8_
LIS
MO
Lm
o1718 p
rote
in
lmo
1718
Q8Y
9D
1_
LIS
MO
Lm
o0601 p
rote
in
lmo
0601
Cell s
urf
ace p
rote
in
Q8Y
4Z
2_
LIS
MO
Pu
tati
ve t
ap
e-m
easu
re [
Bacte
rio
ph
ag
e A
118]
lmo
2287
Q8Y
AF
6_
LIS
MO
Lm
o0170 p
rote
in
lmo
0170
Q8Y
9E
3_
LIS
MO
Pu
tati
ve s
ecre
ted
pro
tein
lm
o0587
Q8Y
4S
2_
LIS
MO
Tra
nsm
em
bra
ne p
rote
in
lmo
2360
Kein
Orth
olo
g g
efu
nden
Q8Y
AJ7
_L
ISM
OL
mo
0127 p
rote
in
lmo
0127
Q8Y
AJ9
_L
ISM
OL
mo
0125 p
rote
in
lmo
0125

8. Anhang
172
Q7A
P94_
LIS
MO
An
tig
en
B
lmaB
lm
o0117
AC
TA
_L
ISM
OA
cti
n a
ssem
bly
-in
du
cin
g p
rote
in
actA
lm
o0204
Q8Y
4Z
9_
LIS
MO
Pro
tein
gp
23 [
Bacte
rio
ph
ag
e A
118]
lmo
2280
Q8Y
4U
9_
LIS
MO
Lm
o2331 p
rote
in
lmo
2331
Q8Y
4M
1_
LIS
MO
Lm
o2416 p
rote
in
lmo
2416
Q8Y
500_
LIS
MO
Ho
lin
[B
acte
rio
ph
ag
e A
118]
lmo
2279
Q8Y
5I3
_L
ISM
OL
mo
2079 p
rote
in
lmo
2079
Q8Y
4M
6_
LIS
MO
Lm
o2410 p
rote
in
lmo
2410
Q8Y
3W
5_
LIS
MO
Pep
tid
og
lycan
an
ch
ore
d p
rote
in (
LP
XT
G m
oti
f)
lmo
2714
Q8Y
AK
0_
LIS
MO
Lm
o0124 p
rote
in
lmo
0124
Q8Y
769_
LIS
MO
Lm
o1432 p
rote
in
lmo
1432
Q8Y
9P
5_
LIS
MO
Pu
tati
ve s
ecre
ted
pro
tein
lm
o0479
Q7A
P60_
LIS
MO
Lm
o1518 p
rote
in
lmo
1518
Q8Y
4V
0_
LIS
MO
Lm
o2330 p
rote
in
lmo
2330
Q8Y
9P
7_
LIS
MO
Pu
tati
ve s
ecre
ted
pro
tein
lm
o0477
Q8Y
4Y
8_
LIS
MO
Majo
r ta
il s
haft
pro
tein
[B
acte
rio
ph
ag
e A
118]
lmo
2291
Q8Y
5I2
_L
ISM
OL
mo
2080 p
rote
in
lmo
2080
Q8Y
3N
0_
LIS
MO
Lm
o2805 p
rote
in
lmo
2805
Q8Y
8Z
2_
LIS
MO
Lm
o0750 p
rote
in
lmo
0750
Q8Y
4W
8_
LIS
MO
Lm
o2311 p
rote
in
lmo
2311
Q8Y
3P
3_
LIS
MO
Lm
o2792 p
rote
in
lmo
2792
Q8Y
6D
5_
LIS
MO
Lm
o1752 p
rote
in
lmo
1752
Q8Y
4Z
6_
LIS
MO
Pro
tein
gp
20 [
Bacte
rio
ph
ag
e A
118]
lmo
2283
Q8Y
4Z
5_
LIS
MO
Pro
tein
gp
19 [
Bacte
rio
ph
ag
e A
118]
lmo
2284





























