Embed Size (px)
Citation preview
LadungstransferDOI: 10.1002/ange.200601623
Distaler Ladungstransport in PeptidenEdward W. Schlag,* Sheh-Yi Sheu, Dah-Yen Yang, Heinrich L. Selzle undSheng Hsien Lin
AngewandteChemie
Stichw�rter:Ladungstransfer · Leitf�higkeit ·Molek�ldynamik · Peptide ·Reaktionsdynamik
E. W. Schlag et al.Aufs�tze
3258 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273

1. Einleitung
Eine der vielen wichtigen Funktionen von Proteinen istdie �bertragung von Informationen mithilfe einer Ladungoder chemischen Ver�nderung �ber betr�chtliche Entfer-nungen innerhalb der Proteinkette, beispielsweise der La-dungstransport durch eine Zellwand zur Aktivierung intra-zellul�rer chemischer Prozesse. Chemische Reaktionen, diean einer Stelle ablaufen, die weit vom Ursprung der Anre-gung entfernt ist, sind eine h�ufige Beobachtung bei Protei-nen und damit von grundlegender Bedeutung; auf atomarerEbene sind sie allerdings noch nicht gut verstanden.[1–17]
Konventionelle Theorien der chemischen Kinetik k2nnennicht direkt auf eine Wirkung �ber große Entfernungen an-gewendet werden, da ein solcher chemischer Transport h�ufigauch eine Ladungswanderung einschließt. Daher ist die Ent-wicklung von Modellen f�r eine distale Wirkung von Inter-esse, um herauszufinden, wie ein Reiz an einer Stelle Mole-k�lbewegungen an einer anderen, entfernten Stelle verur-sacht. Eine solche Wirkung kann als chemische Reaktionbetrachtet werden, die weit entfernt vom Ursprung des Si-gnals oder der Ladung stattfindet. Wir bezeichnen diesenProzess des Transfers von Reaktivit�t (R) und Ladung (C)hier als RC-�bertragung; dies ist ein wichtiger Prozess f�rkomplexere Signal�bertragungen. Ein Problem eines solchenModells ist, dass dissipative Prozesse mit vielen Schwin-gungsfreiheitsgraden entlang des Weges h�ufig eine chemi-sche Reaktivit�t an der entfernten Stelle und damit aucheinen Transport verhindern.[18,19] Wir betrachten hier die RC-�bertragung f�r einfache Peptide, die sich ungeachtet dervielen Freiheitsgrade durch eine ultraschnelle langreichwei-tige �bertragung auszeichnen.
Langreichweitiger Elektronentransfer (ET) ist eingrundlegender Mechanismus in vielf�ltigen biologischenSystemen.[20] Elektron�bertragungen sind direkt oder indirektan vielen biologischen Prozessen beteiligt,[21] z.B. an der
oxidativen Phosphorylierung, der Photosynthese,[22–24] derLeitf�higkeit der DNA-Helix[25] und der aeroben Atmung.[26]
Normalerweise ist an diesen Prozessen ein Metallion-Bio-molek�l-Metallion-System beteiligt, in dem die Metallionenweit voneinander getrennt sind – in einigen F�llen kann derAbstand mehr als 10 D betragen. Isied et al.[27] ver2ffent-lichten 1980 einen Elektronentransfer zwischen zwei Re-doxzentren, der �ber eine Polypeptidbr�cke vermittelt wurde.Gray und Winkler[26] gelang es, die Redoxzentren an einProtein, z.B. an Myoglobin oder Cytochrom c, zu kn�pfen.Dieses modifizierte Donor-Protein-Acceptor-System erm2g-lichte die Untersuchung der Abstands- und Strukturabh�n-gigkeit von ET-Prozessen.[21,28–34]
Welchen Einfluss w�rde das Anbringen einer reinenLadung an einem Peptid, z.B. bei einer lokalisierten Photo-ionisation am C-Terminus, auf die Chemie und die Ladungs-weiterleitung im Peptid zum N-Terminus haben? Ein solchesModell f�r distale Prozesse k2nnte auf verschiedenen Ebenengetestet werden. Zuerst untersuchen wir den Transport derLadung und Reaktivit�t f�r den Fall des isolierten Molek�ls.Anschließend untersuchen wir das m2glicherweise ver�n-
[*] Prof. Dr. E. W. Schlag, Priv.-Doz. Dr. H. L. SelzleInstitut f�r Physikalische und Theoretische ChemieTechnische Universit�t M�nchenLichtenbergstraße 4, 85748 Garching (Deutschland)Fax: (+49)89-289-13389E-Mail: [email protected]: http://www.phys.chemie.tu-muenchen.de/staff/schlag/
Prof. Dr. S.-Y. SheuFaculty of Life ScienceNational Yang-Ming UniversityTaipei (Taiwan)
Dr. D.-Y. Yang, Prof. Dr. S. H. LinInstitute of Atomic and Molecular ScienceAcademia SinicaTaipei (Taiwan)
Biologische Systeme transportieren Ladungen und reaktive Prozesseh"ufig $ber betr"chtliche Entfernungen. Traditionelle Modelle derchemischen Kinetik k)nnen solche extremen distalen Prozesse ge-w)hnlich nicht beschreiben. In diesem Aufsatz betrachten wir einatomares Modell f$r eine distale Informations$bertragung, das spe-ziell f$r Peptide entwickelt wurde. Dabei wird die chemische Reakti-vit"t als Ergebnis distaler Wirkungen betrachtet, die auf einer di-funktionalen Zwei-Stufen-Kinetik, verbunden mit Bewegungen vonPeptiden auf der Sub-Pikosekundenskala beruht. Das difunktionaleModell ergibt einen hocheffizienten Transport von Ladung und Re-aktivit"t $ber eine betr"chtliche Entfernung in einem isolierten Peptid;in einer w"ssrigen Umgebung wird dagegen eine sehr niedrige Effi-zienz gefunden. Dem Modell zufolge findet in einer Peptidumgebungeine ultraschnelle 2bertragung von Ladung und Reaktivit"t $ber einebetr"chtliche Entfernung statt. In einem Protein k)nnen viele solcherDom"nen aktiv sein.
Aus dem Inhalt
1. Einleitung 3259
2. Energetik 3263
3. Rechnungen 3265
4.MD-Simulation und Verteilungder mittleren first passage time 3267
5. Isoliertes System 3268
6. Die w�ssrige Umgebung 3268
7. Sekund�rstruktur 3270
8. Schlussfolgerungen 3270
9. Nachwort 3271
Ladungstransport in PeptidenAngewandte
Chemie
3259Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

derte Verhalten des Prozesses in einem Medium wie Wasser.Hier stellt sich die Frage: Wie beeinflusst das Medium dieInformations�bertragung in Bezug auf die Geschwindigkeitund die Ausbeute? Experimente an einfachen Modellpepti-den haben nun gezeigt, dass der Prozess im isoliertenMolek�laußerordentlich effizient sein kann – hingegen wird f�r dengleichen Prozess in Wasser vorausgesagt, dass er normaler-weise um zwei Gr2ßenordnungen weniger effizient ist.[35–38]
W�nschenswert w�re die Entwicklung eines einfachen Mo-dells, das zumindest f�r Peptide zum Verst�ndnis solcherExtremf�lle f�hrt.
Wir haben eine Reihe von Experimenten an solchen iso-lierten Modellpeptiden durchgef�hrt, in denen wir einehocheffiziente Ladungs�bertragung entlang der Hauptkettedes Peptids und damit einhergehende, langreichweitige che-mische Reaktionen beobachteten.[1–4] Diese Peptide sind zwar
eher klein, repr�sentieren aber wegen ihrer beachtlichen Zahlan Freiheitsgraden eine enorme Menge an mikroskopischenZust�nden und lassen daher in konventionellen Modellen nursehr wenig Reaktivit�t erwarten. Experimente haben gezeigt,dass sich eine solche �bertragung durch den Einbau vonbestimmten Aminos�uren in die Kette in großem Umfangsteuern, ja sich sogar an- und abschalten l�sst. Eine wesent-liche Frage ist nun, wie in bestimmten nichtkonjugierten or-ganischen Systemen eine solche �bertragung zu solch ent-fernten Stellen effizient mithilfe atomarer Bewegungen ge-lingt. Die �bertragung �ber große Distanzen k2nnte einenwichtigen Modellcharakter f�r die Mechanismen biologischerReaktionen haben.
Chemische Reaktionen finden �blicherweise lokal oder inunmittelbarer Nachbarschaft zur Stelle der Anregungstatt.[39,40] Die �bliche mechanistische Theorie zur Erkl�rungder Reaktivit�t bezieht sich lediglich auf lokale Anregungenund schließlich einen Bindungsbruch oder eine andere Bin-dungs�nderung infolge der Tatsache, dass Energie durcheinen statistischen Prozess gekoppelt und auf diese Bindung�bertragen wird. Dieses Modell liegt der Formulierung eines�bergangszustands oder der statistischen unimolekularenTheorie der chemischen Kinetik zugrunde[41] und war Basisder meisten theoretischen Interpretationen der chemischenKinetik seit der ersten Anwendung der Theorie von Rice,Ramsperger, Kassel und Marcus (RRKM-Theorie)[42] durchRabinovitch et al. vor etwa 50 Jahren.[43–45] Solche statisti-schen Prozesse koppeln gew2hnlich an alle Freiheitsgrade derReaktanten. Ein solches Modell der Kopplung an alle Frei-heitsgrade kann nicht auf eine langreichweitige biologischeRC-�bertragung angewendet werden, da die Zahl an Ei-
Edward Schlag, geboren in Los Angeles, pro-movierte bei B. S. Rabinovitch an der Uni-versity of Washington. Von 1969 bis 1971war er Professor an der Northwestern Uni-versity in Evanston, IL, und folgte 1971 demRuf auf den Lehrstuhl f.r Physikalische Che-mie I an die TU M.nchen. Seine Forschungumfasst die zeitliche Verfolgung von Prim5r-prozessen von Molek.len in der Gasphaseund chemische Spektroskopie mit Lasern.1988 entdeckte er langlebige molekulareRydberg-Zust5nde im Kontinuum als Grund-lage f.r eine neue Spektroskopie (ZEKE). Erist Mitglied der Bayerischen Akademie derWissenschaften und der Academia Europea.
Sheh-Yi Sheu ist derzeit Professor an derFaculty of Science, National Yang-Ming Uni-versity (Taipei, Taiwan). Sie hat sich mitBioinformatik, Molek.ldynamiksimulationvon allosterischen Enzymen und Ladungs-transfer befasst. Ihr Forschungsinteresse giltderzeit Ausflussprozessen in Enzymmolek.-len, der Ionenpumpe und der Proteintranslo-kation.
Dah-Yen Yang ist wissenschaftlicher Mitar-beiter am Institute of Atomic and MolecularScience, Academia Sinica (Taipei, Taiwan).Er besch5ftigt sich mit biomolekularen Si-mulationen von Ionenkan5len, molekularenMotoren und Elektronentransfer, Ladungslei-tung von molekularem Draht, Quanten-pumpen, Ionenpumpen sowie Rechnungenzu biomolekularen Systemen.
Heinrich L. Selzle wurde 1944 in Dachaugeboren. Er studierte bis 1969 Physik an derTU M.nchen und promovierte 1971 am In-stitut f.r Physikalische Chemie bei H. Geri-scher. Anschließend arbeitete er als Postdok-torand bei E. W. Schlag an der Northwes-tern University, Evanston. Er kehrte schließ-lich an die TU M.nchen zur.ck, wo er 2000habilitierte und seitdem als Dozent t5tig ist.Zu seinen Forschungsgebieten z5hlten dasStudium der Struktur von molekularen undMetallclustern und ihrer schwachen Wechsel-wirkungen im angeregten Zustand sowie dieZEKE-Spektroskopie.
Sheng Hsien Lin ist wissenschaftlicher Mitar-beiter am Institute of Atomic and MolecularScience (Taipei, Taiwan) und Mitglied derAcademia Sinica. Er promovierte an derUniversity of Utah, Salt Lake City, UT, undarbeitete als Postdoktorand an der ColumbiaUniversity, NY. Seine Arbeitsgruppe hatTheorien zur Frequenzverdopplung, Sum-menfrequenzerzeugung, Resonanz-Raman-Streuung und Dynamik des angeregten Zu-stands (wie strahlungslose Bberg5nge,Schwingungsrelaxation, ultraschneller photo-induzierter Elektronen- und Energietransfer)entwickelt.
E. W. Schlag et al.Aufs�tze
3260 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273

genzust�nden in solch großen Systemen und der damit ver-bundene Phasenraum sogar f�r Peptide astronomisch großwerden. Jegliche lokale Energie w�rde vor der Ankunft ander entfernten Stelle dissipiert werden oder w�rde astrono-mische Zeitspannen f�r die chemische Reaktion von Syste-men mit mehr als zwei oder drei Resten ben2tigen.[46]
Angesichts der Tatsache, dass solche distalen Prozesse f�rviele biologische Systeme experimentell nachgewiesenwurden und auch recht effizient ablaufen, muss f�r ihre Er-kl�rung nach einem anderen Modell gesucht werden. Levin-thal[18, 19] erkannte dieses Problem bei der Proteinfaltung,[47]
bei der ebenfalls zu viele Freiheitsgrade verhindern, dass derProzess in einer endlichen Zeitspanne abl�uft. Zwar wurdenbereits viele Vorschl�ge zur Umgehung dieses Problems ge-macht, die Diskussion ist aber noch nicht beendet.[48–50] Diesevielen Freiheitsgrade f�r Peptide koppeln jedoch nicht beiden hier beteiligten, niedrigen Energien von etwa 200 cm�1.Schwellwerte f�r eine intramolekulare Schwingungsrelaxati-on (IVR) in der Kinetik betragen gew2hnlich mindestens1200 cm�1[51] oder 2200 cm�1[52] und bei niedrigen Energiensogar mehr,[53, 54] wenngleich sich die Situation f�r sehr großeMolek�le mit weichen Schwingungsmoden unterscheidenkann. Die hier vorgeschlagene Kopplung tritt also ein, bevoralle Moden �ber IVR miteinander in Kontakt treten undsomit in einem sehr reduzierten Phasenraum.
Zun�chst betrachten wir die neuesten experimentellenDaten f�r Modellpeptide. Diese Ergebnisse liefern bereitseinen �berzeugenden Beleg daf�r, dass solch ein effizienterAblauf �ber eine gr2ßere Entfernung selbst f�r das isolierteMolek�l existiert, was darauf schließen l�sst, dass es sich umeine rein molekulare Eigenschaft handelt. Wir pr�sentierenein Modell, das die neuen Daten abdecken soll und be-stimmte molekulare Bedingungen vorhersagt, unter denensolche Abl�ufe m2glich oder nicht m2glich sind. Fernerm�ssen wir den beobachteten starken Einfluss der Umgebungauf diesen Prozess erl�utern, da die Effizienz in Wasser starkabnimmt.
Eine typische Theorie zum Verst�ndnis von Leitf�higkeitin Proteinen kann darauf basieren, dieMatrixelemente f�r dieKopplung der Aminos�uren zu berechnen, die in einer un-gef�hr gemittelten Konformation angeordnet sind. SolcheRechnungen wurden f�r ein Modellpeptid (Abbildung 1)durchgef�hrt und zeigen, dass die Energetik zwischen be-nachbarten Positionen normalerweise die �berwindung einerBarriere von etwa 0.4 eV erfordert (Abbildung 2). Bei f�rbiologische Prozesse �blichen Temperaturen ist diese Bar-riere nur schwer zu �berwinden. Dadurch werden die Pro-zesse ineffizienter, wie es in Untersuchungen in w�ssrigemMedium tats�chlich beobachtet wurde. Der sehr leichte La-dungstransport in der Gasphase l�sst sich so jedoch nicht er-kl�ren.
Gem�ß der Marcus-Theorie ist eine ET-Geschwindigkeitproportional zum Produkt des Quadrats der elektronischenKopplungskonstante und des Franck-Condon-Faktors, der aufder treibenden Kraft und der L2sungsmittelreorganisations-energie beruht. In der Marcus-Theorie war zun�chst keinabstandsabh�ngiger Faktor enthalten. Sp�ter wurde dieMarcus-Theorie erweitert,[55] um einen abstandsabh�ngigenFaktor e�b(R�R0) einzuf�gen, wobei b die abstandsabh�ngige
Abklingkonstante, R der Abstand zwischen den Redoxstellenund R0 der Abstand bei der st�rksten Ann�herung zwischenDonor und Acceptor ist. Ein normaler experimenteller b-Wert betr�gt f�r DNA ca. 0.77 D�1,[56, 57] f�r eine a-Helix ca.1.26 D�1 und f�r ein b-Faltblatt ca. 1.00 D�1.[56, 57] Viele Ar-beitsgruppen, wie Beratan, Onuchic et al., f�hrten theoreti-sche Untersuchungen unter Verwendung eines Tunnelmo-dells zur Ermittlung des abstandsabh�ngigen Faktorsdurch.[58–86] Auch ein Superaustauschmodell, das auf nicht-adiabatischem Tunneln von Elektronen basiert, wurde vor-geschlagen.[87–89] Diese Modelle beruhen h�ufig auf einer ge-mittelten Hauptkettenstruktur.
In einer fr�heren Arbeit[11] schlugen wir als Erkl�rung vor,dass Peptide eine einzigartige Molek�leigenschaft aufweisen,die sich daraus ergibt, dass die einzelnen Aminos�ureposi-tionen leichte Rotationen �ber sehr große Winkel (die Ra-machandran-Winkel der Polypeptide) durchlaufen k2nnen.Diese Bewegungen sind durch die nahezu flache Potential-
Abbildung 1. Modelldipeptid f�r die Berechnung der Wechselwirkungzwischen zwei benachbarten Aminos�uren f�r den Ladungstransferentlang einer Peptidkette.
Abbildung 2. Verringerung der Barriere f�r den Ladungstransport zwi-schen benachbarten Aminos�uren (identische Reste) als Funktion derRamachandran-Winkel y und f in Abbildung 1. Die Ladung springtdurch einen nichtadiabatischen Gbergang vom ionischen Zustand 1zum ionischen Zustand 2. Nach diabatischer Transformation betr�gtdie Barriere f�r den Ladungstransfer etwa 0.4 eV. Der Ladungstransferh�ngt tats�chlich von den Ramachandran-Winkeln ab. Details sind inAbbildung 3 dargestellt.Die Ab-initio-Rechnung erfolgte auf der Basisder UHF/3-21G-Methode. jyioni1=elektronische Wellenfunktion mitLadung auf der N-Seite, jyioni2=elektronische Wellenfunktion mitLadung auf der C-Seite.
Ladungstransport in PeptidenAngewandte
Chemie
3261Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
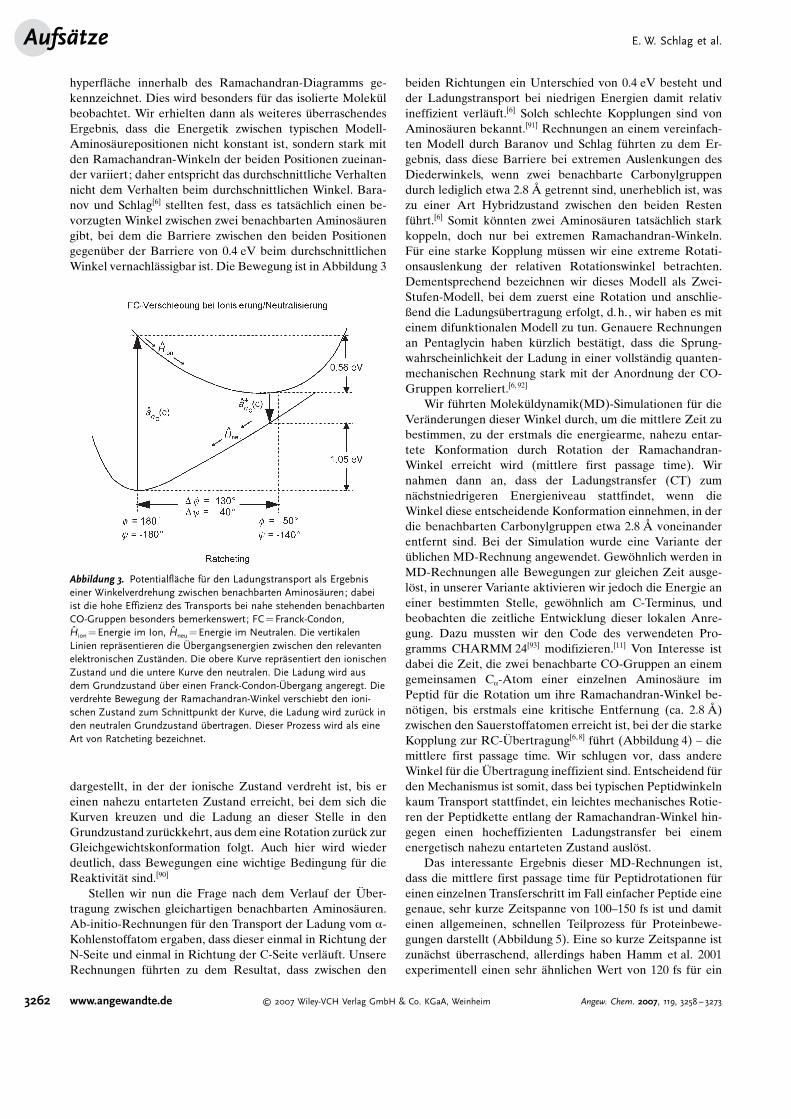
hyperfl�che innerhalb des Ramachandran-Diagramms ge-kennzeichnet. Dies wird besonders f�r das isolierte Molek�lbeobachtet. Wir erhielten dann als weiteres �berraschendesErgebnis, dass die Energetik zwischen typischen Modell-Aminos�urepositionen nicht konstant ist, sondern stark mitden Ramachandran-Winkeln der beiden Positionen zueinan-der variiert; daher entspricht das durchschnittliche Verhaltennicht dem Verhalten beim durchschnittlichen Winkel. Bara-nov und Schlag[6] stellten fest, dass es tats�chlich einen be-vorzugten Winkel zwischen zwei benachbarten Aminos�urengibt, bei dem die Barriere zwischen den beiden Positionengegen�ber der Barriere von 0.4 eV beim durchschnittlichenWinkel vernachl�ssigbar ist. Die Bewegung ist in Abbildung 3
dargestellt, in der der ionische Zustand verdreht ist, bis ereinen nahezu entarteten Zustand erreicht, bei dem sich dieKurven kreuzen und die Ladung an dieser Stelle in denGrundzustand zur�ckkehrt, aus dem eine Rotation zur�ck zurGleichgewichtskonformation folgt. Auch hier wird wiederdeutlich, dass Bewegungen eine wichtige Bedingung f�r dieReaktivit�t sind.[90]
Stellen wir nun die Frage nach dem Verlauf der �ber-tragung zwischen gleichartigen benachbarten Aminos�uren.Ab-initio-Rechnungen f�r den Transport der Ladung vom a-Kohlenstoffatom ergaben, dass dieser einmal in Richtung derN-Seite und einmal in Richtung der C-Seite verl�uft. UnsereRechnungen f�hrten zu dem Resultat, dass zwischen den
beiden Richtungen ein Unterschied von 0.4 eV besteht undder Ladungstransport bei niedrigen Energien damit relativineffizient verl�uft.[6] Solch schlechte Kopplungen sind vonAminos�uren bekannt.[91] Rechnungen an einem vereinfach-ten Modell durch Baranov und Schlag f�hrten zu dem Er-gebnis, dass diese Barriere bei extremen Auslenkungen desDiederwinkels, wenn zwei benachbarte Carbonylgruppendurch lediglich etwa 2.8 D getrennt sind, unerheblich ist, waszu einer Art Hybridzustand zwischen den beiden Restenf�hrt.[6] Somit k2nnten zwei Aminos�uren tats�chlich starkkoppeln, doch nur bei extremen Ramachandran-Winkeln.F�r eine starke Kopplung m�ssen wir eine extreme Rotati-onsauslenkung der relativen Rotationswinkel betrachten.Dementsprechend bezeichnen wir dieses Modell als Zwei-Stufen-Modell, bei dem zuerst eine Rotation und anschlie-ßend die Ladungs�bertragung erfolgt, d.h., wir haben es miteinem difunktionalen Modell zu tun. Genauere Rechnungenan Pentaglycin haben k�rzlich best�tigt, dass die Sprung-wahrscheinlichkeit der Ladung in einer vollst�ndig quanten-mechanischen Rechnung stark mit der Anordnung der CO-Gruppen korreliert.[6, 92]
Wir f�hrten Molek�ldynamik(MD)-Simulationen f�r dieVer�nderungen dieser Winkel durch, um die mittlere Zeit zubestimmen, zu der erstmals die energiearme, nahezu entar-tete Konformation durch Rotation der Ramachandran-Winkel erreicht wird (mittlere first passage time). Wirnahmen dann an, dass der Ladungstransfer (CT) zumn�chstniedrigeren Energieniveau stattfindet, wenn dieWinkel diese entscheidende Konformation einnehmen, in derdie benachbarten Carbonylgruppen etwa 2.8 D voneinanderentfernt sind. Bei der Simulation wurde eine Variante der�blichen MD-Rechnung angewendet. Gew2hnlich werden inMD-Rechnungen alle Bewegungen zur gleichen Zeit ausge-l2st, in unserer Variante aktivieren wir jedoch die Energie aneiner bestimmten Stelle, gew2hnlich am C-Terminus, undbeobachten die zeitliche Entwicklung dieser lokalen Anre-gung. Dazu mussten wir den Code des verwendeten Pro-gramms CHARMM 24[93] modifizieren.[11] Von Interesse istdabei die Zeit, die zwei benachbarte CO-Gruppen an einemgemeinsamen Ca-Atom einer einzelnen Aminos�ure imPeptid f�r die Rotation um ihre Ramachandran-Winkel be-n2tigen, bis erstmals eine kritische Entfernung (ca. 2.8 D)zwischen den Sauerstoffatomen erreicht ist, bei der die starkeKopplung zur RC-�bertragung[6,8] f�hrt (Abbildung 4) – diemittlere first passage time. Wir schlugen vor, dass andereWinkel f�r die �bertragung ineffizient sind. Entscheidend f�rden Mechanismus ist somit, dass bei typischen Peptidwinkelnkaum Transport stattfindet, ein leichtes mechanisches Rotie-ren der Peptidkette entlang der Ramachandran-Winkel hin-gegen einen hocheffizienten Ladungstransfer bei einemenergetisch nahezu entarteten Zustand ausl2st.
Das interessante Ergebnis dieser MD-Rechnungen ist,dass die mittlere first passage time f�r Peptidrotationen f�reinen einzelnen Transferschritt im Fall einfacher Peptide einegenaue, sehr kurze Zeitspanne von 100–150 fs ist und damiteinen allgemeinen, schnellen Teilprozess f�r Proteinbewe-gungen darstellt (Abbildung 5). Eine so kurze Zeitspanne istzun�chst �berraschend, allerdings haben Hamm et al. 2001experimentell einen sehr �hnlichen Wert von 120 fs f�r ein
Abbildung 3. Potentialfl�che f�r den Ladungstransport als Ergebniseiner Winkelverdrehung zwischen benachbarten Aminos�uren; dabeiist die hohe Effizienz des Transports bei nahe stehenden benachbartenCO-Gruppen besonders bemerkenswert; FC=Franck-Condon,Hion=Energie im Ion, Hneu=Energie im Neutralen. Die vertikalenLinien repr�sentieren die Gbergangsenergien zwischen den relevantenelektronischen Zust�nden. Die obere Kurve repr�sentiert den ionischenZustand und die untere Kurve den neutralen. Die Ladung wird ausdem Grundzustand �ber einen Franck-Condon-Gbergang angeregt. Dieverdrehte Bewegung der Ramachandran-Winkel verschiebt den ioni-schen Zustand zum Schnittpunkt der Kurve, die Ladung wird zur�ck inden neutralen Grundzustand �bertragen. Dieser Prozess wird als eineArt von Ratcheting bezeichnet.
E. W. Schlag et al.Aufs�tze
3262 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273
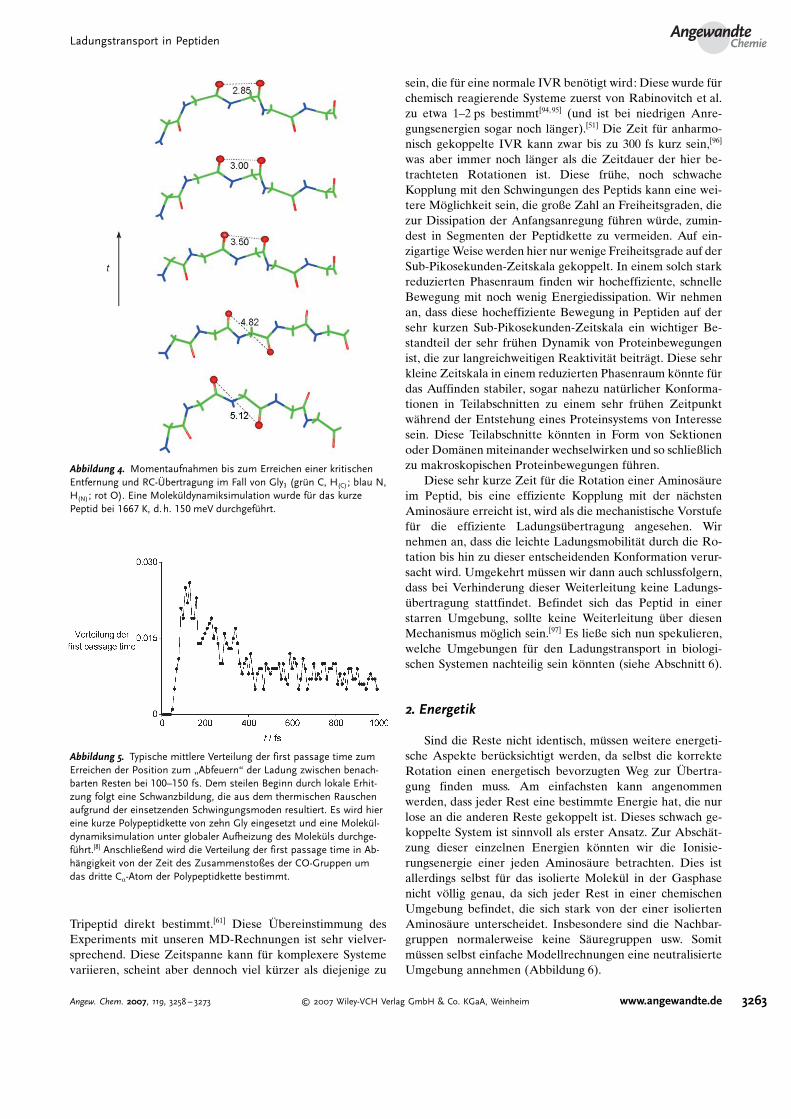
Tripeptid direkt bestimmt.[61] Diese �bereinstimmung desExperiments mit unseren MD-Rechnungen ist sehr vielver-sprechend. Diese Zeitspanne kann f�r komplexere Systemevariieren, scheint aber dennoch viel k�rzer als diejenige zu
sein, die f�r eine normale IVR ben2tigt wird: Diese wurde f�rchemisch reagierende Systeme zuerst von Rabinovitch et al.zu etwa 1–2 ps bestimmt[94,95] (und ist bei niedrigen Anre-gungsenergien sogar noch l�nger).[51] Die Zeit f�r anharmo-nisch gekoppelte IVR kann zwar bis zu 300 fs kurz sein,[96]
was aber immer noch l�nger als die Zeitdauer der hier be-trachteten Rotationen ist. Diese fr�he, noch schwacheKopplung mit den Schwingungen des Peptids kann eine wei-tere M2glichkeit sein, die große Zahl an Freiheitsgraden, diezur Dissipation der Anfangsanregung f�hren w�rde, zumin-dest in Segmenten der Peptidkette zu vermeiden. Auf ein-zigartigeWeise werden hier nur wenige Freiheitsgrade auf derSub-Pikosekunden-Zeitskala gekoppelt. In einem solch starkreduzierten Phasenraum finden wir hocheffiziente, schnelleBewegung mit noch wenig Energiedissipation. Wir nehmenan, dass diese hocheffiziente Bewegung in Peptiden auf dersehr kurzen Sub-Pikosekunden-Zeitskala ein wichtiger Be-standteil der sehr fr�hen Dynamik von Proteinbewegungenist, die zur langreichweitigen Reaktivit�t beitr�gt. Diese sehrkleine Zeitskala in einem reduzierten Phasenraum k2nnte f�rdas Auffinden stabiler, sogar nahezu nat�rlicher Konforma-tionen in Teilabschnitten zu einem sehr fr�hen Zeitpunktw�hrend der Entstehung eines Proteinsystems von Interessesein. Diese Teilabschnitte k2nnten in Form von Sektionenoder Dom�nen miteinander wechselwirken und so schließlichzu makroskopischen Proteinbewegungen f�hren.
Diese sehr kurze Zeit f�r die Rotation einer Aminos�ureim Peptid, bis eine effiziente Kopplung mit der n�chstenAminos�ure erreicht ist, wird als die mechanistische Vorstufef�r die effiziente Ladungs�bertragung angesehen. Wirnehmen an, dass die leichte Ladungsmobilit�t durch die Ro-tation bis hin zu dieser entscheidenden Konformation verur-sacht wird. Umgekehrt m�ssen wir dann auch schlussfolgern,dass bei Verhinderung dieser Weiterleitung keine Ladungs-�bertragung stattfindet. Befindet sich das Peptid in einerstarren Umgebung, sollte keine Weiterleitung �ber diesenMechanismus m2glich sein.[97] Es ließe sich nun spekulieren,welche Umgebungen f�r den Ladungstransport in biologi-schen Systemen nachteilig sein k2nnten (siehe Abschnitt 6).
2. Energetik
Sind die Reste nicht identisch, m�ssen weitere energeti-sche Aspekte ber�cksichtigt werden, da selbst die korrekteRotation einen energetisch bevorzugten Weg zur �bertra-gung finden muss. Am einfachsten kann angenommenwerden, dass jeder Rest eine bestimmte Energie hat, die nurlose an die anderen Reste gekoppelt ist. Dieses schwach ge-koppelte System ist sinnvoll als erster Ansatz. Zur Absch�t-zung dieser einzelnen Energien k2nnten wir die Ionisie-rungsenergie einer jeden Aminos�ure betrachten. Dies istallerdings selbst f�r das isolierte Molek�l in der Gasphasenicht v2llig genau, da sich jeder Rest in einer chemischenUmgebung befindet, die sich stark von der einer isoliertenAminos�ure unterscheidet. Insbesondere sind die Nachbar-gruppen normalerweise keine S�uregruppen usw. Somitm�ssen selbst einfache Modellrechnungen eine neutralisierteUmgebung annehmen (Abbildung 6).
Abbildung 4. Momentaufnahmen bis zum Erreichen einer kritischenEntfernung und RC-Gbertragung im Fall von Gly3 (gr�n C, H(C) ; blau N,H(N); rot O). Eine Molek�ldynamiksimulation wurde f�r das kurzePeptid bei 1667 K, d.h. 150 meV durchgef�hrt.
Abbildung 5. Typische mittlere Verteilung der first passage time zumErreichen der Position zum „Abfeuern“ der Ladung zwischen benach-barten Resten bei 100–150 fs. Dem steilen Beginn durch lokale Erhit-zung folgt eine Schwanzbildung, die aus dem thermischen Rauschenaufgrund der einsetzenden Schwingungsmoden resultiert. Es wird hiereine kurze Polypeptidkette von zehn Gly eingesetzt und eine Molek�l-dynamiksimulation unter globaler Aufheizung des Molek�ls durchge-f�hrt.[8] Anschließend wird die Verteilung der first passage time in Ab-h�ngigkeit von der Zeit des Zusammenstoßes der CO-Gruppen umdas dritte Ca-Atom der Polypeptidkette bestimmt.
Ladungstransport in PeptidenAngewandte
Chemie
3263Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

Weiterhin ist zu ber�cksichtigen, dass die Ionisierungeines bestimmten Restes in der Peptidumgebung andersverl�uft als die Ionisierung der entsprechenden isoliertenAminos�ure, bei der das Elektron bis ins Unendliche entferntwird. Die Ionisierung entlang der Kette verl�uft zu CT-Zu-st�nden, bei denen die Ladung nicht ins Unendliche entferntwird. Diese Zust�nde liegen gew2hnlich etwa 1 eV unterhalbder Standardionisierungsenergien f�r die isolierten Amino-s�uren. Nichtsdestoweniger wird der Transport hier im We-sentlichen durch variierende lokale Energien gesteuert, einModell, das unsere Ergebnisse grob zu erkl�ren scheint. Diesist ein wertvolles Modell f�r den Ladungstransport in solchenPeptidionen und k2nnte sich m2glicherweise auch auf großeMolek�lfragmente, wie sie z.B in einemMassenspektrometerbeobachtet werden, anwenden lassen.
Damit haben wir nun ein difunktionales Modell zur Hand,das den CTentlang der Polypeptidkette vom C-Terminus zumN-Terminus beschreibt. An jedem Ca-Atom gibt es sowohleine N-Seite als auch eine C-Seite. Die Torsionswinkel um dieCa-Drehachse sind in bestimmten Dom�nen innerhalb desPhasenraums (y, f) eingeschr�nkt, der im Ramachandran-Diagramm dargestellt ist. Unterschiedliche Aminos�urepaareder a-Helix und des b-Faltblatts belegen verschiedene Be-reiche im Ramachandran-Diagramm. Wenn die Ladung lokalauf der C-Seite eines Ca-Atoms angeregt wird, springt siedurch den anschließenden O-O-Atomzusammenstoß in derN�he des Ca-Atoms zu dessen nahe gelegener N-Seite. DieserSprungprozess kann weniger als 10 fs beanspruchen. DieTorsionsbewegungen der Carbonylgruppen f�r die y- und f-Winkel ben2tigen jeweils etwa 150 fs, wobei diese Zeitenjedoch in Abh�ngigkeit von den verschiedenen Ausgang-skonformationen variieren und daher nicht miteinander kor-relieren. Daher deuten wir diese Rotationsbewegungen alsein virtuelles Brownsches Teilchen, das sich innerhalb desBereichs mit nahezu freier Bewegung im Ramachandran-Diagramm bewegt.[8] Die O-O-Atome stoßen bei einem be-stimmten Abstand (z.B. bei 2.8 D) zusammen, und das vir-tuelle Brownsche Teilchen erreicht im Ramachandran-Dia-gramm einen festgelegten Punkt, der einen Ausgang zumEntweichen des Brownschen Teilchens definiert. DieserPunkt wird durch die Brownsche Rotationsbewegung er-reicht. So entsteht ein quasi-entarteter Zustand, in dem dannein schneller Ladungstransfer stattfindet. Dieser Ladungs-transfer erfolgt, sobald der C-O-Abstand erreicht ist, imWesentlichen elektronisch unverz2gert auf der Zeitskala derBrownschen Bewegung. Daher entspricht die sequenzielleWeitergabe der Ladung entlang einer Polypeptidkette einer
Folgekombination von 1) Entweichen des Brownschen Teil-chens und 2) Sprung der Ladung.
Bei einem beispielhaften Vergleich der experimentellenBefunde f�r Leu-Leu-Leu-Trp mit Gly-Gly-Gly-Trp beob-achten wir, dass Leu-Leu-Leu-Trp die Ladung von einemionisierten Trp vollst�ndig zum N-terminalen Leu leitet,w�hrend im Fall von Gly-Gly-Gly-Trp �berhaupt keinTransport stattfindet.[1,2] Dies kann einem kleinen Unter-schied der CT-Energien von Leu und Gly zugeschriebenwerden, wobei die Energie f�r Leu etwa 0.2 eV niedriger ist.Da die Energie f�r Trp dazwischen liegt, erm2glicht derAustausch von Trp gegen das energetisch h2her liegende Tyrin beiden F�llen einen vollst�ndigen Transport. Diese beidenPeptide sind nur zwei Vertreter einer betr�chtlichen Zahl anuntersuchten Peptiden.[1,2]
Der leichte Ladungstransport resultiert in einer chemi-schen Reaktion, die an einem von der Ionisierungsstelle weitentfernten Ort stattfindet. Mit anderen Worten erfolgt eine�bertragung sowohl der Reaktivit�t als auch der Ladung. F�rdiese so genannte RC-�bertragung m�ssen zwei Bedingun-gen erf�llt sein: 1) Die ultraschnelle Bewegung der Dieder-winkel hin zum Zustand, in dem die Ladung �bertragen wird,darf nicht behindert werden; 2) die Energetik muss an dieserStelle g�nstig sein. Dies unterscheidet sich von der typischenKinetik lokaler chemischer Reaktionen. Die Anregung er-folgt in unserem System an einem Ende und die chemischeReaktion l�uft als Folge von Ladungstransport am anderenEnde ab.
Welche Rolle spielen nun die normalen, unimolekularenProzesse in Peptiden? Solche Prozesse finden nat�rlich den-noch statt, allerdings auf einer viel gr2ßeren Zeitskala, wie f�rkleinere Peptide in der Gasphase von Lifshitz et al.[46,98]
nachgewiesen wurde. Sie bewiesen, dass sowohl Leu-Tyr alsauch Leu-Leu-Tyr nach IVR normale chemische unimole-kulare Reaktionen eingehen k2nnen, wenngleich viel lang-samer. Die Geschwindigkeitskonstanten liegen dann im vonder Theorie von Rice, Ramsperger, Kassel und Marcus vor-hergesagten Bereich.[42] Das Extrapolieren dieser Zeiten aufunsere gr2ßeren Systeme w�rde zu Reaktionszeiten von ca.0.1 s f�hren, die in unserer Apparatur nicht beobachtetwerden k2nnen und mit strahlendem Zerfall konkurrierenw�rden. Wir nehmen an, dass wir es zum einen mit einemstark verringerten Phasenraum bei kurzen Zeiten und zumanderen mit einem normalen, gr2ßeren Phasenraum nach derIVR zu tun haben. F�r Peptide mit niedrigen Anregungsen-ergien, wie sie in biologischen Systemen gefunden werden,erfolgt vor der IVR eine ultraschnelle RC-�bertragung ent-lang der Kette zu einem energiearmen Rest.[53,99] DieserProzess verl�uft wegen des sehr kleinen Phasenraums �ußersteffizient.
Betrachten wir nun eine Energiefl�che im Peptid, dienicht eben ist, sondern einige ausgepr�gte Energiekonturenaufweist. Als Beispiel soll der gemischte Fall von Leu-Gly-Leu-Trp dienen. Wir beobachten wieder eine �ußerst effizi-ente RC-�bertragung ausgehend vom C-Terminus, doch in-teressanterweise nur bis zum Gly-Rest. Diese energiereichePosition wirkt nun als „Flaschenhals“ f�r die RC-�bertra-gung. Obwohl der N-Terminus die niedrigste Energie in derKette aufweist, ist er nicht der Endpunkt der RC-�bertra-
Abbildung 6. Modellverbindung, die zur Berechnung des IP eines Ami-nos�urerests innerhalb eines Peptids verwendet wurde. y und f sinddie Ramachandran-Winkel.
E. W. Schlag et al.Aufs�tze
3264 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273

gung. Folglich w�hlt dieser Mechanismus nicht den energie-�rmsten Endzustand aus, wie in der normalen statistischenKinetik – vielmehr f�hrt die Engstelle entlang des Wegs zurUnterbrechung des �bertragungsprozesses. Das Experimentzeigt außerdem, dass die RC-�bertragung entlang der Pep-tidkette erfolgt und nicht durch einen anderen Prozess.
Dieser Ablauf �ber große Entfernungen mit einem derartstark reduzierten Phasenraum ist ein Prozess, der vielleichtnur bei Polypeptiden auftritt und durch die ultraschnellen,großamplitudigen Ver�nderungen der Ramachandran-Winkel gegeben ist, wodurch IVR und ebenso lokale elek-tronische Energien �berspielt werden.
Wenn wir diese Denkweise auf Photoionisierungsdatenaus derMassenspektrometrie von Peptiden anwenden, stellenwir fest, dass auch hier Reaktivit�t im Ion �ber betr�chtlicheEntfernungen transportiert wird und so zur Bildung dervielen beobachteten Fragmente f�hrt. Ein statistischer Pro-zess w�re nicht dazu in der Lage, in der Zeitskala eines ty-pischen Massenspektrometers eine derartige, erheblicheFragmentierung großer Ionen so weit entfernt von der An-regungsstelle zu bewirken.[100]
3. Rechnungen
F�r Peptide sind viele Anregungsm2glichkeiten, z.B.durch Redoxreaktionen, vorstellbar, hier allerdings wollenwir den einfachen Prozess der direkten Photoionisierungbetrachten, und das auch nur am C-terminalen Ende desPeptids. Zur Bestimmung der lokalen Energien ermitteltenwir die Ionisierungsenergien der 20 nat�rlichen Aminos�u-ren. Nur vierzehn Werte wurden experimentell bestimmt, f�rdie restlichen sechs Aminos�uren f�hrten wir eine Ab-initio-Rechnung durch (Abbildung 7). Dazu nutzten wir ein DFT-Programm[101] unter Verwendung der B3LYP-Methode undeines 6-31+G*-Basissatzes. Es ergab sich, dass Gly, wie vondem Modell gefordert, tats�chlich 0.2 eV �ber Leu liegt.
Die zun�chst photoionisierte Aminos�ure, hier Trp, ist einSpezialfall, da die Ionisierung an dieser Stelle das Elektronbis ins Unendliche entfernt, w�hrend alle weiteren lokalenIonisierungen CT-Zust�nde sind. Dies zeigt sich bereits beimDimer Leu-Trp, wo die Ladung leicht zum Leu wandert.Außerdem ist dieser Transfer, sogar nach Abschalten desLasers, stabil und offenbar irreversibel, selbst �ber eineDauer von 10 ms.[46]
Die Ionisierung eines Trp wird allein nur �ber den opti-schen �bergang im Benzolteil bei 260 nm hervorgerufen, undes wird eine Ionisierungsenergie erwartet, die der Ionisie-rungsenergie der isolierten Aminos�ure sehr nahe kommt.Diese Ionisierungsenergie ist diejenige Energie, die zur Bil-dung einer positiven Ladung am Molek�l und gleichzeitigenEntfernung des Elektrons ins Unendliche notwendig ist.Unter der Annahme, dass diese Energie n�herungsweise derIonisierungsenergie der isolierten Aminos�ure entspricht, istdie Energetik f�r die beschriebenen Prozesse in Abbildung 7angegeben.
Aus Leu-Trp w�rde Leu-Trp+ entstehen. Es w�re nunvorstellbar, dass die Ladung anschließend von Trp zu Leuspringt. Die daf�r aufgewendete Energie unterscheidet sich
von derjenigen zur Entfernung des Elektrons von einer dieserzwei Positionen, denn nun muss das Elektron nicht bis insUnendliche bewegt werden, sondern lediglich zur anderenPosition. Mithilfe eines einfachen Coulomb-Modells kann dieEnergieeinsparung zu etwa 1 eV berechnet werden; s�mtlicheweiteren Positionen in der Kette nach der ersten sind dannCT-Positionen. Damit haben wir nun den Leu+-Trp-Zustanderreicht.
Wenn wir eine DFT-Rechnung am Tripeptid Leu2Trpdurchf�hren, erhalten wir eine vertikale Ionisierungsenergievon nahezu 7.8 eV, w�hrend die adiabatische Energie fast7.0 eV betr�gt. F�r eine Modellierung m�ssen daher dieprototypischen Ionisierungsenergien der Aminos�uren be-rechnet werden, die in einer Pseudo-Peptidumgebung einge-schlossen sind (Abbildung 6). Diese Energien liegen tat-s�chlich alle etwa 1 eV niedriger als die der isolierten Ami-nos�uren. Dies bedeutet, dass der niedrigste Energiezustandein adiabatischer Zustand ist, der durch den vertikalen Pro-zess, trotz niedrigerer Energie, nicht leicht erreichbar ist.
Als grobe Vereinfachung setzen wir f�r die erste Positiondie vertikale Ionisierungsenergie der isolierten Aminos�ureund f�r alle folgenden Positionen die CT-Energien ein. Wirbetrachten ein Dimer von Leu und Trp (LT) und nehmen an,dass die Wellenl�nge und Intensit�t des Lasers so gew�hltwerden, dass Trp in LT ionisiert wird [Gl. (1)].
LT �hw�!LTþðnÞ ð1Þ
Abbildung 7. Ionisierungspotentiale von Aminos�uren; & experimen-teller Wert, * berechneter Wert (B3LYP/G-31+G*) mit optimierterGeometrie. Entlang der x-Achse sind die polaren, aromatischen, katio-nischen und anionischen Spezies angeordnet.
Ladungstransport in PeptidenAngewandte
Chemie
3265Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

LT+(n) entspricht dem vertikalen Ionisierungszustand,der durch den Franck-Condon-�bergang bestimmt ist. AusAbbildung 7 geht hervor, dass das Energieminimum etwa7.8 eV f�r LT+(n) betr�gt, und aus Abbildung 8, dass die
adiabatische Ionisierungsenergie nur 6.8 eV f�r LT+(a) be-tr�gt (a= adiabatisch). Da LT+(a) nur eine Energie von7.5 eV aufweist (Abbildung 8), kann der CT gem�ß Glei-chung (2) stattfinden.
LTþðnÞ ! LTþðaÞ ð2Þ
Zur Berechnung dieser adiabatischen Energien wurde dieAminos�ure in eine typische Umgebung versetzt (Abbil-dung 6). Auf diese Weise berechneten wir eine Reihe vonadiabatischen Energien f�r die 20 Aminos�uren (Abbil-dung 8). Typischerweise liegen diese Energien alle etwa 1 eVniedriger als die vertikalen Ionisierungsenergien in Abbil-dung 7. Wenn wir nun das Ionisierungspotential (IP) von Trpin Abbildung 7 mit den adiabatischen Energien in Abbil-dung 8 vergleichen, wird deutlich, dass derWert f�r Trp genauzwischen den Werten von Gly und Leu liegt. Daraus kann einLadungstransport zu Leu vorhergesagt werden, nicht jedoch –�bereinstimmend mit unserer Beobachtung – zu Gly. DieAbsorption zum adiabatischen Zustand von Trp (Abbil-dung 8) ist der niedrigste Energiezustand des Systems, weistjedoch f�r ein normales Absorptionsexperiment zu wenigOszillatorst�rke auf. Es ist allerdings anzunehmen, dass durcheine Absorption mit einem kleinen Querschnitt dieser Zu-
stand von Trp im Peptid auch erreicht wird, und dieser istdann der niedrigste Zustand des gesamten Systems. Ausdiesem Zustand ist kein CT m2glich.
Es ist entscheidend, dass der Elektronentransfer (ET) inGleichung (2) vor der IVR stattfindet. Daher sollte der ET inGleichung (2) durch eine Ein-Niveau-ET-Geschwindigkeits-konstante Win beschrieben werden. Ist der ET viel langsamerals die IVR, sodass anstelle eines ET das Schwingungs-gleichgewicht eingestellt wird, erhalten wir die mikrokano-nische ET-Geschwindigkeitskonstante W(E) [Gl. (3)], wobeiE die Anregungsenergie des Systems und Pin(E) die mikro-kanonische Verteilung ist [Gl. (4)]. d(E�Ein) ist die Delta-Funktion und 1i(E) die Zustandsdichte mit der Energie E[Gl. (5)]. Gleichung (3) zeigt, dass IVR die Anregungsener-gie �ber alle Schwingungszust�nde gleicher Energie verteilt.Jeder Zustand ist gleich wahrscheinlich und wird durch Pin(E)beschrieben. Die beobachtete Geschwindigkeit W(E) mussnun alle m2glichen ET-Geschwindigkeitskonstanten Win ein-schließen, die durch ihre mikrokanonische Verteilung Pin(E)bewertet werden. Mit anderen Worten wird IVR die ET-Geschwindigkeit verringern.
WðEÞ ¼X
n
PinðEÞWin ð3Þ
PinðEÞ ¼dðE�EinÞ1iðEÞ
ð4Þ
1iðEÞ ¼X
n
dðE�EinÞ ð5Þ
Diese Schlussfolgerungen f�r die RC-�bertragung sindzwar von allgemeiner Natur, aber es werden dennoch feineUnterschiede vorliegen, wenn die entsprechende Anre-gungsenergie aus Prozessen stammt, die keinem Photoioni-sierungsprozess entsprechen. Es ist zu beachten, dass derR�cktransfer zum adiabatischen Zustand des C-terminalenTrp nach dem Ladungstransfer zum N-terminalen Leu zulangsam verl�uft und daher nicht beg�nstigt ist, da nun derbevorzugte Prozess eine lokale Dissoziation ist.
Der umgekehrte Ladungszustand, der etwa 1 eV unterdem Leu+-Trp-Zustand liegt, f�hrt zu einer beachtlichenEnergiel�cke. Da dies alles Prozesse sind, die innerhalbelektronischer �berg�nge strahlungslos verlaufen, k2nnenwir aus der Theorie der strahlungslosen �berg�nge ableiten,dass sich die Lebensdauer des Leu+-Trp-Zustands etwa mitder Energiel�cke erh2ht,[65,102] entsprechend Gleichung (6),
Wi0 ¼jTfij2�h2
�2pwifw
�1=2
exp��wif
w
�ln
wif
s �ww�1
��s
�ð6Þ
wobei wif die Energiel�cke, s den Huang-Rhys-Faktor, Tfi daselektronische Kopplungsmatrixelement, w die Anregungs-frequenz und w die Durchschnittsfrequenz bezeichnet. EineL�cke von 1 eV f�hrt zu einer Lebensdauer von etwa 100 nsals Untergrenze; auf dieser Zeitskala kann der Leu+-Trp-Zustand fragmentieren. Experimente zu Leu-Tyr zeigen, dassdie Ladung im Dimer an der Leu-Position verbleibt.[46]
Eine detailliertere Dichtefunktionalrechnung f�r Leu2Trpliefert einen vom Torsionswinkel abh�ngigen CT-Prozess, der
Abbildung 8. Ionisierungspotentiale von Aminos�ureresten innerhalbeines Peptids, das aus der Modellverbindung berechnet wurde, in dersich die Aminos�ure in einer Pseudo-Peptidumgebung befindet. DerBasissatz und die Spezies sind die gleichen wie bei Abbildung 7.
E. W. Schlag et al.Aufs�tze
3266 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273
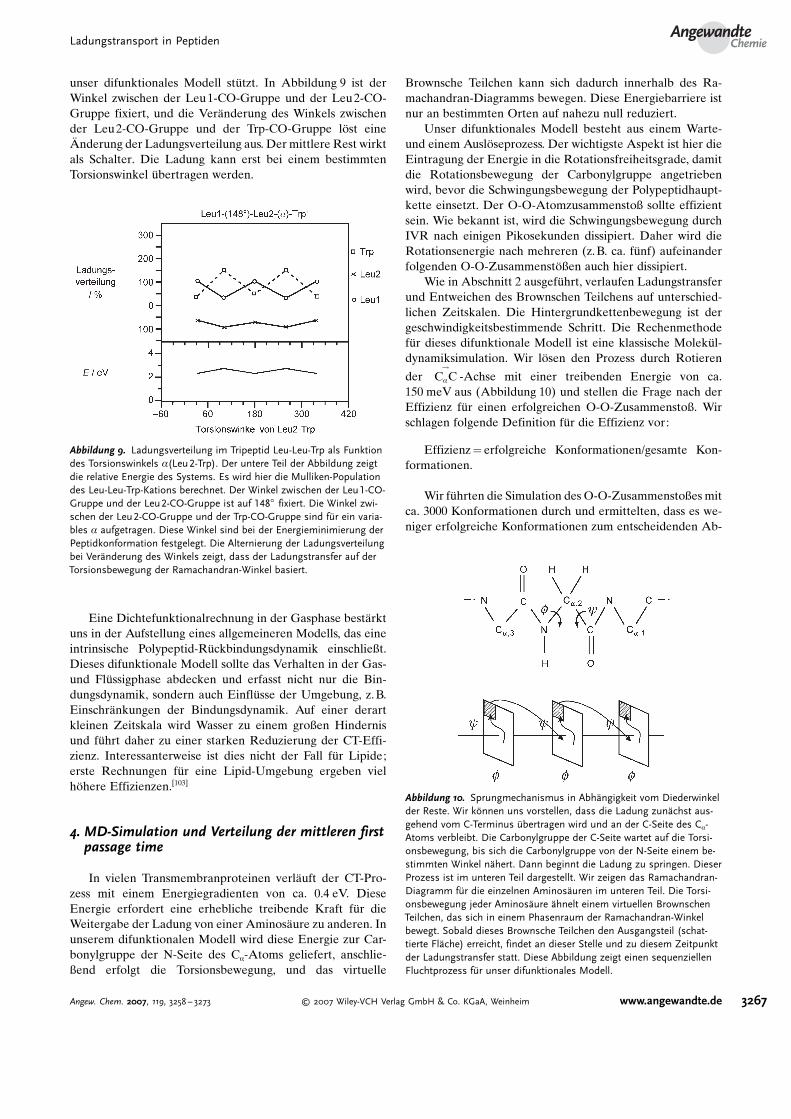
unser difunktionales Modell st�tzt. In Abbildung 9 ist derWinkel zwischen der Leu1-CO-Gruppe und der Leu2-CO-Gruppe fixiert, und die Ver�nderung des Winkels zwischender Leu2-CO-Gruppe und der Trp-CO-Gruppe l2st eineRnderung der Ladungsverteilung aus. Der mittlere Rest wirktals Schalter. Die Ladung kann erst bei einem bestimmtenTorsionswinkel �bertragen werden.
Eine Dichtefunktionalrechnung in der Gasphase best�rktuns in der Aufstellung eines allgemeineren Modells, das eineintrinsische Polypeptid-R�ckbindungsdynamik einschließt.Dieses difunktionale Modell sollte das Verhalten in der Gas-und Fl�ssigphase abdecken und erfasst nicht nur die Bin-dungsdynamik, sondern auch Einfl�sse der Umgebung, z.B.Einschr�nkungen der Bindungsdynamik. Auf einer derartkleinen Zeitskala wird Wasser zu einem großen Hindernisund f�hrt daher zu einer starken Reduzierung der CT-Effi-zienz. Interessanterweise ist dies nicht der Fall f�r Lipide;erste Rechnungen f�r eine Lipid-Umgebung ergeben vielh2here Effizienzen.[103]
4.MD-Simulation und Verteilung der mittleren firstpassage time
In vielen Transmembranproteinen verl�uft der CT-Pro-zess mit einem Energiegradienten von ca. 0.4 eV. DieseEnergie erfordert eine erhebliche treibende Kraft f�r dieWeitergabe der Ladung von einer Aminos�ure zu anderen. Inunserem difunktionalen Modell wird diese Energie zur Car-bonylgruppe der N-Seite des Ca-Atoms geliefert, anschlie-ßend erfolgt die Torsionsbewegung, und das virtuelle
Brownsche Teilchen kann sich dadurch innerhalb des Ra-machandran-Diagramms bewegen. Diese Energiebarriere istnur an bestimmten Orten auf nahezu null reduziert.
Unser difunktionales Modell besteht aus einem Warte-und einem Ausl2seprozess. Der wichtigste Aspekt ist hier dieEintragung der Energie in die Rotationsfreiheitsgrade, damitdie Rotationsbewegung der Carbonylgruppe angetriebenwird, bevor die Schwingungsbewegung der Polypeptidhaupt-kette einsetzt. Der O-O-Atomzusammenstoß sollte effizientsein. Wie bekannt ist, wird die Schwingungsbewegung durchIVR nach einigen Pikosekunden dissipiert. Daher wird dieRotationsenergie nach mehreren (z.B. ca. f�nf) aufeinanderfolgenden O-O-Zusammenst2ßen auch hier dissipiert.
Wie in Abschnitt 2 ausgef�hrt, verlaufen Ladungstransferund Entweichen des Brownschen Teilchens auf unterschied-lichen Zeitskalen. Die Hintergrundkettenbewegung ist dergeschwindigkeitsbestimmende Schritt. Die Rechenmethodef�r dieses difunktionale Modell ist eine klassische Molek�l-dynamiksimulation. Wir l2sen den Prozess durch Rotieren
der CaC!
-Achse mit einer treibenden Energie von ca.150 meV aus (Abbildung 10) und stellen die Frage nach derEffizienz f�r einen erfolgreichen O-O-Zusammenstoß. Wirschlagen folgende Definition f�r die Effizienz vor:
Effizienz= erfolgreiche Konformationen/gesamte Kon-formationen.
Wir f�hrten die Simulation des O-O-Zusammenstoßes mitca. 3000 Konformationen durch und ermittelten, dass es we-niger erfolgreiche Konformationen zum entscheidenden Ab-
Abbildung 9. Ladungsverteilung im Tripeptid Leu-Leu-Trp als Funktiondes Torsionswinkels a(Leu2-Trp). Der untere Teil der Abbildung zeigtdie relative Energie des Systems. Es wird hier die Mulliken-Populationdes Leu-Leu-Trp-Kations berechnet. Der Winkel zwischen der Leu1-CO-Gruppe und der Leu2-CO-Gruppe ist auf 1488 fixiert. Die Winkel zwi-schen der Leu2-CO-Gruppe und der Trp-CO-Gruppe sind f�r ein varia-bles a aufgetragen. Diese Winkel sind bei der Energieminimierung derPeptidkonformation festgelegt. Die Alternierung der Ladungsverteilungbei Ver�nderung des Winkels zeigt, dass der Ladungstransfer auf derTorsionsbewegung der Ramachandran-Winkel basiert.
Abbildung 10. Sprungmechanismus in Abh�ngigkeit vom Diederwinkelder Reste. Wir kNnnen uns vorstellen, dass die Ladung zun�chst aus-gehend vom C-Terminus �bertragen wird und an der C-Seite des Ca-Atoms verbleibt. Die Carbonylgruppe der C-Seite wartet auf die Torsi-onsbewegung, bis sich die Carbonylgruppe von der N-Seite einem be-stimmten Winkel n�hert. Dann beginnt die Ladung zu springen. DieserProzess ist im unteren Teil dargestellt. Wir zeigen das Ramachandran-Diagramm f�r die einzelnen Aminos�uren im unteren Teil. Die Torsi-onsbewegung jeder Aminos�ure �hnelt einem virtuellen BrownschenTeilchen, das sich in einem Phasenraum der Ramachandran-Winkelbewegt. Sobald dieses Brownsche Teilchen den Ausgangsteil (schat-tierte Fl�che) erreicht, findet an dieser Stelle und zu diesem Zeitpunktder Ladungstransfer statt. Diese Abbildung zeigt einen sequenziellenFluchtprozess f�r unser difunktionales Modell.
Ladungstransport in PeptidenAngewandte
Chemie
3267Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

stand gibt als gesamte Konformationen. Das Verh�ltnis gibteine Effizienz an.
Mit dieser Effizienz k2nnen wir durch ein einfaches Ar-gument eine Beziehung zum Abklingfaktor mit dem Abstandherstellen. Zuerst definieren wir die Geschwindigkeitskon-stante kt f�r den CT sowie eine Geschwindigkeitskonstante kb
f�r den Verlust an das Bad. Der Bruchteil der Ladung, dernach n verkn�pften Aminos�uren noch vorhanden ist, kannnach Gleichung (7) ermittelt werden.
an ¼ j kt
kt þ kbjn ð7Þ
Diese Potenzform kann in die exponentielle Form e�bn
�berf�hrt werden, wobei b normalerweise 0.8–1.4 D�1 ist. Dadie L�nge jeder Aminos�ure ca. 3.7 D betr�gt, wird der ab-standsabh�ngige Abklingfaktor als e�bR= e�3.7bn ausgedr�ckt.Wir k2nnen an= en lna anwenden, wobei a= kt/(kt+kb) ist, umGleichung 8 zu erhalten.
b ¼ � ln a3:7
ð8Þ
Wie aus Gleichung (8) hervorgeht, ist a die Effizienz, undwir erhalten den b-Wert des abstandsabh�ngigen Abkling-faktors. Ist der b-Wert klein, ist die CT-Effizienz groß. Einsehr effizienter Transfer erfolgt in DNA, wo der b-Wert ca.0.2 D�1 betr�gt. Das Verh�ltnis kb/kt betr�gt 1:1. Jetzt ver-laufen beide Prozesse gleich schnell. F�r b= 1.4 D�1 erh�ltman kb/kt= 177. Das bedeutet, dass weniger als 1% CT beijedem Schritt dem normalen Wert von b entspricht.
In unserer Simulation wird ein f�r eine Einzelpositions-anregung modifiziertes CHARMM-24-Programm[93] einge-f�hrt, um die in den vorhergehenden Abschnitten beschrie-bene Methode zur lokalen Aufheizung einzusetzen (Abbil-dung 5). Bei der nativen Anfangskonformation beeinflusstdie Rotationsrichtung die mittlere freie Wegl�nge und somitdie mittlere first passage time. Das Modell liefert eine un-verz2gerte treibende Energie zu einer einzelnen Position;diese Energie wird entlang der Polypeptidkette zur n�chstenCa-Position �bertragen. Anschließend folgen MD-Simulatio-nen des CT von der isolierten Gasphase bis hin zum hydra-tisierten System.
5. Isoliertes System
Unser Massenspektrum von kurzen PolypeptidkettenLeunTrp (n= 1–4)[1–3] zeigt nur den Peak des Molek�lions undder N-Termini mit einer Gesamteffizienz von ca. 0.47. Es trittkein weiterer Bindungsbruch bei den dazwischen liegendenResten auf. Daher wird die Effizienz von 0.47 dem erstenLeu-Rest zugeordnet, und andere Reste haben eine Effizienzvon nahezu eins. Diese Effizienz von nahezu eins l�sst dieSchlussfolgerung zu, dass die Energie hoch effizient �bertra-gen wird. Da keine Energiedissipation auftritt, wird die Ro-tationsenergie infolge der Impulserhaltung vollst�ndig auf dien�chste Carbonylgruppe �bertragen (Abbildung 10). DieMethode der lokalen Aufheizung ergibt, dass die verblei-benden Phononen die anschließende Bewegung, die zum
Ladungstransport f�hrt, nicht beeinflussen. Die Ergebnisse inTabelle 1 best�tigen diese hohe Effizienz zwischen benach-barten Aminos�uren und eine ballistische Bewegung im di-funktionalen Modell.
In Tabelle 1 regen wir die C-Seite der Ca-Position vonVal10 f�r das 20mere Polypeptid Mb20 mit einer Anregungs-energie von 150 meVan. Dieses 20mer ist aus der a-Helix desMyoglobinmolek�ls herausgeschnitten. Die Effizienz istselbst an der f�nften Stelle von der Position der lokalenAufheizung (Rest 15) hoch. An Rest 16 sinkt die Effizienzschlagartig ab. Bei Val10 ist der mfp-Peak (mfp=mean firstpassage) sehr stark (Abbildung 11).
6. Die w�ssrige Umgebung
Offensichtlich ist das vorgestellte Modell recht erfolgreichbei der Interpretation des hocheffizienten Transports einerLadung �ber die Ionisierung am C-Terminus des isoliertenPeptidmolek�ls und derenWanderung entlang der Kette. Wiesieht dies nun f�r den Fall der aus �hnlichen Experimenten
Tabelle 1: Effizienz an jeder von der Stelle der lokalen Aufheizung ent-fernten Stelle.[a]
Rest-Nr.[b] Effizienz Rest-Nr.[b] Effizienz
2 0.18 12 0.903 0.0033 13 0.464 0.41 14 0.225 0.29 15 0.226 0.25 16 0.0847 0.40 17 0.118 0.69 18 0.0139 0.85 19 0.025
10 1.0 20 0.2611 0.78
[a] Position der lokalen Aufheizung: Val10; Tlokal=2667 K. THintergrund=
300 K. [b] Mb20= (N-Terminus)Glu1-Asp2-Leu3-Lysn4-Lysn5-Hsd6-Gly7-Val8-Thr9-Val10-Leu11-Thr12-Ala13-Leu14-Gly15-Ala16-Ile17-Leu18-Lysn19-Lysn20(C-Terminus). Lysn=neutrales Lys.
Abbildung 11. Verteilung der mittleren first passage time gegen dieZeit an Val10 f�r das 20mere Mb20. Dies ist die gleiche Peptidkette wiein Tabelle 1. Hier ist ein starker Peak sichtbar, der der Position der lo-kalen Aufheizung zugeordnet wird. Da es zwei Richtungen f�r die Ro-tation der CO-Gruppe gibt, ist die eine die Vorw�rtsbewegung vonniedriger zu hNherer Aminos�urenummer und die andere die R�ck-w�rtsbewegung von hNherer zu niedrigerer Aminos�urenummer. DerEinschub zeigt drei kleine Peaks, von denen (a) der Vorw�rtsbewegungdirekt nach lokaler Aufheizung entspricht, (b) einer R�ckw�rtsbewe-gung und (c) der rekursiven Bewegung von der C- zur N-Seite.
E. W. Schlag et al.Aufs�tze
3268 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273
abgeleiteten, geringeren Leitf�higkeit in Wasser aus? In sol-chen Experimenten wird die negative Ladung gew2hnlich ameinen Ende eines Donor-Acceptor-Redoxkomplexes einge-f�hrt[26,35,36] und am anderen Ende wieder entfernt, wobei sieexponentiell mit der Distanz abnimmt und die Effizienz umetwa zwei Gr2ßenordnungen abf�llt. In Anbetracht dergroßen Bedeutung von Wasser als Medium f�r biologischeProzesse muss dies ebenfalls f�r unseren Fall behandeltwerden.
Die Leitf�higkeit von Proteinen in einem Wassermediumwurde von vielen Gruppen gemessen und ist durch den ab-standsabh�ngigen Abfall der Ladung gekennzeichnet.[35,36]
Dies wird durch die Exponentialfunktion (9) ausgedr�ckt,
Y ¼ A e�bR ð9Þ
wobei b normalerweise 1 D�1 betr�gt (Y=Ausbeute, A=
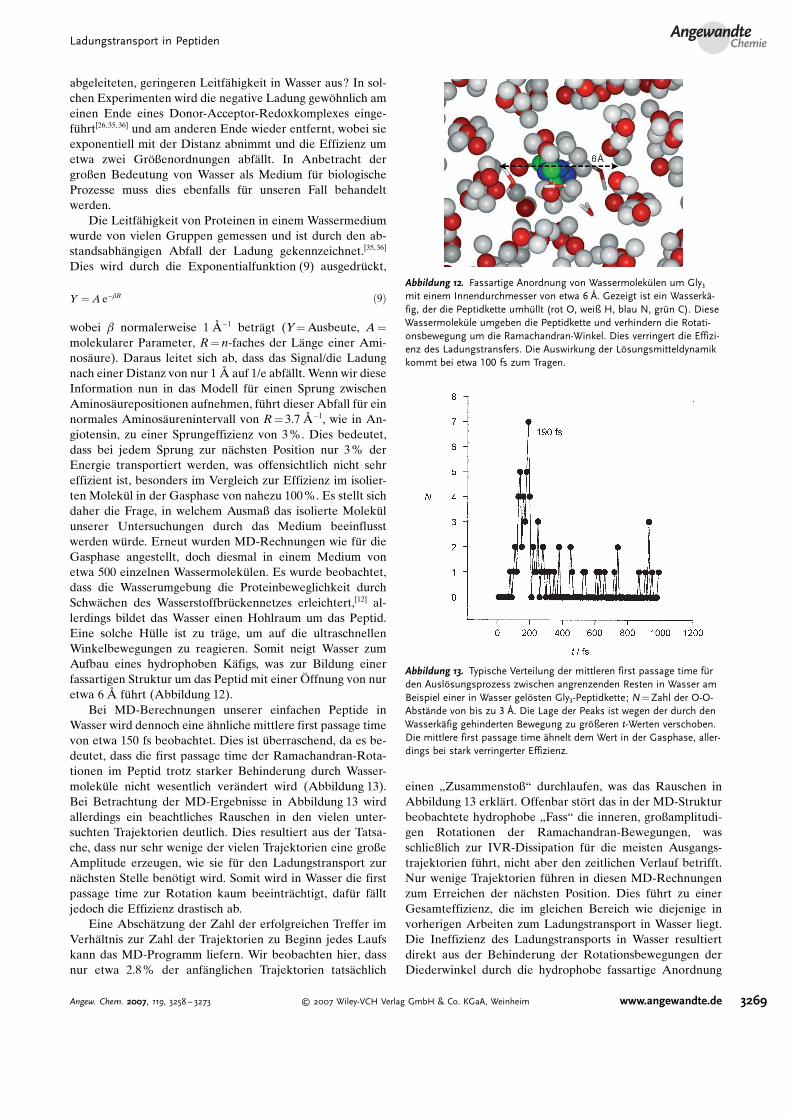
molekularer Parameter, R= n-faches der L�nge einer Ami-nos�ure). Daraus leitet sich ab, dass das Signal/die Ladungnach einer Distanz von nur 1 D auf 1/e abf�llt. Wenn wir dieseInformation nun in das Modell f�r einen Sprung zwischenAminos�urepositionen aufnehmen, f�hrt dieser Abfall f�r einnormales Aminos�urenintervall von R= 3.7 D�1, wie in An-giotensin, zu einer Sprungeffizienz von 3%. Dies bedeutet,dass bei jedem Sprung zur n�chsten Position nur 3% derEnergie transportiert werden, was offensichtlich nicht sehreffizient ist, besonders im Vergleich zur Effizienz im isolier-ten Molek�l in der Gasphase von nahezu 100%. Es stellt sichdaher die Frage, in welchem Ausmaß das isolierte Molek�lunserer Untersuchungen durch das Medium beeinflusstwerden w�rde. Erneut wurden MD-Rechnungen wie f�r dieGasphase angestellt, doch diesmal in einem Medium vonetwa 500 einzelnen Wassermolek�len. Es wurde beobachtet,dass die Wasserumgebung die Proteinbeweglichkeit durchSchw�chen des Wasserstoffbr�ckennetzes erleichtert,[12] al-lerdings bildet das Wasser einen Hohlraum um das Peptid.Eine solche H�lle ist zu tr�ge, um auf die ultraschnellenWinkelbewegungen zu reagieren. Somit neigt Wasser zumAufbau eines hydrophoben K�figs, was zur Bildung einerfassartigen Struktur um das Peptid mit einer Uffnung von nuretwa 6 D f�hrt (Abbildung 12).
Bei MD-Berechnungen unserer einfachen Peptide inWasser wird dennoch eine �hnliche mittlere first passage timevon etwa 150 fs beobachtet. Dies ist �berraschend, da es be-deutet, dass die first passage time der Ramachandran-Rota-tionen im Peptid trotz starker Behinderung durch Wasser-molek�le nicht wesentlich ver�ndert wird (Abbildung 13).Bei Betrachtung der MD-Ergebnisse in Abbildung 13 wirdallerdings ein beachtliches Rauschen in den vielen unter-suchten Trajektorien deutlich. Dies resultiert aus der Tatsa-che, dass nur sehr wenige der vielen Trajektorien eine großeAmplitude erzeugen, wie sie f�r den Ladungstransport zurn�chsten Stelle ben2tigt wird. Somit wird in Wasser die firstpassage time zur Rotation kaum beeintr�chtigt, daf�r f�lltjedoch die Effizienz drastisch ab.
Eine Absch�tzung der Zahl der erfolgreichen Treffer imVerh�ltnis zur Zahl der Trajektorien zu Beginn jedes Laufskann das MD-Programm liefern. Wir beobachten hier, dassnur etwa 2.8% der anf�nglichen Trajektorien tats�chlich
einen „Zusammenstoß“ durchlaufen, was das Rauschen inAbbildung 13 erkl�rt. Offenbar st2rt das in der MD-Strukturbeobachtete hydrophobe „Fass“ die inneren, großamplitudi-gen Rotationen der Ramachandran-Bewegungen, wasschließlich zur IVR-Dissipation f�r die meisten Ausgangs-trajektorien f�hrt, nicht aber den zeitlichen Verlauf betrifft.Nur wenige Trajektorien f�hren in diesen MD-Rechnungenzum Erreichen der n�chsten Position. Dies f�hrt zu einerGesamteffizienz, die im gleichen Bereich wie diejenige invorherigen Arbeiten zum Ladungstransport in Wasser liegt.Die Ineffizienz des Ladungstransports in Wasser resultiertdirekt aus der Behinderung der Rotationsbewegungen derDiederwinkel durch die hydrophobe fassartige Anordnung
Abbildung 12. Fassartige Anordnung von Wassermolek�len um Gly3mit einem Innendurchmesser von etwa 6 Q. Gezeigt ist ein Wasserk�-fig, der die Peptidkette umh�llt (rot O, weiß H, blau N, gr�n C). DieseWassermolek�le umgeben die Peptidkette und verhindern die Rotati-onsbewegung um die Ramachandran-Winkel. Dies verringert die Effizi-enz des Ladungstransfers. Die Auswirkung der LNsungsmitteldynamikkommt bei etwa 100 fs zum Tragen.
Abbildung 13. Typische Verteilung der mittleren first passage time f�rden AuslNsungsprozess zwischen angrenzenden Resten in Wasser amBeispiel einer in Wasser gelNsten Gly3-Peptidkette; N=Zahl der O-O-Abst�nde von bis zu 3 Q. Die Lage der Peaks ist wegen der durch denWasserk�fig gehinderten Bewegung zu grNßeren t-Werten verschoben.Die mittlere first passage time �hnelt dem Wert in der Gasphase, aller-dings bei stark verringerter Effizienz.
Ladungstransport in PeptidenAngewandte
Chemie
3269Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

von Wassermolek�len. Es sollte erneut darauf hingewiesenwerden, dass die Bewegungen der Diederwinkel im Sub-Pi-kosekundenbereich liegen – auf dieser Zeitskala ist die H�lleaus Wassermolek�len starr, da die Bewegungen der Wasser-molek�le selbst auf einer gr2ßeren Zeitskala stattfinden.
Unsere Ergebnisse zeigen, dass Wasser hier zu einerproblematischen Umgebung f�r die Ladungs�bertragungwird. F�r andere Prozesse in der Proteindynamik ist dieWasserumgebung dagegen hilfreich, ja sogar notwendig, dasie dasWasserstoffbr�ckennetzwerk und seine Dynamik starkbeeinflusst.[15] F�r die sehr schnelle RC-�bertragung inWasser betr�gt der vorhergesagte Wert 3% �bertragung voneiner Position zur n�chsten und liegt damit interessanterweiseim Bereich anderer Daten, in denen der b-Wert von ca. 1 D�1
ein bekanntes experimentelles Ergebnis ist.[35, 36] Die hiervorgestellte Arbeit bietet eine alternative Erkl�rung f�r denextremen Abfall der RC-�bertragung in Wasser, wenn manihn mit unseren hocheffizienten Gasphasenergebnissen ver-gleicht. Die Ladungsweiterleitung in biologischen Systemenben2tigt m2glicherweise gesch�tztere und g�nstigere Umge-bungen, z.B. Membranen. Die RC-�bertragung k2nnte ineiner flexibleren Umgebung, z.B. in Lipiden, besser ablau-fen.[103]
7. Sekund�rstruktur
�bertragt man den experimentellen b-Wert in Effizienz-werte, ist das b-Faltblatt etwa dreimal effizienter als die a-Helix. Interessanterweise f�llt ein Vergleich der Effizienzender nat�rlichen a-Helix und des b-Faltblatts jedoch andersaus: Im Ramachandran-Diagramm werden nur sehr kleineBewegungen f�r den Ladungstransport ben2tigt; dies wirddurch die N�he der Gruppen in der a-Helix erleichtert, diesich sehr nah an der Ausl2sungsposition befinden.
Zur Erkl�rung der hohen Effizienz des b-Faltblatts habenwir die Verteilung der first passage time der starren b-Falt-blattstruktur in Azurin untersucht. Wir beobachteten, dassdas solvatisierte b-Faltblatt schw�chere Wasserstoffbr�ckenaufweist als ein isoliertes b-Faltblatt. Im solvatisierten Fallwerden die starken Wechselwirkungen zwischen den Ketteninnerhalb des b-Faltblatts, die im isolierten System durchWasserstoffbr�cken verbunden sind, unterbrochen. Der b-Wert, der durch unsere Methode der lokalen Aufheizung er-halten wurde, betr�gt 1.3 D�1, was mit den experimentellenBefunden f�r Azurin exakt �bereinstimmt.[31]
8. Schlussfolgerungen
Wir analysieren hier Experimente zusammen mit einemtheoretischen Modell f�r einen Vorgang, in dem die Anre-gung an einem Ende des Peptids stattfindet und die resul-tierende Ladung intakt bis zur Reaktion am anderen Endedes Peptids oder bis zur Blockierung in der Kette �bertragenwird. Der Prozess involviert sehr schnelle, leichte Molek�l-bewegungen, die ausschließlich bei Peptiden zu finden sind.Solche Strukturen k2nnten auch zu einer interessanten neuenKlasse von elektronischen Baueinheiten f�hren.[63,104–106]
Wir schlagen vor, dass bei Peptiden die Ver�nderung derDiederwinkel zwischen benachbarten Aminos�uren eine derHauptbewegungen f�r eine ultraschnelle Reaktion und La-dungs�bertragung ist. Diese Bewegungen k2nnten f�r diesehr fr�hen Prozesse in der Proteindynamik von grundle-gender Bedeutung sein. Eine Schwingungskopplung findetauf dieser Zeitskala noch nicht statt, sodass diese langreich-weitige Dynamik resultierend aus dem stark reduziertenPhasenraum mit atypischer Leichtigkeit verl�uft und daherhocheffizient und wenig dissipativ ist. Die sehr schnellen,großamplitudigen Ver�nderungen der Diederwinkel zwi-schen den Aminos�urepositionen sind ein Ergebnis von ver-nachl�ssigbaren Rotationspotentialbarrieren; dies ist ein be-sonderes Merkmal der Bewegungen der Ramachandran-Winkel in Proteinen. An den �ußersten Positionen dieserAmplitudenbewegungen erfolgt die Kopplung der benach-barten Positionen sehr effizient. Hier liegt der Fall einerverzerrten Struktur vor, die energetisch, wie in einem sp3-Hybrid, bevorzugt ist. Wir stellen fest, dass diese �ußerstePosition, und nicht die durchschnittliche Position, f�r die RC-�bertragung verantwortlich ist, da die Barrieren bei mittle-ren Winkeln zwischen den jeweiligen Positionen zu hoch f�reinen effizienten Transport sind.
Des Weiteren muss nat�rlich das Energieprofil entlangder Peptidkette g�nstig sein. Wir schlagen als erste N�herungeine Wechselwirkung voneinander unabh�ngiger Reste mitihren eigenen, inh�renten Energien vor. Diese k2nnen miteinem angen�herten Modell bestimmt werden und sind hiertabellarisch aufgef�hrt (siehe Tabelle 1). Wir f�hren dieEnergie zusammen mit der Ladung am C-Terminus desPeptids ein (Abbildung 7), worauf die Energie auf CT-Zu-st�nde entlang der Kette �bertragen wird (Abbildung 8).Auch eine andere Art der Eintragung von Ladung undEnergie, z.B. in Form eines Redoxsystems, ist hier vorstellbar,solange sie in der Lage ist, die geringe erforderliche Energievon 150 meV zu produzieren.
Umgebungsfaktoren k2nnen diese großamplitudigen Be-wegungen st2ren, was den Transport von Ladung und Reak-tivit�t stark behindern kann; allerdings wird die mittlere firstpassage time offenbar selbst in Wasser nicht wesentlich be-einflusst. Volumenwasser ist f�r diesen sehr schnellen Prozesskein gutes Medium, w�hrend es f�r andere, langsamere Pro-zesse essenziell ist. DiesesModell sagt vorher, dassWasser dieEffizienz des Transports um etwa zwei Gr2ßenordnungenherabsetzt; damit ist das hocheffiziente Gasphasenmodell inEinklang mit experimentellen Beobachtungen in Wasser. DasModell kann eine Ver�nderung beim Signaltransport vonmehr als zwei Gr2ßenordnungen, in Abh�ngigkeit von derUmgebung, relativ gut vorhersagen.
Im Umkehrschluss kann ein Peptid, das nicht rotierenkann, keine gute RC-�bertragung erzielen – jegliche �ber-tragung in einem starren Medium wird sehr ineffizient erfol-gen. Damit stellt sich die Frage, welche Umgebung f�r denLadungstransport in Peptiden g�nstig ist. Wie k�rzlich gezeigtwurde, bieten Lipide eine derartige Umgebung.[103] Eine Re-duzierung des Signal-/Ladungstransports in Proteinen in einerstark einschr�nkenden biologischen Umgebung k2nnte zueiner nennenswerten Unterdr�ckung biologischer Funktio-nen f�hren.
E. W. Schlag et al.Aufs�tze
3270 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273

Das hier beschriebene Modell liefert zusammen mit denbeschriebenen MD-Rechnungen ein Gesamtbild f�r dieChemie einfacher Peptide �ber große Reichweiten; dies istvon besonderem Interesse f�r das Verst�ndnis von lang-reichweitigen Reaktionen, bei denen konventionelle Modelleder Reaktionskinetik versagen. Die grundlegende, �ußerstschnelle RC-�bertragung wird in Experimenten zu Ausbeu-te[1,3] und der damit verbundenen Zeitskala[107] f�r ein iso-liertes Molek�l sowie in den Femtosekundenbewegungeneinfacher Peptide beobachtet. Das Modell erkl�rt die hoheEffizienz in der Gasphase ebenso wie die stark reduzierteEffizienz inWasser, wobei tats�chlich Effizienzwerte erhaltenwerden, wie sie f�r den Elektronentransport von Peptiden inWasser beobachtet wurden. Dies ist ein erstes Modell der RC-�bertragung, die in Peptiden (und allgemein in biologischenSystemen) f�r den Transport von Ladung und Reaktivit�t�ber große Entfernungen verantwortlich ist. Ein solcherProzess l�sst sich kaum mit Theorien zur Kinetik kleinerMolek�le in Einklang bringen.
9. Nachwort
Manifestiert sich dieser schnelle Ladungstransport auch inweiteren Proteinbewegungen? Angesichts der Bedeutungvon Ladungen in ribosomalen Proteinen k2nnte man anneh-men, dass dieses Ph�nomen hier ebenfalls auftritt. DieLadung w�rde dann den Sub-Pikosekunden-Energietransferin einer Dom�ne begrenzter Gr2ße bis hin zu einer Verbin-dungsstelle, z.B. einer Wasserstoffbr�cke, erm2glichen. DieEinstellung des Gleichgewichts innerhalb dieser Dom�new�re somit bis zu dieser Wasserstoffbr�cke sehr schnell.Dieser Prozess w�re f�r eine sehr kleine Zahl von Restenbeg�nstigt, z.B. f�r eine a-Helix, und w�rde auf diese Weisederen Bildung beg�nstigen; aus dieser a-Helix k2nnen sichdann weitere Bewegungen und die Bildung gr2ßerer Dom�-nen mit wiederum neuen Wasserstoffbr�cken ergeben. Es istbekannt, dass die Wasserstoffbr�cke nicht station�r ist, son-dern sich mit einer Durchlasszeit von etwa 10 ps 2ffnet,[15] wasdie Kopplung an die n�chste Dom�ne erm2glicht, die dann zueinem neuen Zustand relaxieren kann. Damit haben wir einSystem von Dom�nen variierender Gr2ße, die sich jeweils ineinem inneren Gleichgewicht befinden und �ber sich 2ff-nende und schließende Durchl�sse mit einer Uffnungszeitvon 10 ps in zuf�lligen Intervallen miteinander koppeln.Diese Dom�nen k2nnen dann großamplitudige strukturelleUmordnungen durchlaufen. Die optimalen langreichweitigenBewegungen solcher Dom�nen finden viel leichter statt alsdie Bewegungen aller Einzelatome,[90,108–111] was zu schnellenGleichgewichten lokaler Strukturen f�hrt, die dann in dielangsameren Ger�stbewegungen einbezogen werden.
Diese Arbeit wurde von NSC und DAAD im Rahmen einesdeutsch-taiwanesischen Programms unterst$tzt. Ein Teil derForschung wurde im Environmental Molecular Sciences La-boratory ausgef$hrt, einer nationalen wissenschaftlichen Be-nutzereinrichtung, die vom Department of Energy6s Office ofBiological and Environmental Research gef)rdert wird undsich am Pacific Northwest National Laboratory befindet.
Großer Dank gilt auch dem Fonds der Chemischen Industrief$r die finanzielle Unterst$tzung. Wir danken den ProfessorenSaykally und Nitzan f$r das Lesen des Manuskripts und denDoktoren Baranov und Schanen sowie Professor Weinkauf f$rzahlreiche Diskussionen.
Eingegangen am 25. April 2006,ver�nderte Fassung am 18. Oktober 2006Online ver2ffentlicht am 20. M�rz 2007
�bersetzt von Dr. Ines Sprung, Edinburgh
[1] R. Weinkauf, P. Schanen, D. Yang, S. Soukara, E. W. Schlag, J.Phys. Chem. 1995, 99, 11255.
[2] R. Weinkauf, P. Schanen, A. Metsala, E. W. Schlag, M. B�rgle,H. Kessler, J. Phys. Chem. 1996, 100, 18567.
[3] F. Remacle, R. D. Levine, E. W. Schlag, R. Weinkauf, J. Phys.Chem. A 1999, 103, 10149.
[4] F. Remacle, R. Weinkauf, D. Steinitz, K. L. Kompa, R. D.Levine, Chem. Phys. 2002, 281, 363.
[5] R. Weinkauf, E. W. Schlag, T. J. Martinez, R. D. Levine, J. Phys.Chem. 1997, 101, 7702.
[6] L. Y. Baranov, E. W. Schlag, Z. Naturforsch. A 1999, 54, 387.[7] F. Remacle, R. D. Levine, Proc. Natl. Acad. Sci. USA 2006, 103,
6793.[8] E. W. Schlag, S. Y. Sheu, D. Y. Yang, H. L. Selzle, S. H. Lin,
Proc. Natl. Acad. Sci. USA 2000, 97, 1068.[9] E. W. Schlag, D. Y. Yang, S. Y. Sheu, H. L. Selzle, S. H. Lin,
P. M. Rentzepis, Proc. Natl. Acad. Sci. USA 2000, 97, 9849.[10] E. W. Schlag, S. Y. Sheu, D. Y. Yang, H. L. Selzle, S. H. Lin, J.
Phys. Chem. B 2000, 104, 7790.[11] S. Sheh Yi, E. W. Schlag, D.-Y. Yang, H. L. Selzle, J. Phys.
Chem. A 2001, 105, 6353.[12] S. Y. Sheu, D. Y. Yang, H. L. Selzle, E. W. Schlag, J. Phys.
Chem. A 2002, 106, 9390.[13] S. Y. Sheu, D. Y. Yang, H. L. Selzle, E. W. Schlag, Eur. Phys. J.
D 2002, 5, 557.[14] E. W. Schlag, S. Y. Sheu, H. L. Selzle, D. Y. Yang, J. Chin.
Chem. Soc. 2006, 53, 265.[15] S. Y. Sheu, D. Y. Yang, H. L. Selzle, E. W. Schlag, Proc. Natl.
Acad. Sci. USA 2003, 100, 12683.[16] R. D. Gorbunov, D. S. Kosov, G. Stock, J. Chem. Phys. 2005,
122, 4904.[17] R. E. Holmin, P. J. Dandliker, J. K. Barton,Angew. Chem. 1997,
109, 2830; Angew. Chem. Int. Ed. Engl. 1997, 36, 2714.[18] C. Levinthal, J. Chim. Phys. 1968, 65, 44.[19] C. Levinthal in Mossbauer Spectroscopy in Biological Systems,
Proceedings of a Meeting Held at Allerton House, Monicello, IL(Hrsg.: J. T. P. De Brunner, E. Munck), University of IllinoisPress, Urbana, 1969, S. 22.
[20] E. Rios, G. Pizarro, E. Stefani, Annu. Rev. Physiol. 1992, 54,109.
[21] C. K. Mathews, K. E. v. Holde, K. G. Ahern, Biochemistry,Bd. 15, Addison Wesley Longman, New York, 2000.
[22] S. L. Mayo, W. R. Ellis, Jr., R. J. Crutchley, H. B. Gray, Science1986, 233, 948.
[23] L. M. Utschig, M. C. Thurnauer, Acc. Chem. Res. 2004, 37, 439.[24] M. Hervas, J. A. Navarro, M. A. D. La Rosa, Acc. Chem. Res.
2003, 36, 798.[25] C. J. Murphy, M. R. Arkin, Y. Jenkins, N. D. Ghatlia, S. H.
Bossmann, N. J. Turro, J. K. Barton, Science 1993, 262, 1025.[26] H. B. Gray, J. R. Winkler, Annu. Rev. Biochem. 1996, 65, 537.[27] R. A.Malak, Z. Gao, J. F.Wishart, S. S. Isied, J. Am. Chem. Soc.
2004, 126, 13888.[28] H. B. Gray, J. Halpern, Proc. Natl. Acad. Sci. USA 2005, 102,
3533.
Ladungstransport in PeptidenAngewandte
Chemie
3271Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

[29] S. S. Skourtis, I. A. Balabin, T. Kawatsu, D. N. Beratan, Proc.Natl. Acad. Sci. USA 2005, 102, 3552.
[30] O. Farver, G. W. Canters, I. van Amsterdam, I. Pecht, J. Phys.Chem. A 2003, 107, 6757.
[31] H. B. Gray, J. R. Winkler, Q. Rev. Biophys. 2003, 36, 341.[32] D. Laage, J. T. Hynes, Science 2006, 311, 832.[33] H. B. Gray, J. R. Winkler, Proc. Natl. Acad. Sci. USA 2005, 102,
3534.[34] B. G. Gerstman, P. P. Chapagain, J. Chem. Phys. 2005, 123,
054901.[35] J. R. Winkler, H. B. Gray, J. Biol. Inorg. Chem. 1997, 2, 399.[36] A. Ponce, H. B. Gray, J. R. Winkler, J. Am. Chem. Soc. 2000,
122, 8187.[37] N. Borovok, A. B. Kotlyar, I. Pecht, L. K. Skov, O. Farver,
FEBS Lett. 1999, 457, 277.[38] O. Farver, J. Zhang, Q. Chi, I. Pecht, J. Ulstrup, Proc. Natl.
Acad. Sci. USA 2001, 98, 4426.[39] S. Glasstone, K. J. Laidler, H. Eyring, The Theory of Rate
Processes, McGraw-Hill Book Company, New York, 1941.[40] K. J. Laidler, Chemical Kinetics, 3. Aufl., Harper & Row, New
York, 1987.[41] K. A. Holbrook, M. J. Pilling, S. H. Robertson, Unimolecular
Reactions, Wiley, New York, 1996.[42] T. Baer, W. L. Hase, Unimolecular Reaction Dynamics: Theory
and Experiments, Oxford University Press, Oxford, 1996.[43] B. S. Rabinovitch, E. W. Schlag, K. B. Wiberg, J. Chem. Phys.
1958, 28, 504.[44] B. S. Rabinovitch, E. Tschukow-Roux, E. W. Schlag, J. Am.
Chem. Soc. 1959, 81, 1081.[45] E. W. Schlag, B. S. Rabinovitch, J. Opt. Soc. Am. 1960, 82, 5996.[46] Y. Hu, B. Hadas, M. Davidovitz, B. Balta, C. Lifshitz, J. Phys.
Chem. A 2003, 107, 6507.[47] N. V. Dokholyan, Curr. Opin. Struct. Biol. 2006, 16, 79.[48] R. Zwanzig, A. Szabo, B. Bagchi, Proc. Natl. Acad. Sci. USA
1992, 89, 20.[49] J. T. Ngo, J. Marks, M. Karplus, in The Protein Folding Problem
and Tertiary Structure Prediction (Hrsg.: J. K. Merz, S. Le-Grand), Birkhauser, Boston, 1994, S. 433.
[50] Y. Zhou, M. Karplus, Nature 1999, 401, 400.[51] M. Gruebele, P. G. Wolynes, Acc. Chem. Res. 2004, 37, 261.[52] J. S. Baskin, L. Banares, S. Pedersen, A. H. Zewail, J. Am.
Chem. Soc. 1996, 118, 11920.[53] G. M. Stewart, J. D. McDonald, J. Chem. Phys. 1983, 78, 3907.[54] T. Kulp, R. Ruoff, G. Stewart, J. D. McDonald, J. Chem. Phys.
1984, 80, 5359.[55] R. A. Marcus, J. Electroanal. Chem. 2000, 483, 2.[56] F. D. Lewis, R. L. Letsinger, M. R. Wasielewski, Acc. Chem.
Res. 2001, 34, 159.[57] R. Langen, I. J. Chang, J. P. Germanas, J. H. Richards, J. R.
Winkler, H. B. Gray, Science 1995, 268, 1733.[58] P. C. P. de Andrade, J. N. Onuchic, J. Chem. Phys. 1998, 108,
4292.[59] J. N. Onuchic, D. N. Beratan, J. Chem. Phys. 1990, 92, 722.[60] D. N. Beratan, J. N. Onuchic, J. N. Hopfield, J. Chem. Phys.
1987, 88, 4488.[61] S. Woutersen, Y. Mu, G. Stock, P. Hamm, Proc. Natl. Acad. Sci.
USA 2001, 98, 11245.[62] S. Y. Sheu, J. Chem. Phys. 2005, 122, 104905.[63] J. M. Seminario, L. Yan, Y. Ma, J. Phys. Chem. A 2005, 109,
9712.[64] M. R. Betancourt, J. Skolnick, J. Mol. Biol. 2004, 342, 635.[65] R. Englman, J. Jortner, Mol. Phys. 1970, 18, 145.[66] J. Gumbart, Y. Wang, A. Aksimentiev, E. Tajkhorshid, K.
Schulten, Curr. Opin. Stuct. Biol. 2005, 15, 423.[67] M. L. Tan, I. Balabin, J. N. Onuchic, Biophys. J. 2004, 86, 1813.
[68] Y. Zhou, C. Oostenbrink, A. Jongejan, W. F. van Gunsteren,W. R. Hagen, S. W. de Leeuw, J. A. Jongejan, J. Comput. Chem.2005, 26, 857.
[69] B. M. Hoffman, L. M. Celis, D. A. Cull, A. D. Patel, J. L. Sei-fert, K. E.Wheeler, J. Wang, J. Yao, I. V. Kurnikov, J. M. Nocek,Proc. Natl. Acad. Sci. USA 2005, 102, 3564.
[70] O. Miyashita, M. Y. Okamura, J. N. Onuchic, Proc. Natl. Acad.Sci. USA 2005, 102, 3558.
[71] E. Yavin, A. K. Boal, E. D. A. Stemp, E. M. Boon, A. L. Liv-ingstone, V. L. OWShea, S. S. David, J. K. Barton, Proc. Natl.Acad. Sci. USA 2005, 102, 3546.
[72] R. H. Goldsmith, L. E. Sinks, R. F. Kelley, L. J. Betzen, W. Liu,E. A. Weiss, M. A. Ratner, M. R. Wasielewski, Proc. Natl.Acad. Sci. USA 2005, 102, 3540.
[73] S. Trebst, M. Troyer, U. H. E. Hansmann, J. Chem. Phys. 2006,124, 174903.
[74] S. H. White, G. von Heijne, Curr. Opin. Struct. Biol. 2005, 15,378.
[75] M. Kouza, M. S. Li, E. P. OWBrian, Jr., C. K. Hu, D. Thirumalai,J. Phys. Chem. A 2006, 110, 671.
[76] G. E. Sims, I. G. Choi, S. H. Kim, Proc. Natl. Acad. Sci. USA2005, 102, 618.
[77] F. J�hnig, Proc. Natl. Acad. Sci. USA 1983, 80, 3691.[78] G. B. Schuster, U. Landman, Top. Curr. Chem. 2004, 236, 139.[79] G. Luo, I. Andricioaci, X. S. Xie, M. Karplus, J. Phys. Chem. B
2006, 110, 9363.[80] A. van der Vaart, M. Karplus, J. Chem. Phys. 2005, 122, 114903.[81] S. Chowdhury, H. Lei, Y. Duan, J. Phys. Chem. 2005, 109, 9073.[82] D. Tsivlin, V. May, Chem. Phys. Lett. 2005, 408, 360.[83] K. W. Plaxco, K. T. Simans, D. Baker, J. Mol. Biol. 1998, 277,
985.[84] M. Karplus, J. A. McCammon, Nat. Struct. Biol. 2002, 9, 646.[85] A. V. Morozov, K. Tsemekhman, D. Baker, J. Phys. Chem. B
2006, 110, 4503.[86] I. H. Lee, S. Y. Kim, J. Lee, Chem. Phys. Lett. 2005, 412, 307.[87] E. G. Petrov, V. May, J. Chem. Phys. 2004, 120, 4441.[88] X. Q. Li, Y. Yan, J. Chem. Phys. 2001, 115, 4169.[89] M. Bixon, J. Jortner, J. Chem. Phys. 1997, 107, 5154.[90] H. Frauenfelder, B. H. McMahon, Ann. Phys. 2000, 9, 655.[91] J. Kim, A. Stuchebrukhov, J. Phys. Chem. B 2000, 104, 8606.[92] M. S. Gutowski, pers2nliche Mitteilung.[93] B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S.
Swaminathan, M. Karplus, J. Comput. Chem. 1983, 4, 187.[94] J. D. Rynbrandt, B. S. Rabinovitch, J. Phys. Chem. 1971, 75,
2164.[95] I. Oref, B. S. Rabinovitch, Acc. Chem. Res. 1979, 12, 166.[96] A. L. Malinovsky, Y. S. Doljikov, A. A. Makarov, N. D. D.
Ogurok, E. A. Ryabov, Chem. Phys. Lett. 2006, 419, 511.[97] F. Polo, S. Antonello, F. F. C. Toniolo, F. Maran, J. Am. Chem.
Soc. 2005, 127, 492.[98] W. Cui, Y. Hu, C. Lifshitz, Eur. Phys. J. D 2002, 5, 565.[99] E. Riedle, T. Weber, U. Schubert, H. J. Neusser, E. W. Schlag, J.
Chem. Phys. 1990, 93, 967.[100] E. W. Schlag, J. Grotemeyer, R. D. Levine, Chem. Phys. Lett.
1992, 190, 521.[101] Gaussian98 (RevisionA.11), M. J. Frisch, G. W. Trucks, H. B.
Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G.Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant,S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C.Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B.Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski,G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K.Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J.Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P.Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T.Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C.Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W.
E. W. Schlag et al.Aufs�tze
3272 www.angewandte.de � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2007, 119, 3258 – 3273

Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S.Replogle, J. A. Pople, Gaussian, Inc., Pittsburgh, PA, 1998.
[102] S. H. Lin, J. Chem. Phys. 1970, 53, 3766.[103] S. Y. Sheu, D. Y. Yang, H. L. Selzle, E. W. Schlag, unver2f-
fentlichte Ergebnisse.[104] G. Maruccio, A. Biasco, P. Visconti, A. Bramanti, P. P. Pompa,
F. Caabi, R. Cingolani, R. Rinaldi, S. Corni, R. Di Felice, E.Molinari, M. P. Verbeet, G. W. Canters, Adv. Mater. 2005, 17,816.
[105] D. Leys, N. S. Scrutton, Curr. Opin. Struct. Biol. 2004, 14, 642.
[106] S. Sek, K. Swiatek, A. Misicka, J. Phys. Chem. B 2005, 109,23121.
[107] L. Lehr, T. Horneff, R. Weinkauf, E. W. Schlag, J. Phys. Chem.A 2005, 109, 8074.
[108] I. N. Berezovsky, E. N. Trifonov, J. Biomol. Struct. Dyn. 2002,20, 5.
[109] C. M. Dobson, Nature 2003, 426, 884.[110] A. R. Panchenko, Z. Luthey-Schulten, P. G. Wolynes, Proc.
Natl. Acad. Sci. USA 1996, 93, 2008.[111] N. Ferguson, A. R. Fersht, Curr. Opin. Struct. Biol. 2003, 13, 75.
Ladungstransport in PeptidenAngewandte
Chemie
3273Angew. Chem. 2007, 119, 3258 – 3273 � 2007 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de






![Extramedulläre Osteosynthesen distaler Femurfrakturen- ein ... › download › pdf › 301531901.pdf · einfachen diaphysären Femurschaftfrakturen gezeigt werden [186]. 2.2.2 Direkte](https://img.pdfslide.org/doc/110x75/60c5341a3ae1e31613726b45/extramedullre-osteosynthesen-distaler-femurfrakturen-ein-a-download-a.jpg)