Embed Size (px)
Citation preview

Dynamik der Zuckertautomerie und ihr Einfluss
auf die Kinetik der MAILLARD-Reaktion
– Katalyse der nicht enzymatischen Bräunung –
vorgelegt von
Diplom-Lebensmittelchemiker
Martin Kaufmann
geboren in Fulda
von der Fakultät III – Prozesswissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
– Dr. rer. nat. –
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender Prof. Dr.-Ing. Frank-Jürgen Methner
Gutachter Prof. Dr. Lothar W. Kroh
Gutachter Prof. Dr. Clemens Mügge
Gutachter Prof. Dr. Dr.-Ing. Thomas Henle
Gutachter Prof. Dr. Ir. MAJS (Tiny) van Boekel
Tag der wissenschaftlichen Aussprache: 01. März 2018
Berlin 2018


I
Kurzzusammenfassung Die MAILLARD-Reaktion wird im Allgemeinen als aminokatalysierte Zuckerabbaureaktion
verstanden. Dabei kondensiert ein primäres bzw. sekundäres Amin mit einem reduzierenden Zucker
und wird im weiteren Reaktionsverlauf zunächst unverändert wieder abgespalten. Gleichzeitig kommt
es zu Veränderungen am Zuckergrundgerüst und zu einer Färbung der Reaktionslösung. Welcher
elementare Reaktionsschritt dabei von der Aminofunktion katalysiert wird, ist bislang jedoch noch
nicht bekannt. Daneben berichten zahlreiche Publikationen von einer beschleunigten
Bräunungsreaktion in Anwesenheit diverser Carbonsäuren. Die Mehrzahl der Untersuchungen von
MAILLARD-Reaktionssystemen basiert auf dem Einsatz von Aminosäuren. Daher konnte bislang nicht
zwischen den jeweils spezifischen Einflüssen der Amino- bzw. der Carboxylfunktion auf den Verlauf
und die Kinetik der nicht enzymatischen Bräunung differenziert werden.
Um diese Frage grundlegend zu klären, wurden NMR-spektroskopische
Strukturuntersuchungen an D-Glucose, D-Tagatose sowie vor allem an Derivaten der D-Fructose
vorgenommen. Darauf aufbauend erfolgte die erstmalige systematische Durchführung selektiver
Sättigungstransfer-NMR-Experimente in Abhängigkeit der Temperatur und des pH-Wertes zur
fundamentalen Bestimmung der jeweiligen Anomerensysteme. Die Interpretation der
NMR-spektroskopischen Daten erlaubt in Kombination mit den aus der Durchführung von
Modellreaktionen erhaltenen Ergebnissen ein detailliertes Verständnis der Mechanismen der frühen
und intermediären Phase der MAILLARD-Reaktion. Die spezifischen katalytischen Einflüsse sowohl der
Amino- als auch der Carboxylfunktion wurden am Beispiel der Dynamik verschiedener
Tautomerisierungsprozesse von Zuckerderivaten erstmals konzeptionell verstanden und in einer
allgemeingültigen Theorie der Katalyse nicht enzymatischer Bräunungsreaktionen zusammengefasst.
Diese beschreibt die einzelnen Schritte der Reaktionskaskade im Wesentlichen auf Grundlage von
aufeinanderfolgenden Prozessen des Protonentransfers. Die eigentliche Katalyse besteht in einer
beschleunigten Dynamik dieses Transfers durch schwache Säuren und deren konjugierte Basen
(allgemeine Säure-Base-Katalyse), der in ausschließlicher Anwesenheit von Lösungsmitteln wie Wasser
oder Ethanol aufgrund deren extremer Säurekonstanten nur ineffizient erfolgt. Da es im Zuge der nicht
enzymatischen Bräunung unter anderem zur Bildung verschiedener Carbonsäuren kommt, handelt es
sich somit sowohl bei der Karamellisierung als auch bei der MAILLARD-Reaktion um autokatalysierte
Reaktionen. Der Einfluss der Aminofunktion ergibt sich demgegenüber durch deren Teilnahme an
kooperativen Wasserstoffbrückenbindungen sowie durch deren Beeinflussung der 𝑝𝐾𝑠-Werte
räumlich benachbarter funktioneller Gruppen.
Es konnte gezeigt werden, dass die milieuabhängige Reaktivität eines reduzierenden Zuckers
mit der Dynamik seiner Ring-Ketten-Tautomerie korreliert. In diesem Zusammenhang wird ein
Parameter präsentiert, der die Berechnung der relativen Reaktivität verschiedener Systeme
reduzierender Zucker erlaubt. Eine Reaktivitätsabschätzung von Zuckern anhand dieses Parameters
steht nicht nur im Einklang mit den im Rahmen dieser Arbeit durchgeführten Experimenten, sondern
auch mit den Beobachtungen zahlreicher weiterer Publikationen. Es zeigt sich damit, dass die
Gültigkeit des Parameters nicht nur auf einfache Modellsysteme beschränkt ist, sondern ebenso für
technologische Zwecke genutzt und potentiell auch auf physiologische Systeme ausgeweitet werden
kann.

II
Abstract The MAILLARD reaction is generally known as amino catalyzed sugar degradation. During this
process, primary and secondary amines condense with the carbonyl function of a reducing sugar.
Subsequently they regenerate via elimination and hydrolysis. Simultaneously, the reducing sugar
becomes chemically modified while the color of the reaction system increases. Up to now, the catalytic
influence of the amino function is not fully understood. Aside, many researchers observed an
acceleration of browning in the presence of carboxylic acids. The majority of analyzed MAILLARD
reaction systems is based on mixtures of reducing sugars and amino acids. Thus, it was not yet possible
to differentiate between the catalytic influence of carboxyl and amino functions on the progress and
the kinetics of non-enzymatic browning reactions.
The present work concerns this question based on NMR spectroscopic investigations
respecting the structures of D-glucose and D-tagatose as well as derivatives of D-fructose.
Subsequently, the respective anomeric systems were analyzed applying selective saturation transfer
NMR experiments in dependence on temperature and pH. The interpretation of the acquired NMR
spectroscopic data in combination with the performance and analysis of model reactions permits a
detailed comprehension of the mechanisms of the early and intermediate stage of MAILLARD reaction.
Examining the dynamics of different tautomeric processes, the specific catalytic influences of amino
as well as carboxyl functions were conceptually understood for the first time. It could be shown that
the observed effects are universally valid. Hence, a novel theory of the catalysis of non-enzymatic
browning is proposed. This theory describes non-enzymatic browning reactions essentially based on
consecutive steps of proton transfer. The catalytic mechanism is due to the accelerated dynamic of
proton transfer processes via weak acids and their conjugated bases (general acid base catalysis). As a
result of their extreme acidity constants, conventional solvents such as water and primary alcohols
catalyze these dynamics only inefficiently. Since carboxylic acids are formed during non-enzymatic
browning reactions, caramelization as well as MAILLARD reaction are autocatalyzed. In contrast, amino
functions influence the kinetics of non-enzymatic browning by participating in the formation of
co-operative hydrogen bond networks as well as by altering the 𝑝𝐾𝑎 values of adjacent functional
groups.
It could be shown that the milieu dependent stability of reducing sugars is correlated with
the dynamic of their anomerization. In that context, a parameter is proposed that allows approximate
calculations of the milieu dependent reactivity of reducing sugars. Calculations of this parameter are
not only consistent with the experimental data the present work is based on, but likewise with the
observations of numerous publications. Thus, the parameter is valid regarding model reaction systems
and can as well be applied for technological as well as physiological purposes.

III

IV
Publikationen Teile dieser Arbeit wurden bereits in wissenschaftlichen Fachzeitschriften als Originalarbeiten publiziert. Diese Veröffentlichungen sind mit * gekennzeichnet und werden im Folgenden nicht mehr zitiert.
Zeitschriftenbeiträge
KAUFMANN, M.; HAASE, P. T.; MÜGGE, C.; KROH, L. W.: Milieu Dependence of Isomeric Composition of
1-Deoxy-D-erythro-hexo-2,3-diulose in Aqueous Solution Determined by High-Resolution NMR
Spectroscopy. Carbohydr. Res. 2012, 364, 15–21.
KAUFMANN, M.; HAASE, P. T.; MÜGGE, C.; KROH, L. W.: Milieu Dependence of Isomeric Composition of
D-arabino-Hexo-2-ulose in Aqueous Solution Determined by High-Resolution NMR Spectroscopy.
J. Agric. Food Chem. 2013, 61, 10220–10224.
WILKER, D.; KAUFMANN, M.; KANZLER, C.; HAASE, P. T.; KEIL, C.; KROH, L. W.: Über Struktur-Wirkungs-
Beziehungen von α-Dicarbonylverbindungen. Lebensmittelchemie. 2014, 68, 143–149.
KAUFMANN, M.; MÜGGE, C.; KROH, L. W.: Theory of the Milieu Dependent Isomerisation Dynamics of
Reducing Sugars applied to D-Erythrose. Carbohydr. Res. 2015, 418, 89–97.*
KAUFMANN, M.; MÜGGE, C.; KROH, L. W.: Kohlenhydratisomerensysteme - Untersuchung mittels
dynamischer NMR-Spektroskopie. Deutsche Lebensmittel-Rundschau (DLR). 2016, 112, 94–98.*
KAUFMANN, M.; MEISSNER, P. M.; PELKE, D.; MÜGGE, C.; KROH, L. W.: Structure–Reactivity Relationship of
AMADORI Rearrangement Products compared to Related Ketoses. Carbohydr. Res. 2016, 428, 87–99.*
WEGENER, S.; KAUFMANN, M.; KROH, L. W.: Influence of L-Pyroglutamic Acid on the Color Formation
Process of Non-Enzymatic Browning Reactions. Food Chem. 2017, 232, 450–454.*
HANSCHEN, F. S.; KAUFMANN, M.; KUPKE, F.; HACKL, T.; KROH, L. W.; ROHN, S.; SCHREINER, M.: Brassica
Vegetables as Sources of Epithionitriles: Novel Secondary Products formed during Cooking. Food Chem.
2018, 245, 564–569.
KAUFMANN, M.; KRÜGER, S.; MÜGGE, C.; KROH, L. W.: Simultaneous formation of 3-deoxy-D-threo-hexo-2-ulose and 3-deoxy-D-erythro-hexo-2-ulose during the degradation of D-glucose derived AMADORI rearrangement products: Mechanistic considerations. Carbohydr. Res. 2018, 458–459, 44–51.*
KAUFMANN, M.; MÜGGE, C.; KROH, L. W.: NMR Analyses of complex D-Glucose Anomerization (submitted).*
KAUFMANN, M.; KRÜGER, S.; MÜGGE, C.; KROH, L. W.: General acid/base Catalysis of Sugar Anomerization (submitted).*
FECHNER, J.; KAUFMANN, M.; HERZ, C.; EISENSCHMIDT, D.; LAMY, E.; KROH, L. W.; HANSCHEN, F. S.: The major Glucosinolate Hydrolysis Product in Rocket (Eruca sativa L.), Sativin, is 1,3-Thiazepane-2-thione: Elucidation of Structure, Bioactivity, and Stability compared to other Rocket Isothiocyanates (submitted).
KAUFMANN, M.; SCHLINGELHOF, X. A.; MÜGGE, C.; KROH, L. W.: Tautomerization of AMADORI Rearrangement Products (in preparation).*

V
Tagungsvorträge
HAASE, P. T.; KAUFMANN, M.; KROH, L. W.:
„D-Glucosone and 1-Deoxyglucosone under MAILLARD-Conditions”
11th International Symposium on the MAILLARD Reaction, Nancy (Frankreich), 16.-20. September 2012.
KAUFMANN, M.; HAASE, P. T.; MÜGGE, C.; KROH, L. W.:
„NMR-spektroskopische Charakterisierung isomerer Zuckerabbauprodukte“
Arbeitstagung des Regionalverbandes Nordost der LChG, Berlin, 28. März 2014.
BAYER, K.; KAUFMANN, M.; HORNEMANN, A.; BECKHOFF, B.; KROH, L. W.:
„Spektroskopische Charakterisierung von Melanoidinen“
Arbeitstagung des Regionalverbandes Nordost der LChG, Berlin, 07. März 2016.
SCHLINGELHOF, X. A.; KAUFMANN, M.; MÜGGE, C.; KROH, L. W.:
„Vergleich der Isomerisierung von D-Fructose-Derivaten mittels dynamischer NMR-Spektroskopie“
Arbeitstagung des Regionalverbandes Nordost der LChG, Berlin, 07. März 2016.
KAUFMANN, M.; MÜGGE, C.; KROH, L. W.:
„Structure and Dynamics of D-Fructose and Related AMADORI Derivatives”
FoodMR 2016, Karlsruhe, 08. Juni 2016.
KAUFMANN, M.; KROH, L. W.:
„Influence of Isomerization on MAILLARD Reaction Kinetics”
Young AGErs Symposium – MAILLARD reaction in Food and in vivo, Dresden, 21-22. Juli 2016.
KAUFMANN, M.; MÜGGE, C.; KROH, L. W.:
„Katalyse der Dynamik von Zuckertautomerien und ihr Einfluss auf die Kinetik der MAILLARD-Reaktion“
46. Deutscher Lebensmittelchemikertag, Würzburg, 25.-27. September 2017.
KAUFMANN, M.; MÜGGE, C.; KROH, L. W.:
„Structure and Dynamics of D-Fructose and Related AMADORI Derivatives”
EuroFoodChem XIX Conference, Budapest (Ungarn), 04.-06. Oktober 2017.

VI
Poster
KAUFMANN, M.; HAASE, P. T.; MÜGGE, C.; KROH, L. W.:
„Untersuchungen zur Struktur und Isomerisierung von α-Dicarbonylverbindungen mittels
hochauflösender NMR-Spektroskopie“
42. Deutscher Lebensmittelchemikertag, Braunschweig, 16.-18. September 2013.
KAUFMANN, M.; HAASE, P. T.; KROH, L. W.; MÜGGE, C.:
„Milieu Dependence of Isomeric Composition of 1-Deoxy-D-erythro-hexo-2,3-diulose in Aqueous
Solution Determined by High-Resolution NMR Spectroscopy”
36th Discussion Meeting of the Magnetic Resonance Spectroscopy division of the German Chemical
Society (GDCh) – 'Advanced Magnetic Resonance - Methods and Applications', Berlin, 29. September
bis 02. Oktober 2014.
SCHALLSCHMIDT, K.; HALAMA, K. M. T.; KAUFMANN, M.; MÜGGE, C.; KROH, L. W.; BECKER, R.; NEHLS, I.:
„Charakterisierung flüchtiger Nitrosothiole“
9. Interdisziplinäres Doktorandenseminar der Bundesanstalt für Materialforschung und –prüfung
(BAM), Berlin, 22.-24. Februar 2015.
KAUFMANN, M.; KROH, L. W.:
„Eins für alles - MARS in der Lebensmittelanalytik“
2. Institutskolloquium des Instituts für Lebensmitteltechnologie und Lebensmittelchemie – „Moderne
Technologien und gesunde Lebensmittel“, Berlin, 20. April 2015.
KAUFMANN, M.; MÜGGE, C.; KROH, L. W.:
„Die Theorie der milieu-abhängigen Dynamik der Isomerisierung reduzierender Zucker angewandt auf
D-Erythrose“
44. Deutscher Lebensmittelchemikertag, Karlsruhe, 14.-16. September 2015.
BAYER, K.; KAUFMANN, M.; HORNEMANN, A.; BECKHOFF, B.; KROH, L. W.:
„Characterisation of Melanoidins and their Metal Complexes applying EPR and FTIR Spectroscopic
Techniques”
9th Conference on "Instrumental Methods of Analysis – Modern Trends and Applications", Kalamata
(Griechenland), 20.-24. September 2015.
KAUFMANN, M.; MEISSNER, P. M.; PELKE, D.; MÜGGE, C.; KROH, L. W.:
„Struktur und Dynamik von D-Fructose und ausgewählten AMADORI-Derivaten“
45. Deutscher Lebensmittelchemikertag, Freising-Weihenstephan, 12.-14. September 2016.

VII
Meiner Familie

VIII
Text aus: ASMUS omnia sua SECUM portans, oder Sämmtliche Werke des Wandsbecker Bothen, 4. Theil, 1. Auflage, Wandsbeck, 1782, Seite 57.

IX
Danksagung Die vorliegende Arbeit entstand am Institut für Lebensmitteltechnologie und
Lebensmittelchemie der Technischen Universität Berlin im Arbeitskreis von Herrn Prof. Dr. Lothar W.
Kroh (Fachgebiet Lebensmittelchemie und Analytik) im Zeitraum von Juli 2013 bis Januar 2018. Die
Durchführung der NMR-spektroskopischen Experimente erfolgte am Institut für Chemie der
HUMBOLDT-Universität zu Berlin in der AG NMR unter der Leitung von Herrn Prof. Dr. Clemens Mügge.
Mein Dank gilt meinem Doktorvater Herrn Prof. Dr. Lothar W. Kroh, der mir den Freiraum
schuf, eigene Forschungsansätze uneingeschränkt zu verfolgen, und mir in wissenschaftlichen
Gesprächen die Motivation gab, dabei auch unkonventionelle Ideen zu entwickeln. Gleichermaßen
ermöglichte er mir die Betreuung wissenschaftlicher Themengebiete, die weit abseits der hier
vorliegenden Arbeit angeordnet sind, sodass ich meine Erfahrungen in vielfältigen Bereichen (nicht
nur) der Lebensmittelchemie sammeln konnte.
Gleichermaßen danke ich meinem zweiten Mentor Herrn Prof. Dr. Clemens Mügge, der mir
das Vertrauen schenkte und mir freien Zugang zu allen verfügbaren NMR-Spektrometern gewährte.
Als mein Lehrmeister der NMR-Spektroskopie unterstützte und stärkte er mich in zahlreichen
fachlichen Gesprächen und vielen Stunden an den Spektrometern.
Besonders erfreut mich die Tatsache, dass die vorliegende Arbeit Schwerpunkte wichtiger
Forschungsthemen meiner beiden Mentoren vereint. Auf diese Weise habe ich einerseits viel aus
ihrem jeweiligen Erfahrungsschatz profitiert und meine Ideen andererseits in zahlreichen Diskussionen
gegenüber spitzfindigen und herausfordernden Fragen zu verteidigen lernen müssen. Vielen Dank
dafür. Das war – nicht nur für mich – immer eine ganz besondere Freude!
Herrn Prof. Dr. Ir. MAJS (Tiny) van Boekel von der Wageningen University & Research
(Niederlande) danke ich für die freundliche Bereitschaft der Begutachtung meiner Dissertation. Dies
freut mich insbesondere, da seine umfangreichen Untersuchungen zur Kinetik der nicht enzymatischen
Bräunung den Grundstein für die hier bearbeitete Fragestellung gelegt haben. Hartelijk bedankt!
Herrn Prof. Dr. Dr.-Ing. Thomas Henle von der Technischen Universität Dresden danke ich für
die kurzfristige Bereitschaft der Begutachtung meiner Dissertation. Ein besonderes Dankeschön gilt
ihm jedoch vor allem für die Unterstützung seiner Doktoranden und Post-Docs bei der Gründung der
YoungAGErs, deren inzwischen internationale Symposien zu einer nachhaltigen Vernetzung des
wissenschaftlichen Nachwuchses mit dem Schwerpunkt der MAILLARD-Reaktion ermöglichen.
Für ihre fachliche und technische Unterstützung danke ich allen Mitarbeitern der
Arbeitskreise von Herrn Prof. Dr. Kroh sowie Herrn Prof. Dr. Mügge. Vor allem ohne die technische
Unterstützung von Beate Beyerlein, Anette Berghäuser und Angela Thiesies wäre es im Laufe der Arbeit
wohl mehr als einmal zu größeren Verzögerungen gekommen. Insbesondere NMR-Spektrometer
haben vor direkten Vorgesetzten zunächst mehr Respekt als vor neuen Doktoranden.
Meinen Kollegen Alexandra Urbisch, Sandra Grebenteuch, Philipp Bruhns, Dr. Martin Doert,
Dr. Paul Haase, Dr. Clemens Kanzler und Steffen Wegener danke ich herzlich für ihre Hilfsbereitschaft
und die nette Arbeitsatmosphäre. Bei gemeinsamen Kaffeerunden und Ausflügen ist mehr als nur eine
Träne geflossen. Vor allem meiner Bürokollegin Alexandra danke ich für die gute gemeinsame Zeit.

X
Daniel Pelke, Philipp Meissner, Kai Halama, Kathrin Bayer, Xenia Schlingelhof, Teodor
Tchipilov, Sophie Krüger, Christoph Mertens und Leon Huder gilt mein Dank für ihre engagierte
Zusammenarbeit im Rahmen ihrer wissenschaftlichen Abschlussarbeiten, Forschungspraktika und
Masterarbeiten sowie als studentische Hilfskräfte. Danke für eure jeweiligen Forschungsbeiträge auch
auf Gebieten jenseits der nicht enzymatischen Bräunung.
Abschließend danke ich meiner Frau Kerstin und meinen Kindern Charlotte, Severin und
Kilian für ihre bedingungslose Unterstützung und die Freude und Abwechslung, die sie mir bereiten.
Gleichermaßen danke ich meinen Eltern und Schwiegereltern für ihren täglichen und nicht
selbstverständlichen Beistand. Die Arbeit steht auf eurem Fundament!

XI

XII
Inhaltsverzeichnis Kurzzusammenfassung ............................................................................................................................. I
Abstract ................................................................................................................................................... II
Publikationen .......................................................................................................................................... IV
Danksagung ............................................................................................................................................ IX
Inhaltsverzeichnis .................................................................................................................................. XII
Abkürzungsverzeichnis ......................................................................................................................... XIV
1 Einleitung .............................................................................................................................................. 1
2 Theoretischer Hintergrund ................................................................................................................... 2
2.1 Die MAILLARD-Reaktion ................................................................................................................... 2
2.1.1 D-Fructose ............................................................................................................................... 3
2.1.1.1 Strukturen und Dynamik der D-Fructose ......................................................................... 3
2.1.1.2 Formen der Isomerisierung ............................................................................................. 6
2.1.2 Die Aminokomponenten ........................................................................................................ 8
2.1.3 AMADORI- und HEYNS-Verbindungen ....................................................................................... 8
2.1.3.1 AMADORI- und HEYNS-CARSON-Umlagerung ....................................................................... 8
2.1.3.2 Strukturen und Dynamik der AMADORI-Verbindungen .................................................. 13
2.1.4 Reaktionen von Derivaten der D-Fructose ........................................................................... 16
2.2 NMR-Spektroskopie ..................................................................................................................... 22
2.2.1 Strukturaufklärung ............................................................................................................... 22
2.2.2 Quantitative NMR-Spektroskopie (qNMR) ........................................................................... 23
2.2.3 Methoden der dynamischen NMR-Spektroskopie (DNMR) ................................................. 26
2.3 Katalyse und mathematische Modellierung der Anomerisierung .............................................. 34
3 Zielstellung ......................................................................................................................................... 40
4 Ergebnisse und Diskussion ................................................................................................................. 42
4.1 Reaktivität von Derivaten der D-Fructose ................................................................................... 42
4.1.1 Kinetik des Abbaus von D-Fructose, Aminosäuren und AMADORI-Verbindungen ................. 42
4.1.2 Kinetik der Bildung von α-Dicarbonylverbindungen ............................................................ 46
4.1.3 Kinetik der Farbentwicklung bei Reaktionen von Derivaten der D-Fructose ....................... 53
4.2 Charakterisierung der Anomerensysteme .................................................................................. 56
4.2.1 Strukturaufklärung der Anomerensysteme.......................................................................... 57
4.2.2 Dynamik der 1,2-Enolisierung .............................................................................................. 68
4.2.3 Zwischenfazit der bisherigen Ergebnisse ............................................................................. 75
4.2.4 Zusammenhang zwischen Anomerisierung und Reaktivität ................................................ 76
4.2.5 Quantitative Zusammensetzung der Anomerensysteme..................................................... 80
4.2.6 Dynamik der Anomerisierung ............................................................................................... 84
5 Zusammenfassung ............................................................................................................................ 101

XIII
6 Experimenteller Teil ......................................................................................................................... 106
6.1 Synthesen der AMADORI-Verbindungen ..................................................................................... 106
6.1.1 Synthese von N-(1-Desoxy-D-fructos-1-yl)-L-alanin ............................................................ 106
6.1.2 Synthese von N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-alanin ................................................. 107
6.1.3 Synthese von N-(1-Desoxy-D-fructos-1-yl)-L-prolin ............................................................ 108
6.1.4 Synthese von N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-prolin .................................................. 109
6.1.5 Synthese von N-(1-Desoxy-D-fructos-1-yl)-glycin ............................................................... 110
6.1.6 Synthese von N-(1-Desoxy-D-fructos-1-yl)-β-alanin ........................................................... 111
6.1.7 Synthese von N-(1-Desoxy-D-fructos-1-yl)-γ-aminobuttersäure ........................................ 112
6.1.8 Regeneration des Ionentauschers ...................................................................................... 113
6.2 pH-konstante Modellreaktionen ............................................................................................... 113
6.2.1 Durchführung und Auswertung der UV/Vis-spektroskopischen Messungen .................... 114
6.2.2 Messung und Auswertung der Abbaukinetiken der D-Fructose......................................... 115
6.2.3 Messung und Auswertung der Abbaukinetiken der Aminosäuren .................................... 116
6.2.4 Messung und Auswertung der Abbaukinetiken der AMADORI-Verbindungen .................... 116
6.2.5 Messung und Auswertung der Kinetiken der α-Dicarbonylverbindungen ......................... 117
6.3 NMR-Spektroskopische Messungen .......................................................................................... 118
6.3.1 Strukturaufklärung ............................................................................................................. 118
6.3.2 Kalibrierung der Temperatur .............................................................................................. 119
6.3.3 Kalibrierung des pD-Wertes ............................................................................................... 120
6.3.4 Quantitative NMR-Spektroskopie (qNMR) ......................................................................... 121
6.3.5 Messung von Enolisierungsraten ....................................................................................... 121
6.3.6 Selektive Sättigungstransfer-NMR-Spektroskopie (SST-NMR) ........................................... 122
6.3.6.1 Frequenz-Sweep-Sättigungstransfer ........................................................................... 123
6.3.6.2 1H-SST-NMR ................................................................................................................. 124
6.3.6.3 13C-SST-NMR ................................................................................................................ 125
6.4 Potentiometrische Bestimmung der 𝑝𝐾𝑠-Werte....................................................................... 125
6.5 Polarimetrische Messungen ...................................................................................................... 126
6.6 Geräte ........................................................................................................................................ 126
6.7 Software .................................................................................................................................... 128
6.8 Chemikalien ............................................................................................................................... 129
Literaturverzeichnis .................................................................................................................................. i
Abbildungsverzeichnis ......................................................................................................................... xxvi
Tabellenverzeichnis .............................................................................................................................. xxx
Schemataverzeichnis ......................................................................................................................... xxxiii
Strukturenverzeichnis......................................................................................................................... xxxv
Anhang .............................................................................................................................................. xxxvi
Curriculum Vitae ................................................................................................................................... lviii

XIV
Abkürzungsverzeichnis 1-DG 1-Desoxyglucoson (1-Desoxy-D-erythro-hexos-2,3-diulose)
1D eindimensional
2D zweidimensional
3-DG 3-Desoxyglucoson (3-Desoxy-D-erythro-hexos-2-ulose)
AAF 2-Aminoacetylfuran
ACuSTiC aus dem Englischen approximated carbohydrate milieu stability time constant
für approximierte milieuabhängige Kohlenhydratstabilitätszeitkonstante
αF α-Furanose
AMV AMADORI-Verbindung
αP α-Pyranose
AS Aminosäure
AU-Programm Automationsprogramm
βF β-Furanose
βP β-Pyranose
BBI aus dem Englischen broadband inverse für Breitband invers
BBO aus dem Englischen broadband observe für Breitband beobachten
CIP CAHN-INGOLD-PRELOG
COSY aus dem Englischen correlation spectroscopy für Korrelationsspektroskopie
CU aus dem Englischen cooling unit für Kühleinheit
DAD aus dem Englischen diode array detector für Diodenarraydetektor
DEPT aus dem Englischen distortionless enhancement by polarization transfer für
Verzerrungsfreie Verstärkung durch Polarisationstransfer
DHHM Dihydrohydroxymaltol (3,5-Dihydroxy-6-methyl-2,3-dihydro-4H-pyran-4-on)
DNMR aus dem Englischen dynamic nuclear magnetic resonance für dynamische
kernmagnetische Resonanz
E Extinktion
EA Elementaranalyse
eq. Aus dem Englischen equivalents für Äquivalente
EU Europäische Union

XV
EXPNO aus dem Englischen experiment number für Experimentnummer
EXSY aus dem Englischen exchange spectroscopy für Austauschspektroskopie
FAla Fructosylalanin (N-(1-Desoxy-D-fructos-1-yl)-L-alanin)
FBala Fructosyl-β-alanin (N-(1-Desoxy-D-fructos-1-yl)-β-alanin)
FGaba Fructosyl-γ-aminobuttersäure (N-(1-Desoxy-D-fructos-1-yl)-γ-aminobuttersäure)
FGly Fructosylglycin (N-(1-Desoxy-D-fructos-1-yl)-glycin)
FID aus dem Englischen free induction decay für freien Induktionsabfall
FP Folgeprodukt
FPro Fructosylprolin (N-(1-Desoxy-D-fructos-1-yl)-L-prolin)
FQSSST aus dem Englischen frequency sweep selective saturation transfer für
Frequenzdurchlaufselektiven Sättigungstransfer
Fru D-Fructose
GC Gaschromatographie
gem geminal
GLUC D-Glucoson (D-arabino-Hexos-2-ulose)
HAF 2-Hydroxyacetylfuran
H/D-Austausch Wasserstoff/Deuterium-Austausch
HILIC aus dem Englischen hydrophilic interaction liquid chromatography für
hydrophile Wechselwirkungsflüssigchromatographie
HMBC aus dem Englischen heteronuclear multi-bond correlation für heteronukleare
Mehrfachbindungskorrelation
HMF Hydroxymethylfurfural (5-Hydroxymethyl-2-furaldehyd)
HMQC aus dem Englischen heteronuclear multi-quantum correlation für
heteronukleare Mehrquantenkorrelation
HPLC aus dem Englischen high performance liquid chromatography für
Hochleistungsflüssigchromatographie
HSQC aus dem Englischen heteronuclear single-quantum coherence für
heteronukleare Einzelquantenkohärenz
H/T-Austausch Wasserstoff/Tritium-Austausch
Int. Integral
IR Infrarot
lb aus dem Englischen line broadening factor für Linienverbreiterungsfaktor

XVI
MW Mittelwert
NMR aus dem Englischen nuclear magnetic resonance für kernmagnetische Resonanz
NOE aus dem Englischen nuclear OVERHAUSER enhancement für Kern-OVERHAUSER
Verstärkung bzw. nuclear OVERHAUSER effect für Kern-OVERHAUSER Effekt
NOESY aus dem Englischen nuclear OVERHAUSER enhancement spectroscopy für
Kern-OVERHAUSER verstärkte Spektroskopie
OPD ortho-Phenylendiamin (1,2-Diaminobenzen)
pI isoelektrischer Punkt
pL aus dem Englischen patch level für Revisionsnummer
PROCNO aus dem Englischen processing number für Prozessierungsnummer bzw.
Bearbeitungsnummer
qNMR aus dem Englischen quantitative nuclear magnetic resonance für quantitative
kernmagnetische Resonanz
RI aus dem Englischen refractive index für Brechungsindex
RP aus dem Englischen reversed phase für Umkehrphase
SET aus dem Englischen set endpoint titration für Titration auf vorgegebenen
Endpunkt
SEXSY aus dem Englischen selective exchange spectroscopy für selektive
Austauschspektroskopie
SIR aus dem Englischen selective inversion recovery für Signalrückentwicklung nach
selektiver Inversion
SST aus dem Englischen selective saturation transfer für selektiver
Sättigungstransfer
STW Standardabweichung
STD aus dem Englischen saturation transfer difference für
Sättigungstransferdifferenz
TOCSY aus dem Englischen total correlated spectroscopy für vollständig korrelierte
Spektroskopie
UV ultraviolett
vA virtuelles Anomer
Vis aus dem Englischen visible für sichtbar
VTU aus dem Englischen variable temperature unit für variable
Temperierungseinheit
z Zentralisomer
EINLEITUNG
- 1 -
1 Einleitung Seit der Entdeckung des Prozesses der Anomerisierung von Zuckern durch die Beobachtung
und Beschreibung der Mutarotation der D-Glucose durch AUGUSTIN-PIERRE DUBRUNFAUT (1846) sind
reduzierende Zucker Bestandteil wissenschaftlicher Forschung und Diskussionen.[1] Dabei sind bei
SciFinder® zum Suchbegriff „Glucose“ bis einschließlich 2016 insgesamt über 1.25 Millionen Einträge
gelistet. Seit der Entdeckung der nicht enzymatischen Bräunung reduzierender Zucker in Anwesenheit
von Aminokomponenten durch LOUIS-CAMILLE MAILLARD (1912)[2] bildet die sogenannte
MAILLARD-Reaktion unter anderem eine Teildisziplin der Kohlenhydratchemie, deren
Publikationszahlen sich um einige Jahrzehnte zeitlich versetzt in etwa parallel zu denen der D-Glucose
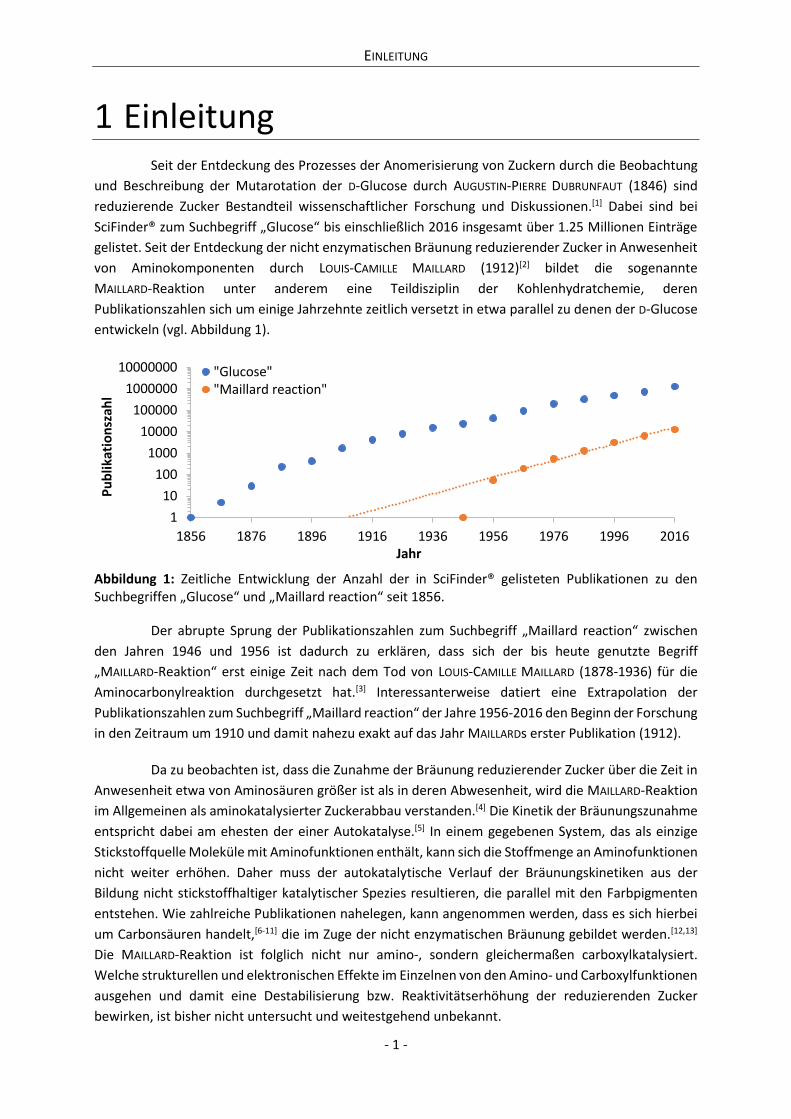
entwickeln (vgl. Abbildung 1).
Abbildung 1: Zeitliche Entwicklung der Anzahl der in SciFinder® gelisteten Publikationen zu den Suchbegriffen „Glucose“ und „Maillard reaction“ seit 1856.
Der abrupte Sprung der Publikationszahlen zum Suchbegriff „Maillard reaction“ zwischen
den Jahren 1946 und 1956 ist dadurch zu erklären, dass sich der bis heute genutzte Begriff
„MAILLARD-Reaktion“ erst einige Zeit nach dem Tod von LOUIS-CAMILLE MAILLARD (1878-1936) für die
Aminocarbonylreaktion durchgesetzt hat.[3] Interessanterweise datiert eine Extrapolation der
Publikationszahlen zum Suchbegriff „Maillard reaction“ der Jahre 1956-2016 den Beginn der Forschung
in den Zeitraum um 1910 und damit nahezu exakt auf das Jahr MAILLARDs erster Publikation (1912).
Da zu beobachten ist, dass die Zunahme der Bräunung reduzierender Zucker über die Zeit in
Anwesenheit etwa von Aminosäuren größer ist als in deren Abwesenheit, wird die MAILLARD-Reaktion
im Allgemeinen als aminokatalysierter Zuckerabbau verstanden.[4] Die Kinetik der Bräunungszunahme
entspricht dabei am ehesten der einer Autokatalyse.[5] In einem gegebenen System, das als einzige
Stickstoffquelle Moleküle mit Aminofunktionen enthält, kann sich die Stoffmenge an Aminofunktionen
nicht weiter erhöhen. Daher muss der autokatalytische Verlauf der Bräunungskinetiken aus der
Bildung nicht stickstoffhaltiger katalytischer Spezies resultieren, die parallel mit den Farbpigmenten
entstehen. Wie zahlreiche Publikationen nahelegen, kann angenommen werden, dass es sich hierbei
um Carbonsäuren handelt,[6-11] die im Zuge der nicht enzymatischen Bräunung gebildet werden.[12,13]
Die MAILLARD-Reaktion ist folglich nicht nur amino-, sondern gleichermaßen carboxylkatalysiert.
Welche strukturellen und elektronischen Effekte im Einzelnen von den Amino- und Carboxylfunktionen
ausgehen und damit eine Destabilisierung bzw. Reaktivitätserhöhung der reduzierenden Zucker
bewirken, ist bisher nicht untersucht und weitestgehend unbekannt.
1
10
100
1000
10000
100000
1000000
10000000
1856 1876 1896 1916 1936 1956 1976 1996 2016
Pu
blik
atio
nsz
ahl
Jahr
"Glucose""Maillard reaction"

THEORETISCHER HINTERGRUND
- 2 -
2 Theoretischer Hintergrund In diesem Kapitel sollen die chemischen und spektroskopischen Grundlagen erläutert
werden, die zum Verständnis der Arbeit relevant sind. Zu diesem Zwecke werden die derzeit
bekannten Strukturen und chemischen Eigenschaften der Zielsubstanzen erörtert und die
bestehenden Möglichkeiten diskutiert, diese gezielt zu untersuchen.
2.1 Die MAILLARD-Reaktion
Der Begriff der MAILLARD-Reaktion geht zurück auf den franko-algerischen Mediziner und
Pharmazeuten LOUIS-CAMILLE MAILLARD, der im Januar 1912 einen Bericht über die Reaktion von
D-Glucose mit Glycin publizierte.[2] Darin beschreibt er seine Beobachtung der nicht enzymatischen
Bräunung. Die später nach ihm benannte Reaktionskaskade läuft in allen Systemen ab, die neben
reduzierenden Zuckern auch Aminokomponenten enthalten. Der Reaktionsverlauf sowie die Kinetik
der MAILLARD-Reaktion sind nicht nur beeinflusst von der Art des Zuckers und der Aminokomponente,
sondern ebenso vom Wassergehalt, der Temperatur und dem pH-Wert.[14] Die MAILLARD-Reaktion
unterscheidet sich damit von der Karamellisierung, bei der Zucker in Abwesenheit von
Aminokomponenten – zumeist bei geringem Wassergehalt – einem Abbau unterliegen.[15,16]
Reaktionen im Sinne der MAILLARD-Reaktion laufen nicht nur – wie allgemein bekannt – beim
thermischen Prozessieren kohlenhydrathaltiger Lebensmittel aller Art ab, sondern ebenso in
physiologischen Systemen, wo sie mit der Genese zahlreicher Krankheiten in Verbindung gebracht
werden.[17] Da die MAILLARD-Reaktion unter physiologischen Bedingungen bspw. im menschlichen
Organismus nur bei geringer Temperatur verläuft und entsprechend langsam ist, handelt es sich
zumeist um Erkrankungen, die erst im fortgeschrittenen Lebensalter in Erscheinung treten. Darunter
zählen unter anderem Arteriosklerose, Diabetes mellitus, Urämie und Morbus ALZHEIMER.[18-21]
Aufgrund ihrer zentralen Rolle in der menschlichen Ernährung und dem menschlichen
Stoffwechsel, beschäftigt sich die Mehrzahl der Publikationen mit dem Reaktionsverhalten von
D-Glucose in MAILLARD-Reaktionssystemen.1 Mit dem Auslaufen der Verordnung (EU) Nr. 1308/2013
des Europäischen Parlaments und des Rates (Europäische Zuckermarktordnung) am 30. September
2017 und dem damit in Zusammenhang stehenden Wegfall der EU-Quoten für Isoglucose2, ist zu
erwarten, dass der Preis für Isoglucose fallen und damit ihre Verwendung in der
Lebensmittelproduktion aus wirtschaftlichen Gründen zunehmen wird.[22] Es ist daher davon
auszugehen, dass der Anteil an D-Fructose in der menschlichen Ernährung innerhalb der Europäischen
Union in den kommenden Jahren steigen wird. Die physiologische Konzentration von D-Fructose im
Blutplasma beträgt nur 1% der Konzentration der D-Glucose.[23] Trotzdem zeigen Untersuchungen der
Glycosylierung von Linsenproteinen menschlicher Augen, dass nur 80 bis 90% der
Proteinveränderungen auf die Reaktion mit D-Glucose zurückzuführen sind. Die übrigen 10 bis 20%
1 2246 Einträge bei SciFinder® zum Suchbegriff „Glucose Maillard“ gegenüber 658 Einträgen zum Suchbegriff
„Fructose Maillard“. Zeitpunkt der Suchanfrage: 06. März 2017.
2 Aus chemischer Sicht handelt es sich bei Isoglucose um ein Stärkehydrolysat, das unter Einwirkung von
Glucoseisomerase teilweise zu D-Fructose isomerisiert wird. Isoglucose ist damit ein Glucose-Fructose- bzw.
Fructose-Glucose-Sirup, der auch unter der Bezeichnung High Fructose Corn Syrup gehandelt wird.

THEORETISCHER HINTERGRUND
- 3 -
resultieren trotz ihrer geringen Plasmakonzentration aus der Reaktion mit D-Fructose.[24] Diese ist
damit unter physiologischen Bedingungen ca. zehnmal so reaktiv wie D-Glucose.[23] Es ist somit
absehbar, dass sich das Auftreten MAILLARD-reaktionsbedingter Alterserscheinungen mittelfristig um
einige Jahre vorverlagern wird. Gleichzeitig ist mit einem Anstieg D-Fructose-assoziierter Erkrankungen
wie der Nichtalkoholischen Fettlebererkrankung (non-alcoholic fatty liver disease) zu rechnen.[25-29]
Darüber hinaus werden auch Unverträglichkeiten wie Fructosemalabsorption in Verbindung mit
intestinaler Fructoseintoleranz[30] sowie Erkrankungen wie die hereditäre Fructoseintoleranz[31]
potentiell an Bedeutung in der Gesellschaft zunehmen. Inhalt dieser Arbeit ist vor dem Hintergrund
dieser Entwicklung die MAILLARD-Reaktion und die Karamellisierung der D-Fructose sowie die
grundlegende Untersuchung der Reaktivität verschiedener D-Fructosederivate. Im Folgenden werden
die derzeit bekannten Eigenschaften der Zielsubstanzen beschrieben.
2.1.1 D-Fructose
D-Fructose 1a gehört zur Klasse der Ketohexosen, zu der neben ihren jeweiligen
L-konfigurierten Enantiomeren auch D-Tagatose 1b, D-Sorbose 1c und D-Psicose 1d zählen (vgl.
Schema 1). D-Fructose wurde erstmals 1847 von AUGUSTIN-PIERRE DUBRUNFAUT bei Untersuchungen der
Vergärung von Zuckern entdeckt und beschrieben.3;[32] Sie ist die in Lebensmitteln am häufigsten
vorkommende Ketose.[33]
Schema 1: Strukturen der Ketohexosen D-Fructose 1a, D-Tagatose 1b, D-Sorbose 1c und D-Psicose 1d.
2.1.1.1 Strukturen und Dynamik der D-Fructose
Aufgrund ihrer Multifunktionalität ist D-Fructose dazu in der Lage, intramolekulare Halbketale
zu bilden, was zur Cyclisierung zu Pyranosen und Furanosen führt. Durch die Cyclisierung wird der
prochirale sp2-hybridisierte Carbonylkohlenstoff chiral und je zwei Epimerenpaare resultieren, die als
α- bzw. β-Anomere bezeichnet werden. Die absolute Konfiguration des α-Anomers ist dabei der
Konfiguration des höchstbezifferten chiralen Kohlenstoffatoms (hier: C-5 mit R-Konfiguration nach
CIP-Nomenklatur) entgegengesetzt. Im Falle des β-Anomers sind die jeweiligen Stereozentren
entsprechend gleichartig konfiguriert. D-Fructose kristallisiert in 2C5-β-pyranoider Struktur.4;[34] Wird
sie gelöst, findet Mutarotation statt, sodass ein Gemisch der Anomere entsteht. Dieses setzt sich aus
3 Nach ihrer Entdeckung wurde D-Fructose aufgrund ihrer optischen Aktivität ab 1860 zunächst als „Levulose“
bzw. „Lävulose“ bezeichnet[358] und bekam ihren bis heute gültigen wissenschaftlichen Namen 1890 durch EMIL
FISCHER.[359] Ihre erstmalige Kristallisation gelang JUNGFLEISCH und LEFRANC 1881.[360]
4 Dieses Konformer ist auch die in der Gasphase vorherrschende Struktur der D-Fructose.[361]
THEORETISCHER HINTERGRUND
- 4 -
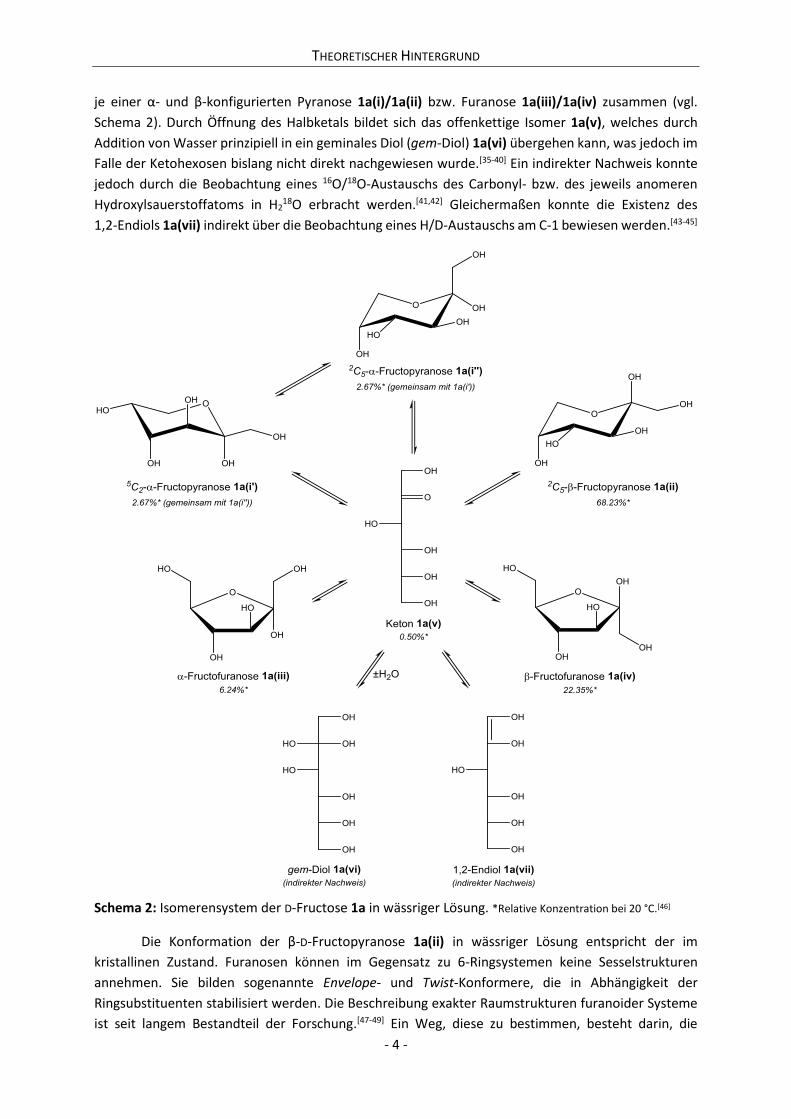
je einer α- und β-konfigurierten Pyranose 1a(i)/1a(ii) bzw. Furanose 1a(iii)/1a(iv) zusammen (vgl.
Schema 2). Durch Öffnung des Halbketals bildet sich das offenkettige Isomer 1a(v), welches durch
Addition von Wasser prinzipiell in ein geminales Diol (gem-Diol) 1a(vi) übergehen kann, was jedoch im
Falle der Ketohexosen bislang nicht direkt nachgewiesen wurde.[35-40] Ein indirekter Nachweis konnte
jedoch durch die Beobachtung eines 16O/18O-Austauschs des Carbonyl- bzw. des jeweils anomeren
Hydroxylsauerstoffatoms in H218O erbracht werden.[41,42] Gleichermaßen konnte die Existenz des
1,2-Endiols 1a(vii) indirekt über die Beobachtung eines H/D-Austauschs am C-1 bewiesen werden.[43-45]
Schema 2: Isomerensystem der D-Fructose 1a in wässriger Lösung. *Relative Konzentration bei 20 °C.[46]
Die Konformation der β-D-Fructopyranose 1a(ii) in wässriger Lösung entspricht der im
kristallinen Zustand. Furanosen können im Gegensatz zu 6-Ringsystemen keine Sesselstrukturen
annehmen. Sie bilden sogenannte Envelope- und Twist-Konformere, die in Abhängigkeit der
Ringsubstituenten stabilisiert werden. Die Beschreibung exakter Raumstrukturen furanoider Systeme
ist seit langem Bestandteil der Forschung.[47-49] Ein Weg, diese zu bestimmen, besteht darin, die

THEORETISCHER HINTERGRUND
- 5 -
exocyclischen Torsionswinkel mit Hilfe der 3J(1H,1H)-Kopplungskonstanten unter Nutzung der
generalisierten KARPLUS-Beziehung zu berechnen.5;[50,51] Diese müssen im Folgenden dazu genutzt
werden, auf die endocyclischen Torsionswinkel zu schließen und damit den Phasenwinkel der
Pseudorotation P und die Puckering-Amplitude τ zu ermitteln. Aufgrund der geringen Energiebarrieren
der Pseudorotation von 5-Ringsystemen, die üblicherweise nahe 0 kJ/mol liegen und Maximalwerte
von rund 20 kJ/mol erreichen,[52] gehen die Rotamere furanoider Strukturen schon meist bei
Raumtemperatur nahezu frei ineinander über. Das geht mit einer temperaturabhängigen Änderung
der 3J(1H,1H)-Kopplungskonstanten einher.[53] Es ist daher anzunehmen, dass die Vorzugskonformation
von monomeren Furanosen keinen Einfluss auf die Reaktivität einfacher Saccharide hat. Dies sollte
jedoch nicht die prinzipielle Notwendigkeit der Konformationsanalyse infrage stellen, da die
Geometrie von Monomeren innerhalb polymerer Strukturen Einfluss auf die Struktur von
Makromolekülen nimmt. Damit ist die Kenntnis des Phasenwinkels der Pseudorotation P und der
Puckering-Amplitude τ bspw. Voraussetzung für die Strukturaufklärung der Nucleinsäuren.[54] Im
weiteren Verlauf dieser Arbeit wird die Pseudorotation der furanoiden Anomere nicht weiter
betrachtet und lediglich von je einem α- und β-furanoiden Anomer gesprochen.
Da bei der Ringinversion pyranoider Strukturen zumeist größere Energiebarrieren zu
überwinden sind, handelt es sich bei 6-Ringsystemen um vergleichsweise starre Strukturen mit
typischen Vorzugskonformationen. Als Beispiel hierfür sei noch einmal die 2C5-β-D-Fructopyranose
1a(ii) genannt. Es ist also davon auszugehen, dass auch im Falle des α-pyranoiden Anomers eine
entsprechende Struktur vorliegt. Hierzu fehlen jedoch verlässliche Literaturwerte. Frühe
Publikationen, die sich mit der Struktur der D-Fructose befassen, gehen rein argumentativ davon aus,
dass die α-Pyranose ebenfalls in Form eines 2C5-Sessels vorliegt.[40] Auch die Autoren jüngerer
Publikationen liefern keine experimentellen Hinweise auf die tatsächliche Struktur, schließen sich
jedoch der bestehenden Vermutung an.[46,55] Diese basiert in erster Linie auf der Betrachtung sterischer
Wechselwirkungen, die im Falle des 5C2-Konformers aufgrund der 1,3-diaxialen Stellung der
Hydroxylfunktionen am C-2 und C-4 stark ausgeprägt sind. Nicht beachtet wird dabei jedoch, dass
dieses Konformer über den anomeren Effekt stabilisiert ist.[56] Theoretische Arbeiten zur Konformation
der α-D-Fructopyranose zeigen, dass die Energiedifferenz der beiden Konformere bei lediglich
2.55 kJ/mol liegt, was der thermischen Energie bei 34 °C entspricht.[57] Dies ist ein Indiz dafür, dass
beide Konformere koexistieren könnten. Ebenfalls dafür sprechen die für das nah verwandte
D-Fructosamin-Hydrochlorid publizierten Kopplungskonstanten der α-Pyranose. Diese sind mit 3J(H-C-5; Ha-C-6) = 3.1 Hz und 3J(H-C-5; Hb-C-6) = 3.0 Hz zu groß für das 2C5-, jedoch zu klein für das 5C2-Konformer.[57,58] Daneben liefern auch ultraschallspektroskopische Messungen Hinweise auf eine 5C2 2C5 Ringinversion.[59-61]
Bei 20 °C bildet 1a(ii) mit 68.23% das Hauptanomer in wässriger Lösung, gefolgt von 1a(iv) mit
22.35%, 1a(iii) mit 6.24%, 1a(i) mit 2.67% und 1a(v) mit 0.50% (vgl. Schema 2).[46] Dass dieses
Gleichgewicht temperaturabhängig ist, wurde bereits im frühen 20. Jahrhundert erkannt.[62] Die
Abhängigkeit des Anomerengleichgewichts von der Konzentration der D-Fructose – und damit
5 Die Anpassung der generalisierten KARPLUS-Beziehung erfordert unter anderem die Zuhilfenahme der 1953
publizierten HUGGINS-Elektronegativitäten,[362] die als Neuberechnung der 1932 von LINUS PAULING eingeführten
Elektronegativitäten zu verstehen sind.[363] Seit 1961 sind diese zwar durch A. L. ALLREDS abermals optimierte
Werte abgelöst,[364] haben dadurch jedoch ihre Gültigkeit für die Anpassung der generalisierten
KARPLUS-Beziehung nicht verloren.

THEORETISCHER HINTERGRUND
- 6 -
gleichermaßen vom Wassergehalt – ist ebenso beschrieben wie dessen Abhängigkeit vom
Lösungsmittel.[55,63-66] Die Ringöffnungs- und Ringschlussraten (im weiteren Verlauf der Arbeit als
Anomerisierungsraten bezeichnet) dieses komplexen Anomerensystems werden bereits seit rund 100
Jahren untersucht. Dazu wurden zunächst polarimetrische Messungen durchgeführt und damit
Mutarotationsraten in Abhängigkeit der Temperatur und des pH-Wertes ermittelt.[67] Dabei handelt es
sich um die Summe der Geschwindigkeitskonstanten für die Ringöffnung von 1a(ii) und der Rate der
Rückreaktion. Die Rate der Rückreaktion entspricht dabei jedoch nicht der Ringschlussrate von 1a(v)
zu 1a(ii), sondern berechnet sich aus den Anomerisierungsraten aller vorhandenen Isomere mit
Ausnahme der Ringöffnungsrate von 1a(ii). In späteren Arbeiten gelang die getrennte Berechnung der
Ringöffnungsraten von 1a(ii) in Abhängigkeit der Temperatur, woraus die Aktivierungsparameter der
Ringöffnung ermittelt wurden.[68] Die Messungen fanden hierbei jedoch in Acetat-Puffer statt, sodass
die Ergebnisse mutmaßlich nicht mit denen in ungepufferter Lösung übereinstimmen. Mit der
Entdeckung der NMR-Spektroskopie durch FELIX BLOCH und EDWARD MILLS PURCELL im Jahre 19466 und
den grundlegenden Arbeiten von HARDEN MCCONNELL[69] sowie STURE FORSÉN und RAGNAR HOFFMAN[70]
wurde die Untersuchung komplexer dynamischer Isomerensysteme im thermodynamischen
Gleichgewicht möglich. Dabei wurde die Dynamik einer Vielzahl von Zuckersystemen insbesondere
durch SNYDER und SERIANNI mittels selektiver Sättigungstransfer-NMR-Spektroskopie untersucht.[71-79]
Zur Dynamik des Anomerensystems der D-Fructose hingegen liegen weiterhin nur wenige
Informationen vor. In der einzig vorhandenen Publikation werden lediglich die Anomere 1a(ii), 1a(iii)
und 1a(iv) untersucht.[80] Die dabei eingesetzten Methoden sind jedoch aus den in Kapitel 2.2.3
diskutierten Gründen nur eingeschränkt für die Untersuchung von dynamischen
Kohlenhydratsystemen nutzbar.
2.1.1.2 Formen der Isomerisierung
Unter Kapitel 2.1.1.1 wurde das dynamische Gleichgewicht der Anomere der D-Fructose 1a
diskutiert. Dabei gehen diese in (protischen) Lösungsmitteln durch intramolekulare Bildung von
Halbketalen sowie Ringinversion schnell ineinander über. Es ist daher unmöglich, eine dauerhafte
Trennung der Anomere vorzunehmen, solange sich diese in Lösung befinden. Die Anomerisierung
sowie deren Elementarschritte – Ringöffnung und Ringschluss – sind damit eine spezielle Form der
Tautomerie (Ring-Ketten-Tautomerie). Dabei bezeichnen Tautomerien nach CONRAD LAAR (1885)
gleichberichtigte Strukturformeln identischer chemischer Verbindungen, die sich durch
intramolekulare Umlagerungen ineinander überführen lassen.[81] Da der Prozess der Tautomerisierung
demnach verschiedene Erscheinungsbilder identischer Verbindungen miteinander verknüpft, sind
Tautomerien zunächst nicht als chemische Reaktionen zu verstehen. Eine wesentliche Eigenschaft der
D-Fructose besteht neben der Anomerisierung (d.h. Ringöffnung und Ringschluss) in einer weiteren
Form der Tautomerie. Hierbei handelt es sich um die Möglichkeit der Enolisierung, die sich aus der
C-H-Acidität α-ständiger Methylen- und Methingruppen in Nachbarschaft von Carbonylfunktionen
ergibt. Damit zeigen sowohl die Protonen am C-1 als auch am C-3 C-H-Acidität. Der Prozess der
Enolisierung führt neben dem 1,2-Endiol auch zum 2,3-Endiol (vgl. Schema 3).
6 Nur 6 Jahre nach der Entdeckung des Effektes der kernmagnetischen Resonanz erhielten FELIX BLOCH und EDWARD
MILLS PURCELL 1952 den Nobelpreis für Physik.[365]
THEORETISCHER HINTERGRUND
- 7 -
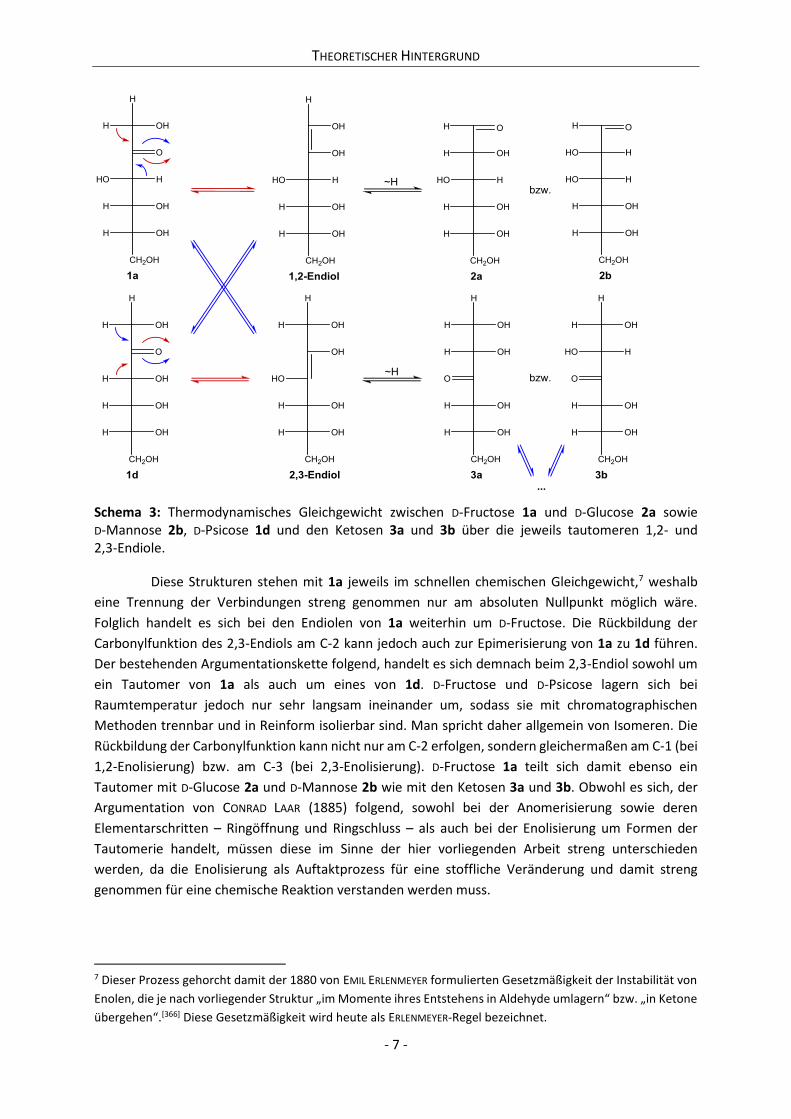
Schema 3: Thermodynamisches Gleichgewicht zwischen D-Fructose 1a und D-Glucose 2a sowie D-Mannose 2b, D-Psicose 1d und den Ketosen 3a und 3b über die jeweils tautomeren 1,2- und 2,3-Endiole.
Diese Strukturen stehen mit 1a jeweils im schnellen chemischen Gleichgewicht,7 weshalb
eine Trennung der Verbindungen streng genommen nur am absoluten Nullpunkt möglich wäre.
Folglich handelt es sich bei den Endiolen von 1a weiterhin um D-Fructose. Die Rückbildung der
Carbonylfunktion des 2,3-Endiols am C-2 kann jedoch auch zur Epimerisierung von 1a zu 1d führen.
Der bestehenden Argumentationskette folgend, handelt es sich demnach beim 2,3-Endiol sowohl um
ein Tautomer von 1a als auch um eines von 1d. D-Fructose und D-Psicose lagern sich bei
Raumtemperatur jedoch nur sehr langsam ineinander um, sodass sie mit chromatographischen
Methoden trennbar und in Reinform isolierbar sind. Man spricht daher allgemein von Isomeren. Die
Rückbildung der Carbonylfunktion kann nicht nur am C-2 erfolgen, sondern gleichermaßen am C-1 (bei
1,2-Enolisierung) bzw. am C-3 (bei 2,3-Enolisierung). D-Fructose 1a teilt sich damit ebenso ein
Tautomer mit D-Glucose 2a und D-Mannose 2b wie mit den Ketosen 3a und 3b. Obwohl es sich, der
Argumentation von CONRAD LAAR (1885) folgend, sowohl bei der Anomerisierung sowie deren
Elementarschritten – Ringöffnung und Ringschluss – als auch bei der Enolisierung um Formen der
Tautomerie handelt, müssen diese im Sinne der hier vorliegenden Arbeit streng unterschieden
werden, da die Enolisierung als Auftaktprozess für eine stoffliche Veränderung und damit streng
genommen für eine chemische Reaktion verstanden werden muss.
7 Dieser Prozess gehorcht damit der 1880 von EMIL ERLENMEYER formulierten Gesetzmäßigkeit der Instabilität von
Enolen, die je nach vorliegender Struktur „im Momente ihres Entstehens in Aldehyde umlagern“ bzw. „in Ketone
übergehen“.[366] Diese Gesetzmäßigkeit wird heute als ERLENMEYER-Regel bezeichnet.

THEORETISCHER HINTERGRUND
- 8 -
2.1.2 Die Aminokomponenten
Zur Untersuchung des Reaktionsverhaltens der D-Fructose 1a im Sinne der Karamellisierung im
Vergleich zu ihrer Reaktivität in MAILLARD-Systemen ist die Auswahl der Aminokomponenten von
entscheidender Bedeutung.[82] Um Gesetzmäßigkeiten des Einflusses der Aminokomponente auf das
Reaktionsverhalten der D-Fructose ableiten zu können, ist es wichtig, chemisch ähnliche Verbindungen
zu untersuchen, die sich etwa strukturell geringfügig voneinander unterscheiden, wie es bei
Homologen der Fall ist. Da aliphatische Aminosäuren von allen niedermolekularen
Stickstoffverbindungen die häufigste Gruppe in Lebensmitteln sind, wurden für diese Arbeit zunächst
L-Alanin 4a und L-Prolin 4b als Reaktionspartner für D-Fructose ausgewählt. Darüber hinaus wurde auch
das Reaktionsverhalten der homologen ω-Aminosäuren Glycin 4c, β-Alanin 4d und
γ-Aminobuttersäure 4e untersucht. Bei den Aminokomponenten (vgl. Schema 4) handelt es sich damit
um aliphatische primäre und sekundäre α- und ω-Aminosäuren, wobei Glycin sowohl in die Gruppe
der α- als auch der ω-Aminosäuren zu zählen ist.
Schema 4: Strukturen der eingesetzten Aminokomponenten L-Alanin 4a, L-Prolin 4b, Glycin 4c, β-Alanin 4d und γ-Aminobuttersäure 4e.
2.1.3 AMADORI- und HEYNS-Verbindungen
Die Kaskade der MAILLARD-Reaktion wurde von JOHN E. HODGE 1953 in eine initiale, eine
intermediäre und eine finale Phase unterteilt.[4] Die initiale Phase beschreibt dabei die
Kondensationsreaktion eines Zuckers mit einer Aminokomponente. Die intermediäre Phase fasst die
Reaktionen zusammen, die zu einem Abbau der Produkte der initialen Phase führen. Die finale Phase
beschreibt schließlich die Bildung stabiler hochmolekularer braun gefärbter Produkte, den
Melanoidinen, aus den Produkten der intermediären Phase. Bei den Abbauprodukten der intermediär
gebildeten Substanzen handelt es sich um Verbindungen, die sich gegenüber ihren Edukten durch eine
erhöhte Reaktivität auszeichnen.[83] Es ist daher davon auszugehen, dass die initialen und
intermediären Schritte der Reaktionskaskade geschwindigkeitsbestimmend für den Verlauf der nicht
enzymatischen Bräunung im Sinne der MAILLARD-Reaktion sind. Demzufolge sollte eine Einflussnahme
auf die Bräunungskinetik durch die Katalyse der ersten und zweiten Phase der MAILLARD-Reaktion
möglich sein. Die vorliegende Arbeit konzentriert sich aufgrund dessen auf die Behandlung der initialen
und intermediären Phase.
2.1.3.1 AMADORI- und HEYNS-CARSON-Umlagerung
Der Begriff der AMADORI-Verbindungen umfasst die Gruppe der (N-substituierten)
1-Desoxy-1-aminoketosen, die sich in der initialen Phase der MAILLARD-Reaktion durch die
Kondensation von reduzierenden Zuckern mit Aminokomponenten bilden. Der Begriff geht zurück auf
mehrere Publikationen des italienischen Pharmazeuten und Chemikers MARIO AMADORI aus den Jahren
1925-1931,[84-86] in denen er unter anderem die Kondensationsreaktion von D-Glucose mit
para-Toluidin und para-Phenitidin beschreibt. Darin diskutiert er die Bildung von „labilen“

THEORETISCHER HINTERGRUND
- 9 -
N-Glucosylaminen und deren hitzeinduzierten Umsatz zu „stabilen“ SCHIFF-Basen. Weiterführende
Untersuchungen von KUHN und DANSI (1936)[87] zeigten jedoch, dass die Hydrierung der mutmaßlichen
SCHIFF-Base nicht zum entsprechenden N-substituierten Glucamin führt, sodass es sich tatsächlich nicht
um die postulierte SCHIFF-Base handeln konnte. KUHN und WEYGAND (1937) formulierten schließlich die
Bildung der N-substituierten 1-Desoxy-1-aminoketose (Isoglucosaminderivate) über die Tautomerie
des N-Glycosylamins und prägten den Begriff der AMADORI-Umlagerung.8;[88] Die so getroffene
Zuordnung des „stabilen“ Isomers des N-Glycosylamins bestätigte sich in vielen weiteren
Untersuchungen.[89-91] Da sich die hier vorliegende Arbeit mit der Bildung und dem Verhalten von
AMADORI-Verbindungen in Lebensmittelsystemen befasst, sei für die Diskussion der in der Literatur
beschriebenen Synthesestrategien auf die entsprechenden Artikel verwiesen.[92-94] Die
mechanistischen Modellvorstellungen der ungerichteten Synthese, wie sie in einfachen
MAILLARD-Reaktionssystemen abläuft, seien im Folgenden am Beispiel der D-Glucose 2a demonstriert.
D-Glucose 2a liegt in wässriger Lösung im thermodynamischen Gleichgewicht – ebenso wie
D-Fructose – als Gemisch mehrerer Anomere vor. Hierbei entfallen bei 30 °C in wässriger Lösung ca.
99.6% der relativen Anomerenkonzentration auf die pyranoiden Anomere.[95] Die folgenden
Mechanismen (Schema 5: Weg a) und Weg b)) werden aufgrund dessen am Beispiel der
Hauptanomere 4C1-α- und -β-D-Glucopyranose vorgestellt. Im ersten Reaktionsschritt kommt es zur
Bildung des „labilen“ N-Glucosylamins 5a.
Schema 5: Möglichkeiten der Bildung eines N-substituierten Glucosylamins 5a sowie der korrespon-dierenden ringoffenen protonierten SCHIFF-Base 5b aus D-Glucose 2a und einer Aminokomponente im sauren pH-Bereich. a) Protonierung des Lactolrings. b) Protonierung der anomeren OH-Funktion.
8 Isoglucosamin wurde als einfachste AMADORI-Verbindung bereits im Geburtsjahr von MARIO AMADORI 1886
erstmals von EMIL FISCHER beschrieben. Die Darstellung erfolgte hierbei jedoch über die Reduktion von
Phenylglucosazon und nicht über die erst 1937 beschriebene AMADORI-Umlagerung.[367]

THEORETISCHER HINTERGRUND
- 10 -
Dabei greift der unprotonierte Aminostickstoff nucleophil am C-1 Atom der D-Glucose an.
Diesbezüglich werden zwei grundsätzliche Reaktionsmechanismen voneinander unterschieden (Weg
a) und b) in Schema 5), die beide einen sauren pH-Wert erfordern. Liegt die 4C1-β-D-Glucopyranose im
sauren Milieu vor, so besteht neben der Protonierung des Lactolrings (Weg a)) auch die Möglichkeit
der Protonierung der anomeren Hydroxylfunktion (Weg b)). Dabei führt die Protonierung des
Lactolsauerstoffes zu einer Spaltung des Halbacetals durch Ringöffnung. Die Protonierung der
anomeren Hydroxylfunktion erzeugt Wasser als gute Abgangsgruppe, wobei das zurückbleibende
Carbeniumion durch die Nachbarschaft zum Ringsauerstoff resonanzstabilisiert ist (vgl. Schema 5).
Anschließend greift der elektroneutrale Stickstoff nucleophil am Carbeniumion an und bildet nach
etwaiger Abspaltung eines Moleküls Wasser das N-substituierte Glucosylamin 5a.[82] Weg a) ist dabei
äquivalent zum Mechanismus, der zur Bildung des gem-Diols der D-Glucose führt. Aus den gezeigten
Reaktionsmechanismen wird der Einfluss des pH-Wertes auf die MAILLARD-Reaktion deutlich. Da der
Zucker protoniert vorliegt, während die basische Aminofunktion elektroneutral ist, kann gefolgert
werden, dass die in Schema 5 gezeigten Mechanismen ein pH-Optimum besitzen müssen. Da die
Bildung des gem-Diols der D-Glucose jedoch nicht nur im Sauren, sondern ebenso im Basischen
erfolgen kann,[96] ist auch hier ein äquivalenter Mechanismus für die Bildung der SCHIFF-Base 5b
denkbar. Dieser geht vom acyclischen Zentralisomer der D-Glucose 2a aus (vgl. Schema 6). Die Bildung
des in den Schemata 5 und 6 gezeigten geminalen Aminols 5c steht demnach in Konkurrenz zur Bildung
des gem-Diols der D-Glucose 2a.
Schema 6: Bildung einer SCHIFF-Base 5b aus D-Glucose 2a und einer Aminokomponente über das gem-Aminol 5c im basischen pH-Bereich. Der gezeigte Mechanismus entspricht dem der Bildung des gem-Diols der D-Glucose 2a aus ihrem acyclischen Zentralisomer.
Alternativ zu den bereits gezeigten Mechanismen wird im Falle von Reaktionen mit
Aminosäuren die Möglichkeit eines konzertierten Mechanismus‘ zur Bildung von 5a diskutiert (vgl.
Schema 7).[82] Auch in diesem Reaktionsschema wird die pH-Abhängigkeit der initialen
MAILLARD-Reaktion deutlich sichtbar, da der Mechanismus von einer ungeladenen Aminosäurespezies
ausgeht, deren relative Konzentration ihr Maximum am isoelektrischen Punkt der Aminosäure zeigt.

THEORETISCHER HINTERGRUND
- 11 -
Schema 7: Konzertierter Mechanismus zur Bildung eines N-substituierten Glucosylamins 5a sowie der
korrespondierenden ringoffenen protonierten SCHIFF-Base 5b aus D-Glucose 2a und einer Aminosäure.
(Frei nach[82])
Zusammenfassend ist die Bildung des N-substituierten Glucosylamins 5a sowie der
korrespondierenden ringoffenen protonierten SCHIFF-Base 5b nur in pH-Bereichen möglich, in denen
die Aminofunktion der reagierenden Aminokomponente nicht protoniert vorliegt. Entsprechend ist
davon auszugehen, dass eine Bildung von 5a und 5b vornehmlich im Basischen stattfindet.
Die eigentliche AMADORI-Umlagerung erfolgt ausgehend von der ringoffenen SCHIFF-Base 5b
durch eine Imin-Enaminol-Aminoketon-Tautomerie hin zur 1-Desoxy-1-aminoketose 6, der
AMADORI-Verbindung.[83,88] Es lassen sich jedoch ebenso Mechanismen formulieren, die direkt vom
N-substituierten Glucosylamin 5a ausgehen (vgl. Schema 8).[97]
Schema 8: Mechanismus der AMADORI-Umlagerung I. (Frei nach[83,88,97])
Im Verlauf dieser Tautomerisierung muss die C-H-Bindung am C-2 gebrochen werden. Dies
kann zum einen – wie von MARIO AMADORI (s.o.) beobachtet – hitzeinduziert erfolgen. Zum anderen ist
eine Steigerung des Dissoziationsbestrebens der Bindung durch die Erhöhung des pH-Wertes in den
basischen Bereich möglich.[98] Schließlich kann die Umlagerung durch Anwesenheit von LEWIS-Säuren,
Mineralsäuren, Carbonsäuren sowie tertiären Aminen katalysiert werden.[90,99-102] Aufgrund dessen
verläuft die AMADORI-Umlagerung mit Aminosäuren autokatalytisch. Alternativ zu den hier gezeigten
weitestgehend akzeptierten Mechanismen der AMADORI-Umlagerung publizierten MICHEEL und DIJONG
(1962) die Möglichkeit eines erneuten nucleophilen Angriffs einer Aminokomponente am

THEORETISCHER HINTERGRUND
- 12 -
Iminokohlenstoff von 5b. Nach anschließender Eliminierung einer Aminokomponente kommt es zur
Ausbildung eines Carbeniumions am C-1. Ein (formaler) Hydridshift vom C-2 zum C-1 und die
Rückbildung der Carbonylfunktion ergeben schließlich 6 (vgl. Schema 9).[103]
Schema 9: Mechanismus der AMADORI-Umlagerung II.[103]
Dieser Mechanismus ist jedoch insofern nicht schlüssig, als es sich bei der Zwischenstufe, die
ein Carbeniumion am C-1 trägt, ohnehin um eine mesomere Grenzform von 5b handelt. Die
Notwendigkeit einer Reaktion mit einer zweiten Aminokomponente ist damit nicht gegeben.
Verbindung 6 unterscheidet sich von 1a lediglich durch den Substituenten am C-1, sodass es
sich bei AMADORI-Verbindungen der D-Glucose 2a (bzw. D-Mannose 2b) um Derivate der D-Fructose 1a
handelt. Analog resultieren aus der Kondensation von D-Fructose 1a mit Aminokomponenten Derivate
der D-Glucose 2a und D-Mannose 2b. Diese werden nach ihrem Entdecker KURT HEYNS zumeist als
HEYNS-Verbindungen 7 bezeichnet.[104-111] Etwa zeitgleich wurden analoge Verbindungen auch von JOHN
F. CARSON beschrieben,[112,113] sodass auch der Begriff der HEYNS-CARSON-Umlagerung für den
entsprechenden Bildungsmechanismus in der Fachliteratur verbreitet ist.[114] Die Kondensation von
D-Fructose mit einer Aminokomponente führt formal betrachtet nicht nur zur
2-Desoxy-2-aminoaldose, sondern erlaubt ebenfalls die Formulierung von
2-Desoxy-2-amino-3-ketosen, die als alternative AMADORI-Verbindungen zu verstehen sind. Hinweise
auf deren Existenz liefern bspw. die Beobachtungen von SUÁREZ et al. (1989).[115]
Bei der Synthese von HEYNS-Verbindungen wurde schon früh erkannt, dass neben den
erwarteten 2-Desoxy-2-aminoaldosen auch 1-Desoxy-1-aminoketosen, d.h. AMADORI-Verbindungen,
als Nebenprodukte anfallen.[109-111] Spätere Arbeiten zeigen, dass diese aus der Reaktion von
HEYNS-Verbindungen mit Aminokomponenten hervorgehen.[116] Dabei kondensiert die
HEYNS-Verbindung 7 zunächst mit der Aminokomponente zu einer SCHIFF-Base.[112] Diese tautomerisiert
im Sinne einer LOBRY-DE-BRUYN-ALBERDA-VAN-EKENSTEIN-Umlagerung[117] und bildet nach hydrolytischer
Spaltung des Imins am C-1 die korrespondierende AMADORI-Verbindung 6 (vgl. Schema 10).

THEORETISCHER HINTERGRUND
- 13 -
Schema 10: Bildung von AMADORI- 6 ausgehend von HEYNS-Verbindungen 7.
AMADORI-Verbindungen primärer Amine sind ihrerseits als sekundäre Amine zu verstehen,
sodass diese als Nucleophile mit einem weiteren Molekül eines reduzierenden Zuckers reagieren
können. Es findet dabei nach erfolgter Kondensation wiederum eine AMADORI-Umlagerung statt, wobei
im Falle der sukzessiven Reaktion zweier Moleküle D-Glucose mit einem primären Amin ein
N,N-Difructosylamin 8 resultiert (vgl. Schema 11). Dieses kristallisiert in Form eines
Morpholinderivates, welches auch als Hauptisomer in wässriger Lösung vorliegt.[118-120]
Schema 11: Bildung eines N,N-Difructosylamins 8 aus der Kondensation einer AMADORI-Verbindung 6 mit D-Glucose 2a. (Frei nach[119,121])
2.1.3.2 STRUKTUREN UND DYNAMIK DER AMADORI-VERBINDUNGEN
AMADORI-Verbindungen kristallisieren in Form ihres 2C5-β-pyranoiden Anomers 6(ii).[122-125] In
wässriger Lösung bilden sie – ebenso wie D-Fructose 1a – verschiedene Halbketale, sodass es zur
Ausbildung eines entsprechenden Anomerensystems kommt. Dabei konnten erwartungsgemäß je
zwei pyranoide 6(i)/6(ii) und furanoide Anomere 6(iii)/6(iv) sowie zum Teil auch ein nicht
hydratisiertes acyclisches 6(v),9 jedoch kein hydratisiertes acyclisches Isomer 6(vi) identifiziert
werden.[126-129] Lediglich in einer Publikation werden hohe Gehalte (rund 10%) des gem-Diols 6(vi) in
wasserfreiem DMSO beschrieben.[130] Diese Ergebnisse beruhen jedoch mutmaßlich auf
fehlinterpretierten NMR-Spektren, wobei es zu einer Verwechslung mit dem α-pyranoiden Anomer
6(i) gekommen sein muss. Das gem-Diol 6(vi) steht, sofern es tatsächlich vorliegt, immer mit seinem
nicht hydratisierten Äquivalent 6(v) im Gleichgewicht. In einem wasserfreien organischen
9 Die Beschreibung des Isomers 6(v) in der Publikation von LI et al. (2014) ist als fehlerhaft zu betrachten, da dem
Carbonylkohlenstoffatom ein Signal mit einer chemischen Verschiebung von 175 ppm zugeordnet wurde. Hierbei
handelt es sich jedoch zweifelsfrei um das Signal eines Carboxylkohlenstoffatoms.[368]

THEORETISCHER HINTERGRUND
- 14 -
Lösungsmittel bedeutet dies, dass es ein Molekül Wasser eliminiert. Durch den resultierenden
Verdünnungseffekt des Wassers im organischen Lösungsmittel ist eine Rückreaktion statistisch
ausgeschlossen, sodass die Konzentration des gem-Diols 6(vi) gegen null konvergiert. Damit muss die
Konzentration von 6(v) in einem wasserfreien organischen Lösungsmittel zwangsläufig größer sein als
die von 6(vi). Da kein Hinweis auf ein acyclisches nicht hydratisiertes Isomer 6(v) beschrieben wurde,
muss es sich um das α-pyranoide Anomer 6(i) handeln. Abgesehen davon ist die Existenz von 6(vi)
durch seine chemische Ähnlichkeit mit einfachen Monosacchariden prinzipiell wahrscheinlich. Da die
Neigung von Ketonen, Hydrate zu bilden, allerdings nur schwach ausgeprägt ist,[131] konnten gem-Diole
von Derivaten der D-Fructose bislang noch nicht direkt nachgewiesen werden.[126,127,132,133] Analog der
D-Fructose wurde die Existenz des Isomers 6(vi) jedoch bereits über die Beobachtung eines 16O/18O-Austauschs des Carbonyl- bzw. des jeweils anomeren Hydroxylsauerstoffatoms in H2
18O
indirekt belegt.[134]
Ähnlich wie bei D-Fructose 1a ist die Diskussion über die Konformation des α-pyranoiden
Anomers noch nicht abgeschlossen. In entsprechenden Publikationen wird die α-Pyranose lediglich
identifiziert, ihre Konformation jedoch nicht diskutiert. Einige Autoren beschreiben sie dabei als 5C2-Konformer 6(i‘),[127,135] andere als 2C5-Konformer 6(i‘‘)[136] und wieder andere stellen beide als
mögliche Konformere dar.[126,128] Darüber hinaus wird in einigen Publikationen der indirekte Nachweis
eines tautomeren 1,2-Enaminols der AMADORI-Verbindungen 6(vii) beschrieben. Der Nachweis besteht
in der Beobachtung der zeitabhängigen Abnahme der Signalintensität der Resonanzen der Protonen
am C-1, die in protischen deuterierten Lösungsmitteln wie Deuteriumoxid einem H/D-Austausch
unterliegen.[126,127] Der H/D-Austausch führt dabei zu einer schwachen lokalen Veränderung der
magnetischen Abschirmung benachbarter Kohlenstoffatome, woraus im Falle der
AMADORI-Verbindungen eine Hochfeldverschiebung der anomeren 13C-NMR-Resonanzen resultiert.[119]
Die Existenz des tautomeren 2,3-Endiols konnte auf diese Weise nicht nachgewiesen werden.[119]
Gleiches zeigen die Ergebnisse von SIMON und HEUBACH (1965), die den H/T-Austausch einer
AMADORI-Verbindung am C-1 und C-3 anhand von Radioaktivitätsmessungen untersuchten.[137] Sie
kommen dabei zu dem Schluss, dass die Enolisierung am C-1 rasch und reversibel ist, während sie sich
am C-3 irreversibel zeigt und zum Abbau der AMADORI-Verbindung durch die Eliminierung des
Aminosubstituenten führt.[137,138] Unter Berücksichtigung aller bisherigen Erkenntnisse zur Struktur der
AMADORI-Verbindungen ergibt sich für wässrige Lösungen in Analogie zu Schema 2 das in Schema 12
gezeigte Tautomerensystem.
Die relativen Konzentrationen der verschiedenen Tautomere sind vergleichbar mit denen der
D-Fructose. 6(ii) bildet mit rund 64-69% das Hauptanomer in wässriger Lösung, gefolgt von 6(iii) und
6(iv) mit je 12-15% und 6(i) mit ca. 5-6%. 6(v) liegt als Minorkomponente zu rund 1-2% vor.[119,127] Der
Einfluss der Stereokonfiguration des Kohlenstoffgerüstes auf die Isomerenverteilung wurde bisher
nicht systematisch untersucht, da AMADORI-Verbindungen zumeist nur auf Basis von D-Glucose 2a
strukturell aufgeklärt sind.[126,127,130,132,133] Lediglich HORVAT et al. (1998) beschreiben die Synthese einer
D-Galactose-basierten AMADORI-Verbindung, bei der es sich folglich um ein Derivat der D-Tagatose 1b
handelt. Dabei wurde die α-Pyranose analog zur D-Tagatose 1b als Hauptanomer identifiziert.[37,139,140]
Es zeigte sich weiterhin sowohl an Kondensationsprodukten aromatischer Amine sowie proteinogener
Aminosäuren mit D-Glucose, dass die Aminokomponente das thermodynamische Gleichgewicht der
Anomere nur marginal beeinflusst.[127,135] Sie stabilisiert die Anomere jedoch durch Ausbildung von
Wasserstoffbrücken zur anomeren Hydroxylfunktion (bei Furanosen) bzw. zur Hydroxylfunktion am
C-3 (bei Pyranosen).[94] Entsprechende Wasserstoffbrücken können über Röntgenstrukturanalysen

THEORETISCHER HINTERGRUND
- 15 -
auch für kristalline AMADORI-Verbindungen nachgewiesen werden.[122-125] Weiterhin zeigen
Untersuchungen an Kondensationsprodukten von Polypeptiden mit D-Glucose, dass die Kettenlänge
des Peptids einen Einfluss auf das Konzentrationsverhältnis von Pyranosen zu Furanosen hat.[130]
Zusätzlich beschreiben ALTENA et al. (1981) eine pH-abhängige Änderung der relativen
Anomerenverteilungen,[126] die in den Arbeiten anderer Forschungsgruppen jedoch nicht gefunden
wurde.[127] Darüber hinaus wurden Signalintensitätsänderungen einzelner AMADORI-Anomere nicht
biologischer Amine in Abhängigkeit der Temperatur registriert.[135] Da diese Beobachtungen bisher
nicht systematisch untersucht wurden, liegen bis heute keine Ergebnisse zu den thermodynamischen
Eigenschaften der Anomerensysteme von AMADORI-Verbindungen vor.
Schema 12: Tautomerensystem D-Fructose-analoger AMADORI-Verbindungen 6 in wässriger Lösung. *Relative Konzentration bei 20-30 °C.[119,127]

THEORETISCHER HINTERGRUND
- 16 -
NMR-Untersuchungen an AMADORI-Verbindungen 6 der D-Glucose 2a zeigen im alkalischen
Milieu eine zunehmende Linienverbreiterung für die Protonenresonanzen der furanoiden Anomere,
die auf eine pH-abhängige Beschleunigung der Mutarotation hinweist. Eine solche beschleunigte
Mutarotation konnte bislang jedoch lediglich unter der Einwirkung von Basen, nicht aber in dem für
die MAILLARD-Reaktion typischen sauren Milieu beobachtet werden.[127] Vielmehr wird die
Äquilibrierung der Anomerensysteme zweier AMADORI-Verbindungen in einer Publikation von DE GRACIA
GARCÍA MARTÍN et al. (1990) als ein langsamer Prozess beschrieben, der erst nach vier Tagen
abgeschlossen ist.10;[141] Eine systematische Untersuchung der Anomerisierung von
AMADORI-Verbindungen ist in der Literatur bislang nicht beschrieben.
2.1.4 REAKTIONEN VON DERIVATEN DER D-FRUCTOSE
Untersuchungen verschiedener MAILLARD-Reaktionssysteme haben gezeigt, dass die
Bräunungskinetik von AMADORI-Verbindungen 6 schneller ist als die entsprechender
HEYNS-Verbindungen 7.[142,143] Gleichermaßen ist – wie auch schon in Kapitel 2.1 beschrieben –
D-Fructose 1a in wässrigen Systemen reaktiver als D-Glucose 2a. Dies ist nicht nur am Beispiel der
Glycosylierung von Proteinen in physiologischen Systemen feststellbar, sondern zeigt sich auch anhand
ihrer jeweiligen Abbaukinetiken in MAILLARD-Modellreaktionen.[144,145] N,N-Difructosylaminoderivate 8
zeigen schließlich im Vergleich zu allen bisher genannten Verbindungen die größte Reaktivität[121] und
folglich auch die höchsten Bräunungsraten.[146]
Da weder reduzierende Zucker noch Aminosäuren und AMADORI- bzw. HEYNS-Verbindungen Strahlung
im sichtbaren Bereich des elektromagnetischen Spektrums absorbieren, muss jegliche Art von
Bräunung aus Reaktionen der verschiedenen Zuckerderivate resultieren. Im Folgenden sollen die
Hauptabbauwege von Derivaten der D-Fructose erörtert werden. Diese stellen sowohl Reaktionen im
Sinne der intermediären Phase der MAILLARD-Reaktion als auch im Sinne der Karamellisierung dar. Wie
bereits in Kapitel 2.1.1.2 beschrieben, ist die Enolisierung nicht nur als Form der Tautomerie, sondern
gleichermaßen als Auftaktprozess für Abbaureaktionen anzusehen. Dies zeigt sich eindrucksvoll in der
in Kapitel 2.1.3.2 beschriebenen Beobachtung der irreversiblen 2,3-Enolisierung von
AMADORI-Verbindungen von SIMON und HEUBACH (1965), die durch eliminative Abspaltung der
Aminokomponente zur Bildung von 1-Desoxy-D-erythro-hexos-2,3-diulose (1-Desoxyglucoson) 9a
führt.11;[137,147,148] Trotz ihres reversiblen Charakters kann jedoch auch die 1,2-Enolisierung in
Abbaureaktionen münden. Hierbei kann es zum einen zur vinylogen β-Eliminierung von Wasser
kommen, woraufhin das intermediäre Imin zur 3-Desoxy-D-erythro-hexos-2-ulose (3-Desoxyglucoson)
9b hydrolysiert wird.[147,149] Dabei wird auch die Bildung der epimeren 3-Desoxy-D-threo-hexos-2-ulose
(3-Desoxygalactoson) 9c beobachtet.[150-153] Der Mechanismus zu deren Bildung geht von 9b aus, das
zunächst ein Molekül Wasser eliminiert und es anschließend unter Konfigurationsumkehr wieder
10 Die Aminosubstituenten der untersuchten AMADORI-Verbindungen tragen hierbei keine Carboxylfunktion.
11 Prinzipiell kann eine β-Eliminierung des 2,3-Endiols eines geeigneten Derivates der D-Fructose auch in Richtung
des C-4 erfolgen, wobei es zur Bildung von 4-Desoxy-2,3-hexosdiulose[181] bzw.
1-Amino-1,4-didesoxy-2,3-hexosdiulose[185] (4-Desoxyglucoson) kommt. Dies ist insbesondere dann zu
beobachten, wenn am C-4 eine glycosidische Bindung vorliegt, sodass im Zuge der β-Eliminierung ein Zucker
abgespalten wird.[369] Im Falle D-Fructose-analoger AMADORI-Verbindungen 6 handelt es sich jedoch bei der
Aminofunktion am C-1 um die beste Abgangsgruppe, sodass lediglich die Bildung von 1-Desoxyglucoson 9a
quantitativ bedeutsam ist.

THEORETISCHER HINTERGRUND
- 17 -
anlagert.[151-153] Zum anderen kann das 1,2-enolisierte Tautomer auch einem schrittweise oxidativen
Abbau unterliegen, wobei nach Hydrolyse D-arabino-Hexos-2-ulose (D-Glucoson) 9d freigesetzt
wird.[154,155] Schließlich kann theoretisch auch eine retro-AMADORI-Umlagerung mit anschließender
hydrolytischer Spaltung zur Freisetzung der korrespondierenden Aldose sowie des
Aminosubstituenten führen.12 Alle genannten Reaktionswege führen damit zur Freisetzung des
unveränderten Aminosubstituenten, der damit definitionsgemäß die Funktion eines Katalysators
einnimmt (vgl. Schema 13).[83,94]
Schema 13: Bildungswege der α-Dicarbonylverbindungen 9a-9d ausgehend von einer AMADORI-Verbindung 6.
Die α-Dicarbonylverbindungen bzw. (Desoxy-) Osone 9a-9d zeichnen sich durch eine
gegenüber reduzierenden Zuckern erhöhte Reaktivität aus.[83] Außerdem bilden sie aufgrund ihrer
zusätzlichen Carbonylfunktion zum Teil außerordentlich komplexe Anomerensysteme.[156-160] (Desoxy-)
Osone werden vorwiegend über retro-Aldolreaktionen, oxidative α-Dicarbonylspaltung, hydrolytische
12 Dieser Reaktionsweg spielt in der Realität allerdings keine Rolle.[108,111]

THEORETISCHER HINTERGRUND
- 18 -
β-Dicarbonylspaltung sowie amininduzierte β-Dicarbonylspaltung abgebaut.[161] Sie gehen jedoch auch
Benzilsäure-Umlagerungen sowie STRECKER-Abbaureaktionen ein.[162,163] Im Zuge dieser Reaktionen
entstehen unter anderem Aldosen sowie α-Dicarbonylverbindungen mit verkürzter Kohlenstoffkette,
Aminozucker, Aldehyde und organische Säuren. Bei einer Vielzahl dieser intermediär gebildeten
Produkte des Zuckerabbaus handelt es sich um reaktive Moleküle. Diese kondensieren im Zuge der
finalen Phase der MAILLARD-Reaktion zu höhermolekularen chromophoren Verbindungen, die zunächst
als Huminstoffe verstanden,[164] später jedoch unter dem Begriff der Melanoidine zusammengefasst
wurden.[165] Aufgrund ihrer braunen Färbung lässt sich der Fortschritt der späten Phase der
MAILLARD-Reaktion durch photometrische Messungen verfolgen.[5,166-169] Dabei wird die Bräunung
zumeist als Extinktion bei einer Wellenlänge von 420 nm angegeben.
Neben den genannten Spaltungs- und Umlagerungsreaktionen können die cyclischen Anomere
von (Desoxy-) Osonen unter Hitzeeinwirkung auch Wasser eliminieren und ggf. aromatisiert werden.
Dabei wird 9a unter anderem zu Dihydrohydroxymaltol (DHHM) 10, Acetylformoin 11 und Isomaltol
12 umgesetzt (vgl. Schema 14).13;[170,171]
Schema 14: Mechanismen der Bildung von Dihydrohydroxymaltol (DHHM) 10, Acetylformoin 11 und
Isomaltol 12 aus den Anomeren des 1-Desoxyglucosons 9a.
Dagegen bildet sich aus 9b insbesondere 5-Hydroxymethylfurfural (HMF) 13. Der in der
Literatur weitverbreitetste Reaktionsmechanismus geht von der Enolisierung von 9b mit
anschließender vinyloger β-Eliminierung von Wasser aus. Nach Cyclisierung des 3,4-ungesättigten
13 Bei den in Schema 14 gezeigten Reaktionsmechanismen sind die in der Publikation von KAUFMANN et al. (2012)
als 1a, 1d, 1e, 1h und 1i bezeichneten Anomere des 1-Desoxyglucosons 9a mutmaßlich
reaktionsverantwortlich.[159] Bei den übrigen Anomeren 1b, 1c, 1f und 1g handelt es sich um die geminalen Diole
der Anomere 1a, 1d und 1h. Da eine Temperaturerhöhung zur Abspaltung von Wasser aus den gem-Diolen
führt,[159] können die in Schema 14 gezeigten Mechanismen auch ausgehend von den gem-Diolen formuliert
werden.

THEORETISCHER HINTERGRUND
- 19 -
Intermediates zum Halbketal erfolgt die Aromatisierung des Heterocyclus‘ durch die wiederholte
Eliminierung eines Wassermoleküls. (vgl. Schema 15 (unterer Reaktionsweg)).[172,173] Mit Blick auf die
in Schema 14 gezeigten Reaktionsmechanismen ist jedoch anzumerken, dass mechanistisch betrachtet
oftmals keine Notwendigkeit besteht, Reaktionen ausgehend von acyclischen Isomeren zu
formulieren. Im Falle der α-Dicarbonylverbindungen 9a-9d liegen die acyclischen Isomere in derart
geringer relativer Konzentration vor, dass ihr direkter Nachweis bis heute aussteht.[156-160] Die
cyclischen Anomere der α-Dicarbonylverbindungen sind demnach die energieärmeren und damit
thermodynamisch stabileren Systeme. Es ist daher anzunehmen, dass die Aktivierungsenergien von
Reaktionen, die unmittelbar von cyclischen Anomeren ausgehen, geringer sind, als solche, die bei
einem cyclischen Anomer starten und durch Ringöffnung zunächst in ein acyclisches Isomer überführt
werden müssen. Somit erscheint es auch hier angemessen, einen alternativen Reaktionsmechanismus
zu formulieren, der von cyclischen Anomeren ausgeht. Zur Formulierung eines solchen
Reaktionsmechanismus bietet es sich an, die Anomere 5g und 5h aus der Publikation von KÖPPER und
FREIMUND (2003) zugrunde zu legen, die bei 27 °C 23% der relativen Anomerenkonzentration
ausmachen.[157] Wie seit langem bekannt, nehmen die relativen Konzentrationen furanoider Anomere
mit steigender Temperatur zu,[39,62,131,174,175] sodass die Konzentration der Anomere 5g und 5h unter
tatsächlichen Reaktionsbedingungen, d.h. unter Hitzeeinwirkung, deutlich erhöht sein muss.
Gleichzeitig ist die Eliminierung von Wasser aus dem gem-Diol bei erhöhter Temperatur – analog dem
Verhalten der Anomere des 1-Desoxyglucosons 9a – wahrscheinlich.[159] Der in Schema 15 (oberer
Reaktionsweg) gezeigte Reaktionsmechanismus basiert auf den obigen Überlegungen und ist zugleich
an den von WEENEN und TJAN (1992) formulierten Mechanismus angelehnt.[176]
Schema 15: Mechanismus der Bildung von 5-Hydroxymethylfurfural (HMF) 13 aus 3-Desoxyglucoson 9b. (Frei nach[172,173,176])
Die Bildung heterocyclischer Substanzen ist prinzipiell nicht nur ausgehend von
α-Dicarbonylverbindungen möglich. Einige solcher O-Heterocyclen wie HMF 13,[173,177,178]
2-Hydroxy- bzw. 2-Aminoacetylfuran (HAF bzw. AAF) 14 und 15, DHHM 10 und Maltol 16[179] können
sich in Abhängigkeit der zumeist sauren Reaktionsbedingungen jeweils auch direkt aus den
Hauptanomeren der D-Fructose 1a bzw. AMADORI-Verbindungen 6 bilden (vgl. Schema 17). Der bisher
diskutierte Mechanismus der Bildung von HAF 14, das als Abbauprodukt verschiedener Zucker (etwa
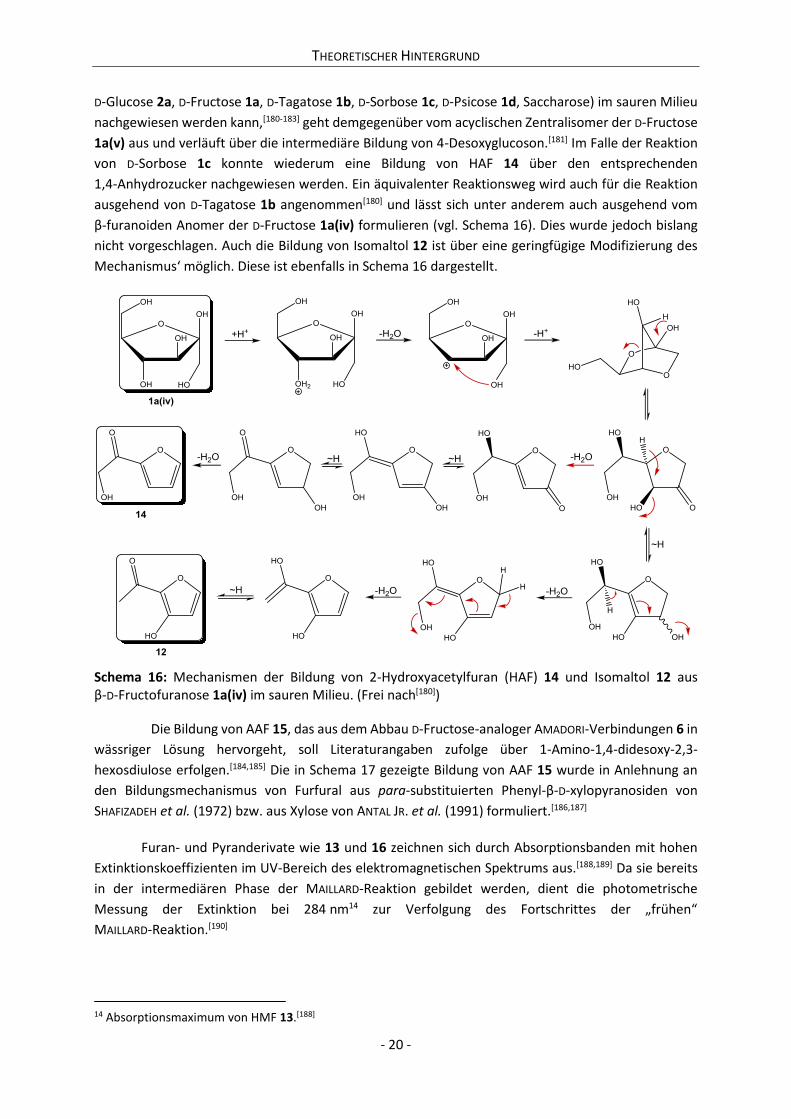
THEORETISCHER HINTERGRUND
- 20 -
D-Glucose 2a, D-Fructose 1a, D-Tagatose 1b, D-Sorbose 1c, D-Psicose 1d, Saccharose) im sauren Milieu
nachgewiesen werden kann,[180-183] geht demgegenüber vom acyclischen Zentralisomer der D-Fructose
1a(v) aus und verläuft über die intermediäre Bildung von 4-Desoxyglucoson.[181] Im Falle der Reaktion
von D-Sorbose 1c konnte wiederum eine Bildung von HAF 14 über den entsprechenden
1,4-Anhydrozucker nachgewiesen werden. Ein äquivalenter Reaktionsweg wird auch für die Reaktion
ausgehend von D-Tagatose 1b angenommen[180] und lässt sich unter anderem auch ausgehend vom
β-furanoiden Anomer der D-Fructose 1a(iv) formulieren (vgl. Schema 16). Dies wurde jedoch bislang
nicht vorgeschlagen. Auch die Bildung von Isomaltol 12 ist über eine geringfügige Modifizierung des
Mechanismus‘ möglich. Diese ist ebenfalls in Schema 16 dargestellt.
Schema 16: Mechanismen der Bildung von 2-Hydroxyacetylfuran (HAF) 14 und Isomaltol 12 aus β-D-Fructofuranose 1a(iv) im sauren Milieu. (Frei nach[180])
Die Bildung von AAF 15, das aus dem Abbau D-Fructose-analoger AMADORI-Verbindungen 6 in
wässriger Lösung hervorgeht, soll Literaturangaben zufolge über 1-Amino-1,4-didesoxy-2,3-
hexosdiulose erfolgen.[184,185] Die in Schema 17 gezeigte Bildung von AAF 15 wurde in Anlehnung an
den Bildungsmechanismus von Furfural aus para-substituierten Phenyl-β-D-xylopyranosiden von
SHAFIZADEH et al. (1972) bzw. aus Xylose von ANTAL JR. et al. (1991) formuliert.[186,187]
Furan- und Pyranderivate wie 13 und 16 zeichnen sich durch Absorptionsbanden mit hohen
Extinktionskoeffizienten im UV-Bereich des elektromagnetischen Spektrums aus.[188,189] Da sie bereits
in der intermediären Phase der MAILLARD-Reaktion gebildet werden, dient die photometrische
Messung der Extinktion bei 284 nm14 zur Verfolgung des Fortschrittes der „frühen“
MAILLARD-Reaktion.[190]
14 Absorptionsmaximum von HMF 13.[188]
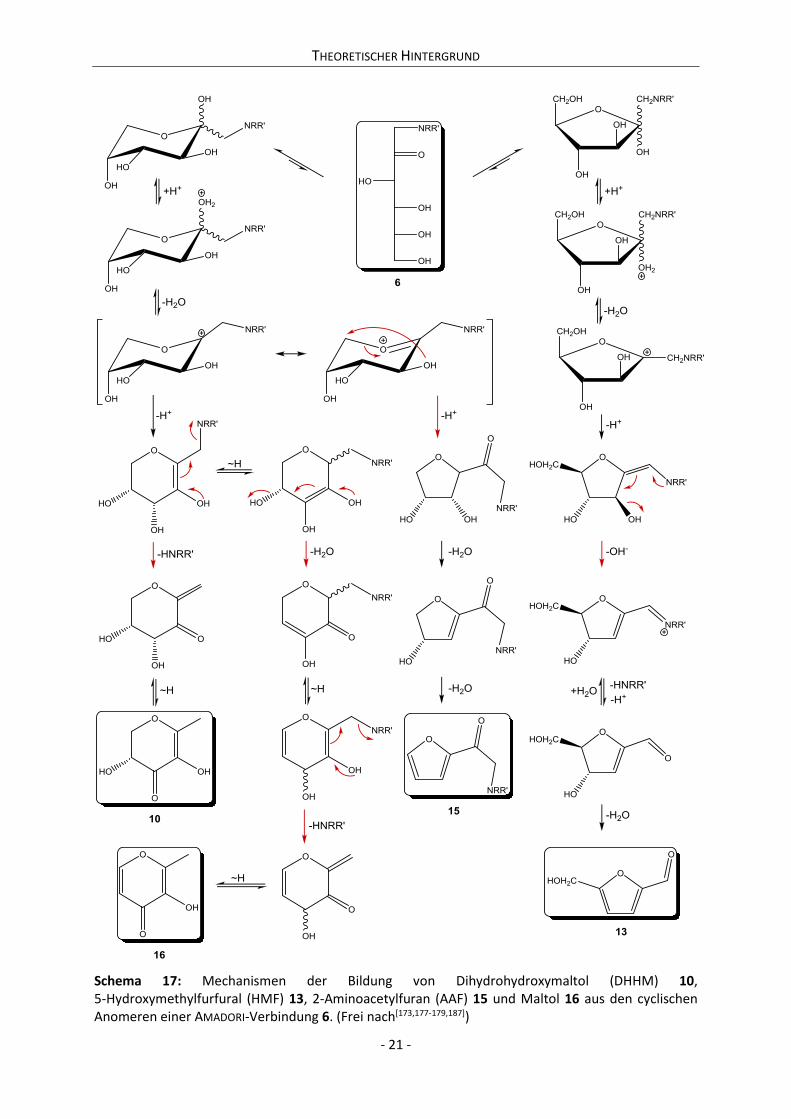
THEORETISCHER HINTERGRUND
- 21 -
Schema 17: Mechanismen der Bildung von Dihydrohydroxymaltol (DHHM) 10, 5-Hydroxymethylfurfural (HMF) 13, 2-Aminoacetylfuran (AAF) 15 und Maltol 16 aus den cyclischen Anomeren einer AMADORI-Verbindung 6. (Frei nach[173,177-179,187])

THEORETISCHER HINTERGRUND
- 22 -
2.2 NMR-Spektroskopie
In diesem Abschnitt werden die für diese Arbeit relevanten Aspekte der kernmagnetischen
Resonanzspektroskopie (Nuclear Magnetic Resonance Spektroskopie; NMR-Spektroskopie)
vorgestellt. Diese unterteilen sich in Methoden der Strukturaufklärung sowie in Methoden der
Quantifizierung und der Analyse kinetischer und dynamischer Prozesse. Dabei werden die allgemeinen
physikalischen Grundlagen des Kernmagnetismus‘ sowie der kernmagnetischen Resonanz als bekannt
vorausgesetzt und es wird lediglich auf die Standardwerke der Fachliteratur verwiesen.[191,192]
2.2.1 STRUKTURAUFKLÄRUNG
Zur vollständigen Strukturaufklärung von Kohlenhydraten in kondensierter Phase stellt die
NMR-Spektroskopie die wichtigste spektroskopische Methode der chemischen Analytik dar.[193,194] Im
Laufe der vergangenen 60 Jahre wurden zahlreiche NMR-Experimente entwickelt, die durch die
Messung verschiedener spektraler Parameter Rückschlüsse auf die räumliche Anordnung von Atomen
innerhalb eines Moleküls und somit auf die geometrische Molekülstruktur zulassen.[195] Dabei zielen
moderne Forschungsansätze neben der Entwicklung immer stärkerer Kryomagnetsysteme,
empfindlicherer Kryoprobenköpfe und leistungsfähiger Shim-Systeme auf eine Vereinfachung von
NMR-Spektren durch Vergrößerung der spektralen Auflösung15;[196-201] sowie auf die Maximierung von
Signal/Rausch-Verhältnissen bei Verkürzung von Messzeiten16 ab.[202,203] Da die Vielzahl entwickelter
Pulsprogramme mit der Zeit unüberschaubar groß geworden ist, werden immer wieder Review-Artikel
publiziert, in denen die Vor- und Nachteile einzelner Sequenzen diskutiert und zweckmäßige
Empfehlungen für spezielle Fragestellungen formuliert werden.[193,194,204] Routinemäßig eingesetzte
NMR-Methoden bei der Strukturaufklärung von Kohlenhydratsystemen sind in erster Linie
eindimensionale 1H- und 13C-NMR-Experimente (1H, 13C und DEPT) sowie zweidimensional
homonuklear (COSY, TOCSY, NOESY, J-resolved) und heteronuklear korrelierte Experimente
(HSQC-TOCSY, HSQC, HMQC, HMBC).[193,194] Dabei finden im Bedarfsfall auch die eindimensionalen
„selektiv“-Varianten der zweidimensionalen NMR-Experimente Anwendung.[205-207] Die aus COSY- und
TOCSY-Experimenten resultierenden Spektren dienen der Ermittlung skalarer
Nachbarschaftsbeziehungen von Wasserstoffatomen und damit zur Identifizierung von
Spinsystemen.[208-210] Im Bereich der chemischen Verschiebung der Resonanzen nicht anomerer
Saccharidprotonen (ca. 3.0 bis 4.2 ppm) zeigen sich in homonuklear-korrelierten 2D-Spektren jedoch
zumeist starke Signalüberlagerungen.[194] Eine erheblich verbesserte Auflösung in der f1-Dimension
wird aufgrund der zumeist deutlich größeren Signaldispersion von X-Kernen durch die Einbeziehung
heteronuklearer Korrelationen im Rahmen von 2D-HSQC-TOCSY-Experimenten erreicht.[211,212]
NOE-Korrelationen spiegeln die Beziehung sich räumlich nahestehender Protonen wider.17 Derartige
Korrelationen treten unter anderem bei 1,3-diaxialer Stellung von Protonen in pyranoiden
15 Die Vergrößerung der spektralen Auflösung wird unter anderem durch vieldimensionale NMR-Experimente
sowie homonukleare Entkopplungstechniken erreicht.
16 Bei der Maximierung von Signal/Rausch-Verhältnissen bei gleichzeitiger Verkürzung von Messzeiten kommt
insbesondere die Verwendung von Radiopulsen im sogenannten ERNST-Winkel zum Einsatz.[239,240]
17 Die Stärke dipolarer Wechselwirkungen, die dem NOE-Effekt zugrunde liegen, nimmt mit der sechsten Potenz
des Abstandes der Dipole ab.[191,370] NOE-Signale können aufgrund dessen für Messungen von Kernabständen in
der Größenordnung bis etwa 5 Å genutzt werden.[371-374]

THEORETISCHER HINTERGRUND
- 23 -
Ringsystemen auf und dienen damit der Konformationsanalyse.[213] HSQC- sowie
HMQC-NMR-Experimente liefern Informationen über direkte Bindungen zwischen X-Kernen (zumeist 13C) und Wasserstoffatomen.[214,215] Dabei lassen sich anhand von 1J(13C,1H)-Kopplungen zusätzliche
Informationen über die Konformation von Zuckeranomeren erhalten, sofern keine 13C-Entkopplung
vorgenommen wird.[157,216-220] HMBC-Experimente wiederum geben Auskunft über 1H-13C-long-range-
Beziehungen.[221] Über entsprechende Korrelationen lässt sich zum einen die Ringgröße cyclischer
Zuckeranomere und zum anderen die Sequenz glycosidischer Bindungen in Oligosacchariden
bestimmen.[159,222] Bezüglich der Strukturaufklärung von Zuckeranomeren haben auch J-resolved-
Experimente einen großen Informationsgehalt. Hierbei werden homonukleare geminale, vicinale und
long-range Kopplungskonstanten gemessen, die Auskunft über exocyclische Torsionswinkel
geben.18;[50,51,223] Daraus lassen sich insbesondere die Konformationen pyranoider Anomere ableiten
sowie pyranoide von furanoiden und bicyclischen Anomeren unterscheiden.
2.2.2 QUANTITATIVE NMR-SPEKTROSKOPIE (QNMR)
NMR-spektroskopische Messungen erlauben nicht nur die Aufklärung geometrischer
Molekülstrukturen, sondern gleichermaßen die Untersuchung kinetischer und dynamischer
Prozesse.[224-226] Grundlage der kinetischen Verfolgung chemischer Reaktionen mittels
NMR-Spektroskopie ist die Möglichkeit der simultanen relativen und absoluten Quantifizierung
mehrerer Analyten innerhalb einer Probe.[227-230] Diese Möglichkeit ist allerdings nur unter sehr
spezifischen Randbedingungen gegeben, die aufgrund dessen möglichst exakt einzuhalten sind. Die
Fachliteratur bietet detaillierte Beschreibungen der Methodik der quantitativen
NMR-Spektroskopie,[231,232] sodass im Folgenden lediglich die für diese Arbeit kritischen Grundlagen
erörtert werden. Bei der üblicherweise genutzten Technik der Puls-FOURIER-Transform
NMR-Spektroskopie werden kurze Radiopulse in der Größenordnung von etwa 10 µs zur Anregung der
Kernspins genutzt. Das Profil der Strahlungsleistung eines solchen Rechteckpulses entspricht einer
𝑠𝑖𝑛𝑐-Funktion.19 Diese Funktion geht unmittelbar aus der FOURIER-Transformation der
Rechteckfunktion hervor und hat bei 𝑥 = 0 ein Maximum und Nullstellen bei 𝑥 = −1 (2𝑡𝑝(90°))⁄
sowie 𝑥 = +1 (2𝑡𝑝(90°))⁄ . Dabei entspricht 𝑡𝑝(90°) der Dauer des 90°-Pulses [s], sodass sich ein
spektrales Anregungsfenster von ∆𝜈 = 1 𝑡𝑝⁄ ergibt. Die Leistung eines Rechteckpulses ist innerhalb
dieses Anregungsfensters demnach nicht konstant, sondern frequenzabhängig.[233] Das hat zur Folge,
dass auch der Anregungswinkel vorhandener Kernspins von ihrer jeweiligen Resonanzfrequenz
abhängt. Eine Pulsdauer von 10 µs entspricht einer Anregungsbreite von 100 kHz. Bei einem 13C-NMR-Experiment verteilen sich die Resonanzen zumeist über einen Verschiebungsbereich von
250 ppm, was bei Messungen an einem 600 MHz-NMR-Spektrometer einem Frequenzfenster von
37.5 kHz gleichkommt. Die Anregungsbreite entspricht damit nur dem rund 2.67-fachen der zu
erwartenden Signaldispersion. 13C-Kerne mit großer Differenz ihrer Resonanzfrequenz zur
Trägerfrequenz erfahren somit nicht die Leistung eines 90°-Pulses und werden entsprechend nicht
18 Zur Abschätzung exocyclischer Torsionswinkel, ausgehend von 3J(1H,1H)-Kopplungskonstanten, steht eine
Reihe von Softwaretools zur Verfügung, die auf den Publikationen von HAASNOOT et al. basieren.[50,51,375-377] Des
Weiteren existieren insbesondere für die Berechnung von Torsionswinkeln von Glycosiden,
Kohlenwasserstoffen, Peptiden und Nukleotiden online-Datenbanken, die auf optimierten KARPLUS-Funktionen
zahlreicher Publikationen der vergangenen vier Jahrzehnte basieren.[378]
19 𝑓(𝑥) = 𝑠𝑖𝑛𝑐(𝑥) =sin(𝑥)
𝑥

THEORETISCHER HINTERGRUND
- 24 -
vollständig angeregt. Die Integrale ihrer entsprechenden Resonanzsignale können somit nicht
zwangsläufig für quantitative Fragestellungen genutzt werden. Betrachtet man jedoch lediglich die
Integrale von Resonanzen mit ähnlichen Frequenzabständen zur Trägerfrequenz, so erfahren die
entsprechenden Kernspins auch eine vergleichbare Anregung, sodass eine quantitative Auswertung
möglich ist. In Bezug auf die quantitative Auswertung von 1H-NMR-Spektren ist ein Arbeiten mit dieser
Einschränkung nicht notwendig. Hier verteilen sich die Resonanzen zumeist über einen
Verschiebungsbereich von rund 12 ppm, was bei Messungen an einem 600 MHz-NMR-Spektrometer
einem Frequenzfenster von 7.2 kHz entspricht. Bei einem Anregungsfenster von 100 kHz erfahren
demnach alle Protonenspins eine vergleichbare Anregung.
Nach erfolgtem 90°-Puls ist der z-Magnetisierungsvektor eines betrachteten Kerns vollständig
in die x,y-Ebene ausgelenkt. Dieser Prozess kehrt sich infolge der longitudinalen Relaxation
(𝑇1-Relaxation) um. Die Beschreibung der Regeneration der z-Magnetisierung in den ursprünglichen
Gleichgewichtszustand erfolgt nach der BLOCH-Gleichung (1).[234]
𝑑𝑀𝑧(𝑡)
𝑑𝑡= −
𝑀𝑧(𝑡) − 𝑀𝑧,𝑒𝑞
𝑇1(𝑖)
⇔𝑀𝑧(𝑡) = 𝑀𝑧,𝑒𝑞 − (𝑀𝑧,𝑒𝑞 −𝑀𝑧(0)) ∙ 𝑒𝑥𝑝 (−𝑡
𝑇1(𝑖)) (1)
Dabei ist 𝑀𝑧(𝑡) die z-Magnetisierung zum Zeitpunkt 𝑡, 𝑀𝑧,𝑒𝑞 die z-Magnetisierung im
Gleichgewichtszustand, 𝑀𝑧(0) die z-Magnetisierung zum Zeitpunkt 𝑡 = 0𝑠, 𝑡 die Zeit [s] und 𝑇1(𝑖)
die
longitudinale Relaxationszeit eines Isomers 𝑖 [s]. Aus Gleichung (1) geht hervor, dass sich die im
Gleichgewichtszustand bestehende z-Magnetisierung zum Zeitpunkt 𝑇1(𝑖)
nach einem 90°-Puls zu
63.2% wiederhergestellt hat.20 Von den in der x,y-Ebene verbleibenden 36.8% der
Gleichgewichtsmagnetisierung gehen innerhalb einer erneuten Wartezeit von 𝑇1(𝑖)
wiederum 63.2% in
z-Richtung über. Nach einer Wartezeit von 5 ∙ 𝑇1(𝑖) ist die ursprüngliche Gleichgewichtsmagnetisierung
auf diese Weise zu 99.3% regeneriert.21 Entsprechend diesen Berechnungen können NMR-Spektren
nur dann quantitativ ausgewertet werden, wenn Recyclingdelays von mindestens 5 ∙ 𝑇1(𝑖) eingehalten
werden. Problematisch dabei ist, dass Relaxationszeiten niemals a priori bekannt sind. Recyclingdelays
müssen daher im Sinne einer worst-case-Betrachtung großzügig abgeschätzt oder 𝑇1-Zeiten
experimentell ermittelt werden. Die experimentelle Bestimmung longitudinaler Relaxationsraten
erfolgt dabei etwa über die Progressive Saturation-Methode oder das Inversion Recovery-
Experiment.22;[235,236] Für das 13C-Isotop liegen 𝑇1-Zeiten üblicherweise im Bereich von 1-40 s.[237] Dabei
beträgt die Messempfindlichkeit des Kerns relativ zur Messempfindlichkeit des 1H-Isotops nur
0.017%.[238] Trotzdem ist die Nutzung der quantitativen 13C-NMR-Spektroskopie für diese Arbeit
20 𝑀𝑧(𝑇1) = (1 −
1
𝑒) ∙ 𝑀𝑧,𝑒𝑞 ≅ 0.632 ∙𝑀𝑧,𝑒𝑞
21 Regeneration der Gleichgewichtsmagnetisierung nach einem 90°-Puls in Abhängigkeit von Vielfachen der
𝑇1-Relaxationszeit, berechnet nach Gleichung (1):
𝑡 1 ∙ 𝑇1 2 ∙ 𝑇1 3 ∙ 𝑇1 4 ∙ 𝑇1 5 ∙ 𝑇1 6 ∙ 𝑇1 7 ∙ 𝑇1
𝑀𝑧(𝑡)/𝑀𝑧,𝑒𝑞 0.632 0.865 0.950 0.982 0.993 0.998 0.999
22 Im Falle dynamischer Anomerensysteme, wie sie im Rahmen dieser Arbeit untersucht werden, liefert das
Inversion Recovery Experiment lediglich effektive 𝑇1-Zeiten, die sich aus einer (teilweisen) Mittelung der
intrinsischen 𝑇1-Zeiten der im chemischen Austausch befindlichen Kerne ergeben.[379]

THEORETISCHER HINTERGRUND
- 25 -
unerlässlich, da hauptsächlich Derivate der D-Fructose untersucht werden. Diese haben keine
anomeren Protonen, sodass eine Vielzahl der Messungen unter Zuhilfenahme der anomeren 13C-NMR-Resonanzen durchgeführt werden muss. Um 13C-NMR-Spektren mit ausreichendem
Signal/Rausch-Verhältnis zu erzielen, ist es je nach Probenkonzentration nötig, hunderte bis tausende
Einzelmessungen zu akkumulieren.[232] Dabei Recyclingdelays von mindestens 5 ∙ 𝑇1 einzuhalten, ist
nicht wirtschaftlich. 13C-NMR-Experimente werden aufgrund dessen üblicherweise unter nicht
quantitativen Bedingungen gemessen. Vielmehr ist man bestrebt, das Signal/Rausch-Verhältnis derart
zu optimieren, dass kurze Messzeiten resultieren. Hierbei bedient man sich einerseits des
ERNST-Winkels[239,240] und andererseits des Kern-OVERHAUSER-Effekts (NOE-Effekt).[241-244] Dieser bewirkt
Intensitätsänderungen einer Kernresonanz bei simultaner Sättigung der Resonanz eines benachbarten
Kerns. Der NOE-Effekt baut sich damit in Abhängigkeit der experimentellen Umsetzung der
Protonen-Breitbandentkopplung im Zuge von 13C-NMR-Experimenten auf. Die Aufbaurate des
NOE-Effektes entspricht dabei der inversen longitudinalen 13C-Relaxationszeit und kann damit als
vergleichsweise langsam betrachtet werden.[245] Die Protonen-Breitbandentkopplung dient damit
nicht nur dem Zweck der Vereinfachung von 13C-NMR-Spektren durch die Beseitigung von
heteronuklearen Spin-Spin-Kopplungen, sondern gleichermaßen einer Verbesserung des
Signal/Rausch-Verhältnisses. In Hinblick auf die 13C-NMR-Spektroskopie verfälscht der heteronukleare
OVERHAUSER-Effekt jedoch die quantitative Spektrenauswertung. Es ist daher notwendig, die
verschiedenen Techniken der Protonen-Breitbandentkopplung näher zu betrachten.
Wie bereits erwähnt, baut sich der NOE-Effekt mit einer der inversen 𝑇1-Zeit entsprechenden
Rate auf.[245] Die Sättigung der Protonen-Übergänge hingegen ist ein schneller Prozess, sodass die auf
der Protonenkopplung basierenden 13C-Multipletts unmittelbar nach dem Einschalten des
Entkopplerkanals kollabieren. Daraus ergeben sich drei mögliche Varianten der Protonen-
Breitbandentkopplung (vgl. Abbildung 2).
Die erste Variante (inverse-gated decoupling) besteht darin, den Entkopplerkanal
ausschließlich während der Detektion des freien Induktionsabfalls (free induction decay, FID)
einzuschalten. Das hat den Kollaps der 13C-Multipletts zur Folge, während sich der NOE-Effekt erst im
Laufe der Akquisitionszeit (Acquisition time) aufbaut und damit keinen Einfluss auf die Amplitude des
FID hat. Die Intensitäten der 13C-NMR-Resonanzen sind somit nicht vom NOE-Effekt verfälscht.
Weiterhin besteht die Möglichkeit der Protonen-Breitbandentkopplung während des
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\zgpg
1
ZE
D11
2
30m 10u D11 DELTA 4u 10u 100m
P1 ph1=[0,2..]
Go loop
30m
MC
MC loop
ZDH 1
PLW 12 PLW 13 PLW 12 PLW 13
C 13
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\zgig
1
ZE
D11
2
30m D1
P1 ph1=[0,2..]
Go loop
30m
MC
MC loop
ZDH 1
PLW 12
C 13
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\zggd
1
ZE
D11
2
30m 4u D1
P1 ph1=[0,2..]
Go loop
30m
MC
MC loop
ZDH 1
PLW 12
C 13
a) b) c)
Abbildung 2: Graphische Darstellung verschiedener Pulsprogramme zur Protonen-Breitbandentkopplung von 13C-NMR-Spektren.* a) inverse-gated decoupling, b) gated decoupling, c) power-gated decoupling. (Der Entkopplerkanal ist jeweils oben und der X-Kernkanal unten abgebildet.) *Die graphischen Darstellungen sind der Software Bruker TopSpin 3.5 pL 2 entlehnt.

THEORETISCHER HINTERGRUND
- 26 -
Relaxationsdelays (gated decoupling).[246] Der Relaxationsdelay liegt in der Regel in der Größenordnung
der 𝑇1-Zeit, sodass sich der NOE-Effekt aufbauen kann. Wird der Entkopplerkanal nun vor Beginn der
Acquisition time ausgeschaltet, beinhalten die resultierenden Spektren durch den NOE-Effekt
intensivierte 13C-Multipletts. Die dritte Variante der Protonen-Breitbandentkopplung besteht
schließlich in einer Kombination der beiden erstgenannten Techniken (power-gated decoupling).[247]
Hieraus ergeben sich 13C-NMR-Spektren mit NOE-verstärkten Singuletts.
Die Durchführung quantitativer 13C-NMR-Experimente ist mit einem hohen Zeitaufwand
verbunden, der jedoch durch verschiedene Maßnahmen stark reduziert werden kann. Die erste
Möglichkeit besteht in der Dotierung der Proben mit paramagnetischen Relaxationsbeschleunigern23
(Paramagnetic Relaxation Enhancement).[248] Diese verkürzen die longitudinalen Relaxationszeiten der 13C-Kerne zum Teil drastisch, sodass auch die Recyclingdelays reduziert werden können. Damit können
unter Einhaltung der quantitativen Messbedingungen im selben Zeitraum mehr FIDs akkumuliert
werden, was zu einem verbesserten Signal/Rausch-Verhältnis führt. Da jedoch die transversale
Relaxationszeit (𝑇2) definitionsgemäß maximal den Wert der longitudinalen Relaxationszeit annehmen
kann, besteht die Gefahr einer Linienverbreiterung der Resonanzsignale.24;[192] Im Falle der
Untersuchung von Kohlenhydraten hat die Methode zudem den Nachteil, dass diese durch organische
Radikale oxidiert werden.[249] Außerdem gehen diverse Monosaccharide koordinative Bindungen mit
Metallionen ein, was zu einer Verschiebung ihrer Anomerengleichgewichte führt.[250,251] In diesem
Zusammenhang wurden auch Einflüsse von Metallionen auf Anomerisierungsraten festgestellt.[252-254]
Aufgrund dessen fiel im Rahmen dieser Arbeit die Wahl auf die alternative Maßnahme der selektiven
Isotopenmarkierung. Durch gezielte vollständige Substitution des 12C-Isotops durch das 13C-Isotop
erhöht sich die Empfindlichkeit gegenüber Messungen bei natürlicher Isotopenhäufigkeit um den
Faktor 90. Um mit Messungen bei natürlicher Häufigkeit dasselbe Signal/Rausch-Verhältnis zu erzielen,
müsste man das 8100-fache an Einzelmessungen akkumulieren. Mit einer angenommenen 𝑇1-Zeit von
20 s ergibt sich ein Recyclingdelay von mehr als 100 s. Um demnach bei natürlicher Häufigkeit dasselbe
Signal/Rausch-Verhältnis zu erlangen wie bei einem Single-Scan-Experiment mit einer Dauer von 100 s
bei vollständiger Isotopenmarkierung, wird eine Messzeit von rund 9.4 Tagen benötigt. Bei der
Untersuchung von Derivaten der D-Fructose bietet es sich dabei an, lediglich das jeweils anomere
Kohlenstoffatom C-2 zu markieren, da dieses im Wesentlichen für NMR-spektroskopische
Untersuchungen genutzt wird. Dies hat den Vorteil, dass es nicht zur Multiplettaufspaltung aufgrund
homonuklearer 13C-Spin-Spin-Kopplungen kommt.
2.2.3 METHODEN DER DYNAMISCHEN NMR-SPEKTROSKOPIE (DNMR)
NMR-spektroskopische Methoden erlauben – wie bereits in Kapitel 2.2.2 angedeutet – die
Untersuchung dynamischer sowie kinetischer molekularer Prozesse. Dabei bezieht sich der Begriff der
Dynamik auf Systeme, die sich im thermodynamischen Gleichgewicht befinden, sodass makroskopisch
betrachtet keine Konzentrationsänderungen einzelner Verbindungen feststellbar sind. Kinetische
Prozesse streben im Gegensatz dazu dem thermodynamischen Gleichgewicht entgegen. Der Prozess
23 Hierbei kann es sich sowohl um organische Radikale[380] als auch um paramagnetische
Übergangsmetallionen[248,381] handeln.
24 Mit den Beziehungen 𝑇1 ≥ 𝑇2 und 𝜈12⁄=
1
𝜋∙𝑇2𝑒𝑓𝑓 folgt für die Linienbreite eines NMR-Signals 𝜈1
2⁄≥
1
𝜋∙𝑇1.[192]

THEORETISCHER HINTERGRUND
- 27 -
der Anomerisierung von Monosacchariden ist aufgrund dessen während der Mutarotation25 kinetisch
und nach vollständiger Anomerenäquilibrierung dynamisch. Die Untersuchung von
Anomerisierungsraten kann daher sowohl auf Grundlage kinetischer als auch dynamischer Messungen
erfolgen. Die verschiedenen Möglichkeiten werden im Folgenden am Anomerensystem der D-Fructose
1a veranschaulicht und diskutiert. Das Anomerensystem ist in Schema 18 dargestellt.
Schema 18: Anomerensystem der D-Fructose 1a. Dabei steht P für Pyranose, F für Furanose und z für das Zentralisomer.
Wie in Kapitel 2.1.1.1 beschrieben, kristallisiert D-Fructose 1a anomerenrein als β-Pyranose
(βP). Wird βP gelöst, bildet sich ein Gleichgewichtszustand mit den übrigen vier Isomeren aus. Die
zeitabhängigen Konzentrationsänderungen der Isomere berechnen sich nach den Gleichungen (2) bis
(6).
𝑑[𝑧]
𝑑𝑡= 𝑘𝛼𝑃,𝑧[𝛼𝑃] + 𝑘𝛽𝑃,𝑧[𝛽𝑃] + 𝑘𝛼𝐹,𝑧[𝛼𝐹] + 𝑘𝛽𝐹,𝑧[𝛽𝐹] − (𝑘𝑧,𝛼𝑃 + 𝑘𝑧,𝛽𝑃 + 𝑘𝑧,𝛼𝐹 + 𝑘𝑧,𝛽𝐹)[𝑧] (2)
𝑑[𝛼𝑃]
𝑑𝑡= 𝑘𝑧,𝛼𝑃[𝑧] − 𝑘𝛼𝑃,𝑧[𝛼𝑃] (3)
𝑑[𝛽𝑃]
𝑑𝑡= 𝑘𝑧,𝛽𝑃[𝑧] − 𝑘𝛽𝑃,𝑧[𝛽𝑃] (4)
𝑑[𝛼𝐹]
𝑑𝑡= 𝑘𝑧,𝛼𝐹[𝑧] − 𝑘𝛼𝐹,𝑧[𝛼𝐹] (5)
𝑑[𝛽𝐹]
𝑑𝑡= 𝑘𝑧,𝛽𝐹[𝑧] − 𝑘𝛽𝐹,𝑧[𝛽𝐹] (6)
Diese bilden ein System homogener linearer Differentialgleichungen der ersten Ordnung und
lassen sich in der Matrixschreibweise wie folgt formulieren (vgl. Gleichung (7)):
𝑑
𝑑𝑡
(
[𝑧]
[𝛼𝑃][𝛽𝑃]
[𝛼𝐹][𝛽𝐹])
=
(
−(𝑘𝑧,𝛼𝑃 + 𝑘𝑧,𝛽𝑃 + 𝑘𝑧,𝛼𝐹 + 𝑘𝑧,𝛽𝐹) 𝑘𝛼𝑃,𝑧 𝑘𝛽𝑃,𝑧 𝑘𝛼𝐹,𝑧 𝑘𝛽𝐹,𝑧
𝑘𝑧,𝛼𝑃 −𝑘𝛼𝑃,𝑧 0 0 0
𝑘𝑧,𝛽𝑃 0 −𝑘𝛽𝑃,𝑧 0 0
𝑘𝑧,𝛼𝐹 0 0 −𝑘𝛼𝐹,𝑧 0
𝑘𝑧,𝛽𝐹 0 0 0 −𝑘𝛽𝐹,𝑧)
∙
(
[𝑧]
[𝛼𝑃][𝛽𝑃]
[𝛼𝐹][𝛽𝐹])
(7)
Das Differentialgleichungssystem hat damit die Form 𝒚′ = 𝑨 ∙ 𝒚. Dieses lässt sich mit Hilfe
eines exponentiellen Ansatzes der Form 𝒚(𝑥) = 𝒅 ∙ 𝑒𝑥𝑝(𝜆 ∙ 𝑥) durch die Berechnung der Eigenwerte
25 Der Wortbedeutung nach beschreibt der Begriff der Mutarotation die zeitabhängige Änderung des
Drehwinkels einer Lösung einer optisch aktiven Substanz.

THEORETISCHER HINTERGRUND
- 28 -
𝜆 der Matrix 𝑨 sowie ihrer entsprechenden Eigenvektoren 𝒅 lösen.[255] Unter der Randbedingung einer
relativen Konzentration der βP von 100% zum Zeitpunkt 𝑡 = 0𝑠 lassen sich geschlossene Ausdrücke
für die jeweiligen Konzentrationen sämtlicher Isomere in Abhängigkeit der Zeit formulieren. Im
Umkehrschluss bedeutet das, dass die Messung der zeitabhängigen Äquilibrierung des
Isomerensystems die Ermittlung sämtlicher Geschwindigkeitskonstanten der Matrix 𝑨 ermöglicht.
Diese Methode beschränkt sich damit nicht nur auf Messungen mittels quantitativer zeitabhängiger
NMR-Spektroskopie, sondern lässt sich auch mit Hilfe chromatographischer Trenntechniken umsetzen.
Hier ist insbesondere die gaschromatographische Analyse (GC) zu nennen.26;[35,55,65,256-260] Dabei haben
allerdings lediglich WERTZ et al. (1981) Berechnungen auf Grundlage von Gleichung (7)
vorgenommen.[259] Problematisch bei der GC-Analytik von Kohlenhydratisomeren ist, dass diese zur
Erhöhung ihrer Flüchtigkeit derivatisiert werden müssen. Hierbei handelt es sich unter anderem um
die Bildung ihrer per-O-Trimethylsilylether oder Essigsäureester in wasserfreien oder auch
wasserhaltigen organischen Lösungsmitteln.[261-263] Der Arbeitsaufwand derartiger Analysen ist
verglichen mit der Durchführung von NMR-Experimenten groß. Daneben ist die Derivatisierung der
anomeren Hydroxylfunktion von Ketosen ein sehr langsamer Prozess.[264-266] Die zu untersuchenden
Isomerensysteme können aufgrund dessen noch während der Derivatisierung anomerisieren.[131] Die
relativen Konzentrationen der Isomere sind somit sowohl von den jeweiligen
Derivatisierungsreagenzien sowie von deren zumeist organischen Lösungsmitteln beeinflusst, sodass
die zu messenden Zielgrößen bereits vor der Messung in einer unvorhersehbaren Weise modifiziert
werden. Demgegenüber erlauben NMR-spektroskopische Experimente die Messung von
Isomerensystemen ohne eine Verfälschung durch ungewollte Einflussfaktoren. Trotzdem ist die
beschriebene Vorgehensweise aus den folgenden Gründen für die Untersuchung der Anomerisierung
von Zuckern prinzipiell wenig geeignet. Wie in den Kapiteln 2.1.1.1 und 2.1.3.2 beschrieben, liegen im
Falle von Derivaten der D-Fructose zumindest fünf Isomere im dynamischen Gleichgewicht vor. Das
zentrale acyclische Isomer dieses Systems (1a(v) bzw. 6(v)) tritt dabei mit einer
Gleichgewichtskonzentration von rund einem Prozent in Erscheinung. Das bedeutet auf Grundlage des
Massenwirkungsgesetzes, dass die Ringschlussrate rund 100-mal größer ist als die Summe aller
Ringöffnungsraten. Weiterhin handelt es sich dabei nicht um das im kristallinen Zustand vorliegende
Isomer, sodass seine anfängliche Konzentration 0% beträgt. Da es den Knotenpunkt des
Isomerisierungsnetzwerkes darstellt, wird es zunächst aus einem großen Reservoir der βP 1a(ii) bzw.
6(ii) gebildet, woraufhin es mit dem 100-fachen seiner Bildungsrate cyclisiert. Es ist daher davon
auszugehen, dass die Gleichgewichtskonzentration des Zentralisomers schnell erreicht ist, während
absolute Konzentrationsänderungen aufgrund der geringen Gleichgewichtskonzentration kaum
messbar sind. Ohne die Möglichkeit einer genauen Messung der zeitlichen Änderung der relativen
Konzentration des Zentralisomers kann eine Berechnung der Anomerisierungsraten auf Grundlage von
Gleichung (7) keine genauen Ergebnisse liefern.27 Wie sich in Kapitel 4.2.6 zeigen wird, sind bereits bei
26 Polarimetrische Messungen bieten keine Grundlage für eine entsprechende Auswertung, da die gemessenen
Drehwinkel als Summenparameter der gewichteten spezifischen Drehwinkel aller vorhandenen Anomere
anzusehen sind. Eine Diskriminierung komplexer Anomerensysteme ist daher nicht möglich. Gleiches gilt für
enzymatische Reaktionen, bei denen anomerenspezifische Enzyme eingesetzt werden.[382] Mittels
infrarotspektroskopischer Messungen (IR) konnten die Hauptanomere der D-Glucose differenziert werden.[383] Es
ist jedoch davon auszugehen, dass diese Methode bei komplexeren Isomerensystemen versagt.
27 Berechnungen auf Grundlage von Gleichung (7) werden in Kapitel 4.2.6 im Rahmen polarimetrischer
Messungen trotzdem noch eine wichtige Rolle spielen.

THEORETISCHER HINTERGRUND
- 29 -
Raumtemperatur die ersten zwei Sekunden nach dem Lösen von D-Fructose 1a für eine Berechnung
der Anomerisierungsraten essentiell. Mit steigender Temperatur verkürzt sich dieses Zeitfenster
entsprechend. Dem entgegen steht, dass eine NMR-Messung bei schneller Arbeitsweise kaum in einem
Zeitraum von weniger als einer Minute nach dem Lösen einer Substanz bewerkstelligt werden kann.
Da für jede Wiederholung eines solchen Experimentes kristalliner Analyt vorhanden sein muss, ist die
praktische Durchführbarkeit neben dem Zeitfaktor auch durch die begrenzte Verfügbarkeit von
Syntheseprodukten beschränkt. Hinzu kommt die Schwierigkeit der definierten Einstellung der
experimentellen Parameter wie dem pH-Wert und der Temperatur. Es bietet sich daher an,
Anomerisierungsraten im Sinne einer Relaxationsmethode zu messen.28 Klassische
Relaxationsmethoden basieren auf einer abrupten Störung des thermodynamischen Gleichgewichts
etwa durch sprunghafte Temperaturänderungen.[267] Die jeweilige Neueinstellung des
Gleichgewichtszustandes wird messtechnisch verfolgt. Die NMR-Spektroskopie bietet in Anlehnung
daran die einzigartige Möglichkeit, Störungen der Gleichgewichtsmagnetisierung zu erzeugen,
während sowohl das thermodynamische Anomerengleichgewicht als auch die Umgebungsparameter
wie Temperatur und pH-Wert unbeeinflusst bleiben. In Bezug auf Zuckersysteme lässt die Analyse der
zeitabhängigen Wiederherstellung des Magnetisierungsgleichgewichtes Rückschlüsse auf die
Anomerisierungsraten zu. Die Wiederherstellung des Gleichgewichtszustandes ist dabei jedoch nicht
nur von den Anomerisierungsraten, sondern gleichermaßen von den longitudinalen Relaxationszeiten
der betrachteten Kerne abhängig. Die zeitabhängige Änderung der z-Magnetisierung in Systemen mit
chemischem Austausch ist durch die von MCCONNELL (1958) erweiterten BLOCH-Gleichungen
gegeben.[69] Für das in Schema 18 gezeigte Isomerensystem ergeben sich die Gleichungen (8) bis (12).29
𝑑𝑀𝑧(𝑧)(𝑡)
𝑑𝑡= −
𝑀𝑧(𝑧)(𝑡) − 𝑀𝑧,𝑒𝑞
(𝑧)
𝑇1(𝑧)
+ 𝑘𝛼𝑃,𝑧 ∙ 𝑀𝑧(𝛼𝑃)(𝑡) + 𝑘𝛽𝑃,𝑧 ∙ 𝑀𝑧
(𝛽𝑃)(𝑡) + 𝑘𝛼𝐹,𝑧 ∙ 𝑀𝑧(𝛼𝐹)(𝑡)
+ 𝑘𝛽𝑃,𝑧 ∙ 𝑀𝑧(𝛽𝐹)(𝑡) − (𝑘𝑧,𝛼𝑃 + 𝑘𝑧,𝛽𝑃 + 𝑘𝑧,𝛼𝐹 + 𝑘𝑧,𝛽𝐹) ∙ 𝑀𝑧
(𝑧)(𝑡)
(8)
𝑑𝑀𝑧(𝛼𝑃)(𝑡)
𝑑𝑡= −
𝑀𝑧(𝛼𝑃)(𝑡) − 𝑀𝑧,𝑒𝑞
(𝛼𝑃)
𝑇1(𝛼𝑃)
+ 𝑘𝑧,𝛼𝑃 ∙ 𝑀𝑧(𝑧)(𝑡) − 𝑘𝛼𝑃,𝑧 ∙ 𝑀𝑧
(𝛼𝑃)(𝑡) (9)
𝑑𝑀𝑧(𝛽𝑃)(𝑡)
𝑑𝑡= −
𝑀𝑧(𝛽𝑃)(𝑡) − 𝑀𝑧,𝑒𝑞
(𝛽𝑃)
𝑇1(𝛽𝑃)
+ 𝑘𝑧,𝛽𝑃 ∙ 𝑀𝑧(𝑧)(𝑡) − 𝑘𝛽𝑃,𝑧 ∙ 𝑀𝑧
(𝛽𝑃)(𝑡) (10)
𝑑𝑀𝑧(𝛼𝐹)(𝑡)
𝑑𝑡= −
𝑀𝑧(𝛼𝐹)(𝑡) − 𝑀𝑧,𝑒𝑞
(𝛼𝐹)
𝑇1(𝛼𝐹)
+ 𝑘𝑧,𝛼𝐹 ∙ 𝑀𝑧(𝑧)(𝑡) − 𝑘𝛼𝐹,𝑧 ∙ 𝑀𝑧
(𝛼𝐹)(𝑡) (11)
28 MANFRED EIGEN, RONALD GEORGE WREYFORD NORRISH und GEORGE PORTER erhielten 1967 den Nobelpreis für Chemie
für die Untersuchung extrem schneller Reaktionen durch Gleichgewichtsstörungen mittels sehr kurzer
Energiepulse (≡ Relaxationsmethode).[384]
29 Dabei ist zu beachten, dass im Rahmen dieser Arbeit off-Resonanzeffekte[385] vernachlässigt werden. Weiterhin
gilt es zu beachten, dass in chemischem Austausch befindliche Stoffsysteme existieren können, bei denen
Kreuzrelaxationsprozesse eine Rolle spielen (vgl. NOE-Effekt). In solchen Fällen sind die in dieser Arbeit genutzten
funktionellen Zusammenhänge entsprechend anzupassen.[243,386-389] In den einschlägigen Publikationen werden
die entsprechenden Terme jedoch zumeist vernachlässigt.[268]

THEORETISCHER HINTERGRUND
- 30 -
𝑑𝑀𝑧(𝛽𝐹)(𝑡)
𝑑𝑡= −
𝑀𝑧(𝛽𝐹)(𝑡) − 𝑀𝑧,𝑒𝑞
(𝛽𝐹)
𝑇1(𝛽𝐹)
+ 𝑘𝑧,𝛽𝐹 ∙ 𝑀𝑧(𝑧)(𝑡) − 𝑘𝛽𝐹,𝑧 ∙ 𝑀𝑧
(𝛽𝐹)(𝑡) (12)
Es handelt sich in diesem Fall um ein System inhomogener linearer Differentialgleichungen der
ersten Ordnung. Ein einfacher Lösungsansatz ergibt sich durch die Bildung der jeweiligen Differenzen
der Ausdrücke zum Zeitpunkt 𝑡 und 𝑡 = ∞. Dies kommt einer Eliminierung der Inhomogenität gleich.
Analog zu Gleichung (7) lässt sich das nun homogene Differentialgleichungssystem in der
Matrixschreibweise vereinfacht darstellen (vgl. Gleichung (13)).
𝑑
𝑑𝑡
(
𝑀𝑧,𝑒𝑞(𝑧) −𝑀𝑧
(𝑧)(𝑡)
𝑀𝑧,𝑒𝑞(𝛼𝑃) −𝑀𝑧
(𝛼𝑃)(𝑡)
𝑀𝑧,𝑒𝑞(𝛽𝑃)
−𝑀𝑧(𝛽𝑃)(𝑡)
𝑀𝑧,𝑒𝑞(𝛼𝐹)
−𝑀𝑧(𝛼𝐹)(𝑡)
𝑀𝑧,𝑒𝑞(𝛽𝐹)
−𝑀𝑧(𝛽𝐹)(𝑡))
= 𝑩 ∙
(
𝑀𝑧,𝑒𝑞(𝑧) −𝑀𝑧
(𝑧)(𝑡)
𝑀𝑧,𝑒𝑞(𝛼𝑃) −𝑀𝑧
(𝛼𝑃)(𝑡)
𝑀𝑧,𝑒𝑞(𝛽𝑃)
−𝑀𝑧(𝛽𝑃)(𝑡)
𝑀𝑧,𝑒𝑞(𝛼𝐹)
−𝑀𝑧(𝛼𝐹)(𝑡)
𝑀𝑧,𝑒𝑞(𝛽𝐹)
−𝑀𝑧(𝛽𝐹)(𝑡))
(13)
Diese Funktion ist vollkommen äquivalent zu Gleichung (7). Lediglich die Matrizen 𝑨 und 𝑩
unterscheiden sich voneinander. Die Matrix 𝑩 beinhaltet dabei im Gegensatz zu 𝑨 neben den
Anomerisierungsraten auch die longitudinalen Relaxationsraten der betrachteten in chemischem
Austausch befindlichen Kerne (vgl. Gleichung (14)). Die Diagonalelemente 𝑅(𝑖) der Matrix 𝑩 sind dabei
gemäß den Gleichungen (15) bis (19) definiert.
𝑩 =
(
−𝑅(𝑧) 𝑘𝛼𝑃,𝑧 𝑘𝛽𝑃,𝑧 𝑘𝛼𝐹,𝑧 𝑘𝛽𝐹,𝑧
𝑘𝑧,𝛼𝑃 −𝑅(𝛼𝑃) 0 0 0
𝑘𝑧,𝛽𝑃 0 −𝑅(𝛽𝑃) 0 0
𝑘𝑧,𝛼𝐹 0 0 −𝑅(𝛼𝐹) 0
𝑘𝑧,𝛽𝐹 0 0 0 −𝑅(𝛽𝐹))
(14)
𝑅(𝑧) =1
𝑇1(𝑧)+ 𝑘𝑧,𝛼𝑃 + 𝑘𝑧,𝛽𝑃 + 𝑘𝑧,𝛼𝐹 + 𝑘𝑧,𝛽𝐹 (15)
𝑅(𝛼𝑃) =1
𝑇1(𝛼𝑃)
+ 𝑘𝛼𝑃,𝑧 (16)
𝑅(𝛽𝑃) =1
𝑇1(𝛽𝑃)
+ 𝑘𝛽𝑃,𝑧 (17)
𝑅(𝛼𝐹) =1
𝑇1(𝛼𝐹)
+ 𝑘𝛼𝐹,𝑧 (18)
𝑅(𝛽𝐹) =1
𝑇1(𝛽𝐹)
+ 𝑘𝛽𝐹,𝑧 (19)
Die allgemeine Lösung von Gleichung (13) erfolgt analog zur Lösung von Gleichung (7).[268-274]
Diese Ausführung soll zeigen, dass die kinetische Betrachtung der Mutarotation mathematisch
gesehen dieselbe Grundlage hat wie die üblicherweise genutzten Experimente der dynamischen
NMR-Spektroskopie. Die in der Literatur beschriebenen Experimente der dynamischen
NMR-Spektroskopie unterscheiden sich demnach offenkundig auch nicht untereinander in ihrer

THEORETISCHER HINTERGRUND
- 31 -
theoretischen Behandlung. Unterschiede ergeben sich lediglich durch die Art der Störung der
Gleichgewichtsmagnetisierung und damit durch die Wahl der Anfangsbedingungen.30 Hierbei
existieren formal gesehen unendlich viele Möglichkeiten. Die in der Literatur am häufigsten
anzutreffenden Varianten sind die (Selective) Exchange-Spectroscopy ((S)EXSY)31 und das Selective
Inversion Recovery-Experiment (SIR-NMR).[275-278] Eine Auswahl sinnvoller Startbedingungen sowie
Möglichkeiten, diese zu optimieren, sind der Literatur zu entnehmen.[70,268] Daneben stehen zur
Auswertung der üblichen Experimente der dynamischen NMR-Spektroskopie nicht kommerzielle
Softwaretools wie EXSYCalc[279] und CIFIT[280] zur Verfügung. Die hier dargestellte Vorgehensweise hat
den Vorteil, dass viele aufeinanderfolgende Messungen mit derselben Probe durchgeführt werden
können. Außerdem ist der jeweilige Nullpunkt einer ausgewählten Anfangsbedingung zeitlich exakter
definiert, als es bei der kinetischen Verfolgung der Anomerisierung der Fall ist. Hier stellt die Kinetik
der Solvatisierung eine gewisse Unschärfe der Startbedingung dar. Trotzdem bleiben die oben
beschriebenen Probleme bestehen, die sich aus der geringen Konzentration des Zentralisomers
ergeben. Hinzu kommt, dass die Matrix 𝑩 neben den Anomerisierungsraten auch die longitudinalen
Relaxationsraten beinhaltet. Diese müssen folglich simultan zur numerischen Berechnung der
Anomerisierungsraten iterativ ermittelt werden. Dementsprechend sind nun 13 anstelle von 8
Koeffizienten zu bestimmen. Dies kann dazu führen, dass der Iterationsprozess aufgrund schlecht
gewählter Startwerte im schlimmsten Falle nicht konvergiert oder aber große Fehlerintervalle für die
Koeffizienten resultieren.
Die meisten in der Literatur beschriebenen Messungen von Anomerisierungsraten diverser
Zuckerderivate erfolgten aus diesen Gründen mittels der selektiven Sättigungstransfer-NMR-
Spektroskopie (SST-NMR).[71-79,281] Die Idee für dieses Experiment besteht darin, die mathematische
Beschreibung des dynamischen Netzwerkes durch geschickte Beeinflussung der Magnetisierung eines
Isomers zu vereinfachen. Im speziellen Falle des in Schema 18 gezeigten Netzwerkes besteht diese
Beeinflussung in der Sättigung der Magnetisierung des anomeren Kohlenstoffatoms des
Zentralisomers. Unter Sättigung ist dabei die Zerstörung der z-Magnetisierung des entsprechenden
Kerns durch schnell aufeinanderfolgende frequenzselektive 90°-Radiopulse zu verstehen. Dies hat zur
Folge, dass 𝑀𝑧(𝑧)(𝑡) zeitlich unabhängig gleich null wird. Das führt mathematisch betrachtet zur
Entkopplung der über Gleichung (8) gekoppelten Differentialgleichungen (9) bis (12). Damit ist die
zeitliche Änderung der z-Magnetisierungen aller nicht zentralen Isomere nur noch abhängig von der
jeweiligen longitudinalen Relaxationszeit 𝑇1(𝑖) sowie der Ringöffnungsrate 𝑘𝑖,𝑧.32 Für ein nicht zentrales
Isomer 𝑖 lässt sich nun der folgende Ausdruck formulieren (vgl. Gleichung (20)).
30 Im Falle der zeitlichen Verfolgung der Mutarotation ist die Startbedingung durch die anfänglich vorhandenen
Konzentrationen der einzelnen Anomere und damit in der Regel durch das im kristallinen Zustand vorliegende
Anomer festgelegt.
31 Die quantitative Auswertung von (S)EXSY-Experimenten liefert weniger vertrauenswürdige Ergebnisse als die
entsprechender SIR-NMR-Experimente, da sie meist nur für eine einzige Mischzeit 𝜏𝑚 durchgeführt werden. Die
Berechnung der Geschwindigkeitskonstanten basiert damit lediglich auf den Daten für 𝑡 = 0𝑠 und 𝑡 = 𝜏𝑚 und
nicht auf einer zeitabhängig gemessenen Spektrenserie. Des Weiteren beansprucht ein zweidimensionales
EXSY-Experiment in Abhängigkeit der Auflösung in der f1-Dimension das bis zu 100-fache der Messzeit eines
SIR-NMR-Experimentes.
32 Zur Bestimmung von Ringöffnungsraten von Zuckeranomeren ist dabei die direkte Detektion des entsprechend
zu sättigenden anomeren Signals des Zentralisomers nicht notwendig. Eine Blindsättigung ist prinzipiell
möglich.[390] Dies ist im Vergleich zu Experimenten wie (S)EXSY und SIR-NMR ein extremer Vorteil.

THEORETISCHER HINTERGRUND
- 32 -
𝑑𝑀𝑧(𝑖)(𝑡)
𝑑𝑡= −
𝑀𝑧(𝑖)(𝑡) − 𝑀𝑧,𝑒𝑞
(𝑖)
𝑇1(𝑖)
− 𝑘𝑖,𝑧 ∙ 𝑀𝑧(𝑖)(𝑡) (20)
Die Lösung dieser Differentialgleichung führt zu Gleichung (21).
𝑀𝑧(𝑖)(𝜏) =𝑀𝑧
(𝑖)(𝜏 = 0𝑠) ∙
(
1+ 𝑇1
(𝑖) ∙ 𝑘𝑖,𝑧 ∙ 𝑒𝑥𝑝(−𝜏 ∙ (1 + 𝑇1
(𝑖)∙ 𝑘𝑖,𝑧)
𝑇1(𝑖) )
1 + 𝑇1(𝑖)∙ 𝑘𝑖,𝑧
)
(21)
Dabei ist 𝜏 die Sättigungsdauer der entsprechenden NMR-Resonanz des Zentralisomers 𝑧. Der
Prozess der Sättigung wird im Folgenden zum Zwecke eines besseren Verständnisses des Experimentes
modellhaft beschrieben. Betrachtet man die z-Magnetisierung eines Kerns im Gleichgewichtszustand
und regt die entsprechende Kernresonanz mit einem 90°-Puls an, so wird der Magnetisierungsvektor
in die x,y-Ebene gedreht. Anschließend finden Relaxationsprozesse statt, die zum einen dazu führen,
dass die z-Magnetisierung ihrem Gleichgewichtszustand entgegenstrebt (longitudinale Relaxation).
Zum anderen kommt es zum zeitabhängigen Verlust der Phasenkohärenz der
Magnetisierungsvektoren in der x,y-Ebene und somit zum Verlust der Quermagnetisierung
(transversale Relaxation). Im rotierenden Koordinatensystem ist die zeitabhängige Änderung der
Quermagnetisierung durch Gleichung (22) gegeben.[192]
𝑑𝑀𝑦′(𝑡)
𝑑𝑡= −
𝑀𝑦′(𝑡)
𝑇2(𝑖)
(22)
Ist 𝑇2(𝑖) relativ zu 𝑇1
(𝑖) kurz, bedeutet dies, dass die Quermagnetisierung aufgehoben ist, bevor
sich die z-Magnetisierung regeneriert hat. Wird nun erneut mit einem 90°-Puls angeregt, werden alle
Magnetisierungsvektoren um 90° gedreht, sodass die Quer- zur z-Magnetisierung wird und umgekehrt.
Dem zuvor beschriebenen Zustand nach ist die z-Magnetisierung nun gleich null und die
Quermagnetisierung geringer als nach dem ersten Puls. Führt man diesen Prozess kaskadenartig fort,
führt dies dazu, dass sowohl die Quer- als auch die z-Magnetisierung ausgelöscht werden (vgl.
Abbildung 3). Sättigung funktioniert demnach effektiv für den Fall, dass 𝑇1(𝑖) ≫ 𝑇2
(𝑖).
Im Falle des chemischen Austauschs gelten für die Relaxationszeiten 𝑇1(𝑖) und 𝑇2
(𝑖) nach
MCCONNELL (1958) die Gleichungen (23) und (24).[69]
1
𝜏1(𝑖)=
1
𝑇1(𝑖)+1
𝜏(𝑖) (23)
1
𝜏2(𝑖)=
1
𝑇2(𝑖)+1
𝜏(𝑖) (24)
Dabei ist 𝜏(𝑖) die inverse Geschwindigkeitskonstante des dynamischen Prozesses (hier: die
Summe aller Ringschlussraten).
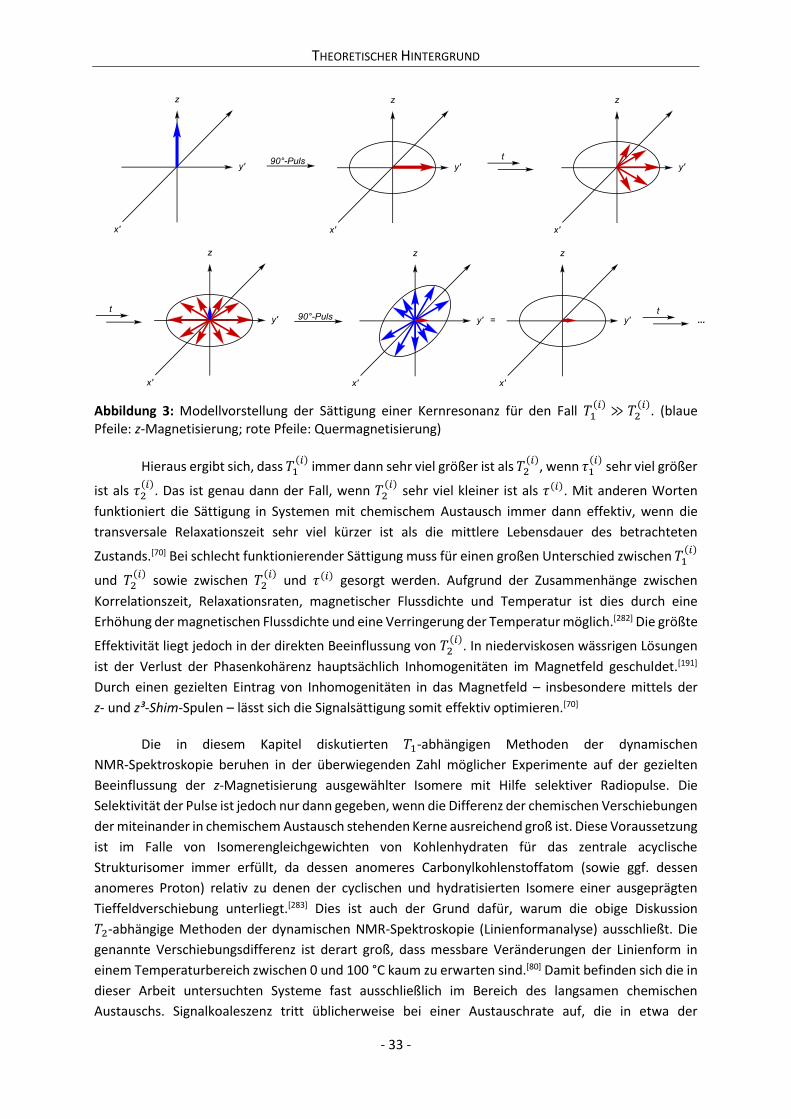
THEORETISCHER HINTERGRUND
- 33 -
Abbildung 3: Modellvorstellung der Sättigung einer Kernresonanz für den Fall 𝑇1(𝑖)≫ 𝑇2
(𝑖). (blaue
Pfeile: z-Magnetisierung; rote Pfeile: Quermagnetisierung)
Hieraus ergibt sich, dass 𝑇1(𝑖)
immer dann sehr viel größer ist als 𝑇2(𝑖)
, wenn 𝜏1(𝑖)
sehr viel größer
ist als 𝜏2(𝑖). Das ist genau dann der Fall, wenn 𝑇2
(𝑖) sehr viel kleiner ist als 𝜏(𝑖). Mit anderen Worten
funktioniert die Sättigung in Systemen mit chemischem Austausch immer dann effektiv, wenn die
transversale Relaxationszeit sehr viel kürzer ist als die mittlere Lebensdauer des betrachteten
Zustands.[70] Bei schlecht funktionierender Sättigung muss für einen großen Unterschied zwischen 𝑇1(𝑖)
und 𝑇2(𝑖)
sowie zwischen 𝑇2(𝑖)
und 𝜏(𝑖) gesorgt werden. Aufgrund der Zusammenhänge zwischen
Korrelationszeit, Relaxationsraten, magnetischer Flussdichte und Temperatur ist dies durch eine
Erhöhung der magnetischen Flussdichte und eine Verringerung der Temperatur möglich.[282] Die größte
Effektivität liegt jedoch in der direkten Beeinflussung von 𝑇2(𝑖)
. In niederviskosen wässrigen Lösungen
ist der Verlust der Phasenkohärenz hauptsächlich Inhomogenitäten im Magnetfeld geschuldet.[191]
Durch einen gezielten Eintrag von Inhomogenitäten in das Magnetfeld – insbesondere mittels der
z- und z³-Shim-Spulen – lässt sich die Signalsättigung somit effektiv optimieren.[70]
Die in diesem Kapitel diskutierten 𝑇1-abhängigen Methoden der dynamischen
NMR-Spektroskopie beruhen in der überwiegenden Zahl möglicher Experimente auf der gezielten
Beeinflussung der z-Magnetisierung ausgewählter Isomere mit Hilfe selektiver Radiopulse. Die
Selektivität der Pulse ist jedoch nur dann gegeben, wenn die Differenz der chemischen Verschiebungen
der miteinander in chemischem Austausch stehenden Kerne ausreichend groß ist. Diese Voraussetzung
ist im Falle von Isomerengleichgewichten von Kohlenhydraten für das zentrale acyclische
Strukturisomer immer erfüllt, da dessen anomeres Carbonylkohlenstoffatom (sowie ggf. dessen
anomeres Proton) relativ zu denen der cyclischen und hydratisierten Isomere einer ausgeprägten
Tieffeldverschiebung unterliegt.[283] Dies ist auch der Grund dafür, warum die obige Diskussion
𝑇2-abhängige Methoden der dynamischen NMR-Spektroskopie (Linienformanalyse) ausschließt. Die
genannte Verschiebungsdifferenz ist derart groß, dass messbare Veränderungen der Linienform in
einem Temperaturbereich zwischen 0 und 100 °C kaum zu erwarten sind.[80] Damit befinden sich die in
dieser Arbeit untersuchten Systeme fast ausschließlich im Bereich des langsamen chemischen
Austauschs. Signalkoaleszenz tritt üblicherweise bei einer Austauschrate auf, die in etwa der

THEORETISCHER HINTERGRUND
- 34 -
Frequenzdifferenz der austauschenden Kerne entspricht.[284] Im Falle von Derivaten der D-Fructose
liegt diese Frequenzdifferenz für 13C-NMR-Spektren bei Messungen an einem 600 MHz-NMR-
Spektrometer bei ca. 15 kHz, weshalb eine Berechnung von Anomerisierungsraten auf Grundlage der
totalen Linienformanalyse nicht angemessen ist. Der Einfluss der Temperatur auf die Linienform spielt
lediglich in Kapitel 4.2.1 in Zusammenhang mit der Strukturaufklärung von Derivaten der D-Fructose
eine Rolle.
2.3 Katalyse und mathematische Modellierung der Anomerisierung
Ein theoretisches Verständnis der Anomerisierung erfordert neben der mechanistischen
Formulierung der Tautomerie auch die mathematische Beschreibung des dynamischen Prozesses, die
im Einklang mit den molekularen Grundlagen steht. Der Prozess der Anomerisierung wurde durch die
Beobachtung der Mutarotation der D-Glucose erstmals im Jahr 1846 von AUGUSTIN-PIERRE DUBRUNFAUT
entdeckt und beschrieben.[1] Der chemische Hintergrund wurde jedoch erst im Verlauf der
darauffolgenden ca. 50 Jahre schrittweise vor allem durch Arbeiten von EMIL FISCHER und CHARLES JOSEPH
TANRET verstanden. THOMAS MARTIN LOWRY (1903) entdeckte schließlich die Abhängigkeit der
Geschwindigkeit der Mutarotation von der Anwesenheit von Säuren und Basen.[285] Diese Abhängigkeit
wurde von CLAUDE SILBERT HUDSON (1907) systematisch untersucht. Er beobachtete dabei, dass die
Mutarotationsrate 𝑘𝑚𝑢𝑡 sowohl mit steigender Protonen- als auch Hydroxylionenkonzentration linear
zunimmt und formulierte die sogenannte katalytische Kettenfunktion (Gleichung (25)).[286]
𝑘𝑚𝑢𝑡 = 𝐴 + 𝐵 ∙ [𝐻+] + 𝐶 ∙ [𝑂𝐻−] (25)
Dabei sind 𝐴, 𝐵 und 𝐶 entsprechende Geschwindigkeitskonstanten. Diese Berechnungsformel
ist rein empirisch und hat keine mechanistische Grundlage. HANS VON EULER et al. (1925)33 sind auf Basis
eigener Vorarbeiten zu der Überzeugung gekommen, dass der Prozess der Mutarotation von ionischen
Spezies der D-Glucose abhängen muss. Entsprechend der jeweiligen 𝑝𝐾𝑠-Werte des Ringsauerstoffs
und der anomeren Hydroxylfunktion bilden sich Kationen und Anionen der D-Glucose, deren
Konzentration vom pH-Wert abhängig ist. Diese ionischen Zuckerspezies sind im Gegensatz zu nicht
ionischen zur spontanen Ringöffnung fähig. Das Minimum der Mutarotationsrate, welches durch die
Nullstelle der ersten Ableitung von Gleichung (25) gegeben ist, entspricht unter Annahme gleicher
spontaner Öffnungsraten des Kations und Anions demnach dem isoelektrischen Punkt (𝑝𝐼) der
D-Glucose. VON EULER et al. (1925, 1926) formulieren schließlich den nach Gleichung (26) gegebenen
Ausdruck zur Beschreibung der Kinetik der Mutarotation.[287,288]
𝑘𝑚𝑢𝑡 = 𝑘0 + 𝑟𝑞2 ∙[𝐻+]
𝐾𝑠1+ 𝑟𝑞1 ∙
𝐾𝑠2[𝐻+]
(26)
Dabei entspricht 𝑘0 der Konstante 𝐴 aus Gleichung (25), 𝑟𝑞𝑖 den spezifischen
Reaktionsfähigkeiten der ionischen Zuckerspezies und 𝐾𝑠1 sowie 𝐾𝑠2 den Säurekonstanten des
Ringsauerstoffatoms und der anomeren Hydroxylfunktion. Es liegt auf der Hand, dass sich
Gleichung (26) und Gleichung (25) nicht unterscheiden, wenn für 𝐴 = 𝑘0, 𝐵 =𝑟𝑞2
𝐾𝑠1 und 𝐶 =
𝑟𝑞1∙𝐾𝑠2
𝐾𝑤 gilt.
Dabei ist 𝐾𝑤 das Ionenprodukt von Wasser. Die von VON EULER et al. aufgestellte Theorie wurde
33 ARTHUR HARDEN und HANS KARL AUGUST SIMON VON EULER-CHELPIN erhielten 1929 den Nobelpreis für Chemie für
ihre Untersuchungen der Zuckerfermentation und fermentativer Enzyme.[391]

THEORETISCHER HINTERGRUND
- 35 -
aufgrund ihrer mathematischen Äquivalenz zur katalytischen Kettenfunktion zunächst nicht beachtet.
Vielmehr erweiterten JOHANNES NICOLAUS BRØNSTED und EDWARD ARMAND GUGGENHEIM (1927) HUDSONS
Funktion von 1907 zur Beschreibung des katalytischen Einflusses von Säuren und Basen.[289] Die
entsprechende Funktion zur Beschreibung der allgemeinen Säure-Base-Katalyse lässt sich in Form von
Gleichung (27) darstellen.
𝑘𝑚𝑢𝑡 = 𝑘0 + 𝑘𝐻 ∙ [𝐻+] + 𝑘𝑂𝐻 ∙ [𝑂𝐻
−] +∑𝑘𝑖 ∙ [𝐻𝐴𝑖]
𝑖
+∑𝑘𝑗 ∙ [𝐵𝑗]
𝑗
(27)
Dabei steht [𝐻𝐴𝑖] für die Konzentration einer Säure und [𝐵𝑗] für die Konzentration einer Base.
Die katalytischen Konstanten 𝑘𝑖 und 𝑘𝑗 hängen dabei nach BRØNSTED und GUGGENHEIM (1927)
exponentiell von den entsprechenden Säure- und Basenkonstanten der Katalysatoren ab.[289] Obwohl
auch diese Gleichung keinerlei mechanistische Grundlage hat, wird sie seit ihrer Formulierung bis
heute fortwährend genutzt.[290-293] Sie findet dabei nicht nur im Kontext der Anomerisierung von
Zuckern, sondern auch in anderen Bereichen der organischen Chemie Anwendung, bei denen säure-
und basenkatalysierte Prozesse eine Rolle spielen.[294] Das ist bemerkenswert, da THOMAS MARTIN LOWRY
ebenfalls 1927 die Theorie ionischer Zuckerspezies von VON EULER et al. (1925, 1926) aufgreift und um
einen entscheidenden Schritt ergänzt.[295] Dieser besteht darin, dass die ionischen Zuckerspezies nach
erfolgter Anomerisierung durch einen Protonentransfer wieder elektroneutral werden müssen. Diese
Erweiterung, die von LOWRY als elektrolytische Theorie der Katalyse bezeichnet wird, impliziert, dass
Anomerisierungsprozesse lediglich in Gegenwart von Ampholyten stattfinden können. Nach dieser
Vorstellung lässt sich der Prozess der Anomerisierung am Beispiel der β-D-Glucopyranose durch
Schema 19 beschreiben.
Schema 19: Mechanismus der Anomerisierung am Beispiel der β-D-Glucopyranose.
Trotz seiner Bemühungen, einen adäquaten Mechanismus für den Prozess der Anomerisierung
zu formulieren, nutzt auch LOWRY die katalytische Kettenfunktion zur mathematischen Beschreibung
der Mutarotation.[295] Im Rahmen der hier vorliegenden Arbeit erfolgt die mathematische
Modellierung der Anomerisierung auf Basis der Grundannahmen von VON EULER und LOWRY. Die
relativen Konzentrationen der verschiedenen ionischen und nicht ionischen Zuckerspezies werden
anhand der Säurekonstanten des Ringsauerstoffs 𝐾𝑠1 und der anomeren Hydroxylfunktion 𝐾𝑠2 des
entsprechenden Zuckers über das Massenwirkungsgesetz berechnet. Die Molenbrüche der vier

THEORETISCHER HINTERGRUND
- 36 -
möglichen Spezies eines reduzierenden Zuckers 𝑍0, 𝑍+, 𝑍− und 𝑍± sind dabei durch die
Gleichungen (28) bis (31) gegeben.
[𝑍0] =[𝐻+] ∙ 𝐾𝑠1
[𝐻+]2 + [𝐻+] ∙ 𝐾𝑠1 + [𝐻+] ∙ 𝐾𝑠2 + 𝐾𝑠1 ∙ 𝐾𝑠2
(28)
[𝑍+] =[𝐻+]2
[𝐻+]2 + [𝐻+] ∙ 𝐾𝑠1 + [𝐻+] ∙ 𝐾𝑠2 + 𝐾𝑠1 ∙ 𝐾𝑠2
(29)
[𝑍−] =𝐾𝑠1 ∙ 𝐾𝑠2
[𝐻+]2 + [𝐻+] ∙ 𝐾𝑠1 + [𝐻+] ∙ 𝐾𝑠2 + 𝐾𝑠1 ∙ 𝐾𝑠2
(30)
[𝑍±] =[𝐻+] ∙ 𝐾𝑠2
[𝐻+]2 + [𝐻+] ∙ 𝐾𝑠1 + [𝐻+] ∙ 𝐾𝑠2 +𝐾𝑠1 ∙ 𝐾𝑠2
(31)
Auf dieser Grundlage gelten nun die folgenden Überlegungen: Mit Verweis auf Schema 19
können weder Ringöffnung noch Ringschluss von nicht ionischen Zuckerspezies ausgehen. Der
pH-abhängige Molenbruch des ungeladenen Zuckers [𝑍0] spielt demnach für
Anomerisierungsprozesse keinerlei Rolle. Weiterhin haben alle ionischen Zuckerspezies spezifische
spontane Ringöffnungsraten 𝑘𝑍+ , 𝑘𝑍− und 𝑘𝑍± . Die messbare Geschwindigkeit der Ringöffnung 𝑘𝑜𝑏𝑠
eines Zuckeranomers berechnet sich demnach mit Hilfe von Gleichung (32).
𝑘𝑜𝑏𝑠 = 𝑘𝑍± ∙ [𝑍±] + 𝑘𝑍+ ∙ [𝑍
+] + 𝑘𝑍− ∙ [𝑍−] (32)
Da davon auszugehen ist, dass der protonierte Ringsauerstoff eines Zuckeranomers eine
extrem starke, die anomere Hydroxylfunktion hingegen eine sehr schwache Säure ist, gilt für den
pH-Bereich 𝑝𝐾𝑠1 ≪ 𝑝𝐻 ≪ 𝑝𝐾𝑠2 und damit für 𝑝𝐻 ≅ 𝑝𝐼, dass der jeweilige Nenner der
Gleichungen (28) bis (31) annähernd gleich [𝐻+] ∙ 𝐾𝑠1 ist. Damit kann Gleichung (32) durch
Gleichung (33) approximiert werden.
𝑘𝑜𝑏𝑠 = 𝑘𝑍± ∙𝐾𝑠2𝐾𝑠1
+ 𝑘𝑍+ ∙[𝐻+]
𝐾𝑠1+ 𝑘𝑍− ∙
𝐾𝑠2[𝐻+]
(33)
Obwohl Gleichung (33) wiederum äquivalent zu den Gleichungen (25) und (26) ist und jeder
dieser Ausdrücke Gleichung (32) im lebensmittelrelevanten pH-Bereich nahezu perfekt approximiert,
bringt insbesondere die Verwendung von Gleichung (25) einige Probleme mit sich. Hier ergeben sich
die Konstanten 𝐴, 𝐵 und 𝐶 als Produkte der jeweiligen spontanen Ringöffnungsraten und der
Säurekonstanten. Eine Analyse der thermodynamischen Aktivierungsparameter auf Grundlage
temperaturabhängiger Messungen von 𝐴, 𝐵 und 𝐶 bringt somit stark fehlerhafte Ergebnisse.
Außerdem hat die Interpretation der Konstanten 𝐵 und 𝐶 unrealistische Konsequenzen. Berechnungen
dieser Konstanten im Falle der D-Glucose 2a führen zu dem Schluss, dass die Katalyse der Mutarotation
durch Hydroxylionen rund 40 000-mal stärker ausgeprägt ist als eine entsprechende
Protonenkatalyse.[286] Eine molekulare Grundlage für derart drastische Unterschiede ist jedoch nicht
gegeben. Gleichermaßen wenig überzeugend ist die Vorstellung einer beliebigen Steigerungsfähigkeit
der Mutarotationsrate durch eine Erhöhung der Protonen- bzw. Hydroxylionenkonzentration, die
sämtliche Approximationen von Gleichung (32) erlauben.

THEORETISCHER HINTERGRUND
- 37 -
Schließlich erfordert die mathematische Beschreibung der Anomerisierung auch die
Beachtung der von BRØNSTED und GUGGENHEIM (1927) beschriebenen Katalyse durch Säuren und Basen.
Gleichung (27) besagt, dass sowohl Säuren als auch Basen die Fähigkeit besitzen, reduzierende Zucker
zur spontanen Ringöffnung zu bewegen. Das Protolysegleichgewicht einer Säure wird nur durch den
pH-Wert, nicht aber durch die Anwesenheit weiterer Säuren beeinflusst. Damit haben katalytisch
wirkende Säuren und Basen auch keinen Einfluss auf die 𝐾𝑠-Werte eines Zuckers und somit auch nicht
auf die Molenbrüche dessen ionischer Spezies. Sie können die Steigerung von Ringöffnungsraten somit
nicht durch eine Veränderung der Ionengleichgewichte erreichen, sondern lediglich durch
Einflussnahme auf deren Dynamik. Das bedeutet, dass Katalysatoren die mittlere Lebensdauer der
ionischen Zuckerspezies verkürzen. Dies geschieht durch einen Ladungsausgleich durch die
Übertragung eines Protons. Dabei bilden ionische Zuckerspezies COULOMB-Paare mit Katalysatoren
entgegengesetzter Ladung (etwa kationische Zuckerspezies in Gegenwart von Carboxylaten).34 Es ist
somit zu erwarten, dass ein Katalysator die Aktivierungsenthalpie der Zuckeranomerisierung erhöht,
da zusätzlich zum Bindungsbruch auch die COULOMB-Anziehung der Ionenpaare überwunden werden
muss. Demgegenüber kommt die beschleunigte Dynamik des Protonentransfers einer Erhöhung der
Stoßrate bzw. der Aktivierungsentropie gleich. Ob der Prozess der Ringöffnung stattfindet, hängt also
davon ab, ob genügend Aktivierungsenergie für die Überwindung der COULOMB-Anziehung und den
erforderlichen Bindungsbruch zur Verfügung steht. Hat ein entsprechendes Molekül zum Zeitpunkt
seiner Ionisierung nicht genügend innere Energie für diesen Prozess, so wird ein Katalysator durch
einen schnellen Protonentransfer für einen Ladungsausgleich sorgen, bevor eine Öffnung des Ringes
erfolgen kann. Das bedeutet, dass nicht die Ionisierung des Zuckers der geschwindigkeitsbestimmende
Schritt der Anomerisierung ist, sondern der Bruch der endocyclischen C-O-Bindung. Diese
Überlegungen werden durch quantenchemische Berechnungen von QIAN (2013) sowie PLAZINSKI et al.
(2015) gestützt.[296,297] Dass der Bruch der endocyclischen C-O-Bindung im Falle einfach geladener
Zuckerionen vor dem abschließenden Protonentransfer erfolgt, legen theoretische Berechnungen von
FENG et al. (2013) nahe.[298] Diese berechneten, dass bereits die Protonierung des Ringsauerstoffatoms
der β-D-Fructopyranose 1a(ii) zum Bruch der C-O-Bindung führt. Außerdem verlängert sich die
C-O-Bindung im Falle der α-D-Glucopyranose durch die Deprotonierung der anomeren
Hydroxylfunktion von 1.41 Å auf 1.55 Å. Als eine Folge daraus ergibt sich, dass die Anomerisierung bei
ausreichend tiefen Temperaturen durch katalytische Säuren und Basen gehemmt wird. Eine solche
Hemmung steht jedoch in Widerspruch zu Gleichung (27), die nur eine Zunahme von
Anomerisierungsraten in Gegenwart von Säuren und Basen erlaubt. Es zeigt sich dadurch, dass auch
diese Gleichung rein empirisch und mechanistisch nicht fundiert ist. Daher wird in dieser Arbeit eine
Erweiterung von Gleichung (32) genutzt, deren Herleitung im Folgenden diskutiert wird.
Der katalytische Einfluss einer Säure besteht den obigen Ausführungen nach in der
Reprotonierung der anionischen Zuckerspezies. Entsprechend dient eine Base der Deprotonierung der
kationischen Zuckerspezies. Folglich katalysiert eine Base im sauren Milieu, während eine Säure im
basischen pH-Bereich katalytisch aktiv ist. Nach BRØNSTED und GUGGENHEIM (1927) steigt die katalytische
Effizienz einer Säure (Base) mit der Zunahme ihrer Stärke.[289] Eine starke Säure (Base) wird jedoch im
basischen (sauren) Milieu, in dem ihr zu katalysierendes Substrat mit hohen Molenbrüchen vorkommt,
34 Für nicht ionische Katalysatoren wie Wasser spielt die COULOMB-Anziehung keine Rolle. Entsprechend ist davon
auszugehen, dass ionische Katalysatoren die Effizienz der Wasserkatalyse verringern, da sie die
Wahrscheinlichkeit von Wechselwirkungen ionischer Zuckerspezies mit elektroneutralen Molekülen
herabsetzen.

THEORETISCHER HINTERGRUND
- 38 -
hauptsächlich in Form ihrer konjugierten Base (Säure) vorliegen, die wiederum katalytisch wenig
effizient ist und kaum Substrat vorfindet. Für eine Abschätzung des katalytischen Einflusses in
Abhängigkeit des pH-Wertes müssen daher die folgenden Fragen beantwortet werden:
1. Wie groß ist die katalytische Effizienz des Katalysators?
2. Zu welchem Prozentsatz liegt die katalytisch aktive Spezies vor?
3. Zu welchem Prozentsatz liegt die zu katalysierende Spezies vor?
Aus den Antworten zu diesen Fragen können einige grundlegende Schlüsse gezogen werden.
Zum einen gibt es für ein gegebenes Zuckeranomer für jeden pH-Wert bei konstanter Temperatur
einen optimalen Katalysator. Zum anderen müssen diejenigen Säuren und Basen den größten Einfluss
auf die Anomerisierung haben, deren 𝑝𝐾𝑠-Werte nahe des pH-Minimums von Gleichung (25) und
damit im Bereich des 𝑝𝐼 des betrachteten Zuckeranomers liegen. Für D-Fructose 1a liegt das Minimum
der Mutarotationsrate je nach Temperatur im Bereich von pH 2.5-6.5[67] und damit in der
Größenordnung der 𝑝𝐾𝑠-Werte gewöhnlicher Carbonsäuren.[299] Dies entspricht auch dem pH-Milieu
von Lebensmitteln.[16] Bezüglich des Einflusses von Aminosäuren auf die Anomerisierung von Zuckern
lässt sich weiterhin folgern, dass der katalytische Effekt der Aminofunktion aufgrund ihres hohen
𝑝𝐾𝑠-Wertes[300] zu vernachlässigen ist. Für stöchiometrische Mischungen von Zuckern 𝑍 und
Monocarbonsäuren 𝑆 bzw. einfachen Aminosäuren sind daher zur mathematischen Modellierung des
Prozesses der Anomerisierung in Anlehnung an Gleichung (32) die Molenbrüche der verschiedenen
ionischen Spezies zu beachten. Da nun insgesamt drei 𝑝𝐾𝑠-Werte relevant sind, existieren 23 = 8
verschiedene Spezies. Bei zwei dieser Spezies ist der Zucker ungeladen, weshalb diese zu ignorieren
sind. Die übrigen Spezies sind die folgenden: 𝑍−𝑆−, 𝑍−𝑆+, 𝑍±𝑆−, 𝑍±𝑆+, 𝑍+𝑆− und 𝑍+𝑆+. (Für eine
Darstellung der entsprechenden molekularen Strukturen siehe Strukturenverzeichnis.) Dabei
bezeichnet 𝑆+ die Säure und 𝑆− ihre konjugierte Base. Die Molenbrüche von 𝑍−𝑆+und 𝑍±𝑆− sowie
von 𝑍±𝑆+ und 𝑍+𝑆− sind in ihrem pH-abhängigen Verlauf jeweils paarweise linear voneinander
abhängig, sodass die spontanen Ringöffnungsraten dieser Spezies nicht differenziert werden können.
Dabei ist davon auszugehen, dass die zwitterionischen Zuckerspezies 𝑍±𝑆− und 𝑍±𝑆+ auch ohne
Einfluss eines Katalysators außerordentlich schnell anomerisieren. Eine Katalyse im Sinne des hier
beschriebenen Modells ist somit nur für die beiden Spezies 𝑍−𝑆+ und 𝑍+𝑆− zu erwarten. Der
Ladungsausgleich der Spezies 𝑍−𝑆− sowie 𝑍+𝑆+ erfolgt dagegen unter Beteiligung des Lösungsmittels.
Die entsprechenden Molenbrüche berechnen sich auf Grundlage des Massenwirkungsgesetzes nach
den Gleichungen (34) bis (37).
[𝑍−𝑆−] =𝐾𝑠1𝐾𝑠2𝐾𝑠3
[𝐻+]3+[𝐻+]2(𝐾𝑠1+𝐾𝑠2+𝐾𝑠3)+[𝐻+](𝐾𝑠1𝐾𝑠2+𝐾𝑠1𝐾𝑠3+𝐾𝑠2𝐾𝑠3)+𝐾𝑠1𝐾𝑠2𝐾𝑠3
(34)
[𝑍−𝑆+] + [𝑍±𝑆−] =[𝐻+](𝐾𝑠1𝐾𝑠2+𝐾𝑠2𝐾𝑠3)
[𝐻+]3+[𝐻+]2(𝐾𝑠1+𝐾𝑠2+𝐾𝑠3)+[𝐻+](𝐾𝑠1𝐾𝑠2+𝐾𝑠1𝐾𝑠3+𝐾𝑠2𝐾𝑠3)+𝐾𝑠1𝐾𝑠2𝐾𝑠3
(35)
[𝑍+𝑆−] + [𝑍±𝑆+] =[𝐻+]2(𝐾𝑠2+𝐾𝑠3)
[𝐻+]3+[𝐻+]2(𝐾𝑠1+𝐾𝑠2+𝐾𝑠3)+[𝐻+](𝐾𝑠1𝐾𝑠2+𝐾𝑠1𝐾𝑠3+𝐾𝑠2𝐾𝑠3)+𝐾𝑠1𝐾𝑠2𝐾𝑠3
(36)
[𝑍+𝑆+] =[𝐻+]3
[𝐻+]3+[𝐻+]2(𝐾𝑠1+𝐾𝑠2+𝐾𝑠3)+[𝐻+](𝐾𝑠1𝐾𝑠2+𝐾𝑠1𝐾𝑠3+𝐾𝑠2𝐾𝑠3)+𝐾𝑠1𝐾𝑠2𝐾𝑠3
(37)

THEORETISCHER HINTERGRUND
- 39 -
Dabei ist 𝐾𝑠3 die Säurekonstante der katalytischen Amino- bzw. Carbonsäure. Die messbare
Geschwindigkeit der Ringöffnung 𝑘𝑜𝑏𝑠 eines Zuckeranomers in Anwesenheit einer katalytischen Säure
berechnet sich schließlich in Analogie zu Gleichung (32) mittels Gleichung (38).
𝑘𝑜𝑏𝑠 = 𝑘𝑍−𝑆− ∙ [𝑍−𝑆−] + 𝑘𝑍−𝑆+/𝑍±𝑆− ∙ ([𝑍
−𝑆+] + [𝑍±𝑆−]) + 𝑘𝑍+𝑆−/𝑍±𝑆+
∙ ([𝑍+𝑆−] + [𝑍±𝑆+]) + 𝑘𝑍+𝑆+ ∙ [𝑍+𝑆+]
(38)
Die Ausführungen dieses Kapitels zeigen, dass eine Bestimmung thermodynamischer
Aktivierungsparameter von Anomerisierungsprozessen nicht korrekt erfolgen kann, solange das
mathematische Modell des dynamischen Prozesses keine molekular-mechanistische Grundlage hat.
Im Rahmen dieser Arbeit soll gezeigt werden, dass die theoretischen Überlegungen, die zu
Gleichung (32) bzw. (38) geführt haben, mit experimentellen Daten im Einklang stehen.

ZIELSTELLUNG
- 40 -
3 Zielstellung Die Reaktionen einfacher reduzierender Zucker und deren Derivate haben entscheidende
Auswirkungen sowohl auf die Qualität von Lebensmitteln als auch auf die Genese verschiedener
altersbedingter Erkrankungen. Ein Verständnis der molekularen Grundlagen der Reaktivität als Basis
der Möglichkeit einer gezielten Beeinflussung von Reaktionen hat in diesem Zusammenhang nicht nur
Bedeutung für den Genusswert prozessierter und gelagerter Lebensmittel, sondern auch für die
Gesundheit und damit für die Qualität des Lebens allgemein. Aus diesem Grund finden sich zahlreiche
Publikationen, in denen der Fokus auf die Untersuchung der relativen Reaktivitäten verschiedener
Zucker gerichtet ist.
Weitverbreitet ist die Auffassung, dass die acyclischen Zentralisomere reduzierender Zucker
für deren Reaktivität verantwortlich sind. Entsprechend gilt die Ringöffnung cyclischer Zuckeranomere
als mutmaßlich geschwindigkeitsbestimmender Schritt chemischer Reaktionen. In Kapitel 2.1 wurde
demgegenüber hervorgehoben, dass die Formulierung der quantitativ bedeutsamen Reaktionen
reduzierender Zucker und deren Derivate vor allem aus ihren cyclischen Hauptanomeren sinnvoll ist
(vgl. dazu Schema 5 und Schema 7 sowie Schema 14 bis 17). Das führt zu der Frage, warum die
Reaktivität von D-Fructose-analogen AMADORI-Verbindungen 6 gegenüber der Reaktivität von
D-Fructose 1a laut den Berechnungen von BRANDS und VAN BOEKEL (1998, 2002) bei 120 °C um das
Zehn- bis Vierzigfache erhöht ist. Da die Anomerensysteme von 1a und 6 in ihrer Qualität und Quantität
nahezu vollständig übereinstimmen, können sich diese lediglich in der Dynamik ihrer Anomerisierung
unterscheiden. Je nach vorherrschenden Milieuparametern liegen die Ringöffnungsraten
verschiedener Zucker bei Temperaturen bis etwa 60 °C in einer Größenordnung von 0.1/s (d.h. 0.1 Hz)
und mehr. Für eine Reaktion erster Ordnung ergibt sich daraus eine Halbwertszeit von weniger als 7 s.
Für den Abbau eines Zuckers im Zuge der MAILLARD-Reaktion werden derartige
Reaktionsgeschwindigkeiten selbst bei Temperaturen über 100 °C bei weitem nicht erreicht. Damit
kann der Prozess der Anomerisierung bzgl. des thermisch induzierten Zuckerabbaus keinesfalls
geschwindigkeitsbestimmend sein.
Vor dem Hintergrund der beschriebenen Widersprüche besteht das Ziel der vorliegenden
Arbeit in der Beantwortung der folgenden Fragen:
1) Welches Anomer eines reduzierenden Zuckers ist verantwortlich für dessen Reaktivität?
2) Existiert eine Korrelation zwischen der Dynamik von Zuckertautomerien – insbesondere der
Ringöffnung – und der Kinetik nicht enzymatischer Bräunungsreaktionen?
3) Wie beeinflussen Aminosäuren die Dynamik der Anomerisierung und die Reaktivität
reduzierender Zucker?
Zur Klärung dieser Fragen ist es zunächst erforderlich, eine geeignete Auswahl an
Zielsubstanzen festzulegen. In Anlehnung an die Arbeiten von BRANDS und VAN BOEKEL (1998, 2002)
erfolgen alle Messungen vorrangig an D-Fructose 1a sowie D-Fructose-analogen
AMADORI-Verbindungen 6. Um den Einfluss von Aminosäuren auf die Dynamik der Anomerisierung
sowie die Reaktivität der Zuckerkomponente zu ermitteln, werden primäre (Glycin 4c, L-Alanin 4a) und
sekundäre (L-Prolin 4b) α-Aminosäuren sowie primäre ω-Aminosäuren (β-Alanin 4d,
γ-Aminobuttersäure 4e) eingesetzt. Diese unterscheiden sich nicht nur strukturell, sondern

ZIELSTELLUNG
- 41 -
insbesondere in ihren elektronischen Eigenschaften, die sich in den 𝑝𝐾𝑠-Werten ihrer funktionellen
Gruppen widerspiegeln. Zur Unterscheidung inter- und intramolekularer Wechselwirkungen erfolgen
alle Messungen an D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der entsprechenden
Aminosäuren 4a-4e im Vergleich zu Messungen an den entsprechenden D-Fructose-analogen
AMADORI-Verbindungen 6a-6e.
Da die Dynamik der Anomerisierung nicht nur temperatur-, sondern vor allem pH-abhängig ist,
besteht der erste Schritt dieser Arbeit in der Ermittlung der Reaktivitäten der Zielsubstanzen in
Abhängigkeit des pH-Wertes bei konstanter Temperatur. Dazu werden Lösungen der Zielsubstanzen
hergestellt und unter kontrollierten Bedingungen zur Reaktion gebracht. Der pH-Wert wird während
aller Modellreaktionen titrimetrisch auf verschiedenen Stufen konstant gehalten. Die Stufen
orientieren sich dabei am Stabilitätsmaximum der untersuchten Zuckerderivate. Dies ermöglicht eine
Differenzierung zwischen säure- und baseninduzierten Reaktionen. Die Ermittlung der Reaktivität
erfolgt anhand dreier Parameter:
1) Messung der zeitabhängigen Konzentrationsänderungen der eingesetzten Edukte
(Abbauraten) mittels HILIC-RI (für D-Fructose 1a) sowie ionenpaarchromatographischer
RP-HPLC-RI (für die Aminosäuren 4a-4e sowie die AMADORI-Verbindungen 6a-6e).
2) Messung der zeitabhängigen Konzentrationsänderungen (Bildungsraten) der
α-Dicarbonylverbindungen 9a-9d nach Derivatisierung mit ortho-Phenylendiamin mittels
RP-HPLC-DAD.
3) Messung der zeitabhängigen Extinktionszunahme (Farbbildungsrate) bei 284 nm
(UV-Bereich) und 420 nm (visueller Bereich) mittels Spektralphotometrie.
Um die erhobenen Daten zur Reaktivität der Zielverbindungen in einen Zusammenhang mit
ihren Anomerensystemen zu bringen, erfolgt parallel dazu deren vollständige NMR-spektroskopische
Charakterisierung. Diese unterteilt sich in die folgenden Punkte:
1) Vollständige Zuordnung der NMR-Signale der Anomere der Zielsubstanzen
(Strukturaufklärung und Identifikation elektronischer Effekte).
2) Bestimmung der relativen Konzentrationen der Anomere der Zielsubstanzen mittels
quantitativer NMR-Spektroskopie in Abhängigkeit der Temperatur.
3) Messung der Geschwindigkeit der reversiblen 1,2-Enolisierung der Zielsubstanzen mittels
zeitabhängiger quantitativer NMR-Spektroskopie in Abhängigkeit des pH-Wertes.
4) Messung der Dynamik der Anomerisierung der Zielsubstanzen mittels dynamischer
NMR-Spektroskopie (SST-NMR) in Abhängigkeit des pH-Wertes und der Temperatur.
Im finalen Schritt der Arbeit werden die erhobenen Daten zur Reaktivität, Struktur und
Dynamik der Anomere der untersuchten Zuckerderivate in einen gemeinsamen molekularen Kontext
gebracht und damit die oben formulierten Fragen beantwortet. Mit Blick auf die übliche Beschreibung
der MAILLARD-Reaktion als aminokatalysierter Zuckerabbau wird der Fokus dabei auf die spezifischen
Einflüsse der Amino- und Carboxylfunktion der eingesetzten Aminosäuren auf die Dynamik und
Reaktivität der Anomere der untersuchten Derivate der D-Fructose gelegt. Auf dieser Basis soll
schließlich ein Mechanismus der grundlegenden katalytischen Prozesse innerhalb der (frühen und
intermediären Phase der) MAILLARD-Reaktion formuliert werden, der die oben formulierten
Widersprüche aufhebt.

ERGEBNISSE UND DISKUSSION
- 42 -
4 Ergebnisse und Diskussion Das folgende Kapitel ist in zwei Abschnitte unterteilt. Der erste Abschnitt befasst sich mit der
Messung der Reaktivitäten der D-Fructose 1a, stöchiometrischen Mischungen der D-Fructose 1a und
den Aminosäuren 4a-4e sowie der D-Fructose-analogen AMADORI-Verbindungen 6a-6e. Im zweiten
Abschnitt erfolgt die strukturelle Charakterisierung der Zielsubstanzen mittels NMR-Spektroskopie.
Diese bildet die Grundlage für die Untersuchung von dynamischen Prozessen der Tautomerensysteme.
Schließlich werden die Zusammenhänge von Struktur, Funktionalität, Dynamik und Kinetik erörtert
und die grundlegenden Einflüsse verschiedener funktioneller Gruppen auf den Verlauf der
MAILLARD-Reaktion diskutiert.Reaktivität von Derivaten der D-Fructose
Zum Vergleich der Reaktivitäten von D-Fructose 1a in An- und Abwesenheit stöchiometrischer
Mengen der Aminosäuren 4a-4e sowie der entsprechenden AMADORI-Verbindungen 6a-6e wurden
wässrige Reaktionslösungen der Analyten unter definierten Bedingungen zur Reaktion gebracht. Diese
Modellreaktionen erfolgten bei einer Temperatur von (90 ± 2) °C auf jeweils sechs konstant
gehaltenen pH-Stufen im Intervall von 1.5 ≤ 𝑝𝐻 ≤ 6.5. Die ablaufenden Reaktionen wurden dabei
durch die zeitabhängige Messung der Konzentrationen der Edukte und ausgewählter
Reaktionsintermediate sowie durch UV/Vis-spektroskopische Messungen des Fortschrittes der nicht
enzymatischen Bräunung verfolgt.
4.1.1 KINETIK DES ABBAUS VON D-FRUCTOSE, AMINOSÄUREN UND AMADORI-VERBINDUNGEN
Die Verfolgung des Abbaus der D-Fructose 1a, der Aminosäuren 4a-4e und der
AMADORI-Verbindungen 6a-6e erfolgte über einen Zeitraum von je etwa sechs Stunden. Dabei wurden
den Reaktionslösungen in regelmäßigen Zeitabständen Proben entnommen und mittels HPLC-RI auf
den jeweiligen Eduktgehalt hin untersucht. Dabei zeigte sich für alle Modellsysteme ein Minimum ihrer
Reaktivität in einem Bereich von pH 2.5-4.5. Die Stabilität der Systeme ist in diesem Bereich so groß,
dass sich der prozentuale Abbau der Edukte in einer Größenordnung befindet, der der
Messunsicherheit der Quantifizierungsmethode entspricht. Die Konzentrations-Zeit-Verläufe der
einzelnen Systeme wurden mit einem Kinetikmodell erster Ordnung angepasst. Für jede der
Messreihen wurde infolge die minimale Geschwindigkeitskonstante berechnet, ab der ein Eduktabbau
sicher erkannt werden kann. Dies ist genau dann der Fall, wenn sich die modellierten Konzentrationen
zu den Zeitpunkten 𝑡𝑆𝑡𝑎𝑟𝑡 und 𝑡𝐸𝑛𝑑𝑒 um mehr als das Dreifache der Wurzel der mittleren quadratischen
Abweichung zwischen gemessenen und modellierten Werten unterscheiden. Da die Streuung der
Messwerte für alle untersuchten Abbaukinetiken ähnlich ist, wurde die Summe des Mittelwertes und
der Standardabweichung aller minimalen Geschwindigkeitskonstanten als Quantifizierungsgrenze für
die Berechnung der Geschwindigkeitskonstanten des Eduktabbaus festgelegt.35 Die entsprechenden
Abbauraten erster Ordnung (angegeben in Hz = 1/s) der D-Fructose 1a, der Aminosäuren 4a-4e sowie
35 Die minimal quantifizierbare Abbaurate der Edukte im Rahmen der durchgeführten Modellreaktionen liegt in
etwa bei 5·10-6 Hz. Für eine Reaktionsdauer von sechs Stunden entspricht dies einer Abnahme der
Eduktkonzentration um rund 10%. Das entspricht einer Streuung der Messwerte von etwa 3.3%. Diese resultiert
größtenteils aus dem Versuchsaufbau. Als Reaktionsgefäß wurde ein 100 mL Sulfierkolben verwendet, in dessen
Hälsen Wasser kondensierte und ggf. wieder nachfloss. Dabei betrug das Volumen des Kondensates ca. 1 mL. Bei
einem Reaktionsvolumen von 50 mL entspricht das bereits einer Schwankung der Konzentrationen von rund 2%.
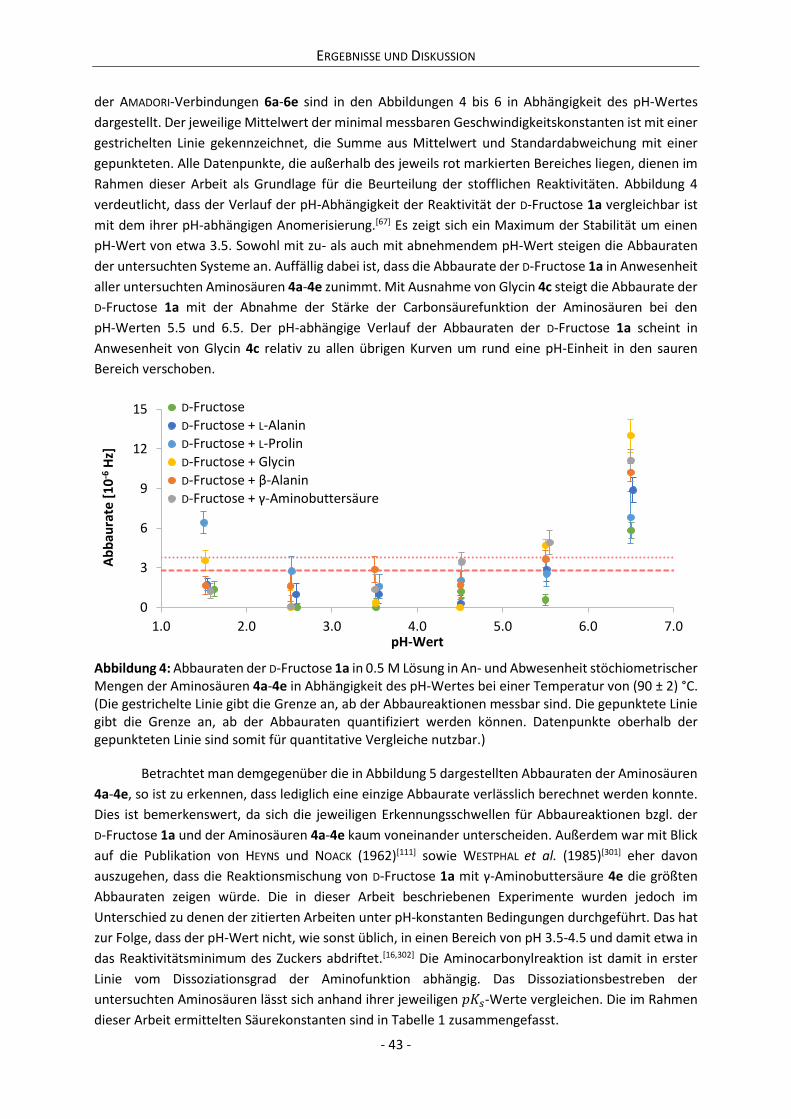
ERGEBNISSE UND DISKUSSION
- 43 -
der AMADORI-Verbindungen 6a-6e sind in den Abbildungen 4 bis 6 in Abhängigkeit des pH-Wertes
dargestellt. Der jeweilige Mittelwert der minimal messbaren Geschwindigkeitskonstanten ist mit einer
gestrichelten Linie gekennzeichnet, die Summe aus Mittelwert und Standardabweichung mit einer
gepunkteten. Alle Datenpunkte, die außerhalb des jeweils rot markierten Bereiches liegen, dienen im
Rahmen dieser Arbeit als Grundlage für die Beurteilung der stofflichen Reaktivitäten. Abbildung 4
verdeutlicht, dass der Verlauf der pH-Abhängigkeit der Reaktivität der D-Fructose 1a vergleichbar ist
mit dem ihrer pH-abhängigen Anomerisierung.[67] Es zeigt sich ein Maximum der Stabilität um einen
pH-Wert von etwa 3.5. Sowohl mit zu- als auch mit abnehmendem pH-Wert steigen die Abbauraten
der untersuchten Systeme an. Auffällig dabei ist, dass die Abbaurate der D-Fructose 1a in Anwesenheit
aller untersuchten Aminosäuren 4a-4e zunimmt. Mit Ausnahme von Glycin 4c steigt die Abbaurate der
D-Fructose 1a mit der Abnahme der Stärke der Carbonsäurefunktion der Aminosäuren bei den
pH-Werten 5.5 und 6.5. Der pH-abhängige Verlauf der Abbauraten der D-Fructose 1a scheint in
Anwesenheit von Glycin 4c relativ zu allen übrigen Kurven um rund eine pH-Einheit in den sauren
Bereich verschoben.
Abbildung 4: Abbauraten der D-Fructose 1a in 0.5 M Lösung in An- und Abwesenheit stöchiometrischer Mengen der Aminosäuren 4a-4e in Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C. (Die gestrichelte Linie gibt die Grenze an, ab der Abbaureaktionen messbar sind. Die gepunktete Linie gibt die Grenze an, ab der Abbauraten quantifiziert werden können. Datenpunkte oberhalb der gepunkteten Linie sind somit für quantitative Vergleiche nutzbar.)
Betrachtet man demgegenüber die in Abbildung 5 dargestellten Abbauraten der Aminosäuren
4a-4e, so ist zu erkennen, dass lediglich eine einzige Abbaurate verlässlich berechnet werden konnte.
Dies ist bemerkenswert, da sich die jeweiligen Erkennungsschwellen für Abbaureaktionen bzgl. der
D-Fructose 1a und der Aminosäuren 4a-4e kaum voneinander unterscheiden. Außerdem war mit Blick
auf die Publikation von HEYNS und NOACK (1962)[111] sowie WESTPHAL et al. (1985)[301] eher davon
auszugehen, dass die Reaktionsmischung von D-Fructose 1a mit γ-Aminobuttersäure 4e die größten
Abbauraten zeigen würde. Die in dieser Arbeit beschriebenen Experimente wurden jedoch im
Unterschied zu denen der zitierten Arbeiten unter pH-konstanten Bedingungen durchgeführt. Das hat
zur Folge, dass der pH-Wert nicht, wie sonst üblich, in einen Bereich von pH 3.5-4.5 und damit etwa in
das Reaktivitätsminimum des Zuckers abdriftet.[16,302] Die Aminocarbonylreaktion ist damit in erster
Linie vom Dissoziationsgrad der Aminofunktion abhängig. Das Dissoziationsbestreben der
untersuchten Aminosäuren lässt sich anhand ihrer jeweiligen 𝑝𝐾𝑠-Werte vergleichen. Die im Rahmen
dieser Arbeit ermittelten Säurekonstanten sind in Tabelle 1 zusammengefasst.
0
3
6
9
12
15
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Ab
bau
rate
[1
0-6
Hz]
pH-Wert
D-FructoseD-Fructose + L-AlaninD-Fructose + L-ProlinD-Fructose + GlycinD-Fructose + β-AlaninD-Fructose + γ-Aminobuttersäure
D-Fructose
D-Fructose + L-Alanin
D-Fructose + L-Prolin
D-Fructose + Glycin
D-Fructose + β-Alanin
D-Fructose + γ-Aminobuttersäure
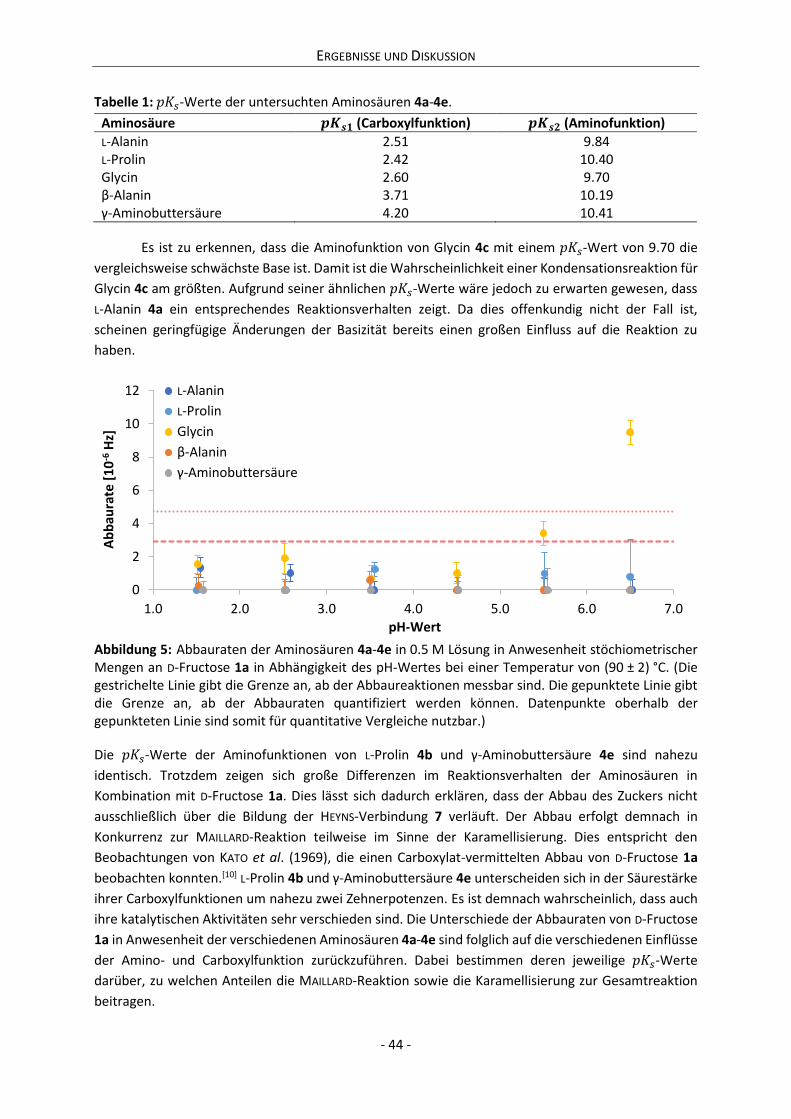
ERGEBNISSE UND DISKUSSION
- 44 -
Tabelle 1: 𝑝𝐾𝑠-Werte der untersuchten Aminosäuren 4a-4e.
Aminosäure 𝒑𝑲𝒔𝟏 (Carboxylfunktion) 𝒑𝑲𝒔𝟐 (Aminofunktion)
L-Alanin 2.51 9.84 L-Prolin 2.42 10.40 Glycin 2.60 9.70 β-Alanin 3.71 10.19 γ-Aminobuttersäure 4.20 10.41
Es ist zu erkennen, dass die Aminofunktion von Glycin 4c mit einem 𝑝𝐾𝑠-Wert von 9.70 die
vergleichsweise schwächste Base ist. Damit ist die Wahrscheinlichkeit einer Kondensationsreaktion für
Glycin 4c am größten. Aufgrund seiner ähnlichen 𝑝𝐾𝑠-Werte wäre jedoch zu erwarten gewesen, dass
L-Alanin 4a ein entsprechendes Reaktionsverhalten zeigt. Da dies offenkundig nicht der Fall ist,
scheinen geringfügige Änderungen der Basizität bereits einen großen Einfluss auf die Reaktion zu
haben.
Abbildung 5: Abbauraten der Aminosäuren 4a-4e in 0.5 M Lösung in Anwesenheit stöchiometrischer Mengen an D-Fructose 1a in Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C. (Die gestrichelte Linie gibt die Grenze an, ab der Abbaureaktionen messbar sind. Die gepunktete Linie gibt die Grenze an, ab der Abbauraten quantifiziert werden können. Datenpunkte oberhalb der gepunkteten Linie sind somit für quantitative Vergleiche nutzbar.)
Die 𝑝𝐾𝑠-Werte der Aminofunktionen von L-Prolin 4b und γ-Aminobuttersäure 4e sind nahezu
identisch. Trotzdem zeigen sich große Differenzen im Reaktionsverhalten der Aminosäuren in
Kombination mit D-Fructose 1a. Dies lässt sich dadurch erklären, dass der Abbau des Zuckers nicht
ausschließlich über die Bildung der HEYNS-Verbindung 7 verläuft. Der Abbau erfolgt demnach in
Konkurrenz zur MAILLARD-Reaktion teilweise im Sinne der Karamellisierung. Dies entspricht den
Beobachtungen von KATO et al. (1969), die einen Carboxylat-vermittelten Abbau von D-Fructose 1a
beobachten konnten.[10] L-Prolin 4b und γ-Aminobuttersäure 4e unterscheiden sich in der Säurestärke
ihrer Carboxylfunktionen um nahezu zwei Zehnerpotenzen. Es ist demnach wahrscheinlich, dass auch
ihre katalytischen Aktivitäten sehr verschieden sind. Die Unterschiede der Abbauraten von D-Fructose
1a in Anwesenheit der verschiedenen Aminosäuren 4a-4e sind folglich auf die verschiedenen Einflüsse
der Amino- und Carboxylfunktion zurückzuführen. Dabei bestimmen deren jeweilige 𝑝𝐾𝑠-Werte
darüber, zu welchen Anteilen die MAILLARD-Reaktion sowie die Karamellisierung zur Gesamtreaktion
beitragen.
0
2
4
6
8
10
12
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Ab
bau
rate
[1
0-6
Hz]
pH-Wert
L-Alanin
L-Prolin
Glycin
β-Alanin
γ-Aminobuttersäure
L-Alanin
L-Prolin
Glycin
β-Alanin
γ-Aminobuttersäure
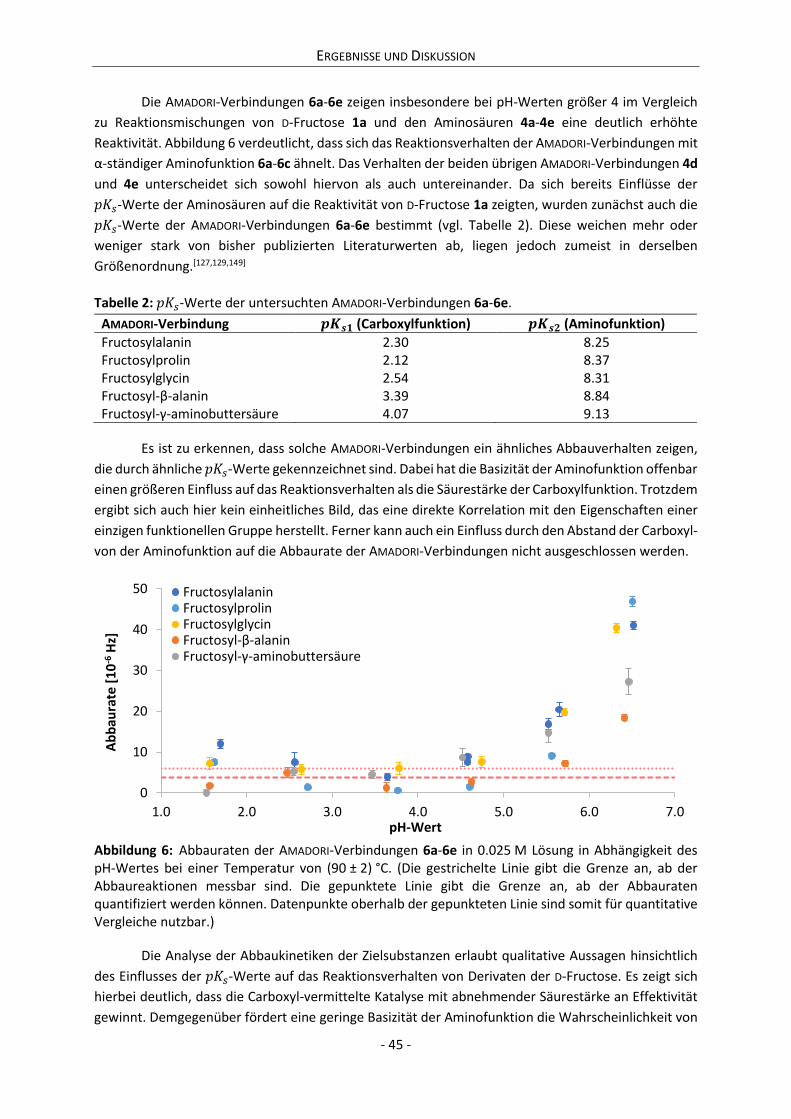
ERGEBNISSE UND DISKUSSION
- 45 -
Die AMADORI-Verbindungen 6a-6e zeigen insbesondere bei pH-Werten größer 4 im Vergleich
zu Reaktionsmischungen von D-Fructose 1a und den Aminosäuren 4a-4e eine deutlich erhöhte
Reaktivität. Abbildung 6 verdeutlicht, dass sich das Reaktionsverhalten der AMADORI-Verbindungen mit
α-ständiger Aminofunktion 6a-6c ähnelt. Das Verhalten der beiden übrigen AMADORI-Verbindungen 4d
und 4e unterscheidet sich sowohl hiervon als auch untereinander. Da sich bereits Einflüsse der
𝑝𝐾𝑠-Werte der Aminosäuren auf die Reaktivität von D-Fructose 1a zeigten, wurden zunächst auch die
𝑝𝐾𝑠-Werte der AMADORI-Verbindungen 6a-6e bestimmt (vgl. Tabelle 2). Diese weichen mehr oder
weniger stark von bisher publizierten Literaturwerten ab, liegen jedoch zumeist in derselben
Größenordnung.[127,129,149]
Tabelle 2: 𝑝𝐾𝑠-Werte der untersuchten AMADORI-Verbindungen 6a-6e.
AMADORI-Verbindung 𝒑𝑲𝒔𝟏 (Carboxylfunktion) 𝒑𝑲𝒔𝟐 (Aminofunktion)
Fructosylalanin 2.30 8.25 Fructosylprolin 2.12 8.37 Fructosylglycin 2.54 8.31 Fructosyl-β-alanin 3.39 8.84 Fructosyl-γ-aminobuttersäure 4.07 9.13
Es ist zu erkennen, dass solche AMADORI-Verbindungen ein ähnliches Abbauverhalten zeigen,
die durch ähnliche 𝑝𝐾𝑠-Werte gekennzeichnet sind. Dabei hat die Basizität der Aminofunktion offenbar
einen größeren Einfluss auf das Reaktionsverhalten als die Säurestärke der Carboxylfunktion. Trotzdem
ergibt sich auch hier kein einheitliches Bild, das eine direkte Korrelation mit den Eigenschaften einer
einzigen funktionellen Gruppe herstellt. Ferner kann auch ein Einfluss durch den Abstand der Carboxyl-
von der Aminofunktion auf die Abbaurate der AMADORI-Verbindungen nicht ausgeschlossen werden.
Abbildung 6: Abbauraten der AMADORI-Verbindungen 6a-6e in 0.025 M Lösung in Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C. (Die gestrichelte Linie gibt die Grenze an, ab der Abbaureaktionen messbar sind. Die gepunktete Linie gibt die Grenze an, ab der Abbauraten quantifiziert werden können. Datenpunkte oberhalb der gepunkteten Linie sind somit für quantitative Vergleiche nutzbar.)
Die Analyse der Abbaukinetiken der Zielsubstanzen erlaubt qualitative Aussagen hinsichtlich
des Einflusses der 𝑝𝐾𝑠-Werte auf das Reaktionsverhalten von Derivaten der D-Fructose. Es zeigt sich
hierbei deutlich, dass die Carboxyl-vermittelte Katalyse mit abnehmender Säurestärke an Effektivität
gewinnt. Demgegenüber fördert eine geringe Basizität der Aminofunktion die Wahrscheinlichkeit von
0
10
20
30
40
50
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Ab
bau
rate
[1
0-6
Hz]
pH-Wert
FructosylalaninFructosylprolinFructosylglycinFructosyl-β-alaninFructosyl-γ-aminobuttersäure

ERGEBNISSE UND DISKUSSION
- 46 -
Kondensationsreaktionen. Ob die beobachtete Zunahme der Reaktivität von AMADORI-Verbindungen
ebenfalls mit einer Abnahme der Basizität korreliert oder mit dem Abstand von Carboxyl- und
Aminofunktion zusammenhängt, kann an dieser Stelle vorerst nicht beantwortet werden.
4.1.2 KINETIK DER BILDUNG VON α-DICARBONYLVERBINDUNGEN
α-Dicarbonylverbindungen gelten als Schlüsselkomponenten der intermediären Phase der
MAILLARD-Reaktion.[303,304] Wie in Schema 13 (Seite 17) gezeigt, gehen diese aus dem Abbau von
AMADORI-Verbindungen 6 hervor, wobei sich analoge Reaktionsmechanismen auch für reine D-Fructose
1a formulieren lassen. D-Glucoson 9d sowie 1- und 3-Desoxyglucoson 9a und 9b gehen dabei unter
Erhalt des Kohlenstoffgerüstes aus den tautomeren Endiolen bzw. Enaminolen von Derivaten der
D-Fructose hervor. Dabei wird in der Literatur ein katalytischer Einfluss von Carbonsäuren auf deren
Bildung diskutiert.[6,10] Es ist aus diesem Grunde von Interesse zu untersuchen, ob ein solcher Einfluss
auch für Aminosäuren nachweisbar ist. In Kapitel 4.1.1 konnte gezeigt werden, dass in den meisten
Fällen keine zeitabhängigen Änderungen der Konzentrationen der untersuchten Aminosäuren im
Rahmen der Modellreaktionen festzustellen waren. Es ist daher davon auszugehen, dass
α-Dicarbonylverbindungen in den untersuchten Reaktionsmischungen der D-Fructose 1a mit den
Aminosäuren 4a-4e hauptsächlich über Reaktionen der Karamellisierung gebildet werden und
Kondensationsreaktionen im Sinne der MAILLARD-Reaktion nur eine untergeordnete Rolle spielen. In
Analogie zu den in Kapitel 2.3 diskutierten Überlegungen zur Katalyse der Anomerisierung ist auch für
die Enolisierung davon auszugehen, dass Aminofunktionen aufgrund ihrer Protonierung im
untersuchten pH-Bereich katalytisch inaktiv sind. Da sich die 𝑝𝐾𝑠-Werte der Carboxylfunktionen der
AMADORI-Verbindungen 6a-6e kaum von denen der entsprechenden Aminosäuren 4a-4e
unterscheiden, ist ein unterschiedliches Bildungsverhalten der α-Dicarbonylverbindungen auf einen
katalytischen Einfluss des Aminostickstoffs zurückzuführen. Der Vergleich der Bildungskinetiken der
genannten α-Dicarbonylverbindungen in Modellreaktionen der AMADORI-Verbindungen 6a-6e sowie
von D-Fructose 1a im Gemisch mit den Aminosäuren 4a-4e gibt daher Auskunft über den Einfluss der
Bindung des Zuckers an die Aminosäure auf die Reaktivität des jeweiligen Systems.
Die Verfolgung der Bildung von D-Glucoson 9d sowie 1- und 3-Desoxyglucoson 9a und 9b im
Verlauf der Modellreaktionen erfolgte über einen Zeitraum von je etwa sechs Stunden. Dabei wurden
den Reaktionslösungen in regelmäßigen Zeitabständen Proben entnommen, mit
ortho-Phenylendiamin zur Bildung stabiler Chinoxalinderivate der α-Dicarbonylverbindungen
derivatisiert und mittels HPLC-DAD auf die jeweiligen Konzentrationen der MAILLARD-Intermediate hin
untersucht. Da diese Intermediate – wie in Kapitel 2.1.4 beschrieben – zahlreiche Folgereaktionen
eingehen, kann der zeitliche Verlauf der jeweiligen Konzentrationen nicht mit einer isolierten
Bildungskinetik erster Ordnung angepasst werden. Vielmehr ist die Reaktionskinetik als ein System von
parallelen Folgereaktionen zu modellieren. Dieses ergibt sich auf Grundlage von Schema 20.
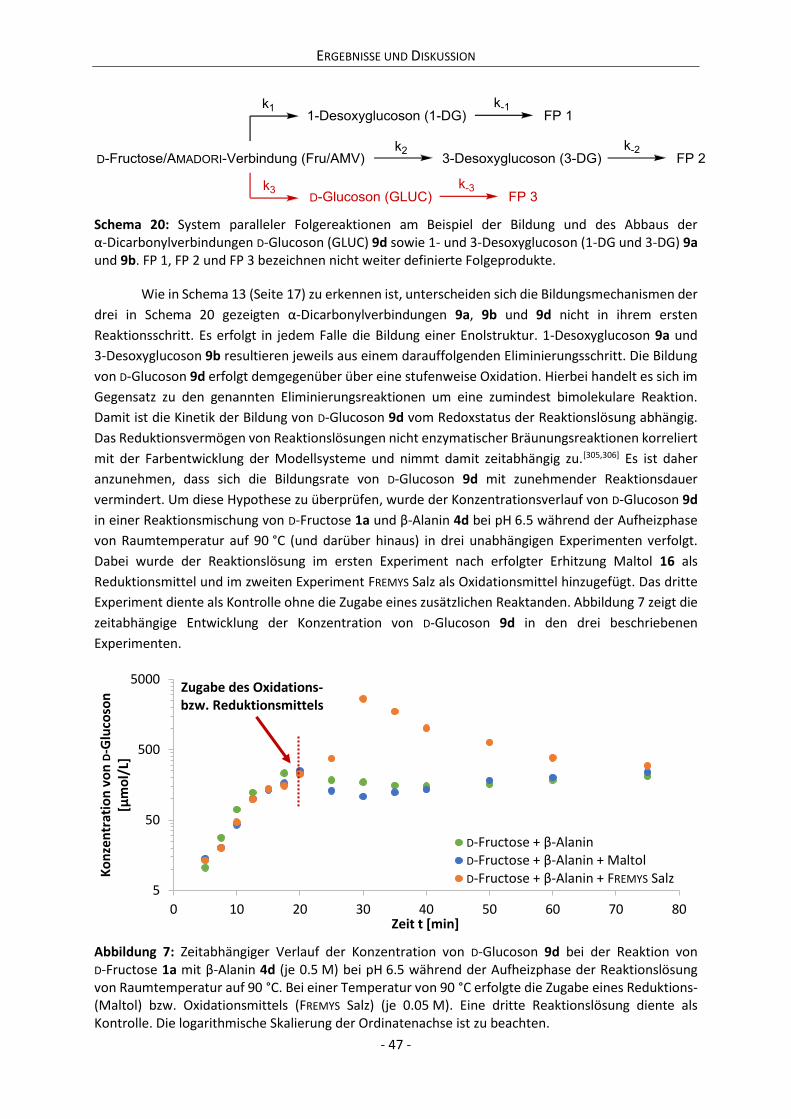
ERGEBNISSE UND DISKUSSION
- 47 -
Schema 20: System paralleler Folgereaktionen am Beispiel der Bildung und des Abbaus der α-Dicarbonylverbindungen D-Glucoson (GLUC) 9d sowie 1- und 3-Desoxyglucoson (1-DG und 3-DG) 9a und 9b. FP 1, FP 2 und FP 3 bezeichnen nicht weiter definierte Folgeprodukte.
Wie in Schema 13 (Seite 17) zu erkennen ist, unterscheiden sich die Bildungsmechanismen der
drei in Schema 20 gezeigten α-Dicarbonylverbindungen 9a, 9b und 9d nicht in ihrem ersten
Reaktionsschritt. Es erfolgt in jedem Falle die Bildung einer Enolstruktur. 1-Desoxyglucoson 9a und
3-Desoxyglucoson 9b resultieren jeweils aus einem darauffolgenden Eliminierungsschritt. Die Bildung
von D-Glucoson 9d erfolgt demgegenüber über eine stufenweise Oxidation. Hierbei handelt es sich im
Gegensatz zu den genannten Eliminierungsreaktionen um eine zumindest bimolekulare Reaktion.
Damit ist die Kinetik der Bildung von D-Glucoson 9d vom Redoxstatus der Reaktionslösung abhängig.
Das Reduktionsvermögen von Reaktionslösungen nicht enzymatischer Bräunungsreaktionen korreliert
mit der Farbentwicklung der Modellsysteme und nimmt damit zeitabhängig zu.[305,306] Es ist daher
anzunehmen, dass sich die Bildungsrate von D-Glucoson 9d mit zunehmender Reaktionsdauer
vermindert. Um diese Hypothese zu überprüfen, wurde der Konzentrationsverlauf von D-Glucoson 9d
in einer Reaktionsmischung von D-Fructose 1a und β-Alanin 4d bei pH 6.5 während der Aufheizphase
von Raumtemperatur auf 90 °C (und darüber hinaus) in drei unabhängigen Experimenten verfolgt.
Dabei wurde der Reaktionslösung im ersten Experiment nach erfolgter Erhitzung Maltol 16 als
Reduktionsmittel und im zweiten Experiment FREMYS Salz als Oxidationsmittel hinzugefügt. Das dritte
Experiment diente als Kontrolle ohne die Zugabe eines zusätzlichen Reaktanden. Abbildung 7 zeigt die
zeitabhängige Entwicklung der Konzentration von D-Glucoson 9d in den drei beschriebenen
Experimenten.
Abbildung 7: Zeitabhängiger Verlauf der Konzentration von D-Glucoson 9d bei der Reaktion von D-Fructose 1a mit β-Alanin 4d (je 0.5 M) bei pH 6.5 während der Aufheizphase der Reaktionslösung von Raumtemperatur auf 90 °C. Bei einer Temperatur von 90 °C erfolgte die Zugabe eines Reduktions- (Maltol) bzw. Oxidationsmittels (FREMYS Salz) (je 0.05 M). Eine dritte Reaktionslösung diente als Kontrolle. Die logarithmische Skalierung der Ordinatenachse ist zu beachten.
5
50
500
5000
0 10 20 30 40 50 60 70 80
Ko
nze
ntr
atio
n v
on
D-G
luco
son
[µ
mo
l/L]
Zeit t [min]
D-Fructose + β-AlaninD-Fructose + β-Alanin + MaltolD-Fructose + β-Alanin + Fremys Salz
Zugabe des Oxidations- bzw. Reduktionsmittels
D-Fructose + β-Alanin D-Fructose + β-Alanin + Maltol D-Fructose + β-Alanin + FREMYS Salz
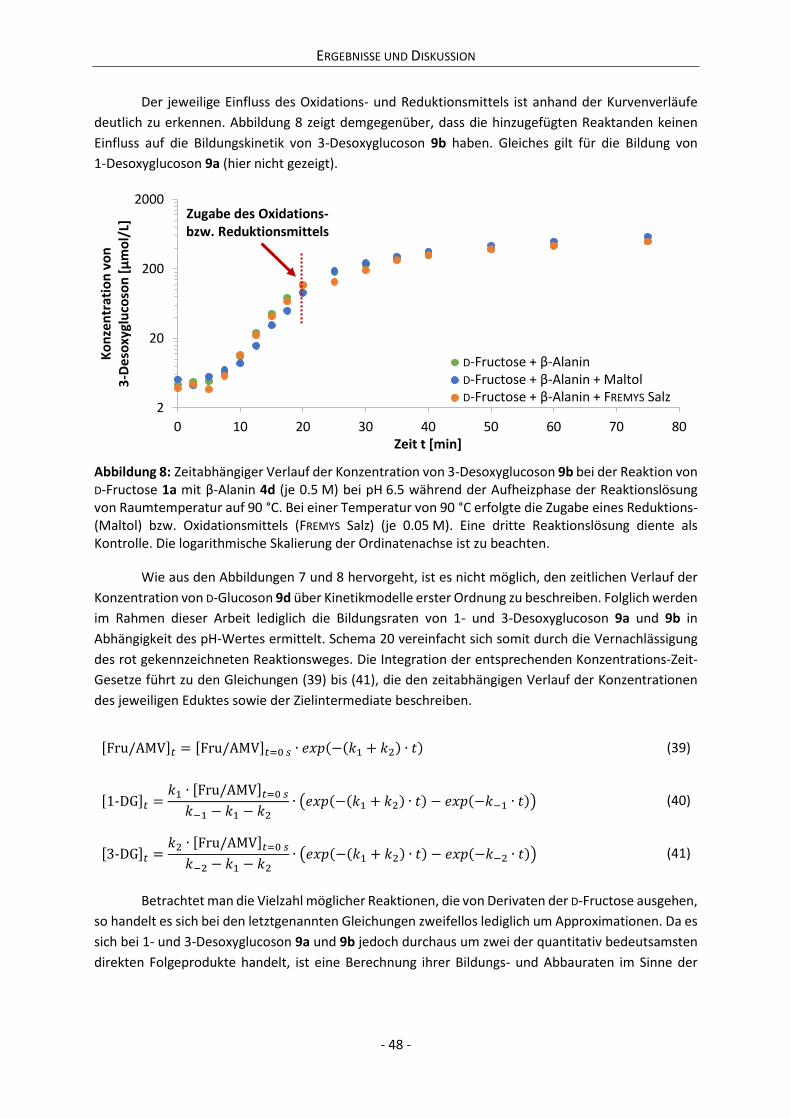
ERGEBNISSE UND DISKUSSION
- 48 -
Der jeweilige Einfluss des Oxidations- und Reduktionsmittels ist anhand der Kurvenverläufe
deutlich zu erkennen. Abbildung 8 zeigt demgegenüber, dass die hinzugefügten Reaktanden keinen
Einfluss auf die Bildungskinetik von 3-Desoxyglucoson 9b haben. Gleiches gilt für die Bildung von
1-Desoxyglucoson 9a (hier nicht gezeigt).
Abbildung 8: Zeitabhängiger Verlauf der Konzentration von 3-Desoxyglucoson 9b bei der Reaktion von D-Fructose 1a mit β-Alanin 4d (je 0.5 M) bei pH 6.5 während der Aufheizphase der Reaktionslösung von Raumtemperatur auf 90 °C. Bei einer Temperatur von 90 °C erfolgte die Zugabe eines Reduktions- (Maltol) bzw. Oxidationsmittels (FREMYS Salz) (je 0.05 M). Eine dritte Reaktionslösung diente als Kontrolle. Die logarithmische Skalierung der Ordinatenachse ist zu beachten.
Wie aus den Abbildungen 7 und 8 hervorgeht, ist es nicht möglich, den zeitlichen Verlauf der
Konzentration von D-Glucoson 9d über Kinetikmodelle erster Ordnung zu beschreiben. Folglich werden
im Rahmen dieser Arbeit lediglich die Bildungsraten von 1- und 3-Desoxyglucoson 9a und 9b in
Abhängigkeit des pH-Wertes ermittelt. Schema 20 vereinfacht sich somit durch die Vernachlässigung
des rot gekennzeichneten Reaktionsweges. Die Integration der entsprechenden Konzentrations-Zeit-
Gesetze führt zu den Gleichungen (39) bis (41), die den zeitabhängigen Verlauf der Konzentrationen
des jeweiligen Eduktes sowie der Zielintermediate beschreiben.
[Fru/AMV]𝑡 = [Fru/AMV]𝑡=0𝑠 ∙ 𝑒𝑥𝑝(−(𝑘1 + 𝑘2) ∙ 𝑡) (39)
[1-DG]𝑡 =𝑘1 ∙ [Fru/AMV]𝑡=0𝑠𝑘−1 − 𝑘1 − 𝑘2
∙ (𝑒𝑥𝑝(−(𝑘1 + 𝑘2) ∙ 𝑡) − 𝑒𝑥𝑝(−𝑘−1 ∙ 𝑡)) (40)
[3-DG]𝑡 =𝑘2 ∙ [Fru/AMV]𝑡=0𝑠𝑘−2 − 𝑘1 − 𝑘2
∙ (𝑒𝑥𝑝(−(𝑘1 + 𝑘2) ∙ 𝑡) − 𝑒𝑥𝑝(−𝑘−2 ∙ 𝑡)) (41)
Betrachtet man die Vielzahl möglicher Reaktionen, die von Derivaten der D-Fructose ausgehen,
so handelt es sich bei den letztgenannten Gleichungen zweifellos lediglich um Approximationen. Da es
sich bei 1- und 3-Desoxyglucoson 9a und 9b jedoch durchaus um zwei der quantitativ bedeutsamsten
direkten Folgeprodukte handelt, ist eine Berechnung ihrer Bildungs- und Abbauraten im Sinne der
2
20
200
2000
0 10 20 30 40 50 60 70 80
Ko
nze
ntr
atio
n v
on
3
-De
soxy
glu
coso
n[µ
mo
l/L]
Zeit t [min]
D-Fructose + β-AlaninD-Fructose + β-Alanin + MaltolD-Fructose + β-Alanin + Fremys Salz
Zugabe des Oxidations- bzw. Reduktionsmittels
D-Fructose + β-Alanin D-Fructose + β-Alanin + Maltol D-Fructose + β-Alanin + FREMYS Salz
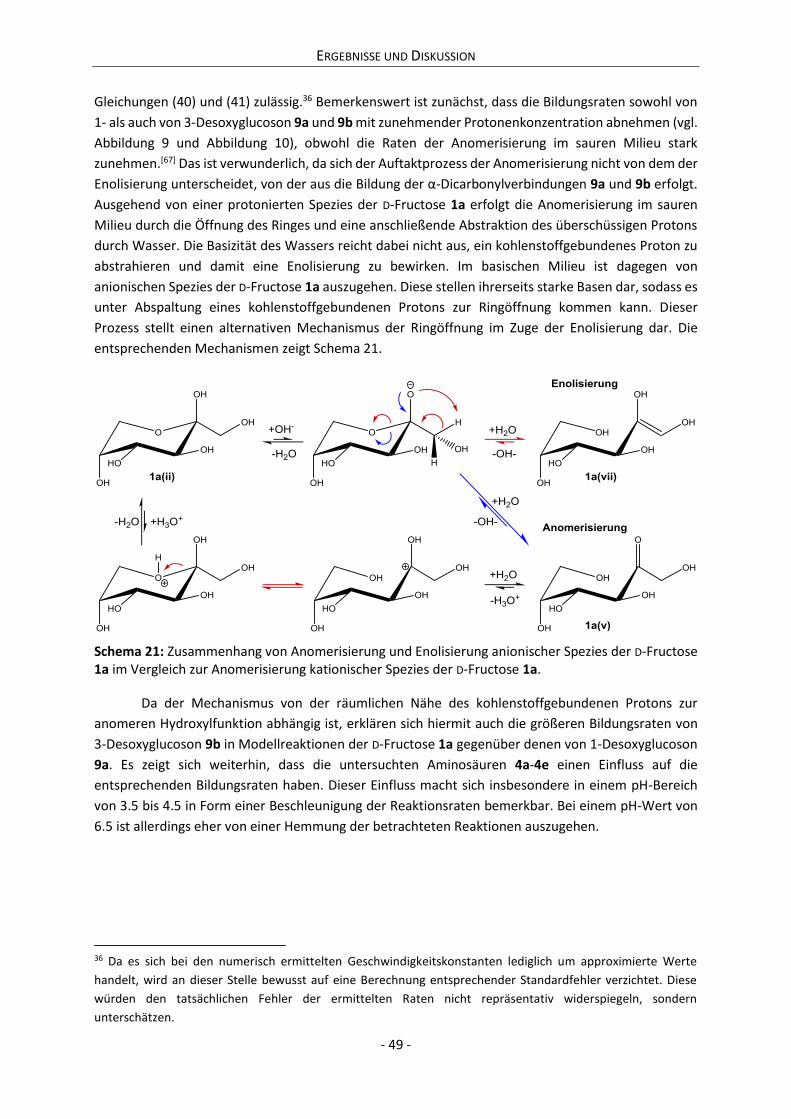
ERGEBNISSE UND DISKUSSION
- 49 -
Gleichungen (40) und (41) zulässig.36 Bemerkenswert ist zunächst, dass die Bildungsraten sowohl von
1- als auch von 3-Desoxyglucoson 9a und 9b mit zunehmender Protonenkonzentration abnehmen (vgl.
Abbildung 9 und Abbildung 10), obwohl die Raten der Anomerisierung im sauren Milieu stark
zunehmen.[67] Das ist verwunderlich, da sich der Auftaktprozess der Anomerisierung nicht von dem der
Enolisierung unterscheidet, von der aus die Bildung der α-Dicarbonylverbindungen 9a und 9b erfolgt.
Ausgehend von einer protonierten Spezies der D-Fructose 1a erfolgt die Anomerisierung im sauren
Milieu durch die Öffnung des Ringes und eine anschließende Abstraktion des überschüssigen Protons
durch Wasser. Die Basizität des Wassers reicht dabei nicht aus, ein kohlenstoffgebundenes Proton zu
abstrahieren und damit eine Enolisierung zu bewirken. Im basischen Milieu ist dagegen von
anionischen Spezies der D-Fructose 1a auszugehen. Diese stellen ihrerseits starke Basen dar, sodass es
unter Abspaltung eines kohlenstoffgebundenen Protons zur Ringöffnung kommen kann. Dieser
Prozess stellt einen alternativen Mechanismus der Ringöffnung im Zuge der Enolisierung dar. Die
entsprechenden Mechanismen zeigt Schema 21.
Schema 21: Zusammenhang von Anomerisierung und Enolisierung anionischer Spezies der D-Fructose 1a im Vergleich zur Anomerisierung kationischer Spezies der D-Fructose 1a.
Da der Mechanismus von der räumlichen Nähe des kohlenstoffgebundenen Protons zur
anomeren Hydroxylfunktion abhängig ist, erklären sich hiermit auch die größeren Bildungsraten von
3-Desoxyglucoson 9b in Modellreaktionen der D-Fructose 1a gegenüber denen von 1-Desoxyglucoson
9a. Es zeigt sich weiterhin, dass die untersuchten Aminosäuren 4a-4e einen Einfluss auf die
entsprechenden Bildungsraten haben. Dieser Einfluss macht sich insbesondere in einem pH-Bereich
von 3.5 bis 4.5 in Form einer Beschleunigung der Reaktionsraten bemerkbar. Bei einem pH-Wert von
6.5 ist allerdings eher von einer Hemmung der betrachteten Reaktionen auszugehen.
36 Da es sich bei den numerisch ermittelten Geschwindigkeitskonstanten lediglich um approximierte Werte
handelt, wird an dieser Stelle bewusst auf eine Berechnung entsprechender Standardfehler verzichtet. Diese
würden den tatsächlichen Fehler der ermittelten Raten nicht repräsentativ widerspiegeln, sondern
unterschätzen.
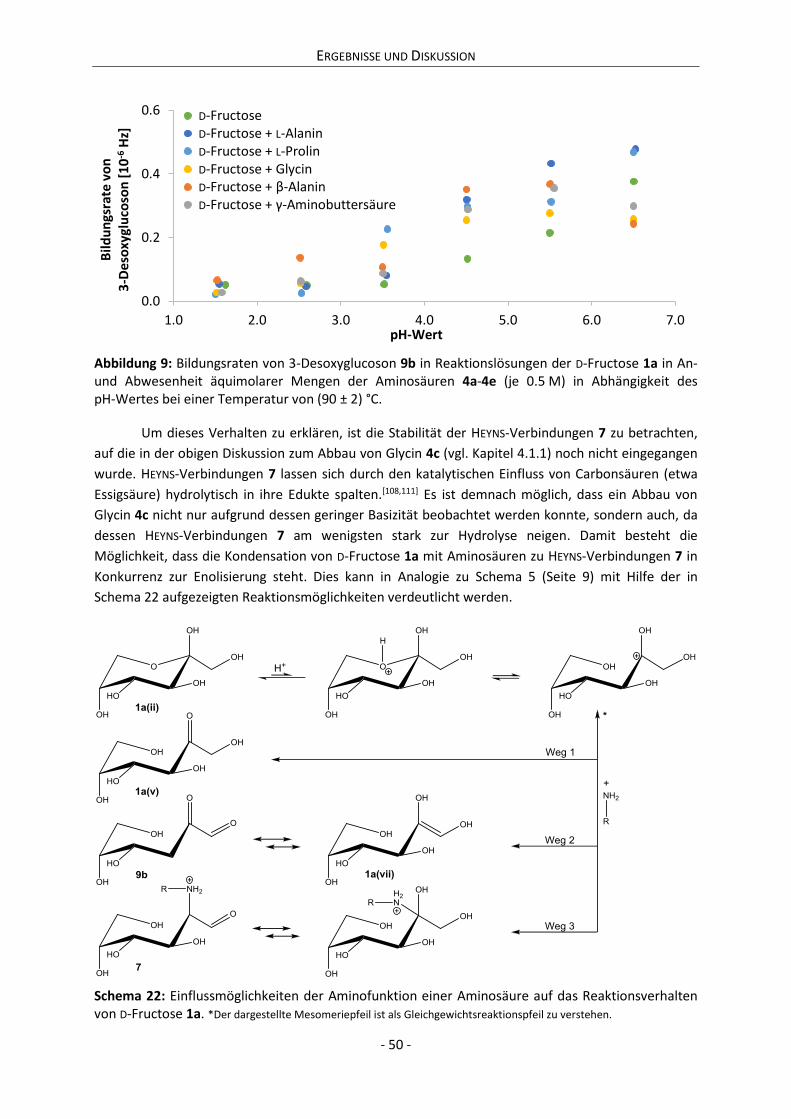
ERGEBNISSE UND DISKUSSION
- 50 -
Abbildung 9: Bildungsraten von 3-Desoxyglucoson 9b in Reaktionslösungen der D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e (je 0.5 M) in Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C.
Um dieses Verhalten zu erklären, ist die Stabilität der HEYNS-Verbindungen 7 zu betrachten,
auf die in der obigen Diskussion zum Abbau von Glycin 4c (vgl. Kapitel 4.1.1) noch nicht eingegangen
wurde. HEYNS-Verbindungen 7 lassen sich durch den katalytischen Einfluss von Carbonsäuren (etwa
Essigsäure) hydrolytisch in ihre Edukte spalten.[108,111] Es ist demnach möglich, dass ein Abbau von
Glycin 4c nicht nur aufgrund dessen geringer Basizität beobachtet werden konnte, sondern auch, da
dessen HEYNS-Verbindungen 7 am wenigsten stark zur Hydrolyse neigen. Damit besteht die
Möglichkeit, dass die Kondensation von D-Fructose 1a mit Aminosäuren zu HEYNS-Verbindungen 7 in
Konkurrenz zur Enolisierung steht. Dies kann in Analogie zu Schema 5 (Seite 9) mit Hilfe der in
Schema 22 aufgezeigten Reaktionsmöglichkeiten verdeutlicht werden.
Schema 22: Einflussmöglichkeiten der Aminofunktion einer Aminosäure auf das Reaktionsverhalten von D-Fructose 1a. *Der dargestellte Mesomeriepfeil ist als Gleichgewichtsreaktionspfeil zu verstehen.
0.0
0.2
0.4
0.6
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Bild
un
gsra
te v
on
3
-De
soxy
glu
coso
n [
10
-6H
z]
pH-Wert
D-FructoseD-Fructose + L-AlaninD-Fructose + L-ProlinD-Fructose + GlycinD-Fructose + β-AlaninD-Fructose + γ-Aminobuttersäure
D-Fructose D-Fructose + L-Alanin D-Fructose + L-Prolin D-Fructose + Glycin D-Fructose + β-Alanin D-Fructose + γ-Aminobuttersäure
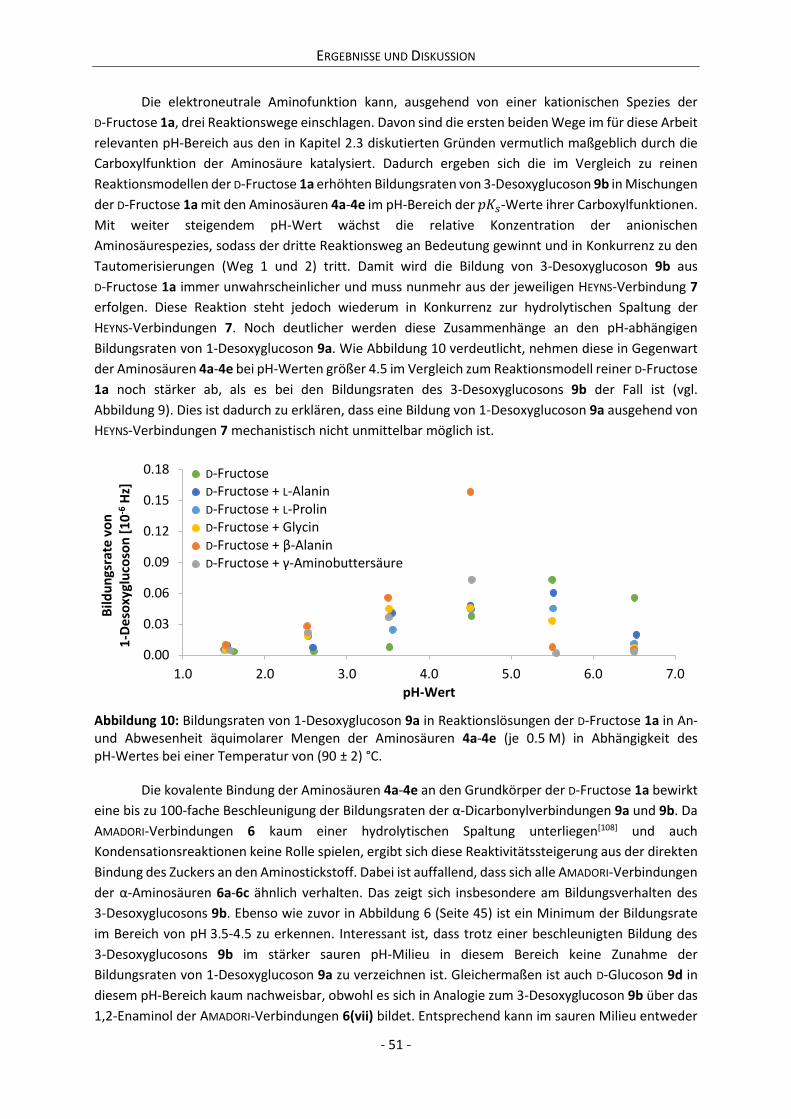
ERGEBNISSE UND DISKUSSION
- 51 -
Die elektroneutrale Aminofunktion kann, ausgehend von einer kationischen Spezies der
D-Fructose 1a, drei Reaktionswege einschlagen. Davon sind die ersten beiden Wege im für diese Arbeit
relevanten pH-Bereich aus den in Kapitel 2.3 diskutierten Gründen vermutlich maßgeblich durch die
Carboxylfunktion der Aminosäure katalysiert. Dadurch ergeben sich die im Vergleich zu reinen
Reaktionsmodellen der D-Fructose 1a erhöhten Bildungsraten von 3-Desoxyglucoson 9b in Mischungen
der D-Fructose 1a mit den Aminosäuren 4a-4e im pH-Bereich der 𝑝𝐾𝑠-Werte ihrer Carboxylfunktionen.
Mit weiter steigendem pH-Wert wächst die relative Konzentration der anionischen
Aminosäurespezies, sodass der dritte Reaktionsweg an Bedeutung gewinnt und in Konkurrenz zu den
Tautomerisierungen (Weg 1 und 2) tritt. Damit wird die Bildung von 3-Desoxyglucoson 9b aus
D-Fructose 1a immer unwahrscheinlicher und muss nunmehr aus der jeweiligen HEYNS-Verbindung 7
erfolgen. Diese Reaktion steht jedoch wiederum in Konkurrenz zur hydrolytischen Spaltung der
HEYNS-Verbindungen 7. Noch deutlicher werden diese Zusammenhänge an den pH-abhängigen
Bildungsraten von 1-Desoxyglucoson 9a. Wie Abbildung 10 verdeutlicht, nehmen diese in Gegenwart
der Aminosäuren 4a-4e bei pH-Werten größer 4.5 im Vergleich zum Reaktionsmodell reiner D-Fructose
1a noch stärker ab, als es bei den Bildungsraten des 3-Desoxyglucosons 9b der Fall ist (vgl.
Abbildung 9). Dies ist dadurch zu erklären, dass eine Bildung von 1-Desoxyglucoson 9a ausgehend von
HEYNS-Verbindungen 7 mechanistisch nicht unmittelbar möglich ist.
Abbildung 10: Bildungsraten von 1-Desoxyglucoson 9a in Reaktionslösungen der D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e (je 0.5 M) in Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C.
Die kovalente Bindung der Aminosäuren 4a-4e an den Grundkörper der D-Fructose 1a bewirkt
eine bis zu 100-fache Beschleunigung der Bildungsraten der α-Dicarbonylverbindungen 9a und 9b. Da
AMADORI-Verbindungen 6 kaum einer hydrolytischen Spaltung unterliegen[108] und auch
Kondensationsreaktionen keine Rolle spielen, ergibt sich diese Reaktivitätssteigerung aus der direkten
Bindung des Zuckers an den Aminostickstoff. Dabei ist auffallend, dass sich alle AMADORI-Verbindungen
der α-Aminosäuren 6a-6c ähnlich verhalten. Das zeigt sich insbesondere am Bildungsverhalten des
3-Desoxyglucosons 9b. Ebenso wie zuvor in Abbildung 6 (Seite 45) ist ein Minimum der Bildungsrate
im Bereich von pH 3.5-4.5 zu erkennen. Interessant ist, dass trotz einer beschleunigten Bildung des
3-Desoxyglucosons 9b im stärker sauren pH-Milieu in diesem Bereich keine Zunahme der
Bildungsraten von 1-Desoxyglucoson 9a zu verzeichnen ist. Gleichermaßen ist auch D-Glucoson 9d in
diesem pH-Bereich kaum nachweisbar, obwohl es sich in Analogie zum 3-Desoxyglucoson 9b über das
1,2-Enaminol der AMADORI-Verbindungen 6(vii) bildet. Entsprechend kann im sauren Milieu entweder
0.00
0.03
0.06
0.09
0.12
0.15
0.18
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Bild
un
gsra
te v
on
1
-De
soxy
glu
coso
n [
10
-6H
z]
pH-Wert
D-FructoseD-Fructose + L-AlaninD-Fructose + L-ProlinD-Fructose + GlycinD-Fructose + β-AlaninD-Fructose + γ-Aminobuttersäure
D-Fructose D-Fructose + L-Alanin D-Fructose + L-Prolin D-Fructose + Glycin D-Fructose + β-Alanin D-Fructose + γ-Aminobuttersäure
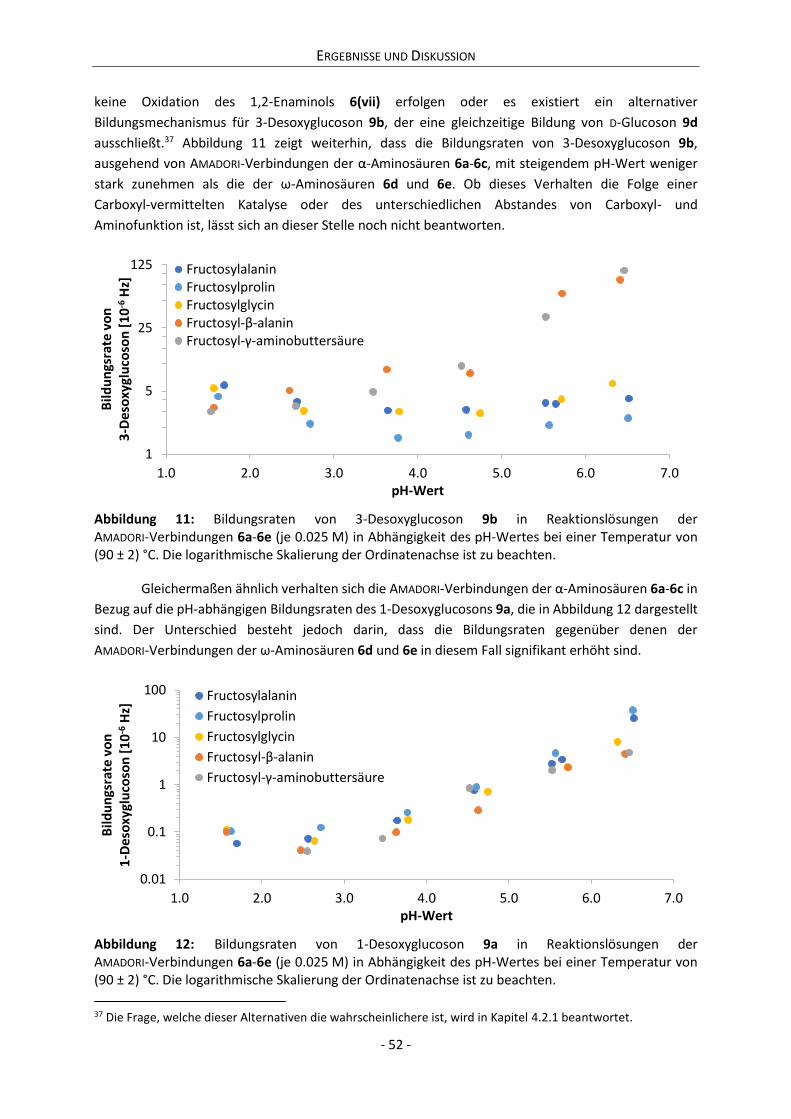
ERGEBNISSE UND DISKUSSION
- 52 -
keine Oxidation des 1,2-Enaminols 6(vii) erfolgen oder es existiert ein alternativer
Bildungsmechanismus für 3-Desoxyglucoson 9b, der eine gleichzeitige Bildung von D-Glucoson 9d
ausschließt.37 Abbildung 11 zeigt weiterhin, dass die Bildungsraten von 3-Desoxyglucoson 9b,
ausgehend von AMADORI-Verbindungen der α-Aminosäuren 6a-6c, mit steigendem pH-Wert weniger
stark zunehmen als die der ω-Aminosäuren 6d und 6e. Ob dieses Verhalten die Folge einer
Carboxyl-vermittelten Katalyse oder des unterschiedlichen Abstandes von Carboxyl- und
Aminofunktion ist, lässt sich an dieser Stelle noch nicht beantworten.
Abbildung 11: Bildungsraten von 3-Desoxyglucoson 9b in Reaktionslösungen der AMADORI-Verbindungen 6a-6e (je 0.025 M) in Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C. Die logarithmische Skalierung der Ordinatenachse ist zu beachten.
Gleichermaßen ähnlich verhalten sich die AMADORI-Verbindungen der α-Aminosäuren 6a-6c in
Bezug auf die pH-abhängigen Bildungsraten des 1-Desoxyglucosons 9a, die in Abbildung 12 dargestellt
sind. Der Unterschied besteht jedoch darin, dass die Bildungsraten gegenüber denen der
AMADORI-Verbindungen der ω-Aminosäuren 6d und 6e in diesem Fall signifikant erhöht sind.
Abbildung 12: Bildungsraten von 1-Desoxyglucoson 9a in Reaktionslösungen der AMADORI-Verbindungen 6a-6e (je 0.025 M) in Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C. Die logarithmische Skalierung der Ordinatenachse ist zu beachten.
37 Die Frage, welche dieser Alternativen die wahrscheinlichere ist, wird in Kapitel 4.2.1 beantwortet.
1
5
25
125
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Bild
un
gsra
te v
on
3
-Des
oxy
glu
coso
n [
10
-6H
z]
pH-Wert
FructosylalaninFructosylprolinFructosylglycinFructosyl-β-alaninFructosyl-γ-aminobuttersäure
0.01
0.1
1
10
100
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Bild
un
gsra
te v
on
1-D
eso
xygl
uco
son
[1
0-6
Hz]
pH-Wert
Fructosylalanin
Fructosylprolin
Fructosylglycin
Fructosyl-β-alanin
Fructosyl-γ-aminobuttersäure

ERGEBNISSE UND DISKUSSION
- 53 -
Das bedeutet, dass die Freisetzung der untersuchten α-Dicarbonylverbindungen 9a und 9b
nicht von denselben Faktoren abhängig ist. Da letztlich nur zwei offensichtliche Einflussfaktoren zur
Diskussion stehen, ist die Bildung einer α-Dicarbonylverbindung vermehrt von einer Carboxylkatalyse
abhängig, während die der anderen sensitiv gegenüber strukturellen Veränderungen ist. Damit
unterscheiden sich die entsprechenden Bildungsmechanismen grundlegender, als es die Analogie der
jeweiligen Enolisierungs- und Elimierungsreaktionen vermuten lässt.
4.1.3 KINETIK DER FARBENTWICKLUNG BEI REAKTIONEN VON DERIVATEN DER D-FRUCTOSE
Im vorigen Kapitel wurde bereits beschrieben, dass α-Dicarbonylverbindungen
Schlüsselintermediate der MAILLARD-Reaktion sind. Dabei ist bekannt, dass sie potente Vorstufen
sowohl von heterocyclischen Verbindungen als auch von Bräunungsprodukten darstellen.[307] Wie in
Kapitel 2.1.4 beschrieben, ist insbesondere die Bildung heterocyclischer Verbindungen auch direkt aus
den cyclischen Anomeren von Derivaten der D-Fructose möglich. Eine Vielzahl derartiger
MAILLARD-Intermediate zeichnet sich durch hohe Extinktionskoeffizienten im UV-Bereich des
elektromagnetischen Spektrums aus. Es ist daher möglich, den Verlauf der frühen sowie intermediären
MAILLARD-Reaktion anhand von Extinktionsmessungen bei einer Wellenlänge von 284 nm zu
verfolgen.[190] Da die Zunahme der Extinktion nicht auf eine einzige Reaktion zurückgeführt werden
kann, ist die entsprechende Kinetik nicht auf Grundlage von Reaktionsmechanismen modellierbar.
Vielmehr hat es sich in diesem Zusammenhang bewährt, entsprechende Kinetiken rein empirisch mit
Hilfe logistischer Verteilungsfunktionen anzupassen.[5,167] Die auf dieser Basis berechneten
Geschwindigkeitskonstanten sind demnach nicht mit denen der hier beschriebenen Abbau- und
Bildungsreaktionen zu vergleichen. Sie dienen lediglich zur Veranschaulichung der Farbentwicklung.
Da Änderungen der Extinktionen von MAILLARD-Reaktionslösungen im UV/Vis-Bereich bereits
auftreten, wenn ein Abbau der Edukte analytisch noch nicht erfassbar ist, handelt es sich hierbei um
eine sehr empfindliche Methode zur Beurteilung der relativen Reaktivitäten verschiedener
Reaktionssysteme. Im Verlauf der nicht enzymatischen Bräunung verschiebt sich die
UV/Vis-Absorption von Reaktionslösungen mehr und mehr in den Bereich größerer Wellenlängen und
damit vom UV- in den visuellen Bereich. Das macht sich in einer Färbung von farblos nach gelb über
braun bis hin zu schwarz bemerkbar. Der Verlauf der fortgeschrittenen MAILLARD-Reaktion kann somit
im visuellen Bereich des elektromagnetischen Spektrums – üblicherweise bei einer Wellenlänge von
420 nm – verfolgt werden.[5,167-169] Die Abbildungen 13 und 14 zeigen, dass die Farbentwicklung von
D-Fructose 1a durch die Anwesenheit der Aminosäuren 4a-4e deutlich beschleunigt ist. Weiterhin
entspricht der pH-Verlauf der Bräunungsraten (Extinktion bei 420 nm) im Wesentlichen dem der
Bildungsraten des 3-Desoxyglucosons 9b. Demgegenüber nehmen die Raten der Extinktionszunahme
bei 284 nm nach Durchlauf eines Minimums mit steigender Protonenkonzentration zu. Das
untermauert die in Kapitel 2.1.4 formulierten Reaktionsmechanismen, in denen die Bildung
heterocyclischer Verbindungen direkt aus den Derivaten der D-Fructose dargestellt ist. Der Einfluss der
Aminosäuren 4a-4e auf die Extinktionszunahme bei 284 nm ist stark ausgeprägt (vgl. Abbildung 13). Je
nach pH-Wert ist die Extinktionszunahme von Reaktionslösungen der D-Fructose 1a in Anwesenheit
der Aminosäuren 4a-4e bei 284 nm rund 5- bis 20-mal schneller als von Lösungen reiner D-Fructose 1a.
Die Art der Aminosäure spielt dabei nur eine untergeordnete Rolle. Für pH 6.5 ergibt sich jedoch
dieselbe Reaktivitätsreihenfolge, die auch für die Abbauraten der D-Fructose 1a beobachtet wurde.
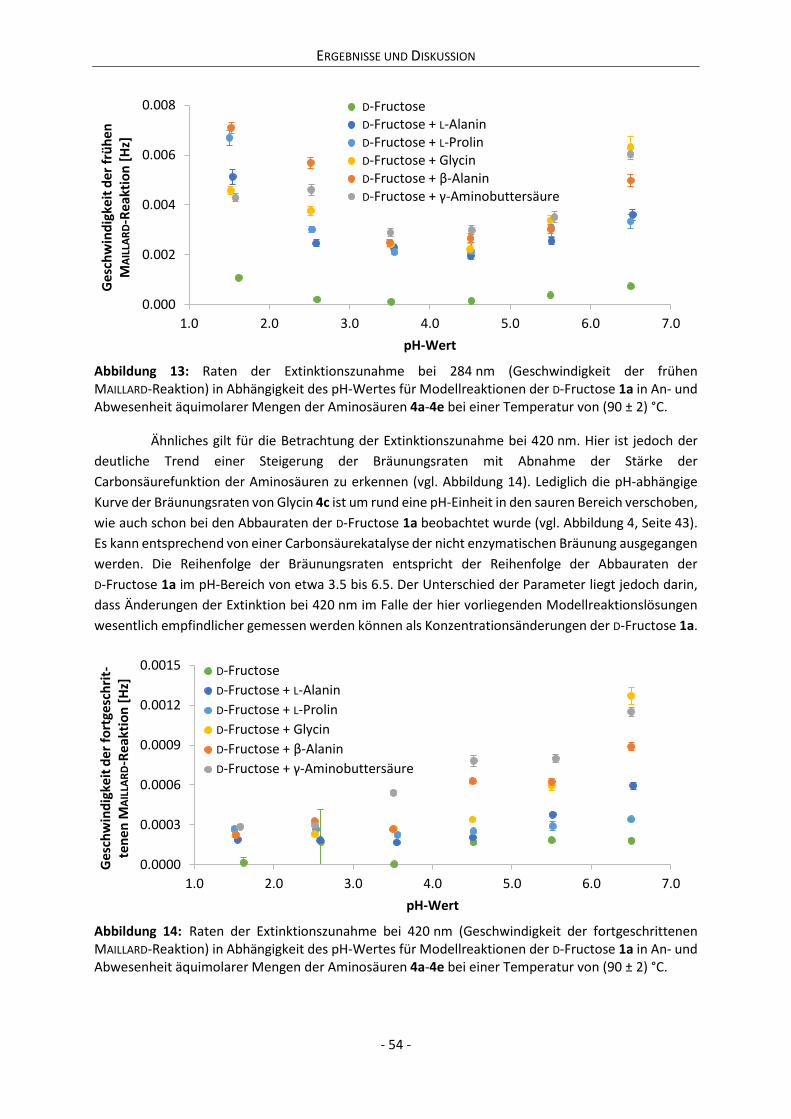
ERGEBNISSE UND DISKUSSION
- 54 -
Abbildung 13: Raten der Extinktionszunahme bei 284 nm (Geschwindigkeit der frühen MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei einer Temperatur von (90 ± 2) °C.
Ähnliches gilt für die Betrachtung der Extinktionszunahme bei 420 nm. Hier ist jedoch der
deutliche Trend einer Steigerung der Bräunungsraten mit Abnahme der Stärke der
Carbonsäurefunktion der Aminosäuren zu erkennen (vgl. Abbildung 14). Lediglich die pH-abhängige
Kurve der Bräunungsraten von Glycin 4c ist um rund eine pH-Einheit in den sauren Bereich verschoben,
wie auch schon bei den Abbauraten der D-Fructose 1a beobachtet wurde (vgl. Abbildung 4, Seite 43).
Es kann entsprechend von einer Carbonsäurekatalyse der nicht enzymatischen Bräunung ausgegangen
werden. Die Reihenfolge der Bräunungsraten entspricht der Reihenfolge der Abbauraten der
D-Fructose 1a im pH-Bereich von etwa 3.5 bis 6.5. Der Unterschied der Parameter liegt jedoch darin,
dass Änderungen der Extinktion bei 420 nm im Falle der hier vorliegenden Modellreaktionslösungen
wesentlich empfindlicher gemessen werden können als Konzentrationsänderungen der D-Fructose 1a.
Abbildung 14: Raten der Extinktionszunahme bei 420 nm (Geschwindigkeit der fortgeschrittenen MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei einer Temperatur von (90 ± 2) °C.
0.000
0.002
0.004
0.006
0.008
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Ge
sch
win
dig
keit
de
r fr
üh
en
M
AIL
LAR
D-R
eak
tio
n [
Hz]
pH-Wert
D-FructoseD-Fructose + L-AlaninD-Fructose + L-ProlinD-Fructose + GlycinD-Fructose + β-AlaninD-Fructose + γ-Aminobuttersäure
0.0000
0.0003
0.0006
0.0009
0.0012
0.0015
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Ges
chw
ind
igke
it d
er f
ort
gesc
hri
t-te
nen
MA
ILLA
RD
-Re
akti
on
[H
z]
pH-Wert
D-Fructose
D-Fructose + L-Alanin
D-Fructose + L-Prolin
D-Fructose + Glycin
D-Fructose + β-Alanin
D-Fructose + γ-Aminobuttersäure
D-Fructose D-Fructose + L-Alanin D-Fructose + L-Prolin D-Fructose + Glycin D-Fructose + β-Alanin D-Fructose + γ-Aminobuttersäure
D-Fructose
D-Fructose + L-Alanin
D-Fructose + L-Prolin
D-Fructose + Glycin
D-Fructose + β-Alanin
D-Fructose + γ-Aminobuttersäure
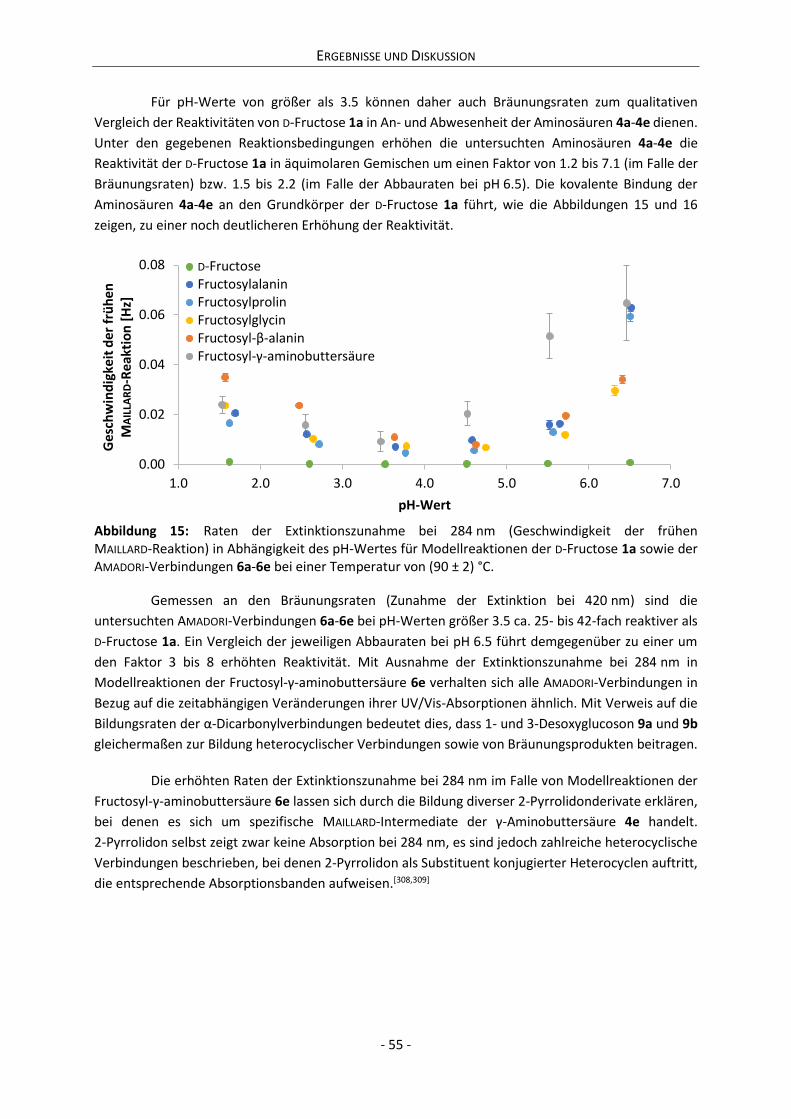
ERGEBNISSE UND DISKUSSION
- 55 -
Für pH-Werte von größer als 3.5 können daher auch Bräunungsraten zum qualitativen
Vergleich der Reaktivitäten von D-Fructose 1a in An- und Abwesenheit der Aminosäuren 4a-4e dienen.
Unter den gegebenen Reaktionsbedingungen erhöhen die untersuchten Aminosäuren 4a-4e die
Reaktivität der D-Fructose 1a in äquimolaren Gemischen um einen Faktor von 1.2 bis 7.1 (im Falle der
Bräunungsraten) bzw. 1.5 bis 2.2 (im Falle der Abbauraten bei pH 6.5). Die kovalente Bindung der
Aminosäuren 4a-4e an den Grundkörper der D-Fructose 1a führt, wie die Abbildungen 15 und 16
zeigen, zu einer noch deutlicheren Erhöhung der Reaktivität.
Abbildung 15: Raten der Extinktionszunahme bei 284 nm (Geschwindigkeit der frühen MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der D-Fructose 1a sowie der AMADORI-Verbindungen 6a-6e bei einer Temperatur von (90 ± 2) °C.
Gemessen an den Bräunungsraten (Zunahme der Extinktion bei 420 nm) sind die
untersuchten AMADORI-Verbindungen 6a-6e bei pH-Werten größer 3.5 ca. 25- bis 42-fach reaktiver als
D-Fructose 1a. Ein Vergleich der jeweiligen Abbauraten bei pH 6.5 führt demgegenüber zu einer um
den Faktor 3 bis 8 erhöhten Reaktivität. Mit Ausnahme der Extinktionszunahme bei 284 nm in
Modellreaktionen der Fructosyl-γ-aminobuttersäure 6e verhalten sich alle AMADORI-Verbindungen in
Bezug auf die zeitabhängigen Veränderungen ihrer UV/Vis-Absorptionen ähnlich. Mit Verweis auf die
Bildungsraten der α-Dicarbonylverbindungen bedeutet dies, dass 1- und 3-Desoxyglucoson 9a und 9b
gleichermaßen zur Bildung heterocyclischer Verbindungen sowie von Bräunungsprodukten beitragen.
Die erhöhten Raten der Extinktionszunahme bei 284 nm im Falle von Modellreaktionen der
Fructosyl-γ-aminobuttersäure 6e lassen sich durch die Bildung diverser 2-Pyrrolidonderivate erklären,
bei denen es sich um spezifische MAILLARD-Intermediate der γ-Aminobuttersäure 4e handelt.
2-Pyrrolidon selbst zeigt zwar keine Absorption bei 284 nm, es sind jedoch zahlreiche heterocyclische
Verbindungen beschrieben, bei denen 2-Pyrrolidon als Substituent konjugierter Heterocyclen auftritt,
die entsprechende Absorptionsbanden aufweisen.[308,309]
0.00
0.02
0.04
0.06
0.08
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Ge
sch
win
dig
keit
de
r fr
üh
en
M
AIL
LAR
D-R
eak
tio
n [
Hz]
pH-Wert
D-FructoseFructosylalaninFructosylprolinFructosylglycinFructosyl-β-alaninFructosyl-γ-aminobuttersäure
D-Fructose Fructosylalanin Fructosylprolin Fructosylglycin Fructosyl-β-alanin Fructosyl-γ-aminobuttersäure
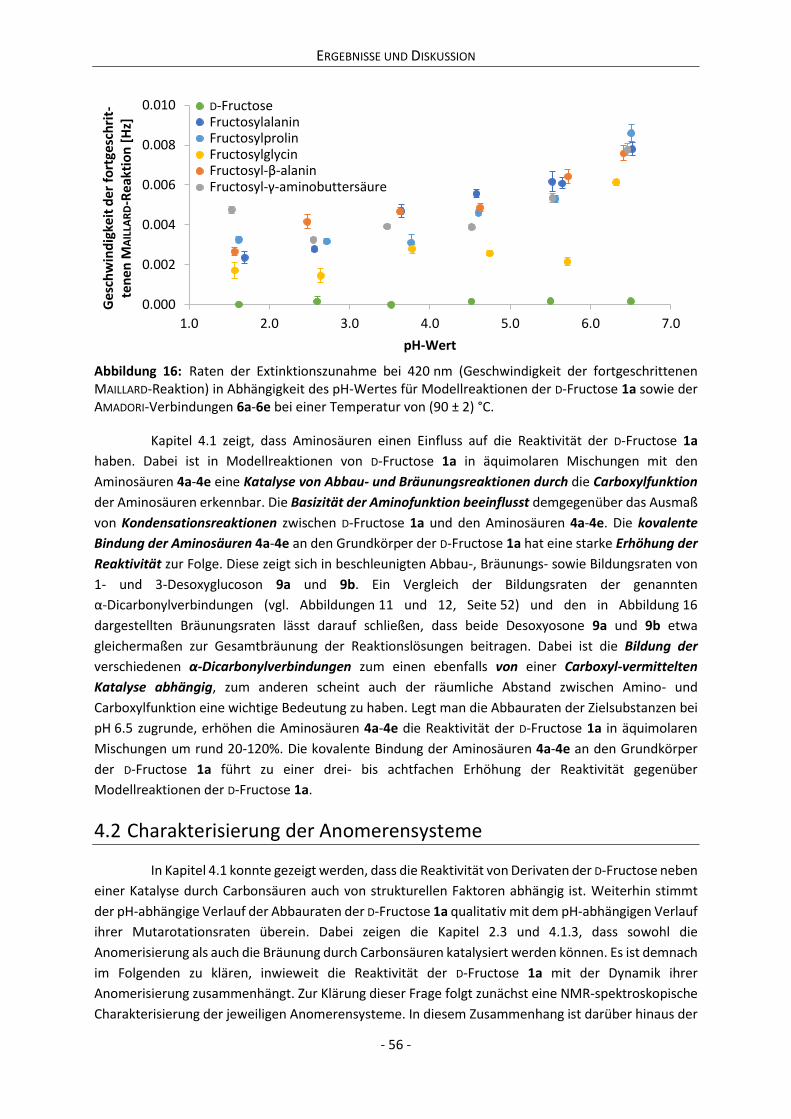
ERGEBNISSE UND DISKUSSION
- 56 -
Abbildung 16: Raten der Extinktionszunahme bei 420 nm (Geschwindigkeit der fortgeschrittenen MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der D-Fructose 1a sowie der AMADORI-Verbindungen 6a-6e bei einer Temperatur von (90 ± 2) °C.
Kapitel 4.1 zeigt, dass Aminosäuren einen Einfluss auf die Reaktivität der D-Fructose 1a
haben. Dabei ist in Modellreaktionen von D-Fructose 1a in äquimolaren Mischungen mit den
Aminosäuren 4a-4e eine Katalyse von Abbau- und Bräunungsreaktionen durch die Carboxylfunktion
der Aminosäuren erkennbar. Die Basizität der Aminofunktion beeinflusst demgegenüber das Ausmaß
von Kondensationsreaktionen zwischen D-Fructose 1a und den Aminosäuren 4a-4e. Die kovalente
Bindung der Aminosäuren 4a-4e an den Grundkörper der D-Fructose 1a hat eine starke Erhöhung der
Reaktivität zur Folge. Diese zeigt sich in beschleunigten Abbau-, Bräunungs- sowie Bildungsraten von
1- und 3-Desoxyglucoson 9a und 9b. Ein Vergleich der Bildungsraten der genannten
α-Dicarbonylverbindungen (vgl. Abbildungen 11 und 12, Seite 52) und den in Abbildung 16
dargestellten Bräunungsraten lässt darauf schließen, dass beide Desoxyosone 9a und 9b etwa
gleichermaßen zur Gesamtbräunung der Reaktionslösungen beitragen. Dabei ist die Bildung der
verschiedenen α-Dicarbonylverbindungen zum einen ebenfalls von einer Carboxyl-vermittelten
Katalyse abhängig, zum anderen scheint auch der räumliche Abstand zwischen Amino- und
Carboxylfunktion eine wichtige Bedeutung zu haben. Legt man die Abbauraten der Zielsubstanzen bei
pH 6.5 zugrunde, erhöhen die Aminosäuren 4a-4e die Reaktivität der D-Fructose 1a in äquimolaren
Mischungen um rund 20-120%. Die kovalente Bindung der Aminosäuren 4a-4e an den Grundkörper
der D-Fructose 1a führt zu einer drei- bis achtfachen Erhöhung der Reaktivität gegenüber
Modellreaktionen der D-Fructose 1a.
4.2 Charakterisierung der Anomerensysteme
In Kapitel 4.1 konnte gezeigt werden, dass die Reaktivität von Derivaten der D-Fructose neben
einer Katalyse durch Carbonsäuren auch von strukturellen Faktoren abhängig ist. Weiterhin stimmt
der pH-abhängige Verlauf der Abbauraten der D-Fructose 1a qualitativ mit dem pH-abhängigen Verlauf
ihrer Mutarotationsraten überein. Dabei zeigen die Kapitel 2.3 und 4.1.3, dass sowohl die
Anomerisierung als auch die Bräunung durch Carbonsäuren katalysiert werden können. Es ist demnach
im Folgenden zu klären, inwieweit die Reaktivität der D-Fructose 1a mit der Dynamik ihrer
Anomerisierung zusammenhängt. Zur Klärung dieser Frage folgt zunächst eine NMR-spektroskopische
Charakterisierung der jeweiligen Anomerensysteme. In diesem Zusammenhang ist darüber hinaus der
0.000
0.002
0.004
0.006
0.008
0.010
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Ge
sch
win
dig
keit
de
r fo
rtge
sch
rit-
ten
en
MA
ILLA
RD
-Re
akti
on
[H
z]
pH-Wert
D-FructoseFructosylalaninFructosylprolinFructosylglycinFructosyl-β-alaninFructosyl-γ-aminobuttersäure
D-Fructose Fructosylalanin Fructosylprolin Fructosylglycin Fructosyl-β-alanin Fructosyl-γ-aminobuttersäure

ERGEBNISSE UND DISKUSSION
- 57 -
Einfluss des Abstandes von Amino- und Carboxylfunktion auf die Bildung der Desoxyosone 9a und 9b
zu untersuchen.
4.2.1 STRUKTURAUFKLÄRUNG DER ANOMERENSYSTEME
Die Strukturaufklärung der Anomerensysteme der D-Fructose 1a sowie der
AMADORI-Verbindungen 6a-6e erfolgte auf Basis der hochauflösenden NMR-Spektroskopie. Da 1H-NMR-Spektren der jeweiligen Analyten im Saccharidbereich (3.4-4.0 ppm)[310] stark überlagert sind
und weder D-Fructose 1a noch deren Derivate 6a-6e ein anomeres Proton tragen, dienten die
jeweiligen anomeren 13C-NMR-Signale als Ankergruppen für die Strukturaufklärung. Über
Korrelationspeaks in HMBC-Spektren wurden diese ihren angrenzenden Spinsystemen zugeordnet. Die
vollständige Aufklärung der Spinsysteme erfolgte auf Basis von COSY- und HSQC-TOCSY-Experimenten.
Kopplungskonstanten wurden mit Hilfe von J-resolved-Experimenten gemessen, auf deren Grundlage
die Konformation der jeweiligen pyranoiden Sesselstrukturen abgeleitet wurde. Die Zuordnung von 1H-13C-Konnektivitäten erfolgte mittels HSQC-Experimenten und die Ringgröße der Zuckeranomere
wurde mit Hilfe von HMBC-Spektren bestimmt.
Alle untersuchten Derivate der D-Fructose zeigen vier Signale im anomeren Bereich der 13C-NMR-Spektren. Darüber hinaus findet sich im Bereich von etwa 205-215 ppm je ein Signal, das dem
Ketokohlenstoffatom des jeweiligen acyclischen Isomers 1a(v) bzw. 6(v) zuzuordnen ist. Die anomeren 13C-NMR-Signale der furanoiden Anomere liegen bei tieferem Feld als die der pyranoiden, wobei das
Signal der β-Furanosen 1a(iv) bzw. 6(iv) bei höherem Feld liegt als das der α-Furanosen 1a(iii) bzw.
6(iii).[311,312] Die vollständige Signalzuordnung steht jeweils im Einklang mit den in den Schemata 2 und
12 gezeigten Anomerensystemen. Da jedoch keinerlei Messung in H218O durchgeführt wurde, konnten
keine Hinweise auf acyclische gem-Diole 1a(vi) bzw. 6(vi) identifiziert werden. Wie bereits in den
Kapiteln 2.1.1.1 und 2.1.3.2 diskutiert wurde, ist die Struktur der α-pyranoiden Isomere 1a(i) und 6(i)
nicht abschließend aufgeklärt. Es zeigen sich vielmehr Hinweise darauf, dass eine schnelle 5C2 2C5
Ringinversion vorliegen könnte. Wenn dies der Fall ist, muss es sich bei dem anomeren 13C-NMR-Signal
der α-Pyranose 1a(i) bzw. 6(i) um ein gemitteltes Signal beider Konformere handeln (schneller
chemischer Austausch). Solange beide Konformere in ähnlichen relativen Konzentrationen
nebeneinander vorliegen, muss eine Verringerung der Temperatur bzw. eine Erhöhung der
magnetischen Flussdichte des Hauptmagnetfeldes demnach zu einer Erhöhung der Linienbreite der
Resonanz des anomeren Kohlenstoffsignals der α-Pyranose 1a(i) bzw. 6(i) führen. Um dies zu testen,
wurden die Linienbreiten aller anomeren 13C-NMR-Signale für D-[2-13C]Fructose 1a sowie
N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-alanin 6a und N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-prolin 6b in
Abhängigkeit der Temperatur gemessen. Es zeigt sich dabei, dass die Linienbreiten mit Ausnahme der
α-Pyranosen 1a(i) und 6(i) für alle anomeren 13C-NMR-Signale nur geringfügig von der Temperatur
abhängig sind. Im Falle der D-Fructose 1a werden die chemischen Verschiebungen der anomeren 13C-NMR-Signale der α- und β-Pyranose 1a(i) und 1a(ii) mit abnehmender Temperatur zunehmend
isochron, weshalb eine exakte Messung der Linienbreiten nicht möglich ist. Qualitativ kann jedoch eine
Zunahme der Linienbreite der entsprechenden Resonanz der α-Pyranose 1a(i) beobachtet werden. Im
Falle der [2-13C]-markierten AMADORI-Verbindungen 6a und 6b ist eine präzise Messung der
entsprechenden Linienbreiten möglich. Diese sind in Abhängigkeit der Temperatur in Tabelle 3
zusammengefasst.
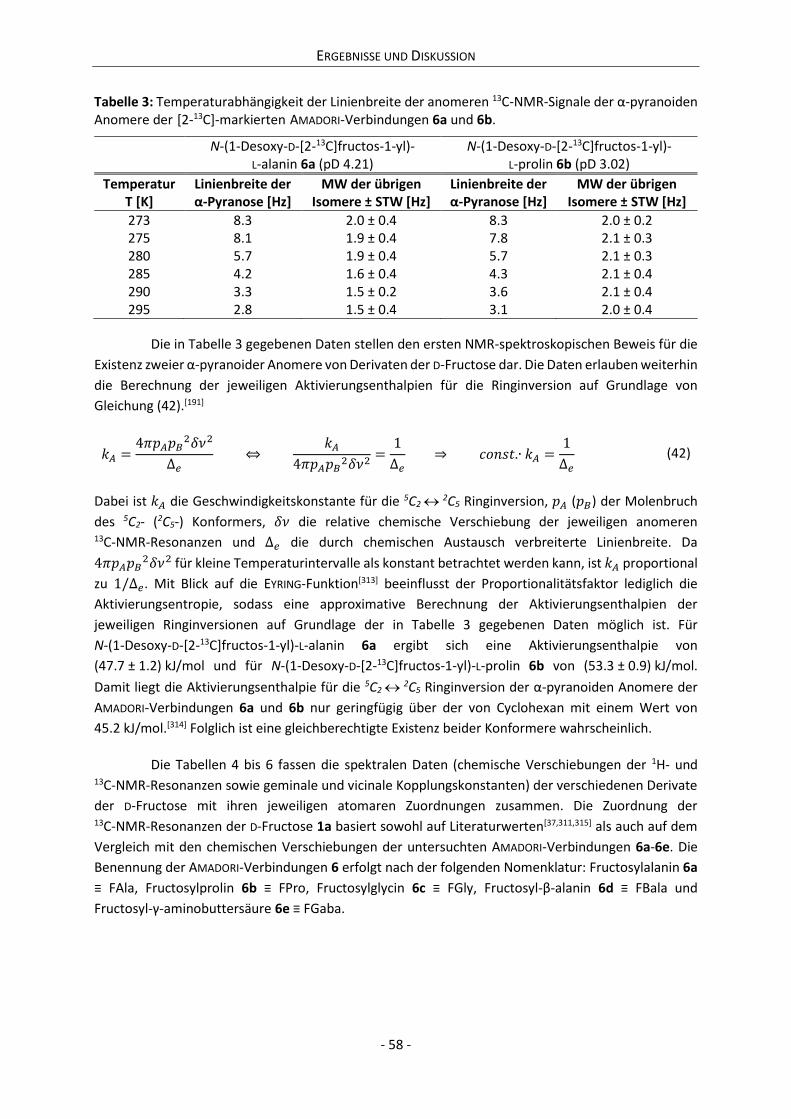
ERGEBNISSE UND DISKUSSION
- 58 -
Tabelle 3: Temperaturabhängigkeit der Linienbreite der anomeren 13C-NMR-Signale der α-pyranoiden Anomere der [2-13C]-markierten AMADORI-Verbindungen 6a und 6b.
N-(1-Desoxy-D-[2-13C]fructos-1-yl)-
L-alanin 6a (pD 4.21) N-(1-Desoxy-D-[2-13C]fructos-1-yl)-
L-prolin 6b (pD 3.02)
Temperatur T [K]
Linienbreite der α-Pyranose [Hz]
MW der übrigen Isomere ± STW [Hz]
Linienbreite der α-Pyranose [Hz]
MW der übrigen Isomere ± STW [Hz]
273 8.3 2.0 ± 0.4 8.3 2.0 ± 0.2 275 8.1 1.9 ± 0.4 7.8 2.1 ± 0.3 280 5.7 1.9 ± 0.4 5.7 2.1 ± 0.3 285 4.2 1.6 ± 0.4 4.3 2.1 ± 0.4 290 3.3 1.5 ± 0.2 3.6 2.1 ± 0.4 295 2.8 1.5 ± 0.4 3.1 2.0 ± 0.4
Die in Tabelle 3 gegebenen Daten stellen den ersten NMR-spektroskopischen Beweis für die
Existenz zweier α-pyranoider Anomere von Derivaten der D-Fructose dar. Die Daten erlauben weiterhin
die Berechnung der jeweiligen Aktivierungsenthalpien für die Ringinversion auf Grundlage von
Gleichung (42).[191]
𝑘𝐴 =4𝜋𝑝𝐴𝑝𝐵
2𝛿𝜈2
Δ𝑒⇔
𝑘𝐴4𝜋𝑝𝐴𝑝𝐵
2𝛿𝜈2=1
Δ𝑒⇒ 𝑐𝑜𝑛𝑠𝑡.∙ 𝑘𝐴 =
1
Δ𝑒 (42)
Dabei ist 𝑘𝐴 die Geschwindigkeitskonstante für die 5C2 2C5 Ringinversion, 𝑝𝐴 (𝑝𝐵) der Molenbruch
des 5C2- (2C5-) Konformers, 𝛿𝜈 die relative chemische Verschiebung der jeweiligen anomeren 13C-NMR-Resonanzen und Δ𝑒 die durch chemischen Austausch verbreiterte Linienbreite. Da
4𝜋𝑝𝐴𝑝𝐵2𝛿𝜈2 für kleine Temperaturintervalle als konstant betrachtet werden kann, ist 𝑘𝐴 proportional
zu 1/Δ𝑒. Mit Blick auf die EYRING-Funktion[313] beeinflusst der Proportionalitätsfaktor lediglich die
Aktivierungsentropie, sodass eine approximative Berechnung der Aktivierungsenthalpien der
jeweiligen Ringinversionen auf Grundlage der in Tabelle 3 gegebenen Daten möglich ist. Für
N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-alanin 6a ergibt sich eine Aktivierungsenthalpie von
(47.7 ± 1.2) kJ/mol und für N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-prolin 6b von (53.3 ± 0.9) kJ/mol.
Damit liegt die Aktivierungsenthalpie für die 5C2 2C5 Ringinversion der α-pyranoiden Anomere der
AMADORI-Verbindungen 6a und 6b nur geringfügig über der von Cyclohexan mit einem Wert von
45.2 kJ/mol.[314] Folglich ist eine gleichberechtigte Existenz beider Konformere wahrscheinlich.
Die Tabellen 4 bis 6 fassen die spektralen Daten (chemische Verschiebungen der 1H- und 13C-NMR-Resonanzen sowie geminale und vicinale Kopplungskonstanten) der verschiedenen Derivate
der D-Fructose mit ihren jeweiligen atomaren Zuordnungen zusammen. Die Zuordnung der 13C-NMR-Resonanzen der D-Fructose 1a basiert sowohl auf Literaturwerten[37,311,315] als auch auf dem
Vergleich mit den chemischen Verschiebungen der untersuchten AMADORI-Verbindungen 6a-6e. Die
Benennung der AMADORI-Verbindungen 6 erfolgt nach der folgenden Nomenklatur: Fructosylalanin 6a
≡ FAla, Fructosylprolin 6b ≡ FPro, Fructosylglycin 6c ≡ FGly, Fructosyl-β-alanin 6d ≡ FBala und
Fructosyl-γ-aminobuttersäure 6e ≡ FGaba.
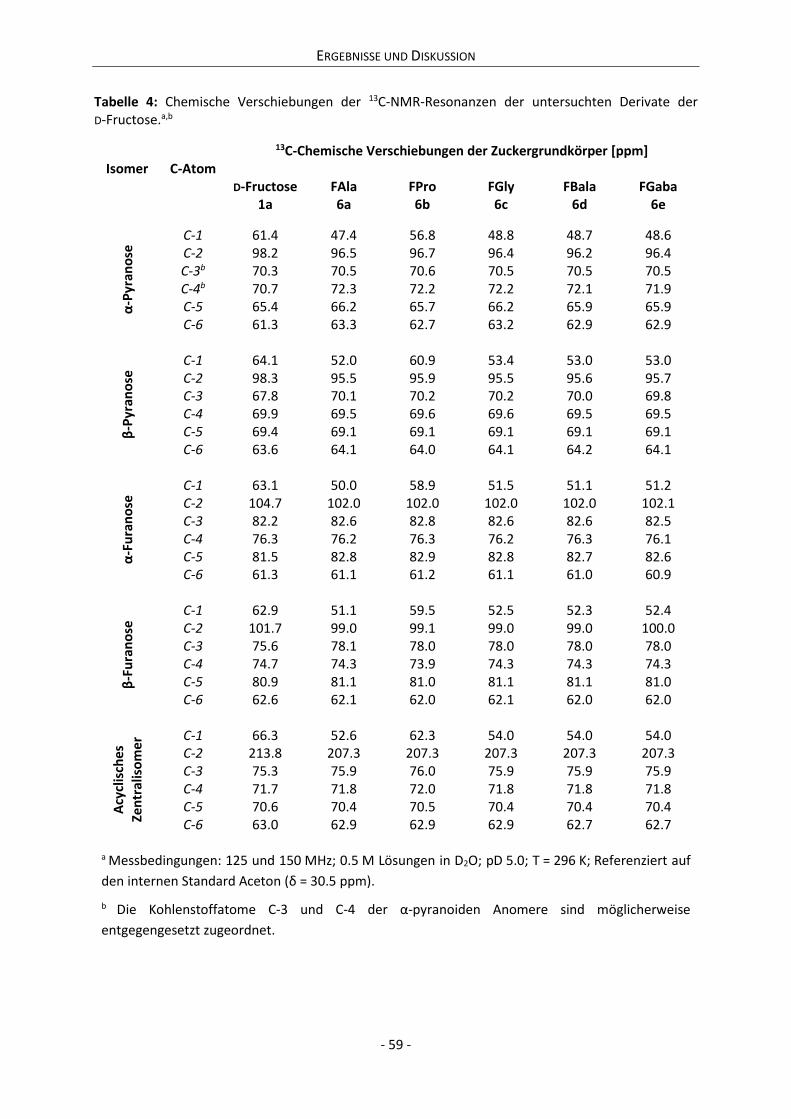
ERGEBNISSE UND DISKUSSION
- 59 -
Tabelle 4: Chemische Verschiebungen der 13C-NMR-Resonanzen der untersuchten Derivate der D-Fructose.a,b
Isomer C-Atom
13C-Chemische Verschiebungen der Zuckergrundkörper [ppm]
D-Fructose FAla FPro FGly FBala FGaba 1a 6a 6b 6c 6d 6e
α-P
yran
ose
C-1 61.4 47.4 56.8 48.8 48.7 48.6 C-2 98.2 96.5 96.7 96.4 96.2 96.4 C-3b 70.3 70.5 70.6 70.5 70.5 70.5 C-4b 70.7 72.3 72.2 72.2 72.1 71.9 C-5 65.4 66.2 65.7 66.2 65.9 65.9 C-6 61.3 63.3 62.7 63.2 62.9 62.9
β-P
yran
ose
C-1 64.1 52.0 60.9 53.4 53.0 53.0 C-2 98.3 95.5 95.9 95.5 95.6 95.7 C-3 67.8 70.1 70.2 70.2 70.0 69.8 C-4 69.9 69.5 69.6 69.6 69.5 69.5 C-5 69.4 69.1 69.1 69.1 69.1 69.1 C-6 63.6 64.1 64.0 64.1 64.2 64.1
α-F
ura
no
se C-1 63.1 50.0 58.9 51.5 51.1 51.2
C-2 104.7 102.0 102.0 102.0 102.0 102.1 C-3 82.2 82.6 82.8 82.6 82.6 82.5 C-4 76.3 76.2 76.3 76.2 76.3 76.1 C-5 81.5 82.8 82.9 82.8 82.7 82.6 C-6 61.3 61.1 61.2 61.1 61.0 60.9
β-F
ura
no
se C-1 62.9 51.1 59.5 52.5 52.3 52.4
C-2 101.7 99.0 99.1 99.0 99.0 100.0 C-3 75.6 78.1 78.0 78.0 78.0 78.0 C-4 74.7 74.3 73.9 74.3 74.3 74.3 C-5 80.9 81.1 81.0 81.1 81.1 81.0 C-6 62.6 62.1 62.0 62.1 62.0 62.0
Acy
clis
ches
Ze
ntr
alis
om
er C-1 66.3 52.6 62.3 54.0 54.0 54.0
C-2 213.8 207.3 207.3 207.3 207.3 207.3 C-3 75.3 75.9 76.0 75.9 75.9 75.9 C-4 71.7 71.8 72.0 71.8 71.8 71.8 C-5 70.6 70.4 70.5 70.4 70.4 70.4 C-6 63.0 62.9 62.9 62.9 62.7 62.7
a Messbedingungen: 125 und 150 MHz; 0.5 M Lösungen in D2O; pD 5.0; T = 296 K; Referenziert auf
den internen Standard Aceton (δ = 30.5 ppm).
b Die Kohlenstoffatome C-3 und C-4 der α-pyranoiden Anomere sind möglicherweise
entgegengesetzt zugeordnet.
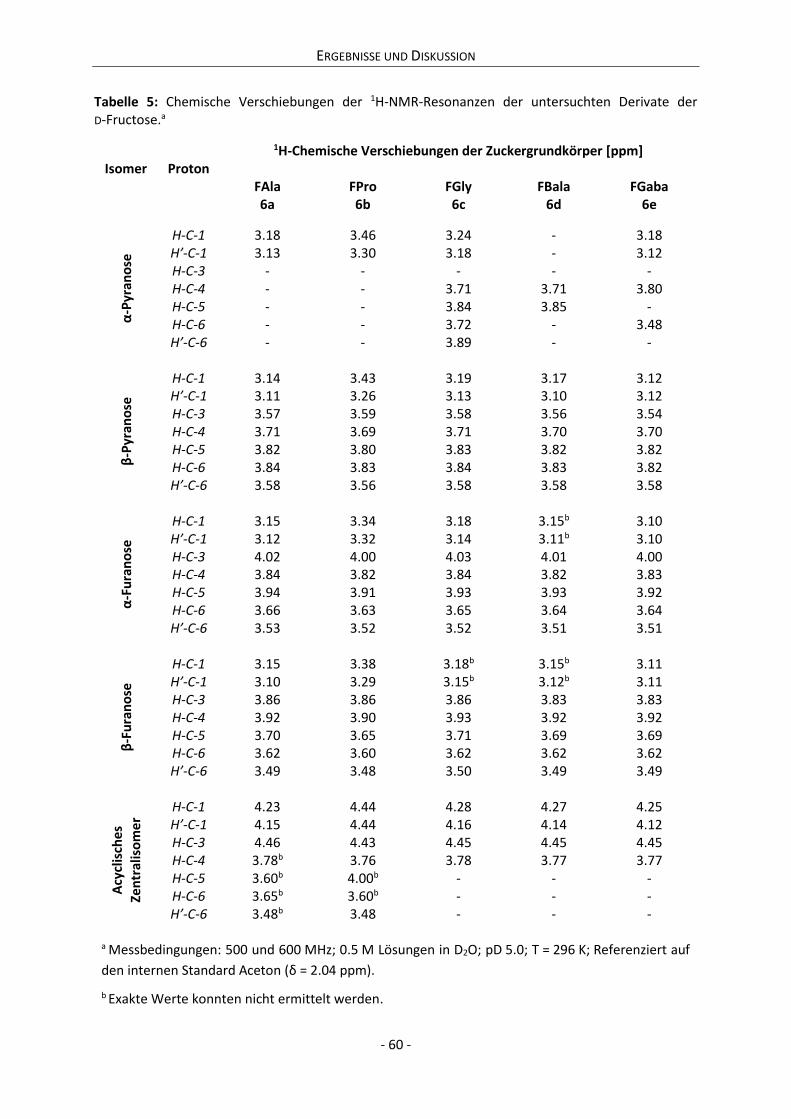
ERGEBNISSE UND DISKUSSION
- 60 -
Tabelle 5: Chemische Verschiebungen der 1H-NMR-Resonanzen der untersuchten Derivate der D-Fructose.a
Isomer Proton
1H-Chemische Verschiebungen der Zuckergrundkörper [ppm]
FAla FPro FGly FBala FGaba 6a 6b 6c 6d 6e
α-P
yran
ose
H-C-1 3.18 3.46 3.24 - 3.18 H’-C-1 3.13 3.30 3.18 - 3.12 H-C-3 - - - - - H-C-4 - - 3.71 3.71 3.80 H-C-5 - - 3.84 3.85 - H-C-6 - - 3.72 - 3.48 H’-C-6 - - 3.89 - -
β-P
yran
ose
H-C-1 3.14 3.43 3.19 3.17 3.12 H’-C-1 3.11 3.26 3.13 3.10 3.12 H-C-3 3.57 3.59 3.58 3.56 3.54 H-C-4 3.71 3.69 3.71 3.70 3.70 H-C-5 3.82 3.80 3.83 3.82 3.82 H-C-6 3.84 3.83 3.84 3.83 3.82 H’-C-6 3.58 3.56 3.58 3.58 3.58
α-F
ura
no
se
H-C-1 3.15 3.34 3.18 3.15b 3.10 H’-C-1 3.12 3.32 3.14 3.11b 3.10 H-C-3 4.02 4.00 4.03 4.01 4.00 H-C-4 3.84 3.82 3.84 3.82 3.83 H-C-5 3.94 3.91 3.93 3.93 3.92 H-C-6 3.66 3.63 3.65 3.64 3.64 H’-C-6 3.53 3.52 3.52 3.51 3.51
β-F
ura
no
se
H-C-1 3.15 3.38 3.18b 3.15b 3.11 H’-C-1 3.10 3.29 3.15b 3.12b 3.11 H-C-3 3.86 3.86 3.86 3.83 3.83 H-C-4 3.92 3.90 3.93 3.92 3.92 H-C-5 3.70 3.65 3.71 3.69 3.69 H-C-6 3.62 3.60 3.62 3.62 3.62 H’-C-6 3.49 3.48 3.50 3.49 3.49
Acy
clis
ches
Ze
ntr
alis
om
er
H-C-1 4.23 4.44 4.28 4.27 4.25 H’-C-1 4.15 4.44 4.16 4.14 4.12 H-C-3 4.46 4.43 4.45 4.45 4.45 H-C-4 3.78b 3.76 3.78 3.77 3.77 H-C-5 3.60b 4.00b - - - H-C-6 3.65b 3.60b - - - H’-C-6 3.48b 3.48 - - -
a Messbedingungen: 500 und 600 MHz; 0.5 M Lösungen in D2O; pD 5.0; T = 296 K; Referenziert auf
den internen Standard Aceton (δ = 2.04 ppm).
b Exakte Werte konnten nicht ermittelt werden.
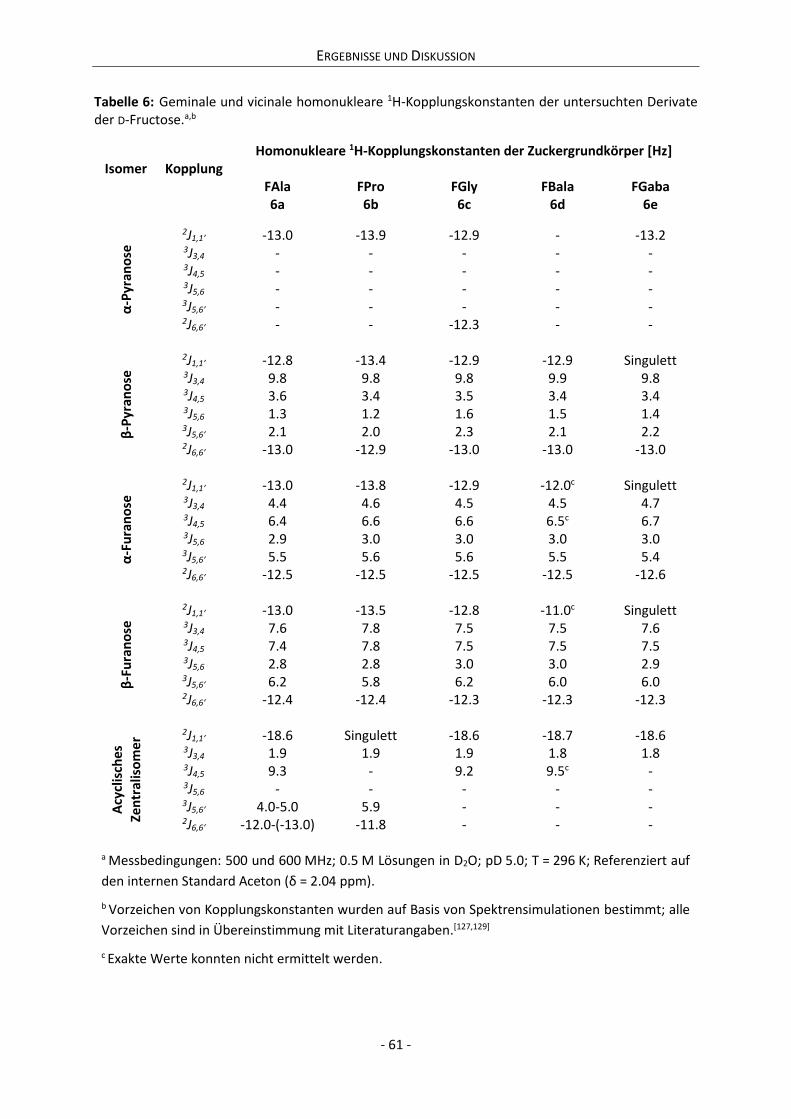
ERGEBNISSE UND DISKUSSION
- 61 -
Tabelle 6: Geminale und vicinale homonukleare 1H-Kopplungskonstanten der untersuchten Derivate der D-Fructose.a,b
Isomer Kopplung Homonukleare 1H-Kopplungskonstanten der Zuckergrundkörper [Hz]
FAla FPro FGly FBala FGaba 6a 6b 6c 6d 6e
α-P
yran
ose
2J1,1’ -13.0 -13.9 -12.9 - -13.2 3J3,4 - - - - - 3J4,5 - - - - - 3J5,6 - - - - - 3J5,6’ - - - - - 2J6,6’ - - -12.3 - -
β-P
yran
ose
2J1,1’ -12.8 -13.4 -12.9 -12.9 Singulett 3J3,4 9.8 9.8 9.8 9.9 9.8 3J4,5 3.6 3.4 3.5 3.4 3.4 3J5,6 1.3 1.2 1.6 1.5 1.4 3J5,6’ 2.1 2.0 2.3 2.1 2.2 2J6,6’ -13.0 -12.9 -13.0 -13.0 -13.0
α-F
ura
no
se
2J1,1’ -13.0 -13.8 -12.9 -12.0c Singulett 3J3,4 4.4 4.6 4.5 4.5 4.7 3J4,5 6.4 6.6 6.6 6.5c 6.7 3J5,6 2.9 3.0 3.0 3.0 3.0 3J5,6’ 5.5 5.6 5.6 5.5 5.4 2J6,6’ -12.5 -12.5 -12.5 -12.5 -12.6
β-F
ura
no
se
2J1,1’ -13.0 -13.5 -12.8 -11.0c Singulett 3J3,4 7.6 7.8 7.5 7.5 7.6 3J4,5 7.4 7.8 7.5 7.5 7.5 3J5,6 2.8 2.8 3.0 3.0 2.9 3J5,6’ 6.2 5.8 6.2 6.0 6.0 2J6,6’ -12.4 -12.4 -12.3 -12.3 -12.3
Acy
clis
ches
Ze
ntr
alis
om
er
2J1,1’ -18.6 Singulett -18.6 -18.7 -18.6 3J3,4 1.9 1.9 1.9 1.8 1.8 3J4,5 9.3 - 9.2 9.5c - 3J5,6 - - - - - 3J5,6’ 4.0-5.0 5.9 - - - 2J6,6’ -12.0-(-13.0) -11.8 - - -
a Messbedingungen: 500 und 600 MHz; 0.5 M Lösungen in D2O; pD 5.0; T = 296 K; Referenziert auf
den internen Standard Aceton (δ = 2.04 ppm).
b Vorzeichen von Kopplungskonstanten wurden auf Basis von Spektrensimulationen bestimmt; alle
Vorzeichen sind in Übereinstimmung mit Literaturangaben.[127,129]
c Exakte Werte konnten nicht ermittelt werden.
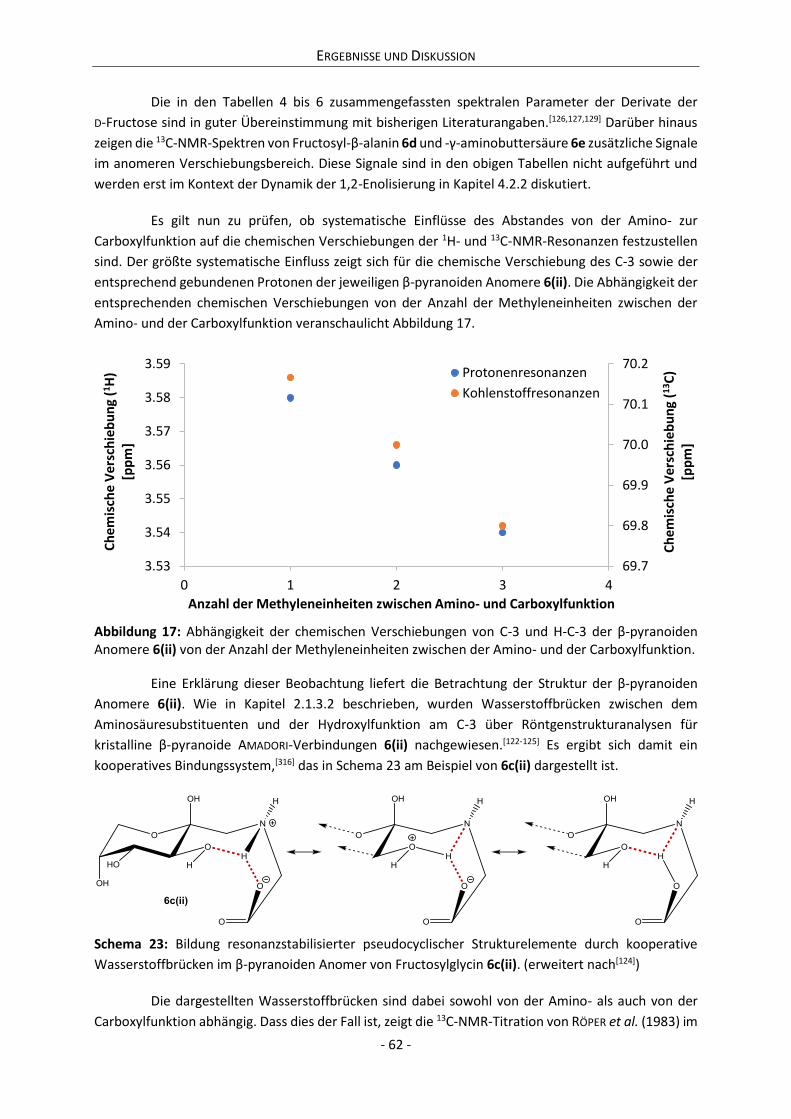
ERGEBNISSE UND DISKUSSION
- 62 -
Die in den Tabellen 4 bis 6 zusammengefassten spektralen Parameter der Derivate der
D-Fructose sind in guter Übereinstimmung mit bisherigen Literaturangaben.[126,127,129] Darüber hinaus
zeigen die 13C-NMR-Spektren von Fructosyl-β-alanin 6d und -γ-aminobuttersäure 6e zusätzliche Signale
im anomeren Verschiebungsbereich. Diese Signale sind in den obigen Tabellen nicht aufgeführt und
werden erst im Kontext der Dynamik der 1,2-Enolisierung in Kapitel 4.2.2 diskutiert.
Es gilt nun zu prüfen, ob systematische Einflüsse des Abstandes von der Amino- zur
Carboxylfunktion auf die chemischen Verschiebungen der 1H- und 13C-NMR-Resonanzen festzustellen
sind. Der größte systematische Einfluss zeigt sich für die chemische Verschiebung des C-3 sowie der
entsprechend gebundenen Protonen der jeweiligen β-pyranoiden Anomere 6(ii). Die Abhängigkeit der
entsprechenden chemischen Verschiebungen von der Anzahl der Methyleneinheiten zwischen der
Amino- und der Carboxylfunktion veranschaulicht Abbildung 17.
Abbildung 17: Abhängigkeit der chemischen Verschiebungen von C-3 und H-C-3 der β-pyranoiden Anomere 6(ii) von der Anzahl der Methyleneinheiten zwischen der Amino- und der Carboxylfunktion.
Eine Erklärung dieser Beobachtung liefert die Betrachtung der Struktur der β-pyranoiden
Anomere 6(ii). Wie in Kapitel 2.1.3.2 beschrieben, wurden Wasserstoffbrücken zwischen dem
Aminosäuresubstituenten und der Hydroxylfunktion am C-3 über Röntgenstrukturanalysen für
kristalline β-pyranoide AMADORI-Verbindungen 6(ii) nachgewiesen.[122-125] Es ergibt sich damit ein
kooperatives Bindungssystem,[316] das in Schema 23 am Beispiel von 6c(ii) dargestellt ist.
Schema 23: Bildung resonanzstabilisierter pseudocyclischer Strukturelemente durch kooperative
Wasserstoffbrücken im β-pyranoiden Anomer von Fructosylglycin 6c(ii). (erweitert nach[124])
Die dargestellten Wasserstoffbrücken sind dabei sowohl von der Amino- als auch von der
Carboxylfunktion abhängig. Dass dies der Fall ist, zeigt die 13C-NMR-Titration von RÖPER et al. (1983) im
69.7
69.8
69.9
70.0
70.1
70.2
3.53
3.54
3.55
3.56
3.57
3.58
3.59
0 1 2 3 4
Ch
emis
che
Ve
rsch
ieb
un
g (1
3C
) [p
pm
]
Ch
emis
che
Ve
rsch
ieb
un
g (1
H)
[pp
m]
Anzahl der Methyleneinheiten zwischen Amino- und Carboxylfunktion
Protonenresonanzen
Kohlenstoffresonanzen

ERGEBNISSE UND DISKUSSION
- 63 -
pH-Bereich von 0.7 bis 11.9.38 In pH-Bereichen oberhalb des 𝑝𝐾𝑠-Wertes der Aminofunktion kommt es
zu einer Hochfeldverschiebung der Resonanz des C-3 des β-pyranoiden Anomers von Fructosylalanin
6a(ii). Gleichermaßen erfährt das Signal im sauren Milieu eine Hochfeldverschiebung.[127] Das
bedeutet, dass die kooperative Bindung zu einer partiellen Protonierung der Hydroxylfunktion am C-3
führt. Damit erhöht sich der negative induktive Effekt der Hydroxylfunktion auf das C-3, woraus dessen
Resonanz nach tiefem Feld verschoben wird. Da sowohl die Erhöhung als auch die Absenkung des
pH-Wertes eine Hochfeldverschiebung bewirken, müssen beide funktionellen Gruppen an der
kooperativen Bindung beteiligt sein. Die Stärke der entsprechenden Wasserstoffbrückenbindung zur
Hydroxylfunktion am C-3 lässt sich demnach an der chemischen Verschiebung der entsprechenden 13C-NMR-Resonanz ablesen. Ausgehend von AMADORI-Verbindungen der α-Aminosäuren 6a-6c führt
jede weitere Methyleneinheit zwischen der Amino- und der Carboxylfunktion zu einer
Hochfeldverschiebung um 0.2 ppm. Da sich der negative induktive Effekt entsprechend auch auf das
H-C-3 auswirkt, ist dies analog auch an den chemischen Verschiebungen der Protonenresonanzen zu
beobachten. Hierbei führt jede weitere Methyleneinheit zu einer Hochfeldverschiebung um 0.02 ppm.
Die Wasserstoffbrückenbindung hat nicht nur Einfluss auf die genannten chemischen
Verschiebungen, sondern beeinflusst auch die Reaktivität der AMADORI-Verbindungen. Der negative
induktive Effekt der Hydroxylfunktion am C-3 bewirkt eine Herabsenkung der Elektronendichte am C-3
bzw. H-C-3. Dies bewirkt die Verschiebung der entsprechenden NMR-Resonanzen nach tiefem Feld.
Damit einher geht jedoch auch eine Zunahme der Polarisierung der C-H-Bindung und damit eine
Erhöhung der C-H-Acidität. Die Dissoziation der C-H-Bindung führt unter Ringöffnung zum tautomeren
2,3-Endiol. Da dieser Prozess, wie in Kapitel 2.1.4 beschrieben, irreversibel ist, hat dies die Eliminierung
der Aminosäure als gute Abgangsgruppe zur Folge und es kommt zur Freisetzung von
1-Desoxyglucoson 9a. Wie Abbildung 12 (Seite 52) zeigt, ist die Bildungsrate von 1-Desoxyglucoson 9a
im pH-Bereich von 2.5 bis 6.5 für die AMADORI-Verbindungen der α-Aminosäuren 6a-6c jeweils größer
als die für die AMADORI-Verbindungen der ω-Aminosäuren 6d und 6e. Diese Beobachtungen lassen sich
demnach auf die molekulare Struktur der jeweils β-pyranoiden Hauptanomere 6(ii) zurückführen.
Damit ist davon auszugehen, dass die Unterschiede der Bildungsraten von 3-Desoxyglucoson 9b auf
einem katalytischen Einfluss der Carboxylfunktionen beruhen. In Schema 21 (Seite 49) wurde der
entsprechende Reaktionsmechanismus und dessen Abhängigkeit von der Anomerisierung bereits
dargestellt. Dieser Mechanismus kann ausgehend von jedem Anomer eines Derivates der D-Fructose
formuliert werden. Im Gegensatz dazu kann eine analoge Bildung von 1-Desoxyglucoson 9a lediglich
aus den jeweils α-konfigurierten Anomeren 1a(i) und 6(i) erfolgen, die jedoch für Derivate der
D-Fructose die Minorkomponenten der Anomerensysteme darstellen.39 Es ist demnach davon
auszugehen, dass 1-Desoxyglucoson 9a vornehmlich unter Einfluss der in Schema 23 dargestellten
Wasserstoffbrücken gebildet wird, während die Bildung von 3-Desoxyglucoson 9b an die
Anomerisierung anionischer Zuckerspezies gekoppelt ist. Diese liegen jedoch nur bei pH-Werten vor,
die höher sind als der jeweilige 𝑝𝐼 des Zuckergrundkörpers. Trotzdem zeigt Abbildung 11 (Seite 52) für
AMADORI-Verbindungen mit α-ständiger Aminofunktion 6a-6c bei einem pH-Wert von ca. 4.0 ein
Minimum der Bildungsraten von 3-Desoxyglucoson 9b. Mit zunehmender Protonenkonzentration
38 RÖPER et al. (1983) führten die 13C-NMR-Titration zum Zwecke einer erleichterten Signalzuordnung durch.
39 Überträgt man diese Gedanken auf D-Glucose 2a, so ergibt sich, dass eine 1,2-Enolisierung und damit die
Bildung von 3-Desoxyglucoson 9b nur ausgehend von β-konfigurierten Anomeren möglich ist. Das erklärt die von
HÄSELER et al. (1998) beobachtete vermehrte Bildung von HMF 13 aus β-D-Glucopyranose unter Bedingungen der
Karamellisierung gegenüber der Bildung von HMF 13 aus α-D-Glucopyranose.[392]

ERGEBNISSE UND DISKUSSION
- 64 -
nehmen auch die Bildungsraten entgegen der oben beschriebenen mechanistischen Vorstellungen
wieder zu, während die entsprechenden Raten der AMADORI-Verbindungen mit nicht α-ständiger
Aminofunktion 6d und 6e stetig abnehmen. Dieses Verhalten lässt sich ebenso auf Grundlage der in
Schema 23 dargestellten mesomeren Grenzstrukturen des kooperativen Bindungssystems erklären.
Wie bereits diskutiert, ist die Hydroxylfunktion am C-3 aufgrund ihrer Teilnahme am kooperativen
Bindungssystem als partiell protoniert zu betrachten. Es ist daher davon auszugehen, dass ihre
vollständige Protonierung leichter erfolgen kann als die der übrigen Hydroxylfunktionen. Es handelt
sich demnach mutmaßlich um die Hydroxylfunktion mit der größten Basizität am Zuckergrundkörper.
Die Protonierung der Hydroxylfunktion führt zur Bildung von Wasser als gute Abgangsgruppe. Die
Stabilisierung des entstehenden Carbeniumions führt zur Bildung von 3-Desoxyglucoson 9b und
3-Desoxygalactoson 9c, ohne dass die Möglichkeit einer Bildung von 1-Desoxyglucoson 9a bzw.
D-Glucoson 9d besteht. Den entsprechenden Reaktionsmechanismus zeigt Schema 24 a).
Schema 24: a) Parallele Bildung von 3-Desoxyglucoson 9b und 3-Desoxygalactoson 9c aus dem β-pyranoiden Anomer von Fructosylglycin 6c(ii) über die Protonierung der Hydroxylfunktion am C-3. b) Bildung von (Z)- und (E)-3,4-Didesoxyglucoson-3-en 17a und 17b aus dem β-pyranoiden Anomer von Fructosylglycin 6c(ii) über die Protonierung der Hydroxylfunktion am C-3.
Erfolgt anstelle der Ringöffnung eine vinyloge β-Eliminierung der anomeren
Hydroxylfunktion, führt der Reaktionsmechanismus zu Derivaten des 4-Pyranons (vgl. Schema 25 a)).
Dazu zählt insbesondere 5-Hydroxy-2-methyl-4H-pyran-4-on (Allomaltol) 18, das in industriellem
Karamell nachgewiesen werden kann.[317] Ein analoger Mechanismus lässt sich darüber hinaus auch
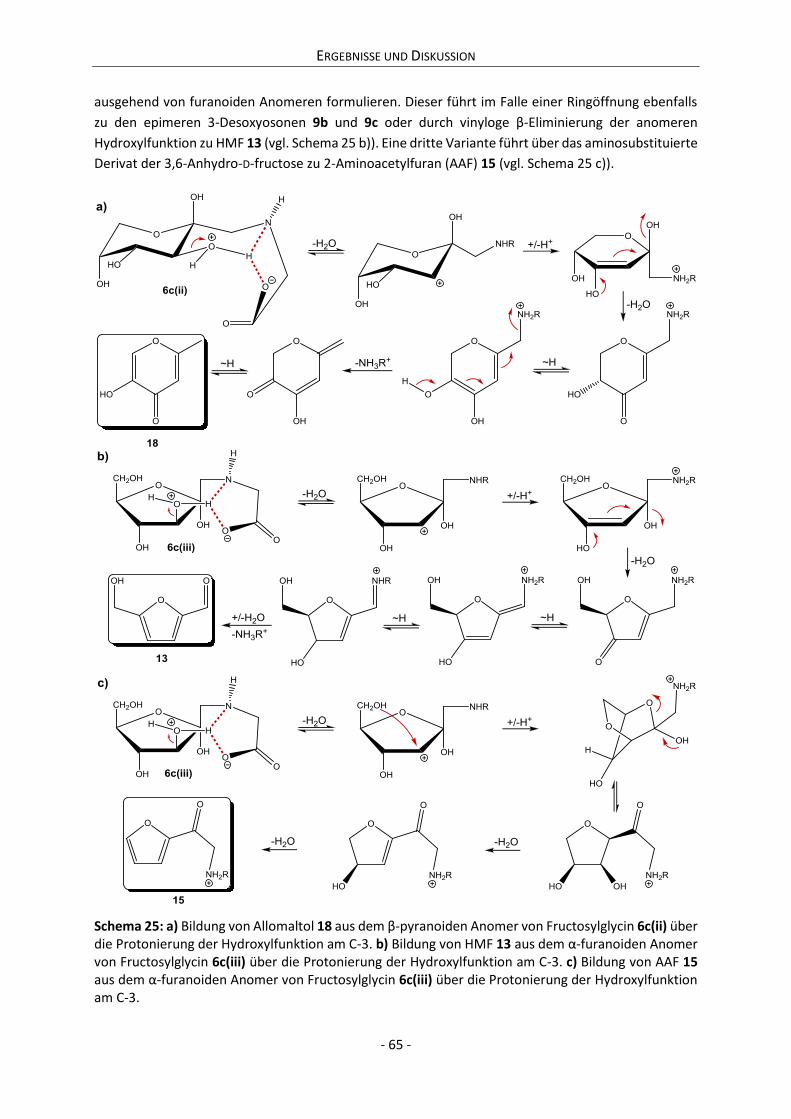
ERGEBNISSE UND DISKUSSION
- 65 -
ausgehend von furanoiden Anomeren formulieren. Dieser führt im Falle einer Ringöffnung ebenfalls
zu den epimeren 3-Desoxyosonen 9b und 9c oder durch vinyloge β-Eliminierung der anomeren
Hydroxylfunktion zu HMF 13 (vgl. Schema 25 b)). Eine dritte Variante führt über das aminosubstituierte
Derivat der 3,6-Anhydro-D-fructose zu 2-Aminoacetylfuran (AAF) 15 (vgl. Schema 25 c)).
Schema 25: a) Bildung von Allomaltol 18 aus dem β-pyranoiden Anomer von Fructosylglycin 6c(ii) über die Protonierung der Hydroxylfunktion am C-3. b) Bildung von HMF 13 aus dem α-furanoiden Anomer von Fructosylglycin 6c(iii) über die Protonierung der Hydroxylfunktion am C-3. c) Bildung von AAF 15 aus dem α-furanoiden Anomer von Fructosylglycin 6c(iii) über die Protonierung der Hydroxylfunktion am C-3.
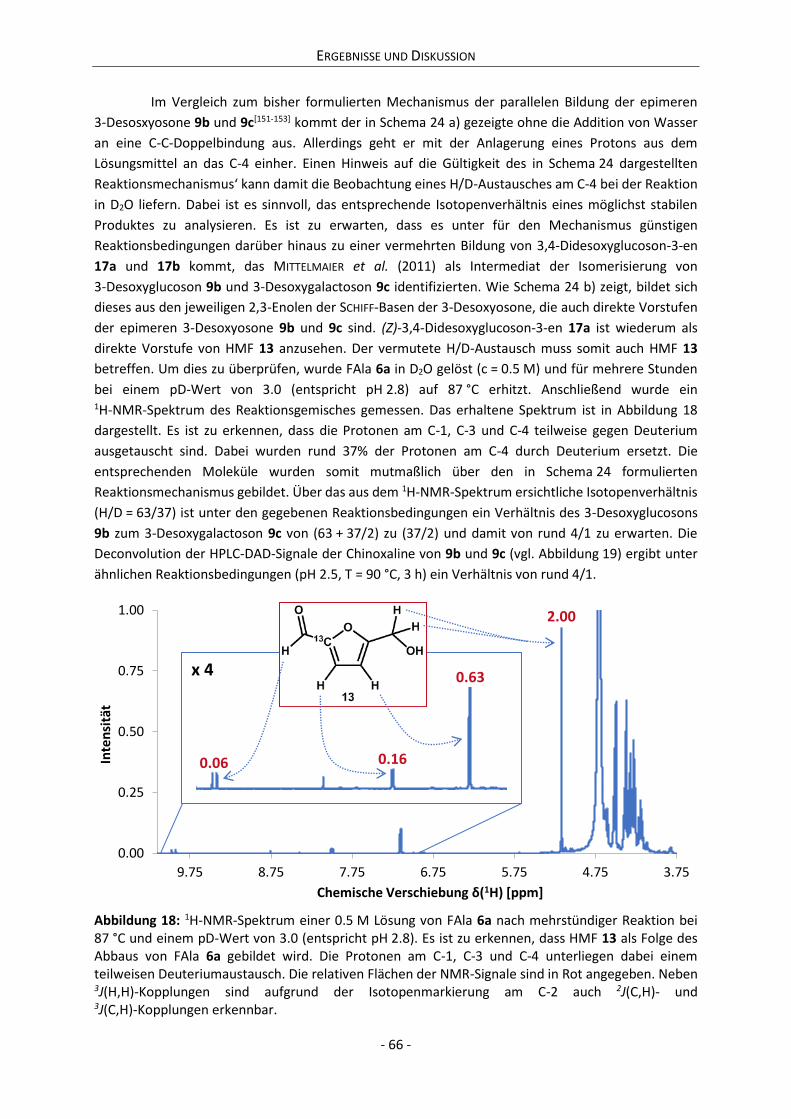
ERGEBNISSE UND DISKUSSION
- 66 -
Im Vergleich zum bisher formulierten Mechanismus der parallelen Bildung der epimeren
3-Desosxyosone 9b und 9c[151-153] kommt der in Schema 24 a) gezeigte ohne die Addition von Wasser
an eine C-C-Doppelbindung aus. Allerdings geht er mit der Anlagerung eines Protons aus dem
Lösungsmittel an das C-4 einher. Einen Hinweis auf die Gültigkeit des in Schema 24 dargestellten
Reaktionsmechanismus‘ kann damit die Beobachtung eines H/D-Austausches am C-4 bei der Reaktion
in D2O liefern. Dabei ist es sinnvoll, das entsprechende Isotopenverhältnis eines möglichst stabilen
Produktes zu analysieren. Es ist zu erwarten, dass es unter für den Mechanismus günstigen
Reaktionsbedingungen darüber hinaus zu einer vermehrten Bildung von 3,4-Didesoxyglucoson-3-en
17a und 17b kommt, das MITTELMAIER et al. (2011) als Intermediat der Isomerisierung von
3-Desoxyglucoson 9b und 3-Desoxygalactoson 9c identifizierten. Wie Schema 24 b) zeigt, bildet sich
dieses aus den jeweiligen 2,3-Enolen der SCHIFF-Basen der 3-Desoxyosone, die auch direkte Vorstufen
der epimeren 3-Desoxyosone 9b und 9c sind. (Z)-3,4-Didesoxyglucoson-3-en 17a ist wiederum als
direkte Vorstufe von HMF 13 anzusehen. Der vermutete H/D-Austausch muss somit auch HMF 13
betreffen. Um dies zu überprüfen, wurde FAla 6a in D2O gelöst (c = 0.5 M) und für mehrere Stunden
bei einem pD-Wert von 3.0 (entspricht pH 2.8) auf 87 °C erhitzt. Anschließend wurde ein 1H-NMR-Spektrum des Reaktionsgemisches gemessen. Das erhaltene Spektrum ist in Abbildung 18
dargestellt. Es ist zu erkennen, dass die Protonen am C-1, C-3 und C-4 teilweise gegen Deuterium
ausgetauscht sind. Dabei wurden rund 37% der Protonen am C-4 durch Deuterium ersetzt. Die
entsprechenden Moleküle wurden somit mutmaßlich über den in Schema 24 formulierten
Reaktionsmechanismus gebildet. Über das aus dem 1H-NMR-Spektrum ersichtliche Isotopenverhältnis
(H/D = 63/37) ist unter den gegebenen Reaktionsbedingungen ein Verhältnis des 3-Desoxyglucosons
9b zum 3-Desoxygalactoson 9c von (63 + 37/2) zu (37/2) und damit von rund 4/1 zu erwarten. Die
Deconvolution der HPLC-DAD-Signale der Chinoxaline von 9b und 9c (vgl. Abbildung 19) ergibt unter
ähnlichen Reaktionsbedingungen (pH 2.5, T = 90 °C, 3 h) ein Verhältnis von rund 4/1.
Abbildung 18: 1H-NMR-Spektrum einer 0.5 M Lösung von FAla 6a nach mehrstündiger Reaktion bei 87 °C und einem pD-Wert von 3.0 (entspricht pH 2.8). Es ist zu erkennen, dass HMF 13 als Folge des Abbaus von FAla 6a gebildet wird. Die Protonen am C-1, C-3 und C-4 unterliegen dabei einem teilweisen Deuteriumaustausch. Die relativen Flächen der NMR-Signale sind in Rot angegeben. Neben 3J(H,H)-Kopplungen sind aufgrund der Isotopenmarkierung am C-2 auch 2J(C,H)- und 3J(C,H)-Kopplungen erkennbar.
0.00
0.25
0.50
0.75
1.00
3.754.755.756.757.758.759.75
Inte
nsi
tät
Chemische Verschiebung δ(1H) [ppm]
2.00
x 4 0.63
0.160.06
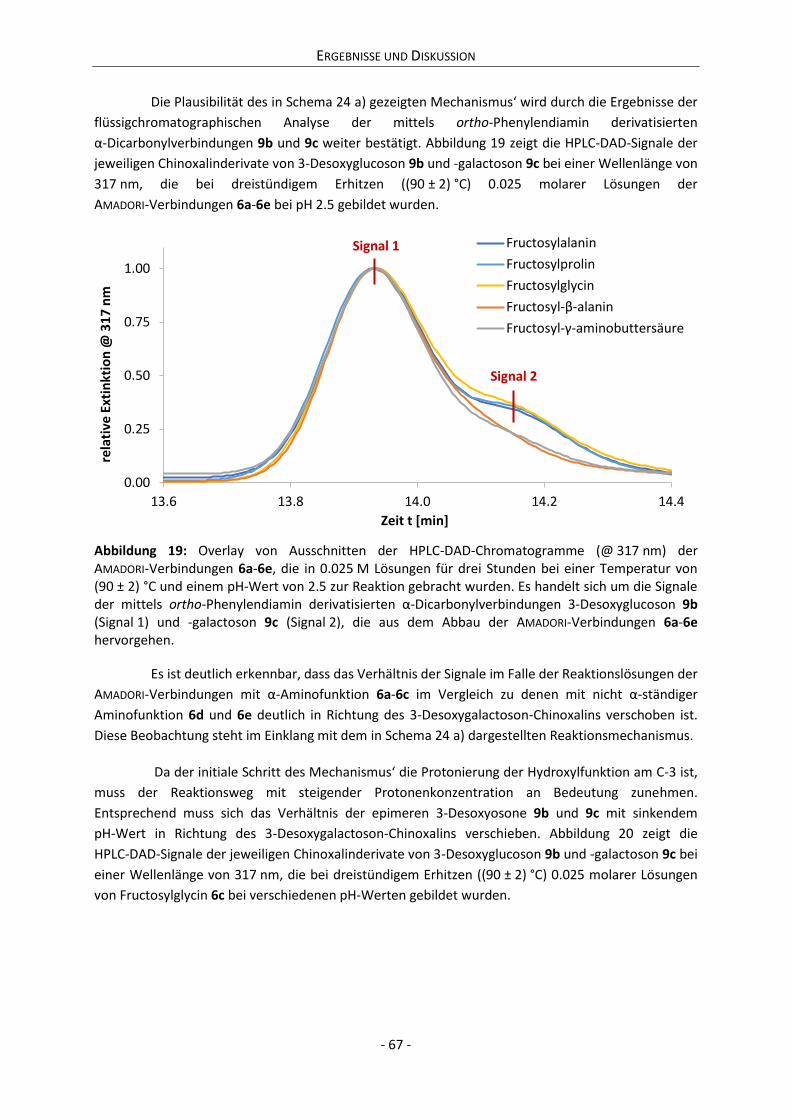
ERGEBNISSE UND DISKUSSION
- 67 -
Die Plausibilität des in Schema 24 a) gezeigten Mechanismus‘ wird durch die Ergebnisse der
flüssigchromatographischen Analyse der mittels ortho-Phenylendiamin derivatisierten
α-Dicarbonylverbindungen 9b und 9c weiter bestätigt. Abbildung 19 zeigt die HPLC-DAD-Signale der
jeweiligen Chinoxalinderivate von 3-Desoxyglucoson 9b und -galactoson 9c bei einer Wellenlänge von
317 nm, die bei dreistündigem Erhitzen ((90 ± 2) °C) 0.025 molarer Lösungen der
AMADORI-Verbindungen 6a-6e bei pH 2.5 gebildet wurden.
Abbildung 19: Overlay von Ausschnitten der HPLC-DAD-Chromatogramme (@ 317 nm) der AMADORI-Verbindungen 6a-6e, die in 0.025 M Lösungen für drei Stunden bei einer Temperatur von (90 ± 2) °C und einem pH-Wert von 2.5 zur Reaktion gebracht wurden. Es handelt sich um die Signale der mittels ortho-Phenylendiamin derivatisierten α-Dicarbonylverbindungen 3-Desoxyglucoson 9b (Signal 1) und -galactoson 9c (Signal 2), die aus dem Abbau der AMADORI-Verbindungen 6a-6e hervorgehen.
Es ist deutlich erkennbar, dass das Verhältnis der Signale im Falle der Reaktionslösungen der
AMADORI-Verbindungen mit α-Aminofunktion 6a-6c im Vergleich zu denen mit nicht α-ständiger
Aminofunktion 6d und 6e deutlich in Richtung des 3-Desoxygalactoson-Chinoxalins verschoben ist.
Diese Beobachtung steht im Einklang mit dem in Schema 24 a) dargestellten Reaktionsmechanismus.
Da der initiale Schritt des Mechanismus‘ die Protonierung der Hydroxylfunktion am C-3 ist,
muss der Reaktionsweg mit steigender Protonenkonzentration an Bedeutung zunehmen.
Entsprechend muss sich das Verhältnis der epimeren 3-Desoxyosone 9b und 9c mit sinkendem
pH-Wert in Richtung des 3-Desoxygalactoson-Chinoxalins verschieben. Abbildung 20 zeigt die
HPLC-DAD-Signale der jeweiligen Chinoxalinderivate von 3-Desoxyglucoson 9b und -galactoson 9c bei
einer Wellenlänge von 317 nm, die bei dreistündigem Erhitzen ((90 ± 2) °C) 0.025 molarer Lösungen
von Fructosylglycin 6c bei verschiedenen pH-Werten gebildet wurden.
0.00
0.25
0.50
0.75
1.00
13.6 13.8 14.0 14.2 14.4
rela
tive
Ext
inkt
ion
@ 3
17
nm
Zeit t [min]
Fructosylalanin
Fructosylprolin
Fructosylglycin
Fructosyl-β-alanin
Fructosyl-γ-aminobuttersäure
Signal 1
Signal 2
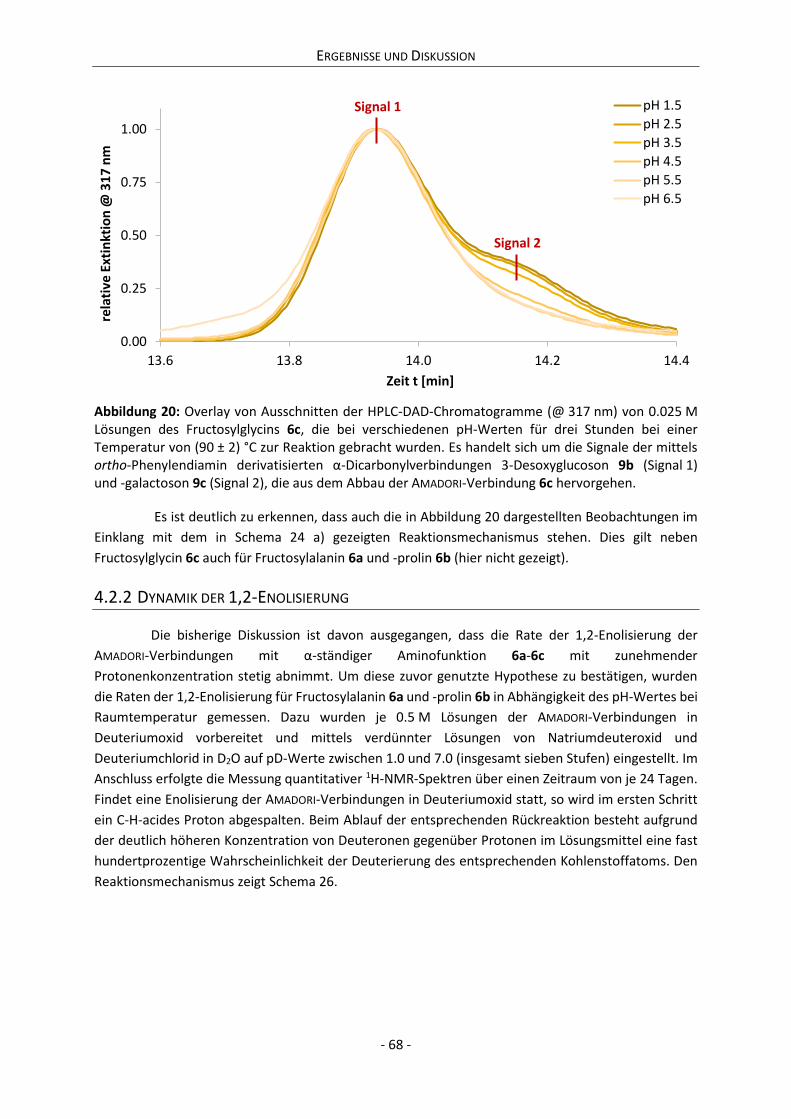
ERGEBNISSE UND DISKUSSION
- 68 -
Abbildung 20: Overlay von Ausschnitten der HPLC-DAD-Chromatogramme (@ 317 nm) von 0.025 M Lösungen des Fructosylglycins 6c, die bei verschiedenen pH-Werten für drei Stunden bei einer Temperatur von (90 ± 2) °C zur Reaktion gebracht wurden. Es handelt sich um die Signale der mittels ortho-Phenylendiamin derivatisierten α-Dicarbonylverbindungen 3-Desoxyglucoson 9b (Signal 1) und -galactoson 9c (Signal 2), die aus dem Abbau der AMADORI-Verbindung 6c hervorgehen.
Es ist deutlich zu erkennen, dass auch die in Abbildung 20 dargestellten Beobachtungen im
Einklang mit dem in Schema 24 a) gezeigten Reaktionsmechanismus stehen. Dies gilt neben
Fructosylglycin 6c auch für Fructosylalanin 6a und -prolin 6b (hier nicht gezeigt).
4.2.2 DYNAMIK DER 1,2-ENOLISIERUNG
Die bisherige Diskussion ist davon ausgegangen, dass die Rate der 1,2-Enolisierung der
AMADORI-Verbindungen mit α-ständiger Aminofunktion 6a-6c mit zunehmender
Protonenkonzentration stetig abnimmt. Um diese zuvor genutzte Hypothese zu bestätigen, wurden
die Raten der 1,2-Enolisierung für Fructosylalanin 6a und -prolin 6b in Abhängigkeit des pH-Wertes bei
Raumtemperatur gemessen. Dazu wurden je 0.5 M Lösungen der AMADORI-Verbindungen in
Deuteriumoxid vorbereitet und mittels verdünnter Lösungen von Natriumdeuteroxid und
Deuteriumchlorid in D2O auf pD-Werte zwischen 1.0 und 7.0 (insgesamt sieben Stufen) eingestellt. Im
Anschluss erfolgte die Messung quantitativer 1H-NMR-Spektren über einen Zeitraum von je 24 Tagen.
Findet eine Enolisierung der AMADORI-Verbindungen in Deuteriumoxid statt, so wird im ersten Schritt
ein C-H-acides Proton abgespalten. Beim Ablauf der entsprechenden Rückreaktion besteht aufgrund
der deutlich höheren Konzentration von Deuteronen gegenüber Protonen im Lösungsmittel eine fast
hundertprozentige Wahrscheinlichkeit der Deuterierung des entsprechenden Kohlenstoffatoms. Den
Reaktionsmechanismus zeigt Schema 26.
0.00
0.25
0.50
0.75
1.00
13.6 13.8 14.0 14.2 14.4
rela
tive
Ext
inkt
ion
@ 3
17
nm
Zeit t [min]
pH 1.5
pH 2.5
pH 3.5
pH 4.5
pH 5.5
pH 6.5
Signal 2
Signal 1

ERGEBNISSE UND DISKUSSION
- 69 -
Schema 26: Mechanismus des Deuteriumaustausches am C-1 von aminosubstituierten Derivaten der D-Fructose.
Nach erfolgter einfacher Deuterierung kann eine erneute Enolisierung ablaufen. Dabei
besteht in etwa dieselbe Wahrscheinlichkeit für die Eliminierung des verbleibenden Protons und die
des Deuterons. Entsprechend ist die Reaktionsgeschwindigkeit von der einfach deuterierten
AMADORI-Verbindung hin zur zweifach deuterierten halb so groß wie von der nicht deuterierten hin zur
einfach deuterierten. Der Prozess führt auf die beschriebene Weise zu einem Intensitätsabfall
entsprechender 1H-NMR-Signale, wobei die Intensität des Restwassersignals (HDO-Signal) in gleichem
Maße zunimmt. In Übereinstimmung mit den Beobachtungen von SIMON und HEUBACH (1965) sowie
MOSSINE et al. (1999) konnte im Rahmen der in dieser Arbeit durchgeführten Experimente keine
reversible 2,3-Enolisierung beobachtet werden.[119,137] Über den Zeitraum von 24 Tagen waren lediglich
Änderungen der Intensitäten der Resonanzen der Protonen am C-1 festzustellen. Zum entsprechenden
Signal trägt sowohl die nicht deuterierte (HH) als auch die einfach deuterierte AMADORI-Verbindung
(HD) bei. Wie Schema 26 verdeutlicht, muss der Modellierung des Prozesses die Kinetik einer
Konsekutivreaktion zugrunde gelegt werden. Dabei ist die zeitabhängige Konzentration der beiden
relevanten Spezies durch die Gleichungen (43) und (44) gegeben.
[𝐻𝐻]𝑡 = [𝐻𝐻]𝑡=0𝑠 ∙ 𝑒𝑥𝑝(−𝑘𝐸𝑛𝑜𝑙 ∙ 𝑡) (43)
[𝐻𝐷]𝑡 = −2 ∙ [𝐻𝐻]𝑡=0𝑠 ∙ {𝑒𝑥𝑝(−𝑘𝐸𝑛𝑜𝑙 ∙ 𝑡) − 𝑒𝑥𝑝 (−1
2∙ 𝑘𝐸𝑛𝑜𝑙 ∙ 𝑡)} (44)
Hierbei gilt es zu beachten, dass der Responsefaktor für die Spezies HH aufgrund der
doppelten Protonenzahl genau zweimal so groß ist wie der für die Spezies HD. Die Intensität der
Protonenresonanz als Funktion der Zeit (𝐼𝑛𝑡. (𝑡)) folgt demnach Gleichung (45).
𝐼𝑛𝑡. (𝑡) = [𝐻𝐻]𝑡 +1
2∙ [𝐻𝐷]𝑡 = [𝐻𝐻]𝑡=0𝑠 ∙ 𝑒𝑥𝑝 (−
1
2∙ 𝑘𝐸𝑛𝑜𝑙 ∙ 𝑡) (45)
Mit Hilfe dieser Funktion erfolgte die nicht lineare Modellierung der Messwerte und damit
die numerische Berechnung der pD-abhängigen Raten der 1,2-Enolisierung. Diese sind in Abbildung 21
graphisch dargestellt und im Anhang tabelliert.
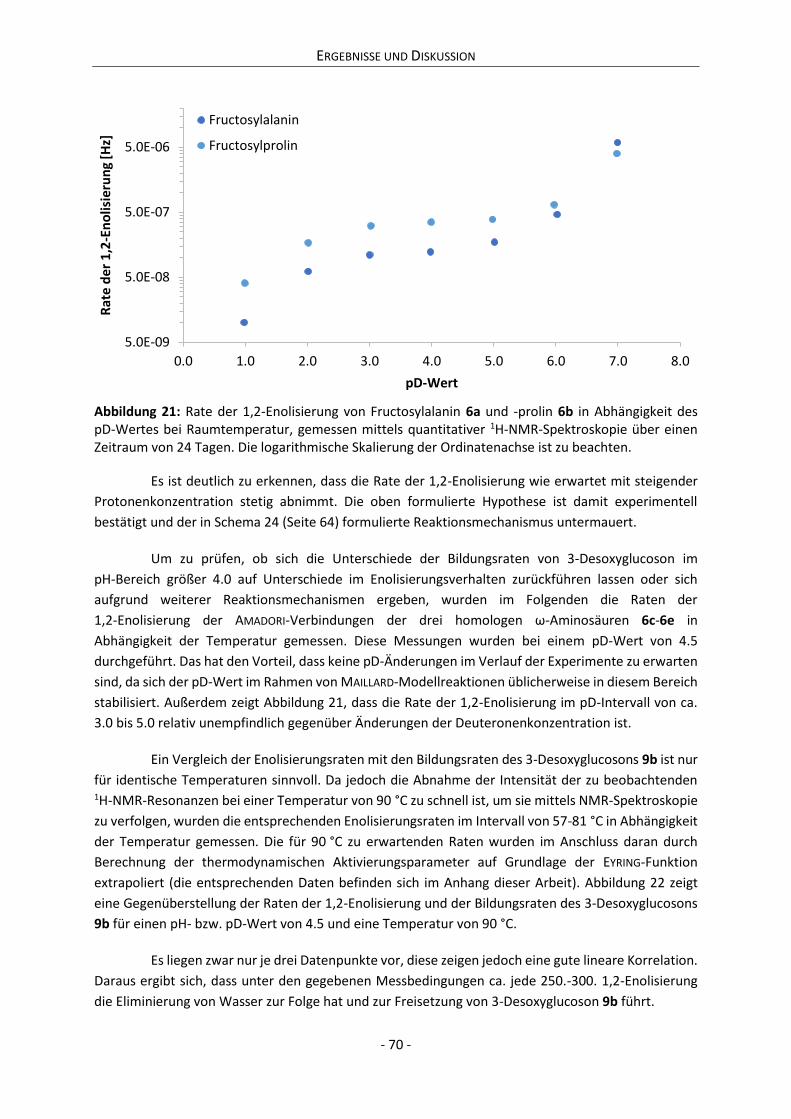
ERGEBNISSE UND DISKUSSION
- 70 -
Abbildung 21: Rate der 1,2-Enolisierung von Fructosylalanin 6a und -prolin 6b in Abhängigkeit des pD-Wertes bei Raumtemperatur, gemessen mittels quantitativer 1H-NMR-Spektroskopie über einen Zeitraum von 24 Tagen. Die logarithmische Skalierung der Ordinatenachse ist zu beachten.
Es ist deutlich zu erkennen, dass die Rate der 1,2-Enolisierung wie erwartet mit steigender
Protonenkonzentration stetig abnimmt. Die oben formulierte Hypothese ist damit experimentell
bestätigt und der in Schema 24 (Seite 64) formulierte Reaktionsmechanismus untermauert.
Um zu prüfen, ob sich die Unterschiede der Bildungsraten von 3-Desoxyglucoson im
pH-Bereich größer 4.0 auf Unterschiede im Enolisierungsverhalten zurückführen lassen oder sich
aufgrund weiterer Reaktionsmechanismen ergeben, wurden im Folgenden die Raten der
1,2-Enolisierung der AMADORI-Verbindungen der drei homologen ω-Aminosäuren 6c-6e in
Abhängigkeit der Temperatur gemessen. Diese Messungen wurden bei einem pD-Wert von 4.5
durchgeführt. Das hat den Vorteil, dass keine pD-Änderungen im Verlauf der Experimente zu erwarten
sind, da sich der pD-Wert im Rahmen von MAILLARD-Modellreaktionen üblicherweise in diesem Bereich
stabilisiert. Außerdem zeigt Abbildung 21, dass die Rate der 1,2-Enolisierung im pD-Intervall von ca.
3.0 bis 5.0 relativ unempfindlich gegenüber Änderungen der Deuteronenkonzentration ist.
Ein Vergleich der Enolisierungsraten mit den Bildungsraten des 3-Desoxyglucosons 9b ist nur
für identische Temperaturen sinnvoll. Da jedoch die Abnahme der Intensität der zu beobachtenden 1H-NMR-Resonanzen bei einer Temperatur von 90 °C zu schnell ist, um sie mittels NMR-Spektroskopie
zu verfolgen, wurden die entsprechenden Enolisierungsraten im Intervall von 57-81 °C in Abhängigkeit
der Temperatur gemessen. Die für 90 °C zu erwartenden Raten wurden im Anschluss daran durch
Berechnung der thermodynamischen Aktivierungsparameter auf Grundlage der EYRING-Funktion
extrapoliert (die entsprechenden Daten befinden sich im Anhang dieser Arbeit). Abbildung 22 zeigt
eine Gegenüberstellung der Raten der 1,2-Enolisierung und der Bildungsraten des 3-Desoxyglucosons
9b für einen pH- bzw. pD-Wert von 4.5 und eine Temperatur von 90 °C.
Es liegen zwar nur je drei Datenpunkte vor, diese zeigen jedoch eine gute lineare Korrelation.
Daraus ergibt sich, dass unter den gegebenen Messbedingungen ca. jede 250.-300. 1,2-Enolisierung
die Eliminierung von Wasser zur Folge hat und zur Freisetzung von 3-Desoxyglucoson 9b führt.
5.0E-09
5.0E-08
5.0E-07
5.0E-06
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0
Rat
e d
er 1
,2-E
no
lisie
run
g [H
z]
pD-Wert
Fructosylalanin
Fructosylprolin
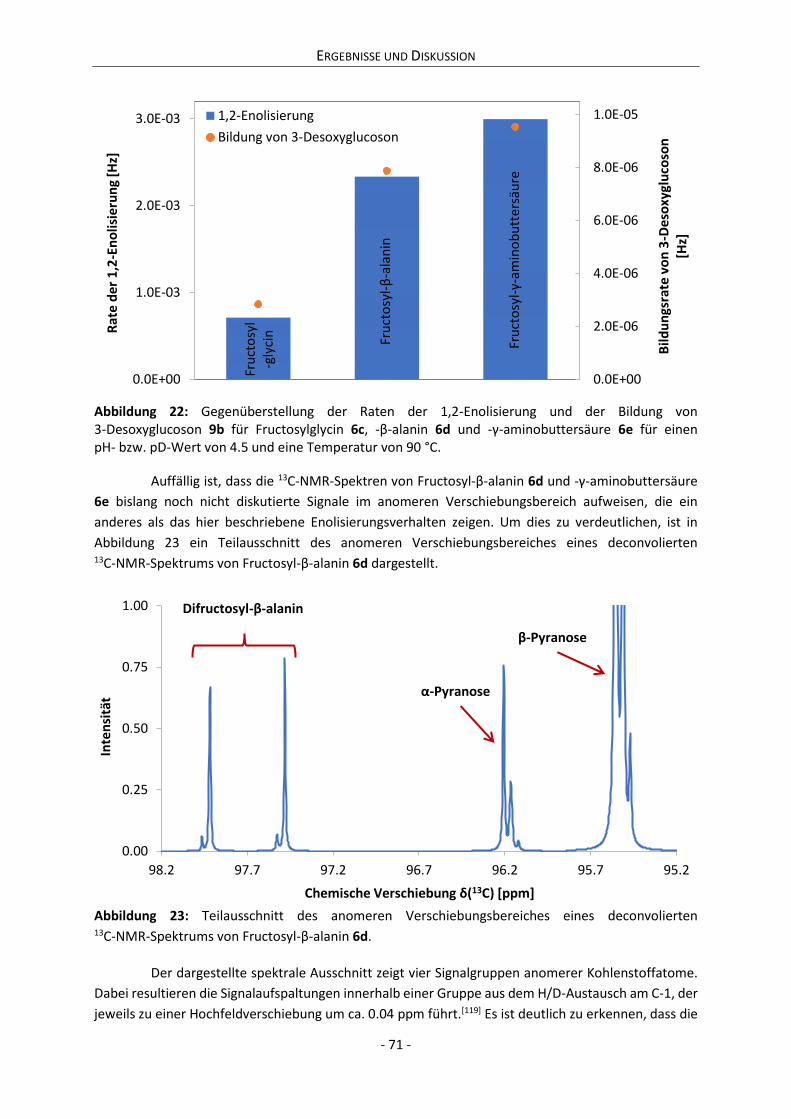
ERGEBNISSE UND DISKUSSION
- 71 -
Abbildung 22: Gegenüberstellung der Raten der 1,2-Enolisierung und der Bildung von 3-Desoxyglucoson 9b für Fructosylglycin 6c, -β-alanin 6d und -γ-aminobuttersäure 6e für einen pH- bzw. pD-Wert von 4.5 und eine Temperatur von 90 °C.
Auffällig ist, dass die 13C-NMR-Spektren von Fructosyl-β-alanin 6d und -γ-aminobuttersäure
6e bislang noch nicht diskutierte Signale im anomeren Verschiebungsbereich aufweisen, die ein
anderes als das hier beschriebene Enolisierungsverhalten zeigen. Um dies zu verdeutlichen, ist in
Abbildung 23 ein Teilausschnitt des anomeren Verschiebungsbereiches eines deconvolierten 13C-NMR-Spektrums von Fructosyl-β-alanin 6d dargestellt.
Abbildung 23: Teilausschnitt des anomeren Verschiebungsbereiches eines deconvolierten 13C-NMR-Spektrums von Fructosyl-β-alanin 6d.
Der dargestellte spektrale Ausschnitt zeigt vier Signalgruppen anomerer Kohlenstoffatome.
Dabei resultieren die Signalaufspaltungen innerhalb einer Gruppe aus dem H/D-Austausch am C-1, der
jeweils zu einer Hochfeldverschiebung um ca. 0.04 ppm führt.[119] Es ist deutlich zu erkennen, dass die
0.0E+00
2.0E-06
4.0E-06
6.0E-06
8.0E-06
1.0E-05
0.0E+00
1.0E-03
2.0E-03
3.0E-03
Bild
un
gsra
te v
on
3-D
eso
xygl
uco
son
[H
z]
Rat
e d
er 1
,2-E
no
lisie
run
g [H
z]1,2-Enolisierung
Bildung von 3-Desoxyglucoson
Fru
cto
syl
-gly
cin Fru
cto
syl-
β-a
lan
in
Fru
cto
syl-
γ-am
ino
bu
tter
säu
re
0.00
0.25
0.50
0.75
1.00
95.295.796.296.797.297.798.2
Inte
nsi
tät
Chemische Verschiebung δ(13C) [ppm]
Difructosyl-β-alanin
β-Pyranose
α-Pyranose

ERGEBNISSE UND DISKUSSION
- 72 -
beiden Gruppen bei höherem Feld je dasselbe Signalmuster zeigen. Dabei hat bei den meisten der
vorliegenden Moleküle noch kein H/D-Austausch stattgefunden. Einige liegen einfach, wenige
zweifach deuteriert vor. Davon abweichend verhalten sich die zwei übrigen Signalgruppen (bei
tieferem Feld). Die Intensitätsverhältnisse innerhalb der jeweiligen Signalgruppen zeigen klar, dass ein
vollständiger H/D-Austausch bei den meisten der entsprechenden Moleküle bereits stattgefunden hat
und nur noch wenige Moleküle einfach deuteriert vorliegen. Das heißt mit anderen Worten, dass
zusätzlich zu den oben beschriebenen Strukturisomeren zumindest ein weiteres vorliegen muss, das
eine stark erhöhte Rate der 1,2-Enolisierung aufweist. Auffällig dabei ist, dass die zwei vorhandenen
anomeren 13C-NMR-Signale ungefähr im Flächenverhältnis von 1:1 vorliegen. Es liegt daher der Schluss
nahe, dass es sich bei dem zusätzlich vorliegenden Molekül um eine zweifach fructosylierte
Aminosäure 8 handelt. Diese zeichnen sich durch eine ca. 7.5-fach erhöhte Rate der 1,2-Enolisierung
sowie besonders hohe Bräunungsraten aus.[119,146] Das lässt vermuten, dass derartige Verbindungen
auch zu einer besonders schnellen Umsetzung zu 3-Desoxyglucoson 9b neigen. Dieses konnte in
HSQC-TOCSY-Spektren der entsprechenden Proben auf Grundlage von Korrelationspeaks der
anomeren Protonen zu deren jeweils direkt gebundenen Kohlenstoffatomen auch zweifelsfrei
nachgewiesen werden. Dabei konnten die Isomere 5a und 5b sowie 5e-5j aus der Publikation von
KÖPPER und FREIMUND (2003) identifiziert werden.[157] Durch Bildung der Volumenintegrale ergibt sich
ein Verhältnis ihrer Konzentrationen von 18:14:3:32:14:11:4:5, was in guter Übereinstimmung mit
Literaturangaben ist.[157] Aus diesen Beobachtungen ergibt sich zum einen die Frage, warum
Difructosylaminosäuren 8 im Rahmen dieser Arbeit nur vergesellschaftet mit AMADORI-Verbindungen
mit nicht α-ständiger Aminofunktion 6d und 6e nachweisbar waren.40 Zum anderen ist zu klären,
warum Difructosylaminosäuren 8 eine gegenüber AMADORI-Verbindungen 6 erhöhte Rate der
1,2-Enolisierung aufweisen.
Der einzige offensichtliche Unterschied zwischen Mono- und Difructosylaminosäuren ist,
dass es sich im ersten Fall um ein sekundäres und im zweiten um ein tertiäres Amin handelt. Betrachtet
man die in den Tabellen 1 (Seite 44) und 2 (Seite 45) aufgeführten 𝑝𝐾𝑠-Werte, so wird deutlich, dass
der Übergang von der Aminosäure 4 zur AMADORI-Verbindung 6 mit einer Verringerung des 𝑝𝐾𝑠-Wertes
der Aminofunktion um ein bis zwei Einheiten verbunden ist. Es ist daher davon auszugehen, dass ein
ähnlicher Effekt beim Übergang von der Mono- zur Difructosylaminosäure zu beobachten ist. HIRSCH et
al. (1995) geben den 𝑝𝐾𝑠-Wert der Aminofunktion von Difructosylglycin 8c mit 5.18 ± 0.01 an.[149] Das
entspricht ausgehend von 6c einer nochmaligen Absenkung des 𝑝𝐾𝑠-Wertes um drei Einheiten
(𝑝𝐾𝑠2(4𝑐) = 9.70, 𝑝𝐾𝑠2(6𝑐) = 8.31, 𝑝𝐾𝑠2(8𝑐) = 5.18). Da die zur Strukturaufklärung nötigen
NMR-Experimente in Lösungen mit einem pD-Wert von 5.0 durchgeführt wurden, bedeutet dies, dass
ein großer Teil der Aminostickstoffatome der Difructosylaminosäuren 8 unter den vorherrschenden
Bedingungen nicht protoniert vorgelegen hat. Damit ist die Katalyse der 1,2-Enolisierung nicht
mehr – wie in Schema 21 (Seite 49) gezeigt – von der ionisierten anomeren Hydroxylfunktion abhängig,
sondern kann direkt über den Aminostickstoff vermittelt erfolgen.41 Die Basizität des
C-1-Substituenten hat damit einen großen Einfluss auf die Rate der 1,2-Enolisierung.
40 Die Klärung dieser Frage erfolgt unterhalb von Abbildung 24.
41 Aufgrund der in Kapitel 2.3 beschriebenen Überlegungen ist in diesem Fall auch von einer Aminokatalyse der
Anomerisierung auszugehen.
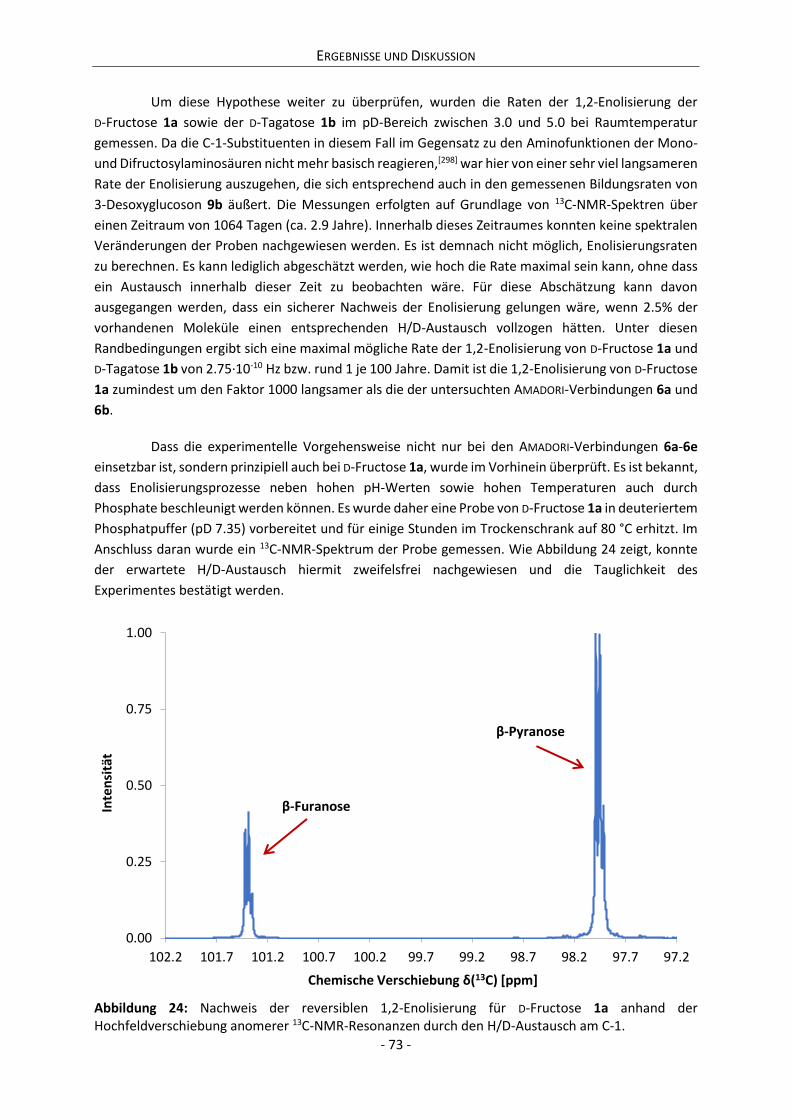
ERGEBNISSE UND DISKUSSION
- 73 -
Um diese Hypothese weiter zu überprüfen, wurden die Raten der 1,2-Enolisierung der
D-Fructose 1a sowie der D-Tagatose 1b im pD-Bereich zwischen 3.0 und 5.0 bei Raumtemperatur
gemessen. Da die C-1-Substituenten in diesem Fall im Gegensatz zu den Aminofunktionen der Mono-
und Difructosylaminosäuren nicht mehr basisch reagieren,[298] war hier von einer sehr viel langsameren
Rate der Enolisierung auszugehen, die sich entsprechend auch in den gemessenen Bildungsraten von
3-Desoxyglucoson 9b äußert. Die Messungen erfolgten auf Grundlage von 13C-NMR-Spektren über
einen Zeitraum von 1064 Tagen (ca. 2.9 Jahre). Innerhalb dieses Zeitraumes konnten keine spektralen
Veränderungen der Proben nachgewiesen werden. Es ist demnach nicht möglich, Enolisierungsraten
zu berechnen. Es kann lediglich abgeschätzt werden, wie hoch die Rate maximal sein kann, ohne dass
ein Austausch innerhalb dieser Zeit zu beobachten wäre. Für diese Abschätzung kann davon
ausgegangen werden, dass ein sicherer Nachweis der Enolisierung gelungen wäre, wenn 2.5% der
vorhandenen Moleküle einen entsprechenden H/D-Austausch vollzogen hätten. Unter diesen
Randbedingungen ergibt sich eine maximal mögliche Rate der 1,2-Enolisierung von D-Fructose 1a und
D-Tagatose 1b von 2.75·10-10 Hz bzw. rund 1 je 100 Jahre. Damit ist die 1,2-Enolisierung von D-Fructose
1a zumindest um den Faktor 1000 langsamer als die der untersuchten AMADORI-Verbindungen 6a und
6b.
Dass die experimentelle Vorgehensweise nicht nur bei den AMADORI-Verbindungen 6a-6e
einsetzbar ist, sondern prinzipiell auch bei D-Fructose 1a, wurde im Vorhinein überprüft. Es ist bekannt,
dass Enolisierungsprozesse neben hohen pH-Werten sowie hohen Temperaturen auch durch
Phosphate beschleunigt werden können. Es wurde daher eine Probe von D-Fructose 1a in deuteriertem
Phosphatpuffer (pD 7.35) vorbereitet und für einige Stunden im Trockenschrank auf 80 °C erhitzt. Im
Anschluss daran wurde ein 13C-NMR-Spektrum der Probe gemessen. Wie Abbildung 24 zeigt, konnte
der erwartete H/D-Austausch hiermit zweifelsfrei nachgewiesen und die Tauglichkeit des
Experimentes bestätigt werden.
Abbildung 24: Nachweis der reversiblen 1,2-Enolisierung für D-Fructose 1a anhand der Hochfeldverschiebung anomerer 13C-NMR-Resonanzen durch den H/D-Austausch am C-1.
0.00
0.25
0.50
0.75
1.00
97.297.798.298.799.299.7100.2100.7101.2101.7102.2
Inte
nsi
tät
Chemische Verschiebung δ(13C) [ppm]
β-Pyranose
β-Furanose
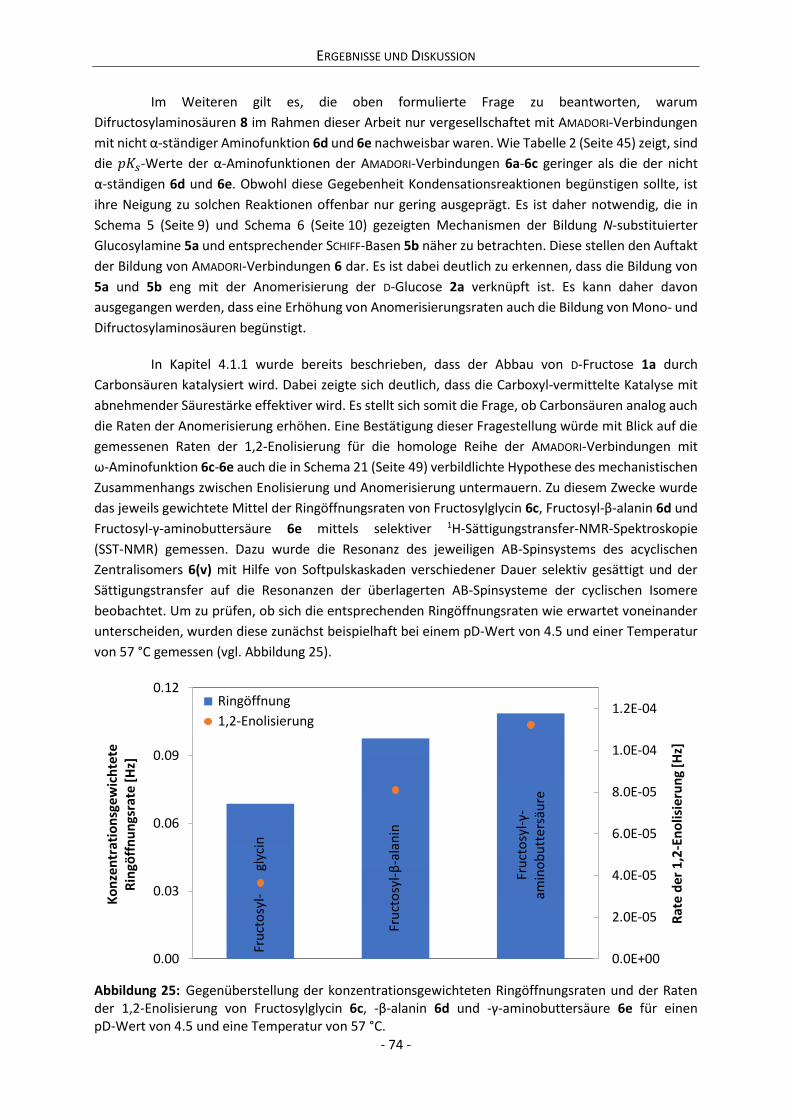
ERGEBNISSE UND DISKUSSION
- 74 -
Im Weiteren gilt es, die oben formulierte Frage zu beantworten, warum
Difructosylaminosäuren 8 im Rahmen dieser Arbeit nur vergesellschaftet mit AMADORI-Verbindungen
mit nicht α-ständiger Aminofunktion 6d und 6e nachweisbar waren. Wie Tabelle 2 (Seite 45) zeigt, sind
die 𝑝𝐾𝑠-Werte der α-Aminofunktionen der AMADORI-Verbindungen 6a-6c geringer als die der nicht
α-ständigen 6d und 6e. Obwohl diese Gegebenheit Kondensationsreaktionen begünstigen sollte, ist
ihre Neigung zu solchen Reaktionen offenbar nur gering ausgeprägt. Es ist daher notwendig, die in
Schema 5 (Seite 9) und Schema 6 (Seite 10) gezeigten Mechanismen der Bildung N-substituierter
Glucosylamine 5a und entsprechender SCHIFF-Basen 5b näher zu betrachten. Diese stellen den Auftakt
der Bildung von AMADORI-Verbindungen 6 dar. Es ist dabei deutlich zu erkennen, dass die Bildung von
5a und 5b eng mit der Anomerisierung der D-Glucose 2a verknüpft ist. Es kann daher davon
ausgegangen werden, dass eine Erhöhung von Anomerisierungsraten auch die Bildung von Mono- und
Difructosylaminosäuren begünstigt.
In Kapitel 4.1.1 wurde bereits beschrieben, dass der Abbau von D-Fructose 1a durch
Carbonsäuren katalysiert wird. Dabei zeigte sich deutlich, dass die Carboxyl-vermittelte Katalyse mit
abnehmender Säurestärke effektiver wird. Es stellt sich somit die Frage, ob Carbonsäuren analog auch
die Raten der Anomerisierung erhöhen. Eine Bestätigung dieser Fragestellung würde mit Blick auf die
gemessenen Raten der 1,2-Enolisierung für die homologe Reihe der AMADORI-Verbindungen mit
ω-Aminofunktion 6c-6e auch die in Schema 21 (Seite 49) verbildlichte Hypothese des mechanistischen
Zusammenhangs zwischen Enolisierung und Anomerisierung untermauern. Zu diesem Zwecke wurde
das jeweils gewichtete Mittel der Ringöffnungsraten von Fructosylglycin 6c, Fructosyl-β-alanin 6d und
Fructosyl-γ-aminobuttersäure 6e mittels selektiver 1H-Sättigungstransfer-NMR-Spektroskopie
(SST-NMR) gemessen. Dazu wurde die Resonanz des jeweiligen AB-Spinsystems des acyclischen
Zentralisomers 6(v) mit Hilfe von Softpulskaskaden verschiedener Dauer selektiv gesättigt und der
Sättigungstransfer auf die Resonanzen der überlagerten AB-Spinsysteme der cyclischen Isomere
beobachtet. Um zu prüfen, ob sich die entsprechenden Ringöffnungsraten wie erwartet voneinander
unterscheiden, wurden diese zunächst beispielhaft bei einem pD-Wert von 4.5 und einer Temperatur
von 57 °C gemessen (vgl. Abbildung 25).
Abbildung 25: Gegenüberstellung der konzentrationsgewichteten Ringöffnungsraten und der Raten der 1,2-Enolisierung von Fructosylglycin 6c, -β-alanin 6d und -γ-aminobuttersäure 6e für einen pD-Wert von 4.5 und eine Temperatur von 57 °C.
0.0E+00
2.0E-05
4.0E-05
6.0E-05
8.0E-05
1.0E-04
1.2E-04
0.00
0.03
0.06
0.09
0.12R
ate
der
1,2
-En
olis
ieru
ng
[Hz]
Ko
nze
ntr
atio
nsg
ewic
hte
te
Rin
göff
nu
ngs
rate
[H
z]
RöG
1,2-Enolisierung
Fru
cto
syl-
glyc
in
Fru
cto
syl-
β-a
lan
in
Fru
cto
syl-
γ-am
ino
bu
tter
säu
re
Ringöffnung
1,2-Enolisierung

ERGEBNISSE UND DISKUSSION
- 75 -
Es zeigt sich eine Zunahme des konzentrationsgewichteten Mittels aller Ringöffnungsraten
mit einer Abnahme der Säurestärke der Carboxylfunktion. Dabei ist die Rate der Anomerisierung
weniger sensitiv gegenüber Änderungen des 𝑝𝐾𝑠-Wertes als die der 1,2-Enolisierung. Darüber hinaus
spielt es für die beobachtete Katalyse offenbar keine merkliche Rolle, wie groß der Abstand zwischen
der Carboxylfunktion und dem Zuckergrundkörper ist. Damit ist es wahrscheinlich, dass
AMADORI-Verbindungen nicht nur ihre eigene Anomerisierung katalysieren, sondern auch die von
vorliegenden reduzierenden Zuckern. Die beobachtete Tendenz der AMADORI-Verbindungen mit
𝑝𝐾𝑠-Werten von mehr als 3.0, mit weiteren Zuckereinheiten zu kondensieren, ist damit einerseits auf
eine katalytische Beschleunigung der Anomerisierung und andererseits auf eine effiziente
AMADORI-Umlagerung (katalysierte 1,2-Enolisierung) zurückzuführen.
4.2.3 ZWISCHENFAZIT DER BISHERIGEN ERGEBNISSE
Die Zusammenfassung der bisherigen Erkenntnisse liefert die Erklärung für die in vielen
Untersuchungen beobachtete erhöhte Reaktivität der basischen Aminosäuren L-Lysin und L-Arginin
gegenüber nicht basischen Aminosäuren.[318,319] Die 𝑝𝐾𝑠-Werte der α-Aminofunktionen der basischen
Aminosäuren (𝑝𝐾𝑠 ≅ 9.0) sind im Vergleich zu denen einfacher neutraler Aminosäuren wie L-Alanin 4a
und Glycin 4c (vgl. Tabelle 1, Seite 44) um fast eine Einheit reduziert.[14] Die in den jeweiligen
Seitenketten gebundenen Aminofunktionen haben demgegenüber 𝑝𝐾𝑠-Werte, die rund zwei bis drei
Einheiten über denen ihrer α-Aminofunktionen liegen.[33] Es ist demnach davon auszugehen, dass die
α-ständigen Aminofunktionen von L-Lysin und L-Arginin bevorzugt Kondensationsreaktionen mit
reduzierenden Zuckern eingehen.42 Dies ist von HEYNS et al. (1967) für die Kondensation von L-Lysin mit
D-Fructose 1a auch entsprechend beobachtet worden,[116] konnte bislang jedoch nicht erklärt werden.
Bei der Kondensation von D-Glucose 2a an die Aminofunktion einer Aminosäure (bspw. 4a-4e) senkt
sich deren 𝑝𝐾𝑠-Wert in der Regel um rund 1.5 Einheiten ab (vgl. hierzu Tabellen 1 und 2, Seiten 44 und
55).43;[129] Das hat drei Konsequenzen. Erstens nähert sich der 𝑝𝐾𝑠-Wert dem 𝑝𝐼 des Zuckers und damit
dem katalytischen Bereich für die Anomerisierung. Zweitens wird die 1,2-Enolisierung durch den
schwach basischen Aminostickstoff katalysiert, was die Bildung von 3-Desoxyglucoson 9b fördert. Und
drittens wird die Kondensation mit einem zweiten Molekül eines reduzierenden Zuckers erleichtert
und die AMADORI-Umlagerung durch nochmalige Herabsetzung der Basizität katalysiert. Durch den
verringerten 𝑝𝐾𝑠-Wert des Aminostickstoffs verstärken sich alle genannten Effekte im
Kondensationsprodukt. Das wiederum führt mutmaßlich zu stark erhöhten Raten der Anomerisierung,
sodass auch die Kondensation der Aminofunktionen der Seitenketten mit reduzierenden Zuckern
begünstigt wird. Aufgrund der zur glycosylierten Aminofunktion α-ständigen Carboxylfunktion ist
darüber hinaus die Ausbildung von Wasserstoffbrücken zur Hydroxylfunktion am C-3 möglich, die
zusätzlich zu einer erhöhten Bildungsrate von 1-Desoxyglucoson 9a führt. Da – wie in den Kapiteln
4.1.2 und 4.1.3 gezeigt – sowohl 1- als auch 3-Desoxyglucoson 9a und 9b maßgeblich zur Bräunung
beitragen, ergibt sich aus der Kombination der beschriebenen Effekte die hohe Bräunungsaktivität des
42 Diese Aussage steht in Widerspruch zu den Untersuchungen von HEYNS und NOACK (1962), die bei ihren
Experimenten keine Bildung von Nα-Fructosyllysin beobachten konnten.[111] Dies liegt jedoch vermutlich an der
genutzten Synthesestrategie. Diese geht vom entsprechenden Hydrochlorid von L-Lysin aus. Die Umsetzung mit
D-Glucose findet dabei nicht im wässrigen Milieu, sondern in organischen Lösungsmitteln statt.
43 Demzufolge entspricht der 𝑝𝐾𝑠-Wert der ε-Aminofunktion der Nε-AMADORI-Verbindung des L-Lysins in etwa
dem 𝑝𝐾𝑠-Wert der α-Aminofunktion des L-Lysins.[149] Der 𝑝𝐾𝑠-Wert der α-Aminofunktion der
Nα-AMADORI-Verbindung des L-Lysins kann demgegenüber mit ungefähr 7.0-7.5 angenommen werden.
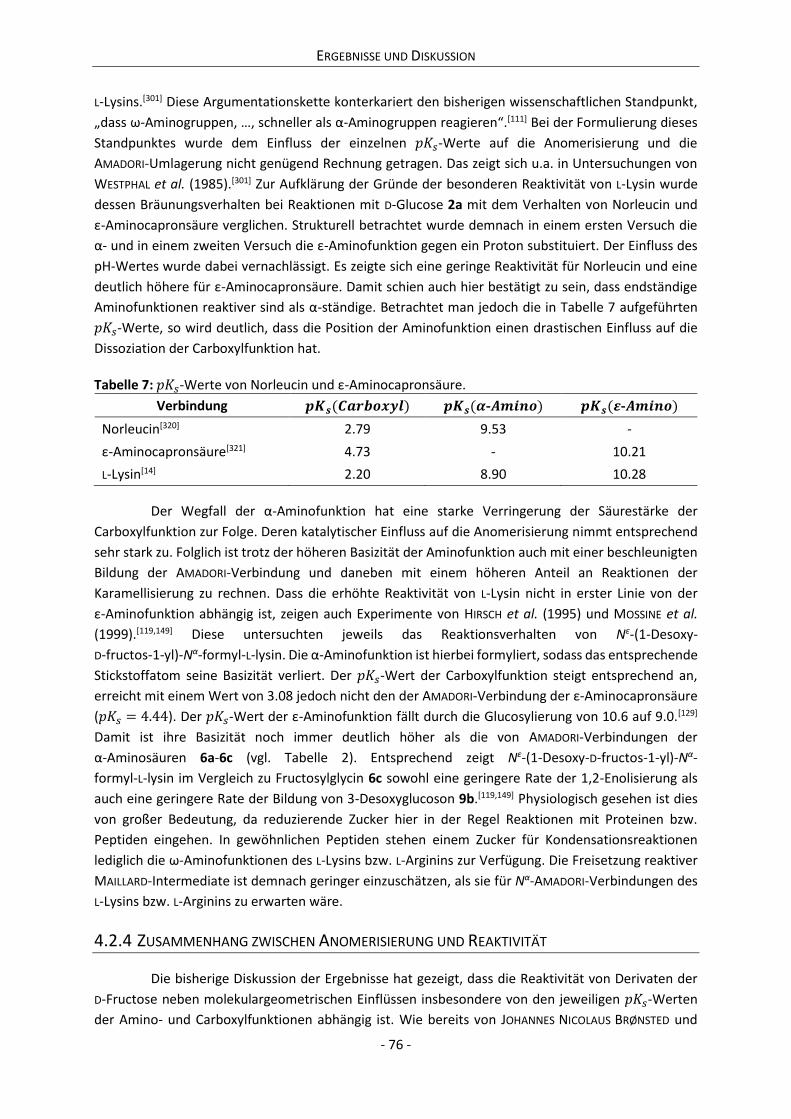
ERGEBNISSE UND DISKUSSION
- 76 -
L-Lysins.[301] Diese Argumentationskette konterkariert den bisherigen wissenschaftlichen Standpunkt,
„dass ω-Aminogruppen, …, schneller als α-Aminogruppen reagieren“.[111] Bei der Formulierung dieses
Standpunktes wurde dem Einfluss der einzelnen 𝑝𝐾𝑠-Werte auf die Anomerisierung und die
AMADORI-Umlagerung nicht genügend Rechnung getragen. Das zeigt sich u.a. in Untersuchungen von
WESTPHAL et al. (1985).[301] Zur Aufklärung der Gründe der besonderen Reaktivität von L-Lysin wurde
dessen Bräunungsverhalten bei Reaktionen mit D-Glucose 2a mit dem Verhalten von Norleucin und
ε-Aminocapronsäure verglichen. Strukturell betrachtet wurde demnach in einem ersten Versuch die
α- und in einem zweiten Versuch die ε-Aminofunktion gegen ein Proton substituiert. Der Einfluss des
pH-Wertes wurde dabei vernachlässigt. Es zeigte sich eine geringe Reaktivität für Norleucin und eine
deutlich höhere für ε-Aminocapronsäure. Damit schien auch hier bestätigt zu sein, dass endständige
Aminofunktionen reaktiver sind als α-ständige. Betrachtet man jedoch die in Tabelle 7 aufgeführten
𝑝𝐾𝑠-Werte, so wird deutlich, dass die Position der Aminofunktion einen drastischen Einfluss auf die
Dissoziation der Carboxylfunktion hat.
Tabelle 7: 𝑝𝐾𝑠-Werte von Norleucin und ε-Aminocapronsäure.
Verbindung 𝒑𝑲𝒔(𝑪𝒂𝒓𝒃𝒐𝒙𝒚𝒍) 𝒑𝑲𝒔(𝜶-𝑨𝒎𝒊𝒏𝒐) 𝒑𝑲𝒔(𝜺-𝑨𝒎𝒊𝒏𝒐)
Norleucin[320] 2.79 9.53 -
ε-Aminocapronsäure[321] 4.73 - 10.21
L-Lysin[14] 2.20 8.90 10.28
Der Wegfall der α-Aminofunktion hat eine starke Verringerung der Säurestärke der
Carboxylfunktion zur Folge. Deren katalytischer Einfluss auf die Anomerisierung nimmt entsprechend
sehr stark zu. Folglich ist trotz der höheren Basizität der Aminofunktion auch mit einer beschleunigten
Bildung der AMADORI-Verbindung und daneben mit einem höheren Anteil an Reaktionen der
Karamellisierung zu rechnen. Dass die erhöhte Reaktivität von L-Lysin nicht in erster Linie von der
ε-Aminofunktion abhängig ist, zeigen auch Experimente von HIRSCH et al. (1995) und MOSSINE et al.
(1999).[119,149] Diese untersuchten jeweils das Reaktionsverhalten von Nε-(1-Desoxy-
D-fructos-1-yl)-Nα-formyl-L-lysin. Die α-Aminofunktion ist hierbei formyliert, sodass das entsprechende
Stickstoffatom seine Basizität verliert. Der 𝑝𝐾𝑠-Wert der Carboxylfunktion steigt entsprechend an,
erreicht mit einem Wert von 3.08 jedoch nicht den der AMADORI-Verbindung der ε-Aminocapronsäure
(𝑝𝐾𝑠 = 4.44). Der 𝑝𝐾𝑠-Wert der ε-Aminofunktion fällt durch die Glucosylierung von 10.6 auf 9.0.[129]
Damit ist ihre Basizität noch immer deutlich höher als die von AMADORI-Verbindungen der
α-Aminosäuren 6a-6c (vgl. Tabelle 2). Entsprechend zeigt Nε-(1-Desoxy-D-fructos-1-yl)-Nα-
formyl-L-lysin im Vergleich zu Fructosylglycin 6c sowohl eine geringere Rate der 1,2-Enolisierung als
auch eine geringere Rate der Bildung von 3-Desoxyglucoson 9b.[119,149] Physiologisch gesehen ist dies
von großer Bedeutung, da reduzierende Zucker hier in der Regel Reaktionen mit Proteinen bzw.
Peptiden eingehen. In gewöhnlichen Peptiden stehen einem Zucker für Kondensationsreaktionen
lediglich die ω-Aminofunktionen des L-Lysins bzw. L-Arginins zur Verfügung. Die Freisetzung reaktiver
MAILLARD-Intermediate ist demnach geringer einzuschätzen, als sie für Nα-AMADORI-Verbindungen des
L-Lysins bzw. L-Arginins zu erwarten wäre.
4.2.4 ZUSAMMENHANG ZWISCHEN ANOMERISIERUNG UND REAKTIVITÄT
Die bisherige Diskussion der Ergebnisse hat gezeigt, dass die Reaktivität von Derivaten der
D-Fructose neben molekulargeometrischen Einflüssen insbesondere von den jeweiligen 𝑝𝐾𝑠-Werten
der Amino- und Carboxylfunktionen abhängig ist. Wie bereits von JOHANNES NICOLAUS BRØNSTED und

ERGEBNISSE UND DISKUSSION
- 77 -
EDWARD ARMAND GUGGENHEIM (1927) beschrieben, trifft das auch auf die Rate der Mutarotation
reduzierender Zucker zu.[289] Diesbezüglich wurde in Schema 19 (Seite 35) gezeigt, dass die
Anomerisierung der Zucker über ionische Spezies erfolgt (vgl. dazu auch das Strukturenverzeichnis).
Dass viele Prozesse, die zur stofflichen Veränderung eines Zuckers führen, gleichermaßen von der
Bildung ionischer Zuckerspezies abhängig sind, wurde im Verlauf der vorliegenden Arbeit an vielen
Stellen erörtert. Dazu zählen Epimerisierungen (Schema 3, Seite 7), die Bildung von N-Glycosiden
(Schema 5, Seite 9 und Schema 6, Seite 10) sowie die AMADORI-Umlagerung (Schema 8, Seite 11), die
Bildung O-heterocyclischer Verbindungen (Schema 17, Seite 21), die Tautomerisierung zu Endiolen und
Enaminolen (Schema 21, Seite 49) sowie die Bildung der epimeren 3-Desoxyosone 9b und 9c
(Schema 24, Seite 64). Reaktionen reduzierender Zucker können demzufolge als Fehler oder
unwahrscheinliche Alternativen der Anomerisierung angesehen werden.44 Dabei handelt es sich bei
den ionischen Spezies, die den Abbau der Verbindung verursachen, nicht zwingend um diejenigen, die
verantwortlich für den Prozess der Anomerisierung sind. Die Deprotonierung der anomeren
Hydroxylfunktion führt zur Ringöffnung und damit zur Anomerisierung, während die Deprotonierung
einer der übrigen Hydroxylfunktionen den Auftakt für eine Abbaureaktion darstellt.
Ausgehend von diesen Überlegungen kann angenommen werden, dass Gleichung (33)
(Seite 36) nicht nur zur Beschreibung der Dynamik der Ringöffnung, sondern ebenso zur Beschreibung
der Abbaukinetik eines reduzierenden Zuckers genutzt werden kann. Die anomeren
Hydroxylfunktionen reduzierender Zucker haben von allen vorhandenen Hydroxylfunktionen die
geringsten 𝑝𝐾𝑠-Werte und damit das größte Bestreben zur Deprotonierung.[298] Betrachtet man
beispielsweise den dritten Summanden aus Gleichung (33), bedeutet dies, dass 𝐾𝑠2 im Falle der
anomeren Hydroxylfunktion von allen vorhanden Hydroxylfunktionen den größten Wert annimmt.
Damit ergibt sich für die Rate der Ringöffnung nahezu unabhängig vom pH-Wert ein konstantes
Vielfaches der Abbaurate des Zuckers. Dabei entspricht das Verhältnis der jeweiligen Raten dem
Verhältnis der Säurekonstanten der betrachteten Hydroxylfunktionen multipliziert mit dem Verhältnis
ihrer spontanen Ringöffnungs- bzw. Abbauraten. Hat die Temperatur schließlich einen ähnlichen
Einfluss auf die spontanen Ringöffnungs- und Abbauraten, ist das beschriebene Verhältnis von
Ringöffnung und Abbau darüber hinaus praktisch temperaturunabhängig.
Auf dieser Basis ergibt sich eine mathematisch begründete Korrelation von Dynamik und
Kinetik. Ein mechanistischer Zusammenhang kann wie folgt hergestellt werden: Die anomere
Hydroxylfunktion stellt nach ihrer Deprotonierung zum Alkoholat eine sehr starke Base dar. Damit ist
ein Protonenshift von weiteren Hydroxylfunktionen hin zum anomeren Alkoholat nicht
unwahrscheinlich. Einen solchen Protonenshift zeigt bspw. Schema 21 (Seite 49). Analog kann auch ein
Protonenshift vom kationischen Ringsauerstoff auf eine Hydroxylfunktion erfolgen.45 Auf dieser
Modellvorstellung beruhen u.a. die in Schema 17 (Seite 21) gezeigten Reaktionsmechanismen.
Weitere mechanistische Verknüpfungen vom Prozess der Anomerisierung mit stofflichen
Veränderungen von Zuckern zeigt Schema 27 am Beispiel der Tautomerisierung verschiedener
Anomere der D-Fructose 1a im Basischen. Entsprechende Reaktionen im sauren Milieu fasst die
Publikation von RASMUSSEN et al. (2014) zusammen.[177]
44 Vergleiche dazu auch die in den Schemata 5 und 6 (Seiten 9 und 10) gezeigte Konkurrenz der Bildung des
gem-Diols der D-Glucose 2a zur Bildung des gem-Aminols 5c.
45 Rechnungen von FENG et al. (2013) zeigen, dass die Protonenaffinität der anomeren Hydroxylfunktionen der
Anomere der D-Fructose 1a sogar geringfügig größer ist als die der entsprechenden Ringsauerstoffatome.[298]

ERGEBNISSE UND DISKUSSION
- 78 -
Bei den in Schema 27 formulierten Abbauprodukten (insbesondere Gycerinaldehyd 19,
Glycerinsäure 20, Glyoxal 21 und Acrolein 22) handelt es sich um repräsentative Intermediate der
MAILLARD-Reaktion.[304,322-326]
Aufgrund der diskutierten Zusammenhänge zwischen dem Prozess der Anomerisierung und
dem Abbau von Zuckern kann davon ausgegangen werden, dass eine Beschleunigung der
Anomerisierung auch zu einer Instabilität des Zuckers im Allgemeinen führt. Demnach handelt es sich
bei dem acyclischen Zentralisomer eines Zuckers nicht – wie üblicherweise angenommen[327,328] – um
dessen reaktionsverantwortliche Komponente. Vielmehr tragen alle Anomere eines Zuckers zur
Reaktivität des Gesamtsystems bei, da jedes Anomer zur Ausbildung ionischer Spezies befähigt ist. Die
Reaktivität eines Zuckers korreliert, dieser Argumentation folgend, mit der Summe der Produkte der
jeweiligen Anomerisierungsraten und der relativen Konzentrationen der Anomere. Für Derivate der
D-Fructose lässt sich diese konzentrationsgewichtete Gesamtanomerisierungsrate 𝑘Σ mit Hilfe von
Gleichung (46) berechnen.
𝑘Σ = [𝛼𝐹] ∙ 𝑘𝛼𝐹,𝑧 + [𝛽𝐹] ∙ 𝑘𝛽𝐹,𝑧 + [𝛼𝑃] ∙ 𝑘𝛼𝑃,𝑧 + [𝛽𝑃] ∙ 𝑘𝛽𝑃,𝑧
+[𝑧] ∙ (𝑘𝑧,𝛼𝐹 + 𝑘𝑧,𝛽𝐹 + 𝑘𝑧,𝛼𝑃 + 𝑘𝑧,𝛽𝐹) = 2 ∙ [𝑧] ∙∑𝑘𝑧,𝑖𝑖
(46)
Dabei bezeichnet 𝑧 das acyclische Zentralisomer, 𝑘𝑖,𝑧 die Ringöffnungsraten und 𝑘𝑧,𝑖 die
Ringschlussraten. Eine Beschleunigung der Anomerisierungsraten führt auch zu einer beschleunigten
Bildung ionischer Zuckerspezies. Die Berechnung der konzentrationsgewichteten
Gesamtanomerisierungsrate 𝑘Σ ermöglicht demnach einen empfindlichen Vergleich der relativen
Reaktivitäten verschiedener Zucker – auch unter Bedingungen bei denen der Ablauf von Reaktionen
selbst noch nicht feststellbar ist. Das gewichtete Mittel der mittleren Lebensdauer der einzelnen
Isomere korreliert folglich mit der milieuabhängigen Stabilität des Zuckers. Zur vergleichenden
Berechnung der Stabilitäten wird auf Grundlage von 𝑘Σ ein neuer Parameter eingeführt. Dieser als
ACuSTiC (Approximated Carbohydrate Milieu Stability Time Constant) bezeichnete Parameter gibt an,
welche Zeit durchschnittlich vergeht, bis jedes Molekül eines Anomerensystems genau einmal ionisiert
wurde. Der ACuSTiC-Wert ergibt sich durch die Lösung des durch Gleichung (47) gegebenen Integrals
und wird somit durch Gleichung (48) beschrieben.
∫𝑘Σ ∙ 𝑑𝑡
𝑡
0
= ∫2 ∙ [𝑧] ∙∑𝑘𝑧,𝑖𝑖
∙ 𝑑𝑡
𝑡
0
= 100% (47)
𝑡 =100%
2 ∙ [𝑧] ∙ ∑ 𝑘𝑧,𝑖𝑖= 𝐴𝐶𝑢𝑆𝑇𝑖𝐶 (48)
In Kapitel 4.2.2 wurde bereits gezeigt, dass das gewichtete Mittel der Ringöffnungsraten von
AMADORI-Verbindungen der ω-Aminosäuren 6c-6e mit deren Raten der 1,2-Enolisierung korreliert, und
dass diese wiederum in direkter Verbindung zu den Bildungsraten von 3-Desoxyglucoson 9b stehen.
Im Folgenden gilt es zu überprüfen, ob die Berechnung von ACuSTiC-Werten eine Abschätzung der
relativen Stabilität verschiedener Zuckersysteme erlaubt. Dazu wurden die relativen Konzentrationen
der Anomere verschiedener reduzierender Zucker in Abhängigkeit der Temperatur und des pH- bzw.
pD-Wertes mittels quantitativer NMR-Spektroskopie bestimmt. Daraufhin erfolgte die Messung der
einzelnen Ringöffnungsraten mittels selektiver Sättigungstransfer-NMR-Spektroskopie (SST-NMR)
unter denselben Bedingungen.
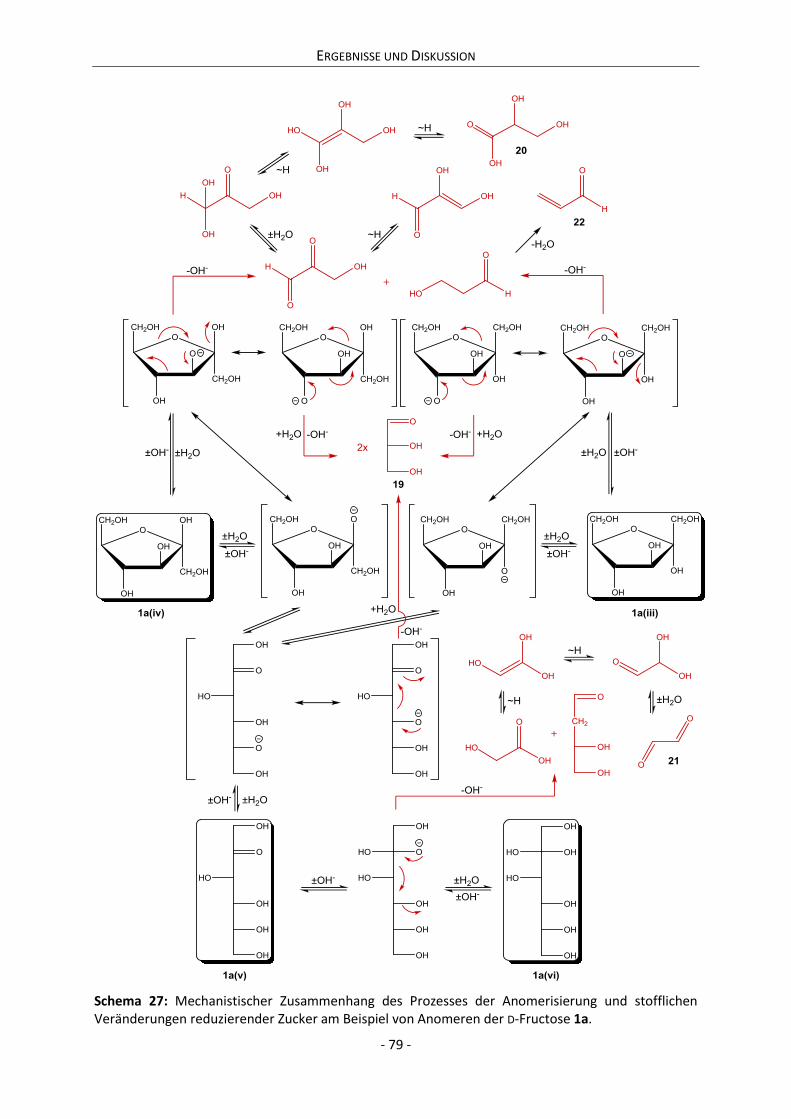
ERGEBNISSE UND DISKUSSION
- 79 -
Schema 27: Mechanistischer Zusammenhang des Prozesses der Anomerisierung und stofflichen Veränderungen reduzierender Zucker am Beispiel von Anomeren der D-Fructose 1a.

ERGEBNISSE UND DISKUSSION
- 80 -
Als Zielsubstanzen wurden D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen an
L-Alanin 4a bzw. L-Prolin 4b sowie die entsprechenden AMADORI-Verbindungen 6a und 6b ausgewählt.
Um eine verlässliche Quantifizierung zu erreichen und daneben die spezifischen Ringöffnungsraten
jedes Anomers ermitteln zu können, erfolgten die NMR-spektroskopischen Messungen auf Grundlage
der anomeren 13C-NMR-Resonanzen. Dafür war es notwendig, mit [2-13C]-markierten
Zuckergrundkörpern zu arbeiten. Um die Allgemeingültigkeit des ACuSTiC-Ansatzes zu überprüfen,
wurden darüber hinaus äquivalente Messungen für D-[2-13C]Tagatose 1b und D-Glucose 2a
durchgeführt. Die Messungen der D-Glucose 2a erfolgten dabei auf Grundlage der anomeren 1H-NMR-Resonanzen.
4.2.5 QUANTITATIVE ZUSAMMENSETZUNG DER ANOMERENSYSTEME
Die Messung der relativen Konzentrationen der Zuckerisomere erfolgte bei Temperaturen
zwischen 27 und 87 °C in einem pD-Intervall von 1.5 bis 6.5. Der Temperaturbereich orientiert sich
dabei am Fest- und Siedepunkt des Lösungsmittels (D2O). Das pD-Intervall wurde dem
lebensmittelrelevanten Bereich angepasst. Da sich der pD-Wert – wie bereits beschrieben – im Verlauf
der nicht enzymatischen Bräunung auf Werte um 4.0 absenkt[16,302] und die zu messenden
Ringöffnungsraten pD-abhängig sind, musste die Messung des pD-Wertes simultan zur Messung der
Anomerisierungsraten erfolgen. Dies ist möglich, da die chemische Verschiebung der
NMR-Resonanzen zahlreicher funktioneller Gruppen pD-abhängig ist. Daneben ist die Signallage
zusätzlich abhängig von der Temperatur.[329] Damit sind die Temperatur und der pD-Wert einer Probe
nahezu in allen zugehörigen NMR-Spektren codiert und können nach geeigneter Kalibrierung
ausgelesen werden. Da die Temperatur der Probe innerhalb des Probenkopfes des
NMR-Spektrometers mit Hilfe von Thermostaten auf etwa 0.1 K genau eingestellt und überwacht
werden kann, ist die Decodierung des pD-Wertes auf Grundlage der HENDERSON-HASSELBALCH-Funktion
für den chemischen Austausch möglich.[330] Es zeigte sich, dass das für die Probenvorbereitung
genutzte Deuteriumoxid jeglicher Hersteller Spuren von Ameisensäure enthielt. Diese hat einen
𝑝𝐾𝑠-Wert von 3.63 in H2O (vgl. Tabelle 12, Seite 97) und damit von 4.13 in D2O.[331] Entsprechend der
HENDERSON-HASSELBALCH-Funktion ist demnach je nach vorherrschender Ionenstärke mit pD-sensitiven
chemischen Verschiebungen der Kernresonanzen der Ameisensäure im pD-Bereich von rund 2.0 bis
6.0 zu rechnen.[332] Dies umfasst nahezu das gesamte pD-Intervall, für das die Messung von
Anomerisierungsraten erfolgen soll. Daneben ist der 𝑝𝐾𝑠-Wert der Ameisensäure im Bereich zwischen
0 und 100 °C kaum temperaturabhängig.[333] Die als Verunreinigung vorhandene Ameisensäure stellt
aufgrund dessen eine gut brauchbare pD-Sonde dar und wurde daher im Rahmen dieser Arbeit als
solche genutzt.
Es zeigte sich für alle untersuchten Systeme reduzierender Zucker sowie von
Zuckerderivaten, dass die relativen Konzentrationen ihrer Anomere im untersuchten Bereich nicht
abhängig vom pD-Wert sind. Demnach wurden die jeweils bei identischer Temperatur gemessenen
relativen Konzentrationen gemittelt. Anschließend wurden die relativen Konzentrationen der
cyclischen Anomere in Relation zur Konzentration des offenkettigen Zentralisomers gesetzt und die
entsprechenden Entropie- und Enthalpiedifferenzen nach Gleichung (49) berechnet.
𝐾 = exp(−∆𝐻 − 𝑇 ∙ ∆𝑆
𝑅 ∙ 𝑇) (49)

ERGEBNISSE UND DISKUSSION
- 81 -
Dabei ist 𝐾 die jeweilige Konstante für das Gleichgewicht zwischen einem cyclischen Anomer
und dem Zentralisomer, ∆𝐻 die Enthalpiedifferenz, ∆𝑆 die Entropiedifferenz, 𝑇 die absolute
Temperatur und 𝑅 die universelle Gaskonstante. Diese Vorgehensweise wurde für (alle Derivate der)
D-Fructose 1a, 6a und 6b sowie D-Tagatose 1b genutzt. Für D-Glucose 2a musste die Berechnung der
thermodynamischen Parameter auf einem anderen Weg erfolgen.
Problematisch bei der Messung der relativen Konzentrationen der Strukturisomere der
D-Glucose 2a ist, dass ihr acyclisches Zentralisomer bei 30 °C lediglich zu rund 0.004% (d.h. 40 ppm)
vorliegt.[95] Ein direkter Nachweis entsprechender 13C-NMR-Resonanzen ist daher nur mit erheblichem
Aufwand möglich. Ein Nachweis zugehöriger 1H-NMR-Resonanzen steht bis heute aus. Die Methode
der NMR-Spektroskopie eignet sich damit nicht für die temperaturabhängige Messung von
Gleichgewichtskonstanten der D-Glucose 2a. Es zeigt sich jedoch, dass sich die relativen
Konzentrationen der einzelnen Anomere der untersuchten Zucker im relevanten Temperaturbereich
in sehr guter Näherung durch quadratische Funktionen beschreiben lassen. Daher wurden 13C-NMR-
basierte Literaturwerte der relativen Konzentrationen des acyclischen Zentralisomers der D-Glucose
2a für verschiedene Temperaturen gesucht[334] und mit Hilfe einer quadratischen Funktion modelliert.
Damit ist die relative Konzentration des acyclischen Zentralisomers in Abhängigkeit der Temperatur
näherungsweise bekannt.
Als weiteres Problem stellte sich heraus, dass bislang keinerlei 1H-NMR-Resonanzen der
furanoiden Anomere der D-Glucose 2a in der Literatur beschrieben sind. Alle bisherigen direkten
NMR-spektroskopischen Messungen der furanoiden Anomere der D-Glucose 2a basieren auf der
Detektion anomerer 13C-NMR-Resonanzen.[95,334,335] Lediglich für 3,6-Anhydro-D-glucose, die in
wässriger Lösung hauptsächlich furanoide Anomere bildet, sind 1H-NMR-Daten verfügbar.[336] Dabei
liegt die 1H-NMR-Resonanz des anomeren Protons des α-Anomers bei tieferem Feld als die des
β-Anomers. Darüber hinaus ist der Unterschied der jeweiligen vicinalen Kopplungskonstanten der
anomeren Protonen markant. Für das α-Anomer beträgt die Kopplungskonstante 4 Hz, für das
β-Anomer hingegen lediglich 1 Hz.[336] Diese Messwerte stehen im Einklang mit der allgemeinen
Beobachtung, dass die 1H-NMR-Resonanz des anomeren Protons des 1,2-cis-Anomers (hier:
α-Anomer) gewöhnlich bei tieferem Feld liegt als die des 1,2-trans-Anomers (hier: β-Anomer).[337] Dies
wird auch als die sogenannte „syn-upfield rule“ bezeichnet.46;[47,73,338] Außerdem liegt die vicinale
Kopplungskonstante des anomeren Protons der 1,2-cis-anomeren Furanose gewöhnlich bei 3-5 Hz und
die der 1,2-trans-anomeren Furanose bei 0-2 Hz.[337] In den 1H-NMR-Spektren der D-Glucose 2a
konnten im Rahmen der vorliegenden Arbeit zwei Resonanzen identifiziert werden, die im anomeren
Bereich des Spektrums liegen und im Einklang mit den oben beschriebenen Literaturangaben stehen.
Die entsprechenden 1H-NMR-Resonanzen zeigt Abbildung 26 für eine Temperatur von 67 °C bei pD 5.0.
Bei Temperaturen unterhalb von 37 °C ist das anomere 1H-NMR-Signal des β-furanoiden
Anomers bei einer Messfrequenz von 600 MHz nicht nachweisbar, da seine Resonanz isochron zu der
des anomeren Protons der α-Pyranose ist. Vermutlich ist dies der Grund dafür, dass bislang keine
entsprechenden Resonanzen in der Literatur beschrieben wurden.
Obwohl sowohl die relative Lage der anomeren Signale der furanoiden Anomere sowie deren
Kopplungskonstanten mit den Erwartungen übereinstimmen, ist dies noch kein Beweis für eine
46 Dabei ist das betrachtete Proton syn-ständig zur funktionellen Gruppe (etwa einer Hydroxylfunktion). Damit
handelt es sich um das 1,2-trans-Anomer.
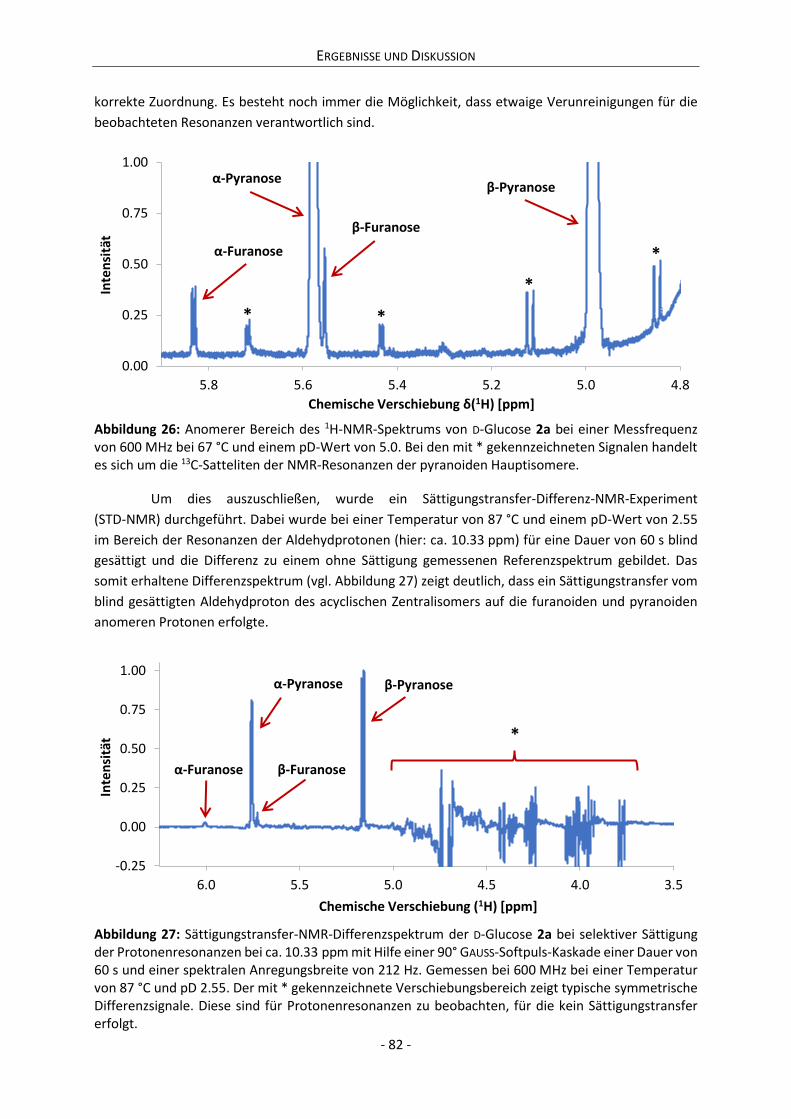
ERGEBNISSE UND DISKUSSION
- 82 -
korrekte Zuordnung. Es besteht noch immer die Möglichkeit, dass etwaige Verunreinigungen für die
beobachteten Resonanzen verantwortlich sind.
Abbildung 26: Anomerer Bereich des 1H-NMR-Spektrums von D-Glucose 2a bei einer Messfrequenz von 600 MHz bei 67 °C und einem pD-Wert von 5.0. Bei den mit * gekennzeichneten Signalen handelt es sich um die 13C-Satteliten der NMR-Resonanzen der pyranoiden Hauptisomere.
Um dies auszuschließen, wurde ein Sättigungstransfer-Differenz-NMR-Experiment
(STD-NMR) durchgeführt. Dabei wurde bei einer Temperatur von 87 °C und einem pD-Wert von 2.55
im Bereich der Resonanzen der Aldehydprotonen (hier: ca. 10.33 ppm) für eine Dauer von 60 s blind
gesättigt und die Differenz zu einem ohne Sättigung gemessenen Referenzspektrum gebildet. Das
somit erhaltene Differenzspektrum (vgl. Abbildung 27) zeigt deutlich, dass ein Sättigungstransfer vom
blind gesättigten Aldehydproton des acyclischen Zentralisomers auf die furanoiden und pyranoiden
anomeren Protonen erfolgte.
Abbildung 27: Sättigungstransfer-NMR-Differenzspektrum der D-Glucose 2a bei selektiver Sättigung der Protonenresonanzen bei ca. 10.33 ppm mit Hilfe einer 90° GAUSS-Softpuls-Kaskade einer Dauer von 60 s und einer spektralen Anregungsbreite von 212 Hz. Gemessen bei 600 MHz bei einer Temperatur von 87 °C und pD 2.55. Der mit * gekennzeichnete Verschiebungsbereich zeigt typische symmetrische Differenzsignale. Diese sind für Protonenresonanzen zu beobachten, für die kein Sättigungstransfer erfolgt.
0.00
0.25
0.50
0.75
1.00
4.85.05.25.45.65.8
Inte
nsi
tät
Chemische Verschiebung δ(1H) [ppm]
β-Furanose
α-Furanose
*
*
*
β-Pyranose
*
α-Pyranose
-0.25
0.00
0.25
0.50
0.75
1.00
3.54.04.55.05.56.0
Inte
nsi
tät
Chemische Verschiebung (1H) [ppm]
β-Furanoseα-Furanose
α-Pyranose β-Pyranose
*
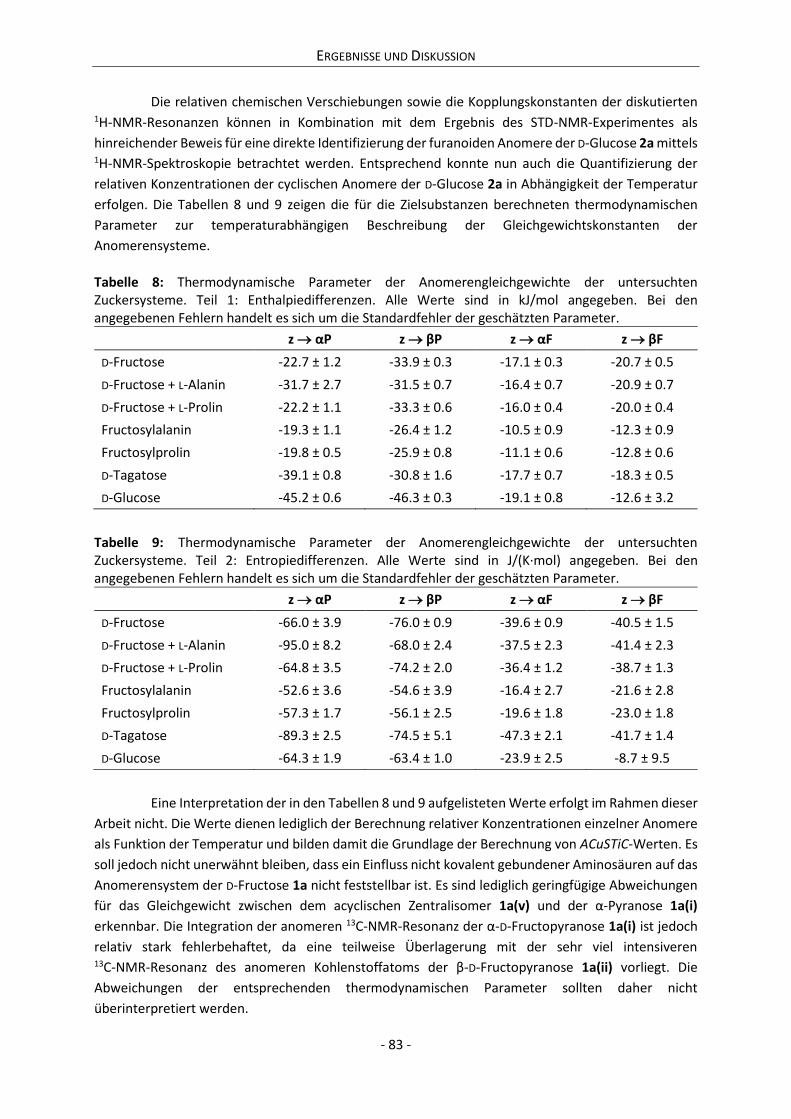
ERGEBNISSE UND DISKUSSION
- 83 -
Die relativen chemischen Verschiebungen sowie die Kopplungskonstanten der diskutierten 1H-NMR-Resonanzen können in Kombination mit dem Ergebnis des STD-NMR-Experimentes als
hinreichender Beweis für eine direkte Identifizierung der furanoiden Anomere der D-Glucose 2a mittels 1H-NMR-Spektroskopie betrachtet werden. Entsprechend konnte nun auch die Quantifizierung der
relativen Konzentrationen der cyclischen Anomere der D-Glucose 2a in Abhängigkeit der Temperatur
erfolgen. Die Tabellen 8 und 9 zeigen die für die Zielsubstanzen berechneten thermodynamischen
Parameter zur temperaturabhängigen Beschreibung der Gleichgewichtskonstanten der
Anomerensysteme.
Tabelle 8: Thermodynamische Parameter der Anomerengleichgewichte der untersuchten Zuckersysteme. Teil 1: Enthalpiedifferenzen. Alle Werte sind in kJ/mol angegeben. Bei den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten Parameter.
z αP z βP z αF z βF
D-Fructose -22.7 ± 1.2 -33.9 ± 0.3 -17.1 ± 0.3 -20.7 ± 0.5
D-Fructose + L-Alanin -31.7 ± 2.7 -31.5 ± 0.7 -16.4 ± 0.7 -20.9 ± 0.7
D-Fructose + L-Prolin -22.2 ± 1.1 -33.3 ± 0.6 -16.0 ± 0.4 -20.0 ± 0.4
Fructosylalanin -19.3 ± 1.1 -26.4 ± 1.2 -10.5 ± 0.9 -12.3 ± 0.9
Fructosylprolin -19.8 ± 0.5 -25.9 ± 0.8 -11.1 ± 0.6 -12.8 ± 0.6
D-Tagatose -39.1 ± 0.8 -30.8 ± 1.6 -17.7 ± 0.7 -18.3 ± 0.5
D-Glucose -45.2 ± 0.6 -46.3 ± 0.3 -19.1 ± 0.8 -12.6 ± 3.2
Tabelle 9: Thermodynamische Parameter der Anomerengleichgewichte der untersuchten Zuckersysteme. Teil 2: Entropiedifferenzen. Alle Werte sind in J/(K·mol) angegeben. Bei den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten Parameter.
z αP z βP z αF z βF
D-Fructose -66.0 ± 3.9 -76.0 ± 0.9 -39.6 ± 0.9 -40.5 ± 1.5
D-Fructose + L-Alanin -95.0 ± 8.2 -68.0 ± 2.4 -37.5 ± 2.3 -41.4 ± 2.3
D-Fructose + L-Prolin -64.8 ± 3.5 -74.2 ± 2.0 -36.4 ± 1.2 -38.7 ± 1.3
Fructosylalanin -52.6 ± 3.6 -54.6 ± 3.9 -16.4 ± 2.7 -21.6 ± 2.8
Fructosylprolin -57.3 ± 1.7 -56.1 ± 2.5 -19.6 ± 1.8 -23.0 ± 1.8
D-Tagatose -89.3 ± 2.5 -74.5 ± 5.1 -47.3 ± 2.1 -41.7 ± 1.4
D-Glucose -64.3 ± 1.9 -63.4 ± 1.0 -23.9 ± 2.5 -8.7 ± 9.5
Eine Interpretation der in den Tabellen 8 und 9 aufgelisteten Werte erfolgt im Rahmen dieser
Arbeit nicht. Die Werte dienen lediglich der Berechnung relativer Konzentrationen einzelner Anomere
als Funktion der Temperatur und bilden damit die Grundlage der Berechnung von ACuSTiC-Werten. Es
soll jedoch nicht unerwähnt bleiben, dass ein Einfluss nicht kovalent gebundener Aminosäuren auf das
Anomerensystem der D-Fructose 1a nicht feststellbar ist. Es sind lediglich geringfügige Abweichungen
für das Gleichgewicht zwischen dem acyclischen Zentralisomer 1a(v) und der α-Pyranose 1a(i)
erkennbar. Die Integration der anomeren 13C-NMR-Resonanz der α-D-Fructopyranose 1a(i) ist jedoch
relativ stark fehlerbehaftet, da eine teilweise Überlagerung mit der sehr viel intensiveren 13C-NMR-Resonanz des anomeren Kohlenstoffatoms der β-D-Fructopyranose 1a(ii) vorliegt. Die
Abweichungen der entsprechenden thermodynamischen Parameter sollten daher nicht
überinterpretiert werden.

ERGEBNISSE UND DISKUSSION
- 84 -
4.2.6 DYNAMIK DER ANOMERISIERUNG
Die Messung der Dynamik der Anomerisierung erfolgte im Rahmen der vorliegenden Arbeit
mit Hilfe der dynamischen NMR-Spektroskopie. Aufgrund ihrer in Kapitel 2.2.3 diskutierten Vorteile
gegenüber anderen Methoden, wurde dabei die selektive Sättigungstransfer-NMR-Spektroskopie
(SST-NMR) eingesetzt. Die Messung der Ringöffnungsraten der Anomere der Ketohexosen 1a und 1b
und deren Derivate 6a und 6b erfolgte unter der Ausnutzung anomerer 13C-NMR-Resonanzen. Im Falle
der D-Glucose 2a wurden wiederum die anomeren Protonenresonanzen zugrunde gelegt. Alle
Messungen erfolgten zwischen 27 und 87 °C in einem pD-Intervall von 1.5 bis 6.5, wobei Ameisensäure
als pD-Sonde genutzt wurde. Die in der Literatur übliche Auswertung der in Abhängigkeit der
Sättigungsdauer veränderten Integrale der beobachteten Kernresonanzen zur Berechnung der
Anomerisierungsraten basiert auf der Linearisierung von Gleichung (21) (Seite 32).[71] Die Messung von
Anomerisierungsraten in Abhängigkeit der Temperatur ermöglicht in einem zweiten Schritt die
Berechnung der thermodynamischen Aktivierungsparameter der Anomerisierung mit Hilfe der
EYRING-Funktion.[313] Im Rahmen der vorliegenden Arbeit erfolgt die Berechnung der
thermodynamischen Aktivierungsparameter im Unterschied dazu unmittelbar aus den Integralen der
beobachteten Kernresonanzen in Abhängigkeit der Sättigungsdauer, der Temperatur und des
pD-Wertes. Dazu wird die EYRING-Funktion in Gleichung (32) (für Modelle ohne Katalysator) bzw.
Gleichung (38) (für Modelle mit Katalysator) und diese wiederum in Gleichung (21) eingesetzt. Die
resultierende Funktion ist durch die thermodynamischen Aktivierungsparameter parametrisiert. Diese
Parameter können mit Hilfe einer nicht linearen Regression numerisch ermittelt werden und dienen
im finalen Schritt der Berechnung von ACuSTiC-Werten für beliebige pD-Werte und Temperaturen
innerhalb der jeweils untersuchten Intervalle. Die numerische Berechnung der thermodynamischen
Aktivierungsparameter der Zielsubstanzen sowie der entsprechenden Fehlerintervalle (Standardfehler
der Schätzwerte) wurde im Rahmen der vorliegenden Arbeit mit Hilfe des XLSTAT 2017
Statistik-Analyse Add-Ins für Microsoft Excel 2016 vorgenommen. Wie den Ausführungen in Kapitel 2.3
zu entnehmen ist, benötigt man für die Berechnungen jedoch die 𝑝𝐾𝑠-Werte der anomeren
Hydroxylfunktionen sowie der Ringsauerstoffatome der im Einzelnen untersuchten Zuckeranomere.
Eine experimentelle Messung entsprechender Dissoziationskonstanten ist hoch problematisch. Eine
NMR-Titration zur Ermittlung der 𝑝𝐾𝑠-Werte der anomeren Hydroxylfunktionen ist zwar prinzipiell
denkbar, jedoch sind die Analyten im entsprechenden pH-Bereich extrem instabil. Bei ihrem Abbau
fällt – wie oben beschrieben – der pH-Wert, sodass dieser im Zuge der Messungen nicht konstant ist.
Weiterhin ist die Rate der Anomerisierung im stark basischen pH-Bereich außerordentlich hoch, sodass
die Signallage der zu beobachtenden anomeren 13C-NMR-Resonanzen nicht mehr nur vom
Gleichgewicht ionischer und nicht ionischer Spezies, sondern gleichermaßen vom chemischen
Austausch der cyclischen Anomere mit dem acyclischen Zentralisomer abhängig ist.[339,340] Die damit
einhergehende Vergrößerung der Linienbreite der NMR-Resonanzen führte in entsprechenden
Experimenten zur Koaleszenz der anomeren 13C-NMR-Signale der cyclischen Anomere.[339,340] Damit ist
selbst die Abschätzung anomerenspezifischer 𝑝𝐾𝑠-Werte auf diese Weise nicht möglich. Im Rahmen
dieser Arbeit wurde daher zunächst das Ziel verfolgt, entsprechende 𝑝𝐾𝑠-Werte mit Hilfe kommerziell
verfügbarer Software (hier: Percepta von ACD/Labs) zu berechnen. Die Algorithmen, die der Software
zugrunde liegen (Bezeichnung: Classic- und GALAS-Algorithmus), erkennen Ether – und damit auch
Ringsauerstoffatome von Zuckeranomeren – jedoch prinzipiell nicht als ionisierbare funktionelle
Gruppen. Folglich ist eine Berechnung der zugehörigen 𝑝𝐾𝑠-Werte ausgeschlossen. Daher wurde die
Literaturrecherche ausgeweitet. Üblicherweise eingesetzte experimentelle Methoden wie
potentiometrische und kalorimetrische Titrationen ermöglichen ebenfalls keine anomerenspezifische

ERGEBNISSE UND DISKUSSION
- 85 -
Bestimmung von 𝑝𝐾𝑠-Werten.[341] Entsprechend wurde die Recherche auf theoretische Berechnungen
von 𝑝𝐾𝑠-Werten fokussiert. Dabei sind jedoch lediglich die Dissoziationskonstanten der
Hydroxylfunktionen (nicht der Ringsauerstoffatome) der quantitativ bedeutsamen Anomere von
D-Glucose 2a und D-Fructose 1a verfügbar.[298] VON EULER et al. (1925, 1926) gingen in ihren Arbeiten
davon aus, dass das pH-Minimum der Mutarotation der D-Glucose 2a an ihrem 𝑝𝐼 liegt. Dies wäre
genau dann der Fall, wenn anionische und kationische Zuckerspezies dieselbe spontane Tendenz zur
Ringöffnung zeigen würden. Die Kenntnis des 𝑝𝐾𝑠-Wertes der anomeren Hydroxylfunktion würde über
die Messung des pH-Minimums der Mutarotationsrate somit die Berechnung des 𝑝𝐾𝑠-Wertes des
Ringsauerstoffatoms erlauben.[287,288] Ob die Prämisse identischer spontaner Ringöffnungsraten dabei
erfüllt ist, muss jedoch infrage gestellt werden. Neben theoretisch berechneten 𝑝𝐾𝑠-Werten liefern
FENG et al. (2013) allerdings auch theoretisch berechnete Protonenaffinitäten. Es zeigt sich dabei, dass
die Protonenaffinität der Ringsauerstoffatome der Anomere der D-Fructose 1a (D-Glucose 2a) in etwa
derjenigen der Hydroxylfunktion von tert-Butanol (Ethanol) entspricht. Frühere Berechnungen von
WOODS et al. (1991) liefern vergleichbare Ergebnisse.[342] Da 𝑝𝐾𝑠-Werte eng mit Protonenaffinitäten
verknüpft sind, wurde im Folgenden mit den 𝑝𝐾𝑠-Werten der genannten Alkohole gerechnet. Diese
liegen für tert-Butanol bei -2.6 und für Ethanol bei -1.94.[343,344] Für β-D-Fructopyranose 1a(ii) liegt der
𝑝𝐾𝑠-Wert der anomeren Hydroxylfunktion bei rund 12.0[298] und das Minimum der Mutarotationsrate
im pH-Bereich zwischen 2.5 und 6.5.[67] Geht man von einer Äquivalenz dieses pH-Intervalls und des 𝑝𝐼
der β-D-Fructopyranose 1a(ii) aus, so würde sich ein 𝑝𝐾𝑠-Wert im Bereich von -7.0 bis 1.0 für den
Ringsauerstoff ergeben. Der 𝑝𝐾𝑠-Wert liegt damit in einem ähnlichen Bereich wie der von tert-Butanol.
Damit ist die spontane Ringöffnungstendenz von anionischen und kationischen Zuckerspezies zwar
nicht zwangsläufig identisch, jedoch in einem ähnlichen Größenbereich.
Für die untersuchten AMADORI-Verbindungen 6a und 6b sowie für D-Tagatose 1b sind keine
Literaturwerte bezüglich der Säurekonstanten der Zuckergrundkörper verfügbar. Für die
Berechnungen der thermodynamischen Aktivierungsparameter der Anomerisierung werden daher die
entsprechenden 𝑝𝐾𝑠-Werte von D-Fructose 1a zugrunde gelegt. Die Dissoziationskonstanten der
Carboxyl- und Aminofunktionen der Aminosäuren 4a und 4b sowie der AMADORI-Verbindungen 6a und
6b sind in den Tabellen 1 (Seite 44) und 2 (Seite 45) zusammengefasst.
Als weiteres Problem ergibt sich, dass Dissoziationskonstanten im Allgemeinen
temperaturabhängig sind.[345-347] Diese Temperaturabhängigkeit ist für die Dissoziationskonstanten der
Zuckergrundkörper unbekannt und wurde im Rahmen der hier vorliegenden Arbeit daher
vernachlässigt. Den Ausführungen dieses Abschnittes ist zu entnehmen, dass die im Rahmen dieser
Arbeit berechneten thermodynamischen Aktivierungsparameter nicht zwangsläufig den tatsächlich
korrekten Werten entsprechen müssen. Im Gegensatz zu früheren Arbeiten (vgl. Kapitel 2.3) basieren
die hier berechneten Parameter zwar auf einem Modell, das molekular-mechanistisch fundiert ist,
jedoch sind die zur Berechnung notwendigen Säurekonstanten und deren Temperaturabhängigkeit nur
unzureichend bekannt. Die in den nachfolgenden Tabellen zusammengefassten thermodynamischen
Aktivierungsparameter sind daher mutmaßlich in einigen Fällen nicht plausibel. Dies ist jedoch mit Blick
auf das verfolgte Ziel der Berechnung von ACuSTiC-Werten irrelevant, da das mathematische Modell
die Raten der Anomerisierung im untersuchten pD- und Temperaturbereich im Rahmen der
angegebenen Fehlertoleranzen exakt beschreibt. Es ist folglich möglich, ACuSTiC-Werte für
Temperaturen im Bereich zwischen 27 und 87 °C in einem pD-Intervall von 1.5 bis 6.5 zu intrapolieren.
Die Tabellen 10 und 11 zeigen die berechneten thermodynamischen Aktivierungsparameter
zur temperatur- und pD-abhängigen Beschreibung der Ringöffnungsraten der untersuchten Anomere.
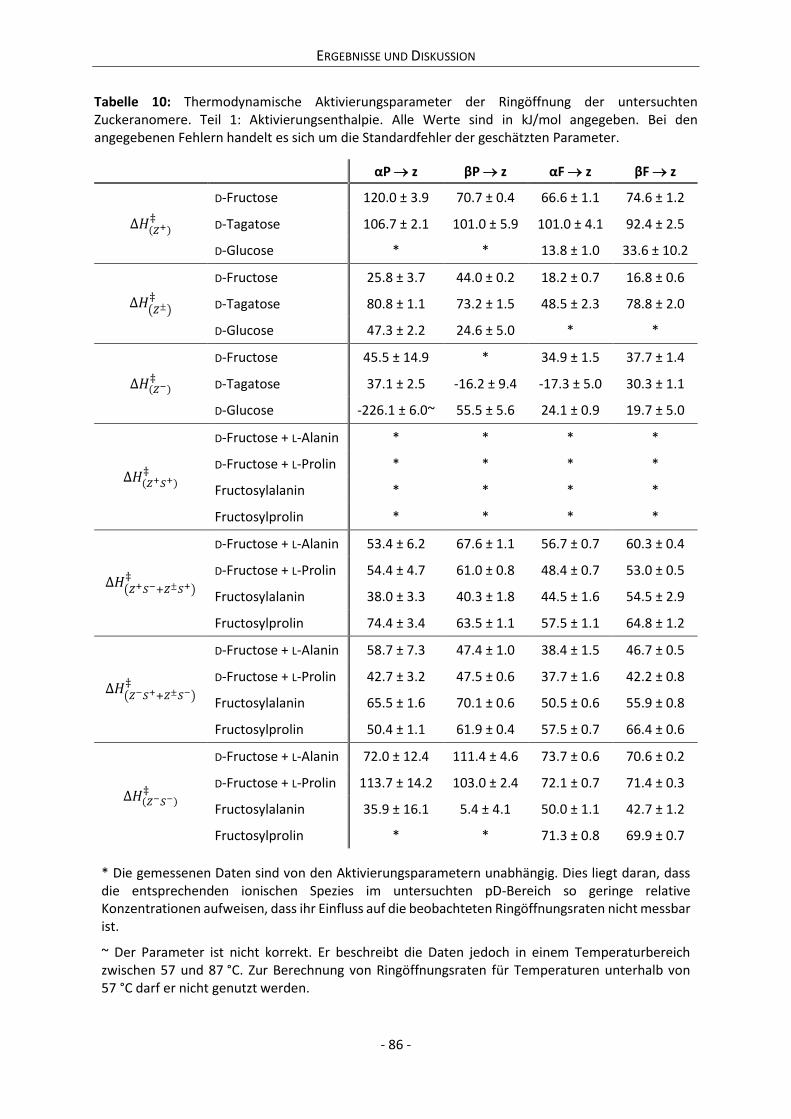
ERGEBNISSE UND DISKUSSION
- 86 -
Tabelle 10: Thermodynamische Aktivierungsparameter der Ringöffnung der untersuchten Zuckeranomere. Teil 1: Aktivierungsenthalpie. Alle Werte sind in kJ/mol angegeben. Bei den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten Parameter.
αP z βP z αF z βF z
∆𝐻(𝑍+)‡
D-Fructose 120.0 ± 3.9 70.7 ± 0.4 66.6 ± 1.1 74.6 ± 1.2
D-Tagatose 106.7 ± 2.1 101.0 ± 5.9 101.0 ± 4.1 92.4 ± 2.5
D-Glucose * * 13.8 ± 1.0 33.6 ± 10.2
∆𝐻(𝑍±)
‡
D-Fructose 25.8 ± 3.7 44.0 ± 0.2 18.2 ± 0.7 16.8 ± 0.6
D-Tagatose 80.8 ± 1.1 73.2 ± 1.5 48.5 ± 2.3 78.8 ± 2.0
D-Glucose 47.3 ± 2.2 24.6 ± 5.0 * *
∆𝐻(𝑍−)‡
D-Fructose 45.5 ± 14.9 * 34.9 ± 1.5 37.7 ± 1.4
D-Tagatose 37.1 ± 2.5 -16.2 ± 9.4 -17.3 ± 5.0 30.3 ± 1.1
D-Glucose -226.1 ± 6.0~ 55.5 ± 5.6 24.1 ± 0.9 19.7 ± 5.0
∆𝐻(𝑍+𝑆+)‡
D-Fructose + L-Alanin * * * *
D-Fructose + L-Prolin * * * *
Fructosylalanin * * * *
Fructosylprolin * * * *
∆𝐻(𝑍+𝑆−+𝑍±𝑆+)
‡
D-Fructose + L-Alanin 53.4 ± 6.2 67.6 ± 1.1 56.7 ± 0.7 60.3 ± 0.4
D-Fructose + L-Prolin 54.4 ± 4.7 61.0 ± 0.8 48.4 ± 0.7 53.0 ± 0.5
Fructosylalanin 38.0 ± 3.3 40.3 ± 1.8 44.5 ± 1.6 54.5 ± 2.9
Fructosylprolin 74.4 ± 3.4 63.5 ± 1.1 57.5 ± 1.1 64.8 ± 1.2
∆𝐻(𝑍−𝑆++𝑍±𝑆−)
‡
D-Fructose + L-Alanin 58.7 ± 7.3 47.4 ± 1.0 38.4 ± 1.5 46.7 ± 0.5
D-Fructose + L-Prolin 42.7 ± 3.2 47.5 ± 0.6 37.7 ± 1.6 42.2 ± 0.8
Fructosylalanin 65.5 ± 1.6 70.1 ± 0.6 50.5 ± 0.6 55.9 ± 0.8
Fructosylprolin 50.4 ± 1.1 61.9 ± 0.4 57.5 ± 0.7 66.4 ± 0.6
∆𝐻(𝑍−𝑆−)‡
D-Fructose + L-Alanin 72.0 ± 12.4 111.4 ± 4.6 73.7 ± 0.6 70.6 ± 0.2
D-Fructose + L-Prolin 113.7 ± 14.2 103.0 ± 2.4 72.1 ± 0.7 71.4 ± 0.3
Fructosylalanin 35.9 ± 16.1 5.4 ± 4.1 50.0 ± 1.1 42.7 ± 1.2
Fructosylprolin * * 71.3 ± 0.8 69.9 ± 0.7
* Die gemessenen Daten sind von den Aktivierungsparametern unabhängig. Dies liegt daran, dass die entsprechenden ionischen Spezies im untersuchten pD-Bereich so geringe relative Konzentrationen aufweisen, dass ihr Einfluss auf die beobachteten Ringöffnungsraten nicht messbar ist.
~ Der Parameter ist nicht korrekt. Er beschreibt die Daten jedoch in einem Temperaturbereich zwischen 57 und 87 °C. Zur Berechnung von Ringöffnungsraten für Temperaturen unterhalb von 57 °C darf er nicht genutzt werden.
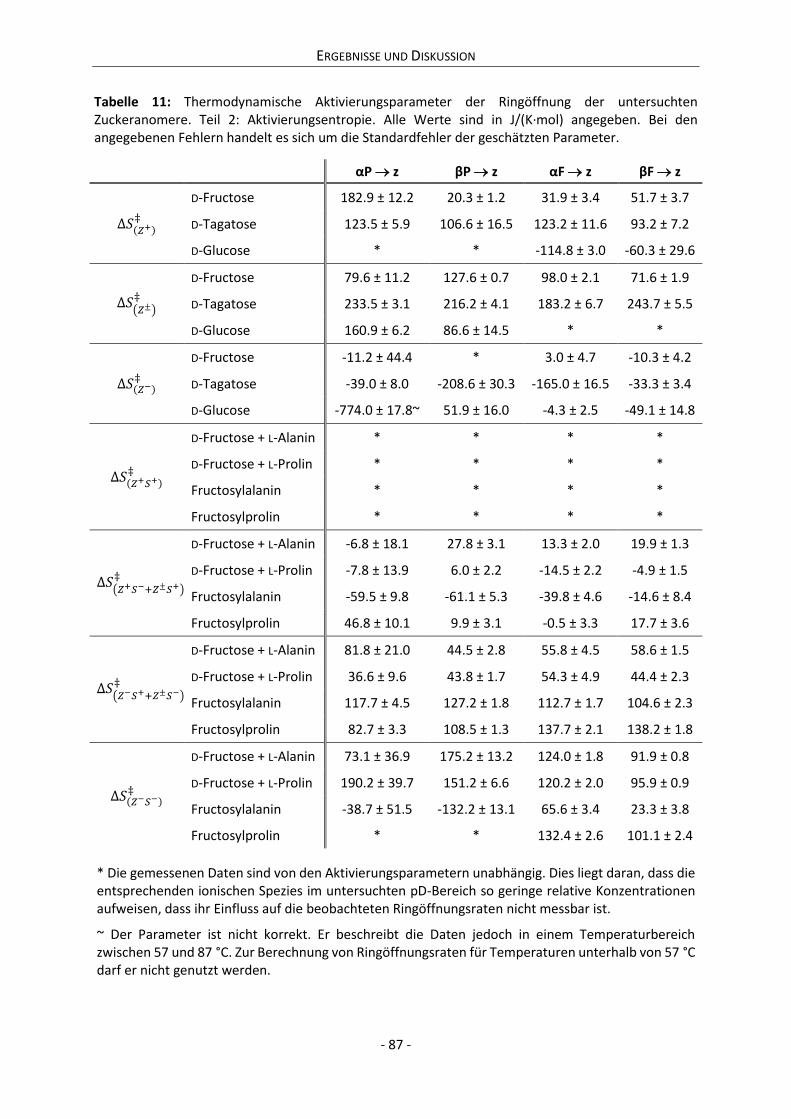
ERGEBNISSE UND DISKUSSION
- 87 -
Tabelle 11: Thermodynamische Aktivierungsparameter der Ringöffnung der untersuchten Zuckeranomere. Teil 2: Aktivierungsentropie. Alle Werte sind in J/(K·mol) angegeben. Bei den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten Parameter.
αP z βP z αF z βF z
∆𝑆(𝑍+)‡
D-Fructose 182.9 ± 12.2 20.3 ± 1.2 31.9 ± 3.4 51.7 ± 3.7
D-Tagatose 123.5 ± 5.9 106.6 ± 16.5 123.2 ± 11.6 93.2 ± 7.2
D-Glucose * * -114.8 ± 3.0 -60.3 ± 29.6
∆𝑆(𝑍±)
‡
D-Fructose 79.6 ± 11.2 127.6 ± 0.7 98.0 ± 2.1 71.6 ± 1.9
D-Tagatose 233.5 ± 3.1 216.2 ± 4.1 183.2 ± 6.7 243.7 ± 5.5
D-Glucose 160.9 ± 6.2 86.6 ± 14.5 * *
∆𝑆(𝑍−)‡
D-Fructose -11.2 ± 44.4 * 3.0 ± 4.7 -10.3 ± 4.2
D-Tagatose -39.0 ± 8.0 -208.6 ± 30.3 -165.0 ± 16.5 -33.3 ± 3.4
D-Glucose -774.0 ± 17.8~ 51.9 ± 16.0 -4.3 ± 2.5 -49.1 ± 14.8
∆𝑆(𝑍+𝑆+)‡
D-Fructose + L-Alanin * * * *
D-Fructose + L-Prolin * * * *
Fructosylalanin * * * *
Fructosylprolin * * * *
∆𝑆(𝑍+𝑆−+𝑍±𝑆+)
‡
D-Fructose + L-Alanin -6.8 ± 18.1 27.8 ± 3.1 13.3 ± 2.0 19.9 ± 1.3
D-Fructose + L-Prolin -7.8 ± 13.9 6.0 ± 2.2 -14.5 ± 2.2 -4.9 ± 1.5
Fructosylalanin -59.5 ± 9.8 -61.1 ± 5.3 -39.8 ± 4.6 -14.6 ± 8.4
Fructosylprolin 46.8 ± 10.1 9.9 ± 3.1 -0.5 ± 3.3 17.7 ± 3.6
∆𝑆(𝑍−𝑆++𝑍±𝑆−)
‡
D-Fructose + L-Alanin 81.8 ± 21.0 44.5 ± 2.8 55.8 ± 4.5 58.6 ± 1.5
D-Fructose + L-Prolin 36.6 ± 9.6 43.8 ± 1.7 54.3 ± 4.9 44.4 ± 2.3
Fructosylalanin 117.7 ± 4.5 127.2 ± 1.8 112.7 ± 1.7 104.6 ± 2.3
Fructosylprolin 82.7 ± 3.3 108.5 ± 1.3 137.7 ± 2.1 138.2 ± 1.8
∆𝑆(𝑍−𝑆−)‡
D-Fructose + L-Alanin 73.1 ± 36.9 175.2 ± 13.2 124.0 ± 1.8 91.9 ± 0.8
D-Fructose + L-Prolin 190.2 ± 39.7 151.2 ± 6.6 120.2 ± 2.0 95.9 ± 0.9
Fructosylalanin -38.7 ± 51.5 -132.2 ± 13.1 65.6 ± 3.4 23.3 ± 3.8
Fructosylprolin * * 132.4 ± 2.6 101.1 ± 2.4
* Die gemessenen Daten sind von den Aktivierungsparametern unabhängig. Dies liegt daran, dass die entsprechenden ionischen Spezies im untersuchten pD-Bereich so geringe relative Konzentrationen aufweisen, dass ihr Einfluss auf die beobachteten Ringöffnungsraten nicht messbar ist.
~ Der Parameter ist nicht korrekt. Er beschreibt die Daten jedoch in einem Temperaturbereich zwischen 57 und 87 °C. Zur Berechnung von Ringöffnungsraten für Temperaturen unterhalb von 57 °C darf er nicht genutzt werden.
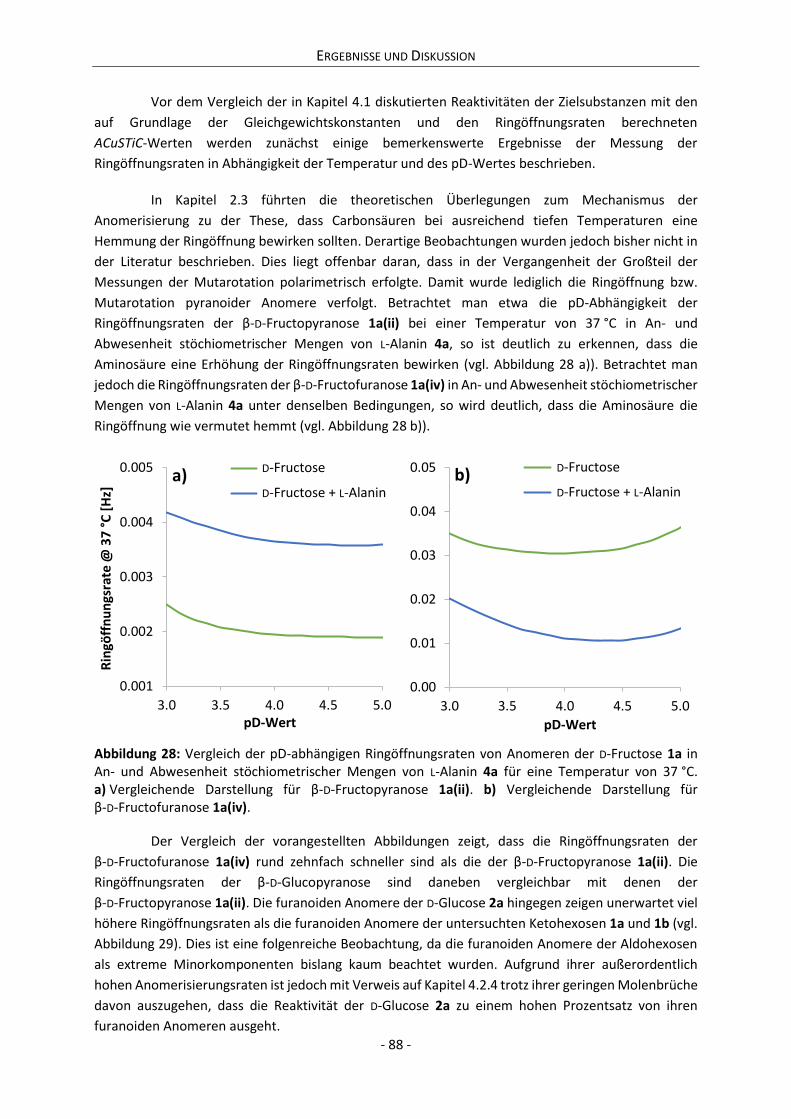
ERGEBNISSE UND DISKUSSION
- 88 -
Vor dem Vergleich der in Kapitel 4.1 diskutierten Reaktivitäten der Zielsubstanzen mit den
auf Grundlage der Gleichgewichtskonstanten und den Ringöffnungsraten berechneten
ACuSTiC-Werten werden zunächst einige bemerkenswerte Ergebnisse der Messung der
Ringöffnungsraten in Abhängigkeit der Temperatur und des pD-Wertes beschrieben.
In Kapitel 2.3 führten die theoretischen Überlegungen zum Mechanismus der
Anomerisierung zu der These, dass Carbonsäuren bei ausreichend tiefen Temperaturen eine
Hemmung der Ringöffnung bewirken sollten. Derartige Beobachtungen wurden jedoch bisher nicht in
der Literatur beschrieben. Dies liegt offenbar daran, dass in der Vergangenheit der Großteil der
Messungen der Mutarotation polarimetrisch erfolgte. Damit wurde lediglich die Ringöffnung bzw.
Mutarotation pyranoider Anomere verfolgt. Betrachtet man etwa die pD-Abhängigkeit der
Ringöffnungsraten der β-D-Fructopyranose 1a(ii) bei einer Temperatur von 37 °C in An- und
Abwesenheit stöchiometrischer Mengen von L-Alanin 4a, so ist deutlich zu erkennen, dass die
Aminosäure eine Erhöhung der Ringöffnungsraten bewirken (vgl. Abbildung 28 a)). Betrachtet man
jedoch die Ringöffnungsraten der β-D-Fructofuranose 1a(iv) in An- und Abwesenheit stöchiometrischer
Mengen von L-Alanin 4a unter denselben Bedingungen, so wird deutlich, dass die Aminosäure die
Ringöffnung wie vermutet hemmt (vgl. Abbildung 28 b)).
Abbildung 28: Vergleich der pD-abhängigen Ringöffnungsraten von Anomeren der D-Fructose 1a in An- und Abwesenheit stöchiometrischer Mengen von L-Alanin 4a für eine Temperatur von 37 °C. a) Vergleichende Darstellung für β-D-Fructopyranose 1a(ii). b) Vergleichende Darstellung für β-D-Fructofuranose 1a(iv).
Der Vergleich der vorangestellten Abbildungen zeigt, dass die Ringöffnungsraten der
β-D-Fructofuranose 1a(iv) rund zehnfach schneller sind als die der β-D-Fructopyranose 1a(ii). Die
Ringöffnungsraten der β-D-Glucopyranose sind daneben vergleichbar mit denen der
β-D-Fructopyranose 1a(ii). Die furanoiden Anomere der D-Glucose 2a hingegen zeigen unerwartet viel
höhere Ringöffnungsraten als die furanoiden Anomere der untersuchten Ketohexosen 1a und 1b (vgl.
Abbildung 29). Dies ist eine folgenreiche Beobachtung, da die furanoiden Anomere der Aldohexosen
als extreme Minorkomponenten bislang kaum beachtet wurden. Aufgrund ihrer außerordentlich
hohen Anomerisierungsraten ist jedoch mit Verweis auf Kapitel 4.2.4 trotz ihrer geringen Molenbrüche
davon auszugehen, dass die Reaktivität der D-Glucose 2a zu einem hohen Prozentsatz von ihren
furanoiden Anomeren ausgeht.
0.001
0.002
0.003
0.004
0.005
3.0 3.5 4.0 4.5 5.0
Rin
göff
nu
ngs
rate
@ 3
7 °
C [
Hz]
pD-Wert
D-Fructose
D-Fructose + L-Alanina)
0.00
0.01
0.02
0.03
0.04
0.05
3.0 3.5 4.0 4.5 5.0
pD-Wert
D-Fructose
D-Fructose + L-Alaninb)D-Fructose
D-Fructose + L-Alanin
D-Fructose
D-Fructose + L-Alanin
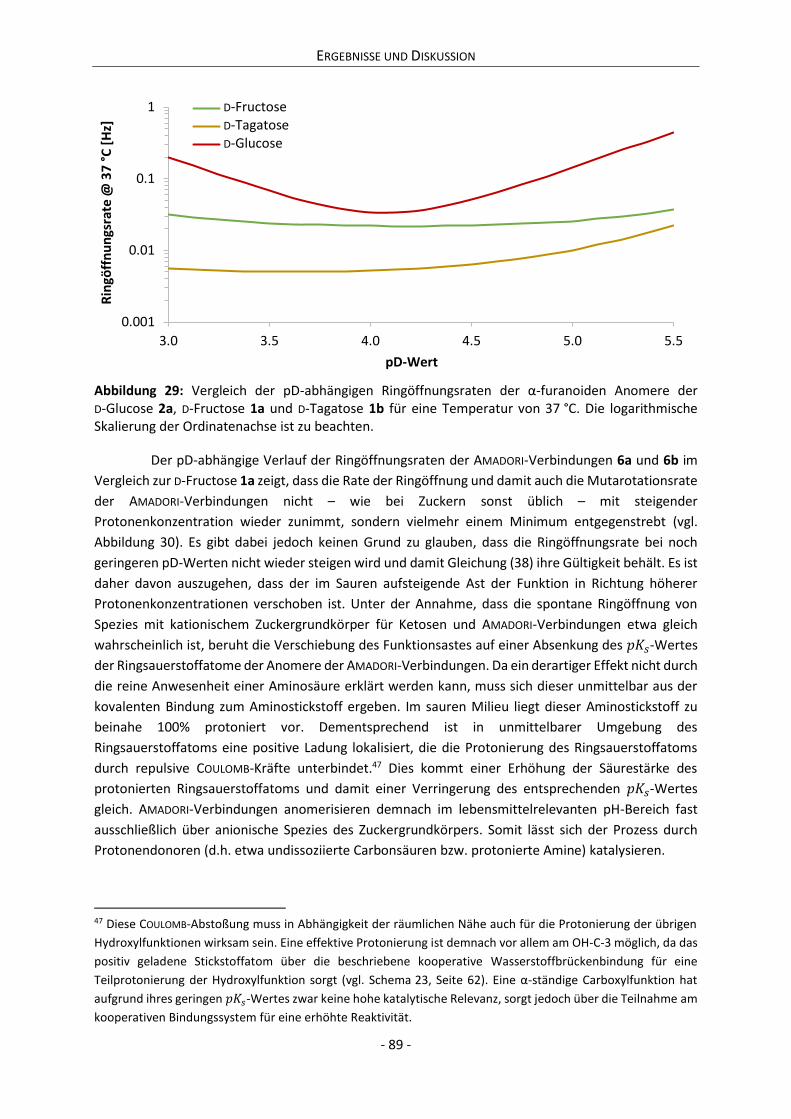
ERGEBNISSE UND DISKUSSION
- 89 -
Abbildung 29: Vergleich der pD-abhängigen Ringöffnungsraten der α-furanoiden Anomere der D-Glucose 2a, D-Fructose 1a und D-Tagatose 1b für eine Temperatur von 37 °C. Die logarithmische Skalierung der Ordinatenachse ist zu beachten.
Der pD-abhängige Verlauf der Ringöffnungsraten der AMADORI-Verbindungen 6a und 6b im
Vergleich zur D-Fructose 1a zeigt, dass die Rate der Ringöffnung und damit auch die Mutarotationsrate
der AMADORI-Verbindungen nicht – wie bei Zuckern sonst üblich – mit steigender
Protonenkonzentration wieder zunimmt, sondern vielmehr einem Minimum entgegenstrebt (vgl.
Abbildung 30). Es gibt dabei jedoch keinen Grund zu glauben, dass die Ringöffnungsrate bei noch
geringeren pD-Werten nicht wieder steigen wird und damit Gleichung (38) ihre Gültigkeit behält. Es ist
daher davon auszugehen, dass der im Sauren aufsteigende Ast der Funktion in Richtung höherer
Protonenkonzentrationen verschoben ist. Unter der Annahme, dass die spontane Ringöffnung von
Spezies mit kationischem Zuckergrundkörper für Ketosen und AMADORI-Verbindungen etwa gleich
wahrscheinlich ist, beruht die Verschiebung des Funktionsastes auf einer Absenkung des 𝑝𝐾𝑠-Wertes
der Ringsauerstoffatome der Anomere der AMADORI-Verbindungen. Da ein derartiger Effekt nicht durch
die reine Anwesenheit einer Aminosäure erklärt werden kann, muss sich dieser unmittelbar aus der
kovalenten Bindung zum Aminostickstoff ergeben. Im sauren Milieu liegt dieser Aminostickstoff zu
beinahe 100% protoniert vor. Dementsprechend ist in unmittelbarer Umgebung des
Ringsauerstoffatoms eine positive Ladung lokalisiert, die die Protonierung des Ringsauerstoffatoms
durch repulsive COULOMB-Kräfte unterbindet.47 Dies kommt einer Erhöhung der Säurestärke des
protonierten Ringsauerstoffatoms und damit einer Verringerung des entsprechenden 𝑝𝐾𝑠-Wertes
gleich. AMADORI-Verbindungen anomerisieren demnach im lebensmittelrelevanten pH-Bereich fast
ausschließlich über anionische Spezies des Zuckergrundkörpers. Somit lässt sich der Prozess durch
Protonendonoren (d.h. etwa undissoziierte Carbonsäuren bzw. protonierte Amine) katalysieren.
47 Diese COULOMB-Abstoßung muss in Abhängigkeit der räumlichen Nähe auch für die Protonierung der übrigen
Hydroxylfunktionen wirksam sein. Eine effektive Protonierung ist demnach vor allem am OH-C-3 möglich, da das
positiv geladene Stickstoffatom über die beschriebene kooperative Wasserstoffbrückenbindung für eine
Teilprotonierung der Hydroxylfunktion sorgt (vgl. Schema 23, Seite 62). Eine α-ständige Carboxylfunktion hat
aufgrund ihres geringen 𝑝𝐾𝑠-Wertes zwar keine hohe katalytische Relevanz, sorgt jedoch über die Teilnahme am
kooperativen Bindungssystem für eine erhöhte Reaktivität.
0.001
0.01
0.1
1
3.0 3.5 4.0 4.5 5.0 5.5
Rin
göff
nu
ngs
rate
@ 3
7 °
C [
Hz]
pD-Wert
D-FructoseD-TagatoseD-Glucose
D-Fructose D-Tagatose D-Glucose
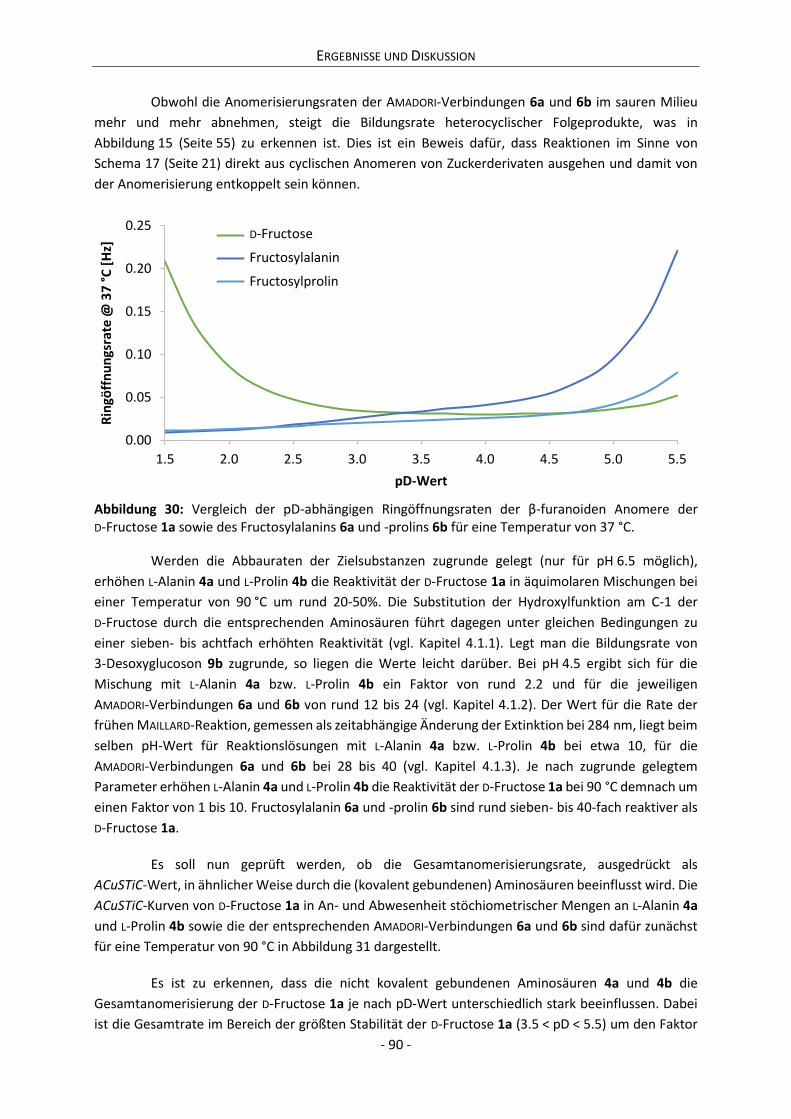
ERGEBNISSE UND DISKUSSION
- 90 -
Obwohl die Anomerisierungsraten der AMADORI-Verbindungen 6a und 6b im sauren Milieu
mehr und mehr abnehmen, steigt die Bildungsrate heterocyclischer Folgeprodukte, was in
Abbildung 15 (Seite 55) zu erkennen ist. Dies ist ein Beweis dafür, dass Reaktionen im Sinne von
Schema 17 (Seite 21) direkt aus cyclischen Anomeren von Zuckerderivaten ausgehen und damit von
der Anomerisierung entkoppelt sein können.
Abbildung 30: Vergleich der pD-abhängigen Ringöffnungsraten der β-furanoiden Anomere der D-Fructose 1a sowie des Fructosylalanins 6a und -prolins 6b für eine Temperatur von 37 °C.
Werden die Abbauraten der Zielsubstanzen zugrunde gelegt (nur für pH 6.5 möglich),
erhöhen L-Alanin 4a und L-Prolin 4b die Reaktivität der D-Fructose 1a in äquimolaren Mischungen bei
einer Temperatur von 90 °C um rund 20-50%. Die Substitution der Hydroxylfunktion am C-1 der
D-Fructose durch die entsprechenden Aminosäuren führt dagegen unter gleichen Bedingungen zu
einer sieben- bis achtfach erhöhten Reaktivität (vgl. Kapitel 4.1.1). Legt man die Bildungsrate von
3-Desoxyglucoson 9b zugrunde, so liegen die Werte leicht darüber. Bei pH 4.5 ergibt sich für die
Mischung mit L-Alanin 4a bzw. L-Prolin 4b ein Faktor von rund 2.2 und für die jeweiligen
AMADORI-Verbindungen 6a und 6b von rund 12 bis 24 (vgl. Kapitel 4.1.2). Der Wert für die Rate der
frühen MAILLARD-Reaktion, gemessen als zeitabhängige Änderung der Extinktion bei 284 nm, liegt beim
selben pH-Wert für Reaktionslösungen mit L-Alanin 4a bzw. L-Prolin 4b bei etwa 10, für die
AMADORI-Verbindungen 6a und 6b bei 28 bis 40 (vgl. Kapitel 4.1.3). Je nach zugrunde gelegtem
Parameter erhöhen L-Alanin 4a und L-Prolin 4b die Reaktivität der D-Fructose 1a bei 90 °C demnach um
einen Faktor von 1 bis 10. Fructosylalanin 6a und -prolin 6b sind rund sieben- bis 40-fach reaktiver als
D-Fructose 1a.
Es soll nun geprüft werden, ob die Gesamtanomerisierungsrate, ausgedrückt als
ACuSTiC-Wert, in ähnlicher Weise durch die (kovalent gebundenen) Aminosäuren beeinflusst wird. Die
ACuSTiC-Kurven von D-Fructose 1a in An- und Abwesenheit stöchiometrischer Mengen an L-Alanin 4a
und L-Prolin 4b sowie die der entsprechenden AMADORI-Verbindungen 6a und 6b sind dafür zunächst
für eine Temperatur von 90 °C in Abbildung 31 dargestellt.
Es ist zu erkennen, dass die nicht kovalent gebundenen Aminosäuren 4a und 4b die
Gesamtanomerisierung der D-Fructose 1a je nach pD-Wert unterschiedlich stark beeinflussen. Dabei
ist die Gesamtrate im Bereich der größten Stabilität der D-Fructose 1a (3.5 < pD < 5.5) um den Faktor
0.00
0.05
0.10
0.15
0.20
0.25
1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5
Rin
göff
nu
ngs
rate
@ 3
7 °
C [
Hz]
pD-Wert
D-Fructose
Fructosylalanin
Fructosylprolin
D-Fructose
Fructosylalanin
Fructosylprolin
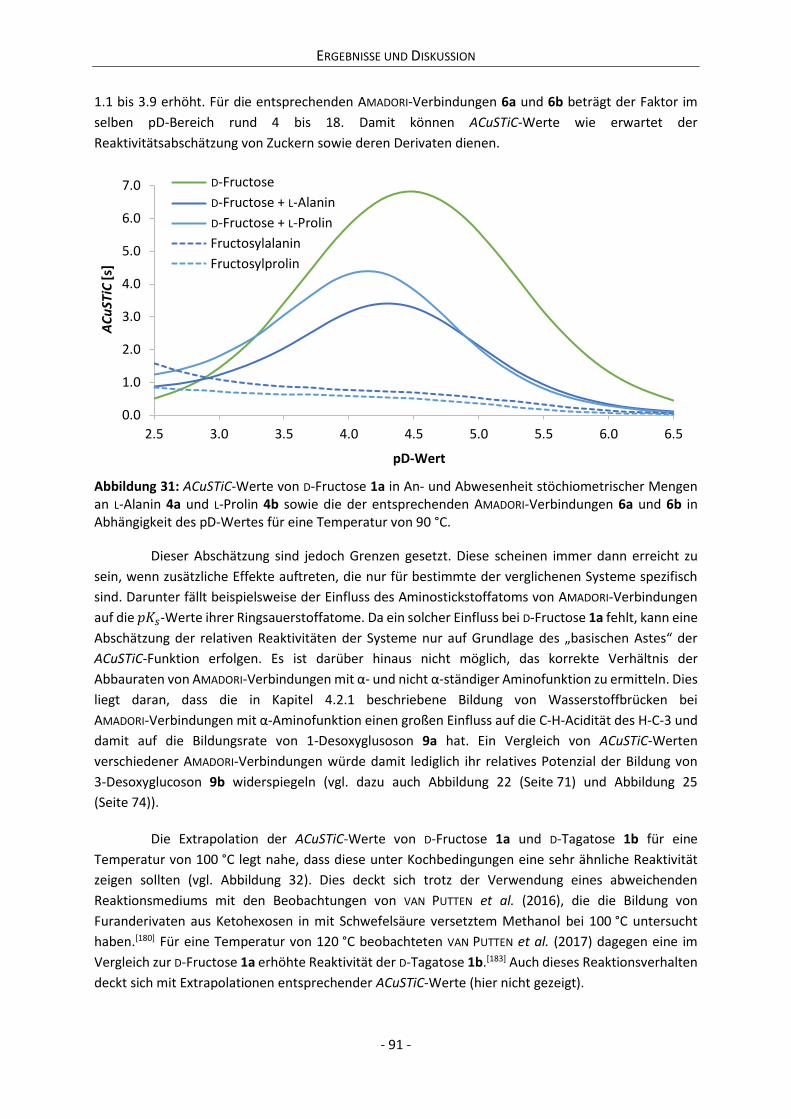
ERGEBNISSE UND DISKUSSION
- 91 -
1.1 bis 3.9 erhöht. Für die entsprechenden AMADORI-Verbindungen 6a und 6b beträgt der Faktor im
selben pD-Bereich rund 4 bis 18. Damit können ACuSTiC-Werte wie erwartet der
Reaktivitätsabschätzung von Zuckern sowie deren Derivaten dienen.
Abbildung 31: ACuSTiC-Werte von D-Fructose 1a in An- und Abwesenheit stöchiometrischer Mengen an L-Alanin 4a und L-Prolin 4b sowie die der entsprechenden AMADORI-Verbindungen 6a und 6b in Abhängigkeit des pD-Wertes für eine Temperatur von 90 °C.
Dieser Abschätzung sind jedoch Grenzen gesetzt. Diese scheinen immer dann erreicht zu
sein, wenn zusätzliche Effekte auftreten, die nur für bestimmte der verglichenen Systeme spezifisch
sind. Darunter fällt beispielsweise der Einfluss des Aminostickstoffatoms von AMADORI-Verbindungen
auf die 𝑝𝐾𝑠-Werte ihrer Ringsauerstoffatome. Da ein solcher Einfluss bei D-Fructose 1a fehlt, kann eine
Abschätzung der relativen Reaktivitäten der Systeme nur auf Grundlage des „basischen Astes“ der
ACuSTiC-Funktion erfolgen. Es ist darüber hinaus nicht möglich, das korrekte Verhältnis der
Abbauraten von AMADORI-Verbindungen mit α- und nicht α-ständiger Aminofunktion zu ermitteln. Dies
liegt daran, dass die in Kapitel 4.2.1 beschriebene Bildung von Wasserstoffbrücken bei
AMADORI-Verbindungen mit α-Aminofunktion einen großen Einfluss auf die C-H-Acidität des H-C-3 und
damit auf die Bildungsrate von 1-Desoxyglusoson 9a hat. Ein Vergleich von ACuSTiC-Werten
verschiedener AMADORI-Verbindungen würde damit lediglich ihr relatives Potenzial der Bildung von
3-Desoxyglucoson 9b widerspiegeln (vgl. dazu auch Abbildung 22 (Seite 71) und Abbildung 25
(Seite 74)).
Die Extrapolation der ACuSTiC-Werte von D-Fructose 1a und D-Tagatose 1b für eine
Temperatur von 100 °C legt nahe, dass diese unter Kochbedingungen eine sehr ähnliche Reaktivität
zeigen sollten (vgl. Abbildung 32). Dies deckt sich trotz der Verwendung eines abweichenden
Reaktionsmediums mit den Beobachtungen von VAN PUTTEN et al. (2016), die die Bildung von
Furanderivaten aus Ketohexosen in mit Schwefelsäure versetztem Methanol bei 100 °C untersucht
haben.[180] Für eine Temperatur von 120 °C beobachteten VAN PUTTEN et al. (2017) dagegen eine im
Vergleich zur D-Fructose 1a erhöhte Reaktivität der D-Tagatose 1b.[183] Auch dieses Reaktionsverhalten
deckt sich mit Extrapolationen entsprechender ACuSTiC-Werte (hier nicht gezeigt).
0.0
1.0
2.0
3.0
4.0
5.0
6.0
7.0
2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5
AC
uST
iC[s
]
pD-Wert
D-Fructose
D-Fructose + L-Alanin
D-Fructose + L-Prolin
Fructosylalanin
Fructosylprolin
D-Fructose
D-Fructose + L-Alanin
D-Fructose + L-Prolin
Fructosylalanin
Fructosylprolin
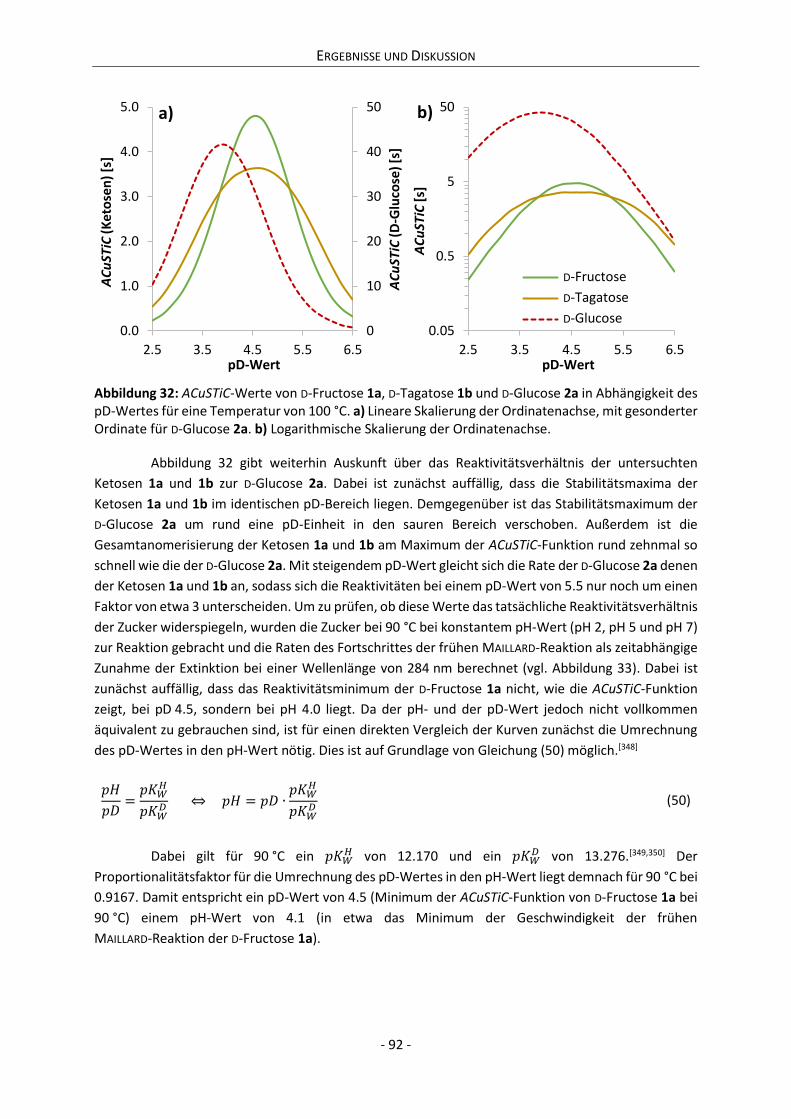
ERGEBNISSE UND DISKUSSION
- 92 -
Abbildung 32: ACuSTiC-Werte von D-Fructose 1a, D-Tagatose 1b und D-Glucose 2a in Abhängigkeit des pD-Wertes für eine Temperatur von 100 °C. a) Lineare Skalierung der Ordinatenachse, mit gesonderter Ordinate für D-Glucose 2a. b) Logarithmische Skalierung der Ordinatenachse.
Abbildung 32 gibt weiterhin Auskunft über das Reaktivitätsverhältnis der untersuchten
Ketosen 1a und 1b zur D-Glucose 2a. Dabei ist zunächst auffällig, dass die Stabilitätsmaxima der
Ketosen 1a und 1b im identischen pD-Bereich liegen. Demgegenüber ist das Stabilitätsmaximum der
D-Glucose 2a um rund eine pD-Einheit in den sauren Bereich verschoben. Außerdem ist die
Gesamtanomerisierung der Ketosen 1a und 1b am Maximum der ACuSTiC-Funktion rund zehnmal so
schnell wie die der D-Glucose 2a. Mit steigendem pD-Wert gleicht sich die Rate der D-Glucose 2a denen
der Ketosen 1a und 1b an, sodass sich die Reaktivitäten bei einem pD-Wert von 5.5 nur noch um einen
Faktor von etwa 3 unterscheiden. Um zu prüfen, ob diese Werte das tatsächliche Reaktivitätsverhältnis
der Zucker widerspiegeln, wurden die Zucker bei 90 °C bei konstantem pH-Wert (pH 2, pH 5 und pH 7)
zur Reaktion gebracht und die Raten des Fortschrittes der frühen MAILLARD-Reaktion als zeitabhängige
Zunahme der Extinktion bei einer Wellenlänge von 284 nm berechnet (vgl. Abbildung 33). Dabei ist
zunächst auffällig, dass das Reaktivitätsminimum der D-Fructose 1a nicht, wie die ACuSTiC-Funktion
zeigt, bei pD 4.5, sondern bei pH 4.0 liegt. Da der pH- und der pD-Wert jedoch nicht vollkommen
äquivalent zu gebrauchen sind, ist für einen direkten Vergleich der Kurven zunächst die Umrechnung
des pD-Wertes in den pH-Wert nötig. Dies ist auf Grundlage von Gleichung (50) möglich.[348]
𝑝𝐻
𝑝𝐷=𝑝𝐾𝑊
𝐻
𝑝𝐾𝑊𝐷 ⇔ 𝑝𝐻 = 𝑝𝐷 ∙
𝑝𝐾𝑊𝐻
𝑝𝐾𝑊𝐷 (50)
Dabei gilt für 90 °C ein 𝑝𝐾𝑊𝐻 von 12.170 und ein 𝑝𝐾𝑊
𝐷 von 13.276.[349,350] Der
Proportionalitätsfaktor für die Umrechnung des pD-Wertes in den pH-Wert liegt demnach für 90 °C bei
0.9167. Damit entspricht ein pD-Wert von 4.5 (Minimum der ACuSTiC-Funktion von D-Fructose 1a bei
90 °C) einem pH-Wert von 4.1 (in etwa das Minimum der Geschwindigkeit der frühen
MAILLARD-Reaktion der D-Fructose 1a).
0
10
20
30
40
50
0.0
1.0
2.0
3.0
4.0
5.0
2.5 3.5 4.5 5.5 6.5
AC
uST
iC(D
-Glu
cose
) [s
]
AC
uST
iC(K
eto
sen
) [s
]
pD-Wert
a)
0.05
0.5
5
50
2.5 3.5 4.5 5.5 6.5
AC
uST
iC[s
]
pD-Wert
D-Fructose
D-Tagatose
D-Glucose
b)
D-Fructose
D-Tagatose
D-Glucose
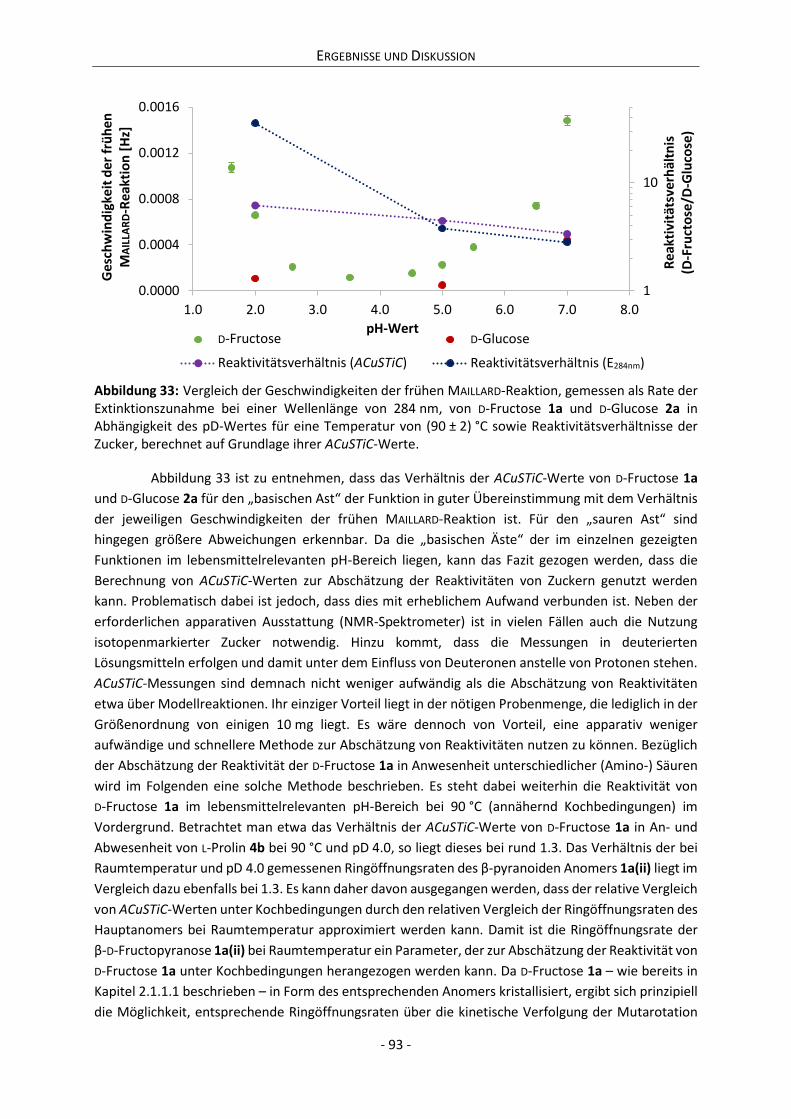
ERGEBNISSE UND DISKUSSION
- 93 -
Abbildung 33: Vergleich der Geschwindigkeiten der frühen MAILLARD-Reaktion, gemessen als Rate der Extinktionszunahme bei einer Wellenlänge von 284 nm, von D-Fructose 1a und D-Glucose 2a in Abhängigkeit des pD-Wertes für eine Temperatur von (90 ± 2) °C sowie Reaktivitätsverhältnisse der Zucker, berechnet auf Grundlage ihrer ACuSTiC-Werte.
Abbildung 33 ist zu entnehmen, dass das Verhältnis der ACuSTiC-Werte von D-Fructose 1a
und D-Glucose 2a für den „basischen Ast“ der Funktion in guter Übereinstimmung mit dem Verhältnis
der jeweiligen Geschwindigkeiten der frühen MAILLARD-Reaktion ist. Für den „sauren Ast“ sind
hingegen größere Abweichungen erkennbar. Da die „basischen Äste“ der im einzelnen gezeigten
Funktionen im lebensmittelrelevanten pH-Bereich liegen, kann das Fazit gezogen werden, dass die
Berechnung von ACuSTiC-Werten zur Abschätzung der Reaktivitäten von Zuckern genutzt werden
kann. Problematisch dabei ist jedoch, dass dies mit erheblichem Aufwand verbunden ist. Neben der
erforderlichen apparativen Ausstattung (NMR-Spektrometer) ist in vielen Fällen auch die Nutzung
isotopenmarkierter Zucker notwendig. Hinzu kommt, dass die Messungen in deuterierten
Lösungsmitteln erfolgen und damit unter dem Einfluss von Deuteronen anstelle von Protonen stehen.
ACuSTiC-Messungen sind demnach nicht weniger aufwändig als die Abschätzung von Reaktivitäten
etwa über Modellreaktionen. Ihr einziger Vorteil liegt in der nötigen Probenmenge, die lediglich in der
Größenordnung von einigen 10 mg liegt. Es wäre dennoch von Vorteil, eine apparativ weniger
aufwändige und schnellere Methode zur Abschätzung von Reaktivitäten nutzen zu können. Bezüglich
der Abschätzung der Reaktivität der D-Fructose 1a in Anwesenheit unterschiedlicher (Amino-) Säuren
wird im Folgenden eine solche Methode beschrieben. Es steht dabei weiterhin die Reaktivität von
D-Fructose 1a im lebensmittelrelevanten pH-Bereich bei 90 °C (annähernd Kochbedingungen) im
Vordergrund. Betrachtet man etwa das Verhältnis der ACuSTiC-Werte von D-Fructose 1a in An- und
Abwesenheit von L-Prolin 4b bei 90 °C und pD 4.0, so liegt dieses bei rund 1.3. Das Verhältnis der bei
Raumtemperatur und pD 4.0 gemessenen Ringöffnungsraten des β-pyranoiden Anomers 1a(ii) liegt im
Vergleich dazu ebenfalls bei 1.3. Es kann daher davon ausgegangen werden, dass der relative Vergleich
von ACuSTiC-Werten unter Kochbedingungen durch den relativen Vergleich der Ringöffnungsraten des
Hauptanomers bei Raumtemperatur approximiert werden kann. Damit ist die Ringöffnungsrate der
β-D-Fructopyranose 1a(ii) bei Raumtemperatur ein Parameter, der zur Abschätzung der Reaktivität von
D-Fructose 1a unter Kochbedingungen herangezogen werden kann. Da D-Fructose 1a – wie bereits in
Kapitel 2.1.1.1 beschrieben – in Form des entsprechenden Anomers kristallisiert, ergibt sich prinzipiell
die Möglichkeit, entsprechende Ringöffnungsraten über die kinetische Verfolgung der Mutarotation
1
10
0.0000
0.0004
0.0008
0.0012
0.0016
1.0 2.0 3.0 4.0 5.0 6.0 7.0 8.0
Re
akti
vitä
tsve
rhäl
tnis
(D-F
ruct
ose
/D-G
luco
se)
Ge
sch
win
dig
keit
de
r fr
üh
en
M
AIL
LAR
D-R
eak
tio
n [
Hz]
pH-WertD-Fructose D-Glucose
Reaktivitätsverhältnis (E284) Reaktivitätsverhältnis (ACuSTiC)
D-Fructose
Reaktivitätsverhältnis (ACuSTiC)
D-Glucose
Reaktivitätsverhältnis (E284nm)
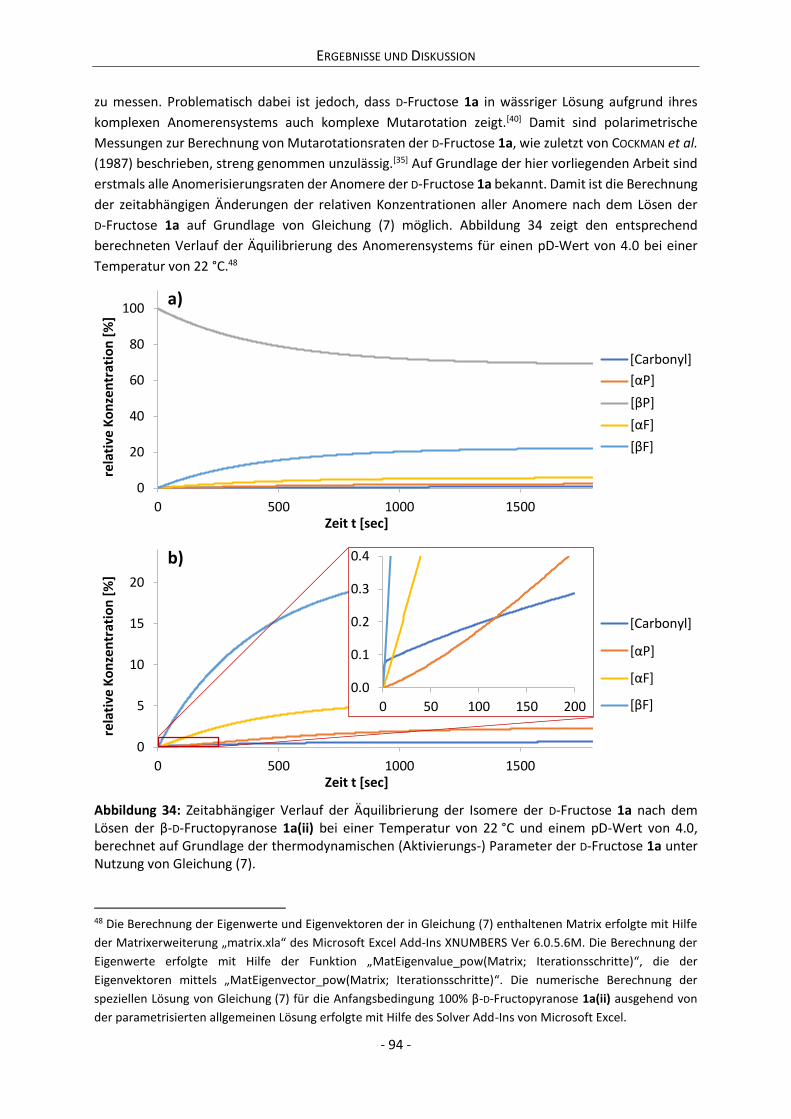
ERGEBNISSE UND DISKUSSION
- 94 -
zu messen. Problematisch dabei ist jedoch, dass D-Fructose 1a in wässriger Lösung aufgrund ihres
komplexen Anomerensystems auch komplexe Mutarotation zeigt.[40] Damit sind polarimetrische
Messungen zur Berechnung von Mutarotationsraten der D-Fructose 1a, wie zuletzt von COCKMAN et al.
(1987) beschrieben, streng genommen unzulässig.[35] Auf Grundlage der hier vorliegenden Arbeit sind
erstmals alle Anomerisierungsraten der Anomere der D-Fructose 1a bekannt. Damit ist die Berechnung
der zeitabhängigen Änderungen der relativen Konzentrationen aller Anomere nach dem Lösen der
D-Fructose 1a auf Grundlage von Gleichung (7) möglich. Abbildung 34 zeigt den entsprechend
berechneten Verlauf der Äquilibrierung des Anomerensystems für einen pD-Wert von 4.0 bei einer
Temperatur von 22 °C.48
Abbildung 34: Zeitabhängiger Verlauf der Äquilibrierung der Isomere der D-Fructose 1a nach dem Lösen der β-D-Fructopyranose 1a(ii) bei einer Temperatur von 22 °C und einem pD-Wert von 4.0, berechnet auf Grundlage der thermodynamischen (Aktivierungs-) Parameter der D-Fructose 1a unter Nutzung von Gleichung (7).
48 Die Berechnung der Eigenwerte und Eigenvektoren der in Gleichung (7) enthaltenen Matrix erfolgte mit Hilfe
der Matrixerweiterung „matrix.xla“ des Microsoft Excel Add-Ins XNUMBERS Ver 6.0.5.6M. Die Berechnung der
Eigenwerte erfolgte mit Hilfe der Funktion „MatEigenvalue_pow(Matrix; Iterationsschritte)“, die der
Eigenvektoren mittels „MatEigenvector_pow(Matrix; Iterationsschritte)“. Die numerische Berechnung der
speziellen Lösung von Gleichung (7) für die Anfangsbedingung 100% β-D-Fructopyranose 1a(ii) ausgehend von
der parametrisierten allgemeinen Lösung erfolgte mit Hilfe des Solver Add-Ins von Microsoft Excel.
0
20
40
60
80
100
0 500 1000 1500
rela
tive
Ko
nze
ntr
atio
n [
%]
Zeit t [sec]
[Carbonyl]
[aPy]
[bPy]
[aFu]
[bFu]
a)
0
5
10
15
20
0 500 1000 1500
rela
tive
Ko
nze
ntr
atio
n [
%]
Zeit t [sec]
[Carbonyl]
[aPy]
[aFu]
[bFu]
b)
[αP]
[αF]
[βF]0.0
0.1
0.2
0.3
0.4
0 50 100 150 200
[αP]
[βP]
[αF]
[βF]

ERGEBNISSE UND DISKUSSION
- 95 -
Eine genauere Analyse der Zeitabhängigkeiten der einzelnen Konzentrationen der
β-D-Fructopyranose 1a(ii) sowie der α- und β-D-Fructofuranose 1a(iii) und 1a(iv) zeigt, dass sich deren
Verlauf mit Kinetikmodellen erster Ordnung anpassen lässt. Hinzu kommt, dass das Verhältnis von
α- zu β-D-Fructofuranose nahezu zeitlich unabhängig konstant ist. Damit ist die Auswertung
polarimetrischer Daten lediglich durch das acyclische Zentralisomer 1a(v) sowie das α-pyranoide
Anomer der D-Fructose 1a(i) verfälscht. Dies zeigt sich insbesondere anhand des zeitlichen
Konzentrationsverlaufes des acyclischen Zentralisomers 1a(v), dessen Konzentration zunächst
sprunghaft ansteigt, um anschließend einer Kinetik erster Ordnung zu folgen. Dies hat zur Folge, dass
eine Extrapolation der relativen Konzentration des acyclischen Zentralisomers zum Zeitpunkt des
Lösens unter Annahme einer einfachen Kinetik erster Ordnung zu dem fälschlichen Schluss führt (vgl.
etwa MAIER et al. (1977)[351]), dass dieses zu einem geringen Prozentsatz bereits im kristallinen Zustand
vorgelegen haben muss.
Das acyclische Zentralisomer 1a(v) sowie das α-pyranoide Anomer der D-Fructose 1a(i)
machen lediglich drei Prozent der in Lösung befindlichen D-Fructose 1a aus. Es ist daher anzunehmen,
dass der Fehler, der bei der Auswertung entsprechender polarimetrischer Daten nach einem
Kinetikmodell erster Ordnung eingegangen wird, in etwa in derselben Größenordnung liegt. Um zu
prüfen, ob eine Auswertung polarimetrischer Daten also tatsächlich zulässig ist, wurde im ersten
Schritt die spezifische Drehung der β-D-Fructopyranose 1a(ii) bestimmt. Bei einer Wellenlänge von
589 nm und einer Temperatur von (22.7 ± 1.0) °C beträgt die spezifische Rotation der
β-D-Fructopyranose 1a(ii) 𝛼𝐷22(𝛽𝑃) = (−130.77 ± 0.52)
°∙𝑚𝐿
𝑑𝑚∙𝑔, was in guter Übereinstimmung mit
Literaturwerten ist.[40,67] Die Multiplikation dieses Wertes mit der relativen Konzentration der
β-D-Fructopyranose 1a(ii) zum Zeitpunkt 𝑡 liefert den Anteil der optischen Rotation des β-pyranoiden
Anomers 1a(ii) an der Gesamtrotation der D-Fructose 1a. Die Differenz der Gesamtrotation und des
Anteils des β-pyranoiden Anomers 1a(ii) ergibt folglich das konzentrationsgewichtete Mittel der
optischen Rotationen aller übrigen Isomere. Um eine entsprechende Differenz bilden zu können,
wurde im zweiten Schritt die Mutarotationskurve der D-Fructose 1a bei (22 ± 1) °C und pH 3.7
(entspricht pD 4.0 bei 22 °C) gemessen. Im dritten Schritt muss ein passendes Kinetikmodell erstellt
werden. Hierbei wird davon ausgegangen, dass das β-pyranoide Anomer 1a(ii) im Sinne von Schema 28
mit einem virtuellen Anomer (vA), bei dem es sich um das konzentrationsgewichtete Mittel der übrigen
Isomere handelt, im dynamischen Gleichgewicht erster Ordnung steht.
Schema 28: Vereinfachtes Modell der Mutarotation der D-Fructose 1a zur Beschreibung polarimetrischer Daten.
Die zugehörigen integrierten Zeitgesetze sind durch die Gleichungen (51) und (52) gegeben.
[𝛽𝑃]𝑡 = 100% ∙(𝑘𝑣𝐴,𝛽𝑃 + 𝑘𝛽𝑃,𝑣𝐴 ∙ 𝑒𝑥𝑝(−(𝑘𝛽𝑃,𝑣𝐴 + 𝑘𝑣𝐴,𝛽𝑃) ∙ 𝑡))
𝑘𝛽𝑃,𝑣𝐴 + 𝑘𝑣𝐴,𝛽𝑃 (51)
[𝑣𝐴]𝑡 =100% ∙ 𝑘𝛽𝑃,𝑣𝐴
𝑘𝛽𝑃,𝑣𝐴 + 𝑘𝑣𝐴,𝛽𝑃∙ (1 − 𝑒𝑥𝑝(−(𝑘𝛽𝑃,𝑣𝐴 + 𝑘𝑣𝐴,𝛽𝑃) ∙ 𝑡)) (52)
Damit ergibt sich für die Zeitabhängigkeit der optischen Rotation Gleichung (53).

ERGEBNISSE UND DISKUSSION
- 96 -
𝛼𝑡 = 𝑙 ∙ 𝑐 ∙ (𝛼𝐷22(𝛽𝑃) ∙ [𝛽𝑃]𝑡 + 𝛼𝐷
22(𝑣𝐴) ∙ [𝑣𝐴]𝑡) =100%∙𝑙∙𝑐
𝑘𝛽𝑃,𝑣𝐴+𝑘𝑣𝐴,𝛽𝑃∙ (𝑘𝑣𝐴,𝛽𝑃 ∙ 𝛼𝐷
22(𝛽𝑃) +
𝑘𝛽𝑃,𝑣𝐴 ∙ (𝛼𝐷22(𝑣𝐴) + (𝛼𝐷
22(𝛽𝑃) − 𝛼𝐷22(𝑣𝐴)) ∙ 𝑒𝑥𝑝(−(𝑘𝛽𝑃,𝑣𝐴 + 𝑘𝑣𝐴,𝛽𝑃) ∙ 𝑡)))
(53)
Dabei ist 𝑙 die Länge der Küvette in dm und 𝑐 die Konzentration der D-Fructose 1a in g/mL.
Ob eine Auswertung polarimetrischer Daten nun auf Grundlage von Schema 28 bzw. Gleichung (53)
legitim ist, kann abgeschätzt werden, indem geprüft wird, ob 𝛼𝐷22(𝑣𝐴), ausgedrückt in Form von
Gleichung (54), zeitunabhängig und damit konstant ist.
𝛼𝐷22(𝑣𝐴) =
𝛼𝑡𝑙 ∙ 𝑐
− 𝛼𝐷22(𝛽𝑃) ∙ [𝛽𝑃]𝑡
[𝑣𝐴]𝑡 (54)
Die entsprechende Berechnung von 𝛼𝐷22(𝑣𝐴) im Sinne von Gleichung (54) ergibt einen Wert
von 𝛼𝐷22(𝑣𝐴) = (−0.51 ± 1.44)
°∙𝑚𝐿
𝑑𝑚∙𝑔. Obwohl die Berechnung dieses Wertes teilweise auf Grundlage
von NMR-spektroskopischen Messungen erfolgte, stimmt der Wert mit dem anhand der
Gleichgewichtsrotation ermittelten in Höhe von 𝛼𝐷22(𝑣𝐴) = (−0.04 ± 0.01)
°∙𝑚𝐿
𝑑𝑚∙𝑔 im Rahmen der
Fehlerintervalle überein. Die konzentrationsgewichtete spezifische Rotation des virtuellen Anomers
der D-Fructose 1a ist damit bei 22 °C etwa gleich null. Gleichung (53) lässt sich entsprechend zu
Gleichung (55) vereinfachen, die lediglich abhängig von der relativen Konzentration der
β-D-Fructopyranose 1a(ii) ist.
𝛼𝑡 =100% ∙ 𝑙 ∙ 𝑐 ∙ 𝛼𝐷
22(𝛽𝑃)
𝑘𝛽𝑃,𝑣𝐴 + 𝑘𝑣𝐴,𝛽𝑃∙ (𝑘𝑣𝐴,𝛽𝑃 + 𝑘𝛽𝑃,𝑣𝐴 ∙ 𝑒𝑥𝑝(−(𝑘𝛽𝑃,𝑣𝐴 + 𝑘𝑣𝐴,𝛽𝑃) ∙ 𝑡)) (55)
Die Berechnung der Ringöffnungsraten 𝑘𝛽𝑃,𝑣𝐴 der β-D-Fructopyranose 1a(ii) ist demnach bei
Raumtemperatur uneingeschränkt durch die nicht lineare Regression polarimetrischer Daten mittels
Gleichung (55) möglich. Die milieuabhängige relative Reaktivität der D-Fructose 1a unter
Kochbedingungen kann folglich anhand einfacher polarimetrischer Messungen innerhalb kürzester
Zeit abgeschätzt werden. Im folgenden Abschnitt wird die Methode der Polarimetrie dazu genutzt, die
pH-abhängige Reaktivität der D-Fructose 1a unter Zusatz verschiedener Katalysatoren abzuschätzen
und gleichermaßen eine Charakterisierung der Katalysatoren vorzunehmen. Dabei wird der Einfluss
verschiedener funktioneller Gruppen auf die Mutarotation der D-Fructose 1a diskutiert und die in
Kapitel 2.3 hergeleitete Theorie der Anomerisierung in der Praxis überprüft.
Es wurde jeweils eine 0.5 M wässrige Lösung des zu untersuchenden Katalysators mit einem
pH-Wert im Bereich von 1.5 bis 6.5 (sechs Stufen) vorbereitet. Diese Lösung wurde anschließend dazu
genutzt, eine stöchiometrische Menge an D-Fructose 1a zu lösen. Die zeitliche Änderung des
Drehwinkels der jeweiligen Lösungen wurde über einen Zeitraum von je etwa einer Stunde
polarimetrisch verfolgt. Zunächst wurden auf diese Weise die Ringöffnungsraten der
β-D-Fructopyranose 1a(ii) in An- und Abwesenheit stöchiometrischer Mengen von L-Alanin 4a und
L-Prolin 4b ermittelt. Ein Vergleich dieser Werte mit den auf Grundlage der SST-NMR-Experimente
berechneten Ringöffnungsraten zeigt eine gute Übereinstimmung (hier nicht gezeigt) und damit eine
ebenso gute Vergleichbarkeit der Methoden. Im nächsten Schritt wurden die entsprechenden
Ringöffnungsraten zusätzlich in Anwesenheit stöchiometrischer Mengen von Glycin 4c, β-Alanin 4d
und γ-Aminobuttersäure 4e ermittelt. Um eine Berechnung der spontanen Ringöffnungsraten der
verschiedenen ionischen Spezies der β-D-Fructopyranose 1a(ii) auf Grundlage von Gleichung (38)
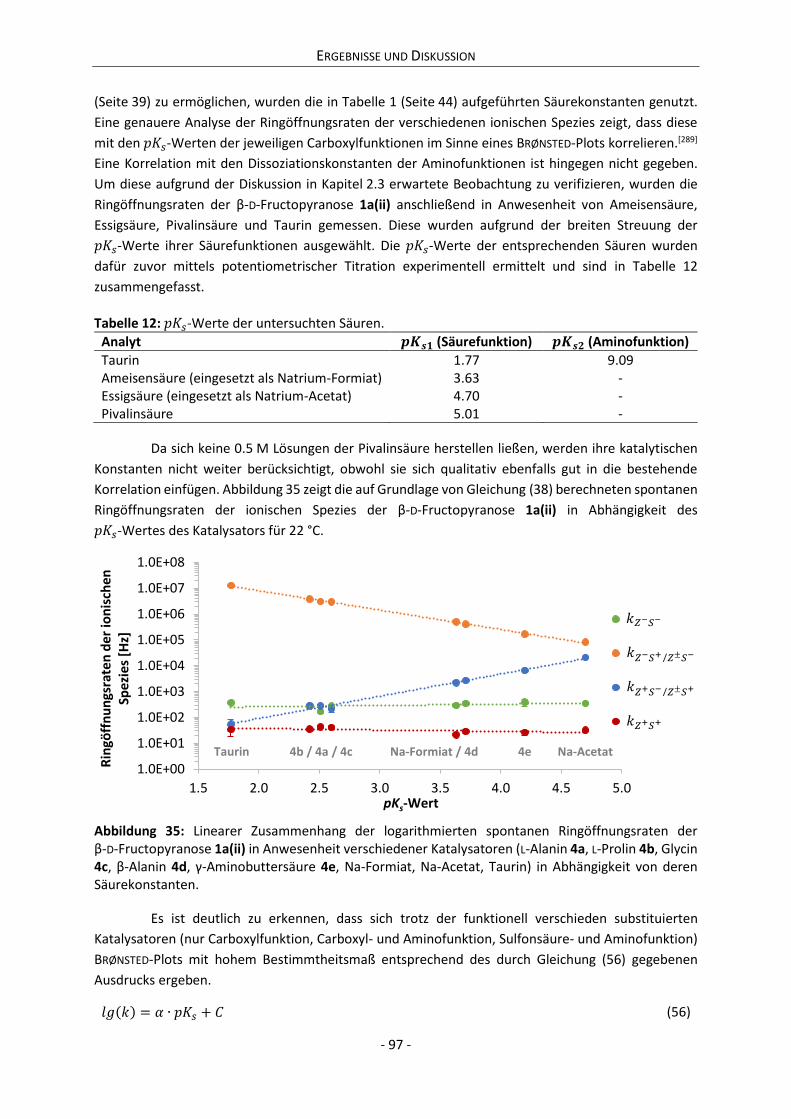
ERGEBNISSE UND DISKUSSION
- 97 -
(Seite 39) zu ermöglichen, wurden die in Tabelle 1 (Seite 44) aufgeführten Säurekonstanten genutzt.
Eine genauere Analyse der Ringöffnungsraten der verschiedenen ionischen Spezies zeigt, dass diese
mit den 𝑝𝐾𝑠-Werten der jeweiligen Carboxylfunktionen im Sinne eines BRØNSTED-Plots korrelieren.[289]
Eine Korrelation mit den Dissoziationskonstanten der Aminofunktionen ist hingegen nicht gegeben.
Um diese aufgrund der Diskussion in Kapitel 2.3 erwartete Beobachtung zu verifizieren, wurden die
Ringöffnungsraten der β-D-Fructopyranose 1a(ii) anschließend in Anwesenheit von Ameisensäure,
Essigsäure, Pivalinsäure und Taurin gemessen. Diese wurden aufgrund der breiten Streuung der
𝑝𝐾𝑠-Werte ihrer Säurefunktionen ausgewählt. Die 𝑝𝐾𝑠-Werte der entsprechenden Säuren wurden
dafür zuvor mittels potentiometrischer Titration experimentell ermittelt und sind in Tabelle 12
zusammengefasst.
Tabelle 12: 𝑝𝐾𝑠-Werte der untersuchten Säuren.
Analyt 𝒑𝑲𝒔𝟏 (Säurefunktion) 𝒑𝑲𝒔𝟐 (Aminofunktion)
Taurin 1.77 9.09 Ameisensäure (eingesetzt als Natrium-Formiat) 3.63 - Essigsäure (eingesetzt als Natrium-Acetat) 4.70 - Pivalinsäure 5.01 -
Da sich keine 0.5 M Lösungen der Pivalinsäure herstellen ließen, werden ihre katalytischen
Konstanten nicht weiter berücksichtigt, obwohl sie sich qualitativ ebenfalls gut in die bestehende
Korrelation einfügen. Abbildung 35 zeigt die auf Grundlage von Gleichung (38) berechneten spontanen
Ringöffnungsraten der ionischen Spezies der β-D-Fructopyranose 1a(ii) in Abhängigkeit des
𝑝𝐾𝑠-Wertes des Katalysators für 22 °C.
Abbildung 35: Linearer Zusammenhang der logarithmierten spontanen Ringöffnungsraten der β-D-Fructopyranose 1a(ii) in Anwesenheit verschiedener Katalysatoren (L-Alanin 4a, L-Prolin 4b, Glycin 4c, β-Alanin 4d, γ-Aminobuttersäure 4e, Na-Formiat, Na-Acetat, Taurin) in Abhängigkeit von deren Säurekonstanten.
Es ist deutlich zu erkennen, dass sich trotz der funktionell verschieden substituierten
Katalysatoren (nur Carboxylfunktion, Carboxyl- und Aminofunktion, Sulfonsäure- und Aminofunktion)
BRØNSTED-Plots mit hohem Bestimmtheitsmaß entsprechend des durch Gleichung (56) gegebenen
Ausdrucks ergeben.
𝑙𝑔(𝑘) = 𝛼 ∙ 𝑝𝐾𝑠 + 𝐶 (56)
1.0E+00
1.0E+01
1.0E+02
1.0E+03
1.0E+04
1.0E+05
1.0E+06
1.0E+07
1.0E+08
1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0
Rin
göff
nu
ngs
rate
n d
er
ion
isch
en
Sp
ezie
s [H
z]
pKs-Wert
Taurin 4b / 4a / 4c Na-Formiat / 4d 4e Na-Acetat
𝑘𝑍−𝑆−
𝑘𝑍−𝑆+/𝑍±𝑆−
𝑘𝑍+𝑆−/𝑍±𝑆+
𝑘𝑍+𝑆+

ERGEBNISSE UND DISKUSSION
- 98 -
Eine negative Steigung (−1 ≤ 𝛼 < 0) der Auftragung der logarithmierten Ringöffnungsraten
der ionischen Zuckerspezies gegen die 𝑝𝐾𝑠-Werte der Katalysatoren liegt laut dem BRØNSTED-Katalyse-
Gesetz bei Säurekatalyse vor. Eine positive Steigung (0 < 𝛼 ≤ 1) ergibt sich entsprechend bei der
Katalyse durch Basen. Abbildung 35 zeigt deutlich, dass die Ringöffnung anionischer Spezies der
D-Fructose 1a durch Säuren katalysiert wird, während die Ringöffnung kationischer Spezies der
Katalyse durch Basen unterliegt.49 Weiterhin ist zu erkennen, dass Säuren (Basen) keinen Einfluss auf
die Ringöffnung kationischer (anionischer) Spezies der D-Fructose 1a haben (𝛼 ≅ 0). Es ist
entsprechend davon auszugehen, dass eine Ringöffnung in diesen Fällen unter dem Einfluss von
Wasser abläuft, welches in jedem Modellsystem im Überschuss vorlag. Tabelle 13 fasst die
BRØNSTED-Parameter der untersuchten Katalyse der Ringöffnung der β-D-Fructopyranose 1a(ii)
zusammen.
Tabelle 13: BRØNSTED-Parameter der Ringöffnungsraten ionischer Spezies der β-D-Fructopyranose 1a(ii) für 22 °C.
Ringöffnungsrate der Zuckerspezies 𝜶 (Steigung) 𝑪 (Achsenabschnitt)
𝑍−𝑆− 0.05 ± 0.04 2.31 ± 0.14 𝑍−𝑆+ -0.75 ± 0.01 8.40 ± 0.04 𝑍+𝑆− 0.86 ± 0.03 0.24 ± 0.11 𝑍+𝑆+ -0.06 ± 0.03 1.69 ± 0.11
Die Frage, warum Wasser – trotz seines extremen Überschusses gegenüber den zugesetzten
Säuren – deren katalytischen Einfluss nicht überdeckt, kann anhand der erhaltenen BRØNSTED-Plots
beantwortet werden. Die 𝑝𝐾𝑠-Werte von Wasser liegen bei 22 °C nach dem Massenwirkungsgesetz
unter Einbeziehung des Ionenproduktes von Wasser bei 22 °C (𝑝𝐾𝑊𝐻(22°𝐶) = 14.0756)[349] und
dessen Dichte bei 22 °C (𝜌(𝐻2𝑂)22°𝐶 = 0.9978𝑔
𝑐𝑚3)[352] bei 𝑝𝐾𝑠1 = −1.74 und 𝑝𝐾𝑠2 = 15.82. Mit
Blick auf eine Extrapolation der BRØNSTED-Plots sind sowohl Hydronium- als auch Hydroxidionen
katalytisch zwar unvergleichbar effektiv, jedoch findet die Katalyse durch Hydroniumionen im
Basischen und die durch Hydroxidionen im Sauren statt, wo die entsprechenden Ionen nur in
geringsten Konzentrationen vorherrschen. Wasser ist demnach in neutralen pH-Bereichen trotz seines
Überschusses ein vergleichsweise ineffizienter Katalysator. Schwache Säuren sind demgegenüber zwar
katalytisch weniger aktiv, die für die Katalyse notwendigen Spezies kommen jedoch in wesentlich
höheren Konzentrationen vor, sodass im Gesamtbild eine effiziente Katalyse resultiert.
Der Schnittpunkt der BRØNSTED-Plots für die saure und basische Katalyse liegt für 22 °C bei
einem 𝑝𝐾𝑠-Wert von 5.07. Für Pivalinsäure ist der katalytische Einfluss der Carboxylfunktion
entsprechend äquivalent zum katalytischen Einfluss ihrer konjugierten Base. Da das Minimum der
Ringöffnungsrate und auch der 𝑝𝐼 der β-D-Fructopyranose 1a(ii) mit einem Wert von rund 4.7 jedoch
bei einem pH-Wert unterhalb von 5.07 liegt, überwiegt der Einfluss der sauren Katalyse. Dies wird
durch die rechtsseitig der Minima liegenden Schultern in Abbildung 36 deutlich.50 Die entsprechenden
49 Ein Einfluss der Säuren bzw. konjugierten Basen auf die zwitterionischen Zuckerspezies sollte nicht gegeben
sein, da diese ohne weiteres Zutun spontan öffnen.
50 Entsprechende Schultern, die aus einer äquivalenten Säurekatalyse resultieren, sind auch in Bezug auf die
Abbauraten von D-Fructose 1a (Abbildung 4, Seite 43), die Bildungsraten von 3-Desoxyglucoson 9b (Abbildung 9,
Seite 50), die Bräunungsraten, gemessen bei 420 nm (Abbildung 14, Seite 54), sowie die Raten der
1,2-Enolisierung (Abbildung 21, Seite 70) erkennbar. Auch im Zuge der Untersuchung der Mutarotation von
6-Desoxy-D-gluco-hepturonsäure durch CAPON und WALKER (1971)[393] sowie der Untersuchung der
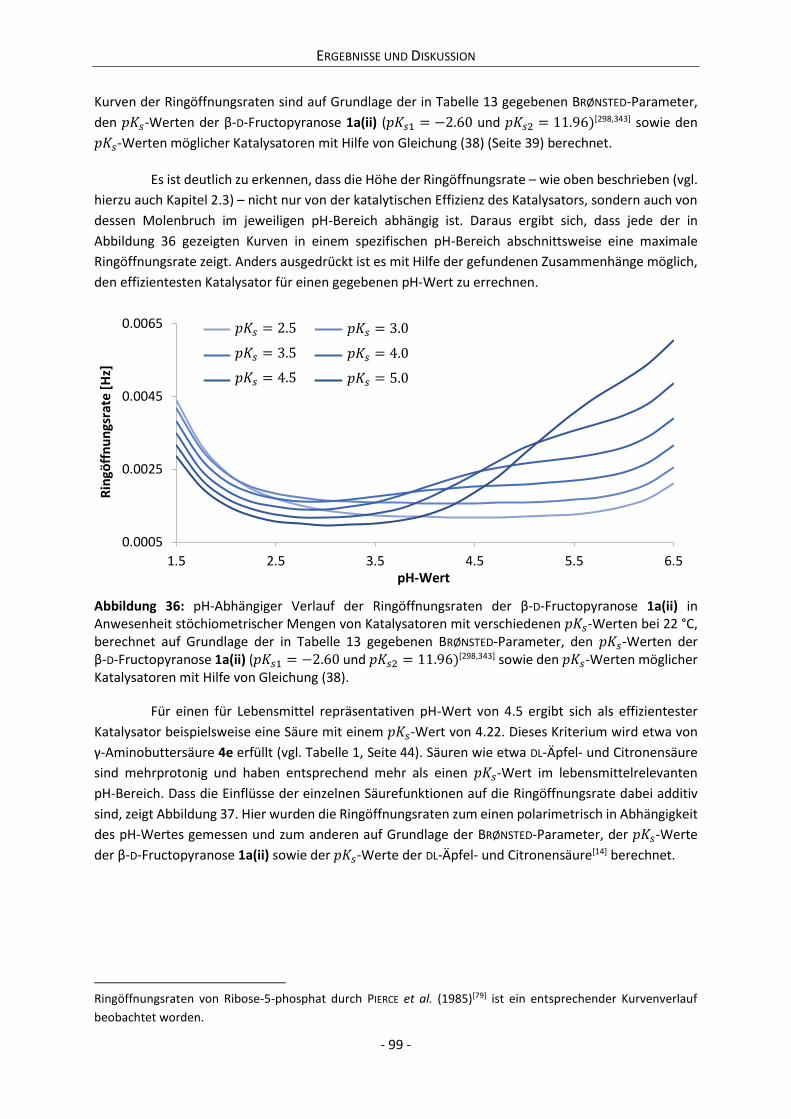
ERGEBNISSE UND DISKUSSION
- 99 -
Kurven der Ringöffnungsraten sind auf Grundlage der in Tabelle 13 gegebenen BRØNSTED-Parameter,
den 𝑝𝐾𝑠-Werten der β-D-Fructopyranose 1a(ii) (𝑝𝐾𝑠1 = −2.60 und 𝑝𝐾𝑠2 = 11.96)[298,343] sowie den
𝑝𝐾𝑠-Werten möglicher Katalysatoren mit Hilfe von Gleichung (38) (Seite 39) berechnet.
Es ist deutlich zu erkennen, dass die Höhe der Ringöffnungsrate – wie oben beschrieben (vgl.
hierzu auch Kapitel 2.3) – nicht nur von der katalytischen Effizienz des Katalysators, sondern auch von
dessen Molenbruch im jeweiligen pH-Bereich abhängig ist. Daraus ergibt sich, dass jede der in
Abbildung 36 gezeigten Kurven in einem spezifischen pH-Bereich abschnittsweise eine maximale
Ringöffnungsrate zeigt. Anders ausgedrückt ist es mit Hilfe der gefundenen Zusammenhänge möglich,
den effizientesten Katalysator für einen gegebenen pH-Wert zu errechnen.
Abbildung 36: pH-Abhängiger Verlauf der Ringöffnungsraten der β-D-Fructopyranose 1a(ii) in Anwesenheit stöchiometrischer Mengen von Katalysatoren mit verschiedenen 𝑝𝐾𝑠-Werten bei 22 °C, berechnet auf Grundlage der in Tabelle 13 gegebenen BRØNSTED-Parameter, den 𝑝𝐾𝑠-Werten der β-D-Fructopyranose 1a(ii) (𝑝𝐾𝑠1 = −2.60 und 𝑝𝐾𝑠2 = 11.96)
[298,343] sowie den 𝑝𝐾𝑠-Werten möglicher Katalysatoren mit Hilfe von Gleichung (38).
Für einen für Lebensmittel repräsentativen pH-Wert von 4.5 ergibt sich als effizientester
Katalysator beispielsweise eine Säure mit einem 𝑝𝐾𝑠-Wert von 4.22. Dieses Kriterium wird etwa von
γ-Aminobuttersäure 4e erfüllt (vgl. Tabelle 1, Seite 44). Säuren wie etwa DL-Äpfel- und Citronensäure
sind mehrprotonig und haben entsprechend mehr als einen 𝑝𝐾𝑠-Wert im lebensmittelrelevanten
pH-Bereich. Dass die Einflüsse der einzelnen Säurefunktionen auf die Ringöffnungsrate dabei additiv
sind, zeigt Abbildung 37. Hier wurden die Ringöffnungsraten zum einen polarimetrisch in Abhängigkeit
des pH-Wertes gemessen und zum anderen auf Grundlage der BRØNSTED-Parameter, der 𝑝𝐾𝑠-Werte
der β-D-Fructopyranose 1a(ii) sowie der 𝑝𝐾𝑠-Werte der DL-Äpfel- und Citronensäure[14] berechnet.
Ringöffnungsraten von Ribose-5-phosphat durch PIERCE et al. (1985)[79] ist ein entsprechender Kurvenverlauf
beobachtet worden.
0.0005
0.0025
0.0045
0.0065
1.5 2.5 3.5 4.5 5.5 6.5
Rin
göff
nu
ngs
rate
[H
z]
pH-Wert
pKs=2.5 pKs=3.0
pKs=3.5 pKs=4.0
pKs=4.5 pKs=5.0
𝑝𝐾𝑠 = 2.5
𝑝𝐾𝑠 = 3.5
𝑝𝐾𝑠 = 4.5
𝑝𝐾𝑠 = 3.0
𝑝𝐾𝑠 = 4.0
𝑝𝐾𝑠 = 5.0
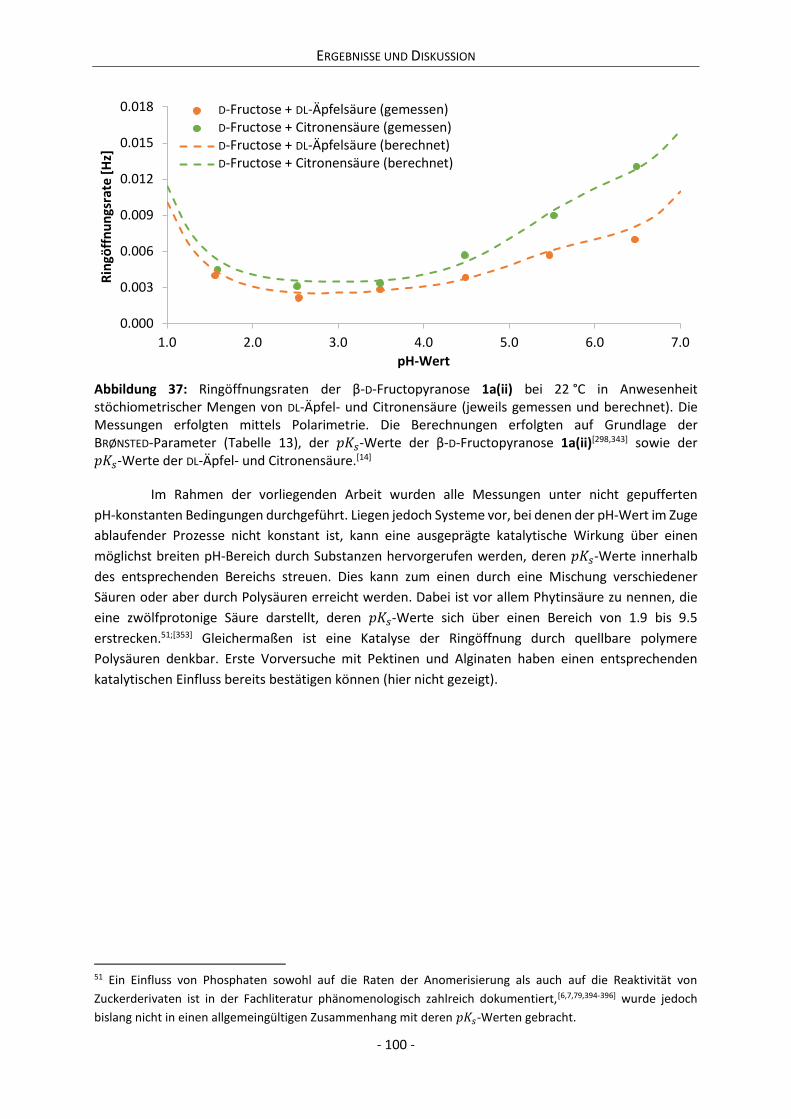
ERGEBNISSE UND DISKUSSION
- 100 -
Abbildung 37: Ringöffnungsraten der β-D-Fructopyranose 1a(ii) bei 22 °C in Anwesenheit stöchiometrischer Mengen von DL-Äpfel- und Citronensäure (jeweils gemessen und berechnet). Die Messungen erfolgten mittels Polarimetrie. Die Berechnungen erfolgten auf Grundlage der BRØNSTED-Parameter (Tabelle 13), der 𝑝𝐾𝑠-Werte der β-D-Fructopyranose 1a(ii)[298,343] sowie der 𝑝𝐾𝑠-Werte der DL-Äpfel- und Citronensäure.[14]
Im Rahmen der vorliegenden Arbeit wurden alle Messungen unter nicht gepufferten
pH-konstanten Bedingungen durchgeführt. Liegen jedoch Systeme vor, bei denen der pH-Wert im Zuge
ablaufender Prozesse nicht konstant ist, kann eine ausgeprägte katalytische Wirkung über einen
möglichst breiten pH-Bereich durch Substanzen hervorgerufen werden, deren 𝑝𝐾𝑠-Werte innerhalb
des entsprechenden Bereichs streuen. Dies kann zum einen durch eine Mischung verschiedener
Säuren oder aber durch Polysäuren erreicht werden. Dabei ist vor allem Phytinsäure zu nennen, die
eine zwölfprotonige Säure darstellt, deren 𝑝𝐾𝑠-Werte sich über einen Bereich von 1.9 bis 9.5
erstrecken.51;[353] Gleichermaßen ist eine Katalyse der Ringöffnung durch quellbare polymere
Polysäuren denkbar. Erste Vorversuche mit Pektinen und Alginaten haben einen entsprechenden
katalytischen Einfluss bereits bestätigen können (hier nicht gezeigt).
51 Ein Einfluss von Phosphaten sowohl auf die Raten der Anomerisierung als auch auf die Reaktivität von
Zuckerderivaten ist in der Fachliteratur phänomenologisch zahlreich dokumentiert,[6,7,79,394-396] wurde jedoch
bislang nicht in einen allgemeingültigen Zusammenhang mit deren 𝑝𝐾𝑠-Werten gebracht.
0.000
0.003
0.006
0.009
0.012
0.015
0.018
1.0 2.0 3.0 4.0 5.0 6.0 7.0
Rin
göff
nu
ngs
rate
[H
z]
pH-Wert
D-Fructose + Äpfelsäure (gemessen)D-Fructose + Citronensäure (gemessen)D-Fructose + Äpfelsäure (berechnet)D-Fructose + Citronensäure (berechnet)
D-Fructose + DL-Äpfelsäure (gemessen) D-Fructose + Citronensäure (gemessen) D-Fructose + DL-Äpfelsäure (berechnet) D-Fructose + Citronensäure (berechnet)

ZUSAMMENFASSUNG
- 101 -
5 Zusammenfassung Die vorliegende Arbeit verfolgte zunächst das Ziel zu klären, ob die Reaktivität eines
reduzierenden Zuckers auf ein spezifisches Anomer zurückgeführt werden kann. Dazu wurden in einem
ersten Schritt verschiedene Reaktionsmechanismen des Zuckerabbaus betrachtet. Die Mehrzahl dieser
Mechanismen weist die Gemeinsamkeit diverser Schritte des Protonentransfers auf. Einen analytisch
einfach zu verfolgenden Prozess, der auf einem mehrstufigen Protonentransfer basiert, stellt die
Ringöffnung bzw. Anomerisierung reduzierender Zucker dar. Unter der Voraussetzung, dass der
Protonentransfer reaktionsunabhängig immer den gleichen Gesetzmäßigkeiten folgt, erlaubt ein
genaues Verständnis des Prozesses der Anomerisierung Rückschlüsse auf den Verlauf der nicht
enzymatischen Bräunung sowohl im Sinne der Karamellisierung als auch im Sinne der
MAILLARD-Reaktion. Daraus wurde die Hypothese abgeleitet, dass die Reaktivität eines reduzierenden
Zuckers durch die Messung seiner Gesamtanomerisierungsrate abgeschätzt werden kann.
Um diese Hypothese zu überprüfen, wurde zunächst ein Modell zur Beschreibung der
Dynamik der Anomerisierung erstellt, das den Prozess nicht nur mathematisch beschreibt, sondern
auch mechanistisch fundiert ist. Anschließend wurden die Gesamtanomerisierungsraten von
D-Glucose 2a, D-Tagatose 1b und D-Fructose 1a sowie D-Fructose-analoger AMADORI-Verbindungen 6a
und 6b mittels selektiver Sättigungstransfer- sowie quantitativer NMR-Spektroskopie in Abhängigkeit
des pH- bzw. pD-Wertes und der Temperatur gemessen.52 In einem parallelen Schritt wurden die
jeweiligen Zucker bzw. Zuckerderivate bei 90 °C und konstanten pH-Werten im Bereich von 1.5 bis 6.5
zur Reaktion gebracht und ihre relativen Reaktivitäten anhand verschiedener Parameter wie etwa den
Abbauraten der Edukte, Bildungsraten von Folgeprodukten sowie Bräunungsmessungen ermittelt. Es
konnte gezeigt werden, dass das Verhältnis der Gesamtanomerisierungsraten der untersuchten
Verbindungen bei 90 °C im lebensmittelrelevanten pH-Bereich (etwa 3 ≤ 𝑝𝐻 ≤ 6) in guter
Übereinstimmung mit dem Verhältnis ihrer Reaktivitäten ist. Dies trifft ebenso auf die für eine
Temperatur von 120 °C extrapolierten Gesamtanomerisierungsraten der D-Fructose 1a sowie der
D-Fructose-analogen AMADORI-Verbindungen 6a und 6b und deren Vergleich mit den von BRANDS und
VAN BOEKEL (1998, 2002) publizierten Reaktivitätsverhältnissen zu. Es ist somit anzunehmen, dass die
Reaktivität eines reduzierenden Zuckers nicht auf ein spezifisches Anomer zurückgeführt werden
kann. Vielmehr resultiert die Reaktivität des Systems primär aus der jeweiligen Ionisierbarkeit der
vorhandenen funktionellen Gruppen.
Die Ringöffnung erfolgt ausgehend von ionisierten Zuckeranomeren. Diese gehen aus einer
Protonierung des Ringsauerstoffatoms bzw. der Deprotonierung der anomeren Hydroxylfunktion
hervor. Bei dem entsprechenden Kation handelt es sich damit um eine starke Säure, beim Anion um
eine starke Base. Diese können eine Protonierung bzw. Deprotonierung der übrigen
Hydroxylfunktionen bewirken (intramolekularer Protonenshift), was die eigentlichen Abbaureaktionen
einleitet. Da es sich bei der anomeren Hydroxylfunktion um diejenige mit der größten Acidität handelt,
ist die Wahrscheinlichkeit ihrer Deprotonierung innerhalb des Moleküls am größten. Abbaureaktionen
reduzierender Zucker sind damit als unwahrscheinliche Alternativen zur Anomerisierung zu
52 In diesem Zusammenhang konnten die anomeren 1H-NMR-Resonanzen der furanoiden Anomere der D-Glucose
2a erstmals direkt und die Protonenresonanz des Aldehydprotons des acyclischen Zentralisomers erstmals
indirekt nachgewiesen werden.

ZUSAMMENFASSUNG
- 102 -
verstehen. Die Dynamik sowohl der Ringöffnung als auch des Ringschlusses steht auf diese Weise mit
der Kinetik nicht enzymatischer Bräunungsreaktionen in enger Verbindung. Bedingungen, die die
Anomerisierung und damit die Dynamik des Protonentransfers fördern, müssen folglich auch zu einer
erhöhten Reaktivität des entsprechenden Zuckers führen.
Um den Einfluss von Aminosäuren auf die Dynamik der Anomerisierung und damit indirekt
auch auf die Reaktivität reduzierender Zucker zu untersuchen, wurde die Rate der Ringöffnung der
β-D-Fructopyranose 1a(ii) im Folgenden in Anwesenheit verschiedener katalytischer Säuren bei 22 °C
in Abhängigkeit des pH-Wertes gemessen. Dabei konnte gezeigt werden, dass die Aminofunktionen
einfacher Aminosäuren im lebensmittelrelevanten pH-Bereich keinen messbaren Einfluss auf die Rate
der Anomerisierung haben. Vielmehr wird der Prozess der Anomerisierung durch Substanzen
katalysiert, die funktionelle Gruppen mit 𝑝𝐾𝑠-Werten im Bereich des isoelektrischen Punktes (𝑝𝐼) des
untersuchten Zuckers tragen. Wie entsprechende BRØNSTED-Plots zeigen, findet eine Katalyse dabei
sowohl säure- als auch basenvermittelt statt (vgl. allgemeine Säure-Base-Katalyse). Für
β-D-Fructopyranose 1a(ii) ist die Effizienz der sauren und basischen Katalyse für einen Katalysator mit
einem 𝑝𝐾𝑠-Wert von 5.07 bei Raumtemperatur identisch. Da jedoch der 𝑝𝐼 des Zuckeranomers mit
rund 4.7 geringfügig darunter liegt, ist die Katalyse der Anomerisierung durch Säuren in der Praxis von
höherer Relevanz als eine Katalyse durch Basen.
Unter der Annahme, dass entsprechende BRØNSTED-Plots auch für Prozesse des
Protonentransfers im Rahmen der nicht enzymatischen Bräunung näherungsweise Gültigkeit haben
(etwa für die Bildung der SCHIFF-Base aus reduzierendem Zucker und Aminosäure, für die sich
anschließende AMADORI-Umlagerung, etc.), ist der Einfluss von schwachen Säuren auf den Abbau
reduzierender Zucker hinreichend genau erklärt. Es verwundert jedoch zunächst, dass
Aminofunktionen mit üblichen 𝑝𝐾𝑠-Werten im Bereich von 9 bis 10 keinen bedeutenden Einfluss auf
Protonentransfers haben, obwohl die MAILLARD-Reaktion allgemein als aminokatalysiert gilt. Der
Aminostickstoff muss demnach einen anderweitigen Effekt ausüben.
Betrachtet man die Raten der 1,2-Enolisierung von D-Fructose 1a sowie D-Fructose-analogen
AMADORI-Verbindungen 6, so wird deutlich, dass die kovalente Bindung zum Aminostickstoff eine
Erhöhung der C-H-Acidität am C-1 zur Folge hat. Die der AMADORI-Umlagerung entsprechende
1,2-Enolsierung ist demnach nicht nur durch schwache Säuren katalysiert und damit durch
Aminosäuren autokatalysiert, sondern auch durch eine Veränderung der C-H-Acidität begünstigt, die
sich aus dem negativen induktiven Effekt des protonierten Aminostickstoffs ergibt. Hierbei handelt es
sich folglich um einen Aspekt der Reaktionskaskade, der aus dem direkten Einfluss des Aminostickstoffs
resultiert.
Die Untersuchung der Bildungskinetik von 1- und 3-Desoxyglucoson 9a und 9b, ausgehend
von den AMADORI-Verbindungen 6a-6e in Abhängigkeit des pH-Wertes, erlaubte weiterhin den Schluss,
dass die C-H-Acidität am C-3 sowie die Protonenaffinität der Hydroxylfunktion am C-3 durch den
Substituenten am C-1 über die Ausbildung kooperativer Wasserstoffbrücken beeinflusst sind. Liegen
entsprechende Wasserstoffbrückenbindungen vor, so wirkt ein verstärkter negativer induktiver Effekt
auf die C-H-Bindung am C-3 und die Bildung von 1-Desoxyglucoson 9a wird erleichtert. Gleichermaßen
ist die Protonierung der OH-C-3-Funktion begünstigt, sodass es unter anderem zur erleichterten
Bildung von 3-Desoxyglucoson 9b, 3-Desoxygalactoson 9c, HMF 13 und AAF 15 im sauren Milieu
kommt.

ZUSAMMENFASSUNG
- 103 -
Der Aminostickstoff der AMADORI-Verbindungen 6 hat weiterhin Einfluss auf die 𝑝𝐾𝑠-Werte
der Ringsauerstoffatome der einzelnen Anomere, sodass eine Katalyse der Ringöffnung durch Basen
weniger wahrscheinlich wird. Das betrifft jedoch nicht die Katalyse durch Säuren im pH-Bereich von
größer als 3.0. Wichtig für den weiteren Verlauf der MAILLARD-Reaktion ist, dass nicht nur der
Aminosubstituent Einfluss auf die 𝑝𝐾𝑠-Werte des Zuckergrundkörpers, sondern dieser gleichermaßen
rückwirkend Einfluss auf den 𝑝𝐾𝑠-Wert der Aminofunktion nimmt. Dieser fällt durch die kovalente
Bindung zum Zucker in der Regel um ca. 1.5 Einheiten. Die 𝑝𝐾𝑠-Werte gewöhnlicher
AMADORI-Verbindungen liegen damit im Bereich von ca. 8 bis 9. Der Anteil der AMADORI-Verbindungen
mit nicht protonierter Aminofunktion ist damit beim selben pH-Wert höher als der ihrer
ursprünglichen Aminosäuren. Damit erhöht sich die Wahrscheinlichkeit einer neuerlichen
Kondensation mit einem zweiten Molekül eines Zuckers. Dabei fällt der 𝑝𝐾𝑠-Wert der Aminofunktion
weiter ab und erreicht den Bereich, in dem deren katalytischer Einfluss bedeutsam wird.
Der Abbau reduzierender Zucker führt in vielen Fällen zur Bildung von Carbonsäuren.53 Dabei
handelt es sich zumeist um schwache Säuren, die entsprechend katalytisch hoch potent sind und
damit ihre eigene Bildung katalysieren. Parallel hierzu kommt es zur Bildung von Bräunungsprodukten,
sodass sich die Kinetik der Farbbildung – wie auch in der vorliegenden Arbeit geschehen – am besten
mit Hilfe einer logistischen Funktion modellieren lässt. Dabei ist die logistische Funktion mathematisch
betrachtet äquivalent zum integrierten Zeitgesetz der Autokatalyse. Folglich unterliegt die MAILLARD-
Reaktion mit fortschreitender Dauer mehr und mehr einer Katalyse im Sinne der Karamellisierung.
Ein von Reaktionsbeginn an beschleunigter Verlauf der MAILLARD-Reaktion sowie der Karamellisierung
im lebensmittelrelevanten pH-Bereich kann daher durch die Zugabe schwacher Säuren wie
Carbonsäuren und Phosphaten – vor allem Phytinsäure – erreicht werden. Als Aminokomponenten
sollten dabei Verbindungen ausgewählt werden, die zumindest eine Aminofunktion mit möglichst
schwacher Basizität – wie etwa L-Lysin – aufweisen. Zusätzlich trägt eine zur Aminofunktion α-ständige
Carboxylfunktion zu einer erhöhten Reaktivität bei, indem sie an kooperativen
Wasserstoffbrückenbindungen teilhat.
Schema 29 stellt die im Rahmen dieser Arbeit gewonnenen Erkenntnisse generalisiert dar,
sodass sich ein umfassendes Bild der frühen und intermediären Phase der MAILLARD-Reaktion –
beginnend bei D-Glucose 2a – ergibt. Der Fokus liegt dabei auf Reaktionsschritten, auf die schwache
Säuren und ihre konjugierten Basen einen katalytischen Einfluss haben (im Schema mit roten Pfeilen
kenntlich gemacht). Gleichermaßen sind die wichtigsten funktionellen Gruppen markiert, für die sich
im Laufe der Reaktion eine Änderung der 𝑝𝐾𝑠-Werte ergibt (mit grünen Pfeilen hervorgehoben).
Quantitative Veränderungen der Anomerensysteme (gelb-orange Pfeile) sowie Folgereaktionen (blaue
Pfeile) sind ebenfalls kenntlich gemacht.
53 Auch die beschriebenen α-Dicarbonylverbindungen sind über ihre Hydrate tautomer zu Hydroxycarbonsäuren
wie etwa Gluconsäure (tautomer zum Hydrat des D-Glucosons 9d). Daneben handelt es sich bei den Endiolen
einiger α-Dicarbonylverbindungen (solche mit Reduktonstruktur, etwa 1-Desoxyglucoson 9a und D-Glucoson 9d)
um vinyloge Carbonsäuren.
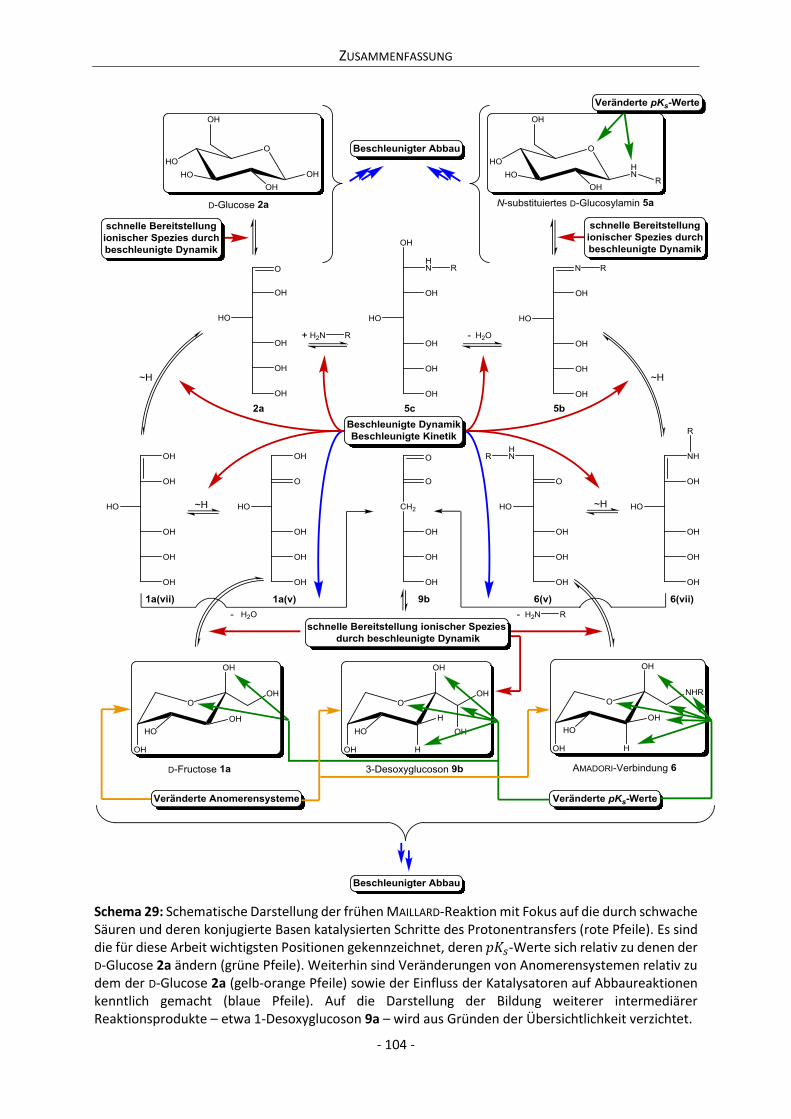
ZUSAMMENFASSUNG
- 104 -
Schema 29: Schematische Darstellung der frühen MAILLARD-Reaktion mit Fokus auf die durch schwache Säuren und deren konjugierte Basen katalysierten Schritte des Protonentransfers (rote Pfeile). Es sind die für diese Arbeit wichtigsten Positionen gekennzeichnet, deren 𝑝𝐾𝑠-Werte sich relativ zu denen der D-Glucose 2a ändern (grüne Pfeile). Weiterhin sind Veränderungen von Anomerensystemen relativ zu dem der D-Glucose 2a (gelb-orange Pfeile) sowie der Einfluss der Katalysatoren auf Abbaureaktionen kenntlich gemacht (blaue Pfeile). Auf die Darstellung der Bildung weiterer intermediärer Reaktionsprodukte – etwa 1-Desoxyglucoson 9a – wird aus Gründen der Übersichtlichkeit verzichtet.

ZUSAMMENFASSUNG
- 105 -
Katalytische Säuren und Basen beschleunigen die Anomerisierung der D-Glucose 2a, indem
sie als Protonendonoren und -akzeptoren wirken und damit einen effektiven Protonentransfer
gewährleisten. Dabei kommt es in wässriger Lösung auch zur beschleunigten Ausbildung des gem-Diols
der D-Glucose und in Anwesenheit von Aminosäuren in Konkurrenz dazu zur Bildung des gem-Aminols
5c. Dieses wird jedoch nicht nur beschleunigt gebildet, sondern gleichermaßen beschleunigt abgebaut.
Dabei kommt es zur Freisetzung von D-Glucose 2a oder alternativ zur Bildung der SCHIFF-Base 5b. Deren
Anomerisierungsdynamik – d.h. die Rate der Cyclisierung zu ihren anomeren N-substituierten
Glycosylaminen 5a – ist ebenso katalysiert wie die 1,2-Enolisierung als Auftakt für die
AMADORI-Umlagerung sowie die Bildung von 3-Desoxyglucoson 9b. Im Vergleich zur reinen
D-Fructose 1a ist die Rate der 1,2-Enolisierung der AMADORI-Verbindungen 6 durch die elektronischen
Effekte des kovalent gebundenen Stickstoffsubstituenten erhöht. Da viele Reaktionen, ebenso wie die
Anomerisierung, auf sukzessivem Protonentransfer beruhen, fördern schwache Säuren und deren
konjugierte Basen nicht nur die Ringöffnung und den Ringschluss von Zuckeranomeren – d.h. die
Bildung und den Zerfall von Halbacetalen bzw. -ketalen –, sondern auch deren Reaktivität im
Allgemeinen.
Die vorliegende Arbeit zeigt, dass sowohl die Karamellisierung als auch die
MAILLARD-Reaktion als konsekutive Säure-Base-Reaktionen verstanden werden können bzw. müssen.
Ihr Verlauf ist damit abhängig von der Dynamik des Protonentransfers, die sich insbesondere durch
schwache Säuren wie Carbonsäuren katalysieren lässt (allgemeine Säure-Base-Katalyse). Daneben
erlaubt die Kenntnis der 𝑝𝐾𝑠-Werte der Struktureinheiten, deren Ionisierung zu Abbaureaktionen
führt, das Ausmaß der entsprechenden Reaktionen im Verhältnis zu anderen Reaktionsmöglichkeiten
abzuschätzen. Da es sich bei der anomeren Hydroxylfunktion sowie dem Ringsauerstoffatom der
Anomere von Zuckerderivaten zumeist um die Struktureinheiten handelt, deren 𝑝𝐾𝑠-Werte am
wenigsten extrem sind, wirken katalytische Einflüsse hier am stärksten.
Die Untersuchung des Einflusses der Molekülgeometrie sowie verschiedener funktioneller
Gruppen auf die entsprechenden 𝑝𝐾𝑠-Werte liefert damit grundlegende Erkenntnisse zum Verlauf der
nicht enzymatischen Bräunung. Im Rahmen der vorliegenden Arbeit konnte gezeigt werden, dass
entsprechende Untersuchungen neben technologisch hoch entwickelten Methoden wie der
NMR-Spektroskopie auch mittels einfachster apparativer Ausstattungen wie etwa manueller
Polarimeter erfolgen können.

EXPERIMENTELLER TEIL
- 106 -
6 Experimenteller Teil Im Folgenden sind die durchgeführten Synthesen, Modellreaktionen, chromatographischen
und spektroskopischen Methoden und Parameter sowie die durchgeführten potentiometrischen und
polarimetrischen Experimente beschrieben. Außerdem sind die verwendeten Geräte,
NMR-Pulsprogramme, Chemikalien sowie die genutzte Software aufgelistet.
6.1 Synthesen der AMADORI-Verbindungen
Die Synthesen der untersuchten AMADORI-Verbindungen 6a-6e erfolgten in Anlehnung an die
von MILLS und HODGE (1976) sowie RÖPER et al. (1983) vorgeschlagenen Methoden.[127,354]
6.1.1 SYNTHESE VON N-(1-DESOXY-D-FRUCTOS-1-YL)-L-ALANIN
Die Synthese von N-(1-Desoxy-D-fructos-1-yl)-L-alanin (Fructosylalanin) 6a erfolgte nach der
Vorschrift von RÖPER et al. (1983).[127]
M [g/mol] eq. n [mmol] m bzw. V
D-Glucose 180.16 4.0 280 50.445 g
L-Alanin 89.09 1.0 70 6.236 g
Natrium-Pyrosulfit 190.11 0.5 35 6.654 g
Methanol/Wasser
(1/1; v/v) 80 mL
Durchführung
6.236 g L-Alanin (70 mmol; 1.0 eq.) werden zusammen mit 50.445 g wasserfreier D-Glucose
(280 mmol; 4.0 eq.) und 6.654 g Natrium-Pyrosulfit (35 mmol; 0.5 eq.) in 80 mL Methanol/Wasser
(1/1; v/v) gelöst und für etwa 4.5 h bis zur leichten Gelbfärbung unter Rückfluss erhitzt. Die
Reaktionskontrolle erfolgt dünnschichtchromatographisch auf Kieselgel 60 unter Nutzung von
n-Butanol/Eisessig/Wasser (4/2/1; v/v/v) als Laufmittel und 5 mM Ninhydrinlösung in Ethanol/Wasser
(9/1; v/v) als Detektionsreagenz.
Aufarbeitung
Nach Abkühlen der Lösung wird diese mit 250 mL Ethanol/Wasser (1/1; v/v) verdünnt. Die
Aufreinigung des Produktes aus dem Filtrat erfolgt säulenchromatographisch an DowexTM 50WX8 (in
der protonierten Form). Dazu wird die Lösung zunächst auf den Kationentauscher aufgezogen.

EXPERIMENTELLER TEIL
- 107 -
Anschließend wird mit Ethanol/Wasser (1/1; v/v) (ca. zehnfaches Säulenvolumen) sowie bidest.
Wasser (ca. zehnfaches Säulenvolumen) gewaschen und mit 0.1 M wässriger ammoniakalischer
Lösung bei einer Tropfgeschwindigkeit von etwa einem Tropfen pro Sekunde eluiert. Die das Produkt
enthaltenden Fraktionen werden vereinigt, unter vermindertem Druck eingeengt und anschließend
gefriergetrocknet. Es ergeben sich 3.82 g Produkt als weißer Feststoff (22% Ausbeute).
Analytische Daten
1H- und 13C-NMR: vergleiche Tabellen 4 bis 6. Elementaranalyse (EA): %N = 5.48, %C = 42.69,
%H = 6.86, %O = 44.97. Summenformel laut EA: C9H17NO7. 𝑝𝐾𝑠1 = 2.30, 𝑝𝐾𝑠2 = 8.25. HPLC-RI
(AMADORI-Methode): 𝑡𝑅 = 16.1𝑚𝑖𝑛.
6.1.2 SYNTHESE VON N-(1-DESOXY-D-[2-13C]FRUCTOS-1-YL)-L-ALANIN
Die Synthese von N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-alanin ([2-13C]Fructosylalanin) 6a
erfolgte nach der Vorschrift von MILLS und HODGE (1976).[354] Die Aufarbeitung erfolgte in Anlehnung
an RÖPER et al. (1983).[127]
M [g/mol] eq. n [mmol] m bzw. V
D-[2-13C]Glucose 181.16 1.0 16.6 3.000 g
L-Alanin 89.09 1.1 18.6 1.660 g
Malonsäure 104.06 0.2 3.9 0.401 g
Methanol 30 mL
Durchführung
1.660 g L-Alanin (19 mmol; 1.1 eq.) werden zusammen mit 3.000 g wasserfreier
D-[2-13C]Glucose (17 mmol; 1.0 eq.) in 30 mL wasserfreiem Methanol weitestgehend gelöst und die
Suspension für 40 min unter Rückfluss erhitzt. Es werden 0.401 g Malonsäure (4 mmol; 0.2 eq.)
hinzugegeben und für weitere 170 min unter Rückfluss erhitzt. Die Reaktionskontrolle erfolgt
dünnschichtchromatographisch auf Kieselgel 60 unter Nutzung von n-Butanol/Eisessig/Wasser (4/2/1;
v/v/v) als Laufmittel und 5 mM Ninhydrinlösung in Ethanol/Wasser (9/1; v/v) als Detektionsreagenz.
Aufarbeitung
Nach Abkühlen der Suspension wird der Bodensatz abfiltriert und verworfen und das Filtrat
mit bidest. Wasser auf ein Gesamtvolumen von 150 mL ergänzt. Die Aufreinigung des Produktes aus
dem Filtrat erfolgt säulenchromatographisch an DowexTM 50WX8 (in der protonierten Form). Dazu
wird die Lösung zunächst auf den Kationentauscher aufgezogen. Anschließend wird mit

EXPERIMENTELLER TEIL
- 108 -
Ethanol/Wasser (1/1; v/v) (ca. zehnfaches Säulenvolumen) sowie bidest. Wasser (ca. dreifaches
Säulenvolumen) gewaschen und schrittweise mit wässriger ammoniakalischer Lösung aufsteigender
Konzentrationen (0.001 M; 0.01 M; 0.1 M) bei einer Tropfgeschwindigkeit von etwa einem Tropfen pro
Sekunde eluiert. Die das Produkt enthaltenden Fraktionen werden vereinigt, unter vermindertem
Druck eingeengt und anschließend gefriergetrocknet. Es ergeben sich 1.221 g Produkt als weißer
Feststoff (26% Ausbeute).
Analytische Daten
1H- und 13C-NMR: vergleiche Tabellen 4 bis 6. Elementaranalyse (EA): %N = 5.04, %C = 39.19,
%H = 7.15, %O = 48.62. Summenformel laut EA: C9H17NO7 · 1.5 H2O.
6.1.3 SYNTHESE VON N-(1-DESOXY-D-FRUCTOS-1-YL)-L-PROLIN
Die Synthese von N-(1-Desoxy-D-fructos-1-yl)-L-prolin (Fructosylprolin) 6b erfolgte nach der
Vorschrift von RÖPER et al. (1983).[127]
M [g/mol] eq. n [mmol] m bzw. V
D-Glucose 180.16 4.0 280 50.445 g
L-Prolin 115.13 1.0 70 8.059 g
Natrium-Pyrosulfit 190.11 0.5 35 6.654 g
Methanol/Wasser
(1/1; v/v) 80 mL
Durchführung
8.059 g L-Prolin (70 mmol; 1.0 eq.) werden zusammen mit 50.445 g wasserfreier D-Glucose
(280 mmol; 4.0 eq.) und 6.654 g Natrium-Pyrosulfit (35 mmol; 0.5 eq.) in 80 mL Methanol/Wasser
(1/1; v/v) gelöst und für etwa 4.5 h bis zur leichten Gelbfärbung unter Rückfluss erhitzt. Die
Reaktionskontrolle erfolgt dünnschichtchromatographisch auf Kieselgel 60 unter Nutzung von
n-Butanol/Eisessig/Wasser (4/2/1; v/v/v) als Laufmittel und 5 mM Ninhydrinlösung in Ethanol/Wasser
(9/1; v/v) als Detektionsreagenz.
Aufarbeitung
Nach Abkühlen der Lösung wird diese mit 250 mL Ethanol/Wasser (1/1; v/v) verdünnt. Die
Aufreinigung des Produktes aus dem Filtrat erfolgt säulenchromatographisch an DowexTM 50WX8 (in
der protonierten Form). Dazu wird die Lösung zunächst auf den Kationentauscher aufgezogen.
Anschließend wird mit Ethanol/Wasser (1/1; v/v) (ca. zehnfaches Säulenvolumen) sowie bidest.

EXPERIMENTELLER TEIL
- 109 -
Wasser (ca. zehnfaches Säulenvolumen) gewaschen und mit 0.1 M wässriger ammoniakalischer
Lösung bei einer Tropfgeschwindigkeit von etwa einem Tropfen pro Sekunde eluiert. Die das Produkt
enthaltenden Fraktionen werden vereinigt, unter vermindertem Druck eingeengt und anschließend
gefriergetrocknet. Es ergeben sich 8.70 g Produkt als weißer Feststoff (42% Ausbeute).
Analytische Daten
1H- und 13C-NMR: vergleiche Tabellen 4 bis 6. Elementaranalyse (EA): %N = 4.76, %C = 44.79,
%H = 7.18, %O = 43.28. Summenformel laut EA: C11H19NO7 · H2O. 𝑝𝐾𝑠1 = 2.12, 𝑝𝐾𝑠2 = 8.37. HPLC-RI
(AMADORI-Methode): 𝑡𝑅 = 14.5𝑚𝑖𝑛.
6.1.4 SYNTHESE VON N-(1-DESOXY-D-[2-13C]FRUCTOS-1-YL)-L-PROLIN
Die Synthese von N-(1-Desoxy-D-[2-13C]fructos-1-yl)-L-prolin ([2-13C]Fructosylprolin) 6b
erfolgte nach der Vorschrift von MILLS und HODGE (1976).[354] Die Aufarbeitung erfolgte in Anlehnung
an RÖPER et al. (1983).[127]
M [g/mol] eq. n [mmol] m bzw. V
D-[2-13C]Glucose 181.16 1.0 5.5 1.000 g
L-Prolin 115.13 1.1 6.2 0.715 g
Malonsäure 104.06 0.3 1.4 0.143 g
Methanol 15 mL
Durchführung
0.715 g L-Prolin (6.2 mmol; 1.1 eq.) werden zusammen mit 1.000 g wasserfreier
D-[2-13C]Glucose (5.5 mmol; 1.0 eq.) in 15 mL wasserfreiem Methanol weitestgehend gelöst und die
Suspension für 40 min unter Rückfluss erhitzt. Es werden 0.143 g Malonsäure (1.4 mmol; 0.3 eq.)
hinzugegeben und für weitere 140 min unter Rückfluss erhitzt. Die Reaktionskontrolle erfolgt
dünnschichtchromatographisch auf Kieselgel 60 unter Nutzung von n-Butanol/Eisessig/Wasser (4/2/1;
v/v/v) als Laufmittel und 5 mM Ninhydrinlösung in Ethanol/Wasser (9/1; v/v) als Detektionsreagenz.
Aufarbeitung
Nach Abkühlen der Suspension wird der Bodensatz abfiltriert und verworfen und das Filtrat
mit bidest. Wasser auf ein Gesamtvolumen von 150 mL ergänzt. Die Aufreinigung des Produktes aus
dem Filtrat erfolgt säulenchromatographisch an DowexTM 50WX8 (in der protonierten Form). Dazu
wird die Lösung zunächst auf den Kationentauscher aufgezogen. Anschließend wird mit
Ethanol/Wasser (1/1; v/v) (ca. zehnfaches Säulenvolumen) sowie bidest. Wasser (ca. dreifaches

EXPERIMENTELLER TEIL
- 110 -
Säulenvolumen) gewaschen und mit 0.1 M wässriger ammoniakalischer Lösung bei einer
Tropfgeschwindigkeit von etwa einem Tropfen pro Sekunde eluiert. Die das Produkt enthaltenden
Fraktionen werden vereinigt, unter vermindertem Druck eingeengt und anschließend
gefriergetrocknet. Mischfraktionen werden abermals einer säulenchromatographischen Trennung
unterzogen. Es ergeben sich 0.541 g Produkt als weißer Feststoff (35% Ausbeute).
Analytische Daten
1H- und 13C-NMR: vergleiche Tabellen 4 bis 6.
6.1.5 SYNTHESE VON N-(1-DESOXY-D-FRUCTOS-1-YL)-GLYCIN
Die Synthese von N-(1-Desoxy-D-fructos-1-yl)-glycin (Fructosylglycin) 6c erfolgte nach der
Vorschrift von RÖPER et al. (1983).[127]
M [g/mol] eq. n [mmol] m bzw. V
D-Glucose 180.16 3.9 268 48.315 g
Glycin 75.07 1.0 68 5.101 g
Natrium-Pyrosulfit 190.11 0.5 34 6.372 g
Methanol 250 mL
Durchführung
5.101 g Glycin (68 mmol; 1.0 eq.) werden zusammen mit 48.315 g wasserfreier D-Glucose
(268 mmol; 3.9 eq.) und 6.372 g Natrium-Pyrosulfit (34 mmol; 0.5 eq.) in 250 mL wasserfreiem
Methanol weitestgehend gelöst und die Suspension für 90 min unter Rückfluss erhitzt. Die
Reaktionskontrolle erfolgt dünnschichtchromatographisch auf Kieselgel 60 unter Nutzung von
n-Butanol/Eisessig/Wasser (4/2/1; v/v/v) als Laufmittel und 5 mM Ninhydrinlösung in Ethanol/Wasser
(9/1; v/v) als Detektionsreagenz.
Aufarbeitung
Nach Abkühlen der Suspension wird der Bodensatz abfiltriert und verworfen und das Filtrat
mit 500 mL bidest. Wasser verdünnt. Die Aufreinigung des Produktes aus dem Filtrat erfolgt
säulenchromatographisch an DowexTM 50WX8 (in der protonierten Form). Dazu wird die Lösung
zunächst auf den Kationentauscher aufgezogen. Anschließend wird mit Ethanol/Wasser (1/1; v/v) (ca.
zehnfaches Säulenvolumen) sowie bidest. Wasser (ca. zehnfaches Säulenvolumen) gewaschen und
schrittweise mit wässriger ammoniakalischer Lösung aufsteigender Konzentrationen (0.001 M;
0.01 M; 0.1 M) bei einer Tropfgeschwindigkeit von etwa einem Tropfen pro Sekunde eluiert. Die das
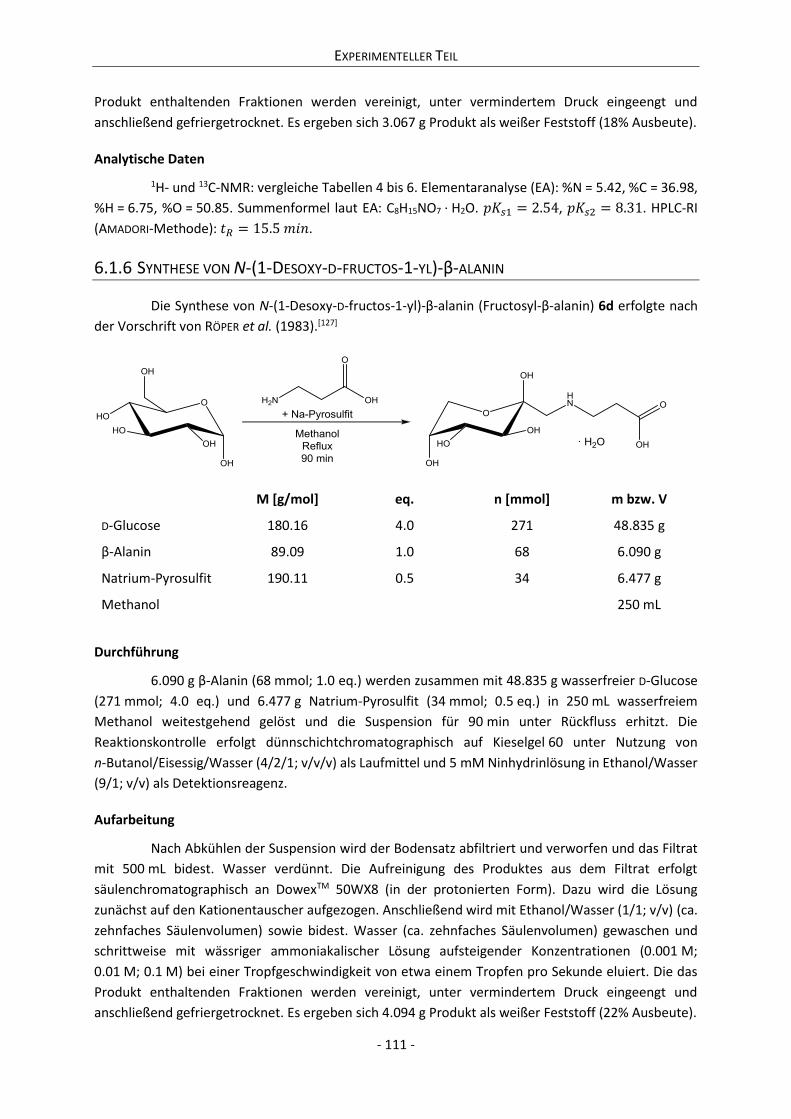
EXPERIMENTELLER TEIL
- 111 -
Produkt enthaltenden Fraktionen werden vereinigt, unter vermindertem Druck eingeengt und
anschließend gefriergetrocknet. Es ergeben sich 3.067 g Produkt als weißer Feststoff (18% Ausbeute).
Analytische Daten
1H- und 13C-NMR: vergleiche Tabellen 4 bis 6. Elementaranalyse (EA): %N = 5.42, %C = 36.98,
%H = 6.75, %O = 50.85. Summenformel laut EA: C8H15NO7 · H2O. 𝑝𝐾𝑠1 = 2.54, 𝑝𝐾𝑠2 = 8.31. HPLC-RI
(AMADORI-Methode): 𝑡𝑅 = 15.5𝑚𝑖𝑛.
6.1.6 SYNTHESE VON N-(1-DESOXY-D-FRUCTOS-1-YL)-β-ALANIN
Die Synthese von N-(1-Desoxy-D-fructos-1-yl)-β-alanin (Fructosyl-β-alanin) 6d erfolgte nach
der Vorschrift von RÖPER et al. (1983).[127]
M [g/mol] eq. n [mmol] m bzw. V
D-Glucose 180.16 4.0 271 48.835 g
β-Alanin 89.09 1.0 68 6.090 g
Natrium-Pyrosulfit 190.11 0.5 34 6.477 g
Methanol 250 mL
Durchführung
6.090 g β-Alanin (68 mmol; 1.0 eq.) werden zusammen mit 48.835 g wasserfreier D-Glucose
(271 mmol; 4.0 eq.) und 6.477 g Natrium-Pyrosulfit (34 mmol; 0.5 eq.) in 250 mL wasserfreiem
Methanol weitestgehend gelöst und die Suspension für 90 min unter Rückfluss erhitzt. Die
Reaktionskontrolle erfolgt dünnschichtchromatographisch auf Kieselgel 60 unter Nutzung von
n-Butanol/Eisessig/Wasser (4/2/1; v/v/v) als Laufmittel und 5 mM Ninhydrinlösung in Ethanol/Wasser
(9/1; v/v) als Detektionsreagenz.
Aufarbeitung
Nach Abkühlen der Suspension wird der Bodensatz abfiltriert und verworfen und das Filtrat
mit 500 mL bidest. Wasser verdünnt. Die Aufreinigung des Produktes aus dem Filtrat erfolgt
säulenchromatographisch an DowexTM 50WX8 (in der protonierten Form). Dazu wird die Lösung
zunächst auf den Kationentauscher aufgezogen. Anschließend wird mit Ethanol/Wasser (1/1; v/v) (ca.
zehnfaches Säulenvolumen) sowie bidest. Wasser (ca. zehnfaches Säulenvolumen) gewaschen und
schrittweise mit wässriger ammoniakalischer Lösung aufsteigender Konzentrationen (0.001 M;
0.01 M; 0.1 M) bei einer Tropfgeschwindigkeit von etwa einem Tropfen pro Sekunde eluiert. Die das
Produkt enthaltenden Fraktionen werden vereinigt, unter vermindertem Druck eingeengt und
anschließend gefriergetrocknet. Es ergeben sich 4.094 g Produkt als weißer Feststoff (22% Ausbeute).

EXPERIMENTELLER TEIL
- 112 -
Analytische Daten
1H- und 13C-NMR: vergleiche Tabellen 4 bis 6. Elementaranalyse (EA): %N = 5.12, %C = 39.90,
%H = 7.09, %O = 47.89. Summenformel laut EA: C9H17NO7 · H2O. 𝑝𝐾𝑠1 = 3.39, 𝑝𝐾𝑠2 = 8.84. HPLC-RI
(AMADORI-Methode): 𝑡𝑅 = 18.7𝑚𝑖𝑛.
6.1.7 SYNTHESE VON N-(1-DESOXY-D-FRUCTOS-1-YL)-γ-AMINOBUTTERSÄURE
Die Synthese von N-(1-Desoxy-D-fructos-1-yl)-γ-aminobuttersäure (Fructosyl-γ-aminobutter-
säure) 6e erfolgte nach der Vorschrift von RÖPER et al. (1983).[127]
M [g/mol] eq. n [mmol] m bzw. V
D-Glucose 180.16 3.9 233 42.048 g
γ-Aminobuttersäure 103.12 1.0 59 6.082 g
Natrium-Pyrosulfit 190.11 0.5 30 5.781 g
Methanol 250 mL
Durchführung
6.082 g γ-Aminobuttersäure (59 mmol; 1.0 eq.) werden zusammen mit 42.048 g wasserfreier
D-Glucose (233 mmol; 3.9 eq.) und 5.781 g Natrium-Pyrosulfit (30 mmol; 0.5 eq.) in 250 mL
wasserfreiem Methanol weitestgehend gelöst und die Suspension für 90 min unter Rückfluss erhitzt.
Die Reaktionskontrolle erfolgt dünnschichtchromatographisch auf Kieselgel 60 unter Nutzung von
n-Butanol/Eisessig/Wasser (4/2/1; v/v/v) als Laufmittel und 5 mM Ninhydrinlösung in Ethanol/Wasser
(9/1; v/v) als Detektionsreagenz.
Aufarbeitung
Nach Abkühlen der Suspension wird der Bodensatz abfiltriert und verworfen und das Filtrat
mit 500 mL bidest. Wasser verdünnt. Die Aufreinigung des Produktes aus dem Filtrat erfolgt
säulenchromatographisch an DowexTM 50WX8 (in der protonierten Form). Dazu wird die Lösung
zunächst auf den Kationentauscher aufgezogen. Anschließend wird mit Ethanol/Wasser (1/1; v/v) (ca.
zehnfaches Säulenvolumen) sowie bidest. Wasser (ca. zehnfaches Säulenvolumen) gewaschen und
schrittweise mit wässriger ammoniakalischer Lösung aufsteigender Konzentrationen (0.001 M;
0.01 M; 0.1 M) bei einer Tropfgeschwindigkeit von etwa einem Tropfen pro Sekunde eluiert. Die das
Produkt enthaltenden Fraktionen werden vereinigt, unter vermindertem Druck eingeengt und
anschließend gefriergetrocknet. Es ergeben sich 8.370 g Produkt als gelblicher Feststoff (48%
Ausbeute).

EXPERIMENTELLER TEIL
- 113 -
Analytische Daten
1H- und 13C-NMR: vergleiche Tabellen 4 bis 6. Elementaranalyse (EA): %N = 4.72, %C = 41.29,
%H = 7.57, %O = 46.42. Summenformel laut EA: C10H19NO7 · 1.5 H2O. 𝑝𝐾𝑠1 = 4.07, 𝑝𝐾𝑠2 = 9.13.
HPLC-RI (AMADORI-Methode): 𝑡𝑅 = 18.1𝑚𝑖𝑛.
6.1.8 REGENERATION DES IONENTAUSCHERS
Zur Regeneration des DowexTM 50WX8 Ionentauschers wird dieser in eine
chromatographische Trennsäule gefüllt, nacheinander mit 3 L bidest. Wasser, 600 mL Ethanol und
wiederum 2 L bidest. Wasser gewaschen und anschließend für 2 h in 1 N Natronlauge gerührt
(Becherglas). Der Ionentauscher wird erneut mit 3 L bidest. Wasser gewaschen und anschließend für
12 h in 2 N Salzsäure gerührt. Abschließend wird der auf diese Weise regenerierte Ionentauscher mit
bidest. Wasser neutral gewaschen.
6.2 pH-konstante Modellreaktionen
Die Abschätzung der Reaktivitäten der Zielsubstanzen erfolgte auf Grundlage von kinetischen
Untersuchungen wässriger Modellreaktionssysteme bei (90 ± 2) °C und konstantem pH-Wert auf sechs
Stufen (pH 1.5, pH 2.5, pH 3.5, pH 4.5, pH 5.5 und pH 6.5).
Durchführung von Modellreaktionen der D-Fructose
Es werden 9.008 g D-Fructose in 80 mL bidest. Wasser gelöst und der pH-Wert mittels
Salzsäure bzw. Natronlauge auf eine der sechs Stufen eingestellt. Die Lösung wird quantitativ in einen
100 mL Maßkolben überführt und mit bidest. Wasser zur Markierung ergänzt. Die
D-Fructosekonzentration der Lösung beträgt 0.5 mol/L. 50 mL dieser Lösung werden in einen 100 mL
Sulfierkolben pipettiert. Dieser ist mit einem Rückflusskühler, einem Temperaturfühler, einer
pH-Elektrode, einer Dosierspitze (Titrationsautomat) und einem Septum mit Kanüle ausgestattet. Die
Lösung wird unter Rückfluss im Ölbad auf 90 °C erhitzt und der pH-Wert mit Hilfe eines
Titrierautomaten auf den Zielwert nachgeregelt. Die Messung der Reaktionszeit wird gestartet, sobald
sowohl Temperatur als auch pH-Wert ihre Zielwerte erreicht haben. Der Reaktionslösung werden mit
Hilfe von Spritzen sodann zeitabhängig Proben (je etwa 1 mL) durch das Septum entnommen. Dabei
sind das derzeit titrierte Volumen, das entnommene Volumen, der pH-Wert sowie die Temperatur zu
protokollieren. Die Probennahme erfolgt über einen Zeitraum von 7 h im Abstand von je 30 min.
Durchführung von Modellreaktionen der D-Fructose in stöchiometrischer Mischung mit
Aminosäuren
Es wird die in Tabelle 14 aufgeführte Masse der jeweiligen Aminosäure in 80 mL bidest.
Wasser gelöst und der pH-Wert mittels Salzsäure bzw. Natronlauge auf eine der sechs Stufen
eingestellt. Es erfolgt die Zugabe von 9.008 g D-Fructose. Die Lösung wird quantitativ in einen 100 mL
Maßkolben überführt und mit bidest. Wasser zur Markierung ergänzt. Die Konzentration der Lösung
an D-Fructose und Aminosäure beträgt jeweils 0.5 mol/L. 50 mL dieser Lösung werden in einen 100 mL
Sulfierkolben pipettiert. Dieser ist mit einem Rückflusskühler, einem Temperaturfühler, einer
pH-Elektrode, einer Dosierspitze (Titrationsautomat) und einem Septum mit Kanüle ausgestattet. Die
Lösung wird unter Rückfluss im Ölbad auf 90 °C erhitzt und der pH-Wert mit Hilfe eines
Titrierautomaten auf den Zielwert nachgeregelt. Die Messung der Reaktionszeit wird gestartet, sobald
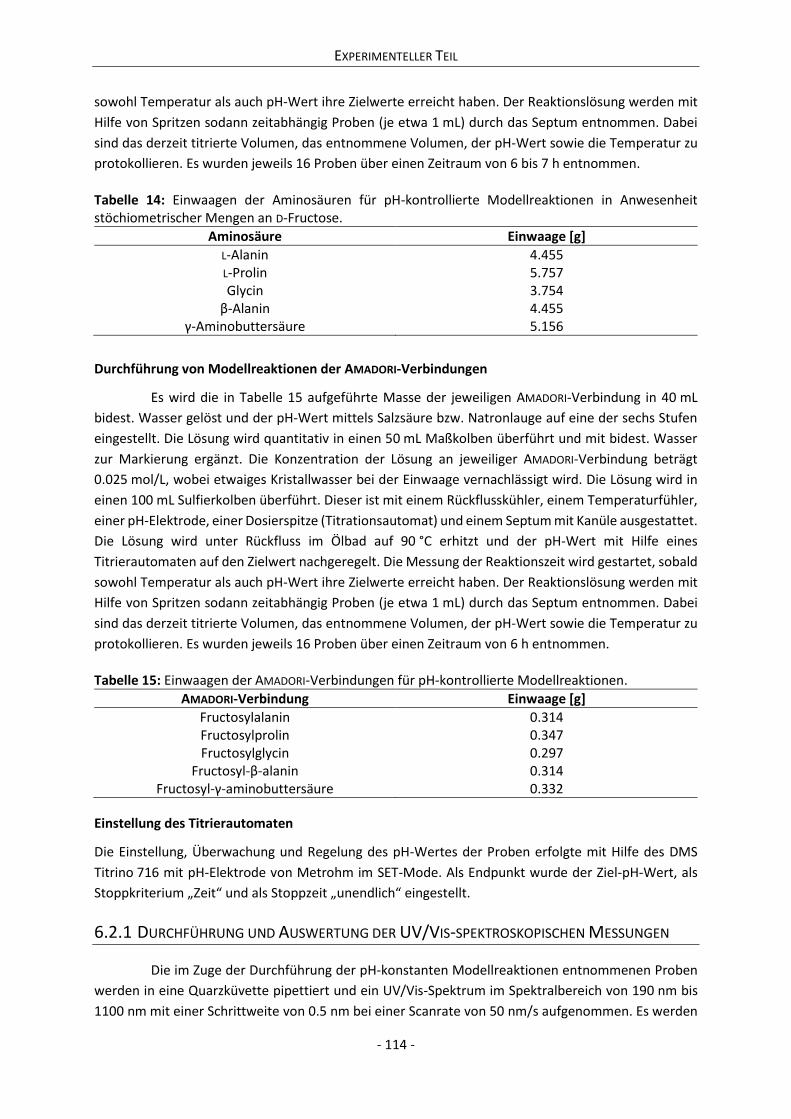
EXPERIMENTELLER TEIL
- 114 -
sowohl Temperatur als auch pH-Wert ihre Zielwerte erreicht haben. Der Reaktionslösung werden mit
Hilfe von Spritzen sodann zeitabhängig Proben (je etwa 1 mL) durch das Septum entnommen. Dabei
sind das derzeit titrierte Volumen, das entnommene Volumen, der pH-Wert sowie die Temperatur zu
protokollieren. Es wurden jeweils 16 Proben über einen Zeitraum von 6 bis 7 h entnommen.
Tabelle 14: Einwaagen der Aminosäuren für pH-kontrollierte Modellreaktionen in Anwesenheit stöchiometrischer Mengen an D-Fructose.
Aminosäure Einwaage [g]
L-Alanin 4.455 L-Prolin 5.757 Glycin 3.754
β-Alanin 4.455 γ-Aminobuttersäure 5.156
Durchführung von Modellreaktionen der AMADORI-Verbindungen
Es wird die in Tabelle 15 aufgeführte Masse der jeweiligen AMADORI-Verbindung in 40 mL
bidest. Wasser gelöst und der pH-Wert mittels Salzsäure bzw. Natronlauge auf eine der sechs Stufen
eingestellt. Die Lösung wird quantitativ in einen 50 mL Maßkolben überführt und mit bidest. Wasser
zur Markierung ergänzt. Die Konzentration der Lösung an jeweiliger AMADORI-Verbindung beträgt
0.025 mol/L, wobei etwaiges Kristallwasser bei der Einwaage vernachlässigt wird. Die Lösung wird in
einen 100 mL Sulfierkolben überführt. Dieser ist mit einem Rückflusskühler, einem Temperaturfühler,
einer pH-Elektrode, einer Dosierspitze (Titrationsautomat) und einem Septum mit Kanüle ausgestattet.
Die Lösung wird unter Rückfluss im Ölbad auf 90 °C erhitzt und der pH-Wert mit Hilfe eines
Titrierautomaten auf den Zielwert nachgeregelt. Die Messung der Reaktionszeit wird gestartet, sobald
sowohl Temperatur als auch pH-Wert ihre Zielwerte erreicht haben. Der Reaktionslösung werden mit
Hilfe von Spritzen sodann zeitabhängig Proben (je etwa 1 mL) durch das Septum entnommen. Dabei
sind das derzeit titrierte Volumen, das entnommene Volumen, der pH-Wert sowie die Temperatur zu
protokollieren. Es wurden jeweils 16 Proben über einen Zeitraum von 6 h entnommen.
Tabelle 15: Einwaagen der AMADORI-Verbindungen für pH-kontrollierte Modellreaktionen.
AMADORI-Verbindung Einwaage [g]
Fructosylalanin 0.314 Fructosylprolin 0.347 Fructosylglycin 0.297
Fructosyl-β-alanin 0.314 Fructosyl-γ-aminobuttersäure 0.332
Einstellung des Titrierautomaten
Die Einstellung, Überwachung und Regelung des pH-Wertes der Proben erfolgte mit Hilfe des DMS
Titrino 716 mit pH-Elektrode von Metrohm im SET-Mode. Als Endpunkt wurde der Ziel-pH-Wert, als
Stoppkriterium „Zeit“ und als Stoppzeit „unendlich“ eingestellt.
6.2.1 DURCHFÜHRUNG UND AUSWERTUNG DER UV/VIS-SPEKTROSKOPISCHEN MESSUNGEN
Die im Zuge der Durchführung der pH-konstanten Modellreaktionen entnommenen Proben
werden in eine Quarzküvette pipettiert und ein UV/Vis-Spektrum im Spektralbereich von 190 nm bis
1100 nm mit einer Schrittweite von 0.5 nm bei einer Scanrate von 50 nm/s aufgenommen. Es werden

EXPERIMENTELLER TEIL
- 115 -
anschließend die Extinktionen bei 284 nm zur Verfolgung der frühen MAILLARD-Reaktion und bei
420 nm zur Verfolgung der fortgeschrittenen MAILLARD-Reaktion extrahiert und gegen die Zeit
aufgetragen. Die Kinetik der Extinktionszunahme wird in Anlehnung an die Arbeiten von VAIKOUSI et al.
(2008, 2009)[5,167] mit einer logistischen Funktion im Sinne einer autokatalytischen Reaktion modelliert.
Die entsprechende Funktion ist durch Gleichung (57) gegeben.
𝐸 =𝐸𝑚𝑎𝑥
1 + (𝐸𝑚𝑎𝑥𝐸0
− 1) ∙ 𝑒𝑥𝑝(−𝑘 ∙ 𝑡) (57)
Dabei ist 𝐸 die gemessene Extinktion, 𝐸𝑚𝑎𝑥 die maximal zu erreichende Extinktion,𝐸0 die
Extinktion zum Zeitpunkt 𝑡 = 0𝑠, 𝑡 die Zeit [s] und 𝑘 die Geschwindigkeitskonstante [Hz]. Die
numerische Berechnung der Parameter 𝐸𝑚𝑎𝑥, 𝐸0 und 𝑘 erfolgte im Rahmen dieser Arbeit mit Hilfe des
XLSTAT 2017 Statistik-Analyse Add-Ins für Microsoft Excel 2016. Bei den angegebenen Fehlern handelt
es sich um die Standardfehler der geschätzten Parameter.
6.2.2 MESSUNG UND AUSWERTUNG DER ABBAUKINETIKEN DER D-FRUCTOSE
Die im Zuge der Durchführung der pH-konstanten Modellreaktionen entnommenen Proben
werden mit Acetonitril/Wasser (40/9; v/v) um den Faktor 50 verdünnt und mittels HILIC-RI gemessen.
Die Parameter der genutzten HPLC-Methode sind in Tabelle 16 gegeben.
Tabelle 16: Parameter der HILIC-RI-Methode zur Quantifizierung von D-Fructose.
Parameter Wert
Laufmittel Acetonitril/Wasser (95/5; v/v) Säule EC 250/3 Nucleodur® HILIC, Macharey Nagel
Flussrate 1.15 mL/min Laufzeit 10 min
Injektionsvolumen 10 µL Temperatur des Säulenofens 35 °C
Detektion und Quantifizierung Brechungsindex
Da im Rahmen der vorliegenden Arbeit lediglich die Abbauraten der D-Fructose von Interesse
sind, erfolgte ihre Quantifizierung nicht in Form von Absolutwerten. Es wurden lediglich relative
Konzentrationsänderungen gemessen und mit einer Reaktionskinetik der ersten Ordnung modelliert.
Die zugrundeliegende Funktion ist durch Gleichung (58) gegeben.
[𝐹𝑟𝑢]𝑡[𝐹𝑟𝑢]𝑡=0𝑠
=𝐼𝑛𝑡(𝑡)
𝐼𝑛𝑡(𝑡 = 0𝑠)= 𝑒𝑥𝑝(−𝑘 ∙ 𝑡) (58)
Dabei ist [𝐹𝑟𝑢]𝑡 die Konzentration der D-Fructose zum Zeitpunkt 𝑡, [𝐹𝑟𝑢]𝑡=0𝑠 die
Konzentration der D-Fructose zum Zeitpunkt 𝑡 = 0𝑠, 𝐼𝑛𝑡(𝑡) das Integral des HPLC-Peaks der D-Fructose
zum Zeitpunkt 𝑡, 𝐼𝑛𝑡(𝑡 = 0𝑠) das Integral des HPLC-Peaks der D-Fructose zum Zeitpunkt 𝑡 = 0𝑠, 𝑡 die
Zeit [s] und 𝑘 die Abbaurate der D-Fructose [Hz]. Die numerische Berechnung der Parameter
𝐼𝑛𝑡(𝑡 = 0𝑠) und 𝑘 erfolgte im Rahmen dieser Arbeit mit Hilfe des XLSTAT 2017 Statistik-Analyse
Add-Ins für Microsoft Excel 2016. Bei den angegebenen Fehlern handelt es sich um die Standardfehler
der geschätzten Parameter.

EXPERIMENTELLER TEIL
- 116 -
6.2.3 MESSUNG UND AUSWERTUNG DER ABBAUKINETIKEN DER AMINOSÄUREN
Die im Zuge der Durchführung der pH-konstanten Modellreaktionen entnommenen Proben
werden mit Hexansulfonsäure (0.1 M in Wasser)/Wasser/Methanol (365/265/292; v/v/v) um den
Faktor 10 verdünnt und die Aminosäuren ionenpaarchromatographisch mittels RP-HPLC-RI gemessen.
Die Parameter der genutzten HPLC-Methode sind in Tabelle 17 gegeben.
Tabelle 17: Parameter der ionenpaarchromatographischen RP-HPLC-RI-Methode zur Quantifizierung der Aminosäuren.
Parameter Wert
Laufmittel Hexansulfonsäure (0.01 M in Wasser)/Methanol (5/2; v/v) Säule EC 250/4.6 Nucleosil® 120-5 C18, Macharey Nagel
Flussrate 1.10 mL/min Laufzeit 5 min
Injektionsvolumen 5 µL Temperatur des Säulenofens 55 °C
Detektion und Quantifizierung Brechungsindex
Da im Rahmen der vorliegenden Arbeit lediglich die Abbauraten der Aminosäuren von
Interesse sind, erfolgte ihre Quantifizierung nicht in Form von Absolutwerten. Es wurden lediglich
relative Konzentrationsänderungen gemessen und mit einer Reaktionskinetik der ersten Ordnung
modelliert. Die zugrundeliegende Funktion ist durch Gleichung (59) gegeben.
[𝐴𝑆]𝑡[𝐴𝑆]𝑡=0𝑠
=𝐼𝑛𝑡(𝑡)
𝐼𝑛𝑡(𝑡 = 0𝑠)= 𝑒𝑥𝑝(−𝑘 ∙ 𝑡) (59)
Dabei ist [𝐴𝑆]𝑡 die Konzentration der Aminosäure zum Zeitpunkt 𝑡, [𝐴𝑆]𝑡=0𝑠 die
Konzentration der Aminosäure zum Zeitpunkt 𝑡 = 0𝑠, 𝐼𝑛𝑡(𝑡) das Integral des HPLC-Peaks der
Aminosäure zum Zeitpunkt 𝑡, 𝐼𝑛𝑡(𝑡 = 0𝑠) das Integral des HPLC-Peaks der Aminosäure zum Zeitpunkt
𝑡 = 0𝑠, 𝑡 die Zeit [s] und 𝑘 die Abbaurate der Aminosäure [Hz]. Die numerische Berechnung der
Parameter 𝐼𝑛𝑡(𝑡 = 0𝑠) und 𝑘 erfolgte im Rahmen dieser Arbeit mit Hilfe des XLSTAT 2017
Statistik-Analyse Add-Ins für Microsoft Excel 2016. Bei den angegebenen Fehlern handelt es sich um
die Standardfehler der geschätzten Parameter.
6.2.4 MESSUNG UND AUSWERTUNG DER ABBAUKINETIKEN DER AMADORI-VERBINDUNGEN
Die im Zuge der Durchführung der pH-konstanten Modellreaktionen entnommenen Proben
werden unverdünnt für die Messung der Konzentrationen der AMADORI-Verbindungen eingesetzt. Die
Quantifizierung der AMADORI-Verbindungen erfolgt ionenpaarchromatographisch mittels RP-HPLC-RI.
Die Parameter der genutzten HPLC-Methode sind in Tabelle 18 gegeben.

EXPERIMENTELLER TEIL
- 117 -
Tabelle 18: Parameter der ionenpaarchromatographischen RP-HPLC-RI-Methode zur Quantifizierung der AMADORI-Verbindungen.
Parameter Wert
Laufmittel Hexansulfonsäure (0.01 M in Wasser)/Methanol (5/2; v/v)
Säulen 1. YMC-Pack ODS-AM AM-303 AM12S05-2546WT 250 x 4.6 mm
T.D. S-5micrometer, 12 nm 2. VDS optilab ODS Hypersil 120 A 5 micrometer 15 x 4.6 mm
Flussrate 0.35 mL/min Laufzeit 25 min
Injektionsvolumen 5 µL Temperatur des Säulenofens 55 °C
Detektion und Quantifizierung Brechungsindex
Da im Rahmen der vorliegenden Arbeit lediglich die Abbauraten der AMADORI-Verbindungen
von Interesse sind, erfolgte ihre Quantifizierung nicht in Form von Absolutwerten. Es wurden lediglich
relative Konzentrationsänderungen gemessen und mit einer Reaktionskinetik der ersten Ordnung
modelliert. Die zugrundeliegende Funktion ist durch Gleichung (60) gegeben.
[𝐴𝑀𝑉]𝑡[𝐴𝑀𝑉]𝑡=0𝑠
=𝐼𝑛𝑡(𝑡)
𝐼𝑛𝑡(𝑡 = 0𝑠)= 𝑒𝑥𝑝(−𝑘 ∙ 𝑡) (60)
Dabei ist [𝐴𝑀𝑉]𝑡 die Konzentration der AMADORI-Verbindung zum Zeitpunkt 𝑡, [𝐴𝑀𝑉]𝑡=0𝑠
die Konzentration der AMADORI-Verbindung zum Zeitpunkt 𝑡 = 0𝑠, 𝐼𝑛𝑡(𝑡) das Integral des HPLC-Peaks
der AMADORI-Verbindung zum Zeitpunkt 𝑡, 𝐼𝑛𝑡(𝑡 = 0𝑠) das Integral des HPLC-Peaks der
AMADORI-Verbindung zum Zeitpunkt 𝑡 = 0𝑠, 𝑡 die Zeit [s] und 𝑘 die Abbaurate der AMADORI-Verbindung
[Hz]. Die numerische Berechnung der Parameter 𝐼𝑛𝑡(𝑡 = 0𝑠) und 𝑘 erfolgte im Rahmen dieser Arbeit
mit Hilfe des XLSTAT 2017 Statistik-Analyse Add-Ins für Microsoft Excel 2016. Bei den angegebenen
Fehlern handelt es sich um die Standardfehler der geschätzten Parameter.
6.2.5 MESSUNG UND AUSWERTUNG DER KINETIKEN DER α-DICARBONYLVERBINDUNGEN
Die im Zuge der Durchführung der pH-konstanten Modellreaktionen entnommenen Proben
werden zur Quantifizierung der α-Dicarbonylverbindungen (3-Desoxyglucoson, 3-Desoxygalactoson,
1-Desoxyglucoson sowie D-Glucoson) mittels ortho-Phenylendiamin (OPD) derivatisiert. Hierzu werden
je 200 µL der Proben mit 200 µL 50 mM OPD-Lösung (54 mg OPD, gelöst in 10 mL Methanol/Wasser
(1/1; v/v)) versetzt und bei Raumtemperatur für mindestens 24 h unter Lichtausschluss gelagert. Die
Quantifizierung der α-Dicarbonylverbindungen als Chinoxalinderivate erfolgt mit Hilfe authentischer
Standards mittels RP-HPLC-DAD. 3-Desoxygalactoson-Chinoxalin wurde mit Hilfe eines authentischen
Standards identifiziert, jedoch als Summenparameter zusammen mit 3-Desoxyglucoson-Chinoxalin
quantifiziert. Die Parameter und der Laufmittelgradient der genutzten HPLC-Methode sind in den
Tabellen 19 und 20 gegeben.
EXPERIMENTELLER TEIL
- 118 -
Tabelle 19: Parameter der RP-HPLC-DAD-Methode zur Quantifizierung der α-Dicarbonylverbindungen.
Parameter Wert
Laufmittel Methanol/Wasser Säule EC 250/4.6 Nucleosil® 120-5 C18, Macharey Nagel
Flussrate 1.0 mL/min Laufzeit 55 min
Injektionsvolumen 8 bis 40 µL Temperatur des Säulenofens 35 °C
Detektion 190 bis 800 nm Quantifizierung 317 nm
Tabelle 20: Laufmittelgradient der RP-HPLC-DAD-Methode zur Quantifizierung der α-Dicarbonyl-verbindungen.
Zeit [min] Anteil Methanol [%] Anteil Wasser [%]
0 20 80 4 20 80
11 33 67 19 65 35 28 100 0 32 0 100 40 30 70 45 20 80
Die Berechnung der Bildungsraten von 1- und 3-Desoxyglucoson erfolgte auf Grundlage der
Gleichungen (40) und (41) (Seite 48). Die numerische Berechnung der Parameter 𝑘1, 𝑘2, 𝑘−1 und 𝑘−2
erfolgte im Rahmen dieser Arbeit mit Hilfe des Solver Add-Ins von Microsoft Excel 2016.
6.3 NMR-Spektroskopische Messungen
Für die NMR-spektroskopischen Messungen wurden jeweils 0.5 M Probelösungen in 750 µL
Deuteriumoxid (D2O) hergestellt (im Falle der Proben der D-Glucose waren die Proben 0.2 M), das
durch Spuren von Ameisensäure verunreinigt war. Die Einstellung des pD-Wertes der Probelösungen
erfolgte durch die Zugabe verdünnter Lösungen von Natrium-Deuteroxid (in D2O) bzw.
Deuteriumchlorid (in D2O). Die Messung des pD-Wertes erfolgte mit Hilfe eines pH-Meters mit
Glaselektrode. Die Kalibrierung erfolgte dabei mit wässrigen Puffern (H2O). Der am pH-Meter
abgelesene Wert wird als pH*-Wert bezeichnet und entspricht nicht dem pD-Wert. Die Umrechnung
des pH*-Wertes in den pD-Wert erfolgt über eine empirisch ermittelte Korrelation, die durch
Gleichung (61) gegeben ist.[348,355,356]
𝑝𝐷 = 𝑝𝐻∗ + 0.44 (61)
6.3.1 STRUKTURAUFKLÄRUNG
Die Durchführung der im Rahmen der Strukturaufklärung der AMADORI-Verbindungen
benötigten NMR-spektroskopischen Experimente erfolgte an nicht isotopenangereicherten
Verbindungen an einem Bruker AVANCE III 500 NMR-Spektrometer (Messfrequenz für Protonen
500.13 MHz) mit einem 5 mm BBO-Probenkopf sowie einem Bruker AVANCE 600 NMR-Spektrometer
(Messfrequenz für Protonen 599.89 MHz) mit einem 5 mm BBI-Probenkopf. Als Akquisitionssoftware
dienten die TopSpin Versionen 1.3, 2.1 und 3.1 von Bruker. Die Auswertung der Spektren erfolgte mit
EXPERIMENTELLER TEIL
- 119 -
den TopSpin Versionen 3.2 pL 6 und 3.5 pL 2. Es wurden Standard Bruker-Pulsprogramme mit
verschiedenen Akquisitionsparametern genutzt. Eine Übersicht über die genutzten Pulsprogramme
sowie repräsentative Akquisitionsparameter gibt Tabelle 21.
Tabelle 21: Übersicht über die genutzten Pulsprogramme und Angabe repräsentativer Akquisitionsparameter.
NMR-Experiment Bruker
Pulsprogramm Anzahl der
Datenpunkte in f1 Anzahl der
Datenpunkte in f2 Anzahl der
Scans 1H zg bzw. zg30 64 k – 32
13C zgpg30 64 k – 15 k
DEPT dept135 64 k – 4 k
COSY cosygpqf 512 4 k 2
NOESY noesygpph 128 2 k 4
HSQC hsqcetgp 512 2 k 32
HSQC-TOCSY hsqcetgpml 512 4 k 32
HMBC hmbcgplpndqf 512 2 k 32
J-resolved jresqf 256 16 k 16
Die angegebenen chemischen Verschiebungen sind auf die jeweiligen 1H- und 13C-NMR-Resonanzen des internen Standards Aceton referenziert (𝛿( 𝐻
1 ) = 2.04𝑝𝑝𝑚bzw.𝛿( 𝐶13 ) =
30.5𝑝𝑝𝑚).
6.3.2 KALIBRIERUNG DER TEMPERATUR
Die Kalibrierung der von der variablen Temperatureinheit (variable temperature unit, VTU;
Bruker BVT 3000 Digital) eingestellten Temperatur innerhalb des NMR-Probenröhrchens erfolgte
anhand der Messung der temperaturabhängigen Differenz der chemischen Verschiebungen der 1H-NMR-Resonanzen der Hydroxyl- und Methylenprotonen von 1,2-Ethandiol.[357] Es wurde dazu eine
kommerzielle Probe von 80% 1,2-Ethandiol in DMSO-d6 verwendet. Die Kalibrierung erfolgte durch die
Messung der Probe bei verschiedenen Solltemperaturen im Bereich von 23 bis 83 °C in Intervallen von
5 K.
Durchführung
Die Solltemperatur wird mit Hilfe des Befehls „edte“ innerhalb der Software eingestellt. Nach
einer Wartezeit von 10 bis 20 min, die für die Einstellung des thermischen Gleichgewichtes benötigt
wird, erfolgt die Messung eines single-scan 1H-NMR-Spektrums. Die Verarbeitung des Spektrums
erfolgt mit einem Linienverbreiterungsfaktor lb von 5 Hz. Anschließend wird die tatsächliche
Temperatur innerhalb der Probe mit Hilfe des Automationsprogramms (AU-Programms) „calctemp“ in
der TopSpin Version 3.2 pL 6 ausgelesen. Die Auftragung der Ist- gegen die Solltemperaturen kann mit
Hilfe einer quadratischen Funktion modelliert werden, die die Daten mit einem Bestimmtheitsmaß von
1.0000 beschreibt.

EXPERIMENTELLER TEIL
- 120 -
6.3.3 KALIBRIERUNG DES PD-WERTES
Die Kalibrierung des pD-Wertes beruht in erster Linie auf der pD-abhängigen chemischen
Verschiebung der 1H-NMR-Resonanz des Methinprotons der Ameisensäure. Um eine Dekodierung des
pD-Wertes aus einem bestehenden 1H-NMR-Spektrum zu ermöglichen, wurde eine 0.5 M Probe der zu
untersuchenden Zielsubstanz in Deuteriumoxid hergestellt (0.2 M Lösungen im Falle der D-Glucose).
Dabei enthielt das verwendete Lösungsmittel in jedem Fall Ameisensäure als Verunreinigung.
Durchführung
Durch Zugabe verdünnter Lösungen von Natrium-Deuteroxid (in D2O) bzw. Deuteriumchlorid
(in D2O) wird ein pD-Wert der Probe im Bereich von 2.0 bis 6.5 eingestellt. Die Messung des pD-Wertes
erfolgt durch die Messung des pH*-Wertes mit Hilfe eines pH-Meters mit Glaselektrode, das zuvor mit
wässrigen Pufferlösungen kalibriert wurde. Der pH*- sowie der pD-Wert werden protokolliert und die
Probe in ein NMR-Probenröhrchen überführt. Die Probe wird innerhalb des NMR-Spektrometers auf
eine vorgegebene Temperatur (hier: 23 °C) temperiert und anschließend ein 1H-NMR-Spektrum der
Probe gemessen. Der Vorgang wird zur Erstellung der Kalibrierfunktion etwa zehnmal wiederholt.
Da die chemische Verschiebung der 1H-NMR-Resonanz des Methinprotons der Ameisensäure
nicht nur pD-, sondern auch in hohem Maße temperaturabhängig ist, muss die pD-Kalibrierung bei
verschiedenen Temperaturen wiederholt werden. Eine andere Möglichkeit besteht in der folgenden
Vorgehensweise. Der pD-Wert der Probe wird auf einen Wert von ca. 2.0 eingestellt. Anschließend
erfolgt die Messung von 1H-NMR-Spektren der Probe bei verschiedenen Temperaturen (hier: 23 bis
83 °C). Die Überlagerung der Spektren zeigt, dass alle gemessenen Protonenresonanzen
temperaturabhängig sind. Eine Überlagerung der in Abhängigkeit des pD-Wertes gemessenen
Spektren zeigt demgegenüber, dass viele der Protonenresonanzen der zu messenden Zuckerderivate
im Bereich von pD 2.0 bis 6.5 keine Abhängigkeit vom pD-Wert zeigen. Der Einfluss der Temperatur
auf die chemische Verschiebung kann demnach korrigiert werden, indem eine NMR-Resonanz des
Zuckerderivates gefunden wird, die eine möglichst ähnliche Temperaturabhängigkeit wie die Resonanz
der Ameisensäure zeigt. Ist die Differenz der Signale temperaturunabhängig konstant, so ist sie
lediglich abhängig vom pD-Wert. Besteht weiterhin eine geringfügige Temperaturabhängigkeit der
Differenz der Frequenzen, so kann diese polynomisch modelliert und herausgerechnet werden. Wurde
ein entsprechendes Paar von Protonenresonanzen identifiziert, erfolgt die Kalibrierung des pD-Wertes
durch die Auftragung der bei verschiedenen pD-Werten gemessenen Differenzen ihrer
Resonanzfrequenzen gegen den pD-Wert der Proben. Die nicht lineare Regression der Daten erfolgt
auf Grundlage von Gleichung (62), die auf Basis der chemischen Verschiebung im schnellen chemischen
Austausch in Kombination mit der HENDERSON-HASSELBALCH-Funktion hergeleitet werden kann.
∆𝜈 =∆𝜈𝑚𝑎𝑥 + ∆𝜈𝑚𝑖𝑛 ∙ 10
(𝑝𝐾𝑠−𝑝𝐷)
1 + 10(𝑝𝐾𝑠−𝑝𝐷) (62)
Dabei ist ∆𝜈𝑚𝑎𝑥 die maximal mögliche Differenz der zugrundeliegenden Frequenzen der
Protonenresonanzen, ∆𝜈𝑚𝑖𝑛 die minimal mögliche Differenz, ∆𝜈 die gemessene Differenz und 𝑝𝐾𝑠 der
𝑝𝐾𝑠-Wert der Ameisensäure in Deuteriumoxid. Dieser wurde im Rahmen der vorliegenden Arbeit
vielfach mit Werten um 4.1 bestimmt. Daraus ergibt sich mit Hilfe der durch MARTIN (1963) gegebenen
Umrechnungsfunktion ein 𝑝𝐾𝑠-Wert der Ameisensäure in H2O von 3.6, der in guter Übereinstimmung

EXPERIMENTELLER TEIL
- 121 -
mit dem im Rahmen dieser Arbeit auf Grundlage potentiometrischer Titrationen ermittelten Wert
ist.[331]
Die Berechnung von pD-Werten auf Grundlage gemessener Frequenzdifferenzen erfolgt
schließlich mit Hilfe von Gleichung (63).
𝑝𝐷 = 𝑝𝐾𝑠 − 𝑙𝑔 (∆𝜈𝑚𝑎𝑥 − ∆𝜈
∆𝜈 − ∆𝜈𝑚𝑖𝑛) (63)
6.3.4 QUANTITATIVE NMR-SPEKTROSKOPIE (QNMR)
Die theoretischen Grundlagen der quantitativen NMR-Spektroskopie wurden in Kapitel 2.2.2
näher erläutert. Die Quantifizierung relativer Anomerenkonzentrationen erfolgte im Rahmen dieser
Arbeit für sämtliche Ketosen auf Grundlage der quantitativen 13C-NMR-Spektroskopie. Dabei kamen in
jedem Fall [2-13C]-markierte Verbindungen zum Einsatz. Im Falle der D-Glucose erfolgte die
entsprechende Quantifizierung auf Grundlage der quantitativen 1H-NMR-Spektroskopie. Diese wurde
darüber hinaus auch für die Messung von Enolisierungsraten genutzt. Alle quantitativen Messungen
erfolgten an einem Bruker AVANCE 600 NMR-Spektrometer (Messfrequenz für Protonen 599.89 MHz)
mit einem 5 mm BBI-Probenkopf. Lediglich ein Teil der Messungen der Enolisierungsraten erfolgte an
einem Bruker AVANCE III 500 NMR-Spektrometer (Messfrequenz für Protonen 500.13 MHz) mit einem
5 mm BBO-Probenkopf. Es wurde in jedem Fall ein Relaxationsdelay von mindestens 5 ∙ 𝑇1 eingehalten.
Die Auswertung der NMR-Spektren erfolgt durch deren Deconvolution. Dabei werden die relevanten
NMR-Resonanzen durch LORENTZ- bzw. GAUSS-Funktionen modelliert und die Integrale der
NMR-Resonanzen durch Integration der Modellfunktionen von −∞ bis +∞ berechnet. Dies erfolgt
über den TopSpin-Befehl „dcon“.
Bei quantitativen 1H-NMR-Experimenten handelt es sich um gewöhnliche Experimente mit
verlängertem Relaxationsdelay. Die Durchführung quantitativer 13C-NMR-Experimente ist im
Folgenden näher beschrieben.
Durchführung quantitativer 13C-NMR-Experimente
Die Messung erfolgt mit dem Bruker Pulsprogramm „zgig“. Dabei handelt es sich um ein
eindimensionales NMR-Experiment mit inverse-gated decoupling. Dabei findet die
Protonenentkopplung der 13C-NMR-Resonanzen ausschließlich während der Akquisitionszeit statt, um
einen Einfluss des Kern-OVERHAUSER-Effektes auf die Signalintensitäten möglichst auszuschließen. Die
Durchführung des Experimentes beginnt mit vier dummy scans. Die nachfolgenden FIDs (etwa 1 bis 16
Einzelexperimente) werden akkumuliert. Die Messung erfolgt mit einer FID-Auflösung von 0.46 Hz pro
Datenpunkt bei einer Gesamtzahl an Datenpunkten von 128 k. Die FOURIER-Transformation erfolgt
nach einem zero filling zu insgesamt 256 k Datenpunkten und der Anwendung einer
Exponentialfunktion mit einem Linienverbreiterungsfaktor lb von 1 Hz.
6.3.5 MESSUNG VON ENOLISIERUNGSRATEN
Die Messung von Enolisierungsraten erfolgte auf Grundlage der NMR-Resonanzen C-H-acider
Protonen. Dazu wurden je 0.5 M Lösungen der Zielsubstanzen in 750 µL Deuteriumoxid hergestellt und
ihre pD-Werte durch Zugabe verdünnter Lösungen von Natrium-Deuteroxid (in D2O) bzw.
Deuteriumchlorid (in D2O) den Vorgaben entsprechend eingestellt.

EXPERIMENTELLER TEIL
- 122 -
Messungen bei Raumtemperatur
Es werden quantitative 1H-NMR-Spektren der vorbereiteten Probelösungen in regelmäßigen
Zeitabständen über einen ausreichend langen Zeitraum gemessen (hier: 24 Tage). Die Verarbeitung
der Spektren erfolgt mit Hilfe des AU-Programms „multicmd“. Die FIDs der Einzelmessungen werden
FOURIER-transformiert sowie phasen- und basislinienkorrigiert. Anschließend werden die
Integrationsgrenzen der relevanten Spektralbereiche festgelegt. Dabei handelt es sich zum einen um
die NMR-Resonanz der austauschenden Protonen und zum anderen um die Resonanz eines nicht
austauschenden Protons. Die Integration des gesamten Datensatzes erfolgt mit Hilfe des
AU-Programms „multi_integ3“. Die von der Software ausgegebene Textdatei wird in ein
Tabellenkalkulationsprogramm importiert und das Integral der NMR-Resonanz der austauschenden
Protonen ins Verhältnis zum entsprechenden Integral des nicht austauschenden Protons gesetzt. Die
nicht lineare Regression der auf diese Weise berechneten relativen Integrale als Funktion der Zeit
erfolgt auf Grundlage von Gleichung (45) (Seite 69).
Temperaturabhängige Messungen
Temperaturabhängige Messungen von Enolisierungsraten erfolgten im Rahmen dieser Arbeit
für die AMADORI-Verbindungen der homologen ω-Aminosäuren. Die Messung der Enolisierungsraten
erfolgte hierbei simultan zur Messung konzentrationsgewichteter Ringöffnungsraten. Das hierfür
geschriebene Pulsprogramm („kfm-double-1hsst“) befindet sich im Anhang dieser Arbeit. Bei dem
resultierenden Spektrum handelt es sich um eine pseudo-2D-Matrix, welche nicht unmittelbar mit
Hilfe des AU-Programms „multi_integ3“ ausgewertet werden kann. Eine zur unter „Messungen bei
Raumtemperatur“ analoge Auswertung ist jedoch möglich, wenn zunächst das AU-Programm „split2D“
auf die pseudo-2D-Matrix angewendet wird. Die eindimensionalen NMR-Experimente, aus denen sich
die pseudo-2D-Matrix zusammensetzt, werden dabei als aufeinanderfolgend nummerierte
„PROCNOs“ innerhalb des „EXPNO“ Dateiordners abgelegt. Da das in TopSpin implementierte
AU-Programm „multicmd“ lediglich die Prozessierung fortlaufend nummerierter „EXPNOs“ erlaubt,
wurde dieses entsprechend für die Prozessierung fortlaufend nummerierter „PROCNOs“ angepasst.
Das resultierende AU-Programm („mpcmd“) befindet sich im Anhang.
6.3.6 SELEKTIVE SÄTTIGUNGSTRANSFER-NMR-SPEKTROSKOPIE (SST-NMR)
Die Messung von anomerenspezifischen Ringöffnungsraten erfolgte im Rahmen der
vorliegenden Arbeit mit Hilfe der selektiven Sättigungstransfer-NMR-Spektroskopie (SST-NMR). Die
Messung der Ringöffnungsraten erfolgte für D-Fructose in An- und Abwesenheit von L-Alanin und
L-Prolin, für D-Tagatose sowie für die AMADORI-Verbindungen Fructosylalanin und -prolin auf Grundlage
ihrer anomeren 13C-NMR-Resonanzen. Dabei kamen in jedem Falle [2-13C]-markierte Verbindungen
zum Einsatz. Im Falle der D-Glucose sowie der AMADORI-Verbindungen der homologen ω-Aminosäuren
wurden die NMR-Resonanzen der Protonen am jeweiligen C-1 ausgenutzt. Alle SST-NMR-Experimente
erfolgten an einem Bruker AVANCE 600 NMR-Spektrometer (Messfrequenz für Protonen 599.89 MHz)
mit einem 5 mm BBI-Probenkopf. Es wurden in jedem Fall quantitative Messbedingungen
(Relaxationsdelay von mindestens 5 ∙ 𝑇1 sowie inverse-gated decoupling) eingehalten. Vor der
Messung der milieuabhängigen Ringöffnungsraten wurden die Proben jeweils für mindestens 12 h bei
der Zieltemperatur gelagert, um sicherzustellen, dass das thermodynamische Anomerengleichgewicht
erreicht wurde. Lediglich im Falle der AMADORI-Verbindungen der homologen ω-Aminosäuren betrug
die Lagerungszeit rund eine Stunde.

EXPERIMENTELLER TEIL
- 123 -
Im Folgenden sind die im Einzelnen genutzten experimentellen Varianten der selektiven
Sättigungstransfer-NMR-Spektroskopie näher beschrieben.
6.3.6.1 FREQUENZ-SWEEP-SÄTTIGUNGSTRANSFER
Im Falle der Messung von Ringöffnungsraten der D-Glucose mit Hilfe der selektiven
Sättigungstransfer-NMR-Spektroskopie besteht das Problem, dass die Frequenz der zu sättigenden
anomeren Protonenresonanz des acyclischen Zentralisomers unbekannt ist. Es muss folglich eine
Blindsättigung erfolgen. Dazu muss jedoch zunächst die (ungefähre) Resonanzfrequenz des
entsprechenden anomeren Protons ermittelt werden. Zu diesem Zweck wurde ein Pulsprogramm
geschrieben („kfm-1hfqssst“; siehe Anhang), welches die Grundlage für den sogenannten
Frequenz-Sweep-Sättigungstransfer bildet. Es handelt sich hierbei um ein pseudo-2D-NMR-
Experiment, das sich aus aufeinanderfolgenden 1D-Experimenten zusammensetzt. Diese Experimente
unterscheiden sich in der Radiofrequenz der zur Sättigung verwendeten 90° GAUSS-Softpuls-Kaskade.
Die Sättigungszeit beträgt dabei für jedes Experiment 45 s und das spektrale Anregungsfenster jeweils
212 Hz. Die Radiofrequenz des selektiven Pulses nimmt von Experiment zu Experiment um jeweils
50 Hz zu. Der Frequenzscan erfolgt dabei im Verschiebungsbereich der 1H-NMR-Resonanzen von
Aldehydprotonen (hier: ca. 9.3 bis 10.5 ppm). Abbildung 38 zeigt ein beispielhaftes
Frequenz-Sweep-Sättigungstransfer-NMR Spektrum einer 200 mM D-Glucoselösung in Deuteriumoxid
bei einer Temperatur von 77 °C und einem pD-Wert von 5.0. Abgebildet ist die Intensität der anomeren 1H-NMR-Resonanz der α-D-Glucofuranose in Abhängigkeit der Frequenz des selektiven Radiopulses.
Der maximale Sättigungstransfer ist für eine Frequenz von 6150 Hz zu erkennen (entspricht einer
chemischen Verschiebung von 10.25 ppm bei einer Spektrometerfrequenz von 599.89 MHz).
Abbildung 38: Frequenz-Sweep-Sättigungstransfer-NMR Spektrum einer 200 mM D-Glucoselösung in Deuteriumoxid bei einer Temperatur von 77 °C und einem pD-Wert von 5.0. Abgebildet ist die Intensität der anomeren 1H-NMR-Resonanz der α-D-Glucofuranose in Abhängigkeit der Frequenz des selektiven Radiopulses. Der maximale Sättigungstransfer ist für eine Frequenz von 6150 Hz zu erkennen (entspricht einer chemischen Verschiebung von 10.25 ppm bei einer Spektrometerfrequenz von 599.89 MHz).
Auf diese Weise lässt sich die chemische Verschiebung einer unbekannten Protonenresonanz
bei einer Messfrequenz von 600 MHz auf ca. 0.1 ppm genau bestimmen. Die Bestimmung der
entsprechend zu sättigenden Protonenresonanz muss nach Änderungen des pD-Wertes und der
Temperatur jeweils neu erfolgen. Bei Kenntnis der Resonanzfrequenz ist die Durchführung eines
regulären Sättigungstransfer-NMR-Experimentes möglich.
6150 Hz (10.25 ppm)
Radiofrequenz [Hz]
Int.

EXPERIMENTELLER TEIL
- 124 -
6.3.6.2 1H-SST-NMR
Zur Messung von Ringöffnungsraten anhand des Sättigungstransfers von
Protonenresonanzen wurden zwei Pulsprogramme geschrieben („kfm-1hsst“ und „kfm-double-1hsst“;
siehe Anhang). Diese unterscheiden sich in der Anzahl der zu sättigenden Protonenresonanzen.
Sättigung einer Protonenresonanz
Im Falle reduzierender Zucker mit anomerem Proton ist die Sättigung der entsprechenden
Resonanzfrequenz hinreichend für die Messung von Ringöffnungsraten. Es kann das Pulsprogramm
„kfm-1hsst“ genutzt werden. Im Rahmen der vorliegenden Arbeit wurde dieses Experiment zur
Messung der anomerenspezifischen Ringöffnungsraten der Anomere der D-Glucose verwendet. Der
Sättigungstransfer wird in Abhängigkeit der Sättigungszeiten im Intervall von 0 bis 73 s für insgesamt
16 Zeiten gemessen. Das spektrale Anregungsfenster des 90° GAUSS-Pulses beträgt 212 Hz. Die
Messung erfolgt mit einer FID-Auflösung von 0.18 Hz pro Datenpunkt bei einer Gesamtzahl an
Datenpunkten von 64 k. Die erhaltene pseudo-2D-Matrix wird nach zero filling FOURIER-transformiert,
phasen- und basislinienkorrigiert. Die Integration der relevanten NMR-Resonanzen erfolgt mit Hilfe
des AU-Programms „decon_t1“. Die Berechnung der Ringöffnungsraten erfolgt numerisch auf
Grundlage von Gleichung (21) (Seite 32).
Sättigung zweier Protonenresonanzen
Im Falle der AMADORI-Verbindungen liegt kein anomeres Proton vor. Die Messung von
Ringöffnungsraten unter Nutzung anomerer NMR-Resonanzen kann daher nur über die 13C-NMR-Spektroskopie erfolgen. Hierzu ist jedoch die Nutzung isotopenmarkierter Verbindungen
notwendig. Alternativ kann eine Messung anomerengewichteter Ringöffnungsraten über die 1H-NMR-Spektroskopie unter Nutzung der NMR-Resonanzen der Protonen am C-1 erfolgen. Hierbei
handelt es sich um ein AB-Spinsystem, dessen Protonenresonanzen sich um rund 75 Hz unterscheiden.
Es kann daher angenommen werden, dass ein selektiver Puls nur zu einer unzureichenden Anregung
beider Protonenresonanzen führen würde. Aufgrund dessen wurde ein zweites Pulsprogramm
geschrieben, bei dem durch die Schaltung zweier aufeinanderfolgender selektiver Pulse
unterschiedlicher Frequenzen beide NMR-Resonanzen des AB-Spinsystems des acyclischen
Zentralisomers alternierend angeregt und damit gesättigt werden („kfm-double-1hsst“). Der
Sättigungstransfer findet dabei auf die entsprechenden Protonenresonanzen der cyclischen Anomere
statt. Diese sind weitestgehend isochron, weshalb die Auswertung des zeitabhängigen
Sättigungstransfers lediglich anomerengewichtete Ringöffnungsraten liefert. Es zeigte sich, dass der
H/D-Austausch im Falle der AMADORI-Verbindungen der homologen ω-Aminosäuren dabei so schnell
ist, dass die Intensitätsabnahme der beobachteten Protonenresonanz nicht nur auf Grundlage des
Sättigungstransfers, sondern zu einem gewissen Anteil auch aufgrund des H/D-Austausches erfolgte.
Entsprechend wurden im Laufe eines pseudo-2D-Experimentes in regelmäßigen Abständen
Einzelexperimente eingeschoben, bei denen keine Sättigung erfolgte. Über die zeitabhängige
Änderung des Integrals ohne Sättigung lassen sich damit Enolisierungsraten ermitteln. Werden die
Integrale der unter Einfluss der Sättigung gemessenen Spektren entsprechend um den Anteil des
H/D-Austausches korrigiert, ergeben sich aus deren Abhängigkeit von der Sättigungsdauer die
anomerengewichteten Ringöffnungsraten. Der Sättigungstransfer wird in Abhängigkeit der
Sättigungszeiten im Intervall von 0 bis 12 s für insgesamt 32 Zeiten gemessen. Das spektrale
Anregungsfenster des 90° GAUSS-Pulses beträgt 60.6 Hz. Die Messung erfolgt mit einer FID-Auflösung

EXPERIMENTELLER TEIL
- 125 -
von 0.16 Hz pro Datenpunkt bei einer Gesamtzahl an Datenpunkten von 64 k. Die erhaltene
pseudo-2D-Matrix wird nach zero filling FOURIER-transformiert, phasen- und basislinienkorrigiert. Die
Integration der relevanten NMR-Resonanzen erfolgt mit Hilfe des AU-Programms „decon_t1“. Die
Berechnung von Ringöffnungsraten erfolgt numerisch auf Grundlage von Gleichung (21) (Seite 32). Die
Berechnung von Enolisierungsraten erfolgt numerisch auf Grundlage von Gleichung (45) (Seite 69).
6.3.6.3 13C-SST-NMR
Zur Messung des Sättigungstransfers von 13C-NMR-Resonanzen wurde ein Pulsprogramm
geschrieben, mit dessen Hilfe die Messung des Sättigungstransfers unter quantitativen Bedingungen
möglich ist („kfm-13csst_nonoe“; siehe Anhang). Dafür erfolgt die Protonenentkopplung der 13C-NMR-Resonanzen mittels inverse-gated decoupling. Eine Protonenentkopplung während der
selektiven Sättigung der anomeren 13C-NMR-Resonanz des acyclischen Zentralisomers der jeweils
untersuchten Ketosen ist nicht notwendig, da die 2J(13C,1H)-Kopplung lediglich im Bereich von rund 1
bis 12 Hz liegt.[191] Soll hingegen die NMR-Resonanz eines nicht quartären Kohlenstoffatoms gesättigt
werden, so kann das oben genannte Pulsprogramm nicht genutzt werden. Dies liegt darin begründet,
dass 1J(13C,1H)-Kopplungen in der Größenordnung von ca. 120 bis 220 Hz liegen.[191] Für die primären
Kohlenstoffatome des Dimethylformamids würde die Nutzung des Pulsprogrammes beispielsweise
dazu führen, dass keinerlei Sättigung erfolgen würde, da der selektive Puls das Zentrum des
Quadrupletts träfe, bei dem jedoch keine 13C-NMR-Resonanz vorliegt. In solchen Fällen kann das
Pulsprogramm auf zweierlei Weise umgeschrieben werden. Entweder es erfolgt – analog zu
„kfm-double-1hsst“ – die alternierende Schaltung selektiver Pulse unterschiedlicher Frequenzen, die
jeweils den Frequenzen der Einzelresonanzen des Multipletts entsprechen. Oder der Entkopplerkanal
für die Protonenentkopplung wird bereits vor Beginn der Sättigung eingeschaltet, sodass das
Multiplett vor Beginn der Sättigung kollabiert. Dies hat allerdings zur Folge, dass der
Kern-OVERHAUSER-Effekt Einfluss auf die Signalintensitäten nimmt. Damit dieser Einfluss für jede
Sättigungsdauer möglichst identisch ist, empfiehlt es sich dabei, den Entkopplerkanal bereits während
des Relaxationsdelays einzuschalten. Entsprechende Pulsprogramme für beide Versuchsvarianten
wurden im Rahmen dieser Arbeit zu Versuchszwecken geschrieben und befinden sich ebenfalls im
Anhang („kfm-13cdosst_nonoe“ und „kfm-13csst“).
Alle im Rahmen dieser Arbeit quantitativ ausgewerteten Experimente des
kohlenstoffbasierten selektiven Sättigungstransfers beruhen auf dem Pulsprogramm
„kfm-13csst_nonoe“. Der Sättigungstransfer wurde in den meisten Fällen in Abhängigkeit der
Sättigungsdauer im Intervall von 0 bis 73 s für insgesamt 25 Zeiten gemessen. Das spektrale
Anregungsfenster des 90° GAUSS-Pulses beträgt 212 Hz. Die Messung erfolgt mit einer FID-Auflösung
von 0.46 Hz pro Datenpunkt bei einer Gesamtzahl an Datenpunkten von 128 k. Die erhaltene
pseudo-2D-Matrix wird nach zero filling FOURIER-transformiert, phasen- und basislinienkorrigiert. Die
Integration der relevanten NMR-Resonanzen erfolgt mit Hilfe des AU-Programms „decon_t1“. Die
Berechnung der Ringöffnungsraten erfolgt numerisch auf Grundlage von Gleichung (21) (Seite 32).
6.4 Potentiometrische Bestimmung der 𝑝𝐾𝑠-Werte
Es wird eine 0.1 M Lösung der zu untersuchenden Substanz in 0.1 M Salzsäure hergestellt.
20 mL dieser Lösung werden in einem schmalen hohen Becherglas mit 25 mL bidest. Wasser versetzt
und mit Hilfe eines Titrationsautomaten mit 0.1 M Natronlauge titriert. Die Messdaten (pH-Wert gegen
EXPERIMENTELLER TEIL
- 126 -
titriertes Volumen der Maßlösung) werden automatisch registriert und als csv-Datei gespeichert. Diese
kann im Anschluss an die Messung mit einem Tabellenkalkulationsprogramm geöffnet werden. Die
erste Ableitung der Titrationskurve zeigt Maxima für die jeweiligen Äquivalenzpunkte und Minima für
die 𝑝𝐾𝑠-Werte. Die Ermittlung der 𝑝𝐾𝑠-Werte erfolgt graphisch.
6.5 Polarimetrische Messungen
Es wird die in Tabelle 22 aufgelistete Masse der entsprechenden (Amino-) Säure in 80 mL
bidest. Wasser gelöst und der pH-Wert (pH 1.5, pH 2.5, pH 3.5, pH 4.5, pH 5.5 oder pH 6.5) mit
verdünnter Salzsäure bzw. Natronlauge möglichst genau eingestellt. Die Lösung wird quantitativ in
einen 100 mL Maßkolben überführt. Es werden 4.504 g D-Fructose in ein Becherglas eingewogen und
unter kräftigem Rühren in 47.5 mL der vorbereiteten Aminosäurelösung bzw. bidest. Wasser gelöst
(ergibt 50 mL Endvolumen und damit eine Konzentration an (Amino-) Säure und D-Fructose von je
0.5 mol/L). Ein Teil dieser Lösung wird rasch in eine Polarimeterküvette überführt und ihre optische
Rotation bei einer Wellenlänge von 589 nm in Abhängigkeit der Zeit über eine Dauer von rund einer
Stunde manuell protokolliert. Der übrige Teil der Lösung wird parallel dazu genutzt, den tatsächlich
resultierenden pH-Wert sowie die Temperatur der Lösung zu messen. Auch diese werden protokolliert.
Die nicht lineare Regression der protokollierten Daten erfolgt auf Grundlage von Gleichung (55)
(Seite 96) mit Hilfe des Solver Add-Ins von Microsoft Excel 2016. Dabei werden die
Geschwindigkeitskonstanten 𝑘𝑣𝐴,𝛽𝑃 und 𝑘𝛽𝑃,𝑣𝐴 numerisch berechnet. Die Summe der beiden
Konstanten stellt die Mutarotationsrate dar. Bei 𝑘𝛽𝑃,𝑣𝐴 handelt es sich um die Ringöffnungsrate der
β-D-Fructopyranose. Diese ist der für die vorliegende Arbeit relevante Parameter.
Tabelle 22: Einwaagen der (Amino-) Säuren für polarimetrische Messungen der Ringöffnungsraten der β-D-Fructopyranose in Anwesenheit stöchiometrischer Mengen an (Amino-) Säuren.
(Amino-) Säure Einwaage [g]
L-Alanin 4.455 L-Prolin 5.757 Glycin 3.754
β-Alanin 4.455 γ-Aminobuttersäure 5.156
Taurin 6.257 Natrium-Formiat 3.401 Natrium-Acetat 4.102
6.6 Geräte
Tabelle 23: Im Verlauf der Arbeit genutzte Geräte.
Gerät Hersteller und Typ
NMR-Spektroskopie
Spektrometer (500 MHz) AVANCE III 500 mit 5 mm BBO Probenkopf mit z-Gradient, Bruker
Spektrometer (600 MHz) AVANCE 600 mit 5 mm BBI Probenkopf mit z-Gradient, Bruker
Variable Temperierungseinheit BVT 3000 DIGITAL, Bruker
Kühleinheit B – CU 05, Bruker

EXPERIMENTELLER TEIL
- 127 -
NMR-Probenröhrchen Norell® ST 500-7 (5 mm)
pH-Meter Five EasyTM FE20, Mettler-ToledoTM
pH-Elektrode BioTrode, Hamilton
Trockenschrank Köttermann® 2715
UV/Vis-Messungen
Spektralphotometer Specord® 200 Plus, Analytik Jena AG
pH-konstante Modellreaktionen
Titrationsautomat DMS Titrino 716 mit pH-Elektrode, Metrohm
pH-Elektrode Unitrode 6.0259.100, Metrohm
Polarimetrie
Polarimeter Polarimeter P1000-LED, A. KRÜSS OPTRONIC
pH-Meter inoLab pH 720 WTW series
pH-Elektrode SenTix® 81 WTW Glaselektrode
HPLC (D-Fructose)
Säule EC 250/3 Nucleodur® HILIC, Macharey Nagel
Pumpe 1100 Series G1311A Quaternary Pump, Agilent
Entgaser DG-2080-53 3-Line degasser, Jasco
Säulenofen Column Blockheater, Jones Chromatography
Detektor 1100 Series G1362A RID, Agilent
Automatischer Probengeber 1050 Series Autosampler, Hewlett Packard
HPLC (Aminosäuren)
Säule EC 250/4.6 Nucleosil® 120-5 C18, Macharey Nagel
Pumpe 1100 Series G1311A Quaternary Pump, Agilent
Entgaser DG-2080-53 3-Line degasser, Jasco
Säulenofen Column Blockheater, Jones Chromatography
Detektor 1100 Series G1362A RID, Agilent
Automatischer Probengeber 1050 Series Autosampler, Hewlett Packard
HPLC (AMADORI-Verbindungen)
Säule
1. YMC-Pack ODS-AM AM-303 AM12S05-2546WT 250 x 4.6 mm T.D. S-5micrometer, 12 nm, YMC 2. ODS Hypersil 120 A 5 micrometer 15 x 4.6 mm, VDS optilab
Pumpe 1100 Series G1311A Quaternary Pump, Agilent
Entgaser DG-2080-53 3-Line degasser, Jasco
Säulenofen Column Blockheater, Jones Chromatography
Detektor 1100 Series G1362A RID, Agilent
Automatischer Probengeber 1050 Series Autosampler, Hewlett Packard
HPLC (α-Dicarbonylverbindungen)
Säule EC 250/4.6 Nucleosil® 120-5 C18, Macharey Nagel
Pumpe 1100 Series G1312A Binary Pump, Agilent
Entgaser 1100 Series G1379A Degasser, Agilent

EXPERIMENTELLER TEIL
- 128 -
Säulenofen 1100 Series G1316A Thermostatted Column Compartment, Agilent
Detektor 1100 Series G1315B DAD, Agilent
Automatischer Probengeber 1100 Series G1313A ALS, Agilent
Bestimmung von 𝑝𝐾𝑠-Werten
Titrationsautomat Automatischer Titrator TitroLine® 5000, SI Analytics
pH-Elektrode A 162 2M-DIN-ID, SI Analytics
Synthesen
Gefriertrocknungsanlage Alpha 2-4, Martin Christ
6.7 Software
Tabelle 24: Im Verlauf der Arbeit genutzte Software.
Software Herausgeber
NMR-Spektroskopie
TopSpin 1.3 Bruker
TopSpin 2.1 Bruker
TopSpin 3.1 Bruker
TopSpin 3.2 pL 6 Bruker
TopSpin 3.5 pL 2 Bruker
Spektralphotometrie
WinAspect Plus Version: 4.1.0.0 Analytik Jena AG
HPLC
ChemStation Rev. A. 10.02 Agilent
Berechnung von 𝑝𝐾𝑠-Werten
Percepta (Testlizenz) ACD/Labs
Tabellenkalkulation
EXCEL 2016 Microsoft
Solver (Add-In für Microsoft Excel) Microsoft
XLSTAT 2017 Version (19.2.01) (Statistik-Analyse Add-In für Microsoft Excel 2016)
Addinsoft
XNUMBERS Ver 6.0.5.6M (Add-In für Microsoft Excel) John Beyer
„matrix.xla“ (Matrixerweiterung des Microsoft Excel Add-Ins XNUMBERS Ver 6.0.5.6M)
Foxes Team
Textverarbeitung
Word 2016 Microsoft
Zeichnen chemischer Strukturen
ChemBioDraw Ultra 14.0 PerkinElmer
Literaturverwaltung
Citavi 5 Swiss Academic Software
EXPERIMENTELLER TEIL
- 129 -
6.8 Chemikalien
Tabelle 25: Im Verlauf der Arbeit genutzte Chemikalien.
Chemikalie Hersteller Reinheit bzw. Gehalt
1,2-Ethandiol in DMSO-d6 (99.9 atom%) Sigma-Aldrich 80%
Aceton Fisher Scientific 99.99%
Acetonitril VWR Chemicals > 99.9%
Acetonitril Carl Roth ≥ 99.9%
β-Alanin Sigma-Aldrich 99%
L-Alanin Carl Roth ≥ 99%
γ-Aminobuttersäure Alfa Aesar 97%
Ammoniaklösung Merck 25%
DL-Äpfelsäure Fluka > 99%
n-Butanol Merck ≥ 99.5%
Citronensäure Carl Roth ≥ 99.5%
Deuteriumchlorid in D2O (99 atom%) Sigma-Aldrich 35%
Deuteriumoxid Sigma-Aldrich 99.9%
Deuteriumoxid Acros organics 99.9%
Deuteriumoxid Chemotrade 99.98%
Deuteriumoxid euriso-top 99.9%
Dinatriumhydrogenphosphat Dihydrat Merck ≥ 99.5%
Dowex® 50WX8 (200 – 400 mesh) Sigma-Aldrich –
Essigsäure VWR Chemicals 100%
Ethanol (vergällt) VWR Chemicals 99%
FREMYS Salz (Kaliumnitrosodisulfonat) Sigma-Aldrich –
D-Fructose (wasserfrei) Carl Roth > 99.5%
D-[2-13C]Fructose (99 atom%) Omicron Biochemicals 99.8%
D-Glucose (wasserfrei) Carl Roth ≥ 99.5%
D-[2-13C]Glucose (99 atom%) Omicron Biochemicals ≥ 99.9%
Glycin Fluka purum
Hexansulfonsäure Dionex 0.10 mol/L
Kaliumdihydrogenphosphat Merck ≥ 99.5%
Malonsäure Sigma-Aldrich 99%
Maltol Sigma-Aldrich ≥ 99%
Methanol VWR Chemicals ≥ 99.8%
Natrium-Acetat Merck ≥ 99%
Natrium-Deuteroxid in D2O (99 atom%) Sigma-Aldrich 30%
Natrium-Formiat Fluka > 99%
Natrium-Pyrosulfit Polskie Odczynniki Chem.-Gliwice, Polen
puriss.
Ninhydrin Fluka > 99%
ortho-Phenylendiamin Fluka ≥ 99%

EXPERIMENTELLER TEIL
- 130 -
Pivalinsäure Sigma-Aldrich 99%
L-Prolin Sigma-Aldrich ≥ 99%
Salzsäure VWR Chemicals 37%
D-Tagatose Sigma-Aldrich ≥ 98.5%
D-[2-13C]Tagatose (99 atom%) Omicron Biochemicals 99.9%
Taurin Sigma-Aldrich ≥ 99%
wässrige Natriumhydroxidlösung Bernd Kraft 0.1 mol/L
wässrige Salzsäurelösung Bernd Kraft 0.1 mol/L
1-Desoxyglucoson-Chinoxalin Synthetisiert und bereitgestellt von den Mitarbeitern des Fachgebietes Lebensmittelchemie und Analytik
(AK Prof. Kroh) des Instituts für Lebensmitteltechnologie und Lebensmittelchemie
der Technischen Universität Berlin
3-Desoxygalactoson-Chinoxalin
3-Desoxyglucoson-Chinoxalin
D-Glucoson-Chinoxalin

LITERATURVERZEICHNIS
i
Literaturverzeichnis [1] DUBRUNFAUT, A. P.: Ann. Chim. Phys. 1846, 18, 99–108.
[2] MAILLARD, L.-C.: Action des Acides Amines sur les Sucres; Formation des Mélanoidines par voie
Méthodique. C. R. Acad. Sci. 1912, 154, 66–68.
[3] HELLWIG, M.; HENLE, T.: Baking, Ageing, Diabetes: A short History of the MAILLARD Reaction.
Angew. Chem., Int. Ed. Engl. 2014, 53, 10316–10329.
[4] HODGE, J. E.: Dehydrated Foods, Chemistry of Browning Reactions in Model Systems. J. Agric.
Food Chem. 1953, 1, 928–943.
[5] VAIKOUSI, H.; KOUTSOUMANIS, K.; BILIADERIS, C. G.: Kinetic Modelling of Non-Enzymatic Browning
of Apple Juice Concentrates differing in Water Activity under Isothermal and Dynamic
Heating Conditions. Food Chem. 2008, 107, 785–796.
[6] RIZZI, G. P.: Role of Phosphate and Carboxylate Ions in MAILLARD Browning. J. Agric. Food
Chem. 2004, 52, 953–957.
[7] DAVIDEK, T.; CLETY, N.; AUBIN, S.; BLANK, I.: Degradation of the AMADORI Compound N-(1-Deoxy-
D-fructos-1-yl)-glycine in Aqueous Model Systems. J. Agric. Food Chem. 2002, 50, 5472–5479.
[8] HERZFELD, J.; RAND, D.; MATSUKI, Y.; DAVISO, E.; MAK-JURKAUSKAS, M.; MAMAJANOV, I.: Molecular
Structure of Humin and Melanoidin via Solid State NMR. J. Phys. Chem. B. 2011, 115, 5741–
5745.
[9] WEDZICHA, B. L.; RIMMER, Y. L.; KHANDELWAL G. D.: Catalysis of MAILLARD Browning by Sorbic Acid.
Lebensm.-Wiss. u.-Technol. 1991, 24, 278–280.
[10] KATO, H.; YAMAMOTO, M.; FUJIMAKI, M.: Mechanisms of Browning Degradation of D-Fructose in
Special Comparison with D-Glucose-Glycine Reaction. Agric. Biol. Chem. 1969, 33, 939–948.
[11] LIVINGSTON, G. E.: Malic Acid—Fructose Reaction. J. Am. Chem. Soc. 1953, 75, 1342–1344.
[12] NOVOTNÝ, O.; CEJPEK, K.; VELÍŠEK, J.: Formation of Carboxylic Acids during Degradation of
Monosaccharides. Czech J. Food Sci. 2008, 26, 117–131.
[13] DAVIDEK, T.; ROBERT, F.; DEVAUD, S.; VERA, F. A.; BLANK, I.: Sugar Fragmentation in the MAILLARD
Reaction Cascade: Formation of Short-Chain Carboxylic Acids by a new Oxidative
α-Ddicarbonyl Cleavage Pathway. J. Agric. Food Chem. 2006, 54, 6677–6684.
[14] BELITZ, H.-D.; GROSCH, W.; SCHIEBERLE, P.: Lehrbuch der Lebensmittelchemie. Springer-Verlag:
Berlin, 2008.
[15] SIMPSON, B. K.: Food Biochemistry and Food Processing, 2nd Edition. Wiley-Blackwell: Oxford,
2012.
[16] KROH, L. W.: Caramelisation in Food and Beverages. Food Chem. 1994, 51, 373–379.
[17] SCHLEICHER, E.: Die Bedeutung der MAILLARD-Reaktion in der menschlichen Physiologie. Z.
Ernährungswiss. 1991, 30, 18–28.

LITERATURVERZEICHNIS
ii
[18] MIYAZAWA, T.; NAKAGAWA, K.; SHIMASAKI, S.; NAGAI, R.: Lipid Glycation and Protein Glycation in
Diabetes and Atherosclerosis. Amino acids. 2012, 42, 1163–1170.
[19] SINGH, R.; BARDEN, A.; MORI, T.; BEILIN, L.: Advanced Glycation End-Products: A Review.
Diabetologia. 2001, 44, 129–146.
[20] ODANI, H.; SHINZATO, T.; MATSUMOTO, Y.; USAMI, J.; MAEDA, K.: Increase in three α,β-Dicarbonyl
Compound Levels in Human Uremic Plasma: Specific in vivo Determination of Intermediates
in Advanced MAILLARD Reaction. Biochem. Biophys. Res. Commun. 1999, 256, 89–93.
[21] SMITH, M. A.; TANEDA, S.; RICHEY, P. L.; MIYATA, S.; YAN, S.-D.; STERN, D.; SAYRE, L. M.; MONNIER, V.
M.; PERRY, G.: Advanced MAILLARD Reaction End Products are associated with ALZHEIMER
Disease Pathology. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 5710–5714.
[22] HAß, M.; BANSE, M.: Die Positionierung Deutschlands in der Internationalisierung der Agrar-
und Ernährungswirtschaft - Das Ende der Zuckerquote 2017: Wie wettbewerbsfähig ist die
deutsche Zuckerwirtschaft? Schriftenreihe der Rentenbank. 2016, 32, 77–108.
[23] GUGLIUCCI, A.: Formation of Fructose-Mediated Advanced Glycation End Products and Their
Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62.
[24] MCPHERSON, J. D.; SHILTON, B. H.; WALTON, D. J.: Role of Fructose in Glycation and Cross-Linking
of Proteins. Biochemistry. 1988, 27, 1901–1907.
[25] BASARANOGLU, M.; BASARANOGLU, G.; BUGIANESI, E.: Carbohydrate Intake and Nonalcoholic Fatty
Liver Disease: Fructose as a Weapon of Mass Destruction. Hepatobiliary Surg. Nutr. 2014, 4,
109–116.
[26] ABDELMALEK, M. F.; SUZUKI, A.; GUY, C.; UNALP-ARIDA, A.; COLVIN, R.; JOHNSON, R. J.; DIEHL, A. M.:
Increased Fructose Consumption is Associated with Fibrosis Severity in Patients with NAFLD.
Hepatology. 2010, 51, 1961–1971.
[27] LOZANO, I.; VAN DER WERF, R.; BIETIGER, W.; SEYFRITZ, E.; PERONET, C.; PINGET, M.; JEANDIDIER, N.;
MAILLARD, E.; MARCHIONI, E.; SIGRIST, S.; DAL, S.: High-fructose and High-Fat Diet-Induced
Disorders in Rats: Impact on Diabetes Risk, Hepatic and Vascular Complications. Nutr. Metab.
2016, 13, 15–27.
[28] TAPPY, L.; LE, K.-A.: Does Fructose Consumption Contribute to Non-Alcoholic Fatty Liver
Disease? Clin. Res. Hepatol. Gastroenterol. 2012, 36, 554–560.
[29] LÍRIO, L. M.; FORECHI, L.; ZANARDO, T. C.; BATISTA, H. M.; MEIRA, E. F.; NOGUEIRA, B. V.; MILL, J. G.;
BALDO, M. P.: Chronic Fructose Intake Accelerates Non-Alcoholic Fatty Liver Disease in the
Presence of Essential Hypertension. J. Diabetes Complications. 2016, 30, 85–92.
[30] LATULIPPE, M. E.; SKOOG, S. M.: Fructose Malabsorption and Intolerance: Effects of Fructose
with and without Simultaneous Glucose Ingestion. Crit. Rev. Food Sci. Nutr. 2011, 51, 583–
592.
[31] MOGOŞ T.; IACOBINI A. E.: A Review of Hereditary Fructose Intolerance. Rom. J. Diabetes Nutr.
Metab. Dis. 2016, 23, 81–85.
[32] DUBRUNFAUT, M.: Sur une Propriété Analytique des Fermentations Alcoolique et Lactique et
sur leur Application à l'Étude des Sucres. Ann. Chim. Phys. 1847, 3, 169–178.

LITERATURVERZEICHNIS
iii
[33] BERG, J. M.; TYMOCZKO, J. L.; STRYER, L.: Biochemistry. 5th ed. W. H. Freeman: Ney York, 2002.
[34] KANTERS, J. A.; ROELOFSEN, G.; ALBLAS, B. P.; MEINDERS, I.: The Crystal and Molecular Structure of
β-D-Fructose, with Emphasis on Anomeric Effect and Hydrogen-Bond Interactions. Acta
Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1977, 33, 665–672.
[35] COCKMAN, M.; KUBLER, D. G.; OSWALD, A. S.; WILSON, L.: The Mutarotation of Fructose and the
Invertase Hydrolysis of Sucrose. J. Carbohydr. Chem. 1987, 6, 181–201.
[36] HORTON, D.; WAŁASZEK, Z.: Tautomeric Equilibria of some Sugars by Partially Relaxed, 13C Pulse
FOURIER-Transform, Nuclear Magnetic Resonance Spectroscopy. Carbohydr. Res. 1982, 105,
145–153.
[37] WOLFF, G. J.; BREITMAIER, E.: C-13 NMR Spectroscopiy Analysis of Keto Form in Aqueous-
Solutions of D-Fructose, L-Sorbose and D-Tagatose. Chem.-Ztg. 1979, 103, 232–233.
[38] ALLERHAND, A.; DODDRELL, D.: Carbon-13 FOURIER Transform Nuclear Magnetic Resonance. VII.
Study of Anomeric Equilibriums of Ketoses in Water by Natural-Abundance Carbon-13
FOURIER Transform Nuclear Magnetic Resonance. D-Fructose and D-Turanose. J. Am. Chem.
Soc. 1971, 93, 2779–2781.
[39] ANGYAL, S. J.; BETHELL, G. S.: Conformational Analysis in Carbohydrate Chemistry. III. The 13C
N.M.R. Spectra of the Hexuloses. Aust. J. Chem. 1976, 29, 1249–1265.
[40] SCHALLENBERGER, R. S.: Intrinsic Chemistry of Fructose. Pure & Appl. Chem. 1978, 50, 1409–
1420.
[41] MODEL, P.; PONTICORVO, L.; RITTENBERG, D.: Catalysis of an Oxygen-Exchange Reaction of
Fructose-1,6-diphosphate and Fructose-1-phosphate with Water by Rabbit Muscle Aldolase.
Biochemistry. 2002, 7, 1339–1347.
[42] MEGA, T. L.; CORTES, S.; VAN ETTEN, R. L.: The Oxygen-18 Isotope Shift in Carbon-13 Nuclear
Magnetic Resonance Spectroscopy. 13. Oxygen Exchange at the Anomeric Carbon of
D-Glucose, D-Mannose, and D-Fructose. J. Org. Chem. 1990, 55, 522–528.
[43] TOPPER, Y. J.; STETTEN, D.: The Alkali-Catalyzed Conversion of Glucose into Fructose and
Mannose. J. Biol. Chem. 1951, 189, 191–202.
[44] SOWDEN, J. C.; SCHAFFER, R.: The Isomerization of D-Glucose by Alkali in D2O at 25°. J. Am. Chem.
Soc. 1952, 74, 505–507.
[45] FEATHER, M. S.: Equilibration of D-Fructose with its 1,2-Enediol. Carbohydr. Res. 1968, 7, 86–
89.
[46] BARCLAY, T.; GINIC-MARKOVIC, M.; JOHNSTON, M. R.; COOPER, P.; PETROVSKY, N.: Observation of the
Keto Tautomer of D-Fructose in D2O using 1H NMR Spectroscopy. Carbohydr. Res. 2012, 347,
136–141.
[47] SERIANNI, A. S.; BARKER, R.: [13C]-Enriched Tetroses and Tetrofuranosides. J. Org. Chem. 1984,
49, 3292–3300.

LITERATURVERZEICHNIS
iv
[48] ALTONA, C.;SUNDARALINGAM, M.: Conformational Analysis of the Sugar Ring in Nucleosides and
Nucleotides. A New Description Using the Concept of Pseudorotation. J. Am. Chem. Soc.
1972, 94, 8205–8212.
[49] MORAVCOVÁ, J.; CAPKOVÁ, J.; STANEK, J.; RAICH, I.: Methyl 5-Deoxy-α and β-D-Xylofuranosides. J.
Carbohydr. Chem. 1997, 16, 1061–1073.
[50] HAASNOOT, C. A. G.; DE LEEUW, F. A. A. M.; ALTONA, C.: The Relationship between Proton-Proton
NMR Coupling Constants and Substituent Electronegativities. Tetrahedron. 1980, 36, 2783–
2792.
[51] HAASNOOT, C. A. G.; DE LEEUW, F. A. A. M.; DE LEEUW, H. P. M.; ALTONA, C.: The Relationship
between Proton-Proton NMR Coupling Constants and Substituent Electronegativities. Org.
Magn. Reson. 1981, 15, 43–52.
[52] WANG, X.; WOODS, R. J.: Insights into Furanose Solution Conformations: Beyond the Two-State
Model. J. Biomol. NMR. 2016, 64, 291–305.
[53] JASEJA, M.; PERLIN, A. S.; DAIS, P.: Two-Dimensional NMR Spectral Study of the Tautomeric
Equilibria of D-Fructose and Related Compounds. Magn. Reson. Chem. 1990, 28, 283–289.
[54] SUNDARALINGAM, M.: Stereochemistry of Nucleic Acids and their Constituents. IV. Allowed and
Preferred Conformations of Nucleosides, Nucleoside mono-, di-, tri-, tetraphosphates,
Nucleic Acids and Polynucleotides. Biopolymers. 1969, 7, 821–860.
[55] FLOOD, A. E.; JOHNS, M. R.; WHITE, E. T.: Mutarotation of D-Fructose in Aqueous-Ethanolic
Solutions and its Influence on Crystallisation. Carbohydr. Res. 1996, 288, 45–56.
[56] ANGYAL, S. J.: The Composition and Conformation of Sugars in Solution. Angew. Chem., Int.
Ed. Engl. 1969, 8, 157–166.
[57] FRENCH, A. D.; DOWD, M. K.; REILLY, P. J.: MM3 Modeling of Fructose Ring Shapes and Hydrogen
Bonding. J. Mol. Struct. – Teochem. 1997, 395–396, 271–287.
[58] MOSSINE, V. V.; BARNES, C. L.; MAWHINNEY, T. P.: Structure of D-Fructosamine Hydrochloride and
D-Fructosamine Hydroacetate. J. Carbohydr. Chem. 2009, 28, 245–263.
[59] POLACEK, R.; KAATZE, U.: Chair–Chair Ring Inversion of D-Fructose Coupled to the Mutarotation
in Aqueous-Ethanolic Solutions. Chem. Phys. Lett. 2001, 345, 93–99.
[60] POLACEK, R.; BEHRENDS, R.; KAATZE, U.: Chair−Chair Conformational Flexibility of
Monosaccharides Linked to the Anomer Equilibrium. J. Phys. Chem. B. 2001, 105, 2894–2896.
[61] POLACEK, R.; STENGER, J.; KAATZE, U.: Chair–Chair Conformational Flexibility, Pseudorotation,
and Exocyclic Group Isomerization of Monosaccharides in Water. J. Chem. Phys. 2002, 116,
2973–2982.
[62] ISBELL, H. S.; PIGMAN, W. W.: Pyranose-Furanose Interconversions with Reference to the
Mutarotations of Galactose, Levulose, Lactulose, and Turanose. J. Res. Natl. Bur. Stand. 1938,
20, 773–798.
[63] SCHNEIDER, B.; LICHTENTHALER, F. W.; STEINLE, G.; SCHIWECK, H.: Distribution of Furanoid and
Pyranoid Tautomers of D-Fructose in Water, Dimethyl Sulfoxide, and Pyridine via 1H NMR

LITERATURVERZEICHNIS
v
Intensities of Anomeric Hydroxy Groups in [D6]DMSO. Liebigs Ann. Chem. 1985, 1985, 2443–
2453.
[64] HYVÖNEN, L.; VARO, P.; KOIVISTOINEN, P.: Tautomeric Equilibria of D-Glucose and D-Fructose:
Polarimetric Measurements. J. Food Sci. 1977, 42, 652–653.
[65] HYVÖNEN, L.; VARO, P.; KOIVISTOINEN, P.: Tautomeric Equilibria of D-Glucose and D-Fructose:
Gas-Liquid Chromatographic Measurements. J. Food Sci. 1977, 42, 654–656.
[66] LICHTENTHALER, F. W.; RÖNNINGER, S.: α-D-Glucopyranosyl-D-fructoses. J. Chem. Soc., Perkin
Trans. 2. 1990, 164, 1489–1497.
[67] NELSON, J. M.; BEEGLE, F. M.: Mutarotation of Glucose and Fructose. J. Am. Chem. Soc. 1919,
41, 559–575.
[68] GRØNLUND, F.; ANDERSEN, B.: Enthalpy and Activation Enthalpy of the Fructose Mutarotation
Determined by Polarimetry. Acta Chem. Scand. 1966, 20, 2663–2666.
[69] MCCONNELL, H. M.: Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 1958, 28,
430–431.
[70] FORSÉN, S.; HOFFMAN, R. A.: Study of Moderately Rapid Chemical Exchange Reactions by Means
of Nuclear Magnetic Double Resonance. J. Chem. Phys. 1963, 39, 2892–2901.
[71] SERIANNI, A. S.; PIERCE, J.; HUANG, S.-G.; BARKER, R.: Anomerization of Furanose Sugars: Kinetics
of Ring-Opening Reactions by Proton and Carbon-13 Saturation-Transfer NMR Spectroscopy.
J. Am. Chem. Soc. 1982, 104, 4037–4044.
[72] SNYDER, J. R.; SERIANNI, A. S.: D-Idose: A One- and Two-Dimensional NMR Investigation of
Solution Composition and Conformation. J. Org. Chem. 1986, 51, 2694–2702.
[73] SNYDER, J. R.; SERIANNI, A. S.: Deoxygenated and Alkylated Furanoses: Thorpe—Ingold Effects
on Tautomeric Equilibria and Rates of Anomerization. Carbohydr. Res. 1991, 210, 21–38.
[74] SNYDER, J. R.; SERIANNI, A. S.: Furanose Ring Anomerization: A Kinetic Study of the
5-Deoxypentoses and 5-O-Methylpentoses. Carbohydr. Res. 1988, 184, 13–25.
[75] SNYDER, J. R.; SERIANNI, A. S.: DL-Apiose Substituted with Stable Isotopes: Synthesis,
N.M.R.-Spectral Analysis, and Furanose Anomerization. Carbohydr. Res. 1987, 166, 85–99.
[76] SNYDER, J. R.; JOHNSTON, E. R.; SERIANNI, A. S.: D-Talose Anomerization: NMR Methods to
Evaluate the Reaction Kinetics. J. Am. Chem. Soc. 1989, 111, 2681–2687.
[77] WU, J.; SERIANNI, A. S.: Ring-Opening Kinetics of the D-Pentofuranuronic Acids. Carbohydr. Res.
1991, 211, 207–217.
[78] WU, J.; SERIANNI, A. S.; VUORINEN, T.: Furanose Ring Anomerization: Kinetic and Thermodynamic
Studies of the D-2-Pentuloses by 13C-N.M.R. Spectroscopy. Carbohydr. Res. 1990, 206, 1–12.
[79] PIERCE, J.; SERIANNI, A. S.; BARKER, R.: Anomerization of Furanose Sugars and Sugar Phosphates.
J. Am. Chem. Soc. 1985, 107, 2448–2456.

LITERATURVERZEICHNIS
vi
[80] GOUX, W. J.: Complex Isomerization of Ketoses: A Carbon-13 NMR Study of the Base-Catalyzed
Ring-Opening and Ring-Closing Rates of D-Fructose Isomers in Aqueous Solution. J. Am. Chem.
Soc. 1985, 107, 4320–4327.
[81] LAAR, C.: Über die Möglichkeit mehrerer Strukturformeln für dieselbe chemische Verbindung.
Ber. Dtsch. Chem. Ges. 1885, 18, 648–657.
[82] WESTPHAL, G.; KROH, L.: Zum Mechanismus der „frühen Phase” der MAILLARD-Reaktion 1. Mitt.
Einfluß der Struktur des Kohlenhydrats und der Aminosäure auf die Bildung des N-Glycosids.
Food / Nahrung. 1985, 29, 757–764.
[83] LEDL, F.; SCHLEICHER, E.: Die MAILLARD-Reaktion in Lebensmitteln und im menschlichen Körper
– neue Ergebnisse zu Chemie, Biochemie und Medizin. Angew. Chem. 1990, 102, 597–626.
[84] AMADORI, M.: Prodotti di Condensazione tra il Glucosio e la para-Fenetidina. Parte I. Atti
Accad. Naz. Lincei, Cl. Sci. Fis., Mat. Nat., Rend. 1925, 2, 337–342.
[85] AMADORI, M.: Atti Accad. Naz. Lincei, Cl. Sci. Fis., Mat. Nat., Rend. 1929, 9, 68–73.
[86] AMADORI, M.: Condensation Products of Glucose with p-Toluidine. Atti Accad. Naz. Lincei, Cl.
Sci. Fis., Mat. Nat., Rend. 1931, 13, 72–77.
[87] KUHN, R.; DANSI, A.: Über eine molekulare Umlagerung von N-Glucosiden. Ber. Dtsch. Chem.
Ges. (A and B Series). 1936, 69, 1745–1754.
[88] KUHN, R.; WEYGAND, F.: Die AMADORI-Umlagerung. Ber. Dtsch. Chem. Ges. (A and B Series).
1937, 70, 769–772.
[89] KUHN, R.; BIRKOFER, L.: Zur Theorie der Mutarotation; die Mutarotation und katalytische
Hydrierung der Glykoside sekundärer Amine. Ber. Dtsch. Chem. Ges. (A and B Series). 1938,
71, 1535–1541.
[90] WEYGAND, F.: Über N-Glykoside, II. Mitteil.: AMADORI-Umlagerungen. Ber. Dtsch. Chem. Ges.
(A and B Series). 1940, 73, 1259–1278.
[91] ZEILE, K.; KRUCKENBERG, W.: Über Phosphorylierungs- und Tritylierungsreaktionen. Ber. Dtsch.
Chem. Ges. (A and B Series). 1942, 75, 1127–1140.
[92] LÓPEZ, M. G.; GRUENWEDEL, D. W.: Synthesis of Aromatic AMADORI Compounds. Carbohydr. Res.
1991, 212, 37–45.
[93] XENAKIS, D.; MOLL, N.; GROSS, B.: Organic Synthesis of AMADORI Rearrangement Products.
Synthesis. 1983, 1983, 541–543.
[94] YAYLAYAN, V. A.; HUYGHUES‐DESPOINTES, A.; FEATHER, M. S.: Chemistry of AMADORI Rearrangement
Products: Analysis, Synthesis, Kinetics, Reactions, and Spectroscopic Properties. Crit. Rev.
Food Sci. Nutr. 1994, 34, 321–369.
[95] ZHU, Y.; ZAJICEK, J.; SERIANNI, A. S.: Acyclic Forms of [1-13C]Aldohexoses in Aqueous Solution:
Quantitation by 13C NMR and Deuterium Isotope Effects on Tautomeric Equilibria. J. Org.
Chem. 2001, 66, 6244–6251.
[96] RITTENBERG, D.; GRAFF, C.: A Comparison of the Rate of Mutarotation and O-18 Exchange of
Glucose. J. Am. Chem. Soc. 1958, 80, 3370–3372.

LITERATURVERZEICHNIS
vii
[97] GOTTSCHALK, A.: Some Biochemically Relevant Properties of N-substituted Fructosamines
derived from Amino-Acids and N-Arylglucosylamines. Biochem. J. 1952, 52, 455–460.
[98] CAPON, B.; ZUCCO, C.: Simple Enols. 2. Kinetics and Mechanism of the Ketonization of Vinyl
alcohol. J. Am. Chem. Soc. 1982, 104, 7567–7572.
[99] HAROHALLY, N. V.; SRINIVAS, S. M.; UMESH, S.: ZnCl2-Mediated Practical Protocol for the Synthesis
of AMADORI Ketoses. Food Chem. 2014, 158, 340–344.
[100] HEYNS, K.; BEHRE, H.; PAULSEN, H.: Darstellung von 1-Desoxy-1-(2-sulfoäthylamino)-D-fructose.
Carbohydr. Res. 1967, 5, 225–228.
[101] YOSHIMURA, J.; FUNABASHI, M.; SIMON, H.: On the Catalysis of the AMADORI Rearrangement.
Carbohydr. Res. 1969, 11, 276–281.
[102] ROSEN, L.; WOODS, J. W.; PIGMAN, W.: Reactions of Carbohydrates with Nitrogenous
Substances. VI. The AMADORI Rearrangement in Methanol. J. Am. Chem. Soc. 1958, 80, 4697–
4702.
[103] MICHEEL, F.; DIJONG, I.: Der Mechanismus der AMADORI-Umlagerung. Justus Liebigs Ann. Chem.
1962, 658, 120–127.
[104] HEYNS, K.; KOCH W.: Über die Bildung eines Aminozuckers aus D-Fructose und Ammoniak. Z.
Naturforsch. B. 1952, 7, 486–488.
[105] HEYNS, K.; MEINECKE, K.-H.: Über Bildung und Darstellung von D-Glucosamin aus Fructose und
Ammoniak. Chem. Ber. 1953, 86, 1453–1462.
[106] HEYNS, K.; EICHSTEDT, R.; MEINECKE, K.-H.: Die Umsetzung von Fructose und Sorbose mit
Ammoniak und Aminen. Chem. Ber. 1955, 88, 1551–1555.
[107] HEYNS, K.; PAULSEN, H.; BREUER, H.: Umsetzung von Fructose mit Aminosäuren zu
Glucosaminosäuren. Angew. Chem. 1956, 68, 334–335.
[108] HEYNS, K.; BREUER, H.; PAULSEN, H.: Darstellung und Verhalten der 2-N-Aminosäure-2-Desoxy-
Glucosen („Glucose-Aminosäuren”) aus Glycin, Alanin, Leucin und Fructose. Chem. Ber. 1957,
90, 1374–1386.
[109] HEYNS, K.; BREUER, H.: Darstellung und Verhalten weiterer N-substituierter 2-Amino-2-desoxy-
aldosen aus D-Fructose und Aminosäuren. Chem. Ber. 1958, 91, 2750–2762.
[110] HEYNS, K.; PAULSEN, H.; SCHROEDER, H.: Die Umsetzung von Ketohexosen mit sekundären
Aminosäuren und sekundären Aminen. Tetrahedron. 1961, 13, 247–257.
[111] HEYNS, K.; NOACK, H.: Die Umsetzung von D-Fructose mit L-Lysin und L-Arginin und deren
Beziehung zu nichtenzymatischen Bräunungsreaktionen. Chem. Ber. 1962, 95, 720–727.
[112] CARSON, J. F.: The Reaction of Fructose with Isopropylamine and Cyclohexylamine. J. Am.
Chem. Soc. 1955, 77, 1881–1884.
[113] CARSON, J. F.: The Reaction of Fructose with Aliphatic Amines. J. Am. Chem. Soc. 1955, 77,
5957–5960.

LITERATURVERZEICHNIS
viii
[114] BALTES, W.; FRANKE, K.: Modelluntersuchungen zur MAILLARD Reaktion. Z. Lebensm. Unters.
Forsch. 1978, 167, 403–409.
[115] SUÁREZ, G.; RAJARAM, R.; ORONSKY, A. L.; GAWINOWICZ, M. A.: Nonenzymatic Glycation of Bovine
Serum Albumin by Fructose (Fructation). Comparison with the MAILLARD Reaction Initiated by
Glucose. J. Biol. Chem. 1989, 264, 3674–3679.
[116] HEYNS, K.; MÜLLER, G.; PAULSEN, H.: Quantitative Untersuchungen der Reaktion von Hexosen
mit Aminosäuren. Justus Liebigs Ann. Chem. 1967, 703, 202–214.
[117] LOBRY VAN TROOSTENBURG DE BRUYN, C. A.; ALBERDA VAN EKENSTEIN, W.: Action des Alcalis sur les
Sucres, II. Transformation Réciproque des Uns dans les Autres des Sucres Glucose, Fructose
et Mannose. Recl. Trav. Chim. Pays-Bas. 1895, 14, 203–216.
[118] ANET, E. F. L. J.: Chemistry of Non-enzymic Browning. VII. Crystalline Di-D-fructose-glycine and
some Related Compounds. Aust. J. Chem. 1959, 12, 280–287.
[119] MOSSINE, V. V.; LINETSKY, M.; GLINSKY, G. V.; ORTWERTH, B. J.; FEATHER, M. S.: Superoxide Free
Radical Generation by AMADORI Compounds: The Role of Acyclic Forms and Metal Ions. Chem.
Res. Toxicol. 1999, 12, 230–236.
[120] MOSSINE, V. V.; BARNES, C. L.; GLINSKY, G. V.; FEATHER, M. S.: Interaction between two C(1)-X-C'(1)
Branched Ketoses: Observation of an Unprecedented Crystalline Spiro-Bicyclic Hemiketal
Tautomer of N,N-Di(1-deoxy-D-fructos-1-yl)-glycine (“Difructose glycine”) having Open Chain
Carbohydrate. Carbohydr. Lett. 1995, 1, 355–362.
[121] FEATHER, M. S.; MOSSINE, V. V.: Correlations between Structure and Reactivity of AMADORI
Compounds in “The MAILLARD Reaction in Foods and Medicine”. The Royal Society of
Chemistry: Cambridge, UK, 1998, 37–42.
[122] MOSSINE, V. V.; BARNES, C. L.; MAWHINNEY, T. P.: The Structure of N‐(1‐Deoxy‐β‐D‐fructopyranos‐
1‐yl)‐L‐proline Monohydrate (“D‐Fructose‐L‐proline”) and N‐(1,6‐Dideoxy‐α‐L‐fructofuranos‐
1‐yl)‐L‐proline (“L‐Rhamnulose‐L‐proline”). J. Carbohydr. Chem. 2007, 26, 249–266.
[123] TARNAWSKI, M.; ŚLEPOKURA, K.; LIS, T.; KULIŚ-ORZECHOWSKA, R.; SZELEPIN, B.: Crystal Structure of
N-(1-Deoxy-β-D-fructopyranos-1-yl)-L-proline — an AMADORI Compound. Carbohydr. Res.
2007, 342, 1264–1270.
[124] MOSSINE, V. V.; GLINSKY, G. V.; BARNES, C. L.; FEATHER, M. S.: Crystal Structure of an AMADORI
Compound, N-(1-Deoxy-β-D-fructopyranos-1-yl)-glycine (“D-Fructose-glycine”). Carbohydr.
Res. 1995, 266, 5–14.
[125] MOSSINE, V. V.; BARNES, C. L.; MAWHINNEY, T. P.: Solubility and Crystal Structure of N-(1-Deoxy-
β-D-fructopyranos-1-yl)-L-histidine Monohydrate (‘D-Fructose-L-histidine’). Carbohydr. Res.
2007, 342, 131–138.
[126] ALTENA, J. H.; VAN DEN OUWELAND, G. A. M.; TEUNIS, C. J.; TJAN, S. B.: Analysis of the 220-MHz,
P.M.R. Spectra of Some Products of the AMADORI and HEYNS Rearrangements. Carbohydr. Res.
1981, 92, 37–49.
[127] RÖPER, H.; RÖPER, S.; HEYNS, K.; MEYER, B.: N.M.R. Spectroscopy of N-(1-Deoxy-D-fructos-1-yl)-
L-Amino Acids (“Fructose-Amino Acids”). Carbohydr. Res. 1983, 116, 183–195.

LITERATURVERZEICHNIS
ix
[128] Tjan, S. B.; van den Ouweland, G. A. M.: PMR Investigation into the Structure of some
N-substituted 1-Amino-1-deoxy-D-fructoses (AMADORI Rearrangement Products).
Tetrahadron. 1974, 30, 2891–2897.
[129] MOSSINE, V. V.; GLINSKY, G. V.; FEATHER, M. S.: The Preparation and Characterization of some
AMADORI Compounds (1-Amino-1-deoxy-D-fructose Derivatives) derived from a series of
Aliphatic ω-Amino Acids. Carbohydr. Res. 1994, 262, 257–270.
[130] JAKAS, A.; HORVAT, Š.: Synthesis and 13C NMR Investigation of novel AMADORI Compounds
(1-Amino-1-deoxy-D-fructose derivatives) related to the Opioid Peptide, Leucine–enkephalin.
J. Chem. Soc., Perkin Trans. 2. 1996, 5, 789–794.
[131] ANGYAL, S. J.: The Composition of Reducing Sugars in Solution. Adv. Carbohydr. Chem.
Biochem. 1984, 42, 15–68.
[132] FERRARI, E.; GRANDI, R.; LAZZARI, S.; MARVERTI, G.; ROSSI, M. C.; SALADINI, M.: 1H, 13C, 195Pt NMR
Study on Platinum(II) Interaction with Sulphur Containing AMADORI Compounds. Polyhedron.
2007, 26, 4045–4052.
[133] NEGLIA, C. I.; COHEN, H. J.; GARBER, A. R.; ELLIS, P. D.; THORPE, S. R.; BAYNES, J. W.: 13C NMR
Investigation of Nonenzymatic Glucosylation of Protein. J. Biol. Chem. 1983, 258, 14279–
14283.
[134] KAPCZYŃSKA, K.; STEFANOWICZ, P.; JAREMKO, Ł.; JAREMKO, M.; KLUCZYK, A.; SZEWCZUK, Z.: The
Efficient Synthesis of Isotopically Labeled Peptide-derived AMADORI Products and their
Characterization. Amino acids. 2011, 40, 923–932.
[135] FUNCKE, W.; KELMER, A.: 13C.N.M.R.-Untersuchungen an Mutarotations-Gleichgewichten von
D-Fructose und 1-Amino-1-desoxy-D-fructose-Derivaten (AMADORI-Verbindungen).
Carbohydr. Res. 1976, 50, 9–13.
[136] GÓMEZ-SÁNCHEZ, A.; DE GRACIA GARCÍA MARTÍN, M.; PASCUAL, C.: Synthesis and Glycosidation of
1-Deoxy-1-[(2,2-diacylvinyl)amino]-D-fructoses. Carbohydr. Res. 1986, 149, 329–345.
[137] SIMON, H.; HEUBACH, G.: Zur Bildung alicyclischer und offenkettiger, stickstoffhaltiger
Reduktone durch Einwirkung sekundärer Aminsalze auf Monosaccharide. Chem. Ber. 1965,
98, 3703–3711.
[138] SIMON, H.; KRAUS, A.: Mechanistische Untersuchungen über Glykosylamine, Zuckerhydrazone,
AMADORI-Umlagerungsprodukte und Osazone. Carbohydr. Chem. 1970, 14, 430–471.
[139] HORVAT, Š.; ROŠČIĆ, M.; VARGA-DEFTERDAROVIĆ, L.; HORVAT, J.: Intramolecular Rearrangement of
the Monosaccharide Esters of an Opioid Pentapeptide. J. Chem. Soc., Perkin Trans. 1. 1998,
5, 909–914.
[140] FREIMUND, S.; HUWIG, A.; GIFFHORN, F.; KÖPPER, S.: Convenient Chemo-Enzymatic Synthesis of
D-Tagatose. J. Carbohydr. Chem. 1996, 15, 115–120.
[141] DE GRACIA GARCÍA MARTÍN, M.; GASCH, C.; GÓMEZ-SÁNCHEZ, A.: Glycosides of 1-Amino-1-deoxy-D-
fructose. Carbohydr. Res. 1990, 199, 139–151.
[142] PILKOVÁ, L.; POKORNÝ, J.; DAVÍDEK, J.: Browning Reactions of HEYNS Rearrangement Products.
Food / Nahrung. 1990, 34, 759–764.

LITERATURVERZEICHNIS
x
[143] POKORNÝ, J.; PILKOVÁ, L.; DAVÍDEK, J.; VALENTOVÁ, H.: Effect of AMADORI Rearrangement Products
on the Non-Enzymic Browning in Model Systems. Food / Nahrung. 1988, 32, 767–776.
[144] BRANDS, C. M. J.; VAN BOEKEL, M. A. J. S.: Kinetic Modeling of Reactions in Heated
Monosaccharide−Casein Systems. J. Agric. Food Chem. 2002, 50, 6725–6739.
[145] VAN BOEKEL, M. A. J. S.; BRANDS, C. M. J.: Heating of Sugar-Casein Solutions: Isomerization and
MAILLARD Reactions in “The MAILLARD Reaction in Foods and Medicine”. The Royal Society of
Chemistry: Cambridge, UK, 1998, 154–159.
[146] ANET, E. F. L. J.: Chemistry of Nonenzymic Browning. X. Difructose-amino Acids as
Intermediates in Browning Reactions. Aust. J. Chem. 1959, 12, 491–496.
[147] VAN BOEKEL, M. A. J. S.: Effect of Heating on MAILLARD Reactions in Milk. Food Chem. 1998, 62,
403–414.
[148] BECK, J.; LEDL, F.; SEVERIN, T.: Formation of 1-Deoxy-D-erythro-2,3-hexodiulose from AMADORI
Compounds. Carbohydr. Res. 1988, 177, 240–243.
[149] HIRSCH, J.; MOSSINE, V. V.; FEATHER, M. S.: The Detection of some Dicarbonyl Intermediates
Arising from the Degradation of AMADORI Compounds (the MAILLARD Reaction). Carbohydr.
Res. 1995, 273, 171–177.
[150] DEGEN, J.; HELLWIG, M.; HENLE, T.: 1,2-Dicarbonyl Compounds in Commonly Consumed Foods.
J. Agric. Food Chem. 2012, 60, 7071–7079.
[151] HELLWIG, M.; DEGEN, J.; HENLE, T.: 3-Deoxygalactosone, a "new" 1,2-Dicarbonyl Compound in
Milk Products. J. Agric. Food Chem. 2010, 58, 10752–10760.
[152] MITTELMAIER, S.; FÜNFROCKEN, M.; FENN, D.; PISCHETSRIEDER, M.: 3-Deoxygalactosone, a new
Glucose Degradation Product in Peritoneal Dialysis Fluids: Identification, Quantification by
HPLC/DAD/MSMS and its Pathway of Formation. Anal. Bioanal. Chem. 2011, 399, 1689–1697.
[153] HELLWIG, M.; NOBIS, A.; WITTE, S.; HENLE, T.: Occurrence of (Z)-3,4-Dideoxyglucoson-3-ene in
Different Types of Beer and Malt Beer as a Result of 3-Deoxyhexosone Interconversion. J.
Agric. Food Chem. 2016, 64, 2746–2753.
[154] KAWAKISHI, S.; TSUNEHIRO, J.; UCHIDA, K.: Autoxidative Degradation of AMADORI Compounds in
the Presence of Copper Ion. Carbohydr. Res. 1991, 211, 167–171.
[155] BAKER, J. R.; ZYZAK, D. V.; THORPE, S. R.; BAYNES, J. W.: Chemistry of the Fructosamine Assay. Clin.
Chem. 1994, 40, 1950–1955.
[156] FREIMUND, S.; BALDES, L.; HUWIG A.; GIFFHORN, F.: Enzymatic Synthesis of D-Glucosone-6-
phosphate (D-arabino-Hexos-2-ulose-6-(dihydrogen-phosphate)) and NMR Analysis of its
Isomeric Forms. Carbohydr. Res. 2002, 337, 1585–1587.
[157] KÖPPER, S.; FREIMUND, S.: The Composition of Keto Aldoses in Aqueous Solution as Determined
by NMR Spectroscopy. Helv. Chim. Acta. 2003, 86, 827–843.
[158] FREIMUND, S.; KÖPPER, S.: The Composition of 2-Keto Aldoses in Organic Solvents as
Determined by NMR Spectroscopy. Carbohydr. Res. 2004, 339, 217–220.

LITERATURVERZEICHNIS
xi
[159] KAUFMANN, M.; HAASE, P. T.; MÜGGE, C.; KROH, L. W.: Milieu Dependence of Isomeric
Composition of 1-Deoxy-D-erythro-hexo-2,3-diulose in Aqueous Solution Determined by
High-Resolution NMR Spectroscopy. Carbohydr. Res. 2012, 364, 15–21.
[160] KAUFMANN, M.; HAASE, P. T.; MÜGGE, C.; KROH, L. W.: Milieu Dependence of Isomeric
Composition of D-arabino-Hexo-2-ulose in Aqueous Solution Determined by High-Resolution
NMR Spectroscopy. J. Agric. Food Chem. 2013, 61, 10220–10224.
[161] SMUDA, M.; GLOMB, M. A.: Fragmentation Pathways during MAILLARD-induced Carbohydrate
Degradation. J. Agric. Food Chem. 2013, 61, 10198–10208.
[162] ZHANG, W.; CARMICHAEL, I.; SERIANNI, A. S.: Rearrangement of 3-Deoxy-D-erythro-hexos-2-ulose
in Aqueous Solution: NMR Evidence of Intramolecular 1,2-Hydrogen Transfer. J. Org. Chem.
2011, 76, 8151–8158.
[163] HOFMANN, T.; MÜNCH, P.; SCHIEBERLE, P.: Quantitative Model Studies on the Formation of
Aroma-Active Aldehydes and Acids by Strecker-Type Reactions. J. Agric. Food Chem. 2000,
48, 434–440.
[164] MAILLARD, L.-C.: Formation de Matières Humiques par Action de Polypeptides sur les Sucres.
C. R. Acad. Sci. 1913, 156, 1159–1160.
[165] HEDGES, J. I.: The Formation and Clay Mineral Reactions of Melanoidins. Geochim. Cosmochim.
Acta. 1978, 42, 69–76.
[166] PEREYRA GONZALES, A.; BURIN, L.; DEL PILAR BUERA, M.: Color Changes during Storage of Honeys
in Relation to their Composition and Initial Color. Food Res. Int. 1999, 32, 185–191.
[167] VAIKOUSI, H.; KOUTSOUMANIS, K.; BILIADERIS, C. G.: Kinetic Modelling of Non-Enzymatic Browning
in Honey and diluted Honey Systems subjected to Isothermal and Dynamic Heating Protocols.
J. Food Eng. 2009, 95, 541–550.
[168] IBARZ, A.; PAGÁN, J.; GARZA, S.: Kinetic Models for Colour Changes in Pear Puree during Heating
at relatively High Temperatures. J. Food Eng. 1999, 39, 415–422.
[169] BAISIER, W. M.; LABUZA, T. P.: MAILLARD Browning Kinetics in a Liquid Model System. J. Agric.
Food Chem. 1992, 40, 707–713.
[170] VOIGT, M.; GLOMB, M. A.: Reactivity of 1-Deoxy-D-erythro-hexo-2,3-diulose: A Key
Intermediate in the MAILLARD Chemistry of Hexoses. J. Agric. Food Chem. 2009, 57, 4765–
4770.
[171] KIM, M.-O.; BALTES, W.: On the Role of 2,3-Dihydro-3,5-dihydroxy-6-methyl-4(H)-pyran-4-one
in the MAILLARD Reaction. J. Agric. Food Chem. 1996, 44, 282–289.
[172] USUI, T.; YANAGISAWA, S.; OHGUCHI, M.; YOSHINO, M.; KAWABATA, R.; KISHIMOTO, J.; ARAI, Y.; AIDA,
K.; WATANABE, H.; HAYASE, F.: Identification and Determination of α-Dicarbonyl Compounds
Formed in the Degradation of Sugars. Biosci., Biotechnol., Biochem. 2007, 71, 2465–2472.
[173] ANTAL JR., M. J.; MOK, W. S. L.; RICHARDS, G. N.: Mechanism of Formation of 5-(Hydroxymethyl)-
2-furaldehyde from D-Fructose and Sucrose. Carbohydr. Res. 1990, 199, 91–109.

LITERATURVERZEICHNIS
xii
[174] ISBELL, H. S.; PIGMAN, W. W.: Note on the Thermal Mutarotation of D-Galactose, L-Arabinose,
and D-Talose. J. Res. Natl. Bur. Stand. 1936, 16, 553–554.
[175] ANGYAL, S. J.: The Composition of Reducing Sugars in Solution: Current Aspects. Adv.
Carbohydr. Chem. Biochem. 1991, 49, 19–35.
[176] WEENEN, H.; TJAN, S. B.: Analysis, Structure, and Reactivity of 3-Deoxyglucosone in “Flavor
precursors”. American Chemical Society: Washington DC, 1992, 217–231.
[177] RASMUSSEN, H.; SØRENSEN, H. R.; MEYER, A. S.: Formation of Degradation Compounds from
Lignocellulosic Biomass in the Biorefinery. Carbohydr. Res. 2014, 385, 45–57.
[178] CARATZOULAS, S.; VLACHOS, D. G.: Converting Fructose to 5-Hydroxymethylfurfural. Carbohydr.
Res. 2011, 346, 664–672.
[179] YAYLAYAN, V. A.; MANDEVILLE, S.: Stereochemical Control of Maltol Formation in MAILLARD
Reaction. J. Agric. Food Chem. 1994, 42, 771–775.
[180] VAN PUTTEN, R.-J.; VAN DER WAAL, J. C.; HARMSE, M.; VAN DE BOVENKAMP, H. H.; DE JONG, E.; HEERES,
H. J.: A Comparative Study on the Reactivity of Various Ketohexoses to Furanics in Methanol.
ChemSusChem. 2016, 9, 1827–1834.
[181] SHAW, P. E.; TATUM, J. H.; BERRY, R. E.: Acid-Catalyzed Degradation of D-Fructose. Carbohydr.
Res. 1967, 5, 266–273.
[182] MILLER, R. E.; CANTOR, S. M.: 2-Hydroxyacetylfuran from Sugars. J. Am. Chem. Soc. 1952, 74,
5236–5237.
[183] VAN PUTTEN, R.-J.; VAN DER WAAL, J. C.; DE JONG, E.; HEERES, H. J.: Reactivity Studies in Water on
the Acid-Catalysed Dehydration of Psicose compared to other Ketohexoses into
5-Hydroxymethylfurfural. Carbohydr. Res. 2017, 446–447, 1–6.
[184] HUBER, B.; LEDL, F.: Formation of 1-Amino-1,4-dideoxy-2,3-hexodiuloses and
2-Aminoacetylfurans in the MAILLARD Reaction. Carbohydr. Res. 1990, 204, 215–220.
[185] HEYNS, K.; HEUKESHOVEN, J.; BROSE, K. H.: Degradation of Fructose Amino Acids to
N-(2-Furoylmethyl)amino Acids. Intermediates in Browning Reactions. Angew. Chem., Int. Ed.
Engl. 1968, 7, 628–629.
[186] SHAFIZADEH, F.; MCGINNIS, G. D.; PHILPOT, C. W.: Thermal Degradation of Xylan and Related
Model Compounds. Carbohydr. Res. 1972, 25, 23–33.
[187] ANTAL JR., M. J.; LEESOMBOON T.; MOK, W. S.; RICHARDS, G. N.: Mechanism of Formation of
2-Furaldehyde from D-Xylose. Carbohydr. Res. 1991, 217, 71–85.
[188] PSARRAS, T.; WILBURN TEFTELLER, J.; ZIMMERMAN, H. K.: Ultraviolet Absorption Spectra of
1-Furfural and of 5-Hydroxymethyl-furfural in Aqueous Buffers. Recl. Trav. Chim. Pays-Bas.
1961, 80, 232–236.
[189] WANG, H.; JENNER, A. M.; LEE, C.-Y. J.; SHUI, G.; TANG, S. Y.; WHITEMAN, M.; WENK, M. R.; HALLIWELL,
B.: The Identification of Antioxidants in Dark Soy Sauce. Free Radical Res. 2007, 41, 479–488.
[190] CHAWLA, S. P.; CHANDER, R.; SHARMA, A.: Antioxidant Properties of MAILLARD Reaction Products
obtained by gamma-Irradiation of Whey Proteins. Food Chem. 2009, 116, 122–128.

LITERATURVERZEICHNIS
xiii
[191] GÜNTHER, H.: NMR Spectroscopy. Third Completely Revised and Updated Edition. Wiley-VCH:
Weinheim, 2013.
[192] FRIEBOLIN, H.: Ein- und zweidimensionale NMR-Spektroskopie. 5. Vollständig überarbeitete
und aktualisierte Auflage. Wiley-VCH: Weinheim, 2013.
[193] BUBB, W. A.: NMR Spectroscopy in the Study of Carbohydrates. Concepts Magn. Reson. 2003,
19A, 1–19.
[194] DUUS, J. Ø.; GOTFREDSEN, C. H.; BOCK, K.: Carbohydrate Structural Determination by NMR
Spectroscopy. Chem. Rev. 2000, 100, 4589–4614.
[195] BRAUN, S.; BERGER, S.: 200 and more basic NMR Experiments. Third edition. Wiley-VCH:
Weinhein, 2004.
[196] DE BEER, T.; VAN ZUYLEN, C. W. E. M.; HÅRD, K.; BOELENS, R.; KAPTEIN, R.; KAMERLING, J. P.;
VLIEGENTHART, J. F. G.: Rapid and Simple Approach for the NMR Resonance Assignment of the
Carbohydrate Chains of an Intact Glycoprotein. Application of Gradient-Enhanced Natural
Abundance 1H-13C HSQC and HSQC-TOCSY to the α-Subunit of Human Chorionic
Gonadotropin. FEBS Lett. 1994, 348, 1–6.
[197] MEYER, N. H.; ZANGGER, K.: Simplifying Proton NMR Spectra by Instant Homonuclear
Broadband Decoupling. Angew. Chem., Int. Ed. Engl. 2013, 52, 7143–7146.
[198] WOODLEY, M.; FREEMAN, R.: "Decoupled" Proton NMR Spectra. J. Magn. Reson., Ser. A. 1994,
109, 103–112.
[199] KAZIMIERCZUK, K.; STANEK, J.; ZAWADZKA-KAZIMIERCZUK, A.; KOŹMIŃSKI, W.: High-Dimensional NMR
Spectra for Structural Studies of Biomolecules. ChemPhysChem. 2013, 14, 3015–3025.
[200] CASTANAR, L.: Pure Shift 1H NMR: What is Next? Magn. Reson. Chem. 2017, 55, 47–53.
[201] MEYER, N. H.; ZANGGER, K.: Boosting the Resolution of 1H NMR Spectra by Homonuclear
Broadband Decoupling. ChemPhysChem. 2014, 15, 49–55.
[202] BECKER, J.; LUY, B.: CLIP-ASAP-HSQC for Fast and Accurate Extraction of One-Bond Couplings
from Isotropic and Partially Aligned Molecules. Magn. Reson. Chem. 2015, 53, 878–885.
[203] SCHULZE-SUNNINGHAUSEN, D.; BECKER, J.; LUY, B.: Rapid Heteronuclear Single Quantum
Correlation NMR Spectra at Natural Abundance. J. Am. Chem. Soc. 2014, 136, 1242–1245.
[204] REYNOLDS, W. F.; ENRÍQUEZ, R. G.: Choosing the Best Pulse Sequences, Acquisition Parameters,
Postacquisition Processing Strategies, and Probes for Natural Product Structure Elucidation
by NMR Spectroscopy. J. Nat. Prod. 2002, 65, 221–244.
[205] DAVIS, D. G.; BAX, A.: Simplification of Proton NMR Spectra by Selective Excitation of
Experimental Subspectra. J. Am. Chem. Soc. 1985, 107, 7197–7198.
[206] INAGAKI, F.; SHIMADA, I.; KOHDA, D.; SUZUKI, A.; BAX, A.: Relayed HOHAHA, a useful Method for
Extracting Subspectra of Individual Components of Sugar Chains. J. Magn. Reson. (1969).
1989, 81, 186–190.
[207] LERNER, L.; BAX, A.: Application of new, High-Sensitivity, 1H-13C-N.M.R.-Spectral Techniques to
the Study of Oligosaccharides. Carbohydr. Res. 1987, 166, 35–46.

LITERATURVERZEICHNIS
xiv
[208] BAX, A.; FREEMAN, R.; MORRIS, G.: Correlation of Proton Chemical Shifts by Two-Dimensional
FOURIER Transform NMR. J. Magn. Reson. (1969). 1981, 42, 164–168.
[209] DAVIS, D. G.; BAX, A.: Assignment of Complex Proton NMR Spectra via Two-Dimensional
Homonuclear HARTMANN-HAHN Spectroscopy. J. Am. Chem. Soc. 1985, 107, 2820–2821.
[210] BRAUNSCHWEILER, L.; ERNST, R. R.: Coherence Transfer by Isotropic Mixing. J. Magn. Reson.
(1969). 1983, 53, 521–528.
[211] DREW, K. N.; ZAJICEK, J.; BONDO, G.; BOSE, B.; SERIANNI, A. S.: 13C-Labeled Aldopentoses: Detection
and Quantitation of Cyclic and Acyclic Forms by Heteronuclear 1D and 2D NMR Spectroscopy.
Carbohydr. Res. 1998, 307, 199–209.
[212] CONG, Y.; LI, W.; LIU, C.; WANG, J.; LI, X.: Application of HSQC-TOCSY to the Analysis of Saponins.
Asian J. Tradit. Med. 2006, 1, 20–24.
[213] LERNER, L. E.: Carbohydrate Structure and Dynamics from NMR in “NMR Spectroscopy and its
Application to Biomedical Research”. Elsevier Science B.V.: Amsterdam, 1996, 313–344.
[214] BAX, A.; GRIFFEY, R. H.; HAWKINS, B. L.: Correlation of Proton and Nitrogen-15 Chemical Shifts
by Multiple Quantum NMR. J. Magn. Reson. (1969). 1983, 55, 301–315.
[215] BODENHAUSEN, G.; RUBEN, D. J.: Natural Abundance Nitrogen-15 NMR by Enhanced
Heteronuclear Spectroscopy. Chem. Phys. Lett. 1980, 69, 185–189.
[216] VUORINEN, T.; SERIANNI, A. S.: 13C-Substituted Pentos-2-uloses: Synthesis and Analysis by 1H- and 13C-N.M.R. Spectroscopy. Carbohydr. Res. 1990, 207, 185–210.
[217] TVAROSKA, I.; TARAVEL, F. R.: Carbon-Proton Coupling Constants in the Conformational Analysis
of Sugar Molecules. Adv. Carbohydr. Chem. Biochem. 1995, 51, 15–61.
[218] BOCK, K.; PEDERSEN, C.: A Study of 13CH Coupling Constants in Pentopyranoses and Some of
their Derivatives. Acta Chem. Scand. B. 1975, 29, 258–264.
[219] BOCK, K.; PEDERSEN, C.: A Study of 13CH Coupling Constants in Hexopyranoses. J. Chem. Soc.,
Perkin Trans. 2. 1974, 3, 293–297.
[220] BOCK, K.; LUNDT, I.; PEDERSEN, C.: Assignment of Anomeric Structure to Carbohydrates through
Geminal 13C-H Coupling Constants. Tetrahedron Lett. 1973, 14, 1037–1040.
[221] BAX, A.; SUMMERS, M. F.: Proton and Carbon-13 Assignments from Sensitivity-Enhanced
Detection of Heteronuclear Multiple-Bond Connectivity by 2D Multiple Quantum NMR. J. Am.
Chem. Soc. 1986, 108, 2093–2094.
[222] VARKI, A.; CUMMINGS, R. D.; ESKO, J. D.; FREEZE, H. H.; STANLEY, P.; BERTOZZI, C. R.; HART, G. W.;
ETZLER, M. E.: Essentials of Glycobiology. 2nd edition. Cold Spring Harbor Laboratory Press:
New York, 2009.
[223] KARPLUS, M.: Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963,
85, 2870–2871.
[224] BAIN, A. D.: Chemical Exchange in NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2003, 43, 63–
103.

LITERATURVERZEICHNIS
xv
[225] BINSCH, G.; KESSLER, H.: The Kinetic and Mechanistic Evaluation of NMR Spectra. New
Analytical Methods. Angew. Chem., Int. Ed. Engl. 1980, 19, 411–428.
[226] HER, C.; ALONZO, A. P.; VANG, J. Y.; TORRES, E.; KRISHNAN, V. V.: Real-Time Enzyme Kinetics by
Quantitative NMR Spectroscopy and Determination of the Michaelis–Menten Constant Using
the Lambert-W Function. J. Chem. Educ. 2015, 92, 1943–1948.
[227] LACHENMEIER, D. W.; HUMPFER, E.; FANG, F.; SCHÜTZ, B.; DVORTSAK, P.; SPROLL, C.; SPRAUL, M.:
NMR-Spectroscopy for Nontargeted Screening and Simultaneous Quantification of Health-
Relevant Compounds in Foods. J. Agric. Food Chem. 2009, 57, 7194–7199.
[228] SPRAUL, M.; LINK, M.; SCHAEFER, H.; FANG, F.; SCHUETZ, B.: Wine Analysis to check Quality and
Authenticity by Fully-Automated 1H-NMR. BIO Web of Conferences. 2015, 5, 02022-p1–p5.
[229] MAZZONI, V.; BRADESI, P.; TOMI, F.; CASANOVA, J.: Direct Qualitative and Quantitative Analysis of
Carbohydrate Mixtures using 13C NMR Spectroscopy: Application to Honey. Magn. Reson.
Chem. 1997, 35, 81–90.
[230] COLOMBO, C.; AUPIC, C.; LEWIS, A. R.; PINTO, B. M.: In Situ Determination of Fructose Isomer
Concentrations in Wine Using 13C Quantitative Nuclear Magnetic Resonance Spectroscopy. J.
Agric. Food Chem. 2015, 63, 8551–8559.
[231] BHARTI, S. K.; ROY, R.: Quantitative 1H NMR spectroscopy. Trends Anal. Chem. 2012, 35, 5–26.
[232] SHOOLERY, J. N.: Some Quantitative Applications of 13C NMR Spectroscopy. Prog. Nucl. Magn.
Reson. Spectrosc. 1977, 11, 79–93.
[233] JACOBSEN, N. E.: NMR Spectroscopy Explained: Simplified Theory, Applications and Examples
for Organic Chemistry and Structural Biology. John Wiley & Sons: Hoboken (New Jersey),
2007.
[234] BLOCH, F.: Nuclear Induction. Phys. Rev. 1946, 70, 460–474.
[235] FREEMAN, R.; HILL, H. D. W.: FOURIER Transform Study of NMR Spin–Lattice Relaxation by
“Progressive Saturation”. J. Chem. Phys. 1971, 54, 3367–3377.
[236] BAIN, A. D.: The Choice of Parameters in an NMR Experiment. Application to the Inversion-
Recovery T1 Method. J. Magn. Reson. (1969). 1990, 89, 153–160.
[237] BECKER, E. D.; SHOUP, R. R.: 13C NMR Spectroscopy: Relaxation Times of 13C and Methods for
Sensitivity Enhancement. Pur Appl. Chem. 1972, 32, 1–4.
[238] HARRIS, R. K.; BECKER, E. D.; CABRAL DE MENEZES, S. M.; GOODFELLOW, R.; GRANGER, P.: NMR
Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts. Magn. Reson.
Chem. 2002, 40, 489–505.
[239] ERNST, R. R.; BODENHAUSEN, G.; WOKAUN, A.: Principles of Nuclear Magnetic Resonance in One
and Two Dimensions. Clarendon Press: Oxford, 1987.
[240] ERNST, R. R.; ANDERSON, W. A.: Application of FOURIER Transform Spectroscopy to Magnetic
Resonance. Rev. Sci. Instrum. 1966, 37, 93–102.
[241] NEUHAUS, D.: Nuclear OVERHAUSER Effect. eMagRes. John Wiley & Sons, 2007.

LITERATURVERZEICHNIS
xvi
[242] ANET, F. A. L.; BOURN, A. J. R.: Nuclear Magnetic Resonance Spectral Assignments from Nuclear
OVERHAUSER Effects. J. Am. Chem. Soc. 1965, 87, 5250–5251.
[243] SOLOMON, I.: Relaxation Processes in a System of Two Spins. Phys. Rev. 1955, 99, 559–565.
[244] ANDERSON, W. A.; FREEMAN, R.: Influence of a Second Radiofrequency Field on High‐Resolution
Nuclear Magnetic Resonance Spectra. J. Chem. Phys. 1962, 37, 85–103.
[245] FREEMAN, R.; HILL, H. D. W.; KAPTEIN, R.: Proton-Decoupled NMR. Spectra of Carbon-13 with the
Nuclear OVERHAUSER Effect Suppressed. J. Magn. Reson. (1969). 1972, 7, 327–329.
[246] FREEMAN, R.; HILL, H. D. W.: Nuclear OVERHAUSER Effect in Undecoupled NMR Spectra of
Carbon-13. J. Magn. Reson. (1969). 1971, 5, 278–280.
[247] LEVY, G. C.; PEAT, I. R.; ROSANSKE, R.: A High Efficiency Broadband Decoupling Scheme. J. Magn.
Reson. (1969). 1975, 18, 205–208.
[248] FREEMAN, R.; PACHLER, K. G. R.; LA MAR, G. N.: Quenching the Nuclear OVERHAUSER Effect in NMR
Spectra of Carbon‐13. J. Chem. Phys. 1971, 55, 4586–4593.
[249] IBERT, M.; MARSAIS, F.; MERBOUH, N.; BRÜCKNER, C.: Determination of the Side-Products Formed
during the Nitroxide-Mediated Bleach Oxidation of Glucose to Glucaric Acid. Carbohydr. Res.
2002, 337, 1059–1063.
[250] ANGYAL, S. J.: Complex Formation Between Sugars and Metal Ions. Pure Appl. Chem. 1973, 35,
131–146.
[251] ANGYAL, S. J.; WHEEN, R. G.: The Composition of Reducing Sugars in Aqueous Solution. Aust. J.
Chem. 1980, 33, 1001–1011.
[252] MITZNER, R.; BEHRENWALD, E.: Die katalytische Wirkung verschiedener Metallionen auf die
Mutarotation von β-D-Mannose. Chem. Inform. Org. Chem. 1971, 2.
[253] MITZNER, R.; BEHRENWALD, E.: Die katalytische Wirkung verschiedener Metallionen auf die
Mutarotation von β-D-Mannose. Z. Chem. 1971, 11, 64–65.
[254] SMIATACZOWA, K.; MAJ, K.; SKURSKI, P.: Mechanism of the Transition-Metal-Catalyzed
Mutarotation Reaction of N-(p-Chlorophenyl)-β-D-glucopyranosylamine in Methanol.
Carbohydr. Res. 2003, 338, 969–975.
[255] JÜNGEL, A.; ZACHMANN, H. G.: Mathematik für Chemiker. 7. aktualisierte und erweiterte
Auflage. Wiley-VCH: Weinheim, 2014.
[256] LE BARC'H, N.; GROSSEL, J. M.; LOOTEN, P.; MATHLOUTHI, M.: Kinetic Study of the Mutarotation of
D-Glucose in Concentrated Aqueous Solution by Gas-Liquid Chromatography. Food Chem.
2001, 74, 119–124.
[257] LEE, C. Y.; ACREE, T. E.; SHALLENBERGER, R. S.: Mutarotation of D-Glucose and D-Mannose in
Aqueous Solution. Carbohydr. Res. 1969, 9, 356–360.
[258] ACREE, T. E.; SHALLENBERGER, R. S.; LEE, C. Y.; EINSET, J. W.: Thermodynamics and Kinetics of
D-Galactose Tautomers during Mutarotation. Carbohydr. Res. 1969, 10, 355–360.

LITERATURVERZEICHNIS
xvii
[259] WERTZ, P. W.; GARVER, J. C.; ANDERSON, L.: Anatomy of a Complex Mutarotation. Kinetics of
Tautomerization of α-D-Galactopyranose and β-D-Galactopyranose in Water. J. Am. Chem.
Soc. 1981, 103, 3916–3922.
[260] CURTIUS, H.-C.; VÖLLMIN, J. A.; MÜLLER, M.: Bestimmung der Mutarotation von
Monosacchariden mittels Gas-Chromatographie. Fres. J. Anal. Chem. 1968, 243, 341–349.
[261] BENTLEY, R.; BOTLOCK, N.: A Gas Chromatographic Method for Analysis of Anomeric
Carbohydrates and for Determination of Mutarotation Coefficients. Anal. Biochem. 1967, 20,
312–320.
[262] SWEELEY, C. C.; BENTLEY, R.; MAKITA, M.; WELLS, W. W.: Gas-Liquid Chromatography of
Trimethylsilyl Derivatives of Sugars and Related Substances. J. Am. Chem. Soc. 1963, 85,
2497–2507.
[263] RUIZ-MATUTE, A. I.; HERNÁNDEZ-HERNÁNDEZ, O.; RODRÍGUEZ-SÁNCHEZ, S.; SANZ, M. L.; MARTÍNEZ-
CASTRO, I.: Derivatization of Carbohydrates for GC and GC–MS Analyses. J. Chromatogr. B.
2011, 879, 1226–1240.
[264] OKUDA, T.; SAITO, S.; HAYASHI, M.: Trimethysilylation and G.L.C.-Mass Spectrometry of
3-Ketoses and 2-Heptuloses. Carbohydr. Res. 1979, 68, 1–13.
[265] SEMENZA, G.; CURTIUS, C.-H.; KOLÍNSKÁ, J.; MÜLLER, M.: Studies on Intestinal Sucrase and
Intestinal Sugar Transport. VI. Liberation of α-Glucose by Sucrase and Isomaltase from the
Glycone Moeity of the Substrates. Biochim. Biophys. Acta, Enzymol. 1967, 146, 196–204.
[266] OKUDA, T.; KONISHI, K.: Trimethylsilylation of Ketoses: Structures and Gas Chromatography of
the Products of Two-step Reactions from Hexuloses and Heptuloses. J. Chem. Soc. D. 1969,
14, 796–797.
[267] EIGEN, M.: Methods for Investigation of Ionic Reactions in Aqueous Solutions with Half-Times
as short as 10–9 sec. Application to Neutralization and Hydrolysis Reactions. Discuss. Faraday
Soc. 1954, 17, 194–205.
[268] BAIN, A. D.; CRAMER, J. A.: Optimal NMR Measurements for Slow Exchange in Two-Site and
Three-Site Systems. J. Phys. Chem. 1993, 97, 2884–2887.
[269] GRASSI, M.; MANN, B. E.; PICKUP, B. T.; SPENCER, C. M.: The Determination of Individual Rates
from Magnetization-Transfer Measurements. J. Magn. Reson. (1969). 1986, 69, 92–99.
[270] MUHANDIRAM, D. R.; MCCLUNG, R. E. D.: Multisite Magnetization Transfer Experiments. J. Magn.
Reson. (1969). 1987, 71, 187–192.
[271] LU, J.; MA, D.; HU, J.; TANG, W.; ZHU, D.: Nuclear Magnetic Resonance Spectroscopic Studies of
Pyridine Methyl Derivatives binding to Cytochrome c. J. Chem. Soc., Dalton Trans. 1998, 13,
2267–2274.
[272] ZOLNAI, Z.; JURANIĆ, N.; VIKIĆ-TOPIĆ, D.; MACURA, S.: Quantitative Determination of
Magnetization Exchange Rate Constants from a Series of Two-Dimensional Exchange NMR
Spectra. J. Chem. Inf. Comput. Sci. 2000, 40, 611–621.
[273] JEENER, J.; MEIER, B. H.; BACHMANN, P.; ERNST, R. R.: Investigation of Exchange Processes by Two‐
Dimensional NMR Spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553.

LITERATURVERZEICHNIS
xviii
[274] VAN LANDEGHEM, M.; HABER, A.; D’ESPINOSE DE LACAILLERIE, J.-B.; BLÜMICH, B.: Analysis of Multisite
2D Relaxation Exchange NMR. Concepts Magn. Reson. 2010, 36A, 153–169.
[275] KUCHEL, P. W.; BULLIMAN, B. T.; CHAPMAN, B. E.: Mutarotase Equilibrium Exchange Kinetics
studied by 13C-NMR. Biophys. Chem. 1988, 32, 89–95.
[276] WILLIAMS, T. J.; KERSHAW, A. D.; LI, V.; WU, X.: An Inversion Recovery NMR Kinetics Experiment.
J. Chem. Educ. 2011, 88, 665–669.
[277] ROBINSON, G.; KUCHEL, P. W.; CHAPMAN, B. E.; DODDRELL, D. M.; IRVING, M. G.: A Simple Procedure
for Selective Inversion of NMR Resonances for Spin Transfer Enzyme Kinetic Measurements.
J. Magn. Reson. (1969). 1985, 63, 314–319.
[278] BAINE, P.; GERIG, J. T.; STOCK, A. D.: An Experimental Study of some NMR Methods for the
Measurement of Slow Exchange Rates. Org. Magn. Reson. 1981, 17, 41–45.
[279] MNOVA, MESTRELAB RESEARCH. [online] http://mestrelab.com/software/mnova/ [Zitat vom
17.03.2017].
[280] MCMASTER CHEMISTRY: BAIN. [online] http://www.chemistry.mcmaster.ca/bain/ [Zitat vom
17.03.2017].
[281] BAUER, H.; BRINKMEIER, A.; BUDDRUS, J.; JABLONOWSKI, M.: Ring-Ketten-Tautomerie von 2-Desoxy-
D-ribose: Vollständige Analyse der Dynamik durch NMR-Linienformsimulation und
Spinsättigungsübertragung. Liebigs Ann. Chem. 1986, 1986, 1804–1807.
[282] BREITMAIER, E.; SPOHN, K.-H.; BERGER, S.: 13C Spin-Lattice Relaxation Times and the Mobility of
Organic Molecules in Solution. Angew. Chem., Int. Ed. Engl. 1975, 14, 144–159.
[283] REICHENBÄCHER, M.; POPP, J.: Strukturanalytik organischer und anorganischer Verbindungen. 1.
Auflage. Teubner Verlag: Wiesbaden, 2007.
[284] BAIN, A. D.; REX, D. M.; SMITH, R. N.: Fitting Dynamic NMR Lineshapes. Magn. Reson. Chem.
2001, 39, 122–126.
[285] LOWRY, T. M.: CXXV.—Studies of Dynamic Isomerism. I. The Mutarotation of Glucose. J. Chem.
Soc., Trans. 1903, 83, 1314–1323.
[286] HUDSON, C. S.: The Catalysis by Acids and Bases of the Mutarotation of Glucose. J. Am. Chem.
Soc. 1907, 29, 1571–1576.
[287] EULER, H.; ÖLANDER, A.; RUDBERG, E.: Zur Theorie der Katalyse I. Kinetik der Mutarotation. Z.
Anorg. Allg. Chem. 1925, 146, 45–68.
[288] EULER, H.; ÖLANDER, A.: Theorie der Katalyse. II. Kinetik der Mutarotation. II. Z. Anorg. Allg.
Chem. 1926, 152, 113–132.
[289] BRØNSTED, J. N.; GUGGENHEIM, E. A.: Contribution to the Theory of Acid and Basic Catalysis. The
Mutarotation of Glucose. J. Am. Chem. Soc. 1927, 49, 2554–2584.
[290] ISBELL, H. S.; FRUSH, H. L.: Mechanisms for the Mutarotation and Hydrolysis of the
Glycosylamines and the Mutarotation of the Sugars. J. Res. Natl. Bur. Stand. 1951, 46, 132–
144.

LITERATURVERZEICHNIS
xix
[291] LIENHARD, G.; ANDERSON, F.: General Acid and General Base Catalysis by the Monoanions of
Dicarboxylic Acids. J. Org. Chem. 1967, 32, 2229–2233.
[292] NIELSEN, H.; SØRENSEN, P. E.: Extended BRØNSTED Plots and Solvent Isotope Effects for the Base
Catalyzed Mutarotation of Glucopyranoses in Water. Acta Chem. Scand. 1984, 38a, 309–326.
[293] DAVIES, C. G. A.; KAANANE, A.; LABUZA, T. P.; MOSCOWITZ, A. J.; GUILLAUME, F.: Evaluation of the
Acyclic State and the Effect of Solvent Type on Mutarotation Kinetics and on MAILLARD
Browning Rate of Glucose and Fructose in “The MAILLARD Reaction in Foods and Medicine”.
The Royal Society of Chemistry: Cambridge, UK, 1998, 166–171.
[294] DAWSON, H. M.; LOWSON, W.: CCCXXVII.—Acid and Salt Effects in Catalysed Reactions. Part XI.
The Hydrolysis of Ethyl Acetate and the Catalytic Catenary. J. Chem. Soc. 1927, 0, 2444–2451.
[295] LOWRY, T. M.: CCCXL.—Studies of Dynamic Isomerism. Part XXV. The Mechanism of Catalysis
by Acids and Bases. J. Chem. Soc. 1927, 0, 2554–2565.
[296] QIAN, X.: Free Energy Surface for BRØNSTED Acid-Catalyzed Glucose Ring-Opening in Aqueous
Solution. J. Phys. Chem. B. 2013, 117, 11460–11465.
[297] PLAZINSKI, W.; PLAZINSKA, A.; DRACH, M.: The Water-Catalyzed Mechanism of the Ring-Opening
Reaction of Glucose. Phys. Chem. Chem. Phys. 2015, 17, 21622–21629.
[298] FENG, S.; BAGIA, C.; MPOURMPAKIS, G.: Determination of Proton Affinities and Acidity Constants
of Sugars. J. Phys. Chem. A. 2013, 117, 5211–5219.
[299] SCHÜRMANN, G.: Modelling pKa of Carboxylic Acids and Chlorinated Phenols. Quant. Struct.-
Act. Relat. 1996, 15, 121–132.
[300] HENCHOZ, Y.; SCHAPPLER, J.; GEISER, L.; PRAT, J.; CARRUPT, P.-A.; VEUTHEY, J.-L.: Rapid Determination
of pKa Values of 20 Amino Acids by CZE with UV and Capacitively Coupled Contactless
Conductivity Detections. Anal. Bioanal. Chem. 2007, 389, 1869–1878.
[301] WESTPHAL, G.; BOCHOW, C.; KROH, L.: Untersuchungen zur MAILLARD-Reaktion. 13. Mitt. Zum
Einfluß der Konstitution der Aminosäure auf den Verlauf der MAILLARD-Reaktion. Food /
Nahrung. 1985, 29, 69–74.
[302] LIU, S.-C.; YANG, D.-J.; JIN, S.-Y.; HSU, C.-H.; CHEN, S.-L.: Kinetics of Color Development, pH
Decreasing, and Anti-Oxidative Activity Reduction of MAILLARD Reaction in Galactose/Glycine
Model Systems. Food Chem. 2008, 108, 533–541.
[303] KROH, L. W.; FIEDLER, T.; WAGNER, J.: α-Dicarbonyl Compounds–Key Intermediates for the
Formation of Carbohydrate-based Melanoidins. Ann. N. Y. Acad. Sci. 2008, 1126, 210–215.
[304] GOBERT, J.; GLOMB, M. A.: Degradation of Glucose: Reinvestigation of Reactive α-Dicarbonyl
Compounds. J. Agric. Food Chem. 2009, 57, 8591–8597.
[305] KANZLER, C.; HAASE, P. T.; KROH, L. W.: Antioxidant Capacity of 1-Deoxy-D-erythro-hexo-2,3-
diulose and D-arabino-Hexo-2-ulose. J. Agric. Food Chem. 2014, 62, 2837–2844.
[306] KANZLER, C.; HAASE, P. T.; SCHESTKOWA, H.; KROH, L. W.: Antioxidant Properties of Heterocyclic
Intermediates of the MAILLARD Reaction and Structural Related Compounds. J. Agric. Food
Chem. 2016, 64, 7829–7837.

LITERATURVERZEICHNIS
xx
[307] HAASE, P. T.; KANZLER, C.; HILDEBRANDT, J.; KROH, L. W.: Browning Potential of C6-α-Dicarbonyl
Compounds under MAILLARD Conditions. J. Agric. Food Chem. 2017, 65, 1924–1931.
[308] TRESSL, R.; KERSTEN, E.; REWICKI, D.: Formation of Pyrroles, 2-Pyrrolidones, and Pyridones by
Heating of 4-Aminobutyric Acid and Reducing Sugars. J. Agric. Food Chem. 1993, 41, 2125–
2130.
[309] TRESSL, R.; KERSTEN, E.; REWICKI, D.: Formation of 4-Aminobutyric Acid specific MAILLARD
Products from [1-13C]-D-Glucose, [1-13C]-D-Arabinose, and [1-13C]-D-Fructose. J. Agric. Food
Chem. 1993, 41, 2278–2285.
[310] HOBLEY, P.; HOWARTH, O.; IBBETT, R. N.: 1H and 13C NMR Shifts for Aldopyranose and
Aldofuranose Monosaccharides. Magn. Reson. Chem. 1996, 34, 755–760.
[311] BOCK, K.; PEDERSEN, C.: Carbon-13 Nuclear Magnetic Resonance Spectroscopy of
Monosaccharides. Adv. Carbohydr. Chem. Biochem. 1983, 41, 27–66.
[312] RITCHIE, R. G. S.; CYR, N.; KORSCH, B.; KOCH, H. J.; PERLIN, A. S.: Carbon-13 Chemical Shifts of
Furanosides and Cyclopentanols. Configurational and Conformational Influences. Can. J.
Chem. 1975, 53, 1424–1433.
[313] EYRING, H.: The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115.
[314] ANET, F. A. L.; ANET, R.: Conformational Processes in Rings in “Dynamic Nuclear Magnetic
Resonance Spectroscopy”. Academic Press, Inc.: New York, USA, 1975, 578–584.
[315] QUE JR, L.; GRAY, G. R.: Carbon-13 Nuclear Magnetic Resonance Spectra and the Tautomeric
Equilibriums of Ketohexoses in Solution. Biochem. 1974, 13, 146–153.
[316] JEFFREY, G. A.; LEWIS, L.: Cooperative Aspects of Hydrogen Bonding in Carbohydrates.
Carbohydr. Res. 1978, 60, 179–182.
[317] PONS, I.; GARRAULT, C.; JAUBERT, J.-N.; MOREL, J.; FENYO, J.-C.: Analysis of Aromatic Caramel. Food
Chem. 1991, 39, 311–320.
[318] AJANDOUZ, E. H.; PUIGSERVER, A.: Nonenzymatic Browning Reaction of Essential Amino Acids:
Effect of pH on Caramelization and MAILLARD Reaction Kinetics. J. Agric. Food Chem. 1999, 47,
1786–1793.
[319] KWAK, E.-J.; LIM, S.-I.: The Effect of Sugar, Amino Acid, Metal Ion, and NaCl on Model MAILLARD
Reaction under pH Control. Amino acids. 2004, 27, 85–90.
[320] NORLEUCINE. [online] https://www.drugbank.ca/drugs/DB04419 [Zitat vom 07.04.2017].
[321] AMINOCAPROIC ACID. [online] https://www.drugbank.ca/drugs/DB00513 [Zitat vom
07.04.2017].
[322] STEVENS, J. F.; MAIER, C. S.: Acrolein: Sources, Metabolism, and Biomolecular Interactions
Relevant to Human Health and Disease. Mol. Nutr. & Food Res. 2008, 52, 7–25.
[323] BECK, J.; LEDL, F.; SENGL, M.; SEVERIN, T.: Formation of Acids, Lactones and Esters through the
MAILLARD Reaction. Z. Lebensm. Unters. Forsch. 1990, 190, 212–216.

LITERATURVERZEICHNIS
xxi
[324] HUYGHUES-DESPOINTES, A.; YAYLAYAN, V. A.: Retro-Aldol and Redox Reactions of AMADORI
Compounds. J. Agric. Food Chem. 1996, 44, 672–681.
[325] HOFMANN, T.; BORS, W.; STETTMAIER, K.: Studies on Radical Intermediates in the Early Stage of
the Nonenzymatic Browning Reaction of Carbohydrates and Amino Acids. J. Agric. Food
Chem. 1999, 47, 379–390.
[326] USUI, T.; YOSHINO, M.; WATANABE, H.; HAYASE, F.: Determination of Glyceraldehyde formed in
Glucose Degradation and Glycation. Biosci., Biotechnol., Biochem. 2007, 71, 2162–2168.
[327] MOSSINE, V. V.; BARNES, C. L.; CHANCE, D. L.; MAWHINNEY, T. P.: Stabilization of the Acyclic
Tautomer in Reducing Carbohydrates. Angew. Chem., Int. Ed. Engl. 2009, 48, 5517–5520.
[328] VAN BOEKEL, M. A. J. S.: Kinetic Aspects of the MAILLARD Reaction. Food / Nahrung. 2001, 45,
150–159.
[329] HALL, L. D.; TALAGALA, S. L.: Mapping of pH and Temperature Distribution using Chemical-Shift-
Resolved Tomography. J. Magn. Reson. (1969). 1985, 65, 501–505.
[330] ACKERMAN, J. J. H.; SOTO, G. E.; SPEES, W. M.; ZHU, Z.; EVELHOCH, J. L.: The NMR Chemical Shift pH
Measurement Revisited: Analysis of Error and Modeling of a pH Dependent Reference. Magn.
Reson. Med. 1996, 36, 674–683.
[331] MARTIN, R. B.: Deuterated Water Effects on Acid Ionization Constants. Science. 1963, 139,
1198–1203.
[332] TYNKKYNEN, T.; TIAINEN, M.; SOININEN, P.; LAATIKAINEN, R.: From Proton Nuclear Magnetic
Resonance Spectra to pH. Assessment of 1H NMR pH Indicator Compound Set for Deuterium
Oxide Solutions. Anal. Chim. Acta. 2009, 648, 105–112.
[333] KIM, M. H.; KIM, C. S.; LEE, H. W.; KIM, K.: Temperature Dependence of Dissociation Constants
for Formic Acid and 2,6-Dinitrophenol in Aqueous Solutions up to 175 °C. J. Chem. Soc.,
Faraday Trans. 1996, 92, 4951–4956.
[334] MAPLE, S. R.; ALLERHAND, A.: Detailed Tautomeric Equilibrium of Aqueous D-Glucose.
Observation of Six Tautomers by Ultrahigh Resolution Carbon-13 NMR. J. Am. Chem. Soc.
1987, 109, 3168–3169.
[335] WILLIAMS, C.; ALLERHAND, A.: Detection of β-D-Glucofuranose in Aqueous Solutions of
D-Glucose. Application of Carbon-13 FOURIER-Transform N.M.R. Spectroscopy. Carbohydr.
Res. 1977, 56, 173–179.
[336] HAYWARD, L. D.; ANGYAL, S. J.: A Symmetry Rule for the Circular Dichroism of Reducing Sugars,
and the Proportion of Carbonyl Forms in Aqueous Solutions thereof. Carbohydr. Res. 1977,
53, 13–20.
[337] KIELY, D. E.; BENZING-NGUYEN, L.: delta-Dicarbonyl Sugars. IV. Oxidation of Carbohydrates with
Chromic Acid. Synthesis of 6-Acetamido-6-deoxy-D-xylo-hexos-5-ulose. J. Org. Chem. 1975,
40, 2630–2634.
[338] ANTEUNIS, M.; DANNEELS, D.: Stereochemical Aspects of Proton Chemical Shifts. III —
Configurational Assignments in Pentacyclic Systems without Recourse to a KARPLUS Equation.
Org. Magn. Reson. 1975, 7, 345–348.

LITERATURVERZEICHNIS
xxii
[339] WIT, G.; KIEBOOM, A. P. G.; VAN BEKKUM, H.: Ionization and Mutarotation of Hexoses in Aqueous
Alkaline Solution as Studied by 13C-NMR Spectroscopy. Tetrahedron Lett. 1975, 16, 3943–
3946.
[340] WIT, G.; KIEBOOM, A. P. G.; VAN BEKKUM, H.: Ionization and Mutarotation of Hexoses in Aqueous
Alkaline Solution as Studied by 13C NMR Spectroscopy. Recl. Trav. Chim. Pays-Bas. 1979, 98,
355–361.
[341] CHRISTENSEN, J. J.; RYTTING, J. H.; IZATT, R. M.: Thermodynamics of Proton Dissociation in Dilute
Aqueous Solution. Part XV. Proton Dissociation from Several Monosaccharides at 10 and
40 °C. J. Chem. Soc. B. 1970, 0, 1646–1648.
[342] WOODS, R. J.; SZAREK, W. A.; SMITH JR., V. H.: The Proton Affinities and Deprotonation Enthalpies
of β-D-Fructopyranose and α-L-Sorbopyranose. Can. J. Chem. 1991, 69, 1917–1928.
[343] DENO, N. C.; TURNER, J. O.: The Basicity of Alcohols and Ethers. J. Org. Chem. 1966, 31, 1969–
1970.
[344] LEE, D. G.; CAMERON, R.: Basicity of Ethanol. Acidity Function for Alcohols. J. Am. Chem. Soc.
1971, 93, 4724–4728.
[345] GILLESPIE, S. E.; OSCARSON, J. L.; IZATT, R. M.; WANG, P.; RENUNCIO, J. A. R.; PANDO, C.:
Thermodynamic Quantities for the Protonation of Amino Acid Amino Groups from 323.15 to
398.15 K. J. Solution Chem. 1995, 24, 1219–1247.
[346] CLARKE, R. G. F.; COLLINS, C. M.; ROBERTS, J. C.; TREVANI, L. N.; BARTHOLOMEW, R. J.; TREMAINE, P. R.:
Ionization Constants of Aqueous Amino Acids at Temperatures up to 250 °C using
Hydrothermal pH Indicators and UV-Visible Spectroscopy. Geochim. Cosmochim. Acta. 2005,
69, 3029–3043.
[347] GUPTA, M.; DA SILVA, E. F.; SVENDSEN, H. F.: Modeling Temperature Dependency of Ionization
Constants of Amino Acids and Carboxylic Acids. J. Phys. Chem. B. 2013, 117, 7695–7709.
[348] KRȨŻEL, A.; BAL, W.: A Formula for Correlating pKa Values Determined in D2O and H2O. J. Inorg.
Biochem. 2004, 98, 161–166.
[349] MARSHALL, W. L.; FRANCK, E. U.: Ion Product of Water Substance, 0–1000 °C, 1–10,000 bars
New International Formulation and its Background. J. Phys. Chem. Ref. Data. 1981, 10, 295–
304.
[350] MESMER, R. E.; HERTING, D. L.: Thermodynamics of Ionization of D2O and D2PO4-. J. Solution
Chem. 1978, 7, 901–912.
[351] MAIER, G. D.; KUSIAK, J. W.; BAILEY, J. M.: The Study of the Mutarotation of D-Glucose,
D-Fructose, and D-Ribose by use of their Circular Dichroism. Carbohydr. Res. 1977, 53, 1–11.
[352] TANAKA, M.; GIRARD, G.; DAVIS, R.; PEUTO, A.; BIGNELL, N.: Recommended Table for the Density
of Water Between 0 °C and 40 °C Based on Recent Experimental Reports. Metrologia. 2001,
38, 301–309.
[353] EVANS, W. J.; MCCOURTNEY, E. J.; SHRAGER, R. I.: Titration Studies of Phytic Acid. J. Am. Oil Chem.
Soc. 1982, 59, 189–191.

LITERATURVERZEICHNIS
xxiii
[354] MILLS, F. D.; HODGE, J. E.: AMADORI compounds: Vacuum Thermolysis of 1-Deoxy-1-L-prolino-
D-fructose. Carbohydr. Res. 1976, 51, 9–21.
[355] BASTARDO, L. A.; MÉSZÁROS, R.; VARGA, I.; GILÁNYI, T.; CLEASSON, P. M.: Deuterium Isotope Effects
on the Interaction Between Hyperbranched Polyethylene Imine and an Anionic Surfactant. J.
Phys. Chem. B. 2005, 109, 16196–16202.
[356] MIKKELSEN, K.; NIELSEN, S. O.: Acidity Measurements with the Glass Electrode in H2O-D2O
Mixtures. J. Phys. Chem. 1960, 64, 632–637.
[357] FINDEISEN, M.; BERGER, S.: 50 and More Essential NMR Experiments. Wiley-VCH: Weinheim,
2014.
[358] BERTHELOT, M. Chimie Organique Fondée sur la Synthèse. Mallet-Bachelier: Paris, 1860, 256.
[359] FISCHER, E.: Reduction der Säuren der Zuckergruppe. II. Ber. Dtsch. Chem. Ges. 1890, 23, 930–
938.
[360] JUNGFLEISCH, E.; LEFRANC, E.: Sur le Lévulose. C. R. Hebd. Seances Acad. Sci. 1881, 93, 547–550.
[361] COCINERO, E. J.; LESARRI, A.; ÉCIJA, P.; CIMAS, Á.; DAVIS, B. G.; BASTERRETXEA, F. J.; FERNÁNDEZ, J. A.;
CASTAÑO, F.: Free Fructose is Conformationally Locked. J. Am. Chem. Soc. 2013, 135, 2845–
2852.
[362] HUGGINS, M. L.: Bond Energies and Polarities. J. Am. Chem. Soc. 1953, 75, 4123–4126.
[363] PAULING, L.: The Nature of the Chemical Bond. IV. The Energy of Single Bonds and the Relative
Electronegativity of Atoms. J. Am. Chem. Soc. 1932, 54, 3570–3582.
[364] ALLRED, A. L.: Electronegativity Values from Thermochemical Data. J. Inorg. Nucl. Chem. 1961,
17, 215–221.
[365] NOBELPRIZE.ORG - THE OFFICIAL WEB SITE OF THE NOBEL PRIZE. [online] http://www.nobelprize.org/
nobel_prizes/physics/laureates/1952/# [Zitat vom 31.05.2017].
[366] ERLENMEYER, E.: Ueber Phenylbrommilchsäure. Ber. Dtsch. Chem. Ges. 1880, 13, 305–310.
[367] FISCHER, E.: Ueber Isoglucosamin. Ber. Dtsch. Chem. Ges. 1886, 19, 1920–1924.
[368] LI, C.; WANG, H.; JUÁREZ, M.; RUAN, E. D.: Structural Characterization of AMADORI
Rearrangement Product of Glucosylated Nα-Acetyl-lysine by Nuclear Magnetic Resonance
Spectroscopy. Int. J. Spectrosc. 2014, 2014, 1–6.
[369] BECK, J.; LEDL, F.; SEVERIN, T.: Formation of Glucosyl-deoxyosones from AMADORI Compounds of
Maltose. Z. Lebensm. Unters. Forsch. 1989, 188, 118–121.
[370] BALEJA, J. D.; MOULT, J.; SYKES, B. D.: Distance Measurement and Structure Refinement with
NOE Data. J. Magn. Reson. (1969). 1990, 87, 375–384.
[371] KALTSCHNEE, L.; KNOLL, K.; SCHMIDTS, V.; ADAMS, R. W.; NILSSON, M.; MORRIS, G. A.; THIELE, C. M.:
Extraction of Distance Restraints from Pure Shift NOE Experiments. J. Magn. Reson. 2016,
271, 99–109.
[372] VRANKEN, W.: A Global Analysis of NMR Distance Constraints from the PDB. J. Biomol. NMR.
2007, 39, 303–314.

LITERATURVERZEICHNIS
xxiv
[373] JONES, C. R.; BUTTS, C. P.; HARVEY, J. N.: Accuracy in Determining Interproton Distances using
Nuclear OVERHAUSER Effect Data from a Flexible Molecule. Beilstein J. Org. Chem. 2011, 7,
145–150.
[374] CID, M.-M.; BRAVO, J.: Structure elucidation in organic chemistry. Wiley-VCH: Weinheim, 2015.
[375] GENERALIZED 3JHH CALCULATION ACC. HAASNOOT ET AL. [online] http://www.stenutz.eu/
conf/haasnoot.php [Zitat vom 13.03.2017].
[376] BALACCO, G.: A Desktop Calculator for the KARPLUS Equation. J. Chem. Inf. Comput. Sci. 1996,
36, 885–887.
[377] BALACCO, G.: Sweet J. [online] http://www.inmr.net/sweetj.html [Zitat vom 14.03.2017].
[378] HETERO- AND HOMONUCLEAR COUPLING CONSTANT CALCULATION. [online] http://www.stenutz.eu/
conf/karplus.html [Zitat vom 14.03.2017].
[379] SPENCER, R. G. S.; FISHBEIN, K. W.: Measurement of Spin–Lattice Relaxation Times and
Concentrations in Systems with Chemical Exchange Using the One-Pulse Sequence. J. Magn.
Reson. 2000, 142, 120–135.
[380] LA MAR, G. N.: Problem with Carbon-13 Intensities in Proton-Decoupled Nuclear Magnetic
Resonance Spectra. Undermining the OVERHAUSER Effect with Free Radicals. J. Am. Chem. Soc.
1971, 93, 1040–1041.
[381] LA MAR, G. N.: The Effect of Paramagnetic Metal Ions on Proton-Decoupled C-13 NMR
Intensities. Chem. Phys. Lett. 1971, 10, 230–232.
[382] GRIMSHAW, C. E.: Direct Measurement of the Rate of Ring Opening of D-Glucose by
Enzymecatalyzed Reduction. Carbohydr. Res. 1986, 148, 345–348.
[383] PARKER, F. S.: Infrared Spectra of Carbohydrates in Water and a New Measure of
Mutarotation. Biochim. Biophys. Acta. 1960, 42, 513–519.
[384] NOBELPRIZE.ORG - THE OFFICIAL WEB SITE OF THE NOBEL PRIZE. [online] http://www.nobelprize.org/
nobel_prizes/chemistry/laureates/1967/ [Zitat vom 16.03.2017].
[385] KINGSLEY, P. B.; MONAHAN, W. G.: Effects of Off-Resonance Irradiation, Cross-Relaxation, and
Chemical Exchange on Steady-State Magnetization and Effective Spin–Lattice Relaxation
Times. J. Magn. Reson. 2000, 143, 360–375.
[386] HOFFMAN, R. A.; FORSÉN, S.: Transient and Steady‐State OVERHAUSER Experiments in the
Investigation of Relaxation Processes. Analogies between Chemical Exchange and Relaxation.
J. Chem. Phys. 1966, 45, 2049–2060.
[387] CAMPBELL, I. D.; DOBSON, C. M.; RATCLIFFE, R. G.; WILLIAMS, R. J. P.: FOURIER Transform NMR Pulse
Methods for the Measurement of Slow-Exchange Rates. J. Magn. Reson. (1969). 1978, 29,
397–417.
[388] MACURA, S.; ERNST, R. R.: Elucidation of Cross Relaxation in Liquids by Two-Dimensional N.M.R.
Spectroscopy. Mol. Phys. 2006, 41, 95–117.
[389] CAMPBELL, I. D.; FREEMAN, R.: Influence of Cross-Relaxation on NMR Spin-Lattice Relaxation
Times. J. Magn. Reson. (1969). 1973, 11, 143–162.

LITERATURVERZEICHNIS
xxv
[390] LEWIS, B. E.; CHOYTUN, N.; SCHRAMM, V. L.; BENNET, A. J.: Transition States for Glucopyranose
Interconversion. J. Am. Chem. Soc. 2006, 128, 5049–5058.
[391] NOBELPRIZE.ORG - THE OFFICIAL WEB SITE OF THE NOBEL PRIZE. [online] https://www.nobelprize.org/
nobel_prizes/chemistry/laureates/1929/ [Zitat vom 23.03.2017].
[392] HÄSELER, J.; BEYERLEIN, B.; KROH, L. W.: Influence of D-Glucose Configuration on the Kinetics of
the Nonenzymatic Browning Reaction in “The MAILLARD Reaction in Foods and Medicine”. The
Royal Society of Chemistry: Cambridge, UK, 1998, 172–177.
[393] CAPON, B.; WALKER, R. B.: Intramolecular Catalysis in the Mutarotation of Aldoses. J. Chem.
Soc. D. 1971, 21, 1323–1324.
[394] LOS, J. M.; SIMPSON, L. B.; WIESNER, K.: The Kinetics of Mutarotation of D-Glucose with
Consideration of an Intermediate Free-Aldehyde Form. J. Am. Chem. Soc. 1956, 78, 1564–
1568.
[395] BAILEY, J. M.; FISHMAN, P. H.; PENTCHEV, P. G.: Anomalous Mutarotation of Glucose 6-phosphate.
An Example of Intramolecular Catalysis. Biochemistry. 2002, 9, 1189–1194.
[396] ZHANG, W.; SERIANNI, A. S.: Phosphate-Catalyzed Degradation of D-Glucosone in Aqueous
Solution is Accompanied by C1-C2 Transposition. J. Am. Chem. Soc. 2012, 134, 11511–11524.

ABBILDUNGSVERZEICHNIS
xxvi
Abbildungsverzeichnis Abbildung 1: Zeitliche Entwicklung der Anzahl der in SciFinder® gelisteten Publikationen zu den
Suchbegriffen „Glucose“ und „Maillard reaction“ seit 1856. ..................................... 1
Abbildung 2: Graphische Darstellung verschiedener Pulsprogramme zur Protonen-
Breitbandentkopplung von 13C-NMR-Spektren.* a) inverse-gated decoupling,
b) gated decoupling, c) power-gated decoupling. (Der Entkopplerkanal ist jeweils
oben und der X-Kernkanal unten abgebildet.) *Die graphischen Darstellungen sind der
Software Bruker TopSpin 3.5 pL 2 entlehnt. ……………………………………………………………………... 25
Abbildung 3: Modellvorstellung der Sättigung einer Kernresonanz für den Fall 𝑇1(𝑖) ≫ 𝑇2
(𝑖). (blaue
Pfeile: z-Magnetisierung; rote Pfeile: Quermagnetisierung) .................................... 33
Abbildung 4: Abbauraten der D-Fructose 1a in 0.5 M Lösung in An- und Abwesenheit
stöchiometrischer Mengen der Aminosäuren 4a-4e in Abhängigkeit des pH-Wertes
bei einer Temperatur von (90 ± 2) °C. (Die gestrichelte Linie gibt die Grenze an, ab
der Abbaureaktionen messbar sind. Die gepunktete Linie gibt die Grenze an, ab der
Abbauraten quantifiziert werden können. Datenpunkte oberhalb der gepunkteten
Linie sind somit für quantitative Vergleiche nutzbar.) .............................................. 43
Abbildung 5: Abbauraten der Aminosäuren 4a-4e in 0.5 M Lösung in Anwesenheit
stöchiometrischer Mengen an D-Fructose 1a in Abhängigkeit des pH-Wertes bei einer
Temperatur von (90 ± 2) °C. (Die gestrichelte Linie gibt die Grenze an, ab der
Abbaureaktionen messbar sind. Die gepunktete Linie gibt die Grenze an, ab der
Abbauraten quantifiziert werden können. Datenpunkte oberhalb der gepunkteten
Linie sind somit für quantitative Vergleiche nutzbar.) .............................................. 44
Abbildung 6: Abbauraten der AMADORI-Verbindungen 6a-6e in 0.025 M Lösung in Abhängigkeit des
pH-Wertes bei einer Temperatur von (90 ± 2) °C. (Die gestrichelte Linie gibt die
Grenze an, ab der Abbaureaktionen messbar sind. Die gepunktete Linie gibt die
Grenze an, ab der Abbauraten quantifiziert werden können. Datenpunkte oberhalb
der gepunkteten Linie sind somit für quantitative Vergleiche nutzbar.) .................. 45
Abbildung 7: Zeitabhängiger Verlauf der Konzentration von D-Glucoson 9d bei der Reaktion von
D-Fructose 1a mit β-Alanin 4d (je 0.5 M) bei pH 6.5 während der Aufheizphase der
Reaktionslösung von Raumtemperatur auf 90 °C. Bei einer Temperatur von 90 °C
erfolgte die Zugabe eines Reduktions- (Maltol) bzw. Oxidationsmittels (FREMYS Salz)
(je 0.05 M). Eine dritte Reaktionslösung diente als Kontrolle. Die logarithmische
Skalierung der Ordinatenachse ist zu beachten. ...................................................... 47
Abbildung 8: Zeitabhängiger Verlauf der Konzentration von 3-Desoxyglucoson 9b bei der Reaktion
von D-Fructose 1a mit β-Alanin 4d (je 0.5 M) bei pH 6.5 während der Aufheizphase
der Reaktionslösung von Raumtemperatur auf 90 °C. Bei einer Temperatur von 90 °C
erfolgte die Zugabe eines Reduktions- (Maltol) bzw. Oxidationsmittels (FREMYS Salz)
(je 0.05 M). Eine dritte Reaktionslösung diente als Kontrolle. Die logarithmische
Skalierung der Ordinatenachse ist zu beachten. ...................................................... 48

ABBILDUNGSVERZEICHNIS
xxvii
Abbildung 9: Bildungsraten von 3-Desoxyglucoson 9b in Reaktionslösungen der D-Fructose 1a in
An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e (je 0.5 M) in
Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C. ........................ 50
Abbildung 10: Bildungsraten von 1-Desoxyglucoson 9a in Reaktionslösungen der D-Fructose 1a in
An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e (je 0.5 M) in
Abhängigkeit des pH-Wertes bei einer Temperatur von (90 ± 2) °C. ........................ 51
Abbildung 11: Bildungsraten von 3-Desoxyglucoson 9b in Reaktionslösungen der
AMADORI-Verbindungen 6a-6e (je 0.025 M) in Abhängigkeit des pH-Wertes bei einer
Temperatur von (90 ± 2) °C. Die logarithmische Skalierung der Ordinatenachse ist zu
beachten. .................................................................................................................. 52
Abbildung 12: Bildungsraten von 1-Desoxyglucoson 9a in Reaktionslösungen der
AMADORI-Verbindungen 6a-6e (je 0.025 M) in Abhängigkeit des pH-Wertes bei einer
Temperatur von (90 ± 2) °C. Die logarithmische Skalierung der Ordinatenachse ist zu
beachten. .................................................................................................................. 52
Abbildung 13: Raten der Extinktionszunahme bei 284 nm (Geschwindigkeit der frühen
MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der
D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e
bei einer Temperatur von (90 ± 2) °C. ....................................................................... 54
Abbildung 14: Raten der Extinktionszunahme bei 420 nm (Geschwindigkeit der fortgeschrittenen
MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der
D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e
bei einer Temperatur von (90 ± 2) °C. ....................................................................... 54
Abbildung 15: Raten der Extinktionszunahme bei 284 nm (Geschwindigkeit der frühen
MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der
D-Fructose 1a sowie der AMADORI-Verbindungen 6a-6e bei einer Temperatur von
(90 ± 2) °C. ................................................................................................................. 55
Abbildung 16: Raten der Extinktionszunahme bei 420 nm (Geschwindigkeit der fortgeschrittenen
MAILLARD-Reaktion) in Abhängigkeit des pH-Wertes für Modellreaktionen der
D-Fructose 1a sowie der AMADORI-Verbindungen 6a-6e bei einer Temperatur von
(90 ± 2) °C. ................................................................................................................. 56
Abbildung 17: Abhängigkeit der chemischen Verschiebungen von C-3 und H-C-3 der β-pyranoiden
Anomere 6(ii) von der Anzahl der Methyleneinheiten zwischen der Amino- und der
Carboxylfunktion. ...................................................................................................... 62
Abbildung 18: 1H-NMR-Spektrum einer 0.5 M Lösung von FAla 6a nach mehrstündiger Reaktion bei
87 °C und einem pD-Wert von 3.0 (entspricht pH 2.8). Es ist zu erkennen, dass HMF
13 als Folge des Abbaus von FAla 6a gebildet wird. Die Protonen am C-1, C-3 und C-4
unterliegen dabei einem teilweisen Deuteriumaustausch. Die relativen Flächen der
NMR-Signale sind in Rot angegeben. Neben 3J(H,H)-Kopplungen sind aufgrund der
Isotopenmarkierung am C-2 auch 2J(C,H)- und 3J(C,H)-Kopplungen erkennbar. ...... 66
Abbildung 19: Overlay von Ausschnitten der HPLC-DAD-Chromatogramme (@ 317 nm) der
AMADORI-Verbindungen 6a-6e, die in 0.025 M Lösungen für drei Stunden bei einer
Temperatur von (90 ± 2) °C und einem pH-Wert von 2.5 zur Reaktion gebracht
wurden. Es handelt sich um die Signale der mittels ortho-Phenylendiamin

ABBILDUNGSVERZEICHNIS
xxviii
derivatisierten α-Dicarbonylverbindungen 3-Desoxyglucoson 9b (Signal 1)
und -galactoson 9c (Signal 2), die aus dem Abbau der AMADORI-Verbindungen 6a-6e
hervorgehen. ............................................................................................................. 67
Abbildung 20: Overlay von Ausschnitten der HPLC-DAD-Chromatogramme (@ 317 nm) von 0.025 M
Lösungen des Fructosylglycins 6c, die bei verschiedenen pH-Werten für drei Stunden
bei einer Temperatur von (90 ± 2) °C zur Reaktion gebracht wurden. Es handelt sich
um die Signale der mittels ortho-Phenylendiamin derivatisierten
α-Dicarbonylverbindungen 3-Desoxyglucoson 9b (Signal 1) und -galactoson 9c
(Signal 2), die aus dem Abbau der AMADORI-Verbindung 6c hervorgehen. .............. 68
Abbildung 21: Rate der 1,2-Enolisierung von Fructosylalanin 6a und -prolin 6b in Abhängigkeit des
pD-Wertes bei Raumtemperatur, gemessen mittels quantitativer 1H-NMR-Spektroskopie über einen Zeitraum von 24 Tagen. Die logarithmische
Skalierung der Ordinatenachse ist zu beachten. ...................................................... 70
Abbildung 22: Gegenüberstellung der Raten der 1,2-Enolisierung und der Bildung von
3-Desoxyglucoson 9b für Fructosylglycin 6c, -β-alanin 6d und -γ-aminobuttersäure 6e
für einen pH- bzw. pD-Wert von 4.5 und eine Temperatur von 90 °C. ..................... 71
Abbildung 23: Teilausschnitt des anomeren Verschiebungsbereiches eines deconvolierten 13C-NMR-Spektrums von Fructosyl-β-alanin 6d. ....................................................... 71
Abbildung 24: Nachweis der reversiblen 1,2-Enolisierung für D-Fructose 1a anhand der
Hochfeldverschiebung anomerer 13C-NMR-Resonanzen durch den H/D-Austausch am
C-1. ............................................................................................................................ 73
Abbildung 25: Gegenüberstellung der konzentrationsgewichteten Ringöffnungsraten und der Raten
der 1,2-Enolisierung von Fructosylglycin 6c, -β-alanin 6d und -γ-aminobuttersäure 6e
für einen pD-Wert von 4.5 und eine Temperatur von 57 °C. .................................... 74
Abbildung 26: Anomerer Bereich des 1H-NMR-Spektrums von D-Glucose 2a bei einer Messfrequenz
von 600 MHz bei 67 °C und einem pD-Wert von 5.0. Bei den mit * gekennzeichneten
Signalen handelt es sich um die 13C-Satteliten der NMR-Resonanzen der pyranoiden
Hauptisomere. ........................................................................................................... 82
Abbildung 27: Sättigungstransfer-NMR-Differenzspektrum der D-Glucose 2a bei selektiver Sättigung
der Protonenresonanzen bei ca. 10.33 ppm mit Hilfe einer 90° GAUSS-Softpuls-
Kaskade einer Dauer von 60 s und einer spektralen Anregungsbreite von 212 Hz.
Gemessen bei 600 MHz bei einer Temperatur von 87 °C und pD 2.55. Der mit *
gekennzeichnete Verschiebungsbereich zeigt typische symmetrische
Differenzsignale. Diese sind für Protonenresonanzen zu beobachten, für die kein
Sättigungstransfer erfolgt. ........................................................................................ 82
Abbildung 28: Vergleich der pD-abhängigen Ringöffnungsraten von Anomeren der D-Fructose 1a in
An- und Abwesenheit stöchiometrischer Mengen von L-Alanin 4a für eine
Temperatur von 37 °C. a) Vergleichende Darstellung für β-D-Fructopyranose 1a(ii).
b) Vergleichende Darstellung für β-D-Fructofuranose 1a(iv).................................... 88
Abbildung 29: Vergleich der pD-abhängigen Ringöffnungsraten der α-furanoiden Anomere der
D-Glucose 2a, D-Fructose 1a und D-Tagatose 1b für eine Temperatur von 37 °C. Die
logarithmische Skalierung der Ordinatenachse ist zu beachten. .............................. 89

ABBILDUNGSVERZEICHNIS
xxix
Abbildung 30: Vergleich der pD-abhängigen Ringöffnungsraten der β-furanoiden Anomere der
D-Fructose 1a sowie des Fructosylalanins 6a und -prolins 6b für eine Temperatur von
37 °C. ......................................................................................................................... 90
Abbildung 31: ACuSTiC-Werte von D-Fructose 1a in An- und Abwesenheit stöchiometrischer
Mengen an L-Alanin 4a und L-Prolin 4b sowie die der entsprechenden
AMADORI-Verbindungen 6a und 6b in Abhängigkeit des pD-Wertes für eine
Temperatur von 90 °C. .............................................................................................. 91
Abbildung 32: ACuSTiC-Werte von D-Fructose 1a, D-Tagatose 1b und D-Glucose 2a in Abhängigkeit
des pD-Wertes für eine Temperatur von 100 °C. a) Lineare Skalierung der
Ordinatenachse, mit gesonderter Ordinate für D-Glucose 2a. b) Logarithmische
Skalierung der Ordinatenachse. ................................................................................ 92
Abbildung 33: Vergleich der Geschwindigkeiten der frühen MAILLARD-Reaktion, gemessen als Rate
der Extinktionszunahme bei einer Wellenlänge von 284 nm, von D-Fructose 1a und
D-Glucose 2a in Abhängigkeit des pD-Wertes für eine Temperatur von (90 ± 2) °C
sowie Reaktivitätsverhältnisse der Zucker, berechnet auf Grundlage ihrer
ACuSTiC-Werte. ......................................................................................................... 93
Abbildung 34: Zeitabhängiger Verlauf der Äquilibrierung der Isomere der D-Fructose 1a nach dem
Lösen der β-D-Fructopyranose 1a(ii) bei einer Temperatur von 22 °C und einem
pD-Wert von 4.0, berechnet auf Grundlage der thermodynamischen (Aktivierungs-)
Parameter der D-Fructose 1a unter Nutzung von Gleichung (7). ............................. 94
Abbildung 35: Linearer Zusammenhang der logarithmierten spontanen Ringöffnungsraten der
β-D-Fructopyranose 1a(ii) in Anwesenheit verschiedener Katalysatoren (L-Alanin 4a,
L-Prolin 4b, Glycin 4c, β-Alanin 4d, γ-Aminobuttersäure 4e, Na-Formiat, Na-Acetat,
Taurin) in Abhängigkeit von deren Säurekonstanten. .............................................. 97
Abbildung 36: pH-Abhängiger Verlauf der Ringöffnungsraten der β-D-Fructopyranose 1a(ii) in
Anwesenheit stöchiometrischer Mengen von Katalysatoren mit verschiedenen
𝑝𝐾𝑠-Werten bei 22 °C, berechnet auf Grundlage der in Tabelle 13 gegebenen
BRØNSTED-Parameter, den 𝑝𝐾𝑠-Werten der β-D-Fructopyranose 1a(ii) (𝑝𝐾𝑠1 = −2.60
und 𝑝𝐾𝑠2 = 11.96)[298,343] sowie den 𝑝𝐾𝑠-Werten möglicher Katalysatoren mit Hilfe
von Gleichung (38). ................................................................................................... 99
Abbildung 37: Ringöffnungsraten der β-D-Fructopyranose 1a(ii) bei 22 °C in Anwesenheit
stöchiometrischer Mengen von DL-Äpfel- und Citronensäure (jeweils gemessen und
berechnet). Die Messungen erfolgten mittels Polarimetrie. Die Berechnungen
erfolgten auf Grundlage der BRØNSTED-Parameter (Tabelle 13), der 𝑝𝐾𝑠-Werte der
β-D-Fructopyranose 1a(ii)[298,343] sowie der 𝑝𝐾𝑠-Werte der DL-Äpfel- und
Citronensäure.[14]..................................................................................................... 100
Abbildung 38: Frequenz-Sweep-Sättigungstransfer-NMR Spektrum einer 200 mM D-Glucoselösung
in Deuteriumoxid bei einer Temperatur von 77 °C und einem pD-Wert von 5.0.
Abgebildet ist die Intensität der anomeren 1H-NMR-Resonanz der α-D-Glucofuranose
in Abhängigkeit der Frequenz des selektiven Radiopulses. Der maximale
Sättigungstransfer ist für eine Frequenz von 6150 Hz zu erkennen (entspricht einer
chemischen Verschiebung von 10.25 ppm bei einer Spektrometerfrequenz von
599.89 MHz). ........................................................................................................... 123

TABELLENVERZEICHNIS
xxx
Tabellenverzeichnis Tabelle 1: 𝑝𝐾𝑠-Werte der untersuchten Aminosäuren 4a-4e. ....................................................... 44
Tabelle 2: 𝑝𝐾𝑠-Werte der untersuchten AMADORI-Verbindungen 6a-6e. ....................................... 45
Tabelle 3: Temperaturabhängigkeit der Linienbreite der anomeren 13C-NMR-Signale der
α-pyranoiden Anomere der [2-13C]-markierten AMADORI-Verbindungen 6a und 6b. .... 58
Tabelle 4: Chemische Verschiebungen der 13C-NMR-Resonanzen der untersuchten Derivate der
D-Fructose.a,b .................................................................................................................. 59
Tabelle 5: Chemische Verschiebungen der 1H-NMR-Resonanzen der untersuchten Derivate der
D-Fructose.a .................................................................................................................... 60
Tabelle 6: Geminale und vicinale homonukleare 1H-Kopplungskonstanten der untersuchten
Derivate der D-Fructose.a,b ............................................................................................. 61
Tabelle 7: 𝑝𝐾𝑠-Werte von Norleucin und ε-Aminocapronsäure. ................................................... 76
Tabelle 8: Thermodynamische Parameter der Anomerengleichgewichte der untersuchten
Zuckersysteme. Teil 1: Enthalpiedifferenzen. Alle Werte sind in kJ/mol angegeben. Bei
den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten
Parameter. ...................................................................................................................... 83
Tabelle 9: Thermodynamische Parameter der Anomerengleichgewichte der untersuchten
Zuckersysteme. Teil 2: Entropiedifferenzen. Alle Werte sind in J/(K·mol) angegeben. Bei
den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten
Parameter. ...................................................................................................................... 83
Tabelle 10: Thermodynamische Aktivierungsparameter der Ringöffnung der untersuchten
Zuckeranomere. Teil 1: Aktivierungsenthalpie. Alle Werte sind in kJ/mol angegeben. Bei
den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten
Parameter. ...................................................................................................................... 86
Tabelle 11: Thermodynamische Aktivierungsparameter der Ringöffnung der untersuchten
Zuckeranomere. Teil 2: Aktivierungsentropie. Alle Werte sind in J/(K·mol) angegeben.
Bei den angegebenen Fehlern handelt es sich um die Standardfehler der geschätzten
Parameter. ...................................................................................................................... 87
Tabelle 12: 𝑝𝐾𝑠-Werte der untersuchten Säuren. ............................................................................ 97
Tabelle 13: BRØNSTED-Parameter der Ringöffnungsraten ionischer Spezies der β-D-Fructopyranose
1a(ii) für 22 °C. ............................................................................................................... 98
Tabelle 14: Einwaagen der Aminosäuren für pH-kontrollierte Modellreaktionen in Anwesenheit
stöchiometrischer Mengen an D-Fructose. .................................................................. 114
Tabelle 15: Einwaagen der AMADORI-Verbindungen für pH-kontrollierte Modellreaktionen. ....... 114
Tabelle 16: Parameter der HILIC-RI-Methode zur Quantifizierung von D-Fructose. ...................... 115

TABELLENVERZEICHNIS
xxxi
Tabelle 17: Parameter der ionenpaarchromatographischen RP-HPLC-RI-Methode zur
Quantifizierung der Aminosäuren. ............................................................................... 116
Tabelle 18: Parameter der ionenpaarchromatographischen RP-HPLC-RI-Methode zur
Quantifizierung der AMADORI-Verbindungen. .............................................................. 117
Tabelle 19: Parameter der RP-HPLC-DAD-Methode zur Quantifizierung der
α-Dicarbonylverbindungen. ......................................................................................... 118
Tabelle 20: Laufmittelgradient der RP-HPLC-DAD-Methode zur Quantifizierung der α-Dicarbonyl-
verbindungen. .............................................................................................................. 118
Tabelle 21: Übersicht über die genutzten Pulsprogramme und Angabe repräsentativer
Akquisitionsparameter. ................................................................................................ 119
Tabelle 22: Einwaagen der (Amino-) Säuren für polarimetrische Messungen der Ringöffnungsraten
der β-D-Fructopyranose in Anwesenheit stöchiometrischer Mengen an (Amino-) Säuren.
...................................................................................................................................... 126
Tabelle 23: Im Verlauf der Arbeit genutzte Geräte. ....................................................................... 126
Tabelle 24: Im Verlauf der Arbeit genutzte Software. .................................................................... 128
Tabelle 25: Im Verlauf der Arbeit genutzte Chemikalien. ............................................................... 129
Tabelle 26: Geschwindigkeitskonstanten des pH-abhängigen Abbaus von D-Fructose 1a in 0.5 M
Lösung in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei
(90 ± 2) °C. .................................................................................................................. xxxvi
Tabelle 27: Geschwindigkeitskonstanten des pH-abhängigen Abbaus der Aminosäuren 4a-4e in
0.5 M Lösung in Anwesenheit äquimolarer Mengen an D-Fructose 1a bei (90 ± 2) °C.
....................................................................................................................................xxxvii
Tabelle 28: Geschwindigkeitskonstanten des pH-abhängigen Abbaus der AMADORI-Verbindungen
6a-6e in 0.025 M Lösung bei (90 ± 2) °C. ................................................................... xxxviii
Tabelle 29: Geschwindigkeitskonstanten der pH-abhängigen Bildung von 1- und 3-Desoxyglucoson
(1-DG und 3-DG) 9a und 9b in 0.5 M Reaktionsmodellen der D-Fructose 1a in An- und
Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei (90 ± 2) °C. .............xxxix
Tabelle 30: Geschwindigkeitskonstanten der pH-abhängigen Bildung von 1- und 3-Desoxyglucoson
(1-DG und 3-DG) 9a und 9b in 0.025 M Reaktionsmodellen der AMADORI-Verbindungen
6a-6e bei (90 ± 2) °C. ........................................................................................................ xl
Tabelle 31: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer
Wellenlänge von 284 nm für 0.5 M Reaktionsmodelle der D-Fructose 1a in An- und
Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei (90 ± 2) °C. ................. xli
Tabelle 32: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer
Wellenlänge von 284 nm, berechnet für 0.5 M Reaktionslösungen der
AMADORI-Verbindungen 6a-6e bei 90 °C auf Grundlage von Messungen basierend auf
0.025 M Reaktionsmodellen der AMADORI-Verbindungen 6a-6e bei (90 ± 2) °C............ xlii

TABELLENVERZEICHNIS
xxxii
Tabelle 33: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer
Wellenlänge von 420 nm für 0.5 M Reaktionsmodelle der D-Fructose 1a in An- und
Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei (90 ± 2) °C. ............... xliii
Tabelle 34: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer
Wellenlänge von 420 nm, berechnet für 0.5 M Reaktionslösungen der
AMADORI-Verbindungen 6a-6e bei 90 °C auf Grundlage von Messungen basierend auf
0.025 M Reaktionsmodellen der AMADORI-Verbindungen 6a-6e bei (90 ± 2) °C. .......... xliv
Tabelle 35: Raten der 1,2-Enolisierung von Fructosylalanin 6a und Fructosylprolin 6b bei
Raumtemperatur in Abhängigkeit des pD-Wertes. ........................................................ xlv
Tabelle 36: Raten und thermodynamische Aktivierungsparameter der 1,2-Enolisierung der
AMADORI-Verbindungen der homologen ω-Aminosäuren 6c-6e bei pD 4.5 in
Abhängigkeit der Temperatur. ....................................................................................... xlv

SCHEMATAVERZEICHNIS
xxxiii
Schemataverzeichnis Schema 1: Strukturen der Ketohexosen D-Fructose 1a, D-Tagatose 1b, D-Sorbose 1c und D-Psicose
1d. ..................................................................................................................................... 3
Schema 2: Isomerensystem der D-Fructose 1a in wässriger Lösung. *Relative Konzentration bei 20 °C.[46]4
Schema 3: Thermodynamisches Gleichgewicht zwischen D-Fructose 1a und D-Glucose 2a sowie
D-Mannose 2b, D-Psicose 1d und den Ketosen 3a und 3b über die jeweils tautomeren
1,2- und 2,3-Endiole. ........................................................................................................ 7
Schema 4: Strukturen der eingesetzten Aminokomponenten L-Alanin 4a, L-Prolin 4b, Glycin 4c,
β-Alanin 4d und γ-Aminobuttersäure 4e. ........................................................................ 8
Schema 5: Möglichkeiten der Bildung eines N-substituierten Glucosylamins 5a sowie der
korrespondierenden ringoffenen protonierten SCHIFF-Base 5b aus D-Glucose 2a und
einer Aminokomponente im sauren pH-Bereich. a) Protonierung des Lactolrings.
b) Protonierung der anomeren OH-Funktion. ................................................................. 9
Schema 6: Bildung einer SCHIFF-Base 5b aus D-Glucose 2a und einer Aminokomponente über das
gem-Aminol 5c im basischen pH-Bereich. Der gezeigte Mechanismus entspricht dem der
Bildung des gem-Diols der D-Glucose 2a aus ihrem acyclischen Zentralisomer. ........... 10
Schema 7: Konzertierter Mechanismus zur Bildung eines N-substituierten Glucosylamins 5a sowie
der korrespondierenden ringoffenen protonierten SCHIFF-Base 5b aus D-Glucose 2a und
einer Aminosäure. (Frei nach[82]) .................................................................................... 11
Schema 8: Mechanismus der AMADORI-Umlagerung I. (Frei nach[83,88,97]) ........................................ 11
Schema 9: Mechanismus der AMADORI-Umlagerung II.[103] .............................................................. 12
Schema 10: Bildung von AMADORI- 6 ausgehend von HEYNS-Verbindungen 7. .................................. 13
Schema 11: Bildung eines N,N-Difructosylamins 8 aus der Kondensation einer AMADORI-Verbindung
6 mit D-Glucose 2a. (Frei nach[119,121])............................................................................. 13
Schema 12: Tautomerensystem D-Fructose-analoger AMADORI-Verbindungen 6 in wässriger Lösung.
*Relative Konzentration bei 20-30 °C.[119,127] .............................................................................. 15
Schema 13: Bildungswege der α-Dicarbonylverbindungen 9a-9d ausgehend von einer
AMADORI-Verbindung 6. .................................................................................................. 17
Schema 14: Mechanismen der Bildung von Dihydrohydroxymaltol (DHHM) 10, Acetylformoin 11 und
Isomaltol 12 aus den Anomeren des 1-Desoxyglucosons 9a. ........................................ 18
Schema 15: Mechanismus der Bildung von 5-Hydroxymethylfurfural (HMF) 13 aus 3-Desoxyglucoson
9b. (Frei nach[172,173,176]) .................................................................................................. 19
Schema 16: Mechanismen der Bildung von 2-Hydroxyacetylfuran (HAF) 14 und Isomaltol 12 aus
β-D-Fructofuranose 1a(iv) im sauren Milieu. (Frei nach[180]) .......................................... 20
Schema 17: Mechanismen der Bildung von Dihydrohydroxymaltol (DHHM) 10,
5-Hydroxymethylfurfural (HMF) 13, 2-Aminoacetylfuran (AAF) 15 und Maltol 16 aus den
cyclischen Anomeren einer AMADORI-Verbindung 6. (Frei nach[173,177-179,187]) ................ 21

SCHEMATAVERZEICHNIS
xxxiv
Schema 18: Anomerensystem der D-Fructose 1a. Dabei steht P für Pyranose, F für Furanose und z
für das Zentralisomer. .................................................................................................... 27
Schema 19: Mechanismus der Anomerisierung am Beispiel der β-D-Glucopyranose. ...................... 35
Schema 20: System paralleler Folgereaktionen am Beispiel der Bildung und des Abbaus der
α-Dicarbonylverbindungen D-Glucoson (GLUC) 9d sowie 1- und 3-Desoxyglucoson (1-DG
und 3-DG) 9a und 9b. FP 1, FP 2 und FP 3 bezeichnen nicht weiter definierte
Folgeprodukte. ............................................................................................................... 47
Schema 21: Zusammenhang von Anomerisierung und Enolisierung anionischer Spezies der D-Fruc-
tose 1a im Vergleich zur Anomerisierung kationischer Spezies der D-Fructose 1a. ...... 49
Schema 22: Einflussmöglichkeiten der Aminofunktion einer Aminosäure auf das Reaktionsverhalten
von D-Fructose 1a. *Der dargestellte Mesomeriepfeil ist als Gleichgewichtsreaktionspfeil zu verstehen.
........................................................................................................................................ 50
Schema 23: Bildung resonanzstabilisierter pseudocyclischer Strukturelemente durch kooperative
Wasserstoffbrücken im β-pyranoiden Anomer von Fructosylglycin 6c(ii). (erweitert
nach[124]) ......................................................................................................................... 62
Schema 24: a) Parallele Bildung von 3-Desoxyglucoson 9b und 3-Desoxygalactoson 9c aus dem
β-pyranoiden Anomer von Fructosylglycin 6c(ii) über die Protonierung der
Hydroxylfunktion am C-3. b) Bildung von (Z)- und (E)-3,4-Didesoxyglucoson-3-en 17a
und 17b aus dem β-pyranoiden Anomer von Fructosylglycin 6c(ii) über die Protonierung
der Hydroxylfunktion am C-3. ........................................................................................ 64
Schema 25: a) Bildung von Allomaltol 18 aus dem β-pyranoiden Anomer von Fructosylglycin 6c(ii)
über die Protonierung der Hydroxylfunktion am C-3. b) Bildung von HMF 13 aus dem
α-furanoiden Anomer von Fructosylglycin 6c(iii) über die Protonierung der
Hydroxylfunktion am C-3. c) Bildung von AAF 15 aus dem α-furanoiden Anomer von
Fructosylglycin 6c(iii) über die Protonierung der Hydroxylfunktion am C-3. ................ 65
Schema 26: Mechanismus des Deuteriumaustausches am C-1 von aminosubstituierten Derivaten
der D-Fructose. ............................................................................................................... 69
Schema 27: Mechanistischer Zusammenhang des Prozesses der Anomerisierung und stofflichen
Veränderungen reduzierender Zucker am Beispiel von Anomeren der D-Fructose 1a.
........................................................................................................................................ 79
Schema 28: Vereinfachtes Modell der Mutarotation der D-Fructose 1a zur Beschreibung
polarimetrischer Daten. ................................................................................................. 95
Schema 29: Schematische Darstellung der frühen MAILLARD-Reaktion mit Fokus auf die durch
schwache Säuren und deren konjugierte Basen katalysierten Schritte des
Protonentransfers (rote Pfeile). Darüber hinaus sind die für diese Arbeit wichtigsten
Positionen gekennzeichnet, deren 𝑝𝐾𝑠-Werte sich relativ zu denen der D-Glucose 2a
ändern (grüne Pfeile). Weiterhin sind Veränderungen von Anomerensystemen relativ zu
dem der D-Glucose 2a (gelb-orange Pfeile) sowie der Einfluss der Katalysatoren auf
Abbaureaktionen kenntlich gemacht (blaue Pfeile). Auf die Darstellung der Bildung
weiterer intermediärer Reaktionsprodukte – etwa 1-Desoxyglucoson 9a – wird aus
Gründen der Übersichtlichkeit verzichtet. ................................................................... 104
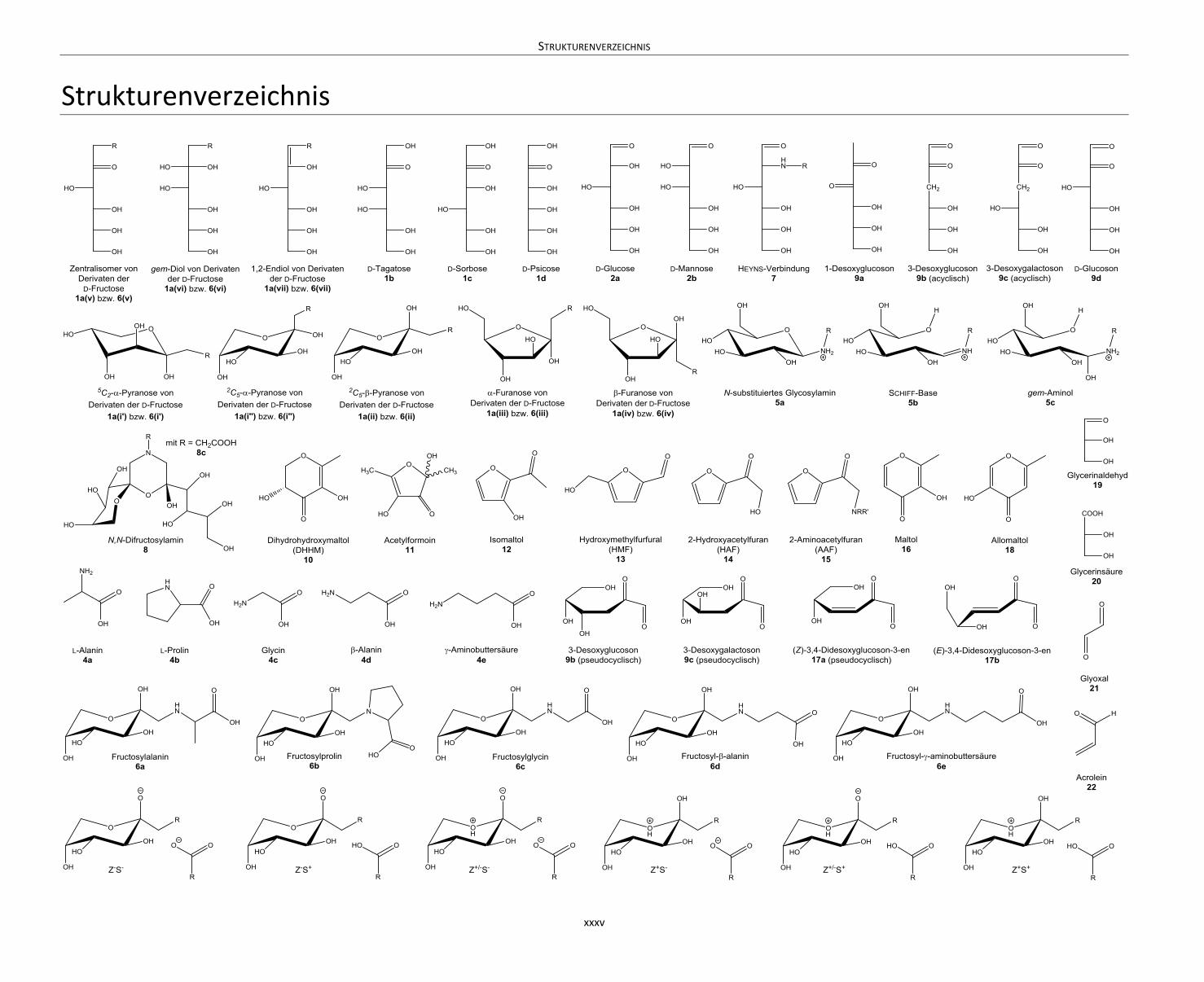
STRUKTURENVERZEICHNIS
xxxv
Strukturenverzeichnis
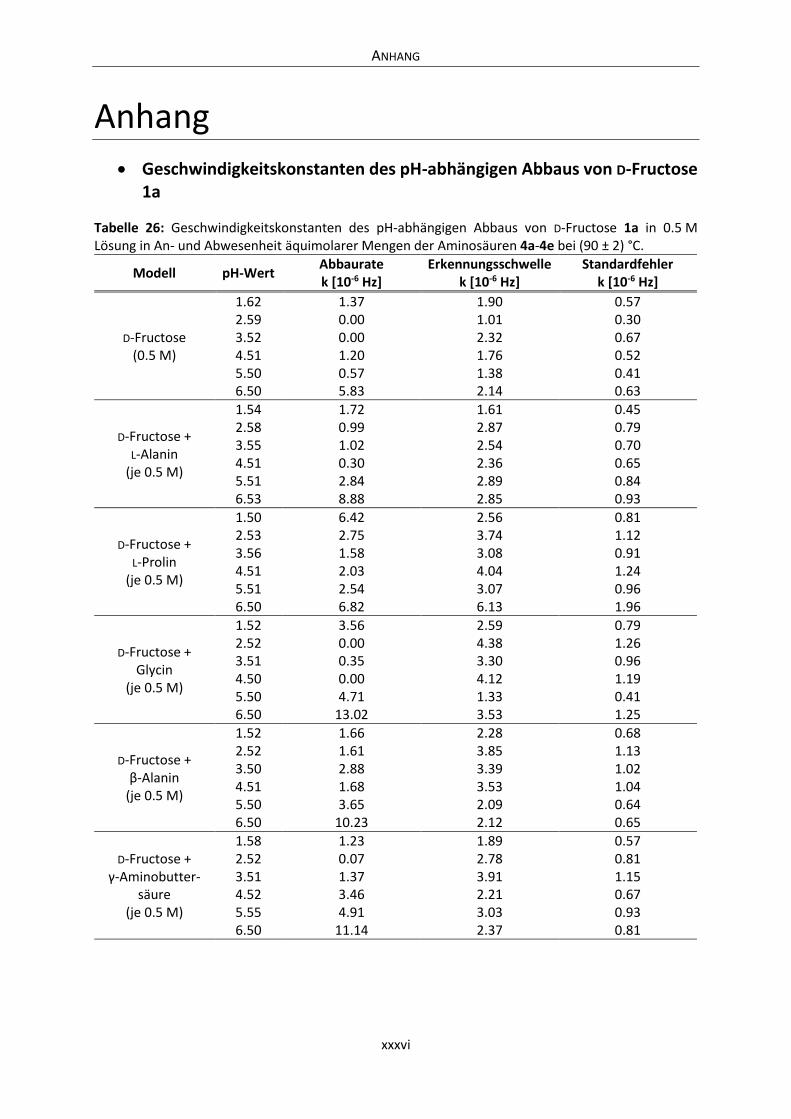
ANHANG
xxxvi
Anhang
• Geschwindigkeitskonstanten des pH-abhängigen Abbaus von D-Fructose 1a
Tabelle 26: Geschwindigkeitskonstanten des pH-abhängigen Abbaus von D-Fructose 1a in 0.5 M Lösung in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei (90 ± 2) °C.
Modell pH-Wert Abbaurate k [10-6 Hz]
Erkennungsschwelle k [10-6 Hz]
Standardfehler k [10-6 Hz]
D-Fructose (0.5 M)
1.62 1.37 1.90 0.57 2.59 0.00 1.01 0.30 3.52 0.00 2.32 0.67 4.51 1.20 1.76 0.52 5.50 0.57 1.38 0.41 6.50 5.83 2.14 0.63
D-Fructose + L-Alanin
(je 0.5 M)
1.54 1.72 1.61 0.45 2.58 0.99 2.87 0.79 3.55 1.02 2.54 0.70 4.51 0.30 2.36 0.65 5.51 2.84 2.89 0.84 6.53 8.88 2.85 0.93
D-Fructose + L-Prolin
(je 0.5 M)
1.50 6.42 2.56 0.81 2.53 2.75 3.74 1.12 3.56 1.58 3.08 0.91 4.51 2.03 4.04 1.24 5.51 2.54 3.07 0.96 6.50 6.82 6.13 1.96
D-Fructose + Glycin
(je 0.5 M)
1.52 3.56 2.59 0.79 2.52 0.00 4.38 1.26 3.51 0.35 3.30 0.96 4.50 0.00 4.12 1.19 5.50 4.71 1.33 0.41 6.50 13.02 3.53 1.25
D-Fructose + β-Alanin
(je 0.5 M)
1.52 1.66 2.28 0.68 2.52 1.61 3.85 1.13 3.50 2.88 3.39 1.02 4.51 1.68 3.53 1.04 5.50 3.65 2.09 0.64 6.50 10.23 2.12 0.65
D-Fructose + γ-Aminobutter-
säure (je 0.5 M)
1.58 1.23 1.89 0.57 2.52 0.07 2.78 0.81 3.51 1.37 3.91 1.15 4.52 3.46 2.21 0.67 5.55 4.91 3.03 0.93 6.50 11.14 2.37 0.81
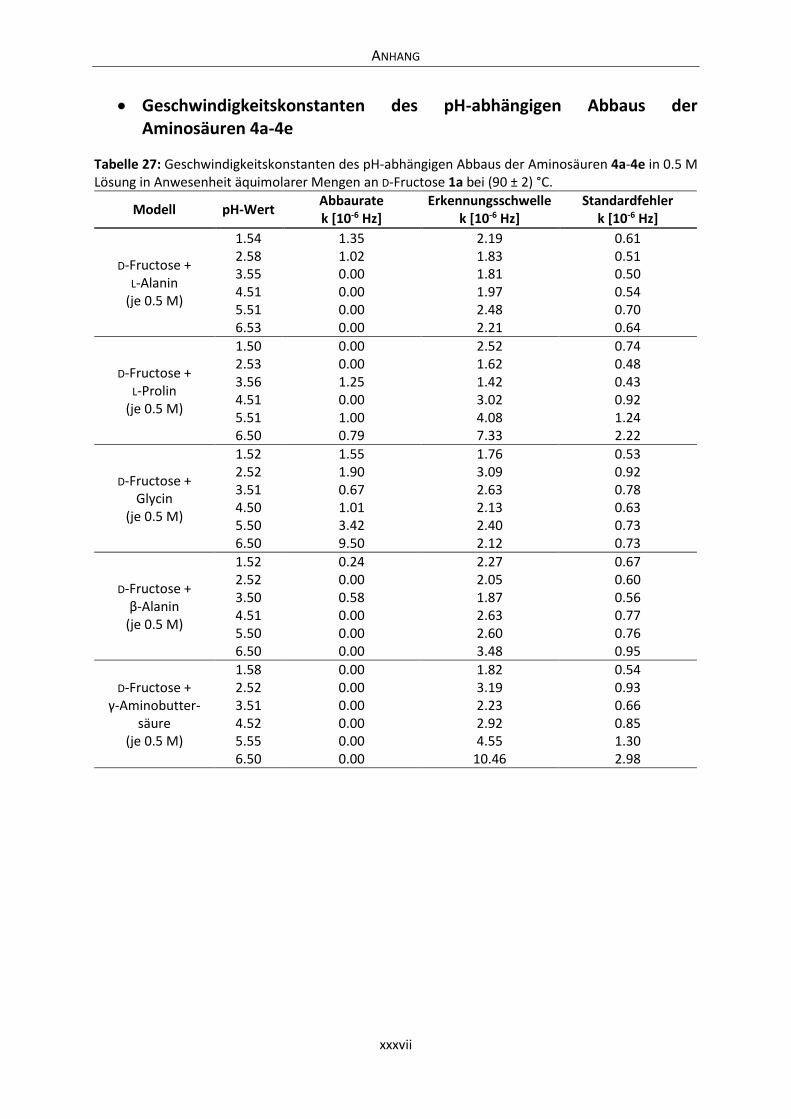
ANHANG
xxxvii
• Geschwindigkeitskonstanten des pH-abhängigen Abbaus der Aminosäuren 4a-4e
Tabelle 27: Geschwindigkeitskonstanten des pH-abhängigen Abbaus der Aminosäuren 4a-4e in 0.5 M Lösung in Anwesenheit äquimolarer Mengen an D-Fructose 1a bei (90 ± 2) °C.
Modell pH-Wert Abbaurate k [10-6 Hz]
Erkennungsschwelle k [10-6 Hz]
Standardfehler k [10-6 Hz]
D-Fructose + L-Alanin
(je 0.5 M)
1.54 1.35 2.19 0.61 2.58 1.02 1.83 0.51 3.55 0.00 1.81 0.50 4.51 0.00 1.97 0.54 5.51 0.00 2.48 0.70 6.53 0.00 2.21 0.64
D-Fructose + L-Prolin
(je 0.5 M)
1.50 0.00 2.52 0.74 2.53 0.00 1.62 0.48 3.56 1.25 1.42 0.43 4.51 0.00 3.02 0.92 5.51 1.00 4.08 1.24 6.50 0.79 7.33 2.22
D-Fructose + Glycin
(je 0.5 M)
1.52 1.55 1.76 0.53 2.52 1.90 3.09 0.92 3.51 0.67 2.63 0.78 4.50 1.01 2.13 0.63 5.50 3.42 2.40 0.73 6.50 9.50 2.12 0.73
D-Fructose + β-Alanin
(je 0.5 M)
1.52 0.24 2.27 0.67 2.52 0.00 2.05 0.60 3.50 0.58 1.87 0.56 4.51 0.00 2.63 0.77 5.50 0.00 2.60 0.76 6.50 0.00 3.48 0.95
D-Fructose + γ-Aminobutter-
säure (je 0.5 M)
1.58 0.00 1.82 0.54 2.52 0.00 3.19 0.93 3.51 0.00 2.23 0.66 4.52 0.00 2.92 0.85 5.55 0.00 4.55 1.30 6.50 0.00 10.46 2.98
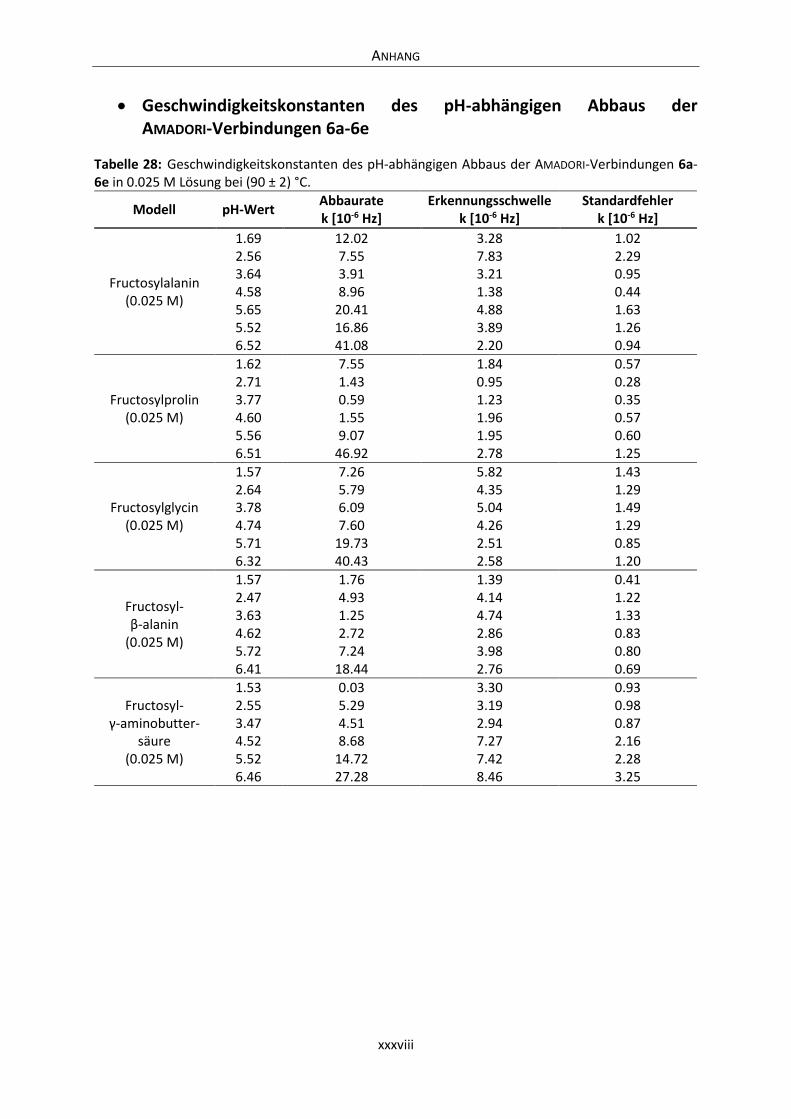
ANHANG
xxxviii
• Geschwindigkeitskonstanten des pH-abhängigen Abbaus der AMADORI-Verbindungen 6a-6e
Tabelle 28: Geschwindigkeitskonstanten des pH-abhängigen Abbaus der AMADORI-Verbindungen 6a-6e in 0.025 M Lösung bei (90 ± 2) °C.
Modell pH-Wert Abbaurate k [10-6 Hz]
Erkennungsschwelle k [10-6 Hz]
Standardfehler k [10-6 Hz]
Fructosylalanin (0.025 M)
1.69 12.02 3.28 1.02 2.56 7.55 7.83 2.29 3.64 3.91 3.21 0.95 4.58 8.96 1.38 0.44 5.65 20.41 4.88 1.63 5.52 16.86 3.89 1.26 6.52 41.08 2.20 0.94
Fructosylprolin (0.025 M)
1.62 7.55 1.84 0.57 2.71 1.43 0.95 0.28 3.77 0.59 1.23 0.35 4.60 1.55 1.96 0.57 5.56 9.07 1.95 0.60 6.51 46.92 2.78 1.25
Fructosylglycin (0.025 M)
1.57 7.26 5.82 1.43 2.64 5.79 4.35 1.29 3.78 6.09 5.04 1.49 4.74 7.60 4.26 1.29 5.71 19.73 2.51 0.85 6.32 40.43 2.58 1.20
Fructosyl-β-alanin
(0.025 M)
1.57 1.76 1.39 0.41 2.47 4.93 4.14 1.22 3.63 1.25 4.74 1.33 4.62 2.72 2.86 0.83 5.72 7.24 3.98 0.80 6.41 18.44 2.76 0.69
Fructosyl-γ-aminobutter-
säure (0.025 M)
1.53 0.03 3.30 0.93 2.55 5.29 3.19 0.98 3.47 4.51 2.94 0.87 4.52 8.68 7.27 2.16 5.52 14.72 7.42 2.28 6.46 27.28 8.46 3.25
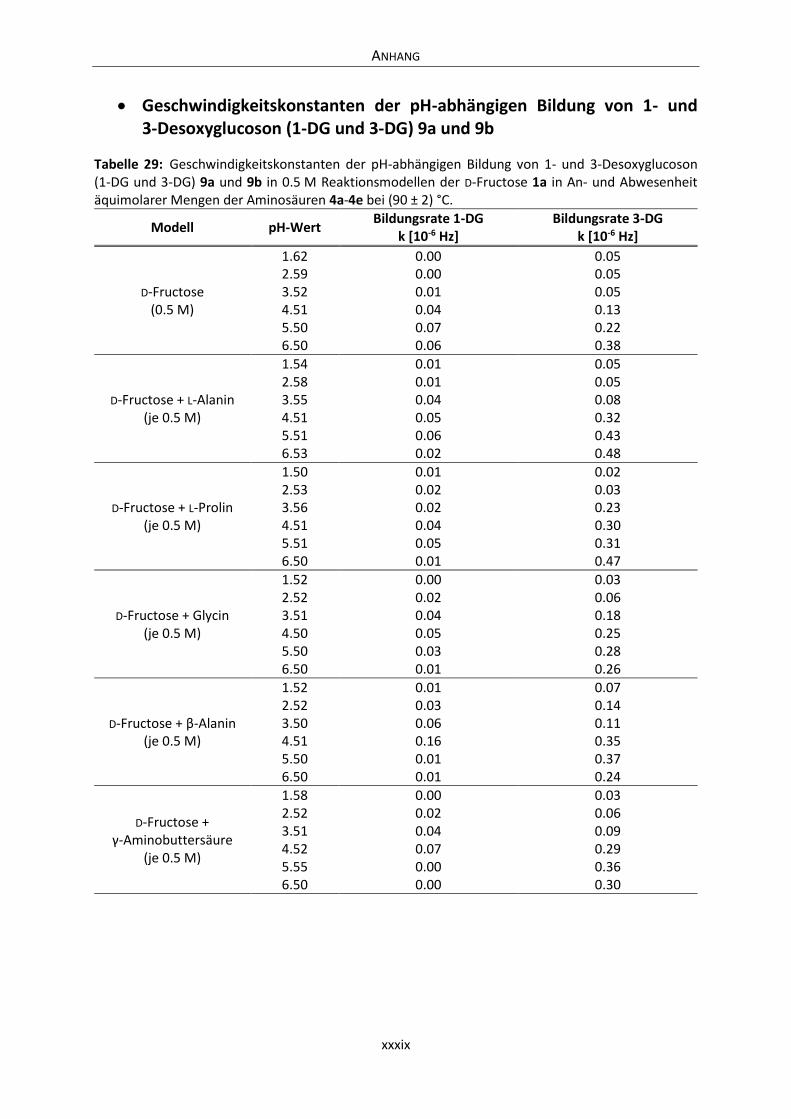
ANHANG
xxxix
• Geschwindigkeitskonstanten der pH-abhängigen Bildung von 1- und 3-Desoxyglucoson (1-DG und 3-DG) 9a und 9b
Tabelle 29: Geschwindigkeitskonstanten der pH-abhängigen Bildung von 1- und 3-Desoxyglucoson (1-DG und 3-DG) 9a und 9b in 0.5 M Reaktionsmodellen der D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei (90 ± 2) °C.
Modell pH-Wert Bildungsrate 1-DG
k [10-6 Hz] Bildungsrate 3-DG
k [10-6 Hz]
D-Fructose (0.5 M)
1.62 0.00 0.05 2.59 0.00 0.05 3.52 0.01 0.05 4.51 0.04 0.13 5.50 0.07 0.22 6.50 0.06 0.38
D-Fructose + L-Alanin (je 0.5 M)
1.54 0.01 0.05 2.58 0.01 0.05 3.55 0.04 0.08 4.51 0.05 0.32 5.51 0.06 0.43 6.53 0.02 0.48
D-Fructose + L-Prolin (je 0.5 M)
1.50 0.01 0.02 2.53 0.02 0.03 3.56 0.02 0.23 4.51 0.04 0.30 5.51 0.05 0.31 6.50 0.01 0.47
D-Fructose + Glycin (je 0.5 M)
1.52 0.00 0.03 2.52 0.02 0.06 3.51 0.04 0.18 4.50 0.05 0.25 5.50 0.03 0.28 6.50 0.01 0.26
D-Fructose + β-Alanin (je 0.5 M)
1.52 0.01 0.07 2.52 0.03 0.14 3.50 0.06 0.11 4.51 0.16 0.35 5.50 0.01 0.37 6.50 0.01 0.24
D-Fructose + γ-Aminobuttersäure
(je 0.5 M)
1.58 0.00 0.03 2.52 0.02 0.06 3.51 0.04 0.09 4.52 0.07 0.29 5.55 0.00 0.36 6.50 0.00 0.30
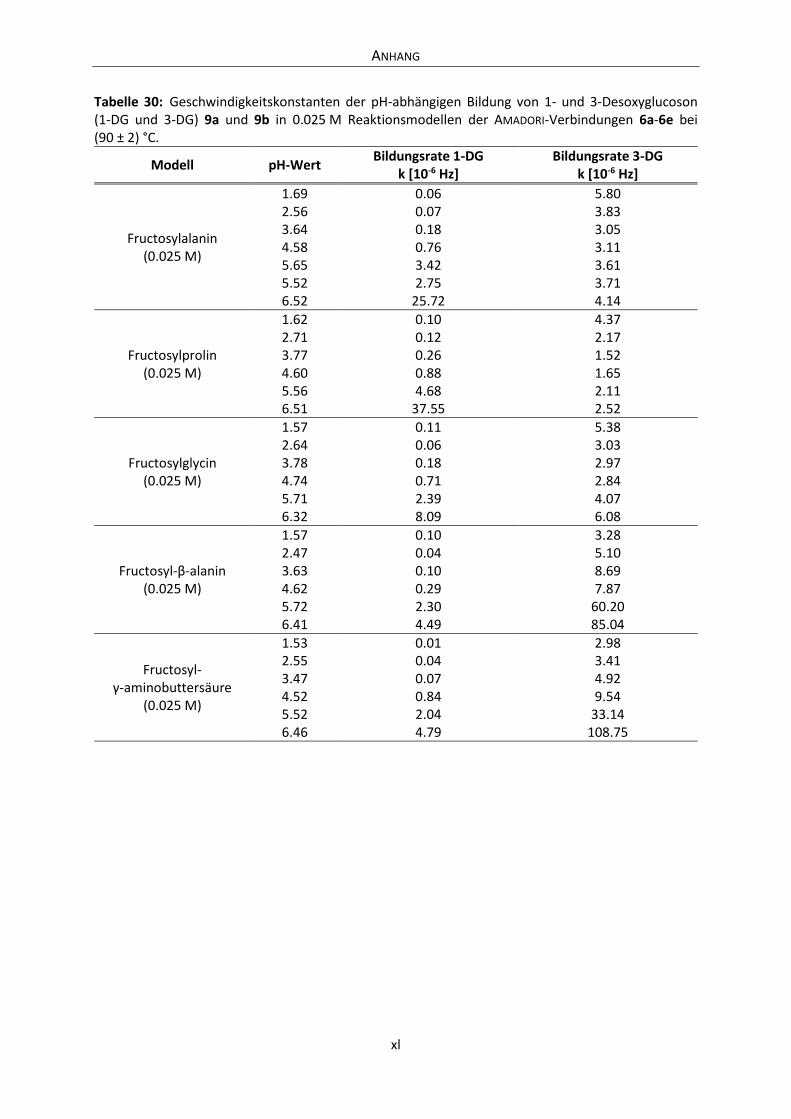
ANHANG
xl
Tabelle 30: Geschwindigkeitskonstanten der pH-abhängigen Bildung von 1- und 3-Desoxyglucoson (1-DG und 3-DG) 9a und 9b in 0.025 M Reaktionsmodellen der AMADORI-Verbindungen 6a-6e bei (90 ± 2) °C.
Modell pH-Wert Bildungsrate 1-DG
k [10-6 Hz] Bildungsrate 3-DG
k [10-6 Hz]
Fructosylalanin (0.025 M)
1.69 0.06 5.80 2.56 0.07 3.83 3.64 0.18 3.05 4.58 0.76 3.11 5.65 3.42 3.61 5.52 2.75 3.71 6.52 25.72 4.14
Fructosylprolin (0.025 M)
1.62 0.10 4.37 2.71 0.12 2.17 3.77 0.26 1.52 4.60 0.88 1.65 5.56 4.68 2.11 6.51 37.55 2.52
Fructosylglycin (0.025 M)
1.57 0.11 5.38 2.64 0.06 3.03 3.78 0.18 2.97 4.74 0.71 2.84 5.71 2.39 4.07 6.32 8.09 6.08
Fructosyl-β-alanin (0.025 M)
1.57 0.10 3.28 2.47 0.04 5.10 3.63 0.10 8.69 4.62 0.29 7.87 5.72 2.30 60.20 6.41 4.49 85.04
Fructosyl-γ-aminobuttersäure
(0.025 M)
1.53 0.01 2.98 2.55 0.04 3.41 3.47 0.07 4.92 4.52 0.84 9.54 5.52 2.04 33.14 6.46 4.79 108.75
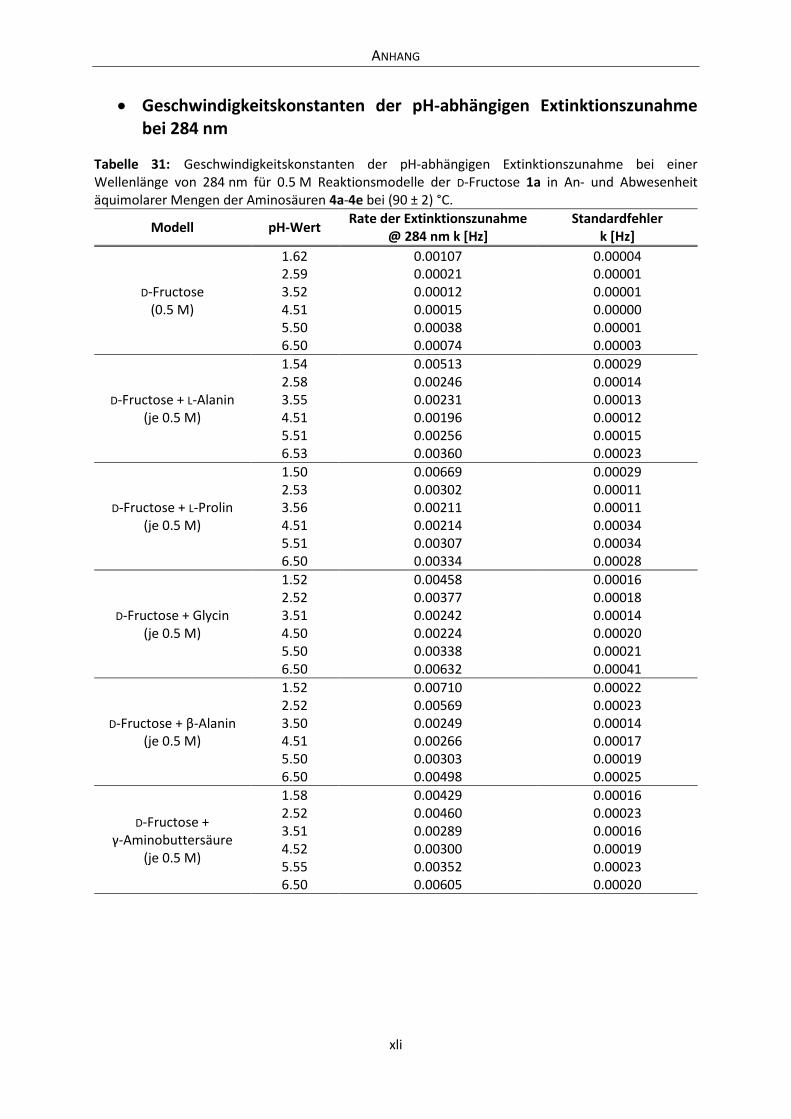
ANHANG
xli
• Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei 284 nm
Tabelle 31: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer Wellenlänge von 284 nm für 0.5 M Reaktionsmodelle der D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei (90 ± 2) °C.
Modell pH-Wert Rate der Extinktionszunahme
@ 284 nm k [Hz] Standardfehler
k [Hz]
D-Fructose (0.5 M)
1.62 0.00107 0.00004 2.59 0.00021 0.00001 3.52 0.00012 0.00001 4.51 0.00015 0.00000 5.50 0.00038 0.00001 6.50 0.00074 0.00003
D-Fructose + L-Alanin (je 0.5 M)
1.54 0.00513 0.00029 2.58 0.00246 0.00014 3.55 0.00231 0.00013 4.51 0.00196 0.00012 5.51 0.00256 0.00015 6.53 0.00360 0.00023
D-Fructose + L-Prolin (je 0.5 M)
1.50 0.00669 0.00029 2.53 0.00302 0.00011 3.56 0.00211 0.00011 4.51 0.00214 0.00034 5.51 0.00307 0.00034 6.50 0.00334 0.00028
D-Fructose + Glycin (je 0.5 M)
1.52 0.00458 0.00016 2.52 0.00377 0.00018 3.51 0.00242 0.00014 4.50 0.00224 0.00020 5.50 0.00338 0.00021 6.50 0.00632 0.00041
D-Fructose + β-Alanin (je 0.5 M)
1.52 0.00710 0.00022 2.52 0.00569 0.00023 3.50 0.00249 0.00014 4.51 0.00266 0.00017 5.50 0.00303 0.00019 6.50 0.00498 0.00025
D-Fructose + γ-Aminobuttersäure
(je 0.5 M)
1.58 0.00429 0.00016 2.52 0.00460 0.00023 3.51 0.00289 0.00016 4.52 0.00300 0.00019 5.55 0.00352 0.00023 6.50 0.00605 0.00020
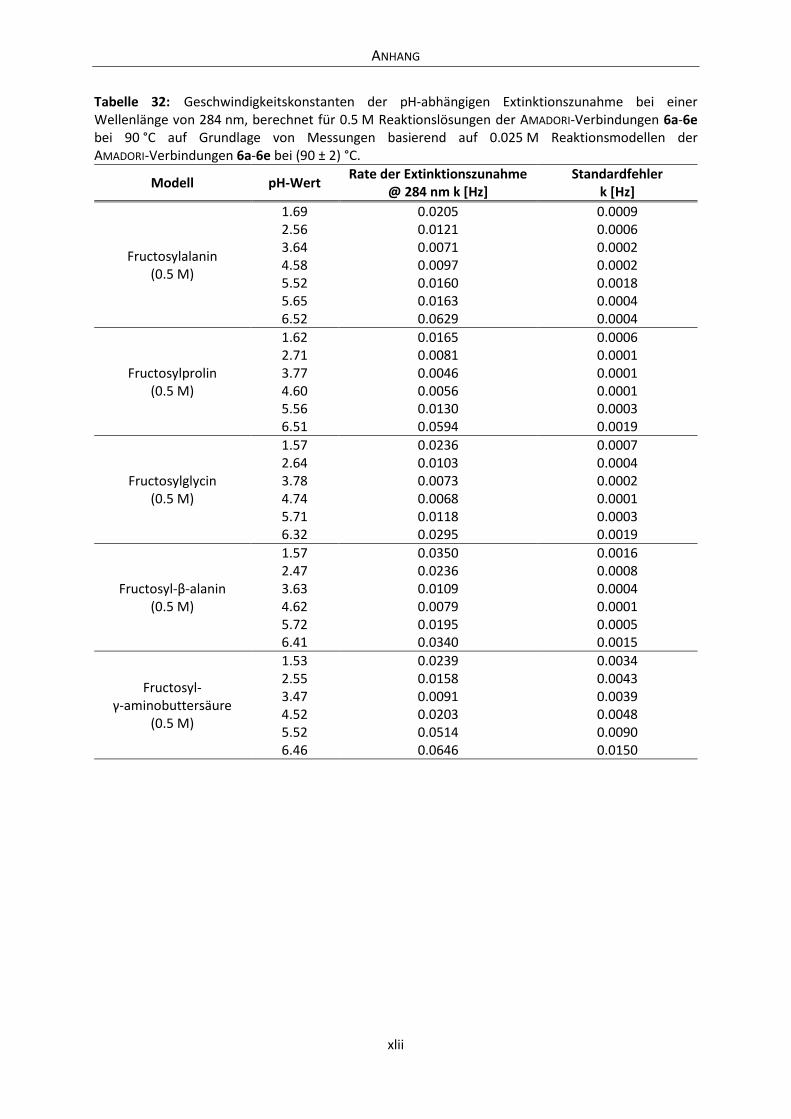
ANHANG
xlii
Tabelle 32: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer Wellenlänge von 284 nm, berechnet für 0.5 M Reaktionslösungen der AMADORI-Verbindungen 6a-6e bei 90 °C auf Grundlage von Messungen basierend auf 0.025 M Reaktionsmodellen der AMADORI-Verbindungen 6a-6e bei (90 ± 2) °C.
Modell pH-Wert Rate der Extinktionszunahme
@ 284 nm k [Hz] Standardfehler
k [Hz]
Fructosylalanin (0.5 M)
1.69 0.0205 0.0009 2.56 0.0121 0.0006 3.64 0.0071 0.0002 4.58 0.0097 0.0002 5.52 0.0160 0.0018 5.65 0.0163 0.0004 6.52 0.0629 0.0004
Fructosylprolin (0.5 M)
1.62 0.0165 0.0006 2.71 0.0081 0.0001 3.77 0.0046 0.0001 4.60 0.0056 0.0001 5.56 0.0130 0.0003 6.51 0.0594 0.0019
Fructosylglycin (0.5 M)
1.57 0.0236 0.0007 2.64 0.0103 0.0004 3.78 0.0073 0.0002 4.74 0.0068 0.0001 5.71 0.0118 0.0003 6.32 0.0295 0.0019
Fructosyl-β-alanin (0.5 M)
1.57 0.0350 0.0016 2.47 0.0236 0.0008 3.63 0.0109 0.0004 4.62 0.0079 0.0001 5.72 0.0195 0.0005 6.41 0.0340 0.0015
Fructosyl-γ-aminobuttersäure
(0.5 M)
1.53 0.0239 0.0034 2.55 0.0158 0.0043 3.47 0.0091 0.0039 4.52 0.0203 0.0048 5.52 0.0514 0.0090 6.46 0.0646 0.0150
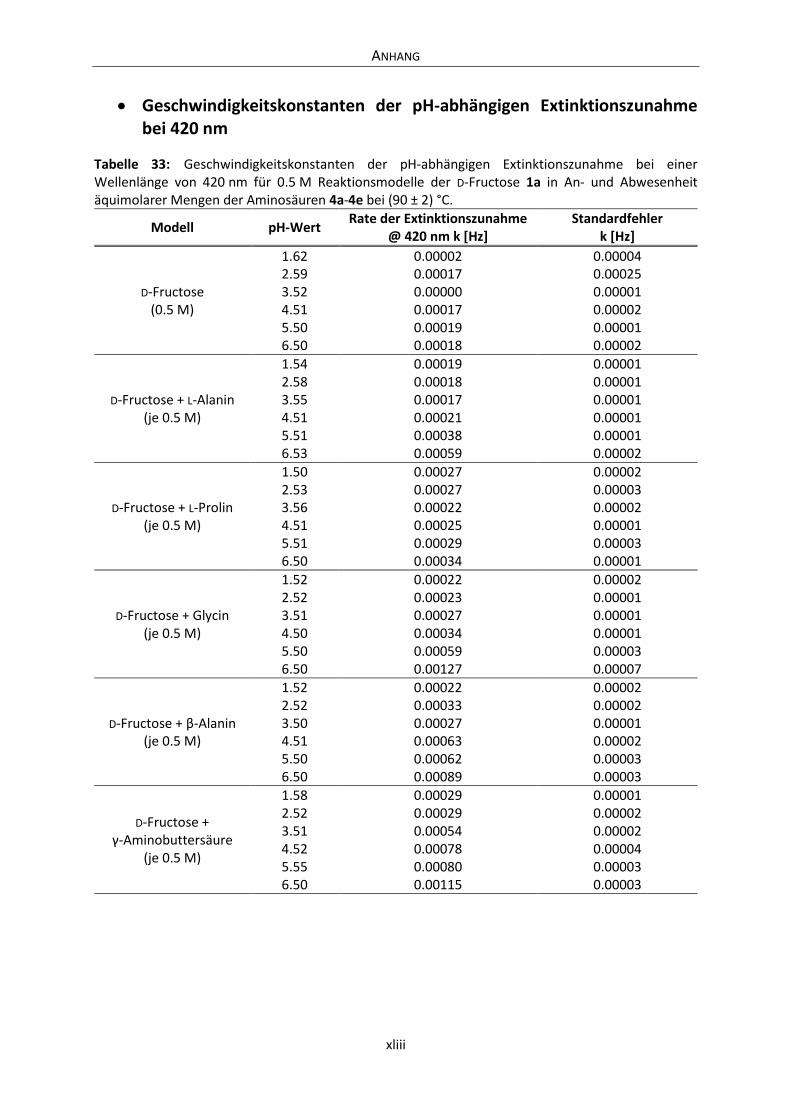
ANHANG
xliii
• Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei 420 nm
Tabelle 33: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer Wellenlänge von 420 nm für 0.5 M Reaktionsmodelle der D-Fructose 1a in An- und Abwesenheit äquimolarer Mengen der Aminosäuren 4a-4e bei (90 ± 2) °C.
Modell pH-Wert Rate der Extinktionszunahme
@ 420 nm k [Hz] Standardfehler
k [Hz]
D-Fructose (0.5 M)
1.62 0.00002 0.00004 2.59 0.00017 0.00025 3.52 0.00000 0.00001 4.51 0.00017 0.00002 5.50 0.00019 0.00001 6.50 0.00018 0.00002
D-Fructose + L-Alanin (je 0.5 M)
1.54 0.00019 0.00001 2.58 0.00018 0.00001 3.55 0.00017 0.00001 4.51 0.00021 0.00001 5.51 0.00038 0.00001 6.53 0.00059 0.00002
D-Fructose + L-Prolin (je 0.5 M)
1.50 0.00027 0.00002 2.53 0.00027 0.00003 3.56 0.00022 0.00002 4.51 0.00025 0.00001 5.51 0.00029 0.00003 6.50 0.00034 0.00001
D-Fructose + Glycin (je 0.5 M)
1.52 0.00022 0.00002 2.52 0.00023 0.00001 3.51 0.00027 0.00001 4.50 0.00034 0.00001 5.50 0.00059 0.00003 6.50 0.00127 0.00007
D-Fructose + β-Alanin (je 0.5 M)
1.52 0.00022 0.00002 2.52 0.00033 0.00002 3.50 0.00027 0.00001 4.51 0.00063 0.00002 5.50 0.00062 0.00003 6.50 0.00089 0.00003
D-Fructose + γ-Aminobuttersäure
(je 0.5 M)
1.58 0.00029 0.00001 2.52 0.00029 0.00002 3.51 0.00054 0.00002 4.52 0.00078 0.00004 5.55 0.00080 0.00003 6.50 0.00115 0.00003
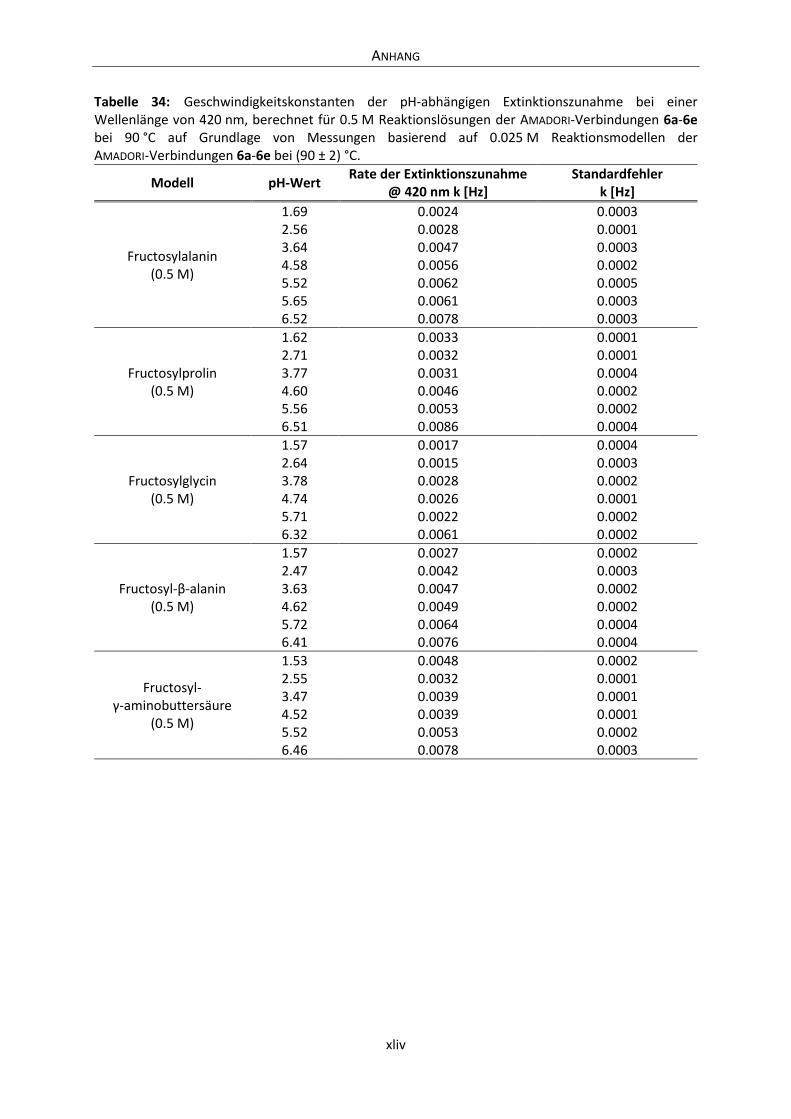
ANHANG
xliv
Tabelle 34: Geschwindigkeitskonstanten der pH-abhängigen Extinktionszunahme bei einer Wellenlänge von 420 nm, berechnet für 0.5 M Reaktionslösungen der AMADORI-Verbindungen 6a-6e bei 90 °C auf Grundlage von Messungen basierend auf 0.025 M Reaktionsmodellen der AMADORI-Verbindungen 6a-6e bei (90 ± 2) °C.
Modell pH-Wert Rate der Extinktionszunahme
@ 420 nm k [Hz] Standardfehler
k [Hz]
Fructosylalanin (0.5 M)
1.69 0.0024 0.0003 2.56 0.0028 0.0001 3.64 0.0047 0.0003 4.58 0.0056 0.0002 5.52 0.0062 0.0005 5.65 0.0061 0.0003 6.52 0.0078 0.0003
Fructosylprolin (0.5 M)
1.62 0.0033 0.0001 2.71 0.0032 0.0001 3.77 0.0031 0.0004 4.60 0.0046 0.0002 5.56 0.0053 0.0002 6.51 0.0086 0.0004
Fructosylglycin (0.5 M)
1.57 0.0017 0.0004 2.64 0.0015 0.0003 3.78 0.0028 0.0002 4.74 0.0026 0.0001 5.71 0.0022 0.0002 6.32 0.0061 0.0002
Fructosyl-β-alanin (0.5 M)
1.57 0.0027 0.0002 2.47 0.0042 0.0003 3.63 0.0047 0.0002 4.62 0.0049 0.0002 5.72 0.0064 0.0004 6.41 0.0076 0.0004
Fructosyl-γ-aminobuttersäure
(0.5 M)
1.53 0.0048 0.0002 2.55 0.0032 0.0001 3.47 0.0039 0.0001 4.52 0.0039 0.0001 5.52 0.0053 0.0002 6.46 0.0078 0.0003
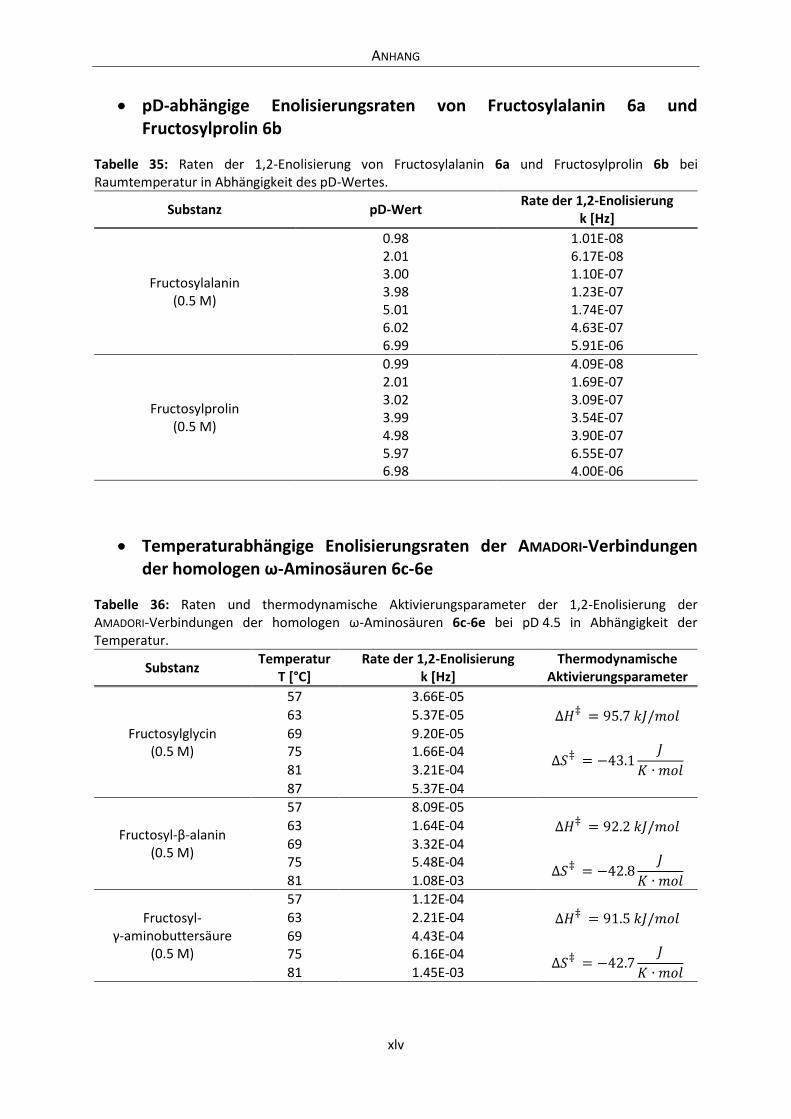
ANHANG
xlv
• pD-abhängige Enolisierungsraten von Fructosylalanin 6a und Fructosylprolin 6b
Tabelle 35: Raten der 1,2-Enolisierung von Fructosylalanin 6a und Fructosylprolin 6b bei Raumtemperatur in Abhängigkeit des pD-Wertes.
Substanz pD-Wert Rate der 1,2-Enolisierung
k [Hz]
Fructosylalanin (0.5 M)
0.98 1.01E-08 2.01 6.17E-08 3.00 1.10E-07 3.98 1.23E-07 5.01 1.74E-07 6.02 4.63E-07 6.99 5.91E-06
Fructosylprolin (0.5 M)
0.99 4.09E-08 2.01 1.69E-07 3.02 3.09E-07 3.99 3.54E-07 4.98 3.90E-07 5.97 6.55E-07 6.98 4.00E-06
• Temperaturabhängige Enolisierungsraten der AMADORI-Verbindungen der homologen ω-Aminosäuren 6c-6e
Tabelle 36: Raten und thermodynamische Aktivierungsparameter der 1,2-Enolisierung der AMADORI-Verbindungen der homologen ω-Aminosäuren 6c-6e bei pD 4.5 in Abhängigkeit der Temperatur.
Substanz Temperatur
T [°C] Rate der 1,2-Enolisierung
k [Hz] Thermodynamische
Aktivierungsparameter
Fructosylglycin (0.5 M)
57 3.66E-05
∆𝐻‡ = 95.7𝑘𝐽/𝑚𝑜𝑙 63 5.37E-05
69 9.20E-05 75 1.66E-04
∆𝑆‡ = −43.1𝐽
𝐾 ∙ 𝑚𝑜𝑙
81 3.21E-04
87 5.37E-04
Fructosyl-β-alanin (0.5 M)
57 8.09E-05
∆𝐻‡ = 92.2𝑘𝐽/𝑚𝑜𝑙 63 1.64E-04
69 3.32E-04 75 5.48E-04
∆𝑆‡ = −42.8𝐽
𝐾 ∙ 𝑚𝑜𝑙
81 1.08E-03
Fructosyl-γ-aminobuttersäure
(0.5 M)
57 1.12E-04
∆𝐻‡ = 91.5𝑘𝐽/𝑚𝑜𝑙 63 2.21E-04
69 4.43E-04 75 6.16E-04
∆𝑆‡ = −42.7𝐽
𝐾 ∙ 𝑚𝑜𝑙
81 1.45E-03

ANHANG
xlvi
• NMR-Pulsprogramme
kfm-double-1hsst
;Double-SST-NMR (01.04.2014) - Kaufmann ;1D 1H selective saturation transfer using selective excitation with a shaped pulse #include <Avance.incl> 1 ze 2 d1 4u pl0:f1 3 (p11:sp11 ph11):f1 4u (p13:sp13 ph13):f1 4u lo to 3 times c d14 pl1:f1 p1 ph1 go=2 ph31 d11 wr #0 if #0 ivc lo to 1 times td1 exit ph1=0 0 ph11=0 2 ph13=0 2 ph31=0 0 ;pl0 : 120dB ;pl1 : f1 channel - power level for pulse (default) ;sp11: f1 channel - power level for first shaped pulse ;sp13: f1 channel - power level for second shaped pulse ;p1 : f1 channel - 90 degree high power pulse ;p11: f1 channel - 90 degree shaped pulse ;p13: f1 channel - 90 degree shaped pulse ;d1 : relaxation delay; 1-5 * T1 ;d14: 4 us ;c: variabel loop counter value (d20=c*(p13+4u))
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\user\kfm-double-1hsst
1
ZE
2
D1
PLW 0
4u
3
P11:sp11 ph11=0,2
4u
P13:sp13 ph13=0,2
4u
VC (1)
PLW 1
D14
P1 ph1=0,0
Go loop
D11
WR0
IF0
TD1 (25)
F1
ANHANG
xlvii
;d20: saturation transfer time ;NS: 2 * n, total number of scans: NS * TD0 ;DS: 0 ;choose p11 and p13 according to desired selectivity ;the flip-angle is determined by the amplitude ;set O1 on resonance on the nucleus that is to be observed (chemical exchange)
kfm-1hsst
;SST-NMR (01.04.2014) - Kaufmann ;1D 1H selective saturation transfer using selective excitation with a shaped pulse #include <Avance.incl> 1 ze 2 d1 4u pl0:f1 3 (p13:sp13 ph13):f1 4u lo to 3 times c d14 pl1:f1 p1 ph1 go=2 ph31 d11 wr #0 if #0 ivc lo to 1 times td1 exit ph1=0 0 ph13=0 2 ph31=0 0 ;pl0 : 120dB ;pl1 : f1 channel - power level for pulse (default) ;sp13: f1 channel - power level for shaped pulse ;p1 : f1 channel - 90 degree high power pulse ;p13: f1 channel - 90 degree shaped pulse ;d1 : relaxation delay; 1-5 * T1
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\user\kfm-1hsst
1
ZE
2
D1
PLW 0
4u
3
P13:sp13 ph13=0,2
4u
VC (1)
PLW 1
D14
P1 ph1=0,0
Go loop
D11
WR0
IF0
TD1 (25)
F1
ANHANG
xlviii
;d14: 4 us ;c: variabel loop counter value (d20=c*(p13+4u)) ;d20: saturation transfer time ;NS: 2 * n, total number of scans: NS * TD0 ;DS: 0 ;choose p13 according to desired selectivity ;the flip-angle is determined by the amplitude ;set O1 on resonance on the nucleus that is to be observed (chemical exchange)
kfm-1hfqssst
;fQSSST-NMR (09.06.2015) - Kaufmann ;1D 1H frequency sweep selective saturation transfer using selective excitation with a shaped pulse #include <Avance.incl> 1 ze 2 d1 fq1:f1 4u pl0:f1 3 (p13:sp13 ph13):f1 4u lo to 3 times 4500 d14 pl1:f1 p1 ph1 go=2 ph31 d11 wr #0 if #0 ivc lo to 1 times td1 exit ph1=0 0 ph13=0 2 ph31=0 0 ;pl0 : 120dB ;pl1 : f1 channel - power level for pulse (default) ;sp13: f1 channel - power level for shaped pulse ;p1 : f1 channel - 90 degree high power pulse
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\user\kfm-1hfqssst
1
ZE
2
FQ1
D1
PLW 0
4u
3
P13:sp13 ph13=0,2
4u
(4500)
PLW 1
D14
P1 ph1=0,0
Go loop
D11
WR0
IF0
TD1 (25)
F1

ANHANG
xlix
;p13: f1 channel - 90 degree shaped pulse ;d1 : relaxation delay; 1-5 * T1 ;d14: 4 us ;NS: 2 * n, total number of scans: NS * TD0 ;DS: 0 ;choose p13 according to desired selectivity ;the flip-angle is determined by the amplitude ;define fq-List ;td1=Summe(fq-List-Eintraege) ;SPOFFS=0
kfm-13csst_nonoe
;SST-NMR (02.04.2014) - Kaufmann ;1D selective 13C saturation transfer using selective excitation with a shaped pulse ;inverse-gated Decoupling (during acquisition) --> elimination of heteronuclear NOE #include <Avance.incl> 1 ze 2 d1 4u pl0:f1 3 (p13:sp13 ph13):f1 4u lo to 3 times c d12 pl1:f1 pl12:f2 p1 ph2 go=2 ph31 cpd2:f2 d11 do:f2 wr #0 if #0 ivc
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\user\kfm-13csst_nonoe
1
ZE
2
D1
PLW 0
4u
3
P13:sp13 ph13=0,2
4u
VC (1)
PLW 1
D12
P1 ph2=0,0
Go loop
D11
WR0
IF0
TD1 (25)
F1
PLW 12
F2

ANHANG
l
lo to 1 times td1 exit ph2=0 0 ph13=0 2 ph31=0 0 ;pl0 : 120dB ;pl1 : f1 channel - power level for pulse (default) ;sp13 : f1 channel - power level for shaped pulse ;pl12: f2 channel - power level for CPD/BB decoupling ;p1 : f1 channel - 90 degree high power pulse ;p13 : f1 channel - 90 degree shaped pulse ;d1 : relaxation delay; 1-5 * T1 ;d11: delay for disk I/O 30m [30 msec] ;d12: delay for power switching 20u [20 usec] ;c: variable loop counter value (d20=c*(p13+4u)) ;d20: saturation transfer time ;NS: 2*n, total number of scans: NS*TD0 ;DS: 0 ;FnMODE: undefined ;cpd2: decoupling according to sequence defined by cpdprg2 ;pcpd2: f2 channel - 90 degree pulse for decoupling sequence ;look at dept135 for decoupling parameters ;define VCLIST
kfm-13cdosst_nonoe
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\user\kfm-13cdosst_nonoe
1
ZE
2
D1
PLW 0
4u
3
P11:sp11 ph11=0,2
4u
P13:sp13 ph13=0,2
4u
VC (1)
PLW 1
D12
P1 ph2=0,0
Go loop
D11
WR0
IF0
TD1 (25)
F1
PLW 12
F2

ANHANG
li
;DOSST-NMR (15.01.2015) - Kaufmann ;1D selective 13C double offset saturation transfer using selective excitation with a shaped pulse ;inverse-gated Decoupling (during acquisition) --> elimination of heteronuclear NOE #include <Avance.incl> 1 ze 2 d1 4u pl0:f1 3 (p11:sp11 ph11):f1 4u (p13:sp13 ph13):f1 4u lo to 3 times c d12 pl1:f1 pl12:f2 p1 ph2 go=2 ph31 cpd2:f2 d11 do:f2 wr #0 if #0 ivc lo to 1 times td1 exit ph2=0 0 ph11=0 2 ph13=0 2 ph31=0 0 ;pl0 : 120dB ;pl1 : f1 channel - power level for pulse (default) ;sp11 : f1 channel - power level for shaped pulse ;sp13 : f1 channel - power level for shaped pulse ;pl12: f2 channel - power level for CPD/BB decoupling ;p1 : f1 channel - 90 degree high power pulse ;p11 : f1 channel - 90 degree shaped pulse ;p13 : f1 channel - 90 degree shaped pulse ;d1 : relaxation delay; 1-5 * T1 ;d11: delay for disk I/O 30m [30 msec] ;d12: delay for power switching 20u [20 usec] ;c: variable loop counter value (d20=c*(p13+4u)) ;d20: saturation transfer time ;NS: 2*n, total number of scans: NS*TD0 ;DS: 0 ;FnMODE: undefined ;cpd2: decoupling according to sequence defined by cpdprg2 ;pcpd2: f2 channel - 90 degree pulse for decoupling sequence ;look at dept135 for decoupling parameters ;define VCLIST
ANHANG
lii
kfm-13csst
;SST-NMR (02.04.2014) - Kaufmann ;1D selective 13C saturation transfer using selective excitation with a shaped pulse ;with power gated decoupling #include <Avance.incl> 1 ze 2 d12 pl13:f2 d1 cpd2:f2 4u pl0:f1 3 (p13:sp13 ph13):f1 4u lo to 3 times c d14 pl1:f1 d12 pl12:f2 p1 ph2 go=2 ph31 d11 wr #0 if #0 ivc lo to 1 times td1 d11 do:f2 exit ph2=0 0 ph13=0 2 ph31=0 0 ;pl0 : 120dB ;pl1 : f1 channel - power level for pulse (default) ;sp13 : f1 channel - power level for shaped pulse
C:\Bruker\TopSpin3.5pl2\exp\stan\nmr\lists\pp\user\kfm-13csst
1
ZE
2
D12 D1
PLW 0
4u
3
P13:sp13 ph13=0,2
4u
VC (1)
PLW 1
D14 D12
P1 ph2=0,0
Go loop
D11
WR0
IF0
TD1 (25)
D11
F1
PLW 13 PLW 12
F2

ANHANG
liii
;pl12: f2 channel - power level for CPD/BB decoupling ;pl13: f2 channel - power level for second CPD/BB decoupling ;p1 : f1 channel - 90 degree high power pulse ;p13 : f1 channel - 90 degree shaped pulse ;d1 : relaxation delay; 1-5 * T1 ;d11: delay for disk I/O 30m [30 msec] ;d12: delay for power switching 20u [20 usec] ;d14: 4u ;c: variable loop counter value (d20=c*(p13+4u)) ;d20: saturation transfer time ;NS: 2*n, total number of scans: NS*TD0 ;DS: 0 ;FnMODE: undefined ;cpd2: decoupling according to sequence defined by cpdprg2 ;pcpd2: f2 channel - 90 degree pulse for decoupling sequence ;define VCLIST
• NMR-AU-Programme
mpcmd
/*** ^^A -*-C++-*- **********************************************/ /* multiprocnocmd 28.09.2015 */ /****************************************************************/ /* Short Description : */ /* Performs multiple commands on increasing procnos. */ /****************************************************************/ /* Keywords : */ /* serial acquisitions, serial processing */ /****************************************************************/ /* Description/Usage : */ /* This AU program performs multiple commands on */ /* increasing procnos. If datasets do not yet exist, the */ /* current dataset and its parameters are copied. If the */ /* data sets already exist, then the commands are */ /* executed as the datasets are. */ /* For the commands 'zg', 'go' or 'xaua' the total */ /* experiment time is estimated and printed out. */ /****************************************************************/ /* Author(s) : */ /* Name : Ulrich Braumann */ /* Organisation : Bruker BioSpin GmbH */ /* Email : [email protected] */ /* Adapted by: : Martin Kaufmann */ /****************************************************************/ /* Name Date Modification: */ /* eub 010810 derived from 'multicmd' */ /* eub 051004 dataset handling changed, VIEWDATA */ /* 12sec delay between acuisiton commands */ /* eub 060502 Delay only for acqu./ formated */

ANHANG
liv
/* eub 060810 Commandlist possible */ /* eub 060816 ExptCalc changed, delay=0 */ /* eub 070911 Abort on autospooling, missing dataset */ /* eub 071025 Processing command with autospooling */ /* possible. */ /* ge 20100127 UXNMR_SAFETY problem with spooler solved */ /****************************************************************/ /* $Id: multicmd,v 1.13 2010/01/27 08:55:12 ge Exp $ */ AUERR = multicmd(curdat); QUIT #include <inc/lcUtil> int multicmd(const char* curdat) { char Answer[PATH_MAX], Question[PATH_MAX], tmpfile[PATH_MAX]; char TempStr[PATH_MAX],TempStr2[PATH_MAX]; char* cp; int i1 = 16; char TSCmd[8][128]; char TSCmdLstDisp[256]; int TSCmdNr = 0; int TSCmdNrMax = -1; char CommandType[128] = "sendgui "; int AcquAllowed = 1; /* Help */ if (i_argc > 2 && strcmp(i_argv[2], "help") == 0) { Proc_err(DEF_ERR_OPT, "Executes up to 1-8 TOPSPIN commands on\n" "subsequent PROCNOs. For example\n" "simply 'zg'\n" "or 'rga' 'zg' 'ef' 'apk'\n" "or more complex like 'rpar PROTON proc'"); return -1; } /* check if started from experiment */ strcpy(tmpfile, ACQUPATH(0)); if (access(tmpfile, F_OK)) { Proc_err(DEF_ERR_OPT, "== ABORT multicmd ==============\n" "Program aborted! Must be started from\n" "an NMR dataset. Change to the first dataset" "and try again."); return -1; }

ANHANG
lv
/* check if autospooling is active */ sprintf(TempStr2,"%s/prop/globals.prop",PathXWinNMRDotXWinNMR()); FindLine(curdat,TempStr2,"AUTO_SPOOL",TempStr); if (strstr(TempStr,"yes")) { strcpy(TempStr,"NORMAL"); GETSTRING( "== WARNING Automatic spooling activated ===\n" "Choose procedure for 'multicmd'\n\n" "(N)ormal: Execute commands immediately\n" "--> No ACQUISITION commands like zg,rga,xaua,... allowed!\n\n" "(S)pool: Send ALL commands to the SPOOLER\n" "-->running acquisition/spooled command will delay new commands)" ,TempStr); if( TempStr[0]=='S' || TempStr[0]=='s') { sprintf(CommandType,"sendgui qu "); } else { AcquAllowed = 0; } } /* Find out how many experiments */ if (i_argc > 2) i1 = atoi(i_argv[2]); else GETINT("Enter number of experiments :", i1) if (i1 <= 0) { Proc_err(DEF_ERR_OPT, "number of experiments must be > 0"); return -1; } /* Input of the commandlist */ strcpy(Answer,"apk"); strcpy(Question,"First command (Any TOPSPIN command)"); for (TSCmdNr = 0; TSCmdNr <= 7; TSCmdNr++) { GETSTRING(Question,Answer); strcpy(Answer,strdlfws(Answer)); if (Answer[0] == 0) break; strcpy(TSCmd[TSCmdNr],Answer); /* Add '_' to front and end to search for whole word */ sprintf(Answer,"_%s_",TSCmd[TSCmdNr]); if ( AcquAllowed==0 && strstr("_zg_go_rga_xaua_ii_",Answer)!=0 )

ANHANG
lvi
{ /* Acquisition command included, abort program */ Proc_err(DEF_ERR_OPT ,"== ABORT multicmd ===============\n" "AU program does not support AUTOMATIC spooling\n" "for acquisition commands like '%s'" ,TSCmd[TSCmdNr]); return -1; } TSCmdNrMax = TSCmdNr; sprintf(Question,"Next command(#%d) or <ENTER> to end ",TSCmdNr+2); Answer[0] = 0; } if (TSCmdNrMax < 0) return 0; /* Create list with the command(s), keep the list short */ TSCmdLstDisp[0] = 0; cp = TSCmdLstDisp; for (TSCmdNr = 0; TSCmdNr <= TSCmdNrMax; TSCmdNr++) { if (strlen(TSCmdLstDisp) <= 36 || TSCmdNr == TSCmdNrMax) cp += sprintf(cp, "[%s] ", TSCmd[TSCmdNr]); else { sprintf(cp, "... [%s]", TSCmd[TSCmdNrMax]); break; } } if (!strcmp(TSCmd[0],"zg") || !strcmp(TSCmd[0],"go") || !strcmp(TSCmd[0],"xaua")) { /* Calculate experiment time for standard acquisition command */ int startProcno = procno; int expTime = 0; TIMES(i1) SETCURDATA expTime += CalcExpTime(); if (loopcount1+1 < i1) { IPROCNO SETCURDATA } END DurationToAscii(TempStr, (double)expTime); /* return to original dataset */ procno = startProcno;

ANHANG
lvii
SETCURDATA if (Proc_err(ERROPT_AK_CAN, "Run acquisition with '%s'\n" "In approx. %s\n" "On dataset '%s' procno(s) %d - %d", TSCmdLstDisp, TempStr, name, procno, procno+i1-1)) return -1; if (strcmp(TSCmd[0],"zg") == 0) strcat(TSCmd[0], " yes"); /* override UXNMR_SAFETY if set */ } else { if (Proc_err(ERROPT_AK_CAN, "Run %d command(s)\n%s\n" "On dataset '%s' procno(s) %d - %d", TSCmdNrMax+1, TSCmdLstDisp, name, procno, procno+i1-1)) return -1; } /* Loop with execution of commands */ TIMES(i1) if (TSCmdNrMax > 0) Proc_err(ERROPT_AK_NO, "On dataset '%s' %d\nRunning command(s)\n%s", name, procno, TSCmdLstDisp); for (TSCmdNr = 0; TSCmdNr <= TSCmdNrMax; TSCmdNr++) { (void)sprintf(tmpfile,"running '%s' on procno # %d", TSCmd[TSCmdNr], procno); Show_status(tmpfile); /* use 'sendgui' to allow all types of commends */ sprintf(TempStr,"%s %s",CommandType,TSCmd[TSCmdNr]); XCMD(TempStr); if (AUERR) { /* stop processing of THIS dataset if one command fails */ sleep(2); break; } } if (loopcount1+1 < i1) { IPROCNO SETCURDATA } END Proc_err(ERROPT_AK_NO, "--- multicmd finished ---"); return 0; }

CURRICULUM VITAE
lviii
Curriculum Vitae
Der Lebenslauf ist in der online-Version aus Gründen des Datenschutzes nicht enthalten.

CURRICULUM VITAE
lix
Der Lebenslauf ist in der online-Version aus Gründen des Datenschutzes nicht enthalten.

CURRICULUM VITAE
lx
Der Lebenslauf ist in der online-Version aus Gründen des Datenschutzes nicht enthalten.

NOTIZEN
lxi

NOTIZEN
lxii












![O Morphological, thermal and bioactivity evaluation of O ... · matrix or coatings of polymer matrix are alternatives to improve these limitations[7]. Calcium phosphate bioceramics](https://img.pdfslide.org/doc/110x75/60220a16e4b99421dd6582ff/o-morphological-thermal-and-bioactivity-evaluation-of-o-matrix-or-coatings.jpg)
















