Embed Size (px)
Citation preview

B ~ A BENCZ~: Ein neuer Apparat zur Stickstoffbestimmung. 125
- [11] SCI~LESING~R, H . J . u . H . B . VAN VALKENBURGtt" J . Amer. Chem. Soc. .53, 1212 (1931); vgl. diese Z. 91, 17 (1933). - [12] MOLLER, ~{. : Kern. Maanedsblad 18, 138 (1937). - [13] BENT, ~I. E. u. C. L. FRENCH: J . Amer. Chem. Soc. 63, 568 (1941). - [14] EDMONDS, M. U. N. BIRNBAU_'Cf : J . Amer. Chem. See. 68, 1471 (1941). - [15] PETERS, Ch. A. u. 'Ch. L. FRENCH: Ind. eng. Chem. Anal. Edit . 13, 604 (1941). - [16] STOKES, It . N. u. J . 1~. CAIN: J . Amer. Chem. Soc. 29, 409, 443 (1907); vgI. diese Z. 58, 224 (1919). - [17] PHILIP, J. I-[. U. BRAI~ILEY : J. chem. See. London103, I, 795 (1913); vgl. diese Z. 73,309 (1928). - [18] B~ATTAe~ARYA, A. K. u. N.!~. DHAa: J . Indian. Chem. See. 6, 197 (1929). - [19] BttATTACItARYA, A. ]Q u. N. l~. DI~Atl: J . physic. Chem. 8~, 653 (1931~. - [20] BHATTACm~aYA, A. X. u. N. I%. DHAR: Z. anorg. Chem.~209, 123 (1932). - [21] EM~ETT, W. G.: J . chem. See. London 1927 I I , 2060 - [22] B E ~ I G , M. u. I t . HIaseH~ICLLER: Diese Z. 92, 5 (1933). - [23] TATLOCK, 1% R. : J . Soc. Chim. Ind. 6, 279, 352 (1887); vgl. diese Z. 28, 700 (1889). - [24] v. X~LER, H. U. G. LuNcE : Z. angew. Chem. 1894, 670); vgl . diese Z. 52, 159 (1913). - [25] ANDaEWS, L.: Chem. News 70, 165 (1894). - [26] STEINI~XUSEa, K. u. X. GINSBERG: Eiese Z. 11)4, 385 (1936). - [27] THIEL, A.: Bet. dtsch, chem. Ges. 71, 756 (1938). - [28] FAaY, Ch. u. E. TASSILY : Bull. Soc. Ph~rm. Bordeaux 19, 11 (1912). - [29] GeT,]:S, F: Z. Ungers. Nahr .u . Gen.Mittel 27, 676 (1914); vgh diese Z. 61, 300 (1922). - [30] WILLSTXTTER, 1~.: Ber. dtseh, chem. Ges. 53, I152 (1920); vgl. diese Z. 62, 3(;8 (1923); 67,461 (1925/26). - [31]WALKaa, W.B . : Analyst. 50, 279 (1925); vgl. diese Z. 67, 462 (1925/26). - [32] v. D. VLUGT, L. S. : Chem. Weekblad 25,495 (1928). - [33] SAC~Ea, J. F. : Farbenchem. 2, 120 (1931). - [34] DE B~OUCK~E L . u . A, E. GILLET: Bull. Soc. chim. Belg. 4- o, 28I (1933); vgI. diese Z. 11)7, 366 (1936). - [35] KosLEn, M. u. H. v. H~LBAN : Helv. Chimiea Aeta 2-0, 1395 (1939). - [36] LELTGEBEL, W. u.~G. MuHs: 3/Ietallwirtsehaft -00, 1089 (1941),
Prof. Dr. WII.HEL~ ]-IACKER, (22c) KSnigswinter, Kronprinzenstrage 12.
Aus der ung. chem. Landesanstalt, Abe. ffir Biochemie und Vitamine, Budapest.
E i n n e u e r A p p a r a t z u r S t i c k s t o f f b e s t i r n m u n g nach KJELDAHL.
Von
B~LA BE~CZE.
~lit 2 Textabbildungen.
(Eingegangen am 3. Mai 1948.)
Die KJELDAHLSehe S t i c k s t o f f b e s t i m m u n g h a t im Lau fe der Zei t sehr
v ie le )~ndernngen e r f a h r e n und f a s t jedes L a b o r a t o r i u m bes i t z t h e u t e se inen
e igenen A p p a r a t , seine e igene Arbe i t swe i se u n d besonde re Kuns tg r i f f e , die sozusagen auf die N a c h k o m m e n s c h a f t v e r e r b t werden-
Der w e i t a u s b e k a n n t e s t e u n t e r den v e r s c h i e d e n e n A p p a r a t e n i s t der n a c h
d e m WAG~R-PARNAs-P r inz ip [ i ] au fgebau te Wasse rdampfdes t i l t a t i ons -
a p p a r a t , der s ich zur A u s f i i h r u n g sowohl yon Makro- als a u e h y o n Mikro- Ztsch~t. f. anal. Chem. Bd. 129. 9

126 B~LA BEh~C ZE :
bestimmungen eignet. Der Apparat hat den Vorzug, dal~ eine Destillation in einigen Minuten ausgef~ihrt werden kann. Die Handhabung ist jedoch einigermaf~en umstiindlich, auch benOtigt der Apparat stiindige 0ber- wachung, welcher Umstand unter dem Gesichtspunkt der Zeitersparnis be- senders bei Serienbestimmungen unerwiinscht ist. Ein ~weiterer Nachteil
ist~ da~ das Wasser, welches sich
a . t)
Abb. 1.
wfihrend der Destillation im Sammel- gef~B kondensiert, das Volumen der Vorlagefliissigkeit bei den einzelnen Destillation~n in ungleiehartigem Mate vermehrt; dadurch wird der Endpunkt der Titration unscharf und verfinderlich.
Der im folgende n besehriebene Apparat vermeidet diese Naehteile. Bei der Ausftihrung stiitzte ich reich
auf die Apparate yon HI~SBERG [2] und L ~ G [3], welche in biochemischen and ~rztlichen Laboratorien zu Reststickstoff-, Carbamid-, Aeeton- and Milchs~urebestimmungen verwendet werden.
Der Destillierapparat (Abb. 1) besteht aus dem retor~en~hnlichen Destillierk61b- eher~ a mit Ansatz c und aus dem zylinderf6rmigen, zum Auff~ngen des Destilla~s dienenden Teii h. Bei dem zu Mikrobestimmungen dienenden Appara~ fa~ der refer- tenarfige Teil a e~w~ 35ml, der Ansatzteil c etw~ 10 mlund der Vorlageteil b etwa, 30 ml. Man bringt die sticksto/]haltige sa ure Fliissigkelt in das KSlbchen a, die KJ E L B ~ H L- .Lauge in das ~om letzteren oollstiindig getrennte AnsatzkSlbchen c und die Vorlage- ]liissigkeit in das zylindex]Srmige Ge]h~ b. Die 0ffnungsbreite des App~rates ist so gew~hlt (25 ram), daft mit gentigend ~usgezogener Pipette bei einiger Llbung das Einbringen der Flfissigkeiten leicht ausgefiihr~ werden kann. Die Teile a und b sind mit guten Sehliffen versehen, sie kSnnen mit Spiralfedern verbunden werden, wodureh der Apparag a uch gegen etw~igen ~berdruck gesichert ist. Zum Appar~t gehSrt noch ein doppelter Wasserbeh$1ter, dessert beide Teile voneinander vollkommen ge- r sind. Diese Teile sind zur W/~rmeisolierung mit Asbestplaften versehen. Die DestillierkSlbchen werden dutch sogenannfe ,,Tulpen" so in der Wanne befestigt, dab der Tell a yon siedendem Wasser vollkommen bedeckt wird; der Vorlugeteil b wird irt d~s Kiihlwasser get~ueh~.
Der Apparat ist leieht za handhaben. Er hat aufierdem den Vorteil, daft man Serienbestimmungen ohne besondere l)ber~vaehang aus/iihren kann, da die Destillation im geschlossenen System aasge/iihrt ~,ird, Verluste also aasge- schlossen sind. Das Volumen der r Fliissigkeit kann sich ~'iihrend der Destillation nur ~enig ~ermehren, daher ist der Umschlag beim Titrieren immer schar/ und der Endp~nkt der Titration kann sich nieht iindern.
Das Volumen des Auffanggefal~es ist so gewahlt, dal~ die Rtiektitration in dem Geffil~ selbst erfolgen kann. Das vereinfaeht die Durchfiihrung der Analyse bedeutend. Die K01behen sind mit ,,Normalschliff" versehen, daher k(innen KSlbehen verschiedener GriSl3e miteinander verbunden
Ein neuer Apparat zm" Stiekstoffbesbimmung. 127
werden, was wiederum fi~r die allgemeine Anwendbarkeit des Apparates yon Bedeutung ist.
Die Destitlierk6lbehen sind vet dem Gebraueh zu kalibrieren, da die Destillationsdauer yon der Gr6fte der K61bchen abhiingig ist. Die Destilla- tionsdauer ist aul]erdem eine Funktion der Menge der Destillierfliissigkeit, der angewandten Stiekstoffmenge und des Temperaturunterschiedes zwi- schen den Wasserb~idern. Die Kalibrierung der KOlbehen wurde folgender- mal~en ausgefi~hrt :
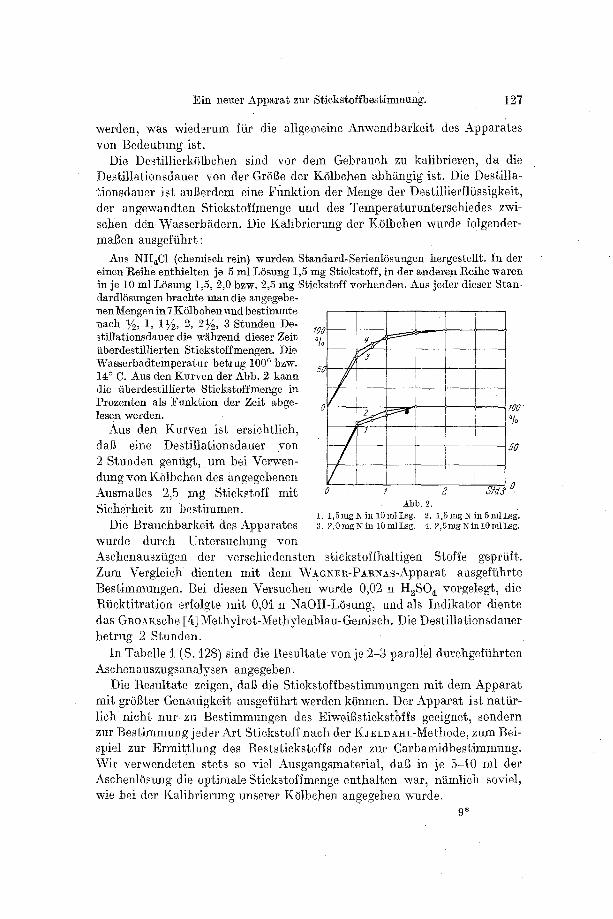
Aus NH~C1 (chemisch rein) wurden Sfandard-Serient6sungen hergestellt. In der einen l~eihe enthielten je 5 ml LSsung 1,5 mg Stickstoff, in der anderen t~eihe waren in je 10 ml LSsung 1,5, 2,0 bzw. 2,5 mg Stickstoff vorhanden. Aus jeder dieser Stun- dardlSsungen brachte man die angegebe- nenMengen in 7 KSlbchen und bestimmte nach ~2, 1, 1�89 2, 2~, 3 S~nnden De- stillationsdauer die w/~hrend dieser Zeit iiberdestillierten Stickstoffmengen. Die Wasserbadtemperatur betrug 100 ~ bzw. 14 ~ C. Aus den Kurven der Abb. 2 kann die iiberdestillierte Stickstoffmenge in Prozenten als Funk~ion der Zeit abge- lesen werden.
Aus den Kurven ist ersichtlich, dab eine Destil~ationsdauer yon 2 Stunden geniigt, um bei Verwen- dung -:on K01behen des angegebenen Ausmaftes 2,5 rag Stickstoff mit Sieherheit zu bestimmen.
Die Branchbarkeit des Apparates wurde dureh Untersnchung yon
! ~<00! ~
I
! ~ ~ r / ~
i o l 2 Stgd o
Abb. 2. 1. 1,5mgiXil110mlLsg. 2. 1,5mg]N=il~5mlLsg'. 3. 2,0mgNin 10mlLsg. 4.2,5mglN'inl0mlLsg.
100"
Asehenausziigen der versehiedensten stickstoffhaltigen Stoffe gepriift. Zum Vergleich dienten mit dem WAG~R-PAR~As-Apparat ausgeftihrte Bestimmungen. Bei diesen Versuohen wurde 0,02 n H2SO ~ vorgelegt, die Riicktitration erfolgte mit 0,01 n NaOH-L6sung, und als Indikator diente das GRoAKsohe [4~1Methylrot-Methylenblau-Oemisch. Die Destillationsdauer betrug 2 Stunden.
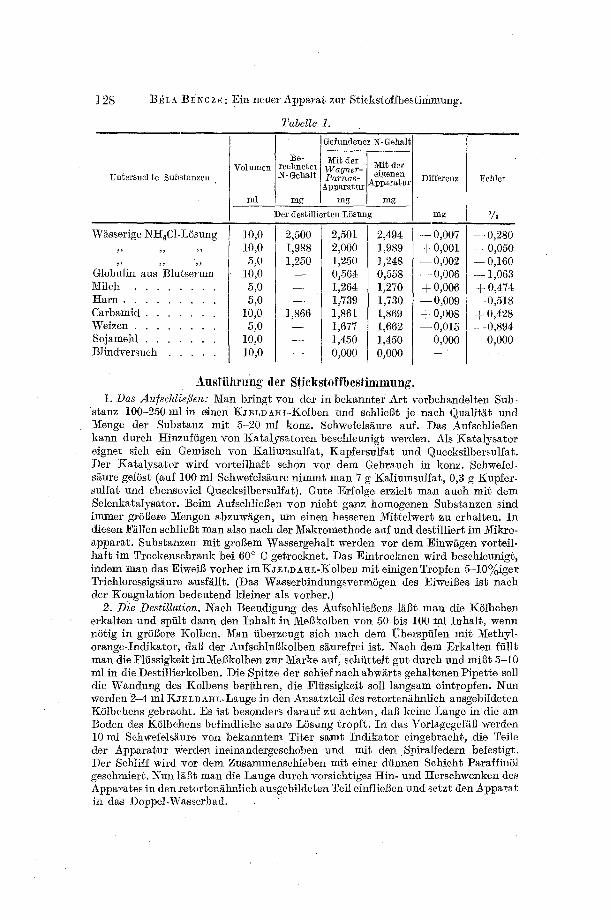
In Tabelle i (S. 128) sind die Resultate yon je 2-3 parallel durchgefiihrten Aschenauszugsanalysen angegeben.
Die t{esultate zeigen, dal3 die Sti@stoffbestimmungen mit dem Apparat mit grSftter Genanigkeit ausgefiihrt werden k0nnen. Der Apparat ist natiir- lich nicht nur zu Best immungen des Eiweil3stickstbffs geeignet, sondern zur Bestimmung jeder Art Stickstoff nach der KJELDAHL-Methode, zum Bei- spiel zur Ermittlung des l~eststickstoffs oder zur Carbamidbestimmung. Wit verwendeten stets so viel Ausgangsmaterial, daft in ie 5M0 ml der AschenlSsung die optimale Stickstoffmenge enthalten war, namlich soviel, wie bei der Kalibrierung unserer K01behen angegebeu wurde.
9*
]28 ]~1~ L A ~B E N C Z E: Ein neuer Appara~ zur Stickstoffbestimmung.
Tabelle 1.
Untersuchte Substanzen
Wgsserige NHaC1-L5sung
Globulin aus Blutserum Milch . . . . . . . . H A I T I . . . . . . . . .
Carbamid . . . . . . . Weizen . . . . . . . . Sojamehl . . . . . . . Blindversuch . . . . .
Volumen Gefunden_ er_ N-Gehalt :Be- I ~it der ~Iit der rechneter ~r .
N-Gehalt PaThos elgenen ' ~ - Apparatur IApparatm"
ml ~ mg ~ rng mg Der destillierten LSsun
10,0 10,0 5,0
10,0 5,0 5,0
10,0 5,0
10,0 10,0
2,500 1,988 1,250
1,866
Differenz
mg
2,501 2,000 1,250 0,564 1,264 1,.739 1,861 1,677 1,450 0,000
2,494 - - 0,007 1,989 + 0,001 1,248 - -0 ,002 0,558 - - 0,006 1,270 + 0,006 1,730 --0,009 1,869 + 0,008 1;662 - -0 ,015 1,450 0,000 0,0O0 - -
Fehler
%
- - 0,280 d- 0,050 - - 0,160 - - 1,063 + 0,474 - -0 ,518 + 0,428 - - 0,894
0,000
A u s f i i h r u n g der S t i c k s t o f f b e s i i m m u n g .
1. Das Au]schllefien: Man bringt yon der in bekannter Ar t vorbehandelten Sub- s tanz 100-250 ml in oinen KJnLDA~L-Ko]ben und schlieBt je nach Qualitg~ und Menge der Substanz mit 5-20 ml konz. Schwefe]sgure auf. ])as AufschlieBen kann durch Hinzufiigen yon Katalysatoren bescMeunig~ werden. Als Katalysator eignet sich ein Gemiseh yon Kaliumsulfat, Kupfersulfat und Quecksilbersulfat. Der Katalysator wird vortei lhaf t schon vor dem Gebrauch in konz. Schwefel- sgure gelSs~ (auf 100 ml Schwefdsgure n immt man 7 g Kaliumsulfat , 0,3 g Kupfer- sulfat und ebensoviel Quecksilbersulfat). Gute Erfolge erzielt man auch mit dem Selenkatalysator. Beim Aufsch]ieBen yon nicht ganz homogenen Substanzen sind immer grSBere Mengen abzuwggen, um einen besseren Mittelwert zu erhal~en. In diesen F~llen schlieBt man also nach der Makromethode auf und des~illier~ im Mikro- apparat. Substanzen mi t groBem Wassergehalt werden vor dem Einwggen vorteil- haft im Trockenschrank bei 60 ~ C getrocknet. Das Eintrocknen wird beschleunig~, indem man das Eiweig vorher im KJELDA~L-Kolben mit einigen Tropfen 5-10%iger Trichloressigsgure ausf~llt. (Das WasserbindungsvermSgen des EiweiBes ist nach der Koagulation bedeutend kleiner als vorher.)
2. Die Destillatlon. Nach Beendigung des AufschlieBens lgBt man die KSlbchen erkalten und spiil~ dann den Inhal t in MeBkolben yon 50 bis 100 ral Inhalt , wenn nStig in gr5Bere Kolben. Man tiberzeugt sich nach dem ~)berspiilen mi t Methyl- orange-Indikator, dal~ der AufschluSkolben sgurefrei ist. Nach dem Erkal ten fiillt man die Flfissigkeit imMel3kolben zur Marke auf, sctf i i t tdt gut durch und mi8t 5-10 ml in die Destillierkolben. Die Spitze der schief nach abwgrts gehal~enen Pipette soll die Wandung des Kolbens beriihren, die Fliissigkeit soll langsam eintropfen. Nun werden 2-4 ml KJ E L~ A H L-Lauge in den Ansatzteil des retortenghnlich ausgebildeten K61bchens gebracht. Es is~ besonders darauf zu achten, dab keine Lauge in die am Boden des K61bchens befindliche saute LSsung tropft . In das VorlagegJ~fl werden 10 ml Schwefelsgure yon bekanntem Titer saint Indikator eingebrach~, d ie Teile der Appara~ur werden ineinandergeschoben und mit den Spiralfedern befestigt. Der Schliff wird vor dem Zusammenschieben mit einer diinnen Schicht Paraffin51 geschmiert. Nun IgBt man die Lauge dutch vorsichtiges Hin- und Herschwenken des Apparates in den retortenghnlich ausgebildeten Teil einflieBen und setzt den Apparat in das Doppel.Wasserbad.

HORST B6H~E : Uber die quant.itative Bestimmung yon SenfSlen. 129
3. Das Titrieren. Nach Beendigung der Destillation hebt man die K61behen ~us dem Wasserbad her~as, bringt sie durch Abkiihlen mi~ Leitungswasser auf Zimmer- temperatur und t i t r ier t d~s Destillat in dem Vorlageteil mit genau eingestellter Natronl~uge. Start in SehwefelsS~ure kann men d~s Destillat ~ueh in 3~ Bor- sEure ~tfff~ngen. In diesem :F~ll t i t r ier t man unmittelbar mit 0,02 n Sehwefels~ure ~anter Verwendung des Indik~tors n~ch GROAK [4] bzw. T_~SEIRO [5] (i-Vlethylrog- Nethylenblau). Es is(~ also keine genau eingestellte Natronlauge nStig; man hat bloB den S~ureverbraueh der 3%igen Borsgurel6sung in einem Blindversueh festzustellen.
Sehrifttum.
[1] Diese Z. 11/, 26I (1938). - [2] Klin. Wchsehr. 22, 792 (1935). - [3] Klin. Wehschr. 26, 913 (1939). - [4] Bioehem. Z. 244, 294 (1931). - [5] Vgl. diese Z. 114, 278 (1938).
Dr. Bg~x ] ~ c z ~ , Orsz~gos Chemiai Int@zet, Budapes~ II, Keleti K~roly u. 24.
Aus dem Pharmazeutisch-ehemischen Inst i tut der Universitat ~arburg a. d. Lahn*.
Ober die quantitative B e s t i m m u n g yon Senf61en. Von
]-[OtlST BSHME
Mitbearbeitet yon Gerhard EPDING und Martha FLIES..
(Eingegangen am 17. M~irz 1948.)
Im Rahmen einer gr61~eren Unte r suehung h a t t e n wir Interesse an e iner le icht und q u a n t i t a t i v durchf t ihrbaren Methode zur Bes t immung yon Sen/glen, insbesondere neben den isomeren Rhodaniden. Die gebrauch- ! iehste Methode naeh J. GADAM~ 1, die auf der Umsetzung des dureh An- !agerung yon A m m o n i a k gebi ldeten Thioharns tof fes mi t S i lbe rn i t ra t be- ruht , g ibt im al lgemeinen zu hohe Wer te . Wi r haben daher die Umse tzung der SenfOle mi t Wa.ssersto//peroxyd in alkalischer Ldst~ng unte r sueh t . R. KITAS~RX 2 ha t n~m]ieh in mehreren Arbe i ten gezeigt, dab Verbindungen mi t einer S-C-Doppelb indung un te r diesen Bedingungen in die entspreehen- den Carbonylverb indungen t ibergefi ihrt werden, wfihrend der abgespal tene Sehwefel wel te r zum Sulfa t - Ion oxyd ie r t wird
- ) / C = S + 2 0 H ~ + 4 H 2 0 ~ " = C - - 0 + 5 H 2 0 + S 0 ~ " .
* Die experimentellen Arbeiten wurden zum grS/~ten Teil im Pharmazeutisehen Insti tut der Universitgt Berlin durehgeffihrt und in der Diplomarbeit G. EPD~NG, Berlin 1941, und der Dissertation NI. FLIES, Berlin 1944 , niedergelegt.
1 Arch. Pharm. igg, 110 (1899). "~ J. pharm. Soe. Japan 5'4, 1 (1934).