Embed Size (px)
Citation preview
MakrocyclensyntheseDOI: 10.1002/ange.200503988
Formtreue Makrocyclen: Strukturen und Synthesen ausArylen- und Ethinylen-BausteinenWei Zhang und Jeffrey S. Moore*
AngewandteChemie
Stichw�rter:Cyclooligomerisierungen · Dynamischekovalente Chemie · FormtreueMakrocyclen · Kreuzkupplun-gen · Metathese
J. S. Moore und W. ZhangAufs�tze
4524 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

1. Einleitung
Formtreue Arylenethinylen-Makrocyclen finden seit ei-nigen Jahren ein großes Interesse wegen ihrer m�glichenVerwendung als große supramolekulare Bausteine f�r p-Stapelungen, stabile nanopor�se Feststoffe und tubul#refl�ssige Phasen.[1–11] Im Unterschied zu konformativ flexiblenRingen weisen formtreue Makrocyclen regelm#ßige Wieder-holungseinheiten mit nur wenigen Konformationsfreiheits-graden auf. Durch ihr starres Ger�st ergeben sich großeMolek�loberfl#chen, die sich zu geordneten Strukturen or-ganisieren. Die Ethinylengruppe ist aus mehreren Gr�ndenein geeigneter Baustein f�r diese Strukturen: 1) Sie ist in derLage, an ausgedehnten Elektronenkonjugationen teilzuneh-men; 2) ihre einfache lineare Geometrie f�hrt zu vorhersag-baren Strukturen; 3) sie besteht ausschließlich aus C-Atomen, was insbesondere f�r die Herstellung von kohlen-stoffreichen Materialien interessant ist (Abbildung 1);[8,9]
4) sie ist durch eine Vielzahl von Synthesen leicht zug#ng-lich.[11, 12] Formtreue Makrocyclen verf�gen �ber eine Innen-und eine Außenseite, die spezifisch funktionalisiert werdenk�nnen. Alle diese Eigenschaften machen formtreue Mak-
rocyclen aus Arylen- und Ethinylen-Bausteinen zu attraktiven Strukturenf�r organische Materialien.[13]
Eine entscheidende Rolle bei derSynthese von Arylenethinylen-Ma-krocyclen spielen Kohlenstoff-Koh-lenstoff-Kupplungen (Abbildung 2).Die Makrocyclensynthese profitierteganz erheblich von den methodischen
Fortschritten bei Kreuzkupplungen zwischen sp- oder sp2-hybridisierten Kohlenstoffatomen.[12] Dazu z#hlen die palla-diumkatalysierte Alkinvariante der Heck-Reaktion,[14] die inVerbindung mit Kupfersalzen als Sonogashira-Kreuzkupp-lung bekannt ist,[15–19] die Negishi-Kreuzkupplung[20] zwischenArylhalogeniden und Alkinylzinkhalogeniden, die Homo-kupplung zwischen terminalen Alkinen nach Glaser[21–32] unddie Cu-vermittelte Castro-Stephens-Kupplung.[33] DieseKupplungsreaktionen tolerieren eine Vielzahl von funktio-nellen Gruppen, ergeben meist hohe Ausbeuten und k�nnenauf zahlreiche Vorstufen zur�ckgreifen. Im Zuge der Fort-schritte bei der Alkinmetathese[34] wurde diese Art vonKohlenstoff-Kohlenstoff-Verkn�pfung auch zur Synthese vonEthinylenverbindungen angewendet.
Die Strategien zur Synthese von Makrocyclen k�nnen invier Hauptkategorien unterteilt werden (Abbildung 3):1) Cyclooligomerisierungen,[35] 2) intramolekularer Ring-schluss von a,w-difunktionalisierten Oligomeren, 3) inter-molekulare Kupplung zwischen zwei oder mehreren oligo-meren Fragmenten mit anschließender unimolekularer Cyc-lisierung, 4) templatvermittelte Cyclisierung von Fragmen-ten. Sind an der Cyclooligomerisierung (Strategie 1) irrever-sible Reaktionen beteiligt, z.B. Kreuzkupplungen, soverringert das Problem des „Produkt-Overshooting“ dietheoretische Ausbeute erheblich; Einzelheiten hierzu werdenin Abschnitt 2.1 besprochen. Die Strategien 2 und 3 z#hlen zuden h#ufigsten Kreuzkupplungsans#tzen bei der Makrocyc-
[*] Dr. W. Zhang, Prof. J. S. MooreDepartment of Chemistry and Materials Science & EngineeringUniversity of Illinois at Urbana-Champaign600 South Mathews Avenue, Urbana, IL 61801 (USA)Fax: (+1)217-244-8024E-mail: [email protected]
Formtreue Arylenethinylen-Makrocyclen mit ihren besonderenStrukturen und Eigenschaften werden f�r m gliche Anwendungen inder supramolekularen Chemie und den Materialwissenschaften in-tensiv erforscht. In diesem Aufsatz betrachten wir neuere Beispiele f�rdie Synthese von Makrocyclen durch Kreuzkupplungen (Sonogashi-ra: Arylacetylen-Makrocyclen, Glaser: Aryldiacetylen-Makrocyclen)und Methoden der dynamischen kovalenten Chemie. Ein Nachteil derKupplungsmethoden ist die kinetisch kontrollierte Produktverteilung,da ein erheblicher Anteil der Oligomere �ber die erw�nschte Cyclen-gr ße hinausw1chst („Overshooting“). Bessere Ergebnisse wurden inj�ngster Zeit mithilfe der dynamischen kovalenten Strategien auf derGrundlage reversibler Metathesereaktionen erzielt, die formtreueMakrocyclen in nur einem Reaktionsschritt liefern. MechanistischeUntersuchungen belegen, dass diese Form der Makrocyclenbildungein thermodynamisch kontrollierter Prozess ist. Verbleibende pr1pa-rative Probleme betreffen die Synthese von spezifisch funktionali-sierten Strukturen sowie gr ßeren und komplexeren zweidimensio-nalen und dreidimensionalen Molek�len.
Aus dem Inhalt
1. Einleitung 4525
2. Kinetisch kontrollierte Synthesevon formtreuen Makrocyclen 4527
3. Thermodynamisch kontrollierteSynthese von Arylenethinylen-Makrocyclen 4536
4. Schlussfolgerungen undAusblick 4545
Abbildung 1. Zwei- und dreidimensionales Kohlenstoffnetzwerk ausdrei- bzw. vierfach substituierten Ethinylen-Monomeren. SchwarzePunkte repr@sentieren Monomereinheiten.
Formtreue MakrocyclenAngewandte
Chemie
4525Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

lensynthese und werden in den Abschnitten 2.2 bzw. 2.3 be-handelt. Der templatvermittelten Makrocyclensynthese(Strategie 4) widmen wir uns in Abschnitt 2.4.
Methoden der dynamischen kovalenten Chemie (DCC)beruhen auf reversiblen Reaktionen, die unter Gleichge-
wichtsbedingungen ablaufen. Durcheine reversible Reaktionsf�hrung be-steht die M�glichkeit, die chemischen„Defekte“, die bei kinetisch kontrol-lierten Prozessen auftreten, zu beseiti-gen. Auf DCC beruhende Synthesensowie theoretische Studien zur ther-modynamischen Stabilit#t der erzeug-ten Makrocyclen werden in Abschnitt 3beschrieben. Da schon mehrere Iber-sichten zu verwandten Themen er-schienen sind,[1,3–11] wollen wir uns hierauf die j�ngsten Entwicklungen bei derSynthese von Arylenethinylen-Makro-cyclen beschr#nken.
Zur Synthese von Makrocyclensteht zwar eine Reihe von Kreuzkupp-lungen zur Auswahl, diese f�hrenjedoch zu kinetisch kontrollierten Pro-duktverteilungen, was insbesondere beiCyclooligomerisierungen problema-tisch ist. Die Bildung von unerw�nsch-ten Spezies (z.B. von h�heren Oligo-meren, die die Gr�ße des gew�nschtenMakrocyclus �berschreiten) kann
wegen der Irreversibilit#t der Reaktionen nicht korrigiertwerden. Selektivere Produktverteilungen erh#lt man im All-gemeinen durch intramolekularen Ringschluss oder inter-molekulare Kupplung von Oligomeren. Diese erfordernjedoch meist zahlreiche Syntheseschritte oder, im Falle derRingschlussreaktionen, verd�nnte L�sungen (< 1 mm),sodass die Gesamtausbeuten oft niedrig sind.[5, 7, 36]
Jeffrey Moore wurde 1962 bei Joliet in Illi-nois geboren. Er studierte Chemie an derUniversit$t von Illinois (B.S. 1984) und pro-movierte dort bei Samuel Stupp in Material-wissenschaften (1989). Nach einer NSF-Stelle am Caltech bei Robert Grubbs undForschungen an der Universit$t von Michi-gan in Ann Arbor kehrte er 1993 an die Uni-versit$t von Illinois zur3ck und 3bernahmdie William H. and Janet G. Lycan-Professurf3r Chemie und Materialwissenschaften.Seine Forschungen betreffen molekulareSelbstorganisation, strukturkontrollierte Ma-
kromolek3le und Foldamere, stimuliresponsive Materialien und selbsthei-lende Polymere.
Wei Zhang wurde 1977 in China geboren.Nach dem Chemiestudium in Peking (B.S.2000) und Forschungen an der Universit$tvon Utah wechselte er 2001 an die Universi-t$t von Illinois, wo er 2005 bei Prof. JeffreyMoore seine Promotion abschloss. Seine For-schungen betrafen die Entwicklung einesMolybd$nkatalysators f3r die Alkinmetathe-se und dessen Anwendung zur Synthese vonformtreuen Makrocyclen und konjugiertenPolymeren. Seit M$rz 2006 ist er Postdocam Massachusetts Institute of Technologybei Prof. Timothy M. Swager.
Abbildung 2. Aufbau von Kohlenstoff-Kohlenstoff-Bindungen ausgehend von Ethinylengruppen.
Abbildung 3. Bbersicht Cber Cyclisierungsstrategien: a) Cyclooligome-risierung, b) intramolekulare Cyclisierung, c) bimolekulare Kupplung/unimolekulare Cyclisierung, d) templatvermittelte Cyclisierung.
J. S. Moore und W. ZhangAufs�tze
4526 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

Mit Methoden der dynamischen kovalentenChemie[34,37–42] zur Synthese vonMakrocyclen konnten jedochsehr gute Ausbeuten erzielt werden. Berichtet wurde �ber dieSynthese von makrocyclischen Verbindungen,[43] molekularenKapseln[44] und verzahnten Strukturen.[45] Der Erfolg dieserStrategie beruht auf dem Energiel�ckenprinzip, auf das inAbschnitt 3.2.3 n#her eingegangen wird.
Ob es bei thermodynamisch kontrollierten Prozessen ge-lingt, von einer Vielzahl m�glicher Produkte eine einzigebestimmte Spezies zu erzeugen, h#ngt von den relativenfreien Energien der beteiligten Spezies ab. Die Energien derProduktspezies definieren die Energielandschaft, ein Begriff,den man h#ufig im Zusammenhang mit der Proteinfaltungfindet,[46] der aber auch bei anderen thermodynamisch kon-trollierten Prozessen, bei denen eine Vielzahl m�glicherStrukturen zur Auswahl steht, verwendet wird. Bei der Cyc-looligomerisierung kommen als m�gliche Produkte sowohlcyclische als auch lineare Oligomere unterschiedlicher Gr�ßeinfrage. Falls eine große Energiel�cke zwischen dem er-w�nschten Makrocyclus und anderen m�glichen Makrocyc-len vorliegt, kann mithilfe von DCC das gew�nschte Produktin hoher Ausbeute erzeugt werden, selbst wenn zu Beginn derReaktion die unerw�nschten Produkte kinetisch beg�nstigtsind (Abbildung 4). Bei Systemen, die aus konformativ fle-xiblen Bausteinen aufgebaut sind, dominieren gew�hnlichentropische Faktoren, und meist �berwiegt der kleinste Mak-rocyclus (cyclisches Monomer oder cyclisches Dimer). Hin-gegen sind bei formtreuen Makrocyclen kleine Ringe wegender hohen Winkelspannung oft benachteiligt, sodass sich dieinteressante M�glichkeit bietet, große Ringe thermodyna-misch kontrolliert zu erzeugen.
2. Kinetisch kontrollierte Synthese von formtreuenMakrocyclen
2.1. Cyclooligomerisierung2.1.1. Cyclisierung von Monomeren des AB- und AA-Typs
Homokupplungen (z.B. die Glaser-Reaktion) undKreuzkupplungen haben bei der Synthese von Arylenethi-nylen-Makrocyclen breite Anwendung gefunden. Da dieseReaktionen irreversibel sind, kann die Bildung unerw�nsch-ter Bindungen nicht korrigiert werden, und die Produktver-teilung ist kinetisch bestimmt. Einer der gr�ßten Nachteilebei kinetisch kontrollierten Cyclooligomerisierungen bestehtdarin, dass die Oligomere, die durch Kupplung von difunk-tionellen Monomeren wie Alkinylhalogenarenen entstehen,�ber die gew�nschte Gr�ße des Makrocyclus „hinausschie-ßen“ k�nnen. Ein nachtr#gliches K�rzen der Oligomere istnicht m�glich. Die Auswirkungen dieses „Overshooting“ aufdie Makrocyclen-Synthese k�nnen durch Simulation derReaktionskinetik analysiert werden. Im Folgenden wollen wirSimulationen von Cyclotrimerisierungen und Cyclohexame-risierungen diskutieren.[47]
Man kann zwei Arten von Monomeren betrachten, jenachdem, ob zur Cyclooligomerisierung eine Homokupplungoder eine Kreuzkupplung angewendet wird. Ein difunktio-nelles Monomer AB, das zwei unterschiedliche funktionelleGruppen A und B enth#lt (wobei A nur mit B reagierenkann), geht typischerweise eine Kreuzkupplung ein, wohin-gegen ein difunktionelles Monomer AA, das zwei identischefunktionelle Gruppen A enth#lt (wobei A mit A reagiert),durch Homokupplung reagiert. In Abbildung 5 ist der Re-
aktionsweg der Cyclotrimerisierung gezeigt, und Tabelle 1enth#lt die relevanten Oligomere Mi sowie die cyclischen C3-Produkte. Die Monomere k�nnen schrittweise wachsen (M1
! M2), bis lineare Trimere (M3) oder h�here Oligomereentstanden sind. In der Simulation wird der g�nstigste Fallangenommen, wonach alle linearen Trimere sofort, d.h. ohne
Abbildung 4. Energielandschaft einer Cyclooligomerisierung: Mit zu-nehmendem Umsatz entstehen grDßere Oligomere, die sich in cycli-sche Produkte umwandeln. Die Ringspannung und entropische Fakto-ren bestimmen die bevorzugte RinggrDße, wobei Makrocyclen mit we-niger Monomereinheiten entropisch begCnstigt sind. Ob ein Makrocy-clus gespannt oder spannungsfrei ist, h@ngt von den Bindungswinkelnund der Konformation der Monomere ab. Die EnergielCcke ist dieEnergiedifferenz zwischen dem stabilsten und dem zweitstabilstenProdukt. Von der EnergielCcke h@ngt es ab, ob unter reversiblen Bedin-gungen ein einziges Produkt erhalten werden kann.
Abbildung 5. Reaktionswege bei der Cyclotrimerisierung aus AB- oderAA-Monomeren.
Tabelle 1: MDgliche bimolekulare Kombinationen bei der Cyclotrimeri-sierung von AB- oder AA-Monomeren unter irreversiblen Bedingungen.Da angenommen wird, dass sich M3 sofort in C3 umwandelt, erscheintkeine M3-Spezies in der Tabelle. Mx bezeichnet hDhere Oligomere als M3.
Reaktant M1 M2 Mx
M1 M2 C3 Mx
M2 C3 Mx Mx
Mx Mx Mx Mx
Formtreue MakrocyclenAngewandte
Chemie
4527Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
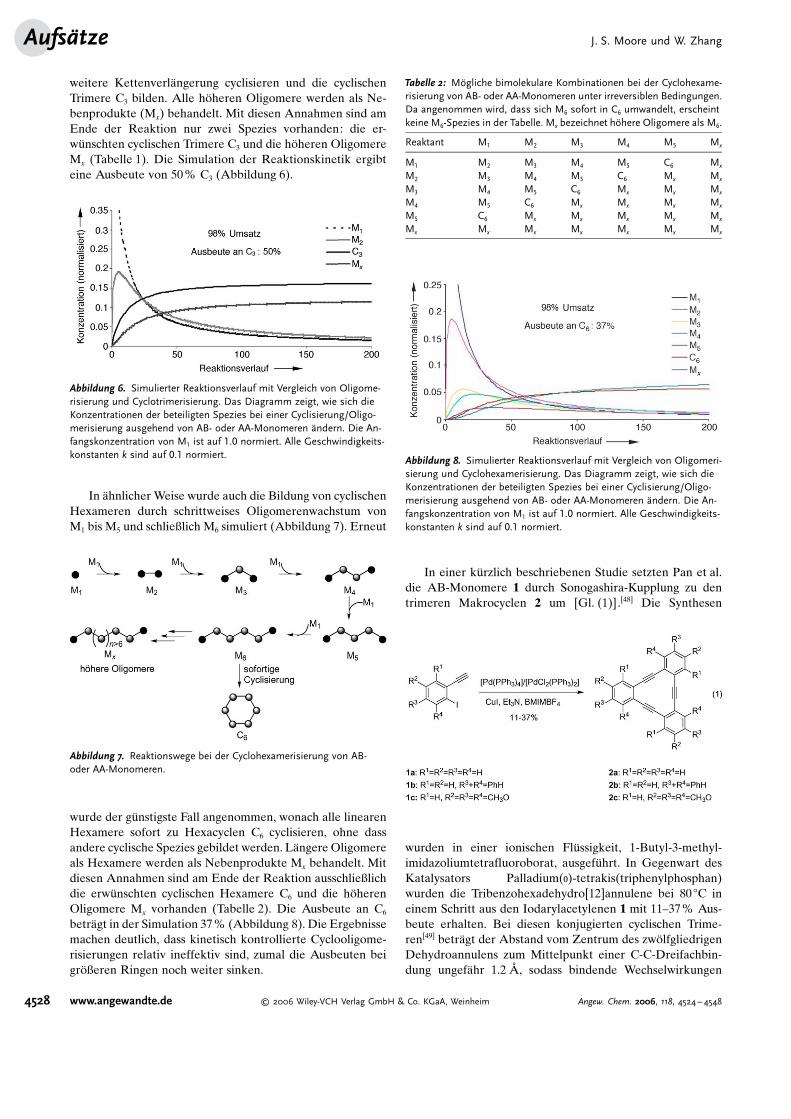
weitere Kettenverl#ngerung cyclisieren und die cyclischenTrimere C3 bilden. Alle h�heren Oligomere werden als Ne-benprodukte (Mx) behandelt. Mit diesen Annahmen sind amEnde der Reaktion nur zwei Spezies vorhanden: die er-w�nschten cyclischen Trimere C3 und die h�heren OligomereMx (Tabelle 1). Die Simulation der Reaktionskinetik ergibteine Ausbeute von 50% C3 (Abbildung 6).
In #hnlicher Weise wurde auch die Bildung von cyclischenHexameren durch schrittweises Oligomerenwachstum vonM1 bis M5 und schließlich M6 simuliert (Abbildung 7). Erneut
wurde der g�nstigste Fall angenommen, wonach alle linearenHexamere sofort zu Hexacyclen C6 cyclisieren, ohne dassandere cyclische Spezies gebildet werden. L#ngere Oligomereals Hexamere werden als Nebenprodukte Mx behandelt. Mitdiesen Annahmen sind am Ende der Reaktion ausschließlichdie erw�nschten cyclischen Hexamere C6 und die h�herenOligomere Mx vorhanden (Tabelle 2). Die Ausbeute an C6
betr#gt in der Simulation 37% (Abbildung 8). Die Ergebnissemachen deutlich, dass kinetisch kontrollierte Cyclooligome-risierungen relativ ineffektiv sind, zumal die Ausbeuten beigr�ßeren Ringen noch weiter sinken.
In einer k�rzlich beschriebenen Studie setzten Pan et al.die AB-Monomere 1 durch Sonogashira-Kupplung zu dentrimeren Makrocyclen 2 um [Gl. (1)].[48] Die Synthesen
wurden in einer ionischen Fl�ssigkeit, 1-Butyl-3-methyl-imidazoliumtetrafluoroborat, ausgef�hrt. In Gegenwart desKatalysators Palladium(0)-tetrakis(triphenylphosphan)wurden die Tribenzohexadehydro[12]annulene bei 80 8C ineinem Schritt aus den Iodarylacetylenen 1 mit 11–37% Aus-beute erhalten. Bei diesen konjugierten cyclischen Trime-ren[49] betr#gt der Abstand vom Zentrum des zw�lfgliedrigenDehydroannulens zum Mittelpunkt einer C-C-Dreifachbin-dung ungef#hr 1.2 N, sodass bindende Wechselwirkungen
Abbildung 6. Simulierter Reaktionsverlauf mit Vergleich von Oligome-risierung und Cyclotrimerisierung. Das Diagramm zeigt, wie sich dieKonzentrationen der beteiligten Spezies bei einer Cyclisierung/Oligo-merisierung ausgehend von AB- oder AA-Monomeren @ndern. Die An-fangskonzentration von M1 ist auf 1.0 normiert. Alle Geschwindigkeits-konstanten k sind auf 0.1 normiert.
Abbildung 7. Reaktionswege bei der Cyclohexamerisierung von AB-oder AA-Monomeren.
Tabelle 2: MDgliche bimolekulare Kombinationen bei der Cyclohexame-risierung von AB- oder AA-Monomeren unter irreversiblen Bedingungen.Da angenommen wird, dass sich M6 sofort in C6 umwandelt, erscheintkeine M6-Spezies in der Tabelle. Mx bezeichnet hDhere Oligomere als M6.
Reaktant M1 M2 M3 M4 M5 Mx
M1 M2 M3 M4 M5 C6 Mx
M2 M3 M4 M5 C6 Mx Mx
M3 M4 M5 C6 Mx Mx Mx
M4 M5 C6 Mx Mx Mx Mx
M5 C6 Mx Mx Mx Mx Mx
Mx Mx Mx Mx Mx Mx Mx
Abbildung 8. Simulierter Reaktionsverlauf mit Vergleich von Oligomeri-sierung und Cyclohexamerisierung. Das Diagramm zeigt, wie sich dieKonzentrationen der beteiligten Spezies bei einer Cyclisierung/Oligo-merisierung ausgehend von AB- oder AA-Monomeren @ndern. Die An-fangskonzentration von M1 ist auf 1.0 normiert. Alle Geschwindigkeits-konstanten k sind auf 0.1 normiert.
J. S. Moore und W. ZhangAufs�tze
4528 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

zwischen den p-Ethinylengruppen und einem Ibergangs-metall m�glich sind. Ein Metallzentrum kann dabei entwederinnerhalb oder außerhalb der Ebene des Makrocyclus koor-dinieren. Die strukturellen Merkmale und Leitf#higkeitsei-genschaften solcher Ibergangsmetallkomplexe sind erforschtworden.[50]
Komatsu und Mitarbeiter verwendeten ebenfalls eineCyclooligomerisierungsstrategie, um das trimere Hexadehy-dro[18]annulen 4 in einem Schritt durch kupfervermittelteoxidative Kupplung des AA-Monomers 1,2-Diethinylaren 3herzustellen [Gl. (2)].[51] Unter Eglinton-Bedingungen wurde
der Makrocyclus 4 mit 38% Ausbeute erhalten. Die Be-handlung von 4 mit einem großen Iberschuss an Cerammo-niumnitrat (CAN) ergab das p-Benzochinon-Derivat 5.Dieses enth#lt sowohl p-Elektronendonor- als auch Elektro-nenacceptorgruppen und kann als Push-pull-System be-trachtet werden,[52] allerdings wiesen UV/Vis-spektroskopi-sche Messungen und Cyclovoltammetrie (CV) auf einen nurgeringen intramolekularen Ladungstransfer hin.
Die erste Synthese von hexameren Arylenethinylen-Makrocyclen aus AB-Monomeren liegt schon 30 Jahrezur�ck. In einer klassischen Arbeit stellten damals Staab undNeunhoeffer den hexameren Phenylenethinylen-Makrocy-clus 7 durch eine sechsfache Stephens-Castro-Kupplung desKupfersalzes von m-Iodphenylacetylen 6 in 4.6% Ausbeuteher [Gl. (3)].[53] Diese Methode war ein fr�hes Beispiel f�r dieAnwendung einer Cyclooligomerisierungsstrategie zur Her-stellung von formtreuen Makrocyclen. Das Monomer w#chstzu offenkettigen Oligomeren, und die Cyclisierung verl#uft inKonkurrenz zur Kettenverl#ngerung. Die geringe Ausbeuteist wahrscheinlich auf das Overshooting-Problem und m�gli-cherweise auch auf die schlechte L�slichkeit der wachsendenOligomere zur�ckzuf�hren.
Die Simulationen und experimentellen Ergebnisse bele-gen, dass die Ausbeute des gew�nschten Makrocyclus mit
zunehmender Ringgr�ße stark abf#llt. Aus diesem Grund istdie irreversible Cyclooligomerisierung keine sehr geeigneteMethode zur Synthese von Makrocyclen mit vielen Mono-mereinheiten.
2.1.2. Cyclooligomerisierung von Monomeren des A2- und B2-Typs
In Abbildung 9 sind die Reaktionswege dargestellt, diezur Bildung von Cyclohexameren aus einemMonomerenpaarA2 + B2 f�hren. Zur Simulation der Reaktionskinetik
wurden dieselben Annahmen zugrunde gelegt, die im vorigenAbschnitt f�r die AB-Szenarien genannt wurden: Sobald sichein lineares Hexamer AM6B gebildet hat, cyclisiert es zu C6,und es werden keine anderen cyclischen Spezies erzeugt;h�here Oligomere als das Hexamer werden als Nebenpro-dukte behandelt. Am Ende der Reaktion liegen zweiHauptprodukte vor: die erw�nschten cyclischen HexamereC6 und h�here Oligomere (Tabelle 3, Mx umfasst AMxB,AMxAund BMxB).Wie erwartet, f�hrten die Simulationen zu#hnlichen Ergebnissen wie bei den AB-Monomeren. Die Si-mulation ergab eine Ausbeute an C6 von 36% (Abbil-dung 10). Auch in diesem Fall beeintr#chtigt die Bildung vonh�heren Oligomeren die Ausbeute der Makrocyclensynthese.
Iyoda et al. synthetisierten Arylenethinylen-Makrocyclendurch eine palladiumkatalysierte Sonogashira-Kupplung vondrei A2- und drei B2-Monomereinheiten [Gl. (4)].[54] Bei derdirekten Umsetzung von o-Diiodbenzol-Derivaten 8 (A2-Monomer) mit Acetylengas (B2-Monomer) in Gegenwart von[PdCl2(PPh3)2] und CuI bei 60 oder 40 8C entstanden die
Abbildung 9. Reaktionswege bei der Oligomerisierung und Cyclo-hexamerisierung von A2- und B2-Monomeren.
Formtreue MakrocyclenAngewandte
Chemie
4529Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
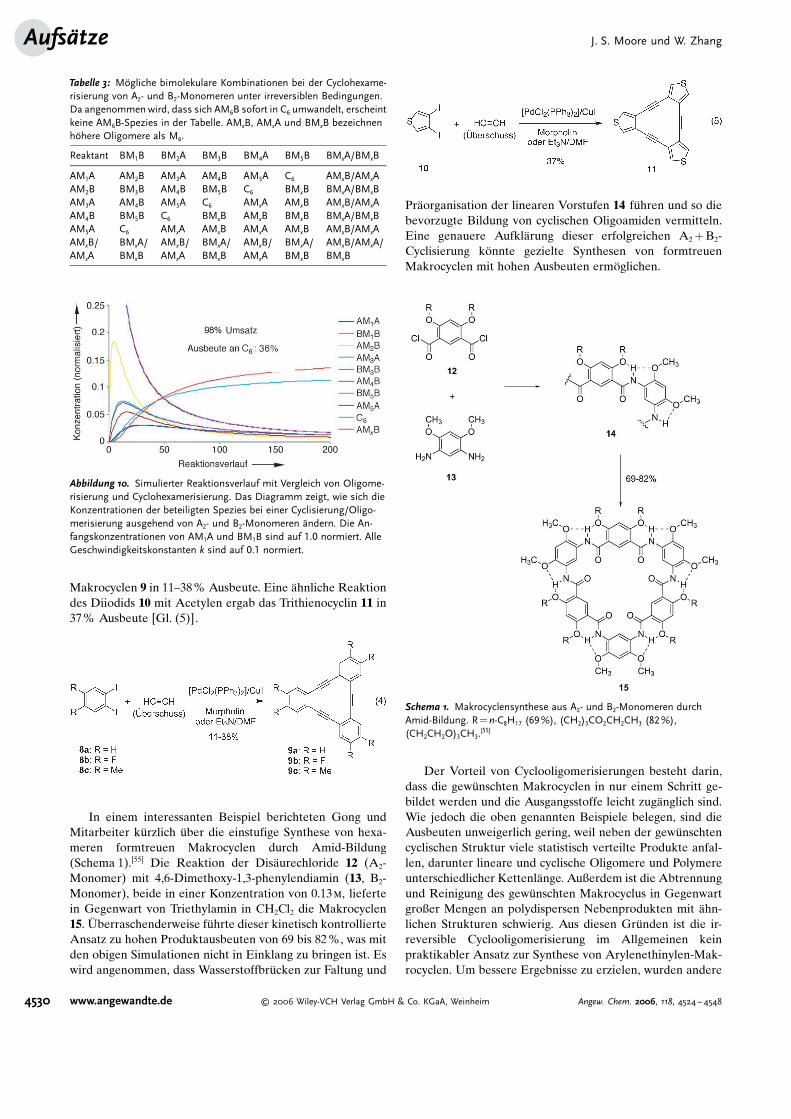
Makrocyclen 9 in 11–38% Ausbeute. Eine #hnliche Reaktiondes Diiodids 10 mit Acetylen ergab das Trithienocyclin 11 in37% Ausbeute [Gl. (5)].
In einem interessanten Beispiel berichteten Gong undMitarbeiter k�rzlich �ber die einstufige Synthese von hexa-meren formtreuen Makrocyclen durch Amid-Bildung(Schema 1).[55] Die Reaktion der Dis#urechloride 12 (A2-Monomer) mit 4,6-Dimethoxy-1,3-phenylendiamin (13, B2-Monomer), beide in einer Konzentration von 0.13m, liefertein Gegenwart von Triethylamin in CH2Cl2 die Makrocyclen15. Iberraschenderweise f�hrte dieser kinetisch kontrollierteAnsatz zu hohen Produktausbeuten von 69 bis 82%, was mitden obigen Simulationen nicht in Einklang zu bringen ist. Eswird angenommen, dass Wasserstoffbr�cken zur Faltung und
Pr#organisation der linearen Vorstufen 14 f�hren und so diebevorzugte Bildung von cyclischen Oligoamiden vermitteln.Eine genauere Aufkl#rung dieser erfolgreichen A2+B2-Cyclisierung k�nnte gezielte Synthesen von formtreuenMakrocyclen mit hohen Ausbeuten erm�glichen.
Der Vorteil von Cyclooligomerisierungen besteht darin,dass die gew�nschten Makrocyclen in nur einem Schritt ge-bildet werden und die Ausgangsstoffe leicht zug#nglich sind.Wie jedoch die oben genannten Beispiele belegen, sind dieAusbeuten unweigerlich gering, weil neben der gew�nschtencyclischen Struktur viele statistisch verteilte Produkte anfal-len, darunter lineare und cyclische Oligomere und Polymereunterschiedlicher Kettenl#nge. Außerdem ist die Abtrennungund Reinigung des gew�nschten Makrocyclus in Gegenwartgroßer Mengen an polydispersen Nebenprodukten mit #hn-lichen Strukturen schwierig. Aus diesen Gr�nden ist die ir-reversible Cyclooligomerisierung im Allgemeinen keinpraktikabler Ansatz zur Synthese von Arylenethinylen-Mak-rocyclen. Um bessere Ergebnisse zu erzielen, wurden andere
Tabelle 3: MDgliche bimolekulare Kombinationen bei der Cyclohexame-risierung von A2- und B2-Monomeren unter irreversiblen Bedingungen.Da angenommen wird, dass sich AM6B sofort in C6 umwandelt, erscheintkeine AM6B-Spezies in der Tabelle. AMxB, AMxA und BMxB bezeichnenhDhere Oligomere als M6.
Reaktant BM1B BM2A BM3B BM4A BM5B BMxA/BMxB
AM1A AM2B AM3A AM4B AM5A C6 AMxB/AMxAAM2B BM3B AM4B BM5B C6 BMxB BMxA/BMxBAM3A AM4B AM5A C6 AMxA AMxB AMxB/AMxAAM4B BM5B C6 BMxB AMxB BMxB BMxA/BMxBAM5A C6 AMxA AMxB AMxA AMxB AMxB/AMxAAMxB/AMxA
BMxA/BMxB
AMxB/AMxA
BMxA/BMxB
AMxB/AMxA
BMxA/BMxB
AMxB/AMxA/BMxB
Abbildung 10. Simulierter Reaktionsverlauf mit Vergleich von Oligome-risierung und Cyclohexamerisierung. Das Diagramm zeigt, wie sich dieKonzentrationen der beteiligten Spezies bei einer Cyclisierung/Oligo-merisierung ausgehend von A2- und B2-Monomeren @ndern. Die An-fangskonzentrationen von AM1A und BM1B sind auf 1.0 normiert. AlleGeschwindigkeitskonstanten k sind auf 0.1 normiert.
Schema 1. Makrocyclensynthese aus A2- und B2-Monomeren durchAmid-Bildung. R=n-C8H17 (69%), (CH2)3CO2CH2CH3 (82%),(CH2CH2O)3CH3.
[55]
J. S. Moore und W. ZhangAufs�tze
4530 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548
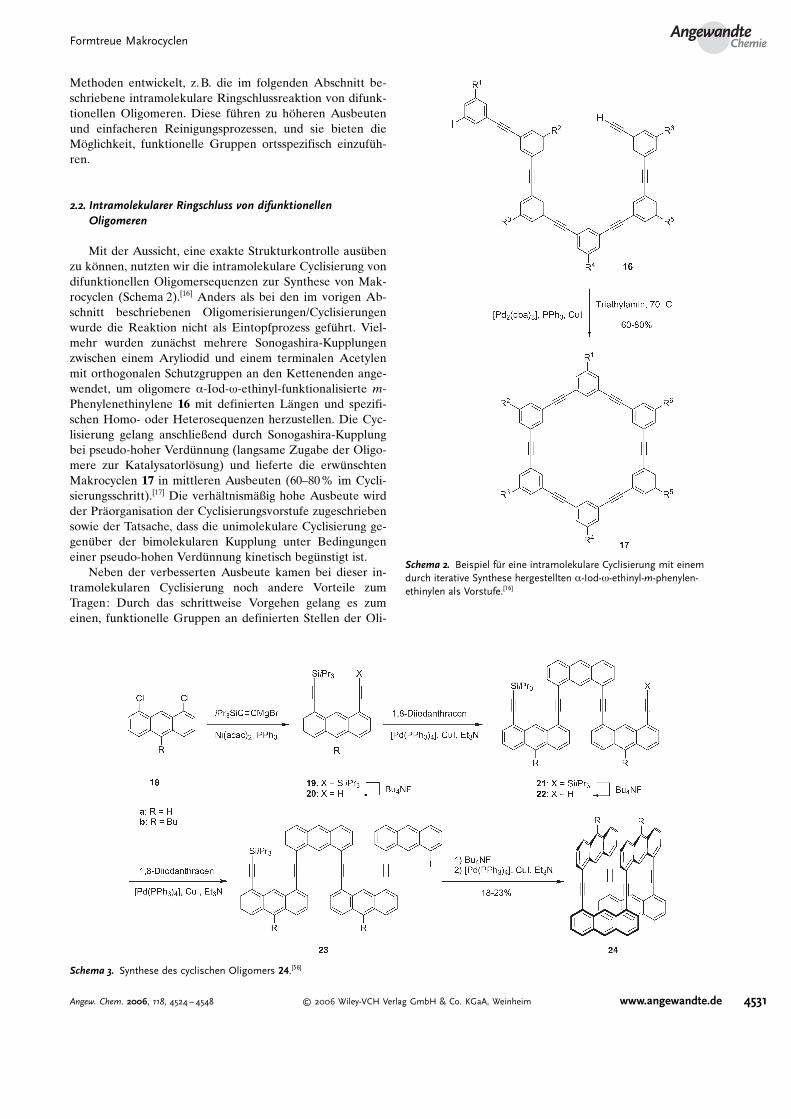
Methoden entwickelt, z.B. die im folgenden Abschnitt be-schriebene intramolekulare Ringschlussreaktion von difunk-tionellen Oligomeren. Diese f�hren zu h�heren Ausbeutenund einfacheren Reinigungsprozessen, und sie bieten dieM�glichkeit, funktionelle Gruppen ortsspezifisch einzuf�h-ren.
2.2. Intramolekularer Ringschluss von difunktionellenOligomeren
Mit der Aussicht, eine exakte Strukturkontrolle aus�benzu k�nnen, nutzten wir die intramolekulare Cyclisierung vondifunktionellen Oligomersequenzen zur Synthese von Mak-rocyclen (Schema 2).[16] Anders als bei den im vorigen Ab-schnitt beschriebenen Oligomerisierungen/Cyclisierungenwurde die Reaktion nicht als Eintopfprozess gef�hrt. Viel-mehr wurden zun#chst mehrere Sonogashira-Kupplungenzwischen einem Aryliodid und einem terminalen Acetylenmit orthogonalen Schutzgruppen an den Kettenenden ange-wendet, um oligomere a-Iod-w-ethinyl-funktionalisierte m-Phenylenethinylene 16 mit definierten L#ngen und spezifi-schen Homo- oder Heterosequenzen herzustellen. Die Cyc-lisierung gelang anschließend durch Sonogashira-Kupplungbei pseudo-hoher Verd�nnung (langsame Zugabe der Oligo-mere zur Katalysatorl�sung) und lieferte die erw�nschtenMakrocyclen 17 in mittleren Ausbeuten (60–80% im Cycli-sierungsschritt).[17] Die verh#ltnism#ßig hohe Ausbeute wirdder Pr#organisation der Cyclisierungsvorstufe zugeschriebensowie der Tatsache, dass die unimolekulare Cyclisierung ge-gen�ber der bimolekularen Kupplung unter Bedingungeneiner pseudo-hohen Verd�nnung kinetisch beg�nstigt ist.
Neben der verbesserten Ausbeute kamen bei dieser in-tramolekularen Cyclisierung noch andere Vorteile zumTragen: Durch das schrittweise Vorgehen gelang es zumeinen, funktionelle Gruppen an definierten Stellen der Oli-
Schema 2. Beispiel fCr eine intramolekulare Cyclisierung mit einemdurch iterative Synthese hergestellten a-Iod-w-ethinyl-m-phenylen-ethinylen als Vorstufe.[16]
Schema 3. Synthese des cyclischen Oligomers 24.[56]
Formtreue MakrocyclenAngewandte
Chemie
4531Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

gomersequenz einzuf�hren. Damit ist es m�glich, eine vor-gegebene Struktur gezielt herzustellen. Eine kontrollierteEinf�hrung von ortho-, meta- und para-Phenylenethinylen-Fragmenten in die Oligomersequenz erlaubt es außerdem,Makrocyclen mit unterschiedlichen geometrischen Formen zuerzeugen. Ein zweiter Vorteil dieses Vorstufenansatzes be-steht darin, dass er mit einer Vielzahl von Kupplungsmetho-den kombiniert werden kann.[18,28–32]
Toyota et al. setzten eine sequenzielle Kupplung/Desily-lierung ein, um die makrocyclischen 1,8-Anthrylenethinylene24 zu synthetisieren (Schema 3).[56] Ausgehend von einemdichlorierten Anthracen 18 wurde das monosilylierte 1,8-Di-ethinylanthracen 20 durch eine Ni-katalysierte Kupplung[57]
und anschließende partielle Desilylierung hergestellt. DieSonogashira-Kupplung von 20 mit 1,8-Diiodanthracen undanschließende Desilylierung lieferten das Trimer 22, das miteinem Iberschuss an 1,8-Diiodanthracen zum Tetramer 23,der Vorstufe des cyclischen tetrameren Zielprodukts, gekup-pelt wurde. Desilylierung und Kupplung von 23 wurden ineinem Eintopfprozess in THF ausgef�hrt, wobei 23% (24a)und 18% (24b) Ausbeute erzielt wurden. Die Lage der Ab-sorptionsmaxima von acyclischem 23a und cyclischem 24a istfast identisch, allerdings weist 24a das schw#chere molareAbsorptionsverm�gen auf.[58] 24a zeigt eine intensive undbreite Emission bei lmax= 478 nm, wohingegen die acycli-schen Oligomere relativ scharfe und strukturierte Bandenaufweisen. Diese charakteristischen Spektren sind den p-p-Wechselwirkungen zwischen den Anthracenchromophorenzuzuordnen. Die Synthese von 24 k�nnte zu einer allgemei-nen Methode zur gezielten Herstellung von aromatischenVerbindungen mit vielf#ltigen Molek�lstrukturen ausgear-beitet werden, indem man den Oligomerisierungsgrad unddie Art der aromatischen Bausteine variiert („panel/rodmethod“).
Yoshida und Mitarbeiter verwendeten die Methode derintramolekularen Cyclisierung zur Synthese von m-Cyclinen(28) (Schema 4).[59] Die intermolekulare Kupplung des ter-minalen Acetylens 25 mit dem Aryliodid 26 und der an-schließende Austausch der N,N-Dialkyltriazengruppe gegeneine Iodgruppe durch Reaktion mit Methyliodid[60] liefertedie gew�nschte lineare Vorstufe 27. Diese wurde bei hoherVerd�nnung durch intramolekulare Kupplung in das er-w�nschte Cyclin 28 �berf�hrt. Die Azamakrocyclen 28 habenDurchmesser von ca. 1 nm und weisen starke lichtemittie-rende Eigenschaften auf.[30, 61] Interessanterweise kann derMakrocyclus 28b zwei Molek�le [CuII(hfac)2] (hfac=1,1,1,5,5,5-Hexafluor-2,4-pentandion) koordinieren. Der ent-stehende pentakoordinierte CuII-Komplex ist �berraschen-derweise stark fluoreszierend, sodass es lohnend erscheint,auch andere Ibergangsmetallkomplexe der m-Cycline 28 alsm�gliche Lumineszenzmaterialien zu untersuchen.
Haley et al. nutzten diese Vorstufenstrategie, um die Se-lektivit#tsunterschiede zwischen Pd- und Cu-katalysiertenCyclisierungen zu untersuchen (Schema 5).[62] Zun#chstwurde das Polyin 30 durch eine vierfache Kreuzkupplung desDiins 29 mit 1,2,4,5-Tetraiodbenzol aufgebaut. Um zu unter-suchen, ob entweder Bis[15]annulene 31 (durch Cyclisierungder meta-st#ndigen Diin-Einheiten) oder Bis[14]annulene 32(durch Cyclisierung der ortho-st#ndigen Diin-Einheiten) er-
zeugt werden k�nnen, wurde die Vorstufe unter Glaser-Be-dingungen mit dem Katalysator Cu(OAc)2 umgesetzt. Hier-bei wurde 31 als das einzige nennenswerte cyclische Produktin einer Ausbeute von 70% isoliert. Mit einem Pd-Katalysa-tor hingegen wurde 32 in 84% Ausbeute erhalten. Die Se-lektivit#t wird damit erkl#rt, dass bei der Cu- (A) und der Pd-vermittelten Reaktion (B) unterschiedliche Metallacetylid-Intermediate entstehen (Schema 6). Es wird angenommen,dass das Cu-haltige Intermediat vor der reduktiven Elimi-nierung eine pseudo-trans-Konfiguration einnimmt,[63] diemanche Systeme in eine hochgradig gespannte Konfigurationzwingt, um eine Homokupplung einzugehen. Pd-Komplexesind andererseits in der Lage, Liganden auf sehr flexible undvielf#ltige Weise zu koordinieren, sodass sowohl trans- alsauch cis-Konfigurationen m�glich sind.
2.3. Intermolekulare Eintopfkupplung mit anschließenderunimolekularer Cyclisierung
Der intramolekulare Ringschluss bietet hohe Ausbeutenund beste M�glichkeiten der Strukturkontrolle. Wegen der
Schema 4. Synthese der m-Cycline28 : a) [PdCl2(PPh3)2] , CuI, Et3N/THF, 53% (27aa), 62% (27ba);b) 1. I2, CH2ClCH2Cl, 2. KOH,MeOH, CHCl3, 3. [PdCl2(PPh3)2] ,CuI, Et3N/THF; Ausb. <5% (28a,drei Stufen), 35% (28b, dreiStufen); c) Na, BuOH, CHCl3, 95%(28c).[59]
J. S. Moore und W. ZhangAufs�tze
4532 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

arbeitsintensiven und zeitaufw#ndigen Synthese und Reini-gung der Vorstufen ist diese Methode aber zur Herstellunggroßer Produktmengen wenig geeignet. Aus diesem Grundsuchte man nach einer verbesserten Strategie, die die Vorteileder beiden bisher beschriebenen Ans#tze vereint und derenNachteile vermeidet.[23, 28,64–66] Es wurde entdeckt, dass zwei
oligomere Vorstufen mit entsprechender Kettenl#nge undgeeigneten Endgruppen eine intermolekulare Kupplung mitanschließender intramolekularer Cyclisierung in einer Ein-topfreaktion eingehen k�nnen, wobei die erw�nschten Ma-krocyclen in mittleren Ausbeute entstehen. Obwohl dieAusbeuten in diesem Schritt meist geringer sind als bei derintramolekularen Oligomercyclisierung, liegt die Gesamt-ausbeute oft h�her, da weniger Syntheseschritte zum Aufbauder Vorstufen erforderlich sind.
H�ger und Enkelmann verwendeten diese Strategie zurSynthese des formtreuen amphiphilen Makrocyclus 37,dessen polare (in der THP-gesch�tzten Form) und nichtpo-lare Funktionalit#ten wechselnde Anordungen bilden k�nnen(Schema 7).[23] Die palladiumkatalysierte Kupplung von 33mit 3,5-Diiodtoluol ergab nach Abspaltung der SchutzgruppeTriisopropylsilylacetylen (TIPS) das Tetrain 34, das mit 3-Brom-5-iodtoluol eine weitere Kupplung zum Dibromid 35einging. Eine Sonogashira-Kupplung von 35 mit Trimethyl-silylacetylen (TMSA) lieferte nach Abspaltung der TMS-Gruppe die Bisacetylenvorstufe 36, die bei hoher Verd�n-nung durch Eglinton-Glaser-Kupplung zum THP-gesch�tztenMakrocyclus 37 in 45% Ausbeute umgesetzt wurde. Durchs#urekatalysierte Abspaltung der Schutzgruppe von 37 ent-stand 38 mit nahezu quantitativer Ausbeute. Der Mak-rocyclus 38 ist das erste Beispiel einer amphiphilen form-treuen Struktur. Eine relativ freie Rotation um die Diphe-nylacetyleneinheiten f�hrt dazu, dass sich entweder ein hy-drophiler oder ein hydrophober Innenraum bilden kann.
Mit einer #hnlichen Methode synthetisierten Cho et al.einen mit Dibutylamino- und Butyloxygruppen funktionali-sierten Phenylenethinylen-Makrocyclus (Schema 8).[67,68] DasDiiodid 40 wurde durch Kupplung von 1-Butoxy-4-dibutyl-amino-2,6-diiodbenzol mit 39 im Verh#ltnis 2:1 erhalten. DieReaktion von 40 mit TMSA und die anschließende Abspal-tung der TMS-Gruppe unter basischen Bedingungen liefertendas terminale Acetylen 41, das mit 40 in einer Sonogashira-Kupplung zum gew�nschten Makrocyclus 42 umgesetztwurde (15% Ausbeute). Bei Untersuchungen der photophy-sikalischen Eigenschaften wurde nachgewiesen,[67] dass 42eine Zweiphotonenabsorption eingeht. Die Donorgruppenam Makrocyclus erh�hen die langwelligen Absorptionsma-xima und vergr�ßern die Stokes-Verschiebung. Generellwerden in polareren L�sungsmitteln ebenfalls gr�ßereStokes-Verschiebungen beobachtet, was darauf hinweist, dassim angeregten Zustand des Makrocyclus 42 ein erheblicherLadungstransfer stattfindet.
K�rzlich stellten Baxter und Mitarbeiter einen neuenDehydroannulen-Makrocyclus 47 vor, der ein konjugiertesspiralf�rmiges Ger�st aus heterocyclischen Thiophen-(Elektronendonoren) und Pyridineinheiten (Elektronenac-ceptoren) aufweist (Schema 9).[69] Eine Sonogashira-Kupp-lung von 44 mit 2 Rquivalenten 43 und die anschließendeDesilylierung ergaben das Dialkin 46 in 88% Ausbeute.Dieses Oligomer wurde durch Eglinton-Galbraith-Ethin-kupplung bei mittlerer Verd�nnung und mit einem Iber-schuss an Cu2(OAc)4 cyclisiert, wobei der Makrocyclus 47 in46% Ausbeute entstand. Es wurde festgestellt, dass dieserMakrocyclus f�r spezielle Metallionen wie AgI als ein selek-tiver F#llungs- und Fluoreszenzsensor fungieren kann[70–72]
Schema 5. a) 1,2,4,5-Tetraiodbenzol, [Pd(PPh3)4] , CuI, iPr2NH, THF,40 8C; b) Bu4NF, MeOH, THF; dann Cu(OAc)2, Pyridin, 60 8C;c) Bu4NF, MeOH, THF; dann [PdCl2(PPh3)2] , CuI, I2, iPr2NH, THF,50 8C; d) Bu4NF, MeOH, THF; dann [PdCl2(dppe)], CuI, I2, iPr2NH,THF, 50 8C.[62]
Schema 6. Vorgeschlagene Metallacetylid-Intermediate bei der Cu-ver-mittelten (A) und der Pd-katalysierten Bildung von Diacetylenen (B).[63]
Formtreue MakrocyclenAngewandte
Chemie
4533Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
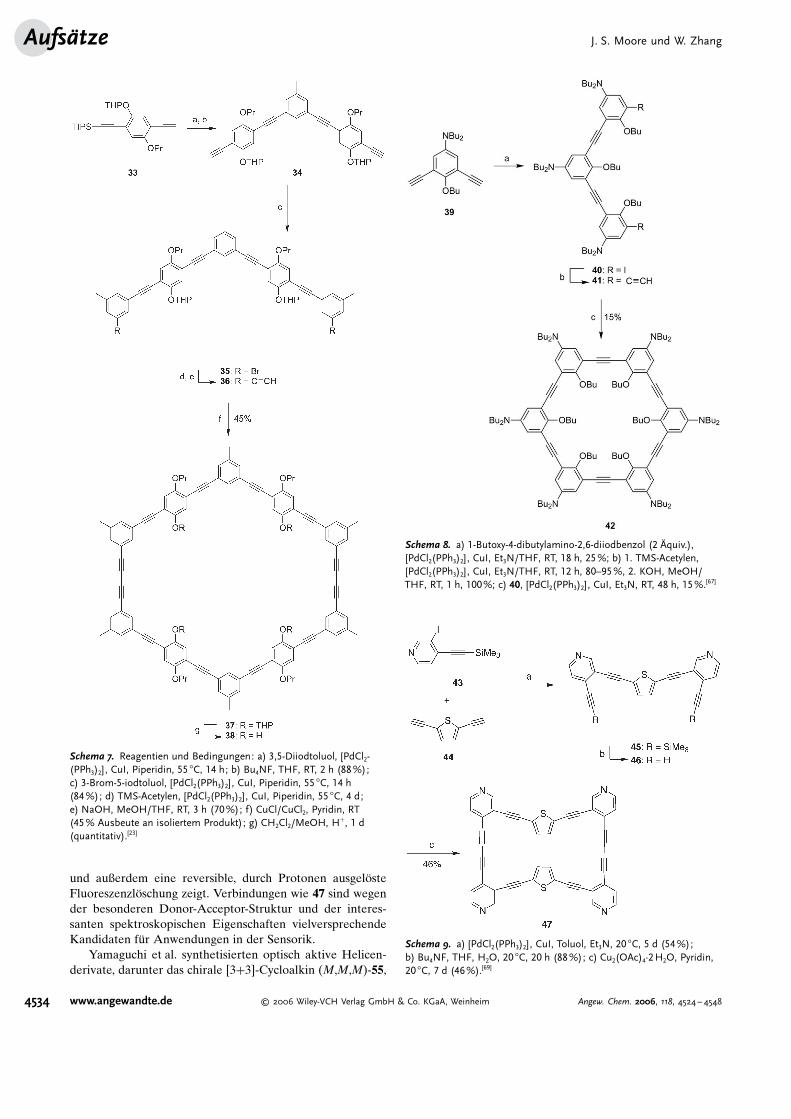
und außerdem eine reversible, durch Protonen ausgel�steFluoreszenzl�schung zeigt. Verbindungen wie 47 sind wegender besonderen Donor-Acceptor-Struktur und der interes-santen spektroskopischen Eigenschaften vielversprechendeKandidaten f�r Anwendungen in der Sensorik.
Yamaguchi et al. synthetisierten optisch aktive Helicen-derivate, darunter das chirale [3+3]-Cycloalkin (M,M,M)-55,
Schema 7. Reagentien und Bedingungen: a) 3,5-Diiodtoluol, [PdCl2-(PPh3)2] , CuI, Piperidin, 55 8C, 14 h; b) Bu4NF, THF, RT, 2 h (88%);c) 3-Brom-5-iodtoluol, [PdCl2(PPh3)2] , CuI, Piperidin, 55 8C, 14 h(84%); d) TMS-Acetylen, [PdCl2(PPh3)2] , CuI, Piperidin, 55 8C, 4 d;e) NaOH, MeOH/THF, RT, 3 h (70%); f) CuCl/CuCl2, Pyridin, RT(45% Ausbeute an isoliertem Produkt); g) CH2Cl2/MeOH, H+, 1 d(quantitativ).[23]
Schema 8. a) 1-Butoxy-4-dibutylamino-2,6-diiodbenzol (2 Oquiv.),[PdCl2(PPh3)2] , CuI, Et3N/THF, RT, 18 h, 25%; b) 1. TMS-Acetylen,[PdCl2(PPh3)2] , CuI, Et3N/THF, RT, 12 h, 80–95%, 2. KOH, MeOH/THF, RT, 1 h, 100%; c) 40, [PdCl2(PPh3)2] , CuI, Et3N, RT, 48 h, 15%.[67]
Schema 9. a) [PdCl2(PPh3)2] , CuI, Toluol, Et3N, 20 8C, 5 d (54%);b) Bu4NF, THF, H2O, 20 8C, 20 h (88%); c) Cu2(OAc)4·2H2O, Pyridin,20 8C, 7 d (46%).[69]
J. S. Moore und W. ZhangAufs�tze
4534 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

das aus drei (M)-Helicenen und drei m-Phenyleneinheitenbesteht (Schema 10).[73–75] Der Dialdehyd (M)-48wurde in dasBis(dibromolefin) �berf�hrt und anschließend mit einemIberschuss an n-Butyllithium behandelt, wobei das Diace-tylen (M)-49 in 99% Ausbeute entstand. Dieses wurde mit1.1 Rquivalenten n-Butyllithium in 62% Ausbeute in daseinfach gesch�tzte Acetylen (M)-50 umgewandelt. EineSonogashira-Kupplung von (M)-50 mit 5 Rquivalenten desBis(triflat)benzoes#ureesters 51 ergab (M)-52, das mit (M)-49gekuppelt wurde. Anschließend wurde zur trimeren Vorstufe(M,M,M)-53 desilyliert. Die Cyclisierung von 53 mit Diiod-benzol 54 bei hoher Verd�nnung ergab die [3+3]-Cycloalkine(M,M,M)-55 und (M,M,M)-56 in 54 bzw. 40% Ausbeute. Eszeigte sich, dass der Makrocyclus 55 (R=H) in CHCl3 undBenzol ein starkes und selektives dimolekulares Aggregatbildet.[75] Die Aggregation wird den p-p-Wechselwirkungeninnerhalb des nichtplanaren p-Elektronensystems des Heli-cens zugeschrieben. Untersuchungen an Derivaten von(M,M,M)-55 zeigen, dass Spezies mit flexiblen Linkern starkeintramolekulare Aggregate bilden, wohingegen sich Speziesmit starren und linearen Linkern zu dimolekularen Aggre-gaten anordnen.[74] Ausgehend von (M,M,M)-56 wurdenweitere [3+3]-Cycloalkine (R=OTf, ONf, OAc, OH) syn-thetisiert. Dabei wurde festgestellt, dass Derivate mit elek-tronenziehenden Substituenten st#rkere Aggregate bilden alssolche mit elektronenschiebenden Substituenten.[73]
2.4. Templatvermittelte Cyclisierung
Die im vorigen Abschnitt beschriebene Methode derCyclisierung gezielt entworfener Oligomere f�hrte zu deut-lich verbesserten Makrocyclenausbeuten, in manchen F#llenließ die Leistungsf#higkeit der Synthese aber noch zu w�n-schen �brig. Eine weitere Zerlegung der Cyclisierungsvor-
stufen in drei oder mehr Bausteine erwies sich als erfolgloserAnsatz, da geringere Ausbeuten resultierten.[24] Auch unterstarken Verd�nnungsbedingungen lassen sich Oligomerisie-rungen nur teilweise verhindern, außerdem sind großeMengen an L�sungsmittel erforderlich. Um die intramole-kulare Kupplung weiter zu beg�nstigen, d.h. das Verh#ltnisvon cyclischen zu linearen Produkten zu erh�hen, wurdedaher eine templatvermittelte Cyclisierungsstrategie entwor-fen.
Sanders und Anderson waren die ersten, die eine Tem-platmethode zur Synthese von formtreuen Makrocyclen ein-setzten (Schema 11).[76,77] Zun#chst wurde das relativ kleineBisacetylen 57 unter oxidativen Bedingungen cyclisiert,wobei erwartungsgem#ß ein Gemisch von Makrocyclen un-terschiedlicher Gr�ße entstand. F�hrte man die Cyclisierungjedoch in Gegenwart eines geeigneten Templats aus, etwa des4,4’-Bipyridyls 58 oder des Terpyridyls 59, so konnte das cyc-lische Dimer 60 oder das cyclische Trimer 61 in 70% bzw.50% Ausbeute isoliert werden. Die Selektivit#t dieser Re-aktionen wird darauf zur�ckgef�hrt, dass sich die Vorstufenum das als Templat fungierende Pyridylderivat anordnen.
Marsella et al. synthetisierten die thiophenhaltigen Ary-lenethinylen-Makrocyclen 65a–c durch Sonogashira-Kupp-lung des Dialkins 62 mit dem Diiodid 63 (Schema 12).[78] Dieelektronischen Wechselwirkungen zwischen den ArH- undArF-Einheiten
[79] wurden genutzt, um einen Templateffekt zuerzielen. An der Reaktion ist das lineare Intermediat 64 be-teiligt, das eine entscheidende Rolle bei der Makrocyclen-bildung spielt, indem es sowohl als Substrat als auch alsTemplat fungiert. Wie erwartet, wird der Makrocyclus 65c(X=H, Y=F), bei dem die p-p-Stapelung am st#rksten be-g�nstigt ist, mit der h�chsten Ausbeute erhalten (30% f�r 65cgegen�ber 12% und 10% f�r 65a und 65b). Dies war daserste Beispiel f�r eine Synthese von formtreuenMakrocyclen,bei der die elektrostatischen Wechselwirkungen zwischen
Schema 10. a) PPh3, CBr4, CH2Cl2; b) nBuLi, THF; c) nBuLi, Me3SiCl, THF; d) 51, [Pd2(dba)3], CuI, Mes3P, PPh3, Bu4NI, NEt3, DMF; e) 49,[Pd2(dba)3], CuI, Mes3P, PPh3, Bu4NI, NEt3, DMF; f) Bu4NF, THF; g) 54, [Pd2(dba)3], CuI, Mes3P, Bu4NI, NEt3, DMF, Toluol (40–54%).[73,75]
Formtreue MakrocyclenAngewandte
Chemie
4535Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

einer Perfluorphenyl- und einer Phenyleinheit genutztwurden, um einen nichtkovalenten Templateffekt zu erzielen.
Anstatt einer nichtkovalenten Metallkoordination oderelektrostatischen Wechselwirkung nutzten H�ger et al. dieEsterbindung in ihrer Templatsynthese von Phenylenethiny-len-Makrocyclen (Schema 13).[7,24,26] Zun#chst wurde diepolare Alkoholfunktion im Diin 66 als Benzoat 67 gesch�tzt.Wenn 67 unter Verd�nnungsbedingungen der intermoleku-laren Cyclisierung unterworfen wurde, entstand ein Gemischaus haupts#chlich polymeren Produkten zusammen mit cy-clischem Trimer in nur 20–25% Ausbeute. Die Verteilungfolgt in etwa der erwarteten Statistik (siehe vorigen Ab-schnitt). Das templatgebundene Hexaacetylen 68 hingegen,
das ebenfalls aus 66 hergestellt wurde, ergab ein Produkt, daszu mehr als 95% aus dem templatgebundenen cyclischenTrimer 69 bestand. Durch basenkatalysierte Spaltung derEsterbindungen wurde der gew�nschte Makrocyclus 70 dannin nahezu quantitativer Ausbeute erhalten.
Das Konzept der templatvermittelten Cyclisierung be-steht darin, dass alle reaktiven Komponenten in der Reakti-onsmischung in einer geringen Konzentration vorliegen(entsprechend den Bedingungen bei hoher Verd�nnung), bisauf eine hohe lokale Konzentration an terminalen Acetyle-nen (wegen der Pr#organisation der Reaktionsvorstufen),wodurch der Cyclisierungsprozess stark erleichtert wird. Esbesteht die M�glichkeit, die Gr�ße des Makrocyclus �ber dieWahl des Templats zu steuern.[76, 77] Kovalent bindendeTemplate erfordern normalerweise die Einf�hrung speziellerfunktioneller Gruppen in das Substrat und damit zus#tzlicheSyntheseschritte f�r deren Anbringung und Abspaltung. Sogesehen sollten nichtkovalente Template, die reversibelbinden und leicht wieder entfernt werden k�nnen, meist diebessere Alternative sein.
3. Thermodynamisch kontrollierte Synthese vonArylenethinylen-Makrocyclen
Der wesentliche Nachteil der Kupplungsstrategien be-steht darin, dass die Produktverteilung kinetisch bestimmt istund unerw�nschte Bindungsbildungen nicht korrigiertwerden k�nnen. Die Bildung h�herer Oligomere (Over-shooting) ist ein zus#tzliches Problem, das die Ausbeute angew�nschtem Produkt mindert. Meist m�ssen erheblicheAnstrengungen unternommen werden, um die Bildung vonunerw�nschten Oligomeren zu verhindern.
In den vergangenen Jahren wurden Methoden auf derGrundlage von thermodynamisch kontrollierten, nichtkova-lenten Metallkoordinationswechselwirkungen eingef�hrt, umformtreue metallgebundene Makrocyclen in hohen Ausbeu-ten herzustellen.[80] Mit Methoden der dynamischen kova-lenten Chemie (DCC)[37,38] (Iminmetathese[39] und Alkinme-tathese[34,40–42]) gelang z.B. die Synthese von formtreuenArylenethinylen-Makrocyclen in hohen Ausbeuten. Auf-
Schema 11. Synthese der Porphyrinoligomere 60 und 61 unter Verwendung von nichtkovalenten Templaten.[76, 77]
Schema 12. Synthese der thiophenhaltigen Makrocyclen 65. Reaktions-bedingungen: [PdCl2(PPh3)2] , CuI, iPr2NH, THF, 80 8C.[78]
J. S. Moore und W. ZhangAufs�tze
4536 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548
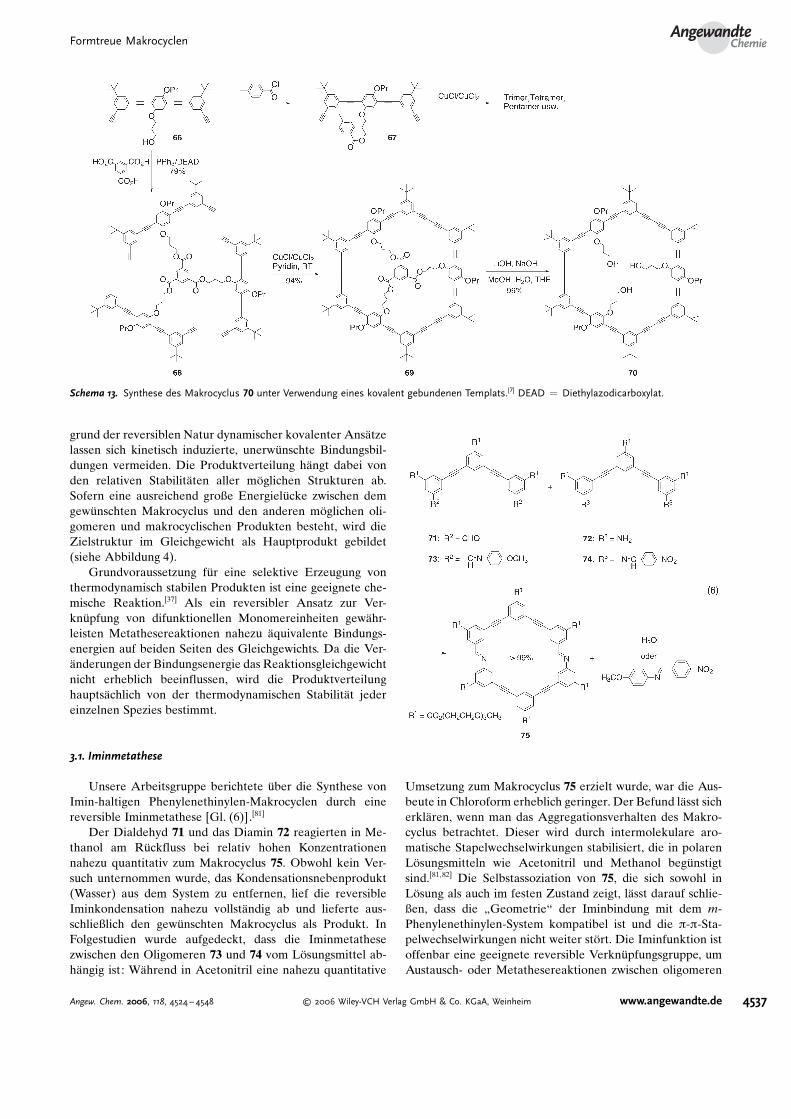
grund der reversiblen Natur dynamischer kovalenter Ans#tzelassen sich kinetisch induzierte, unerw�nschte Bindungsbil-dungen vermeiden. Die Produktverteilung h#ngt dabei vonden relativen Stabilit#ten aller m�glichen Strukturen ab.Sofern eine ausreichend große Energiel�cke zwischen demgew�nschten Makrocyclus und den anderen m�glichen oli-gomeren und makrocyclischen Produkten besteht, wird dieZielstruktur im Gleichgewicht als Hauptprodukt gebildet(siehe Abbildung 4).
Grundvoraussetzung f�r eine selektive Erzeugung vonthermodynamisch stabilen Produkten ist eine geeignete che-mische Reaktion.[37] Als ein reversibler Ansatz zur Ver-kn�pfung von difunktionellen Monomereinheiten gew#hr-leisten Metathesereaktionen nahezu #quivalente Bindungs-energien auf beiden Seiten des Gleichgewichts. Da die Ver-#nderungen der Bindungsenergie das Reaktionsgleichgewichtnicht erheblich beeinflussen, wird die Produktverteilunghaupts#chlich von der thermodynamischen Stabilit#t jedereinzelnen Spezies bestimmt.
3.1. Iminmetathese
Unsere Arbeitsgruppe berichtete �ber die Synthese vonImin-haltigen Phenylenethinylen-Makrocyclen durch einereversible Iminmetathese [Gl. (6)].[81]
Der Dialdehyd 71 und das Diamin 72 reagierten in Me-thanol am R�ckfluss bei relativ hohen Konzentrationennahezu quantitativ zum Makrocyclus 75. Obwohl kein Ver-such unternommen wurde, das Kondensationsnebenprodukt(Wasser) aus dem System zu entfernen, lief die reversibleIminkondensation nahezu vollst#ndig ab und lieferte aus-schließlich den gew�nschten Makrocyclus als Produkt. InFolgestudien wurde aufgedeckt, dass die Iminmetathesezwischen den Oligomeren 73 und 74 vom L�sungsmittel ab-h#ngig ist: W#hrend in Acetonitril eine nahezu quantitative
Umsetzung zum Makrocyclus 75 erzielt wurde, war die Aus-beute in Chloroform erheblich geringer. Der Befund l#sst sicherkl#ren, wenn man das Aggregationsverhalten des Makro-cyclus betrachtet. Dieser wird durch intermolekulare aro-matische Stapelwechselwirkungen stabilisiert, die in polarenL�sungsmitteln wie Acetonitril und Methanol beg�nstigtsind.[81,82] Die Selbstassoziation von 75, die sich sowohl inL�sung als auch im festen Zustand zeigt, l#sst darauf schlie-ßen, dass die „Geometrie“ der Iminbindung mit dem m-Phenylenethinylen-System kompatibel ist und die p-p-Sta-pelwechselwirkungen nicht weiter st�rt. Die Iminfunktion istoffenbar eine geeignete reversible Verkn�pfungsgruppe, umAustausch- oder Metathesereaktionen zwischen oligomeren
Schema 13. Synthese des Makrocyclus 70 unter Verwendung eines kovalent gebundenen Templats.[7] DEAD = Diethylazodicarboxylat.
Formtreue MakrocyclenAngewandte
Chemie
4537Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

m-Phenylenethinylenen verschiedener Gr�ßen und Ger�st-strukturen zu erm�glichen.[39]
MacLachlan et al. verwendeten einen #hnlichen Ansatz,um formtreue konjugierte Makrocyclen zu synthetisieren(Schema 14).[83–85] Der Makrocyclus 78 wurde durch Cyclo-
oligomerisierung von 3,6-Diformylcatechol (76) mit demPhenylendiamin 77 in 70% Ausbeute hergestellt. Es wurdeangenommen, dass die selektive Erzeugung des Makrocyclus78 aus der Reversibilit#t der Iminkondensation resultiert, diedaf�r sorgt, dass bei der Reaktion das thermodynamisch be-g�nstigte, makrocyclische [3+3]-Kondensationsprodukt ge-bildet werden kann. Die Reaktion ist ein Beispiel f�r eineCyclooligomerisierung von A2- mit B2-Monomeren unter re-versiblen Bedingungen.
Um aufzuzeigen, dass das Energiel�ckenprinzip f�r diehohe Ausbeute des [3+3]-Produkts ausschlaggebend ist,haben wir einfache Rechnungen zur thermodynamischenStabilit#t der drei m�glichen makrocyclischen Iminmetathe-seprodukte 78, 79 und 80 durchgef�hrt (Abbildung 11). Wirbetrachteten hierzu die Produktverteilung vom [2+2]-Tetra-cyclus 79 (2 Rquiv. 76 + 2 Rquiv. 77) bis hin zum [4+4]-Octacyclus 80 (4 Rquiv. 76 + 4 Rquiv. 77) und verglichen die
Stabilit#ten dieser Spezies mit der des [3+3]-Hexacyclus 78.Die Entropie kann bei der Cyclooligomerisierung eine ent-scheidende Rolle spielen und beg�nstigt tendenziell diejeni-gen Makrocyclen, die die geringste Zahl an Monomerein-heiten haben. Entropie#nderungen zwischen Makrocyclenk�nnen mit einem halbquantitativen Modell ermitteltwerden,[86] w#hrend Enthalpie#nderungen aus Molek�lme-chanikrechnungen zug#nglich sind (Tabelle 4).[87] Anhand derEntropie- und Enthalpiewerte kann die freie Enthalpie(Gibbs-Energie) der Makrocyclen (n= 0, 1, 2) im Vergleichzum Hexacyclus 78 ermittelt werden [Gl. (7), (8)]. Die
2 cycl: Trimer Ð 3 cycl: Dimer ð7Þ
4 cycl: Trimer Ð 3 cycl: Tetramer ð8Þ
Rechnungen zeigen, dass der [3+3]-Hexacyclus 78 stabiler alsder kleinere Tetracyclus 79 ist, weil er wegen der geringerenWinkelspannung enthalpisch beg�nstigt ist. Gegen�ber demgr�ßeren Octacyclus 80 ist der Hexacyclus 78 dagegen en-tropisch beg�nstigt. Somit ist 78 von allen bei der Iminme-tathese erzeugten Makrocyclen am meisten beg�nstigt.Rechnungen dieser Art k�nnten von großem Wert f�r dieVorhersage reversibler Cyclooligomerisierungen sein.
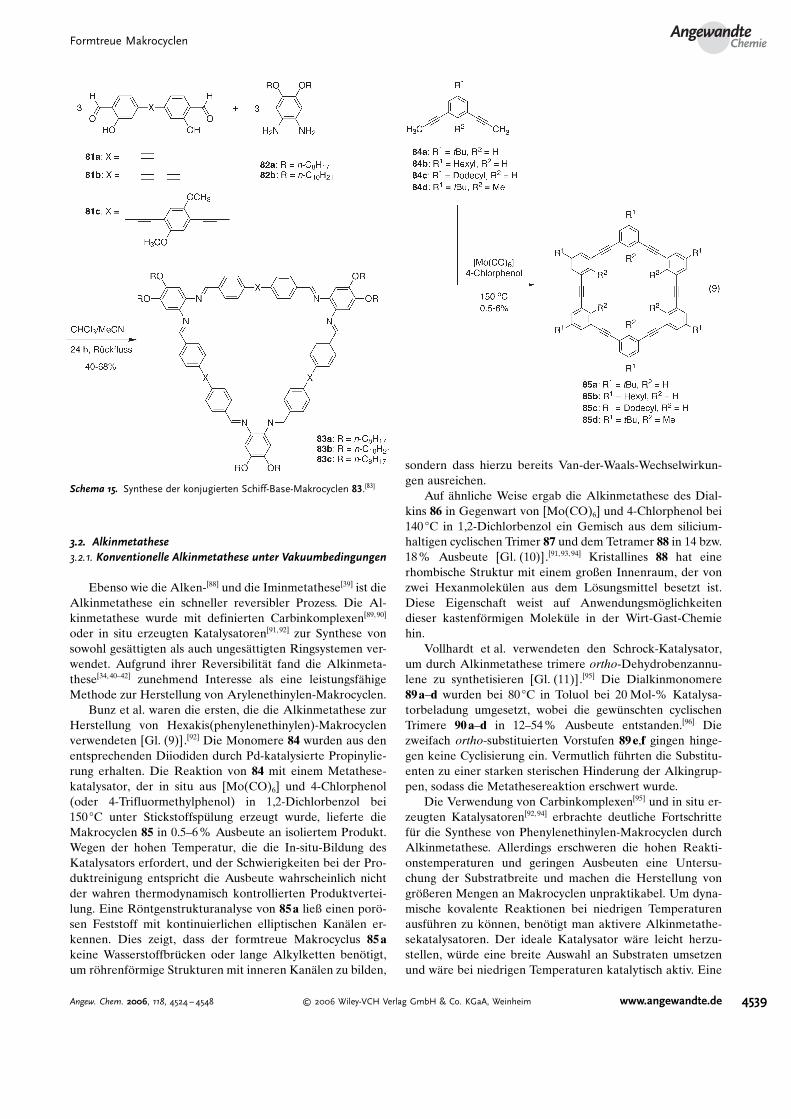
MacLachlan et al. fanden, dass sich der Makrocyclus 78wie ein Kronenether verh#lt, wobei die nach innen gerichte-ten phenolischen Sauerstoffatome kleine Kationen im Zen-trum des Makrocyclus koordinieren.[85] Allerdings ist derDurchmesser relativ klein, sodass der Makrocyclus selbst inKomplexen mit Ibergangsmetallen eine nichtplanare Kon-formation einnimmt. Um große, flache Makrocyclen aufzu-bauen, die sich zu Nanor�hren anordnen k�nnen, verwen-deten MacLachlan und Mitarbeiter die Bausteine 81a–c, dieentweder durch Pd-katalysierte Sonogashira-Kupplung oderdurch Cu-katalysierte oxidative Kupplungmit demDiamin 82in einer Iminkondensation zu den Makrocyclen 83a–c in 40–68% Ausbeute umgesetzt wurden (Schema 15).[83] DurchEinf�hrung von Phenylenethinylen-Abstandhaltern in dasMakrocyclenger�st gelingt es, große planare Makrocyclen miteinstellbaren Durchmessern herzustellen. Die Makrocyclenk�nnen unterschiedliche Metalle komplexieren und so l�sli-che lumineszierende Komplexe bilden, die als supramoleku-lare Materialien und auf Metall-Ligand-Koordination rea-gierende Sensoren genutzt werden k�nnten.
Schema 14. Synthese des konjugierten Schiff-Base-Makrocyclus 78.[85]
Abbildung 11. MDgliche makrocyclische Produkte der Iminmetathese.
Tabelle 4: Onderung der Gibbs-Energien DG in den Reaktionen (7) und(8).
Reaktion DH[kcalmol�1]
DS[kcalmol�1K�1]
DG[kcalmol�1] (363 K)
7 16.8 0.029 6.28 0.39 �0.015 6.0
J. S. Moore und W. ZhangAufs�tze
4538 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548
3.2. Alkinmetathese3.2.1. Konventionelle Alkinmetathese unter Vakuumbedingungen
Ebenso wie die Alken-[88] und die Iminmetathese[39] ist dieAlkinmetathese ein schneller reversibler Prozess. Die Al-kinmetathese wurde mit definierten Carbinkomplexen[89,90]
oder in situ erzeugten Katalysatoren[91,92] zur Synthese vonsowohl ges#ttigten als auch unges#ttigten Ringsystemen ver-wendet. Aufgrund ihrer Reversibilit#t fand die Alkinmeta-these[34, 40–42] zunehmend Interesse als eine leistungsf#higeMethode zur Herstellung von Arylenethinylen-Makrocyclen.
Bunz et al. waren die ersten, die die Alkinmetathese zurHerstellung von Hexakis(phenylenethinylen)-Makrocyclenverwendeten [Gl. (9)].[92] Die Monomere 84 wurden aus denentsprechenden Diiodiden durch Pd-katalysierte Propinylie-rung erhalten. Die Reaktion von 84 mit einem Metathese-katalysator, der in situ aus [Mo(CO)6] und 4-Chlorphenol(oder 4-Trifluormethylphenol) in 1,2-Dichlorbenzol bei150 8C unter Stickstoffsp�lung erzeugt wurde, lieferte dieMakrocyclen 85 in 0.5–6% Ausbeute an isoliertem Produkt.Wegen der hohen Temperatur, die die In-situ-Bildung desKatalysators erfordert, und der Schwierigkeiten bei der Pro-duktreinigung entspricht die Ausbeute wahrscheinlich nichtder wahren thermodynamisch kontrollierten Produktvertei-lung. Eine R�ntgenstrukturanalyse von 85a ließ einen por�-sen Feststoff mit kontinuierlichen elliptischen Kan#len er-kennen. Dies zeigt, dass der formtreue Makrocyclus 85akeine Wasserstoffbr�cken oder lange Alkylketten ben�tigt,um r�hrenf�rmige Strukturen mit inneren Kan#len zu bilden,
sondern dass hierzu bereits Van-der-Waals-Wechselwirkun-gen ausreichen.
Auf #hnliche Weise ergab die Alkinmetathese des Dial-kins 86 in Gegenwart von [Mo(CO)6] und 4-Chlorphenol bei140 8C in 1,2-Dichlorbenzol ein Gemisch aus dem silicium-haltigen cyclischen Trimer 87 und dem Tetramer 88 in 14 bzw.18% Ausbeute [Gl. (10)].[91,93,94] Kristallines 88 hat einerhombische Struktur mit einem großen Innenraum, der vonzwei Hexanmolek�len aus dem L�sungsmittel besetzt ist.Diese Eigenschaft weist auf Anwendungsm�glichkeitendieser kastenf�rmigen Molek�le in der Wirt-Gast-Chemiehin.
Vollhardt et al. verwendeten den Schrock-Katalysator,um durch Alkinmetathese trimere ortho-Dehydrobenzannu-lene zu synthetisieren [Gl. (11)].[95] Die Dialkinmonomere89a–d wurden bei 80 8C in Toluol bei 20 Mol-% Katalysa-torbeladung umgesetzt, wobei die gew�nschten cyclischenTrimere 90a–d in 12–54% Ausbeute entstanden.[96] Diezweifach ortho-substituierten Vorstufen 89e,f gingen hinge-gen keine Cyclisierung ein. Vermutlich f�hrten die Substitu-enten zu einer starken sterischen Hinderung der Alkingrup-pen, sodass die Metathesereaktion erschwert wurde.
Die Verwendung von Carbinkomplexen[95] und in situ er-zeugten Katalysatoren[92,94] erbrachte deutliche Fortschrittef�r die Synthese von Phenylenethinylen-Makrocyclen durchAlkinmetathese. Allerdings erschweren die hohen Reakti-onstemperaturen und geringen Ausbeuten eine Untersu-chung der Substratbreite und machen die Herstellung vongr�ßeren Mengen an Makrocyclen unpraktikabel. Um dyna-mische kovalente Reaktionen bei niedrigen Temperaturenausf�hren zu k�nnen, ben�tigt man aktivere Alkinmetathe-sekatalysatoren. Der ideale Katalysator w#re leicht herzu-stellen, w�rde eine breite Auswahl an Substraten umsetzenund w#re bei niedrigen Temperaturen katalytisch aktiv. Eine
Schema 15. Synthese der konjugierten Schiff-Base-Makrocyclen 83.[83]
Formtreue MakrocyclenAngewandte
Chemie
4539Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
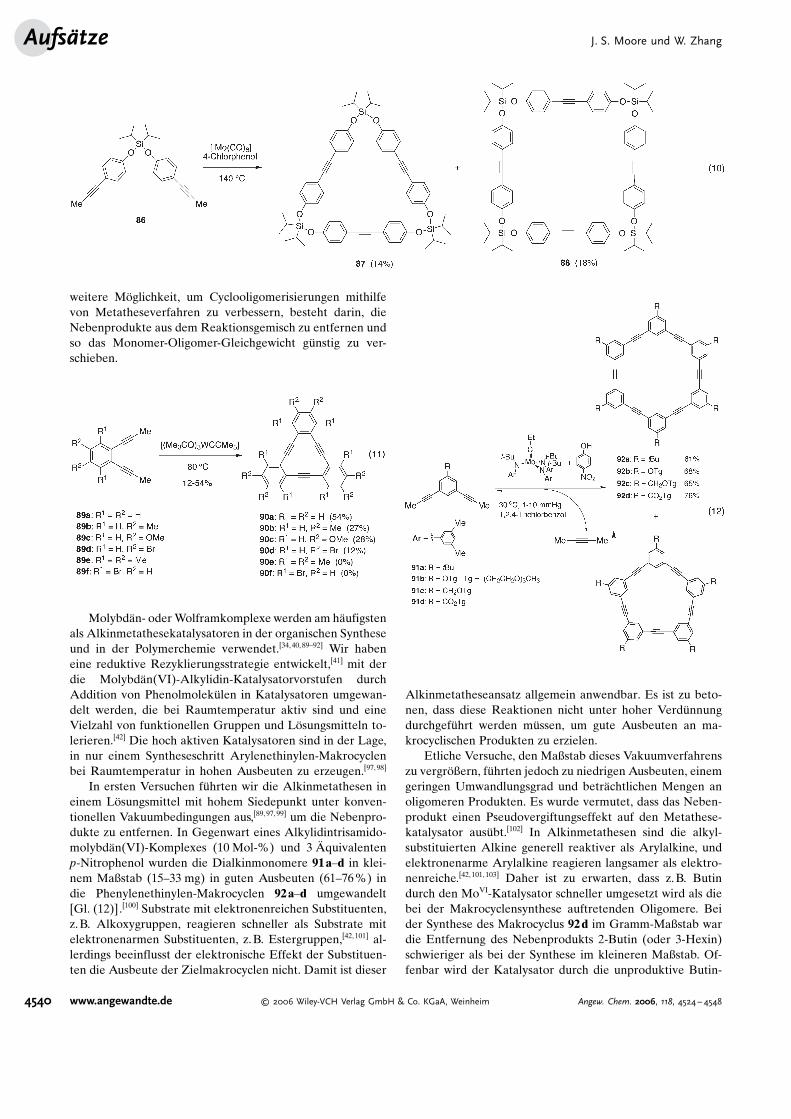
weitere M�glichkeit, um Cyclooligomerisierungen mithilfevon Metatheseverfahren zu verbessern, besteht darin, dieNebenprodukte aus dem Reaktionsgemisch zu entfernen undso das Monomer-Oligomer-Gleichgewicht g�nstig zu ver-schieben.
Molybd#n- oderWolframkomplexe werden am h#ufigstenals Alkinmetathesekatalysatoren in der organischen Syntheseund in der Polymerchemie verwendet.[34,40,89–92] Wir habeneine reduktive Rezyklierungsstrategie entwickelt,[41] mit derdie Molybd#n(VI)-Alkylidin-Katalysatorvorstufen durchAddition von Phenolmolek�len in Katalysatoren umgewan-delt werden, die bei Raumtemperatur aktiv sind und eineVielzahl von funktionellen Gruppen und L�sungsmitteln to-lerieren.[42] Die hoch aktiven Katalysatoren sind in der Lage,in nur einem Syntheseschritt Arylenethinylen-Makrocyclenbei Raumtemperatur in hohen Ausbeuten zu erzeugen.[97,98]
In ersten Versuchen f�hrten wir die Alkinmetathesen ineinem L�sungsmittel mit hohem Siedepunkt unter konven-tionellen Vakuumbedingungen aus,[89, 97,99] um die Nebenpro-dukte zu entfernen. In Gegenwart eines Alkylidintrisamido-molybd#n(VI)-Komplexes (10 Mol-%) und 3 Rquivalentenp-Nitrophenol wurden die Dialkinmonomere 91a–d in klei-nem Maßstab (15–33 mg) in guten Ausbeuten (61–76%) indie Phenylenethinylen-Makrocyclen 92a–d umgewandelt[Gl. (12)].[100] Substrate mit elektronenreichen Substituenten,z.B. Alkoxygruppen, reagieren schneller als Substrate mitelektronenarmen Substituenten, z.B. Estergruppen,[42, 101] al-lerdings beeinflusst der elektronische Effekt der Substituen-ten die Ausbeute der Zielmakrocyclen nicht. Damit ist dieser
Alkinmetatheseansatz allgemein anwendbar. Es ist zu beto-nen, dass diese Reaktionen nicht unter hoher Verd�nnungdurchgef�hrt werden m�ssen, um gute Ausbeuten an ma-krocyclischen Produkten zu erzielen.
Etliche Versuche, den Maßstab dieses Vakuumverfahrenszu vergr�ßern, f�hrten jedoch zu niedrigen Ausbeuten, einemgeringen Umwandlungsgrad und betr#chtlichen Mengen anoligomeren Produkten. Es wurde vermutet, dass das Neben-produkt einen Pseudovergiftungseffekt auf den Metathese-katalysator aus�bt.[102] In Alkinmetathesen sind die alkyl-substituierten Alkine generell reaktiver als Arylalkine, undelektronenarme Arylalkine reagieren langsamer als elektro-nenreiche.[42,101,103] Daher ist zu erwarten, dass z.B. Butindurch den MoVI-Katalysator schneller umgesetzt wird als diebei der Makrocyclensynthese auftretenden Oligomere. Beider Synthese des Makrocyclus 92d im Gramm-Maßstab wardie Entfernung des Nebenprodukts 2-Butin (oder 3-Hexin)schwieriger als bei der Synthese im kleineren Maßstab. Of-fenbar wird der Katalysator durch die unproduktive Butin-
J. S. Moore und W. ZhangAufs�tze
4540 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

metathese so stark in Anspruch genommen, dass er f�r diegew�nschte Kettenverl#ngerung oder Cyclisierung nichtmehr ausreichend zur Verf�gung steht. Letztlich wird derKatalysator durch die Polymerisation des NebenproduktsButin desaktiviert, wenn dessen Konzentration zu hochwird.[42] Diese Ergebnisse zeigen auf, dass dialkylsubstituierteAlkine als Nebenprodukte vermieden oder wirksam entferntwerden sollten, wenn die Metathese von elektronenarmenArylsubstraten mit einem hohen Umsetzungsgrad realisiertwerden soll.
3.2.2. F&llungsvermittelte Alkinmetathese
Um den Vergiftungseffekt zu vermeiden, �berlegten wiruns, dass ein schlecht l�sliches, weniger reaktives Diarylace-tylen wirksam aus dem Reaktionsgemisch entfernt werdenk�nnte, sodass die Metathesegleichgewicht hin zum ge-w�nschten Makrocyclus verschoben wird. Statt der Ver-dampfung im Vakuum sollte daher eine F#llung als treibendeKraft dienen. Wir fanden, dass die Metathese der Benzoyl-biphenyl-substituierten Monomere 93a,b in Gegenwart einesMolybd#n(VI)-propylidin-Katalysators bei 30 8C die Makro-cyclen 94a,b in hohen Ausbeuten und mit geringen Mengenan Pentacyclen liefert [Gl. (13)].[97] Das Diarylacetylen-Ne-
benprodukt ist in vielen L�sungsmitteln extrem schwer l�s-lich, z.B. betr#gt seine L�slichkeit in CCl4 bei 25 8C nur 1.09 T10�6 molL�1. Somit l#sst sich das Nebenprodukt dieser Me-tathese durch F#llung sehr wirksam entfernen. Bei optimier-ten Bedingungen gelang die Synthese von 5.68 g 94a in einemSchritt in 77% Ausbeute. Dies war die erste erfolgreicheSynthese von Hexakisphenylenethinylen-Makrocyclen ingr�ßerem Maßstab.
Die Ergebnisse verdeutlichen die klaren Vorteile desF#llungsansatzes gegen�ber dem konventionellen Vakuum-ansatz. Es sind keine zus#tzlichen Schritte zur Entfernung desNebenproduktes erforderlich, denn dieses f#llt sofort aus undbeeintr#chtigt somit nicht die Aktivit#t des Katalysators. DieF#llung des Nebenprodukts dient somit nicht nur als trei-bende Kraft f�r die Metathesereaktion, sie verhindert au-ßerdem die Pseudovergiftung des Katalysators. Außerdeml#sst sich das Produkt durch einfaches Abfiltrieren und an-schließendes Einengen der L�sung isolieren.
Die allgemeine Anwendbarkeit der F#llungsstrategiewurde durch die erfolgreiche Synthese des quadratisch ge-formten Makrocyclus 96[104] aus dem Dialkin 95 best#tigt[Gl. (14)].[97,105] Als einziges Produkt wurde der Tetracyclus
96 beobachtet, der in 84% Ausbeute isoliert wurde. Zumweiteren Beleg wurde der dreieckf�rmige Makrocyclus 98 ineinem Schritt aus dem Monomer 97 in 86% Ausbeute her-gestellt [Gl. (15)],[106] Die Ausbeute ist deutlich h�her als bei
Kreuzkupplungsreaktionen [2 in Gl. (1), 9 in Gl. (4)]. AlleReaktionen wurden in CCl4 bei 30 8C in abgeschlossenenReaktionsgef#ßen durchgef�hrt. Es ist vorstellbar, dass die
Formtreue MakrocyclenAngewandte
Chemie
4541Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de
reversible f#llungsvermittelte Alkinmetathese einen Zugangzu weiteren zweidimensionalen oder dreidimensionalenArylenethinylen-Strukturen sowie alkinverbr�ckten Oligo-meren und Polymeren bietet.
3.2.3. Thermodynamische Stabilit&t von Arylenethinylen-Makro-cyclen (Energiel(ckenprinzip) und Cyclooligomerenvertei-lung
Eine Reihe von Experimenten wurde durchgef�hrt, umdas mechanistische Prinzip hinter der selektiven Bildung vonArylenethinylen-Makrocyclen durch Alkinmetathese aufzu-kl#ren und um zu belegen, dass die Reaktion wirklich ther-modynamisch kontrolliert ist.[102] Einen Beweis daf�r, dassalle Schritte der Makrocyclenbildung reversibel sind, erhieltman, indem man ein vorab synthetisiertes Phenylenethiny-lenpolymer unter Alkinmetathesebedingungen umsetzte[Gl. (16)]. Im Gelpermeationschromatogramm der Reakti-
onsmischung war zu erkennen, dass sich das Polymer in k�r-zere Oligomere mit dem Hexacyclus 94a als Hauptproduktumgewandelt hatte.
Einen zus#tzlichen Nachweis erlangte man durch einAustauschexperiment, bei dem ein 1:1-Gemisch aus zweiPhenylenethinylen-Makrocyclen (94a und 100) der Meta-these unterworfen wurde [Gl. (17)]. Im MALDI-Massen-spektrum wurden mehrere makrocyclische Austauschpro-dukte mit verschiedenen Kombinationen an Tg- und tBu-substituierten Seitenketten beobachtet, was ein weiteres Indizf�r die Reversibilit#t der Makrocyclenbildung ist.
In einem weiteren Austauschexperiment wurde ein 2:1-Gemisch aus den Makrocyclen 98 und 102 unter Metathese-bedingungen umgesetzt [Gl. (18)] und anschließend mit FD-MS analysiert.[106] Es wurde ein tetramerer Makrocyclus be-obachtet, vermutlich mit der Struktur 103 (m/z 1200), derzwei ortho-disubstituierte Einheiten 98 (m/z 900) und zweimeta-disubstituierte Einheiten 102 (m/z= 1800) aufweist.Alle Befunde deuten darauf hin, dass sich die Phenylenethi-
nylen-Hexacyclen unter diesen Bedingungen thermodyna-misch kontrolliert bilden.
Theoretische Studien zur thermodynamischen Stabilit#tformtreuer Makrocyclen lieferten weitere Hinweise darauf,dass die beobachtete Produktverteilung der Gleichgewichts-verteilung entspricht. Die Verteilung der Produkte vom Tet-racyclus bis zum Octacyclus [Gl. (13)] im Vergleich mit demHexacyclus wurde berechnet,[86,87] wobei aus den Entropie-und Enthalpiewerten zun#chst die Gibbs-Energien der Ma-krocyclen mit n= 4, 5, 7, 8 ermittelt wurden (siehe Tabelle 5und Diagramm in Abbildung 12 sowie Gleichungen (19)–(22)). Die Rechnungen belegen, dass der Hexacyclus bei
J. S. Moore und W. ZhangAufs�tze
4542 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548
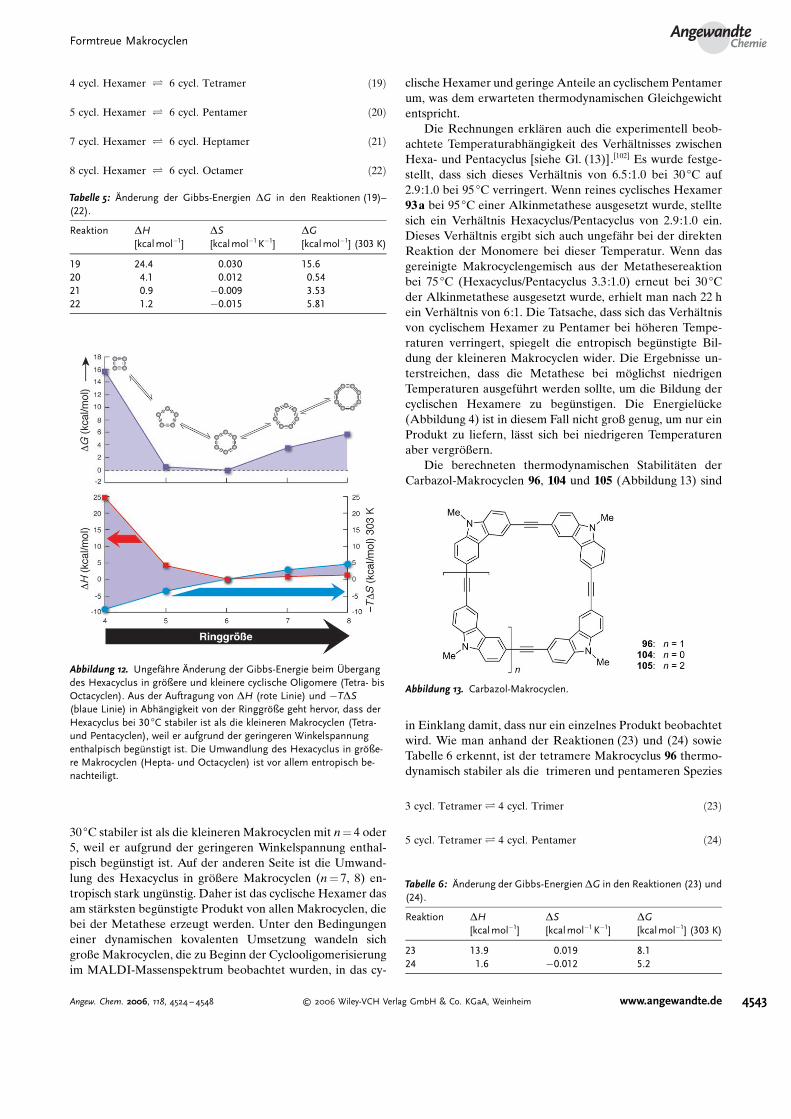
4 cycl: Hexamer Ð 6 cycl: Tetramer ð19Þ
5 cycl: Hexamer Ð 6 cycl: Pentamer ð20Þ
7 cycl: Hexamer Ð 6 cycl: Heptamer ð21Þ
8 cycl: Hexamer Ð 6 cycl: Octamer ð22Þ
30 8C stabiler ist als die kleineren Makrocyclen mit n= 4 oder5, weil er aufgrund der geringeren Winkelspannung enthal-pisch beg�nstigt ist. Auf der anderen Seite ist die Umwand-lung des Hexacyclus in gr�ßere Makrocyclen (n= 7, 8) en-tropisch stark ung�nstig. Daher ist das cyclische Hexamer dasam st#rksten beg�nstigte Produkt von allen Makrocyclen, diebei der Metathese erzeugt werden. Unter den Bedingungeneiner dynamischen kovalenten Umsetzung wandeln sichgroße Makrocyclen, die zu Beginn der Cyclooligomerisierungim MALDI-Massenspektrum beobachtet wurden, in das cy-
clische Hexamer und geringe Anteile an cyclischem Pentamerum, was dem erwarteten thermodynamischen Gleichgewichtentspricht.
Die Rechnungen erkl#ren auch die experimentell beob-achtete Temperaturabh#ngigkeit des Verh#ltnisses zwischenHexa- und Pentacyclus [siehe Gl. (13)].[102] Es wurde festge-stellt, dass sich dieses Verh#ltnis von 6.5:1.0 bei 30 8C auf2.9:1.0 bei 95 8C verringert. Wenn reines cyclisches Hexamer93a bei 95 8C einer Alkinmetathese ausgesetzt wurde, stelltesich ein Verh#ltnis Hexacyclus/Pentacyclus von 2.9:1.0 ein.Dieses Verh#ltnis ergibt sich auch ungef#hr bei der direktenReaktion der Monomere bei dieser Temperatur. Wenn dasgereinigte Makrocyclengemisch aus der Metathesereaktionbei 75 8C (Hexacyclus/Pentacyclus 3.3:1.0) erneut bei 30 8Cder Alkinmetathese ausgesetzt wurde, erhielt man nach 22 hein Verh#ltnis von 6:1. Die Tatsache, dass sich das Verh#ltnisvon cyclischem Hexamer zu Pentamer bei h�heren Tempe-raturen verringert, spiegelt die entropisch beg�nstigte Bil-dung der kleineren Makrocyclen wider. Die Ergebnisse un-terstreichen, dass die Metathese bei m�glichst niedrigenTemperaturen ausgef�hrt werden sollte, um die Bildung dercyclischen Hexamere zu beg�nstigen. Die Energiel�cke(Abbildung 4) ist in diesem Fall nicht groß genug, um nur einProdukt zu liefern, l#sst sich bei niedrigeren Temperaturenaber vergr�ßern.
Die berechneten thermodynamischen Stabilit#ten derCarbazol-Makrocyclen 96, 104 und 105 (Abbildung 13) sind
in Einklang damit, dass nur ein einzelnes Produkt beobachtetwird. Wie man anhand der Reaktionen (23) und (24) sowieTabelle 6 erkennt, ist der tetramere Makrocyclus 96 thermo-dynamisch stabiler als die trimeren und pentameren Spezies
3 cycl: Tetramer Ð 4 cycl: Trimer ð23Þ
5 cycl: Tetramer Ð 4 cycl: Pentamer ð24Þ
Tabelle 5: Onderung der Gibbs-Energien DG in den Reaktionen (19)–(22).
Reaktion DH[kcalmol�1]
DS[kcalmol�1K�1]
DG[kcalmol�1] (303 K)
19 24.4 0.030 15.620 4.1 0.012 0.5421 0.9 �0.009 3.5322 1.2 �0.015 5.81
Abbildung 12. Ungef@hre Onderung der Gibbs-Energie beim Bbergangdes Hexacyclus in grDßere und kleinere cyclische Oligomere (Tetra- bisOctacyclen). Aus der Auftragung von DH (rote Linie) und �TDS(blaue Linie) in Abh@ngigkeit von der RinggrDße geht hervor, dass derHexacyclus bei 30 8C stabiler ist als die kleineren Makrocyclen (Tetra-und Pentacyclen), weil er aufgrund der geringeren Winkelspannungenthalpisch begCnstigt ist. Die Umwandlung des Hexacyclus in grDße-re Makrocyclen (Hepta- und Octacyclen) ist vor allem entropisch be-nachteiligt.
Abbildung 13. Carbazol-Makrocyclen.
Tabelle 6: Onderung der Gibbs-Energien DG in den Reaktionen (23) und(24).
Reaktion DH[kcalmol�1]
DS[kcalmol�1K�1]
DG[kcalmol�1] (303 K)
23 13.9 0.019 8.124 1.6 �0.012 5.2
Formtreue MakrocyclenAngewandte
Chemie
4543Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

104bzw. 105. Dieser Befund stimmt mit unserer Beobachtung�berein, dass ausschließlich der Tetracyclus 96 gebildet wird.
Auf #hnlicheWeise wurden die Catechol-Makrocyclen 98,106 und 107 untersucht (Abbildung 14, Reaktionen (25) und(26), Tabelle 7). Den Rechnungen zufolge ist trimeres 98
2 cycl: Trimer Ð 3 cycl: Dimer ð25Þ
4 cycl: Trimer Ð 3 cycl: Tetramer ð26Þ
thermodynamisch stabiler als dimeres 106 und tetrameres107. Auch dieses Ergebnis stimmt mit unserer vorherigenBeobachtung �berein, dass sich ausschließlich das cyclischeTrimer 98 bildet. Die Ergebnisse verdeutlichen die Bedeu-tung des Energiel�ckenprinzips f�r die selektive Bildungeines makrocyclischen Produkts.
3.2.4. +berlegungen zur Synthese von formtreuen und nicht-formtreuen Makrocyclen durch DCC
Makrocyclen mit flexiblen Verkn�pfungen in ihremGer�st haben weitgehend unabh#ngig von ihrer Gr�ße meistsehr #hnliche Winkelspannungen. Durch diese Flexibilit#tf#llt der enthalpische Beitrag zur thermodynamischen Stabi-lit#t weniger ins Gewicht, weshalb die Entropie zur ent-scheidenden Triebkraft f�r die Produktbildung wird. Entro-pische Faktoren beg�nstigen im Allgemeinen Makrocyclenmit geringerer Anzahl an Monomereinheiten. Dies erkl#rt,warum sich Ringschlussmetathesen sehr gut zur Herstellungvon monomeren cyclischen Produkten eignen.[107]
Ein Beispiel ist die selektive und quantitative Synthesedes makrocyclischen Ethylenisophthalat-Dimers 110, �berdie Nagahata und Mitarbeiter berichteten [Gl. (27)].[108] Iso-phthals#uredimethylester (108) geht mit einem RquivalentEthylenglycoldiacetat (EGA; 109) in Gegenwart von Natri-umethoxid bei 65 8C eine Umesterung ein, wobei das cycli-
sche dimere Produkt 110 (ein Dimer aus zwei AB-Mono-mereinheiten) in nahezu quantitativer Ausbeute entsteht(97.6%). Die thermodynamischen Stabilit#ten der Ethylen-isophthalat-Makrocyclen, die auf die im vorigen Abschnittbeschriebene Weise berechnet wurden, sind in Tabelle 8aufgef�hrt [Reaktionen (28) und (29)]. Aus den Rechnungen
3 cycl: Dimer Ð 2 cycl: Trimer ð28Þ
4 cycl: Dimer Ð 2 cycl: Tetramer ð29Þ
l#sst sich vorhersagen, dass der dimere Makrocyclus 110haupts#chlich aus entropischen Gr�nden thermodynamischstabiler ist als trimeres 111 und tetrameres 112.
Auf #hnliche Weise wurde der dimere Alkoxyamin-Mak-rocyclus 115 (ein Dimer aus zwei AB-Monomereinheiten) mitflexiblen Verkn�pfungen direkt aus demAlkoxyamin 113 undAdipoylchlorid (114) bei hoher Verd�nnung hergestellt[Gl. (30)].[109] Die Cyclodepolymerisation[110] des Poly(alk-oxyamins) 116 bei 125 8C lieferte als Hauptbestandteil eben-falls 115, was auf die thermodynamische Stabilit#t des Dimersgegen�ber anderen Makrocyclen unter den gegebenen Re-aktionsbedingungen hinweist. Vermutlich hat die bevorzugteBildung von 115 ebenfalls entropische Ursachen.[111]
Aus diesen Beispielen kann geschlossen werden, dassdimere oder (wenn die Verkn�pfungen ausreichend lang sind)monomere Makrocyclen mit flexiblen Verkn�pfungen unteroptimierten Bedingungen in hohen Ausbeuten erhaltenwerden k�nnen. Anders als bei formtreuen Makrocyclen, wojedes Monomer ein Enthalpieinkrement beisteuert, h#ngt die
Abbildung 14. Catechol-Makrocyclen.
Tabelle 7: Onderung der Gibbs-Energien DG in den Reaktionen (25) und(26).
Reaktion DH[kcalmol�1]
DS[kcalmol�1K�1]
DG[kcalmol�1] (303 K)
25 57.2 0.026 49.226 0.42 �0.014 4.6
Tabelle 8: Onderung der Gibbs-Energien DG in den Reaktionen (28) und(29).
Reaktion DH[kcalmol�1]
DS[kcalmol�1K�1]
DG[kcalmol�1] (303 K)
28 �0.53 �0.018 5.629 0.10 �0.014 9.5
J. S. Moore und W. ZhangAufs�tze
4544 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

thermodynamische Stabilit#t von nicht-formtreuen Makro-cyclen kaum von der Enthalpie ab. Somit ist nicht zu erwar-ten, dass große Makrocyclen mit nicht-formtreuen Strukturendurch reversible Prozess erzeugt werden k�nnen.
3.2.5. Grenzen der thermodynamisch kontrollierten Cyclooligo-merisierungsmethoden
Den Methoden der thermodynamisch kontrollierten Cyc-looligomerisierung sind gewisse Grenzen gesetzt: Erstens istder Ansatz f�r die Synthese von gespannten, enthalpisch sehrung�nstigen Molek�len nicht praktikabel. Zweitens m�ssendie wachsenden Oligomere l�slich bleiben, damit derGleichgewichtszustand erreicht werden kann. Um dies zuverdeutlichen, wurde der Versuch unternommen, durch f#l-lungsvermittelte Alkinmetathese tetramere Carbazol-Mak-rocyclen herzustellen, die an den Stickstoffatomen mit ter-ti#ren Alkylcarboxylatgruppen funktionalisiert sind.[105] Diedrei Dialkinmonomere 117a–c, die mit tert-Butylcarboxylat,1,1-Dimethyloctylcarboxylat bzw. 1,1-Dimethyltridecylcarb-oxylat funktionalisiert sind, wurden den Standardmetathese-bedingungen ausgesetzt [Gl. (31)]. 117a und 117b wurdennahezu quantitativ in unl�sliche Produkte umgewandelt. Es
wurde angenommen, dass es sich bei den unl�slichen Pro-dukten um intermedi#re Oligomere und die Diarylacetylen-Nebenprodukte handelt. Im Unterschied dazu lief die Meta-these des Monomers 117c in 1,2,4-Trichlorbenzol bei 30 8Cbereitwillig ab, und der tetramere Makrocyclus 118c wurde inhoher Ausbeute erhalten.
Dieses Beispiel zeigt, wie wichtig es ist, dass l#ngereOligomere – und insbesondere diejenigen, die �ber die er-w�nschte Makrocyclenl#nge hinausschießen – l�slich bleiben.Alle intermedi#ren Verbindungen sollten w#hrend der Re-aktion gel�st bleiben, damit sich die thermodynamisch sta-bilen Produkte im Verlauf der reversiblen Alkinmetathesedurchsetzen k�nnen. Wie das Beispiel von Gleichung (31)zeigt, ist nie klar vorauszusehen, welche Stoffe gel�st bleibenund welche eher ausfallen. Eine schlechte L�slichkeit inter-medi#rer Oligomere ist ein ernstes Problem bei der Synthesel#ngerer und komplexerer zwei- und dreidimensionaler Mo-lek�lstrukturen.
Ein weiteres Problem bei Cyclooligomerisierungen be-trifft die spezifische Funktionalisierung. Die Energiel�ckezwischen Makrocyclen mit identischen Monomereinheitenund solchen mit Comonomereinheiten ist vermutlich oft zuklein, als dass die Bildung einer bestimmten Struktur be-g�nstigt w#re. Abgesehen von der variablen Zusammenset-zung ist die Herstellung von Makrocyclen, die mit einembestimmten Baustein spezifisch funktionalisiert sind, unterreversiblen Bedingungen ebenfalls problematisch. EineL�sung dieser Probleme erfordert die Entwicklung von neuenStrategien.
4. Schlussfolgerungen und Ausblick
In den letzten zwei Jahrzehnten waren bez�glich derSynthese und supramolekularen Chemie von formtreuenArylenethinylen-Makrocyclen erhebliche Fortschritte zuverzeichnen. Die Entwicklung von vielseitigen und effizien-ten Synthesemethoden erm�glicht heute einen leichtenZugang zu diesen Makrocyclen sowie eine systematischeAnalyse ihrer Eigenschaften. Bei den irreversiblen Cyclooli-gomerisierungen sind die Ausbeuten an Makrocyclen nor-malerweise durch Bildung h�herer Oligomere begrenzt(„Overshooting“). Durch wiederholende (konvergente/di-
vergente) Kupplungen l#sstsich die Gr�ße der Makro-cyclen und die Anordnungfunktioneller Gruppen steu-ern. Synthesemethodenunter Verwendung vonTemplaten und pr#organi-sierten Vorstufen erfordernnormalerweise weniger Syn-theseschritte, was zu hohenGesamtausbeuten f�hrt unddie Herstellung von funk-tionalisierten Makrocyclenunter kinetisch kontrollier-ten Bedingungen stark er-leichtert.
Formtreue MakrocyclenAngewandte
Chemie
4545Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

Metathesereaktionen unter thermodynamisch kontrol-lierten Bedingungen zeichnen sich durch leicht zug#nglicheAusgangsverbindungen und hohe Produktausbeuten aus. DasEnergiel�ckenprinzip, dessen Anwendung hier f�r die Ma-krocyclensynthese diskutiert wurde, findet auch bei anderendynamischen kovalentenMethoden breiten Einsatz. Die hocheffiziente, einstufige Synthese von Makrocyclen mithilfe derf#llungsvermittelten Alkinmetathese kann den Weg zu einerVielzahl von zwei- oder dreidimensionalen Arylenethinylen-Strukturen sowie alkinverbr�ckten Oligomeren und Poly-meren ebnen. Wegen ihrer einfachen Synthese und der F#-higkeit zur Selbstorganisation sind Arylenethinylen-Makro-cyclen vielversprechende Bausteine f�r supramolekulareStrukturen wie r�hrenf�rmige Kan#le, diskotische Fl�ssig-kristalle, molekulare elektronische und optische Bauele-mente und mikroskopische Reaktoren.
Die Arbeiten in unserer Arbeitsgruppe am Frederick SeitzMaterials Research Laboratory an der Universit1t von Illinoisin Urbana-Champaign werden durch die National ScienceFoundation und die Division of Materials Science des U.S.Department of Energy unterst�tzt.
Eingegangen am 9. November 2005Online ver�ffentlicht am 13. Juni 2006
Ibersetzt von Katrin Harder, Birkenstein
[1] S. H�ger,Angew. Chem. 2005, 117, 3872;Angew. Chem. Int. Ed.2005, 44, 3806.
[2] S. H�ger, Chem. Eur. J. 2004, 10, 1320.[3] D. Zhao, J. S. Moore, Chem. Commun. 2003, 807.[4] Y. Yamaguchi, Z. Yoshida, Chem. Eur. J. 2003, 9, 5430.[5] C. Grave, A. D. Schl�ter, Eur. J. Org. Chem. 2002, 3075.[6] F. Diederich, Chem. Commun. 2001, 219.[7] S. H�ger, J. Polym. Sci. Part A 1999, 37, 2685.[8] U. H. F. Bunz, Y. Rubin, Y. Tobe, Chem. Soc. Rev. 1999, 28, 107.[9] M. M. Haley, J. J. Pak, S. C. Brand, Top. Curr. Chem. 1999, 201,
81.[10] J. S. Moore, Acc. Chem. Res. 1997, 30, 402.[11] J. K. Young, J. S. Moore inModern Acetylene Chemistry (Hrsg.:
P. J. Stang, F. Diederich), VCH, Weinheim, 1995, Kap. 12.[12] K. Sonogashira, Comprehensive Organic Synthesis, Bd. 3
(Hrsg.: B. M. Trost, I. Fleming), Pergamon, Oxford, 1991,S. 521.
[13] K�rzlich berichteten Yaghi und Mitarbeiter �ber die Synthesevon kovalenten organischen Ger�sten (COFs) durch Konden-sation von Phenyldiborons#ure und Hexahydroxytriphenylen.Die hoch kristallinen Produkte bilden por�se graphitischeSchichten, die laut Pulverr�ntgenstrukturanalyse entwederversetzt oder ekliptisch angeordnet sind (A. P. CUtV, A. I.Benin, N. W. Ockwig, M. OWKeeffe, A. J. Matzger, O. M. Yaghi,Science 2005, 310, 1166).
[14] H. A. Dieck, F. R. Heck, J. Organomet. Chem. 1975, 93, 259.[15] a) K. Sonogashira, Y. Tohda, N. Hagihara, Tetrahedron Lett.
1975, 16, 4467; b) L. Cassar, J. Organomet. Chem. 1975, 93, 253.[16] J. S. Moore, J. Zhang, Angew. Chem. 1992, 104, 873; Angew.
Chem. Int. Ed. Engl. 1992, 31, 922.[17] J. Zhang, D. J. Pesak, J. L. Ludwick, J. S. Moore, J. Am. Chem.
Soc. 1994, 116, 4227.[18] S. Lahiri, J. L. Thompson, J. S. Moore, J. Am. Chem. Soc. 2000,
122, 11315.[19] U. H. F. Bunz, Chem. Rev. 2000, 100, 1605.
[20] a) A. O. King, N. Okukado, E. Negishi, J. Chem. Soc. Chem.Commun. 1977, 683; b) A. O. King, E. Negishi, F. J. Villani, Jr.,A. Silveira, Jr., J. Org. Chem. 1978, 43, 358; c) E. Negishi, Acc.Chem. Res. 1982, 15, 340.
[21] a) G. Eglinton, A. R. Galbraith, J. Chem. Soc. 1959, 889;b) O. M. Behr, G. Eglinton, A. R. Galbraith, R. A. Raphael, J.Chem. Soc. 1960, 3614; c) D. OWKrongly, S. R. Denmeade, M. Y.Chiang, R. Breslow, J. Am. Chem. Soc. , 1985, 107, 5544.
[22] Ibersicht �ber Acetylenkupplungen: P. Siemsen, R. C. Living-ston, F. Diederich, Angew. Chem. 2000, 112, 2740; Angew.Chem. Int. Ed. 2000, 39, 2632.
[23] S. H�ger, V. Enkelmann, Angew. Chem. 1995, 107, 2917;Angew. Chem. Int. Ed. Engl. 1995, 34, 2713.
[24] S. H�ger, A. D.Meckenstock, H. Pellen, J. Org. Chem. 1997, 62,4556.
[25] S. H�ger, A. D. Meckenstock, S. M�ller, Chem. Eur. J. 1998, 4,2423.
[26] S. H�ger, A. D. Meckenstock, Chem. Eur. J. 1999, 5, 1686.[27] S. H�ger, K. Bonrad, A. Mourran, U. Beginn, M. M�ller, J. Am.
Chem. Soc. 2001, 123, 5651.[28] Y. Tobe, N. Utsumi, K. Kawabata, K. Naemura, Tetrahedron
Lett. 1996, 37, 9325.[29] Y. Tobe, N. Utsumi, A. Nagano, K. Naemura, Angew. Chem.
1998, 110, 1347; Angew. Chem. Int. Ed. 1998, 37, 1285.[30] Y. Tobe, A. Nagano, K. Kawabata, M. Sonoda, K. Naemura,
Org. Lett. 2000, 2, 3265.[31] A. Nomoto, M. Sonoda, Y. Yamaguchi, T. Ichikawa, K. Hirose,
Y. Tobe, J. Org. Chem. 2006, 71, 401.[32] Y. Tobe, N. Utsumi, K. Kawabata, A. Nagano, K. Adachi, S.
Araki, M. Sonoda, K. Hirose, K. Naemura, J. Am. Chem. Soc.2002, 124, 5350.
[33] C. E. Castro, R. D. Stephens, J. Org. Chem. 1963, 28, 2163.[34] Ibersichten zur Alkinmetathese: a) A. F�rstner, P. W. Davies,
Chem. Commun. 2005, 2307; b) R. R. Schrock, A. H. Hoveyda,Angew. Chem. 2003, 115, 4740; Angew. Chem. Int. Ed. 2003, 42,4592; c) R. R. Schrock, Chem. Rev. 2002, 102, 145; d) A.F�rstner in Handbook of Metathesis, Bd. 2 (Hrsg.: R. H.Grubbs), Wiley-VCH, Weinheim, 2003, Kap. 2.12; e) U. H. F.Bunz, Acc. Chem. Res. 2001, 34, 998.
[35] Die Bildungen von Porphyrinen und Calixarenen durch Pyrrol-bzw. Phenol-Aldehyd-Kondensation sind Beispiele f�r Cyclo-oligomerisierungen; siehe: a) A. Ghosh, Angew. Chem. 2004,116, 1952; Angew. Chem. Int. Ed. 2004, 43, 1918; b) C. D. Gut-sche, D. E. Johnston, Jr., D. R. Stewart, J. Org. Chem. 1999, 64,3747; c) B. Dhawan, S.-I. Chen, C. D. Gutsche, Makromol.Chem. 1987, 188, 921; d) A. D. Adler, F. R. Longo, W. Shergalis,J. Am. Chem. Soc. 1964, 86, 3145.
[36] V. Prautzsch, S. Ibach, F. J. V�gtle, J. Inclusion Phenom.Macrocyclic Chem. 1999, 33, 427.
[37] S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders,J. F. Stoddart, Angew. Chem. 2002, 114, 938; Angew. Chem. Int.Ed. 2002, 41, 898.
[38] J.-M. Lehn, Chem. Eur. J. 1999, 5, 2455.[39] a) B. M. Trost, Science 1991, 254, 1471; b) K. Oh, K.-S. Jeong,
J. S. Moore, Nature 2001, 414, 889; c) K. Oh, K.-S. Jeong, J. S.Moore, J. Org. Chem. 2003, 68, 8397; d) T. Nishinaga, A. Ta-natani, K. Oh, J. S. Moore, J. Am. Chem. Soc. 2002, 124, 5934;e) D. Zhao, J. S. Moore, Macromolecules 2003, 36, 2712.
[40] a) R. R. Schrock, D. N. Clark, J. Sancho, J. H.Wengrovius, S. M.Rocklage, S. F. Pedersen, Organometallics 1982, 1, 1645;b) L. G. McCullough, R. R. Schrock, J. Am. Chem. Soc. 1984,106, 4067; c) L. G. McCullough, R. R. Schrock, J. C. Dewan,J. C. Murdzek, J. Am. Chem. Soc. 1985, 107, 5987; d) J. M.Blackwell, J. S. Figueroa, F. H. Stephens, C. C. Cummins, Or-ganometallics 2003, 22, 3351; e) Y.-C. Tsai, P. L. Diaconescu,C. C. Cummins, Organometallics 2000, 19, 5260; f) C. E. La-plaza, A. L. Odom, W. M. Davis, C. C. Cummins, J. D. Prota-
J. S. Moore und W. ZhangAufs�tze
4546 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548

siewics, J. Am. Chem. Soc. 1995, 117, 4999; g) A. Mortreux, M.Blanchard, J. Chem. Soc. Chem. Commun. 1974, 786; h) N. G.Pschirer, U. H. F. Bunz, Tetrahedron Lett. 1999, 40, 2481.
[41] W. Zhang, S. Kraft, J. S. Moore, Chem. Commun. 2003, 832.[42] W. Zhang, S. Kraft, J. S. Moore, J. Am. Chem. Soc. 2004, 126,
329.[43] a) D. J. Brunelle, E. P. Boden, T. G. Shannon, J. Am. Chem. Soc.
1990, 112, 2399; b) S. J. Rowan, P. A. Brady, J. K. M. Sanders,Angew. Chem. 1996, 108, 2283; Angew. Chem. Int. Ed. Engl.1996, 35, 2143; c) S. J. Rowan, J. K. M. Sanders, J. Org. Chem.1998, 63, 1536; d) S. J. Rowan, D. G. Hamilton, P. A. Brady,J. K. M. Sanders, J. Am. Chem. Soc. 1997, 119, 2578.
[44] a) S.-W. Tam-Chang, J. S. Stehouwer, J. Hao, J. Org. Chem.1999, 64, 334; b) S. Ro, S. J. Rowan, A. R. Pease, D. J. Cram,J. F. Stoddart, Org. Lett. 2000, 2, 2411.
[45] a) D. G. Hamilton, N. Feeder, S. J. Teat, J. K. M. Sanders,New J.Chem. 1998, 22, 1019; b) D. G. Hamilton, J. E. Davies, L. Prodi,J. K. M. Sanders, Chem. Eur. J. 1998, 4, 608; c) D. G. Hamilton,M. Montalti, L. Prodi, M. Fontani, P. Zanello, J. K. M. Sanders,Chem. Eur. J. 2000, 6, 608.
[46] a) J. N. Onuchic, H. Nymeyer, A. E. Garcia, J. Chahine, N. D.Socci,Adv. Protein Chem. 2000, 53, 87; b) D. J. Brockwell, D. A.Smith, S. E. Radford, Curr. Opin. Struct. Biol. 2000, 10, 16.
[47] Die Simulationen wurden mit dem Mechanism-Based KineticsSimulator ausgef�hrt: http://www.stolaf.edu/depts/chemistry/courses/toolkits/126/js/kinetics/index.htm.
[48] Y. Li, J. Zhang, W. Wang, Q. Miao, X. She, X. Pan, J. Org.Chem. 2005, 70, 3285.
[49] C. Eickmeier, H. Junga, A. J. Matzger, F. Scherhag, M. Shim,K. P. C. Vollhardt, Angew. Chem. 1997, 109, 2194; Angew.Chem. Int. Ed. Engl. 1997, 36, 2103.
[50] a) J. D. Ferrara, C. Tessier-Youngs, W. J. Youngs, J. Am. Chem.Soc. 1985, 107, 6719; b) W. J. Youngs, J. D. Kinder, J. D. Brad-shaw, C. A. Tessier, Organometallics 1993, 12, 2406; c) J. D.Ferrara, A. Djebli, C. Tessier-Youngs, W. J. Youngs, J. Am.Chem. Soc. 1988, 110, 647.
[51] T. Nishinaga, Y. Miyata, N. Nodera, K. Komatsu, Tetrahedron2004, 60, 3375.
[52] P. N. Prasad, D. J. Williamsm, Introduction to Nonlinear OpticalEffects in Molecules and Polymers, Wiley, New York, 1991,Kap. 7, S. 132.
[53] H. A. Staab, K. Neunhoeffer, Synthesis 1974, 424.[54] M. Iyoda, A. Vorasingha, Y. Kuwatani, M. Yoshida, Tetrahe-
dron Lett. 1998, 39, 4701.[55] a) L. Yuan, W. Feng, K. Yamato, A. R. Sanford, D. Xu, H,
Guo, B. Gong, J. Am. Chem. Soc. 2004, 126, 11120; die gleicheArbeitsgruppe berichtete k�rzlich �ber #hnliche Synthesen vontetrameren aromatischen Sulfonamid-Makrocyclen mit Alk-oxyseitenketten in Ausbeuten von 38 – 51%: b) L. He, Y. An, L.Yuan, K. Yamato, W. Feng, O. Gerlitz, C. Zheng, B. Gong,Chem. Commun. 2005, 3788 – 3790.
[56] S. Toyota, M. Goichi, M. Kotani,Angew. Chem. 2004, 116, 2298;Angew. Chem. Int. Ed. 2004, 43, 2248.
[57] H. E. Katz, J. Org. Chem. 1989, 54, 2179.[58] Zu Beispielen von hypsochromen Effekten, die in aromati-
schen Makrocyclen und Polymeren mit p-p-Wechselwirkungenbeobachtet wurden, siehe: a) F. V�gtle, Cyclophane Chemistry,Wiley, Chichester, 1993, S. 77; b) S. Dumitrescu, M. Grigoras,C. I. Simionescu, Eur. Polym. J. 1983, 19, 1137.
[59] Y. Yamaguchi, S. Kobayashi, S. Miyamura, Y. Okamoto, T.Wakamiya, Y. Matsubara, Z. Yoshida,Angew. Chem. 2004, 116,370; Angew. Chem. Int. Ed. 2004, 43, 366.
[60] J. S. Moore, E. J. Weinstein, Z. Y. Wu, Tetrahedron Lett. 1991,32, 2465.
[61] a) O. Henz, D. Lentz, A. D. Schl�ter, Chem. Eur. J. 2000, 6,2362; b) O. Henz, D. Lentz, A. Schater, P. Franke, A. D.Schl�ter, Chem. Eur. J. 2002, 8, 357; c) S. Kobayashi, Y. Yama-
guchi, T. Wakamiya, Y. Matsubara, K. Sugimoto, Z. Yoshida,Tetrahedron Lett. 2003, 44, 1469.
[62] J. A. Marsden, J. J. Miller, M. M. Haley, Angew. Chem. 2004,116, 1726; Angew. Chem. Int. Ed. 2004, 43, 1694.
[63] F. Bohlmann, H. Sch�nowsky, E. Inhoffen, G. Grau,Chem. Ber.1963, 96, 794.
[64] Z. Y. Wu, J. S. Moore, Angew. Chem. 1996, 108, 320; Angew.Chem. Int. Ed. Engl. 1996, 35, 297.
[65] Z. Y.Wu, S. Lee, J. S. Moore, J. Am. Chem. Soc. 1992, 114, 8730.[66] V. Hensel, K. L�tzow, J. Jacob, K. Gessler, W. Saenger, A. D.
Schl�ter,Angew. Chem. 1997, 109, 2768;Angew. Chem. Int. Ed.Engl. 1997, 36, 2654.
[67] O. S. Pyun, W. Yang, M.-Y. Jeong, S. H. Lee, K. M. Kang, S.-J.Jeon, B. R. Cho, Tetrahedron Lett. 2003, 44, 5179.
[68] Ein #hnlicher Ansatz wurde k�rzlich genutzt, um einen Donor-Acceptor-substituierten Phenylenethinylen-Makrocyclus zusynthetisieren: B. Traber, T. Oeser, R. Gleiter, Eur. J. Org.Chem. 2005, 1283.
[69] P. N. W. Baxter, J. Org. Chem. 2004, 69, 1813.[70] P. N. W. Baxter, Chem. Eur. J. 2003, 9, 2531.[71] P. N. W. Baxter, Chem. Eur. J. 2002, 8, 5250.[72] P. N. W. Baxter, J. Org. Chem. 2001, 66, 4170.[73] H. Sugiura, Y. Takahira, M. Yamaguchi, J. Org. Chem. 2005, 70,
5698.[74] Y. Saiki, K. Nakamura, Y. Nigorikawa, M. Yamaguchi, Angew.
Chem. 2003, 115, 5348; Angew. Chem. Int. Ed. 2003, 42, 5190.[75] K. Nakamura, H. Okubo, M. Yamaguchi, Org. Lett. 2001, 3,
1097.[76] H. L. Anderson, J. K. M. Sanders, Angew. Chem. 1990, 102,
1478; Angew. Chem. Int. Ed. Engl. 1990, 29, 1400.[77] D. W. J. McCallien, J. K. M. Sanders, J. Am. Chem. Soc. 1995,
117, 6611.[78] M. J. Marsella, Z.-Q. Wang, R. J. Reid, K. Yoon, Org. Lett.
2001, 3, 885.[79] Ausf�hrungen zur Aryl-Perfluoraryl-Wechselwirkung (ArH-
ArF) als zuverl#ssiges supramolekulares Synthon im Kristall-Engineering: a) G. W. Coates, A. R. Dunn, L. M. Henling, J. W.Ziller, E. B. Lobkovsky, R. H. Grubbs, J. Am. Chem. Soc. 1998,120, 3641; b) G. W. Coates, A. R. Dunn, L. M. Henling, D. A.Dougherty, R. H. Grubbs, Angew. Chem. 1997, 109, 290;Angew. Chem. Int. Ed. Engl. 1997, 36, 248.
[80] Ibersichten �ber metallgebundene formtreue Makrocyclen:a) M. Fujita, M. Tominaga, A. Hori, B. Therrien, Acc. Chem.Res. 2005, 38, 369; b) S. R. Seidel, P. J. Stang, Acc. Chem. Res.2002, 35, 972; c) S. Leininger, B. Olenyuk, P. J. Stang, Chem.Rev. 2000, 100, 853.
[81] D. Zhao, J. S. Moore, J. Org. Chem. 2002, 67, 3548.[82] D. J. Hill, J. S. Moore, Proc. Natl. Acad. Sci. USA 2002, 99, 5053.[83] a) A. J. Gallant, J. K.-H. Hui, F. E. Zahariev, Y. A. Wang, M. J.
MacLachlan, J. Org. Chem. 2005, 70, 7936; b) C. Ma, A. Lo, A.Abdolmaleki, M. J. MacLachlan, Org. Lett. 2004, 6, 3841.
[84] A. J. Gallant, B. O. Patrick, M. J. MacLachlan, J. Org. Chem.2004, 69, 8739.
[85] A. J. Gallant, M. J. MacLachlan,Angew. Chem. 2003, 115, 5465;Angew. Chem. Int. Ed. 2003, 42, 5307.
[86] M. Mammen, E. I. Shakhnovich, J. M. Deutch, G. M. Whitesi-des, J. Org. Chem. 1998, 63, 3821; die Entropiewerte f�r diemethoxysubstituierten Makrocyclen sind gesch#tzt.
[87] Die Bildungsw#rmen der methoxysubstituierten Makrocyclenwurden mit dem Programm Spartan (Version 4.0; Wavefunc-tion, Irvine) auf AM1-Niveau berechnet.
[88] Ibersichten: a) R. H. Grubbs, Tetrahedron 2004, 60, 7117;b) T. M. Trnka, R. H. Grubbs, Acc. Chem. Res. 2001, 34, 18;c) S. J. Connon, S. Blechert, Angew. Chem. 2003, 115, 1944;Angew. Chem. Int. Ed. 2003, 42, 4592; d) S. T. Diver, A. J.Giessert, Chem. Rev. 2004, 104, 1317; e) A. F�rstner, Angew.
Formtreue MakrocyclenAngewandte
Chemie
4547Angew. Chem. 2006, 118, 4524 – 4548 � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.angewandte.de

Chem. 2000, 112, 3140; Angew. Chem. Int. Ed. 2000, 39, 3012;siehe auch Lit. [34b].
[89] K, Weiss, A. Michel, E. M. Auth, U. H. F. Bunz, T. Mangel, K.M�llen, Angew. Chem. 1997, 109, 522; Angew. Chem. Int. Ed.Engl. 1997, 36, 506.
[90] a) A. F�rstner, O. Guth, A. Rumbo, G. Seidel, J. Am. Chem.Soc. 1999, 121, 11108; b) A. F�rstner, C. Mathes, C. W. Leh-mann, J. Am. Chem. Soc. 1999, 121, 9453.
[91] a) L. Kloppenburg, D. Jones, U. H. F. Bunz, Macromolecules1999, 32, 4194; b) N. G. Pschirer, W. Fu, R. D. Adams, U. H. F.Bunz, Chem. Commun. 2000, 87.
[92] P.-H. Ge, W. Fu, W. A. Herrmann, E. Herdtweck, C. Campana,R. D. Adams, U. H. F. Bunz, Angew. Chem. 2000, 112, 3753;Angew. Chem. Int. Ed. 2000, 39, 3607.
[93] Rhnlich wie bei der Synthese des Makrocyclus 85 spiegeln diehier angegebenen Ausbeuten an isoliertem Produkt wahr-scheinlich nicht die tats#chliche thermodynamisch kontrollierteProduktverteilung wider.
[94] Zu #hnlichen siliciumhaltigen Makrocyclen siehe: a) F. Q. Liu,G. Harder, T. D. Tilley, J. Am. Chem. Soc. 1998, 120, 3271;b) S. S. H. Mao, F. Q. Liu, T. D. Tilley, J. Am. Chem. Soc. 1998,120, 1193.
[95] O. S. Miljanic, K. P. C. Vollhardt, G. D. Whitener, Synlett 2003,29.
[96] Die angegebenen Ausbeuten spiegeln m�glicherweise nicht dietats#chliche thermodynamisch kontrollierte Produktverteilungwider.
[97] W. Zhang, J. S. Moore, J. Am. Chem. Soc. 2004, 126, 12796.[98] Der gleiche Ansatz wurde auch zur Synthese von konjugierten
Arylenethinylen-Polymeren mit hohen Molmassen bei Raum-temperatur angewendet, siehe: W. Zhang, J. S. Moore, Ma-cromolecules 2004, 37, 3973.
[99] a) A. F�rstner, G. Seidel, Angew. Chem. 1998, 110, 1758;Angew. Chem. Int. Ed. 1998, 37, 1734.
[100] In diesen F#llen wird eine geringe Menge an pentamerenMakrocyclen als haupts#chliches Nebenprodukt erhalten.
[101] J. H. Greudenberger, R. R. Schrock, M. R. Churchill, A. L.Rheingold, J. W. Ziller, Organometallics 1984, 3, 1563.
[102] W. Zhang, J. S. Moore, J. Am. Chem. Soc. 2005, 127, 11863.[103] a) R. R. Schrock, Acc. Chem. Res. 1986, 19, 342; b) J. H. Wen-
grovius, J. Sancho, R. R. Schrock, J. Am. Chem. Soc. 1981, 103,3932.
[104] Der tetramere Carbazol-Makrocyclus 96 wurde erstmals durcheine Kreuzkupplung in 14.3% Ausbeute synthetisiert: S.Maruyama, H. Hokari, T. Wada, H. Sasabe, Synthesis 2001,1794.
[105] Ein weiterer 1,1-Dimethyltridecylcarboxylat-funktionalisierterCarbazol-Makrocyclus wurde k�rzlich durch f#llungsvermit-telte Alkinmetathese hergestellt: W. Zhang, H. M. Cho, J. S.Moore, Org. Synth. , zur Ver�ffentlichung eingereicht.
[106] W. Zhang, S. M. Brombosz, J. L. Mendoza, J. S. Moore, J. Org.Chem. 2005, 70, 10198.
[107] a) A. Deiters, S. F. Martins, Chem. Rev. 2004, 104, 2199; b) A.F�rstner,Angew. Chem. 2003, 115, 3706;Angew. Chem. Int. Ed.2003, 42, 3582.
[108] R. Nagahata, J. Sugiyama, Y. Nakao, M. Asai, K. Takeuchi,Macromolecules 2003, 36, 2582.
[109] G. Yamaguchi, Y. Higaki, H. Otsuka, A. Takahara, Macromo-lecules 2005, 38, 6316.
[110] Zu entropisch angetriebenen Cyclodepolymerisationen, siehe:a) A. Ben-Haida, I, Baxter, H. M. Colquhoun, P. Hodge, F. H.Kohnke, D. J. Williams, Chem. Commun. 1997, 1533; b) I.Baxter, A. Ben-Haida, H. M. Colquhoun, P. Hodge, F. H.Kohnke, D. J. Williams, Chem. Commun. 1998, 2213; c) B.-H.Lim, S.-H. Kwon, E.-C. Kang, H. Park, H.-W Lee, W.-G. Kim, J.Polym. Sci. Part A 2003, 41, 881; d) P. Hodge, React. Funct.Polym. 2001, 48, 15; e) P. Hodge, Z. Yang, A. Ben-Haida, C. S.McGrail, J. Mater. Chem. 2000, 10, 1533; f) A. J. Hall, P. Hodge,C. S. McGrail, J. Rickerby, Polymer 2000, 41, 1239.
[111] Iber die bevorzugte Bildung von cyclischen Dimeren mit fle-xiblen Bindungen durch Alkenmetathese wurde ebenfalls be-richtet: A. B. Smith III, S. A. Kozmin, C. M. Adams, D. V.Paone, J. Am. Chem. Soc. 2000, 122, 4984.
J. S. Moore und W. ZhangAufs�tze
4548 www.angewandte.de � 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. 2006, 118, 4524 – 4548





























