Embed Size (px)
Citation preview

1
FP 26
Pulverdiffraktometrie
Raum 420 (Nord) Universität Augsburg Institut für Physik
Lehrstuhl für Festkörperchemie
Stand: Oktober 2014

2
Inhaltsverzeichnis
1. Einleitung............................................................................................................................. 3
2. Bragg̍sche-Gleichung ......................................................................................................... 4
3. Pulverdiffraktometer ............................................................................................................ 5
3.1 Röntgenröhre...................................................................................................................... 6
3.2 Monochromatoren, Blenden und Soller-Blenden .............................................................. 7
3.3 Detektor.............................................................................................................................. 8
4. Pulverdiffraktogramm ......................................................................................................... 9
5. Probenvorbereitung ........................................................................................................... 10
6. Hoch-und Tieftemperaturkammer ..................................................................................... 11
7. Rietveld-Methode .............................................................................................................. 12
8. Ziel des Versuchs............................................................................................................... 15
9. Versuchsdurchführung ....................................................................................................... 15
9.1 Probenpräparation und Messung..................................................................................... 15
9.2 Qualitative Phasenanalyse ............................................................................................... 15
9.3 Bestimmung der Gitterkonstanten und Raumgruppe von Pulverdaten ............................ 16
9.4 Quantitative Phasenanalyse ............................................................................................. 16
10. Literatur ............................................................................................................................. 16

3
1. Einleitung
Mit der Entdeckung der Röntgenstrahlung Ende 1895 durch W. C. Röntgen hat ein neues Zeitalter für Festkörperforscher begonnen. Sehr schnell wurden die zahlreichen Anwendungsmöglichkeiten der Röntgenstrahlung erkannt. Die ersten durchgeführten Experimente durch M. von Laue, W. Friedrich und P. Knipping haben bewiesen, dass Röntgenstrahlen eine Wellennatur vorweisen. Zudem wurde gezeigt, dass die Kristalle gitterartig, 3-dimensional periodisch aufgebaut sind. Schon 1913 wurden die ersten Kristallstrukturen von NaCl, ZnS und KCl bestimmt. Im Jahr 1915 haben Debye und Scherrer die erste Pulverbeugungsaufnahme -mittels eines heute noch nach ihnen benannten Verfahrens- durchgeführt.
Die Röntgenstrahlen bilden einen Teil des elektromagnetischen Spektrums und umfassen Wellenlängen von ca. 10-8 bis 10-13 m. Die Abgrenzung zu den beiden benachbarten Gebieten der UV-Strahlung und der γ-Strahlen ist nicht streng gesetzt. Die gute Wechselwirkung zwischen Röntgenstrahlung und Kristall erklärt sich aus ähnlichen Dimensionen. Beispielsweise entspricht die Wellenlänge der Cu-Kα-Strahlung dem C-C Abstand in Diamant. Für Röntgenbeugungsuntersuchungen werden die Wellenlänge von 0.2 bis 2.5 Å verwendet.
Heutzutage gehört Pulverdiffraktometrie zu einer Routinemethode zur Charakterisierung von kristallinen Materialen. Mit Hilfe von Pulverröntgenbeugung können kristalline Proben identifiziert (qualitative Phasenanalyse) und quantifiziert (quantitative Phasenanalyse) werden. Zudem kann die Kristallstruktur der Verbindungen gelöst werden. Diese Methode wird in ganz unterschiedlichen Einsatzgebieten, wie zum Beispiel der Baustoffindustrie (Zusammensetzung vom Zement, Homogenität, Teilchengröße), Pharmaindustrie (Strukturaufklärung von APIs, Homogenität, Temperatur- und Feuchtigkeitsstabilität von Arzneien, Polymorphie), Kriminaltechnik (Spurensuche, Identifikation von Drogen und Drogenverunreinigungen), Halbleiterindustrie (Textur, Spannung), Stahlindustrie (Homogenität, Zusammensetzung, Textur) und zur Untersuchung von sehr dünnen Schichten (Dicke, Rauigkeit, Zusammensetzung), verwendet.

4
2. Braggˈsche-Gleichung
Röntgendiffraktometrie basiert auf einer Formel, die für die Erklärung der Röntgenbeugung durch W. L. Bragg im Jahr 1912 geliefert worden ist. Bragg führte die Röntgenbeugung auf eine selektive Reflexion an einer Netzebenenschar zurück. Der Röntgenstrahl wird an einer Netzebene reflektiert, dabei sind Ein- und Ausfallwinkel gleich. Da die Röntgenstrahlung eine energiereiche Strahlung ist, dringt sie auch in den Kristall ein, so dass die zweite als auch die folgenden Netzebenen mit der gleichen Intensität bestrahlt werden wie die oberste Netzebene. Es kommt zu Interferenzen zwischen den an den verschiedenen Netzebenen reflektierten Röntgenstrahlen. Diese können sich nur dann zu einem messbaren Effekt aufaddieren, wenn die Gangunterschiede der Strahlen ganzzahlige Vielfache der Wellenlänge der angewendeten Strahlung sind. Der Gangunterschied errechnet sich wie folgt:
�λ = 2���� sin θ
λ = Wellenlänge dhkl = Netzebenenabstand θ = Beugungswinkel
Abb. 1 Beugungsvorgang an einer Netzebenenschar nach W. L. Bragg. Der untere Strahl legt einen längeren Weg zurück: Gangunterschied = 2dsinθ.
Der Netzebenenabstand in einem Gitter kann nach folgenden Gleichungen berechnet werden:
• kubisches Kristallsystem:
• tetragonales Kristallsystem:
• orthorhombisches Kristallsystem:
• hexagonales Kristallsystem:
2
222
2
1
a
lkh
d
++=
2
2
2
22
2
1
c
l
a
kh
d++=
2
2
2
2
2
2
2
1
c
l
b
k
a
h
d++=
2
2
2
22
2 3
41
c
l
a
khkh
d+++=

5
• monoklines Kristallsystem:
• triklines Kristallsystem:
a, b, c – Gitterkonstanten α, β, γ – Winkel h k l – Miller Indices
3. Pulverdiffraktometer
Moderne Pulverdiffraktometer werden als geschlossene Einheiten gebaut, die Beugungsaufnahmen automatisch durchführen. Das Diffraktometer besteht aus Röntgengenerator, Röntgenröhre, Probenhalter und Detektor. Das am weitesten verbreitete Pulverdiffraktometer ist das Bragg-Brentano Diffraktometer. Das Aufbauprinzip ist Folgendes: ein ebenes, pulverförmiges Präparat befindet sich in der Mitte des Messkreises, der Brennfleck der Röntgenröhre und die Eintrittsblende des Detektors befinden sich am Rand des Fokussierungskreises. Es gibt zwei mögliche Messgeometrien: θ - 2θ-Geometrie: Detektor bewegt sich mit bestimmter Geschwindigkeit entlang des Messkreises, das Präparat wird mit halber Geschwindigkeit bewegt und Interferenzen werden nacheinander registriert. θ - θ-Geometrie: die Röntgenröhre und der Detektor bewegen sich entlang des Messkreises und das Präparat befindet sich an fester Position (z. B. für Messungen von flüssigen Proben).
Abb. 2 Siemens 3003TT Pulverdiffraktometer: Gehäuse (links), innerhalb des Gerätes (rechts).
ββ
ββ 222
2
2
2
22
2
2 sin
cos2
sinsin
1
ac
hl
c
l
b
k
a
h
d+++=
)coscoscos2coscoscos1/(
)coscos(cos2
sin)coscos(cos
2
sin)coscos(cos
2
sin
1
222
22
2
22
2
22
2
2
γβαγβα
γβαγ
βγαβ
αγβα
+−−−
−++−++−+=
ab
hk
c
l
ac
hl
b
k
bc
kl
a
h
d

6
3.1 Röntgenröhre
Die meist verwendeten Röntgenröhren sind aus Glas, Keramik oder Metall hergestellt und vakuumdicht abgeschmolzen. Die Röntgenröhre enthält einen Wolframglühdraht (Kathode), der auf 1500-2300 °C erhitzt wird und eine gekühlte Anode, die aus Cu, Cr, Fe, Mo oder Ag hergestellt ist. Durch die angelegte Hochspannung werden die aus der Kathode ausgehenden Elektronen stark beschleunigt und auf das Anodenmaterial geschossen. Die kinetische Energie der Elektronen wird bei der Abbremsung in der Anode überwiegend in Wärme umgesetzt (ca. 99.5 %), der Rest in Strahlung, die durch Be-Austrittfenster gezielt nach außen durchgehen kann.
Abb. 3 Eine schematische Darstellung (links) und das Foto (rechts) von einer Röntgenröhre.1
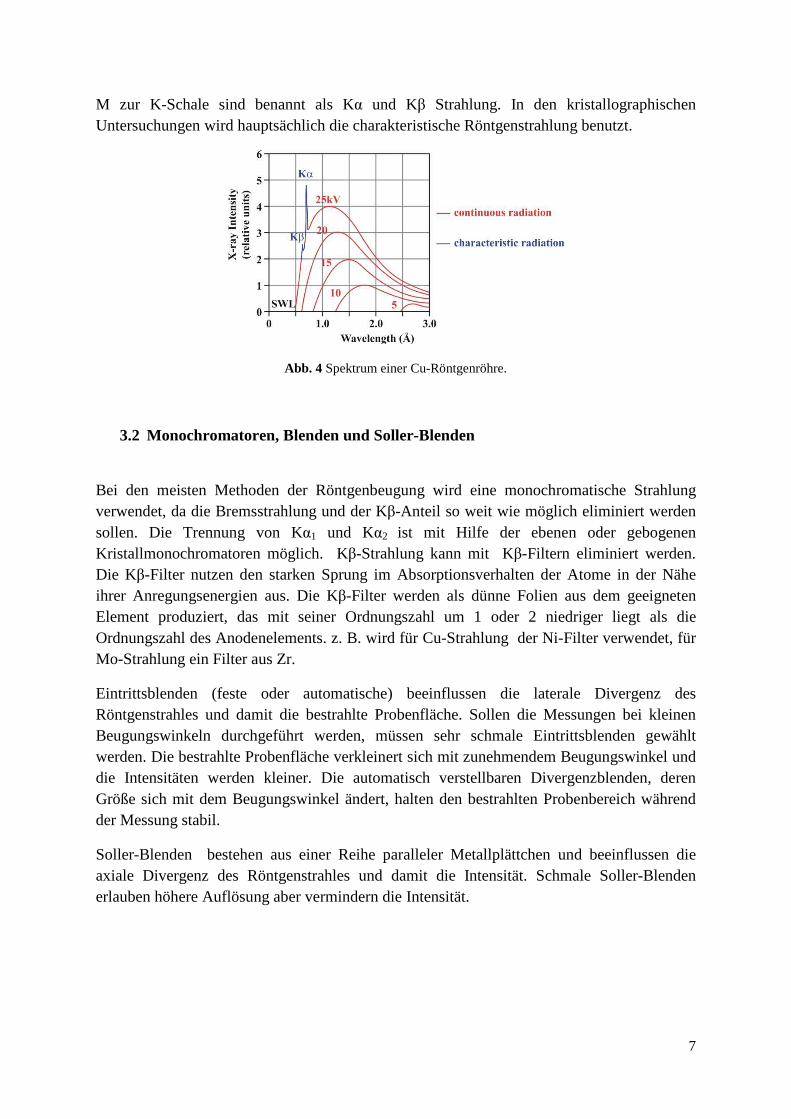
Das typische X-ray Spektrum, erzeugt durch eine Röntgenröhre, ist auf Abb. 4 dargestellt. Das Spektrum besteht aus einzelnen, intensiven Linien, bezeichnet als charakteristische Linien (charakteristische Röntgenstrahlung), die auf dem kontinuierlichen Untergrund überlagert sind. Der kontinuierliche Teil des Spektrums entsteht durch die Elektronen, die rasch und unvorhersehbar abgebremst werden, manche sofort, andere stufenweise. Die Wellenlänge und Intensität der entstehenden Röntgenstrahlung ist abhängig von der angelegten Hochspannung. Diese Strahlung (Bremsstrahlung) ist in der Röntgendiffraktometrie meistens unerwünscht. Die Photonen mit der höchsten Energie (Strahlung mit der längsten Wellenlänge) werden durch die Elektronen emittiert, die durch das Target rasch abgebremst werden. In diesem Fall wird die ganze kinetische Energie des Elektrons in die Energie des Photons umgewandelt. Die drei charakteristischen Linien sind ziemlich intensiv und ergeben sich aus Elektronenübergängen von höheren zu niedrigeren Energieniveaus. Ein Elektron wird beim Aufprall von beschleunigten Elektronen in der Röhre aus einem Energieniveau ausgeschlagen. Die freie Stelle wird durch ein Elektron aus einem höheren Energieniveau aufgefüllt. Die Energiedifferenz zwischen verschiedenen Energieniveaus im Atom ist charakteristisch für das Element. Die Übergänge zwischen L und
1 Fundamentals of powder diffraction and structural characterization of materials, V. Pecharsky, P. Zavalij, Springer 2005
7
M zur K-Schale sind benannt als Kα und Kβ Strahlung. In den kristallographischen Untersuchungen wird hauptsächlich die charakteristische Röntgenstrahlung benutzt.
Abb. 4 Spektrum einer Cu-Röntgenröhre.
3.2 Monochromatoren, Blenden und Soller-Blenden
Bei den meisten Methoden der Röntgenbeugung wird eine monochromatische Strahlung verwendet, da die Bremsstrahlung und der Kβ-Anteil so weit wie möglich eliminiert werden sollen. Die Trennung von Kα1 und Kα2 ist mit Hilfe der ebenen oder gebogenen Kristallmonochromatoren möglich. Kβ-Strahlung kann mit Kβ-Filtern eliminiert werden. Die Kβ-Filter nutzen den starken Sprung im Absorptionsverhalten der Atome in der Nähe ihrer Anregungsenergien aus. Die Kβ-Filter werden als dünne Folien aus dem geeigneten Element produziert, das mit seiner Ordnungszahl um 1 oder 2 niedriger liegt als die Ordnungszahl des Anodenelements. z. B. wird für Cu-Strahlung der Ni-Filter verwendet, für Mo-Strahlung ein Filter aus Zr.
Eintrittsblenden (feste oder automatische) beeinflussen die laterale Divergenz des Röntgenstrahles und damit die bestrahlte Probenfläche. Sollen die Messungen bei kleinen Beugungswinkeln durchgeführt werden, müssen sehr schmale Eintrittsblenden gewählt werden. Die bestrahlte Probenfläche verkleinert sich mit zunehmendem Beugungswinkel und die Intensitäten werden kleiner. Die automatisch verstellbaren Divergenzblenden, deren Größe sich mit dem Beugungswinkel ändert, halten den bestrahlten Probenbereich während der Messung stabil.
Soller-Blenden bestehen aus einer Reihe paralleler Metallplättchen und beeinflussen die axiale Divergenz des Röntgenstrahles und damit die Intensität. Schmale Soller-Blenden erlauben höhere Auflösung aber vermindern die Intensität.

8
Abb. 5 Divergenzblende2 und Soller-Blende.3
3.3 Detektor
Der ortsempfindliche Detektor (PSD, Position Sensitive Detector) ist ein linearer Detektor. Er kann die Intensität vom gebeugten Strahl an verschiedenen Punkten gleichzeitig messen. Dadurch wird eine Messung deutlich schneller im Vergleich zum ganz normalen Proportionalzählrohr. Der typische Detektor ist mit Edelgasen (Argon, Krypton, Xenon mit Stabilisierungszusätzen aus verschiedenen organischen Dämpfen und Halogenen) gefüllt. Eine hohe Spannung wird zwischen den Kathode (Detektorbody) und Anode (zentraler Draht) angelegt. Wenn ein Photon durch das Fenster des Detektors eindringt, wird dieses durch das Gas absorbiert. Dieses ionisiert Xe-Atome und produziert positiv geladene Ionen und Elektronen. Die entstanden Elektronen werden in Richtung der Anode beschleunigt und dort entladen. Der sich daraus ergebene Strom wird an zwei Stellen der Anode gemessen und die Zahl der Stromimpulse ist proportional zur Zahl der absorbierten Photonen. Durch den Zeitunterschied zwischen zwei Messungen desselben Entladungsimpulses ist es möglich die Position der Entladung zu detektieren. Vorausgesetz die Länge der Anode ist bekannt.
Abb. 6 Schema eines PSD Detektors.4
2 Bruker 3 http://pd.chem.ucl.ac.uk/pdnn/inst1/optics1.htm 4 Fundamentals of powder diffraction and structural characterization of materials, V. Pecharsky, P. Zavalij, Springer 2005

9
4. Pulverdiffraktogramm
Das gemessene Pulverdiffraktogramm enthält zahlreiche Informationen über eine gemessene Probe (siehe Abb. 7).
Abb. 7 Informationen, die das Pulverdiffraktogramm enthält.5
Das Pulverdiffraktogramm ist für jede Substanz charakteristisch und kann daher, ähnlich wie ein Fingerabdruck, für die Identifizierung verwendet werden. Jede Substanz behält auch in einem Gemisch ihr eigenes Diagramm, wobei Linien verschiedener Substanzen zusammenfallen und sich aufaddieren können. Für qualitative Phasenanalyse hat ICDD die Röntgendaten aller untersuchten Substanzen in der PDF-Kartei gesammelt und als Datenbank zusammengestellt. Die Gesamtdaten sind in einen anorganischen, organischen und organo-metallischen Teil gegliedert. Als weitere Funktionen sind eine Unterrichtshilfe sowie Daten häufig auftretender Phasen, Daten von Mineralien, Metallen, sowie Daten forensischer Materialen erhältlich. Die PDF-Kartei enthält für jede Substanz alle dhkl-Werte, die dazugehörenden Intensitäten, sowie eine Anzahl weiterer Größen. Es gibt verschiedene Programme, die die PDF-Datenbank zur qualitativen Phasenanalyse verwenden (z.B. Analyze (Seifert), X’Pert HighScore (PANalytical), EVA (Bruker), Match). Bei der Identifizierung mit Hilfe der Analyze-Software sucht das Programm im ersten Schritt die drei stärksten Linien, wenn dabei eine Übereistimmung in der PDF-Kartei vorhanden ist, prüft das 5 Powder Diffraction, Theory and Practice R. Dinnebier et. all; RSCPublishing, 2008

10
Programm ob Restlinien vorhanden sind. Angaben über die chemische Zusammensetzung der Probe können die Suchzeiten stark verkürzen.
5. Probenvorbereitung
Die Pulver sollten eine optimale Korngröße im Bereich 1-10 µm haben. Die Zerkleinerung eines Pulvers ist durch Mörsern oder Mahlen möglich. Eine Erwärmung der Probe sollte dabei vermieden werden, um eventuellen Phasenumwandlungen oder chemischen Reaktionen (eine Oxidation durch den Luftsauerstoff) vorzubeugen. Das Präparat wird in eine Vertiefung des Probenträgers gegeben und die Oberfläche wird vorsichtig mit dem Objektträger angedrückt bzw. glattgestrichen. Wenn die Form der Partikel anisotrop (plättchen- oder nadelförmig) ist, haben die Partikel eine Tendenz Vorzugsorientierung einzunehmen. Beispielsweise richten sich Plättchen gern parallel zur Probenoberfläche aus. Die Textur hat einen großen Einfluss auf die gemessenen Intensitäten (manche Reflexe sind extrem verstärkt, manche stark abgeschwächt). Um Vorzugsorientierung zu reduzieren, wird das Präparat während der Messung rotiert oder seitlich in den Probeträger eingefüllt.
Abb. 8 Verschiedene Probenträger (a-c) und Glättung der Oberfläche mit dem Objektträger (d).

11
6. Hoch-und Tieftemperaturkammer
Zur Untersuchung der Stabilität einer Verbindung oder zur Charakterisierung von Phasenumwandlungen bei bestimmten Temperaturen oder Drücken im Festkörper werden Hoch,- Tieftemperatur, und Druckkammern verwendet. Das sind abgeschlossene Gehäuse, in denen die Probe erhitzt oder gekühlt werden kann. Der Durchgang der Röntgenstrahlen wird durch dünne Kunststofffolien oder Berrylliumbleche gewährleistet. Es ist auch möglich, die Atmosphäre im Proberaum zu kontrollieren oder diese zu evakuieren. Dadurch kann beim Aufheizen eine mögliche Oxidation der Probe verhindert werden, indem der Probenraum mit reinem Stickstoff oder Helium gefüllt wird. Tieftemperaturkammern werden mit flüssigem oder verdampfendem Stickstoff verwendet.
Abb. 9 Hoch- (oben) und Tieftemperaturkammern (unten).6
6 Anton Paar (anton-paar.com); HTK 1200N; TTK 450

12
7. Rietveld-Methode
Ende der sechziger Jahre hat H. M. Rietveld eine Methode entwickelt, in der alle Messpunkte eines Pulverdiffraktogramms mit analytischen Funktionen beschrieben wird. Die Funktionsparameter werden im Verfeinerungsprozess mit Hilfe der Methode der kleinsten Quadrate simultan angepasst, bis die beste mögliche Übereinstimmung zwischen gemessenem und einem anhand eines Strukturmodells, sowie von Beugungseffekten, Instrumenten- und Probeneffekten berechneten Pulverdiffraktogramms erreicht ist. Die überlappenden Reflexe werden in ihre Anteile zugehörend zu den verschieden Bragg-Reflexen aufgeteilt.
Die Rietveld-Verfeinerung benötigt ein Strukturmodell. Als Strukturmodel kann das Model von einer strukturell verwandten Substanz gewählt werden oder das Strukturmodel wird ab initio von Pulverdaten erhalten. Dafür braucht man gute chemische und physikalische Voruntersuchungen.
Das Pulverdiffraktogramm eines polykristallinen Materials kann als Überlagerung individueller Reflexprofile betrachtet werden. Die integrierte Fläche jedes Reflexes ist proportional der Bragg-Intensität Ik, wobei k für die Millerschen Indizes hkl steht.
Es werden Intensitätswerte yoi an Punkten i der Aufnahme in digitaler Form in Folge von mehreren tausend gleich großen Schritten (step scan) gemessen, die sich zu Bragg-Intensitäten Ik summieren. Den beobachteten Intensitäten yoi werden berechnete Intensitäten yci am Messpunkt i
gegenübergestellt. Die berechneten Intensitäten yci für jeden Messpunkt werden aus dem Strukturmodell durch Aufsummieren der Beiträge innerhalb eines bestimmten Bereiches benachbarten Reflexe und dem Untergrund berechnet:
yci - berechnete Intensität s - Skalierungsfaktor lp - Lorenz-, Polarisationsfaktor mk - Multiplizitätsfaktor Fk - Strukturfaktor f - Funktion für das gewählte Reflexprofil Tk - Faktor für bevorzugte Orientierung (Texturfaktor) A - Absorptionsfaktor ybi - Untergrundintensität am i-ten Messpunkt
Die berechneten Werte yci werden an gemessene Intensitätswerte nach der Methode kleinster Fehlerquadrate angepasst. Die Anpassung wird an mehreren tausend Datenpunkte yoi gleichzeitig gemacht. Die Größe Sy die im least square Verfeinerungsprozess minimiert wird, ist durch die Summe über alle Datenpunkte definiert.
bikkikk
kci yATfFmlpsy +⋅⋅Θ−Θ⋅⋅⋅= ∑ )22(2

13
wi - Gewichtungsfaktor yoi - gemessene Intensität am i-ten Messpunkt yci - berechnete Intensität für den Messpunkt i
Die verfeinerbaren Modellparameter enthalten Atompositionen, Temperaturfaktoren und Besetzungszahlen, sowie Parameter für den Untergrund, für Gitterkonstanten, für geometrische und Instrumenteigenschaften, für Probenabweichungen, wie Fehler in der Probenhöhe, für amorphe Anteile und für profilverbreiternde Anteile wie Kristallitgröße und Gitterstörungen. Mehrere Phasen können simultan verfeinert werden, und vergleichende Analysen der verschiedenen Skalierungsfaktoren sind eine der besten Methoden zur Durchführung quantitativer Analysen. Während einer Rietveld-Verfeinerung werden die Parameter so verändert, dass die Funktion Sy ein Minimum erreicht, d. h. die beste Übereistimmung zwischen berechnetem und gemessenem Profil erreicht ist. Diese Übereistimmung hängt stark vom Auffinden des globalen Minimums und der Wahl des richtigen Strukturmodells ab. Bei der Verfeinerungsstrategie ist die Reihenfolge der verfeinerbaren Parameter wichtig:
1. Skalierungsfaktor SF 2. Untergrund 3. Nullpunktsfehler 4. Gitterkonstanten 5. Atomparametern x, y, z 6. Parameter U, V, W (2θ –abhängige Halbwertsbreite) 7. Temperaturfaktoren 8. Besetzungsfaktoren 9. Vorzugsorientierung
Zum Abschluss sollen die Übereistimmungsfaktoren möglichst niedrige Werte haben. Das Strukturmodell soll richtig im Sinne der Kristallchemie sein, es soll eine Übereistimmung des Strukturmodells mit der Elementaranalyse sein, die Temperaturfaktoren sollen positiven Werte haben; zusätzliche Berechnungszyklen bewirken keine erheblichen Veränderungen des Strukturmodells.
Übereinstimmungsfaktoren:
Ik – die integrierten Intensitäten des Reflexes K
2)( cioii iy yywS −=∑
∑
∑ −=
21
)(
21
)(2
1
)(
)(
)()(
obsk
calckobsk
F
I
IIR
∑∑ −
=oi
cioiP y
yyR
∑∑ −
=)(
)()(
obsk
calckobsk
B I
IIR

14
Zur Beurteilung der Verfeinerung sind die numerischen Kriterien und die graphische Darstellung, z. B. Differenzdarstellungen zwischen gemessenen und berechneten Profilen sehr wichtig. Bei der Verfeinerung eröffnet eine graphische Darstellung von gemessenen, berechneten und Differenzdaten die Möglichkeit, Fehler beim Skalierungsfaktor, beim Niveau oder Form der Untergrundmodellierung sowie bei den Gitterkonstanten oft auf den ersten Blick zu erkennen. Man kann leicht Phasenverunreinigungen erkennen und wie weit das Strukturmodell fehlerhaft ist. Außerdem hilft die graphische Darstellung beim Erkennen von Profilabweichungen, wie unkorrekt berechnete Ausläufer der Reflexprofile, unkorrigierte Asymmetrie, anisotrope Profilverbreiterung oder die Wahl einer unpassenden Profilfunktion.
Die meist verwendeten Programme für Rietveld-Verfeinerung:
Jana2006, RIETAN, FULLPROF, EXPO 2013, GSAS, DBWS, XRS-82, TOPAS, DASH

15
8. Ziel des Versuchs
Das Ziel des Versuchs ist das Beherrschen von theoretischen und vor allem praktischen Grundlagen der Pulverdiffraktometrie. Im Praktikumsversuch soll ein Gemisch aus drei unbekannten Phasen mittels Röntgenbeugung eindeutig identifiziert und charakterisiert werden. Die wichtigsten Schritte, die praktisch im Versuch bei der Analyse einer kristallinen Probe angewendet werden, umfassen: Probenaufbereitung, Messen eines Pulverdiffraktogramms, Zuordnung aller gemessenen Peaks unter Zuhilfenahme einer Datenbank (qualitative Phasenanalyse), Durchführung einer Rietveld-Verfeinerung der Messdaten und Bestimmung des Massenanteil der drei Phasen in der Mischung.
9. Versuchsdurchführung
Sie erhalten vier Proben, drei gängige Minerale und ein Pulvergemisch aus drei kristallinen Substanzen an denen Sie eine qualitative und eine quantitative Phasenanalyse (Pulvergemisch) durchführen sollen.
9.1 Probenpräparation und Messung
In dem Versuch sollen vier Pulverdiffraktogramme aufgenommen werden. Dazu müssen alle 4 Proben für die Messung am Pulverdiffraktometer Seifert TT3003 vorbereitet werden. Mörsern Sie die Proben, geben Sie die Substanzen auf den Probenträger und montieren Sie diesen im Diffraktometer. Bereiten Sie ein Messprogram mit den geeigneten Messparametern vor und starten Sie die Messungen.
9.2 Qualitative Phasenanalyse
Gleichen Sie die gemessenen Pulverdiffraktogramme mittels entsprechender Software (Analyze, Seifert) mit den Einträgen einer Datenbank (Powder Diffraction File, ICSD) ab. Diskutieren Sie mit Ihrem Betreuer, ob die gefundenen Ergebnisse sinnvoll sind und identifizieren Sie eindeutig die drei Minerale in Ihren Proben.

16
9.3 Bestimmung der Gitterkonstanten und Raumgruppe von Pulverdaten
Bestimmen Sie für eine vom Betreuer ausgewählte Probe die Gitterkonstanten (Fullprof Package) und Raumgruppe (Checkcell).
9.4 Quantitative Phasenanalyse
Für die quantitative Phasenanalyse benötigen Sie strukturelle Daten ihrer Substanz. Diese können Sie in einer Strukturdatenbank (ICSD, FIZ-Karlsruhe) finden. Ermitteln Sie für die quantitative Phasenanalyse nach der Rietveld-Methode (Jana2006) den prozentualen Massenanteil für jede einzelne Substanz im Ihrem Gemisch.
10. Literatur
- Röntgenpulverdiffraktometrie, R. Allmann, Springer 2003
- Röntgenstrukturanalyse und Rietveldmethode, H. Krischner, B. Koppelhuber-Bitschnau, Vieweg 1994
- Moderne Röntgenbeugung, L. Spieß et al., Vieweg + Tübner Verlag, 2009
- Principles and Applications of Powder Diffraction, A. Clearfield et al., WILEY 2008
- Powder Diffraction, Theory and Practice R. Dinnebier et al., RSCPublishing 2008

![Allgemeine Sicherheitsunterweisung · [ und eine extra Unterweisung notwendig ] radioaktive Stoffen Röntgenstrahlung Laser Spannungen über 60V Elektrostatik Hitze chemische Reaktionen](https://img.pdfslide.org/doc/110x75/5b9f5b4509d3f25b318cf822/allgemeine-sicher-und-eine-extra-unterweisung-notwendig-radioaktive-stoffen.jpg)