Embed Size (px)
Citation preview

Gen- und verfahrenstechnische Ansätze zur
Optimierung von Aspergillus niger als
Expressionsplattform
vorgelegt von
Dipl.-Ing. Franziska Wanka
geb. in Berlin
von der Fakultät III - Prozesswissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktorin der Ingenieurwissenschaften
- Dr.-Ing. -
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. rer. nat. Roland Lauster
Gutachterin: Prof. Dr.-Ing. Vera Meyer
Gutachter: Prof. Dr. rer. nat. Peter Neubauer
Gutachter: Dr. rer. nat. Manfred Gossen
Tag der wissenschaftlichen Aussprache: 11.05.2016
Berlin 2016

„Ich hielt es für besser, etwas zu leisten, als nichts zu versuchen, weil man nicht alles leisten
kann.“
Alexander von Humboldt

Die experimentellen Arbeiten für diese kumulative Dissertationsschrift wurden von Juni 2011
bis Januar 2016 unter der Leitung von Prof. Dr.-Ing. Vera Meyer in der Abteilung
Angewandte und Molekulare Mikrobiologie (Institut für Biotechnologie) der Technischen
Universität Berlin durchgeführt.
Zum Erlernen der praktischen Durchführung der Retentostat-Kultivierung (Kapitel 5)
besuchte ich für vier Wochen die Arbeitsgruppe Molecular Microbiology & Biotechnology
von Dr. Arthur Ram an der Leiden Universiteit (Niederlande).
Des Weiteren konnte ich einen vierwöchigen Forschungsaufenthalt in der Abteilung
Molecular and applied Biocatalysis von Dr. Ayelet Fishman am Technion-Israel Institute of
Technology in Haifa (Israel) durch die Förderung der Deutschen-Technion Gesellschaft
absolvieren.
Franziska Wanka, Berlin, den 10.03.2016

- 4 -
Inhaltsverzeichnis
Zusammenfassung 5
Abstract 6
Kapitel 1 Einleitung 7
1.1 Einordnung und Charakterisierung von Aspergillus spp. 8
1.2 Aspergillus niger im Vergleich zu anderen häufig genutzten Zellfabriken 10
1.3 Die Bedeutung von Aspergillus niger als Expressionsplattform 11
1.4 Strategien zur Produktivitätssteigerung von Aspergillus niger 16
1.4.1 Induzierbare Promotoren und Expressionssysteme 16
1.4.2 Kultivierungsbedingungen 18
1.4.3 Morphologie 21
1.4.4 Fermentationsstrategien zur Produktion 22
1.5 Ziel und Gliederung der Arbeit 26
Kapitel 2 Aspergillus: A Cell Factory with Unlimited Prospects 28
Kapitel 3 Engineering of Aspergillus niger for the production of secondary metabolites 92
Kapitel 4 Tet-on, or Tet-off, that is the question: Advanced Conditional Gene
Expression in Aspergillus
121
Kapitel 5 Highly active promoters and native secretion signals for protein production
during extremely low growth rates in Aspergillus niger
154
Diskussion und Ausblick 179
Epilog I 184
Publikationen 188
Patente 189
Posterbeiträge 189
Stipendien und Preise 190
Danksagung 191
Referenzen 192

- 5 -
Zusammenfassung
Aspergillus niger besitzt die Fähigkeit, große Mengen an Säuren und Proteinen zu
synthetisieren und in die Umwelt auszuscheiden. Diese besondere Eigenschaft wird
biotechnologisch genutzt, um sowohl Plattformchemikalien als auch industrielle Enzyme
herzustellen. Trotz beeindruckender Stoffwechselleistungen, gibt es weiterhin
Forschungsbedarf, um die Produktausbeuten von A. niger mit Hilfe von gen- und
verfahrenstechnischer Methoden zu optimieren.
Diese Arbeit fokussierte sich daher zum einen auf gentechnische Ansätze, um synthetische
Genschalter, wie Tetracyclin-abhängige Systeme (Tet-on und -off), an A. niger zu adaptieren
und ihre Anwendbarkeit zu evaluieren. Es konnte erfolgreich gezeigt werden, dass
pharmakologisch interessante Sekundärmetabolite, wie das nicht-ribosomale
Cyclodepsipeptid Enniatin, in A. niger unter Regulierung des Tet-on-Systems in wirtschaftlich
relevanten Mengen (g/l) in Fed-Batch-Fermentationen produziert werden können. Des
Weiteren gelang es, eine genomisch stabile Variante des Tet-off-Systems für A. niger zu
etablieren. Eine besondere Herausforderung stellte hierbei die Validierung der am besten
geeignetsten Transaktivatorvariante und deren transkriptionelle Expressionsstärke dar. Die
Verwendung eines Promotors mit moderater Aktivität ermöglichte eine stabile Expression des
toxischen Transaktivators im Genom von A. niger. Die funktionale und quantitative Analyse
und Validierung des neuen Tet-off-Systems wurde anhand dreier Gene durchgeführt. Somit
stehen nun stabile und induzierbare Genschalter zur Verfügung, die zukünftig zur Aufklärung
und Optimierung metabolischer Stoffwechselwege in Aspergillus beitragen werden.
Zum anderen war es das Ziel dieser Arbeit, autoinduzierbare Promotoren zu ermitteln, die bei
geringer Wachstumsrate aktiv sind, um die kontinuierliche Perfusionsfermentation zur
Produktion extrazellulärer heterologer Proteine in A. niger nutzbar zu machen. Es wurden mit
Hilfe von Transkriptomanalysen zwei geeignete Promotoren (anafp, hfbD) identifiziert, deren
Promotoraktivitäten mit einem intrazellulären Reportergen (Luziferase) und ebenso mit einem
heterologen extrazellulären, antifungal wirkenden Protein (AFP) in 14-tägigen
Perfusionskultivierungen validiert. Beide Promotoren unterscheiden sich bezüglich der
Expressionsstärke und des temporären Verlaufes, wobei der anafp-Promotor eine sehr hohe
aber peakartige und der hfbD-Promotor eine konstante aber moderate Expression vermittelt.
Ebenso konnten die jeweiligen Sekretionssignalsequenzen der anafp und hfbD Gene ihre
Funktionalität durch erfolgreiche Sekretion des heterologen Proteins unter Beweis stellen. Die
systematische Analyse und Anpassung neuer autoinduzierbarer Promotoren an die
Perfusionskultivierung von A. niger legt somit eine geeignete molekulargenetische Grundlage
für zukünftige kontinuierliche Prozesse in der Pilzbiotechnologie.

- 6 -
Abstract
Aspergillus niger has the ability to synthesise large amounts of acids and proteins, and to
secrete these into the environment. This particular property is used in biotechnology to
produce platform chemicals and industrial enzymes. Despite impressive metabolic activities,
there is still a need for research to optimise product yields of A. niger using genetic and
engineering approaches.
This work therefore focuses on using genetic approaches to adapt synthetic gene switches,
such as tetracycline-dependent systems (Tet-on and -off), and evaluate their applicability in A.
niger. It was successfully demonstrated that pharmacologically interesting secondary
metabolites, such as the non-ribosomal cyclodepsipeptide enniatin under the control of the
Tet-on system can be produced in economically relevant amounts (g/l) in fed-batch
cultivation of A. niger. Furthermore, there has been success in establishing a genetically stable
variant of the Tet-off system in A. niger. A particular challenge hereby is the validation of the
most appropriate transactivator variant and their transcriptional expression strength. The use
of a promoter with moderate activity enabled stable expression of the toxic transactivator in
the genome of A. niger. The functional and quantitative analysis and validation of the new
Tet-off system was carried out based on three genes. Stable and inducible gene switches are
thus now available which will contribute to the elucidation and optimization of metabolic
pathways in Aspergillus in the future.
Secondly, the objective of this work was to determine autoinducible promoters that are active
at low growth rates, in order to achieve the continuous perfusion for the production of
extracellular heterologous proteins in A. niger. Two suitable promoters (anafp, hfbD) were
identified with the aid of transcriptomic data, and their promoter activities were validated with
an intracellular reporter (luciferase) and a heterologous extracellular antifungal protein (AFP)
in 14 days of perfusion cultivations. The two promoters examined differed in regards to
expression level and temporary activity, whereas the anafp promoter results in a very high but
peak-like result, and the hfbD promoter a constant, yet moderate expression. Likewise, the
respective secretion signal sequences of anafp and hfbD genes proved their functionality by
successful secretion of the heterologous protein. Systematic analysis and adaptation of new
autoinducible promoters for perfusion cultivation of A. niger thus create an appropriate
molecular genetic basis for future continuous processes in fungal biotechnology.

- 7 -
Kapitel 1
Einleitung

- 8 -
Abbildung. 1: A. niger im Fokus der
zeitgenössischen Kunst.
Öl auf Leinwand, F. Wanka, 2012, 30 x 40 cm
1.1 Einordnung und Charakterisierung von Aspergillus spp.
Nach der mykologischen Geschichte war der italienische Priester und Biologe Micheli 1729
der Erste, der den Gießkannenschimmel unter dem Mikroskop zu sehen bekam. Dieser
erinnerte ihn durch die sporentragende Struktur an Aspergill, ein sakrales Gerät zur
Weihwasserverteilung, daher gab er dem Schimmelpilz den Namen Aspergillus (Ainsworth,
1976). Seitdem beschäftigen sich Mykologen bis heute mit der Systematik der Gattung
Aspergillus, 1926 wurde die erste Unterteilung der bis dahin bekannten 350 Arten in Gruppen
veröffentlicht (Thom & Church, 1926), seit 1985 wird die Gattung in Untergattungen und
Sektionen eingeteilt (Samson & Pitt, 1985). Im neuesten Aspergillus Buch, herausgegeben
von Machida and Gomi (2010), wird darauf hingewiesen, dass Aspergillus weniger eine
Gattung ist, sondern eher ein Formtaxon, es ist also aus morphologischen Gründen
zusammengehörig und nicht aus stammesgeschichtlichen. Die Nomenklatur ist bis heute recht
unübersichtlich und wird stetig aktualisiert, nur die anamorphe (asexuelle) Form darf als
Aspergillus bezeichnet werden, die teleomorphe (sexuelle) Form, welche eher selten
vorkommt, muss je nach Art einer sexuellen Gattung zugeordnet werden (Geiser et al., 2008).
Alle Aspergilli sind multizelluläre Organismen, die aus langen fadenförmigen Zellen,
bezeichnet als Hyphen, aufgebaut sind. Die
Hyphe ist in Kompartimente unterteilt durch
sogenannte Septa, eine Querwand mit
Septenporus, der die Diffusion von Nährstoffen,
Proteinen und sogar Zellorganellen innerhalb
der Hype reguliert. Ein Netzwerk aus Hyphen
wird Myzel genannt. Bei der asexuellen
Fortpflanzung entsteht aus der Hyphe ein
Konidiophor (Abb. 1), welcher aus einem
Vesikel überzogen von Metulae mit Phialiden
besteht. Die kegelförmigen Phialiden sind
spezialisiert auf die Produktion von asexuellen
Sporen, die in langen Ketten abgeschnürt
werden und sich aufgrund ihrer Hydrophobizität
in der Luft verbreiten können, bis sie auf
neuem Nährsubstrat wieder zu einem Myzel
auskeimen.

- 9 -
Die industrielle Nutzung von Aspergillus niger, dem schwarzen Gießkannenschimmel,
begann bereits 1860 mit der Herstellung von Gallussäure durch die Fermentation von
Tanninen aus Pflanzengallen (Lockwood & Moyer, 1938). Im Jahr 1919 startete die
Massenproduktion von Zitronensäure mit A. niger, lizenziert durch Pfizer (Bennett, 1998).
Heute ist A. niger weiterhin der bedeutendste Produzent von Zitronensäure und darüber
hinaus von zahlreichen Enzymen (z.B. α-Amylase, Cellulase, Pectinase etc. (Kapitel 2)) und
zunehmend auch für heterologe Proteine (z.B. Chymosin (Dunn-Coleman et al., 1991),
humanes Interleukin-6 (Broekhuijsen et al., 1993), Mangan-Peroxidase (Conesa et al., 2000))
und Sekundärmetaboliten, z.B. Enniatin (Kapitel 3). Daneben ist auch Aspergillus oryzae von
industrieller Relevanz als Produzent von Kojisäure sowie von kommerziellen Enzymen, wie
Protease, Lipase und Amylase (Kapitel 2). Mit A. terreus wird Itaconsäure und Lovastatin, ein
Lipidsenker, von Merck seit 1987 erfolgreich produziert (Alberts, 1988).
Abbildung 2 zeigt die Anzahl der Publikationen der gängigsten Aspergillus Arten zum
aktuellen Zeitpunkt im Vergleich zu den letzten 10 und 20 Jahren, hauptsächlich sind die
Artikel im Bereich der Grundlagen- als auch in der angewandten Forschung erschienen.
Ersichtlich wird, dass A. niger als Spezies am häufigsten Gegenstand wissenschaftlicher
Publikationen ist. Innerhalb von 10 Jahren haben sich die Publikationen jeweils ungefähr
verdoppelt, wie bei A. niger oder auch bei dem Modellorganismus für eukaryotische
Zellbiologie A. nidulans zu sehen ist. Eine Ausnahme stellt die Anzahl der Artikel über den
humanpathogenen A. fumigatus dar, diese hat sich von 2006 bis 2016 vervierfacht.
Abbildung 2: Vergleich der Popularität von Aspergillus spp. anhand der Anzahl von
veröffentlichten Artikeln bis 1996, 2006 und 22.02.2016 (ermittelt mit Google Scholar).
0
200000
400000
600000
800000
A. niger A. oryzae A. fumigatus A. flavus A. nidulans A. terreus
Anza
hl
der
Art
ikel
1996
2006
2016
- 10 -
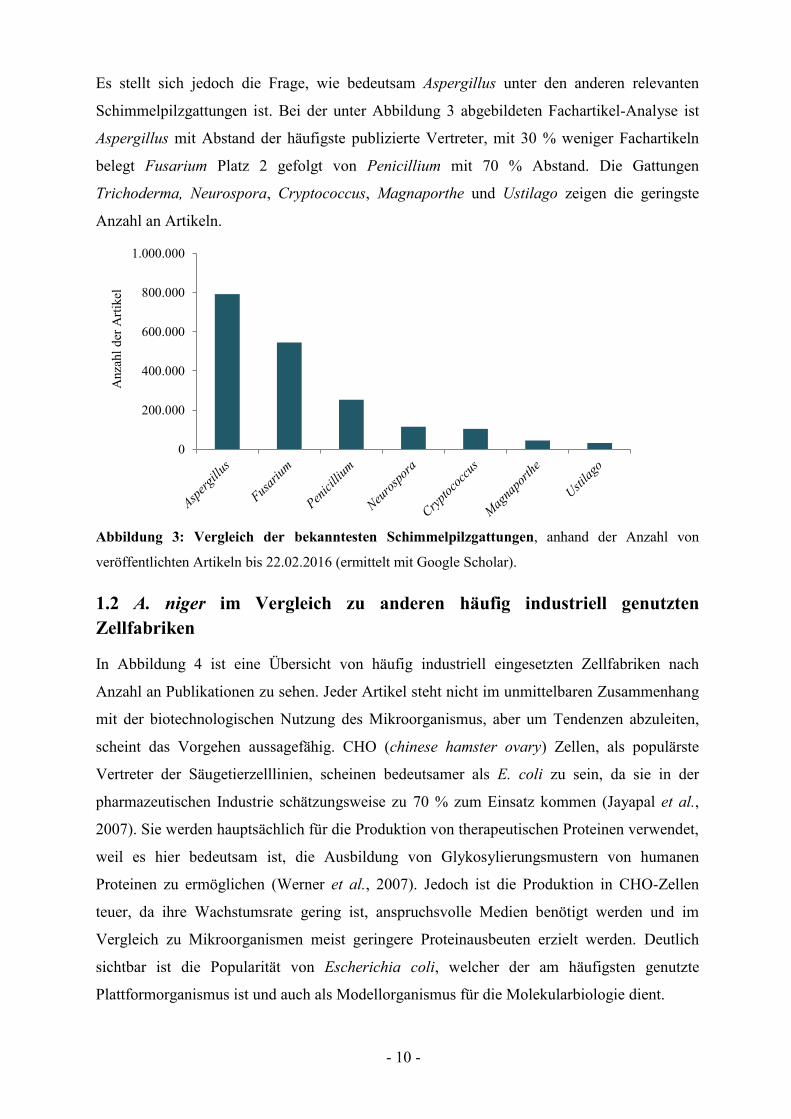
Es stellt sich jedoch die Frage, wie bedeutsam Aspergillus unter den anderen relevanten
Schimmelpilzgattungen ist. Bei der unter Abbildung 3 abgebildeten Fachartikel-Analyse ist
Aspergillus mit Abstand der häufigste publizierte Vertreter, mit 30 % weniger Fachartikeln
belegt Fusarium Platz 2 gefolgt von Penicillium mit 70 % Abstand. Die Gattungen
Trichoderma, Neurospora, Cryptococcus, Magnaporthe und Ustilago zeigen die geringste
Anzahl an Artikeln.
Abbildung 3: Vergleich der bekanntesten Schimmelpilzgattungen, anhand der Anzahl von
veröffentlichten Artikeln bis 22.02.2016 (ermittelt mit Google Scholar).
1.2 A. niger im Vergleich zu anderen häufig industriell genutzten
Zellfabriken
In Abbildung 4 ist eine Übersicht von häufig industriell eingesetzten Zellfabriken nach
Anzahl an Publikationen zu sehen. Jeder Artikel steht nicht im unmittelbaren Zusammenhang
mit der biotechnologischen Nutzung des Mikroorganismus, aber um Tendenzen abzuleiten,
scheint das Vorgehen aussagefähig. CHO (chinese hamster ovary) Zellen, als populärste
Vertreter der Säugetierzelllinien, scheinen bedeutsamer als E. coli zu sein, da sie in der
pharmazeutischen Industrie schätzungsweise zu 70 % zum Einsatz kommen (Jayapal et al.,
2007). Sie werden hauptsächlich für die Produktion von therapeutischen Proteinen verwendet,
weil es hier bedeutsam ist, die Ausbildung von Glykosylierungsmustern von humanen
Proteinen zu ermöglichen (Werner et al., 2007). Jedoch ist die Produktion in CHO-Zellen
teuer, da ihre Wachstumsrate gering ist, anspruchsvolle Medien benötigt werden und im
Vergleich zu Mikroorganismen meist geringere Proteinausbeuten erzielt werden. Deutlich
sichtbar ist die Popularität von Escherichia coli, welcher der am häufigsten genutzte
Plattformorganismus ist und auch als Modellorganismus für die Molekularbiologie dient.
0
200.000
400.000
600.000
800.000
1.000.000
Anza
hl
der
Art
ikel

- 11 -
Weitere prokaryotische Zellfabriken wie Bacillus subtilis, bekannt für die Herstellung von
Vitamin B2 und Corneybacterium glutamicum, als Produzent für Glutaminsäure, zeigen
deutlich weniger Publikationen. A. niger zeigt fast so viele Treffer wie die Hefe
Saccharomyces cerevisiae, die als eukaryotischer Modellorganismus bekannt ist und im
Lebensmittelbereich, z.B. bei der Brot-, Bier- und Weinherstellung, eine große Bedeutung hat.
Hingegen ergibt Picha pastoris wesentlich weniger Treffer, obwohl diese Hefe auch häufig
zum Einsatz für die rekombinante Proteinherstellung kommt (Cereghino & Cregg, 2000).
Abbildung 4: Vergleich häufig industriell genutzter Zellfabriken, anhand der Anzahl von
veröffentlichten Artikeln bis 22.02.2016 (ermittelt mit Google Scholar).
Je nach den Anforderungen des jeweiligen biotechnologischen Produktes sollte der
Expressionsorganismus ausgewählt werden, zum jetzigen Zeitpunkt gibt es jedoch nicht die
eine optimale Zellfabrik, die alle anderen ersetzt.
1.3 Die Bedeutung von Aspergillus niger als Expressionsplattform
Der filamentöse Pilz Aspergillus niger ist ein sehr effizienter Saprobiont, da er eine hohe
Sekretionskapazität für verschiedene hydrolytische Enzyme besitzt, um sich von
extrazellulären organischen Substanzen zu ernähren. Diese hohe Sekretionsleistung von
Hydrolasen, wie z.B. Cellulase (Dashtban et al., 2009), Glucoamylase (Withers et al., 1998),
Lipase (Mahadik et al., 2002), Pektinase (Acuna-Arguelles et al., 1995), zusammen mit
seinen vielseitigen primären und sekundären Metaboliten macht A. niger zu einer
herausragenden Expressionsplattform. In den letzten Jahren wurde A. niger auch ein
zunehmend geschätzter Wirt für die Überexpression von heterologen Proteinen (Kapitel 2,
Fleissner and Dersch, 2010; Lubertozzi and Keasling, 2009; Punt et al., 2002) .
0
1.000.000
2.000.000
3.000.000
Anza
hl
der
Art
ikel

- 12 -
In Tabelle 1 ist eine Übersicht dargestellt, welche Vor-und Nachteile A. niger als heterologer
Proteinproduzent besitzt und wo Verbesserungsbedarf besteht. A. niger produziert unter
anderem eine große Menge an endogenen Proteasen, die insbesondere bei der heterologen
Proteinproduktion problematisch werden können (Punt et al., 2002), nur wenige rekombinante
Proteine werden nicht abgebaut, z.B. das kleine kompakte antifungale Protein von A.
giganteus (Kapitel 5). Daher beschäftigen sich Wissenschaftler schon seit über 40 Jahren
damit, Protease-defiziente Stämme zu entwickeln (Cohen, 1977). Einige Fortschritte wurden
zwar schon erreicht (siehe 1.4.2), aber von dem Ziel des proteasefreien Stammes, wenn es
denn physiologisch überhaupt möglich ist, ist man noch einige Forschungsprojekte entfernt
(Tab.1).
Schnellere und präzisere gentechnische Modifikationen werden durch kontinuierliche neue
Entwicklungen ermöglicht, die dazu beitragen A. niger besser zu erforschen und noch
effizienter als Expressionswirt zu nutzen. Mittlerweile kann man die notwendigen
Genkonstrukte in E. coli dank Aqua Cloning (Beyer et al., 2015) innerhalb von einem Tag
herstellen. Durch die Hochdurchsatz-Sequenzierungstechnologien stehen für A. niger bereits
drei annotierte Genome zur Verfügung, hierbei handelt es sich um zwei Wildtypstämme
ATCC1015, starker Zitronensäureproduzent (sequenziert vom US Department for Energy,
Joint Genome Institute; Andersen et al., 2011) und NRRL3 (sequenziert von der US Firma
Integrated Genomics) sowie um einen Proteinproduzenten CBS513.88 (sequenziert von der
niederländischen Firma DSM; Pel et al., 2007). Durch die Kenntnis der Genome wurden bzw.
werden Realisierungen von gezielten gentechnischen Veränderungen wesentlich vereinfacht.
Ein großer Vorteil von A. niger ist, dass die zielgerichtete Integration von externer DNA ins
Genom durch die PEG (Polyethylenglycol)-vermittelte Protoplastentransformation sehr gut
funktioniert (Meyer et al., 2010) und die resultierenden Stämme im Regelfall genetisch stabil
bleiben (Tab. 1), es sei denn es werden Sequenzen für toxische Proteine eingeführt (siehe
Kapitel 4). Wenn man den Prozess der Stammerstellung allerdings mit Prokaryonten bzw.
Hefen vergleicht, wäre es wünschenswert, die Transformationseffizienz weiter zu erhöhen.
Mit dem ΔkusA Stamm, welcher eine nicht-homologe Integration ins Genom verringert,
konnte die homologe Rekombinationsfrequenz bis auf 95 % erhöht werden, wenn 1000 bp als
Flanken benutzt wurden (Meyer et al., 2007). Jedoch zeigten Untersuchungen auch, dass
durch die Inaktivierung des NHEJ (Non-homologous end-joining)-Weges Stämme entstehen,
die anfälliger für Mutagene sind, da der DNA-Doppelstrangbruch-Reparaturmechanismus
beeinflusst wird (Meyer et al., 2007). Die Deletion von Genen wurde durch die Split Marker
Methode (Arentshorst et al., 2015) beziehungsweise durch das CRISPR-Cas9-System
(Clustered Regularly Interspaced Short Palindromic Repeats), welches auf einer RNA

- 13 -
gesteuerten Mutagenese beruht (Nødvig et al., 2015), vereinfacht. Vom CRISPR-Cas9-
System erhofft man sich, dass sich genetische Variationen mit weniger Aufwand in Genome
einführen lassen und dass man zukünftig sogar gezielt innerhalb einer Transformation
verschiedene, parallel angesteuerte Integrationsorte verändern kann. Nach einer erfolgreichen
A. niger Transformation entstehen heterokaryontische Zellen, die mehrere, genetisch
unterschiedliche Zellkerne enthalten, eine zweifach wiederholte Vereinzelung der Sporen ist
unumgänglich um homokaryontische Zellen, die nur aus genetisch identischen Nuclei
bestehen, zu erhalten. Diese Aufreinigung zum homokaryontischen Myzel benötigt mehr als
sechs Tage und lässt sich nicht verkürzen, dennoch wäre es eine Verbesserung die Anzahl an
Falsch-Positiven Transformanten zu reduzieren, weil dadurch weniger Transformanten
analysiert werden müssen und somit der Aufwand geringer wird. Des Weiteren wäre es
erstrebenswert, den Prozess nach der Aufreinigung bis hin zur Southern-Analyse zu
automatisieren (Tab.1), z.B. durch die Realisierung der Anzucht als auch DNA Extraktion im
Mikrotiterplattenformat. Eine Silica-basierte Aufreinigungsmethode für genomische DNA im
Mikrotiterformat, welche auch für Pilze funktionieren soll, ist bereits beschrieben
(Elphinstone et al., 2003). Es wurde jedoch noch nicht gezeigt, ob die Menge als auch die
Qualität der extrahierten DNA ausreichend für die Southern-Analyse ist.
Tabelle 1: Eigenschaften von A. niger als heterologer Proteinproduzent (nach Archer 1994).
Vorteile Nachteile Forschungsbedarf
Natürlicher Produzent
sekretorischer Proteine
Proteasen im Kulturüberstand
können das Zielprotein abbauen
Protease-defiziente
Stämme
Stabile Integration von Fremd-
DNA ins Genom
Geringe
Transformationseffizienz,
zeitliche Aufwendung für
Stammerstellung (inkl.
Klonierung und Southern Blot)
>20 Tage
Effiziente
gentechnische
Methoden (vermutlich
durch CRISPR-Cas9
gelöst)
N- und O-Glykosylierungen
von Proteinen
Ähnlich, aber nicht identisch zu
humanen Systemen
Humane
Glykosylierungsmuster
Einfache Medienbedingungen
Hohe Wachstumsrate
Wenig kontaminations-
anfällig
Viele Produkte mit GRAS-
Status, industrielle Akzeptanz

- 14 -
A. niger verfügt über viele verschiedene Promotoren, die eine große Breite an
Expressionsprofilen ermöglichen, diese sind im Kapitel 2 zusammengefasst und werden durch
die in 1.4.3 beschriebenen induzierbaren Expressionssysteme ergänzt. Mittlerweile
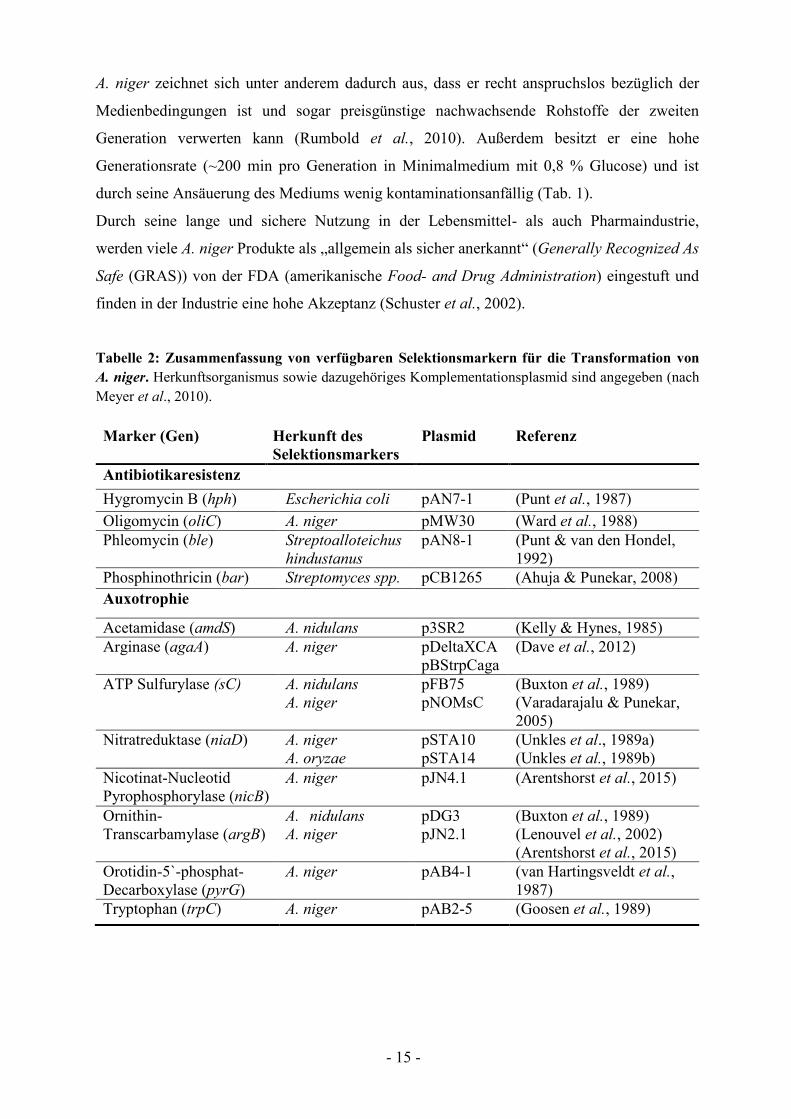
gibt es für A. niger eine recht große Auswahl an Selektionsmarkern (Tab. 2), wobei es sich bei
den auxotrophen Markern pyrG, niaD und sC um bidirektionale Marker handelt, da sie die
Selektion sowohl auf Anwesenheit als auch auf Abwesenheit des funktionalen Genproduktes
ermöglichen. Durch die Anwendung des Cre-LoxP-Systems kann man die zur Verfügung
stehenden Marker recyceln (Mizutani et al., 2012) und Stämme mit komplexen Schaltkreisen
aus Gen-Überexpression, verringerter Expression oder Deletion erstellen.
Um heterologe oder homologe Proteine zu sekretieren, arbeitet man häufig mit einem
Fusionsprotein, wie der stark sekretierten Glucoamylase, diese wird N-terminal an das
rekombinante Protein fusioniert (Kapitel 2; Epilog I, Projekt 1 & 3; Cullen et al., 1987). Eine
andere Möglichkeit ist, ausschließlich die Sekretionssignalsequenz von einem sehr gut
sekretierten Protein zu nutzen, wobei es sich hier in der Regel um homologe Proteine handelt
(van den Hondel et al., 1991). In Kapitel 5 konnten zwei neue Sekretionssequenzen, sowohl
vom einem antifungalen Protein (ANAFP) als auch von einem Hydrophobin-ähnlichen
Protein (HFBD) aus A. niger, verifiziert und validiert werden.
A. niger ist in der Lage, sekretierte Proteine durch N-Glykosylierung und O-Glykosylierung
posttranslational zu modifizieren und somit Glykoproteine herzustellen (Kapitel 2). Eine
Herausforderung dabei ist, dass die Struktur der N-Glykane spezies-spezifisch ist und aus
unterschiedlichen Monosacchariden besteht. Pilze bauen hauptsächlich Mannose ein,
hingegen ist das menschliche Glykosylierungsmuster komplexer und besteht zusätzlich aus N-
Acetylglucosamin, Galactose, Fucose und Sialinsäure. Filamentöse Pilze unterscheiden sich
von einzelligen Hefen, da sie zwei Typen von α-1,2-Mannosidasen besitzen. Die α-1,2-
Mannosidase aus dem endoplasmatischen Retikulum kann alle vier Mannose-Reste von dem
Oligosaccharid entfernen, dadurch entsteht eine vielversprechende Ausgangsstruktur, die
ähnlich zu denen im Menschen gefunden Glycoproteinen ist (Deshpande et al., 2008). Erste
Versuche wurden unternommen um in filamentösen Pilzen komplexe Glycane an funktionelle
Proteine zu synthetisieren, bspw. gelang in A. oryzae der erfolgreiche N-Acetylglucosamin-
Transfer (Kasajima et al., 2006). Ein wesentliches Ziel ist das Glykosylierungsmuster von A.
niger noch besser zu verstehen und dahingehend zu verändern, dass das menschliche
Glykosylierungsmuster nachgestellt werden kann (Tab. 1; Anyaogu and Mortensen, 2015;
Kainz et al., 2008; Ward et al., 2004).
- 15 -
A. niger zeichnet sich unter anderem dadurch aus, dass er recht anspruchslos bezüglich der
Medienbedingungen ist und sogar preisgünstige nachwachsende Rohstoffe der zweiten
Generation verwerten kann (Rumbold et al., 2010). Außerdem besitzt er eine hohe
Generationsrate (~200 min pro Generation in Minimalmedium mit 0,8 % Glucose) und ist
durch seine Ansäuerung des Mediums wenig kontaminationsanfällig (Tab. 1).
Durch seine lange und sichere Nutzung in der Lebensmittel- als auch Pharmaindustrie,
werden viele A. niger Produkte als „allgemein als sicher anerkannt“ (Generally Recognized As
Safe (GRAS)) von der FDA (amerikanische Food- and Drug Administration) eingestuft und
finden in der Industrie eine hohe Akzeptanz (Schuster et al., 2002).
Tabelle 2: Zusammenfassung von verfügbaren Selektionsmarkern für die Transformation von
A. niger. Herkunftsorganismus sowie dazugehöriges Komplementationsplasmid sind angegeben (nach
Meyer et al., 2010).
Marker (Gen) Herkunft des
Selektionsmarkers
Plasmid Referenz
Antibiotikaresistenz
Hygromycin B (hph) Escherichia coli pAN7-1 (Punt et al., 1987)
Oligomycin (oliC) A. niger pMW30 (Ward et al., 1988)
Phleomycin (ble) Streptoalloteichus
hindustanus
pAN8-1
(Punt & van den Hondel,
1992)
Phosphinothricin (bar) Streptomyces spp. pCB1265 (Ahuja & Punekar, 2008)
Auxotrophie
Acetamidase (amdS) A. nidulans p3SR2 (Kelly & Hynes, 1985)
Arginase (agaA) A. niger pDeltaXCA
pBStrpCaga
(Dave et al., 2012)
ATP Sulfurylase (sC) A. nidulans
A. niger
pFB75
pNOMsC
(Buxton et al., 1989)
(Varadarajalu & Punekar,
2005)
Nitratreduktase (niaD) A. niger
A. oryzae
pSTA10
pSTA14
(Unkles et al., 1989a)
(Unkles et al., 1989b)
Nicotinat-Nucleotid
Pyrophosphorylase (nicB)
A. niger
pJN4.1 (Arentshorst et al., 2015)
Ornithin-
Transcarbamylase (argB)
A. nidulans
A. niger
pDG3
pJN2.1
(Buxton et al., 1989)
(Lenouvel et al., 2002)
(Arentshorst et al., 2015)
Orotidin-5`-phosphat-
Decarboxylase (pyrG)
A. niger
pAB4-1
(van Hartingsveldt et al.,
1987)
Tryptophan (trpC) A. niger pAB2-5 (Goosen et al., 1989)

- 16 -
1.4 Strategien zur Produktivitätssteigerung von Aspergillus niger
Die Produktivität der Synthese von Enzymen, organischen Säuren und Sekundärmetaboliten
in A. niger hängt von verschiedenen Faktoren ab, z.B. von der transkriptionellen Aktivität des
eingesetzten Promotors, den verwendeten Kultivierungsbedingungen, der Morphologie sowie
von der eingesetzten Fermentationsstrategie. Um die Proteinexpression zu steigern, ist der
erste zu optimierende Ansatzpunkt immer die Transkription. Sollten heterologe Gene
eingebracht werden, ist darauf zu achten, dass der Codongebrauch für A. niger ausreichend
gut ist und gegebenenfalls eine Codon-Optimierung vorgenommen werden muss. Des
Weiteren kann man aus einer ganzen Reihe an Promotoren (Übersicht in Kapitel 2 (Tab. 3))
und Expressionssystemen (detailliert unter 1.4.1 erläutert) wählen, um homologe oder
heterologe Proteine zu überexprimieren. Im Unterkapitel 1.4.2 wird beschrieben, welche
Kultivierungsbedingungen einen großen Einfluss auf die Produktausbeute haben, dabei wird
insbesondere auf die Medienzusammensetzung, Osmolarität, Gelöstsauerstoff- und
Kohlenstoffdioxid-Konzentration, Temperatur und den pH-Wert eingegangen. Des Weiteren
wird über die optimale Morphologie von A. niger unter 1.4.3 diskutiert und welche
Auswirkungen sie auf den Produkttiter ausübt. In Kapitel 1.4.4 sind die verschiedenen
Fermentationsstrategien, die hauptsächlich für A. niger angewandt werden beschrieben und
ihre jeweiligen Vor- und Nachteile diskutiert.
1.4.1 Induzierbare Promotoren und Expressionssysteme
Man unterscheidet die autoinduzierbaren wachstumsabhängigen Promotoren von den
induzierbaren und meistens ebenso metabolismusabhängigen Promotoren (Kapitel 2).
Beispiele für wachstumsabhängige Promotoren sind der konstitutive Glyceraldehyd-3-
phosphat-Dehydrogenase-Promotor (PgpdA), der sofort nach dem Auskeimen angeschaltet
wird (Punt et al., 1990), der Citrat-Synthase-Promotor (PcitA), der mit dem Beginn der
Zitronensäureproduktion aktiviert wird (Dave & Punekar, 2011) sowie die anafp- und hfbD-
Promotoren, die bei geringen Wachstumsraten (µ<0.008-0.0001 h-1
) die Transkription der
Zielgene starten (Kapitel 5). Bei den induzierbaren aber meist metabolismusabhängigen
Promotoren handelt es sich um Promotoren wie den Glucoamylase-Promotor (PglaA) (Fowler
et al., 1990), der durch Stärke, Glucose oder Maltose aktiviert wird oder den Exo-Inulinase-
Promotor (inuE) (Yuan et al., 2008), der sich durch Inulin und Saccharose induzieren lässt.
Die induzierbaren sind den wachstumsabhängigen Promotoren überlegen, da die
Proteinexpression besser gesteuert werden kann. Jedoch ist man mit diesen klassischen
induzierbaren Promotoren, die aus metabolischen Genen (wie Amylase, Invertase, Xylanase)

- 17 -
abgeleitet wurden, auf spezielle Fermentationsmedien angewiesen. Meistens können
günstigere komplexe Medien nicht genutzt werden, weil definierte Spezialmedien bestimmte
Signalwege triggern müssen, damit die Promotoren aktiv sind. Daher versucht man
metabolismusunabhängige, besser regulierbare Expressionssysteme für Aspergillus zu
entwickeln. Es ist sinnvoll eine Vielfalt von regulatorischen Systemen in A. niger zu
etablieren, um simultan aber unabhängig, die Expression von verschiedenen Genen zu
regulieren bzw. auszubalancieren. Es wurde bereits viel Aufwand betrieben um für
Aspergillus induzierbare, dichte und justierbare Expressionssysteme zu entwickeln. Der durch
Thiaminzugabe reprimierbare thiA-Promotor wurde in A. oryzae und A. nidulans erfolgreich
evaluiert, unter alkalischen Bedingungen zeigt der Promotor allerdings keine Repression
(Shoji et al., 2005). Das induzierbare menschliche Estrogenrezeptor-System (hERα)
funktioniert metabolismusunabhängig und sehr sensitiv auf estrogene Substanzen (z.B.
Diethylstilbestrol) in A. nidulans und A. niger (Pachlinger et al., 2005). Abhängig vom
Konstrukt zeigt sich bei der Induktion eine starke Expression, aber eine relativ hohe
Basalexpression ohne estrogene Substanz oder nur eine schwache Expression im aktivierten
Zustand, aber eine geringe Basalexpression im deaktivierten Zustand.
Das erste Expressionssystem, welches vielversprechende Ergebnisse in Aspergillus lieferte,
war das Tet-on-System. Bei diesem wird die Transkription durch Hinzufügen des Tetracyclin
Derivates Doxycyclin (Dox) dosisabhängig aktiviert, es wurde für A. fumigatus etabliert
(Vogt et al., 2005) und für A. niger weiterentwickelt (Meyer et al., 2011; Kapitel 2, 3, 4). Das
optimierte Tet-on-System fand großen Anklang innerhalb der Aspergillus-
Forschergemeinschaft, das Plasmid wurde 24-mal versandt und die Publikation bereits 33-mal
zitiert (Stand 21.2.16). Es wurde genutzt, um Gene zu charakterisieren (Meyer et al., 2011;
Ouedraogo et al., 2011) und die intrazelluläre Lokalisation von Proteinen in A. niger zu
studieren (mit Hilfe von GFP (grünfluoreszierendes Protein)) (Steiger et al., 2016). Ebenso
konnten durch Überexpression putativer Transkriptionsfaktoren neue Gencluster in A.
fumigatus aktiviert und hierbei neue Sekundärmetabolite identifiziert werden (Macheleidt et
al., 2015; Kalb et al., 2015). In Studien mit A. fumigatus wurden native Promotoren durch das
Tet-on-System ersetzt (Dichtl et al., 2012; Samantaray et al., 2013), ebenso bei der
Charakterisierung von einem Produkt des Nichtribosomalen-Peptidsynthase-ähnlichen Gens
aus A. terreus (Sun et al., 2016). In einem anderen Ansatz wurde parallel zur Integration des
zu untersuchenden Gens im Tet-on-System das Wildtyp-Allel deletiert um eine genaue
quantitative Charakterisierung des Transkriptes vorzunehmen (Meyer et al., 2011; Kwon et
al., 2014). In Kapitel 3 ermöglichte das Tet-On-System die Expression der Enniatinsynthetase

- 18 -
aus Fusarium oxysporum während der exponentiellen Wachstumsphase zu induzieren, dies
führte zu hohen Produktmengen des Sekundärmetaboliten Enniatin.
Auch zum Knock down von Genen wurde das Tet-on-System verwendet, in dem eine
Antisense-RNA des Zielgens exprimiert wurde (Wartenberg et al., 2012).
Abhängig von der Funktion des Genes kann es für Funktionsstudien von Vorteil sein über ein
Tet-off-System zu verfügen, insbesondere bei Virulenzstudien im Tiermodell. Aus diesem
Grund wurde das Tet-off-System auch für A. niger adaptiert (Kapitel 4).
1.4.2 Kultivierungsbedingungen
Verschiedene Kultivierungsbedingungen haben einen großen Einfluss auf die Produktivität
von A. niger. Eine bedeutende Rolle spielt zum Beispiel das Kulturmedium, die Osmolarität,
die Gelöstsauerstoff- und Kohlenstoffdioxid-Konzentration, die Temperatur, der pH-Wert und
die Kultivierungsart (Schüttelkolben, Festbett- oder submerse Kultivierung).
Die im Kulturmedium enthaltene Kohlenstoff- oder Stickstoffquelle reguliert das
Transkriptom und damit ebenso das Proteom. Abhängig vom eingesetzten Kohlenstoff
werden z.B. verschiedene amylolytische Enzyme unterschiedlich stark sekretiert. Der
transkriptionelle Regulator amyR, welcher durch Stärke und deren Abbauprodukte induziert
wird, vermittelt die Expression von amylolytischen Enzymen (Petersen et al., 1999; van Kuyk
et al., 2012). Hingegen wirkt der transkriptionelle Repressor creA reprimierend auf die
Expression dieser Stärke abbauenden Gene, wenn leicht zu metabolisierende
Kohlenstoffquellen (z.B. Glucose) anwesend sind (Dowzer & Kelly, 1991). Ein weiteres
Beispiel für die Kohlenstoffregulation ist die Induktion des Transkriptionsfaktor xlnR durch
Xylose, der die Expression von xylanolytischen und cellulolytischen Genen aktiviert (De
Vries et al., 1999; Van Peij et al., 1998), wobei creA auch hier als Repressor von Xylanase
bei Anwesenheit von Glucose eine Rolle spielt (Pinaga et al., 1994). Wenn man einen
metabolismusabhängigen Promotor verwendet, ist auch auf die Zusammensetzung des
Mediums zu achten, z.B. sollte für die Aktivierung des glaA-Promotors Stärke, Maltose,
Maltodextrin oder Glucose als Kohlenstoffquelle genutzt werden. Bedingt durch den
verwendeten Stamm und das gewünschte Produkt ist es ratsam, verschieden kombinierte
Kohlenstoff- und Stickstoffquellen für eine Produktivitätssteigerung auszutesten. Bei der
Biosynthese von Fumagillin in A. fumigatus konnte eine 15-fache Steigerung durch eine
Kombination aus Xylan, Mannose und L-Glutaminsäure erreicht werden (Yang et al., 2003).
Verschiedene Glucose- und Stickstoffkonzentrationen haben Einfluss auf die Morphologie
eine Reduktion von Glucose führt z.B. zu kürzeren Hyphenfilamenten mit höherer
Verzweigungsrate (Papagianni et al., 1999).

- 19 -
Spurenelemente wie Mangan, Eisen, Zink, Kupfer und Molybdän sind unentbehrlich für das
Wachstum von A. niger und für hohe Produkttiter. Besonders oft wurde im letzten
Jahrhundert der Einfluss verschiedener Metalle, wie z.B. Kupfer, Zink und Eisen (Majolli &
Aguirre, 1999) und Salze, wie z.B. Calciumchlorid (Pera & Callieri, 1999) auf die
Zitronensäureproduktion untersucht. Die Abwesenheit von Mangan führt zu einem hohen
metabolischen Fluss durch die Glykolyse, daraus resultiert eine hohe
Zitronensäurekonzentration (Habison et al., 1979). Zudem wurde herausgefunden, dass die
Spurenelemente auch die Morphologie beeinflussen, Calciumchlorid induziert eine
pelletartige Morphologie, hingegen führt Eisen (III)-chlorid zum dispergierten Myzelium
(Colin et al., 2013; 1.4.3).
Durch die Zugabe von Natriumchlorid kann die Osmolarität variiert werden, mit 3,1 osmol/kg
zeigte ein Fructofuranosidase produzierender A. niger Stamm eine 18-fache spezifische
Produktivitätssteigerung, wobei die Biomassekonzentration um ~40 % reduziert wurde
aufgrund der Bildung von kleineren fungalen Pelletstrukturen (Wucherpfennig et al., 2011).
Die kritische Menge von gelöstem Sauerstoff (dissolved oxygen tension, DOT, 210 mbar ~
100 %) ist für die Produktbildung höher als für das Wachstum, bei der
Zitronensäureproduktion konnte als Minimalwert für DOT 25 mbar ermittelt werden, während
die Ausbeute zwischen 40-150 mbar stieg (Kubicek et al., 1980). Einen Einfluss von
Sauerstoff auf die Morphologie von A. niger wurde unter Extrembedingungen analysiert, eine
Sauerstoffanreicherung führt zu oxidativem Stress (Li et al., 2008) während bei einem DOT
annähernd 0 % sich eine verringerte Verzweigungsintensität einstellt und mehr Vakuolen pro
Hyphe zu erkennen sind (Rahardjo et al., 2005). Über 3 % gelöste Kohlenstoffdioxidmenge
im Zulaufgas führt zur Abnahme von Biomasse und Zitronensäurekonzentration und ist
ebenso mit morphologischen Änderungen verbunden (McIntyre & McNeil, 1997).
A. niger toleriert Temperaturen zwischen 6 und 47 °C, als Kultivierungstemperatur wird am
häufigsten 30 °C gewählt. Die optimale Temperatur für die heterologe Proteinexpression ist
oftmals abhängig davon, um welches Produkt es sich handelt, z.B. wurde im Kapitel 4
gezeigt, dass bei 26 °C in A. niger mehr Enniatin produziert wird als bei 30 °C, vermutlich,
weil die Enniatinsynthetase bei 26 °C ihr Aktivitätsoptimum hat (Zocher & Kleinkauf, 1978).
Bei der Produktion von Orchatoxin in A. niger führte die Kultivierung bei 15 °C sogar zur
höchsten Ausbeute (Leong et al., 2006). Eine geringere Temperatur führt im Allgemeinen zu
einer langsameren Wachstumsrate, die vorteilhaft für die Überexpression von extrazellulären
Proteinen ist wie in Trichoderma reesei ermittelt wurde (Pakula et al., 2005).
Außergewöhnlich tolerant zeigt sich A. niger auch gegenüber einem weiten pH Bereich von
1,5-9,8, er ist somit in der Lage sowohl im stark sauren als auch basischen Milieu zu leben.

- 20 -
Der extrazelluläre pH-Wert hat einen großen Einfluss auf die Fermentation von Aspergillus,
Zitronensäure wird besser bei geringerem pH-Wert (<3) produziert, während man für die
Produktion von Oxal- und Gluconsäure einen höheren pH-Wert (5-8) wählt (Andersen et al.,
2009). Der pH-Wert beeinflusst die Ladung der Sporenoberfäche, ein basischer pH kann
dadurch Sporenaggregationen induzieren und Einfluss auf die Morphologie nehmen (Grimm
et al., 2005). Des Weiteren hat der pH-Wert einen großen Einfluss auf die extrazelluläre
Proteaseaktivität, bei geringerem pH (z.B. pH 3) werden sehr viele saure Proteasen induziert,
hingegen findet bei pH 6 weniger proteolytischer Abbau statt (Donnell et al., 2001). Für den
sequenzierten CBS 513.88 Stamm wurden 198 Proteine ermittelt, die im proteolytischen
Abbau involviert sind (Pel et al., 2007). Mittels UV-Bestrahlung wurde AB4.1 mutagenisiert
und auf ein Defizit in der extrazellulären Proteaseproduktion selektioniert. Der Mutant
AB1.13 zeigt eine reduzierte Proteaseaktivität von 98 % durch eine Mutation im Regulator für
extrazelluläre Proteasegene (prtT), dadurch sind Aspergillopepsin A und B deaktiviert
(Mattern et al., 1992; Punt et al., 2008). Der A. niger Stamm D15 (Gordon et al. 2000), UV-
mutagenisiert aus AB1.13 zeigt eine nochmals reduziertere Proteaseaktivität, verursacht durch
eine Mutation im laeA Gen, welches vermutlich ein Methyltransferasedomain-Protein codiert,
das für die Zitronensäureproduktion essentiell zu sein scheint (Tab. 3; Niu et al., 2015). Durch
die fehlende Zitronensäurebildung wird das Medium von D15 Kulturen weniger angesäuert
und somit werden die bei einem geringen pH-Wert induzierten Proteasen nicht exprimiert.
Wie in Tabelle 3 dargestellt, konnte mit der Mutante D15 die Proteaseaktivität bereits um 90
% reduziert werden im Vergleich zum Wildtypstamm und stellt damit zukünftig einen
vielsprechenden Wirtsstamm für die heterologe Proteinexpression unter kontrollierten pH-
Bedingungen dar.
Tabelle 3: Relative Proteaseaktivität und extrazellulärer pH-Wert nach 120 Stunden
Kultivierung im Schüttelkolben. Die Proteaseaktivität wurde mit dem P-Check Assay (Protease-
Detektionskit von Jena BioScience) bestimmt und wird in Prozent im Vergleich zum Wildtyp N402
angegeben. Für die Experimente wurden pyrG+ Stämme in Minimalmedium (pH 6.5) kultiviert. (aus
Niu et al., 2015)
Stamm Relevanter Genotyp pH-Wert
Relative
Proteaseaktivität
N402 (CBS513.88 Derivat) WT 5.3 100 %
AB1.13 prtT- 4.8 48 %
D15 prtT-, laeA
- 6.8 10 %

- 21 -
Eine weitere wichtige Bedingung für eine hohe Produktivität stellt die Kultivierungsart dar.
Abhängig davon, ob eine Kultivierung im Schüttelkolben, Festbett- oder Batch-Reaktor
stattfindet, variieren unter gleichen Mediumsbedingungen das Transkriptom und das daraus
resultierende Proteom (Oda et al., 2006; Gamarra et al., 2010; de Oliveira & de Graaff, 2011).
Kontrollierbare und reproduzierbare Bedingungen bzgl. Temperatur, pH-Wert,
Gelöstsauerstoff-Konzentration und Morphologie erreicht man nur in Bioreaktor
Kultivierungen. Hier können wiederum verschiedene Fermentationsstrategien gefahren
werden, die in Kapitel 1.4.4 behandelt werden.
1.4.3 Morphologie
Die makroskopische Morphologie in submersen Kultivierungen kann zwischen kompakten
kugelförmigen Pellets bis hin zum dispergierten Myzel (Abb.5) variieren. Sie wird im
Bioreaktor oder Schüttelkolben durch verschiedene Prozessparameter beeinflusst, unter
Kapitel 1.4.2 wurden schon Medienzusammensetzung, Begasung, Temperatur sowie pH-Wert
beschrieben. Weitere nennenswerte Einflüsse sind Rührerdrehzahl bzw. Schüttelfrequenz
(Papagianni, 2004) sowie das Inokulum (Sporenanzahl (Papagianni & Mattey, 2006), das
Alter der Sporen (Colin et al., 2013)) als auch Medienzusätze wie Mikropartikel (Driouch et
al., 2010).
Abbildung 5: Verschiedene morphologische Formen. A) Pelletform, 38 h Batch mit
Reaktormedium im Wellen durchmischten Einwegreaktor (CELL-tainer®, BIOTECH BV) (T. Kurt,
TU Berlin) B) dispergiertes Myzel, 44 h Batch mit Reaktormedium im Rührreaktor (BioFlo3000,
New Brunswick Scientific, NJ). In beiden Kultivierungen wurde N402, ein Wildtypstamm von A.
niger, verwendet.
Die optimale Morphologie von filamentösen Pilzen ist vom gewünschten Produkt abhängig
und muss für jeden Prozess neu evaluiert werden (Grimm et al., 2005). Bei der
Zitronensäureproduktion geht man davon aus, dass die Pelletform (Abb. 5A) produktiver ist
(Bodie et al, 1994) hingegen wird bei der Proteinproduktion meistens die dispergierte Form
(Abb. 5C) bevorzugt (Gyamerah et al., 2002). Beide Extreme zeigen Vor- und Nachteile,
B A

- 22 -
Pellets sind im kompakten Inneren nicht ausreichend mit Nährstoffen versorgt, während ein
stark dispergiertes Myzel zu einer unerwünscht hohen Viskosität (Nicht-Newton`sches Fluid)
führt, welche ebenso den Nährstofftransfer limitiert. Bei der Pelletform geht man davon aus,
dass sie durch die Agglomeration von Sporen (Dynesen & Nielsen, 2003) initiiert wird. Um
die meist uneinheitliche Pelletgröße zu kontrollieren, kann man Mikropartikel wie Talkum
(Magnesiumsilikat) einsetzen, welche die Pelletbildung, bspw. in
Schüttelkolbenkultivierungen, reduzieren. Die zugegebenen Partikel sorgen dafür, dass sich
die auskeimenden Sporen nicht aneinander lagern so dass sich kleine und weniger kompakte
Strukturen bilden (Kapitel 2 Abb. 3), die eine höhere Proteinproduktivität zeigen (Kapitel 3,
(Driouch et al., 2010)).
Bei der sogenannten Mikromorphologie einer Hyphe unterscheidet man zwischen linearen
Filamenten bis hin zu stark verzweigten Strukturen. Bekannt ist, dass die
Verzweigungsintensität abnimmt, wenn die Konzentration von Sauerstoff zu gering ist
(Rahardjo et al., 2005) oder das Rühren verringert wird (Amanullah et al., 2002). Bisher ist
noch nicht eindeutig klar, ob eine höhere Verzweigungsrate auch zu mehr Proteinsekretion
führt. Es gibt Studien, die das in A. oryzae bestätigen (Te Biesebeke et al., 2005, Amanullah
et al., 2002) und andere Veröffentlichungen, die keinen Zusammenhang in A. oryzae und A.
niger finden (Müller et al., 2002; Kwon et al., 2013).
1.4.4 Fermentationsstrategien zur Produktion
In dem biotechnologischen Prozess ist die Fermentation definiert als die enzymatische
Umwandlung organischer Stoffe zu wertvollen Produkten mit Hilfe von Mikroorganismen
(oder Zellkulturen) in Bioreaktoren (Enfors & Häggström, 2000). Jedoch kommt es immer
wieder zu Missverständnissen, da in der Mikrobiologie bzw. Biochemie oft die ursprüngliche
Definition von Fermentation als Gärung, das heißt biotische Reaktion unter
Sauerstoffausschluss, verstanden wird, wie sie Louis Pasteur 1850 prägte. Daher gibt es
Bioverfahrenstechniker/innen die dafür plädieren von Kultivierung zu sprechen um
Missverständnisse vorzubeugen und darauf hinzuweisen, dass die mikrobielle Kultivierung
ein aktiver Prozess ist, in dem die durchführende Person eine signifikante Rolle spielt.
Bioreaktorkultivierungen erlauben eine erweiterte Kontrolle vom Wachstum und stellen ein
definiertes Umfeld zur Verfügung mit denen sich konditionale Phänotypen adäquat
untersuchen lassen. Ebenso spielt die Fermentation besonders in der pharmazeutischen und
chemischen Industrie für die Produktion von gewünschten Substanzen eine große Rolle. Die
hohe Steigerung der Zitronensäure Ausbeute seit 1919 steht im engen Zusammenhang mit

- 23 -
einer steten Optimierung der Fermentation von A. niger. Anfangs wurde A. niger in
sogenannten Festbett-Reaktoren kultiviert, dabei werden die Organismen als
Oberflächenkulturen angezogen und das Medium strömt über sie hinweg, wobei die
Durchströmung relativ inhomogen und der Sauerstoffeintrag limitiert ist. Seit 1932 wird A.
niger in submersen Verfahren kultiviert (Kluyver and Perquin, 1932), da höhere
Produktionsraten erzielt werden konnten (Bodie et al., 1994).
In den letzten 100 Jahren wurden verschiedene Kultivierungskonzepte, abhängig vom
gewünschten Produkt und vom eingesetzten Mikroorganismus, etabliert. Industriell eingesetzt
werden Batch, Fed-Batch und die kontinuierlichen Chemostat- und Perfusionsverfahren (Abb.
5) (Liese et al. 2006).
In der Batch-Fermentation wird das Nährmedium im Reaktor vorgelegt, es müssen darin alle
Nährstoffe in ausreichender Konzentration für die gewünschte Menge an Biomasse- und
Produktbildung enthalten sein. Während der frühen Phase der Fermentation erreicht die
spezifische Wachstumsrate einen konstanten Wert (µmax) durch den exponentiellen Anstieg
der Biomasse. Dadurch, dass die Substratkonzentration im Reaktor verbraucht wird, nimmt
die Wachstumsrate nach der exponentiellen Phase ab, hingegen steigt die
Biomassekonzentration weiter langsam an, dann spricht man vom limitierten Wachstum.
Anschließend folgt die stationäre Phase, bei der die Geschwindigkeit der Vermehrung und des
Absterbens der Hyphenzellen gleich ist und daher die Biomassekonzentration konstant bleibt
bis die Absterbephase erreicht ist, wobei die Absterberate exponentiell zunimmt und damit
einhergehend die Biomasse abnimmt. Für die meisten Expressions- bzw.
Morphologieanalysen sind die durchgeführten Batch-Kultivierungen in A. niger Glucose-
limitiert (0,8 %) (Kapitel 3; 4; Andersen et al., 2011; Kwon et al., 2013; Meyer et al., 2009).
Lediglich wenn größere Produktausbeuten für weiterführende Untersuchungen benötigt
werden, wird mit höheren Zuckerkonzentrationen (bspw. 5 %) und einem Komplettmedium
gearbeitet (Epilog I, Projekt 3).
Beim Fed-Batch startet man mit einem Batch im geringeren Volumen im Reaktor und
nachdem die exponentielle Phase beendet ist, werden weiter kontinuierlich Nährstoffe
zugegeben bis das maximale Reaktorvolumen erreicht ist. Die Biomassekonzentration steigt
proportional zur Zufütterungsrate und dadurch wird die Produktbildungsrate kontrolliert. Die
Zufütterungsrate kann konstant sein, stufenweise oder exponentiell steigend. Durch diese
kontrollierte Zufütterung vermeidet man den sogenannten “Überflussmetabolismus”, der im
klassischen nicht Kohlenstoff-limitierten Batch dazu führen kann, dass das Substrat für die
Bildung ungewollter Nebenprodukte verwendet wird (siehe Epilog I, Projekt 3). Weitere

- 24 -
Vorteile sind, dass beim Fed-Batch sehr hohe Zelldichten erreicht werden können, die zu
einem gesteigerten Produktertrag führen oder dazu beitragen, dass wachstumsgekoppelte
Produkte in hohen Menge exprimiert werden können. Der Fed-Batch Modus wird
hauptsächlich genutzt um Proteine zu produzieren (Pel et al., 2007; Jacobs et al., 2009) oder
auch Sekundärmetabolite (Kapitel 3).
Abb. 6: Schematische Übersicht von verschiedenen submersen Bioreaktor Kultursystemen. Die
runden Partikel symbolisieren A. niger in seiner für den Prozess optimierten Makromorphologie. In A)
ist die Batch-Kultivierung dargestellt, die im gleichbleibenden Volumen gefahren wird. Die
Zeichnung B) zeigt den Fed-Batch, der mit einem Batch von geringerem Volumen startet als
vergleichsweise in A) und nach der exponentiellen Phase eine kontinuierliche Zufütterung impliziert.
In C) ist eine kontinuierliche Kultivierung dargestellt (Chemostat), die ebenfalls mit einem Batch
startet, hierbei wird jedoch während der exponentiellen Phase auf konstanten Zulauf und Ablauf
umgestellt, so dass eine gewünschte Wachstumsrate erreicht wird. D) Die Perfusionskultivierung
funktioniert ähnlich wie die Chemostat-Kultivierung, nur dass der Abfluss durch eine
Zellrückhaltungsmembran fließt, so dass es zu einer Anreicherung der Biomasse im Reaktor kommt.
Bei der kontinuierlichen Kultivierung (Chemostat) startet man gleichfalls mit einem Batch
wobei schon während der exponentiellen Phase auf einen kontinuierlichen Fluss umgestellt
wird, so dass das Reaktorvolumen durch einen kontinuierlichen Mediumszufluss und einen
Kulturbrüheabfluss konstant gehalten wird. Diese Art der Kultivierung ermöglicht es, einen
steady state zu erreichen, der dazu genutzt werden kann optimale Wachstumsraten für
bestimmte Produkte zu ermitteln bzw. die Kultur länger in der optimalen Produktsynthese zu
halten als bspw. bei einer Batch-Fermentation möglich ist. In der Industrie kommt die
Chemostate-Fermentation eher selten zum Einsatz, da sie durch den hohen
Mediumsdurchfluss recht kostenintensiv ist. Am häufigsten wird der Chemostat zur
Untersuchung physiologischer Zustände angewandt, wie z.B. Glucoseaufnahme, Wachstum,
Einfluss verschiedener Zucker, Sekretionsstress (Carlsen et al., 1996; Jørgensen et al., 2009;
Kwon et al., 2012).

- 25 -
Die letzte große Innovation in der Bioverfahrenstechnik stellt die Änderung zu
kontinuierlichen Verfahren mit Zellrückhaltungssystemen dar, welche auch Perfusion-,
Retentostat- oder Recycling-Kultivierung genannt werden (Kapitel 5; Jørgensen et al., 2010;
van Verseveld et al., 1991). Das Volumen der Kultur bleibt konstant, nur die Biomasse steigt
an bis zu ihrem maximalen Wert, wobei die zugeführten Nährstoffe nur noch ausreichen, um
den Erhaltungsstoffwechsel der Zellen sicherzustellen. Das Ziel ist der konventionellen
chemischen Industrie zu folgen und die Prozesse kontinuierlich zu gestalten (Schmid et al.,
2001), um Kosten zu sparen. Bisher sind jedoch erst ca. 11 % der industriellen
biotechnologischen Fermentationen kontinuierlich (Liese et al. 2006). 2015 hat die FDA das
erste Mal ein neues Therapeutikum (Orkambi, ein Medikament gegen zystische Fibrose der
Firma Vertex), welches in kontinuierlicher Fermentation produziert wurde, zugelassen. Die
Entwicklung in Richtung kontinuierlicher Herstellungsverfahren wird von der FDA
wohlwollend unterstützt. Ein besonderer Vorteil wird darin gesehen, dass die Produktqualität
in Echtzeit überwacht werden kann und dass die Glykosylierungsmuster konsistener innerhalb
des Prozesses sind als bspw. in Fed-Batch-Fermentationen. Der größte Vorteil für den
Anwender ist, dass sich die Raum-Zeit-Ausbeuten bei der Perfusionsfermentation, z.B. bei der
Produktion von Mucin-1 in CHO Zellen um das 30-fache im Vergleich zu der Batch-
Fermentation (Link et al., 2004), erhöhen lassen. Die weiteren Vorteile und Nachteile werden
im Kapitel 5 bzw. im Abschnitt Diskussion und Ausblick beschrieben.

- 26 -
1.5 Ziel und Gliederung der Arbeit
Das Ziel dieser Dissertationsarbeit ist mit gen- und verfahrenstechnischen Ansätzen das
Potential von Aspergillus niger als Expressionsplattform aufzuzeigen und gezielt weiter
auszuschöpfen. Zum einen sollten neue Expressionssysteme entwickelt und evaluiert werden,
die eine metabolismusunabhängige bzw. autoinduzierbare Produktion von industriell
relevanten Proteinen und Metaboliten ermöglichen. Zum anderen war es das Ziel, neue
innovative Fermentationsstrategien anzuwenden und durch Fermentationszusätze bereits im
Schüttelkolben den Ertrag zu steigern.
Kapitel 1 liefert eine allgemeine Einleitung in die Arbeit, hierbei wird die Gattung
Aspergillus eingeführt und die Vor- und Nachteile von A. niger als Expressionsplattform
diskutiert. Im Weiteren wird auf die angewandten Strategien zur Produktivitätssteigerung
eingegangen und somit spezifisch in die Themen der publizierten Arbeiten aus Kapitel 3 - 5
eingeführt.
Kapitel 2 besteht aus einer umfassenden Literaturarbeit zum Thema: „Aspergillus: A Cell
Factory with Unlimited Prospects“. Hier wird speziell auf die relevanten industriell
hergestellten Produkte eingegangen und die zur Verfügung stehenden molekulargenetischen
Methoden werden erläutert, um die rekombinante Proteinexpression in A. niger auf
Transkriptom-, Translations- und Sekretionsebene zu verbessern. Des Weiteren werden
systembiologische Analysen diskutiert, die Aspergillus als Säure- und Proteinproduzenten,
sowie als Sekundärmetabolitproduzenten beleuchten.
In Kapitel 3 wird die heterologe Expression einer Nichtribosomalen-Peptidsynthetase ESYN
mit Hilfe des Tet-on-Systems beschrieben. Durch Induktion in der frühen exponentiellen
Phase werden hohe Enniatinmengen erreicht. Schüttelkolbenerträge konnten durch einen
Design-of Experiments Ansatz um den Faktor 950 gesteigert werden. Durch Fed-Batch-
Kultivierungen im 5 l Maßstab gelang es schlussendlich 4,5 g/l Enniatinausbeute zu erzielen.
Bei der Etablierung eines neuen Tet-off Expressionssystems für A. niger, dargestellt in
Kapitel 4, hat sich gezeigt, dass es notwendig ist der Codongebrauch von den verschiedenen
etablierten Transaktivatoren (tTA, tTA2, tTA2S) für A. niger zu analysieren, wobei sich die
Versionen für Säugetierzellen wesentlich besser eigneten als die für E .coli. Des Weiteren
stellte sich heraus, dass die richtig gewählte Promotorstärke für das System von elementarer
Bedeutung für die Stabilität des integrierten Konstrukts im Genom ist. Der Transaktivator
tTA2S wirkt bei zu starker Expression toxisch auf den Pilz und initiiert dadurch einen starken
negativen Selektionsdruck. Nach Verwendung eines moderaten Promotors gelang es, ein
stabiles Tet-off-System zu erstellen und mit verschiedenen Genen zu validieren. Durch diese

- 27 -
Erkenntnisse konnte das ursprüngliche Tet-on-System bezüglich höherer genetischer Stabilität
verbessert werden.
Im abschließenden Kapitel 5 werden zwei neue Promotoren, Panafp und PhfbD, die bei
geringen Wachstumsraten aktiv sind, durch intra- und extrazelluläre Reporterproteine
evaluiert, um eine heterologe Proteinexpression während einer Perfusionskultivierung zu
ermöglichen. Zusätzlich wurden die Sekretionssignalsequenzen der beiden Gene für eine
Sekretion von heterologen Proteinen validiert.
Die Arbeit schließt mit einer zusammenfassenden Diskussion der erzielten Ergebnisse und
einem weiterführenden Ausblick ab. Im Epilog I werden alle nicht publizierten Projekte kurz
erläutert und die hier gewonnenen wichtigsten Erkenntnisse diskutiert.

- 28 -
Kapitel 2
Aspergillus: A Cell Factory with Unlimited Prospects

- 29 -
Aspergillus: A Cell Factory with Unlimited Prospects
Markus R.M. Fiedler, Benjamin M. Nitsche, Franziska Wanka and Vera Meyer*
Berlin University of Technology,
Institute of Biotechnology,
Department of Applied and Molecular Microbiology,
Gustav-Meyer-Allee 25,
13355 Berlin,
Germany
Tel: +49 (0)30 31472825
Fax: +49 (0)30 31472922
*Corresponding author email: [email protected]

- 30 -
Abstract
The genus Aspergillus covers a diverse group of filamentous fungi including industrially
important species like A. niger, A. oryzae, A. awamori, A. sojae and A. terreus. Species of
this genus have been exploited in large scale industrial production processes for almost 100
years. As microbial cell factories, filamentous fungi are outstanding with respect to their
tolerance of extreme cultivation conditions, their ability to grow on plant biomass, their high
secretion capacities and versatile secondary metabolism. The array of Aspergillus products
includes bulk chemicals, enzymes for food and feed processing, homologous and
recombinant proteins as well as bioactive compounds. This chapter aims at providing a
comprehensive overview of the advances made during the last decade to further establish and
improve Aspergilli as industrial production platforms. It starts with a description of the
molecular genetics toolbox that has been developed for rational strain improvement, followed
by various genetic strategies that have been applied to improve production of heterologous
proteins including optimization of transcription, translation, secretory fluxes, product
degradation and morphology. The second part of this chapter provides an overview of omics
tools established for Aspergilli and highlights recent omics studies on Aspergillus as producer
of organic acids, plant polysaccharide degrading enzymes and secondary metabolites. Finally,
the future prospects of Aspergillus as a cell factory are discussed.
Introduction
The kingdom of fungi covers a large and diverse group of lower eukaryotes which includes
about 100,000 known species and presumably a million yet to be described and characterized
(Hawksworth 1991). Fungi range from unicellular (yeasts) to multicellular organisms
(filamentous fungi) and are diverse in morphology, physiology and ecology. Among the
group of filamentous fungi, the genus Aspergillus is of considerable importance for industrial
biotechnology. Their ability to grow on rather simple and inexpensive substrates as well as
their natural capacity to secrete high amounts of hydrolytic proteins into the environment
combined with its ability to synthesize and secrete various organic acids have attracted
considerable interest to exploit them as production organisms in biotechnology and food
industry. Important industrial production hosts include A. niger, A. awamori (a subspecies of
the Aspergillus section Nigri (Perrone et al. 2011)), A. oryzae, A. sojae and A. terreus. In
general, most members of the genus Aspergillus are saprophobic and are of vital importance
for nutrient cycling and the function of ecosystems. However, few Aspergilli are pathogenic

- 31 -
causing detrimental effects on plants and humans such as A. flavus, A. parasiticus and A.
fumigatus.
Aspergilli exploited at an industrial scale, have a long history of safe use and many of their
products have acquired the GRAS status meaning that they are generally regarded as safe by
the American Food and Drug Administration (Table 1).
Table 1. Selected examples of industrially important compounds produced by Aspergilli.
Product Host Company
Organic acids
Citric acid A. niger Adcuram, ADM, Anhui BBCA Biochemical,
Cargill, Jungbunzlauer, Gadot Biochemical
Industries, Iwata Chemical Co. Ltd, Tate & Lyle
Itaconic acid A. terreus Itaconix, Shandong Kaison Biochemical,
Qingdao Langyatai Group
Kojic acid A. oryzae Chengdu Jinkai Biology Engineering Industry,
MHC INDUSTRIAL CO., LTD., Sansyo
Pharmaceutical Co. Ltd, Wuxi syder Bio-products
Co. Ltd
Enzymes
α-Amylase A. oryzae Amano Enzyme Co. Ltd., Biocon, DSM,
Novozymes, Dupont IB, Novo Nordisk, Hunan
Hong-Ying-Xiang Bio-Chemistry, Shin Nihon
Chemical Co. Ltd.
Arabinase A. niger DSM, Shin Nihon Chemical Co. Ltd.
Asparaginase A. niger,
A. oryzae
DSM, Novozymes
Catalase A. niger DSM, Dupont IB, Novozymes,
Shin Nihon Chemical Co. Ltd.
Cellulase A. niger Biocon, DSM, Dyadic, Genencor INT, Haihang
Industry, Shin Nihon Chemical Co. Ltd., TNO
Chymosin A. niger Christian Hansen
β-Galactosidase
A. niger,
A. oryzae
Amano Enzyme Co. Ltd., DSM, Dupont IB,
Genencor INT, Novozymes, Shin Nihon
Chemical Co. Ltd.
Glucoamylase A. niger Amano Enzyme Co. Ltd., Cangzhou
Kangzhuang Chemical, DSM, Dyadic,
Novozymes, Dupont IB, Shandong Longda Bio-
Products
Glucose oxidase A. niger,
A. oryzae
Amano Enzyme Co. Ltd., DSM, Dupont IB,
Dyadic, Novozymes
Hemicellulase A. niger Amano Enzyme Co. Ltd., BASF, Biocon, DSM,
Dupont IB, Genencor INT, Novozymes, Shin
Nihon Chemical Co. Ltd
Lactoferrin A. niger DSM, Agennix
Lipase A. niger,
A. oryzae
DSM, Dupont IB, Novozymes, Novo Nordisk
Pectinase A. niger Biocon, DSM, Dupont IB, Novozymes,
Shandong Longda Bio-Products

- 32 -
Phytase A. niger,
A. oryzae
BASF, DSM, Novozymes, TNO
Proteases (Acid,
Neutral, Alkaline)
A. niger,
A. oryzae,
A. saitoi
Amano Enzyme Co. Ltd., DSM, Novozymes,
Mitsubishi Foods Co. Ltd., Shin Nihon Chemical
Co. Ltd.
Tannase A. oryzae,
A. ficuum
ASA Spezialenzyme GmbH, Biocon, Kikkoman
Corp.
Secondary metabolites
Fumagillin A. fumigatus Merck
Lovastatin A. terreus Biocon, Merck
After (Ward 2011, Meyer 2008)
The groundwork for Aspergillus as microbial cell factory was laid at the dawn of the
twentieth century that was accompanied by advances in microbiology, biochemistry and
fermentation technology. The pioneering works of Jokichi Takamine (production of amylase
from Japanese koji mold, Aspergillus oryzae, 1894), James Currie (development of fungal
fermentation for citric acid production, 1917) and Alexander Fleming (discovery of penicillin
production by Penicillium notatum, 1928) stimulated scientists to further explore fungal
metabolic capacities and, moreover, triggered engineers to develop large-scale production
processes for filamentous fungi. The findings of James Curie, for example, led to the
establishment of the first industrial scale production process with a filamentous fungus by
Pfizer already in 1919. Improvements of fungal capacities to produce metabolites of interest
were, however, mainly restricted to classical mutagenesis techniques followed by tedious
selection strategies. New classical genetic techniques such as (para) sexual processes and
protoplast fusion became available around the 1950’s and further advanced the productivity
of industrial processes. The birth of molecular biology in 1941 with the demonstration of the
‘one gene – one enzyme’ relationship in the filamentous fungus Neurospora crassa (Beadle
and Tatum 1941) and the development of recombinant DNA technologies for filamentous
fungi, shown for the first time in 1979 for N. crassa (Case et al. 1979), has finally
revolutionised Aspergillus biotechnology. Since then it became possible to obtain insights
into the molecular basis of product formation and to improve traditional fungal fermentations
by rational genetic engineering approaches, not only allowing production of homologous
proteins but also production of proteins from non-fungal origin. For example, Novozymes has
been the first company in the world which commercialised a recombinant lipase using A.
niger as production host in 1984. Nowadays, the growing demand for industrial enzymes and
organic aicds is met by Aspergilli (Table 1), which can be cultivated in large-scale stirred
tank reactors reaching volumes up to 300,000 litres (Elander 2003). The advantage of
Aspergilli over other microbial cell factories of bacterial or yeast origin is that they can

- 33 -
tolerate extreme cultivation conditions covering a broad spectrum of pH (2-10), temperature
(10-50°C), salinity (0-34%) and water activity (0.6-1) (Meyer et al. 2011b). As they are also
able to efficiently degrade plant-derived polysaccharides such as starch, cellulose,
hemicellulose, pectin and inulin, the importance of Aspergilli and its hydrolytic enzymes
might even rise in the near future. For example, the efficiency of the saccharification process
of second-generation feedstock used for bioethanol production might become improved by A.
niger derived (hemi)cellulases (Rumbold et al. 2009, 2010, Pel et al. 2007, de Souza et al.
2011).
The challenge for current and future strain development programs aiming at full exploitation
of Aspergilli as multi-purpose expression platform is the full understanding of molecular cell
biology of these hosts, the identification of pathway limitations and the substantiate
prediction of beneficial metabolic engineering strategies. The aim of this chapter is to explore
the possibilities and limitations of Aspergillus as a cell factory for the production of platform
chemicals, proteins and pharmaceuticals. We review the progress made during in recent years
to implement new molecular genetic engineering tools for rational strain improvements and
discuss current technologies for the determination and evaluation of transcriptomic,
proteomic and metabolomics data from different industrial Aspergilli. We highlight
representative systems biology approaches which have uncovered some key players and
regulatory mechanisms involved in protein secretion and the formation of primary and
secondary metabolites. We also summarise the current knowledge of compartmentalized
product biosynthesis as well as transport and traffic phenomena in Aspergillus as this is key
to fully understand the link between product formation, secretion and morphology in these
versatile expression hosts.
The molecular genetic toolbox for Aspergillus
The basis for every rational experimental approach that applies genetic modification in any
organism is a well-equipped molecular toolbox including suitable vectors, selection markers
and transformation protocols. Although several plasmids have been found in bacteria, yeast
and filamentous fungi such as N. crassa, no naturally occurring plasmids are present in
Aspergillus (Griffiths 1995). Nevertheless, it has been shown that artificial plasmids with
replication sites targeting the Aspergillus replication machinery are able to autonomously
replicate in Aspergilli and distribute during mitosis. The introduction of the autonomous
maintenance in Aspergillus (AMA1) sequence from a genomic library of A. nidulans resulted
in plasmids which displayed autonomous replication properties similar to plasmids with

- 34 -
homologous sequences used in S. cerevisiae (Verdoes et al. 1994b, Khalaj et al. 2007, Gems
et al. 1991, Carvalho et al. 2010). However, the mycelium is heterogenic even under selection
pressure, tolerating hyphae devoid of plasmid without showing any phenotype or growth
defects (Aleksenko and Clutterbuck 1997). In addition, AMA1-based plasmids can get lost
during long-term cultivation, especially under non-selective pressure. Due to these reasons,
protein overexpression approaches mainly target the genome of Aspergilli. However, AMA1-
based vectors carrying auxotrophic (pyrG) or dominat (hygB) markers are very helpful for
complementation approaches of gene deletion mutants (Carvalho et al. 2010).
Several selection markers are available for genetic modification of Aspergillus. Well
established are nutritional and auxotrophic markers like argB, pyrG, pyrE, trpC, amdS and
niaD as well as antibiotic resistance markers based on hygromycin B (hygB) and phleomycin
(phle) resistance (Meyer et al. 2010b). The advantage of using pyrG or pyrE as selection
marker is that pyrG- or pyrE
- strains can easily be obtained by direct selection on 5’-
fluoroorotic acid medium without any mutagenic treatment. Another advantage of these
markers is that they can be used repeatedly, i.e. after integration of a pyrG- or pyrE-
containing plasmid into the genome, transformants can be cured from pyrG or pyrE by
cultivating them on FOA plates. The amdS gene can be used, much like pyrG and pyrE, as a
bidirectional marker by counterselecting for the loss of amdS with media containing the
antimetabolite fluoroacetamide (FAA). Detailed protocols for obtaining pyrG- pyrE
- or amdS
-
recipient strains of A. niger have recently been published (Meyer et al. 2010b).
Other versatile selection markers have recently been added to this collection. For example,
three mutant alleles encoding subunits of a succinate dehydrogenase (sdhB, sdhC, sdhD) have
been isolated from a carboxin resistant strain of A. oryzae. One of them (sdhB) has been
shown to provide resistance to carboxin in A. parasiticus (Shima et al. 2009). Another
auxotrophic marker for A. niger is based on the sC gene encoding an ATP-sulfurylase
homologous to the Saccharomyces cerevisisae MET3 gene. Complementation with a fully
functional copy of the sC gene from a wild type A. niger strain conferes cysteine prototrophy
(Varadarajalu and Punekar 2005). Finally, an efficient selection system targeting arginine
catabolism of Aspergillus has most recently been established. An A. niger strain deficient in
arginase (agaA) and hence unable to grow on arginine as the sole nitrogen source can
efficiently be complemented with an arginase expression plasmid (Dave et al. 2012).
Counterselection of bi-directional markers is an effective approach to re-use selection
markers in Aspergilli. As discussed above, counterselection with antimetabolites such as
FOA and FAA allow the re-use of markers such as pyrG or amdS. The concept behind is that
the antimetabolite used is intracellularly converted into a toxic substance when an intact copy

- 35 -
of the marker gene pyrG or amdS is present in the genome and actively expressed. Such a
selection pressure allows the isolation of natural mutants which accumulate loss-of-function
mutations within pyrG or amdS and/or the isolation of induced mutants which have lost the
selection marker due to homologous recombination between direct repeats flanking pyrG or
amdS, respectively (Meyer et al. 2010b, Carvalho et al. 2010). An alternative system for
marker recycling has been developed for filamentous fungi by adaptation of the cre/loxP
system from the bacteriophage P1. Here, the marker gene is flanked by 34 bp long DNA
sequences (loxP), which become specifically recognized by the recombinase Cre. This
enzyme efficiently catalyses the excision of any DNA sequence located between both loxP
sites, which was successfully demonstrated in A. nidulans, A. fumigatus and A. oryzae
(Krappmann et al. 2005, Forment et al. 2006, Mizutani et al. 2012) .
For gene targeting approaches in Aspergillus, two challenges have to be met. Firstly, any
recombinant DNA has to pass the cell wall and cell membrane of the recipient strain.
Secondly, the DNA introduced has to effectively targeted to genetic locus intended. Several
transformation techniques have been established to transform Aspergilli, among which
polyethylene glycol (PEG)-mediated transformation of protoplasts is the most frequently used
method. Other transformation methods such as Agrobacterium tumefaciens – mediated
transformation (Michielse et al. 2008, de Groot et al. 1998), electroporation or biolistic
transformation have been established as well, although their transformation efficiencies are
not as high compared to PEG-mediated transformation of protoplasts (Meyer et al. 2003,
Meyer 2008). Detailed protocols for PEG-mediated transformation of A. niger providing
step-by-step instructions and helpful advices on how to avoid potential pitfalls have recently
been published (Arentshorst et al. 2012, Meyer et al. 2010b). Basically, young mycelium is
incubated with cell wall degrading enzymes or enzymes mixtures including Lysing Enzyme®
from Trichoderma harzianum, chitinase from Streptomyces griseus or β-glucuronidase from
Helix pomatia to degrade the cell wall releasing protoplasts (de Bekker et al. 2009).
Protoplasts are suspended in buffers with high salt or sugar concentrations (0.7- 1.2 M
sucrose, sorbitol or NaCl), in order to protect them from burst due to osmotic imbalance.
DNA uptake of the cells is mediated by calcium chloride (10-50 mM) in combination with
high concentrations of PEG. Although PEG-mediated transformation of cells is a well-
established method for bacteria, yeast and filamentous fungi since the 1980s, the actual
mechanism on how PEG enables DNA to enter the cell membrane has remained cryptic for
almost three decades. Recently, it was uncovered that high concentrations of PEG mediate
the attachment of dissolved DNA to cells and protoplasts of S. cerevisiae and facilitate the
uptake of DNA via endocytosis (Kawai et al. 2010, Zheng et al. 2005). However, compared

- 36 -
to transformation efficiencies obtained with Escherichia coli or S. cerevisiae, the
transformation efficiencies of Aspergilli, and in general of filamentous fungi, are
considerably lower, reaching only about 10-100 transformants per µg DNA (Fincham 1989).
A second challenge, when working with Aspergilli is their low frequency of homologous
recombination, which hampered efficient functional gene analyses for a long time. Only after
determining that disruption of the non-homologous end-joining (NHEJ) pathway in N. crassa
resulted in homologous recombination frequencies up to 100% (Ninomiya et al. 2004),
respective mutants were established in various filamentous fungi, thereby allowing genome-
wide functional genomics studies to become feasible (see Kück and Hoff 2010, Meyer 2008).
In brief, the NHEJ pathway is a eukaryotic mechanism which bridges broken DNA ends by
the joint activities of the Ku heterodimer (Ku70/Ku80-protein complex) and the DNA ligase
IV-Xrcc4 complex (Dudásová et al. 2004, Krogh and Symington 2004). In eukaryotes, the
NHEJ pathway competes with another repair mechanism, the homologous recombination
(HR) pathway, which mediates interaction between homologous DNA sequences, whereas
the NHEJ pathway ligates double-strand breaks without the requirement of any homology
(Shrivastav et al. 2008). By deleting either ku70, ku80 or lig4 genes, the HR frequency is
dramatically increased in Aspergilli (Table 2). Another advantage of this high efficiency of
gene targeting is that essential genes can easily be identified by the so called heterokaryon
rescue technique as shown for A. nidulans and A. niger (Nayak et al. 2006, Carvalho et al.
2010). However, several studies have shown that inactivation of the NHEJ pathway makes
fungal strains vulnerable to DNA damaging conditions thus increasing their sensitivity
towards UV, X-ray or chemical mutagens (Meyer et al. 2007a, Malik et al. 2006, Kito et al.
2008, Snoek et al. 2009). To eliminate the risk that NHEJ deficiency influences or obscures
phenotypic analyses, A. nidulans and A. niger strains were established being transiently
silenced in NHEJ, and respective strains have proven to perform as efficient as constitutive
silenced NHEJ strains with respect to gene targeting (Carvalho et al. 2010, Nielsen et al.
2008).

- 37 -
Table 2. Homologous recombination frequencies in NHEJ defective mutants of different Aspergilli
Species
Length of
homologous
sequence (bp)
Homologous
recombination
frequency (%)
Reference
A. fumigatus
100
500
1000
1500
2000
(75)a
(84)a
96
96
95
(Krappmann et al.
2006)
A. nidulans
500
1000
2000
89
92
90
(Nayak et al. 2006)
A. niger
100
200
500
1000
1500
18
33
88
95
98
(Meyer et al. 2007a)
A. sojae
500
1000
1400
2000
14.3
71
75
87
(Takahashi et al.
2006)
Neurospora crassa
100
500
1000
10
91
100
(Ninomiya et al.
2004)
a Frequencies given in brackets might not be significant due to low numbers of obtained
transformants
In summary, the molecular genetic toolbox for Aspergillus has considerably been extended in
recent years and provides the research community with versatile tools to genetically modify
this genus in a rational and user-specified way.
Genetic strategies to improve Aspergillus as protein producer
Aspergilli are extraordinary in their ability to secrete high amounts of proteins into the
environment. Concentrations up to 20 g/l culture medium are no peculiarities for a wide
range of host specific proteins (Finkelstein 1987). Secreted fungal proteins like amylases,
lipases or proteases are produced for a wide range of applications including laundry,
biorefinery pre-processing or food industry (Table 1 and (Fleissner and Dersch 2010)). Their
outstanding secretion capacity in case of homologous fungal proteins, their ability to post-
translationally modify proteins and their long history of safe use fostered attempts to produce
proteins of non-fungal origin in Aspergilli. Unfortunately, respective expression levels were
considerably lower compared to homologous proteins (Conesa et al. 2001). This phenomenon
is still one of the major challenges in Aspergillus biotechnology, although some progress has

- 38 -
been made in the last decade. Successful and combined approaches include (i) the use of
strong, constitutive or inducible promoters, (ii) carrier protein approaches, i.e. a genetic
fusion of the heterologous protein of interest with a homologous protein (‘carrier protein’)
which is secreted by Aspergillus, (iii) increased expression of chaperones and foldases by
tackling the unfolded protein response (UPR) and the endoplasmatic reticulum associated
degradation (ERAD) pathway and (iv) down-regulation of proteases secreted by Aspergillus.
We will briefly summarise these strategies here, and also recommend that readers the review
Lubertozzi and Keasling 2009, Ward 2011, Meyer 2008, Fleissner and Dersch 2010 for more
detailed information.
Improving recombinant protein expression by optimising transcription
To produce homologous and heterologous proteins in Aspergilli, different promoter systems,
i.e. growth-dependent or inducible, have been established and validated (Table 3) (Fleissner
and Dersch 2010). In general, inducible promoters are superior to growth-dependent
promoters because they offer more flexibility for controlled protein production. For example,
protein expression can be induced after cells reach the exponential growth phase at an
optimum level thus lowering any potential toxic side effects to the host.
Among the growth-dependent promoters, the glyceraldehyde-3-phosphate dehydrogenase
(gpdA) promoter is the most widely used in Aspergilli. Other successfully promoters include
the adhA and tpiA promoter of A. nidulans (Upshall et al. 1987), the pkiA promoter of the A.
niger (Storms et al. 2005), the gdhA promoter of A. awamori (Moralejo et al. 1999) and the
hlyA promoter from A. oryzae which has been shown to be highly active under solid state
fermentations. Recently, the citrate synthase citA promoter of A. niger has been evaluated
showing high expression under different carbon sources such as glucose, sucrose, starch,
glycerol, acetate or molasses (Dave and Punekar 2011). Furthermore, Blumhoff and
colleagues selected from transcriptomic data of A. niger six novel growth-dependent
promoters (mbfA, coxA, srpB, tvdA, mdhA, manB), which display different activation levels,
thus being potentially applicable for fine-tuned protein expression when low or medium
levels of expression are aimed for (Blumhoff et al. 2012).

- 39 -
Table 3. Established and new promoters applicable for recombinant protein overexpression in Aspergillus
Species Promote
r
Gene function Inducible Remark Reference
A. awamori exlA Xylanase
Yes High expression level
Inductor: D-xylose
Repressor: glucose
(Gouka et al. 1996)
gdhA Glutamate-
dehydrogenase
No Nitrogen-and growth-dependent (Cardoza et al. 1998)
glaA Glucoamylase Yes High expression level
Inductor: starch, maltose,
maltodextrin, glucose
(Ward et al. 1995)
A. fumigatus niiA Nitrite reductase Yes Nitrogen-dependent
Repressor: ammonium
(Hu et al. 2007)
A. nidulans alcA Alcohol
dehydrogenase I
Yes Medium expression level
Inductor: ethanol,
ethylmethylketone
Repressor: glucose
(Gwynne et al. 1989)
gpdA Glycerinaldehyd
-3- phosphat
dehydrogenase
No High expression level
Growth-dependent
(Punt et al. 1990)
A. niger catR Catalase Yes Inductor: H2O2 (Sharma et al. 2012)
citA Citrate synthase No High expression level (Dave and Punekar 2011)
glaA Glucoamylase Yes High expression level
Inductor: starch, maltose,
maltodextrin, glucose
Repressor: xylose, sucrose
Leaky
(Fowler et al. 1990)
HERα Human estrogen
receptor-
expression
system
Yes Tunable expression
Inductor: estrogen
Tight but weak
(Pachlinger et al. 2005)

- 40 -
inuE Exoinulinase Yes Inductor: inulin and sucrose (Yuan et al. 2008)
mbfA Predicted
multiprotein
bridging factor 1
No High expression level (Blumhoff et al. 2012)
pkiA Pyruvate kinase No High expression level (Storms et al. 2005)
sucA ß-fructo-
furanosidase
Yes High expression level
Inductor: sucrose, inulin
Tight
(Roth and Dersch 2010)
Tet-On Tetracycline-
expression
system
Yes Tunable expression
Inductor: Doxycycline
Tight
(Meyer et al. 2011a)
tvdA Predicted
transport
docking protein
No Medium expression level (Blumhoff et al. 2012)
A. oryzae amyA Alpha-amylase Yes High expression level
Inductor: starch, maltose
(Tada et al. 1991)
hlyA Hemolysin No High expression level
No glucose repession
(Bando et al. 2011)
sodM Manganese
superoxid
dismutase
Yes Controllable with H2O2 (Ishida et al. 2004)
tef1 Translation
elongation factor
No Growth-dependent
No glucose repression
(Kitamoto et al. 1998)
thiA Thiamine Yes Tunable expression
Inductor: Thiamine
Inactive under alkaline pH
(Shoji et al. 2005)

- 41 -
Among the inducible promoter systems, the starch inducible glaA and the ethanol inducible
alcA promoters are most widely used. The gene glaA encodes a α-glucoamylase of A. niger
which is induced by starch, maltose and repressed in the presence of xylose (Fowler et al.
1990). Activation of glaA is mediated via the transcriptional regulator AmyR, which controls
the expression of amylolytic genes upon growth on starch or maltose as carbon sources
(Vinck et al. 2011, Vongsangnak et al. 2009). The alcohol dehydrogenase (alcA) promoter of
A. nidulans, becomes strongly activated by ethanol (Nikolaev et al. 2002, Felenbok 1991),
which is mediated by the AlcR transcription factor and depends on acetaldehyde as co-inducer
(Nikolaev et al. 2002). Expression of both the glaA and the alcA gene are repressed by the
catabolite repressor CreA in the presence of an easily metabolizable carbon source like
glucose (Prathumpai et al. 2004). CreA binds competitively to both promoters, thereby
hindering accession of AlcR or AmyR to their promoter binding sites. This competitive
inhibition was shown to become bypassed by introducing multiple copies of AlcR binding
sites to the alcA promoter (Gwynne et al. 1989). In addition, the number of AlcR molecules
expressed can increase the activation level of the alcA promoter (Panozzo et al. 1997).
Other inducible promoter systems include the xylose inducible exlA promoter (Gouka et al.
1996), the sucrose and inulin inducible sucA promoter of A. niger, repressible by glucose or
maltose (Roth and Dersch 2010), the inulin inducible inuE promoter of A. niger, repressible
by glucose (Yuan et al. 2008) and the hydrogen peroxide and calcium carbonate inducible
catR promoter of A.niger (Sharma et al. 2012). Most of the inducible promoter systems
described above are metabolism-dependent; hence, the choice of the growth medium is
limited. In order to overcome this constraint, different efforts were undertaken to establish and
validate metabolism-independent inducible promoter systems for the genus Aspergillus: the
thiamine promoter system (PthiA) in A. oryzae (Shoji et al. 2005), the human estrogen
receptor (hER ) system in A. nidulans and A. niger (Pachlinger et al. 2005), and an system
based on the Escherichia coli tetracycline-resistance operon (Tet-On) in A. fumigatus (Vogt et
al. 2005) and A. niger (Meyer et al. 2011a). All systems drive gene expression in a inducer-
dependent manner, however the hERα promoter system is either leaky but strong or tight and
weak, whereas the PthiA system is inactive under alkaline conditions. The most promising
expression system is the Tet-On system, which is tight under non-induced conditions, and
responds within minutes after inducer addition and mediates high gene expression levels,
which are comparable to levels of the gpdA promoter in A. niger (Meyer et al. 2011a). The
Tet-On system is composed of the reverse transactivator rtTA2S-M2 which is under control of
the gpdA promoter, thus being constitutively expressed. It can only bind to the tetO operator
sequence which are located upstream of a minimal promoter (Pmin) after addition of

- 42 -
doxycycline (DOX). Consequently it activates transcription of the gene of interest in a DOX-
dependent manner (Fig. 1). Most importantly, expression levels of the gene of interest can be
fine-tuned in a user-specified manner: the amount of DOX added to medium and the copy
number of the GOI expression cassette present in the genome of A. niger are causal to the
amount of protein produced. In addition, the spectrum of time-dependent induction can also
vary between hours (one copy, low amount of DOX, low amount of protein product) to only
few minutes (multiple copies, high DOX, high amount of protein product) (Meyer et al.
2011a). These characteristics make the Tet-On system superior to the other inducible systems
mentioned above and suggest that this system can be used as a general and excellent tool for
user-defined protein overexpression in Aspergillus.
Fig. 1: Schematic representation of the Tet-On expression system for A. niger (Meyer et al. 2011a).
The constitutively expressed reverse transactivator (rtTA) is unable to bind to the operator sequence in
the absence of DOX (the system is OFF). However, in the presence of DOX, it undergoes a
conformational change upon binding to doxycycline (DOX). Consequently, binding of rtTA to the
tetracycline operator, of which seven copies are present (tetO7) is activated and expression of the gene
of interest (GOI) becomes induced (the system is ON). A prerequisite for induction of gene expression
is the presence of a minimal promoter sequence which is located downstream of tetO7.
Improving recombinant protein expression by optimising translation
Production rates of recombinant proteins do not only depend on the amount of transcript
produced but also on the amount of transcript translated and channelled through the secretory
pathway before becoming released into the cultivation broth. Codon bias has been known for
a long time to be an important factor for efficient protein expression in pro- and eukaryotes.
Studies carried out in E. coli have suggested that the level of every tRNA differs according to
the distribution of the corresponding codon among highly and lowly expressed genes. In
highly expressed genes, favoured codons correlate with high levels of the corresponding
tRNAs (Ikemura 1981), which was also shown for S. cerevisiae (Ikemura 1982). The codon

- 43 -
usage of N. crassa (Whittle et al. 2012, Edelmann and Staben 1994) and A. nidulans (Lloyd
and Sharp 1991) have recently been revisited and compared with 10 other food-related
filamentous fungi, including A. niger, A. oryzae and A. terreus. This comparative study
revealed that preferred nucleotides at the third position of a codon are pyrimidines (Chen et al.
2012). In case that a purine is present at the wobble position, guanine is the favoured
nucleotide. That the presence/absence of abundant/rare codons can indeed affect protein
translation has been proven for filamentous fungi two decades ago. It has been shown that the
insertion of three rare codons into the highly expressed glutamate dehydrogenase of N. crassa
decreases the protein level to 30% of the native gene while the transcription level was
unaffected (Kinnaird et al. 1991). Several studies have confirmed this observation and higher
product levels were indeed obtained by codon-optimization in filamentous fungi (Nelson et al.
2004, Cardoza et al. 2003). For instance, the rate of a recombinant aequorin expressed in N.
crassa has increased 45 fold compared to the native gene (Nelson et al. 2004) and expression
of a codon-optimised synthetic gene encoding an industrial relevant alpha-glucan
phosphorylase in A. niger has been proved successful (Koda et al. 2005).
Other studies have shown that the codon usage can have an impact on polyadenylation and
mRNA stability. For example, an optimal codon usage reduced the premature adenylation of a
heterologous endoglucanase in A. oryzae, thus preventing the accumulation of abbreviated
mRNA (Sasaguri et al. 2008). Likewise, optimization of the codon usage improved the
mRNA stability of a heterologously expressed mite allergen in A. oryzae. The observed half-
life of the codon optimized recombinant protein was tripled to 43 minutes compared to its
native mRNA, which is comparable to the half-life of the mRNA of the highly expressed glaA
gene from A. oryzae (Tanaka et al. 2012). Most recent codon usage analyses of eight different
Aspergilli (including the industrial strains A. niger, A. oryzae and A. terreus) made use of
publically available genomic and transcriptomic data and confirmed that synonymous codon
usage bias is indeed associated with expression levels in the analyzed species, since in all
cases genes coding for ribosomal proteins and other highly expressed genes (e.g. translation
elongation factors, enzymes of the Krebs cycle, tubulin) clustered together in correspondence
analyses. This data was used to define, for each species, a set of “optimal codons” for highly
expressed genes (Iriarte et al. 2012).
In addition to the optimisation of single codons, adaptation of codon pairs according to the
host genomic distribution has been shown to influence mRNA folding stability and the
accuracy and speed of the translation elongation process in S. cerevisiae (Kahali et al. 2011).
Most recently, van Peij and co-workers have adapted this approach for A. niger by comparing
the codon and codon pair usage of highly secreted endogenous and heterologous genes with

- 44 -
those of poorly secreted proteins. Based on this data, the codon pair usage of poorly secreted
proteins was redesigned, thereby considerably increasing the production yields, while
maintaining the biological activity of the proteins produced (van Peij et al. 2012). Finally,
machine learning algorithms were used to identify from to a large set of homologous (600)
and heterologous (2,000) fungal genes (all being expressed from a standardized expression
cassette at a specific genomic locus) those relevant DNA and protein sequence features which
correlate with high/low protein production level in A. niger. Using this approach, van den
Berg and colleagues uncovered that the amino-acid composition of the protein sequence is
most predictive and that, for both homologous and heterologous gene expression, the same
features are important: tyrosine and asparagine positively correlate with high-level
production, whereas methionine and lysine composition contribute to unsuccessful production
(van den Berg et al. 2012). The trained classifier algorithm that can be used as predictor for
low/high protein level expression has been made available online at
http://bioinformatics.tudelft.nl/hipsec.
Beside codon bias/codon pair optimisation, other strategies have been developed to increase
protein production. In general, multiple copies of an expression construct do usually increase
protein production levels (Verdoes et al. 1993, 1994a, Cardoza et al. 2003, Meyer et al.
2011a); however, this positive correlation is limited. For example, transcription factor titration
and/or positional effects such as insertion of the expression cassette into silenced genomic
regions or inactivation of surrounding genes which might cause pleiotropic effects, can
interfere with protein expression levels (Verdoes et al. 1994c, 1994a, Kelly and Hynes 1987) .
Although manifold rational genetic engineering strategies have been developed, such as
codon bias / codon pair optimization, optimised expression cassettes with suitable promoters
and multiple integration events, protein overexpression in Aspergillus remains an art and no
generic solutions are available. However, changing the perspective from a protein-oriented to
a host-oriented view might open new possibilities for substantial improvements. This means
that it is not only important to adapt the characteristics of the protein of interest to the host’s
requirements but it is also mandatory to truly understand the capacities of Aspergillus to
express and channel any protein of interest through the secretory pathway and at which
cellular burden this is accomplished. Basically, induced overexpression of any recombinant
protein in Aspergillus has to compete with the natural secretion capacity of homologous
proteins thus being at the cost of other cellular processes or even at the cost of other
homologous secretory proteins important for normal growth. In agreement, RNAi-mediated
silencing of abundantly secreted endogenous amylases has been shown to improve the
expression yield of recombinant chymosin in A. oryzae and silencing of an endogenous fungal

- 45 -
cellobiohydrolase in Trichoderma reesei increased the amount of secreted lipase several fold
compared to non-silenced strains (Nemoto et al. 2009, Qin et al. 2012). Hence, it is crucial to
identify and understand all homeostatic control mechanisms, which modulate the secretory
flux of Aspergillus so that the fungus can flexibly adapt its cellular capacities to the artificially
induced burden and to its own cellular needs for fast growth.
Improving recombinant protein expression by optimising the secretory flux
An inventory of the Aspergillus secretion pathway
In general, Aspergillus proteins and enzymes destined for secretion into the extracellular
space have to follow the secretory route (Fig. 2). Their journey starts with protein
translocation into the lumen of the endoplasmic reticulum (ER) via a translocon that forms a
channel through the ER (Römisch 1999). At the ER, several ER resident chaperons and
foldases including the binding protein (BipA), protein disulfide isomerase (PdiA) and
calnexin (ClxA) assist secretory proteins in their folding (Määttänen et al. 2010). Many
secretory proteins also become glycosylated, via attaching oligosaccharides to their
asparagine residues (N-glycosylation) or serine and threonine residues (O-glycosylation).
After correct folding and glycosylation, secretory proteins are packed into vesicles and
transported to and through the Golgi complex via coatamer protein complex vesicles (COPII)
and are then delivered to the Spitzenkörper, which is localised at the hyphal tip, via a
microtubule-mediated transport. At the Spitzenkörper, secretory vesicles become translocated
to actin cables and transported to the plasma membrane, where the vesicles release their cargo
into the extracellular space. Empty vesicles become recycled by endocytosis which takes
place at subapical regions of the hyphal tip and are transported back to the Golgi to become
reloaded with new cargo (Taheri-Talesh et al. 2008). Hence, protein secretion in Aspergillus
should be viewed as a concerted and balanced activity of exo- and endocytotic mechanisms.
High protein flux through the ER or expression of heterologous proteins in Aspergillus can
result in the accumulation of misfolded proteins, which is recognized by a quality control
system known as ER-associated degradation (ERAD). This control mechanism guides
misfolded proteins to the cytosol where they become degraded by the proteasome (Fig. 2). In
addition, another control mechanism becomes induced, named the unfolded protein response
(UPR). The purpose of UPR is to refold misfolded proteins via induced expression of
chaperones and foldases (Geysens et al. 2009). For this purpose, the UPR transcription factor
HacA becomes activated via splicing of an unconventional 20-nt intron out of the hacA
mRNA. This subsequently facilitates translation of the hacA mRNA and formation of HacA,

- 46 -
which in turn induces transcription of a number of UPR target genes including bipA and pdiA
(Mulder et al. 2004). Hence, both ERAD and UPR are key for the function of the secretory
pathway – control mechanisms which are not only in present filamentous fungi but also in
yeast and mammals (Kaufman 1999, Yoshida et al. 2001, Saloheimo et al. 2003, Saloheimo
and Pakula 2012).
Fig. 2: The secretion pathway in Aspergillus.
Improving secretory protein fluxes by manipulating quality control systems
There are several bottlenecks for the efficient production of recombinant proteins in
Aspergillus. These are due to limitations within the secretory pathway (Nyyssönen and
Keränen 1995, Jeenes et al. 1994, Gouka et al. 1997). Cullen and co-workers developed a
protein carrier system for A. niger by genetically fusing the N-terminus of the abundantly
secreted protein glucoamylase GlaA to the heterologous target protein chymosin (Cullen et al.
1987). They have shown that the fusion partner facilitated production of the recombinant
protein by (i) increasing the translocation rates into the ER thus improving folding, maturation
and stability (Bermúdez-Humarán et al., Le Loir et al. 2001) and (ii) masking the
heterologous protein product thereby preventing proteolytic degradation (Gouka et al. 1997).
To date, the GlaA protein is the most commonly used secretion carrier in A. niger and several
proteins have been successfully produced using this approach (Martin et al. 2003, Ward et al.
1990, 1995, 2004). In addition to GlaA, other homologous and heterologous secretion carriers
have been successfully used as carrier in different Aspergillus species, e.g. the α-amylase
sequence of A. oryzae (Nakajima et al. 2006, Korman et al. 1990). The fusion carrier gets

- 47 -
removed in the Golgi network prior to secretion when a Lys-Arg or Arg-Arg doublet is
inserted between the carrier and the recombinant protein. These motifs are specifically
recognized in the trans-Golgi network by the KexB protease, a furin-type endoprotease
member of the kexin protease family, which removes the carrier protein (Jalving et al. 2000,
Contreras et al. 1991, Punt et al. 2003). However, processing rates depend on the localization
of the protease recognition site within the three dimensional structure of the fusion protein
and can thus be inefficient in case that the site is inaccessible (Spencer et al. 1998).
When Aspergillus is forced to overexpress a heterologous protein, the ER protein folding
capacity becomes overloaded, resulting in delayed or incorrect protein folding (Guillemette et
al. 2007) and induction of the UPR (Kaufman et al. 2002, Punt et al. 1998, Wang et al. 2003).
Hence, numerous studies have been conducted on Aspergilli in order to increase the level of
chaperons and foldases to promote the synthesis of correctly folded heterologous proteins. For
example, overexpression of the chaperone BipA in A. awamori resulted in a 2- to 2.5 fold
increase of the sweet plant protein thaumatin (Lombraña et al. 2004). Similarly, insertion of
multiple copies of the pdiA gene also led to an increased amount of thaumatin in A. awamori
(Moralejo et al. 2001). In another approach, Valkonen and co-workers constitutively induced
the UPR in A. awamori by removing the 20 bp intron of hacA leading to a 2.8 fold and 7.6
fold increase of a bovine chymosin and Trametes vesicolor laccase production, respectively
(Valkonen et al. 2003). However, whether recombinant protein production can be improved
by up-regulation of the UPR is largely dependent on the target protein. For example,
overexpression of BipA in A. awamori did not increase cutinase production (van Gemeren et
al. 1998) and overexpression of PdiA did not improve expression levels of hen egg white
lysozyme in A. niger (Ngiam et al. 2000).
To systematically explore the role of the UPR in protein secretion and the role of HacA as its
central regulator, the transcriptome of A. niger expressing a constitutive active form of HacA
(HacACA
), thus mimicking an overload of the ER with misfolded proteins, was compared with
the transcriptome of a strain expressing the wild type copy of HacA (Carvalho et al. 2012).
Several genes related to ER translocation, protein folding, protein glycosylation, vesicle-
mediated transport between organelles, exo- and endocytosis and the ERAD pathway were
up-regulated in the (HacACA
) strain, including well known HacA targets such as bipA, pdiA,
tigA and prpA, suggesting that HacA is a master regulator coordinating many processes
within the secretory pathway. However, many genes involved in transcription and translation
as well as in central catabolic pathways, including the AmyR regulon responsible for starch
degradation, were down-regulated. This phenomenon, earlier reported for A. niger and the
filamentous fungus Trichoderma reesei, and known as the REpression under Secretion Stress

- 48 -
(RESS) phenomenon (Pakula et al. 2003, Al-Sheikh et al. 2004), suggests that Aspergillus and
other filamentous fungi respond to the protein overload in the secretion pathway by reducing
the expression of genes encoding (unimportant) secretory proteins on the one hand and by
increasing the folding capacity of the cell on the other hand. In any case, global mechanisms
for energy generation and cell development seem to become arrested probably to prevent the
entry and overload of newly synthesized proteins into the already “clogged” ER. Decreasing
the metabolic activity reduces the influx of new proteins into the overloaded ER and extends
the residence time of misfolded proteins within the ER to become successfully refolded.
The ERAD pathway is closely linked to the UPR and has been studied for a long time in S.
cerevisiae. In case that correct folding of secretory proteins fails although expression of
foldases and chaperones has been up-regulated by the UPR, misfolded proteins become
targeted to the ERAD pathway (Fig. 2). In brief, protein degradation is initiated by the
removal of an 1,2-α-mannose residue from the protein by the specific 1,2-α-mannosidase
Msn1p in S. cerevisiae the respective homolog of which is MsnA in A. niger (Gonzalez et al.
1999, Tremblay and Herscovics 1999, Carvalho et al. 2011). Subsequently, the protein
becomes retrotranslocated from the ER to the cytoplasm through the Sec61p translocon or the
Der1p (DerA) translocon (Schäfer and Wolf 2009, Goder et al. 2008)). Thereafter, the protein
becomes ubiquitinylated by different complexes consisting of different proteins such as Hrd3p
(HrdC) and Doa1p (DoaA) (Kostova et al. 2007). Mif1p (MifA) transports the labelled protein
to the 26S proteasome (van Laar et al. 2001), where it becomes degraded in an ATP-
dependent manner (Fischer et al. 1994). Carvalho and co-workers recently investigated the
role of the ERAD pathway in A. niger and its potential link to the UPR. UPR-inducing
conditions were achieved by induced expression of heterologous proteins from bacterial,
metazoan and human origin or due to treatment of A. niger with dithiothreitol (DTT) or
tunicamycin. These stress conditions were applied to different A. niger strains, a wild type
strain or mutant strains carrying a single deletion in one of the ERAD components MnsA,
DerA, DoaA, HrdC and MifA, respectively (Carvalho et al. 2011) Forced expression of
heterologous proteins in A. niger indeed triggered up-regulation of BipA, PdiAdiA, DerA and
HrdC in the wild type background strain, thus proving a close link between the UPR and
ERAD pathway in A. niger. Surprisingly, deletion of none of the ERAD proteins MnsA,
DerA, DoaA, HrdC or MifA increased the susceptibility of A. niger towards DTT and
tunicamycin which are both known to induce expression of the UPR genes bipA and pdiA. In
agreement, deletion of ERAD components did not result in any apparent phenotype (except
for ΔdoaA) and did not impair the faith of the heterologous protein. These observations pose
the question, whether protein degradation of misfolded proteins is mediated by the ERAD

- 49 -
pathway in A. niger, or whether this might be accomplished by an alternative, yet unknown
mechanism, e.g. by the activity of ER-resident proteases as discussed for mammals
(Evnouchidou et al. 2009, Carvalho et al. 2011). Taken together, the data from A. niger
suggest that both the UPR and the ERAD pathways are cellular responses to forced protein
overexpression and/or ER stress; however, induction of the UPR seems to be sufficient to
maintain ER homeostasis, whereas the ERAD pathway is of minor importance for efficient
protein production and secretion.
Improving secretory protein fluxes by targeting protein glycosylation
In eukaryotic organisms, most of the secretory proteins become glycosylated during their
passage through the secretory pathway. Congurently, Aspergilli are able to perform both N-
and O-glycosylation which are accomplished in the ER and the Golgi apparatus. In contrast to
homologous proteins, heterologous proteins often fail to become properly N-glycosylated.
Protein glycosylation, however, is an essential step for effective protein secretion because it
increases the solubility of the yet unstructured polypeptides (Helenius and Aebi 2004). A
detailed overview of the N-glycosylation pathway performed in A. fumigatus and a
comparison to the pathways present yeasts and mammals was recently given (Jin 2012). In
brief, a lipid anchored Glc3Man9GlcNAc2 oligosaccharide is synthesized at the ER lumen in
several consecutive steps via a series of glycosyltransferases, which are either located in the
cytoplasm or the ER (Fig. 2). The oligosaccharide is subsequently transferred to secretory
proteins within the ER by the activity of an oligosaccharyltransferase complex (OST in S.
cerevisiae). Thereafter, two of the three glucose molecules are removed by glucosidase I and
II and the glycoprotein binds to the calcium-binding proteins calnexin/calreticulin. After
correct folding, the last glucose residue is cleaved off by glucosidase II, causing the
dissociation of the glycoprotein from calnexin/calreticulin. In case that the protein did not fold
properly, an UDP-glucose:glycoprotein glucosyltransferase (UGT), a key enzyme of the ER
quality control, reintegrates again one glucose molecule to the misfolded protein, thereby
causing retention of the glycoprotein and thus preventing its premature exit from the ER
(Ruddock and Molinari 2006). If the protein is correctly folded, the glycoprotein is
transported to the Golgi, where additional modifications take place, including the removal of
mannose residues and the addition of other monosaccharides (Jin 2012).
With the assumption that incorrect protein glycosylation decreases heterologous protein
production, several studies have been conducted in Aspergillus. For example, overexpression
of calnexin in A. niger resulted in a fourfold increase of the secretion of manganese
peroxidase from Phanerochaete chrysosporium (Conesa et al. 2002). In another study, a

- 50 -
poorly used glycosylation site within bovine chymosin was genetically modified,
subsequently leading to a doubling of the protein secreted by A. niger (van den Brink et al.
2006). In conclusion, the introduction of an additional glycosylation site between the GlaA
carrier and prochymosin considerably increased the amount of secreted fusion protein in A.
niger (van den Brink et al. 2006).
Improving secretory capacities of Aspergillus by establishing alternative secretion routes
So far, this chapter has dealt with the production of secretory proteins channelled through the
classical secretory pathway. However, many proteins and enzymes of industrial interest are
intracellular proteins but their production is commercially not vital due to very high
downstream processing cost. These enzymes would have, however, realistic potential for
industrial applications. To address this limitation, an alternative, artificial secretion route,
designated peroxicretion, was established and validated in A. niger (Sagt et al. 2009). The
idea behind this was that any intracellular (homologous or heterologous) protein is targeted at
peroxisomes by genetically fusing its C-terminus to the peroxisome import signal (SKL). In
addition, peroxisomes were decorated with Golgi-derived v-SNAREs, enabling peroxisomes
to be transported to the hyphal tip, where they should fuse with the plasma membrane thus
releasing their content into the culture broth. As a proof of concept, the fate of the green
fluorescent protein (GFP) tagged with the SKL motif was studied in A. niger (Sagt et al.
2009). As 55% of total GFP was found extracellularly suggested that peroxicretion might
indeed be a feasible alternative secretion route. This conclusion was corroborated by the fact
that addition of C2-ceramide, causing enrichment of t-SNAREs, which are important for v-
SNARE pin formation prior to membrane fusion (Bonifacino and Glick 2004), increased the
amount of secreted protein even further. However, increasing the amount of peroxisomes did
not increase the amount of protein released into the medium (Sagt et al. 2009). These
observations imply that rerouting of proteins is in general possible in Aspergillus but this
requires more understanding before becoming feasible for industrial production of
intracellular heterologous proteins.
Improving recombinant protein expression by preventing product degradation
According to their saprophytic lifestyle, Aspergilli are capable of secreting vast amounts of
different hydrolytic enzymes including proteases and glycan hydrolases required for the
degradation of dead organic matter (Lopes et al. 2011, van den Hombergh et al. 1997).
Although this high secretion capability makes Aspergilli as outstanding expression platforms,

- 51 -
parallel secretion of proteases limits productivity as the protein of interest is likely to become
degraded extracellularly. About 200 proteases have been predicted in the genome of A. niger
CBS513.88, of which approximately 50 have a signal peptide sequence, thus being potentially
secreted (Pel et al. 2007). Consequently, deactivation of single proteases is probably not
sufficient to prevent proteolytic degradation as this will be accomplished by other secreted
proteases. Hence, efforts have been undertaken to deactivate multiple proteases in
Aspergillus. For example, double disruption of the proteases TppA and PepE increased the
production of human lysozyme in A. oryzae (Jin et al. 2007). Through selection marker
recycling, even quintuple (Yoon et al. 2009) and tenfold deletion (Yoon et al. 2011) of
proteases have been achieved in A. oryzae leading to considerable further increases of the
production yields for human lysozyme and bovine chymosin.
Already 20 years ago, a UV mutant of A. niger was isolated (AB1.13) which secreted about
80% less proteases than the wild type strain (Mattern et al. 1992). The underlying genotype
remained unclear until complementation approaches identified the mutated gene to encode for
a fungal Gal4-type Zn(2)-Cys(6) transcription factor designated PrtT (Punt et al. 2008). Most
interestingly, no PrtT orthologues are present in any other non-Aspergillus (or related) species
and also not in A. nidulans. In all Aspergillus species carrying a prtT orthologue, the gene is
clustered with AmyR amylolytic gene cluster (Punt et al. 2008). Whereas the point mutated
version of the prtT gene of the A. niger AB1.13 mutant (causing a change of a conserved
leucin at position 112 to prolin) considerably decreased protease expression, complete
deletion of the prtT gene in A. fumigatus not only lowered protease activities but also
adversely affected expression of genes involved in iron uptake, ergosterol synthesis and
secondary metabolism, suggesting that PrtT fulfils multiple cellular functions for Aspergillus
(Hagag et al. 2012).
Improving recombinant protein expression by optimising filamentous morphology
Polarised growth and secretion is a defining attribute of the lifestyle of filamentous fungi.
Highly polarised hyphae send out branches from apical or lateral regions thereby forming a
dense cellular meshwork, the mycelium (Fig. 3, A-B). Long-distance transport of RNA,
proteins, vesicles and organelles along cytoskeletal tracks ensures that new cell material is
exclusively added to the growth zone at the hyphal tip. Thesetransport processes are also a
prerequisite for polarised protein secretion (Harris 2008, Torralba et al. 1998, 1996, Harris
2006). Although a link between protein production and the abundance of actively growing
hyphal tips has been proposed for a long time in Aspergillus (Wösten et al. 1991, Gordon et
al. 2000), only contradictory results have been reported so far. An increase in the number of

- 52 -
hyphal tips has been reported to improve protein secretion in some cases but not in all. Thus,
no generally accepted model can be used as basis for rationally optimising the morphology of
filamentous fungi with respect to protein secretion and their rheological behaviour in a
bioreactor.
Basically, Aspergilli and all other filamentous fungi can form two different
macromorphologies during submerged growth. They grow either as dense mycelial
aggregates, so called pellets, which get formed when hyphae branch out at a high frequency
(Fig. 3, C-E). Alternatively, they grow as freely dispersed mycelium, a result of low
branching frequencies. Fungal macromorphologies affect the productivity of Aspergillus
cultivation and the preferred morphology would consist of highly branched dispersed mycelia.
Whereas the formation of pellets is less desirable because of the high proportion of biomass in
a pellet that does not contribute to product formation, long, unbranched hyphae tend to
entangle and are also sensitive to shear forces in the reactor. Lysis of hyphae and the
subsequent release of intracellular proteases have thus a negative effect on protein production.
Fungal macromorphologies can be controlled by environmental conditions including pH level,
amount of spore inoculum, power input, osmolarity and the presence of shear stress provoking
micro particles (Wucherpfennig et al. 2010, 2011) . The addition of talc micro particles to the
cultivation medium, for example, affects pellet size and densities allowing the identification
of the best performing macromorphology for a certain process (Fig 3, C-E). Lowering pellet
size has been shown to lead to a 4 and 9 fold increase of fructofuranosidase and glucoamylase
production, respectively, in A. niger (Driouch et al. 2012a).
Fig. 3: Macromorphologies of A. niger. A, B: Young hyphae of A. niger cultivated in liquid medium.
Cell wall and septa were stained with calcoflour white to visualize septa (arrows) and newly formed
branched (stars). C-E: A. niger cultivated in shake flask cultures with increasing concentration of talc
micro particles (C: 0 g/l, D: 2.5 g/l, E: 10 g/l).

- 53 -
However, to systematically improve the morphological features of filamentous fungi in
industrial processes, much more basic knowledge is required to obtain a deeper insight into
the molecular networks regulating fungal morphology. To understand the connection between
the processes of polarized growth and secretion in the industrially important fungus A. niger,
genome-wide expression profiling studies were used to predict and identify signalling
molecules and networks involved in these processes (Jørgensen et al. 2010, Jacobs et al. 2009,
Meyer et al. 2009, 2007b). For example, the transcriptomic fingerprint of apically branched
hyphae of A. niger uncovered that the stage for the formation of two new branches is set by
increased activity of different signalling pathways including TORC2 signalling, phospholipid
signalling, cell wall integrity signalling and calcium signalling (Meyer et al. 2009, 2010a). In
addition, functional genomics approaches uncovered cellular protagonists coordinating these
processes in A. niger, such as the Rho GTPase RacA, the polarisome componenent SpaA, the
TORC2 component RmsA and the transcription factor RlmA (Meyer et al. 2008, 2009, 2010a,
Damveld et al. 2005, Kwon et al. 2011). Future studies are necessary to inventory the full set
of genes and proteins defining the morphology of Aspergillus and to disclose the key players
and their embedment in different signalling pathways which drive growth, morphology and
secretion of Aspergillus.
The genomics toolbox for Aspergillus
During the last decade, the genomes of many industrial or medical relevant Aspergillus
species have been sequenced and published (Table 4). Numerous sequencing projects are on-
going as well and are expected to extend the current collection of Aspergillus genomes
(http://www.fgsc.net/Aspergillus and (Andersen and Nielsen 2009)). Genomic resources have
initiated multiple new research activities in the Aspergillus research community including
comparative genomics, functional genomics and systems biology approaches, aiming at a
comprehensive understanding of Aspergilli and at a descriptive and predictive modelling of
their growth and behaviour. Establishing such models follows four progressive steps, defining
a cycle: (i) definition of an experimental set-up to investigate the organism of choice under
specific conditions; (ii) application of different omics methods including genomics,
transcriptomics, proteomics and/or metabolomics, producing large amounts of data; (iii)
mining of these data by bioinformatics tools to gain information; and (iv) using the
information obtained to set up a model. At this point, phase one starts again in order to
validate or refine the model in terms of its predictive function (Aldridge et al. 2006).
Although the field of systems biology for filamentous fungi and Aspergilli is developing fast
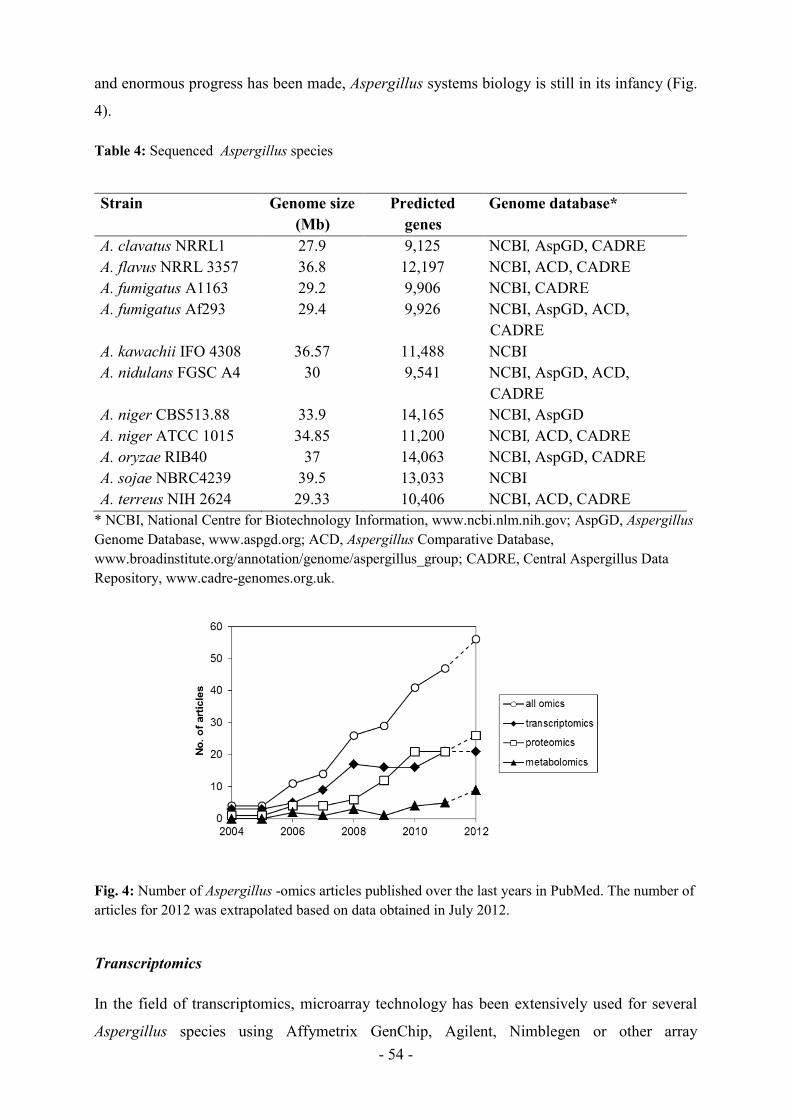
- 54 -
and enormous progress has been made, Aspergillus systems biology is still in its infancy (Fig.
4).
Table 4: Sequenced Aspergillus species
Strain Genome size
(Mb)
Predicted
genes
Genome database*
A. clavatus NRRL1 27.9 9,125 NCBI, AspGD, CADRE
A. flavus NRRL 3357 36.8 12,197 NCBI, ACD, CADRE
A. fumigatus A1163 29.2 9,906 NCBI, CADRE
A. fumigatus Af293 29.4 9,926 NCBI, AspGD, ACD,
CADRE
A. kawachii IFO 4308 36.57 11,488 NCBI
A. nidulans FGSC A4 30 9,541 NCBI, AspGD, ACD,
CADRE
A. niger CBS513.88 33.9 14,165 NCBI, AspGD
A. niger ATCC 1015 34.85 11,200 NCBI, ACD, CADRE
A. oryzae RIB40 37 14,063 NCBI, AspGD, CADRE
A. sojae NBRC4239 39.5 13,033 NCBI
A. terreus NIH 2624 29.33 10,406 NCBI, ACD, CADRE
* NCBI, National Centre for Biotechnology Information, www.ncbi.nlm.nih.gov; AspGD, Aspergillus
Genome Database, www.aspgd.org; ACD, Aspergillus Comparative Database,
www.broadinstitute.org/annotation/genome/aspergillus_group; CADRE, Central Aspergillus Data
Repository, www.cadre-genomes.org.uk.
Fig. 4: Number of Aspergillus -omics articles published over the last years in PubMed. The number of
articles for 2012 was extrapolated based on data obtained in July 2012.
Transcriptomics
In the field of transcriptomics, microarray technology has been extensively used for several
Aspergillus species using Affymetrix GenChip, Agilent, Nimblegen or other array

- 55 -
technologies (Andersen and Nielsen 2009). Only recently, first studies applying RNA
sequencing (RNA-seq) have been published for this genus (Yu et al. 2011, Wang et al. 2010,
Coradetti et al. 2012, Gibbons et al. 2012). In comparison to microarray analysis, RNA-seq
offers higher reliability when analysing lowly expressed genes and the possibility to
absolutely quantify transcript amount (Wang et al. 2009). Most recently, de Bekker and co-
workers published a protocol on how to use laser pressure catapulting to isolate RNA of
single cells from A. niger hyphae (de Bekker et al. 2011a), opening up a new door towards
single cell (transcript)omics analyses.
Transcriptomic data analysis requires several evaluation steps including background
correction, normalization and identification of co-expressed or differentially expressed genes.
Several commercial and open source programs like R/Bioconductor (Gentleman et al. 2004)
are available for transcriptomic data analysis. After identification of subset of co-expressed or
differentially expressed genes, enrichment analysis of functional annotations, including Gene
Ontology (GO), functional categories (FunCat), Pfam domain and KEGG pathway
annotations, has been shown to strongly facilitate omics data analysis (Pel et al. 2007, Nitsche
et al. 2011, 2012). Among the numerous tools that can be used for enrichment analysis of
functional annotations are Blat2GO (http://www.blast2go.de/b2ghome) (Conesa et al. 2005),
GSEA (http://www.broadinstitute.org/gsea/index.jsp) (Subramanian et al. 2005) and FetGOat
(http://www.broadinstitute.org/fetgoat/index.html) (Nitsche et al. 2011) which was recently
released as an online tool for enrichment analysis providing GO annotation for A. nidulans, A.
niger, A .fumigatus, A. terreus, A. flavus, A. oryzae, A. clavatus and N. fischeri.
Proteomics
Proteomics research for filamentous fungi is a relatively new, though fast growing branch of
fungal systems biology. Recently, a comprehensive review of the latest developments in the
proteomics field of filamentous fungi has been released (de Oliveira and de Graaff 2011).
Several intra- and extracellular proteomes have been published for different Aspergillus
species based on two dimensional gel electrophoresis (2D-GE), 2D-gel electrophoresis
coupled with mass spectrometry (MS) or based on liquid chromatography (LC) coupled with
mass spectrometry (LC-MS/MS) methods. Most of the MS based methods applied in
proteomics make use of selective labelling of peptides and proteins with stable isotopes, e.g.
2H,
13C,
15N or
18O. The methods can be classified into two categories: (i) absolute
quantification methods, where known amounts of labelled peptide or protein standards are
mixed with the sample prior to MS analysis to estimate the protein concentrations or ii)
relative quantification methods, where all proteins of a single biological sample are labelled

- 56 -
and afterwards compared to the unlabelled proteins of a reference sample (de Oliveira and de
Graaff 2011). For labelling of the whole proteome, different in vivo and in vitro methods have
been developed. Cells can be grown in 15
N-medium (Krijgsveld et al. 2003) or in medium
containing one or two labelled essential amino acids (stable isotope labeling by amino acids,
SILAC) (Ong et al. 2002). In vitro methods cover the incorporation of heavy 18
O during
tryptic digestion of proteins or linkage of isobaric tags for relative and absolute quantitation
(iTRAQ) to all proteins (Adav et al. 2010). In addition to different labelling methods,
advanced methods for protein extraction and purification have been developed, even allowing
the purification of single organelles and determination of their proteome. One such example
has been given for microbodies of P. chrysogenum (Kiel et al. 2009).
Metabolomics
Even less metabolomics studies have been published for Aspergilli. Metabolomics and flux
analysis, describe the identification and quantification of all metabolites of a cell per time and
the flux of metabolites through different catabolic and anabolic pathways. Metabolomics and
flux analysis are suitable for identifying bottlenecks that can be targeted by genetic
modification approaches or alternative feeding strategies to improve product formation.
Classical chromatographic methods (gas chromatography, LC) coupled with MS are applied
to detect a wide range of intracellular metabolites (Oldiges et al. 2007). For metabolomics
analyses, short sampling times followed by an immediate stop of any cellular metabolic
activity (quenching) are crucial. As the metabolome of a cell responds within subseconds to
changes in the environment (de Koning and van Dam 1992), extremely fast sampling is
mandatory which can usually be accomplished by automatic samplers. Cell samples become
quenched by an immediate transfer to methanol (de Koning and van Dam 1992), liquid
nitrogen (Hajjaj et al. 1998) or perchloric acid (Shryock et al. 1986). Subsequently,
metabolites can be quantified by isotope dilution MS (IDMS) (Hintenberger et al. 1955),
whereby cell extracts labelled with 13
C are widely applied as internal standards (Wu et al.
2005).
The first metabolic models for A. niger and A. oryzae have been established in 2008 by using
bibliome and genomic information of both strains (Vongsangnak et al. 2008, Andersen et al.
2008). In case of A. niger, a network has been constructed that comprises 1,190 biochemical
reactions from 52 enzyme complexes including a total of 1,045 metabolites distributed across
the cytosol, mitochondria and the extracellular space. It covers the central carbon metabolism,
catabolic pathways, 115 different carbon and 23 different nitrogen sources as well as anabolic
pathways and de novo assembled pathways such as the ergosterol synthesis pathway.

- 57 -
Additionally, secretion rates of the endogenous genes glucoamylase and alpha-amylase have
been modelled to allow predictions of theoretical yields. The model was further validated by
predicting yields of organics acids including oxalic acid, citric acid and gluconic acid.
Applying 13
C metabolic flux analysis, Driouch and co-workers compared the fluxes through
the central carbon metabolism of an A. niger strain overproducing fructofuranosidase and its
parental wild type strain (Driouch et al. 2012b). It was found that the carbon flux in the
overproducing strain was redirected from the Krebs cycle towards the pentose phosphate
pathway, probably to supply the cells with a surplus of NADPH important for anabolic
pathways. By simulating the optimal fluxes in silico and comparing them to the in vivo fluxes,
different candidate genes were identified (including enzymes of the Krebs cycle and
gluconate synthesis), which might be promising target genes for rational metabolic
engineering approaches. In addition, the metabolic model for A. niger (Andersen et al. 2008)
was used and applied to correlate fluxes of independent pathways to biomass and product
formation (flux mode analysis) and thereby the production of fructofuranosidase was
predicted when A. niger is cultivated on different carbon sources (Melzer et al. 2009).
Bioinformatics
Biofinformatic tools for the statistical analysis of omics data are a prerequisite for effective
data interpretation and integration. The development of such tools for Aspergillus is an on-
going process. Recently, a step-by-step protocol on how to dissect microarray data from
Aspergillus by using open source bioinformatics was published (Nitsche et al. 2012). These
instructions combined with the Aspergillus FetGOat gene set enrichment tool, is freely
accessible via the Broad Institute's website (http://www.broadinstitute.org/fetgoat/index.html)
(Nitsche et al. 2011), gives guidance for systematic analyses of transcriptomic data obtained
for Aspergillus. Moreover, the web-based toolbox “BiotMet” (www.sysbio.se/BioMet/)
allows the integration of genomics, transcriptomics and metabolomics data from A. niger and
A. oryzae with their genome scale metabolic models (Cvijovic et al. 2010). Another tool,
MEMOSys (MEtabolic MOdel research and development System), a bioinformatics platform
for genome-scale metabolic models (https://memosys.i-med.ac.at/MEMOSys/home.seam),
has been launched recently. It allows the management, storage, and development of metabolic
models for A. niger, A. oryzae, A. nidulans and other publically available models for non-
Aspergillus species (Pabinger et al. 2011). Finally, the first web-based modelling tool FAME
(Flux Analysis and Modeling Environment, http://f-a-m-e.org/) which combines the creation,
edition, simulation and visualisation of stoichiometric models of A. niger, A. oryzae and A.
nidulans in a single program, was recently published (Boele et al. 2012).

- 58 -
A major challenge in omics data analysis is the fact that the datasets can be error-prone due to
technical irreproducibility (e.g. cultivations of Aspergillus in shake flasks, long sampling
times, target degradation, inaccurate measurements) or due to biological variations caused by
hyphal heterogeneity in Aspergillus populations (Levin et al. 2007, Vinck et al. 2005, de
Bekker et al. 2011b, Vinck et al. 2011). Hence, statistical analyses for handling and modeling
errors as well as internal controls are crucial to obtain legitimate interpretations from high-
throughput omics data.
An omics view on Aspergillus as acid and protein producer
For A. niger, strains have evolved that are both excellent producers of organic acids and
protein secretors. To understand the genotypic traits that are beneficial for protein secretion
and/or organic acid production, the genomic landscape of two industrial predecessor strains of
A. niger have been dissected (Andersen et al. 2011). The genome of the acidogenic strain
ATCC 1015 was compared with the genome of the GlaA-overproducing strain CBS513.88.
This comparison revealed a high amount of single nucleotide polymorphisms (SNPs),
insertions/deletions of up to 200 kb, differences in transposon populations, a set of about 400-
500 unique genes in both strains and an inversion of an entire chromosomal arm. Thirty-seven
proteins involved in transcriptional regulation were predicted to be non-functional in CBS
315.88. All gene encodings for proteins that display differences in amino acid composition
were mapped to the metabolic network of A. niger. Differences were found in biosynthetic
pathways for proline, aspartate, asparagine, tryptophan and histidine as well as in the electron
transport chain and the Krebs cycle. Transcriptomic comparison of both strains, cultivated
under identical growth conditions, revealed that approximately 6,000 genes are differentially
expressed in both strains. ATCC 1015 showed a higher expression of genes involved in the
alternative oxidative pathway, whereas metabolic pathway genes involved in glycolysis and
TCA cycle, tRNA synthases as well as genes of the entire biosynthetic pathways of threonine,
serine, and tryptophan were induced in CBS513.88, amino acids which are required for
efficient GlaA production as they are overrepresented in the GlaA protein sequence.
It has further been shown that the UPR is not exclusively activated in response to artificially
induced protein overexpression in Aspergillus. When an A. niger wild type strain is cultivated
at the same specific growth rate on two different carbon sources - xylose or maltose - it
secretes about three times more (glyco)proteins on maltose compared to xylose.
Transcriptomic analyses disclosed that the higher secretion level on maltose is permitted by
up-regulation of more than 90 genes related to protein secretion including UPR target genes
(Ferreira de Oliveira et al. 2011, Jørgensen et al. 2009). Furthermore, up-regulation of the

- 59 -
UPR genes hacA and pdiA is also evident, when A. niger is cultivated at specific growth rates
approaching zero (Jørgensen et al. 2010). These observations suggest that the UPR should be
viewed as a general homeostatic control mechanism of A. niger, which not only gets activated
under ER stress provoked by artificially forced protein overexpression, but also when A. niger
has to modulate its secretory flux in order to flexibly adapt its cellular capacities and needs to
changes in the environmental conditions.
An omics view on Aspergillus as plant polysaccharide degrader
The use of second-generation feedstocks, i.e. the use of non-starch and non-edible plant
polysaccharides as carbon sources for microbial biotechnology, requires the availability of
complex mixtures of enzymes for the complete conversion of plant polymers such as
lignocelluloses, xylan, pectin, inulin and other non-starch polymers to fermentable
monosaccharides. To evaluate the potential of Aspergillus for polysaccharide degradation, the
starch-, pectin- and inulin-degrading enzyme network of A. niger was uncovered using
genomics and transcriptomics approaches (Martens-Uzunova et al. 2006, Yuan et al. 2006,
Martens-Uzunova and Schaap 2009). Using RNA-Seq, the degradation potential of A. niger
was studied when cultivated on wheat straw as a model lignocellulosic substrate and during
carbon starvation condition. The data obtained supports the very interesting models that a
subset of polymer-degrading enzymes is expressed and secreted by A. niger, which acts as
scouts to test which polymers are available in the environment. The liberation of inducing
sugars from actually present polymers subsequently induces the expression of hydrolases,
whereby their respective mRNA species can account for up to 20% of the total mRNA
(Delmas et al. 2012).
To obtain a comprehensive and even graphical overview on the complete carbohydrate-active
enzyme network of A. niger, a bibliomic and genomic survey was recently undertaken, where
data from 203 articles on the structure and degradation of 16 different types of plant
polysaccharides was compiled and combined with a list of all 188 known or predicted
carbohydrate-active enzymes from A. niger (Andersen et al. 2012). This data was combined
with transcriptomic analyses on three monosaccharides and three complex carbohydrates,
which identified enzymes being cross-induced and/or acting in a concerted manner on these
carbon sources.

- 60 -
An omics view on Aspergillus as secondary metabolite producer
Not only do filamentous fungi have high secretion capacities making them outstanding
industrial production hosts, but genome sequencing have revealed that they have a highly
versatile secondary metabolism of which the vast majority has yet remained unexplored. As
incidentally discovered by Alexander Fleming in 1928, fungal secondary metabolites
constitute a valuable source for potent antibiotics. In light of increasing microbial resistances
to antibiotics, it is of utmost importance to continue exploring this natural treasure chest. Only
recently, the World Health Organization has forecasted a disaster, should the current number
of newly acquired antibiotic resistances continue to increase without a corresponding
improvement in the exploration of new antibiotic drugs and their approval (Cooper and Shlaes
2011).
What are secondary metabolites? They have been generally described as structurally diverse
natural products of low-molecular weight that often exert bioactivity (Keller et al. 2005).
Their synthesis is restricted to specific tissues or phases during growth and development.
Although secondary metabolites are extremely versatile, they are synthesized from a rather
small pool of primary metabolic precursors including short-chain carboxylic acids, amino
acids and terpenes (Keller et al. 2005). Secondary metabolites are produced by
microorganisms as well as higher eukaryotes such as algae, plants and animals (Hoffmeister
and Keller 2007). It has been estimated that more than 40% of the known microbial secondary
metabolites originate from fungal species (Brakhage and Schroeckh 2011, Lazzarini et al.
2001). The roles of secondary metabolites in microbial habitats are not well understood and
opposing views on their evolution and natural functions have emerged (Firn and Jones 2003).
Among the fungal secondary metabolites identified to date are, potent pharmaceutical agents
that are clinically applied like the well-known beta-lactam antibiotics (penicillin and
cephalosporin) as well as immunosuppressive (cyclosporine A), cholesterol-lowering
(lovastatin), anticancer (taxol) and antifungal (griseofulvin) compounds (Meyer 2008) . In
contrast to many natural products with beneficial bioactivities, several fungal secondary
metabolites are toxic and/or carcinogenic to humans and animals, e.g. aflatoxin, fumonisin B1
and ochratoxin A (Frisvad et al. 2007, Nielsen et al. 2009). In field infection and post-harvest
contamination of food and feed, potent mycotoxin producing fungi such as A. flavus and A.
niger have been suggested as the main risk (Keller et al. 2005), thus emphasizing the
necessity of strict guidelines for monitoring of food and feed.
The availability of fungal genome sequences has made ‘genome-mining’ for new secondary
metabolic genes by bioinformatics feasible and has revealed that the genes are physically
clustered, which is in contrast to fungal primary metabolic genes and even secondary

- 61 -
metabolite genes from plants (Sanchez et al. 2012). Aspergilli have been estimated to
typically carry 30-40 secondary metabolite clusters (Brakhage and Schroeckh 2011). Besides
large multidomain enzymes including polyketide synthases (PKSs) and nonribosomal peptide
synthetases (NRPSs), secondary metabolite clusters often encode transporters, oxidases,
hydroxylases and regulatory proteins that are generally thought to be cluster-specific
(Brakhage and Schroeckh 2011). The large number of fungal secondary metabolite clusters
has exceeded the number of known and expected secondary metabolites by far. However,
genome-wide transcriptomic studies have shown that most secondary metabolite clusters are
silent under standard laboratory conditions and consequently their corresponding putative
secondary metabolites are unknown (Brakhage and Schroeckh 2011, Pel et al. 2007). These
silent clusters are also referred to as ‘orphan’ or ‘cryptic’ and have initiated many approaches
to explore this fungal treasure chest as resource for new potential pharmacologically or
biologically active compounds.
Several excellent reviews have been published describing these approaches (Gross 2007,
Hertweck 2009, Brakhage and Schroeckh 2011, Sanchez et al. 2012). The most
straightforward and powerful approach is referred to as the “one strain many compounds”
(OSMAC) strategy which, instead of genetically modifying organisms, simply aims at
changing secondary metabolic profiles by altering the cultivation conditions, including C-, N-,
P-sources and concentrations, pH, water activity, oxygen availability, light, temperature and
specific growth rates (Bide 2002). The OSMAC strategy has for example been applied to
demonstrate the toxigenic potential of A. niger. Despite the fact that A. niger has been
considered to be nontoxic under industrial conditions (Schuster et al. 2002), genome
sequencing revealed two secondary metabolite clusters sharing homology to those required
for the synthesis of the mycotoxins fumonisin and ochratoxin. Both clusters have been shown
to be silent under standard cultivation conditions (Pel et al. 2007), while several studies have
confirmed the general toxigenic potential of A. niger, in particular as a contaminant of food
and feed, by cultivation under non-standard conditions, e.g. on substrates with low water
activity (Frisvad et al. 2007) or at specific growth rates near zero (Jørgensen et al. 2010,
2011). In another OSMAC approach, A. nidulans was cultivated in different single nutrient-
limited (N, C and P) chemostat cultures at low specific growth rates to induce silent PKS
genes. This approach resulted in an induction of the two PKSs encoding genes orsA and
mdpG under N-limitation, the detection of several known secondary metabolites under N-
and P-limitations as well as the identification of three novel secondary metabolites, namely
sanghaspirodin A and B as well as pre-shamixanthone (Sarkar et al. 2012).

- 62 -
Although the biological function of secondary metabolites is unknown and remain a matter of
speculation, it has been generally believed that they play a role in intermicrobial
communication and niche securement (Rohlfs et al. 2007, Kobayashi and Crouch 2009). This
has motivated testing the hypothesis that co-cultivation between Aspergillus and soil bacteria
- which share the same habitat in nature - might trigger the synthesis of certain secondary
metabolites. Indeed, an early example of this OSMAC approach was the discovery of the
antibiotic pestalone from co-cultivation of a marine fungus (Pestaltia sp.) with an unidentified
Gram-negative bacterium (Cueto et al. 2001). This strategy has been recently been refined by
combining microarray analysis and systematic co-cultivation of A. nidulans with 58
actinomycetes (Schroeckh et al. 2009). Surprisingly, only one of the screened actinomycetes
(Sreptomyces hygroscopicus) specifically induced secondary metabolism genes. Subsequent
transcriptome and metabolome analyses led to the identification of an induced PKS gene
cluster a required for orsellinic acid synthesis.
Alternative approaches for studying secondary metabolite clusters and biosynthetic pathways
involve molecular genetic manipulation and subsequent comparative metabolic analysis of
wild-type and mutant strains. One example for a successfully applied knock out strategy is the
targeted deletion of six randomly selected NRPS encoding genes in A. nidulans and
comparative metabolic profiling. Thus approach led to the identification of five secondary
metabolites, emericellamide A, C-F (Chiang et al. 2008). Interestingly, only one of the six
gene deletions resulted in an altered metabolic profile under laboratory conditions, implying
that the other five clusters were silent. Hence, a combinatorial approach of the OSMAC
strategy to analyse the generated mutants is more promising. Another knock-out project has
systematically targeted 32 genes putatively encoding PKSs in A. nidulans and identified two
austinol meroterpenoids and improved the understanding of the biosynthetic routes for
arugosins and violaceols (Nielsen et al. 2011).
The concept of OSMAC includes that environmental pH as well as carbon and nitrogen
sources do influence fungal secondary metabolite production. Three wide-domain
transcriptional regulators, PacC, CreA and AreA, are known to mediate the adaption of fungi
to these environmental conditions (Peñalva et al. 2008, Etxebeste et al. 2010) and have indeed
been proven to either induce or repress secondary metabolite production in a metabolite-
dependent manner (Hoffmeister and Keller 2007). Another approach relates to the observation
that many secondary metabolite clusters contain potential pathway-specific transcription
factors. This was first discovered for the aflatoxin biosynthesis cluster in A. nidulans which
contains the alfR gene encoding a cluster specific C(6) zinc finger transcription factor that
specifically recognizes a palindromic motif found upstream of most aflatoxin biosynthesis

- 63 -
genes. Deletion of aflR or additional aflR copies have been shown to abolish or induce the
transcription of the aflatoxin cluster genes, respectively (Hoffmeister and Keller 2007). The
existence of cluster-specific regulators was further demonstrated for the apd gene cluster of A.
nidulans which was awakened from its silent transcriptomic state simply by conditional
overexpression of its regulatory gene apdR leading to the production of the two new PKS-
NRPS hybrids aspyridone A and B (Bergmann et al. 2007).
The analysis of classical developmental mutants of A. nidulans being defective in alleles of
the fluffy genes fluG, flbA or fadA have disclosed that developmental regulation and
secondary metabolism are closely linked via a shared G-protein signalling pathway (Calvo et
al. 2002). Mutants affected in sterigmatocystin biosynthesis but with negligible
developmental defects have been isolated and studied, whereby laeA (loss of aflR expression)
has been identified to complement one of the mutants. LaeA acts as a global regulatory
protein of secondary metabolism. Expression of secondary metabolic clusters including
penicillin, lovastatin and stergimatocystin is blocked or induced by deletion or induction of
laeA, respectively. The laeA gene encodes a nuclear protein that shares homology with
methyltransferases suggesting that its global regulatory mechanism is mediated by chromatin
remodelling (Bok and Keller 2004). LaeA has not been identified in yeast and the
identification of the velvet complex in A. nidulans indicates a mechanistic link between light-
induced fungal differentiation by VeA and secondary metabolism by LaeA (Bayram et al.
2008).
The discovery of LaeA as a global regulator of secondary metabolism suggests an upper-
hierarchy, epigenetic level of regulation through chromatin remodelling. Two principally
distinct chromatin forms exist for genomic DNA, i.e. a tightly packed and hence
transcriptionally silenced chromatin (heterochromatin), and a less dense packed form, which
allows higher transcriptional activities (euchromatin). The structure and packaging of
chromatin is determined by histones and their post-translational modifications which
comprises more than hundred distinct modifications including acetylation, methylation,
phosphorylation and sumoylation (Brakhage and Schroeckh 2011, Rando 2012). It is
generally thought that heterochromatin and gene silencing are associated with
hypoacetylation. Accordingly, it has been shown in A. nidulans that deletion of the hdaA gene
encoding the major histone deacetylase resulted in increased production of sterigmatocystin
and penicillin. This induction was specific for subtelomeric secondary metabolite clusters.
Through a chemical epigenetic approach with the histone deacetylase inhibitor TSA, this
epigenetic regulatory mechanism has been demonstrated to be also present in other
filamentous fungi including Penicillium and Fusarium (Shwab et al. 2007). Further evidence

- 64 -
for a general regulatory mechanism of secondary metabolite cluster expression by histone (de-
)acetylation in filamentous fungi was provided by another chemical epigenetic approach,
where A. niger was treated with suberoylanilidehydrozamic acid (SAHA), which lead to the
discovery of the new fungal metabolite nygerone A (Henrikson et al. 2009).
Beside chemical approaches, molecular genetic approaches are also powerful tools to access
the potential of fungi as secondary metabolite producers. Using such approaches, it was
shown that besides acetylation, methylation and sumoylation of histones also affect the
secondary metabolic landscape of A. nidulans. For example, deletion of the cclA genes, which
encodes a subunit of the COMPASS complex that catalyses H3K4 methylation led to the
identification of F9775A, F9775B, monodicyphenon, emodin and derivatives thereof.
Deletion of the sumO gene encoding a small ubiquitin-like modifier (SUMO) protein in A.
nidulans resulted in a strong induction of asperthecin production which allowed the
identification of its biosynthetic genes. Furthermore, synthesis of austinol/dehydroaustinol
and sterigmatocystin have been reported to be severely reduced in ΔsumO strains (Szewczyk
et al. 2008). Taken together, different chemical and molecular OSMAC approaches have
shown that Aspergilli can be considered a valuable sources for new medically interesting
compounds – the discovery of many new secondary metabolites can be expected in the near
future.
Conclusion: A glimpse into the future of Aspergillus as cell factory
Aspergilli are natural producers and secretors of enzymes, primary and secondary metabolites.
All these products have an enormous commercial potential - a potential, which has, so far,
only partly been tapped. On the one hand, new Aspergillus enzymes will play a key role for
the implementation of secondary feedstocks in microbial biotechnology. On the other hand,
interesting fungal secondary metabolites have the potential to become new commercial
bestsellers: compounds including NRPS as new antibacterials to fight pathogenic bacteria but
also isoprenoids, which are interesting as neutraceuticals or aroma compounds, lipopeptides
and small proteins applicable as new antifungals and poly-unsaturated fatty acids and lipids
with high application potential as food additives or fuel feedstocks.
Life sciences are currently moving from descriptive, qualitative sciences to predictive,
quantitative sciences. For the Aspergillus research community, where systems and synthetic
biology are still in their infancy, it will be a formidable task to analyse, understand, control
and manipulate these hosts for targeted production of any compound of interest. The tight-
linking of experiment and modelling requires a multi-disciplinary collaboration with

- 65 -
biologists, mathematicians, physicians, chemists, engineers, and statisticians. Hence, the new
generation of Aspergillus researchers should be well trained on fungal physiology and
genetics, extended by training in genomics, transcriptomics, proteomics and metabolomics.
These experimental expertise should be combined with mathematical modelling, fermentation
skills and industrial know-how to address fundamental and applied questions.
References
Adav, S.S., A.A. Li, A. Manavalan, P. Punt and S.K. Sze. 2010. Quantitative iTRAQ
Secretome Analysis of Aspergillus niger Reveals Novel Hydrolytic Enzymes research articles.
Journal of Proteome Research. 9: 3932-3940.
Al-Sheikh, H., A.J. Watson, G.A. Lacey, P.J. Punt, D.A. MacKenzie, D.J. Jeenes, T. Pakula,
M. Penttilä, M.J.C. Alcocer and D.B. Archer. 2004. Endoplasmic reticulum stress leads to the
selective transcriptional downregulation of the glucoamylase gene in Aspergillus niger.
Molecular Microbiology. 53:1731-42.
Aldridge, B.B., J.M. Burke, D.A. Lauffenburger and P.K. Sorger. 2006. Physicochemical
modelling of cell signalling pathways. Nature Cell Biology. 8:1195-203.
Aleksenko, A. and A.J. Clutterbuck. 1997. Autonomous plasmid replication in Aspergillus
nidulans: AMA1 and MATE elements. Fungal Genetics and Biology. 21:373-87.
Andersen, M.R., M. Giese, R.P. de Vries and J. Nielsen. 2012. Mapping the polysaccaride
degradation potential of Aspergillus niger. BMC Genomics. 13:313.
Andersen, M.R. and J. Nielsen. 2009. Current status of systems biology in Aspergilli. Fungal
Genetics and Biology. 46:180-90.
Andersen, M.R., M.L. Nielsen and J. Nielsen. 2008. Metabolic model integration of the
bibliome, genome, metabolome and reactome of Aspergillus niger. Molecular Systems
Biology. 4:178.
Andersen, M.R., M.P. Salazar, P.J. Schaap, P.J.I. van de Vondervoort, D. Culley, J. Thykaer,
J.C. Frisvad, K.F. Nielsen, R. Albang, K. Albermann, R.M. Berka, G.H. Braus, S.A. Braus-
Stromeyer, L.M. Corrochano, Z. Dai, P.W.M. van Dijck, G. Hofmann, L.L. Lasure, J.K.
Magnuson, H. Menke, M. Meijer, S.L. Meijer, J.B. Nielsen, M.L. Nielsen, A.J.J. van Ooyen,
H.J. Pel, L. Poulsen, R.A. Samson, H. Stam, A. Tsang, J.M. van den Brink, A. Atkins, A.

- 66 -
Aerts, H. Shapiro, J. Pangilinan, A. Salamov, Y. Lou, E. Lindquist, S. Lucas, J. Grimwood,
I.V. Grigoriev, C.P. Kubicek, D. Martinez, N.N.M.E. van Peij, J.A. Roubos, J. Nielsen and
S.E. Baker. 2011. Comparative genomics of citric-acid-producing Aspergillus niger ATCC
1015 versus enzyme-producing CBS 513.88. Genome Research. 21:885-97.
Arentshorst, M., A.F.J. Ram and V. Meyer. 2012. Using non-homologous end-joining-
deficient strains for functional gene analyses in filamentous fungi. Methods in Molecular
Biology. 835:133-50.
Bando, H., H. Hisada, H. Ishida, Y. Hata, Y. Katakura and A. Kondo. 2011. Isolation of a
novel promoter for efficient protein expression by Aspergillus oryzae in solid-state culture.
Applied Microbiology and Biotechnology. 92:561-9.
Bayram, O., S. Krappmann, M. Ni, J.W. Bok, K. Helmstaedt, O. Valerius, S. Braus-
Stromeyer, N.-J. Kwon, N.P. Keller, J.-H. Yu and G.H. Braus. 2008. VelB/VeA/LaeA
complex coordinates light signal with fungal development and secondary metabolism.
Science. 320:1504-6.
Beadle, G.W. and E.L. Tatum. 1941. Genetic control of biochemical reactions in Neurospora.
Proceedings of the National Academy of Sciences of the United States of America. 27:499-
506.
de Bekker, C., O. Bruning, M.J. Jonker, T.M. Breit and H. A. B. Wösten. 2011a. Single cell
transcriptomics of neighboring hyphae of Aspergillus niger. Genome Biology. 12:R71.
de Bekker, C., G.J. van Veluw, A. Vinck, L.A. Wiebenga and H.A.B. Wösten. 2011b.
Heterogeneity of Aspergillus niger microcolonies in liquid shaken cultures. Applied and
Environmental Microbiology. 77:1263-7.
de Bekker, C., A. Wiebenga, G. Aguilar and H.A.B. Wösten. 2009. An enzyme cocktail for
efficient protoplast formation in Aspergillus niger. Journal of Microbiological Methods.
76:305-6.
van den Berg, B.A., M.J.T. Reinders, M. Hulsman, L. Wu, J. Herman, H.J. Pel, J.A. Roubos
and D. de Ridder. 2012. Exploring sequence characteristics to high-level protein production in
Aspergillus niger. PloS one. 7(10)

- 67 -
Bergmann, S., J. Schümann, K. Scherlach, C. Lange, A.A. Brakhage and C. Hertweck. 2007.
Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans.
Nature Chemical Biology. 3:213-7.
Bermúdez-Humarán, L.G., N.G. Cortes-Perez, Y. Le Loir, A. Gruss, C. Rodriguez-Padilla, O.
Saucedo-Cardenas, P. Langella and R. Montes de Oca-Luna. 2003. Fusion to a carrier protein
and a synthetic propeptide enhances E7 HPV-16 production and secretion in Lactococcus
lactis. Biotechnology Progress. 19:1101-4.
Bide, H.B. 2002. Big effects from small changes: possible ways to explore nature’s chemical
diversity. Chembiochem. 3:619-27.
Blumhoff, M., M.G. Steiger, H. Marx, D. Mattanovich and M. Sauer. 2012. Six novel
constitutive promoters for metabolic engineering of Aspergillus niger. Applied Microbiology
and Biotechnology.
Boele, J., B.G. Olivier and B. Teusink. 2012. FAME, the Flux Analysis and Modeling
Environment. BMC Systems Biology. 6:8.
Bok, J.W. and N.P. Keller. 2004. LaeA , a Regulator of Secondary Metabolism in Aspergillus
spp. Eukaryotic Cell 3:527-535.
Bonifacino, J.S. and B.S. Glick. 2004. The mechanisms of vesicle budding and fusion. Cell.
116:153-66.
Brakhage, A.A. and V. Schroeckh. 2011. Fungal secondary metabolites - strategies to activate
silent gene clusters. Fungal Genetics and Biology. 48:15-22.
van den Brink, H.J.M., S.G. Petersen, H. Rahbek-Nielsen, K. Hellmuth and M. Harboe. 2006.
Increased production of chymosin by glycosylation. Journal of Biotechnology. 125:304-10.
Calvo, A.M., R.A. Wilson, J.W. Bok, P. Nancy and N.P. Keller. 2002. Relationship between
Secondary Metabolism and Fungal Development. Microbiol Mol Biol Rev 66: 447-59
Cardoza, R.E., S. Gutiérrez, N. Ortega, A. Colina, J. Casqueiro and J.F. Martín. 2003.
Expression of a synthetic copy of the bovine chymosin gene in Aspergillus awamori from
constitutive and pH-regulated promoters and secretion using two different pre-pro sequences.
Biotechnology and Bioengineering. 83:249-59.

- 68 -
Cardoza, R.E., F.J. Moralejo, S. Gutiérrez, J. Casqueiro, F. Fierro and J.F. Martín. 1998.
Characterization and nitrogen-source regulation at the transcriptional level of the gdhA gene
of Aspergillus awamori encoding an NADP-dependent glutamate dehydrogenase. Current
Genetics. 34:50-9.
Carvalho, N.D., T.R. Jørgensen, M. Arentshorst, B.M. Nitsche, C.A. van den Hondel, D.B.
Archer and A.F. Ram. 2012. Genome-wide expression analysis upon constitutive activation of
the HacA bZIP transcription factor in Aspergillus niger reveals a coordinated cellular
response to counteract ER stress. BMC Genomics. 13:350.
Carvalho, N.D.S.P., M. Arentshorst, M.J. Kwon, V. Meyer and A.F.J. Ram. 2010. Expanding
the ku70 toolbox for filamentous fungi: establishment of complementation vectors and
recipient strains for advanced gene analyses. Applied Microbiology. 87:1463-1473.
Carvalho, N.N.D.S.P., M. Arentshorst, R. Kooistra, H. Stam, C.M.J. Sagt, C.A.M.J.J. van den
Hondel and A.F.J. Ram. 2011. Effects of a defective ERAD pathway on growth and
heterologous protein production in Aspergillus niger. Applied Genetics and Molecular
Biotechnology. 89:357-373.
Case, M.E., M. Schweizer, S.R. Kushner and N.H. Giles. 1979. Efficient transformation of
Neurospora crassa by utilizing hybrid plasmid DNA. Proceedings of the National Academy
of Sciences of the United States of America. 76:5259-63.
Chen, W., T. Xie, Y. Shao and F. Chen. 2012. Genomic characteristics comparisons of 12
food-related filamentous fungi in tRNA gene set, codon usage and amino acid composition.
Gene. 497:116-24.
Chiang, Y., E. Szewczyk, T. Nayak and A. Davidson. 2008. Molecular Genetic Mining of the
Aspergillus Secondary Metabolome: Discovery of the Emericellamide Biosynthetic Pathway.
Chemistry Biology. 15:527-532.
Conesa, a, P.J. Punt, N. van Luijk and C.A. van den Hondel. 2001. The secretion pathway in
filamentous fungi: a biotechnological view. Fungal Genetics and Biology. 33:155-71.
Conesa, A., S. Götz, J.M. García-Gómez, J. Terol, M. Talón, M. Robles, S. Gotz, J.M. Garcia-
Gomez and M. Talon. 2005. Blast2GO: a universal tool for annotation, visualization and
analysis in functional genomics research. Bioinformatics. 21:3674-6.

- 69 -
Conesa, A., D. Jeenes, D.B. Archer, C.A.M.J.J. van den Hondel and P.J. Punt. 2002. Calnexin
overexpression increases manganese peroxidase production in Aspergillus niger. Applied and
Environmental Microbiology. 68:846-51.
Contreras, R., D. Carrez, J.R. Kinghorn, C.A. van den Hondel and W. Fiers. 1991. Efficient
KEX2-like processing of a glucoamylase-interleukin-6 fusion protein by Aspergillus nidulans
and secretion of mature interleukin-6. Nature Biotechnology. 9:378-81.
Cooper, M.A. and D. Shlaes. 2011. Fix the antibiotics pipeline. Nature. 472:32.
Coradetti, S.T., J.P. Craig, Y. Xiong, T. Shock, C. Tian and N.L. Glass. 2012. Conserved and
essential transcription factors for cellulase gene expression in ascomycete fungi. Proceedings
of the National Academy of Sciences. 109:7397-402.
Cueto, M., P. Jensen, C. Kauffman, W. Fenical, E. Lobkovsky and J. Clardy. 2001. Pestalone,
a new antibiotic produced by a marine fungus in response to bacterial challenge. J. Nat. Prod.
1:1444-1446.
Cullen, D., G.L. Gray, L.J. Wilson, K.J. Hayenga, M.H. Lamsa, M.W. Rey, S. Norton and
R.M. Berka. 1987. Controlled Expression and Secretion of Bovine Chymosin in Aspergillus
Nidulans. Nature Biotechnology. 5:369-376.
Cvijovic, M., R. Olivares-Hernández, R. Agren, N. Dahr, W. Vongsangnak, I. Nookaew, K.R.
Patil and J. Nielsen. 2010. BioMet Toolbox: genome-wide analysis of metabolism. Nucleic
Acids Research. 38:W144-9.
Damveld, R.A., M. Arentshorst, A. Franken, P. a vanKuyk, F.M. Klis, C. a M.J.J. van den
Hondel, A.F.J. Ram, A. Patricia and C.A.M.J.J.V.D. Hondel. 2005. The Aspergillus niger
MADS-box transcription factor RlmA is required for cell wall reinforcement in response to
cell wall stress. Molecular microbiology. 58:305-319.
Dave, K., M. Ahuja, T.N. Jayashri, R.B. Sirola and N.S. Punekar. 2012. A novel selectable
marker based on Aspergillus niger arginase expression. Enzyme and Microbial Technology.
51:53-8.
Dave, K. and N.S. Punekar. 2011. Utility of Aspergillus niger citrate synthase promoter for
heterologous expression. Journal of Biotechnology. 155:173-7.

- 70 -
Delmas, S., S.T. Pullan, S. Gaddipati, M. Kokolski, S. Malla, M.J. Blythe, R. Ibbett, M.
Campbell, S. Liddell, A. Aboobaker, G. a. Tucker and D.B. Archer. 2012. Uncovering the
Genome-Wide Transcriptional Responses of the Filamentous Fungus Aspergillus niger to
Lignocellulose Using RNA Sequencing. PLoS Genetics. 8:e1002875.
Driouch, H., R. Hänsch, T. Wucherpfennig, R. Krull and C. Wittmann. 2012a. Improved
enzyme production by bio-pellets of Aspergillus niger: targeted morphology engineering
using titanate microparticles. Biotechnology and Bioengineering. 109:462-71.
Driouch, H., G. Melzer and C. Wittmann. 2012b. Integration of in vivo and in silico metabolic
fluxes for improvement of recombinant protein production. Metabolic Engineering. 14:47-58.
Dudásová, Z., A. Dudás and M. Chovanec. 2004. Non-homologous end-joining factors of
Saccharomyces cerevisiae. FEMS Microbiology Reviews. 28:581-601.
Edelmann, S.E. and C. Staben. 1994. A Statistical Analysis of Sequence Features within
Genes from Neurospora crassa. Experimental Mycology. 18:70-81.
Elander, R.P. 2003. Industrial production of beta-lactam antibiotics. Applied Microbiology
and Biotechnology. 61:385-92.
Etxebeste, O., U. Ugalde and E.A. Espeso. 2010. Adaptative and developmental responses to
stress in Aspergillus nidulans. Current protein & peptide science. 11:704-18.
Evnouchidou, I., A. Papakyriakou and E. Stratikos. 2009. A new role for Zn(II)
aminopeptidases: antigenic peptide generation and destruction. Current Pharmaceutical
Design. 15:3656-70.
Felenbok, B. 1991. The ethanol utilization regulon of Aspergillus nidulans: the alcA-alcR
system as a tool for the expression of recombinant proteins. Journal of Biotechnology. 17:11-
7.
Ferreira de Oliveira, J.M.P., M.W.J. van Passel, P.J. Schaap and L.H. de Graaff. 2011.
Proteomic Analysis of the Secretory Response of Aspergillus niger to D-Maltose and D-
Xylose. PloS one. 6:e20865.
Fincham, J.R. 1989. Transformation in fungi. Microbiological Reviews. 53:148-70.

- 71 -
Finkelstein, D.B. 1987. Improvement of enzyme production in Aspergillus. Antonie van
Leeuwenhoek. 53:349-352.
Firn, R.D. and C.G. Jones. 2003. Natural products? a simple model to explain chemical
diversity. Natural Product Reports. 20:382.
Fischer, M., W. Hilt, B. Richter-Ruoff, H. Gonen, A. Ciechanover and D.H. Wolf. 1994. The
26S proteasome of the yeast Saccharomyces cerevisiae. FEBS Letters. 355:69-75.
Fleissner, A. and P. Dersch. 2010. Expression and export: recombinant protein production
systems for Aspergillus. Applied Microbiology and Biotechnology. 87:1255-70.
Forment, J.V., D. Ramón and A.P. MacCabe. 2006. Consecutive gene deletions in Aspergillus
nidulans: application of the Cre/loxP system. Current Genetics. 50:217-24.
Fowler, T., R.M. Berka, M. Ward, K. Dave and N.S. Punekar. 1990. Regulation of the glaA
gene of Aspergillus niger. Current Genetics. 18:537-545.
Frisvad, J.C., J. Smedsgaard, R.A. Samson, T.O. Larsen and U. Thrane. 2007. Fumonisin B2
production by Aspergillus niger. Journal of Agricultural and Food chemistry. 55:9727-32.
van Gemeren, I.A., A. Beijersbergen, C.A. van den Hondel and C.T. Verrips. 1998.
Expression and secretion of defined cutinase variants by Aspergillus awamori. Applied and
Environmental Microbiology. 64:2794-9.
Gems, D., I.L. Johnstone and a J. Clutterbuck. 1991. An autonomously replicating plasmid
transforms Aspergillus nidulans at high frequency. Gene. 98:61-7.
Gentleman, R.C., V.J. Carey, D.M. Bates, B. Bolstad, M. Dettling, S. Dudoit, B. Ellis, L.
Gautier, Y. Ge, J. Gentry, K. Hornik, T. Hothorn, W. Huber, S. Iacus, R.A. Irizarry, F. Leisch,
C. Li, M. Maechler, A.J. Rossini, G. Sawitzki, C. Smith, G. Smyth, L. Tierney, J.Y. Yang and
J. Zhang. 2004. Bioconductor: open software development for computational biology and
bioinformatics. Genome Biol. 5:R80.
Geysens, S., G. Whyteside and D.B. Archer. 2009. Genomics of protein folding in the
endoplasmic reticulum, secretion stress and glycosylation in the Aspergilli. Fungal Genetics
and Biology. 46:S121-S140.

- 72 -
Gibbons, J.G., A. Beauvais, R. Beau, K.L. McGary, J.-P. Latgé and A. Rokas. 2012. Global
transcriptome changes underlying colony growth in the opportunistic human pathogen
Aspergillus fumigatus. Eukaryotic Cell. 11:68-78.
Goder, V., P. Carvalho and T.A. Rapoport. 2008. The ER-associated degradation component
Der1p and its homolog Dfm1p are contained in complexes with distinct cofactors of the
ATPase Cdc48p. FEBS Letters. 582:1575-80.
Gonzalez, D.S., K. Karaveg, A.S. Vandersall-Nairn, A. Lal and K.W. Moremen. 1999.
Identification, expression, and characterization of a cDNA encoding human endoplasmic
reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in
mammalian Asn-linked oligosaccharide biosynthesis. The Journal of Biological Chemistry.
274:21375-86.
Gordon, C.L., V. Khalaj, A.F. Ram, D.B. Archer, J.L. Brookman, a P. Trinci, D.J. Jeenes,
J.H. Doonan, B. Wells, P.J. Punt, C. a van den Hondel and G.D. Robson. 2000.
Glucoamylase::green fluorescent protein fusions to monitor protein secretion in Aspergillus
niger. Microbiology. 146:415-26.
Gouka, R.J., J.G. Hessing, P.J. Punt, H. Stam, W. Musters and C.A. Van den Hondel. 1996.
An expression system based on the promoter region of the Aspergillus awamori 1,4-beta-
endoxylanase A gene. Applied Microbiology and Biotechnology. 46:28-35.
Gouka, R.J., P.J. Punt and C.A. van den Hondel. 1997. Glucoamylase gene fusions alleviate
limitations for protein production in Aspergillus awamori at the transcriptional and (post)
translational levels. Applied and Environmental Microbiology. 63:488-97.
Griffiths, a J. 1995. Natural plasmids of filamentous fungi. Microbiological Reviews. 59:673-
85.
de Groot, M.J., P. Bundock, P.J. Hooykaas and A.G. Beijersbergen. 1998. Agrobacterium
tumefaciens - mediated transformation of filamentous fungi. Nature Biotechnology. 16:839-
42.
Gross, H. 2007. Strategies to unravel the function of orphan biosynthesis pathways: recent
examples and future prospects. Applied Microbiology and Biotechnology. 75:267-77.

- 73 -
Guillemette, T., N.N.M.E. van Peij, T. Goosen, K. Lanthaler, G.D. Robson, C.A.M.J.J. van
den Hondel, H. Stam and D.B. Archer. 2007. Genomic analysis of the secretion stress
response in the enzyme-producing cell factory Aspergillus niger. BMC Genomics. 8:158.
Gwynne, D.I., F.P. Buxton, S.A. Williams, A.M. Sills, J.A. Johnstone, J.K. Buch, Z.M. Guo,
D. Drake, M. Westphal and R.W. Davies. 1989. Development of an expression system in
Aspergillus nidulans. Biochemical Society transactions. 17:338-40.
Hagag, S., P. Kubitschek-Barreira, G.W.P. Neves, D. Amar, W. Nierman, I. Shalit, R. Shamir,
L. Lopes-Bezerra and N. Osherov. 2012. Transcriptional and Proteomic Analysis of the
Aspergillus fumigatus ΔprtT Protease-Deficient Mutant. PLoS ONE. 7:e33604.
Hajjaj, H., P.. Blanc, G. Goma and J. François. 1998. Sampling techniques and comparative
extraction procedures for quantitative determination of intra- and extracellular metabolites in
filamentous fungi. FEMS Microbiology Letters. 164:195-200.
Harris, S.D. 2008. Branching of fungal hyphae: regulation, mechanisms and comparison with
other branching systems. Mycologia. 100:823-832.
Harris, S.D. 2006. Cell polarity in filamentous fungi: shaping the mold. International Review
of Cytology. 251:41-77.
Hawksworth, D.L. 1991. The fungal dimension of biodiversity: magnitude, significance, and
conservation. Mycological Research. 95:641-655.
Helenius, A. and M. Aebi. 2004. Roles of N-linked glycans in the endoplasmic reticulum.
Annual Review of Biochemistry. 73:1019-49.
Henrikson, J.C., A.R. Hoover, P.M. Joyner and R.H. Cichewicz. 2009. A chemical
epigenetics approach for engineering the in situ biosynthesis of a cryptic natural product from
Aspergillus niger. Organic & Biomolecular Chemistry. 7:435-8.
Hinterberger H. 1956. In: Electromagnetically Enriched
Isotopes and Mass Spectrometry (Smith, M. L., ed.) Butterworth,
London, pp, 177-189.
Hertweck, C. 2009. Hidden biosynthetic treasures brought to light. Nature Chemical Biology.
5:450-2.

- 74 -
Hoffmeister, D. and N.P. Keller. 2007. Natural products of filamentous fungi: enzymes,
genes, and their regulation. Natural Product Reports. 24:393-416.
van den Hombergh, J.P., P.J. van de Vondervoort, L. Fraissinet-Tachet and J. Visser. 1997.
Aspergillus as a host for heterologous protein production: the problem of proteases. Trends in
Biotechnology. 15:256-63.
Hu, W., S. Sillaots, S. Lemieux, J. Davison, S. Kauffman, A. Breton, A. Linteau, C. Xin, J.
Bowman, J. Becker, B. Jiang and T. Roemer. 2007. Essential gene identification and drug
target prioritization in Aspergillus fumigatus. PLoS Pathogens. 3:e24.
Ikemura, T. 1981. Correlation between the abundance of Escherichia coli transfer RNAs and
the occurrence of the respective codons in its protein genes: a proposal for a synonymous
codon choice that is optimal for the E. coli translational system. Journal of Molecular
Biology. 151:389-409.
Ikemura, T. 1982. Correlation between the abundance of yeast transfer RNAs and the
occurrence of the respective codons in protein genes. Differences in synonymous codon
choice patterns of yeast and Escherichia coli with reference to the abundance of isoaccepting
transfer RNAs. Journal of Molecular Biology. 158:573-97.
Iriarte, A., M. Sanguinetti, T. Fernández-Calero, H. Naya, A. Ramón and H. Musto. 2012.
Translational selection on codon usage in the genus Aspergillus. Gene. 506:98-105.
Ishida, H., Y. Hata, A. Kawato, Y. Abe and Y. Kashiwagi. 2004. Isolation of a novel
promoter for efficient protein production in Aspergillus oryzae. Bioscience, Biotechnology,
and Biochemistry. 68:1849-57.
Jacobs, D.I., M.M.A. Olsthoorn, I. Maillet, M. Akeroyd, S. Breestraat, S. Donkers,
R.A.M.V.D. Hoeven, C.A.M.J.J.V.D. Hondel, R. Kooistra, T. Lapointe, H. Menke, R.
Meulenberg, M. Misset, W.H. Müller, N.N.M.E.V. Peij, A. Ram, S. Rodriguez, M.S. Roelofs,
J.A. Roubos, M.W.E.M.V. Tilborg, A.J. Verkleij, H.J. Pel, H. Stam and C.M.J. Sagt. 2009.
Effective lead selection for improved protein production in Aspergillus niger based on
integrated genomics. Fungal Genetics and Biology. 46:S141-S152.
Jalving, R., P.J. van de Vondervoort, J. Visser and P.J. Schaap. 2000. Characterization of the
kexin-like maturase of Aspergillus niger. Applied and Environmental Microbiology. 66:363-
8.

- 75 -
Jeenes, D.J., D.A. Mackenzie and D.B. Archer. 1994. Transcriptional and post-transcriptional
events affect the production of secreted hen egg white lysozyme by Aspergillus niger.
Transgenic Research. 3:297-303.
Jin, C. 2012. Protein Glycosylation in Aspergillus fumigatus Is Essential for Cell Wall
Synthesis and Serves as a Promising Model of Multicellular Eukaryotic Development.
International Journal of Microbiology. 2012:654251.
Jin, F.J., T. Watanabe, P.R. Juvvadi, J.-ichi Maruyama, M. Arioka and K. Kitamoto. 2007.
Double disruption of the proteinase genes, tppA and pepE, increases the production level of
human lysozyme by Aspergillus oryzae. Applied Microbiology and Biotechnology. 76:1059-
68.
Jørgensen, T.R., T. Goosen, C.A.M.J.J.V.D. Hondel, A.F.J. Ram and J.J.L. Iversen. 2009.
Transcriptomic comparison of Aspergillus niger growing on two different sugars reveals
coordinated regulation of the secretory pathway. BMC Genomics. 10:44.
Jørgensen, T.R., K.F. Nielsen, M. Arentshorst, J. Park, C.A. van den Hondel, J.C. Frisvad and
A.F. Ram. 2011. Submerged conidiation and product formation by Aspergillus niger at low
specific growth rates are affected in aerial developmental mutants. Applied and
Environmental Microbiology. 77:5270-7.
Jørgensen, T.R., B.M. Nitsche, G.E. Lamers, M. Arentshorst, C. A. van den Hondel and A.F.
Ram. 2010. Transcriptomic insights into the physiology of Aspergillus niger approaching a
specific growth rate of zero. Applied and Environmental Microbiology. 76:5344-55.
Kahali, B., S. Ahmad and T.C. Ghosh. 2011. Selective constraints in yeast genes with
differential expressivity: codon pair usage and mRNA stability perspectives. Gene. 481:76-82.
Kaufman, R.J. 1999. Stress signaling from the lumen of the endoplasmic reticulum:
coordination of gene transcriptional and translational controls. Genes & Development.
13:1211-1233.
Kaufman, R.J., D. Scheuner, M. Schröder, X. Shen, K. Lee, C.Y. Liu and S.M. Arnold. 2002.
The unfolded protein response in nutrient sensing and differentiation. Nature Reviews.
Molecular Cell Biology. 3:411-21.

- 76 -
Kawai, S., W. Hashimoto and K. Murata. 2010. Transformation of Saccharomyces cerevisiae
and other fungi: methods and possible underlying mechanism. Bioengineered Bugs. 1:395-
403.
Keller, N.P., G. Turner and J.W. Bennett. 2005. Fungal secondary metabolism - from
biochemistry to genomics. Nature reviews. Microbiology. 3:937-47.
Kelly, J.M. and M.J. Hynes. 1987. Multiple copies of the amdS gene of Aspergillus nidulans
cause titration of trans-acting regulatory proteins. Current Genetics. 12:21-31.
Khalaj, V., H. Eslami, M. Azizi, N. Rovira-Graells and M. Bromley. 2007. Efficient
downregulation of alb1 gene using an AMA1-based episomal expressionofRNAi construct in
Aspergillus fumigatus. FEMS Microbiology Letters. 270:250-254.
Kiel, J.A.K.W., M.A. van den Berg, F. Fusetti, B. Poolman, R.A.L. Bovenberg, M. Veenhuis
and I.J. van der Klei. 2009. Matching the proteome to the genome: the microbody of
penicillin-producing Penicillium chrysogenum cells. Functional & Integrative Genomics.
9:167-84.
Kinnaird, J.H., P.A. Burns and J.R. Fincham. 1991. An apparent rare-codon effect on the rate
of translation of a Neurospora gene. Journal of Molecular Biology. 221:733-6.
Kitamoto, N., J. Matsui, Y. Kawai, A. Kato, S. Yoshino, K. Ohmiya and N. Tsukagoshi.
1998. Utilization of the TEF1-alpha gene (TEF1) promoter for expression of
polygalacturonase genes, pgaA and pgaB, in Aspergillus oryzae. Applied Microbiology and
Biotechnology. 50:85-92.
Kito, H., T. Fujikawa, A. Moriwaki, A. Tomono, M. Izawa, T. Kamakura, M. Ohashi, H.
Sato, K. Abe and M. Nishimura. 2008. MgLig4, a homolog of Neurospora crassa Mus-53
(DNA ligase IV), is involved in, but not essential for, non-homologous end-joining events in
Magnaporthe grisea. Fungal Genetics and Biology. 45:1543-51.
Kobayashi, D.Y. and J.A. Crouch. 2009. Bacterial/Fungal interactions: from pathogens to
mutualistic endosymbionts. Annual Review of Phytopathology. 47:63-82.
Koda, A., T. Bogaki, T. Minetoki and M. Hirotsune. 2005. High expression of a synthetic
gene encoding potato alpha-glucan phosphorylase in Aspergillus niger. Journal of Bioscience
and Bioengineering. 100:531-7.

- 77 -
de Koning, W. and K. van Dam. 1992. A method for the determination of changes of
glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH.
Analytical Biochemistry. 204:118-23.
Korman, D.R., F.T. Bayliss, C.C. Barnett, C.L. Carmona, K.H. Kodama, T.J. Royer, S.A.
Thompson, M. Ward, L.J. Wilson and R.M. Berka. 1990. Cloning, characterization, and
expression of two alpha-amylase genes from Aspergillus niger var. awamori. Current
Genetics. 17:203-12.
Kostova, Z., Y.C. Tsai and A.M. Weissman. 2007. Ubiquitin ligases, critical mediators of
endoplasmic reticulum-associated degradation. Seminars in Cell & Developmental biology.
18:770-9.
Krappmann, S., O. Bayram and G.H. Braus. 2005. Deletion and allelic exchange of the
Aspergillus fumigatus veA locus via a novel recyclable marker module. Eukaryotic cell.
4:1298-307.
Krappmann, S., C. Sasse and G.H. Braus. 2006. Gene targeting in Aspergillus fumigatus by
homologous recombination is facilitated in a nonhomologous end- joining-deficient genetic
background. Eukaryotic Cell. 5:212-5.
Krijgsveld, J., R.F. Ketting, T. Mahmoudi, J. Johansen, M. Artal-Sanz, C.P. Verrijzer, R.H.A.
Plasterk and A.J.R. Heck. 2003. Metabolic labeling of C. elegans and D. melanogaster for
quantitative proteomics. Nature Biotechnology. 21:927-31.
Krogh, B.O. and L.S. Symington. 2004. Recombination proteins in yeast. Annual Review of
Genetics. 38:233-71.
Kwon, M.J., M. Arentshorst, E.D. Roos, C. a M.J.J. van den Hondel, V. Meyer and A.F.J.
Ram. 2011. Functional characterization of Rho GTPases in Aspergillus niger uncovers
conserved and diverged roles of Rho proteins within filamentous fungi. Molecular
Microbiology. 79:1151-67.
Kück, U. and B. Hoff. 2010. New tools for the genetic manipulation of filamentous fungi.
Applied Microbiology and Biotechnology. 86:51-62.

- 78 -
van Laar, T., A.J. van der Eb and C. Terleth. 2001. Mif1: a missing link between the unfolded
protein response pathway and ER-associated protein degradation? Current Protein & Peptide
Science. 2:169-90.
Lazzarini, A., L. Cavaletti, G. Toppo and F. Marinelli. 2001. Rare genera of actinomycetes as
potential producers of new antibiotics. Antonie van Leeuwenhoek. 79:399-405.
Levin, A.M., R.P. de Vries, A. Conesa, C. de Bekker, M. Talon, H.H. Menke, N.N.M.E. van
Peij and H. a B. Wösten. 2007. Spatial differentiation in the vegetative mycelium of
Aspergillus niger. Eukaryotic Cell. 6:2311-22.
Lloyd, A.T. and P.M. Sharp. 1991. Codon usage in Aspergillus nidulans. Molecular &
General Genetics. 230:288-94.
Le Loir, Y., S. Nouaille, J. Commissaire, L. Brétigny, A. Gruss and P. Langella. 2001. Signal
peptide and propeptide optimization for heterologous protein secretion in Lactococcus lactis.
Applied and Environmental Microbiology. 67:4119-27.
Lombraña, M., F.J. Moralejo, R. Pinto and J.F. Martín. 2004. Modulation of Aspergillus
awamori thaumatin secretion by modification of bipA gene expression. Applied and
Environmental Microbiology. 70:5145-52.
Lopes, F.C., L.A.D.E. Silva, D.M. Tichota, D.J. Daroit, R.V. Velho, J.Q. Pereira, A.P.F.
Corrêa and A. Brandelli. 2011. Production of Proteolytic Enzymes by a Keratin-Degrading
Aspergillus niger. Enzyme Research. 2011:487093.
Lubertozzi, D. and J.D. Keasling. 2009. Developing Aspergillus as a host for heterologous
expression. Biotechnology Advances. 27:53-75.
Malik, M., K.C. Nitiss, V. Enriquez-Rios and J.L. Nitiss. 2006. Roles of nonhomologous end-
joining pathways in surviving topoisomerase II-mediated DNA damage. Molecular Cancer
Therapeutics. 5:1405-14.
Martens-Uzunova, E.S. and P.J. Schaap. 2009. Assessment of the pectin degrading enzyme
network of Aspergillus niger by functional genomics. Fungal Genetics and Biology. 46 Suppl
1:S170-S179.

- 79 -
Martens-Uzunova, E.S., J.S. Zandleven, J.A. Benen, H. Awad, H.J. Kools, G. Beldman, A.G.
Voragen, J.A. van den Berg and P.J. Schaap. 2006. A new group of exo-acting family 28
glycoside hydrolases of Aspergillus niger that are involved in pectin degradation. Biochem J.
400:43-52.
Martin, J.A., R.A. Murphy and R.F.G. Power. 2003. Cloning and expression of fungal
phytases in genetically modified strains of Aspergillus awamori. Journal of Industrial
Microbiology & Biotechnology. 30:568-76.
Mattern, I.E., J.M. van Noort, P. van den Berg, D.B. Archer, I.N. Roberts and C.A. van den
Hondel. 1992. Isolation and characterization of mutants of Aspergillus niger deficient in
extracellular proteases. Molecular & General Genetics. 234:332-6.
Melzer, G., M.E. Esfandabadi, E. Franco-Lara and C. Wittmann. 2009. Flux Design: In silico
design of cell factories based on correlation of pathway fluxes to desired properties. BMC
Systems Biology. 3:120.
Meyer, V. 2008. Genetic engineering of filamentous fungi--progress, obstacles and future
trends. Biotechnology Advances. 26:177-85.
Meyer, V., M. Arentshorst, A. El-Ghezal, A.-C. Drews, R. Kooistra, C.A.M.J.J. van den
Hondel and A.F.J. Ram. 2007a. Highly efficient gene targeting in the Aspergillus niger kusA
mutant. Journal of Biotechnology. 128:770-5.
Meyer, V., M. Arentshorst, S.J. Flitter, B.M. Nitsche, M.J. Kwon, C.G. Reynaga-Peña, S.
Bartnicki-Garcia, C. a M.J.J. van den Hondel and A.F.J. Ram. 2009. Reconstruction of
signaling networks regulating fungal morphogenesis by transcriptomics. Eukaryotic cell.
8:1677-91.
Meyer, V., M. Arentshorst, C.A.M.J.J. van den Hondel and A.F.J. Ram. 2008. The polarisome
component SpaA localises to hyphal tips of Aspergillus niger and is important for polar
growth. Fungal Genetics and Biology 45:152-64.
Meyer, V., R. a Damveld, M. Arentshorst, U. Stahl, C. a M.J.J. van den Hondel and A.F.J.
Ram. 2007b. Survival in the presence of antifungals: genome-wide expression profiling of
Aspergillus niger in response to sublethal concentrations of caspofungin and fenpropimorph.
The Journal of Biological Chemistry. 282:32935-48.

- 80 -
Meyer, V., S. Minkwitz, T. Schütze, C.A.M.J.J.V.D. Hondel and A.F.J. Ram. 2010a. The
Aspergillus niger RmsA protein. A node in a genetic network? Communicative & Integrative
Biology. 3:195-197.
Meyer, V., D. Mueller, T. Strowig and U. Stahl. 2003. Comparison of different transformation
methods for Aspergillus giganteus. Current Genetics. 43:371-7.
Meyer, V., A.F.J. Ram and P.J. Punt. 2010b. Genetics, Genetic Manipulation, and
Approaches to Strain Improvement of Filamentous Fungi. In: Manual of Industrial
Microbiology and Biotechnology, 3rd Editio. NY: Wiley, p 318-329.
Meyer, V., F. Wanka, J. van Gent, M. Arentshorst, C.A.M.J.J. van den Hondel and A.F.J.
Ram. 2011a. Fungal gene expression on demand: an inducible, tunable, and metabolism-
independent expression system for Aspergillus niger. Applied and Environmental
Microbiology. 77:2975-83.
Meyer, V., B. Wu and A.F.J. Ram. 2011b. Aspergillus as a multi-purpose cell factory: current
status and perspectives. Biotechnology Letters. 33:469-76.
Michielse, C.B., P.J.J. Hooykaas, C.A.M.J.J. van den Hondel and A.F.J. Ram. 2008.
Agrobacterium-mediated transformation of the filamentous fungus Aspergillus awamori.
Nature Protocols. 3:1671-8.
Mizutani, O., K. Masaki, K. Gomi and H. Iefuji. 2012. Modified Cre-loxP Recombination in
Aspergillus oryzae by Direct Introduction of Cre Recombinase for Marker Gene Rescue.
Applied and Environmental Microbiology. 78:4126-33.
Moralejo, F.J., R.E. Cardoza, S. Gutierrez and J.F. Martin. 1999. Thaumatin production in
Aspergillus awamori by use of expression cassettes with strong fungal promoters and high
gene dosage. Applied and Environmental Microbiology. 65:1168-74.
Moralejo, F.J., A.J. Watson, D.J. Jeenes, D.B. Archer and J.F. Martín. 2001. A defined level
of protein disulfide isomerase expression is required for optimal secretion of thaumatin by
Aspegillus awamori. Molecular Genetics and Genomics. 266:246-53.
Mulder, H.J., M. Saloheimo, M. Penttilä and S.M. Madrid. 2004. The transcription factor
HACA mediates the unfolded protein response in Aspergillus niger, and up-regulates its own
transcription. Molecular Genetics and Genomics. 271:130-40.

- 81 -
Määttänen, P., K. Gehring, J.J.M. Bergeron and D.Y. Thomas. 2010. Protein quality control in
the ER: the recognition of misfolded proteins. Seminars in Cell & Developmental Biology.
21:500-11.
Nakajima, K.-ichiro, T. Asakura, J.-ichi Maruyama, Y. Morita, H. Oike, A. Shimizu-Ibuka, T.
Misaka, H. Sorimachi, S. Arai, K. Kitamoto and K. Abe. 2006. Extracellular production of
neoculin, a sweet-tasting heterodimeric protein with taste-modifying activity, by Aspergillus
oryzae. Applied and Environmental Microbiology. 72:3716-23.
Nayak, T., E. Szewczyk, C.E. Oakley, A. Osmani, L. Ukil, S.L. Murray, M.J. Hynes, S.A.
Osmani and B.R. Oakley. 2006. A versatile and efficient gene-targeting system for
Aspergillus nidulans. Genetics. 172:1557-66.
Nelson, G., O. Kozlova-Zwinderman, A.J. Collis, M.R. Knight, J.R.S. Fincham, C.P. Stanger,
A. Renwick, J.G.M. Hessing, P.J. Punt, C.A.M.J.J. van den Hondel and N.D. Read. 2004.
Calcium measurement in living filamentous fungi expressing codon-optimized aequorin.
Molecular Microbiology. 52:1437-50.
Nemoto, T., J.-ichi Maruyama and K. Kitamoto. 2009. Improvement of heterologous protein
production in Aspergillus oryzae by RNA interference with alpha-amylase genes. Bioscience,
Biotechnology, and Biochemistry. 73:2370-3.
Ngiam, C., D.J. Jeenes, P.J. Punt, C.A. Van Den Hondel and D.B. Archer. 2000.
Characterization of a foldase, protein disulfide isomerase A, in the protein secretory pathway
of Aspergillus niger. Applied and Environmental Microbiology. 66:775-82.
Nielsen, J.B., M.L. Nielsen and U.H. Mortensen. 2008. Transient disruption of non-
homologous end-joining facilitates targeted genome manipulations in the filamentous fungus
Aspergillus nidulans. Fungal Genetics and Biology 45:165-70.
Nielsen, K.F., J.M. Mogensen, M. Johansen, T.O. Larsen and J.C. Frisvad. 2009. Review of
secondary metabolites and mycotoxins from the Aspergillus niger group. Analytical and
Bioanalytical Chemistry. 395:1225-42.
Nielsen, M.L., J.B. Nielsen, C. Rank, M.L. Klejnstrup, D.K. Holm, K.H. Brogaard, B.G.
Hansen, J.C. Frisvad, T.O. Larsen and U.H. Mortensen. 2011. A genome-wide polyketide
synthase deletion library uncovers novel genetic links to polyketides and meroterpenoids in
Aspergillus nidulans. FEMS Microbiology Letters. 321:157-66.

- 82 -
Nikolaev, I., M. Mathieu, P. van de Vondervoort, J. Visser and B. Felenbok. 2002.
Heterologous expression of the Aspergillus nidulans alcR-alcA system in Aspergillus niger.
Fungal Genetics and Biology. 37:89-97.
Ninomiya, Y., K. Suzuki, C. Ishii and H. Inoue. 2004. Highly efficient gene replacements in
Neurospora strains deficient for nonhomologous end-joining. Proceedings of the National
Academy of Sciences of the United States of America. 101:12248-53.
Nitsche, B.M., J. Crabtree, G.C. Cerqueira, V. Meyer, A.F.J. Ram, J.R. Wortman and A. Fj.
2011. New resources for functional analysis of omics data for the genus Aspergillus. BMC
Genomics. 12:486.
Nitsche, B.M., A.F.J. Ram and V. Meyer. 2012. The use of open source bioinformatics tools
to dissect transcriptomic data. Methods in Molecular Biology (Clifton, N.J.). 835:311-31.
Nyyssönen, E. and S. Keränen. 1995. Multiple roles of the cellulase CBHI in enhancing
production of fusion antibodies by the filamentous fungus Trichoderma reesei. Current
Genetics. 28:71-9.
Oldiges, M., S. Lütz, S. Pflug, K. Schroer, N. Stein and C. Wiendahl. 2007. Metabolomics:
current state and evolving methodologies and tools. Applied Microbiology and
Biotechnology. 76:495-511.
de Oliveira, J.M.P.F. and L.H. de Graaff. 2011. Proteomics of industrial fungi: trends and
insights for biotechnology. Applied Microbiology and Biotechnology. 89:225-37.
Ong, S.-E., B. Blagoev, I. Kratchmarova, D.B. Kristensen, H. Steen, A. Pandey and M. Mann.
2002. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate
approach to expression proteomics. Molecular & Cellular Proteomics. 1:376-86.
Pabinger, S., R. Rader, R. Agren, J. Nielsen and Z. Trajanoski. 2011. MEMOSys:
Bioinformatics platform for genome-scale metabolic models. BMC Systems Biology. 5:20.
Pachlinger, R., R. Mitterbauer, G. Adam and J. Strauss. 2005. Metabolically independent and
accurately adjustable Aspergillus sp. expression system. Applied and Environmental
Microbiology. 71:672-8.

- 83 -
Pakula, T.M., M. Laxell, A. Huuskonen, J. Uusitalo, M. Saloheimo and M. Penttilä. 2003.
The effects of drugs inhibiting protein secretion in the filamentous fungus Trichoderma
reesei. Evidence for down-regulation of genes that encode secreted proteins in the stressed
cells. The Journal of Biological Chemistry. 278:45011-20.
Panozzo, C., V. Capuano, S. Fillinger and B. Felenbok. 1997. The zinc binuclear cluster
activator AlcR is able to bind to single sites but requires multiple repeated sites for synergistic
activation of the alcA gene in Aspergillus nidulans. The Journal of Biological Chemistry.
272:22859-65.
van Peij, N.N.M.E., A. Los, J. Bakhuis, L. Wu, J.-M. van der Laan, M.M.A. Olsthoorn, J..
Roubos and H.J. Pel. 2012. Novel approaches for solving bottlenecks and improving
recombinant protein production by Aspergillus. In: The 9th International Aspergillus Meeting.
Marburg.
Pel, H.J., J.H. de Winde, D.B. Archer, P.S. Dyer, G. Hofmann, P.J. Schaap, G. Turner, R.P.
de Vries, R. Albang, K. Albermann, M.R. Andersen, J.D. Bendtsen, J.A.E. Benen, M. van den
Berg, S. Breestraat, M.X. Caddick, R. Contreras, M. Cornell, P.M. Coutinho, E.G.J. Danchin,
A.J.M. Debets, P. Dekker, P.W.M. van Dijck, A. van Dijk, L. Dijkhuizen, A.J.M. Driessen, C.
D’Enfert, S. Geysens, C. Goosen, G.S.P. Groot, P.W.J. de Groot, T. Guillemette, B.
Henrissat, M. Herweijer, J.P.T.W. van den Hombergh, C.A.M.J.J. van den Hondel, R.T.J.M.
van der Heijden, R.M. van der Kaaij, F.M. Klis, H.J. Kools, C.P. Kubicek, P.A. van Kuyk, J.
Lauber, X. Lu, M.J.E.C. van der Maarel, R. Meulenberg, H. Menke, M.A. Mortimer, J.
Nielsen, S.G. Oliver, M. Olsthoorn, K. Pal, N.N.M.E. van Peij, A.F.J. Ram, U. Rinas, J.A.
Roubos, C.M.J. Sagt, M. Schmoll, J. Sun, D. Ussery, J. Varga, W. Vervecken, P.J.J. van de
Vondervoort, H. Wedler, H.A.B. Wösten, A.-P.P. Zeng, A.J.J. van Ooyen, J. Visser, H. Stam,
M.A. van den Berg, Hondel, P.A. VanKuyk and H.A. Wosten. 2007. Genome sequencing and
analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nature Biotechnology.
25:221-31.
Perrone, G., G. Stea, F. Epifani, J. Varga, J.C. Frisvad and R.A. Samson. 2011. Aspergillus
niger contains the cryptic phylogenetic species A. awamori. Fungal Biology. 115:1138-50.
Peñalva, M. a, J. Tilburn, E. Bignell and H.N. Arst. 2008. Ambient pH gene regulation in
fungi: making connections. Trends in Microbiology. 16:291-300.

- 84 -
Prathumpai, W., M. McIntyre and J. Nielsen. 2004. The effect of CreA in glucose and xylose
catabolism in Aspergillus nidulans. Applied Microbiology and Biotechnology. 63:748-53.
Punt, P.J., M.A. Dingemanse, A. Kuyvenhoven, R.D.M. Seede, P.H. Peuwels and C. a M.J.J.
van den Hondel. 1990. Functional elements in the promoter region of the Aspergillus nidulans
gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene. 93:101-109.
Punt, P.J., A. Drint-Kuijvenhoven, B.C. Lokman, J.A. Spencer, D. Jeenes, D.A. Archer and
C.A.M.J.J. van den Hondel. 2003. The role of the Aspergillus niger furin-type protease gene
in processing of fungal proproteins and fusion proteins. Evidence for alternative processing of
recombinant (fusion-) proteins. Journal of Biotechnology. 106:23-32.
Punt, P.J., I.A. van Gemeren, J. Drint-Kuijvenhoven, J.G. Hessing, G.M. van Muijlwijk-
Harteveld, A. Beijersbergen, C.T. Verrips and C.A. van den Hondel. 1998. Analysis of the
role of the gene bipA, encoding the major endoplasmic reticulum chaperone protein in the
secretion of homologous and heterologous proteins in black Aspergilli. Applied Microbiology
and Biotechnology. 50:447-54.
Punt, P.J., F.H.J. Schuren, J. Lehmbeck, T. Christensen, C. Hjort and C.A.M.J.J. van den
Hondel. 2008. Characterization of the Aspergillus niger prtT, a unique regulator of
extracellular protease encoding genes. Fungal Genetics and Biology. 45:1591-9.
Qin, L.-N., F.-R. Cai, X.-R. Dong, Z.-B. Huang, Y. Tao, J.-Z. Huang and Z.-Y. Dong. 2012.
Improved production of heterologous lipase in Trichoderma reesei by RNAi mediated gene
silencing of an endogenic highly expressed gene. Bioresource Technology. 109:116-22.
Rando, O.J. 2012. Combinatorial Complexity in Chromatin Structure and Function:
Revisiting the Histone Code. Current Opinion in Genetics & Development. 22:148-155.
Rohlfs, M., M. Albert, N.P. Keller and F. Kempken. 2007. Secondary chemicals protect
mould from fungivory. Biology Letters. 3:523-5.
Roth, A.H.F.J. and P. Dersch. 2010. A novel expression system for intracellular production
and purification of recombinant affinity-tagged proteins in Aspergillus niger. Applied
Microbiology and Biotechnology. 86:659-70.
Ruddock, L.W. and M. Molinari. 2006. N-glycan processing in ER quality control. Journal of
Cell Science. 119:4373-80.

- 85 -
Rumbold, K., H.J.J. van Buijsen, V.M. Gray, J.W. van Groenestijn, K.M. Overkamp, R.S.
Slomp, M.J. van der Werf and P.J. Punt. 2010. Microbial renewable feedstock utilization: a
substrate-oriented approach. Bioengineered Bugs. 1:359-66.
Rumbold, K., H.J.J. van Buijsen, K.M. Overkamp, J.W. van Groenestijn, P.J. Punt and M.J.
van der Werf. 2009. Microbial production host selection for converting second-generation
feedstocks into bioproducts. Microbial Cell Factories. 8:64.
Römisch, K. 1999. Surfing the Sec61 channel: bidirectional protein translocation across the
ER membrane. Journal of Cell Science. 112 ( Pt 2:4185-91.
Sagt, C.M.J., P.J. ten Haaft, I.M. Minneboo, M.P. Hartog, R. a Damveld, J.M. van der Laan,
M. Akeroyd, T.J. Wenzel, F. a Luesken, M. Veenhuis, I. van der Klei and J.H. de Winde.
2009. Peroxicretion: a novel secretion pathway in the eukaryotic cell. BMC Biotechnology.
9:48.
Saloheimo, M. and T.M. Pakula. 2012. The cargo and the transport system: secreted proteins
and protein secretion in Trichoderma reesei (Hypocrea jecorina). Microbiology (Reading,
England). 158:46-57.
Saloheimo, M., M. Valkonen and M. Penttilä. 2003. Activation mechanisms of the HAC1-
mediated unfolded protein response in filamentous fungi. Molecular Microbiology. 47:1149-
61.
Sanchez, J.F., A.D. Somoza, N.P. Keller and C.C.C. Wang. 2012. Advances in Aspergillus
secondary metabolite research in the post-genomic era. Natural Product Reports. 29:351-71.
Sarkar, A., A.N. Funk, K. Scherlach, F. Horn, V. Schroeckh, P. Chankhamjon, M.
Westermann, M. Roth, A.A. Brakhage, C. Hertweck and U. Horn. 2012. Differential
expression of silent polyketide biosynthesis gene clusters in chemostat cultures of Aspergillus
nidulans. Journal of Biotechnology. 160:64-71.
Sasaguri, S., J.-ichi Maruyama, S. Moriya, T. Kudo, K. Kitamoto and M. Arioka. 2008.
Codon optimization prevents premature polyadenylation of heterologously-expressed
cellulases from termite-gut symbionts in Aspergillus oryzae. The Journal of General and
applied Microbiology. 54:343-51.

- 86 -
Schroeckh, V., K. Scherlach, H.-W. Nützmann, E. Shelest, W. Schmidt-Heck, J. Schuemann,
K. Martin, C. Hertweck and A. a Brakhage. 2009. Intimate bacterial-fungal interaction
triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proceedings of the
National Academy of Sciences of the United States of America. 106:14558-63.
Schuster, E., N. Dunn-Coleman, J.C. Frisvad and P.W.M. Van Dijck. 2002. On the safety of
Aspergillus niger-a review. Applied Microbiology and Biotechnology. 59:426-35.
Schäfer, A. and D.H. Wolf. 2009. Sec61p is part of the endoplasmic reticulum-associated
degradation machinery. The EMBO Journal. 28:2874-84.
Sharma, R., M. Katoch, N. Govindappa, P.S. Srivastava, K.N. Sastry and G.N. Qazi. 2012.
Evaluation of the catalase promoter for expressing the alkaline xylanase gene (alx) in
Aspergillus niger. FEMS microbiology letters. 327:33-40.
Shima, Y., Y. Ito, S. Kaneko, H. Hatabayashi, Y. Watanabe, Y. Adachi and K. Yabe. 2009.
Identification of three mutant loci conferring carboxin-resistance and development of a novel
transformation system in Aspergillus oryzae. Fungal Genetics and Biology. 46:67-76.
Shoji, J.Y., J. Maruyama, M. Arioka and K. Kitamoto. 2005. Development of Aspergillus
oryzae thiA promoter as a tool for molecular biological studies. FEMS Microbiology Letters.
244:41-6.
Shrivastav, M., L.P. De Haro and J.A. Nickoloff. 2008. Regulation of DNA double-strand
break repair pathway choice. Cell Research. 18:134-47.
Shryock, J.C., R. Rubio and R.M. Berne. 1986. Extraction of adenine nucleotides from
cultured endothelial cells. Analytical Biochemistry. 159:73-81.
Shwab, E.K., J.W. Bok, M. Tribus, J. Galehr, S. Graessle and N.P. Keller. 2007. Histone
deacetylase activity regulates chemical diversity in Aspergillus. Eukaryotic Cell. 6:1656-64.
Snoek, I.S.I., Z.A. van der Krogt, H. Touw, R. Kerkman, J.T. Pronk, R.A.L. Bovenberg, M.A.
van den Berg and J.M. Daran. 2009. Construction of an hdfA Penicillium chrysogenum strain
impaired in non-homologous end-joining and analysis of its potential for functional analysis
studies. Fungal Genetics and Biology. 46:418-26.

- 87 -
de Souza, W.R., P.F. de Gouvea, M. Savoldi, I. Malavazi, L.A. de Souza Bernardes, M.H.S.
Goldman, R.P. de Vries, J.V. de Castro Oliveira and G.H. Goldman. 2011. Transcriptome
analysis of Aspergillus niger grown on sugarcane bagasse. Biotechnology for Biofuels. 4:40.
Spencer, J.A., D.J. Jeenes, D.A. MacKenzie, D.T. Haynie and D.B. Archer. 1998.
Determinants of the fidelity of processing glucoamylase-lysozyme fusions by Aspergillus
niger. European Journal of Biochemistry / FEBS. 258:107-12.
Storms, R., Y. Zheng, H. Li, S. Sillaots, A. Martinez-Perez and A. Tsang. 2005. Plasmid
vectors for protein production, gene expression and molecular manipulations in Aspergillus
niger. Plasmid. 53:191-204.
Subramanian, A., P. Tamayo, V.K. Mootha, S. Mukherjee, B.L. Ebert, M.A. Gillette, A.
Paulovich, S.L. Pomeroy, T.R. Golub, E.S. Lander and J.P. Mesirov. 2005. Gene set
enrichment analysis: a knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545-15550.
Szewczyk, E., Y.-M. Chiang, C.E. Oakley, A.D. Davidson, C.C.C. Wang and B.R. Oakley.
2008. Identification and characterization of the asperthecin gene cluster of Aspergillus
nidulans. Applied and Environmental Microbiology. 74:7607-12.
Tada, S., K. Gomi, K. Kitamoto, K. Takahashi, G. Tamura and S. Hara. 1991. Construction of
a fusion gene comprising the Taka-amylase A promoter and the Escherichia coli beta-
glucuronidase gene and analysis of its expression in Aspergillus oryzae. Molecular & General
Genetics. 229:301-6.
Taheri-Talesh, N., T. Horio, L. Araujo-Bazan, X. Dou, E.A. Espeso, M.A. Pen, S.A. Osmani
and B.R. Oakley. 2008. The Tip Growth Apparatus of Aspergillus nidulans. Molecular
Biology of the Cell. 19:1439 -1449.
Takahashi, T., T. Masuda and Y. Koyama. 2006. Enhanced gene targeting frequency in ku70
and ku80 disruption mutants of Aspergillus sojae and Aspergillus oryzae. Molecular Genetics
and Genomics. 275:460-70.
Tanaka, M., M. Tokuoka, T. Shintani and K. Gomi. 2012. Transcripts of a heterologous gene
encoding mite allergen Der f 7 are stabilized by codon optimization in Aspergillus oryzae.
Applied Microbiology and Biotechnology. 96: 1275-1282

- 88 -
Torralba, S., A.M. Pedregosa, J.R. De Lucas, M.S. Diaz, I.F. Monistrol and L. F. 1996. Effect
of the microtubule inhibitor methyl benzimidazol-2-yl carbamate (MBC) on production and
secretion of enzymes in Aspergillus nidulans. Mycological Research. 11:1375-1382.
Torralba, S., M. Raudaskoski, A M. Pedregosa and F. Laborda. 1998. Effect of cytochalasin A
on apical growth, actin cytoskeleton organization and enzyme secretion in Aspergillus
nidulans. Microbiology. 144 ( Pt 1:45-53.
Tremblay, L.O. and A. Herscovics. 1999. Cloning and expression of a specific human alpha
1,2-mannosidase that trims Man9GlcNAc2 to Man8GlcNAc2 isomer B during N-glycan
biosynthesis. Glycobiology. 9:1073-8.
Upshall, A., A.A. Kumar, M.C. Bailey, M.D. Parker, M.A. Favreau, K.P. Lewison, M.L.
Joseph, J.M. Maraganore and G.L. McKnight. 1987. Secretion of Active Human Tissue
Plasminogen Activator from the Filamentous Fungus Aspergillus nidulans. Nature
Biotechnology. 5:1301-1304.
Valkonen, M., M. Ward, H. Wang, M. Penttilä and M. Saloheimo. 2003. Improvement of
foreign-protein production in Aspergillus niger var. awamori by constitutive induction of the
unfolded-protein response. Applied and environmental microbiology. 69:6979-86.
Varadarajalu, L.P. and N.S. Punekar. 2005. Cloning and use of sC as homologous marker for
Aspergillus niger transformation. Journal of Microbiological Methods. 61:219-24.
Verdoes, J.C., A.D. van Diepeningen, P.J. Punt, A.J. Debets, A.H. Stouthamer and C.A. van
den Hondel. 1994a. Evaluation of molecular and genetic approaches to generate glucoamylase
overproducing strains of Aspergillus niger. Journal of Biotechnology. 36:165-75.
Verdoes, J.C., P.J. Punt, P. van der Berg, F. Debets, A.H. Stouthamer and C.A. van den
Hondel. 1994b. Characterization of an efficient gene cloning strategy for Aspergillus niger
based on an autonomously replicating plasmid: cloning of the nicB gene of A. niger. Gene.
146:159-65.
Verdoes, J.C., P.J. Punt, J.M. Schrickx, H.W. van Verseveld, A.H. Stouthamer and C.A. van
den Hondel. 1993. Glucoamylase overexpression in Aspergillus niger: molecular genetic
analysis of strains containing multiple copies of the glaA gene. Transgenic Research. 2:84-92.

- 89 -
Verdoes, J.C., P.J. Punt, A.H. Stouthamer and C.A. van den Hondel. 1994c. The effect of
multiple copies of the upstream region on expression of the Aspergillus niger glucoamylase-
encoding gene. Gene. 145:179-87.
Vinck, A., C. de Bekker, A. Ossin, R.A. Ohm, R.P. de Vries and H.A.B. Wösten. 2011.
Heterogenic expression of genes encoding secreted proteins at the periphery of Aspergillus
niger colonies. Environmental Microbiology. 13:216-25.
Vinck, A., M. Terlou, W.R. Pestman, E.P. Martens, A.F. Ram, C. a M.J.J. van den Hondel
and H.A.B. Wösten. 2005. Hyphal differentiation in the exploring mycelium of Aspergillus
niger. Molecular Microbiology. 58:693-9.
Vogt, K., R. Bhabhra, J. Rhodes and D.S. Askew. 2005. Doxycycline-regulated gene
expression in the opportunistic fungal pathogen Aspergillus fumigatus. BMC Microbiology.
11:1-11.
Vongsangnak, W., P. Olsen, K. Hansen, S. Krogsgaard and J. Nielsen. 2008. Improved
annotation through genome-scale metabolic modeling of Aspergillus oryzae. BMC genomics.
9:245.
Vongsangnak, W., M. Salazar, K. Hansen and J. Nielsen. 2009. Genome-wide analysis of
maltose utilization and regulation in Aspergilli. Microbiology. 155:3893-902.
Wang, B., G. Guo, C. Wang, Y. Lin, X. Wang, M. Zhao, Y. Guo, M. He, Y. Zhang and L.
Pan. 2010. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA
sequencing. Nucleic Acids Research. 38:5075-87.
Wang, H., J. Entwistle, E. Morlon, D.B. Archer, J.F. Peberdy, M. Ward and D.J. Jeenes. 2003.
Isolation and characterisation of a calnexin homologue, clxA, from Aspergillus niger.
Molecular Genetics and Genomics. 268:684-91.
Wang, Z., M. Gerstein and M. Snyder. 2009. RNA-Seq: a revolutionary tool for
transcriptomics. Nature Reviews. Genetics. 10:57-63.
Ward, M., C. Lin, D.C. Victoria, B.P. Fox, J.A. Fox, D.L. Wong, H.J. Meerman, J.P. Pucci,
R.B. Fong, M.H. Heng, N. Tsurushita, C. Gieswein, M. Park and H. Wang. 2004.
Characterization of humanized antibodies secreted by Aspergillus niger. Applied and
Environmental Microbiology. 70:2567-76.

- 90 -
Ward, M., L.J. Wilson, K.H. Kodama, M.W. Rey and R.M. Berka. 1990. Improved
Production of Chymosin in Aspergillus by Expression as a Glucoamylase-Chymosin Fusion.
Nature Biotechnology. 8:435-440.
Ward, O.P. 2011. Production of recombinant proteins by filamentous fungi. Biotechnology
Advances. 30:1119-1139.
Ward, P.P., C.S. Piddington, G.A. Cunningham, X. Zhou, R.D. Wyatt and O.M. Conneely.
1995. A system for production of commercial quantities of human lactoferrin: a broad
spectrum natural antibiotic. Biotechnology. 13:498-503.
Whittle, C.A., Y. Sun and H. Johannesson. 2012. Genome-Wide Selection on Codon Usage at
the Population Level in the Fungal Model Organism Neurospora crassa. Molecular Biology
and Evolution.
Wu, L., M.R. Mashego, J.C. van Dam, A.M. Proell, J.L. Vinke, C. Ras, W.A. van Winden,
W.M. van Gulik and J.J. Heijnen. 2005. Quantitative analysis of the microbial metabolome by
isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal
standards. Analytical Biochemistry. 336:164-71.
Wucherpfennig, T., T. Hestler and R. Krull. 2011. Morphology engineering - Osmolality and
its effect on Aspergillus niger morphology and productivity. Microbial Cell Factories. 10:58.
Wucherpfennig, T., K.A. Kiep, H. Driouch, C. Wittmann, R. Krull, C. Cordes, H. Horn, I.
Kampen, A. Kwade, T.R. Neu and B. No. 2010. Morphology and rheology in filamentous
cultivations. Advances in Applied Microbiology. 72:89-136.
Wösten, H. a B., S.M. Moukha, J.H. Sietsma and J.G. Wessels. 1991. Localization of growth
and secretion of proteins in Aspergillus niger. Journal of General Microbiology. 137:2017-23.
Yoon, J., S. Kimura, J. Maruyama and K. Kitamoto. 2009. Construction of quintuple protease
gene disruptant for heterologous protein production in Aspergillus oryzae. Applied
Microbiology and Biotechnology. 82:691-701.
Yoon, J., J. Maruyama and K. Kitamoto. 2011. Disruption of ten protease genes in the
filamentous fungus Aspergillus oryzae highly improves production of heterologous proteins.
Applied microbiology and biotechnology. 89:747-59.

- 91 -
Yoshida, H., T. Matsui, a Yamamoto, T. Okada and K. Mori. 2001. XBP1 mRNA is induced
by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription
factor. Cell. 107:881-91.
Yu, J., N.D. Fedorova, B.G. Montalbano, D. Bhatnagar, T.E. Cleveland, J.W. Bennett and
W.C. Nierman. 2011. Tight control of mycotoxin biosynthesis gene expression in Aspergillus
flavus by temperature as revealed by RNA-Seq. FEMS Microbiology Letters. 322:145-9.
Yuan, X.-L., C. Goosen, H. Kools, M.J.E.C. van der Maarel, C.A.M.J.J. van den Hondel, L.
Dijkhuizen and A.F.J. Ram. 2006. Database mining and transcriptional analysis of genes
encoding inulin-modifying enzymes of Aspergillus niger. Microbiology. 152:3061-73.
Yuan, X.-L., J.A. Roubos, C.A.M.J.J. van den Hondel and A.F.J. Ram. 2008. Identification of
InuR, a new Zn(II)2Cys6 transcriptional activator involved in the regulation of inulinolytic
genes in Aspergillus niger. Molecular Genetics and Genomics. 279:11-26.
Zheng, H.-Z., H.-H. Liu, S.-X. Chen, Z.-X. Lu, Z.-L. Zhang, D.-W. Pang, Z.-X. Xie and P.
Shen. 2005. Yeast transformation process studied by fluorescence labeling technique.
Bioconjugate Chemistry. 16:250-4.
http://bioinformatics.tudelft.nl/hipsec.
http://www.fgsc.net/Aspergillus
http://www.blast2go.de/b2ghome
http://www.broadinstitute.org/gsea/index.jsp
http://www.broadinstitute.org/fetgoat/index.html
http://www.broadinstitute.org/fetgoat/index.html
www.sysbio.se/BioMet/
https://memosys.i-med.ac.at/MEMOSys/home.seam
http://f-a-m-e.org/
www.ncbi.nlm.nih.gov
www.aspgd.org
www.broadinstitute.org/annotation/genome/aspergillus_group
www.cadre-genomes.rg.uk

- 92 -
Kapitel 3
Engineering of Aspergillus niger for the production of secondary metabolites

- 93 -
Engineering of Aspergillus niger for the production of secondary
metabolites
Lennart Richter1,*
and Franziska Wanka2,*
, Simon Boecker1,2
, Dirk Storm2, Tutku Kurt
2,
Özlem Vural2, Roderich Süßmuth
1, § and Vera Meyer
2, §
1
Institute of Chemistry, Department of Biological Chemistry, Berlin University of Technology,
Straße des 17. Juni 124, 10623 Berlin, German 2
nstitute of Biotechnology, Department Applied and Molecular Microbiology, Berlin
University of Technology, Gustav-Meyer-Allee 25, 13355 Berlin, Germany
* These authors equally contributed to this work
§ Corresponding authors:
Prof. Vera Meyer, Berlin University of Technology, Institute of Biotechnology, Department
Applied and Molecular Microbiology, Gustav-Meyer-Allee 25, D-13355 Berlin, Germany,
Phone: +49-30-31472827, Fax : +49-30-31472922, [email protected]
Prof. Roderich Süßmuth, Berlin University of Technology, Institute of Chemistry,
Department of Biological Chemistry, Strasse des 17. Juni 124, D- 10623 Berlin, Germany,
Phone: +49-30-314 78774, Fax : +49-30-314 79651, [email protected]
Keywords:
Aspergillus niger, secondary metabolite, nonribosomal peptide synthetase, enniatin,
heterologous gene expression
Email addresses:

- 94 -
Abstract
Background: Filamentous fungi can each produce dozens of secondary metabolites which are
attractive as therapeutics, drugs, antimicrobials, flavour compounds and other high-value
chemicals. Furthermore, they can be used as an expression system for eukaryotic proteins.
Application of most fungal secondary metabolites is, however, so far hampered by the lack of
suitable fermentation protocols for the producing strain and/or by low product titers. To
overcome these limitations, we report here the engineering of the industrial fungus
Aspergillus niger to produce high titers (up to 4,500 mg • l-1
) of secondary metabolites
belonging to the class of nonribosomal peptides.
Results: For a proof-of-concept study, we heterologously expressed the 351 kDa
nonribosomal peptide synthetase ESYN from Fusarium oxysporum in A. niger. ESYN
catalyzes the formation of cyclic depsipeptides of the enniatin family, which exhibit
antimicrobial, antiviral and anticancer activities. The encoding gene esyn1 was put under
control of a tunable bacterial-fungal hybrid promoter (Tet-on) which was switched on during
early-exponential growth phase of A. niger cultures. The enniatins were isolated and purified
by means of reverse phase chromatography and their identity and purity proven by tandem
MS, NMR spectroscopy and X-ray crystallography. The initial yields of 1 mg • l-1
of enniatin
were increased about 950 fold by optimizing feeding conditions and the morphology of A.
niger in liquid shake flask cultures. Further yield optimization (about 4.5 fold) was
accomplished by cultivating A. niger in 5 l fed batch fermentations. Finally, an autonomous A.
niger expression host was established, which was independent from feeding with the enniatin
precursor D-2-hydroxyvaleric acid D-Hiv. This was achieved by constitutively expressing a
fungal D-Hiv dehydrogenase in the esyn1-expressing A. niger strain, which used the
intracellular ɑ-ketovaleric acid pool to generate D-Hiv.
Conclusions: This is the first report demonstrating that A. niger is a potent and promising
expression host for nonribosomal peptides with titers high enough to become industrially
attractive. Application of the Tet-on system in A. niger allows precise control on the timing of
product formation, thereby ensuring high yields and purity of the peptides produced.
Background
Recent genome mining efforts have uncovered that the genomes of filamentous fungi encode
an unexpected rich repertoire of low-molecular-weight compounds with commercial
relevance. These natural products known as secondary metabolites include nonribosomal
peptides, polyketides and lipopeptides, which have pharmacological implications. Isoprenoids

- 95 -
are interesting for the food industry as nutraceuticals or aroma compounds and poly-
unsaturated fatty acids or lipids, can potentially be commercialized as biofuels. The natural
product portfolio of filamentous fungi is thus impressive and emphasizes their great potential
to become multi-purpose expression platforms in biotechnology. However, most of the genes
involved in secondary metabolism pathways are not expressed under standard laboratory or
industrial conditions and/or are present in intractable filamentous fungi, which prevents
application of these natural products [1, 2, 3]. Different strategies based on molecular and
epigenetics factors as well as cultivation methods have thus been undertaken to awaken these
silent genes [4, 5]. In brief, secondary metabolite (SM) production is under control of
complex regulatory gene networks and involves intricate multi-step biosynthetic machineries,
as well as major reorganization of primary metabolic fluxes to redirect cellular metabolic
resources towards their biosynthesis. SM expression is naturally linked with starvation-
induced developmental processes leading to (a)sexual spore formation [6, 7, 8]. These
processes can easily be tracked and even induced during bioreactor cultivations by adjusting
low growth rates [9, 10].
The advent of synthetic biology opens new avenues to express any SM gene of interest in a
filamentous fungal host which is easily tractable by genetic engineering. For example, the
geodin gene cluster of Aspergillus terreus was recently reconstituted in A. nidulans and the
penicillin cluster of P. chrysogenum was completely rewired and expressed as a polycistronic
gene cluster under control of a single xylose-inducible promoter in A. nidulans [11, 12, 13]
Another system for A. nidulans is based on expression of any fungal SM gene of interest
under control of an alcohol-inducible promoter and includes methods for deletion entire A.
nidulans SM gene clusters. This approach is especially interesting as it eliminates production
of the most abundant A. nidulans SMs, thus reducing the SM background and facilitating
purification of the heterologously expressed SMs [14]. None of the inducible promoters used
so far is tunable, carbon source-independent and tight under non-induced conditions. This,
however, poses limitations in their use, especially when the switch in the carbon source
affects changes in the primary metabolic fluxes which should provide precursors for
heterologous SM production. This limitation, however, can be overcome by applying an
artificial expression system based on the Tet-on system, which was established and
systematically evaluated for use in A. niger but is functional in many other filamentous fungi
[15, 16, 17]. The Tet-on system is a tunable bacterial-fungal hybrid expression system, which
becomes induced upon addition of the tetracyclin-derivative doxycycline (Dox). Importantly,
the inducing power depends on the Dox concentration applied and reaches expression levels

- 96 -
which can compete with the strength of one of the strongest promoters known for filamentous
fungi, the gpdA promoter from the glycolytic pathway [15].
We chose the industrial fungus A. niger as the expression host to determine whether or not the
Tet-on system can be applied to produce high amounts of fungal SMs. Although the genome
of A. niger carries the pptA gene encoding the key biosynthetic enzyme of fungal SM
pathways (a 4’-phosphopantetheinyl transferase responsible for posttranslational activation of
nonribosomal peptide synthetases and polyketide synthases [18]), A. niger has so far only
been exploited as expression platform for large-scale production of organic acids, proteins and
enzymes [19]. Most importantly, the Tet-on system allows free choice over the timing of
product formation as it can be switched on at any time during cultivation of A. niger [15]. We
decided to induce heterologous SM expression during the exponential growth phase of
A. niger due to two reasons. First, a maximum of ATP and primary metabolism intermediates
are available during exponential growth phase, hence very high SM yields could supposedly
be achievable. Second, endogenous SMs of A. niger become mainly expressed during carbon-
starvation, i.e. during post-exponential growth phase [9, 10, 20]. Hence, heterologous SM
production can be decoupled from homologous SM production and the A. niger cultures could
largely be kept SM background-free.
For the proof-of-concept study, we decided to express the enniatin synthetase ESYN from
Fusarium oxysporum in A. niger. Enniatin is a mixture of nonribosomal peptides and belong
to the group of cyclic depsipeptides [21] which are mainly produced by the genus Fusarium
(for reviews see [22, 23]). Enniatin is synthesized by the multifunctional enzyme ESYN,
which uses three D-hydroxycarboxylic acids and three L-amino acids as precursors and
requires the cofactors ATP and S-adenosylmethionine (Fig. 1, [24, 25]). ESYNs from various
Fusarium species use different amino acid precursors and display relaxed substrate
specificities, which results in a wide spectrum of naturally occurring enniatins (Fig. 2). After
the first isolation of enniatin in 1947 [26], at least 29 naturally occurring derivatives were
isolated from Fusaria.
Enniatin features antimicrobial [27], antiviral [28], cytotoxic [29] and phytotoxic [30] effects.
For example, fusafungin, which is a mix of three derivatives (enniatin A, B and C), is a
bactericide acting against gram-positive and gram-negative bacteria and is used as a topical
agent for the treatment of respiratory infections [31]. The mode of action of enniatin is mainly
linked to its ionophoric activity. It is known that enniatin B forms complexes with cations in
the ratio 1:1, 2:1 or 2:3 and complexes K+, Ca
2+, Na
+, Mg
2+ and Li
+ [32], thereby forming
cation-selective pores in biomembranes [33, 34]. Additionally, the bioactivity of enniatins can
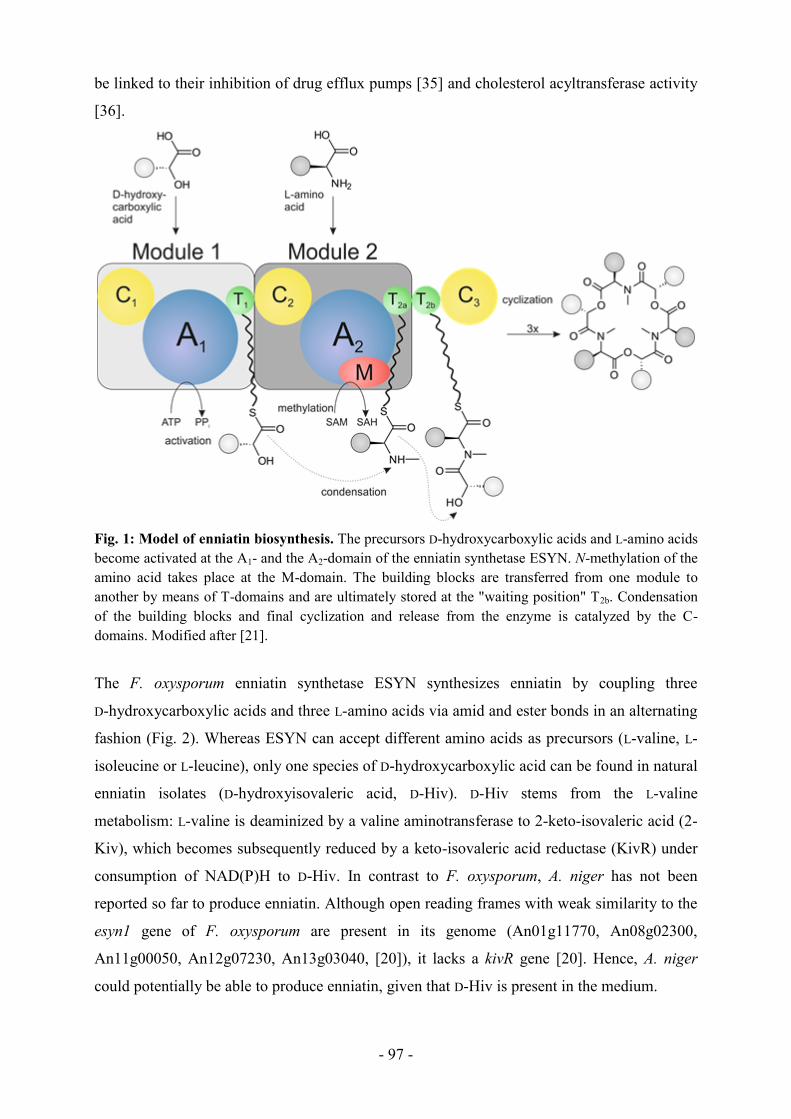
- 97 -
be linked to their inhibition of drug efflux pumps [35] and cholesterol acyltransferase activity
[36].
Fig. 1: Model of enniatin biosynthesis. The precursors D-hydroxycarboxylic acids and L-amino acids
become activated at the A1- and the A2-domain of the enniatin synthetase ESYN. N-methylation of the
amino acid takes place at the M-domain. The building blocks are transferred from one module to
another by means of T-domains and are ultimately stored at the "waiting position" T2b. Condensation
of the building blocks and final cyclization and release from the enzyme is catalyzed by the C-
domains. Modified after [21].
The F. oxysporum enniatin synthetase ESYN synthesizes enniatin by coupling three
D-hydroxycarboxylic acids and three L-amino acids via amid and ester bonds in an alternating
fashion (Fig. 2). Whereas ESYN can accept different amino acids as precursors (L-valine, L-
isoleucine or L-leucine), only one species of D-hydroxycarboxylic acid can be found in natural
enniatin isolates (D-hydroxyisovaleric acid, D-Hiv). D-Hiv stems from the L-valine
metabolism: L-valine is deaminized by a valine aminotransferase to 2-keto-isovaleric acid (2-
Kiv), which becomes subsequently reduced by a keto-isovaleric acid reductase (KivR) under
consumption of NAD(P)H to D-Hiv. In contrast to F. oxysporum, A. niger has not been
reported so far to produce enniatin. Although open reading frames with weak similarity to the
esyn1 gene of F. oxysporum are present in its genome (An01g11770, An08g02300,
An11g00050, An12g07230, An13g03040, [20]), it lacks a kivR gene [20]. Hence, A. niger
could potentially be able to produce enniatin, given that D-Hiv is present in the medium.

- 98 -
The main objective of this study was to determine whether A. niger is a suitable expression
host for high-level production of fungal nonribosomal peptides. We therefore put the esyn1
gene from F. oxysporum under control of the Dox-inducible Tet-on system and expressed it
heterologously in A. niger to produce the enniatin as a model nonribosomal peptide. We
optimized the production conditions using a design-of-experiment (DOE) approach which
addressed medium composition, D-Hiv feeding conditions and Dox concentration. We
furthermore optimized the cultivation conditions by establishing batch and fed batch cultures
for an esyn1 expressing A. niger strain. Finally, we engineered an autonomous enniatin B
producing A. niger strain which is independent from D-Hiv feeding.
Fig. 2: Amino acid composition and methylation pattern of the enniatin family. Enniatins are
composed of three D-hydroxycarboxylic acids and three L-amino acids. The structural diversity is
defined by the incorporation of different L-amino acids (R1-R3), which can be valine (iPr), leucine
(iBu) or isoleucine (sBu). L-amino acids can be methylated (Me, R4-R6). Modified after [22].
Results
Heterologous expression of the esyn1 gene in A. niger
The esyn1 gene of F. oxysporum was integrated in plasmid pVG2.2 [15] to give plasmid
pDS4.2, which comprises all three components of the Tet-on system: PgpdA::rtTA2S-M2 for
constitutive expression of the transactivator rtTA, tetO7::Pmin::esyn1, which mediates esyn1
expression in a Dox-dependent manner (note that rtTA is only able to bind to its operator
sequence tetO7 when bound to Dox, [15]) and the pyrG* cassette, necessary for selection and
targeting of the system to the pyrG locus of A. niger (Additional Files, Fig. 1). As recipient
strain, the protease-negative (prtT-) and uracil-auxotroph (pyrG
-) strain AB1.13 [37] was
used. Uridine-prototroph transformants were selected and screened via PCR and Southern
analysis for the presence of single or multiple pDS4.2 copies in the genome of A. niger

- 99 -
(Additional Files, Fig. 1 and data not shown). Ten pDS4.2-carrying transformants were
selected and cultivated in liquid shake flask cultures in the presence or absence of Dox.
Controls were an A. niger wild type strain (strain N402), the original producer F. oxysporum
(strain ETH1536) and an A. niger strain harboring a single copy of the esyn1-free plasmid
pVG2.2 at the pyrG locus (strain VG5.1). After cultivation, enniatin was isolated from the
biomass and the culture supernatant by means of ethyl acetate extraction. The amount of
enniatin produced was determined and quantified by HPLC-MS. Among the transformants,
strain DS3.1, which carried a single copy of pDS4.2 at the pyrG locus, produced the highest
amount of enniatin (about 1 mg • l-1
, Fig. 3). Only minute amounts of enniatin were detectable
in the control strains N402 and VG5.1 and all pDS4.2 carrying strains in the absence of Dox,
verifying that the expression system is tight under non-induced conditions. The m/z values
and retention time of enniatin isolated from the different A. niger transformants were equal to
those extracted from the natural enniatin producer F. oxysporum (Fig. 4 and data not shown).
Several derivatives could be detected and characterized by tandem MS, amongst them
enniatin A, A1, B and B1 (data not shown). However, because the standard used was a mixture
of enniatin A, B, and C isolated from F. oxysporum, the exact ratio and amount of the enniatin
variants could not be determined. However, full MS-scans of the standard and DS3.1 samples
showed that the enniatin composition was similar to F. oxysporum (Fig. 4).
Fig. 3: Screening for the best enniatin producing strain. 5 x 106
spores/ml were cultivated in 20 ml
complete medium for 40 h. Expression of the esyn1 gene was induced after 16 h of cultivation time
using 20 µg/ml Dox. From each strain, enniatin was purified from biomass and supernatant samples
and the overall enniatin concentration harvested is indicated.

- 100 -
Fig. 4: Analysis of enniatins produced in strain DS3.1. A) HR-LCMS chromatogram of purified
enniatin B. A segment from the mass spectrum shows [M+H+] of enniatin B with the characteristic
isotope pattern is shown. The main peak can be assigned to enniatin B. Minor impurities can be
detected at retention time 11.8 min and 12.2 min. B) HR-LCMS average mass spectrum. As example,
the mass spectrum of purified enniatin B is shown. The H+, NH4
+, Na
+ adducts of enniatin B can be
observed. The sample contains small amounts of enniatin B1. Samples were measured on an ESI-
Orbitrap-MS. C) ESI-HRMS/MS spectrum obtained with a LTQ Orbitrap XL apparatus using direct
injection and applying a collision energy of 12 eV. The moiety highlighted in green represents the L-

- 101 -
valine and the moiety highlighted in red represents D-Hiv incorporated in the enniatin B structure. For
the fragments m/z values were calculated. The calculated m/z value for the [C27H47N2O8]+ fragment is
527.33269 and the m/z value observed was 527.33270. For the [C22H39N2O6]+ fragment, the calculated
m/z value was 427.28026 and the m/z value observed was 427.28021. The calculated m/z value for the
[C16H28NO5]+
fragment was 314.19620 and the m/z value measured was 314.19638. The calculated m/z
value for the [C11H20NO3]+ fragment was 214.14377 and the m/z value observed was 214.14392.
Optimization of enniatin production
In order to identify the optimum condition for high yield production of enniatin, a design-of-
experiment approach was followed using the statistical software program MODDE (see
experimental part). The following parameters were varied in 20 ml shake flask cultures of the
esyn1-expressing strain DS3.1: (i) medium composition (minimal medium, complete medium,
Fusarium defined medium) [38], (ii) L-valine, L-leucine, L-isoleucine supplementation (0, 10,
20 mM), (iii) D-Hiv supplementation (0, 5, 10, 50 mM), (iv) glucose concentration (1, 2.5,
5%), (v) temperature (26°C, 30°C), (vi) cultivation time (24, 36, 48, 92 h) and (vii) Dox
concentration (0, 5, 10, 20 µg/ml). The parameters which mainly affected enniatin yields were
Dox and D-Hiv (data not shown) and the best cultivation medium identified contained 20 mM
D-Hiv, 20 mM of one of the amino acids and 10 μg/ml Dox. This medium composition
improved the enniatin yield by a factor of 200 to 200 mg ∙ l-1
(Fig. 5), whereby most of the
enniatin could be extracted from biomass samples after 92 h of cultivation. Remarkably, the
enniatin yield was further increased about 4.75-fold by increasing the glucose concentration to
5% and by adding talcum to the DS3.1 cultures (Fig. 5). As reported recently, the addition of
microparticles to liquid cultures of A. niger reduces the diameter of macromorphological
pellets to only a few hundred micrometers. This in turn considerably improves uptake rates of
nutrients and oxygen and increases the metabolic activity of A. niger [39]. Taken together, the
final enniatin yield was 950 mg · l−1 culture broth (corresponding to 0.04 g · g−
1 dry weight
biomass).

- 102 -
Fig. 5: Optimization of the enniatin yield. 5 x 106
spores /ml of strain DS3.1 were cultivated in 20
ml shake flask cultures containing complete medium with varying composition. Induction of esyn1
expression was performed in all media with 10 µg/ml Dox. Selected results are exemplarily shown: (1)
0 mM L-valine / L-isoleucine / L-leucine, 1% glucose, 0 g/l talcum, 40 h cultivation time, 30°C
cultivation temperature. (2) 10 mM L-valine / L-isoleucine / L-leucine, 1 % glucose, 0 g/l talcum, 10
mM D-Hiv, 92 h cultivation time, 26°C cultivation temperature. (3) 10 mM L-valine / L-isoleucine / L-
leucine, 2.5% glucose, 2.5 g/l talcum, 10 mM D-Hiv, 92 h cultivation time, 26°C cultivation
temperature. (4) 20 mM L-valine / L-isoleucine / L-leucine, 5% glucose, 10 g/l talcum, 10 mM D-Hiv,
92 h cultivation time, 26°C cultivation temperature. The total enniatin concentration (black bars) and
biomass concentration (grey bars) is given. Data from biological triplicates are shown. Microscopic
pictures of DS3.1 pellets are shown. Bar, 500 μm.
Modulation of the enniatin product spectrum by targeted supplementation with amino
acids
In order to determine the spectrum of enniatin variants, which can be heterologously
synthesized by A. niger, the precursor amino acids L-valine, L-leucine and L-isoleucine were
added to DS3.1 cultures either individually or in combination. After cultivation for 72 h, the
profile of the synthesized enniatins was determined by HPLC-MS. When no or all three
amino acids were added at the same concentration, the main products were enniatin B, B1 and
B4 and enniatins A, A1, A2, C, E and F were only present in trace amounts (Figure 6A). The
latter fraction increased, when L-leucine or L-isoleucine were added and reached almost 50%
of total enniatin, when both amino acids were supplemented each at 20 mM. When the
cultivation medium was supplemented with L-valine only, the main product was enniatin B
(87%). Interestingly, enniatin variants containing D-lactate moieties were also synthesized by
strain DS3.1 (Fig. 6B), which have so far not been observed in F. oxysporum. This suggests
that the precursor repertoire which can be used by ESYN is broader in A. niger compared to
F. oxysporum.

- 103 -
Fig. 6: Spectrum of enniatin species produced by strain DS3.1. A) Different enniatin variants were
produced depending on the amino acids fed. The final concentration of L-valine, L-leucine L-isoleucine
in the cultivation medium is given in mM. B) Structures of enniatin species produced in strain DS3.1.
Isolation and analytics of enniatin B
The identity and purity of enniatin produced by strain DS3.1 was confirmed by 1H-NMR-,
13C-NMR-, IR-, MS-, MS/MS- and X-ray analysis (Additional Files, Fig. 2 and data not
shown). From a 1 liter shake flask culture (20 mM D-Hiv, 20 mM L-valine, 5% glucose, 10
μg/ml Dox, 10 g/l talcum), 800 mg enniatin could be purified by preparative HPLC, which
corresponded to an enniatin yield of 0.04 g · g−1 dry weight biomass. The majority of enniatin
was isolated from the biomass. After repetitive crystallization, a total of 675 mg enniatin were
obtained which had a dark yellow to brownish color (data not shown). The NMR spectra were

- 104 -
identical to the ones published in literature [32, 40] and proved that the crystals were pure
enniatin B as summarized as follows: 1H-NMR spectrum: (400.1 MHz, CDCl3) δ = 5.11 (d,
3JH,H = 8.7 Hz, 3 H), 4.49 (d,
3JH,H = 9.7 Hz, 3 H), 3.11 (s, 9 H), 2.32_2,21 (m, 6 H), 1.05 -
0.86 ppm (m, 36 H). 13
C-NMR spectrum: (100.6 MHz, CDCl3) δ = 170.25, 169.31, 75.67,
63.20, 33.26, 29.91, 27.92, 20.42, 19.34, 18.72, 18.50 ppm. IR spectrum (Neat): υ =
2963.6_2873.4 (C-H, CH3 and CH), 1736.1 (C=O, ester), 1660.9 (C=O, amide), 1183.6 (C-H,
isopropyl) 1011.0 (CO, ɑ-hydroxycarboxylic acid). ESI-HRMS spectrum: m/z calculated for
[C33H57N3O9+Na]+: 662.39870; found: 662.39859; ESI-HRMS/MS: m/z calculated for
[C27H47N2O8]+: 527.33269; found: 527.33221, m/z calculated for [C22H39N2O6]
+: 427.28026;
found: 427.27988, m/z calculated for [C16H28NO5]+: 314.19620; found: 314.19614, m/z
calculated for [C11H20NO3]+: 214.14377; found: 214.14375. The masses of the daughter ions
are due to cleavages at the ester and amide bonds (m/z = 527.33, 427.28, 314.20, 214.14) as
described by [41].
The X-ray crystallographs demonstrated that the crystals had no impurities and were a
complex of enniatin B with Na+ ions, whereby one Na
+ ion was located in the center of an
enniatin B molecule (Additional Files, Fig. 2). As a result, the adjoining molecule from the
next layer in the crystal is not located on the same axis but is shifted to the side. Thus,
sandwich structures of enniatin B with the Na+ ions were not formed.
Production of enniatin B by batch and fed batch bioreactor cultivation
In order to obtain high enniatin yields under controlled conditions in bioreactors, 5 l batch
cultivations of strain DS3.1 were performed using a defined fermentation medium. This
medium had a pH of 3 and was balanced as such, that glucose was the growth-limiting
nutrient (final concentration 0.8 %; see Materials and Methods). Note that the low pH of the
medium and the use of ammonia as nitrogen source ensures dispersed morphology of A. niger
during bioreactor cultivation with no need for adding microparticles [42]. After the culture
reached the early exponential growth phase (corresponding to 1 g biomass dry weight · l−1
culture broth after about 14-16 h post inoculation), production of enniatin was induced by the
addition of 10 μg/ml Dox, 20 mM D-Hiv and 20 mM L-valine, respectively. In two
independent runs, the maximal specific growth rate achieved was 0.24 h-1
and the biomass
concentration peaked at 4.2 g · kg−1 culture broth after about 26 h post inoculation (Fig. 7A).
During exponential growth, pH 3 was maintained by the addition of 1 M NaOH, which has
been shown to linearly correlate with the biomass accumulation and reflecting ammonium
uptake during balanced growth [9, 43]. The end of the exponential growth phase was detected
by an increase of the dissolved oxygen signal (data not shown), after which the cell mass

- 105 -
decreased by nearly 50 % (Fig. 7A). Importantly, the levels of CO2 and O2 in the exhaust gas
clearly indicated that the cultures were still metabolically active, even 100 hours after
depletion of the carbon source (data not shown). As recently demonstrated, carbon starvation
of A. niger during submerged cultivation results in secondary growth by carbon recycling
leading to a gradual transition from old to young hyphae [9]. Enniatin levels determined for
selected time points demonstrated that enniatin B was mainly produced after carbon source
depletion (i.e. after about 55 h post inoculation) and reached a maximum value of 0.29 g · g−1
dry weight biomass after about 110 h of cultivation (Fig. 7A).
Finally, one fed batch cultivation was performed to increase biomass concentration and
thereby enniatin B yield. After the culture reached the late exponential growth phase
(corresponding to 4 g biomass · kg −1 culture broth after about 18 h post inoculation),
expression of the esyn1 gene was induced by feeding with 5% glucose, 10 μg/ml Dox, 20 mM
D-Hiv and 20 mM L-valine. To ensure that the esyn1 gene was continuously expressed at
highest level, 10 μg/ml Dox were added every 4-7 h resulting in a final Dox concentration of
90 μg/ml. As depicted in Figure 7B, this fermentation protocol ensured a specific growth rate
of 0.15 h-1
and the biomass concentration reached 24.9 g · kg−1 culture broth after about 66 h
post inoculation. Enniatin B production started immediately after Dox induction and reached a
maximum of 4.5 g · kg−1 culture broth after 66 h of cultivation (corresponding to 0.18 g · g−
1
dry weight biomass, Fig. 7B). Interestingly, linear accumulation of biomass in the fed batch
run was paralleled by linear accumulation of enniatin B (Fig. 7B), suggesting that the
dynamics of enniatin B production under the Tet-on system displays primary metabolite
kinetics.
Establishment of an autonomous A. niger production strain
As mentioned above, A. niger cannot produce enniatin autonomously as its genome lacks the
kivR gene encoding the D-Hiv generating enzyme ɑ-ketoisovalerate reductase. In order to
engineer an autonomous enniatin production strain which is independent from D-Hiv feeding,
strain DS3.1 was transformed with the kivR gene from F. oxysporum (E-KivR), which was put
under control of the constitutive gpdA promoter (see Materials and Methods). Three
transformants carrying multi-copy integrations of the PgpdA::kivR construct (strains ÖV4.3,
ÖV4.10, ÖV4.11, data not shown) were analyzed for enniatin production using 20 ml shake
flask cultures. After induction with a final concentration of 10 μg/ml Dox and
supplementation of 20 mM L-valine, the enniatin yield in all three transformants reached 0.03

- 106 -
g · g−1
dry weight biomass after 92 h of cultivation (data not shown), which is about 75 % of
the enniatin yield of strain DS3.1 when fed with 20 mM D-Hiv and 20 mM L-valine.
Discussion
The fungal kingdom of approximately 1.5 million species exhibits a huge reservoir of
secondary metabolites that span a broad variety of structurally and chemically diverse natural
products. This reservoir has and will increasingly have a considerable potential impact on
human welfare. The need to identify and produce novel bioactive fungal products goes far
beyond antimicrobials and includes the requirement for novel drugs for various human health
problems ranging from different cancers to neurodegenerative diseases, which are particularly
emerging in aging societies. The newly emerged fungal ‘omics’ era and the advent of systems
and synthetic biology provide innovative concepts and ideas to harness this untapped potential
and to produce novel bioactive compounds from fungi on an industrial scale. The gene(s) or
gene cluster(s) of interest can be of fungal or even (non)fungal origin and respective
expression constructs can be plugged into fungal chassis strains allowing high expression
levels. The gene(s) of interest will be expressed under the control of synthetic promoters and
the product repertoire, the timing of product formation and productivity can be optimized
using metabolic engineering strategies.
The aim of this study was to explore the potential of A. niger to become an expression system
for secondary metabolites from other organisms. A. niger is so far being used in
biotechnology as cell factory for the production of organic acids and secreted proteins [1].
For the proof-of-concept study, the cyclic depsipeptide enniatin was chosen. Heterologous
production of cyclic depsipeptides such as beauvericin has been achieved in microbial host
strains but only with very low yields, ranging from 3 mg · l−1 in Escherichia coli to 100 mg
· l−1 in Saccharomyces cerevisiae [44, 45]. Heterologous expression of the F. oxysporum
esyn1 gene in E. coli was accomplished as well; however, only minute amounts were
produced (1 mg · l−1; own unpublished data). Although chemical synthesis of enniatin is in
principle possible, it can by far not fulfill the requirements for an efficient large-scale drug
production process since its synthesis is too complex and very cost-intensive [46]. The
pharmaceutical relevance of enniatin stimulated studies to use homologous fungal hosts such
as F. sambucinum for enniatin production ([47] [48]). Although high amounts of enniatin
were produced (1.7 g · l−1), these surface cultures lasted up to 5 weeks. Submerged shake
flask cultivation of a randomly mutagenized F. oxysporum strain (strain ETH 1536) which

- 107 -
included additional amino acid feeding resulted in the highest enniatin titer reported so far (5
g · l−1 after 96 h of cultivation, [38]).
Here, we demonstrate that heterologous expression of the esyn1 gene under control of the Tet-
on system in A. niger allows enniatin production rates which are considerably higher than
ever reported for a heterologous host. 4.5 g · l−1
have been reached after 66 h of a fed batch
cultivation of strain DS3.1, a yield which is sufficient for rapid scale-up, biological testing
and commercial production. The yields which can be achieved with A. niger nearly reach the
titer of the original production strain F. oxysporum. Two explanations might explain why A.
niger is well suited for heterologous enniatin (and other nonribosomal peptides) production.
First, A. niger possesses an endogenous PPTase, which is key for the posttranslational
activation of NRPS [18]. Second, the ability to synthesize secondary metabolites is conserved
in filamentous fungi. Their secondary metabolites are adaptive traits that have been subjected
to natural selection during evolution. Although their occurrence apparently reflects particular
life style and survival strategies and differ among fungal species, multiple secondary
metabolic pathways are prevalent in each filamentous fungus. The existence of these
pathways predestines filamentous fungi as hosts for heterologous fungal secondary metabolite
production.
The genome of A. niger harbors five open reading frames (An01g11770, An08g02300,
An11g00050, An12g07230, An13g03040), which display a weak similarity to the esyn1 gene
of F. oxysporum [20]. Analysis of their expression profiles using published transcriptomics
data from A. niger cultures subjected to carbon-limited growth in batch, chemostat and
retentostat bioreactor fermentations [9, 49, 50] revealed that An01g11770, An11g00050,
An12g07230 and An13g03040 are silent under these conditions. An08g02300 though is
expressed at low levels, with mean expression values of 10% or less when compared with the
actin gene (An15g00560; data not shown). Hence, A. niger is likely to have the metabolic
pathways and flexibility to synthesize enniatin with an esyn1 gene being either of endogenous
or exogenous origin. However, as it does not comprise the kivR gene encoding a D-Hiv
dehydrogenase, feeding with D-Hiv or heterologous expression of a kivR gene is key to obtain
high enniatin levels with A. niger as shown in this study. It has to be mentioned that
An11g09950 shares high similarity with the D-Hiv dehydrogenase from Gibberella intermedi.
An11g09950 is a predicted 2-dehydropantoate 2-reductase catalyzing a similar reaction as
KivR, which is the reduction of 2-dehydropantoate to the D-hydroxycarboxylic acid D-
pantoate under consumption of NADPH [51]. Due to the relatively high degree of similarity
of both enzymes and their substrates, it might be conceivable that An11g09950 could accept
ɑ-ketoisovalerate as substrate to synthesize D-Hiv. This would explain why enniatin is present

- 108 -
in minute amounts in strain DS3.1 cultures when not fed with D-Hiv. In any case,
establishment of an autonomous A. niger strain being independent of D-Hiv feeding will
considerably reduce the cost of the fermentation process. Our data clearly demonstrated that
heterologous expression of the kivR gene from F. oxysporum rendered A. niger autonomous
with respect to D-Hiv feeding and allowed high level enniatin production.
Different cultivation protocols were run in this study to heterologously produce enniatin in
strain DS3.1. The specific yields of enniatin accomplished in shake flask cultures were 0.04 g
· g−1 dry weight biomass (20 ml and 1 l cultures) and 0.18 g · g−
1 dry weight biomass during
the fed batch cultivation. In these cultivations, 5% glucose served as carbon source. The
dynamics of enniatin B production clearly displayed primary metabolite kinetics in the fed
batch run (Fig. 7B), proving that the Tet-on system can ensure high level esyn1 expression
during exponential growth. The highest specific enniatin yield, however, was observed for the
batch cultivation (0.29 g · g−1 dry weight biomass), which used only 0.8 % glucose as carbon
source. Interestingly, the enniatin production followed secondary metabolite kinetics although
the Tet-on system was switched on early during exponential growth phase (Fig. 7A). Carbon-
induced starvation has clearly been demonstrated to induce a plethora of secondary
metabolites in A. niger [9, 50]. It is thus conceivable that the transition to the post-exponential
growth phase might have caused a metabolic shift in A. niger which favored secondary
metabolite production in general and enniatin production in particular. The overall activated
secondary metabolite machinery of A. niger might have strongly supported enniatin
expression, e.g. by providing additional carbon and nitrogen due to autophagy [9], by
ensuring a higher amino acid pool and/or by increasing the stability of the esyn1 transcript or
the ESYN protein. Future studies are necessary to understand these processes; clearly, only
systems-level insights will help to elucidate the molecular mechanisms behind.

- 109 -
Fig 7: Submerged batch and fed batch cultivation of strain DS3.1. A) Biomass (open symbols) and
enniatin accumulation (closed symbol) of two batch cultivations are shown. B) Biomass (open
symbol) and enniatin accumulation (closed symbol) of a fed batch cultivation are shown.
Conclusions
This is the first report demonstrating that A. niger is a potent expression host for
nonribosomal peptide synthesis. The strong inducibility of the Tet-on system combined with
controlled bioreactor cultivation allowed the production of enniatin with yields which are high
enough to become industrially relevant.

- 110 -
Methods
Strains, media and molecular techniques
Aspergillus strains used in this study are given in Table 1. Strains were grown on minimal
medium (MM) [52] containing 1 % (w • v-1
) glucose and 0.1 % (w • v-1
) casamino acids or on
complete medium (CM), containing 0.5 % (w • v-1
) yeast extract in addition to MM. When
required, plates were supplemented with uridine (10 mM). Transformation of A. niger and
fungal chromosomal DNA isolation was performed as described [53]. All molecular
techniques were carried out as described earlier [54].
The coding sequence of ESYN was PCR-amplified from a fosmid library of F. oxysporum
ETH 1536 and ligated into the PmeI-linearized plasmid pVG2.2 (PgpdA::rtTA::TcgrA-
tetO7::Pmin::TtrpC-pyrG*, [15]). The resulting plasmid was named pDS4.2. The kivR gene
from F. oxysporum ETH 1536 was PCR-amplified and ligated into the expression vector
pNOM102 [55] via NcoI restriction. Thereby, the ß-glucuronidase gene was replaced by kivR.
The resulting plasmid was named pÖV4.1. The protease-deficient A. niger strain AB1.13 was
transformed with pDS4.2 using its uracil-auxotrophy for selection [56]. Strain DS3.1 was co-
transformed with plasmid pÖV4.1 and the selection plasmid p3SR2, which expresses
acetamidase (amdS) as selection marker. Transformation of A. niger and fungal chromosomal
DNA isolation was performed as described [53]. All molecular techniques were carried out as
described earlier [54].
Table 1: Strains used in this study.
Strain Relevant genotype Source
Fusarium oxysporum
ETH1536 wild type [58]
Aspergillus niger
N402 wild type [59]
AB1.13 pyrG-, prtT
- [37]
VG5.1 pyrG+
, ∆kusA (transformed with pVG2.2; single copy) [15]
DS3.1 pyrG+, prtT
-, esyn1 (transformed with pDS4.2, single copy) this work
ÖV4.3 pyrG+, prtT
-, esyn1, EkivR (transformed with pÖV2.3;
multi copy)
this work

- 111 -
ÖV4.10 pyrG+, pyrtT
-, esyn1, EkivR (transformed with pÖV2.3;
multi copy)
this work
ÖV4.11 pyrG+, prytT
-, esyn1, EkivR (transformed with pÖV2.3;
multi copy)
this work
Optimization of enniatin production
Optimum cultivation conditions for enniatin production were identified using the statistical
software program MODDE 9.1 (Umetrics). The screening experiments were performed in
20 ml of CM which were inoculated with 5 x 106 spores · ml −
1 of strain DS3.1. All
cultivations were performed at 30°C and 250 rpm. After 16 h (~1 g dry weight · l−1), enniatin
expression was induced by the addition of different concentrations of Dox. Cultures were
harvested after 24 h by filtration and defined amounts of biomass and supernatant were used
to isolate enniatin. Several cycles of optimization were performed which included the
parameters cultivation time, concentration of inductor and D-Hiv, type of the cultivation
medium, temperature, concentration of L-Val, L-Leu, and L-Ile, concentration of carbon
source and talcum concentration (-350 mesh). As optimum cultivation condition was
eventually identified: CM containing 5% glucose and 10 g/l talcum, temperature 26°C,
addition of 20 mM D-Hiv, 20 mM L-Val and 10 µg/ml Dox after 16 h of cultivation, total
cultivation time 92 h.
Extraction and purification of enniatin
Defined amounts of DS3.1 supernatant and biomass cultures were extracted with ethyl
acetate. Extracts were centrifuged, dried over Na2SO4 and the solvent evaporated. Samples
were dissolved in methanol, diluted if necessary and the enniatin concentration determined by
HPLC-MS analysis. The HPLC-MS measurements for quantification were performed on an
ESI-Triple-Quadrupol-MS, 6460 Series (Agilent Technologies) in multiple reaction
monitoring mode. The utilized column was an Eclipse Plus C18, 2.1x50 mm column (Agilent
Technologies) and the mobile phases were H2O + 0.1% formic acid (A) and acetonitrile +
0.1% formic acid (B). The injection volume was set to 2 μl and the flow rate was 0.3 ml/min.
The m/z value for the precursor ion was set to 640.4 (m/z of [enniatin B H+] - adduct) and for
the fragment ions to 527.4 as quantifier, 427.3 and 196.2 as qualifier. For every set of
measurements, a new calibration curve was made using enniatin isolated from F. oxysporum
as an external standard. Peak areas were determined by manual integration using masshunter
workstation quantitative analysis (Agilent).

- 112 -
For purifying enniatin by recrystallization, the crystals were resolved in a minimal amount of
hot ethyl acetate. Acetonitrile was slowly added until clear crystals started to appear. The
mother liquor was decanted and the crystals were washed several times with acetonitrile.
Enniatin obtained from crystallization was applied to preparative HPLC (1100 series, Agilent
Technologies) running isocratically 70% methanol, containing 0.1 % formic acid on a C18-
column (Grom-Sil 120 ODS-5 ST, 10 µm, 250 x 20 mm, Grace).
Identification and characterization of enniatin B
1H-NMR and
13C-NMR spectra of enniatin B were recorded on a Bruker Avance 400 NMR-
spectrometer. The signals of the non-deuterated solvent rests were used as standards.
Chemical shifts are given in δ-units (ppm) relative to the solvent signal. IR spectra were
recorded on a Jasco FT-IR 4100 spectrometer. High-resolution mass-spectrometry (HRMS)
using ESI-technique was performed on a LTQ Orbitrap XL apparatus. Data for the single-
crystal structure determination of enniatin B were collected on an Oxford-Diffraction
Xcalibur diffractometer, equipped with a CCD area detector Sapphire S and a graphite
monochromator utilizing MoKɑ radiation (λ = 0.71073 Å). Suitable crystals were attached to
glass fibers using per-fluoropolyalkylether oil and transferred to a goniostat where they were
cooled to 150 K for data collection. The software packages used were CrysAlis CCD for data
collection, CrysAlis Pro for cell refinement and data reduction.
Bioreactor cultivation
Submerged cultivations were performed with 6.6 liter BioFlo3000 bioreactors (New
Brunswick Scientific, NJ, USA) and a detailed description of the fermentation settings was
previously given [50]. In brief, glucose-limited batch cultivation was initiated by inoculation
of 5 l (kg) fermentation medium with conidial suspension of strain DS3.1 to give 109 conidia
l-1
. Glucose was sterilized separately from the fermentation medium and final concentration
was 0.8 % (w • v-1
). Temperature of 30°C and pH 3 were kept constant, the latter by computer
controlled addition of 2 M NaOH or 1 M HCl, respectively. Acidification of the culture broth
was used as an indirect growth measurement [57]. When the culture reached the early
exponential growth phase (corresponding to 1 g biomass dry weight · kg-1
), Dox (10 μg/ml), D-
Hiv (20 mM) and L-Val (20 mM) were added.
The fed batch cultivation was started with 4 l fermentation medium. Induction of the Tet-on
system with Dox and addition of feeding medium (FM, 0.046 l • h-1
) was started when the
culture reached the late exponential growth phase. FM is composed of fermentation medium

- 113 -
with 5 % glucose, 0.5 % YE, 0.1 % casamino acids, 20 mM D-Hiv and 20 mM L-valine.
Every 4-7 h, 10 μg/ml of Dox were added.
Authors' contributions
DS, ÖV, LR and FW performed the cloning experiments, SB, TK, ÖV and LR carried out the
chemical analyses, TK and FW performed the batch and fed batch experiments. SB, DS, FW,
LR, TK, ÖV, RS and VM designed the experiments and interpreted the results. FW, LR, RS
and VM drafted the manuscript and were involved in writing the manuscript. All authors read
and approved the final manuscript.
Acknowledgements
This project was partly funded by the Marie Curie International Training Network QuantFung
(FP7-People-2013-ITN, Grant 607332) and supported by the Cluster of Excellence “Unifying
Concepts in Catalysis” funded by the Deutsche Forschungsgemeinschaft DFG and
coordinated by the TU Berlin.
References
1. Meyer V, Wu B, Ram AFJ: Aspergillus as a multi-purpose cell factory: current status
and perspectives. Biotechnol Lett 2011, 33:469–76.
2. Lubertozzi D, Keasling JD: Developing Aspergillus as a host for heterologous
expression. Biotechnol Adv 2009, 27:53–75.
3. Brakhage AA: Regulation of fungal secondary metabolism. Nat Rev Micro 2013, 11:21–
32.
4. Brakhage AA, Schroeckh V: Fungal secondary metabolites - strategies to activate silent
gene clusters. Fungal Genet Biol 2011, 48:15–22.
5. Wiemann P, Keller NP: Strategies for mining fungal natural products. J Ind Microbiol
Biotechnol 2014, 41:301–313.
6. Amare MG, Keller NP: Molecular mechanisms of Aspergillus flavus secondary
metabolism and development. Fungal Genet Biol 2014, 66:11–18.

- 114 -
7. Wiemann P, Guo CJ, Palmer JM, Sekonyela R, Wang CCC, Keller NP: Prototype of an
intertwined secondary-metabolite supercluster. Proc Natl Acad Sci U S A 2013,
110:17065–70.
8. Yin WB, Reinke AW, Szilágyi M, Emri T, Chiang YM, Keating AE, Pócsi I, Wang CCC,
Keller NP: bZIP transcription factors affecting secondary metabolism, sexual
development and stress responses in Aspergillus nidulans. Microbiology 2013, 159:77–88.
9. Nitsche BM, Jørgensen TR, Akeroyd M, Meyer V, Ram AFJ: The carbon starvation
response of Aspergillus niger during submerged cultivation: insights from the
transcriptome and secretome. BMC Genomics 2012, 13:380.
10. Jørgensen TR, Nitsche BM, Lamers GE, Arentshorst M, van den Hondel CA, Ram AF:
Transcriptomic insights into the physiology of Aspergillus niger approaching a specific
growth rate of zero. Appl Environ Microbiol 2010, 76:5344–55.
11. Nielsen MT, Nielsen JB, Anyaogu DC, Holm DK, Nielsen KF, Larsen TO, Mortensen
UH: Heterologous reconstitution of the intact geodin gene cluster in Aspergillus nidulans
through a simple and versatile PCR based approach. PLoS One 2013, 8:e72871.
12. Yaegashi J, Oakley BR, Wang CCC: Recent advances in genome mining of secondary
metabolite biosynthetic gene clusters and the development of heterologous expression
systems in Aspergillus nidulans. J Ind Microbiol Biotechnol 2014, 41:433–442.
13. Unkles SE, Valiante V, Mattern DJ, Brakhage AA: Synthetic biology tools for
bioprospecting of natural products in eukaryotes. Chem Biol 2014, 21:502–8.
14. Chiang YM, Oakley CE, Ahuja M, Entwistle R, Schultz A, Chang SL, Sung CT, Wang
CCC, Oakley BR: An efficient system for heterologous expression of secondary
metabolite genes in Aspergillus nidulans. J Am Chem Soc 2013, 135:7720–31.
15. Meyer V, Wanka F, van Gent J, Arentshorst M, van den Hondel CAMJJ, Ram AFJ:
Fungal gene expression on demand: an inducible, tunable, and metabolism-independent
expression system for Aspergillus niger. Appl Environ Microbiol 2011, 77:2975–83.
16. Helmschrott C, Sasse A, Samantaray S, Krappmann S, Wagener J: Upgrading fungal
gene expression on demand: improved systems for doxycycline-dependent silencing in
Aspergillus fumigatus. Appl Environ Microbiol 2013, 79:1751–4.

- 115 -
17. Wartenberg D, Vödisch M, Kniemeyer O, Albrecht-Eckardt D, Scherlach K, Winkler R,
Weide M, Brakhage AA: Proteome analysis of the farnesol-induced stress response in
Aspergillus nidulans - The role of a putative dehydrin. J Proteomics 2012, 75:4038–49.
18. Jørgensen TR, Park J, Arentshorst M, van Welzen AM, Lamers G, vanKuyk PA, Damveld
RA, van den Hondel CAM, Nielsen KF, Frisvad JC, Ram AFJ: The molecular and genetic
basis of conidial pigmentation in Aspergillus niger. Fungal Genet Biol 2011, 48:544–53.
19. Meyer V: Genetic engineering of filamentous fungi - progress, obstacles and future
trends. Biotechnol Adv 2008, 26:177–85.
20. Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries
RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JAE, van den Berg M,
Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EGJ, Debets
AJM, Dekker P, van Dijck PWM, van Dijk A, Dijkhuizen L, Driessen AJM, d’Enfert C,
Geysens S, Goosen C, Groot GSP, et al.: Genome sequencing and analysis of the versatile
cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 2007, 25:221–31.
21. Süssmuth R, Müller J, von Döhren H, Molnár I: Fungal cyclooligomer depsipeptides:
from classical biochemistry to combinatorial biosynthesis. Nat Prod Rep 2011, 28:99–124.
22. Sy-Cordero AA, Pearce CJ, Oberlies NH: Revisiting the enniatins: a review of their
isolation, biosynthesis, structure determination and biological activities. J Antibiot
(Tokyo) 2012, 65:541–9.
23. Firáková S, Proksa B, Sturdíkova M: Biosynthesis and biological activity of enniatins.
Pharmazie 2007, 62:563–568.
24. Krause M, Lindemann A, Glinski M, Hornbogen T, Bonse G, Jeschke P, Thielking G,
Gau W, Kleinkauf H, Zocher R: Directed biosynthesis of new enniatins. J Antibiot (Tokyo)
2001, 54:797–864.
25. Feifel SC, Schmiederer T, Hornbogen T, Berg H, Süssmuth RD, Zocher R: In vitro
synthesis of new enniatins: probing the alpha-D-hydroxy carboxylic acid binding pocket
of the multienzyme enniatin synthetase. Chembiochem 2007, 8:1767–70.
26. Gäumann E, Roth S, Etlinger L, Plattner PA, Nager U: Enniatin, ein neues, gegen
Mykobaktieren wirksames Antibiotikum. Experientia 1947, III:202–203.

- 116 -
27. Jayasinghe L, Abbas HK, Jacob MR, Herath WHMW, Nanayakkara NPD: N-Methyl-4-
hydroxy-2-pyridinone analogues from Fusarium oxysporum. J Nat Prod 2006, 69:439–42.
28. Mckee TC, Bokesch HR, McCormick JL, Rashid MA, Spielvogel D, Gustafson KR,
Alavanja MM, Cardellina JH, Boyd MR: Isolation and characterization of new anti-HIV
and cytotoxic leads from plants, marine, and microbial organisms. J Nat Prod 1997,
60:431–438.
29. Dornetshuber R, Heffeter P, Kamyar MR, Peterbauer T, Berger W, Lemmens-Gruber R:
Enniatin exerts p53-dependent cytostatic and p53-independent cytotoxic activities
against human cancer cells. Chem Res Toxicol 2007, 20:465–73.
30. Herrmann M, Zocher R, Haese A: Effect of disruption of the enniatin synthetase gene
of virulence of Fusarium avenaceum. Am Phytopathol Soc 1996, 9:226–232.
31. Levy D, Bluzat A, Seigneuret M, Rigaud JL: Alkali cation transport through liposomes
by the antimicrobial fusafungine and its constitutive enniatins. Biochem Pharmacol 1995,
50:2105–2107.
32. Ovchinnikow YA, Ivanov VT, Evstratov A V, Mikhaleva II, Bystrov VF, Portnova SL,
Balashova T, Meshcheryakova E, Tulchinsky VM: The enniatin ionophores. Conformation
and ion binding properties. Int J Pept Protein 1974, 6:465–498.
33. Kamyar M, Rawnduzi P, Studenik CR, Kouri K, Lemmens-Gruber R: Investigation of
the electrophysiological properties of enniatins. Arch Biochem Biophys 2004, 429:215–23.
34. Tonshin AA, Teplova VV, Andersson MA, Salkinoja-Salonen MS: The Fusarium
mycotoxins enniatins and beauvericin cause mitochondrial dysfunction by affecting the
mitochondrial volume regulation, oxidative phosphorylation and ion homeostasis.
Toxicology 2010, 276:49–57.
35. Hiraga K, Yamamoto S, Fukuda H, Hamanaka N, Oda K: Enniatin has a new function
as an inhibitor of Pdr5p, one of the ABC transporters in Saccharomyces cerevisiae.
Biochem Biophys Res Commun 2005, 328:1119–25.
36. Miyazaki A; Kanome T, Watanabe T: Inhibitors of acyl-coenzyme a: cholesterol
acyltransferase. Curr Drug Targets Cardiovasc Haematol Disord 2005, 5: 463–9.

- 117 -
37. Mattern IE, van Noort JM, van den Berg P, Archer DB, Roberts IN, van den Hondel CA:
Isolation and characterization of mutants of Aspergillus niger deficient in extracellular
proteases. Mol Gen Genet 1992, 234:332–6.
38. Madry N, Zocher R, Kleinkauf H: Enniatin Production by Fusarium oxysporum in
chemically defined media. Appl Microbiol Biotechnol 1983, 17:75–79.
39. Driouch H, Sommer B, Wittmann C: Morphology engineering of Aspergillus niger for
improved enzyme production. Biotechnol Bioeng 2010, 105:1058–68.
40. Zhukhlistova NE: X-ray crystal structure of the complex of enniatin B with KNCS.
Crystallogr Reports 2002, 47:433–442.
41. Burmeister HR, Plattner RD: Enniatin production by Fusarium tricinctum and its
effect on germinating wheat seeds. Phytopathology 1987, 77:1483–1487.
42. Jørgensen TR, Goosen T, Hondel CAMJJ Van Den, Ram AFJ, Iversen JJL:
Transcriptomic comparison of Aspergillus niger growing on two different sugars reveals
coordinated regulation of the secretory pathway. BMC Genomics 2009, 10:44.
43. Hrdlicka PJ, Sørensen AB, Poulsen BR, Ruijter GJG, Visser J, Iversen JJL:
Characterization of nerolidol biotransformation based on indirect on-line estimation of
biomass concentration and physiological state in batch cultures of Aspergillus niger.
Biotechnol Prog 2004, 20:368–76.
44. Matthes D, Richter L, Müller J, Denisiuk A, Feifel SC, Xu Y, Espinosa-Artiles P,
Süssmuth RD, Molnár I: In vitro chemoenzymatic and in vivo biocatalytic syntheses of
new beauvericin analogues. Chem Commun (Camb) 2012, 48:5674–6.
45. Yu D, Xu F, Zi J, Wang S, Gage D, Zeng J, Zhan J: Engineered production of fungal
anticancer cyclooligomer depsipeptides in Saccharomyces cerevisiae. Metab Eng 2013,
18:60–8.
46. Hu DX, Bielitza M, Koos P, Ley SV: A total synthesis of the ammonium ionophore,
(−)-enniatin B. Tetrahedron Lett 2012, 53:4077–4079.
47. Audhya TK, Russell DW: Spectrophotometric determination of enniatin a and
valinomycin in fungal extracts by ion complexation. Anal Lett 1973, 6:265–274.

- 118 -
48. Audhya TK, Russell DW: Production of enniatin A. Can J Microbiol 1973, 19:1051–
1054.
49. Kwon MJ, Jørgensen TR, Nitsche BM, Arentshorst M, Park J, Ram AFJ, Meyer V: The
transcriptomic fingerprint of glucoamylase over-expression in Aspergillus niger. BMC
Genomics 2012, 13:701.
50. Jørgensen TR, Nitsche BM, Lamers GE, Arentshorst M, van den Hondel CA, Ram AF:
Transcriptomic insights into the physiology of Aspergillus niger approaching a specific
growth rate of zero. Appl Environ Microbiol 2010, 76:5344–55.
51. King HL, Dyar RE, Wilken R: Ketopantoyl lactone and acid reductases. J Biol Chem
1974, 249:4689–4695.
52. Bennett JW, Lasure LL: More gene manipulations in fungi. In New York Acad Press;
1991:441–457.
53. Meyer V, Ram A, Punt P: Genetics, genetic manipulation and approaches to strain
improvement of filamentous fungi. In Man Ind Microbiol Biotechnol 3rd ed NY Wiley;
2010:318–329.
54. Sambrook J, Russell DW: Molecular cloning: A laboratory manual. In New York Cold
Spring Habour Lab Press; 2001:2344.
55. Punt PJ, Dingemanse MA, Kuyvenhoven A, Soede RDM, Pouwels PH, van den Hondel
CAMJJ: Functional elements in the promoter region of the Aspergillus nidulans gpdA
gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 1990, 93:101–109.
56. Punt PJ, van den Hondel CA: Transformation of filamentous fungi based on
hygromycin B and phleomycin resistance markers. Methods Enzymol 1992, 216:447–57.
57. Iversen JJL, Thomsen JK, Cox RP: On-line growth measurements in bioreactors
bytitrating metabolic proton exchange. Appl Environ Microbiol 1994, 42:256–262.
58. Zocher R, Keller U, Kleinkauf H: Enniatin synthetase, a novel type of multifunctional
enzyme catalyzing depsipeptide synthesis in Fusarium oxysporum. Biochemistry 1982,
21:43–8.

- 119 -
59. Bos CJ, Debets AJM, Swart K, Huybers A, Kobus G, Slakhorst SM: Genetic analysis
and the construction of master strains for assignment of genes to six linkage groups in
Aspergillus niger. Curr Genet 1988, 14:437–443.
Additional files
Additional file 1
Figure S1 Southern analysis of A. niger transformants. To confirm homologous integration of the
constructs at the A. niger pyrG locus, genomic DNAs of selected transformants were restricted with
NcoI and subjected to Southern hybridization using pyrG as a probe. Strain N402 served as a wild type
control. (A, B) The expected fragment size for the wild type pyrG is 3.1 kb. For a single-copy
integration of construct pDS4.2 at pyrG, two signals are expected (12.2 kb, 8.1 Kb). For a tandem-
copy integration of construct pDS4.2 at pyrG, three signals are expected (12.2 kb, 8.1 Kb, 17.2 kb).

- 120 -
Additional file 2
Figure 2S COSY NMR-spectrum and crystal structure of enniatin B. (A) COSY NMR-spectrum
of enniatin B. The COSY spectrum of enniatin B was recorded on a Bruker Avance 400 NMR-
spectrometer.
(B) Crystal structure of enniatin B: Data for the single-crystal structure determination of enniatin B
were collected on an Oxford-Difraction Xcalibur difractometer, equipped with a CCD area detector
Sapphire S and a graphite monochromator utilizing MoKα radiation (λ=0.71073 Å). Suitable crystals
were attached to glass fibers using per-fluoropolyalkylether oil and transferred to a goniostat. The
sample was cooled to 150K for data collection. Software packages used: CrysAlis CCD fordata
collection, CrysAlis Pro for cell refinement and data reduction. The conformation of a single enniatin
B molecule is shown. Enniatin B cocrystalized with sodium.
- 121 -
Kapitel 4
Tet-on, or Tet-off, that is the question: Advanced Conditional
Gene Expression in Aspergillus

- 122 -
Tet-on, or Tet-off, that is the question:
Advanced Conditional Gene Expression in Aspergillus
Franziska Wanka1, Timothy Cairns
1, Simon Boecker
1, Christian Berens
2, Anna Happel
3,
Xiaomei Zheng4, Jibin Sun
4, Sven Krappmann
3 and Vera Meyer
1, §
1Institute of Biotechnology, Department Applied and Molecular Microbiology, Berlin
University of Technology, Gustav-Meyer-Allee 25, 13355 Berlin, Germany 2Institute of Molecular Pathogenesis, Friedrich-Loeffler-Institut, Jena, Germany
3Mikrobiologisches Institut - Klinische Mikrobiologie, Immunologie und Hygiene,
Universitätsklinikum Erlangen, Friedrich-Alexander-Universität (FAU) Erlangen-Nürnberg,
Erlangen, Germany 4Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin 300308,
People’s Republic of China
§
Corresponding author:
Prof. Vera Meyer, Berlin University of Technology, Institute of Biotechnology, Department
Applied and Molecular Microbiology, Gustav-Meyer-Allee 25, D-13355 Berlin, Germany,
Phone: +49-30-31472827, Fax : +49-30-31472922, [email protected]
Keywords:
Aspergillus, Tet-on, Tet-off, conditional expression, heterologous gene expression, genetic
engineering
Email addresses:

- 123 -
Abstract
In Aspergillus, controlled gene expression is often achieved using the reverse tetracycline-
controlled transactivator (rtTA) dependent Tet-on system, whereby transcription is titratably
activated by addition of the tetracycline derivative doxycycline. The complementary Tet-off
system utilises the tetracycline-controlled transactivator (tTA) component to quantitatively
reduce gene expression. In this study, we utilized a synthetic biological approach to engineer
highly optimized Tet-off conditional expression systems in Aspergillus niger and Aspergillus
fumigatus. Steps for delivery of these tools include utilizing codon optimized cassette
components, testing several promoters for improved genetic stability and validating two
modified luciferase reporters for highly accurate measurements of gene expression. The Tet-
off cassettes developed in this study enable facile and quantitative functional analysis, as
validated by Tet-off analysis of genes involved in chitin synthesis and cell wall polarity in A.
niger, and para-aminobenzoic acid synthesis in A. fumigatus. We also used a racAG18V
dominant allele to demonstrate that Tet-off in A. niger enables gene over-expression and
downregulation in a single isolate. Additionally, we used the improved luciferase reporters to
show that the Tet-off cassette in A. niger enables quantification of gene oscillations. In order
to demonstrate that synthetic biological approaches developed here are broadly applicable to
engineering transcriptional circuits in filamentous fungi, we used our strategy for improving
cassette stability by promoter replacement in the A. niger Tet-on system, which resulted in a
modified Tet-on cassette with higher stability in recipient genomes.

- 124 -
Introduction
The design and reengineering of fungal systems at the molecular level has generated
numerous improved tools for basic and applied research. Such synthetic biological approaches
include reengineering a prokaryotic serine recombinase for site-specific marker recycling
(Hartmann et al., 2010), the CRISPR-Cas9 system for genome editing (Vyas et al., 2015)
(Nødvig et al., 2015), generation of RNA interference vectors for gene knock-down (Skowyra
and Doering, 2012) and an impressive range of inducible promoter systems from bacteria
(Meyer et al., 2011) and other kingdoms (Hörner and Weber, 2012). Such conditional
expression systems, where transcript abundance is controlled by experimental parameters, are
a vital tool for characterisation of essential genes, which are unable to be analysed by deletion
strategies. Moreover, titratable systems allow quantitative control of gene expression, which
might be an important technique for deciphering complex phenotypes by enabling tightly
controlled native, reduced or over-expression levels. One such tool is the tetracycline
inducible system, commonly referred to as Tet-on, whereby transcription is titratably
activated by addition of the highly stable tetracycline derivative doxycycline (Dox) to growth
media (Fig. 1A). In this system, a reverse tetracycline-controlled transactivator (rtTA)
requires a Dox ligand for DNA binding to rtTA responsive operator sequences (tetO). rtTA is
constitutively expressed and a gene of interest placed under control of a minimal promoter
next to several tetO elements, which enable activation of transcription in the presence of Dox.
In a seminal study, Vogt et al. established conditional gene expression in Aspergillus
fumigatus (Vogt et al., 2005). We have optimised this system and demonstrated that Tet-on is
titratable in the model organism Aspergillus niger, with the level of gene induction
proportional to the concentration of Dox added to the media (Meyer et al., 2011). The
minimal promoter of the Tet-on system, which controls the gene of interest has
low/undetectable basal rates of expression in the absence of inducers. Accordingly this is a
popular and versatile tool for gene functional characterisation in a variety of model and
pathogenic fungi. For example, a simple application of the Tet-on system for analysis of
uncharacterised gene (cfrX) in A. niger was to quantitatively elevate transcript levels using
Dox and assess strain phenotypes with/without induction (Meyer et al., 2011). For natural
product genome mining, Macheleidt and colleagues used Tet-on over-expression of a putative
transcription factor to activate an A. fumigatus secondary metabolite cluster, which resulted in
biosynthesis of a novel compound in induced strains (Macheleidt et al., 2015). Alternatively,
replacing the native promoter of a gene with the Tet-on cassette can be used for gene
functional analysis. For example, the Rho GTPase rho1 in the pathogenic mould A. fumigatus

- 125 -
was demonstrated to be an essential gene by replacement of the native rho1 promoter with the
Tet-on cassette, which resulted in a mutant isolate which could not grow in the absence of
Dox (Dichtl et al., 2012). An alternative strategy is to place genes of interest under control of
the Tet-on system and subsequently delete the wild-type allele, enabling characterisation of
gene function by quantitative transcript downregulation by reducing Dox in growth media, an
approach validated using the A. niger γ-actin encoding gene (Meyer et al., 2011). An
innovative application in A. nidulans by Wartenberg and colleagues used Tet-on to express
antisense RNA for a gene encoding a putative dehydrin (Wartenberg et al., 2012). This
enabled mRNA knock-down and functional characterisation of this mutant, which
demonstrated a role of the product of this gene in stress resistance of dormant conidia
(Wartenberg et al., 2012). Thus, the Tet-on system is a versatile tool that can be used for
multiple applications in a variety of fungi.
An alternative conditional expression system, named Tet-off, utilises the tetracycline-
controlled transactivator (tTA), in which tTA binding to tetO is prevented by tetracycline,
enabling quantitative reduction of gene expression (Fig. 1B). In the diploid yeast Candida
albicans, a high throughput approach in which one allele was deleted and the other placed
under control of the Tet-off system enabled analysis of 1152 genes, of which 567 were
demonstrated as essential for growth following Tet-off mediated down regulation (Roemer et
al., 2003). In the corn smut Ustilago maydis, Tet-off replacement of the native promoter for a
mating regulatory transcription factor enabled Dox controlled abolishment of sex in vitro
(Zarnack et al., 2006). In addition to demonstrating the utility of the Tet-off system, this latter
study highlights the numerous engineering steps that are required to develop highly optimised
conditional expression systems in fungi, which included obviating early polyadenylation of
the tetR gene by codon optimisation and decreasing basal activity of the Tet promoter by
removal of enhancer elements in U. maydis (Zarnack et al., 2006). For the Tet-on system in A.
fumigatus, further development entailed removal of repetitive sequences that were resulting in
intramolecular recombination and loss of the cassette from recipient genomes (Helmschrott et
al., 2013). These studies demonstrate the importance of synthetic biological approaches which
are necessary to deliver optimal functionality of conditional expression systems.

- 126 -
Figure 1: Design of the Tet-expression systems. (A) In the Tet-on system gene transcription is
reversibly turned on by the addition of Dox. It forms a complex with the constitutive expressed
transcription factor rtTA2S-M2, thereby inducing association of rtTA2
S-M2 protein to its operator
binding site tetO7. As reporter gene, behind the minimal promoter of gpdA (Pmin), different luciferase
versions were used. (B) The Tet-off system works in opposite direction, through addition of Dox the
gene transcription is reversibly turned off, because the antibiotic induced dissociation of tTA2S from
tetO7.
The objective of this study was to develop, optimise and validate a functional Tet-off system
in the model genus Aspergillus, represented by the industrially important model organism A.
niger and the pulmonary pathogen A. fumigatus. We describe a synthetic biological approach
in which the Tet-on cassette was reengineered to a titratable, stable, tightly regulated Tet-off
conditional expression system in both organisms. These inducible downregulation systems
significantly expand the toolkit of Aspergillus spp. and provide an engineering framework for
adapting any given promoter system in filamentous fungi.

- 127 -
Results
Engineering of the Tet-off system in Aspergillus niger
The fully functional A. niger Tet-on cassette was optimized previously and is encoded on
plasmid pVG4.1 (Meyer et al., 2011). This vector also enables A. niger transformation using a
pyrG auxotrophic marker (Meyer et al., 2011). Given that promoter and terminator sequences
are common to both Tet-on and Tet-off conditional expression systems (Fig. 1), it was
reasoned that simple exchange of the pVG4.1 reverse transactivator (rtTA2S-M2) with a
transactivator (tTA) would yield a derivative vector encoding a functional Tet-off cassette.
Accordingly, a tTA sequence was PCR amplified from p473 (Vogt et al., 2005), which was
used to replace the entire rtTA2S-M2 coding sequence in pVG4.1 to give plasmid pMA247.
This vector also contains a luciferase reporter encoded by the mluc gene under control of the
minimal promoter so that functionality of the cassette can be validated. This vector was
transformed into A. niger strain AB4.1, to give strain MA241.1, expressing a single copy of
the putative Tet-off cassette at the pyrG locus. However, in microtiter assays this isolate
demonstrated very low mluc expression as measured by LCPS values (data not shown). Strain
SB1.16, which contains multiple integrations of the putative Tet-off expression system at
random loci, did not sufficiently improve mluc expression (data not shown).
Hence, a transactivator optimized for mammals (tTAS2, see Material and methods) was used
to replace the rtTA2S-M2 sequence in pVG4.1, which gave derivative plasmid pFW9.1.
Transformation of A. niger with this plasmid generated strain FW13.11, which expressed a
single copy of the Tet-off modified cassette at the pyrG locus. In microtiter assays, this isolate
demonstrated strong luciferase activity following 8 hours growth, which was not observed in
growth media supplemented with various concentrations of Dox, indicating successful Tet-off
mediated downregulation of this gene (Fig. 2A). Interestingly, non-induced FW13.11 shows
higher luciferase values than the Tet-on expressing strain VG8.27 following induction with 20
µg/ml Dox (Fig. 2A). Importantly, induction of the Tet-off cassette with higher concentrations
of Dox resulted in faster downregulation of luciferase, indicating this system is titratable (Fig.
2A). Bioreactor cultivation of Tet-off expressing strain FW13.11, pictured in Fig 2B, shows
that downregulation using the expression system works on a large scale and with low
concentrations of Dox induction (5 µg/ml). These data also demonstrate high expression of
the mluc gene results in slower growth rates of A. niger, an effect which is abolished by Tet-
off mediated downregulation of this gene (Fig. 2B). During development and testing the Tet-
off system, we observed that not all PCR and Southern blot confirmed A. niger transformants
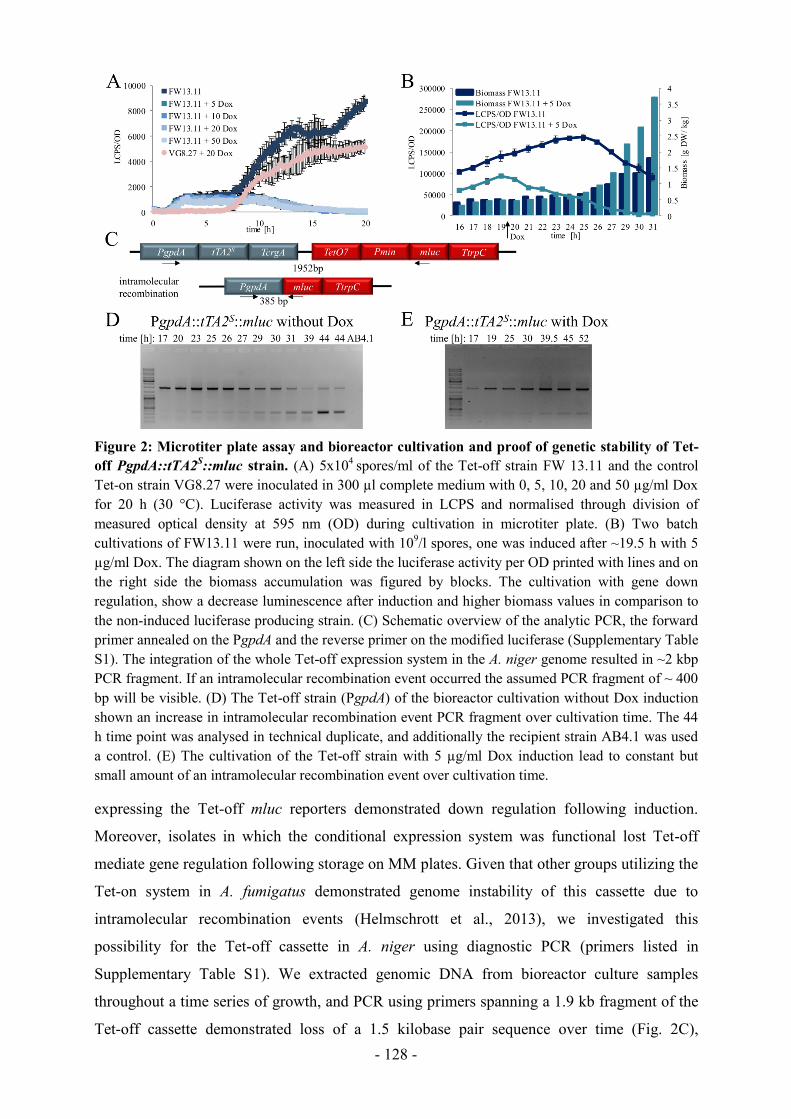
- 128 -
Figure 2: Microtiter plate assay and bioreactor cultivation and proof of genetic stability of Tet-
off PgpdA::tTA2S::mluc strain. (A) 5x10
4 spores/ml of the Tet-off strain FW 13.11 and the control
Tet-on strain VG8.27 were inoculated in 300 µl complete medium with 0, 5, 10, 20 and 50 µg/ml Dox
for 20 h (30 °C). Luciferase activity was measured in LCPS and normalised through division of
measured optical density at 595 nm (OD) during cultivation in microtiter plate. (B) Two batch
cultivations of FW13.11 were run, inoculated with 109/l spores, one was induced after ~19.5 h with 5
µg/ml Dox. The diagram shown on the left side the luciferase activity per OD printed with lines and on
the right side the biomass accumulation was figured by blocks. The cultivation with gene down
regulation, show a decrease luminescence after induction and higher biomass values in comparison to
the non-induced luciferase producing strain. (C) Schematic overview of the analytic PCR, the forward
primer annealed on the PgpdA and the reverse primer on the modified luciferase (Supplementary Table
S1). The integration of the whole Tet-off expression system in the A. niger genome resulted in ~2 kbp
PCR fragment. If an intramolecular recombination event occurred the assumed PCR fragment of ~ 400
bp will be visible. (D) The Tet-off strain (PgpdA) of the bioreactor cultivation without Dox induction
shown an increase in intramolecular recombination event PCR fragment over cultivation time. The 44
h time point was analysed in technical duplicate, and additionally the recipient strain AB4.1 was used
a control. (E) The cultivation of the Tet-off strain with 5 µg/ml Dox induction lead to constant but
small amount of an intramolecular recombination event over cultivation time.
expressing the Tet-off mluc reporters demonstrated down regulation following induction.
Moreover, isolates in which the conditional expression system was functional lost Tet-off
mediate gene regulation following storage on MM plates. Given that other groups utilizing the
Tet-on system in A. fumigatus demonstrated genome instability of this cassette due to
intramolecular recombination events (Helmschrott et al., 2013), we investigated this
possibility for the Tet-off cassette in A. niger using diagnostic PCR (primers listed in
Supplementary Table S1). We extracted genomic DNA from bioreactor culture samples
throughout a time series of growth, and PCR using primers spanning a 1.9 kb fragment of the
Tet-off cassette demonstrated loss of a 1.5 kilobase pair sequence over time (Fig. 2C),

- 129 -
indicating the Tet-off cassette is not genetically stable due to a intramolecular recombination
event which was observed to increase throughout cultivation (Fig. 2D). The cultivation with
Dox induction in Fig. 2E shows that this event occurs in a small but consistent subset of the
population. Given that there is 176 bp sequence homology between the minimal gpdA
promoter and constitutive gpdA promoter, we reasoned that this was resulting in
intramolecular recombination and loss of functionality in A. niger.
Accordingly, we decided to replace the constitutive gpdA promoter, which in vector pFW9.1
originates from A. nidulans. Firstly, we utilised the A. niger gpdA promoter (gpdAn), with the
rationale that this gene would offer similar expression levels to the A. nidulans orthologue
encoded in the Tet-off cassette. This was verified to lack homologous sequences to the
minimal gpdA promoter as determined by DNA alignment analysis (data not shown). Initial
experiments using a gpdAn promoter resulted in a functional Tet-off system, but ~15% of
transformants demonstrated loss functionality as measured by luciferase activity in the
presence of Dox (data not shown). By PCR amplification of the Tet-off cassette from these
isolates and subsequent sequencing we demonstrate this is due to another recombination event
between this promoter and the crgA terminator (primers describe in Supplementary Table S1).
In order to identify a further constitutive promoter which lacks any sequence homology to
other regions of the cassette, we interrogated an in-house compendium of microarray data for
genes which have comparable transcriptional profiles to gpdAn. This analysis yielded one
gene, which we term fraA, encoding a putative ribosomal subunit (An16g04690) which
demonstrated comparable transcriptional profiles with gpdAn in a variety of experiments (data
not shown). Exchange of the constitutive gpdA promoter with the fraA promoter resulted in a
stable strain as repeated purifications of transformants on MM plates did not result in loss of
function of the expression system, and evaluation of southern blots demonstrated no
detectable intramolecular recombination event (data not shown). In microtiter plate assays, the
resulted new Tet-off strain FW25.35 demonstrated titratable downregulation of luciferase
after induction with Dox, but the strain is about 50 % less active in the absence of induction
than the established Tet-on system (VG8.27) induced with 20 µg/ml Dox (Fig. 3A). Batch
cultivations in a bioreactor demonstrated that biomass accumulation was comparable under
both inducing and non-inducing conditions (Fig. 3B), which was a notable improvement when
compared to isolate FW13.11, which demonstrated reduced growth under inducing conditions
(Fig. 2B). Using the PfraA Tet-off system, gene downregulation occurred in the presence of 5
µg/ml Dox, and we observed rapid decrease in reporter luminescence after induction (< 11
minutes, Fig. 3B). Figure 3D and 3E demonstrate that in both bioreactor cultivations with and
without inducer there was no intramolecular recombination event as determined by diagnostic
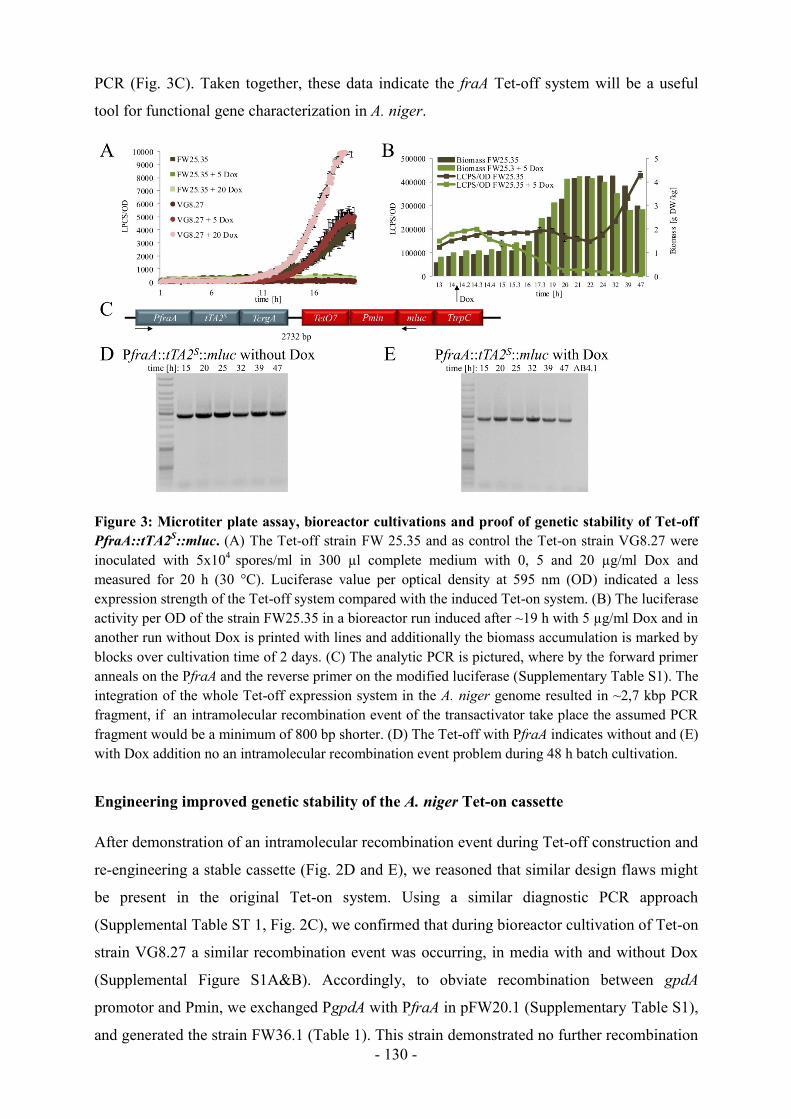
- 130 -
PCR (Fig. 3C). Taken together, these data indicate the fraA Tet-off system will be a useful
tool for functional gene characterization in A. niger.
Figure 3: Microtiter plate assay, bioreactor cultivations and proof of genetic stability of Tet-off
PfraA::tTA2S::mluc. (A) The Tet-off strain FW 25.35 and as control the Tet-on strain VG8.27 were
inoculated with 5x104
spores/ml in 300 µl complete medium with 0, 5 and 20 µg/ml Dox and
measured for 20 h (30 °C). Luciferase value per optical density at 595 nm (OD) indicated a less
expression strength of the Tet-off system compared with the induced Tet-on system. (B) The luciferase
activity per OD of the strain FW25.35 in a bioreactor run induced after ~19 h with 5 µg/ml Dox and in
another run without Dox is printed with lines and additionally the biomass accumulation is marked by
blocks over cultivation time of 2 days. (C) The analytic PCR is pictured, where by the forward primer
anneals on the PfraA and the reverse primer on the modified luciferase (Supplementary Table S1). The
integration of the whole Tet-off expression system in the A. niger genome resulted in ~2,7 kbp PCR
fragment, if an intramolecular recombination event of the transactivator take place the assumed PCR
fragment would be a minimum of 800 bp shorter. (D) The Tet-off with PfraA indicates without and (E)
with Dox addition no an intramolecular recombination event problem during 48 h batch cultivation.
Engineering improved genetic stability of the A. niger Tet-on cassette
After demonstration of an intramolecular recombination event during Tet-off construction and
re-engineering a stable cassette (Fig. 2D and E), we reasoned that similar design flaws might
be present in the original Tet-on system. Using a similar diagnostic PCR approach
(Supplemental Table ST 1, Fig. 2C), we confirmed that during bioreactor cultivation of Tet-on
strain VG8.27 a similar recombination event was occurring, in media with and without Dox
(Supplemental Figure S1A&B). Accordingly, to obviate recombination between gpdA
promotor and Pmin, we exchanged PgpdA with PfraA in pFW20.1 (Supplementary Table S1),
and generated the strain FW36.1 (Table 1). This strain demonstrated no further recombination
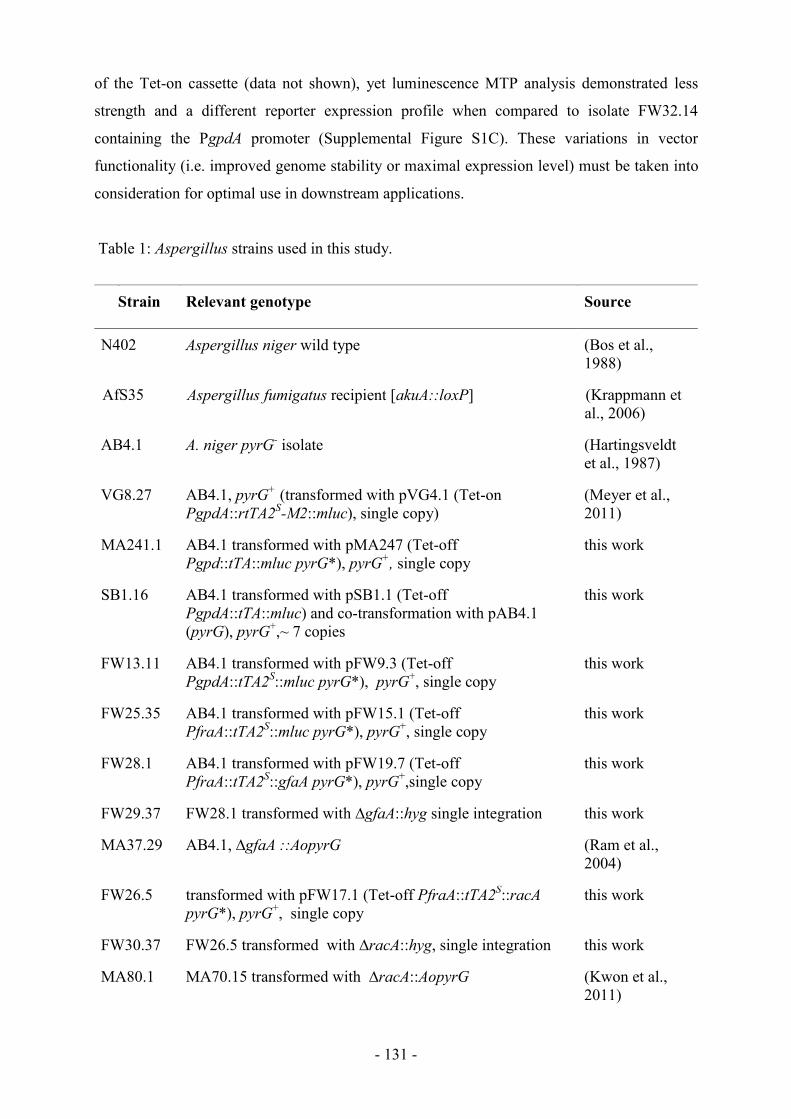
- 131 -
of the Tet-on cassette (data not shown), yet luminescence MTP analysis demonstrated less
strength and a different reporter expression profile when compared to isolate FW32.14
containing the PgpdA promoter (Supplemental Figure S1C). These variations in vector
functionality (i.e. improved genome stability or maximal expression level) must be taken into
consideration for optimal use in downstream applications.
Table 1: Aspergillus strains used in this study.
Strain Relevant genotype Source
N402 Aspergillus niger wild type (Bos et al.,
1988)
AfS35 Aspergillus fumigatus recipient [akuA::loxP] (Krappmann et
al., 2006)
AB4.1 A. niger pyrG- isolate (Hartingsveldt
et al., 1987)
VG8.27 AB4.1, pyrG+ (transformed with pVG4.1 (Tet-on
PgpdA::rtTA2S-M2::mluc), single copy)
(Meyer et al.,
2011)
MA241.1 AB4.1 transformed with pMA247 (Tet-off
Pgpd::tTA::mluc pyrG*), pyrG+, single copy
this work
SB1.16 AB4.1 transformed with pSB1.1 (Tet-off
PgpdA::tTA::mluc) and co-transformation with pAB4.1
(pyrG), pyrG+,~ 7 copies
this work
FW13.11 AB4.1 transformed with pFW9.3 (Tet-off
PgpdA::tTA2S::mluc pyrG*), pyrG
+, single copy
this work
FW25.35 AB4.1 transformed with pFW15.1 (Tet-off
PfraA::tTA2S::mluc pyrG*), pyrG
+, single copy
this work
FW28.1 AB4.1 transformed with pFW19.7 (Tet-off
PfraA::tTA2S::gfaA pyrG*), pyrG
+,single copy
this work
FW29.37 FW28.1 transformed with ∆gfaA::hyg single integration this work
MA37.29 AB4.1, ∆gfaA ::AopyrG (Ram et al.,
2004)
FW26.5 transformed with pFW17.1 (Tet-off PfraA::tTA2S::racA
pyrG*), pyrG+, single copy
this work
FW30.37 FW26.5 transformed with ∆racA::hyg, single integration this work
MA80.1 MA70.15 transformed with ∆racA::AopyrG (Kwon et al.,
2011)

- 132 -
FW27.7 AB4.1 transformed with pFW18.1 (Tet-off
PfraA::tTA2S::racA
G18V pyrG*), pyrG
+, single copy
this work
FW31.14 FW27.7 transformed with ∆racA::hyg, single integration this work
MA61.24 AB4.1 transformed with PinuE::racAG18V (Kwon et al.,
2011)
FW35.1 AB4.1 transformed with pAB4.1 (pyrG), pyrG+, single copy this work
AfS191 AfS35 transformed with pSK606 (A. fum. Tet-on::pabaA )
HpaI fragment replacing the endogenous pabaA promoter
region
this work
XM1.7 AB4.1 transformed with pXM1.1 (Tet-on PgpdA::rtTA2S-
M2::luc pyrG*), pyrG+, single copy
this work
FW32.14 AB4.1 transformed with pFW20.1 (Tet-on PgpdA::rtTA2S-
M2::luc-PEST pyrG*), pyrG+, single copy
this work
FW33.23 AB4.1 transformed with pFW21.8 (Tet-off
PfraA::tTA2S::luc-PEST pyrG*), pyrG
+, single copy
this work
FW36.1 AB4.1 transformed with pFW22.1 (Tet-On PfraA::rtTA2S-
M2::luc-PEST pyrG*), pyrG+, single copy
this work
Validation of the Tet-off system in A. niger
In order to validate that the Tet-off inducible expression system developed in this study will
enable gene functional characterization, we utilised a strategy where genes which mediate
easily detectable phenotypes in A. niger were placed under control of the Tet-off system,
followed by deletion of the wild-type allele. In these isolates, Tet-off mediated transcript
downregulation should result in comparable phenotypes to previously published null isolates.
Firstly, we substituted the coding sequence for the mluc reporter in the fraA Tet-off system
with gene gfaA, which encodes a glutamine: fructose- 6- phosphate amidotransferase (Ram et
al., 2004). This gene is responsible for the first step in chitin synthesis and null isolates are
unable to grow on media without exogenous supplementation of glucosamine, which is the
metabolite produced by the enzyme gfaA (Ram et al., 2004). Additionally, the RhoGTPase
racA coding sequence was cloned into fraA Tet-off. Deletion of this gene results in reduced
colony sporulation and a hyphal hyperbranching phenotype in simple growth assays (Kwon et
al., 2011).
In order to demonstrate that the Tet-off system can also be used for gain-of-function studies
under non-induced conditions, we cloned the dominant activation allele of the racA
RhoGTPase racAG18V
into fraA Tet-off. racAG18V
confers a clavate germling phenotype when

- 133 -
over-expressed in A. niger (Kwon et al., 2011). We therefore hypothesised that by deletion of
the wild-type racA in a PfraA::tTA2S::racA
G18V background, this single isolate can be used for
both gain-of-function and loss-of-function analysis during non-inducing and inducing growth
conditions respectively, thus proving a further technique for use with the Tet-off system.
Accordingly, A. niger strains expressing gfaA, racA, racAG18V
under Tet-off control at the
pyrG locus were generated (Table 1). These Tet-off expression strains were used as recipient
isolates in which the respective native gene was deleted using a split marker approach. We
used ∆gfaA and ∆racA isolates as positive controls for the predicted growth deficiencies of
Tet-off downregulation strains (Table 1). All isolates were phenotypically screened on solid
media +/- Dox induction (Fig. 4). For all strains macroscopic colony morphology was
assessed, and for analysis of racA mutant isolates cell morphology was analysed
microscopically to determine apolar growth phenotypes.
For strain FW29.37 (PfraA:tTA2S::gfaA, ∆gfaA) growth on MM plates was identical to the
control isolate FW35.1 (Fig. 4A). Induction of gfaA Tet-off by supplementation of the growth
media with more than 5 µg/ml Dox resulted in complete absence of growth in FW29.37 (10-
103 spores), which is consistent with loss of chitin synthesis due to gfaA down-regulation. The
gfaA knock out strain was unable to grow on MM or MM+Dox plates, but supplementation
with 10 mg/ml glucosamine enabled growth of this isolate. The plate assay also shows rescue
of FW29.37 growth on MM+Dox plates through addition of glucosamine. Down-regulation of
gene gfaA was titratable with various Dox concentrations as determined by colony growth on
plate assays (Fig. 4A).
Growth of strain FW30.37 (PfraA::tTA2S::racA, ∆racA) was identical to control isolate
FW35.1 on MM (Fig. 4 B). Tet-off downregulation resulted in radial colony growth which
was indistinguishable from that of a ∆racA strain but with notably less spores than the control
isolate (Fig. 4 B). Microscopic inspection revealed a clear hyberbranching phenotype in both
∆racA and following Tet-off downregulation in strain FW30.37 (Fig. 4 C).
In FW31.14 (PfraA::tTA::racAG18V
, ∆racA) dominant activation of RacAG18V
under non-
inducing conditions resulted in growth inhibition due to the previously documented actin mis-
localisation defect in this over-expression strain (Fig. 4D and 4E). Following Tet-off
downregulation, FW31.14 demonstrated reduced growth rates and hyphal hyperbranching
consistent with loss of racA function. These data collectively demonstrate that Tet-off is a
versatile tool for both gain-of-function and quantitative downregulation studies in A. niger.

- 134 -
Figure 4: Phenotypical growth assay and microscopic analysis of gfaA, racA and racAG18V
under
control of the new Tet-off (PfraA) in A. niger. Minimal medium plates (MM) and a serial spore
dilution (10, 102, 10
3) were used. (A) Tet-off mediate down regulation of the gfaA gene (FW29.37)
encoding glutamine fructose-6-phosphate amidotransferase. Deletion of gfaA is lethal (MA29.37) but
can be rescued by addition of glucosamine (GA). (B) The Rho GTPase racA integrated in the Tet-Off
system (FW30.37) shows a down regulation with low Dox concentrations (1 µg/ml), which was a
comparable phenotype to the knock out strain (MA80.1), with reduced diameter and spore formation.
(C) The strains grown on coverslips in MM for microscopy. Microscopic studies confirmed that
FW30.37 showed hyperbranching similar to the knock out mutant (MA80.1) under induced conditions
in the Tet-off system, and under uninduced conditions a phenotype comparable with the wild type
(FW35.1). Scale bar, 10 µm. (D) The strain FW31.14 included the dominant activation allele racAG18V
,
without Dox the overexpression of racAG18V
confers a lethal clavate germling phenotype similar to the
control strain MA61.24, which shown the overexpression phenotype only on saccharose medium
plates (SM) because of the used inuE promoter. Under induced conditions, the downregulation of

- 135 -
racAG18V
in the Tet-off system introduced reduced macroscopic growth rates and hyphal
hyperbranching consistent with loss of racA function as in B. (E) The clavate germling overexpression
phenotype of the Tet-off racAG18V
system looked similar to the induced control strain MA61.24 in
microscopic analysis with a magnification of 40x and also the hyperbranching took place under
induced condition similar to C. Scale bar, 10 µm.
Conditional gene silencing in A. fumigatus by an alternative Tet-off module
In a parallel effort, the established Tet-on system as validated in the human-pathogen
A. fumigatus was remodeled to a Tet-off version. Initial attempts after replacing the rtTA2S-
M2 sequence in the established Tet-on module of pVG4.1 (Meyer et al., 2011) by a formerly
validated tTA sequence (Vogt et al., 2005) (Gossen and Bujardt, 1992) were unsuccessful,
presumably due to toxicity this transactivator when expressed at high levels in the host cell.
Accordingly, we made use of the recently modified version of the Tet-on system (Helmschrott
et al., 2013), in which rtTA2S-M2 expression is driven by the tpiA promoter. Moreover, we
used a synthetic tTA2 transactivator that might be tolerated in A. fumigatus at higher
intracellular concentrations (Baron et al., 1997) (Urlinger et al., 2000). The resulting Tet-off
module of plasmid pSK606 was then used to assemble a conditional promoter replacement
(Hu et al., 2007) cassette in order to target the pabaA gene in A. fumigatus (Fig. 5A).
Respective recombinant strains were screened for their growth behavior in the presence and
absence of Dox with respect to para-aminobenzoic acid (PABA) necessity, a vitamin K
precursor that is formed by the action of the pabaA-encoded PABA synthetase (Fig. 5B).
Inoculation of the Tet-off::pabaA strain AfS191 on solid culture medium lacking PABA
revealed that the presumed auxotrophy depends on the presence of the inducer, as the isolate
was unable to grow significantly when Dox was supplemented at a concentration of 50 μg/ml
and this conditional auxotrophy could rescued by the presence of PABA.

- 136 -
Figure 5: Conditional Tet-off promoter replacement in A. fumigatus. (A) Schematic outline of the
A. fumigatus pabaA locus and the conditional promoter replacement allele carrying the functional Tet-
off module, in which expression of the doxycycline-responsive trans-activator (tTA2) is driven by the
tpiA promoter. Transcription of the pabaA gene is initiated from a minimal promoter comprising tetO
sequences that the tTA2 factor binds to in the absence of the tetracycline derivative Dox; the
pyrithiamine resistance-conferring ptrA gene is used as selection marker after genetic recombination.
(B) Growth phenotype of the Tet-off::pabaA strain AfS191 in dependency of Dox and
supplementation of para-aminobenzoic acid (PABA) compared to its wild-type progenitor and an
auxotrophic pabaAΔ deletion strain. Indicated amounts of conidia were spotted on Aspergillus minimal
culture medium (AMM) in the presence or absence of supplements and growth was monitored after
three days of incubation at 37 °C. A conditional requirement for PABA is evident for the conditional
promoter replacement strain AfS191, demonstrating functionality of the Tet-off system.
Application of the Tet-on and Tet-off system for induced gene oscillations
In order to conduct gene oscillatory studies in Aspergillus spp., we decided to test two
modified luciferase reporters, including a fungal codon optimized version luc (Gooch et al.,
2008) and a reporter encoding a proline, glutamic acid, serine, and threonine (PEST) protein
degradation sequence luc-PEST (Cesbron et al., 2013). We reasoned that these modifications
would increase fluorescent intensity and decrease luciferase half-life, which is essential for
accurate measurement of gene expression during oscillations. We firstly used the Tet-on
system [4] to compare both luc and luc-PEST with the conventionally used mluc (for
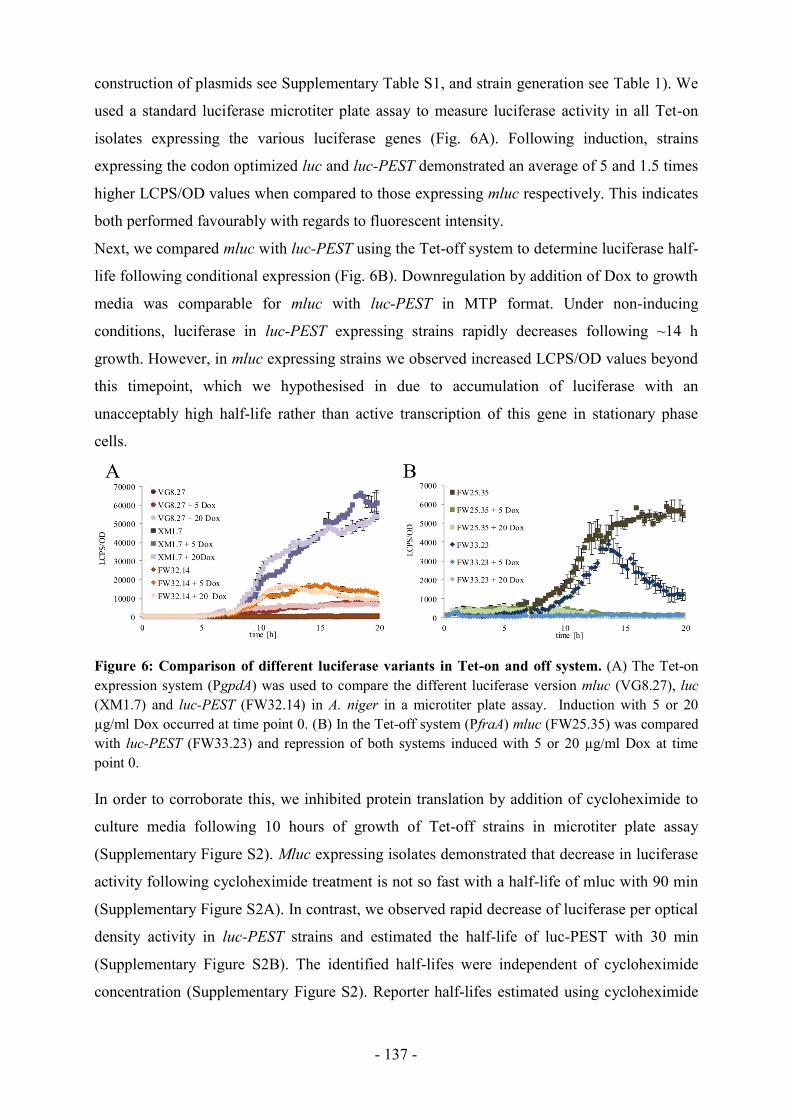
- 137 -
construction of plasmids see Supplementary Table S1, and strain generation see Table 1). We
used a standard luciferase microtiter plate assay to measure luciferase activity in all Tet-on
isolates expressing the various luciferase genes (Fig. 6A). Following induction, strains
expressing the codon optimized luc and luc-PEST demonstrated an average of 5 and 1.5 times
higher LCPS/OD values when compared to those expressing mluc respectively. This indicates
both performed favourably with regards to fluorescent intensity.
Next, we compared mluc with luc-PEST using the Tet-off system to determine luciferase half-
life following conditional expression (Fig. 6B). Downregulation by addition of Dox to growth
media was comparable for mluc with luc-PEST in MTP format. Under non-inducing
conditions, luciferase in luc-PEST expressing strains rapidly decreases following ~14 h
growth. However, in mluc expressing strains we observed increased LCPS/OD values beyond
this timepoint, which we hypothesised in due to accumulation of luciferase with an
unacceptably high half-life rather than active transcription of this gene in stationary phase
cells.
Figure 6: Comparison of different luciferase variants in Tet-on and off system. (A) The Tet-on
expression system (PgpdA) was used to compare the different luciferase version mluc (VG8.27), luc
(XM1.7) and luc-PEST (FW32.14) in A. niger in a microtiter plate assay. Induction with 5 or 20
µg/ml Dox occurred at time point 0. (B) In the Tet-off system (PfraA) mluc (FW25.35) was compared
with luc-PEST (FW33.23) and repression of both systems induced with 5 or 20 µg/ml Dox at time
point 0.
In order to corroborate this, we inhibited protein translation by addition of cycloheximide to
culture media following 10 hours of growth of Tet-off strains in microtiter plate assay
(Supplementary Figure S2). Mluc expressing isolates demonstrated that decrease in luciferase
activity following cycloheximide treatment is not so fast with a half-life of mluc with 90 min
(Supplementary Figure S2A). In contrast, we observed rapid decrease of luciferase per optical
density activity in luc-PEST strains and estimated the half-life of luc-PEST with 30 min
(Supplementary Figure S2B). The identified half-lifes were independent of cycloheximide
concentration (Supplementary Figure S2). Reporter half-lifes estimated using cycloheximide
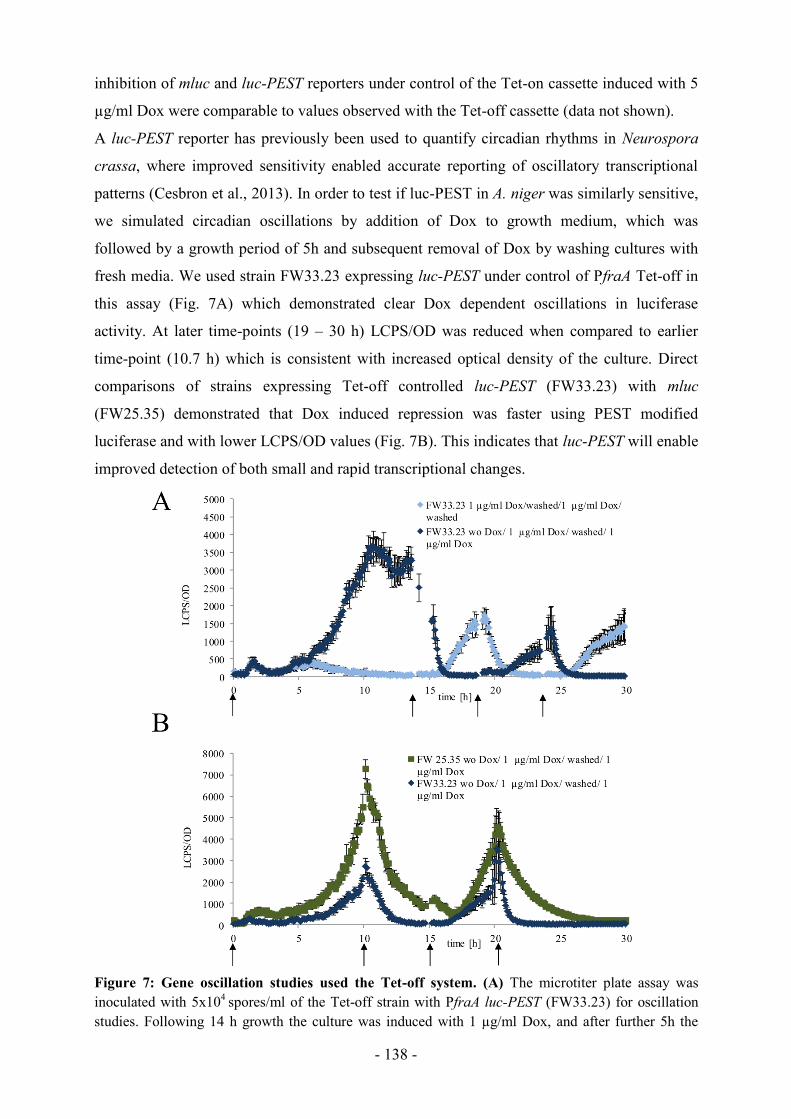
- 138 -
inhibition of mluc and luc-PEST reporters under control of the Tet-on cassette induced with 5
µg/ml Dox were comparable to values observed with the Tet-off cassette (data not shown).
A luc-PEST reporter has previously been used to quantify circadian rhythms in Neurospora
crassa, where improved sensitivity enabled accurate reporting of oscillatory transcriptional
patterns (Cesbron et al., 2013). In order to test if luc-PEST in A. niger was similarly sensitive,
we simulated circadian oscillations by addition of Dox to growth medium, which was
followed by a growth period of 5h and subsequent removal of Dox by washing cultures with
fresh media. We used strain FW33.23 expressing luc-PEST under control of PfraA Tet-off in
this assay (Fig. 7A) which demonstrated clear Dox dependent oscillations in luciferase
activity. At later time-points (19 – 30 h) LCPS/OD was reduced when compared to earlier
time-point (10.7 h) which is consistent with increased optical density of the culture. Direct
comparisons of strains expressing Tet-off controlled luc-PEST (FW33.23) with mluc
(FW25.35) demonstrated that Dox induced repression was faster using PEST modified
luciferase and with lower LCPS/OD values (Fig. 7B). This indicates that luc-PEST will enable
improved detection of both small and rapid transcriptional changes.
Figure 7: Gene oscillation studies used the Tet-off system. (A) The microtiter plate assay was
inoculated with 5x104
spores/ml of the Tet-off strain with PfraA luc-PEST (FW33.23) for oscillation
studies. Following 14 h growth the culture was induced with 1 µg/ml Dox, and after further 5h the

- 139 -
wells were washed with fresh medium to enable gene expression. Following 5 h further growth, we
induced the culture again. For comparison the same experiment was started directly with induction
through 1 µg/ml Dox. (B) To show the advantage of luc-PEST, FW25.35 (mluc) was used in microtiter
plate assay for comparison. After 10 h growth both cultures were induced with 1µg/ml Dox, after 5h
the cultures were washed and let grown again for 5 h and after further 5 h induced the last time. The
LCPS per OD values indicated that FW33.23 show a faster decrease after gene repression and enable
vales closed to zero.
Discussion
The rational redesign of fungal genetic circuits has huge promise for industrial applications
and fundamental research. Such optimised systems might enable production of a greater
variety of bioactive products at higher yields by experimentally and genetically tractable
fungi. For characterisation of gene function in basic research, applications of reengineered
conditional expression systems have enabled interrogation of multiple attributes of fungal
biology, including gene essentiality (Roemer et al., 2003), secondary metabolism (Skowyra
and Doering, 2012) and mating (Zarnack et al., 2006). In this study we deliver a functional
Tet-off conditional expression system in A. niger and A. fumigatus which are model
/industrially important and pathogenic moulds respectively.
In A. niger we re-engineered the established Tet-on cassette to a functional Tet-off conditional
expression system by sequential molecular modifications which included: (i) replacement of
the rtTA2S-M2 with a codon optimised tTA2S; (ii) testing several promoters for improved
genetic stability of the cassette and (iii) validating two modified luciferase reporters for highly
accurate measurements of transcription.
During validation experiments, poor genetic stability of both Tet-off and Tet-on cassettes was
identified. Previous studies have demonstrated that the strength of promoter has an influence
on the stability of the expression system, where an overexpressed transactivator interacts with
a variety of essential components of the transcriptional machinery, which can be deleterious to
cell metabolism, a phenomena described as squelching (Gill and Ptashne, 1988). To improve
stability of the Tet-off cassette, we exchanged the gpdA promoter with the fraA promoter, so
that an intramolecular recombination event was undetectable in various laboratory cultures.
Importantly, this Tet-off system maintained titratable downregulation of a luciferase reporter.
In order to demonstrated the utility of the new Tet-off expression system for gene functional
analysis, we could confirmed roles of gfaA and racA in chitin biosynthesis and regulation of
polar growth respectively. Thus, distinct processes which include fungal metabolism and
signalling cascade components can be assessed using the Tet-off system described in this
study. We also used a dominant racAG18V
allele to prove that the Tet-off cassette will enable

- 140 -
both gain-of-function and conditional gene downregulation in a single strain background.
Concomitant gene over-expression and downregulation enables comprehensive
characterisation of gene function, yet has previously required time-consuming generation of
multiple mutant isolates. That a single background can be used for both experimental
approaches greatly enhances the available toolkit for gene functional analysis in Aspergillus
spp.
Probing the cellular function of gene products in the context of fungal virulence is a valuable
and promising application of conditional gene expression in Aspergillus. Successful
establishment of the Tet-on system in A. fumigatus made such studies possible, in which the
expression of distinct genes during infection could be manipulated by Dox feeding of the
inoculated animals (our unpublished results). Accordingly by addressing the role of
presumably essential genes during pathogenesis of aspergillosis, the conditional promoter
replacement approach employing the Tet-on or Tet-off system is valid and supportive in
defining novel targets of antifungal therapy. When titrating the minimal amount of Dox that
would result in auxotrophy in the Tet-off::pabaA strain AfS191, concentrations of 3 μg/ml
turned out to be sufficient when inoculating 103 conidia (data not shown), prompting
successful application of the Tet-off system in infection series where concentrations of 0.2%
in the drinking water of susceptible mice are routinely applied.
With regards to modification of luciferase reporters for optimal measurement of gene
expression, we tested the fully codon optimised luciferase (luc) (Gooch et al., 2008) and short
half-life luc-PEST (Cesbron et al., 2013). Using a gene oscillation approach in A. niger with
the Tet-off system, we demonstrate approximately 5 times higher luciferase values using luc,
which is therefore a useful tool for experiments which assess activity of lower activity
promoters. With regards to improved sensitivity, our results determined the average half-life
of luc-PEST as 30 mins, which is comparable to the half-life published in Neurospora crassa
(Cesbron et al., 2013) . In contrast, the conventionally used mluc (Morgan et al., 2003)
demonstrated an extended half-life of 90 min with cycloheximide microtiter plate
determination. Faster degradation of luciferase reporters presented here will enable more
accurate measurements of conditional expression systems in future synthetic biological
applications.
Conclusions
In this study we engineered a titratable Tet-off system in A. niger and A. fumigatus. This
conditional expression system enabled gene functional analysis as determined by quantitative

- 141 -
downregulation of racA and gfaA in A. niger and pabaA in A. fumigatus. These data provide
proof of principle that this tool will be useful for assessing essential genes in both these
organisms. Using a racAG18V
dominant activation allele in A. niger, we were able to confirm
that the Tet-off conditional expression system enabled downregulation and overexpression in
a single isolate, an approach which obviates experimentally costly generation of multiple
mutant strains. We conducted several quality control experiments in which genetic stability of
the Tet-off encoding cassette was maximised by replacement of a gpdA promoter with a fraA
promoter. Accordingly, the Tet-off system is a versatile and robust tool for gene functional
analysis in industrially important and pathogenic Aspergilli. Expansion of this synthetic
biological approach enabled improvement of genetic stability in the previously published A.
niger Tet-on cassette, demonstrating that tools and techniques described in this study can
broadly be applied to engineering transcriptional circuits in filamentous fungi. Finally, in
order to conduct gene oscillatory studies, we describe two improved luciferase reporters
which can be used for accurate measurement of gene transcription in Aspergillus spp.
Material and methods
Cloning
The Gibson assembly method was utilized for plasmid construction (Gibson et al., 2009).
Briefly, PCR products or restriction endonuclease digested DNA fragments to be recombined
were designed with 20 over-lapping base pair regions to facilitate homologous recombination
(all plasmid constructions are listed in Supplementary Table S1). 5 µl DNA fragments were
mixed with 15 µl Gibson master mix consisting of 4 µl 5x isothermal reaction buffer (25 %
PEG-8000, 500 mM Tris-HCl pH 7.5, 50 mM MgCl2, 50 mM DTT, 1 mM of each four
dNTPs and 5 mM NAD), 0.08 units T5 exonuclease (Epicentre), 0.5 units Q5 DNA
polymerase (NEB) and 80 units Taq DNA ligase (NEB), made up to 15 µl with sterile water.
Samples were incubated at 50 °C for 60 minutes, before 3 minutes cooling at room
temperature, subsequent 3 minute incubation on ice, after which 3 µl aliquots were
transformed into chemically competent Escherichia coli strain TOP 10. Bacteria were grown
in LB medium supplemented with ampicillin at 50 µg/ml where appropriate.
Construction of Tet-off cassettes
Several versions of the tTA were used throughout this study. Initial experiments utilized the
tTA gene provide from p473 (Vogt et al., 2005), which was then cloned into the Tet-On

- 142 -
system of pVG4.1 plasmid (Meyer et al., 2011) (all plasmid constructions are listed in
Supplementary Table S1). In subsequent work we used the commercially available tTA2 S
codon optimized for eukaryotic translation (pTet-Off® Advanced Vector, Clontech).
To validate the functionality of other promoters instead of gpdA, 5’ upstream regions from
two genes were rationally selected based on comparable gene expression profiles to gpdAn
transcripts from an in-house compendium of A. niger microarray experiments (gpdAN
An16g01830, fraA An16g04690). In both cases we choose 1000 bp in front of the annotated
gene as promoter region.
To determine whether the new Tet-off system allowed gene function assays, the gene racA
(An11g10030), racAG18V
(dominant activation of racA) and gfaA (An18g06820) were used to
verify the concept. The reporter gene mluc was PmeI restriction digested from pFW15.1 and
replaced by DNA sequences encoding one of the above genes using Gibson cloning (racA:
pFW17.1, racAG18V
: pFW18.1, gfaA: pFW19.7), in detail see Supplementary Table S1.
A Tet-off module based on the pCH008 construct of Wagener and co-workers (Helmschrott et
al., 2013) was generated by replacing the rtTA2S-M2-encoding sequence by the synthetic tTA2
sequence isolated from the plasmid pUHT61-1 (Urlinger et al., 2000) by sequence and
ligation independent cloning (SLIC) using the Seamless Cloning system of Life Technologies.
A fragment of the resulting plasmid pSK606 was then used to assemble a conditional
promoter replacement cassette targeting the pabaA locus of A. fumigatus, using PCR
amplicons covering 1.5 kb of the 5' region and the coding sequence together with the 3'
region. A 7 kb HpaI fragment from this conditional promoter replacement vector pSK607 was
used for transformation of the A. fumigatus recipient strain AfS35.
Furthermore both luc-PEST (1879 bp) and luc (1741 bp) DNA encoding sequences were
amplified by Q5-polymerase from pFH62 (Cesbron et al., 2013) and Gibson cloned into the
recently used Tet-on system (pVG4.1) at PmeI restriction locus. luc-PEST was also cloned
into the Tet-off plasmid using this approach (pFW15.1). Additionally, the published Tet-on
cassette (pVG4.1) was also optimised with the new fraA promotor (Tab.1) and luc-PEST
resulting in pFW22.1.
A. niger transformation
A. niger transformation protocols, selection procedures, fungal chromosomal DNA isolation,
diagnostic PCR and Southern analyses were performed as described in Meyer et al., 2010.
We observed that expression of the Tet-off reporter cassettes resulted in slow growth of
positive clones during transformation. Accordingly, transformation media was supplemented
with Dox (1-5 µg/ml) in order to prevent heterokaryotic colonies overgrowing positive

- 143 -
transformants. In the case of the transformation with pFW18.1 (Tet-off with racAG18V
) it was
necessary to add Dox in the transformation and purification plates, because the overexpressed
racAG18V
transformant is non-viable.
A split marker approach enabled directed deletion of the target genes gfaA and racA at
endogenous loci, with a hygromycin resistance gene (from pAN7.1 (Punt et al., 1987)) used as
a selectable marker. Following transformation, agar plates were supplemented with 200 µg/ml
and 100 µg/ml hygromycin in subsequent purification plates (Arentshorst et al., 2015).
In order to confirm single cassette integration in the recipient genome at the pyrG locus,
transformant genomic DNA was restriction endonuclease digested and probed using a DIG
labelled DNA amplicon, which was a homologous sequence to 538 bp in AnpyrG* at 3` of the
Tet-off construct (described in detail in Supplementary Table S1). Two independent
restriction endonucleases were used for confirmation of each strain. Similarly, for
confirmation of gfaA and racA gene deletion events, two DIG labelled DNA probes were used
which targeted either the promoter or terminator region of each gene respectively
(Supplementary Table S1).
Strains and culture conditions
Aspergillus strains used in this study are given in Table 1. In all instances where A. niger was
modified with derivative plasmids of pVG4.1, we used AB4.1 (Table 1) as recipient strain
using its uracil-auxotrophy for selection. This enabled comparison to established Tet-On
strains (Meyer et al. 2011) and AB4.1 may show a better genomic stability in comparison to
NHEJ-inactivated strains (Zhang et al., 2011). A. niger strains were routinely grown on
minimal medium (MM) containing 1 % glucose, 1 x ASP+N (50 x ASP+N: 3.5 M NaNO3,
550 mM KH2PO4, 350 mM KCl, pH5.5), 2 mM MgSO4 and 1x trace elements solution
(modified from composition given by Vishniac and Santer, 1957, 1000 x trace elements
solution: 10 g of EDTA, 4.4 g of ZnSO4·7H2O, 1.01 g of MnCl2·4H2O, 0.32 g of CoCl2
·6H2O, 0.315 g of CuSO4 ·5H2O, 0.22 g of (NH4)6Mo7O24·4H2O, 1.47 g of CaCl2·2H2O, and
1 g of FeSO4·7H2O ) or on complete medium (CM) consisting of MM supplemented with 0.5
% yeast extract and 0.1 % casamino acids. For the growth assay we used saccharose medium
plates (SM) composed of the same ingredients as MM, except 1 % saccharose replacing 1 %
glucose. For growth on solid plates, media was supplemented with 1.5 % agar.
All bacterial and fungal strains were routinely stored at −80 °C in 50 % (v/v) glycerol. For
short term storage of fungal strains, spores were suspended in physiological sodium chloride
solution and kept at 4 °C.

- 144 -
Bioreactor cultivation
Bioreactor cultivation was conducted as described previously (Jørgensen et al., 2010). Briefly,
glucose-limited batch cultivations were performed with 5 l reactor minimal medium
containing the following 22.5 g of NH4Cl, 7.5 g of KH2PO4, 2.5 g of KCl, 2.5 g of
MgSO4·7H2O, 5 ml of trace metal solution (described above) and 0.8 % glucose, with pH
adjusted to 3. The 5 l bioreactor cultivation was inoculated with a spore density of 109
/liter in
BioFlo3000 bioreactors (6.6 liter, New Brunswick Scientific, NJ, USA). Temperature of 30°
C and pH 3 through computer controlled addition of 2 M NaOH or 1 M HCl were kept
constant. The addition of base was used as an indirect growth measurement, after
consumption of ~12,5 ml 2M NaOH (correlated with 1 gbiomass dryweight/kg, early
exponential phase), we induced or repressed our expression systems Tet-On and Tet-off with
5 µg/ml Dox.
Luciferase Measurement in Microtiter plate
Luciferase reporter activity was measured in microtiter ViewPlates (96 white with transparent
bottom, from Perkin Elmer) using a Victor3X plate reader. Two different types of
luminescence assays to evaluate the conditional expression systems were performed. In the
first type of assay, the strains were grown directly in the microtiter plates for ~20 h. In the
second assay, strains were grown in bioreactor cultivation, after which aliquots were taken
and measured in microtiter plates. For microtiter growth, wells were inoculated with
4x105sp/ml, 70 µl luciferin solution (diluted with CM to 1.4 mM, Promega), Dox were stated
(ranging from 0 – 20 µg/ml) and a final volume of 300 µl made up by addition of CM
medium. For every condition, triplicate biological replicates were performed. The
measurement protocol determines luminescence (LCPS) and optical densitiy (OD) values at
595 nm. For determination of the half-life of the different luciferase proteins (from mluc, luc,
luc-PEST) cycloheximide in a final concentrations of 10, 20, 30 and 100 µg/ml was added 10h
after inoculation of the respective strains. For oscillation studies, we used the PfraA Tet-off
system mluc and luc-PEST. Microtiter samples were inoculated with spores 10h in media
without Dox. Then cultures were induced with 1 µg/ml Dox, and after 5h the inoculated Dox
was removed by washing MTP wells (through discarding the used CM, addition of fresh CM,
pipette mixing, centrifugation of the plates 5min 1000 x g followed by exchange with fresh
CM plus luciferin solution). After a further 5 h the cultures were induced again. For
bioreactor growth assays, triplicate biological replicates were conducted and LCPS and OD
measured as described above. In this experiment 230 µl samples were mixed with 70 µl

- 145 -
luciferin (diluted with reactor minimal medium to 1.4 mM) and directly measured in
Victor3X.
Phenotypical growth assays on plates
A dilution series of A. niger spores (10, 102, 10
3) and A. fumigatus spores (10, 10
2, 10
3, 10
4,
105) of indicated strains (listed in Table 1) were spotted on either MM, SM or AMM plates,
supplemented where indicated with 10 mg/ml glucosamine, 1/ 10/ 50 µg/ml Dox
concentrations or 50 µg/ml para-aminobenzoic acid. Plates with A. niger strains were
incubated at 30 °C and A. fumigatus plates at 37 °C for 3 days. All plates were photographed
to visualize macroscopic colony morphology.
Microscopy
Two coverslips were disinfected and placed onto the bottom of a small Petri Dish, after which
5 ml of liquid MM or SM supplemented with 0.003 % yeast extract and 0 or 50 µg/ml Dox
were added. Petri dishes were inoculated with 106 spores of A. niger strains and incubated for
7 h at 30 °C. Coverslips with adherent germlings were placed upside down on an object slide
and analysed by microscopy. Light microscopic pictures (using DIC settings) were captured
with a 40x objective using a Leica DMI5000 CS equipped with a Leica DFC365 FX camera
and processed with GIMP 2.8 afterwards.
Authors' contributions
FW, SK and VM designed the study. FW, SB, AH, CB and XM performed the experiments.
FW, TC, SK, and VM interpreted the results and were involved in writing the manuscript. All
authors read and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge Swantje Lenz for the analysis of transcriptomic data
of A. niger to identify suitable promoters. We are grateful to Johanna Piepjohn, as well Mark
Arentshorst, and Michaela Dümig for their excellent technical assistance.

- 146 -
References
Arentshorst, M., Niu, J. and Ram, A.F.J., 2015. Efficient Generation of Aspergillus niger
Knock Out Strains by Combining NHEJ Mutants and a Split Marker Approach, in: Genetic
Transformation Systems in Fungi, Fungal Biology. 263–272. doi:10.1007/978-3-319-02904-7
Baron, U., Gossen, M., Bujard, H., 1997. Tetracycline-controlled transcription in eukaryotes:
Novel transactivators with graded transactivation potential. Nucleic Acids Res. 25, 2723–
2729. doi:10.1093/nar/25.14.2723Bos, C.J., Debets, A.J.M., Swart, K., Huybers, A., Kobus,
G., Slakhorst, S.M., 1988. Genetic analysis and the construction of master strains for
assignment of genes to six linkage groups in Aspergillus niger. Curr. Genet. 14, 437–443.
doi:10.1007/BF00521266
Cesbron, F., Brunner, M., Diernfellner, A.C.R., 2013. Light-dependent and circadian
transcription dynamics in vivo recorded with a destabilized luciferase reporter in Neurospora.
PLoS One 8, 1–6. doi:10.1371/journal.pone.0083660
Dichtl, K., Helmschrott, C., Dirr, F., Wagener, J., 2012. Deciphering cell wall integrity
signalling in Aspergillus fumigatus: Identification and functional characterization of cell wall
stress sensors and relevant Rho GTPases. Mol. Microbiol. 83, 506–519. doi:10.1111/j.1365-
2958.2011.07946.x
Gibson, D.G., Young, L., Chuang, R.-Y., Venter, J.C., Hutchison, C. a, Smith, H.O., 2009.
Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6,
343–345. doi:10.1038/nmeth.1318
Gill G. and Ptashne M., 1988. Negative effect of the transcriptional activator GAL4. Nature
334, 721–724. doi:10.1038/334721a0
Gooch, V.D., Mehra, A., Larrondo, L.F., Fox, J., Touroutoutoudis, M., Loros, J.J., Dunlap,
J.C., 2008. Fully codon-optimized luciferase uncovers novel temperature characteristics of the
Neurospora clock. Eukaryot. Cell 7, 28–37. doi:10.1128/EC.00257-07
Gossen, M., Bujardt, H., 1992. Tight control of gene expression in mammalian cells by
tetracycline-responsive promoters. Cell Biol. 89, 5547–5551. doi:10.1073/pnas.89.12.5547

- 147 -
Hartingsveldt, W. Van, Mattern, I.E., Zeijl, C.M.J. Van, Pouwels, P.H., 1987. Development of
a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol gen.
Genet 206, 71–75. doi: 10.1007/BF00326538
Hartmann, T., Dümig, M., Jaber, B.M., Szewczyk, E., Olbermann, P., Morschhäuser, J.,
Krappmann, S., 2010. Validation of a self-excising marker in the human pathogen Aspergillus
fumigatus by employing the βrec/six site-specific recombination system. Appl. Environ.
Microbiol. 76, 6313–6317. doi:10.1128/AEM.00882-10
Helmschrott, C., Sasse, A., Samantaray, S., Krappmann, S., Wagener, J., 2013. Upgrading
fungal gene expression on demand: improved systems for doxycycline-dependent silencing in
Aspergillus fumigatus. Appl. Environ. Microbiol. 79, 1751–4. doi:10.1128/AEM.03626-12
Hörner, M., Weber, W., 2012. Molecular switches in animal cells. FEBS Letters 586, 2084–
2096. doi:10.1016/j.febslet.2012.02.032
Hu, W., Sillaots, S., Lemieux, S., Davison, J., Kauffman, S., Breton, A., Linteau, A., Xin, C.,
Bowman, J., Becker, J., Jiang, B., Roemer, T., 2007. Essential gene identification and drug
target prioritization in Aspergillus fumigatus. PLoS Pathog. 3, e24.
doi:10.1371/journal.ppat.0030024
Jørgensen, T.R., Nitsche, B.M., Lamers, G.E., Arentshorst, M., van den Hondel, C. a, Ram,
A.F., 2010. Transcriptomic insights into the physiology of Aspergillus niger approaching a
specific growth rate of zero. Appl. Environ. Microbiol. 76, 5344–55.
doi:10.1128/AEM.00450-10
Krappmann, S., Sasse, C., Braus, G.H., 2006. Gene Targeting in Aspergillus fumigatus by
Homologous Recombination Is Facilitated in a Nonhomologous End- Joining-Deficient
Genetic Background Gene Targeting in Aspergillus fumigatus by Homologous Recombination
Is Facilitated in a Nonhomologous End- Join. Eukaryot. Cell 5, 212–215.
doi:10.1128/EC.5.1.212
Kwon, M.J., Arentshorst, M., Roos, E.D., Van Den Hondel, C. a M.J.J., Meyer, V., Ram,
A.F.J., 2011. Functional characterization of Rho GTPases in Aspergillus niger uncovers
conserved and diverged roles of Rho proteins within filamentous fungi. Mol. Microbiol. 79,
1151–1167. doi:10.1111/j.1365-2958.2010.07524.x

- 148 -
Macheleidt, J., Scherlach, K., Neuwirth, T., Schmidt-Heck, W., Straßburger, M., Spraker, J.,
Baccile, J. a., Schroeder, F.C., Keller, N.P., Hertweck, C., Heinekamp, T., Brakhage, A. a.,
2015. Transcriptome analysis of cyclic AMP-dependent protein kinase A-regulated genes
reveals the production of the novel natural compound fumipyrrole by Aspergillus fumigatus.
Mol. Microbiol. 96, 148–162. doi:10.1111/mmi.12926
Meyer, V., Ram, A., Punt, P., 2010. Genetics, genetic manipulation and approaches to strain
improvement of filamentous fungi, in: Manual of Industrial Microbiology and Biotechnology.
3rd Ed. NY: Wiley. 318–329. doi: 10.1128/9781555816827.ch22
Meyer, V., Wanka, F., van Gent, J., Arentshorst, M., van den Hondel, C. a M.J.J., Ram,
A.F.J., 2011. Fungal gene expression on demand: an inducible, tunable, and metabolism-
independent expression system for Aspergillus niger. Appl. Environ. Microbiol. 77, 2975–83.
doi:10.1128/AEM.02740-10
Morgan, L.W., Greene, A. V., Bell-Pedersen, D., 2003. Circadian and light-induced
expression of luciferase in Neurospora crassa. Fungal Genet. Biol. 38, 327–332.
doi:10.1016/S1087-1845(02)00562-5
Nødvig, C.S., Nielsen, J.B., Kogle, M.E., Mortensen, U.H., 2015. A CRISPR-Cas9 System
for Genetic Engineering of Filamentous Fungi. PLoS One 10, e0133085.
doi:10.1371/journal.pone.0133085
Punt, P.J., Oliver, R.P., Dingemanse, M.A., Pouwels, P.H., Handel, C.A.M.J.J. van den, 1987.
Transformation of Aspergillus based on the hygromycin B resistance marker from
Escherichia coli. Gene 56, 117–124. doi:10.1016/0378-1119(87)90164-8
Ram, a. F.J., Arentshorst, M., Damveld, R. a., vanKuyk, P. a., Klis, F.M., van den Hondel, C.
a M.J.J., 2004. The cell wall stress response in Aspergillus niger involves increased
expression of the glutamine: Fructose-6-phosphate amidotransferase-encoding gene (gfaA)
and increased deposition of chitin in the cell wall. Microbiology 150, 3315–3326.
doi:10.1099/mic.0.27249-0
Roemer, T., Jiang, B., Davison, J., Ketela, T., Veillette, K., Breton, A., Tandia, F., Linteau,
A., Sillaots, S., Marta, C., Martel, N., Veronneau, S., Lemieux, S., Kauffman, S., Becker, J.,
Storms, R., Boone, C., Bussey, H., 2003. Large-scale essential gene identification in Candida

- 149 -
albicans and applications to antifungal drug discovery. Mol. Microbiol. 50, 167–181.
doi:10.1046/j.1365-2958.2003.03697.x
Skowyra, M.L., Doering, T.L., 2012. Host-Fungus Interactions. Methods Mol Biol. 845, 165–
186. doi:10.1007/978-1-61779-539-8
Urlinger, S., Baron, U., Thellmann, M., Hasan, M.T., Bujard, H., Hillen, W., 2000. Exploring
the sequence space for tetracycline-dependent transcriptional activators: novel mutations yield
expanded range and sensitivity. Proc. Natl. Acad. Sci. U. S. A. 97, 7963–8.
doi:10.1073/pnas.130192197
Vishniac, W., Santer, M., 1957. The Thiobacilli. Bacteriological Reviews. 21, 195-213.
Vogt, K., Bhabhra, R., Rhodes, J.C., Askew, D.S., 2005. Doxycycline-regulated gene
expression in the opportunistic fungal pathogen Aspergillus fumigatus. BMC Microbiol. 5, 1.
doi:10.1186/1471-2180-5-1
Vyas, V.K., Barrasa, M.I., Fink, G.R., 2015. A Candida albicans CRISPR system permits
genetic engineering of essential genes and gene families. Sci. Adv. 1, e1500248–e1500248.
doi:10.1126/sciadv.1500248
Wartenberg, D., Vödisch, M., Kniemeyer, O., Albrecht-Eckardt, D., Scherlach, K., Winkler,
R., Weide, M., Brakhage, A. a, 2012. Proteome analysis of the farnesol-induced stress
response in Aspergillus nidulans-The role of a putative dehydrin. J. Proteomics 75, 4038–49.
doi:10.1016/j.jprot.2012.05.023
Zarnack, K., Maurer, S., Kaffarnik, F., Ladendorf, O., Brachmann, A., Kämper, J.,
Feldbrügge, M., 2006. Tetracycline-regulated gene expression in the pathogen Ustilago
maydis. Fungal Genet. Biol. 43, 727–738. doi:10.1016/j.fgb.2006.05.006
Zhang, J., Mao, Z., Xue, W., Li, Y., Tang, G., Wang, A., Zhang, Y., Wang, H., 2011. Ku80
gene is related to non-homologous end-joining and genome stability in Aspergillus niger.
Curr. Microbiol. 62, 1342–1346. doi:10.1007/s00284-010-9853-5

- 150 -
Supplementary Table S1: Expression vectors and split markers designed and used in this study.
Plasmid/ split
marker (SM)
Description Source
pVG4.1 Tet-on PgpdA::rtTA2S-M2::TcrgA::TetO7::Pmin::mluc::Ttrpc (Meyer et al.,
2011)
p473 synthetic tTA transactivator (Vogt et al.,
2005)
pMA247 Exchange rtTA with tTA from p473 in pVG4.1, by digestion with
EcoRI-BamHI in 2 steps, because of the extra BamHI site in
pVG4.1.
Southern probe for pyrG was amplified with
5`TCTCGCGCAGAAGCACAACT and
3`GCAGCCTGCACCGGATCG.
This work
pSB1.1 pMA247 was digested with AscI (flanked pyrG*), the linear
DNA without the pyrG* sequence ligated again.
This work
pTet-off®
Advanced
Vector
synthetic tTA2S transactivator Clontech
pFW9.3 pVG4.1 was digested with BamHI (2 restrictions sites) and
EcoRI, two linear fragments without rtTA-M2 was cloned
together with cutted tTA2S
from pTet-off® Advanced Vector
(BamHI/ EcoRI) in a three-way ligation.
To proof in the genome the recombination event between PgpdA
and Pmin 5` TTCCTGCTCTCCCCACCAG (in PgpdA) and 3`
TGTCCACCTCGATATGTGCA (in mluc) were used.
This work
pFW15.1 pFW9.3 was digested with EcoRI and BspLUII, the backbone
was used for Gibson cloning. And also the promoter of fraA
(1000 bp in front of An16g04690) was amplified from AB4.1
genome with 5` CCCTCGGCTGGTCTGTCTTA and 3`
agtctagacatggtgaattcTTTGGCGGTTTGTTGCTGGC and
additionally a PCR part which was amplified with 5`
TTTTGCTGGCCTTTTGCTCA and 3`
taagacagaccagccgagggAAGCTTATCGATACCGTCGA from
pFW9.3 as template.
To proof in the genome the recombination event between PfraA
and Pmin 5` CCCTCGGCTGGTCTGTCTTA (in PfraA) and 3`
TGTCCACCTCGATATGTGCA (in mluc) were used.
This work
pAT1.11 pFW9.3 was digested with EcoRI and BspLUII, the backbone
was used for Gibson cloning. And also the Syer of gpdAn (1000
bp in front of An16g01830) was amplified from AB4.1 genome
with 5`TAAGAATGGGGAAGGCGAAG and 3`
cagtctagacatggtgaattcTGTTTAGATGTGTCTATGTG and
additionally a PCR part which was amplified with 5`
TTTTGCTGGCCTTTTGCTCA and 3`
cttcgccttccccattcttaAGCTTATCGATACCGTCGAC from
pFW9.3 as template.
To proof in the genome the recombination event between
PgpdAn and Pmin 5` TAAGAATGGGGAAGGCGAAG and 3`
TGTCCACCTCGATATGTGCA (in mluc) were used.
This work
pFW17.1 With PmeI cut out the backbone (8138 bp) of FW15.1 and
amplified racA with 5`
gacatcaccgtttaaacaccATGGCCACTGGTCCAGCT and 3`
tcggcatctactgtttaaacCTACAGAATCACGCATTTCTTGTTCT
from AB4.1 genome and Gibson-cloned together.
This work

- 151 -
pFW18.1 With PmeI cut out the backbone (8138 bp) of FW15.1 and
amplified racAG18V
with 5`
gacatcaccgtttaaacaccATGGCCACTGGTCCAGCT and 3`
tcggcatctactgtttaaacCTACAGAATCACGCATTTCTTGTTCT
from MA61.24 genome and Gibson-cloned together.
This work
pAN7.1 PgpdA::hph (hygromycin resistence)::Ttrpc (Punt et al.,
1987)
SM 1 and
SM2 for
∆racA
Construction of 2 split markers for the deletion of racA. For split
marker 1 we amplified GOI 5` of racA with
5`AGCAGCAGCAGCAACACTAA and 3`
ccagaaagagtcaccggtcaGTCGAATTGAGGCGAGG from
AB4.1genome and the hygromycin resistance with
5`aagccgctgctggaattgGGCTCTGAGGTGCAGTGGAT and
cgatggataattgtgccgtgTTGGGTGTTACGGAGCATTCA from
pAN7.1. Following we fusioned GOI 5` and hph together with a
PCR with 5`ACCTGTCCAGTGGCTATCTT and 3´
GAAATTGCCGTCAACCAA. With the same approach we
constructed the splitmarker 2, we amplified GOI 3` part with 5`
acgagactgaggaatccgctGCCAAACCGAAGAACAAGAA and 3`
CAACTACGACCGCATGAAGA from AB4.1 genome and
fusioned it together with hph in a PCR with
5`AGAGCCTGACCTATTGCATCT and 3`
CAACTACGACCGCATGAAGA . By recombination of the two
parts of the selection marker and homologous integration of the
cassette in the genome, a successful gene deletion mutant can be
obtained. Probe for the promoter was amplified with 5` ACCTGTCCAGTGGCTATCTTT and
3`ccagaaagagtcaccggtcaGTCGAATTGAGGCGAGGG and for
the terminator with 5` acgagactgaggaatccgctGCCAAACCGAAGAACAAGAA and 3` CAACTACGACCGCATGAAGA.
This work
pFW19.1 With PmeI cut out the backbone (8138 bp) of FW15.1 and
amplified racA with 5`
gacatcaccgtttaaacaccATGTGGTATGTATGGCTCCAAAG and
3` gtcggcatctactgtttaaacGCTCTCTATTCAACAGTAACCGAC
from AB4.1 genome and Gibson-cloned together.
This work
SM 1 and
SM2 for
∆gfaA
Construction of 2 split markers for the deletion of gfaA. For
splitmarker 1 we amplified GOI 5` of gfaA with
5`AGCAGGTCACCACTACCATC and 3`
caattccagcagcggctATGTGATTACTCGGAGGCGT from AB4.1
genome and the hygromycin with
5`aagccgctgctggaattgGGCTCTGAGGTGCAGTGGAT and
3`cgatggataattgtgccgtgTTGGGTGTTACGGAGCATTCA from
pAN7.1. Following we fusioned GOI 5` and hph together with a
PCR with 5`AGCAGGTCACCACTACCATC and
3`GGCGTCGGTTTCCACTATC. With the same approach we
constructed the splitmarker 2, we amplified GOI 3` part with
5`acacggcacaattatccatcgGTGGGCACGAGACTGGGA and
3`ATCTGGGAAGCCGCGTATAA from AB4.1 genome and
fusioned it together with hph in a PCR with
5`AAAGTTCGACAGCGTCTCC and
3`ATCTGGGAAGCCGCGTATAA. By recombination of the
two parts of the selection marker and homologous integration of
the cassette in the genome, a successful gene deletion mutant can
be obtained. Probe for the promoter was amplified with
5`AGCAGGTCACCACTACCATC and 3`
caattccagcagcggctATGTGATTACTCGGAGGCGT and for the
This work

- 152 -
terminator with 5`
acacggcacaattatccatcgGTGGGCACGAGACTGGGA and 3`
ATCTGGGAAGCCGCGTATAA.
pCH008 Tet-on module ptpiA::rtTA2
S-M2::cgrA
t-tetO-
pmin (Helmschrott et
al., 2013)
pUHT61-1 synthetic tTA2 transactivator (Urlinger et al.,
2000)
pSK606 Tet-off module ptpiA::tTA2::cgrA
t-tetO-
pmin: 800 bp PCR
amplicon with tTA2 sequence from pUHT61-1 assembled with
540 bp BstBI/XbaI fragment from pCH008 carrying tpiA
promoter assembled in SpeI/PstI vector pCH008 backbone
This work
pSK607 Tet-off::pabaA conditional promoter replacement cassette:
assembly of 1.5 kb 5' pabaA flanking region, 4.1 kb SfiI
fragment of pSK606, and 1.5 kb pabaA cds and 3' flanking
region in pUC19
This work
pXM1.1 With PmeI cut out the backbone (7818 bp) of pVG4.1 and luc
sequence (1741 bp) was amplified from pFH62 (Cesbron et al.
2013). Primer 5`
ttgagcagacatcaccgtttaaacaccATGGAGGACGCCAAGAACA
and
3`ccggtcggcatctactgtttaaacttaGAGCTTGGACTTGCCGCCCT
were used, with a stop codon encoded in the 3’ region of primer.
Backbone and PCR product were fusioned by Gibson cloning.
This work
pFW20.1 With PmeI cut out the backbone (7818bp) of pVG4.1 and luc-
PEST sequence (1879 bp) was amplified from pFH62 (Cesbron
et al. 2013) with primers 5`
ttgagcagacatcaccgtttaaacaccATGGAGGACGCCAAGAACA
and 3`
atcccggtcggcatctactgtttaaacTTAGACGTTGATCCTGGCGCT
and fusioned by Gibson cloning.
This work
pFW21.8 With PmeI cut out the backbone (8138bp) of pVG15.1 and luc-
PEST sequence (1879 bp) was amplified as describe above (see
pFW20.1) and fusioned by Gibson cloning.
This work
pFW22.1 pFW20.1 was digested with EcoRI and BspLUII, the backbone
(8589 bp) was used for Gibson cloning together with the same
two PCR products as for construction of pFW15.1.
To proof in the genome the recombination event between PfraA
and Pmin 5` CCCTCGGCTGGTCTGTCTTA (in PfraA) and 3`
CTCGAAGTACTCGGCGTAGG (in luc-PEST) were used.
This work
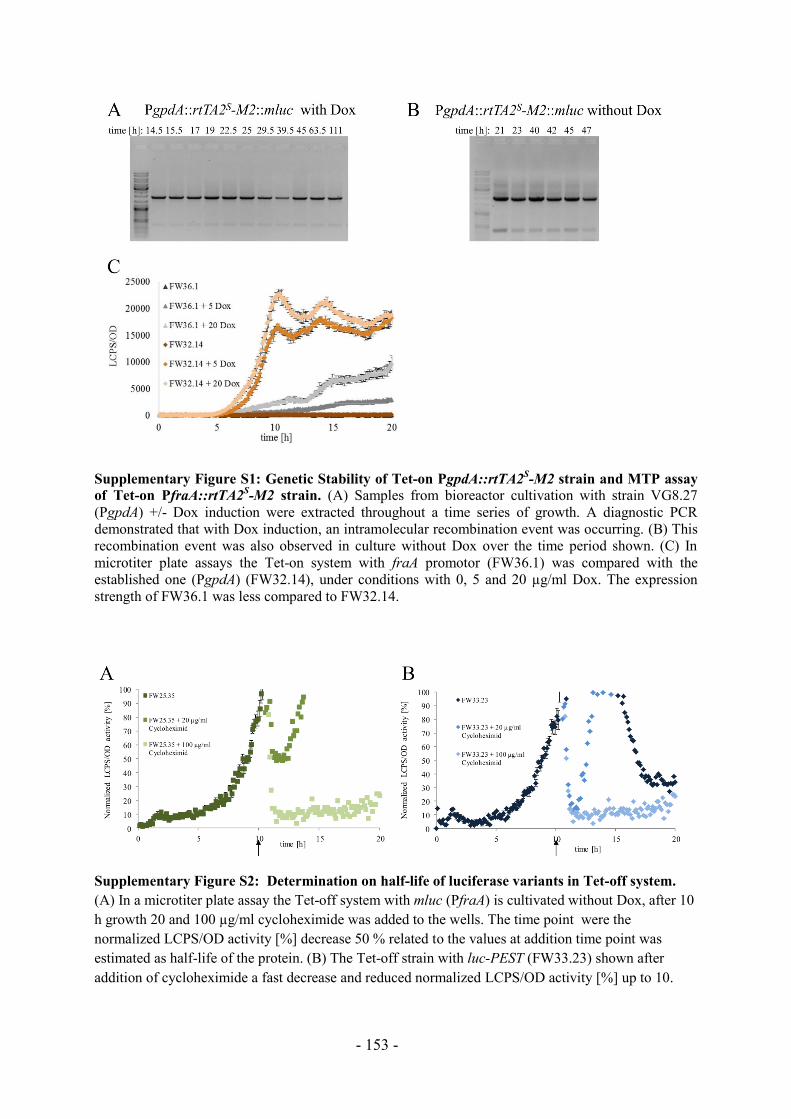
- 153 -
Supplementary Figure S1: Genetic Stability of Tet-on PgpdA::rtTA2S-M2 strain and MTP assay
of Tet-on PfraA::rtTA2S-M2 strain. (A) Samples from bioreactor cultivation with strain VG8.27
(PgpdA) +/- Dox induction were extracted throughout a time series of growth. A diagnostic PCR
demonstrated that with Dox induction, an intramolecular recombination event was occurring. (B) This
recombination event was also observed in culture without Dox over the time period shown. (C) In
microtiter plate assays the Tet-on system with fraA promotor (FW36.1) was compared with the
established one (PgpdA) (FW32.14), under conditions with 0, 5 and 20 µg/ml Dox. The expression
strength of FW36.1 was less compared to FW32.14.
Supplementary Figure S2: Determination on half-life of luciferase variants in Tet-off system.
(A) In a microtiter plate assay the Tet-off system with mluc (PfraA) is cultivated without Dox, after 10
h growth 20 and 100 µg/ml cycloheximide was added to the wells. The time point were the
normalized LCPS/OD activity [%] decrease 50 % related to the values at addition time point was
estimated as half-life of the protein. (B) The Tet-off strain with luc-PEST (FW33.23) shown after
addition of cycloheximide a fast decrease and reduced normalized LCPS/OD activity [%] up to 10.

- 154 -
Kapitel 5
Highly active promoters and native secretion signals for protein production
during extremely low growth rates in Aspergillus niger

- 155 -
Highly active promoters and native secretion signals for protein production
during extremely low growth rates in Aspergillus niger
Franziska Wanka1, Mark Arentshorst
2, Timothy Cairns
1, Thomas Jørgensen
3, Arthur F.J.
Ram2, Vera Meyer
1
1 Berlin University of Technology, Institute of Biotechnology, Department Applied and
Molecular Microbiology, Gustav-Meyer-Allee 25, 13355 Berlin, Germany
2 Leiden University, Institute of Biology Leiden, Department Molecular Microbiology and
Biotechnology, Sylviusweg 72, 2333 BE Leiden, The Netherlands
3 Protein Expression, Novo Nordisk, Novo Nordisk Park, 2760 Måløv, Denmark
*Address correspondence to: Prof. Dr.-Ing. Vera Meyer
Department Applied and Molecular Microbiology
Institute of Biotechnology
Berlin University of Technology
Gustav-Meyer-Allee 25
13355 Berlin
Germany
Email: [email protected]
Keywords: perfusion cultivation, zero growth rate, antifungal protein, hydrophobin, promoter,
Aspergillus niger
Email addresses:

- 156 -
Abstract
Background: The filamentous ascomycete Aspergillus niger is used in many industrial
processes for the production of enzymes and organic acids by batch and fed-batch cultivation.
An alternative technique is continuous cultivation, which promises improved yield and
optimized pipeline efficiency.
Results: In this work, we have used perfusion (retentostat) cultivation to validate two promoters that
are suitable for A. niger continuous cultivation of industrially relevant products. Firstly, promoters of
genes encoding either an antifungal protein (Panafp) or putative hydrophobin (PhfbD) were confirmed
as active throughout retentostat culture by assessing mRNA and protein levels using a luciferase
(mluc) reporter system. This demonstrated the anafp promoter mediates a high but temporally
variable expression profile, whereas the hfbD promoter mediates a semi-constant, moderate-
to-high protein expression during retentostat culture. In order to assess whether these
promoters were suitable to produce heterologous proteins during retentostat cultivation, the
secreted antifungal protein (AFP) from Aspergillus giganteus, which has many potential
biotechnological applications, was expressed in A. niger during retentostat cultivation.
Additionally, this assay was used to concomitantly validate that native secretion signals
encoded in anafp and hfbD genes can be harnessed for secretion of heterologous proteins. Afp
mRNA and protein abundance were comparable to luciferase measurements throughout
retentostat cultivation, validating the use of Panafp and PhfbD for perfusion cultivation.
Finally, a gene encoding the highly commercially relevant thermal hysteresis protein (THP)
was expressed in this system, which did not yield detectable protein.
Conclusion: Both hfbD and anafp promoters are suitable for production of useful products in
A. niger during perfusion cultivation. These findings provide a platform for further
optimisations for high production of heterologous proteins with industrial relevance.
Background
An inherent component of the saprophytic lifestyle of the filamentous mould A. niger is the
ability to secrete large amounts of enzymes into its environment, which has been harnessed in
biotechnological pipelines for the efficient production of platform chemicals and industrial
proteins. In recent years, improved morphological [1], genetic [2], metabolic [3], and systems
biological tools [4, 5] offer improved efficiency and tractability of A. niger in industrial
applications.
However, innovations specifically tailored to improving bioprocess strategies have been
limited. Currently, approximately 90 % of industrial biotechnological cultivations rely on
batch or fed-batch culture [6], which is often inefficient as organisms have short periods of

- 157 -
high product biosynthesis, and there is considerable manufacture downtime for technical
reasons, such as equipment sterilization. Additionally, fed-batch or batch cultivation can
result in inconsistent product quality (e.g. multiple glycosylation variants) because of the
disparities in medium environment [7].
An alternative and potentially useful strategy for biotechnological manufacture is continuous
processing. Chemostats, in which fresh medium is continually added to a bioreactor, and
effluent containing metabolite products, used medium, and microbial biomass continually
removed, enables steady state microbial growth. Accordingly, optimal growth rates for
product biosynthesis can be maintained, and the period of product biosynthesis increased
when compared to batch cultivation [8].
A modification of conventional chemostat cultivation is termed perfusion or retentostat
cultivation, in which microbial biomass is retained in the bioreactor. Consequently, in
retentostat cultivation, microbial biomass increases to a maximum biomass, after which
available nutrients are sufficient for maintenance of cell viability, and growth rates approach
zero. Additionally, perfusion cultivation has several advantages to conventional steady state
chemostat cultivation. Firstly, extremely low microbial growth may increase available
metabolic energy for product biosynthesis, thus potentially improving product yield.
Secondly, many microbial secondary metabolite products are only produced during phases of
low or zero growth, and accordingly novel products or those previously recalcitrant to batch
or fed-batch cultivation might be amenable to retentostat biosynthesis. Another advantage of
this cell retention cultivation mode is the continuous removal of toxic or growth inhibitory
products and/or the production of unstable products, which cannot remain stable in a batch or
fed-batch culture due to inherent sensitivities to proteases or other degradative enzymes.
Additionally, this kind of cultivation enables continuous product monitoring and prompt
downstream processing of secreted metabolites or enzymes. A major advantage is the high
productivity in small-scale bioreactors, which save money, space, and allow an easier scale up
process. Accordingly, expanding the cultivation tool-kit of A. niger to include perfusion
cultivation is an important goal in biotechnology.
Currently, a significant technical challenge to the development of efficient A. niger perfusion
cultivation is the absence of suitable promoter systems. For example, conventionally used
promoters for high expression in industrial systems (e.g. the glucoamylase promoter PglaA)
show prohibitively low expression activity at growth rates which are close to zero [9].
Discovery of promoters with high activity at ultralow growth rates is a prerequiste for A. niger
retentostat cultivation of useful products. Several other factors for optimal promoter

- 158 -
functionality include activity in the absence of an inducer for simple recovery of desired
product from culture medium, and continual promoter activity over a maximal time-period.
The objective of the study was to identify and validate A. niger promoters suitable for
retentostat cultivation, and provide proof of principle for retentostat biosynthesis of
heterologous proteins with potential biotechnological applications. Accordingly, from a
previous transcriptomic analysis of A. niger retentostat culture [9], we rationally selected
promoter regions of two genes, one encoding the A. niger antifungal protein (ANAFP;
An07g01320), and the other encoding a putative hydrophobin (HFBD; An08g09880), both of
which had high transcript abundance and strong supporting evidence of gene transcription
during low A. niger growth [9]. Using a luciferase reporter for highly accurate readouts of
promoter activity, we validated that both these promoters resulted in high heterologous gene
transcription and protein translation during retentostat culture. In order to provide proof of
principle that: (i) this platform can be used for cultivation of heterologous proteins with
biotechnological applications, and (ii) native secretion signals encoded in these genes are
useful for protein secretion during cultivation, both promoters and native secretion signals
were used to express a gene encoding an A. giganteus antifungal protein, which has promising
applications as a novel therapeutic in agriculture and the clinic. Lastly, a gene encoding the
highly commercially relevant thermal hysteresis protein (THP) from Choristoneura
fumiferana isoform 337 [10] was expressed during perfusion culture, which was unsuccessful
for protein production, which we hypothesise is due to poor codon usage of this gene or
proteolytic degradation.
Results and discussion
Establishment of zero growth active promoters in A. niger
We have previously interrogated global transcriptional changes of A. niger wild type strain
N402 during retentostat cultivation, which identified a striking transcriptional increase for
genes encoding putatively secreted, small molecular weight, cysteine-rich proteins during
cultivation relative to batch cultivation controls [9]. Accordingly, promoters from these genes
were hypothesized to be good candidates for maintaining high expression of heterologous
genes during retentostat cultivation. From this subset we rationally selected promoters
upstream of: (i) a gene encoding the A. niger antifungal protein (An07g01320), which has
been previously characterized as both expressed and translated during periods of low A. niger
growth [9], [11], and (ii) a gene encoding a putative hydrophobin (An08g09880), for which

- 159 -
expression levels from global transcriptional profiling were validated by Northern blot probe,
thus giving high confidence of expression throughout retentostat culture [9].
To validate the functionality of the promoter regions for production of heterologous proteins,
we generated DNA cassettes that replaced coding sequences for An08g09880 or An07g01320
at the native genomic locus with one of three genes (Fig. 1). DNA cassettes with the mluc
gene encoding a luciferase protein were used for facile intracellular reporters of heterologous
gene transcription and translation (Fig. 1A). Additionally, in order to demonstrate retentostat
cultivation using these promoters could produce proteins of industrial relevance, AFP and
THP encoding genes were also cloned into An07g01320 or An08g09880 loci. These DNA
cassettes were also designed to validate whether native signal sequence of ANAFP or HFBD
encoding genes can be harnessed for efficient secretion of heterologous proteins. Accordingly,
DNA cassettes pFW4.4 (pEN1) and pFW2.52 (pEN2) contained codons encoding 34 and 22
amino acids of the predicted native secretion signal of An07g01320 and An08g09880
respectively at the N-terminus of the protein of interest (Fig. 1 B, C).
Figure 1: Schematic diagram depicting plasmids utilized to test activity of promoters during
extremely low A. niger growth. For plasmids pCH8.1, pFW4.4 and pEN1, the promoter for the
antifungal protein from A. niger (Panafp) and the corresponding terminator (Tanafp) were used. In the
other three plasmids (pPK4.1, pFW2.52, pEN2,) the promoter PhfbD and terminator ThfbD are
utilized. These ~1000 bp promoter and terminator regions ensure homologous integration of
exogenous DNA cassettes at the respective native locus via a double crossover event. In all plasmids,
termination of transcription of the gene of interest was attained using the terminator of tryptophan
synthase of Aspergillus nidulans, Ttrpc. All plasmids utilized the short version of pyrG (spyrGAO) for
selection of transformants, with exception of pFW2.52, which encoded the hygromycin resistance
gene (hygR). A) For facile intracellular reporting of promoter activity, plasmids CH8.1 and PK4.1
encode the modified luciferase gene mluc [16]. B) Plasmids FW4.4 and FW2.52 contain the afp gene
encoding the A. giganteus antifungal protein and additionally the signal sequence for secretion of the
respective genes (SSanafp and SShfbD). C) For expression of the thermal hysteresis protein gene from

- 160 -
the spruce budworm C. fumiferana, the codon optimized (for A. niger) thp gene was utilized in
plasmids EN1 and EN2.
In order to accurately assay biosynthesis of A. giganteus AFP at the hfbD locus, it was first
necessary to delete the gene encoding A. niger AFP (An07g01320) as this polypeptide would
likely be co-purified with heterologously expressed AFP (MA170.27, Table 1). This Δanafp
isolate was then used as a recipient strain for heterologous expression of A. giganteus AFP
following transformation with pFW2.52, with this latter transformation utilizing a
hygromycin gene as selection marker (Fig. 1 B). All other plasmids utilized the short version
of orotidine-5'-decarboxylase gene from Aspergillus oryzae (spyrGAO) as a selection marker.
All strains used in this study are described in Table 1.
Table 1: Aspergillus strains used in this study.
Retentostat cultivation
A.niger isolates expressing DNA cassettes (Table 1) were cultivated under retentostat
conditions for 13 days in 5 L bioreactors. These data were compared to wild type progenitor
isolate N402 grown under perfusion cultivation conditions [9]. Biomass and specific growth
rates (µ) were calculated throughout the time series of cultivation at 12 hour intervals (Fig. 2).
After five days (+/- 12 hours), specific growth rate of all fungal isolates approached zero
strain genotype source
MDH18894 A. giganteus wild type [12]
N402 A. niger wild type [13]
AB4.1 A. niger pyrG- isolate [14]
PK2.9 AB4.1 transformed with pCH8.1 (Panafp::mluc), pyrG+ This work
PK4.3 AB4.1 transformed with pPK4.1 (PhfbD::mluc), pyrG+ This work
FW23.7 AB4.1 transformed with pFW4.4 (Panafp:: SSanafp::afp),
pyrG+
This work
MA170.27 AB4.1 transformed with PCR product of
pCH3.3(Panafp::spyrGAO::Tanafp) → Δanafp, pyrG+
This work
FW6.6 MA170.27 transformed with pFW2.52 (PhfbD::SShfbD::afp),
hygR+, pyrG
+
This work
MA237 AB4.1 transformed with pEN1 (Panafp::SSanafp::thp), pyrG+ This work
MA238 AB4.1 transformed with pEN2 (PhfbD::SShfbD::thp), pyrG+ This work
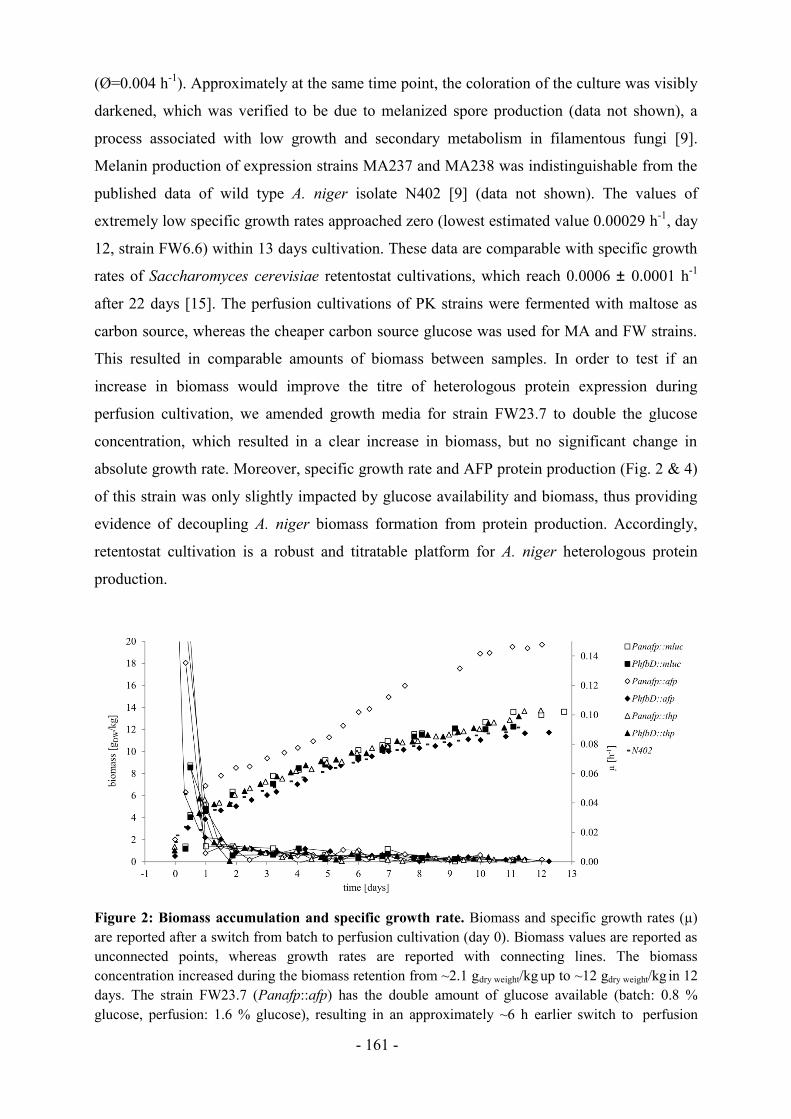
- 161 -
(Ø=0.004 h-1
). Approximately at the same time point, the coloration of the culture was visibly
darkened, which was verified to be due to melanized spore production (data not shown), a
process associated with low growth and secondary metabolism in filamentous fungi [9].
Melanin production of expression strains MA237 and MA238 was indistinguishable from the
published data of wild type A. niger isolate N402 [9] (data not shown). The values of
extremely low specific growth rates approached zero (lowest estimated value 0.00029 h-1
, day
12, strain FW6.6) within 13 days cultivation. These data are comparable with specific growth
rates of Saccharomyces cerevisiae retentostat cultivations, which reach 0.0006 ± 0.0001 h-1
after 22 days [15]. The perfusion cultivations of PK strains were fermented with maltose as
carbon source, whereas the cheaper carbon source glucose was used for MA and FW strains.
This resulted in comparable amounts of biomass between samples. In order to test if an
increase in biomass would improve the titre of heterologous protein expression during
perfusion cultivation, we amended growth media for strain FW23.7 to double the glucose
concentration, which resulted in a clear increase in biomass, but no significant change in
absolute growth rate. Moreover, specific growth rate and AFP protein production (Fig. 2 & 4)
of this strain was only slightly impacted by glucose availability and biomass, thus providing
evidence of decoupling A. niger biomass formation from protein production. Accordingly,
retentostat cultivation is a robust and titratable platform for A. niger heterologous protein
production.
Figure 2: Biomass accumulation and specific growth rate. Biomass and specific growth rates (µ)
are reported after a switch from batch to perfusion cultivation (day 0). Biomass values are reported as
unconnected points, whereas growth rates are reported with connecting lines. The biomass
concentration increased during the biomass retention from ~2.1 gdry weight/kg up to ~12 gdry weight/kg
in 12
days. The strain FW23.7 (Panafp::afp) has the double amount of glucose available (batch: 0.8 %
glucose, perfusion: 1.6 % glucose), resulting in an approximately ~6 h earlier switch to perfusion

- 162 -
cultivation, and a final biomass after 12 days of ~20 gdry weight/kg. The right diagram axis shows strain
specific growth rate, which decreased within two days of changing cultivation conditions and reached
values close to zero. For each strain one single bioreactor run was performed. N402 data taken from
[9].
Analysis of mRNA and luminescence levels of a luciferase reporter demonstrate high
activity of anafp and hfbD promoters during perfusion cultivation
For comprehensive analysis of anafp and hfbD promoter activity throughout retentostat
cultivation, we measured both mRNA and protein abundance for an intracellular reporter
gene, mluc [16], using Northern blot probes and luciferase activity, respectively. This
experiment highlighted a general and significant technical challenge for estimating mRNA
transcript abundance during ultralow fungal growth, i.e. the absence of constitutively active
housekeeping genes for normalization of mRNA levels for genes of interest. During
retentostat culture, transcripts from actin, histone 2B (H2B), 18S and 28S ribosomal encoding
genes were tested using Northern blot probes, which demonstrated a clear reduction in
transcript abundance throughout the experiment as exemplarily shown in Figure 3A, 4A and
5, an observation supported by global transcriptional profiling as previously reported for A.
niger perfusion cultivations [9]. Appropriate reference genes for mRNA normalization for
industrially important fungi is highly complex and dependent on the experimental context
[17]. Accordingly, we report mRNA abundance at each time point normalized to the highest
measurement throughout the experiment, reasoning that this will enable interpretation of
relative temporal changes of mRNA abundance throughout this experiment.
mRNA levels demonstrated that mluc transcripts were highest for both anafp and hfbD
promoters at day 4 (Fig. 3A), at which time the specific growth rates of strains PK2.9 and
PK4.3 were approaching zero (0.007 h-1
, respectively). Encouragingly, this suggested that
decoupling biomass accumulation and heterologous protein expression had been achieved in
this system.
This was broadly supported by analysis of protein expression, which for strain PK2.9 Panafp
showed a peak in luciferase activity at day 4 (1,200,000 luminescent counts per second
(LCPS)/ optical density (OD),) which dropped to relatively lower, but still detectable
luciferase values (34,000 LCPS/OD) by day 8 (Fig. 3B). In strain PK4.3 with PhfbD,
luciferase values were highest at day 7 (400,000 LCPS/OD), and were detectable at 40,000
LCPS/OD by day 11 (Fig. 3B). While these data demonstrate promoters anafp and hfbD are
not constitutively active throughout retentostat culture, greater LCPS/OD values were
achieved in this system than during conventional batch cultivation of A. niger strain
expressing mluc under control of a titratable, tetracycline inducible expression system (at 41
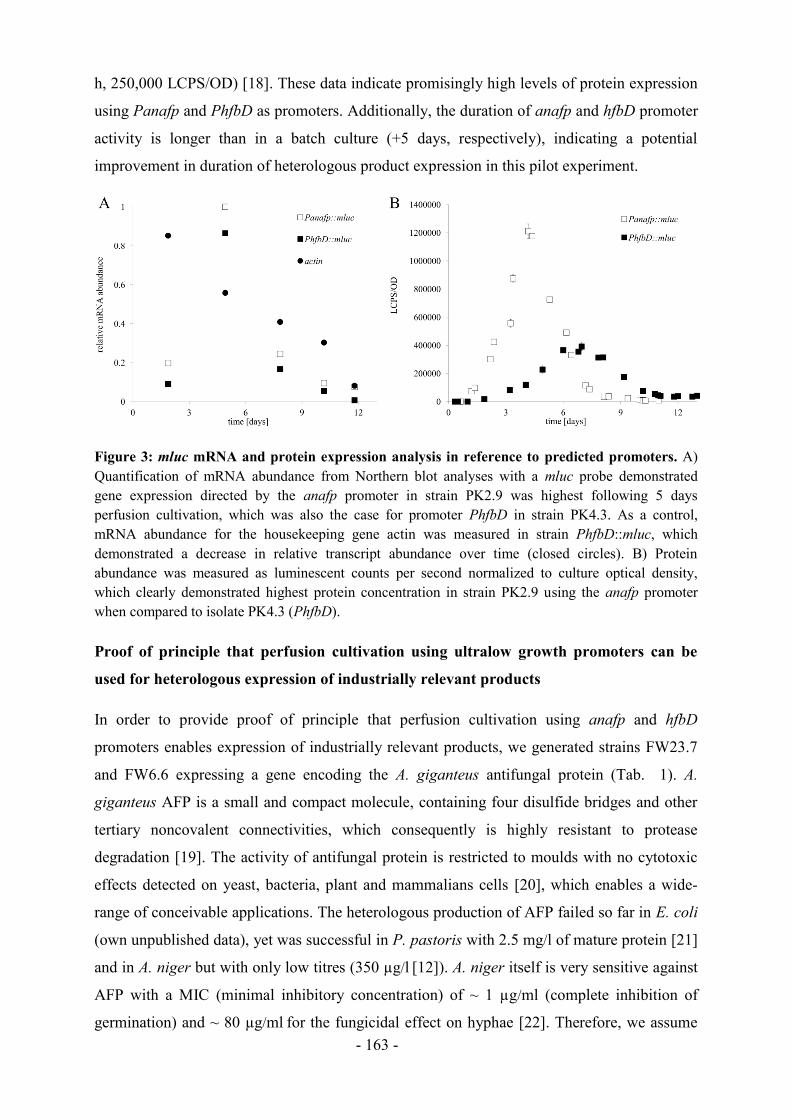
- 163 -
h, 250,000 LCPS/OD) [18]. These data indicate promisingly high levels of protein expression
using Panafp and PhfbD as promoters. Additionally, the duration of anafp and hfbD promoter
activity is longer than in a batch culture (+5 days, respectively), indicating a potential
improvement in duration of heterologous product expression in this pilot experiment.
Figure 3: mluc mRNA and protein expression analysis in reference to predicted promoters. A)
Quantification of mRNA abundance from Northern blot analyses with a mluc probe demonstrated
gene expression directed by the anafp promoter in strain PK2.9 was highest following 5 days
perfusion cultivation, which was also the case for promoter PhfbD in strain PK4.3. As a control,
mRNA abundance for the housekeeping gene actin was measured in strain PhfbD::mluc, which
demonstrated a decrease in relative transcript abundance over time (closed circles). B) Protein
abundance was measured as luminescent counts per second normalized to culture optical density,
which clearly demonstrated highest protein concentration in strain PK2.9 using the anafp promoter
when compared to isolate PK4.3 (PhfbD).
Proof of principle that perfusion cultivation using ultralow growth promoters can be
used for heterologous expression of industrially relevant products
In order to provide proof of principle that perfusion cultivation using anafp and hfbD
promoters enables expression of industrially relevant products, we generated strains FW23.7
and FW6.6 expressing a gene encoding the A. giganteus antifungal protein (Tab. 1). A.
giganteus AFP is a small and compact molecule, containing four disulfide bridges and other
tertiary noncovalent connectivities, which consequently is highly resistant to protease
degradation [19]. The activity of antifungal protein is restricted to moulds with no cytotoxic
effects detected on yeast, bacteria, plant and mammalians cells [20], which enables a wide-
range of conceivable applications. The heterologous production of AFP failed so far in E. coli
(own unpublished data), yet was successful in P. pastoris with 2.5 mg/l of mature protein [21]
and in A. niger but with only low titres (350 µg/l [12]). A. niger itself is very sensitive against
AFP with a MIC (minimal inhibitory concentration) of ~ 1 µg/ml (complete inhibition of
germination) and ~ 80 µg/ml for the fungicidal effect on hyphae [22]. Therefore, we assume

- 164 -
that the retentostat mode is a useful cultivation, due to the high dilution of the secreted toxic
products, and the fact that the chosen promoters are not active during germination or
exponential growth phase.
In order to confirm that mRNA decrease of housekeeping genes over the period of zero
growth conditions was not due to an artefact of Northern blot analysis, we measured mRNA
transcript abundance of h2B (Fig. 4A) and gpdA (data not shown) genes with qRT-PCR,
which confirmed mRNA transcript of these genes decreased throughout perfusion
fermentation. mRNA measurements of afp transcripts in FW23.7 (Panafp::afp) and FW6.6
(PhfbD::afp) using qRT-PCR revealed comparable temporal expression patterns to mluc,
indicating promoter activity was consistent between the different expression cassettes (Fig.
4A). In effluent extracted throughout perfusion cultivation, AFP protein was detectable using
an anti-AFP antibody at a maximum of 0.065 mgAFP/gDW (using promoter anafp) and 0.03
mgAFP/gDW (using promoter hfbD) estimated in comparison to the positive control of purified
A. giganteus AFP (see Methods section). The calculated highest AFP production rate of
FW23.7 (Panafp) it was 0.8 mg/l/d at day 5 (µ = 0.004 h-1
), and for FW6.6 (PhfbD) was 0.08
mg/l/d from day 3 until 4, which corresponds to µ = 0.006 h-1
. These differences may be due
to improved functionality of the Panafp promoter, increased production of AFP under higher
glucose concentrations used in growth media for strain FW23.7, or a combination of these
factors.
In order to validate that heterologous expression of AFP using A. niger resulted in protein
with potential biotechnological applications, heterologously produced AFP from strain FW6.6
(day 9) was purified via FPLC and tested for antifungal activity. 94 % growth inhibition of A.
niger was achieved using 10 µg/ml heterologously expressed AFP, which is comparable to
AFP purified from A. giganteus (100 % growth inhibition using 10 µg/ml) [22]. These data
demonstrate that heterologously produced AFP is biologically active. The AFP yield using A.
niger perfusion cultivation was 650 µg/l (FW23.7, day 5), which is a notable improvement
when compared to previous AFP production using A. niger in shake flask culture (~350 µg/l)
[12]. While shake flask production is not suitable for industrial applications, these data
suggest that further optimization of perfusion fermentation is warranted. Elsewhere, groups
using P. pastoris for AFP production have achieved yields of 2.5 mg/l [21]. However, all
heterologous expression attempts are low in comparison to the homologous production host A.
giganteus (30 - 40 mg/l), indicating further optimisation of all current systems is required for
commerical use of AFP. Nevertheless, the significantly increased AFP yield reported here
when compared to conventional purifications demonstrates perfusion cultivation is a
promising technique which warrants future optimisation.

- 165 -
Figure 4: afp mRNA and protein expression analysis in reference to predicted promoters. A) afp
mRNA levels, pictured in diamonds, were measured using qRT-PCR throughout a time period of
perfusion cultivation from strain FW23.7 (Panafp::afp) and FW6.6 (PhfbD::afp). Reported standard
deviation resulted from a minimum of two and maximum of three measurements per sample. These
data support temporal expression profiles reported for mluc gene, with afp expression highest at day 5
for both Panafp and PhfbD. As a control the housekeeping gene h2B was measured in strain
PhfbD::afp, which demonstrated a decrease in relative transcript abundance over time (closed circles).
B) Protein abundance was quantified using Western blot (anti-AFP primary antibody) which
demonstrated that Panafp produced maximal 0.06 mgAFP/gdry weight correspond to 0.65 mgAFP/l. Standard
deviation was estimated through three Western blots per sample.
Taken together, these data are the first example of heterologous protein expression during
perfusion cultivation using a filamentous fungal cell factory, and validate that promoters
anafp and hfbD are sufficiently active for heterologous gene expression during ultralow
growth. In A. niger, refinement and expansion of the promoter tool-kit has been an ongoing
effort for nearly 30 years, with over 15 constitutive or inducible systems described [23].
Accordingly, discovery of two promoters that are functional for protein production during A.
niger perfusion cultivation is an important step which will facilitate future optimization of this
technique. With regards to use of native secretion signals anafp and hfbD, A. niger is a useful
cultivation system due to its ability to secrete post-translationally processed, active
recombinant proteins into cultivation media at high concentrations. Protein signal peptides are
therefore of critical importance as they enable translocation of useful enzymes through
cellular secretory machinery. Our data demonstrate that the anafp and hfbD signal sequences
encoded in pFW4.4 and pFW2.52 enable secretion of heterologous protein during ultralow
growth in perfusion cultivation.
Perfusion cultivation of THP protein from Choristoneura fumiferana
In order to test whether non Aspergillus spp. derived proteins with existing technological
applications could be heterologously expressed using the perfusion cultivation platform, we

- 166 -
generated strains expressing the THP protein from Choristoneura fumiferana isoform 337
[10]. This protein inhibits growth and recrystallisation of ice, lowers freezing temperature,
increases melting temperature, is used in the food industry to improve storage of frozen
products, and has potential applications in tissue preservation for organ transplants [24]. Fig.
5 depicts relative mRNA abundance as measured by Northern blot analysis of strain MA237
(Panafp::thp) and MA238 (PhfbD::thp) with thp probes, which show comparable temporal
expression profiles as those described for mluc and afp genes.
However, throughout perfusion cultivation, THP was not detected in the effluent. The
enrichment through a cooling finger [25] suggested that no active THP was secreted in the
medium, as the ice layer did not show the expected cloudiness from previously reported ice-
THP interactions, and further analysis in SDS-PAGE did not demonstrate protein bands of
predicted molecular mass (data not shown). To exclude the possibility of THP remaining
intracellular, proteins were extracted from mycelium and protein samples analysed by SDS-
PAGE which did not reproducibly result in a band of expected molecular mass. Where
putative bands were detected, samples were further analysed by
HPLC-ESI-MS, but these could not be confirmed as THP. Western blots with the mycelium
extracts using rabbit anti-THP antiserum [26] demonstrated a non-specific band of high
molecular mass (data not shown). We speculate that the THP protein, which is natively
expressed in lymph, is not suited for an extracellular expression in A. niger, or alternatively,
the secreted THP degraded through instability problems due to pH or protease sensitivity. It is
possible that further optimizations of the strain such as the use of protease-negative isolates
[27], or modification of medium pH, may result in detectable THP protein, which could be
tested in future experiments but is outside of the objectives of this work. We therefore
conclude that the codon-optimized THP is not amenable to extracellular expression in A.
niger.
Figure 5: thp mRNA analysis in reference to predicted promoters. A) Northern blot analysis of
mRNA extracted from strains MA237 (Panafp::thp) and MA238 (PhfbD::thp) during a time-course of

- 167 -
perfusion fermentation are shown, with probes for THP mRNA (upper panel) and actin mRNA
(middle panel). The exposure time was 6 hours. These data demonstrate thp mRNA is expressed in
this system using both anafp and hfbD promoters. The lower panel demonstrates representative
decrease of 18S and 28S rRNA on Northern blot membranes throughout the experiment time course,
which we hypothesize is due to decrease in cellular metabolism. B) Quantification of mRNA
abundance from Northern blot results of thp (A) in strain MA237 (Panafp::thp) and strain MA238
(PhfbD::thp). These data suggest highest mRNA expression after 4 days of perfusion cultivation in
isolate MA237, and approximately constitutive expression between days 4 until day 12 in isolate
MA238.
Comparison of both promoters and future optimisations
In general, measurements of mRNA and protein abundance throughout this study demonstrate
that gene expression using the antifungal protein promoter throughout perfusion fermentation
results in higher mRNA and translated protein at low growth rates (µ = 0.008 - 0.003 h-1
)
when compared to use of the hydrophobin promoter. While strains expressing genes under
control of the antifungal promoter were highest at day 5, values approach zero at the later time
periods, and gene expression is down-regulated to levels observed during batch fermentation.
In contrast, the hydrophobin promoter results in a lower, yet semi-constitutive expression
profile, with mRNA and protein abundance for mluc and afp genes remaining approximately
constant between days 4 and 10 (µ = 0.007 - 0.001 h-1
).
Analyses of protein abundance for mluc and afp genes expressed using both hfbD and afp
promoters suggests that ultralow A. niger growth achieved following ~10 days retentostat
culture results in reduced protein concentration, which we hypothesize is due to a reduction of
the energy supply necessary for protein translation and processing. Consequently, for optimal
protein biosynthesis, it may be necessary to keep absolute growth rates above a certain
threshold, for example µ > 0.001 h-1
for strains using PhfbD to drive gene expression. This is
supported by zero growth studies in multiple microorganisms, which suggests that calorie-
restricted conditions lead to down-regulation of genes involved in protein synthesis and
scarcity of ATP [28]. We conclude therefore that efficient production of biotechnology
products during zero growth rates may not be possible. One possibility for future optimization
would be to work with a promoter specific feeding rate to enable a constant glucose
concentration/growth rate in the bioreactor, which may result in more efficient protein
expression. Another potential improvement to this protocol would the use of non-sporulating
A. niger, e.g. the deletion mutant flbA or brlA [29], to maximise available energy for protein
synthesis, because produced biomass after day 4/5 consists of around 20 % melanin [9], and
the process of sporulation itself has been shown to inhibt protein secretion [29].

- 168 -
Conclusions
This study demonstrates that A. niger is suited for perfusion cultivation of heterologous
proteins, and describes an experimental platform for future optimization studies of this
technique. Using the well-established intracellular luciferase (mluc) system, promoters of two
genes were validated as highly active during carbon-starvation mode of chemostat culture.
Heterologous expression of the industrially relevant AFP provided the first proof of principle
that perfusion cultivation at ultralow growth is possible in A. niger. Moreover, it validated that
secretion signals of proteins ANAFP (An07g01320) and HFBD (An08g09880) enable
efficient secretion of heterologous proteins into the culture media, a prerequisite for
successful perfusion cultivation. The advancement of more efficient cultivation methods and
associated molecular tools is an important step towards future utilization of fungal continuous
expression systems in industrial biotechnology.
Methods
Construction of the plasmids and cloning.
Plasmid design for A. niger transformation followed a strategy whereby ~1000 bp of 5’ and 3’
flanking regions of genes (anafp An07g01320, hfbD An08g09880) facilitated gene
replacement by a double cross over event in the recipient strain AB4.1 or MA170.27
(plasmids maps made available on request). To analyse the expression profile of the two
promoters, mluc [16] was chosen as an intracellular reporter gene, with a trpC terminator
followed in the 3` orientation by the short version of pyrG from Aspergillus oryzae as a
selection marker (Fig.1). Further constructs for extracellular expression using anafp contain
additional signal sequences for secretion (presequence) and prosequence, whereas the hfbD
gene lacks a prosequence and accordingly only encodes a presequence. In the case of anafp,
the signal sequence, termed SSanafp, is encoded by 102 bp of DNA [20]. For efficient
secretion, a codon encoding a leucine amino acid was introduced immediately after the signal
peptide, which is absent in exogenous A. giganteus AFP but consistent with the initial amino
acid in mature A. niger AFP [30]. The signal sequence for gene hfbD, termed SShfbD consists
of a 66 bp DNA region [31], which for maximal secretion encodes an additional serine as the
first amino acid of heterologously expressed proteins. A gene encoding the thermal hysteresis
protein (THP) from Choristoneura fumiferana isoform 337 (AF263009) [32] was codon
optimised for gene expression in A. niger and obtained from GeneArt™, Germany. The afp
encoding gene from A. giganteus MDH 18894 was amplified with PCR from genomic DNA

- 169 -
and cloned immediately downstream of leucine and serine codons in anafp or hfbD regions
respectively. All cloning procedures utilised conventional endonuclease digestion and
ligation, after which reactions were transformed into competent Escherichia coli strain
TOP10. DNA constructs were verified through restriction endonuclease analysis and
sequencing.
A. niger transformation, strains and molecular techniques.
The transformation of A. niger isolates AB4.1 and MA170.27 and subsequent selection
procedures for pyrG+ or hygR
+ transformants respectively were performed using recently
described protocols [33]. Fungal chromosomal DNA isolation was extracted from
transformants following two rounds of selection purification, and positive transformants were
confirmed with diagnostic PCR and Southern Blot analysis for single integration of the
plasmids in the recipient strain. Depending on integration locus of the plasmid, DIG labelled
DNA probes were used in the Southern blot, which have a homologous sequence in Panafp
(5`:AGTACGACGAACTGCCGATA ,3`:AGTCGCTGAGATGTCGTTCA, product size:
510 bp) or in PhfbD (5`:GAGGCTGTGTATTTGGCGAG,
3`:CCTCTCATTACAGGCGGGAT, product size 498 bp). To confirm the absence of ectopic
integration of DNA constructs in the genome, at least two different restriction enzymes were
used in independent Southern blots. The A. niger strains used in this study are shown in Table
1. MA170.27 carried a deletion of the anafp gene, introduced through a double crossover with
a PCR product of pCH3.3 (Panafp::spyrGAO::Tanafp) (5`: TTCCCCTGCTCCTTAGGCAG,
3`: AATTTCGACTTGGTGGTTAG, product size: 4.053 kbp). Strains were routinely
cultivated on minimal medium (MM) plates containing 1x ASP + N (50x ASP + N: 3.5 M
NaNO3, 550 mM KH2PO4, 350 mM KCl, pH 5.5), 2 mM MgSO4, 1x trace elements solution
(modified from composition given by Vishniac and Santer [34], consisting of 1000x trace
elements solution (10 g of EDTA, 4.4 g of ZnSO4·7H2O, 1.01 g of MnCl2·4H2O, 0.32 g of
CoCl2·6H2O, 0.315 g of CuSO4·5H2O, 0.22 g of (NH4)6Mo7O24·4H2O, 1.47 g of CaCl2·2H2O,
and 1 g of FeSO4·7H2O)), 1.5% agar, and 1% glucose. Alternatively, complete medium (CM)
plates were used for routine culture, consisting of MM supplemented with 1% yeast extract
and 0.5% casamino acids.
RNA Extraction
For RNA extractions from frozen bioreactor mycelium, samples were flash frozen in liquid
nitrogen and ground using a pestle and mortar. The RNA was extracted with TRIzol® reagent

- 170 -
(Invitrogen) and analysed for quality in a spectrophotometer at an absorbance of 260 nm. For
northern blot analyses, 5 µg of RNA was used for glyoxal RNA gel electrophoresis and after
capillary blotting by means of radioactive hybridization probes the expression of mluc, thp,
actA (An15g00560), and h2B (An11g11310). 32
P-labeled primers used for the generation of
the probes can be found in the Additional File 1. For visualization and relative quantification
of northern blots, the image processing program (ImageJ) was used. A semi-quantitative
calculation of relative mRNA abundance was used. Firstly, the highest probe intensity
observed throughout the experiment was calculated. Next, this value was used to normalize all
other probe intensities, by dividing all probe intensities by the highest value. For the
transcription level investigation of the afp expressing strains, qRT-PCR was used. RNA was
extracted as describe above and afterwards the quality was also checked by glyoxal RNA gel
electrophoresis. High quality RNA with a ratio of 260/280 nm of 2, estimated with the
spectrometer, was used for further analysis. 10 μg RNA per time point were treated with
DNAse (DNA-free™ Kit Applied Biosystems). The amount of RNA was estimated with an 1-
Step Kit (qPCRBIO SYGreen, PCR Biosystems), whereby cDNA synthesis takes place
directly followed by quantification with SYBR Green PCR in the Stratagene Mx3005P cycler
via ct (cycle threshold) value calculations [35]. Primers are listed in Additional file 1. For
normalization of relative mRNA abundance, all ct values throughout the experiment were
subtracted by the lowest ct value observed (corresponding to the mRNA transcript for
Panafp::afp at day 5), which provided values of mRNA abundance relative to this time point.
Perfusion cultivation cultures
The bioreactors BioFlo3000 (New Brunswick Scientific, NJ) with a working volume of 5 litre
was inoculated with 5∙109
A. niger spores. For preparation of inoculums fresh conidia were
harvested from CM plates in sterile sodium chloride solution (0.05 % Tween 80, 0.9 % NaCl)
to avoid spore aggregation, which was filtered through sterile Miracloth (CalBiochem). 20
litre reactor medium consists of 90 g NH4Cl, 30 g KH2PO4, 10 g KCl, 10 g MgSO4·7H2O, 20
ml modified trace metal solution at pH 3. As carbon source for the batch phase, 1 litre of 80
g/l maltose was added for the cultivation of the mluc expressing strains (PK2.9, PK4.3), or
glucose for all strains with extracellular expressed reporter genes. For retentostat phase 0.01
% polypropylene glycol and 1 litre of 160 g/l respective C-source were added to 20 l bottles
for each run. The cultivation of FW23.7 contained double the amount of glucose in batch and
retentostat phase. During the cultivation a temperature of 30 °C was supported through a
temperature sensor in combination with heat exchanger. In addition, cooling tubing was fixed

- 171 -
around the reactor above the working volume to avoid fungal growth in the head plate. A pH
of 3 was maintained with a glass electrode (405-DPAS, Mettler Toledo) in connection with
computer controlled addition of 2 M NaOH. Sterile air was added with a gas flow of 1 l/min
through a sparger, with an optical oxygen sensor (InPro®6000, Mettler Toledo) which
monitored measured oxygen saturation to a level of at least 71 %. In the bioreactor run with
double glucose concentration, minimum 54 % dissolved oxygen was maintained.
Conidial germination occurred 6 hours after inoculation, which was enabled by addition of
0.003 % yeast extract in reactor media. To prevent the dispersal of the hydrophobic spores in
the bioreactor headspace, the stirrer speed was limited to 250 rpm and air supplementation
limited to the bioreactor head space. After6 hours, 0.01% polypropylene glycol was added to
bioreactor and rpm increased to 750. The air flow was disconnected from the headspace to
aerate the culture through the ring sparger. The batch phase was changed to continuous
cultivation after a consumption of 24 ml 2M NaOH at a concentration of approximately 2.1
gdry weight kg-1
biomass according to Iversen et al. [36]. The retentostat cultivation mode
commenced with a feeding flowrate of 0.125 l h-1
(correspond to a dilution rate of 0.025 h-1
),
and installation of a special cell retention device which pumped out used medium [9]. Stable
culture mass was achieved by monitoring retentostat culture by weight using an analytical
balance. Broth samples for biomass, RNA or MLUC protein analyses were taken at 12 h
intervals, with a maximum extraction of 100 g culture broth in 24 hours in order to minimize
impact on the cultivation. The sample preparation for determination of biomass concentration
and RNA analysis was conducted as described in Jørgensen et al., 2010 [9]. For analysis of
extracellular AFP or THP protein producing strains, 1.5 litres of effluent was collected from
the retention device every 12 h.
Reporter protein analysis
Broth samples from the cultivation with strains PK2.9 and PK4.3 were used for the
determination of promoter activity using the luciferase reporter protein. Bioluminescence
measurements were made in triplicate, with 130 µl of sample and 70 µl luciferin substrate mix
(1.4 mM luciferin) in 96-well microtiter plates. The luminescent counts per second (LCPS) at
537 nm and the optical density (OD) at 595 nm were measured using a Victor3TM
Perkin
Elmer reader. Melanin extraction was done with hot NaOH and measured at 425 nm as
described previously [9], [37].
For determination of AFP protein concentration, the effluent was concentrated from 100 ml
with Centrifugal Filter Units (with cut off above 3 kDa, Amicon®
Ultra) and eluted in a

- 172 -
volume of ~100 µl, after which samples were loaded on 16 % Tricine-SDS-PAGE gels and
analysed via western blot (anti-AFP primary antibody, IPK Gatersleben), which were
calibrated using a positive control of purified AFP from A. giganteus [22]. To evaluate
heterologous AFP for antifungal bioactivity, a MIC (minimal inhibitory concentration) assay
in microtiter plates was conducted, in which 100 µl 2xYPG medium (0.6 % yeast extract, 2 %
bacto peptone, 4 % glucose, pH 4.5), 10 µl of 105 spores/ml A. niger N402 and 90 µl purified
10 µg/ml AFP in FPLC buffer (0.05 M NaAc, 0.1 M NaCl) were combined [38]. For every
condition three replicates were prepared and growth rates determined using OD measurements
at 600 nm after 48 hours incubation at 30 °C, and the mean and standard deviations were then
calculated for each treatment.
For MA237 and MA238 strains containing the thp expression cassette, the effluent medium
was investigated for antifreeze activity with a self-made cooling finger as previously
described [25]. Additionally, determination of THP concentration was attempted using
Centrifugal Filter Units (with cut off above 3 kDa, Amicon®
Ultra), lyophilisation, different
precipitations (TCA, acetone), or dialysis. Lyophilized biomass samples were grinded using
pestle and mortar, and mycelium dissolved with protein extraction buffer (3.3 ml 0.5 M
Na2HPO4, 6.6 ml 0,5 M NaH2PO4, 0.2 ml 0.5 M EDTA, 20 l 100 mM PMSF, 1 ml 10 %
SDS, pH 7.0). This protein solution and the different concentrated effluent were investigated
using Tricine-SDS-PAGEs (16 %) [39], and Western blot with rabbit anti-THP primary
antibody [26]. HPLC-ESI-MS analysis was conducted using an Orbitrap XL hybrid mass
spectrometer (Thermo Fisher Scientific) coupled to a HPLC system 1200 (Agilent
Technologies) as described by Jungmann et al. 2014 [40]. For SDS-PAGE separated proteins,
gel spots on the expected height were digested (Trypsin In-Gel Digestion Kit, Thermo
Scientific).
List of abbreviations
A.niger: Aspergillus niger
AFP: antifungal protein from Aspergillus giganteus
bp: base pair
cDNA: complementary DNA
CM: complete medium
ct: cycle treshold
DNA: deoxyribonucleic acid
EDTA: ethylenediaminetetraacetic acid

- 173 -
FPLC: fast protein liquid chromatography
gDW/kg: gram dry weight per kilogram culture broth
HPLC-ESI-MS: high performance liquid chromatography-electrospray ionization-mass
spectrometric
hygR: hygromycin resistance gene
LCPS/OD: luminescent counts per second per optical density
gpdA: glyceraldehyde-3-phosphate dehydrogenase from A. nidulans
MIC: minimal inhibitory concentration
mluc: modified luciferase
MM: minimal medium
mRNA: messenger ribonucleic acid
Panafp: promoter of a gene encoding an antifungal protein in Aspergillus niger
PCR: polymerase chain reaction
PglaA: promoter of a gene encoding glucoamylase in Aspergillus niger
PhfbD: promoter of gene encoding a putative hydrophobin in Aspergillus niger
PMSF: phenylmethylsulfonyl fluoride
pyrG: orotidine-5'-decarboxylase gene from Aspergillus oryzae
qRT-PCR: quantitative reverse transcriptase PCR
SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis
SSanafp and SShfbD: signal sequence for secretion of the respective genes
TCA: trichloroacetic acid
THP: Thermal hysteresis protein from Choristoneura fumiferana isoform 337
TtrpC: the terminator of tryptophan synthase of Aspergillus nidulans
µ: specific growth rate
Declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable

- 174 -
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its
additional files.
Competing interests
The authors declare that they have no competing interests.
Funding
This project was partly funded by the Marie Curie Integration grant to VM (CIG 303684) and
supported by the Kluyver Centre for Genomics of Industrial Fermentation which is part of the
Netherlands Genomics Initiative/ Netherlands Organization for Scientific Research.
Authors' contributions
VM, FW, TJ and AR designed the study. FW and MA performed the experiments. FW, TC
and VM interpreted the results and were involved in writing the manuscript. All authors read
and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge Caroline Heiderich, Pim Kuizenga and Elizabeth
Norton for technical support. We are grateful to Imgard Schäffl for the analysis of AFP at
mRNA and protein level.
References
1. Krull R, Wucherpfennig T, Esfandabadi ME, Walisko R, Melzer G, Hempel DC, Kampen
I, Kwade A, Wittmann C: Characterization and control of fungal morphology for
improved production performance in biotechnology. J Biotechnol 2013, 163:112–123.
2. Nødvig CS, Nielsen JB, Kogle ME, Mortensen UH: A CRISPR-Cas9 System for Genetic
Engineering of Filamentous Fungi. PLoS One 2015, 10:1–18.
3. Oldiges M, Lütz S, Pflug S, Schroer K, Stein N, Wiendahl C: Metabolomics: Current
state and evolving methodologies and tools. Appl Microbiol Biotechnol 2007, 76:495–511.

- 175 -
4. Andersen MR, Nielsen J: Current status of systems biology in Aspergilli. Fungal Genet
Biol 2009, 46:180–190.
5. Meyer V, Fiedler M, Nitsche B, King R: The Cell Factory Aspergillus Enters the Big
Data Era: Opportunities and Challenges for Optimising Product Formation. Adv
Biochem Eng Biotechnol 2015, 149:91–132.
6. Liese A, Seelbach K, Wandrey C: Industrial Biotransformations. Volume 5. Wiley-VCH;
2006.
7. Pacis E, Yu M, Autsen J, Bayer R, Li F: Effects of cell culture conditions on antibody N-
linked glycosylation-what affects high mannose 5 glycoform. Biotechnol Bioeng 2011,
108:2348–2358.
8. Withers JM, Swift RJ, Wiebe MG, Robson GD, Punt PJ, Van den Hondel CAMJJ, Trinci
APJ: Optimization and stability of glucoamylase production by recombinant strains of
Aspergillus niger in chemostat culture. Biotechnol Bioeng 1998, 59:407–418.
9. Jørgensen TR, Nitsche BM, Lamers GE, Arentshorst M, van den Hondel CAMJJ, Ram AF:
Transcriptomic insights into the physiology of Aspergillus niger approaching a specific
growth rate of zero. Appl Environ Microbiol 2010, 76:5344–55.
10. Doucet D, Tyshenko MG, Davies PL, Walker VK: A family of expressed antifreeze
protein genes from the moth, Choristoneura fumiferana. Eur J Biochem 2002, 269:38–46.
11. Meyer V, Wedde M, Stahl U: Transcriptional regulation of the Antifungal Protein in
Aspergillus giganteus. Mol Genet Genomics 2002, 266:747–57.
12. Wnendt S, Ulbrich N, Stahl U: Molecular cloning, sequence analysis and expression of
the gene encoding an antifungal-protein from Aspergillus giganteus. Curr Genet 1994,
25:519–23.
13. Bos CJ, Debets AJM, Swart K, Huybers A, Kobus G, Slakhorst SM: Genetic analysis
and the construction of master strains for assignment of genes to six linkage groups in
Aspergillus niger. Curr Genet 1988, 14:437–443.

- 176 -
14. Van Hartingsveldt W, Mattern I, van Zeijl C, Pouwels P, van den Hondel CAMJJ:
Development of a homologous transformation system for Aspergillus niger based on the
pyrG gene. Mol Gen Genet 1987, 206:71–5.
15. Boender LGM, van Maris AJA, de Hulster EAF, Almering MJH, van der Klei IJ,
Veenhuis M, de Winde JH, Pronk JT, Daran-Lapujade P: Cellular responses of
Saccharomyces cerevisiae at near-zero growth rates: transcriptome analysis of anaerobic
retentostat cultures. FEMS Yeast Res 2011, 11:603–20.
16. Morgan LW, Greene A V., Bell-Pedersen D: Circadian and light-induced expression of
luciferase in Neurospora crassa. Fungal Genet Biol 2003, 38:327–332.
17. Llanos A, François JM, Parrou J: Tracking the best reference genes for RT-qPCR data
normalization in filamentous fungi. BMC Genomics 2015, 16:71.
18. Meyer V, Wanka F, van Gent J, Arentshorst M, van den Hondel CAMJJ, Ram AFJ:
Fungal gene expression on demand: an inducible, tunable, and metabolism-independent
expression system for Aspergillus niger. Appl Environ Microbiol 2011, 77:2975–83.
19. Lacadena J, Martínez del Pozo a, Gasset M, Patiño B, Campos-Olivas R, Vázquez C,
Martínez-Ruiz a, Mancheño JM, Oñaderra M, Gavilanes JG: Characterization of the
antifungal protein secreted by the mould Aspergillus giganteus. Arch Biochem Biophys
1995, 324:273–281.
20. Meyer V: A small protein that fights fungi: AFP as a new promising antifungal agent
of biotechnological value. Appl Microbiol Biotechnol 2008, 78:17–28.
21. López-García B, Moreno AB, San Segundo B, De los Ríos V, Manning JM, Gavilanes JG,
Martínez-del-Pozo Á: Production of the biotechnologically relevant AFP from Aspergillus
giganteus in the yeast Pichia pastoris. Protein Expr Purif 2010, 70:206–210.
22. Theis T, Wedde M, Meyer V, Stahl U: The Antifungal Protein from Aspergillus
giganteus Causes Membrane Permeabilization. 2003, 47:588–593.
23. Fiedler M, Nitsche B, Wanka F, Meyer V: Aspergillus: A Cell Factory with Unlimited
Prospects. In Appl Microb Eng; 2013:1–51.

- 177 -
24. Venketesh S, Dayananda C: Properties, potentials, and prospects of antifreeze
proteins. Crit Rev Biotechnol 2008, 28:57–82.
25. Kuiper MJ, Lankin C, Gauthier SY, Walker VK, Davies PL: Purification of antifreeze
proteins by adsorption to ice. Biochem Biophys Res Commun 2003, 300:645–648.
26. Tyshenko MG, d’Anjou M, Davies PL, Daugulis AJ, Walker VK: Challenges in the
expression of disulfide bonded, threonine-rich antifreeze proteins in bacteria and yeast.
Protein Expr Purif 2006, 47:152–61.
27. Punt PJ, Schuren FHJ, Lehmbeck J, Christensen T, Hjort C, van den Hondel CAMJJ:
Characterization of the Aspergillus niger prtT, a unique regulator of extracellular
protease encoding genes. Fungal Genet Biol 2008, 45:1591–9.
28. Ercan O, Bisschops MMM, Overkamp W, Jørgensen TR, Ram AF, Smid EJ, Pronk JT,
Kuipers OP, Daran-Lapujade P, Kleerebezem M: Physiological and transcriptional
responses of different industrial microbes at near-zero specific growth rates. Appl
Environ Microbiol 2015, 81:5662–5670.
29. Krijgsheld P, Nitsche BM, Post H, Levin AM, Müller WH, Heck AJR, Ram AFJ, Altelaar
AFM, Wösten HAB: Deletion of flbA results in increased secretome complexity and
reduced secretion heterogeneity in colonies of Aspergillus niger. J Proteome Res 2013,
12:1808–1819.
30. Gun LD, Shin SY, Maeng CY, Jin ZZ, Kim KL, Hahm KS: Isolation and
characterization of a novel antifungal peptide from Aspergillus niger.
BiochemBiophysResCommun 1999, 263:646–651.
31. Asgeirsdóttir S, Halsall J, Casselton L: Expression of two closely linked hydrophobin
genes of Coprinus cinereus is monokaryon-specific and down-regulated by the oid-1
mutation. Fungal Genet Biol 1997, 22:54–63.
32. Doucet D, Tyshenko M, Kuiper M, Graether S, Sykes B, Daugulis A, Davies P, Walker
V: Structure-function relationships in spruce budworm antifreeze protein revealed by
isoform diversity. Eur J Biochem 2000, 267:6082–8.
33. Meyer V, Ram AFJ, Punt P: Genetics, genetic manipulation and approaches to strain
improvement of filamentous fungi. In Man Ind Microbiol Biotechnol 3rd ed NY Wiley;
2010:318–329.
34. Vishniac W, Santer M: The thiobacilli. Bacteriol Rev 1957, 21:195–213.

- 178 -
35. Pfaffl MW: A new mathematical model for relative quantification in real-time RT-
PCR. Nucleic Acids Res 2001, 29:e45.
36. Iversen JJL, Thomsen JK, Cox RP: On-line growth measurements in bioreactors
bytitrating metabolic proton exchange. Appl Environ Microbiol 1994, 42:256–262.
37. de Cássia R Goncalves R, Pombeiro-Sponchiado SR: Antioxidant activity of the
melanin pigment extracted from Aspergillus nidulans. Biol Pharm Bull 2005, 28:1129–
1131.
38. Hagen S, Marx F, Ram AF, Meyer V: The antifungal protein AFP from Aspergillus
giganteus inhibits chitin synthesis in sensitive fungi. Appl Environ Microbiol 2007,
73:2128–34.
39. Schägger H: Tricine–SDS-PAGE. Nat Protoc 2006, 1:16–22.
40. Jungmann NA, Krawczyk B, Tietzmann M, Ensle P, Süssmuth RD: Dissecting Reactions
of Nonlinear Precursor Peptide Processing of the Class III Lanthipeptide Curvopeptin.
2014. J. Am. Chem. Soc. 2014, 136: 15222−15228.
41. Wanka F, Cairns T, Boecker S, Berens C, Happel A, Zheng X, Sun J, Krappmann S,
Meyer V: Tet-on, or Tet-off, that is the question: Advanced conditional gene expression
in Aspergillus. Fungal Genet Biol 2016, 89: 72-83.
Additional materials
Additional file 1: Design of Northern and qRT-PCR probes.
Northern
probe
Digestion
product
size [bp]
mluc pMA247 [41] with PmeI 1667
thp pEN2 with XbaI and MluI 780
Forward primer Reverse primer
act ATCTCCCGTGTCGACATGG GCGGTGGACGATCGAGG 656
qRT-PCR
probe
gpdA AGGGCACCATCGAGACCTAC TGGGTGGTGAAGACACCAGT 145
h2B CTCGAAACTTGCCGCTTACA ACTTCGTGACAGCCTTGGTG 131
afp TGGCAAATGCTACAAGAAGG AGCAGTAGCACTTCCCCTTG 150

- 179 -
Diskussion und Ausblick
In der industriellen Biotechnologie, ist Aspergillus niger eine viel genutzte
Expressionsplattform für organische Säuren, Enzyme und heterologe Proteine. Um die
Produktivität zu steigern und auch A. niger als Zellfabrik für neuartige Produkte, wie
Sekundärmetabolite oder glykosylierte Therapeutika zu etablieren, ist es wichtig weiterhin
neue molekulare Methoden für ein genetic engineering von pilzlichen Systemen zu
entwickeln und neueste Fermentationsverfahren für A. niger auf Anwendbarkeit zu testen.
Für die Schimmelpilzforschung ist die erfolgreiche Adaption von induzierbaren
metabolismusunabhängigen Expressionssystemen von großer Bedeutung, sie vereinfachen
Genfunktionsstudien und können Produkterträge wesentlich verbessern (Kapitel 1.4.1). Ein
wichtiger Meilenstein war die Anpassung des Tetracyclin kontrollierten Systems (Gossen &
Bujard, 1992) an A. fumigatus (Vogt et al., 2005) und die weiterführende Verbesserung des
Tet-on-Systems in A. niger (Meyer et al., 2011). Im Kapitel 3 konnte gezeigt werden, dass
durch die Anwendung des Tet-on-Systems und die Induktion bei 1 gTrockengewicht/kg Biomasse
ein sehr hoher spezifischer Ertrag an antibiotisch wirkendem Enniatin von 0,29
gEnniatin/gTrockengewicht nach 110 h in der Batch-Fermentation (0,8 % Glucose) erzielt wurde. Die
erreichte Enniatinmenge von 4,5 g/l in 66 h Fed-Batch-Fermentation (5 % Glucose) entspricht
der höchsten publizierten Enniatinmenge im heterologen Wirt und ist fast annährend so hoch
wie die des mutagenisierten Originalorganismus F. oxysporum, welcher in 96 h bis zu 5 g/l
produziert (Madry et al., 1983). Für eine höhere Ausbeute an Enniatin B in der A. niger Fed-
Batch-Kultivierungen ist es nötig mehr vom limitierenden D-Hiv zu zufüttern, denn der
berechnete theoretisch maximale Enniatinertrag aus der eingesetzten Menge an D-Hiv wird zu
85 % erreicht, der Verlust könnte z.B. durch die Aufreinigung erklärt werden. Ein
vielversprechender Ansatz wäre daher die Fed-Batch-Kultivierung bzgl. kontinuierlicher
Glucose- und D-Hiv-Zufütterung zu optimieren, um die spezifische Enniatinausbeute pro
Biomasse zu steigern, dadurch wird es realisierbar sein F. oxysporum in seinem
Enniatingehalt zu übertreffen. A. niger tritt somit ins Rampenlicht als potenzieller Produzent
für Sekundärmetabolite, im Speziellen von Antibiotika, deren effiziente Produktion gerade
heutzutage, wo jahrelang nicht in die Entwicklung von Antibiotika investiert wurde, von
enormer Wichtigkeit ist. Weiterführend wird es für die Enniatin B Produktion von Interesse
sein, die metabolischen Flüsse vollständig zu analysieren und so zu regulieren, dass mehr
verfügbares Valin, Adenosintriphosphat (Energieträger) und S-Adenosylmethionin
(Methyldonor) vorhanden ist, um eine Maximierung der Enniatinausbeute zu erreichen. Die
autonome D-Hiv Produktion, durch die Einführung der Keto-Isovalerat-Reductase (kivR) ins
A. niger Genom, ist der Schlüssel um hohe Enniatinausbeuten kosteneffizient zu generieren

- 180 -
und sollte trotz erster Erfolge (Kapitel 3) weiterhin optimiert werden. Mit dem Design of
Experiments Ansatz könnten die optimale Dox- und Valinkonzentrationen ermittelt werden.
Weiterführend wäre auch eine polycistronische Expression des esynI und kivR Gens unter der
Regulation des Tet-on-Systems interessant anstatt der vorgenommenen mehrfachen
Integration von kivR unter der Regulation von PgpdA in das Genom des
Enniatinproduziereres DS3.1. Eine zusätzliche Verbesserung des Enniatin-produzierenden A.
niger-Stammes wäre die Überexpression von einem geeigneten Membran lokalisierten ABC-
Transporter (ATP binding cassette) zum Ausschleusen des Produktes. Es wurde in einem
Penicillin produzierenden A. nidulans-Stamm erstmals gezeigt, dass die Sekretion von
heterologen Sekundärmetaboliten über einen ABC-Transporter vermittelt wird, der durch das
Gen atrD codiert wird (Andrade et al., 2000). Bei einer Analyse der Nichtribosomalen-
Peptid-Synthetasen in A. nidulans wurde festgestellt, dass einige ABC-Transporter nah an
diese Cluster assoziiert sind und daher wahrscheinlich eine Rolle bei ihrer Sekretion spielen
(von Döhren, 2009). Insgesamt ist jedoch noch wenig über die pilzlichen ABC-Transporter,
die in der Sekretion von Sekundärmetaboliten involviert sind, bekannt (Martín et al., 2005;
Kovalchuk & Driessen, 2010). Ebenfalls ist es denkbar, die in Autophagie involvierten Gene
atg1 oder atg8 zu deletieren um den Autolyseprozess zu beschleunigen (Nitsche et al., 2012)
und somit einen kostenintensiven Zellaufschluss bei der Aufreinigung zu umgehen. Wenn
Enniatin im Überstand vorliegt, könnte durch den Einsatz von makroporösen Polystyrol-Resin
(X-5)-Absorptionsmittel Enniatin gebunden und somit die Effizienz des Prozesses erhöht
werden (Xu et al., 2009).
Um einen regulierbaren Knock down von Genen zu erreichen wurde bisher in Aspergillus die
RNA-Interferenz genutzt. Jedoch konnte nur eine 90 % metabolismusabhängige
Verminderung der Genexpression erreicht werden, welche in einem unvollständigen
Knockout-Phänotyp resultiert (Yamada et al., 2007). Das Ziel der Etablierung des Tet-off-
System für Aspergillus ist eine quantitative Reduktion der Genexpression eines Zielgens zu
erreichen, so dass alle Aktivitäten im Bereich 0-100 % angesteuert werden können. Bei der
Konstruktion des Tet-off-Systems für A. niger (Kapitel 4) wurde die entscheidende
Bedeutung eines optimalen Expressionsniveaus vom Transaktivator (tTA2S) nachgewiesen.
Eine zu hohe Menge an Transaktivatorprotein führt in A. niger zu einer Squelching-Reaktion,
die toxische Auswirkungen auf den Organismus hat (Gill & Ptashne, 1988). Wenn der
Transaktivator zu stark transkribiert wird, schützt der Mikroorganismus sich selbst indem die
Gensequenz aus dem Genom durch Rekombination deletiert wird, welches instabile Stämme
zur Folge haben kann. Um diesen toxischen Effekt zu verhindern, wurde der vorgeschaltete
gpdA Promotor gegen einen schwächeren aber ebenso konstitutiven Promotor (PfraA) im Tet-

- 181 -
on/-off ausgetauscht. Das Ergebnis waren stabile Stämme aber auch eine über 50 % geringere
Expression des Zielgenes. Somit stellt sich die Frage, ob es zu Limitationen bei
Genfunktionsstudien im neukonstruierten Tet-on und Tet-off-System kommen kann. Da
mitunter Gene sehr stark exprimiert werden können, ist die Expressionsstärke im aktivierten-
Zustand des Systems möglicherweise nicht ausreichend um den Wildtyp-Zustand zu
erzeugen. Untersuchungen mit dem hoch exprimierten Gen für die Glutamin-Fructose-6-
Phosphat-Amidotransferase (gfaA) im verbesserten Tet-off zeigten jedoch keine
Einschränkungen (Kapitel 4). Das optimierte stabilere Tet-on (Kapitel 4) wird sich
wahrscheinlich weniger gut für die Produktion von z.B. hohen Mengen Enniatin (Kapitel 3)
eignen, wenn die Transkriptmenge des Transaktivator bzw. der ESYN limitierend für die
Enniatinproduktion sein sollte. Die Ergebnisse verdeutlichen, dass es wichtig ist, die richtige
Balance zu finden. Es muss je nach Fragestellung abgewogen werden, ob ein genetisch
stabiles aber schwächeres, oder genetisch instabiles aber starkes Expressionssystem besser
geeignet ist. Diese Problematik wird sich bei allen ähnlich zum Tet-System aufgebauten
Expressionssystemen zeigen, sollte uns aber nicht daran hindern weitere Expressionssysteme
für die Nutzung in A. niger zu entwickeln. Es ist unerlässlich an regulierbaren und
metabolismusunabhängigen Systemen zu arbeiten, damit es zukünftig möglich ist komplexe
Netzwerkregulationen zu erstellen, um auch in Aspergillus den Ansätzen der synthetischen
Biologie folgen zu können. Denn zweifelsohne ist es für weitere Produktivitätssteigerungen
vonnöten metabolische Stoffwechselwege möglichst genau aufeinander abzustimmen
(Keasling, 2008). Nach der erfolgreichen Etablierung vom Tet-on System, wurde ein neues
Expressionssystem, basierend auf den regulatorischen Elementen von A. terreus Terrein
Gencluster, in A. niger publiziert. In dem System wird der Transkriptionsfaktor TerR durch
einen metabolismusabhängigen amyB-Promotor exprimiert, der durch Bindungsstellen den
dahinterliegenden TetA-Promotor induziert und so zur Expression eines Reportergenes in A.
niger führt (Gressler et al., 2015). Als Mehrwert kann lediglich eine höhere Expression (>2x)
im Vergleich zum amyB-Promotor festgestellt werden, eine Induzierung ist weiterhin nur über
die Kohlenstoffquelle (Glucose, Maltose) möglich. Für die Erweiterung der induzierbaren
Genregulationssysteme in A. niger wären die Streptogramin- und Makrolid-basierenden
Genschalter interessant (siehe Tab. 4), da sie sehr ähnlich zum etablierten Tetracyclin
Expressionssystem aufgebaut sind und in Säugetierzellen ein exzellentes
Regulationsverhalten zeigen (Fussenegger et al., 2000; Weber et al., 2002). Wenn man auf
antibiotische Genschalter verzichten möchte, stehen noch biologische Induktoren zur
Verfügung, wie bei dem Cuminsäure-(4-Isopropylbenzosäure) (Mullick et al., 2006) oder
Biotin-induzierbaren Expressionssystem (Weber et al., 2009) (Tab. 4).

- 182 -
Weniger sinnvoll erscheinen folgende Expressionssysteme für den Einsatz in Aspergillus i)
Temperatur induzierbare Genregulation (TIGR) (Weber et al., 2003), weil dadurch der
Metabolismus von A. niger stark beeinflusst wird, ii) Licht (bestimmte Wellenlänge)
induzierbare Genregulation (Bacchus & Fussenegger, 2012), weil sich die technische
Umsetzung in der Fermentation schwierig gestaltet und davon auszugehen ist, dass der
Metabolismus, z.B. durch den lichtabhängigen Regulator Velvet A (Spröte & Brakhage, 2007;
Tisch & Schmoll, 2010), beeinflusst wird.
Tabelle 4: Übersicht über weitere vielversprechende induzierbare Expressionssysteme, die sich
für eine Anpassung in Aspergillus eignen könnten.
Expressionssysteme Eigenschaften Publikation
Streptogramin (z.B.
Pristinamycin, Pip-on/off)
Pip-off-System zeigt geringere
Hintergrundexpression und höhere
Induktionsraten als das Tet-off
(Fussenegger et
al., 2000)
Makrolid (z.B.
Erythromycin, ETR-on/off)
exzellente regulatorische Charakteristika,
kompatibel mit Tet-on/off und Pip-on/off
(Weber et al.,
2002)
Cuminsäure (on/off) relativ schwache Expression und hohe
Basalexpression im Vergleich zu Pip- on/off
(Mullick et al.,
2006)
Biotin (on) Repression eventuell nicht möglich, da Biotin
auch im A. niger Stoffwechsel vorkommt
(Weber et al.,
2009)
Ein weiterer Ansatz zur zukünftigen schnelleren Regulierung der Expressionssysteme könnte
die Integration des Auxin-induzierbaren Degron (AID) sein. Basierend auf einer
Ubiquitinierung kann man das Proteinlevel kontrollieren und damit die zeitliche Verzögerung
resultierend aus der Promotormodulation verhindern (Karlsson et al., 2012). Jedoch konnte in
Kapitel 4 gezeigt werden, dass durch das simplere Anhängen der
Proteindegradierungssequenz PEST eine kurze Halbwertszeit des Reportergenes Luziferase
(30 min) und damit eine akkuratere Genexpressionsmessung erzielt werden konnte.
Interessant wäre es zu testen, ob man mit Hilfe von IRES (interne ribosomale Eintrittsstelle)
zwei Gene bicistronisch coexprimieren kann (Martin et al., 2006) oder ob sich durch 2A-
Peptid-Sequenzen sogar mehrere Proteine polycistronisch in A. niger prozessieren lassen
(Unkles et al., 2014).
Ein weiterer Ansatz ist durch verfahrenstechnische Methoden eine höhere Produktivität von
A. niger zu ermöglichen. Der Trend geht in Richtung kontinuierliche Perfusionsfermentation,
deren Vorteile umfassend in Kapitel 5 diskutiert wurden. Es gelang in dieser Arbeit, die Basis
für die Perfusionskultivierung mit A. niger durch die Evaluierung von geeigneten Promotoren
zu legen. Ziel war es, das möglichst viel vom eingesetzten Kohlenstoff in das sekretierte

- 183 -
Produkt gelangt und nicht in den Aufbau von Biomasse. Des Weiteren sollte die Kultivierung
über einen möglichst langen Zeitraum gefahren werden, daher wurden Promotoren benötigt,
die bei einer geringen Wachstumsrate hohe transkriptionelle Aktivitäten zeigen. In der Arbeit
ist es gelungen die Promotoren Panafp und PhfbD zu evaluieren, die bei abnehmender
Wachstumsrate aktiv sind und ein gleichbleibendes Transkriptom- und Translationslevel mit
verschiedenen intra- und extrazellulären Reportergenen aufweisen. Es wurde jedoch auch
gezeigt, dass die Wachstumsrate nicht unter <0,001 h-1
sinken darf, um effizient mRNA zu
bilden und Proteine zu synthetisieren bzw. sekretieren. Auch andere Studien, die
Nullwachstum in verschiedenen Mikroorganismen untersuchen, kommen zu der Erkenntnis,
dass kohlenstofflimitierte Konditionen zur Reduktion der Genexpression von
Translationsmaschinerie, ribosomaler Proteine und Aminoacyl-tRNA führen und es an ATP
fehlt (Boender et al., 2011; Ercan et al., 2015; Overkamp et al., 2015).
Um zukünftig die Perfusionsfermentation für die effiziente extrazelluläre Proteinproduktion in
A. niger zu nutzen, wäre es sinnvoll im sogenannten Glucose-Auxostat Modus zu arbeiten.
Dabei wird in Echtzeit die Glucosekonzentration im Reaktor gemessen und über eine
regulierbare Flussrate konstant gehalten (Konstantinov et al., 1996). Wenn bspw. der
Promotor von anafp zur Proteinproduktion verwendet werden sollte, wäre es ratsam, nach
Erhalt von 1 gTrockengewicht/ kg in der Batch-Fermentation auf Perfusionskultivierung mit einer
optimalen Wachstumsrate von 0,004 h-1
(Kapitel 5) umzustellen. Dies sollte theoretisch eine
kontinuierliche Kultivierung von über 30 Tage mit sehr hohen Produktionsraten ermöglichen.
Es wäre interessant die Biomasseprobe eines späteren Zeitpunktes der durchgeführten
Perfusionskultivierungen von A. niger (~Tag 12, µ<0,0001 h-1
) auf die transkriptionelle
Expression und das Metabolom zu untersuchen. In vorherigen transkriptionellen Analysen
von Tag 8 der Perfusionsfermentation wurden im Vergleich zu der Batchphase mindestens 14
mögliche Sekundärmetabolitcluster gefunden (Jørgensen et al., 2010, SA9). Allerdings
könnten sich diese Erkenntnisse über die Expression bspw. toxischer Metabolite negativ auf
die Zulassung des Perfusionsprozesses für die Produktion in A. niger auswirken.
Des Weiteren bleibt zu überprüfen, ob es zu Sterilitätsproblemen durch die lange
Kultivierungszeit und die technisch und Material anspruchsvolle Fermentation kommen kann.
In der kontinuierlichen monoklonalen Antikörperproduktion in Zellkultur wurde eine 6-fach
höhere Anzahl an kontaminierten Fermentationen im Vergleich zum Fed-Batch festgestellt
(Pollock et al., 2013). Ebenso stehen noch Untersuchungen bzgl. der genetischen Stabilität
von A. niger Stämmen in der Perfusionskultivierung aus. Man geht allerdings davon aus, dass
der Mutationsdruck nicht so hoch wie in Chemostat-Kultivierungen ist, wo eine Selektion auf
angepasste Mutanten stattfindet (Withers et al., 1994).

- 184 -
Epilog I
In diesem Abschnitt möchte ich die wissenschaftlichen Projekte und Ergebnisse aufführen,
die noch nicht publiziert sind.
1. Projekt: Expression und Sekretion von Alkoholdehydrogenasen aus E. coli in A. niger
(In Kooperation mit Dr. Jochen Schmidt und Prof. Volker Sieber, Lehrstuhl für Chemie
Biogener Rohstoffe von der TU München, praktische Durchführung hauptsächlich Ulrike
Obst (Diplomarbeit) mit Assistenz von Anna Maria Groß (Bachelorarbeit), 9 Monate
Projektzeit, 2013)
Alkoholdehydrogenasen sind von großer Bedeutung, sie katalysieren die reversible
Oxidation von Alkoholen zu Ketonen oder Aldehyden, welche essentiell bei der Produktion
von Biokraftstoffen der zweiten Generation oder von Chemikalien (z.B. 1,3-Propandiol)
sind. Es wurden zwei unterschiedliche Alkoholdehydrogenasen AdhZ2 und AdhZ3
ausgewählt, wobei zum einen zusätzliche Mutationen eingeführt wurden (durchgeführt an
der TU München), die zu einem eukaryotischen Glykosylierungsmuster und somit zu einer
Stabilitätsverbesserung für die Produktion in A. niger führen sollten (van den Brink et al.,
2006) und zum anderen wurden beide Alkoholdehydrogenasegene für A. niger codon-
optimiert (Tokuoka et al., 2008). Um die Aufreinigung zu erleichtern, wurde N-terminal an
alle sechs Alkoholdehydrogenase-Varianten ein His-Tag (6-Histidine) angefügt. Des
Weiteren wurden N-terminal die ersten 514 Aminosäuren der Glucoamylase (inklusive
KEX2-Schnittstelle) als Sekretionshelfer, abgeleitet von dem am häufigsten sekretierten
Enzym Glucoamylase aus A. niger (Gordon et al., 2000), fusioniert (Kapitel 1.3). Die
insgesamt sechs verschiedenen Konstrukte wurden in einen Tet-on-Vektor (pVG2.2, Kapitel
3) kloniert und die erhaltenen Plasmide anschließend in den Protease-defizienten A. niger
Stamm AB1.13 (Kapitel 3) transformiert. Nach Kultivierung der Stämme konnte kein
sekretiertes Protein mit His-Tag im Überstand gefunden werden. Jedoch zeigt das
Proteinlysat der extrahierten Biomasse im Western Blot mit Anti-His-Tag-Antikörper eine
spezifische Bande bei ~90 kDA, die dem Gesamtkonstrukt von dem Fusionsprotein
Glucoamylase bis zum Ende der jeweiligen ADH entspricht. Eine quantitative Aussage bzgl.
der verschiedenen Varianten ist nicht möglich. Anschließende Untersuchungen der TU
München ergaben keine Enzymaktivität im Überstand, eine Untersuchung der extrahierten
Biomasse wurde nicht vorgenommen, weil zu viele andere Enzyme mit NAD(P)H Aktivität
in A. niger vorhanden sind. Die Untersuchungen sollten fortgeführt werden, sinnvoll wäre es
mit Nickel-Affinitätschromatographie das Protein aus dem Überstand zu konzentrieren

- 185 -
bzw.extrahierte His-Proteine aus der Biomasse zu reinigen und bzgl. ihrer Aktivität zu
untersuchen.
Es ist schwierig ein prokaryotisches intrazellulares Enzym in einem eukaryotischen
Organismus extrazellulär zu sekretieren. Verschiedene Gründe für die nicht sekretierten
Alkoholdehydrogenasen sind vorstellbar. Möglicherweise ist die Sekretionssequenz nicht
optimal, die KEX2-Schnittstelle oder der His-Tag bzw. sein Spacer, aber am
Wahrscheinlichsten ist ein Zusammenhang mit A. niger Proteasen zu vermuten, die das
Zielprotein verdauen bevor der Nachweis gelingt. Abschließende Diskussion dazu unter
Projekt 4.
2. Projekt: Evaluierung der EnBase® Technologie für die Kultivierung von A. niger
(In Kooperation mit Prof. Neubauer, FG Bioverfahrenstechnik, hauptsächlich durchgeführt
von Ulrike Will (Bachelorarbeit), 3 Monate Projektzeit, 2014)
Das EnBase®-System ist ein entwickeltes Kultivierungssystem für E. coli und Hefen, dass
durch die enzymatische Degradierung von Stärke eine Art Fed-Batch-Kultivierung im
kleinen Maßstab ermöglicht (Panula-Perälä et al., 2008). Dieses System führt zu einem
langsameren aber kontrollierten Wachstum, welches Overflow-Metabolismus unterbindet,
hingegen Genexpressionen und Ausbeuten steigert und zukünftige Scale up-Prozeduren
verkürzen soll. Um in A. niger den Stärkeabbau ebenso durch das System kontrollieren zu
können, erfolgte zuerst die Erstellung eines AmyR-deletierten A. niger-Stammes, der somit
eine Deaktivierung der Glycosidasen bewirkt (Yuan et al., 2008; van Kuyk et al., 2012).
Anschließend wurde die Evaluierung des EnBase®-System im Mikrotiterplatten und
Schüttelkolbenformat durchgeführt, wobei in allen Versuchen ebenfalls der A. niger-
Ausgangsstamm mit intaktem AmyR-Gen als Kontrolle mitgeführt wurde. Ziel war, die
Effizienz der EnBase® Technologie gegenüber herkömmlichem Komplettmedium
(äquivalente Glucosemenge 3 %) und die Notwendigkeit der AmyR-Deletion im Pilz anhand
der gebildeten Biotrockenmasse und des Genexpressionlevels vom Reportergen Luziferase
zu evaluieren. Die Ergebnisse führten zu dem Schluss, dass ein amyR–
Stamm generiert
werden muss, damit die Degradierung der Stärke kontrolliert abläuft und sich Fed-Batch
Bedingungen ergeben. Mit diesem A. niger-Stamm und der optimalen Enzymkonzentration
(aus dem EnBase®-System) konnten Fed-Batch Bedingungen erreicht werden. Jedoch
konnte keine höheren Biomasse- und Genexpressionswerte im Vergleich zum
Komplettmedium nachgewiesen werden. Abschließende Diskussion dazu unter Projekt 3.

- 186 -
3. Projekt: Prozesstechnische Optimierung von A. niger als Produzent von
Sekundärmetaboliten
(Hauptsächlich durchgeführt von Aike Tappe (Bachelorarbeit), 3 Monate Projektzeit, 2015)
Aufbauend auf Kapitel 3 wurden weitere Untersuchungen, wie man die Enniatin-Ausbeute
steigern kann, durchgeführt. Im Schüttelkolben wurde mit dem Enniatin produzierenden
Stamm DS3.1 die optimale Induktionszeit für das Tet-on-System bestimmt. Dabei zeigte
sich, dass nach 8 h mit 0,053 gEnniatin/gTrockengewicht der optimale Zeitpunkt zum Induzieren des
Systems erreicht ist. Bisher wurde aus Gründen des Zeitmanagements nach 16 h induziert,
jedoch war der Wert mit 0,031 gEnniatin/gTrockengewicht sogar etwas geringer im Vergleich zum
Zeitpunkt 0 h mit 0,037 gEnniatin/gTrockengewicht. Wie im Kapitel 3 dargestellt, wurde die höchste
Enniatin-Ausbeute in Fed-Batch-Kultivierungen (Batch-Phase: 4 l Fermentationsmedium
mit 0,8 % Glucose; durch Zufütterung von 1.5 l Feedingmedium: 255 g Glucose (absolute
Glucosekonzentration im Reaktor: 5 %), 127.5 g Hefeextrakt, 255 ml Casaminosäuren) mit
4.5 gEnniatin/l (0,18 gEnniatin/gTrockengewicht) erreicht. Die Ergebnisse dieses Projektes lassen
schlussfolgern, dass durch eine Batch-Kultivierung mit 5 l Komplexmedium mit 5 %
Glucose eine Ertragssteigerung bzgl. der Enniatinausbeute in gEnniatin/l um weitere 25 %
erreicht werden kann. A. niger zeigte sich weitestgehend unempfindlich gegenüber hohen
Substratkonzentrationen, was die somit aufwendigere Fed-Batch-Kultivierung überflüssig
macht. Das Ergebnis und auch die Erkenntnisse von Projekt 2 stimmen mit anderen
Publikationen überein, in denen gezeigt wurde, dass A. niger sehr hohe
Glucosekonzentrationen toleriert und somit die Biomasse und der Ertrag, bezogen auf Liter
oder Biomasse, erhöht werden kann. Bei Tests auf festen Agarmedium stieg bis zu einer
Glucosekonzentration von 120 g/l die Biomasse (Larralde-Corona et al., 1997), in
Flüssigkulturen wurde die maximale Produktausbeute zwischen 5-12 % Glucose erreicht
(Fontana et al., 2005). Wenn E. coli unter aeroben Wachstumsbedingungen aus Glucose
Acetat oder Lactat produziert (Basan et al., 2015), spricht man vom sogenannten Overflow-
Metabolismus. Bei Hefe ist dieser als „Crabtree Effekt“ bekannt, der in Ethanol und Acetat
resultiert (Postma et al., 1989). A. niger produziert in geringen Mengen auch diese
Nebenprodukte, aber bisher wurden sie erst zum Ende der Kultivierung nachgewiesen und
man nimmt an, dass sie aus der Sauerstofflimitierung bei hoher Biomassekonzentration
resultieren. In A. niger würde man die Herstellung von Zitronensäure unter sauren
Bedingungen und die hohe Oxal-/Gluconsäureproduktion im alkalischen Bereich bei der
Produktion rekombinanter Proteine als Überflussmetabolite benennen (Papagianni, 2007).
Wenn man den Basenverbrauch beider genannten Fermentationen vergleicht, werden im
Batch ~105 ml 2 M Natriumhydroxid verbraucht hingegen im Fed-Batch nur ~47 ml, das

- 187 -
bedeutet, dass ein Überflussmetabolismus in Richtung Zitronensäureproduktion bei
dauerhaft hoher Glucosekonzentration stattfindet. Wenn die Wirtschaftlichkeit zukünftig im
Fokus stehen sollte, muss ermittelt werden wieviel Glucose benötigt wird, um die maximale
Biomassekonzentration im Reaktor zu erreichen. Die Ausbeuten sollten pro eingesetztem
Gramm Glucose kalkuliert und die Zitronensäureproduktion als Überflussprodukt
weiterführend untersucht werden.
4. Projekt: Expression und Sekretion einer Laccase aus Trametes versicolor in A. niger
(In Kooperation mit der Bundesanstalt für Materialforschung und -prüfung, hauptsächlich
durchgeführt von Martina Matzke (PhD Thesis), >12 Monate, 2014)
Für die Entwicklung eines antikörperbasierten Nachweisverfahrens war die hochreine
Laccaseherstellung des Weißfäulepilzes grundlegend. Zum einen wurde als Basis die
extrahierte genomische DNA und zum anderen die aus der extrahierten RNA
umgeschriebene cDNA aus T. versicolor verwendet. Für eine vereinfachte Aufreinigung
wurde der His-Tag sowohl C- als auch N-terminal an die Laccase kloniert, so dass insgesamt
mit vier verschiedenen Varianten gearbeitet wurde. Um die Sekretion zu ermöglichen,
wurde das Glucoamylase-Fusionsprotein (siehe Projekt 1) vor die verschiedenen Konstrukte
in das Tet-on-System kloniert und anschließend in AB1.13 transformiert. Nach
verschiedenen Kultivierungen im Schüttelkolben mit Proteaseinhibitoren und gepufferten
pH-Wert von 6, wurde der Überstand durch Nickelaffinitätschromatographie und mit
anderen Konzentrierungsverfahren (Lyophilisierung, Fällung, zentrifugale Filtereinheiten)
gereinigt, jedoch konnte bisher keine Laccase im Überstand gefunden werden. Die
extrahierten Proteine aus der Biomasse zeigten im Western Blot mit einem Anti-His-Tag-
Antikörper Banden auf der Höhe des gesamten Konstruktes, also mit nicht abgespaltenem
Fusionsprotein. Die Ergebnisse sind sehr ähnlich zu Projekt 1. In anderen Projekten der
Arbeitsgruppe wurden Variationen in dem Glucoamlyase-Fusionsprotein (499
Aminosäuren), in den Basen für den His-Tag oder des Spacers vorgenommen. Jedoch
konnte damit das Problem nicht gelöst werden. Auch im Austausch mit anderen
Arbeitsgruppen kristallisiert sich heraus, dass die Sekretion von heterologen Proteinen stark
durch den Abbau von Proteasen limitiert ist. Mittlerweile wird zwar vielfach der Stamm
D15 genutzt, der das Medium weniger ansäuert und somit weniger Proteasen exprimiert,
aber vollkommen frei von proteolytischem Abbau sind die Kultivierungen nicht (detailliert
im Kapitel 1.4.2). Die hohe Sekretionsleistung ist A. niger größtes Potential gegenüber den
anderen, häufig genutzten biotechnologisch relevanten Mikroorganismen und daher ist es
von großer Relevanz zukünftig den Abbau der produzierten Proteine zu verhindern.

- 188 -
Publikationen
Diese kumulative Dissertationsschrift basiert auf folgenden Publikationen:
Die Reihenfolge der entsprechenden Kapitel ist chronologisch nach dem Erscheinungsdatum
der Publikation geordnet.
Kapitel 2: Fiedler MRM, Nitsche BM, Wanka F, Meyer V: Aspergillus. A Cell Factory with
Unlimited Prospects. Applications of Microbial Engineering. CRC Press 2013. 1-51 pp. doi:
10.1201/b15250-2
Kapitel 3: Richter L*, Wanka F*, Boecker S, Storm D, Kurt T, Vural Ö, Süßmuth R, Meyer
V. Engineering of Aspergillus niger for the production of secondary metabolites. Fungal
Biology and Biotechnology 2014, 1:4, doi: 10.1186/s40694-014-0004-9
Kapitel 4: Wanka F, Cairns T, Boecker S, Berens C, Happel A, Zheng X, Sun J, Krappmann
S, Meyer V. Tet-on, or Tet-off, that is the question: Advanced Conditional Gene Expression
in Aspergillus. Fungal Genetics and Biology 2016, 89, 72-83, doi: 10.1016/j.fgb.2015.11.003
Kapitel 5: Wanka F, Arentshorst M, Cairns T, Jørgensen
T, Ram AFJ, Meyer
V. Highly active
promoters and native secretion signals for protein production during extremely low growth
rates in Aspergillus niger. Submitted to Microbial Cell Factories 04.03.2016
* geteilte Erstautorschaft
Tabelle 5: Arbeitsanteil von F. Wanka an den Publikationen der vorliegenden
kumulativen Schrift.
Publikation Konzept Durchführung und Auswertung
der Versuche
Verfassen der
Publikation
Fiedler et al. 2013 20 % 20 %
Richter*, Wanka*
et al. 2014 40 % 50 % 40 %
Wanka et al. 2016
FGB 80 % 85 % 70 %
Wanka et al. 2016 70 % 80 % 70 %
* geteilte Erstautorschaft

- 189 -
Patente
Europäische Patentanmeldung 14 160 821.6, „Filamentöse Pilze als Wirtsorganismen für die
Produktion von Sekundärmetaboliten“, 20.03.2014
Internationale Patentanmeldung PCT/EP 2015/055978, „Method for obtaining microbial
secondary metabolite or a derivative thereof by heterologous expression of at least one
synthetase of said secondary metabolite in a filamentous fungi“, 20.03.2015, Veröffentlichung
am 24.09.2015 unter WO 2015/140315 A2. Zusätzlich wurde “Process for producing
chimeric cyclooligodepsipeptides in filamentous fungi” am 3.12. 2015 unter WO 2015/140315
A3 veröffentlicht.
Posterbeiträge
Wanka F., Fiedler M., van Gent J., Arentshorst M., van den Hondel C., Ram A., Meyer V.
Fungal gene expression on demand: An inducible, tuneable and metabolism-independent
expression system for Aspergillus niger.
2011, VAAM (Vereinigung für Allgemeine und Angewandte Mikrobiologie), Marburg
Wanka F., Arentshorst M., Jørgensen T., Ram A., Meyer V.
Novel promoters for increased protein expression during high-cell density continuous
cultivation of Aspergillus niger.
2013, RPP7 (Konferenz zur rekombinanten Proteinproduktion), Laupheim
Wanka F., Arentshorst M., Jørgensen T., Ram A., Meyer V.
Novel promoters for increased protein expression during high-cell density continuous
cultivation of Aspergillus niger.
2013, DECHEMA Jahrestagung (Gesellschaft für Chemische Technik und Biotechnologie),
Frankfurt
Wanka F., Boecker S., Arentshorst M., Ram A., Meyer V.
Gene silencing on demand: Establishment of the Tet-off system for Aspergillus niger.
2014, ECFG 12 (Europäische Konferenz für pilzliche Genetik), Sevilla (Spanien)
Wanka F., Arentshorst M., Ram A., Meyer V.
Gene silencing on demand: Establishment of the Tet-off system for Aspergillus niger.
2014, VAAM, Dresden
Wanka F., Meyer V.
Choose On or Off: A regulatable gene expression system for filamentous fungi.
2015, FGC (Konferenz für pilzliche Genetik), Asilomar (USA)

- 190 -
Wanka F., Meyer V.
Tet-off system –A quantitative control of gene expression in Aspergillus niger.
2015, MBF (Molekulare Biologie für Pilze, Fachgruppe der VAAM), Berlin
Wanka F., Arentshorst M., Jørgensen T., Ram A., Meyer V.
Highly active promoters for protein production during extremely low growth rates in
Aspergillus niger.
2016, ECFG 13, Paris (Frankreich)
Stipendien und Preise
Stipendium für einen vierwöchigen Aufenthalt am Technion in Haifa (Israel) von der DTG
(Deutsche Technion-Gesellschaft), 2014
Stipendium für die Kongressreise zur FGC in die USA vom DAAD (Deutscher Akademischer
Austauschdienst), 2015
Gewinner des Novozymes Best Student Poster Award für das Poster mit dem Titel „Choose
On or Off: A regulatable gene expression system for filamentous fungi“ beim 12. Aspergillus
meeting in der USA, 2015

- 191 -
Danksagungen
Obwohl nur ein Name auf dem Deckel der Arbeit steht, ist diese Dissertation gewiss nicht
eine einzelne Leistung und daher möchte ich meinen Dank gegenüber allen Menschen
ausdrücken, die Anteil genommen und mir geholfen haben, dieses Ziel zu erreichen.
Zuerst möchte ich Vera danken, ohne ihre Beharrlichkeit und Überzeugungskraft hätte ich
mich möglicherweise gar nicht für den Weg der Promotion und des Studiums der
Schimmelpilze entschieden. Ich danke dir für Dein Vertrauen, Deine Geduld und Deine
positive Sicht der Dinge.
Markus, meinem Leidensgenossen, danke ich sehr für den wissenschaftlichen Austausch, die
Unterstützung und die vielen geteilten kulinarischen Highlights. Ich bin äußerst dankbar, dass
Benjamin, unser kurzweiliger Post Doc, das Bioreaktorlabor aufgebaut hat, sonst hätte ich
wesentlich weniger zu tun gehabt . Sehr, sehr großer Dank geht auch an die frischeren
Doktoranden: den lustigen Norman, der fleißigen und hilfsbereiten Tabea und dem stets gut
gelaunten Simon. I would like to thank Charlie, for the fact that you joined our lab and helped
me a lot with more than proof reading my English. Der holländischen Muttergruppe danke
ich, weil ich von Mark viel Praktisches gelernt habe, Arthur immer tolle Ideen hat und Cees
der witzigste Prof. ist. Meinen Studenten Dirk, Simon, Tutku, Özlem, Ulrike O., Ulrike W.,
Aike, Johanna und Irmi danke ich für ihre Arbeit und ihren Einsatz. Bedanken möchte ich
mich bei Rita, die mir durch ihre super geführte Spülküche viel Arbeit abgenommen hat,
ebenso bei Birgit für die tadellose Unterstützung in den Praktika und bei Roslin für die
Englisch Stunden bzw. Hilfe. Ich möchte mich bei allen Kollegen bedanken für die schönen
gemeinsamen Mittags- oder Kaffeepausen, die vielen Leckereien, die einzigartigen
Betriebsausflüge, die glühweinlastigen Weihnachtsfeiern, die leckeren Neujahrsfrühstücke,
die wilden Jubiläumsfeiern, die grillreichen BSc Abschlussfeste aka Tag der BT, die
organisationsreichen sehr langen Nächte der Wissenschaft, das unterhaltsame Berlin-Leiden
Treffen, unsere eigene weltbeste MBF Konferenz ... ohne diese ganzen Höhepunkte wären die
5 Jahre nicht so schnell vergangen. In besonderer Erinnerung bleiben mir diverse
Nebenprojekte wie die Apfelweinproduktion, die Garnelenaufzucht und das Chinchilla-
Verdauungsprojekt, auch die romantischen Sonnenuntergänge in der botanischen Glaskuppel
und die zahlreichen Kongresse an tollen Orten haben die PhD Zeit wesentlich aufgewertet.
Meinen Freunden Tine R., Franzi, Melli, Flavia, Inge, Ise, Flo, Tine D., Rici, Tom und allen
anderen danke ich fürs Zuhören und Erden. Und zu guter Letzt danke ich meinen Eltern,
Omas`s und allen die mich stets unterstützt haben und zum Gelingen der Arbeit beigetragen
haben. Eure Franzi

- 192 -
Referenzen
ACUNA-ARGUELLES, M.E., GUTIIERREZ-ROJAS, M., VINIEGRA-GONZALEZ, G. & FAVELA-
TORRES, E. 1995. Production and properties of three pectinolytic activities produced by
Aspergillus niger in submerged and solid-state fermentation. Appl Microbiol Biotechnol, 43,
808–814.
AHUJA, M. & PUNEKAR, N.S. 2008. Phosphinothricin resistance in Aspergillus niger and its
utility as a selectable transformation marker. Fungal Genetics and Biology, 45, 1103–1110,
AINSWORTH, G.C. 1976. Introduction to the History of Mycology, Cambridge University
Press., 359 pp
ALBERTS, A.W. 1988. Discovery, biochemistry and biology of lovastatin. Am J Cardiol., 62,
10 – 15
AMANULLAH, A., CHRISTENSEN, L.H., HANSEN, K., NIENOW, A.W. & THOMAS, C.R. 2002.
Dependence of morphology on agitation intensity in fed-batch cultures of Aspergillus oryzae
and its implications for recombinant protein production. Biotechnology and Bioengineering,
77, 815–826
ANDERSEN, M.R., LEHMANN, L. & NIELSEN, J. 2009. Systemic analysis of the response of
Aspergillus niger to ambient pH. Genome Biology, 10, R47
ANDERSEN, M.R., SALAZAR, M.P., SCHAAP, P.J., VONDERVOORT, P.J.I. VAN DE, CULLEY, D.,
THYKAER, J., FRISVAD, J.C., … BAKER, S.E. 2011. Comparative genomics of citric-acid-
producing Aspergillus niger ATCC 1015 versus enzyme-producing. 21, 885–897
ANDRADE, A.C., VAN NISTELROOY, J.G., PEERY, R.B., SKATRUD, P.L. & DE WAARD, M.A.
2000. The role of ABC transporters from Aspergillus nidulans in protection against cytotoxic
agents and in antibiotic production. Molecular & general genetics, 263, 966–977
ANYAOGU, D.C. & MORTENSEN, U.H. 2015. Manipulating the glycosylation pathway in
bacterial and lower eukaryotes for production of therapeutic proteins. Current Opinion in
Biotechnology, 36
ARENTSHORST, M., NIU, J. & RAM, A.F.J. 2015. Efficient Generation of Aspergillus niger
Knock Out Strains by Combining NHEJ Mutants and a Split Marker Approach. In Genetic
Transformation Systems in Fungi, Fungal Biology. 263–272
BACCHUS, W. & FUSSENEGGER, M. 2012. The use of light for engineered control and
reprogramming of cellular functions. Current Opinion in Biotechnology, 23, 695–702
BASAN, M., HUI, S., OKANO, H., ZHANG, Z., SHEN, Y., WILLIAMSON, J.R. & HWA, T. 2015.
Overflow metabolism in Escherichia coli results from efficient proteome allocation. 528
BENNETT, J.W. 1998. Mycotechnology: The role of fungi in biotechnology. Journal of
Biotechnology, 66, 101–107

- 193 -
BEYER, H.M., GONSCHOREK, P., SAMODELOV, S.L., MEIER, M., WEBER, W. & ZURBRIGGEN,
M.D. 2015. AQUA cloning: A versatile and simple enzyme-free cloning approach. PLoS
ONE, 10
BODIE, E., BOWER, B., BERKA, R. & DUNN-COLEMAN, N. 1994. Economically important
organic acid and enzyme products. Prog Ind Microbiol, 29, 561–602.
BOENDER, L.G.M., VAN MARIS, A.J.A., DE HULSTER, E.A., ALMERING, M.J.H., VAN DER
KLEI, I.J., VEENHUIS, M., DE WINDE, J.H., PRONK, J.T. & DARAN-LAPUJADE, P. 2011.
Cellular responses of Saccharomyces cerevisiae at near-zero growth rates: transcriptome
analysis of anaerobic retentostat cultures. FEMS yeast research, 11, 603–620
BROEKHUIJSEN, M.P., MATTERN, I.E., CONTRERAS, R., KINGHORN, J.R. & VAN DEN HONDEL,
C.A.M.J.J. 1993. Secretion of heterologous proteins by Aspergillus niger: Production of
active human interleukin-6 in a protease-deficient mutant by KEX2-like processing of a
glucoamylase-hIL6 fusion protein. Journal of Biotechnology, 31, 135–145
BUXTON, F.P., GWYNNE, D.I. & DAVIES, R.W. 1989. Cloning of a new bidirectionally
selectable marker for Aspergillus strains. Gene, 84, 329–334
CARLSEN, M., NIELSEN, J.B. & VILLADSEN, J. 1996. Growth and a amylase production by
Aspergillus oryzae during continuous cultivations. J Biotechnol, 45, 81–93.
CEREGHINO, J.L. & CREGG, J.M. 2000. Heterologous protein expression in the methylotrophic
yeast Pichia pastoris. FEMS Microbiology Reviews, 24, 45–66.
COHEN, B. 1977. The proteases of Aspergilli. In Smith, J. & Pateman, J., eds. Genetics and
Physiology of Aspergillus. Academic Press, New York, 71–75
COLIN, V.L., BAIGORÍ, M.D. & PERA, L.M. 2013. Tailoring fungal morphology of Aspergillus
niger MYA 135 by altering the hyphal morphology and the conidia adhesion capacity:
biotechnological applications. AMB Express, 3, 1–13
CONESA, A., VAN DEN HONDEL, C.A.M.J.J. & PUNT, P.J. 2000. Studies on the Production of
Fungal Peroxidases in Aspergillus niger Studies on the Production of Fungal Peroxidases in
Aspergillus niger. 66, 3016–3023
CULLEN, D., GRAY, G.L., WILSON, L.J., HAYENGA, K.J., MICHAEL, H., REY, M.W., NORTON,
S. & BERKA, R.M. 1987. Controlled expression and secretion of bovine chymosin in
Aspergillus nidulans. Nature Biotechnology, 5, 369–376
DASHTBAN, M., SCHRAFT, H. & QIN, W. 2009. Fungal Bioconversion of Lignocellulosic
Residues ; Opportunities & Perspectives. Int. J. Biol. Sci., 5, 578–595
DAVE, K., AHUJA, M., JAYASHRI, T.N., SIROLA, R.B. & PUNEKAR, N.S. 2012. A novel
selectable marker based on Aspergillus niger arginase expression. Enzyme and microbial
technology, 51, 53–58
DAVE, K. & PUNEKAR, N.S. 2011. Utility of Aspergillus niger citrate synthase promoter for
heterologous expression. Journal of Biotechnology, 155, 173–177

- 194 -
DE OLIVEIRA, J.M.P.F. & DE GRAAFF, L.H. 2011. Proteomics of industrial fungi: trends and
insights for biotechnology. Applied microbiology and biotechnology, 89, 225–237
DE VRIES, R.P., VISSER, J. & DE GRAAFF, L.H. 1999. CreA modulates the XlnR-induced
expression on xylose of Aspergillus niger genes involved in xylan degradation. Research in
Microbiology, 150, 281–285
DESHPANDE, N., WILKINS, M.R., PACKER, N. & NEVALAINEN, H. 2008. Protein glycosylation
pathways in filamentous fungi. Glycobiology, 18, 626–6374.
DICHTL, K., HELMSCHROTT, C., DIRR, F. & WAGENER, J. 2012. Deciphering cell wall
integrity signalling in Aspergillus fumigatus: Identification and functional characterization of
cell wall stress sensors and relevant Rho GTPases. Molecular Microbiology, 83, 506–519
DONNELL, D.O., WANG, L., XU, J., RIDGWAY, D., GU, T. & MOO-YOUNG, M. 2001. Enhanced
heterologous protein production in Aspergillus niger through pH control of extracellular
protease activity. Biochemical Engineering Journal, 8, 187–193
DOWZER, C.E.A. & KELLY, J.M. 1991. Analysis of mutations in the creA gene involved in
carbon catabolite repression in Aspergillus nidulans. Molecular and Cellular Biology, 11,
5701–5709
DRIOUCH, H., SOMMER, B. & WITTMANN, C. 2010. Morphology engineering of Aspergillus
niger for improved enzyme production. Biotechnology and bioengineering, 105, 1058–1068
DUNN-COLEMAN, N.S., BLOEBAUM, P., BERKA, R.M., BODIE, E., ROBINSON, N.,
ARMSTRONG, G., WARD, M., PRZETAK, M., CARTER, G.L. & LACOST, R. 1991. Commercial
levels of chymosin production by Aspergillus. Biotechnology (N Y), 9, 976–981
DYNESEN, J. & NIELSEN, J. 2003. Surface hydrophobicity of Aspergillus nidulans
conidiospores and its role in pellet formation. Biotechnology Progress, 19, 1049–1052
ELPHINSTONE, M.S., HINTEN, G.N., ANDERSON, M.J. & NOCK, C.J. 2003. An inexpensive and
high-throughput procedure to extract and purify total genomic DNA for population studies.
Molecular Ecology Notes, 3, 317–320
ENFORS, S. & HÄGGSTRÖM, L. 2000. Bioprocess Technolgy Fundamentals and Applications,
Stockholm, Sweden, 355 pp.
ERCAN, O., BISSCHOPS, M.M.M., OVERKAMP, W., JØRGENSEN, T.R., RAM, A.F., SMID, E.J.,
PRONK, J.T., KUIPERS, O.P., DARAN-LAPUJADE, P. & KLEEREBEZEM, M. 2015. Physiological
and transcriptional responses of different industrial microbes at near-zero specific growth
rates. Applied and Environmental Microbiology, 81, 5662–5670
FLEISSNER, A. & DERSCH, P. 2010. Expression and export: recombinant protein production
systems for Aspergillus. Applied microbiology and biotechnology, 87, 1255–1270
FONTANA, R.C., SALVADOR, S. & DA SILVEIRA, M.M. 2005. Influence of pectin and glucose
on growth and polygalacturonase production by Aspergillus niger in solid-state cultivation.
Journal of Industrial Microbiology and Biotechnology, 32, 371–377

- 195 -
FOWLER, T., BERKA, R.M. & WARD, M. 1990. Regulation of the glaA gene of Aspergillus
niger. Current Genetics, 18, 537–545
FUSSENEGGER, M., MORRIS, R.P., FUX, C., RIMANN, M., VON STOCKAR, B., THOMPSON, C.J.
& BAILEY, J.E. 2000. Streptogramin-based gene regulation systems for mammalian cells.
Nature biotechnology, 18, 1203–1208
GAMARRA, N.N., VILLENA, G.K. & GUTIERREZ-CORREA, M. 2010. Cellulase production by
Aspergillus niger in biofilm, solid-state, and submerged fermentations. Applied Microbiology
and Biotechnology, 87, 545–551
GEISER, D.M., SAMSON, R.A. & VARGA, J. 2008. A review of molecular phylogenetics in
Aspergillus and prospects for a robust genus-wide phylogeny. In Varga, K. & Samson, R.A.,
eds. Aspergillus in the genomic era. 17–32
GILL, G. & PTASHNE, M. 1988. Negative effect of the transcriptional activator GAL4. Nature,
334, 721–724
GOOSEN, T., VAN ENGELENBURG, F., DEBETS, F., SWART, K., BOS, K. & VAN DEN BROEK, H.
1989. Tryptophan auxotrophic mutants in Aspergillus niger: Inactivation of the trpC gene by
cotransformation mutagenesis. Molecular & General Genetics, 219, 282–288
GORDON, C.L., KHALAJ, V., RAM, A.F.J., ARCHER, D.B., BROOKMAN, J.L., TRINCI, A.P.J.,
JEENES, D.J., DOONAN, J.H., WELLS, B. & PUNT, P.J. 2000. Glucoamylase :: green fluorescent
protein fusions to monitor protein secretion in Aspergillus niger. Microbiology, 146, 415–426
GOSSEN, M. & BUJARD, H. 1992. Tight control of gene expression in mammalian cells by
tetracycline-responsive promoters. Cell Biology, 89, 5547–5551
GRESSLER, M., HORTSCHANSKY, P., GEIB, E. & BROCK, M. 2015. A new high-performance
heterologous fungal expression system based on regulatory elements from the Aspergillus
terreus terrein gene cluster. Frontiers in Microbiology, 6
GRIMM, L.H., KELLY, S., KRULL, R. & HEMPEL, D.C. 2005. Morphology and productivity of
filamentous fungi. Applied Microbiology and Biotechnology, 69, 375–384
GYAMERAH, M., MERICHETTI, G., ADEDAYO, O., SCHARER, J.M. & MOO-YOUNG, M. 2002.
Bioprocessing strategies for improving hen egg-white lysozyme (HEWL) production by
recombinant Aspergillus niger HEWL WT-13-16. Applied Microbiology and Biotechnology,
60, 403–407
HABISON, A., KUBICEK, C. & RÖHR, M. 1979. Phosphofructokinase as a regulatory enzyme in
citric acid producing Aspergillus niger. FEMS microbiology letters, 5, 39–42
HELMSCHROTT, C., SASSE, A., SAMANTARAY, S., KRAPPMANN, S. & WAGENER, J. 2013.
Upgrading fungal gene expression on demand: improved systems for doxycycline-dependent
silencing in Aspergillus fumigatus. Applied and environmental microbiology, 79, 1751–1754
JACOBS, D.I., OLSTHOORN, M.M.A., MAILLET, I., AKEROYD, M., BREESTRAAT, S., DONKERS,
S., VAN DER HOEVEN, R.A., … SAGT, C.M.J. 2009. Effective lead selection for improved
protein production in Aspergillus niger based on integrated genomics. Fungal Genetics and
Biology :, 46, 141–152

- 196 -
JAYAPAL, K.P., WLASCHIN, K.F., HU, W.-S. & YAP, M.G.S. 2007. Recombinant Protein
Therapeutics from CHO Cells — 20 Years and Counting. CHO Consortium, SBE Specia, 40–
47.
JØRGENSEN, T.R., GOOSEN, T., VAN DEN HONDEL, C.A.M.J.J., RAM, A.F.J. & IVERSEN, J.J.L.
2009. Transcriptomic comparison of Aspergillus niger growing on two different sugars
reveals coordinated regulation of the secretory pathway. BMC Genomics, 10
JØRGENSEN, T.R., NITSCHE, B.M., LAMERS, G.E., ARENTSHORST, M., VAN DEN HONDEL, C. &
RAM, A.F. 2010. Transcriptomic insights into the physiology of Aspergillus niger
approaching a specific growth rate of zero. Applied and environmental microbiology, 76,
5344–5355
KAINZ, E., GALLMETZER, A., HATZL, C., NETT, J.H., LI, H., SCHINKO, T., PACHLINGER, R., …
STRAUSS, J. 2008. N-glycan modification in Aspergillus species. Applied and Environmental
Microbiology, 74, 1076–1086
KALB, D., HEINEKAMP, T., LACKNER, G., SCHARF, D.H., DAHSE, H.M., BRAKHAGE, A.A. &
HOFFMEISTER, D. 2015. Genetic Engineering activates biosynthesis of aromatic fumaric acid
amides in the human pathogen Aspergillus fumigatus. Applied and Environmental
Microbiology, 81, 1594–1600.
KARLSSON, M., WEBER, W. & FUSSENEGGER, M. 2011. De novo design and construction of
an inducible gene expression system in mammalian cells. In Methods in Enzymology. Elsevier
Inc., 239–253
KARLSSON, M., WEBER, W. & FUSSENEGGER, M. 2012. Design and Construction of Synthetic
Gene Networks in Mammalian Cells. In Weber, W. & Fussenegger, M., eds. Synthetic Gene
Networks: Methods and Protocols, Methods in Molecular Biology. 287–300
KASAJIMA, Y., YAMAGUCHI, M., HIRAI, N., OHMACHI, T. & YOSHIDA, T. 2006. In Vivo
Expression of UDP- N -Acetylglucosamine: α-3-D-Mannoside β-1,2- N -
Acetylglucosaminyltransferase I (GnT-1) in Aspergillus oryzae and Effects on the Sugar
Chain of α-Amylase. Bioscience, Biotechnology and Biochemistry, 70, 2662–2668
KEASLING, J. 2008. Synthetic biology for synthetic chemistry. ACS chemical biology, 3, 64–
76
KELLY, J.M. & HYNES, M.J. 1985. Transformation of Aspergillus niger by the amdS gene of
Aspergillus nidulans. The EMBO journal, 4, 475–479
KLUYVER, A. & PERQUIN, L. 1932. Zur Methodik der Schimmelstoffwechseluntersuchung.
Biochem Z, 266, 68–81
KONSTANTINOV, K.B., TSAI, Y., MOLES, D. & MATANGUIHAN, R. 1996. Control of long-term
perfusion Chinese hamster ovary cell culture by glucose auxostat. Biotechnology Progress,
12, 100–109
KOVALCHUK, A. & DRIESSEN, A.J.M. 2010. Phylogenetic analysis of fungal ABC
transporters. BMC genomics, 11

- 197 -
KUBICEK, C.P., ZEHENTGRUBER, O., EL-KALAK, H. & RÖHR, M. 1980. Regulation of citric
acid production by oxygen: Effect of dissolved oxygen tension on adenylate levels and
respiration in Aspergillus niger. Applied Microbiology and Biotechnology, 9, 101–11
KWON, M.J., ARENTSHORST, M., FIEDLER, M., DE GROEN, F.L.M., PUNT, P.J., MEYER, V. &
RAM, A.F.J. 2014. Molecular genetic analysis of vesicular transport in Aspergillus niger
reveals partial conservation of the molecular mechanism of exocytosis in fungi. Microbiology,
160, 316–329
KWON, M.J., JØRGENSEN, T.R., NITSCHE, B.M., ARENTSHORST, M., PARK, J., RAM, A.F.J. &
MEYER, V. 2012. The transcriptomic fingerprint of glucoamylase over-expression in
Aspergillus niger. BMC Genomics, 13
KWON, M.J., NITSCHE, B.M., ARENTSHORST, M., JØRGENSEN, T.R., RAM, A.F.J. & MEYER,
V. 2013. The Transcriptomic Signature of RacA Activation and Inactivation Provides New
Insights into the Morphogenetic Network of Aspergillus niger. 8
LARRALDE-CORONA, C.P., LOPEZ-ISUNZA, F. & VINIEGRA-GONZALES, G. 1997.
Morphometric evaluation of the specific growth rate of Aspergillus niger grown in agar plates
at high glucose levels. Biotechnology and bioengineering, 56, 287–294
LENOUVEL, F., VAN DE VONDERVOORT, P.J.I. & VISSER, J. 2002. Disruption of the Aspergillus
niger argB gene: A tool for transformation. In Current Genetics. 425–432
LEONG, S. L L., HOCKING, A.D. & SCOTT, E.S. 2006. Effect of temperature and water activity
on growth and ochratoxin A production by Australian Aspergillus carbonarius and A. niger
isolates on a simulated grape juice medium. International Journal of Food Microbiology, 110,
209–216
LI, Q., HARVEY, L.M. & MCNEIL, B. 2008. Oxygen enrichment effects on protein oxidation,
proteolytic activity and the energy status of submerged batch cultures of Aspergillus niger B1-
D. Process Biochemistry, 43, 238–243
LINK, T., BÄCKSTRÖM, M., GRAHAM, R., ESSERS, R., ZÖRNER, K., GÄTGENS, J., BURCHELL,
J., TAYLOR-PAPADIMITRIOU, J., HANSSON, G.C. & NOLL, T. 2004. Bioprocess development
for the production of a recombinant MUC1 fusion protein expressed by CHO-K1 cells in
protein-free medium. Journal of Biotechnology, 110, 51–62
LOCKWOOD, L. & MOYER, A. 1938. The production of chemicals by filamentous fungi. The
Botanical Review, 4, 140-164
LUBERTOZZI, D. & KEASLING, J.D. 2009. Developing Aspergillus as a host for heterologous
expression. Biotechnology Advances, 27, 53–75
MACHELEIDT, J., SCHERLACH, K., NEUWIRTH, T., SCHMIDT-HECK, W., STRAßBURGER, M.,
SPRAKER, J., BACCILE, J. A., … BRAKHAGE, A. A. 2015. Transcriptome analysis of cyclic
AMP-dependent protein kinase A-regulated genes reveals the production of the novel natural
compound fumipyrrole by Aspergillus fumigatus. Molecular Microbiology, 96, 148–162
MACHIDA, M. AND GOMI, K. 2010. Aspergillus. Molecular Biology and Genomics, Caister
Academic Press, 238 pp

- 198 -
MADRY, N., ZOCHER, R. & KLEINKAUF, H. 1983. Microbiology and Enniatin Production by
Fusarium oxysporum in Chemically Defined Media. Applied Microbiology and
Biotechnology, 17, 75–79
MAHADIK, N.D., PUNTAMBEKAR, U.S., BASTAWDE, K.B., KHIRE, J.M. & GOKHALE, D. V.
2002. Production of acidic lipase by Aspergillus niger in solid state fermentation. Process
Biochemistry, 38, 715–721
MAJOLLI, M. & AGUIRRE, S. 1999. Effect of trace metals on the cell morphology, enzymic
activity and citric acid production in a strain of Aspergillus wentii. Rev. Argent. Microbiol.,
31, 65–71
MARTÍN, J.F., CASQUEIRO, J. & LIRAS, P. 2005. Secretion systems for secondary metabolites:
How producer cells send out messages of intercellular communication. Current Opinion in
Microbiology, 8, 282–293
MARTIN, P., ALBAGLI, O., POGGI, M.C., BOULUKOS, K.E. & POGNONEC, P. 2006.
Development of a new bicistronic retroviral vector with strong IRES activity. BMC
Biotechnology, 6
MATTERN, I.E., VAN NOORT, J.M., VAN DEN BERG, P., ARCHER, D.B., ROBERTS, I.N. & VAN
DEN HONDEL, C.A.M.J.J. 1992. Isolation and characterization of mutants of Aspergillus niger
deficient in extracellular proteases. Molecular & General Genetics, 234, 332–336
MCINTYRE, M. & MCNEIL, B. 1997. Dissolved carbon dioxide effects on morphology,
growth, and citrate production in Aspergillus niger A60. Enzyme and Microbial Technology,
20, 135–142
MEYER, V., ARENTSHORST, M., EL-GHEZAL, A., DREWS, A.-C., KOOISTRA, R., VAN DEN
HONDEL, C.A.M.J.J. & RAM, A.F.J. 2007. Highly efficient gene targeting in the Aspergillus
niger kusA mutant. Journal of Biotechnology, 128, 770–775
MEYER, V., ARENTSHORST, M., FLITTER, S.J., NITSCHE, B.M., KWON, M.J., REYNAGA-PENA,
C.G., BARTNICKI-GARCIA, S., VAN DEN HONDEL, C.A.M.J.J. & RAM, A.F.J. 2009.
Reconstruction of Signaling Networks Regulating Fungal Morphogenesis by Transcriptomics.
Eukaryotic Cell, 8, 1677–1691
MEYER, V., RAM, A. & PUNT, P. 2010. Genetics, genetic manipulation and approaches to
strain improvement of filamentous fungi. In Manual of industrial microbiology and
biotechnology. 3rd ed. NY: Wiley. 318–329.
MEYER, V., WANKA, F., VAN GENT, J., ARENTSHORST, M., VAN DEN HONDEL, C.A.M.J.J. &
RAM, A.F.J. 2011. Fungal gene expression on demand: an inducible, tunable, and metabolism-
independent expression system for Aspergillus niger. Applied and environmental
microbiology, 77, 2975–2983
MIZUTANI, O., MASAKI, K., GOMI, K. & IEFUJI, H. 2012. Modified Cre-loxP recombination in
Aspergillus oryzae by direct introduction of Cre recombinase for marker gene rescue. Applied
and Environmental Microbiology, 78, 4126–4133

- 199 -
MÜLLER, C., MCINTYRE, M., HANSEN, K. & NIELSEN, J. 2002. Metabolic Engineering of the
Morphology of Aspergillus oryzae by Altering Chitin Synthesis. 68, 1827–1836
MULLICK, A., XU, Y., WARREN, R., KOUTROUMANIS, M., GUILBAULT, C., BROUSSAU, S.,
MALENFANT, F., … MASSIE, B. 2006. The cumate gene-switch: a system for regulated
expression in mammalian cells. BMC Biotechnology, 6
NITSCHE, B.M., JØRGENSEN, T.R., AKEROYD, M., MEYER, V. & RAM, A.F.J. 2012. The
carbon starvation response of Aspergillus niger during submerged cultivation: insights from
the transcriptome and secretome. BMC Genomics, 13.
NIU, J., ARENTSHORST, M., NAIR, P.D.S., DAI, Z., BAKER, S., FRISVAD, J.C., NIELSEN, K.F.,
PUNT, P.J. & RAM, A.F.J. 2016. Identification of a Classical Mutant in the Industrial Host
Aspergillus niger by Systems Genetics: LaeA Is Required for Citric Acid Production and
Regulates the Formation of Some Secondary Metabolites. G3:Genes,Genomes,Genetics, 6,
193–204
NØDVIG, C.S., NIELSEN, J.B., KOGLE, M.E. & MORTENSEN, U.H. 2015. A CRISPR-Cas9
System for Genetic Engineering of Filamentous Fungi. Plos One, 10
ODA, K., KAKIZONO, D., YAMADA, O., IEFUJI, H., AKITA, O. & IWASHITA, K. 2006.
Proteomic Analysis of Extracellular Proteins from Aspergillus oryzae Grown under
Submerged and Solid-State Culture Conditions Proteomic Analysis of Extracellular Proteins
from Aspergillus oryzae Grown under Submerged and Solid-State Culture Conditi. Applied
and environmental microbiology, 72, 3448–3457
OUEDRAOGO, J.P., HAGEN, S., SPIELVOGEL, A., ENGELHARDT, S. & MEYER, V. 2011.
Survival strategies of yeast and filamentous fungi against the antifungal protein AFP. The
Journal of biological chemistry, 286, 13859–13868
OVERKAMP, W., ERCAN, O., HERBER, M., MARIS, A.J.A. VAN, KLEEREBEZEM, M. &
KUIPERS, O.P. 2015. Physiological and cell morphology adaptation of Bacillus subtilis at
near-zero specific growth rates : a transcriptome analysis. 17, 346–363
PACHLINGER, R., MITTERBAUER, R., ADAM, G. & STRAUSS, J. 2005. Metabolically
independent and accurately adjustable Aspergillus sp. expression system. Applied and
Environmental Microbiology, 71, 672–678
PAKULA, T.M., SALONEN, K., UUSITALO, J. & PENTTILÄ, M. 2005. The effect of specific
growth rate on protein synthesis and secretion in the filamentous fungus Trichoderma reesei.
Microbiology, 151, 135–143
PANULA-PERÄLÄ, J., SIURKUS, J., VASALA, A., WILMANOWSKI, R., CASTELEIJN, M.G.,
NEUBAUER, P., ŠIURKUS, J., … NEUBAUER, P. 2008. Enzyme controlled glucose auto-delivery
for high cell density cultivations in microplates and shake flasks. Microbial Cell Factories, 7
PAPAGIANNI, M. 2007. Advances in citric acid fermentation by Aspergillus niger:
Biochemical aspects, membrane transport and modeling. Biotechnology Advances, 25, 244–
263

- 200 -
PAPAGIANNI, M. 2004. Fungal morphology and metabolite production in submerged mycelial
processes. Biotechnology Advances, 22, 189–259
PAPAGIANNI, M. & MATTEY, M. 2006. Morphological development of Aspergillus niger in
submerged citric acid fermentation as a function of the spore inoculum level. Application of
neural network and cluster analysis for characterization of mycelial morphology. Microbial
cell factories, 5
PAPAGIANNI, M., MATTEY, M. & KRISTIANSEN, B. 1999. The influence of glucose
concentration on citric acid production and morphology of Aspergillus niger in batch and
culture. Enzyme and Microbial Technology, 25, 710–717
PEL, H.J., DE WINDE, J.H., ARCHER, D.B., DYER, P.S., HOFMANN, G., SCHAAP, P.J., TURNER,
G., … STAM, H. 2007. Genome sequencing and analysis of the versatile cell factory
Aspergillus niger CBS 513.88. Nature biotechnology, 25, 221–231
PERA, L.M. & CALLIERI, D.A. 1999. Influence of calcium on fungal growth and citric acid
production during fermentation of a sugarcane molasses-based medium by a strain of
Aspergillus niger. World Journal of Microbiology and Biotechnology, 15, 647–649
PETERSEN, K.L., LEHMBECK, J. & CHRISTENSEN, T. 1999. A new transcriptional activator for
amylase genes in Aspergillus. Molecular and General Genetics, 262, 668–676
PINAGA, F., FERNANDEZ-ESPINAR, M.T., VALLES, S. & RAMON, D. 1994. Xylanase
Production in Aspergillus nidulans: Induction and Carbon Catabolite Repression. FEMS
Microbiology Letters, 115, 319–324
POLLOCK, J., HO, S. V. & FARID, S.S. 2013. Fed-batch and perfusion culture processes:
Economic, environmental, and operational feasibility under uncertainty. Biotechnology and
Bioengineering, 110, 206–219
POSTMA, E., VERDUYN, C., SCHEFFERS, W.A. & VAN DIJKEN, J.P. 1989. Enzymic Analysis of
the Crabtree Effect in Glucose-Limited Chemostat Cultures of Saccharomyces cerevisiae.
Applied and environmental microbiology, 55, 468–477
PUNT, P.J., DINGEMANSE, M.A., KUYVENHOVEN, A., SOEDE, R.D.M., POUWELS, P.H. & VAN
DEN HONDEL, C.A.M.J.J. 1990. Functional elements in the promoter region of the Aspergillus
nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene, 93, 101–109
PUNT, P.J., OLIVER, R.P., DINGEMANSE, M.A., POUWELS, P.H. & VAN DEN HONDEL,
C.A.M.J.J. 1987. Transformation of Aspergillus based on the hygromycin B resistance marker
from Escherichia coli. Gene, 56, 117–124
PUNT, P.J., SCHUREN, F.H.J., LEHMBECK, J., CHRISTENSEN, T., HJORT, C. & VAN DEN
HONDEL, C.A.M.J.J. 2008. Characterization of the Aspergillus niger prtT, a unique regulator
of extracellular protease encoding genes. Fungal Genetics and Biology, 45, 1591–1599
PUNT, P.J., VAN BIEZEN, N., CONESA, A., ALBERS, A., MANGNUS, J. & VAN DEN HONDEL, C.
2002. Filamentous fungi as cell factories for heterologous protein production. Trends in
Biotechnology, 20, 200–206

- 201 -
PUNT, P.J. & VAN DEN HONDEL, C.A.M.J.J. 1992. Transformation of filamentous fungi based
on hygromycin B and phleomycin resistance markers. Methods in enzymology, 216, 447–457
RAHARDJO, Y., SIE, S., WEBER, F., TRAMPER, J. & RINZEMA, A. 2005. Effect of low oxygen
concentrations on growth and alpha-amylase production of Aspergillus oryzae in model solid-
state fermentation systems. Biomol Eng, 21, 163–172
ROSCHE, B., LI, X.Z., HAUER, B., SCHMID, A. & BUEHLER, K. 2009. Microbial biofilms: a
concept for industrial catalysis? Trends in biotechnology, 27, 636–643
RUMBOLD, K., VAN BUIJSEN, H.J.J., GRAY, V.M., VAN GROENESTIJN, J.W., OVERKAMP, K.M.,
SLOMP, R.S., VAN DER WERF, M.J. & PUNT, P.J. 2010. Microbial renewable feedstock
utilization: A substrate-oriented approach. Bioengineered Bugs, 1, 359–366
SAMANTARAY, S., NEUBAUER, M., HELMSCHROTT, C. & WAGENER, J. 2013. Role of the
guanine nucleotide exchange factor Rom2 in cell wall integrity maintenance of Aspergillus
fumigatus. Eukaryotic cell, 12, 288–298
SAMSON, R.A. & PITT, J.I. 1985.. Advances in Penicillium and Aspergillus systematics,
Plenum Press, New York, 483 pp
SCHMID, A, DORDICK, J.S., HAUER, B., KIENER, A, WUBBOLTS, M. & WITHOLT, B. 2001.
Industrial biocatalysis today and tomorrow. Nature, 409, 258–268
SCHUSTER, E., DUNN-COLEMAN, N., FRISVAD, J.C. & VAN DIJCK, P.W.M. 2002. On the
safety of Aspergillus niger-a review. Applied microbiology and biotechnology, 59, 426–435
SHOJI, J.Y., MARUYAMA, J.I., ARIOKA, M. & KITAMOTO, K. 2005. Development of
Aspergillus oryzae thiA promoter as a tool for molecular biological studies. FEMS
Microbiology Letters, 244, 41–46
SPRÖTE, P. & BRAKHAGE, A.A. 2007. The light-dependent regulator velvet A of Aspergillus
nidulans acts as a repressor of the penicillin biosynthesis. Archives of Microbiology, 188, 69–
79
STEIGER, M.G., PUNT, P.J., RAM, A.F., MATTANOVICH, D. & SAUER, M. 2016. Characterizing
MttA as a mitochondrial cis-aconitic acid transporter by metabolic engineering. Metabolic
Engineering
SUN, W., GUO, C. & WANG, C.C.C. 2016. Characterization of the product of a nonribosomal
peptide synthetase- like ( NRPS-like ) gene using the doxycycline dependent Tet-on system in
Aspergillus terreus. Fungal Genetics and Biology 89, 84-88
TE BIESEBEKE, R., RECORD, E., VAN BIEZEN, N., HEERIKHUISEN, M., FRANKEN, A., PUNT,
P.J. & VAN DEN HONDEL, C.A.M.J.J. 2005. Branching mutants of Aspergillus oryzae with
improved amylase and protease production on solid substrates. Applied Microbiology and
Biotechnology, 69, 44–50
THOM, C. & CHURCH, M. 1926. The Aspergilli, Williams & Wilkins Comp., Baltimore 272 pp

- 202 -
TISCH, D. & SCHMOLL, M. 2010. Light regulation of metabolic pathways in fungi. Applied
Microbiology and Biotechnology, 85, 1259–1277
TOKUOKA, M., TANAKA, M., ONO, K., TAKAGI, S., SHINTANI, T. & GOMI, K. 2008. Codon
Optimization Increases Steady-State mRNA Levels in Aspergillus oryzae Heterologous Gene
Expression. Applied and environmental microbiology, 74, 6538–6546
UNKLES, S.E., CAMPBELL, E.I., CARREZ, D., GRIEVE, C., CONTRERAS, R., FIERS, W., VAN DEN
HONDEL, C.A.M.J.J. & KINGHORN, J.R. 1989a. Transformation of Aspergillus niger with the
homologous nitrate reductase gene. Gene, 78, 157–166
UNKLES, S.E., CAMPBELL, E.I., DE RUITER-JACOBS, Y.M.J.T., BROEKHUIJSEN, M., MACRO,
J.A., CARREZ, D., CONTRERAS, R., VAN DEN HONDEL, C.A.M.J.J. & KINGHORN, J.R. 1989b.
The development of a homologous transformation system for Aspergillus oryzae based on the
nitrate assimilation pathway: A convenient and general selection system for filamentous
fungal transformation. Molecular & General Genetics, 218, 99–104
UNKLES, S.E., VALIANTE, V., MATTERN, D.J. & BRAKHAGE, A.A. 2014. Synthetic biology
tools for bioprospecting of natural products in eukaryotes. Chemistry & Biology, 21, 502–508
URLINGER, S., BARON, U., THELLMANN, M., HASAN, M.T., BUJARD, H. & HILLEN, W. 2000.
Exploring the sequence space for tetracycline-dependent transcriptional activators: novel
mutations yield expanded range and sensitivity. PNAS, 97, 7963–7968
VAN DEN BRINK, H. (J. ) M., PETERSEN, S.G., RAHBEK-NIELSEN, H., HELLMUTH, K. &
HARBOE, M. 2006. Increased production of chymosin by glycosylation. Journal of
Biotechnology, 125, 304–310
VAN DEN HONDEL, C.A.M.J.J., PUNT, P.J. & VAN GORCOM, R.F.M. 1991. Heterologous gene
expression in filamentous fungi. In Lasure, W.B. and L.L., ed. More Gene Manipulations in
Fungi. Academic Press, New York, 396–428.
VAN HARTINGSVELDT, W., MATTERN, I., VAN ZEIJL, C., POUWELS, P. & VAN DEN HONDEL, C.
1987. Development of a homologous transformation system for Aspergillus niger based on
the pyrG gene. Molecular & General Genetics, 206, 71–75
VAN KUYK, P. A., BENEN, J. A E., WÖSTEN, H. A B., VISSER, J. & DE VRIES, R.P. 2012. A
broader role for AmyR in Aspergillus niger: Regulation of the utilisation of D-glucose or D-
galactose containing oligo- and polysaccharides. Applied Microbiology and Biotechnology,
93, 285–293
VAN PEIJ, N.N.M.E., VISSER, J. & DE GRAAFF, L.H. 1998. Isolation and analysis of xlnR,
encoding a transcriptional activator co-ordinating xylanolytic expression in Aspergillus niger.
Molecular Microbiology, 27, 131–142
VAN VERSEVELD, H.W., METWALLY, M., EL SAYED, M., OSMAN, M., SCHRICKX, J.M. &
STOUTHAMER, A H. 1991. Determination of the maximum product yield from glucoamylase-
producing Aspergillus niger grown in the recycling fermentor. Antonie van Leeuwenhoek, 60,
313–323
VARADARAJALU, L.P. & PUNEKAR, N.S. 2005. Cloning and use of sC as homologous marker
for Aspergillus niger transformation. Journal of microbiological methods, 61, 219–224,

- 203 -
VOGT, K., BHABHRA, R., RHODES, J.C. & ASKEW, D.S. 2005. Doxycycline-regulated gene
expression in the opportunistic fungal pathogen Aspergillus fumigatus. BMC Microbiology, 5
VOIGT, C.A. 2006. Genetic parts to program bacteria. Current Opinion in Biotechnology, 17,
548–557
VON DÖHREN, H. 2009. A survey of nonribosomal peptide synthetase (NRPS) genes in
Aspergillus nidulans. Fungal Genetics and Biology, 46, 45–52
WARD, M., LIN, C., VICTORIA, D.C., FOX, B.P., FOX, J.A., WONG, D.L., MEERMAN, H.J., …
WANG, H. 2004. Characterization of humanized antibodies secreted by Aspergillus niger.
Applied and Environmental Microbiology, 70, 2567–2576
WARD, M., WILSON, L., CARMONA, C. & TURNER, G. 1988. The oliC3 gene of Aspergillus
niger: isolation , sequence and use as a selectable marker for transformation. Curr Genet, 14,
37–42
WARTENBERG, D., VÖDISCH, M., KNIEMEYER, O., ALBRECHT-ECKARDT, D., SCHERLACH, K.,
WINKLER, R., WEIDE, M. & BRAKHAGE, A. A. 2012. Proteome analysis of the farnesol-
induced stress response in Aspergillus nidulans-The role of a putative dehydrin. Journal of
Proteomics, 75, 4038–4049
WEBER, W., FUX, C., DAOUD-EL BABA, M., KELLER, B., WEBER, C.C., KRAMER, B.P.,
HEINZEN, C., AUBEL, D., BAILEY, J.E. & FUSSENEGGER, M. 2002. Macrolide-based transgene
control in mammalian cells and mice. Nature biotechnology, 20, 901–907
WEBER, W., LIENHART, C., DAOUD-EL BABA, M. & FUSSENEGGER, M. 2009. A biotin-
triggered genetic switch in mammalian cells and mice. Metabolic Engineering, 11, 117–124
WEBER, W., MARTY, R.R., LINK, N., EHRBAR, M., KELLER, B., WEBER, C.C., ZISCH, A.H.,
HEINZEN, C., DJONOV, V. & FUSSENEGGER, M. 2003. Conditional human VEGF-mediated
vascularization in chicken embryos using a novel temperature-inducible gene regulation
(TIGR) system. Nucleic acids research, 31, e69
WERNER, R.G., KOPP, K. & SCHLUETER, M. 2007. Glycosylation of therapeutic proteins in
different production systems. Acta paediatrica. Supplementum, 96, 17–22
WITHERS, J., WIEBE, M., ROBSON, G. & TRINCI, A. 1994. Development of Morphological
Heterogeneity in Glucose-Limited Chemostat Cultures of Aspergillus oryzae. Mycological
Research, 98, 95–100
WITHERS, J.M., SWIFT, R.J., WIEBE, M.G., ROBSON, G.D., PUNT, P.J., VAN DEN HONDEL,
C.A.M.J.J. & TRINCI, A.P.J. 1998. Optimization and stability of glucoamylase production by
recombinant strains of Aspergillus niger in chemostat culture. Biotechnology and
Bioengineering, 59, 407–418
WUCHERPFENNIG, T., HESTLER, T. & KRULL, R. 2011. Morphology engineering-osmolality
and its effect on Aspergillus niger morphology and productivity. Microbial cell factories, 10
XU, L.-J., LIU, Y.-S., ZHOU, L.-G. & WU, J.-Y. 2009. Enhanced beauvericin production with
in situ adsorption in mycelial liquid culture of Fusarium redolens Dzf2. Process Biochemistry,
44, 1063–1067

- 204 -
YAMADA, O., IKEDA, R., OHKITA, Y., HAYASHI, R., SAKAMOTO, K. & AKITA, O. 2007. Gene
Silencing by RNA Interference in the Koji Mold Aspergillus oryzae. Biosci. Biotechnol.
Biochem., 71, 138–144
YANG, W., KIM, W., FANG, A. & DEMAIN, A.L. 2003. Carbon and Nitrogen Source Nutrition
of Fumagillin Biosynthesis by Aspergillus fumigatus. Current Microbiology, 46, 275–279
YUAN, X.L., VAN DER KAAIJ, R.M., VAN DEN HONDEL, C. A M.J.J., PUNT, P.J., VAN DER
MAAREL, M.J.E.C., DIJKHUIZEN, L. & RAM, A.F.J. 2008. Aspergillus niger genome-wide
analysis reveals a large number of novel alpha-glucan acting enzymes with unexpected
expression profiles. Molecular Genetics and Genomics, 279, 545–561
ZOCHER, R. & KLEINKAUF, H. 1978. Biosynthesis of Enniatin B: Partial Purification and
Characterization of the Synthesizing Enzyme and Studies of the Biosynthesis. Biochemical
and biophysical research communications, 81, 1162–1167




























