Embed Size (px)
Citation preview

JWSTUS LIEBIGS ANNALEN DER CHEMIE
S07. Band
[Mitteilungen aus d. Chem. Institut der Unversitat Frankfurt a. M.]
Haftfestigkeit organischer Reste. IX') ; von Julius 21. Braun und Rohert illichaelis.
(Eingelaufen am 29. August 1933.)
Die im folgenden beschriebenen, sich an friihere Unter- suchungsreihen anschliefienden Versuche galten der Prufung von zwei Fragen:
1. I n dem MaSe als die Bindungsfestigkeit von Resten an N und S wachst, nehmen auch die Unterschiede in den Bindungsfestigkeiten ab: wahrend Athyl und n-Propyl einer- seits, n-Propyl und Isopropyl andererseits so verschieden fest am N in tertiaren Basen haften, daB vom erstgenannten Paar dnrch Bromcyan BUT Athyl, vom letztgenannten nur n-Propyl abgelost wird, wird bei gleichzeitiger Anwesenheit von n-Butyl und Isobutyl neben Butylbromid in kleiner Menge Isobntylbromid nnd bei gleichzeitiger Gegenwart von n-Heptyl und n-Oktyl neben vie1 Heptylbromid etwas Oktyl- bromid gebildet.
Die Frage, die wir nun zu losen versnchten, war, ob auch bei Resten mit sehr gem'nger Bindnngsfestigkeit die scharfe Differenzierung aufhbrt ? Wir gingen zu diesem Zweck von den zwei durch besonders lockere Bindung charakterisierten Resten 2):
aus und veranderten jeden in einer nicht allzu einschnei- denden Weise: I durch Ersatz von CH,O durch C,H,O
CHaO@). CeH,CH*- (I) und CeH,(p). C6HdCH2- (11)
l) VIII. Mitteilung: A. 490, 189 (1931). Vgl. die unten gegebene Zusammenstellung.
Annalen der Chemie. 607. Band. 1

2 v. Braun und Michael i s ,
[C,H,O(p). C8H4CHz- (III)], I1 durch Hydrierung der end- standigen Phenylgruppe zur Cyclohexylgruppe einerseits [C,H,,(p) . C,H,. CH,- (IV)] und durch para-Angliederung eines weiteren Phenylrestes [C,H,(p). C,H,(p).C,H,.CH,- (V)] andererseits. Der Vergleich von I und 111 fuhrte experimen- teller Schwierigkeiten wegen zu keinem ganz durchsichtigen Resultat, dagegen ergab der Vergleich von I1 sowohl mi t IV als auch mit V, dalj diese drei sehr locker an den N gebundenen Reste sich einander in der Haftfestigkeit nahern: sie werden neheneinander - genau so wie Heptyl und Oktyl - abgelost, wobei allerdings zu bemerken ist, dab der Biphenyl-benzylrest in groflerem Umfang als der Phenyl-benzylrest aus dem Molekul austritt. Was also fur das eine Ende der Haftfestigkeitsreihe gilt, gilt auch fur das andere, und eindeutige klare Verhaltnisse bietet nur das dazwischenliegende Gebiet, dessen weiterer Ausbau aus diesem Grunde besonders verlockend erscheint.
2. I n der Haftfestigkeitsreihe (vgl. Tabelle auf S. 4) zeigt sich, dafi in den Fallen, in denen in der Benzylgruppe die Verschiebung eines Substituenten aus der para-, in die meta- und dann in die ortho-Stellung durchgefiihrt worden ist - das war der Fall bei CH,, C1 und F - bei CH, und C1 eine sprungweise im Sinne dieser Verschiebung zu Tage tretende Erhohung der Haft,festigkeit eintritt; nur beim Fluor, dessen eigenartiges von anderen Halogenen abweichendes Verhalten mehrfach schon bei chemischen Umsetzungen beobachtet worden ist, ist eine gleichsinnige Zunahme der Bindungsfestigkeit nur andeutungsweise vor- handen: das hangt damit zusammen, daB die Einfuhrung von F in den Benzylrest dessen Haftfestigkeit iiberhaupt nicht merklich beeinflufit, so dad p, m und o-Fluorbenzyl vom N mit etwa gleicher Leichtigkeit wie Benzyl abgelost werden. W e werden aber - so lautete die Frage - sich andere ,,normale" Substituenten der Benzylgruppe verhalten? Wird auch hier, wie bei CH, und C1, sich die Zunahme der Bin- dungsfestigkeit bei der Verschiebung aus der p- in die m- und o-Stellung zeigen? Zur Beantwortung dieser Frage zogen wir die durch Phenyl, Athyl, Brom und Jod substi-

Huffest igkei t oryunischer Reste. I X . 3
tuierten Benzylreste heran. In der Phenyl- und Athylbenzyl- reihe begnugten wir uns mit der Verschiebung aus der para- in die ortho-Stellung und konnten die vorhin auf- geworfene Frage dahin beantworten, da13 in der Tat diese Verschiebung eine Verstarkung der Bindung hervorruft : denn die Reste ortho-Athylbenzyl C,H,(o).C,H,.CH,- und ortho-Phenyl-benzyl C,H,(o) . C6H4CH2-- riicken i n bezug auf ihre Haftfestigkeit in die Nahe des Benzyls, dem sie ungefahr gleich werden; sie erreichen aber noch nicht die Haftfestigkeit des o-Methylbenzyls, ebenso wie p-Phenyl- und p-Athylbenzyl hinter dem p-Methylbenzyl zuruckbleiben. Es sei ubrigens bemerkt, daD auch der o-Vinylbenzylrest CH,=CH(o). C,H,CH,--, den wir bei dieser Qelegenheit in den Kreis der Untersuchung zogen, dem o-Athylbeuzyl gleicht: das ist verstandlich, denn nur die zur Bindungs- stelle an N oder S p,y-standige Doppelbindung (z. B. im Allyl) bewirkt eine Auflockerung gegenuber einem ge- sattigten Rest.
Beim Brom und Jod untersuchten wir die Verhaltnisse voll- standiger, denn wir zogen auch die meta-substituierten Reste heran und wir konnten eine vollstandjge 'ijbereinstimmung mit den Ergebnissen in der Methyl- und Chlorreihe erzielen: die meta-substituierten Reste nehmen eine Mlttelstellung zwischen den lockerer sitzenden para- und den am festesten sitzenden ortho-Resten ein.
Mit dieser Peststellung war die eingangs aufgeworfene Frage beantwortet, es schlod sich an sie aber gleich eine weitere an. I n der Reihe der p-halogenierten Benzylreste nimmt, wie die oben angefuhrte Reihe zeigt, die Haftfestig- keit mit steigendem Atomgewicht des Halogens zu; wie werden nun die Verhaltnisse in der meta- una ortho-Halogen- reihe liegen? Die Antwort auf diese Frage fie1 sehr ein- deutig und in Ubereinstimmung mit der para-Reihe Bus: m-Brombenzyl nimmt eine scharf definierte mittlere Stellung zwischen m-Chlor- und m-Jodbenzyl, o-Brombenzyl findet ebenso eindeutig seinen Platz zwischen o-Chlor- und o-Jod- benzyl.
Uberblickt man die beiden Resultate, die sich am ein- 1*

4 v. Braun und Michae l i s ,
fachsten in das folgende Schema bringen lassen (die Pfeile bedeuten Zunahme der Haftfestigkeit):
1. p.C1 1 4. m.C1 I 7. o.C1 --f-t --++
2. p.Br [ 5. m.Br I 8. o.Br --++ --+-t 3 . p . J Y 6 . m . J Y 9 . o . J .
_ _ _ f - 3
1 so ersieht man, daS durch sie noch eine dritte Frage auf- geworfen wird: ist die Zunahme der Haftfestigkeit, von einem bestimmten Punkt ausgehend, groder in der horizon- talen oder in der vertikalen Richtung, mi t anderen Worten, ist fur die Haftfestigkeit eines durch Halogen substituierten Benzylrestes mehr die Natur des Halogens oder dessen Stellung mabgebend? Auch diese Frage lieB sich durch passend synthetisiertes Material beantworten, und zwar fie1 die Antwort dahin aus, daB es die Stellung der Halogene ist, die den HaupteinfluB ausubt; denn fur die Haftfestig- keiten der neuen halogenhaltigen Benzylreste ergab sich eine Reihenfolge, die den Ziffern 1-9 entspricht and in der die engere Zusammengehorigkeit der drei para-, der drei meta- und der drei ortho-substituierten Badikale zum Ausdruck kommt. Urn diese ganze Neunergruppe in der gesam ten Haftfestigkeitsreihe richtig einzuordnen, muBte noch das Verhaltnis zum o-Methylbenzyl, dem einzigen von den bisher untersuchten Benzylresten, der fester als C,H,CH,- am N haftet, festgestellt werden. Der Vergleich mit p-Chlor- benzyl ergab, daS der letztere fester gebunden ist und so ergibt sich fur die ganze bisher untersnchte Reihe der nahezu 25 Benzylabkommlinge die Reihenfolge (B=C,H,. CH,-).
CH, (m)B - I HB-
FB-
CH2=CH(0)B- CSH&O)B- CH,(o). B- Cl(p) . B- I
I C6HdOP- Br(p).B- 1 J(p)B- 1 Cl(m).B- I Br.(m).B- 1 J(m).B- I Cl(o)B- I
Br(o).B- I J(o).B- I . . . . I CH,.

Haftfestigheit organischer Reste. IX. 5
Man ersieht aus ihr, duf3 auch bei Kohlenwasserstoff- substituenten der Benzylgruppe (Phenyl, Athyl, Methyl) sich das gleiche Gesetz wie bei den Halogenen zeigt, d. h. bei chernisch analogen Substituenten die Stellung mehr als die Masse die Haftfestigkeit bestimmt, und es wird nicht ohne Interesse sein, dieses Gesetz auch noch in anderen Fallen auf seine Bultigkeit zu prufen.
Beschreibung der Versuche
I. D e r p -Athoxy- , p -Cyclohexyl - u n d p - B i p h e n y l - b enz y l r e s t.
Fur den Vergleich des p-Methoxy- und p-Athoxybenzyl- restes stellten wir aus dem bereits bekannten p-Xethoxy- benzylmethylamin und dem p-k’2ho~ybenzyZchlorid die tertiare Base CH,O(p).C,H,CH, .N(CHJ.CH,. C,H,OC,HS(p) her.
p-Athoxybenzaldehyd laBt sich mit konz. waSriger Natronlauge unter Zusatz von Alkohol bequem disproportionieren. Die Reaktion ist nach 2-stundigem Erwarmen auf dem Wasserbad zu Ende; man verdiinnt mit Wasser, athert das 01 aus und fraktioniert, wobei es unter 12 mm bei 134-135 iibergeht. Der Geruch des p-~tholl;y~en;tyZalkohoL ist schwacher als der des Anisalkohols.
0,1350 g Subst.: 0,3522 g CO,, 0,0924 g H,O. C9HI%O2 Ber. C 71,02 H 7,95 Gef. C 71,14 H 7,134.
Das zugeharige, durch Einleiten von HC1-Gas in die eisgekuhlte Benzol- oder kherl6sung des Alkohols gewonnene ChZorid siedet als wasserhelle, etwas stechend riechende Fliissigkeit bei 95-100 O/0,8 mm ; erwies sich aber bei mehrfacher Darstellung etwas zu chlorarm. Es wurde mit 2 Mol. Anisylmethylamin durch 4-stundiges Erwarmen auf dem Wasserbade umgesetzt , die basischen Produkte in der iiblichen Weise herausgearbeitet und fraktioniert. Bei 120-125°/14mm destilliert Anisyl-methylamin, bei 220-225 O, die gemischte tertiare Base als wasser- belles 01.
(z. T. mitbearbeitet von F. F i s c h e r und R. Mur jahn) .
0,0940 g Subst.: 4,l ccm N (23O, 765 mm).
Mit Bromcyan erwarmt sich die Base schwach. C,,H,,O*N Ber. N 4,91 Gef. N 5,06.
Nach kurzem Stehen auf dem Wasserbade wurde mit Ather versetzt, nach AbgieSen von der dickoligen nicht krystallisierenden Fallung mit verdunnter Saure ausgeschiittelt, der Ather abdestilliert und der bromhaltige Riick-
’) T i f f e n e a u , Bull. SOC. Chim., [4] 9, 825 (1911).

6 v. B r a u n und Jlichaelis,
stand mit Trimethylamin versetzt. Das gebildete dickiilige quartare Produkt lieB leider keine Reinigung und keine Entscheidung dariiber zu, in welchem Umfang an seiner Bildung die beiden Bromide CH,O. . C,H,CH,Br und C,H,OC,H,CH,Br beteiligt sind. Die iilige Beschaffen- heit la& vermuten, daB es nicht die Methoxyverbindung allein ist.')
Fur die Untersuchung des CycZohezyZ- und des Biphenyl- benzyl-Restes war es erforderlich, die zwei tertiaren Basen: C,H,, .C,H,CH,N(CH,).CH,.C,H,.C,H, und C,H,.C,H,.C,H,. , CH, .N(CH,). CH, . C,H,. C,H, darzustellen; wir konnten sie mit Hilfe des Phenylbenzylmethylamins HN(CH,). CH, .C,H,. . C,H6 und des Cyclohexylbenzylchlorids C,H,, . C,H,CH,CI bzw. Biphenylbenzylbromids C,H,. C,H,. C,H,CH,Br ,) er- halten.
Das Phenylbenxylrnethylanzi7E haben seinerzeit v. B r a u n s ) und E n g e l aus dem p-Biphenylaldehyd durch Kondensation mit Methyl- amin und Reduktion erbalten. Bequemer erhalt man es aus dem p-Phenylbenzylchlorid (v. Br., I. u. N., a. a. 0.) durch Erwarmen mit NH,CH, in Benzol. Neben dem sekundaren Amin entsteht in kleiner Menge die sehr dickolige tertiare Base (C,H,. C,H,. CH,XNCH,, die unter 0,8 mm bei 255O siedet und sich rnit JCH, zii einem langsam krystallisierenden Jodmethylat vom Schmelzp. 205O vereinigt. Man kann auch - und wir haben diesen Weg zur Kontrolle eingeschlagen - f u r die Darstellung von C,H,, . C,H,. CH, .N(CH,) . CH,. C,H,. C,H, von dem Chlorid C,H, . C,H,CH,Cl und der Base C6Hll. C,H4. CH,. NHCH, ausgehen. Cyclohexylbenzylchlorid und NH,CH, (in benzolischer Losung) liefern bei mehrstundigem Erwarmen auf looo auch neben der durch ihr schwer in Wasser losliches Chlorhydrat charakterisierten sekundaren Base etwas tertiare. Die erstere siedet um 165O/11 mm als farbloses 01.
32,7 rng Subst.: 1,85 ccm N (19O, 753 mm) C,,H,,N Ber. N 6,89 Gef. N 6,55.
Sie liefert a d e r dem Chlorhydrat vorn Schmelzp. 186O ein gut charak- terisiertes Pt- Salx vom Zersetzungspuukt 215 O. Die tertiiire Base destilliert unter 0,4 mm um 240°, erstarrt allmahlich, schmilzt bei 52O (C,,H,,N Ber. N 4,03 Gef. N 4,OO) und I%& sich durch das feste, in Wasser schwer losliche Jodmethylat vom Schmelep. 196 O charakterisieren.
Auf den zwei moglichen Wegen erhalt man nun (durch 3-stundiges Erwarmen des betreffenden Chlorids mit 2 Mol.
l) Vgl. A. 490, 198 (1931). ,) v . B r a u n , I r m i s c h u. N e l l e s , B. 6G, 1471 (1933). ,) A. 436, 312 (1924).

Haftfestigkeit organischer Reste. IX, 7
der betreffenden sekundaren Base in etwas Benzol) das- selbe gemischte tertiare Amin. Es siedet bei 230 bis 240°/0,3 mm 81s gelbes dickes 01, das ziemlich schnell kry- stallisiert und bei 52-53 O schmilzt.
30,4 mg Subst.: 97,6 mg CO,, 23,6 mg H,O. - 41,l mg Subst.: 1,40 ccm N (20°, 758 mm).
C,,H,,N Ber. C 87,74 H 8,46 N $79 Gef. ,, 87,56 ,, 8,69 ,, 3,96.
Pikrat un d Corhydrat sind Blig. Gut krystallisiert aus Methanol erhiilt man aber das Jodmethylat (Ber. J 24,83 Gef. J 24,92), das bei 200° schmilzt und mit den zwei oben erwahnten Jodmethylaten vom Schmelzp. 205O und 196O Depressionen auf 165-170° gibt.
Biphenylbeneylbromid und Phenylbenzylmethylamin setzten wir wegen der Schwerlijslichkeit des Bromids in den meisten Lijsungs- mitteln in Trichlorbeneol um. Nach 5-stundigem Erwarmen auf dem Wasserhade ist die Reaktion noch unvollstandig und man setzt sie zweckmaBig noch 3 Stunden bei 130° fort. Man macht salzsauer, treibt das Losungsmittel mit Wasserdampf ab, filtriert das beim Er- kalten sich ziemlich vollstandig abscheidende Gemisch der Salze der Basen, lost es heiB in Pyridin, fallt mit Ammoniak und wascht den Niederschlag mit Alkohol aus, wobei die tertiare Base rein zuriick- bleibt. Sie ist schwer in Ather und Alkohol lijslich, leicht in heiBem Benzol. Der Schmelzpunkt liegt bei 186O, der dee Chlwhydrats bei 2450.
29,l mg Subst.: 0,85 ccm N (19O, 765 mm). C.33H39N Ber. N 3,20 Gef. N 3,44.
Setzt man C y c b h e ~ y E b e n x y l - p h e ~ y ~ ~ e n ~ ~ ~ - ~ e t h y l a mit BrCN auf dem Wasserbade 3/4 Stunden um, fallt mit.Ather, wobei sich eine geringe Menge eines allmahlich fest werdenden 01s abscheidet, schuttelt mit verdiinnter H,SO, durch, trocknet und fraktioniert, so erhalt man ein von 120-130°/0,3 mm, dann nach einem kleinen Zwischenlauf von 150-160 O ubergehendes Destillat, dessen letzte Teile ziemlich schnell erstarren. Man versetzt das Ganze mit benzolischem Trimethylamin, filtriert nach einigem Stehen den farblosen NiederschIag (A) ab und fraktioniert das Filtrat, wobei das meiste um 160° ubergeht und teil- weise in der Vorlage erstarrt. Der auf Ton abgepreBte feste Teil (Schmelzp. 67-70 O) nahert sich in der Zusammensetzung dem H-Brmeren Cyanamid C,H, .C,H,. CH, .N(CH,)CN (Ber. C,,H,,N, C 81,03 H 6,35 Gef. C 80,34 H 6,62), der olige, der erst nach Wochen zu krystalli- sieren beginnt, dem H-reicheren C,H,, . C,H,CH,. N(CH3)CN (Ber. C,,H,,N, C, 78,88 H 8,84 Gef. C 79,57 H S,25). Beide sind der Menge nach etwa gleich. Dem entspricht, daE A (Schmelzp. 170-200°) in der Zusammensetzung zwischen den aus C,H,. C,H,CH,Br und C,H,, . C,H,. . CR,Br hervorgegangenen quartlren Bromiden steht (Gef. C 62,21 H 7,51.

8 v. Braun und Michaelis,
Das Biphenyl-bemxylhaltige tertiare Amin setzten wir mit BrCN in Benzollijsung um (etwa 1 Stunde!. Nach Ausfallen eines geringen Niederschlags mit Ather, wurden Ather und Benzol verjagt und der feste Ruckstand 3 Stunden im Rohr mit benzolischem Trimethylamin auf looo erwarmt. Der abgeschiedene Krystallbrei (A) wurde gut mit Ather ausgewaschen, schmolz bei 225-245O und erwies sich als im wesentlichen aus C,H,C,H,C,H, . CH,Br hervorgegangenes quartires Salz(C,,H,NBr Ber. Br 20,94 Gef. Br 21,83; C,,H,,NBr Ber. Br 26,14). Dementsprechend stellte die im Filtrat von A, enthaltene Br-freie Substme, die sich destillierbar erwies, im wesentlichen das Cyaiaamid der Phelzylbmxyl-Reihe dsr: Unter 0,7 mm destillierte bei 18O-2OO0 die Hauptmenge, die nach dem Festwerden von 50° bis 160° schmolz und bei der Analyse C 81,75 H 6,59 und N 11,5 Proc. lieferte, dann folgte ein kleiner Nachlauf, mit dern Siedep. 160-175" (C 84,22 H 6,14 N 10,05). Fiir C,H,C,H,CH,N(CH,)CN berechnet sich C 81,08 H 6,30 N 12,61, f u r C,H,.C,H,.C,H4.CH2.N(CH3)CN C 84,56 H 6,OP N 9,39.
11. D e r o-Vinyl- , o-Athyl- u n d o - P h e n y l b e n z y l r e s t . Die Versuche dieses Abschnittes betrafen die Synthese
und das Verhalten der 3 tertiaren Basen: 1. C& (0). C,H,. CH,. N (CH,) CH, . C,H,, 11. C,H, (0) . C,H4. GH, . N (CH,) . CH, . C,H,
und 111. C,H, (0). C,H,. CR,. N(CH3). CH,. C,H4. CH = CH, (0).
F u r die Synthese von I und I1 diente Benzylmethyl- amin und die bei unseren fruheren Versuchen I) dargestellten Bromide: C,H6 (o)C,H,CH,Br und CaH5(o). C,H,CH,Br; fur 111: o-Vinylbenzylbromid und o-Athylbenzylmethylamin.
o-Athylben~ylnzethylami@a wurde in der ublichen Weise aus o-Athyl-
Siedep. 92,,O. 0,1248 g Subst.: 0,3683 g GO,, 0,1142 g H,O.
beneylbromid und benzolischem Methylamin bei 100 O gewonnen.
Cl0Hl6N Ber. C 80,53 H 10,06 Gef. C 80,49 H 10,39.
Das Pikrat und Chlorhydrst fallen in gtherischer Lijsung erst ijlig aus, werden aber beim Reiben feet. Das hygroskopische Chlorhydrat schmilzt bei 108O, das gut aus Alkohol krystallisierende Pikrat bei 139 O.
Die drei tertiiiren Basen gewannen wir in gleicher Weise aus 2 Mol. des betreffenden sekundaren Amins und 1 Mol des Bromids durch Vermischen bei 0 O, kurzes Stehenlassen in der Kiilte, mehr- stundiges Erwarmen auf 100 und gewijhnliche weitere Aufarbeitung.
I) A. 468, 258 (1929); B. 50 45 (1917).

Hafttfestigkeit organischer Reste. LK 9
o-Phenylbenxyl-benxyl-methylamin (I) siedet unter 12 mm bei 223 O
als wasserhelles, nicht krystallisierendes 01, dessen Chlorhydrat sehr hygroskopisch iet und dessen in der Kalte aus den Romponenten ge- bildetes Jodmethylat, das sich leicht in Alkohol lost, bei 163 O schmilzt.
0,1080 g Subst.: 0,3484 g CO,, 0,0705 g H,O.
o-Athylbenxyl-benxyl-methylamin (11) zeigt den Siedep. 173,,O. 0,1113 g Subst.: 0,3473 g CO,, 0,0962 g H,O.
CiTHaiN Ber. C 85,35 H 8,78 Gef. C 85,lO H 9,06.
Der Schmelzpunkt des ChZorhydrats liegt bei 174O, des in der Ealte
o-~lhyZben~yl-o-vi~yZbelzxyl-methyZamin (111). Farbloses 01 vom
0,1037 g Subst.: 0,3260 g CO,, 0,0792 g H,O.
Schmelzpunkt des Chlorhydrats 138 0, des kalt hergestellten, in
Ber. C 87,81 H 7,31 Gef. C 88,Ol H 7,13. C21H2,Br
gebildeten, in Alkohol leicht liislichen Jodmethylats bei 154 O.
Siedep. 195--196O, 13 mm.
C,,H,,N Ber. C 86,03 H 8,67 Gef. C 85,76 H 8,54.
Alkohol leicht lijslichen Jodmethylats 164O.
Die Umsetzung mit Bromcyan wurde in allen drei Fallen durch Vermischen in der Kalte, viertelstiindiges Er- warmen auf dem Wasserbade, Zusatz von Ather (zur Aus- fallung der kleinen Mengen von quartaren Verbindungen), Ausscliutteln mit verd. Saure, Destillation des aus zwei Bromiden und zwei Cyanamiden bestehenden Atherinhalts (A), Zusatz von (CH,),N und Analyse des gefallten quartaren Bromidgemisches (B) ansgefiihrt. Zur Kontrolle diente der Vergleich des Schmelzpunktes von B mit den Schmelz- pnnkten der reinen quartaren Bromide und die Analyse des Cy anamidgemisches.
o-Phenylbennxyl-benxyl-methylamin. A = Siedep. 144-230,,0.
0,1024 g Subst.: 0,0662 g AgBr. C,H,(o) .C,H,CH,.N(CH,),Br.
Ber. Br 26,14 Schmelzp. 205O Gemisch 182-210@. C,HS.CH, .N(CH,),Br.
Ber. Br 34,?8 Schmelzp. 232O Gemisch 182-210°. Qef. Br 27,52.
B = Schmelzp. 183-209O.
o-Athy lbenxyl- bennxyl-methylamin. A = Siedep. 105-165,,0
0,1111 g Subst.: 0,0870 g AgBr. CaH,(o). C,H,CH,. N (CHJ,Br.
B = Sehmelzp. 197-212O.
Ber. Br 31,OO Schmelzp. 2200 Gemisch 192-210'.
10 v. B r a u n und M i c h a e l i s ,
C,H,CH, .N (CH3),Br. Ber. Br 34,78 Schmelzp. 232' Gemisch 192-210°. Gef. Br 33,63.
o- Athylbelzxyl-o-vilzylben~yl-methyl~~~.
0,0806 g Subst.: 0,1654 g CO,, 0,0549 g H,O. A = Siedep. 100-180,,o.
C2H, (0). C,H,CH, .N (CH,),Br. Ber. C 55,81 H 7,75 Schmelzp. 220' Gemisch 205-217O. C,H, (0) C,H4CH,. N(CEI&Br. Ber. C 56,25 H 7,03 Schmelzp. 217O Gemisch 205-217O. Gef. C 55,95 H 7,61.
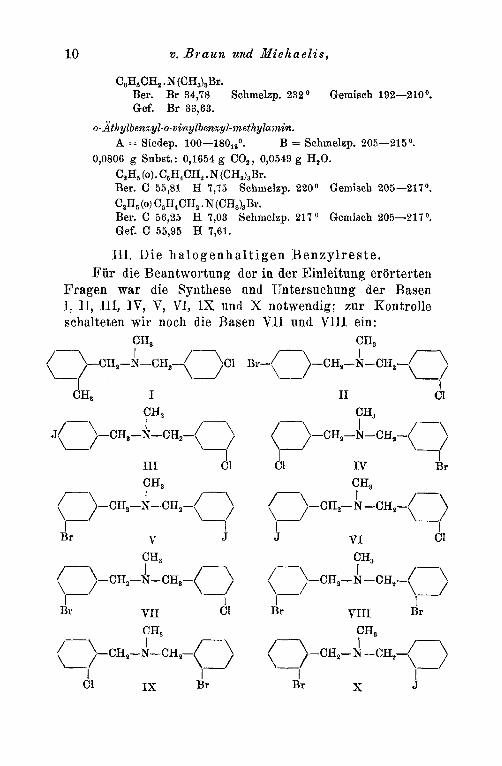
111. Die h a1 o g e n h a1 t i gen B enz y l r e s t e. Fur die Beantwortung der in der Einleitung erorterten
Fragen war die Synthese und Untersuchung der Basen I, 11, 111, IV, V, VI, IX und X notwendig; zur Kontrolle schalteten wir noch die Basen V I I und VIII ein:
CH3 I I-\ -L/ /~-CH,--W--CH,-/---\C~ Br---/ \-cH,--N-cH,
B = Schmelzp. 205-215O.
- CHS I
\-I LJ I
CHS I I1 c1 CH3 CH3
J/ \-CH,-N-CH, I -\ I--\ 1 \J-CHS-N-CH2- /--\ 1
111 c1 IV Br I
c1
I
1 \-I
CH, CH, I-\ r \ - C H 2 - N -CH,--/ CH2-N- CH2- ' \->
I \-.-I \-/
c1 I
Br V J VI I I
J
CH8 CH, /-'-\-cH2-N-.cHa-/--\ I /--\-cH,-N--CH,--/-\ I
/p\-CHa-N-CHg- I /-\ ~ - L X , - N - C H , - I \-(
\-I \-I \-/ \-/ I
Br I
Br VII Br VIII I I
c1
CH, CH3
\ / \-/ I J
I c1 I X Br Br x
I-

Haftfestigkeit organischer Reste. IX. 11
Zu ihrer Synthese diente eine Reihe passend gewahlter, im Benzolkern substituierter Benzylbromide, die samtlich bekannt waren (und zwar waren es: o-Methyl-, o-Chlor-, o-Jod, m-Chlor nnd m-Brombenzylbromid), die mit Methyl- amin in die sekundaren Basen I bis V verwandelt wurden. CH3(0)* C6H4CH,NHCH,, Cl(o)C,H4CH2NHCH3, J(o)C,H,CH,NHCH,
I I1 I11
Cl(m)C,H,CH,NHCH, Br(m)C,H,CH,NHCH, IV v
Von diesen gelangte man durch Umsetzung rnit den entsprechend substituierten, bereits bekannten Benzyl- bromiden zu den Basen 1-10.
Die Umsetzung mit Methylamin geschah durch mehrstiindiges Erliitzen mit benzolischem Methylamin (2 bis 3 Mol) in der Wasser- badkanone. Die Ausbeute von sekundzren Basen betrug unter diesen UmstPnden meist nicht mehr ale 30 Proc., da gleichzeitig quartiire Verbindungen und durch Destillation leicht abtrennbare tertiare Basen sich bilden. Wir gehen auf die letzteren im folgenden der Raum- ersparnis wegen nicht naher ein.
o-MethyZbe~xyl-rnethyZanzin (I). Siedep. 100-1021,0 (Siedep. des Di-o-methylbenzyl-methylamins 182,,O). (Rer. C,H,,N N 10,37, Clef. N 10,30.) Chlorhydrat (leicht loslich in Wasser und Alkohol) Schmelzp. 1504 Pikrat Schmelzp. 129 O.
o-ChZorbennyl-nzeth~lumi~ (11). Siedep. 98-100,00 (Siedep. des Di- o-chlorbenzylmethylamins 200,,3. (Ber. C,HloNCI C122,83, Gef. C122,75.) Chlorhydrat (leicht in Wasser und Alkohol liislich), Schmelzp. 135O, Pikrat Schmelzp. 120O.
o-Jodbenxyl-methylamin (111). Siedep. 130-132100 (C,H,,NJ Ber. N 5,66, Gef. N 5,75). Chlorhydrat (leicht liislich) Schmelzp. 180°, Pikrat Schmelzp. 150O.
m- Chlorbenxyl-methylamin (IV). Siedep. 122--124,: (Siedep. des Di-m-chlorbenzylmethylamins 198,,O). (C,H,,NCl Ber. N 9,00, Gef. N 8,81). Chlorhydrat Schmelzp. 170°, Pikrat Schmelep. 128O, beide leicht liislich.
m-Brombenxyl-methylamilE (V). Siedep. 12ZI4O (C,H,,NBr Ber. N 7,00, Gef. N 7,06). Chlorhydrat Schmelzp. 147O, Pikrat Schmelzp. 135O.
Die Darstellung der tertiiiren Basen 1 bis 10 geschah durch Zusammenbringen von 2 Mol. der sekundaren Base mit 1 Mol des Bromids, wobei schon in der KLlte eine Umsetzung sich bemerkbar machte, einstiindiges Erwarmen auf dem Wasserbade, Isolierung der basischen Produkte

12 v. B r a u n und M i c h a e l i s ,
und Trennung der sekundaren Base von der tertiaren durch fraktionierte Destillation. Zur vijlligen Reinigung wurden die tertiaren Basen noch einmal destilliert. Die Ausbenten sind gut. Die neuen Basen sind bis auf eine fliissig.
o-Methylbenxyl-p-ehlorbenxyl- methylamin (1) , schwach gelbes 01. Siedep. 180-184O (C,,H,,NCl Ber. N 5,39, Cef. N 5,31). Chlorhydrat (leicht loslich) Schmelzp. 170 O, Jodmethylat (in der Kalte hergestellt) Schmelzp. 215O (schwer lijslich in Alkohol); das Pikrat ist 81ig.
0- Chlorbenxyl-o-brombenxyl~methylami?~ (S), sehr dickes 01. Siedep. 1500,,0 (C,,H,,NClBr Ber. N 4,32, Gef. N 4,50). Chlorhydrat (leicht liislich) Schmelzp. 160°, Pikrat 129 O.
o-Chlorbenxyl-m-jodbenzyl-methylamin (6). Siedep. 2000,,0. Nach langerem Abkiihlen wird die Base fest und schmilzt nach Abpressen auf Ton bei 55-56'' (C,,H,,NCIJ Ber. N 3,76, Gef. N 3,92). Chlor- hydrat Schmelzp. 195O, Pikrat Schmelzp. 123".
o-Jodbenxyl- o - brornbenxyl-methylami* (10). Siedep. 200-2030,,0. (C,,R,,NBrJ Ber. N 3,36, Gef. N 3,36). Schmelzpunkt des in Alkohol leicht loslichen Chlorhydrats 200°; das Pikrat ist iiIig.
m-Chlorbenxyl-p-brombenxyl-methylam& (2). Siedep. 186-1900,50 (C,,H,,NClBr Ber. N 4,32, Gef. N 4,51). Das Pikrat ist olig, das Chlorhydrat schmilzt bei 187O.
Sehr z5he Fliissigkeit vom Siedep. 210,,60 (C,,H,,NClJ Ber. N 3,76, Gef. N 3,90). Chlor- hydrat Schmelzp. 203 O, Pikrat erst 61ig, nach mehrmaligem Umfkllen aus Alkohol-Ather fest, Schmelzp. 110O.
m-Chlorbenzyl-m-brombenxyl-methylamin (4). Siedepnnkt 1800,,o (C,,H,,NClBr Ber. N 4,32, Gef. N 4,39). Chlorhydrat (in Wasser nicht ganz leicht liislich) Schmelzp. 210°, Pikrat Schmelzp. 147O.
m - Brombenxyl-m-jodbenxyl- methylamin (5). Siedepunkt 2000,,0 (C,,H,,NBrJ Ber. N 3,36, Gef. N 3,41). Chlorhydrat Schmelzp. 194O, Pikrat 140O.
m- Brombenxyl-o-chlorbenxyl -methylamin (7) . Siedepunkt 185,,, (C,,H,,NClBr Ber. N 4,24, Gef. N 4,32). Chlorhydrat leicht liislich Schmelzp. 1984 Pikrat Schmelzp. 1339
m-Brombennyl-o- brombemyl-methylamin (8). Siedep. 198-2000,~ (C,,H,,NBr, Ber. N 3,79, Gef. N 3,92). Chlorhydrat Schmelzp. 183; Pikrat ist iilig.
m- Chlorbenxyl-p-jodbenxyl-methylamin (3).
Die Umsetzung der tertiaren Basen m i t Bromcyan geschah gleichmalig durch Vermischen der Komponenten bei Oo, kurzes Stehenlassen bei Zimmertemperatur, halb- stundiges Erwarmen auf dem Wasserbade, Zusatz von Ather, Abgiefien von der meist geringen oligen Fallung, Ausschutteln mit verd. H,SO,, Trocknen uber CaC1, und

HaftfestigReit organischer Reste. IX 13
Destillation des Atherinhalts, wobei bei der Spaltung ge- bildetes niedriger siedendes Benzylbromid (Aj vom hBher siedenden Cyanamid (B) meist gut getrennt werden konnte. B wurde durch Analyse, A durch Siedepunkt, Schmelzpunkt und das Vereinigungsprodukt mit Trimethylamin identifiziert. I n allen 10 Fallen war im Gegensatz zu den im Abschnitt I und I1 beschriebenen Versuchen die Reaktion bis auf Base 6 (vgl. unten) voZZig eindeutig.
Base 1. A: Siedep. 104-1061,0. Schmelzpunkt des Additions- produktes an (CH,),N 200-202° (Mischprobe mit CH,(oK&H,CH,N (CH,),Br : 201 O).
B: Siedep. 165-167 O (farblose Flussigkeit). 0,1471 g Subst.: 0,1191 g AgCl.
Cl(p).C,H,CH,N(CH,)CN = C,H,N,Cl Ber. C1 19,70 Gef. C1 20,03. Base 2. A: Siedep. 120-125,, O. Sehmel~p.62~ von Br(p). C,B,CH,Br.
B: Siedep. 180,,o. 0,1598 g Subst.: 0,12,96 g AgC1.
Cl(m).C,H,CH,N(CH,)CN = C,H,N,Cl Ber. C1 19,61 Gef. C1 20,06. Base 3. A: Siedep. 15Ol4O, Schmelzp. 79O von J(p)C,H,CH&r.
0,1433 g Subst.: 0,1157 g AgC1.
Base 4. A: Siedep. 110-114,: = C1 (m) C,H,CH,Br B: Siedep. 180-184'.
0,1664 g Subst.: 0,1411 g AgBr.
B: Siedep. 160-170110.
Cl(m).C,H,N(CH,)CN = CeH,N,C1 Ber. C1 19,70 Gef. C1 19,97.
Br(m)C,H,CH,N(CH,)CN = CeHeN2Br Ber. Br 35,52 Gef. Br 36,03. Base 5. A: Siedep.122-1301,0,Schmelzp.410 von Br(m)C,H,CH,Br.
B: Siedep. etwa 160,,2 fliiseig. 0,1634 g Subst.: 0,1454 g AgJ.
J(m)C,H,CH,N(CH,)CN = C,H,N,J Ber. J 47,76 Gef. J 48,lO. Base 6. A: Siedep. 140-150°, Schmelzp. 50° von J(m)C,,H,CH,Br. I)
B: Siedep.185-19O0. 0,1371 g Subst.: 0,1116 g AgCI.
Cl(o)C,H,.CH,.N(CH,)CN = C,H,N,Cl Ber. C1 19,70 Gef. C1 20,14.
1) Einige wenige unterhalb 140 iibergehende Tropfen lieferten mit (CH,),N ein qaartiires Bromid vom Schmelzp. 179--183O, das keine Depression rnit CL(Q)C,H,CH, .N(CH&BY (Sehmelzp. 183 O, wohl aber rnit J(m)C,H,CH,N(CH,),Br (Schmelzp. 206O) zeigte, ao dal3 bei 6 offen- bar in winziger Menge auch die Ablosung des o-Chlorbenzylrestes erfolgt.

14 v. Braun ,
Base 7. A: Siedep.120-1261,0 (C7H,Br, Ber. Br 64,OO Gef. Br 64,32). B: Siedep.160-1661,0.
0,1614 g Subst.: 0,1309 g AgC1. Cl(o)C,H,CH,N(CH,)CN = C,HgN,C1 Ber. C1 19,70 Gef. 20,07.
Base 8. A: Siedep.125-1301,0, Schmelzp. 41 O von Br(m)C,H,CH,Br. B: Siedep.180-184°.
0,1379 g Subst.: 0,1159 g AgBr. BrC,H,CH,N(CH,)CN = C,H,NPBr Ber. Br 35,53 Gef. Br 35,77.
Base 9. A: Siedep. 118-122150, Schmelzpunkt des Additions- produktes an (CH,),N 180- 181O; Mischprobe mit Cl(o)C,H,CH,N(CH,),Br 180-183 O.
B: Siedep. 180-1851,0' 0,1611 g Subst.: 0,1358 g AgBr.
Br(o).C,H,CB,N(CH,)CN = C,H,N,Br Ber. Br 35,53 Gef. Br 35,87. Base 10. A: Siedep. 125-1301,0,Schmelzp.30~ von Br(o).C,H4CH,Br.
B: Siedep.205-210,,0 bleibt flussig. 0,1471 g Subst.: 0,1306 g AgJ.
J(o)C,H,CH,.N(CH,)CN = C,H,N,.J Ber. J 47,76 Gef. J 47,99.
f5ber den Zerfall basischer und phenolischer Diphenylmethanderivate
und die Syntliese optisch aktiver aromatischer Verbindungen.
11. M i t t e i l u n g ; von Julius v. Braun.
(Mitbearbeitet von E r n s t An ton , W e r n e r H a e n s e l , G e r - h a r d I r m i s c h , R o b e r t Michae l i s u. Wilhelml 'euffer t . )
(Eingelaufen am 2. September 1933.)
Die im folgenden mitgeteilten Versuche sind zur wei- t e rm Abrundung und Klarung einiger bereits in der I. &lit- teilnng I) untersuchter Fragen angestellt worden. Es wurde damals u. a. gezeigt, daD die aus offenen und cyclischen
l) A. 47'2, 1 (1929).




















![Untersuchung zur Leistungsfähigkeit von Biofilmverfahren ... · ³] organischer Gesamttrockensubstanzgehalt (sessil und suspendiert) im Reaktor bezogen auf das Reaktorvolumen OV](https://img.pdfslide.org/doc/110x75/5e1d2b0087f9c466593a1feb/untersuchung-zur-leistungsfhigkeit-von-biofilmverfahren-organischer-gesamttrockensubstanzgehalt.jpg)








