Embed Size (px)
Citation preview

SS 2007
HauptseminarSynthetische Biologie
Department Physik der LMU
Friedrich Simmel

Ziele
• selbstständiges Einarbeiten in wissenschaftliche (Primär-) Literatur (Anleitung ca. 1 h)
• Konzeption eines wissenschaftlichen Vortrags:
- Gestaltung von Vortrags-Folien - Zeitplanung (45 min Vortrag, 10 min Diskussion)- wissenschaftlicher Vortragsstil (Anleitung ca. 1 h)
• Überblick über ein hochaktuelles Forschungsgebiet, das auch in unserer Arbeitsgruppe bearbeitet wird

Organisatorisches
• Themenvergabe
• 1-2 Vorbesprechungen (nach pers. Absprache): (a) Literatur und (b) Vortrag
• Ort + Zeit: Seminarraum N110, Altbau Physik, Mittwoch um 15 Uhr ct

Synthetische Biologie: Überblick
1. Allgemeine Gesichtspunkte
1.1 Netzwerkmotive in biochemischen Schaltkreisen
1.2 Reduzierte und minimale Genome
1.3 Was benötigt man für eine künstliche Zelle ?
1.4 Selbstreplikatoren

Synthetische Biologie: Überblick
1. Allgemeine Gesichtspunkte
1.1 Netzwerkmotive in biochemischen Schaltkreisen
1.2 Reduzierte und minimale Genome
1.3 Was benötigt man für eine künstliche Zelle ?
1.4 Selbstreplikatoren
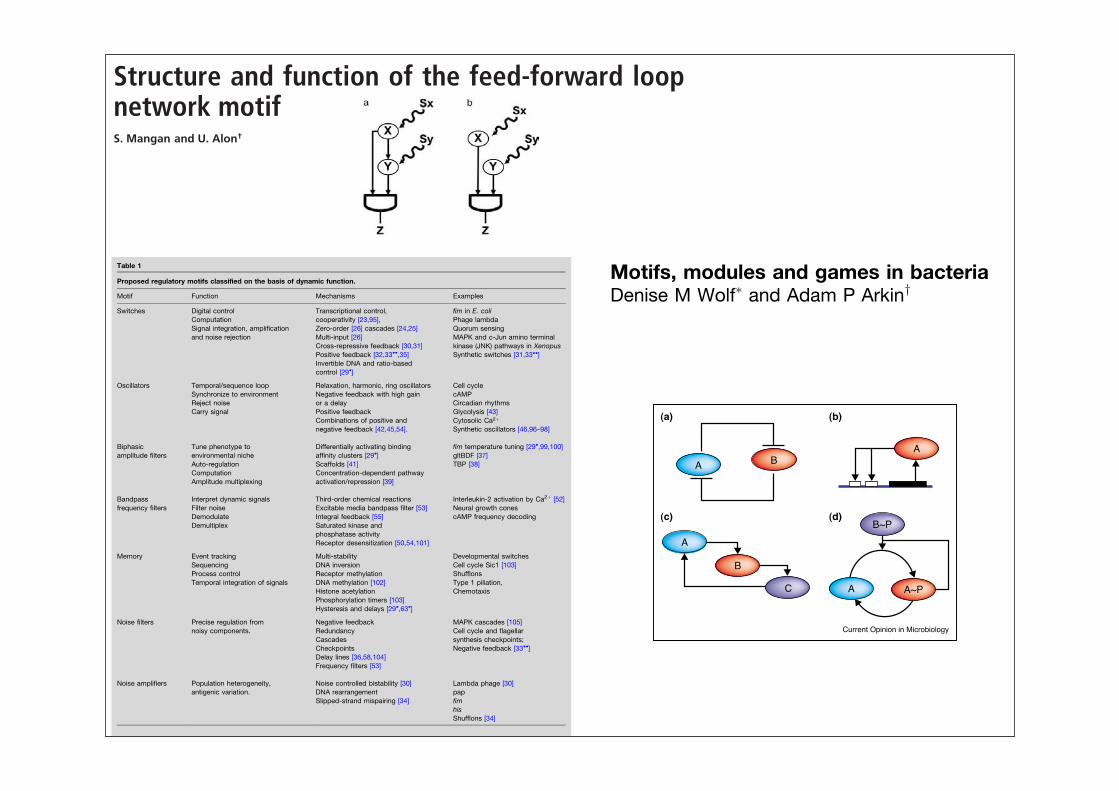
Structure and function of the feed-forward loopnetwork motifS. Mangan and U. Alon†
Departments of Molecular Cell Biology and Physics of Complex Systems, Weizmann Institute of Science, Rehovot 76100, Israel
Edited by Arnold J. Levine, Institute for Advanced Study, Princeton, NJ, and approved August 25, 2003 (received for review June 22, 2003)
Engineered systems are often built of recurring circuit modulesthat carry out key functions. Transcription networks that regulatethe responses of living cells were recently found to obey similarprinciples: they contain several biochemical wiring patterns,termed network motifs, which recur throughout the network. Oneof these motifs is the feed-forward loop (FFL). The FFL, a three-genepattern, is composed of two input transcription factors, one ofwhich regulates the other, both jointly regulating a target gene.The FFL has eight possible structural types, because each of thethree interactions in the FFL can be activating or repressing. Here,we theoretically analyze the functions of these eight structuraltypes. We find that four of the FFL types, termed incoherent FFLs,act as sign-sensitive accelerators: they speed up the response timeof the target gene expression following stimulus steps in onedirection (e.g., off to on) but not in the other direction (on to off).The other four types, coherent FFLs, act as sign-sensitive delays. Wefind that some FFL types appear in transcription network databasesmuch more frequently than others. In some cases, the rare FFLtypes have reduced functionality (responding to only one of theirtwo input stimuli), which may partially explain why they areselected against. Additional features, such as pulse generation andcooperativity, are discussed. This study defines the function of oneof the most significant recurring circuit elements in transcriptionnetworks.
Cells contain networks of biochemical transcription interac-tions. These networks have evolved to perform information-
processing functions (1, 2). The inputs to the network, such asexternal nutrients and stresses, affect the activity of transcriptionfactor proteins. The transcription factors bind regulatory regionsof specific genes and activate or repress their transcription. As aresult, cell processes are modulated to fit the environmentalconditions. Transcription networks can be described as directedgraphs, in which the nodes are genes (3–12). Directed edgesrepresent transcription interactions, where a transcription factorencoded by one gene modulates the transcription rate of thesecond gene.
It is of interest to understand the dynamic behavior oftranscription networks (2, 3, 5, 7–10). It was recently found thatthese networks contain significantly recurring wiring patternstermed ‘‘network motifs’’ (6, 11, 12). Network motifs are pat-terns that occur in the network far more often than in random-ized networks with the same degree sequence (6, 11). Thetranscription networks of the bacterium Escherichia coli (6, 11)and the yeast Saccharomyces cerevisiae (11, 12) were found tocontain the same small set of highly significant motifs. Thesignificance of these structures raises the question of whetherthey have specific information-processing roles in the network.If they do, they might be used to understand the networkdynamics in terms of elementary computational building blocks.
One of the most significant network motifs in both E. coli andyeast is the feed-forward loop (FFL) (6, 11). The FFL iscomposed of a transcription factor X, which regulates a secondtranscription factor Y (Fig 1a). X and Y both bind the regulatoryregion of target gene Z and jointly modulate its transcriptionrate. The FFL has two input signals, the inducers, Sx and Sy,which are small molecules, protein partners, or covalent modi-
fications that activate or inhibit the transcriptional activity of Xand Y (Fig. 1a). The FFL has three transcription interactions.Each of these can be either positive (activation) or negative(repression). There are therefore eight possible structural con-figurations of activator and repressor interactions (6) (Tables 1and 2). Four of these configurations are termed ‘‘coherent’’(Table 1): the sign of the direct regulation path (from X to Z)is the same as the overall sign of the indirect regulation path(from X through Y to Z) (6). The other four structures aretermed ‘‘incoherent’’ (Table 2): the signs of the direct andindirect regulation paths are opposite.
The effects of transcription factors X and Y are integrated atthe promoter region of gene Z. The level of Z expression ismodulated according to the concentrations of X and Y tran-scription factors bound to their inducers. This modulation isdescribed by the cis-regulatory input function of gene Z (7, 13,14). Common examples of cis-regulatory input functions includeAND-like gates, in which both X and Y are needed to expressZ, and OR-gate logic in which either X or Y is sufficient toexpress Z.
Here we use mathematical modeling to study the function ofthe eight FFL structural configurations, with AND- and OR-gatelogic. This work extends our previous study that was limited toonly one FFL type with three activators and AND logic (6). Wefind that incoherent FFLs can serve as a novel mechanism foraccelerating the expression of the target genes. Both coherentand incoherent FFL behavior is sign sensitive: they accelerate or
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: FFL, feed-forward loop.†To whom correspondence should be addressed. E-mail: [email protected].
© 2003 by The National Academy of Sciences of the USA
Fig. 1. (a) FFL. Transcription factor X regulates transcription factor Y, andboth jointly regulate Z. Sx and Sy are the inducers of X and Y, respectively. Theaction of X and Y is integrated at the Z promoter with a cis-regulatory inputfunction (7, 14), such as AND or OR logic. (b) Simple regulation of Z by Xand Y.
11980–11985 ! PNAS ! October 14, 2003 ! vol. 100 ! no. 21 www.pnas.org"cgi"doi"10.1073"pnas.2133841100
Structure and function of the feed-forward loopnetwork motifS. Mangan and U. Alon†
Departments of Molecular Cell Biology and Physics of Complex Systems, Weizmann Institute of Science, Rehovot 76100, Israel
Edited by Arnold J. Levine, Institute for Advanced Study, Princeton, NJ, and approved August 25, 2003 (received for review June 22, 2003)
Engineered systems are often built of recurring circuit modulesthat carry out key functions. Transcription networks that regulatethe responses of living cells were recently found to obey similarprinciples: they contain several biochemical wiring patterns,termed network motifs, which recur throughout the network. Oneof these motifs is the feed-forward loop (FFL). The FFL, a three-genepattern, is composed of two input transcription factors, one ofwhich regulates the other, both jointly regulating a target gene.The FFL has eight possible structural types, because each of thethree interactions in the FFL can be activating or repressing. Here,we theoretically analyze the functions of these eight structuraltypes. We find that four of the FFL types, termed incoherent FFLs,act as sign-sensitive accelerators: they speed up the response timeof the target gene expression following stimulus steps in onedirection (e.g., off to on) but not in the other direction (on to off).The other four types, coherent FFLs, act as sign-sensitive delays. Wefind that some FFL types appear in transcription network databasesmuch more frequently than others. In some cases, the rare FFLtypes have reduced functionality (responding to only one of theirtwo input stimuli), which may partially explain why they areselected against. Additional features, such as pulse generation andcooperativity, are discussed. This study defines the function of oneof the most significant recurring circuit elements in transcriptionnetworks.
Cells contain networks of biochemical transcription interac-tions. These networks have evolved to perform information-
processing functions (1, 2). The inputs to the network, such asexternal nutrients and stresses, affect the activity of transcriptionfactor proteins. The transcription factors bind regulatory regionsof specific genes and activate or repress their transcription. As aresult, cell processes are modulated to fit the environmentalconditions. Transcription networks can be described as directedgraphs, in which the nodes are genes (3–12). Directed edgesrepresent transcription interactions, where a transcription factorencoded by one gene modulates the transcription rate of thesecond gene.
It is of interest to understand the dynamic behavior oftranscription networks (2, 3, 5, 7–10). It was recently found thatthese networks contain significantly recurring wiring patternstermed ‘‘network motifs’’ (6, 11, 12). Network motifs are pat-terns that occur in the network far more often than in random-ized networks with the same degree sequence (6, 11). Thetranscription networks of the bacterium Escherichia coli (6, 11)and the yeast Saccharomyces cerevisiae (11, 12) were found tocontain the same small set of highly significant motifs. Thesignificance of these structures raises the question of whetherthey have specific information-processing roles in the network.If they do, they might be used to understand the networkdynamics in terms of elementary computational building blocks.
One of the most significant network motifs in both E. coli andyeast is the feed-forward loop (FFL) (6, 11). The FFL iscomposed of a transcription factor X, which regulates a secondtranscription factor Y (Fig 1a). X and Y both bind the regulatoryregion of target gene Z and jointly modulate its transcriptionrate. The FFL has two input signals, the inducers, Sx and Sy,which are small molecules, protein partners, or covalent modi-
fications that activate or inhibit the transcriptional activity of Xand Y (Fig. 1a). The FFL has three transcription interactions.Each of these can be either positive (activation) or negative(repression). There are therefore eight possible structural con-figurations of activator and repressor interactions (6) (Tables 1and 2). Four of these configurations are termed ‘‘coherent’’(Table 1): the sign of the direct regulation path (from X to Z)is the same as the overall sign of the indirect regulation path(from X through Y to Z) (6). The other four structures aretermed ‘‘incoherent’’ (Table 2): the signs of the direct andindirect regulation paths are opposite.
The effects of transcription factors X and Y are integrated atthe promoter region of gene Z. The level of Z expression ismodulated according to the concentrations of X and Y tran-scription factors bound to their inducers. This modulation isdescribed by the cis-regulatory input function of gene Z (7, 13,14). Common examples of cis-regulatory input functions includeAND-like gates, in which both X and Y are needed to expressZ, and OR-gate logic in which either X or Y is sufficient toexpress Z.
Here we use mathematical modeling to study the function ofthe eight FFL structural configurations, with AND- and OR-gatelogic. This work extends our previous study that was limited toonly one FFL type with three activators and AND logic (6). Wefind that incoherent FFLs can serve as a novel mechanism foraccelerating the expression of the target genes. Both coherentand incoherent FFL behavior is sign sensitive: they accelerate or
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: FFL, feed-forward loop.†To whom correspondence should be addressed. E-mail: [email protected].
© 2003 by The National Academy of Sciences of the USA
Fig. 1. (a) FFL. Transcription factor X regulates transcription factor Y, andboth jointly regulate Z. Sx and Sy are the inducers of X and Y, respectively. Theaction of X and Y is integrated at the Z promoter with a cis-regulatory inputfunction (7, 14), such as AND or OR logic. (b) Simple regulation of Z by Xand Y.
11980–11985 ! PNAS ! October 14, 2003 ! vol. 100 ! no. 21 www.pnas.org"cgi"doi"10.1073"pnas.2133841100
Motifs, modules and games in bacteriaDenise M Wolf! and Adam P Arkiny
Global explorations of regulatory network dynamics,
organization and evolution have become tractable thanks to
high-throughput sequencing and molecular measurement of
bacterial physiology. From these, a nascent conceptual
framework is developing, that views the principles of regulation in
term of motifs, modules and games. Motifs are small, repeated,
and conserved biological units ranging from molecular domains
to small reaction networks. They are arranged into functional
modules, genetically dissectible cellular functions such as the
cell cycle, or different stress responses. The dynamical
functioning ofmodules defines the organism’s strategy to survive
in a game, pitting cell against cell, and cell against environment.
Placing pathway structure and dynamics into an evolutionary
context begins to allow discrimination between those physical
and molecular features that particularize a species to its
surroundings, and those that provide core physiological function.
This approach promises to generate a higher level understanding
of cellular design, pathway evolution and cellular bioengineering.
AddressesDepartments of Bioengineering and Chemistry, University of California,Physical Biosciences Division, Lawrence Berkeley National Laboratory,Howard Hughes Medical Institute, 1 Cyclotron Road, MS 3-144,Berkeley, CA 94720, USA!e-mail: [email protected]: Adam P Arkine-mail: [email protected]
Current Opinion in Microbiology 2003, 6:125–134
This review comes from a themed issue onCell regulationEdited by Andree Lazdunski and Carol Gross
1369-5274/03/$ – see front matter! 2003 Elsevier Science Ltd. All rights reserved.
DOI 10.1016/S1369-5274(03)00033-X
AbbreviationsJNK c-Jun amino-terminal kinaseMAPK mitogen activated protein kinaseTBP TATA-binding protein
IntroductionWhole-genome/high-throughput techniques open ques-tions about entire organismal function and make feasiblecomparisons of the behavior of different organisms andtheir mutants. The number of computational tools used toperform and quantify these comparisons has multiplied[1–11]. This new fare is generating a more complete viewof cellular function, by exposing and investigating theextensive networks of interconnections amongst cellularcomponents and processes.
Analysis and simulation of network dynamics can verifythat all the data on a particular pathway are consistent; itcan test and generate hypotheses about network struc-ture, the fundamental operating principles governingnetwork function and the role of feedback and proteinmodifications. It can also predict the effects of mutation,environmental perturbation and pharmaceutical actions[12]. Topological analyses look for metrics and patterns ofinterconnections across and between networks [13",14,15,16"]. Evolutionary analysis on the level of networksand pathways is also now possible, together with moretraditional physiological and molecular evolutionaryinvestigations. Dynamics, topology and evolution areall interconnected, because evolutionary forces constraindynamics, and the functional imperatives of dynamicscanalize topology. Moreover, investigations into thesetopics provide clues on network decomposition (theidentification of functionally significant subnetworks suchas motifs and modules or other, yet to be discovered,organizational units besides operons and regulons) [17].
Network-oriented approaches have extended questionsof similarity and design far beyond the level of singlegenes and proteins, to how networks translate perturba-tions into dynamical behavior of the cell, how they are thesame and different across many different species, andwhy behavior is different in one species from that inanother, despite a good deal of network homology.
In this review, we organize recent work on these networktopics into a framework for thinking about how intracel-lular networks regulate cellular behavior and why theydo it the way they do. The framework is built on theconcepts of motifs, modules and games.
MotifsCellular regulation is achieved through the complex net-work of interactions among biochemicals and cellularstructures. The challenge to understanding the dynamicfunction of these networks, composed of perhaps tens ofthousands of reactions among thousands of distinct che-mical species, lies in this very complexity. It is thereforeimportant to find ways of simplifying the description ofthese networks to facilitate analysis. One such attempt isin the identification of motifs (small, repeated, perhapsevolutionarily conserved regulatory subnetworks, classifi-able on the basis of function, architecture, dynamics, orbiochemical process) [17–19]. Regulatory motifs proposedto date, with the help of mathematical systems theory andcomplementary experiments, include switches, ampli-tude filters, oscillators, frequency filters, noise filters andamplifiers, combinatorial logic, homeostats, rheostats, logic
125
www.current-opinion.com Current Opinion in Microbiology 2003, 6:125–134
gates and memory elements (Table 1; [18,20]). Wedescribe just a few of these examples below.
SwitchesRegulatory switches enable cells to respond to environ-mental or intercellular signals with an all-or-nothingresponse. Switches control eukaryotic development(e.g. vulvar development in Caenorhabditis elegans) andmany bacterial stress responses (e.g. alternative meta-bolic pathways, pili expression, sporulation and compe-tence). Switches can be memory-less, like a doorbell
(Figure 1a), or multistable, like a light switch. Theyare randomly triggered or tightly controlled, and mani-fested by single cells or populations, as in quorum sensing[21,22]. Elementary memory-less switching mechanismsinclude the cooperative activation or repression of geneexpression [23]; cascade ultrasensitivity, arising in mito-gen-activated protein kinase (MAPK) cascades [24,25];multi-input cascades, as found in glycolysis [26]; zero-order ultrasensitivity, postulated for futile cycles operat-ing near saturation [27] and observed in the formate/lactic dehydrogenase cycle [28]; and ratio-controlled
Table 1
Proposed regulatory motifs classified on the basis of dynamic function.
Motif Function Mechanisms Examples
Switches Digital control Transcriptional control,cooperativity [23,95],
fim in E. coliComputation
Zero-order [26] cascades [24,25]Phage lambda
Signal integration, amplificationand noise rejection Multi-input [26]
Quorum sensing
Cross-repressive feedback [30,31]MAPK and c-Jun amino terminalkinase (JNK) pathways in XenopusSynthetic switches [31,33!!]Positive feedback [32,33!!,35]
Invertible DNA and ratio-basedcontrol [29!]
Oscillators Temporal/sequence loop Relaxation, harmonic, ring oscillators Cell cycleSynchronize to environment Negative feedback with high gain
or a delaycAMP
Reject noisePositive feedback
Circadian rhythmsCarry signal
Combinations of positive andnegative feedback [42,45,54].
Glycolysis [43]Cytosolic Ca2"
Synthetic oscillators [46,96–98]
Biphasicamplitude filters
Tune phenotype toenvironmental niche
Differentially activating bindingaffinity clusters [29!]
fim temperature tuning [29!,99,100]
Auto-regulation Scaffolds [41]gltBDF [37]
Computation Concentration-dependent pathwayactivation/repression [39]
TBP [38]
Amplitude multiplexing
Bandpassfrequency filters
Interpret dynamic signals Third-order chemical reactions Interleukin-2 activation by Ca2" [52]Filter noise Excitable media bandpass filter [53] Neural growth conesDemodulate Integral feedback [55] cAMP frequency decodingDemultiplex Saturated kinase and
phosphatase activityReceptor desensitization [50,54,101]
Memory Event tracking Multi-stability Developmental switchesSequencing DNA inversion Cell cycle Sic1 [103]Process control Receptor methylation ShufflonsTemporal integration of signals DNA methylation [102] Type 1 piliation,
Histone acetylation ChemotaxisPhosphorylation timers [103]Hysteresis and delays [29!,63!]
Noise filters Precise regulation fromnoisy components.
Negative feedback MAPK cascades [105]Redundancy Cell cycle and flagellar
synthesis checkpoints;CascadesNegative feedback [33!!]Checkpoints
Delay lines [36,58,104]Frequency filters [53]
Noise amplifiers Population heterogeneity,antigenic variation.
Noise controlled bistability [30] Lambda phage [30]DNA rearrangement papSlipped-strand mispairing [34] fim
hisShufflons [34]
126 Cell regulation
Current Opinion in Microbiology 2003, 6:125–134 www.current-opinion.com
activation, characterized by differential activation of aprocess by two competing regulatory proteins, as found inthe network controlling the probability of type 1 piliexpression in Escherichia coli [29!].
Unlike memory-less switches, bistable switches are hys-teretic and so can be ‘set’ (possibly irreversibly) to an ‘on’or ‘off’ state by the transient application of a stimulus(Figure 1b). Bistable switching motifs, particularlyimportant to developmental and transformational pro-cesses, include cross-repressive feedback loops withcooperativity, as found in the lambda phage [30] andsynthetically constructed inE. coli [31]; positive feedback
with cooperativity, as seen in c-Jun amino-terminalkinase (JNK) circuits [32] and tested in the syntheticyeast switch [33!!]; site specific DNA inversion, found innetworks controlling surface structures like pili and fla-gella [29!,34]; andmanymemory-less switching architec-tureswrapped in feedback (for example, the autocatalyticultrasensitive MAPK cascade in Xenopus oocytes [35])(Figure 2). Bistable switches are thought to control devel-opmental and transformative processes because of theirability to ‘remember’ a stimulus and maintain a stateindefinitely. Memory-less switches, however, are likelyto serve as signal-thresholding components in larger sys-tems, or control processes requiring reversible on/offcontrol. Even in reversible control, however, a smallamount of hysteresis can prevent ‘switching chatter’,rapid, unproductive cycling between ‘on’ and ‘off ’ statestriggered by intracellular noise [29!,36].
Biphasic amplitude filtersA biphasic amplitude filter is a device that amplifies aninput signal only if it is within a specific range, therebyallowing a process to be triggered by a particular envir-onmental or intracellular condition. In theory, the serialconnection of two oppositely oriented switches canimplement a biphasic response; however, recent analyseshave uncovered alternative mechanisms. One proposedmechanism for biphasic control involves multiple DNA-binding sites with differential affinities and regulatoryeffects (Figure 3a). This motif was thought to tune type 1
Figure 1
0
Outp
ut sig
nal
Outp
ut sig
nal
Low responselevel
High responselevel
Going up
Coming down
0.5 1 1.5 2 2.5 3 3.5 4 4.5 5
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
Input signal
0.5 1 1.5 2 2.5 3 3.5 4 4.5 50
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1
Input signal
Current Opinion in Microbiology
(a)
(b)
Memory-less (a) and bistable (b) switches. (a) A memory-less switch canbe ‘on’ or ‘off’ depending on the level of the input signal, but cannotbe ‘set’ by the transient application of a stimulus. (b) Bistable switchesare hysteretic, meaning that different stimulus-response curves aregenerated depending on whether the system begins in the ‘on’ or ‘off’state. These systems have memory, as a transient input stimulus canpotentially ‘set’ a bistable switch to an ‘on’ or ‘off’ state.
Figure 2
Current Opinion in Microbiology
AB
A
A
B~P
A~P
(a) (b)
(c) (d)
A
B
C
Bistable switching mechanisms. Bistable switching mechanisms include(a) cross-repressive feedback with cooperativity, in which A inhibits Bcooperatively and B inhibits A [30,31]; (b) cooperativeauto-activation of gene expression, for example if gene product Aactivates its own expression in a cooperative manner; (c) ultrasensitivecascades with feedback as postulated for a MAPK cascade switch inXenopus oocytes [35]; and (d) zero-order sensitivity with feedback, forexample in auto-catalyzed phosphorylation/dephosphorylation reactioncycles operating near saturation.
Motifs, modules and games in bacteria Wolf and Arkin 127
www.current-opinion.com Current Opinion in Microbiology 2003, 6:125–134

Synthetische Biologie: Überblick
1. Allgemeine Gesichtspunkte
1.1 Netzwerkmotive in biochemischen Schaltkreisen
1.2 Reduzierte und minimale Genome
1.3 Was benötigt man für eine künstliche Zelle ?
1.4 Selbstreplikatoren
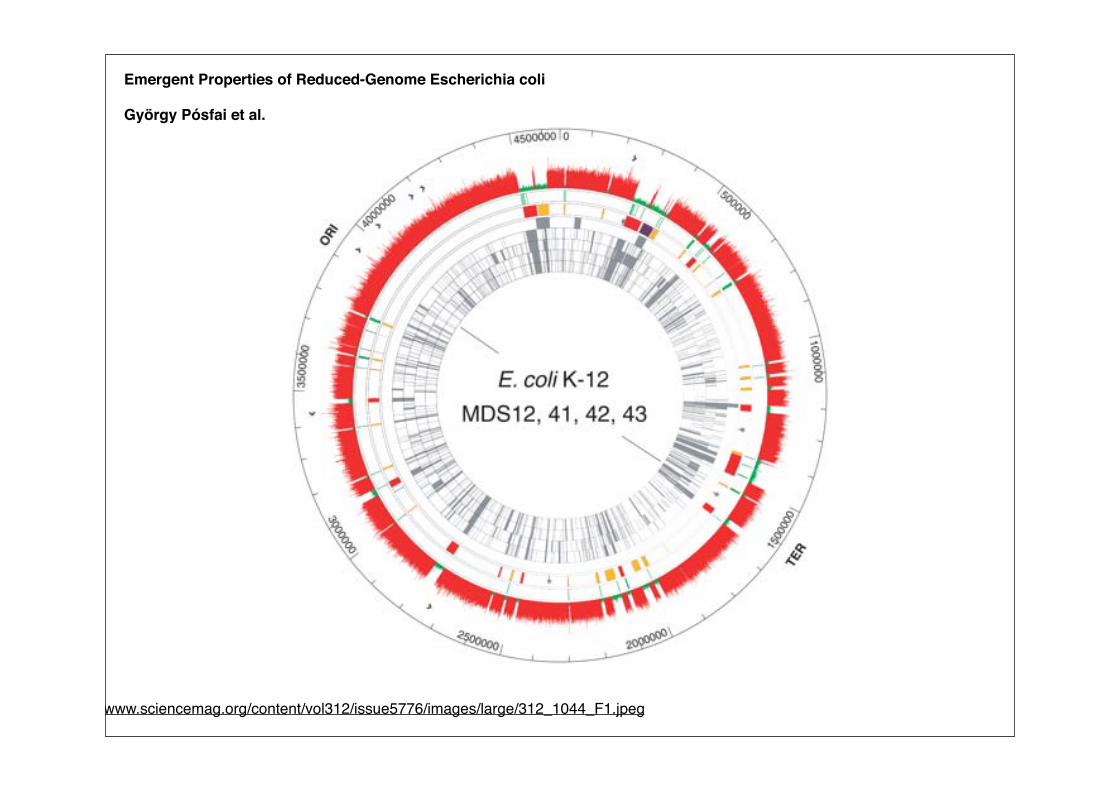
Emergent Properties of Reduced-Genome Escherichia coli
György Pósfai et al.
http://www.sciencemag.org/content/vol312/issue5776/images/large/312_1044_F1.jpeg
Essential Bacillus subtilis genesK. Kobayashia, S. D. Ehrlichb,c, A. Albertinid, G. Amatid, K. K. Andersene, M. Arnaudf, K. Asaig, S. Ashikagah, S. Aymerichi,P. Bessieresj, F. Bolandk, S. C. Brignelll, S. Bronm, K. Bunain, J. Chapuisb, L. C. Christianseno, A. Danchinp,M. Debarbouillef, E. Dervynb, E. Deuerlingq, K. Devinee, S. K. Devinee, O. Dreesenp, J. Erringtonr, S. Fillingeri,S. J. Fosterk, Y. Fujitas, A. Galizzid, R. Gardanf, C. Eschevinsm, T. Fukushimat, K. Hagau, C. R. Harwoodl, M. Heckerv,D. Hosoyaw, M. F. Hullop, H. Kakeshitan, D. Karamatax, Y. Kasaharaa, F. Kawamurah, K. Kogah, P. Koskiy, R. Kuwanaz,D. Imamuraw, M. Ishimaruw, S. Ishikawat, I. Ishios, D. Le Coqi, A. Massonaa, C. Mauelx, R. Meimam, R. P. Melladobb,A. Moirk, S. Moriyaa, E. Nagakawas, H. Nanamiyah, S. Nakaia, P. Nygaardo, M. Oguracc, T. Ohananq, M. O’Reillye,M. O’Rourkek, Z. Pragail, H. M. Pooleyx, G. Rapoportf, J. P. Rawlinsr, L. A. Rivasbb, C. Rivoltax, A. Sadaieu, Y. Sadaieg,M. Sarvasy, T. Satow, H. H. Saxildo, E. Scanlane, W. Schumannq, J. F. M. L. Seegersaa, J. Sekiguchit, A. Sekowskap,S. J. Seroraa, M. Simondd, P. Stragierdd, R. Studerx, H. Takamatsuz, T. Tanakacc, M. Takeuchiw, H. B. Thomaidesr,V. Vagnerb, J. M. van Dijlm, K. Watabez, A. Wipatl, H. Yamamotot, M. Yamamotos, Y. Yamamotos, K. Yamanen, K. Yataee,K. Yoshidas, H. Yoshikawau, U. Zuberv, and N. Ogasawaraa
aGraduate School of Information Science, Nara Institute of Science and Technology, Nara 630-0101, Japan; bGenetique Microbienne, Institut National de laRecherche Agronomique, 78530 Jouy en Josas, France; dGenetica e Microbiologia, Universita di Pavia, 1 via Ferrata, 27100 Pavia, Italy; eGenetics, SmurfitInstitute, Trinity College, Dublin 2, Ireland; fBiochimie Microbienne, Institut Pasteur, 25 Rue du Dr. Roux, 75015 Paris, France; gFaculty of Science, SaitamaUniversity, Saitama 338-8570, Japan; hCollege of Science, Rikkyo (St. Paul’s) University, Tokyo 171-8501, Japan; iGenetique Moleculaire et Cellulaire,Institut National de la Recherche Agronomique–Centre National de la Recherche Scientifique–Institut National Agronomique Paris-Grignon, 78850Thiverval-Grignon, France; jMathematiques Informatique Genomes, Institut National de la Recherche Agronomique, 78530 Jouy en Josas, France;kMolecular Biology and Biotechnology, University of Sheffield, Sheffield S10 2TN, United Kingdom; lCell and Molecular Bioscience, Newcastle UniversityMedical School, Framlington Place, Newcastle upon Tyne NE2 4HH, United Kingdom; mGenetics, Groningen Biomolecular Sciences and BiotechnologyInstitute, 9750 AA, Haren, The Netherlands; nInstitute of Biological Sciences, University of Tsukuba, Ibaraki 305-8572, Japan; oBiological Chemistry, Instituteof Molecular Biology, Solvgade 83, 1307 K, Copenhagen, Denmark; pGenetique des Genomes Bacteriens, Institut Pasteur, Unite de Recherche Associee,Centre National de la Recherche Scientifique 2171, 75015 Paris, France; qInstitute of Genetics, Bayreuth University, D-95440 Bayreuth, Germany; rSirWilliam Dunn School of Pathology, Oxford University, Oxford OX1 3RE, United Kingdom; sFaculty of Life Science and Biotechnology, Fukuyama University,Hiroshima 729-0292, Japan; tFaculty of Textile Science and Technology, Shinshu University, Nagano 386-8564, Japan; uDepartment of Bioscience, TokyoUniversity of Agriculture, Tokyo 156-8502, Japan; vInstitute for Microbiology, Ernst-Moritz-Arndt-University, D-17487 Greifswald, Germany; wDepartmentof International Environmental and Agricultural Science, Tokyo University of Agriculture and Technology, Tokyo 183-8509, Japan; xInstitut de Genetiqueet de Biologie Microbiennes, CH-1005 Lausanne, Switzerland; yNational Public Health Institute, 00300, Helsinki, Finland; zFaculty of Pharmaceutical Sciences,Setsunan University, Osaka 573-0101, Japan; aaInstitut de Genetique et Microbiologie, Centre National de la Recherche Scientifique Unite Mixte deRecherche 8621, Universite Paris-Sud, 91405 Orsay Cedex, France; bbCentro Nacional de Biotecnologıa, Campus de la Universidad Autonoma,Cantoblanco, 28049 Madrid, Spain; ccSchool of Marine Science and Technology, University of Tokai, Shizuoka 424-8610, Japan; ddInstitut deBiologie Physico-Chimique, 75005 Paris, France; and eeRadioisotope Center, National Institute of Genetics, Shizuoka 411-8540, Japan
Communicated by Richard M. Losick, Harvard University, Cambridge, MA, January 27, 2003 (received for review November 10, 2002)
To estimate the minimal gene set required to sustain bacterial lifein nutritious conditions, we carried out a systematic inactivation ofBacillus subtilis genes. Among !4,100 genes of the organism, only192 were shown to be indispensable by this or previous work.Another 79 genes were predicted to be essential. The vast majorityof essential genes were categorized in relatively few domains ofcell metabolism, with about half involved in information process-ing, one-fifth involved in the synthesis of cell envelope and thedetermination of cell shape and division, and one-tenth related tocell energetics. Only 4% of essential genes encode unknownfunctions. Most essential genes are present throughout a widerange of Bacteria, and almost 70% can also be found in Archaeaand Eucarya. However, essential genes related to cell envelope,shape, division, and respiration tend to be lost from bacteria withsmall genomes. Unexpectedly, most genes involved in the Emb-den–Meyerhof–Parnas pathway are essential. Identification of un-known and unexpected essential genes opens research avenues tobetter understanding of processes that sustain bacterial life.
The definition of the minimal gene set required to sustain aliving cell is of considerable interest. The functions specified
by such a set are likely to provide a view of a ‘‘minimal’’ bacterialcell. Many functions should be essential in all cells and could beconsidered as a foundation of life itself. The determination of therange of essential functions in different cells should revealpossible solutions for sustaining life. Computational and exper-imental research has previously been carried out to define aminimal protein-encoding gene set. An upper-limit estimate ofa minimal bacterial gene set was obtained from the sequence ofthe entire Mycoplasma genitalium genome, which contains only!480 genes (1). A computational approach, based on theassumption that essential genes are conserved in the genomes of
M. genitalium and Haemophilus influenzae, led to a description ofa smaller set of some 260 genes (2). More recently, an experi-mental approach involving high-density transposon mutagenesisof the H. influenzae genome led to a much higher estimate of!670 putative essential genes (3), whereas transposon mutagen-esis of two mycoplasma species led to an estimate of 265–360essential genes (4). Another experimental approach using anti-sense RNA to inhibit gene expression led to the identification ofsome 150 essential genes in Staphylococcus aureus (5). However,these approaches have limitations. Computation is likely tounderestimate the minimal gene set because it takes into accountonly those genes that have remained similar enough during thecourse of evolution to be recognized as true orthologues.Transposon mutagenesis might overestimate the set by misclas-sification of nonessential genes that slow down the growthwithout arresting it but can also miss essential genes that toleratetransposon insertions (3, 6). Finally, the use of antisense RNAis limited to the genes for which an adequate expression of theinhibitory RNA can be obtained in the organism under study.
To obtain an independent and possibly more reliable estimateof a minimal protein-encoding gene set for bacteria, we system-atically inactivated Bacillus subtilis genes. B. subtilis was chosenbecause it is one of the best studied bacteria (7) and is a modelfor low-G"C Gram-positive bacteria, which include both deadlypathogens, such as Bacillus anthracis, and bacteria widely used infood and industry, such as lactococci and bacilli. Because theessentiality of a gene depends on the conditions under which theorganism is propagated, we used an environment likely to beoptimal for B. subtilis and thus carried out inactivation on a
cTo whom correspondence should be addressed. E-mail: [email protected] [email protected].
4678–4683 ! PNAS ! April 15, 2003 ! vol. 100 ! no. 8 www.pnas.org"cgi"doi"10.1073"pnas.0730515100
Essential Bacillus subtilis genesK. Kobayashia, S. D. Ehrlichb,c, A. Albertinid, G. Amatid, K. K. Andersene, M. Arnaudf, K. Asaig, S. Ashikagah, S. Aymerichi,P. Bessieresj, F. Bolandk, S. C. Brignelll, S. Bronm, K. Bunain, J. Chapuisb, L. C. Christianseno, A. Danchinp,M. Debarbouillef, E. Dervynb, E. Deuerlingq, K. Devinee, S. K. Devinee, O. Dreesenp, J. Erringtonr, S. Fillingeri,S. J. Fosterk, Y. Fujitas, A. Galizzid, R. Gardanf, C. Eschevinsm, T. Fukushimat, K. Hagau, C. R. Harwoodl, M. Heckerv,D. Hosoyaw, M. F. Hullop, H. Kakeshitan, D. Karamatax, Y. Kasaharaa, F. Kawamurah, K. Kogah, P. Koskiy, R. Kuwanaz,D. Imamuraw, M. Ishimaruw, S. Ishikawat, I. Ishios, D. Le Coqi, A. Massonaa, C. Mauelx, R. Meimam, R. P. Melladobb,A. Moirk, S. Moriyaa, E. Nagakawas, H. Nanamiyah, S. Nakaia, P. Nygaardo, M. Oguracc, T. Ohananq, M. O’Reillye,M. O’Rourkek, Z. Pragail, H. M. Pooleyx, G. Rapoportf, J. P. Rawlinsr, L. A. Rivasbb, C. Rivoltax, A. Sadaieu, Y. Sadaieg,M. Sarvasy, T. Satow, H. H. Saxildo, E. Scanlane, W. Schumannq, J. F. M. L. Seegersaa, J. Sekiguchit, A. Sekowskap,S. J. Seroraa, M. Simondd, P. Stragierdd, R. Studerx, H. Takamatsuz, T. Tanakacc, M. Takeuchiw, H. B. Thomaidesr,V. Vagnerb, J. M. van Dijlm, K. Watabez, A. Wipatl, H. Yamamotot, M. Yamamotos, Y. Yamamotos, K. Yamanen, K. Yataee,K. Yoshidas, H. Yoshikawau, U. Zuberv, and N. Ogasawaraa
aGraduate School of Information Science, Nara Institute of Science and Technology, Nara 630-0101, Japan; bGenetique Microbienne, Institut National de laRecherche Agronomique, 78530 Jouy en Josas, France; dGenetica e Microbiologia, Universita di Pavia, 1 via Ferrata, 27100 Pavia, Italy; eGenetics, SmurfitInstitute, Trinity College, Dublin 2, Ireland; fBiochimie Microbienne, Institut Pasteur, 25 Rue du Dr. Roux, 75015 Paris, France; gFaculty of Science, SaitamaUniversity, Saitama 338-8570, Japan; hCollege of Science, Rikkyo (St. Paul’s) University, Tokyo 171-8501, Japan; iGenetique Moleculaire et Cellulaire,Institut National de la Recherche Agronomique–Centre National de la Recherche Scientifique–Institut National Agronomique Paris-Grignon, 78850Thiverval-Grignon, France; jMathematiques Informatique Genomes, Institut National de la Recherche Agronomique, 78530 Jouy en Josas, France;kMolecular Biology and Biotechnology, University of Sheffield, Sheffield S10 2TN, United Kingdom; lCell and Molecular Bioscience, Newcastle UniversityMedical School, Framlington Place, Newcastle upon Tyne NE2 4HH, United Kingdom; mGenetics, Groningen Biomolecular Sciences and BiotechnologyInstitute, 9750 AA, Haren, The Netherlands; nInstitute of Biological Sciences, University of Tsukuba, Ibaraki 305-8572, Japan; oBiological Chemistry, Instituteof Molecular Biology, Solvgade 83, 1307 K, Copenhagen, Denmark; pGenetique des Genomes Bacteriens, Institut Pasteur, Unite de Recherche Associee,Centre National de la Recherche Scientifique 2171, 75015 Paris, France; qInstitute of Genetics, Bayreuth University, D-95440 Bayreuth, Germany; rSirWilliam Dunn School of Pathology, Oxford University, Oxford OX1 3RE, United Kingdom; sFaculty of Life Science and Biotechnology, Fukuyama University,Hiroshima 729-0292, Japan; tFaculty of Textile Science and Technology, Shinshu University, Nagano 386-8564, Japan; uDepartment of Bioscience, TokyoUniversity of Agriculture, Tokyo 156-8502, Japan; vInstitute for Microbiology, Ernst-Moritz-Arndt-University, D-17487 Greifswald, Germany; wDepartmentof International Environmental and Agricultural Science, Tokyo University of Agriculture and Technology, Tokyo 183-8509, Japan; xInstitut de Genetiqueet de Biologie Microbiennes, CH-1005 Lausanne, Switzerland; yNational Public Health Institute, 00300, Helsinki, Finland; zFaculty of Pharmaceutical Sciences,Setsunan University, Osaka 573-0101, Japan; aaInstitut de Genetique et Microbiologie, Centre National de la Recherche Scientifique Unite Mixte deRecherche 8621, Universite Paris-Sud, 91405 Orsay Cedex, France; bbCentro Nacional de Biotecnologıa, Campus de la Universidad Autonoma,Cantoblanco, 28049 Madrid, Spain; ccSchool of Marine Science and Technology, University of Tokai, Shizuoka 424-8610, Japan; ddInstitut deBiologie Physico-Chimique, 75005 Paris, France; and eeRadioisotope Center, National Institute of Genetics, Shizuoka 411-8540, Japan
Communicated by Richard M. Losick, Harvard University, Cambridge, MA, January 27, 2003 (received for review November 10, 2002)
To estimate the minimal gene set required to sustain bacterial lifein nutritious conditions, we carried out a systematic inactivation ofBacillus subtilis genes. Among !4,100 genes of the organism, only192 were shown to be indispensable by this or previous work.Another 79 genes were predicted to be essential. The vast majorityof essential genes were categorized in relatively few domains ofcell metabolism, with about half involved in information process-ing, one-fifth involved in the synthesis of cell envelope and thedetermination of cell shape and division, and one-tenth related tocell energetics. Only 4% of essential genes encode unknownfunctions. Most essential genes are present throughout a widerange of Bacteria, and almost 70% can also be found in Archaeaand Eucarya. However, essential genes related to cell envelope,shape, division, and respiration tend to be lost from bacteria withsmall genomes. Unexpectedly, most genes involved in the Emb-den–Meyerhof–Parnas pathway are essential. Identification of un-known and unexpected essential genes opens research avenues tobetter understanding of processes that sustain bacterial life.
The definition of the minimal gene set required to sustain aliving cell is of considerable interest. The functions specified
by such a set are likely to provide a view of a ‘‘minimal’’ bacterialcell. Many functions should be essential in all cells and could beconsidered as a foundation of life itself. The determination of therange of essential functions in different cells should revealpossible solutions for sustaining life. Computational and exper-imental research has previously been carried out to define aminimal protein-encoding gene set. An upper-limit estimate ofa minimal bacterial gene set was obtained from the sequence ofthe entire Mycoplasma genitalium genome, which contains only!480 genes (1). A computational approach, based on theassumption that essential genes are conserved in the genomes of
M. genitalium and Haemophilus influenzae, led to a description ofa smaller set of some 260 genes (2). More recently, an experi-mental approach involving high-density transposon mutagenesisof the H. influenzae genome led to a much higher estimate of!670 putative essential genes (3), whereas transposon mutagen-esis of two mycoplasma species led to an estimate of 265–360essential genes (4). Another experimental approach using anti-sense RNA to inhibit gene expression led to the identification ofsome 150 essential genes in Staphylococcus aureus (5). However,these approaches have limitations. Computation is likely tounderestimate the minimal gene set because it takes into accountonly those genes that have remained similar enough during thecourse of evolution to be recognized as true orthologues.Transposon mutagenesis might overestimate the set by misclas-sification of nonessential genes that slow down the growthwithout arresting it but can also miss essential genes that toleratetransposon insertions (3, 6). Finally, the use of antisense RNAis limited to the genes for which an adequate expression of theinhibitory RNA can be obtained in the organism under study.
To obtain an independent and possibly more reliable estimateof a minimal protein-encoding gene set for bacteria, we system-atically inactivated Bacillus subtilis genes. B. subtilis was chosenbecause it is one of the best studied bacteria (7) and is a modelfor low-G"C Gram-positive bacteria, which include both deadlypathogens, such as Bacillus anthracis, and bacteria widely used infood and industry, such as lactococci and bacilli. Because theessentiality of a gene depends on the conditions under which theorganism is propagated, we used an environment likely to beoptimal for B. subtilis and thus carried out inactivation on a
cTo whom correspondence should be addressed. E-mail: [email protected] [email protected].
4678–4683 ! PNAS ! April 15, 2003 ! vol. 100 ! no. 8 www.pnas.org"cgi"doi"10.1073"pnas.0730515100
unknown role. The last two genes, pdhA and odh, encodesubunits of pyruvate and 2-oxoglutarate dehydrogenase, respec-tively; growth of the mutants could be restored by addition to LBof the metabolites (acetate and succinate, respectively) relatedto the activity of the proteins they encode.Unknown. The last category groups 11 essential genes for which wewere unable to suggest a role in cell physiology. Biochemicalfunctions, a protease and a hydrolase of the metallo-!-lactamasesuperfamily, can be suggested for products of two gene, ydiC andykqC. One gene, yneS, encodes a putative membrane protein,and another, ymdA, encodes a protein with an HD domain ofmetal-dependent phosphohydrolases, whereas three, yloQ, yqjK,and ywlC, encode proteins with recognizable signatures, anATP!GTP-binding site, a metallo-!-lactamase motif, and aputative RNA-binding motif, respectively. Four genes, yacA,ydiB, ylaN, and yqeI, have no easily recognizable features.
Conservation of Essential Genes. The average level at which ho-mologues of essential B. subtilis genes are present in bacteria israther high (approaching 80%), one-fourth being found in allbacteria and three-fourths in at least 75% (Fig. 1 Upper). Theaverage is !36% in Eucarya and Archaea, but some 20% of thegenes are nevertheless present in all 18 organisms we analyzed(Fig. 1 Upper). About one-third of the genes are found in all threekingdoms of life, and a further one-third are shared betweenBacteria and either Archaea or Eucarya (Fig. 1 Lower).
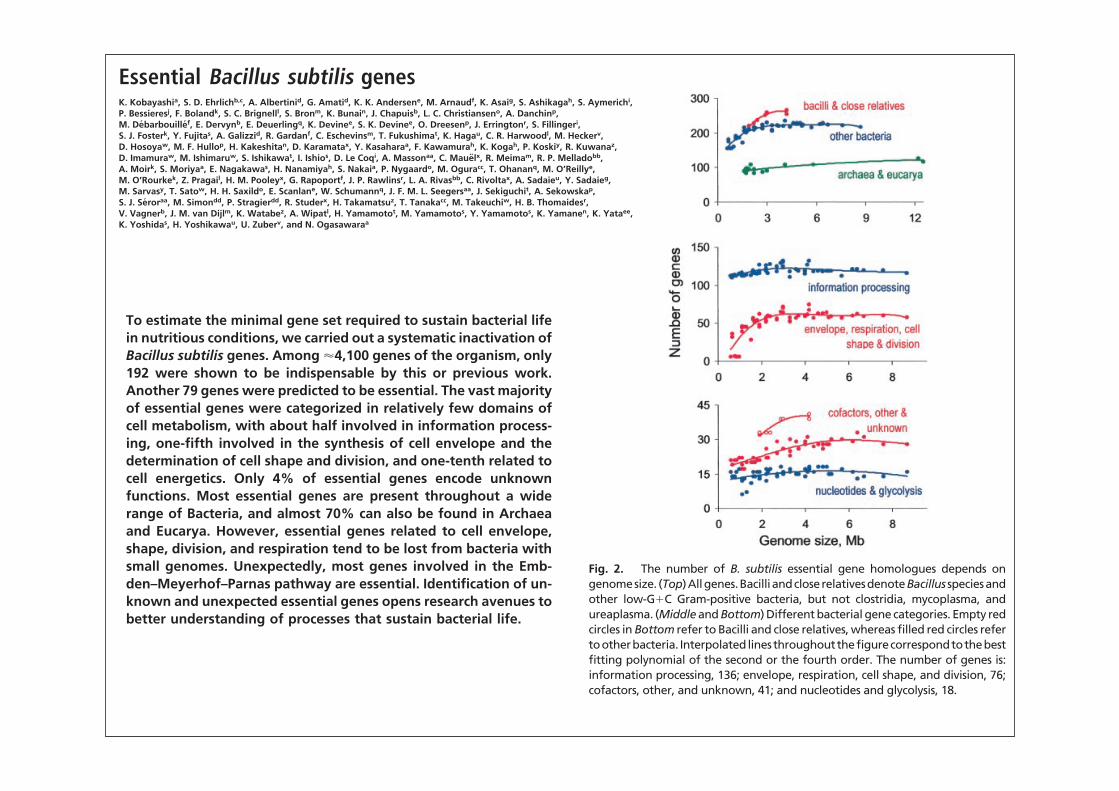
The number of B. subtilis essential gene homologues presentin an organism depends on at least two parameters: phylogeneticproximity to B. subtilis and genome size (Fig. 2 Top). The highestnumber is found in bacilli and close relatives, having genomes of
"3 Mb (highlighted in red). Other bacteria with genomes of asimilar size have, on average, slightly "80% of the B. subtilisessential gene homologues. This proportion drops to 57% withdecreasing bacterial genome size, indicating progressive loss ofessential genes. Archaea and Eucarya maintain, on average, 36%of the essential gene homologues, with the proportion varyingbetween 33% and 44% almost linearly with genome size. Inbacteria, gene loss occurs mainly in three categories (cell enve-lope, shape and division, and respiratory pathways) and to alower extent in three other categories (cofactor synthesis, otherprocesses, and unknown functions). In contrast, informationprocessing, glycolysis, and nucleotide synthesis genes are largelyretained (Fig. 2 Middle and Bottom).
Phylogenetic profiling of essential B. subtilis genes is summarizedin Fig. 3. Organisms were grouped into four classes and orderedwithin each class on the basis of the number of essential genehomologues they share with B. subtilis, placing the organisms withfewest conserved genes at the right of each class. Genes weregrouped in categories and ordered by abundance among all bac-teria, which placed the less abundant genes at the bottom of eachcategory. A number of general features are easily discernible fromthis analysis. (i) The five top categories are composed of genespresent in "80% of Bacteria and at least 40% of Eucarya andArchaea, with the exception of RNA synthesis, which is less wellconserved in the last two kingdoms. (ii) The next two categories,DNA metabolism and cell shape and division, contain genes
Fig. 2. The number of B. subtilis essential gene homologues depends ongenomesize. (Top)Allgenes.Bacilli andcloserelativesdenoteBacillus speciesandother low-G#C Gram-positive bacteria, but not clostridia, mycoplasma, andureaplasma. (Middle and Bottom) Different bacterial gene categories. Empty redcircles in Bottom refer to Bacilli and close relatives, whereas filled red circles refertootherbacteria. Interpolated linesthroughoutthefigurecorrespondtothebestfitting polynomial of the second or the fourth order. The number of genes is:information processing, 136; envelope, respiration, cell shape, and division, 76;cofactors, other, and unknown, 41; and nucleotides and glycolysis, 18.
Fig. 3. Phylogenetic profiling of essential genes. The 271 B. subtilis geneswere grouped in 266 clusters. Only one gene, yhdL, which encodes a possibleanti-sigma protein, had no orthologues in the database and is not presentedhere. Each line and column corresponds to individual gene and organism,respectively. Presence and absence of a gene is indicated by a black and whitesquare, respectively. The list of genes and organisms is given in Table 5, whichis published as supporting information on the PNAS web site and the orderingis described in the text.
4682 " www.pnas.org!cgi!doi!10.1073!pnas.0730515100 Kobayashi et al.

Synthetische Biologie: Überblick
1. Allgemeine Gesichtspunkte
1.1 Netzwerkmotive in biochemischen Schaltkreisen
1.2 Reduzierte und minimale Genome
1.3 Was benötigt man für eine künstliche Zelle ?
1.4 Selbstreplikatoren
REVIEW
Towards synthesis of a minimal cell
Anthony C Forster1,* and George M Church2,*
1 Department of Pharmacology and Vanderbilt Institute of Chemical Biology,Vanderbilt University Medical Center, Nashville, TN, USA and
2 Department of Genetics, Harvard Medical School, Boston, MA, USA* Corresponding authors. AC Forster, Department of Pharmacology, VanderbiltUniversity Medical Center, 23rd Ave.S. at Pierce, Nashville, TN 37232, USA.Tel.: ! 1 615 936 3112; Fax: ! 1 615 936 5555;E-mail: [email protected] or GM Church, Department of Genetics,Harvard Medical School, 77 Avenue Louis Pasteur, NBR 238, Boston,MA 02115, USA. Tel.: ! 1 617 432 1278; Fax: ! 1 617 432 6513
Received 7.5.06; accepted 26.7.06
Construction of a chemical system capable of replicationand evolution, fed only by small molecule nutrients, is nowconceivable. This could be achieved by stepwise integrationof decades of work on the reconstitution of DNA, RNA andprotein syntheses from pure components. Such a minimalcell project would initially define the components sufficientfor each subsystem, allow detailed kinetic analyses andlead to improved in vitro methods for synthesis ofbiopolymers, therapeutics and biosensors. Completionwould yield a functionally and structurally understoodself-replicating biosystem. Safety concerns for synthetic lifewill be alleviated by extreme dependence on elaboratelaboratory reagents and conditions for viability. Ourproposed minimal genome is 113kbp long and contains151 genes. We detail building blocks already in place andmajor hurdles to overcome for completion.Molecular Systems Biology 22 August 2006;doi:10.1038/msb4100090Subject Categories: synthetic biologyKeywords: cell; genome;minimal; RNA; self-replication; translation
Overview
‘How far can we push chemical self-assembly?’
This questionwas posed recently as one of the big 25 questionsin science for the next 25 years (Service, 2005). Nowadays, bigquestions often are addressed by big experimental efforts. Butbefore embarking on a big project, it is helpful to get specific.What push in chemical self-assembly might be most worth-while and practical? Self-assembly in vitro of viruses andthe ribosome, achieved decades ago, taught us some of theprinciples assumed to be used in general by cells (Lewin,2004). For example, self-assembly occurs in a definitesequence and is generally energetically favored, obviatingthe need for enzymes and an energy source. Assembling sometype of cell (i.e. a self-replicating, membrane-encapsulatedcollection of biomolecules) would seem to be the next major
step, yet detailed plans have not been published. Here, weattempt to outline the synthesis of a minimal cell containingthe core cellular replication machinery, review the pertinentliterature and highlight gaps in knowledge that need filling.
Utility
Synthesizing a minimal cell will advance knowledge ofbiological replication. Many hypotheses in replication and itssubsystems can only be tested in such a synthetic biologyproject. The meaning of ‘synthetic’ (from Greek sunthesis, toput together) discussed here bypasses the current reliance ofsynthetic biology on cells or macromolecular cell products: theaim is to put together an organism from small molecules alone.The simplest approach for creating an artificial cell may beby evolving an RNA polymerase made exclusively of RNA(Szostak et al, 2001) to replace all protein components ofin vitro replicating and evolving systems (e.g. to replace Qbreplicase; Mills et al, 1967). But in comparison with a purifiedprotein-based system, it is neither guaranteed to arrive soonernor tell usmore. A protein-based systemwill connect with, andreveal more about, existing biological systems. Life, like amachine, cannot be understood simply by studying it and itsparts; it must also be put together from its parts. Along the wayto synthesizing a cell, we might discover new biochemicalfunctions essential for replication, unsuspected macromole-cular modifications or previously unrecognized patterns ofcoordinated expression.How good a model would an artificial, protein-based,
minimal cell be for natural cells? The only cellular alternativeis a perturbed natural cell, an incredibly complex system evenfor the simplest of cells. A much simpler purified system basedon a real cell would thus be easier to model and understand.It could certainly answer questions that cannot be answeredin vivo or in crude extracts, such aswhichmacromolecules andmacromolecular modifications are sufficient for subsystemfunction. However, even the simplest minimal cell would stillbe highly complex; so its construction and study would befacilitated by substituting some of the necessary subsystemswith simpler analogs. Should the simpler in vitro model turnout to be a poor model for the more complex in vivo system,one could always construct a more complex in vitro systemthat may better reflect in vivo.Synthesizing a cell will also lead to new applications.
Purified biochemical systems already offer major advantages,such as the polymerase chain reaction (PCR) and in vitrotranscription. A better understanding and manipulation of allcellular replication subsystems (molecular biology’s tool kit)should spin off new technologies. For example, in vitrogenome replication may be useful for replicating very largesegments of DNA with high fidelity. Combined in vitrotranscription, RNA processing and RNA modification wouldallow preparation of rRNAs and tRNAs with defined modifica-tions to test the roles of the modifications, andmodified tRNAs
& 2006 EMBO and Nature Publishing Group Molecular Systems Biology 2006 msb4100090-E1
Molecular Systems Biology (2006) doi:10.1038/msb4100090& 2006 EMBO and Nature Publishing Group All rights reserved 1744-4292/06www.molecularsystemsbiology.com000045
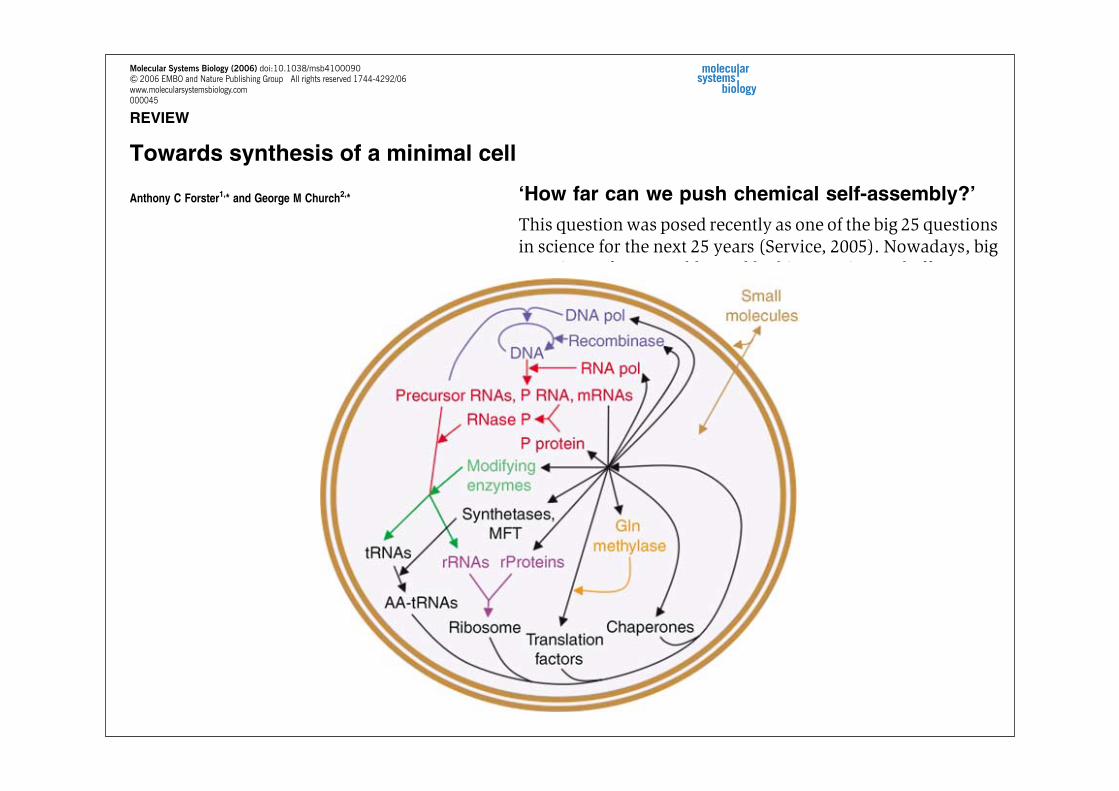
fission autocatalytically (Szostak et al, 2001), so membraneproteins may be dispensable. Polysaccharides should also bedispensable. If the simplest and best-characterized examplesof DNA, RNA and protein synthesis are selected, if translationof all codons is enabled for generalizability and if efficiencyand accuracy are not compromised, then this leads to themacromolecules and pathways of Figure 1.A detailed list of the gene products in the hypothetical
synthetic minimal cell of Figure 1 is shown in Table I (leftcolumn). This list overlaps with a computational model ofminimal cell genes largely derived from a minimal organism,M. genitalium (Tomita et al, 1999; Supplementary Table S4),but differs by omitting enzymes for synthesizing smallmolecules (e.g. lipids and glycolysis substrates) and byincluding DNA replication, RNA processing, RNA modifica-tion, extra tRNAs to decode the whole genetic code, someadditional essential translation components and chaperones.It should be emphasized that Table I is a working model onlyand that strict adherence will likely hamper progress.Examples of omitted, potentially stimulatory genes are givenbelow and in Supplementary Table S1. Conversely, examplesof included, potentially dispensable genes may be gleaned bycomparison with the streamlinedMycoplasma genome (Fraseret al, 1995; Table I, middle column; Supplementary Table S2).Several conclusions can be drawn from the provisional list
of genes selected for a minimal cell, most of which areattractive when contemplating an MCP. In genomic terms, the
list is very short, containing only 151 genes and 113 kbp. Allof the genes are derived from E. coli and its bacteriophages(except for the hammerhead RNA from a plant virus; Forsterand Symons, 1987), implying that the individual subsystemswill be compatible. In contrast to lists derived by comparativegenomics or genetic approaches, the biochemically based listdoes not contain any genes of unknown function or challen-ging membrane proteins; so it is close to a fully understood,accurately replicating ‘platform’ for life. The few known gapsconstitute only about seven genes, all of which are predictedto be for RNA modification (Table I, bold in the left column).From the viewpoint of structural biology, courtesy of recentbreakthroughs in ribosome structure determination (Diaconuet al, 2005; Ogle and Ramakrishnan, 2005), significant three-dimensional information is lacking for only 3% of theproducts: a few RNA modification proteins and aminoacyl-tRNA synthetases (Table I, right column). While some of thestates and complexes remain to be solved at high resolution,a draft three-dimensional structure for any replicating systemis a major milestone in the history of biology.
Tools
Genes for an MCP could be synthesized using either natural orunnatural gene sequences as starting points. Using naturalgene sequences, genes can be readily synthesized by PCR, andlarge cloned operons of essential genes can be fused usingsynthetic linkers and homologous recombination. However,gene synthesis by cloning and PCR will soon be moreexpensive than raw synthesis from synthetic oligo-deoxyribonucleotides (oligos). The latter also allows unnatur-al sequences, such as versions with altered codon bias toadjust mRNA secondary structures (Tian et al, 2004).Scalability and cost limitations of established methods forgene synthesis from synthetic oligos are now being overcomeby oligo synthesis on chips followed by PCR amplificationand error correction (Carr et al, 2004; Richmond et al, 2004;Tian et al, 2004; Zhou et al, 2004).
Biochemical subsystems
Several biochemical subsystems are required to synthesize aminimal cell, and they are reviewed here. For each subsystem,possible examples from natural systems will be compared,gaps in knowledge will be identified and diagnostic anddebugging strategies to fill the gaps will be suggested. Mindfulof the goal of integration of the subsystems, emphasis is placedon subsystems that are homologous and that operate understandard physiological conditions.
Genome replication
In principle, the genetic material for an MCP could be eitherDNA or RNA. Although an RNA genome has the advantageof obviating genes for DNA replication, the challenges ofpreventing inhibitory double-stranded RNA structures andreplicative mutations in artificial RNA genomes (Mills et al,1967) are unsolved. So the genetic material for an MCP shouldbe DNA.
Figure 1 A minimal cell containing biological macromolecules and pathwaysproposed to be necessary and sufficient for replication from small moleculenutrients. The macromolecules are all nucleic acid and protein polymers and areencapsulated within a bilayer lipid vesicle. The small molecules (brown) diffuseacross the bilayer. The macromolecules are ordered according to the pathways inwhich they are synthesized and act. They are colored by biochemical subsystemas follows: blue!DNA synthesis, red!RNA synthesis and cleavage, green-RNA modification, purple!ribosome assembly, orange!post-translationalmodification and black!protein synthesis. MFT!methionyl-tRNAfMeti formyl-transferase. The system could be bootstrapped with DNA, RNA polymerase,ribosome, translation factors, tRNAs, MTF, synthetases, chaperones and smallmolecules.
Towards synthesis of a minimal cellAC Forster and GM Church
& 2006 EMBO and Nature Publishing Group Molecular Systems Biology 2006 msb4100090-E3
REVIEW
Towards synthesis of a minimal cell
Anthony C Forster1,* and George M Church2,*
1 Department of Pharmacology and Vanderbilt Institute of Chemical Biology,Vanderbilt University Medical Center, Nashville, TN, USA and
2 Department of Genetics, Harvard Medical School, Boston, MA, USA* Corresponding authors. AC Forster, Department of Pharmacology, VanderbiltUniversity Medical Center, 23rd Ave.S. at Pierce, Nashville, TN 37232, USA.Tel.: ! 1 615 936 3112; Fax: ! 1 615 936 5555;E-mail: [email protected] or GM Church, Department of Genetics,Harvard Medical School, 77 Avenue Louis Pasteur, NBR 238, Boston,MA 02115, USA. Tel.: ! 1 617 432 1278; Fax: ! 1 617 432 6513
Received 7.5.06; accepted 26.7.06
Construction of a chemical system capable of replicationand evolution, fed only by small molecule nutrients, is nowconceivable. This could be achieved by stepwise integrationof decades of work on the reconstitution of DNA, RNA andprotein syntheses from pure components. Such a minimalcell project would initially define the components sufficientfor each subsystem, allow detailed kinetic analyses andlead to improved in vitro methods for synthesis ofbiopolymers, therapeutics and biosensors. Completionwould yield a functionally and structurally understoodself-replicating biosystem. Safety concerns for synthetic lifewill be alleviated by extreme dependence on elaboratelaboratory reagents and conditions for viability. Ourproposed minimal genome is 113kbp long and contains151 genes. We detail building blocks already in place andmajor hurdles to overcome for completion.Molecular Systems Biology 22 August 2006;doi:10.1038/msb4100090Subject Categories: synthetic biologyKeywords: cell; genome;minimal; RNA; self-replication; translation
Overview
‘How far can we push chemical self-assembly?’
This questionwas posed recently as one of the big 25 questionsin science for the next 25 years (Service, 2005). Nowadays, bigquestions often are addressed by big experimental efforts. Butbefore embarking on a big project, it is helpful to get specific.What push in chemical self-assembly might be most worth-while and practical? Self-assembly in vitro of viruses andthe ribosome, achieved decades ago, taught us some of theprinciples assumed to be used in general by cells (Lewin,2004). For example, self-assembly occurs in a definitesequence and is generally energetically favored, obviatingthe need for enzymes and an energy source. Assembling sometype of cell (i.e. a self-replicating, membrane-encapsulatedcollection of biomolecules) would seem to be the next major
step, yet detailed plans have not been published. Here, weattempt to outline the synthesis of a minimal cell containingthe core cellular replication machinery, review the pertinentliterature and highlight gaps in knowledge that need filling.
Utility
Synthesizing a minimal cell will advance knowledge ofbiological replication. Many hypotheses in replication and itssubsystems can only be tested in such a synthetic biologyproject. The meaning of ‘synthetic’ (from Greek sunthesis, toput together) discussed here bypasses the current reliance ofsynthetic biology on cells or macromolecular cell products: theaim is to put together an organism from small molecules alone.The simplest approach for creating an artificial cell may beby evolving an RNA polymerase made exclusively of RNA(Szostak et al, 2001) to replace all protein components ofin vitro replicating and evolving systems (e.g. to replace Qbreplicase; Mills et al, 1967). But in comparison with a purifiedprotein-based system, it is neither guaranteed to arrive soonernor tell usmore. A protein-based systemwill connect with, andreveal more about, existing biological systems. Life, like amachine, cannot be understood simply by studying it and itsparts; it must also be put together from its parts. Along the wayto synthesizing a cell, we might discover new biochemicalfunctions essential for replication, unsuspected macromole-cular modifications or previously unrecognized patterns ofcoordinated expression.How good a model would an artificial, protein-based,
minimal cell be for natural cells? The only cellular alternativeis a perturbed natural cell, an incredibly complex system evenfor the simplest of cells. A much simpler purified system basedon a real cell would thus be easier to model and understand.It could certainly answer questions that cannot be answeredin vivo or in crude extracts, such aswhichmacromolecules andmacromolecular modifications are sufficient for subsystemfunction. However, even the simplest minimal cell would stillbe highly complex; so its construction and study would befacilitated by substituting some of the necessary subsystemswith simpler analogs. Should the simpler in vitro model turnout to be a poor model for the more complex in vivo system,one could always construct a more complex in vitro systemthat may better reflect in vivo.Synthesizing a cell will also lead to new applications.
Purified biochemical systems already offer major advantages,such as the polymerase chain reaction (PCR) and in vitrotranscription. A better understanding and manipulation of allcellular replication subsystems (molecular biology’s tool kit)should spin off new technologies. For example, in vitrogenome replication may be useful for replicating very largesegments of DNA with high fidelity. Combined in vitrotranscription, RNA processing and RNA modification wouldallow preparation of rRNAs and tRNAs with defined modifica-tions to test the roles of the modifications, andmodified tRNAs
& 2006 EMBO and Nature Publishing Group Molecular Systems Biology 2006 msb4100090-E1
Molecular Systems Biology (2006) doi:10.1038/msb4100090& 2006 EMBO and Nature Publishing Group All rights reserved 1744-4292/06www.molecularsystemsbiology.com000045

Synthetische Biologie: Überblick
1. Allgemeine Gesichtspunkte
1.1 Netzwerkmotive in biochemischen Schaltkreisen
1.2 Reduzierte und minimale Genome
1.3 Was benötigt man für eine künstliche Zelle ?
1.4 Selbstreplikatoren
!"#$%&'!"#$ !""#( )*+ ,- . /&0#123!"2345678 9:;"( <2=>?@, /4ABC4A:( -DD, ,?E>2?--FGD,GD-G,- H ,F+@DI+@DGD !"#
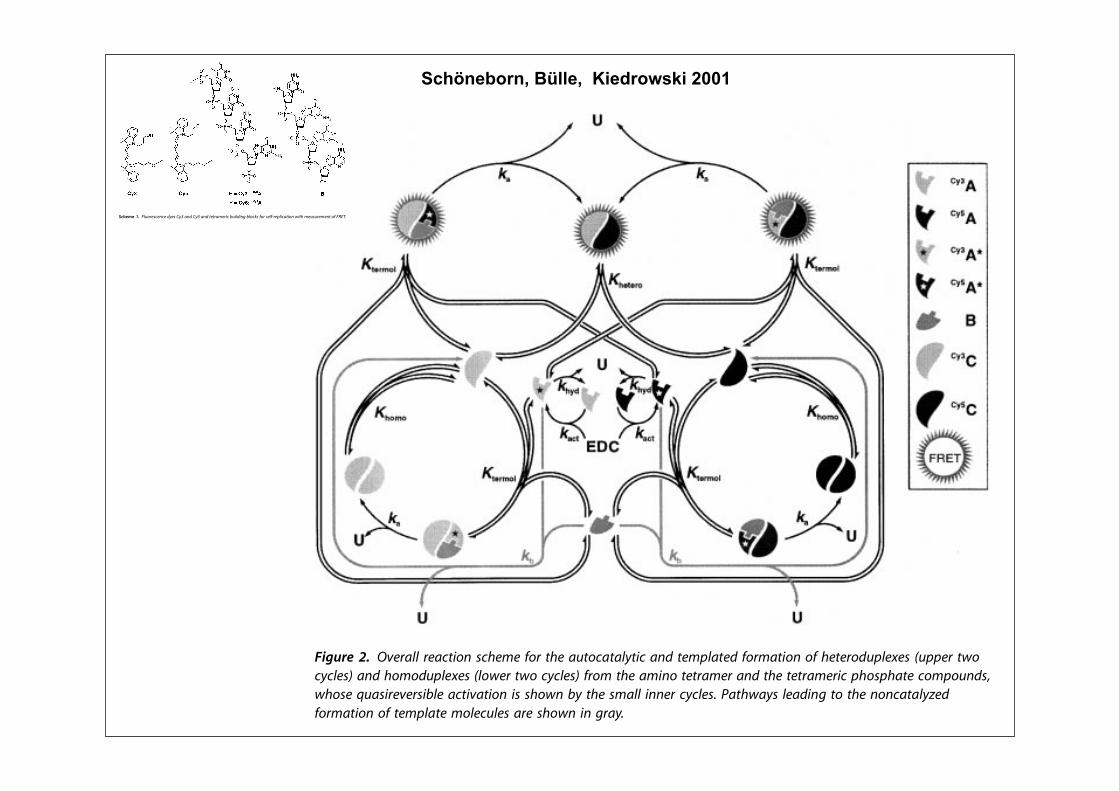
!"#$% $&' $()"* '+ ,-./ "#)"012"3$ &"0" 45001"6 '7$8 93 $:"+10*$ $()"% ;'$: <(=$ 536 <(>$ &"0" 5??'&"6 $' 0"54$ &1$: $"$052"0% 13 $:" 5;*"34" '+ $"2)?5$" @,1A70" BC8 /:" +'025$1'3 '+ $:"
:"$"0'67)?"# &5* 2'31$'0"6 'D"0BE :'70* 536 0"*7?$"6 610"4$?( 13 5*1A2'165? A0'&$: 470D" &:'*" 131$15?6"D"?')2"3$ +'??'&* 5 )505;'?5@,1A70" EC8 /:" 4'34"3$05$1'3 '+ '4F$52"0* *(3$:"*1G"6 &5* 6"$"0213"6610"4$?( +0'2 $:" 6"40"5*" '+ ,-./6'3'0 5;*'0;534" 5+$"0 45?1;05$1'3&1$: 5 *52)?" 13 &:14: $:" '4$52"04'34"3$05$1'3 50'*" +0'2 HIIJ 4'3FD"0*1'3 '+ $:" $"$052"0*8 /:" *"4'36$()" '+ "#)"012"3$ 13D'?D"6 K13"$142'31$'013A '+ $:" +'025$1'3 '+ <(>&@+0'2 L7*$ <(>$ 536 %C 13 $:" )0"*"34"'+ D501'7* 131$15? 4'34"3$05$1'3* '+M"#$"035?N $"2)?5$" <(=&!8 ,1A70"* >5536 ; 6")14$ $:" $12" 4'70*"* +'0+'025$1'3 '+ $:" :"$"0'67)?"# 536$:" 45?47?5$"6 $'$5? 4'34"3$05$1'3 '+3"&?( +'02"6 <(>&% 0"*)"4$1D"?(8 /:"3'3?13"50 470D"F+1$$13A '+ ,1A70" >&5* 5A513 )"0+'02"6 ;( 2"53* '+'70 O12,1$ )0'A052% 536 $:" 0"54$1'32'6"? *:'&3 13 .P75$1'3* @EC Q @HHC&5* $5K"3 13$' 544'73$8/:" 05$" 4'3*$53$* J54$ 536 J:(6 13
.P75$1'3* @EC 536 @>C &"0" +1#"6 $'$:" D5?7"* 6"$"0213"6 ;( $:" HR !S-
*)"4$0'*4')( K13"$14 *$76(8 .P75$1'3* @TC 536 @UC 4'2)01*" $:"0"D"0*1;?" +'025$1'3 '+ $"02'?"47?50 4'2)?"#"* 536 $:" 100"D"0F*1;?" 4:"2145? ?1A5$1'3 "D"3$* &1$:13 $:" ?5$$"0 4'2)?"#"*8 V*?'3A 5* 0"?15;?" $:"02'6(35214 65$5 +'0 $"02'?"47?50 4'2)?"#"*
$%&'() !* 'K45766 547LMA*B NLC4:4 O*5 MC4 7PM*L7M76QMAL 7BR M4:S67M4R O*5:7MA*B *O C4M45*RPS64T4N UPSS45 MV*LQL64NW 7BR C*:*RPS64T4N U6*V45 MV* LQL64NW O5*: MC4 7:AB* M4M57:45 7BR MC4 M4M57:45AL SC*NSC7M4 L*:S*PBRN(VC*N4 XP7NA54K45NA;64 7LMAK7MA*B AN NC*VB ;Q MC4 N:766 ABB45 LQL64N+ Y7MCV7QN 647RAB8 M* MC4 B*BL7M76QZ4RO*5:7MA*B *O M4:S67M4 :*64LP64N 754 NC*VB AB 857Q+
$%&'() +* [A:4 L*P5N4 O*5 MC4 7PM*L7M76QMAL O*5:7MA*B *O C4M45*RPS64T4N O5*: 7:ATMP54 *O M4M57:45AL ;PA6RAB8 ;6*LJN !QE,( !Q@,( 7BR -( .,\,\-( 54NS4LMAK46QW AB MC4S54N4BL4 *O #<!+ [C4 S*ABMN V454 R45AK4R O5*: O6P*54NL4BL4 :47NP54:4BMN 7BR7SS5*TA:7M4R ;Q 7 MC4*54MAL76 LP5K4 ;7N4R *B MC4 547LMA*B :*R46 NC*VB AB]A8P54 - 7BR MC4 57M4 S757:4M45N 4NMA:7M4R ;Q MC4 ^A:]AM S5*857: O5*: R7M7NC*VB AB ]A8P54 E+ [C4 PSS45 LP5K4 54S54N4BMN MC4 M*M76 L*BL4BM57MA*BN *O 766*LM7:45N O*5:4R+ !*BRAMA*BN\ ?D !$ !QE,( ?D !$ !Q@,( _D !$ -( D+-$ #<!( D+,$"#Y#^` S" F+E@( /! ,@ "!+
$%&'() /* [A:4 L*P5N4 *O MC4 O*5:7MA*B *O P547 0 U^LC4:4 -W RP5AB8 MC4 547LMA*B*O M4M57:45AL SC*NSC7M4 %PS[!!9S U_EE !$ N*6PMA*BW AB MC4 S54N4BL4 *O #<!( MC4L*BL4BM57MA*BN *O VCALC 754 8AK4B AB MC4 OA8P54+ [C4 4TS45A:4BM76 L*BL4BM57MA*BNUS*ABMNW V454 R4M45:AB4R O5*: MC4 V4662N4S757M4R ," )$a NA8B76 *O MC4 4MCQ6 !"E85*PS+ [C4 547LMA*BN V454 L755A4R *PM 7M ,@ "! AB 7 ;POO454R N*6PMA*B L*BM7ABAB8D+,$ -2b?2U-2CQR5*TQ4MCQ6W2,2SAS457ZABQ6c4MC7B4NP6O*BAL 7LAR U"#Y#^W AB "-'G<-'G!<E!) UF-\_\-DW 7M S"! F+E@+ [C4 MC4*54MAL76 MA:4 L*P5N4N ULP5K4NW V454 R45AK4RO5*: ^A:]AM L76LP67MA*BN ;7N4R *B MC4 547LMA*B :*R46 8AK4B AB MC4 M4TM+ [C4O*66*VAB8 S757:4M45N V454 R4M45:AB4R O5*: MC4N4 4TS45A:4BMN\ K7LM!E+@@U"D+D-W# ,D$E( KCQR! >+--U"D+EDW# ,D$?( 7BR KSS! E+DEU"D+,,W#,D$- $$, N$,+
!"#$%&'!"#$ !""#( )*+ ,- . /&0#123!"2345678 9:;"( <2=>?@, /4ABC4A:( -DD, ,?E>2?--FGD,GD-G,- H ,F+@DI+@DGD !"#
!"#$% &'()#* #*+# )+, -(# ./0(1-.-#$+!2 34# 0+'+3(!$%5 6+'+3(!$%&'()#* 7"-+8$%, +!,( 4-7.'!$.8('. %(80!./ ,",#.8, )*.'.0'(74%# $-*$3$#$(- $, +- $,,4.2 ,4%*+, %'(,,1%+#+!"#$% ,",#.8, (' '.+%1#$(- -.#)('9, )$#* + 84!#$#47. (:+4#(%+#+!"#$% (' %'(,,1%+#+!"#$%:..73+%9 !((0,5;<= >().?.'2 ./0(1-.-#$+! &'()#* *+, 3..- 7.1,%'$3.7 +, #*. 0'.'.@4$,$#. :('0(,,$3!. ./0.'$8.-#+! +00'(+%*.,+$8$-& #( '.+!$A. 8(!.%4!+' .?(!41#$(- 3+,.7 (- + B+')$-$+- C,4'1?$?+! (: #*. :$##.,#D 8(7.!5;E= F.1%.-#!"2 ./0(-.-#$+! &'()#* )+,'.+!$A.7 $- #*. !+3('+#('" 3"8.+-, (: + ,#.0)$,. '.0!$%+#$(-0'(%.74'. G%+!!.7 H6FIJB;K= L #*+#$-?(!?., #*. %(?+!.-# +##+%*8.-#(: #.80!+#., #( ,4':+%.,5 M(,#'.%.-#!"2 #*.('.#$%+! .?$7.-%. )+,:(4-7 #*+# #*. -(-,#.0)$,. G+41#(-(8(4,L %(40!$-& (: %*'(8+#(&'+0*" +-7 +4#(%+#+!"#$% ,"-1#*.,$, 8+" !.+7 #( B+')$-$+- ,.!.%#$(- +-72 8('. &.-.'+!!",0.+9$-&2 #( #*. .?(!4#$(- (: .?(!?+3$!$#" (: + %*.8$%+! ,",#.82.?.- $- #*. 0'.,.-%. (: 0'(74%# $-*$3$#$(-5;NO= J!#.'-+#$?.,#'+#.&$., #( .,#+3!$,* #*. .?(!?+3$!$#" 8+" 3. 3+,.7 (- #'+?.!$-&)+?.,2;NN= ,#'4%#4'+!!" 4-,#+3!. #.80!+#.,2;NP= (' 8$-$8+! '.0!$1%+,.,5;NQ= R*$%*.?.' ,#'+#.&" #( $80!.8.-# #*$, %*+!!.-&$-& &(+!)$-, $- #*. :4#4'.2 $# %+- 3. :('.,..- #*+# '+0$7 #.%*-$@4., :('8(-$#('$-& #*. %*.8$%+! 9$-.#$%, $- + 0+'+!!.! :+,*$(- +'. + 8+S('0'.'.@4$,$#.5 J, ,4%*2 *$&*10'.,,4'. !$@4$7 %*'(8+#(&'+0*"G>6TUL +-7 VMF ,0.%#'(,%(0" 9$-.#$%,2 3" )*$%* 8(,# (: #*.,",#.8, ,( :+' 7.,%'$3.7 *+?. 3..- ,#47$.72 +'. $- + ,.-,. #((!+3('$(4,5 J8(-& ,.?.'+! %(-%.$?+3!. #.%*-$@4.,2 #*(,. 3+,.7(- :!4('.,%.-%.2 :!4('.,%.-%. @4.-%*$-&2 +-7 :!4('.,%.-%.'.,(-+-%. .-.'&" #'+-,:.' GWFIXL;NY= +'. ,40.'$(' 74. #( #*.$'*$&* ,.-,$#$?$#"5 W(' ./+80!.2 :!4('.,%.-%. @4.-%*$-& 4-7.'!$.,#*. #.%*-$@4. (: 8(!.%4!+' 3.+%(-, #*+# *+, ,$&-$:$%+-#!",$80!$:$.7 #*. @4+-#$:$%+#$(- (: 0'(74%# +80!$:$%+#$(- 74'$-&#*. 0(!"8.'+,. %*+$- '.+%#$(-5;NZ= X*. 4#$!$A+#$(- (: :!4('.,%.-%[email protected]%*$-& +-7 WFIX *+, +!,( 3..- '.0('#.7 $- 9$-.#$% ,#47$.,;N[=
(: (#*.' 3$(('&+-$% 8(7.! ,",#.8,2 +8(-& )*$%* '$3(A"8.'.+%#$(-,;N<= +-7 %(83$-+#('$+! +00'(+%*.,;NE= +'. $-%!47.75
R. *.'. '.0('# (- #*. (-!$-. 8(-$#('$-& (: %*.8$%+!'.0!$%+#$(- (: ,.!:1%(80!.8.-#+'" (!$&(-4%!.(#$7., 3" 8.+-,(: WFIX5 H%*.8., N +-7 P ,*() #*. 34$!7$-& 3!(%9, +-7 #*. 3+,$%'.+%#$(-, $- (4' ,#47"5 X*. #)( :!4('.,%.-%. 7".,2 7(-(' U"Q +-7+%%.0#(' U"Z2 ).'. $-#'(74%.7 +, Z! !+3.!, #( &$?. #*. #.#'+8.',U"Q$ +-7 U"Z$ 3" ,#+-7+'7 ,(!$710*+,. ,"-#*.,$,5 \- #*. 0'.,.-%.(: #*. )+#.'1,(!43!. )!1GQ17$8.#*"!+8$-(0'(0"!L1)1.#*"!%+'3(17$$8$7. GIBUL2 #*. Q!10*(,0*+#. &'(40, (: U"Q$ +-7 U"Z$ +'.+%#$?+#.7 +, #*. '.,0.%#$?. $,(4'.+ +774%#, U"Q$% +-7 U"Z$% +-7#*.- '.+%# )$#* #*. Z!1+8$-( &'(40 (: #.#'+8.' & #( "$.!7 #*.%(''.,0(-7$-& (%#+8.', U"Q' +-7 U"Z'2 )*$%* 3.+' + %.-#'+! Q!2Z!10*(,0*('+8$7+#. !$-9+&. GH%*.8. PL5 J77$#$(-+!!"2 #*. (%#+8.'
U"Z'!2 )*$%* *+, + %.-#'+! Q!2Z!10*(,0*(7$.,#.' !$-9+&. )+,,"-#*.,$A.7 +, +- C./#.'-+!D #.80!+#.2 #( 8$8$% #*. .::.%# (: #*.$-#.'-+!!" ,"-#*.,$A.7 #.80!+#. U"Z'5
X*. '+#$(-+!. 3.*$-7 #*. ,"-#*.,$, (: #)( 7$::.'.-#!" !+3.!.7#.#'+8.', )$#* #*. ,+8. ,[email protected]%. $, 4-7.',#((7 $- #*. %(-#./#(: #*.$' '.+%#$(-,5 \: 3(#* U"Q$ +-7 U"Z$ +'. +!!().7 #( '.+%# )$#*#.#'+8.' &2 (-. ./0.%#, #*. ,$84!#+-.(4, :('8+#$(- (: #*.(%#+8.', U"Q' +-7 U"Z'5 ](#* (%#+8.', +'. ,.!:1%(80!.8.-#+'"+-72 #*4,2 ,*(4!7 7$8.'$A. #( "$.!7 #*. *(8(8(!.%4!+' 740!./.,2GU"Q'LP +-7 GU"Z'LP 2 +, ).!! +, #*. *.#.'(8(!.%4!+' 740!./ U"Q' ^U"Z'GW$&4'. NL5 X*. !+##.' %(80!./ ,*(4!7 3. + WFIX1+%#$?. ,0.%$.,3.%+4,.2 :(' + %(-:('8+#$(- (: #*. ]1BVJ #"0.2 #*. 7$,#+-%.3.#)..- 7(-(' U"Q +-7 +%%.0#(' U"Z $, ./0.%#.7 #( 3. 3.!()Z -82 +3(?. )*$%* #*. .::$%$.-%" (: WFIX 7'(0, #( ZO_ (: $#,#*.('.#$%+! ?+!4. :(' #*. U"Q `U"Z %(40!.5;NK= \- +-" %+,.2 #*..::$%$.-%" (: WFIX,*(4!7 ,%+!. !$-.+'$!" )$#* #*. %(-%.-#'+#$(- (:#*. %(80!./ U"Q' ^U"Z'5 W4'#*.'8('.2 #*. 8+/$8+! %(-%.-#'+#$(-(: #*$, %(80!./ $, +%*$.?+3!.2 $: #*. %(80!./., GU"Q'LP +-7 GU"Z'LP*+?. .@4+! #*.'8(7"-+8$% ,#+3$!$#$.,5 \: ,(2 #*. *.#.'(8(!.%4!+'740!./ ,*(4!7 '.+%*a:(' ,#+#$,#$%+! '.+,(-,a#)$%. #*. %(-%.-1#'+#$(-, (: #*. *(8(8(!.%4!+' (-.,5 X*$, $, ./+%#!" #*. '.+,(-)*" ). ,.!.%#.7 #*. U"Q `U"Z %(40!. $-,#.+7 (: (#*.' 7".,G,4%* +, :!4('.,%.$- ` '*(7+8$-.L5 U"Q +-7 U"Z (-!" 7$::.' 3" +,$-&!. U!U 3(-7 $- #*. %*'(8(0*('. +-72 #*4,2 +'. ./0.%#.7 #(*+?. ?.'" ,$8$!+' $-:!4.-%., (- #*. %(80!./., :('8.75 \-7..72 +0!(# (: #*. '.%$0'(%+! bc18.!#$-& #.80.'+#4'., J8 +&+$-,# #*.!(&+'$#*8 (: #*. (!$&(-4%!.(#$7. %(-%.-#'+#$(- G,.. H400('#$-&\-:('8+#$(-L %(-:$'8.7 #*+# #*. #*.'8(7"-+8$% 7+#+ (: #*.*(8(8(!.%4!+' 740!./., +'. 3+'.!" 7$,#$-&4$,*+3!.5 \- #*.:(!!()$-&2 ). 4,.7 +- +?.'+&. ?+!4. (: !9"!Y[5K 9d8(!!N5W'(8 #*.,. 7+#+ $# :(!!(), #*+#2 4-7.' #*. %(-7$#$(-, (: #*. ,.!:1'.0!$%+#$(- ./0.'$8.-#,2 #*. +,,(%$+#$(- %(-,#+-# K*(8( :(' #*.*(8(8(!.%4!+' 740!./., $, Q5P# NOE T8(!!N +# NZ "U2 )*.'.+,K*.#.'("P K*(8( :(' ,"88.#'" '.+,(-,5
$%&'(' #) L6M*54NO4BO4 PQ4N !QE 7BP !Q@ 7BP R4R57:45AO ;MA6PAB8 ;6*OSN T*5 N46T254U6AO7RA*B VARC :47NM54:4BR *T LW#J+
Schöneborn, Bülle, Kiedrowski 2001

Synthetische Biologie: Überblick
2. Künstliche Schaltkreise in Bakterien
2.1 Der „Repressilator“
2.2 Quorum sensing und Sender-Empfänger-Systeme
2.3 Ein Chemostat
2.4 Raumzeitliche Effekte
2.5 Rauschen in Gennetzwerken

Synthetische Biologie: Überblick
2. Künstliche Schaltkreise in Bakterien
2.1 Der „Repressilator“
2.2 Quorum sensing und Sender-Empfänger-Systeme
2.3 Ein Chemostat
2.4 Raumzeitliche Effekte
2.5 Rauschen in Gennetzwerken
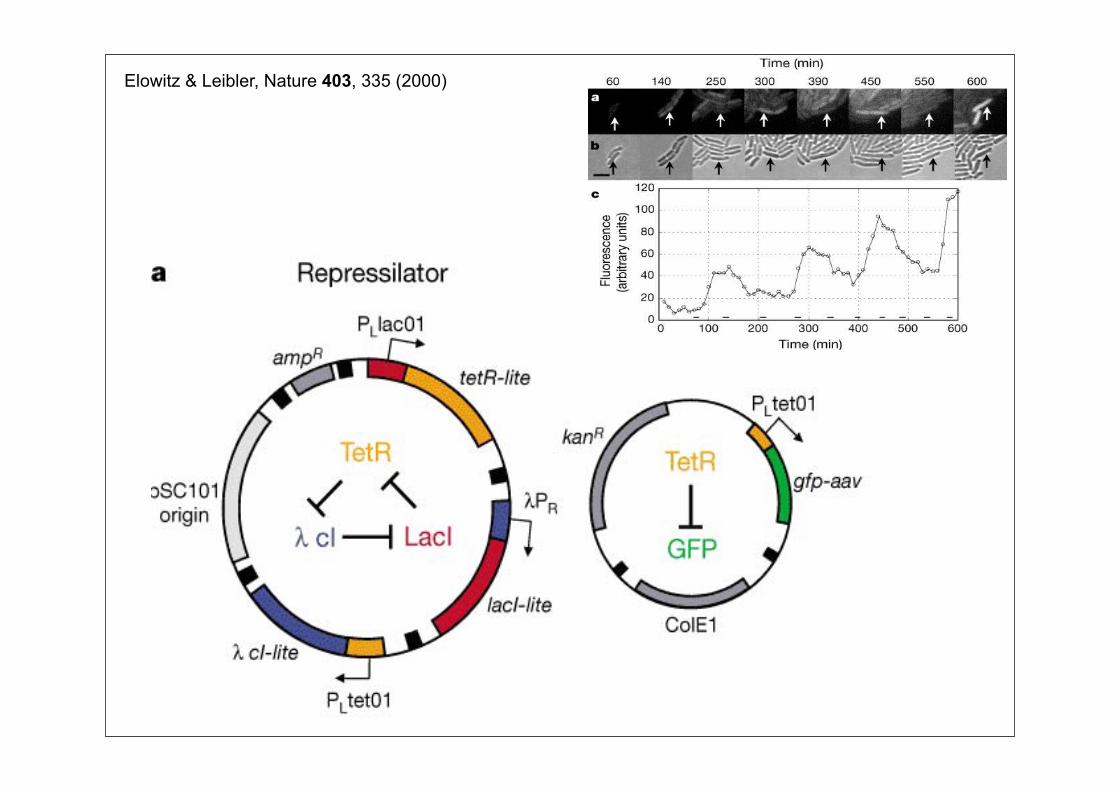
Elowitz & Leibler, Nature 403, 335 (2000)

Synthetische Biologie: Überblick
2. Künstliche Schaltkreise in Bakterien
2.1 Der „Repressilator“
2.2 Quorum sensing und Sender-Empfänger-Systeme
2.3 Ein Chemostat
2.4 Raumzeitliche Effekte
2.5 Rauschen in Gennetzwerken
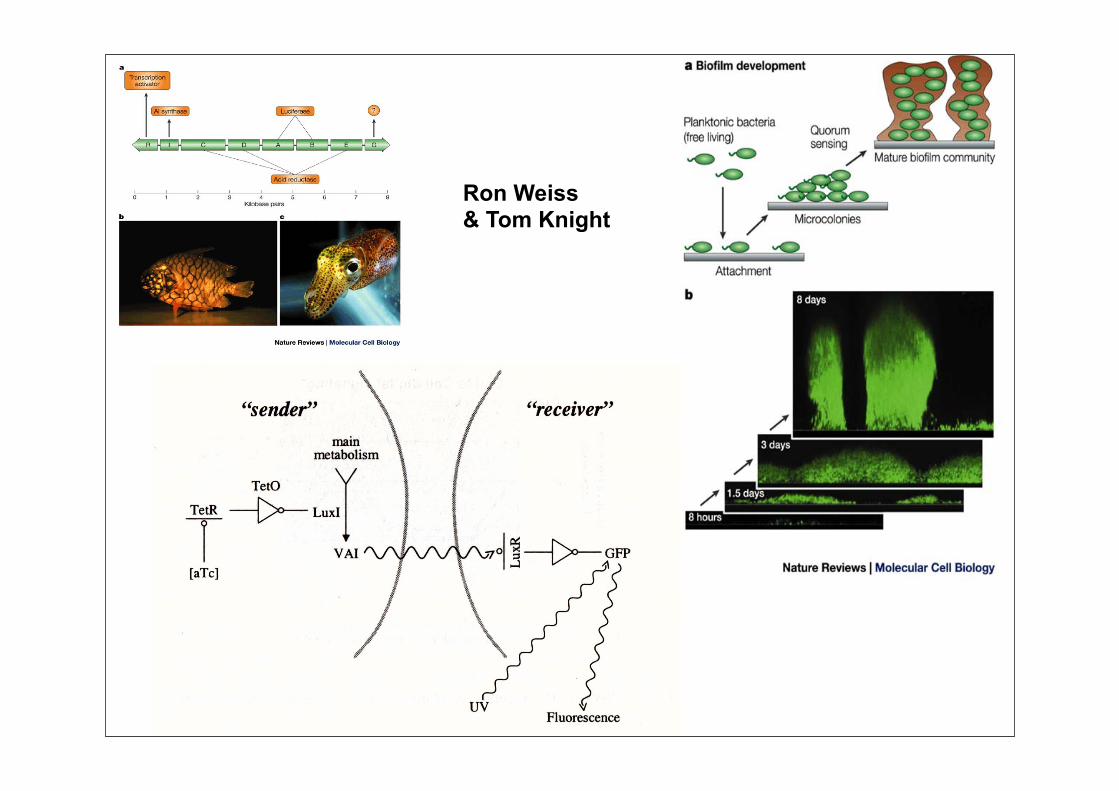
Ron Weiss& Tom Knight

Synthetische Biologie: Überblick
2. Künstliche Schaltkreise in Bakterien
2.1 Der „Repressilator“
2.2 Quorum sensing und Sender-Empfänger-Systeme
2.3 Ein Chemostat
2.4 Raumzeitliche Effekte
2.5 Rauschen in Gennetzwerken
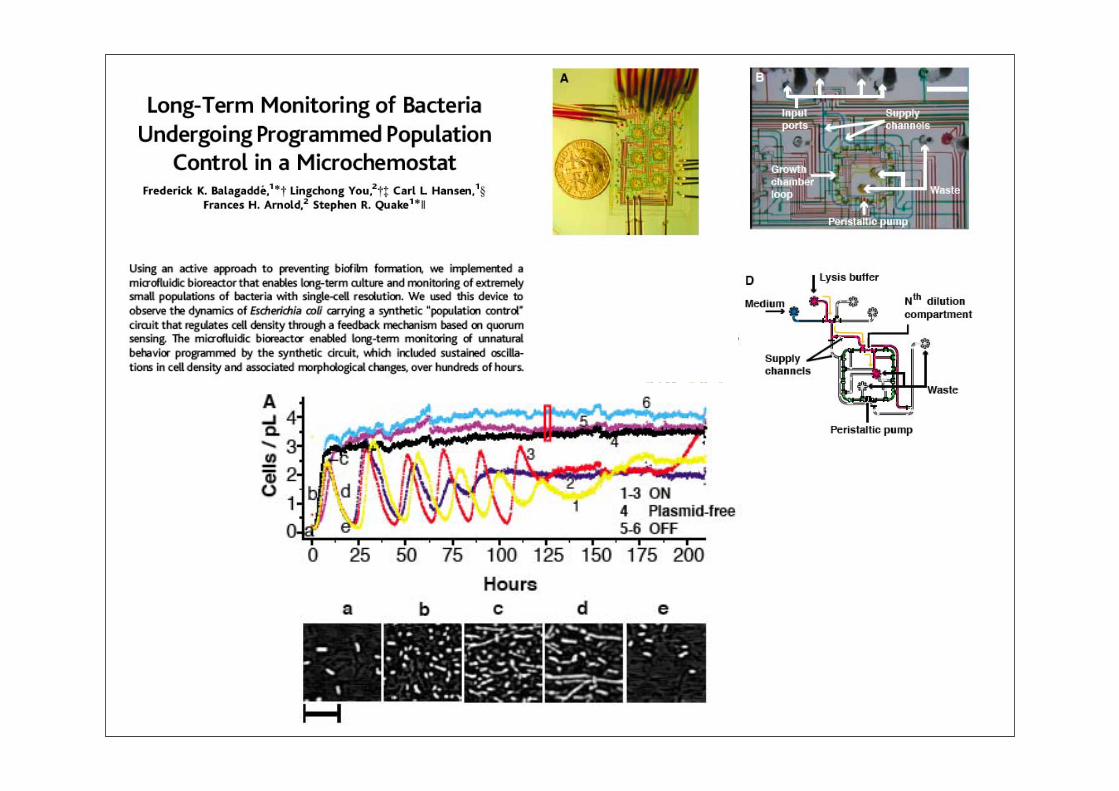

Synthetische Biologie: Überblick
2. Künstliche Schaltkreise in Bakterien
2.1 Der „Repressilator“
2.2 Quorum sensing und Sender-Empfänger-Systeme
2.3 Ein Chemostat
2.4 Raumzeitliche Effekte
2.5 Rauschen in Gennetzwerken
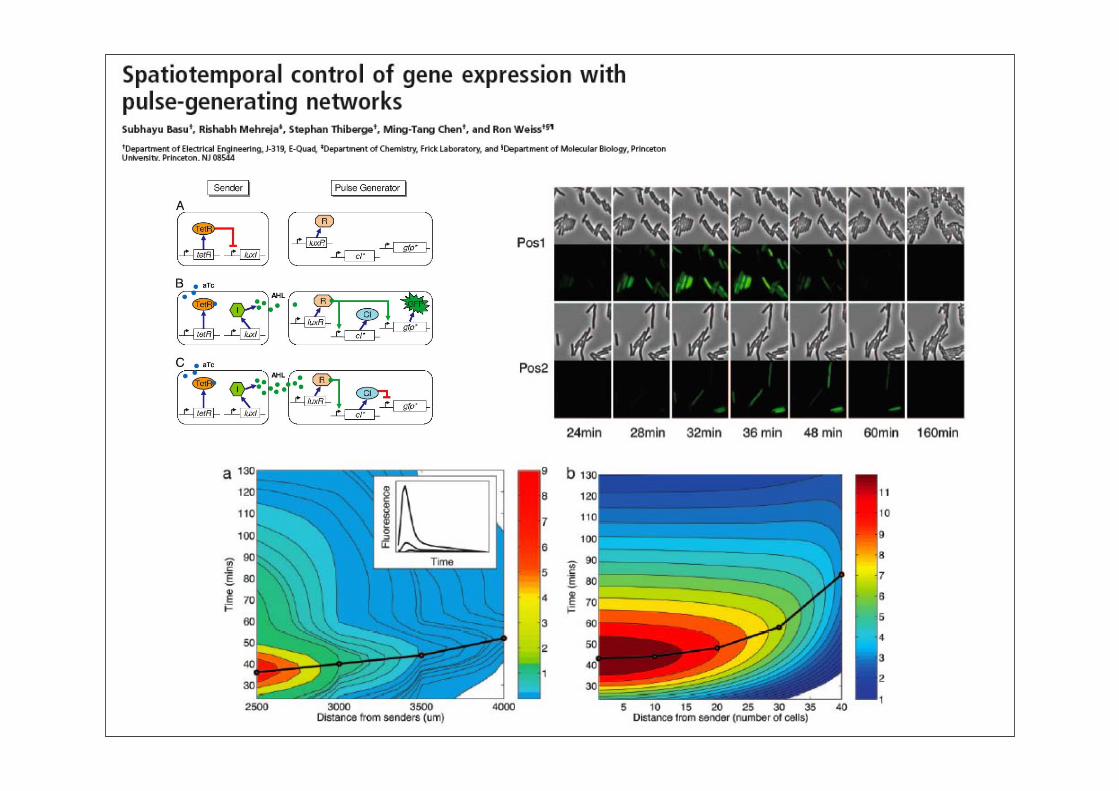

Synthetische Biologie: Überblick
2. Künstliche Schaltkreise in Bakterien
2.1 Der „Repressilator“
2.2 Quorum sensing und Sender-Empfänger-Systeme
2.3 Ein Chemostat
2.4 Raumzeitliche Effekte
2.5 Rauschen in Gennetzwerken
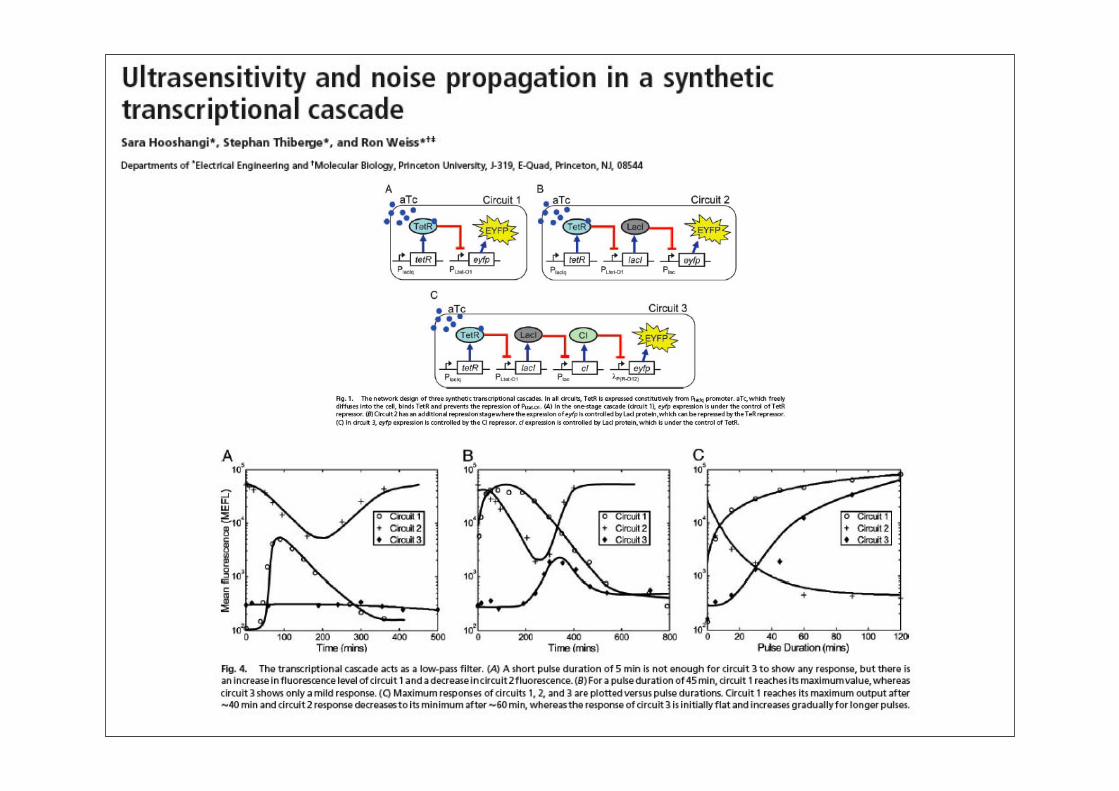

Synthetische Biologie: Überblick
3. In vitro Schaltkreise
3.1 Ein bistabiles Transkriptionssystem
3.2 Genexpression auf dem Chip
3.3 Auf dem Weg zur künstlichen Zelle
3.4 Künstliche „Entwicklungsbiologie“
3.5 Logikschaltkreise mit DNA

Synthetische Biologie: Überblick
3. In vitro Schaltkreise
3.1 Ein bistabiles Transkriptionssystem
3.2 Genexpression auf dem Chip
3.3 Auf dem Weg zur künstlichen Zelle
3.4 Künstliche „Entwicklungsbiologie“
3.5 Logikschaltkreise mit DNA
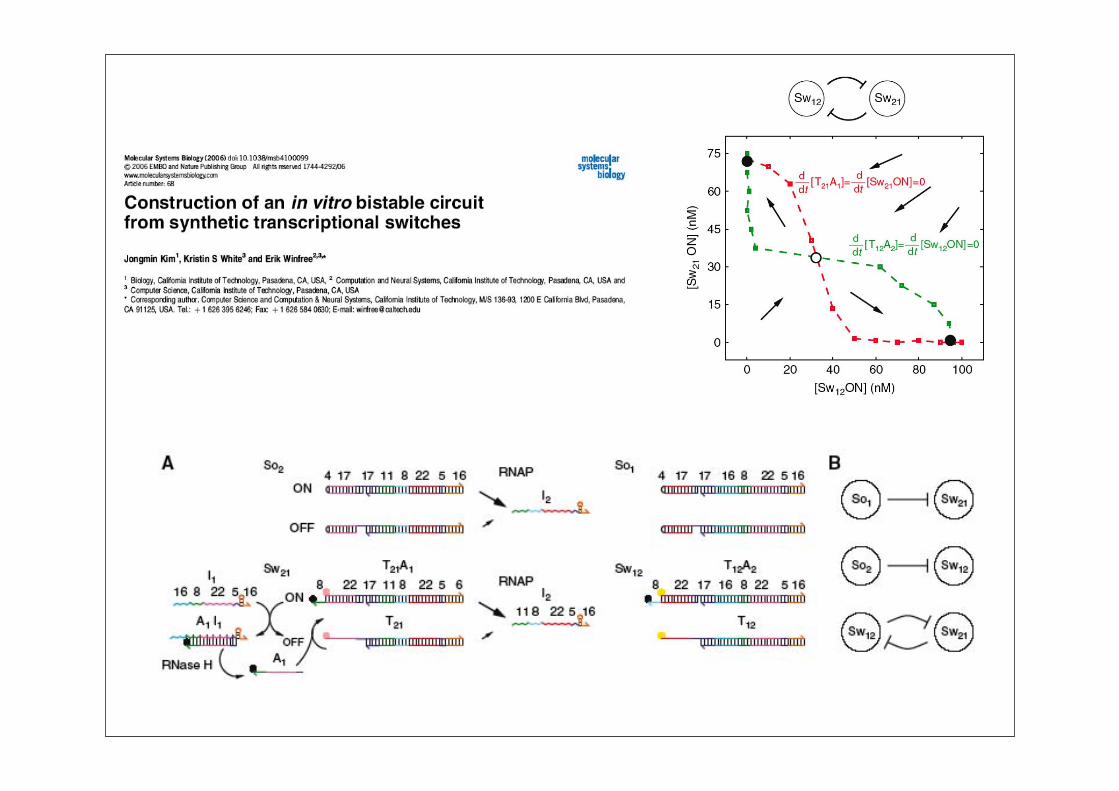

Synthetische Biologie: Überblick
3. In vitro Schaltkreise
3.1 Ein bistabiles Transkriptionssystem
3.2 Genexpression auf dem Chip
3.3 Auf dem Weg zur künstlichen Zelle
3.4 Künstliche „Entwicklungsbiologie“
3.5 Logikschaltkreise mit DNA
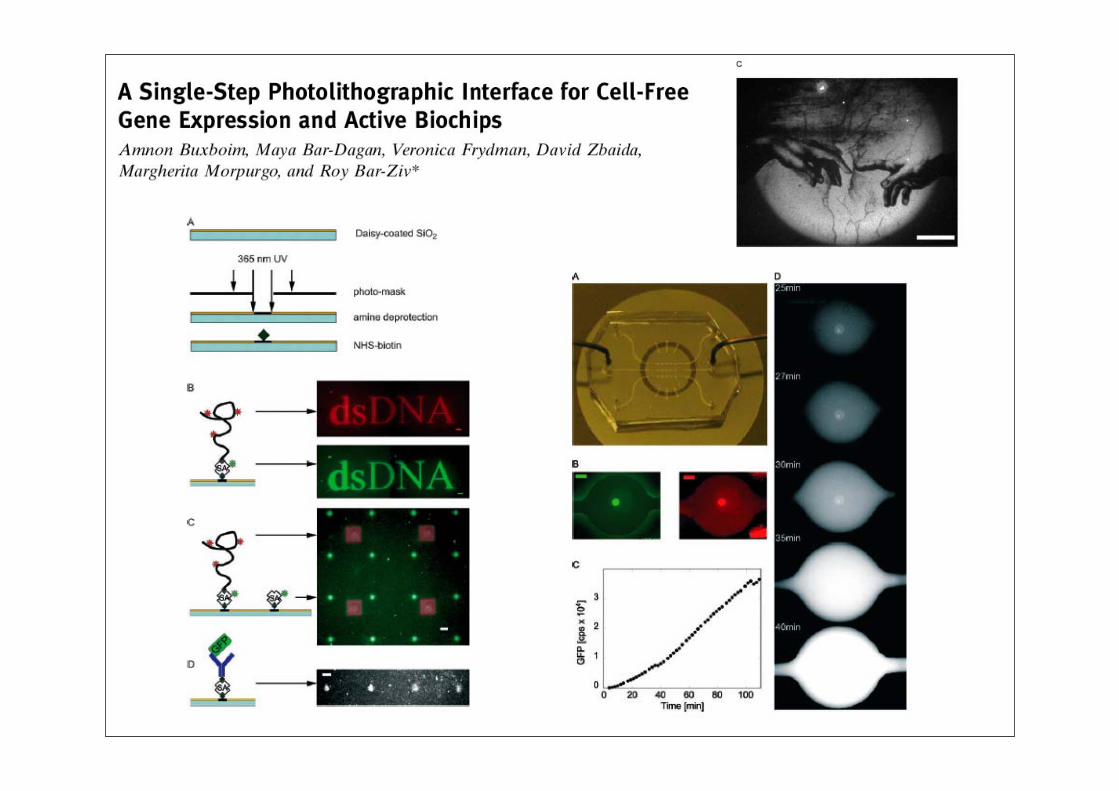

Synthetische Biologie: Überblick
3. In vitro Schaltkreise
3.1 Ein bistabiles Transkriptionssystem
3.2 Genexpression auf dem Chip
3.3 Auf dem Weg zur künstlichen Zelle
3.4 Künstliche „Entwicklungsbiologie“
3.5 Logikschaltkreise mit DNA
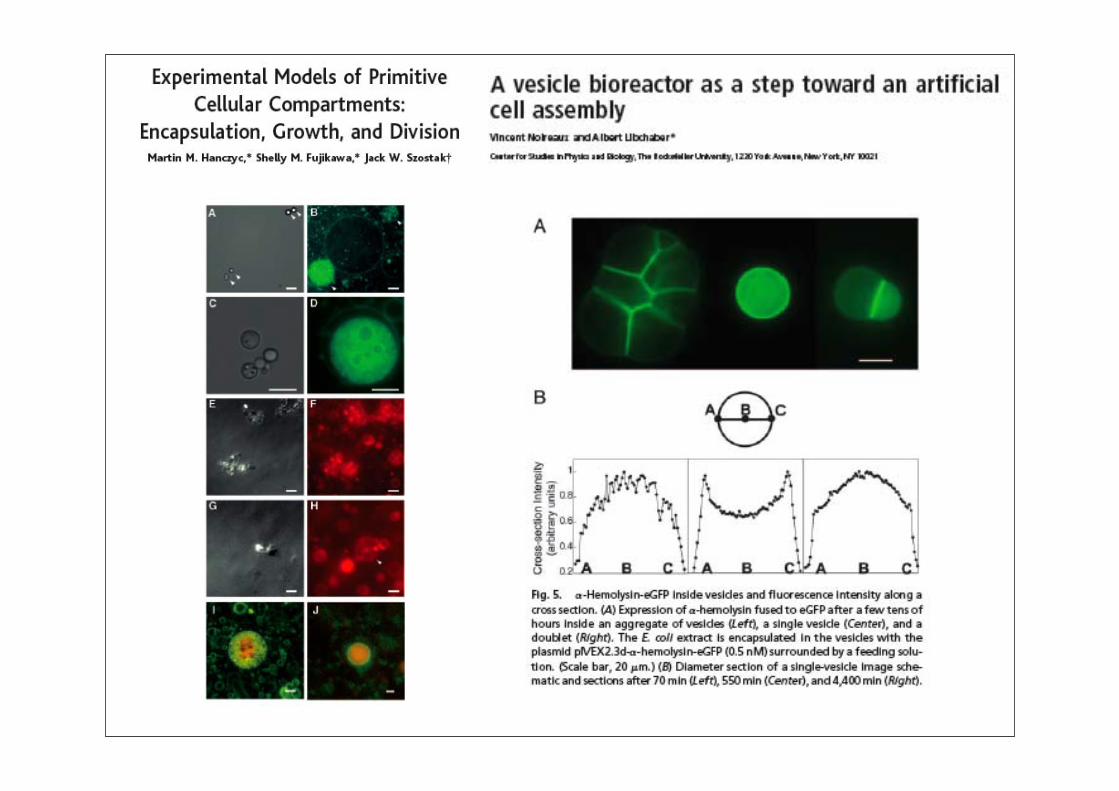

Synthetische Biologie: Überblick
3. In vitro Schaltkreise
3.1 Ein bistabiles Transkriptionssystem
3.2 Genexpression auf dem Chip
3.3 Auf dem Weg zur künstlichen Zelle
3.4 Künstliche „Entwicklungsbiologie“
3.5 Logikschaltkreise mit DNA
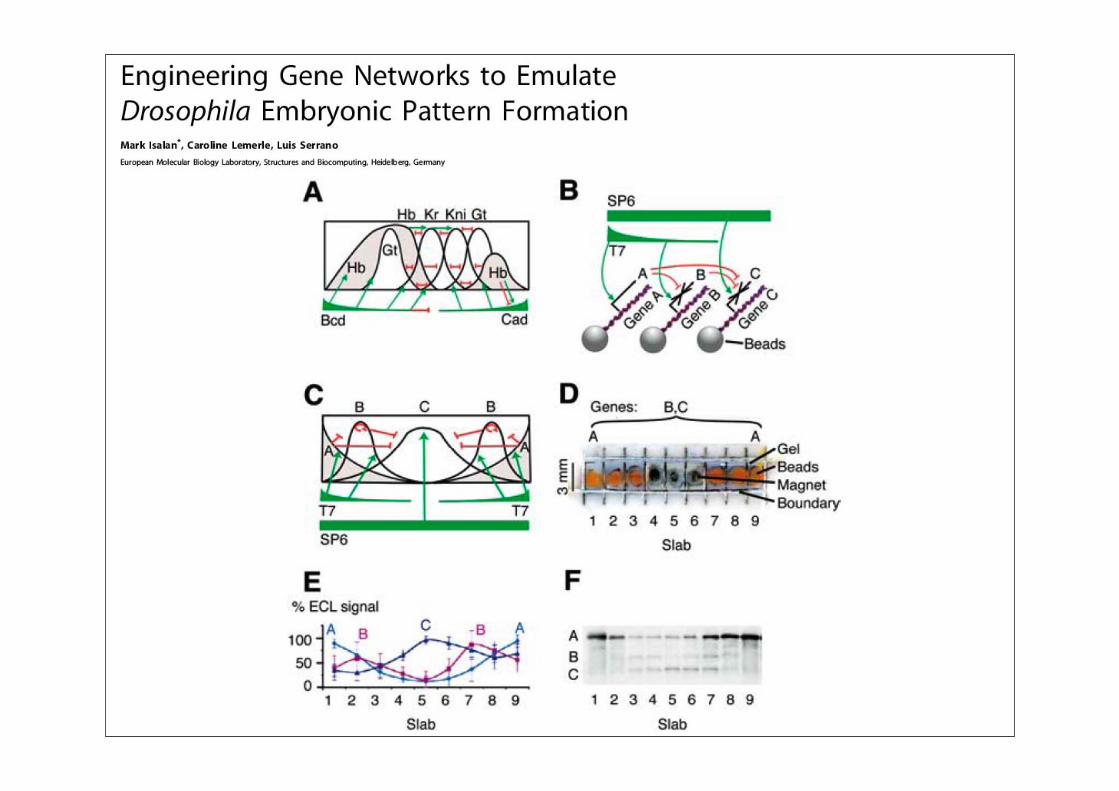

Synthetische Biologie: Überblick
3. In vitro Schaltkreise
3.1 Ein bistabiles Transkriptionssystem
3.2 Genexpression auf dem Chip
3.3 Auf dem Weg zur künstlichen Zelle
3.4 Künstliche „Entwicklungsbiologie“
3.5 Logikschaltkreise mit DNA


09.05. Einführung (Simmel)
16.05. Selbstreplikatoren (Simmel)
23.05. Künstliche Zellen (Meyer)
06.06. Quorum Sensing ()
Themenauswahl

13.06. Biochemische Oszillatoren ()
20.06. Transkriptionsschalter (Otten)
04.07. Logikgatter aus DNA ()
11.07.
18.07.
Themenauswahl