Embed Size (px)
Citation preview

6
1 Zusammenfassung
Im Folgenden soll ein Einblick in die wesentlichen biologischen Systeme gegeben
werden, die im Zusammenhang mit der Manipulation von G-Protein-gekoppelter
Rezeptoren (GPCR) in dieser Arbeit von Bedeutung sind. Es stellt sich die Frage,
wie die hohe Spezifität der Signaltransduktion in biologischen System gewährleis-
tet werden kann. Der beobachteten hohen Spezifität der Signaltransduktion steht
eine limitierte Anzahl der an der Signalkaskade beteiligten Interaktionspartner ge-
genüber.
In dieser Arbeit wird das System des G-Protein aktivierten einwärtsgleichrichten-
den Kaliumkanals (GIRK) an Vorhofmyozyten untersucht. Über die Aktivierung des
G-Protein koppelnden M2 Rezeptors durch Azetylcholin und konsekutiver Öffnung
des Kaliumkanals gehört dieser Signalweg zu einem der wichtigen physiologi-
schen, parasympathischen Regelkreisläufe des Herzen. Neben der parasympathi-
schen Aktivierung der GIRKs durch Azetylcholin, ist Adenosin über den A1 Rezep-
tor ein physiologischer Aktivator der GIRKs. Beide Signalwege sind in Kardiomyo-
zyten in vivo exprimiert und zeigen eine bisher nicht geklärte Interaktion.
Da ein wesentlicher Ergebnisanteil elektrophysiologische Daten beinhaltet, wird in
einem ersten Teil ein Überblick über die Familie der Kaliumkanäle gegeben, mit
besonderem Bezug auf die kardiale Physiologie und Pathophysiologie. Eine be-
sondere Rolle spielt dabei die Kanalfamilie der einwärtsgleichrichtenden Kalium-
kanäle, da sie als Zieleffektor für die Manipulation der G-Protein koppelnden Re-
zeptoren genutzt werden. An ihnen werden beispielhaft Fragen nach Spezifität der
Signaltransduktion durch GPCRs, sowie speziellere Fragestellungen wie Desensi-
tisierungsmechanismen demonstriert. Die vegetative Regulation des Herzens und
mögliche pathophysiologische und therapeutische Zusammenhänge stellen den
Rahmen der Arbeit dar.
1.1 Physiologische und biochemische Grundlagen von Kaliumkanälen
K+ Kanäle sind an einer Vielzahl von biologischen Vorgängen beteiligt, so z.B. an
der Neurotransmitterfreisetzung, Insulinsekretion, renalen Regulation der Elektro-
lythomeostase und in der Muskelkontraktion. In erregbaren Zellen sind Kali-
umkanäle mitverantwortlich für die Erregbarkeit, auf der einen Seite durch die Sta-

7
bilisation des Membranruhepotentials, auf der anderen Seite durch ihre Möglich-
keit, Aktionspotentiale zu terminieren, bzw. zu modifizieren. Diese beiden Eigen-
schaften sind von großer physiologischer und pathophysiologischer Relevanz in
der Erregungsleitung am Herzmuskel.
Kaliumkanäle können aufgrund ihrer Sekundärstruktur in drei große morphologi-
sche Familien eingeteilt werden. Dabei richtet sich ihre Zuteilung nach der Anzahl
der α-helikalen Transmembranhelices (TM) und nach der Anzahl der kaliumselek-
tiven Poren (P):
- 6TM-1P-Kanäle
- 4TM-2P-Kanäle
- 2TM-1P-Kanäle
Die Kaliumselektivität wird durch ein hochkonserviertes Sequenzmotiv in der Po-
rendomäne vermittelt. Die Aminosäuresequenz Gly-Tyr-Gly (Gly-Phe-Gly in eag,
Kir 6.1, Kir 6.2) in der H5 Region der 6-TM-1P, bzw. in der P-loop Region bei 2
TM-P Kanäle, determiniert die Kaliumselektivität (Doyle et al., 1998). So zeigen
z.B. Mutanten mit einer GY-Deletion die charakteristisch verminderte Ionenselek-
tivtät eines CNG-Kanals (cylic nucleotide gated). Im Folgenden wird ein kurzer
Überblick über die einzelnen K+ Kanäle gegeben.
Zur Familie der 6TM-1P Kanäle gehören mehrere Untergruppen. Eine große
Gruppe stellt die der spannungsabhängigen K+-Kanäle dar. Dabei zählen die Kv-
Familie (auch Shaker-Kanäle nach Drosophilamutanten), eag (ether-à-go-go) und
KCNQ1 zu den klassischen spannungsabhängigen Kanälen. CNG und HCN-
Kanäle zeigen hingegen eine geringere Spannungsabhängigkeit und sind eher
unselektive Kationenkanäle, werden jedoch zusätzlich durch zyklische Nukleotide
reguliert. Die Spannungsabhängigkeit, Aktivierungs- und Inaktivie-
rungsgeschwindigkeit variieren in dieser Gruppe von Kanälen stark, sie zeigen je-
doch auch strukturelle Gemeinsamkeiten (Nerbonne, 2000). Der Aktivierungsme-
chanismus ist mit einer Konformationsänderung im „spannungssensitiven“ S4
Segment verbunden (Mannuzzu et al., 1996), die zu einer Öffnung der kaliumse-
lektiven Pore führt. Die Inaktivierung der Kanäle beruht auf verschiedenen Me-
chanismen. Während in einer Gruppe (z.B. Kv. 1.4 ) über einen sogenannten
„ball and chain“ Mechanismus die Kanalpore über eine N-terminale Domäne des
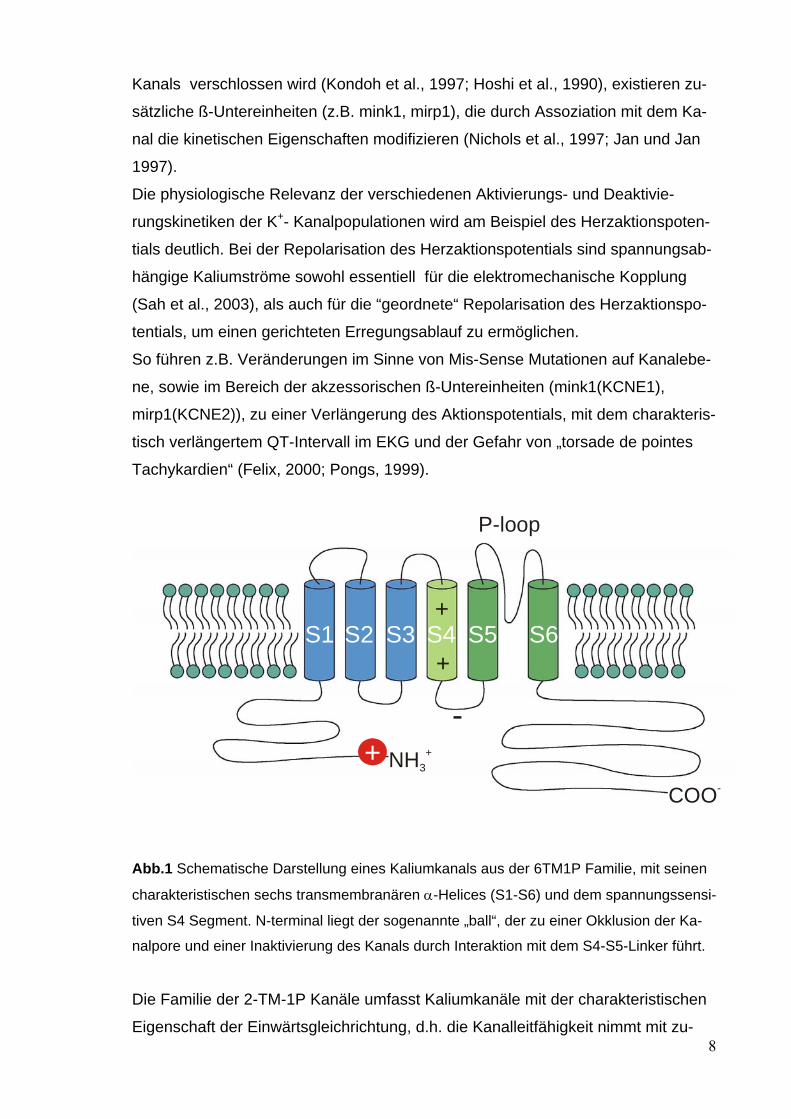
Kanals verschlossen wird (Kondoh et al., 1997; Hoshi et al., 1990), existieren zu-
sätzliche ß-Untereinheiten (z.B. mink1, mirp1), die durch Assoziation mit dem Ka-
nal die kinetischen Eigenschaften modifizieren (Nichols et al., 1997; Jan und Jan
1997).
Die physiologische Relevanz der verschiedenen Aktivierungs- und Deaktivie-
rungskinetiken der K+- Kanalpopulationen wird am Beispiel des Herzaktionspoten-
tials deutlich. Bei der Repolarisation des Herzaktionspotentials sind spannungsab-
hängige Kaliumströme sowohl essentiell für die elektromechanische Kopplung
(Sah et al., 2003), als auch für die “geordnete“ Repolarisation des Herzaktionspo-
tentials, um einen gerichteten Erregungsablauf zu ermöglichen.
So führen z.B. Veränderungen im Sinne von Mis-Sense Mutationen auf Kanalebe-
ne, sowie im Bereich der akzessorischen ß-Untereinheiten (mink1(KCNE1),
mirp1(KCNE2)), zu einer Verlängerung des Aktionspotentials, mit dem charakteris-
tisch verlängertem QT-Intervall im EKG und der Gefahr von „torsade de pointes
Tachykardien“ (Felix, 2000; Pongs, 1999).
S1 S2 S3 S4 S5 S6
P-loop
NH +3
COO-
+
+
+
+-
Abb.1 Schematische Darstellung eines Kaliumkanals aus der 6TM1P Familie, mit seinen
charakteristischen sechs transmembranären α-Helices (S1-S6) und dem spannungssensi-
tiven S4 Segment. N-terminal liegt der sogenannte „ball“, der zu einer Okklusion der Ka-
nalpore und einer Inaktivierung des Kanals durch Interaktion mit dem S4-S5-Linker führt.
8
Die Familie der 2-TM-1P Kanäle umfasst Kaliumkanäle mit der charakteristischen
Eigenschaft der Einwärtsgleichrichtung, d.h. die Kanalleitfähigkeit nimmt mit zu-

9
aliumka-
n Ami-
da
nehmender Depolarisation ab. Bisher sind 7 Familien bekannt, die mit Kir (K+ in-
ward rectifying) 1.x-7.x klassifiziert werden. 1993 konnte erstmals durch Kubo die
cDNA für den Kir 2.1 isoliert werden, im gleichen Jahr erfolgte die Klonierung des
Kir 1.1 durch Ho und Mitarbeiter (Yamada et al.,1998). Kir-Kanäle sind wie die 6-
TM-1P Kanäle Tetramere, die sich entweder aus identischen Untereinheiten (Ho-
motetramere) oder aus verwandten Untereinheiten (Heterotetramere) zusammen-
setzen. Strukturell bestehen die Untereinheiten aus zwei α-helikalen Trans-
membrandomänen (mit M1 und M2 bezeichnet) und der kaliumselektiven, poren-
formenden Region H5 mit dem schon beschriebenen charakteristischen kaliumse-
lektiven Gly-Tyr-Gly Motiv. C und N Terminus liegen zytoplasmatisch. Das 2-TM-
1P Motiv kann strukturell und funktionell mit der S5-P-S6 Domäne der 6-TM-1P
Kanäle verglichen werden. Durch Mutageneseuntersuchung von Kir-Kanälen
konnten verschiedene Kanaldomänen identifiziert werden, die Einfluß auf die Ein-
wärtsgleichrichtung haben. Ein wichtiger Anteil an der Einwärtsgleichrichtung wird
durch negativ geladene Aminosäuren (z.B. Asparagin 173 bei Kir 3.x) in der M2
Domäne (Stanfield et al., 1994) und durch verschiedene anionische Aminosäuren
des C-terminalen Abschnittes der Untereinheiten (Taglialatela et al., 1995; Kubo
und Murata., 2001) vermittelt. Durch die Interaktion von Mg++ Ionen und Polyami-
nen (z.B. Spermin) mit diesen Aminosäuren der Kanalpore ergibt sich eine span-
nungsabhängige Blockade der Kanalpore bei Membranpotentialen, die positiver
als das Kaliumgleichgewichtspotential sind (Ficker et al., 1994; Wible et al., 1994).
Es blieb lange ungeklärt, wie diese verschiedenen und auf den ersten Blick to-
pograpisch ungeordneten Aminosäurereste die Einwärtsgleichrichtung bestimmen
konnten. Nishida und MacKinnon gaben im Jahre 2002 in Kristallisationsexperi-
menten ein Modell der tertiären Struktur von einwärtsgleichrichtenden K
nälen. Sie beschreiben in ihrem Modell neben der transmembranären Pore mit
dem bekannten Kaliumselektivitätsfilter zusätzlich eine zytoplasmatische Pore, die
durch den C-Terminus gebildet wird. Die meisten identifizierten anionische
nosäurereste projizieren sich auf den inneren Rand der zytoplasmatischen Pore
und bilden somit ein Interaktionsinterface für die kationischen Polyamine (Nishi
und MacKinnon, 2002).
Eine besondere Gruppe stellen die Unterfamilien Kir 3.x und Kir 6.x dar, da ihre
Regulation in der 3.x Familie zusätzlich an βγ Untereinheiten von heterotrimeren

10
G-Proteinen gebunden, oder in der 6.x Familie durch ihre ATP-Sensitivität direkt
an den Zellmetabolismus gekoppelt ist.
Die Familie der ATP-sensitiven Kaliumkanäle sind Heterooctamere von Sulfonyl-
harnstoffrezeptoren (SUR) und von Kir 6.x-Untereinheiten mit einer 1:1 Stöchio-
metrie. Die Kir 6.x Untereinheiten formen mit ihrer 2-TM-1P-Topologie die Kanal-
pore. Weder Sulfonylharnstoffrezeptoren, noch Kir 6.x formen funktionelle Kanäle,
erst die Heteromultimere stellen funktionelle Kanäle dar (Babenko et al., 1998).
Als wichtige regulatorische Metabolite sind Magnesium gebundene Nukleotide
sowie ATP bekannt. Mg++-gebundene Nukleotide haben über den SUR Rezeptor
einen stimulatorischen Effekt auf den IKATP, während ATP über die direkte Bin-
dung an die porenbildende Untereinheit Kir 6.x einen inhibitorischen Effekt vermit-
telt (Baukrowitz und Fakler, 2000). Über diese entgegengesetzte Regulation ist es
möglich, den Status der zellulären Energieversorgung abzuschätzen. Der am Bes-
ten verstandene Kanaltyp ist der SUR1/Kir6.2, der in den ß-Zellen des Pankreas
über seinen Schluss und nachfolgende Depolarisation die Insulinsekretion vermit-
telt.
Eine zweite Gruppe (Kir 3.x) von Kanälen aus der 2-TM-1P Familie ist hauptsäch-
lich an die Aktivierung durch βγ Untereinheiten von heterotrimeren G-Proteinen
gekoppelt.
Man findet neben der Kir 3.x Nomenklatur auch die Bezeichnung GIRK 1-5 (G-
protein activated inward rectifying K+ channels) für die einzelnen Untereinheiten.
Funktionelle Kanäle werden aus Tetrameren gebildet. Die Komposition der Unter-
einheiten unterscheidet sich in verschiedenen Geweben. In neuronalem Gewebe
scheinen Heterotetramere aus GIRK1 und GIRK 2/3 vorzuherrschen, während die
kardiale Kanalpopulation eine GIRK 1/4 Komposition aufweist (Jelacic et al., 2000;
Fleischmann et al., 2004; Dobrzynski et al., 2001; Karschin et al., 1996). Während
GIRK4 funktionelle Homotetramere in der Membran bilden kann und diese mögli-
cherweise physiologisch vorkommen (Corey and Clapham, 1998; Bender et al.,
2001), konnte gezeigt werden, dass GIRK1 keine funktionellen Kanäle in der
Membran bildet und zur Membranlokalisation eine zusätzliche GIRK-Untereinheit
benötigt (Kennedy et al., 1998). Die Kanalkomposition beeinflusst die biophysikali-
schen Eigenschaften der funktionellen Kanäle in Bezug auf ihre kinetischen Ei-
genschaften, Desensitisierung und Größe des induzierbaren Stroms (Silvermann
et al., 1996). Die charakteristische porenformende Domäne (H5) zeigt zwischen

11
den Untereinheiten große Sequenzhomologie. Die zytosolisch gelegenen C- und
N-Termini sind in Sequenz und Länge jedoch unterschiedlich (Yamada e al.,
1998). Die für die Kanalaktivierung wichtigen βγ Bindungsstellen sind C- und N-
terminal lokalisiert. Die GIRK1 Untereinheit besitzt im Vergleich zu GIRK 4 eine C-
terminale zusätzliche βγ Bindungsstelle (Huang et al., 1997, Krapivinsky et al.,
1998). Corey et al. beschreiben eine 1:1 Bindungsstöchiometrie zwischen GIRK-
Untereinheit und βγ-UE (Corey und Clapham, 2001).
Weiterhin existiert am C-Terminus eine Bindungsstelle für PIP2 und Na+. Sowohl
für PIP2 als auch für Na+ ist ein aktivierender, regulatorischer Einfluss auf die Ka-
näle bekannt (Petit-Jacques et al., 2001). Aus Untersuchungen mit Chimeren von
GIRK4 und Kir2.1 ist für PIP2 ist eine Bindungsstelle C-terminal lokalisiert worden,
zusätzliche soll Natrium über Neutralisierung eines Aspartatrestes in der Nähe der
PIP2 Bindungsstelle die Bindungswahrscheinlichkeit für PIP2 an die GIRK Unter-
einheit erhöhen (Zhang et al., 1999, Ho and Murrell-Lagnado,1999). Es bleibt zur
Zeit unklar, in wiefern Natrium und PIP2 essentielle Kofaktoren für die βγ Bindung
sind oder „nur“ eine regulatorische Funktion übernehmen.
Während die meisten Ergebnisse durch heterologe Expressionsysteme gewonnen
wurden, konnte auch an Kardiomyozyten gezeigt werden, dass durch eine Deple-
tion von PIP2 durch den PLC Signalweg eine Hemmung des IK(ACh) möglich ist
(Meyer et al., 2001).
Die im heterologen Expressionssystem beobachtete hochgradige Natriumabhän-
gigkeit von GIRK Kanälen (Shui et al., 1996) konnte in Kardiomyozten an endoge-
nen GIRK Kanälen nicht nachvollzogen werden. Erst bei einer Überexpression
z.B. von GIRK 4 Untereinheiten ergab sich ein natriumabhäniger Hintergrundstrom
(eigene Daten, nicht publiziert).

Abb.2 Schematische Darstellung einer GIRK4 (Kir 3.4) Untereinheit. Mit M1 und M2 sind
die transmembranären Domänen bezeichnet. (–) Zeichen kennzeichnen die negativ ge-
ladenen Aminosäuren, die die Einwärtsgleichrichtung vermitteln, insbesondere Asparagin
in Position 173. Die Aminosäuresequenz im Bereich der PIP2 und Natrium Bin-
dungsstellen ist gesondert dargestellt. Das Aspartat (D) in Position 223 vermittelt eine
natriumabhängige Modulation, Arginin (R) 218 und 228, sowie Isoleucin (I) 229 sind kriti-
sche Aminosäuren für die PIP2 Bindung. N-terminal ist an Position 34-86 eine βγ-
Bindungsstelle lokalisiert, C-terminal an der Position 318-374. (modifiziert aus Mark und
Herlitze, 2000)
1.2 Kardiale G-Protein gekoppelte Rezeptoren
Die für die Aktivierung von GIRK-Kanälen nötigen Rezeptoren fallen in die große
Gruppe der „G-protein-coupled-receptors“ (GPCRs). GPCR sind eine der größten
Rezeptorfamilien der Vertebraten. Zwischen 1000 und 2000 Gene (ca. 1% des ge-
samten Genoms) kodieren für diese Proteine (Ji et al., 1998). GPCR spielen eine
Rolle bei der Erkennung und der Signalübermittlung durch Licht, Duftstoffe, Nukle-
otide, Aminosäuren, Peptide und Proteine. Der Aufbau der Rezeptoren ist prinzi-
12
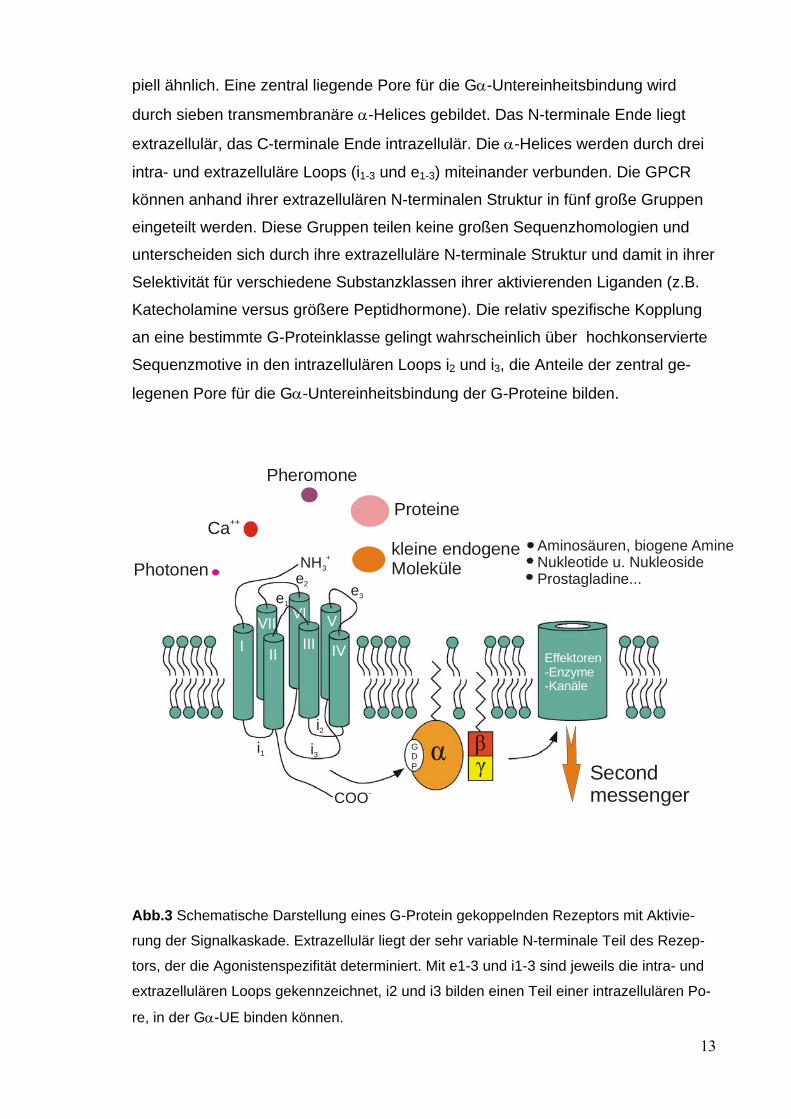
piell ähnlich. Eine zentral liegende Pore für die Gα-Untereinheitsbindung wird
durch sieben transmembranäre α-Helices gebildet. Das N-terminale Ende liegt
extrazellulär, das C-terminale Ende intrazellulär. Die α-Helices werden durch drei
intra- und extrazelluläre Loops (i1-3 und e1-3) miteinander verbunden. Die GPCR
können anhand ihrer extrazellulären N-terminalen Struktur in fünf große Gruppen
eingeteilt werden. Diese Gruppen teilen keine großen Sequenzhomologien und
unterscheiden sich durch ihre extrazelluläre N-terminale Struktur und damit in ihrer
Selektivität für verschiedene Substanzklassen ihrer aktivierenden Liganden (z.B.
Katecholamine versus größere Peptidhormone). Die relativ spezifische Kopplung
an eine bestimmte G-Proteinklasse gelingt wahrscheinlich über hochkonservierte
Sequenzmotive in den intrazellulären Loops i2 und i3, die Anteile der zentral ge-
legenen Pore für die Gα-Untereinheitsbindung der G-Proteine bilden.
α βγ
I IIIII IV
VVIVII
i1
i2i3
e1
e2 e3
GDP
NH +3
COO-
Secondmessenger
Effektoren-Enzyme-Kanäle
Ca++
Photonen
Pheromone
kleine endogeneMoleküle
Aminosäuren, biogene AmineNukleotide u. NukleosideProstagladine...
Proteine
Abb.3 Schematische Darstellung eines G-Protein gekoppelnden Rezeptors mit Aktivie-
rung der Signalkaskade. Extrazellulär liegt der sehr variable N-terminale Teil des Rezep-
tors, der die Agonistenspezifität determiniert. Mit e1-3 und i1-3 sind jeweils die intra- und
extrazellulären Loops gekennzeichnet, i2 und i3 bilden einen Teil einer intrazellulären Po-
re, in der Gα-UE binden können.
13

14
Durch extrazelluläre Signale wie Hormone, Neurotransmitter und sensorische Sti-
muli werden GPCR (G-protein-coupled receptors) aktiviert und führen über eine
Konformationsänderung zur Aktivierung von G-Proteinen (Gether, 2000).
G-Proteine gehören zu der Familie der GTP- Hydrolasen. Diese Familie schließt
sowohl die „kleinen“ G-Proteine als einzelne Polypeptide wie RAS und Elongati-
onsfaktoren ein, als auch heterotrimere G-Proteine, die sich aus drei Untereinhei-
ten (α,β ,γ) zusammensetzen. Bis heute sind 20 α, 5 β und 13 γ Untereinheiten
identifiziert worden. Die α Untereinheiten werden auf Basis von Se-
quenzhomologien und funktionellen Eigenschaften in die vier Gruppen α s,i,o,q und
q/13 unterteilt. Die Suffixe s und i ergeben sich aus der Fähigkeit, die Adenylat-
zyclase zu stimulieren oder inhibieren. Eine pharmakologische Möglichkeit zur
Unterscheidung von Gi und Gs besteht durch die ADP-Ribosylierung und Inakti-
vierung von Gi durch Pertussis-Toxin, bzw. Aktivierung von Gs durch Cholera-
Toxin. Gq- α-Untereinheiten stimulieren primär den Phospholipase C Signalweg.
Die βγ Untereinheiten sind funktionelle Monomere und unterscheiden sich in ihrer
Untereinheitskomposition. Als Effektorproteine sind Adenylatzyclasen, Phospholi-
pasen, Rezeptorkinasen und Ionenkanäle bekannt (Mesters et al., 2001).
Die Aktivierung eines G-Proteins erfolgt über die Agonisten Bindung an einen
GPCR, daraufhin erfolgt die Aktivierung des G-Protein Zyklus. Mit der Aktivierung
des G-Proteins durch z.B. Agonistenbindung am Rezeptor, wird an der α Unter-
einheit das GDP gegen GTP ausgetauscht. Nach diesem Austausch können die
GTP-bindende α Untereinheit und die βγ Untereinheit dissoziieren und jeweils un-
abhängig voneinander als „second messenger“ fungieren. Durch die intrinsische
GTPase Aktivität der α Untereinheit erfolgt der Austausch von GTP nach GDP und
schließt mit der Reassoziation der Untereinheiten den G-Protein Zyklus (Mesters
et al., 2001).

+
+
Pi GTP
GTP
GDPGDP
RGS
β
β
βα
α
α
γ
γγ
EffektorenEffektoren
GPCR Aktivierung
GIRK
Phospholipase C (ß )
Ca Kanäle (N/P)Adenylatzyclase (I)
Phospholipase A
ß-ARKinase
MAP-Kinasen
2
2
++
(+)
(+)
(+)(+)
(+)
(-)
(-)
Gαs
Gα12
Gαq
Gαi
Adenylatzyclase (alle)
Adenylatzyclase (I,V,VI)Adenylatzyclase (I)
Phospholipase C (ß ,ß )1 3
Phospolipase A2Na+/H+ Austauscher
cGMP-Phosphodiesterase
(+)(+)
(+)
(+)(+)
(+)
(-)
Abb.4 Schematische Darstellung des G-Proteinzyklus: Das G-Protein liegt in Abwesen-
heit eines Liganden am GPRC in inaktiver GDP gebundener Form vor. Nach Rezeptor
Aktivierung erfolgt der Austausch von GDP gegen GTP an der α-Untereinheit, mit folgen-
der Dissoziation der α und βγ Untereinheit. Durch eine intrinsische GTPase Aktivität
schließt sich der G-Proteinzyklus. Durch sog. RGS-Proteine kann dieser Schritt beschleu-
nigt werden. Beispielhaft sind Effektoren der α und βγ Untereinheit aufgezählt (modifiziert
nach Mesters, 2001)
1.3 Vegetative Regulation des Herzen durch A1 und M2 Rezeptoren
In die vegetative Regulation des Herzen sind verschiedene GPCR und G-Proteine
involviert. Die parasympathische Regulation wird zu einem Großteil durch den M2
Rezeptor vermittelt. Er gehört in die Familien der muskarinergen Azetylcholin-
Rezeptoren.
15

16
Azetycholin-Rezeptoren wurden bereits 1914 durch Sir Henry Dale in zwei phar-
makologische Klassen eingeteilt. Die Unterteilung orientierte sich an der Aktivier-
barkeit durch Nikotin oder das Alkaloid Muskarin. Die muskarinergen Acetylcholin-
rezeptoren gehören zu der Klasse der GPCR. Fünf verschiedene Rezeptoren die-
ser Klasse wurden bisher identifiziert (M1-5). Sie unterscheiden sich in der Primär-
struktur, Gewebelokalisation, Funktion sowie in ihrer Kopplung an die G-Protein-
Kaskade. Während M1, M3 und M5 Rezeptoren über Gq/11 an den DAG/IP3 Signal-
weg koppeln, interagieren M2 und M4 Rezeptoren mit G-Proteinen der Klasse Gi/o
und hemmen die Adenylatzyclase (Caulfield und Birdsall, 1998). Der dominierende
kardiale Rezeptortyp der muskarinergen Rezeptoren ist der Gi-koppelnde M2 Re-
zeptor. Er vermittelt maßgeblich die vegetativen Reize durch den Nervus vagus
und damit die typischen negativ chrono-, dromo- und inotropen Effekte am Herzen
(Yamada et al., 2002). Es zeigt sich eine regional unterschiedliche Verteilung des
Rezeptors mit einer verstärkten Expression im Bereich der Vorhöfe und dort mit
einer topographischen Assoziation zu GIRK-Kanälen. Die Expression anderer
muskarinerger Rezeptoren scheint sehr speziesspezifisch zu sein und im Herzen
großen lokalen Unterschieden zu unterliegen (Krejci und Tucek 2002; Wang et al.,
2001).
Ein weiterer kardial exprimierter Gi koppelnder GPCR ist der purinerge Adenosin
Rezeptor (A1). Burnstock klassifizierte die purinergen Rezeptoren in zwei Grup-
pen: P1 und P2 Rezeptoren (Burnstock, 1980). Dabei orientierte er sich an der
Affinität der Rezeptoren für Adenosin oder dessen Nukleotide (AMP,ADP,ATP).
P1 Rezeptoren haben die größte Affinität für Adenosin, die kleinste für ATP, die
P2 Rezeptoren verhalten sich entgegengesetzt. Man unterscheidet vier Adenosin-
rezeptoren, A1, A2a/b und A3-Rezeptoren. Die Kopplung an G-Proteine ist unter-
schiedlich. A1 koppelt an Gi/o, A2a/b an Gs und A3 Rezeptoren an Gi/o und Gq/11
(Fredholm et al., 2001).
Koronarer Blutfluss, Nocizeption, extrapyramidale Motorik und immunologische
Modulation sind nur ein paar physiologische und pathophysiologische Zusammen-
hänge, bei denen Adenosinrezeptoren von Relevanz sind.
Am Herzen ist der A1 Rezeptor die dominierende Klasse purinerger Rezeptoren.
A1 Rezeptoren vermitteln analog zum M2 Rezeptor in supraventrikulärem Gewebe,
durch eine Gi-vermittelte Aktivierung von einwärtsgleichrichtenden Kaliumkanälen,
einen negativ chronotropen und dromotropen Effekt, (Kurachi et al., 1986). Kli-
nisch wird diese Wirkung von Adenosin (Adrekar®) bei der Behandlung und Dia-

17Aktionspotential zu kommen und konsekutiv zu einem negativ inotropen Effekt
gnostik von supraventrikulären Tachykardien und AV-Knoten Reentrytachykardien
genutzt (Shen und Kurachi, 1995). A2a Rezeptoren und A3-Rezeptoren wurden im
Ventrikel verschiedener Spezies nachgewiesen (Fredholm et al., 2001, Marala et
al., 1998). A2a-Rezeptoren können mit ihrer Gs-Kopplung adrenerge, zum A1-
Rezeptor antagonistische Effekte vermitteln (Norton et al., 1999). Außerdem ha-
ben sie einen regulatorischen Einfluß auf den koronaren Blutfluss (Hein et al.,
1999).
Sowohl A1 als auch M2 Rezeptor aktivieren GIRK–Kanäle durch ßγ-Untereinheiten
von heterotrimeren G-Proteinen am Herzen.
Bereits seit 1921 ist bekannt, dass nach Stimulation des Nervus vagus, der da-
mals als „Vagusstoff“ bezeichnete Transmitter zu einer Bradykardie führt (Loewi,
1921). 1955 wiesen Hutter und Trautwein eine Steigerung des Kaliumeffluxes
nach vagaler Stimulation nach (Hutter und Trautwein, 1955). Der Mechanismus
blieb lange Zeit unklar, nicht zuletzt dadurch, dass βγ Untereinheiten von hetero-
tetrimeren G-Proteinen lange Zeit nur als Membrananker verstanden wurden und
ihnen keine weitere funktionale Bedeutung zugemessen wurde. Der essentielle
Zusammenhang zwischen GIRK-Kanalaktivierung durch βγ Untereinheiten wurde
erst 1987 durch Logothetis beschrieben (Logothetis et al., 1987).
Mit ihrer starken Einwärtsgleichrichtung und ihrer großen Leitfähigkeit negativ vom
Kaliumgleichgewichtspotential wirkt sich eine Aktivierung von GIRK-Kanälen je
nach kardialer Zielstruktur negativ chronotrop, dromotrop und inotrop aus. Die Ex-
pression von GIRK Kanälen ist in kardialem Gewebe relativ ubiquitär, zeigt jedoch
eine relativ höhere Expression im Vorhofgewebe und im supraventrikulären
Erregungsleitungssystem. In Knock-out Studien an Mäusen konnte gezeigt wer-
den, dass beide Untereinheiten, sowohl GIRK1 als auch GIRK4, für eine physiolo-
gische Frequenzmodulation essentiell sind (Stengel et al., 2000, Fisher et.al.,
2004, Bettahi et al., 2002). Es werden mehrere Signaltransduktionswege in Ab-
hängigkeit der kardialen Zielstruktur diskutiert. Im Bereich des supraventrikulären
Gewebes und im Reizleitungsgewebe erscheint eine direkte Signaltransduktion
und Hyperpolarisation über die Aktivierung von GIRK Kanälen über βγ
Untereinheiten von heteromultimeren G-Proteinen der Klasse Gi wahrscheinlich
(Yamada, 2002). Auf ventrikulärer Ebene scheint es durch den oben be-
schriebenen hyperpolarisierenden Effekt zu einer Verkürzung des

18
konsekutiv zu einem negativ inotropen Effekt durch eine veränderte Calcium Ho-
meostase (Dobrzynski et al., 2002).
Neben diesen „direkten“ Wirkungen von Acetylcholin existiert ein zweiter, an
cAMP gebundener Signalweg. Über die inhibitorische Wirkung der α-Untereinheit
des aktivierten G-Proteins kommt es zu einer Abnahme der intrazellulären cAMP
Konzentration. Damit antagonisiert dieser Signalweg die adrenerge Stimulation
(Brodde et al., 2001). Die Adenylatzyklase stellt somit einen Konvergenzpunkt der
parasympathischen und sympathischen Regulation dar.
1.4 Desensitisierung und Interferenzen des IK(ACh) Signalweges
A1 und M2 Rezeptoren konvergieren somit in gleichen Zielstrukturen über die glei-
che Signalkaskade auf das gleiche Zielprotein, den GIRK 1/4 Kanal. Es ist schon
länger bekannt, dass eine Interferenz zwischen diesen beiden Rezeptoren besteht
(Wellner-Kienitz et al., 2000). Des weiteren sind Interferenzen im Sinne einer hete-
rologen Desensitisierung, zwischen GPCRs an Kardiomyozyten bekannt. Für den
Azetylcholin induzierten Kaliumstrom (IK(ACh)) sind verschiedenste Mechanismen
einer Desensitisierung beschrieben.
Boyett zeigte 1987 erstmals an isolierten Sinusknoten von Kaninchen einen
schnellen Wirkverlust von Azetylcholin und damit erstmals eine Desensitisierung
für den damals noch unbekannten Signaltransduktionsmechanismus (Boyett und
Roberts, 1987).
Der Terminus Desensitisierung umfasst mehrere Mechanismen, die alle zu einer
Reduktion des Kaliumstroms führen, aber in Hinsicht auf zeitlichen Ablauf, Größe
und Mechanismus sehr unterschiedlich sind. Es kann zwischen homologer De-
sensitisierung und heterologer Desensitisierung unterschieden werden. Eine ho-
mologe Desensitisierung ist rezeptorspezifisch und nur aktivierte Rezeptoren und
Signalkomponenten werden deaktiviert. Unter heterologer Desensitisierung wird
eine verminderte Antwort auf einen Agonisten durch eine Aktivierung eines ande-
ren Rezeptorklasse verstanden. Insbesondere wenn zwei Signalwege konver-
gieren oder gleiche Effektormoleküle haben, kann es zur Deaktivierung von Re-
zeptoren oder Signalwegskomponenten kommen (Bünemann et al., 1999).
Für die homologe Desensitsierung von IK(ACh) auf Rezeptorebene werden zwei
Mechanismen diskutiert.

19
Im Bereich von Sekunden bis Minuten erfogt die Desensitisierung über G-Protein
gekoppelte Rezeptorkinasen (GRK´s), die durch Phosphorylierung des Rezeptors
zu deren Inaktivierung führen (Shui et al., 2002; Zang und Boyett, 1992; Büne-
mann und Horsey, 1999). Parallel zu diesem Phosphorylierungsprozess erfolgt die
Bindung an Arrestin. Arrestin verhindert die erneute Kopplung von G-Proteinen an
den GPRC und markiert den Rezeptor für die Endozytose, die über Stunden die
Rezeptordichte vermindert (Shui et al., 2001). Über diesen Mechanismus erfolgt
über die Phosphorylierung eine Desensitisierung über Minuten, über die Endozy-
tose über Stunden bis Tage.
Ein von der Desensitisierung zu unterscheidender Mechanismus ist eine Inhibition,
die auf einer Verringerung der Offenwahrscheinlichkeit der Kanäle beruht. Meyer
et al. konnten zeigen, dass über Gq gekoppelte Rezeptoren eine Inhibition des
IK(ACh) möglich ist. Gq koppelnde Rezeptoren, wie α1 oder ET1 Rezeptoren, füh-
ren über die Aktivierung der PLC zur Hydrolyse von PIP2, das als ein wichtiger
Kofaktor für die Aktivierbarkeit der GIRK-Kanäle bekannt ist (Meyer et al., 2001,
Kobrinsky et al., 2000).
Eine Besonderheit des voll aktivierten IK(ACh) ist eine initiale sehr schnell desensi-
tisierende Komponente des Stroms im Sekundenbereich. Diese schnelle Desensi-
tisierung wurde erstmals von Kurachi beschrieben (Kurachi et al., 1987). Da diese
desensitisierende Komponente unabhängig vom aktivierenden Rezeptor ist und
sogar über Gs koppelnde Rezeptoren auslösbar ist (Wellner-Kienitz et al., 2001
und 2003), wurde ein rezeptorunabhängiger Mechanismus der Signalkaskade an-
genommen. Die schnelle Desensitisierung scheint eine bestimmte Anzahl von
funktionellen Rezeptoren bzw. eine bestimmte Anzahl an aktivierten G-Proteinen
zu benötigen, um dieses Verhalten zu zeigen.
Bei Überexpression des endogen nur gering exprimierten A1 Rezeptors zeigt der
induzierbare Strom gleiche elektrophysiologische Eigenschaften, inklusive schnel-
ler Desensitisierung, wie bei Aktivierung über den endogenen in hoher Dichte
exprimierter M2 Rezeptor (Wellner-Kienitz et al., 2000).

Abb 5. Orginalaufzeichnung eines durch eine sättigende Konzentration von Azetylcholin
induzierten Kaliumstroms an einer Vorhofmyozyte. Es zeigt sich die charakteristische
schnelle, homologe Desensitisierung binnen von Sekunden (rot unterlegt)
Es wurden verschiedene Hypothesen zur Erklärung dieser akuten oder ultra-
schnellen Desensitisierung aufgestellt.
Chuang et al. beschreiben über ein kinetisches Modell, dass der Nukleotid-
austausch am G-Protein und die Hydrolyse des GTP zu dieser Art Desensitisie-
rung führen kann (Chunag et al., 1998; Leaney et al., 2004). In diesen Arbeiten
wurde daher RGS-Proteinen (regulator of G-protein signaling) eine Rolle bei dem
beschriebenen Desensitisierungsprozess zugeschrieben. RGS-Proteine interagie-
ren mit Gα Untereinheiten und beschleunigen den geschwindigkeitsbestimmenden
Schritt der GTP-Hydrolyse und damit die Inaktivierung des G-Proteins (Doupnik et
al., 1997; Herlitze et al., 1999; Hollinger und Hepler 2002). In eigenen Versuchs-
reihen konnte durch Überexpression von RGS Proteinen keine Zunahme der
schnellen Desensitisierung beobachtet werden, lediglich in der Auswaschphase
des Agonisten kam es zu einer beschleunigten Deaktivierung.
Auf Kanalebene konnte gezeigt werden, dass GIRK4 Homotetramere die schnelle
Desensitisierung nicht zeigen, was zu der Vermutung führte, dass die Kanalunter-
einheitskomposition bestimmend für die kinetischen Eigenschaften ist (Bender et
al., 2001).
20
Ein neuer Ansatz für die Erklärung der schnellen Desensitisierung geht von einer
subsarkolemmalen Kaliumakkumulation mit einer daraus folgenden Verminderung
der treibenden Kraft für den Kaliumefflux aus (Bender et al., 2004).
Zur Zeit existiert noch kein allgemein akzeptiertes Konzept für das Phänomen der
schnellen Desensitisierung. Die verschiedenen experimentellen Bedingungen (z.B.
Zelllinien versus Primärkultur) und die nicht eindeutige Nomenklatur in Bezug auf
die schnelle Desensitisierung, erschwert die Einordnung mancher Ergebnisse. So
wird z.B. die Desensitisierung innerhalb der ersten Minuten von einigen Autoren
als schnell bezeichnet. Die in dieser Arbeit verwendete Nomenklatur bezeichnet
mit „schnell“ die Desensitisierung binnen von Sekunden.
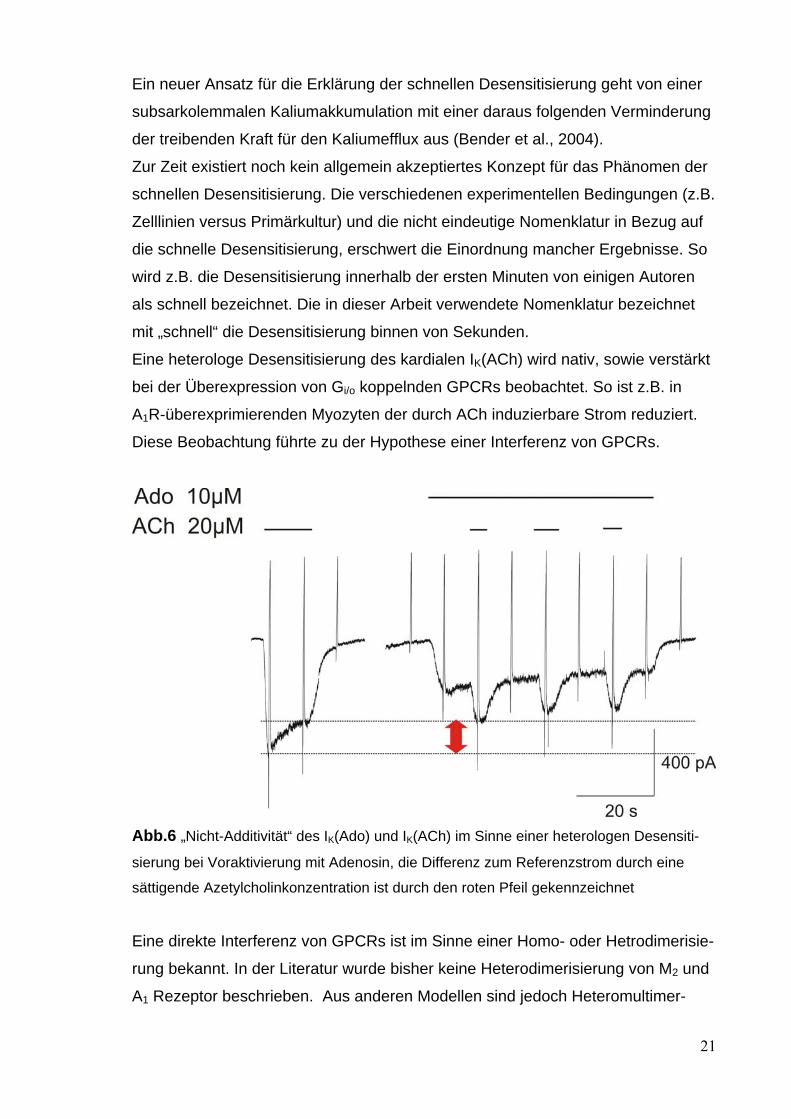
Eine heterologe Desensitisierung des kardialen IK(ACh) wird nativ, sowie verstärkt
bei der Überexpression von Gi/o koppelnden GPCRs beobachtet. So ist z.B. in
A1R-überexprimierenden Myozyten der durch ACh induzierbare Strom reduziert.
Diese Beobachtung führte zu der Hypothese einer Interferenz von GPCRs.
Abb.6 „Nicht-Additivität“ des IK(Ado) und IK(ACh) im Sinne einer heterologen Desensiti-
sierung bei Voraktivierung mit Adenosin, die Differenz zum Referenzstrom durch eine
sättigende Azetylcholinkonzentration ist durch den roten Pfeil gekennzeichnet
Eine direkte Interferenz von GPCRs ist im Sinne einer Homo- oder Hetrodimerisie-
rung bekannt. In der Literatur wurde bisher keine Heterodimerisierung von M2 und
A1 Rezeptor beschrieben. Aus anderen Modellen sind jedoch Heteromultimer-
21

22
bildungen von Rezeptoren bekannt, die die Kopplung eines Rezeptors an seinen
Signalweg verändern bzw. unterbrechen können (z.B. Barnes, 2006).
Eine weitere Hypothese, die Interaktionen zwischen GPCR zu erklären versucht,
beschäftigt sich mit dem Mechanismus der Kopplung des Rezeptors an die G-
Protein Signalkaskade. Es besteht die Vorstellung, dass GPCRs um G-Proteine
konkurieren könnten.
Diese Vorstellung wirft zusätzlich die Frage nach der Spezifität der Kopplung eines
Rezeptors an einen bestimmten G-Protein Pool und der räumlichen Organisation
auf. Für die Aktivierung von GIRK Kanälen durch GPCRs konnte bisher keine
hochspezifische Assoziation, der an der Signaltransduktion teilnehmenden Protei-
ne nachgewiesen werden. Die bisherigen Befunde legen eher eine gegenteilige
Interpretation nahe, nämlich einen relativ unspezifischen Signaltransduktionsme-
chanismus. Der C-Terminus der α Untereinheit von G-Proteinen wird als Interface
für die Kopplung an den Rezeptor diskutiert. Hier findet sich z.B. bei Gαi die ADP-
Ribosylierungsstelle durch Pertussistoxin (Cabrera-Vera et al., 2003). Antikörper
gegen diese C-terminale Region der α Untereinheit blockieren die Signaltransduk-
tion. In einem dominant negativen Versuchsansatz konnte mit Hilfe von Minige-
nen, durch Expression des funktionsdefizienten C-Terminus von Gαi eine Inhibie-
rung des IK(ACh) nachgewiesen werden (Gilchrist et al., 1999). Ähnliche Ergeb-
nisse wurden durch Leaney et al. berichtet. Chimeren aus Gi und Gs α-UE mit ei-
nem C-terminalen Gs-Anteil, konnten einen primär Gi koppelnden Signalweg in
einen Gs koppelnden Signalweg konvertieren (Leaney et al., 2000). Des weiteren
kann die Aktivierung der GIRK Kanäle durch verschiedene βγ -UE Kompositionen
erfolgen (Clapham und Neer; 1997, Wickman et al., 1994). Es konnte ebenfalls
gezeigt werden, dass keine feste Assoziation an βγ Untereinheiten von Gαi-
Untereinheit besteht. Gs koppelnde β oder H2-Rezeptoren führen z.B. zu einer Ak-
tivierung von GIRK-Kanälen (Wellner-Kienitz et al., 2001, Wellner-Kienitz et al.,
2003). Diese „Unspezifität“ spricht nicht nur für eine mögliche Interferenz von Re-
zeptoren, sie ist vielmehr Voraussetzung für ein mögliches Kompetitionsmodell.
Hinsichtlich der Interaktion von Rezeptoren und G-Protein werden zur Zeit zwei
Modelle diskutiert:
Das „collision model“ geht davon aus, dass Rezeptor und G-Protein frei in der Zell-
membran diffundieren können und bei Rezeptoraktivierung eine „zufälliges“ Zu-
sammentreffen von GPCR und G-Protein die Signalkaskade aktiviert. Geschwin-

23
digkeitsbestimmend in diesem Modell sind damit Rezeptor und G-Proteindichte in
der Membran.
Das Precoupling-Model hingegen geht davon aus, dass präformierte Komplexe
von Rezeptor, G-Protein und akzessorischen, regulativen Proteinen in der Plas-
mamembran vorliegen. In diesem Model ist der geschwindigkeitsbestimmende
Schritt die Kinetik der Aktivierung der Signalkaskade, z.B initial die GTPase Aktivi-
tät des G-Proteins.
Beide Modelle werden z.Z. noch diskutiert. Für das „precoupling Modell“ sprechen
die schon lange bekannten sogenannten „Caveolae“ in der Plasmamenbran. Das
intergrale Membranprotein Caveolin formt mit Cholesterinderivaten sogenannte
Mikrodomänen, die mit Signaltransduktionsmolekülen angereichert sind (Okamoto
et al., 1998). Es wurde z.B. ein „GIRK1-Signalkomplex“ beschrieben, in dem GIRK
1, G-Protein und G-Protein abhängige Rezeptorkinase kolokalisiert waren (Nikolov
und Nikolova 2004). Auf der anderen Seite führten Hein und Mitarbeiter kürzlich
den Nachweis in FRET-Experimenten, dass durch Erhöhung der Konzentration an
α Untereinheiten die Aktivierungskinetik verkürzt werden konnte, was für das „col-
lision coupling“ Modell sprechen würde (Hein et al., 2006). Feron et al. zeigten
eine agonisteninduzierte Translokation des M2 Rezeptors in caveolinreiche Plas-
mamembranabschnitte (Feron et al., 1997), dieser Befund würde unter Umstän-
den eine Mischung beider Konzepte darstellen.
1.5 Zusammenfassung und Diskussion der experimentellen Ergebnisse
GIRK Kanäle sind eine der wichtigsten Zielproteine der vagalen Innervation in kar-
dialem, supraventrikulären Gewebe. Die Aktivierung erfolgt über G-Protein akti-
vierende Rezeptoren und die konsekutive Aktivierung von Pertussis Toxin sensiti-
ven G-Proteinen der Klasse Gi/o. Als der stärkste physiologische Aktivator ist Aze-
tylcholin via den Azetylcholinrezeptor (M2) bekannt. Neben diesem klassischen
Mediator sind andere, wie Adenosin und Sphingolipide bekannt. Während Azetyl-
cholin als klassischer Neurotransmitter des Parasympathikus die vegetative Regu-
lation z.B. von kardialer Chrono- und Dromotropie vermittelt, wird Adenosin als
metabolisches Feedback nach Ischämie oder als Modulator bei starker sympathi-
scher Innervation verstanden. Adenosin und der zugehörige A1-Rezeptor haben
im Rahmen des Konzeptes der ischämischen Präkonditionierung an Interesse hin-

24
sichtlich klinischer Implikationen und einer möglichen Gentherapie gewonnen
(Dougherty et al., 1998).
Um mögliche klinische und gentherapeutische Ansätze für die Zukunft zu evaluie-
ren, ist die genaue Kenntnis der molekularen Abläufe der Aktivierung der Signal-
kaskade nötig, insbesondere wenn wie im Falle von M2- und A1-Rezeptor, eine
relativ kurze Konvergenz des Signalweges auf das Zielprotein, den GIRK-Kanal,
besteht.
Fragestellung der vorliegenden Arbeit ist die Spezifität der Signalwege in Hinblick
auf den Einfluss verschiedener Konstellationen von Rezeptorentypen und Expres-
sionsstärken von Rezeptoren auf den Endeffektor GIRK Kanal. Dabei soll auch ein
besonderes Augenmerk auf die induzierten Kaliumströme mit Hinblick auf Aktivie-
rungseigenschaften und Desensitisierung gerichtet werden.
Aus mehreren Vorarbeiten ist bekannt, dass GIRK-Ströme durch verschiedene G-
Protein gekoppelte Rezeptoren aktiviert werden können. Durch Überexpression
des Gi/o koppelnden A1 Rezeptor konnte eine Interferenz mit dem „nativen“ M2
Signalweg gezeigt werden, im Sinne einer verminderten Antwort auf Azetylcholin.
Ähnliche Ergebnisse wurden auch durch die Überexpression von nicht Gi/o kop-
pelnden Rezeptoren, z.B. ß-adrenergen und H2 -Rezeptoren beobachtet. Dies
führte zu der Vermutung, dass eine Überexpression zu einem Spezifitätsverlust
der Signalkaskade und zu einer Kompetition um freie G-Proteine führt (Wellner-
Kienitz et al., 2000 & 2003).
Der 5-HT1A Rezeptor für Serotonin ist ein in der Vorhofkardiomyozyte nicht expri-
mierter Rezeptor und koppelt vornehmlich an Gi/o. Durch seine Überexpression
konnten somit die vorherigen Ergebnisse bezüglich der Interaktion zweier nativer
Gi/o koppelnder Rezeptoren (A1 und M2 Rezeptor) mit einem nativ nicht expre-
mierten Rezeptor der gleichen Klasse reevaluiert werden.
Es zeigte sich erneut ein Zusammenhang zwischen Expressionsstärke des 5-HT1A
Rezeptors und Abnahme des maximal induzierbaren K+ Stroms durch Azetylcho-
lin. Morphologisch zeigte sich ein „desensitisierter“ Azetylcholinstrom, wie bei Ex-
position mit nicht sättigenden ACh Konzentrationen oder nach Antagonisten Expo-
sition. Diese Ergebnisse bestätigten zunächst die Hypothese der Kompetition der
beiden Rezeptoren um einen bestehenden G-Protein Pool und war deckungs-

25
gleich mit Vorexperimenten, bei denen pharmakologisch die M2 Rezeptordichte
vermindert worden war und eine Korrelation zwischen maximaler Stromdichte,
bzw. Aktivierungszeit und M2 Rezeptordichte postuliert worden war (Bünemann et
al., 1997).
In einem Antisense-Ansatz sollte die Hypothese geprüft werden, ob eine Vermin-
derung der M2 Rezeptorendichte einen Einfluss auf den durch Adenosin induzier-
ten K+ Strom hat. Es konnte bestätigt werden, dass eine verminderte M2-Rezeptor
Expression zu einer vergrößerten Adenosin Antwort führt. Zurückgreifend auf die
Hypothese einer Kompetition um einen für beide Rezeptoren fixen G-Protein-Pool,
wurde eine negative Korrelation zwischen den jeweils induzierbaren GIRK-
Strömen durch die konkurrierenden Rezeptoren vermutet. Überraschend zeigte
sich jedoch eine eindeutig positive Korrelation der K+ Ströme, sowohl in der Grup-
pe mit der artifiziellen 5-HT1A Rezeptor Überexpression, als auch im M2 Antisense
Ansatz. In der nativen Kontrollgruppe ergab sich keine Korrelation zwischen
IK(ACh) und IK (Ado). Eine pharmakologische Desensitisierung des M2 Rezeptors
durch Carbachol konnte die zuvor beobachtete positive Korrelation zwischen
IK(ACh) und IK(Ado) nicht zeigen, was ebenfalls ein Argument gegen die „einfache“
Kompetitionshypothese war.
Vielmehr deuten die Befunde auf einen neuen „cross-talk“ zwischen Gi/o koppeln-
den Rezeptoren hin, der von der Expressionsstärke der GPCRs abhängig ist. Die
Expressionsniveaus von M2, 5-HT1A und A1 Rezeptoren beeinflussen möglicher-
weise gegenseitig die mögliche maximale GIRK-Kanal Aktivierung. Es zeigte sich
weiterhin, dass ein reiner Kompetitionsmechanismus nicht ausreichend als Erklä-
rung zu sein scheint.
Ein weiterer Aspekt des maximal induzierbaren K+ Stroms ist die bei GIRK-
Kanälen bekannte Desensitisierung des Kaliumstromes. Während die Mechanis-
men der Langzeitdesensitisierung und „schnellen“ Desensitisierung im Minutenbe-
reich gut bekannt sind, stellt die ultraschnelle Desensitisierung von GIRK Strömen
weiterhin ein noch nicht geklärtes Phänomen dar. Phänomenologisch ist seit lan-
gem eine ca. 30%ige Desensitisierung des maximal aktivierten GIRK-Stromes im
Bereich von Sekunden bekannt. Diese Desensitisierung erfordert eine maximale
GIRK Kanalaktivierung und damit eine vorherige starke G-Proteinaktivierung durch
z.B. eine hohe Rezeptordichte. An der nativen Zelle zeigt sich diese Abhängigkeit
zwischen Anzahl aktivierter GPRCs und Desensitisierung am IK(ACh). Eine sätti-
gende Agonistenkonzentration induziert eine schnelle Desensitisierung, eine ge-

26
ringere Konzentration an ACh zeigt eine schwächere oder keine Desensitisierung.
Aus Voruntersuchungen ist bekannt, dass durch die Manipulation der Rezeptor-
dichte von nativ gering, oder gar nicht exprimierten Rezeptoren, ein morphologisch
gleicher GIRK-K+ Strom induziert werden kann. Die spezifische Kopplung des Re-
zeptors an eine G-Proteinklasse scheint dabei zweitrangig zu sein, auch Gq/11 und
Gs koppelnde Rezeptoren sind in der Lage, große GIRK-Ströme mit der charakte-
ristischen Desensitisierung zu induzieren. Dieser Befund legt einen rezeptorunab-
hängigen, bzw. einen nicht M2 - oder A1-Rezeptor exklusiven Mechanismus nahe.
Im Rahmen der Untersuchung der Interferenz von Gi/o koppelnden Rezeptoren
zeigte sich bei der adenoviralen Überexpression des A1 Rezeptors neben der zu-
vor bekannten Linksverschiebung der Dosiswirkungsbeziehung und der bekannten
charakteristischen Morphologie eines maximal aktivierten GIRK-Stroms, eine bis-
her noch nicht beobachtete Rebound-Aktivierung, direkt nach der schnellen De-
sensitisierung in der Auswaschphase des Agonisten.
Die beobachtete Desensitisierung war außerdem schneller und betraf einen pro-
zentual größeren Anteil des maximalen Stroms. In der weiteren Untersuchung die-
ses Phänomens konnte gezeigt werden, dass es weniger eine Reaktivierung des
IK(Ado) war, als vielmehr ein Wegfall einer adenosinabhängigen Hemmung.
In Kardiomyozyten, in denen durch GTP-γ-S oder eine sättigende Konzentration
von Azetylcholin ein Großteil der GIRK Kanäle voraktiviert war, zeigte sich eine
durch Adenosin induzierbare, konzentrationsabhängige Inhibition des GIRK Stro-
mes, damit erscheint die Inhibition G-Protein unabhänigig.
Bei der Überexpression von GIRK 4 Homomultimeren konnte die Adenosin emp-
findliche Inhibition nicht nachgewiesen werden.
Bei einer isolierten GIRK 4 Überexpression ist ein Verlust der schnellen Kompo-
nente der Desensitisierung aus Voruntersuchungen bereits bekannt (Bender et al.,
2001). Insgesamt sind diese Beobachtungen deckungsgleich mit bekannten Be-
obachtungen der schnellen Desensitisierung des nativen IK(ACh):
Auf der einen Seite die Konzentrationsabhängigkeit der Desensitisierung, und auf
der anderen Seiten das Fehlen der Desensitisierung bei Überexpression von
GIRK 4 Untereinheiten.
Durch eine M2 Rezeptor vermittelte GIRK Aktivierung mit einer hohen sättigenden
Konzentration von Azetylcholin konnte eine ähnliche, wenn auch kleinere Re-
boundaktivierung nachgewiesen werden, bei gleichzeitig ausgeprägter schneller
Desensitisierung, was nahe legen würde, dass den Phänomenen der schnellen

27
Desensitisierung und der G-Protein unabhängige Inhibition bei A1-Rezeptor Über-
expression ein ähnlicher Mechanismus zu grunde liegt.
In wieweit die „G-proteinunabhängige Inhibiton“ durch A1-Rezeptor Überexpressi-
on und die schnelle Desensitisierung des IK(ACh) eine nur koinzidentiell ähnliche
Morphologie und Eigenschaften haben, bleibt zu diskutieren. Zumindest ist aus
der Literatur eine ähnliche G-proteinunabhängige Inhibierung für den α2 Rezeptor
in Zusammenhang mit GIRK 1/2 Heteromultimeren beschrieben worden (Leaney
et al., 2004).
Es ist bisher nicht geklärt, ob dieses Phänomen auf Grund der geringen Expressi-
on des A1 Rezeptors an der nativen Kardiomyozyte eine physiologische Relevanz
bei Gesunden hat.
Zusammenfassend ist zu bemerken, dass die Manipulation des Expressionsni-
veaus Gi/o koppelnder Rezeptoren an Kardiomyozyten vielfältigen Einfluss auf Ak-
tivierung und Desensitisierung von GIRK Strömen hat. Es konnte gezeigt werden,
dass Gi/o-koppelnde GPCRs untereinander interferieren und sich gegenseitig bei
der GIRK Aktivierung beeinflussen. Des weiteren zeigt sich bei der Überexpressi-
on des A1 Rezeptors ein neuer G-Protein unabhängiger Signalweg, der Parallelen
zur schnellen Desensitisierung des IK(ACh) hat. Dieser Befund könnte ein Hinweis
auf eine direkte Protein-Protein Interaktion von GIRK-Kanal und Rezeptor sein.
Beide Befunde zeigen mögliche Protein-Protein Interaktionen, die durch Kompeti-
tionsmodelle alleine, wie „precoupling“, „collision coupling“ oder vorbekannte De-
sensitisierungsmechanismen, nicht erklärt werden können.
Ferner muss durch diese Ergebnisse in Frage gestellt werden, in wieweit pharma-
kologische oder gentherapeutische Manipulation nicht ähnliche Effekte auf die
Signaltransduktion von GPCR haben. Die Überexpression des A1 Rezeptors wird
z.B. als möglicher Therapieansatz im Rahmen der ischämischen Präkonditionie-
rung diskutiert (Murry et al., 1986), da eine solche Überexpression die gezeigten
dramatischen Veränderungen in der Signaltransduktion bedingen würde, ist es
letzlich fraglich, in wieweit dieser Therapieansatz überhaupt möglich ist.
















![Kapitel 7) HORMONE 1 [Kompatibilitätsmodus]biochemie-trainings-camp.de/stoff/h1/igandengesteurter_kanal.pdf · G-Protein-gekoppelter Kanal!! Dieses Protein liegt im inaktiven Zustand](https://img.pdfslide.org/doc/110x75/5e04a9c0d53a6263c03aa53b/kapitel-7-hormone-1-kompatibilittsmodusbiochemie-trainings-campdestoffh1igandengesteurterkanalpdf.jpg)











