Embed Size (px)
Citation preview

KOLLOIDMODELLE ZUR ILLUSTRATION BIOLOGISCHER VORGANGE
IV. DER KONZENTRATIONSEFFEKT BEI KOAZERVATKETTEN
yon H. G. BUNGENBERG DE JONG ul~d R. ~. WESTERKAMP (Arts dem biochemischen Institut der Universit~t Leiden)
Mit 6 Textfiguren
Eingegangen am 20. Juli 1936
1. Einleitung: Wahl des Koazervattypus
Die einfachsten Modelle ffir das Zustandekommen bioelektrischer Poten- r so die ,,01ketten" nach B e u t n e r und die Membranpotentiale nach M i c h a e l i s u. a. unterscheiden sich de facto durch den Zustand des zwischen den beiden Elektrolytl5sungen eingeschalteten KSrpers (einer nieht mit Wasser mischbaren organischen Fliissigkeit, bzw. einer starren Kolloidmembran).
Die an diesen Modellen anknfipfenden Theorien nehmen auBerdem jeweils verschiedene Vorg/inge zum Ausgangspunkt der Betrachtung, die Rolle der Verteilungsquotienten einerseits, die Diffusionsbehinderung der Ioncn infolge dcr Porenwandladung andererseits.
Die Verwendung von Koazervaten (kolloidreichen Fliissigkeiten) der Bio- kolloide fiberbriickt gewisserma6en bereits den erstgenannten Gegensatz. Ein Koazervat ist ja als eine mit dem Dispersionsmittel nicht mischbare Flfissigkeit, als ,,O1" im Sinne B e u t n e r s zu betrachten. Weiter kSnnte man ein Koazervat ebenfalls als ein von Poren durchsetztes System, als (fliissige) Membran auf- fassen, wenn mar~ in Betracht zieht, dab es nicht nur reich an Kolloidsubstanz~ sondern a,uch an Dispersionsmittel ist. Im Koazervat sind ja die unter sich frei bewegliehen kinetischen Einheiten der Biokolloide durch Dispersionsmittel getrennt. Es erschien daher wfinschenswcrt zu untersuchen; ob Koazervat- kettcn (0lketten, in denen ein Koazervat das 01 bildet)eioe E.M.K. aufweisen.
Diese Problemstelhmg erscheint in ihrer Formulierung neu. Es soll gleich hier hinzugeffigt werden, dab bei den weiteren beaehtenswerten Modellen fiir die Entstehung bioelektrischer StrSme : die sog. EiweiBketten (M o n d, D eu t s e h), der zwischengesehaltete ]YIembrankOrper mSglicherweise in einigen F/~llen die ~qatur eines wasserarmen Koazervats besaB 1).
Wie friiher berichtet, ist es auf verschiedene Weise m5glich eine Ko- azervation der Bi0kolloide zu erzielen. :Nach dem !V[eehanismus der zur Ko-.
z) ,,Gerbsaure Gelatine" ist vielleieht als ein sehr wasserarmes ,,einfaehes Koazervat" anzuspreehen; desgleichen handelt es sieh bei den zu einer Schieht zusammen zentrifu- gierten Niederschl~gen der Globulinfraktionen vielleieht um wasserarme Autokomplex- koazervate.

Kolloidmodelie zur Illustration biologischer Vorggnge. IV 33
aze rva t ion f i ihrenden Vorg/inge kann m a n 1) zwei H a u p t g r u p p e n unte rsche iden: die einfache und die Komplexkoaze rva t ion . Bei der e infachen K o a z e r v a t i o n spiel t die Ladung der Biokolloide keine ausschlaggebende Rolle , w/~hrend es gerade Ladungsgegens/~tze sind, die zur K o m p l e x k o a z e r v a t i o n fi ihren und die
' i r a en t s t andenen K o a z e r v a t e rha l ten bleiben. Die K o m p l e x k 0 a z e r v a t i o n seheint uns biologisch besonders bedeutungs-
voll, da es m6glich is t viele Analogien zwischen diesen K o a z e r v a t e n und der lebendigen Subs tanz aufzuzeigen. E in K o m p l e x k o a z e r v a t als ,,~1" in der t ( o a z e r v a t k e t t e zu benutzen war in te ressan t um zu erfahren, ob m a n mi t dessen Mithi lfe Modelle fiir die b ioelekt r isehen Po ten t i a l e verwirkl iehen kann. Von den zwei U n t e r g r u p p e n der K o m p l e x k o a z e r v a t i o n : K o m p l e x k o a z e r v a t i o n im engeren Sinn und Au tokompiexkoaze rva t ion , w/~hlten wir die erstere. Es war vorauszusehen, dab n~m]ieh bei Verwendung der A u t o k o m p l e x k o a z e r v a t i o n die Verh/i l tnisse weniger i ibersicht l ieh sein k6nnten . I n der vor l iegenden Unter - suchung beschr/s wi t uns ausschlieBlieh auf den sog. , ,Konzent ra t ions- e f fekt" , wozu wir das K o a z e r v a t im U-Rohr mi t L6sungen desselben Neut ra l - salzes verschiedener K o n z e n t r a t i o n t iberschichte ten.
2. Methodik
a) W a h l d e s b e n u t z t e n K o m p l e x k o a z e r v a t s
Die Komplexkoazervation positiver Gelatine mit negativem arabischen Gummi wurde friiher eingehend untersueht2). Jedoch eignet sich dieses System fiir den Aufbau yon Koazervatketten weniger, da es durch ziemlich geringe Salzkonzentrationen bereits aufgehoben wird (vOllige Aufhebung z. B. bereits mit 50 Milli~quiv. KC1). Wir w~hlten daher die I{ombination: positive Gelatine - - negative Nueleinsi~ure, die erst bei wesent- lich h5heren Konzentrationen durch ~eutralsalze (z. B. ungef~hr 200 Milli~quiv. KC1) auf- gehoben wird. ])as hier auftretende Komplexkoazervat ist weniger wasserreich als das erstgenannte, jedoch noch geniigend flfissig, um einen zusammenh~ngenden und die Glas- wand gut benetzenden Flfissigkeitsschicht in dem U-ROhrchen zu erhalt.en. Die Verwendung yon Gelatine als Kolloidkomponente in dem Komplexkoazervat bringt mit sich, dal~ zur Vermeidung yon Gelatinisierungserscheinungen die gesamte Untersuchung bei genfigend holier Temperatur (410 C) ausgefiihr$ werden mul~.
b) B e r e i t u n g d e r k o a z e r v i e r t e n S y s t e m e
Fiir die unter 3. und 4. aufgeffihrten Versuche werden die koazervierten Systeme durch Mischen yon 2 isohydrischen Stammsolen in verschiedenen Mengenverhi~ltnissen erhalten.
Die beiden Stammsole waren wie folgt l~ergestcllt: l p r o z . N u c i e i n s ~ u r e s o l . 4 g Na-Nucleinat aus I-Iefe (Merck) wird in 300 ccm
koehendem dest. Wasser gelSst und nach Abkfihlen bin 350 ccm aufgefiillt. An Hand yon pI-I-Titrationskurven wird dieses Sol durch Zufiigen yon Essigs~urelOsung und Wasser auf das gewiinschte pH und ein Endvolumen yon 400 ccm gebracht.
1) Vgl. Sammelreferat fiber Koazervation: H. G. B u n g e n b e r g de J o n g , diese Zeitschr. 15, 110 (1932).
3) It . G. ]3ungenberg de J o n g und W. A. L. D e k k e r , Kolloid Beihefte, 48, 143 (1935); 48, 213 (1936).
Protoplasma. XXVII 3

34 B u n g e n b e r g de J o n g und W e s t e r k a m p
2 p r o z . p o s i t i v e s G e l a t i n e s o l . Hochwertige Gelatine der Leim- and Gelatine- fabrik Delft in Delft (Holland) wird naeh L o e b gereinigt. 8 g dieses Produktes werden in dest. Wasser gelSst, auf ein Volumen yon 350 cem gebracht und wie oben ein End- volumen von 400 cem bei gewfinsehtem pH erhalten.
Die in Abschni t t 5. und 6. verwendeten koazervierten Systeme werden, wie sp~iter noch in diesen angegeben wird auf einfaehere Weise bereitet.
c) E i n f i i l l e n d e r U - R 6 h r c h e n
I n ein gl/isernes U-Rohr yon best immter Form (Fig. 1A) werden 20 ccm koazer- vierte Mischung der beiden Stammsole eingefiillt. Um das l~iedersinken und Zu-
sammenfliel]en der Koazervat-
th
A 8
Pig. 1
C
tropfen zu einer mSglichst ho- mogenen Schicht zu beschleu- nigen wurde d~s U-RShrchen in einen grol]en Zentrifugenbecher mi t Sand yon maximal 70 o C gebracht. Nach 10 Minuten Zentrifugieren bei 3000 Um- drehungen pro Minute ist die Temperatur auf ungef/ihr 250 C gesunken. Das Koazervat, das sich nunmehr in dem gebogenen Teil des U-Rohres gesammelt hat , ist dann, selbst wenn es relativ viel Gelatine enth/~lt, noch stets fltissig. ] )as U-RShr- chen blieb hemach noch un- gef~hr 12 Stunden in einem Thermosta ten yon 410 C.
Nur die l~6hrchen sind brauehbar, deren Koazervatschichten groB genug sind um die Bucht des U-Rohrs aufzufiillen und genfigend homogenisiert (durchsichtig).. Einge- schlossene kleine Vakuolen, dureh die die Koazervatschicht etwas getriibt erscheinen kann, schaden nicht.
Ohne das R6hrchen dem Thermostaten zu entnehmen, werden die fast klaren O[:er- schichten mi t dem in Fig. 1B abgebildeten Appara t abgehebert. Falls der Appara t gleich lange Kapillare d gleichen ])urchschnit ts hat , so ist man imstand bei Eintauchen der Ka- pillaren in ein und dieselbe L6sung durch Ansaugen an dem mi t einem Gummiscblauch mi t a verbundenen Mundstiick per Zeiteinheit gleiche Flfissigkeitsmengen in die beiden Behal ter b zu bef6rdern.
Da sowohl die Dimensionen der Kapillaren, s0wie die Durchmesser der beiden Schenkel des U-Robrs nur ann&hernd gleich sind, besteht dennoch beim Abhebern der Oberschichten mit diesem Appara t die M6glichkeit, dab die Menisken in den Schenkeln n icht gleich rasch sinken.
])er dadurch im U-Rohr verursachte hydrostatische Druckunterschied s t rebt nach Ausgleich, was zu einer Verschiebung der Koazerv~tschicht ffihrt, mi t der Gefahr, dieselbe aus der Bucht zu dr/~ngen, bzw. sie aufzuwirbeln. Aus diesem Grund m u ] der Schlauch zwischen a und Mundstfick lang genug sein, um gut beobaehten zu k6nnen und aui]erdem die beiden 9 em langen Schlauchstiicke bei c so beschaffen sein, d~B man bequem ab- quetschen und damit die beiderseitige Aufsauggeschwindigkeit willkiirlich regeln kann.

Kolloidmodelle zur I l lustrat ion biologiseher Vorg~nge. IV 35
Die letzten Reste der Oberschicht k6nnen mi t dem eben beschriebenen Appara t n ich t ent fernt werden; sie miissen mit Hilfe yon Watteb~usehen, die an besonderen Hal te rn befestigt sind, entfernt werden. Is t die gesamte Obersehieht entfernt, so befindet sich in dem U-Rohr allein noch die Koazervatsehicht , die als dieke , ,Membran" die beiden Sehenkel der U-R6hre voneinander scheidet. Nunmehr mfissen zu beiden Seiten der , ,Membran" die Salzl6sungen eingeffillt, werden, was mi t demselben Apparat , der zum Abhebern der Oberschichten gebraueht wurde, geschieht. Zu diesem Zweek werden die beiden oberen Beh~lter durch e m i t den ben6tigten Elektrolytl6sungen gef011t. Man laBt zun~chst aus den Kapil lalen erst vorsichtig die Salzl6sungen tropfenweise langs der U-Rohrwandung zufliel]en bis die kapillaren Teile des U-Rohres geftillt sind. 5Nun kann man, ohne eine Zerst6rung der Grenzschicht befiirchten zu mfissen, mi t gr613erer Ge- schwindigkeit einfiillen. Wenn man nun die beiden Salzl6sungen mi t umpolarisierbaren Elektroden ableitet, so erhal t man ein Element, dessen E .M.K. yon dem Konzentrat ions- untersehied der Elektrolytl6sungen, yon den Elektrolyten selbst und yon der Ar t der , ,Membran" abhhngt.
d) d i e M c s s u n g d e r E . M . K
Die Potentiale werden mit der tiblichen Poggendorfschen Aufstellung mit Hilfe eines Students Potentolneters und eines Spiegelgalvanometers von L e e d s und N o r t h r u p gemessen. Die Systeme waren ziemlich gut lei tend und das Galvanometer zur Messung gentigend empfindlich. Das Ableiten gesehah mi t ges~ttigt~n Kalomelelektroden und Agar- hebern, deren best immte Form aus Fig. 1 C ersiehtlieh ist. Sie bestehen aus den Glasteilen a und b, die mi t einem ziemlich dicken Gummischlaueh mite inander verbunden sind. Das System wurde mit einer 3proz. AgarlOsung in ges~ttigtem KC1 aufgefiillt. Der unters te Tell yon a muf~ gut in die Sehenkel tier verwendeten U-Rohre einffihrbar sein. Die Heber wurden w~hrend des Versuehes dutch einen Stf i tzapparat festgehalten. Sie waren, jeder fiir sich, dutch einen Meehanismus, der dem der Eintauchst~be eines Keleri- meters ahnlich ist, vert ikal b~weglich. Die Heberst i i tzpunkte waren welter um eine verti- kale Achse drehbar. Die Is waren mi t Rollen an einer Schiene derar t befestigt, da{~ sie leicht yon einem U-Rohr zmn n~tchsten gebracht werden konnten. Die U-Rohre, wie oben besehrieben hergestellt, h~ngen, dutch einen Hal ter fixiert, im Thermo- staten.
Die Heberschiene wurde so hoeh angebraeht , daI~ die Teile a der Heber beim hOchsten Stand nieht an die SalzlSsungen in dem U-Rohr heranreichen. Die Glasgloeken yon a werden mi t den Elektrolytl6sungen geftillt, in die sie getaucht werden sollen.
Nun werden unter genauer Beobachtung die beiden Heber durch Drehen so fief gestellt, bis die Glasgloeken vollkommen untergetaucht sind. Es muB dabei beaehte t werden, dal~ das fltissige Koazervat du tch hydrostatisehe Druckunterschiede in seiner Ruhestel lung nieht gestSrt wird. Die Teile b der Heber werden nun in GefAl]e mi t ge- sat t igter KC1-LSsung verbracht , in welche aueh die Mtindungsrohre der Kalomelelek~roden eintauchen.
I s t die Messung beendet, so kSnnen die Heber naeh kurzem Absptilen flit die nachste Messung gebraucht werden. E~ ist besser die Heber naeh der Messung schnell zu entfernen, da die MSgliehkeit besteht, dab sich du tch alas aus den Hebern diffun- dierende KC1 die ElektrolytlSsung verandert .
MiBt man das Potent ia l unmit te lbar naeh dem Herstellen des Elements, so ist in den meisten FAllen das Gleiehgewieht noch nicht erreicht. Letzteres geschieht ziemlich rasch (in etwa 5 Minuten ) und erhalt sich dann lange Zeit konstant . Stellt sich das Gleichgewicht nicht innerhalb 10 Minuten ein, so bleibt das Potent ia l inkons tan t ; steigend oder fallend, doch niemals abweehselnd.
3*

36 B u n g e n b e r g de J o n g und W e s t e r k a m p
Aus praktisehen Erw~gungen nahmen wir als definitives Potential dasjenige an, welches naeh 30 Minuten gemessen wurde. Unterdessen wurde kontrolliert, ob das Po- tential nach dieser Zeit konstant war. Falls dies nicht der Fall war, wurde es wie folgt angegeben: ~ bedeutet steigend, 4 fallend.
3 . D i e P o l u n g d e r K o a z e r v a t k e t t e u n d d a s Z e i c h e n d e r e l e k t r o p h o r e t i s c h e n
L a d u n g d e r K o a z e r v a t t r o p f e n
Es wurden bei drei verschiedenen p H - W e r t e n Mischungsreihen der un te r sich isohydr ischen S tammsole ( l p roz . Nucleins/~ure bzw. 2proz . Gelat inel vgl. oben) berei te t , die Gemische in U-Rohre eingeffillt , die Trennung der Schichten t en durch Zentr i fugieren beschleunigt und nach En t fe rnen der Oberschichten d ie E . M . K . der K o a z e r v a t k e t t e n :
0,01 n KC1 [ K o m p l e x k o a z e r v a t [ 0,1 n KC1 gemessen.
Das zur Hers te l lung der verschiedenen K o a z e r v a t e benu~zte Mischungs- verh/~ltnis der beiden isohydr ischen Sole, ausgedrf ickt in Volumprozen ten des
T a b e l l e I
Polung und U pH der beiden Mischungsverh~ltnisse E.M.K. der Elektrophoresegeschwindig- isohydrischen der beiden Sole in Ketten 1) in keit (in willkiirlich
Stammsole Vol.-% Gelatinesol Millivolt gew~hlte Einheiten)
3,50
3,75
80 70 60 50 40 30 20
82,5 80 75 70 50 30 20
80 75 70 60 40 20
- - i(?)
-- 5,8 -~- I,I
8,I -~ 11,5 -~ 14,5
15,2
- - 5 ,8
- - 5 ,5
- - 3 , 5
- - 0,5 Jr 12,5 -~- 16,7
17
- - 2
1,5 + 4,8 -~ 12 § 17 ~- 19,5
228 ~- 98,5
- - 108
~- 151
~- 67 • 0 - - 204 - - 295 - - 305
1) In dieser und folgenden Tabellen ist die Polung wie folgt angegeben: ~ bedeutet: die schw~ehere Salzl6sung ist jeweils positiv gegeniiber der konzentrierteren, - - bedeutet: die schw~ehere LSsung ist gegentiber der konzentrierteren negativ.

Kolloidmodelle zur Illustration biologischer Vorg~nge. IV 37
Gelatinesols, ist in Spalte 2 der Tabelle I angegeben. Spalte 3 gibt Aufschlu$ fiber die gemessene E.M.K. uud Spalte 4 fiber die Elektrophoresegeschwindig- keit 1) (in willkiirlich gew~thlten Mal~einheiten ausgedriiekt) der suspendierten Koazervattropfen.
Die Elektrophoresemessungen wurden mit Hilfe einer mit wasserdurch- str6mtem Blechgeh~use auf 41 0 C erw~rmten Kfivette durchgefiihrt. Die dabei sieh in apparativer Hinsicht ergebenden 20 Schwierigkeiten (Weichwerden der Kitt- re.v0# ~ ~ substanz und dadurch Durchbiegen der ~ - ~ ~ Kiivette was Undiehtwerden derselben zur +z NNkx ~ X~ ;=r Folge hatte) beschr/tnkten die Zahl der durehfiihrbaren Messungen. Aus diesem ~=s, so~ kk Grund konnte die Untersuehungsreihe bei 0 pH 4,0 nieht mehr durchgeffihrt werden. \ Diese, sp/~ter allerdings iiberwundenen ~ fi=s, z5
I Sehwierigkeiten, waren zu der Zeit als - z ! I
diese Arbeit entstand (1932) noeh nicht _ _ / i behoben. Wir konnten daher leider diese u ~ - ~ i seinerzeit geplanten systematischen Par- -zoo i alleluntersuchungen nicht weiter dureh- ~ I fiihren. -1oo ~ I ~
Die in Fig. 2 zusammengesteilten Ergebnisse sind : 1. Die in einer isohydrischen Mischungs-
reihe entstandenen Komplexkoazervate ~#~=3,75
zeigen im allgemeinen den ,,Konzen- PM 350
trationseffekt"; letzterer fehlt allein ~ z0 ~ ~0 00 100 bei e inem Misehungsverh/~ltnis. go/umzzozente &latinesol
2. Die Polung der Koazervatkette h/~ngt Fig. 2 mit dem Zeiehen der elektrophore- tischen Ladung des Komplexkoazervats zusammen. Falls das Koazervat negativ geladen ist, verh~Llt sieh die verdiinntere KC1-LSsung gegenfiber der konzentrierteren positiv. Ist das Koazervat dagegen positiv geladen, so trifft das Umgekehrte zu. Beim Umladungspunkt des Koazervates ist die E.M.K. der Kette null.
3. Die Gr6f~e der E.M.K. der Kette w~ehst in der Umgebung des Umladungs- punktes mit zunehmender elektrophoretischer Ladung der Koazervattropfen: Die drei oberen und die zwei unteren Kurven zeigen einen nahezu par- allelen Verlauf.
4. Die positiv geladenen Koazervate weisen bei gleieher e]ektrophoretischer Ladung gegeniiber den negativ geladenen einen niedrigeren Absolutwert der E.M.K. der Kette auf.
1) Ffir ausfiihrliche Einzelheiten der MeBmethode vgl. H. G. Bungenberg de Jong und P. H. Teunissen, Rec. Tray. Chim. ~4, 460 (1935).
38 Bungenberg de Jong und Westerkamp
Bezfiglieh 4. vermuten wir einen Zusammenhang mit dem Wassergehalt des Koazervats. Es sind die elektrophoretisch positiven Koazervate (d. h.
diejenigen, die reicher an Gelatine sind) auf- ~ : / / ! ~ fallend flfissiger und damit wasserreieher als
] die elektrophoretiseh negativen. m.
i 4. Abhgngigkeit der E.M.K. der Koazervat- kette yon den Konzentrationen der b e i d e n
J0 K C I - L i i s u n g e n
Die folgenden Versuche wurden mit einem Komplexkoazervat angestellt, das durch
2o Mischen von 3 Volumina 2proz. Gelatinesol (pH ~ 3,7) mit 7 Volumina 1 proz. Nuclein- s~uresol (pH ~ 3,7) erhalten war. Bei diesem
10 Misehungsverh/~ltnis zeigt die Koazervat- kette: 0,01 n KC1 [ Koazervat I 0,1 n KC1 eine E.M.K. yon 14,8 Millivolt.
Ffir den Fall, dal,~ man mit demselben 0 0,~ r r 2,o z,5 J,0 Koazervat die Ket te : 0,001 n KC11Koazervat [
/~ c1 0,01 n KC1 mil?t, erh/~lt man jedoeh mit Fig, 3 18 Millivolt einen h6heren Wert. Zur ein-
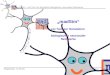
gehenderen Untersuehung dieses Befundes wurde eine Reihe yon Koazervatketten gemessen, wobei die Konzentration der einen KC1-L6sung stets 1 Milli/~quiv. betrug, wghrend die der anderen variiert wurde (vgl. Tab. II). Die Werte der E.M.K. dieser Ket ten sind in Fig. 3 als Funktionen der Logarithmen der KCl-Konzentrationsverh/~ltnisse aufgetragen.
T a b e l l e I I
Konzentrationsverhaltnis C~'C 1 der KC1-L6sungen log C2 E.M.K. in
�9 C1 Millivolt (C1 = 1 Milli~quiv. p. L.)
1 : 1 3 , 3 : 1
10 : 1
33 : 1
100 : 1 250 : 1
1000 : 1
0 -t- 0,5 0,52 @ 9,6 1,00 + 18 !,52 ~ 27,7 2,00 -4- 33,2 2,40 -~ 34,5 $ 3,00 -~ 37 $
Diese Kurve sollte ihren Anfang in dem Sehnittpunkt des Koordinaten- 0 2 systems nehmen, da bei symmetrischer Ableitung (log cl = 0) die E.M.K. der
Ket te null sein muB. Die in Wirklichkeit gefundene E.M.K. yon 0,5 Millivolt muB Versuehsfehlern zugeschrieben werden. Um diesen Betrag k6nnten also auch die fibrigen Werte der E.M.K. fehlerhaft variieren. Die erhaltene Kurve

Kolloidmodelle zur Illustration biologischer Vorg/inge. IV 39
zeigt zun/~chst ein geradliniges Stiick bis zu dem Wert : log c2 ---- 1,7, um von e 1
hier an merklich abzuweiehen. Innerhalb des Konzentrationsbereiches yon 1 bis 50 Milli~quiv. KC1 gilt
somit die Beziehung : E.M.K. = k �9 log c~. Es mii•te daher bei tJbersehich- cx
tung des Koazervats mit zwei KC1-L5sungen der Konzentrationen 3 und 30 Millii~quiv. dieselbe E.M.K. erhalten werden wie bei Verwendung zweier Kon- zentrationen von 1 und 10 Millii~quiv. KC1, was in der Tat durch einen Kon- trollversuch best~tigt werden konnte. Ebenfalls miii~te man die bei Verwen- dung yon Konzentrationen zu 10 und 100 Milli~quiv. KC1 zu erwartende E.M.K. berechnen kSnnen aus den E.M.K. der Ketten mit 1 und 100, bzw. 1 und l0 Milli/iquiv. KC1. Der in Wirklichkeit gefundene Wert yon 14,8 Milli- volt kommt der Differenz yon 33,2 und 18,0 Millivolt tats/~ehlich sehr nahe. Bei den hSchsten untersuchten KC1-Konzentrationen erreichten die E.M.K. der Kette keine konstanten Werte; sie nehmen mit der Zeit langsam und fortw/~hrend ab (angegeben mit +). Es treten nunmehr aber aueh an der Grenzfl/tche Ko- azervat :konz. KC1-LSsung makroskopiseh siehtbare ~nderungen auf. So hellt sich das sonst etwas trtibe Koazervat auf, w/ihrend sieh gleichzeitig die scharfe Trennungsfl/tche verwiseht.
Da aber eine 1 n KC1-LSsung das Komplexkoazervat bereits vSllig auf- hebt, so diirften die beobachteten Anderungen an der Trennungsfl/~ehe auf der aufhebenden Wirkung der starken KC1-Konzentration beruhen. Kleinere Kon- zentrationen (z. B. 100 und 250 Milli/tquiv.) haben noch keine vSllige Aufhebung zur Folge. Die Koazervate werden jedoch in ihrem inneren Zustand bereits merklich beeinflu6t, was sieh u. a. dureh Zunahme ihres Wassergehaltes zeigt.
Aus diesem Grund iiberraseht es nicht, wenn die Proportionalit/~t zwi-
schen E.M.K. und log c2 fiir hShere Konzentrationen ihre Giiltigkeit nieht C 1
beibeh/~lt.
5. Ionenvalenz und Konzentrationseffekt
Da fiir die in diesem und dem folgenden Abschnitt beschriebenen Ver- suche stets das gleiche negativ geladene Komplexkoazervat bei gleichem pI-I und gleiehem Mischungsverh/~ltnis der beiden Kolloide benutzt werden sollte, wurde yon nun an folgende Vereinfachung seiner Darstellung befolgt:
11,2 g Na-Nucleinat in dest. Wasser gelSst und 1050 ecru aufgefiillt und 9,6 g Gelatine analog in 450 ccm wurden miteinander gemischt. Diese 1,5 1 Fltissigkeitsmenge wurde in 5 Portionen ~ 300 eem im Eissehrank aufbewahrt. Fiir jede Versuehsreihe wurde 1 Portion yon 300 ccm erw/~rmt und mit 20 ccm 0,5 n Essigs/~ure versetzt. Mit dem so erhaltenen koazervierten System konnten 16 U-RShren besehickt werden. Mit mehr als 16 U-R5hren gleichzeitig zu arbeiten liel~ unsere Methodik nieht zu.
Die eben gegebene Vorschrift fiihrt zu dem gleiehen Verh/iltnis Gelatine : Nucleinat wie es fiir die Untersuchung des Konzentrationseffektes yon KC1 im vorhergehenden Abschnitt zur Anwendung kam. Das pH der Oberschichten
40 Bungenberg de Jong und Wes te rkamp
in den U-RThren betrug nach Stehen fiber Naeht im Thermostaten 3,80 und s t immt somit mit dem Wert des vordem verwendeten isohydrischen Sols (pH = 3,75) praktisch fiberein. Es handelt sich also im folgenden stets um ein negativ geladenes Komplexkoazervat und damit zusammenh/~ngend wird
50
re.Volt
o,,s
r / / /
,i
! \ ! \
[ ,~-1
~,o f,5 ;,o ~5
Fig. 4
3,o
bei der Untersuchung des Konzentrations- effektes die verdfinntere SalzlSsung gegen- fiber der konzentrierten positiv gefunden.
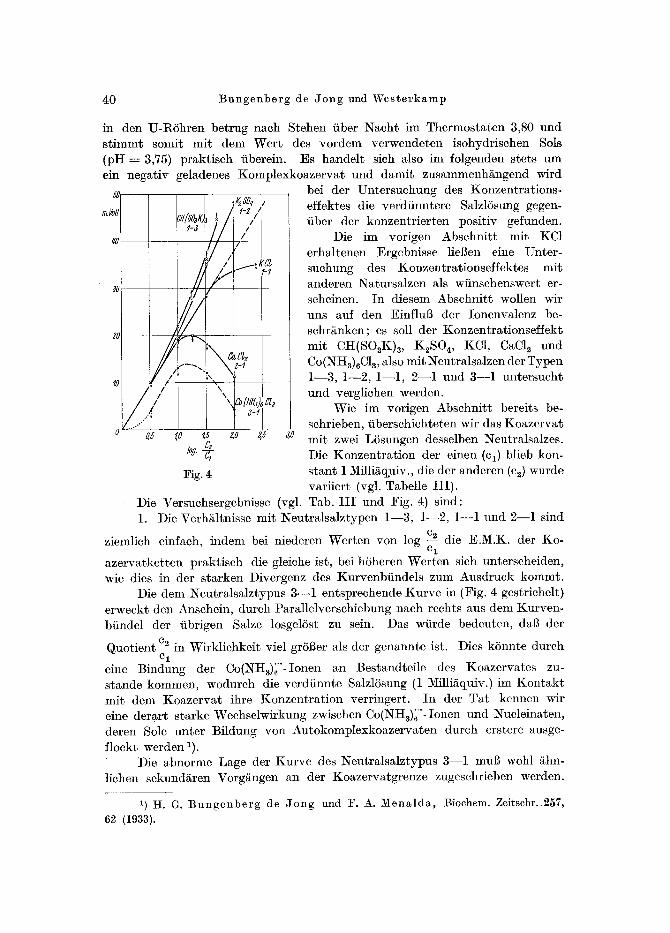
Die im vorigen Abschnitt mit KCt erhaltenen Ergebnisse lieBen eine Unter- suchung des Konzentrationseffektes mit anderen Natursalzen als wfinschenswert er- seheincn. In diesem Abschnitt wollen wir uns auf den EinfluB der Ionenvalenz be- schr/s es soll der Konzentrationseffekt mit CH(SOsK)a, K2SO 4, KC1, CaCls und Co(NH3)6C13, also mit Neutralsalzen der Typen 1--3, 1--2, l - - l , 2--1 und 3--1 untersucht und vergliehen werden.
Wie im vorigen Abschnitt bereits be- schrieben, fiberschichteten wir das Koazervat mit zwei LTsungen desselben Neutralsalzes. Die Konzentration der einen (el) blieb kon- stant 1 Milli/~%uiv., die der anderen (c2) wurde variiert (vgl. Tabelle I I I ) .
Die Versuehsergebnisse (vgl. Tab. I I I und Fig. 4) sind: 1. Die Verh/iltnisse mit ~eutralsalztypen 1--3, 1--2, 1--1 und 2--1 sind
ziemlieh einfaeh, indem bei niederen Werten yon log c~ die E.M.K. der Ko- cl
azervatketten praktisch die gleiehe ist, bei hTheren Werten sich unterscheiden, wie dies in der starken Divergenz des Kurvenbfindels zum Ausdruck kommt.
Die dem Neutralsalztypus 3--1 entsprechende Kurve in (Fig. 4 gestrichelt) erweckt den Anschein, durch Parallelverschiebung naeh rechts aus dem Kurven- bfindel der fibrigen Salze losgelSst zu sein. Das wfirde bedeuten, da[3 der
Quotient % in Wirklichkeit viel grSfter als der genannte ist. Dies kTnnte durch C 1
eine Bindung der Co(~qtt3)~"-Ionen an Bestandteile des Koazervates zu- stande kommen, wodurch die verdiinnte SalzlTsung (1 Milli~quiv.) im Kontak t mit dem Koazervat ihre Konzentrat ion verringert. In der Tat kennen wir cine derart starke Weehselwirkung zwisehen Co(NH~)~"-Ionen und Nueleinaten, deren Sole unter Bildung yon Autokomplexkoazervaten durch erstere ausge- floekt werden 1).
Die abnorme Lage der Kurve des ~eutralsalztypus 3--1 mul~ wohl /~hn- lichen sekund/~ren Vorg/ingen an der Koazervatgrenze zugesehrieben werden.
1) H. G. Bungenberg de Jong und F. A. Menalda, Biochem. Zeitschr. 257, 62 (1933).
Kolloidmodelle zur Illustration biologischer Vorg~nge. IV 41
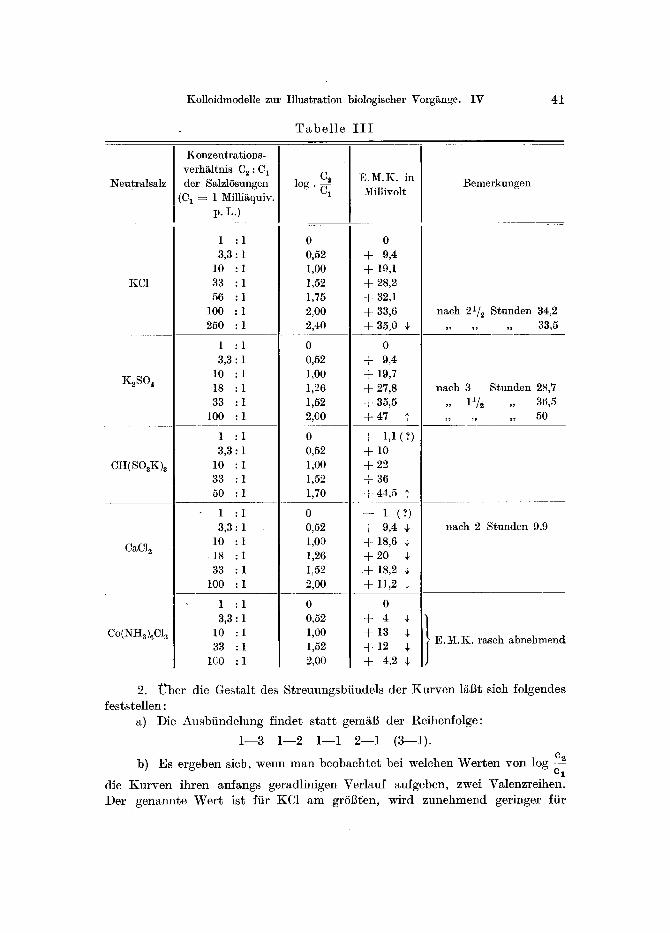
T a b e l l e I I I
Neutrals~lz
Konzentrations- verh~ltnis C 2 : C1 I
I der SalzlSsungen I [ (C~ = 1 Milliaquiv. I
C2 log. E.M.K. in Millivolt
Bemerkungen
KC1
K~SO4
CH(SOaK)2
CaC12
Co(NHa )6Cla
1 :1 3,3 : 1
10 :1 33 : l 56 : l
100 : 1 250 : 1
1 :1 3,3:1
10 :1 18 :1 33 : 1
100 : 1
1 :1 3,3 : 1
10 :1 33 :1 50 :1
1 : ] 3,3 : 1
10 :1 18 :1 33 : 1
100 : 1
1 :1 3,3:1
10 :1 33 :1
100 : 1
0 0,52 1,00 1,52 1,75 2,00 2,40
0 0,52 1,00 1,26 1,52 2,00
O 0,52 1,00 1,52 1,70
0 0,52 1,00 1,26 1:52 2,00
0 0,52
0 + 9,4 + 19,1 q- 28,2 + 32,1 + 33,6 + 35~0 4,
0 q- 9,4 q- 19,7 + 27,8 + 35,5 q- 47
+ 1,1(?) § 10
22 + 36 + 44,5
- - 1 (?) + 9 , 4 r 4- 18,6 4 + 20 4, + 18,2 4, +11 , 2
0 -~- 4 4.
nach 21/2 Stunden 34,2 . . . . . . 33,5
nach 3 Stunden 28,7 ,, 11/2 ,, 36,5 . . . . . . 50
nach 2 Stunden 9,9
1,00 1,52 2,00
+ 13 4, + 12 4' + 4,24.
E. M. K. rasch abnehmend
2. U b e r die Ges ta l t des S t r e u u n g s b i i n d e l s der K u r v e n ls sich fo lgendes fes t s te l l en :
a) Die A u s b f i n d e l u n g f inder s t a r t gem~l] der Re ihenfo lge :
1 - - 3 1 - - 2 1 - - 1 2 - - 1 (3 - -1 ) .
b) Es e rgeben sich, w e n n m a n b e o b a c h t e t bei we lchen W e r t e n y o n log c-~2 e l
die K u r v e n i h r e n an fangs ge rad l in igen Ver lauf aufgeben , zwei Va lenz re ihen . Der g e n a n n t e W e r t is t fiir KC1 a m grSl~t~en, wi rd z u n e h m e n d ger inger fi ir

42 Bungenberg de Jong und Westerkalnp
K2SO 4 und CH(SOsK)a , aber ebenso zunehmend geringer ffir CaC12 und Co(NHs)6C1 a. Demnach bestehen nachfolgende Beziehungen:
1--3 < 1--2 < 1--1, 3--1 < 2--1 < 1--1.
c) Wird ein konstanter Endwert der E.M.K. nicht erreicht, so /~ndert sich dieser mit der Zeit in derselben Richtung, nach der die Abbiegung der Kurve yon dem geradlinigen Verlauf erfolgt.
Das merkwfirdigste Ergebnis der vorliegenden Versuche ist wohl, dab der Konzentrationseffekt im Prinzip bei gegebenem Koazervat und gegebenem ptI yon der Wahl des Neutralsalzes wenig abh/~ngig zu sein scheint. Man ver- gleiche die praktisch gleichen Werte der E.M.K. fiir KC1, K2SO 4 und CaC12
bei c2 = 3,33. Man mug bier jedoch mit Nachdruek darauf hinweisen, dab C 1
keine Neutralsalzkonzentrationen benutzt werden diirfen, die merkliche Ande- rungen des urspriinglichen inneren Zustandes des Komplexkoazervates zustande bringen.
Treten bei h6heren Konzentrationen Abweichungen yon der einfachen C 2 Beziehung: E.M.K. = k �9 log cl auf, so wcisen die in 2a und 2b angefiihrten
Regelm/~Bigkeiten in dieser Abweichung deutlich auf J~nderungen des ursprfing- lichen Zustandes des Komplexkoazervates an der Grenze: Koazervat - - h6her konzentrierte Salzl6sung bin.
Die sog. fortlaufende Valenzreihe: (Relative Negativierung) 1--3, 1--2, 1--1, 2--1, 3--1 (Relative Positi-
vierung) t r i t t bei der Beeinflussung der elektrophoretischen Ladung der Komplex- koazervattropfen durch Neutralsalze auf2).
Andererseits gelten ffir die Abschw~chung der gegcnseitigen Anziehung der beiden Kolloide im Komplexkoazervat dutch Neutralsalze, die bei ge- ntigendem AusmaB bis zur vSlligen Aufhebung des Koazervats ffihren kann, zwei gesonderte Valenzreihenl):
1--3 > 1--2 > 1--1 (Valenzreihe der Anionen), 3--1 > 2--1 > 1--1 (Valenzreihe der Kationen).
6. Die spezifischen Einfliisse der einwertigen Kationen und Anionen
Mit dem gleichen im vorhergehenden Abschnitt beschriebenen Koazerv~t und glcicher Methodik verglichen wir in Versuchsreihen die Konzentrations- effekte einiger NeutrMsalze des Typus 1--1 miteinander (vgl. Tab. IV, V und VI).
Es zeigte sich, dal~ KC1, KBr und K J (Tab. IV) nahezu den gleichen Konzentrationseffekt geben. Dies gilt jedoch nicht ffir KC1, NaC1 und LiC1 (Tab. V und Fig. 5), deren Kurven bereits in ihrem geradlinigen Tell die Reihen- folge Li, Na, K zeigcn. Bei dem Vergleich yon KC1, KNOa und KCNS (Tab. VI
und Fig. 6) sind die, bei den niedrigeren Werten von log c2 erhaltenen E.M.K. e 1
nicht s eh r befriedigend, was wir auf Versuchsfehler zurfickffihren m6chten.
1) H. G. Bungenberg de Joug und W. A. L. Dekker , ~. a. O.
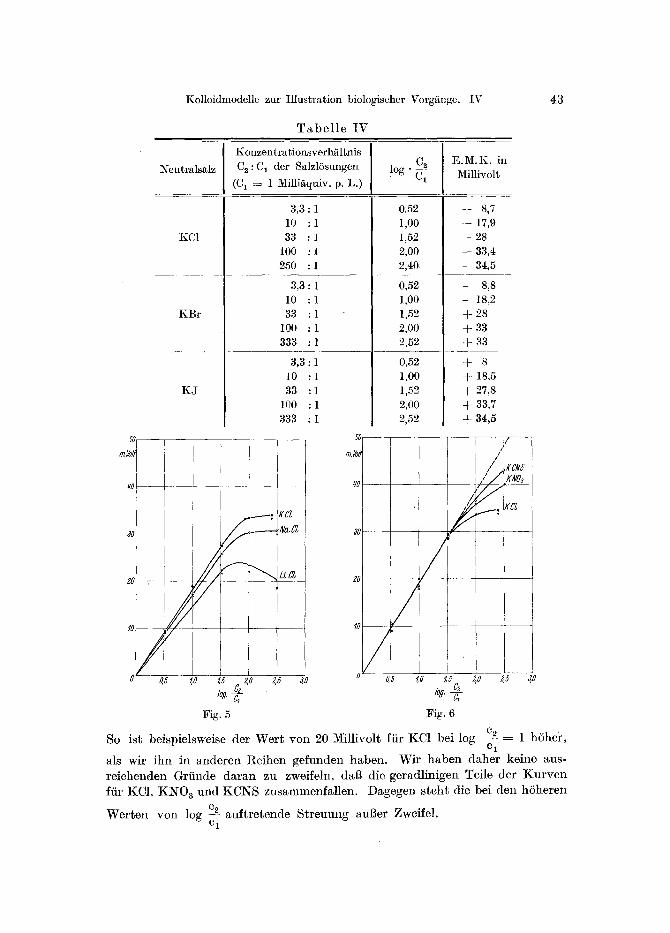
Kolloidmodelle zur Illustration biologischer Vorg~nge. IV 43
T a b e l l e IV
j r ~ _ _
q8
30
23
Konzentrationsverh~ltnis Neutralsalz C~:C1 der SalzlSsungen log" C~ E.M.K. in
(C z = 1 Millii~quiv. p.L.) . C1 Millivolt
KC1
KBr
KJ
/
o,5 q,o
3,3:1 10 :1 33 : 1
100 : 1
250 : 1
3,3:1 10 :1 33 : 1
100 : 1
333 : 1
3,3 : 1 10 :1 33 : 1
100 : 1
333 : 1
0,52 1 ,00
1,52 2,00 2,40
0,52 1,00 1,52
2,00 2,52
0,52 1,00 1,52 2,00 2,52
5~
m.Vo/
?
I _ _ _ _ L _ _
/
-~- 8,7 ~- 17,9 + 28 + 33,4 @ 34,5
+ 8,8 _a 18,2 + 28 @ 33 -+ 33
4- 8 zc 18,5 -c 27,8 ~- 33,7 -~ 34,5
i I i I / /(s
, F,o__~
gcz
/ !
I
0 - - r 2,o 2,5 s,o o,5 r ~,5g~ 2,8 log. ~ I+ G
Fig. 5 Fig. 6
So ist beispielsweise der Wer t von 20 Millivolt fiir KC1 bei log
g5 3,0
cA = 1 h6hei ~, 0 1
als wir ihn in anderen Reihen gefunden haben. Wir haben daher keine aus- reichenden Grtinde daran zu zweifeln, da6 die geradlinigen Teile der K u r v e n fiir KCI, KNO 3 und KCNS zusammenfal len. Dagegen s teht die bei den hSheren
Wer ten yon log cA auf t re tende S t r e u u n g au6er Zweifel. C 1
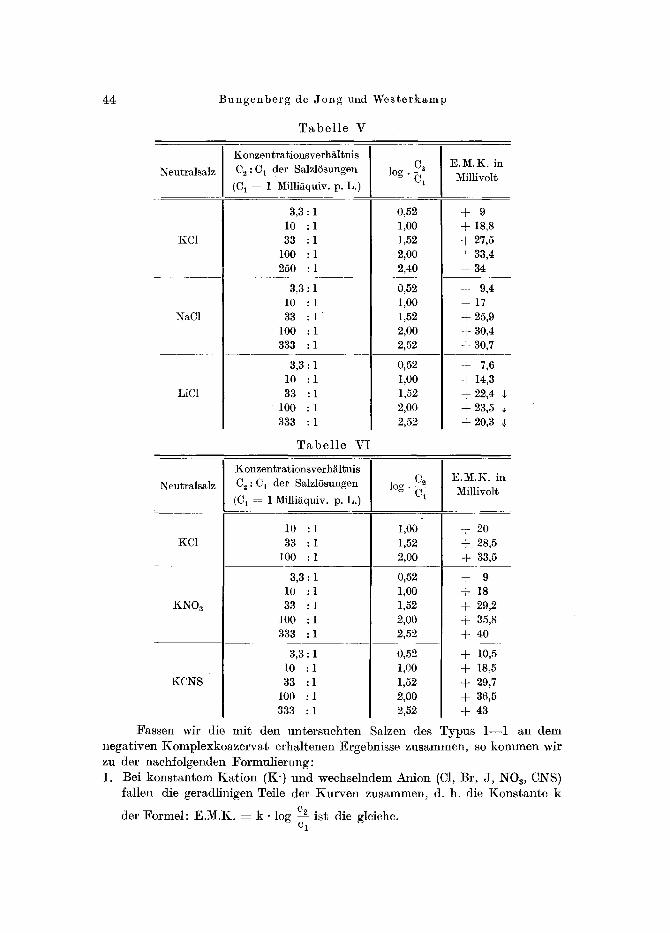
44 B u n g e n b e r g de J o n g und W e s t e r k a m p
T a b e l l e V
Neutralsalz
KC1
NaC1
LiC1
KonzentrationsverhMtnis C~ : C1 der Salzl6sungen
(C1 = 1 Millii~quiv. p. L.)
3,3 : 1 I0 :1 33 : 1
100 : 1 250 : 1
3,3:1 10 : 1 33 : 1
100 : 1 333 : 1
3 ,3 :1 10 :1 33 :1
100 : l 333 : 1
C~ log �9 C~
0,52 1,00 1,52 2,00 2,40
0,52 1,00 1,52 2,00 2,52
0,52 1,00 1,52 2,00 2,52
E .M.K. in Millivolt
+ 9 + 18,8 + 27,5 + 33,4 + 34
-~- 9,4 + 17 + 25,9 + 30,4 + 30,7
+ 7,6 -I 14,3 + 22,4 3̀ + 23,5 3, + 20,3 3̀
T a b e l l e V I
Konzentr~tionsverh/Htnis NeutrMsalz C 2 : C 1 der SalzlSsungen log �9 C2 E.M.K. in
(C 1 = 1 Milli/~quiv. p .L.) ~ Millivolt
10 : 1 1~00 + 20 KC1 33 : 1 ],52 + 28,5
100 : 1 2,00 + 33,5
KN03
KCNS
3,3:1 10 :1 33 : 1
100 : 1 333 : 1
3,3 : 1 10 :1 33 : 1
100 : 1
0,52 1,00 1,52 2,00 2,52
0,52 1,00 1,52 2,00
+ 9 + 18 + 29,2 + 35,8 + 40
+ 10,5 + 18,5 + 29,7 + 36,5
333 : 1 2,52 + 43
F a s s e n wir die m i t d e n u n t e r s u c h t e n lalzen des T y p u s 1 - - 1 an d e m
n e g a t i v e n K o m p l e x k o a z e r v a t e r h a l t e n e n Ergebn i s se z u s u m m e n , so k o m m e n wi r zu der n a c h f o l g e n d e n F o r m u l i e r u n g :
1. Be i k o n s t a n t e m K a t i o n (K ' ) u n d w e c h s e l n d e m A n i o n (C1, Br , J , NOa, CNS) fa l len die ge rad l in igen Tei le de r K u r v e n z u s a m m e n , d. h. die K o n s t a n t e k
der F o r m e l : E . M . K . z k �9 log c2 i s t die gleiche. C 1

Kolloidmodelle zur Illustration biologischer Vorg~nge. IV 45
2. Bei konstantem Anion (C1) und wechselndem Kat ion (Li, Na, K) ist ftir die geradlinigen Teile der Kurven die Konstante k der Formel : E.M.K. =
C 2 k �9 log ~ jedesmal eine andere.
3. Bei den Werten yon log cA, die eine Abweichung yon der Formel: E.M.K. e 1
= k �9 log cA ergeben, tritt , entsprechend der Stellung der Ionen in der ]yo- cl
tropen I~eihe: Li, Na, K und C1, NO3, CNS, Ausbiindelung ein. Die unter 3. genannten Ionenfolgen werden durch neuere, z. T. noch
nicht ver6ffentliehten Untersuchungen versts Es handelt sich bier um die Reihenfolgen der NeutrMsalze hinsichtlich der Umladungskonzentrationen yon Biokolloiden. So ergibt sich aus den Messungen yon P. H. T e u n i s s e n 1) ftir Na-Nucleinat, dal~ die Umladungskonzentration in der Reihenfolge: LiC1, NaC1, KC1 gr6ger wird. Fiir positive Gelatine land L. v a n Z i j p in diesem Laboratorium (noeh nieht ver6ffentlieht), dab die Umladungskonzentrat ion in der Reihenfolge KCNS, NN03, KC1 zunimmt.
Bei der Einwirkung eines NeutrMsalzes auf den Ladungszustand eines Komplexkoazervates wirken nun aber dessen beide Ionen getrennt, und zwar die Kationen auf den negativ, die Anionen auf den positiv geladenen Kolloid- komponenten.
Hi~lt man also das Anion bei variierendem Nation konstant, so wird bei gleicher Salzkonzentration die positive Ladung der Gelatinekomponente ent- spreehend erniedrigt , die negative Ladung der Nueleins/~urekomponente aber zunehmend in der Reihenfolge K, Na, Li erniedrigt.
Das hat zur Folge, dag die negative elektrophoretische Ladung des Komp]exkoazervats (die den Charakter einer algebrMschen Summe hat) bei gleicher Salzkonzentration in der Reihenfolge KC1, NaC1, LiC1 abnimmt.
Wo die E.M.K. der Noazervatkette , und damit bereits jeder der beiden Einzelpotentialspriinge: Koazervat--Salzl6sung mit der GrSge der elektro- phoretischen Ladung des Koazervats (Absehnitt 3) parallel geht, mug der Potentialsprung an der Grenze Koazervat - - h6her konzentrierte SMzl6sung ebenfMls in der Reihenfolge: I(C1, NaC1, LiC1 abnehmen. Dementsprechend wird auch die E.M.K. der Koazervatket te in derselben Reihenfo]ge abnehmen ~ miissen, was in der Tat in der Ausbiindelung der Kurve yon Fig. 5 ersicht- lich ist.
Analog 1/~gt sich aus der Anordnung: KC1, KN03 und KCNS fiir zu- nehmende Umladungskonzentrationen der positiven Gelatine die in Fig. 6 zu beobaehtende Reihenfolge : KCNS, KN03, KC1 ableiten. Es ergibt sich daraus, dab bei gleieher SMzkonzentration die negative elektrophoretisehe Ladung des Komplexkoazervats in der Reihenfolge: KC1, KNOa, KCNS zunehmen mug2). Aus diesem Grund ist die in Fig. 6 auftretende Ausbiindelung der Kurven auch zu erwarten.
1) Vgl. P. H. Teunis.sen, Dissertation, Leiden, 1936. 3) Die im Text erw/~hnten Ladungsab- und -zunahmen sind nieht auf den ursprting-
lichen Zustand bezogen, sondern bezeiehnen bei einer gegebenen NeutrMsalzkonzentration

46 B u n g e n b e r g de J o n g und W e s t e r k a m p
7. Einige Messungen an Iietten: 0,1 n Salzl~sung !Koazervatl 0,01 n SalzlSsung
Neben den oben besehriebenen Ke t ten sind in Doppelversuchen auch noch einige Xe t t en hergestellt worden, in denen das Koazerva t einerseits, 0,01 n, andererseits mit 0,1 n NeutralsalzlSsung iiberschichtet wurde.
Nach der iiblichen Wartezei t yon 30 Minuten erhielten wir folgende Mittel- werte der E.M.K. in Millivolt:
K2S04 -- -4- 28,3 KCNS ~-- d- 17,8 KC1 ~ -4- 14,8 LiC1 ~ d- 10,1 CaCl~ -- 6,4. Es ist beaehtenswert , daft diese gefundenen Werte fiir die E.M.K. denen
nahekommen, die erhalten werden, wenn man den Wert E.M.K. fiir log c~ cl
---- 1 von dem fiir log c2 _-- 2 (die zusamnlengehSrigen Wer te kSnnen jeweils C 1
der vorangegangenen Tabelle en tnommen werden) subtrahiert . Die sieh hier ergebende u m g e k e h r t e P o l u n g der Koazerva tke t te mit
CaC]~ bildet eine Stiitze ftir die yon uns gegebene Auslegung der Kurvenaus- biindelung.
Der Einzelpotentialsprung (~): SalzlSsung-Koazervat wird best immt dureh: a) d i e Konzent ra t ion der SalzlSsung, und b) den elektrophoretischen Ladungszus tand der Koazervatgrenze. Bei zunehmendem a wird ~ entsprechend einer logarithmischen Formel
immer zunehmen miissen. Dies gilt jedoch nur so lange, als a auf b praktiseh keinen Einflul~ hat.
I n dem vorliegenden Fall eines negat iven Komplexkoazervates fiihrt nun aber CaC12 zu einer Positivierung, d. h. zu einer Abnahme der negativen elektro- phoretischen Ladung.
Es ist also durchaus zu erwarten, dal~ der Einzelpotentialsprung: CaC12- LSsung - - Koazerva t bei steigender CaCl~-Konzentration zuerst zunimmt, sehlieftlieh ein Maximum erreicht um naehher wieder abzunehmen. (Fiir den Fall, dalt der Umladungspunkt des Koazervats mit CaC12 erreicht werden kSnnte, wgre , sogar = 0 und wiirde bei noch hSheren Konzentra t ionen das umgekehrte Zeiehen annehmen.)
Aus dieser ~Tber]egung folgt, dab es eine Reihe von jeweils zwei zu- sammengehSrenden CaC12-Konzentrationen gibt, ftir die , jedesmal denselben Wer t hat. Wiirde man sieh mit je einem solchen Paar CaC12-LSsungen Ko- azervatket ten herstellen, so wiirden die E.M.K. ~ 0 sein. Dieser Fall wiirde u. a., entsprechend unseren Ergebnissen fiir eine Ke t t e : 0,004 n CaCI 2 K o -
die Folge der LadungsgrSl3e. In den FAllen yon Neutralsalzen des Typus 1--1, deren Kurvenabbiegungen von dem urspriinglieh gemdlinigen Verlauf dem Sinn nach gleich sind, bedeutet dies, dal~, bezogen auf den urspriinglichen Ladungszustand, immer I~ositivierung auftritt. Letztere nimmt in der l~eihenfolge: KCNS, KN03, KC1, NaC1 zu. Bei den Neu- trulsalzen des Typus 2--1 und 3--1 ist die Positivierung wesentlich stgrker, wghrend Neutralsalze des Typus 1--2 und 1--3, bezogen auf den ursprtinglichen Ladungszust~nd, eine Negativierung bewirken. Dies zeigt sieh auf Grund der Tatsache, dab der Sinn der Xurvenabbiegung, demjenigen der Neutralsalztypen 1--1, 2--1 und 3--1 gerade entgsgen- gesetzt ist (vgl. Fig. 4).

Kolloidmodelle zur Illustration biologischer Vorg~nge. IV 47
e 2 azervat I 0,1 n CaCI~ zutreffen. (Der Punkt ftir log ~ ----- 0,60 der aufsteigenden
% CaCl~-Kurve liegt in Fig. 4 ebenso hoch wit der ffir log Cl .-- 2,0 auf dem
absteigen4en Tell derselben. Die entsprechenden Absolutwerte von c 2 sind daher, da c 1 = 1 Milli/~quiv., 4 und 100.)
Auf Grund des Sachverhaltes erhellt, dab dig Ket te : 0,01 n CaC12 IKoazervat 0,1 n CaCl~ gegenfiber den beiden folgenden Ket ten: 0,001 CaC12 IKoazervat 0,1 n CaCl~ und 0,001 n CaC12 tKoazervat[ 0,01 n CaClz eine ent- gegengesetzte Polung aufweisen muB.
8. Diskussion der Versuchsergebnisse
Zuniichst zeigen die Versuchsergebnisse, dab bei Ketten, in denen ein Komplexkoazervat zwei LSsungen ein und desselben Neutra]sMzes verschiedener Konzentrat ion trennt, in der Tat ein Konzentrationseffekt auftritt . Obwohl die hSchsten der yon uns beobachteten E.M.K. nur ein Drittel des theoretisch m6glichen MaximMwertes erreichen (bei einem Konzentrationsverhi~ltnis der KC1-LSsungen yon 1 : 10 finden wir nur 18--19 Millivolt), so erscheint uns die genannte Feststellung doch biologisch bedeutungsvoll. Besonders auch deshalb, da die stoffliche Zusammensetzung dieser mir Wasser nicht mischbaren , ,Membranen" den Grenzschichten der lebenden Substanz weit n/~her steht, Ms die in den be- kanntesten einfachen Modellen gebr/~uchlichen ,,Membranen" (z. B. organischen Fliissigkeiten wie SMicylaldehyd, bei de~l 01ketten oder Cellulosennitrat bei den starren Membranen).
In dieser Hinsicht besteht zwischen den Koazervatket ten und den sog. ,,EiweiBketten" (Mond, D e u t s c h ) ein verwandtschaftlicher Zusammenhang. Letztere, wie die hier studierten Koazervatket ten bilden auch insofern ein mit den Protololasmamembranen der lebenden Substanz besser vergleichbares Modell, da sie gleichfMls im Prinzip keine invarianten Systeme darstellen.
Die Koazervatket ten sind stark abh/~ngig vom pH und erleiden bei ge- wissen Abs01utwerten der NeutrMsMzkonzentrationen Anderungen ihres inneren Zustandes.
Die durch nicht zu hohe Salzkonzentrationen herbeigeffihrten _4nderungen ffihren noch zu definierten Zust/~nden des Koazervats und damit zu definierten E.M.K. Bei noch hSheren Konzentrationen ist dies jedoch nicht mehr der Fall. Die definierte Koazervatgrenze geht verloren und die E.M.K. ~ndern sich im Lauf der Zeit st/~ndig.
Wir wollen uns nun noch mit der Frage nach dem Mechanismus des Zustandkommens der PotentiMsloriinge an der Koazervatgrenze besch/~ftigen. Jede Theorie sol] natfirlich bef/~higt sein, den auffallenden Zusammenhang zwischen Konzentrationseffekt und elektrophoretischem Ladungszustand des Koazervats zu erkl~ren. Die Tatsachen, die ffir diesen Zusammenhang direkt (1. u. 2.) oder indirekt (3. u. 4.) sprechen, seien nochmals kurz aufgeffihrt: 1. t)olung der Koazervatket te und elektrophoretisches Zeichen des Komplex-
koazervats (Abschnitt 3).

48 Bungenberg de Jong und Westerk~mp
2. Ansteigen des Absolutwertes der E.M.K. der Koazervatkette mit steigendem Absolutwert der elektrophoretischen Ladung des Koazervats (Abschnitt 3).
3. Auftreten der ,,fortlaufenden Valenzreihe": 1--3, 1--2, l - - l , 2--1, 3--1
in der Kurvenausbiindeluug bei hSheren Werten des Ausdrucks log e~ e 1
(Abschnitt 5). 4. Desgleichen Auftreten der geihenfolgen: Li, Na, K bzw. C1, NO3, CNS
bei Neutralsalzen vom Typus 1--1 {Absehnitt 6). Fiir die Erkl/~rung des Konzentrationseffektes scheint eben durch diesen
Zusammenhang die Auffassung yon M i e h a e l i s selbstverstgndlich: FaBt man das Koazervat als fltissige Membran auf, so ist fiir das Anion, fulls die Membran- looren negativ, fiir das Kation, fulls die Membranporen positiv geladen sind, eine Diffusionserschwerung zu erwarten.
Es ergibt sich dann, dug die verdiinntere Salzl6sung einer konzentrierteren gegentiber loositiv sein mug, falls das Koazervat negativ geladen ist und dab bei positiv geladenem Koazervat gerade entgegengesetzte Verh~ltnisse bestehen. Diese Folgerung steht in der Tat im Einklang mit unseren Versuchsergebnissen. Von diesem Gedankengang ausgehend, k6nnen auch die restliehen drei, oben gen~nnten Punkte qualitativ erkl/~rt werden.
Es ist dies um so beachtenswerter, d a bei unserer Membran der ver- wiekeltere Full vorliegt, dab sie aus eifiem feinsten Mosaik zweier verschiedener Membranbestandteile mit entgegengesetzten Ladungen, den positiv und negativ geladenen Kolloidteilchen besteht. Dennoch bestimmt das Vorzeichen ihrer algebraischen Summe (als Zeiehen der elektrophoretischen Ladung) im Sinne yon M i c h a e l i s die Polung der Koazervatkette.
Was nun die quantitative Seite anbetrifft, wiirde ebenfalls l)berein- stimmung mit der Theorie bestehen, falls gem/tg der bekannten Formel:
u - - v RT u - k v F lg c2, die E.M.K. bei einem gegebenen Neutralsalz und
el
gegebener Membran pr0portional log e~ w/~re. el
In der Tat trifft innerhalb eines gr6geren Bereiehes der Konzentrationen bei Neutralsalzen vom Typus 1--1 diese Proportionalitgt zu (Abschnitt 4 und 6).
Die bei h6heren Konzentrationen auftretenden Abweichungen bedeuten, da hierbei eine Anderung des urspriinglichen Koazervats stattfindet, keine Einschr~nkung bzgl. unserer Auffassung der genannten Proportionalit/~t. Eine
e~ Koazervat/~nderung finder bereits bei kleinen Werten yon - - s tat t fulls die
el Kationen- bzw. Anionenvalenz steigt, sodag die Beziehung innerhalb eines weir kleineren Bereiehes Giiltigkeit besitzt und experimentell bei den Neutral- .salzen vom Typus 3--1, 2--1 und 1--3 tiberhaupt nicht mehr erfagt werden :konnte.
Die yon M i e h a e l i s angegebene Theorie fiir das Auftreten des Konzen- trationseffektes ist demnach imstand viele unserer Ergebnisse zu erkl/~ren.
Demgegeniiber sei festgestellt, dab aber aueh vom Standpunkt der :Phasengrenzpotentiale ( B e u t n e r) die Ergebnisse ebenfalls erkl/~rt werden kSnnen.

Kolloidmodelle zur Illustration biologischer Vorg/~nge. IV 49
Hierzu ist allgemein nStig klarzustellen, da$ das elektrophoretisch negative Koazervat als ,,saures 0l" , das ungeladene Koazervat als ,,neutrales 01" und das elektrophoretisch positive Koazervat als ,,basisches ()l" betrachtet werden kann.
Das in den letzten Jahren fortgeschrittene eingehende Studium der Komplexkoazervation 1) erm6glicht es, diese schematiseh als eine doppelte Um- setzung zweier Kolloidelektrolyte z. B.
Gelat" C1- -~- Na" A r a b - --> [Gelat ' A r a b - ] ~- Na 'C1- aufzufassen, wobei sich die beiden entgegengesetzt geladenen Kolloidionen, zu- sammen mit Wasser, als entmischte Schieht abseheiden. Obwohl das Koazervat bei ~quivalenter Mischung der beiden Kolloide die formelle Zusammensetzung eines Kolloid-Kolloidsalzes hat, darf man dieses doch nieht mit einem, bei einer gewShnlichen doppelten Umsetzung sich ausscheidenden ,,unl6slichen Salz" identifizieren. Es handelt sich hier um eine Phase, in der die beiden Kolloid- ionen selbst, neben Wasser als frei vorhanden vorzustellen sind. Diese Auf- fassung wird noch dadurch unterstiitzt, dalt es gelungen ist, vSllig analoge Entmisehungen yon KristalloidlSsungen aufzufinden 3).
Das in der gegebenen Formulierung flit unsere Betrachtung besonders wiehtige ist, dal] die Gegenionen selbst im Prinzip nieht mit in das Koazervat gehen, sondern sich in der w/isserigen Sehicht anh/s
Das bei ~tquivalenter Mischung entstandene Koazervat ist elektrophoretiseh ungeladen und kann als ,,neutrales 01" betrachtet werden. Bei anderen Mischungsverh/s nimmt dieses neutrale Koazervat noeh gewisse Mengen des fiberschfissig vorhandenen Kolloidelektrolyten auf, wobei das Koazervat das Ladungszeichen des entsprechenden Kolloidions erh/~lt. Die in einer iso- hydrischen Mischungsreihe auftretenden drei Typen yon Komplexkoazervaten k6nnen somit folgendermal~en schematisiert werden:
[Gelat" A r a b - -~ Na' A r a b - ] elektrophoretiseh negativ; ,,sautes (~1" [ Gelat" Arab - ] ,, neutral; ,,neutrales ()1" [ Gelat" A r a b - ~- Gelat" C1-] ,, positiv; ,,basisches 01".
Eben dieser aufgenommene Kolloidelektrolyt ist es, der im Sinn der B e u t n e r schen "~berlegungen das Auftreten des Konzentrationseffektes er- mSglicht. Von den drei Ionenarten, die ein elektrophoretiseh geladenes Komplex- koazervat also im Prinzip enth/ilt, sind die beiden Kolloidionen 4) sehwierig, das Kristalloidion dagegen dureh gleichnamige Kristalloidionen leicht auswechselbar. Dies wird an der Trennungsfl/~che des Koazervats in den Koazervatket ten
1) H.G.Bungenberg de Jong und W. A. L.Dekker , Kolloidbeihefte 48, 213 (1936). 2) H. R. K r u y t und H. G. Bungenberg de Jong. Prec. Royal Aead. Amster-
dam 38, 714 (1935). 8) Bei den ersten Versuchen mit den hier beschriebenen Koazervatketten heberten
wir die 0berschichten ab, stellten durch Aufl6sen yon festem KC1 zwei verschiedene Konzentrationen her und iiberschichteten das Koazervat neuerdings ,nit diesen Obersehichten. Die folgenden Messungen ergaben ftir die E.M.K. nur geringe Werte. Dies ist v611ig verst/~ndlieh, da ja die Obersehichten bereits Neutralsalz, das bei der ,,doppelten Um- setzung" entstanden war, enthielten.
1) In unserem Fall lese man in den oben gegebenen Schemata Nuelein- anstatt Arab--. Protoplasma. XXVII 4

50 Bungenberg de Jong und Wes te rkamp
stattfinden und damit die fiir den Konzentrationseffekt n6tige Vorbedingung erftillt sein. Die kurze prinzipielle Erkl/~rung yore Standpunkt der Phasen- grenzpotentiale aus m6ge geniigen, da die entspreehende Erkl/~rung samtlicher Versuchsergebnisse verst/~ndlieherweise zu welt fiihren wtirde.
Endlieh sei noch bemerkt, dab der ziemlieh niedere Absolutwert der E.M.K. unserer Koazervatket ten wohl mit deren relativem Wasserreiehtum zusammenh~tngt, woftir wir in Abschnitt 3 (vgl. bier unter 4) einen ttinweis fanden. Die welt h6here E.M.K. gebenden ()lketten bzw. stark eingetrockneten Kolloidmembranen von M i e h a e l i s sind gleichfalls wasserarme Gebilde.
Somit ist zu erwarten, dal3, falls sie nur geniigend wasserarm sind, aueh Komplexkoazervate gleichfalls h6here E.M.K. beim Konzentrationseffekt zeigen. Der hSehste bisher gemesscne Konzentrationseffekt fiir eine Ket te aus zwei KC1-LSsungen aus einem Konzentrationsverh~tltnis 1 : l0 und einem aus positivem Clupein und negativem Lecithin hergestellten wasserarmen Koazervat betrug 32 Millivolt. Wasserarme Koazervate bereiten jedoeh in experimenteller Hinsieht erhebliche Sehwierigkeiten, besonders was deren Homogenisierung zu einer kompakten, die beiden Salzl6sungen wirklieh trennenden und dabei die Glaswandung gut benetzenden Sehieht betrifft. Sie eignen sieh daher nicht zu systematischen Untersuchungen, um die analogen Gesetzm/il~igkeiten der in dieser Arbeit behandelten Fglle zu studieren.
Zusammenfassung
1. Ketten, bei denen sich zwischen zwei L5sungen des gleichen Neutralsalzes, jedoch verschiedener Konzentrat ion (z. B. 0,01 u. 0,1 n KC1) ein aus positiver Gelatine und negativer Nucleins/~ure bestehendes ](omlolex- koazervat befindet, zeigen im allgemeinen einen Konzentrationseffekt.
2. Die Polung der Koazervatket te h~ngt mit dem Zeichen der elektro- phoretischen Ladung des=Komplexkoazervats zusammen. Falls das Ko- azervat negativ geladen ist, verhMt sich die verdiinntere KC1-L6sung gegeniiber der konzentriertereu positiv. Is t das Koazervat dagegen positiv geladen, so trifft das Umgekehrte zu. Beim Umladungspunkt des Ko- azervats ist die E.M.K. der Ket te null.
3. Die GrSl3e der E.M.K. der Ket te w/~chst in der Umgebung des Umladungs- lounktes mit zunehmender elektrophoretischer Ladung der Koazervat- tropfen.
4. Um den Konzentrationseffekt mit KC1, sowie mit weiteren Neutralsalzen eingehender zu untersuehen, besehr/tnken wir uns auf Ketten, die stets dasselbe negetiv geladene Komplexkoazervat als Bestandteil enthielten und in denen die niedrigere Konzentration (el) der Salzl6sungen 1 Milli/~quiv. be- trug. Nit Hilfe dieser Anordnung wurde gelunden:
5. Innerhalb des Konzentrationsbereiehes von 1--50 Milli~quiv. KC1 (fiir %)
gilt die Beziehung : E.M.K. = k �9 log e 2 Bei hSheren Konzentrationen el
ftir e~ nimmt die E.M.K. um weniger zu als der Formel entsprieht. 6. Beim Vergleieh des Konzentrationseffektes yon CH(SOaK)~, K2S04, KC1,
CaCI.~ und Co(NH~)~C1 a ergab sieh, dab die E.M.K. bei niederen Werten

Kolloidmodelle zur Illustration biologischer Vorg/~nge. IV 51
des Ausdrucks log c2 fiir die vier erstgenannten Salze nahezu gleich ist cl
und somit der Faktor k der logarithmischen Formel nahezu denselben Wert hat. Das abweichende VerhaIten yon Co(NH3)BC1 a mug wohl auf eine stSrende Nebenreaktion (Fiillung) an der Koazervatgrenze zuriickge- ffihrt werden.
7. Der Giiltigkeitsbereich der logarithmischen Formel verkleinert sich um so mehr, je hSher bei konstant gehaltenem monovalenten Anion die Valenz des Kations ist bzw. je hSher bei konstant gehaltenem monovalenten Kation die Valenz des Anions ist. Es besteht also folgende Reihe:
1--1 > 1--2 > 1--3 und 1--1 > 2--1 (> 3--1). 8. Die Neutralsalztypen ordnen sich nach Richtung und GrSBe der Ab-
weichungen der logarithmischen Formel bei hSheren Werten des Ausdrucks
c2 in der nachstehenden Reihenfolge: e 1
1 - - 3 1 - - 2 1 1 2 - - 1 (3 - -1 ) . 9. Bei vergleichendem Studium des Konzentrationseffektes einiger Neutral-
salze yore Typus 1--1 ergab sich: a) In dem fiir die logarithmische Formel gfiltigen Konzentrationsbereich
sind die Werte k ffir KC1, KBr. K J, KNOa, KCNS einander praktiscL gleich, ffir KC1, NaC1 und LiC1 wenig voneinander verschieden.
e 2 b) Die bei hSheren Werten yon - - zu beobachtende Abweichung ver- C 1
grSBert sich in der Reihenfolge: KCNS, KNOa, KC1, NaC1, LiC1. 10. Aus den Versuchsergebnissen glauben wit schlieBen zu diirfen, dab der
Potentialsprung : NeutralsalzlSsung-Komplexkoazervat a) yon der Konzentration der SalzlSsung, b) von dem elektrophoretischen Ladungszustand des Koazervats abh/s Die einfache unter 5. dieser Zusammenfassung angegebene Formel hat also nut innerhalb eines Konzentrationsbereiches, bei dem a noch keine merkliche Xnderung yon b herbeifiihrt, Gf i l t igke i t . Sobald aber eine Beeinflussung yon a auf b stattfindet, kSnnen die uns hierffir bekannten Gesetzmi~l~igkeiten eine Erkl~rung abgeben ffir die unter 7 , 8. und 9. dieser Zusammenfassung aufgefiihrten I~eihenfolgen.
11. Zu dem Mechanismus des Zustandekommens der Potentialspriinge an der Koazervatgrenze erscheint uns auf Grund unserer Ausffihrungen die yon M i c h a e l i s gegebene Erkli~rung ffir das Auftreten des Konzentrations- effektes (Diffusionserschwerung ffir das mit der Membranpore gleichartig geladene Ion) gangbar.
12. Die Ergebnisse scheinen uns aber auch yore Standpunkt der Phasengrenz- potentiale nach B e u t n e r erkl/~rbar zu sein. In diesem Zusammenhang wird erl/~utert, dab das elektrophoretisch negative Koazervat als ,,sautes O1 , d a s ungeladene Koazervat als ,,neutrales C)I" und das elektrophoretisch positive Koazervat als ,,basisches ()l betrachtet werden kann.
4*