Embed Size (px)
Citation preview

Fallvorstellung 5 Süd Tagesklinik
Medizinische Klinik Abteilung IILunchseminar 06.11.2007

Patient M. F., �, 44 Jahre
12/05Vorstellung zur Einholung einer Zweitmeinung (bislang in Stuttgart in hämatologischer Betreuung)

Patient M. F., �, 44 Jahre
Symptome:- seit Monaten abnehmende Leistungsfähigkeit, Belastungsdyspnoe-5/05 fieberhafter Infekt:Blutuntersuchung beim Hausarzt: auffallende Panzytopenie (Leukos1900/µl, Hb 4,9 g/dl (!), Thrombos7000/µl (!)). Normales Diff.-Blutbild.

�auswärtige hämatologische Abklärung:
KM-Punktion 25.05.: Punctio siccaKM-Zytologie (30.05.05):„Hyperzelluläres KM, Megakaryozyten nachweisbar. G:E-Verhältnis zugunsten der Granulopoeseverschoben. Hier Nachweis von toxischen Veränderungen/Granulationsanomalien. Vermehrt Blasten (5-10%), ohne Nachweis von Auerstäbchen. Reifungsgestörte Erythropoese mit Doppelkernigkeit und Mitosen. 30% reifzellig erscheinende Lymphozyten, teils herdförmig betont. Vermehrt Plasmazellen (6%).“

KM-Zytologie (30.05.05):„Hyperzelluläres KM, Megakaryozyten nachweisbar.
G:E-Verhältnis zugunsten der Granulopoeseverschoben. Granulopoese mit toxischen Veränderungen/Granulationsanomalien. Vermehrt Blasten (5-10%), ohne Nachweis von Auerstäbchen. Reifungsgestörte Erythropoese mit Doppelkernigkeit und Mitosen. 30% reifzelligerscheinende Lymphozyten, teils herdförmig betont. Vermehrt Plasmazellen (6%).“
1. Dysplasiezeichen in zwei Zelllinien bei �10% der Zellen. Blastenvermehrung 5-10% ohne Auerstäbchen.
�V. a. MDS RAEB-1
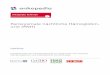
Besprochene Therapiestrategie in Stuttgart
MDS RAEB I bei jungem PatientPrognose-Score IPSS: intermediär I(KM-Blasten 5-10%, 2-3 Zelllinien betroffen, normaler Karyotyp: medianes Überleben: 3,3 Jahre)
2 Geschwister: 1 Schwester HLA-ident.�familiäre allogene PBSZT (einzig kurativer
Ansatz)

Patient M. F., �, 44 Jahre
•skeptisch gegenüber allogener PBSZT•vorerst rein supportive Therapie:
5/05 bis 12/05:regelmäßige Transfusionstherapie2 LAE alle 2 Wochen (+ Fe2+Chelator)1 PTK alle 1-4 Wochen

Zweitmeinung bei uns
KM-Zytologie:„Hyperzelluläres KM, Megakaryozyten nachweisbar. G:E-Verhältnis
zugunsten der Granulopoese verschoben. Granulopoese mit toxischen Veränderungen/Granulationsanomalien. Vermehrt Blasten (5-10%), ohne Nachweis von Auerstäbchen. Reifungsgestörte Erythropoese mit Doppelkernigkeit und Mitosen. 30% reifzellig erscheinende Lymphozyten, teils herdförmig betont. Vermehrt Plasmazellen (6%).“
2. Lymphozytose 30(-40)% und Plasmozytose im KM.
� ???

�Referenzzentrum für Lymphknotenpathologie Würzburg, Prof. Müller-Hermelink:
KM-Histologie (2/06):•Bestätigung des MDS•geringe Plasmozytose, polyklonal, als reaktiv zu werten•deutliche KM-Lymphozytose:immunhistochemisch:CD57 pos, CD8 pos., Perforin pos.,CD 56 neg. molekulargenetisch:Nachweis von Monoklonalität;Amplifikation des Rearrangements der Gamma-Kette des T-Zell-Rezeptors

�KM-Infiltration durch malignes Non-Hodgkin-Lymphom der T-Zell-Reihe
�am ehesten
T-LGL-Leukämie

LGL-Leukämie (I)
Large granular lymphocyte leukemiaDefinition:Syndrom einer chronischen, teilweise zyklischen Neutropenie mit Vermehrung zirkulierender granulierter großer Lymphozyten, definiert über Morphologie und CD57-PositivitätPB-Konzentration: > 500/µl(Norm: 223±99/µl)

LGL-Leukämie (II)
• Infiltration von Knochenmark (in 70%), Leber und Milz
• Unterscheidung zwischen reaktiver und klonaler (neoplastischer) LGL-Lymphozytose oft schwierig
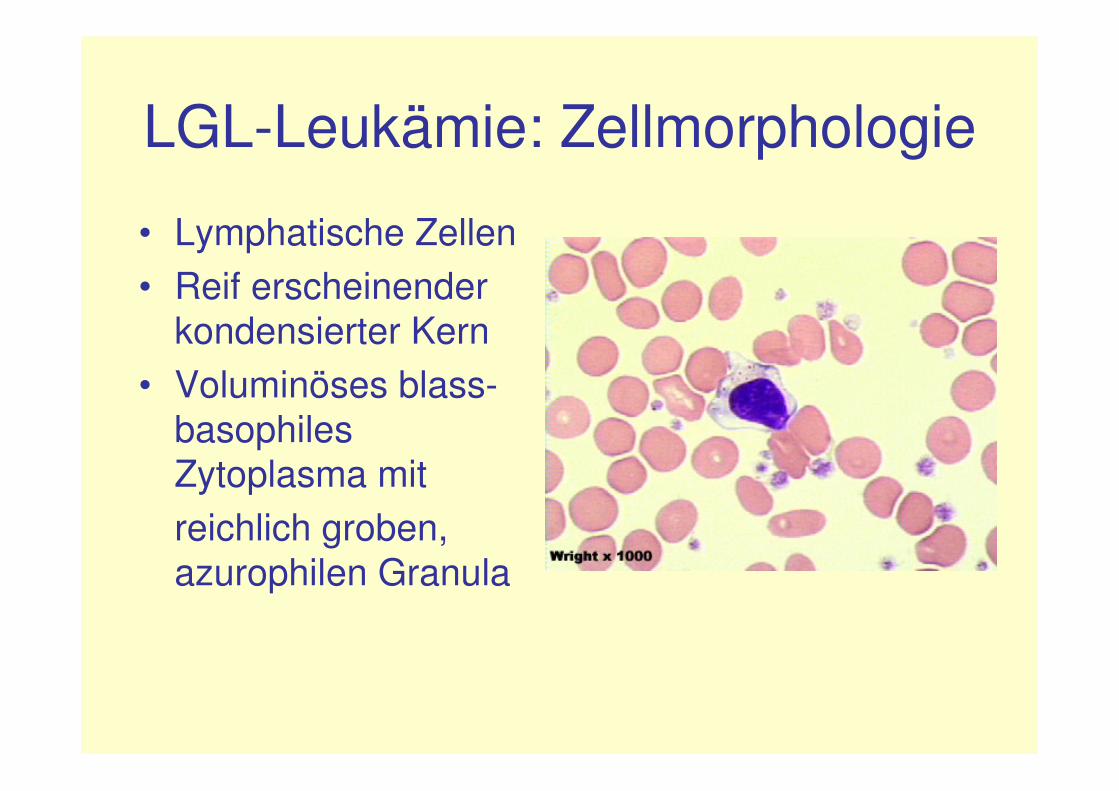
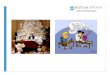
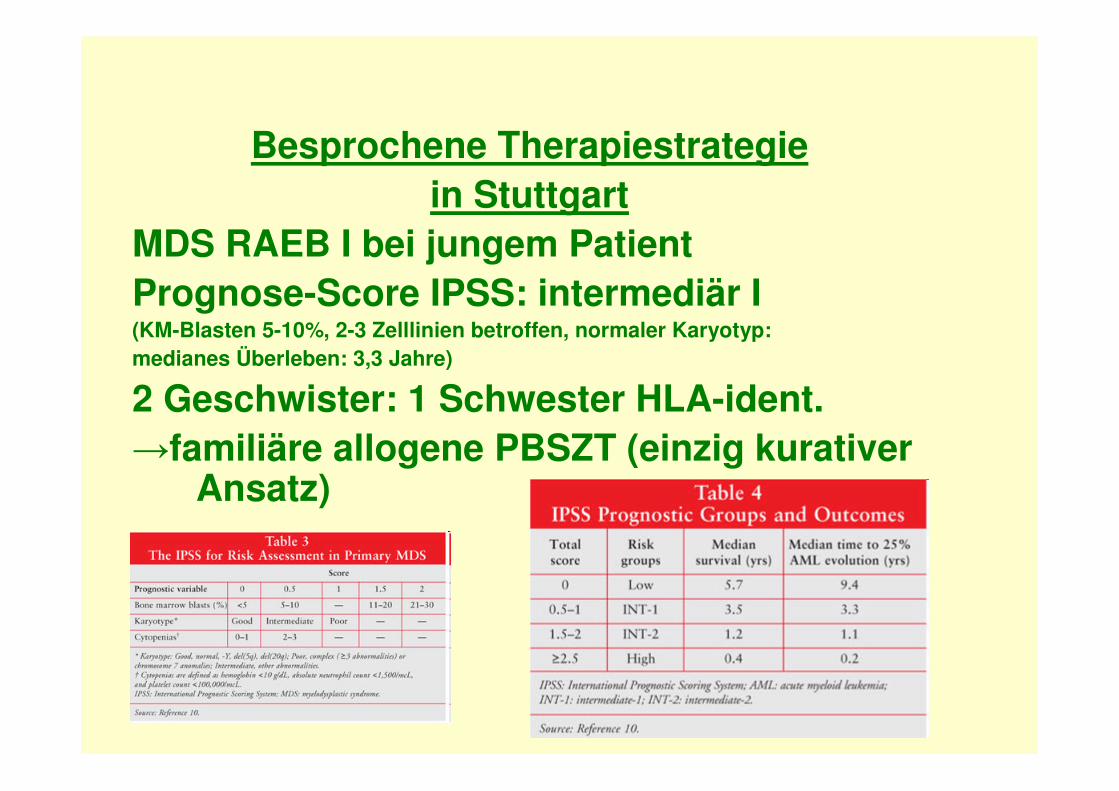
LGL-Leukämie: Zellmorphologie
• Lymphatische Zellen• Reif erscheinender
kondensierter Kern• Voluminöses blass-
basophiles Zytoplasma mitreichlich groben, azurophilen Granula
LGL-Leukämie: Zellmorphologie
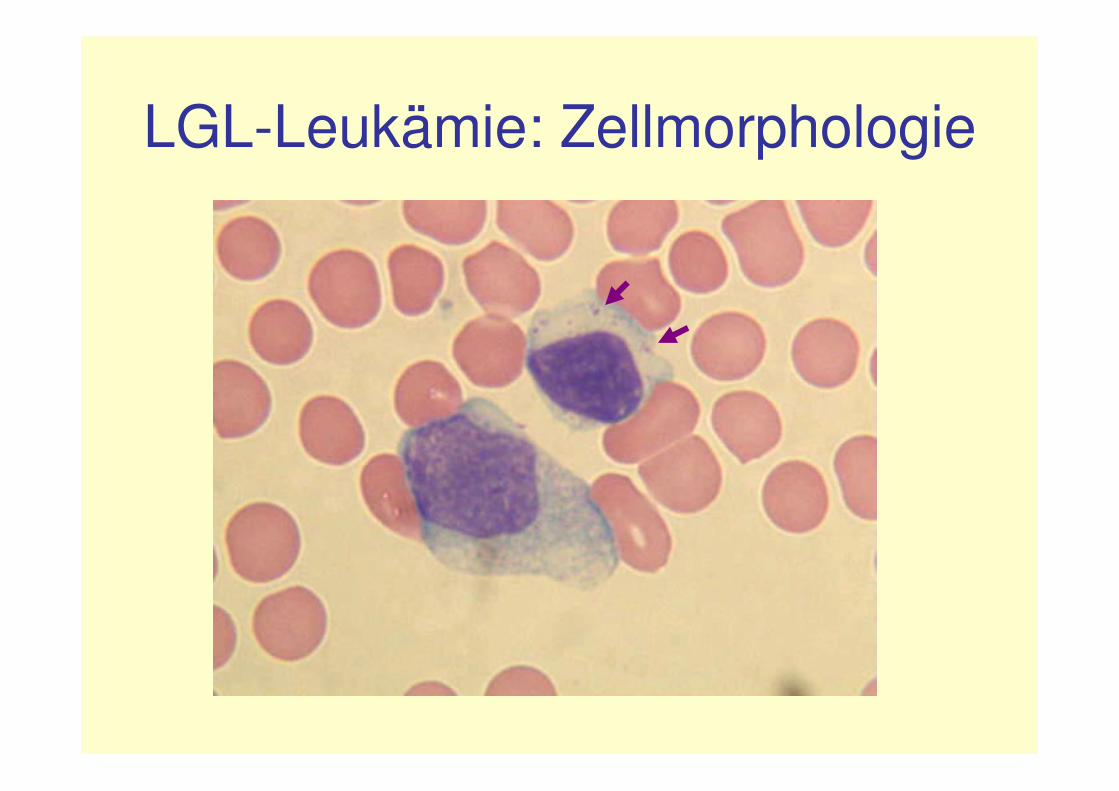
LGL-Leukämie: Zellmorphologie

T-LGL-Leukämie: Klinik (I)• Erkrankung des höheren Lebensalters (Median
60 Jahre)• Verhältnis �:� beträgt 1:1• gekennzeichnet durch Folgen der Neutropenie:
Fieber, rezidivierende bakterielle Infekte(besonders Haut, obere Atemwege, perirektal)
• B-Symptomatik (20-30%)• geringe bis mäßige Hepatosplenomegalie
(20-50%)

LGL-Leukämie: Klinik (II)
• bei bis zu 40% findet sich eineBegleiterkrankung
• sehr häufig eine seropositiverheumatoide Arthritis
• somit deutliche Überlappung mit Felty-Syndrom (rheumatoide Arthritis, Neutropenie, Splenomegalie)

LGL-Leukämie: Ätiologie• nicht eindeutig geklärt• evtl. zentrale pathogenetische Rolle von
Zytokinen:proliferationsfördernder Effekt auf CD3+ LGL-Zellen von IL-12 und IL-15(Kukita et al., Br J Haematol. 2002 Nov;119(2):467-74)
• Apoptoseresistenz einer ursprünglichphysiologisch aktivierten polyklonalenzytotoxischen T-Lymphozytenpopulation(Nagata et al., Cell 1997;88:355-365)

T-LGL-Leukämie:Hämatologische Merkmale
charakteristische Blutbildveränderungen:Lymphozytose (> 4000/µ) mit Vermehrung der LGL-Zellen (10-15%: > 400/µ; meist > 2000/µl)
• Neutropenie (< 1500/µl: 80%, < 500/µl: 45%)• Anämie mit Hb < 11 g/dl (50%)• Thrombopenie < 150 000/µl (20%)
Serologie:• oft polyklonale Hypergammaglobulinämie,
zirkulierende Immunkomplexe, pos. Rheumafaktoroder ANF
• oft AK gegen neutrophile Granulozyten oderThrombozyten

T-LGL-Leukämie: Prognose
• medianes Überleben: > 10 Jahre• limitierend: Infektionen als Folge der
Neutropenie• 70% der Patienten benötigen im Verlauf
eine Therapie:Indikation: Neutropenie, Anämie, symptomatische Splenomegalie

T-LGL-Leukämie:Therapie(I)Medikamentös-Konservative Therapie• kaum Daten aus klinischen Studien• keine etablierte Standardtherapie• postulierte Wirksamkeit einer
immunsuppressiven Therapie
Case reports/kleine Fallserien über:1. niedrig dosiertes Methotrexat2. Fludarabin, Ciclosporin, Cyclophosphamid
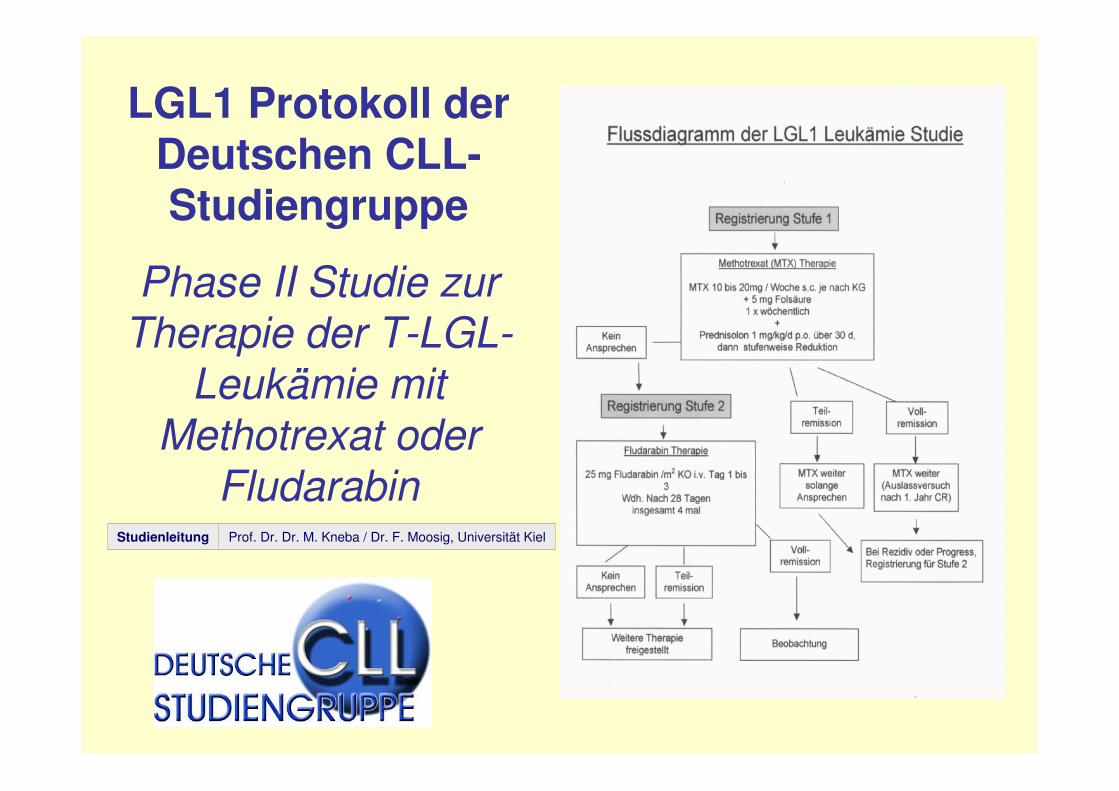
LGL1 Protokoll der Deutschen CLL-Studiengruppe
Phase II Studie zur Therapie der T-LGL-
Leukämie mit Methotrexat oder
FludarabinProf. Dr. Dr. M. Kneba / Dr. F. Moosig, Universität KielStudienleitung

Patient M. F., �, 44 Jahre
Infektkomplikationen4/06:•Periproktitischer Abszess mit transsphinktärer Fistelbildung•operative Spaltung 6/06:Erysipel rechte Wange

Patient M. F., �, 44 Jahre7/06:studienanaloger Therapieversuch mit Methotrexat + Prednison
-3-9/07
2,5 � absetzen2/07
7,5 � 51/07
15 � 1012/06
201511/06
3010/06
40109/06
507,58/06
Prednisolon (mg)täglich p.o.
Methotrexat (mg)1x pro Woche s.c.
(+ 5mg Folsäure am Folgetag)
MTX wurde 9/07 wegen progredienter Hepatotoxizität abgesetzt
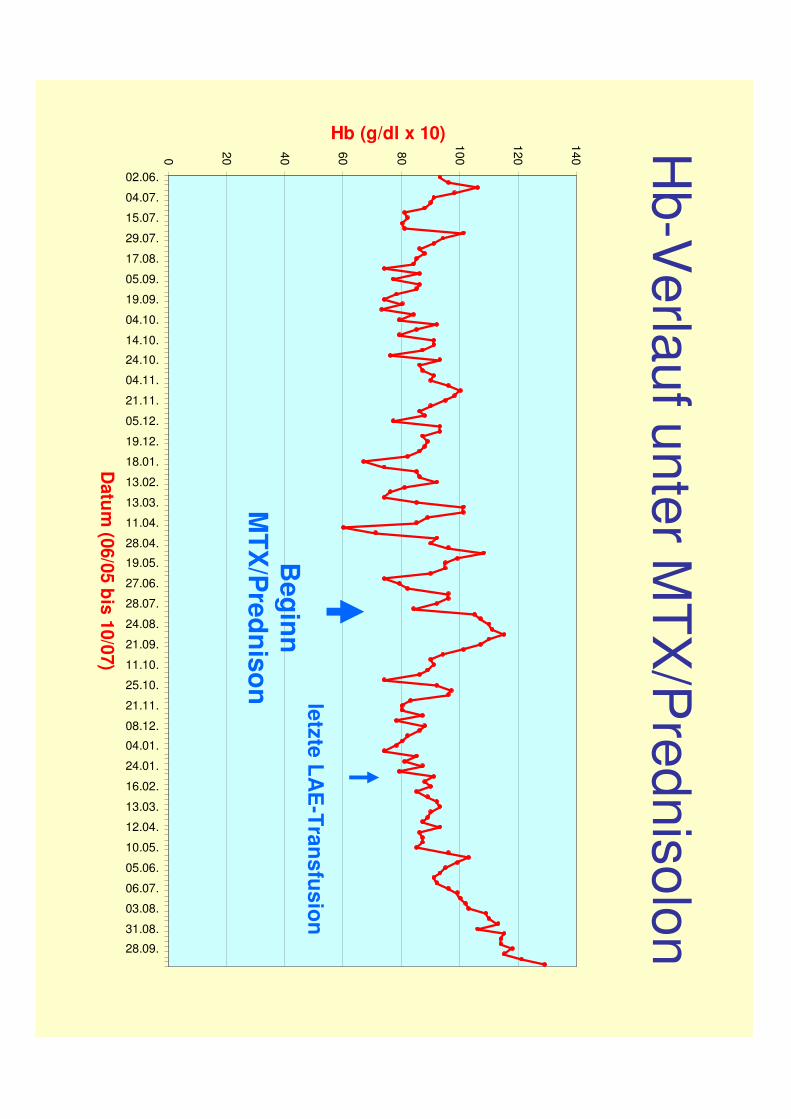
Hb-V
erlaufunter MTX
/Prednisolon
0 20 40 60 80
100
120
140
02.06.
04.07.
15.07.
29.07.
17.08.
05.09.
19.09.
04.10.
14.10.
24.10.
04.11.
21.11.
05.12.
19.12.
18.01.
13.02.
13.03.
11.04.
28.04.
19.05.
27.06.
28.07.
24.08.
21.09.
11.10.
25.10.
21.11.
08.12.
04.01.
24.01.
16.02.
13.03.
12.04.
10.05.
05.06.
06.07.
03.08.
31.08.
28.09.
Datum
(06/05 bis 10/07)
Hb (g/dl x 10)
Beginn
MTX
/Prednison
letzte LAE
-Transfusion
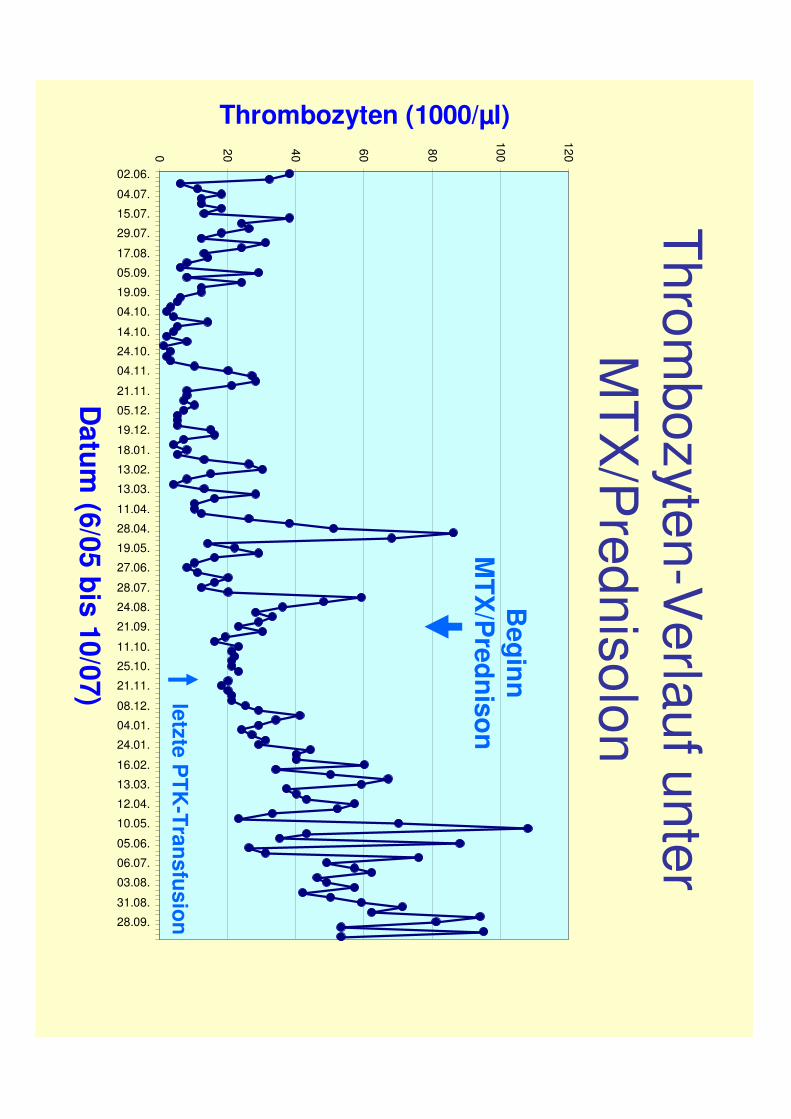
Thrombozyten-V
erlaufunter M
TX/P
rednisolon
0 20 40 60 80
100
120
02.06.
04.07.
15.07.
29.07.
17.08.
05.09.
19.09.
04.10.
14.10.
24.10.
04.11.
21.11.
05.12.
19.12.
18.01.
13.02.
13.03.
11.04.
28.04.
19.05.
27.06.
28.07.
24.08.
21.09.
11.10.
25.10.
21.11.
08.12.
04.01.
24.01.
16.02.
13.03.
12.04.
10.05.
05.06.
06.07.
03.08.
31.08.
28.09.
Datum
(6/05 bis 10/07)
Thrombozyten (1000/µl)
Beginn
MTX
/Prednison
letzte PTK
-Transfusion
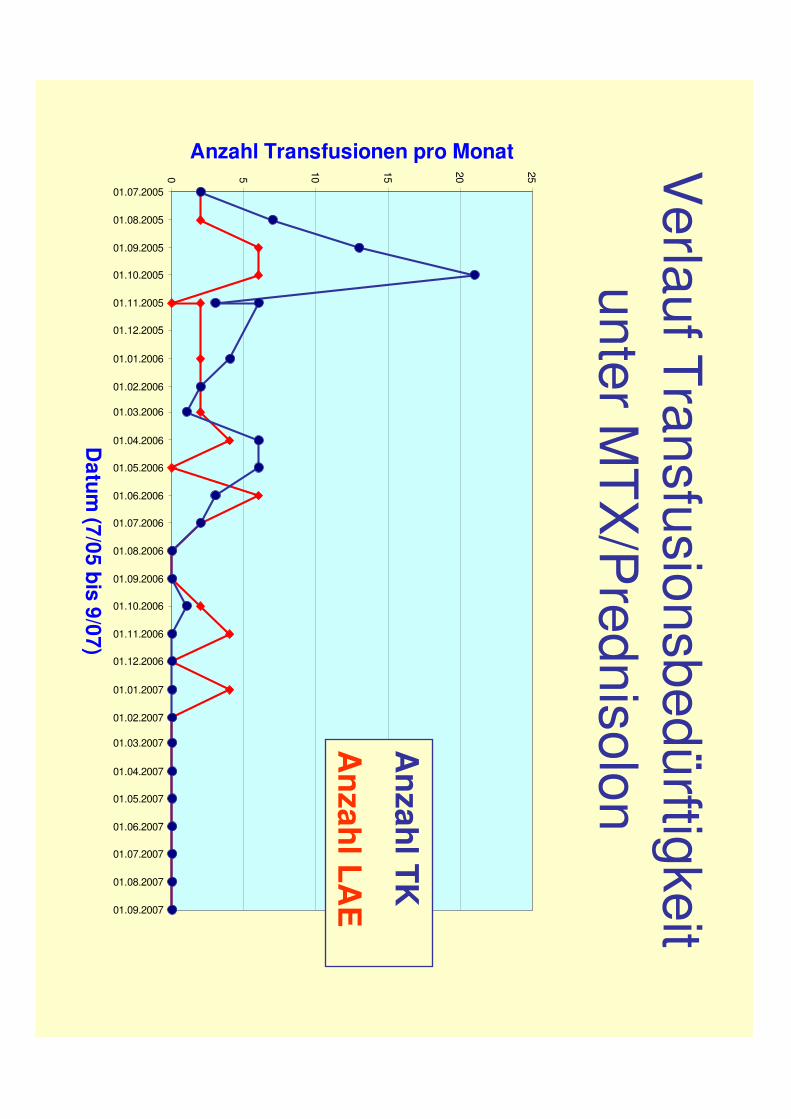
Verlauf Transfusionsbedürftigkeit
unter MTX
/Prednisolon
0 5 10 15 20 25
01.07.2005
01.08.2005
01.09.2005
01.10.2005
01.11.2005
01.12.2005
01.01.2006
01.02.2006
01.03.2006
01.04.2006
01.05.2006
01.06.2006
01.07.2006
01.08.2006
01.09.2006
01.10.2006
01.11.2006
01.12.2006
01.01.2007
01.02.2007
01.03.2007
01.04.2007
01.05.2007
01.06.2007
01.07.2007
01.08.2007
01.09.2007
Datum
(7/05 bis 9/07)
Anzahl Transfusionen pro Monat
Anzahl TK
Anzahl LA
E

Patient M. F., �, 44 Jahre
erneute Problematik:ab 1/07:• steigende LDH-Werte (von initial 400-
1000 U/l auf 1400-2000 U/l)• Haptoglobin <7 mg/dl, freies Hb im
Plasma 14 mg/dl• Hämoglobin im Urin ++• keine Fragmentozyten
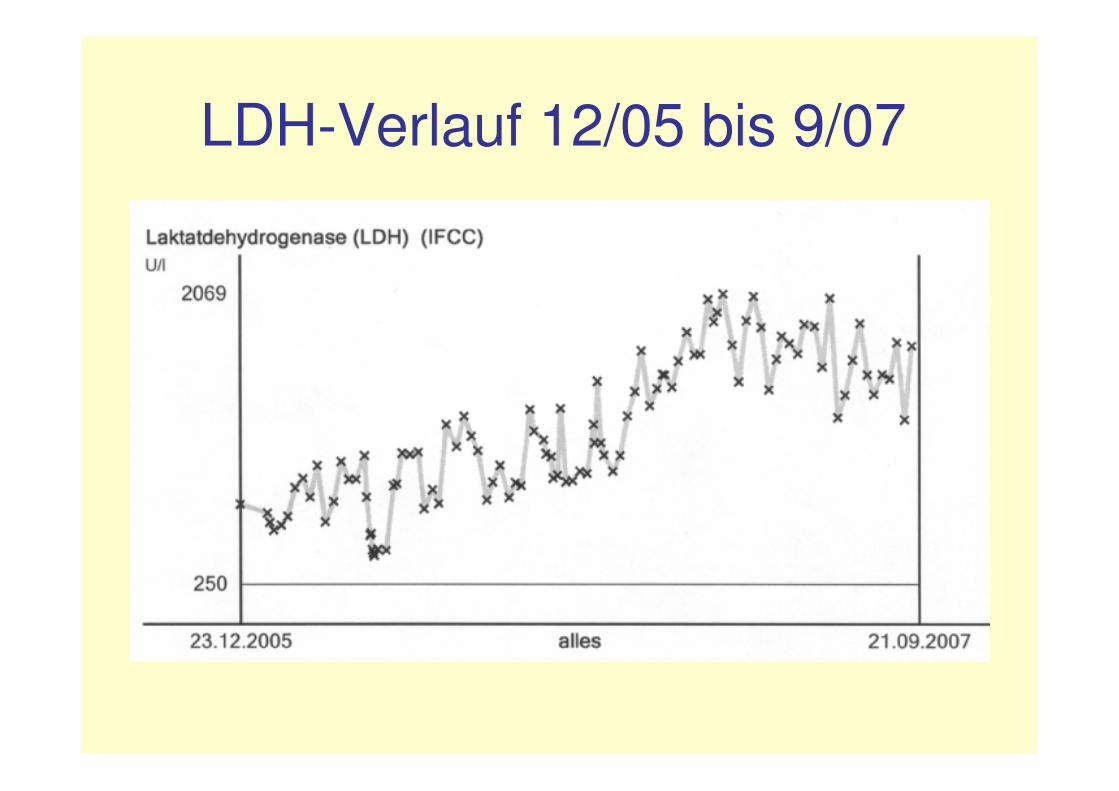
LDH-Verlauf 12/05 bis 9/07

Patient M. F., �, 44 Jahre
�Hämolysesituation (neu! �erworben)
kein Nachweis einer antikörper-vermitteltenHämolyse (durch Auto- oder Allo-Ak)
Diagnostik auf PNH positiv!

Paroxysmale nächtliche Hämoglobinurie (PNH)
• Erstbeschreibung 1882 von Strübing und 1911 von Marchiafava
• einzige Form einer erworbenenkorpuskulären hämolytischen Anämie
• klonale Erkrankung der pluripotentenhämatopoetischen Stammzelle(alle 3 Zellreihen betroffen)

Paroxysmale nächtliche Hämoglobinurie (PNH)
• Pathogenese:somatische (erworbene) Mutation des X-chromosomalen PIG-A-Gens(Phosphatidylinositolglykan-Klasse A)
� Genprodukt notwendig für die Synthese von GPI (Glykosylphosphatidylinositol):Ankermolekül für verschiedeneMembranproteine bzw. Oberflächenmoleküle

Paroxysmale nächtliche Hämoglobinurie (PNH)
• zwei derartige GIP-verankerte Moleküle schützen die Erythrozyten vor verstärkter Komlementlyse:
1. DAF (decay accelerating factor), CD 55
2. MIRL (membrane inhibitor of reactive lysis), CD 59
• neutrophile Granulozyten: Fehlen der alkalischenGranulozytenphosphatase

Paroxysmale nächtliche Hämoglobinurie (PNH): Klinik
Zwei Phasen:1. korpuskuläre hämolytische Anämie, v. a.
nachts auftretend (geringfügiger pH-Abfall des Blutes verstärkt Membran-instabilität); Eisenmangel; häufig atypische Thromboembolien (50% derPNH-Todesfälle)
2. Erschöpfung der Stammzellen, zunehmendeAplasie, teilweise Übergang in AML
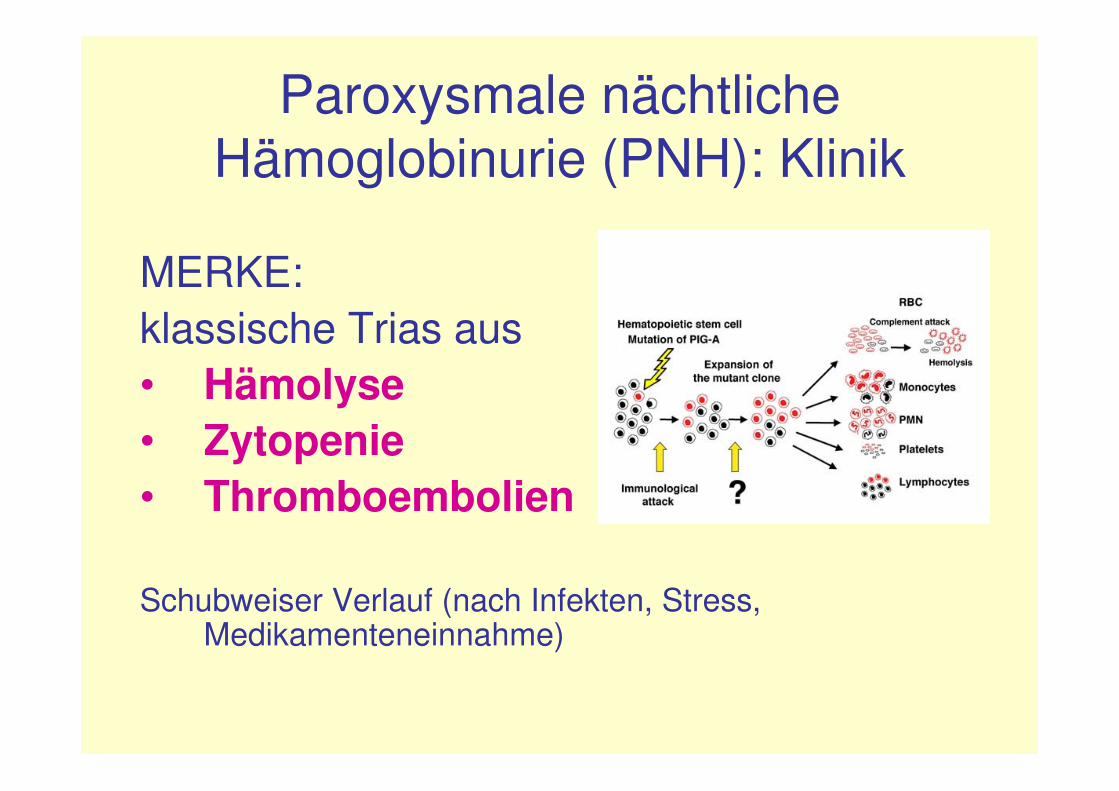
Paroxysmale nächtliche Hämoglobinurie (PNH): Klinik
MERKE:klassische Trias aus• Hämolyse• Zytopenie• Thromboembolien
Schubweiser Verlauf (nach Infekten, Stress, Medikamenteneinnahme)

Paroxysmale nächtliche Hämoglobinurie (PNH): Klinik
Symptome:1. colafarbener Morgenurin (selten)2. Cephalgien, abdominelle Schmerzen, WS-
Syndrom, erektile Dysfunktion3. Neutro- und/oder Thrombopenie, chronische
hämolytische Anämie4. Eisenmangelanämie5. Hepatosplenomegalie6. (atypische) Thrombo-
embolien

Paroxysmale nächtliche Hämoglobinurie (PNH):Diagnostik(I)
Goldstandard:Durchflusszytometrie zur Negativdarstellungder GPI-verankerten Proteine CD59, CD55 und CD24 mittels fluoreszeinmarkierterAntikörper.-an Neutrophilen, Erythrozyten, (Monozyten, Retikulozyten) in Heparinblut
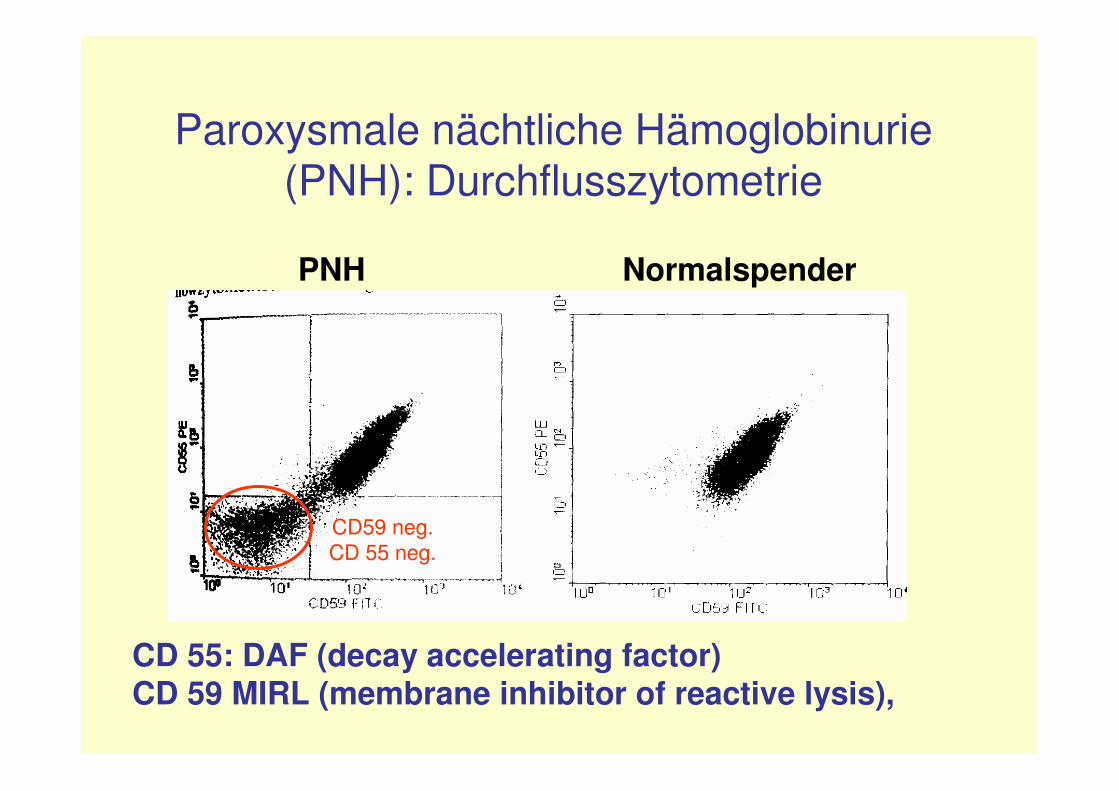
Paroxysmale nächtliche Hämoglobinurie(PNH): Durchflusszytometrie
CD 55: DAF (decay accelerating factor)CD 59 MIRL (membrane inhibitor of reactive lysis),
PNH Normalspender
CD59 neg. CD 55 neg.

Paroxysmale nächtliche Hämoglobinurie (PNH):Diagnostik(II)
•Molekulargenetischer Nachweis des PIG-A-Gens•Zuckerwassertest und Säurehämolysetest(Ham-Test) sind nicht mehr notwendig
A Serum Normalspender
B Serum Normalspender, angesäuert, keine Hämolyse
C Serum PNH-Patient
D Serum PNH-Patient, angesäuert, Hämolyse: Messung freies Hb

Paroxysmale nächtliche Hämoglobinurie (PNH):Therapie (I)
•Allogene Stammzelltransplantation einzigekausale Therapie. Problem: hohe Mortalität(40%)Biol Blood Marrow Transplant 2003;9:689-97, Hämatologie Uniklinik Leipzig
•Androgene bei KM-Hypoplasie•evtl. Versuch mit Prednison bei aktiverHämolyse•Transfusion von gewaschenen Erythrozyten(Elimination von Komplementkomponenten)
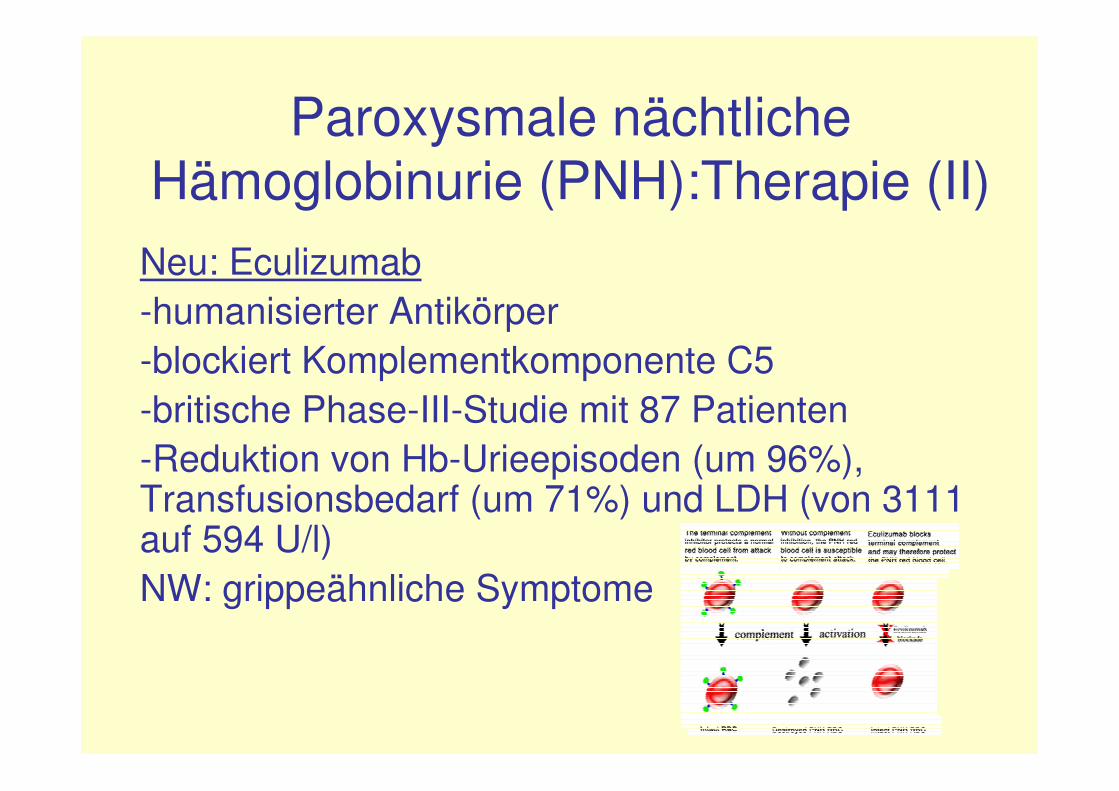
Paroxysmale nächtliche Hämoglobinurie (PNH):Therapie (II)
Neu: Eculizumab-humanisierter Antikörper-blockiert Komplementkomponente C5-britische Phase-III-Studie mit 87 Patienten-Reduktion von Hb-Urieepisoden (um 96%), Transfusionsbedarf (um 71%) und LDH (von 3111 auf 594 U/l)NW: grippeähnliche Symptome

Zusammenfassung
44jähriger Patient mit1. Myelodysplastischem Syndrom (MDS)2. T-LGL-Leukämie3. Paroxysmaler nächtlicher Hämoglobinurie
(PNH)
Mögliche Erklärung dieser Kombination?
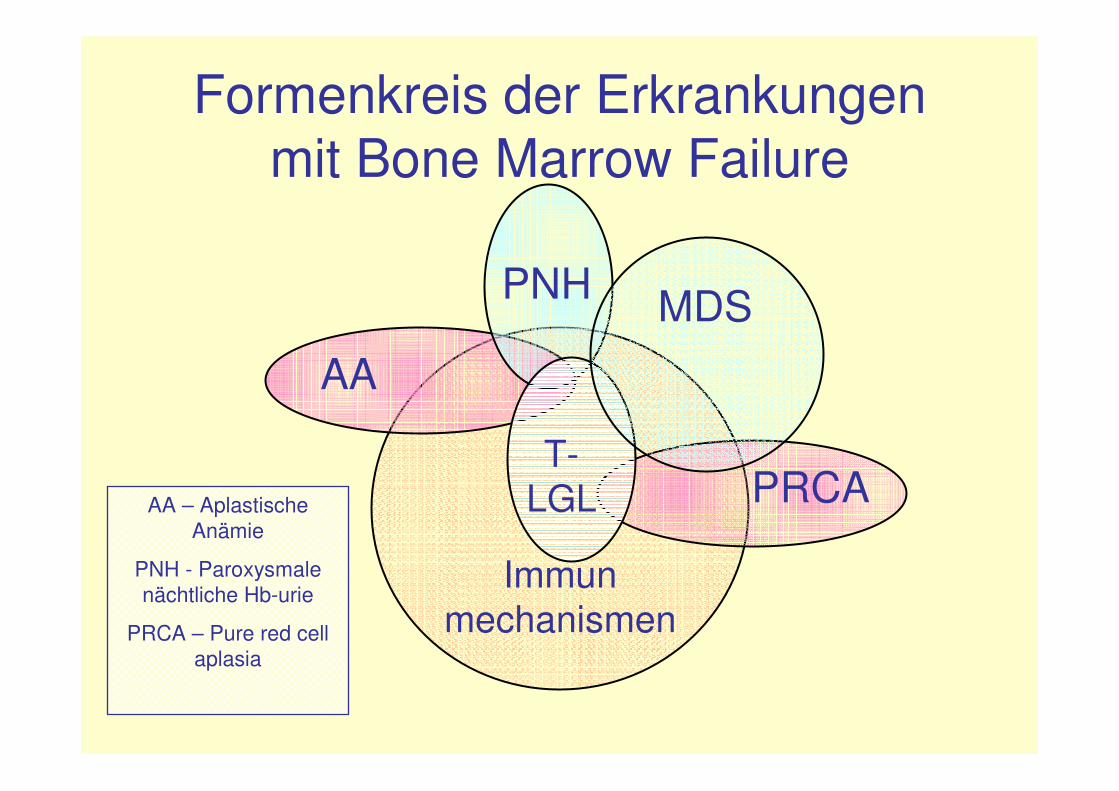
Formenkreis der Erkrankungenmit Bone Marrow Failure
AA
PNH MDS
T-LGL
Immunmechanismen
PRCAAA – AplastischeAnämie
PNH - Paroxysmalenächtliche Hb-urie
PRCA – Pure red cellaplasia

Zusammenhänge der verschiedenen Bone marrow failure-Syndrome (I)
• bei 10 -18 % der MDS-Patienten kann einPNH-Klon detektiert werden (Maciejewski et al.,B J Haematology 2001; Iwanaga et al., B J Haematology 1998 )
• von 100 untersuchten MDS-Patientenwiesen 9 zusätzlich T-LGL-Charakteristikaauf (Sauntharajah et al., B J Haematology 2001)
• bei 4/24 PNH-Patienten Detektion einesmonoklonalen T-LGL-Klons (Risitano et al.,
Leukemia 2005)

Zusammenhänge der verschiedenen Bone marrow failure-Syndrome (II)
Hypothese:bei Patienten mit MDS, AA, PNH und T-LGL autoimmune Pathogenese und Nachweis eines oligoklonalen TCR-Repertoires
�Entwicklung immundominanter Clonotypes�klinisches Bild einer LGL-Leukämie oder
PNH(Maciejewski et al.,B J Haematology 2001, Folia Histochem Cytobiol 2007)

Patient M. F., �, 44 Jahre
•engmaschiges Monitoring•evtl. erneuter immunsuppressiverTherapieansatz (Ciclosporin; ATG)•bei Entwicklung eines Hochrisiko-MDSfamiliäre allogene Stammzelltransplantation