Embed Size (px)
Citation preview

Netzartig strukturierte Oberflächen aus
präkeramischen Polymeren
Der Technischen Fakultät der
Friedrich-Alexander-Universität Erlangen-Nürnberg
zur
Erlangung des Doktorgrades
D O K T O R - I N G E N I E U R
vorgelegt von
Michael Woiton
aus Erlangen


Als Dissertation genehmigt
von der Technischen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung: 16.07.2014
Vorsitzende des Promotionsorgans: Prof. Dr.-Ing. habil. Marion Merklein
Gutachter: Prof. Dr. Christoph J. Brabec
Prof. Dr. Michael Scheffler


„Hohe Kontingenz von Ereignissen bedeutet, dass alles, was ist,
auch anders sein könnte.“
Niklas Luhmann


Inhaltsverzeichnis
I
Inhaltsverzeichnis
Inhaltsverzeichnis ............................................................................................................ I
Abkürzungsverzeichnis ................................................................................................. V
Kurzzusammenfassung .................................................................................................. 1
Abstract ............................................................................................................................ 3
1 Einleitung und Zielsetzung ................................................................................. 5
2 Stand der Technik ............................................................................................... 7
2.1 Einsatzgebiete für strukturierte keramische Beschichtungen ...................... 7
2.2 Strukturierungsmechanismen beim Beschichten ......................................... 9
2.2.1 Strukturierung durch Entmischung in Mehrphasensystemen ...................... 9
2.2.1.1 Entmischung in Polymer-Lösungsmittel-Systemen .................................. 11
2.2.2 Strukturierte Beschichtungen auf der Basis von Polymeren-Lösungen .... 14
2.2.3 Entstehung von Oberflächenstrukturen durch Entnetzung ........................ 15
2.2.4 Beeinflussung von Polymerbeschichtungen durch Füllstoffe ................... 16
2.2.5 Strukturbildung durch Konvektion ............................................................ 18
2.3 Technisch relevante Verfahren zur Herstellung poröser Keramiken ........ 19
2.4 Präkeramische Polymere ........................................................................... 20
2.5 Theorie der Löslichkeitsparameter ............................................................ 22
2.5.1 Definition der Löslichkeitsparameter ........................................................ 23
2.5.2 Anwendung der Löslichkeitstheorie von Hansen auf Polymere ............... 25
2.5.3 Bestimmung der Löslichkeitsparameter mittels Löslichkeitsversuchen ... 26
2.5.4 Bestimmung der Löslichkeitsparameter anhand der intrinsischen
Viskosität ................................................................................................... 27
2.6 Beschichtungsverfahren............................................................................. 28
2.6.1 Tauchbeschichten ...................................................................................... 28
2.6.2 Schleuderbeschichten ................................................................................ 29
2.6.3 Automatisches Filmziehen ........................................................................ 31
2.7 Rheologische Eigenschaften keramischer Schlicker ................................. 32

Inhaltsverzeichnis
II
3 Experimentelles Vorgehen ................................................................................ 33
3.1 Verwendete Rohstoffe ............................................................................... 33
3.1.1 Polymere .................................................................................................... 33
3.1.2 Lösungsmittel ............................................................................................. 34
3.1.3 Substrate ..................................................................................................... 35
3.1.4 Füllstoffe .................................................................................................... 36
3.2 Bestimmung der Löslichkeitsparameter .................................................... 36
3.3 Bestimmung der Mischbarkeiten in den verschiedenen Systemen ............ 38
3.4 Herstellung der Schichten .......................................................................... 38
3.5 Herstellung von füllstofffreien Beschichtungen ........................................ 40
3.6 Herstellung der Beschichtungen mittels Schleuderbeschichten und
Filmziehen .................................................................................................. 41
3.6.1 Schleuderbeschichten ................................................................................. 41
3.6.2 Filmziehen .................................................................................................. 41
3.7 Charakterisierung der Beschichtungen ...................................................... 41
4 Ergebnisse .......................................................................................................... 43
4.1 Ungefüllte Systeme auf Basis präkeramischer Polymere PMS
und PMPS .................................................................................................. 43
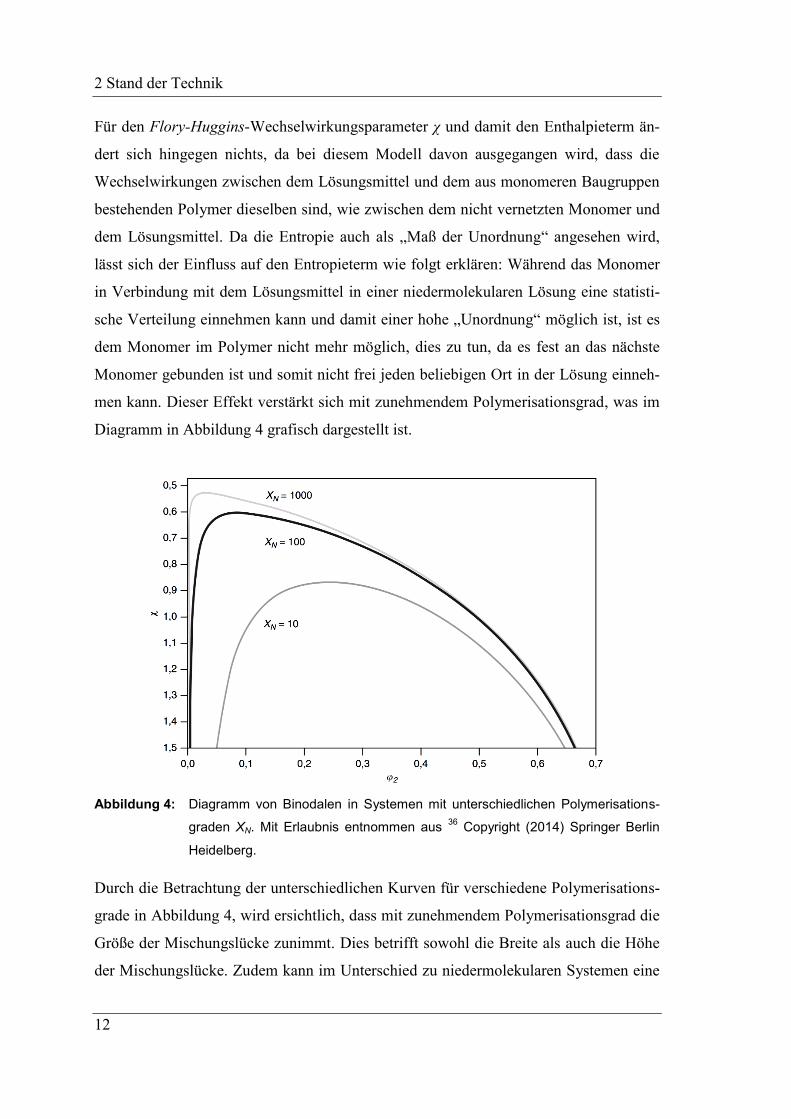
4.1.1 Hansen Löslichkeitsparameter der präkeramischen Polymere .................. 43
4.1.2 Phasendiagramme der Mischbarkeiten von
PMS – PMPS – MTES – MeOH ................................................................ 52
4.1.3 Beschichtungen mit verschiedenen Zusammensetzungen von
PMS und PMPS und deren Einfluss auf die Strukturbildung .................... 53
4.1.4 Der Einfluss des Methanolgehalts auf die Mischbarkeit im
Dreiphasensystem von PMS – PMPS – MTES ......................................... 56
4.1.5 Charakterisierung von PMS und PMPS mittels Dynamischer
Differenzkalorimetrie (DSC) ..................................................................... 58

Inhaltsverzeichnis
III
4.2 Füllstoffhaltige Beschichtungen auf Basis präkeramischer Polymere ...... 58
4.2.1 Einfluss des Methanolanteils auf die Strukturbildung ............................... 60
4.2.2 Ersatz von Methanol als Nichtlösungsmittel ............................................. 61
4.2.3 Beeinflussung des Systems durch das Lösungsmittel MTES .................... 62
4.2.4 Einfluss des Verhältnisses von PMS zu PMPS und der
Polymerkonzentration ................................................................................ 63
4.2.5 Einfluss der Füllstoffpartikel auf die Strukturbildung ............................... 65
4.2.5.1 Variation des Füllstoffgehalts .................................................................... 65
4.2.5.2 Einfluss der Füllstoffkorngröße ................................................................. 66
4.2.5.3 Variation des Füllstoffmaterials ................................................................ 67
4.2.6 Einfluss des Substratmaterials auf die Beschichtungen ............................ 68
4.2.7 Reduzierung auf die minimal möglichen Komponenten im System ......... 68
4.2.8 Zeitlicher Ablauf der Strukturbildung ....................................................... 70
4.2.9 Verwendbarkeit des Systems für verschiedene Beschichtungsverfahren.. 73
4.2.9.1 Tauchbeschichten ...................................................................................... 73
4.2.9.2 Schleuderbeschichten ................................................................................ 73
4.2.9.3 Automatisches Filmziehen ........................................................................ 75
4.2.10 Variation der Schichtdicke und ihr Einfluss auf die Porengröße .............. 78
4.2.11 Verhalten der Beschichtung auf dreidimensionalen Strukturen am
Beispiel von Rohren .................................................................................. 80
5 Diskussion .......................................................................................................... 83
5.1 Ungefüllte Systeme auf Basis präkeramischer Polymere PMS
und PMPS .................................................................................................. 83
5.1.1 Hansen Löslichkeitsparameter der Polysiloxane ....................................... 83
5.1.2 Mischbarkeitsdiagramme im System PMS – PMPS – MTES – MeOH.... 86
5.1.3 Beschichtungen mit verschiedenen Zusammensetzungen von
PMS und PMPS und deren Einfluss auf die Strukturbildung .................... 86
5.2 Zusammenfassung der wesentlichen Erkenntnisse in ungefüllten
Systemen .................................................................................................... 88
5.3 Gefüllte Systeme ....................................................................................... 89

Inhaltsverzeichnis
IV
6 Zusammenfassung und Ausblick ..................................................................... 99
7 Literaturverzeichnis ........................................................................................ 103
8 Abbildungsverzeichnis .................................................................................... 113
9 Tabellenverzeichnis ......................................................................................... 119
Lebenslauf ......................................................................................................................... i
Wissenschaftliche Veröffentlichungen ......................................................................... iii
Danksagung...................................................................................................................... v

Abkürzungsverzeichnis
V
Abkürzungsverzeichnis
Viskosität
Scherspannung
Fl Viskosität der Flüssigkeit
inh Inhärenten Viskosität
rel Relative Viskosität
A Fläche
AFM Atomic Force Microscopy
Al2O3 Aluminiumoxid
B4C Borcarbid
c Konzentration
COMP i Zusammensetzung
D Schergeschwindigkeit
DIN Deutsche Industrie Norm
DSC Differential Scanning Calorimetry
EDX Energiedispersive Röntgenanalyse
Ei Kohäsive Energie
F Kraft
g Erdbeschleunigung
H Wasserstoff
h0 Rackelabstand zur Grundplatte
HSP Hansen-Löslichkeitsparameter
L Rackelbreite
LCST Lower Critical Solution Temperature
MeOH Methanol
MTES Methyltriethoxysilan
Mw Massen gemittelte molare Masse
NaCl Natriumchlorid
PDCs Polymer Derived Ceramics
PMMA Polymethylmetacrylat

Abkürzungsverzeichnis
VI
PMPS Polymethylphenylvinylsiloxane
PMS Polymethylsiloxan
PP Polypropylen
PS Polystyrol
R Allgemeine Gaskonstante
R Organischer Rest
Ra Interaktionsradius des Polymeres
RED Relativer Abstand
REM Rasterelektronenmikroskopie
Ro Relativer Abstand
s Schichtdicke
s0 Ausgangsschichtdicke
Si Silizium
Si3N4 Siliziumnitrid
SiC Siliziumcarbid
SiO2 Siliziumdioxid
T Absolute Temperatur
t Zeit
TiC Titancarbid
tpol Durchlaufzeit für das Lösungsmittel
tsol Durchlaufzeit für das Polymer
U Verfahrgeschwindigkeit
UCST Upper Critical Solution Temperature
V Molare Volumen
v0 Ziehgeschwindigkeit
VLS Vapor-Liquid-Solid Mechanismus
WC Wolframcarbid
XN Polymerisationsgrad
ZAE Bayerisches Zentrum für Angewandten Energieforschung
γ Oberflächenspannung der Flüssigkeit
δD Disperser-Anteil der Hansen-Löslichkeitsparameter
ΔGm
Gibbs’sche freie Enthalpie der Mischung

Abkürzungsverzeichnis
VII
ΔH Verdampfungsenthalpie
δH Wasserstoffbrückenbindungs-Anteil der HSP
ΔHm
Mischungsenthalpie
δiD Hansen-Löslichkeitsparameter-Anteil für Polymere
δiS Hansen-Löslichkeitsparameter-Anteil für Lösungsmittel
δP Polarer-Anteil der Hansen-Löslichkeitsparameter
ΔP Druckunterschied
ΔSm
Mischungsentropie
δT Hildebrand- oder absoluter Löslichkeitsparameter
ρ Dichte
υ Kinematische Viskosität
φi Volumenbrüche
χ Flory-Huggins-Wechselwirkungsparameter
ω Umdrehungsgeschwindigkeit

Abkürzungsverzeichnis
VIII

Kurzzusammenfassung
1
Kurzzusammenfassung
In dieser Arbeit wird eine selbststrukturierende Beschichtung auf der Basis präkerami-
scher Polymere untersucht. Ziel ist es dabei, sowohl die verwendeten Rohstoffe, als
auch Beschichtungen mit und ohne Füllstoffe zu untersuchen und zu charakterisieren.
Ebenso sollen die strukturbildenden Mechanismen aufgeklärt werden.
Als präkeramische Polymere kamen zwei kommerziell erhältliche Polymere zum Ein-
satz, dabei handelt es sich um zwei Polysiloxane, nämlich Polymethylsiloxan (PMS)
und Polymethylphenylvinylsiloxane (PMPS). Von beiden Polymeren wurden die Han-
sen-Löslichkeitsparameter (HSP) nach drei unterschiedlichen Methoden bestimmt. Da-
bei handelte es sich um zwei Methoden, die auf Lösungsversuchen in unterschiedlichen
Lösungsmitteln beruhen und eine dritte, bei der die HSP anhand der intrinsischen Vis-
kosität in verschiedenen Lösungsmitteln bestimmt werden. Es konnte gezeigt werden,
dass alle drei Methoden zu gut übereinstimmenden Ergebnissen führen. Die dabei ge-
messenen HSP für die Polymere betragen gemittelt: δD = 16,8 MPa1/2
± 0,1,
δP = 3,3MPa1/2
± 1,0, δH = 5,0 MPa1/2
± 1,1 mit einem Radius von 4 MPa1/2
± 0,9 für
PMS und für PMPS δD = 17,7 MPa1/2
± 0,7, δP = 8,2 MPa1/2
± 1,3, δH = 6,4 MPa1/2
± 0,9
mit einem Radius von 9,2 MPa1/2
± 0,8. Anhand der ermittelten HSP und des vier Pha-
sensystems PMS – PMPS – MTES – MeOH lässt sich zeigen, dass Methanol (MeOH)
für beide Polymere als „Nichtlösungsmittel“ bezeichnet werden kann und dass eine In-
kompatibilität der beiden Polymere vorliegt. Die Rolle von Methyltriethoxysilan
(MTES) konnte hingegen nicht eindeutig anhand der für MTES berechneten HSP be-
stimmt werden.
Anhand der Ergebnisse dieser Arbeit konnte für füllstofffreie Beschichtungen gezeigt
werden, dass die Strukturbildung hierbei durch eine Polymer/Polymer Entmischung
erfolgt. Hierbei bildet PMS die Matrix und PMPS die Inseln. Während MTES für die
Strukturbildung in füllstofffreien Beschichtungen zwingend als Lösungsmittel erforder-
lich ist, ist Methanol nicht notwendig und hat lediglich in hohen Konzentrationen einen
geringen Einfluss auf die Morphologie der Beschichtung.
In gefüllten Beschichtungen wurden die Grenzen für die einzelnen Komponenten der
Beschichtung wie Methanol, Füllstoffe, MTES, Polymere usw. ermittelt, innerhalb wel-
cher die Strukturbildung funktioniert. Aufgrund der ermittelten Grenzen konnten das

Kurzzusammenfassung
2
System auf drei für die Strukturbildung notwendigen Komponenten reduziert werden.
Lediglich Methanol, der Füllstoff SiC und ein kleiner Anteil eines der beiden Polymere
von lediglich 2,5 vol. % sind für die Strukturbildung notwendig.
Aufgrund der Untersuchungen wird daher davon ausgegangen das die Strukturbildung
in gefüllten Systemen wie folgt abläuft: Zu Beginn der Beschichtung sind die Füllstoff-
partikel durch die Scherkräfte während des Rührens und des Beschichtungsvorganges
gleichmäßig verteilt. Sobald die Beschichtung keinen Scherkräften mehr ausgesetzt ist,
beginnt sich das Methanol von den Partikeln zu trennen, so dass erste Methanol Tropfen
entstehen, welche solange anwachsen, bis das Methanol vollständig verdunstet ist. Der
geringe Polymeranteil ist als Binder notwendig, um die trockene Beschichtung zu stabi-
lisieren.
Zudem konnte gezeigt werden, dass die polymeren Beschichtungen unter Beibehaltung
ihrer Morphologie durch thermisches Umsetzen in keramische Beschichtungen über-
führt werden können. Ebenfalls wurde gezeigt, dass die Struktur mit unterschiedlichen
Beschichtungsverfahren wie Tauchbeschichten, Schleuderbeschichten und dem automa-
tischen Filmziehverfahren gleichermaßen aufgebracht werden kann und ebenso auf
planaren wie komplexen dreidimensionalen Substraten, wie beispielsweise auf Rohren
oder keramischen Schäumen, funktioniert.
Als eine einfache Möglichkeit, die Porengröße der Beschichtung gezielt einzustellen,
konnte die definierte Einstellung der Schichtdicke der Beschichtung durch unterschied-
liche Ziehgeschwindigkeiten bei der Tauchbeschichtung dargestellt werden.

Abstract
3
Abstract
This thesis describes the self-assembly of coatings based on preceramic polymers. The
aims were to characterize the raw materials and the coatings with and without filler ma-
terial. Finally, the mechanisms behind the self-assembly were revealed.
Two commercially available preceramic polymers (both polysiloxanes) have been used:
polymethylsilsesquioxane (PMS) and polymethylphenylsilsesquioxane (PMPS). The
Hansen solubility parameters (HSP) for both polymers were determined by three differ-
ent methods. Two of the methods are based on solubility tests with different solvents.
The third method derived the HSP based on the intrinsic viscosity in different solvents.
The determined HSPs values, when applying the three different methods, agreed well.
The mean HSP values for the polymers are: δD = 16.8 MPa1/2
± 0.1, δP = 3.3 MPa1/2
±
1.0, δH = 5.0 MPa1/2
± 1.1 with a radius of 4 MPa1/2
± 0.9 for PMS and for PMPS δD =
17.7 MPa1/2
± 0.7, δP = 8.2 MPa1/2
± 1.3, δH = 6.4 MPa1/2
± 0.9 with a radius 9.2 MPa1/2
± 0.8. Methanol (MeOH) was proven to be non-solvent for the two polymers, based on
the derived HSP values and the four phase system (PMS – PMPS – MTES – MeOH).
Furthermore, the two polymers are incompatible. The solubility behaviour of Methyltri-
ethoxysilan (MTES) could not be clarified by the calculated HSP of MTES.
It could be shown for coatings without filler material that the self-assembly occurs via a
polymer/polymer separation. The polymer/polymer separation yields PMS as the matrix
and PMPS as islands. MTES was found to be crucial as solvent for self-assembly of
filler-free coatings. Methanol was not found to be crucial for such a self-assembly. Only
at very high concentrations, methanol showed a small effect on the morphology of the
coating.
As a next step, coatings with filler material were investigated. Certain limits for single
components of the coating such as methanol, fillers, MTES, polymers, etc. were identi-
fied. Within these limits self-assembly did occur. Accordingly, the three components
necessary for self-assembly could be identified: methanol, SiC as filler material and a
small amount of one of the two polymers (2.5 vol. %).
Based on the detailed experiments and analysis, the following explanation for the self-
assembly in filled systems is proposed. At the beginning of the coating, the filler parti-
cles are evenly distributed because of the shear forces during stirring and during the

Abstract
4
coating process. Methanol starts to separate from the particles when no more shear forc-
es act on the coating. This results in initial methanol drops, which are growing until
methanol is completely evaporated. A small content of polymer is necessary as binder
to stabilize the dry coating.
Furthermore, it could be shown that polymeric coatings could be transferred to ceramic
coatings by thermal treatment. Importantly, the precursor morphology stayed the same
during the thermal process. The structures could be realized by different coating tech-
niques such as dip coating, spin coating or doctor blade. Next to planar substrates, the
coating also works for complex three-dimensional substrates, for example tubes or ce-
ramic foams.
Finally an easy way to tailor the pore size of the coating was found. To do so, the fact
was utilized that the pore size depends strongly on the coating thickness. The film
thickness, again, depends on the withdrawal speed, which can be set within reasonable
limits. Accordingly, the pore size can be tuned by the film thickness.

1 Einleitung und Zielsetzung
5
1 Einleitung und Zielsetzung
Der stetige technische Fortschritt bringt immer neue An- und Herausforderungen an die
verwendeten Materialien mit sich. Aus diesem Grund gewinnen die keramischen Werk-
stoffe wegen ihrer vielfältigen und teils einzigartigen Eigenschaften sowie des Einsatzes
neuer Herstellungs- und Verarbeitungsverfahren immer mehr an Bedeutung. Eine zu
dieser Klasse gehörende Werkstoffgruppe sind die Carbide, zu denen unter anderem
Siliziumcarbid (SiC) zählt. Ursprünglich vor allem wegen ihrer hohen Härte auch in
Verbindung mit der hohen Temperaturbeständigkeit vorwiegend für Schleifmittel und
Feuerfestmaterialien von großem Interesse, gewinnt es heute in Kombination mit hoher
spezifischer Oberfläche immer mehr an Bedeutung für den gezielten Einsatz in neuen
Bereichen wie: Katalyse, Filtration, Verbrennung (Porenbrenner), in Medizinprodukten
und im elektrochemischen Bereich.1
Der Begriff Carbide wurde 1796 erstmals durch A.F. de Fourcroy als Analogie zu Sul-
fiden und Phosphiden verwendet.2 Heutzutage sind eine Vielzahl an Carbiden bekannt,
dazu zählen außer dem bereits genannten SiC auch TiC, WC, und B4C, um nur einige
wichtige zu nennen. Die heute üblichen Syntheseverfahren für diese Stoffklasse ist die
carbothermische Reduktion3–5
oder die Verwendung von Precursor-Materialien6–8
auch
„Polymer derived Ceramics (PDCs)“ genannt. Vor allem im Bereich der PDCs wurden
in den letzten Jahren große Fortschritte gemacht. Bei den dabei verwendeten Precurso-
ren handelt es sich um organische Verbindungen, welche bereits in großen Mengen die
in der daraus abgeleiteten Keramik enthaltenen Elemente beinhalten. Dabei handelt es
sich beispielsweise um siliziumorganische Polymere, die bei vergleichsweise niedrige-
ren Temperaturen zu SiC umgesetzt werden können. Anders als es beispielsweise bei
der carbothermische Reduktion im sogenannten Acheson-Prozess9 der Fall ist, bei dem
Temperaturen von >1800 °C benötigt werden. Da bereits eine große Zahl der Precurso-
ren im industriellen Maßstab hergestellt werden und gleichzeitig die Umsetzungstempe-
ratur gesenkt werden kann, ist damit eine teils erhebliche Kostenreduzierung möglich.
Gleichzeitig ermöglicht der Einsatz von Precursoren auch eine große Anzahl an neuen
Anwendungen, da sowohl mit der Auswahl dieser, gezielt die Eigenschaften der fertigen
Produkte gesteuert werden können, als auch zusätzlich neue Herstellungsrouten möglich

1 Einleitung und Zielsetzung
6
sind, da beispielsweise etablierte Verfahren aus der Kunststofftechnik zur Formgebung
eingesetzt werden können.
Im Jahr 2009 wurde eine Beschichtung10
erstmals beschrieben, die sich auf der Basis
eines Schlickers zur Herstellung von keramischen Folien11
bei der Zugabe von Metha-
nol selbst strukturiert. Ziel dieser Arbeit ist es, die selbststrukturierende Beschichtung
genauer zu charakterisieren und ein erstes Modell der Strukturbildung zu erstellen. Da-
für werden die verwendeten Rohstoffe charakterisiert und Faktoren, die die Strukturen
beeinflussen, untersucht. Zusätzlich sollen die Rahmenbedingungen unter denen die
Beschichtung Strukturen bildet, erforscht werden.
Bei den Rohstoffen soll insbesondere die Löslichkeit der Polymere in Form der Hansen-
Löslichkeitsparameter bestimmt werden, sowie die Löslichkeit der im System verwen-
deten Stoffe untereinander. Zudem sollen füllstofffreie Beschichtungen hergestellt und
charakterisiert werden. Ein besonderes Interesse gilt dabei dem Methanol, da dies der
Hauptunterschied zum Schlicker zur Herstellung keramischer Folien ist.
In gefüllten Systemen sollen die Bedingungen unter denen die Struktur entsteht er-
forscht werden. Dazu werden die Grenzen bestimmt, in denen das System Strukturen
bildet und der Effekt der einzelnen Komponenten auf die Strukturbildung erforscht.
Zudem soll der Ersatz einzelner Rohstoffe insbesondere von Methanol getestet werden.
Hierfür sollen unterschiedliche Lösungsmittel mit Hilfe der zuvor bestimmten Parame-
ter ausgesucht und auf die Eignung zur Strukturbildung hin getestet werden. Auch der
Füllstoff soll genauer untersucht werden. Hier ist insbesondere von Interesse, welche
Parameter erfüllt sein müssen, damit die Strukturbildung erfolgt. Auch der Einfluss des
beschichteten Substrates auf die Strukturbildung soll untersucht werden. Zudem sollen
Parameter untersucht werden, mit deren Hilfe es möglich ist, die Strukturbildung gezielt
zu steuern, um beispielsweise eine definierte Maschenweite einzustellen.
Am Ende der Untersuchungen sollen die Rahmenbedingungen unter denen die Struk-
turbildung funktioniert, geklärt und ein erstes Modell des strukturbildenden Mechanis-
mus aufgestellt sein. Zudem wird ein erster Ausblick auf mögliche zukünftige Anwen-
dungen gegeben, insbesondere im Bezug zu denen, die in der Motivation genannten
wurden.

2 Stand der Technik
7
2 Stand der Technik
2.1 Einsatzgebiete für strukturierte keramische Beschichtungen
Im Jahr 2009 wurde am ZAE Bayern eine netzartige strukturierte keramische Beschich-
tung auf der Basis präkeramischer Polymere erstmals beschrieben.10
Diese Beschich-
tung, auf der Basis eines Schlickers zur Herstellung von keramischen Folien,11
struktu-
riert sich bei der Zugabe von Methanol selbst. Die in Abbildung 1 dargestellte Be-
schichtung, zeigt die Beschichtung sowohl im grünen Zustand (links) nach dem Aushär-
ten bei 110 °C, als auch im gesinterten Zustand (rechts) nach dem Sintern bei 1100 °C.
Zu erkennen ist, dass sich die polymeren Strukturen gut in keramische überführen las-
sen, ohne dass sich die Morphologie dabei verändert.
Abbildung 1: Lichtmikroskopische Aufnahmen mit SiC gefüllter Beschichtungen, links nach
dem Aushärten bei 110 °C und rechts nach dem Sintern unter Stickstoff bei
1100 °C.12
Eine derartige Beschichtung ist von großem Interesse, da eine große Anzahl von An-
wendungen eine gezielte Einstellung der spezifischen Oberfläche bzw. der Porosität
erfordert. Dazu zählen z.B. die Filtration13
und auch Membrane14
. Ein interessantes Ein-
satzgebiet hier sind beispielsweise Rußpartikelfilter in Dieselfahrzeugen, zur Minderung
der Feinstaubbelastung der Luft. Dafür werden Filter benötigt, die einen mittleren Po-
rendurchmesser von 10-20 µm haben und gleichzeitig die hohen Temperaturen im Ab-
gasstrang und während des Ausbrennens der angelagerten Rußpartikel überstehen.13
Zudem konnte gezeigt werden, dass das zusätzliche Aufbringen einer 20 µm dicken
SiC-Membran die Filtereffizienz steigern kann.15
Um die Temperatur von 600 °C wäh-

2 Stand der Technik
8
rend des Ausbrennens zu senken, ist es zudem sinnvoll, die Strukturen mit katalytisch
aktiven Substanzen zu beschichten.16
Auch für Solarabsorber sind sehr ähnliche Anfor-
derungen gefragt. Bei sogenannten Solarturmkraftwerken wird das Sonnenlicht mittels
Spiegeln auf eine kleine Fläche auf einem Turm konzentriert. An dieser Stelle werden
spezielle Materialien benötigt. Zum einen müssen diese bei hohen Temperaturen korro-
sionsbeständig an Luft sein und zum anderen das Sonnenlicht gut absorbieren. Auch die
Temperaturwechselbeständigkeit spielt hier ebenso eine große Rolle. Dabei ist die Poro-
sität der Absorber eine wesentliche Eigenschaft, da kalte Luft durch die Absorber ge-
saugt wird, um diese zu erhitzen, um sie zur Dampferzeugung zu nutzen, wobei über die
Porosität der Strömungswiderstand und der Wärmeübergang eingestellt werden kann.17
Porenbrenner sind ein weiteres interessantes Beispiel, bei denen sowohl die Porosität als
auch die Materialeigenschaften eine wichtige Rolle spielen.18
Bei Untersuchungen für
Porenbrenner hat sich herausgestellt, dass SiC der bester Kompromiss im Vergleich zu
anderen Materialien in Bezug auf Wärmeleitfähigkeit, Temperaturwechselbeständigkeit,
Korrosionsbeständigkeit und Arbeitstemperatur ist.19
Auch für katalytische Prozesse
sind die eben genannten Eigenschaften von Interesse.20
So konnte beispielsweise von
Pham-Huu gezeigt werden, dass die Fischer-Tropsch-Synthese besser auf SiC- als auf
Al2O3-Strukturen funktioniert.21
Zusätzlich ist in Bezug auf Katalyse von Interesse, so-
genannte hierarchische Strukturen zu erzeugen. Hierbei handelt es sich um Strukturen,
bei denen eine feine in eine grobe Struktur eingebracht wird, um beispielsweise die
Oberfläche weiter zu vergrößern. Ein Beispiel hierfür sind keramische Schäume auf die
mit Hilfe des VLS-Mechanismus sogenannte Nanowires aufgebracht wurden22
, um die
katalytisch aktive Oberfläche gezielt zu vergrößern und gleichzeitig katalytisch aktive
Materialien einzubringen. Auch im klassischen Bereich der Feuerfestmaterialien werden
heute teils neue Herausforderungen an die Materialien gestellt. Ein Beispiel hierfür sind
Beschichtungen welche sich für die Transpirationskühlung in Gasturbinen23,24
eignen.
Im Gegensatz zu klassischen Beschichtungen zum Schutz der Turbinen25
, die möglichst
nicht gasdurchlässig sind, ist für die Transpirationskühlung eine gezielte Einstellung der
Porosität und somit der Gasdurchlässigkeit notwendig, da kalte Luft zur Kühlung aus
der Turbinenschaufel durch die Beschichtung strömen muss. Gleichzeitig muss die Be-
schichtung den rauen Bedingungen in einer Gasturbine stand halten können.26
Aus der
Literatur sind weitere Anwendungen bekannt, für die die oben genannte Beschichtung

2 Stand der Technik
9
in Frage kommen könnte. Dazu zählen medizinische Anwendungen wie Knochenim-
plantate27
oder auch die Gasspeicherung,28,29
und elektrochemische Anwendungen wie
Lithiumionenbatterien30,31
, Superkondensatoren32,33
und Brennstoffzellen34,35
.
2.2 Strukturierungsmechanismen beim Beschichten
2.2.1 Strukturierung durch Entmischung in Mehrphasensystemen
Die Strukturbildung durch Entmischung ist für Polymersysteme ein seit langem bekann-
tes Phänomen und wissenschaftlich gut untersucht und daher Bestandteil vieler grund-
legender Artikel und Monographien über Polymere. Daher ist das folgende Kapitel den
Monographien „Polymere: Synthese, Eigenschaften und Anwendungen“36
und „Poly-
mer Physics“37
inhaltlich entnommen.
Für das Verständnis von Mischungen soll zunächst die Gibbs-Helmholz-Gleichung (1)
betrachtet werden, mit deren Hilfe sich eine grundlegende Aussage über die Mischbar-
keit von Systemen machen lässt:
(1)
Dabei ist ΔGm
die Gibbs’sche Freie Enthalpie der Mischung, ΔHm die Mischungsenthal-
pie, ΔSm die Mischungsentropie und T ist die absolute Temperatur. Der Index m be-
schreibt dabei, dass es sich um eine Mischung handelt. Es gilt für Werte ΔGm < 0: das
System mischt sich bzw. ist mischbar, für ΔGm > 0: das System entmischt sich bzw. ist
nicht mischbar. Der Grenzfall ΔGm = 0, bei dem sich Mischungsenthalpie und Mi-
schungsentropie aufheben, wird Φ-Bedingung genannt.
Für ideale niedermolekulare Systeme lässt sich die Gleichung (1) auch wie folgt aus-
drücken:
( ⏟
⏟
) (2)
Dabei ist R die allgemeine Gaskonstante, φ1 und φ2 die Volumenbrüche der jeweiligen
Stoffe und χ der Flory-Huggins-Wechselwirkungsparameter. Da die Volumenbrüche
von Mischungen immer kleiner 1 sind, ergibt sich, dass der Entropieterm immer negativ
ist, was in sich schlüssig ist, da beim Mischen die Entropie als „Maß für die Unord-
nung“ stets zunimmt und das Mischen somit begünstigt. Für χ gilt: je kleiner der Wert,
2 Stand der Technik
10
desto kleiner der Beitrag des Enthalpieterms, was ebenfalls schlüssig ist, da ein großer
Wert für χ auf einen stark endothermen Mischungsvorgang hinweisen würde.
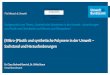
In Abbildung 2 ist der Verlauf der freien Mischungsenthalpie in Abhängigkeit der Zu-
sammensetzung für unterschiedliche Wechselwirkungsparameter χ dargestellt. Wie be-
reits durch Gleichung (2) beschrieben, ist ersichtlich, dass niedrige Wechselwirkungspa-
rameter die Mischung begünstigen und daher zu stabilen Mischungen führen. Für kleine
Werte von χ gilt, dass der Verlauf stets links gekrümmt ist. Für Werte von χ > 2 beginnt
die Kurve in der Mitte rechts gekrümmt zu sein. Dies bedeutet, dass Mischungen rechts
und links existieren, die eine niedrige Mischungsenthalpie aufweisen und das System
beginnt daher instabil zu werden.
Abbildung 2: Diagramm des Verlaufes der freien Mischungsenthalpie für Gemische mit nie-
dermolekularen Substanzen, dargestellt als Funktion der Zusammensetzung bei
verschiedenen Wechselwirkungsparametern. Mit Erlaubnis entnommen aus 36
Copyright (2014) Springer Berlin Heidelberg.
Durch die Linie für χ = 3 ist ersichtlich, dass Mischungen, mit φ = 0,5 nicht stabil sind.
Ein Sonderfall ergeben hier Mischungen die zwischen dem Minimum und dem Wende-
punkt der Kurve liegen. Da die Kurve hier ebenfalls links gekrümmt ist, sind Mischun-
gen in diesem Bereich metastabil. Dies bedeutet, dass zur Entmischung eine Aktivie-
rungsenergie benötigt wird, die vergleichbar mit der Keimbildungsenergie ist. Mit die-
ser Erkenntnis kann nun das Diagramm in Abbildung 2 in das Diagramm in Abbildung
3 überführt werden. Durch das Verbinden der Wendepunkte für unterschiedliche χ

2 Stand der Technik
11
ergibt sich die Spinodale. Unterhalb dieser ist das System instabil und es liegen zwei
separate Phasen vor. Durch das Verbinden der Minima für unterschiedliche χ ergibt sich
die Binodale. Oberhalb dieser ist das System stabil und es liegt eine gemeinsame Phase
vor. Der Bereich zwischen Spinodale und Binodale ist metastabil. Der Punkt an dem
sich beide treffen ist der kritische Punkt.
Abbildung 3: Verlauf von Spinodale und Binodale in niedermolekularen Systemen. Mit Er-
laubnis entnommen aus 36
Copyright (2014) Springer Berlin Heidelberg.
Aufgrund der idealen Annahmen, z.B. gleiches Molvolumen, zeigen niedermolekularen
Systeme einen symmetrischen Verlauf. Daher spielt es hier auch keine Rolle, wenn die
Indizes in den Gleichung (2) vertauscht werden.
2.2.1.1 Entmischung in Polymer-Lösungsmittel-Systemen
Durch die Betrachtung von Polymerlösungen nach Flory und Huggins ändert sich unter
weiterhin idealisierten Annahmen, (z.B. das Monomer als kleinste Baueinheit des Po-
lymers und das Lösungsmittel haben die selbe Größe), dass das Polymer feste nicht
trennbare Verbindungen hat. Dies führt dazu, dass die mögliche Entropiezunahmen
kleiner ist. Um diesem Effekt gerecht zu werden, muss Gleichung (2) um XN, den Poly-
merisationsgrad, im Entropieterm erweitert werden, wodurch sich Gleichung (3) ergibt.
( ) (3)
2 Stand der Technik
12
Für den Flory-Huggins-Wechselwirkungsparameter χ und damit den Enthalpieterm än-
dert sich hingegen nichts, da bei diesem Modell davon ausgegangen wird, dass die
Wechselwirkungen zwischen dem Lösungsmittel und dem aus monomeren Baugruppen
bestehenden Polymer dieselben sind, wie zwischen dem nicht vernetzten Monomer und
dem Lösungsmittel. Da die Entropie auch als „Maß der Unordnung“ angesehen wird,
lässt sich der Einfluss auf den Entropieterm wie folgt erklären: Während das Monomer
in Verbindung mit dem Lösungsmittel in einer niedermolekularen Lösung eine statisti-
sche Verteilung einnehmen kann und damit einer hohe „Unordnung“ möglich ist, ist es
dem Monomer im Polymer nicht mehr möglich, dies zu tun, da es fest an das nächste
Monomer gebunden ist und somit nicht frei jeden beliebigen Ort in der Lösung einneh-
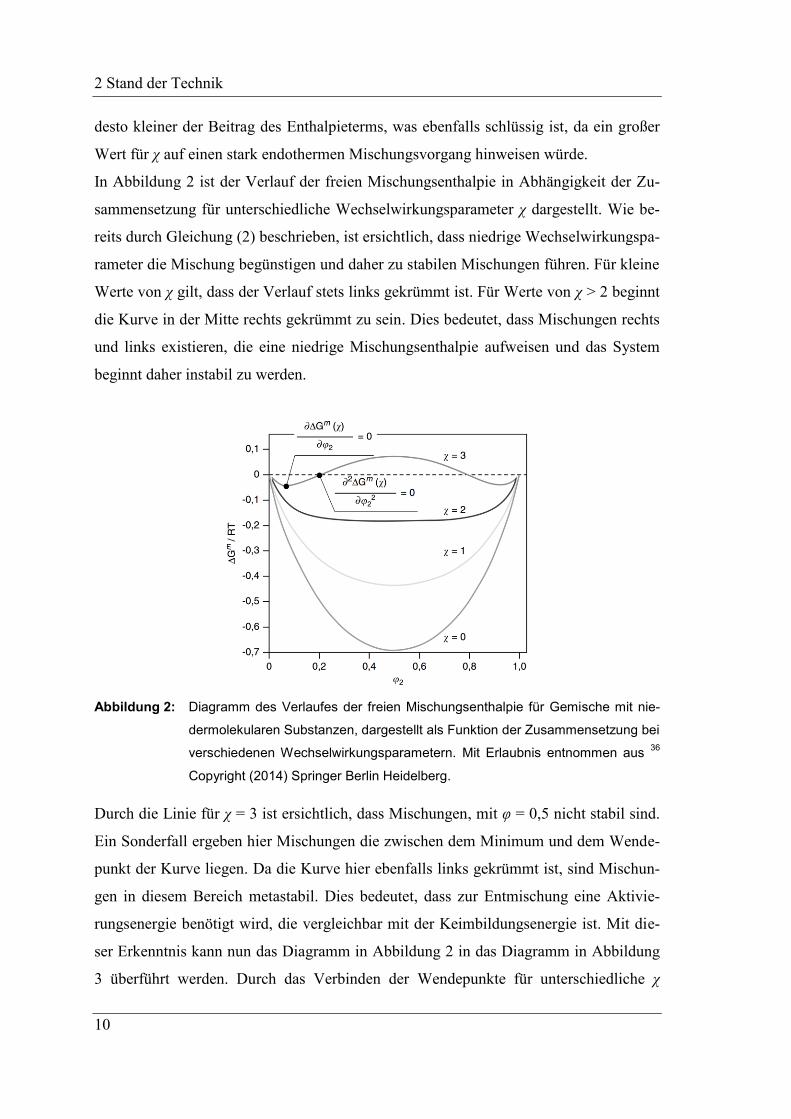
men kann. Dieser Effekt verstärkt sich mit zunehmendem Polymerisationsgrad, was im
Diagramm in Abbildung 4 grafisch dargestellt ist.
Abbildung 4: Diagramm von Binodalen in Systemen mit unterschiedlichen Polymerisations-
graden XN. Mit Erlaubnis entnommen aus 36
Copyright (2014) Springer Berlin
Heidelberg.
Durch die Betrachtung der unterschiedlichen Kurven für verschiedene Polymerisations-
grade in Abbildung 4, wird ersichtlich, dass mit zunehmendem Polymerisationsgrad die
Größe der Mischungslücke zunimmt. Dies betrifft sowohl die Breite als auch die Höhe
der Mischungslücke. Zudem kann im Unterschied zu niedermolekularen Systemen eine

2 Stand der Technik
13
Asymmetrie beobachtet werden: der kritische Punkt verschiebt sich mit zunehmendem
Polymerisationsgrad immer weiter zur lösungsmittelreichen Seite des Phasendiagram-
mes. Dies bedeutet auch, dass nicht, wie bei niedermolekularen Systemen, das Vertau-
schen der Indizes keine Bedeutung hat.
Daher lässt sich festhalten, dass Polymere mit einem höheren Polymerisationsgrad
schlechter löslich sind als solche mit einem niedrigen. Dies rührt daher, dass die Entro-
pie, welche die Mischung fördert, für solche Polymere niedriger ist und daher bereits
ein kleinerer Wechselwirkungsparameter als in niedermolekularen Systemen zur Entmi-
schung führt.
Weiter ist festzuhalten, dass die hier getroffenen Annahmen idealisiert sind. So gilt die-
ses Modell nur für Lösungen, bei denen die Funktionseinheiten (Lösungsmittelmolekül,
Polymermonomer usw.) gleich groß sind und auch keine Größenänderung durch das
Mischen erfolgt. Auch ist es nur für nichtpolare Stoffe definiert, da beispielsweise Was-
serstoffbrückenbindungen nicht berücksichtigt werden können. Dennoch ist das Modell
gut geeignet, um die komplexen Vorgänge in Polymerlösungen zu erläutern und ver-
ständlich darzustellen.
Aufgrund der komplexen Vorgänge in Polymermischungen, und da sowohl der Entro-
pieterm als auch der Enthalpieterm stark von der Temperatur abhängen, treten in Poly-
mer-Lösungsmittel-Systemen unterschiedlichste Mischungslücken auf. So gibt es Mi-
schungslücken die mit UCST (Upper Critical Solution Temperature) bezeichnet werden,
bei der es eine obere kritische Temperatur gibt, oberhalb welcher das System vollstän-
dig mischbar ist. Es existieren Systeme, bei denen es eine Temperatur gibt unterhalb der
das System vollständig mischbar ist und die Mischungslücke oberhalb dieser Tempera-
tur liegt; diese werden mit LCST (Lower Critical Solution Temperature) bezeichnet.
Auch Kombinationen aus beiden sind bekannt. So kann es Systeme geben, die sowohl
eine UCST als auch eine LCST aufweisen oder aber auch solche, bei denen sich beide
überschneiden, so dass nur Lösungen rechts und links der Mischungslücke existieren.
Auch sind Systeme bekannt, bei denen die Mischungslücke flächig in der Mitte des
Phasendiagrammes liegt, so dass es Lösungen oberhalb, unterhalb, rechts und links der
Mischungslücke gibt.

2 Stand der Technik
14
2.2.2 Strukturierte Beschichtungen auf der Basis von Polymeren-Lösungen
Aus der Literatur ist eine Vielzahl von selbststrukturierenden Beschichtungen auf der
Basis von Polymeren bekannt. Der hierbei häufigste Mechanismus ist die Entmischung
zweier unterschiedlicher Polymere. In Abbildung 5 sind Rasterkraftmikroskopische-
Aufnahmen (AFM) einer Polystyrol/ Polymethylmetacrylat (PS/PMMA) Schicht, die
aus einer Toluol Lösung mittels Schleuderbeschichten auf einer Goldoberfläche herge-
stellt wurden, dargestellt. Die dabei entstanden Entmischungsstrukturen unterscheiden
sich lediglich in ihrer Schichtdicke, die durch unterschiedliche Rotationsgeschwindig-
keiten von 2000 min-1
bis 10000 min-1
hergestellt wurden. Die Strukturen zeigen dunkle
Inseln aus PS, umgeben von einer hellen Matrix aus PMMA. Die Morphologie der Be-
schichtung ändert sich mit abnehmender Schichtdicke von Abbildung 5 (a) mit 140 nm
über (b) mit 105 nm und (c) mit 95 nm nach (d) mit 80 nm nicht. Die Größe der Domä-
nen nimmt gleichsam mit der Schichtdicke ab.38
Abbildung 5: AFM-Aufnahmen (26 µm x 28 µm) einer Schichtdickenreihe einer PS/PMMA
Lösung auf einer Goldoberfläche hergestellt mittels Schleuderbeschichten. Mit
Erlaubnis entnommen aus 38
Copyright (1997) American Chemical Society.
Aus der Literatur ist eine Vielzahl weiterer Polymersysteme bekannt, die ähnliche
Strukturen aufweisen. So lassen sich strukturierte, dünne Schichten mit Dicken von < 1

2 Stand der Technik
15
µm aus deuteriertem Polystyrol und Poly-p-Methylstyrol aus Toluol-Lösungen39
, aus
Polystyrol und Polyvinylmethylether in Toluol-Lösungen40
, aus Polyethylenterephtalat
und Styrolbutadien Copolymere in Toluol- und Hexafluoroisopropanol-Lösungen41
, aus
deuteriertem Polystyrol und Polybutadien in Toluol-Lösungen42
und aus Polystyrol und
Polymethylmetacrylat in Toluol-, Tetrahydrofuran- oder Methylethylketon-Lösungen38
erzeugen. Hierbei führt die Verdunstung des Lösungsmittels zur Phasenseparation der
beteiligten Polymere, die insel- oder netzartige Strukturen bilden. Die Ausbildung der
Strukturen hängt von der Unverträglichkeit der Polymere zueinander, dem Polymer-
mengenverhältnis, der Verdunstungsrate des Lösungsmittels und der Dicke der Schicht
ab.38–40
2.2.3 Entstehung von Oberflächenstrukturen durch Entnetzung
Entnetzung ist ein bekannter Mechanismus, der zur Bildung von Strukturen in Be-
schichtungen führen kann. Bei der Entnetzung handelt es sich um einen gut untersuch-
ten Vorgang, da dieser in der Lack- und Farbenindustrie eine große Rolle spielt, um
fehlerfreie Beschichtungen herstellen zu können.
Für die Entstehung von Strukturen durch Entnetzung sind drei verschiedene Mechanis-
men bekannt:43–45
a) spinodale Undulation/Fluktuation der Filmdicke, die durch Wechselwirkungen
an beiden Grenzflächen (zum Substrat und zur Oberfläche) ausgelöst wird,
b) homogene oder thermische Nukleation, bei der die thermische Aktivierung groß
genug ist, um die Barriere zur Nukleation zu überwinden und
c) heterogene Nukleation an Defekten (Blasen, Risse, Partikel), die zu zufällig ver-
teilten, runden Löchern gleicher Größe führt.
Die spinodale Undulation und die homogene oder thermische Nukleation können nur in
hochreinen Systemen auftreten und dort auch nur bis zu einer bestimmten Filmdicke,
die vom zur Beschichtung verwendeten Material sowie den Substrateigenschaften ab-
hängt. Oberhalb dieser Schichtdicke oder bei Verunreinigungen findet heterogene Nuk-
leation statt.43–45

2 Stand der Technik
16
In Abbildung 6 ist der schematische Verlauf der Entnetzung eines dünnen, flüssigen
Filmes, wie er von Stange beschrieben wird, dargestellt.46
Die Entnetzung beginnt mit
der Entstehung von Keimen. Dieser Schritt wird auch Nukleation genannt und ist in
Abbildung 6 a dargestellt. Im weiteren Verlauf entstehen Löcher, welche zunehmend
wachsen, dargestellt in Abbildung 6 b und c. Die Entnetzung ist mit dem vollständigen
Vereinigen der Löcher abgeschlossen. Bei vollständiger Entnetzung liegt eine zellulare
Struktur vor, die von kleinen Tropfen auf den Grenzen gebildet wird und in Abbildung
6 d dargestellt ist.
Abbildung 6: Schematischer Ablauf der Strukturentstehung währende der Entnetzung eines
dünnen flüssigen Filmes. Mit Erlaubnis entnommen aus 46
Copyright (1997)
American Chemical Society
2.2.4 Beeinflussung von Polymerbeschichtungen durch Füllstoffe
Da in dieser Arbeit sowohl füllstoffhaltige als auch füllstofffreie Beschichtungen be-
trachtet werden, soll in diesem Abschnitt auf den Einfluss von Füllstoffen auf struktu-
rierte Beschichtungen eingegangen werden. In der Literatur ist eine Vielzahl von Effek-
ten durch Füllstoffe auf die Strukturierung von Polymerbeschichtungen beschrieben
worden.47–50
In Polystyrol/Polypropylen-Systemen kann die Zugabe von SiO2-Nanopartikeln die
Phasenseparation verlangsamen. Dieser Effekt wird darauf zurückgeführt, dass sich die

2 Stand der Technik
17
Partikel an den Phasengrenzen der Polymere sammeln und somit die Vereinigung der
Tröpfchen behindern.47
Bei der Zugabe von Schichtsilikatpartikeln zu einer Polystyrol-Polyvinylmethylether-
Mischung wurde ein Einfluss der Partikelgröße auf die Entmischung beobachtet. Die
Zugabe von großen Partikeln mit einem Durchmesser von 10 µm führt zu einer Be-
schleunigung der Entmischung und damit zu größeren Entmischungsstrukturen in der
Beschichtung. Während hingegen die Zugabe kleinerer Partikel mit einem Durchmesser
von 1µm die Phasenseparation verlangsamte und die Strukturen in der Beschichtung
verkleinerte. Gleichzeitig konnte auch ein Einfluss des Volumenanteils der Silikate mit
einem Durchmesser von 1µm auf die Entmischung beobachtet werden. Mit zunehmen-
dem Silikatanteil steigt die Anzahl der Domänen, bei gleichzeitiger Abnahme ihrer
Größe.48
Simulationen mit sich entmischenden binären Polymersystemen, bei denen die Partikel
vorwiegend von einer Phase benetzt werden, haben gezeigt, dass sich die dabei bilden-
den Muster wie in Abbildung 7 dargestellt verhalten. Dabei hat sich gezeigt, dass sich
mit der Art und der Anzahl der Partikel, die Morphologie der Entmischungsstrukturen
gezielt beeinflussen lässt.49
Während zunächst in Abbildung 7 a) und b) die zu erwar-
tende Struktur gebildet wurde, bei der die Phase mit dem geringeren Volumenanteil
(graue Phase) die Inseln bildet und die Phase mit dem höheren Volumenanteil (schwar-
ze Phase) ein Netzwerk formte, bildete in Abbildung 7 c die graue Phase, die die Parti-
kel beinhaltet, ein Netzwerk aus. Dieser Effekt wird auf den Anstieg der Viskosität mit
steigendem Partikelgehalt zurückgeführt, da dieser Effekt sich auch in ungefüllten Sys-
temen, bei denen sich die Phasen in der Viskosität unterscheiden, beobachtet werden
kann.49,50

2 Stand der Technik
18
Abbildung 7: Darstellung von Simulationsergebnis für ein entmischendes binäres System mit
mobilen runden Partikeln für unterschiedliche Zeiten und unterschiedliche Parti-
kelzahlen a) 144 Partikel, b) 289 Partikel und c) 400 Partikel. Mit Erlaubnis ent-
nommen aus 49
Copyright (2006) by The American Physical Society.
Entnetzungsstrukturen werden ebenfalls durch Füllstoffe beeinflusst. Die Entnetzung
von Polystyrol- und Polybutadien-Schichten wird durch Zusatz von Fulleren-
Nanopartikeln gehemmt, die sich an die Substratoberfläche anlagern und die Polymer-
Substrat-Wechselwirkung beeinflussen. Zudem kann der Zusatz von Nanopartikeln die
Entnetzung von dünnen Polymerschichten in der Weise verzögern, dass durch Partikel-
Polymer-Anziehung eine lokale Erhöhung der Viskosität erfolgt. Die Verwendung grö-
ßerer Partikel führt im beschrieben System dagegen zu spontaner Entnetzung.51,52
2.2.5 Strukturbildung durch Konvektion
Die Strukturierung von Filmen durch Konvektion wurde bereits 1900 von Benard expe-
rimentell und 1916 von Rayleigh theoretisch beschrieben.53
Die dabei entstehenden
Strukturen werden durch sogenannte Benard-Konvektionszellen erzeugt, die in Abbil-
dung 8 schematisch dargestellt sind. Die Konvektionszellen entstehen in diesem Bei-
spiel durch eine Temperaturdifferenz zwischen dem beschichteten Substrat und der
Oberfläche der Beschichtung. Teile der flüssigen Beschichtung werden hierbei am Sub-
strat erhitzt, wodurch sie sich ausdehnen und die Dichte abnimmt. Dadurch entsteht eine
Auftriebskraft vom Substrat weg, hin zur Oberfläche der Beschichtung. An der Oberflä-
che der Beschichtung kühlt die Beschichtung wieder ab, wodurch sie sich zusammen-

2 Stand der Technik
19
zieht und die Dichte dadurch zunimmt, was zu einem Absinken führt. Da der Prozess im
Kreislauf abläuft, können sich sogenannte Konvektionswalzen bilden, die sich wiede-
rum in einzelnen Zellen befinden können.
Abbildung 8: Schematische Darstellung einer Benard-Konvektionszelle entnommen aus 54
Aus der Literatur ist bekannt, dass durch diesen Effekt sowohl sehr regelmäßige hexa-
gonale Strukturen, ähnlich derer von Bienenwaben,55
als auch unregelmäßige,56
die den
in dieser Arbeit beschrieben Struktur ähneln, entstehen können.
Die Temperaturdifferenz kann hierbei durch verschieden Effekte zustande kommen. Der
ursprüngliche, durch Benard beschriebene Fall, wird durch gezieltes Heizen des Sub-
strates hervorgerufen.57
Jedoch sind auch Fälle bekannt, bei denen die Temperaturdiffe-
renz durch die Verdunstungskälte des Lösungsmittels an der Oberfläche der Beschich-
tung entsteht.56,58
Sind Füllstoffe in der Beschichtung enthalten, so werden diese bevorzugt an den Au-
ßenseiten der Konvektionszellen angelagert, was zu porösen Strukturen nach dem Ver-
dampfen des Lösungsmittels führt.54
2.3 Technisch relevante Verfahren zur Herstellung poröser
Keramiken
Da es sich in dieser Arbeit um ein neues Verfahren zur Herstellung strukturierter bzw.
poröser Beschichtungen handelt, soll in diesem Abschnitt ein kurzer Überblick über
heute bekannte Verfahren gegeben werden, um diese Verfahren mit dem neunen Ver-
fahren, entwickelt im Laufe dieser Arbeit, besser vergleichen zu können.
Für die Herstellung poröser Beschichtungen mit gezielt steuerbarer Porosität sind meh-
rere Verfahren bekannt. Die bei weitem häufigste Methode ist die Verwendung von
Platzhaltern. Dafür werden in die Beschichtung gezielt Platzhaltermaterialien einge-

2 Stand der Technik
20
bracht, welche anschließen während des Sinterns ausgebrannt werden. Mit der gezielten
Auswahl der Platzhaltermaterialien kann die Porengröße und Porenform definiert einge-
stellt werden. Auch das Steuern von offener und geschlossener Porosität ist mit dem
Volumenanteil der Platzhaltermaterialien einstellbar.19
Ein Beispiel hierfür ist eine mik-
roporöse Kalziumphosphatbeschichtung auf einem Zirkoniumoxidträgermaterial27
, bei
der die Porosität durch die Zugabe von PMMA und Stärke als Platzhaltermaterial er-
zeugt wurde. Ein weiteres Beispiel ist eine Anodenbeschichtung für eine Direktkohlen-
stoffbrennstoffzelle59
, bei der die Porosität der Anodenbeschichtung durch die Zugabe
von Kohlenstoff erzeugt wurde.
Eine weitere, häufig verwendete Methode zur Herstellung poröser Keramiken ist das
sogenannte Replikaverfahren: Hierbei werden zumeist polymere Strukturen, häufig PU-
Schäume mit einer Suspension, auch Schlicker genannt, aus keramischen Füllstoffen,
Bindern und weiteren Bestandteilen beschichtet und anschließend gesintert, wobei der
polymere Schaum ausgebrannt wird und die Keramik die ursprüngliche Form beibe-
hält.19
Ein auf diesem Weg häufig hergestelltes Produkt sind keramische Schäume für
Porenbrenner60
oder Filter für Metallschmelzen61
.
Ein weiterer Weg ist das direkte Schäumen keramischer Schlicker. Hierfür gibt es zwei
unterschiedliche Wege: das Aufschäumen des flüssigen Schlickers durch die Zugabe
von Gas62
oder das Erzeugen der Gasblasen durch im Schlicker ablaufende Reaktionen
währende der thermischen Umsetzung63
, welche die Gasblasen erzeugen.
Zudem sind weitere Verfahren zur Erzeugung von porösen Strukturen bekannt, dazu
zählen das erzeugen der Porosität durch Gefriertrocknung64
oder das Umsetzen natürli-
cher Strukturen wie Holz65
in Keramiken.19
2.4 Präkeramische Polymere
Carbide, Nitride oder Boride werden zum gegenwärtigen Zeitpunkt meist durch car-
bothermische Reduktion der entsprechenden Metalloxide oder durch Reaktion der sie
aufbauenden Elemente hergestellt.7,66
Für diese Prozesse sind jedoch sehr hohe Tempe-
raturen, je nach System, von über 1600 °C notwendig und daher mit einem hohen Ener-
gieaufwand verbunden. Der häufig verwendete Achenson-Prozess zur Herstellung von
SiC zum Beispiel benötigt Temperaturen von über 2000 °C bei einem geringen Umset-
zungsgrad von lediglich 10 % bis 15 %.9,66

2 Stand der Technik
21
Ein weiterer Weg zu Herstellung von nichtoxidischen Keramiken ist der Weg über prä-
keramische Polymere.67
Bei präkeramischen Polymeren handelt es sich im weitesten
Sinne um organische Verbindungen, die in ihrem chemischen Verhalten denen von Oli-
gomeren ähneln. Je nach ablaufender Vernetzungsreaktion erfolgt eine Umwandlung in
Duroplaste, Elastomere oder Thermoplaste. In ihrer Oligomer-bzw. Polymerform treten
die meisten präkeramischen Polymere als Pulver oder hochviskose Flüssigkeiten auf.
Aufgrund ihres chemischen Aufbaus reagieren einige Prekursoren mehr oder minder
stark mit Sauerstoff bzw. dem in der Luft enthaltenen Wasser, wodurch die Haltbarkeit
in Abhängigkeit von der umgebenden Atmosphäre eingeschränkt wird. Des Weiteren
sind präkeramische Polymere z.T. stark temperaturreaktiv. Dies ist sowohl bei der La-
gerung als auch bei der Verarbeitung zu beachten. Eine Übersicht über präkeramische
Polymere und deren Nomenklatur ist in Abbildung 9 gegeben. Die organischen Reste R
können hierbei z.B. H-, Alkyl-, Hydroxyl-, Ethoxy-, Aryl- oder Arenylgruppen sein.6,66
Abbildung 9: Übersicht über die verschiedenen Siliziumpolymere und deren Namensgebung.
Mit Erlaubnis entnommen aus 6 © 2000 WILEY-VCH Verlag GmbH, Weinheim,
Fed. Rep. of Germany
Eine häufig eingesetzte Gruppe präkeramischer Polymere sind die Polysiloxane, da die-
se bereits seit vielen Jahren bekannt sind und industriell hergestellt werden. Diese wer-
den weiter unterscheiden in linearvernetzte Polysiloxane mit der Baugruppe [-R2SiO-]n
und quervernetzte Polysilsesquioxane mit der Baugruppe [-RSiO1,5-]n. Polysilsesquio-

2 Stand der Technik
22
xane die kommerziell erhältlich sind, haben überwiegend Methyl- und/oder Phenyl-
gruppen an das Si-Atom gebunden.66,68
Die Vernetzung von Siloxanen findet durch Polykondensation, Additionsreaktionen
oder Radikalreaktionen statt. Bei kommerziellen Siloxanen findet die Vernetzung
hauptsächlich durch Silanol-Silanol-Kondensation statt.8 Für die Vernetzung sind in der
Regel Temperaturen zwischen 150 °C und 250 °C oder Katalysatoren notwendig. Als
Katalysatoren kommen häufig Metallsalze wie Kobalt-, Blei- und Zinksaltze zum Ein-
satz.66,69
Ab etwa 500 °C beginnt die Zersetzung der Polymere und die Bildung der ke-
ramischen Phasen. Die Umsetzung ist bei etwa 1000 °C abgeschlossen. Bei der Ver-
wendung von Polysiloxanen entstehen Siliziumoxicarbid-Keramiken. Die Vernetzung
sowie die Pyrolyse unter unterschiedlichen Atmosphären wie Argon- oder Stickstoffat-
mosphäre, sowie die dabei entstehenden Phasen sind Gegenstand zahlreicher wissen-
schaftlicher Veröffentlichungen.6–9,11,66–79
2.5 Theorie der Löslichkeitsparameter
Um die Löslichkeitsparameter zu verstehen, sollte zunächst Löslichkeit an sich definiert
werden. Unter Löslichkeit wird in der Regel die Menge eines Stoffes, die sich in einer
bestimmten Menge eines anderen Stoffes löst, verstanden. Damit ist gemeint in welcher
Menge sich die Atome, Moleküle oder Ionen des Stoffes A gleichmäßig in der bestimm-
ten Menge des Stoffes B verteilen. Ein einfaches Beispiel hierfür ist das lösen von
Kochsalz (NaCl) in Wasser, wobei sich der Feststoff NaCl bis zu einer bestimmten
Menge als Ionen Na+ und Cl
- in der Flüssigkeit Wasser löst. Vergleiche hierzu auch
Kapitel 2.2.1 in dem die Thermodynamischen Bedingungen beschrieben werden, unter
denen eine Löslichkeit bzw. Mischbarkeit vorliegt.
Löslichkeitsparameter finden seit vielen Jahren in der Lack- und Farbenindustrie ihren
Einsatz.80
Heute gibt es noch viele weitere Einsatzgebiete: organische Halbleiter81
,
Elektrospinnen82
sowie in der Beschichtungsindustrie im Allgemeinen83
. Eine einfache
Art „Löslichkeitsparameter“ ist ein einfacher Merksatz, der heißt: „Gleiches löst Glei-
ches“84
, der Vielen bekannt sein dürfte. Damit ist gemeint, dass sich Stoffe mit gleichen
Eigenschaften ineinander lösen jedoch nicht in Stoffen mit anderen Eigenschaften. Ein
bekanntes Beispiel hierfür ist Zucker, der sich im polaren Lösungsmittel Wasser löst
jedoch nicht im unpolaren Lösungsmittel Benzin. Doch ist damit nicht genau definiert,

2 Stand der Technik
23
was „Gleich“ ist und es gibt auch keine entsprechenden Tabellenwerke. Daher wurde
1950 von Hildebrand und Scott ein auf Zahlenwerten basierender Parameter etabliert,
der auf der Kohäsive-Energiedichte beruht und daher der Kraft entspricht, die notwen-
dig ist, um alle Moleküle zu trennen.85
Dieser Parameter ist jedoch nur für die Bestim-
mung der Löslichkeit unpolare Stoffe geeignet. Daher wurden im Laufe der Zeit weitere
Parameter eingeführt. Eines der bekannten Systeme ist hierbei das von Hansen 197983
eingeführte System der Hansen-Löslichkeits-Parameter (HSP), welches den Parameter
nach Hildebrand weiter aufteilt und dadurch auch für polare Stoffe angewendet werden
kann.
2.5.1 Definition der Löslichkeitsparameter
Der erste Löslichkeitsparameter wurde von Hildebrand und Scott eingeführt, dabei han-
delt es sich um den sogenannten Hildebrand- bzw. absoluter Löslichkeitsparameter δT,
der die Wurzel aus der Kohäsive-Energiedichte c ist. Bei der Kohäsive-Energiedichte
eines Materials handelt es sich um die Energie, die notwendig ist, um alle Atome oder
Moleküle dieses Materials vollständig voneinander zu trennen. Die Kohäsive-
Energiedichte wird nach Gleichung (4) berechnet, indem von der Verdampfungsenthal-
pie ΔH das Produkt aus der Gaskonstante R und Temperatur T abgezogen wird und die
Differenz daraus durch das molare Volumen V geteilt wird. Da diese auf der Messung
der Verdampfungsenthalpie beruht, ist dies eine direkte Messung der Kräfte, die die
Atome oder Moleküle eines Stoffes zusammenhalten. Dieser Parameter ist jedoch nur
für unpolare Stoffe geeignet. Daher wurde der absoluter Löslichkeitsparameter von
Hansen in drei Parameter aufgeteilt, die den Einsatz auch für polare Stoffe zulässt.86
√ √
√
(4)
Die Hansen-Löslichkeitsparameter ergeben sich durch das Aufteilen der Energie, die für
den Zusammenhalt der Atome und Moleküle verantwortlich ist, nach Gleichung (5).
Hierfür werden die einzelnen Anteile für den dispersen Anteil ED, den polaren Anteil EP
und den Wasserstoffbrückenanteil EH gebildet und anschließend durch das molare Vo-
lumen V geteilt, woraus sich Gleichung (6) und (7) ergeben:

2 Stand der Technik
24
oder:
Dabei gilt für den dispersen Anteil δD:
√
(8)
für den polaren Anteil δP:
√
(9)
und für den Wasserstoffbrückenanteil δH:83
√
(10)
Für Lösungsmittel ist eine große Datenbasis an experimentell bestimmten HSP in der
Literatur vorhanden.80
Ursprünglich wurden die HSP von Lösungsmitteln mit Hilfe der
sogenannten Homomorphen-Methode bestimmt. Dabei wird zuerst der Hildebrand-
Löslichkeitsparameter des zu bestimmenden Lösungsmittels über dessen Verdamp-
fungsenthalpie bestimmt. Anschließend wird von diesem der Anteil des nicht polaren
Homomorphs des zu bestimmenden Lösungsmittels abgezogen (z. B. ist n-Butan der
nicht polare Homomorph von N-Butylalkohol). Dies ergibt die Summe des polaren und
des Wasserstoffbrückenanteils. Hansen teilte diese Summe durch Versuch und Irrtum
unter Verwendung mehrerer Lösungsmittel in die jeweiligen Anteile auf, bis diese em-
pirisch am besten das jeweilige Dipolmoment wiederspiegelten. Heutzutage werden der
disperse Anteil meist mit Hilfe des Brechungsindex, der polare Anteil mit Hilfe des
(5)
(6)
(7)

2 Stand der Technik
25
Dipolmomentes und der Wasserstoffbrückenanteil mit Hilfe der Gruppen-
Beitragsmethode bestimmt.86
2.5.2 Anwendung der Löslichkeitstheorie von Hansen auf Polymere
Die HSP von Polymeren sind in der Literatur nicht so vollständig erfasst, wie die der
Lösungsmittel. Insbesondere für die in dieser Arbeit verwendeten Polymere auf Silizi-
umbasis besteht eine große Lücke. Daher müssen diese bestimmt werden. Besser jedoch
funktioniert die Methode, bei der die HSP anhand der Verdampfungsenthalpie bestimmt
werden, bei Polymeren nicht, da diese sich bereits vor dem Verdampfen zersetzen.80
Zur Bestimmung der HSP von Polymeren gibt es mehrere Möglichkeiten. Eine Methode
besteht darin, die HSP mit Hilfe der Gruppen-Beitragsmethode zu bestimmen. Hierfür
wird jedoch der Anteil, den die jeweilige Gruppe des Polymers zur Kohäsive-
Energiedichte beiträgt, benötigt. Da die in dieser Arbeit verwendeten Polymere jedoch
siliziumbasiert sind, fehlen die entsprechenden Gruppen bei den gängigen Methoden zur
Berechnung der HSP.80,87
Eine weitere Methode die HSP theoretisch zu bestimmen,
beruht auf COSMO RS.88
Diese Methode ist vor allem für die Berechnung kleiner Mo-
leküle wie Lösungsmittel und Pharmazeutika bekannt.89,90
Für Polymere hingegen müs-
sen diese Methoden noch verbessert werden. Neueste Fortschritte auf diesem Gebiet
zeigen bereits vielversprechende Ergebnisse.91,92
Zudem liefern die heute bekannten
Methoden meist abweichende HSP zu denen, die experimentell bestimmt wurden.80
Milliman hat beispielsweise für Siliziumpolymere gezeigt, dass theoretisch berechnete
HSP zu weniger brauchbaren Ergebnissen führen, als die experimentellen HSP für die-
selben Polymere.93
Daher wird es als notwendig erachtet, die HSP für die in dieser Ar-
beit verwendeten Polymere experimentell zu bestimmen. Dafür gibt es mehrere Metho-
den. Eine indirekte Methode besteht darin, die Polymere mit einer großen Anzahl an
Lösungsmitteln, in einer für das vorliegende Problem relevanten Weise in Kontakt zu
bringen. Hierfür eignen sich zum Beispiel Lösungsansätze, bei denen das Polymer in
einer bestimmten Konzentration in verschiedenen Lösungsmitteln zu lösen versucht und
beurteilt wird, ob dieses sich darin löst oder nicht.83
Eine weitere Methode besteht in der
Bestimmung der inhärenten Viskosität. Hierbei wird davon ausgegangen, dass das Po-
lymer die größte Interaktion mit dem Lösungsmittel zeigt, das den HSP des Polymers

2 Stand der Technik
26
am besten entspricht und somit auch die inhärente Viskosität bei diesem Lösungsmittel
am größten ist.94
2.5.3 Bestimmung der Löslichkeitsparameter mittels Löslichkeitsversuchen
Eine von Hansen bereits 1969 beschriebene Methode zur Bestimmung von HSP für
Polymere basiert darauf, das zu bestimmende Polymer in Kontakt mit einer Vielzahl
von Lösungsmitteln zu bringen.83
Dabei wird davon ausgegangen, dass Lösungsmittel
mit HSP nahe derer des zu bestimmenden Polymers mit diesem besser interagieren, als
solche, die weit davon entfernt liegen. Anhand verschiedener Parameter, wie der voll-
ständigen Lösung einer bestimmten Konzentration, visueller Beurteilung der Schwel-
lung, Volumen- oder Gewichtszunahme und weiteren, werden die verwendeten Lö-
sungsmittel in zwei Gruppen eingeteilt: in Lösungsmittel und in Nichtlösungsmittel. Mit
Hilfe eines Computerprogramms oder graphisch können anhand dieser Einteilung, die
HSP des Polymers bestimmt werden. Hierfür werden die HSP der Lösungsmittel in 3D-
Diagramme eingetragen, unter Verwendung des dispersen, polaren und Wasserstoffbrü-
ckenanteils als Achsen. Untersuchungen haben hierbei gezeigt, dass wenn der disperse
Anteil hierbei verdoppelt wird, sich nahezu kugelförmige Bereiche für die Löslichkeit
ergeben. Daher wird zur Bestimmung der HSP der Polymere anschließend eine Kugel in
das Diagramm konstruiert, welche alle Lösungsmittel einschließt womit alle Nichtlö-
sungsmittel außerhalb dieser Kugel liegen. Das Zentrum der Kugel gibt hierbei die HSP
des zu bestimmenden Polymers wieder, während das Volumen der Kugel der Bereich
ist, in dem die Lösungsmittel liegen, mit denen das Polymer interagiert und in der Regel
als Interaktionsradius Ra angegeben wird.80,83,95
Mit Hilfe von Gleichung (11) kann der
Abstand Ro zwischen zwei Materialien bestimmt werden, wie zum Beispiel der zwi-
schen einem Lösungsmittel und einem Polymer. Der Faktor vier in dieser Gleichung
ergibt sich hierbei aus der Doppelung des dispersen Anteiles.
Mit Hilfe des Abstandes Ro und dem ermittelten Interaktionsradius Ra eines Polymers,
kann nun mit Gleichung (12) der relative Abstand RED bestimmt werden.
√ ( ) ( ) ( ) (11)

2 Stand der Technik
27
Hierbei gilt: ist der relative Abstand RED gleich 0 so haben die beiden verglichenen
Materialien die gleichen HSP, ist RED kleiner gleich 1 interagieren die Materialien mit-
einander, da sie innerhalb der Kugel bzw. auf der Oberfläche der Kugel liegen und ist
RED größer als 1 interagieren sie nicht, da sie außerhalb der Kugel liegen.
Mit Hilfe der in der Literatur vorhanden Daten für Lösungsmittel HSP80
und Gleichung
(11) und (12) kann somit ermittelt werden, welche Lösungsmittel mit einem Material
interagieren. Somit ist zum Beispiel die Wahl neuer Lösungsmittel für einen gegebenen
Prozess möglich, die beispielsweise eine besseren Prozess ermöglichen oder die Um-
weltverträglichkeit verbessern.
2.5.4 Bestimmung der Löslichkeitsparameter anhand der intrinsischen Viskosität
Eine weitere experimentelle Methode zur Bestimmung der HSP beruht auf der Bestim-
mung der intrinsischen Viskosität für verschiedene Lösungsmittel. Bei dieser Methode
wird davon ausgegangen, dass Lösungsmittel, die HSP nahe derer der Polymere haben,
eine höhere intrinsische Viskosität zeigen, also solche, die eine große Differenz zu den
HSP der Polymere aufweisen.80,96
Van Dyk hat gezeigt, dass die intrinsische Viskosität
auch durch die inhärente Viskosität bei einer Konzentration von 5 g l-1
ersetzt werden
kann, was den Messaufwand deutlich verringert, da nur eine Konzentration pro Lö-
sungsmittel gemessen werden muss und nicht mehrere zur Bestimmung des Nulldurch-
ganges, wie bei der intrinsischen Viskosität.96,97
Segarceanun hat die mathematischen Grundlagen für die Gleichungen (13), (14) und
(15) gelegt, mit deren Hilfe aus den gemessenen inhärenten Viskositäten die einzelnen
Anteile der HSP des jeweilige Polymeres bestimmt werden können.94
Hierfür wird die
Summe der Produkte aus dem jeweiligen Anteil der HSP des Lösungsmittels δDS i, δPS i,
und δHS i und der zugehörigen inhärenten Viskosität inh i multipliziert und durch die
Summe der inhärenten Viskosität geteilt.
(12)

2 Stand der Technik
28
∑ ∑
(13)
∑ ∑
(14)
∑ ∑
(15)
Bei dieser Methode kann mit Hilfe von Gleichung (11) der Radius der Kugel bestimmt
werden, indem zum einen die mit Gleichungen (13), (14) und (15) gewonnen HSP für
das Polymer und zum anderen nacheinander die HSP der verwendeten Lösungsmittel
eingesetzt werden. Ein als gutes ermitteltes Lösungsmittel mit dem größten Abstand zu
den HSP des Polymers ergibt dabei den Radius.
2.6 Beschichtungsverfahren
2.6.1 Tauchbeschichten
Das Tauchbeschichtungsverfahren ist ein Verfahren, bei dem ein Substrat durch Eintau-
chen in eine Flüssigkeit und anschließendes Herausziehen unter definierten Bedingun-
gen beschichtet wird. Damit steht ein Beschichtungsprozess zur Verfügung, der mit
vergleichsweise geringem technischem Aufwand betrieben werden kann. Diese Metho-
de liefert in hohem Maße homogene Schichten und kann dadurch auch in der Produkti-
on großflächiger Bauteile eingesetzt werden.98
Nachteilig ist, dass gerade bei komple-
xen Formteilen große Flüssigkeitsmengen benötigt werden. Neben flachen Trägermate-
rialien können bedarfsweise auch Hohlkörper, Röhren, Fasern und Schäume mit ver-
tretbarem Aufwand beschichtet werden.99–101
In Abbildung 10 ist der Tauchbeschich-
tungsprozess schematisch dargestellt. Dabei wird das Substrat mit einer definierten Ge-
schwindigkeit in den Schlicker eingetaucht. Nach einer definierten Haltezeit, wird es
mit einer fest definierten Geschwindigkeit wieder herausgezogen. Das verwendete Lö-
sungs- bzw. Suspensionsmittel verdunstet, wodurch sich der Schlickerfilm zu verfesti-
gen beginnt. Ein wichtiger Aspekt sind die im Beschichtungsraum vorherrschenden
Bedingungen wie Temperatur und Luftfeuchtigkeit. Diese haben Einfluss auf die vor-

2 Stand der Technik
29
herrschende Konvektion bzw. das Verdampfen und somit auch auf die Trocknungszeit.
Um eine Rissbildung zu vermeiden, sind demnach eine kontrollierte Atmosphäre und
eine entsprechende Trocknungszeit notwendig.99–106
Abbildung 10: Schematische Darstellung des Tauchbeschichtungsprozesses. Die Pfeile ent-
lang des Substrates stellen die Strömungsrichtung der Luft dar. Mit Erlaubnis
aus 102
entnommen und übersetzt.
Bereits 1942 haben Landau und Levich den theoretischen Zusammenhang zwischen der
Schichtdicke s und den Beschichtungsbedingungen, wie der Ziehgeschwindigkeit v0, der
Viskosität der Flüssigkeit Fl, der Dichte der Flüssigkeit ρ, der Erdbeschleunigung g,
und der Oberflächenspannung der Flüssigkeit γ mathematisch beschrieben (vgl. Glei-
chung (16)):101,106
Mit Hilfe von Gleichung (16) ist es möglich, die Schichtdicke der nicht getrockneten
Beschichtung zu berechnen. Es ist jedoch nicht direkt möglich, die Schichtdicke nach
dem Trocknen zu berechnen. Zudem ergibt sich aus der Gleichung, dass mit Hilfe der
Ziehgeschwindigkeit die Schichtdicke gezielt beeinflusst werden kann. Höhere Ziehge-
schwindigkeiten führen nach Gleichung (16) zu dickeren Schichten, sofern die übrigen
Beschichtungsbedingungen konstant gehalten werden.
2.6.2 Schleuderbeschichten
Eine ebenfalls technisch ausgereifte Beschichtungsmethode ist das Schleuderbeschich-
ten. Hierbei wird das Substrat mittels Vakuum auf einem Drehteller fixiert. Die Entste-
( )
( )
(16)

2 Stand der Technik
30
hung des Films läuft nach Scriven in vier Phasen ab.100
Die vier Phasen der Schichtent-
stehung sind in Abbildung 11 schematisch dargestellt. Der Beschichtungsprozess be-
ginnt mit dem Auftragen des Beschichtungssystems, wobei dieses sowohl auf das noch
ruhende als auch auf das bereits rotierende Substrat erfolgen kann. Wichtig ist in diesem
Zusammenhang, dass wesentlich mehr Material auf das Substrat aufgetragen werden
muss, als zur Realisierung der Schicht notwendig ist. Dann wird das Substrat in eine
Drehbewegung versetzt. Dies entspricht der Hochschleuderphase, bei der das Beschich-
tungssystem radial nach außen getrieben wird, wodurch eine Verteilung auf dem Sub-
strat erreicht wird. In der anschließenden Abschleuderphase wird überschüssiges Mate-
rial entfernt. Schließlich verdunstet das Lösungsmittel und hinterlässt einen festen
Film.100
Abbildung 11: Schematische Darstellung der Schichtentwicklung während des Schleuderbe-
schichtens.100
Meyerhofer hat 1978 die Schichtentstehung dabei mit Gleichung (17) beschrieben:
Die Schichtdicke s wird aus der Ausgangsschichtdicke s0, der Umdrehungsgeschwin-
digkeit ω, der kinematischen Viskosität υ und der Zeit t nach Gleichung (17)
berechnet.107
Ein entscheidender Parameter beim Schleuderbeschichten ist die Umdre-
hungsgeschwindigkeit, da sie vor allem Einfluss auf die auftretenden Zentrifugalkräfte
√
(17)

2 Stand der Technik
31
ausübt, die für die Verteilung des Schlickers verantwortlich sind. Die Dicke des Films
entsteht durch die Wechselwirkung zwischen der Zentrifugalkraft und der Trocknungs-
zeit. Mit einsetzender Verdunstung des Lösungsmittels und beginnender Trocknung
steigt die Viskosität des Schlickers und der Einfluss der Zentrifugalkraft reduziert sich.
Ab diesem Zeitpunkt wird die Filmdicke nicht mehr von der Endrotationsgeschwindig-
keit bestimmt.100,107
2.6.3 Automatisches Filmziehen
Das automatische Filmziehverfahren, im Folgenden auch als Rakeln bezeichnet, kann
als eine spezielle Form des „doctor blade“-Beschichtens angesehen werden.108
Dabei
wird eine geringe Menge Schlicker vor einer sich in einem Schlitten befindlichen Klin-
ge gegeben. Substrat und Klinge werden relativ zueinander bewegt, so dass der Schli-
cker sich auf dem Substrat verteilt bzw. zwischen Trägermaterial und Klinge austritt,
wodurch sich eine dünne Schicht ausbildet. Die Klinge ist dabei in ihrer Höhe verstell-
bar. Somit lassen sich verschiedene Schichtdicken erzeugen. Während bzw. nach dem
Beschichtungsvorgang verdunstet das Lösungsmittel und es bildet sich eine stabile
Schicht aus.108–111
Nach Mistler und Twiname ist für die Erzeugung von Schichten beim Rakeln u.a. die
Viskosität des Beschichtungsmediums von Bedeutung, da sie einen erheblichen Einfluss
auf das Fließverhalten ausübt. Während die Klinge über dem Substrat bewegt wird, be-
stimmt die Viskosität das Maß des Schlickerflusses. Niedrigviskose Schlicker verteilen
sich demnach schneller und können dem sich bewegenden Schlitten lediglich eine ver-
gleichsweise kleine Kraft entgegenbringen. Neben der Viskosität hat auch die Ge-
schwindigkeit des Schlittens einen wichtigen Einfluss auf die Verteilung des Schlickers.
Von ihr geht die Verteilung des Schlickers aus und sie bestimmt die auf den Schlicker
einwirkende Kraft.
Der Klingenabstand ist die wichtigste Einflussgröße bei der Erzeugung der Schichtdi-
cke. Parameter wie Viskosität und Geschwindigkeit haben also vor allem Einfluss auf
das Ausbreitungsverhalten des Schlickers, wohingegen der Klingenabstand die Höhe
definiert.108–111
Durch Chou wurde eine Gleichung (21) beschrieben mit deren Hilfe sich die nasse
Filmdicke s berechnen lässt:112

2 Stand der Technik
32
Dabei ist: ρ die Dichte der Flüssigkeit, ρ‘ die Dichte des nassen Filmes, h0 der Abstand
zwischen dem Rakel und der Platte, ΔP der Druckunterschied, die Viskosität der
Flüssigkeit, U die Verfahrgeschwindigkeit des Rakel und L die Breite des Rakel senk-
recht zur Verfahrrichtung.
2.7 Rheologische Eigenschaften keramischer Schlicker
Für die Betrachtung der rheologischen Eigenschaften von keramischen Schlickern ist
die Viskosität eine entscheidenden Größe. Wobei die Viskosität ein Maß für die Rei-
bung der in der Flüssigkeit bewegten Teilchen darstellt. Die Viskosität ist dabei nach
Gleichung (19) als das Verhältnis von Scherspannung zu Schergeschwindigkeit D
definiert:113
Wobei die Scherspannung nach Gleichung (20) das Verhältnis der Kraft F, die benö-
tigt wird, um zwei parallele Platten gegeneinander zu verschieben, zu Fläche A ist:
Die Viskosität ist eine Materialeigenschaft und hängt in Falle von keramischen Schli-
ckern von der Zusammensetzung ab. Einen großen Einfluss haben dabei die Füllstoff-
partikel. Mit zunehmendem Füllstoffgehalt steigt auch die Viskosität an. Auch die
Korngröße der Füllstoffe hat dabei einen Einfluss. Mit abnehmender Korngröße, bei
gleichem Füllstoffgehalt und Material, steigt die Viskosität ebenfalls an. Dies liegt da-
ran, dass kleiner Partikel im Verhältnis zu ihrem Volumen eine größere Oberfläche ha-
ben und die Reibung in der Flüssigkeit dadurch steigt.113
Flüssigkeiten die einen linearen Zusammenhang zwischen Scherspannung und Scherge-
schwindigkeit aufweisen, nennt man Newton’sche Flüssigkeiten. Im Falle von Schli-
(
) (18)
(19)
(20)

3 Experimentelles Vorgehen
33
ckern, kann es dazu kommen, das durch die Ausrichtung von z.B. plättchenförmigen
Partikeln bei der Erhöhung der Schergeschwindigkeit die Scherspannung abnimmt, da
die Partikel so leichter aneinander abgeleiteten werden können. Dieses Verhalten wird
als strukturviskos oder scherverdünnend bezeichnet. Auch das gegenteilige Verhalten
ist bekannt und wird als dilatantes oder scherverfestigendes Verhalten bezeichnet und
tritt beispielsweise bei hohen Feststoffgehalten auf, bei denen mit zunehmender Scher-
geschwindigkeit die Wechselwirkungen zwischen den Partikeln zunehmen. Auch sind
von der Einwirkzeit der Scherrate abhängige Reaktionen bekannt. So werden Schlicker,
die sich in Abhängigkeit von der Zeit verdünnend verhalten als thixotrop und sich ver-
festigende als rheoplex bezeichnet. Die Ursachen hierfür sind ähnlich den zuvor be-
schrieben.113
3 Experimentelles Vorgehen
3.1 Verwendete Rohstoffe
3.1.1 Polymere
Als Polymere wurden zwei kommerziell erhältliche Polysilsesquioxane, ein Polyme-
thylsiloxan (PMS, Silres® MK, allgemeine Formel: [CH3SiO1.5]n, Mw = 6600 g mol-1
)
und ein Polymethylphenylvinylsiloxane (PMPS, Silres® H62 C,
[(C6H5)0.44(CH3)0.24(C2H3)0.16H0.16SiO1.5]n , Mw = 1200 g mol-1
), beide von der Firma
Wacker-Chemie GmbH, München verwendet. Zur Vernetzung der Polymere wurden
zwei Katalysatoren eingesetzt: Ölsäure (C18H34O2; Merck KGaA, Darmstadt, Deutsch-
land) dient der Vernetzung bei Raumtemperatur unter 100 °C und Aluminiumacetylace-
tonat (C15H21AlO6, Merck KGaA, Darmstadt) katalysiert die Vernetzung des PMS bei
Temperaturen ab etwa 100 °C. Die Vernetzung von PMPS und Überführung in ein
Thermoset läuft ab etwa 80 °C platin-katalysiert ab. Der dazu notwendige Katalysator
ist Bestandteil des kommerziell erhältlichen PMPS.

3 Experimentelles Vorgehen
34
3.1.2 Lösungsmittel
Für die Beschichtungen wurden zwei Lösungsmittel verwendet, Methanol (ROTI-
PURAN® ≥99.9 %, [CH3OH], Carl Roth GmbH + Co. KG, Karlsruhe) und Methyl-
triethoxysilan (MTES, [(C2H5O)3SiCH3], Wacker-Chemie GmbH, München). Zur Er-
mittlung der Hansen Löslichkeitsparameter wurde eine weitere Reihe Lösungsmittel
verwendet, diese können Tabelle 1 entnommen werden. Die Lösungsmittel wurden im-
mer in der höchsten kommerziell erhältlichen Reinheit bezogen.
Tabelle 1: Lösungsmittel zur Ermittlung der Löslichkeitsparameter nach Hansen
Lösungsmittel Summenformel Hersteller
1,3-Propandiol C3H8O2 ABCR GmbH & Co. KG
1,4-Dichlorbutan C4H8Cl Merck KGaA
N-Methyl-2-pyrrolidon C5H9NO Alfa Aesar GmbH & Co. KG
1-Methylnaphthalin C11H10 Merck KGaA
2-Butanol C4H10O Sigma-Aldrich Chemie GmbH
2-Butanonoxim C4H9NO k. A.
2-Propanol C3H8O Honeywell
Aceton C3H6O Honeywell
Benzoylchlorid C7H5ClO Alfa Aesar GmbH & Co. KG
Butanon C4H8O Carl Roth GmbH & Co. KG
Cyclopentan C5H10 Merck KGaA
Diacetonalkohol C6H12O2 Alfa Aesar GmbH & Co. KG
Dibutylmaleat C12H20O4 Sigma-Aldrich Chemie GmbH
Diethylcarbonat C5H10O3 Merck KGaA
Diethyleneglycol C4H10O3 Carl Roth GmbH & Co. KG
Diglykolmonobutyletheracetat C10H20O4 Sigma-Aldrich Chemie GmbH
Dimethylaminoethanol C4H11NO Sigma-Aldrich Chemie GmbH
Dipropylamin C6H15N Merck KGaA
Essigsäure C2H4O2 Carl Roth GmbH & Co. KG
Ethanol C2H7O Th. Geyer GmbH & Co. KG
Ethylencyanhydrin C3H5NO Merck KGaA
Furfurylalkohol C5H6O2 Merck KGaA
Inden C9H8 Merck KGaA
Isophoron C9H14O Merck KGaA
Mesitylen C9H12 Merck KGaA
Mesityloxid C6H10O Merck KGaA
Methanol CH4O Carl Roth GmbH & Co. KG
Methylphenylsulfid C7H8S k. A.
Methyltriethoxysilan C7H18O3Si Wacker-Chemie GmbH
Monoethanolamin C2H7NO Sigma-Aldrich Chemie GmbH
n-Hexan C6H14 Merck KGaA
Nitroethan C2H5NO2 Alfa Aesar GmbH & Co. KG
Ölsäure C18H34O2 Merck KGaA
Tetrahydronaphthalin C10H12 Merck KGaA
Wasser H2O ZAE Bayern

3 Experimentelles Vorgehen
35
3.1.3 Substrate
Als Referenzsubstrat wurde ein foliengegossenes Aluminiumoxid (Keral 96, KERA-
FOL Keramische Folien GmbH, Eschenbach) mit einer Stärke von 0,5 mm eingesetzt.
Zur Charakterisierung des Einflusses der Substratmaterialien auf die Struktur der Be-
schichtungen wurden weitere Werkstoffe verwendet, siehe Tabelle 2.
Gitter und Rohre wurden hierbei zur Untersuchung des Einflusses komplexer dreidi-
mensionaler Geometrien herangezogen. Insbesondere im Falle der Rohre ist eine geziel-
te Untersuchung dieser Strukturen möglich, da sowohl das Verhalten auf konvexen und
konkaven Substraten, als auch im Inneren von Strukturen untersucht werden kann. Zu-
dem ist die Variation der Radien leicht möglich.
Tabelle 2: Zur Beschichtung verwendete Substrate
Substrat Material Firma
Keral 96 Al2O3 Kerafol
Stahl 1.4876 X10NiCrAlTi32 20H Zapp AG
Bandstahl Stahl HMC H.Meyer & Co. Spezialstahl GmbH
Kupferblech Cu k. A.
Alu dick Al k. A.
Alu mittel Al Merck KGaA
Alu dünn Al k. A.
Objektträger Kalk-Natron-Glas Carl Roth GmbH + Co. KG
Si-Wafer Si k. A.
Gitterstruktur Al k. A.
Al2O3 Tapes Al2O3-SiOC-Keramik ZAE Bayern
SiC Tapes SiC-SiOC-Keramik ZAE Bayern
Rohre Al2O3 Friatec AG

3 Experimentelles Vorgehen
36
3.1.4 Füllstoffe
Als Referenzfüllstoff, soweit nicht anders angegeben, wurde ein Siliziumcarbidpulver
(SM07, SiC, d50 1,3-1,7 µm, ESK-SIC GmbH, Frechen-Grefrath) eingesetzt. Um den
Einfluss von Werkstoff und Partikelgröße auf die Schichtstruktur zu untersuchen, wur-
den weitere Korngrößen und Werkstoffe verwendet, siehe Tabelle 3.
Tabelle 3: Verwendete Füllstoffe und Partikelgrößen
Bezeichnung Material d50 Firma
[µm]
NF25 SiC 0,67 ESK-SIC GmbH
NF15 SiC 0,87 ESK-SIC GmbH
NF10 SiC 1,06 ESK-SIC GmbH
SM07 SiC 1,41 ESK-SIC GmbH
F1500 SiC 2,22 ESK-SIC GmbH
F1200 SiC 4,55 ESK-SIC GmbH
F1000 SiC 5,41 ESK-SIC GmbH
F800 SiC 8,42 ESK-SIC GmbH
CT530SG Al2O3 1,72 Almatis GmbH
Silgrain HQ Si 3,84 Elkem
KS10 C 6,20 TIMCAL Deutschland GmbH
AlFP11010 Al 6,86 Sigma-Aldrich Chemie GmbH
Siliziumnitrid Si3N4 k. A. k. A.
Bornitrid BN k. A. k. A.
Borcarbid B4C k. A. k. A.
Quarzgut SiO2 k. A. k. A.
Glas Glas k. A. k. A.
Kupferoxid CuO k. A. k. A.
Magnesiumoxid MgO k. A. k. A.
Zirkoniumoxid ZrO2 k. A. k. A.
3.2 Bestimmung der Löslichkeitsparameter
Die Löslichkeitsparameter der Polymere wurden mit zwei unterschiedlichen Methoden
ermittelt. Die erste Methode beruht auf Lösungsversuchen, wobei hierbei zwei unter-
schiedliche Berechnungsmethoden zum Einsatz kamen. Bei der zweiten Methode wer-
den die HSP mit Hilfe der inhärenten Viskosität bestimmt.
Für die Lösungsversuche wurden Mischungen von 50 vol. % Polymer und 50 vol. %
Lösungsmittel mit Hilfe eines Magnetrührers hergestellt. Als geeignete Lösungsmittel
wurden solche definiert, bei denen die Mischung vollständig klar wurde und blieb; diese
Lösungsmittel wurden zur Berechnung mit „1“ markiert. Lösungsmittel, bei denen die
Mischung nicht vollständig klar blieb, wurden als Nichtlösungsmittel deklariert und zur

3 Experimentelles Vorgehen
37
Berechnung mit „0“ gekennzeichnet. Die HSP wurden anhand dieser Einteilung zum
einen mit der Software HSPiP von Hansen80
und zum anderen mit Hilfe eines von Gha-
ragheizi114
beschriebenen Algorithmus in Matlab (The MathWorks, Inc., Natick, MA,
USA) berechnet. Die Kugeldiagramme in den Abbildungen wurden ebenso mit Matlab
erstellt.
Als weitere Methode zur Bestimmung der Löslichkeitsparameter nach Segarceanu94
diente die Bestimmung der inhärenten Viskosität mittels Ubbelohde-Viskosimetrie.
Hierfür wurde zuerst die Durchlaufzeit nach DIN 51562115
für das reine Lösungsmittel
tsol bestimmt und anschließend die Durchlaufzeit tsol+pol für Lösungsmittel und Polymer
in einer Konzentration von c = 5 g l-1
ermittelt. Als Lösungsmittel wurden nur solche
verwendet, die in den Lösungsversuchen als geeignet mit „1“ klassifiziert wurden. Mit
Hilfe von Gleichung (21) wurde aus den gemessenen Durchlaufzeiten die relative Vis-
kosität rel bestimmt:
(21)
Aus der relativen Viskosität kann mit Hilfe von Gleichung (22) die inhärente Viskosität
inh berechnet werden, die Konzentration c muss hierbei in g dl-1
zur Berechnung her-
angezogen werden:
(22)
Mit Hilfe der Gleichungen (13), (14) und (15) können aus den gemessenen inhärenten
Viskositäten die einzelnen Anteile der HSP des jeweiligen Polymers bestimmt werden.
Hierfür wird die Summe der Produkte aus den jeweiligen Anteilen der HSP des Lö-
sungsmittels δDS i, δPS i, und δHS i mit der zugehörigen inhärenten Viskosität inh i, durch
die Summe der inhärenten Viskositäten der Lösungsmittel geteilt:

3 Experimentelles Vorgehen
38
∑ ∑
(13)
∑ ∑
(14)
∑ ∑
(15)
3.3 Bestimmung der Mischbarkeiten in den verschiedenen
Systemen
Zur Aufklärung der Mischbarkeit im Vier-Phasen-System Methanol, Methyltriethoxy-
silan (MTS), Polymethylsiloxan (PMS) und Polymethylphenylsiloxan (PMPS) wurden
Mischungen mit einer Abstufung von 16,67 vol. %, über einen großen Zusammenset-
zungsbereich hergestellt. Es konnten dabei keine Proben mit einer PMS-Konzentration
größer als 50 vol. % hergestellt werden, da PMS als festes Pulver in solch hohen Kon-
zentrationen unter Raumtemperatur nicht im System löslich ist. Die Komponenten wur-
den in einem Becherglas mit Hilfe eines Magnetrührers für 15 Minuten vermischt und
anschließend im Ultraschallbad für 10 Minuten weiter homogenisiert. Die Mischbarkeit
der Proben wurde anhand ihrer Trübung beurteilt, wobei nur vollständig klare Mischun-
gen als mischbar gewertet wurden.
Zudem wurden DSC-Untersuchungen (Q1000, TA Instruments, New Castle, Vereinigte
Staaten) zur Ermittlung der Glasübergangstemperaturen zur Bestimmung der Mischbar-
keit der reinen Polymere PMS und PMPS sowie Mischungen der beiden Polymere
durchgeführt.
3.4 Herstellung der Schichten
Die Herstellung der Schichten erfolgte im Allgemeinen nach dem nachstehenden Ver-
fahren. Hierbei wurde darauf geachtet, dass die Schritte möglichst exakt eingehalten
wurden, um Einflüsse bei der Herstellung der Schlicker bzw. der Schichten möglichst

3 Experimentelles Vorgehen
39
gering zu halten. Für einzelne Versuche wurden einzelne Komponenten weggelassen,
die Zeiten wurden jedoch weiter eingehalten.
Zuerst wurden PMPS und MTES für 10 min mit Hilfe eines Magnetrührers verrührt.
Anschließend wurde PMS zugegeben und vollständig durch weitere 2 h rühren aufge-
löst. Danach wurde Methanol zugegeben und für weitere 30 min homogenisiert. Im An-
schluss an die Methanolzugabe wurde der Füllstoff zugegeben und für 15 min gerührt.
Zum Schluss wurden die Katalysatoren Ölsäure und Aluminiumacetylacetonat zugege-
ben. Nach vollständiger Zugabe aller Ausgangssubstanzen wurde der Schlicker durch
weiteres Rühren auf dem Magnetrührer über 18 h vollständig homogenisiert. Zum Auf-
lösen eventuell noch vorhandener Agglomerate wurde der Schlicker vor dem Beschich-
tungsprozess für 10 min bei höchster Stufe im Ultraschallbad homogenisiert und nach
Entnahme wiederholt für 3 min aufgerührt.
Beim anschließenden Tauchbeschichtungs-Prozess (Tauchbeschichter, Eigenbau ZAE
Bayern) wurden die Substrate mit einer Geschwindigkeit von 4,7 mm s1-
eingetaucht,
für 1 s im Schlicker gehalten und mit einer Geschwindigkeit von 23,1 mm s-1
aus dem
Schlicker gezogen. Zwischen jedem Beschichtungsvorgang wurde der Schlicker für 2
min gerührt. Die angegebenen Geschwindigkeiten und Haltezeiten wurden bei jedem
Versuch konstant gehalten. Nach dem Tauchbeschichten wurden die Proben bei 30 °C
im Trockenschrank (TR 120, Nabertherm GmbH, Lilienthal, Deutschland) über 24 h
getrocknet und anschließend für weitere 24 h bei 110 °C vernetzt.
Ausgewählte Proben wurden im Rohrofen (LOSIC, HTM Reetz GmbH, Berlin,
Deutschland) in Stickstoffatmosphäre pyrolysiert. In der ersten Stufe wurde bis 400 °C
mit 1 K min-1
aufgeheizt und für 1 h gehalten, danach bis 800 °C wiederum mit 1 K
min-1
und ebenso für 1 h gehalten. Anschließend wurde bis zur Maximaltemperatur von
1100 °C mit 2 K min-1
geheizt und für 2 h gehalten. Das Abkühlen auf Raumtemperatur
erfolgte mit 3 K min-1
.
Alle füllstoffhaltigen Schichten wurden, soweit nicht anders angegeben, nach der oben
angegebenen Verfahrensweise und mit der Zusammensetzung wie in Tabelle 4 angege-
ben, hergestellt. Wurden einzelne Komponenten variiert, ist das im jeweiligen Abschnitt
angegeben. Die restlichen Komponenten wurden stets im gleichen Verhältnis zueinan-
der belassen.

3 Experimentelles Vorgehen
40
Tabelle 4: Referenzzusammensetzung für füllstoffhaltige Systeme
Ausgangsstoff Menge
[vol %]
PMPS 9,2
MTES 8,1
PMS 9,2
Methanol 53,8
SiC SM07 18,1
Ölsäure 1,1
Al(acac)3 0,6
3.5 Herstellung von füllstofffreien Beschichtungen
Zur Untersuchung ungefüllter Systeme wurden vier Grundzusammensetzungen COMP
n, vgl. Tabelle 5, so gewählt, dass COMP 1 nahe der Zusammensetzung der Referenz-
zusammensetzung in gefüllten Systemen lagt, bei der PMS, PMPS und MTES etwa im
selben Verhältnis enthalten sind. Die weiteren drei Zusammensetzungen wurden so ge-
wählt, dass sie COMP 1, welche exakt in der Mitte des Dreiphasensystem PMS – PMPS
– MTES liegt, gleichmäßig umgeben. Mit jeder Grundzusammensetzung wurden
Schichten mittels Tauchbeschichtung hergestellt und untersucht. Die Herstellung der
Schlicker und Beschichtungen erfolgte hierbei analog wie unter 3.4 beschrieben, jedoch
wurden dabei der Füllstoff und das Methanol zunächst weggelassen.
Zusätzlich wurde Zusammensetzung COMP 1 in Schritten von 10 vol. % mit Methanol
verdünnt, um den Einfluss von Methanol auf die Strukturbildung zu untersuchen. Hier-
für wurde von der Stammlösung COMP1 mit Methanol eine Verdünnungsreihe herge-
stellt. Von der Verdünnungsreihe wurden ebenfalls Beschichtungen hergestellt und un-
tersucht.
Für ausgewählte Zusammensetzungen, insbesondere für solche, die für Beschichtungen
in Frage kamen, wurden UV/VIS spektroskopische Untersuchungen (Cary 500 Scan,
Varian Inc., Palo Alto, USA) bei einer Wellenlänge von 550nm durchgeführt. Als Be-
zugslinie der Transmission aus den UV/VIS-Untersuchungen diente die Grundzusam-
mensetzung ohne Methanol.

3 Experimentelles Vorgehen
41
Tabelle 5: Zusammensetzung für ungefüllte Beschichtungssysteme
System PMS PMPS MTES
[vol %] [vol %] [vol %]
COMP 1 33,3 33,3 33,3
COMP 2 60,0 20,0 20,0
COMP 3 20,0 60,0 20,0
COMP 4 20,0 20,0 60,0
3.6 Herstellung der Beschichtungen mittels Schleuderbeschichten
und Filmziehen
3.6.1 Schleuderbeschichten
Neben dem beschriebenen Tauchbeschichten wurde als weitere Beschichtungsmethode
das Schleuderbeschichten angewandt. Auch hierfür wurde ein Spincoater verwendet,
der vom ZAE Bayern selbst konstruiert wurde. Basis der Beschichtung war auch hier
die Referenzzusammensetzung.
In diesem Zusammenhang wurde der Einfluss der Erdrotationsgeschwindigkeit unter-
sucht. Die Rotationsdauer betrug dabei 60 s. Die Rotationsgeschwindigkeit an sich wur-
de bei gleichbleibender Dauer im Bereich von 1000 s-1
in 1000er Schritten auf 6000 s-1
erhöht.
3.6.2 Filmziehen
Als weitere Beschichtungsmethode kam ein automatisches Filmziehverfahren (Rakeln)
zur Anwendung. Hierfür wurde das automatische Filmziehgerät (ZAA 2300, Zehntner
GmbH, Sissach, Schweiz) eingesetzt. Als Schlickeransatz wurde wieder die Referenz-
zusammensetzung gewählt. Im Rahmen dieser Versuchsreihe wurde ein Applikator mit
einer Breite von 60 mm verwendet. Die Ziehgeschwindigkeit wurde von 10 mm s-1
bis
99 mm s-1
in 10 mm s-1
Schritten bei einer Spaltbreite von 500 µm variiert. Zudem wur-
de bei einer Ziehgeschwindigkeit von 40 mm s-1
die Spaltbreite von 10 µm bis 3000 µm
logarithmisch variiert.
3.7 Charakterisierung der Beschichtungen
Zur Charakterisierung der Beschichtungen wurden lichtmikroskopische Aufnahmen
(Carl Zeiss MicroImaging GmbH, Jena) in verschiedenen Vergrößerungen, unter der

3 Experimentelles Vorgehen
42
Verwendung von Objektiven mit verschiedenen Brennweiten von 2,5x, 10x, 20x, 50x
und 100x angefertigt. Die Charakterisierung erfolgte nach dem Vernetzen bei 110 °C.
Die Aufnahmen wurden im Auflicht mit dem Kontrastverfahren Dunkelfeld erstellt. Zur
Erzielung einer besseren Tiefenschärfe wurden Stapelaufnahmen verschiedener Schär-
feebenen erstellt, die mit Hilfe der Software AxioVisio (V 4.8.1, Carl Zeiss MicroIma-
ging GmbH, Jena) zu einem Bild zusammengesetzt wurden. Die Porengrößenanalyse
erfolgte mit der Software AxioVisio anhand der Kontrastunterschiede in den Mikro-
skopaufnahmen.
Zur Bestimmung der Schichtdicke wurden die verwendeten Al2O3-Referenzsubstrate
beim Zuschneiden mittels Laser (PowerLine E 30, ROFIN-SINAR Technologies, Inc.,
Plymouth, USA) zusätzlich in der Hälfte gelasert (Sollbruchstelle), so dass sie nach dem
Aushärten durch gezieltes Brechen geteilt werden konnten. Zum Messen der Schichtdi-
cke wurden anschließend lichtmikroskopische Aufnahmen vom Querschnitt mit 10x
Objektiv erstellt und mit der Software AxioVisio vermessen.
Zusätzlich wurden rasterelektronenmikroskopische Aufnahmen (REM) (JSM 6400, Jeol
Ltd., Tokio, Japan oder ULTRA PLUS, Carl Zeiss AG, Jena, Deutschland) und ener-
giedispersive Röntgenanalysen (EDX) angefertigt. Dazu wurden ausgewählte polymere
und pyrolysierte Proben mit Gold besputtert.

4 Ergebnisse
43
4 Ergebnisse
4.1 Ungefüllte Systeme auf Basis präkeramischer Polymere
PMS und PMPS
4.1.1 Hansen Löslichkeitsparameter der präkeramischen Polymere
Die Hansen Löslichkeitsparameter wurden anhand von drei Methoden bestimmt. Zwei
Methoden beruhen auf Löslichkeitsexperimenten, bei denen die Lösungsmittel in zwei
Gruppen eingeteilt wurden. Anhand dieser Aufteilung wurden die HSP nach zwei Be-
rechnungsmethoden nach Hansen80
und Gharagheizi114
bestimmt. Die dritte Methode
beruht auf der Bestimmung der inhärenten Viskosität mittels Ubbelohde-Viskosimetrie
und der Berechnung der HSP aus diesen Viskositäten nach Segarceanu94
.
In Tabelle 6 sind die verwendeten Lösungsmittel und ihre zugehörigen HSP dargestellt.
Auch dargestellt ist deren Einteilung in Lösungsmittel markiert mit „1“ und Nichtlö-
sungsmittel markiert mit „0“, für das jeweilige Polymer PMS oder PMPS. Einige der
verwendeten Lösungsmittel können jedoch nicht für die Berechnung herangezogen
werden; dies hat verschiedene Gründe. Einige der Lösungsmittel interagieren chemisch
mit den verwendeten Polymeren und sind mit „a“ in der Tabelle gekennzeichnet. Im
Falle von PMPS gestaltet sich die Einteilung als schwierig, da PMPS eine hochviskose
Flüssigkeit ist, die eine leicht gelbliche Eigenfärbung und zusätzlich eine leichte Trü-
bung aufweist. Daher wurden im Falle von PMPS Dibutylmaleat, Inden und Tetrahyd-
ronaphthalin nicht zur Berechnung herangezogen, da nicht eindeutig sicherzustellen
war, dass die Mischungen keine Trübung aufweisen.
Diethylcarbonat in Mischung mit PMS ergibt zunächst eine klare Lösung, welche je-
doch nach einigen Minuten beginnt sich einzutrüben. Dies ist ein Hinweis auf eine
chemische Reaktion. Daher wurde auch Diethycarbonat nicht zur Berechnung der HSP
von PMS herangezogen. Dies gilt auch für Dipropylamin, welches als Mischung mit
PMS nach kurzer Zeit aushärtet, was ein Zeichen dafür ist, dass Dipropylamin ein Ver-
netzungsmittel für PMS ist.
Methyltriethoxysilan, in der Tabelle mit „b“ gekennzeichnet, ist für beide Polymere
PMS und PMPS, ein gutes Lösungsmittel. Da die HSP für MTES jedoch nicht in der in
der Hansen Software enthaltenen Liste der Lösungsmittel enthalten sind und diese daher
mithilfe dieser Software berechnet wurden, kann nicht sichergestellt werden, dass die

4 Ergebnisse
44
HSP für MTES richtig sind. Daher wurde auch im Falle von MTES darauf verzichtet,
dieses für die Berechnung heranzuziehen.
Im Falle von Ölsäure (mit „c“ gekennzeichnet) ist bekannt, dass diese als Vernetzungs-
hilfsmittel bei niedrigen Temperaturen fungiert, daher wurde auch Ölsäure nicht zur
Berechnung der HSP der beiden Polymere PMS und PMPS herangezogen.
Methanol konnte, wie aus der Tabelle ersichtlich ist, als Nichtlösungsmittel für beide
Polymere identifiziert werden und ist demensprechend mit „0“ in der Tabelle gekenn-
zeichnet.
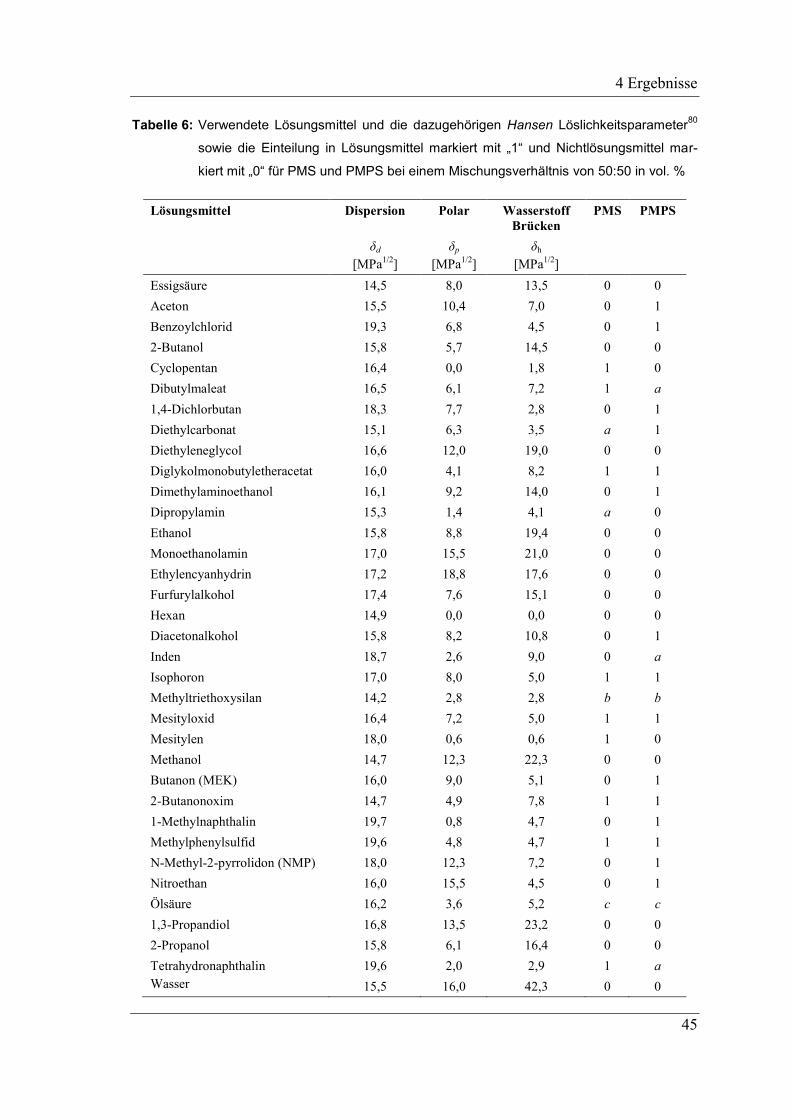
4 Ergebnisse
45
Tabelle 6: Verwendete Lösungsmittel und die dazugehörigen Hansen Löslichkeitsparameter80
sowie die Einteilung in Lösungsmittel markiert mit „1“ und Nichtlösungsmittel mar-
kiert mit „0“ für PMS und PMPS bei einem Mischungsverhältnis von 50:50 in vol. %
Lösungsmittel Dispersion Polar Wasserstoff
Brücken
PMS PMPS
δd
[MPa1/2
]
δp
[MPa1/2
]
δh
[MPa1/2
]
Essigsäure 14,5 8,0 13,5 0 0
Aceton 15,5 10,4 7,0 0 1
Benzoylchlorid 19,3 6,8 4,5 0 1
2-Butanol 15,8 5,7 14,5 0 0
Cyclopentan 16,4 0,0 1,8 1 0
Dibutylmaleat 16,5 6,1 7,2 1 a
1,4-Dichlorbutan 18,3 7,7 2,8 0 1
Diethylcarbonat 15,1 6,3 3,5 a 1
Diethyleneglycol 16,6 12,0 19,0 0 0
Diglykolmonobutyletheracetat 16,0 4,1 8,2 1 1
Dimethylaminoethanol 16,1 9,2 14,0 0 1
Dipropylamin 15,3 1,4 4,1 a 0
Ethanol 15,8 8,8 19,4 0 0
Monoethanolamin 17,0 15,5 21,0 0 0
Ethylencyanhydrin 17,2 18,8 17,6 0 0
Furfurylalkohol 17,4 7,6 15,1 0 0
Hexan 14,9 0,0 0,0 0 0
Diacetonalkohol 15,8 8,2 10,8 0 1
Inden 18,7 2,6 9,0 0 a
Isophoron 17,0 8,0 5,0 1 1
Methyltriethoxysilan 14,2 2,8 2,8 b b
Mesityloxid 16,4 7,2 5,0 1 1
Mesitylen 18,0 0,6 0,6 1 0
Methanol 14,7 12,3 22,3 0 0
Butanon (MEK) 16,0 9,0 5,1 0 1
2-Butanonoxim 14,7 4,9 7,8 1 1
1-Methylnaphthalin 19,7 0,8 4,7 0 1
Methylphenylsulfid 19,6 4,8 4,7 1 1
N-Methyl-2-pyrrolidon (NMP) 18,0 12,3 7,2 0 1
Nitroethan 16,0 15,5 4,5 0 1
Ölsäure 16,2 3,6 5,2 c c
1,3-Propandiol 16,8 13,5 23,2 0 0
2-Propanol 15,8 6,1 16,4 0 0
Tetrahydronaphthalin 19,6 2,0 2,9 1 a
Wasser 15,5 16,0 42,3 0 0

4 Ergebnisse
46
Die mit dem Ubbelohde-Viskosimeter bestimmten Durchflusszeiten und die berechne-
ten inhärenten Viskosität sind in Tabelle 7 dargestellt. Für die Messung der Viskosität
wurden nur die in Tabelle 6 mit „1“ für das jeweilige Polymer markierten Lösungsmittel
mit einer Konzentration von 5 g l-1
verwendet. Die angegebenen Durchflusszeiten sind
die direkt nach DIN 51562115
gemessenen.
Zu erkennen ist, dass im Falle von PMS Dimethylaminoethanol die höchste und Me-
thylphenylsulfid die niedrigste inhärente Viskosität aufzeigt. Im Falle von PMPS zeigen
die Lösungen mit Inden die höchste und die mit N-Methyl-2-pyrrolidon die niedrigste
inhärente Viskosität. Der Theorie zufolge sollte somit Dimethylaminoethanol für PMS
und Inden für PMPS die Lösungsmittel aus der Reihe der untersuchten Lösungsmittel
sein, deren HSP den HSP des jeweiligen Polymers am nähesten kommen. Umgekehrt
sollte somit Methylphenylsulfid für PMS und N-Methyl-2-pyrrolidon für PMPS die
HSP haben, die am weitesten von denen der Polymere entfernt sind.
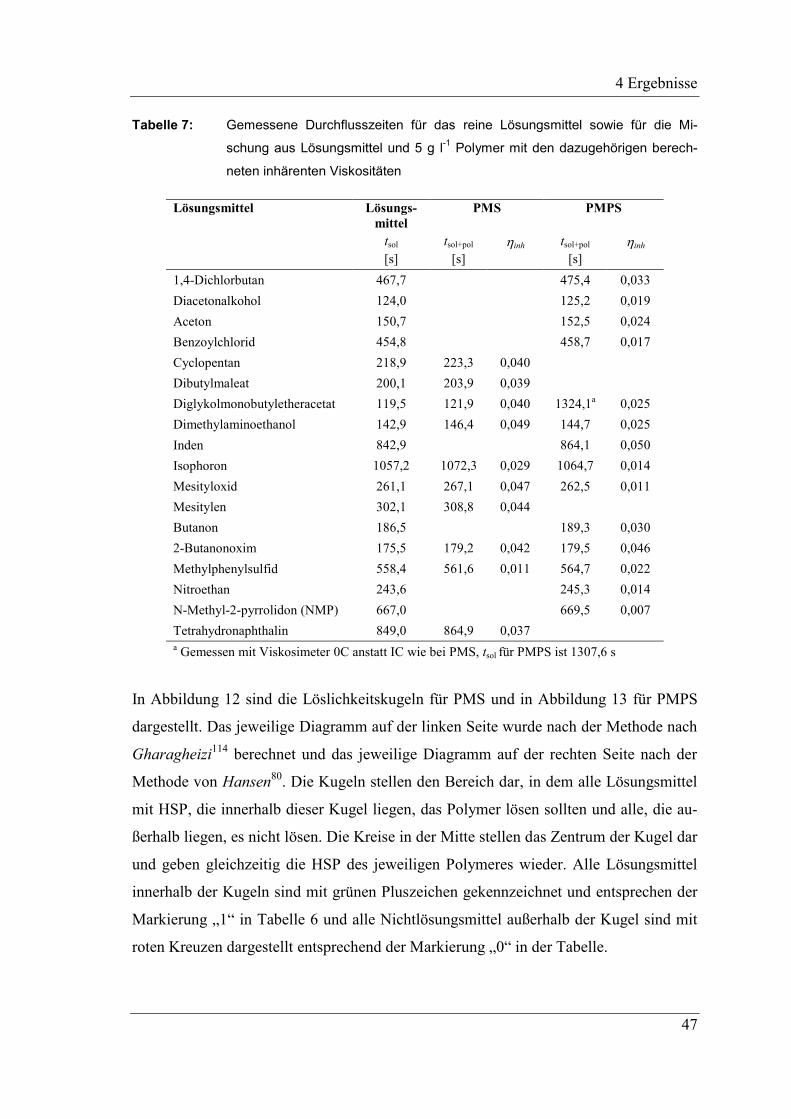
4 Ergebnisse
47
Tabelle 7: Gemessene Durchflusszeiten für das reine Lösungsmittel sowie für die Mi-
schung aus Lösungsmittel und 5 g l-1
Polymer mit den dazugehörigen berech-
neten inhärenten Viskositäten
Lösungsmittel Lösungs-
mittel
PMS PMPS
tsol
[s]
tsol+pol
[s]
inh tsol+pol
[s]
inh
1,4-Dichlorbutan 467,7 475,4 0,033
Diacetonalkohol 124,0 125,2 0,019
Aceton 150,7 152,5 0,024
Benzoylchlorid 454,8 458,7 0,017
Cyclopentan 218,9 223,3 0,040
Dibutylmaleat 200,1 203,9 0,039
Diglykolmonobutyletheracetat 119,5 121,9 0,040 1324,1a 0,025
Dimethylaminoethanol 142,9 146,4 0,049 144,7 0,025
Inden 842,9 864,1 0,050
Isophoron 1057,2 1072,3 0,029 1064,7 0,014
Mesityloxid 261,1 267,1 0,047 262,5 0,011
Mesitylen 302,1 308,8 0,044
Butanon 186,5 189,3 0,030
2-Butanonoxim 175,5 179,2 0,042 179,5 0,046
Methylphenylsulfid 558,4 561,6 0,011 564,7 0,022
Nitroethan 243,6 245,3 0,014
N-Methyl-2-pyrrolidon (NMP) 667,0 669,5 0,007
Tetrahydronaphthalin 849,0 864,9 0,037
a Gemessen mit Viskosimeter 0C anstatt IC wie bei PMS, tsol
für PMPS ist 1307,6 s
In Abbildung 12 sind die Löslichkeitskugeln für PMS und in Abbildung 13 für PMPS
dargestellt. Das jeweilige Diagramm auf der linken Seite wurde nach der Methode nach
Gharagheizi114
berechnet und das jeweilige Diagramm auf der rechten Seite nach der
Methode von Hansen80
. Die Kugeln stellen den Bereich dar, in dem alle Lösungsmittel
mit HSP, die innerhalb dieser Kugel liegen, das Polymer lösen sollten und alle, die au-
ßerhalb liegen, es nicht lösen. Die Kreise in der Mitte stellen das Zentrum der Kugel dar
und geben gleichzeitig die HSP des jeweiligen Polymeres wieder. Alle Lösungsmittel
innerhalb der Kugeln sind mit grünen Pluszeichen gekennzeichnet und entsprechen der
Markierung „1“ in Tabelle 6 und alle Nichtlösungsmittel außerhalb der Kugel sind mit
roten Kreuzen dargestellt entsprechend der Markierung „0“ in der Tabelle.
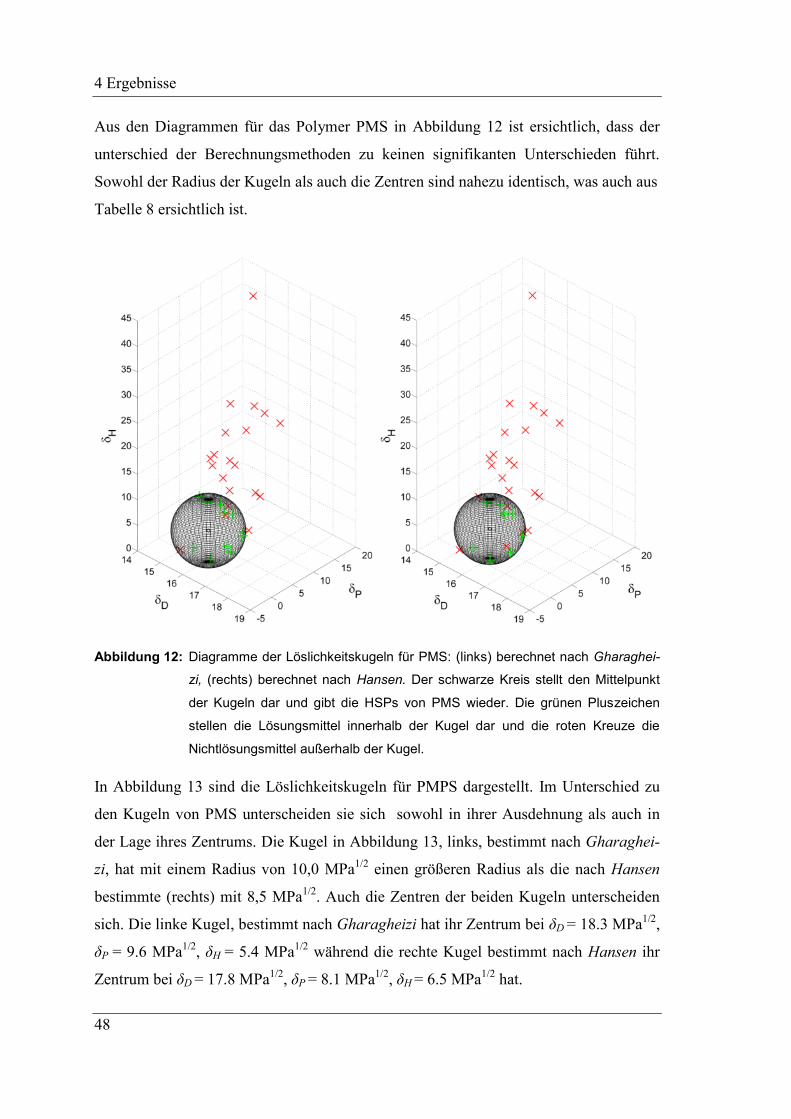
4 Ergebnisse
48
Aus den Diagrammen für das Polymer PMS in Abbildung 12 ist ersichtlich, dass der
unterschied der Berechnungsmethoden zu keinen signifikanten Unterschieden führt.
Sowohl der Radius der Kugeln als auch die Zentren sind nahezu identisch, was auch aus
Tabelle 8 ersichtlich ist.
Abbildung 12: Diagramme der Löslichkeitskugeln für PMS: (links) berechnet nach Gharaghei-
zi, (rechts) berechnet nach Hansen. Der schwarze Kreis stellt den Mittelpunkt
der Kugeln dar und gibt die HSPs von PMS wieder. Die grünen Pluszeichen
stellen die Lösungsmittel innerhalb der Kugel dar und die roten Kreuze die
Nichtlösungsmittel außerhalb der Kugel.
In Abbildung 13 sind die Löslichkeitskugeln für PMPS dargestellt. Im Unterschied zu
den Kugeln von PMS unterscheiden sie sich sowohl in ihrer Ausdehnung als auch in
der Lage ihres Zentrums. Die Kugel in Abbildung 13, links, bestimmt nach Gharaghei-
zi, hat mit einem Radius von 10,0 MPa1/2
einen größeren Radius als die nach Hansen
bestimmte (rechts) mit 8,5 MPa1/2
. Auch die Zentren der beiden Kugeln unterscheiden
sich. Die linke Kugel, bestimmt nach Gharagheizi hat ihr Zentrum bei δD = 18.3 MPa1/2
,
δP = 9.6 MPa1/2
, δH = 5.4 MPa1/2
während die rechte Kugel bestimmt nach Hansen ihr
Zentrum bei δD = 17.8 MPa1/2
, δP = 8.1 MPa1/2
, δH = 6.5 MPa1/2
hat.
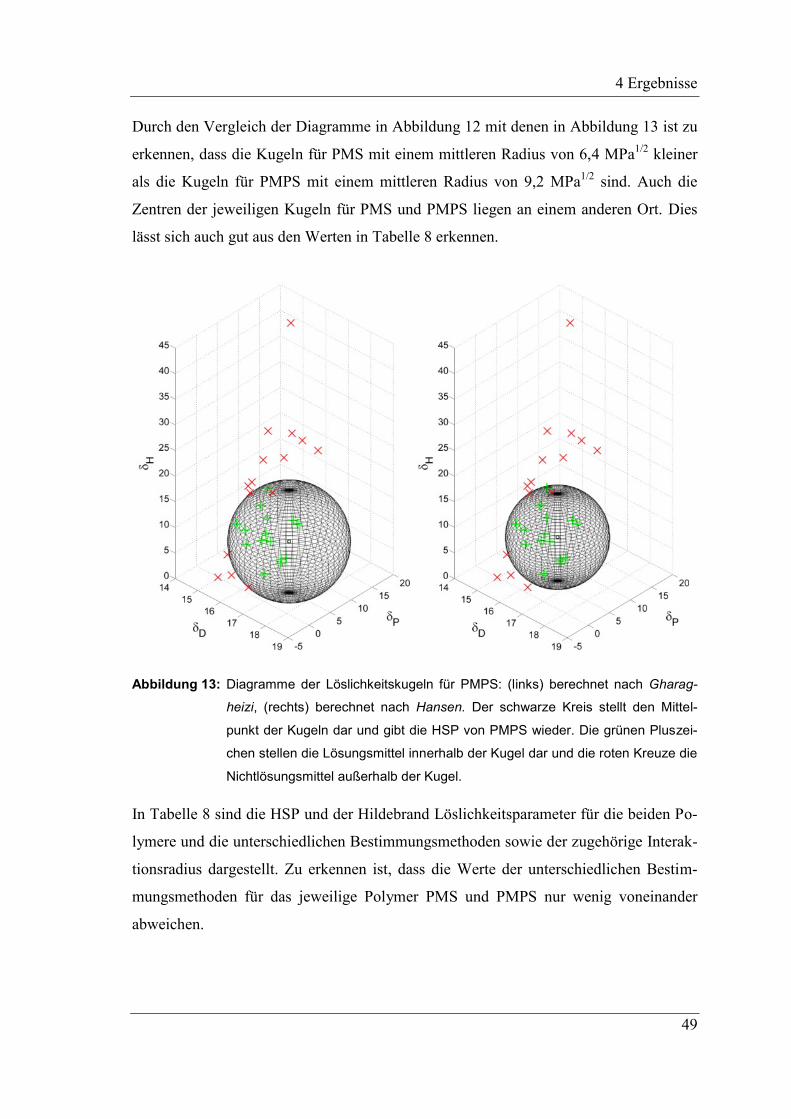
4 Ergebnisse
49
Durch den Vergleich der Diagramme in Abbildung 12 mit denen in Abbildung 13 ist zu
erkennen, dass die Kugeln für PMS mit einem mittleren Radius von 6,4 MPa1/2
kleiner
als die Kugeln für PMPS mit einem mittleren Radius von 9,2 MPa1/2
sind. Auch die
Zentren der jeweiligen Kugeln für PMS und PMPS liegen an einem anderen Ort. Dies
lässt sich auch gut aus den Werten in Tabelle 8 erkennen.
Abbildung 13: Diagramme der Löslichkeitskugeln für PMPS: (links) berechnet nach Gharag-
heizi, (rechts) berechnet nach Hansen. Der schwarze Kreis stellt den Mittel-
punkt der Kugeln dar und gibt die HSP von PMPS wieder. Die grünen Pluszei-
chen stellen die Lösungsmittel innerhalb der Kugel dar und die roten Kreuze die
Nichtlösungsmittel außerhalb der Kugel.
In Tabelle 8 sind die HSP und der Hildebrand Löslichkeitsparameter für die beiden Po-
lymere und die unterschiedlichen Bestimmungsmethoden sowie der zugehörige Interak-
tionsradius dargestellt. Zu erkennen ist, dass die Werte der unterschiedlichen Bestim-
mungsmethoden für das jeweilige Polymer PMS und PMPS nur wenig voneinander
abweichen.
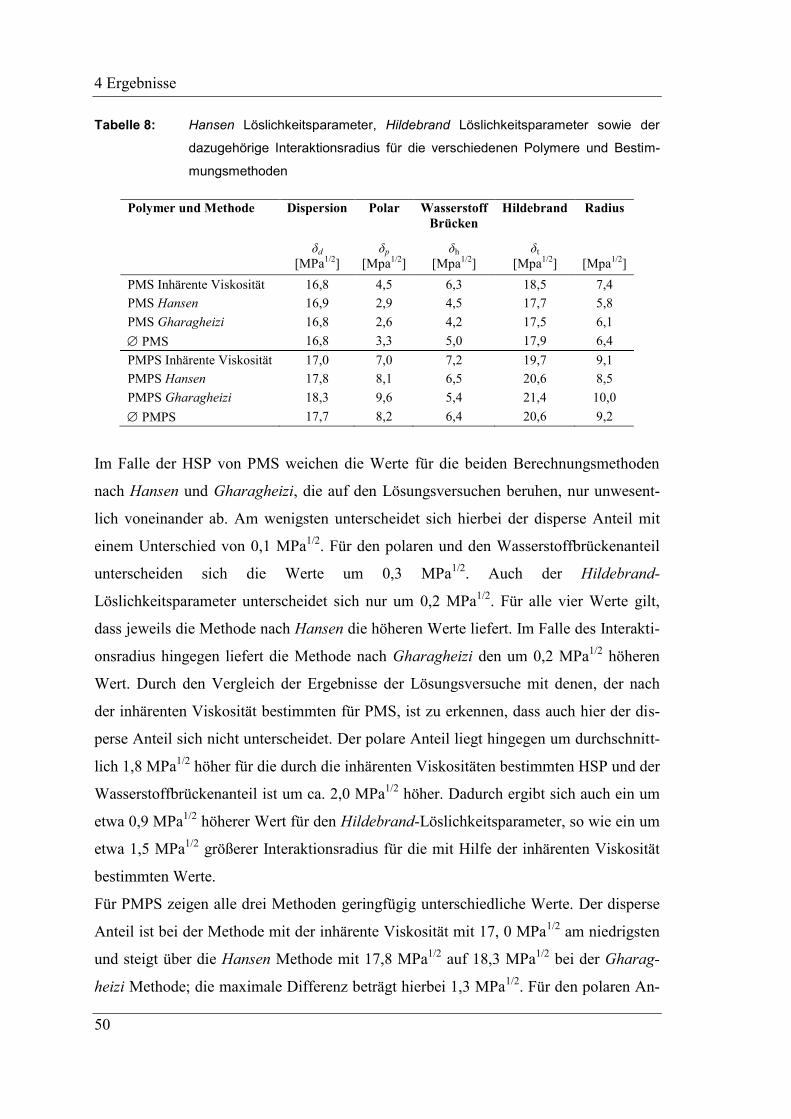
4 Ergebnisse
50
Tabelle 8: Hansen Löslichkeitsparameter, Hildebrand Löslichkeitsparameter sowie der
dazugehörige Interaktionsradius für die verschiedenen Polymere und Bestim-
mungsmethoden
Polymer und Methode Dispersion Polar Wasserstoff
Brücken
Hildebrand Radius
δd
[MPa1/2
]
δp
[Mpa1/2
]
δh
[Mpa1/2
]
δt
[Mpa1/2
]
[Mpa1/2
]
PMS Inhärente Viskosität 16,8 4,5 6,3 18,5 7,4
PMS Hansen 16,9 2,9 4,5 17,7 5,8
PMS Gharagheizi 16,8 2,6 4,2 17,5 6,1
PMS 16,8 3,3 5,0 17,9 6,4
PMPS Inhärente Viskosität 17,0 7,0 7,2 19,7 9,1
PMPS Hansen 17,8 8,1 6,5 20,6 8,5
PMPS Gharagheizi 18,3 9,6 5,4 21,4 10,0
PMPS 17,7 8,2 6,4 20,6 9,2
Im Falle der HSP von PMS weichen die Werte für die beiden Berechnungsmethoden
nach Hansen und Gharagheizi, die auf den Lösungsversuchen beruhen, nur unwesent-
lich voneinander ab. Am wenigsten unterscheidet sich hierbei der disperse Anteil mit
einem Unterschied von 0,1 MPa1/2
. Für den polaren und den Wasserstoffbrückenanteil
unterscheiden sich die Werte um 0,3 MPa1/2
. Auch der Hildebrand-
Löslichkeitsparameter unterscheidet sich nur um 0,2 MPa1/2
. Für alle vier Werte gilt,
dass jeweils die Methode nach Hansen die höheren Werte liefert. Im Falle des Interakti-
onsradius hingegen liefert die Methode nach Gharagheizi den um 0,2 MPa1/2
höheren
Wert. Durch den Vergleich der Ergebnisse der Lösungsversuche mit denen, der nach
der inhärenten Viskosität bestimmten für PMS, ist zu erkennen, dass auch hier der dis-
perse Anteil sich nicht unterscheidet. Der polare Anteil liegt hingegen um durchschnitt-
lich 1,8 MPa1/2
höher für die durch die inhärenten Viskositäten bestimmten HSP und der
Wasserstoffbrückenanteil ist um ca. 2,0 MPa1/2
höher. Dadurch ergibt sich auch ein um
etwa 0,9 MPa1/2
höherer Wert für den Hildebrand-Löslichkeitsparameter, so wie ein um
etwa 1,5 MPa1/2
größerer Interaktionsradius für die mit Hilfe der inhärenten Viskosität
bestimmten Werte.
Für PMPS zeigen alle drei Methoden geringfügig unterschiedliche Werte. Der disperse
Anteil ist bei der Methode mit der inhärente Viskosität mit 17, 0 MPa1/2
am niedrigsten
und steigt über die Hansen Methode mit 17,8 MPa1/2
auf 18,3 MPa1/2
bei der Gharag-
heizi Methode; die maximale Differenz beträgt hierbei 1,3 MPa1/2
. Für den polaren An-

4 Ergebnisse
51
teil zeigt sich das gleiche Verhalten. Der Wert steigt von 7,0 MPa1/2
bei der inhärenten
Viskosität über 8,1 MPa1/2
bei der Hansen Methode bis 9,6 MPa1/2
bei der Gharagheizi
Methode. Hierbei zeigt sich die größte Differenz überhaupt von 2,6 MPa1/2
. Für den
Wasserstoffbrückenanteil hingegen zeigt sich ein umgekehrtes Verhalten. Hier fällt der
Wert von 7,2 MPa1/2
bei der inhärenten Viskosität über 6,5 MPa1/2
bei der Hansen Me-
thode auf 5,4 bei der Gharagheizi Methode, wobei die maximale Differenz 1,8 MPa1/2
beträgt. Der Hildebrand-Löslichkeitsparameter steigt wiederum von der inhärenten Vis-
kosität mit 19,7 MPa1/2
über die Hansen Methode mit 20,6 MPa1/2
bis zu 21,4 MPa1/2
bei der Gharagheizi Methode an; die maximale Differenz beträgt hierbei 1,7 MPa1/2
.
Der Interaktionsradius hingegen zeigt ein völlig abweichendes Verhalten. Hier zeigt die
Methode nach Gharagheizi den größten Radius mit 10,0 MPa1/2
, währende die Methode
bestimmt durch die inhärente Viskosität den kleinsten Radius mit 8,5 MPa1/2
liefert und
die Methode nach Hansen mit 8,5 MPa1/2
dazwischen liegt, wobei die maximale Diffe-
renz 1,5 MPa1/2
beträgt.
Durch den Vergleich der HSP der Polymere PMS und PMPS untereinander, ist zu er-
kennen, dass auch hier der disperse Anteil sehr ähnliche Werte aufweist. Die mittlere
Differenz beträgt hierbei 0,9 MPa1/2
. Die größte mittlere Differenz ergibt sich beim po-
laren Anteil mit 4,9 MPa1/2
. Die mittlere Differenz für den Wasserstoffbrückenanteil
liegt mit 1,4 MPa1/2
dazwischen. Für die Radien liegt die mittlere Differenz bei 2,8
MPa1/2
. Im Mittel unterscheidet sich der Hildebrand-Löslichkeitsparameter um 2,7
MPa1/2
. Durch den mit Hilfe von Gleichung (5) ermittelten Abstand der durchschnittli-
chen HSP der Polymere zueinander, ergibt sich ein Abstand von 5,4 MPa1/2
, was kleiner
als der kleinste gemessene Radius von 5,8 MPa1/2
ist. Dies ist gleichbedeutend damit,
dass die jeweiligen Zentren der Polymere in diesem Fall innerhalb der jeweiligen Kugel
des anderen Polymers liegen. Der Vergleich der Ergebnisse der einzelnen Methoden
ergibt, dass der kleinste Abstand mit 2,7 MPa1/2
mit der Methode, bei der die HSP mit
Hilfe der inhärenten Viskosität bestimmt worden ist, festgestellt wurde. Der größte Ab-
stand ergibt sich bei der Methode nach Gharagheizi von 7,7 MPa1/2
. Im letzteren Fall
bedeutet dies, dass das Zentrum und somit die HSP von PMPS außerhalb aller bestimm-
ten Radien für PMS liegt. Jedoch liegt das Zentrum der PMS Kugel innerhalb aller be-
stimmten Radien für PMPS.

4 Ergebnisse
52
4.1.2 Phasendiagramme der Mischbarkeiten von PMS – PMPS – MTES – MeOH
In Abbildung 14 sind die Mischbarkeitsdiagramme für die vier Komponenten MTES,
PMS, PMPS und Methanol dargestellt. Die blauen Rauten (♦) stellen die nicht mischba-
re Zusammensetzungen dar, die roten Kreuze (x) die mischbaren. Der Abstand zwi-
schen den einzelnen Punkten in den Diagrammen beträgt 16,67 vol. %. Jedes der vier
Diagramme entspricht einem unterschiedlichen Methanol-Anteil: im Diagramm oben
links enthalten alle Zusammensetzungen 0 vol. % Methanol, im Weiteren wird die Me-
thanolkonzentration ebenfalls in 16,67 vol. % Schritten von Abbildung 14 a) (0 vol. %)
über 16,67 vol. % Abbildung 14 b) und 33,33 vol. % Abbildung 14 c) auf 50 vol. %
Abbildung 14 d) erhöht. Die schwarzen Linien trennen den Bereich der mischbaren Zu-
sammensetzungen von den nicht mischbaren Zusammensetzungen.12
Aus den Diagrammen ist zu erkennen, dass im Zweiphasensystem PMPS – PMS, darge-
stellt durch die Basislinie im Diagramm mit 0 vol. % Methanol, keine mischbaren Zu-
sammensetzungen gefunden wurden. Dies gilt ebenso für die beiden Zweiphasensyste-
me PMPS – Methanol (linke untere Ecken der vier Diagramme) und PMS – Methanol
(rechte untere Ecken der vier Diagramme), bei denen ebenfalls keine mischbaren Zu-
sammensetzungen gefunden wurden. Das Zweiphasensystem MTES – PMS (rechte
Linie im Diagramm mit 0 vol. % Methanol) zeigt Mischbarkeit im Bereich von 0 vol. %
– 50 vol. % PMS und ab einem Gehalt von 66,67 vol. % PMS keine Mischbarkeit mehr.
Die Zweiphasensysteme MTES – PMPS (linke Linie im Diagramm mit 0 vol. % Me-
thanol) und MTES – Methanol (obere Ecke der vier Diagramme) sind hingegen über
den kompletten untersuchten Bereich von 0 vol. % bis 100 vol. % mischbar. Zudem ist
zu erkennen, dass mit zunehmendem Methanol-Anteil der Bereich abnimmt, in dem
mischbare Proben hergestellt werden können. Dies beginnt unten rechts im PMS-Eck,
von dem aus sich der Bereich der nicht mischbaren Proben mit zunehmendem Metha-
nol-Anteil ausbreitet. Zusätzlich ist ersichtlich, dass es im gesamten Vierphasensystem
keine Zusammensetzung gibt, die mischbar ist und gleichzeitig kein MTES enthält. Da-
her wird aus den Mischungsdiagrammen ersichtlich, dass MTES zwingend als Lö-
sungsmittel erforderlich ist. Das lässt sich einerseits daran erkennen, dass die linken
äußeren Linien, die das Phasendiagram PMPS – Methanol – PMS darstellen und somit
kein MTES enthalten, keine mischbaren Zusammensetzungen aufweisen. Andererseits

4 Ergebnisse
53
nimmt mit zunehmendem Methanolgehalt, verbunden mit einer Abnahme des MTES-
Gehaltes, die Mischbarkeit ab.12
Abbildung 14: Mischbarkeitsdiagramme für die vier Komponenten MTES, PMS, PMPS und
Methanol. Die blauen Rauten (♦) stellen nicht mischbare, die roten Kreuze (x)
mischbare Zusammensetzung dar. Der Methanol-Anteil nimmt mit dem Dia-
gramm von oben links a) mit 0 vol. % in 16,67 vol. % Schritten auf 50 vol. % im
Diagramm unten rechts d) zu. Der Abstand zwischen den einzelnen Punkten
beträgt ebenfalls 16,67 vol. %. Die schwarzen Linien trennen den Bereich der
mischbaren Zusammensetzungen vom Bereich der nicht mischbaren Zusam-
mensetzungen.12
4.1.3 Beschichtungen mit verschiedenen Zusammensetzungen von PMS und PMPS und deren Einfluss auf die Strukturbildung
In Abbildung 15 sind lichtmikroskopische Aufnahmen von Beschichtungen auf Alumi-
niumoxidsubstraten der Zusammensetzung COMP 1 bis COMP 4 (Tabelle 5) darge-

4 Ergebnisse
54
stellt. Zu erkennen ist, dass die Veränderung der Zusammensetzung zu Veränderungen
in der Schichtstruktur führt. COMP 1 zeigt eine Struktur, bei der vergleichsweise viele
große „Inseln“ von einer Matrix umgeben sind, in der nur wenige und sehr kleine „In-
seln“ zu erkennen sind. Bei COMP 2 hingegen sind unregelmäßige Strukturen zu er-
kennen (Erhebungen). In der Matrix sind hierbei wenige kleine, gut zu identifizierende
„Inseln“ eingebettet, die Gebirgsketten mit Vulkanen ähneln. COMP 3 hingegen zeigt
eine ähnliche Struktur wie COMP1 (große „Inseln“ sind von kleinen „Inseln“ umge-
ben). Der Unterschied zu COMP 1 besteht darin, dass die in der Matrix eingebetteten,
kleinen „Inseln“ größer sind, als jene in COMP 1. Das Verhältnis von „Insel“-Fläche zu
Matrixfläche scheint bei Proben aus COMP 3 größer zu sein, als bei COMP 1. Die Auf-
nahmen von COMP 4 zeigen eine gleichmäßigere Struktur, bei der viele ähnlich große
„Inseln“ gleichmäßig in die Matrix eingebettet sind.12
Durch den Vergleich der Größe und der Anzahl der „Inseln“, wird deutlich, dass Proben
mit einem höheren Anteil an PMPS mehr bzw. größere „Inseln“ aufweisen. COMP 3
mit dem größten Anteil an PMPS weist deutlich mehr und größere „Inseln“ auf, als die
Probe COMP 2 (niedrigster PMPS-Anteil). Dies gilt auch für den Vergleich von COMP
3 zu COMP 1 und COMP 4.12
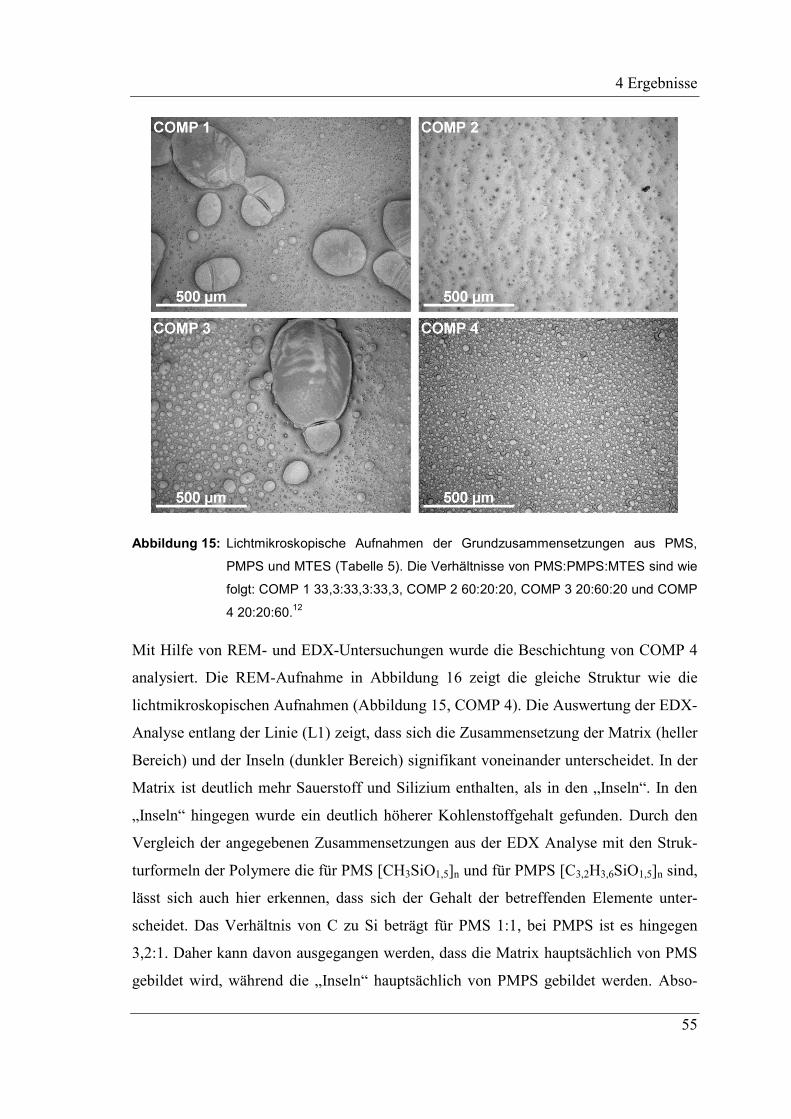
4 Ergebnisse
55
Abbildung 15: Lichtmikroskopische Aufnahmen der Grundzusammensetzungen aus PMS,
PMPS und MTES (Tabelle 5). Die Verhältnisse von PMS:PMPS:MTES sind wie
folgt: COMP 1 33,3:33,3:33,3, COMP 2 60:20:20, COMP 3 20:60:20 und COMP
4 20:20:60.12
Mit Hilfe von REM- und EDX-Untersuchungen wurde die Beschichtung von COMP 4
analysiert. Die REM-Aufnahme in Abbildung 16 zeigt die gleiche Struktur wie die
lichtmikroskopischen Aufnahmen (Abbildung 15, COMP 4). Die Auswertung der EDX-
Analyse entlang der Linie (L1) zeigt, dass sich die Zusammensetzung der Matrix (heller
Bereich) und der Inseln (dunkler Bereich) signifikant voneinander unterscheidet. In der
Matrix ist deutlich mehr Sauerstoff und Silizium enthalten, als in den „Inseln“. In den
„Inseln“ hingegen wurde ein deutlich höherer Kohlenstoffgehalt gefunden. Durch den
Vergleich der angegebenen Zusammensetzungen aus der EDX Analyse mit den Struk-
turformeln der Polymere die für PMS [CH3SiO1,5]n und für PMPS [C3,2H3,6SiO1,5]n sind,
lässt sich auch hier erkennen, dass sich der Gehalt der betreffenden Elemente unter-
scheidet. Das Verhältnis von C zu Si beträgt für PMS 1:1, bei PMPS ist es hingegen
3,2:1. Daher kann davon ausgegangen werden, dass die Matrix hauptsächlich von PMS
gebildet wird, während die „Inseln“ hauptsächlich von PMPS gebildet werden. Abso-
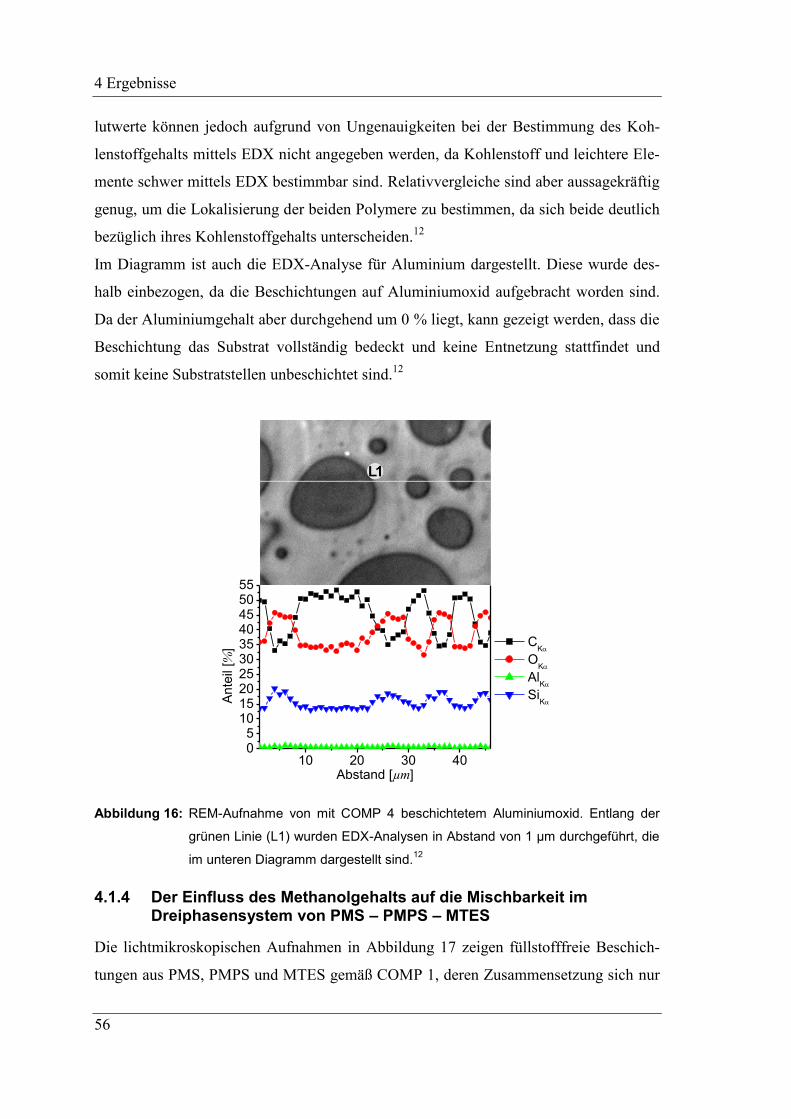
4 Ergebnisse
56
lutwerte können jedoch aufgrund von Ungenauigkeiten bei der Bestimmung des Koh-
lenstoffgehalts mittels EDX nicht angegeben werden, da Kohlenstoff und leichtere Ele-
mente schwer mittels EDX bestimmbar sind. Relativvergleiche sind aber aussagekräftig
genug, um die Lokalisierung der beiden Polymere zu bestimmen, da sich beide deutlich
bezüglich ihres Kohlenstoffgehalts unterscheiden.12
Im Diagramm ist auch die EDX-Analyse für Aluminium dargestellt. Diese wurde des-
halb einbezogen, da die Beschichtungen auf Aluminiumoxid aufgebracht worden sind.
Da der Aluminiumgehalt aber durchgehend um 0 % liegt, kann gezeigt werden, dass die
Beschichtung das Substrat vollständig bedeckt und keine Entnetzung stattfindet und
somit keine Substratstellen unbeschichtet sind.12
Abbildung 16: REM-Aufnahme von mit COMP 4 beschichtetem Aluminiumoxid. Entlang der
grünen Linie (L1) wurden EDX-Analysen in Abstand von 1 µm durchgeführt, die
im unteren Diagramm dargestellt sind.12
4.1.4 Der Einfluss des Methanolgehalts auf die Mischbarkeit im Dreiphasensystem von PMS – PMPS – MTES
Die lichtmikroskopischen Aufnahmen in Abbildung 17 zeigen füllstofffreie Beschich-
tungen aus PMS, PMPS und MTES gemäß COMP 1, deren Zusammensetzung sich nur
10 20 30 4005
10152025303540455055
CK
OK
AlK
SiKA
nte
il [%
]
Abstand [µm]
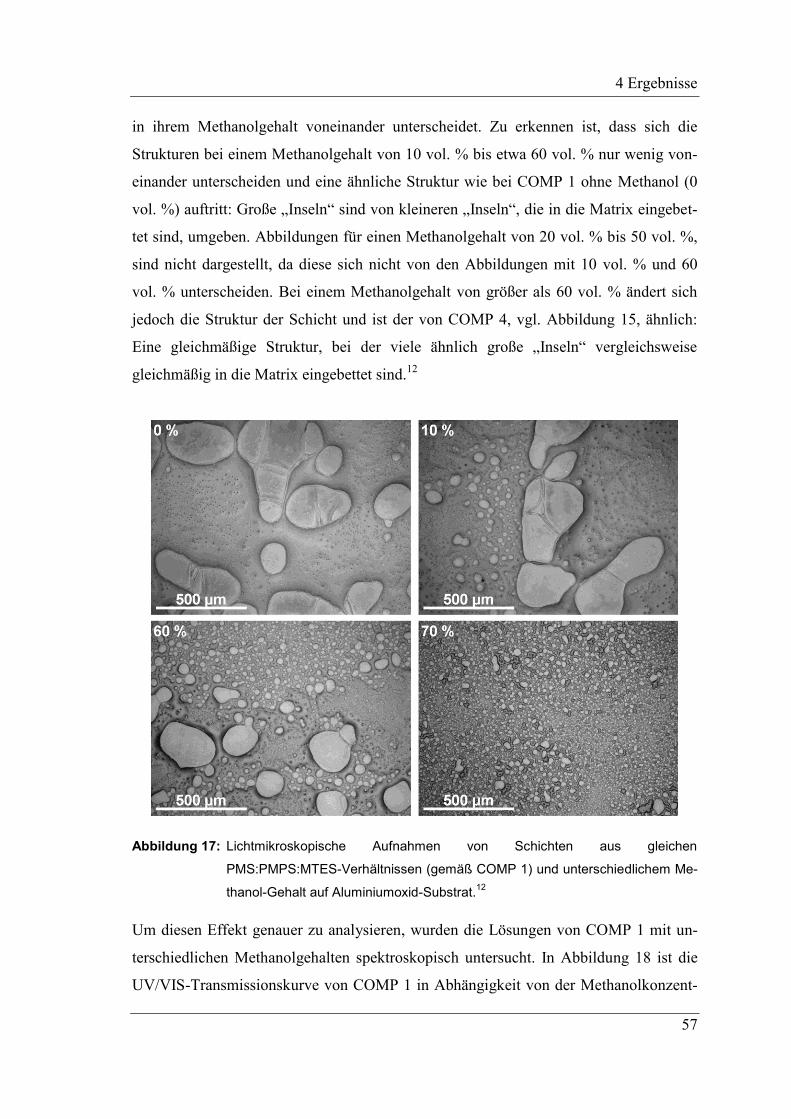
4 Ergebnisse
57
in ihrem Methanolgehalt voneinander unterscheidet. Zu erkennen ist, dass sich die
Strukturen bei einem Methanolgehalt von 10 vol. % bis etwa 60 vol. % nur wenig von-
einander unterscheiden und eine ähnliche Struktur wie bei COMP 1 ohne Methanol (0
vol. %) auftritt: Große „Inseln“ sind von kleineren „Inseln“, die in die Matrix eingebet-
tet sind, umgeben. Abbildungen für einen Methanolgehalt von 20 vol. % bis 50 vol. %,
sind nicht dargestellt, da diese sich nicht von den Abbildungen mit 10 vol. % und 60
vol. % unterscheiden. Bei einem Methanolgehalt von größer als 60 vol. % ändert sich
jedoch die Struktur der Schicht und ist der von COMP 4, vgl. Abbildung 15, ähnlich:
Eine gleichmäßige Struktur, bei der viele ähnlich große „Inseln“ vergleichsweise
gleichmäßig in die Matrix eingebettet sind.12
Abbildung 17: Lichtmikroskopische Aufnahmen von Schichten aus gleichen
PMS:PMPS:MTES-Verhältnissen (gemäß COMP 1) und unterschiedlichem Me-
thanol-Gehalt auf Aluminiumoxid-Substrat.12
Um diesen Effekt genauer zu analysieren, wurden die Lösungen von COMP 1 mit un-
terschiedlichen Methanolgehalten spektroskopisch untersucht. In Abbildung 18 ist die
UV/VIS-Transmissionskurve von COMP 1 in Abhängigkeit von der Methanolkonzent-
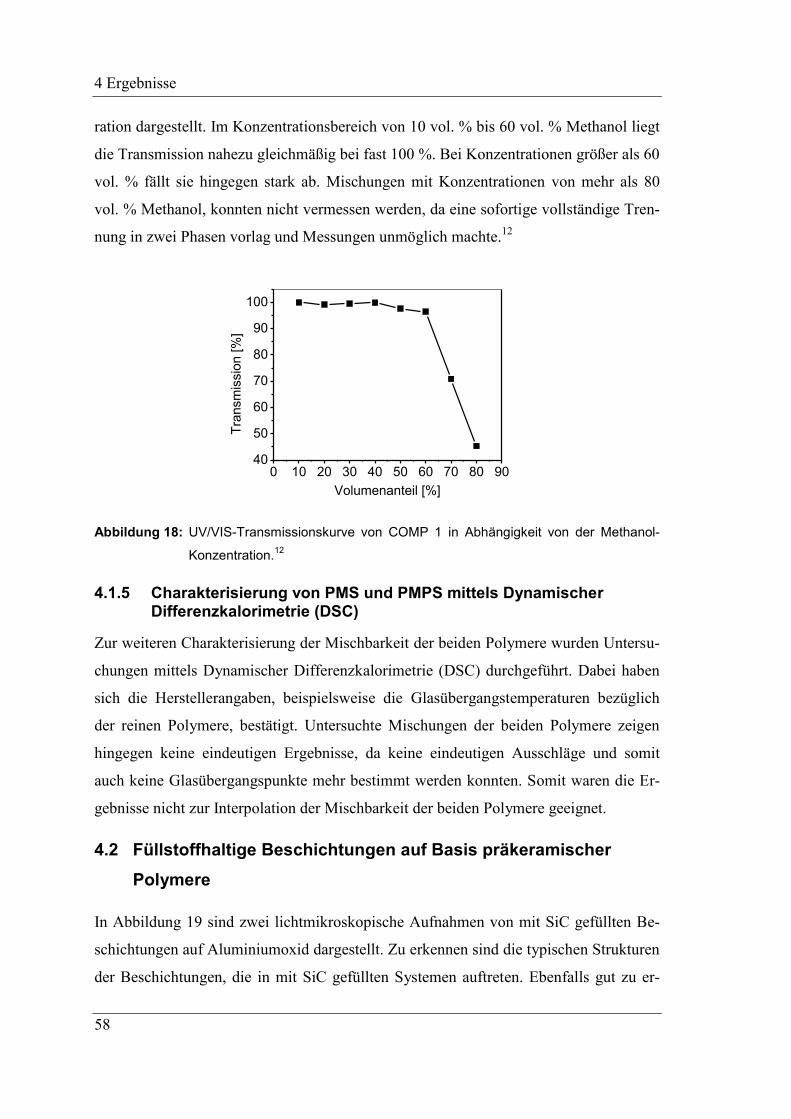
4 Ergebnisse
58
ration dargestellt. Im Konzentrationsbereich von 10 vol. % bis 60 vol. % Methanol liegt
die Transmission nahezu gleichmäßig bei fast 100 %. Bei Konzentrationen größer als 60
vol. % fällt sie hingegen stark ab. Mischungen mit Konzentrationen von mehr als 80
vol. % Methanol, konnten nicht vermessen werden, da eine sofortige vollständige Tren-
nung in zwei Phasen vorlag und Messungen unmöglich machte.12
Abbildung 18: UV/VIS-Transmissionskurve von COMP 1 in Abhängigkeit von der Methanol-
Konzentration.12
4.1.5 Charakterisierung von PMS und PMPS mittels Dynamischer Differenzkalorimetrie (DSC)
Zur weiteren Charakterisierung der Mischbarkeit der beiden Polymere wurden Untersu-
chungen mittels Dynamischer Differenzkalorimetrie (DSC) durchgeführt. Dabei haben
sich die Herstellerangaben, beispielsweise die Glasübergangstemperaturen bezüglich
der reinen Polymere, bestätigt. Untersuchte Mischungen der beiden Polymere zeigen
hingegen keine eindeutigen Ergebnisse, da keine eindeutigen Ausschläge und somit
auch keine Glasübergangspunkte mehr bestimmt werden konnten. Somit waren die Er-
gebnisse nicht zur Interpolation der Mischbarkeit der beiden Polymere geeignet.
4.2 Füllstoffhaltige Beschichtungen auf Basis präkeramischer
Polymere
In Abbildung 19 sind zwei lichtmikroskopische Aufnahmen von mit SiC gefüllten Be-
schichtungen auf Aluminiumoxid dargestellt. Zu erkennen sind die typischen Strukturen
der Beschichtungen, die in mit SiC gefüllten Systemen auftreten. Ebenfalls gut zu er-
0 10 20 30 40 50 60 70 80 9040
50
60
70
80
90
100
Tra
nsm
issio
n [%
]
Volumenanteil [%]

4 Ergebnisse
59
kennen ist, dass sich die bei 110 °C ausgehärteten polymeren Beschichtungen gut in
keramische Beschichtungen durch sintern bei 1100 °C in Stickstoffatmosphäre umwan-
deln lassen und dabei die Struktur der Beschichtung ohne große Veränderung beibehal-
ten wird.12
Abbildung 19: Lichtmikroskopische Aufnahmen von mit SiC gefüllter Beschichtungen, links
nach dem Vernetzten bei 110 °C und rechts nach dem Sintern unter Stickstoff
bei 1100 °C.12
Die REM-Aufnahmen in Abbildung 20 zeigen die gleichen Beschichtungen nach dem
aushärten bei 110 °C wie in Abbildung 19 links, bei deutlich erhöhter Vergrößerung.
Hierbei ist zu erkennen, dass sich in gefüllten Systemen eine Porenstruktur ausbildet,
bei der die Füllstoffpartikel gut erkennbar in den Stegen lokalisierbar sind, wie auch am
Boden der Poren. Die ausgehärteten Polymere befinden sich ebenso in den Stegen, da
bei der Aufnahme mittels REM die Partikel am Boden der Poren, wenn diese mit Poly-
mer gefüllt wären, nicht sichtbar wären. Wie bereits bei den ungefüllten Systemen,
wurden auch die gefüllten Systeme mittels EDX untersucht. Dabei hat sich gezeigt, dass
sowohl im Bereich der Poren, als auch auf den Stegen, die Zusammensetzung gleich
und das Substrat vollständig bedeckt ist.12
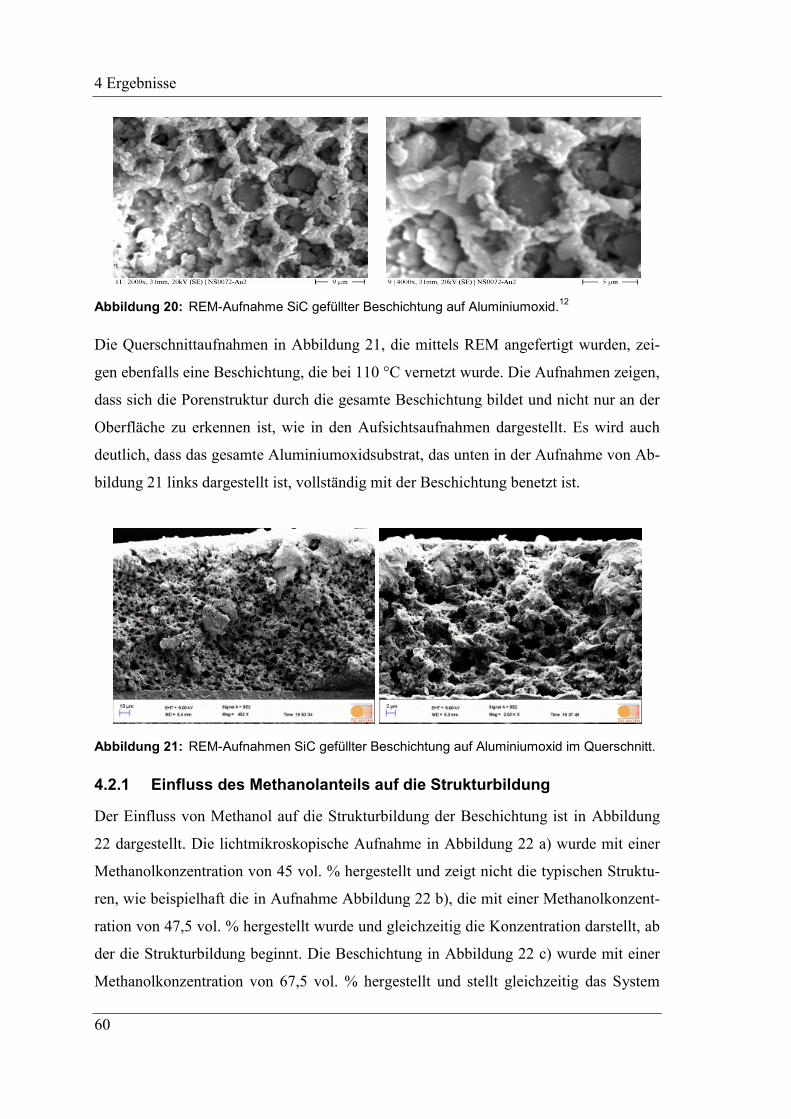
4 Ergebnisse
60
Abbildung 20: REM-Aufnahme SiC gefüllter Beschichtung auf Aluminiumoxid.12
Die Querschnittaufnahmen in Abbildung 21, die mittels REM angefertigt wurden, zei-
gen ebenfalls eine Beschichtung, die bei 110 °C vernetzt wurde. Die Aufnahmen zeigen,
dass sich die Porenstruktur durch die gesamte Beschichtung bildet und nicht nur an der
Oberfläche zu erkennen ist, wie in den Aufsichtsaufnahmen dargestellt. Es wird auch
deutlich, dass das gesamte Aluminiumoxidsubstrat, das unten in der Aufnahme von Ab-
bildung 21 links dargestellt ist, vollständig mit der Beschichtung benetzt ist.
Abbildung 21: REM-Aufnahmen SiC gefüllter Beschichtung auf Aluminiumoxid im Querschnitt.
4.2.1 Einfluss des Methanolanteils auf die Strukturbildung
Der Einfluss von Methanol auf die Strukturbildung der Beschichtung ist in Abbildung
22 dargestellt. Die lichtmikroskopische Aufnahme in Abbildung 22 a) wurde mit einer
Methanolkonzentration von 45 vol. % hergestellt und zeigt nicht die typischen Struktu-
ren, wie beispielhaft die in Aufnahme Abbildung 22 b), die mit einer Methanolkonzent-
ration von 47,5 vol. % hergestellt wurde und gleichzeitig die Konzentration darstellt, ab
der die Strukturbildung beginnt. Die Beschichtung in Abbildung 22 c) wurde mit einer
Methanolkonzentration von 67,5 vol. % hergestellt und stellt gleichzeitig das System
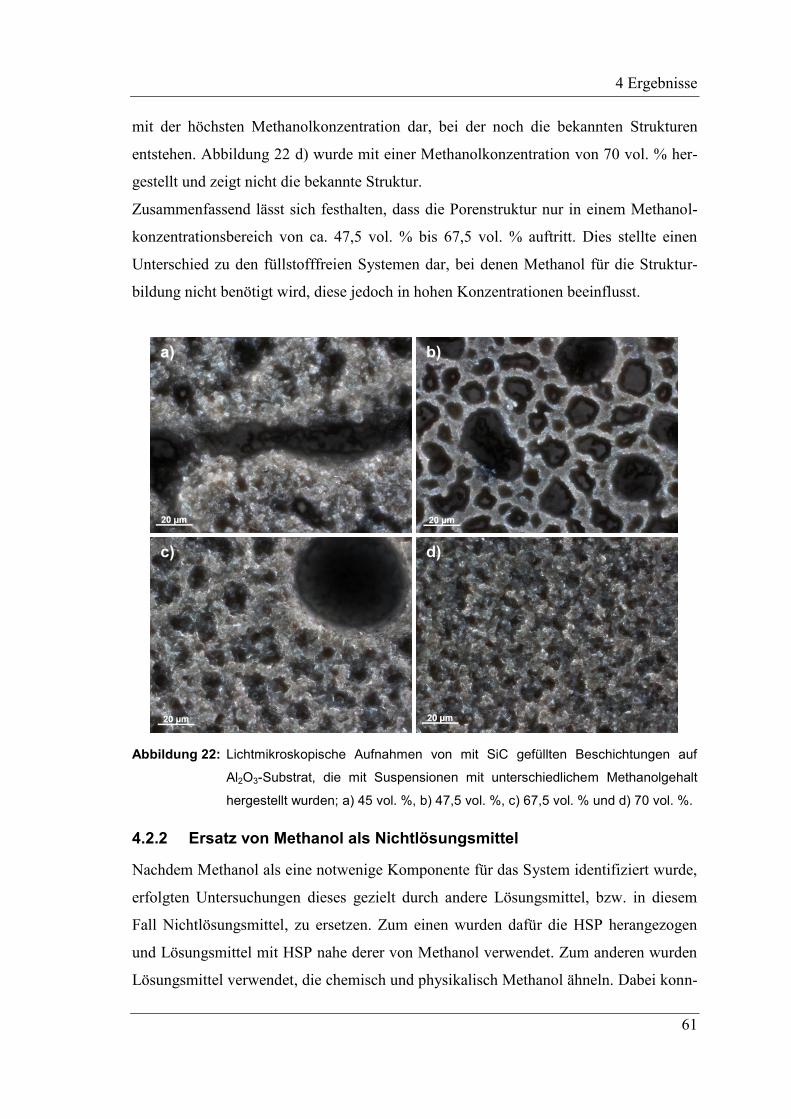
4 Ergebnisse
61
mit der höchsten Methanolkonzentration dar, bei der noch die bekannten Strukturen
entstehen. Abbildung 22 d) wurde mit einer Methanolkonzentration von 70 vol. % her-
gestellt und zeigt nicht die bekannte Struktur.
Zusammenfassend lässt sich festhalten, dass die Porenstruktur nur in einem Methanol-
konzentrationsbereich von ca. 47,5 vol. % bis 67,5 vol. % auftritt. Dies stellte einen
Unterschied zu den füllstofffreien Systemen dar, bei denen Methanol für die Struktur-
bildung nicht benötigt wird, diese jedoch in hohen Konzentrationen beeinflusst.
Abbildung 22: Lichtmikroskopische Aufnahmen von mit SiC gefüllten Beschichtungen auf
Al2O3-Substrat, die mit Suspensionen mit unterschiedlichem Methanolgehalt
hergestellt wurden; a) 45 vol. %, b) 47,5 vol. %, c) 67,5 vol. % und d) 70 vol. %.
4.2.2 Ersatz von Methanol als Nichtlösungsmittel
Nachdem Methanol als eine notwenige Komponente für das System identifiziert wurde,
erfolgten Untersuchungen dieses gezielt durch andere Lösungsmittel, bzw. in diesem
Fall Nichtlösungsmittel, zu ersetzen. Zum einen wurden dafür die HSP herangezogen
und Lösungsmittel mit HSP nahe derer von Methanol verwendet. Zum anderen wurden
Lösungsmittel verwendet, die chemisch und physikalisch Methanol ähneln. Dabei konn-
a) b)
c) d)

4 Ergebnisse
62
ten mit keinem weiteren Lösungsmittel reproduzierbar netzartige Strukturen hergestellt
werden. Mit Ethanol, welches der chemisch nächste Verwandte zu Methanol ist, wurden
zusätzlich Versuche durchgeführt, bei denen die Temperatur beim Beschichten erhöht
wurde, um die Verdunstung von Ethanol zu beschleunigen, da ein wesentlicher Unter-
schied von Ethanol zu Methanol im Dampfdruck bei gleicher Temperatur liegt. Die Be-
schichtungsreihe hat nicht zu netzartig strukturierten Oberflächen geführt. Auch alle
nach den HSP ausgewählten Lösungsmittel haben keinen Erfolg gezeigt. Dabei ist fest-
zuhalten, dass Methanol eine exponierte Lage innerhalb der HSP hat und es keine Lö-
sungsmittel gibt, die HSP sehr nahe denen von Menthol haben.
4.2.3 Beeinflussung des Systems durch das Lösungsmittel MTES
Im Abschnitt 4.1 (Ungefüllte Systeme auf Basis präkeramischer Polymere
PMS und PMPS) konnte gezeigt werden, dass MTES als Lösungsmittel zwingend er-
forderlich für beide Polymere ist. Daher wurde im Folgenden ebenso der Einfluss von
MTES auf gefüllte Beschichtungen untersucht. Hierfür wurden Schlicker mit unter-
schiedlichen MTES-Gehalten, und daraus resultierende Beschichtungen, hergestellt.
Der MTES-Gehalt wurde dabei von 0 vol. % in 5 vol. % Schritten erhöht. In Abbildung
23 sind die daraus resultierenden Beschichtungen dargestellt. Auch Beschichtungen, bei
denen kein MTES verwendet wurde, siehe Abbildung 23 a), zeigen die netzartigen
Strukturen. Die Struktur entsteht dabei bis zu einer MTES-Konzentration von 25 vol. %
(nicht dargestellt). Erst aber bei einer Konzentration von 30 vol. % in Abbildung 23 d)
und größer entsteht die Struktur nicht mehr. Des Weiteren lässt sich beobachten, dass
mit zunehmender MTES-Konzentration von Abbildung 23 a) nach c) auch die Poren-
größe zunimmt. Außerdem ist ersichtlich, dass zwei Generationen von Poren entstehen.
Hierbei wachsen die großen Poren, die bereits in Abbildung 23 a) zu erkennen sind, auf
Kosten der kleineren Poren. Dies erfolgt solange, bis, wie in Abbildung 23 c) darge-
stellt, keine kleineren Poren mehr erkannt werden können.
Zusammenfassend lässt sich festhalten, dass die Strukturbildung in gefüllten Systemen
auch ohne MTES erfolgen kann und bis zu einer maximalen MTES-Konzentration von
25 vol. % auftritt.

4 Ergebnisse
63
Abbildung 23: Lichtmikroskopische Aufnahmen von mit SiC gefüllten Beschichtungen auf
Al2O3-Substrat, die mit Suspensionen mit unterschiedlichen MTES-Gehalten
hergestellt wurden; a) 0 vol. %, b) 10 vol. %, c) 20 vol. % und d) 30 vol. %.
4.2.4 Einfluss des Verhältnisses von PMS zu PMPS und der Polymerkonzentration
Bereits zuvor wurde in dieser Arbeit im Abschnitt 4.1.3 der Einfluss des Polymerver-
hältnisses auf die Strukturbildung in ungefüllten Systemen untersucht, wobei sich ge-
zeigt hat, dass dort eine Polymer/Polymer-Entmischung für die Strukturbildung verant-
wortlich ist. Daher wurde der gleiche Ansatz bei gefüllten Systemen verfolgt und Be-
schichtungen mit unterschiedlichen Polymerverhältnissen hergestellt. In Abbildung 24
sind lichtmikroskopische Aufnahmen von Beschichtungen mit unterschiedlichen Poly-
mergehalten dargestellt. Abbildung 24 a) zeigt eine Beschichtung, bei der nur PMPS als
Polymer in der Zusammensetzung verwendet wurde und die damit nur eines der beiden
Polymere enthält. Dasselbe gilt für Abbildung 24 d), bei der nur PMS verwendet wurde.
Abbildung 24 b) weist ein Polymerverhältnis von PMPS:PMS von 7:3 und Abbildung
24 c) von 3:7 auf. Zu erkennen ist, dass die Porengröße mit zunehmendem PMS Gehalt
von Abbildung 24 a) nach Abbildung 24 d) abnimmt. Diese Ergebnisse deuten darauf
a) b)
c) d)

4 Ergebnisse
64
hin, dass nur ein Polymer ausreichend ist, um die bekannten Strukturen zu erhalten und
dass das Polymerverhältnis einen Einfluss auf die Porengröße hat.
Abbildung 24: Beschichtungen mit unterschiedlichen Polymerverhältnissen. Abbildung a) zeigt
die lichtmikroskopische Aufnahme einer Beschichtung mit reinem PMPS, Abbil-
dung d) eine mit reinem PMS. Das PMS/PMPS-Verhältnis von Probe b) betrug
3:7 und das der Probe c) 7:3.
Zusätzlich wurde untersucht, in welchem Polymer-Konzentrationsbereich sich Be-
schichtungen herstellen lassen, bei denen sich die netzartige Struktur ausbildet. Hierfür
wurden wie bei der Referenzzusammensetzung das Polymerverhältnis von PMS:PMPS
bei 1:1 belassen, lediglich der Gesamtgehalt der Polymere wurde in 5 vol. % Schritten
stufenweise erhöht, der Bereich zwischen 0 vol. % und 5 vol. % wurde zusätzlich in
kleinere Schritte unterteilt. Hierbei hat sich gezeigt, dass sich bereits ab einer Polymer-
konzentration von jeweils 2,5 vol. % PMS und PMPS die netzartigen Strukturen ausbil-
den. Mit zunehmender Polymerkonzentration nimmt hierbei auch die Porengröße der
Struktur zu. Die Struktur bildet sich bis zu einer Gesamtpolymerkonzentration von 30
a) b)
d) c)

4 Ergebnisse
65
vol. % aus. Ab einer Konzentration von 35 vol. % lässt sich hingegen keine Strukturbil-
dung mehr beobachten.
4.2.5 Einfluss der Füllstoffpartikel auf die Strukturbildung
Da die größte Änderung von den füllstofffreien Systemen zu den füllstoffhaltigen Sys-
temen folglich der Füllstoff ist, soll in diesem Abschnitt der Einfluss des Füllstoffes auf
die Strukturbildung untersucht werden. Insbesondere von Interesse ist hierbei die Art
des Füllstoffmaterials, die Korngröße und der Volumenanteil des Füllstoffes im Schli-
cker.
4.2.5.1 Variation des Füllstoffgehalts
Wie bereits für Methanol, die Polymere und MTES geschehen, soll auch für den Füll-
stoff SiC, im speziellen für den am häufigsten verwendeten Füllstoff SiC SM07, nach-
gewiesen werden, in welchen Mengen er verwendet werden kann, um zu den bekannten
Strukturen zu führen. Hierfür wurden Schlicker hergestellt, die sich um 2,5 vol. % im
SiC Gehalt voneinander unterscheiden und daraus Beschichtungen mittels Tauchbe-
schichten hergestellt.
Abbildung 25 a) zeigt eine Beschichtung , die mit einem Schlicker hergestellt wurde,
der 10 vol. % SiC als Füllstoff enthält. Hierbei ist zu erkennen, dass die bekannten
Strukturen nicht auftreten. Es sind große Poren zu erkennen, die als präparationsbeding-
te Luftblasen interpretiert werden. Dies gilt auch für Beschichtungen, die mit einem
Volumenanteil von kleiner als 10 vol. % SiC hergestellt wurden. Erst ab einem SiC-
Gehalt von 12,5 vol. %, in Abbildung 25 b) dargestellt, entstehen netzartige Strukturen.
Die in Abbildung 25 c) dargestellte Beschichtung mit einem SiC-Gehalt von 17,5 vol.
% ist nahe der verwendeten Referenzzusammensetzung, bei der der SiC-Gehalt 18,1
vol. % beträgt. Die Strukturen entstehen bis zu einem maximalen SiC-Gehalt von 25
vol. %, die in Abbildung 25 d) dargestellt sind. Beschichtungen mit Schlickern, deren
SiC-Gehalt 25 vol. % übersteigt, konnten nicht hergestellt werden. Da hierbei aufgrund
der Zugabe des Füllstoffes, die Viskosität derart ansteigt, dass eine weitere Zugabe von
Füllstoff nicht möglich ist.
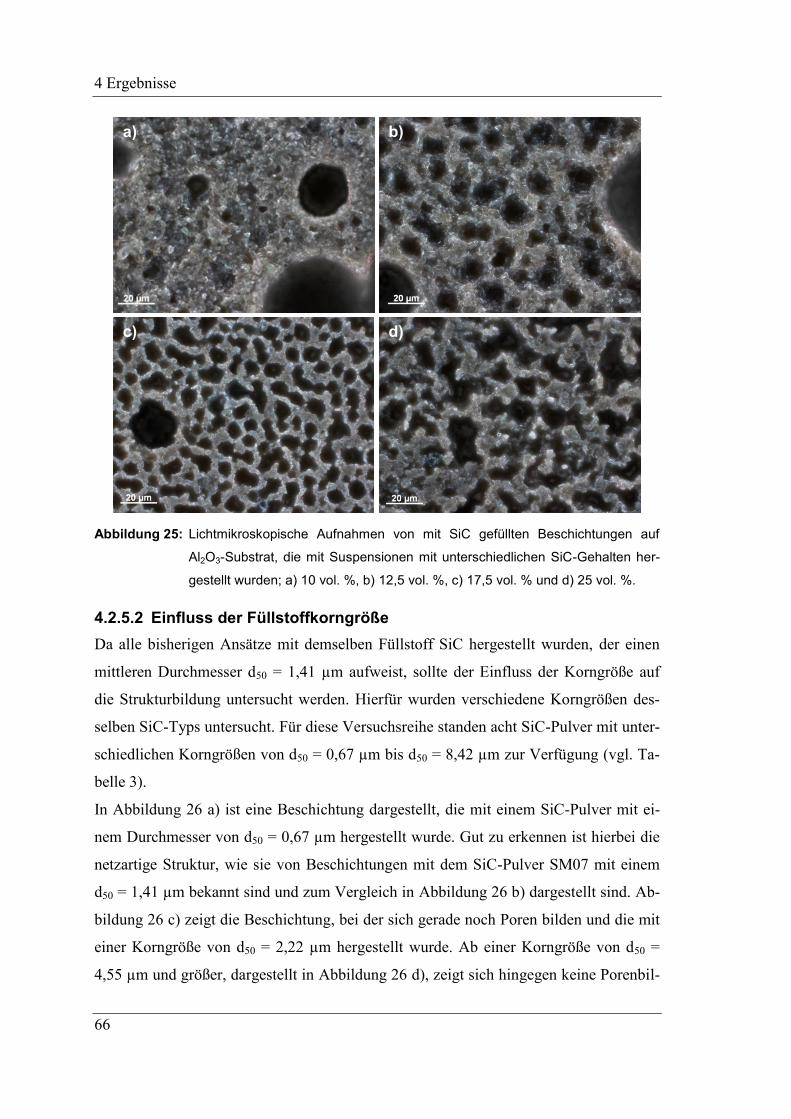
4 Ergebnisse
66
Abbildung 25: Lichtmikroskopische Aufnahmen von mit SiC gefüllten Beschichtungen auf
Al2O3-Substrat, die mit Suspensionen mit unterschiedlichen SiC-Gehalten her-
gestellt wurden; a) 10 vol. %, b) 12,5 vol. %, c) 17,5 vol. % und d) 25 vol. %.
4.2.5.2 Einfluss der Füllstoffkorngröße
Da alle bisherigen Ansätze mit demselben Füllstoff SiC hergestellt wurden, der einen
mittleren Durchmesser d50 = 1,41 µm aufweist, sollte der Einfluss der Korngröße auf
die Strukturbildung untersucht werden. Hierfür wurden verschiedene Korngrößen des-
selben SiC-Typs untersucht. Für diese Versuchsreihe standen acht SiC-Pulver mit unter-
schiedlichen Korngrößen von d50 = 0,67 µm bis d50 = 8,42 µm zur Verfügung (vgl. Ta-
belle 3).
In Abbildung 26 a) ist eine Beschichtung dargestellt, die mit einem SiC-Pulver mit ei-
nem Durchmesser von d50 = 0,67 µm hergestellt wurde. Gut zu erkennen ist hierbei die
netzartige Struktur, wie sie von Beschichtungen mit dem SiC-Pulver SM07 mit einem
d50 = 1,41 µm bekannt sind und zum Vergleich in Abbildung 26 b) dargestellt sind. Ab-
bildung 26 c) zeigt die Beschichtung, bei der sich gerade noch Poren bilden und die mit
einer Korngröße von d50 = 2,22 µm hergestellt wurde. Ab einer Korngröße von d50 =
4,55 µm und größer, dargestellt in Abbildung 26 d), zeigt sich hingegen keine Porenbil-
a) b)
c) d)

4 Ergebnisse
67
dung mehr. Es ist eine Anordnung der SiC-Körner nur noch annahmeweise zu beobach-
ten. Dies lässt sich auch für Beschichtungen feststellen, die mit einer Korngröße größer
als d50 = 4,55 µm hergestellt wurden und hier nicht abgebildet sind. Ebenso lässt sich
erkennen, dass mit zunehmender Korngröße von Abbildung 26 a) nach c) die Porengrö-
ße entgegengesetzt der Korngröße abnimmt.
Es lässt sich festhalten, dass die bekannte Porenstruktur bereits ab der kleinsten ver-
wendeten Korngröße von d50 = 0,67 µm bis zu einer Korngröße von d50 = 2,22 µm aus-
gebildet werden.
Abbildung 26: Lichtmikroskopische Aufnahmen von mit SiC gefüllten Beschichtungen auf
Al2O3-Substrat, die mit Suspensionen mit unterschiedlichen SiC-Korngrößen
hergestellt wurden; a) d50 = 0,67 µm, b) d50 = 1,41 µm, c) d50 = 2,22 µm und d)
d50 = 4,55 µm.
4.2.5.3 Variation des Füllstoffmaterials
Im vorangegangenen Abschnitt wurde bereits eine mögliche Eigenschaft, nämlich die
Korngrößevon Partikeln und Systemen in denen Füllstoffe verwendet werden, behan-
delt. Bei der Korngröße handelt es sich gleichzeitig, um eine der wenigen Eigenschaften
b)
c) d)
a)

4 Ergebnisse
68
von Partikeln, die vergleichsweise leicht erfasst werden kann. Jedoch hat auch die Par-
tikeloberfläche Einfluss auf solche Systeme. Jedoch sind hier die messtechnischen Me-
thoden zur Charakterisierung der Partikeloberfläche weit eingeschränkter. Daher wurde
im Folgenden zunächst die Art des Füllstoffes geändert und dabei die Korngröße und
sonstige Parameter möglichst gleich gehalten. Um den Einfluss der Art des Füllstoffs zu
ermitteln, wurde eine Versuchsreihe mit der Referenzzusammensetzung und variieren-
der Füllstoffart (Aluminium, Aluminiumoxid, Bohrnitrid, Bohrcarbid, Glas, Graphit,
Kupferoxid, Manganoxid, Zirkoniumoxid, Quarzgut, Silizium, Siliziumnitrid und Sili-
ziumcarbid; vgl. Tabelle 3) durchgeführt. Die Bildung der Netzstrukturen wurde in Ge-
genwart von nur drei Füllstoffen beobachtet: Si, SiC und Si3N4.
Zudem wurde versucht, die Oberflächeneigenschaften von Al2O3-Pulvern als Füllstoffe
so zu beeinflussen, dass deren Einsatz ebenso zu netzartig strukturierten Oberflächen
führt. Sowohl das ausheizen der Partikel zwischen 120 °C und 900 °C und die damit
verbundene Veränderung der OH-Gruppenbelegung der Oberfläche, als auch das Be-
schichten der Partikel mit PMPS vor dem Einsatz, haben dabei zu keinem Erfolg ge-
führt.
4.2.6 Einfluss des Substratmaterials auf die Beschichtungen
Zur Klärung des Einflusses des Substratmaterials auf die Struktur der Beschichtung
wurden unterschiedliche Substratmaterialien untersucht. Dazu gehörten Alumini-
umoxid, polymerabgeleitete keramische Folien, Glas, verschiedene Kunststoffe, Poly-
tetrafluorethylen, Kupfer, Aluminium, Weißblech, FeCrAl-Legierungen und Silizium-
wafer. Dabei hat sich gezeigt, dass sich die Beschichtungen nahezu fehlerfrei auf allen
getesteten Substratmaterialien aufbringen lassen. Die bekannten Strukturen bildeten sich
dabei auf allen Substraten aus. Ihre Morphologie ändert sich dabei nur geringfügig.
4.2.7 Reduzierung auf die minimal möglichen Komponenten im System
Zur Klärung, welche Komponenten im System notwendig sind, um eine strukturierte
Beschichtung zu erhalten, wurden die Komponenten der Schlickerzusammensetzung
soweit wie möglich reduziert. Aus Abschnitt 4.2.4 (Einfluss des Verhältnisses von PMS
zu PMPS und der Polymerkonzentration) ist bekannt, dass nur eines der beiden Polyme-
re notwendig ist. Ebenso ist aus dieser Versuchsreihe bekannt, dass bereits eine geringe
Menge an Polymer von 5 vol. % ausreichend ist. Daher wurde für den folgenden Ver-

4 Ergebnisse
69
such nur PMPS als Polymer verwendet und mit einer Startkonzentration von 5 vol. %
begonnen. Auch ist aus der Versuchsreihe (4.2.3 Beeinflussung des Systems durch das
Lösungsmittel MTES) bekannt, dass MTES nicht zwingend als Lösungsmittel für die
Strukturbildung in gefüllten Systemen erforderlich ist. Daher wurde im Folgenden auch
auf MTES als Lösungsmittel verzichtet. Dies ist jedoch nur möglich, da PMPS eine
hochviskose Flüssigkeit ist, die sich daher mit Methanol und dem Füllstoff mischen
lässt. Aus vorangegangenen Versuchen war bereits bekannt, dass die Vernetzungskata-
lysatoren Ölsäure und Aluminiumacetylacetonat für die Strukturbildung nicht benötigt
werden. So wurde als Ausgangszusammensetzung ein Schlicker hergestellt, bei dem das
Methanol:SiC-Verhältnis gleich ist wie in der Referenzzusammensetzung, der jedoch
nur 5 vol. % PMPS enthält.
Abbildung 27 zeigt eine Versuchsreihe mit Beschichtungen, bei der der PMPS-Gehalt
variiert wurde. Es sollte herausgefunden werden, ob Strukturen entstehen, wenn das
Polymer vollständig weggelassen wird, bzw. ab welchem Polymergehalt Strukturen
entstehen. Die Beschichtung, dargestellt in Abbildung 27 a), hergestellt ohne PMPS,
zeigt keine netzartigen Strukturen, auch in Abbildung 27 b), die eine Beschichtung
zeigt, die mit 1 vol. % PMPS hergestellt wurde, zeigt keine netzartigen Strukturen. Erst
ab einer Polymerkonzentration von 2,5 vol. % PMPS in Abbildung 27 c), sind netzarti-
ge Strukturen zu erkennen, die mit einer Polymerkonzentration von 5 vol. % PMPS in
Abbildung 27 d) noch deutlicher ausgeprägt sind.
Damit lässt sich festhalten, dass für die Bildung netzartiger Strukturen in gefüllten Sys-
temen nur ein Polymer in einer geringen Konzentration von 2,5 vol. % und der Füllstoff
sowie Methanol notwendig sind.

4 Ergebnisse
70
Abbildung 27: Lichtmikroskopische Aufnahmen von mit SiC gefüllten Beschichtungen auf
Al2O3-Substrat, die mit Suspensionen hergestellt sind, die nur Methanol, SiC
und PMPS enthalten. Das Methanol:SiC Verhältnis entspricht dem im Refe-
renzansatz, der PMPS-Gehalt variiert a) 0 vol. %, b) 1 vol. %, c) 2,5 vol. % und
d) 5 vol. %.
4.2.8 Zeitlicher Ablauf der Strukturbildung
Um die Entstehung der Struktur besser zu verstehen, wurde versucht, die Beschichtung
vor dem Trocknen, direkt nach dem Tauchbeschichten, unter das Mikroskop zu bringen
und einen Film der Porenentstehung aufzunehmen. Da es nicht möglich ist, mit einem
herkömmlichen Mikroskop, während des Beschichtens, ein scharfgestelltes Bild zu er-
halten, ist es auf diesem Weg auch nicht möglich, bereits die ersten Sekunden der Struk-
turentstehung zu beobachten. Daher beginnen die Aufnahmen in Abbildung 28, die
Ausschnitte aus diesem Film darstellen, erst nach 15 s. In dieser ersten Abbildung sind
bereits die ersten kleinen Poren und auch deutlich größere Poren, die sich als dunkle
Stellen zeigen, zu erkennen. Im weiteren Verlauf zwischen 15 s und 54 s ist zu erken-
nen, dass die Poren stetig größer werden. Dies erfolgt, indem sich die kleinen Poren zu
immer größeren Poren verbinden, indem die Stege aufbrechen und sich die Porenflä-
a) b)
c) d)

4 Ergebnisse
71
chen somit addieren. So ist auch zu beobachten, dass aus anfänglich vielen nahezu run-
den, unregelmäßig geformte Poren entstehen, je nachdem in welche Richtung die Po-
renvergrößerung erfolgt. In diesem Zusammenhang konnte auch die Entstehung der
zweiten Generation von Poren beobachtet werden. Nach 67 s beginnt die Beschichtung
schnell zu trocknen, was bedeutet, dass das Methanol fast vollständig verdunstet. Dies
wird dadurch sichtbar, dass Spiegelungen durch sich in den großen Poren bildende Me-
nisken entstehen. Die bei 67 s sichtbare beginnende Trocknung ist nach weiteren 14 s,
also nach einer Gesamtzeit von 81 s, vollständig abgeschlossen. Der Moment, in dem
die Trocknung abgeschlossen ist, ist geleichbedeutend mit dem Zeitpunkt, an dem die
netzartigen Strukturen ihre endgültige Form angenommen haben. Daher lässt sich fest-
halten: Ab Beginn der Beschichtung wachsen die Poren und mit vollständigem Ver-
dampfen des Methanols ist die Strukturbildung abgeschlossen.
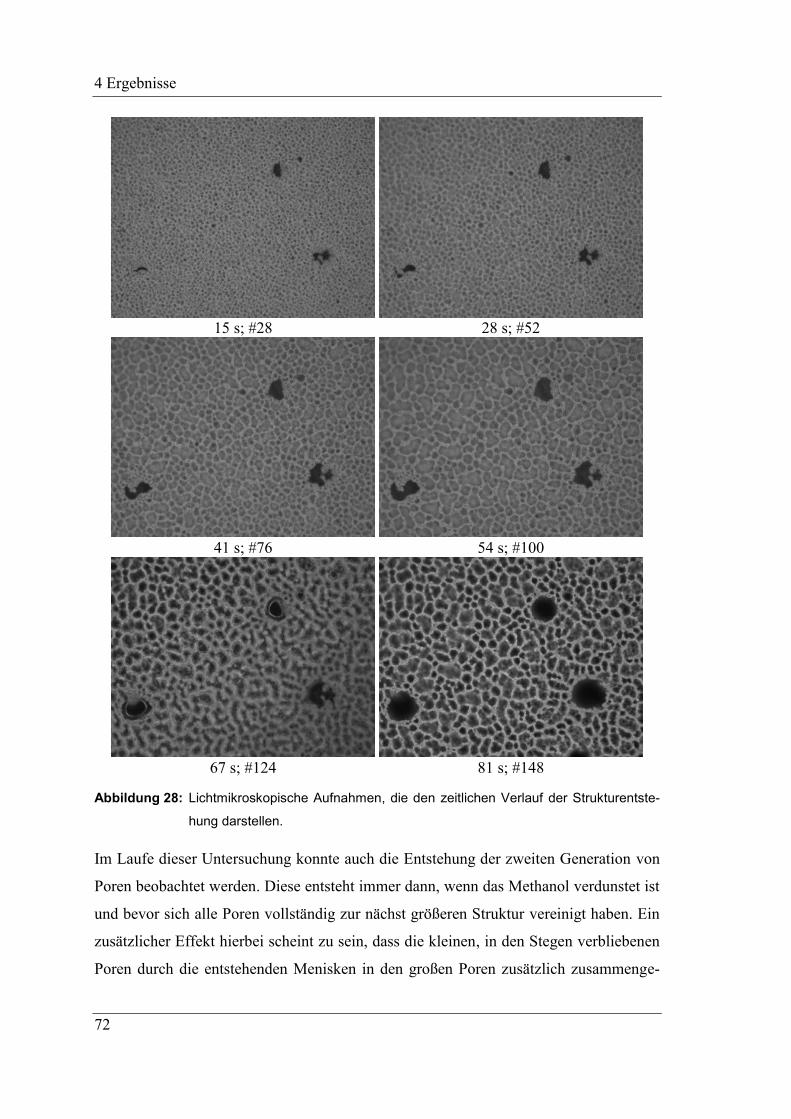
4 Ergebnisse
72
15 s; #28
28 s; #52
41 s; #76 54 s; #100
67 s; #124
81 s; #148
Abbildung 28: Lichtmikroskopische Aufnahmen, die den zeitlichen Verlauf der Strukturentste-
hung darstellen.
Im Laufe dieser Untersuchung konnte auch die Entstehung der zweiten Generation von
Poren beobachtet werden. Diese entsteht immer dann, wenn das Methanol verdunstet ist
und bevor sich alle Poren vollständig zur nächst größeren Struktur vereinigt haben. Ein
zusätzlicher Effekt hierbei scheint zu sein, dass die kleinen, in den Stegen verbliebenen
Poren durch die entstehenden Menisken in den großen Poren zusätzlich zusammenge-

4 Ergebnisse
73
drückt werden und somit deutlich kleiner erscheinen, als die größere Genration, die
hierdurch zusätzlich wächst.
4.2.9 Verwendbarkeit des Systems für verschiedene Beschichtungsverfahren
Im Laufe der Arbeit wurden unterschiedlichste Beschichtungsverfahren getestet, mit
denen die Beschichtung aufgebracht werden kann und bei der die bekannte Struktur
entsteht. Hierbei hat sich gezeigt, dass bei allen getesteten Verfahren die Strukturbil-
dung auftritt. Getestet wurden unter anderem Tauchbeschichten, Schleuderbeschichten,
Rakeln, manuelles Auftragen (Hand/Pinsel) und das Beschichten von Schäumen mittels
Tauchen und anschließendem Ausschleudern.
4.2.9.1 Tauchbeschichten
Das Tauchbeschichten ist eine sehr einfache Beschichtungsmethode und zeigt gute Re-
produzierbarkeit, bezüglich der Schichtdicken der Beschichtungen. Die Schichtdicke
lässt sich dabei gut über die Ziehgeschwindigkeit beim Herausziehen des Substrates
einstellen. Aus diesen Gründen ist es am besten, zur Herstellung der hier gezeigten
Schichten geeignet. Aufgrund der Einfachheit sind alle gezeigten Beschichtungen mit
diesem Verfahren entstanden, ausgenommen der Beschichtungen, die in den folgenden
beiden Abschnitten für Schleuderbeschichten und Rakeln gezeigt werden.
4.2.9.2 Schleuderbeschichten
Das Schleuderbeschichten hat sich insbesondere zur Herstellung dünner Beschichtun-
gen als geeignet gezeigt. Es wurde daher insbesondere dafür genutzt, möglichst dünne
Beschichtungen herzustellen und zu charakterisieren. Da bei dieser Versuchsreihe mög-
lichst kleine Strukturen erzeugt werden sollten, wurde hierfür ein Füllstoff mit einem
kleineren mittleren Durchmessers gewählt. Im Referenzansatz wird SM07 mit einem d50
Wert von 1,41 µm verwendet und in dieser Versuchsreihe NF25 mit einem d50 Wert von
0,67 µm. Abbildung 29 a) Aufsicht und b) Querschnitt zeigt eine Beschichtung, die mit
einer Endrotationsgeschwindigkeit von 6000 1 min-1
hergestellt wurde, die gleichzeitig
die dünnste hergestellte Beschichtung in dieser Arbeit darstellt. Die Beschichtung hat,
wie in Abbildung 29 b) zu erkennen ist, eine Dicke von rund 8,9 µm und zeigt dabei
eine Porengröße von einigen wenigen Mikrometern (Abbildung 29 a)). Mit abnehmen-
der Endrotationsgeschwindigkeit steigt die Schichtdicke an. Exemplarisch ist hierfür

4 Ergebnisse
74
eine Beschichtung in Abbildung 29 c) Aufsicht und d) Querschnitt, die mit einer Endro-
tationsgeschwindigkeit von 2000 1 min-1
hergestellt wurde, dargestellt. Zu erkennen ist
hierbei, dass sowohl die Schichtdicke deutlich auf rund 15,6 µm zugenommen hat
(Abbildung 29 d)) als auch, dass die Poren dieser Beschichtung deutlich angewachsen
sind, wie Abbildung 29 c) zeigt. Die Entstehung größerer Poren bei dickeren Beschich-
tungen lässt sich hierbei über den gesamten hergestellten Bereich beobachten und wird
im Abschnitt 4.2.10 (Variation der Schichtdicke und ihr Einfluss auf die Porengröße)
genauer behandelt.
Beim Schleuderbeschichten treten Schwierigkeiten auf, wenn es darum geht, die Be-
schichtung sehr gleichmäßig aufzutragen, da aufgrund der sehr dünnen Schichten und
der hohen Luftströmung, durch die teils hohen Rotationsgeschwindigkeit, diese sehr
schnell trocknen. Daher eignet sich das Schleuderbeschichten nur für kleine, planare
Substrate. Somit lässt sich festhalten, dass sich auf kleinen, planaren Substraten mittels
Schleuderbeschichten sehr dünne Beschichtungen herstellen lassen, die dann eine sehr
feine Porenstruktur aufweisen.

4 Ergebnisse
75
Abbildung 29: Mittels Schleuderbeschichten hergestellte Beschichtungen, bei denen der Füll-
stoff NF25 mit einem d50 = 0,67 µm verwendet wurde. Die Endrotationsge-
schwindigkeit für die Aufsicht a) und den Querschnitt b) betrug 6000 1 min-1
und
für die Aufsicht c) und den Querschnitt d) 2000 1 min-1
.
4.2.9.3 Automatisches Filmziehen
Ein weiteres angewandtes Beschichtungsverfahren ist das sogenannte Rakelverfahren.
Hierbei sollte untersucht werden, ob auch eigenständige keramische Folien hergestellt
werden können und ob diese dann ebenfalls die netzartigen Strukturen aufweisen. Hier-
für wird eine speziell vorbeschichtete Kunststofffolie mit Schlicker beschichtet, von der
nach dem Aushärten bei 110 °C die Beschichtung abgelöst werden kann und somit eine
eigenständige keramische Folie entsteht. Die netzartigen Strukturen entstehen bereits ab
der geringsten gewählten Einstellung für die Spaltbreite am Rakel von 10 µm und sind
bis zur maximalen Spaltbreite von 3000 µm zu beobachten. Dabei hat sich gezeigt, dass
sich nicht alle Beschichtungen von der Kunststofffolie ablösen lassen. Ablösbar sind
Beschichtungen, die mit einer Spaltbreite ab 250 µm hergestellt worden sind. In Abbil-
dung 30 sind beispielhaft zwei Folien nach dem Aushärten bei 110 °C und dem Ablösen
von der Kunststofffolie dargestellt. Abbildung 30 a) zeigt die Aufsicht auf die dünnst
a) b)
c) d)

4 Ergebnisse
76
mögliche, abgelöste Folie, die mit einer Spaltbreite von 250 µm hergestellt wurde. Zu
erkennen sind die typischen Strukturen, wie sie bereits vom Tauchbeschichten und vom
Schleuderbeschichten bekannt sind. Abbildung 30 b) zeigt den Querschnitt derselben
Folie, die hierbei gemessene Dicke der Folie beträgt 118 µm. Gut zu erkennen ist, dass
die Porenstruktur auch hierbei durch die gesamte Folie geht. Abbildung 30 c) zeigt die
Aufsicht einer Folie, die mit einer Spaltbreite von 2500 µm hergestellt wurde. Auch hier
ist die bekannte Struktur zu erkennen, jedoch zeigt sich hierbei wieder die Ausbildung
zweier Generationen von Poren, große Poren und kleinere, die in den Stegen zu erken-
nen sind. Im Vergleich zu Abbildung 30 a) zeigt sich das wie bereits beim Schleuderbe-
schichten beobachtete Phänomen, dass mit zunehmender Schichtdicke auch die Poren-
größe zunimmt. In Abbildung 30 d) ist ein Querschnitt derselben Folie wie Abbildung
30 c) dargestellt. Die gemessene Schichtdicke dieser Folie beträgt 485 µm und ist damit
etwa vier Mal dicker als die mit einer zehn Mal kleineren Spaltbreite hergestellten Folie
in Abbildung 30 b). Auch hier ist im Querschnitt (Abbildung 30 d)) gut zu beobachten,
dass die Porenstruktur durch die gesamte Folie geht und ebenfalls zwei Generationen
von Poren aufweist.
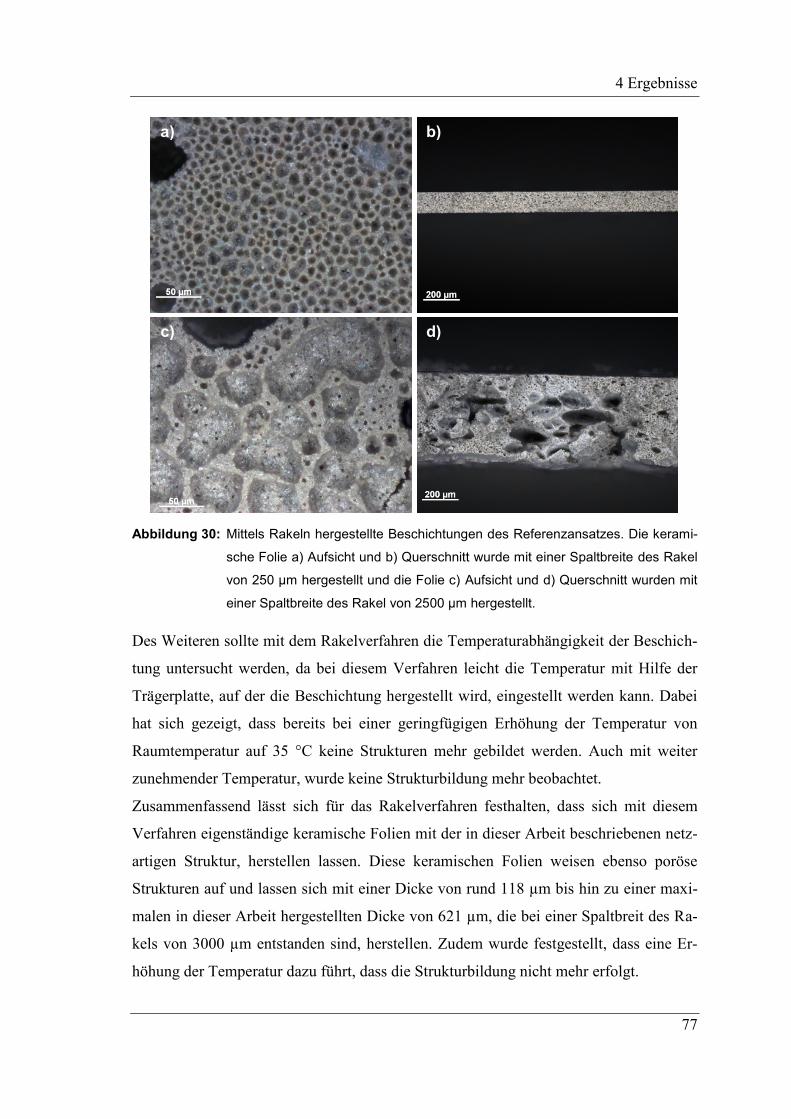
4 Ergebnisse
77
Abbildung 30: Mittels Rakeln hergestellte Beschichtungen des Referenzansatzes. Die kerami-
sche Folie a) Aufsicht und b) Querschnitt wurde mit einer Spaltbreite des Rakel
von 250 µm hergestellt und die Folie c) Aufsicht und d) Querschnitt wurden mit
einer Spaltbreite des Rakel von 2500 µm hergestellt.
Des Weiteren sollte mit dem Rakelverfahren die Temperaturabhängigkeit der Beschich-
tung untersucht werden, da bei diesem Verfahren leicht die Temperatur mit Hilfe der
Trägerplatte, auf der die Beschichtung hergestellt wird, eingestellt werden kann. Dabei
hat sich gezeigt, dass bereits bei einer geringfügigen Erhöhung der Temperatur von
Raumtemperatur auf 35 °C keine Strukturen mehr gebildet werden. Auch mit weiter
zunehmender Temperatur, wurde keine Strukturbildung mehr beobachtet.
Zusammenfassend lässt sich für das Rakelverfahren festhalten, dass sich mit diesem
Verfahren eigenständige keramische Folien mit der in dieser Arbeit beschriebenen netz-
artigen Struktur, herstellen lassen. Diese keramischen Folien weisen ebenso poröse
Strukturen auf und lassen sich mit einer Dicke von rund 118 µm bis hin zu einer maxi-
malen in dieser Arbeit hergestellten Dicke von 621 µm, die bei einer Spaltbreit des Ra-
kels von 3000 µm entstanden sind, herstellen. Zudem wurde festgestellt, dass eine Er-
höhung der Temperatur dazu führt, dass die Strukturbildung nicht mehr erfolgt.
a) b)
c) d)

4 Ergebnisse
78
4.2.10 Variation der Schichtdicke und ihr Einfluss auf die Porengröße
Abbildung 31 zeigt Aufnahmen von Referenzbeschichtungen auf Aluminiumoxid, die
mit unterschiedlichen Ziehgeschwindigkeiten hergestellt worden sind und die sich somit
in der Schichtdicke unterscheiden. Die Ziehgeschwindigkeit nimmt dabei von a) mit ca.
28,6 mm s-1
nach i) mit ca. 2,2 mm s-1
ab. Dabei lässt sich erkennen, dass mit abneh-
mender Ziehgeschwindigkeit bzw. Schichtdicke sich auch die Porengröße von Abbil-
dung 31 a) nach i) verringert.
Abbildung 31: Lichtmikroskopische Aufnahmen von Referenzbeschichtungen hergestellt mit
unterschiedlichen Ziehgeschwindigkeiten von 2,5 bis 30 mm s-1
.
Das Diagramm in Abbildung 32 zeigt die Schichtdicke in Abhängigkeit von der Ziehge-
schwindigkeit. Dabei zeigt sich, dass sich das System wie nach Landau-Levic beschrie-
ben verhält. Mit zunehmender Ziehgeschwindigkeit nimmt auch die Schichtdicke zu.
An den Fehlerbalken ist zu erkennen, dass die Reproduzierbarkeit des Tauchbeschich-
tens sehr gut gegeben ist. Die Schichtdicke nimmt hierbei von 25,7 µm bei einer Zieh-
a)
i) h) g)
f) e) d)
c) b)
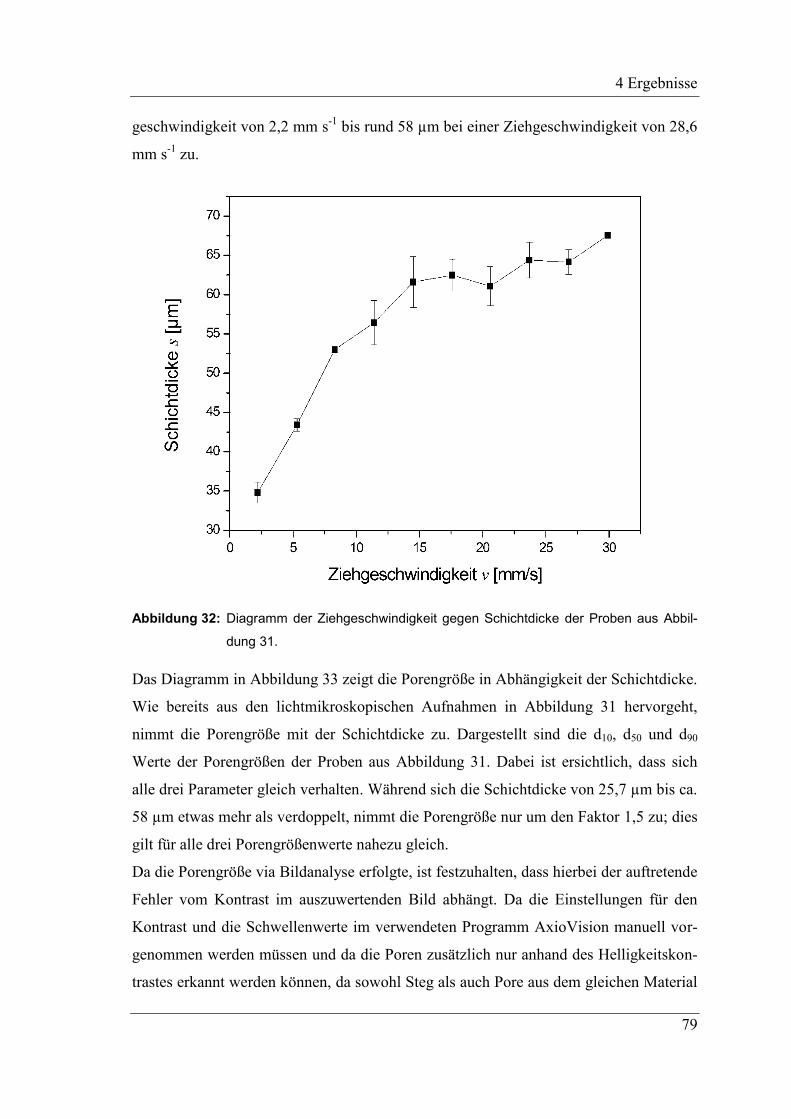
4 Ergebnisse
79
geschwindigkeit von 2,2 mm s-1
bis rund 58 µm bei einer Ziehgeschwindigkeit von 28,6
mm s-1
zu.
Abbildung 32: Diagramm der Ziehgeschwindigkeit gegen Schichtdicke der Proben aus Abbil-
dung 31.
Das Diagramm in Abbildung 33 zeigt die Porengröße in Abhängigkeit der Schichtdicke.
Wie bereits aus den lichtmikroskopischen Aufnahmen in Abbildung 31 hervorgeht,
nimmt die Porengröße mit der Schichtdicke zu. Dargestellt sind die d10, d50 und d90
Werte der Porengrößen der Proben aus Abbildung 31. Dabei ist ersichtlich, dass sich
alle drei Parameter gleich verhalten. Während sich die Schichtdicke von 25,7 µm bis ca.
58 µm etwas mehr als verdoppelt, nimmt die Porengröße nur um den Faktor 1,5 zu; dies
gilt für alle drei Porengrößenwerte nahezu gleich.
Da die Porengröße via Bildanalyse erfolgte, ist festzuhalten, dass hierbei der auftretende
Fehler vom Kontrast im auszuwertenden Bild abhängt. Da die Einstellungen für den
Kontrast und die Schwellenwerte im verwendeten Programm AxioVision manuell vor-
genommen werden müssen und da die Poren zusätzlich nur anhand des Helligkeitskon-
trastes erkannt werden können, da sowohl Steg als auch Pore aus dem gleichen Material
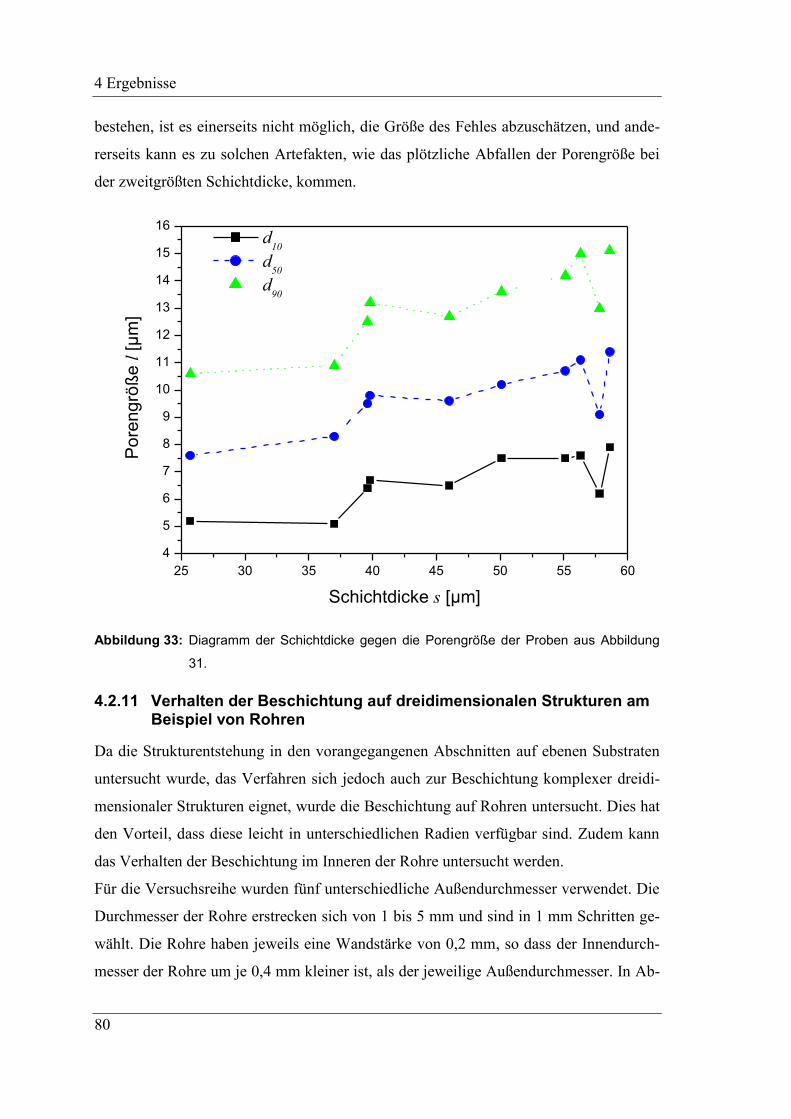
4 Ergebnisse
80
bestehen, ist es einerseits nicht möglich, die Größe des Fehles abzuschätzen, und ande-
rerseits kann es zu solchen Artefakten, wie das plötzliche Abfallen der Porengröße bei
der zweitgrößten Schichtdicke, kommen.
Abbildung 33: Diagramm der Schichtdicke gegen die Porengröße der Proben aus Abbildung
31.
4.2.11 Verhalten der Beschichtung auf dreidimensionalen Strukturen am Beispiel von Rohren
Da die Strukturentstehung in den vorangegangenen Abschnitten auf ebenen Substraten
untersucht wurde, das Verfahren sich jedoch auch zur Beschichtung komplexer dreidi-
mensionaler Strukturen eignet, wurde die Beschichtung auf Rohren untersucht. Dies hat
den Vorteil, dass diese leicht in unterschiedlichen Radien verfügbar sind. Zudem kann
das Verhalten der Beschichtung im Inneren der Rohre untersucht werden.
Für die Versuchsreihe wurden fünf unterschiedliche Außendurchmesser verwendet. Die
Durchmesser der Rohre erstrecken sich von 1 bis 5 mm und sind in 1 mm Schritten ge-
wählt. Die Rohre haben jeweils eine Wandstärke von 0,2 mm, so dass der Innendurch-
messer der Rohre um je 0,4 mm kleiner ist, als der jeweilige Außendurchmesser. In Ab-
25 30 35 40 45 50 55 60
4
5
6
7
8
9
10
11
12
13
14
15
16
d10
d50
d90
Po
ren
grö
ße
l [µ
m]
Schichtdicke s [µm]

4 Ergebnisse
81
bildung 34 sind die mittels Tauchbeschichten beschichteten Rohre dargestellt. Zu er-
kennen ist hierbei, insbesondere bei dem Rohr mit einem Durchmesser von 4 mm, dass
diese nach dem Beschichten am unteren Ende mit einem Tropfen verschlossen sind.
Abbildung 34: Fotografie von beschichteten Aluminiumoxidrohren mit unterschiedlichen
Durchmessern von 1 bis 5 mm, beschichtet nach dem Referenzverfahren, aus-
gehärtet bei 110 °C
Abbildung 35 zeigt die Beschichtung auf der Außenseite der Rohre bei drei exempla-
risch gewählten Durchmessern von a) d = 1 mm, b) d = 3 mm, c) d = 5 mm und zum
Vergleich auf einem planaren Substrat. Zu erkennen ist hierbei, dass sich die Strukturen
in allen vier Fällen nur geringfügig voneinander unterscheiden. Der Unterschied liegt
hierbei im selben Bereich, wie er auch bei planaren Substraten auftritt. Auf der Innen-
seite der Rohre war hingegen keine netzartige Struktur der Beschichtung zu erkennen.
Daher wurde der Versuch wiederholt mit dem Unterschied, dass bei der zweiten Ver-
suchsreihe, der Tropfen der die Rohre verschließt, unmittelbar nach dem Tauchbe-
schichten und somit vor dem Trocknen der Schicht entfernt wurde. Dabei war zu be-
obachten, dass in diesem Fall auch auf der Innenseite der Rohre die netzartigen Struktu-
ren auftreten und sich dabei nicht wesentlich von der Struktur auf der Außenseite unter-
scheiden.
5 mm
4 mm
3 mm
2 mm
1 mm

4 Ergebnisse
82
Abbildung 35: Lichtmikroskopische Aufnahmen von Beschichtungen auf Aluminiumoxidrohren
mit unterschiedlichen Durchmessern: a) d = 1 mm, b) d = 3 mm, c) d = 5 mm
und eine Referenzbeschichtung auf planarem Aluminiumoxid d).
a) b)
c) d)

5 Diskussion
83
5 Diskussion
5.1 Ungefüllte Systeme auf Basis präkeramischer Polymere
PMS und PMPS
5.1.1 Hansen Löslichkeitsparameter der Polysiloxane
Die Bestimmung der HSP mit den beiden Methoden auf Grundlage der Lösungsversu-
che, sowie der Methode auf Grundlage der inhärenten Viskosität, ergeben gemittelte
HSP von δD = 16,8 MPa1/2
± 0,1, δP = 3,3 MPa1/2
± 1,0, δH = 5,0 MPa1/2
± 1,1 mit einem
Radius von 4 MPa1/2
± 0,9 für PMS und für PMPS δD = 17,7 MPa1/2
± 0,7, δP = 8,2
MPa1/2
± 1,3, δH = 6,4 MPa1/2
± 0,9 mit einem Radius von 9,2 MPa1/2
± 0,8. Eine Auffäl-
ligkeit der Untersuchungen ist, dass die Interaktionskugeln von PMPS durchweg größer
sind als die von PMS. Dies ist wahrscheinlich auf die geringere molare Masse von
PMPS mit Mw = 1200 g mol-1
gegenüber PMS mit Mw = 6600 g mol-1
zurückzuführen,
denn je kleiner die molare Masse, umso höher ist die Löslichkeit.116
Dies ist auch mit
Hilfe von Gleichung (3) erklärbar und ist auf den verminderten Anstieg der Entropie bei
höheren molaren Massen beim Mischen zurückzuführen (vergleiche Kapitel 2.2.1).36
Ebenso wurde gezeigt, dass die HSP der Polymere so weit auseinander liegen, dass im
Falle der Methode nach Gharagheizi sogar das Zentrum von PMPS außerhalb der Kugel
von PMS liegt und somit als „Nichtlösungsmittel“ für PMS gilt. Da zusätzlich insbe-
sondere die polaren, aber auch die Wasserstoffbrückenanteile der Polymere voneinander
abweichen, ist anhand der HSP davon auszugehen, dass die Polymere nicht vollständig
miteinander mischbar sind.
Eine weitere Auffälligkeit der Untersuchung ist, dass MTES, das für beide Polymere
experimentell als gutes Lösungsmittel identifiziert wurde und auch in anderen Anwen-
dungen als Lösungsmittel für die beiden Polymere PMS und PMPS eingesetzt wird10–12
,
gerade noch innerhalb der Kugeln von PMS liegt. Im Falle von PMPS liegt MTES so-
gar, für die zwei Methoden, die mit Hilfe der Löslichkeitsversuche die HSP bestimmen,
außerhalb der Kugeln, weswegen MTES anhand der HSP bei diesen Methoden als
Nichtlösungsmittel für PMPS gilt. Dies kann mehrere Gründe haben, zum einen sind die
HSP von MTES nicht in der Datenbank der Hansen-Software enthalten und sind somit
nicht experimentell bestimmt, sondern konnten nur anhand der Molekülstruktur von der
Hansen-Software berechnet werden. Da aber die Berechnung der HSP von MTES auf

5 Diskussion
84
vergleichenden Methoden basiert und in der Datenbank nur wenige siliziumbasierte
Lösungsmittel enthalten sind, kann die Berechnung somit zu fehlerhaften Ergebnissen
führen. Das zeigt sich auch dadurch, dass sich die HSP von MTES mit jeder neuen
Softwareversion und der damit wachsenden Datenbank leicht ändern. Eine weitere
Möglichkeit, warum MTES außerhalb der Kugel liegt, könnte darin begründet sein, dass
die bestimmten HSP der Polymere, insbesondere der disperse Anteil, nicht ganz exakt
sind. Dies kann daher rühren, dass die verwendeten Lösungsmittel im dispersen Anteil
in einem Bereich von 14,5 bis 19,7 MPa1/2
liegen und somit auch über den berechneten
von MTES mit 14,2 MPa1/2
. Auch hier ist es so, dass die verwendeten Lösungsmittel
rein organischer Natur und nicht siliziumbasiert sind, wie die untersuchten Polymere
oder MTES. Der Einsatz von Lösungsmitteln mit niedrigerem dispersem Anteil bzw.
solchen, die ebenfalls auf Silizium basieren, könnte daher dazu führen, dass die HSP der
Polymere zu einem etwas niedrigeren dispersen Anteil verschoben werden. Dies lässt
sich auch dadurch erkennen, dass bei der Methode basierend auf der inhärenten Viskosi-
tät, bei der die Qualität der Lösungsmittel gewertet wird, der disperse Anteil für beide
Polymere am niedrigsten ist und MTES für diese Methode innerhalb der Kugel liegt.
Die Verwendung von Lösungsmitteln mit einem sehr niedrigen dispersen Anteil oder
solchen, die auf Silizium basieren, ist jedoch zum gegenwärtigen Zeitpunkt nur schwer
möglich, da nur sehr wenige in der Datenbank vorhanden sind. Dieser Umstand hat je-
doch nur einen geringen Einfluss auf den praktischen Nutzen der HSP der Polymere, da
der Großteil der in der Datenbank vorhandenen Polymere organischer Natur sind und
im Bereich der verwendeten Lösungsmittel liegen. Daher sollten alle Lösungsmittel in
der Datenbank, die innerhalb der Kugeln der Polymere liegen, diese auch lösen. Somit
ist ein Hauptnutzen der HSP, nämlich die Auswahl passender Lösungsmittel, weiterhin
gegeben. Jedoch ist es auch möglich, dass es bestimmte Lösungsmittel gibt, insbesonde-
re solche die einen niedrigen dispersen Anteil haben und außerhalb der Kugel liegen,
die Polymere ebenso lösen.
Durch den Vergleich der angewendeten Methoden miteinander ist festzustellen, dass
alle drei Methoden sehr ähnliche Werte liefern. Der Unterschied der beiden Berech-
nungsmethoden, die auf den gleichen experimentellen Daten der Lösungsversuche be-
ruhen, begründet sich in den unterschiedlichen verwendeten Algorithmen. Bei der Han-
sen-Software wird eine „quality-of-fit function“, auch „desirability function“ genannt,

5 Diskussion
85
eingesetzt. Während das Programm von Gharagheizi das Downhill-Simplex-Verfahren
einsetzt. Zu den Methoden ist weiter festzuhalten, dass die Methoden beruhend auf den
Lösungsversuchen einfacher und schneller durchzuführen sind. Hier wird keine kom-
plexe Laborausrüstung wie das Ubbelohde-Viskosimeter benötigt, wie es z.B. im Falle
der Methode, bei der die inhärente Viskosität bestimmt wird, notwendig ist. Da aber alle
drei Methoden sehr ähnliche Werte liefern und für die Bestimmung der inhärenten Vis-
kosität nur Lösungsmittel verwendet werden können, die als gute Lösungsmittel durch
Lösungsversuche ermittelt wurden und somit die Lösungsversuche auch hierfür durch-
geführt werden müssen, ist daher zu empfehlen, nur dann aufwendigere Bestimmungs-
methoden, wie die intrinsische Viskosität zu verwenden, wenn die nach den Lösungs-
versuchen bestimmten HSP für den konkreten Anwendungsfall nicht ausreichend genug
sind, bzw. die durch die Lösungsversuche ermittelten HSP nicht zum Ziel führen.
Ein Vergleich mit aus der Literatur bekanntem HSP für siliziumbasierte Polymere zeigt,
dass diese in ähnlichen Größenordnungen liegen, vergleiche Tabelle 9.80,93,117
Die dabei
auftretenden geringen Abweichungen können verschiedene Ursachen haben: Zum einen
unterscheiden sich die HSP je nach Messmethode und gewählten Bedingungen bei de-
nen die Messung durchgeführt wird, wie auch die Untersuchungen in dieser Arbeit ge-
zeigt haben. Daher ist ein genauer Vergleich der HSP nur dann sinnvoll, wenn die Be-
stimmung unter den gleichen Bedingungen durchgeführt wurde. Zudem sind die Poly-
mere sowohl chemisch als auch physikalisch nicht exakt identisch, was ebenso zu Ab-
weichungen führt.
Tabelle 9: Hansen Löslichkeitsparameter, Hildebrand Löslichkeitsparameter sowie der
dazugehörige Interaktionsradius für verschiedene Siliziumpolymere.
Polymer Dispersion Polar Wasserstoff
Brücken
Hildebrand Radius
δd
[MPa1/2
]
δp
[Mpa1/2
]
δh
[Mpa1/2
]
δt
[Mpa1/2
]
[Mpa1/2
]
PMS 16,8 3,3 5,0 17,9 6,4
PMPS 17,7 8,2 6,4 20,6 9,2
MS081393
16,9 5,7 7,2 19,3 4,3
MS081493
16,3 8,3 8,7 20,3 7,0
Poly(methyl ethyl siloxane)117
16,2 1,4 4,1 16,9 -
Polysilicone80
17,2 3,0 3,0 - 8,0
Vinyl Silane80
16,4 3,7 4,5 - 10,0

5 Diskussion
86
5.1.2 Mischbarkeitsdiagramme im System PMS – PMPS – MTES – MeOH
Aus den Mischungsdiagrammen in Abbildung 14 wird ersichtlich, dass MTES zwin-
gend als Lösungsmittel erforderlich ist. Das lässt sich einerseits daran erkennen, dass
alle unteren Außenlinien der vier Phasendiagramme, die zusammen das Phasendiagram
PMPS – MeOH – PMS darstellen und somit kein MTES enthalten, keine mischbaren
Zusammensetzungen aufweisen. Zudem ist ersichtlich, dass nur im oberen MTES-
reichen Teil der Diagramme mischbare Zusammensetzungen existieren. Andererseits
nimmt gleichzeitig mit zunehmendem Methanolgehalt, verbunden mit einer Abnahme
des MTES-Gehaltes, die Mischbarkeit im gesamten untersuchten System ab.
Methanol hingegen bewirkt den gegenteiligen Effekt zu MTES, was sich dadurch zeigt,
dass methanolreiche Proben keine Mischbarkeit zeigen. Dies gilt insbesondere für die
binären Phasensysteme PMS–MeOH und PMPS–MeOH, in denen keine mischbare Zu-
sammensetzung gefunden wurde. Somit ist Methanol kein Lösungsmittel für beide Po-
lymere und Mischungen der beiden Polymere. Dies steht im Einklang mit den HSP der
Polymere, bei denen Methanol außerhalb beider Interaktionskugeln liegt.
Ebenso konnten keine mischbaren Zusammensetzungen im Zweiphasensystem der Po-
lymere PMS – PMPS gefunden werden, was ein deutliches Zeichen dafür ist, dass die
Polymere nicht mischbar sind.
Aus den Phasendiagrammen kann somit abgelesen werden, dass MTES zwingend als
Lösungsmittel erforderlich ist, um die beiden Polymere miteinander zu mischen, wäh-
rend Methanol als Nicht-Lösungsmittel für die beiden Polymere identifiziert werden
konnte. Außerdem konnte gezeigt werden, dass keine Mischbarkeit der Polymere bei
Raumtemperatur gegeben ist.
5.1.3 Beschichtungen mit verschiedenen Zusammensetzungen von PMS und PMPS und deren Einfluss auf die Strukturbildung
Die Struktur in ungefüllten Systemen besteht aus Inseln die von einer Matrix umgege-
ben sind, wobei das Substrat vollständig bedeckt ist. Ebenso hat sich gezeigt, dass die
Inseln und die Matrix sich deutlich in ihrer Zusammensetzung unterscheiden und somit
aus verschiedenen Polymeren bestehen. Die Veränderung der Zusammensetzung, hat
einen direkten Einfluss auf die entstehenden Strukturen. Methanol hingegen zeigt in
weiten Bereichen keinen Einfluss auf die Strukturbildung; erst in hohen Konzentratio-

5 Diskussion
87
nen wird diese beeinflusst. Ebenso ist Methanol nicht notwendig für die Strukturbil-
dung.
Da das Substrat vollständig beschichtet ist und sich die Inseln in ihrer gemessenen Zu-
sammensetzung derart unterscheiden, dass PMPS die Inseln und PMS die Matrix bildet,
kann davon ausgegangen werden, dass die Strukturbildung auf dem Prozess der Entmi-
schung der beiden Polymere beim Verdampfen des Lösungsmittels MTES beruht. Dies
steht im Einklang mit den vorangegangenen Untersuchungen, bei denen sich gezeigt
hat, dass die Polymere nicht mischbar sind und MTES zwingend für Mischungen erfor-
derlich ist. Die Ausbildung von PMS als Matrix und PMPS als Inseln steht zudem im
Einklang mit der gängigen Theorie.50
Diese besagt, dass die höherviskose Phase, in die-
sem Fall PMS, dass bei Raumtemperatur ein festes Pulver ist, die Matrix bildet und die
niederviskose Phase die Inseln bildet, die in diesem Fall PMPS ist, das bei Raumtempe-
ratur eine honigartige Flüssigkeit mit einer Viskosität von 1 Pas ist.
Ein besonderes Interesse bei den Untersuchungen ungefüllter Beschichtungen gilt dem
Methanol, da Methanol in gefüllten Systemen ein notwendiger Bestandteil der Zusam-
mensetzung ist, um Strukturen zu erhalten. Bei der Herstellung und Untersuchung unge-
füllter Beschichtungen hat sich jedoch gezeigt, dass Methanol für die Strukturbildung
keine Rolle spielt und lediglich die zwei Polymere PMS und PMPS, sowie ein Lö-
sungsmittel, in diesem Fall MTES, für die Strukturbildung erforderlich sind. Es hat sich
ferner gezeigt, dass Methanol nur in sehr hohen Konzentrationen von größer als 60 vol.
% einen Einfluss auf die Strukturbildung ausübt und dass Lösungen mit einem Anteil
von größer als 80 vol. % Methanol nicht zur Beschichtung geeignet sind, da diese be-
reits entmischt vorliegen. Der Einfluss von Methanol in hohen Konzentrationen zeigt
sich dadurch, dass die Struktur der Beschichtung gleichmäßiger wird und die Inseln
kleiner werden. Dieser Einfluss kann zwei Ursachen haben, zum einen hat sich gezeigt,
dass Mischungen mit einem Methanolgehalt von größer als 60 vol. % bereits eintrüben,
was auf eine beginnende Entmischung hinweist und mittels UV/VIS-Spektroskopie
nachgewiesen wurde. Zum anderen nimmt mit zunehmender Methanolkonzentration die
Dicke der trockenen Beschichtung ab, da weniger Polymer im nassen Film enthalten
und somit die Trockenfilmdicke niedriger ist. Aus der Literatur ist bekannt, dass mit
abnehmender Schichtdicke die Strukturgröße wie hier beschrieben ebenfalls abnimmt38
.
Ein weiterer Hinweis zu Bestätigung dieser Theorie ist, dass dasselbe Verhalten zu be-

5 Diskussion
88
obachten ist, wenn die MTES Konzentration erhöht wird, was denselben Effekt auf die
Schichtdicke und somit auch auf die Struktur ausübt.
5.2 Zusammenfassung der wesentlichen Erkenntnisse in
ungefüllten Systemen
Es konnte gezeigt werden, dass alle drei Bestimmungsmethoden der HSP zu verwertba-
ren Ergebnissen führen. Für ungefüllte Systeme konnte zudem geklärt werden, dass die
Strukturierung auf dem Prozess der Entmischung der Polymere PMS und PMPS beruht.
Der hierzu führende Teilprozess ist die Verdunstung des Lösungsmittels MTES. Dies
wird ebenso durch die ermittelten Mischungsdiagramme bestätigt. Ebenso konnte ge-
klärt werden, dass PMS die Matrix und PMPS die Inseln der Entmischungsstrukturen
bildet. Methanol wurde für beide Polymere als „Nichtlösungsmittel“ identifiziert und ist
für die Strukturbildung in ungefüllten Systemen nicht zwingend notwendig, es zeigt nur
in hohen Konzentrationen einen Einfluss auf die Strukturbildung.

5 Diskussion
89
5.3 Gefüllte Systeme
REM-Untersuchungen haben gezeigt, dass es sich bei der Struktur in gefüllten Syste-
men um Poren und nicht, wie in ungefüllten Systemen, um Inseln des einen Polymers,
eingebettet in einer Matrix des zweiten Polymers, handelt. Das zeigt sich dadurch, dass
die Poren in REM-Aufnahmen als solche deutlich zu erkennen sind und ebenso die SiC-
Partikel am Boden dieser erkannt werden können. Zudem ist kein Unterschied in der
Zusammensetzung einer EDX-Analyse zwischen Poren und den Stegen zu erkennen,
was zeigt, dass beide aus demselben Material bestehen. Ein Beweis hierfür ist zudem,
dass die Strukturbildung auch dann noch funktioniert, wenn nur eines der beiden Poly-
mere im System vorhanden ist und es somit keine Polymer/Polymer-Entmischung ge-
ben kann. Somit muss der Strukturbildungsmechanismus in gefüllten Systemen ein an-
derer sein, als die Polymer/Polymer-Entmischung in ungefüllten Systemen.
Ein weiterer großer Unterschied zu den ungefüllten Systemen ist, dass Methanol zwin-
gend für die Strukturbildung erforderlich ist. Dies zeigt sich dadurch, dass die Beschich-
tung auf einem bereits bekannten Ansatz zur Herstellung von keramischen Folien ba-
siert11
, der ohne Methanol zu dichten Schichten ohne die hier gezeigten Strukturen
führt. Untersuchungen zum Ersatz von Methanol haben zudem gezeigt, dass Methanol
nicht einfach durch ein anderes Lösungsmittel ersetzt werden kann. Methanol sollte
dabei anhand seiner bekannten Eigenschaften wie HSP, Dampfdruck und chemischer
Verwandtschaft ersetzt werden. Dies ist ein Hinweis darauf, dass nicht nur einzelne Ei-
genschaften von Methanol, wie dessen HSP, Dampfdruck, Viskosität und das Benet-
zungsverhalten eine Rolle spielen, sondern es sich um die Kombination dieser Eigen-
schaften von Methanol handelt. Denn es konnte kein anderes Lösungsmittel gefunden
werden, mit dem die Struktur reproduzierbar herstellbar ist. Zum anderen haben Unter-
suchungen in Bezug auf den Methanolgehalt der verwendeten Schlicker gezeigt, dass
die Strukturen nur in Anwesenheit von Methanol entstehen und auch dann nur in einem
relativ engen Konzentrationsbereich von 47,5 vol. % bis 67,5 vol. %. Daher lässt sich
festhalten, dass Methanol für gefüllte Systeme eine entscheidende Komponente für den
strukturbildenden Mechanismus ist.
Für das Lösungsmittel MTES, das in ungefüllten Systemen zwingend als Lösungsmittel
erforderlich ist, hat sich in gefüllten Systemen gezeigt, dass die Bildung netzartiger

5 Diskussion
90
Strukturen in einem MTES-Konzentrationsbereich von 0 - 25 vol. % erfolgt. Daraus ist
ersichtlich, dass die Bildung netzartiger Strukturen, anders als in ungefüllten Systemen,
auch ohne MTES erfolgt.
Ein entscheidender Unterschied zu ungefüllten Systemen ist der Füllstoff. Aus diesem
Grund ist auch von Interesse wie sich der Füllstoff im System verhält. Als erstes wurde
hierfür untersucht, in welchen Mengen der Füllstoff notwendig ist, damit sich die netz-
artigen Strukturen ausbilden. Dabei hat sich gezeigt, dass die Strukturen sich erst ab
einer Füllstoffmenge von 12,5 vol. % SiC ausbilden und sich von dieser Menge an bis
zur maximal produzierbaren Menge von 25 vol. % kontinuierlich ausbilden. Es besteht
durchaus die Möglichkeit, dass die Strukturbildung auch bei Mengen von größer als 25
vol. % funktioniert, allerdings ist die Herstellung von Schlickern und vor allem auch
von Beschichtungen aus diesen nur noch sehr erschwert bis gar nicht möglich, da mit
zunehmendem Füllstoffgehalt die Viskosität des Schlickers stark zunimmt.
Eine relativ einfache Methode zur Charakterisierung von Füllstoffpartikeln ist die
Korngröße der Partikel. Untersuchungen hierzu haben gezeigt, dass auch die Korngröße
einen Einfluss auf die Strukturbildung hat. Hierfür wurde das gleiche SiC Pulver einge-
setzt, lediglich dessen mittlerer Durchmesser d50 variiert. Dabei hat sich gezeigt, dass
sich von der kleinsten eingesetzten Korngröße mit einem d50 Wert von 0,67 µm bis zu
einem d50 Wert von 2,22 µm die netzartigen Strukturen ausbilden. Korngrößen darüber
zeigen keine porösen Strukturen mehr.
Auch die Art und die Oberflächeneigenschaften der Partikel spielen eine Rolle für die
Strukturbildung. Hierfür wurde der standardmäßig verwendete Füllstoff SiC durch eine
Vielzahl anderer Füllstoffmaterialien ersetzt. Dabei hat sich gezeigt, dass die Herstel-
lung von Beschichtungen mit netzartiger Struktur nur mit drei Arten von Füllstoffparti-
keln, nämlich SiC, Si und Si3N4, erfolgt. Alle anderen verwendeten Füllstoffmaterialien
haben keine strukturierten Beschichtungen gezeigt. Da die Korngröße hierbei nach
Möglichkeit stets gleich derer des verwendeten SiC war, müssen die Oberflächeneigen-
schaften bzw. die Oberflächenchemie der Partikel eine entscheidende Rolle spielen.
Daher wurden vom Projektpartner an der Universität Magdeburg zusätzliche Untersu-
chungen zur Charakterisierung der Partikeleigenschaften vorgenommen. Dabei konnten
lediglich zwei Messgrößen einen Hinweis auf die notwendigen Oberflächeneigenschaf-
ten der Füllstoffpartikel geben, zum einen ist eine Oberflächenspannungen des Schli-

5 Diskussion
91
ckers zwischen etwa 20 mN m-1
und 22 mN m-1
erforderlich, die nur mit den drei funk-
tionierenden Füllstoffarten SiC, Si und Si3N4 erzielt werden konnte. Zudem hat sich
gezeigt, dass der pH-Wert der verwendeten Partikel in Wasser gelöst ca. 5,5 sein sollte.
Alle anderen untersuchten Materialien hatten einen höheren pH-Wert (z.B. Al2O3 = 8,7;
Glaspulver = 8 - 10). Weitere Untersuchungen bezüglich der OH-Gruppenbelegung, die
an einem externen Messlabor durchgeführt worden sind, zeigen ebenso keinen messba-
ren Nachweis auf die Strukturbildung. Im Rahmen dieser Arbeit ausgeheiztes Al2O3 und
die damit verbundene Reduzierung der OH-Gruppenbelegung von Al2O3 hat zu keinen
Erfolg geführt. Zudem wurde versucht, die Oberfläche von Al2O3 vor dem Verwenden
für den Schlicker zu verändern, indem diese mit PMPS beschichtet wurde. Auch dies
hat nicht dazu geführt, dass mit Al2O3 als Füllstoff netzartig strukturierte Beschichtun-
gen hergestellt werden konnten.
Somit lässt sich festhalten, dass die Art des Füllstoffes eine entscheidende Rolle für die
Entstehung von netzartig strukturierten Beschichtungen spielt, da diese nur mit den
Füllstoffmaterialien SiC, Si und Si3N4 und auch nur in einem engen Konzentrationsbe-
reich von 12,5 vol. % bis ca. 25 vol. % hergestellt werden können. Auch die Korngröße
spielt dabei eine Rolle, sie muss zwischen d50 Werten von 0,67 µm und 2,22 µm liegen.
Der Einfluss der Partikeloberfläche hingegen konnte nicht vollständig geklärt werden.
Der Schlicker, in dem die Partikel eingesetzt werden, muss eine Oberflächenspannung
zwischen etwa 20 mN m-1
und 22 mN m-1
aufweisen, damit poröse Strukturen entste-
hen. Während für die OH-Gruppenbelegung kein Einfluss gefunden werden konnte. Es
hat sich jedoch gezeigt, dass der pH-Wert der in Wasser gelösten Partikel bei etwa 5,5
liegen muss. Alle weiteren getesteten Füllstoffe haben einen höheren pH-Wert von grö-
ßer als 8 gezeigt. Es wird vermutet, dass dafür die Art und Stärke der Hydroxylgruppen
verantwortlich ist: Während es sich bei Si-OH-Gruppen (die auf SiC-, Si- und Si3N4-
Pulvern zu finden sind) um Brønsted-acide Gruppen handelt, sind die Al-OH-Gruppen
(auf Al2O3) Lewis-acid Gruppen. Dies ist augenscheinlich im Widerspruch zu silikati-
schen Glasfüllstoffen, die ebenfalls über Si-OH-Gruppen verfügen. Hierbei muss jedoch
in Betracht gezogen werden, dass durch die Präsenz von Alkali- und Erdalkaliionen als
Netzwerkwandler in Gläsern ein anderer pH-Wert-Bereich vorliegt, was anhand der
Messungen des pH-Wertes der in Wasser gerührten Pulver bei ca. 8 liegen.

5 Diskussion
92
Zur Klärung des Einflusses des Substratmaterials auf die Strukturbildung und der Mög-
lichkeit von Entnetzung als strukturbildendem Mechanismus wurden Untersuchungen
an verschieden Substraten durchgeführt. Hierfür wurden vom Projektpartner in Magde-
burg verschiedene Substrate untersucht. Dabei wurden die Oberflächenenergien der
Substrate und der Kontaktwinkel des Beschichtungsschlickers auf den Substraten ge-
messen, sowie der Einfluss gezielter Oberflächenveränderung der Substrate bestimmt.
Dabei hat sich herausgestellt, dass aus diesen Oberflächeneigenschaften kein direkter
Einfluss des Substrates auf den Strukturbildungsmechanismus nachweisbar ist. Nur
kleine Veränderungen in der Morphologie der Struktur sind als Einfluss des Substratma-
terials nachweisbar. Aber auch hierfür konnte keine direkte Ableitung aus den Oberflä-
cheneigenschaften auf die Morphologie nachgewiesen werden. Einzig die Rauheit der
Substrate, die auch die Schichtdicke beeinflusst, welche wiederum die Porengröße
beeinflusst, wurde nachgewiesen.
Im Rahmen der hier vorliegenden Arbeit konnte wiederum gezeigt werden, dass alle
verwendeten Substrate vollständig beschichtet sind und somit eine Entnetzung der Be-
schichtung vom Substrat als Strukturbildungsmechanismus ausgeschlossen werden
kann. Dies wird auch dadurch bestätigt, dass Querschnittsaufnahmen zeigen, dass die
Beschichtung die komplette Substratoberfläche besetzt und an keiner Stelle Ablösungen
zu beobachten sind. Zudem ist die Strukturgröße, sprich die Porengröße der Beschich-
tung, in aller Regel deutlich kleiner als die Schichtdicke. Das bedeutet, dass die Struktur
mehrere Ebenen in der Beschichtung einnimmt und somit unabhängig vom den Grenz-
flächen Substrat/Beschichtung oder Beschichtung/Atmosphäre durch die gesamte Be-
schichtung entsteht. Somit lassen sich Entnetzung und auch die Strukturbildung durch
Konvektion als strukturbildender Mechanismus sowohl für ungefüllte als auch für ge-
füllte Systeme ausschließen.
Die zuvor diskutierten Ergebnisse haben gezeigt, dass die Strukturbildung auch dann
funktioniert, wenn einzelne Komponenten nicht im System sind. Dies gilt insbesondere
für MTES und die Vernetzungskatalysatoren Ölsäure und Aluminiumacetylacetonat,
ohne die die Strukturbildung problemlos erfolgt. Auch gezeigt wurde, dass nur eines der
beiden Polymere vorhanden sein muss, so entsteht die Struktur wahlweise mit PMS
oder PMPS. Auf Grundlage der durchgeführten Untersuchungen, konnten die Grenzen,
innerhalb welcher die Strukturbildung erfolgt, für die einzelnen Komponenten des Sys-

5 Diskussion
93
tems ermittelt werden.. Diese Grenzen sind für die einzelnen Komponenten in Tabelle
10 aufgelistet.
Tabelle 10: Grenzen für die einzelnen Komponenten innerhalb der die Strukturbildung in
gefüllten Beschichtungen funktioniert.
Komponente Anteil
[vol %]
Methanol 47,5 – 67,5
Füllstoff 12,5 – 25
Korngröße (SiC) 0,67 – 2,22 [µm]
MTES 0 – 25
Polymere 2,5 – 30
Polymerverhältnis PMS:PMPS beliebig
Füllstoffmaterial Si, SiC, Si3N4
Vernetzungskatalysatoren nicht notwendig
So war es möglich das System von sieben Komponenten auf drei zu reduzieren. Ledig-
lich der Füllstoff SiC, das „Nichtlösungsmittel“ Methanol und eines der Polymere sind
für die Bildung netzartiger Strukturen erforderlich. Durch weiteres reduzieren der Kom-
ponenten im System konnte nachgewiesen werden, dass für eine Strukturbildung nur
SiC, Methanol und, im Vergleich zum Referenzansatz, ein kleiner Anteil von 2,5 vol. %
PMPS erforderlich sind, um die Strukturen zu bilden. Da zudem gezeigt werden konnte,
dass die Substrate vollständig benetzt sind, konnte Entnetzung als Mechanismus ausge-
schlossen werden. Da die Struktur zudem mit kleinen, übereinander liegenden Poren
durch die komplette Beschichtung geht, kann die Strukturbildung durch Konvektion
ebenso ausgeschlossen werden, da in dem genannten Fall einzelne Poren durch die ge-
samte Beschichtung vom Substrat bis zur Oberfläche gehen müssten (vgl. Kapitel 2.2.5)
und nicht, wie im vorliegenden Fall, schaumartig viele kleine Poren in mehreren Ebe-
nen vorliegen dürften.
Aufgrund der gezeigten Ergebnisse wird daher davon ausgegangen, dass im System
Füllstoff-Methanol-Polymer, das Polymer den Füllstoff benetzt, da das Polymer zum
einen nicht vollständig mit dem Methanol mischbar ist und zum anderen vom Projek-
partner in Magdeburg anhand der Kontaktwinkel gezeigt werden konnte, dass das Po-
lymer das Füllstoffmaterial benetzt. Die in der Beschichtung gleichzeitig stattfindende
Entmischung der Suspension von mit Polymer benetzten Partikeln und dem Methanol,
führt dazu, dass die Strukturen gebildet werden. In Abbildung 36 ist schematisch darge-
stellt, wie die Bildung netzartiger Strukturen abläuft. Abbildung 36 a) zeigt die Be-

5 Diskussion
94
schichtung direkt nach dem Auftragen. Hier sind Methanol und Füllstoffpartikel weit-
gehend gleichmäßig verteilt, während davon ausgegangen wird, dass sich das Polymer
bereits um die Füllstoffpartikel angelagert hat. Die gleichmäßige Verteilung der Partikel
wird hierbei durch Scherkräfte während des Rührens des Schlickers bis kurz vor der
Beschichtung bewirkt. Abbildung 36 b) zeigt die Beschichtung kurz nach dem Be-
schichten. Die bei der Entmischung freiwerdenden Methanol-Tröpfchen führen zur Ent-
stehung von Strukturen und durch Verschmelzen einzelner Tröpfchen zum Wachstum
der späteren Poren. Gleichzeitig entspricht dies dem Bereich zwischen 15 s und 54 s,
wie er in Abbildung 28 dargestellt ist. In diesem Bereich wachsen die Poren durch Zu-
sammenschluss. Durch das weitere Verdampfen des Methanols entsteht, wie in Abbil-
dung 36 c) und Abbildung 28 nach 67 s dargestellt, Menisken in den Poren, die insbe-
sondere bei großen Poren dazu führen, dass diese zusätzlich geweitet werden. Mit dem
vollständigen Verdampfen des Methanols, dargestellt in Abbildung 36 d) und Abbil-
dung 28 nach 81 s, ist das Porenwachstum abgeschlossen und die netzartigen Strukturen
haben ihre endgültige Form angenommen. Weitere Prozessschritte, wie Aushärten und
Sintern führen zu keiner weiteren Änderung der Struktur. Die Benetzung der Füllstoff-
partikel mit Polymeren ermöglicht zum einen, dass die Partikel aneinander abgleiten
können und sich somit bewegen können und zum anderen, dass die Strukturen nach
dem Verdampfen des Methanols stabilisiert werden, indem das Polymer die Rolle einer
Art Binder einnimmt.

5 Diskussion
95
Abbildung 36: Schematische Darstellung der Porenentstehung.
Mit diesem Modell lässt sich die Abhängigkeit der Porengröße von der Schichtdicke
folgendermaßen erklären: Bei gleichen Beschichtungsbedingungen, gleicher Zusam-
mensetzung des Schlickers, Temperatur, Luftdruck, Luftfeuchte usw. und somit gleicher
Verdunstungsrate für das Methanol, erfolgt die Trocknung der Beschichtung bei größe-
ren Schichtdicken und damit höherem Volumen pro Fläche langsamer. Daher bleibt bei
dicken Schichten mehr Zeit für die Verschmelzung der entmischten Methanol-
Tröpfchen, wodurch die Poren größer werden.
Auch das Phänomen, dass in den Rohren keine Strukturen entstehen, wenn diese auf
einer Seite verschlossen sind, lässt sich mit diesem Modell erklären: Dadurch dass die
Rohre auf einer Seite verschlossen sind, kann der Methanoldampf nicht zügig entwei-
chen, was zu einer methanolgesättigten Atmosphäre in den Rohren führt. Dies wiede-
rum führt dazu, dass das Methanol nur sehr langsam verdampfen kann und die Zeit bis
zur vollständigen Trocknung der Beschichtung im Inneren der Rohre stark zunimmt.
Das führt wiederum dazu, dass die Poren solange wachsen, bis die Beschichtung nahezu
vollständig entmischt ist und somit keine Strukturen zum Zeitpunkt des Trocknens mehr
vorhanden sind.
a) b)
c) d)
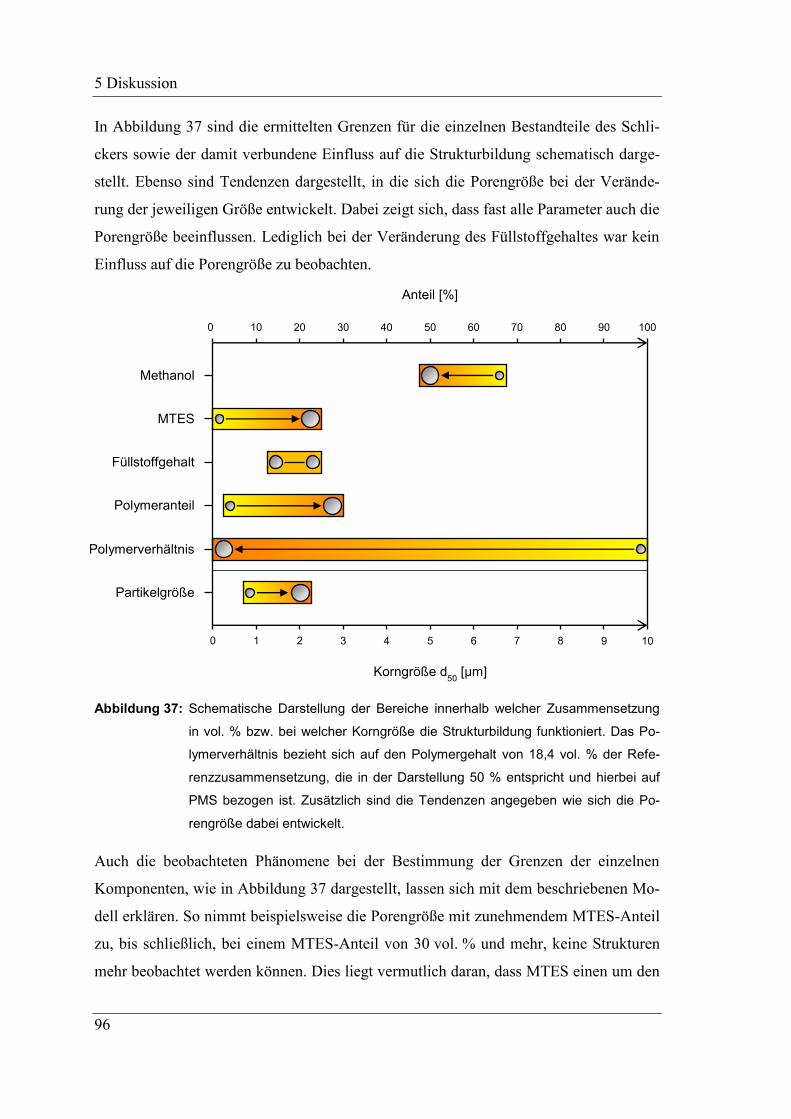
5 Diskussion
96
In Abbildung 37 sind die ermittelten Grenzen für die einzelnen Bestandteile des Schli-
ckers sowie der damit verbundene Einfluss auf die Strukturbildung schematisch darge-
stellt. Ebenso sind Tendenzen dargestellt, in die sich die Porengröße bei der Verände-
rung der jeweiligen Größe entwickelt. Dabei zeigt sich, dass fast alle Parameter auch die
Porengröße beeinflussen. Lediglich bei der Veränderung des Füllstoffgehaltes war kein
Einfluss auf die Porengröße zu beobachten.
Abbildung 37: Schematische Darstellung der Bereiche innerhalb welcher Zusammensetzung
in vol. % bzw. bei welcher Korngröße die Strukturbildung funktioniert. Das Po-
lymerverhältnis bezieht sich auf den Polymergehalt von 18,4 vol. % der Refe-
renzzusammensetzung, die in der Darstellung 50 % entspricht und hierbei auf
PMS bezogen ist. Zusätzlich sind die Tendenzen angegeben wie sich die Po-
rengröße dabei entwickelt.
Auch die beobachteten Phänomene bei der Bestimmung der Grenzen der einzelnen
Komponenten, wie in Abbildung 37 dargestellt, lassen sich mit dem beschriebenen Mo-
dell erklären. So nimmt beispielsweise die Porengröße mit zunehmendem MTES-Anteil
zu, bis schließlich, bei einem MTES-Anteil von 30 vol. % und mehr, keine Strukturen
mehr beobachtet werden können. Dies liegt vermutlich daran, dass MTES einen um den
Methanol
MTES
Füllstoffgehalt
Polymeranteil
Polymerverhältnis
Partikelgröße
0 10 20 30 40 50 60 70 80 90 100
Anteil [%]
1 0 2 3 4 5 6 7 8 9 10
Korngröße d50
[µm]

5 Diskussion
97
Faktor 10 niedrigeren Dampfdruck von 13,3 hPa gegenüber dem von Methanol mit 129
hPa hat. Dadurch wird die Trocknung verlangsamt und die Poren haben länger Zeit zu
wachsen, bis schließlich bei einer MTES-Konzentration von 30 vol. % die Trocknungs-
zeit so groß wird, dass, wie in den verschlossenen Rohren, keine Strukturen mehr beo-
bachtet werden können. Gleichzeitig verringert ein erhöhter Anteil des Lösungsmittels
MTES die Viskosität. Durch die verringerte Viskosität nimmt die Beweglichkeit der
Füllstoffe zu und die Tröpfchenbildung und Vereinigung wird beschleunigt, was den
Effekt des Porenwachstums zusätzlich verstärkt. Dieser Effekt der Viskositätsverände-
rung lässt sich auch bei der Variation des Polymerverhältnisses beobachten, dort steigt
mit zunehmendem PMS-Anteil die Viskosität an, wodurch die Poren kleiner werden.
Die bereits beschriebenen ermittelten Einflussgrößen neben der Zusammensetzung sind
in Abbildung 38 schematisch dargestellt. Die Schichtdicke ist dabei ein Parameter mit
dem sich, wie bereits zuvor beschrieben, die Porengröße relativ leicht steuern lässt. Di-
ckere Schichten führen hierbei zu größeren Poren. Auch für die Viskosität konnte der
Einfluss auf die Porengröße gezeigt werden. Mit zunehmender Viskosität werden die
Poren kleiner, jedoch lässt sich mit der Viskosität nicht ohne Weiteres die Porengröße
gezielt einstellen, da mit ihr auch immer weitere Parameter variiert werden. Denn zum
einen ist die Änderung der Viskosität beispielsweise mit der Änderung der Zusammen-
setzung möglich, diese hat jedoch, wie bereits beschrieben, ebenso einen Einfluss auf
die Porengröße. Zum anderen geht mit der Veränderung der Viskosität auch eine Ver-
änderung der Schichtdicke einher, wie mit den Gleichungen (16) (17) und (18) zur Be-
rechnung der Schichtdicke für die verschiedenen Beschichtungsverfahren beschreiben
wird. Auch die Verdunstungsrate hat einen Einfluss auf die Schichtdicke. Mit zuneh-
mender Verdunstungsrate und der damit verbundenen schnelleren Trocknung nimmt die
Porengröße ab. Auch hier ist eine direkte Einflussnahme auf die Porengröße nur schwer
möglich, da die Verdunstungsrate stark vom Dampfdruck des verwendeten Lösungsmit-
tels abhängt und zum gegenwärtigen Zeitpunkt kein anderes Lösungsmittel außer Me-
thanol gefunden wurde, mit dem die Strukturbildung funktioniert.

5 Diskussion
98
Abbildung 38: Schematische Darstellung der gefundenen Einflussparameter Schichtdicke,
Viskosität und Verdunstungsrate und der damit verbundene Einfluss auf die Po-
rengröße.
Auch die Umgebungsbedingungen wie Temperatur, Luftfeuchte, Luftdruck usw. haben
einen großen Einfluss auf die Strukturbildung. Es hat sich gezeigt, dass bei exakt der
gleichen Zusammensetzung und Herstellungsweise der Beschichtungen, bei der Herstel-
lung an unterschiedlichen Tagen, auch Veränderungen in der Morphologie der Be-
schichtungen auftreten. Dies zeigt sich zum einen darin, dass dieselbe Beschichtung an
einem Tag nur eine Porengeneration zeigt und an einem anderen Tag zwei Porengenera-
tionen vorliegen, und zum anderen darin, dass die Porengrößen unterschiedlich ausfal-
len. Die Grenzen innerhalb der die Beschichtung Strukturen zeigt, lassen sich hingegen
sehr gut reproduzieren.
Der Vorteil gegenüber anderen bekannten Methoden zu Erzeugung poröser Beschich-
tungen liegt vor allem in der Einfachheit der Methode, bei der gleichzeitigen Möglich-
keit, die Porengröße zu steuern und der zusätzlichen Option komplexe dreidimensionale
Substrate zu beschichten. Während die meisten anderen Methoden einen speziellen Pro-
zessschritt notwendig machen, bei dem die Porosität erzeugt wird, ist das bei der hier
aufgezeigten Methode nicht notwendig. Zum Beispiel ist eine sehr häufig verwendete
Methode zum Erzeugen von porösen Beschichtungen mit gezielt eingestellter Porengrö-
ße die Verwendung von Platzhaltermaterialien, die in einem speziellen Prozessschritt,
z.B. durch Ausbrennen, wieder entfernt werden müssen. Das Ausbrennen bedeutet je-
doch zusätzliche Haltezeiten beim Sintern, was wiederum den Energiebedarf erhöht.19
dünn
hoch niedrig
dick Schichtdicke
Viskosität
Verdunstungsrate

6 Zusammenfassung und Ausblick
99
6 Zusammenfassung und Ausblick
In dieser Arbeit wurde eine selbststrukturierende Beschichtung auf der Basis präkerami-
scher Polymere untersucht. Dabei wurden sowohl die verwendeten Rohstoffe, als auch
Beschichtungen mit und ohne Füllstoffe untersucht und charakterisiert.
Als präkeramische Polymere kamen zwei kommerziell erhältliche Polymere zum Ein-
satz, dabei handelt es sich um zwei Polysiloxane, nämlich Polymethylsiloxan (PMS)
und Polymethylphenylvinylsiloxane (PMPS). Von beiden Polymeren wurden die Han-
sen-Löslichkeitsparameter (HSP) nach drei unterschiedlichen Methoden bestimmt. Da-
bei handelte es sich um zwei Methoden, die auf Lösungsversuchen in unterschiedlichen
Lösungsmitteln beruhen und einer dritten, bei der die HSP anhand der intrinsischen
Viskosität mit verschieden Lösungsmittel bestimmt wurden. Es konnte gezeigt werden,
dass alle drei Methoden zu gut übereinstimmenden Ergebnisse führen. Die dabei ge-
messenen HSP für die Polymere betragen gemittelt: δD = 16,8 MPa1/2
± 0,1, δP = 3,3
MPa1/2
± 1,0, δH = 5,0 MPa1/2
± 1,1 mit einem Radius von 4 MPa1/2
± 0,9 für PMS und
für PMPS δD = 17,7 MPa1/2
± 0,7,δP = 8,2 MPa1/2
± 1,3, δH = 6,4 MPa1/2
± 0,9 mit einem
Radius von 9,2 MPa1/2
± 0,8. Anhand der ermittelten HSP, lässt sich zeigen, dass Me-
thanol (MeOH) für beide Polymere als „Nichtlösungsmittel“ bezeichnet werden kann
und dass eine Nichtmischbarkeit der beiden Polymere vorliegt. Der Einfluss von Me-
thyltriethoxysilan (MTES) konnte hingegen nicht eindeutig anhand der für MTES be-
rechneten HSP bestimmt werden.
Zudem wurde die Mischbarkeit im Vierphasensystem PMS – PMPS – MTES – MeOH
untersucht und entsprechende Mischungsdiagramme aufgestellt. Dabei konnte MTES
als Lösungsmittel für beide Polymere identifiziert werden, auch Methanol wurde durch
diese Untersuchungen als „Nichtlösungsmittel“ eindeutig identifiziert. Zudem konnte
die Nichtmischbarkeit der beiden Polymere PMS und PMPS gezeigt werden.
Anhand der ermittelten Ergebnisse konnte für füllstofffreie Beschichtungen gezeigt
werden, dass die Strukturbildung hierbei durch eine Polymer/Polymer Entmischung
erfolgt. Hierbei bildet PMS die Matrix und PMPS die Inseln. Während MTES für die
Strukturbildung in füllstofffreien Beschichtungen zwingend als Lösungsmittel erforder-
lich ist, ist Methanol nicht notwendig und hat lediglich in hohen Konzentrationen einen
geringen Einfluss auf die Morphologie der Beschichtung.

6 Zusammenfassung und Ausblick
100
In gefüllten Beschichtungen wurden die Grenzen für die einzelnen Komponenten des
Systems ermittelt, innerhalb welcher die Strukturbildung funktioniert (vgl. Tabelle 10).
Aufgrund der ermittelten Grenzen konnte das System auf drei für die Strukturbildung
notwendigen Komponenten reduziert werden: Methanol, der Füllstoff SiC und ein klei-
ner Anteil eines der beiden Polymere von lediglich 2,5 vol. % sind für die Strukturbil-
dung notwendig. Diese Ergebnisse zeigen auch, dass in gefüllten Systemen ein anderer
Mechanismus als in ungefüllten Systemen für die Strukturbildung verantwortlich ist, da
sowohl Methanol notwendig ist, als auch nur eines der beiden Polymere ausreicht und
daher die Polymer/Polymer Entmischung als strukturbildender Mechanismus nicht in
Frage kommt. Durch Untersuchungen mit unterschiedlichen Substratmaterialien konnte
zudem Entnetzung vom Substrat als strukturbildendem Mechanismus ausgeschlossen
werden. Versuchsreihen mit unterschiedlichen Füllstoffmaterialien haben gezeigt, dass
sich von den untersuchten Füllstoffen nur SiC, Si und Si3N4 als geeignet für die Struk-
turbildung erwiesen haben. Der Ersatz von Methanol durch andere Lösungsmittel hat
nicht zu strukturierten Beschichtungen geführt. Die Beobachtung des zeitlichen Ablaufs
der Strukturbildung unter dem Mikroskop hat gezeigt, dass sich während des Trocknens
der Beschichtung kleine mit Methanol gefüllte Tropfen solange zu immer größeren
Tropfen vereinigen, bis die Trocknung abgeschlossen ist. Aufgrund der Untersuchungen
wird daher davon ausgegangen, dass die Strukturbildung in gefüllten Systemen wie
folgt abläuft: Zum Beginn sind die Füllstoffpartikel durch die Scherkräfte während des
Rührens und bei der Beschichtung gleichmäßig verteilt. Sobald die Beschichtung keinen
Scherkräften mehr ausgesetzt ist, beginnt sich das Methanol von den Partikeln zu tren-
nen, so dass erste Methanoltropfen entstehen, die solange anwachsen, bis das Methanol
vollständig verdunstet ist. Der geringe Polymeranteil ist als Binder notwendig, um die
trockene Beschichtung zu stabilisieren.
Zudem konnte gezeigt werden, dass die polymeren Beschichtungen unter Beibehaltung
ihrer Morphologie durch Sintern in keramische Beschichtungen überführt werden kön-
nen. Ebenfalls wurde gezeigt, dass die Strukturbildung bei der Anwendung unterschied-
lichen Beschichtungsverfahren wie Tauchbeschichten, Schleuderbeschichten und dem
automatischen Filmziehverfahren gleichermaßen funktioniert und ebenso auf planaren
wie komplexen dreidimensionalen Strukturen, wie beispielsweise auf Rohren oder ke-
ramischen Schäumen, realisierbar ist.

6 Zusammenfassung und Ausblick
101
Als eine einfache Möglichkeit die Porengröße der Beschichtung gezielt einzustellen,
konnte die deferierte Einstellung der Schichtdicke der Beschichtung identifiziert wer-
den. Dies wurde durch unterschiedliche Ziehgeschwindigkeiten bei der Tauchbeschich-
tung erreicht.
Um den strukturbildenden Mechanismus zukünftig noch besser zu verstehen, um bei-
spielsweise neue Rohstoffe zu verwenden oder zur gezielten Entwicklung von Produk-
ten, erscheint es sinnvoll, den Einfluss der Füllstoffe noch besser zu verstehen. Eine
hierfür geeignete Methode könnte die Messung des Zetapotentials sein, da mit Hilfe
dessen sowohl das Verhalten der Partikel in der Suspension, als auch zueinander besser
verstanden werden kann.
Nachdem durch die in dieser Arbeit durchgeführten Arbeiten weitgehend geklärt ist,
wie und unter welchen Bedingungen die Strukturierung der Beschichtung funktioniert
und zudem gezeigt werden konnte, wie die Porosität definiert eingestellt werden kann,
ist zukünftig vor allem die gezielte Untersuchung zur Verwendung der Beschichtung für
spezielle Anwendungen von Interesse. In weiterführenden Untersuchungen konnte ge-
zeigt werden, dass es möglich ist, auf die Beschichtung Zeolithe aufwachsen zu lassen.
Für die Herstellung der Beschichtung die zur Zeolithkristallisation verwendet wurde,
wurde als Füllstoff sowohl Silizium als auch Siliziumcarbid in einem Verhältnis von
Si:SiC von 3:1 verwendet.118
Die Zeolithkristallisation wurde dabei nach Schoeman
durchgeführt.119
Der Vorteil der Verwendung der netzartig strukturierten Beschichtung
liegt hierbei zum einen in der Kostenreduktion durch die Verwendung von kostengüns-
tigen Methoden, wie der Tauchbeschichtung, und zum anderen darin, dass durch die
offene Porosität der Beschichtung der Stofftransport sowohl für die Kristallisation der
Zeolithe, als auch bei deren späterer Verwendung gut möglich ist.
Ein weiteres spannendes Anwendungsgebiet könnte das Feld der Transpirationskühlung
von Gasturbinen sein. Insbesondere deshalb, weil sehr ähnliche Beschichtungen, aber
ohne die Struktur, bereits gezeigt haben, dass sie sich für den Einsatz unter den Bedin-
gungen, wie sie in Gasturbinen herrschen, eignen.120
Für diesen Zweck ist vor allem die
Permeabilität der Beschichtung eine wichtige Größe,23,24
die hierfür in Zukunft noch
untersucht werden muss.
Da die Untersuchungen weiter gezeigt haben, dass es möglich ist, SiC als Füllstoff ganz
oder zumindest in großen Teilen zu ersetzen, ergeben sich weitere Anwendungsgebiete.

6 Zusammenfassung und Ausblick
102
So sollte es möglich sein, die große, durch die Struktur zur Verfügung gestellte Oberflä-
che gleichzeitig mit katalytisch aktiven Substanzen zu versehen. Dadurch können wie-
derum Kosten gespart werden, da zusätzliche Schritte zur Oberflächenvergrößerung
oder zur Einbringung von katalytisch aktiven Substanzen, wie beispielsweise durch den
VLS-Mechanismus erzeugte,121,122
wegfallen können.
Auch der Einsatz als Beschichtungen für Knochenimplantate ist ein mögliches Anwen-
dungsgebiet, dessen nähere Untersuchung sicher interessant ist, da zum einen, wie be-
reits erwähnt, unterschiedliche Füllstoffe verwendet werden können, und somit die für
das Anwachsen des Knochens an die Knochenimplantate wichtigen Kalziumphosphate
eingebracht werden können, als auch die notwendige Porosität vorhanden ist.27
Für die-
sen Zweck ist die zukünftige Untersuchung der Beschichtung hinsichtlich ihrer Bio-
kompatibilität notwendig. Hierbei könnte auch von Interesse sein, dass die Siliziumpo-
lymere, die hier verwendet werden, beim Sintern an Luft zu SiO2 umgesetzt werden
können,123
das weitgehend biologisch kombatibel124
ist.
Auch für weitere Anwendungen wie sie in der Motivation genannt wurden, könnte die
Beschichtung von Interesse sein, dazu zählen Rußpartikelfilter in Dieselfahrzeugen,15
Solarabsorber in Solarturmkraftwerken17
oder, aufgrund der realisierbaren hierarchi-
schen Struktur, auch für die Gasspeicherung.28,29

7 Literaturverzeichnis
103
7 Literaturverzeichnis
1. Borchardt, L. et al. Preparation and application of cellular and nanoporous
carbides. Chem. Soc. Rev. 41, 5053–67 (2012).
2. Fourcroy, A. de. Encyclopédie Methodique. (1796).
3. Weimer, A. W. et al. Kinetics of Carbothermal Reduction Synthesis of Boron
Carbide. J. Am. Ceram. Soc. 75, 2509–2514 (1992).
4. Martin, H.-P., Ecke, R. & Müller, E. Synthesis of nanocrystalline silicon carbide
powder by carbothermal reduction. J. Eur. Ceram. Soc. 18, 1737–1742 (1998).
5. Gotoh, Y. et al. Synthesis of titanium carbide from a composite of TiO2
nanoparticles/methyl cellulose by carbothermal reduction. Mater. Res. Bull. 36,
2263–2275 (2001).
6. Greil, P. Polymer Derived Engineering Ceramics. Adv. Eng. Mater. 2, 339–348
(2000).
7. Narula, C. K. Ceramic Precursor Technology and its Applications. 350 (CRC
Press, 1995).
8. Baney, R., Itoh, M., Sakakibara, A. & Suzuki, T. Silsesquioxanes. Chem. Rev.
95, 1409–1403 (1995).
9. Gupta, G. S., Vasanth Kumar, P., Rudolph, V. R. & Gupta, M. Heat-transfer
model for the acheson process. Metall. Mater. Trans. A 32, 1301–1308 (2001).
10. Stern, E., Heyder, M. & Scheffler, F. Micropatterned Ceramic Surfaces by
Coating with Filled Preceramic Polymers. J. Am. Ceram. Soc. 92, 2438–2442
(2009).
11. Cromme, P., Scheffler, M. & Greil, P. Ceramic Tapes from Preceramic Polymers.
Adv. Eng. Mater. 4, 873–877 (2002).
12. Woiton, M. et al. Self-assembled microstructured polymeric and ceramic
surfaces. J. Eur. Ceram. Soc. 31, 1803–1810 (2011).
13. Adler, J. Ceramic Diesel Particulate Filters. Int. J. Appl. Ceram. Technol. 2, 429–
439 (2005).
14. Ciora, R. J. et al. Preparation and reactive applications of nanoporous silicon
carbide membranes. Chem. Eng. Sci. 59, 4957–4965 (2004).

7 Literaturverzeichnis
104
15. Oki, H., Karin, P. & Hanamura, K. Visualization of Oxidation of Soot
Nanoparticles Trapped on a Diesel Particulate Membrane Filter. SAE Int. J.
Engines 4, 515–526 (2011).
16. Fino, D. Diesel emission control: Catalytic filters for particulate removal. Sci.
Technol. Adv. Mater. 8, 93–100 (2007).
17. Fend, T. High porosity materials as volumetric receivers for solar energetics. Opt.
Appl. XL, (2010).
18. Ortona, A. et al. Aging of reticulated Si-SiC foams in porous burners. Adv. Appl.
Ceram. 109, 246–251 (2010).
19. Scheffler, M. & Colombo, P. Cellular Ceramics. 670 (Wiley-VCH Verlag GmbH
& Co. KGaA, 2005). doi:10.1002/3527606696
20. Twigg, M. V. & Richardson, J. T. Fundamentals and Applications of Structured
Ceramic Foam Catalysts. Ind. Eng. Chem. Res. 46, 4166–4177 (2007).
21. Lacroix, M. et al. Silicon carbide foam composite containing cobalt as a highly
selective and re-usable Fischer–Tropsch synthesis catalyst. Appl. Catal. A Gen.
397, 62–72 (2011).
22. Scheffler, M., Greil, P., Berger, A., Pippel, E. & Woltersdorf, J. Nickel-catalyzed
in situ formation of carbon nanotubes and turbostratic carbon in polymer-derived
ceramics. Mater. Chem. Phys. 84, 131–139 (2004).
23. Lugscheider, E. et al. Electron beam-physical vapor deposition - thermal barrier
coatings on laser drilled surfaces for transpiration cooling. Surf. Coatings
Technol. 133-134, 49–53 (2000).
24. Polezhaev, J. The transpiration cooling for blades of high temperatures gas
turbine. Energy Convers. Manag. 38, 1123–1133 (1997).
25. Clarke, D. R., Oechsner, M. & Padture, N. P. Thermal-barrier coatings for more
efficient gas-turbine engines. MRS Bull. 37, 891–898 (2012).
26. Arai, M. & Suidzu, T. Porous Ceramic Coating for Transpiration Cooling of Gas
Turbine Blade. J. Therm. Spray Technol. 22, 690–698 (2013).
27. Yang, J.-Z. et al. Micro-porous calcium phosphate coatings on load-bearing
zirconia substrate: Processing, property and application. Ceram. Int. null, (2013).
28. Oschatz, M. et al. A cubic ordered, mesoporous carbide-derived carbon for gas
and energy storage applications. Carbon N. Y. 48, 3987–3992 (2010).

7 Literaturverzeichnis
105
29. Zampieri, a. et al. Biomorphic Cellular SiSiC/Zeolite Ceramic Composites: From
Rattan Palm to Bioinspired Structured Monoliths for Catalysis and Sorption. Adv.
Mater. 17, 344–349 (2005).
30. Magasinski, A. et al. High-performance lithium-ion anodes using a hierarchical
bottom-up approach. Nat. Mater. 9, 461–461 (2010).
31. Kasavajjula, U., Wang, C. & Appleby, A. J. Nano- and bulk-silicon-based
insertion anodes for lithium-ion secondary cells. J. Power Sources 163, 1003–
1039 (2007).
32. Rose, M. et al. Hierarchical micro- and mesoporous carbide-derived carbon as a
high-performance electrode material in supercapacitors. Small 7, 1108–17
(2011).
33. Korenblit, Y. et al. High-rate electrochemical capacitors based on ordered
mesoporous silicon carbide-derived carbon. ACS Nano 4, 1337–44 (2010).
34. Zhu, Q., Zhou, S., Wang, X. & Dai, S. Controlled synthesis of mesoporous
carbon modified by tungsten carbides as an improved electrocatalyst support for
the oxygen reduction reaction. J. Power Sources 193, 495–500 (2009).
35. Schröder, U. Discover the possibilities: microbial bioelectrochemical systems
and the revival of a 100-year–old discovery. J. Solid State Electrochem. 15,
1481–1486 (2011).
36. Koltzenburg, S., Maskos, M. & Nuyken, O. Polymere: Synthese, Eigenschaften
und Anwendungen. (Springer Berlin Heidelberg, 2014). doi:10.1007/978-3-642-
34773-3
37. Rubinstein, M. Polymer physics. 444 (Oxford Univ. Press, 2003).
38. Walheim, S., Böltau, M., Mlynek, J., Krausch, G. & Steiner, U. Structure
Formation via Polymer Demixing in Spin-Cast Films. Macromolecules 30, 4995–
5003 (1997).
39. Müller-Buschbaum, P., Gutmann, J. S. & Stamm, M. Influence of Blend
Composition on Phase Separation and Dewetting of Thin Polymer Blend Films.
Macromolecules 33, 4886–4895 (2000).
40. Reich, S. & Cohen, Y. Phase separation of polymer blends in thin films. J.
Polym. Sci. Polym. Phys. Ed. 19, 1255–1267 (1981).
41. Schmidt, U., Hild, S., Ibach, W. & Hollricher, O. Characterization of Thin
Polymer Films on the Nanometer Scale with Confocal Raman AFM. Macromol.
Symp. 230, 133–143 (2005).

7 Literaturverzeichnis
106
42. Sung, L., Karim, A., Douglas, J. & Han, C. Dimensional Crossover in the Phase
Separation Kinetics of Thin Polymer Blend Films. Phys. Rev. Lett. 76, 4368–
4371 (1996).
43. Jacobs, K. & Herminghaus, S. Oberflächenphysik: Strukturbildung in dünnen
Filmen: Wie perlt eine Flüssigkeit von einer Unterlage ab? Phys. J. 55, 35–40
(1999).
44. Seemann, R., Herminghaus, S. & Jacobs, K. Gaining control of pattern formation
of dewetting liquid films. J. Phys. Condens. Matter 13, 4925–4938 (2001).
45. Seemann, R., Herminghaus, S. & Jacobs, K. Dewetting Patterns and Molecular
Forces: A Reconciliation. Phys. Rev. Lett. 86, 5534–5537 (2001).
46. Stange, T. G., Evans, D. F. & Hendrickson, W. A. Nucleation and Growth of
Defects Leading to Dewetting of Thin Polymer Films. Langmuir 13, 4459–4465
(1997).
47. Zhang, Q., Yang, H. & Fu, Q. Kinetics-controlled compatibilization of
immiscible polypropylene/polystyrene blends using nano-SiO2 particles.
Polymer (Guildf). 45, 1913–1922 (2004).
48. Yurekli, K., Karim, A., Amis, E. J. & Krishnamoorti, R. Influence of Layered
Silicates on the Phase-Separated Morphology of PS−PVME Blends.
Macromolecules 36, 7256–7267 (2003).
49. Araki, T. & Tanaka, H. Wetting-induced depletion interaction between particles
in a phase-separating liquid mixture. Phys. Rev. E 73, 061506 (2006).
50. Tanaka, H. M. Spinodal Decomposition of Binary Fluid Mixtures with Viscosity
Difference. Prog. Theor. Phys. 101, 863–873 (1999).
51. Luo, H. & Gersappe, D. Dewetting Dynamics of Nanofilled Polymer Thin Films.
Macromolecules 37, 5792–5799 (2004).
52. Barnes, K. A. et al. Suppression of Dewetting in Nanoparticle-Filled Polymer
Films. Macromolecules 33, 4177–4185 (2000).
53. Rayleigh, Lord. LIX. On convection currents in a horizontal layer of fluid, when
the higher temperature is on the under side. Philos. Mag. Ser. 6 32, 529–546
(1916).
54. Lindblad, N. R. Overcoat for imaging members. (1992).
55. Koschmieder, E. in Adv. Chem. Phys (eds. Prigogine, I. & Rice, S. A.) 177–210
(John Wiley & Sons, 2009).

7 Literaturverzeichnis
107
56. Suresh Singh, R., Grimes, C. A. & Dickey, E. C. Fabrication of nanoporous TiO
2 films through Benard-Marangoni convection. Mater. Res. Innov. 5, 178–184
(2002).
57. Rogers, J. L., Schatz, M. F., Bougie, J. L. & Swift, J. B. Rayleigh-Bénard
Convection in a Vertically Oscillated Fluid Layer. Phys. Rev. Lett. 84, 87–90
(2000).
58. Sullivan, T. S., Liu, Y. & Ecke, R. E. Turbulent solutal convection and surface
patterning in solid dissolution. Phys. Rev. E 54, 486–495 (1996).
59. Desclaux, P. et al. On the Catalytic Activity of Ceria for Direct Carbon
Conversion in a SOFC. Zeitschrift für Phys. Chemie 130722000303001 (2013).
doi:10.1524/zpch.2013.0422
60. Vogt, U. F., Györfy, L., Herzog, A., Graule, T. & Plesch, G. Macroporous silicon
carbide foams for porous burner applications and catalyst supports. J. Phys.
Chem. Solids 68, 1234–1238 (2007).
61. Gray, T. J. & Pryor, M. J. Ceramic foam filter. (1976).
62. Gonzenbach, U. T., Studart, A. R., Tervoort, E. & Gauckler, L. J. Ultrastable
particle-stabilized foams. Angew. Chem. Int. Ed. Engl. 45, 3526–30 (2006).
63. Colombo, P., Gambaryan-Roisman, T., Scheffler, M., Buhler, P. & Greil, P.
Conductive Ceramic Foams from Preceramic Polymers. J. Am. Ceram. Soc. 84,
2265–2268 (2004).
64. Yoon, B.-H., Lee, E.-J., Kim, H.-E. & Koh, Y.-H. Highly Aligned Porous Silicon
Carbide Ceramics by Freezing Polycarbosilane/Camphene Solution. J. Am.
Ceram. Soc. 90, 1753–1759 (2007).
65. Haas, D., Fey, T. & Greil, P. Biomorphous Metal-Ceramic-Composites with
High Coefficient of Friction. Adv. Eng. Mater. 9, 892–897 (2007).
66. Zeschky, J. Dissertation - Keramikschäume aus gefüllten Polysilsesquioxanen,
Universität Erlangen-Nürnberg. 205 (2004).
67. Klose, T. Dissertation - Mikro- und mesoporöses Siliciumcarbid aus
siliciumorganischen Precursoren, Technische Universität Freiberg. 120 (2001).
68. Kipping, F. CCXXII.—Organic derivatives of silicon. Part XV. The
nomenclature of organic silicon compounds. J. Chem. Soc. Trans. 101, 2106
(1912).
69. Noll, W. Chemistry and Technology of Silicones. 716 (Elsevier, 1968).

7 Literaturverzeichnis
108
70. Scheffler, M., Bordia, R., Travitzky, N. & Greil, P. Development of a rapid
crosslinking preceramic polymer system. J. Eur. Ceram. Soc. 25, 175–180
(2005).
71. Scheffler, M. et al. Pyrolytic decomposition of preceramic organo polysiloxanes.
Ceram. Trans. 115, 239–250 (2001).
72. Renlund, G. M., Prochazka, S. & Doremus, R. H. Silicon oxycarbide glasses:
Part II. Structure and properties. J. Mater. Res. 6, 2723–2734 (2011).
73. Zhang, H. & Pantano, C. G. Synthesis and Characterization of Silicon
Oxycarbide Glasses. J. Am. Ceram. Soc. 73, 958–963 (1990).
74. Singh, A. K. & Pantano, C. G. The Role of Si-H Functionality in Oxycarbide
Glass Synthesis. MRS Proc. 271, 795 (1992).
75. Soraru, G. D., Campostrini, R., Maurina, S. & Babonneau, F. Gel Precursor to
Silicon Oxycarbide Glasses with Ultrahigh Ceramic Yield. J. Am. Ceram. Soc.
80, 999–1004 (2005).
76. Soraru, G. D., Dallapiccola, E. & D’Andrea, G. Mechanical Characterization of
Sol-Gel-Derived Silicon Oxycarbide Glasses. J. Am. Ceram. Soc. 79, 2074–2080
(1996).
77. Belot, V., Corriu, R. J. P., Leclercq, D., Mutin, P. H. & Vioux, A. Silicon
oxycarbide glasses with low O/Si ratio from organosilicon precursors. J. Non.
Cryst. Solids 176, 33–44 (1994).
78. Esquivias, L., Babonneau, F., Bois, L. & Livage, J. Silicon oxycarbides via sol-
gel route: characterization of the pyrolysis process. J. Non. Cryst. Solids 147,
280–284 (1992).
79. Wild, M. J. & Buhler, P. On the phase composition of polymethylsiloxane
derived ceramics. J. Mater. Sci. 33, 5441–5444 (1998).
80. Hansen, C. M. Hansen Solubility Parameters: A User’s Handbook, Second
Edition. 544 (CRC Press, 2007).
81. Machui, F., Abbott, S., Waller, D., Koppe, M. & Brabec, C. J. Determination of
Solubility Parameters for Organic Semiconductor Formulations. Macromol.
Chem. Phys. 212, 2159–2165 (2011).
82. Haas, D., Heinrich, S. & Greil, P. Solvent control of cellulose acetate nanofibre
felt structure produced by electrospinning. J. Mater. Sci. 45, 1299–1306 (2009).
83. Hansen, C. M. The Universality of the Solubility Parameter. Ind. Eng. Chem.
Prod. Res. Dev. 8, 2–11 (1969).

7 Literaturverzeichnis
109
84. Rinnbauer, M., Stein, G. & Peterseim, V. in 1203–1268 (Springer Berlin
Heidelberg, 2012). doi:10.1007/978-3-642-16173-5_3
85. Hildebrand, J. H. & Scott, R. L. Solutions of Nonelectrolytes. Annu. Rev. Phys.
Chem. 1, 75–92 (1950).
86. Buckley-Smith, M. The Use of Solubility Parameters to Select Membrane
Materials for Pervaporation of Organic Mixtures. (2006).
87. Krevelen, D. W. Van. Properties of Polymers. 898 (Elsevier Science Pub Co,
1990).
88. Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach
to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 99,
2224–2235 (1995).
89. Járvás, G., Quellet, C. & Dallos, A. Estimation of Hansen solubility parameters
using multivariate nonlinear QSPR modeling with COSMO screening charge
density moments. Fluid Phase Equilib. 309, 8–14 (2011).
90. Stefanis, E. & Panayiotou, C. A new expanded solubility parameter approach.
Int. J. Pharm. 426, 29–43 (2012).
91. Panayiotou, C. Polymer–polymer miscibility and partial solvation parameters.
Polymer (Guildf). 54, 1621–1638 (2013).
92. Panayiotou, C. Redefining solubility parameters: the partial solvation parameters.
Phys. Chem. Chem. Phys. 14, 3882–908 (2012).
93. Milliman, H. W., Boris, D. & Schiraldi, D. A. Experimental Determination of
Hansen Solubility Parameters for Select POSS and Polymer Compounds as a
Guide to POSS–Polymer Interaction Potentials. Macromolecules 45, 1931–1936
(2012).
94. Segarceanu, O. & Leca, M. Improved method to calculate Hansen solubility
parameters of a polymer. Prog. Org. Coatings 31, 307–310 (1997).
95. Burke, J. in AIC B. Pap. Gr. Annu. (ed. C Jensen) 13–58 (The Book and Paper
Group of the American Institute for Conservation, 1984).
96. Bustamante, P., Navarro-Lupión, J. & Escalera, B. A new method to determine
the partial solubility parameters of polymers from intrinsic viscosity. Eur. J.
Pharm. Sci. 24, 229–237 (2005).
97. Mark, J. E. Polymer Data Handbook: On-line access to full text available with
purchase; instructions in book. 1040 (Oxford University Press, USA, 1999).

7 Literaturverzeichnis
110
98. Dislich, H. & Hussmann, E. Amorphous and crystalline dip coatings obtained
from organometallic solutions: Procedures, chemical processes and products.
Thin Solid Films 77, 129–140 (1981).
99. Brinker, C. J., Hurd, A. J., Schunk, P. R., Frye, G. C. & Ashley, C. S. Review of
sol-gel thin film formation. J. Non. Cryst. Solids 147-148, 424–436 (1992).
100. Scriven, L. E. Physics and Applications of DIP Coating and Spin Coating. MRS
Proc. 121, 717 (2011).
101. Fehringer, G. Herstellung von Schichten aus Nanopartikeln über das Dip-
Coating-Verfahren mit wässrigen Suspensionen , über atmosphärisches
Plasmaspritzen und über Elektroschmelzsprühen. 167 (2007).
102. Puetz, J. & Aegerter, M. A. in Sol-Gel Technol. Glas. Prod. Users (eds. Aegerter,
M. A. & Mennig, M.) 37–48 (Springer US, 2004). doi:10.1007/978-0-387-
88953-5
103. Liquid Film Coating. (Springer Netherlands, 1997). doi:10.1007/978-94-011-
5342-3
104. Jeffrey Brinker, C. & Hurd, A. J. Fundamentals of sol-gel dip-coating. J. Phys.
III 4, 1231–1242 (1994).
105. Brinker, C. J., Frye, G. C., Hurd, A. J. & Ashley, C. S. Fundamentals of sol-gel
dip coating. Thin Solid Films 201, 97–108 (1991).
106. Levich, B. & Landau, L. Dragging of liquid by a plate. Acta Physiochim U.R.S.S.
52, (1942).
107. Meyerhofer, D. Characteristics of resist films produced by spinning. J. Appl.
Phys. 49, 3993 (1978).
108. Williams, J. Doctor-blade process. Treatise Mater. Sci. … 9, 173–198 (1976).
109. Hotza, D. & Greil, P. Review: aqueous tape casting of ceramic powders. Mater.
Sci. Eng. A 202, 206–217 (1995).
110. Mistler, R. E. & Twiname, E. R. Tape Casting: Theory and Practice. 298
(Wiley-American Ceramic Society, 1999).
111. Hyatt, E. P. Making thin, flat ceramics ― a review. Am. Ceram. Soc. Bull. 65,
637–638 (1986).
112. Chou, Y. T., Ko, Y. T. & Yan, M. F. Fluid Flow Model for Ceramic Tape
Casting. J. Am. Ceram. Soc. 70, C–280–C–282 (1987).

7 Literaturverzeichnis
111
113. Salmang, H. & Scholze, H. in Keramik (ed. Telle, R.) 527–689 (Springer Berlin
Heidelberg, 2007). doi:10.1007/978-3-540-49469-0
114. Gharagheizi, F. New procedure to calculate the Hansen solubility parameters of
polymers. J. Appl. Polym. Sci. 103, 31–36 (2007).
115. Normenausschuss Materialprüfung. DIN 51562: Viskosimetrie - Messung der
kinematischen Viskosität mit dem Ubbelohde-Viskosimeter. Dtsch. Inst. für
Normung e. V. (2002).
116. Scott, R. L. The Thermodynamics of High Polymer Solutions. V. Phase
Equilibria in the Ternary System: Polymer 1—Polymer 2—Solvent. J. Chem.
Phys. 17, 279 (1949).
117. Baney, R., Voigt, C. & Mentele, J. in Struct. Relationships Polym. (eds. Harrys,
F. & Seymour, R.) (1977).
118. Scheffler, F. Persönliche Mitteilung. (2012).
119. Persson, A. E., Schoeman, B. J., Sterte, J. & Otterstedt, J.-E. Synthesis of stable
suspensions of discrete colloidal zeolite (Na, TPA)ZSM-5 crystals. Zeolites 15,
611–619 (1995).
120. Devapal, D., Sebastain, T. V., Prabhakaran, P. V. & Packirisamy, S. Thermal
Barrier Coating on Metallic Substrates by Preceramic Route. Mater. Sci. Forum
710, 786–791
121. Vakifahmetoglu, C., Colombo, P., Carturan, S. M., Pippel, E. & Woltersdorf, J.
Growth of One-Dimensional Nanostructures in Porous Polymer-Derived
Ceramics by Catalyst-Assisted Pyrolysis. Part I: Iron Catalyst. J. Am. Ceram.
Soc. 93, 959–968 (2010).
122. Vakifahmetoglu, C., Pippel, E., Woltersdorf, J. & Colombo, P. Growth of One-
Dimensional Nanostructures in Porous Polymer-Derived Ceramics by Catalyst-
Assisted Pyrolysis. Part I: Iron Catalyst. J. Am. Ceram. Soc. 93, 959–968 (2010).
123. Ohl, C. et al. Novel Open-Cellular Glass Foams for Optical Applications. J. Am.
Ceram. Soc. 94, 436–441 (2011).
124. Thimm, B. W., Unger, R. E., Neumann, H.-G. & Kirkpatrick, C. J.
Biocompatibility studies of endothelial cells on a novel calcium phosphate/SiO2-
xerogel composite for bone tissue engineering. Biomed. Mater. 3, 015007 (2008).

7 Literaturverzeichnis
112

8 Abbildungsverzeichnis
113
8 Abbildungsverzeichnis
Abbildung 1: Lichtmikroskopische Aufnahmen mit SiC gefüllter Beschichtungen,
links nach dem Aushärten bei 110 °C und rechts nach dem Sintern unter
Stickstoff bei 1100 °C.12
......................................................................... 7
Abbildung 2: Diagramm des Verlaufes der freien Mischungsenthalpie für Gemische
mit niedermolekularen Substanzen, dargestellt als Funktion der
Zusammensetzung bei verschiedenen Wechselwirkungsparametern. Mit
Erlaubnis entnommen aus 36
Copyright (2014) Springer Berlin
Heidelberg. ............................................................................................ 10
Abbildung 3: Verlauf von Spinodale und Binodale in niedermolekularen Systemen.
Mit Erlaubnis entnommen aus 36
Copyright (2014) Springer Berlin
Heidelberg. ............................................................................................ 11
Abbildung 4: Diagramm von Binodalen in Systemen mit unterschiedlichen
Polymerisationsgraden XN. Mit Erlaubnis entnommen aus 36
Copyright
(2014) Springer Berlin Heidelberg. ...................................................... 12
Abbildung 5: AFM-Aufnahmen (26 µm x 28 µm) einer Schichtdickenreihe einer
PS/PMMA Lösung auf einer Goldoberfläche hergestellt mittels
Schleuderbeschichten. Mit Erlaubnis entnommen aus 38
Copyright
(1997) American Chemical Society. ..................................................... 14
Abbildung 6: Schematischer Ablauf der Strukturentstehung währende der Entnetzung
eines dünnen flüssigen Filmes. Mit Erlaubnis entnommen aus 46
Copyright (1997) American Chemical Society .................................... 16
Abbildung 7: Darstellung von Simulationsergebnis für ein entmischendes binäres
System mit mobilen runden Partikeln für unterschiedliche Zeiten und
unterschiedliche Partikelzahlen a) 144 Partikel, b) 289 Partikel und c)
400 Partikel. Mit Erlaubnis entnommen aus 49
Copyright (2006) by The
American Physical Society. .................................................................. 18
Abbildung 8: Schematische Darstellung einer Benard-Konvektionszelle entnommen
aus 54
..................................................................................................... 19

8 Abbildungsverzeichnis
114
Abbildung 9: Übersicht über die verschiedenen Siliziumpolymere und deren
Namensgebung. Mit Erlaubnis entnommen aus 6 © 2000 WILEY-VCH
Verlag GmbH, Weinheim, Fed. Rep. of Germany ................................ 21
Abbildung 10: Schematische Darstellung des Tauchbeschichtungsprozesses. Die Pfeile
entlang des Substrates stellen die Strömungsrichtung der Luft dar. Mit
Erlaubnis aus 102
entnommen und übersetzt. ......................................... 29
Abbildung 11: Schematische Darstellung der Schichtentwicklung während des
Schleuderbeschichtens.100
..................................................................... 30
Abbildung 12: Diagramme der Löslichkeitskugeln für PMS: (links) berechnet nach
Gharagheizi, (rechts) berechnet nach Hansen. Der schwarze Kreis stellt
den Mittelpunkt der Kugeln dar und gibt die HSPs von PMS wieder.
Die grünen Pluszeichen stellen die Lösungsmittel innerhalb der Kugel
dar und die roten Kreuze die Nichtlösungsmittel außerhalb der Kugel.
48
Abbildung 13: Diagramme der Löslichkeitskugeln für PMPS: (links) berechnet nach
Gharagheizi, (rechts) berechnet nach Hansen. Der schwarze Kreis stellt
den Mittelpunkt der Kugeln dar und gibt die HSP von PMPS wieder.
Die grünen Pluszeichen stellen die Lösungsmittel innerhalb der Kugel
dar und die roten Kreuze die Nichtlösungsmittel außerhalb der Kugel.
49
Abbildung 14: Mischbarkeitsdiagramme für die vier Komponenten MTES, PMS,
PMPS und Methanol. Die blauen Rauten (♦) stellen nicht mischbare,
die roten Kreuze (x) mischbare Zusammensetzung dar. Der Methanol-
Anteil nimmt mit dem Diagramm von oben links a) mit 0 vol. % in
16,67 vol. % Schritten auf 50 vol. % im Diagramm unten rechts d) zu.
Der Abstand zwischen den einzelnen Punkten beträgt ebenfalls 16,67
vol. %. Die schwarzen Linien trennen den Bereich der mischbaren
Zusammensetzungen vom Bereich der nicht mischbaren
Zusammensetzungen.116
........................................................................ 53
Abbildung 15: Lichtmikroskopische Aufnahmen der Grundzusammensetzungen aus
PMS, PMPS und MTES (Tabelle 5). Die Verhältnisse von

8 Abbildungsverzeichnis
115
PMS:PMPS:MTES sind wie folgt: COMP 1 33,3:33,3:33,3, COMP 2
60:20:20, COMP 3 20:60:20 und COMP 4 20:20:60.116
...................... 55
Abbildung 16: REM-Aufnahme von mit COMP 4 beschichtetem Aluminiumoxid.
Entlang der grünen Linie (L1) wurden EDX-Analysen in Abstand von 1
µm durchgeführt, die im unteren Diagramm dargestellt sind.116
.......... 56
Abbildung 17: Lichtmikroskopische Aufnahmen von Schichten aus gleichen
PMS:PMPS:MTES-Verhältnissen (gemäß COMP 1) und
unterschiedlichem Methanol-Gehalt auf Aluminiumoxid-Substrat.116
. 57
Abbildung 18: UV/VIS-Transmissionskurve von COMP 1 in Abhängigkeit von der
Methanol-Konzentration.12
................................................................... 58
Abbildung 19: Lichtmikroskopische Aufnahmen von mit SiC gefüllter
Beschichtungen, links nach dem Vernetzten bei 110 °C und rechts nach
dem Sintern unter Stickstoff bei 1100 °C.116
........................................ 59
Abbildung 20: REM-Aufnahme SiC gefüllter Beschichtung auf Aluminiumoxid.116
.. 60
Abbildung 21: REM-Aufnahmen SiC gefüllter Beschichtung auf Aluminiumoxid im
Querschnitt. ........................................................................................... 60
Abbildung 22: Lichtmikroskopische Aufnahmen von mit SiC gefüllten
Beschichtungen auf Al2O3-Substrat, die mit Suspensionen mit
unterschiedlichem Methanolgehalt hergestellt wurden; a) 45 vol. %, b)
47,5 vol. %, c) 67,5 vol. % und d) 70 vol. %. ....................................... 61
Abbildung 23: Lichtmikroskopische Aufnahmen von mit SiC gefüllten
Beschichtungen auf Al2O3-Substrat, die mit Suspensionen mit
unterschiedlichen MTES-Gehalten hergestellt wurden; a) 0 vol. %, b)
10 vol. %, c) 20 vol. % und d) 30 vol. %. ............................................. 63
Abbildung 24: Beschichtungen mit unterschiedlichen Polymerverhältnissen.
Abbildung a) zeigt die lichtmikroskopische Aufnahme einer
Beschichtung mit reinem PMPS, Abbildung d) eine mit reinem PMS.
Das PMS/PMPS-Verhältnis von Probe b) betrug 3:7 und das der Probe
c) 7:3. .................................................................................................... 64
Abbildung 25: Lichtmikroskopische Aufnahmen von mit SiC gefüllten
Beschichtungen auf Al2O3-Substrat, die mit Suspensionen mit

8 Abbildungsverzeichnis
116
unterschiedlichen SiC-Gehalten hergestellt wurden; a) 10 vol. %, b)
12,5 vol. %, c) 17,5 vol. % und d) 25 vol. %. ....................................... 66
Abbildung 26: Lichtmikroskopische Aufnahmen von mit SiC gefüllten
Beschichtungen auf Al2O3-Substrat, die mit Suspensionen mit
unterschiedlichen SiC-Korngrößen hergestellt wurden; a) d50 = 0,67
µm, b) d50 = 1,41 µm, c) d50 = 2,22 µm und d) d50 = 4,55 µm. ............. 67
Abbildung 27: Lichtmikroskopische Aufnahmen von mit SiC gefüllten
Beschichtungen auf Al2O3-Substrat, die mit Suspensionen hergestellt
sind, die nur Methanol, SiC und PMPS enthalten. Das Methanol:SiC
Verhältnis entspricht dem im Referenzansatz, der PMPS-Gehalt variiert
a) 0 vol. %, b) 1 vol. %, c) 2,5 vol. % und d) 5 vol. %. ........................ 70
Abbildung 28: Lichtmikroskopische Aufnahmen, die den zeitlichen Verlauf der
Strukturentstehung darstellen. ............................................................... 72
Abbildung 29: Mittels Schleuderbeschichten hergestellte Beschichtungen, bei denen
der Füllstoff NF25 mit einem d50 = 0,67 µm verwendet wurde. Die
Endrotationsgeschwindigkeit für die Aufsicht a) und den Querschnitt b)
betrug 6000 1 min-1
und für die Aufsicht c) und den Querschnitt d)
2000 1 min-1
. ......................................................................................... 75
Abbildung 30: Mittels Rakeln hergestellte Beschichtungen des Referenzansatzes. Die
keramische Folie a) Aufsicht und b) Querschnitt wurde mit einer
Spaltbreite des Rakel von 250 µm hergestellt und die Folie c) Aufsicht
und d) Querschnitt wurden mit einer Spaltbreite des Rakel von 2500
µm hergestellt. ....................................................................................... 77
Abbildung 31: Lichtmikroskopische Aufnahmen von Referenzbeschichtungen
hergestellt mit unterschiedlichen Ziehgeschwindigkeiten von 2,5 bis 30
mm s-1
. ................................................................................................... 78
Abbildung 32: Diagramm der Ziehgeschwindigkeit gegen Schichtdicke der Proben aus
Abbildung 31. ........................................................................................ 79
Abbildung 33: Diagramm der Schichtdicke gegen die Porengröße der Proben aus
Abbildung 31. ........................................................................................ 80

8 Abbildungsverzeichnis
117
Abbildung 34: Fotografie von beschichteten Aluminiumoxidrohren mit
unterschiedlichen Durchmessern von 1 bis 5 mm, beschichtet nach dem
Referenzverfahren, ausgehärtet bei 110 °C .......................................... 81
Abbildung 35: Lichtmikroskopische Aufnahmen von Beschichtungen auf
Aluminiumoxidrohren mit unterschiedlichen Durchmessern: a) d = 1
mm, b) d = 3 mm, c) d = 5 mm und eine Referenzbeschichtung auf
planarem Aluminiumoxid d). ................................................................ 82
Abbildung 36: Schematische Darstellung der Porenentstehung. .................................. 95
Abbildung 37: Schematische Darstellung der Bereiche innerhalb welcher
Zusammensetzung in vol. % bzw. bei welcher Korngröße die
Strukturbildung funktioniert. Das Polymerverhältnis bezieht sich auf
den Polymergehalt von 18,4 vol. % der Referenzzusammensetzung, die
in der Darstellung 50 % entspricht und hierbei auf PMS bezogen ist.
Zusätzlich sind die Tendenzen angegeben wie sich die Porengröße
dabei entwickelt. ................................................................................... 96
Abbildung 38: Schematische Darstellung der gefundenen Einflussparameter
Schichtdicke, Viskosität und Verdunstungsrate und der damit
verbundene Einfluss auf die Porengröße. ............................................. 98

8 Abbildungsverzeichnis
118

9 Tabellenverzeichnis
119
9 Tabellenverzeichnis
Tabelle 1: Lösungsmittel zur Ermittlung der Löslichkeitsparameter nach Hansen ... 34
Tabelle 2: Zur Beschichtung verwendete Substrate ................................................... 35
Tabelle 3: Verwendete Füllstoffe und Partikelgrößen ................................................ 36
Tabelle 4: Referenzzusammensetzung für füllstoffhaltige Systeme .......................... 40
Tabelle 5: Zusammensetzung für ungefüllte Beschichtungssysteme ......................... 41
Tabelle 6: Verwendete Lösungsmittel und die dazugehörigen Hansen
Löslichkeitsparameter80
sowie die Einteilung in Lösungsmittel markiert
mit „1“ und Nichtlösungsmittel markiert mit „0“ für PMS und PMPS bei
einem Mischungsverhältnis von 50:50 in vol. % ...................................... 45
Tabelle 7: Gemessene Durchflusszeiten für das reine Lösungsmittel sowie für die
Mischung aus Lösungsmittel und 5 g l-1
Polymer mit den dazugehörigen
berechneten inhärenten Viskositäten ......................................................... 47
Tabelle 8: Hansen Löslichkeitsparameter, Hildebrand Löslichkeitsparameter sowie
der dazugehörige Interaktionsradius für die verschiedenen Polymere und
Bestimmungsmethoden ............................................................................. 50
Tabelle 9: Hansen Löslichkeitsparameter, Hildebrand Löslichkeitsparameter sowie
der dazugehörige Interaktionsradius für verschiedene Siliziumpolymere. 85
Tabelle 10: Grenzen für die einzelnen Komponenten innerhalb der die Strukturbildung
in gefüllten Beschichtungen funktioniert. ................................................. 93

9 Tabellenverzeichnis
120

Lebenslauf
i
Lebenslauf
1988 – 1997 Grund- und Hauptschule Herzogenaurach
1997 – 2000 Ausbildung zum Ver- und Entsorger bei der Stadt Erlangen
2000 – 2003 Berufstätigkeit als Ver- und Entsorger bei der Stadt Erlangen
und der Stadt Nürnberg
2003 – 2005 Weiterbildung zum Staatlich geprüften Umweltschutztechniker
und Fachhochschulreife an der LGA Fachschule Nürnberg
2005 – 2009 Studium an der Georg-Simon-Ohm-Hochschule Nürnberg im
Diplomstudiengang Werkstofftechnik
2006 – 2008 Werkstudent bei der Siemens AG Erlangen
2007 Praktisches Studiensemester bei der Firma Robert Bosch
GmbH Gerlingen, Corporate Sector Research and Advance
Engineering, Applied Research – Materials
2009-2013 Wissenschaftlicher Mitarbeiter mit dem Ziel der Promotion am
ZAE Bayern (Universität Erlangen-Nürnberg), Abschluss vo-
raussichtlich 7/2014
seit 5/2014 Wissenschaftlicher Mitarbeiter an der Friedrich-Alexander-
Universität Erlangen-Nürnberg

ii

Wissenschaftliche Veröffentlichungen
iii
Wissenschaftliche Veröffentlichungen
M. Woiton, J. Gutierrez, M. Heyder, M. Scheffler, E. Stern; Porous Polymer Derived Ceramic
Surface Layers from Demixing Processes; 34th International Conference and Exposition on
Advanced Ceramics and Composites; Daytona Beach USA; 2010
M. Woiton, M. Heyder, A. Laskowsky, E. Stern, M. Scheffler, C. J. Brabec; Netlike Structured
Surfaces from Preceramic Polymers; HT-CMC7 - 7th International Conference on High Tem-
perature Ceramic Materials and Composites; Bayreuth Germany; 2010
M. Woiton, M. Heyder, A. Laskowsky, E. Stern, M. Scheffler, C. J. Brabec; Self-Assembled Mi-
crostructured Polymeric and Ceramic Surfaces; Journal of the European Ceramic Society; Vol.
31 pp 1803-1810 2011
A. Laskowsky, M. Woiton, M. Heyder, E. Stern, M. Scheffler; Polymere und keramische Netz-
strukturen auf planaren Substraten; 2. Sitzung des DGM-Fachausschusses Zellulare Werkstof-
fe; Berlin Germany; 2011
P. Desclaux, M. Rzepka, E. Stern, M. Woiton, U. Stimming, R. Hempelmann; Direct Conversion
of Carbon in a HighTemperature Fuel Cell Using Ceria as Catalyst; International Workshop on
Electrocatalysis, Saarbrücken Germany; 2012
M. Woiton, M. Heyder, A. Laskowsky, E. Stern, M. Scheffler, C. J. Brabec; Porous Coatings for
Complex 3D-StructuresFormed by Self-Assemblage; 36th International Conference and Exposi-
tion on Advanced Ceramics and Composites; Daytona Beach USA; 2012
A. Laskowsky, P. Thiem, S. Cung, M. Woiton, M. Heyder, E. Stern, M. Scheffler; Cellular ceram-
ics from demixing processes – interactions between substrates and preceramic polymer coating
systems; CELLMAT 2012 Cellular Materials; Dresden Germany; 2012
P. Desclaux, H.C. Schirmer, M. Woiton, E. Stern, M. Rzepka; Influence of the Carbon/Anode
Interaction on Direct Carbon Conversion in a SOFC; International Journal of Electrochemical
Science; Vol. 8 2013
A. Laskowsky, A.-K. Schuldt, M. Woiton, M. Heyder, E. Stern, M. Scheffler; Structured ceramic
surfaces by preceramic polymer demixing processes; IOP Conference Series: Materials Sci-
ence and Engineering; Vol. 47; 2013
P. Desclaux, M. Rzepka, E. Stern, M. Woiton, U. Stimming, R. Hempelmann; On the Catalytic Activity of Ceria for Direct Carbon Conversion in a SOFC; Zeitschrift für Physikalische Chemie; 2013

iv

Danksagung
v
Danksagung
Ein wichtiger Teil einer Arbeit, wie ich sie hier abgegeben habe, sind die Menschen, die
durch ihr Engagement, ihren Einsatz und ihre Hilfe und Inspiration einen wichtigen Teil
zum Gelingen beigetragen haben. An dieser Stelle möchte ich mich dafür ganz herzlich
bedanken.
Ein großer Dank gilt hierbei gleichermaßen meinen beiden Betreuern Prof. Dr.
Christoph J. Brabec und Prof. Dr. Michael Scheffler, denn ohne deren Unterstützung
und Hilfe wäre diese Dissertation nicht zu realisieren gewesen. Ich kann mich glücklich
schätzen, gleich zwei so gute Betreuer gefunden zu haben.
Einen ganz besonderen Dank möchte ich meiner Gruppenleiterin und Mentorin Dr. Ed-
da Stern aussprechen. Sie war es, die die ersten Schritte zu diesem Thema gemacht und
mich dazu ermutigt hat, es in Angriff zu nehmen. Zudem hatte Sie stets ein offenes Ohr
für meine Fragen und hat immer wieder neue Anregungen geliefert. Auch hat sie mich
immer wieder motiviert, wofür ich ihr sehr dankbar bin.
Natürlich möchte ich mich auch bei den vielen anderen Helfern bedanken, ohne deren
Einsatz und Mithilfe bei Messungen, im Umgang mit neuen Geräten oder bei der Ein-
führung neuer Methoden und vielem vielem mehr, die hier vorliegende Arbeit nur
schwer machbar gewesen wäre. Ein großer Dank an: Madeleine Heyder, Alexandra
Laskowsky, Astrid Kidzun, Dr. Andreas Vetter, Florian Machui, Philip Hofmann, Mi-
chael Ehrl, Stefan Schön, Fabian Peichl, Felix Hoga, Tom Engl, David Mbodep, Anna
Lloyd, Tobias Zehnder, Prof. Dr. Franziska Scheffler, Clemens Kowalli, Vladimir Ga-
zuz, Stephan Wittmann, Maik Hessmann, Edeltraud Völkel, Dr. Matthias Wiener, Urs
Bogner, Peter Kubis Frank Fecher, Jens Adams, Jonas Bachmann, Thomas Swonke, Dr.
Tobias Fey, und Anita Metter.
Auch bei meinem privaten Umfeld möchte ich mich sehr herzlich bedanken, denn ohne
den Rückhalt, die Unterstützung aber auch die Ablenkung der Familie und der Freunde,
kurz ohne liebe Menschen, die einem den Rücken frei halten (und auch mal den Kopf
waschen) ist die Realisierung eines solchen Projektes unmöglich.
Auch der DFG möchte ich für die finanzielle Unterstützung danken.