Embed Size (px)
Citation preview

Phosphanchalkogenide und ihre Metallkomplexe. II. Komplexe einigerGold(I)-Halogenide mit Diphosphanmonochalkogeniden [1]Phosphane Chalcogenides and their Metal Complexes. II. Gold(I) Halide Complexes of someDiphosphane Monochalcogenides
Christina Taouss und Peter G. JonesInstitut fur Anorganische und Analytische Chemie, Technische Universitat Braunschweig,Postfach 3329, D-38023 Braunschweig, Germany
Reprint requests to Prof. Dr. P. G. Jones. E-mail: [email protected]
Z. Naturforsch. 2014, 69b, 25 – 48 / DOI: 10.5560/ZNB.2014-3273Received October 9, 2013
We report the crystal structures of 19 gold(I) complexes with diphosphane monochalcogenideligands, general formula (PP)EAuHal (PP = diphosphane, E = chalcogen, Hal = halogen). ForPP = bis(diphenylphosphano)methane (dppm): 1, dppmOAuCl as its diisopropyl ether solvate; 2,dppmOAuI as its dichloromethane solvate; 3, dppmSAuCl as its dichloromethane hemisolvate; 4,dppmSAuI; 5, dppmSeAuBr as a 1:1 adduct with dppmSe2; 6, dppmSeAuI. Compound 3 is iso-typic to the known structure of dppmSeAuCl. Most of the structures have an E–P···P–Au torsionangle close to zero, which, especially for the larger E atoms, facilitates a close intramolecular ap-proach of E and Au. The packing of 1–3 involves hydrogen bonds from the methylene hydrogens.For PP = 1,2-bis(diphenylphosphano)ethane (dppe): 7, dppeOAuBr and 8, dppeOAuI, both as theirhemihydrates; 9, dppeSAuBr; 10, dppeSAuI. All four compounds crystallise with two independentmolecules of the gold complex, between which short Au···Au contacts are observed; the moleculesof 7 and 8 are further linked by hydrogen bond systems [. . .P=O···H–O–H···O=P. . .]. Compounds7 and 8 form an isotypic series with their known chloride analogue, as do 9 and 10. For PP = 1,2-bis(diphenylphosphano)benzene (dppbz): 11, dppbzOAuCl; 12, dppbzOAuBr; 13, dppbzSAuBr; 14,dppbzSAuI, 16, dppbzSeAuBr and 17, dppbzSeAuI, all as their isotypic dichloromethane disolvates;15, dppbzSeAuCl (isotypic to 13). The absolute E–P···P–Au torsion angles are all in the range50 – 70◦; short intramolecular E···Au contacts are observed. The packing of 11–17 shows few no-table short contacts. For PP = cis-1,2-bis(diphenylphosphano)ethene (cisdppen): 18, cisdppenSAuCl;19, cisdppenSAuBr. Both compounds crystallise with two independent molecules, and in both casesthe molecules display considerable differences in configuration. The packing of 18 involves shortH···Cl and that of 19 short H···S contacts. For PP = 1,8-bis(diphenylphosphano)naphthalene (dppn),an inversion-symmetric gold(III) complex (dppnSeAu)2Cl2 20, with a central four-membered ringAu2Se2, was obtained instead of the expected isomeric gold(I) derivative by formal insertion of goldinto a P–Se bond; it crystallised as a deuterochloroform hexasolvate.
Key words: Gold, Diphosphane, Chalcogenide
Einleitung
Phosphanchalkogenide sind gut zur Koordinationan Goldzentren geeignet. So sind in der Litera-tur einige Gold(I)-Halogenide mit Phosphansulfidenund -seleniden bekannt, bei denen das Gold an dasChalkogen koordiniert. Diese Komplexe der Form(R3PE)AuHal (E = Chalkogen, Hal = Cl, Br) konnendurch Umsetzung von Phosphanchalkogeniden R3PEmit anderen Gold(I)-Komplexen (L′)AuHal erhaltenwerden, wobei L′ ein schwach koordinierender undsomit leicht zu ersetzender Ligand ist (z. B. Tetrahy-
drothiophen (tht) oder Me2S). Alternativ konnendie Gold(I)-Bromide auch durch Halogenaustauschaus den entsprechenden Chloriden erhalten wer-den [2 – 10]. Phosphanoxide hingegen sind zu harteDonoren, um an Gold(I)-Zentren zu koordinieren.
Auf die gleiche Weise konnen auch zweikernigeGold(I)-Halogenide mit Diphosphandisulfiden syn-thetisiert werden [11]. Diese sind bisher allerdingsnicht kristallographisch untersucht worden.
Auch Diphosphanmonochalkogenide, uber die wirin Teil I dieser Reihe berichtet haben [1], sind zur Ko-ordination an Gold(I)-Halogenide geeignet. So erhiel-
© 2014 Verlag der Zeitschrift fur Naturforschung, Tubingen · http://znaturforsch.com

26 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
ten Schmidbaur et al. im Jahr 1990 aus der Reaktionvon dppmSe mit (CO)AuCl zunachst einen einkerni-gen Gold(I)-Komplex, bei dem das Gold an dasPhosphoratom des Liganden koordiniert. Ein weiteresAquivalent (CO)AuCl fuhrte zur Bildung eines zwei-kernigen Komplexes mit einem zweiten, an das Selen-atom koordinierenden Goldchlorid [12]. Die Kristall-struktur des einkernigen Komplexes wurde zwei Jahrespater in diesem Arbeitskreis bestimmt [13]. Bisheute sind nicht viele Gold(I)-Komplexe mit Diphos-phanmonochalkogeniden bekannt, und nur von sehrwenigen (manchen als Zufallsprodukten) wurde uberKristallstrukturen berichtet [14 – 17].
Die hier berichtete kristallographische Analyse derGold(I)-Komplexe 1–19 wurde im Rahmen von Un-tersuchungen zur Halogenierung dieser Verbindungendurchgefuhrt, uber die wir bereits in einer Kommu-nikation berichtet haben [18]. Der Gold(III)-Komplex20 wurde uberraschenderweise bei dem Versuch, einenweiteren Gold(I)-Komplex zu synthetisieren, erhaltenund ebenfalls kristallographisch analysiert.
Ergebnisse und Diskussion
dppmEAuHal [dppm = Bis(diphenylphosphano)-methan; E = O, Hal = Cl (1), I (2); E = S, Hal = Cl(3), I (4); E = Se, Hal = Br (5), I (6)]
Tabelle 1 fasst einige Strukturparameter vonVerbindungen der Form dppmEAuHal zusammen.
Es ist auffallig, dass es sich bei funf der acht (ein-schl. der zwei bereits bekannten) Strukturen dieserKomplexe um Solvate und bei einer weiteren (5,siehe unten) um ein unerwartetes Addukt handelt. DerLosungsmittelgehalt fuhrte dazu, dass die Elemen-taranalysen z. T. unzufriedenstellend waren. Die hierbestimmte Struktur des dppmSAuCl (3) ist isotyp zuder bekannten Struktur des dppmSeAuCl [13], was
P···P Au···E P–C–P P–Au–Hal E–P···P–AudppmOAuCl 1 3,052(2) 4,247(3) 114,2(2) 176,20(3) 94,0(1)dppmOAuBr [15] 3,07 3,274(4) 115,0(2) 178,53(4) 8,3(2)dppmOAuI 2 3,055(2) 3,226(4) 114,0(2) 177,70(3) 10,9(2)dppmSAuCl 3 3,1157(6) 3,3162(5) 117,02(9) 176,76(2) 9,20(2)dppmSAuI 4 a 3,066(3) 3,438(2) 113,2(3) 177,06(6) −59,71(8)
3,111(3) 3,169(2) 115,9(3) 176,48(6) −3,03(8)dppmSeAuCl [13] 3,12 3,277(1) 117,0(5) 175,2(1) 8,3dppmSeAuBr 5 3,132(2) 3,2194(4) 118,1(2) 172,35(2) −7,91(3)dppmSeAuI 6 3,078(4) 3,434(2) 114,4(5) 177,76(7) −57,45(9)
a Zwei unabhangige Molekule.
Tabelle 1. Ausgewahlte Ab-stande [A], Winkel [◦] undTorsionswinkel [◦] in dppm-EAuHal.
den einzigen Fall der Isotypie bei den acht Struk-turen darstellt; beide kristallisieren als Dichlormethan-Hemisolvate in der monoklinen Raumgruppe P21/c.Die Iodide dppmSAuI (4) und dppmSeAuI (6)kristallisieren monoklin primitiv mit sehr ahnlichenAchsenlangen; die Raumgruppen sind aber unter-schiedlich (Pn bzw. P21/n; siehe unten). Im Folgendenwerden die Kristallstrukturen genauer beschrieben.
Das dppmOAuCl (1) kristallisiert in der Raum-gruppe Pbca mit einem Molekul des Komplexesund einem geordneten Molekul Diisopropyletherin der asymmetrischen Einheit (Abb. 1a). DieBindungslangen und -winkel liegen alle im erwartetenBereich, wobei die Phosphoratome tetraedrischund das Goldatom linear koordiniert sind (bei denanderen Strukturen wird das nicht extra kommen-tiert). Das Goldchlorid und der Sauerstoff sind soangeordnet, dass sie auf unterschiedlichen Seitenaus der P–C–P-Ebene herausstehen (Au–P2–C1–P155,9(2)◦, O–P1–C1–P2 50,1(2)◦). Der TorsionswinkelO–P1···P2–Au betragt 93,97 (9)◦ und ist damitwesentlich großer als im analogen Bromid und Iodid.Es gibt also keine intramolekulare Gold-Sauerstoff-Wechselwirkung; der Abstand Au···O ist sehr groß(4,246(2) A).
Bei den dppmE-Derivaten sind die Torsionswinkelentlang der P1···P2-Achse unterschiedlich. Bezeich-net man freie Elektronenpaare, deren Richtungen manabschatzen kann [1], als ,,X“, so ist bei der Stamm-verbindung dppm der Torsionswinkel X1–P1···P2–X2etwa 75◦ [19]. Die Struktur von dppmO ist noch un-bekannt, obwohl wir viel Zeit investiert haben, brauch-bare Kristalle zu bekommen [20]. Bei dppmS [1] unddppmSe [21] betragen die absoluten TorsionswinkelE–P1···P2–X 60 bzw. 12◦. Bei denjenigen Goldkom-plexen, die entsprechende Werte E–P1···P2–Au um0◦ aufweisen, sind nicht nur AuHal und E, sondernauch zwei Ringe, einer an jedem Phosphoratom, etwa
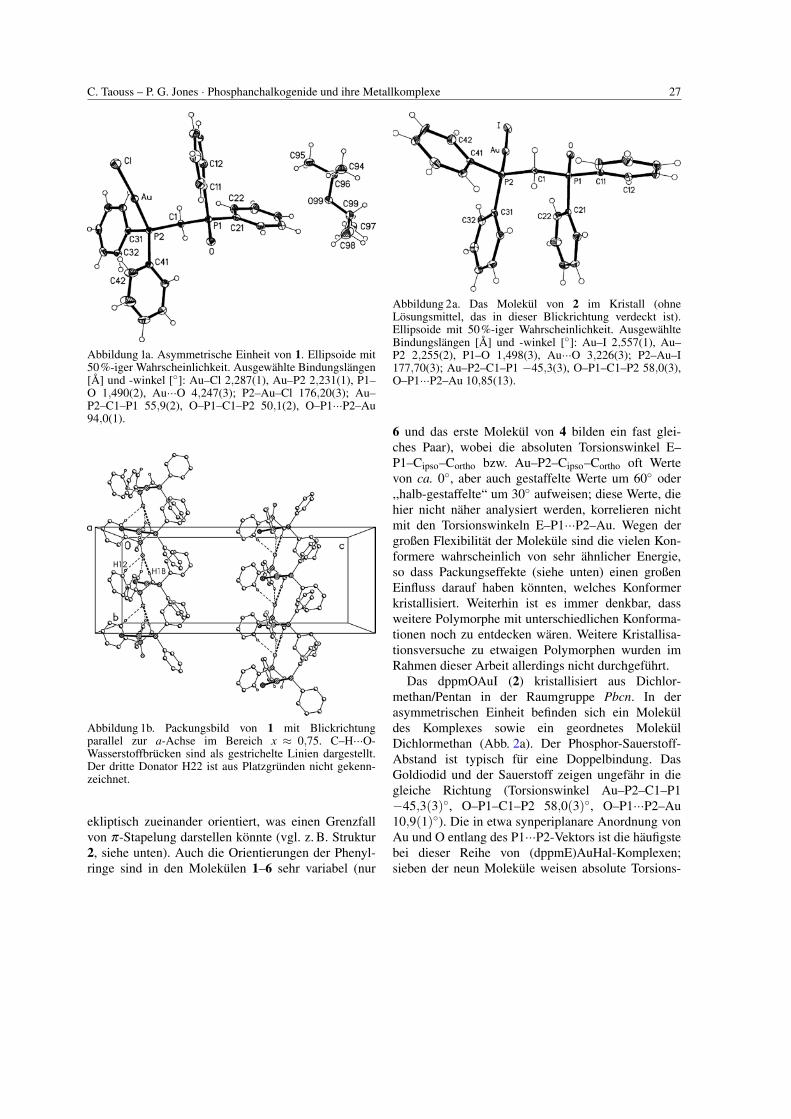
C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 27
Abbildung 1a. Asymmetrische Einheit von 1. Ellipsoide mit50%-iger Wahrscheinlichkeit. Ausgewahlte Bindungslangen[A] und -winkel [◦]: Au–Cl 2,287(1), Au–P2 2,231(1), P1–O 1,490(2), Au···O 4,247(3); P2–Au–Cl 176,20(3); Au–P2–C1–P1 55,9(2), O–P1–C1–P2 50,1(2), O–P1···P2–Au94,0(1).
Abbildung 1b. Packungsbild von 1 mit Blickrichtungparallel zur a-Achse im Bereich x ≈ 0,75. C–H···O-Wasserstoffbrucken sind als gestrichelte Linien dargestellt.Der dritte Donator H22 ist aus Platzgrunden nicht gekenn-zeichnet.
ekliptisch zueinander orientiert, was einen Grenzfallvon π-Stapelung darstellen konnte (vgl. z. B. Struktur2, siehe unten). Auch die Orientierungen der Phenyl-ringe sind in den Molekulen 1–6 sehr variabel (nur
Abbildung 2a. Das Molekul von 2 im Kristall (ohneLosungsmittel, das in dieser Blickrichtung verdeckt ist).Ellipsoide mit 50%-iger Wahrscheinlichkeit. AusgewahlteBindungslangen [A] und -winkel [◦]: Au–I 2,557(1), Au–P2 2,255(2), P1–O 1,498(3), Au···O 3,226(3); P2–Au–I177,70(3); Au–P2–C1–P1 −45,3(3), O–P1–C1–P2 58,0(3),O–P1···P2–Au 10,85(13).
6 und das erste Molekul von 4 bilden ein fast glei-ches Paar), wobei die absoluten Torsionswinkel E–P1–Cipso–Cortho bzw. Au–P2–Cipso–Cortho oft Wertevon ca. 0◦, aber auch gestaffelte Werte um 60◦ oder,,halb-gestaffelte“ um 30◦ aufweisen; diese Werte, diehier nicht naher analysiert werden, korrelieren nichtmit den Torsionswinkeln E–P1···P2–Au. Wegen dergroßen Flexibilitat der Molekule sind die vielen Kon-formere wahrscheinlich von sehr ahnlicher Energie,so dass Packungseffekte (siehe unten) einen großenEinfluss darauf haben konnten, welches Konformerkristallisiert. Weiterhin ist es immer denkbar, dassweitere Polymorphe mit unterschiedlichen Konforma-tionen noch zu entdecken waren. Weitere Kristallisa-tionsversuche zu etwaigen Polymorphen wurden imRahmen dieser Arbeit allerdings nicht durchgefuhrt.
Das dppmOAuI (2) kristallisiert aus Dichlor-methan/Pentan in der Raumgruppe Pbcn. In derasymmetrischen Einheit befinden sich ein Molekuldes Komplexes sowie ein geordnetes MolekulDichlormethan (Abb. 2a). Der Phosphor-Sauerstoff-Abstand ist typisch fur eine Doppelbindung. DasGoldiodid und der Sauerstoff zeigen ungefahr in diegleiche Richtung (Torsionswinkel Au–P2–C1–P1−45,3(3)◦, O–P1–C1–P2 58,0(3)◦, O–P1···P2–Au10,9(1)◦). Die in etwa synperiplanare Anordnung vonAu und O entlang des P1···P2-Vektors ist die haufigstebei dieser Reihe von (dppmE)AuHal-Komplexen;sieben der neun Molekule weisen absolute Torsions-
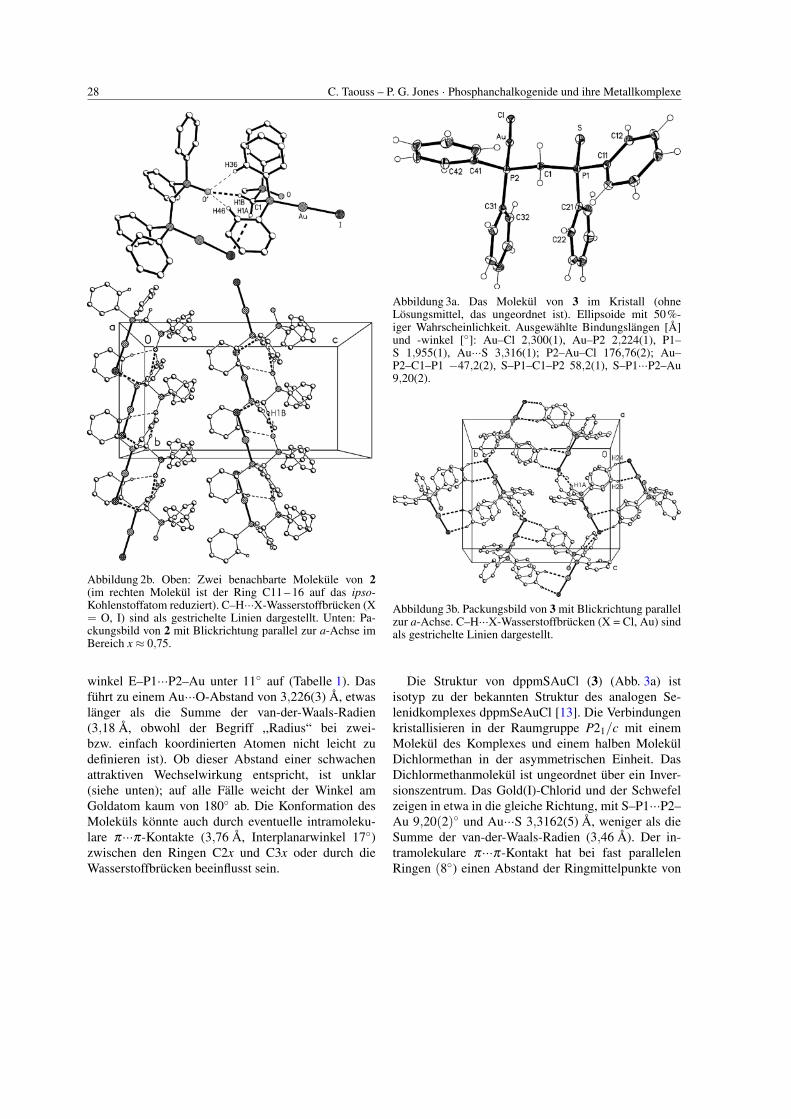
28 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
Abbildung 2b. Oben: Zwei benachbarte Molekule von 2(im rechten Molekul ist der Ring C11 – 16 auf das ipso-Kohlenstoffatom reduziert). C–H···X-Wasserstoffbrucken (X= O, I) sind als gestrichelte Linien dargestellt. Unten: Pa-ckungsbild von 2 mit Blickrichtung parallel zur a-Achse imBereich x≈ 0,75.
winkel E–P1···P2–Au unter 11◦ auf (Tabelle 1). Dasfuhrt zu einem Au···O-Abstand von 3,226(3) A, etwaslanger als die Summe der van-der-Waals-Radien(3,18 A, obwohl der Begriff ,,Radius“ bei zwei-bzw. einfach koordinierten Atomen nicht leicht zudefinieren ist). Ob dieser Abstand einer schwachenattraktiven Wechselwirkung entspricht, ist unklar(siehe unten); auf alle Falle weicht der Winkel amGoldatom kaum von 180◦ ab. Die Konformation desMolekuls konnte auch durch eventuelle intramoleku-lare π···π-Kontakte (3,76 A, Interplanarwinkel 17◦)zwischen den Ringen C2x und C3x oder durch dieWasserstoffbrucken beeinflusst sein.
Abbildung 3a. Das Molekul von 3 im Kristall (ohneLosungsmittel, das ungeordnet ist). Ellipsoide mit 50%-iger Wahrscheinlichkeit. Ausgewahlte Bindungslangen [A]und -winkel [◦]: Au–Cl 2,300(1), Au–P2 2,224(1), P1–S 1,955(1), Au···S 3,316(1); P2–Au–Cl 176,76(2); Au–P2–C1–P1 −47,2(2), S–P1–C1–P2 58,2(1), S–P1···P2–Au9,20(2).
Abbildung 3b. Packungsbild von 3 mit Blickrichtung parallelzur a-Achse. C–H···X-Wasserstoffbrucken (X = Cl, Au) sindals gestrichelte Linien dargestellt.
Die Struktur von dppmSAuCl (3) (Abb. 3a) istisotyp zu der bekannten Struktur des analogen Se-lenidkomplexes dppmSeAuCl [13]. Die Verbindungenkristallisieren in der Raumgruppe P21/c mit einemMolekul des Komplexes und einem halben MolekulDichlormethan in der asymmetrischen Einheit. DasDichlormethanmolekul ist ungeordnet uber ein Inver-sionszentrum. Das Gold(I)-Chlorid und der Schwefelzeigen in etwa in die gleiche Richtung, mit S–P1···P2–Au 9,20(2)◦ und Au···S 3,3162(5) A, weniger als dieSumme der van-der-Waals-Radien (3,46 A). Der in-tramolekulare π···π-Kontakt hat bei fast parallelenRingen (8◦) einen Abstand der Ringmittelpunkte von
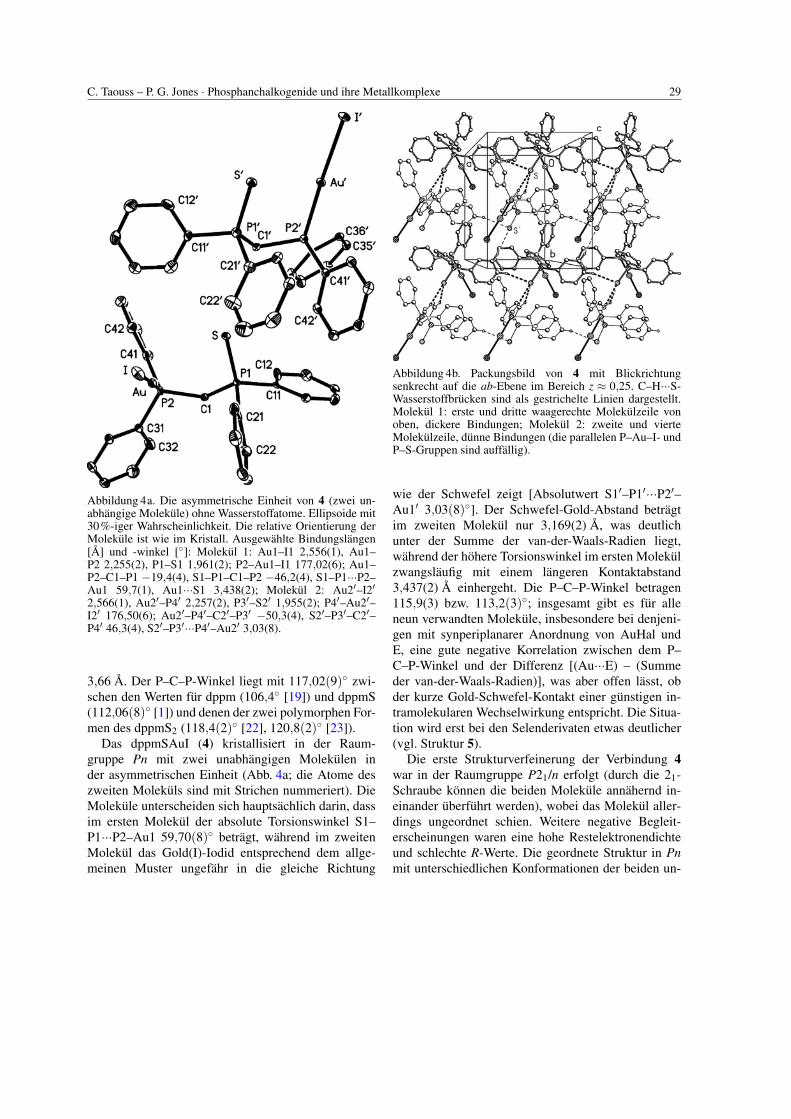
C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 29
Abbildung 4a. Die asymmetrische Einheit von 4 (zwei un-abhangige Molekule) ohne Wasserstoffatome. Ellipsoide mit30%-iger Wahrscheinlichkeit. Die relative Orientierung derMolekule ist wie im Kristall. Ausgewahlte Bindungslangen[A] und -winkel [◦]: Molekul 1: Au1–I1 2,556(1), Au1–P2 2,255(2), P1–S1 1,961(2); P2–Au1–I1 177,02(6); Au1–P2–C1–P1 −19,4(4), S1–P1–C1–P2 −46,2(4), S1–P1···P2–Au1 59,7(1), Au1···S1 3,438(2); Molekul 2: Au2′–I2′
2,566(1), Au2′–P4′ 2,257(2), P3′–S2′ 1,955(2); P4′–Au2′–I2′ 176,50(6); Au2′–P4′–C2′–P3′ −50,3(4), S2′–P3′–C2′–P4′ 46,3(4), S2′–P3′···P4′–Au2′ 3,03(8).
3,66 A. Der P–C–P-Winkel liegt mit 117,02(9)◦ zwi-schen den Werten fur dppm (106,4◦ [19]) und dppmS(112,06(8)◦ [1]) und denen der zwei polymorphen For-men des dppmS2 (118,4(2)◦ [22], 120,8(2)◦ [23]).
Das dppmSAuI (4) kristallisiert in der Raum-gruppe Pn mit zwei unabhangigen Molekulen inder asymmetrischen Einheit (Abb. 4a; die Atome deszweiten Molekuls sind mit Strichen nummeriert). DieMolekule unterscheiden sich hauptsachlich darin, dassim ersten Molekul der absolute Torsionswinkel S1–P1···P2–Au1 59,70(8)◦ betragt, wahrend im zweitenMolekul das Gold(I)-Iodid entsprechend dem allge-meinen Muster ungefahr in die gleiche Richtung
Abbildung 4b. Packungsbild von 4 mit Blickrichtungsenkrecht auf die ab-Ebene im Bereich z ≈ 0,25. C–H···S-Wasserstoffbrucken sind als gestrichelte Linien dargestellt.Molekul 1: erste und dritte waagerechte Molekulzeile vonoben, dickere Bindungen; Molekul 2: zweite und vierteMolekulzeile, dunne Bindungen (die parallelen P–Au–I- undP–S-Gruppen sind auffallig).
wie der Schwefel zeigt [Absolutwert S1′–P1′···P2′–Au1′ 3,03(8)◦]. Der Schwefel-Gold-Abstand betragtim zweiten Molekul nur 3,169(2) A, was deutlichunter der Summe der van-der-Waals-Radien liegt,wahrend der hohere Torsionswinkel im ersten Molekulzwangslaufig mit einem langeren Kontaktabstand3,437(2) A einhergeht. Die P–C–P-Winkel betragen115,9(3) bzw. 113,2(3)◦; insgesamt gibt es fur alleneun verwandten Molekule, insbesondere bei denjeni-gen mit synperiplanarer Anordnung von AuHal undE, eine gute negative Korrelation zwischen dem P–C–P-Winkel und der Differenz [(Au···E) – (Summeder van-der-Waals-Radien)], was aber offen lasst, obder kurze Gold-Schwefel-Kontakt einer gunstigen in-tramolekularen Wechselwirkung entspricht. Die Situa-tion wird erst bei den Selenderivaten etwas deutlicher(vgl. Struktur 5).
Die erste Strukturverfeinerung der Verbindung 4war in der Raumgruppe P21/n erfolgt (durch die 21-Schraube konnen die beiden Molekule annahernd in-einander uberfuhrt werden), wobei das Molekul aller-dings ungeordnet schien. Weitere negative Begleit-erscheinungen waren eine hohe Restelektronendichteund schlechte R-Werte. Die geordnete Struktur in Pnmit unterschiedlichen Konformationen der beiden un-
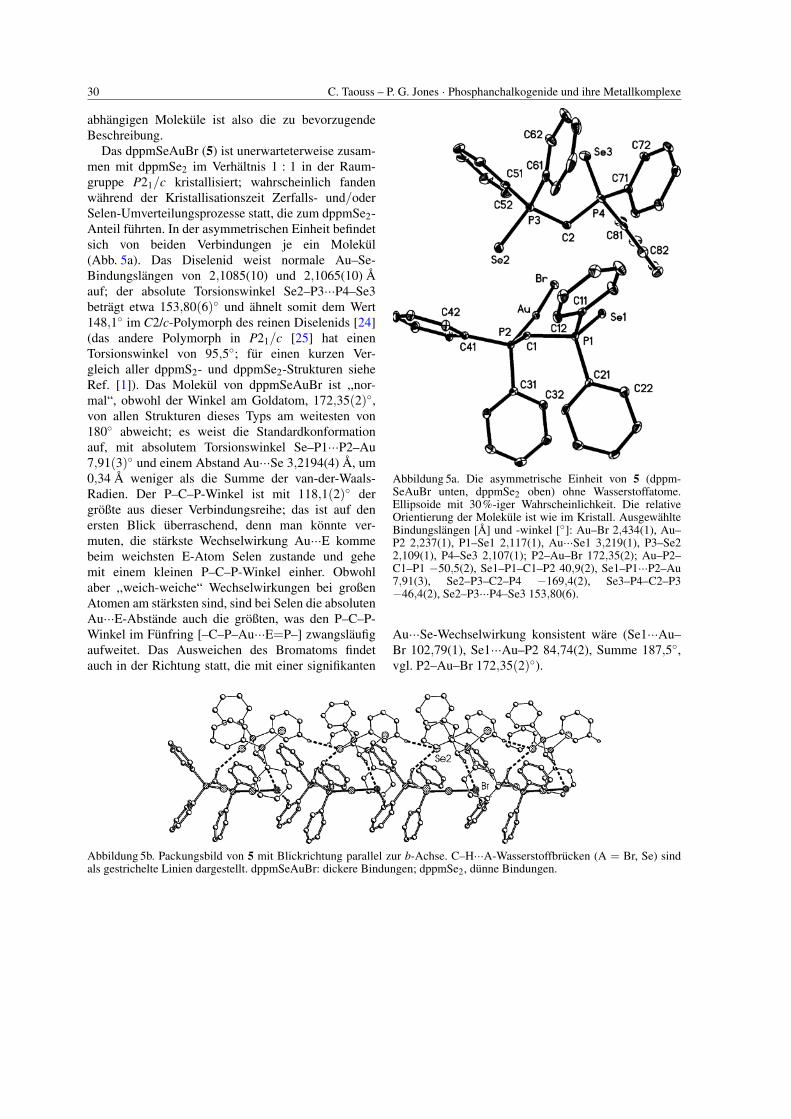
30 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
abhangigen Molekule ist also die zu bevorzugendeBeschreibung.
Das dppmSeAuBr (5) ist unerwarteterweise zusam-men mit dppmSe2 im Verhaltnis 1 : 1 in der Raum-gruppe P21/c kristallisiert; wahrscheinlich fandenwahrend der Kristallisationszeit Zerfalls- und/oderSelen-Umverteilungsprozesse statt, die zum dppmSe2-Anteil fuhrten. In der asymmetrischen Einheit befindetsich von beiden Verbindungen je ein Molekul(Abb. 5a). Das Diselenid weist normale Au–Se-Bindungslangen von 2,1085(10) und 2,1065(10) Aauf; der absolute Torsionswinkel Se2–P3···P4–Se3betragt etwa 153,80(6)◦ und ahnelt somit dem Wert148,1◦ im C2/c-Polymorph des reinen Diselenids [24](das andere Polymorph in P21/c [25] hat einenTorsionswinkel von 95,5◦; fur einen kurzen Ver-gleich aller dppmS2- und dppmSe2-Strukturen sieheRef. [1]). Das Molekul von dppmSeAuBr ist ,,nor-mal“, obwohl der Winkel am Goldatom, 172,35(2)◦,von allen Strukturen dieses Typs am weitesten von180◦ abweicht; es weist die Standardkonformationauf, mit absolutem Torsionswinkel Se–P1···P2–Au7,91(3)◦ und einem Abstand Au···Se 3,2194(4) A, um0,34 A weniger als die Summe der van-der-Waals-Radien. Der P–C–P-Winkel ist mit 118,1(2)◦ dergroßte aus dieser Verbindungsreihe; das ist auf denersten Blick uberraschend, denn man konnte ver-muten, die starkste Wechselwirkung Au···E kommebeim weichsten E-Atom Selen zustande und gehemit einem kleinen P–C–P-Winkel einher. Obwohlaber ,,weich-weiche“ Wechselwirkungen bei großenAtomen am starksten sind, sind bei Selen die absolutenAu···E-Abstande auch die großten, was den P–C–P-Winkel im Funfring [–C–P–Au···E=P–] zwangslaufigaufweitet. Das Ausweichen des Bromatoms findetauch in der Richtung statt, die mit einer signifikanten
Abbildung 5b. Packungsbild von 5 mit Blickrichtung parallel zur b-Achse. C–H···A-Wasserstoffbrucken (A = Br, Se) sindals gestrichelte Linien dargestellt. dppmSeAuBr: dickere Bindungen; dppmSe2, dunne Bindungen.
Abbildung 5a. Die asymmetrische Einheit von 5 (dppm-SeAuBr unten, dppmSe2 oben) ohne Wasserstoffatome.Ellipsoide mit 30%-iger Wahrscheinlichkeit. Die relativeOrientierung der Molekule ist wie im Kristall. AusgewahlteBindungslangen [A] und -winkel [◦]: Au–Br 2,434(1), Au–P2 2,237(1), P1–Se1 2,117(1), Au···Se1 3,219(1), P3–Se22,109(1), P4–Se3 2,107(1); P2–Au–Br 172,35(2); Au–P2–C1–P1 −50,5(2), Se1–P1–C1–P2 40,9(2), Se1–P1···P2–Au7,91(3), Se2–P3–C2–P4 −169,4(2), Se3–P4–C2–P3−46,4(2), Se2–P3···P4–Se3 153,80(6).
Au···Se-Wechselwirkung konsistent ware (Se1···Au–Br 102,79(1), Se1···Au–P2 84,74(2), Summe 187,5◦,vgl. P2–Au–Br 172,35(2)◦).

C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 31
Abbildung 6a. Das Molekul von 6 im Kristall. Ellipsoide mit50%-iger Wahrscheinlichkeit. Ausgewahlte Bindungslangen[A] und -winkel [◦]: Au–I 2,563(1), Au–P2 2,256(3), P1–Se 2,115(3); P2–Au–I 177,76(7); Au–P2–C1–P1 −18,5(6),Se–P1–C1–P2 −44,3(6), Se–P1···P2–Au 57,45(9), Au···Se3,434(2).
Abbildung 6b. Packungsbild von 6 mit Blickrichtung senk-recht auf die ab-Ebene im Bereich z ≈ 0,25. C–H···Se-Wasserstoffbrucken sind als gestrichelte Linien dargestellt.
Das dppmSeAuI (6) kristallisiert in der RaumgruppeP21/n mit einem Molekul in der asymmetrischen Ein-heit (Abb. 6a). Im Gegensatz zum analogen Chlo-rid [13] liegt keine synperiplanare Konformation vor,sondern der Torsionswinkel Se–P1···P2–Au betragt57,45(9)◦, analog dem ersten Molekul von dppmSAuI4. Der Selen-Gold-Abstand von 3,4336(11) A ist etwa0,1 A kurzer als die Summe der van-der-Waals-Radien,was auch beim großeren Torsionswinkel und einemSe–Au–I-Winkel nahe 180◦ auf eine intramolekulare
Wechselwirkung zwischen den beiden Atomen hin-deuten konnte.
Bei den Molekulpackungen von 1–6 hatten wirerwartet, die leicht sauren Wasserstoffatome derMethylengruppe konnten eine Rolle als Wasser-stoffbrucken-Donatoren spielen, denn diese Eigen-schaft vom dppm-Molekul und ahnlichen Liganden istin der Literatur bekannt [26]. Tatsachlich bilden dieMolekule von 1 Strange parallel zur kurzen b-Achse(Abb. 1b), in denen jedes Sauerstoffatom vom benach-barten, uber den b-Gleitspiegeloperator verwandtenMolekul drei H-Brucken akzeptiert (ein dreifach gega-beltes System); die Brucke vom Methylenwasser-stoffatom H1B ist mit nur 2,18 A [27] extremkurz, wahrend die anderen Brucken von ortho-Wasserstoffatomen der Ph2PO-Gruppe ausgehen undetwa 2,5 A lang sind. Es gibt vier Strange pro El-ementarzelle. Ahnliche Strange, aber mit dreifachenS-Akzeptoren, wurden bei dppmS beobachtet [1].Auch die Diisopropylether-Molekule von 1 akzep-tieren zwei C–H···O-Brucken; diese sind aber wegender Ubersichtlichkeit in Abb. 1b nicht dargestellt.
Die Molekulpackung von 2 (Abb. 2b) ahnelt invielerlei Hinsicht der von 1. Beide Zellen sind or-thorhombisch, mit in etwa gleich langen a- und b-Achsen (die langere c-Achse von 1 ist vielleichtdem großeren Losungsmittelmolekul zuzuschreiben).Auch bei 2 bilden sich parallel zur kurzen b-Achsevier Molekulstrange pro Zelle; im Strang benach-barte Molekule sind uber die b-Gleitspiegelebenemiteinander verwandt. Das Sauerstoffatom akzeptiertwieder drei C–H···O-Brucken, von denen die uber einMethylenwasserstoffatom gebildete Brucke mit 2,19 Asehr kurz ist; die anderen Brucken gehen im Gegen-satz zu 1 von ortho-Wasserstoffatomen der Ph2PAuI-Gruppe aus. Die Orientierung der Molekule in denStrangen von 1 und 2 ist ebenfalls unterschiedlich, undbei 2 ist auch das zweite Methylenwasserstoffatom aneiner Brucke beteiligt, namlich zum Iodatom desselbenNachbarmolekuls (H···I 3,03 A, C–H···I 157◦).
Bei der Packung von Verbindung 3 geht derkurzeste Kontakt wieder von einem Methylenwasser-stoffatom aus; H1A···Cl 2,62 A. Der Doppelkontakt(H24, H25)···(Cl, Au), der die Molekule zu inversions-symmetrischen Paaren (Abb. 3b) verbindet, ist auchauffallig, selbst wenn die Abstande (2,96, 3,10 A)lang sind. Vor einiger Zeit berichteten wir uber einenExtremfall mit H···Au 2,76 und H···Cl 2,89 A [28].Eine Suche der Cambridge-Datenbank [29] ergab 49

32 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
solche Treffer mit H···Au < 3,2 und H···Cl < 2,95 A.Das Schwefelatom ist an keinen kurzen Kontaktenbeteiligt.
Die Molekule von Verbindung 4 bilden Schichtenparallel zur ab-Ebene, in denen die Hauptkontakte C–H···S-Brucken sind. Das Schwefelatom von Molekul1, mit Torsionswinkel S1–P1···P2–Au1 59,70(8)◦, istetwas exponierter und kann drei Brucken bilden (ein-schließlich der kurzen Brucke H1A′···S, 2,70 A); S′,ekliptisch zu Au′, bildet nur zwei wesentlich langereBrucken mit Langen 2,92, 2,97 A.
Die Packungen von 4 und 6 sind sehr ahnlich; in derStruktur von 6 (Raumgruppe P21/n) gibt es jedoch nurein unabhangiges Molekul, so dass die waagerechtenMolekulzeilen im Packungsbild (Abb. 6b) nicht wiebei 4 alternieren, sondern alle symmetrieaquivalentsind. Das Se-Atom, analog zu S1 bei 4, akzep-tiert drei Wasserstoffbrucken, die allerdings lang sind(2,89 – 3,06 A).
Bei Verbindung 5 setzt sich das Packungsmusteraus neun C–H···A-Brucken < 3,10 A (A = Br, Se)zusammen, was zu einem sehr unubersichtlichenGesamtbild fuhrt. Vier der funf kurzeren Brucken (<2,85 A), von denen je zwei vom Bromatom bzw. vonSe2 akzeptiert werden und zwei von Methylenwasser-stoffatomen ausgehen, bilden gemeinsam Strangeparallel zur c-Achse (Abb. 5b). Ein Au···Se2-Kontaktvon 3,8066(4) A, der als Grenzfall einer ,,weich-weich“-Wechselwirkung angesehen werden kann, istin den Abb. 5a und 5b implizit zu erkennen. DiesesPackungsbild zeigt jedoch, dass die Ansichtsweise beiPackungsbildern im Allgemeinen subjektiv werdenkann. Dance [30] hat darauf hingewiesen, dass diekurzesten und auffalligsten Kontakte nicht unbedingtdie energetisch wichtigsten fur den Zusammenhaltder Struktur sein mussen; vielmehr kann sich dieGesamtpackungsenergie großtenteils aus der Summeeiner Vielzahl schwacherer Wechselwirkungen(Dispersionswechselwirkungen usw.) zusammenset-zen. In diesem Manuskript haben wir tatsachlicheher die kurzesten Kontakte diskutiert (und sind z. B.auf eine Vielzahl C–H···π-Wechselwirkungen nicht
P–C–C–P E–P···P–Au Au···Au′
dppeOAuBr 7 165,7(2), 169,0(2) 167,87(12), 165,71(12) 3,2362(3)dppeOAuI 8 166,01(13), 169,68(13) 166,51(8), 165,81(8) 3,2745(3)dppeSAuBr 9 166,09(12), 170,93(13) 176,30(3), 176,58(3) 3,2666(2)dppeSAuI 10 166,23(12), 171,30(13) 176,65(3), 176,76(3) 3,3386(2)
a Alle Strukturen mit zwei unabhangigen Molekulen.
Tabelle 2 . Ausgewahlte Torsionswinkel[◦] und Au···Au-Kontakte [A] indppeEAuHala.
Abbildung 7a. Asymmetrische Einheit von 7 ohne Wasser-stoffatome (außer am Wassermolekul). Ellipsoide mit 30%-iger Wahrscheinlichkeit. Ausgewahlte Bindungslangen [A]und -winkel [◦]: Au–Br 2,4120(4), Au–P2 2,2508(11), P1–O1,490(3); P2–Au–Br 173,26(3); P1–C1–C2–P2 165,7(2),O–P1–C1–C2 −61,1(3), Au–P2–C2–C1 60,8(3), O–P1···P2–Au 167,87(12); Au′–Br′ 2,4141(4), Au′–P2′
2,2466(10), P1′–O′ 1,482(3); P2′–Au′–Br′ 172,27(3);P1′–C1′–C2′–P2′ 169,0(2), O′–P1′–C1′–C2′ −62,8(3),Au′–P2′–C2′–C1′ 56,6(3), O′–P1′···P2′–Au′ 165,71(12);Au···Au′ 3,2362(3).
naher eingegangen), aus der praktischen Uberlegung,dass die phanomenologische Beschreibung einesPackungsbilds so leichter und ubersichtlicher wird.
dppeEAuHal [dppe = 1,2-Bis(diphenylphosphano)-ethan; E = O, Hal = Br (7), I (8); E = S, Hal = Br (9),I (10)]
Tabelle 2 fasst einige Strukturparameter derVerbindungen dppeEAuHal zusammen. DieKristallstrukturen der Chloridkomplexe sind bereits
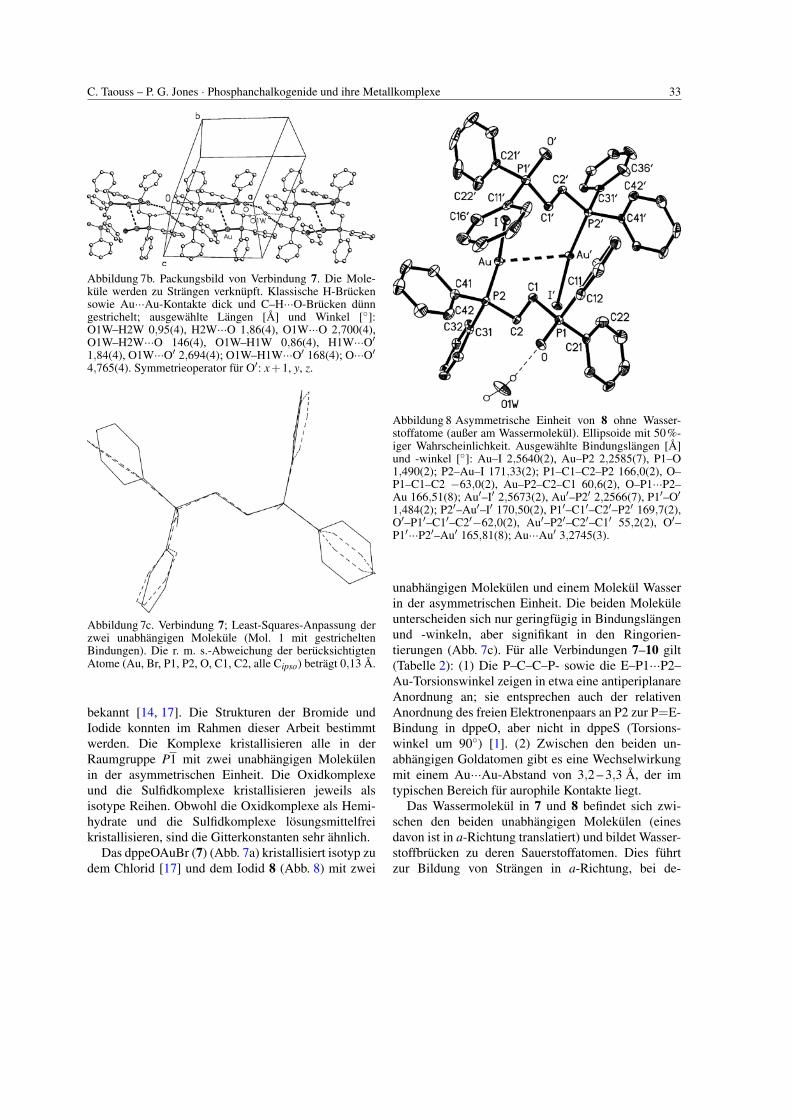
C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 33
Abbildung 7b. Packungsbild von Verbindung 7. Die Mole-kule werden zu Strangen verknupft. Klassische H-Bruckensowie Au···Au-Kontakte dick und C–H···O-Brucken dunngestrichelt; ausgewahlte Langen [A] und Winkel [◦]:O1W–H2W 0,95(4), H2W···O 1,86(4), O1W···O 2,700(4),O1W–H2W···O 146(4), O1W–H1W 0,86(4), H1W···O′1,84(4), O1W···O′ 2,694(4); O1W–H1W···O′ 168(4); O···O′4,765(4). Symmetrieoperator fur O′: x+1, y, z.
Abbildung 7c. Verbindung 7; Least-Squares-Anpassung derzwei unabhangigen Molekule (Mol. 1 mit gestricheltenBindungen). Die r. m. s.-Abweichung der berucksichtigtenAtome (Au, Br, P1, P2, O, C1, C2, alle Cipso) betragt 0,13 A.
bekannt [14, 17]. Die Strukturen der Bromide undIodide konnten im Rahmen dieser Arbeit bestimmtwerden. Die Komplexe kristallisieren alle in derRaumgruppe P1 mit zwei unabhangigen Molekulenin der asymmetrischen Einheit. Die Oxidkomplexeund die Sulfidkomplexe kristallisieren jeweils alsisotype Reihen. Obwohl die Oxidkomplexe als Hemi-hydrate und die Sulfidkomplexe losungsmittelfreikristallisieren, sind die Gitterkonstanten sehr ahnlich.
Das dppeOAuBr (7) (Abb. 7a) kristallisiert isotyp zudem Chlorid [17] und dem Iodid 8 (Abb. 8) mit zwei
Abbildung 8. Asymmetrische Einheit von 8 ohne Wasser-stoffatome (außer am Wassermolekul). Ellipsoide mit 50%-iger Wahrscheinlichkeit. Ausgewahlte Bindungslangen [A]und -winkel [◦]: Au–I 2,5640(2), Au–P2 2,2585(7), P1–O1,490(2); P2–Au–I 171,33(2); P1–C1–C2–P2 166,0(2), O–P1–C1–C2 −63,0(2), Au–P2–C2–C1 60,6(2), O–P1···P2–Au 166,51(8); Au′–I′ 2,5673(2), Au′–P2′ 2,2566(7), P1′–O′
1,484(2); P2′–Au′–I′ 170,50(2), P1′–C1′–C2′–P2′ 169,7(2),O′–P1′–C1′–C2′−62,0(2), Au′–P2′–C2′–C1′ 55,2(2), O′–P1′···P2′–Au′ 165,81(8); Au···Au′ 3,2745(3).
unabhangigen Molekulen und einem Molekul Wasserin der asymmetrischen Einheit. Die beiden Molekuleunterscheiden sich nur geringfugig in Bindungslangenund -winkeln, aber signifikant in den Ringorien-tierungen (Abb. 7c). Fur alle Verbindungen 7–10 gilt(Tabelle 2): (1) Die P–C–C–P- sowie die E–P1···P2–Au-Torsionswinkel zeigen in etwa eine antiperiplanareAnordnung an; sie entsprechen auch der relativenAnordnung des freien Elektronenpaars an P2 zur P=E-Bindung in dppeO, aber nicht in dppeS (Torsions-winkel um 90◦) [1]. (2) Zwischen den beiden un-abhangigen Goldatomen gibt es eine Wechselwirkungmit einem Au···Au-Abstand von 3,2 – 3,3 A, der imtypischen Bereich fur aurophile Kontakte liegt.
Das Wassermolekul in 7 und 8 befindet sich zwi-schen den beiden unabhangigen Molekulen (einesdavon ist in a-Richtung translatiert) und bildet Wasser-stoffbrucken zu deren Sauerstoffatomen. Dies fuhrtzur Bildung von Strangen in a-Richtung, bei de-
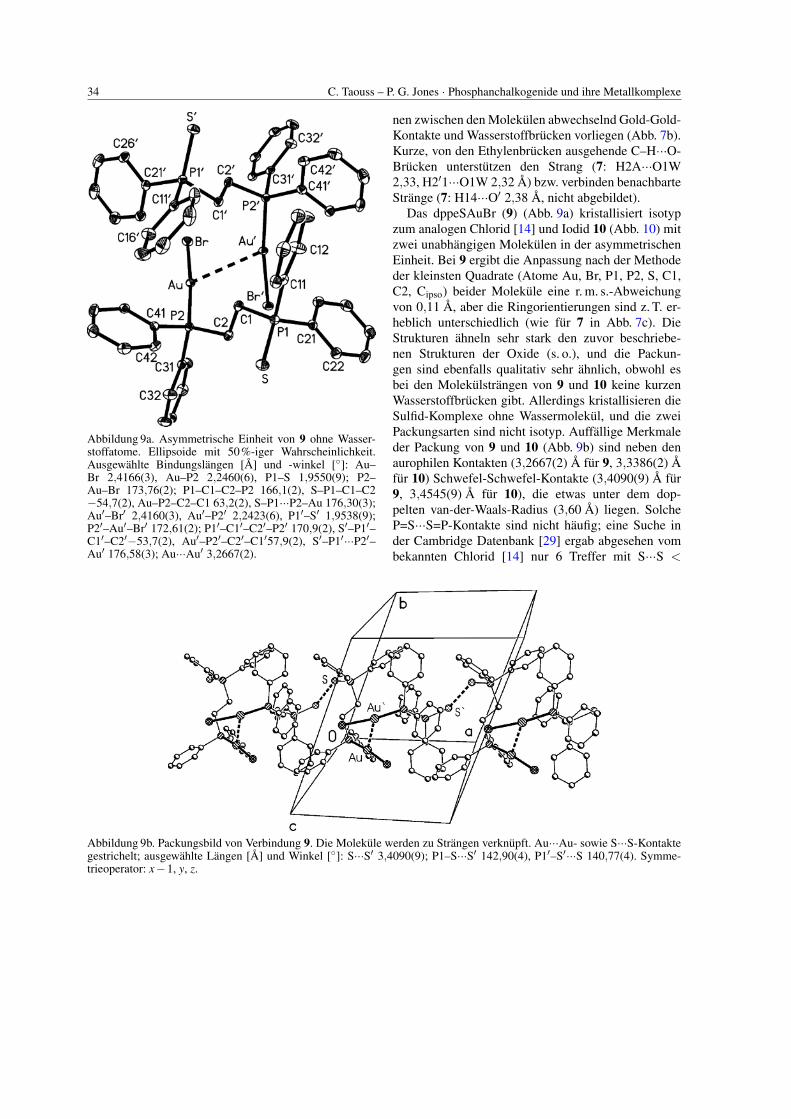
34 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
Abbildung 9a. Asymmetrische Einheit von 9 ohne Wasser-stoffatome. Ellipsoide mit 50%-iger Wahrscheinlichkeit.Ausgewahlte Bindungslangen [A] und -winkel [◦]: Au–Br 2,4166(3), Au–P2 2,2460(6), P1–S 1,9550(9); P2–Au–Br 173,76(2); P1–C1–C2–P2 166,1(2), S–P1–C1–C2−54,7(2), Au–P2–C2–C1 63,2(2), S–P1···P2–Au 176,30(3);Au′–Br′ 2,4160(3), Au′–P2′ 2,2423(6), P1′–S′ 1,9538(9);P2′–Au′–Br′ 172,61(2); P1′–C1′–C2′–P2′ 170,9(2), S′–P1′–C1′–C2′−53,7(2), Au′–P2′–C2′–C1′57,9(2), S′–P1′···P2′–Au′ 176,58(3); Au···Au′ 3,2667(2).
Abbildung 9b. Packungsbild von Verbindung 9. Die Molekule werden zu Strangen verknupft. Au···Au- sowie S···S-Kontaktegestrichelt; ausgewahlte Langen [A] und Winkel [◦]: S···S′ 3,4090(9); P1–S···S′ 142,90(4), P1′–S′···S 140,77(4). Symme-trieoperator: x−1, y, z.
nen zwischen den Molekulen abwechselnd Gold-Gold-Kontakte und Wasserstoffbrucken vorliegen (Abb. 7b).Kurze, von den Ethylenbrucken ausgehende C–H···O-Brucken unterstutzen den Strang (7: H2A···O1W2,33, H2′1···O1W 2,32 A) bzw. verbinden benachbarteStrange (7: H14···O′ 2,38 A, nicht abgebildet).
Das dppeSAuBr (9) (Abb. 9a) kristallisiert isotypzum analogen Chlorid [14] und Iodid 10 (Abb. 10) mitzwei unabhangigen Molekulen in der asymmetrischenEinheit. Bei 9 ergibt die Anpassung nach der Methodeder kleinsten Quadrate (Atome Au, Br, P1, P2, S, C1,C2, Cipso) beider Molekule eine r. m. s.-Abweichungvon 0,11 A, aber die Ringorientierungen sind z. T. er-heblich unterschiedlich (wie fur 7 in Abb. 7c). DieStrukturen ahneln sehr stark den zuvor beschriebe-nen Strukturen der Oxide (s. o.), und die Packun-gen sind ebenfalls qualitativ sehr ahnlich, obwohl esbei den Molekulstrangen von 9 und 10 keine kurzenWasserstoffbrucken gibt. Allerdings kristallisieren dieSulfid-Komplexe ohne Wassermolekul, und die zweiPackungsarten sind nicht isotyp. Auffallige Merkmaleder Packung von 9 und 10 (Abb. 9b) sind neben denaurophilen Kontakten (3,2667(2) A fur 9, 3,3386(2) Afur 10) Schwefel-Schwefel-Kontakte (3,4090(9) A fur9, 3,4545(9) A fur 10), die etwas unter dem dop-pelten van-der-Waals-Radius (3,60 A) liegen. SolcheP=S···S=P-Kontakte sind nicht haufig; eine Suche inder Cambridge Datenbank [29] ergab abgesehen vombekannten Chlorid [14] nur 6 Treffer mit S···S <

C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 35
Abbildung 10. Die asymmetrische Einheit von 10 ohneWasserstoffatome. Ellipsoide mit 50%-iger Wahrschein-lichkeit. Ausgewahlte Bindungslangen [A] und -winkel[◦]: Au–I 2,5722(2), Au–P2 2,2598(6), P1–S 1,9526(9);P2–Au–I 172,45(2); P1–C1–C2–P2 166,2(2), S–P1–C1–C2−54,1(2), Au–P2–C2–C1 62,7(2), S–P1···P2–Au 176,65(3);Au′–I′ 2,5685(2), Au‘–P2′ 2,2569(6), P1′–S′ 1,9543(9);P2′–Au′–I′ 171,29(2), P1′–C1′–C2′–P2′ 171,3(2), S′–P1′–C1′–C2′–53,5(2), Au′–P2′–C2′–C1′ 57,8(2), S′–P1′···P2′–Au′ 176,76(3); Au···Au′ 3,3386(2).
3,6 A. Bei den Oxid-Komplexen 7 und 8 betragtder Abstand zwischen den Sauerstoffatomen benach-barter Molekule etwa 4,8 A; in dieser ,,Lucke“ kannalso ein Wassermolekul mitkristallisieren und Wasser-stoffbrucken bilden, was bei den großeren Schwefel-atomen von 9 und 10 nicht der Fall ist. Das Wasserscheint die Packung der Molekule also wenig zu be-einflussen.
Im Gegensatz zu den Komplexen mit dppm gibt esbei den Komplexen mit dppe also keine intramoleku-laren Chalkogen-Gold-Wechselwirkungen, sondernaurophile Wechselwirkungen zwischen verschiedenenMolekulen, die zu Molekulstrangen fuhren. DiesesPackungsmuster scheint hier so vorteilhaft zu sein,dass es in allen sechs Strukturen vorliegt.
dppbzEAuHal [dppbz = 1,2-Bis(diphenylphosphano)-benzol; E = O, Hal = Cl (11), Br (12); E = S, Hal =Br (13), I (14); E = Se, Hal = Cl (15), Br (16), I (17)]
Tabelle 3 fasst einige Strukturparameter derVerbindungen dppbzEAuHal zusammen. Das dppbz-
Tabelle 3. Ausgewahlte Abstande [A], Winkel [◦] und Tor-sionswinkel [◦] in dppbzEAuHal.
Au···E P–Au–Hal P–C–C–P E–P···P–AudppbzOAuCl 11 3,051(5) 179,00(6) 4,9(7) −69,8(2)dppbzOAuBr 12 2,992(2) 179,10(3) 15,6(4) 53,6(1)dppbzSAuBr 13 3,1479(10) 171,77(3) −2,7(5) 66,28(4)dppbzSAuI 14 3,2304(13) 169,27(3) 2,0(6) 69,85(6)dppbzSeAuCl 15 3,2147(4) 172,01(3) −4,4(4) 65,13(3)dppbzSeAuBr 16 3,2428(5) 169,92(3) −0,7(6) 67,78(4)dppbzSeAuI 17 3,2162(3) 168,36(2) −1,0(4) 67,05(3)
Gerust unterliegt einigen geometrischen Ein-schrankungen, die bei dppe nicht gelten. Erstens istdas P–C–C–P-Ruckgrat starr, mit einem P···P-Abstandum 3,4 – 3,5 A. Zweitens kann der E–P···P–Au-Torsionswinkel keine Werte um 0◦ aufweisen (undsomit konnen die Vektoren P1–E und P2–Au–Halnicht parallel sein), sonst kame es zu Ringkol-lisionen; tatsachlich liegen diese Torsionswinkelbis auf Struktur 12 (53,6◦) alle im absoluten Be-reich 60 – 70◦. Diese Faktoren fuhren dazu, dassdie Molekulstrukturen 11–17 bei Weitem nichtso konformativ unterschiedlich ausfallen wie diedppm-Strukturen 1–6. Einige der Strukturen sindisotyp zueinander; zum einen sind das dppbzSAuI(14), dppbzSeAuBr (16) und dppbzSeAuI (17), diealle mit zwei Aquivalenten Dichlormethan in derRaumgruppe P21/n kristallisieren. Außerdem sind dieStrukturen von dppbzSAuBr (13) und dppbzSeAuCl(15) in P21/c isotyp. Bei den Gitterkonstanten lassensich allerdings Ahnlichkeiten zwischen allen funfStrukturen 13–17 erkennen. Der einzige großereUnterschied ist, dass die ersten drei StrukturenDichlormethan-Solvate sind und eine langere c-Achsehaben.
Das dppbzOAuCl (11) kristallisiert in der Raum-gruppe P21/c mit einem Molekul in der asym-metrischen Einheit (Abb. 11). Das Sauerstoffatomder P=O-Gruppe liegt erheblich außerhalb der zen-tralen Ringebene, die AuCl-Gruppe ist auf der an-deren Seite zu finden (O–P1–C11–C12 −23,5(6),Au–P2–C12–C11 −57,7(5)◦). Der Au···O-Abstandliegt mit 3,051(5) A unter der Summe der van-der-Waals-Radien (3,18 A) der beiden Atome. Es konntealso eine schwache Gold-Sauerstoff-Wechselwirkunggeben, auch wenn die Linearitat am Goldatom nichtdavon beeinflusst wird. Die intramolekularen KontakteO···P2 und O···C51 sind mit 2,941(5) bzw. 2,881(8) Aebenfalls kurz.
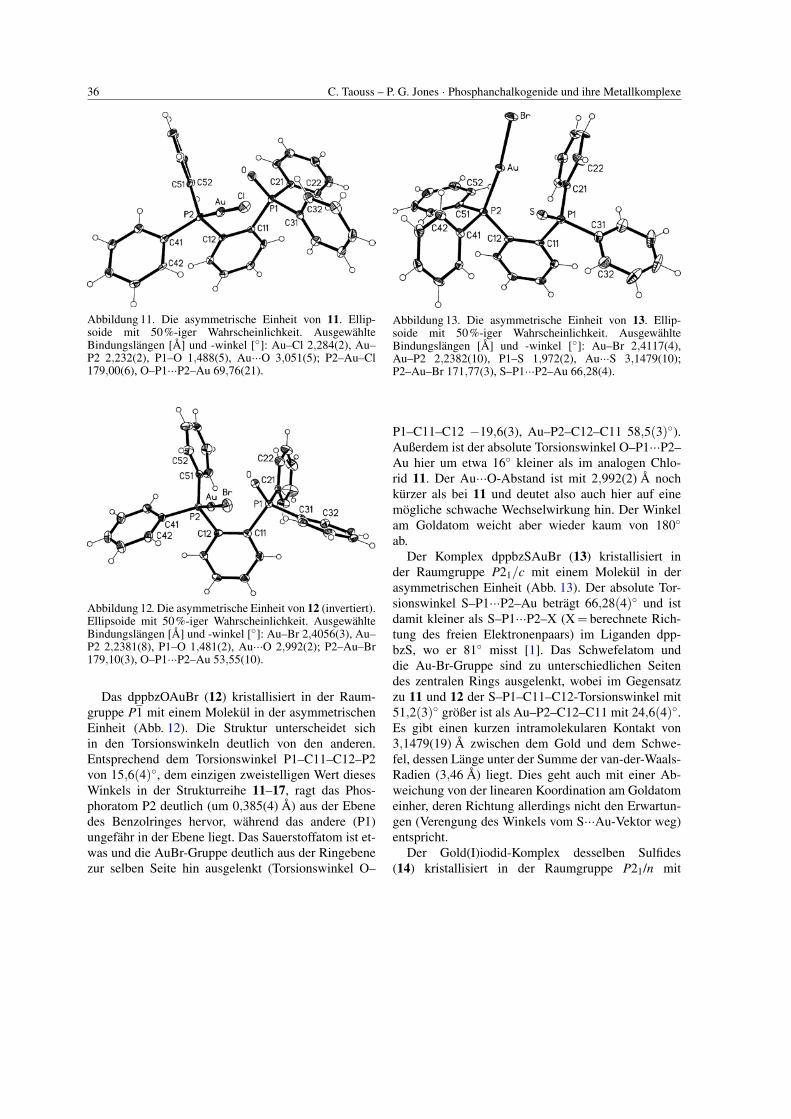
36 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
Abbildung 11. Die asymmetrische Einheit von 11. Ellip-soide mit 50%-iger Wahrscheinlichkeit. AusgewahlteBindungslangen [A] und -winkel [◦]: Au–Cl 2,284(2), Au–P2 2,232(2), P1–O 1,488(5), Au···O 3,051(5); P2–Au–Cl179,00(6), O–P1···P2–Au 69,76(21).
Abbildung 12. Die asymmetrische Einheit von 12 (invertiert).Ellipsoide mit 50%-iger Wahrscheinlichkeit. AusgewahlteBindungslangen [A] und -winkel [◦]: Au–Br 2,4056(3), Au–P2 2,2381(8), P1–O 1,481(2), Au···O 2,992(2); P2–Au–Br179,10(3), O–P1···P2–Au 53,55(10).
Das dppbzOAuBr (12) kristallisiert in der Raum-gruppe P1 mit einem Molekul in der asymmetrischenEinheit (Abb. 12). Die Struktur unterscheidet sichin den Torsionswinkeln deutlich von den anderen.Entsprechend dem Torsionswinkel P1–C11–C12–P2von 15,6(4)◦, dem einzigen zweistelligen Wert diesesWinkels in der Strukturreihe 11–17, ragt das Phos-phoratom P2 deutlich (um 0,385(4) A) aus der Ebenedes Benzolringes hervor, wahrend das andere (P1)ungefahr in der Ebene liegt. Das Sauerstoffatom ist et-was und die AuBr-Gruppe deutlich aus der Ringebenezur selben Seite hin ausgelenkt (Torsionswinkel O–
Abbildung 13. Die asymmetrische Einheit von 13. Ellip-soide mit 50%-iger Wahrscheinlichkeit. AusgewahlteBindungslangen [A] und -winkel [◦]: Au–Br 2,4117(4),Au–P2 2,2382(10), P1–S 1,972(2), Au···S 3,1479(10);P2–Au–Br 171,77(3), S–P1···P2–Au 66,28(4).
P1–C11–C12 −19,6(3), Au–P2–C12–C11 58,5(3)◦).Außerdem ist der absolute Torsionswinkel O–P1···P2–Au hier um etwa 16◦ kleiner als im analogen Chlo-rid 11. Der Au···O-Abstand ist mit 2,992(2) A nochkurzer als bei 11 und deutet also auch hier auf einemogliche schwache Wechselwirkung hin. Der Winkelam Goldatom weicht aber wieder kaum von 180◦
ab.Der Komplex dppbzSAuBr (13) kristallisiert in
der Raumgruppe P21/c mit einem Molekul in derasymmetrischen Einheit (Abb. 13). Der absolute Tor-sionswinkel S–P1···P2–Au betragt 66,28(4)◦ und istdamit kleiner als S–P1···P2–X (X = berechnete Rich-tung des freien Elektronenpaars) im Liganden dpp-bzS, wo er 81◦ misst [1]. Das Schwefelatom unddie Au-Br-Gruppe sind zu unterschiedlichen Seitendes zentralen Rings ausgelenkt, wobei im Gegensatzzu 11 und 12 der S–P1–C11–C12-Torsionswinkel mit51,2(3)◦ großer ist als Au–P2–C12–C11 mit 24,6(4)◦.Es gibt einen kurzen intramolekularen Kontakt von3,1479(19) A zwischen dem Gold und dem Schwe-fel, dessen Lange unter der Summe der van-der-Waals-Radien (3,46 A) liegt. Dies geht auch mit einer Ab-weichung von der linearen Koordination am Goldatomeinher, deren Richtung allerdings nicht den Erwartun-gen (Verengung des Winkels vom S···Au-Vektor weg)entspricht.
Der Gold(I)iodid-Komplex desselben Sulfides(14) kristallisiert in der Raumgruppe P21/n mit
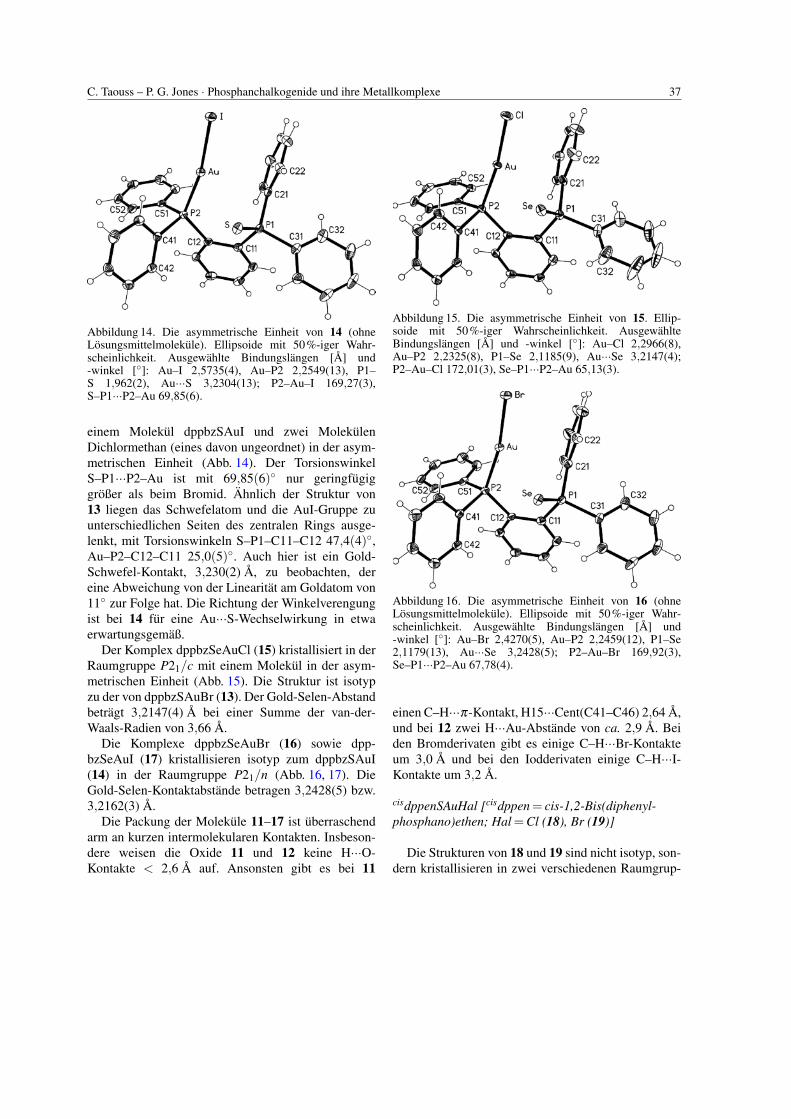
C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 37
Abbildung 14. Die asymmetrische Einheit von 14 (ohneLosungsmittelmolekule). Ellipsoide mit 50%-iger Wahr-scheinlichkeit. Ausgewahlte Bindungslangen [A] und-winkel [◦]: Au–I 2,5735(4), Au–P2 2,2549(13), P1–S 1,962(2), Au···S 3,2304(13); P2–Au–I 169,27(3),S–P1···P2–Au 69,85(6).
einem Molekul dppbzSAuI und zwei MolekulenDichlormethan (eines davon ungeordnet) in der asym-metrischen Einheit (Abb. 14). Der TorsionswinkelS–P1···P2–Au ist mit 69,85(6)◦ nur geringfugiggroßer als beim Bromid. Ahnlich der Struktur von13 liegen das Schwefelatom und die AuI-Gruppe zuunterschiedlichen Seiten des zentralen Rings ausge-lenkt, mit Torsionswinkeln S–P1–C11–C12 47,4(4)◦,Au–P2–C12–C11 25,0(5)◦. Auch hier ist ein Gold-Schwefel-Kontakt, 3,230(2) A, zu beobachten, dereine Abweichung von der Linearitat am Goldatom von11◦ zur Folge hat. Die Richtung der Winkelverengungist bei 14 fur eine Au···S-Wechselwirkung in etwaerwartungsgemaß.
Der Komplex dppbzSeAuCl (15) kristallisiert in derRaumgruppe P21/c mit einem Molekul in der asym-metrischen Einheit (Abb. 15). Die Struktur ist isotypzu der von dppbzSAuBr (13). Der Gold-Selen-Abstandbetragt 3,2147(4) A bei einer Summe der van-der-Waals-Radien von 3,66 A.
Die Komplexe dppbzSeAuBr (16) sowie dpp-bzSeAuI (17) kristallisieren isotyp zum dppbzSAuI(14) in der Raumgruppe P21/n (Abb. 16, 17). DieGold-Selen-Kontaktabstande betragen 3,2428(5) bzw.3,2162(3) A.
Die Packung der Molekule 11–17 ist uberraschendarm an kurzen intermolekularen Kontakten. Insbeson-dere weisen die Oxide 11 und 12 keine H···O-Kontakte < 2,6 A auf. Ansonsten gibt es bei 11
Abbildung 15. Die asymmetrische Einheit von 15. Ellip-soide mit 50%-iger Wahrscheinlichkeit. AusgewahlteBindungslangen [A] und -winkel [◦]: Au–Cl 2,2966(8),Au–P2 2,2325(8), P1–Se 2,1185(9), Au···Se 3,2147(4);P2–Au–Cl 172,01(3), Se–P1···P2–Au 65,13(3).
Abbildung 16. Die asymmetrische Einheit von 16 (ohneLosungsmittelmolekule). Ellipsoide mit 50%-iger Wahr-scheinlichkeit. Ausgewahlte Bindungslangen [A] und-winkel [◦]: Au–Br 2,4270(5), Au–P2 2,2459(12), P1–Se2,1179(13), Au···Se 3,2428(5); P2–Au–Br 169,92(3),Se–P1···P2–Au 67,78(4).
einen C–H···π-Kontakt, H15···Cent(C41–C46) 2,64 A,und bei 12 zwei H···Au-Abstande von ca. 2,9 A. Beiden Bromderivaten gibt es einige C–H···Br-Kontakteum 3,0 A und bei den Iodderivaten einige C–H···I-Kontakte um 3,2 A.
cisdppenSAuHal [cisdppen = cis-1,2-Bis(diphenyl-phosphano)ethen; Hal = Cl (18), Br (19)]
Die Strukturen von 18 und 19 sind nicht isotyp, son-dern kristallisieren in zwei verschiedenen Raumgrup-
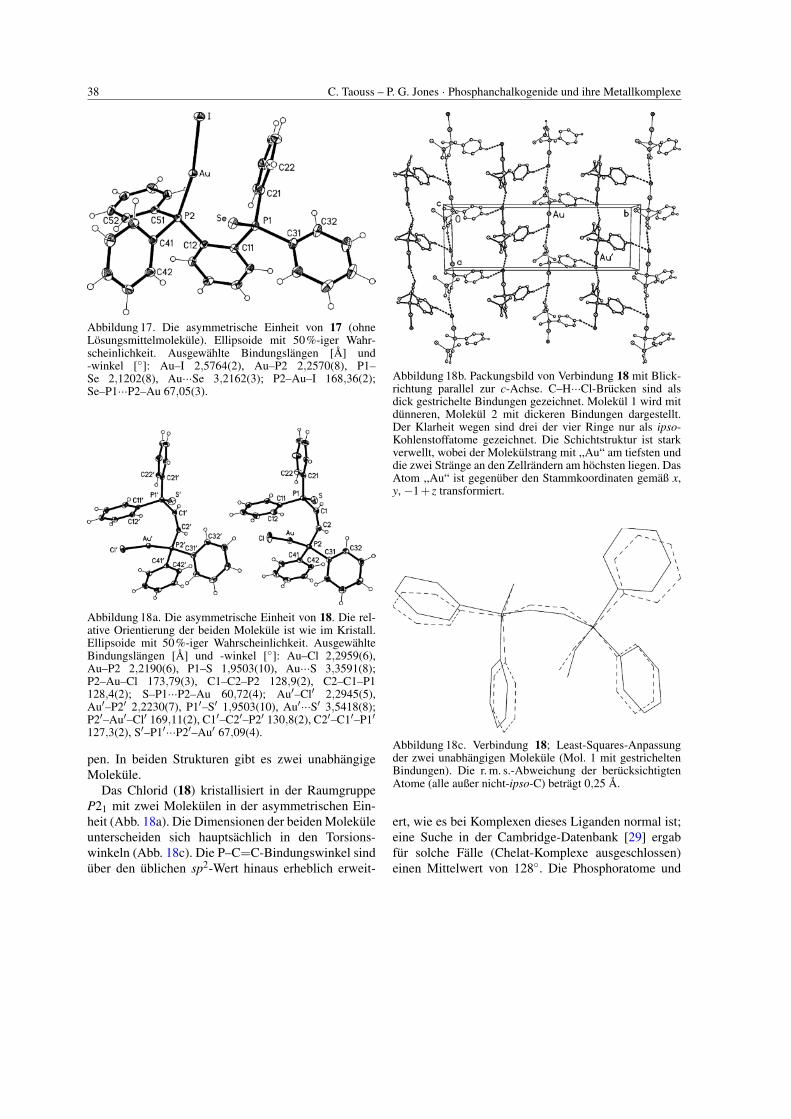
38 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
Abbildung 17. Die asymmetrische Einheit von 17 (ohneLosungsmittelmolekule). Ellipsoide mit 50%-iger Wahr-scheinlichkeit. Ausgewahlte Bindungslangen [A] und-winkel [◦]: Au–I 2,5764(2), Au–P2 2,2570(8), P1–Se 2,1202(8), Au···Se 3,2162(3); P2–Au–I 168,36(2);Se–P1···P2–Au 67,05(3).
Abbildung 18a. Die asymmetrische Einheit von 18. Die rel-ative Orientierung der beiden Molekule ist wie im Kristall.Ellipsoide mit 50%-iger Wahrscheinlichkeit. AusgewahlteBindungslangen [A] und -winkel [◦]: Au–Cl 2,2959(6),Au–P2 2,2190(6), P1–S 1,9503(10), Au···S 3,3591(8);P2–Au–Cl 173,79(3), C1–C2–P2 128,9(2), C2–C1–P1128,4(2); S–P1···P2–Au 60,72(4); Au′–Cl′ 2,2945(5),Au′–P2′ 2,2230(7), P1′–S′ 1,9503(10), Au′···S′ 3,5418(8);P2′–Au′–Cl′ 169,11(2), C1′–C2′–P2′ 130,8(2), C2′–C1′–P1′
127,3(2), S′–P1′···P2′–Au′ 67,09(4).
pen. In beiden Strukturen gibt es zwei unabhangigeMolekule.
Das Chlorid (18) kristallisiert in der RaumgruppeP21 mit zwei Molekulen in der asymmetrischen Ein-heit (Abb. 18a). Die Dimensionen der beiden Molekuleunterscheiden sich hauptsachlich in den Torsions-winkeln (Abb. 18c). Die P–C=C-Bindungswinkel sinduber den ublichen sp2-Wert hinaus erheblich erweit-
Abbildung 18b. Packungsbild von Verbindung 18 mit Blick-richtung parallel zur c-Achse. C–H···Cl-Brucken sind alsdick gestrichelte Bindungen gezeichnet. Molekul 1 wird mitdunneren, Molekul 2 mit dickeren Bindungen dargestellt.Der Klarheit wegen sind drei der vier Ringe nur als ipso-Kohlenstoffatome gezeichnet. Die Schichtstruktur ist starkverwellt, wobei der Molekulstrang mit ,,Au“ am tiefsten unddie zwei Strange an den Zellrandern am hochsten liegen. DasAtom ,,Au“ ist gegenuber den Stammkoordinaten gemaß x,y, −1+ z transformiert.
Abbildung 18c. Verbindung 18; Least-Squares-Anpassungder zwei unabhangigen Molekule (Mol. 1 mit gestricheltenBindungen). Die r. m. s.-Abweichung der berucksichtigtenAtome (alle außer nicht-ipso-C) betragt 0,25 A.
ert, wie es bei Komplexen dieses Liganden normal ist;eine Suche in der Cambridge-Datenbank [29] ergabfur solche Falle (Chelat-Komplexe ausgeschlossen)einen Mittelwert von 128◦. Die Phosphoratome und

C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 39
die Kohlenstoffe der Ethylenbrucke liegen in bei-den Molekulen ungefahr in einer Ebene, mit Tor-sionswinkeln P1–C1–C2–P2 von 5,7(4) bzw. 4,8(4)◦.Im ersten Molekul liegt auch das Goldchlorid indieser Ebene, wahrend das Schwefelatom daraus her-vorsteht (Torsionswinkel Au–P2–C2–C1 −2,6(3), S–P1–C1–C2 63,5(3)◦). Im zweiten Molekul steht dasGoldatom etwas außerhalb der Ebene der P–C=C–P-Einheit, wahrend das Schwefelatom in die an-dere Richtung aus der Ebene ragt (Au′–P2′–C2′–C1′ 13,0(3), S′–P1′–C1′–C2′ 55,8(3)◦). Bei P···P-Abstanden um 3,6 A betragen die TorsionswinkelS–P1···P2–Au 60,72(4)◦ im ersten Molekul und67,09(4)◦ im zweiten. Durch die unterschiedlichenTorsionswinkel der beiden Molekule ergibt sich einesignifikante Differenz des Gold-Schwefel-Abstands,der im ersten Molekul mit 3,3591(8) A noch unter derSumme der van-der-Waals-Radien liegt, im zweitenjedoch mit 3,5418(8) A etwas daruber liegt. Die et-was großere Verengung des Winkels am Goldatom imersten Molekul, um 11◦ in eine Richtung weg vomS···Au-Vektor, ware mit einer schwachen Wechsel-wirkung konsistent.
Das Bromid (19) kristallisiert in der RaumgruppeP21/c mit ebenfalls zwei Molekulen in der asym-metrischen Einheit (Abb. 19a). Im Gegensatz zu 18weisen die Molekule untereinander – und auch zu 18– große Konformationsunterschiede auf (die WinkelC2–C1–P1 sind mit 129,4(6), 133,6(6)◦ auch sig-nifikant unterschiedlich). Die Phosphoratome liegenerwartungsgemaß wieder in einer Ebene mit der zen-tralen Doppelbindung (Torsionswinkel P1–C1–C2–P23,6(1), 2,2(1)◦), aber weder das Goldbromid nochder Schwefel liegen in dieser Ebene. Im erstenMolekul ragen sie auf entgegengesetzten Seiten ausder Ebene heraus (S–P1–C1–C2 38,0(9), Au–P2–C2–C1 35,3(9)◦) und im zweiten Molekul auf dersel-ben Seite (S′–P1′–C1′–C2′ 22,6(9)◦, Au′–P2′–C2′–C1′ −78,3(9)◦). Eine Anpassung nach der Methodeder kleinsten Quadrate beider Molekule (Abb. 19c)macht deutlich, dass nur die Atome C1, C2, P1, P2, S,C11 und C22 in etwa konstante Positionen einnehmen.Die intramolekularen Au···S-Abstande, 3,423(3) und3,589(3) A, sind etwas großer als im Chlorid, unddie Bindungswinkel am Goldatom weichen kaum von180◦ ab, was nicht fur signifikante Schwefel-Gold-Wechselwirkungen spricht.
Die Packung von 18 (Abb. 18b) ist durch kurzeC–H···Cl-Wechselwirkungen gepragt. Vier der funf
Abbildung 19a. Die asymmetrische Einheit von 19 ohnePhenyl-H-Atome. Die relative Orientierung der beidenMolekule ist wie im Kristall. Ellipsoide mit 50%-iger Wahrscheinlichkeit. Ausgewahlte Bindungslangen [A]und -winkel [◦]: Au–Br 2,4127(9), Au–P2 2,237(2),P1–S 1,957(3), Au···S 3,423(3); P2–Au–Br 179,15(3),C1–C2–P2 129,4(6), C2–C1–P1 128,8(6); S–P1···P2–Au74,31(10); Au′–Br′ 2,4130(9), Au′–P2′ 2,238(2), P1′–S′
1,946(3), Au′···S′ 3,589(3); P2′–Au′–Cl′ 178,91(6), C1′–C2′–P2′ 133,6(6), C2′–C1′–P1′ 130,4(6); S′–P1′···P2′–Au′
40,35(11).
kurzesten sind H1···Cl 2.56, H1′···Cl′ 2.64 (jeweilsuber a-Translation, bilden Ketten aus Molekul 1bzw. Molekul 2), H44′···Cl 2.69, H44···Cl′ 2.79 A(jeweils uber die 21-Schraubenachse, verknupfen dieKetten zu gewellten Schichten). Auch drei C–H···πum 2,8 A und zwei C–H···S-Kontakte mit 2,8 und2,9 A werden beobachtet. Bei 19 sind die auffallig-sten Kontakte H1···S′ und H1′···S (2,64, 2,72 A), dievon H2···Au′ und H2′···Au (3,18, 3,08 A) begleitetsind (Abb. 19b). Zwei dieser Kontakte liegen inner-halb der asymmetrischen Einheit und konnen im-plizit in Abb. 19a erkannt werden; die anderen bei-den kommen uber b-Translation zustande, so dass dieMolekule alternierend zu Ketten parallel zur b-Achseverknupft werden. Sieben H···Br-Kontakte im Bereich2,96 – 3,10 A und ein sehr kurzer C–H···π-Abstand,H44′···Cent(C31′–36′) 2,67 A, tragen auch zur Pa-ckung bei.

40 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
Abbildung 19b. Packungsbild von Verbindung 19 mit Blick-richtung senkrecht auf die bc-Ebene. C–H···S-Brucken sindals dick und C–H···Au-Kontakte als dunn gestrichelte Liniengezeichnet. Molekul 1 wird mit dunneren, Molekul 2 mitdickeren Bindungen dargestellt. Der Klarheit wegen sind dieRinge nur als ipso-Kohlenstoffatome gezeichnet.
Abbildung 19c. Verbindung 19; Least-Squares-Anpassungder zwei unabhangigen Molekule (Mol. 1 mit gestricheltenBindungen). Die r. m. s.-Abweichung der berucksichtigtenAtome (C1, C2, C11, C21, P1, P2 S) betragt 0,19 A.
(dppnSeAu)2Cl2 (20) (dppn = 1,8-Bis(diphenyl-phosphano)naphthalen)
Auch mit dem Naphthalen-verbruckten Ligan-den dppnSe wurde versucht, entsprechende Gold(I)-Halogenidkomplexe zu synthetisieren. Dazu wurde derLigand bei Raumtemperatur in Dichlormethan mitthtAuCl umgesetzt. Die Losung wurde sofort schwarz,was zunachst auf einen Zerfall zu metallischem Goldhinzudeuten schien; genauere Untersuchungen zeigten
jedoch, dass die Losung keine Feststoffe enthielt. NachKristallisation stellte sich heraus, es war nicht dererwartete Gold(I)-Komplex, sondern eine Gold(III)-Verbindung mit gleicher Stochiometrie entstanden,formal nach Insertion eines Goldatoms in die P–Se-Bindung (Abb. 20). Das Selen wurde zwar vom Phos-phoratom abgespalten, ist aber immer noch Teil desMolekuls. Uber eine ahnliche (reversible) oxidative In-sertion von Gold(I)-Einheiten in eine P–Te-Bindungwurde bereits von Chivers et al. berichtet [31]; wir be-dauern, diese Arbeit in Teil 1 nicht zitiert zu haben. ImFolgenden wird die Kristallstruktur naher beschrieben.
Der Gold(III)-Komplex kristallisiert in der Raum-gruppe P1 mit einem halben Molekul des Kom-plexes und drei Molekulen Chloroform in der asym-metrischen Einheit. Fur das Gold wird aufgrundder Farbe des Komplexes und der quadratisch-planaren Koordinationsgeometrie die Oxidationsstufe+III angenommen. Es koordiniert an die beiden Phos-phoratome eines dppn-Liganden und an zwei Selen-atome, mit denen es einen Au2Se2-Vierring bildet. DasSelen kann formal als Selenid-Ion mit der Oxidations-stufe –II betrachtet werden. Insgesamt ergibt sich alsofur das Kation eine zweifach positive Ladung. Es liegtauf einem Inversionszentrum, welches sich in der Mittedes Au2Se2-Vierringes befindet.
1,8-Disubstituierte Naphthalenderivate sind in derRegel durch ungunstige sterische Wechselwirkun-gen der Substituenten verzerrt [32]. In diesem Fallist das Ringgerust noch fast planar (mittlere Ab-weichung 0,03 A), und die Substituenten sind nurmaßig aus der Ringebene ausgelenkt (P1 um 0,35,P2 um 0,16 A, zu unterschiedlichen Seiten), wohlwegen der chelatisierenden Natur des Liganden. DieWinkel in der Naphthalen-,,Bucht“ sind aber er-heblich aufgeweitet (C8A–C1–P1 126,0(9), C8A–C8–P2 126,4(8), C1–C8A–C8 126,4(10)◦), und dieBindungslangen C1–C8A und C8–C8A sind trotz dermaßigen Genauigkeit signifikant – uber das normaleMaß fur Naphthalene hinaus – verlangert (zu 1,478(15)bzw. 1,441(15) A).
Das Chlorid-Anion ist von den drei Dichlormethan-Molekulen umgeben, welche Wasserstoffbrucken mitH···Cl-Abstanden 2,31 bis 2,42 A dazu ausbilden. DieC–H···Cl-Winkel liegen etwas uber 160◦. Vor kurzemfanden wir das gleiche Aggregat bei einem Ruthenium-Komplex [33]. Eine Suche in der Cambridge Daten-bank [29] ergab 60 Treffer, bei denen ein Chlorid-Anion von 3 bis 6 Chloroform-Molekulen koordiniert
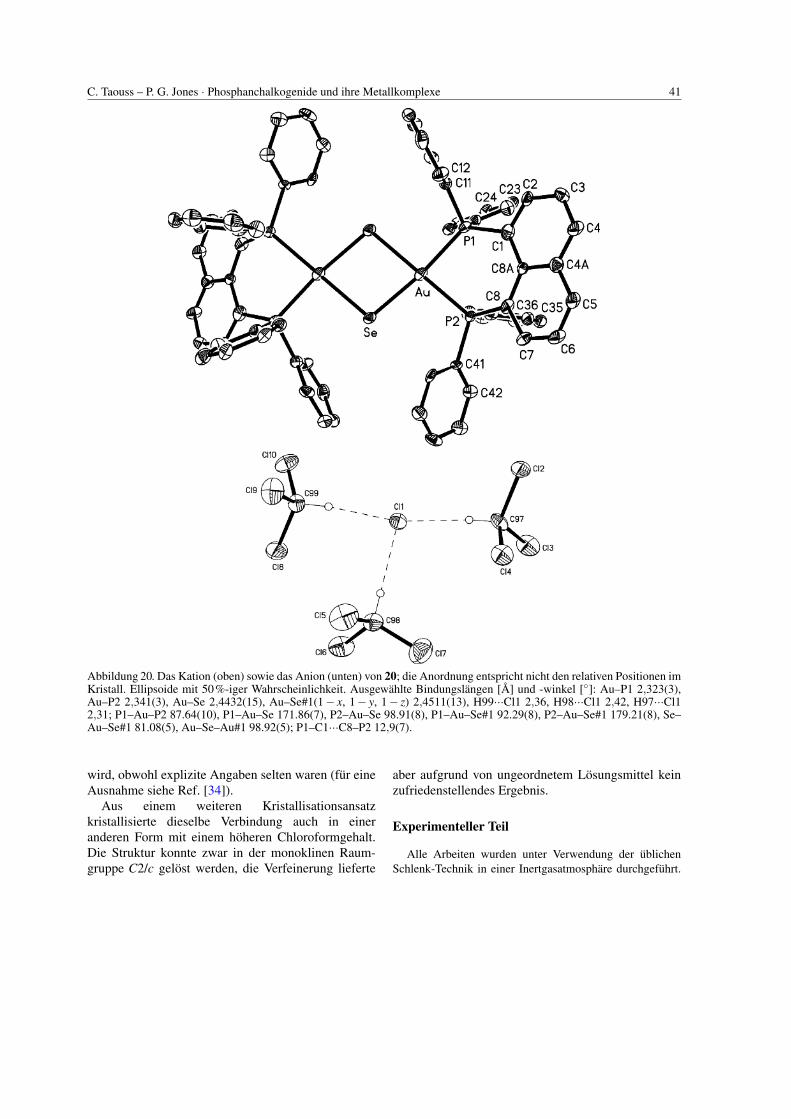
C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 41
Abbildung 20. Das Kation (oben) sowie das Anion (unten) von 20; die Anordnung entspricht nicht den relativen Positionen imKristall. Ellipsoide mit 50%-iger Wahrscheinlichkeit. Ausgewahlte Bindungslangen [A] und -winkel [◦]: Au–P1 2,323(3),Au–P2 2,341(3), Au–Se 2,4432(15), Au–Se#1(1− x, 1− y, 1− z) 2,4511(13), H99···Cl1 2,36, H98···Cl1 2,42, H97···Cl12,31; P1–Au–P2 87.64(10), P1–Au–Se 171.86(7), P2–Au–Se 98.91(8), P1–Au–Se#1 92.29(8), P2–Au–Se#1 179.21(8), Se–Au–Se#1 81.08(5), Au–Se–Au#1 98.92(5); P1–C1···C8–P2 12,9(7).
wird, obwohl explizite Angaben selten waren (fur eineAusnahme siehe Ref. [34]).
Aus einem weiteren Kristallisationsansatzkristallisierte dieselbe Verbindung auch in eineranderen Form mit einem hoheren Chloroformgehalt.Die Struktur konnte zwar in der monoklinen Raum-gruppe C2/c gelost werden, die Verfeinerung lieferte
aber aufgrund von ungeordnetem Losungsmittel keinzufriedenstellendes Ergebnis.
Experimenteller Teil
Alle Arbeiten wurden unter Verwendung der ublichenSchlenk-Technik in einer Inertgasatmosphare durchgefuhrt.

42 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
Als Schutzgas diente Stickstoff, welcher uber Phosphor-pentoxid/Silicagel (SICAPENT; E. Merck) und BTS-Kupferkatalysator-Kolonnen (BASF, bei T = 100 ◦C) vomRestsauerstoff befreit und getrocknet wurde. Uber die Syn-these der Liganden dppmE, dppeE, dppbzE und dppnEwurde bereits berichtet [1]. Die Verbindungen thtAuCl undthtAuBr wurden nach der Literatur synthetisiert [35].
cis-1,2-Bis(diphenylphosphano)ethenmonosulfid (cisdppenS)
cisdppen (1,25 g, 3,15 mmol) und Schwefel (50 mg,1,56 mmol) werden in Toluol (ca. 50 mL) zwei Stun-den refluxiert. Anschließend wird das Losungsmittel imVakuum entfernt. Die entstandene Produktmischung auscisdppen und cisdppenS wird saulenchromatographisch miteinem Dichlormethan/Pentan-Gemisch uber Alox getrennt.– C26H22P2S (428,47): ber. C 72,88, H 5,18, S 7,48, gef. C72,03, H 5,05, S 7,13. – 1H-NMR (200 MHz, CDCl3): δ =7,91 – 7,04 (m). – 31P-NMR (200 MHz, CDCl3): δ = 32,0(d, J = 18 Hz, R3P=S), −26,6 (d, J = 18 Hz, R3P).
[Bis(diphenylphosphano)methanmonoxid]chloridogold(I)(dppmOAuCl, 1)
Zu einer Losung von dppmO (300 mg, 0,75 mmol) inDichlormethan (ca. 20 mL) wird unter Ruhren eine Losungvon thtAuCl (240 mg, 0,75 mmol) in Dichlormethan (ca.20 mL) gegeben. Es wird zwei Stunden bei Raumtem-peratur geruhrt. Anschließend wird das Losungsmittel imVakuum entfernt und der Ruckstand im Vakuum getrocknet.Umkristallisation aus Dichlormethan und Diisopropyletherliefert zur Rontgenstrukturanalyse geeignete Einkristalle inForm von farblosen Tafeln. – C25H22 AuClOP2 (632,81):ber. C 47,45, H 3,50, gef. C 47,48, H 3,77. – 1H-NMR(200 MHz, CDCl3): δ = 7,8 – 7,5 (m, 8H, o-C6H5), 7,5 – 7,2(m, 12H, m/p-C6H5), 3,49 (t, 2H, J = 12 Hz, CH2). – 31P-NMR [36] (200 MHz, CDCl3): δ = 25,2 (s, P=O), 17,6 (s,P–Au); keine P–P-Kopplung detektierbar.
[Bis(diphenylphosphano)methanmonoxid]iodidogold(I)(dppmOAuI, 2)
Zu einer Losung von dppmOAuCl (200 mg, 0,32 mmol)in Dichlormethan (ca. 20 mL) wird eine Losung vonKaliumiodid (262 mg, 1,58 mmol) in Wasser (ca. 20 mL)gegeben. Es wird uber Nacht bei Raumtemperatur geruhrtund anschließend werden die Phasen getrennt. Die wass-rige Phase wird dreimal mit Dichlormethan extrahiert. Dievereinigten organischen Phasen werden dreimal mit Wassergewaschen und dann uber Magnesiumsulfat getrocknet. DasLosungsmittel wird im Vakuum entfernt und der weiße Ruck-stand im Vakuum getrocknet. Kristalle fur die Einkristall-strukturanalyse werden erhalten durch Diffusion von Pen-tan in eine Dichlormethanlosung. – 1H-NMR (200 MHz,CDCl3): δ = 7,7 – 7,5 (m, 8H, o-C6H5), 7,5 – 7,2 (m, 12H,
m/p-C6H5), 3,47 (t, 2H, J = 12 Hz, CH2). – 31P-NMR(200 MHz, CDCl3): δ = 24,5 (d, J = 3 Hz, P=O), 23,0 (d,J = 3 Hz, P–Au).
[Bis(diphenylphosphano)methanmonosulfid]chloridogold(I)(dppmSAuCl, 3)
Synthese analog zu 1, Kristallisation aus Dichlormethanund Pentan. – C25H22AuClP2S (648,87): ber. (3 · 0,5CH2Cl2): C 44,30, H 3,35, S 4,64, gef. C 44,44, H 3,39,S 4,67. – 1H-NMR (200 MHz, CDCl3): δ = 7,8 – 7,4 (m,8H, o-C6H5), 7,4 – 7,1 (m, 12H, m/p-C6H5), 3,68 (t, 2H,J = 12 Hz, CH2). – 31P-NMR (200 MHz, CDCl3): δ = 36,7(d, J = 15 Hz, P=S), 17,2 (d, J = 15 Hz, P–Au).
[Bis(diphenylphosphano)methanmonosulfid]iodidogold(I)(dppmSAuI, 4)
Synthese analog zu 2, Kristallisation aus Dichlormethanund Pentan. – C25H22AuIP2S (740,33): ber. C 40,56, H 3,00,S 4,33, gef. C 40,09, H 3,16, S 3,99. – 1H-NMR (200 MHz,CDCl3): δ = 7,76 – 7,49 (m, 8H, o-C6H5), 7,43 – 7,19 (m,12H, m/p-C6H5), 3,65 (t, 2H, J = 12 Hz, CH2). – 31P-NMR(200 MHz, CDCl3): δ = 37,2 (d, J = 19 Hz, P=S), 21,9 (d,J = 19 Hz, P–Au).
[Bis(diphenylphosphano)methanmonoselenid]bromido-gold(I) (dppmSeAuBr, 5)
Zu einer Losung von dppmSe (300 mg, 0,69 mmol) inDichlormethan (ca. 20 mL) wird unter Ruhren eine Losungvon thtAuBr (251 mg, 0,69 mmol) in Dichlormethan (ca.20 mL) gegeben. Es wird 30 Minuten bei Raumtemper-atur geruhrt. Dann wird das Losungsmittel im Vakuumentfernt und der Ruckstand zweimal umkristallisiert ausDichlormethan und Pentan. – C25H22AuBrP2Se (740,22):ber. C 40,56, H 3,00, gef. C 41,33, H 3,57. – 1H-NMR(400 MHz, CDCl3): δ = 7,73 – 7,50 (m, 8H, o-C6H5),7,38 – 7,19 (m, 12H, m/p-C6H5), 3,85 (dd, 2H, J = 12 Hz,J′ = 13 Hz, CH2). – 31P-NMR (400 MHz, CDCl3): δ = 28,8(d, JPP = 22 Hz, JPSe = 362 Hz, P=Se), 20,6 (d, JPP = 22 Hz,P–Au).
[Bis(diphenylphosphano)methanmonoselenid]iodidogold(I)(dppmSeAuI, 6)
Synthese analog zu 2. – C25H22AuBrP2Se (787,22): ber.C 38,14, H 2,82, gef. C 38,03, H 3,06. – 1H-NMR (200 MHz,CDCl3): δ = 7,82 – 7,54 (m, 8H, o-C6H5), 7,48 – 7,25 (m,12H, m/p-C6H5), 3,86 (dd, 2H, J = 12 Hz, J′ = 13 Hz,CH2). – 31P-NMR (200 MHz, CDCl3): δ = 29,6 (d, JPP =27 Hz, JPSe = 370 Hz, P=Se), 22,8 (d, JPP = 27 Hz, P–Au).
[1,2-Bis(diphenylphosphano)ethanmonoxid]bromidogold(I)(dppeOAuBr, 7)
Synthese analog zu 5, Kristallisation aus Dichlormethanund Pentan. – 1H-NMR (200 MHz, CDCl3): δ = 7,77 – 7,39

C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 43
1·(C3H7)2O 2·CH2Cl2 3 · 1/2 CH2Cl2Summenformel C31H36AuClO2P2 C26H24AuCl2IOP2 C25.5H23AuCl2P2SMr 734,95 809,16 691,31Kristallgroße [mm3] 0,28× 0,10× 0,02 0,18× 0,10× 0,04 0,38× 0,12× 0,03T [K] 100(2) 100(2) 100(2)λ [A] 0,71073 0,71073 0,71073Kristallsystem orthorhombisch orthorhombisch monoklinRaumgruppe Pbca Pbcn P21/ca [A] 19,0953(4) 22,6613(4) 9,7352(3)b [A] 10,9614(3) 11,2746(2) 18,0348(4)c [A] 28,9363(7) 20,9925(4) 14,8520(5)α [◦] 90 90 90β [◦] 90 90 105,725(4)γ [◦] 90 90 90V [A3] 6056,7(3) 5363,52(17) 2510,01(13)Z 8 8 4Dber. [g cm−3] 1,61 2,00 1,83µ(MoKα ) [mm−1] 5,1 7,0 6,3Durchlassigkeiten 1,0 – 0,433 1,0 – 0,736 1,0 – 0,486F(000) [e] 2912 3072 13402θmax [◦] 52,7 56,6 55,8Gemessene Reflexe 141 020 211 249 100 132Unabh. Reflexe 6191 6625 5976Rint 0,073 0,081 0,040Verfeinerte Parameter 338 298 293R(F) [F > 4 σ(F)]a 0,0241 0,0305 0,0129wR(F2)a (alle Reflexe) 0,0463 0,0709 0,0277x(Flack) – – –GoF (F2)b 1,07 0,97 0,93∆ρfin (max/min) [e A−3] 1,09/−1,18 4,23/−0,84 0,59/−0,52
Tabelle 4 . Kristallstrukturda-ten fur 1–3.
4 5·dppeSe2 6Summenformel C25H22AuIP2S C50H44AuBrP4Se3 C25H22AuIP2SeMr 740,29 1282,49 787,19Kristallgroße [mm3] 0,10× 0,06× 0,03 0,20× 0,15× 0,10 0,06× 0,05× 0,04T [K] 100(2) 100(2) 100(2)λ [A] 0,71073 0,71073 0,71073Kristallsystem monoklin monoklin monoklinRaumgruppe Pn P21/c P21/na [A] 9,0420(18) 21,5166(6) 9,1056(6)b [A] 11,741(2) 13,4494(3) 11,6979(9)c [A] 23,300(5) 16,6830(5) 22,8993(16)α [◦] 90 90 90β [◦] 99,86(3) 104,959(4) 95,433(7)γ [◦] 90 90 90V [A3] 2437,1(8) 4664,2(2) 2428,2(3)Z 4 4 4Dber. [g cm−3] 2,02 1,83 2,15µ(MoKα ) [mm−1] 7,5 6,5 9,0Durchlassigkeiten 1,0 – 0,845 1,0 – 0,886 1,0 – 0,928F(000) [e] 1400 2480 14722θmax [◦] 54,96 55,76 50,04Gemessene Reflexe 68199 102273 34440Unabh. Reflexe 11181 11105 4274Rint 0,048 0,054 0,128Verfeinerte Parameter 535 532 271R(F) [F > 4 σ(F)]a 0,0273 0,0250 0,0394wR(F2)a (alle Reflexe) 0,0436 0,0458 0,0592x(Flack) 0,039(4) – –GoF (F2)b 0,87 0,87 0,72∆ρfin (max/min) [e A−3] 2,03/−1,31 2,33/−1,81 2,01/−1,54
a R(F) = Σ ‖ Fo|− |Fc ‖ /Σ|Fo|; wR(F2) = [Σ{w(F2o −F2
c )2}/Σ{w(F2o )2}]0,5; w−1 = σ 2(F2
o )+(aP)2 +bPmit P = [F2
o +2F2c ]/3, a und b sind vom Programm gewahlte Konstanten; b GoF = [Σ{w(F2
o −F2c )2}/(n−
p)]0,5 mit n Daten und p verfeinerten Parametern.
Tabelle 5 . Kristallstrukturda-ten fur 4–6.

44 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
7·1/2H2O 8·1/2H2O 9Summenformel C26H25AuBrO1.5P2 C26H25AuIO1.5P2 C26H24AuBrP2SMr 700,28 747,27 707,33Kristallgroße [mm3] 0,38× 0,04× 0,02 0,45× 0,20× 0,06 0,35× 0,10× 0,04T [K] 100(2) 100(2) 100(2)λ [A] 0,71073 0,71073 0,71073Kristallsystem triklin triklin triklinRaumgruppe P1 P1 P1a [A] 11,5162(4) 11,6920(4) 11,1680(2)b [A] 12,8669(4) 12,7967(4) 12,8856(3)c [A] 18,7300(6) 18,8950(6) 19,6572(3)α [◦] 85,972(3) 86,073(4) 85,3145(16)β [◦] 77,419(4) 77,792(4) 76,2437(16)γ [◦] 65,634(4) 65,870(5) 65,450(2)V [A3] 2466,86(14) 2521,17(14) 2498,80(8)Z 4 4 4Dber. [g cm−3] 1,89 1,97 1,88µ(MoKα ) [mm−1] 7,7 7,2 7,7Durchlassigkeiten 1,0 – 0,746 1,0 – 0,241 1,0 – 0,328F(000) [e] 1348 1420 13602θmax [◦] 52,7 55,0 55,8Gemessene Reflexe 81604 81821 107657Unabh. Reflexe 10091 11554 11912Rint 0,079 0,041 0,045Verfeinerte Parameter 576 576 559R(F) [F > 4 σ(F)]a 0,0243 0,0175 0,0187wR(F2)a (alle Reflexe) 0,0323 0,0315 0,0376x(Flack) – – –GoF (F2)b 0,70 0,90 1,04∆ρfin (max/min) [e A−3] 1,38/−1,01 0,97/−1,02 0,82/−0,56
Tabelle 6 . Kristallstrukturda-ten fur 7–9.
10 11 12Summenformel C26H24AuIP2S C30H24AuClOP2 C30H24AuBrOP2Mr 754,32 694,85 739,31Kristallgroße [mm3] 0,35× 0,15× 0,05 0,20× 0,13× 0,03 0,20× 0,08× 0,03T [K] 100(2) 100(2) 100(2)λ [A] 0,71073 0,71073 0,71073Kristallsystem triklin monoklin triklinRaumgruppe P1 P21/c P1a [A] 11,3252(3) 8,8503(3) 10,3777(2)b [A] 13,0277(3) 29,9641(8) 10,9440(2)c [A] 19,7458(5) 9,8577(3) 14,3798(2)α [◦] 85,355(2) 90 108,5883(14)β [◦] 76,542(3) 96,890(3) 94,3061(14)γ [◦] 64,563(3) 90 116,5968(19)V [A3] 2558,05(11) 2595,30(14) 1337,67(4)Z 4 4 2Dber. [g cm−3] 1,96 1,78 1,84µ(MoKα ) [mm−1] 7,2 5,9 7,1Durchlassigkeiten 1,0 – 0,406 1,0 – 0,672 1,0 – 0,597F(000) [e] 1432 1352 7122θmax [◦] 55,8 52,7 55,8Gemessene Reflexe 120171 87901 64805Unabh. Reflexe 12179 5304 6379Rint 0,040 0,078 0,062Verfeinerte Parameter 559 306 316R(F) [F > 4 σ(F)]a 0,0159 0,0466 0,0209wR(F2)a (alle Reflexe) 0,0322 0,0890 0,0384x(Flack) – – –GoF (F2)b 0,94 1,18 0,86∆ρfin (max/min) [e A−3] 0,83/−0,58 2,40/−2,35 1,10/−0,97
a R(F) = Σ ‖ Fo|− |Fc ‖ /Σ|Fo|; wR(F2) = [Σ{w(F2o −F2
c )2}/Σ{w(F2o )2}]0,5; w−1 = σ 2(F2
o )+(aP)2 +bPmit P = [F2
o +2F2c ]/3, a und b sind vom Programm gewahlte Konstanten; b GoF = [Σ{w(F2
o −F2c )2}/(n−
p)]0,5 mit n Daten und p verfeinerten Parametern.
Tabelle 7 . Kristallstrukturda-ten fur 10–12.

C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 45
13 14·2CH2Cl2 15Summenformel C30H24AuBrP2S C32H28AuCl4IP2S C30H24AuClP2SeMr 755,37 972,21 757,81Kristallgroße [mm3] 0,30× 0,13× 0,01 0,20× 0,20× 0,05 0,35× 0,15× 0,02T [K] 100(2) 100(2) 100(2)λ [A] 0,71073 0,71073 0,71073Kristallsystem monoklin monoklin monoklinRaumgruppe P21/c P21/n P21/ca [A] 20,2650(2) 20,5332(3) 20,4012(3)b [A] 7,70598(8) 7,70840(10) 7,65230(10)c [A] 17,55551(17) 21,2946(3) 17,5700(4)α [◦] 90 90 90β [◦] 94,0852(9) 92,871(2) 92,877(2)γ [◦] 90 90 90V [A3] 2734,53(5) 3366,24(8) 2739,50(8)Z 4 4 4Dber. [g cm−3] 1,84 1,92 1,84µ(MoKα ) [mm−1] 7,1 5,8 6,9Durchlassigkeiten 1,0 – 0,444 1,0 – 0,553 1,0 – 0,434F(000) [e] 1456 1864 14562θmax [◦] 52,7 55,8 56,6Gemessene Reflexe 63124 136749 177513Unabh. Reflexe 5580 7993 6777Rint 0,066 0,056 0,071Verfeinerte Parameter 316 378 316R(F) [F > 4 σ(F)]a 0,0219 0,0336 0,0218wR(F2)a (alle Reflexe) 0,0447 0,0721 0,0471x(Flack) – – –GoF (F2)b 0,89 1,13 0,94∆ρfin (max/min) [e A−3] 1,00/−0,52 1,37/−1,79 1,24/−0,79
Tabelle 8 . Kristallstrukturda-ten fur 13–15.
16·2CH2Cl2 17·2CH2Cl2 18Summenformel C32H28AuBrCl4P2Se C32H28AuCl4IP2Se C26H22AuClP2SMr 972,12 1019,11 660,85Kristallgroße [mm3] 0,20× 0,15× 0,15 0,35× 0,15× 0,08 0,25× 0,16× 0,16T [K] 100(2) 100(2) 100(2)λ [A] 0,71073 0,71073 0,71073Kristallsystem monoklin monoklin monoklinRaumgruppe P21/n P21/n P21
a [A] 20,0970(4) 20,3895(3) 9,2311(2)b [A] 7,6984(2) 7,77550(10) 27,1780(3)c [A] 21,2768(3) 21,3598(3) 9,9750(2)α [◦] 90 90 90β [◦] 92,696(2) 93,803(2) 99,7407(15)γ [◦] 90 90 90V [A3] 3288,19(12) 3378,89(8) 2466,48(8)Z 4 4 4Dber. [g cm−3] 1,96 2,00 1,78µ(MoKα ) [mm−1] 7,2 6,8 6,3Durchlassigkeiten 1,0 – 0,433 1,0 – 0,400 1,0 – 0,568F(000) [e] 1864 1936 12802θmax [◦] 58,3 58,3 56,6Gemessene Reflexe 149647 216538 141281Unabh. Reflexe 8837 9085 12250Rint 0,045 0,048 0,037Verfeinerte Parameter 383 364 559R(F) [F > 4 σ(F)]a 0,0345 0,0247 0,0146wR(F2)a (alle Reflexe) 0,0875 0,0602 0,0296x(Flack) – – −0,0090(19)GoF (F2)b 1,05 1,07 1,01∆ρfin (max/min) [e A−3] 1,99/−1,82 2,22/−2,35 0,99/−0,75
a R(F) = Σ ‖ Fo|− |Fc ‖ /Σ|Fo|; wR(F2) = [Σ{w(F2o −F2
c )2}/Σ{w(F2o )2}]0,5; w−1 = σ 2(F2
o )+(aP)2 +bPmit P = [F2
o +2F2c ]/3, a und b sind vom Programm gewahlte Konstanten; b GoF = [Σ{w(F2
o −F2c )2}/(n−
p)]0,5 mit n Daten und p verfeinerten Parametern.
Tabelle 9 . Kristallstrukturda-ten fur 16–18.

46 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
19 20·6CDCl3Summenformel C26H22AuBrP2S C74H52D6Au2Cl20P4Se2Mr 705,31 2337,98Kristallgroße [mm3] 0,15× 0,05× 0,05 0,20× 0,02× 0,01T [K] 100(2) 100(2)λ [A] 0,71073 0,71073Kristallsystem monoklin triklinRaumgruppe P21/c P1a [A] 12,4446(8) 9,974(2)b [A] 12,3897(6) 13,760(3)c [A] 32,813(2) 15,679(3)α [◦] 90 95,70(3)β [◦] 99,713(7) 97,32(3)γ [◦] 90 95,08(3)V [A3] 4986,8(5) 2112,5(7)Z 8 1Dber. [g cm−3] 1,88 1,84µ(MoKα ) [mm−1] 7,7 5,0Durchlassigkeiten 1,0 – 0,752 1,0 – 0,743F(000) [e] 2704 11282θmax [◦] 52,7 50,1Gemessene Reflexe 140909 68021Unabh. Reflexe 10198 7450Rint 0,135 0,186Verfeinerte Parameter 299 460R(F) [F > 4 σ(F)]a 0,0436 0,0687wR(F2)a (alle Reflexe) 0,0718 0,1695x(Flack) – –GoF (F2)b 0,81 1,04∆ρfin (max/min) [e A−3] 2,04/−1,17 3,55/−1,72
a R(F) = Σ ‖Fo|−|Fc ‖ /Σ|Fo|; wR(F2) = [Σ{w(F2o −F2
c )2}/Σ{w(F2o )2}]0,5; w−1 = σ2(F2
o )+(aP)2 +bP mit P = [F2
o + 2F2c ]/3, a und b sind vom Programm gewahlte Konstanten; b GoF = [Σ{w(F2
o −F2
c )2}/(n− p)]0,5 mit n Daten und p verfeinerten Parametern.
Tabelle 10. Kristallstrukturda-ten fur 19–20.
(m, 20H, C6H5), 2,80 – 2,36 (m, 4H, CH2). – 31P-NMR(200 MHz, CDCl3): δ = 35,8 (d, JPP = 59 Hz, P–Au), 31,7(d, JPP = 59 Hz, P=O).
[1,2-Bis(diphenylphosphano)ethanmonoxid]iodidogold(I)(dppeOAuI, 8)
Synthese analog zu 2, Kristallisation aus Dichlormethanund Diisopropylether. – C25H22AuBrP2Se (787,22): ber. C42,30, H 3,28, gef. C 40,54, H 4,00 (Reste H2O vorhanden).– 1H-NMR (200 MHz, CDCl3): δ = 7,70 – 7,32 (m, 20H,C6H5), 2,74 – 2,31 (m, 4H, CH2). – 31P-NMR (200 MHz,CDCl3): δ = 39,6 (d, JPP = 59 Hz, P–Au), 31,9 (d, JPP =59 Hz, P=O).
[1,2-Bis(diphenylphosphano)ethanmonosulfid]bromido-gold(I) (dppeSAuBr, 9)
Synthese analog zu 5, Kristallisation aus Dichlormethanund Pentan. – C26H24AuBrP2S (707,35): ber. C 44,15, H3,42, S 4,53, gef. C 40,96, H 3,74, S 4,21 (Reste CH2Cl2und H2O). – 1H-NMR (200 MHz, CDCl3): δ = 7,80 – 7,31
(m, 20H, C6H5), 2,75 – 2,48 (m, 4H, CH2). – 31P-NMR(200 MHz, CDCl3): δ = 44,2 (d, JPP = 63 Hz, P=S), 34,6(d, JPP = 63 Hz, P–Au).
[1,2-Bis(diphenylphosphano)ethanmonosulfid]iodidogold(I)(dppeSAuI, 10)
Synthese analog zu 2, Kristallisation aus Dichlormethanund Diisopropylether. – C26H24AuIP2S (754,35): ber. C41,40, H 3,21, S 4,25, gef. C 40,03, H 4,31, S 3,12 (Rest H2Ovorhanden). – 1H-NMR (200 MHz, CDCl3): δ = 7,79 – 7,30(m, 20H, C6H5), 2,76 – 2,48 (m, 4H, CH2). – 31P-NMR(200 MHz, CDCl3): δ = 44,2 (d, JPP = 63 Hz, P=S), 38,3(d, JPP = 63 Hz, P–Au).
[1,2-Bis(diphenylphosphano)benzolmonoxid]chlorido-gold(I) (dppbzOAuCl, 11)
Der zur Strukturbestimmung verwendete Einkristallwurde vermutlich nach Einwirkung von Luftsauerstoff auseiner Losung von dppbzSeAuCl (15) in Dichlormethan er-halten.

C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe 47
[1,2-Bis(diphenylphosphano)benzolmonoxid]bromido-gold(I) (dppbzOAuBr, 12)
Synthese analog zu 5, Kristallisation aus Dichlormethanund Pentan. – 1H-NMR (200 MHz, CDCl3): δ = 7,53 – 7,24(m). – 31P-NMR (200 MHz, CDCl3): δ = 36,8 (d, JPP =6 Hz), 32,5 (d, JPP = 6 Hz).
[1,2-Bis(diphenylphosphano)benzolmonosulfid]bromido-gold(I) (dppbzSAuBr, 13)
Synthese analog zu 5, Kristallisation aus Deutero-Chloroform und Pentan. – 1H-NMR (200 MHz, CDCl3): δ =7,70 – 7,09 (m). – 31P-NMR (200 MHz, CDCl3): δ = 44,5(d, JPP = 17 Hz, P=S), 26,8 (d, JPP = 17 Hz, P–Au).
[1,2-Bis(diphenylphosphano)benzolmonosulfid]iodido-gold(I) (dppbzSAuI, 14)
Synthese analog zu 2, Kristallisation aus Dichlormethanund Pentan. – C30H24AuIP2S (802,40): ber. C 44,91, H 3,01,S 4,00, gef. C 42,57, H 3,62, S 3,69 (Rest CH2Cl2). – 1H-NMR (200 MHz, CDCl3): δ = 7,64 – 7,00 (m). – 31P-NMR(200 MHz, CDCl3): δ = 44,5 (d, JPP = 18 Hz, P=S), 28,0(d, JPP = 18 Hz, P–Au).
[1,2-Bis(diphenylphosphano)benzolmonoselenid]chlorido-gold(I) (dppbzSeAuCl, 15)
Synthese analog zu 1, Kristallisation aus Dichlormethanund Pentan. – C30H24AuClP2Se (757,84): ber. C 47,55, H3,19, gef. C 47,75, H 3,44. – 1H-NMR (200 MHz, CDCl3):δ = 7,69 – 6,98 (m). – 31P-NMR (200 MHz, CDCl3): δ =34,7 (d, JPP = 21 Hz, JPSe = 358 Hz, P=Se), 22,6 (d, JPP =21 Hz, P–Au).
[1,2-Bis(diphenylphosphano)benzolmonoselenid]bromido-gold(I) (dppbzSeAuBr, 16)
Synthese analog zu 5, Kristallisation aus Dichlormethanund Pentan. – C30H24AuBrP2Se (802,29): ber. C 44,91, H3,02, gef. C 44,75, H 3,20. –1H-NMR (200 MHz, CDCl3):δ = 7,76 – 7,06 (m). – 31P-NMR (200 MHz, CDCl3): δ =34,6 (d, JPP = 23 Hz, JPSe = 357 Hz, P=Se), 23,0 (d, JPP =23 Hz, P–Au).
[1,2-Bis(diphenylphosphano)benzolmonoselenid]iodido-gold(I) (dppbzSeAuI, 17)
Synthese analog zu 2, Kristallisation aus Dichlormethanund Pentan. – C30H24AuIP2Se (849,29): ber. C 42,43, H2,85, gef. C 41,10, H 3,13. –1H-NMR (200 MHz, CDCl3):δ = 7,68 – 7,00 (m). – 31P-NMR (200 MHz, CDCl3): δ =34,1 (d, JPP = 27 Hz, JPSe = 351 Hz, P=Se), 22,4 (d, JPP =27 Hz, P–Au).
[cis-1,2-Bis(diphenylphosphano)ethenmonosulfid]chlorido-gold(I) (cisdppenSAuCl, 18)
Synthese analog zu 1, Kristallisation aus Dichlormethanund Pentan. – 1H-NMR (200 MHz, CDCl3): δ = 7,78 – 7,10(m). – 31P-NMR (200 MHz, CDCl3): δ = 29,9 (d, JPP =23 Hz, P=S), 14,2 (d, JPP = 23 Hz, P–Au).
[cis-1,2-Bis(diphenylphosphano)ethenmonosulfid]bromido-gold(I) (cisdppenSAuBr, 19)
Synthese analog zu 5, Kristallisation aus Deutero-Chloroform und Pentan. – C26H22AuBrP2S (705,34): ber. C44,27, H 3,14, S 4,55, gef. C 44,21, H 3,32, S 4,41. – 1H-NMR (200 MHz, CDCl3): δ = 7,86 – 7,14 (m). – 31P-NMR(200 MHz, CDCl3): δ = 30,0 (d, JPP = 23 Hz, P=S), 15,8(d, JPP = 23 Hz, P–Au).
(dppnSeAu)2Cl2 (20)
Zu einer Losung von dppnSe (500 mg, 0,87 mmol) inDichlormethan (ca. 25 mL) wird unter Ruhren eine Losungvon thtAuCl (279 mg, 0,87 mmol) in Dichlormethan (ca.15 mL) gegeben. Es wird zehn Minuten bei Raumtemper-atur geruhrt. Dann wird das Losungsmittel im Vakuum ent-fernt und der schwarze Ruckstand aus Dichlormethan undPentan umkristallisiert. Kristalle fur die Strukturlosung wur-den aus einer gesattigten Losung in Deuterochloroform er-halten. – C68H52Au2Cl2P4S2 (1615,79): ber. C 50,55, H3,24, gef. C 46,07, H 3,57 (Rest CDCl3 vorhanden). – MS(NSI): m/z(%) = 773,0 (100) [(dppnSeAu)2]+, 1581,0 (27)[(dppnSeAu)2+Cl]+). – 1H-NMR (200 MHz, CDCl3): δ =7,68 – 7,00 (m). – 31P-NMR (200 MHz, CDCl3): δ = 13,7 s.
Rontgenstrukturanalysen
Die Kristalle wurden aus der Mutterlauge entnom-men, in Inertol prapariert und darin auf Glasfaden mon-tiert. Die Beugungsdaten wurden mit monochromatisierterMoKα -Strahlung auf einem Oxford Diffraction XcaliburE Diffraktometer bei 100 K aufgenommen. Die Struktur-modelle wurden anisotrop gegen F2 mit dem ProgrammSHELXL-97 [37] verfeinert. Die Wasserstoffatome wurdenauf geometrisch idealisierte Lagen gesetzt und per Reiter-modell verfeinert. Daten zu den Strukturbestimmungen sindin den Tabellen 4 – 10 zu finden, Ellipsoidbilder der asym-metrischen Einheiten in Abb. 1 – 20 (ggf. mit Zusatz ,,a“,Packungsbilder/Molekulvergleiche sind ,,b“ und/oder ,,c“).Ausnahmen und spezielle Aspekte: Die Wasserstoffatome derWassermolekule bei 7 und 8 wurden frei verfeinert. Bei 11mussten die Atome C53 und C54 isotrop verfeinert werden.Bei 19 wurden wegen der schwachen Daten alle Kohlen-stoffatome isotrop verfeinert. Bei 3 war das Dichlormethan-molekul uber ein Inversionszentrum ungeordnet; bei 14, 16,und 17 war je eines der zwei Dichlormethanmolekule unge-ordnet.

48 C. Taouss – P. G. Jones · Phosphanchalkogenide und ihre Metallkomplexe
CCDC 962176 – 962195 enthalten die beim CambridgeCrystallographic Data Centre hinterlegten Kristallstruktur-
daten fur Verbindungen 1–20 (in derselben Reihenfolge).Anforderung: www.ccdc.cam.ac.uk/data request/cif.
[1] Teil I: C. Taouss, P. G. Jones, Z. Naturforsch. 2013,68b, 860 – 870.
[2] I. M. Keen, J. Chem. Soc. 1965, 5751 – 5752.[3] P. G. Jones, E. Bembenek, J. Crystallogr. Spectrosc.
Res. 1992, 22, 397 – 401.[4] S. Ahmad, M. N. Akhtar, A. A. Isab, A. R. Al-Arfaj,
M. S. Hussain, J. Coord. Chem. 2000, 51, 225 – 234.[5] M. S. Hussain, A. A. Isab, J. Chem. Crystallogr. 2000,
30, 731 – 735.[6] S. Ahmad, A. A. Isab, H. P. Perzanowski, M. S. Hus-
sain, M. N. Akhtar, Trans. Met. Chem. 2002, 27,177 – 183.
[7] M. S. Hussain, J. Crystallogr. Spectrosc. Res. 1986, 16,91 – 99.
[8] M. S. Hussain, E. O. Schlemper, Acta Crystallogr.1987, C43, 450 – 453.
[9] M. S. Hussain, A. A. Isab, Z. Kristallogr. – New Cryst.Struct. 2001, 216, 479.
[10] M. S. Hussain, A. A. Isab, A. Saeed, A. R. Al-arfaj, Z.Kristallogr. – New Cryst. Struct. 2001, 216, 629.
[11] M. Preisenberger, A. Bauer, H. Schmidbaur, Chem. Ber.1997, 130, 955 – 958.
[12] H. Schmidbaur, J. E. von Eschenbach, O. Kumberger,G. Muller, Chem. Ber. 1990, 123, 2261 – 2265.
[13] P. G. Jones, C. Thone, Acta Crystallogr. 1992, C48,2114 – 2116.
[14] S. M. Aucott, P. Bhattacharyya, H. L. Milton, A. M. Z.Slawin, J. D. Woollins, New J. Chem. 2003, 27,1466 – 1469.
[15] M. L. Williams, S. E. Boyd, S. P. C. Dunstan, D. L.Slade, P. C. Healy, Acta Crystallogr. 2003, E59, m768–m770 (Acetonitril-Monosolvat).
[16] M. L. Williams, D. L. Slade, S. E. Boyd, P. C. Healy,Acta Crystallogr. 2005, E61, m30–m32.
[17] Z.-L. Xie, Z.-N. Chen, Acta Crystallogr. 2006, E62,m2277–m2278 (Hemihydrat).
[18] C. Taouss, P. G. Jones, Dalton Trans. 2011, 40,11687 – 11689.
[19] H. Schmidbaur, G. Reber, A. Schier, F. E. Wagner,G. Muller, Inorg. Chim. Acta 1988, 147, 143 – 150.
[20] Es ist uns in den letzten Wochen gelungen, die Struk-tur des dppmO zu bestimmen; daruber wird an an-derer Stelle berichtet (C. Taouss, P. G. Jones, in Vor-bereitung).
[21] R. Colton, B. F. Hoskins, P. Panagiotidou, Aust.J. Chem. 1987, 40, 1909 – 1912.
[22] C. J. Carmalt, A. H. Cowley, A. Decken, Y. G. Lawson,N. C. Norman, Acta Crystallogr. 1996, C52, 931 – 933.
[23] R. Thirumoorthi, T. Chivers, Eur. J. Inorg. Chem. 2012,3061 – 3069.
[24] B. Ahrens, P. G. Jones, Acta Crystallogr. 1997, C53,1852 – 1854.
[25] P. J. Carroll, D. D. Titus, J. Chem. Soc., Dalton Trans.1977, 824 – 829.
[26] P. G. Jones, B. Ahrens, Chem. Commun. 1998,2307 – 2308.
[27] Wasserstoffbrucken in dieser Veroffentlichung wur-den nicht nach T. Steiner, Acta Crystallogr. 1998,B54, 456 – 463, auf C–H 1,08 A ,,normalisiert“; nor-malisierte H···A-Abstande waren kurzer als die hiervorgestellten.
[28] E. M. Barranco, O. Crespo, M. C. Gimeno, P. G. Jones,Inorg. Chem. 2000, 39, 680 – 687.
[29] F. H. Allen, Acta Crystallogr. 2002, B58, 380 – 388.[30] I. Dance, New J. Chem. 2003, 27, 22 – 27.[31] D. J. Eisler, S. D. Robertson, T. Chivers, Can. J. Chem.
2009, 87, 39 – 46.[32] Fur eine Diskussion dieser Effekte s. z. B. A. Karacar,
M. Freytag, H. Thonnessen, J. Omelanczuk, P. G.Jones, R. Bartsch, R. Schmutzler, Z. Anorg. Allg.Chem. 2000, 626, 2361 – 2372.
[33] A. R. Petrov, T. Bannenberg, C. G. Daniliuc, P. G. Jo-nes, M. Tamm, Dalton Trans. 2011, 40, 10503 – 10512.
[34] X. Xie, R. E. McMarley, Inorg. Chem. 1997, 36, 4011 –4016.
[35] S. Ahrland, K. Dreisch, B. Noren, A. Oskarsson, Mat.Chem. Phys. 1993, 35, 281 – 289.
[36] Fur 31P-NMR-Parameter der freien Liganden sieheRef. [1]. Die chemische Verschiebung des PIII-Atoms nimmt beim Komplexieren erwartungsgemaßzu, wahrend sich die P-P-Kopplungskonstanten un-terschiedlich verhalten (bei den dppm- und dppbz-Derivaten werden sie kleiner, bei den dppe-Derivatengroßer). Die P-Se-Kopplungskonstanten unterscheidensich im Schnitt um weniger als 10 Hz von denen derfreien Liganden.
[37] G. M. Sheldrick, Acta Crystallogr. 2008, A64, 112 –122.












![Metallkomplexe mit biologisch wichtigen Liganden, LXVIII ... · R. Bergs, K. Sünkel, W. Beck 2429 Notiz / Note Metallkomplexe mit biologisch wichtigen Liganden, LXVIII[1] Metallorganische](https://img.pdfslide.org/doc/110x75/5d66bbe188c9937b6e8bd214/metallkomplexe-mit-biologisch-wichtigen-liganden-lxviii-r-bergs-k-suenkel.jpg)







![Metallkomplexe funktioneller Isoeyanide, VI [1]zfn.mpdl.mpg.de/data/Reihe_B/37/ZNB-1982-37b-1044.pdf · 2018. 2. 9. · Metallkomplexe funktioneller Isoeyanide, VI [1] Tosylmethyl-,](https://img.pdfslide.org/doc/110x75/60b34e1c6326ed06121b3e8d/metallkomplexe-funktioneller-isoeyanide-vi-1zfnmpdlmpgdedatareiheb37znb-1982-37b-1044pdf.jpg)








