Embed Size (px)
DESCRIPTION
Proseminar Bioinformatik: Theoretical Analysis of Protein-Protein-Interactions. Scoring Functions. Silke Ruzek. 22.Juni.2004. Überblick. Einleitung 1.Modell: Paar Potentiale zur Bewertung möglicher Docking-Komplexe 2.Modell: Kombination von Scoring Funktionen Fazit - PowerPoint PPT Presentation
Citation preview

1
Proseminar Bioinformatik:Theoretical Analysis of Protein-Protein-Interactions
Scoring Functions
Silke Ruzek
22.Juni.2004

2
Überblick Einleitung
1.Modell: Paar Potentiale zur Bewertung
möglicher Docking-Komplexe
2.Modell: Kombination von Scoring Funktionen
Fazit
Diskussion

3
Einleitung Ziel von Protein-Protein-Docking: Bestimmung der
detaillierten Struktur des Komplexes ausgehend von den ungebundenen Komponenten
Docking Methoden verwenden rigid-body-Strategien und erzeugen eine große Anzahl von Docking-Komplex-Kandidaten
Aus vielen möglichen Konformationen sollen die stabilsten Komplexe ausgewählt werden
Scoring Funktionen bewerten Konformationen unter Berücksichtigung der chemischen Aktivität zwischen den Molekülen und der Größe der Kontaktfläche

4
Modell 1: Paar Potentiale Residue-Residue-Paar Potentiale sind ein einfaches Modell um die Vielfachheit der Interaktionen zwischen Residuen darzustellen
Anwendung zur Identifizierung des nativen Protein- Komplexes aus der Menge der möglichen Konformationen
Ein Komplex ist nahe an der nativen Konformation, wenn der RMSD der C -Atome im Interface kleiner als 2.5Å ist

5
Modell 1: Paar PotentialeMethode: 4 Typen von Paar-Potentialen
• basierend auf C -Atomen von Residuen• basierend auf allen Atomen von Residuen• basierend auf allen Seitenketten-Atomen• Atom-level Potential Jedem Atom in jeder Residue wird einer von 40
Atomtypen zugewiesen
Paar c existiert zwischen Residue/ Atomtyp i und j wenn die Atome der beiden Residuen/ die Atomtypen innerhalb einer Cutoff-Distanz liegen
ji,

6
Modell 1: Paar Potentiale
3 Datensätze werden getestet:
Interdomän-Paarungen nicht-redundanter Heterodimere
11 Strukturen mit RMSD von 2.5Å oder besser
Interdomän-Paarungen nicht-redundanter Homodimere
23 Strukturen mit RMSD von 2.5Å oder besser
Intramolekulare Residuen-Paarungen nicht-homologer
Protein-Domänen
385 Domänen mit RMSD von 2.5Å oder besser Für jede Superfamilie der Klassen α,β,α/β,α+β aus SCOP wurde die beste Struktur genommen

7
Modell 1: Paar PotentialeGenerierung des Potentials: Bestimmung der Anzahl der Paarungen zwischen
jedem Residuen-Typ Kalkulation der erwarteten Anzahl von Paaren
zwischen Residue i und j:
Nur Werte größer 5 werden berücksichtigt
• Mole-fraction-MethodeN
n*
N
n*Ce ji
i,j
• Contact-fraction-MethodeC
c*
C
c*Ce ji
i,j
20
1nN
20
1cC
20
1cc
i ii ij i,ji ji n,n =Vorkommen jeder Residue

8
Modell 1: Paar PotentialePaar-Potential-Matrix: Score für jedes Paar entspricht dem log von
tatsächlicher Anzahl durch erwartete Anzahl
Dadurch ergibt sich der
Gesamtscore als Summe
der individuellen Scores
)ji,
eji,
clog(ij,
scoreji,
score

9
Modell 1: Paar PotentialeBewertung der Docking-Komplex: Für jeden Komplex wird ein Gesamtscore durch
Addieren der Scores der Paare, die das Interface des Komplexes bilden, berechnet.
Zusatzbedingung:
Minimum relative surface accessibility (MRSA)
Residuen,die nicht zugänglich sind können
ausgeschlossen werden Komplexe werden nach ihren Scores geordnet und die
Position des nativen Komplexes in der Liste wird
bestimmt

10
Modell 1: Paar PotentialeTestsysteme: 5 Enzym-Inhibitor-Systeme und 4 Antikörper-Antigen-Systeme
Parameter: Distance-Cutoff: - C -Atome: 5 -15 Å in 1Å Schritten - sonst : 4 -10 Å in 1Å Schritten MRSA : 0, 5, 20%
Nur Komplexe aus mindestens 20 Paaren werden berücksichtigt
11
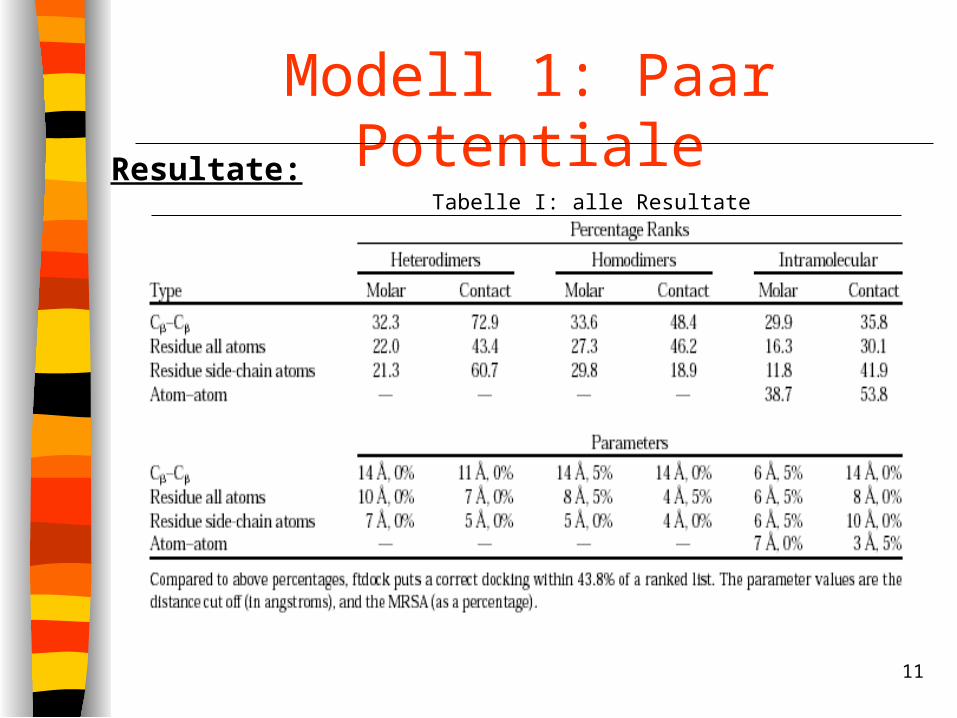
Modell 1: Paar PotentialeResultate:
Tabelle I: alle Resultate

12
Modell 1: Paar Potentiale
Tabelle II : Einordnung der korrekten StrukturResultate:
Enzym-Inhibitor Systeme
Antikörper-Antigen Systeme
13
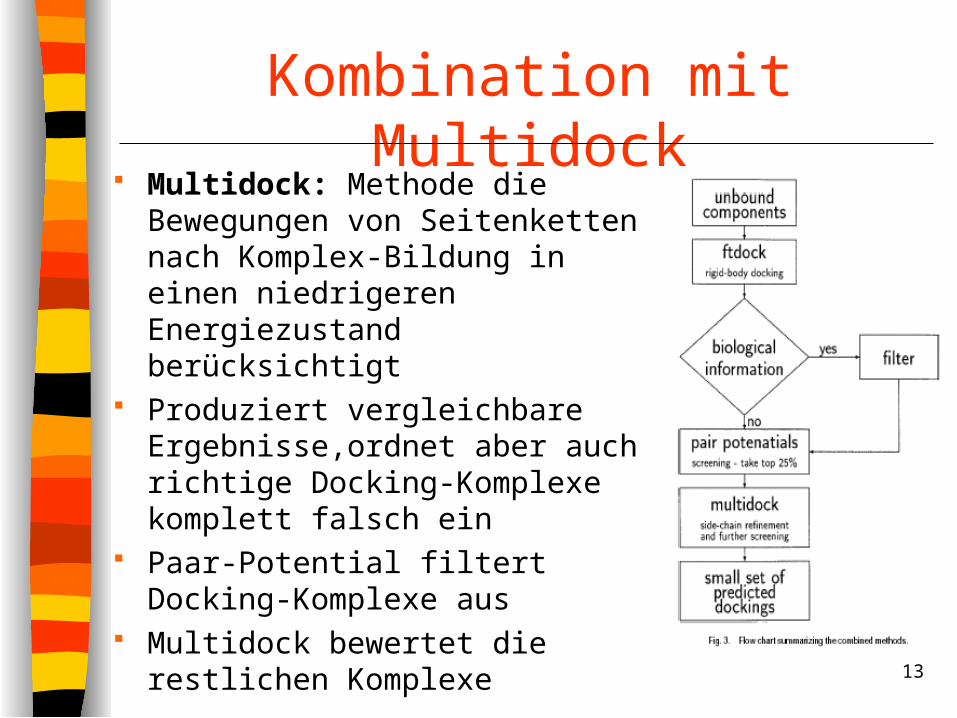
Kombination mit Multidock Multidock: Methode die
Bewegungen von Seitenketten nach Komplex-Bildung in einen niedrigeren Energiezustand berücksichtigt
Produziert vergleichbare Ergebnisse,ordnet aber auch richtige Docking-Komplexe komplett falsch ein
Paar-Potential filtert Docking-Komplexe aus
Multidock bewertet die restlichen Komplexe
14
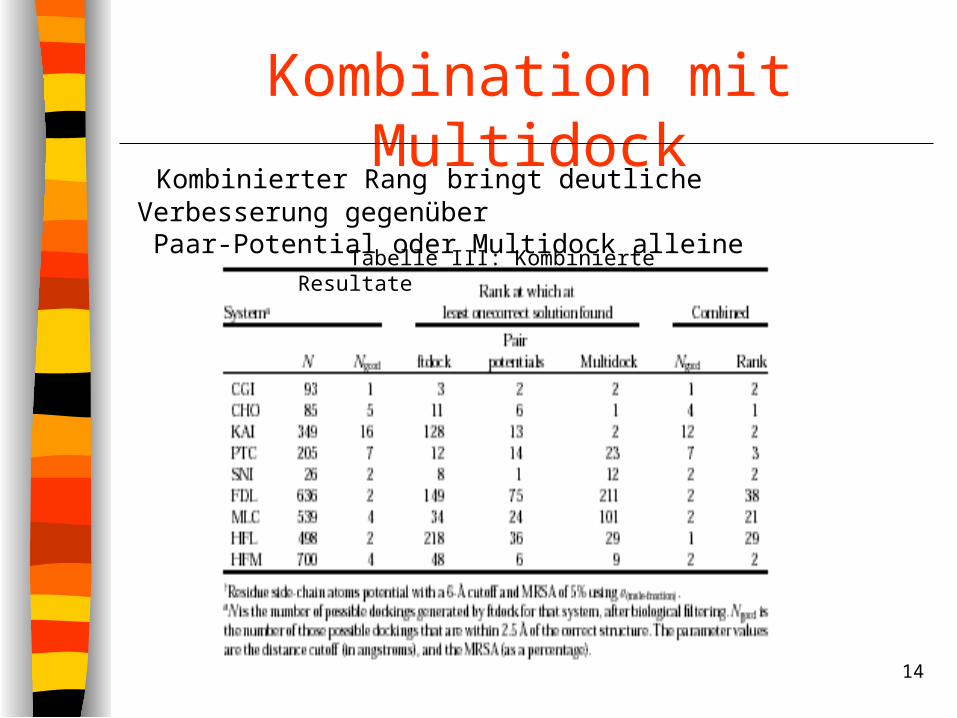
Kombination mit Multidock
Tabelle III: Kombinierte Resultate
Kombinierter Rang bringt deutliche Verbesserung gegenüber Paar-Potential oder Multidock alleine

15
Fazit Paar Potentiale können die stabile Docking-
Konformation aus einer Liste von möglichen Komplexen auswählen
Ein korrektes Docking wird am oberen Ende der Hit-Liste plaziert
Beste Resultate durch intermolekulare Paarungen in einem Datensatz nicht-homologer Protein-Domänen
Paar Potentiale können eine Liste mit möglichen Komplexen verkleinern, bevor detailliertere Prozeduren angewendet werden können
Paar Potentiale ordnen auch falsch-positive Komplexe hoch ein, bewerten aber ein korrektes Docking nicht komplett falsch

16
Modell 2: Kombination von Scoring Funktionen
Struktur-basierte Potentiale werden zum Filtern, Rescoring und Einordnen der ausgewählten Konformationen verwendet.
• Atomic Contact Potential (ACP)• Residue Potential Score (RPScore)
Kombination der Potentiale mit Energie-Termen Anwendung auf
• 13 Protein-Test-Sets• Docking-Resultate von 10 Paaren
ungebundener Proteine

17
Motivation Frühere Studie: Kombinierten Potentialen aus
elektrostatischen, Desolvations- und van der Waals-Energien liefern bessere Ergebnisse
Entwicklung des Residue-Potential-Scores (Paar Potential) erlaubt Verbesserung der bisherigen Ergebnisse
ACP konnte durch Hinzufügen von elektrostatischem und van der Waals Term verbessert werden
Test mehrerer kombinierter Potentiale auf Verbesserungen

18
ACP Atomic Contact Potential Freie Energie die benötigt wird um Atom-Wasser-
Kontakte durch Atom-Atom-Kontakte zu ersetzten. ACP-Energien wurden für 18 Atom-Typen aus 89
nicht-homologen Proteinstrukturen abgeleitet
:ACP-Energie der wechselwirkenden Atome
i und j , wobei alle Paare mit weniger als 6Å
Abstand beachtet werden
ij ije
ACPΔG
ije

19
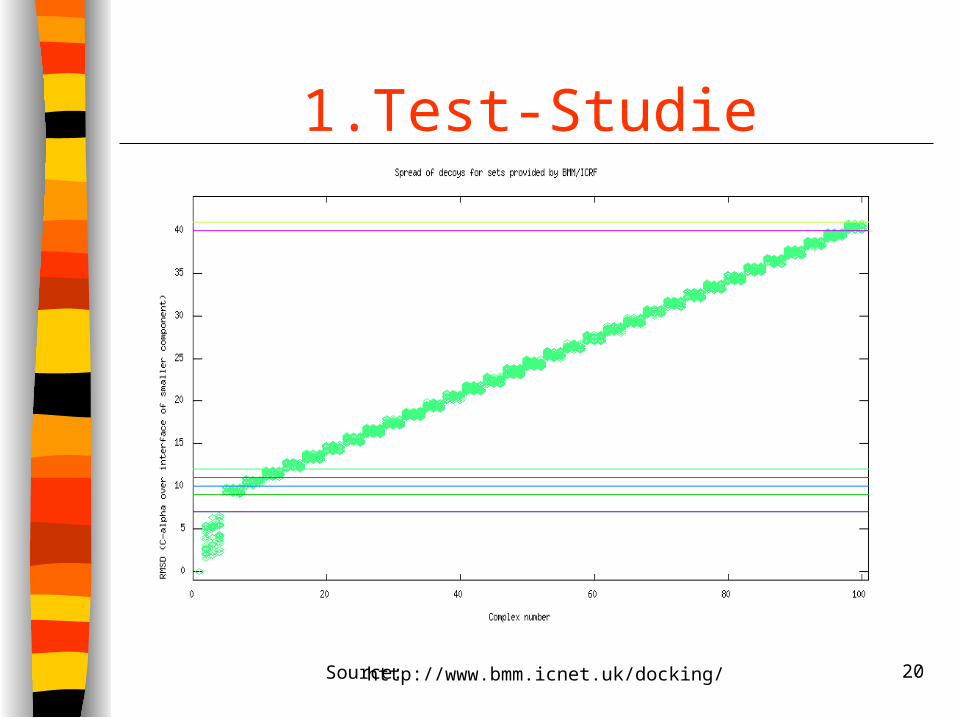
1.Test-StudieMethode:
Trainings-Sets aus 99 Konformationen für 13 Komplexe generiert mit FTDock
Minimierung der Konformationen in CHARMM Bewertung der Testsets mit 16 verschiedenen
Scoring Funktionen
20
1.Test-Studie
Source: http://www.bmm.icnet.uk/docking/

21
1.Test-Studie
Bewertung der Scoring-Funktionen durch• Ø RMSD der 10 top-plazierten Strukturen• Anzahl der Strukturen innerhalb 10Å RMSD der
nativen Struktur (Hits) • Rang des ersten Hits • RMSD der best-bewertesten Struktur
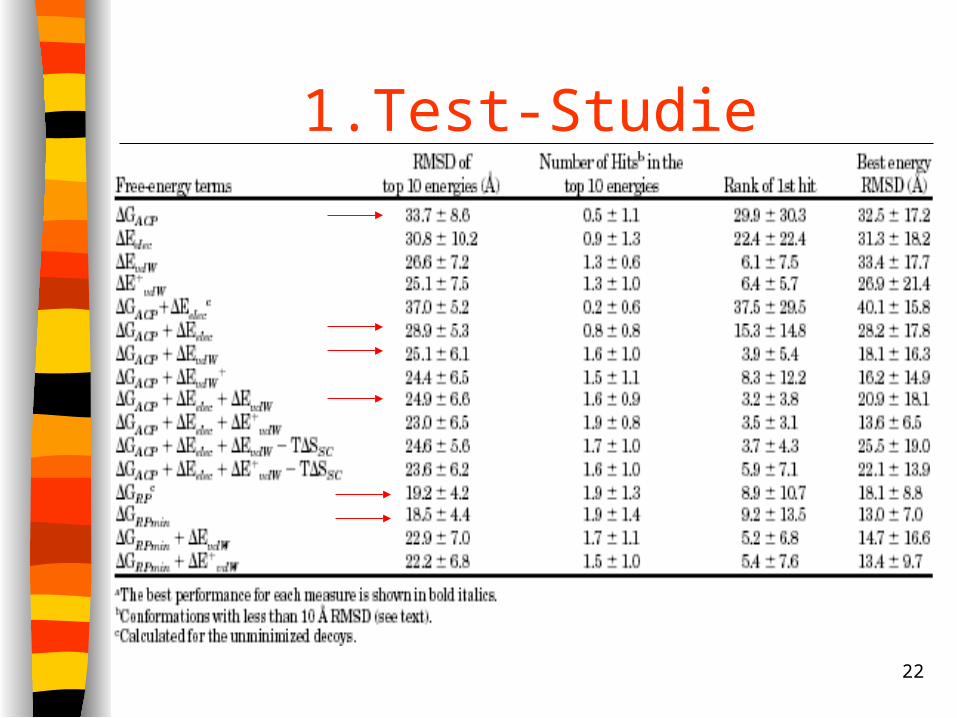
22
1.Test-Studie
23
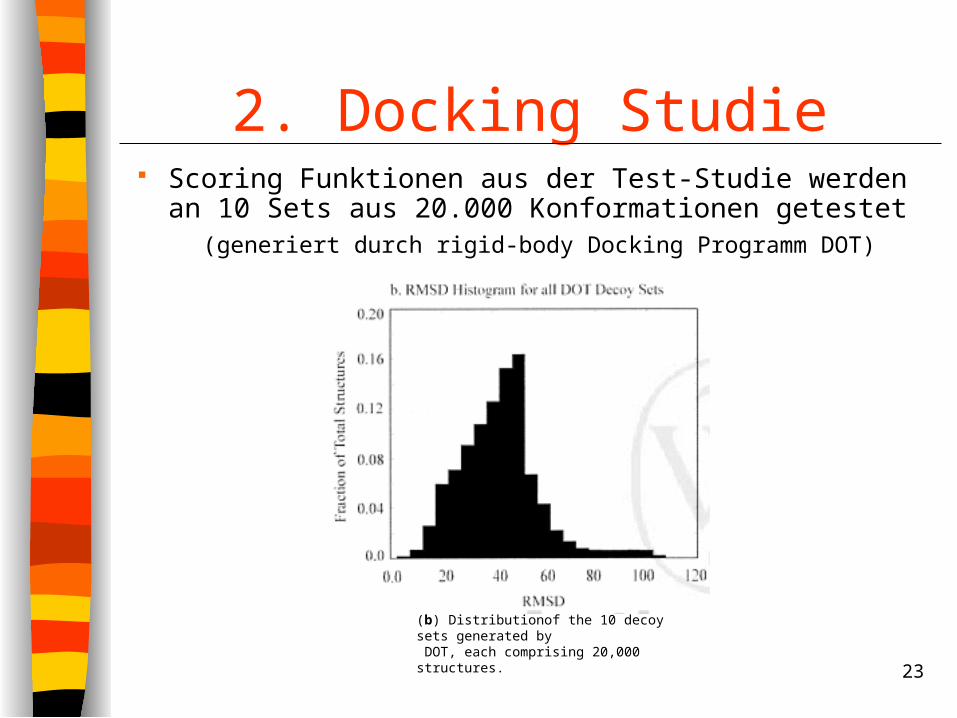
2. Docking Studie Scoring Funktionen aus der Test-Studie werden an
10 Sets aus 20.000 Konformationen getestet (generiert durch rigid-body Docking Programm DOT)
(b) Distributionof the 10 decoy sets generated by DOT, each comprising 20,000 structures.

24
2.Docking Studie
Anwenden von zwei verschiedenen Filtern
1. ACP+Elec Filter
500 Strukturen mit niedrigsten ΔG Werten
1500 Strukturen mit niedrigsten ΔE Werten
Kalkulierung von ΔE berücksichtigt die
strukturellen Unsicherheiten beim Docking besser
2. RPScore Filter
500 Strukturen mit niedrigsten ΔG Werten
ACP
Elec
RP
Elec
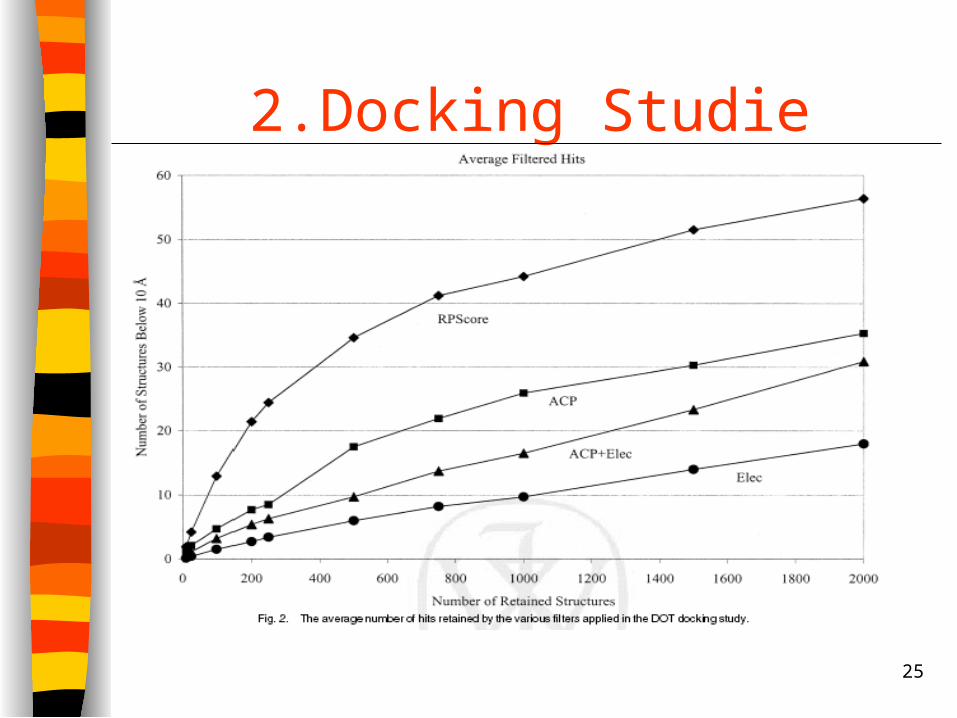
25
2.Docking Studie

26
2.Docking StudieFilterleistung in Abhängigkeit von der Ladung der Komplexe

27
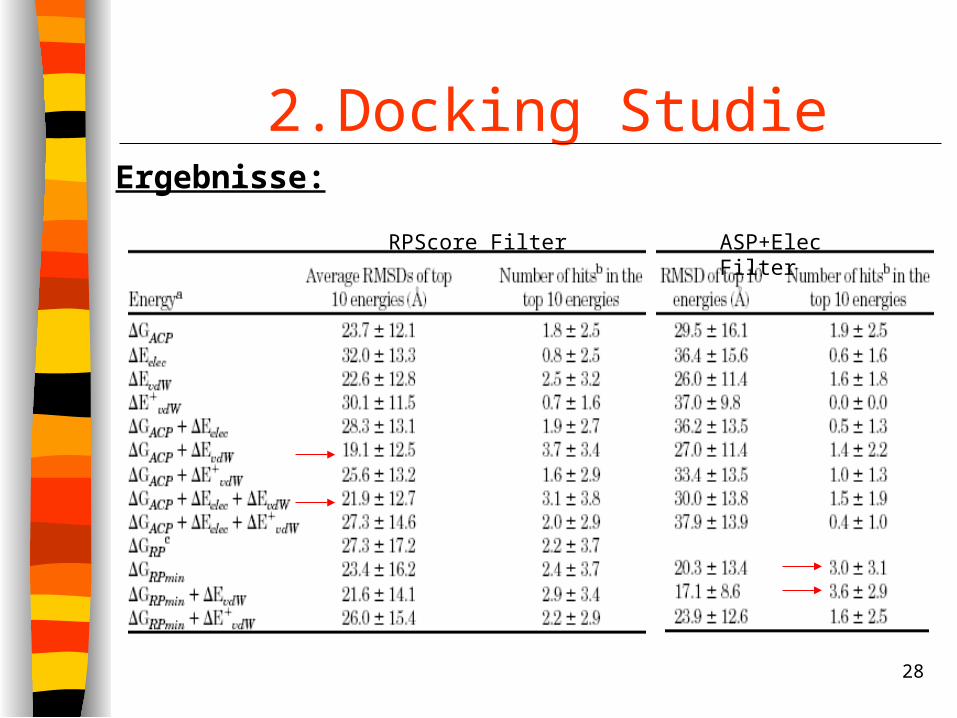
2.Docking Studie Minimierung der gefilterten Strukturen in CHARMM Pro Komplex entstehen 2 Test-Sets Rescoring der Test-Sets anaolg zur ersten Studie
28
2.Docking Studie
RPScore Filter ASP+Elec Filter
Ergebnisse:

29
Fazit Verfeinerung der Strukturen durch
Energieminimierung Zusätzlicher van der Waals Term verbessert die
Einordnung der Konformationen ΔG findet beim Filtern die meisten Hits und ist
besonders für geladene Komplexe geeignet Beste Strategien: RPScore Filter + ACP Scoring Funktion ACP+Elec Filter + RPScore Scoring Funktion Durch Kombination von mehreren Potentialen in
mulitstage postprocessing Prozeduren besseres Auffinden von möglichen stabilen Komplexen möglich
RP

30
Referenz
Moont, G., Gabb, H.A., and Sternberg, M.J.E., (1999) Proteins, 35, 364-373. Use of Pair Potentials Across Protein Interfaces in Sreening Predicted Docked Complexes.
Zhang, C., Vasmatzis, G., Cornette, J.L., and DeLisi, C., (1997) J. Mol. Biol., 267, 707-726. Determination of Atomic Desolvation Energies from the Structures of Crystallized Proteins.
Murphy, J., Gatchell, D.W., Prasad, J.C., and Vajda, S., (2003) Proteins, 53, 840-854. Combination of Scoring Functions Improves Discrimination in Protein-Protein Docking.





























