Embed Size (px)
Citation preview

Quantitative Auswertung von Papier- und D0nnschicht-Chromato- grammen mit Hilfe eines Kathodenstrahlpolarographen
Quantitative Evaluation of Paper and Thin Layer Chromatography by means of a Cathode-Ray Polarograph
Analyse quantitative en chromatographie sur papier et en couche mince a I'aide d'un polarographe a rayon cathodique
W. Huber Badische Anilin- & Soda-Fabrik AG, Ammoniaklaboratorium, D-67 Ludwigshafen/Rhein
Summary: The sensitivity of spectrophotometry for evaluation of eluted substances from TL- and paperchro- matograms is not very high. Furthermore, a blank often occurs. Polarography, especially cathode - ray polaro- graphy, increases the sensitivity by the factor 10 - 100 without a significant blank, produced by the adsorption layer. The determinations can be made from a single spot, even with side components. Isolation of the sub- stances is carried out by the suction of the layer from TLC - plates into a small column and elution from it, or the cutting of the paper and digesting with a suitable solvent. The polarograms are adjusted on the screen of the cathode - ray tube and, after optimal adjustment, recorded (one polarogram in 2 sec., all 7 sec. ).
The evaluation is performed graphically. Three examp- les show the applicability of the method:
1. Determination of dinitrophenylhydrazones from aldehydes as an example for hydrophobic compounds, separated by TLC;
2. Determination of trimellitic acid, as an example for a hydrophilic compound, separated by PC;
3. Determination of N, N'-dimethylurea, as an example for a hydrophilic compound, which must be conver- ted into a polarographic active derivative.
Zusammenfassung: Die Spektrophotometrie zeigt bei der Auswertung eluierter Substanzen aus Papier- und Diinn- schichtchromatogrammen h~iufig eine unbefriedigende Empfindlichkeit. Dazu kommt eine oft stdrende Unter- grundabsorption. Die Anwendung der Polarographie, spe- zieU der Kathodenstrahlpolarographie, bringt eine Erhd- hung der Empfindlichkeit urn den Faktor 10 bis 100 ohne einen bemerkbaren, aus der Sorptionsschicht stammenden
Blindwert. Die Bestimmungen kdnnen auch bei Neben- komponenten an einem einzelnen Fleck durchgefdart wer- den. Die Isolierung erfolgt bei Diinnschichtchromato- grammen dutch Absaugen der Schicht in ein kurzes S~iul- chert mit anschliet~ender Elution, bei Papierchromato- grammen durch Ausschneiden und Digerieren mit einem geeigneten Ldsungsmittel. Die Polarogramme werden auf dem Kathodenschirm optimal eingestellt und dann mit einem SchneUschreiber registriert (alle 7 sek. ein Polaro- gramm von 2 sek. Dauer). Die Auswertung erfolgt gra- phisch. An drei Beispielen wird die Anwendbarkeit der Methode gezeigt:
1. Bestimmung yon Dinitrophenylhydrazonen von Alde- hyden als Beispiel fiir hydrophobe Komponenten, diinnschichtchromatographisch getrennt;
2. Bestimmung yon Trimelliths~iure, als Beispiel einer sehr hydrophilen Substanz, papierehromatographisch ge- trennt;
3, Bestimmung von N,N'-Dimethyi.harnstoff, als Beispiel einer hydrophilen Substanz, die vor der Bestimmung in ein polarographisch aktives Derivat 0bergeftihrt wet- den mut~.
Sommaire: La sensibilit6 de la spectrophotometrie est souvent insuffisante pour l'analyse quantitative des sub- stances sdparCes par chromatographic sur papier ou en cou- che mince. De plus, il se produit souvent une absorption de fond g~nante. L'utilisation de la polarographie, en particu- lier de la polarographie h rayon cathodique, perment d'augmenter la sensibilitr dans un rapport de 10/1 100 sans que pour cela on puisse observer un signal de fond dfi h la couche absorbante. Les dosages peuvent ~tre effectuCs sur une seule tache m~me pour des traces. La rr des substances est obtenue, dans le cas de la chromatographic en couche mince, par aspiration de la partie de la couche
212 Chromatographia 1, 1968 Original

correspondante dans une colonne courte et 61ution subs~- quente. Pour la chromatographie sur papier on proc~de par d6coupage et extraction darts un solvant appropri& Apr~s r~glage de l'appareil pour obtenir un polarogramme con- venable sur l'6cran cathodique, on proc~de hun enregistre- ment rapide (toutes les 7 secondes un polarogramme d'un dur~e de 2 sec). L'~valuation quantitative est effectu6e graphiquement. Trois exemples nous montrent les possibi- lit~s d'application de cette m6thode:
1. Dosage des dinitropMnylhydrazones de divers ald6hydes, comme exemple de la s6paration de substances hydro- phobes par chromatographie en couche mince.
2. Dosage d'acide trimellitique, comme exemple de la s6paration de substances hydrophiles par chrolnato- graphie sur papier.
3. Dosage de la N,N' dim6thylur~e illustrant le cas de sub- stances hydrophiles qui avant dosage doivent ~tre trans- form6es en un d6riv6 utilisable en polarographie.
Zur quantitativen Auswertung von Papier- und Dtinn- schicht-Chromatogrammen werden bis jetzt praktisch aus- schlieBlich optisehe Methoden benutzt. Dabei wird ent- weder direkt gemessen, oder indirekt, nach Abl6sung der Substanz aus der Schieht, die Menge in der gewonnenen L6sung bestimmt. L~ber den Vergleich der beiden Metho- den ist sicheflich das letzte Wort noch nicht gesprochen, doch wird man sagen k6nnen, dafS man sich bei der indi- rekten Methode physikaliseh auf tragfiihigerem Grund be- findet.
Die indirekte optische Methode leidet - unter anderem - an dem Nachteil, daf~ das Signal/Rausch-Verhiiltnis nicht allzu giinstig liegt, oder anders ausgedrtiekt, daf~ bei relativ m~giger Empfindlichkeit mehr oder wertiger schwankende Blindwerte auftreten. Dies gilt besonders f'tir Substanzen, die im UV gemessen werden mtissen. Die erzielbare Ge- nauigkeit wird dadurch eingeschr~nkt, au~erdem ist es in der Regel notwendig, Strichchromatogramme herzustellen, da bei Punktchromatogrammen die Substanzmenge nicht ausreicht.
Es erschien daher lohnend, nach empfindlicheren Be- stimmungsmethoden zu suchen. Hierzu bietet sich die Polarographie, insbesondere in der Ausfiihrungsform der rasch arbeitenden Kathodenstrahlpolarographie an. Die Empfindlichkeit Iiegt dabei gr6genordnungsm~l~ig um den Faktor 10 bis 100 h6her, wiihrend ein Blindwert in einem interessierenden MeBbereich in der Regel gar nicht beobachtet wird. Erkauft wird diese hohe Spezifit~it na- tiirlich mit der Beschr~nkung der Methode auf polaro- graphisch aktive Substanzen. Allerdings kann eine grol~e Anzahl yon inaktiven Substanzen in aktive Derivate iiber- gef'tihr t werden.
Sowohl reversible als auch irreversible Systeme k6nnen mit guter Empfindlichkeit untersucht werden. Die Auf- nahme eines Polarogramms dauert 2 sek., w~ihrend denen eine Potentialdifferenz yon 500 mV durehlaufen wird.
Alle 7 sek. wird das Polarogramm reproduziert. Die Syn- chronisation zwischen Tropfenfall und Spannungsanstieg erfolgt durch einen Abklopfmechanismus. Ein exakter Vergleieh der Empfindlichkeit zwischen konventioneller Polarographie und Kathodenstrahlpolarographie ist nicht m6glich, da Adsorptionsvorg~nge art der Tropfelektrode eine RoUe spielen k6nnen.
Besonders ausgepr~gt ist dieser Effekt bei der Pikrins~ure, die bei Derivativschaltung mit extremer Empfindlichkeit (10 -9 g/nd) nachweisbar ist. Dabei tritt eine ausgepfiigte Querempfindlichkeit gegentiber Fremdkomponenten auf. Auch im Normalfall ist jedoch die Empfindlichkeit der Kathodenstrahlpolarographie wesentlich h6her ( ~ 10 fach), als die der konventionellen Polarographie, Dazu kommt eine wesenflich raschere Arbeitsweise, da die Aufnahme eines Polarogramms -nach vollzogener Enfliiftung - nicht wesenflich liinger dauert als eine Extinktionsmessung.
Als untere Grenze der Substanzmenge, die bei normaler Arbeitsweise noch ein gut auswertbares Polarogramm er- gibt, kann man etwa 1/lg ansetzen. Ausnahmen nach oben und unten kommen vor. Immerhin liegt diese Grenze so niedrig, da~ es in der Regel keine Schwierigkeiten berei- tet, in Punktchromatogrammen auch noch Nebertkompo- nenten mit optimaler Genauigkeit zu bestimmen.
Diese ist im wesentlichen gegeben durch die Fehler bei der Dosierung und bei der Endbestimmung. Sic liegen etwa bei +-- 1% relativ. Dabei wird selbstverst~ndlich vor- ausgesetzt, dat~ sich die Substanz unzersetzt restlos von der Schicht abl6sen lhBt. Die Methode ist somit bei An- wendung eines nicht zu strengen Mal~stabs ftir Gehaltsbe- stimmungen geeignet. Sic wird seit etwa drei Jahren rou- tinem~iNg angewendet.
Arbeitstechnik
Dosiemng auf die Trennschieht
Die Auftragung erfolgt mR Ham~tonspritzen, die mit einer festeinstellbaren Dosiervorrichtung versehen sind. Aut~erdem kann die automatische Dosiervorrichtung ver- wendet werden, die auf Knopfdruck 1/50 des Spritzen- irthalts auf einmal zugibt. Die Spitze der Kantile ist waag- recht abgeschliffen und mut~ in Kontakt mit der Schicht stehen. Um Beseh~digungen der Schicht zu vemleiden, spannt man am besten die Spritze lest ein und nahert die Schicht der Spitze durch langsames Anheben mit einer geeigneten Hebevorriehtung.
Isolierung der Substanz yon der Trennschicht
Die Substanzen sind in der Regel ohne Bespriihung beim Betrachten unter UV-Lampe (366 m~t und 254 mg) zu identifizieren. AndernfaUs ist ein Leitchromatogramm anzufertigen.
Bei Papierchromatogrammen wird der Fleck mit Bleistift angezeichnet und ausgeschnitten. Das Papier wird in Stiick- chen von ca. 3 nun Kantenl~nge geschnitten und diese mit der polarographischen Grundl6sung digeriert.
Chromatographia 1, 1968 Original 213

Bei Diinnschichtchromatogrammen wird der Fleck eben- falls angezeichnet. Anschliefiend wird die Schicht mit einem Absaugeger~t in eine Mikros~ule gesaugt (Fig. 1). Mit wenigen Tropfen eines geeigneten Elutionsmittds wird dann die Substanz in ein Bechergl/ischen eluiert.
5 /13
V2A-Netz
tO crn
Fig. 1 Aspiration unit
Absaugger~it
Syst6me d'aspiration
Bei der Isolierung yon kleinen Substanzmengen ist es wesentlich, daft die Abgrenzung des Flecks in gleicher Gr6f~e erfolgt, wie bei grot~en Substanzmengen, auch wenn der Fleck ldeiner erscheint. Die Unterschiede in der Flek- kengr6fie sind nur scheinbar und werden dutch die relativ hoch liegende optische Nachweisgrenze vorget/iuscht. Unter gleichen Bedingungen (z.B. gleicher Rf-Wert) ist aber die wahre Fleckengr6fie yon der Substanzmenge nicht abh~ngig (unter der Voraussetzung, dat~ keine/3ber- s~ttigungserscheinungen auftreten). Es sind daher immer etwa gleichgrot~e Flecken zu isolieren.
Zur Kontrolle, ob die Substanz ohne Verluste zurtickge- wonnen werden kann, gibt man eine bekannte Menge auf die Schicht und eluiert sofort wieder ohne Entwicklung. Bei einem Vergleich mit direkt in die L6sung gegebener Substanz lassen sich die Vefluste bestimmen und evtl. als Korrekturglied erfassen. Als Verluste kommen irreversible Adsorption an der Schicht sowie Fliichtigkeit in Frage.
Polarographische Aufnahme
Es wurde der Davis - Differential - Kathodenstrahl - Polaro- graph (1) der Fa. Southern Analytical Limited verwen- det. Die Einrichtung zur Differentialpolarographie wurde nicht benutzt. Das Ger~t war mit einem Schnellsehreiber HKS/S 120 1) zur Registrierung ausgestattet. Ein Polaro- gramm wird in 2 sek. geschrieben, die Einstellzeit fiber
l) Firma Dr. W. H6fler, Maschinen und Geriitebau, Ettlingen/Baden.
die ganze Papierbreite betr/igt 0,08 sek. Die Kombination der Anzeigeger/Re Kathodenschirm - Schreiber bringt ein Optimum in Bezug auf Schnelligkeit und Genauigkeit. Die Grobjustierung des Polarogramms ist mit Hilfe des Kathodensehirms raseh durchgeflihrt, worauf sofort die Dokumentation auf dem Schreiber erfolgen kann (zweck- m~f~igerweise mehrfach, unter Mittelung der Werte). Die zeitaufwendige Feinjustierung auf dem Kathodenschirm, die auch FehlerqueUe sein kann, entf'aUt, da auf dem Schreiberstreifen grapkisch ausgewertet werden kann. Die Synchronisation zwischen Schreiber und Ger~it scheint auch bei scharfen Stromspitzen einwandfrei zu sein, wenn das Registriersystem nicht zu sehr ged~impft wird. Dadurch kommt es allerdings zu mechanischen OsziUationen des Schreibers, die aber die Auswertung nicht emstlich be- tfindern.
Die normale polarographische ZeUe fafit 5 ml L6sung, wobei gegen Bodenquecksilber gemessen wird. Dies bringt den Nachteil mit sich, daft die Stromspitzenpotentiale nut mangelhaft definiert sind. Das ist bei der Kathodenstrahl- polarographie besonders unangenehm, weil dieEmpffmd- lichkeit der Bestimmung yon der Lage des Startpotentials abh/ingig ist (Schwankungsbreite ca. -+ 5 % relativ). Die Ursaehe liegt in tier verschiedenen Gr6t~e des Quecksilber- tropfens. Um den Effekt zu eliminieren, wird nicht auf ein definiertes Startpotential eingestellt, sondern das Stromspitzenpotential auf eine definierte SteUe des Ka- thodenschirms (z.B. genau die Mitte) gesetzt. Aufierdem besteht natiidich die M6glichkeit der Zugabe eines inneren Standards nach der ersten Aufnahme. Von dieser M6glich- keit wird sehr h~iufig Gebrauch gemacht, da der Arbeits- aufwand sehr gering ist. Man gibt wenige/A einer ent- sprechenden L6sung mit einer Hamilton-Spritze zu (ein- tauchende Spitze), enfl0ftet erneut und nimmt ein zwei- tes Polarogramm direkt tiber das erste auf. Der Verdiin- nungsfehler kann meist vernachl/issigt werden. In dieser Weise kann eine ganze Eiehkurve aufgenommen werden.
Das Ger~t bietet die M6gliehkeit, sowohl Normal- als aueh Derivativpolarogramme aufzunehmen. Wegen der besseren Grundlinie wird meist vonder zweiten M6glichkeit Ge- brauch gemacht, zumal die Empfindlichkeit trotzdem aus- reichend ist und die Linearit~t der Anzeige nicht beein- tr/ichtigt wird. In manchen Fallen vorkommende Abwei- chungen seheinen auf Adsorptionseffekten an der Tropf- elektrode zu beruhen und nicht auf M/ingeln im elek- trischen Teil der Anlage. Wie schon oben erw/thnt, zeigt die Kathodenstrahlpolarographie eine ausgesprochene Querempfindlichkeit, die z.Zt. eingehend untersucht wird. Dies ist ein Mangel gegentiber der konventioneUen Polaro- graphie, der aber dadurch wettgemacht wird, daft die An- wendung eines inneren Standards sehr einfach ist.
Die Auswertung der erhaltenen Polarogramme bietet keine besonderen Schwierigkeiten, sofern keine Oberlagemngen auftreten (bei der Auswertung yon Chromatogrammen nieht zu erwarten). In alien F~llen werden Peakh6hen ge- messen. Dazu wird bei der Normalmethode zur Konstruk- tion der Grundlinie eine Tangente angelegt, w/ihrend bei der Derivativschaltung entweder eine Sehne konstruiert
214 Chromatographia 1, 1968 Original
4
0,5 Sek.
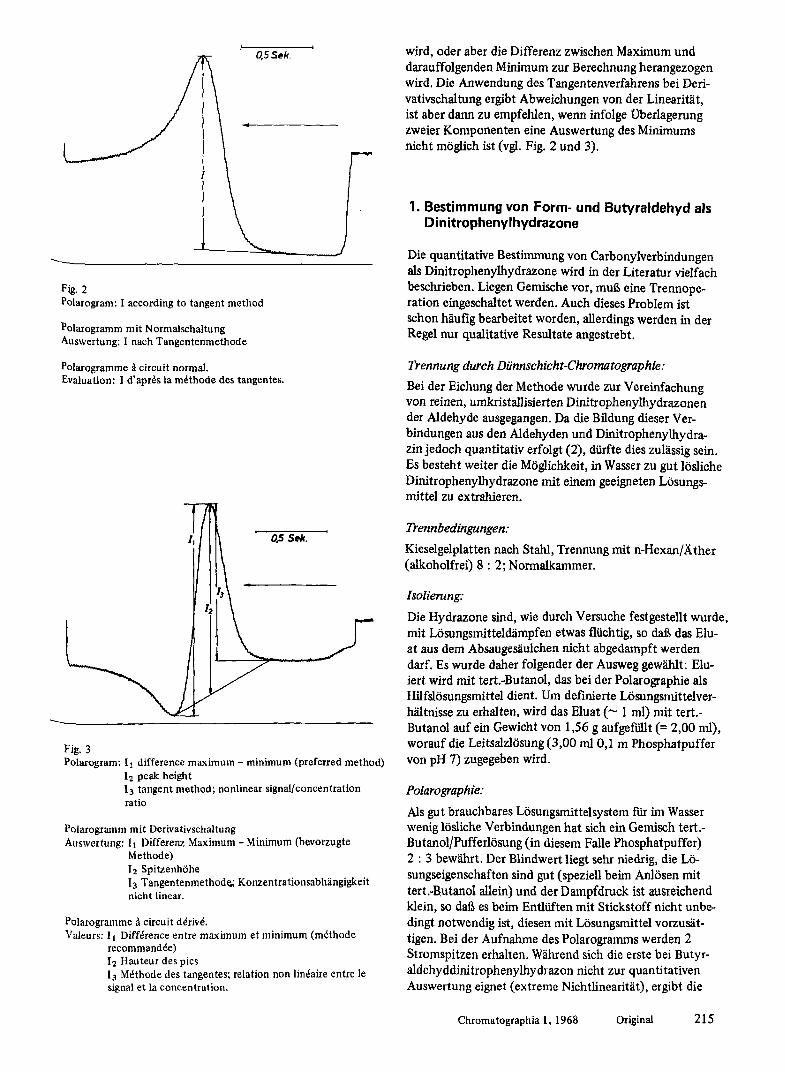
C..- I Fig. 2 Polarogram: I according to tangent method
Polarogramm mit Normalschaltung Auswertung: I nach Tangentenmethode
Polarogramme ~1 circuit normal. Evaluation: I d'apr6s la m6thode des tangentes.
Ij 5 Sek.
Fig. 3 Polarogram: Ix difference maximum - minimum (preferred method)
I2 peak height 13 tangent method; nonlinear signal/concentration ratio
Polarogramm mit Derivativschaltung Auswertung: 11 Differenz Maximum -Minimum (bevorzugte
Methode) I2 Spitzenh~he I3 Tangentenmethode.; Konzentrationsabhiingigkeit nicht lineal
Polarogramme A circuit d~riv6. Vaieurs: 11 Diff&ence entre maximum et minimum (m6thode
recommand6e) 12 Hauteur des pics 13 M6thode des tangentes; relation non lin6aire entre le signal et ia concentration.
wird, oder aber die Differenz zwisehen Maximum und darauffolgenden Minimum zur Berechnung herangezogen wird. Die Anwendung des Tangentenverfahrens bei Deri- vativsehaltung ergibt Abweiehungen yon der Linearit~t, ist aber dann zu empfehlen, wenn infolge Obeflagerung zweier Komponenten eine Auswertung des Minimums nieht m6glieh ist (vgl. Fig. 2 und 3).
I. Bestimmung von Form- und Butyraldehyd als Oinitrophenylhydrazone
Die quantitative Bestimmung von Carbonylverbindungen als Dinitrophenylhydrazone wird in der Literatur vielfaeh beschrieben. Liegen Gemisehe vor, muf~ eine Trennope- ration eingesehaltet werden. Aueh dieses Problem ist schon h~ufig bearbeitet worden, allerdings werden in der Regel nur qualitative Resultate angestrebt.
Trennung durch Diinnschicht-Chromatographie:
Bei der Eichung der Methode wurde zur Vereinfachung yon reinen, umkristal/isierten Dinitrophenyhhydrazonen tier Aldehyde ausgegangen. Da die Bildung dieser Ver- bindungen aus den Aldehyden und Dinitrophenylhydra- zin jedoeh quantitativ erfolgt (2), dtirfte dies zul~tssig sein. Es besteht weiter die M6glichkeit, in Wasser zu gut 16sliehe Dinitrophenylhydrazone mit einem geeigneten L6sungs- mittel zu extrahieren.
Trennbedingungen:
Kiesdgelplatten naeh Stahl, Trennung mit n-Hexan/Ather (alkoholfrei) 8 : 2; Normalkammer.
Isolierung:
Die Hydrazone sind, wie durch Versuehe festgestellt wurde, mit LOsungsmitteldiimpfen etwas fliiehtig, so dat~ das Elu- at aus dem Absauges/iutehen nicht abgedampft werden darf. Es wurde daher folgender der Ausweg gewiffdt: Elu- iert wird mit tert.-Butanol, das bei der Polarographie als Hilfsl6sungsmittel client. Um definierte L6sungsmittelver- h/iltnisse zu erhalten, wird das Eluat (~ 1 ml) mit tert.- Butanol auf ein Gewieht yon 1,56 g aufgefdlt (= 2,00 ml), worauf die LeitsalzlOsung (3,00 ml 0,1 m Phosphatpuffer von pH 7) zugegeben wird.
Polarographie:
Als gut brauchbares L6sungsmittelsystem for im Wasser wenig 16sliche Verbindungen hat sich ein Gemisch tert.- Butanol/Puffefl6sung (in diesem Falle Phosphatpuffer) 2 : 3 bew~ihrt. Der Blindwert liegt sehr niedrig, die L6- sungseigenschaften sind gut (speziell beim Anl6sen mit tert.-Butanol aUein) und der Dampfdruck ist ausreichend klein, so dat~ es beim Entliiften mit Stickstoff nicht unbe- dingt notwendig ist, diesen mit L6sungsmittel vorzus~t- tigen. Bei der Aufnahme des Polarogramms werden 2 Stromspitzen erhalten. Wiihrend sich die erste bei Butyr- aldehyddinitrophenylhydrazon nicht zur quantitativen Auswertung eignet (extreme Nichtlinearitiit), ergibt die
Chromatographia 1, 1968 Original 215

zweite Spitze bei - 1,06 V bei beiden Verbindungen line- are Eichkurven (Derivativsehaltung). Die Elution mit tert.- Butanol erfolgt bei Formaldehyddinitrophenylhydrazon quantitativ, bei Butyraldehyddinitrophenylhydrazon re- produzierbar und mengenunabhiingig zu 94,0 %. Die Standardabweichung betmg bei 12 Bestimmungen und Mengen yon 5 - 30 tag s = -+ 0,6 %. Bei der Auswertung kommt es mit einer Wahrscheinlichkeit von etwa 10 - 20 % zu Ausreifiern, die offenbar yon einer mangelhaften Iso- liemng der Substanz herriihren, da stets zu tiefe Werte ge- funden werden.
2. Bestimmung von Trimelliths~ure
Die polar0graphische Bestimmung ohne vorhergegangene Trennung wurde beschrieben (3). Eine vorhergehende Trennung erh6ht die Spezifit/it. Eine optische Messung im UV ergibt wegen zu geringer Absorption unbefriedi- gende Resultate. Bei dem verwendeten L6sungsmittelsys- tem ergibt nur Trimesinsiiure denselben Rf-Wert, w/ihrend die iibrigen Benzolpolycarbons~uren nieht st6ren. Trime- sinsfiure ist aber polarographisch nicht aktiv.
Trennung:
Papierchromatographiseh an Schleieher und Seh'till 2043b; L6sungsmittelsystem Propanol/1,5 n NHa, 72 : 28. Die S/iuren mtissen als Ammoniumsalze vodiegen (evil. Ionen- austausch bei vorhergegangener Verseifung). Rf-Wert ca. 0,06. Die Flecke werden im UV-Lieht angezeiehnet (254 mp, Durcldicht).
Bestimmung:
Die ausgesehnittenen Flecke werden mit 5,00 ml 0,1 n H2 SO4 30 Min. digeriert, die L6sung wird polarographiert.
Bedingungen: Derivativschaltung (RC-Glied 50 m sek.), Stromspitzenpotential ca. - 1,45 V.
Die mit Trimellith~ure direkt erhaltene Eiehkurve ist nicht linear. Zwischen 10 und 50 tag betfligt jedoch die Abweichung yon der Linearit/it nur 4 % relativ, so dat~ eine Korrektur unschwierig m6glich ist. Unterhalb 10 tag bis 0,5 tag nimmt die Empfindliehkeit stark ab (bis auf etwa 114 des urspriingliehen Wertes), so da~ die Genauig- keit der Bestimmungen erheblich sinkt. Die Substanz wird aus dem Papier nur zu 97,7 % zuriickgewonnen. Man er- h/ilt unter Beriicksichtigung dieses Faktors eine Standard- abweichung s = +- 0,7 % (I 1 Bestimmungen mit 15 - 50 tag TrimeUiths/iure).
3. Bestimmung yon N, N' - Dimethylharnstoff
Vide Alkylharnstoffe mit der Gruppierung - CO - NHR k6nnen polarographiseh nach Oberfiihrung in die Nitroso- verbindung bestimmt werden. Eine ausfiihdiehe Arbeit tiber dieses Thema soil an anderer Stelle erscheinen. In Gegenwart anderer Alkylhamstoffe mut~ eine Trennung durchgefiihrt werden, was papierehromatographisch ge- schehen kann. Da die Substanz im UV nicht sichtbar ist, mul~ ein Leitehromatogramm angefertigt werden.
Papierchromatographische Trennung:
200 pl einer L6sung mit max. 0,5 % Dimethylharnstoff in Wasser werden als Strich auf 25 cm breitem Normal- papier aufgetragen (GorbachbOrette). Am Rand des Strei- fens wird Hatz gelassen ftir ein Punkt-Leitchromatogramm. Die Trennung erfolgt aufsteigend mit n-Butanol, wasser- ges~ittigt (ca. 17 Std., H6he ~ 35 cm). Das Papier wird getrocknet und der Streifen mit dem Leitchromatogramm der Liinge nach abgesehnitten. Die Farbentwicklung er- folgt dureh Sprfihen mit Diacetylmonoxim (600 mg wet- den in 50 ml H20 gel6st und mit 2n H2SO4 auf 100 ml aufgefiillt). Beim Erwiirmen auf 110 ~ erfolgt Rotf~ir- bung. In H6he dieses Flecks wird der Quere nach ein Streifen aus dem Papier ausgeschnitten, der den Dimethyl- harnstoff enth/ilt.
Polarographische Bestimmung:
Der Papierstreffen wird in kleine Sttickchen geschnitten und mit 20,0 ml Wasser digeriert. Das Wasser wird ab- ffltriert und aus dem Filtrat 10,0 ml in ein 50 ml Mef$- k61bchen abpipettiert. Nach KiiMung im Eisbad gibt man 5 ml Eisessig und I ml 1,5 %ige NatriumnitritlSsung zu und liifit 10 Min. bei Zimmertemperatur stehen. Danach werden 5 ml 0,6 %ige AmidosuffonsiiurelSsung und 5 ml n Salzsaure zugegeben und zur Marke aufgeftillt. Die LO- sung wird sofort polarographisch untersucht, da die Dinit- rosoverbindung nieht vOllig stabil ist. Zur Eiehung werden Mengen yon 0,1 - 1 mg Dimethylharnstoff in gleicher Weise nitrosiert und polarographiseh bestimmt. Das Strom- spitzenpotential liegt bei - 0,72 V. Die N/iherungsstandard- abweichung (6 Werte) ist Sw = +- 2,8 %.
FOr die Ausftihrung der Versuche danke ich den Herren K. Tremmel, B. Riedle, E. FrShlke, K. Nagel und V. Fischer-
Literatur
[1] Davis, 11. M., Seaborn, J.E., Advances in Polarography, Pergamon, London, New York, Paris (1961) Vol. 2, S. 239.
[2] Siggia, S., Quantitative Organic Analysis, Wiley, New York, London 3raedition (1963), S. 92.
[3] Knappe, E., Werdehausen, A., Z. anal. Chem. 208, 284 (1965).
Received: March 9, 1968 Accepted: March 14, 1968
216 Chromatographia 1, 1968 Original





























