Embed Size (px)
Citation preview

C. Groß1 · I. Haußer2 · A. von der Wense3 · C. Langner1 · W. Simoens4 · O. Bau5 · R. Rompel6
W. Meyer1 · J. Rüschoff1
1 Institut für Pathologie,3 Kinderklinik,4 Frauenklinik6 Hautklinik des Klinikum Kassel, Akademisches Lehrkrankenhaus der Philipps-Universität Marburg2 Hautklinik der Ruprecht-Karls-Universität Heidelberg5 Frauenklinik Kreiskrankenhaus Eschwege
Restriktive Dermopathie*
O-förmig offenen Mund, tiefsitzendenOhren und schmächtiger Sattelnase.Dazu kommen multiple Gelenkkon-trakturen. Die Schwangerschaft wirdhäufig durch Polyhydramnion (>85%)und vorzeitigen Blasensprung kompli-ziert. Überdies bestehen in der Regel ei-ne intrauterine Wachstumsretardie-rung und deutlich verminderte Kinds-bewegungen im Sinne einer fetalenAkinesie [4, 17, 21, 23, 28, 32].
Kasuistik
Anamnese und Verlauf
Stationäre Aufnahme einer 29 Jahre al-ten Zweitgravida und Erstpara – einätiologisch unklarer Abort war vorauf-gegangen – mit Polyhydramnion 5 Tagepräpartal wegen vorzeitiger Wehen undFiebers. Keine Konsanguinität der El-tern. Der Mutter waren keine vermin-derten Kindsbewegungen aufgefallen.Nach Induktion der Lungenreifungdurch Betamethason, spontanem Bla-sensprung und unaufhaltsamer WehenGeburt eines 1230 g schweren hypotro-phen weiblichen Frühgeborenen ausSchädellage in der vollendeten 31. SSW.Relativ kurze Nabelschnur.Ausgedehnteglatte Rißwunde der Haut in der vorde-
Die Perinatalpathologie hat in denletzten Jahren eine zunehmende Zahlbislang unbekannter letal verlaufenderSyndrome hervorgebracht. Ihre Identi-fikation ist insbesondere für die Neo-natologen und Gynäkologen von Be-deutung, da in ihren Händen die Ver-antwortung zur genetischen Beratungliegt. Unter diesen Erkrankungen sindDermatosen jedoch selten. Hierzu ge-hört das Krankheitsbild der restrikti-ven Dermopathie (RD), das eine erst inder jüngeren Zeit definierte kongenitaleHauterkrankung mit autosomal rezes-sivem Vererbungsmuster darstellt. Ge-schaffen wurde der Terminus 1986 vonWitt et al. [32], nachdem bereits mehre-re Fälle unter verschiedenen Namen,wie z.B. Aplasia cutis congenita [2, 15,26] oder letale Ichthyosis [22], in der Li-teratur veröffentlicht worden waren[16]. Gleichzeitig erfolgte damit eineeindeutige Abgrenzung dieser Entitätvon anderen mit fetaler Akinesie/Hy-pokinesie Sequenz einhergehendenKrankheiten [18]. In der deutschen Li-teratur beschrieb 1929 als erster Dr. Tas-silo Antoine von der Universitäts-Frau-enklinik Wien [1] diese Erkrankung mitdem Titel:„Ein Fall von allgemeiner, an-geborener Hautatrophie“ [14], als zwei-ter 1938 Dr. Wilhelm Wepler von derUniversität Göttingen unter dem The-ma: „Zur Frage allgemeiner Hypoplasieder Haut“ [31]. Insgesamt finden sichbis 1998 33 Fallvorstellungen [23]. Typi-sche morphologische Befunde sind ei-ne extrem straffe glänzende glatte Hautmit durchscheinenden Blutgefäßen undEinrissen unter der Geburt oder beiFlexionsversuchen [6]. Folge der zustraffen Haut sind eine charakteristischfixierte Gesichtsmorphe mit zu kleinem
Der Pathologe 6·99 | 365
FallberichtPathologe1999 · 20:365–370 © Springer-Verlag 1999
Zusammenfassung
Die restriktive Dermopathie ist eine seltene
autosomal rezessive letal verlaufende Geno-
dermatose. Die Straffheit der dünnen, durch-
scheinenden, leicht einreißenden Haut be-
wirkt intrauterin Die „Fetale Akinesie bzw.
Hypokinesie Deformation Sequenz“ (FADS),
eine charakteristische Fazies mit fixierter Ge-
sichtsmorphe und O-förmig offenem Mund
sowie Gelenkkontrakturen (Arthrogryposis).
Dem Polyhydramnion folgt die stets vor-
zeitige Geburt etwa in der 31. SSW sowie
pulmonale Hypoplasie und respiratorische
Insuffizienz.Wir berichten über einen weite-
ren Fall eines Frühgeborenen mit den typi-
schen klinisch-morphologischen Stigmata,
beschreiben die licht- und elektronenmikro-
skopischen Befunde und diskutieren das
Leiden vor dem Hintergrund der vorliegen-
den Literatur.
Schlüsselwörter
Restriktive Dermopathie · Autosomal
rezessive Genodermatose · Fetale
Akinesie/Hypokinesie-Deformationssequenz ·
Arthrogryposis · Polyhydramnion
* Herrn Prof. Dr. med.W.Wepler, ehemals Leiter
des Instituts für Pathologie Kassel, als
Erstbeschreiber des Krankheitsbildes in der
pathologisch-anatomischen Fachliteratur
zum 90. Geburtstag gewidmet
Dr. C. GroßInstitut für Pathologie, Klinikum Kassel,
Mönchebergstraße 41–43, D-34125 Kassel&/fn-block:&bdy:

C. Groß · I. Haußer · A. von der Wense ·
C. Langner · W. Simoens · O. Bau · R. Rompel ·
W. Meyer · J. Rüschoff
Restrictive dermopathy
Summary
Restrictive dermopathy is a rare, fatal,
autosomal recessive, congenital skin disease.
Rigidity of translucent thin skin, which is
thus highly vulnerable and tears, spontane-
ously causes intra-uterine fetal akinesia or
hypokinesia deformation sequence (FADS),
characteristic dysmorphic facies with fixed
open mouth in O position, and generalized
joint contractures (arthrogryposis). Poly-
hydramnios and pulmonary hypoplasia are
distinctive manifestations, leading to respi-
ratory insufficiency and premature delivery
at about 31 weeks of gestation.We report on
a case of a prematurely born infant who
presented with the typical morphological
features and describe the light- and
electronmicroscopical findings as described
in the literature.
Key words
Restrictive dermopathy · Autosomal
recessive genodermatosis · Fetal
akinesia/hypokinesia deformation sequence ·
Arthrogryposis · Polyhydramnios
deutlich reduzierten Tonofilamenten[32]. Dilatation der interzellulären Räu-me bevorzugt in den basalen und sup-rabasalen Keratinozytenlagen. Gele-gentlicher Mangel an Desmosomen. Inder Dermis sehr wenige, winzige Frag-mente von elastischen Fasern (Abb. 3b),die zum subkutanen Fettgewebe hinimmer kleiner werden. Relativ dünneKaliber der Kollageneinzelfibrillen inirregulärer, auffallend wenig kompak-ter Anordnung. Dazwischen deutlich„degenerierte“ Fibroblasten mit ge-buchteten Kernen und Zytoplasmava-kuolen (Abb. 3c).
Sektionsergebnis
Keine Mißbildungen der inneren Orga-ne. Die Autopsie bestätigte den klini-schen Befund einer respiratorischenInsuffizienz infolge Lungenhypoplasiemit zahlreichen hyalinen Membranen.
Plazenta
770 g schwere für die 31. SSW deutlichübergewichtige Frühgeborenenplazen-ta (Normal: P10 230 g, P50 335 g, P90 450 g)mit diffuser Zottenreifungsarretierung,fokalen morphologischen Zeichen dereingeschränkten Perfusionskapazitätund eben beginnendem Amnioninfek-tionssyndrom [29].
Diskussion
Das Krankheitsbild der RD zeigt socharakteristische klinische Befunde,daß – sofern man mit dem Krankheits-bild vertraut ist – die Verdachtsdiagno-se sofort gestellt werden kann. Die bis1998 nur sehr geringe Zahl von 33 in derWeltliteratur publizierten Fällen könntemöglicherweise in der Unkenntnis be-gründet liegen [23]. Während die Enti-tät erst 1986 von Witt et al. [32] geschaf-fen wurde, hatte bereits 1929 T. Antoine[1] die wesentlichen Kriterien darge-stellt: „Die Hauptveränderung zeigtaber die Haut... . Am Schädel ist sie engund fast unverschieblich auf der Unter-lage, so daß ... das Gesicht einen mas-kenartigen Eindruck macht. Es siehtaus, als ob die Haut zu kurz wäre undjeden Augenblick irgendwo einreißenmüßte... . Der Mund ständig offen... .“
Zusätzlich zu den bereits beschrie-benen Hautveränderungen, dem charak-teristischen fixierten fazialen Ausdruck –
ren Halsfalte (Abb. 1a).Dünne glänzendestraff gespannte Haut mit durchschei-nenden Blutgefäßen sowie initial deut-liche, später rückläufige Hyperkeratoseder Fußsohlen und Handinnenflächen;Hypertelorismus; Hypoplasie der Lider,Fehlen der Wimpern; sehr kleiner O-förmig offener Mund, unbeweglicheLippen; kurze flache Sattelnase; tief an-setzende Ohrmuscheln; Mikrognathie;multiple Beugekontrakturen vornehm-lich der großen, aber auch der Finger-gelenke, ausgeprägt reduzierte Spon-tanmotorik, keine Mimik (Abb. 1a). ImRöntgenbefund Hypoplasie beider Kla-vikel (Abb. 1b). Wegen primärer Atem-insuffizienz Intubation und Beatmungnach 5 Lebensminuten.Wenige Stundenpostnatal Naht der Rißwunde am Halsin Vollnarkose. Entnahme von Haut-biopsien zur histologischen und elek-tronenmikroskopischen Untersuchung.Nach Diagnosestellung keine Auswei-tung der Intensivtherapie aufgrund derinfausten Prognose. Tod am 20. Lebens-tag infolge globaler Ateminsuffizienztrotz IPPV-Beatmung.
Hautbefunde
In der Übersicht fällt eine deutliche Re-duktion der Dermisdicke auf, die höch-stens 4mal so breit ist wie die Epider-mis, während die Dicke im Normalfalldas 5–8fache beträgt [32]. Die stärkereVergrößerung zeigt eine intakte Schich-tung der Epidermis und leichte Hyper-keratose. Auffallend ein vollständigerVerlust der Reteleisten sowie eine deut-liche Verminderung der Hautanhangs-gebilde, wobei die vorhandenen Haar-follikel zudem hypoplastisch, Haar-muskeln nicht erkennbar sind (Abb. 2a).Diagnostisch entscheidend in der Der-mis sind der Mangel an elastischen Fa-sern und dicht gepackte, zur Hautober-fläche parallel ausgerichtete kollageneLamellen, woraus ein sehnenartigesMuster resultiert (Abb. 2b). Weitgehendunauffälliges subkutanes Fettgewebemit regulär gestalteten elastischen Fa-sern.
Elektronenmikroskopie
Meist orthokeratotische leicht abschil-fernde Hornschicht der Epidermis. ImStratum granulosum ausgesprochengrobe fleckig verteilte Keratohyalingra-nula (Abb. 3a) ohne Verbindung zu den
| Der Pathologe 6·99
Fallbericht
366
Pathologe1999 · 20:365–370 © Springer-Verlag 1999
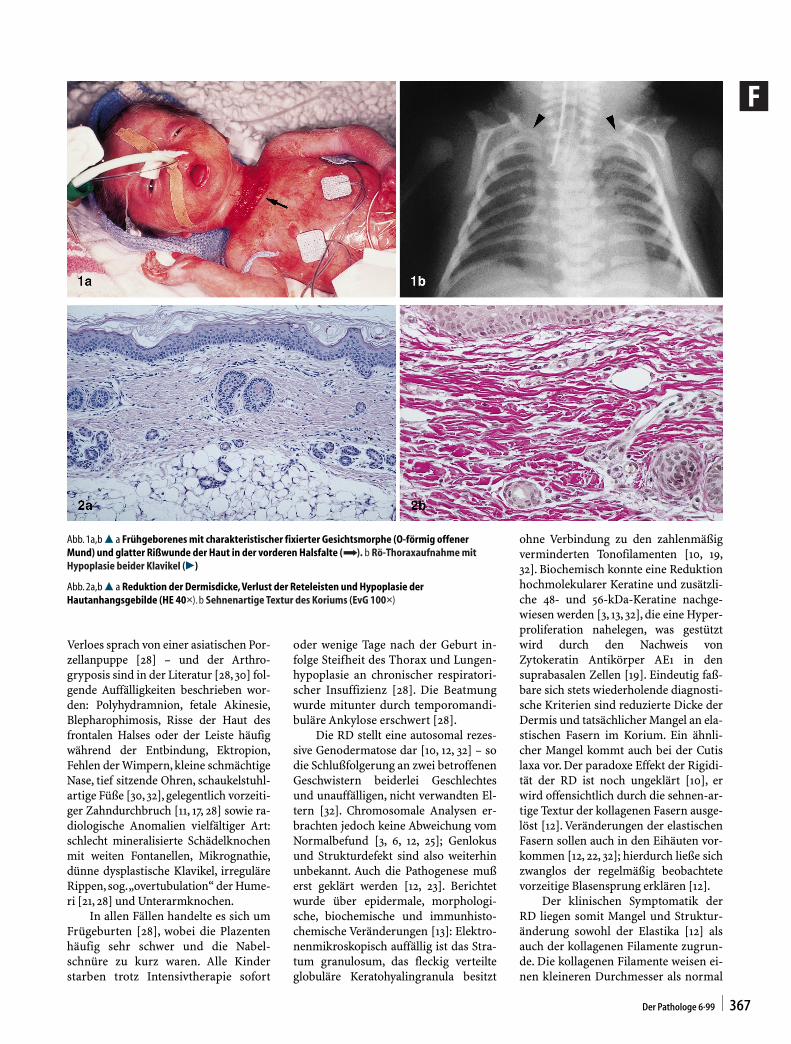
Verloes sprach von einer asiatischen Por-zellanpuppe [28] – und der Arthro-gryposis sind in der Literatur [28, 30] fol-gende Auffälligkeiten beschrieben wor-den: Polyhydramnion, fetale Akinesie,Blepharophimosis, Risse der Haut desfrontalen Halses oder der Leiste häufigwährend der Entbindung, Ektropion,Fehlen der Wimpern, kleine schmächtigeNase, tief sitzende Ohren, schaukelstuhl-artige Füße [30, 32], gelegentlich vorzeiti-ger Zahndurchbruch [11, 17, 28] sowie ra-diologische Anomalien vielfältiger Art:schlecht mineralisierte Schädelknochenmit weiten Fontanellen, Mikrognathie,dünne dysplastische Klavikel, irreguläreRippen, sog.„overtubulation“ der Hume-ri [21, 28] und Unterarmknochen.
In allen Fällen handelte es sich umFrügeburten [28], wobei die Plazentenhäufig sehr schwer und die Nabel-schnüre zu kurz waren. Alle Kinderstarben trotz Intensivtherapie sofort
ohne Verbindung zu den zahlenmäßigverminderten Tonofilamenten [10, 19,32]. Biochemisch konnte eine Reduktionhochmolekularer Keratine und zusätzli-che 48- und 56-kDa-Keratine nachge-wiesen werden [3, 13, 32], die eine Hyper-proliferation nahelegen, was gestütztwird durch den Nachweis vonZytokeratin Antikörper AE1 in densuprabasalen Zellen [19]. Eindeutig faß-bare sich stets wiederholende diagnosti-sche Kriterien sind reduzierte Dicke derDermis und tatsächlicher Mangel an ela-stischen Fasern im Korium. Ein ähnli-cher Mangel kommt auch bei der Cutislaxa vor. Der paradoxe Effekt der Rigidi-tät der RD ist noch ungeklärt [10], erwird offensichtlich durch die sehnen-ar-tige Textur der kollagenen Fasern ausge-löst [12]. Veränderungen der elastischenFasern sollen auch in den Eihäuten vor-kommen [12, 22, 32]; hierdurch ließe sichzwanglos der regelmäßig beobachtetevorzeitige Blasensprung erklären [12].
Der klinischen Symptomatik derRD liegen somit Mangel und Struktur-änderung sowohl der Elastika [12] alsauch der kollagenen Filamente zugrun-de. Die kollagenen Filamente weisen ei-nen kleineren Durchmesser als normal
oder wenige Tage nach der Geburt in-folge Steifheit des Thorax und Lungen-hypoplasie an chronischer respiratori-scher Insuffizienz [28]. Die Beatmungwurde mitunter durch temporomandi-buläre Ankylose erschwert [28].
Die RD stellt eine autosomal rezes-sive Genodermatose dar [10, 12, 32] – sodie Schlußfolgerung an zwei betroffenenGeschwistern beiderlei Geschlechtesund unauffälligen, nicht verwandten El-tern [32]. Chromosomale Analysen er-brachten jedoch keine Abweichung vomNormalbefund [3, 6, 12, 25]; Genlokusund Strukturdefekt sind also weiterhinunbekannt. Auch die Pathogenese mußerst geklärt werden [12, 23]. Berichtetwurde über epidermale, morphologi-sche, biochemische und immunhisto-chemische Veränderungen [13]: Elektro-nenmikroskopisch auffällig ist das Stra-tum granulosum, das fleckig verteilteglobuläre Keratohyalingranula besitzt
Der Pathologe 6·99 | 367
Abb. 1a,b m a Frühgeborenes mit charakteristischer fixierter Gesichtsmorphe (O-förmig offenerMund) und glatter Rißwunde der Haut in der vorderen Halsfalte (➡). b Rö-Thoraxaufnahme mitHypoplasie beider Klavikel (c)
Abb. 2a,b m a Reduktion der Dermisdicke, Verlust der Reteleisten und Hypoplasie derHautanhangsgebilde (HE 40×). b Sehnenartige Textur des Koriums (EvG 100×)
F
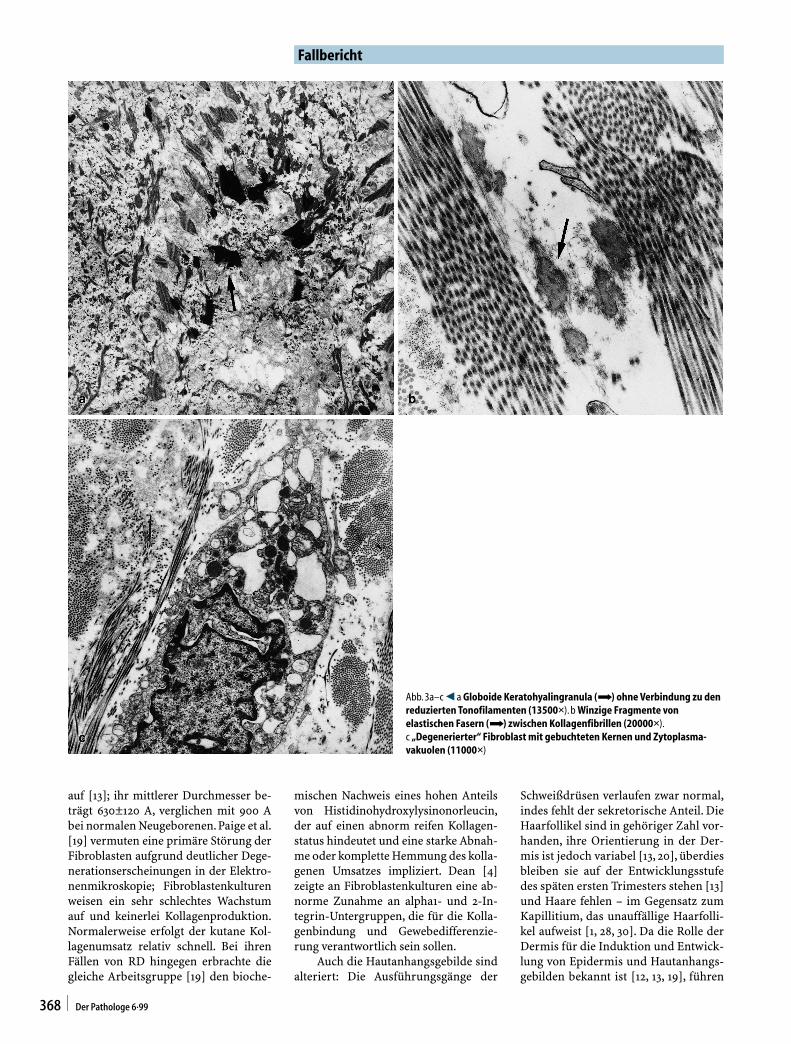
auf [13]; ihr mittlerer Durchmesser be-trägt 630±120 A, verglichen mit 900 Abei normalen Neugeborenen.Paige et al.[19] vermuten eine primäre Störung derFibroblasten aufgrund deutlicher Dege-nerationserscheinungen in der Elektro-nenmikroskopie; Fibroblastenkulturenweisen ein sehr schlechtes Wachstumauf und keinerlei Kollagenproduktion.Normalerweise erfolgt der kutane Kol-lagenumsatz relativ schnell. Bei ihrenFällen von RD hingegen erbrachte diegleiche Arbeitsgruppe [19] den bioche-
Schweißdrüsen verlaufen zwar normal,indes fehlt der sekretorische Anteil. DieHaarfollikel sind in gehöriger Zahl vor-handen, ihre Orientierung in der Der-mis ist jedoch variabel [13, 20], überdiesbleiben sie auf der Entwicklungsstufedes späten ersten Trimesters stehen [13]und Haare fehlen – im Gegensatz zumKapillitium, das unauffällige Haarfolli-kel aufweist [1, 28, 30]. Da die Rolle derDermis für die Induktion und Entwick-lung von Epidermis und Hautanhangs-gebilden bekannt ist [12, 13, 19], führen
mischen Nachweis eines hohen Anteilsvon Histidinohydroxylysinonorleucin,der auf einen abnorm reifen Kollagen-status hindeutet und eine starke Abnah-me oder komplette Hemmung des kolla-genen Umsatzes impliziert. Dean [4]zeigte an Fibroblastenkulturen eine ab-norme Zunahme an alpha1- und 2-In-tegrin-Untergruppen, die für die Kolla-genbindung und Gewebedifferenzie-rung verantwortlich sein sollen.
Auch die Hautanhangsgebilde sindalteriert: Die Ausführungsgänge der
| Der Pathologe 6·99
Fallbericht
368
Abb. 3a–c b a Globoide Keratohyalingranula (➡) ohne Verbindung zu denreduzierten Tonofilamenten (13500×). b Winzige Fragmente vonelastischen Fasern (➡) zwischen Kollagenfibrillen (20000×).
c „Degenerierter“ Fibroblast mit gebuchteten Kernen und Zytoplasma-vakuolen (11000×)

dermale Strukturanomalien wahr-scheinlich zur gestörten Interaktionzwischen Dermis und Epidermis.
Insgesamt sind die Veränderungender Epidermis, die in den ersten Publi-kationen besonders hervorgehobenwurden [32], offensichtlich von unter-geordneter Bedeutung, sie müssen alssekundäre Folge der dermalen patholo-gischen Veränderungen aufgefaßt wer-den. Bedeutsam ist die Reduktion derDermis und die lichtmikroskopischeparallele Anordnung der kollagenenFasern als konstantes Phänomen! Dadie histologischen Veränderungen beiden betroffenen Kindern nicht immergleich kräftig ausgeprägt sind und esauch in ein und demselben Fall zu ört-lich unterschiedlicher Ausprägung desKrankheitsbildes kommt, sind mituntermultiple Hautbiopsien zur Diagnose-findung erforderlich [30]. Selbst nachder Geburt zeigt die Haut keine Rei-fungstendenzen, sie wird eher zuneh-mend gespannt und rissig [28].
Hinsichtlich des subkutanen Fett-gewebes berichtet ein Teil der Autoren[20, 28] über unreife Adipozyten, wäh-rend andere keine wesentlichen struk-turellen Abweichungen [1, 10] nachwei-sen konnten.
Folge der Rigidität der Haut sindfetale Akinesie und Gelenkkontraktu-ren – Erscheinungen, die auch unterdem Oberbegriff „Fetale Akinesie De-formation Sequenz“ (FADS) bzw.„PenaShokeir Phänotyp“ zusammengefaßtwerden [4, 7, 9, 10, 17, 30, 32]. Damit sollbetont werden, daß die ungehinderteintrauterine Bewegung für die normalefetale Entwicklung erforderlich ist –wie das auch an Tierversuchen nachge-wiesen werden konnte [30]. WeitereFolgen der rigiden Haut sind die intra-uterine Wachstumsretardierung [21],die pulmonale Hypoplasie durch einenweitgehend starren Thorax und dashäufig entstehende Polyhydramnion,das durch unzureichendes Verschluk-ken von Amnionflüssigkeit erklärtwird, sowie die oft zu kurze Nabel-schnur [12, 32].
Darüber hinaus wurde über einemangelhafte oder irreguläre Minerali-sation der medialen Klavikelabschnitte[21, 30], über eine übermäßige Beto-nung des tubulären Aspektes der lan-gen Röhrenknochen (sog. „overtubula-tion“) und über weitere Skelettverände-rungen berichtet [21]. Die Ursache die-
drome“, dem Sclerema neonatorumvorkommt, kann jedoch durch typischeklinische und histologische sowie ultra-strukturelle Besonderheiten abge-grenzt werden [5, 28, 30]. Im Gegensatzzur Sklerodermie ist die Abwesenheitder elastischen Fasern differentialdia-gnostisch von Bedeutung [10, 11]. Kon-trakturen und Immobilität sieht manauch bei der Harlekin-Ichthyosis; histo-logisch imponieren jedoch dicke Kera-tinkrusten mit vorzeitiger Reifung undPapillomatose [5, 10]; Beeinträchtigun-gen der fetalen Bewegung und kurzeNabelschnur kommen auch hier vor.
Andere Formen, die ebenfalls miteiner fetalen Akinesie einhergehen –wie das zerebro-okulo-fazio-skeletaleSyndrom und das Neu-Laxova-Syn-drom – sind Folge eines primären ZNS-Defekts, verbunden mit Mikrozephalieoder Verlust der Vorderhörner desRückenmarks [7, 9, 10, 18]. Ähnliche Be-funde wie bei RD konnten in einem iso-lierten Fall des M. Gaucher Typ II asso-ziiert mit Ichthyosis erhoben werden:Hierbei zeigte sich eine verdickte Hautmit offenem Mund und fixierten Extre-mitäten; jedoch bestimmten typischeGaucher-Zellen das histologische Bild[9, 24].
Fazit für die Praxis
RD ist eine eigenständige, leicht vonanderen kongenitalen Hauterkrankungenabzugrenzende Genodermatose. DieKenntnis dieses Krankheitsbildes ist wich-tig, um bei gesicherter Diagnose ange-sichts der infausten Prognose auf eineweitergehende Intensivtherapie zu ver-zichten sowie die betroffenen Familien zuberaten, da das Risiko ein weiteres Kindmit demselben Krankheitsbild zu zeugen,25% beträgt.
Wir danken Frau I. Segschneider für die her-vorragende technische Assistenz bei derHerstellung der elektronenmikroskopischePräparate.
Addendum
Nach Einreichen des Manuskripts hatdie Mutter ein weiteres weibliches Früh-geborenes mit demselben Krankheits-bild in der 30. SSW nach vorzeitigemBlasensprung zur Welt gebracht, welcheswenige Stunden nach der Geburt ver-starb (Platzentagewicht 900 g!).
ser Störungen läßt sich nicht einfachauf der Basis reduzierter Kindsbewe-gungen oder Knochenkompressiondurch die gestraffte Haut erklären [9],möglicherweise ist sie Folge eines überdas bisher Bekannte hinaus reichendenDefekts des Kollagenmetabolismus [19,20, 21].
Eine pränatale Diagnostik ist bis-lang nicht etabliert. Eindeutige histolo-gische Veränderungen treten erst nachder 20.–22. SSW auf [8, 10, 17, 21], wenndie mesenchymale Entwicklung derDermis und die Keratinisation der Epi-dermis sich dem Ende zuneigt [26, 27].Hamel und Mitarbeiter berichten übereinen Fall, in dem fetale Hautbiopsienin der 20. SSW einen falsch-negativenBefund ergaben [8, 10]. Erst nach der 21.SSW entwickelt sich das Polyhydramni-on, erscheinen die Kindsbewegungenreduziert [26, 27]. Deshalb ist es wenigwahrscheinlich, daß immunhistoche-mische oder biochemische Verände-rungen (AE1 Nachweis in den supraba-salen Zellen der Epidermis, Abnahmeder hochmolekularen Keratine, Auftre-ten von niedermolekularen) als früh-zeitige Marker für die pränatale Dia-gnostik dienen könnten [3, 8, 10]. DerDifferenzierungsarrest muß zwischender 20. und 29. SSW liegen [4]. Ein kon-tinuierlich offener Mund war das auf-fallendste Merkmal in der pränatalenSonographie (31. SSW) und könnte – sovan der Stege et al. [25] – ebenso wie dasPolyhydramnion und die Abnahme derfetalen Bewegung ein Hinweis für dieErkrankung sein. Solange ein spezifi-scher Gendefekt nicht identifiziert ist,sind in betroffenen Familien sorgfältigeUltraschalluntersuchungen indiziert[30]. Dagegen kann in nicht betroffenenFamilien ein fixiert offener Mund ledig-lich als Indiz für eine Hauterkrankunggewertet werden [25]. Obwohl verschie-dene andere Syndrome eine verdickteHaut einschließen, gibt es unter Be-rücksichtigung sowohl des klinischenals auch des histologischen Befundeskeine tatsächliche Differentialdiagnose[28, 32]: Die Aplasia cutis congenita hatzwar ähnliche Aspekte, betrifft indesnur ganz bestimmte Hautareale [5, 28]und weist gewöhnlich ausgedehnte Epi-dermisablösungen unterhalb der Basal-membran auf. Die ungewöhnliche Rigi-dität der Haut bei RD, die auch bei Ich-thyosis congenita, dem „stiff skin syn-drome“, dem „Parana hard skin syn-
Der Pathologe 6·99 | 369

Literatur1. Antoine T (1929) Ein Fall von allgemeiner,
angeborener Hautatrophie.Monatsschr Geburtsh Gynäkol 81:276–283
2. Carmi R, Sofer S, Karplus M, Ben-Yakar Y,
Mahler D, Zirkin H, Bar-Ziv J (1982) Aplasiacutis congenita in two sibs discordant forpyloric atresia. Am J Med Genet 11:319–328
3. Dale BA, Holbrook KA,Witt DR,Toriello HV
(1987) Abnormal keratinization in restric-tive dermopathy. Curr Probl Dermatol
17:45–51
4. Dean JCS, Gray ES, Stewart KN, Brown T,
Lloyd DJ, Smith NC, Pope FM (1993)
Restricitive dermopathy: a disorder of skindifferentiation with abnormal integrinexpression. Clin Genet 44:287–291
5. Dehner LP, Kaye V (1992) The skin.In: Stocker JT, Dehner LP (eds) Pediatric
pathology. Lippincott, Philadelphia, p 1135
6. Gillerot Y, Koulischer L (1987) Letter to theeditor: restrictive dermopathy.Am J Med Genet 27:239–240
7. Hall JG (1986) Invited editorial comment:analysis of pena shokeir phenotype.Am J Med Genet 25:99–117
8. Hamel BCJ, Happle R, Steylen PM, Kollée LAA,
Schuurmans Stekhoven JH, Nijhuis JG,
Rauskolb R, Anton-Lambrecht I (1992)
False-negative prenatal diagnosis ofrestrictive dermopathy. Am J Med Genet
44:824–826
9. Hammond E, Donnenfeld AE (1995) Fetalakinesia. Obstet Gynecol Surv 50:240–249
10. Happle R, Schuurmans Stekhoven JH,
Hamel BCJ, Kollée LAA, Nijhuis JG,
Anton-Lambrecht I, Steijlen PM (1992)
Restrictive dermopathy in two brothers.Arch Dermatol 128:232–235
11. Van Hoestenberghe M-R, Legius E,
Vandevoorde W, Eykens A, Jaeken J,
Eggermont E, Devos R, de Wolf-Peeters C,
Fryns J-P (1990) Restrictive dermopathywith distinct morphological abnormali-ties. Am J Med Genet 36:297–300
24. Sherer DM, Metlay LA, Sinkin RA, Mongeon C,
Lee RE,Woods JR (1993) Congenitalichthyosis with restrictive dermopathyand gaucher disease: a new syndromewith associated prenatal diagnostic andpathology findings. Obstet Gynecol
81:842–844
25. Van der Stege JG, van Straaten HLM,
van der Wal AC, van Eyck J (1997) Restrictivedermopathy and associated prenatalultrasound findings: case report.Ultrasound Obstet Gynecol 10:140–141
26. Toriello HV, Higgins JV,Waterman DF (1983)
Autosomal-recessive aplasia cutiscongenita – report of two affected sibs.Am J Med Genet 15:153–156
27. Toriello HV (1986) Invited editorialcomment: restrictive dermopathy andreport of another case. Am J Med Genet
24:625–629
28. Verloes A, Mulliez N, Gonzales M, Laloux F,
Hermanns-Lê T, Piérard GE, Koulischer L (1992)
Restrictive dermopathy, a letal form ofarthogryposis multiplex with skin andbone dysplasias: three new cases andreview of the literature. Am J Med Genet
43:539–547
29. Vogel M (1992) Atlas der morphologischenPlazentadiagnostik. Springer, Berlin
Heidelberg New York (Anhang)
30. Welsh KM, Smoller BR, Holbrook KA, Johnston K
(1992) Restrictive dermopathy.Arch Dermatol 128:228–231
31. Wepler W (1938) Zur Frage allgemeinerHypoplasie der Haut. Beitr Path Anat
101:457–469
32. Witt DR, Hayden MR, Holbrook KA, Dale BA,
Baldwin VJ,Taylor GP (1986) Restrictivedermopathy: a newly recognizedautosomal recessive skin dysplasia.Am J Med Genet 24:631–648
12. Hoffmann R, Lohner M, Böhm N, Leititis J,
Helwig H (1993) Restrictive dermopathy:a lethal congenital skin disorder.Eur J Pediatr 152:95–98
13. Holbrook KA, Dale BA,Witt DR, Hayden MR,
Toriello HV (1987) Arrested epidermalmorphogenesis in three newborn infantswith a fatal genetic disorder (Restrictivedermopathy). J Invest Dermatol 88:330–339
14. Lenz W, Meschede D (1993) Historical noteon restrictive dermopathy and report oftwo new cases. Am J Med Genet
47:1235–1237
15. Leschot NJ,Treffers PE, Becker-Bloemkolk MJ,
Van Zanten S, De Groot WP,Verjaal M (1980)
Severe congenital skin defects in anewborn. Eur J Obstet Gynecol Reprod Biol
10:381–388
16. Lowry RB, Machin GA, Morgan K, Mayock D,
Marx L (1985) Congenital contractures,edema, hyperkeratosis, and intrauterinegrowth retardation: a fatal syndrome inhutterite and mennonite kindres.Am J Med Genet 22:531–543
17. Mau U, Kendziorra H, Kaiser P, Enders H (1997)
Restrictive dermopathy: report undreview. Am J Med Genet 71:179–185
18. Mok Q, Curley R,Tolmie JL, Marsden RA,
Patton MA, Davies EG (1990) Restrictivedermopathy: a report of three cases.J Med Genet 27:315–319
19. Paige DG, Lake BD, Bailey AJ, Ramani P,
Harper JI (1992) Restrictive dermopathy:a disorder of fibroblasts. Br J Dermatol
127:630–634
20. Piérard-Franchimont C, Piérard GE,
Hermanns-Lê T, Arrese Estrada J,Verloes A,
Mulliez N (1992) Dermatopathologicalaspects of restrictive dermopathy. J Pathol
167:223–228
21. Reed MH, Chudley AE, Kroeker M,Wilmot DM
(1993) Restrictive dermopathy.Pediatr Radiol 23:617–619
22. Schnur RE, Ashmead J, Kelley RI (1985)
A lethal ichthyosis variant witharthrogryposis (Abstract). Am J Hum Genet
37:A76
23. Sillevis Smitt JH, van Asperen CJ, Niessen CM,
Beemer FA, van Essen AJ, Hulsman RFHJ,
Oranje AP, Steijlen PM,Wesby-van Swaay E
(1998) Restrictive dermopathy. Report of12 cases. Arch Dermatol 134:577–579
| Der Pathologe 6·99
Fallbericht
370
![Luftnot in der Palliativmedizin.ppt [Schreibgeschützt] · Ursachen von Luftnot • Obstruktive Ursachen – Asthmoide Bronchitis, Stenosen, Tumoren • Restriktive Ursachen – Intrapulmonal](https://img.pdfslide.org/doc/110x75/5e205289af108517021548ca/luftnot-in-der-schreibgeschtzt-ursachen-von-luftnot-a-obstruktive-ursachen.jpg)






![Luftnot in der Palliativmedizin.ppt [Schreibgeschützt]€¦ · – Asthmoide Bronchitis, Stenosen, Tumoren • Restriktive Ursachen – Intrapulmonal (Fibrose, Atelektase, Pneumonie)](https://img.pdfslide.org/doc/110x75/605f76e6b633d170a31b2246/luftnot-in-der-schreibgeschtzt-a-asthmoide-bronchitis-stenosen-tumoren-a.jpg)

