Embed Size (px)
Citation preview

“Retinoic acid signaling after nerve injury”
Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der Rheinisch-
Westfälischen Technischen Hochschule Aachen zur Erlangung des akademischen Grades
einer Doktorin der Naturwissenschaften genehmigte Dissertation
vorgelegt von
Diplom-Biologin Kirsten Schrage aus Hagen (NRW)
Berichter: PD DR. Jörg Mey
Prof. Dr. Hermann Wagner
Tag der mündlichen Prüfung: 01.12.2005
Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.

Table of contents
1 Introduction
1.1 Peripheral nerve lesions – Axon - Schwann cell interactions 1
1.2 Retinoic acid and axonal regeneration 3
1.3 CNS lesions – Spinal cord injuries 5
1.4 Retinoic acid signaling after spinal cord injury 8
1.5 The retinoic acid signaling system 8
1.6 Goals 11
2 Schwann cells are targets of retinoic acid signaling
2.1 Abstract 12
2.2 Materials and methods 13
2.2.1 Preparation of Schwann cell primary cultures from newborn
rat sciatic nerves 13
2.2.2 Western blotting 14
2.2.3 Determination of protein distribution 16
2.2.4 RNase Protection Assay 17
2.2.5 Cytokine treatment 19
2.3 Results
2.3.1 Molecules of the retinoic acid signaling cascade are present
in Schwann cell primary cultures of newborn rats 21
2.3.2 RA treatment induces translocation of retinoid X receptors and
CRABP-II into the nucleus 22
2.3.3 Autoregulatory functions of retinoid signal transduction
in Schwann cells 25
2.3.4 Cytokine expression in Schwann cells 27
2.3.5 Molecular mechanisms of RA-/cytokine interactions
in Schwann cells 29
2.4 Discussion
2.4.1 Intracellular (trans-)location of RA signaling components 33
2.4.2 Autoregulatory functions of RA 35
2.4.3 RA – cytokine interactions 36

3 Reactive microglia/macrophages and neurons are targets of retinoic acid
signaling after spinal cord injury
3.1 Abstract 40
3.2 Materials and methods
3.2.1 Animal experimentation 41
3.2.2 Western blotting 42
3.2.3 Immunohistochemistry 42
3.2.4 Quantification of the intracellular localization of retinoid receptors 43
3.3 Results
3.3.1 Behavioral effects of spinal cord contusion 44
3.3.2 Morphological changes after spinal cord injury 44
3.3.3 RARα and all RXRs are present in the rat spinal cord 46
3.3.4 SCI causes a small reduction in the amount of retinoid
receptor proteins 46
3.3.5 Cellular distribution of retinoid receptors in the adult spinal cord 48
3.3.6 Nuclear localization of retinoid receptors in
reactive microglia/macrophages 50
3.3.7 Retinoid receptor expression in neurons 52
3.3.8 Reactive astrocytes in the glial scar are immunoreactive
for retinoid receptors 52
3.3.9 Retinoid receptor distribution in oligodendrocytes 55
3.4 Discussion
3.4.1 Cellular targets of retinoic acid signaling after spinal cord contusion 56
3.4.2 Intracellular translocation of retinoid receptors 57
3.4.3 Possible physiological functions of RA after spinal cord injury 58
4 General discussion 60
5 Summary 62
6 Bibliography 63

Abbreviations
ANOVA analysis of variance APS ammoniumpersulfate Ara C cytosine arabinosid BBB Basso, Beattie, Bresnahan BCA bicinchoninic acid BDNF brain-derived neurotrophic factor BMP bone morphogenetic protein BSA bovine serum albumin cAMP cyclic adenosine mono-phosphate CNPase 2´,3´-cyclic nucleotide 3´-phosphodiesterase CNS central nervous system CNTF ciliary neurotrophic factor COX cyclooxygenase CRABP cellular retinoic acid binding protein CRBP cellular retinol binding protein Cyp26 cytochrome P450-oxidase, number 26 DAPI 4’,6-diamidino-2-phenylindole dihydrochloride DEPC diethyl-pyrocarbonat DMEM Dulbecco´s Modified Eagle´s Medium DMSO dimethyl sulfoxide DNA desoxyribonucleic acid dpo days post operation DRG dorsal root ganglia ECL enhanced chemiluminiscence ELISA Enzyme-Linked Immunosorbent Assay erbB erythroblastosis gene B Erk extracellular-signal-regulated kinases FCS fetal calf serum GACU guanine, adenine, cytosine, uracil GAP growth associated protein GAPDH glycerinaldehyde-3-phosphate dehydrogenase GDNF glial cell line-derived neurotrophic factor GFAP glial fibrillary acidic protein GFP green fluorescent protein GM-CSF granulocyte macrophage colony stimulating factor gp glycoprotein HeLa cells cells from the cervix of Henrietta Lacks HEPES N-2-Hydroxyethylpiperazine-N´-2ethanesulfonic acid HRP horseradish peroxidase IFN interferon Ig immune globuline IL interleukin IL-6Rα interleukin-6 receptor α Jak Janus kinase LIF leukemia inhibitory factor Lt lymphotoxin MAG myelin-associated glycoprotein MAP2 microtubule associated protein 2 MAPK mitogen-activated protein kinase

MIF macrophage inhibitory factor NC nitrocellulose NCAM neural cell adhesion molecule N-CoR nuclear receptor corepressor NF-κB nuclear factor-kappa B NGF nerve growth factor NGFI nerve growth factor-inducible NGS normal goat serum NLS nuclear localization sequence NT neurotrophin NYU New York University OD optic density P0 myelin protein zero p75 protein 75 PBS phosphate buffered saline PC12 rat phaeochromocytoma PFA para-formaldehyde PKA cAMP-dependent protein kinase PMSF phenylmethylsulfonylfluorid PNS peripheral nervous system PPAR peroxisomal proliferator-activated receptor RA retinoic acid RALDH retinaldehyde dehydrogenase RAR retinoic acid receptors RARE retinoic acid responsive elements Ras rat sarcoma viral oncogene homolog RNA ribonucleic acid ROLDH retinol dehydrogenase RPA ribonuclease protection assay RT room temperature RXR retinoid X receptors SCI spinal cord injury SDS-PAGE sodium dodecyl sulfate – polyacrylamide gel electrophoresis SEM standard error of mean SMAD vertebrate homologues of Sma and Mad of Xenopus SMRT silencing mediator for retinoid and thyroid hormone receptors SOCS suppressors of cytokine signaling SSC cells stromal stem cells Stat signal transducer and activator of transcription TEMED N,N,N´,N´-tetramethylethylenediamine ΤGFβ transforming growth factor β TNF tumor necrosis factor TR thyroid hormone receptor Trk tyrosin kinase VDR vitamin D receptor

1
1 Introduction
1.1 Peripheral nerve lesions - Axon-Schwann cell interactions
Neurons of the peripheral nervous system (PNS) are able to regenerate after nerve injury. This
is in contrast to neurons of the central nervous system (CNS; Ramón y Cajal, 1914). Crushing
or cutting a peripheral nerve triggers a well characterized cascade of cellular and molecular
events, collectively described as Wallerian degeneration, throughout the distal extent of that
nerve. This process involves a number of phases in which the distal portions of all affected
axons, irrespective of size and modality, degenerate and disappear. Within two days
associated myelin sheaths are degraded. Recruited macrophages invade the distal segment and
remove axonal and myelin debris. Schwann cells proliferate showing a maximum after 3 – 4
days leading to steadily increasing numbers up to day 14 after lesion (Oaklander, 1988). The
de-differentiated daughter cells line up within each basal lamina tube to form the bands of
Büngner and become permissive for regeneration. Proximal to the site of injury, axons give
rise to one or more sprouts, each of which is tipped by a growth cone (Ramón y Cajal, 1928).
For axonal regeneration to be successful, growth cones must first reach the distal nerve stump,
which is even possible when they have to cross a gap between the proximal and distal nerve
stumps. Growth cones then contact the basal lamina, and Schwann cells that re-differentiate
after a second burst of proliferation induced by ingrowing axons enwrap the ingrowing axon
forming a new myelin sheath (Fawcett and Keynes, 1990; Fig. 1.1).
Schwann cells have long been known to be central players in Wallerian degeneration and
subsequent regeneration. A differentiated axon-associated Schwann cell is a polarized cell
with a basal, abaxonal surface apposed to a basal lamina (which it secretes), an apical surface
apposed to an axon or group of axons, and lateral surfaces tipped with microvilli, which
interdigitate with adjacent Schwann cells. Functionally, they have been defined almost
entirely in terms of their reciprocal relationships with the axons they ensheathe. Axonal
signals, whether acting by direct contact or diffusible molecules, regulate the expression of
many Schwann cell genes and control both proliferation and differentiation of these cells
(Bolin and Shooter, 1993; Jessen and Richardson, 2001; Lemke and Chao, 1988; Maurel and
Salzer, 2000; Thomson et al., 1993). The relative contributions of the Schwann cells and their
basal laminae have been evaluated in vivo by comparing regeneration in cellular versus a-
cellular nerves: PNS axons will regenerate also in a-cellular nerve grafts, but regeneration is
better in nerves that contain living Schwann cells (Sketelj et al., 1989).

2
Fig. 1.1: Changes in peripheral nerve during Wallerian degeneration. a: Schwann cells surrounded by a basal lamina enwrap an uninjured axon. b: A few days after injury, the axon and its myelin sheath degenerate distal to the lesion site, Schwann cells proliferate and macrophages invade the distal nerve stump to phagocytose myelin debris. c: A few weeks later, the regenerating axon has been re-myelinated by Schwann cells. Jessen, 2001. After axotomy this tightly regulated relationship between axons and Schwann cells is
disrupted. Acutely denervated Schwann cells downregulate transcripts of myelin-specific
proteins, like myelin-associated glycoprotein (MAG), myelin basic protein, protein P0,
periaxin, peripheral myelin protein 22kD, plasmolipin, and the fatty acid-binding protein P2
(Gillen et al., 1997; Mitchell et al., 1990; Willison et al., 1988). They also reduce the
expression of connexin 32, a gap junction protein which forms reflexive contacts within
individual myelinating Schwann cells at paranodes and incisures (Arroyo and Scherer, 2000;
Chandross, 1998), and caveolin-1, a putative regulator of signal transduction and/or
cholesterol transport in myelinating Schwann cells (Nishio et al., 2002). De-differentiated
proliferating Schwann cells upregulate gene expression of the low affinity neurotrophin
receptor p75 (Heumann et al., 1987a; Heumann et al., 1987b; Taniuchi et al., 1986), GAP-43
(Curtis et al., 1992), nerve growth factor (NGF; Matsuoka et al., 1991), brain-derived
neurotrophic factor (BDNF; Korsching, 1993; Meyer et al., 1992), the neuregulin receptors
erbB2, erbB3 and erbB4 (Cohen et al., 1992; Li et al., 1997a; Li et al., 1998), N-CAM and L1
(Jessen et al., 1987; Martini and Schachner, 1988). They increase the expression of laminin
(Kuecherer-Ehret et al., 1990), tenascin (Martini et al., 1990), fibronectin (Lefcort et al.,
1992), the neuropoietic cytokines leukemia inhibitory factor (LIF) and interleukin-6 (IL-6;
Kurek et al., 1996), and netrin-1, a secreted protein which influences growth cone and axon

3
guidance (Madison et al., 2000). Gene expression of the gap junction protein connexin 43,
which may mediate junctional coupling in de-differentiated Schwann cells and so facilitate
the rapid diffusion of signaling molecules between cells downstream of the lesion site
(Chandross, 1998) and TGFβ-1 (Scherer et al., 1993) is also upregulated by proliferating
Schwann cells. These changes in gene expression mean that acutely denervated Schwann cells
are in a reactive, axon-responsive state in the first weeks after injury. Re-innervated Schwann
cells cease dividing, downregulate expression of the molecules listed above and revert to an
axon-associated phenotype.
1.2 Retinoic acid and axonal regeneration
Schwann cells are the most important modulators of axonal regeneration. They are the main
source of trophic factors that promote regeneration after peripheral nerve injury. It is still
unknown how these trophic factors themselves are modulated. My hypothesis is that retinoic
acid (RA), a transcriptional regulator of gene expression, whose importance in embryonic
development is well established (Ross et al., 2000), is one of the factors that control Schwann
cell reactions after nerve injury. All necessary components of the RA signal transduction
pathway are detectable in mammalian peripheral nerves. Among the molecular targets that are
known to be expressed by Schwann cells and regulated by RA in various cell culture systems
unrelated to the nervous system (Mey, 2001) are neuropoietic cytokines, neurotrophins and
members of the transforming growth factor-β (TGFβ) family. They all belong to a diverse
group of proteins with small molecular weight (6-26 kDa), secreted by all cell types that
participate in traumatic interactions after nerve injury (Zhao and Schwartz, 1998), and they
are known to be regulated early after sciatic nerve crush or transection (Gillen et al., 1997).
The neurotrophin family consists of four members in mammals: NGF, BDNF, neurotrophin
(NT)-3 and NT-4 (Ip and Yancopoulos, 1996; Korsching, 1993; Lewin and Barde, 1996;
Snider, 1994). Each neurotrophin binds selectively to high-affinity neurotrophin receptors,
TrkA (tyrosin kinase), TrkB and TrkC and non-selectively to the low-affinity neurotrophin
receptor p75 (Ip and Yancopoulos, 1996). Upon binding their cognate neurotrophin(s), Trk
receptors dimerize and are autophosphorylated via their intracellular kinase domain; this, in
turn, activates various intracellular signaling pathways including Ras, phosphatidylinositol 3-
kinase and phospholipase Cγ (Segal and Greenberg, 1996; Skaper and Walsh, 1998).
In mammals, the TGFβ family is comprised of three isoforms termed TGFβ1, -β2 and -β3.
They initiate their cellular action by binding to heterodimeric receptors type I and type II,
with intrinsic serine/threonine kinase activity (Wrana et al., 1994). The TGF receptors

4
phosphorylate SMAD transcription factors, which translocate from the cytoplasm to the
nucleus (Heldin et al., 1997).
Ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF) and IL-6 belong to the
neuropoietic cytokines that share the common membrane receptor subunit gp130. Their
receptor complex contains gp130 plus one or two more specific receptor subunits. Receptors
signal through the Jak-Stat pathway, in which cytoplasmic STAT proteins translocate into the
nucleus and act as transcription factors (Ip and Yancopoulos, 1996).
Schwann cells synthesize NGF (Heumann et al., 1987a; Heumann et al., 1987b), BDNF
(Funakoshi et al., 1993), GDNF (Iwase et al., 2005), all three TGF-βs (Mews and Meyer,
1993), CNTF (Sendtner et al., 1992b), LIF (Curtis et al., 1993b) and IL-6 (Grothe et al.,
2000). After peripheral nerve lesion their expression is differentially regulated: NGF mRNA
is upregulated early in Schwann cells after axotomy, while BDNF expression is induced
rather late. It has been suggested that NGF promotes sprouting, but not axonal regeneration
(Diamond et al., 1992a, b; Gloster and Diamond, 1992). BDNF, NT-3 and NT-4 support the
survival of motoneurons in culture, after neonatal axotomy and in animal models of motor
neuron disease (Elliott and Snider, 1996; Oppenheim, 1996). The level of TGF-β1 mRNA
increases, whereas the expression of TGF-β3 mRNA falls during Wallerian degeneration. It
has been suggested that increases in TGF-β expression in the distal nerve stump enhance
axonal regeneration by promoting a non-myelinating Schwann cell phenotype as well as
macrophage infiltration (Jessen and Richardson, 2001). LIF mRNA expression increases after
axotomy in the distal nerve (Sendtner et al., 1992). LIF is retrogradely transported in the
axons and functions as a neurotrophic factor for sensory and motoneurons (Matsuoka et al.,
1997). CNTF is downregulated after sciatic nerve lesion, but is released from damaged cells
and exerts a neurotrophic effect on motoneurons (Curtis et al., 1993; Sendtner et al., 1992).
IL-6 and IL-6Rα are also upregulated after axotomy distal to the injury (Bolin et al., 1995; Ito
et al., 1998; Reichert et al., 1996), and IL-6 seems to promote axonal regeneration of motor
axons (Hirota et al., 1996).
The way how RA exerts its regulatory functions after nerve lesion may involve different
mechanisms. 1. They could include changes in expression of RA signaling molecules: CRBP-
I and CRABP-II are strongly upregulated after sciatic nerve injury. Its transcript levels rose
10- and 15-fold (Zhelyaznik et al., 2003). The expression of RARα, RARβ and RXRα rose
after injury (Zhelyaznik and Mey, 2005). 2. RA synthesis is increased: in a transgenic reporter
mouse a local activation of RA responsive elements was induced by sciatic nerve crush and
RALDH2 was found to be biologically active (Zhelyaznik et al., 2003). 3. Modifications in

5
the intracellular localization of CRABP-II which transports RA into the nucleus (Budhu and
Noy, 2002; Dong et al., 1999) and of retinoid receptors which act as transcription factors, may
directly influence the transcriptional activity of RA.
1.3 CNS lesions - Spinal cord injuries
Although CNS axons have an intrinsic capacity to regenerate in vivo (David and Aguayo,
1981; Li et al., 1997b) or in vitro (Naumann et al., 1992; Schubert et al., 2005), if provided
with the correct environment, several factors account for the regenerative failure seen in the
adult CNS. Two circumstances unique to the CNS block regeneration: 1. The glial scar
presents a physical, impenetrable barrier to regeneration and contains several inhibitory
molecules associated with the extracellular matrix such as chondroitin sulphate proteoglycans
(Lepore and Fischer, 2005; McKeon et al., 1991). 2. In the white matter, a regenerating axon
will encounter many myelin-associated molecules expressed on the membrane surface or
released after damage that can inhibit regeneration (Fig.1.2). These include Nogo (Caroni and
Schwab, 1988; Chen et al., 2000), MAG (McKerracher et al., 1994; Mukhopadhyay et al.,
1994) and oligodendrocyte-myelin glycoprotein (Wang et al., 2002; Kottis et al., 2002).
Fig. 1.2: Among several factors that prevent regeneration in the CNS environment the glial scar and myelin associated molecules are the most prominent. Modified after Spencer, 2003.

6
Spinal cord injuries (SCI) can occur due to a number of different insults, generally classified
as a-traumatic and traumatic injuries. A-traumatic SCI mostly result from tumors exerting
continuous pressure on the cord, but can also be caused by vascular disturbances or infections
(Pelser and van Gijn, 1993; Svensson et al., 1993). In traumatic SCI, the cord is subjected to
mechanical stress. Here, the resultant injury is dependent upon the intensity and direction of
the force, and the vertebral level the force is applied to. Lesion can result in either paraplegia
(paralysis of the lower body) or quadriplegia (paralysis of the whole body from the neck
down), depending on whether the injury was received in the thoracic/lumbar region or neck
region of the spinal column. The most common type of traumatic SCI is a spinal cord
contusion injury. It is mostly caused by motor vehicle accidents, diving into shallow water or
sport accidents. It is induced by a relatively short pressure on, or stretching of the cord (Tator
and Edmonds, 1979) most often at cervical and thoracal levels of C5 – C6 (~60 %) and T12 –
L1 (~25 %; Tator, 1983).
After a CNS lesion, two phases of injury can be distinguished: (1) the primary damage
consists of the mechanical tissue destruction. (2) It initiates a cascade of events that further
harm the spinal cord and are mainly caused by local interruption of the blood supply (Dumont
et al., 2001; Schwab and Bartholdi, 1996). This secondary damage leads to further extension
of the lesion, followed by cyst and glial scar formation (Fig.1.3).
Fig. 1.3: Rat spinal cords after injury. The epicenter of the lesion can be detected as early as 15 min after injury (asterisk). Necrosis associated with it progressed up to 3 days after injury (double asterisks). Reduction of the necrosis was observed at 7 and 14 days after injury. Yan, 1999.

7
The ultimate goal in the treatment of SCI is the complete protection of the spinal cord and its
functions after injury (acute phase), and the restoration of lost functions (subacute and chronic
phase). In the acute phase, research focuses on reduction of secondary damage, thereby
minimizing the loss of function, with neuroprotective agents. Of these only the synthetic
corticosteroid methylprednisolone, which has to be given at high dosage within 8 hours after
injury to improve neurological outcome, is in use clinically (Bracken and Shepard, 1998;
Schwab and Bartholdi, 1996). It is known to reduce lipid peroxidation, lessen edema and
inflammation, lower excitatory amino acid release, and inhibit tumor necrosis factor-α
(TNFα) expression and nuclear factor-kappa B (NF-κB) activation (Oudega et al., 1999). In
the subacute and chronic phase, stimulation of axonal outgrowth is the goal of medical
research: Since complete disruption of the cord is rare after SCI, bridges of nerve tissue that
connect regions above and below the lesion often persist, and neurons often exhibit an
initiative regenerative response, as evidenced by an upregulation of immediate early genes,
cytosceletal proteins and the growth associated protein B50 (GAP43). However, ultimately
this regenerative response fails and is followed by a downregulation of growth-associated
genes, leaving the neurons in an atrophic state (Linda et al., 1993; Nacimiento et al., 1995;
Schwab and Bartholdi, 1996; Theriault et al., 1992). Strategies to promote re-growth and
restore functions therefore involve multiple approaches like bridges that form growth-
permissive scaffolds within the lesion site, blocking of inhibitory molecules and
pharmacological stimulation with axonal growth promoting substances. Implantation of
pieces of peripheral nerve (Cheng et al., 1996), fetal tissue (Bregman et al., 2002), olfactory
ensheathing glia (Boyd et al., 2003; Santos-Benito and Ramón-Cueto, 2003) and Schwann
cell bridges (Bunge, 2001) have been assessed. Enzymes were delivered to overcome
inhibitory factors that prevent the formation of or degrade chondroitin sulphate proteoglycans
as they accumulate near the site of injury (Bradbury et al., 2002; Yick et al., 2000). In order to
enable the axons to grow through an inhibitory milieu, surface receptors on growing nerve
fibers that respond to inhibitory factors were blocked by antibodies to the inhibitory factors
(Schwab, 2002) or fragments thereof (Fiedler et al., 2002) or by introducing competitive
antagonists (GrandPré et al., 2002),. Transplants are often tested in conjunction with the
neurotrophins BDNF and neurotrophin 3 (NT-3). These neurotrophins have been shown to
increase axonal growth into transplants (Bregman et al., 2002; Jones et al., 2001; Murray et
al., 2002) and to promote the growth of regenerating axons from transplants into the spinal
cord (Bamber et al., 2001; Oudega and Hagg, 1999). These data demonstrate CNS plasticity
after trauma and give hope for future treatments of spinal cord lesions.

8
1.4 Retinoic acid signaling after spinal cord injury
After spinal cord injury, microglia and macrophages clear debris from degenerating cells and
myelin and secrete a number of inflammatory cytokines. Due to these functions microglia has
a decisive influence on the medical outcome after spinal cord injury (Kiefer et al., 2001; Yang
et al., 2005; Batchelor et al., 2004; Gomes-Leal et al., 2004), and differences in the
inflammatory reaction between CNS and PNS have been suggested to be responsible for the
different growth promoting properties of these environments (Lazarov-Spiegler et al., 1998;
Slovodov et al., 2000; Stoll et al., 2002).
The putative relevance of the RA signaling system for CNS injury rests on three kinds of ob-
servations: (1) A subpopulation of proliferating cells that express the proteoglycan NG-2 as
well as RALDH2 were specifically observed in the vicinity of a spinal cord contusion, and
RA-synthesizing activity by those enzymes increased locally after the lesion (Mey et al.,
2005). (2) Corcoran, Maden and coworkers (2002a) found a strong correlation between
axonal regeneration and the expression of RARβ2: Embryonic mouse spinal cord tissue
extended neurites after application of RA in vitro and this correlated with the expression of
RARβ2, whereas tissue from adult mouse spinal cord did not regenerate neurites and did not
show an upregulation of RARβ2. Adult spinal cord tissue which was transfected to over-
express RARβ2 showed prolific neurite outgrowth in vitro. (3) In retinoid-deficient adult rats,
motoneurons showed an accumulation of intracellular neurofilaments, vacuoles and an
increase in astrocytosis, associated with motoneuron disease. These effects were ascribed to
the failed activation of RARα (Corcoran et al., 2002b).
1.5 The retinoic acid signaling system
Retinoic acid (RA) is the bioactive metabolite of vitamin A (retinol) which acts on cells to
establish or change the pattern of gene activity. Vitamin A that is obtained from the diet is
stored in the liver in the form of retinyl esters (Blomhoff, 1994). For release, the esters are
hydrolyzed to retinol, which is released into the bloodstream for transport within the body
bound to plasma retinol-binding protein. Cells which require RA take up retinol and convert it
to RA through the action of two types of enzymes, retinol dehydrogenases (ROLDH) and
retinaldehyde dehydrogenases (RALDH; Duester, 1996; Napoli, 1996). In addition, there are
cytochrome P450 enzymes known as CYP26 which break down all-trans-RA to 4-oxo-RA,
4-hydroxy-RA and 18-hydroxy-RA (Abu-Abed et al., 1998; White et al., 1996; White et al.,
1997) or 5,8-epoxy-RA (Fujii et al., 1997). Most of these breakdown products are thought to
be inactive metabolites on their way to being excreted.

9
Cellular retinol binding proteins (CRBP-I and -II) and cellular retinoic acid binding proteins
(CRABP-I and -II) bind retinol (CRBP-I), retinol and retinaldehyde (CRBP-II), and RA
(CRABP-I and –II), and are members of the fatty acid-binding protein super-family. CRBPs
facilitate retinol uptake into the target cell and direct the intracellular metabolism of retinol to
retinyl esters (storage form) or RA (only CRBP-I) (Napoli, 1996). CRABPs are hypothesized
to solubilize and protect RA in the aqueous cytosol and to act as shuttles to move lipophilic
RA to various subcellular compartments. Both CRABP-I and -II are found mainly in the
cytosol but have also been reported to be present in the nucleus (Gaub et al., 1998). With 74%
sequence identity the two CRABP isoforms are highly homologous and are also extremely
conserved between species. The CRABPs belong to the family of intracellular lipid-binding
proteins, which comprises small (~15 kDa) proteins that bind to a variety of lipophilic ligands
(Noy, 2000). They are thought to regulate the ability of RA to bind to its receptors and
thereby alter gene transcription (Delva et al., 1999; Dong et al., 1999; Yamamoto et al.,
1998). Kinetic studies of the movement of RA to RAR show that CRABP-I is a passive
vehicle, binding and releasing its ligand depending on concentration gradients. CRABP-I
appears to decrease cellular responses to RA by catalyzing its degradation and thereby
lowering active intracellular RA concentrations (Boylan and Gudas, 1992). In contrast,
CRABP-II appears to increase RA-mediated gene transcription and sensitizes cells to the
effects of RA (see 2.4).
The biological activity of RA is mediated by interaction with retinoid receptors, which are
ligand-activated transcription factors within the nuclei of RA-sensitive cells. They form part
of the gene super-family including the steroid hormone receptors. There are two classes of
these transcription factors, the retinoic acid receptors (RAR) and retinoid X receptors (RXR),
each of which consist of three subtypes, (α, β and γ) with several isoforms of each of these six
genes, formed by differential promoter usage (Chambon, 1996). This complexity gives rise to
numerous receptor combinations as RAR exist as heterodimers with RXR. RXR can
heterodimerize with numerous other nuclear receptor proteins including thyroid hormone
receptors (TR), vitamin D receptor (VDR), and peroxisomal proliferator-activated receptor
(PPAR; Bugge et al., 1992; Kliewer et al., 1992). Activation of the RAR-RXR heterodimer is
controlled by RAR because RXR-selective ligands do not induce gene transcription
(Kurokawa et al., 1994). However, once an RAR ligand is bound, RXR ligands can increase
transcriptional efficiency of the RAR-RXR heterodimer (Minucci et al., 1997). As with all
nuclear receptors, the RAR and RXR contain a DNA-binding and a ligand-binding domain
and recognize consensus sequences known as RA response elements (RARE) which are often

10
present in the upstream promoter sequences of RA-responsive genes. RAR bind all-trans RA
with high affinity and 9-cis-RA with lower affinity, whereas RXR bind 9-cis-RA only
(Soprano et al., 2004). The effects of Vitamin A and its derivatives on gene transcription are
presumed to be mediated largely by all-trans RA with RAR because 9-cis-RA has not been
detected in vivo (Mic et al., 2003).
In the absence of RA, a corepressor is bound to the RAR, preventing gene transcription via
RAREs. When RA binds to RARs, the corepressor is released and replaced with a coactivator,
resulting in the induction of gene transcription. These corepressors and coactivators mediate
their effects by recruiting proteins that regulate the acetylation of histones surrounding the
DNA that contain the RARE. Thus, RAR corepressor proteins recruit histone deacetylase
complexes to remove the acetyle groups from the lysine residues of histones and prevent gene
transcription. In contrast, RAR coactivator proteins recruit histone acetyl transferases to
acetylate these lysine residues, unwind DNA and facilitate gene transcription (reviewed by
Wei, 2003). The major corepressors are nuclear receptor corepressor (N-CoR) and silencing
mediator for retinoid and thyroid hormone receptors (SMRT; Chen and Evans, 1995;
Piedrafita and Pfahl, 1999). Upon binding of RA, the ligand-binding domain of the RAR
changes shape and releases the corepressors. Coactivators are then recruited to the RAR
(Piedrafita and Pfahl, 1999). The RA signaling cascade is summarized in Fig. 1.4.
Fig. 1.4: Pathways for the synthesis and mechanism of action of retinoic acid in the context of nerve regeneration. Modified after Mey, 2001.

11
1.6 Goals
The molecular adjustments of cells after peripheral nerve and spinal cord lesions have been
described in detail. Yet, the intracellular signals that regulate these processes are not
understood as well. Recent evidence indicates that RA is involved in these processes, which
raises the question of the physiological role of RA in the injured nervous system.
As mentioned above RA mediates its biological activity through interaction with retinoid
receptors, which function as transcription factors. The first aim of this work was to investigate
their expression, intracellular distribution and differential regulation in the context of nerve
injury.
The cellular and molecular functions of RA are still not known in this context. Possible
transcriptional targets of RA are cytokines that promote regeneration after nerve injury. I
tested whether the expression patterns of injury-related cytokines and neurotrophins or the
activation of cytokine signaling cascades were altered by RA.
As Schwann cells are the most important modulators of Wallerian degeneration and
subsequent regeneration in the PNS, I used Schwann cell primary cultures of newborn rats to
investigate cellular effects of RA in vitro. In the CNS spinal cord contusion injuries were
performed. Here, special attention was paid to neurons, because of the neurotrophic effects of
RA, and to microglia/macrophages, because anti-inflammatory activity has been ascribed to
RA in different contexts (Sjöberg et al., 1992).
The experiments concerning injury-related processes after 1. peripheral and 2. central nerve
lesions are outlined in separate chapters. Each chapter is introduced by an abstract followed
by the particular materials and methods, results and discussion. A comparison of RA signaling
after central and after peripheral nerve injury is made in a subsequent general discussion.

12
2 Schwann cells are targets of retinoic acid signaling
2.1 Abstract
It is likely that the transcriptional activator retinoic acid (RA) is a modulator of lesion induced
signaling in the adult PNS. To test whether Schwann cells are targets of RA, primary cultures
from sciatic nerves of newborn rats were investigated with Western blotting. I first showed
that crucial components of the RA signal transduction cascade (retinoic acid receptors,
retinoid X receptors, RALDH2, Cyp26A1, CRABP-II) occur in Schwann cells. To discover
whether RA affects Schwann cell physiology, I then analyzed the intracellular distribution of
RA signaling molecules via immunohistochemistry. Protein levels of retinoid receptors and
CRABP-II were investigated after RA exposure. The spatial distribution of RXRs and
CRABP-II changed from a cytosolic to a nuclear localization after RA treatment and CRABP-
II was downregulated by RA. To identify RA-regulated genes in Schwann cells I investigated
the expression of trauma-related cytokines and neurotrophins with the RNase protection assay
and tested whether RA influences their signaling pathways. The following cytokines were
expressed by Schwann cell primary cultures: ßNGF, BDNF, GDNF, CNTF, TGFβ1, TGFβ2,
TGFβ3, IFNβ, IFNγ and MIF. Although in various other cell culture systems unrelated to the
nervous system RA has been reported to regulate these cytokines, I did not see any
differential regulation by RA in Schwann cells. RA did not activate intracellular messenger
cascades of neuropoietic cytokines or neurotrophins either.
While my results are still in accordance with the hypothesis that Schwann cells are targets of
RA transcriptional activity after peripheral nerve injury, the precise physiological function of
RA in Schwann cells remains to be discovered.

13
2.2 Materials and methods
2.2.1 Preparation of Schwann cell primary cultures from newborn rat sciatic nerves
Primary cultures of Schwann cells were obtained from newborn (postnatal day 0 - 2) Sprague-
Dawley rats under sterile conditions. Rats were decapitated with scissors and immersed into
70% ethanol for sterilization. Sciatic nerves were dissected from the dorsal side (Fig. 2.1) and
freed from connective tissue. Nerves of six rats were combined into one petri dish (Greiner, 6
cm ∅) containing 5 ml medium [Dulbecco´s Modified Eagle´s Medium (DMEM, Life
Technologies) with 2 U/ml Penicillin and 2 µg/ml Streptomycin (Sigma)]. Nerves were then
cut up into small pieces with a scalpel and the pieces were collected with a syringe into a 15
ml tube containing 10 ml medium. After centrifugation for 5 min at 130 g the obtained pellet
was digested for 1 h at 37°C in 5 ml DMEM containing 0.06% collagenase (Life
Technologies) and 2.5% trypsin (Sigma). The enzymatic reaction was stopped by adding 5 ml
DMEM containing 10% fetal calf serum (FCS, Life Technologies). Tissue was again spun
down at 130 g for 5 min and the pellet was resuspended in 1 ml serum containing DMEM.
Fig. 2.1: Sciatic nerves were dissected after incisions through skin and muscle.
To gain a single cell suspension, tissue was triturated with small gauge canules (∅ 0.7 mm
and 0.4 mm). After final resuspension of cells in 5 ml medium and centrifugation cells were
plated in uncoated T25-culture flasks with 5 ml serum containing DMEM. Fibroblast growth
was reduced by adding 10 µM cytosine arabinoside (Ara C, Sigma), a cytostatic drug, after 24
hrs for 7 days. Medium was changed every 3 days. After reaching confluency (about 3
months) remaining fibroblasts (about 10%) had to be eliminated via a complement lysis:
adherent cells were washed 3 times with sterile phosphate buffered saline (PBS) and then

14
trypsinated. The serum protease trypsin disrupts peptide chains of amino acid residues of L-
arginine and L-lysine. Cells were incubated for about 2 min with 1.5 ml of a 0.25%
trypsine/EDTA-solution at 37°C and cell disruption was controlled under a microscope.
Again, serum terminated the enzymatic reaction. The cell suspension was transferred into 8
ml FCS containing medium and centrifuged. Pellet was resuspended in 1 ml of fibroblast-
binding monoclonal anti-mouse Thy 1.1 antibody (Sigma M7898), diluted 1/40 in PBS, and 1
ml DMEM and incubated at 37°C for 30 min. Lyophilized baby rabbit complement (Linaris
CL 3441), dissolved in 1 ml sterile water was added for another 30 min. Complement proteins
bind to Thy 1.1 antibody labeled fibroblasts and induce the formation of pores in the cell
membrane and by this lysis of the tagged cells. Lysis was blocked by adding serum containing
medium. Pure Schwann cells were centrifuged, resuspended in 5 ml DMEM containing 10%
FCS, 2 µM forskolin (ICN Biomedicals) and 100 µg/ml bovine pituitary extract (Invitrogen)
and cultured in poly-L-lysine (Sigma) coated (200µg/ml in sterile water) T25-culture dishes.
Confluent Schwann cells were expanded on poly-L-lysine coated T75-culture dishes with
DMEM containing 10% FCS and 2 µM forskolin. Forskolin activates the adenylyl cyclase,
thus raises the level of cyclic adenosine mono-phosphate (cAMP). During experiments cells
were kept in medium without forskolin 24 hrs before RA treatment. Confirmation of Schwann
cell identity was given by immunohistochemical staining with the S-100 antibody (Sigma).
2.2.2 Western blotting
For immunoblotting Schwann cells were treated with of 1, 10 and 100 nM all-trans RA
(Sigma). In all experiments RA was diluted in dimethyl sulfoxide (DMSO, Sigma) and
applied in 10 µl of a 1000-fold stock solution to 10 ml culture medium. Only final
concentrations are mentioned below. Cells treated only with DMSO (also 10 µl in 10 ml
medium) served as control. After 24 hrs and 48 hrs cells were trypsinized and the pellet was
washed with PBS to remove serum constituents. Schwann cells were triturated in 50 µl
hypotonic buffer (20 mM HEPES, pH 4.4) containing 1% Triton X-100 (Serva) for break-
down of cell membranes, and protease inhibitors 1 mM PMSF (Serva), 1 µM leupeptin
(Sigma), 1% aprotinin (Sigma). They were kept on ice for one hour and after 20 min
centrifugation at 12,000 g at 4°C the content of soluble protein extracts was measured by
bicinchoninic acid protein assay kit (BCA, Sigma). In this test alkaline Cu(II) is reduced to
Cu(I) by proteins depending on their concentration. Cu(I) forms a purple complex with
bicinchoninic acid with a maximum of absorbance at 562 nm. The extinction at 562 nm is
proportional to the protein concentration. With bovine serum albumin (BSA, Serva) protein

15
standards were generated of 0.1, 0.2, 0.4, 0.8, 1.4, 2.0 and 3.2 µg/µl. Schwann cell extracts
were diluted 1/10 in hypotonic buffer and to every sample/standard 500 µl of 1x 4% Cu2SO4
(Sigma) and 50x BCA were added. After incubation at 37°C for 30 min extinction was
measured with the ELISA-reader (Bio-Tec Instruments) at 630 nm. Protein amounts were
calculated by linear regression of measured extinctions of protein standards. The test was
performed to apply equal amounts of protein concentrations in the subsequent gel
electrophoresis.
Proteins were separated with SDS-PAGE (sodium dodecyl sulfate – polyacrylamide gel
electrophoresis) according to their size under denaturating conditions (Mini-Protean 3 Cell,
BIO-RAD). A discontinuous buffer system was used, where gel and electrode reservoirs
contained different buffer ions. With use of two different gel systems the compression of
protein samples in a thin starting band at the end of the stacking gel followed by fine
separation within the resolving gel was achieved. The 12% resolving gel [2.5 ml 1.5 mM Tris-
HCl pH 8.8, 4.0 ml acrylamide/bis (30% T, 2.67% C, Sigma), 3.5 ml aqua dest., 100 µl SDS
(10%, Serva), 60 µl ammoniumpersulfate (APS, 10%, Serva), 5 µl TEMED, (VWR)] and
afterwards the 5% stacking gel [1.2 ml 0.5 mM Tris-HCl pH 6.8, 0.5 ml acrylamide/bis (30%
T, 2.67% C), 3.3 ml aqua dest., 50 µl SDS (10%), 30 µl APS (10%), 10 µl TEMED]
polymerized 1 h each. Before the gel was loaded with protein samples and molecular weight
standards (Precision Plus ProteinTM, BIO-RAD), the samples were denaturated by heating for
3 min with 2% SDS and 5% 2-mercaptoethanol. Electrophoresis lasted at 15 mA/gel about 1 h
in the stacking gel and with 30 mA/gel about 45 min in the resolving gel. Electrophoresis
buffer (pH 8.3) contained 25 mM Tris, 192 mM glycine and 0.1% SDS.
To expose separated proteins to antibody labeling, they were transferred onto a nitrocellulose
(NC) membrane by tank blotting. The transfer occurred in a gel cassette (Mini Trans-Blot
electrophoretic transfer cell, BIO-RAD) in transfer buffer (25 mM tris, 192 mM glycine, pH
8.3) at a power of 35 W for 2 hrs or 4 W overnight. The protein transfer was checked by
incubation with Ponceau-S solution (3% Ponceau-S, 30% TCA, 30% sulfosalicilic acid) for 5
min at room temperature (RT).
For antibody staining of the blotted proteins an enhanced chemiluminiscence reaction was
performed (ECL; Amersham). ECL is a light emitting, non-radioactive method for the
detection of immobilized specific antigens with horseradish peroxidase (HRP) labeled
antibodies. Chemical excitation is the effect of HRP catalyzed oxidation of luminol under
alcaline conditions and the presence of chemical enhancers such as phenols. The maximum
light emission occurs at a wavelength of 428 nm and can be detected with X-ray films.

16
Non-specific binding sites were blocked with 10% BSA in Tris buffered saline/Tween 20
buffer (TBS-T; 20 mM Tris, 0.8% NaCl, 0.1% Tween 20, pH 7.6) for 10 min (polyclonal
antibody) or 1 h (monoclonal antibody) at RT on a laboratory shaker. Then, the NC
membrane was washed 3 times in TBS-T for 15 sec. The following primary antibodies were
used: polyclonal rabbit serum anti RARα (dilution 1/500, Santa Cruz sc-551), RARβ (1/500,
sc-552), RXRα (1/500, sc-553), RXRβ (1/500, sc-831) and RXRγ (1/500, sc-555) as well as
mouse monoclonal anti RARγ (sc-7387) and CRABP-I (1/1000, Affinity BioReagents, MA3-
813). The CRABP-II monoclonal antibody (1/1000) was kindly provided by Pierre Chambon,
Strasbourg. RALDH-2 (gift from Peter McCaffery, Boston) and Cyp26A1 (Alpha Diagnostic)
were detected with a rabbit antiserum (1/2000). All antibodies were diluted in TBS-T/1%
normal goat serum (NGS, Sigma). Incubation was done 1 h at RT on a rocker. After washing
3 times short and once for 10 min with TBS-T, NC membrane was incubated for 15 min with
peroxidase conjugated secondary antibodies goat anti-rabbit IgG (1/5000) and goat anti-
mouse IgG (1/10000, Sigma A6154, A3682). Secondary antibodies were diluted in TBS-T
plus 5 % gelatine (Amersham). The washing procedure was repeated and immunoreactivity
was detected via ECL (equal volumes of detection solutions from ECL kit), exposure on an
X-ray film (Hyperfilm ECL) for 1 to 30 min (depending on the intensity of the signal) and use
of digital image analysis system (LTF) to measure the optical density of protein bands. For
statistics logarithms of immunoreactivity values were analyzed with ANOVA and post-hoc
Dunnett´s test.
2.2.3 Determination of protein distribution
For immunofluorescence staining Schwann cells were subcultured in 24-well-plates on poly-
L-lysine coated glass cover slips (VWR). After reaching confluency they were treated with 10
nM all-trans-RA or with DMSO for 5, 15, 30, 60 min and 19 hrs. Afterwards, the culture
medium was removed and cells were fixed in 1 ml/well of 4% para-formaldehyde (PFA,
Fluka) for 15 min and washed with TBS 3 times for 5 min. To avoid background staining
cells were blocked for 90 min with 300 µl/well of 2% normal goat serum NGS in TBS and
0,4% Triton X-100 for permeabilization. Schwann cells were incubated overnight at 4°C with
primary antibodies (300 µl/well) against RALDH-2, Cyp26A1 (1/200), CRABP-I and -II
(1/1000), RARα, β, γ and RXRα, β, γ (1/100), also diluted in 1% NGS and 0,04% Triton X-
100. After three washes in TBS (5 min each), cells were incubated for 60 min with 300
µl/well of Alexa FlourTM 546 goat anti rabbit (1/1000) and goat anti mouse (1/2000) IgGs
(MoBiTec). Following three washes with TBS (5 min each), Schwann cell covered slips were
17
embedded upside down in Fluoprep (Merieux) on a slide. Cells were analyzed by
fluorescence microscopy using a Zeiss axiophot fluorescent microscope (20x objective for
cell counting and 100x objective for pictures). Pictures were made with Zeiss Axiovision
software. For quantitative categorization according to the intracellular localization of the
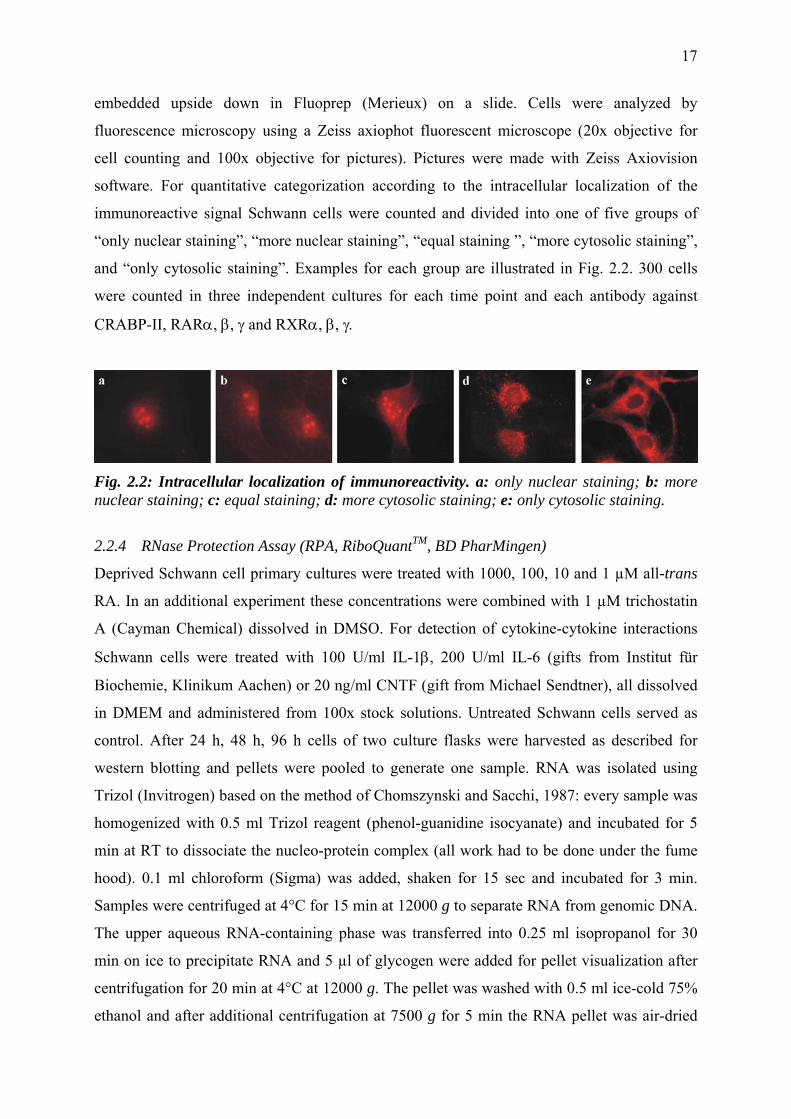
immunoreactive signal Schwann cells were counted and divided into one of five groups of
“only nuclear staining”, “more nuclear staining”, “equal staining ”, “more cytosolic staining”,
and “only cytosolic staining”. Examples for each group are illustrated in Fig. 2.2. 300 cells
were counted in three independent cultures for each time point and each antibody against
CRABP-II, RARα, β, γ and RXRα, β, γ.
Fig. 2.2: Intracellular localization of immunoreactivity. a: only nuclear staining; b: more nuclear staining; c: equal staining; d: more cytosolic staining; e: only cytosolic staining.
2.2.4 RNase Protection Assay (RPA, RiboQuantTM, BD PharMingen)
Deprived Schwann cell primary cultures were treated with 1000, 100, 10 and 1 µM all-trans
RA. In an additional experiment these concentrations were combined with 1 µM trichostatin
A (Cayman Chemical) dissolved in DMSO. For detection of cytokine-cytokine interactions
Schwann cells were treated with 100 U/ml IL-1β, 200 U/ml IL-6 (gifts from Institut für
Biochemie, Klinikum Aachen) or 20 ng/ml CNTF (gift from Michael Sendtner), all dissolved
in DMEM and administered from 100x stock solutions. Untreated Schwann cells served as
control. After 24 h, 48 h, 96 h cells of two culture flasks were harvested as described for
western blotting and pellets were pooled to generate one sample. RNA was isolated using
Trizol (Invitrogen) based on the method of Chomszynski and Sacchi, 1987: every sample was
homogenized with 0.5 ml Trizol reagent (phenol-guanidine isocyanate) and incubated for 5
min at RT to dissociate the nucleo-protein complex (all work had to be done under the fume
hood). 0.1 ml chloroform (Sigma) was added, shaken for 15 sec and incubated for 3 min.
Samples were centrifuged at 4°C for 15 min at 12000 g to separate RNA from genomic DNA.
The upper aqueous RNA-containing phase was transferred into 0.25 ml isopropanol for 30
min on ice to precipitate RNA and 5 µl of glycogen were added for pellet visualization after
centrifugation for 20 min at 4°C at 12000 g. The pellet was washed with 0.5 ml ice-cold 75%
ethanol and after additional centrifugation at 7500 g for 5 min the RNA pellet was air-dried

18
and re-dissolved in 40 µl diethyl-pyrocarbonat (DEPC, Sigma) treated water. Before freezing
RNA was heated for 10 min at 55°C. To estimate the RNA content in the sample the
extinction of the RNA sample was measured photometrically (Hitachi) at 260 nm (OD260)
after 1/100 dilution of extracted RNA. The concentration of total RNA [µg/ml] was calculated
as follows: OD260 x 40 µg/ml x 100 (dilution 1/100).
The subsequent ribonuclease protection assay was performed in the university´s radiation
safety because of the use of the nuclid P33 ([α33P]UTP, Hartmann Analytic, SCF-210, 110
TBq/mmol, 3000 Ci/mmol). The RNase protection assay (RPA) is a highly sensitive and
specific method for the detection and simultaneous quantification of several mRNA species in
a single sample of total RNA. Template sets contain linearized plasmid DNA templates, each
of which represents a single RNA-species, possesses a specific size and can be used for the T7
RNA polymerase-directed synthesis of [α33P]-labeled anti-sense probes. Probes hybridized
with target mRNA encoding βNGF, BDNF, GDNF, CNTF, NT-3 and NT-4 (rNT, Cat. #
45028P) and IFNβ, TNFβ, GM-CSF, TGFβ1, TGFβ3, TGFβ2, Ltβ, TNFα, MIF and IFNγ
(rCK-1, Cat. # 45027P), and two housekeeping gene products, L32 ribosomal protein and
glycerinaldehyde-3-phosphate dehydrogenase (GAPDH). Housekeeping genes are expressed
in almost every cell type and encode for constantly used proteins. The RPA protocol
according to the instruction manual of the RiboQuantTM In Vitro Transcription Kit
(Pharmingen) is briefly described below:
For synthesis of radioactive labeled RNA antisense-probes via T7 RNA Polymerase the
following substances were used: a template set (containing plasmid DNA fragments under
control of the T7 bacteriophage promoter), RNasin protease for digestion of RNases, GACU
(nucleotids for RNA generation), dithiothreitol for reduction of proteins and radioactive
[α33P]UTP. The reaction was terminated after 1 h by adding template digesting DNase for 30
min. Spared probes were purified via phenol/chloroform-isoamyl alcohol and precipitated
with ammonium acetate and 100% ethanol at –70°C for 30 min. Antisense probes were
hybridized with target mRNA in hybridization buffer overnight. On the following day non-
hybridized probes and other single-stranded RNA were digested adding RNase A which was
inhibited after 45 min via proteinase K for 15 min. Protected hybrids were again purified and
precipitated at –70°C for 30 min and separated via electrophoresis on a denaturing 5%
polyacrylamide gel. Probes of housekeeping genes (L32, GAPDH) were used to compare the
levels of individual mRNA species between samples and served as control for integrity of the
RNA in the RPA procedure. Dilutions (1/10, 1/100) of the probe set were used as size
markers. These unprotected probe bands migrate slower than their appropriate protected
19
bands, e.g. the unprotected probe of βNGF has a nucleotide size of 351 and that of the
protected probe is 322 (see table 2.1). This is due to flanking sequences in the (unprotected)
probe that are not protected by mRNA. Samples of rat control RNA and yeast tRNA served as
positive and negative control, respectively. Gel was dried in a gel dryer (BIO-RAD) under
vacuum for 1.5 hrs at 75°C and was then exposed to an X-ray film in an autoradiography
cassette for at least three days at –70°C.
Table 2.1: Templates with corresponding unprotected and protected nucleotide (nt) sizes.
template unprotected probe (nt)
protected probe (nt)
template unprotected probe (nt)
protected probe (nt)
βNGF 351 322 TGFβ1 285 256
BDNF 315 286 TGFβ3 255 226
GDNF 282 253 TGFβ2 231 202
CNTF 255 226 Ltβ 210 181
NT-3 231 202 TNFα 189 160
NT-4 213 184 MIF 171 142
IFNβ 390 361 IFNγ 158 127
TNFβ 351 322 L32 141 112
GM-CSF 324 295 GAPDH 126 97
2.2.5 Cytokine treatment
Deprived Schwann cell primary cultures were treated with 10 nM all-trans RA for 3 h, 6 h, 12
h, 24 h and 48 h for cytokine treatment; for neurotrophin treatment 48 h. Medium was
changed and neuropoietic cytokines [20 ng/ml CNTF, 200 U/ml IL-6 or IL-6 plus soluble IL-
6 receptor α-subunit (1 µg/ml; gifts from Institut für Biochemie, Klinikum Aachen)] or
neurotrophins [20 ng/ml NGF (Sigma, N8133) or 20 ng/ml BDNF (Sigma, B3795)] were
added for 30 min.
After protein isolation, discontinuous SDS-PAGE and Western blot (see 2.3.2)
phosphorylated and unphosphorylated Stat3 or Erk1/Erk2, respectively, were detected with
rabbit antisera, diluted 1/1000 (Stat3: #S21320, Transduction Laboratories; Phospho-Stat3:
#9131, polyclonal p44/42 MAP Kinase: #9102 and polyPhospho-p44/42 MAP Kinase: #9101,
Cell signaling technology). Peroxidase-conjugated secondary antibodies goat anti-rabbit IgG
were diluted 1/5000. Immunoreactivity was recorded with ECL and compared in RA-treated
and untreated Schwann cells. The best way of confirming equal amounts of applied protein is
stripping (complete removal of primary and secondary antibodies from the membrane) after

20
antibody staining against phospho Stat3 and phospho Erk1/Erk2 and reprobing membranes
with antibodies against unphosphorylated Stat3 or Erk1/Erk2. According to the
manufacturer´s protocol membranes should be stored wet-wrapped in a refrigerator after each
immunodetection and stripped before re-use. In my experience immunoreactivity after
stripping was much weaker than without, so I decided to use a different membrane for
antibody staining against Stat3 and Erk1/Erk2. Later, after I had completed these experiments,
we found out that signals can be improved by stripping membranes immediately after
finishing the immunodetection protocol.
21
2.3 Results
2.3.1 Molecules of the retinoic acid signaling cascade are present in Schwann cell primary
cultures of newborn rats
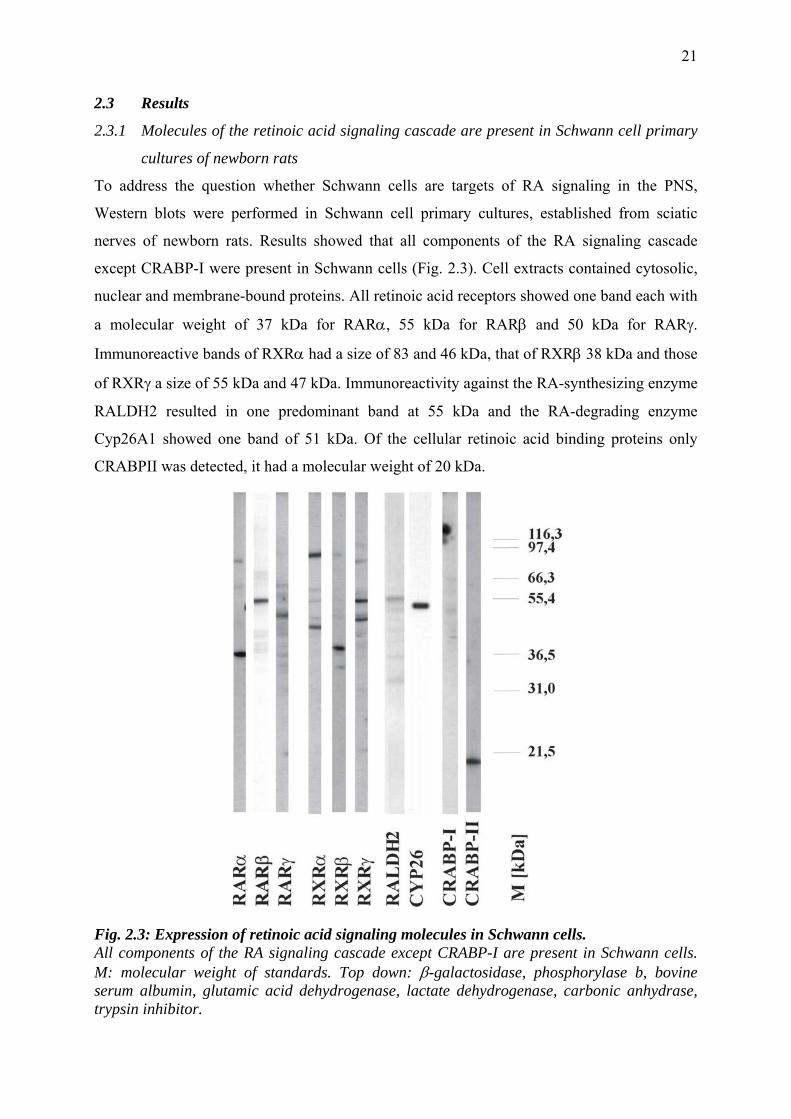
To address the question whether Schwann cells are targets of RA signaling in the PNS,
Western blots were performed in Schwann cell primary cultures, established from sciatic
nerves of newborn rats. Results showed that all components of the RA signaling cascade
except CRABP-I were present in Schwann cells (Fig. 2.3). Cell extracts contained cytosolic,
nuclear and membrane-bound proteins. All retinoic acid receptors showed one band each with
a molecular weight of 37 kDa for RARα, 55 kDa for RARβ and 50 kDa for RARγ.
Immunoreactive bands of RXRα had a size of 83 and 46 kDa, that of RXRβ 38 kDa and those
of RXRγ a size of 55 kDa and 47 kDa. Immunoreactivity against the RA-synthesizing enzyme
RALDH2 resulted in one predominant band at 55 kDa and the RA-degrading enzyme
Cyp26A1 showed one band of 51 kDa. Of the cellular retinoic acid binding proteins only
CRABPII was detected, it had a molecular weight of 20 kDa.
Fig. 2.3: Expression of retinoic acid signaling molecules in Schwann cells. All components of the RA signaling cascade except CRABP-I are present in Schwann cells. M: molecular weight of standards. Top down: β-galactosidase, phosphorylase b, bovine serum albumin, glutamic acid dehydrogenase, lactate dehydrogenase, carbonic anhydrase, trypsin inhibitor.
22
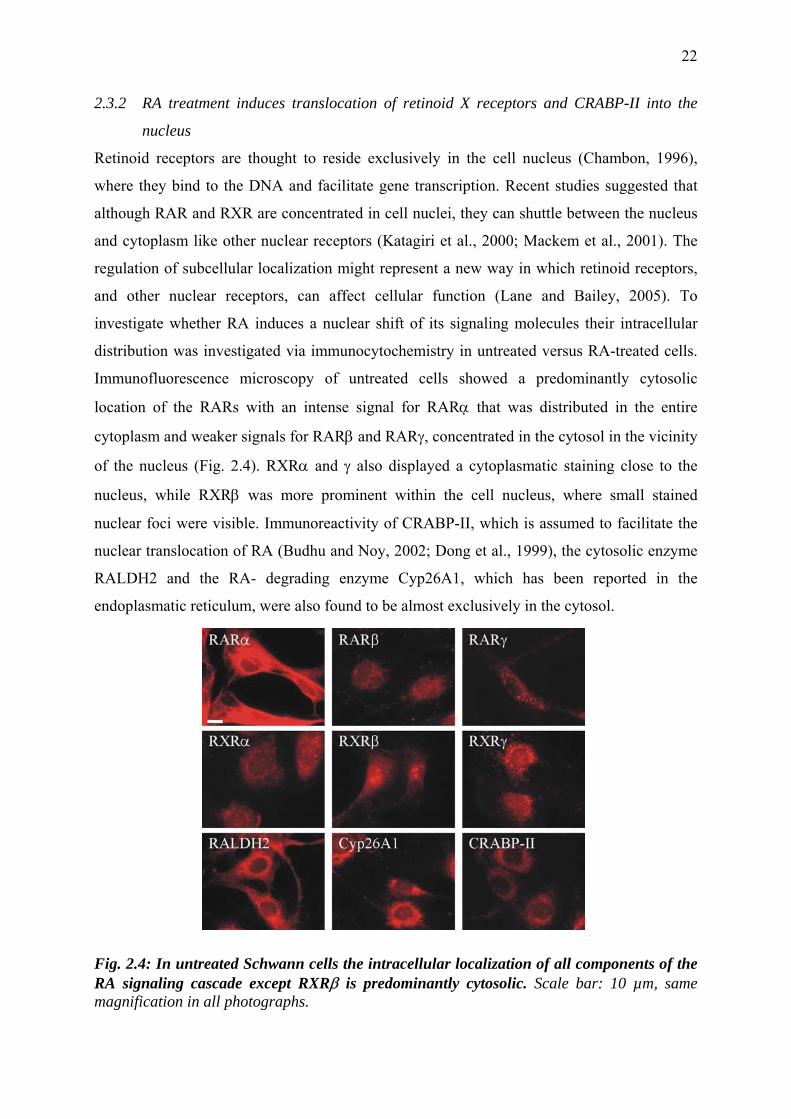
2.3.2 RA treatment induces translocation of retinoid X receptors and CRABP-II into the
nucleus
Retinoid receptors are thought to reside exclusively in the cell nucleus (Chambon, 1996),
where they bind to the DNA and facilitate gene transcription. Recent studies suggested that
although RAR and RXR are concentrated in cell nuclei, they can shuttle between the nucleus
and cytoplasm like other nuclear receptors (Katagiri et al., 2000; Mackem et al., 2001). The
regulation of subcellular localization might represent a new way in which retinoid receptors,
and other nuclear receptors, can affect cellular function (Lane and Bailey, 2005). To
investigate whether RA induces a nuclear shift of its signaling molecules their intracellular
distribution was investigated via immunocytochemistry in untreated versus RA-treated cells.
Immunofluorescence microscopy of untreated cells showed a predominantly cytosolic
location of the RARs with an intense signal for RARα that was distributed in the entire
cytoplasm and weaker signals for RARβ and RARγ, concentrated in the cytosol in the vicinity
of the nucleus (Fig. 2.4). RXRα and γ also displayed a cytoplasmatic staining close to the
nucleus, while RXRβ was more prominent within the cell nucleus, where small stained
nuclear foci were visible. Immunoreactivity of CRABP-II, which is assumed to facilitate the
nuclear translocation of RA (Budhu and Noy, 2002; Dong et al., 1999), the cytosolic enzyme
RALDH2 and the RA- degrading enzyme Cyp26A1, which has been reported in the
endoplasmatic reticulum, were also found to be almost exclusively in the cytosol.
Fig. 2.4: In untreated Schwann cells the intracellular localization of all components of the RA signaling cascade except RXRβ is predominantly cytosolic. Scale bar: 10 µm, same magnification in all photographs.
23
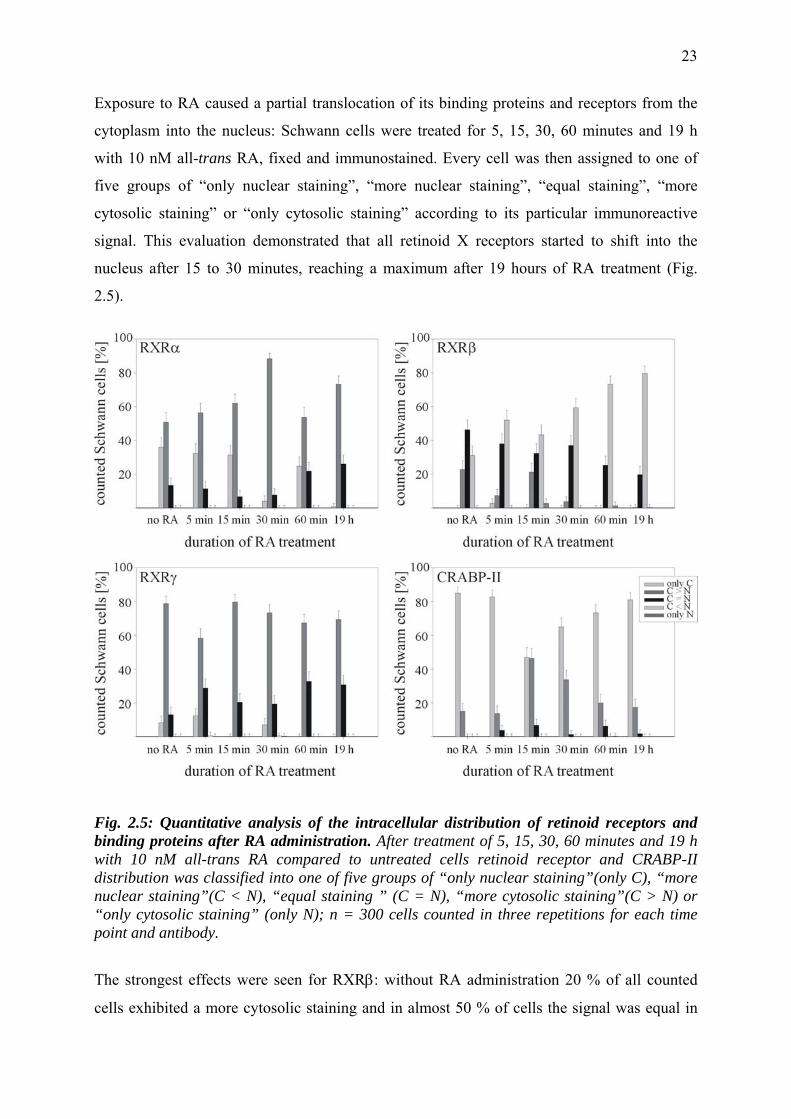
Exposure to RA caused a partial translocation of its binding proteins and receptors from the
cytoplasm into the nucleus: Schwann cells were treated for 5, 15, 30, 60 minutes and 19 h
with 10 nM all-trans RA, fixed and immunostained. Every cell was then assigned to one of
five groups of “only nuclear staining”, “more nuclear staining”, “equal staining”, “more
cytosolic staining” or “only cytosolic staining” according to its particular immunoreactive
signal. This evaluation demonstrated that all retinoid X receptors started to shift into the
nucleus after 15 to 30 minutes, reaching a maximum after 19 hours of RA treatment (Fig.
2.5).
Fig. 2.5: Quantitative analysis of the intracellular distribution of retinoid receptors and binding proteins after RA administration. After treatment of 5, 15, 30, 60 minutes and 19 h with 10 nM all-trans RA compared to untreated cells retinoid receptor and CRABP-II distribution was classified into one of five groups of “only nuclear staining”(only C), “more nuclear staining”(C < N), “equal staining ” (C = N), “more cytosolic staining”(C > N) or “only cytosolic staining” (only N); n = 300 cells counted in three repetitions for each time point and antibody.
The strongest effects were seen for RXRβ: without RA administration 20 % of all counted
cells exhibited a more cytosolic staining and in almost 50 % of cells the signal was equal in
24
cytoplasm and nucleus; 30 % showed a more nuclear staining. After 19 h 20 % of counted
cells exhibited an equal staining of cytoplasm and nucleus and 80 % a more nuclear signal.
CRABP-II was located almost exclusively in the cytoplasm in untreated cells. After RA
treatment it migrated into the nucleus with a maximum after 15 minutes, while after 30
minutes it was again predominantly in the cytoplasm. Typical immunoreactivity data are
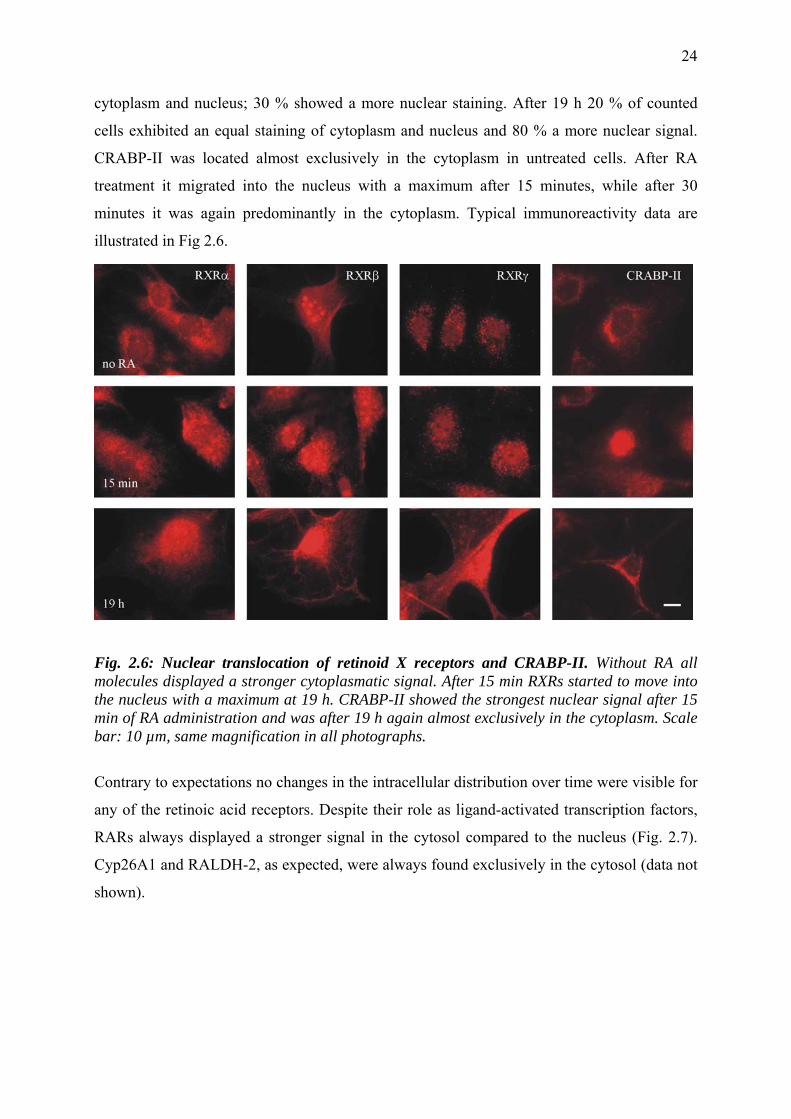
illustrated in Fig 2.6.
Fig. 2.6: Nuclear translocation of retinoid X receptors and CRABP-II. Without RA all molecules displayed a stronger cytoplasmatic signal. After 15 min RXRs started to move into the nucleus with a maximum at 19 h. CRABP-II showed the strongest nuclear signal after 15 min of RA administration and was after 19 h again almost exclusively in the cytoplasm. Scale bar: 10 µm, same magnification in all photographs.
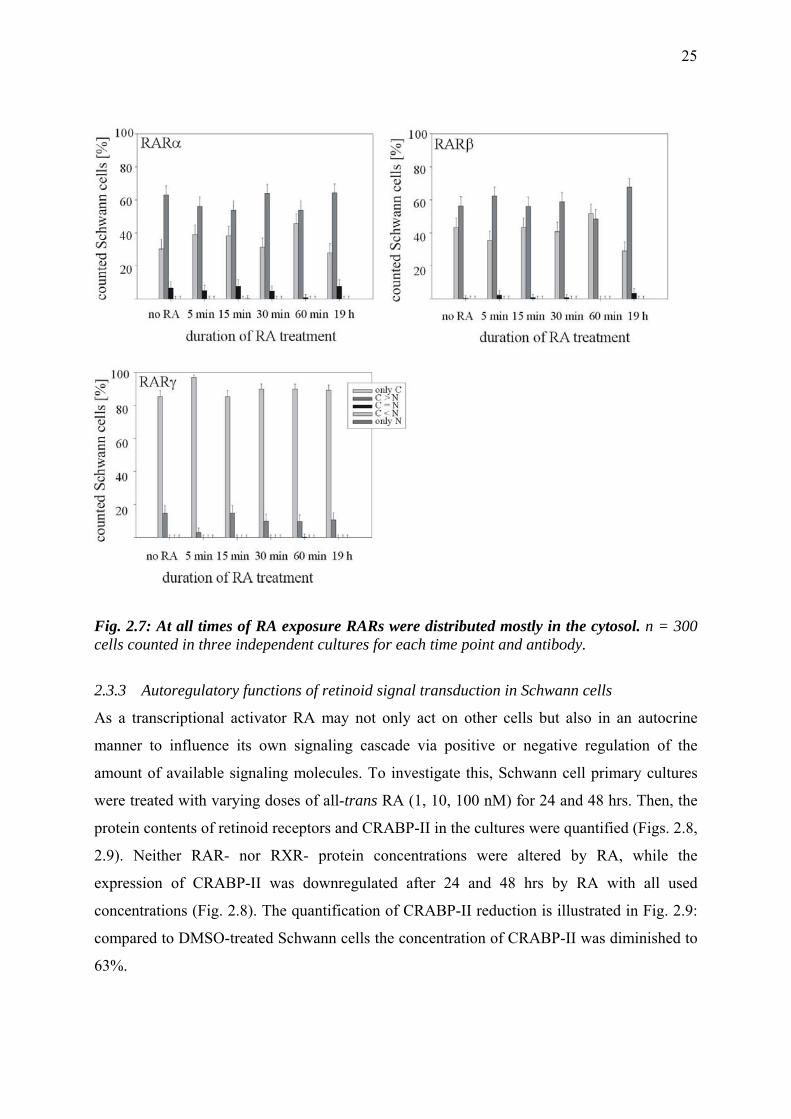
Contrary to expectations no changes in the intracellular distribution over time were visible for
any of the retinoic acid receptors. Despite their role as ligand-activated transcription factors,
RARs always displayed a stronger signal in the cytosol compared to the nucleus (Fig. 2.7).
Cyp26A1 and RALDH-2, as expected, were always found exclusively in the cytosol (data not
shown).
25
Fig. 2.7: At all times of RA exposure RARs were distributed mostly in the cytosol. n = 300 cells counted in three independent cultures for each time point and antibody.
2.3.3 Autoregulatory functions of retinoid signal transduction in Schwann cells
As a transcriptional activator RA may not only act on other cells but also in an autocrine
manner to influence its own signaling cascade via positive or negative regulation of the
amount of available signaling molecules. To investigate this, Schwann cell primary cultures
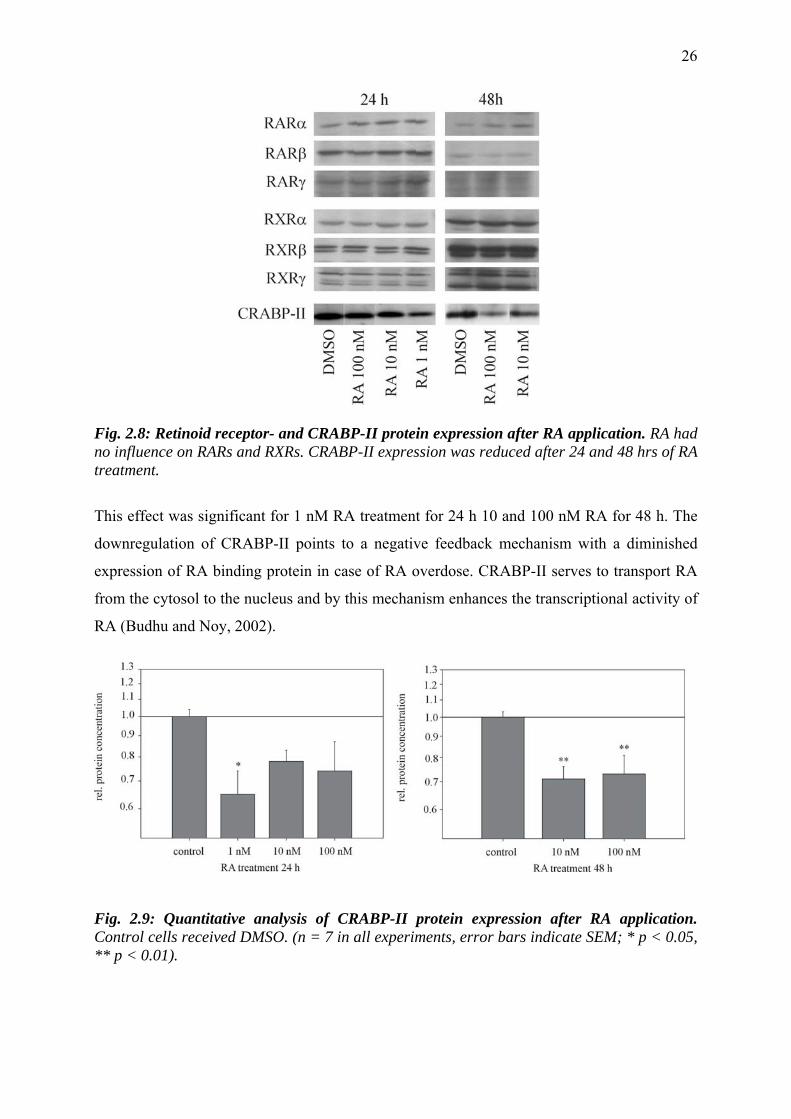
were treated with varying doses of all-trans RA (1, 10, 100 nM) for 24 and 48 hrs. Then, the
protein contents of retinoid receptors and CRABP-II in the cultures were quantified (Figs. 2.8,
2.9). Neither RAR- nor RXR- protein concentrations were altered by RA, while the
expression of CRABP-II was downregulated after 24 and 48 hrs by RA with all used
concentrations (Fig. 2.8). The quantification of CRABP-II reduction is illustrated in Fig. 2.9:
compared to DMSO-treated Schwann cells the concentration of CRABP-II was diminished to
63%.
26
Fig. 2.8: Retinoid receptor- and CRABP-II protein expression after RA application. RA had no influence on RARs and RXRs. CRABP-II expression was reduced after 24 and 48 hrs of RA treatment.
This effect was significant for 1 nM RA treatment for 24 h 10 and 100 nM RA for 48 h. The
downregulation of CRABP-II points to a negative feedback mechanism with a diminished
expression of RA binding protein in case of RA overdose. CRABP-II serves to transport RA
from the cytosol to the nucleus and by this mechanism enhances the transcriptional activity of
RA (Budhu and Noy, 2002).
Fig. 2.9: Quantitative analysis of CRABP-II protein expression after RA application. Control cells received DMSO. (n = 7 in all experiments, error bars indicate SEM; * p < 0.05, ** p < 0.01).

27
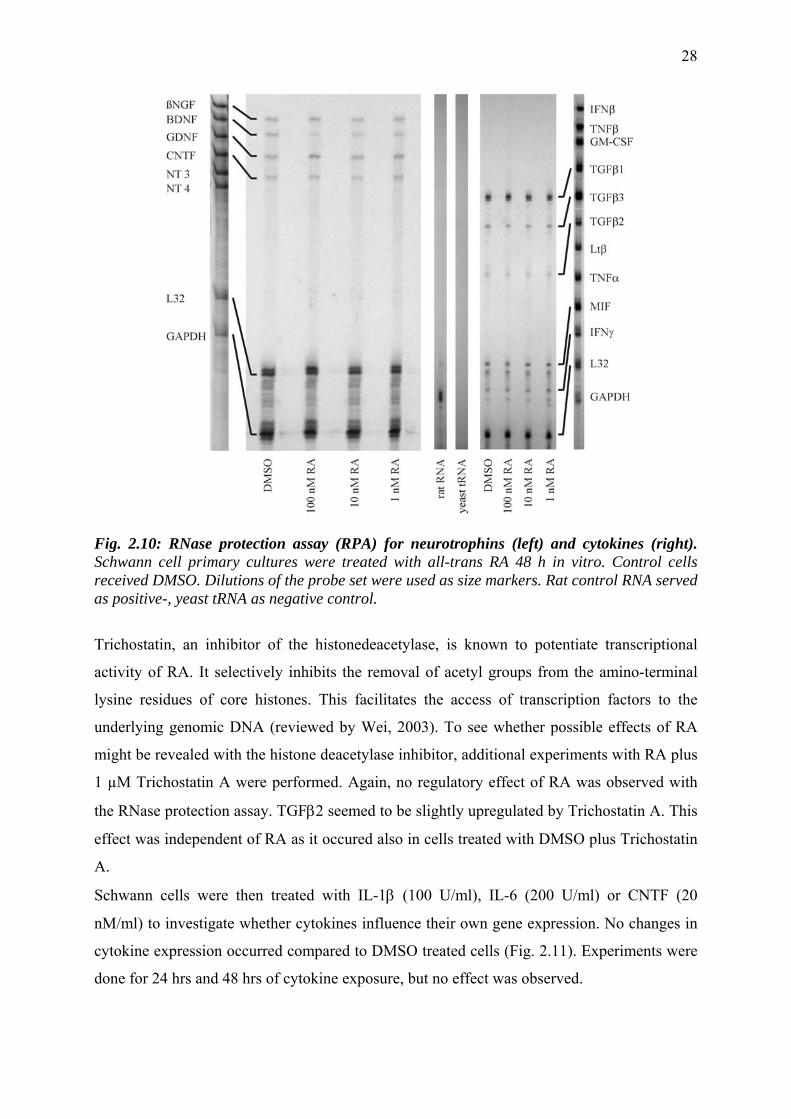
2.3.4 Cytokine expression in Schwann cells
From the RA-effect on intracellular distribution of RXRs and CRABP-II and from
observations after sciatic nerve injury in vivo (Zhelyaznik et al., 2003) I expected that RA
would regulate the expression of lesion-induced genes in Schwann cells. Since neuropoietic
cytokines or neurotrophins serve as mediators in neuroglial interactions and are differentially
regulated after peripheral nerve injury, I investigated whether their expression in Schwann
cells was altered by RA. Therefore Schwann cells were treated with 1, 10 and 100 nM all-
trans RA for 24 h, 48 h and 96 h. I investigated their cytokine expression with ribonuclease
protection assay. The RNase protection assay (RPA) is a highly sensitive and specific method
for the simultaneous detection and quantification of several gene transcripts out of one mRNA
extract. The results showed that ßNGF, BDNF, GDNF, CNTF, TGFβ1, TGFβ2, TGFβ3,
IFNβ, IFNγ and MIF were expressed in Schwann cell primary cultures, while the following
cytokine transcripts could not be detected: NT3, NT4, TNFα, TNFβ, GM-CSF and Ltβ (Figs.
2.10, 2.11). While unprotected probe bands migrated slower than their corresponding
protected bands of target mRNA, the identity of every protected band can clearly be
determined due to the its own spatial position and nucleotide size and those of unprotected
probes (outlined in table 2.1). After RA-treatment of 24, 48 and 96 hrs no differential
regulation of these cytokines was visible. Experiments were done for neurotrophins: 24h, n =
4; 48 h, n = 2; 96 h, n = 1; for cytokines: 24 h, n = 1; 48 h, n = 1; 96 h, n = 1. IFNβ is
expressed in Schwann cells as demonstrated in Fig. 2.11, but on a very low level.
28
Fig. 2.10: RNase protection assay (RPA) for neurotrophins (left) and cytokines (right). Schwann cell primary cultures were treated with all-trans RA 48 h in vitro. Control cells received DMSO. Dilutions of the probe set were used as size markers. Rat control RNA served as positive-, yeast tRNA as negative control.
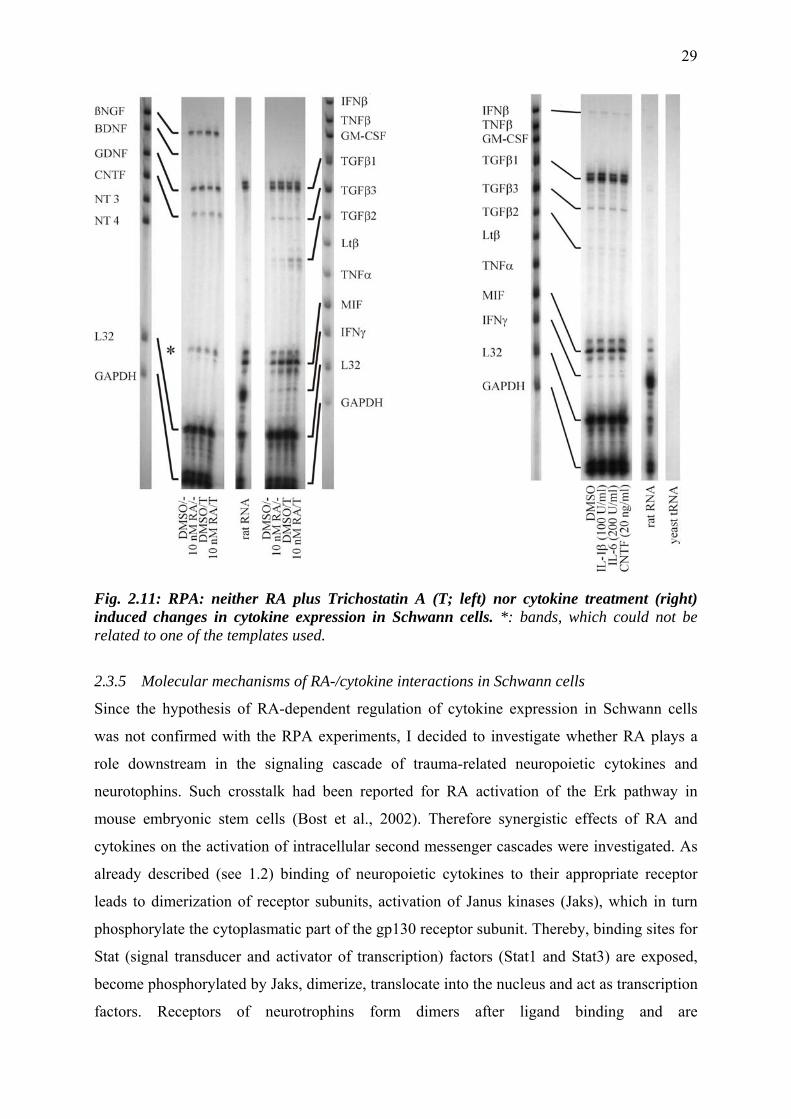
Trichostatin, an inhibitor of the histonedeacetylase, is known to potentiate transcriptional
activity of RA. It selectively inhibits the removal of acetyl groups from the amino-terminal
lysine residues of core histones. This facilitates the access of transcription factors to the
underlying genomic DNA (reviewed by Wei, 2003). To see whether possible effects of RA
might be revealed with the histone deacetylase inhibitor, additional experiments with RA plus
1 µM Trichostatin A were performed. Again, no regulatory effect of RA was observed with
the RNase protection assay. TGFβ2 seemed to be slightly upregulated by Trichostatin A. This
effect was independent of RA as it occured also in cells treated with DMSO plus Trichostatin
A.
Schwann cells were then treated with IL-1β (100 U/ml), IL-6 (200 U/ml) or CNTF (20
nM/ml) to investigate whether cytokines influence their own gene expression. No changes in
cytokine expression occurred compared to DMSO treated cells (Fig. 2.11). Experiments were
done for 24 hrs and 48 hrs of cytokine exposure, but no effect was observed.
29
Fig. 2.11: RPA: neither RA plus Trichostatin A (T; left) nor cytokine treatment (right) induced changes in cytokine expression in Schwann cells. *: bands, which could not be related to one of the templates used.
2.3.5 Molecular mechanisms of RA-/cytokine interactions in Schwann cells
Since the hypothesis of RA-dependent regulation of cytokine expression in Schwann cells
was not confirmed with the RPA experiments, I decided to investigate whether RA plays a
role downstream in the signaling cascade of trauma-related neuropoietic cytokines and
neurotophins. Such crosstalk had been reported for RA activation of the Erk pathway in
mouse embryonic stem cells (Bost et al., 2002). Therefore synergistic effects of RA and
cytokines on the activation of intracellular second messenger cascades were investigated. As
already described (see 1.2) binding of neuropoietic cytokines to their appropriate receptor
leads to dimerization of receptor subunits, activation of Janus kinases (Jaks), which in turn
phosphorylate the cytoplasmatic part of the gp130 receptor subunit. Thereby, binding sites for
Stat (signal transducer and activator of transcription) factors (Stat1 and Stat3) are exposed,
become phosphorylated by Jaks, dimerize, translocate into the nucleus and act as transcription
factors. Receptors of neurotrophins form dimers after ligand binding and are

30
autophosphorylated via an intracellular kinase domain. This activates several intracellular
signal transduction pathways, including the Ras/Raf cascade, which leads to phosphorylation
of the MAP kinase (mitogen-activated protein kinase, MAPK) that in turn activates further
transcription factors. Thus the status of phosphorylation of Stat factors and MAP kinase is
indicative of gene activation by neuropoietic cytokines or neurotrophins.
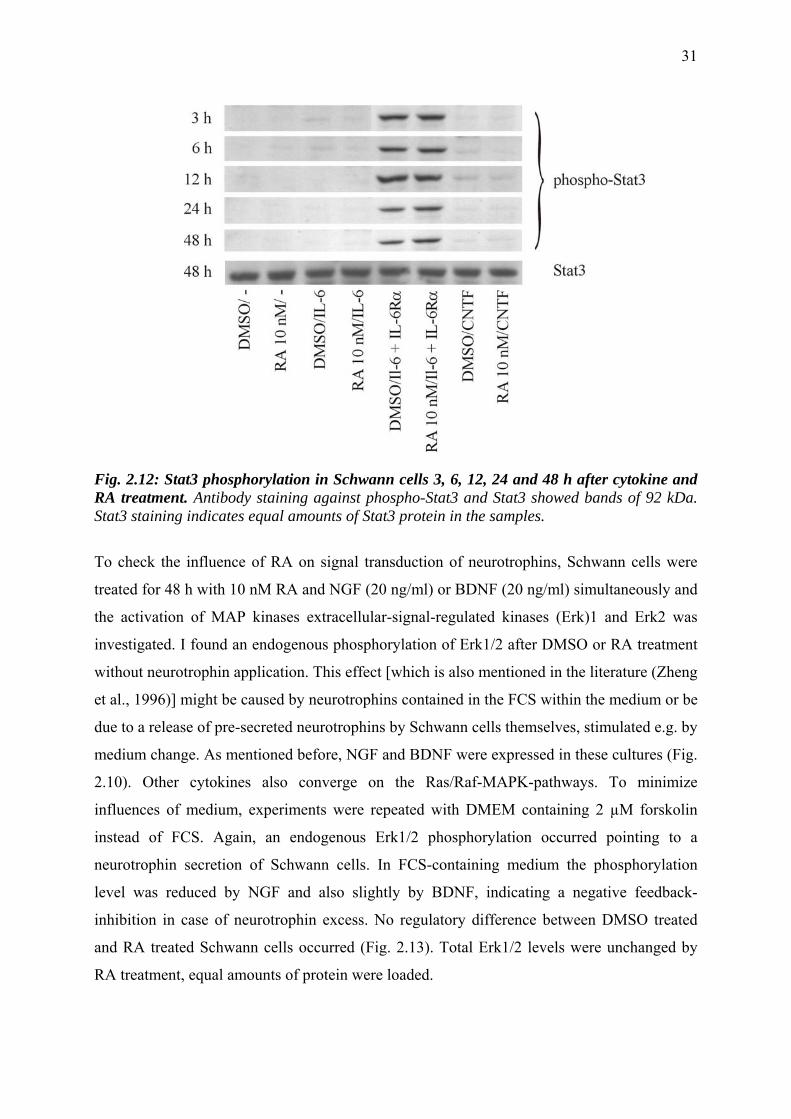
I treated Schwann cell primary cultures for 3 h, 6 h, 12 h, 24 h and 48 h with 10 nM RA and,
after change of medium (to exclude cytokine induction via RA), with CNTF (20 ng/ml), IL-6
(200 U/ml) and IL-6 plus soluble gp80, the IL-6 receptor subunit (1 µg/ml) for 30 min. Most
extracellular signals typically activate signal transduction pathway within 30 min, and a
change in phosphorylation status can be observed. Adding of soluble gp80 normally prevents
IL-6 binding to endogenous gp80 and subsequent Stat-activation. As expected, Schwann cells
treated without cytokine did not show any Stat3 phosphorylation. Since the medium was
changed before cytokine treatment the effect of endogenous RA-induced cytokines can be
excluded and therefore no Stat3 phosphorylation was expected. IL-6 and CNTF treatment did
not induce phosphorylation of Stat3, neither in DMSO treated control cells nor in combination
with RA. Application of IL-6 plus soluble IL-6 receptor subunit in contrast resulted in Stat3
phosphorylation. This seems to be due to binding of gp80-bound IL-6 to the gp130 subunit
which forms the functional IL-6 receptor complex. It seems also that the gp80 subunit, as well
as LIFR subunit (needed for CNTF binding together with CNTFR subunit, which is known to
be expressed in Schwann cell primary cultures) are not expressed on the surface of Schwann
cells as neither IL-6 alone nor CNTF induce a phosphorylation of Stat3. No regulation was
seen after RA treatment at any measured time point (Fig. 2.12). Stat3 staining was also not
altered by RA, which indicates that RA does not regulate Stat3 expression.
31
Fig. 2.12: Stat3 phosphorylation in Schwann cells 3, 6, 12, 24 and 48 h after cytokine and RA treatment. Antibody staining against phospho-Stat3 and Stat3 showed bands of 92 kDa. Stat3 staining indicates equal amounts of Stat3 protein in the samples.
To check the influence of RA on signal transduction of neurotrophins, Schwann cells were
treated for 48 h with 10 nM RA and NGF (20 ng/ml) or BDNF (20 ng/ml) simultaneously and
the activation of MAP kinases extracellular-signal-regulated kinases (Erk)1 and Erk2 was
investigated. I found an endogenous phosphorylation of Erk1/2 after DMSO or RA treatment
without neurotrophin application. This effect [which is also mentioned in the literature (Zheng
et al., 1996)] might be caused by neurotrophins contained in the FCS within the medium or be
due to a release of pre-secreted neurotrophins by Schwann cells themselves, stimulated e.g. by
medium change. As mentioned before, NGF and BDNF were expressed in these cultures (Fig.
2.10). Other cytokines also converge on the Ras/Raf-MAPK-pathways. To minimize
influences of medium, experiments were repeated with DMEM containing 2 µM forskolin
instead of FCS. Again, an endogenous Erk1/2 phosphorylation occurred pointing to a
neurotrophin secretion of Schwann cells. In FCS-containing medium the phosphorylation
level was reduced by NGF and also slightly by BDNF, indicating a negative feedback-
inhibition in case of neurotrophin excess. No regulatory difference between DMSO treated
and RA treated Schwann cells occurred (Fig. 2.13). Total Erk1/2 levels were unchanged by
RA treatment, equal amounts of protein were loaded.

32
Fig. 2.13: Phosphorylation of Erk1 (phospho-p42 MAPK) and Erk2 (phospho-p44MAPK) in Schwann cells after treatment with RA and neurotrophins. Antibody staining against phospho-Erk1 and Erk1 showed bands of 42 kDa, against phospho-Erk2 and Erk2 of 44 kDa. Schwann cells were deprivated in DMEM containing 10 % FCS or 2 µM forskolin.

33
2.4 Discussion
2.4.1 Intracellular (trans-)location of RA signaling components
It has been suggested that RA participates in the physiological processes after nerve injury. In
this work I investigated whether Schwann cells are putative targets of RA in the PNS. With
Western blotting I found that all components of RA signaling except cellular retinoic acid
binding protein (CRABP)-I were present in Schwann cells. This finding is in agreement with
data of sciatic nerves where CRABP-I gene transcripts but not protein expression were
detected (Zhelyaznik et al., 2003; Schrage, 2001). Immunofluorescence microscopy showed a
predominantly cytosolic location of retinaldehyde dehydrogenase RALDH2, the RA-
degrading enzyme CYP26A1, the retinoid receptors RARα, β, γ, RXRα, γ, and CRABP-II. In
contrast, RXRβ was more concentrated in the cell nucleus. After RA treatment all retinoid X
receptors showed a nuclear shift after 15 to 30 minutes, reaching a maximum after 19 hours.
The strongest effects were seen for RXRβ. CRABP-II migrated into the nucleus with a
maximum at 15 minutes, but after 30 minutes CRABP-II was again localized predominantly
in the cytoplasm. No change in the intracellular distribution was visible for any of the retinoic
acid receptors. Studies of the subcellular localization of CRABP-II revealed that it is cytosolic
in the absence of its ligand and that it translocates into the nucleus upon treatment with RA.
CRABP-II interacts with RAR in a ligand-dependent fashion forming a short-lived
intermediate complex (Dong et al., 1999) that facilitates RA channeling to the receptor and by
this enhances the transcriptional activity of RA, especially when cellular levels of either RA
or RAR are limiting (Budhu and Noy, 2002). It was also shown that CRABP-II binds RARα
and RXRα also in a ligand independent manner in mammalian cells (Bastie et al., 2001).
CRABP-II mediates its nuclear localization by using a nuclear localization sequence (NLS)
manifested in its folded state. It does not possess an NLS in its primary structure which
normally serve as signature for subcellular targeting (Sessler and Noy, 2004). Thus, CRABP-
II appears to be a transcriptional regulator involved in RA signaling.
In contrast, retinoid receptors are thought to be localized exclusively in cell nuclei, working as
ligand activated transcription factors. However, in Schwann cell primary cultures RARs
always displayed a stronger signal in the cytosol compared to the nucleus. Recent studies
suggested that RARs can shuttle between the nucleus and cytoplasm like other nuclear
receptors. This was shown using chimeras consisting of RAR and glucocorticoid receptors
(Mackem et al., 2001). Similarly, Maruvada and others demonstrated that approximately 20 %
of RAR-green fluorescent protein (GFP) chimeras were cytosolic but moved rapidly to the
nucleus on binding all-trans-RA (Maruvada et al., 2003). In vitamin A-deficient rats, the

34
RARα subcellular localization in germ cells changed from mainly nuclear to more
cytoplasmatic following retinol depletion (Akmal et al., 1998). In accordance with these
findings Braun and others (2002) discovered that RA induced the nuclear translocation of
RARα within 30 min in mouse Sertoli cell lines and that long term exposure increased
transcription of the receptor gene resulting in increased RARα protein levels. These findings
are in agreement with the predominant cytosolic location of RARs found in untreated cells.
The observation that in my experiments receptors were never found exclusively in the nucleus
and that even after RA treatment no nuclear shift was detectable might be explained as
follows: 1. The translocation of RARs constitutes a regulatory mechanism for RA-facilitated
gene transcription. This would be prevented through cytosolic location of RARs. Forskolin,
contained in the cell culture medium, activates the cAMP-dependent protein kinase (PKA) via
activating the adenylyl cyclase and elevating the cAMP level. It might be one factor that
provokes the cytoplasmatic location of RARs even after RA application. Dibutyryl cAMP and
follicle stimulating hormone inhibit the nuclear localization of RARα (Braun et al., 2002; Hu
and Gudas, 1990; Xiao et al., 1996). However, in Schwann cells it was recently shown that
forskolin had no direct influence on the localization of retinoid receptors (Weßels, 2005). 2.
Although RAR displayed a more cytosolic signal, there was always a substantial proportion
located in the nucleus. This proportion may be sufficient to mediate gene transcription
forming heterodimers with translocating RXRs. As I found transcriptional activity after RA
treatment, namely the downregulation of CRABP-II, this explanation is more likely.
A regulation of nuclear localization for RXRs, as observed for RXRα, -β, and -γ in Schwann
cells has also been shown by others in different cell culture systems: NGF induced the
phosphorylation of Ser 105 of the orphan receptor NGFI-B in PC12 cells. Using nuclear
export signals within NGFI-B this resulted in a translocation of the NGFI-B:RXR heterodimer
complex out of the nucleus and lead to a reduced transcriptional activity of RXR:RAR
heterodimers (Katagiri et al., 2000b). The trafficking of ligand-bound retinoid receptors to the
nucleus, where they can induce gene expression, represents a new way in which RAR and
RXR can affect cellular function. The time course of receptor translocation with a maximum
at 19 h and of CRABP-II within 15 min indicates that RA increases CRABP-II-facilitated
receptor nuclear localization followed by de novo protein synthesis of RXRs, as thereafter
CRABP-II is found mainly in the cytoplasm. In Sertoli cells such a positive feedback loop for
RARα is suggested in the presence of RA. RA triggers a relatively rapid nuclear localization
of RARα, leading to enhanced transcriptional activity of RARα, and this in turn causes
increased expression of endogenous RARα (Braun et al., 2000). After retinol injection into

35
vitamin A-deficient rats the RARα protein level was elevated within 4 hrs (Akmal et al.,
1998b). For this reason, I investigated the effect of RA on the expression of retinoid receptors
and binding proteins in Schwann cells.
2.4.2 Autoregulatory functions of RA
Some of the target genes for retinoids are RARs themselves, in particular the RARß gene,
whose expression is controlled by two promoters (Zelent et al., 1989). The promoter that
regulates the expression of RARß2 gene is strongly induced by RA in different cell types (de
The et al., 1990; Sucov et al., 1990). RA-responsive elements have been identified in the
regulatory regions of several genes involved in retinoid signaling including RARα2, RARβ2,
RARγ2, CRBP-I and CRABP-II (Piedrafita and Pfahl, 1999). The induction of RARβ is
thought to play an important role in growth arrest and morphological differentiation of
embryonal carcinoma cells, human breast cancer cells or neuroblastoma cells (Faria et al.,
1999; Giannini et al., 1997; Liu et al., 1996). The protein expression of retinoid receptors was
unaffected by RA in Schwann cell primary cultures. Gene expression data of Schwann cells
showed that mainly RARβ and slightly RARγ were induced after treatment with 100 nM RA
(Zhelyaznik, 2003). At a concentration of 10 nM RA (which I used during experiments) only
RARβ gene expression was slightly increased. After sciatic nerve injury gene expression data
of retinoid receptors showed an upregulation of all RARs and RXRα which was confirmed on
the protein level only for RARα and RXRα. In the mouse CNS the protein localization of
several retinoid receptors did not correlate with the distribution of the corresponding RNA
transcripts (Krezel et al., 1999). These findings suggest that 1. not all elevated genes are
translated into comparable levels of protein, 2. that other cell types than Schwann cells must
be involved in RA signaling after sciatic nerve injury and 3. that the doses of applied RA
seem to play an important role.
CRABP-II was significantly downregulated after 24 and 48 h. This might represent a
regulatory mechanism for the responsiveness of Schwann cells to RA. The loss of expression
of CRABP-II is correlated with resistance of mammary carcinoma cell lines to RA-induced
growth arrest (Jing et al., 1997), and the expression of a CRABP-II antisense construct in
stromal stem cells results in reduced sensitivity to RA-induced cell-cycle arrest (Vo and
Crowe, 1998). Interestingly, the expression of CRABP-II is upregulated in cells that
synthesize relatively large amounts of RA (Bucco et al., 1997; Yamamoto et al., 1998; Zheng
et al., 1996), suggesting that an increased physiological need for RA also requires the
presence of higher CRABP-II levels which, in turn, will allow for efficient and rapid delivery
36
of the hormone to RA. A negative feedback inhibition in case of RA overdose might be
possible, too.
2.4.3 RA – cytokine interactions
There are some hints that cytokines participating in traumatic reactions of the nervous system
are regulated in Schwann cells via RA, as we found with RT-PCR that RA downregulated the
expression of CNTF in Schwann cell primary cultures to 63 % of control level (Johann et al.,
2003). To test whether RA regulates the expression of other cytokines in Schwann cells I
performed RNase protection assays and detected NGF, BDNF, GDNF, CNTF, TGFβ1,
TGFβ2, TGFβ3, IFNβ, IFNγ and MIF gene transcripts. An RA-dependent regulation of some
of the cytokines detected in other culture systems is recorded and summarized in table 2.2:
RA induces the transcription of NGF, GDNF, members of the TGFβ family and MIF and
inhibits CNTF expression. In Schwann cell primary cultures, no differential regulation of
these cytokines was seen after RA-treatment of 24, 48 and 96 hrs with the RPA.
Table 2.2: Regulation of cytokines by RA.
trophic factor effect of RA on expression
cell system/tissue references
NGF ↑ ↑
Schwann cells primary culture PC12 cells mouse L cells
present work (Scheibe and Wagner, 1992) (Wion et al., 1987)
BDNF
Schwann cells primary culture
present work
GDNF
↑
Schwann cells primary culture Schwann cells primary culture
present work (Kinameri and Matsuoka, 2003)
CNTF
↓
Schwann cells primary culture Schwann cells primary culture
present work (Johann et al., 2003)
TGFβ1,2,3
↑ ↑
Schwann cells primary culture PC12 cells embryonic testis
present work (Cosgaya et al., 1997) (Cupp et al., 1999)
IFNβ
Schwann cells primary culture
present work
IFNγ
Schwann cells primary culture
present work
MIF
↑
Schwann cells primary culture murine embryonic stem cells
present work (Sarkar and Sharma, 2002)
The contradictory results of CNTF and GDNF upregulation in Schwann cells might be due to
the different methods used. RT-PCR which was used for detection is an amplifying method
and therefore more sensitive than the RPA. TGFβ data were confirmed with ELISA of
supernatant of Schwann cell primary cultures: Cells secreted TGFβ but no effect of RA was
visible (Mey, unpublished observations). Also no influence of IL-6, CNTF or the pro-

37
inflammatory cytokine IL-1β on cytokine expression in Schwann cells could be detected. This
was described in the literature for other cell culture systems (Gross et al., 1993; Kagechika et
al., 1997; Lindholm et al., 1987; Zitnik et al., 1994). Possibly, cytokines expressed in
Schwann cells regulate cytokine expression in other cell types, e.g. during Wallerian
degeneration: axotomy-induced production of TNFα and IL-1α and also delayed IL-1β
expression in Schwann cells induce IL-6 and GM-CSF production in fibroblasts. Additionally
they induce recruited macrophages to produce the pro-inflammatory cytokines IL-6, TNFα,
IL-1α, and IL-1β and the anti-inflammatory cytokine IL-10, which in turn downregulates the
production of all inflammatory cytokines and itself in all non-neuronal cells (Shamash et al.,
2002). I cannot completely exclude the possibility that the RPA method was too insensitive to
detect RA-or IL-1-induced changes in gene expression. The absence of any effect of IL-1 is
disconcerting.
Since no RA-dependent regulation of cytokine expression in Schwann cells was detectable
with the RPA experiments, I investigated whether RA plays a role downstream in the
signaling cascade of trauma-related neuropoietic cytokines and neurotophins, leading to a
phosphorylation of transcription factors and subsequent gene transcription. The mechanism by
which RA can cause this activation is still unknown. There are several steps in the cascade at
which RA may exert an influence (table 2.3): For example it changes the expression of
cytokine receptors: in the signaling cascade of neuropoietic cytokines RA inhibits IL-6-
dependent signaling in B lymphocytes via downregulation of both membrane and soluble Il-
6R (IL-6 agonists) and the increased production of soluble gp130, an IL-6 antagonist (Zancai
et al., 2004). Another level of interference displays the modulation of suppressors of cytokine
signaling (SOCS), that negatively regulate Jak and Stat pathways. SOCS3 is induced by RA
to inhibit the Jak/Stat pathway in astrocytes (Choi et al., 2005), although no RARE sequences
in the promoter regions of SOCS3 have been identified to date. RA is also able to affect the
expression and/or the activity of Stat factors in some cellular systems, as retinoids exerted
their anti-proliferative activity by regulation of Stat1 and Stat2 (Dimberg et al., 2000;
Matikainen et al., 1998). Another signaling pathway that can be affected by RA is the
regulation of the MAP-kinase, which is downstream of neurotrophins. RA upregulates low-
and high-affinity NGF receptors in immature chick sympathetic neurons (Haskell et al.,
1987). RARα specific retinoids reduce epidermal growth factor receptor expression/activity
which results in reduced activation of Erk1/2 kinase (Sah et al., 2002). In HL-60 cells RA
increased a MEK-dependent increase in the amount of phosphorylated Raf (Yen and
Varvayanis, 2000). It is also likely that RA regulates the synthesis of intermediate proteins
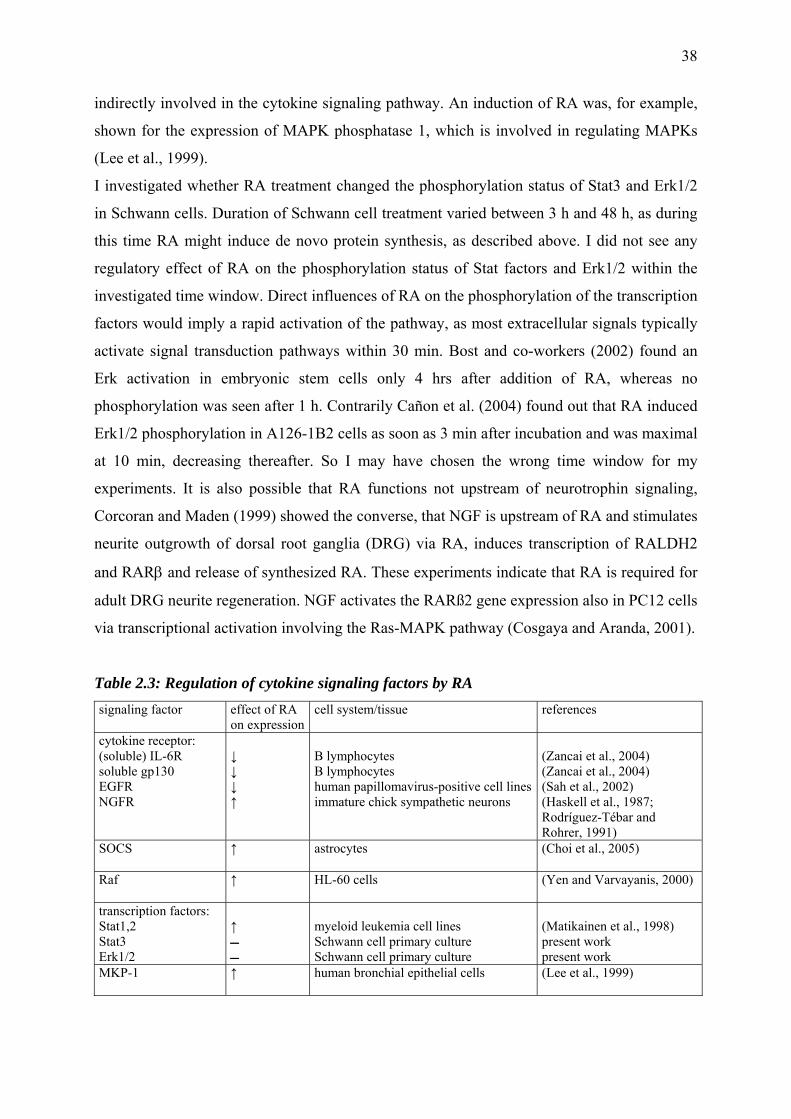
38
indirectly involved in the cytokine signaling pathway. An induction of RA was, for example,
shown for the expression of MAPK phosphatase 1, which is involved in regulating MAPKs
(Lee et al., 1999).
I investigated whether RA treatment changed the phosphorylation status of Stat3 and Erk1/2
in Schwann cells. Duration of Schwann cell treatment varied between 3 h and 48 h, as during
this time RA might induce de novo protein synthesis, as described above. I did not see any
regulatory effect of RA on the phosphorylation status of Stat factors and Erk1/2 within the
investigated time window. Direct influences of RA on the phosphorylation of the transcription
factors would imply a rapid activation of the pathway, as most extracellular signals typically
activate signal transduction pathways within 30 min. Bost and co-workers (2002) found an
Erk activation in embryonic stem cells only 4 hrs after addition of RA, whereas no
phosphorylation was seen after 1 h. Contrarily Cañon et al. (2004) found out that RA induced
Erk1/2 phosphorylation in A126-1B2 cells as soon as 3 min after incubation and was maximal
at 10 min, decreasing thereafter. So I may have chosen the wrong time window for my
experiments. It is also possible that RA functions not upstream of neurotrophin signaling,
Corcoran and Maden (1999) showed the converse, that NGF is upstream of RA and stimulates
neurite outgrowth of dorsal root ganglia (DRG) via RA, induces transcription of RALDH2
and RARβ and release of synthesized RA. These experiments indicate that RA is required for
adult DRG neurite regeneration. NGF activates the RARß2 gene expression also in PC12 cells
via transcriptional activation involving the Ras-MAPK pathway (Cosgaya and Aranda, 2001).
Table 2.3: Regulation of cytokine signaling factors by RA signaling factor effect of RA
on expression cell system/tissue references
cytokine receptor: (soluble) IL-6R soluble gp130 EGFR NGFR
↓ ↓ ↓ ↑
B lymphocytes B lymphocytes human papillomavirus-positive cell linesimmature chick sympathetic neurons
(Zancai et al., 2004) (Zancai et al., 2004) (Sah et al., 2002) (Haskell et al., 1987; Rodríguez-Tébar and Rohrer, 1991)
SOCS
↑
astrocytes
(Choi et al., 2005)
Raf
↑ HL-60 cells (Yen and Varvayanis, 2000)
transcription factors: Stat1,2 Stat3 Erk1/2
↑
myeloid leukemia cell lines Schwann cell primary culture Schwann cell primary culture
(Matikainen et al., 1998) present work present work
MKP-1 ↑
human bronchial epithelial cells (Lee et al., 1999)

39
In summary, I demonstrated that the RA signaling cascade is present in Schwann cells and
that RA increased the nuclear translocalization of retinoid receptors and CRABP-II. I did not
detect any impact of RA on cytokine signaling. Therefore the biological significance of
retinoid receptor expression in Schwann cells remains to be discovered.

40
3 Reactive microglia/macrophages and neurons are targets of retinoic acid signaling
after spinal cord injury
3.1 Abstract
Inflammatory reactions after spinal cord injury are accompanied by local synthesis of the
transcriptional activator retinoic acid (RA). RA exerts its effects by binding to nuclear
receptors (RAR) which heterodimerize with retinoid X receptors (RXR), and these then act as
ligand-activated transcription factors. To identify possible cellular targets of RA after CNS
injury the cellular distribution and protein levels of all retinoid receptors in the rat spinal cord
were investigated. Changes in these parameters were monitored at 4, 7, 14 and 21 days after
contusion injury. Immunoreactivity of RARα, RXRα, RXRβ and RXRγ but not of RARβ and
RARγ was detected. In the non-lesioned spinal cord, immunoreactivity of RARα, RXRα,
RXRβ and RXRγ was localized in the cytosol of neurons, of RXRα and RXRβ in astrocytes,
and of RARα, RXRα and RXRγ in some oligodendrocytes. After contusion injury RARα and
all RXR appeared in the cell nuclei of reactive microglia/macrophages. This nuclear staining
began at 4 days, was most prominent at 7 and 14 days and had decreased at 21 days after
injury. A similar change in intracellular distribution was also observed for the RARα, RXRα
and RXRβ staining in neurons situated around the border of the contusion. The injury-induced
subcellular translocation was strongest for RARα, while the intracellular distribution of RXRγ
did not change. Astrocytes and oligodendrocytes were also immunoreactive for retinoid
receptors. These observations suggest that RA participates as a signal in inflammatory
reactions after CNS injury.
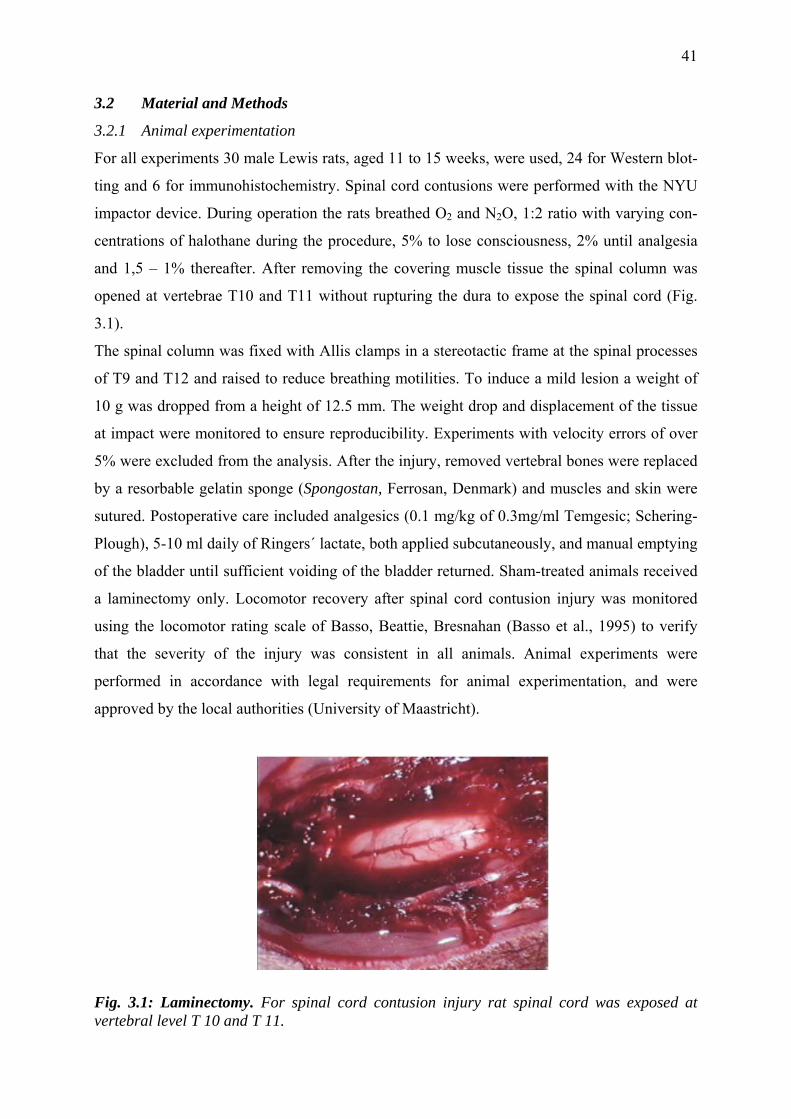
41
3.2 Material and Methods
3.2.1 Animal experimentation
For all experiments 30 male Lewis rats, aged 11 to 15 weeks, were used, 24 for Western blot-
ting and 6 for immunohistochemistry. Spinal cord contusions were performed with the NYU
impactor device. During operation the rats breathed O2 and N2O, 1:2 ratio with varying con-
centrations of halothane during the procedure, 5% to lose consciousness, 2% until analgesia
and 1,5 – 1% thereafter. After removing the covering muscle tissue the spinal column was
opened at vertebrae T10 and T11 without rupturing the dura to expose the spinal cord (Fig.
3.1).
The spinal column was fixed with Allis clamps in a stereotactic frame at the spinal processes
of T9 and T12 and raised to reduce breathing motilities. To induce a mild lesion a weight of
10 g was dropped from a height of 12.5 mm. The weight drop and displacement of the tissue
at impact were monitored to ensure reproducibility. Experiments with velocity errors of over
5% were excluded from the analysis. After the injury, removed vertebral bones were replaced
by a resorbable gelatin sponge (Spongostan, Ferrosan, Denmark) and muscles and skin were
sutured. Postoperative care included analgesics (0.1 mg/kg of 0.3mg/ml Temgesic; Schering-
Plough), 5-10 ml daily of Ringers´ lactate, both applied subcutaneously, and manual emptying
of the bladder until sufficient voiding of the bladder returned. Sham-treated animals received
a laminectomy only. Locomotor recovery after spinal cord contusion injury was monitored
using the locomotor rating scale of Basso, Beattie, Bresnahan (Basso et al., 1995) to verify
that the severity of the injury was consistent in all animals. Animal experiments were
performed in accordance with legal requirements for animal experimentation, and were
approved by the local authorities (University of Maastricht).
Fig. 3.1: Laminectomy. For spinal cord contusion injury rat spinal cord was exposed at vertebral level T 10 and T 11.

42
3.2.2 Western blotting
After survival periods of 4, 7, 14 and 21 days rats were killed through decapitation. A 30 mm
piece of spinal cord was removed and divided into sections 10 mm central, rostral and caudal
to the lesion. Every piece was triturated in 4 µl/mg of 20 mM HEPES buffer with 1% Triton
X-100 and protease inhibitors 1 mM PMSF, 1 µM leupeptin, 1% aprotinin. After 20 min
centrifugation at 13,000 g at 4°C the content of soluble protein extracts was measured with
the bicinchoninic acid protein assay. Proteins were separated with discontinuous SDS-PAGE
and transferred onto a nitrocellulose membrane and stained with Ponceau-S. We used rabbit
antisera binding specifically to the RA-receptor RARα or retinoid X receptors (RXRα, -β, -γ),
all diluted 1/500. To test the specifity of the RARα antiserum, it was blocked with RARα
peptide (Santa Cruz, sc-551P): This was diluted 1/50 and pre-incubated with RARα antiserum
2 hrs before use. Detection of immunoreactivity was based on peroxidase conjugated
secondary antibodies goat anti-rabbit IgG (1/5000) and goat anti-mouse IgG (1/10000),
enhanced chemiluminiscence and a digital image analysis system. When evaluating the
Western blots I used exposure times between 30 sec and 30 min to determine the linear range
of chemiluminiscence output. For each time point 3 animals receiving a SCI were compared
to 3 sham-operated animals. For statistics the immunoreactivity values after spinal cord
contusion were normalized using the averaged data from sham-operated animals with the
same period of survival. In the quantification experiments, Ponceau-S staining of all proteins
was used as control when comparing the strengths of specific immunoreactive signals.
Logarithms of corrected data were analyzed with ANOVA and post-hoc Dunnett’s test.
3.2.3 Immunohistochemistry
For immunohistochemistry rats were sacrificed using an overdose of 0.8 ml pentobarbital so-
dium (Narcoren, Merial GmbH) after the same survival periods as described above. Rats were
perfused with 4% PFA, a 10 mm piece of lesioned spinal cord was removed and postfixed
over night, again in 4% PFA. For cryoprotection the tissue was equilibrated in 15% and 25%
sucrose for 24 hrs each and then quick-frozen in isopentane. On a cryostat, the spinal cord
was sectioned parasagittally in 10 µm slices and dry-mounted on ice-cold, gelatin-coated
slides. To avoid background staining, sections were blocked for 60 min with 3% NGS and 1%
Triton X-100 for permeabilization. Sections were incubated overnight at 4°C with
combinations of one primary antibody against a retinoid receptor and one antibody used as a
cell type marker. Primary antibodies were directed against RARα, RXRα, RXRβ, (diluted
1/100), RXRγ (1/75), all polyclonal, and monoclonal antibodies against GFAP (1/2000,

43
Sigma), CNPase (1/1000, Sigma), MAP2 (1/500, Sigma) and ED1 (1/600, Serotec).
Secondary antibodies were Alexa Flour488 goat anti rabbit (1/1000) and Alexa Flour594 goat
anti mouse (1/500) IgGs. Cell nuclei were visualized with 4’,6-diamidino-2-phenylindole
dihydrochloride (DAPI, Sigma D9564). Sections were analyzed with fluorescence microscopy
using a Zeiss axiophot microscope. Again, pre-incubation with antigenic peptide allowed to
test the specifity of RARα staining in the tissue.
3.2.4 Quantification of the intracellular localization of retinoid receptors
I evaluated the intracellular distribution of RARα, RXRα, RXRβ and RXRγ in neurons, ED1-
positive microglia/macrophages, astrocytes and oligodendrocytes in spinal cord sections from
non-operated and lesioned rats at 4, 7, 14 and 21 dpo. DAPI-stained cells were identified by
their morphology and location in the spinal cord, and their identity was verified with
immunoreactivity for the markers mentioned above. A quantitative assessment was made for
neurons and activated microglia/macrophages: for every double labeling condition, three
sections, separated by 100 µm, were photographed (40 x objective, Zeiss Axiovision
software). From these photographs 20 ED1- and MAP2-positive cells were randomly selected
within (macrophages) or close and ventral to the epicenter of the injury (motoneurons). The
fluorescence values of staining against the retinoid receptor (Alexa Fluor488) of equal sized
areas within the nucleus and within the cytoplasm were measured with NIH image software
and the quotient was calculated for every cell. The position of the cell nucleus was determined
with DAPI, the cell body with Alexa Fluor594 counterstaining (ED1, MAP2). Dependence of
the nuclear/cytosolic staining ratio on time after injury was evaluated with ANOVA.
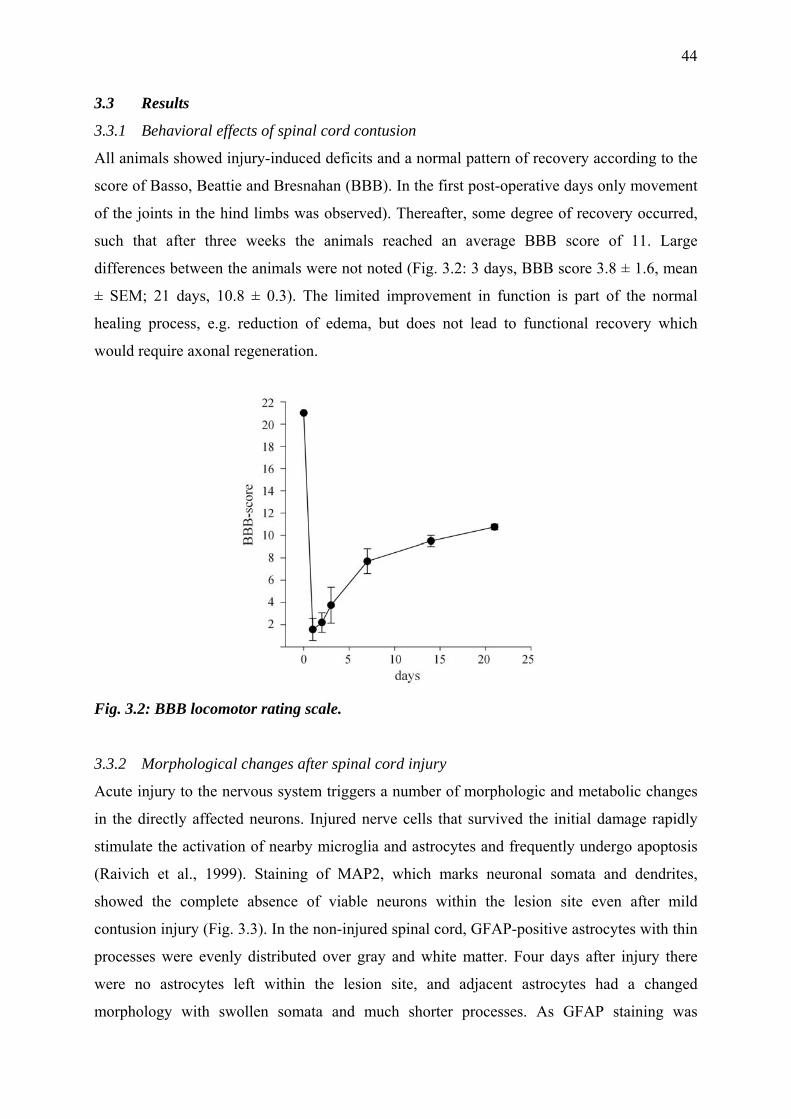
44
3.3 Results
3.3.1 Behavioral effects of spinal cord contusion
All animals showed injury-induced deficits and a normal pattern of recovery according to the
score of Basso, Beattie and Bresnahan (BBB). In the first post-operative days only movement
of the joints in the hind limbs was observed). Thereafter, some degree of recovery occurred,
such that after three weeks the animals reached an average BBB score of 11. Large
differences between the animals were not noted (Fig. 3.2: 3 days, BBB score 3.8 ± 1.6, mean
± SEM; 21 days, 10.8 ± 0.3). The limited improvement in function is part of the normal
healing process, e.g. reduction of edema, but does not lead to functional recovery which
would require axonal regeneration.
Fig. 3.2: BBB locomotor rating scale.
3.3.2 Morphological changes after spinal cord injury
Acute injury to the nervous system triggers a number of morphologic and metabolic changes
in the directly affected neurons. Injured nerve cells that survived the initial damage rapidly
stimulate the activation of nearby microglia and astrocytes and frequently undergo apoptosis
(Raivich et al., 1999). Staining of MAP2, which marks neuronal somata and dendrites,
showed the complete absence of viable neurons within the lesion site even after mild
contusion injury (Fig. 3.3). In the non-injured spinal cord, GFAP-positive astrocytes with thin
processes were evenly distributed over gray and white matter. Four days after injury there
were no astrocytes left within the lesion site, and adjacent astrocytes had a changed
morphology with swollen somata and much shorter processes. As GFAP staining was
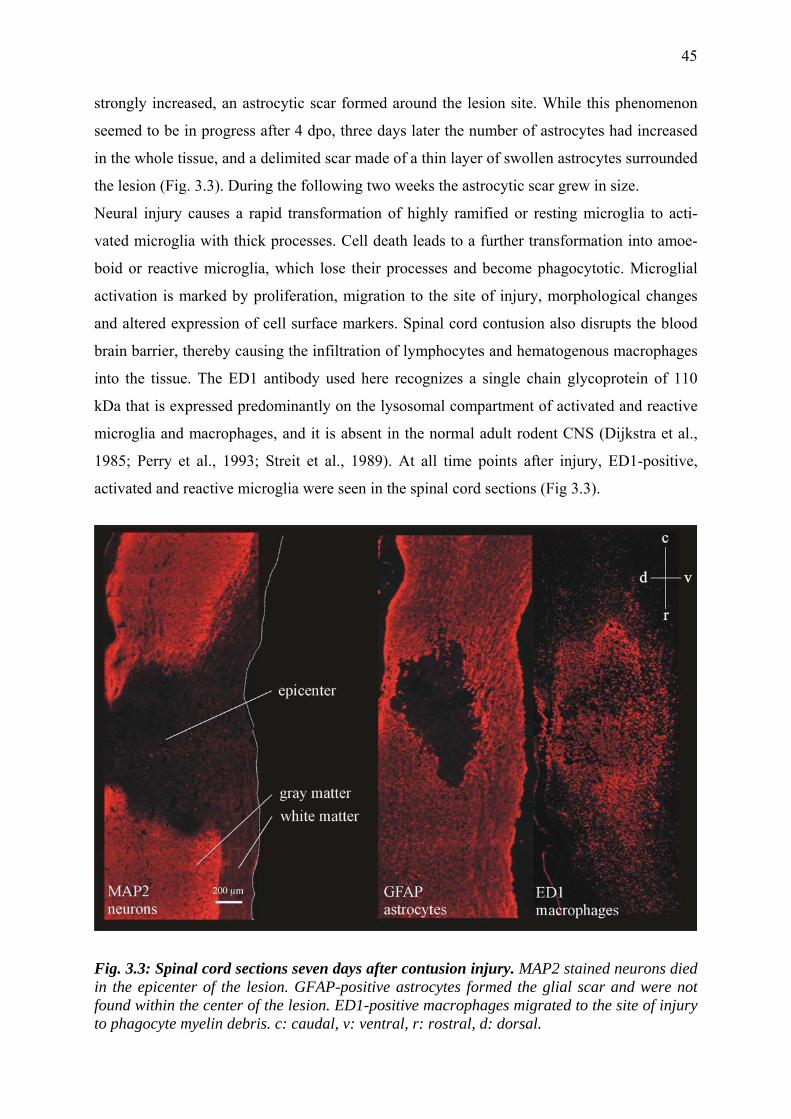
45
strongly increased, an astrocytic scar formed around the lesion site. While this phenomenon
seemed to be in progress after 4 dpo, three days later the number of astrocytes had increased
in the whole tissue, and a delimited scar made of a thin layer of swollen astrocytes surrounded
the lesion (Fig. 3.3). During the following two weeks the astrocytic scar grew in size.
Neural injury causes a rapid transformation of highly ramified or resting microglia to acti-
vated microglia with thick processes. Cell death leads to a further transformation into amoe-
boid or reactive microglia, which lose their processes and become phagocytotic. Microglial
activation is marked by proliferation, migration to the site of injury, morphological changes
and altered expression of cell surface markers. Spinal cord contusion also disrupts the blood
brain barrier, thereby causing the infiltration of lymphocytes and hematogenous macrophages
into the tissue. The ED1 antibody used here recognizes a single chain glycoprotein of 110
kDa that is expressed predominantly on the lysosomal compartment of activated and reactive
microglia and macrophages, and it is absent in the normal adult rodent CNS (Dijkstra et al.,
1985; Perry et al., 1993; Streit et al., 1989). At all time points after injury, ED1-positive,
activated and reactive microglia were seen in the spinal cord sections (Fig 3.3).
Fig. 3.3: Spinal cord sections seven days after contusion injury. MAP2 stained neurons died in the epicenter of the lesion. GFAP-positive astrocytes formed the glial scar and were not found within the center of the lesion. ED1-positive macrophages migrated to the site of injury to phagocyte myelin debris. c: caudal, v: ventral, r: rostral, d: dorsal.
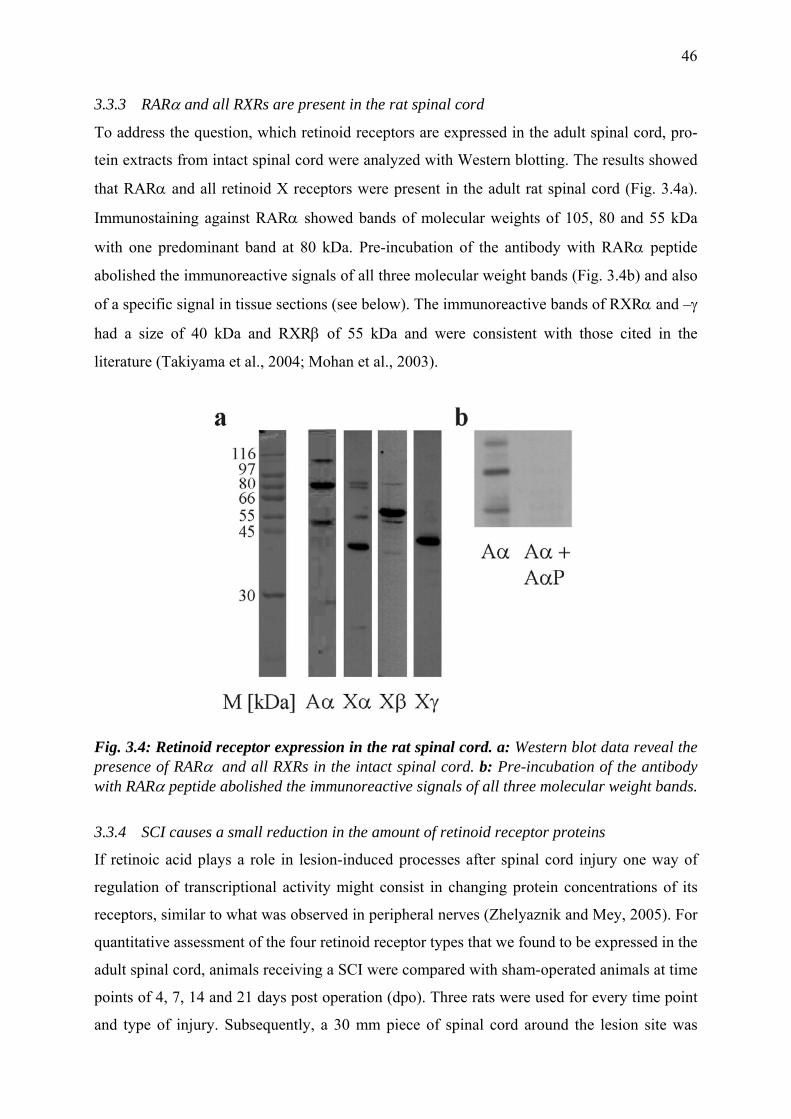
46
3.3.3 RARα and all RXRs are present in the rat spinal cord
To address the question, which retinoid receptors are expressed in the adult spinal cord, pro-
tein extracts from intact spinal cord were analyzed with Western blotting. The results showed
that RARα and all retinoid X receptors were present in the adult rat spinal cord (Fig. 3.4a).
Immunostaining against RARα showed bands of molecular weights of 105, 80 and 55 kDa
with one predominant band at 80 kDa. Pre-incubation of the antibody with RARα peptide
abolished the immunoreactive signals of all three molecular weight bands (Fig. 3.4b) and also
of a specific signal in tissue sections (see below). The immunoreactive bands of RXRα and –γ
had a size of 40 kDa and RXRβ of 55 kDa and were consistent with those cited in the
literature (Takiyama et al., 2004; Mohan et al., 2003).
Fig. 3.4: Retinoid receptor expression in the rat spinal cord. a: Western blot data reveal the presence of RARα and all RXRs in the intact spinal cord. b: Pre-incubation of the antibody with RARα peptide abolished the immunoreactive signals of all three molecular weight bands.
3.3.4 SCI causes a small reduction in the amount of retinoid receptor proteins
If retinoic acid plays a role in lesion-induced processes after spinal cord injury one way of
regulation of transcriptional activity might consist in changing protein concentrations of its
receptors, similar to what was observed in peripheral nerves (Zhelyaznik and Mey, 2005). For
quantitative assessment of the four retinoid receptor types that we found to be expressed in the
adult spinal cord, animals receiving a SCI were compared with sham-operated animals at time
points of 4, 7, 14 and 21 days post operation (dpo). Three rats were used for every time point
and type of injury. Subsequently, a 30 mm piece of spinal cord around the lesion site was

47
dissected and divided into three segments of equal size, central, rostral and caudal to the
lesion. Semi-quantitative analysis of Western blots revealed a moderate but significant
reduction in the total amount of RARα and RXRβ central to the lesion at 4, 7 and 14 dpo and
in the caudal part at 4 dpo (Figs. 3.5, 3.6a,c).
Fig. 3.5: Seven days after contusion injury: the total amount of RARα, RXRβ and RXRγ was reduced after spinal cord injury (sci) central and caudal to the lesion site compared to sham operated animals (control: C). RXRα protein level decreased rostral and in the central part of the lesion.
After 21 days, central and caudal of the lesion site the protein levels had returned to baseline.
In the rostral segment at 21 dpo, an increase of RARα and RXRβ appeared. The amount of
RXRα (Fig. 3.6b) was reduced significantly at 4 dpo central and caudal to the lesion, at 7 dpo
rostral and central (see also Fig. 3.5) and at 14 dpo in the caudal segment. RXRγ was
significantly downregulated only 21 days after injury in each of the three segments of the cord
(Fig. 3.6d). These changes may have occurred because of a downregulation of retinoid
receptors within surviving cells or because of degeneration of RAR/RXR expressing cell
types. This issue was resolved with immunohistochemical analysis.
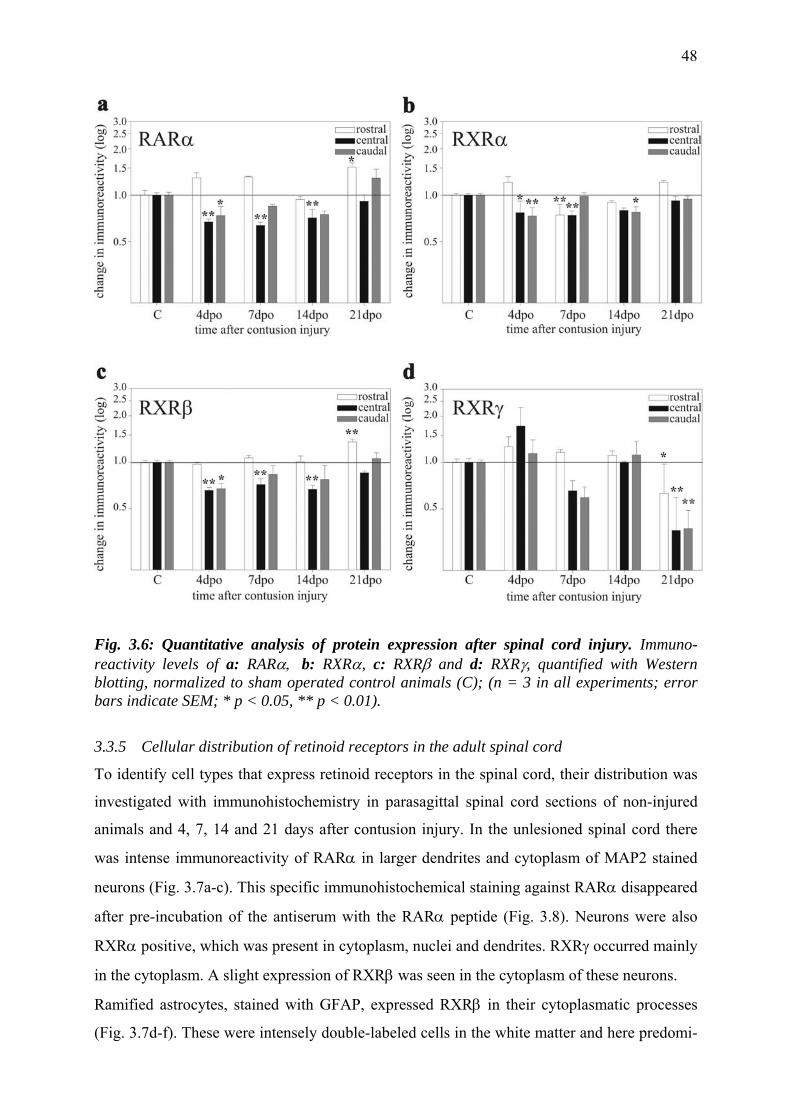
48
Fig. 3.6: Quantitative analysis of protein expression after spinal cord injury. Immuno-reactivity levels of a: RARα, b: RXRα, c: RXRβ and d: RXRγ, quantified with Western blotting, normalized to sham operated control animals (C); (n = 3 in all experiments; error bars indicate SEM; * p < 0.05, ** p < 0.01).
3.3.5 Cellular distribution of retinoid receptors in the adult spinal cord
To identify cell types that express retinoid receptors in the spinal cord, their distribution was
investigated with immunohistochemistry in parasagittal spinal cord sections of non-injured
animals and 4, 7, 14 and 21 days after contusion injury. In the unlesioned spinal cord there
was intense immunoreactivity of RARα in larger dendrites and cytoplasm of MAP2 stained
neurons (Fig. 3.7a-c). This specific immunohistochemical staining against RARα disappeared
after pre-incubation of the antiserum with the RARα peptide (Fig. 3.8). Neurons were also
RXRα positive, which was present in cytoplasm, nuclei and dendrites. RXRγ occurred mainly
in the cytoplasm. A slight expression of RXRβ was seen in the cytoplasm of these neurons.
Ramified astrocytes, stained with GFAP, expressed RXRβ in their cytoplasmatic processes
(Fig. 3.7d-f). These were intensely double-labeled cells in the white matter and here predomi-
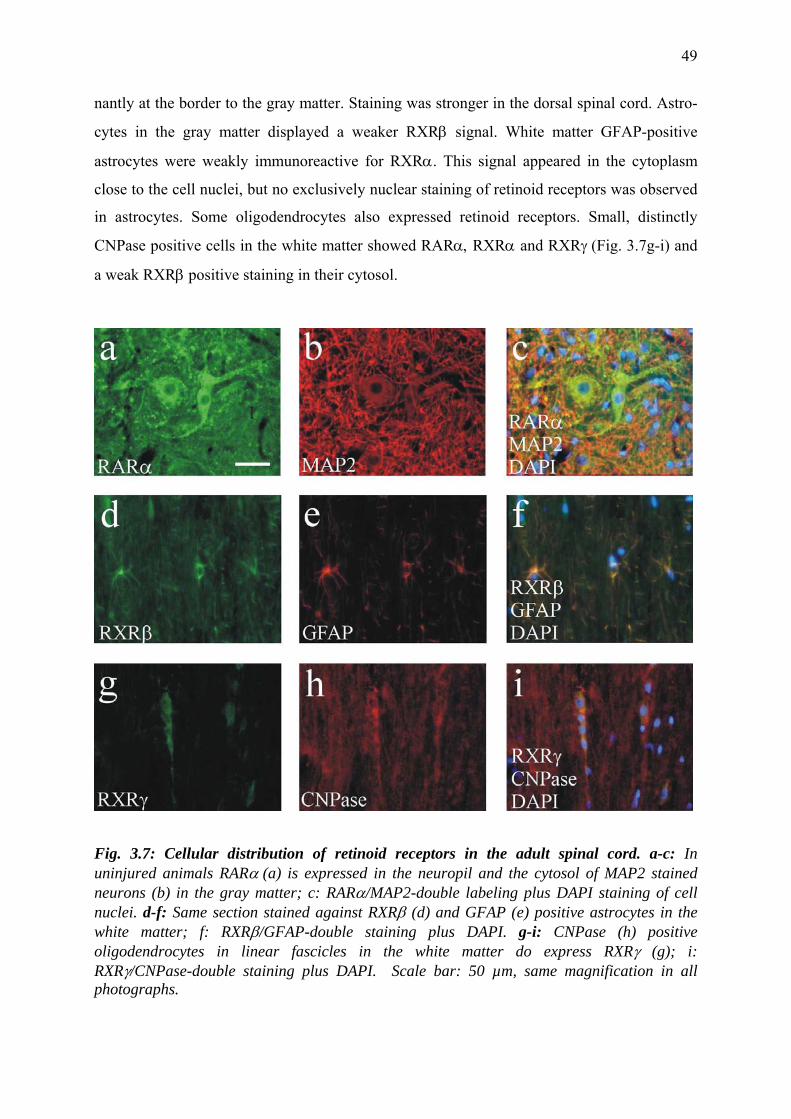
49
nantly at the border to the gray matter. Staining was stronger in the dorsal spinal cord. Astro-
cytes in the gray matter displayed a weaker RXRβ signal. White matter GFAP-positive
astrocytes were weakly immunoreactive for RXRα. This signal appeared in the cytoplasm
close to the cell nuclei, but no exclusively nuclear staining of retinoid receptors was observed
in astrocytes. Some oligodendrocytes also expressed retinoid receptors. Small, distinctly
CNPase positive cells in the white matter showed RARα, RXRα and RXRγ (Fig. 3.7g-i) and
a weak RXRβ positive staining in their cytosol.
Fig. 3.7: Cellular distribution of retinoid receptors in the adult spinal cord. a-c: In uninjured animals RARα (a) is expressed in the neuropil and the cytosol of MAP2 stained neurons (b) in the gray matter; c: RARα/MAP2-double labeling plus DAPI staining of cell nuclei. d-f: Same section stained against RXRβ (d) and GFAP (e) positive astrocytes in the white matter; f: RXRβ/GFAP-double staining plus DAPI. g-i: CNPase (h) positive oligodendrocytes in linear fascicles in the white matter do express RXRγ (g); i: RXRγ/CNPase-double staining plus DAPI. Scale bar: 50 µm, same magnification in all photographs.

50
Fig. 3.8: Immunohistochemical staining against RARα is abolished by pre-incubating the antiserum with RARα peptide. Microglia/macrophages (a, b) and neurons (c, d) were stained with RARα antiserum (a, c) or RARα antiserum pre-incubated with RARα peptide (b, d). Arrows in d) point to neurons (MAP2 positive), which are now devoid of staining and darker than non-specific background in gray matter. Scale bar for a, b marks 20 µm, scale bar for c, d 50 µm.
3.3.6 Nuclear localization of retinoid receptors in reactive microglia/macrophages
Activated microglia was distributed peripheral to the lesion site (Fig. 3.9a), and amoeboid
reactive microglia/macrophages were close to and within the epicenter of the lesion (Fig.
3.9b,c). Four days after injury a weak signal of RARα immunoreactivity was seen in the
nuclei and cytoplasm of these ED1 positive reactive monocytes within the lesion site (Fig.
3.9e). Three days later, at 7 dpo, a strong signal was present in the nuclei while the cytosolic
staining was attenuated (Fig. 3.9f). At 14 dpo the nuclear staining had become even more
intense (Fig. 3.9g). One week later, 21 dpo, it had decreased to a low level (Fig. 3.9h). In
contrast, RXRα and RXRβ were found predominantly in the cytoplasm of reactive
microglia/macrophages at 4 dpo (Fig. 3.9i,m), where the RXR staining did not distinguish cell
nuclei from the surrounding cytoplasm. After 7 (Fig. 3.9j,n) and 14 days (Fig. 3.9k,o) the
receptors had shifted almost completely into the nucleus. Subsequently RXRα and -β proteins
appeared to have returned to their initial distribution, because after 21 days the signals were
again seen in the cytoplasm (Fig. 3.9l,p). Immunoreactivity of RXRγ was present at 4 dpo in
the cytoplasm (Fig. 3.9q), while the nuclei were almost devoid of staining. Nuclear staining
was visible at 7 and 14 dpo (Fig. 3.9r,s), but less prominent than that of RARα, RXRα and
RXRβ. This pattern changed to a weak cytoplasmatic staining at 21 dpo (Fig. 3.9t). Fig. 3.11a
presents a quantification of these observations: At 4 dpo the nuclear and cytosolic signals of
all four receptors were about equal. A translocation of RARα into the nuclei of macrophages
caused the ratio of nuclear/cytosolic staining to increase. This ratio reached 2-3 at 7 and 14
dpo then it declined to about 1.4 at 21 dpo. Similar, albeit less dramatic changes are apparent
for the RXRs. In summary, all expressed retinoid receptors showed a shift from a
cytoplasmatic to a nuclear localization 7 to 14 days after injury and a subsequent decline of
the nuclear immunoreactivity (see also Fig. 3.12). These observations, which pertain to ED1
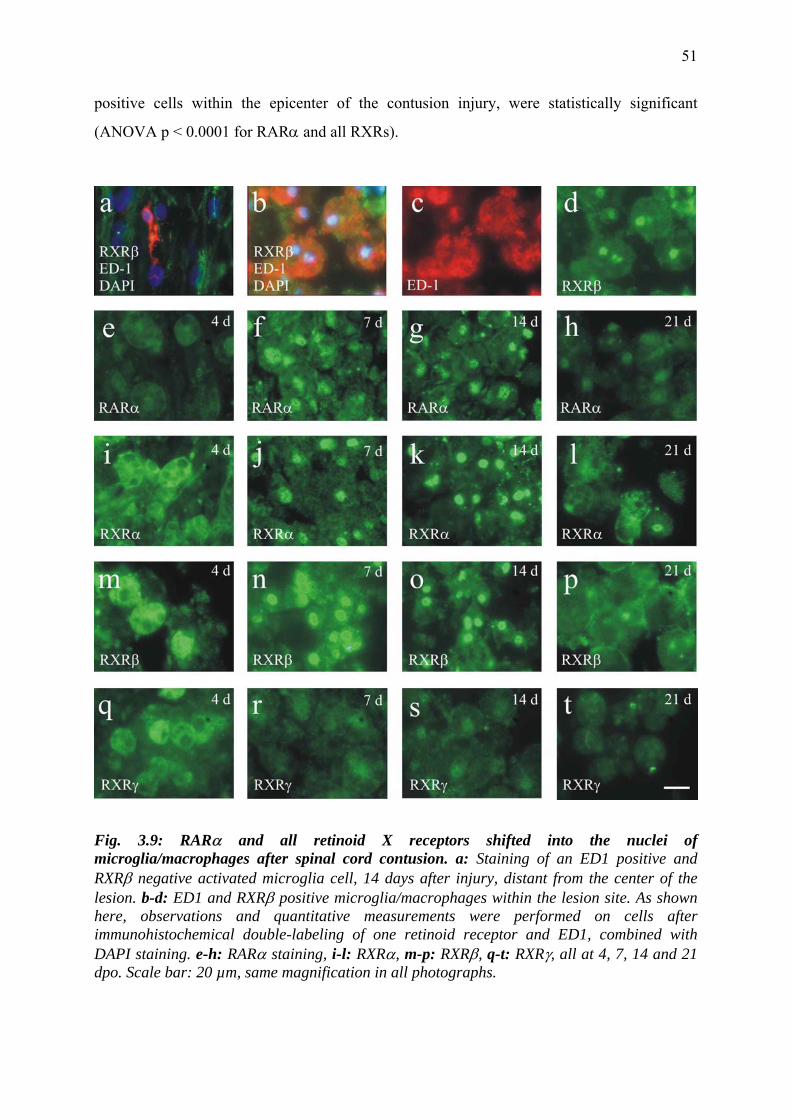
51
positive cells within the epicenter of the contusion injury, were statistically significant
(ANOVA p < 0.0001 for RARα and all RXRs).
Fig. 3.9: RARα and all retinoid X receptors shifted into the nuclei of microglia/macrophages after spinal cord contusion. a: Staining of an ED1 positive and RXRβ negative activated microglia cell, 14 days after injury, distant from the center of the lesion. b-d: ED1 and RXRβ positive microglia/macrophages within the lesion site. As shown here, observations and quantitative measurements were performed on cells after immunohistochemical double-labeling of one retinoid receptor and ED1, combined with DAPI staining. e-h: RARα staining, i-l: RXRα, m-p: RXRβ, q-t: RXRγ, all at 4, 7, 14 and 21 dpo. Scale bar: 20 µm, same magnification in all photographs.

52
3.3.7 Retinoid receptor expression in neurons
Our investigation of the neuronal localization of retinoid receptors in neurons therefore
focused on cells in the ventral spinal cord adjacent to the epicenter of the injury. After 4 days
there was strong RARα immunoreactivity predominantly in the cytoplasm and bigger
dendrites of neurons positive for MAP2 rostral and distal to the lesion site (Fig. 3.10a,b).
Three days later, at 7 dpo, this signal appeared also within some cell nuclei (Fig. 3.10c,d). At
14 dpo RARα had shifted almost completely into the nuclei (Fig. 3.10e,f), while after 21 dpo
it was again located predominantly in the cytoplasm and bigger dendrites (Fig. 3.10g,h). An
RXRα signal was present in the cytoplasm, in bigger and smaller dendrites and to a lesser
extent in neuronal nuclei at 4 dpo. Dendrites were stained more strongly with the antibody
against RXRα than that against RARα. Seven days post operation the dendritic staining had
decreased, and it was almost gone after 14 dpo. In contrast, the nuclear staining increased
during this time. After 21 days the RXRα signal was weak and mostly cytoplasmatic. The
neuronal staining of RXRβ was in general rather faint, it decreased with time and changed
from predominantly cytosolic with no distinct dendritic staining at 4 dpo to a more nuclear lo-
cation at 7 and 14 dpo, and then was again mostly cytosolic. The intracellular distribution of
RXRγ changed less than was observed for the other receptors (Fig. 3.11b). The intensity of
staining, indicative for the overall amount of receptor protein, decreased from 4 to 7 dpo and
recovered again after 14 and 21 days. In comparison with macrophages, the intracellular
distribution of retinoid receptors in neurons always displayed a stronger cytoplasmatic signal.
Yet, a transient shift of all detected retinoid receptors towards the nucleus did occur
(ANOVA, p < 0.0001 for RARα and all RXRs). This lesion-induced change was most
pronounced at 7dpo. For the quantification of these observations motoneurons, identified by
their size, morphology and location in the ventral horn, were selected. While it is possible that
the changed retinoid distribution is part of physiological changes that may be detrimental or
supportive to the further fate of these cells, they exhibited no cytological signs of necrosis or
apoptosis. The data are shown in Fig. 3.11b and summarized in Fig. 3.12.
3.3.8 Reactive astrocytes in the glial scar are immunoreactive for retinoid receptors
While in unoperated animals and still at 4 dpo RARα was not present in astroglia, this
receptor was upregulated in some astrocytes of animals that survived 7 days after injury. The
receptor protein was discernible in nuclei and cytoplasm, but not in the ramifications of
astrocytes in gray or white matter. RXRα, in contrast, exhibited a weak staining at all the time
points after injury that we looked at. It was present in the cytoplasm of swollen astrocytes
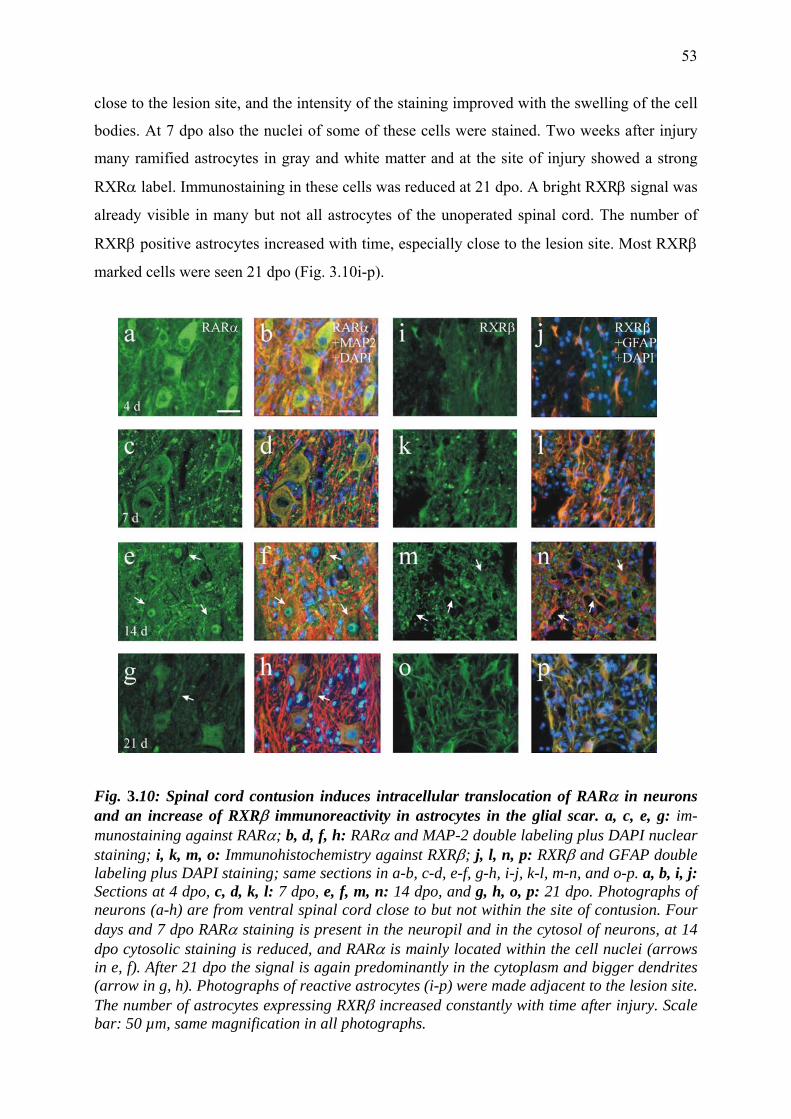
53
close to the lesion site, and the intensity of the staining improved with the swelling of the cell
bodies. At 7 dpo also the nuclei of some of these cells were stained. Two weeks after injury
many ramified astrocytes in gray and white matter and at the site of injury showed a strong
RXRα label. Immunostaining in these cells was reduced at 21 dpo. A bright RXRβ signal was
already visible in many but not all astrocytes of the unoperated spinal cord. The number of
RXRβ positive astrocytes increased with time, especially close to the lesion site. Most RXRβ
marked cells were seen 21 dpo (Fig. 3.10i-p).
Fig. 3.10: Spinal cord contusion induces intracellular translocation of RARα in neurons and an increase of RXRβ immunoreactivity in astrocytes in the glial scar. a, c, e, g: im-munostaining against RARα; b, d, f, h: RARα and MAP-2 double labeling plus DAPI nuclear staining; i, k, m, o: Immunohistochemistry against RXRβ; j, l, n, p: RXRβ and GFAP double labeling plus DAPI staining; same sections in a-b, c-d, e-f, g-h, i-j, k-l, m-n, and o-p. a, b, i, j: Sections at 4 dpo, c, d, k, l: 7 dpo, e, f, m, n: 14 dpo, and g, h, o, p: 21 dpo. Photographs of neurons (a-h) are from ventral spinal cord close to but not within the site of contusion. Four days and 7 dpo RARα staining is present in the neuropil and in the cytosol of neurons, at 14 dpo cytosolic staining is reduced, and RARα is mainly located within the cell nuclei (arrows in e, f). After 21 dpo the signal is again predominantly in the cytoplasm and bigger dendrites (arrow in g, h). Photographs of reactive astrocytes (i-p) were made adjacent to the lesion site. The number of astrocytes expressing RXRβ increased constantly with time after injury. Scale bar: 50 µm, same magnification in all photographs.

54
No intracellular shift towards a nuclear localization was apparent. Compared to the controls,
RXRγ was upregulated 4 dpo in the cytoplasm of astrocytes in the white matter and around
the lesion site, and after 7 dpo the receptor was seen also in the nuclei. At 14 dpo RXRγ was
found in astrocytes in the gray matter adjacent to the lesion site. After 21 days the RXRγ
signal had again decreased. These observations of retinoid receptor expression in GFAP
positive astrocytes are summarized in Fig. 3.12.
Fig. 3.11: Quantitative analysis of the intracellular receptor distribution in microglia and neurons. a: In microglia all retinoid receptors translocated into the nucleus with a peak at 14 dpo for RARα, RXRβ and RXRγ and at 7 dpo for RXRα. This intracellular translocation was most prominent for RARα where the ratio of nuclear to cytosolic signal rose from 1.1 to 2.7. Changes were significant for RARα, and all RXRs (ANOVA, p < 0.0001) b: In neurons, RARα, RXRα and RXRβ signals shifted to the nucleus with a maximum at 14 dpo (ANOVA, p < 0.0001). Again, RARα showed the strongest nuclear/cytosolic signal ratio. The intracellular distribution of RXRγ changed very little. Error bars indicate SEM, n = 20 cells in all cases.

55
3.3.9 Retinoid receptor distribution in oligodendrocytes
In the intact spinal cord CNPase staining of gray and white matter was used to mark oli-
godendrocytes and their processes. The pattern of staining did not change much at 4 dpo.
Seven and 14 days after injury the CNPase staining was more intense, and the structural dif-
ferences between gray and white matter myelin were better distinguishable. Already at 4 dpo
a lot of myelin debris, distributed within the entire lesion site, was partially phagocytosed by
microglia. After 21 dpo this remaining CNPase staining was again weaker and the lesion site
was almost cleared of myelin debris.
All analyzed retinoid receptors were co-expressed with CNPase in a pattern that resembled
that of myelin. The signal of all retinoid receptors increased over time, at 7 and 14 dpo, and
declined 21 days after lesion. CNPase immunoreactive structures that also carried RARα,
RXRα and RXRγ occurred in linear fascicles in the white matter, yet most oligodendrocyte
cell bodies were not immunoreactive for the retinoid receptors. When present, the
cytoplasmatic signal of RARα and RXRα did not change over time whereas RXRγ
translocated into the nucleus of some oligodendrocytes between 7 and 14 days after injury
(Fig. 3.12). RXRβ was not visible in oligodendrocyte cell bodies.
Fig. 3.12: Intracellular localization of retinoid receptors. Schematic representation of the distribution (cytoplasm, nucleus) and strength (white = none, gray = average, dark gray = strong) of the immunoreactive signals in macrophages/microglia, neurons, scar-forming astrocytes and oligodendrocytes. n.d.: no ED1 staining detected.

56
3.4 Discussion
In this study I found that spinal cord contusion injury in the adult rat causes an intracellular
translocation of retinoid receptors RARα, RXRα, RXRβ and RXRγ in macrophages and
neurons. The time course of these changes, predominantly 7-14 days after injury, suggests an
involvement of retinoic acid signaling in late inflammatory processes. Retinoid receptors were
also expressed in astroglia and oligodendrocytes, and the staining patterns in these cells were
similarly affected by the injury.
Western blotting results showed that RARα and all RXR isoforms were also present in the
unlesioned spinal cord. Quantitative assessment of the retinoid receptor proteins at 4, 7, 14
and 21 days after spinal cord injury showed a moderate reduction in the total amount of
RARα, RXRα and RXRβ central and caudal to the lesion at 4, 7 and 14 dpo. After 21 days
the protein levels had returned to baseline. RXRγ was significantly reduced only 21 days after
injury in each of the three segments of the cord.
3.4.1 Cellular targets of retinoic acid signaling after spinal cord contusion
Immunohistochemical assessment revealed that the reduction in the total amount of retinoid
receptor protein was due to the degeneration of RAR/RXR expressing cell types and not to the
downregulation of RARα or RXRs in surviving cells: As observed previously, the mild
contusion injury (Basso et al., 1995) produced an expanded area of secondary damage,
resulting in tissue destruction at the epicenter of the lesion, where all neurons had died, while
a peripheral rim of white matter tracts was spared. Oligodendrocytes underwent apoptosis in
those white matter tracts that contained degenerating axons. Glial cell death is maximal 8 dpo
(Shuman et al., 1997), and even oligodendrocytes located at considerable distance from the
injury died of apoptosis (Crowe et al., 1997). Astrocytes proliferated and produced a network
of interdigitated processes that encapsulate the lesion, but within the lesion cavity I did not
detect any GFAP positive astrocytes. Neurons and oligodendrocytes expressed all four
retinoid receptor types in the uninjured spinal cord and at all measured time points after
injury. The loss of neurons at the site of the lesion explains the reduction of RARα, RXRα
and RXRβ in the tissue samples taken from the lesion site, and the spreading apoptotic death
of oligodendrocytes probably that in the caudal segment. Although MAP2 positive
motoneurons that expressed retinoid receptors in their nuclei did not show cytological signs of
death, I cannot exclude the possibility that the receptor translocation forms an early part of a
cell death pathway. Whatever the function, neurons appear to be targets of retinoid signaling
after spinal cord injury.

57
The compensating increase of all retinoid receptors at later time points might be due to the
upregulation of all retinoid receptors in reactive microglia, which strongly proliferated and
invaded the lesion cavity in great numbers. I conclude from these observations that
macrophages are also targets of RA signaling. This interpretation is also in accordance with
recent experiments by Dheen et al. (2005), who demonstrated anti-inflammatory effects of
brain microglia primary cultures activated with LPS or β-amyloid peptide.
3.4.2 Intracellular translocation of retinoid receptors
The standard model of RA-dependent gene regulation holds that RAR/RXR heterodimers
bind to RARE on the DNA where, in the presence of corepressors, they inhibit gene tran-
scription. Retinoid receptors are thought to reside exclusively in the cell nucleus (Braun et al.,
2002a; Chambon, 1996). In contrast, we found that in the non-injured spinal cord retinoid
receptors were located in the cytoplasm of neurons and glia cells. We saw a transfer of RARα
and RXRs to the cell nuclei only after contusion injury. While this constitutes a contradiction
with the original concept, other instances of cytosolic distribution of retinoid receptors have
recently been reported, e.g. in Sertoli cells (Braun et al., 2002a, b). Cañón and co-workers
report non-genomic effects of RA like CREB- and ERK-phosphorylation in PC12 cells
(Cañón et al., 2004), which might reside in the cytosol.
The injury-induced subcellular translocation of retinoid receptors may either be a
consequence of RA exposure or may represent a RA-independent, regulatory mechanism that
renders these cells responsive to RA. Is RA the trigger of subcellular translocation? It was
recently reported that spinal cord injury, performed as in the present study, resulted in an
increase of local enzyme activity of RALDH2, presumably leading to local synthesis of all-
trans RA. In accordance with the intracellular shift or RARα, the strongest increase of
RALDH2 activity occurred 7 to 14 dpo (Mey et al., 2005). Since RXRs can be heterodimeric
partners of other nuclear receptors [VDR, TR, PPAR; (Chambon, 1996)] their translocation
may also be caused by transcriptional activators other than RA, i.e. the ligands of these
receptors. Thyroid hormone, for instance, may be involved in the responses to nerve injury
(Barakat-Walter, 1999). In another context, TRβ was found to be partially in the cytoplasm
and transferred to the nucleus upon T3 binding (Zhu et al., 1998). Co-transfected retinoid X
receptor markedly decreased the rapid shuttling of TRβ between nuclear and cytoplasmatic
compartments in HeLa cells by maintaining them unliganded within the nucleus (Baumann et
al., 2001). Also RARs exhibited reduced shuttling in the presence of RXRs suggesting that

58
heterodimerization plays a major role in maintaining the normal cellular distribution of RARs
(Maruvada et al., 2003; Prüfer et al., 2000).
Alternatively, are other factors responsible for a nuclear shift of RARα and RXRs? Injury-
related factors that are not ligands of the nuclear hormone receptor super-family may cause
the intracellular shift of RARα or RXR proteins. In the liver, pro-inflammatory cytokines, e.g.
IL-1β, influence the subcellular localization and DNA-binding of RARα:RXRβ heterodimers
(Denson et al., 2000; Ghose et al., 2004). In the spinal cord, cytokines that lead to a transfer of
retinoid receptors into the nucleus could thereby induce the responsiveness of the affected
cells to RA. The DNA-binding domains of RARs, RXRs and other hormone receptors can
serve as nuclear export signals. (Black et al., 2001) and it was also shown that the DNA
binding domain of RXRs contains a nuclear localization sequence and mediates binding to
import receptors (Prüfer and Barsony, 2002).
3.4.3 Possible physiological functions of RA after spinal cord injury
The time course of retinoid signaling suggests that RA does probably not initiate the
inflammatory reaction or axonal sprouting because these changes occur earlier. Based on
receptor expression in target cells it is tempting to conclude that RA may fulfill
neuroprotective functions. This has already been shown in vitro in different contexts,
including mouse DRG neurons (Corcoran et al., 2000; Corcoran and Maden, 1999),
embryonic spinal cord of humans and mice (Quinn and De Boni, 1991), and chick retinal
ganglion cells (Mey and Rombach, 1999). In vivo, RA increased trkA mRNA levels of
sympathetic neurons during development, which is needed to acquire dependency on the
survival factor NGF (Holst et al., 1997).
In astrocytes, RA might suppress the astroglial inflammatory response. For instance, it has re-
cently been discovered that RA inhibited IFN-γ-activated Jak/Stat pathways, probably via the
expression of SOCS3 (Choi et al., 2005). In addition to degeneration of their motoneurons
retinoid deficient rats show an increase in astrocytosis which was found to be a consequence
of the lack of RARα (Corcoran et al., 2002b). Besides, astrocytes have been suggested to
regulate the concentration of RA through oxidation of retinol to the biologically active RA,
which then gives trophic support to spinal cord neurons in vitro (Wuarin et al., 1990).
In macrophages, which were found to be the main targets of RA signaling after SCI, potent
anti-inflammatory effects of RA have also been reported. In peritoneal macrophages, RA in-
hibited almost completely the production of tumor necrosis factor, nitric oxide (Sjöberg et al.,
1992), the lipopolysaccharid-induced production of prostaglandin E2 and expression of COX-

59
2 protein, which take part in pro-inflammatory reactions as well (Kim et al., 2004). In
activated rat microglia the RA-induced reduction of the expression of TNFα, and inducible
nitric oxide synthase was correlated with enhanced expression of RARβ and TGF-β1 and
inhibited translocation of and NF-κB (Dheen et al., 2004). Therefore, RA might contribute to
the termination of inflammatory response in the CNS.
In summary, these experiments demonstrate that the RA signaling system participates in the
physiological responses to SCI: They show that activated microglia in their phagocytotic
stage are the most prominent targets of this signaling pathway. In addition, neurons, scar
forming astrocytes and oligodendrocytes express retinoid receptors. The intracellular
distribution of several of these receptors indicates a RA- or cytokine-mediated translocation
from the cytosol into the nucleus, which is triggered by a contusion injury. RA signaling may
thus provide a novel target for therapeutic interventions of inflammatory processes in the
CNS.

60
4 General discussion
Nerve injury leads to a series of molecular and cellular responses which are fundamentally
different in the peripheral and central nervous system. In the PNS they can cause a successful
regenerative response and functional recovery. This is among other factors a consequence of
the cell types expressed in each of these compartments of the nervous system (CNS:
astrocytes, oligodendrocytes, microglia; PNS: Schwann cells, macrophages, fibroblasts) and
the different intercellular signaling pathways between these cell types.
With respect to retinoic acid signaling a number of similarities and differences are observed
after injury in the central and peripheral nervous system. In both cases, RA synthesis was
catalyzed by RALDH2, while RALDH1 and –3 seem not involved. A small lesion-induced
increase in immunoreactivity or enzyme activity of RALDH2 was seen, which was stronger
after spinal cord contusion (Mey et al., 2005) than after sciatic nerve injury (Zhelyaznik et al.,
2003). In the spinal cord as well as in the sciatic nerve RARα and all RXRs were expressed.
After peripheral nerve lesion, nuclear translocation of RARα and RXRα in macrophages and
Schwann cells was also visible, and as in the spinal cord this activity appeared much later than
the initial inflammatory response (Zhelyaznik and Mey, 2005 and unpublished observations).
Different from the sciatic nerve I saw no increase in the expression levels of retinoid receptors
in the CNS (Zhelyaznik and Mey, 2005). Sources of RA-synthesis in the spinal cord were
oligodendrocytes, meningeal cells and, after injury, NG-2 expressing cells (Mey et al., 2005),
which proliferate and are thought to differentiate mostly to oligodendrocytes (Aguirre et al.,
2004; Nishiyama et al., 2002). In the PNS the RA-releasing cells have not yet been identified,
although RALDH2 staining suggests Schwann cells to be likely candidates.
Even though my immunohistochemical studies of RARα expression point to likely targets of
this signaling pathway after injury, at this time the physiological functions of RA are not
resolved. Besides its putative neuroprotective and anti-inflammatory role RA might be
involved in the regulation of glial re-differentiation after injury. After sciatic nerve lesion
CRBP-I and CRABP-II expression was upregulated in the first days followed by a
downregulation concomitantly with the re-differentiation of Schwann cells during subsequent
regeneration. This downregulation did not occur in degenerating transected nerves
(Zhelyaznik et al., 2003). Thus RA might regulate the state of Schwann cell differentiation. In
cell culture, RA affects monocyte differentiation (Kreutz et al., 1998). OLN93 cells, which
represent a model for immature oligodendrocytes, express RALDH2 and are able to
synthesize RA (Mey and Hammelmann, 2000). These cells are concurrently able to respond to

61
RA: gene expression of LIF was 3-fold upregulated in OLN93 cells after RA treatment (Mey
and Henkes, 2002). It might therefore also be possible that RA promotes cell differentiation of
oligodendrocytes. In addition, my experiments indicate that astrocytes may belong to the
targets of RA as well. For instance, RA has been reported to induce the release of NGF from
cortical astrocytes in vitro (Ahlemeyer et al., 2000). Such effects on transcription of cytokine
and neurotrophin genes were investigated in this work for glia of the PNS, i.e. Schwann cells.
Although I monitored the expression of many inflammatory and regeneration-associated
cytokines, no such function was discovered.
The discrepancy between reported regulatory properties of RA in other cell culture systems
and their non-regulation in Schwann cell primary cultures might be due to the fact that other
contributing factors are involved. For example, treatment of cultured Schwann cells with a
combination of bone morphogenetic protein-2 (BMP2) and RA dramatically induced GDNF-
mRNA, while BMP2 or RA alone had no effect (Kinameri and Matsuoka, 2003).

62
5 Summary
Experiments with sciatic nerve lesions and spinal cord contusions have demonstrated that
retinoic acid is involved in the physiological reactions after PNS and CNS injuries. The aim
of this thesis was to identify the cellular targets of injury-related RA signals in the peripheral
and central nervous system and to investigate the functional significance of RA in this
context.
I discovered that crucial components of the RA signal transduction cascade (retinoic acid
receptors, retinoid X receptors, RALDH2, Cyp26A1, CRABP-II) were present in Schwann
cell primary cultures isolated from newborn rats. After RA treatment the spatial distribution
of RXRs and CRABP-II but not from RARs changed from a cytosolic to a nuclear
localization, and CRABP-II was downregulated via RA. In the search for possible genetic
targets of RA signaling I detected that NGF, BDNF, GDNF, CNTF, TGFβ1, TGFβ2, TGFβ3,
IFNβ, IFNγ and MIF were expressed in Schwann cells. No differential regulation via RA or
activation of intracellular signaling cascades of neuropoietic cytokines or neurotrophins was
seen.
Spinal cord contusion injuries were performed to identify possible cellular targets of RA after
damage of the CNS. Cellular distribution and protein levels of all retinoid receptors in the rat
spinal cord were investigated 4, 7, 14 and 21 days after injury. I detected immunoreactivity of
RARα, RXRα, RXRβ and RXRγ. In the non-lesioned spinal cord, immunoreactivity of
RARα, RXRα, RXRβ and RXRγ was localized in the cytosol of neurons, of RXRα and
RXRβ in astrocytes, and of RARα, RXRα and RXRγ in some oligodendrocytes. After
contusion injury a transient nuclear shift of RARα and of all RXR was visible in reactive
microglia/macrophages. This nuclear staining, already seen at 4 days, was most prominent at
7 and 14 days after injury. A similar change in intracellular distribution was also observed for
the RARα, RXRα and RXRβ staining in neurons situated around the border of the contusion.
The injury-induced subcellular translocation was strongest for RARα, while the intracellular
distribution of RXRγ did not change.
While the identification of the cellular targets of RA signaling confirm the hypothesis of an
involvement of RA in nerve injury-related processes, the functional significance of RA
signaling in Schwann cells remains to be elucidated. In the CNS it seems likely that RA
fulfills neuroprotective functions and contributes to the termination of the inflammatory
response. A complete understanding of these processes might help to develop strategies to
heal neurodegenerative diseases and traumatic injuries.

63
6 Bibliography
Abu-Abed S, Beckett B, Chiba H, Chithalen JV, Jones G, Metzger D, Chambon P, Petkovich M (1998) Mouse P450RAI (CYP26) expression and retinoic acid-inducible retinoic acid metabolism in F9 cells are regulated by retinoic acid receptor gamma and retinoid X receptor alpha. J Biol Chem 273: 2409-2415.
Aguirre AA, Chittajallu R, Belachew S, Gallo V (2004) NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J Cell Biol 165: 575-589.
Ahlemeyer B, Huhne R, Krieglstein J (2000) Retinoic acid potentiated the protective effect of NGF against staurosporine-induced apoptosis in cultured chick neurons by increasing the trkA protein expression. J Neurosci Res 60: 767-778.
Akmal KM, Dufour JM, Vo M, Higginson S, Kim KH (1998) Ligand-dependent regulation of retinoic acid receptor-α in rat testis: in vivo response to depletion and repletion of vitamin A. Endocrinology 139: 1239-1248.
Arroyo EJ, Scherer SS (2000) On the molecular architecture of myelinated fibers. Histochem Cell Biol 113: 1-18.
Bamber NI, Li H, Lu X, Oudega M, Aebischer P, Xu XM (2001) Neurotrophins BDNF and NT-3 promote axonal re-entry into the distal host spinal cord through Schwann cell-seeded mini-channels. Eur J Neurosci 13: 257-268.
Barakat-Walter I (1999) Role of thyroid hormones and their receptors in peripheral nerve regeneration. J Neurobiol 40: 541-559.
Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12: 1-21.
Bastie J-N, Despouy G, Balitrand N, Rochette-Egly C, Chomienne C, Delva L (2001) The novel co-activator CRABP-II binds to RARα and RXRα via two nuclear receptor interacting domains and does not require the AF-2 'core'. FEBS Lett 507: 67-73.
Batchelor PE, Porritt MJ, Martinello P, Parish CL, Liberatore GT, Donnan GA, Howells DW (2004) Macrophages and microglia produce local trophic gradients that stimulate axonal sprouting toward but not beyond the wound edge. Mol Cell Neurosci 21: 436-453.
Baumann CT, Maruvada P, Hager GL, Yen PM (2001) Nuclear cytoplasmic shuttling by thyroid hormone receptors. J Biol Chem 276: 11237-11245.
Black BE, Holaska JM, Rastinejad F, Paschal BM (2001) DNA binding domains in diverse nuclear receptors function as nuclear export signals. Curr Biol 11: 1749-1758.
Blomhoff R (1994) Overview of vitamin A metabolism and function. In: Vitamin A in health and disease (Blomhoff R, ed), pp 1-35. New York: Marcel Dekker.
Bolin LM, Shooter EM (1993) Neurons regulate Schwann cell genes by diffusable molecules. J Cell Biol 123: 237-243.

64
Bolin LM, Verity AN, Silver JE, Shooter EM, Abrams JS (1995) Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. J Neurochem 64: 850-858.
Bost F, Caron L, Marchetti I, Dani C, Marchand-Brustel Y, Binetruy B (2002) Retinoic acid activation of the ERK pathway is required for embryonic stem cell commitment into the adipocyte lineage. Biochem J 361: 621-627.
Boyd JG, Skihar V, Kawaja M, Doucette R (2003) Olfactory ensheathing cells: historical perspective and therapeutic potential. Anat Rec 271B: 49-60.
Boylan JF, Gudas L (1992) The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells. J Biol Chem 267: 21486-21491.
Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, Fehlings MG, Herr DL, Hitchon PW, Marshall LF, Nockels RP, Pascale V, Perot PL Jr, Piepmeier J, Sonntag VK, Wagner F, Wilberger JE,Winn HR,Young W (1998) Methylprednisolone or tirilazad mesylate administration after acute spinal cord injury: 1-year follow up. J Neurosurg 89: 699-706.
Bradbury EJ, Moon LDF, Popat RJ, King VR, Bennet GS, Patel PN (2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416: 636-640.
Braun KW, Tribley WA, Griswold MD, Kim KH (2000) Follicle-stimulating hormone inhibits all-trans-retinoic acid-induced retinoic acid receptor alpha nuclear localization and transcriptional activation in mouse Sertoli cell lines. J Biol Chem 275: 4145-4151.
Braun KW, Vo M-N, Kim KH (2002) Positive regulation of retinoic acid receptor alpha by protein kinase C and mitogen-activated protein kinase in sertoli cells. Biol Reprod 67: 29-37.
Bregman BS, Coumans JV, Dai HN, Kuhn PL, Lynskey J, McAtee M (2002) Transplants and neurotrophic factors increase regeneration and recovery of function after spinal cord injury. Prog Brain Res 137: 257-273.
Bucco RA, Zheng WL, Davis JT, Sierra-Rivera E, Osteen KG, Chaudhary AK, Ong DE (1997) Cellular retinoic acid-binding protein (II) presence in rat uterine epithelial cells correlates with their synthesis of retinoic acid. Biochemistry 36: 4009-4014.
Budhu AS, Noy N (2002) Direct channeling of retinoic acid between cellular retinoic acid-binding protein II and retinoic acid receptor sensitizes mammary carcinoma cells to retinoic acid-induced growth arrest. Mol Cell Biol 22: 2632-2641.
Bugge TH, Pohl J, Lonnoy O, Stunnenberg HG (1992) RXRα, a promiscuous partner of retinoic acid and thyroid hormone receptors. EMBO J 11: 1409-1418.
Bunge MB (2001) Bridging areas of injury in the spinal cord. Neuroscientist 7: 325-339.
Cañon E, Cosgaya JM, Scsucova S, Aranda A (2004) Rapid effects of retinoic acid on CREB and ERK phosphorylation in neuronal cells. Mol Biol Cell 15: 5583-5592.
Caroni P, Schwab ME (1988) Antibody against myelin-associated inhibitor of neurite growth neutralizes non-permissive substrate properties of CNS white matter. Neuron 1: 85-96.

65
Chambon P (1996) A decade of molecular biology of retinoic acid receptors. FASEB J 10: 940-954.
Chandross K (1998) Nerve injury and inflammatory cytokines modulate gap junctions in the peripheral nervous system. Glia 24: 21-31.
Chen J, Evans RM (1995) A transcriptional corepressor that interacts with nuclear hormone receptors. Nature 377: 454-457.
Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME (2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403: 434-439.
Cheng H, Cao Y, Olson L (1996) Spinal cord repair in adult paraplegic rats: partial restoration of hind limb function. Science 273: 510-513.
Choi W-H, Ji K-A, Jeon S-B, Yang M-S, Kim H, Min K, Shong M, Jou I, Joe E-H (2005) Anti-inflammatory roles of retinoic acid in rat brain astrocytes: suppression of interferon-γ-induced JAK/STAT phosphorylation. Biochem Biophys Res Com 329: 125-131.
Chomszynski P, Sacchi N (1987) Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156-159.
Cohen JA, Yachnis AT, Arai M, Davies JG, Scherer SS (1992) Expression of the neu proto-oncogene by Schwann cells during peripheral nerve development and Wallerian degeneration. J Neurosci Res 31: 622-634.
Corcoran J, Maden M (1999) Nerve growth factor acts via retinoic acid synthesis to stimulate neurite outgrowth. Nat Neurosci 2: 307-308.
Corcoran J, Shroot B, Pizzey J, Maden M (2000) The role of retinoic acid receptors in neurite outgrowth from different populations of embryonic mouse dorsal root ganglia. J Cell Sci 113: 2567-2574.
Corcoran J, So PL, Barber RD, Vincent KJ, Mazarakis ND, Mitrophanous KA, Kingsman SM, Maden M (2002a) Retinoic acid receptor beta2 and neurite outgrowth in the adult mouse spinal cord in vitro. J Cell Sci 115: 3779-3786.
Corcoran J, So PL, Maden M (2002b) Absence of retinoids can induce motoneuron disease in the adult rat and a retinoid defect is present in motoneuron disease patients. J Cell Sci 115: 4735-4741.
Cosgaya JM, Perona R, Aranda A (1997) Retinoic acid induces secretion of transforming growth factors by PC12 pheochromocytoma cells. Oncogene 14: 579-587.
Cosgaya JM, Aranda A (2001) Nerve growth factor activates the RARβ2 promoter by a Ras-dependent mechanism. J Neurochem 76: 661-670.
Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3: 240.

66
Cupp AS, Dufour JM, Kim G, Skinner MK, Kim KH (1999) Action of retinoids on embryonic and early postnatal testis development. Endocrinol 140: 2343-2352.
Curtis R, Stewart HJS, Hall SM, Wilkin GP, Mirsky R, Jessen KR (1992) GAP-43 is expressed by non-myelin-forming Schwann cells of the peripheral nervous system. J Cell Biol 116: 1455-1464.
Curtis R, Adryan K, Zhu Y, Harkness P, Lindsay R, DiStephano P (1993) Retrograde axonal transport of ciliary neurotrophic factor is increased by peripheral nerve injury. Nature 365: 253-255.
David S, Aguayo A (1981) Axonal regeneration into peripheral nervous system "bridges" after central nervous system injury in adult rats. Science 214: 931-933.
de The H, Vivanco-Ruiz MM, Tiollais P, Stunnenberg H, Dejean A (1990) Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 343: 177-180.
Delva L, Bastie J-N, Rochette-Egly C, Kraiba R, Balitrand G, Despouy G, Chambon P, Chomienne C (1999) Physical and functional interactions between cellular retinoic acid binding proteins II and the retinoic acid-dependent nuclear complex. Mol Cell Biol 19: 7158-7167.
Denson LA, Auld KL, Schiek DS, McClure MH, Mangelsdorf DJ, Karpen SJ (2000) Interleukin-1β suppresses retinoid trans-activation of two hepatic transporter genes involved in bile formation. J Biol Chem 275: 8835-8843.
Dheen ST, Jun Y, Yan Z, Tay SSW, Ling EA (2005) Retinoic acid inhibits expression of TNFα and iNOS in activated rat microglia. Glia 50: 21-31.
Diamond J, Foerster A, Holmes M, Coughlin M (1992a) Sensory nerves in adult rats regenerate and restore sensory function to the skin independently of endogenous NGF. J Neurosci 12: 1467-1476.
Diamond J, Holmes M, Coughlin M (1992b) Endogenous NGF and nerve impulses regulate the collateral sprouting of sensory axons in the skin of the adult rat. J Neurosci 12: 1454-1466.
Dijkstra CD, Dopp EA, Joling P, Kraal G (1985) The heterogeneity of mononuclear phagocytes in lymphoid organs: distinct macrophage subpopulations in the rat recognized by monoclonal antibodies ED1, ED2 and ED3. Immunology 54: 589-599.
Dimberg A, Nilsson K, Oberg F (2000) Phosphorylation-deficient Stat1 inhibits retinoic acid-induced differentiation and cell cycle arrest in U-937 monoblasts. Blood 96: 2870-2878.
Dong D, Ruuska SE, Levinthal DJ, Noy N (1999) Distinct roles for cellular retinoic acid binding proteins I and II and regulating signaling by retinoic acid. J Biol Chem 274: 23695-23698.

67
Duester G (1996) Involvement of alcohol dehydrogenase, short chain dehydrogenase/reductase, aldehyde dehydrogenase, and cytochrome p450 in the control of retinoid signaling by activation of retinoic acid synthesis. Biochemistry 35: 12221-12227.
Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS (2001) Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol 24: 254-264.
Elliott JL, Snider WD (1996) Motor neuron growth factors. Neurology 47: S47-S53.
Faria TN, Mendelsohn C, Chambon P, Gudas LJ (1999) The targeted disruption of both alleles of RARß2 in F9 cells results in the loss of retinoic acid-associated growth arrest. J Biol Chem 274: 26783-26788.
Fawcett JW, Keynes RJ (1990) Peripheral nerve regeneration. Ann Rev Neurosci 13: 43-60.
Fiedler M, Horn C, Bandtlow C, Schwab ME, Skerra A (2002) An engineered IN-1 F(ab) fragment with improved affinity for the Nogo-A axonal growth inhibitor permits immunochemical detection and shows enhanced neutralizing activity. Protein Eng 15: 931-941.
Fujii H, Sato T, Kaneko S, Gotoh O, Fujii-Kuriyama Y, Osawa K, Kato S, Hamada H (1997) Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J 16: 4163-4173.
Fukunaka K, Saito T, Wataba K, Ashihara K, Ito E, Kudo R (2001) Changes in expression and subcellular localization of nuclear retinoic acid receptors in human endometrial epithelium during the menstrual cycle. Mol Hum Reprodr 7: 437-446.
Funakoshi H, Frisén J, Barbany G, Timmusk T, Zachrisson O, Verge O, Persson H (1993) Differential expression of mRNAs for neurotrophins and their receptors after axotomy of the sciatic nerve. J Cell Biol 123: 455-465.
Gaub M-P, Lutz Y, Ghyselinck NB, Scheuer I, Pfister V, Chambon P, Rochette-Egly C (1998) Nuclear detection of cellular retinoic acid binding proteins I and II with new antibodies. J Histochem Cytochem 46: 1103-1111.
Ghose R, Zimmerman TL, Thevananther S, Karpen SJ (2004) Endotoxin leads to rapid subcellular re-localization of hepatic RXRα: A novel mechanism for reduced hepatic gene expression in inflammation. Nuclear Receptor 2: 4.
Giannini G, Dawson MI, Zhang X, Thiele CJ (1997) Activation of three distinct RXR/RAR heterodimers induces growth arrest and differentiation of neuroblastoma cells. J Biol Chem 272: 26693-26701.
Gillen C, Korfhage C, Müller HW (1997) Gene expression in nerve regeneration. Neuroscientist 3: 112-122.
Gloster A, Diamond J (1992) Sympathetic nerves in adult rats regenerate normally and restore pilomotor function during an anti-NGF treatment that prevents their collateral sprouting. J Comp Neurol 326: 363-374.

68
Gomes-Leal W, Corkill DJ, Freire MA, Picanço-Diniz CW, Perry VH (2004) Astrocytosis, microglia activation, oligodendrocyte degeneration, and pyknosis following acute spinal cord injury. Exp Neurol 190: 456-467.
GrandPré T, Li S, Strittmatter SM (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417: 547-551.
Gross V, Villiger PM, Zhang B, Lotz M (1993) Retinoic acid inhibits interleukin-1-induced cytokine synthesis in human monocytes. J Leukocyte Biol 54: 125-132.
Grothe C, Heese K, Meisinger C, Wewetzer K, Kunz D, Cattini P, Otten U (2000) Expression of interleukin-6 and its receptor in the sciatic nerve and cultured Schwann cells: relation to 18-kD fibroblast growth factor-2. Brain Res 885: 172-181.
Haskell BE, Stach RW, Werrbach-Perez K, Perez-Polo JR (1987) Effect of retinoic acid on nerve growth factor receptors. Cell Tissue Res 247: 67-73.
Heldin CH, Miyazono K, ten Dijke P (1997) TGF-ß signaling from cell membrane to nucleus through SMAD proteins. Nature 390: 465-471.
Heumann R, Korsching S, Bandtlow C, Thoenen H (1987a) Changes of nerve growth factor synthesis in nonneuronal cells in response to sciatic nerve transection. J Cell Biol 104: 1623-1631.
Heumann R, Lindholm D, Bandtlow C, Meyer M, Radecke MJ, Misko TP, Shooter EM, Thoenen H (1987b) Differential regulation of mRNA encoding nerve growth factor and its receptor in rat sciatic nerve during development, degeneration, and regeneration: role of macrophages. Proc Natl Acad Sci USA 84: 8735-8739.
Hirota H, Kiyama H, Kishimoto T, Taga T (1996) Accelerated nerve regeneration in mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J Exp Med 183: 2627-2634.
Holst Av, Lefcort F, Rohrer H (1997) TrkA expression levels of sympathetic neurons correlate with NGF-dependent survival during development and after treatment with retinoic acid. Eur J Neurosci 9: 2169-2177.
Hu L, Gudas LJ (1990) Cyclic AMP analogs and retinoic acid influence the expression of retinoic acid receptor alpha, beta, and gamma mRNAs in F9 teratocarcinoma cells. Mol Cell Biol 10: 391-396.
Ip NY, Yancopoulos GD (1996) The neurotrophins and CNTF: two families of collaborative neurotrophic factors. Ann Rev Neurosci 19: 491-515.
Ito Y, Yamamoto M, Li M, Doyu M, Tanaka F, Mutch T, Mitsuma T, Sobue G (1998) Differential temporal expression of mRNAs for ciliary neurotrophic factor (CNTF), leukemia inhibitory factor (LIF), interleukin-6 (IL-6), and their receptors (CNTFRα, LIFRβ, IL-6Rα and gp130) in injured peripheral nerves. Brain Res 793: 321-327.
Iwase T, Jung CG, Bae H, Zhang M, Soliven B (2005) Glial cell line-derived neurotrophic factor-induced signaling in Schwann cells. J Neurochem [epub ahead of print].

69
Jessen KR, Mirsky R, Morgan L (1987) Myelinated, but not unmelinated axons, reversibly down-regulate N-CAM in Schwann cells. J Neurocytol 16: 681-688.
Jessen, K. R. and Richardson, W. D. Glial cell development. 2. 2001. Oxford, New York, Oxford University Press. Molecular and cellular neurobiology. Davies, R. W., Collingridge, G. L., and Hunt, S. P. Ref Type: Serial (Book,Monograph)
Jing Y, Waxman S, Mira-Y-Lopez R (1997) The cellular retinoic acid binding protein II is a positive regulator of retinoic acid signaling in breast cancer cells. Cancer Res 57: 1668-1672.
Johann V, Jeliaznik N, Schrage K, Mey J (2003) Retinoic acid downregulates the expression of ciliary neurotrophic factor (CNTF) in rat Schwann cells. Neurosci Lett 339: 13-16.
Jones LL, Oudega M, Bunge MB, Tuszynski MH (2001) neurotrophic factors, cellular bridges, and gene therapy for spinal cord injury. J Physiol (London) 533: 83-89.
Kagechika H, Kawachi E, Fukasawa H, Saito G, Iwanami N, Umemiya H, Hashimoto Y, Shudo K (1997) Inhibition of IL-1-induced IL-6 production by synthetic retinoids. Biochem Biophys Res Commun 231: 243-248.
Katagiri Y, Takeda K, Yu Z-X, Ferrans VJ, Ozato K, Guroff G (2000) Modulation of retinoid signalling through NGF-induced nuclear export of NGFI-B. Nat Cell Biol 2: 435-440.
Kiefer R, Kieseier BC, Stoll G, Hartung H-P (2001) The role of macrophages in immune-mediated damage to the peripheral nervous system. Progr Neurobiol 64: 109-127.
Kim B-H, Kang K-S, Lee Y-S (2004) Effect of retinoids on LPS-induced COX-2 expression and COX-2 associated PGE2 release from mouse peritoneal macrophages and TNFα release from rat peripheral blood mononuclear cells. Toxicol Lett 150: 191-201.
Kinameri E, Matsuoka I (2003) Autocrine action of BMP2 regulates expression of GDNF-mRNA in sciatic Schwann cells. Mol Brain Res 117: 221-227.
Kliewer S, Umesono K, Mangelsdorf DJ, Evans RM (1992) Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signaling. Nature 355: 446-449.
Korsching S (1993) The neurotrophic factor concept - a reexamination. J Neurosci 13: 2739-2748.
Kottis V, Thibault P, Mikol D, Xiao ZC, Zhang R, Dergham P, Braun PE (2002) Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J Neurochem 82: 1566-1569.
Krezel W, Kastner P, Chambon P (1999) Differential expression of retinoid receptors in the adult mouse central nervous system. Neurosci 89: 1291-1300.
Kreutz M, Fritsche J, Ackermann U, Krause SW, Andreesen R (1998) Retinoic acid inhibits monocyte to macrophage survival and differentiation. Blood 91: 4796-4802.

70
Kuecherer-Ehret A, Graeber MB, Edgar D, Thoenen H, Kreutzberg GW (1990) Immunoelectron microscopic localization of laminin in normal and regenerating mouse sciatic nerve. J Neurocytol 19: 101-109.
Kurek JB, Austin L, Cheema SS, Bartlett PF, Murphy M (1996) Up-regulation of leukemia inhibitory factor and interleukin-6 in transected sciatic nerve and muscle following denervation. Neuromuscular Disorders 6: 105-114.
Kurokawa R, DiRenzo J, Boehm M, Sugarman J, Gloss B, Rosenfeld MG, Heyman RA, Glass CK (1994) Regulation of retinoid signaling by receptor polarity and allosteric control of ligand binding. Nature 371: 528-531.
Lane MA, Bailey SJ (2005) Role of retinoid signaling in the adult brain. Progr Neurobiol 75: 275-293.
Lazarov-Spiegler O, Rapalino O, Agranow G, Schwartz M (1998) Restricted inflammatory reaction in the CNS: a key impediment to axonal regeneration? Mol Med Today 4: 337-342.
Lee HY, Sueoka N, Hong WK, Mangelsdorf DJ, Claret FX, Kurie JM (1999) All-trans-retinoic acid inhibits Jun N-terminal kinase by increasing dual-specifity phosphatase activity. Mol Cell Biol 19: 1973-1980.
Lefcort F, Venstrom K, McDonald JA, Reichardt LF (1992) Regulation of expression of fibronectin and its receptor alpha 5 beta 1, during development and regeneration of peripheral nerve. Development 116: 767-782.
Lemke G, Chao M (1988) Axons regulate Schwann cell expression of the major myelin and NGF receptor genes. Development 102: 499-504.
Lepore AC, Fischer I (2005) Lineage-restricted neural precursors survive, migrate, and differentiate following transplantation into the injured adult spinal cord. Exp Neurol 194: 230-242.
Lewin GR, Barde Y-A (1996) Physiology of the neurotrophins. Ann Rev Neurosci 19: 289-317.
Li H, Terenghi G, Hall SM (1997a) The effects of delayed re-innervation on the expression of c-erbB receptors by chronically denervated rat Schwann cells in vivo. Glia 20: 333-347.
Li Y, Field PM, Raisman G (1997b) Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science 277: 2000-2002.
Li H, Wigley C, Hall SM (1998) Chronically denervated rat Schwann cells respond to GGF in vitro. Glia 24: 290-303.
Linda H, Piehl F, Dagerlind A, Verge VMK, Arvidsson U, Cullheim S, Risling M, Ulfhake B, Hokfelt T (1993) Expression of GAP-43 mRNA in the adult mammalian spinal cord under normal conditions and after different types of lesions, with special reference to motoneurons. Exp Brain Res 91: 284-295.

71
Lindholm D, Heumann R, Meyer M, Thoenen H (1987) Interleukin-1 regulates synthesis of nerve growth factor in non-neuronal cells of rat sciatic nerve. Nature 330: 658-659.
Liu Y, Lee MO, Wang HG, Li Y, Hashimoto Y, Klaus M, Reed JC, Zhang X (1996) Retinoic acid receptor beta mediates the growth-inhibitory effect of retinoic acid by promoting apoptosis in human breast cancer cells. Mol Cell Biol 16: 1138-1149.
Mackem S, Baumann CT, Hager GL (2001) A glucocorticoid/retinoic acid receptor chimera that displays cytoplasmic/nuclear translocation in response to retinoic acid. A real time sensing assay for nuclear receptor ligands. J Biol Chem 276: 45501-45504.
Madison RD, Zomorodi A, Robinson GA (2000) Netrin-1 and peripheral nerve regeneration in the adult rat. Exp Neurol 161: 563-570.
Martini R, Schachner M (1988) Immunoelectron microscopic localization of neural cell adhesion molecules (L1, N-CAM, and myelin associated glycoprotein) in regenerating adult mouse sciatic nerve. J Cell Biol 106: 1735-1746.
Martini R, Schachner M, Faissner A (1990) Enhanced expression of the extracellular matrix moleculeJ1/tenascin in the regenerating adult mouse sciatic nerve. J Neurocytol 19: 601-616.
Maruvada P, Baumann CT, Hager G, Yen PM (2003) Dynamic shuttling and intranuclear mobility of nuclear hormone receptors. J Biol Chem 278: 12425-12432.
Matikainen S, Lehtonen A, Sareneva T, Julkunen I (1998) Regulation of IRF and STAT gene expression by retinoic acid. Leuk Lymphoma 30: 63-71.
Matsuoka I, Meyer M, Thoenen H (1991) Cell-type-specific regulation of nerve growth factor synthesis in non-neuronal cells: comparison with other cell types. J Neurosci 11: 3165-3177.
Matsuoka I, Nakane A, Kurihara K (1997) Induction of LIF-mRNA by TGF-beta 1 in Schwann cells. Brain Res 776: 170-180.
Maurel P, Salzer JL (2000) Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J Neurosci 20: 4635-4645.
McKeon RJ, Schreiber RC, Rudge JS, Silver J (1991) Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci 11: 3398-3411.
McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE (1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13: 805-811.
Mews M, Meyer M (1993) Modulation of Schwann cell phenotype by TGFß-1 - inhibition of P0 messenger RNA expression and downregulation of the low affinity NGF receptor. Glia 8: 208-217.
Mey J, Rombach N (1999) Retinoic acid increases BDNF-dependent regeneration of chick retinal ganglion cells in vitro. Neuroreport 10: 3573-3577.

72
Mey J, Hammelmann S (2000) OLN-93 oligodendrocytes synthesize all-trans retinoic acid in vitro. Cell Tissue Res 302: 49-58.
Mey J (2001) Retinoic acid as a regulator of cytokine signaling after nerve injury. Z Naturforsch C 56c: 163-176.
Mey J, Henkes L (2002) Retinoic acid enhances leukemia inhibitory factor expression in OLN-93 oligodendrocytes. Cell Tissue Res 310: 155-161.
Mey J, Morasutti D, Brook G, Liu R-H, Zhang Y-P, Koopmans G, McCaffery P (2005) Retinoic acid synthesis by a population of NG-2 positive cells in the injured spinal cord. Eur J Neurosci 21: 1555-1568.
Meyer M, Matsuoka I, Wetmore C, Olson L, Thoenen H (1992) Enhanced synthesis of brain-derived neurotrophic factor in the lesioned peripheral nerve: different mechanisms are responsible for the regulation of BDNF and NGF mRNA. J Cell Biol 119: 45-54.
Mic FA, Molotkov A, Benbrook DM, Duester G (2003) Retinoid activation of retinoic acid receptor but not retinoid X receptor is sufficient to rescue lethal defect in retinoid synthesis. Proc Natl Acad Sci USA 100: 7135-7140.
Minucci S, Leid M, Toyama R, Saint-Jeannet J-P, Peterson VJ, Horn V, Ishmael JE, Bhattacharyya N, Dey A, Dawid IB, Ozato K (1997) Retinoid X receptor (RXR) within the RXR-retinoic acid receptor heterodimer binds to its ligand and enhances retinoid-dependent gene activation. Mol Cell Biol 17: 644-655.
Mitchell LS, Griffith IR, Morrison S, Barrie JA, Kirkham A, McPhilmey K (1990) Expression of myelin protein gene transcripts by Schwann cells of regenerating nerve. J Neurosci 27: 125-135.
Mohan M, Thirumalapura N, Malayer J (2003) Bovine cumulus-granulosa cells contain biologically active retinoid receptors that can respond to retinoic acid. Reprod Biol Endocrinol 1: 104.
Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT (1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13: 757-767.
Murray M, Kim D, Liu Y, Tobias C, Tessler A, Fischer I (2002) Transplantation of genetically modified cells contributes to repair and recovery from spinal injury. Brain Res Rev 40: 292-300.
Nacimiento W, Sappok T, Brook GA, Toth L, Oestreicher AB, Gispen WH, Noth J, Kreutzberg GW (1995) B-50 (GAP43) in the rat spinal cord caudal to hemisection: lack of intrasoinal sprouting by dorsal root axons. Neurosci Lett 194: 13-16.
Napoli JL (1996) Retinoic acid biosynthesis and metabolism. FASEB J 10: 993-1001.
Naumann T, Peterson GM, Frotscher M (1992) Fine structure of rat septohippocampal neurons: II. A time course analysis following axotomy. J Comp Neurol 325: 219-242.

73
Nishio Y, Nishihira J, Ishibashi T, Kato H, Minami A (2002) Role of macrophage migration inhibitory factor (MIF) in peripheral nerve regeneration: Anti-MIF antibody induce delay of nerve regeneration and the apoptosis of Schwann cells. Mol Med 8: 509-520.
Nishiyama A, Watanabe M, Yang Z, Bu J (2002) Identity, distribution, and development of polydendrocytes: NG2-expressing glial cells. J Neurocytol 31: 437-455.
Noy N (2000) Retinoid-binding proteins: mediators of retinoid action. Biochem J 348: 481-495.
Oaklander AL, Spencer PS (1988) Cold blockade of axonal transport activates premitotic activity of Schwann cells and Wallerian degeneration. J Neurochem 50: 490-496.
Oppenheim RW (1996) Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron 17: 195-197.
Oudega M, Hagg T (1999a) Neurotrophins promote regeneration of sensory axons in the adult rat spinal cord. Brain Res 818: 431-438.
Oudega M, Vargas CG, Weber AB, Kleitman, N, Bunge MB (1999b) Long-term effects of methylprednisolone following transection of adult rat spinal cord. Eur J Neurosci 11: 2453-2464.
Pelser H, van Gijn J (1993) Spinal infarction. A follow-up study. Stroke 24: 896-898.
Perry VH, Matyszak MK, Fearn S (1993) Altered antigen expression of microglia in the aged rodent CNS. Glia 7: 60-67.
Piedrafita FJ, Pfahl M (1999) Nuclear retinoid receptors and mechanisms of action. In: Retinoids: The Biochemical and Molecular Basis of Vitamin A an Retinoid Action pp 153-184. Springer, Berlin.
Prüfer K, Racz A, Lin GC, Barsony J (2000) Dimerization with retinoid X receptors promotes nuclear localization and subnuclear targeting of vitamin D receptors. J Biol Chem 275: 41114-41123.
Prüfer K, Barsony J (2002) Retinoid X receptor dominates the nuclear import and export of the unliganded vitamin D receptor. Mol Endocrinol 16: 1738-1751.
Quinn SDP, De Boni U (1991) Enhanced neuronal regeneration by retinoic acid of murine dorsal root ganglia and of fetal murine and human spinal cord in vitro. In Vitro Cell Dev Biol 27A: 55-62.
Raivich G, Bohatschek M, Kloss CUA, Werner A, Jones LL, Kreutzberg GW (1999) Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Rev 30: 77-105.
Ramón y Cajal S (1914) Estudios sobre la degeneración y regeneración del sistema nervioso. Madrid: Imprenta hihos de Nicolás Moya.
Ramón y Cajal S. Degeneration and regeneration of the nervous system. May, R.M. 1928. Oxford, Oxford university press. Ref Type: Serial (Book, Monograph)

74
Reichert F, Levitzky R, Rotshenker S (1996) Interleukin 6 in intact and injured mouse peripheral nerves. Eur J Neurosci 8: 530-535.
Rodríguez-Tébar A, Rohrer H (1991) Retinoic acid induces NGF-dependent survival response and high-affinity binding NGF receptors in immature chick sympathetic neurons. Development 112: 813-820.
Ross S, McCaffery P, Dräger UC, De Luca LM (2000) Retinoids in embryonal development. Physiol Rev 80: 1021-1054.
Sah JF, Eckert RL, Chandraratna RA, Rorke EA (2002) Retinoids suppress epidermal growth factor-associated cell proliferation by inhibiting epidermal growth factor receptor-dependent Erk1/2 activation. J Biol Chem 277: 9728-9735.
Santos-Benito FF, Ramón-Cueto A (2003) Olfactory ensheathing glia transplantation: a therapy to promote repair in the mammalian central nervous system. Anat Rec 271B: 77-85.
Sarkar SA, Sharma RP (2002) Expression of selected apoptosis related genes, MIF, IGIF and TNF alpha, during retinoic acid-induced neural differentiation in murine embryonic stem cells. Cell Struct Funct 27: 99-107.
Scheibe RJ, Wagner JA (1992) Retinoic acid regulates both expression of the nerve growth factor receptor and sensitivity to nerve growth factor. J Biol Chem 267: 17611-17616.
Scherer SS, Kamholz J, Jakowlew SB (1993) Axons modulate the expression of transforming growth factor betas in Schwann cells. Glia 8: 265-276.
Schrage K (2001) Regulation der Retinsäure-Signaltransduktion nach Verletzung des Ischiasnervs der Ratte. Aachen: Diplomarbeit.
Schubert KO, Naumann T, Schnell O, Zhi Q, Steup A, Hofmann H-D, Kirsch M (2005) Activation of STAT3 signaling in axotomized neurons and reactive astrocytes after fimbria-fornix transection. Exp Brain Res in press.
Schwab ME, Bartholdi D (1996) Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev 76: 319-370.
Schwab ME (2002) Increasing plasticity and functional recovery of the lesioned spinal cord. Prog Brain Res 137: 351-359.
Segal RA, Greenberg ME (1996) Intracellular signaling pathways activated by neurotrophic factors. Ann Rev Neurosci 19: 463-489.
Sendtner M, Stöckli KA, Thoenen H (1992b) Synthesis and localization of ciliary neurotrophic factor in the sciatic nerve of the adult rat after lesion and during regeneration. J Cell Biol 118: 139-148.
Sessler, R. J. and Noy, N. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Division of nutritional sciences . 2004. Ref Type: Magazine Article

75
Shamash S, Reichert F, Rotshenker S (2002) The cytokine network of Wallerian degeneration: tumor necrosis factor-α, interleukin-1α, and interleukin-1β. J Neurosci 22: 3052-3060.
Shuman SL, Bresnahan JC, Beattie MS (1997) Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res 50: 798-808.
Sjöberg M, Vennström B, Forrest D (1992) Thyroid hormone receptors in chick retinal development: differential expression of mRNAs for α and N-terminal variant β receptors. Development 114: 39-47.
Skaper SD, Walsh FS (1998) Neurotrophic molecules: strategies for designing effective therapeutic molecules in neurodegeneration. Mol Cell Neurosci 12: 179-193.
Sketelj J, Bresjanac M, Popovic M (1989) Rapid growth of regenerating axons across segments of sciatic nerve devoid of Schwann cells. J Neurosci Res 24: 153-162.
Slovodov U, Reichert F, Mirski R, Rotshenker S (2000) Distinct inflammatory stimuli induce different patterns of myelin phagocytosis and degradation in recruited macrophages. Exp Neurol 167: 401-409.
Snider WD (1994) Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell 77: 627-638.
Soprano DR, Qin P, Soprano KJ (2004) Retinoic acid receptors and cancers. Annu Rev Nutr 24: 201-221.
Stoll G, Jander S, Schroeter M (2002) Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol 513: 87-113.
Streit WJ, Graeber MB, Kreutzberg GW (1989) Expression of Ia antigen on perivascular and microglial cells after sublethal and lethal motor neuron injury. Exp Neurol 105: 115-126.
Sucov HM, Murakami KK, Evans RM (1990) Charcterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc Natl Acad Sci USA 87: 5392-5396.
Svensson LG, Crawford ES, Hess KR, Coselli JS, Safi HJ (1993) Experience with 1509 patients undergoing thoracoabdominal aortic operations. J Vasc Surg 17: 357-368.
Takiyama Y, Miyokawa N, Sugawara A, Kato S, Ito K, Sato K,Oikawa N, Kobayashi H, Kimura S, Tateno M (2004) Decreased expression of retinoid X receptor isoforms in human thyroid carcinomas. J Clin Endocrinol Metabol 89 : 5851-5861.
Taniuchi M, Clark HB, Johnson EM (1986) Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci USA 83: 4094-4098.
Tator CH, Edmonds VE (1979) Acute spinal cord injury: analysis of epidemiological factors. Can J Surg 22: 575-578.

76
Tator CH (1983) Spine-spinal cord relationships in spinal cord trauma. In: Clinical Neurosurgery (Weiss MH, ed), Baltimore: Williams &Wilkins.
Theriault E, Tetzlaff W, Tator CH (1992) Elevated gene expression in the red nucleus after spinal cord compression injury. Neuroreport 3: 559-562.
Thomson CE, Griffith IR, McCullough MC, Kyriakides E, Barrie JA, Montague P (1993) In vitro studies of axonally-regulated Schwann cell genes during Wallerian degeneration. J Neurocytol 22: 590-602.
Vo HP, Crowe DL (1998) Transcriptional regulation of retinoic acid binding protein II modulates RA mediated tumor cell proliferation and invasion. Anticancer Res 18: 217-224.
Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z (2002) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417: 941-944.
Wei L-N (2003) Retinoid receptors and their co-regulators. Ann Rev Pharmacol Toxicol 43: 47-72.
Weßels I (2005) Einfluss proinflammatorischer Zytokine auf die intrazelluläre Verteilung von Retinoid-Rezeptoren. Aachen: Bachelorarbeit.
White JA, Guo YD, Baetz K, Beckett-Jones B, Bonasoro J, Hsu KE, Dilworth FJ, Jones G, Petkovic M (1996) Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J Biol Chem 271: 29922-29927.
White JA, Beckett-Jones B, Guo YD, Dilworth FJ, Bonasoro J, Jones G, Petkovic M (1997) cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450 (CYP26). J Biol Chem 272: 18538-18541.
Willison HJ, Trapp BD, Bacher JD, Quarles RH (1988) The expression of myelin-associated glycoprotein in regenerating cat sciatic nerve. Brain Res 444: 10-16.
Wion D, Houlgatte R, Barbot N, Barrand P, Dicou E, Brachet P (1987) Retinoic acid increases the expression of NGF gene in mouse L cells. Biochem Biophys Res Com 149: 510-514.
Wrana JL, Attisano L, Wiester R, Ventura F, Massague J (1994) Mechanism of activation of the TGF-ß receptor. Nature 370: 341-347.
Wuarin L, Sidell N, De Vellis J (1990) Retinoids increase perinatal spinal cord neuronal survival and astroglial differentiation. Int J Dev Neurosci 8: 317-326.
Xiao Y, Desai D, Quick TC, Niles RM (1996) Control of retinoic acid receptor expression in mouse melanoma cells by cyclic AMP. J Cell Physiol 167: 413-421.
Yamamoto M, Dräger UC, Ong DE, McCaffery P (1998) Retinoid-binding proteins in the cerebellum and choroid plexus and their relationship to regionalized retinoic acid synthesis and degradation. Eur J Biochem 257: 344-350.

77
Yang L, Jones NR, Blumbergs PC, Heuvel VD, Moore EJ, Manavis J, Sarvestani GT, Gabriel MN (2005) Severity-dependent expression of pro-inflammatory cytokines in traumatic spinal cord injury in the rat. J Clin Neurosci 12: 276-284.
Yen A, Varvayanis S (2000) Retinoic acid increases amount of phosphorylated RAF; ectopic expression of cFMS reveals that retinoic acid-induced differentiation is more strongly dependent on ERK2 signaling than induced GO arrest is. In Vitro Cell Dev Biol Anim 36: 249-255.
Yick LW, Wu W, So KF, Yip HK, Shum DK (2000) Chondroitinase ABC promotes axonal regeneration of Clarke´s neurons after spinal cord injury. Neuroreport 11: 1063-1067.
Zancai P, Cariati R, Quaia M, Guidoboni M, Rizzo S, Boiocchi M, Dolcetti R (2004) Retinoic acid inhibits IL-6-dependent but not constitutive Stat3 activation in Epstein-Barr virus-immortalized B lymphocytes. Int J Oncol 25: 345-355.
Zelent A, Krust A, Petkovich M, Kastner P, Chambon P (1989) Cloning of murine alpha and beta retinoic acid receptors and a novel receptor gamma predominantly expressed in skin. Nature 339: 714-717.
Zhao B, Schwartz JP (1998) Involvement of cytokines in normal CNS development and neurological diseases: recent progress and perspectives. J Neurosci Res 52: 7-16.
Zhelyaznik N, Schrage K, McCaffery P, Mey J (2003) Activation of retinoic acid signaling after sciatic nerve injury: upregulation of cellular retinoid binding proteins. Eur J Neurosci 18: 1033-1040.
Zhelyaznik N (2003) Retinoic acid signaling after peripheral nerve injury. Aachen: Doktorarbeit.
Zhelyaznik N, Mey J (2005) Expression of retinoic acid receptors and retinoid X receptors after sciatic nerve injury. Neuroforum 11: Suppl. 381A.
Zheng WL, Bucco RA, Schmitt MC, Wardlaw SA, Ong DE (1996) Localization of cellular retinoic acid-binding protein (CRABP) II and CRABP in developing rat testis. Endocrinology 137: 5028-5035.
Zhu X, Hanover JA, Hager GL, Cheung S-Y (1998) Hormone-induced translocation of thyroid hormone receptors in living cells visualized using a receptor green fluorescent protein chimera. J Biol Chem 273: 27058-27063.
Zitnik RJ, Kotloff RM, Latifpour J, Zheng T, Whiting NL, Schwalb J, Elias JA (1994) Retinoic acid inhibition of IL-1-induced IL-6 production by human lung fibroblasts 2260. J Immunol 152: 1419-1427.

78
Danksagung
Ich möchte mich bei allen Mitgliedern des Instituts für Biologie II bedanken. Die herzliche
und unkomplizierte Arbeitsatmosphäre hat wesentlich dazu beigetragen, daß ich mich im
Institut so wohl gefühlt habe. Mein besonderer Dank gilt meinem Doktorvater PD Dr. Jörg
Mey für sein Engagement bei meiner Betreuung aber auch für die Freiheit, selbstständig
arbeiten zu können. Seine positive und unkonventionelle Einstellung und seine konstruktive
Kritik haben mich immer motiviert und viel zu meiner beruflichen Entwicklung beigetragen.
Prof. Dr. Hermann Wagner danke ich für die Übernahme des Koreferats, seinem Interesse an
meinem beruflichen Werdegang und seiner Hilfe bei organisatorischen Problemen. PD Dr.
Harald Luksch möchte ich für seine Hilfsbereitschaft bei der Bewältigung spontan
auftretender praktischer Probleme danken. Helga Gaube gebührt ein großes Dankeschön für
die Englischkorrektur meiner Dissertation. Guido Koopmans hat mich während meines
Aufenthalts in Maastricht unter seine Fittiche genommen. Dafür danke ich ihm ganz herzlich.
Meinen langjährigen Bürokollegen Ilona Vollpracht-Crijns und Benedikt Mönig danke ich für
die vielen lustigen Momente, die unseren Laboralltag erfrischt haben und auch mal auf meine
Kosten gingen :-). Meinen Freundinnen Nina Zhelyaznik, Verena Johann, Sarah Golz, Liliane
Henkes und Katrin Vonderschen möchte ich für die schönen und witzigen gemeinsamen
Stunden während der Arbeit und in den „Regenerations“phasen danach danken. Meinem
Freund Wolf danke ich für sein Dasein. Mein größter Dank geht an meine Familie für ihre
Liebe und immerwährende Unterstützung.

Lebenslauf Persönliche Daten Name Kirsten Ingrid Schrage Anschrift Mozartstrasse 24 – 26, 52064 Aachen Telefon 0241/4015760 email [email protected] 19.11.1972 Geburtsort Hagen (NRW) Familienstand ledig Staatsangehörigkeit deutsch Ausbildung 1979-1983 Besuch der Gemeinschafts-Grundschule Hagen-Helfe 1983-1992 Besuch des Gymnasiums Hildegardis-Schule Hagen 10.1992-01.1994 Studium der Medizin an der RWTH Aachen 10.1994-01.2001 Studium der Biologie an der RWTH Aachen 12.1999-01.2001 Diplomarbeit am Lehrstuhl für Zoologie und Tierphysiologie
der RWTH Aachen mit dem Thema „Regulation der Retinsäure-Signaltransduktion nach Verletzung des Ischiasnerven der Ratte“
02.2001-12.2005 Promotion am Lehrstuhl für Zoologie und Tierphysiologie der RWTH Aachen mit dem Thema „Retinoic acid signaling after nerve injury“
Auszeichnungen/Stipendien Springorum-Denkmünze 2001 der RWTH Aachen 10.2003-07.2005 Abschlußstipendium der Graduiertenförderung der RWTH
Aachen 03.2004-06.2004 Marie Curie Training Site an der European Graduate School of
Neuroscience, EURON Veröffentlichungen V. Johann, N. Jeliaznik, K. Schrage and J. Mey. Retinoic acid downregulates the expression of ciliary neurotrophic factor (CNTF) in rat Schwann cells. Neurosci. Lett. 339 (2003): 13-16. N. Zhelyaznik, K. Schrage, P. McCaffery and J. Mey. Activation of retinoic acid signaling after sciatic nerve injury: upregulation of cellular retinoid binding proteins. Eur. J. Neurosci. 18 (2003): 1033-1040. K. Schrage, G. Koopmans, E. A. J. Joosten and J. Mey. Reactive microglia, macrophages and neurons are targets of retinoic acid signaling after spinal cord injury. Eur. J. Neurosci., in press.













![Vorlesung Zellbiologie Teil Biologie: Evolution – Zellbiologie – Entwicklung Institut für Biologie II Jörg Mey [ppt-Folien ohne Copyright-Abb.] Jörg Mey](https://img.pdfslide.org/doc/110x75/55204d6949795902118bff2a/vorlesung-zellbiologie-teil-biologie-evolution-zellbiologie-entwicklung-institut-fuer-biologie-ii-joerg-mey-ppt-folien-ohne-copyright-abb-joerg-mey.jpg)















