Embed Size (px)
Citation preview

N.G. Anderson: The Development of Fast Analyzers 271
13. Burtis, C. A., Johnson, W. F., Attrill, J. E., Scott, C.D., Cho, N., Anderson, N.G.: Clin. Chem. 17, 686--695 (1971).
14. :Burtis, C.A., Johnson, W.F., Mailen, J.C., Attrill, J. E.: Clin. Chem. 18, 433 (1972).
15. Burtis, C. A., Mailen, J. C., Johnson, W. F., Scott, C. D. Tiffany, T. O., Anderson, N. G. : Clin. Chem. 18, 753--761 (1972).
16. Fabiny, D.L., Ertingshausen, G. : Clin. Chem. 17, 696--700 (1971).
17. Hatcher, D. W., Anderson, N. G.: Am. J. Clin. Pathol. 52. 645--650 (/969).
18. Jansen, J. M., Jr.: Clin. Chem. 16, 515 (1970). 19. Kelley, M. T., Jansen, M. T.: Clin. Chem. 17, 701--706
(1971). 20. Kinney, T. D., Melville, R. S. (compilers) : The mechani-
zation, automation, and increased effectiveness of the clinical laboratory. Status report by the research committee on automation in medical sciences of the National Institute of General Medical Sciences, DttEW Publication No. (NIH 72-145), 1971.
21. Kinney, T. D., Melville, R. S. : Lab. Investigation 20, 382 (1969).
22. Maclin, E.: Clin. Chem. 17, 707--714 (1971).
23. Mashburn, D. N., Stevens, R. H., Willis, D. D., Elrod, L.H., Anderson, N.G.: Anal. Biochem. 85, 98--112 (1970).
24. Proceedings of the third annual symposium on high resolution analyses and advanced concepts for the clinical laboratory, March 11--12, 1971, Oak Ridge National Laboratory, Oak Ridge, Tennessee. Clin. Chem. 17, 685--821 (1971).
25. Purdy, W. C., Melville, R. S. : Anal. Chem. 42 (12), 32A (1970).
26. Scott, C. D., Attrill, J. E., Anderson, N. G.: Proc. Soe. Exptl. Biol. Med. 125, 181 (1967).
27. Scott, C. D., Mailen, J. C. : Clin. Chem. 18, 749-- 752 (1972). 28. Spragg, S. P., Goodman, R. F.: Ann. N. Y. Acad. Sei.
164, 294--305 (1969). 29. Tiffany, T.O., Jansen, J.M., Burtis, C.A., 0verton,
J. B., Scott, C. D.: Clin. Chem. 18, 829--840 (1972). 30. Tiffany, T.O., Johnson, G.F., Chilcote, M.E. : Clin.
Chem. 17, 715--720 (1971).
Dr. N. G. Anderson Oak Ridge National Laboratory P.O. Box X Oak Ridge, Tenn. 37830 U.S.A.
Z. Anal. Chem. 261,271 280 (1972) �9 by Springer-Verlag 1972
Schnelle Photometrie komplizierter biochemischer Mehrkomponentensysteme
D. W. Lfibbers und t~. Wodick
Max-Planck-Institut fiir Arbeitsphysiologie, Dortmund
Eingegangen am 2.Juni 1972
Rapid Photometry o] Complicated Biochemical Multi-Component Systems. After discussing general problems concerning appara tus and measuring conditions the analysis of mul t i -component systems in homogeneous solutions (multiplicative colour mixture) and of those with inhomogeneous distribution (additive colour mixture) is described. Analysis of nueleotides and guinea pig brain and determination of the degree of oxygena- t ion of haemoglobin are presented as examples.
Zusammen/assung. Eine Ubersieht fiber die schnelle photometrische Analyse komplizierter bioehemiseher Mehrkomponentensysteme wird gegeben. Zun~ehst werden apparat ive Probleme und Megbedingungen be- handelt. Anschliegend werden die Auswertverfahren ffir Mehrstoffsysteme in homogener LSsung (multi- plikative Farbmischung) und ffir solche mit inhomogener Verteilung (additive Farbmischung) besehrieben. Als praktisehe Anwendungsbeispiele dienen die Analyse yon Nucleot idsummenspektren und Meerschweinchen- gehirnreflexionspektren sowie die Best immung des Oxigenierungsgrades yon Hgmoglobin.
Untersuehung komplizierter Mehrkomponentensysteme; Photometr ie / Bioehemisehe Analyse; Analyse addi- tiver und multiplikativer Farbmisehungen / Methode kleinster Quadrate.

272 Z. Anal. Chem., Band 261, Heft 4/5 (1972)
Komplexe t{eaktionen yon Mehrkomponentensyste- men lassen sich h~ufig nut am intakten System unter- suchen, da geakt ionsmeehanismus und I{eaktions- kinetik oft schon durch die Auftrennung des Systems stark ver/~ndert werden. I m folgenden soll gezeigt werden, dag die photometrisehe Methode besonders gut ffir eine solehe Analysenaufgabe geeignet ist. Es ist m6glieh, den Informationsgehalt der photometri- sehen Messungen dabei optimal auszunutzen, auBer- dem unbekannte St6rfunktionen durch eine N/~he- rungslSsung mit zu berficksiehtigen. Kompliziertere biochemisehe Systeme lassen sich erfassen, wenn ein neuartiges Invar iantenverfahren angewandt wird.
I m folgenden werden zun~ehst die photometrisehen MeBbedingungen ftir die Anwendung der Mehrkom- ponentenanalyse diskutiert und dann die Auswert- verfahren dargestellt [20, 22].
1. Photometer fiir die Mehrkomponentenanalyse
Um eine Mehrkomponentenanalyse durchfiihren zu k6nnen, mtissen Spektralphotometer mit hoher Re- produzierbarkeit und groBer Genauigkeit eingesetzt werden. Die Photometer sollten eine digitale Daten- erfassung sowohl der Wel]enlgnge als auch der Ex- t inktion m5glich maehen, damit die Auswertung nieht zu langwierig und zu ungenau wird.
Die hSehste Genauigkeit kann mit Zweistrahlver- fahren erreicht werden [11,12], wenn die optimale Megbedingung ,,g]eiehe Lichtstrahlen - - gleiehe Optik - - g]eieher Photomultiplier" fiir Mel3strahl und Vergleiehsstrahl m5gliehst gut verwirklieht ist. Die maximale Megfrequenz ist bei Zweistrahlverfahren durch den Umsehalter bzw. durch die Strahlen- trennung gegeben.
Die Umschaltung zwisehen den beiden Lichtwegen ge- schieht meistens mechanisch. Sie kann durch einen beweg- lichen Spiegel oder dureh Bewegung der K/ivette bzw. des Photomultipliers oder durch einen entsprechend gefiihrten Lichtleiter bewirkt werden. Eine andere M6glichkeit der Signaltrennung wird durch Modulation der beiden Liehtwege mit verschiedenen Frequenzen erreicht [34]. Dann ist es m6glich, beide Signale durch selektive Verst~rker zu trennen. Soweit uns bekannt wurde, ist dieses an sich sehr gute Ver- fahren bisher nur bei relativ niedrigen Frequenzen angewandt worden (Thews, Liibbers, 1955:450 Hz/1800 Hz). Man kann bei geeigneter optischer Abbildung und mit Unterbrecher- scheiben, die im Vakuum laufen, sicher sehr viel h6here Frequenzen erreichen. Die Lichtmodulation l~l~t sich auch durch rotierende Polarisationsfilter, Strahlversetzung, durch schwingende St~be (z.B. Nickel, Saiteli oder Kristalle) er- reiehen. Aul3erdem sind Kerr-Zellen oder i~hnliche An- ordnungen geeignet; sie arbeiten jedoch mit einem sehr hohen Liehtverlust. Alle zuletzt genannten technischen MSglich- keiten habeli aber bisher noch zu keiner ausgereiften Kon- struktion gefiihrt.
Soweit wie wir fibersehen, ist z.Z. die hSchste Um- schaltfrequenz beim I~apidspektrometer der Kieler Howaldts-Werke [16,25] mit 25 kIIz verwirklicht. Bei diesem Geriit schaltet ein 2 • m m groBer Schwingspiegel, der in seiner Eigenfrequenz schwingt, die Lichtstrahlen um.
Die Wiederholungsfrequenz ffir die Spektren ist beim Rapidspektrometer durch einen im Parallel-Strahlengang des Monoehromators angebrachten Planspiegels yon 50 • 70 mm vorgegeben, der als Torsionsschwinger aufgebaut ist. Seine Eigenfrequenz kann je naeh Konstruktion zwischen 25 und 200 Hz betragen, d.h. fiir ein Spektrum werden 20--2,5 ms benStigt. Das Ger~t der Kieler Howaldts-Werke benStigt 10 ms fiir ein Spektrum.
I-I/ilt man die strikte Forderung ,,gleiche Licht- quelle - - gleiche Optik - - gleiehe Liehtempf~nger" nieht ein, so bieten sieh mehrere konstruktive M6g- liehkeiten, um Spektren schnell hintereinander durch- zumessen : Man kann fiir MeB- und Vergleiehsstrahlen 2 Photomultiplier benutzen. Dadurch h/~ngt die Registriergeschwindigkeit nut noeh yon der Wieder- holungsgesehwindigkeit des Spektrums und yon den Eigensehaften des Photomultipliers ab. Grabowski u. Koszewski [8] benutzen den Leuchtfleek eines Oszillo- graphensehirms als Lichtquelle. Die Ablenkge- schwindigkeit des Elektronenstrahls kann natfirlich sehr hoeh gemaeht werden. Jedoch ist die Wieder- holungsgesehwindigkeit dureh die Abklingzeit bzw. die Naehleuehtdauer des Leuehtphosphors begrenzt. Zur Zeit betr/igt die l~egistrierdauer ffir ein Spektrum 100 ~zs. Die Giite der Methode h/~ngt yon der Qualit/~t der Leuehtphosphore ab.
Verziehtet man auf die stgndige Kontrolle gegen einen Vergleichsstrahl, so kann in einfachster Form das gesamte Spektrum gleiehzeitig durch eine photo- graphische Aufnahme erfaBt werden. Die not- wendigen Belichtungszeiten liegen in der GrSBen- ordnung yon mehreren Sekunden. Interessiert man sieh nur fiir bestimmte spektrale Bereiche, so k6nnen s ta t t des photographischen Films mehrere Multiplier gleiehzeitig nebeneinander geordnet werden, wie es yon Hagenah [13] angegeben wurde. Bei diesem Auf- bau ist die zeitliehe Aufl6sung nur noeh yon den Eigenschaften des Photomultipliers abhgngig. Das photometrische Bild des Spektrums kann start photo- graphiseh auch mit einem Vidicon [5,24] festgehalten werden. Moehizuki u. Mitarb. haben die 2 H~lften eines Vidieons getrennt benutzt, die eine zur Messung des MeBstrahls, die andere ffir den Vergleichsstrahl. Jedoch sind die Photosehiehten nie ganz homogen. Die Wiederholungsfrequenz lag zwisehen 10 und 0,05 ms. Die kurzen Zeiten k6nnen Mlerdings nicht voll fiir die Messung genutzt werden, da die photo-

D.W. Lfibbers und R. Wodick: Sehnelle Photometric komplizierter bioehemischer l~Iehrkomponentensysteme 273
empflndliche Schicht einem plStzlichen Wechsel hell- dunkel nur verzSgert folgt. Auch die Wellenl/ingen- auflSsung ist noch begrenzt, da die Einzelelemente der Photoschicht noch relativ grob sind. Eine wesent- liche Verbesserung brachte die Weiterentwieklung yon Bridoux u. Delhaye [5], die zus/~tzlich vor dem Vidicon einen Bildverst/~rker einfiigten. Dadurch er- hielten sic eine Signalverst/irknng yon 105-10 s und Aufnahmezeiten fiir ein Spektrum yon 10 ns. Dieses Photometer wurde zur Messung yon Raman-Spektren eingesetzt (UF 76, Ultra fast spectrograph, Cod- berg, SA).
Zuletzt sei noch das Rapid Scanning Spectro- meter 502 yon der Fa. OCLI Inst ruments erw/~hnt. Es ist ein Einstrahlger/it, bei dem die Spektren- ablenkung durch die Bewegung yon Winkelspiegeln hervorgerufen wird [1, 7]. Da mehrere Winl~elspiegel auf einer rotierenden Scheibe angebracht sind, er- geben sich Spektrenwiederholfrequenzen yon 1 bis 100 ms.
Letzten Endes liegt die Zeitbegrenzung ffir optisch analytische Messungen beim Photoempf/inger. I m ultravioletten und im sichtbaren Bereich ist der Photomultiplier mit seinen hintereinandergeschalte- ten Dynoden der beste Empf/~nger. Seine Grenz- frequenz liegt zwischen 100 MHz und 1 GHz. Seine Empfindliehkeit h/s yon den Eigenschaften der Photokathode ab. Man erstrebt eine mSglichst hohe Quantenausbeute. Sie ist bei guten Schichten im Ultravioletten nahe bei 1, im Siehtbaren nahe bei 0,3. Ffir das nahe Infrarot gibt es neuerdings Schiehten (GaAs-Cs20; InAsP-Cs20 ) mit / ihnlich grol3en Quan- tenausbeuten. I m weiteren Infrarot nehmen die Quantenausbeuten sehr stark ab, so dab dort zweek- m/~l~igerweise andere Photoempfiinger eingesetzt werden sollten.
Die Umwandlung der Photonen in Elektronen ist ein statistischer ProzeB, der einer Poisson-Statistik folgt. Durch ihn wird der Signalstrom I a des Photo- multipliers mit einer statistischen Komponente ver- sehen, die als Rauschantefl bezeiehnet wird. Wie aus der Poisson-Statistik folgt, ist der Signalrauseh- strom Isr
= g l /2e Is, d 2" (1)
nnd damit das Signat-Rauschstromverh/fltnis
Is~ = V I a I,--~ _ _ 2 e AF g2 (2)
A 2' ist die Bandbreite des zu fibertragenden Signals, Isr
g i s t die Verst/~rkung. Man sieht, dab ~ klein wird,
wenn I a groB und zJ 2" klein werden. Man sollte also
18 z. Anal. Chem., Bd. 261
die Bandbreite bei schnellen Reaktionen so klein wie mSglich w/~hlen.
Au te r diesem Signalstromrausehen gibt es noch ein Dunkelstromrauschen. Dies hat mehrfaehe Ur- sachem Die zwei wichtigsten sind:
1. Das thermisehe Freisetzen yon Elektronen.
Dieser Rauschanteil h~ng~ yon der Temperatur und yon der geometrisehen Ausdehnung der Sehieht ab. Er kann durch Verkleinerung der Sehieht und dutch Abkiihlen des Photomultipliers erheblieh reduziert werden. Die notwendige Kfihltemperatur h~ngt yon der Wellenl~nge des Signalliehtes ab. Im siehtbaren Bereieh geniigt eine Abkiihlung auf ca. --20~ um das Dunkelstromrausehen bei Zimmertempera- ~ttr auf 20/0 zu verkleinern [4].
2. Das Freisetzen yon parasit/@en Elektronen aus dem Glaskolben oder den Dynoden.
Dieser Rausehantefl l~Bt sieh dureh einen sorgf~ltigen Aufbau des Photomultipliersystems und dutch Anlegen einer Suppressionsspannung an den Kolben des Photomultipliers verkleinern. Aullerdem daft die Betriebsspannung des Photo- multipliers nieht zu hoch gew~hlt werden.
Da sowohl das Signalrausehen als aueh die 2 wichtigsten Teile des Dunkelstromrausehens stati- stisehe Prozesse sind [6], kann durch geeignete Mittel- wertbfldung eine erhebliche Verbesserung des Signal] Rauschverh/iltnisses erreicht werden. Mittelwert- bfldung kann auch durch h/iufige Wiederholung der Reaktion vorgenommen werden.
2. Photometrisehe Analyse yon Mehrkomponentensystemen in homogenen L~sungen (multiplikative Farbmischung)
Bei der photometrischen Messung wird im einfachsten Falle das Verh~ltnis des einfallenden Liehtes L o zum ausfallenden Lieht L 1 gemessen:
L1 L~ = LoTs; Lo = T1. (3)
T 1 wird als Transmission bezeiehnet. Wird eine zweite Kfivette hinf~r die erste gestellt, so ist das ausfallende Lieht L z.
L2 = L1T2 = LoT1T~ = LoTses. (4)
Die Gesamttransmission Tges ist also gleich dem Produkt der Einzeltransmissionen.
Diese Art der Komponentenmisehung wird als multiplikative Mischung bezeiehnet. Aus den photo- metriseh gemessenen Transmissionswerten lassen sich die Konzentrationen der Komponenten nach dem Bouguer-Lambert-Beersehen Gesetz berechnen.
Es ist genau genommen ein Grenzgesetz, das nut fiir sehr verdiinnte LSsungen gilt, in denen der EinfluB yon mole- kularen Wechselwirkungen und von Breehungsindexunter- schieden vernachliissigt werden kann. Es ist 72 = 10-E1, (5)

274 Z. Anal. Chem., Band 261, Heft 4/5 (1972)
wobei E als Extinktion bezeichnet wird.
E1 = ~(~)z~d. (6)
e (2) ist der molare Extinktionskoeffizient bei der Wellenl~nge 2, x die Konzentration der Substanz und d die Sehichtdieke der Kfivette. Nach G1. (4) ist
L~ = Lo 10-~ ~0-~, = L~ 1O-W, + ~,). (7)
Die Gesamtextinktion ist also bei multiplikativer Misehung gleich der S u m m e der Einzelextinktionen.
Zeiehne$ man bei der Messung einer Substanz ihren molaren Exbinktionskoeffizienten fiber der Wellenl~nge auf, so erh~lt man das molare Extinktionsspektrum der Sub- stanz n: ~(~).
Ein Mehrkomponentensystem y (~) l~l]~ sich daher folgen- dermal]en beschreiben:
y(~) = ~ ( ~ ) + ~ ( ~ ) + .. . . + x.~.(~) . (8)
Steigt die Zahl der Komponenten, so mull die Zahl der unabh~ngigen Gleichungen mindestens der Zahl der Kom- ponenten entsprechen.
Um die MeBgenauigkeit zu erh6hen, ist es bei Mehr- komponentensystemen zweckm~i~ig, die Zahl der geInessenen Wellenl~ngen zu erh6hen, um alle zur Verffigung stehenden Informationen zu erfassen. In der Literatur sind eine Reihe yon entsprechenden Auswerteverfahren bekannt [2,3,9,26, 29--33,38].
Bei unseren Untersuchungen fiber Mehrkomponenten- systeme, die wir gemeinsam mit I~iesel [14,15] durchgeffihrt haben, sind wit yon einem Ansatz ausgegangen, der eine kontinuierliche on-line-Analyse erm6glicht. Versuchen wir aus den Spektren der Komponenten das Summenspektrum zu rekonstruieren, so wird das nich~ ganz gelingen: Es bleibt ein bestimmter Fehler ~ fibrig.
y(2)-- ~ x~@v(2)=y(A). (9)
Um eine mSglichst gute l~bereinstimmung zwischen dem gemessenen und dem zusammengesetzten Spektrum zu er- halten, fordern wir nach GauB, dab die Konzentration der Komponenten so gew~hlt werden soll, dab das Quadrat des l~ehlers (~)~ ein Minimum wird. l~fihrt man diesen Ansatz dutch, so ergibt sich ffir die gesuchten Konzentrationen xv
x~ = ~ y(~)g~(~)d~,
x~ = f y(2)g~().)d,~, (10)
x ~ = f y(A)gv(,~)d2. &
g~(Z) sind charakteristische Funktionen, die sich aus den molaren Extlnktionsspektren der Komponenten in dem ge- wiinschten Intervall ffir jede Komponente berechnen lassen [18].
Das Resultat (10) zeigt, dab es relativ einfach mSglich ist, eine on-line-Analyse yon Mehrkompo- nentensystemen durchzUffihren: Es muB nur eine Multiplikation zwischen der gemessenen und den
eharakteristischen Funktionen sowie eine Integration fiber das Produkt kontinuierlieh durehgeffihrt werden. Fiir das einfaehe System oxigeniertes-desoxige- niertes H~moglobin konnten wir mit Niesel diesen Ansatz mit einem Analogreehner sehon vor l~ngerer Zeit verifizieren.
Alle bisher genannten Veffahren der Mehrkompo- nentenanalyse haben 2 sehwerwiegende Naehtefle:
1. Es wird bei ihnen nieht berfieksiehtigt, dab der Informationsgehalt der einzelnen Komponenten- spektren bei den versehiedenen Wellenl~ngen sehr untersehiedlieh ist. Man kann z.B. damit reehnen, dab der Informationsgehalt im Bereieh einer eharak- teristisehen Absorptionsbande groB, in der Naehbar- sehaft der Bande in der Regel kleinist. Dieser Naehteil kann beseitigt werden, wenn man, wie wir gemeinsam mit Wodiek [18] zeigen konnten, eine Gewichts/unk- tion einfiihrt, die dem unterschiedliehen Informations- gehalt Reehnung tr~gt. Wodiek [35] konnte nach- weisen, dab es ffir ein bestimmtes Mehrkomponenten- system eine optimale Gewiehtsfunktion gibt. Dureh die Einbeziehung einer solehen Gewiehtsfunktion l~Bt sieh die Analysengenauigkeit der Mehrkomponenten- analyse erheblieh steigern. Werden die Gewichtsfunk- tionen als ~ ( 2 ) bezeiehnet, so wird aus der urspriinglichen Formel (10)
2~ ~z x, = f y(~)g,(~)a,(~)d2) = f y (~ )GJ~)d2 . (11)
ha ~1
2. Der zweite Nachteil der bisherigen Verfahren ist, dab sie voraussetzen, dab alle Komponenten und ihre Spektren bekannt sind. Ist dies nieht der Fall, so wird das unbekannte Spektrum -- im folgenden als StSrspektrum s bezeichnet -- so auf die bekannten Spektren verteilt, dab die Minimalbedingung erffillt ist. Dadurch kann die Mehrkomponentenanalyse so erheblieh gestSrt werden, dab sie nieht mehr an- zuwenden ist. Eine gewisse Kontrolle ist dadureh mSglieh, dab man gemessenes und berechnetes Spek- t rum gemeinsam aufzeiehnet und sieht, ob sie ge- ntigend gut zur Deekung kommen. Man kann aueh die Gr6Be der Restfl~che erreehnen und versuehen, bestimmte Kriterien zu entwiekein, die angeben, unter welchen Bedingungen eine Ubereinstimmung der Spektren anerkarm~ werden soll. Diese Verfahren mildern, aber beseitigen den prinzipiellen Fehler nicht. Sieht man sieh das Gleichungssystem mit einer StSrkomponente s an,
xl ~1 (21) + x2 ~2 (21) + sl = E (2~)

D.W. Liibbers and R. Wodiek: Schnelle Photometrie komplizierter bioehemiseher Mehrkomponentensysteme 275
so erkennt man, dab jede neue Messung eine neue StSrkomponente enthiilt: Daher kann ein solches Gleiehungssystem prinzipiell nicht gelSst werden, wenn nichts fiber Sx, s2, s s . . . sn bekannt ist. Wit konnten korzlich zeigen [21], dal~ man hier weiter- kommen kann, wenn die Annahme gemacht wird, da$ sich das Spektrum der StSrkomponente als Polynom darstellen l~$t. W~hlt man die Spektral- bereiche nieht zu groG, so genfigt h/~ufig schon ein Polynom 2.Grades zur Beschreibung der StSrfunk- tion.
s (4) = do A- dx2 -f- d~2 ~. (13)
Praktisch wird damit arts einem Zweikomponenten- system ein Ffinfkomponentensystem.
y (4) = x1~91 (~) -~- x2~o 2 (4) -~- d o ~- dl~ -~ d ~ ~. (14)
Durch geschiekte Wahl der Auswertungsbereiche kSnnen mi t diesem Verfahren erstmals such Ge- mische untersucht werden, deren Zusammensetzung nur teilweise bekarmt ist. Alle unbekannten Kom- ponenten werden dabei gemeinsam ermittelt. Eine weitere Differenzierung der , ,StSrfunktion" ohne zu- s/~tzliche Information ist allerdings nicht mehr mSglieh.
Bevor eine Mehrkomponentcnanalyse durchgeffihrt wird, mug man immer prfifen, ob sich das in ~rage stehende System ffir eine solche Analyse eignet. Dies 1/~Bt sieh entweder theoretisch oder expcrimentell da- durch abkl/~ren, dab man die Komponenten mit dem for die Analyse vorgesehenen Ger/it durchmiBt und ermittelt, ob die gewfinschten Konzentrationsunter- schiede erfaBt werden kSnnen.
Allgemein kann gesagt werden, dab die Analysen- genauigkeit und die Eindeutigkeit mi t Zunahme der Komponentenzahl deutlich abnimmt.
Die statistischen Fehler, die bei der Messung auf- treten, kSnnen durch die Zahl der MeBpunkte pro Spektrum und durch Methoden der Mittelwerts- bildung bek/impft werden. In beiden F/illen ist die Verkleinerung des statistischen Fehlers etwa pro- portional der Quadratwurzel aus der Zahl der Mes- sungen, d.h. bei 1000 1Vfessungen ca. 30lath. Bei etwa gleichem statistischem Fehler im gesamten Megbereich bringt die Einfiihrung der Gewichtsfunk- tion eine deutliehe Verbesserung der Genauigkeit. Sie ist besonders wirksam bei grol3en statistischen Fehlern.
Gef/ihrlich ffir die Mehrkomponentenanalyse kSn- nen systematische Fehler werden: Ein systematischer Fehler der Wellenl/ingenwerte kann die Analyse er- heblich stSren. Werden jedoch alle Spektren mlt dem gleiehen Photometer aufgenommen, so kommt es nur
18"
auf die Reproduzierbarkeit und nicht auf die Ab- solutgenauigkeit der Wellenl/ingenwerte an. Die Ab- solutwerte sind erst dann yon Bedeutung, wenn man mit Spektren arbeitet, die aus der Literatur ent- nommen werden. I s t die Kennlinie des Logarithmier- ghedes keine Gerade, so k6nnen aueh dadurch er- hebliche systema~isehe Fehler entstehen. Man mu$ daher in diesem Falle die Mehrkomponentenanalyse mit entsprechend korrigierten Spektren vornehmen.
Die beschriebene Mehrkomponentenanalyse 1/~$t sich unserer Erfahrung nach nur rationell durch- ffihren, wenn die MeBwerte durch eine Datenerfas- sungsanlage automatisch aufgezeichnet werden. Da- bei rout eine eindeutige Zuordnung der Mel~werte ge- w/~hrleistet sein. Wir haben mit einer Datenerfas- sungsanlage gearbeitet, die entsprechend unseren Anforderungen yon der 1%. Carl Zeiss, 0berkochen, zu dem Photometer DRM 21 angefertigt wurde 1. Die Reproduzierbarkeit der Wellenl/~ngenwerte betrug im Bereich yon 215--340 nm =k 0,5/~. Bis zur Extink- tion yon ca. 1,0 OE schwankte die Extinkt ion nur im Bereieh yon 10-40E. Die Linearit/~t ist deutlieh schlechter. Wenn nStig, haben wir sie auf Grund einer Eichkurve korrigiert.
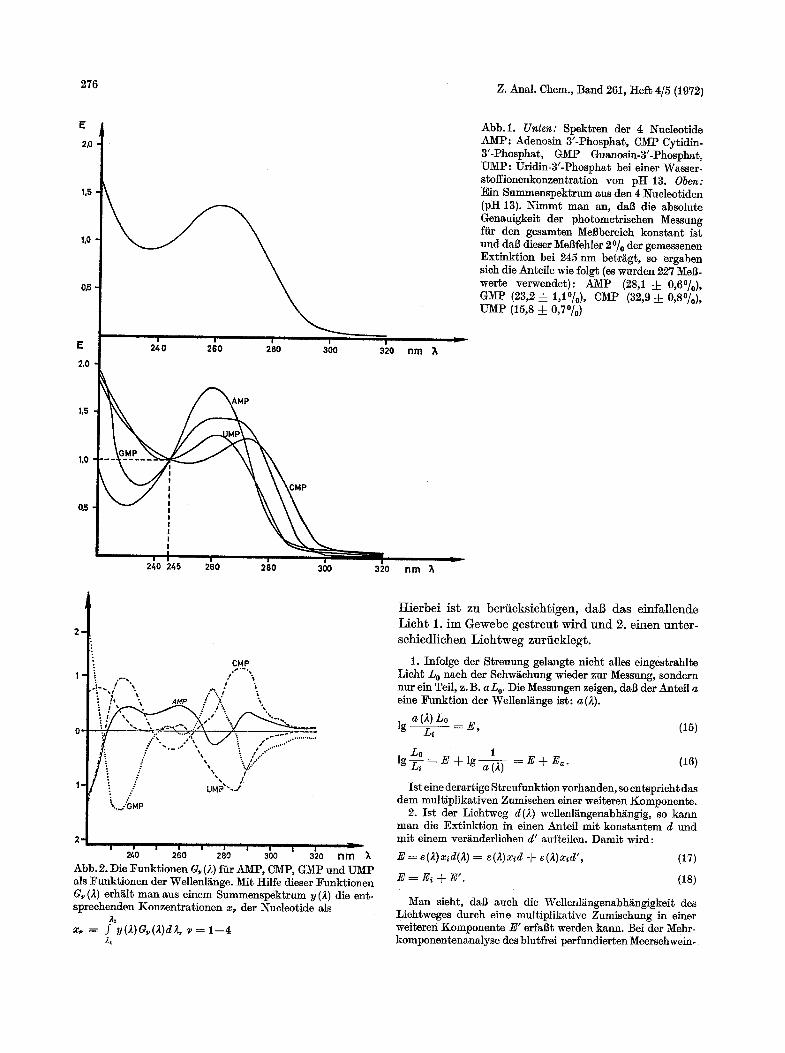
Dieses Analysenverfahren wurde in Modellunter- suchungen gemeinsam mit Schneider an Nucleotid- mischungen getestet. Wir haben Nucleotidmischungen gew/ihlt, da die Nucleotide ein relativ/~hnliches Spek- t rum (Abb. 1) ohne charakteristische Banden haben.
Alle Messungen wurden bei pH 13 durchgefiihrt. Ffir die Analyse wurden zun/~ehst LSsungen der einzelnen Kom- ponenten mit einem maximalen Extinktionswert yon 0,80 in einer 0,1 M KOH bei einer Schiehtdicke yon 1 cm her- gestellt. Diese LSsungen dienten einmal zur Aufnahme der Grundspektren und zweitens zur Herstellung der zu analy- sierenden Misehungen. Das Photometer stand in einem auf 22~ temperierten Raum. Gemessen und auf Loehstreffen gestanzt wurde der Wellenli~ngenbereieh yon 340--215 rim. Das entsprieht bei dem verwendeten Gerat etwa 1300 ge- stanzten B15eken, zusammengesetzt aus 2- und Extinktions- wert. 1~fir die Messung eines Spek~rums werden ca. 7 rain benStigt. Abb.2 zeigt die Gewiehtsfunktionen ffir ein Ge- miseh, wie es in Abb. 1 dargestellt ist.
Als Beispiel fiir unsere Resultate sei eine Seehskompo- nentenanalyse der Monophosphate yon Adenosin, Cytidin, Guanosin, Thymidin, Uridin und aul~erdem Xanthin auf- gefiihr~. Bei 2 Komponenten versehiedenster Kombinationen war der Fehler • 0,36% , bei 3 Komponenten • 0,76%, bei 4Komponenten i l,75~ Die fehlenden Komponenten konnten in tier iiberwiegenden Zahl der Fi~lle als eindeu~ig fehlend identifiziert werden.
Wir haben alas Verfahren auBerdem anf Reflexions- spektren vom Meerschweinchengehirn angewandt.
1 Herrn Franke aus der Fa. Zeiss sei ffir seine Hilfe be- sonders gedankt.
E
2,0
1,5
1,0
0,5
E
2,o
i i i i 240 260 280 300
1,5
1,0
0.5
276 Z. Anal. Chem., Band 261, Heft 4/5 (1972)
=;,o ~ =~o ~o ~o
t ! C M P
~- / ", !
ol i " i".. "~ / / - -,. \ . ... / ".. o . .
:" ~ ~ /,.-': / L. \$"
: , /
1- / UHP " - J
~-.../GHP
2-
240 260 2 0 300 3 0 nm k Abb. 2. Die Funktionen G~ (4) ffir AMP, CMP, GMP und UMP als Funk~ionen der WeIlenl~nge. Mit Hilfe dieser Funktionen G~ (2) erh~ilt man aus einem Summenspektrum y (2) die ent- spreehenden Konzentrationen x~ der Nueleotide als
22
x~ = f y(2)G~(2)d4, v = 1--4
i 320
3t, o
nm
Abb.1. Unten: Spektren der 4 Nueleotide AMP: Adenosin 3'-Phospha$, CMP Cytidin- 3'-Phosphat, G/~IP Guanosin-3'-Phospha~, UMP: Uridin-3'-Phosphat bei einer Wasser- stoffionenkonzentration yon pH 13. Oben: Ein Summenspek~rum aus den 4 Nueleotiden (pH 13). Nimmt man an, dab die absolute Genauigkei~ der photemetrisehen Messung fiir den gesamten MeBbereieh konstant ist and dab dieser Mel3fehler 2% tier gemessenen Extink~ion bei 245 nm be~r~gt, so ergaben sieh die Anteile wie folgt (es wurden 227 MeB- werte verwendet): AMP (28,1 :{: 0,60/0), GMP (23,2 -~ 1,1% ), CMP (32,9 __ 0,8%), T2~P (15,8 • 0,7%)
D-
n m
Hierbei is t zu beriieksichtigen, dab das einfallende Licht 1. im Gewebe gestreut wird u n d 2. e inen unter- schiedlichen Lichtweg zurticklegt.
1. Infolge der Streuung gelangte nieht alles eingestrahlte Lich~ L o nach der Schw~ehung wieder zur Messung, sondern nur ein Tell, z. B. a L o. Die l~Iessungen zeigen, dai] der Anteil a eine Funktion der Wellenl~nge ist: a(4).
a (2) Lo lg L~ = E, (15)
Lo 1 l g ~ - - - - E + I g a(,~) - - E - ~ E ~ " (16)
Ist eine derartige Streufunktion vorhanden, so entsprieh~ das dem multiplikativen Zumischen einer weiteren Komponente.
2. Ist der Liehtweg d(2) wellenl~ngenabh~ngig, so kann man die Extinktion in einen Anteil mi~ konstantem d und mi~ einem ver~nderliehen d' aufteilen. Damit wird:
E = s(~)x~d(~) = e().)x~d .-{- e() .)x~d' , (17)
E = E~ + E ' . (18)
Man sieht, dab aueh die Wellenl~ngenabh~ngigkei~ des Liehtweges dutch eine multiplikative Zumisehung in einer weiteren Komponente E" erfaflt werden kann. Bei der Mehr- komponentenanalyse des blu~frei perfundierten Meerschwein.
D.W. Liibbers und R. Wodick: Selmelle Photometrie komplizierter bioehemischer Mehrkomponentensysteme 277
,I . . . . . ~ " ' '~C% ~ +
_
\ , , _ 6 t O - -
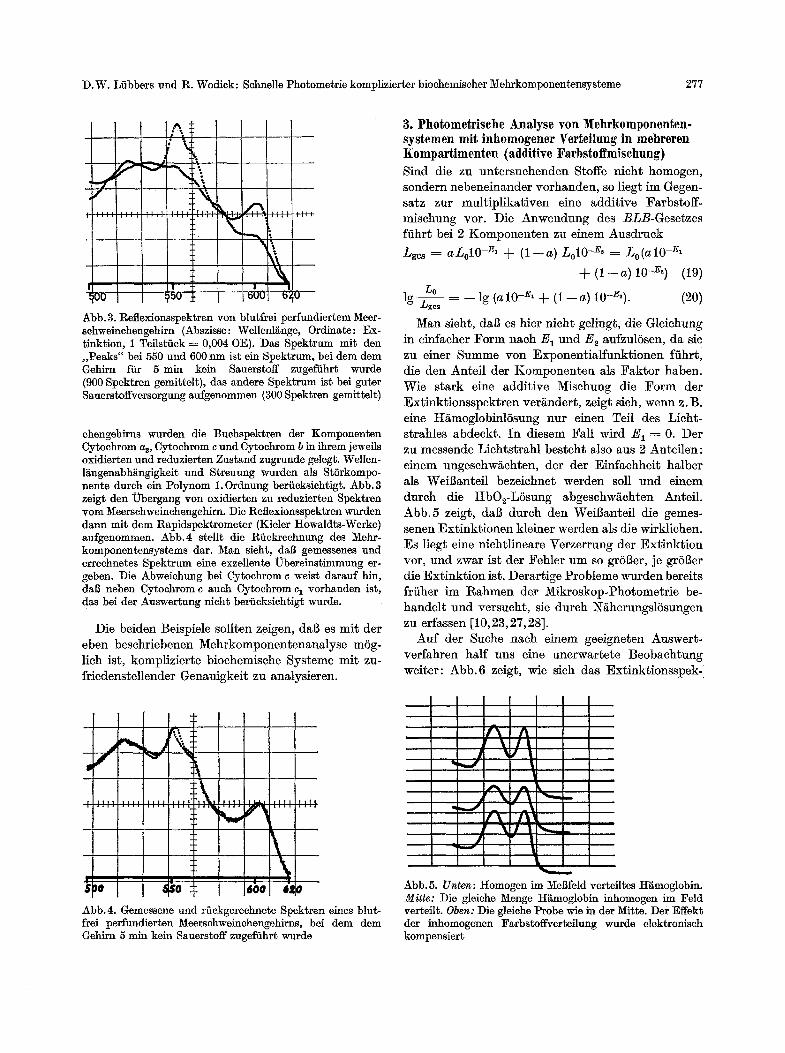
Abb. 3. Reflexionsspektren yon blutfrei 1)erfundiertem Meer- sehweinchengehirn (Abszisse: Wellenl~nge, Ordinate: Ex- tinktion, 1 Teilstiiek = 0,004 OE). Das Spektrum mit den ,,Peaks" bei 550 und 600 nm ist ein Spektmm, bei dem dem Gehim ffir 5 rain kein Sauerstoff zugefiihr~ wurde (900 Spektren gemittelt), das andere Spektrum is~ bei guter Sauerstoffversorgung aufgenommen (300 Spek~ren gemittelt)
ehengehirns wurden die Buehspektren der Komponenten Cytochrom a~, Cytoehrom c und Cy~chrom b in ihrem jeweils oxidierten und reduzierten Zustand zugrunde geIegk Wellen- l~ngenabh~ngigkeit und Streuung wurden als StSrkompo- nente durch ein Polynom 1. Ordnung beriicksichtigt. Abb. 3 zeigt den Ubergang yon oxidierten zu reduzierten Spektren vom Nfeerschweinchengehim. Die Reflexionsspektren wurden dann mit dem Rapidspek~rometer (Kieler Howaldts-Werke) aufgenommen. Abb.4 stellt die Rfickreelmtmg des Mehr- komponentensystems dar. Man sieht, dal~ gemessenes und errechnetes Spektrum eine exzellente ~bereins~immung er- geben. Die Abweichung bei Cytoehrom r weist darauf hin, dul~ neben Cy~ehrom c aueh Cytoehrom c~ vorhanden ist, das bei der Auswer~ung nicht beriicksiehtigt wurde.
Die beiden Beispiele sollten zeigen, da~ es mit der eben beschriebenen Mehrkomponentenanalyse m6g- lich ist, komplizierte biochemische Systeme mit zu- friedenstellender Genauigkeit zu analysieren.
3. Photometrische Analyse yon Mehrkomponenten- systemen mit inhomogener Verteilung in mehreren Kompartimenten (additive Farbstoffmischung) Sind die zu untersuehenden Stoffe nicht h0mogen, sondern nebeneinander vorhanden, so liegt im Gegen- satz zur multiplika~iven eine additive Farbstoff- mischung vor. Die Anwendung des BLB-Gesetzes
ffihrt bei 2 Komponenten zu einem Ausdruck
Lges ~ aL010 -E' -~ ( l - - a ) L010 -E~ ~-- L0(al0 -El
~- ( l - - a ) 10 -~) (19)
L0 lg-Lgr ---- -- lg (a 10 -~' ~- (1 - -a ) 10-E,). (20)
Man sieht, dab es hier nich~ gelingt, die Gleichung in einfacher Form nach E 1 und E 2 aufzulSsen, da sie zu einer Summe yon Exponentialfunktionen ffihrt, die den Anteil der Komponenten als Faktor haben. Wie stark eine additive Mischung die Form der Ex~inktionsspektren ver~ndert, zeigt sieh, werm z.B. eine tt~moglobialSsung nur einen Teil des Licht- strahles abdeekt. In diesem Fall wird E 1 ----- 0. Der zu messende Lichtstrahl besteht also aus 2 Anteilen: einem ungeschw~chten, der der Einfaehheit halber als Weil~anteil bezeichnet werden soll und einem durch die tIbO2-LSsung abgeschw~chten Anteil. Abb.5 zeigt, dal~ dutch den WeiB~ntefl die gemes- senen Extinktionen kleiner werden als die wirk]ichen. Es liegt eine nichtlineare Verzerrung der Extinktion vor, und zwar ist der Fehler um so grSi~er, je grSl~er die Extinktion ist. Derartige Probleme wurden bereits friiher im Rahmen der Mikroskop-Photometrie be- handelt und versucht, sie durch N~herungslSsungen zu erfassen [10, 23,27, 28].
Auf der Suche nach einem geeigneten Auswert- verfahren half uns eine unerw~rtete Beobachtung welter: Abb.6 zeigt, wie sich das Extinktionsspek- I
J f ~
,1~ t t , l , | , s . J
E 0
Abb.4. Gemessene und rSekgerechnete Spek~ren eines blut- frei perfundierten Meersehweinehengehirns, bei dem dem Gehirn 5 rain kein Sauerstoff zugeffihr~ wurde
A , A '-'/ 'Jl
Ok k
k l t I ~ . . _ - |
Abb. 5. Unten" Homogen im l~Iegfeld vert~ilt;es H~moglobim Mitre: Die gleiehe Menge Hgmoglobin inhomogen im Feld verteilt. Oben: Die gleiehe Probe wie in der Mitte. Der Effek• der inhomogenen Farbst~ffverteilung wurde elektroniseh kompensiert
278 Z. Anal. Chem., Band 261, Heft 4/5 (1972)
Extinktion
2~ ~ r u n g s g r a d o , , , = , o , i
40 60 80 lO0 %
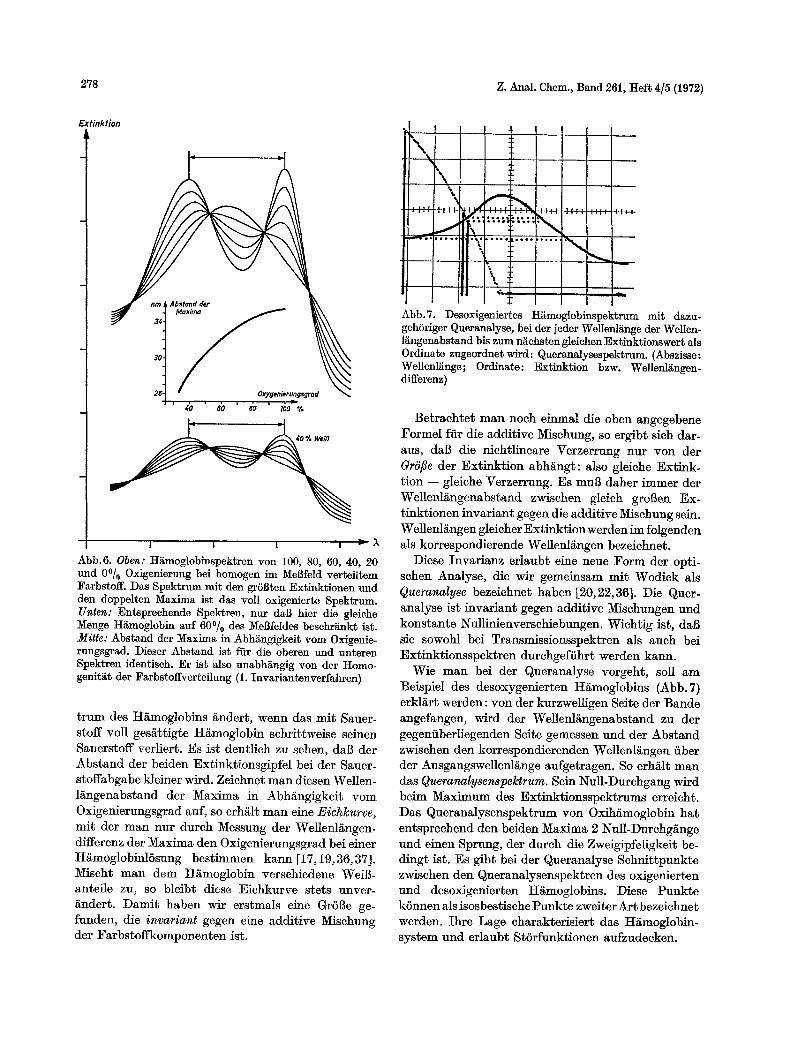
Abb.6. Obey: H~Lmoglob]~spek~ren yon 100, 80, 60, 40, 20 und 0~ Oxigenierung bei homogen im Mel]feld vert~iltem Farbstoff. Das Spektrum mit den grSl]ten Extinktionen und den doppelten Maxima ist das roll oxigenierte Spektrum. Unten: Entspreehende Spektren, nur da] hier die gleiche Menge H~moglobin auf 60~ des Me]feldes besehr~nkt ist. Mitte: Abstand der Maxima in Abh~ngigkeit yore Oxigenie- rungsgrad. Dieser Abstand ist fiir die oberen und unteren Spektren identiseh. Er is~ also unabh~ngig yon der Homo- genit~t der Farbstoffver~eilung (1. Invari~ntenverfahren)
t rum des H/imoglobins ander~, wenn das mit Sauer- stoff voll ges/~ttigte H/~moglobin sehrittweise seinen Sauerstoff verliert. Es ist deutlieh zu sehen, da[3 der Abstand der beiden Extinktionsgipfel bei der Sauer- stoffabgabe kleiner wird. Zeiehnet man diesen Wellen- langenabstand der Maxima in Abh~ngigkeit vom Oxigenierungsgrad auf, so erh~it man eine Eiehkurve, mit der man nur dureh Messung der Wellenl/~ngen- differenz der Maxima den Oxigenierungsgrad bei einer H~moglobinlSsung bestimmen kann [17,19, 36,37]. Mischt man dem 17I~moglobin verschiedene Weil~- antefle zu, so bleib~ diese Eiehkurve stets unver- ~ndert. Damit haben wir erstmals eine Gr61~e ge- funden, die invariant gegen eine additive Misehung der Farbstoffkomponenten ist.
\ \
\ , i , t
i o ~ ~ m g t ,
e a P l l I o o e ~ o t o * o l ~
\ II! =:
| l l
Abb.7. Desoxigeniertes H~moglobinspektrum mi~ dazuo gehSriger Queranalyse, bei der jeder Wellenl~nge der Wellen- l~ngenabstand bis zum n~ehsten gleiehen Extinktionswert als Ordinate zugeordnet wird: Queranalysespektrum. (Abszisse: Wellenl~nge; Ordinate: Extinktion bzw. Wellenl~ngen- differenz)
Betraehtet man noeh einmal die oben angegebene Formel fiir die additive Misehung, so ergibt sieh dar- aus, dal3 die niehtlineare Verzerrung nur yon der Gr6[3e der Extinktion abh~ngt: also gleiehe Extink- tion -- gleiehe Verzerrung. Es mul3 daher immer der Wellenlangenabstand zwischen gleieh gro•en Ex- tinktionen invariant gegen die additive Misehung sein. Wellenlangen gleieher Extinktion werden im folgenden als korrespondierende Wellenls bezeiehnet.
Diese Invarianz erlaubt ehle neue Form der opti- sehen Analyse, die wir gemeinsam mit Wodiek als Queranalyse bezeiehnet haben [20, 22,36]. Die Quer- analyse ist invariant gegen additive Misehungen und konstante Nullinienversehiebungen. Wiehtig ist, da~ sie sowohl bei Transmissionsspektren als aueh bei Extinktionsspektren durehgefiihrt werden kann.
Wie man bei der Queranalyse vorgeht, soll am Beispiel des desoxygenierten HamogIobins (Abb.7) erkl~rt werden: yon der kurzwelligen Seite der Bande angefangen, wird der Wellenlangenabstand zu der gegenfiberliegenden Seite gemessen und der Abstand zwisehen den korrespondierenden Wellenl/~ngen fiber der Ausgangswellenlange aufgetragen. So erh/~lt man das Queranalysenspektrum. Sein Null-Durehgang wird beim Maximum des Extinktionsspektrums erreieht. Das Queranalysenspektrum yon Oxih/~moglobin hat entspreehend den beiden Maxima 2 Null-Durehg/~nge und einen Sprung, der dureh die Zweigipfeligkeit be- dingt ist. Es gibt bei der Queranalyse Sehnittpunkte zwisehen den Queranalysenspektren des oxigenierten und desoxigenierten H~moglobins. Diese Punkte kSnnen als isosbestische Punkte zweiter Art bezeiehnet werden. Ihre Lage eharakterisiert das H~moglobin- system und erlaubt StSrfunktionen aufzudeeken.
D.W. Liibbers und R. Wodick: Schnelle Photometrie komplizierter biochemischer Mehrkomponentensysteme 279
t (E fa),g)
0 0,2 0,4 0,6 0o$ 1,00xygenterungsgrod g
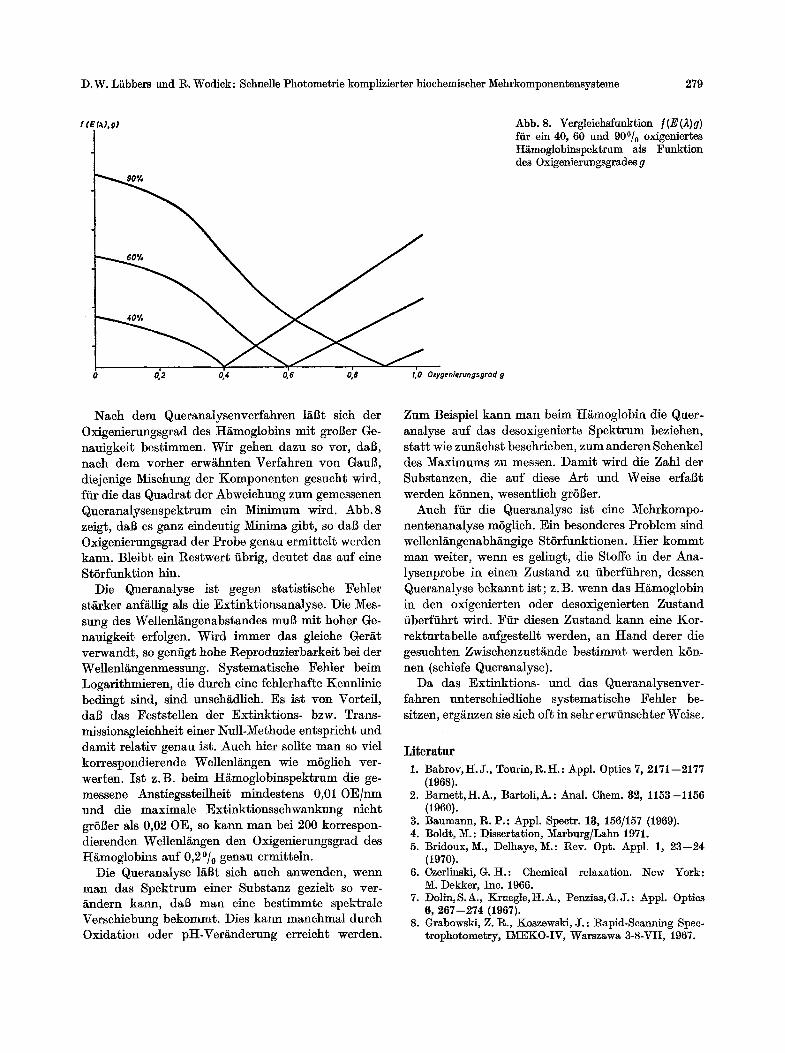
Abb. 8. Vergleichsfunktion t(E(,~)g) fiir ein 40, 60 und 90~ oxigeniert~s H~moglobinspektrum als Funktion des Oxigenierungsgrades g
Nach dem Queranalysenverfahren I/~l~t sich der Oxigenierungsgrad des H/imoglobins mit groBer Ge- nauigkeit bestimmen. Wir gehen dazu so vor, daB, naeh dem vorher erw~hnten Verfahren yon Gau$, diejenige Misehung der Komponenten gesucht wird, ftir die das Quadrat der Abweiehung zum gemessenen Queranalysenspektrum ein Minimum wird. Abb.8 zeig$, da$ es ganz eindeutig Minima gibt, so da$ tier Oxigenierungsgrad der Probe genau ermittelt werden kann. Bleibt ein Restwert iibrig, deutet das auf eine StSrfunktion bin.
Die Queranalyse ist gegen statistisehe Fehler st/~rker anfs als die Extinktionsanalyse. Die Mes- sung des Wellenl/ingenabstandes mug mit hoher Ge- nauigkeit erfolgen. Wird immer das gleiehe Ger/tt verwandt, so gentigt hohe Reproduzierbarkeit bei der Wellenl/~ngenmessung. Systematische Fehler beim Logarithmieren, die durch eine fehlerhafte Kennlinie bedingt sind, sind unseh/~diieh. Es ist yon Vortefl, dab alas Feststellen tier Extinktions- bzw. Trans- missionsgleiehheit einer Null-Methode entsprieht und damit relativ genau ist. Aueh hier sollte man so viel korrespondierende Wellenl/~ngen wie m6glieh ver- werten. Ist z.B. beim H/~moglobinspektrum die ge- messene Anstiegssteilheit mindestens 0,01 0E/rim und die maximale Extinktionssehwankung nicht gr6$er als 0,02 OE, so kann man bei 200 korrespon- dierenden Wellenl/ingen den Oxigenierungsgrad des H/~moglobins auf 0,2 ~ genau ermitteln.
Die Queranalyse lil~t sieh aueh anwenden, wenn man das Spektrum einer Substanz gezielt so ver- /indern kann, dal~ man eine bestimmte spektrale Versehiebung bekommt. Dies kann manehmal (lurch Oxidation oder pH-Ver/inderung erreieht werden.
Zum Beispiel kann man beim H/~moglobin die Quer- analyse auf das desoxigenierte Spektrum beziehen, start wie zun/~ehst beschrieben, zum anderen Schenkel des Maximums zu messen. Damit wird die Zahl der Substanzen, die auf diese Art und Weise erfaflt werden kSnnen, wesentlich grSBer.
Aueh fiir die Queranalyse ist eine Mehrkompo- nentenanalyse mSglieh. Ein besonderes Problem sind wellenl/ingenabh/~ngige StSrfunktionen. Hier kommt man weiter, wenn es gelingt, die Stoffe in der Ana- lysenprobe in einen Zustand zu iiberffihren, dessen Queranalyse bekannt ist; z.B. wenn das H/~moglobin in den oxigenierten oder desoxigenierten Zustand fiberfiihr~ wird. Ffir diesen Zustand kann eine Kor- rekturtabelle aufgestellt werden, an Hand derer die gesuehten Zwisehenzust/~nde bestimmt werden kSn- nen (sehiefe Queranalyse).
Da das Extinktions- und das Queranalysenver- fahren untersehiedliche systematisehe Fehler be- sitzen, erg/~nzen sie sieh oft in sehr erwfinsehter Weise.
Literatur I. Babrov, H.J., Tourin, R. H. : Appl. Optics 7, 2171 --2177
(1968). 2. Barnett, H.A., Bartoli, A.: Anal. Chem. 32, 1153--1156
(1960). 3. Baumann, R. P.: Appl. Speetr. 18, 156/157 (1969). 4. Boldt, M.: Dissertation, Marburg/Lahn 1971. 5. Bridoux, M., Delhaye, M.: Rev. Opt. Appl. 1, 23--24
(1970). 6. Czerlinski, G.H.: Chemical relaxation. New York:
M. Dekker, Inc. 1966. 7. Dolin, S.A., Krueglv, H.A., Penzias, G.J.: Appl. Optics
6, 267--274 (1967). 8. Grabowski, Z. R., Koszewski, J. : Rapid-Scanning Spcc-
trophotometry, L~EKO-IV, Warszawa 3-8-VII, 1967.

280 Z. Anal. Chem., Band 261, Heft 4/5 (1972)
9. Guschlbauer,W., Riehards, E.G., Beurling, K., Adams,A, Fresco, J. R.: Biochemistry 4, 964--975 (1965).
10. Ifutten, H.: Pfliigers Arch. 805, 177--189 (1969). 11. Kortiim, G.: Colorimetrie, Photometrie und Spektro-
metric, 4. Aufl. Berlin-GSttingen-ifeidelbcrg: Springer 1962.
12. Kortfim, G.: Reflexionsspektroskopie. Berlin-Heidel- berg-New York: Springer 1969.
13. Laqua, K., Hagenah, W. D.: Critical review of methods and results of time-resolved spectroscopy--Prec. X. Col- loquium Spectroscopicum Interna%ionale, p. 91. Edit. by E. R. Lippineott and )/f. Margoshcs. Washington: Spartan Books 1963.
14. Liibbers, D. W. : Ifabilitation, Kiel 1956. 15. Liibbers, D.W., Niesel, W.: Naturwissenschaften 44,
59/60 (1957). 16. Liibbers, D. W., Nicsel, W.: Pfliigers Arch. Ges. Physiol.
268, 286--295 (1959). 17. Lfibbers, D.W., Piroth, D., Wodick, R.: Naturwissen-
schaSten 57, 42 (1970). 18. Liibbers, D. W., Wodick, R. : Appl. Optics 8, 1055--1062
(1969). 19. Liibbei~, D.W., Wodick, I~.: Na%urwissensehaften 58,
321 (1971). 20. Liibbers, D. W., Wodick, R. : Umsehau 1971, 486--491. 21. Liibbers, D.W., Wodick, R.: NaturwissenschaSten (ira
Druck). 22. Liibbers, D. W., Wodiek, R. : Modern Teehnol. Physiol.
Sei. (im Druek). 23. Mendelsohn, M. L.: Absorption eythophotometry: Com-
parative methodology for heterogenous objects, and the two-wavelength method. In: Introduction to quantita-
rive cy~ochemistry. G. L. Wied, ed. New York-London: Academic Press 1966.
24. Moehizuki, M., Saito, Y., Akiyama, S.: Rapid scanning spectrophotometer, Digest of the 6th International Con- ference on Me & BE 1965, 622--623.
25. Niesel, W., Lfibbers, D. W., Schneewolf, D., Richter, J., Bottieher, W.: Rev. Sei. Instrum. 85, 578--581 (1964).
26. Opfell, J. B., Sage, B. If. : Ind. Engg. Chem. 50,803--806 (1958).
27. Ornstein, L. : Lab. Invest. 1, 251--265 (1952) 28. Pateau, K.: Chromosoma 5, 341 (1952). 29. Pratt, A.W., Toal, J.N., Rushizky, G.W., Sobcr, H.A.:
Biochemistry 8, 1831--1837 (1964). 30. Reid, J. C., Pratt, A.W.: Biophys. Res. Communica-
tions 8, 337--342 (1960). 31. Richards, E. G.: European J. Biochem. 4, 256--264 (1968). 32. Rogoff, M.: Ann. N. Y. Acad. Sci. 69, 27--37 (1957). 33. Stcrnberg, g.C., Stfllo, If. S., Schwendeman, R. If. : Anal.
Chem. 82, 84--90 (1960). 34. Thews, G., Liibbers, D. W. : Z. Angew. Phys. 7, 325--331
(1955). 35. Wodick, R. : Dissertation, Nfarburg/Lahn 1968. 36. Wodiek, R.: Dissertation, Marburg/Lahn 1971. 37. Wodick, R., Liibbers, D. W.: Pfliigers Arch. 819, R60
(1970). 38. Zsehcilc, F. P., Murray, If. C., Baker, G. A., Peddieord,
R. G.: Anal. Chem. 84, 1776--1780 (1962).
ProS. Dr. D. W. Lfibbers Max-Planck-Institut flit Arbeitsphysiologie D-4600 Dortmund, Rheinlanddamm 201 Bundesrepublik Deutschland
Z. Anal. Chem. 261,280--286 (1972) �9 by Springer-Verlag 1972
Ein Polarimeter zur Messung schneller chemischer Reaktionen*
Gernot Hanisch
Biozentrum der Universit~t Basel
Gundolf Beier
Institut fiir Reehtsmedizin der Universit~t Miinchen
Eingegangen am 16. Mai 1972
Polarimeter ]or the Measurement ol Fast Chemical Reactions. A polarimeter for the measurement of fast changes in optical rotat ion is described. The optical sys tem is of the double beam t y p e . Optical rota t ion is t ransformed into a signal, which is directly proport ional to ro ta t ion and is used for its compensation. The compensat ion method eliminates the influence of changes in optical densi ty which m a y parallel the change in optical rotat ion, The t ime constant of the ins t rument is about 20 ~s. The resolution is limited by the shot noise and amounts at this t ime constant to about =J= 15 millidegrees.
* Dicsc Arbeit wurde am Max-Planek-Institut Stir Eiweill-und Lederforsehung in Nfiinchen durehgeSiihrt.