Embed Size (px)
Citation preview

Z. Anal. Chem. 260, 177--184 (1972) 177 �9 by Springer-Verlag 1972
Spurenanreicherung durch partielles LSsen der Matrix* Ein schnelles und einfaches Verfahren zur Spurenanalyse y o n R e i n s t z i n k
E. JACKWEI~TH, E. DSmNG, J. LOHlVlAIr und G. SCH~u
Institut fiir Spektroehemie und Angewandte Spektroskopie, Dortmund
Eingegangen am 14. Januar 1972
Trace Enrichment by Partial Dissolution o/the Matrix. A Rapid and Simple Method/or Trace Analysis o/High. Purity Zinc. Trace elements such as Bi, Cd, Pb, Co, Ni, Sn, T1, In, Ag, Au, and Pd which are present as impuri- ties in high-purity zinc are almost quantitatively enriched, ff the zinc is coated with a thin layer of mercury before dissolution in hydrochloric acid up to a small residue. The "endpoint" of the dissolution can be indicated potentiometrically. The trace elements remain in the residue coated by the mercury and are effectively enriched. Samples of zinc of at least 100 g can be used without loss of the trace elements. The addition of 1--5~ of mercury based on the zinc is sufficient for the amalgamation, and has practically no effect on the rate of dissolution.
For analysis of the concentrated trace elements, mercury spheres tha t still contain some mg of Zn are dissolv- ed in nitric acid. After separation of the mercury, the concentrated elements are determined photometrically, polarographically and spectrochemically with AAS or DC-arc spectroscopy.
Zusammen/assung. Aus reinstem Zinkmetall wird eine Reihe yon Spurenelementen, wie Bi, Cu, Cd, Pb, Co, Ni, Sn, T1, In, Ag, Au und Pd praktisch quanti tat iv angereichcrt, wenn man das zur Analyse eingesetzte Metall mit einer dfinnen Schicht Quecksflber iiberzieht und anschliel]end bis auf cinen geringen Rest in Salzsaure 15st. Der , ,Endpunkt" des LSsevorgangs ist potentiometrisch indizierbar. Die Spurenelemente werden in dem yon Quecksilber eingehallten Metallrackstand festgehalten und darin angereichert. Zinkeinwaagen bis zu mindestens 100 g kSnnen ohne Verlust an Spurenelementen eingesetzt werden. Zum Verquicken des Zinks geniigt ein Quecksilberzusatz yon 1--5~ des eingewogenen Probematerials, wobei die LSsegeschwindigkeit durch den Zusatz nur wenig beeintrgchtigt wird.
Zur Analyse des Spurenkonzentrats werden die noch einige Milligramm Zn enthaltenden Quecksilberkugeln in Salpetersaure gelSst. Die angereicherten Elemente werden nach Abtrennung des Quecksilbers photometrisch oder polarographisch sowie spektroehemisch im Gleichstrombogen bzw. mit Hilfe der Atomabsorption be- stimmt.
1. Einleitung In einer kfirzlich erschienenen Arbeit haben wit fiber ein neues Verfahren zur Anreicherung yon Edel- metallspuren in Reinstqueeksflber berichtet [5]. Danach kSnnen in Quecksflbermetall als Spuren- verunreinigung enthaltene Metalle Gold und Palla- dium in einfaeher Weise angereiehert werden, indem man die Quecksilbereinwaage bis auf einen Rest yon wenigen Milligramm in Salpeters/~ure 15st. Der Rack- stand enth/flt das gesamte Gold und Palladium der Einwaage.
Im Zusammenhang damit entstand die Frage, ob diese unkompliziertc Art der Spurenanreicherung nieht auch far die Analyse anderer Reinstmetalle und
* tterrn Prof. Dr. H. Speeker zum 60. Geburtstag gewid- met.
12 Z. Anal. Chem., Bd. 260
zur quantitativen Abtrennung weiterer Spuren- elemente anwendbar ist. Nach einer alteren Arbeit von Seith u. Herrmann kann man den spektro- chemischcn Nachweis einer Reihe yon Elementen in Zink verbessern, wenn man die stabchenfSrmige Zink- elektrode vor dem Abfunken einige Zeit in verdfinnte Salzsaure taucht [12]. Dabei 15st sich ein Tell des Zinks auf, und auf der Oberflache der Elektrode bleiben die Metalle zurfick, die edler sind als Zink.
Unsere ersten Versuche, diese Beobachtung zu einem quantitativen chemischen Anreicherungs- verfahren fiir die Zinkanalyse auszubauen, scheiter- ten jedoch: Die Spurenelemente sammeln sich beim LSsen yon Zink in Salzsaure nur in Form eines locke- ren Belages an der Metalloberfliiche an. Dieser Spu- renbelag wird bei fortschreitender AuflSsung des Zinks

178 Z. Anal. Chem., Band 260, Heft 3 (1972)
durch die Einwirkung yon Wasserstoffbl/isehen abgehoben, verteilt sich dann in Form feinster Par- tikel und 15st sieh, besonders in Gegenwar~ yon Luftsauerstoff, in der Saute zum Tefl mit auf. Der Vorgang ist sehr stark yon Zufiillen abh/ingig und fiir eine quanti tat ive Analyse, zumindest in dieser ein- faehen Versuehsanordnung, nieht verwendbar.
Diese Sehwierigkeiten bei der Spurenanreieherung aus Reinstzink, die vor allem wegen des sehleeht anhaftenden Spurenbelages auf dem LSseriiekstand der Zinkeinwaage entstehen, lassen sieh nun einfaeh dadureh beheben, da$ man das Zink vor dem Auf- 16sen in Salzs/iure dureh Zusatz eines Queeksilber- trSpfchens mit einem dtinnen Queeksilberfilm fiber- zieht: Der Film 15st und verbindet den Spuren- riiekstand mit noeh ungel6sten Resten des Matrix- elementes, so da$ meehaniseh bedingte Verluste nieht mehr auftreten. Wie unsere Arbeit zeigen soll, ist unter diesen Bedingungen eine reproduzierbare Spurenanreieherung selbst dann mSglieh, wenn man Zinkeinwaagen yon mehr als 100 g verwendet und das Analysenmaterial bis auf einen Rest yon wenigen Milligramm in Salzs/iure aufl6st. Ober die Ergebnisse entspreehender Versuehe zur Spurenanalyse aueh anderer Materialien als Zink werden wir sp/iter beriehten (s. aueh [4]).
2. Allgemeine ~berlegungen zur Spurenanreieherung dureh partielles Liisen der Matrix
Tab.1 enth/ilt die Ergebnisse yon Versuchen, bei denen Zinkstiicke yon 10 g mit je 200 mg Quecksilber- metal1 fiberzogen und ansehlieBend in konz. Salzs/iure bis auf einen Zinkrest yon 10--50 mg gel6st wurden. Die als Verunreinigung zu wenigen p p m enthaltenen bzw. zu Eiehzwecken dureh Zementation auf der Oberfl/iehe niedergesehlagenen Spurenelemente wer- den entspreehend den Angaben der Tabelle im LSse- riiekstand angereiehert, innerhalb der Analysen- streuungen also praktisch quantitativ. Man erh/ilt damit allein dureh weitgehendes AuflSsen des Proben- materials in Gegenwart yon Queeksflber, ohne zus/itz- lithe Trennoperation, Anreieherungsfaktoren yon 103 oder sogar 104. ErfaBt wird dabei, wie die Tabelle ausweist, u.a. eine ganze Reihe yon Spurenelementen, die fiir die Reinheitspriifung des Zinks yon Interesse sein kSnnen.
Die Frage, ob ein Metall yon einem Oxydations- mittel angegriifen wird oder nieht, h/ingt wesentlieh yon den Redoxlootentialen der Reaktionspartner ab. Alle bei unseren Versuehen im L6serfiekstand ange- reicherten Spurenelemente sind ihrem Normalpoten- tial nach positiver als das Matrixelement Zink (Tab. 1).
Tabelle 1
Spuren- E ~ [Volt] LSsliehkeit in aus 10 g Zn ~ elemen~ Me Hg bei Raum- Me angereichert
temperatur [Gew.-6/6]"
Au + 1,50 0,13 > 95 Pd + 0,987 0,06 > 95 Ag -~ 0,799 0,03 > 95 Cu + 0,337 0,002 90 Bi + 0,215 1,4 > 95 Pb -- 0,126 1,5 > 95 Sn -- 0,136 0,9 > 95 bTi -- 0,250 2 �9 10 -6 > 95 Co --0,277 < 1.10 -6 > 95 T1 -- 0,336 43 > 95 In -- 0,342 57 > 95 Cd -- 0,403 5 > 95 Fe -- 0,440 < 5 �9 10 -~ etwa 40 Ga -- 0,53 etwa 1 etwa 80
Zn -- 0,763 2 --
a Naeh [6,11,13].
Das tats~ichliche Potential der in Quecksilber gel6sten Spuren, yon dem ihr Verhalten gegenfiber Salzs/iure abh/~ngt, ist in der Regel jedoeh sehr versehieden yon den tabellierten Normalpotentialen der reinen Metalle.
Das elektrochemisehe Verhalten eines Amalgams wird dureh mehrere GrSl3en bestimmt. Hierzu geh6- ren die Aktivit/it des Metalls in Queeksflber sowie die der zugeh5rigen Metallionen in der umgebenden w/iBrigen LSsung. Sofern intermetallische Verbin- dungen gebfldet werden, spielen aueh die Bindungs- parameter eine wiehtige Rolle.
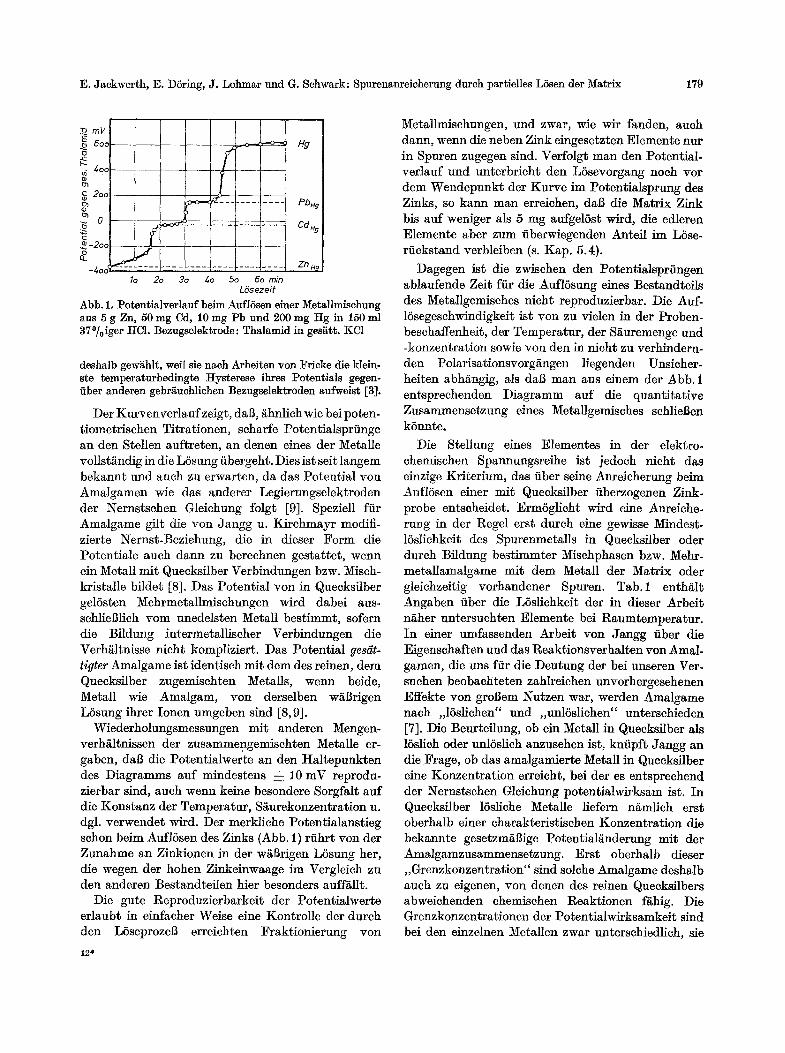
In unseren Versuehen werden die Potentialwerte in s tarkem MaBe dureh die hohe Chloridkonzentration der umgebenden LSsung beeinfluBt, welehe die Aktivi- t/it an freien Metallionen maneher Elemente zugun- sten der Bildung wenig dissoziierter Chloride oder yon Chlorokomplexen klein h/ilt. Die Potential- /inderung einer , ,amalgierten" Metallmisehung im Verlaufe der MetallauflSsung in Salzs/iure gibt einen ersten Einbliek in das chemisehe Verhalten der einzel- nen Bestandtefle beim Anreicherungsproze$ (Abb. 1).
Hierzu haben wit eine Mischung aus 5 g Zink, 50 rag Cad- mium and 10 mg Blei mit 200 mg Quecksilber verquiekt, das Metall mit 150 ml konz. Salzsiure versetzt und auf etwa 70~ erw~rmt. Der Potentialverlauf des vom Quecksilber umsehlossenen Metalls wurde auf eine ,,Thalamid"-Elektrode in gesgttigt~r KaliumehloridlSsung bezogen, wobei das Potential der Metallmischung durch eine amalgamierte Platinspitze indiziert wurde. Die Thalamid-Elektrode wurde
E. Jackwerth, E. DSring, J. Lohmar und G. Schwark: Spurenanreicherung dureh partielles LSsen der 5fatrix 179
mV E 6oc
o5 4 0 c - (9 c~
c 2oc
g -d o
~ -2oc
j /
. . . . . . . . !1 --- - P b,vg
~o~ ~:~ C d Hg I
1o 20 30 40 50 60 min L6sezeff
Abb. 1, Potentialverlauf beim Aufl6sen einer Metallmischung aus 5 g Zn, 50 nag Cd, 10 mg Pb und 200 mg Hg in 150 ml 37~ HC1. Bezugselektrode: Thalamid in ges~tt. KC1
deshalb gews weft sie naeh Arbeiten yon Fricke die klein- ste temperaturbedingte Hysterese ihres Potentials gegen- fiber anderen gebr~uchlichen Bezugselektroden aufweist [3].
Der Kurvenverlauf zeigt, daB, i~hnlich wie bei poten- tiometrischen Titrationen, seharfe Potentialsprfinge an den Stellen auftreten, an denen eines der Metalle vollst~ndig in die LSsung fibergeht. Dies ist seit langem bekannt und auch zu erwarten, da das Potential yon Amalgamen wie das anderer Legierungselektroden der I~ernstsehen Gleiehung folgt [9]. Speziell ffir Amalgame gilt die yon Jangg u. Kirchmayr modifi- zierte I~ernst-Beziehung, die in dieser Form die Potentiale auch dann zu berechnen gestattet, wenn ein Metall mit Quecksflber Verbindungen bzw. Misch- kristalle bildet [8]. Das Potential yon in Quecksilber gelSsten Mehrmetallmischungen wird dabei aus- schlieBlieh yore unedelsten Metall bestimmt, sofern die Bildung intermetalliseher Verbindungen die Verh/~ltnisse nicht kompliziert. Das Potential gesdit- tigter Amalgame ist identisch mit dem des reinen, dem Quecksilber zugemischten Metalls, wenn beide, Metall wie Amalgam, yon derselben w/tBrigen L5sung ihrer Ionen umgeben shad [8, 9].
Wiederholungsmessungen mit anderen Mengen- verh/iltnissen der zusammengemischten Metalle er- gaben, dab die Potentialwerte an den Haltepunkten des Diagramms auf mindestens ~: 10 mV reprodu- zierbar shad, auch wenn keine besondere Sorgfalt auf die Konstanz der Temperatur, S/iurekonzentration u. dgl. verwendet wird. Der merkliehe Potentialanstieg sehon beim AuflSsen des Zinks (Abb. 1) rfihrt yon der Zunahme an Zinkionen in der w/~Brigen LSsung her, die wegen der hohen Zinkeinwaage im Vergleich zu den anderen Bestandteflen bier besonders auff/~llt.
Die gute Reproduzierbarkeit der Potentialwerte erlaubt in einfacher Weise eine Kontrolle der dureh den L6seprozeB erreichten Fraktionierung yon
12"
Metallmisehungen, und zwar, wie wir fanden, aueh dann, wenn die neben Zink eingesetzten Elemente nur in Spuren zugegen sind. Verfolgt man den Potential- verlauf und unterbrieht den LSsevorgang noch vor dem Wendepunkt der Kurve im Potentialsprung des Zinks, so kann man erreichen, dab die Matrix Zink bis auf weniger als 5 mg aufgelSst wird, die edleren Elemente aber zum fiberwiegenden Anteil im LSse- rfiekstand verbleiben (s. Kap. 5.4).
Dagegen ist die zwischen den Potentialsprfingen ablaufende Zeit ffir die AuflSsung eines Bestandtefls des Metallgemisehes nieht reproduzierbar. Die Auf- 15segesehwindigkeit ist yon zu vielen in der Proben- besehaffenheit, der Temperatur, der S~uremenge und -konzentration sowie yon den in nieht zu verhindern- den Polarisationsvorg/~ngen liegenden Unsieher- heiten abh~ngig, als dab man aus einem der Abb. 1 entspreehenden DiagTamm auf die quantitative Zusammensetzung eines Metallgemisches schlieBen kSnnte.
Die Stellung eines Elementes in der elektro- ehemisehen Spannungsreihe ist jedoeh nicht das einzige Kriterium, das fiber seine Anreieherung beim AuflSsen einer mit Quecksilber fiberzogenen Zink- probe entscheidet. ErmSglieht wird eine Anreiche- rung in der Regel erst dureh eine gewlsse Mindest- 15sliehkeit des Spurenmetalls in Queeksilber oder dureh Bildung bestimmter Misehphasen bzw. Mehr- metallamalgame mit dem Metall der Matrix oder gleichzeitig vorhandener Spuren. Tab. 1 enth/~lt Angaben fiber die LSsliehkeit der in dieser Arbeit n~her untersuehten Elemente bei Raumtemperatur. In einer umfassenden Arbeit yon Jangg fiber die Eigensehaften und das Reaktionsverhalten yon Amal- gamen, die uns ffir die Deutung der bei unseren Ver- suchen beobachteten zahlreiehen unvorhergesehenen Effekte yon groBem Nutzen war, werden Amalgame nach ,,15slichen" und ,,unlSslichen" untersehieden [7]. Die Beurteilung, ob ein Metall in Queeksilber als 15slich oder unlSslich anzusehen ist, knfipft Jangg an die Frage, ob das amalgamierte Metall in Queeksflber eine Konzentration erreicht, bei der es entspreehend der Nernstschen Gleichung potentialwirksam ist. In Quecksilber 15sIiehe Metalle liefern n/~mlieh erst oberhalb einer charakteristischen Konzentration die bekannte gesetzm/iBige Potential~nderung mit der Amalgamzusammensetzung. Erst oberhalb dieser ,,Grenzkonzentration" sind solche Amalgame deshalb auch zu eigenen, yon denen des relnen Queeksflbers abweiehenden ehemischen Reaktionen f/~hig. Die Grenzkonzentrationen der Potentialwirksamkeit shad bei den einzelnen Metallen zwar untersehiedlieh, sie

180 Z. Anal. Chem., Band 260, Heft 3 (1972)
liegen aber innerhalb eines Konzentrationsbereiches yon etwa 10 -4 his 10 -~ Gew.-~ [1,2,6,8]. Unterhalb tier Grenzkonzentration eines Metalls wird ein wenig definiertes Misehpotential und sehlieBlieh das Potential des Queeksilbers gemessen: solehe fremd- metallarmen Amalgame reagieren infolgedessen ehe- miseh wie reines Queeksilber, d.h., die zu Spuren- mengen enthaltenen Metalle gehen selbst keine Umsetzungen mehr ein.
Das Vorhandensein einer unteren Grenze fiir die Gfiltigkeit der Nernst-Gleiehung bei Amalgamen hat ffir die Spurenanreieherung naeh dem bier besehrie- benen Verfahren insofern Bedeutung, als der naeh dem partiellen L6sen yon Reinstzink im Quecksilber- Rfiekstand verbleibende Spurenelement- Gehalt gerade in dem Konzentrationsbereieh liegen kann, in welehem das Potential des Amalgams sieh dem des reinen Queeksilbers nahert und die Reaktionsf/~higkeit der Analysenprobe gegenfiber Salzs/~ure nur noeh gering ist. Solange ein Rest yon Zink im LSserfiekstand enthalten ist, der deutlieh oberhalb dieser Konzen- trationsgrenze liegt, bestimmt alas Zink als unedelstes Metall der Probe zuniiehst allein das Potential und damit alas Reaktionsverhalten gegenfiber Salzs/~ure. Dies vor allem bewirkt die Anreieherung der Spuren- elemente, die edier shad als Zink. Abet aueh dann, wenn der Zinkgehalt im LSserfiekstand bei weiterem AuflSsen der Probe so welt absinkt, dab Zink das Potential des Amalgams nieht mehr beeinfluBt, daft man erwarten, dab die angereieherten Spurenelemente wegen der geringen Reaktionsfiihigkeit sehr verdfinn- ter Amalgame gar nieht oder nut langsam yon der S/~ure aus dem Rfiekstand herausgelSst werden. Allerdings wird die Oxydation der einzelnen Spuren- elemente entspreehend ihrem Potential dutch den Luftsauerstoff unter Umsti~nden wesentlieh mit- bestimmt. Ebenfalls beeinfluBt alas Entstehen inter- metalliseher Verbindungen oder die Bildung yon Misehkristallen zwisehen Spuren und Matrix das AuflSsungsverhalten der Spurenelemente vor allem gegen Ende des Anreieherungsprozesses.
Eigene Versuehe zeigten, dab Spuren Cd, In, Pb und T1 sehr bald in LSsung gehen, wenn das restliehe Zink dutch Koehen des Riiekstandes mit Salzs/iure herausgel6st wird. Die Menge der im Spurenkonzen- trat enthaltenen Elemente Bi, Co, Cu und Iqi wird dagegen kaum ver~ndert.
Sehliefllieh werden manehe in Quecksilber unl6s- liche Metalle vor dem Aufl6sen in S/s aueh dadurch gesehfitzt, dab die im Rfiekstand angesammelten Partikel yon Queeksilber benetzt und mit einem dieh- ten Film iiberzogen werden.
3. Erl~uterungen zur Spurenanreieherung in Reinstzink und Arbeitsvorsehrift
Folgende allgemeine Arbeitsbedingungen haben sich ffir die Spurenanreieherung aus Reinstzink-Materia- lien bewi~hrt: Das zur Analyse eingesetzte Proben- material sollte aus einem Stfiek oder aus wenigen groben Zinkstfiekehen bestehen. Bei sieh einzeln auflSsenden Sp/inen besteht die Gefahr, dab die mit einem Zinkteilehen in LSsung gegangenen Spuren- ante fie nieht mehr vollst/~ndig an anderen noeh unge15st vorhandenen Zinkspiinen abgesehieden (zementiert) und angereiehert werden. Zerspantes Probenmaterial kann aber leieht zu diehten Tabletten verprel3t werden, bei denen diese Schwierigkeiten nieht mehr auftreten. Bei Tabletten, die aus 10 g Zinksp/~nen erhalten wurden (~ 20 mm, PreBdruek 10 t), waren die Streuungen der AnalysenmeBwerte kaum versehieden yon denen kompakter Stfieke des Materials. Zinkgranalien yon etwa 5 mm ~, die wir fiir unsere Versuche hiiufig verwendet haben, werden yon dem wenigen zugesetzten Queeksflber bereits so gut verklebt, dab Schwierigkeiten beim Aufl6sen ebenfalls nieht entstehen.
Da Zinkeinwaagen von 100 g und mehr ohne An- reieherungsverluste eingesetzt werden kSnnen, 1/~13t sich die zur Analyse verwendete Zinkmenge dem Spurengehalt des Materials und dem Naehweis- vermSgen der Bestimmungsverfahren fiber einen weiten Bereieh anpassen.
Die zum L6sen bestimmte Salzs/iure mug sorg- f~ltig gereinigt sein, da ein Teil der als Verunreinigung enthaltenen Sehwermetallspuren aufdem amalgamier- ten Zink zementiert und mit angereiehert werden kann. Die LSsegeschwindigkeit des Zinks wird dureh die Bedeekung mit Queeksflber zwar beeintr/iehtigt, sie ist aber immer noch so hoeh, dab ein Stfick von 10 g Zink in etwa 1 h in LSsung geht.
Die weitere Verarbeitung des aus noch etwa 100 mg Zinkmetall, dem unverbraueht erhaltenen Queck- silber und den angereieherten Spurenelementen bestehenden L6serfiekstands wird dureh das Analy- senverfahren bestimmt. Fiir die in dieser Arbeit ausgewiihlten photometrisehen undpolarographischen Bestimmungsmethoden ist eine sorgf/~ltige Abtren- hung des Queeksilbers erforderlich, da Quecksilber- salze in der im Rfickstand vorhandenen Menge die Analyse empfindlieh st6ren. Aueh fiir die spektro- chemisehe Analyse des Spurenkonzentrats wurde das Queeksilber abgetrennt. Dagegen kann bei der Spu- renbestimmung mit Hilfe der Atomabsorptions- Spektrometrie auf die Entfernung des Quecksilbers unter Umst/~nden verziehtet werden, wenn dessen

E. Jaekwerth, E. DTring, J. Lohmar und G. Sehwark: Spurenanreieherung dnreh partielles LTsen der Matrix 181
Einflul~ auf die Absorption der Spuren bei der Eiehung des Verfahrens berficksichtigt wird.
Bei Verwenden einer Aeetylen-Luft-Flamme als Absorptionsvolumen beobachteten wir jedoeh mehr- faeh die Bildung yon Queeksilberaeetylid in der aus der Zerst/iuberkammer abfliel]enden ProbenlTsung, so dab eine vorherige Abtrennung des Queeksilbers unbedingt notwendig ist, wenn Acetylen als Brenngas verwendet wird.
Zur Abtrennung wurde das Queeksilber dureh Zusatz yon Ameisens/~ure selektiv zum Metall redu- ziert und die iiberstehende LTsung der Spurenelemen- te dekantiert. Ein mel~barer Verlust unedlerer Metallspuren dureh Mitreil]en tri t t bei der Ausf/illung des Queeksflbers nieht t in [10]. Dagegen werden Gold und Palladium sowie (zu unreproduzierbaren Anteflen) Sflberspuren zusammen mit dem Queek- silber reduziert und ausgef/illt. Die ausgeschiedenen Edelmetalle werden dabei vollst/indig yore Queek- silber aufgenommen. Eine quantitative Abseheidung aueh der Silberspuren wird mit Aseorbins/~ure als Reduktionsmittel erreieht. Allerdings erschweren der Uberschul3 und die Oxydationsprodukte derAseorbin- s~ure die Analyse der in der fiberstehenden LTsung angereieherten unedleren Elemente.
Arbeitsvorschri/t ])as eingewogene Zink (in der Regel 5--25 g) wird mit 1--5~ der Einwaage an Quecksilbermetall versetzt. 1VIan gibt einige Tropfen verd. Salzs~ure hinzu, wobei sieh das Metall vollst~ndig mit einem diinnen Queeksilberfilm fiber- zieht.
Nach Zusatz yon 50 ml 37~ Salzs~ure wird das Zink unter Erw~rmen auf tier Heizplatte gelTst. Wenn der LTse- prozess durch S~ureverbrauch und Ansteigen der Zink- ehlorid-Konzentration langsamer wird, dekantierg man die fiberstehende LTsung und ersetzt sie dureh frische Salzs~ure. ])ieser Vorgang wird wiederholt, bis der LTseriickstand nur noch etwa 100--200 mg Zink enth~lt. ])iese Menge erkennt man mit etwas ~bung daran, dab der zuriiekbleibende Queeksilbertropfen noch nieht ganz die ~ul]ere Form des reinen Quecksilbers angenommen hat.
Zur Abtrennung des Queeksilbers wird der MetaIlriick- stand mit Wasser frei yon Chlorid gewasehen, dann in 5 ml 65~ Salpeters~ure aufgelSst 1. Die LSsung wird bis auf etwa i ml eingedampft. Iqaeh dem Verdiinnen mit Wasser auf etwa 30 ml werden 5 ml unverdiinnte Ameisensiiure zugesetzt. Naeh kurzem ErwSrmen fiillt das Quecksilber feinverteilt als Metalt aus. ])ureh Kochen der Suspension fliel3en die QueeksilbertrSpfehen zusammen und die fiber- stehende LTsung kann leicht dekantiert werden.
1 Sollen angereicherte Spuren Zinn bestimmt werden, so mug anstelle der Salpeters~ure Schwefels~ure verwendet werden. Das Zinn wird ansehliel3end selektiv aus jodid- haltiger LTsung mit Benzol extrahiert und nach Reextrak- tion mit einem geeigneten Reagens, z.B. H~matein, photo~ metrisch bestimmt.
Naeh Abdekantieren vom Quecksilber und Nachwaschen mit einigen Millilitern Wasser wird die klare w/~llrige LTsung zur Troekne eingedampft. Der Rfickstand wird unter leieh- tern Erw/~rmen mit 5 ml 2,5 M Salzs~ure aufgenommen und im Meflkolben zu 25,0 ml Wasser aufgefiillt. Aliquote Teile der L5sung werden zur photometrisehen bzw. polarogra- phischen Bestimmung der Spurenelemente verwendet.
Zur Bestimmung der Spuren dureh Atomabsorption wird unmittelbar die vom Quecksilber dekantierte und mit Wasser auf 20,0 ml aufgeffillte LTsung verwendct. Die Probenvorbereitung ffir die Spurenbestimmung im GIeieh- strombogen ist in Kap. 5.4. besehrieben.
Zur Analyse der im ausgefiillten Queeksilber en~haltenen Edelmetallspuren wird das Metall in etwa 10 ml KTnigs- wasser gelSst. Die LTsung wird bis auf etwa 1--2 ml (nicht zur Troekne !) abgeraueht.
Der Rfickstand wird im Mel]kolben zu 25,0 ml mit Wasser aufgefiillt. Aliquote Teile der LSsung werden zur Bestim- mung yon Au und Pd verwendet. Soll Ag bestimmt werden, so mul~ das dureh Reduktion mit Aseorbins~ure (s. oben) erhaltene Queeksilbermetall in Salpeters~ure anstelle yon KTnigswasser aufgelTst werden.
4. Zur Eichung des Verfahrens
Die Vollsti~ndigkeit der Anreicherung sowie der Aus- schinB systematiseher Fehler bei den yon uns benutz- ten Bestimmungsverfahren wurde mit analysiertem Probenmaterial, soweit vorhanden, fiberprfift. Sofern Zinkmetall mit bekannten Spurengehalten zur Kon- trolle des Analysenverfahrens und der Arbeits- methodik nicht zur Verffigung stand, lieferten Reinst- zink-Einwaagen, denen Spurenelemente in definierter Menge zugesetzt und durch Zementation auf der Oberfl~che niedergeschlagen wurden, einen geeigne- ten Ersatz. Nach unseren Erfahrungen werden die naeh Zementation auf der Oberfliiche des Zinks anhaftenden und die im Innern homogen verteilten Spurenelemente beim AuflTsen des Zinks in gleicher Weise in dem yon Quecksflber umschlossenen LTse- rfiekstand angereiehert.
Zur Zementation werden die Spuren zu wenigen ml in 1 M salzsaurer LSsung der Zinkeinwaage zugesetzt. Das Probegut wird leicht erw~rmt und zum Teil eingedampft, oder man l~iI]t es 1--2 hunter gelegentlichem Umsehwenken stehen. Naeh ~berziehen des Metalls mit Queeksilber werden die Proben entspreehend der angegebenen Arbeitsvorsehrift weiter behandelt.
5. Bestimmung der angereicherten Spurenelemente
Aus den im Spurenkonzentrat angereicherten Spuren (Tab. l) haben wir einige uns wichtig erscheinende Elemente ausgew/ihlt und sie unter routinemiil3igen Arbeitsbedingungen mit versehiedenen Analysen- verfahren bestimmt. Ftir Zinksorten mit Spuren- verunreinigungen im unteren ppm-Bereieh bietet sich vor allem die Atomabsorptions-Spektrometrie

182 Z. Anal. Chem., Band 260, Heft 3 (1972)
wegen ihrer unkomplizierten Arbeitsweise an (Kap. 5.1). Daneben haben wir zum Vergleieh einen Tell der Spurenelemente mit photometrischen und polaro- graphischen Verfahren in aliquoten Teilen des Spuren- konzentrats analysiert (Kap. 5.2, 5.3). Ein besseres NaehweisvermSgen erreieht man durch die spektro- chemische Analyse des ungeteilten Konzentrats im Gleiehstrombogen. Hierzu ist allerdings eine zusitz- fiche Aufarbeitung des LSseriickstandes erforderlieh (Kap. 5.4).
5.1. Bestimmung yon Cd, Cu, Ni und Pb dutch Atomabsorption
In Vorversuchen wurde kein merklicher EinfluB yon Zink und Queeksilber in den im L6serfiekstand zu erwartenden Mengen auf die Absorption der Spuren- elemente Cd, Cu, Ni und Pb gefunden, wenn mi~ einer Acetylen-Luft-Flamme gearbeitet wird. In der ki l teren Propan-Luft -Flamme erh~lt man dagegen eine merkliche Beeinflussung der Absorption durch beide Matrixelemen~e. Wegen der in Kap. 3 besehrie- benen Bildung yon Queeksilberaeetylid w~hrend der Spurenbestimmung in Luft-Acetylen haben wir vor der Analyse das Queeksflber jedoch aus der L6sung des Spurenkonzentrats abgetrennt (s. Kap. 3).
Als GerEt wurde das Atomabsorptions-Spektrometer Modell 1000 der Fa. Varian-Techtron mit den zugeh6rigen Hohlkathodenlampen benutzt. Analysenlinien: Cd: 228,8 nm; Cu: 324,8nm; I~i: 232,0nm; Pb: 217,0nm. Gasgemiseh: Acetylen/Luft. Statistische Daten s. Tab.2.
5.2. Photometrische Bestimmung von Cu, Bi, Ni, A u und Pd
Die photometrisehe Bestimmung yon Cu, Bi und Ni ist in der naeh Abtrennung des Quecksilbers erhal- tenen ProbelSsung ohne Sehwierigkeiten durchffihr- bar. Dasselbe gilt fiir Au und Pd, die naeh den yon uns kfirzlieh besehriebenen Verfahren [5] photo- metriseh neben dem Queeksilber des LSseriickstandes best immt werden. Ffir Au wurde dazu der Farb- komplex mit Rhodamin B verwendet, Pd wurde als (PdJ4) 2- bestimmt. Die folgenden Angaben besehr~n- ken sieh auf wenige I)aten zur Charakterisierung der photometrischen Verfahren ffir die yon uns aus dem Spurenkonzentrat ausgewihlten Elemente Cu, Bi und Ni. Die Arbeitsvorschrfften fiir die Bestimmung yon Pd und Au sind der Arbeit [5] zu entnehmen. S~atistische Da~en s. Tab. 2.
Photometrische Bestimmung yon Gu mit Natriumdi~ithyl- dithio~rbamidat (NaDDTC). 5,0 ml Probenl6sung (s. Kap. 3) werden mit 2 M Aeetatpufferl6sung pH 4,6 auf diesen
pH-Wert gebraeht und mit Wasser auf 25 ml verdfinnt. Nach Zusatz yon 2 ml l~ NaDDTC-LSsung wird mit 20,0 ml Chloroform extrahiert. Der Extrakt wird photometriert.
Photometer Eppendoff, Filter Hg 436, 2 cm-Kiivetten. Photometrische Be~timmung yon Bi mit NaDDTC. 5,0 ml
ProbenlSsung (s. Kap. 3) werden mit 10 ml 10~ Natron- lauge sowie 20 ml 10~ NatriumcyanidISsung versetzt und mit Wasser auf etwa 50 ml verdiinnt. Nach Zusatz yon 2 ml l~ NaDDTC-L6sung wird mit 3 Portionen Chloro- form (10,0 ml, 10 ml, 5 ml) je 1 min extrahiert. Die vereinig- ten Extrakte werden mit Chloroform zu 25,0 ml erg~inzt und photometriert.
Photometer Eppendorf, Filter Hg 366, 2 em-Kiivetten. Photometrische Bestimmung yon Ni mit Diacetyldioxim/
NaDDTC. 5,0 ml ProbenlSsung (s. Kap. 3) werden mi~ Wasser auf etwa 40 ml verdiinnt. Nach Zusatz yon 1 ml 10~ Hydroxylammoniumchlorid-LSsung, 1 ml 25~ AmmoniaklSsung sowie 1 ml l~ DiaeetyldioximlSsung in Methanol wird 3 rain mit 20 ml Chloroform extrahiert. Die abgetrennte organische Phase wird mit 15 ml 2~ Salzsiure 1 min reextrahiert. Die salzsaure L6sung wird mit 10ml Tetraehlorkohlenstoff 1 min gewaschen, dann mit 2ml 25~ AmmoniaklSsung sowie 1 ml 0,05~ NaDDTC-LSsung versetzt und mit 20,0ml Tetraehlor- kohlensteff 3 min extrahiert. Der Extrakt wird photometrier~. Photometer Eppendorf, Filter Hg 334, 2 cm-Kiivetten.
5.3. Polarographische Bestimmung von Cd und Pb
5,0 ml ProbenlSsung (s. Kap. 3) werden mit 2 ml 37~ Salzsgure abgeraucht. Der Rfiekstand wird mit 5,0 ml 0,5 M Salzs~ure aufgenommen. Die L6sung wird in die mit Queek- silber als Anode beschiekte Polarographiezelle geffillt. Nach 3 rain I)urehleiten yon gereinigtem Stickstoff werden die Stromspitzen yon Pb (--0,40 V) sowio Cd (--0,63V) gemessen.
Davis-Differential-Kathodenstrahlpolarograph A 1660 (Southern Analytical Ltd.) in Einzellenanordnung.
Statistisehe Daten s. Tab. 2.
5.4. Spektrochemische Be~timmung yon Bi, Cd, Go, In, N i und Tl
Der entsprechend der Arbeitsvorschrfft (Kap. 3) nach partiellem L6sen yon Zink erhaltene Rfickstand besteht neben den angereicherten Spuren aus dem eingesetzten Quecksilber sowie aus weehselnden Mengen Zink. Selbst wenn das Queeksilber, wie besehrieben, daraus abgetrennt wird, ist der Ein- dampfrfiekstand der Probenl6sung noch so gro6, daI3 man nicht das gesamte Spurenkonzentrat in einer einzigen spektroehemischen Bestimmung im Gleieh- strombogen einsetzen kann. Das Zinksalz im Ein- dampfrficks~and mach~ auch dadureh Schwierig- keiten, dab es stark hygroskopisch ist, und dab die Mengen selbst je naeh Grad der vorangegangenen Anreieherung zwischen 50 und 200 mg Zink schwan- ken. Aueh bei sorgfi~ltigem Arbeiten sind diese Sehwankungen nicht zu vermeiden. In einer Reihe

E. gaekwerth, E. DSring, J. Lohmar und G. Schwark: Spuronanreieherung durch partielles LSsen der matrix 183
von Versuchen haben Mr uns deshalb zun/ichst bemfiht, das restliche Zink aus dem Salzrfickstand durch einc selektive Extraktion zu entfernen und yon den Spuren zu trennen. Es gelang uns jedoch nicht, ein Verfahren zu finden, welches nur das Zink oder nur die zur Analyse interessanten Spurenelemente erfal~t.
Eine genfigende Abtrennung des Zinks gelingt jedoeh, wenn man das Potential beim LSsen der mit Quecksilber fiberzogenen Zinkeinwaage gegen Ende der Reaktion kontrolliert und den LSsevorgang nicht schon, wie in der Arbeitsvorschrift angegeben ist, nach Abschiitzen des Restgehaltes auf etwa 100 mg Zink im L6serfickstand unterbricht. Entsprechend dem Diagramm der Abb. 1 mil3t man in dem Augen- blick, in dem Zink aus einem gesi~ttigten Zink- amalgam vollst/~ndig in LSsung geht, einen Potential- sprung yon etwa 1 V. Gegen eine Thalamid-Bezugs- elektrode (Schott, Mainz) gemessen, /indert sich das Potential yon etwa - - 4 0 0 mV des AmMgams bis -~ 600 mV des reinen Quecksi]bers. Dabei k6nnen die gleichzeitig enthaltenen Spuren anderer Elemente, je naeh Menge, den Kurvenanstieg etwas beeinflussen, ohne in der Regel selbst schon ausgebfldete Stufen zu erzeugen. Je reiner das zur Analyse eingesetzte Zink- material ist, desto sch/~rfer ist der Potentialsprung beim AuflSsen des restlichen Zinks aus dem L6se- rfickstand.
Der LSseprozeB muB aber unterbrochen werden, bevor alles Zink aufgel6st ist und die etwas edleren Spurenelemente naehfolgend in L6sung gehen. Die Reaktion wird beendet, wenn das zun/iehst praktisch konstante Potential yon - - 4 0 0 mV anzusteigen beginnt, - - 3 0 0 mV aber noch nieht fiberschritten sind. Eine Uberpriifung der Anreieherung zeigte, dab die Spurenelemente bis dahin noeh praktiseh voll- st/indig neben 1--5 mg Zink ira Quecksflber enthalten sind. Den notwendigen geringen Zinkgehalt im Rfiekstand kann man bei jeder Analyse leieht am Vorhandensein der Zinklinie bei 3282,33 ~ im Spek- trum des Spurenkonzentrats naehweisen und den Erfolg der Anreieherung dadureh kontrollieren. Is t diese Linie nur sehr sehwaeh oder gar nieht erkennbar, so muB man damit reehnen, dab ein Tefl der Spuren zusammen mit dem Zink in L6sung gegangen ist. Naeh Abtrennung des Queeksilbers entsprechend der Arbeitsvorschrift bleibt naeh dem Eindampfen der LSsung ein nur noeh geringer Salzrfickstand fibrig.
Versuche, diesen Spurenrfickstand quantitativ dureh Eindampfen der w/iBrigen LSsung fiber Spektralkohle odor einer Misehung aus KC1 und Spektralkohle zu ge~nnen, erbrachten nut wenig
reproduzierbare AnalysenergebIfisse: Der Eindampf- rfickstand halter so fest an der Gef/~Bwandung, dab mit dem Spatel nur eine teilweise Entfernung gelingt. Wir haben deshalb die Spuren Pb, Cu, Cd, Bi, Co, In, Ni und T1 zusammen mit dem noch vorhandenen Zink als Di/~thyldithioearbamidate mit Chloroform extrahiert und den organisehen Extrakt fiber eine Mischung yon KC1/Spektralkohle, der als Bezugs- elemente Ge und Pd zugegeben waren, eingedampft. tIierbei erh/~lt man einen leicht aus dem Glasgef/~B zu entfernenden Rfiekstand, der vollst/indig in eine Kohlebeeher-Elektrode fibergefiihrt und im Gleich- strombogen abgebrannt werden kann.
Arbeitsvorschri]t. Der nach der Anroicherungsvorschrift (Kap. 3) erhaltene LSseriickstand aus 100--200 mg Zink uud dem eingesetzten Queeksilber wird mit 25 ml 37~ Salz- s~uro vorsetzt und mit einer amalgamierten Platinspitze in Kontakt gebracht. Unter loiehtem Erw~rmen der LSsung wird das gegen eine ges~ttigto Thalamid-Elektrode gemossene Potential (Kniek-pH-Meter) beobachtet. Es betr/~gt zu- n/ichst etwa -- 400 mV. Man unterbrieht die Reaktion durch rasches Verdtinnen der LSsung mit Wasser, worm das Potential ansteigt und einen Wert yon etwa --300 mV erreicht. Anschliel3end wird die ~otallkugel chloridfrei gewaschen und das Quecksilber daraus abgetrennt (Kap. 3).
Die quecksilberfreie LSsung wird bis auf etwa 5 ml ein- geengt, in einen 100 ml-Schfitteltriehter iibergefiihrt und mit Wasser auf etwa 30 ml erg~nzt. Naoh Zusatz yon 25 ml 40~ NatriumacetatlSsung sowie 5 ml 2~ Natrium- di~thyldithiocarbamidat-LSsung wird 2mal mit jo 10ml, dann mit 5 ml Chloroform je 1 min extrahier~. Die organischon Extrakte werden durch Filterflockenmasse in ein breites 50 ml-Bechorglas filtriert und auf etwa 5 ml eingeengt. Gehen die Gehalte einzelner Spuronelemento fiber den MeB- bereich des spektroehemischen Verfahrens hinaus, so worden die Extrakte zu 25,0 ml mit Chloroform erg~nzt und ali. quote Teile zur Analyse verwendet.
Naeh Zusatz yon 80 mg Kaliumehlorid-Spektralkohle- Mischung (a) wird vorsiehtig zur Troekno oingedampft. Das Bocherglas wird anschliel3end im Trookenschrank 15 min bei 110--120~ orhitzt, um letzte anhaftende Feuchtigkeit zu entfernon. Das Probegut wird mit einem Gtasstab aus dora Becherglas entfornt, im AehatmSrsor zerrieben und voll- st~ndig in die Becherelektrode gestopft. Die Elektroden werden zur Verbesserung des Abbrandverhaltens vor dem Zfinden des Bogens 15 see kurzgesehlossen und vorgeheizt. Wiehtig ist, dab dor Bogen unmittelbar nach dem Abbren- nen des Probeguts (,,Umsohlagon" der Farbe des Bogens) gelSscht wird, um die Intensit~t des Bandenspektrums goring zu halten.
Die Photoplatten wurden geeieht und der Untergrund wurde korrigiert. Die statistisehen Daten sind in Tab.2 zusammengostellt.
a) Kaliumchlorld-Spektralkohle-Mischung. Kaliumchlorid (Suprapur, Merck) und Spektralkohlepulver (RWB, Rings- dorff) worden im Verh~Itnis 1:1 gemiseht. Als Bezugs- elemente werden Go (als GoOd) und Pd [als (NH~)2PdCI~] dureh Heruntermisehen zugegeben und zwar 10 ~g Ge sowie 2 ~g Pd pro 80 mg der Mischung.

184 Z. Anal. Chem., Band 260, Heft 3 (1972)
Tabelle 2
Spuren- Gehalt Bestimmungsverfahren rel. element [~ Me] Standard- Me abwei-
chung
Au 4,0 �9 10 -5 Photometrie mit Rhod- amin B 0,092
Pd 1,4.10 -4 Photometrie als [PdJ4]2- 0,034 Cu 2,0.10 -a Photometrie mit NaI)DTC 0,028
3,7.10 -4 Atomabsorption 0,026 Bi 1,6 �9 10 -4 Photometrie mit NaDDTC 0,058
1,5 �9 10 -~ Gleichstrombogen 0,11 Pb 2,0" 10 -a Impulspolarographie 0,014
1,6" 10 -a Atomabsorption 0,018 Ni 1,0.10 -a Photometrie mit Diacetyl-
dioxim/I~aDDTC 0,035 1,1 �9 10 -4 Atomabsorption 0,021 7,5- 10 -~ Gleichstrombogen 0,18
Cd 8,0.10 -s Impulspolarographie 0,076 7,2- 10 -6 Atomabsorption 0,038 7,0.10 -5 Gleichstrombogen 0,048
T1 1,3" 10 -5 Gleichstrombogen 0,14 Co 8,0.10 -6 Gleichstrombogen 0,11 In 2,0" 10 -e Gleichstrombogen 0,081
A pparative A ngaben zur spektrochemi~chen Bestimmung Spektrograph. 3,4 m Ebert-Spek~rograph,
Beugunfsgitter. 590 Str./mm, Gitterfl~che 137)<57 ram, Gesamtstrichzahl 77000, Messung in 2. Ordnung.
(J//nungszahl. 1 : 35. Spaltbreite. 20 ~z. Lichtquelle. Gleichstrombogen 10 A in Luft. Elektroden-
abstand 9 ram. Brennzeit bis zum Abbrand des Proben- materials etwa 5,5 rain.
Ele~roden. Gestopfte Graphit-Elektroden (RingsdorffRW 0076) ~ 6 ram, Qualit~t RWO, Bohrung 4,5 )<4,5 ram, Probe als Anode, Gegenelektrode Kohle RW II, ~ 6 ram.
Strahlungsemp/dnger. Photoplatten Agfa-Gevaert 34B50. Analysen- und Bezugslinien (in A). Bi 3067,72-- Ge 3039,06;
In 3256,09--Ge 3269,49; Cd 3261,06--Ge 3269,49; Ni 3414,77--Pd 3421,24; Co 3453,50--Pd 3421,24; T1 3519,24-- Pd 3421,24.
6. S t a t i s t i s c h e D a t e n
Die in Tab. 2 zusammengefaBten W e r t e fiir die rela- t ive S t anda rdabwe ichung wurden un te r EinsehluB
des gesamten Anreieherungs- und jewefligen Bes t im- mungsver fahrens gewonnen. I r a FaUe der photo- met r i schen bzw. po la rographischen Veffahren wurden en t sp rechend den Arbe i t svorsehr f f ten a l iquote Teile der aus Zinke inwaagen yon je 25 g gewonnenen Spu- r enkonzen t r a t e verwendet . Den D a t e n fiir die Be- s t i m m u n g mi t Hilfe der A tomabso rp t ions -Spek t ro - me t t l e sowie der spekt roehemisehen B e s t i m m u n g im Gle iehs t rombogen l iegen E inwaagen yon je 10 g Zink zugrunde.
Die ffir die Bogenana lyse e rmi t t e l t e Naehweis- grenze fiir die yon uns q u a n t i t a t i v be s t immte n Ele- men te be t r~g t (in Mik rog ramm) :
Cd: 0,7 ; Co: 0,06; Ni : 0,8; B i : 0,3; T1: 0,3; I n : 0,03.
Wir danken der Deutsehen Forschungsgemeinschaft und dem Fond der chemisehen Industrie fiir finanzielle Unter- stfitzung.
L i t e r a t u r
1. Erdey-Grfiz, T., Szarvas, P. : Z. Physik. Chem. (A) 177, 277 (1936).
2. -- V~zsonyi-Zilahy, A.: Z. Physik. Chem. (2~) 177, 292 (1936).
3. Fricke, H. K. : Beitr. Angew. Glasforsch. 175. Stuttgart: Wiss. Yerlagsges. 1960.
4. J a c k w e r t h , E. : diese Z. 256, 128 (1971). 5. -- Kulok, A.: diese Z. 257, 28 (1971). 6. Jangg, G., Bach, H.: Quecksilber und Amalgammetall-
urgie. In: G. Eger: Handbueh der r Elektro- chemie, 1. Tell, 2. Aufl. Leipzig: Akad. Verlagsges. 1961.
7. - - -- Metall 19, 442 (1965). 8. -- Kirehmayr, H. : Z. Chem. 3, 47 (1963). 9. Kortiim, G.: Lehrbuch der Elektrochemie. Weinheim
(Bergstr.): Verlag Chemie 1957. 10. Meyer, J. : diese Z. 219, 147 (1966). 11. Neeb, R.: Inverse Polarographie und Voltammetrie,
Weinheim (Bergstr.): Verlag Chemie 1969. 12. Seith, W., Herrmann, J . : Spektrochim. Acta 1, 548
(1941). 13. Ullmanns Encyklop~die d. techn. Chemie, 3. Aufl.
Miinchen-Berlin: Urban & Schwarzenberg 1963.
Priv.-Doz. Dr. E. Jackwerth Institut fiir Spektrochemie und angewandte Spektroskopie ])-4600 Dortmund, Bunsen-Kirchhoff-StraBe 11 Deutschland























![]Matrix Tablet](https://img.pdfslide.org/doc/110x75/577ccf211a28ab9e788ef4c9/matrix-tablet.jpg)





