Embed Size (px)
Citation preview

Studien
zur Asymmetrischen
Vinylogen Mukaiyama-Aldolreaktion
Von der Naturwissenschaftlichen Fakultät
der Gottfried Wilhelm Leibniz Universität Hannover
zur Erlangung des Grades
Doktor der Naturwissenschaften
– Dr. rer. nat. –
genehmigte Dissertation
von
Dipl.-Chem. Serkan Simsek
geboren am 27.06.1981 in Hannover
2009

Die vorliegende Arbeit wurde in der Zeit von Mai 2006 bis Juli 2009 unter der Anleitung von
Herrn Prof. Dr. Markus Kalesse am Institut für Organische Chemie der Gottfried Wilhelm
Leibniz Universität Hannover angefertigt.
Hiermit erkläre ich, dass die vorliegende Dissertation selbständig verfasst und alle benutzten
Hilfsmittel sowie eventuell zur Hilfeleistung herangezogenen Institutionen vollständig
angegeben wurden.
Diese Dissertation wurde nicht schon als Diplom- oder ähnliche Prüfungsarbeit verwendet.
Hannover, den 31.07.2009
Referent: Prof. Dr. Markus Kalesse Korreferent: Prof. Dr. Andreas Kirschning Tag der Promotion: 16.09.2009

Kurzfassung
Studien zur Asymmetrischen Vinylogen Mukaiyama-Aldolreaktion
Schlagworte: Vinyloge MUKAIYAMA -Aldolreaktion, Aymmetrische Synthese,
chirale Lewis-Säuren, Oxazaborolidine, Polyketide, Streptogramine
Polyketide stellen heute eine der wichtigsten Naturstoffklassen dar. Die strukturelle
Komplexität und die daraus resultierenden vielfältigen biologischen Aktivitäten dieser
Verbindungen waren in den letzten Jahrzehnten Anreiz für die synthetische organische
Chemie zur Totalsynthese vieler unterschiedlicher Polyketide.
Stereoselektive Aldolreaktionen sind hierfür die wichtigsten Synthesemethoden. Allerdings
ermöglichen diese nur den Aufbau von 1,3-funktionalisierten Bausteinen. Durch die vinyloge
Erweiterung der Aldolreaktion in einer vinylogen MUKAIYAMA -Aldolreaktion (VMAR)
können in einem Syntheseschritt γ-Hydroxy-α,β-ungesättigte Carbonylverbindungen generiert
werden. Diese 1,5-funktionalisierten Intermediate können durch einfache Transformationen in
größere Polyketidfragmenten umgewandelt werden, wodurch diese Methode immer mehr an
Bedeutung für die Polyketidsynthese gewinnt. So wurden in den letzten Jahren
unterschiedliche Methoden zur stereoselektiven Reaktionsführung der vinylogen
MUKAIYAMA -Aldolreaktion entwickelt. Diese gewährleisten jedoch nur eine eingeschränkte
Anwendung in der Polyketidsynthese, da sie nur auf wenige, für die Polyketidsynthese eher
unbedeutende Substrate beschränkt sind. Für eine breite Anwendung dieser Reaktion in der
Polyketidsynthese bedarf es deswegen der Entwicklung neuer Methoden.
Im Rahmen dieser Dissertation wurden unterschiedliche chirale Lewis-Säuren auf ihr
Potential in der asymmetrischen vinylogen MUKAIYAMA -Aldolreaktion mit γ-methylierten
Ketenacetalen untersucht. Hier erwiesen sich Oxazaborolidine als besonders effektiv, um die
Reaktion in hohen Stereoselektivitäten durchzuführen.
Die neu entwickelten asymmetrische vinyloge MUKAIYAMA -Aldolreaktionen, wurden
anschließend in der Formalsynthese von Streptogramin Naturstoffen angewandt.

Abstract
Studies on Asymmetric Vinylogous MUKAIYAMA -Aldolreaction
Keywords: vinylogous MUKAIYAMA -Aldolreaction, Asymmetric Synthesis,
chiral Lewis-acids, Oxazaborolidines, Polyketides, Streptogramines
Today the polyketide class of natural products represents a great variety of structures with
manifold biological activities. For this reason a lot of research groups work on the total
synthesis on different polyketide derived natural products.
Stereoselective aldolreactions are one of the most important methods for synthesis of
polyketides. However these high stereoselective methods allow only the construction of
1,3-functionalised fragments. Through the vinylogous extension of the aldolreaction
in a vinylogous MUKAIYAMA -aldolreaction (VMAR), γ-hydroxy-α,β-unsaturated carbonyl-
compounds can be generated in one step. These 1,5-functionalised versatile intermediates can
easily be transformed to larger polyketidfragments with simple reactions, that is why this
reaction becomes more important in total synthesis of polyketides. In the last years different
approaches were described to control the vinylogous MUKAIYAMA -aldolreaction in high
enantioselectivities. However previous methods are limited to few substrates which are rather
unimportant for the synthesis of polyketides. Especially aliphatic aldehydes are a unmet
challenge in this reaction.
In the course of this thesis different chiral Lewis-acids were tested in the asymmetric
vinylogous MUKAIYAMA -aldolreaction on terminal substituted ketene acetals. Herein the use
of Oxazaborolidines for the asymmetric vinylogous MUKAIYAMA -aldolreaction provided an
efficient access to chiral building blocks for polyketide synthesis.
Additionally the new developed enantioselective method was used in the formal synthesis of
Streptogramin natural products.

Danksagung
Für die Überlassung des Interessanten Themas, die intensive Betreuung während der
Doktorarbeit und die ständig vorhandene Diskussionsbereitschaft möchte ich mich bei
meinem Doktorvater Prof. Dr. Markus Kalesse herzlich bedanken.
Herrn Professor Dr. Andreas Kirschning danke ich für die sehr freundliche Übernahme des
Korreferates.
Beim VMAR-Team Melanie Horzella und Yi Su danke ich für die gute Zusammenarbeit und
die langen wissenschaftlichen Gespräche über die VMAR und darüber hinaus.
Weiterhin bedanke ich mich bei allen ehemaligen und jetzigen Mitgliedern des Arbeitskreises
Kalesse für die gute Arbeitsatmosphäre und die immerwährende Hilfsbereitschaft.
Ferner möchte ich auch allen Mitgliedern des Arbeitskreises Kirschning, Boysen, Duddeck
und Butenschön danken.
Den Mitarbeitern der Spektroskopie danke ich für ihre Hilfsbereitschaft, vor allem Monika
Rettstadt und Dagmar Körtje.
Besonders möchte ich mich auch bei Michael Richter und Tobias Brodmann für das
sorgfältige Korrekturlesen dieser Arbeit und die tolle Arbeitsatmosphäre bedanken.
Der größte Dank gebührt aber meinen Eltern für die sehr große Unterstützung in allen
möglichen Bereichen während des Studiums und speziell während der letzten Zeit, die
wesentlich zum Gelingen dieser Arbeit beigetragen hat.

Meinen Eltern

Inhaltsverzeichnis
Abkürzungsverzeichnis
Allgemeine Vorbemerkung
1. Einleitung 1
1.1 Polyketide als wichtige Quellen für neue Wirkstoffe 1
1.2 Stereoselektive Methoden zur Polyketidsynthese 3
1.2.1 Die Aldolreaktion 3
1.2.1.1 Die EVANS-Variante 4
1.2.1.2 Die PATERSON-Variante 5
1.2.3 Die MUKAIYAMA -Aldolreaktion 6
1.3 Die Vinyloge MUKAIYAMA -Aldolreaktion 8
1.3.1 Die Vinyloge Erweiterung der Aldolreaktion 8
1.3.2 Relevante Varianten der VMAR 11
1.3.2.1 Die CAMPAGNE-Variante 11
1.3.2.2 Die KALESSE-Variante 13
1.3.2.3 Die DENMARK-Variante 15
1.3.2.4 Die KOBAYASHI-Variante 16
2. Aufgabenstellung 18
3. Studien zur Entwicklung neuer VMAR-Methoden 20
3.1 Biphenyl-Borane als chirale Lewis-Säuren für die VMAR 20
3.2 BINOL-Liganden in der asymmetrischen VMAR 24
3.2.1 3,3’-substituierte BINOL-Liganden 24
3.2.2 BINOL-Trimethylborat Komplex nach KECK 27
3.2.3 Schiff’sche Base-Liganden nach CARREIRA 28
3.3 Bisoxazolin-Liganden in der asymmetrischen VMAR 30
3.4 Chirale Ammoniumsalze als potentielle Lewis-Säuren für die VMAR 32
3.5 Oxazaborolidine als potentielle chirale Lewis-Säuren für die VMAR 34
3.5.1 N-Ts-Aminosäure abgeleitete Oxazaborolidinone 34
3.5.2 Oxazaborolidinon vermittelte VMAR am γ-unsubstituiertem Ketenacetal 42
3.5.3 Weinsäure abgeleite Acyloxyborane 50
3.5.4 Aminoalkohol basierende Oxazaborolidine 53

Inhaltsverzeichnis
4. Studien zur Anwendung der neuen VMAR-Methode 62
4.1 Die VMAR als Schlüsselschritt in der Synthese von Streptograminen 62
4.2 Retrosynthese des Polyketidfragments von Madumycin II auf Basis 65
der neuen VMAR-Methoden
4.3 Synthetische Arbeiten zum Polyketidfragment von Madumycin II 67
4.3.1 Erste Route zur Synthese des Polyketidfragments 67
4.3.2 Zweite Route zur Synthese des Polyketidfragments 69
4.3.3 Verknüpfung von West- und Ostfragment 74
4.3.3.1 STILLE-Kupplung 74
4.3.3.2 SUZUKI-Kupplung 75
4.3.3.3 HECK-Kupplung 76
5. Zusammenfassung und Ausblick 78
6. Experimenteller Teil 82
6.1 Allgemeine Arbeitshinweise 82
6.2 Allgemeine Arbeitsvorschriften 83
6.3 Versuchsbeschreibung zu Kapitel 3 85
6.4 Versuchsbeschreibung zu Kapitel 4 119
7. Literaturverzeichnis 131
Spektrenanhang

Abkürzungsverzeichnis
°C Grad Celsius
Å Angström
Abb. Abbildung
ATP Adenosin-5‘-triphosphat
AXB Acyloxyboran
Bn Benzyl
Bu Butyl
BuLi Butyllithium
bzw. Beziehungsweise
CAN Cerammoniumnitrat
CSA Camphersulfonsäure
d dublett (NMR)
δ Chemische Verschiebung
d.h. das heißt
DDQ 2,3-Dichlor-5,6-dicyano-1,4- benzochinon
de Diastereomerenüberschuss
DET Diethyltartrat
DiBAl-H Di-isobutylalluminiumhydrid
DIPP Diisopropylphenol
DMAP 4-Dimehtylaminopyridin
DMF Dimethylformamid
DMPU 1,3-Dimethyl-3,4-5,6-tetrahydro- 2-(1H)-Pyrimidion
ee Enantiomerenüberschuss
eq Äquivalente
ESI Elektrospray-Ionisation
Et Ethyl
Et2O Diethylether
et al. et alli
EtCN Propionitril
EtOAc Essigsäureethylester
Eu(hfc)3 Tris-[3-(heptafluorpropyl hydroxymethylen)-(+) camphorato]-Europium
g Gramm
GC Gaschromatographie
h Stunde
HMDS Hexamethyldisilazan
Hz Hertz
Ipc Isopinocamphenyl
i-Pr Isopropyl
i-PrOH Isopropanol
J Kopplungskonstante
KA Ketenacetal
L Liter
LS Lewis-Säure
LDA Lithiumdiisopropylamid
NADP Nicotinsäureamid-Adenin- Dinukleotid-Phosphat
M molar
m multiplett (NMR)
Me Methyl
MeOH Methanol
MHz Megahertz
min Minuten
mL Milliliter
MS Molsieb
MTPA Methoxy-a-(trifluormethyl)- phenylessigsäure
Nr. Nummer
NMR Kernresonanzspektroskopie
OXI Oxazaborolidin
OXB Oxazaborolidinon
PE Petrolether
PMB para-Methoxybenzyl
PMP para-Methoxyphenyl
ppm parts per million
Pr Propyl
PrCN Butyronitril
Rf Retentionsfaktor
s singulett (NMR)
SAR Struktur-Aktivitäts-Beziehung
RT Raumtemperatur
t triplett (NMR)
TBDPS tert-Butyldiphenylsilyl
TBS tert-Butyldimethylsilyl
Tf Trifluormethylsulfonyl
THF Tetrahydrofuran
TMS Trimethylsilyl
TPPB Tris(pentafluorphenyl)boran
Ts para-Toluolsulfonyl
VMAR Vinyloge MUKAIYAMA - Aldolreaktion
z.B. zum Beispiel

Allgemeine Vorbemerkung
Im Verlauf der Arbeit werden zur Darstellung der absoluten Stereochemie Keile verwendet,
die relative Stereochemie wird durch Balken beschrieben,
R1 R2OH
R1 R2OH
absoluteStereochemie
relativeStereochemie
während Striche benutzt werden, wenn die Konfiguration nicht bekannt ist.
R1 R2OH
unbekannteStereochemie

Einleitung
- 1 -
1. EINLEITUNG
1.1 Polyketide als wichtige Quellen für neue Wirkstoffe
Polyketide stellen heute eine der größten und bedeutendsten Naturstoffklassen dar. Es handeln
sich hierbei um Sekundärmetaboliten, die typischerweise in Pflanzen und Mikroorganismen
vorkommen und eine hohe biologische Aktivität aufweisen. Die instabile und empfindliche
Struktur der Polyketide führt allerdings dazu, dass bei der Isolierung ein großer Teil dieser
Verbindungen zersetzt werden. Dennoch sind heute mehr als 10.000 Polyketide bekannt,
wovon bisher nur für 1% eine pharmakologische Wirkung nachgewiesen werden konnte.
Wegen ihres hohen Wirkungsspektrums sind sie für die pharmazeutische Industrie von
großem Interesse. Einer der wichtigsten Anwendungsgebiete der Polyketide sind Wirkstoffe
als Zytostatika, Immunosuppressiva und Antibiotika. [1]
Einige der bekanntesten Beispiele für pharmakologisch aktive Polyketide stellen z.B. Erythro-
mycin (1), Epothilon B (2) und Rapamycin (3) dar (Abb. 1). Hierbei handelt es sich um
Verbindungen, die bereits als Medikamente verwendet werden. Während Erythromycin (1)
Epothilon B 2
N
S
O
O
MeO
OH O
OHO
O
O
OH
O
O
HO OH
OOH
OMe
Me
Me
OMe
NMe
Me
Me
Erythromycin 1
Rapamycin 3
O
O
NO
OH
HOO
O
OH
O
OH
MeO
OMe
OMe
Abbildung 1. Wichtige Polyketid-Naturstoffe für neue Wirkstoffe.

Einleitung
- 2 -
und Rapamycin (3) in Streptomyceten vorkommen und antibakteriell [2] oder imunsupressiv [3]
wirken, wird Epothilon B (2) aus dem Myxobakterium Sorangium Cellulosum isoliert
und in der Krebstherapie eingesetzt. [4] Seit 2007 wird das Epothilon-Derivat Ixabepilone als
neues Medikament unter dem Namen Ixempra® zur Behandlung von Brustkrebs weiträumig
eingesetzt. Ein Charakteristikum der Polyketid-Naturstoffklasse ist die komplexe Polyol-
Kernstruktur mit sich wiederholenden 1,3-Diol-Mustern. Obwohl sich viele Polyketide
strukturell stark voneinander unterscheiden, erfolgt ihre Biosynthese nach identischem
Mechanismus. Durch biosynthetische Studien ist bewiesen, dass die komplexe Polyol-
Kernstruktur in der Natur durch multifunktionale Enzyme, sogenannte Polyketidsynthasen
synthetisiert wird. [5] Durch Verwendung von kleinen Carboxylaten, wie Acetat oder Pro-
pionat, die als Thioester am Acylträgerprotein (ACP) gebunden sind, wird die Kernstruktur
eines Polyketids schrittweise um weitere zwei Kohlenstoffatome verlängert. Dieses erfolgt
durch enzymatische decarboxylierende Claisen-Kondensation (Schema 1). Das resultierende
β-Keto-Thioester Intermediat wird durch NADPH zum entsprechenden β-Hydroxy-Thioester
reduziert. Da die Proteine die Orientierung der Thioester zueinander fixieren, erfolgt der
ACP
S O
O
OH
S O
R
KS
-CO2
ACP
S O
O
R
KR
NADPH NADP
ACP
S O
OH
R
SH
KS
S O
OH
R
KS
OH
TE
HO O
OH
R
DH
ACP
S O
R
OH
TE
HO O
R Schema 1. Biosynthese der Polyol-Kernstruktur durch Polyketidsynthasen (ACP = Acylträgerprotein,
KS = Ketosynthaseprotein, KR = Ketoreduktase, DH = Dehydratase, TE = Thioesterase).[5]

Einleitung
- 3 -
Aufbau sämtlicher Stereozentren in hohen Stereoselektivitäten. Anschließend wird der
β-Hydroxy-Thioester auf eine Ketosynthaseprotein (KS) transferiert, wodurch der Einbau
einer weiteren C2-Einheit ermöglicht wird. Neben 1,3-Diol-Mustern weisen Polyketide
ebenfalls eine hohe Anzahl an Doppelbindungen auf. Diese resultieren bei der Biosynthese
ebenfalls aus den β-Hydroxy-Thioestern. Unter der Einwirkung von Dehydratase (DH) wird
durch Wasserabspaltung die Doppelbindung generiert. Mittels Wiederholung dieser
Sequenzen wird die erwünschte Polyolstruktur aufgebaut. Ist die erwünschte Struktur erzeugt,
erfolgt der Kettenabbruch mit der Thioesterase (TE), wodurch die Verbindung als freie Säure
vom Protein abgespalten wird. Durch weitere Enzyme kann das Molekül zusätzlich noch
weiteren Modifizierungen wie Epoxidierungen oder Glykosidierungen unterzogen werden.
Einer der wichtigsten Aufgabenstellungen der organischen Synthesechemie ist die
Entwicklung nicht-enzymatischer asymmetrischer Reaktionen, die inspiriert durch die Natur
ebenso hoch selektiv verlaufen. Zum einen ist es wichtig neue Methoden zu etablieren die
einen schnellen synthetischen Zugang zu biologisch aktiven Naturstoffen ermöglichen, zum
anderen können hierdurch umfangreichere Derivatisierungsversuche für SAR-(Struktur-
Aktivitäts-Beziehung)-Studien durchgeführt werden, welche für die Aufklärung der Wirk-
mechanismen von Naturstoffen unerlässlich sind. Zusätzlich erlaubt die Synthese von
Derivaten unerwünschte Nebenwirkungen eines biologisch aktiven Stoffes zu reduzieren und
somit ihr Wirkprofil zu optimieren. Insofern ist heute die Entwicklung neuer asymmetrischer
Synthesemethoden ein wichtiges Forschungsgebiet, um einen möglichst direkten und
ökonomischen Zugang zu einer Vielzahl von Naturstoffderivaten zu erhalten.
1.2 Stereoselektive Methoden zur Polyketidsynthese
1.2.1 Die Aldolreaktion
Die gekreuzte Aldolreaktion stellt heute eine der wichtigsten Methoden zur Darstellung von
Polyketidbausteinen dar. Die erstmals zeitgleich von WURTZ [6] und BORODIN
[7] im
19. Jahrhundert beschriebene Reaktion ermöglicht den Zugang zu β-Hydroxy-Carbonyl-
Verbindungen und erlaubt so den Aufbau von 1,3-Diol-Mustern (Schema 2). Durch geeignete
Substrate können hierdurch bis zu zwei neuen Stereozentren aufgebaut werden. Seit ihrer
ersten Beschreibung wurden viele Konzepte zum Verständnis und zur Stereokontrolle
der Aldolreaktion entwickelt. So konnte man anhand des von ZIMMERMANN und TRAXLER
postulierten Übergangszustands für die Aldolreaktion ein allgemeines Verständnis bezüglich

Einleitung
- 4 -
H
O NaOH, H2O
RT, 50%
OH
H
O
4 5
2
Schema 2. Erste beschriebene Aldolreaktion mit Acetaldehyd als Substrat.[6,7] der Diastereoselektivität der Reaktion erlangen und so gezielt anti- bzw. syn-Aldolprodukte
generieren. [8] Die bedeutenden Fortschritte, die in den letzten 30 Jahren in der stereo-
selektiven Aldolreaktion sowie ihrer Variationen erzielt wurden, revolutionierten die Natur-
stoffchemie und öffneten den Weg zur Totalsynthese von einer Vielzahl von biologisch
aktiven Naturstoffen. [9] So hielt 1956 WOODWARD die Synthese von Erythromycin (1) mit den
damaligen Synthesemethoden aufgrund seiner komplexen Polyol-Struktur für unmöglich. [10]
Schließlich sollte er 25 Jahre später von einer erfolgreichen Totalsynthese berichten, in
der die damals neu entwickelten stereoselektiven Aldol-Methoden eine wesentliche
Rolle spielten. [11] Seither stehen für den linearen Aufbau und die Erweiterung von
Polyketidfragmenten eine Vielzahl von asymmetrischen Aldolreaktionen zur Verfügung. [12]
1.2.1.1 Die EVANS-Variante
Die am weitesten etablierte Methode für den stereoselektiven Aufbau von Polyketid-
fragmenten stellt zweifelsfrei die von EVANS entwickelte und durch chirale Auxiliare
kontrollierte Aldolreaktion dar. [13] Als chirales Auxiliar wird ein Hilfsreagenz bezeichnet,
welches kovalent an das prochirale Edukt gebunden wird. Im Übergangszustand der Reaktion
kann dieses chirale Auxiliar seine stereochemische Information auf das Produkt übertragen
und dadurch Chiralität induzieren. Dieses geschieht gewöhnlich, durch Abschirmung einer
Seite des Edukts durch sterisch anspruchsvolle Gruppen des Auxiliars, so dass diese für eine
mögliche chemische Reaktion unzugänglich gemacht wird. Bei der EVANS-Aldolreaktion
werden aus Aminosäuren abgeleitete Oxazolidin-2-on-Derivate als chirale Auxiliare
verwendet, die oftmals aus Phenylalanin bzw. Valin dargestellt werden. Die hohen
Stereoselektvitäten, die bei dieser Reaktion erzielt werden können, resultieren aus der Bildung
eines starr fixierten Komplexes zwischen Lewis-Säure und den Reaktanden. Die
Reorganisation der Bindungen erfolgt nach ZIMMERMANN und TRAXLER über einen
sechsgliedrigen zyklischen Übergangszustand. Hierbei wird eine Anordnung bevorzugt, in
dem 1,3-diaxiale Wechselwirkungen vermieden und die Dipolmomente von Enolat- und
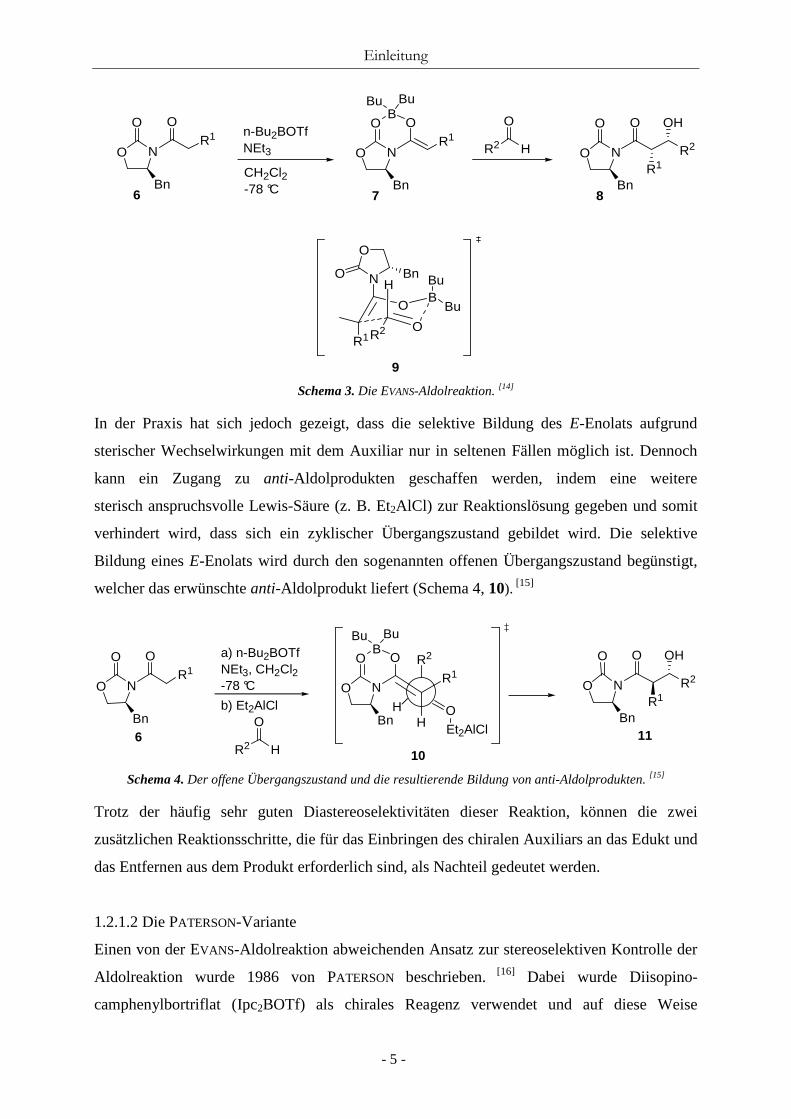
Oxazolidinon-Sauerstoff in entgegengesetzte Richtungen zeigen (Schema 3, 9). [14]
Entsprechend dem postuliertem Übergangszustand von ZIMMERMANN und TRAXLER
resultieren aus den gebildeten E- oder Z-Enolaten die entsprechenden anti- bzw. syn-Aldol-
produkte.
Einleitung
- 5 -
O N
Bn
OR1
O
6
n-Bu2BOTfNEt3
CH2Cl2-78 °C
O N
Bn
OR1
O
7
BBu Bu
R2 H
O
O N
Bn
O O
8
R2
R1
OH
O
BO
R1
N H
R2
O
BnO
Bu
Bu
9
Schema 3. Die EVANS-Aldolreaktion. [14]
In der Praxis hat sich jedoch gezeigt, dass die selektive Bildung des E-Enolats aufgrund
sterischer Wechselwirkungen mit dem Auxiliar nur in seltenen Fällen möglich ist. Dennoch
kann ein Zugang zu anti-Aldolprodukten geschaffen werden, indem eine weitere
sterisch anspruchsvolle Lewis-Säure (z. B. Et2AlCl) zur Reaktionslösung gegeben und somit
verhindert wird, dass sich ein zyklischer Übergangszustand gebildet wird. Die selektive
Bildung eines E-Enolats wird durch den sogenannten offenen Übergangszustand begünstigt,
welcher das erwünschte anti-Aldolprodukt liefert (Schema 4, 10). [15]
O N
Bn
OR1
O
6
a) n-Bu2BOTfNEt3, CH2Cl2-78 °C O N
Bn
O O
10
BBu Bu
R2 H
O
O N
Bn
O O
11
R2
R1
OH
b) Et2AlCl
R1
H
R2
OH
Et2AlCl
Schema 4. Der offene Übergangszustand und die resultierende Bildung von anti-Aldolprodukten. [15]
Trotz der häufig sehr guten Diastereoselektivitäten dieser Reaktion, können die zwei
zusätzlichen Reaktionsschritte, die für das Einbringen des chiralen Auxiliars an das Edukt und
das Entfernen aus dem Produkt erforderlich sind, als Nachteil gedeutet werden.
1.2.1.2 Die PATERSON-Variante
Einen von der EVANS-Aldolreaktion abweichenden Ansatz zur stereoselektiven Kontrolle der
Aldolreaktion wurde 1986 von PATERSON beschrieben. [16] Dabei wurde Diisopino-
camphenylbortriflat (Ipc2BOTf) als chirales Reagenz verwendet und auf diese Weise

Einleitung
- 6 -
ebenfalls hohe Stereoselektivitäten erzielt. Die Carbonylverbindung wird in dieser Variante
mit tertiären Aminen enolisiert und durch das chirale Bor-Reagenz abgefangen. Hierdurch
wird am Enolat eine chirale Umgebung erzeugt, und durchläuft ähnlich der EVANS-Aldol-
reaktion, einen zyklischen, fixierten Übergangszustand. Ausschlaggebend für die entstehende
Diastereoselektivität ist die Verminderung der Wechselwirkung zwischen den Ipc-Resten und
dem R2-Enolat-Rest im Übergangszustand (Schema 5). Es kommt zur einer matched-
missmatched-Situation, wobei das matched-Paar 13a bevorzugt gebildet wird. Im Gegensatz
zur EVANS-Variante in der anti- sowie syn-Aldolprodukte generiert werden können, liefert die
PETERSON-Aldolreaktion, wegen der bevorzugten Ausbildung von Z-Enolaten, lediglich
R1R2
OB(-)-Ipc2 CH2Cl2, -15 °C
R3 H
O
O
BO
R1R3
R2
Me
Me
Me
Me
MeMe
12
13a matched
O
BO
R1
R3
Me
Me
Me
Me
MeMe
13b missmatched
R2
R2
O OH
R3
R1
14
Schema 5. Die PATERSON-Aldolreaktion. [16]
syn- konfigurierte Aldolprodukte. Ein weiterer Nachteil ist die limitierte Anwendbarkeit
dieser Reaktion, da sie auf ein kleines Eduktspektrum beschränkt ist. So resultieren
beispielsweise bei Methylketonen und sterisch gehinderten Aldehyden nur moderate
Stereoselektivitäten. Dennoch findet die PETERSON-Variante häufig Verwendung in der
Naturstoffsynthese, da hier mehrere Syntheseschritte wie das Einfügen und die Abspaltung
von Auxiliaren wegfallen.
1.2.2 Die MUKAIYAMA -Aldolreaktion
MUKAIYAMA et al. konnten 1973 zeigen, dass Silylenolether unter der Einwirkung von Lewis-
Säuren mit Aldehyden zu Aldolprodukten reagieren. [17] Seither ist sie eine häufig angewandte
Methode zur diastereoselektiven C-C-Verknüpfung in der Naturstoffsynthese. [18] Der genaue
Reaktionsablauf ist bislang nicht abschließend aufgeklärt. Angenommen wird jedoch, dass die

Einleitung
- 7 -
Reaktion nicht über einen zyklischen Übergangszustand verläuft und ihr keine
Transmetallierung zugrunde liegt. Vorliegende 1,2- sowie 1,3- asymmetrischer Induktionen
waren der Grund vielseitiger Diskussionen über einen möglichen azyklischen Übergangs-
zustand. [19] Durch Einwirkung von Chelatisierungsprozessen konnten diese Annahmen weiter
gestärkt werden. Gewöhnlich koordiniert die Lewis-Säure an der Carbonylverbindung und
leitet durch die Aktivierung den nucleophilen Angriff ein (Schema 6, 16). Dieses wird als
Aldehydaktivierung bezeichnet. Eine weitere Form der Aktivierung ist die Aktivierung des
Silylenolethers durch Übergangsmetalle wie Palladium oder Kupfer. Hier findet eine
Transmetallierung statt, wodurch ein delokalisierter Enolat-Palladium-Komplex 18 entsteht.
Dieser aktive Komplex ist ausreichend reaktiv, um im weiteren Reaktionsverlauf unaktivierte
Aldehyde nucleophil anzugreifen.
OTMS
15
TiCl4, RCHOCH2Cl2, -78 °C
PdCl2(P2L), AgOTf
DMF, H2O, RTRCHOR
OTiCl4
H
OTMSOH
R
OO Pd
PRPR
RH
O
OH
R
O
16 18
17 17
Schema 6. Allgemeiner Mechanismus der MUKAIYAMA-Aldolreaktion (links : Aldehyd-Aktivierung; rechts : Enolat-Aktivierung).
Anfängliche Studien zu enantioselektiven Varianten durch chirale Reagenzien wurden Mitte
der 80er Jahre veröffentlicht. Ähnlich wie bei der Aldolreaktion, wurden die ersten Versuche
über die Auxiliar-Methode mittels chiraler Silylketenacetalen durchgeführt. Ein Beispiel
hierfür ist die Variante nach GENNARI, in der durch Verwendung von N-Methyl-Ephedrin-
Derivaten hohe Enantioselektivitäten erzielt werden konnten (Schema 7). [20]
NMe2
Ph
O OTMS
19
TiCl4, CH2Cl2-78 °C, 60%
O
O OHNMe2
Ph
20 84% de
i-PrCHO,
Schema 7. Auxiliar kontrollierte MUKAIYAMA-Aldolreaktion nach GENNARI. [20]
Präparative Fortschritte in der Chemie führten ebenfalls zur Entwicklung chiraler Lewis-
Säuren. Im Gegensatz zu den reinen anorganischen Lewis-Säuren sind die Lewis-Säuren mit
einem organischen Anteil meist deutlich stärker hydrolyseempfindlich und können nur in
Einzelfällen gelagert werden, weswegen sie oftmals in situ generiert werden müssen. Ein
wichtiges Anwendungsgebiet der chiralen Lewis-Säuren ist die asymmetrische MUKAIYAMA -
Aldolreaktion. REETZ versuchte als einer der Ersten die MUKAIYAMA -Aldolreaktion durch
Einsatz von chiralem Rhodium-Komplex 22 stereoselektiv zu katalysieren (Schema 8). [21]

Einleitung
- 8 -
TMSO
MeO
PPh2
PPh2
Rh
Ph2P
Ph2P
ClO4-
5 mol%
PhCHO, CH2Cl2, RT, 75% MeO
O OH
Ph
23 12% ee21
22
Schema 8. MUKAIYAMA-Aldolreaktion mit chiralen Lewis-Säuren nach REETZ. [21] Obwohl anfänglich unbrauchbare Enantioselektivitäten erzielt wurden, konnten diese Studien
die klaren Vorteile der Stereokontrolle durch chirale Lewis-Säuren aufzeigen. So kann in
dieser Variante eine deutlich höhere Anzahl an Substraten verwendet werden. Inspiriert
durch diese Studien arbeiteten in den letzten zwei Jahrzenten viele renommierte
Arbeitsgruppen an der Entwicklung leistungsfähiger chiraler Lewis-Säure-Katalysatoren und
deren Anwendung in der MUKAIYAMA -Aldolreaktion. Ein breites Spektrum an
unterschiedlichen Methoden, die zu hohen Selektivitäten führen, war das Ergebnis dieser
Bemühungen. [22] Hierbei zählt die Titan-katalysierte MUKAIYAMA -Aldolreaktion nach
KECK [23] sicherlich zu den populärsten Beispielen, in der das chirale Binaphtyl-Derivat 25 zur
Stereokontrolle verwendet wird (Schema 9).
TMSO
t-BuS
O
OTi
PhCHO, 4Å MS
CH2Cl2, 0 °C 90%
20 mol%
24
25
t-BuS
O
Ph
OH
26 97% ee
Oi-Pr
Oi-Pr
Schema 9. Chirale Titan-Lewis-Säure von KECK in der asymmetrischen Mukaiyama-Reaktion. [23] Obwohl mittlerweile viele Methoden existieren, besteht immer noch ein großer Bedarf an
weiteren Entwicklungen, die einen effizienten und stereoselektiven Zugang zu
Polyketidbausteinen ermöglichen.
1.3 Die Vinyloge MUKAIYAMA -Aldolreaktion
1.3.1 Die Vinyloge Erweiterung der Aldolreaktion
In den vorangegangenen Kapiteln wurde die Relevanz der Polyketidsynthese für die
Entwicklung neuer potentieller Medikamente dargestellt. Stereoselektive Aldol- oder Aldol

Einleitung
- 9 -
ähnliche Reaktionen sind hierfür die wichtigsten Synthesemethoden. Einige der populärsten
Vertreter wurden in Kapitel 1.2 diskutiert. Diese führen jedoch nur zu 1,3-funktionalisierten
Fragmenten. Ständig verfeinerte Isolations- und Analysemethoden ermöglichen den Zugang
zu deutlich größeren und biologisch aktivieren Naturstoffen. Eine Totalsynthese solcher
Verbindungen über gewöhnliche 1,3-Funktionalisierung ist jedoch ein mühsames
Unterfangen. Deswegen ist die heutige Synthesechemie bemüht, neue effizientere Methoden
zu entwickeln, die den Zugang zu größeren Polyol-Fragmenten in äquivalenten Selektivitäten
gewährleisten.
1935 veröffentlichte FUSON das Konzept der Vinylogie. [24] Darin wird die Übertragung von
elektronischen Effekten durch ein konjugiertes π-System beschrieben. Resultierend kann
durch eine vinyloge Erweiterung, die nucleophile oder elektrophile Eigenschaft einer
funktionalen Gruppe erweitert und somit 1,5-funktionalisierte Synthesebausteine in einem
Schritt generiert werden. Dieses wird entsprechend als vinyloge Aldolreaktion bezeichnet.
Dabei resultieren abhängig vom verwendetem Enolat, δ-hydroxy-β-keto- oder δ-hydroxy-α,β-
ungesättigte Carbonylverbindungen (Schema 10).
O
R1R3CHO
R3
OH
R1
O
R2
O O
R1 R2R2R3
OH O O
R1
R2
γ -Hydroxy-α,β -ungesättige Carbonylverbindung
γ -Hydroxy-β -ketoCarbonylverbindung
R3CHO
Schema 10. FUSON‘s Prinzip der Vinylogie an Dienolaten (rechts) und Dienolethern (links).
Besonders δ-hydroxy-α,β-ungesättigte Carbonylverbindungen stellen interessante Inter-
mediate für die Synthese komplexer Polyketide dar. Die unterschiedlichen funktionellen
Gruppen in diesen Bausteinen erlauben eine große Anzahl an nachfolgenden Trans-
formationen (Schema 11). So können durch einfache Dihydroxylierung, Epoxidierung oder
weiteren Methoden in kurzen Schritten größere 1,3-Polyole mit beliebigen Substitutions-
mustern synthetisiert werden. Des Weiteren können mit diesen Bausteinen ebenfalls sehr
leicht Tetrahydropyranstrukturen konstruiert werden.
Für die Synthese von δ-hydroxy-α,β-ungesättigte Carbonylverbindungen wurden zuvor
mehrere Stufen benötigt. Eine der häufig verwendeten Routen hierfür ist die Ozonolyse von
Homoallylalkoholen mit anschließender Olefinierung (Schema 12). Eine modernere Variante
stellt die Kreuzmetathese zwischen Acrylaten mit Homoallylalkoholen dar. Die Metathese
zwischen diesen beiden Substraten ist jedoch nur bedingt einsetzbar und erfordert
substratspezifische Optimierungen. So steht die vinyloge Aldolreaktion als einzige und

Einleitung
- 10 -
R3
OH
R1
O
R2
Epoxidierung
R3
OH
R1
O
R2
O
R3
OH
R1
O
R2
OH
OH
Dihydroxylierung
R3
OH
R1
O
R2
OH
R4
Epoxidierung + Cuprat Addition
R3
O
R1
O
R2
O
R
Acetalisierung
R3
OH
R1
O
R2
Hydrierung
R3
OH
R1
O
R2
R4
Cuprat Addition
R3
R3O
OHR2
OH
R1
O
Epoxidierung + Intramolekularer Nucleophile Angriff
Schema 11. Mögliche Transformationen von γ-Hydroxy-α,β-ungesättigten Carbonylverbindungen. effizienteste Methode zur Darstellung von δ-hydroxy-α,β-ungesättigten Carbonylver-
bindungen in einem Syntheseschritt.
O
R2R1
OH
R2R1
OH
R3
O
R1
O
H
1) Crotylborierung
2) Ozonolyse
3) Olefinierung
R1
OH
R2
1) Crotylborierung 2) Metathese
1) vinyloge Aldolreaktion
Schema 12. Moderne Routen zur Synthese von δ-hydroxy-α,β-ungesättigte Carbonylverbindungen. Trotz ihrer Vorteile ist die stereoselektive Kontrolle dieser Reaktionen eine deutlich größere
Problemstellung als in gewöhnlichen Aldolreaktionen. Neben den üblichen Heraus-
forderungen wie Enantio- oder Diastereoselektivität, muss hier zusätzlich die
Regioselektivität kontrolliert werden. So kann ein nucleophiler Angriff an der α-Position oder
γ-Position erfolgen (Schema 13). SCHLESSINGER [25] und RHATKE
[26] veranschaulichten in
ihren Studien bezüglich der Regioselektivität von Metalldienolaten mit Alkylhalogeniden,
eine starke Affinität für die unerwünschte α-Alkylierung.
Durch Verwendung von Silyldienolethern in einer MUKAIYAMA -Typ Reaktion wird die
Regioselektivität zugunsten des γ-Produkts verschoben. Dieses wird dementsprechend als
vinyloge MUKAIYAMA -Aldolreaktion (VMAR) bezeichnet. Diese Tatsache kann durch

Einleitung
- 11 -
R1
OH
R2
OO
R1αγ
R2CHO
R2
OOH
R1
γ -Produkt
α -ProduktMeX
O
R1
Me
Gemisch
Hauptprodukt
M
Schema 13. Regioselektive Herausforderungen durch die vinyloge Erweiterung von Enolaten.
die Betrachtung der HOMO-Koeffizienten der Molekülorbitale verstanden werden (Abb. 2).
Hiernach weisen Metalldienolate am α-C-Atom hohe HOMO-Werte auf, wodurch ein
nucleophiler Angriff von dieser Position bevorzugt wird. Silyldienolether hingegen weisen
auf ein HOMO-Maximum am γ-C-Atom hin, was zur erwünschten γ-Alkylierung führt. [27, 28a]
OMe
OLi
OMe
OTMS0.22
0.23
0.09
0.30
0.130.04
0.310.28
27 28
Abbildung 2. Vergleich der HOMO-Koeffizienten vom Metalldienolat 27 und Silyldienolether 28. [28a]
Angeregt durch die besseren Regioselektivitäten wurden seit Anfang der 90er Jahre die ersten
Studien zur stereoselektiven Synthese von δ-hydroxy-α,β-ungesättigten Carbonyl-
verbindungen über die vinylogen MUKAIYAMA -Aldolreaktion veröffentlicht. [28] Viele
verschiedene Ansätze zur Stereokontrolle über Auxiliare oder über chirale Lewis-Säuren,
wurden hierfür untersucht. Diese weisen zwar hohe Stereoselektivitäten auf, sind bisher nur
für eine beschränkte Anzahl von Substraten anwendbar. Als besondere Herausforderung gilt
die VMAR mit Dienolethern, speziell terminal substituierten, da hier in den meisten
Versuchen nur geringe Stereoselektivitäten erzielt werden konnten. Gerade diese Substrate
würden jedoch eine enorme Bereicherung für die Polyketidsynthese darstellen.
Im weiteren Verlauf dieser Arbeit sollen die einzigen vorhandenen Methoden dargestellt
werden, die eine stereoselektive Addition mit terminal substituierten Dienolaten ermöglichen.
1.3.2 Relevante Varianten der VMAR
1.3.2.1 Die CAMPAGNE-Variante
Als einer der ersten Arbeitsgruppen veröffentlichten CAMPAGNE et al. ihre Ergebnisse zur
asymmetrischen VMAR mit γ-methyl substituierten Dienolethern. [29] Dabei wurden chirale

Einleitung
- 12 -
Kupfer-Komplexe, welche zuvor von CARREIRA [30] entwickelt und bereits erfolgreich an der
VMAR mit Dienolaten untersucht wurden, eingesetzt. Sie katalysierten hiermit die VMAR
am Silyl-Ketenacetal 29 in hohen Stereoselektivitäten. Durch diese Enolat-Aktivierte Variante
konnte sowohl das erwünschte VMAR-Produkt 31, als auch das entsprechende ungesättigte
Lacton 30 isoliert werden, wobei nur das Lacton in akzeptablen Stereoselektivitäten gebildet
wird (Tab. 1). Das Verhältnis beider Produkte sowie die Enantioselektivitäten hängen dabei
Tabelle 1. VMAR nach CAMPAGNE. [29]
OEt
OTMS O
O
R OEt
O
R
OTMS
+
CuF(S)-tolBINAP10 mol% THF, RT
RCHO
29 30 31
Nr. Aldehyd Ausbeute (%) 30+31 Verhältnis 30/31 ee (%) 30
1 Benzaldehyd 85 86/14 87
2 2-Napthaldehyd 95 80/20 85 3 (E)-Zimtaldehyd 60 70/30 82
4 2-Furfural 60 50/50 86 stark vom verwendeten Aldehyd ab. In den zahlreichen Beispielen wurde jedoch das
Lacton 30 als Hauptprodukt isoliert. Das resultierende Produktgemisch lässt sich durch den
postulierten Mechanismus erklären, der allerdings noch nicht vollständig aufgeklärt ist
(Schema 14). Hiernach bildet der chirale Kupfer-Komplex mit dem Ketenacetal 29 das
Kupfer-Enolat 32. Das aktivierte Enolat 32 reagiert mit dem zugesetzten Aldehyd,
wodurch es zur Generierung des Kupfer-Alkoxids 33 kommt, welches weiter zum
offenkettigen γ-VMAR- Produkt 31 reagiert. Das Kupfer-Alkoxid 33 kann seinerseits
in einer Folgereaktion mit dem Ketenacetal 29 eine Transmetallierung eingehen,
R H
O
OEt
OTMS
R
OOEt
OCuLn
29
3233
R
O
O
30
[CuF2-(S)-tol-binap]OEt
OCuLn
29 31
Schema 14. Vorgeschlagener Mechanismus für die Kupfer-Katalysierten VMAR nach CAMPAGNE. [29f]

Einleitung
- 13 -
wodurch es zur spontanen Zyklisierung kommt, indem das ungesättigte Lacton 30 entsteht
und das aktivierte Kupfer-Enolat 32 regeneriert wird. Hohe Stereoselektivitäten und
Ausbeuten, die mit dem CAMPAGNE-Protokoll erzielt werden können, etablierten diese zur
wichtigen Methode für die angewandte Naturstoffchemie, wodurch sie bereits in einigen
Totalsynthesen Anwendung fand. [29f] Das Anwendungsspektrum dieser Methode ist jedoch
aufgrund der entstehenden Produktgemische stark begrenzt.
1.3.2.2 Die KALESSE-Variante
In vorherigen Arbeiten wurde bereits die VMAR in unserem Arbeitskreis ausführlich
untersucht. Inspiriert durch die Forschungsergebnisse von PATERSON et al. [31], konnte eine
weitere Variante zur Substratkontrollierten VMAR mit von Estern abgeleiteten Ketenacetalen
etabliert werden. So konnten in Studien zur Totalsynthese unterschiedlicher Polyketide [32]
erfolgreich gezeigt werden, dass durch Einwirkung von Tris(pentafluorophenyl)boran (TPPB)
als sterisch anspruchsvolle Lewis-Säure im Gegensatz zur Bortrifluoretherat oder weiteren
Titan-Lewis-Säuren deutlich höhere Ausbeuten und Stereoselektivitäten resultieren
(Schema 15). [33] In sämtlichen Beispielen mit den Ketenacetalen 35, 38 und 41 konnte das
Felkin-Produkt als Hauptprodukt synthetisiert werden. Dabei führt wie erwartet das 3,4-Z-
Ketenacetal 38 zum syn-Felkin Produkt.
OTBSO OHTBSO
OMe
O
36 >95% de
B(C6F5)
i-PrOH, Et2O −78 °C, 67%
OMe
OTBS
3534
OTBSO OHTBSO
OMe
O
39 >95% de
B(C6F5)
i-PrOH, Et2O −78 °C, 76%
OMe
OTBS
3837
OH
OMe
O
42 50% de
B(C6F5)
i-PrOH, Et2O −78 °C, 69%
OMe
OTBS
41
O
40
OTBSO OHTBSO
OMe
O
36 50% de
BF3.Et2O
i-PrOH, Et2O −78 °C, 59%
OMe
OTBS
3534
Schema 15. VMAR mit TPPB nach KALESSE. [33]
Essentiell für diese Variante ist dabei der Zusatz von Additiven wie Isopropanol.
Veröffentlichte Arbeiten von CARREIRA konnten zeigen, dass Silylkationen bereits
ausreichende Lewis-Säure Eigenschaften aufweisen um Aldehyde für MUKAIYAMA - oder
MUKAIYAMA -ähnliche Reaktionen zu aktivieren. [34] Dieses führt schließlich dazu, dass in
Einleitung
- 14 -
einigen MUKAIYAMA -Reaktionen eine durch freie Silylkationen hervorgerufene Neben-
reaktion eintritt, welche in deutlich geringeren Stereoselektivitäten verläuft (Schema 16).
OMe
OTBS
38
OTBS
OMe
O
46 <60% de
B(C6F5)3
O
40
O
43
R3Si+
SiR3+
O
44
B(C6F5)3
OMe
OTBS
38
OH
OMe
O
45 >95% de
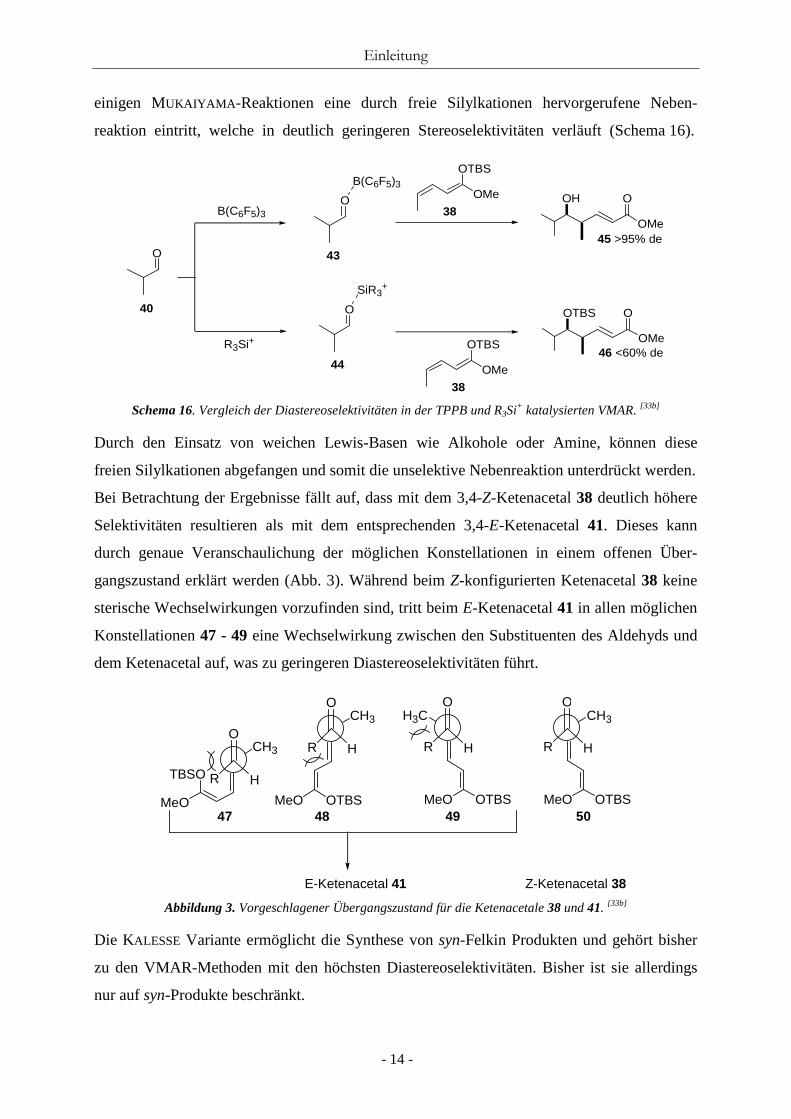
Schema 16. Vergleich der Diastereoselektivitäten in der TPPB und R3Si+ katalysierten VMAR. [33b]
Durch den Einsatz von weichen Lewis-Basen wie Alkohole oder Amine, können diese
freien Silylkationen abgefangen und somit die unselektive Nebenreaktion unterdrückt werden.
Bei Betrachtung der Ergebnisse fällt auf, dass mit dem 3,4-Z-Ketenacetal 38 deutlich höhere
Selektivitäten resultieren als mit dem entsprechenden 3,4-E-Ketenacetal 41. Dieses kann
durch genaue Veranschaulichung der möglichen Konstellationen in einem offenen Über-
gangszustand erklärt werden (Abb. 3). Während beim Z-konfigurierten Ketenacetal 38 keine
sterische Wechselwirkungen vorzufinden sind, tritt beim E-Ketenacetal 41 in allen möglichen
Konstellationen 47 - 49 eine Wechselwirkung zwischen den Substituenten des Aldehyds und
dem Ketenacetal auf, was zu geringeren Diastereoselektivitäten führt.
CH3
O
HR
MeO
TBSO
CH3
O
HR
MeO OTBS
H3CO
HR
OTBSMeO
CH3
O
HR
OTBSMeO
E-Ketenacetal 41 Z-Ketenacetal 38
47 48 49 50
Abbildung 3. Vorgeschlagener Übergangszustand für die Ketenacetale 38 und 41. [33b] Die KALESSE Variante ermöglicht die Synthese von syn-Felkin Produkten und gehört bisher
zu den VMAR-Methoden mit den höchsten Diastereoselektivitäten. Bisher ist sie allerdings
nur auf syn-Produkte beschränkt.

Einleitung
- 15 -
1.3.2.3 Die DENMARK-Variante
Als einer der wertvollsten Arbeiten zur vinylogen MUKAIYAMA -Aldolreaktion sind die
Studien von DENMARK et al. anzusehen. Hier wurden chirale Phosphoramide zur Enolat-
aktivierung verwendet. [35] Das dimere Phosphoramid wurde mit Siliziumtetrachlorid
umgesetzt, was zu dem pentakoordinierten Katalysator 52 (Schema 17) führt. Durch
Interaktion der Lewis-Base mit Silizium, ist die Aktivität der Lewis-Säure entsprechend nach
GUTMANN [36] erhöht, wodurch höhere Ausbeuten und Stereoselektivitäten resultieren.
DENMARK untersuchte den entwickelten Katalaysator auf seine Stereoselektivität an
unterschiedlichen Diensystemen sowie Aldehyden. Die in Tabelle 2 gezeigten Ergebnisse
verdeutlichen, dass mit dem vom DENMARK entwickelten Katalysator sogar mit
γ-methylierten Ketenacetalen hohe Dia- und Enantioselektivitäten resultieren.
Si
Cl
ClClN
N
Me
MeP
O
NMe
NN
Me
Me
PO
NMe
52
NN
Me
Me
PO
NMe
NMe
51
PN
ONMe
Me
SiCl4
Cl
Schema 17. Penta-koordinierter Katalysator von DENMARK. [35]
In sämtlichen Beispielen wurde hierbei das anti-konfigurierte VMAR-Produkt isoliert.
Allerdings ist das Anwendungsspektrum dieser Methode nur auf ungesättigte Aldehyde
beschränkt. Bei Verwendung von aliphatischen Aldehyden kann mit der DENMARK-Variante
keine Produktbildung beobachtet werden (Tab. 2, Nr. 3, 6, 8).
Tabelle 2. VMAR nach DENMARK. [35]
R1 H
O
OR2
OTBS
OR2
O
R1
OH
+(R,R)-52
CH2Cl2, -78 °C
Nr. Aldehyd R² Ausbeute (%) de (%) ee (%)
1 Benzaldehyd Ot-Bu 85 82 87
2 (E)-Zimtaldehyd Ot-Bu 95 60 85 3 dihydro-Zimtaldehyd Ot-Bu - - - 4 Benzaldehyd t-Bu 92 99 89 5 (E)-Zimtaldehyd t-Bu 71 99 82 6 dihydro-Zimtaldehyd t-Bu - - - 7 Benzaldehyd ON(CH2CH2)2O 98 94 78 8 dihydro-Zimtaldehyd ON(CH2CH2)2O - - -

Einleitung
- 16 -
Obwohl diese Variante nur auf ungesättigte Aldehyde beschränkt ist, fand sie Verwendung in
einigen Totalsynthesen von Polyketiden. [37]
1.3.2.4 Die KOBAYASHI-Variante
Im Jahr 2004 konnte KOBAYASHI et al. von einer weiteren bedeutenden Möglichkeit zur
stereoselektiven Reaktionsführung der VMAR berichten. [38] Chirale N,O-Silyl-Ketenacetale
reagieren hier unter Einwirkung von Lewis-Säuren mit Aldehyden durch eine 1,7- oder 1,6,7-
Induktion in hohen Stereoselektivitäten. Als chirale Quelle wird das bekannte EVANS-Auxiliar
(siehe Kapitel 1.2.1.1) verwendet. Einige Beispielreaktionen sind in Schema 18 gezeigt. Hier
wird ersichtlich, dass mit α-methylierten Ketenacetalen 53 und 55 hohe Stereoselektivitäten
erzielbar sind. Die VMAR mit α-unsubstituierten Ketenacetal 58 hingegen, liefert mit dieser
Methode nur moderate Stereoselektivitäten.
N O
OTBSO
O TiCl4
CH2Cl2, -78 °C, 99%
OH O
N
O
40 55 56 >99% de
N O
OTBSO
TiCl4
CH2Cl2, -78 °C, 99%
OH O
N
O
58 59 60% de
O
57
N O
OTBSO
O TiCl4
CH2Cl2, -78 °C, 99%
OH O
N
O
40 53 54 >99% de
Schema 18. Auxiliar-Kontrollierte VMAR nach KOBAYASHI. [38]
Diese Beobachtung kann anhand von KOBAYASHI’s postuliertem offenen Übergangszustand
begründet werden. Bei Betrachtung der Anordnung in der NEWMAN-Projektion ist zu
erkennen, dass im Fall des α-unsubstituiertem Ketenacetals 58 der Aldehyd-Rest
entgegengesetzt zu dem chiralen Auxiliar positioniert ist (Abb. 4, 61). Hierdurch ist eine
chirale Induktion nur noch bedingt effektiv. Eine Methylgruppe in α-Position führt durch
Verminderung der sterischen Wechselwirkung zu einer Anordnung, in dem der Aldehyd und
das Auxiliar-Rest auf der gleichen Seite stehen 60. Dieses ermöglicht eine optimale Stereo-
kontrolle durch das Auxiliar und führt so zu hohen Stereoselektivitäten.

Einleitung
- 17 -
MeO
HR
OTBSAuxiliarN
60
Me
TiCl4
H HO
RH
61
TiCl4
Me
OTBSAuxiliarN
Abbildung 4. Postulierter offener Übergangszustand für die Ketenacetale 55 und 58. [38a]
Die KOBAYASHI-Variante gehört unumstritten zur wichtigsten Methode, um α-methylierte
Ketenacetale stereoselektiv in einer VMAR umzusetzen. KOBAYASHI wendete diese Methode
bereits erfolgreich in einigen seiner Naturstoffsynthesen an und konnte dadurch nochmals das
Potential seiner Methode untermauern.

Aufgabenstellung
- 18 -
2. AUFGABENSTELLUNG
Das steigende Bedürfnis nach neuen potentiellen Wirkstoffen stellt insbesondere die Poly-
ketidsynthese in den Vordergrund, weswegen ein enormer Bedarf an neuen Synthese-
methoden besteht, welche einen schnellen und effizienten Zugang zu größeren
Polyketidbausteinen ermöglichen. In den vorherigen Kapiteln dieser Arbeit wurde die
steigende Bedeutung der vinylogen MUKAIYAMA -Aldolreaktion (VMAR) für die Natur-
stoffchemie diskutiert, welche eine hervorragende Möglichkeit zur Synthese von γ-Hydroxy-
α,β-ungesättigten Carbonylverbindungen in einem Syntheseschritt darstellt. Diese hoch
funktionalisierten Bausteine spielen in der Polyketidsynthese eine wichtige Rolle, da sie durch
einfache Transformationen schnell in größere Polyol-Einheiten überführt werden können.
Angeregt durch die synthetische Bedeutung dieser Methode, haben viele Forschungsgruppen
versucht, enantioselektive Varianten dieser Reaktion zu etablieren. Die hohen stereoselektiven
Herausforderungen der VMAR waren bisher die Hauptgründe, dass nur wenige
asymmetrische Methoden mit begrenzten Anwendungsmöglichkeiten bekannt sind. Speziell
die enantioselektive Reaktionsführung mit γ-alkylierten Ketenacetalen erwies sich hier als
besonders schwierig. Die in Kapitel 1.3 vorgestellten Methoden sind die einzigen, die eine
hoch enantioselektive VMAR mit γ-methylierten Ketenacetalen ermöglichen, die aber
hauptsächlich nur zu anti-konfigurierten Produkten führen. Zudem ist das Anwendungs-
spektrum dieser Methoden nur auf eine geringe Anzahl von Substraten beschränkt. Besonders
die aliphatischen Aldehyde sind hier nur geringfügig einsetzbar. Allerdings sind gerade diese
Substrate essentiell für die Polyketidsynthese. Wie bedeutend die VMAR mit aliphatischen
Aldehyden ist, soll an einigen Polyketid-Antibiotika verdeutlicht werden (Abb. 5).
Die hervorgehobenen Bausteine könnten leicht durch eine syn-selektive VMAR ausgehend
von Isobutyraldehyd (40) und den Ketenacetalen 38 oder 41 synthetisiert werden, welche
jedoch mit den bestehenden Methoden nicht in brauchbaren Enantioselektivitäten
durchgeführt werden kann.
Im Rahmen der vorliegenden Arbeit sollte eine weitere asymmetrische VMAR-Variante für
γ-methylierte Ketenacetale etabliert werden, die den Einsatz von aliphatischen Aldehyden
ermöglicht. Primärziel war dabei die Entwicklung einer syn-selektiven VMAR. Dieses sollte
unter Verwendung verschiedener literaturbekannter, sowie neuer chiraler Lewis-Säuren
erreicht werden.
Als Ausgangsverbindungen für die Studien zur asymmetrischen VMAR wurden Isobutyr-

Aufgabenstellung
- 19 -
O OH
O
OH OH OH OH OH OH
HO
O
O
OH OH OH OH OH OH
O
OH
OH
OH
O
O
O
HN
N
O
OH
N
O
O
Virginiamycin M1 63Mycotoxin A 62
Roflamycoin 64
O
O
O
HN
HN
O
OH
N
O
Madumycin II 65
OH
OH
OMe
O
O
OMe
OTBS
+
VMAR
40
38
66
oder OMe
OTBS
41
Abbildung 5. VMAR-Bausteine in unterschiedlichen Polyketid-Antibiotika.
aldehyd (40) und die γ-methylierten Ketenacetale 38 und 41 gewählt, da hier bereits das
entsprechende VMAR-Produkt 66 einen interessanten Baustein für eine Vielzahl von
Naturstoffen darstellt. Die Bestimmung der Enantiomerenüberschüsse der isolierten VMAR-
Produkte sollten mittels chiraler Lanthanoid Shift-Reagenzien erfolgen und diese zusätzlich
mit gängigen instrumentellen Methoden abgeglichen werden.
Um die VMAR in erwünschten Enantioselektivitäten zu kontrollieren, sollten neben
verschiedenen chiralen Lewis-Säuren auch Reaktionsparameter wie Temperatur, als auch
Lösungsmittel variiert und zusätzlich Reagenzien zum Abfangen der Siliziumkationspezies
verwendet werden. Zudem sollten neben den γ-methylierten Ketenacetalen 38 und 41 auch
weitere Ketenacetale wie das γ-unsubstituierte und das α-methylierte Ketenacetal in die
Studien miteinbezogen werden.
Nach einer erfolgreichen Entwicklung der syn-selektiven VMAR mit hohen Enantio-
selektivitäten, sollte diese anschließend in der Synthese der in Abbildung 5 dargestellten
Polyketide angewendet werden.

Studien zur Entwicklung neuer VMAR-Methoden
- 20 -
3. STUDIEN ZUR ENTWICKLUNG NEUER VMAR-M ETHODEN
3.1 Biphenyl-Borane als chirale Lewis-Säuren für die VMAR
Die Untersuchung der asymmetrischen vinylogen MUKAIYAMA -Aldolreaktion begann mit der
Synthese der benötigten 3,4-Z- bzw. 3,4-E-Ketenacetale 38 und 41. Ausgehend von
kommerziell erhältlichen trans-3- bzw. trans-2-Pentensäuremethylester (67) und (68)
konnten diese durch Deprotonierung mit Lithiumdiisopropylamin in Gegenwart von
Dimethylpropylenharnstoff (DMPU) und anschließendes Abfangen vom Esterenolat mit
TBS-Chlorid in hohen Ausbeuten synthetisiert werden (Schema 19). [39] DMPU dient hierbei
als Chelatbildner zur Freisetzung des Esterenolat-Anions, wodurch erst die Silylierung
ermöglicht wird. Mit dieser Methode konnten ebenfalls für das 3,4-Z-Ketenacetal 38 hohe
Isomerenverhältnisse von >20:1 bezüglich der 3,4-Z- Konfiguration erzielt werden.
OMe
O
67
OMe
O
68
LDA, DMPUTBSCl
THF, -78 °C
LDA, DMPUTBSCl
THF, -78 °C
OMe
OTBS
41 93%
OMe
OTBS
38 89%
Schema 19. Darstellung der 3,4-E- und 3,4-Z-Ketenacetale 38 und 41. Nachdem die benötigten Ketenacetale erfolgreich synthetisiert wurden, sollten anfänglich
chirale Triarylborane zur chiralen Induktion eingesetzt werden. Triarylborane wurden in
unserem Arbeitskreis bereits erfolgreich in der diastereoselektiven VMAR eingeführt (siehe
Kapitel 1.3.2.2). Die positiven Ergebnisse die mit Triarylboranen erlangt wurden, führten zu
der Annahme, dass chiral modifizierte Arylborane ebenfalls fähig sind, die VMAR
enantioselektiv zu kontrollieren.
Als einer der ersten Arbeitsgruppen berichteten PIERS et al. über chirale Arylborane in der
asymmetrischen Synthese. [40] Sie entwickelten unterschiedliche axial chirale mono-
1,1’-Binaphtyl substituierte Borane. Diese wurden in einer Allylstannylierung eingesetzt,
welche eine verwandte Form der VMAR darstellt (Tab. 3). Obwohl nur geringe
Stereoselektivitäten erzielt wurden, konnten diese Studien zeigen, dass chirale Arylborane
durchaus fähig sind, Aldol-ähnliche Reaktionen stereoselektiv zu steuern. Wie auch
schon in den Untersuchungen vom Arbeitskreis KALESSE beobachtet, führten sterisch

Studien zur Entwicklung neuer VMAR-Methoden
- 21 -
anspruchsvollere Borane zu höheren Stereoselektivitäten.
Tabelle 3. Katalytische asymmetrische Allylstannylierung nach PIERS.
[40]
OSnBu3
LS (5 mol%)
CH2Cl2, -78 °C
OH
73
7475
BF2 B(C6F5)2 BF2Me
B(C6F5)2Me
69 70 71 72
Nr. LS Ausbeute (%) ee (%)
1 69 90 4
2 70 92 8 3 71 93 15
4 72 90 26 Auf Grundlage dieser Veröffentlichungen sollten im Rahmen dieser Arbeit weitere axial
chirale Arylborane synthetisiert und in der VMAR getestet werden. Im Gegensatz zu PIERS
sollten statt mono-Biaryl di- oder tri-Biaryl substituierte Borane synthetisiert werden, aus
denen im Idealfall höhere Enantioselektivitäten resultieren könnten. Als Aryl-Gruppe sollten
anfänglich Biphenyl-Systeme konstruiert werden. Die erste erfolgreiche Synthese von
achiralen Tri-Biphenyl Boranen wurde von WITTIG aus einfachen Bipehnylhalogeniden
beschrieben. [41] Die Einführung der axialen Chiralität in Biaryl-Systemen erfolgt gewöhnlich
über Umkristallisation der entsprechenden Biarylamine mit chiralen Substanzen wie z.B.
Weinsäure. Die Darstellung des Biarylamins beginnt mit der Synthese der jeweiligen
Arylamine, in diesem Fall wird vom kommerziell erhältlichen 3-Nitrotoluol (76) ausgegangen
(Schema 20). Dieses wird an Position zwei iodiert und das erhaltene Iodid 77 in einer
ULLMANN -Kupplung umgesetzt. Anschließende Reduzierung der Nitro-Gruppen ergibt das
erwünschte Biphenylamin 78 in hohen Ausbeuten. Das racemische Biphenylamin 78 wurde
im nächsten Schritt in die optisch aktiven Vertreter überführt. Die Enantiomerentrennung
erfolgte nach mehrmaliger Umkristallisation von Biphenylamin 78 aus Weinsäure in heißem
Ethanol. [42] Hier konnten erst nach acht- bis neunmaligen Umkristallisationsversuchen der
Literaturdrehwert erreicht werden, was zu einem deutlichen Substanzverlust führte. Daher
wurde im weiteren Verlauf der Studien auf die Enantiomerentrennung verzichtet und die
Synthese mit achiralem Bausteinen fortgeführt. Anschließend erfolgt durch Diazotierung und
Substitution mit Kaliumiodid die Synthese vom achiralem Biphenyliodid 79. Nachdem der
Grundbaustein synthetisiert wurde, konnte die Synthese zu neuen Biphenyl-Boranen

Studien zur Entwicklung neuer VMAR-Methoden
- 22 -
NO2
76
a) H2SO4, NaNO3 0 °C
b) KI, RT 93%
NO2
77
Ia) Cu, DMF
160 °CNH2
78
b) H2, Pd/C 89%
NH2
(+)-WeinsäureEtOH 4%
NH2
NH2 a) H2SO4, NaNO3 0 °C
b) KI, RT 67%
I
79
Ia) n-BuLi, MeOH -78 °C
b) HCl
80
I
(+)-78
Schema 20. Darstellung vom Biphenyl-Baustein für neue Triaryl-Borane.
nach dem WITTIG-Protokoll erprobt werden. Das Iodid 80, wurde hierfür in den
entsprechenden Grignard überführt und mit Bortrifluoretherat zu dem resultierenden Tri-
Biphenyl-Boran 81 umgesetzt (Schema 21). Neben dem erwünschten Tri-substituirten Boran,
traten auch die mono- bzw. di-substituierte Borane als Nebenprodukte auf, so dass eine
Aufreinigung durch Umkristallisation notwendig wurde. Obwohl von WITTIG beschrieben,
konnte das Boran 81 durch Umkristallisation in Hexan nicht im erwünschten Reinheitsgrad
erhalten werden. Weitere Versuche zur Umkristallisation in unterschiedlichen Lösungsmitteln
blieben hier zunächst erfolglos. Schließlich konnte durch Sublimation und anschließender
Umkristallisation in Hexan die Substanz in befriedigendem Reinheitsgrad erhalten werden.
80
Ia) Mg, Et2Ob) BF3
.Et2O, Toluol
120 °C 38%3 B
81 Schema 21. Darstellung vom Tri-5,5’-Methyl-Biphenyl-Boran 81.
Nachdem eine erfolgreiche Route für Biphenyl-Borane erarbeitet wurde, sollten verschiedene
Substitutionsmuster synthetisiert werden. Ausgehend von den Biphenyl-Bausteinen 79 und 80
war die Darstellung der Borane in Abbildung 6 das nächste Syntheseziel, wobei ähnliche
Strukturen aus den Arbeiten von REETZ bekannt sind. [43] Allerdings konnten die gewünschten
Studien zur Entwicklung neuer VMAR-Methoden
- 23 -
Produkte weder nach WITTIG oder nach REETZ durch Verwendung von Dichlorphenylboran
bzw. Dicyclohexylchlorboran oder Bortribromid dargestellt werden. Ferner wurden in
sämtlichen Versuchen Zersetzungsprodukte beobachtet.
BB
BrBBBr2
82 83 84 85 Abbildung 6. Weitere Potentielle Arylborane für die VMAR.
Nachdem die Versuche, weitere Borane zu synthetisieren erfolglos blieben, wurde das einzig
synthetisierte Boran 81 an der VMAR untersucht (Tab. 4). Hierbei wurden die beiden
dargestellten Ketenacetale 38 und 41 (KA) durch Umsetzung mit Isobutyraldehyd (40)
Tabelle 4. Studien zur asymmetrische VMAR mit Tri-5,5’-Methyl-Bipheny-Boran 81.
O OH
40 66
OMe
OTBS
OMe
O
B
81R1
R2
R1 = H, R2 = Me 38R1 = Me, R2 = H 41
Nr. KA Lsg konz (mmol/mL) eq (81) T (°C) Ausbeute (%) 1 41 Et2O 0.2 0.2 -78 - 2 38 Et2O 0.2 0.2 -78 - 3 41 Et2O 0.5 0.2 -78 - 4 38 Et2O 0.5 0.2 -78 - 5 41 Et2O 0.2 0.2 -78 - 6 38 Et2O 0.2 0.2 -78 - 7 41 Et2O 0.5 1.0 -78 - 8 38 Et2O 0.5 1.0 -78 - 9 41 Et2O 1.0 1.0 -40 - 10 41 Et2O 0.5 1.0 -25 - 11 41 Et2O 0.5 1.0 -93 - 12 38 CH2Cl2 0.5 0.5 -78 - 13 38 CH2Cl2 0.5 0.5 -93 - 14a 41 Et2O 0.2 0.5 -78 - 15 41 THF 0.5 0.5 -78 - 16b 38 Et2O 0.5 0.5 -78 - 17c 38 Et2O 0.5 0.5 -78 -
a) Reaktion mit Benzaldehyd (73). b) Aldehyd und KA über 30 min zur LS zugetropft. c) LS über 30 min zur Reaktion zugetropft.

Studien zur Entwicklung neuer VMAR-Methoden
- 24 -
getestet. Die Testreaktionen wurden analog der TPPB-katalysierten VMAR durchgeführt,
indem der Aldehyd und das Boran vorgelegt und langsam das Ketenacetal hinzugetropft wird.
Die ersten Reaktionen blieben erfolglos und es konnte keine Produktbildung nachgewiesen
werden. Auch durch die Erhöhung der Konzentrationen und der Äquivalenten des Borans
führte nicht zu einer Produktbildung. Ebenso wenig konnte das gewünschte Produkt nicht
durch Variation des Lösungsmittels und der Reaktionsbedingungen erhalten werden (Tab. 4,
Nr. 12-17). Ein Problem bei der Umsatzkontrolle war, dass beide Startmaterialien nicht per
Dünnschichtchromatographie detektiert werden konnte. Daher konnten Ursachenbegründun-
gen wie Substratzersetzungen oder Nebenreaktionen nicht ohne weiteres getroffen werden.
Testreaktionen mit detektierbarem Benzaldehyd (73) zeigte zumindest, dass der Aldehyd
während der Reaktion nicht Aufgebraucht wird (Tab. 4, Nr. 14).
Da die Versuche mit Tri(5,5’-methyl-biphenyl)Boran 81 zu keiner Produktbildung führten,
wurde die Synthese von weiteren Biphenyl-Boranen sowie ihre Anwendung in der VMAR
vorläufig eingestellt.
3.2 BINOL-Liganden in der asymmetrischen VMAR
3.2.1 3,3’-substituierte BINOL-Liganden
Seit ihrer erstmaligen Anwendung Anfang der 80er Jahre durch NOYORI, [44] gehören die axial
chiralen Binaphthole (BINOL) zu den wichtigsten chiralen Liganden für die asymmetrische
Synthese. [45] Aufgrund der einfachen Zugänglichkeit und ihrer vielfältigen Einsatz-
möglichkeiten in unterschiedlichen enantioselektiven Reaktionen, hat ihre Anwendung
eine schnelle Entwicklung genommen. So erstreckt sich ihr Anwendungsspektrum
von enantioselektiven Cyclopropanierungen-[46] über DIELS-ALDER-[47] und MUKAIYAMA -
Aldolreaktionen. [48]
Infolgedessen fanden die BINOL-Liganden auch Verwendung in der VMAR. Umfangreiche
Studien zur asymmetrischen VMAR von SATO zeigten, dass sich der Titan-(VI)-BINOL
Komplex 25, welcher zuvor von KECK et al. entwickelt und bereits in MUKAIYAMA -
Aldolreaktionen Verwendung fand (siehe Kapitel 1.2.2), sich durch seine hohe
Regioselektivität auszeichnet (Schema 22, oben). [49] In Reaktionen mit Benzaldehyd (73)
und dem Dienolat 86 unter Einwirkung des BINOL-Katalysators 25 konnte das γ-VMAR-
Produkt 87 in hohen Enantioselektivitäten isoliert werden. Des Weiteren wurde im Rahmen
der Diplomarbeit von HAßFELD dieser BINOL-Komplex in der asymmetrischen VMAR mit
unterschiedlichen Ketenacetalen eingesetzt. [50] Mit dem γ-unsubstituierten Ketenacetal 35

Studien zur Entwicklung neuer VMAR-Methoden
- 25 -
wurde ein Enantiomerenüberschuss von 80% ee für das γ-VMAR-Produkt 88
nachgewiesen (Schema 22, unten). In Reaktionen mit den γ-methyl-substituierten Keten-
acetalen 38 und 41 wurden hingegen keine ausgeprägten Enantioselektivitäten beobachtet.
OO
Ti
O OTBS
OMe
25
CH2Cl2, -78 °C - RT
OH O
OMe
40 35 8880% ee
Oi-Pr
Oi-Pr
OO
Ti
O O
OTMS
25
THF, -78 °C - RT
OH O
O
73 86 87
88% ee
Oi-Pr
Oi-Pr
O O
Schema 22. Ti-(VI)-BINOL Komplex 25 in der VMAR nach SATO (oben) und KALESSE (unten). [49, 50]
Die oben dargestellten Beispiele zeigen, dass BINOL-Komplexe möglicherweise fähig sind,
die VMAR mit aliphatischen Aldehyden in hohen Selektivitäten zu katalysieren.
Ermutigt durch diese Tatsache sollten weitere modifizierte BINOL-Komplexe in der
asymmetrischen VMAR getestet werden. Hierzu wurden zuerst die in Abbildung 7
dargestellten BINOL-Liganden verwendet.
O
OTi
Oi-Pr
Oi-Pr
O
OTi
Oi-Pr
Oi-Pr
i-Pr
i-Pr
i-Pr
i-Pr
i-Pr
i-Pr
O
OTi
Cl
Cl
i-Pr
i-Pr
i-Pr
i-Pr
i-Pr
i-Pr
O
Si(Ph)3
OTi
Oi-Pr
Oi-Pr
Si(Ph)3
89 90 91 92 Abbildung 7. Weitere modifizierte BINOL-Katalysatoren für die VMAR.
Durch die gezielte Wahl unterschiedlicher Reste an der 3,3‘-Position sollten durch wachsende
sterische Wechselwirkungen höhere Enantioselektivitäten resultieren. Wie auch beim
BINOL-Komplex 25 konnten die Liganden käuflich erworben und die erwünschten
chiralen Komplexe in situ mit Titan-(IV)-isopropoxid in refluxierendem Dichlomethan

Studien zur Entwicklung neuer VMAR-Methoden
- 26 -
hergestellt werden. Die Darstellung des Katalysators 92 ging hingegen von
Diisopropoxytitan-(IV)-dichlorid aus, welches zuvor aus Titan-(IV)-isopropoxid durch
Zugabe von Titantetrachlorid bei Raumtemperatur generiert wurde.
Nach dem KECK-Protokoll erfolgte die Aktivierung des Isobutyraldehyds (40) durch die
BINOL-Katalysatoren für die Reaktion mit dem 3,4-E-41 bzw. 3,4-Z-Ketenacetal 38. Wie
auch von HAßFELD berichtet, wurde in sämtlichen Reaktionen das γ-VMAR-Produkt 66 in
moderaten Ausbeuten erhalten, wobei die Bestimmung der Enantiomerenüberschüsse durch
chirale Shift-Messung mit Eu(hfc)3 erfolgte.[51] Aus den in Tabelle 5 aufgelisteten Ergebnissen
ist ersichtlich, dass durch die Anwendung der BINOL-Katalysatoren 89-92 in der vinylogen
MUKAIYAMA -Aldolreaktion nur Enantioselektivitäten bis zu 32% ee resultieren. Dieses
Ergebnis konnte jedoch nur mit dem BINOL-Katalysator 90 erreicht werden. Die VMAR mit
den übrigen BINOL-Systemen hingegen führten zu deutlich geringeren Enantioselektivitäten.
Im Verlauf der Experimente erwies sich das 3,4-Z-Ketenacetal 38 als Zielführendes Substrat,
da hier in sämtlichen Reaktionen höhere Stereoselektivitäten im Vergleich zu dem 3,4-E-
Tabelle 5. VMAR mit BINOL-Katalysatoren unter verschiedenen Bedingungen.
O OH
40 66
OMe
OTBS
OMe
OLS 10 mol%
CH2Cl2, 36 h
R1 = H, R2 = Me 38R1 = Me, R2 = H 41
R1
R2
Nr.a KA LS Additiv T (°C) Ausbeute (%) de (%) ee (%)b
1 41 89 - -30 34 44 17
2 38 89 - -30 28 77 26
3 41 90 - -30 29 41 24
4 38 90 - -30 22 76 32
5 41 91 - -30 27 45 15
6 38 91 - -30 24 70 21
7 41 92 - -30 21 26 <5
8 38 92 - -30 24 59 <5
9 38 90 - -40 8 77 32
10 38 90 Et2O -30 19 75 32
11 38 90 i-PrOH -30 13 74 28
12cc 38 90 - -30 61 76 31
13d 38 90 - -30 30 72 29 a) 1 eq Aldehyd und 1.7 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) 100 mol% LS. d) Reaktion in THF.
Ketenacetal 41 erzielt werden konnten. Darüber hinaus wurden verschiedene Reaktions-
bedingungen erprobt, um die Stereoselektivität der VMAR zu verbessern. Bei Verwendung
niedrigere Reaktionstemperaturen sowie stöchiometrischen Mengen des Katalysators 90
konnte jedoch kein Anstieg der Selektivitäten registriert werden (Tab. 5, Nr. 12).

Studien zur Entwicklung neuer VMAR-Methoden
- 27 -
Der Einfluss des Lösungsmittels sollte nur auf den Umsatz beschränkt bleiben. Um eine
Aldehyd-Aktivierung durch freie Silylkationen auszuschließen (siehe Kapitel 1.3.2.2) sind in
einigen Versuchen Additive wie Isopropanol und Diethylether in äquivalenter Menge
zugesetzt worden. Hier blieb die Selektivität ebenfalls unverändert. Die oben
zusammengefassten Ergebnisse zeigen, dass diese Katalysator-Typen in der Lage sind, die
VMAR stereoselektiv zu kontrollieren, woraufhin im Verlauf weitere BINOL-Katalysatoren
untersucht wurden.
3.2.2 BINOL-Trimethylborat Komplex nach K ECK
Zeitgleich zu unseren Untersuchungen mit den BINOL-Komplexen veröffentlichte KECK eine
weitere interessante Modifikation vom BINOL-Komplex. KECK konstruierte den zweifach
BINOL-substituierten Titan-Komplex 95, welchen er zusätzlich mit Trimethylborat versetzte.
Obwohl der Einfluss des Trimethylborats ungeklärt ist, konnten KECK et al. mit diesem
Katalysatorsystem die VMAR zwischen 3-(Siloxy)-propenal 93 und dem α-methyliertem
Ketenacetal 94 in hervorragenden Regio- sowie Enantioselektivitäten katalysieren (Schema
23). [52] Die Experimente von KECK blieben allerdings nur auf das Ketenacetal 94 beschränkt.
OO
Ti
O OTMS
StBu
95 OH O
StBu
9394
9697%, 92% ee
OO
TBDPSOB(OMe)3, MS 4Å
Et2O, RT TBDPSO
Schema 23. VMAR nach KECK mit 3-(siloxy)-propenal 93 und BINOL-Komplex 95. [52]
Ermutigt durch die Ergebnisse von KECK, wurde der Katalysator 95 auch in unseren Studien
verwendet (Tab. 6). Die ersten Versuche wurden nach dem KECK-Protokoll durchgeführt,
wobei jedoch nur moderate Selektivitäten von bis zu 27% ee (Tab 6, Nr. 1) mit dem 3,4-Z-
Ketenacetal 38 resultierten. Zusätzlich wurde ein geringer Anteil vom α-VMAR-Produkt 97
gebildet. Durch Variation der Reaktionstemperatur konnte eine Erhöhung der Enantio-
selektivitäten auf bis zu 53% ee erzielt werden (Tab. 6, Nr. 6). Entscheidend war es, die
Reaktion bei -30 °C für sechs Stunden und anschließender langsamer Erwärmung auf
-20 °C durchzuführen. Die Variation des Lösungsmittels zeigte im Gegensatz zum KECK-
Protokoll nur Auswirkung auf die Ausbeute, nicht aber auf die Enantioselektivität der
Reaktion. Diethylether erwies sich als Lösungsmittel, mit dem die höchsten Ausbeuten erlangt
werden konnten. Darüber hinaus wurde ein kleiner, unbedeutender Einfluss bei der

Studien zur Entwicklung neuer VMAR-Methoden
- 28 -
Tabelle 6. VMAR mit BINOL-Katalysatoren 95 unter verschiedenen Bedingungen.
O OH
40 66
OMe
OTBS
OMe
O95, B(OMe)3
MS 4Å, 48 h OMe
OOH
9738
Nr.a Lsg Additiv T (°C) 66 : 97 Ausbeute 66 (%) de (%) ee (%)b
1 Et2O - -30 � 0 92 : 8 67 94 27
2 Et2O i-PrOH -30 � 0 91 : 9 52 94 30
3 Et2O DIPP -30 � 0 92 : 8 59 94 27
4 CH2Cl2 - -30 � 0 92 : 8 37 94 27
5 Toluol - -30 � 0 80 : 20 26 81 25
6 Et2O i-PrOH -30 95 : 5 10 >95 53
7 Et2O i-PrOH -30 � -20 94 : 6 27 >95 50
8 Et2O i-PrOH -30 � -10 95 : 5 53 >95 41
9 Et2O i-PrOH -40 94 : 6 3 >95 53
10 Et2O - -30 � -20 94 : 6 51 >95 49
11 THF i-PrOH -30 � -20 94 : 6 13 94 49
12c Et2O i-PrOH -30 � -20 81 : 19 50 42 11 a) 1 eq Aldehyd, 1.7 eq KA, 20 mol% 95. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)2. c) Rkt. mit KA-41. Verwendung von Isopropanol als Additiv auf die Enantioselektivitäten beobachtet. Wie
auch zuvor beobachtet, resultieren mit dem 3,4-E-Ketenacetal 41 deutlich geringere
Selektivitäten (Tab. 6, Nr. 12). Da mit dem KECK-System ebenfalls nur unbefriedigende
Enantioselektivitäten resultierten, wurden keine weiteren Experimente durchgeführt
3.2.3 Schiff’sche Base-Liganden nach CARREIRA
Als Ergebnis der ausführlichen Studien von CARREIRA et al., wurde 1994 ein weiteres
interessantes Katalysator-System veröffentlicht. [53] Ausgehend von 2-Amino-2’-hydroxy-
binapthyl-Liganden (NOBIN) entwickelten sie den Schiff’sche Base/Titan-(IV)-Komplex 99,
welcher sich in der MUKAIYAMA -Aldolreaktionen durch seine hohen Enantioselektivitäten
bewährt hat. Ein Jahr später gelang die Erweiterung auf Dienolate unter Beibehaltung der
Enantioselektivitäten (Schema 24). [54] Im Gegensatz zu anderen Katalysatorsystemen
können sogar mit dem CARREIRA-Katalysator die VMAR an α-alkin substituierte Aldehyde in
hohen Stereoselektivitäten durchgeführt werden. Infolgedessen fand dieser Katalysator auch
Verwendung in der Naturstoffsynthese von Macrolactin und Dihydroxy Vitamin D3. [55]
Die hohen Selektivitäten sowie das breite Substratspektrum dieser Methode war Anlass, diese
ebenfalls in der VMAR zwischen Isobutyraldehyd (40) und den γ-methylierten
Ketenacetalen zu verwenden (Tab. 7).
Die Experimente hierfür wurden analog zu dem CARREIRA-Protokoll durchgeführt.
Neben dem Katalysator 99 fand auch der Katalysator 101 der 1. Generation Verwendung in

Studien zur Entwicklung neuer VMAR-Methoden
- 29 -
N
O
98
O Br
t-Bu
Ti
O OO
t-Bu
t-Bu
O O
OTMS
i-Pr3Si
O
99
a) 2,6-Lutidin, Et2O, 0 °C
b)10 % TFA/THF
O O
O
i-Pr3Si
OH
86 10080%, 89% ee
3 mol%
Schema 24. CARREIRA’S Schiff’sche Base/Titan-(IV)-Komplex 99 in der VMAR. [54]
den Studien. Letzterer ergab jedoch deutlich geringere Regio- und Enantioselektivtäten von
nur 22% ee. Im Gegensatz hierzu, wurden mit dem Katalysator 99 der 2. Generation
ausschließlich das erwünschte γ-VMAR-Produkt 66 isoliert und dies mit einer Enantio-
selektivität von 44% ee für das 3,4-Z-Ketenacetal 38. Des Weiteren konnte auch in diesen
Tabelle 7. VMAR mit BINOL-Katalysatoren unter verschiedenen Bedingungen.
O OH
40 66
R2OMe
OTBS
OMe
OLS 5 mol%, 2,4-Lutidin
Et2O
N
OO Br
t-Bu
Ti
O OO
t-Bu
t-Bu99
N
OO Br
t-Bu
Ti
Oi-Pr
101
Oi-Pr
OMe
OOH
97R1 = H, R2 = Me 38R1 = Me, R2 = H 41
R!
Nr.a KA LS T (°C) 66: 97 Ausbeute 66 (%) de (%) ee (%)b
1 41 101 0 89 : 11 63 60 11 2 38 101 0 87 : 13 67 80 19 3 41 101 -15 90 : 10 46 60 19 4 38 101 -15 90 : 10 43 80 22 5 38 101 -30 90 : 10 25 80 22
6c 38 101 -15 90 : 10 33 80 22
7 41 99 0 99 : 1 47 63 31 8 38 99 0 99 : 1 55 88 44 9 41 99 -15 99 : 1 35 63 40
10 38 99 -15 99 : 1 32 88 58 11 38 99 -30 99 : 1 9 88 59
12c 38 99 -15 99 : 1 22 88 31
13d 38 99 -15 99 : 1 38 88 58 a) 1 eq Aldehyd und 1.7 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) 100 mol% LS. d) Reaktion in THF.

Studien zur Entwicklung neuer VMAR-Methoden
- 30 -
Systemen eine Verbesserung der Stereoselektivitäten durch Erniedrigung der Reaktions-
temperatur beobachtet werden. So resultierten die höchsten Selektivitäten bei -15 °C mit
58% ee. Tiefere Temperaturen resultierten jedoch in niedrigeren Ausbeuten. Durch den
Zusatz von Additiven sowie der Einsatz einer stöchiometrischen Menge an Katalysator sollten
identische Stereoselektivitäten liefern.
Da auch mit diesem BINOL-Katalysator die VMAR in geringen Enantioselektivitäten unter
60% ee ergab, wurden im Verlauf die Experimente mit diesen Liganden abgebrochen.
3.3 Bisoxazolin-Liganden in der asymmetrischen VMAR
Neben den BINOL-Liganden gewannen in den letzten 20 Jahren auch die C2-symmetrischen
Bisoxazolin-Liganden an Bedeutung für die asymmetrische Synthese. [56] Aufgrund der
einfachen Zugänglichkeit aus β-Aminoalkoholen und ihrer vielseitigen Einsatzmöglichkeiten
fanden sie in unterschiedlichen metallkatalysierten Reaktionen Verwendung. Beispiele für
Reaktionen mit hohen Enantoselektivitäten sind Cyclopropanierungen, [57] DIELS-ALDER-
Reaktion [58] sowie MUKAIYAMA -Aldolreaktionen. [59]
EVANS et al. konnten als erste von einer erfolgreichen Anwendung der Bisoxazolin-Liganden
in der asymmetrischen VMAR berichten. Sie nutzten die von ihnen entwickelten Kupfer-
Bisoxazolin-Liganden 104 als chirale Lewis-Säure für die VMAR zwischen Benzyloxyacet-
aldehyd 102 mit dem Dienolat 103 (Schema 25). [60] In den ersten Arbeiten konnten die
Aldolprodukte in sehr guten Ausbeuten und Enantioselektivitäten von bis zu 97% ee isoliert
werden, weswegen diese bereits Anwendung in einigen Totalsynthesen fand. [61] In weiteren
Untersuchungen berichtete EVANS über eine erfolgreiche Durchführung an Ketonen wie z.B.
BnOH
O
OMe
TMSO OTMSBnO
OtBu
OH O O
+
NN
OO
N
Ph Ph
Cu
2 SbF6-2+
a) 10 mol%, -78 °C, CH 2Cl2
b) 1N HCl, THF
104
102 103 10590%, 97% ee
Schema 25. Vinyloge Mukaiyama Aldolreaktion nach EVANS mit Bisoxazolin-Liganden. [60]
Methylpyruvaten als Elektrophile, wobei gleichwertige Enantioselektivitäten erreicht
werden konnten.
Diese ersten erfolgreichen Resultate sprechen für das Potential von chiralen Bisoxazolin-

Studien zur Entwicklung neuer VMAR-Methoden
- 31 -
Liganden in der vinylogen MUKAIYAMA -Aldolreaktion. Daher wurde der Kupfer-Bisoxazolin-
Komplex 104 in der VMAR eingesetzt und bezüglich seiner Selektivität untersucht. Der
benötigte Bisoxalin-Ligand wurde hierfür käuflich erworben. Die Resultate sind in Tabelle 8
zusammengestellt. Die Reaktionen mit dem Komplex 104 wurden mit Isobutyraldehyd (40)
und den γ-methylierten Ketenacetalen 38 bzw. 41 nach dem EVANS-Protokoll durchgeführt.
Dieses führte allerdings nur zu moderaten Ausbeuten sowie Enantioselektivitäten unter
35% ee (Tab. 8, Nr. 1, 2) für das γ-VMAR-Produkt 66. Variation der Reaktionstemperatur
sowie der Einsatz von Additiven blieben ebenfalls vergebens.
Tabelle 8. VMAR mit BINOL-Katalysatoren unter verschiedenen Bedingungen.
O OH
40 66
R2OMe
OTBS
OMe
O10 mol%
CH2Cl2, -78 °C
NN
OO
N
Ph Ph
Cu
2 SbF6-2+
104
R1 = H, R2 = Me 38R1 = Me, R2 = H 41
R1
Nr.a KA Additiv γ : α Ausbeute (%) de (%) ee (%)b
1 41 - 99 : 1 63 64 21 2 38 - 99 : 1 46 86 32 3c 38 - 99 : 1 25 86 32 4 38 i-PrOH 99 : 1 33 71 27 5 38 Et2O 99 : 1 44 76 32
6 38 DIPP 99 : 1 30 70 22
7d 38 - 99 : 1 52 80 12 8e 38 - 99 : 1 39 84 64
a) 1 eq Aldehyd und 1.7 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) Reaktion bei -93 °C. d) 100 mol% 104. e) Reaktion mit (PMBO)-Milschsäurealdehyd 106.
Da mit diesem Katalysatorsystem für zweifach koordinierende Aldehyde deutlich höhere
Selektivitäten resultieren, wurde in einer Testreaktion ebenfalls der PMB geschützte
Milchsäurealdehyd 106 eingesetzt. Dieser lieferte wie erwartet, vergleichsweise höhere aber
dennoch unbefriedigende Enantioselektivitäten von 64% ee (Tab. 8, Nr. 8). Da dieses Substrat
jedoch nicht im Interessenbereich der anfänglichen Studien lag, wurde auf weitere
Optimierungsversuche für dieses Substrat verzichtet.
Die Verwendung von Bisoxazolin-Liganden ergaben wie bei den BINOL-Liganden wenig
zufriedenstellende Ergebnisse. Aus diesem Grund wurden weitere Lewis-Säuren untersucht.

Studien zur Entwicklung neuer VMAR-Methoden
- 32 -
3.4 Chirale Ammoniumsalze als potentielle Lewis-Säuren für die VMAR
Neben BINOL- und Bisoxazolin-Liganden, wurden parallel weitere literaturbekannte chirale
Lewis-Säuren in der VMAR getestet. So fanden auch chirale Ammoniumsalze auf
Chinchonidin-Basis Verwendung in unseren Studien. Chirale Ammoniumsalze (AS) aus
Chinchonidinen weisen eine hohe Lewis-Säure Eigenschaft auf. [62] Durch gezielte Wahl des
Gegenanions können hohe Stereoselektivitäten erzielt werden. Erstmals konnten COREY
et al. zeigen, dass diese leistungsfähige Katalysatoren für MUKAIYAMA -Aldolreaktionen
darstellen. [63] Chirale Ammoniumsalze wurden als erste von CAMPAGNE et al. in vinylogen
MUKAIYAMA -Aldolreaktionen eingesetzt. CAMPAGNE’s Untersuchungen diesbezüglich
wurden mit dem α-methyliertem Ketenacetal 107 und Isobutyraldehyd (40) durchgeführt
(Schema 26). [64] In den beschrieben Versuchen konnte ausschließlich nur das γ-VMAR-
Produkt 109 in quantitativen Ausbeuten isoliert werden. Die hohe Regioselektivität ist hierbei
sicherlich auf die zusätzliche Methylgruppe in α-Position zurückzuführen. Im Gegensatz zur
Ausbeute und der Regioselektivität waren die Enantioselektivitäten mit nur 30% ee allerdings
wenig zufriedenstellend.
O
OMe
OTMS
Ot-Bu
OH O+ THF, RT, 24 h
108
40 107 10999%, 30% ee
NHO
N
OH
Schema 26. VMAR nach CAMPAGNE mit chiralem Ammoniumsalz (AS) 108. [64]
Durch die bereits veröffentlichten Ergebnisse und die leichte Zugänglichkeit der chiralen
Ammoniumsalze, untersuchten wir diese in der VMAR mit den γ-methyl substituierten
Ketenacetalen 38 und 41. Durch eine geeignete Wahl der Reaktionsbedingungen wie
Temperatur oder Zusatz von Additiven, sind hier höhere Enantioselektivitäten denkbar.
Hierfür wurden anfänglich ausgehend von kommerziell erhältlichen Chinchonidin (110)
sowie den entsprechenden Benzyl- sowie Anthracenyl-Halogeniden durch refluxieren in
Aceton die entsprechenden Ammoniumsalze 111 und 112 synthetisiert. Anschließend erfolgte
durch Verwendung von Anionaustauscher Amberlyst A-26 der Ionenaustausch zu 113 bzw.
114 (Schema 27). [65]

Studien zur Entwicklung neuer VMAR-Methoden
- 33 -
110
NHO
N
R-X, ∆Aceton
Amberlyst A-26
R = benzyl, 72% 111
NHO
N
R
X
R = anthracenyl, 57% 112R = benzyl, 87% 113
NHO
N
R
OH
R = anthracenyl, 82% 114
MeOH
Schema 27. Synthese der chiralen Ammoniumsalze (AS) 113 und 114.
Die synthetisierten chiralen Salze wurden auf Grundlage des CAMPGANE-Protokolls in der
VMAR eingesetzt. Allerdings konnten in sämtlichen Experimenten auch unter Variation der
Reaktionsbedingungen das VMAR-Produkt nicht isoliert werden, woraufhin die Experimente
mit den chiralen Ammmoniumsalzen abgebrochen wurden (Tab. 9).
Tabelle 9. Reaktionsbedingungen der VMAR mit chiralen Ammoniumsalzen (AS) 113 und 114.
O OH
40 66
OMe
OTBS
OMe
OAS
NHO
N
OH
NHO
N
OH
113 114
38
Nr.a AS AS (mol %) Lsg T (°C) Ausbeute (%)
1 113 10 CH2Cl2 0 - 2b 113 100 CH2Cl2 0 -
3 113 100 THF 0 -
4 113 100 THF 0� RT -
5 113 100 CH2Cl2 0� RT -
6 113 100 CH2Cl2 -16 -
7 114 100 CH2Cl2 0� RT -
8 114 10 CH2Cl2 -16 -
9 114 100 CH2Cl2 0 - 10 114 10 CH2Cl2 0 - 11c 113 100 CH2Cl2 0 - 12c 114 100 CH2Cl2 0 -
a) 1 eq Aldehyd und 2.5 eq KA. b) 4.0 eq KA. c) Reaktion mit KA-41.

Studien zur Entwicklung neuer VMAR-Methoden
- 34 -
3.5 Oxazaborolidine als potentielle chirale Lewis-Säuren für die VMAR
3.5.1 Oxazaborolidinone auf Basis von N-Ts-Aminosäuren
Chirale α-Aminosäuren stellen heute die wichtigsten Bausteine zur Darstellung von chiralen
Reagenzien und Naturstoffen dar. Der kostengünstige Zugang veranlasste viele renommierte
Arbeitsgruppen in den letzten zwei Jahrzehnten an der Entwicklung leistungsfähiger chiraler
Reagenzien auf Basis von Aminosäuren und ihrer erfolgreichen Anwendung in der modernen
organischen Synthese. So entstanden eine Vielzahl von unterschiedlichen Ansätzen, in denen
Aminosäuren zur chiralen Induktion eingesetzt wurden. [66] Als einer der wertvollsten
Beispiele für auf Aminosäuren basierenden chiralen Reagenzien zählen die sogenannten
Oxazaborolidinone (OXB) (Abb. 8).
NB
O
R2 O
O2S
R1
R3
Abbildung 8. Allgemeine Struktur von Oxazaborolodinonen (OXB).
Die zeitgleich von HELMCHEN
[67] und YAMAMOTO [68] Anfang der 90er Jahre entwickelten
Oxazaborolidinone aus N-sulfonierten Aminosäuren fanden erstmals Verwendung als chirale
Lewis-Säuren in DIELS-ALDER-Reaktionen (Schema 28).
NB
O
O
O2S
HR
R = 2,4,6-(iPr)-C6H2
O
115 116 CHO
Me
118
NB
O
O
O2S
HPh
117
CHO
Me
11858%, 72% ee54%, 52% ee
CH2Cl2, -78 °C, 19 h
10 mol%
CH2Cl2, -78 °C, 6 h
119
20 mol%
Schema 28. Oxazaborolodinone in DIELS-ALDER-Reaktion (HELMCHEN : rechts, YAMAMOTO : links). [67, 68]
Beide Arbeitsgruppen setzten Oxazaborolidinone als Katalysator für die DIELS-ALDER-
Reaktion zwischen Cyclopentadien (116) und Crotonaldehyd (115) ein und erzielten
ausreichende Ausbeuten und Stereoselektivitäten. Die Darstellung der Oxazaborolidinone
erfolgt in situ durch Umsatz der Aminosäuren mit den entsprechenden Boranen. HELMCHEN
et al. verwendeten (S)-Valin für die Oxazaborolidinone, wodurch im Gegensatz zu
YAMAMOTO ausgeprägtere Enantioselektivitäten erzielt werden konnten. Ermutigt durch diese
Studien arbeiteten zahlreiche Forschungsgruppen an der Optimierung der OXB-Systeme,

Studien zur Entwicklung neuer VMAR-Methoden
- 35 -
wodurch unterschiedlich modifizierte OXB-Systeme entwickelt und untersucht wurden.
Deshalb wurden in kurzer Zeit zahlreiche C-C-Verknüpfungsreaktionen mit den OXB-
Systemen beschrieben, wobei KIYOOKA ‘s Studien zeigten, dass OXB-Systeme aus
N-tosyliertem Valin in der Lage sind, die MUKAIYAMA -Aldolreaktion stereoselektiv zu
kontrollieren. [69] In der Totalsynthese von Filipin III wurden ebenfalls hervorragende
Stereoselektivitäten mit dem Dienolat 103 beobachtet (Schema 29). [70]
NB
O
O
Ts
H121
100 mol%
CH2Cl2, -78 °COMe
OTMS
120 103 122 65%, 92% de
TMSOOTBS
n-C5H11
O
O
O
OTBS
n-C5H11
O
O
OMe
O OOH
Schema 29. MUKAIYAMA-Aldolreaktion nach KIYOOKA mit N-Ts-Valin abgeleitetem OXB 121. [70]
Abbildung 9 stellt das von KIYOOKA postulierte, allgemein akzeptierte Modell für den
Übergangszustand 123 der OXB katalysierten MUKAIYAMA -Aldolreaktion dar, der sogleich
die bevorzugte Konfiguration erklärt. Bei der Kombination von Aminosäure und Boran bildet
sich ein planarer fünfgliedriger Ring. Der Isopropyl-Rest steht dabei oberhalb der Ringebene.
Die Tosyl-Gruppe hingegen befindet sich unterhalb der Ringebene. Der Aldehyd koordiniert
am Bor-Atom und wird durch zusätzliche Wasserstoffbrückenbindungen fixiert. Durch diese
Anordnung wird folglich die re-Seite abgeschirmt und ein si-Seitenangriff vom Nucleophil
begünstigt. [71]
N BO
O
TsH
O
R
HNu
si - Angriff
123 Abbildung 9. KIYOOKA-Modell. [71]
YAMAMOTO veröffentlichte eine weitere bedeutende Modifikation der OXB-Systeme. Dabei
wurden die erstmals von COREY [72] beschriebenen auf N-Ts-Tryptophan basierenden OXB-
Liganden verwendet und daraus unterschiedlich B-Aryl-Substituierte OXB-Katalysatoren
synthetisiert. [73] Durch die zusätzliche sterische Hinderung der Aryl-Reste konnten in
MUKAIYAMA -Aldolreaktionen deutlich höhere Stereoselektivitäten erzielt werden, als mit

Studien zur Entwicklung neuer VMAR-Methoden
- 36 -
O OH
73126
OTMS 10 mol%
Ph EtCN, -78 °C, 3hPh
O
91%, 93% ee124
NB
OO
Ts
Ph
125
HN
Schema 30. MUKAIYAMA-Aldolreaktion nach KIYOOKA mit N-Ts-Tryptophan abgeleitetem OXB. [ 73]
dem KIYOOKA-Katalysator (Schema 30). Im Gegensatz zum Valin-System konnte
YAMAMOTO in Experimenten mit den Tryptohphan-Systemen die entgegengesetzte
Konfiguration für das neu gebildete Stereozentrum nachweisen. Auf dieser Grundlage wurde
ein erweitertes Modell 127 für das Tryptophan-OXB-System postuliert (Abb. 10). Wie auch
im KIYOOKA-Modell liegt ein planarer Fünfring vor, bei dem die Substituenten oberhalb,
sowie unterhalb der Ebene angeordnet sind. Durch eine zusätzliche π,π-Wechselwirkung
mit dem Indol-Substituenten nimmt der Aldehyd bevorzugt eine Anordnung oberhalb des
Ringes an, wodurch nur ein re-Seitenangriff erfolgen kann. Zudem resultiert durch die
π,π-Wechselwirkung eine stärkere Fixierung des Aldehyds im Übergangszustand, was die
höheren Stereoselektivitäten erklärt.
Nu
re - Angriff
π,π-WW
O
NB
O
HN
O
RH
Ts
127 Abbildung 10. Yamamoto-Modell. [73]
Im Rahmen der Diplomarbeit von HAßFELD wurden in unserem Arbeitskreis erstmals Oxaza-
borolidinone in der asymmetrischen VMAR mit γ-methylierten Ketenacetalen 38 verwendet
(Schema 31). [50] Es konnte mit dem von KIYOOKA benutzten Oxazaborolidinon 121 eine
γ-selektive Addition von 38 an Isobutyraldehyd (40) beobachtet werden. Die
Enantioselektivitäten dieser Reaktion wurden von HAßFELD allerdings nicht bestimmt. In
Versuchen mit YAMAMOTO ‘s Oxazaborolidinon 125 konnte hingegen keine Produktbildung
nachgewiesen werden.
Speziell HAßFELD’s Beispiel hat gezeigt, dass Oxazaborolidinone in der Lage sind, die
VMAR in hohen Stereoselektivitäten zu katalysieren.

Studien zur Entwicklung neuer VMAR-Methoden
- 37 -
O
40
60 mol%
CH2Cl2, -78 °C, 17h
121
38
NB
O
O
Ts
H
OMe
OTBS
OMe
OOH
66 67% de Schema 31. Erste Experimente zur VMAR mit Oxazaborolidinonen im Arbeitskreis KALESSE. [50]
Wie in den vorherigen Beispielen gezeigt, kann durch geeignete Modifikationen der Oxaza-
borolidinone und zusätzlicher Optimierung der Versuchsbedingungen die Effizienz und die
Selektivität der Reaktion gesteigert werden. Um dieses zu untersuchen, wurden im Verlauf
der Dissertation Oxazaborolidinone aus unterschiedlichen Aminosäuren in der VMAR
eingesetzt. Zunächst wurden die benötigten tosylierten Aminosäuren nach SCHOTTEN-
BAUMAN -Bedingungen [74] synthetisiert. Anschließend wurden diese in der VMAR mit den
bekannten Substraten erprobt. Die anfänglichen Studien wurden mit N-Tosyl-Valin 128 und
N-Tosyl-tert-Leucin 129 abgeleiteten Oxazaborolidinonen nach dem KIYOOKA-Protokoll
durchgeführt (Tab. 10). Neben unterschiedlichen Aminosäuren kamen auch verschiedene
Borane zum Einsatz. Die Generierung der Oxazaborolidinone erfolgte hierbei in situ durch
Reaktion der N-tosylierten Aminosäuren mit den entsprechenden Boranen in Dichlormethan
bei Raumtemperatur.
Die ersten Versuche zeigten, dass die in der MUKAIYAMA -Aldolreaktion sehr wirksamen
Katalysatoren 121 und 133 in der VMAR mit den Testsubstraten nur moderate Ausbeuten und
keine Enantioselektivitäten liefert, so dass ausschließlich nur Racemate erhalten wurden.
Zudem zeigte sich, dass sowohl die Regio- als auch die Diastereoselektivität deutlich von dem
eingesetzten Ketenacetal abhängt. Während in Reaktionen zwischen Isobutyraldehyd (40) und
dem 3,4-Z-Ketenacetal 38 ausschließlich das γ-VMAR-Produkt 66 gebildet wurde, war bei
Reaktionen mit 3,4-E-Ketenacetal 41 auch ein geringer Anteil des α-VMAR-Produktes 97
vorzufinden. Darüberhinaus lieferten die Reaktionen nur eine geringe Diastereoselektivität,
die in Abhängigkeit der Bor-Substituenten nur geringfügig variierte. Hierbei lieferten die
Phenyl-substituierten Oxazaborolidinone 132 bzw. 135 die höchsten Diastereoselektivitäten
(Tab. 10, Nr. 14, 18, 19, 27, 29).
Des Weiteren wurde die VMAR bei tieferen Temperaturen sowie mit Additiven durchgeführt
(Tab. 10, Nr. 21-24), was allerdings zu keiner signifikanten Steigerung der Diastereo-
selektivität führte.
KIYOOKA et al. stellten in ihren Arbeiten einen Einfluss des verwendeten Lösungsmittels auf

Studien zur Entwicklung neuer VMAR-Methoden
- 38 -
Tabelle 10. VMAR mit Oxazaborolidinonen aus Valin und tert.-Leucin.
O OH
40 66
R2OMe
OTBS
OMe
O
CH2Cl2, -78 °C
NB
O
O
Ts NB
O
O
Ts
OXB
R = H 121 Cl 130 Br 131 Ph 132
OMe
OH O
97
R1
R1 = H, R2 = Me 38R1 = Me, R2 = H 41
R R
R = H 133 Cl 134 Ph 135
Nr.a KA OXB Additiv 66 : 97 Ausbeute 66 (%) de (%) ee (%)b
1 41 121 - 2 : 1 7 13 0 2 38 121 - 99 : 1 9 50 0 3 41 130 - 3 : 1 14 26 0 4 38 130 - 99 : 1 24 50 0 5 41 131 - 3 : 1 10 24 0 6 38 131 - 99 : 1 22 49 0 7 41 132 - 3 : 1 21 40 0 8 38 133 - 99 : 1 35 50 0 9 41 133 - 2 : 1 5 13 0
10 38 133 - 99 : 1 10 50 0 11 41 134 - 3 : 1 14 27 0 12 38 134 - 99 : 1 24 50 0 13 41 135 - 3 : 1 25 40 0 14 38 135 - 99 : 1 31 77 0 15 38 121 i-PrOH - - - - 16 38 130 i-PrOH 99 : 1 4 50 0 17 38 131 i-PrOH 99 : 1 8 74 0 18 38 132 i-PrOH 99 : 1 20 77 0
19 38 132 Et2O 99 : 1 32 77 0
20 41 132 Et2O 3 : 1 5 40 0
21c 38 121 - - - - -
22c 38 130 - 99 : 1 10 50 0
23c 38 131 - 99 : 1 8 47 0
24c 38 132 - 99 : 1 19 70 0
25d 38 121 - 99 : 1 6 50 0 26d 38 130 - 99 : 1 12 53 0 27d 38 132 - 99 : 1 26 77 0 28d 38 133 - 99 : 1 5 50 0 29d 38 135 - 99 : 1 23 77 0
a) 1 eq Aldehyd und 1.7 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) Reaktion bei -93 °C. d) Reaktion in THF.
die Enantioselektivität der VMAR fest und konnten durch Verwendung von THF eine erhöhte
Enantioselektivität nachweisen. [70] Eine Erklärung hierfür liefert das Modell 136 in
Abbildung 11. Im Übergangszustand wird der Oxazaborolidinon-Aldehyd-Komplex von
Tetrahydrofuran-Molekülen umgeben, die durch Wasserstoffbrückenbindungen koordinieren.
Der Komplex wird dadurch stärker fixiert, wodurch höhere Stereoselektivitäten resultieren.
Im Gegensatz zu KIYOOKA ‘s Beobachtungen, konnte in Reaktionen mit Tetrahydrofuran als

Studien zur Entwicklung neuer VMAR-Methoden
- 39 -
Lösungsmittel allerdings keine Erhöhung der Stereoselektivitäten registriert werden.
(Tab. 10, Nr. 25-29).
NB
O
O
HTs
O
R
HO
HTHF
THF
THF
136 Abbildung 11. Übergangszustand in THF. [70]
Da die Oxazaborolidinone aus Valin und tert-Leucin nur geringe Stereoselektivitäten
lieferten, wurden im weiteren von N-Tosyl-Phenylalanin 137 und N-Tosyl-Tryptophan 138
abgeleiteten Oxazaborolidinone aus Phenylalanin und Tryptophan verwendet (Tab. 11).
Zusätzlich wurden ebenfalls unterschiedlich substituierte Borane eingesetzt. Erstmals konnten
in diesen Reaktionen das VMAR-Produkt 66 in enantiomerenangereicherter Form isoliert
werden.
So konnte bei Verwendung der n-Butyl-substituierten Borane 141 und 146 bei -78 °C ein
Enantiomerenüberschuss von 22% ee ermittelt werden (Tab. 11, Nr. 3, 10). Tryptophan
scheint wegen der höheren Diastereoselektivität eine Sonderstellung einzunehmen. Durch die
Wahl aromatischer Bor-Reste konnten Enantioselektivitäten von bis zu 42% ee erzielt
werden, wobei B-Phenyl-substituierte Oxazaborolidinone höhere Ausbeuten lieferten. Die
zusätzliche Verwendung von Isopropanol als Additiv ergab bei Verwendung des von
Tryptophan abgeleiteten B-Phenyl-Oxazaborolidinon 125 weiterhin einen leichten Anstieg der
Enantioselektivitäten auf 52% ee (Tab 11, Nr. 15). Durch Versuche bei tieferen Temperaturen
konnten keine gesteigerten Stereoselektivitäten erzielt werden. Zudem führte der Einsatz von
überstöchiometrischen Mengen des Katalysators sogar zu einem Abfall der
Enantioselektivitäten (Tab. 11, Nr. 30).
Die geeignete Wahl der Additive kann ausschlaggebend für höhere Enantiomeren-
überschüsse sein, weswegen auch unterschiedliche Additive auf ihre Wirkung untersucht
werden sollten. Neben den bekannten Additiven, wie Diethylether oder Isopropanol, welche
sich in vorherigen Arbeiten in unserem Arbeitskreis als besonders wirkungsvoll erwiesen
haben, wurden zusätzlich unterschiedlich substituierte Amine, Alkohole sowie Ether als
Additive eingesetzt (Tab. 11, Nr. 19-26). Bei Betrachtung der Ergebnisse fällt auf, dass

Studien zur Entwicklung neuer VMAR-Methoden
- 40 -
Insbesondere aromatische Additive zu unerwartet geringeren Enantioselektivitäten führten.
Tabelle 11. VMAR mit Oxazaborolidinonen aus Phenylalanin und Tryptophan.
O OH
40 66
R2OMe
OTBS
OMe
O
-78 °C
NB
O
O
TsNB
O
O
Ts
HN
OXB
R = H 139 Cl 140 n-Bu 141 Ph 142 3,5-(CF3)-Ph 143
R1 = H, R2 = Me 38R1 = Me, R2 = H 41
R1OH
97
OMe
O
R = H 144 Cl 145 n-Bu 146 Ph 125 3,5-(CF3)-Ph 147
R R
Nr.a KA OXB Additiv 66 : 97 Ausbeute 66 (%) de (%) ee (%)b
1 38 139 - 99 : 1 11 50 0 2 38 140 - 99 : 1 26 47 0 3 38 141 - 99 : 1 33 50 22 4 38 142 - 99 : 1 38 50 42 5 38 143 - 99 : 1 13 50 42 6 41 142 - 3 : 1 27 33 20 7 41 141 - 3 : 1 20 33 8 8 38 144 - 99 : 1 8 50 0 9 38 145 - 99 : 1 18 50 0 10 38 146 - 99 : 1 23 66 22 11 38 125 - 99 : 1 38 82 42 12 38 147 - 99 : 1 11 82 42 13 38 145 i-PrOH 99 : 1 8 50 0 14 38 146 i-PrOH 99 : 1 10 66 27 15 38 125 i-PrOH 99 : 1 18 82 52 16 38 147 i-PrOH 99 : 1 - - - 17 38 142 i-PrOH 99 : 1 19 50 42 18 38 141 i-PrOH 99 : 1 24 48 22 19 38 125 DIPP 99 : 1 30 50 38
20 38 125 Et2O 99 : 1 32 82 42
21 38 125 Pyridin 99 : 1 19 45 22 22 38 125 2,6-Lutidin 99 : 1 22 42 23 23 38 125 PhOMe 99 : 1 35 80 29 24 38 125 PhOH 99 : 1 31 44 30 25 38 125 t-BuOH 99 : 1 37 80 42 26 38 125 (CF3)2COH 99 : 1 12 80 50
27c 41 147 i-PrOH 99 : 1 8 47 24
28c 38 142 - 99 : 1 19 70 0
29c 38 125 - 99 : 1 6 50 0
30d 38 125 i-PrOH 99 : 1 30 82 33
31d 38 142 i-PrOH 99 : 1 28 50 29 a) 1 eq Aldehyd und 1.7 eq KA 100 mol% OXB. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)2. c) Reaktion bei -93 °C. d) 200 mol% OXB.
Dies könnte durch das zusätzlich in die Reaktion eingebrachte π-System verursacht werden,

Studien zur Entwicklung neuer VMAR-Methoden
- 41 -
welches die für die Selektivität wichtige π,π-Wechselwirkung zwischen Aldehyd und
Oxazaborolidinon beeinträchtigt und somit zu verminderten Enantioselektvitäten führt. Durch
diese Experimente konnte die besondere Stellung von Isopropanol als Additiv
verdeutlicht werden, da es als einziges Additiv eine Steigerung der Enantioselektivitäten bei
gleichbleibenden Ausbeuten ermöglicht.
Da durch einen Wechsel der Aminosäure erste Enantiomerenüberschüsse resultierten, lag es
nahe weitere Aminosäuren zu testen. HARADA et al. berichteten 2001 erstmals von Threonin
abgeleiteten Oxazaborolidinonen, welche zu hervorragenden Enantioselektivitäten in der
MUKAIYAMA -M ICHAEL-Reaktion führten. [75] Ein bedeutender Vorteil von Threonin als
Baustein für Oxazaborolidinone, ist die freie Alkohol-Funktion die eine Veresterung mit
unterschiedlichen Gruppen ermöglicht und somit den Zugang zu weiteren Katalysator-
Derivaten gewährleistet. Die benötigten Threonin-Derivate wurden nach der in Schema 32
abgebildeten Syntheseroute dargestellt. Zuerst erfolgte eine N-Tosylierung zu 149 gefolgt von
einer säurekatalysierten Veresterung mit Benzylalkohol zum Monoester 150. Dieser wurde
anschließend mit unterschiedlichen Carbonsäurechloriden in Gegenwart von Pyridin in
Dichlormethan zu den Diestern 151 und 152 umgesetzt und durch Hydrierung der
Benzylgruppen entschützt.
HO
O OH
NH2
Ts-Cl, 1M NaOH
Et2O, RT, 76% HO
O OH
NHTsBnO
O OH
NHTs
OH
p-TsOH, ToluolReflux, 91%
148 149 150
Pyridin, CH2Cl2RT, 77-86 %
BnO
O O
NHTsR = phenyl 77% 151
R
O
Pd/C, H2
HO
O O
NHTs
R
O
99%
Cl R
O
R = biphenyl 86% 152R = phenyl 77% 153R = biphenyl 86% 154
Schema 32. Darstellung von Threonin-Derivaten für neue Oxazaborolidinone. Analog zu HARADA ’s Arbeiten kamen hierfür aromatsiche Substituenten zum Einsatz.
Sämtliche durchgeführten Reaktionen erfolgten entsprechend der Literatur in guten
Ausbeuten. [76]
Die auf diese Weise synthetisierten Threonin-Derivate 153 und 154 wurden wie gewohnt
in situ in Dichlormethan mit Dichlorphenylboran zu den entsprechenden Oxazaborolidinionen

Studien zur Entwicklung neuer VMAR-Methoden
- 42 -
umgesetzt und in der VMAR mit dem γ-methylierten Ketenacetalen 38 und 41 untersucht.
Die Ergebnisse in Tabelle 12 zeigen, dass die VMAR mit den Threonin-abgeleiteten
Oxazaborolidinonen 155 und 156 nahezu gleichwertige Enantioselektivitäten lieferten, wie
das Oxazaborolidinon 125. Ebenso konnten diese nur durch den Einsatz von Isopropanol als
Additiv erzielt werden.
Tabelle 12. Oxazaborolidinon vermittelte VMAR mit Threonin-Derivaten.
O OH
40 66
OMe
OTBS
OMe
O
CH2Cl2-78 °C
NB
O
O
Ts
O
155
OXB 100 mol%
O
NB
O
O
Ts
O
O
156
38
Ph Ph
Nr.a OXB Additiv γ : α Ausbeute (%) de (%) ee (%)b
1 155 - 99 : 1 11 80 34 2 155 i-PrOH 99 : 1 26 82 47 3 156 - 99 : 1 33 80 34 4 156 i-PrOH 99 : 1 38 82 47 5c 155 i-PrOH 99 : 1 13 82 47 6c 156 i-PrOH 99 : 1 27 82 47
a) 1 eq Aldehyd und 1.7 eq KA 100 mol% OXB. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) Reaktion bei -93 °C.
Weiterführende Experimente wurden im Rahmen der Masterarbeit von SU durchgeführt,
allerdings führten auch sterisch anspruchsvoll substituierte Threonin-Derivate nicht zum
gewünschten Erfolg. [77]
3.5.2 Oxazaborolidinon vermittelte VMAR am γγγγ-unsubstituiertem Ketenacetal
Während der Studien mit Oxazaborolidinonen an γ-methyl substituierten Ketenacetalen,
arbeitete HORZELLA parallel im Rahmen der Diplomarbeit an der Entwicklung der
asymmetrischen vinylogen MUKAIYAMA -Aldolreaktion für das γ-unsubstituierte Keten-
acetal 35. Nach den ersten positiven Ergebnissen mit den Oxazaborolidinonen, sollten diese
auch an γ-unsubstituierten Ketenacetalen 35 getestet werden. So erreichte HORZELLA unter
den in Tabelle 11 (Nr. 15) dargestellten Bedingungen, erstmals Enantioselektivitäten von über
90% ee (Schema 33). Die Verwendung des von Tryptophan abgeleiteten Oxazaborolidinons

Studien zur Entwicklung neuer VMAR-Methoden
- 43 -
125 und Isopropanol als Additiv lieferte akzeptable Ausbeuten und Enantioselektivitäten von
96% ee für das VMAR-Produkt 88. [78]
Da jedoch in mehrmaligen Versuchen deutlich geringere Ausbeuten (27-32%) sowie
Enantioselektivitäten (72-78% ee) erzielt wurden und somit das vielversprechende Ergebnis
nicht reproduziert werden konnte, mussten eingehende Untersuchungen für die Erklärung der
beobachteten schlechteren Ergebnisse durchgeführt werden.
Da die Ergebnisse mit dem von Tryptophan abgeleiteten Oxazaborolidinon 125 die einzigen
waren, die nicht reproduziert werden konnten, lag es nahe, die Ursache hierfür am
synthetisierten N-Ts-Tryptophan 138 zu suchen.
O OH
4035
OMe
OTBS
OMe
O
BuCN, -88 °C
NB
OO
Ts
88 54%, 96% ee
125
HN
i-PrOH
50 mol%
Ph
Schema 33. Oxazaborolidinon vermittelte VMAR mit unsubstituiertem Ketenacetal 35. [78]
Im Gegensatz zu den restlichen Aminosäuren erfolgte die Aufreinigung des
N-Ts-Tryptophans 138 nicht durch Umkristallisation sondern über Säulenchromatographie.
Obwohl Schmelzpunkt sowie spektroskopische Daten auf keine Verunreinigung hinwiesen,
wurde als erstes versucht, die säulenchromatographische Aufreinigung zu optimieren.
Zusätzlich wurden Chargen von unterschiedlichen Anbietern getestet. Allerdings blieben
sämtliche Versuche hierzu erfolglos. Schließlich sollten weitere Überlegungen unter
Berücksichtigung der Experimente mit aromatischen Additiven die Ursache erfolgreich
ausmachen. Hier war das für die Aufreinigung der Aminosäure als Eluat verwendete Toluol
Hauptursache für die geringen Stereoselektivitäten. Es zeigte sich, dass bereits geringe Spuren
von Toluol in der Reaktion die Stereoselektivitäten drastisch minimieren (Abb. 12). Daher
wurde das synthetisierte N-Ts-Tryptophan 138 im Verlauf mehrmals mit Dichlormethan
redestilliert, um das Toluol zu entfernen. Mit dem toluolfreien Liganden konnten die
vielversprechenden Ergebnisse reproduziert und die Enantioselektivität sogar auf 98% ee
gesteigert werden.
Nach dem Erreichen zufriedenstellender Enantioselektivitäten sollte im Weiteren versucht
werden, die Ausbeute der Reaktion zu erhöhen. Bei genauer Beobachtung der Reaktion zeigte
sich, dass während der Reaktion ein schwer zu erkennender farbloser Niederschlag ausfällt,

Studien zur Entwicklung neuer VMAR-Methoden
- 44 -
Abbildung 12. Einfluss von Toluol auf die Enantioselektivität.
welcher beim Zutropfen des Aldehyds zum Oxazaborolidinon entsteht. Da der ausfallende
Niederschlag vermutlich durch Polymerisation des aktvierten Aldehyds entsteht, sollte dieses
durch gemeinsames Zutropfen des Aldehyds und des Oxazaborolidinons verhindert werden.
Der auf diese Weise resultierende Aldehyd-OXB-Komplex müsste nach seiner Bildung sofort
weiter mit dem Nucleophil reagieren, wodurch die Polymerisation unterdrückt werden kann.
In vorangegangenen Experimenten wurde der Isobutyraldehyd (40) zusammen mit dem
Ketenacetal 35 zugetropft. Dabei sind unterschiedliche Zutropfgeschwindigkeiten sowie
weitere Reaktionsparameter variiert worden (Tab. 13). Durch die Verwendung einer
stöchiometrischen Menge an OXB konnte erstmals bei -94 °C eine Ausbeute von 64%
erreicht werden (Tab. 13, Nr. 1). Versuche mit substöchiometrischer sowie
überstöchiometrischer Menge an OXB führten zu deutlich geringeren Ausbeuten, wobei nur
eine minimale Abnahme der Enantioselektivität beobachtet wurde. Durch Variation der
Reaktionskonzentrationen auf 0.28 M konnte die Ausbeute zusätzlich auf 75% erhöht
werden (Tab. 13, Nr. 12).
Um die OXB katalysierte VMAR jedoch für die Synthesechemie attraktiver zu gestalten,
wurde versucht die Reaktionstemperatur der VMAR auf -78 °C zu erhöhen mit gleich-
bleibenden Ausbeuten und Stereoselektivitäten zu gewährleisten. Durch Erniedrigung der
Reaktionskonzentration sowie der Zugabe der Reaktanden über einen Zeitraum von zehn
Minuten konnte dieses erreicht werden (Tab. 13, Nr. 18), wobei eine 75%ige Ausbeute und
eine Enantioselektivität von 98% ee erzielt werden konnte.
Nachdem die Reaktionsbedingungen für die Oxazaborolidinon 125 vermittelten VMAR
optimiert worden sind, sollte die Verwendung weiterer Borane erprobt werden. Das besonders
aus YAMAMOTO ‘s Veröffentlichungen vielversprechende Boran 147, führte hier allerdings
Toluol
Toluol
98% ee72% ee78% ee
Toluol frei

Studien zur Entwicklung neuer VMAR-Methoden
- 45 -
Tabelle 13. Optimierung der VMAR am unsubstituierten Ketenacetal 35.
O OH
4035
OMe
OTBS
OMe
O
PrCN
NB
OO
Ts
Ph
88
125
HN
i-PrOH
Nr. OXB (mol%)
OXB-Lsg (mL)
Substrat-Lsg (mL)
∆t Substrat (min)
T (°C) Ausbeute (%) ee (%)
1 100 2 0.5 5 -94 64 98 2 50 2 0.5 5 -94 53 97 3 30 2 0.5 5 -94 46 96 4 150 2 0.5 5 -94 51 98 5 100 2.5 0.5 5 -94 44 98 6 100 1.5 0.5 5 -94 71 98 7 50 1.5 0.5 5 -94 62 97 8 50 1.5 1 5 -94 66 98 9 50 1.5 2 5 -94 50 97
10 100 1 0.5 5 -94 60 98 11 100 1.5 0.5 1 -94 48 98 12 100 1.5 0.5 20 -94 74 98 13 50 1.5 0.5 30 -78 55 97 14 100 1.5 0.5 30 -78 64 98 15 100 1.5 0.5 60 -78 42 98 16 100 1.5 0.5 90 -78 35 98 17 100 2 0.5 30 -78 44 98 18 100 2 0.5 10 -78 75 98 19 100 2.5 0.5 10 -78 68 98 20 50 2 0.5 10 -78 63 97
21a 100 2 0.5 10 -78 74 98
22b 100 2 0.5 10 -78 73 98 23c 100 2 0.5 10 -78 74 98
a) 1 eq Aldehyd und 1.2 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) Rkt. in EtCN. d) 2.0 eq KA.
nur zu äquivalenten Enantioselektivitäten wie das Oxazaborolidinon 125 (Tab. 14, Nr. 2).
Tabelle 14. VMAR katalysiert durch unterschiedlich B-substituierter Oxazaborolidinone
O OH
40 35
OMe
OTBS
OMe
O
BuCN, -78 °C
NB
OO
Ts
R
88
HN
100 mol%, i-PrOH
R = n-Bu 146 3,5-(CF3)-Ph 147
Nr. OXB Ausbeute (%) ee (%)
1 146 34 73 2 147 41 98

Studien zur Entwicklung neuer VMAR-Methoden
- 46 -
Um den Einfluss der Tosyl-Schutzgruppe im OXB-System zu untersuchen, wurden die in
Abbildung 13 dargestellten Oxazaborolidinone mit verschiedenen Amino-Schutzgruppen in
der VMAR getestet. Hierbei handelt es sich um kommerziell zugängliche N-geschützte
Tryptophane. Die Reaktionen mit diesen Oxazaborolidinonen führte jedoch zu keinem
Umsatz, was für die essentielle Bedeutung der Tosylschutzgruppe spricht.
NB
O
O
Ph
HN
Me NB
O
O
Ph
HN
Cbz NB
O
O
Ph
HN
Fmoc
157 158 159 Abbildung 13. Unterschiedliche N-geschützte Oxazaborolidinone.
Um die Leistungsfähigkeit der Oxazaborolidinon-katalysierten VMAR mit 125 zu bestimmen,
sind im Verlauf unterschiedliche verzweigte, aliphatische und aromatische Aldehyde sowie
reaktive Ketone aus Pyruvaten eingesetzt worden (Tab. 15). In den meisten Fällen konnten
hohe Enantioselektivitäten festgestellt werden, besonders bemerkenswert waren hierbei die
erzielten Enantioselektivitäten bei Verwendung von zyklischen und langkettigen-
aliphatischen Aldehyden (Tab. 15, Nr. 1-3). Unter Betrachtung der bekannten VMAR
Varianten in denen aliphatische Aldehyde nur bedingt eingesetzt werden können, stellen diese
Tabelle 15. Untersuchung der VMAR an unterschiedlich verzweigten Carbonylverbindungen.
R
O
R
OH
35
OMe
OTBS
OMe
O
BuCN, -78 °C, 3h
NB
OO
Ts
Ph
125
HN
i-PrOH100 mol%
Nr.a Carbonyl- verbindung
Ausbeute (%) α-Aldolprodukt
Ausbeute (%) γ-Aldolprodukt
ee (X)b γ-Aldolprodukt
1 Pivalinaldehyd <1 160 70 99c
2d Valeral 4 161 65 94e
3 Cyclohexanal <1 162 80 96e
4d Benzaldehyd 9 163 60 83e
5d E-Zimtaldehyd 11 164 66 80e
6 2-Furfural 13 165 63 76e
7f Methylpyruvat - - -
8f Methylbenzoylformat - - - a) 1 eq Aldehyd und 1.2 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) Zusätzlich bestimmt durch chirale GC am TBS-geschütztem Produkt. d) zusätzlich 4 % racemisches TBS-Produkt.
e) Zusätzlich bestimmt durch MOSHER-Methode. f) Reaktion bei -78 und -50°C durchgeführ t.

Studien zur Entwicklung neuer VMAR-Methoden
- 47 -
Ergebnisse eine bedeutende Erweiterung für die Polyketidsynthese dar. Der Umsatz
ungestättigter Aldehyde lieferte einen geringen Anteil des α-VMAR-Produktes und zudem
einen 3-5%igen Anteil racemischen TBS-geschützten γ-Produktes, dessen Bildung jedoch
durch die Erhöhung der zugesetzten Menge Isopropanol von 1.2 eq auf 1.7 eq unterdrückt
werden konnte. Insgesamt jedoch verliefen die Reaktionen mit ungesättigten Aldehyden in
geringeren Enantioselektivitäten.
In Versuchen mit Pyruvatsäureestern konnte kein Umsatz beobachtet werden, woraufhin
höhere Reaktionstempraturen gewählt wurden, die ebenfalls kein positives Ergebnis lieferten.
Die absolute Stereochemie der neu gebildeten Alkoholfunktion wurde durch die MOSHER-
Methode bestimmt. [79] Dabei wurde das VMAR-Produkt 161 mit (R)-(-)- bzw. (S)-(+)-α-
Methoxy-(trifluormethyl)-phenylessigsäurechlorid, Triethylamin und 4-Dimethylamino-
pyridin in die entsprechenden MOSHER-Ester 161-(S)-MTPA und 161-(R)-MTPA umgesetzt.
Der Vergleich der beiden 1H-NMR- Spektren und der Ermittlung der ∆δ-Werte deutete auf
eine (S)-Konfiguration des neu gebildeten Stereozentrums für die Verbindung 161 hin
(Abb. 14).
O
H
MeO
O
O
(R)
CF3
PhOMe
3.69
6.74
5.76
2.48
0.86
1.641.30
O
H
MeO
O
O
(S)
CF3
OMePh
3.71
6.85
5.86
2.53
0.81
1.601.25
OMTPA
H
MeO
O
+0.02
+0,11
+0.1
+0.05
-0.05
-0.04-0.05
(S)
161-(S)-MTPA ∆ δ(SR)= δ(S) - δ(R)161-(R)-MTPA Abbildung 14. Untersuchung der absoluten Konfiguration durch die MOSHER-Methode.
Um das Anwendungsspektrum zu erweitern, wurden im Verlauf der Studien, die für die
Polyketidsynthese wertvollen α-chiralen Aldehyde in der neu entwickelten VMAR-Variante
eingesetzt. Hier sollte die matched-missmatched Situation untersucht und gleichzeitig der
Einfluss von Schutzgruppen untersucht werden. Dazu wurden die vom Roche-Ester bzw. von
Milchsäure abgeleiteten Aldehyde eingesetzt (Schema 34). Als Schutzgruppen kamen die in
der Naturstoffsynthese häufig eingesetzten TBS- sowie die chelatisierende PMB-
Schutzgruppen zum Einsatz.
Es zeigte sich, dass beim Roche-Ester im matched-Fall, sowohl höhere Ausbeuten als auch
gesteigerte Selektivitäten beobachtet werden konnten. Dabei lieferten die TBS-Ether in beiden
Fällen ausgeprägtere Stereoselektivitäten von bis zu 94% ee. Die Verwendung der PMB-
Schutzgruppe lieferte hingegen in sämtlichen Experimenten Enantioselektivitäten von unter

Studien zur Entwicklung neuer VMAR-Methoden
- 48 -
80% ee. Beim Milchsäure-Aldehyd wurden im missmatched-Fall für keine der beiden
Schutzgruppen brauchbare Selektivitäten und Ausbeuten verzeichnet.
Die Betrachtung der Ergebnisse, deutet auf eine offensichtliche Bevorzugung des Felkin-
Produkts hin. Besonders bei den eingesetzten Milchsäurealdehyden, sind deutliche Abnahmen
der Ausbeute und Stereoselektivitäten zum anti-Felkin-Produkt erkennbar.
ORO
(L)-12535
ORO
O
RO
O
RO
OHRO
OMe
O
OHRO
OMe
O
OH
ROOMe
O
OH
ROOMe
O
R = TBS 37R = PMB 166
R = TBS 37R = PMB 166
R = TBS 171R = PMB 172
R = TBS 175, 11%, 56% eeR = PMB 176, 46%, 61% ee
(D)-12535
(L)-12535
(D)-12535
R = TBS 167, 65%, 94% eeR = PMB 168, 61%, 79% ee
R = TBS 169, 64%, 90% eeR = PMB 170, 56%, 77% ee
R = TBS 173, 59%, 90% eeR = PMB 174, 50%, 68% ee
R = TBS 171R = PMB 172
Schema 34. Untersuchung der VMAR an α-chiralen Aldehyden.
Da hier eines der beiden möglichen Produkte bevorzugt gebildet wird, wurde versucht eine
kinetische Racematspaltung an α-substituierten Aldehyden mit dem Oxazaborolidinon 125
durchzuführen. Dieses könnte den gleichzeitigen Aufbau von zwei Stereozentren und
zusätzlich die Synthese mit einfachen Bausteinen ermöglichen. Daher wurden die in Schema
35 dargestellten Silylether in der VMAR eingesetzt. Studien in diesem Bereich zeigten, dass
in α-Position sterisch anspruchsvolle Substituenten benötigt werden, um akzeptable
OTBSO
(L)-12535OHTBSO
OMe
O
167, 67%, 1:1 dr
O
OTBS
(L)-12535OH
OTBS
OMe
O
173, 53%, 4:1 dr
O
OTBDPS
(L)-12535OH
OTBDPS
OMe
O
178, 50%, 6:1 dr
( )-37
( )-171
( )-177 Schema 35. Versuche zur kinetischen Racematspaltung.

Studien zur Entwicklung neuer VMAR-Methoden
- 49 -
Diastereoselektivitäten zu erzielen. Hier konnte mit dem TBDPS-geschützten Aldehyd 177 in
die höchste Selektivität von 6:1 für das Felkin-Produkt erzielt werden.
Die anfänglichen Experimente konnten zeigen, dass mit der neuen VMAR-Methode
prinzipiell kinetischen Racematspaltungen möglich sind. Allerdings wurden die Studien im
Rahmen dieser Arbeit nicht weiter vertieft. Sicherlich ist durch weitere Optimierung der
Versuchsbedingungen, besonders durch die richtige Wahl der α-Substituenten ein besseres
Ergebnis zu erwarten.
Nachdem wir die Leistungsfähigkeit der Oxazaborolidinon 125 vermittelten VMAR am
unsubstituierten Ketenacetal 35 verdeutlichen konnten, wurden diese Ergebnisse von uns
erfolgreich publiziert [80] und nach kürzester Zeit von weiteren Forschungsgruppen in der
Naturstoffchemie angewendet.
Als einer der ersten wendete die renommierte Arbeitsgruppe um SMITH die neue VMAR-
Methode in ihrer Totalsynthese von (+)-Psymberin (179) an. [81] Dieses konnte nochmals die
Notwendigkeit neuer VMAR-Methoden für die Polyketidsynthese verdeutlichen, speziell
solche, die auf aliphatische Aldehyden anwendbar sind. In ihrer Synthese benutzten SMITH
et al. die neue VMAR-Methode für das Grundgerüst vom Tetrahydopyran-Fragment 180
(Schema 36). Sie konnten durch die Umsetzung vom TBS-geschütztem Aldehyd 183 mit dem
Ketenacetal 35, den gewünschten γ-Hydroxy-α,β-ungesättigte Ester 182 in hervorragenden
Enantioselektivtäten von >99% ee erhalten. Dieser wurde anschließend selektiv epoxidiert
und nach einigen weiteren Transformationen in das Tetrahydropyran-Fragment 180 überführt.
O
OOMe
CO2Me
OTBS
HH O OH OTBS O
OMeO
TBSO OH O
OMe
TBSO180
181 182
OTBS
OMe
18335
125, i-PrOH
PrCN, -78 °C
66%, >99% ee
OMe
OHNH
O OMe
O
OH
OHO
O
HO
OH
H
H
(+)-Psymberin 179
O
Schema 36. Die VMAR als Schlüsselschritt für das Tetahydropyran-Fragment 180 von (+)-Psymberin (179). [81]

Studien zur Entwicklung neuer VMAR-Methoden
- 50 -
Neben den unsubstituierten Ketenacetal 35, ist eine Anwendung mit dem α-methylierten
Ketenacetal 184 und dem Dien 185 ebenfalls interessant für die Polyketidsynthese. Die
Experimente sind für die neue VMAR-Methode insofern bedeutend, da durch zusätzliche
sterische Hinderung in der α-Position ein positiver Einfluss auf die Regioselektivität möglich
werden könnte. Diese währe besonders wichtig für die VMAR mit ungesättigten Aldehyden,
da hier in sämtlichen Beispielen ein geringer Anteil des α-Alkylierungsprodukts beobachtet
werden konnte.
Um die Methode an α-methylierten Diensystemen zu untersuchen, wurde das Ketenacetal 184
und das Dien 185 analog zu den vorherigen Ketenacetalen aus Tiglinsäuremethylester
bzw. Tiglinaldehyd synthetisiert und durch Umsatz mit verschiedenen Aldehyden getestet
(Tab. 16).
Tabelle 16. Untersuchung der VMAR am a-methylierten Ketenacetal 184 und Dien 185.
R
O
R
OH
R2
OR1
R
O
PrCN, -78 °C, 3h
NB
OO
Ts
Ph
125
HN
100 mol%, i-PrOH
R = OMe = H
R1 = TBS, R2 = OMe 184R1 = TMS, R2 = H 185
Nr.a Aldehyd Nucleophil γ : α Ausbeute (%) γ-Aldolprodukt
ee (%)b γ-Aldolprodukt
1 Isobutyraldehyd 184 99 : 1 109 45 80 2 Valeral 184 99 : 1 186 48 68 3 Cyclohexanal 184 99 : 1 187 41 80 4 Benzaldehyd 184 99 : 1 188 38 44 5 Isobutyraldehyd 185 - - - 6 Valeral 185 - - -
a) 1 eq Aldehyd und 1.2 eq KA. b) bestimmt durch die MOSHER-Methode.
Aus den Ergebnissen von Tabelle 16 ist zu erkennen, dass die zusätzliche Methylgruppe in
α-Position zu geringeren Ausbeuten und Enantioselektivitäten führt. In der VMAR zwischen
Isobutyraldehyd (40) und Ketenacetal 184 konnten Enantioselektivitäten von lediglich 80% ee
verzeichnet werden. Die übrigen Aldehyde führten zu deutlich geringeren Enantio-
selektivitäten. Hier ist jedoch zu erwähnen, dass die Erwartung einer Unterdrückung der
α-Alkylierung in der Reaktion mit Benzaldehyd bestätigt wurde.
3.5.3 Weinsäure abgeleite Acyloxyborane
Durch Verwendung von Oxazaborolidinonen in der VMAR konnten erste positive Ergebnisse

Studien zur Entwicklung neuer VMAR-Methoden
- 51 -
erzielt werden. Da mit diesen chiralen Lewis-Säuren noch nicht das volle Potential
ausgeschöpft wurde, sollten weitere Oxazaborolidin-Derivate in die Studien miteinbezogen
werden. Eine Variante der Oxazoborolidin-Familie stellen die ebenfalls von YAMAMOTO
entwickelten auf Weinsäure basierenden Acyloxyborane (AXB) dar. [82] Diese fielen
besonders durch ihre hohe Reaktivität und Stereoselektvität auf. In YAMAMOTO ‘s
Untersuchungen zeigte sich, dass eine intramolekulare Wasserstoffbrücke zwischen der
Carbonsäure und dem Alkoxysauerstoffatom existiert, wodurch eine zusätzliche Aktivierung
der Lewis-Säure resultiert, was zu einer höheren Reaktivität führt. Dieses wird als Brønsted-
Säure Aktivierung der Lewis-Säure bezeichnet. Weiter konnte durch NOE-Experimente die
Ursache für die hohen Selektivitäten nachgewiesen werden. Diese resultieren aus der
π-Stapelung des Dimethoxyphenylringes und des koordinativ gebundenen Aldehyds, was zu
einer wirksamen Abschirmung des Aldehyds führt.
O
OB
OO
O
RH
OO
H
F3C
CF3
O
MeO
MeO
189 Abbildung 15. Übergangszustand nach YAMAMOTO für Acyloxyborane (AXB). [82]
Des Weiteren konnte SATO in seinen Arbeiten zeigen, dass mit Acyloxyboranen die VMAR
am Dienolat 86 höhere Stereoselektivitäten erzielt werden können als beim Einsatz von
Oxazaborolidinonen. [83]
Aus diesem Grund lag es nahe, Acyloxyboran-Katalysatoren auch in Reaktionen mit den
γ-methyl-substituierten Ketenacetalen 38 bzw. 41 zu untersuchen.
Im Gegensatz zu den Oxazaborolidinonen erfolgt die Synthese der Acyloxyboran Vorläufern
in mehren Stufen und beginnt mit kommerziell erhältlichen 2,6-Dihydroxybenzoesäure (190)
(Schema 37). Nach der Veresterung mit Methyliodid erfolgt die Alkylierung der
freien Alkoholgruppen mit Halogenalkanen. Durch Verseifung der Ester-Funktion und
anschließender Trifluoressigsäureanhydrid vermittelten Veresterung mit Dibenzyltartrat 196
werden die Weinsäurederivate isoliert. Eine Benzylentschützung durch Palladium-
katalysierten Hydrierung unter Wasserstoffatmosphäre führte schließlich zu den gewünschten

Studien zur Entwicklung neuer VMAR-Methoden
- 52 -
HO OH
HO O
190
K2CO3, MeI
DMF, RT, 91%HO OH
MeO O
191
K2CO3, RI
DMF, RT, 91%RO OR
MeO O
R = Me 87% 192 = i-Pr 85% 193
RO OR
HO O
R = Me 98% 194 = i-Pr 91% 195
KOH, MeOH∆CO2Bn
OH
CO2Bn
HO
196
a) TFA, CH2Cl2, 0 °C
b) Pd/C, H2, EtOAc
CO2HCO2H
OHO
OOR
OR
R = Me 78% 197 = i-Pr 70% 198
Schema 37. Darstellung von Weinsäure-Derivaten für Acyloxyborane (AXB). Liganden 197 und 198. Sämtliche durchgeführten Reaktionen erfolgten hier entsprechend der
Literatur in guten Ausbeuten.
Die dargestellten Weinsäureliganden wurden in situ mit den entsprechenden Boronsäuren zu
chiralen Lewis-Säuren umgesetzt und in der VMAR mit den bekannten Testsubstraten
eingesetzt (Tab. 17).
Erstmals resultierten hier, bei Verwendung von Acyloxyboranen Enantioselektivitäten von
über 70% ee für das Ketenacetal 38. Interessanteweise führte allein das 3,5-Tri-
fluormethylphenyl-substituierte Boran 204 zu einem Enantiomerenüberschuss von 78% ee
(Tab. 17, Nr. 19). Die entsprechende Reaktion mit Phenyl-substituierten Boran 203 resultierte
in leicht verminderten Enantioselektivitäten von 70% ee (Tab. 17, Nr. 10). Wie bereits von
YAMAMOTO beschrieben, liefen die Reaktionen insgesamt mit dem Diisopropoxyphenyl-
substituierten Liganden 198 in höheren Enantioselektivitäten als mit dem entsprechenden
Dimethoxyphenyl-substituierten Liganden 197.
Auch die Variation der Reaktionstemperatur sowie die Verwendung von Additiven, sollten
zu keiner signifikanten Veränderung der Stereoselektivitäten führen.
Im weiteren Verlauf wurden die Studien mit Acyloxyboranen ebenfalls von SU im Rahmen
der Masterarbeit fortgeführt. Unterschiedlich verzweigte aromatische sowie aliphatische
substitituierte Acyloxyborane, sollten jedoch deutlich niedrigere Enantioselektivitäten
aufweisen als das Acyloxyboran 204.

Studien zur Entwicklung neuer VMAR-Methoden
- 53 -
Tabelle 17. Untersuchung der VMAR mit Acyloxyboranen
O OH
40 66R2
OMe
OTBS
OMe
O
OB
O
OO
HO2C
O
i-PrO
Oi-PrR = H 199 = Ph 200 = 3,5-(CF3)-Ph 201
AXB
OB
O
OO
HO2C
O
MeO
OMe
R1 = H, R2 = Me 38R1 = Me, R2 = H 41
R1
R R
R = H 202 = Ph 203 = 3,5-(CF3)-Ph 204
Nr.a KA AXB T (°C) Additiv γ : α Ausbeute (%) de (%) ee (%)b
1 38 202 -78 - - - - -
2 38 202 -93 - - - - -
3 38 202 -50 - - - - -
4 41 202 -78 - - - - -
5 41 202 -93 - - - - -
6 41 202 -50 - - - - -
7 38 203 -78 - 99 : 1 24 77 70
8 38 203 -93 - 99 : 1 16 77 70
10 38 203 -78 i-PrOH 99 : 1 18 77 70
11 41 203 -78 - 99 : 1 20 42 36
12 41 203 -93 - 99 : 1 13 50 36
12 41 203 -78 i-PrOH 99 : 1 8 42 36
14 38 204 -78 - 99 : 1 19 80 74
15 38 204 -93 - 99 : 1 11 80 74
17 38 204 -78 i-PrOH 99 : 1 16 80 74
18 41 204 -78 - 99 : 1 22 46 40
19c 38 204 -78 i-PrOH 99 : 1 25 80 78
20 38 199 -78 - 99 : 1 - -
21 38 200 -78 - 99 : 1 28 77 59
22 38 201 -78 - 99 : 1 24 80 63 a) 1 eq Aldehyd, 1.5 eq KA, 50 mol% OXB. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) 100 mol% OXB.
3.5.4 Aminoalkohol basierende Oxazaborolidine
Bisherige Erfolge mit dieser Lewis-Säure-Klasse, speziell letzteres mit den Acyloxyboranen,
veranlasste uns, weitere Variationen der Oxazaborolidinone zu untersuchen. Eine der
wichtigsten und häufig verwendeten Vertreter dieser Lewis-Säure-Klasse stellen die von
COREY entwickelten und von Prolinol abgeleiteten Oxazaborolidine (OXI) dar. Diese fielen
besonders im Vergleich mit den restlichen Varianten durch hervorragende Stereoselektivitäten

Studien zur Entwicklung neuer VMAR-Methoden
- 54 -
in der asymmetrischen Synthese auf. [84] Die Ursache hierfür liegt im bizyklischen-System,
wodurch ein deutlich stärker fixierter Übergangszustand durchlaufen wird.
BOECKMAN et al. berichteten 2006 in ihren Syntheseuntersuchungen zu (-)-Rasfonin von einer
erfolgreichen Anwendung unterschiedlicher Oxazaborolidinen in der VMAR mit dem Siloxy-
Furan 205 (Schema 38). [85] Bemerkenswerte Ausbeuten sowie Stereoselektivitäten
resultierten hierbei durch Katalyse mit dem Brönstedt-Säure aktivierten Oxazaborolidin 207.
OTMSO
O
NB
O
PhPh
o-tolTfO 48 mol%
OTMS
OO
205 206 208
CH2Cl2, -78 °C
83%, >95% de
207
Schema 38. VMAR nach BOECKMAN mit Brønsted-Säure aktivierten Oxazaborolidin 207. [85]
In Anlehnung an das COREY-Modell [86] postulierte BOECKMANN für die Reaktion den in
Abbildung 16 dargestellten Übergangszustand 209, in dem ein si-Seitenangriff begünstig ist.
NB
O
Me
Me
HO
R
HMe
Me
OTMSO
TfO
209 Abbildung 16. Übergangszustand für die VMAR nach BOECKMAN. [86]
Aufgrund dieses wichtigen Beispiels der von Prolinol abgeleiteten Oxazaborolidinen wurden
die Bemühungen in diese Richtung verstärkt. Die Experimente wurden zunächst
mit den kommerziell erhältlichen Boranen 210-212 und dem 3,4-Z-Ketenacetal 38
durchgeführt (Tab. 18).
Erste Versuche mit dem Oxazaborolidin 212 führte erstmals zu hervorragende Enantio-
selektivitäten von über 80% ee für das Ketenacetal 38. Ohne weitere Optimierungsschritte
erhielt man mit dem B-Phenyl-substituierten Oxazaborolidin 212 bei zufriedenstellenden
Ausbeuten einen Enantiomerenüberschuss von 83%. Die Diastereoselektivitäten lagen
ebenfalls im sehr zufriedenstellenden Bereich von 95% de.

Studien zur Entwicklung neuer VMAR-Methoden
- 55 -
Tabelle 18. Prolinol abgeleitete Oxazaboroldine an der VMAR mit dem Ketenacetal 38.
NB
O
PhPh
H
NB
O
PhPh
Me
NB
O
PhPh
Ph210 211 212
O OH
4066
OMe
OTBS
OMe
OOXI 50 mol%
38
CH2Cl2, -78 °C, 14 h
Nr.a OXI γ : α Ausbeute (%) de (%) ee (%)b
1 210 - - - -
2 210 - - - -
3 212 99 :1 67 95 83
4c 212 99 :1 70 95 83 a) zusätzlich 4 % racemisches TBS-Produkt, b) bestimmt durch chirale Shift-Messung
mit (+)-Eu(hfc)3 und MOSHER-Methode, c) OXI in situ generiert mit Dichlorphenylboran.
Interessanterweise führten die oftmals in asymmetrischen Reduktionen eingesetzten
Oxazaborolidine 210 und 211 zu keinem Umsatz. Um die Qualität des gekauften
Oxazaborolidins 212 zu prüfen, wurde in einem zusätzlichen Experiment das Oxazaborolidin
212 analog zu den Oxazaborolidinonen aus Dichlorphenylboran und Diphenylprolinol 213,
welches aus Prolinol dargestellt wurde, in situ generiert. Dieser Versuch führte wie
angenommen zum selben Resultat (Tab. 18, Nr. 4).
Da mit dem B-Phenyl-substituierten Oxazaborolidin 212 erstmals hohe Stereoselektivitäten
erreicht werden konnten, lag es nahe, diesen weiter zu modifizieren und auf seine Wirkung zu
untersuchen (Tab. 19). Zur Modifikation wurden unterschiedlich substituierte Aromaten
gewählt. Zudem kamen für zusätzliche Aktivierung des Katalysators verschiedene Brønsted-
sowie Lewis-Säuren zum Einsatz. Sämtliche Reaktionen wurden hier analog der
Oxazaborolidinon vermittelten VMAR durchgeführt. Wie erhofft führte die zusätzliche
Protonierung des Prolinol-Stickstoffs durch Brønsted-Säuren zu einer signifikanten
Steigerung der Dia- und Enantioselektivitäten.
Interessanterweise spielte wiederholt Isopropanol als Additiv eine wesentliche Rolle für die
Stereoselektivitäten. Während in Experimenten mit Brønsted-Säuren aktivierten Oxaza-
borolidinen ohne Zusatz von Additiven die TBS-geschützten VMAR-Produkte in nahezu
identischen Selektivitäten resultierten, wurde durch den Zusatz von äquivalenter Menge an
Isopropanol der entsprechende freie Alkohol 66 mit leicht erhöhten Diastereo- und Enantio-
selektivitäten erhalten (Tab. 19, Nr. 4-8). Insbesondere Trifluormethansulfonsäure
konnte durch eine höhere Enantioselektivität von 90% ee eine Sonderstellung einnehmen

Studien zur Entwicklung neuer VMAR-Methoden
- 56 -
Tabelle 19. Brønsted- und Lewis-Säure aktivierte Oxazaborolidine
NB
O
RO OH
4066
OMe
OTBS
OMe
OOXI 50 mol %
38
-78 °C, CH 2Cl2
R'
R'R'
R'
X
Nr.a R R’ X Additiv γ : α Ausbeute (%) de (%) ee (%)b
1 o-Tol - - 214 - 99 : 1 60 95 83 2 Ph Me - 215 - 99 : 1 60 - 83 3 Ph - OTf 216 - 99 : 1 63 >95 83c
4 Ph - OTf 216 i-PrOH 99 : 1 70 >95 90 5 Ph Me OTf 217 i-PrOH 99 : 1 62 >95 90 6 o-Tol - OTf 218 i-PrOH 99 : 1 54 >95 90 7 o-Tol Me OTf 219 i-PrOH 99 : 1 52 >95 90 8 Ph - NTf2 220 i-PrOH 99 : 1 68 >95 86 9 Ph - NTf2 220 - 99 : 1 58 >95 80c
10 Ph - AlBr3 221 i-PrOH 99 : 1 54 >95 83 11 Ph - SnCl4 222 i-PrOH 99 : 1 74 >95 82
a) 1 eq Aldehyd, 1.5 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3 und MOSHER-Methode. c) TBS-geschütztes Produkt. (Tab. 19, Nr. 4). Die Lewis-Säure aktivierten Oxazaborolidine zeigten neben geringfügig
verbesserten Diastereoselektivitäten keine bedeutenden Veränderungen. Variationen am
Substitutionsmuster der Aromat-Reste zeigten ebenfalls keine signifikanten Unterschiede in
den Stereoselektivitäten.
Parallel zu den Untersuchungen mit den von Prolinol abgeleiteten Oxazaborolidinen sind
weitere Aminoalkohole untersucht worden. Hierfür wurden die Aminoalkohole Valinol und
Phenylalaninol verwendet, welche zuvor von YAMAMOTO entwickelt und erfolgreich in
DIELS-ALDER-Reaktionen eingesetzt werden konnten. [87] Die in Tabelle 20 aufgelisteten
Ergebnisse zeigen, dass mit diesen Aminoalkoholen deutlich geringere Stereoselektivitäten in
der VMAR resultieren. Prolinol scheint hier durch das bizyklische System und die daraus
resultierende höhere Fixierung, wie in vielen asymmetrischen Reaktionen auch in der VMAR
eine Sonderstellung einzunehmen.
Da die VMAR mit dem vom Prolinol abgeleiteten B-Phenyl-substituiertem Oxazaborolidin
216 in sehr guten Stereoselektivitäten resultierte, sollte als nächstes in weiteren Experimenten
der Umsatz der Reaktion gesteigert werden. Aus diesem Grund wurden Reaktionszeiten,
Zutropfzeiten sowie weitere Reaktionsparameter variiert (Tab. 21).

Studien zur Entwicklung neuer VMAR-Methoden
- 57 -
Tabelle 20. Valinol und Phenylalaninol abgeleitete Oxazaborolidine in der VMAR.
NB
O
PhPh
Ph
O OH
4066
OMe
OTBS
OMe
OOXI, i-PrOH
38
-78 °C, 1h, CH 2Cl2
NB
O
PhPh
Ph
RRX X
R = propyl 223 = napthyl 224 = isopropyl 225
R = propyl 226 = napthyl 227 = isopropyl 228
Nr.a OXI X γ : α Ausbeute 66 (%) de (%) ee (%)b
1 223 - 99 : 1 53 >95 68
2 224 - 99 : 1 58 >95 69
3 225 - 99 : 1 42 >95 70
4 223 SnCl4 99 : 1 47 >95 70
5 224 SnCl4 99 : 1 62 >95 76
6 225 SnCl4 99 : 1 61 >95 78
7 223 OTf 99 : 1 53 >95 72
8 224 OTf 99 : 1 58 >95 77
9 225 OTf 99 : 1 42 >95 79
10 226 - 99 : 1 43 >95 72
11 227 - 99 : 1 39 >95 72
12 228 - 99 : 1 48 >95 74
13 226 SnCl4 99 : 1 53 >95 70
14 227 SnCl4 99 : 1 58 >95 78
15 228 SnCl4 99 : 1 42 >95 77
16 226 OTf 99 : 1 53 >95 74
17 227 OTf 99 : 1 58 >95 80
18 228 OTf 99 : 1 42 >95 80 a) 1 eq Aldehyd, 1.2 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3.
In diesen Versuchen konnte allerdings beobachtet werden, dass längere Reaktionszeiten die
Ausbeuten drastisch verringern. Vermutlich stellen die sauren Reaktionsbedingungen den
Grund für die Zersetzung des säurelabilen VMAR-Produktes 66 dar. Durch Verkürzung der
Reaktionszeit auf eine Stunde erhöhte sich wie vermutet die Ausbeute auf 78%.
Darüberhinaus konnte durch eine alternative Aufarbeitung mit 1M NaHCO3 die Ausbeute
nochmals auf 85% verbessert werden (Tab. 21, Nr. 11).
Für die Bestimmung der absoluten Stereochemie der beiden neu gebildeten Stereozentren
wurde zunächst die absolute Konfiguration der Alkohol-Funktion durch die MOSHER-
Methode bestimmt. Analog zu Kapitel 3.5.2 wurde das VMAR-Produkt 66 mit (R)-(-)- bzw.
(S)-(+)-α-Methoxy-(trifluormethyl)-phenylessigsäurechlorid in die entsprechenden MOSHER-

Studien zur Entwicklung neuer VMAR-Methoden
- 58 -
Tabelle 21. Optimierung der VMAR am Methyl-substituierten Ketenacetal 38.
NB
O
PhPh
Ph
216O OH
4066
OMe
OTBS
OMe
O i-PrOH
38
-78 °C, CH 2Cl2
TfO
Nr.a 216 (mol%) Reaktionszeit (h) ∆t Substrat (min) Ausbeute (%) de (%) ee (%)b
1 100 2 10 64 >95 90
2 20 2 10 53 >95 85
3c 50 2 10 47 >95 90
4 50 14 10 33 >95 90
5 50 6 10 44 >95 90
6 50 0.5 10 62 >95 90
7 50 1 10 78 >95 90
8 50 1 30 66 >95 90
9 50 1 5 32 >95 90
10d 50 1 10 65 >95 90
11e 50 1 10 85 >95 90
12f 50 1 10 78 >95 90 a) 1 eq Aldehyd, 1.2 eq KA. b) bestimmt durch chirale Shift-Messung mit (+)-Eu(hfc)3. c) Rkt. bei -94 °C. d) 1.8 eq KA. e) gequencht mit 2M NaHCO3-Lsg. f) gequencht mit Phosphor Puffer pH = 7.
Ester 66-(S)-MTPA und 66-(R)-MTPA überführt und aus den 1H-NMR-Spektren beider
Verbindungen die ∆δ-Werte ermittelt. Hier ergab sich für die Alkohol-Funktion eine (R)-
Konfiguration (Abb. 17).
O
H
Me
MeO
O
O
(R)
CF3
PhOMe
3.71
6.81
5.82
2.67
1.89O
H
Me
MeO
O
O
(S)
CF3
OMePh
3.70
6.79
5.79
2.66
0.86 OMTPA
H
Me
MeO
O
-0.01
-0,02
-0.03
-0.01
+0.01
(R)
∆δ(SR)= δ(S) - δ(R)66-(R)-MTPA
Me
Me
Me
Me Me
Me1.90
0.94
0.86
0.98
0.85
0.85
-0.04
+0.01+0.01
66-(S)-MTPA Abbildung 17. Untersuchung der absoluten Konfiguration von 66 anhand der MOSHER-Methode.
Anschließend erfolgte die Bestimmung der relativen Konfiguration der beiden Stereozentren.
Hierfür wurde der Alkohol 66 zunächst mittels Palladiumkatalyse hydriert und anschließend
unter Säurekatalyse in das Lacton 229 überführt (Schema 39). Durch einen Vergleich mit den
spektroskopischen Literaturdaten für die identischen syn- [88] bzw. anti- [89] Produkte konnte
die syn-Konfiguration für das VMAR-Produkt 66 ermittelt werden.

Studien zur Entwicklung neuer VMAR-Methoden
- 59 -
OH
OMe
O
66
1)Pd/ C, H2, EtOAc2)CSA, CH2Cl2
O
O
229
82%
Schema 39. Bestimmung der relativen Konfiguration durch Überführung von 66 in das Lacton 229.
Als nächstes wurde die VMAR mit OXI 216 auf das 3,4-E-Ketenacetal 41 angewendet. Wie
zuvor mit anderen chiralen Lewis-Säuren beobachtet resultierten hier deutlich geringere
Stereoselektivitäten und Ausbeuten.
NB
O
PhPh
Ph
216O OH
40 66
OMe
OTBS
OMe
Oi-PrOH
41
-78 °C, 1h, CH 2Cl2
TfO 50 mol%
12%, 54% de, 85% ee
OMe
OOH
97
6%
Schema 40. VMAR mit dem Oxazaborolidin 216 am Ketenacetal 41.
Interessanterweise ergab die VMAR mit dem E-Ketenacetal 41 und dem Oxazaborolidin 216
ebenfalls das syn-Produkt 66 (Schema 40).
Die Anwendbarkeit dieser Methode wurde überprüft, indem die optimierten VMAR-
Bedingungen auf eine Reihe unterschiedlicher Aldehyde angewendet wurde (Tab. 22).
Tabelle 22. Untersuchung der VMAR an unterschiedlich verzweigten Aldehyden.
NB
O
PhPh
Ph
216
R
O
R
OH
OMe
OTBS
OMe
Oi-PrOH
38
CH2Cl2, -78 °C, 1h
TfO 50 mol%
Nr.a Aldehyd Ausbeute (%) α-Aldolprodukt
Ausbeute (%) γ-Aldolprodukt
de (%) γ-Aldolprodukt
ee (%)b
γ-Aldolprodukt 1 Pivalinaldehyd <1 230 37 95 90
2d Valeral 3 231 76 92 92
3 Cyclohexanal <1 232 84 95 85
4d Benzaldehyd 23 233 53 54 83
5d E-Zimtaldehyd 24 234 38 51 80
6 2-Furfural 11 235 51 71 76 a) 1 eq Aldehyd, 1.2 eq KA. b) bestimmt durch die MOSHER-Methode

Studien zur Entwicklung neuer VMAR-Methoden
- 60 -
Hierbei ergab sich ein ähnliches Bild wie bei der Oxazaborolidinon vermittelten VMAR
(siehe Kapitel 3.5.2). Einzig die aliphatischen Aldehyde lieferten, im Gegensatz zu den
ungesättigten Aldehyden, verwendbare Ausbeuten und Stereoselektivitäten. Somit stellt diese
Variante der vinylogen MUKAIYAMA -Aldolreaktion eine wertvolle Ergänzung zu der
bestehenden Methode von DENMARK dar, welche ausschließlich mit ungesättigten Aldehyden
das anti-Diastereomer ergibt.
Das von Prolinol abgeleitete Oxazaborolidin 216 wies im Vergleich zum Oxazaborolidinon
125 an γ-methyl-substituierten Ketenacetalen deutlich höhere Stereoselektivitäten auf,
weswegen die Reaktionen mit der chiralen Lewis-Säure 216 an den γ-unsubstituierten
Ketenacetalen 35 und 184 sowie dem Dien 185 wiederholt werden sollten. Speziell die
VMAR am α-methylierten Ketenacetal 184 mit Oxazaborolidinon 125 lieferte
unbefriedigende Stereoselektivitäten, woraufhin diese im weiteren Verlauf optimiert werden
sollte. Hierfür kamen wieder die bekannten Aldehyde zum Einsatz (Tab. 23).
Tabelle 23. Untersuchung der VMAR mit dem Oxazaboroldin 216 mit unterschiedlichen Nucleophilen.
R
O
R
OH
R2
OR3
R2
O
R1 R1CH2Cl2, 1h, -78 °C
NB
O
PhPh
PhOTf 50 mol%
i-PrOH
R1 = H, R2 = OMe, R3 = TBS 35R1 = Me, R2 = OMe, R3 = TBS 184R1 = Me, R2 = TMS, R3 = H 185
R = H, MeR = H, OMe
216
Nr.a Aldehyd Dien Ausbeute (%) α-Aldolprodukt
Ausbeute (%) γ-Aldolprodukt
ee (%)b
γ-Aldolprodukt
1 Isobutyraldehyd 35 <1 88 71 99 2 Pivalinaldehyd 35 <1 160 21 99 3 Valeral 35 3 161 64 98 4 Cyclohexanal 35 <1 162 80 98 5 Benzaldehyd 35 11 163 53 54 6 E-Zimtaldehyd 35 18 164 62 48 7 2-Furfural 35 7 165 49 63 8 Isobutyraldehyd 184 <1 109 42 57 9 Valeral 184 <1 186 44 48
10 Benzaldehyd 184 <1 188 47 40 11 Isobutyraldehyd 185 - - - 12 Valeral 185 - - -
a) 1 eq Aldehyd, 1.2 eq KA. b) bestimmt durch die MOSHER-Methode. Im direkten Vergleich mit dem auf Tryptophan basierenden Oxazaborolidinon 125 führt das
Oxazaborolidin 216 bei aliphatischen Aldehyden zu nahezu identischen Ergebnissen.

Studien zur Entwicklung neuer VMAR-Methoden
- 61 -
Auffällig ist, dass die chirale Lewis-Säure 216 auf ungesättigte Aldehyde und das
α-substituierten Ketenacetal 184 stärker empfindlich reagiert. Hier wurde in beiden Fällen
deutlich geringere Stereoselektivität beobachtet als beim Oxazaborolidinon 125. Wie zuvor
auch konnte mit dem Dien 185 kein Umsatz beobachtet werden.

Studien zur Anwendung der neuen VMAR-Methode
- 62 -
4. STUDIEN ZUR ANWENDUNG DER NEUEN VMAR-M ETHODE
4.1 Die VMAR als Schlüsselschritt in der Synthese von Streptograminen
Nachdem eine neue VMAR-Methode auf Basis von Oxazaborolidinen erfolgreich für das
3,4-Z- und γ-unsubstituierte Ketenacetal 35 und 38 in hohen Stereoselektivitäten entwickelt
wurde, sollte im nächsten Abschnitt der Dissertation diese erstmals in der Synthese von
Polyketiden angewendet werden. Dadurch sollte in einer praktischen Anwendung das
Potential der neuen vinylogen MUKAIYAMA -Aldolreaktion bei der Synthese komplexer
Polyketide untermauert werden.
Inwieweit die neue VMAR-Methode die Polyketidsynthese vereinfachen kann, soll am
Beispiel von Madumycin II (65) verdeutlicht werden (Abb. 18). Madumycin II (65) gehört zu
der Naturstoffklasse der Streptrogramine, welche erstmals in den 60er Jahren entdeckt
wurden. [90] Diese interessanten Naturstoffe besitzen die Fähigkeiten die Proteinbiosynthese
zu verhindern, indem sie die Bakterien-Ribosomen blockieren. Aus diesem Grund finden sie
heute Verwendung als Antibiotika. [91]
HN
OHO
O
O O
O
NX
Y
Antibiotikum X Y
N
O
NH
O
N
O
S
N(C2H5)2
O
O
O
OH
O
Virginiamycin M2 63
Madumycin II 65
Dalfopristin 236
NO
HN
ONH
O
O
NH
XO
N
ON
O
HO
Antibiotikum X
Pristinamycin I 237
Quinupristin 238
NO
O
NO
O
S N
Abbildung 18. Streptogramin Antibiotika (links : Typ A; rechts : Typ B).
Die Streptogramine werden hauptsächlich von der Bakteriengattung der Streptomyceten
produziert, aus denen sie als Kombination von zwei unterschiedlichen Strukturtypen isoliert

Studien zur Anwendung der neuen VMAR-Methode
- 63 -
werden können, die entsprechend als Streptogramin-A bzw. Streptogramin-B Gruppe
bezeichnet werden. Die Streptogramine des Typs-A bestehen aus 23-gliedrigen
polyungesättigten Makrolactonen und stellen eine Kombination aus Polyketiden und Peptiden
dar. Wichtige Vertreter hierfür sind Virginiamycin M2 (63) und Madumycin II (65). Diese
sind strukturell stark miteinander verwandt, unterscheiden sich jedoch durch die eingebaute
Aminosäure und der funktionellen Gruppe am C-16-Atom. Zusätzlich ist hier noch das
semisynthetische Dalfopristin (236) zu nennen.
Streptogramine der Gruppe B hingegen beinhaltet eine aus reinen Peptidlacton-
Verknüpfungen aufgebaute Hauptstruktur, die aus sechs bis sieben Aminosäuren aufgebaut
ist. Ein besonderes Merkmal dieser Verbindungen ist der über Threonin am Ringerüst
gebundene 3-Hydroxy-picolinsäurerest. Bekannte Vertreter dieser Gruppe sind
Pristiniaymycin (237) und das semisynthetische Quinupristin (238).
Eine Kombination aus 70:30 (Dalfopristin:Quinupristin) der semisynthetischen
Streptogramine wird heute unter dem Namen Synercid® in der Humanmedizin zur
Bekämpfung von gram-positiven Bakterien eingesetzt, welche eine Vancomycin- und
Methacillin-Resistenz aufweisen.
Durch ausführliche SAR-Studien konnte Sanofi-Aventis kürzlich das semisynthetische
Virginiamycin-Derivat RPR132552A (239) entwickeln (Abb. 19). [92] Dieses wird zurzeit als
eines der Hauptkomponenten des neuen Medikaments XRP 2868 in klinischen Studien
untersucht, die sich gerade in der klinschen Phase II befindet. Bisher veröffentlichte Studien
weisen für diese Substanz eine um circa vierfach höhere Potenz als Synercid® auf. [93] Diese
aktuellen Forschungsergebnisse zeigen nochmals, dass trotz langjähriger Forschung an den
Streptograminen, das volle Potential dieser Naturstoffklasse noch nicht gänzlich ausgeschöpft
wurde. Aus diesem Grund sind weitere SAR-Studien sinnvoll und könnten eventuell zu noch
wirkungsvolleren Substanzen führen.
HN
OHO
O
O O
O
N
F
N
RPR132552A239
Abbildung 19. Neues Virginiamycin-Derivat RPR132552X (239).

Studien zur Anwendung der neuen VMAR-Methode
- 64 -
Die interessante komplexe Struktur der Streptogramin-A Klasse und ihre Bedeutung
als wirksame Antibiotika veranlasste viele Arbeitsgruppen zur strategischen Entwicklung
einer Totalsynthese. Seither wurden hierzu viele weitere Totalsynthesen sowie
Formalsynthesen veröffentlicht. [94] Nach vielen Bemühungen konnte im Jahr 1996
MEYERS [95] erstmals von einer erfolgreichen Totalsynthese von Madumycin II
(65) berichten. Zeitgleich wurde ebenfalls die erste Totalsynthese von Virginiamycin M2 (63)
von SCHLESSINGER [96] veröffentlicht. Ein Jahr später konnte eine weitere Madumycin II (65)
Synthese von GOUSH [97] publiziert werden.
In diesen publizierten Synthesen wurden mehr als zwanzig lineare Stufen benötigt. Die
Stereozentren wurden dabei durch gewöhnliche asymmetrische Aldolreaktionen oder Aldol-
ähnliche Reaktionen eingeführt. Dieses zeigt, dass eine Synthese dieser Naturstoffklasse mit
gewöhnlichen 1,3 Verknüpfungsmethoden über 20 lineare Reaktionsschritte benötigt
2001 berichtete CAMPAGNE et al. von einer erfolgreichen Anwendung der VMAR in der
Formalsynthese von Streptogramin des Typs-A. [98] Diese Arbeit verdeutlicht, inwieweit die
VMAR eine Synthese verkürzen und infolgedessen effizienter gestallten kann. In der
Synthese wurde der in Kapitel 1.3.2.1 vorgestellte chirale Kupfer-Komplex verwendet und
der ungesättigte Aldehyd 240 mit dem aus Acetoacetat abgeleiteten Dienolat 86 in einer
vinylogen Aldolreaktion umgesetzt (Schema 41), wobei hohe Ausbeuten und Stereo-
selektivitäten von bis zu 81% ee erzielt werden konnten.
O CuF(S)-tol-BINAP10 mol% THF, RT
BocHN
OTMS
OO
86
240
OH
BocHN
O
O
O
241
14BocHNOH
O
O
N
2429
MeO
O
23
80%, 81% ee
Schema 41. Anwendung der VMAR in der Formalsynthese von Streptogramin Antibiotika .[98]

Studien zur Anwendung der neuen VMAR-Methode
- 65 -
Durch weitere Syntheseschritte konnte das C-9-C-24-Grundgerüst 242 der Streptogramin-A
Vertreter in nur zehn linearen Stufen aufgebaut werden. Für die Synthese von Madumycin II
wird eine zusätzliche Stufe für die selektive Reduktion des C-16-Ketons benötigt.
Da dieses Fragment allerdings nicht den vollständigen Polyketidteil von Streptograminen
darstellt und mit den damaligen VMAR-Methoden eine selektive Durchführung nur mit dem
Dienolat 86 möglich war, musste der restliche Polyketidteil durch gewöhnliche Aldol-
reaktionen aufgebaut werden. Darüberhinaus bietet die Route mit dem Dienolat 86 nur
bedingte Möglichkeiten zur Derivatisierung.
Durch Verwendung der neuen, durch Oxazaborolidin vermittelten VMAR-Methode, könnte
in relativ wenigen Synthesestufen der gesamte Polyketidteil von Madumycin II aufgebaut
werden. Im Verlauf der Dissertation wurde versucht, die neue VMAR-Methode in der
Synthese des Polyketidteils von Madumycin II (65) anzuwenden, um so das hohe Potential
der neuen VMAR-Methode zu verdeutlichen.
4.2 Retrosynthese des Polyketidfragments von Madumycin II auf Basis
der neuen VMAR-Methoden
Die retrosynthetischen Überlegungen bezüglich des Polyketidfragments 243 sollten,
entsprechend der heutigen Anforderungen, auf möglichst vielseitig verwendbaren und aus
einfachen Bausteinen aufgebauten Untereinheiten zurückzuführen sein. Hierfür wurde in der
retrosyntetischen Analyse, auf Basis der zuvor neu entwickelten VMAR-Methode, eine
alternative Formalsynthese des Polyketidfragments 243 von Madumycin II (65) entwickelt
(Schema 42).
Hierfür wurden Zwei Syntheserouten erarbeitet, die sich einzig am Schnittpunkt des
Polyketidfragments 243 unterscheiden. Analog zu MEYER’s Totalsynthese soll in der ersten
Route das Fragment 244 an C-11 durch eine Amidbindung verknüpft werden, wobei die
benötigte Aminofunktion aus dem entsprechenden Nitril 245 hervorgehen soll.
In der zweiten Route soll der retrosynthetische Schnitt alternativ an C-8 erfolgen und eine
Palladiumkatalysierte Kreuzkupplung zum Aufbau des Fragments 243 verwendet werden. Die
letztere Route stellt insofern eine interessante Möglichkeit dar, da in bisherigen Streptogramin
Totalsynthesen eine solche Verknüpfung keine Anwendung fand.
Hier sollen dabei die neuen Oxazaborolidin vermittelten VMAR-Methoden, als
Schlüsselschritt für die beiden resultierenden Untereinheiten 246 und 247 dienen.
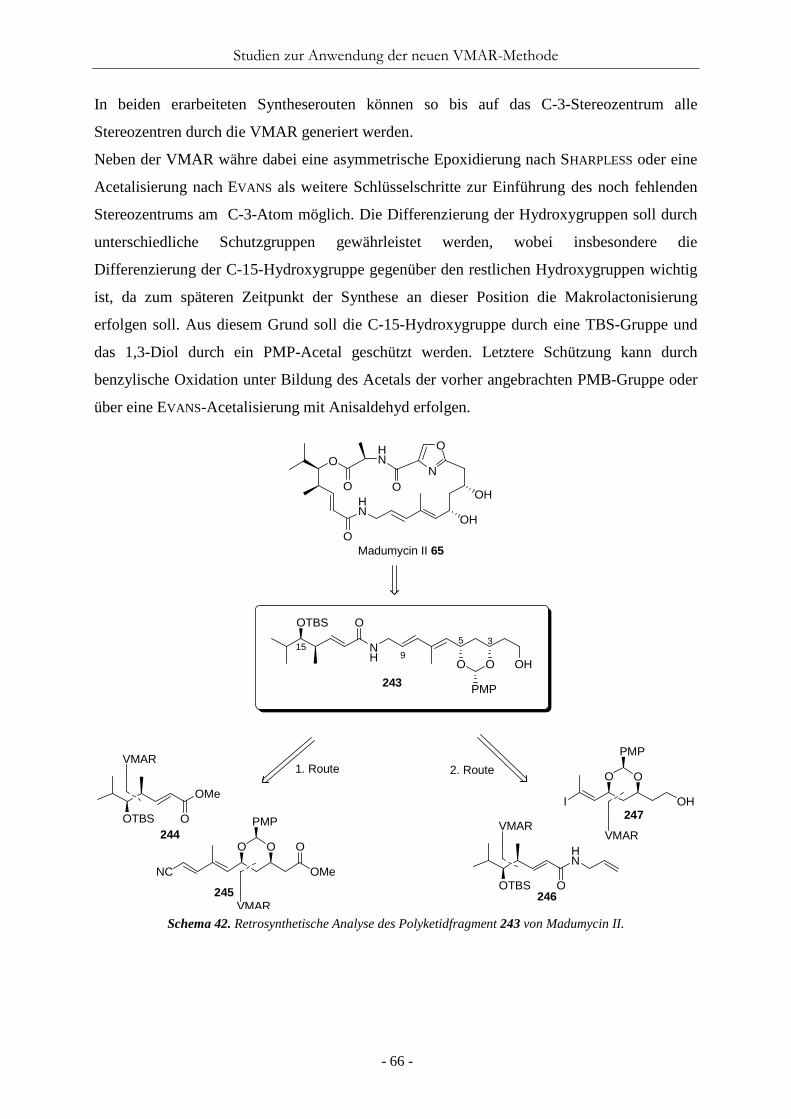
Studien zur Anwendung der neuen VMAR-Methode
- 66 -
In beiden erarbeiteten Syntheserouten können so bis auf das C-3-Stereozentrum alle
Stereozentren durch die VMAR generiert werden.
Neben der VMAR währe dabei eine asymmetrische Epoxidierung nach SHARPLESS oder eine
Acetalisierung nach EVANS als weitere Schlüsselschritte zur Einführung des noch fehlenden
Stereozentrums am C-3-Atom möglich. Die Differenzierung der Hydroxygruppen soll durch
unterschiedliche Schutzgruppen gewährleistet werden, wobei insbesondere die
Differenzierung der C-15-Hydroxygruppe gegenüber den restlichen Hydroxygruppen wichtig
ist, da zum späteren Zeitpunkt der Synthese an dieser Position die Makrolactonisierung
erfolgen soll. Aus diesem Grund soll die C-15-Hydroxygruppe durch eine TBS-Gruppe und
das 1,3-Diol durch ein PMP-Acetal geschützt werden. Letztere Schützung kann durch
benzylische Oxidation unter Bildung des Acetals der vorher angebrachten PMB-Gruppe oder
über eine EVANS-Acetalisierung mit Anisaldehyd erfolgen.
HN
OHO
O
O O
O
N
OH
HN
Madumycin II 65
OOTBS
HN
O O
OH
PMP
I
246
247
243
1. Route 2. Route
VMARVMAR
OOTBS244
VMAR
OMe
NC OMe
OO O
PMP
VMAR245
15 NH
O
9
5 3
OH
OTBS
O O
PMP
Schema 42. Retrosynthetische Analyse des Polyketidfragment 243 von Madumycin II.

Studien zur Anwendung der neuen VMAR-Methode
- 67 -
4.3 Synthetische Arbeiten zum Polyketidfragment von Madumycin II
4.3.1 Erste Route zur Synthese des Polyketidfragments
Die synthetischen Studien zur ersten Route beschäftigten sich mit der Darstelllung des
östlichen Fragments 245 und begann mit der Umsetzung des kommerziell erhältlichen Ethyl-
3-methyl-4-oxocrotonate (248) mit dem Diethylcyanomethylphosphonat (249) in einer
WITTIG-HORNER-Reaktion nach dem NAKANISHI -Protokoll zum Ester 250 (Schema 43). [99]
Hier wurde mit Natriumhydrid bei Raumtemperatur eine E/Z-Selektivität von 4:1 zugunsten
des erwünschten E-Olefins erzielt. Die Isomere konnten jedoch leicht durch Chromatographie
voneinander getrennt werden. Eine alternativ durchgeführte Vorschrift bei niedriger
Temperatur (0°C) und verschiedener Base (n-BuLi) führte dabei zu starken Einbußen
bezüglich der Ausbeute ohne eine registrierbare Verbesserung der E/Z-Selektivität.
Anschließend erfolgte die DiBAl-H Reduktion des Esters 250 zum Allylalkohol, an dem sich
eine Braunstein Oxidation anschloss und so der Aldehyd 251 erhalten werden konnte.
O
NC251
O CO2Et
248
NaH, THF
RT, 16 h, 88%
P(OEt)2NC
O
249 O
NC
250
OEt
E/Z 4:1
1) DiBAl-H, CH2Cl2 -78 °C, 1 h
2) MnO2, CH2Cl2 RT, 12 h 79% (2 Stufen)
Schema 43. Anwendung der VMAR in der Formalsynthese von Streptogramin Antibiotika.
Der synthetisierte Aldehyd 251 wurde im weiteren Verlauf in einer durch Oxazaborolidinon
vermittelten VMAR mit dem Ketenacetal 35 eingesetzt (Schema 44). Für die benötigte
Stereochemie musste das N-Ts-D-Tryptophan verwendet werden. Es wurde nur das
gewünschte γ-Regioisomer in guter Ausbeute von 70% isoliert. Im Gegensatz zur
Regioselektivität führte jedoch die Enantioselektivität (71% ee) dieser Reaktion nur zu einem
bedingt befriedigenden Ergebnis.
O
NC251
OH
35
OMe
OTBS
OMe
O
NB
OO
Ts
PhHN
125
EtCN, i-PrOH, -78 °C
NC
70%, 71% ee252
Schema 44. Enantioselektive VMAR von Ketenacetal 35 und Aldehyd 251.

Studien zur Anwendung der neuen VMAR-Methode
- 68 -
Aus diesem Grund wurde die Reaktion mit dem Prolinol abgeleitetem Oxazaborolidin 216
wiederholt, was allerdings zu geringeren Enantioselektivitäten von 60% ee führte.
Die erstmals 1993 von EVANS vorgestellte Methode zur diastereoselektiven Acetalisierung
von δ-Hydroxy-α,β-ungesättigten Carbonylverbindungen mit aromatischen Aldehyden zählt
heute zu den wichtigsten Methoden zur Darstellung von 1,3-Diolen und sollte im nächsten
Reaktionsschritt ihre Anwendung finden. [100] In Gegenwart katalystischer Mengen einer Base
kommt es zur Bildung eines Halbacetalalkoxids, welches das Michael-System nucleophil
angreift und somit zum 1,3-syn-Acetal führt. Diese Transformation ermöglicht neben dem
stereoselektiven Aufbau einer neuen Hydroxyfunktion, die zusätzliche Umfunktionalisierung
durch eine Schutzgruppe, was zu einer Einsparung an nachfolgenden Reaktionsschritten führt.
Der aus der vinylogen MUKAIYAMA -Aldolreaktion erhaltene Ester 252 wurde in einer
EVANS-Acetalisierung mit Benzaldehyd (73) umgesetzt (Tabelle 24).
In ersten Versuchen konnte allerdings kein Umsatz festgestellt werden. Aus diesem Grund
wurden verschiedene Reaktionsbedingungen untersucht und unterschiedliche Basen sowie
Temperaturen hierfür getestet. Diese Versuche konnten ebenfalls nicht zum erwünschten
Ergebnis führen. Entweder erfolgte kein Umsatz oder eine Zersetzung des Substrats 252,
wobei die Zersetzung bei Temperaturen über 40 °C oder einer Basenkonzentration von über
50 mol% eintrat.
Tabelle 24. Experimente zur Acetalisierung von 252.
OH
OMe
O
NC252
Base, THF
O
73
O
OMe
O
NC253
O
Ph
Nr. T (°C) Base Base (mol%) konz (mmol/mL) Ausbeute (%) 1 0 KOt-Bu 30 0.1 - a 2 10 KOt-Bu 30 0.1 - a 3 RT KOt-Bu 30 0.1 - a 4 40 KOt-Bu 30 0.1 - a 5 50 KOt-Bu 30 0.1 - b 6 0 KOt-Bu 60 0.1 - a 7 -20 KHMDS 30 0.1 - a 8 0 KHMDS 30 0.1 - a 9 RT KHMDS 30 0.1 - b 10 0 KOt-Bu 30 0.5 - a
11 0 KOt-Bu 30 1.0 - a 12 0 KOt-Bu 30 5.0 - a 13 10 KOt-Bu 30 5.0 - b
a) kein Umsatz b) Zersetzung des Substrats.

Studien zur Anwendung der neuen VMAR-Methode
- 69 -
Da kein Erfolg dieser Umsetzung zu erkennen war, erschien eine Abbrechung der
Acetalisierungsversuche als sinnvoll.
Da in dieser Route bereits mit der VMAR nicht die gewünschten Stereoselektivitäten erreicht
wurden und die Experimente zur Acetalisierung trotz Variation der Reaktionsbedingungen
keinen Umsatz zeigten, wurde auf weitere Versuche zum Aufbau des Stereozentrums am
C-3-Atom verzichtet und diese Route schließlich nicht weiter verfolgt.
4.3.2 Zweite Route zur Synthese des Polyketidfragments
Analog zur ersten Route begannen die synthetischen Bemühungen ebenfalls mit der östlichen
Hemisphäre 247 des Polyketidteils 243. Die C-7-C-8-Verknüpfung sollte über eine
Übergangsmetallkatalysierte Kreuzkupplung beider Fragmente erfolgen. Daher musste die
VMAR an einem Aldehyd durchgeführt werden, der die benötigten funktionellen Merkmale
für eine Kreuzkupplung aufweist oder zumindest leicht funktionalisiert werden kann.
Für die anfänglichen Arbeiten wurde hierfür das Vinyliodid 254 und das Vinylstannan 255 als
geeignete Aldehyde angesehen und in der VMAR umgesetzt (Schema 45). Die VMAR mit
diesen Substraten verlief jedoch in schlechten Ausbeuten und Stereoselektivitäten, so dass
diese Substrate für die VMAR nicht weiter in Frage kamen. In beiden Reaktionen konnte ein
zusätzliches Zersetzungsprodukt isoliert werden, was die geringen Ausbeuten erklären konnte.
I
O
254
OH
35
OMe
OTBS
OMe
O
NB
OO
Ts
PhHN
125
EtCN, i-PrOH, -78 °C
I
9%, 66% ee256
Bu3Sn
O
255
OH
35
OMe
OTBS
OMe
O
NB
OO
Ts
PhHN
125
EtCN, i-PrOH, -78 °C
Bu3Sn
7%, 54% ee257
Schema 45. Versuche zur enantioselektiven VMAR mit dem und Vinyliodid 254 und Vinylstannan 255.
Nach den negativen Ergebnissen mit olefinischen Aldehyden war nun die Anwendung der
neuen VMAR-Methode an propargylischen Aldehyden von Interesse. Diese können ebenfalls
in einigen Kreuzkupplungsvarianten eingesetzt oder alternativ für weitere Varianten leicht in
olefinische Vertreter überführt werden. Daher wurde als nächstes das 2-Butinal (258) in der

Studien zur Anwendung der neuen VMAR-Methode
- 70 -
Oxazaborolidinon vermittelten VMAR eingesetzt (Schema 46). Im Gegensatz zu den
vorherigen Experimenten zeigte diese Reaktion jedoch keinen Umsatz.
O
258
OH
35
OMe
OTBS
OMe
O
NB
OO
Ts
PhHN
125
EtCN, i-PrOH, -78 °C
259
Schema 46. Enantioselektive VMAR mit 2-Butinal (258). Das besonders propargylische Aldehyde eine große Herausforderung in stereoselektiven
Aldol- oder Aldol ähnlichen Reaktionen darstellen, ist bereits aus vielen Literaturbeispielen
bekannt. Im Jahr 1972 berichtete NICHOLAS erstmals über eine Addition von Dicobalt-
octacarbonylen an Alkinen, unter Bildung von Dicobalthexacarbonyl-Alkin-Komplexen mit
interessanten Eigenschaften (Schema 47). [101] Die Geometrie vom organischem Rest in
diesen Komplexen gleicht dabei stark die eines Z-Olefins. Ein wichtiges Merkmal dieser
Komplexe ist die chemische Inertheit gegenüber vielen Reaktionsbedingungen, wodurch sie
als Schutzgruppen für Alkine fungieren können. So erlauben diese Komplexe die selektive
Umsetzung von Olefinen in Gegenwart von Alkinen. Zusätzlich ermöglichen diese Komplexe
durch Mesomerie die Stabilisierung von Kationen in Propargylstellung, wodurch sie wichtige
Vorläufer für die NICHOLAS- und PAUSON-KHAND-Reaktionen sind.
R R'Co2(CO)8
-CO2
R R'
Co(CO)3(OC)3Co Schema 47. Allgemeine Schema von Dicobalthexacarbonyl-Alkin-Komplexen.
Weiter konnte NICHOLAS zeigen, dass durch Komplexierung von propargylischen Aldehyden
mit Dicobaltoctacarbonylen, die asymmetrische Crotylborierung in den gewohnten
Stereoselektivitäten durchgeführt werden kann (Schema 48). [102] Seither sind einige Beispiele
für die Anwendung der Dicobalthexacarbonyl-Alkin-Komplexe in Aldol- oder Aldol
ähnlichen Reaktionen beschrieben, welche mit propargylischen Aldehyden Probleme
aufweisen. Beispiele der Cobalt-Komplexe an vinylogen MUKAIYAMA -Aldolreaktionen sind
bisher nur von KOBAYASHI’s arbeiten bekannt. [38d]
O
(OC)3Co Co(CO)3OMe
B(Ipc)2 BF3.Et2O
THF, -78 °C(OC)3Co Co(CO)3
HO
OMe
260 261 262 Schema 48. Asymmetrische Crotylborierung am Cobalt-Alkin-Komplex 260 nach NICHOLAS.

Studien zur Anwendung der neuen VMAR-Methode
- 71 -
Angetrieben durch die positiven Literaturbeispiele wurden die Untersuchungen der VMAR
mit dem Alkin-Komplex fortgeführt. Der benötigte Cobalt-komplexierte Aldehyd 260 konnte
leicht aus 2-Butindiethylacetal (263) in hohen Ausbeuten gewonnen werden (Schema 49).
O
(OC)3Co Co(CO)3260
OEt
OEt
263
a) Co2(CO)8, CH2Cl2, RT
b) Amberlyst 15, Aceton, RT
82%
Schema 49. Asymmetrische Crotylborierung am Cobalt-Alkin-Komplex 260.
Nach der erfolgreichen Synthese des Aldehyds 260 wurde dieser anschließend in der Oxaza-
borolidinon vermittelten VMAR eingesetzt (Schema 50).
OH
35
OMe
OTBS
OMe
O
NB
OO
Ts
PhHN
125
a) EtCN, i-PrOH, -78 °C
259
O
(OC)3Co Co(CO)3260
b) CAN, Aceton, -78 °C
71%, 94% ee
Schema 50. Asymmetrische Crotylborierung am Cobalt-Alkin-Komplex (260). Erstaunlicherweise konnten in dieser Reaktion, ohne weitere Optimierungsversuche
hervorragende Enantioselektivitäten über 90% ee erzielt werden. Um sicher zu gehen, dass
mit dem Cobalt-komplexiertem Aldehyd die identische Stereochemie wie in den anderen
Beispielen resultiert, wurde der Ester 259 mit der MOSHER-Methode untersucht. Wie aus den
in Abbildung 20 dargestellten ∆δ-Werten hervorgeht, wurde auch mit diesem Substrat das
identische Stereoisomer gebildet.
O
OMe
O
H
O
(S)
OMe
CF3
Ph
3.7012
5.8008
6.79122.6340
1.8440
O
OMe
O
H
O
(R)
Ph
CF3
OMe
3.7158
5.9074
6.88762.7031
1.8132
O
OMe
(S)
OMTPA
H
-0.0146
-0.1066
-0.096-0.0691
+0.0307
259-(S)-MTPA 259-(R)-MTPA
Abbildung 20. Bestimmung der absoluten Konfiguration über die MOSHER-Methode.

Studien zur Anwendung der neuen VMAR-Methode
- 72 -
Im nachfolgenden Schritt wurde der synthetisierte Ester 259 in einer EVANS-Acetalisierung
eingesetzt. Erstmals konnte mit dem Ester 259 ein Umsatz mit Benzaldehyd (73) beobachtet
werden. Allerdings war die erreichte 11%ige Ausbeute sowie die Diastereoselektivität (1:1)
nicht befriedigend (Tabelle 25, Nr. 1), weswegen eingehende Optimierungsversuche durch
Variation der Reaktionsparametern sowie den Aldehyden durchgeführt wurden. Diese sollten
letztlich erfolglos bleiben und die Ausbeute sowie Diastereoselektivitäten nicht gesteigert
werden.
Da die EVANS-Acetalisierung wiederholt nicht die erwünschten Ergebnisse lieferte, wurde
diese Transformation für den Aufbau des C-3-Stereozentrums nicht weiter verfolgt.
Im weiteren Verlauf sollte das C-3-Stereozentrum über eine SHARPLESS-Epoxidierung
generiert werden. Hierzu erfolgte zuerst die Schützung der entstandenen Hydroxygruppe des
Esters 259 mit p-Methoxybenzyltrichloracetimidat 265 (Schema 51). Standardbedingungen
mit Camphersulfonsäure oder p-Toluolsulfonsäure lieferte hier zu unerwartet geringen
Ausbeuten von unter 15%. Eine WILLIAMSON -Ethersynthese mit Natriumhydrid und
p-Methoxybenzylchlorid führte hingegen zur Zersetzung des Substrats.
Tabelle 25. Experimente zur Acetalisierung von 259.
OH
OMe
O
259
Base, THFO
OMe
O
264
O
R
Aldehyd
Nr. Aldehyd T (°C) Base Base (mol%) konz (mmol/mL) Ausbeute (%) dr 1 Benzaldehyd 0 KOt-Bu 30 0.1 9 1:1 2 Benzaldehyd 0 KHMDS 30 0.1 9 1:1 3 Anisaldehyd 0 KOt-Bu 60 0.1 6 1:1 4 Anisaldehyd 10 KOt-Bu 30 0.1 5 1:1 5 p-Nitrobenzaldehyd 0 KOt-Bu 30 0.1 11 1:1 6 p-Nitrobenzaldehyd 10 KOt-Bu 30 0.1 8 1:1 7 Benzaldehyd 0 KOt-Bu 30 0.05 6 1:1 8 Benzaldehyd 0 KOt-Bu 30 0.5 11 1:1 9 Benzaldehyd 0 KOt-Bu 30 1.0 10 1:1 10 Benzaldehyd -10 KOt-Bu 30 1.0 7 1:1 11 Benzaldehyd 0 NaOt-Bu 30 0.5 10 1:1 12a Benzaldehyd 0 KOt-Bu 30 0.5 8 1:1 13b Benzaldehyd 0 KOt-Bu 30 0.5 10 1:1 14 Benzaldehyd 0 KOt-Bu 50 0.5 10 1:1 15 Benzaldehyd 0 KOt-Bu 10 0.5 4 1:1
a) Reaktion in Toluol b) Reaktion in Et2O. Schließlich wurde unter Verwendung von BF3·Et2O als Lewis-Säure bei -78 °C eine Methode
mit angemessenen Ausbeuten (83%) etabliert. [103] Anschließend erfolgte die Reduktion des
Esters 266 zum Allylalkohol 267 mit DiBAl-H.

Studien zur Anwendung der neuen VMAR-Methode
- 73 -
OH
OMe
O
259
BF3.Et2O
-78 °C, 83%
CH2Cl2
MeO
CCl3
NH
265
OPMB
OMe
O
266
DibAl-HCH2Cl2
-78 °C, 92%
OPMB
OH
267
Schema 51. Darstellung des Allylalkohols 267.
Die SHARPLESS-Epoxidierung des Allylalkohols 267 verlief im nächsten Schritt mit
außerordentlich hohen Ausbeuten von 82% und Diastereoselektivitäten von >90% de
(Schema 52). [104] Eine Diasteroselektive Öffnung des Epoxids mit Red-Al führte
anschließend zum 1,3-Diol 269, das bereits die gesamte im Zielfragment vorhandene
Stereochemie besitzt. Die C-3-Hydroxygruppe des 1,3-Diols 269 wurde durch benzylische
Oxidation der PMB-Schutzgruppe mit DDQ unter Wasserausschluss durch die Bildung des
p-Methoxybenzylidenketals 270 geschützt. Die abschließende Funktionalisierung zum
Vinyliodid führte schließlich zum Ostfragment 247. Dieses erfolgte durch eine
Hydrostannylierung und nachfolgender Substitution des Zinnorganyls mit Iod. [105] In Analog
durchgeführten Experimenten, indem das Vinyliodid 247 über eine Hydrozirkonierung
generiert werden sollte, blieben hingegen erfolglos.
OPMB
OH
267
(-)-DET, t-BuOOH, Ti(Oi-Pr)4, MS 4Å
PMBO
OH
268
O
CH2Cl2, -20 °C, 82% >90% de
Red-Al, CH2Cl2-5 °C, 92%
PMBO
OH
269
OH
DDQ,CH2Cl2
0 °C, 67%
O
OH
270
O
PMPa) HSnBu3, Pd(PPh3)2Cl2 Benzol, 78%
O
OH
247
O
PMP
Ib) I2, CH2Cl2 0 °C, 77%
Schema 52. Darstellung des Ostfragments 247.
Nach der erfolgreichen Synthese des Ostfragments 247 wurden Arbeiten an der Darstellung
des Westfragments 246 durchgeführt. Die Synthese des Westfragments 246 begann mit dem
in Kapitel 3.5.4 beschriebenen VMAR-Produkt 66, das bereits alle Stereozentren für das
Westfragment beinhaltet. Durch eine kurze Synthesesequenz mit TBS-Schützung der C-15-
Hydroxygruppe, gefolgt von einer Amidierung nach WEINREB mit Trimethylaluminium, [106]
konnte das Westfragment 246 in hohen Ausbeuten erhalten werden (Schema 53). Um
unterschiedliche Kreuzkupplungsstrategien anzuwenden wurde parallel hierzu auch das
entsprechende Propargylamid 272 dargestellt.

Studien zur Anwendung der neuen VMAR-Methode
- 74 -
OH
OMe
O TBSOTf2,6-Lutidin, CH2Cl2
0 °C, 94%
OTBS
OMe
O OTBS
NH
OMe3Al, RNH2CH2Cl2
RT
R = Allyl 246 72% = Propargyl 272 88%
66 271
R
Schema 53. Darstellung des Westfragments 246.
4.3.3 Verknüpfung von West- und Ostfragment
Im Folgenden wurden verschiedene Kreuzkupplungsstrategien für die beiden synthetisierten
Fragmente untersucht. Als potentielle Kupplungsreaktionen kamen etablierte Methoden nach
HECK, SUZUKI sowie STILLE in Frage.
4.3.3.1 STILLE-Kupplung
Als eine der leistungsfähigsten Methoden zur Verknüpfung olefinischer Bindungen hat sich in
den letzen Jahren die Palladiumkatalysierte Kreuzkupplung von Tetraorganostannanen nach
STILLE bewährt. [107] Trotz der hohen Giftigkeit der Organostannane war die große
Toleranz gegenüber funktionellen Gruppen sowie die neutralen Reaktionsbedingungen die
entscheidenden Aspekte, welche zum Erfolg dieser Transformation beigetragen haben.
Insbesondere die milden Reaktionsbedingungen der STILLE-Kupplung waren für uns Grund
genug, um diese in unseren Studien aufzunehmen.
Hierfür musste das alternative Westfragment 272 zuerst in das Tetraorganostannan
umgewandelt werden, was wie zuvor über eine palladiumkatalysierte Hydrostannylierung mit
Tributylzinnhydrid erfolgen sollte. Die Anwendung dieses Verfahrens führte jedoch
unerwartet zum falschen Regioisomer 273 in moderaten Ausbeuten (Schema 54). Das
identische Produkt ergab sich ebenfalls durch eine radikalische Variante mit Azobisisobutyro-
nitril. Als alternative Variante zur Hydrostannylierung des Alkins kam die Methode über
Stannyl-Kupraten in Frage. [108] Allerdings zeigte diese Reaktion nach Literaturbekannter
Durchführung bei -78 °C keinen Umsatz. Die Erhöhung der Reaktionstemperatur führte
hingegen zur Zersetzung des Substrats.
OTBS
NH
O
272
Pd(PPh3)2Cl2,HSnBu3, Benzol oder
AIBN, HSnBu3
OTBS
NH
O
273SnBu3
< 19%
Schema 54. Versuche zur Hydrostannylierung von 272.
Aufgrund der Erfolglosen Hydrostannylierung des Alkins 272 die das falsche Regioisomer

Studien zur Anwendung der neuen VMAR-Methode
- 75 -
lieferten, wurden auf weitere Versuche zur Hydrostannylierung verzichtet.
4.3.3.2 SUZUKI-Kupplung
Neben der STILLE-Kupplung gehört heute die SUZUKI-Kupplung zu den wichtigsten
Palladiumkatalysierten Kreuzkupplungen. [109] In Gegenwart einer Base können hierdurch
Organoboronverbindungen nicht nur mit Vinyl- oder Arylhalogeniden, sondern auch mit
Alkylhalogeniden verknüpft werden. Die hohe Anwendungsbreite und die resultierende hohen
E/Z-Selektivitäten dieser Reaktion waren bedeutende Eigenschaften, die viele Arbeitsgruppen
in den letzten Jahrzehnten häufig dazu bewegte diese Verknüpfung an komplexen Synthese-
bausteinen anzuwenden.
Deswegen sollte auch hier die SUZUKI-Reaktion für die Kupplung beider Fragmente
angewendet werden. Die hierfür benötigte Borspezies 275 konnte erfolgreich über eine
Hydroborierung mit Catecholboran (274) erhalten werden (Schema 55). [110]
OTBS
NH
O
272
OTBS
NH
O
275
BOH
OHCH2Cl2
OB
OH
274
73%
Schema 55. Hydroborierung von 272 mit Catecholboran (274).
In Kupplungsversuchen nach dem SUZUKI-Protokoll konnten allerdings trotz verschiedener
Reaktionsbedingungen kein Umsatz beobachtet werden (Tabelle 26). In sämtlichen
Experimenten resultierte hier nur die Zersetzung beider Fragmente.
Tabelle 26. Experimente zur SUZUKI-Kupplung.
OTBS
NH
O
275
BOH
OH
O
OH
O
PMP
247OTBS
NH
O
243O O
PMP
OH
I
Nr. Reaktionsbedingungen Ausbeute
1 Pd(OAc)2, PPh3, Cs2CO3, DMF, RT -a
2 TlOEt, Pd(PPh3)4, DMF, RT -a
3 Pd(PPh3)4, K2CO3, DMF, RT
-a
a) Zersetzung von 247 und 275.

Studien zur Anwendung der neuen VMAR-Methode
- 76 -
Da in dieser Reaktion ebenfalls keine erfolgreiche Kupplung erzielt werden konnte, erschien
das Abbrechen der Experimente zur SUZUKI-Reaktion als sinnvoll. Stattdessen wurden
weitere Strategien zur Kupplung verfolgt.
4.3.3.3 HECK-Kupplung
Die HECK-Reaktion gehört heute neben der STILLE und SUZUKI-Kupplung zu einer der
populärsten Übergangsmetallkatalysierten Kreuzkupplungen. [111] Aryl- bzw. Vinylhalogenide
und neuerdings auch entsprechende Triflate werden unter basischen Reaktionsbedingungen
und einer Palladiumspezies mit terminalen Olefinen gekuppelt. Hierbei sind sowohl intra- als
auch intermolekulare Beispiele bekannt. Ein besonderer Vorteil dieser Reaktion ist die hohe
Toleranz gegenüber Funktionalitäten bei den olefinischen Kupplungspartnern.
Aus diesem Grund sollte als nächstes die HECK-Reaktion für die Kupplung des Ost- und
Westfragments angewendet werden. Beide Fragmente konnten hierfür unverändert eingesetzt
werden.
Zunächst kamen hierfür die Phosphinfreie Bedingung nach JEFFEREY [112] zum Einsatz.
(Tabelle 27, Nr. 1). Diese führte jedoch zur Zersetzung des Vinyliodids 247, wobei das Olefin
unverändert reisoliert werden konnte
Tabelle 27. Experimente zur HECK-Kupplung.
OTBS
NH
O
246
O
OH
O
PMP
247OTBS
NH
O
243O O
PMP
OH
I
Nr. Reaktionsbedingungen Ausbeute
1 Pd(OAc)2, Bu4NBr, Cs2CO3, NEt3, DMF, RT -a
2 Pd(PPh3)4, NEt3, MeCN, 70 °C -b
3 Pd(OAc)2, P(o-tol)3, NEt3,
MeCN, 70 °C -b
4 Pd(OAc)2, P(o-tol)3,
Bu4NBr, Cs2CO3, NEt3, DMF, RT
-a
5 Pd(PPh3)2Cl2, NEt3, DMF, RT -a
a) Zersetzung von 247 b) Zersetzung von 246 und 247.

Studien zur Anwendung der neuen VMAR-Methode
- 77 -
Auch die Verwendung einiger weiterer Bedingungen unter Variation der Palladiumquelle und
der Reaktionstemperatur führte nicht zum gewünschten Kupplungsprodukt 243. Insbesondere
höhere Reaktionstemperaturen führten zu einer zusätzlichen Zersetzung des Olefins 246
(Tabelle 27, Nr. 2 und 3). Diese ersten Kupplungsversuche zeigten die hohe Empfindlichkeit
der Substrate auf, besonders die des Vinyliodids 247.
1991 veröffentlichte JEFFEREY [113] eine weitere Phopshinfreie Bedingung für HECK-
Reaktionen, mit der auch die Kupplung von Allylalkoholen ermöglicht wird. In unserem
Arbeitskreis konnte sich diese milde Reaktionsbedingung bereits bei der Synthese komplexer
Naturstoffe als zielführende Methode bewähren. [114]
Unter Verwendung von Silber(I)acetat und Palladium(II)acetat nach JEFFREY konnte erstmals
eine Produktbildung beobachtet werden (Schema 56). Die Aufreinigung des isolierten
Produkts konnte jedoch nicht im gewünschten Reinheitsgrad erzielt werden. Erst eine HPLC-
Aufreinigung erlaubte die Interpretation der analytischen Daten, wobei hiernach immer noch
gewisse Verunreinigungen vorhanden waren. Auswertung von 1H-NMR und HRMS-ESI
lieferten eindeutige Indizien für das gewünschte Zielprodukt 243 auf, so dass von einer
erfolgreichen Kupplung zum Zielprodukt 243 in ungefähr 32% ausgegangen werden kann.
So erlaubt die von uns erarbeite Route auf Basis der neuen VMAR-Methoden die schnelle
Synthese des vollständigen Polyketidteils von Madumycin II in nur neun linearen Stufen.
OTBS
NH
O
246
O
OH
247
O
PMP
I
OTBS
NH
O
243O O
PMP
Pd(OAc)2, AgOAc
DMF, RT
32%
OH
Schema 56. Erfolgreiche HECK-Kupplung unter den JEFFEREY-Bedingungen zum Zielfragment 243.
Weitere Optimierungen der erfolgreichen HECK-Kupplung, um das Zielfragment 243 in
höheren Umsatz und steigender Reinheit zu synthetisieren, konnten im Rahmen dieser Arbeit
allerdings nicht mehr durchgeführt werden.

Zusammenfassung und Ausblick
- 78 -
5. ZUSAMMENFASSUNG UND AUSBLICK
Im Rahmen der vorliegenden Dissertation wurden Studien zur asymmetrischen vinylogen
MUKAIYAMA -Aldolreaktion durchgeführt. Die aus dieser Reaktion resultierenden δ-Hydroxy-
α,β-ungesättigte Carbonylverbindungen stellen wertvolle Bausteine für den effizienten und
ökonimischen Aufbau von größeren Polyketidfragmenten dar. Die ständig wachsende
Bedeutung dieser Reaktion für die Polyketidsynthese verleitete uns nach neuen Methoden zu
suchen, die eine enantioselektive Reaktionsführung der VMAR ermöglicht. In den Studien lag
der Schwerpunkt in erster Linie an der VMAR mit γ-methylsubstituierten Ketenacetalen 38
und 41, da die bisher veröffentlichten Varianten nur ein eingeschränktes Anwendungs-
spektrum für eine kleine Anzahl von Substraten umfasst. Hierfür wurde eine Reihe von
unterschiedlichen chiralen Lewis-Säuren dargestellt und ihr Potential in der VMAR zwischen
Isobutyraldehyd (40) und den γ-methylierten Ketenacetalen 38 und 41 untersucht.
Anhand der Ergebnisse der durchgeführten Experimente konnte festgestellt werden, dass mit
sämtlichen chiralen Lewis-Säuren das γ-Aldolprodukt als Hauptregioisomer gebildet wird. Im
Falle des 3,4-Z-Ketenacetals 38 konnten hierbei im Vergleich zum 3,4-E-Ketenacetal 41
höherere Stereoselektivitäten beobachtet werden.
Im Verlauf der Studien zeigten besonders Oxazaborolidine ein hohes Potential auf, um die
VMAR in guten bis sehr guten Stereoselektivitäten zu katalysieren. So konnten in dieser
Arbeit durch Verwendung unterschiedlich aufgebauter Oxazaborolidine erfolgreich neue
asymmetrische VMAR-Varianten etabliert werden.
Durch den Einsatz von Oxazaborolidinonen, die sich von N-Tosyl-Aminosäuren ableiten,
wurden anfänglich nur moderate Ausbeuten sowie Stereoselektivitäten unter 52% ee
bezüglich der VMAR zwischen Isobutyraldehyd (40) und den γ-methylierten Ketenacetalen
38 und 41 beobachtet. Modifikationen der chiralen Lewis-Säure sowie Veränderungen der
Reaktionsparametern konnten allerdings keine Steigerung der Ausbeute und der
Enantioselektivität bewirken.
Die anfänglichen interessanten Ergebnisse mit Oxazaborolidinonen führten jedoch dazu, dass
zunächst weniger komplexe Ketenacetale als Edukte in der VMAR untersucht wurden.
In Zusammenarbeit mit HORZELLA konnte durch Verwendung des, von Tryptophan
abgeleiteten Oxazaborolidinons 125 erfolgreich eine Methode zur asymmetrischen VMAR am
γ-unsubstituiertem Ketenacetal 35 entwickelt werden, in denen Enantioselektivitäten bis
99% ee erreicht wurden (Schema 57). Als besonders wichtig für die hohen Enantio-

Zusammenfassung und Ausblick
- 79 -
selektivitäten in dieser Methode erwies sich der Einsatz von Isopropanol zum Abfangen der
am Edukt befindlichen Siliziumspezies. Ein besonderer Vorteil dieser neu etablierten
Methode ist die Anwendbarkeit auf aliphatische Aldehyde unter Beibehaltung der Stereo-
selektvitäten, wodurch sie eine wertvolle Ergänzung zu den bisherigen Varianten darstellt. Ein
weiterer Vorteil ist der kostengünstige, sowie schnelle Zugang zu der benötigten chiralen
Lewis-Säure. Angetrieben durch die klaren Vorteile, wurde diese Methode deshalb in kurzer
Zeit von renommierten Arbeitsgruppen in ihren Totalsynthesen angewendet.
R
O
R
OH
35
OMe
OTBS
OMe
O
NB
OO
Ts
PhHN
125
R = Alkyl, Aryl
EtCN, i-PrOH, -78 °C
R = Alkyl, Aryl
bis 99% ee
Schema 57. Erste etablierte VMAR mit Oxazaboroldinon 125 am γ-unsubstituiertem Ketenacetal 35.
In ausführlichen Experimenten mit variierten Oxazaborolidin-Systemen konnte das Potential
dieser VMAR-Methode für das Primärziel erweitert werden.
Hier konnten die aus Aminoalkoholen abgeleiteten Oxazaborolidine die erwünschten
Stereoselektvitäten liefern. Durch Verwendung des auf Prolinol basierenden Oxazaborolidins
216 konnte erstmals eine asymmetrische Variante für das Ketenacetal 38 etabliert werden
(Schema 58). Hier führte das Oxazaborolidin 216 speziell an aliphatischen Aldehyden zu
hervorragenden Stereoselektivitäten von bis zu >95 de und 92% ee für das entsprechende,
syn-konfigurierte Produkt. Modifikationen des Oxazaborolidins 216 zeigten hingegen keine
signifikanten Veränderungen der Stereoselektivitäten. Die Reaktion mit dem 3,4-E-
Ketenacetal 41 resultierte interessanterweise ebenfalls in dem syn-konfiguriertem Produkt,
dies allerdings mit deutlich geringeren Stereoselektivitäten.
In Kombination mit DENMARK’s und KOBAYASHI’s Arbeiten ermöglicht diese neue Variante
eine bedeutende Ergänzung für die Synthese komplexer Polyketidbausteine.
R
O
R
OH
38
OMe
OTBS
OMe
O
R = Alkyl, Aryl
CH2Cl2, i-PrOH
R = Alkyl, Aryl
bis >95 de, 92% ee
-78 °C
NB
O
PhPh
PhTfO
216
Schema 58. Zweite etablierte VMAR mit Oxazaboroldinon 216 am γ-methylierten Ketenacetal 38.

Zusammenfassung und Ausblick
- 80 -
Die beiden neu etablierten VMAR-Methoden sollten im weiteren Verlauf der Dissertation für
die Synthese des Polyketidteils 243 von Madumycin II angewendet werden, um so zusätzlich
das Potential der neuen Methoden in der Polyketidsynthese zu untermauern. Eine alternative
Formalsynthese dieses antibakteriellen Naturstoffs, währe zudem für zukünftige SAR-Studien
sehr bedeutend. Zu diesem Zweck wurden verschiedene retrosynthetische Strategien
erarbeitet, von denen der in Schema 59 gezeigte, das Erfolg führende war. Der Polyketidteil
243 wurde hier formell in das westliche 246 und das östliche 247 Fragment zerlegt, wobei für
die Darstellung beider Verbindungen die neu entwickelten VMAR-Methoden als
Schlüsselschritt dienen sollten.
11
OOTBS
HN
O O
OH
PMP
I
246 247
243
NH
O
9
5 3
OH
OTBS
O O
PMP
Schema 59. Der Polyketidteil 243 von Madumycin II und das westliche 246 sowie östliche 247 Fragment.
Die Synthese dieser komplexen Fragmente konnte aufgrund der Vorzüge der VMAR in
wenigen Reaktionsstufen durchgeführt werden (Schema 60). Während dieser Bemühungen
konnte erfolgreich das Anwendungsspektrum der VMAR auf Dicobalthexacarbonyl-
maskierten Aldehyden erweitert werden. Erfolgreiche vinyloge MUKAIYAMA -Aldolreaktionen
sind mit diesen interessanten Substraten bisher nur wenig bekannt, was in naher Zukunft die
Bedeutung der neuen VMAR-Methoden enorm steigern könnte.
OMe
OOH2 Stufen
NH
OOTBS
66 246
O
(CO)3Co Co(CO)3
260
OHOPMB3 Stufen
OHO O
PMP
I
247
O4 Stufen
268
Schema 60. Synthese des östlichen 247 sowie dem westlichen 246 Fragment.

Zusammenfassung und Ausblick
- 81 -
Im abschließenden Syntheseschritt sollten die Fragmente 246 und 247 durch Übergangs-
metallkatalysierte Kreuzkupplungen miteinander verknüpft werden. Populäre Palladium-
katalysierte Kreuzkupplungen wurden hierfür getestet. Durch eine HECK-Reaktion nach dem
JEFFERY-Protokoll konnte eine erfolgreiche Kupplung durchgeführt werden (Schema 61).
Allerdings bedarf diese Kupplung noch an Optimierungen, da das Polyketidfragment 243
nicht in erwünschter Reinheit und Ausbeute isoliert werden konnte. Dennoch zeigten unsere
Studien, die klaren Vorzüge der neuen VMAR-Methoden in der Synthese komplexer
Naturstoffe.
OTBS
NH
O
246
O
OH247
O
PMP
I
Pd(OAc)2, AgOAc
DMF, RT34%243
NH
O
OH
OTBS
O O
PMP
Schema 61. HECK-Kupplung zum Zielfragment 243.
In zukünftigen Arbeiten gilt es die HECK-Kupplungen zwischen beiden Fragmenten zu
Optimieren, um höhere Ausbeuten und Reinheitsgrade zu gewährleisten. Alternativ könnten
hierfür auch weitere Kreuzkupplungen nach NEGISHI oder HIYAMA untersucht werden. Nach
der Etablierung einer effizienten Kupplung könnte diese Syntheseroute für SAR-Studien
eingesetzt werden.
Ein weiteres wichtiges Ziel ist die Entwicklung einer asymmetrischen VMAR an α,γ-methyl-
substituierten Ketenacetalen, da hier mit den neuen Methoden nur unbefriedigende Stereo-
selektvititäten erzielt werden konnten. Ein Schwerpunkt sollte auf einer syn-selektiven
Addition anliegen, da die KOBAYASHI-Variante bereits den Zugang zum anti-konfigurierten
Produkten gewährleistet und durch eine neue Variante hervorragend ergänzt werden kann.
Eine VMAR mit diesen Ketenacetalen ist in Kombination mit der in unserem Arbeitskreis neu
etablierten Kupfer-katalysierten intramolekularen Hydrierung von δ-Hydroxy-α,β-
ungesättigten Aldehyden, welche in ausgezeichneten Diastereoselektvitäten zu Lactolen
führen, besonders interessant.

Experimenteller Teil
- 82 -
6. EXPERIMENTELLER TEIL
6.1 Allgemeine Arbeitshinweise
Alle Reaktionen mit luft- oder feuchtigkeitsempfindlichen Reagenzien wurden in
ausgeheizten Glasgefäßen unter Inertgasatmosphäre (Stickstoff) durchgeführt. Spritzen und
Kanülen wurden bei 80 °C getrocknet und mit Inertgas gespült.
Verwendete Chemikalien
Lösungsmittel sind nur destilliert eingesetzt worden. Absolute Lösungsmittel sind nach den
bekannten Vorschriften getrocknet worden. THF wurde über Natrium/Benzophenon in einer
Argonatmosphäre, Et2O über Natrium und DCM über CaH2 in einer Stickstoffatmosphäre
destilliert. EtCN, BuCN und DMF wurden als absolutes Lösungsmittel vom Hersteller Acros
bezogen.
Reaktionen wurden unter Stickstoffatmosphäre durchgeführt. Bei allen Experimenten wurde,
sofern nicht anders angegeben, ein Magnetrührer verwendet.
Säulenchromatographien wurden mit Kieselgel der Firma Merck (Korngröße 40-63 µm) bei
leichtem Überdruck durchgeführt. Das verwendete Solvens ist jeweils angegeben.
Dünnschichtchromatographien wurden mit DC Aluminiumfolien Kieselgel 60 der Firma
Merck durchgeführt. Die Indikation erfolgte mit Hilfe von UV-Licht (λ = 254 nm) und
Färbereagentien wie Vanillin-, Cer- oder DNPH-Tauchreagenzien.
Instrumentelle Analytik
NMR – Spektroskopie: 1H- und 13C-NMR Spektren wurden mit dem Gerät Avance 400 der
Firma Bruker aufgenommen. Die Angabe der chemischen Verschiebung wurde in δ ppm, die
Kopplungskonstante J in Hz angegeben. Die Zuordnung der Signale wurde zum Teil mit Hilfe
zweidimensionaler NMR-spektroskopischer Methoden (1H-1H-COSY) vorgenommen.
NMR-Spektren wurden mit Hilfe der jeweiligen Lösungsmittelsignale kalibriert:
CDCl3 1H 7.24 ppm 13C 77.0 ppm
MeOH-d4 1H 3.35 ppm 13C 49.0 ppm

Experimenteller Teil
- 83 -
Für die Signale wurden folgende Abkürzungen verwendet: s = Singulett, d = Dublett, t=
Triplett, q = Quartett, m = Multiplett, dd = Dublett von Dubletts, ddd = Dublett von Dubletts
von Dubletts, dt = Dublett von Tripletts, br s = breites Singulett, br d = breites Dublett.
Massenspektren wurden mit dem Gerät Micromass LCT der Firma Waters aufgenommen.
Injektionen für das Elektronenspray-Verfahren (ESI) erfolgten im Loop-Modus in einer
HPLC-Anlage der Firma Waters (Alliance 2695).
Drehwerte [α] wurden mit einem Polarimeter des Typs Perkin-Elmer 341 bestimmt. Die
Messungen erfolgten mit absoluten Chloroform der Firma Merck bei einer Wellenlänge von
λ = 589.3 nm in einer 1 mL Quarzglaszelle. Die Drehwerte [α] wurden in Grad ° und die
Konzentrationen c in g/100 mL angegeben.
Chirale Gaschromatogramme wurden mit dem Gerät HP 5890 II der Firma Hewlett-
Packard unter Verwendung einer Säule vom Typ Hydrodex-β PM capillary column (50 m,
0.25 mm, 723370, Machery-Nagel) mit Wasserstoff als Trägergas und einem Flammen-
ionisationsdetektor gemessen. Die Messung erfolgte bei konstantem Wasserstoffstrom, wobei
die Messung bei 50 °C gestartet wurde.
Chirale Shiftmessungen mit Eu(hfc)3 (Aldrich) wurden mit 10.0 mg Substanz und 18.0 mg
Eu(hfc)3 in 0.7 mL CDCl3 durchgeführt. Als Referenz wurde hierfür die entsprechende
racemische Verbindung dargestellt und gemessen. Anschließend wurde die Enantiomeren
angereicherte Verbindung eingesetzt. Die Bestimmung des Enantiomerenüberschusses
erfolgte durch Integration der Peaks im 1H-NMR.
6.2 Allgemeine Arbeitsvorschriften
AAV 1: N-Tosylierung von Aminosäuren
Zu einer Lösung der entsprechenden Aminosäure (83.9 mmol) in 1M NaOH (150 mL) wird
bei 0 °C p-Toluolsulfonsäurechlorid (17.6 g, 92.3 mmol) in Methyl-tert-Butylether (150 mL)
addiert. Die Mischung wird über Nacht bei RT gerührt. Die Phasen werden getrennt und
die wässrige Phase zweimal mit Methyl-tert-Butylether gewaschen und mit konz HCl
angesäuert. Die ausfallende N-Tosylierte Aminosäure wird filtriert und unter Vakuum
getrocknet.

Experimenteller Teil
- 84 -
AAV 2: VMAR mit Oxazaborolidinon 125 auf Tryptophan Basis
N-Ts-L-Tryptophan (197 mg, 0.55 mmol) wird in CH2Cl2 (5 mL) aufgenommen und mit
62 µL (0.55 mmol) Dichlorphenylboran versetzt. Die Suspension wird 1 h bei RT gerührt.
Das Lösungsmittel wird unter Stickstoffatmosphäre entfernt. Der bleibende Rückstand wird in
2 mL EtCN gelöst und auf -78 °C abgekühlt. Eine Mischung aus dem entsprechendem
Aldehyd (0.55 mmol), Ketenacetal (0.66 mmol) und absoluten Isopropanol (0.66 mmol) in
0.5 mL EtCN wird über 10 min addiert. Die Reaktionsmischung wird 1 h bei -78 °C gerührt
und anschließend mit 5 M NaHCO3-Lsg. gequencht. Die Phasen werden getrennt und die
wässrige Phase mit Methyl-tert-Butylether (3 x 5 mL) extrahiert. Die vereinigten organischen
Phasen werden über MgSO4 getrocknet, filtriert und eingeengt. Nach Aufreinigung durch
Flash-Chromatographie wird der erwünschte γ-Hydroxy-α,β-ungesättigte Ester erhalten.
AAV 3: VMAR mit Oxazaborolidin 216 auf Prolinol Basis
Zu einer Lösung von α,α-Diphenyl-L-prolinol (70 mg, 0.275 mmol) in CH2Cl2 (2.5 mL) wird
31 µL (0.275 mmol) Dichlorphenylboran zugegeben. Das Gemisch wird 1 h bei RT gerührt.
Das Lösungsmittel wird unter Stickstoffatmosphäre entfernt. Der Rückstand wird in 2 mL
CH2Cl2 aufgenommen und auf -78 °C abgekühlt. Triflourmethansulfonsäure (22 µL, 0.247
mmol) in 0.1 mL CH2Cl2 wird langsam zugegeben und für 15 min gerührt. Anschließend wird
eine Mischung aus dem entsprechenden Aldehyd (0.55 mmol), Ketenacetal (0.66 mmol) und
absoluten Isopropanol (0.66 mmol) in 0.5 mL CH2Cl2 über 10 min addiert. Die
Reaktionsmischung wird 1 h bei -78 °C gerührt und anschließend mit 1 M NaHCO3-Lsg.
gequencht. Die Phasen werden getrennt und die wässrige Phase mit CH2Cl2 (3 x 5 mL)
extrahiert. Die vereinigten organischen Phasen werden über MgSO4 getrocknet, filtriert und
das Lösungsmittel am Rotationsverdampfer eingeengt. Nach Aufreinigung durch Flash-
Chromatographie wird der erwünschte γ-Hydroxy-α,β-ungesättigte Ester erhalten.
AAV 4: Synthese der Mosher-Ester
Zu einer Lösung aus dem entsprechendem γ-Hydroxy-α,β-ungesättigtem Ester (29 µmol) in
CH2Cl2 (1 mL) wird bei RT nacheinander Triethylamin (32 µL, 232 µmol), 4-Dimethyl-
aminopyridin (5 mg, 40.6 µmol) und Mosher-Chlorid (21 µL, 116 µmol) gegeben. Nach 1 h
wird die Lösung mit EtOAc (10 mL) verdünnt. Die organische Phase wird nacheinander mit
gesättigter NaHSO4-Lsg. (3 x 5 mL), 2M NaOH-Lsg. (5 mL), gesättigter NaHCO3-Lsg. (2 x
5 mL) und mit gesättigter NaCl-Lsg. (5 mL) gewaschen und über MgSO4 getrocknet, filtriert

Experimenteller Teil
- 85 -
und am Rotationsverdampfer eingeengt. Nach säulenchromatographischer Aufreinigung wird
der erwünschte Mosher-Ester erhalten.
AAV 5: TBS-Schützung von Alkoholen mit TBSOTf
Der entsprechende Alkohol (0.5 mmol) wird in CH2Cl2 (5 mL) gelöst und auf 0 °C abgekühlt.
Nacheinander wird der Lösung langsam 2,6-Lutidin (0.12 mL, 1.0 mmol) und TBSOTf
(0.14 mL, 0.6 mmol) zugesetzt und die Reaktionsmischung 1 h gerührt. Anschließend wird
die Reaktion langsam auf RT erwärmt und mit gesättigter NaHCO3-Lsg. gequencht. Die
wässrige Phase wird mit CH2Cl2 extrahiert (3 x 5 mL). Die vereinigten organischen Phasen
werden über MgSO4 getrocknet, filtriert und am Rotationsverdampfer eingeengt. Nach
säulenchromatographischer Reinigung wird der erwünschte Silylether erhalten.
6.3 Versuchsbeschreibungen zu Kapitel 3
3,4-E-Ketenacetal 41
OMe
O
[114.14]
LDA, TBSClTHF, -78 °C
OMe
OTBS
[228.40]
Diisopropylamin (7 mL, 49.4 mmol) wird in THF (100 mL) auf 0 °C gekühlt. Zu der Lösung
wird langsam n-BuLi (2.5 M in Hexan, 19.8 mL, 49.4 mmol) zugetropft. Die Mischung wird
20 min bei 0 °C gerührt und anschließend auf -78 °C gekühlt. Nacheinander werden über
20 min DMPU (8 mL, 66.2 mmol), trans-3-Pentensäuremethylester (5 g, 43.8 mmol) und
TBS-Chlorid (10 g, 66.2 mmol) in THF (15 mL) zugegeben. Das Reaktionsgemisch wird
nach 1 h auf RT erwärmt und für weitere 2 h gerührt. Anschließend wird durch Zugabe von
gesättigter NaHCO3-Lsg. (50 mL) gequencht. Die Phasen werden getrennt und die wässrige
Phase mit Petrolether extrahiert. Die vereinigten organischen Phasen werden nochmals mit
gesättigter NaHCO3-Lsg. (5 x 100 mL) gewaschen. Anschließend wird über MgSO4
getrocknet und im Vakuum eingeengt. Durch Destillation (Sdp. 58 °C / 1 mbar) erhält man
das erwünschte E-Ketenacetal (8.9 g, 38.9 mmol, 88%) als farbloses Öl.
Hauptprodukt (1,2-Z-41): 1H-NMR (400 MHz, CDCl3) δ 6.15-6.05 (m, 1H), 5.26 (ddq,
J = 15.3, 6.7, 0.6 Hz, 1H), 4.42 (d-artig, J = 10.2 Hz, 1H), 3.52 (s, 3H), 1.68 (ddd, J = 6.7,
1.6, 0.3 Hz, 3H), 0.96 (s, 9H), 0.17 (s, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 158.2, 127.5,
119.2, 80.8, 55.2, 26.2, 19.0, 18.6, -4.1 ppm.

Experimenteller Teil
- 86 -
3,4-Z-Ketenacetal 38
OMe
O
[114.14]
LDA, TBSClTHF, -78 °C
OMe
OTBS
[228.40]
Diisopropylamin (7 mL, 49.4 mmol) wird in THF (100 mL) auf 0 °C gekühlt. Zu der Lösung
wird langsam n-BuLi (2.5 M in Hexan, 19.8 mL, 49.4 mmol) zugetropft. Die Mischung wird
20 min bei 0 °C gerührt und anschließend auf -78 °C gekühlt. Nacheinander werden über
20 min DMPU (8 mL, 66.2 mmol), trans-2-Pentensäuremethylester (5 g, 43.8 mmol) und
TBS-Chlorid (10 g, 66.2 mmol) in THF (15 mL) zugegeben. Das Reaktionsgemisch wird
nach 1 h auf RT erwärmt und für weitere 2 h gerührt. Anschließend wird durch Zugabe von
gesättigter NaHCO3-Lsg. (50 mL) gequencht. Die Phasen werden getrennt und die wässrige
Phase mit Petrolether extrahiert. Die vereinigten organischen Phasen werden nochmals mit
gesättigter NaHCO3-Lsg. (5 x 100 mL) gewaschen. Anschließend wird über MgSO4
getrocknet und im Vakuum eingeengt. Durch Destillation (Sdp. 59 °C / 1 mbar) erhält man
das erwünschte Z-Ketenacetal (9.4 g, 41.1 mmol, 94%) als farbloses Öl.
Hauptprodukt (1,2-Z-38): 1H-NMR (400 MHz, CDCl3) δ 6.09 (tq-artig, J = 10.8, 1.8 Hz, 1H),
5.03 (ddq, J = 10.8, 6.8, 1.0 Hz, 1H), 4.58 (dd, J = 10.8, 1.0 Hz, 1H), 3.58 (s, 3H), 1.64 (dd,
J = 6.8, 1.7 Hz, 3H), 0.95 (s, 9H), 0.17 (s, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 159.6,
125.5, 116.9, 76.7, 55.3, 26.1, 19.0, 13.2, -4.1 ppm.
Ketenacetal 35
OMe
O
[100.12]
LDA, TBSClTHF, -78 °C
OMe
OTBS
[214.38]
Diisopropylamin (7 mL, 49.4 mmol) wird in THF (100 mL) auf 0 °C gekühlt. Zu der Lösung
wird langsam n-BuLi (2.5 M in Hexan, 19.8 mL, 49.4 mmol) zugetropft. Die Mischung wird
20 min bei 0 °C gerührt und anschließend auf -78 °C gekühlt. Nacheinander werden über
20 min DMPU (8 mL, 66.2 mmol), Crotonsäuremethylester (4.4 g, 43.8 mmol) und TBS-
Chlorid (10 g, 66.2 mmol) in THF (15 mL) zugegeben. Das Reaktionsgemisch wird nach 1 h
auf RT erwärmt und für weitere 2 h gerührt. Anschließend wird durch Zugabe von gesättigter
NaHCO3-Lsg. (50 mL) gequencht. Die Phasen werden getrennt und die wässrige Phase mit
Petrolether extrahiert. Die vereinigten organischen Phasen werden nochmals mit gesättigter
NaHCO3-Lsg. (5 x 100 mL) gewaschen. Anschließend wird über MgSO4 getrocknet und im

Experimenteller Teil
- 87 -
Vakuum eingeengt. Durch Destillation (Sdp. 59 °C / 1 mbar) erhält man das erwünschte
Ketenacetal (8.2 g, 38.1 mmol, 87%) als farbloses Öl.
Hauptprodukt (1,2-Z-35): 1H-NMR (400 MHz, CDCl3) δ 6.53-6.41 (m, 1H), 4.82 (dd,
J = 17.2, 2.1 Hz, 1H), 4.57 (dd, J = 10.2, 2.1 Hz, 1H), 4.45 (d, J = 10.4 Hz, 1H), 3.52 (s, 3H),
0.89 (s, 9H), 0.11 (s, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 160.5, 134.2, 108.6, 81.9,
56.5, 27.5, 19.6, -2.3 ppm.
2-Methyl-Ketenacetal 184
OMe
O
[114.14]
LDA, TBSClTHF, -78 °C
OMe
OTBS
[228.40]
Diisopropylamin (7 mL, 49.4 mmol) wird in THF (100 mL) auf 0 °C gekühlt. Zu der Lösung
wird langsam n-BuLi (2.5 M in Hexan, 19.8 mL, 49.4 mmol) zugetropft. Die Mischung wird
20 min bei 0 °C gerührt und anschließend auf -78 °C gekühlt. Nacheinander werden über
20 min DMPU (8 mL, 66.2 mmol), Tiglinsäuremethylester (5.0 g, 43.8 mmol) und
TBS-Chlorid (10.0 g, 66.2 mmol) in THF (15 mL) zugegeben. Das Reaktionsgemisch wird
nach 1 h auf RT erwärmt und für weitere 2 h gerührt. Anschließend wird durch Zugabe von
gesättigter NaHCO3-Lsg. (50 mL) gequencht. Die Phasen werden getrennt und die wässrige
Phase mit Petrolether extrahiert. Die vereinigten organischen Phasen werden nochmals mit
gesättigter NaHCO3-Lsg. (5 x 100 mL) gewaschen. Anschließend wird über MgSO4
getrocknet und im Vakuum eingeengt. Durch Destillation (Sdp. 59 °C / 1 mbar) erhält man
das erwünschte Ketenacetal (9.4 g, 41.1 mmol, 94%) als farbloses Öl.
Hauptprodukt (1,2-Z-184): 1H-NMR (400 MHz, CDCl3) δ 6.74 (dd, J = 17.5, 11.0 Hz, 1H),
4.83 (dd, J = 17.4, 1.6 Hz, 1H), 4.76 (dd, J = 10.8, 1.6 Hz, 1H), 3.52 (s, 3H), 1.61 (s, 3H),
0.89 (s, 9H), 0.12 (s, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 156.3, 136.9, 110.2, 100.2,
60.7, 28.3, 20.7, 13.3, -1.8 ppm.
2-Methyl-Dien 185
O
[84.11]
Et3N, TMSCl, ZnCl2
H
OTMS
[156.29]
840 mg Tiglinaldehyd (10 mmol) in Benzol (50 mL) wird langsam zu einer Suspension von

Experimenteller Teil
- 88 -
Zinkchlorid (100 mg, 0.7 mmol) in Triethylamin (1.3 mL) zugetropft. Nach 1 h wird langsam
1.3 g (12 mmol) Trimethylchlorsilan zugegeben und die Mischung über Nacht bei RT gerührt.
Anschließend werden 10 ml Diethylether hinzugefügt und die Suspension filtriert. Das Filtrat
wird am Rotationsverdampfer eingeengt und der Rückstand nochmals mit 10 ml Petrolether
versetzt und wiederholt filtriert und das Filtrat eingeengt. Durch Destillation (Sdp. 47 °C /
1 mbar) erhält man das erwünschte Dien 185 (922 mg, 5.9 mmol, 59%) als farbloses Öl. 1H-NMR (400 MHz, CDCl3) δ 6.39 (s, 1H), 6.29 (dd, J = 17.1, 10.6 Hz, 1H), 1.77 (d, J = 1.4
Hz, 3H), 0.20 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3) δ 141.2, 137.0, 118.9, 108.3, 8.7,
0.5 ppm.
2-Iodo-3-nitrotoluol 77
NO2a) H2SO4, NaNO2, 0°Cb) KI,RT
NO2
I
[137.13] [263.03]
2-Methyl-6-nitroanilin (25 g, 165 mmol) wird in Essigsäure gelöst (400 ml). Zu dieser Lösung
wird langsam bei 0 °C eine Mischung aus Natriumnitrit (16.5 g, 235 mmol) in H2SO4
(60 ml) zugetropft. Hierbei sollte die Reaktionstemperatur 5 °C nicht übersteigen.
Anschließend wird die Mischung für 20 min bei RT gerührt. Harnstoff (16 g, 279 mmol)
werden langsam zugegeben und für weitere 10 min gerührt. Zu der Lösung wird anschließend
Kaliumiodid (34 g, 230 mmol) in Wasser (200 ml) zugetropft. Des Weiteren werden zuletzt
noch 7 g Natriumbisulphit in Portionen zugegeben. Der ausfallende gelbe Niederschlag wird
filtriert und unter Vakuum getrocknet. Es wurden 37.2 g (141 mmol, 85%) des erwünschten
Produkts als gelber Feststoff erhalten.
Smp.: 59-64 °C; 1H-NMR (400 MHz, CDCl3) δ 7.45-7.27 (m, 3H), 2.59 (s, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 145.6, 132.8, 129.1, 122.2, 92.8, 30.1, 30.1 ppm; HRMS-ESI
[M+Na+] berechnet für C7H6O2N2I1Na: 285.9341, gefunden: 285.9407.

Experimenteller Teil
- 89 -
(+)-2,2’-Dimethyl-binapthylamin 78
a) Cu, DMFb) Pd/C, H2
NH2
NH2NO2
I
[263.03]
[212.29]
2-Iodo-3-nitrotoluol 77 (17.5 g, 75 mmol), Kupferpulver (20 g, 315 mmol) und DMF
(125 ml) werden zusammengegeben und refluxiert. Nach 3 h werden zusätzlich 10 g
Kupferpulver hinzugefügt und für weitere 2 h refluxiert. Die Mischung wird abgekühlt und
filtriert. Dem Filtrat wird 200 ml Methyl-tert-Butylether zugegeben und mit 3 x 100 ml
Wasser extrahiert. Die organische Phase wird über MgSO4 getrocknet, eingeengt und unter
Vakuum getrocknet. Der dunkel gelbe Feststoff wird in 100 ml EtOAc gelöst und mit 100 mg
Pd/C (10%ig) versetzt. Die Suspension wird unter Wasserstoffatmosphäre gerührt. Nach
Beendigung der Reaktion wird die Suspension über Kieselgur filtriert und im Vakuum
eingeengt. Der dunkel gelbe Rückstand (7 g, 33.2 mmol, 89%) wird in 100 ml heißem Ethanol
gelöst. Eine heiße Lösung von 12 g L-Weinsäure (80 mmol) in Ethanol (20 ml) wird
zugegeben und die Mischung abgekühlt. Der ausfallende Niederschlag wird wieder in heißem
Ethanol gelöst und die Kristallisation 5-6 wiederholt. Es wurden 318 mg (1.5 mmol, 4%) des
erwünschten Produkts als weißer Feststoff erhalten.
[α]20D = +118.6 (c 1.0 Pyridin); Smp.: 156-162 °C; 1H-NMR (400 MHz, CDCl3) δ 7.11 (t,
J = 8.0 Hz, 2H), 6.75 (d, J = 8.0 Hz, 2H), 6.67 (d, J = 8.0 Hz, 2H), 3.53 (br s, 4H), 2.0 (s, 6H)
ppm; 13C-NMR (400 MHz, CDCl3) δ 148.1, 138.8, 135.6, 131.9, 129.1, 122.8, 20.3 ppm;
HRMS-ESI [M+H+] berechnet für C14H17O2N2: 213.1392, gefunden: 213.1390.
2,2’-Dimethyl-binapthyliodid 79
a) H2SO4, NaNO2, 0 °Cb) KI, RT I
INH2
NH2
[212.29] [433.05]
2,2’-Dimethyl-binapthylamin 78 (5 g, 24 mmol) wird in H2SO4 gelöst (30 ml). Zu dieser
Lösung wird langsam bei 0 °C eine Mischung aus Natriumnitrit (3.5 g, 68 mmol) in H2SO4
(60 ml) zugetropft. Die Mischung wird bei RT für 20 min gerührt. Harnstoff (2 g, 68 mmol)

Experimenteller Teil
- 90 -
werden anschließend zugegeben und für weitere 10 min gerührt. Danach wird langsam eine
Lösung aus Kaliumiodid (3 g, 68 mmol) in Wasser (50 ml) zugegeben und anschließend in
Portionen 1 g Natriumbisulfit addiert. Die Reaktion wird durch Zugabe von Wasser gequencht
und mit Methyl-tert-Butylether extrahiert (3 x 100 mL). Die vereinigten organischen Phasen
werden über MgSO4 getrocknet, filtriert und eingeengt. Die Reinigung erfolgt über Flash-
Chromatographie (PE/EE 6:1). Es wurde 5 g (11.5 mmol, 48%) des erwünschten Produkts als
gelblicher Feststoff erhalten.
Smp.: 124-127 °C; 1H-NMR (400 MHz, CDCl3) δ 7.65-7.11 (m, 6H), 2.0 (s, 6H) ppm; 13C-NMR (400 MHz, CDCl3) δ 148.1, 138.6, 135.6, 131.9, 128.4, 100.8, 20.3 ppm; HRMS-
ESI [M+Na+] berechnet für C14H12I2Na: 456.8926, gefunden: 456.8903.
2,2’-Dimethyl-binapthyliodid 80
a) n-BuLi, -78 °Cb) HClI
I
[433.05]
I
[308.15]
2,2’-Dimethyl-binapthyliodid 79 (600 mg, 1.4 mmol) in THF (5 ml) wird auf -78 °C gekühlt.
Der Lösung wird langsam n-BuLi (2.5 M in Hexan, 0.56 ml, 1.4 mmol) zugetropft und die
Mischung 2 h gerührt. Anschließend wird vorsichtig 1 M HCl (2 mL) zugetropft und die
Reaktion auf RT erwärmt. Zu der Lösung wird Wasser zugegeben und mit Methyl-tert-
Butylether extrahiert (3 x 10 mL). Die vereinigten organischen Phasen werden über MgSO4
getrocknet, filtriert und eingeengt. Die Reinigung erfolgt über Flash-Chromatographie (PE/EE
8:1). Man erhält das Monoiodid (357 mg, 1.18 mmol, 83%) als farblosen Feststoff.
Smp.: 124-127 °C; 1H-NMR (400 MHz, CDCl3) δ 7.60-7.05 (m, 7H), 2.0 (s, 3H), 1.99 (s, 3H)
ppm; 13C-NMR (400 MHz, CDCl3) δ 148.1, 138.6, 135.6, 131.9, 129.1, 102.8, 20.3,
19.2 ppm; HRMS-ESI [M+Na+] berechnet für C14H13I1Na: 330.9960, gefunden: 330.9793.

Experimenteller Teil
- 91 -
Tris-(2,2’-dimethyl-binapthyl)-6-boran 81
I BF3.Et2O B
[308.15]
[554.57]
Zu einer Suspension von Magnesiumspäne (39 mg, 1.6 mmol) in 5 ml Et2O wird bei 0 °C
langsam eine Lösung aus 2,2’-Dimethyl-binapthyliodid 80 500 mg (1.6 mmol) in Et2O
(10 mL) zugetropft. Die Reaktion wird solange bei RT gerührt bis sich das Magnesium löst.
Anschließend wird das Lösungsmittel entfernt und nacheinander 10 ml Toluol und 0.35 ml
BF3.Et2O (0.2 mL, 1.60 mmol) zugegeben und 5 h refluxiert. Unter Stickstoffatmosphäre wird
vorsichtig die Reaktionsmischung filtriert und das Filtrat eingeengt. Die Aufarbeitung erfolgt
durch Sublimation, gefolgt von einer Umkristallisation in Heptan. Man erhält das Boran
(112 mg, 0.2 mmol, 38%) als weißen Feststoff. 1H-NMR (400 MHz, CDCl3) δ 7.60-7.05 (m, 7H), 2.0 (s, 3H), 1.99 (s, 3H) ppm; 13C-NMR
(100 MHz, CDCl3) δ 148.1, 138.6, 135.6, 131.9, 129.1, 102.8, 20.3, 19.5 ppm.
N-Ts-L-Tryptophan 138
[204.23]
Et3N, TsClTHF
[358.401]
HN
CO2HH2N
HN
CO2HTsHN
2 g (9.8 mmol) L-Tryptophan wird in einem Gemisch aus H2O / THF 9:1 (20 mL) und
Triethylamin (3.4 mL, 18.9 mmol) gelöst. Die Lösung wird auf 0 °C abgekühlt, mit
p-Toluolsulfonsäurechlorid (1.86 g, 9.8 mmol) in THF (18 mL) versetzt und bei RT für 2 h
gerührt. Das Gemisch wird mit Methyl-tert-Butylether gewaschen, die wässrige Phase mit
1M HCl (10 mL) angesäuert und mit EtOAc extrahiert. Die vereinigten organischen Phasen
werden über MgSO4 getrocknet, filtriert und am Rotationsverdampfer eingeengt. Nach
Aufreinigung durch Flash-Chromatographie (Toluol/MeOH 8:1) und Redestillation mit
CH2Cl2 (3 x 20 mL) wird die erwünschte N-Tosylierte Aminosäure (2.89, 8.17 mmol, 84%)
als weißer Feststoff erhalten.

Experimenteller Teil
- 92 -
[α]20D = -41.6 (c 1.0 EtOH); Rf = 0.10 (Toluol/MeOH 8:1); 1H-NMR (400 MHz, MeOH-d4)
δ 7.40 (dd, J = 15.0, 8.2 Hz, 3H), 7.28 (d, J = 8.2 Hz, 1H), 7.09-6.63 (m, 5H), 4.04 (dd,
J = 13.7, 8.2 Hz, 1H), 3.19 (dd, J = 14.7, 5.5 Hz, 1H), 2.97 (dd, J = 14.7, 8.2 Hz, 1H),
2.30 (s, 3H) ppm; 13C-NMR (400 MHz, MeOH-d4) δ 175.3, 144.2, 138.6, 137.9, 130.2,
128.5, 127.7, 124.8, 122.2, 119.7, 119.1, 112.2, 110.3, 58.0, 30.0, 21.5 ppm; HRMS-ESI
[M+H+] berechnet für C18H19O4N2S1: 359.1066, gefunden: 359.1069.
N-Ts-L-Valin 128
[271.33]
CO2HTsHN
Die Verbindung wurde nach AAV 1 synthetisiert. Es wurden 17.2 g (63.4 mmol, 75%) von
N-Ts-L-Valin 128 als weißes Pulver erhalten.
[α]20D = +25.6 (c 1.0 EtOH); Smp: 151-153 °C; 1H-NMR (400 MHz, CDCl3) δ 7.72 (d,
J = 8.3 Hz, 2H), 7.27 (d, J = 8.3 Hz, 2H), 5.18 (d, J = 9.9 Hz, 1H), 3.78 (dd, J = 9.9, 4.7 Hz,
1H), 2.41 (s, 3H), 2.09 (m, 1H), 0.95 (d, J = 6.8 Hz, 3H), 0.86 (d, J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 176.5, 143.9, 136.5, 129.6, 127.3, 60.6, 31.4, 21.6, 19.0,
17.2 ppm; HRMS-ESI [M-H+] berechnet für C12H16O4N1S1: 270.0861, gefunden: 270.0936.
N-Ts-L-Phenylalanin 137
[319.38]
CO2TsHN
Die Verbindung wurde nach AAV 1 synthetisiert. Es wurden 19.2 g (60.1 mmol, 72%) von
N-Ts-L-Phenylalanin 137 als weißes Pulver erhalten.
[α]20D = -5.6 (c 1.0 EtOH); Smp: 157-160 °C; 1H-NMR (400 MHz, CDCl3) δ 7.56 (d,
J = 7.7 Hz, 2H), 7.19 (m, 5H), 7.06 (d, J = 3.4 Hz, 2H), 5.17 (s, 1H), 4.18 (d, J = 7.1 Hz, 1H),
2.94 (dq, J = 7.0, 6.5, 14.1 Hz, 2H), 2.39 (s, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 172.7,
143.2, 137.1, 135.9, 129.6, 129.5, 128.2, 127.0, 126.8, 56.7, 38.8, 21.4 ppm; HRMS-ESI
[M-H+] berechnet für C16H16O4N1S1: 318.0819, gefunden: 318.0802.

Experimenteller Teil
- 93 -
N-Ts- L-tert-Leucin 129
[285.36]
CO2HTsHN
Die Verbindung wurde nach AAV 1 synthetisiert. Es wurden 1.2 g (4.2 mmol, 63%) von
N-Ts-L-tert-Leucin 129 als weißes Pulver erhalten.
[α]20D = +35.6 (c 2.0 EtOH); Smp: 210-214 °C; 1H-NMR (400 MHz, DMSO-d6) δ 7.65 (d,
J = 8.4 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 3.38 (s, 1H), 2.36 (s, 3H), 0.90 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3) δ 177.2, 142.8, 134.2, 128.7, 127.2, 60.0, 31.4, 22.8 ppm;
HRMS-ESI [M-H+] berechnet für C13H18O4N1S1: 284.1032, gefunden: 284.1073.
N-Ts-L-Threonin 149
[273.31]
CO2HTsHN
OH
Die Verbindung wurde nach AAV 1 synthetisiert. Es wurden 17.0 g (62.2 mmol, 74%) von
N-Ts- L-Threonin 149 als weißes Pulver erhalten.
[α]20D = -14.2 (c 2.0 MeOH); Smp: 143-145 °C; 1H-NMR (400 MHz, Aceton-d6) δ 7.74 (m,
2H), 7.36 (m, 2H), 6.24 (br d, J = 13.0 Hz, 1H), 4.21 (qt, J = 7.0 Hz, 1 H), 3.84 (dd, J = 7.0,
12.8 Hz, 1H), 2.41 (s, 3H), 1.19 (d, J = 7.0 Hz, 3H) ppm; 13C-NMR (100 MHz, Aceton-d6)
δ 170.6, 165.9, 144.0, 139.5, 134.1, 131.0, 130.6, 129.4, 128.0, 72.3, 60.4, 21.5, 20.9,
17.3 ppm; HRMS-ESI [M-H+] berechnet für C11H14O5N1S1: 272.0625, gefunden: 272.0600.
L-(+)-Dibenzyltartrat 196
[150.02]
HO
CO2HHO
CO2H
OH
200 °C
[330.11]
HO
CO2BnHO
CO2Bn
Eine Mischung aus L-(+)-Weinsäure (10 g, 66.6 mmol) und Benzylalkohol (28.8 g,
266.4 mmol) wird unter einer Dean-Stark Apparatur bei 200 °C gerührt. Nach 1 h wird die
Temperatur auf 225 °C erhöht und für eine weitere Stunde gerührt. Die Mischung wird

Experimenteller Teil
- 94 -
anschließend auf RT gebracht und das Rohprodukt durch Säulenchromatographie (EtOAc/PE
1:4) aufgereinigt. Es wird das erwünschte (+)-Dibenzyltartrat (12.5 g, 38 mmol, 57%) als
weißer Feststoff erhalten.
[α]20D = +16.6 (c 1.5 CHCl3); Smp: 60-61 °C; Rf = 0.15 (EtOAc/PE 1:4); 1H-NMR (400 MHz,
CDCl3) δ 7.36 (s, 10H), 5.28 (dd, J = 20.0, 12.0 Hz, 4H), 4.62 (d, J = 6.9 Hz, 2H), 3.20 (m,
2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 171.5, 128.7, 72.2, 68.2 ppm; HRMS-ESI [M+Na+]
berechnet für C18H18O6Na: 353.1025, gefunden: 353.1078.
2,6-Diisopropoxybenzoesäuremethylester 193
[168.04]
O OMe
OHHOK2CO3, DMF, RT
[252.31]
O OMe
Oi-Pri-PrOi-PrI
Zu einem Gemisch aus 9.7 g (57.7 mmol) Methyl-2,6-Dihydroxybenzoat 191 und 150 ml
absolutem DMF werden 22.3 g (160 mmol) Kaliumcarbonat und 15.7 ml (180 mmol)
2-Iodpropan zugegeben und die Reaktionsmischung 48 h bei RT gerührt. Die Lösung
wird mit 1M HCl versetzt und mit Methyl-tert-Butylether extrahiert. Die vereinigten
organischen Phasen werden mit gesättigter NaCl-Lsg. gewaschen und über MgSO4 getrocknet
und das Lösungsmittel am Rotationsverdampfer entfernt. Die Reinigung erfolgt durch Flash-
Chromatographie (PE/EtOAc 4:1). Es wird das erwünschte Produkt (7.9 g, 31.3 mmol, 54%)
als weißer Feststoff erhalten.
Smp: 60-61 °C; Rf = 0.27 (EtOAc/PE 1:4); 1H-NMR (400 MHz, CDCl3) δ 7.20 (t, J = 8.4 Hz,
1H), 6.53 (d, J = 8.2 Hz, 2H), 4.51 (m, 2H), 3.87 (s, 3H), 1.32 (d, J = 6.4 Hz, 12H) ppm; 13C-NMR (100 MHz, CDCl3) δ 171.4, 157.2, 134.7, 101.9, 69.1, 46.5, 25.3 ppm; HRMS-ESI
[M+Na+] berechnet für C14H20O4Na: 275.1254, gefunden: 275.1236.
2,6-Diisopropoxybenzoesäure 195
[252.31]
O OMe
Oi-Pri-PrO
[238.12]
O OH
Oi-Pri-PrOKOH, 80 °C
Kaliumhydroxid (20 g, 356 mmol) wird in einem Gemisch aus 90 ml MeOH und 10 ml H2O

Experimenteller Teil
- 95 -
gelöst. Die Lösung wird mit Methyl-2,6-Diisopropoxybenzoat 193 (7.9 g, 31.3 mmol)
versetzt, auf 80 °C erwärmt und über Nacht bei dieser Temperatur gerührt. Die Lösung
wird mit 1M HCl versetzt und mit Methyl-tert-Butylether extrahiert. Die vereinigten
organischen Phasen werden über MgSO4 getrocknet und das Lösungsmittel am
Rotationsverdampfer entfernt. Der Rückstand wird mit Hexan gewaschen und getrocknet. Es
wird das erwünschte 2,6-Diisopropoxybenzoat 195 (7.4 g, 31.2 mmol, 99%) als weißer
Feststoff erhalten.
Smp: 101-103 °C; 1H-NMR (400 MHz, CDCl3) δ 7.28 (t, J = 8.8 Hz, 1H), 6.55 (d, J = 8.4 Hz,
2H), 4.52 (m, 2H), 1.32 (d, J = 6.4 Hz, 12H) ppm; 13C-NMR (100 MHz, CDCl3) δ 171.4,
157.2, 134.7, 101.9, 69.1, 25.3 ppm; HRMS-ESI [M+Na+] berechnet für C13H17O4Na:
236.1048, gefunden: 236.1047.
2,6-Diisopropoxybenzoesäure-L-(+)-tartratester 198
[238.12]
O OH
Oi-Pri-PrO
[370.35]
O O
Oi-Pri-PrO1) TFAA, RT
HOCO2Bn
CO2Bn
OH CO2H
OH
HO2C
2) H2, Pd/C
Zu einer Suspension aus 1.2 g (5.03 mmol) 2,6-Diisopropoxybenzoesäure 195 und 1.66 g
(5.03 mmol) L-(+)-Dibenzyltartrat 196 in 25 ml absolutem Benzol werden 3.1 ml (5.53 mmol)
Trifluoressigsäureanhydrid bei RT tropfenweise zugegeben. Anschließend wird 30 min bei
RT gerührt. Es werden zusätzliche 1.5 mL (2.75 mmol) Trifluoressigsäureanhydrid zugegeben
und die Reaktion mit gesättigter NaHCO3-Lsg. (20 mL) gequencht. Die Phasen werden
getrennt und die wässrige Phase mit Methyl-tert-Butylether (3 x 20 mL) extrahiert. Die
vereinigten organischen Phasen werden über MgSO4 getrocknet und das Lösungsmittel am
Rotationsverdampfer entfernt. Die Reinigung erfolgt durch Flash-Chromatographie
(PE/EtOAc 3:1). Das farblose Öl wird in 10 ml EtOAc gelöst und mit 50 mg 10 % Pd/C
versetzt. Die Reaktionsmischung wird unter Wasserstoffatmosphäre über Nacht gerührt. Die
Mischung wird über Kieselgur filtriert und das Filtrat am Rotationsverdampfer eingeengt. Das
erwünschte Produkt (753 mg, 2.03 mmol, 41%) wird als weißer Feststoff erhalten.
[α]20D = -25.9 (c 1.0 EtOH); Smp: 181-182 °C; 1H-NMR (400 MHz, CDCl3) δ 7.16 (t,
J = 8.2 Hz, 1H), 6.46 (d, J = 7.8 Hz, 2H), 5.72 (d, J = 1.4 Hz, 1H), 4.73 (d, J = 1.4 Hz, 1H),
4.49 (m, 2H), 1.25 (m, 12H) ppm; 13C-NMR (100 MHz, CDCl3) δ 171.4, 167.2, 165.7, 158.8,

Experimenteller Teil
- 96 -
103.7, 74.1, 71.5, 56.1 ppm; HRMS-ESI [M-H-] berechnet für C17H21O9: 369.1186, gefunden:
369.1124.
2,6-Methoxybenzoesäure-L-(+)-tartratester 197
[182.05]
O OH
OMeMeO
[314.06]
O O
OMeMeO1) TFAA, RT
HOCO2Bn
CO2Bn
OH CO2H
OH
HO2C
2) H2, Pd/C
Zu einer Suspension aus 2 g (10.1 mmol) 2,6-Methoxybenzoesäure 194 und 3.37 g
(10.1 mmol) L-(+)-Dibenzyltartrat 196 in 25 ml absolutem Benzol werden 5.6 ml (10.2 mmol)
Trifluoressigsäureanhydrid bei RT tropfenweise zugegeben. Anschließend wird 30 min bei
RT gerührt. Es werden zusätzliche 1.5 mL (2.75 mmol) Trifluoressigsäureanhydrid zugegeben
und die Reaktion mit gesättigter NaHCO3-Lsg. (20 mL) gequencht. Die Phasen werden
getrennt und die wässrige Phase mit Methyl-tert-Butylether (3 x 20 mL) extrahiert. Die
vereinigten organischen Phasen werden über MgSO4 getrocknet und das Lösungsmittel am
Rotationsverdampfer entfernt. Die Reinigung erfolgt durch Flash-Chromatographie
(PE/EtOAc 3:1). Das farblose Öl wird in 10 ml EtOAc gelöst und mit 50 mg 10 % Pd/C
versetzt. Die Reaktionsmischung wird unter Wasserstoffatmosphäre über Nacht gerührt. Die
Mischung wird über Kieselgur filtriert und das Filtrat am Rotationsverdampfer eingeengt. Das
erwünschte Produkt (1.93 g, 6.16 mmol, 61%) wird als weißer Feststoff erhalten.
[α]20D = -35.9 (c 1.0 EtOH); Smp: 192-193 °C; 1H-NMR (400 MHz, CDCl3) δ 7.35 (t,
J = 8.5 Hz, 1H), 6.68 (d, J = 8.5 Hz, 2H), 5.74 (d, J = 2.0 Hz, 1H), 4.50 (d, J = 2.0 Hz, 1H),
3.78 (s, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 171.9, 168.4, 165.2, 158.8, 132.5, 105.0,
74.1, 71.5, 56.4 ppm; HRMS-ESI [M-H+] berechnet für C13H13O9Na: 313.0560, gefunden:
313.0533.

Experimenteller Teil
- 97 -
O-Benzoyl-N-tosyl-L-Threonin 151
[363.42]
CO2BnTsHN
OH
Cl
O
Pyridin, CH2Cl20 °C - RT
[377.41]
CO2HTsHN
O
O
N-Tosyl-L-threoninbenzylester 150 (1 g, 2.75 mmol) wird in Pyridin (5 mL) gelöst.
Anschließend wird bei 0 °C eine Lösung aus Benzoylchlorid (0.36 mL, 3.08 mmol) in
Dichlormethan (5 mL) über 30 min tropfenweise hinzugefügt und die Reaktionsmischung bei
RT über Nacht gerührt. Die Reaktionsmischung wird mit Wasser (30 mL) gewaschen und die
wässrige Phase mit EtOAc (3 x 50 ml) extrahiert. Die vereinigten organischen Phasen werden
mit 1M HCl (100 mL) sowie gesättigter NaHCO3-Lsg. (100 ml) gewaschen, über MgSO4
getrocknet, filtriert und das Lösungsmittel unter reduziertem Druck entfernt. Das Rohprodukt
wird durch Säulenchromatographie (EtOAc/PE 1:4) aufgereinigt. Das Zwischenprodukt
(1.22 g, 2.61 mmol, 94 %; Rf = 0.35 EtOAc/PE 1:3) wird in EtOAc (5 mL) gelöst und mit
10% Pd/C (20 mg) versetzt. Die Suspension wird unter Wasserstoffatmosphäre über Nacht bei
RT gerührt. Die Reaktionsmischung wird über Kieselgur filtriert und das Lösungsmittel unter
reduziertem Druck entfernt. Es wird das erwünschte Produkt (985 mg, 2.61 mmol, 94%) als
weißes Pulver erhalten.
[α]20D = -31.2 (c 1.0 EtOH); Smp: 186-187 °C; 1H-NMR (400 MHz, Aceton-d6) δ 8.04 (m,
2H), 7.80 (m, 2H), 7.63 (m, 1H), 7.47 (m, 2H), 7.38 (m, 2H), 7.10 (br d, J = 9.4 Hz, 1H),
5.39 (dq, J = 6.5, 4.6 Hz, 1H), 4.42 (dd, J = 9.4, 4.6 Hz, 1H), 2.37 (s, 3H), 1.42 (d, J = 6.5 Hz,
3H) ppm; 13C-NMR (100 MHz, Aceton-d6) δ 169.4, 164.9, 143.0, 138.5, 133.0, 130.0, 129.5,
129.4 128.4, 126.9, 70.7, 58.8, 20.5, 15.1 ppm; HRMS-ESI [M-H+] berechnet für
C18H18O6N1S1: 376.0855, gefunden: 376.0901.
O-Biphenyl-N-tosyl- L-Threonin 152
[363.42]
CO2BnTsHN
OH Pyridin, CH2Cl20 °C - RT
[453.12]
CO2HTsHN
O
OCl
O
N-Tosyl-L-threonin-benzylester 150 (1 g, 2.75 mmol) wird in Pyridin (5 mL) gelöst.
Anschließend wird bei 0 °C eine Lösung aus 4-Phenylbenzoylchlorid (667 mg, 3.08 mmol) in

Experimenteller Teil
- 98 -
CH2Cl2 (5 mL) über 30 min tropfenweise hinzugefügt und die Reaktionsmischung bei RT
über Nacht gerührt. Die rosafarbene Reaktionsmischung wird mit Wasser (30 mL) gewaschen
und die wässrige Phase mit EtOAc (3 x 50 ml) extrahiert. Die vereinigten organischen Phasen
werden mit 1M HCl (100 mL) sowie gesättigter NaHCO3-Lsg. (100 ml) gewaschen, über
MgSO4 getrocknet, filtriert und das Lösungsmittel unter reduziertem Druck entfernt. Das
Rohprodukt wird durch Säulenchromatographie (EtOAc/PE 1:5) aufgereinigt. Das zwischen
Produkt (787 mg, 1.76 mmol, 64 %; Rf = 0.75 EtOAc/PE 2:3) wird in EtOAc (5 mL) gelöst
und mit 10% Pd/C (20 mg) versetzt. Die Suspension wird unter Wasserstoffatmosphäre über
Nacht bei RT gerührt. Die Reaktionsmischung wird über Kieselgur filtriert und das
Lösungsmittel unter reduziertem Druck entfernt. Es wird das erwünschte Produkt (630 mg,
1.76 mmol, 68%) als weißes Pulver erhalten.
[α]20D = -21.9 (c 1.2 EtOH); Smp: 156-159 °C; 1H-NMR (400 MHz, Aceton-d6): δ 8.10 (d,
J = 8.5 Hz, 2H), 7.70-7.85 (m, J = 8.5, 2.4 Hz, 6H), 7.35-7.50 (m, J = 8.5, 2.4 Hz, 5H),
5.60 (dd, J = 6.5, 4.6 Hz, 1H), 4.30 (d, J = 4.6 Hz, 1H), 2.40 (s, 3H), 1.45 (d, J = 6.5 Hz, 3H)
ppm; 13C-NMR (100 MHz, Aceton-d6): δ 179.2, 164.3, 157.4, 143.0, 138.5, 131.1, 129.3,
127.1, 113.2, 104.0, 71.0, 59.4, 55.5, 20.5, 16.6 ppm; HRMS-ESI [M-H+] berechnet für
C24H22O6N1S1: 452.1168, gefunden: 452.1167.
5-(R)-Hydroxy-6-methyl-2-heptensäuremethylester 88
OMe
OOH
[172.22]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 71 mg (0.41 mmol, 75%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+17.7 (c 1.3, CHCl3); Rf = 0.29 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.00
(dt, J = 15.4, 7.6 Hz, 1H), 5.90 (dt, J = 15.4, 1.1 Hz, 1H), 3.71 (s, 3H), 3.54-3.46 (m, 1H),
2.45-2.35 (m, 1H), 2.33-2.24 (m, 1H), 1.69-1.65 (m, 1H), 0.93 (dd, J = 6.7, 1.5 Hz, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 167.8, 146.2, 123.2, 75.3, 51.5, 37.1, 33.4, 18.7, 17.2 ppm;
HRMS-ESI [M+MeCN+Na+] berechnet für C11H19O3N1Na: 236.1263, gefunden: 236.1261.

Experimenteller Teil
- 99 -
5-(R)-Hydroxy-6-dimethyl-2-heptensäuremethylester 160
OMe
OOH
[186.24]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 62 mg (0.33 mmol, 61%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+34.3 (c 1.1, CHCl3); Rf = 0.40 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.02
(dt, J = 15.4, 6.5 Hz, 1H), 5.90 (dt, J = 15.7, 1.0 Hz, 1H), 3.71 (s, 3H), 3.35 (dd, J = 10.6,
2.4 Hz, 1H), 2.45-2.39 (m, 1H), 2.21-2.12 (m, 1H), 0.91 (s, 9H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 166.8, 147.5, 122.9, 78.3, 51.5, 34.9, 35.0, 34.9, 25.6 ppm; HRMS-ESI
[M+MeCN+Na+] berechnet für C12H21O3N1Na: 250.1419, gefunden: 250.1415.
5-(S)-Hydroxy-2-nonensäuremethylester 161
OMe
OOH
[186.24]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 72 mg (0.39 mmol, 70%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+13.3 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.97
(dt, J = 15.4, 7.5 Hz, 1H), 5.88 (dt, J = 15.7, 1.4 Hz, 1H), 3.77-3.73 (m, 1H), 3.71 (s, 3H),
2.43-2.26 (m, 2H), 2.34-2.27 (m, 1H), 1.49-1.27 (m, 6H), 0.88 (t, J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 167.6, 147.8, 125.4, 76.1, 52.6, 42.2, 38.3, 28.8, 23.1,
14.2 ppm; HRMS-ESI [M+MeCN+Na+] berechnet für C12H21O3N1Na: 250.1419, gefunden:
250.1412.
5-(R)-Cyclohexyl-5-hydroxy-2-pentensäuremethylester 162
OMe
OOH
[212.28]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 92 mg (0.44 mmol, 80%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.

Experimenteller Teil
- 100 -
[α]20D =
+43.3 (c 1.0, CHCl3); Rf = 0.42 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.99
(dt, J = 15.4, 7.5 Hz, 1H), 5.89 (dt, J = 15.7, 1.4 Hz, 1H), 3.71 (s, 3H), 3.52-3.46 (m, 1H),
2.44-2.37 (m, 1H), 2.34-2.27 (m, 1H), 1.82-1.62 (m, 5H), 1.38-0.93 (m, 6H) ppm; 13C-NMR
(100 MHz, CDCl3) δ 166.8, 146.3, 123.2, 74.4, 51.5, 43.3, 37.1, 29.1, 27.8, 26.4, 26.1,
26.0 ppm; HRMS-ESI [M+MeCN+Na+] berechnet für C14H23O3N1Na: 276.1576, gefunden:
276.1579.
5-(R)-Benzyl-5-hydroxy-2-pentensäuremethylester 163
OMe
OOH
[206.23]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 68 mg (0.33 mmol, 60%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+28.2 (c 1.2, CHCl3); Rf = 0.33 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.37-
7.25 (m, 5H), 6.95 (dt, J = 14.7, 7.5 Hz, 1H), 5.89 (dt, J = 15.7, 1.4 Hz, 1H), 4.81 (t,
J = 6.1 Hz, 1H), 3.70 (s, 3H), 2.70-2.57 (m, 2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.7,
144.9, 143.4, 128.6, 128.0, 125.7, 123.6, 73.1, 51.5, 41.8 ppm; HRMS-ESI [M+MeCN+Na+]
berechnet für C14H17O3N1Na: 270.1106, gefunden: 270.1108.
5-(R)-Benzyl-5-hydroxy-2-pentensäuremethylester 164
OMe
OOH
[232.27]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 70 mg (0.34 mmol, 63%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+1.3 (c 1.0, CHCl3); Rf = 0.30 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.36-
7.22 (m, 5H), 6.99 (dt, J = 15.7, 7.5 Hz, 1H), 6.60 (d, J = 15.7 Hz, 1H), 6.21 (dd, J = 16.0,
6.8 Hz, 1H), 5.93 (dt, J = 15.7, 1.4 Hz, 1H), 4.42 (q, J = 12.6, 6.5 Hz, 1H), 3.71 (s, 3H),
2.52 (t, J = 6.8 Hz, 2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.7, 144.5, 136.2, 131.2,
130.8, 128.6, 127.9, 126.5, 123.8, 71.5, 51.5, 40.1 ppm; HRMS-ESI [M+MeCN+Na+]
berechnet für C16H19O3N1Na: 296.1262, gefunden: 296.1272.

Experimenteller Teil
- 101 -
5-(R)-Furyl-5-hydroxy-2-pentensäuremethylester 165
OMe
OOH
[196.19]O
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 71 mg (0.36 mmol, 66%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+16.0 (c 1.1, CHCl3); Rf = 0.35 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.32
(dt, J = 2.0, 1.0 Hz, 1H), 6.90 (dt, J = 14.7, 7.5 Hz, 1H), 6.27 (dd, J = 3.0, 1.7 Hz, 1H),
6.20 (dd, J = 4.0, 0.7 Hz, 1H), 5.87 (dt, J = 15.4, 1.4 Hz, 1H), 4.77 (q, J = 11.3, 6.5 Hz, 1H),
3.65 (s, 3H), 2.70 (dt, J = 7.2, 1.4 Hz, 2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.6, 155.3,
144.1, 142.3, 123.8, 110.3, 106.5, 66.4, 51.5, 38.2 ppm; HRMS-ESI [M+MeCN+Na+]
berechnet für C12H15O4N1Na: 260.0899, gefunden: 260.0902.
7-(OTBS)-6-(R)-Methyl-5-(R)-hydroxy-2-heptensäuremethylester 167
OMe
OOH
[302.48]
TBSO
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 110 mg (0.37 mmol, 69%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+16.5 (c 1.0, CHCl3); Rf = 0.38 (PE/EtOAc 5:1); 1H-NMR (400 MHz, CDCl3) δ 6.98
(dt, J = 15.4, 7.5 Hz, 1H), 5.88 (dt, J = 15.7, 1.7 Hz, 1H), 3.97-3.92 (m, 1H), 3.76 (dd,
J = 10.2, 3.8 Hz, 1H), 3.70 (s, 3H), 3.64 (dd, J = 9.9, 5.5 Hz, 1H), 2.45-2.37 (m, 1H),
2.31-2.25 (m, 1H), 1.74-1.69 (m, 1H), 0.91 (d, J = 7.2 Hz, 1H), 0.87 (s, 9H), 0.05 (s, 6H)
ppm; 13C-NMR (100 MHz, CDCl3) δ 166.8, 146.3, 122.8, 73.6, 68.2, 51.4, 38.4, 37.2, 25.8,
18.1, 10.1, -5.6, -5.7 ppm; HRMS-ESI [M+H+] berechnet für C15H31O4Si: 303.1992,
gefunden: 303.1999.
7-(OTBS)-6-(R)-Methyl-5-(R)-hydroxy-2-heptensäuremethylester 169
OMe
OOH
[302.48]
TBSO
Die Verbindung wurde nach AAV 2 mit N-Ts-D-Tryptophan synthetisiert. Es wurden 104 mg

Experimenteller Teil
- 102 -
(0.34 mmol, 63%) des erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
-13.1 (c 1.5, CHCl3); Rf = 0.38 (PE/EtOAc 5:1); 1H-NMR (400 MHz, CDCl3) δ 6.98
(dt, J = 15.4, 7.0 Hz, 1H), 5.89 (dt, J = 15.7, 1.4 Hz, 1H), 3.78 (dd, J = 10.2, 4.2 Hz, 1H),
3.70 (s, 3H), 3.69-3.65 (m, 1H), 3.55 (dd, J = 10.2, 7.8 Hz, 1H), 2.48-2.41 (m, 1H), 2.38-
2.30 (m, 1H), 1.75-1.69 (m, 1H), 0.87 (s, 9H), 0.83 (d, J = 7.2 Hz, 1H), 0.05 (s, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.8, 146.2, 122.9, 75.6, 68.4, 51.4, 39.0, 38.0, 25.8, 18.1,
13.4, -5.6, -5.7 ppm; HRMS-ESI [M+H+] berechnet für C15H31O4Si: 303.1992, gefunden:
303.1997.
6-(S)-(OTBS)-5-(R)-hydroxy-2-heptensäuremethylester 173
OMe
OOH
[288.45]
OTBS
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 90 mg (0.30 mmol, 56%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+16.1 (c 1.1, CHCl3); Rf = 0.39 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.99
(dt, J = 15.7, 7.2 Hz, 1H), 5.90 (dt, J = 15.7, 1.7 Hz, 1H), 3.80-3.74 (m, 1H), 3.70 (s, 3H),
3.65-3.59 (m, 1H), 2.30 (dt, J = 7.2, 1.7 Hz, 2H), 1.09 (d, J = 6.5 Hz, 3H), 0.86 (s, 9H),
0.06 (s, 3H), 0.05 (s, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.7, 145.8, 123.8, 74.0,
70.9, 51.4, 34.9, 25.7, 17.9, 17.5, -4.4, -4.9 ppm; HRMS-ESI [M+Na+] berechnet für
C14H28O4Si1Na: 311.1655, gefunden: 311.1655.
7-(OTBS)-6-(R)-methyl-5-(R)-hydroxy-2-heptensäuremethylester 168
OMe
OOH
[308.36]
PMBO
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 103 mg (0.33 mmol, 61%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
+8.1 (c 1.0, CHCl3); Rf = 0.38 (PE/EtOAc 5:1); 1H-NMR (400 MHz, CDCl3) δ 7.24
(d, J = 8.5 Hz, 2H), 7.03 (dt, J = 15.4, 7.5 Hz, 1H), 6.89 (d, J = 8.5 Hz, 2H), 5.09 (dt,
J = 15.7, 1.4 Hz, 1H), 4.45 (s, 2H), 3.81 (s, 3H), 3.71 (s, 3H), 3.69-3.65 (m, 1H), 3.60 (dd,

Experimenteller Teil
- 103 -
J = 9.4, 4.0 Hz, 1H), 3.45 (dd, J = 9.2, 7.8 Hz, 2H), 2.49-2.40 (m, 1H), 2.37-2.30 (m, 1H),
1.92-1.83 (m, 1H), 0.90 (d, J = 7.2 Hz 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.7, 159.3,
146.1, 129.3, 126.9, 122.9, 113.8, 75.1, 74.6, 73.1, 55.2, 51.3, 37.8, 37.7, 13.4 ppm; HRMS-
ESI [M+Na+] berechnet für C17H24O4Na: 331.3587, gefunden: 331.3594.
7-(OTBS)-6-(R)-methyl-5-(R)-hydroxy-2-heptensäuremethylester 170
OMe
OOH
[308.36]
PMBO
Die Verbindung wurde nach AAV 2 mit N-Ts-D-Tryptophan synthetisiert. Es wurden 95 mg
(0.31 mmol, 56%) des erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D =
-2.1 (c 1.0, CHCl3); Rf = 0.38 (PE/EtOAc 5:1); 1H-NMR (400 MHz, CDCl3) δ 7.24
(d, J = 8.5 Hz, 2H), 7.03 (dt, J = 15.4, 7.5 Hz, 1H), 6.89 (d, J = 8.5 Hz, 2H), 5.09 (dt,
J = 15.7, 1.4 Hz, 1H), 4.45 (s, 2H), 3.81 (s, 3H), 3.71 (s, 3H), 3.69-3.65 (m, 1H), 3.60 (dd,
J = 9.4, 4.0 Hz, 1H), 3.45 (dd, J = 9.2, 7.8 Hz, 2H), 2.49-2.40 (m, 1H), 2.37-2.30 (m, 1H),
1.92-1.83 (m, 1H), 0.90 (d, J = 7.2 Hz 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.7, 159.3,
146.1, 129.3, 126.9, 122.9, 113.8, 75.1, 74.6, 73.1, 55.2, 51.3, 37.8, 37.7, 13.4 ppm; HRMS-
ESI [M+Na+] berechnet für C17H24O4Na: 331.3587, gefunden: 331.3628.
5-(R)-(OTBS)-6-Methyl-2-heptensäuremethylester 88(OTBS)
OMe
OOTBS
[286.48]
Die Verbindung wurde nach AAV 5 synthetisiert. Es wurden 26 mg (90.2 µmol, 78%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-5.7 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 7:1); 1H-NMR (400 MHz, CDCl3) δ 6.95
(dt, J = 15.4, 7.5 Hz, 1H), 5.82 (dt, J = 15.7, 1.4 Hz, 1H), 3.70 (s, 3H), 3.53 (q, J = 10.9,
5.8 Hz, 1H), 2.30 (dt, J = 7.5, 1.4, 2H), 1.68-1.66 (m, 1H), 0.87 (s, 9H), 0.85 (dd, J = 6.8,
4.1 Hz, 6H), 0.01 (s, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.9, 146.9, 122.5, 75.8,
51.4, 36.8, 33.1, 25.8, 18.2, 17.5, -4.4, -4.6 ppm; HRMS-ESI [M+MeCN+Na+] berechnet für
C11H19O3N1Na: C15H30O3Si1Na: 309.4716, gefunden: 309.4769.

Experimenteller Teil
- 104 -
5-(R)-(OTBS)-6-Dimethyl-2-heptensäuremethylester 160(OTBS)
OMe
OOTBS
[300.50]
Die Verbindung wurde nach AAV 5 synthetisiert. Es wurden 62 mg (0.33 mmol, 61%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
+34.7 (c 1.1, CHCl3); Rf = 0.40 (PE/EtOAc 7:1); 1H-NMR (400 MHz, CDCl3) δ 7.01
(dt, J = 15.1, 7.8 Hz, 1H), 5.79 (dt, J = 15.4, 1.4 Hz, 1H), 3.71 (s, 3H), 3.39 (q, J = 10.8,
4.4 Hz, 1H), 2.45-2.40 (m, 1H), 2.32-2.26 (m, 1H), 0.87 (s, 9H), 0.86 (s, 9H), 0.01 (s, 3H),
0.00 (s, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.9, 148.6, 121.8, 79.2, 51.4, 36.7,
36.2, 26.4, 26.0, 18.2, -3.6, -4.2 ppm; HRMS-ESI [M+Na+] berechnet für C16H32O3Si1Na:
323.2018, gefunden: 323.2011.
5-(R)-Hydroxy-2,6-methyl-2-heptensäuremethylester 109
OMe
OOH
[186.24]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 46 mg (24.7 µmol, 45%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-9.7 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.84
(dt, J = 7.2, 1.4 Hz, 1H), 3.71 (s, 3H), 3.50 (q, J = 12.6 Hz, 1H), 2.39-2.26 (m, 2H), 1.84 (s,
3H), 1.70 (dqq, J = 6.8, 6.8, 5.6 Hz, 1H), 0.92 (d, J = 6.8 Hz, 6H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 168.4, 138.9, 129.5, 75.8, 51.7, 33.6, 33.4, 18.8, 17.2, 12.7 ppm; HRMS-ESI
[M+Na+] berechnet für C10H18O3Na: 209.1153, gefunden: 209.1168.
5-(R)-Cyclohexyl-5-hydroxy-2-methyl-2-pentensäuremethylester 187
OMe
OOH
[226.31]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 56 mg (22.5 µmol, 41%) des
erwünschten Produkts als farbloses Öl erhalten.

Experimenteller Teil
- 105 -
[α]20D =
-4.7 (c 1.0, CHCl3); Rf = 0.37 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.84
(dt, J = 7.2, 1.4 Hz, 1H), 3.69 (s, 3H), 3.50 (q, J = 12.6, 5.4 Hz, 1H), 2.38-2.24 (m, 2H),
1.82 (s, 3H), 1.72 (br s, 1H), 1.63 (br d, J = 11.9 Hz, 2H), 1.25-0.93 (m, 6H) ppm; 13C-NMR
(100 MHz, CDCl3) δ 168.4, 139.2, 129.3, 75.2, 51.7, 43.3, 33.5, 29.1, 27.8, 26.4, 26.1, 25.9,
12.6 ppm; HRMS-ESI [M+Na+] berechnet für C13H22O3Na: 229.3017, gefunden: 229.2998.
5-(R)-Benzyl-5-hydroxy-2-methyl-2-pentensäuremethylester 188
OMe
OOH
[220.26]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 46 mg (20.9 µmol, 38%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-7.1 (c 1.0, CHCl3); Rf = 0.33 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.36
(d, J = 4.4 Hz, 4H), 7.33-7.27 (m. 1H), 6.84 (dt, J = 7.2, 1.4 Hz, 1H), 4.80 (t, J = 5.80 Hz,
1H), 3.71 (s, 3H), 2.72-2.55 (m, 2H), 1.79 (s, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 168.4,
143.7, 137.9, 129.7, 128.4, 127.7, 125.6, 73.3, 51.7, 38.4, 12.5 ppm; HRMS-ESI [M+Na+]
berechnet für C13H16O3Na: 242.0997, gefunden: 242.1081.
5-(S)-Hydroxy-2-methyl-2-nonensäuremethylester 186
OMe
OOH
[200.14]
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 52 mg (26.4 µmol, 48%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-3.4 (c 1.0, CHCl3); Rf = 0.36 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.84
(dt, J = 7.2, 1.4 Hz, 1H), 3.72 (q, J = 12.3, 6.8 Hz, 1H), 3.70 (s, 3H), 2.36-2.26 (m, 2H),
1.83 (s, 3H), 1.50-1.25 (m, 6H), 0.88 (t, J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 168.5, 138.4, 129.6, 71.1, 51.7, 36.9, 36.7, 27.8, 22.6, 14.0, 12.6 ppm; HRMS-ESI
[M+Na+] berechnet für C11H20O3Na: 223.1310, gefunden: 223.1376.

Experimenteller Teil
- 106 -
5-(R)-(Mosher-ester(S))-2-nonensäuremethylester 161-(S)-MTPA
OMe
OO
[402.40]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 7 mg (17.4 µmol, 60%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
+34.0 (c 0.7, CHCl3); Rf = 0.31 (PE/EtOAc 8:1); 1H-NMR (400 MHz, CDCl3) δ 7.51-
7.45 (m, 2H), 7.39-7.33 (m, 3H), 6.87 (dt, J = 15.5, 7.4 Hz, 1H), 5.89 (dt, J = 15.6, 1.3 Hz,
1H), 5.21-5.14 (m, 1H), 3.71 (s, 3H), 3.51 (s, 3H), 2.53 (dt, J = 7.4, 1.3 Hz, 2H), 1.66-1.53
(m, 2H), 1.26-1.23 (m, 4H), 0.81 (t, J = 6.9 Hz, 3H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 166.3, 166.2, 143.1, 129.6, 128.3, 127.4, 127.3, 124.3, 75.5, 51.6, 36.4, 33.1, 26.8,
22.3, 13.8 ppm; HRMS-ESI [M+Na+] berechnet für C20H25O5F3Na: 425.1552, gefunden:
425.1560.
5-(R)-(Mosher-ester(R))-2-nonensäuremethylester 161-(R)-MTPA
OMe
OO
[402.40]
(R)
O
F3C OMePh
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 8 mg (19.4 µmol, 66%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-2.1 (c 0.8, CHCl3); Rf = 0.31 (PE/EtOAc 8:1); 1H-NMR (400 MHz, CDCl3) δ 7.50-
7.48 (m, 2H), 7.38-7.36 (m, 3H), 6.77 (dt, J = 15.5, 7.4 Hz, 1H), 5.76 (dt, J = 15.6, 1.3 Hz,
1H), 5.21-5.15 (m, 1H), 3.69 (s, 3H), 3.51 (s, 3H), 2.49-2.45 (m, 2H), 1.70-1.58 (m, 2H),
1.32-1.27 (m, 4H), 0.86 (t, J = 6.9 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.3, 166.2,
142.8, 129.6, 128.4, 127.4, 127.3, 124.3, 75.4, 51.5, 36.3, 33.1, 27.3, 22.3, 13.9 ppm; HRMS-
ESI [M+Na+] berechnet für C20H25O5F3Na: 425.1552, gefunden: 425.1578.
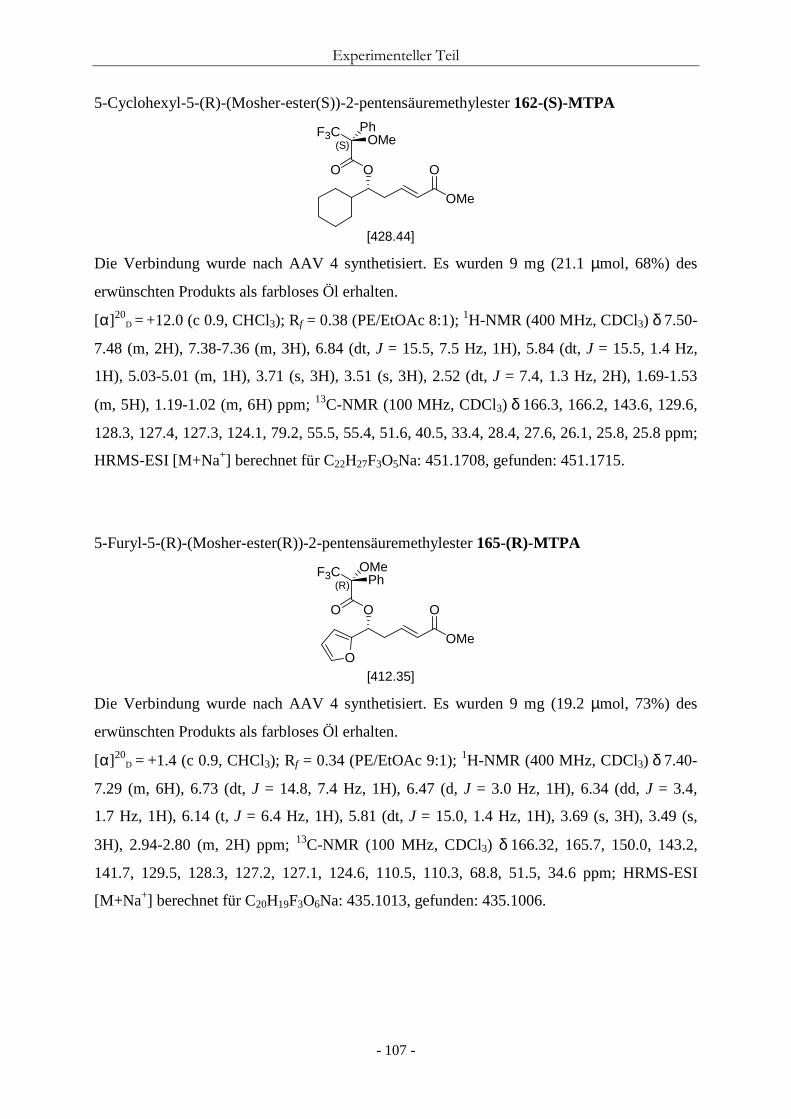
Experimenteller Teil
- 107 -
5-Cyclohexyl-5-(R)-(Mosher-ester(S))-2-pentensäuremethylester 162-(S)-MTPA
OMe
OO
[428.44]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (21.1 µmol, 68%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
+12.0 (c 0.9, CHCl3); Rf = 0.38 (PE/EtOAc 8:1); 1H-NMR (400 MHz, CDCl3) δ 7.50-
7.48 (m, 2H), 7.38-7.36 (m, 3H), 6.84 (dt, J = 15.5, 7.5 Hz, 1H), 5.84 (dt, J = 15.5, 1.4 Hz,
1H), 5.03-5.01 (m, 1H), 3.71 (s, 3H), 3.51 (s, 3H), 2.52 (dt, J = 7.4, 1.3 Hz, 2H), 1.69-1.53
(m, 5H), 1.19-1.02 (m, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.3, 166.2, 143.6, 129.6,
128.3, 127.4, 127.3, 124.1, 79.2, 55.5, 55.4, 51.6, 40.5, 33.4, 28.4, 27.6, 26.1, 25.8, 25.8 ppm;
HRMS-ESI [M+Na+] berechnet für C22H27F3O5Na: 451.1708, gefunden: 451.1715.
5-Furyl-5-(R)-(Mosher-ester(R))-2-pentensäuremethylester 165-(R)-MTPA
OMe
OO
[412.35]
(R)
O
F3C OMePh
O
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (19.2 µmol, 73%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
+1.4 (c 0.9, CHCl3); Rf = 0.34 (PE/EtOAc 9:1); 1H-NMR (400 MHz, CDCl3) δ 7.40-
7.29 (m, 6H), 6.73 (dt, J = 14.8, 7.4 Hz, 1H), 6.47 (d, J = 3.0 Hz, 1H), 6.34 (dd, J = 3.4,
1.7 Hz, 1H), 6.14 (t, J = 6.4 Hz, 1H), 5.81 (dt, J = 15.0, 1.4 Hz, 1H), 3.69 (s, 3H), 3.49 (s,
3H), 2.94-2.80 (m, 2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.32, 165.7, 150.0, 143.2,
141.7, 129.5, 128.3, 127.2, 127.1, 124.6, 110.5, 110.3, 68.8, 51.5, 34.6 ppm; HRMS-ESI
[M+Na+] berechnet für C20H19F3O6Na: 435.1013, gefunden: 435.1006.

Experimenteller Teil
- 108 -
5-Benzyl-5-(R)-(Mosher-Ester(R))-2-pentensäuremethylester 163-(R)-MTPA
OMe
OO
[422.39]
(R)
O
F3C OMePh
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 8 mg (18.9 µmol, 65%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
+11.4 (c 0.8, CHCl3); Rf = 0.36 (PE/EtOAc 9:1); 1H-NMR (400 MHz, CDCl3) δ 7.38-
7.29 (m, 10H), 6.72 (dt, J = 15.6, 7.2 Hz, 1H), 6.00 (q, J = 8.2, 5.2 Hz, 1H), 5.76 (dt, J = 15.6,
1.4 Hz, 1H), 3.68 (s, 3H), 3.40 (s, 3H), 2.86-2.79 (m, 1H), 2.72-2.67 (m, 1H) ppm; 13C-NMR
(100 MHz, CDCl3) δ 166.2, 165.8, 142.2, 137.9, 129.5, 128.9, 128.7, 128.6, 128.3, 127.3,
127.2, 126.8, 124.5, 55.4, 51.5, 38.6 ppm; HRMS-ESI [M+Na+] berechnet für C22H21F3O5Na:
445.1238, gefunden: 445.1233.
7-Benzyl-5-(R)-(Mosher-Ester(S))-2,6-heptensäuremethylester 164-(S)-MTPA
OMe
OO
[448.43]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (20.2 µmol, 71%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
+10.2 (c 0.9, CHCl3); Rf = 0.30 (PE/EtOAc 9:1); 1H-NMR (400 MHz, CDCl3) δ 7.46
(d, J = 7.2 Hz, 2H), 7.39-7.30 (m, 6H), 6.89 (dt, J = 15.7, 8.2 Hz, 1H), 6.63 (d, J = 16.0 Hz,
1H), 6.03 (dd, J = 16.0, 7.5 Hz, 1H), 5.92 (dt, J = 15.7, 1.3 Hz, 1H), 5.72 (q, J = 13.3, 6.5 Hz,
1H), 3.71 (s, 3H), 3.53 (s, 3H), 2.76-2.63 (m, 2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.3,
165.7, 142.4, 135.5, 134.9, 129.6, 128.7, 128.5, 128.4, 127.3, 127.2, 126.8, 124.6, 124.5,
75.6, 55.7, 51.5, 51.6, 37.1 ppm; HRMS-ESI [M+Na+] berechnet für C24H23F3O5Na:
417.1395, gefunden: 417.1391.

Experimenteller Teil
- 109 -
5-(R)-(Mosher-ester(S))-2,6-methyl-2-heptensäuremethylester 109-(S)-MTPA
OMe
OO
[402.40]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (21.8 µmol, 75%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-9.7 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.50-
7.48 (m, 2H), 7.38-7.36 (m, 3H), 6.61 (dt, J = 7.2, 1.4 Hz, 1H), 5.05 (q, J = 12.6, 5.3 Hz, 1H),
3.69 (s, 3H), 3.50 (s, 3H), 2.53-2.37 (m, 2H), 2.00-1.90 (m, 1H), 1.74 (s, 3H), 0.92 (d, J = 6.8,
1.7 Hz, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 168.0, 166.3, 136.2, 130.2, 129.5, 128.3,
127.4, 127.5 80.1, 51.8, 31.1, 30.0, 18.4, 17.4, 12.5 ppm; HRMS-ESI [M+Na+] berechnet für
C20H15F3O5Na: 425.1552, gefunden: 425.1602.
5-Cyclohexyl-5-(R)-(Mosher-ester(S))-2-methyl-2-pentensäuremethylester 187-(S)-MTPA
OMe
OO
[442.46]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 8 mg (18 µmol, 62%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-9.7 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.50-
7.48 (m, 2H), 7.38-7.36 (m, 3H), 6.60 (dt, J = 7.2, 1.4 Hz, 1H), 5.04 (q, J = 12.6, 5.3 Hz, 1H),
3.69 (s, 3H), 3.50 (s, 3H), 2.50-2.38 (m, 2H), 2.00-1.90 (m, 1H), 1.74 (s, 3H), 1.69-1.53 (m,
5H), 1.19-1.02 (m, 6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 168.0, 166.3, 136.4, 130.92,
129.5, 128.3, 127.5, 127.4, 79.7, 51.7, 40.8, 30.0, 28.8, 27.9, 26.2, 25.9, 25.8, 12.5 ppm;
HRMS-ESI [M+Na+] berechnet für C23H29F3O5Na: 465.1865, gefunden: 465.1823.

Experimenteller Teil
- 110 -
5-Benzyl-5-(R)-(Mosher-ester(S))-2-methyl-2-pentensäuremethylester 188-(S)-MTPA
OMe
OO
[436.42]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 10 mg (23.5 µmol, 81%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-9.7 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.40-
7.20 (m, 10H), 6.59 (dt, J = 7.2, 1.4 Hz, 1H), 6.08 (q, J = 12.6, 5.3 Hz, 1H), 3.69 (s, 3H),
3.40 (s, 3H), 2.90-2.79 (m, 1H), 2.70-2.60 (m, 1H), 1.66 (s, 3H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 168.0, 165.9, 138.3, 135.2, 129.5, 128.8, 128.7, 128.6, 128.3, 126.8, 70.0, 51.8,
38.7, 35.3, 12.5 ppm; HRMS-ESI [M+Na+] berechnet für C23H23F3O5Na: 459.1395, gefunden:
459.1300.
5-(S)-(Mosher-ester(S))-2-methyl-2-nonensäuremethylester 186-(S)-MTPA
OMe
OO
[416.43]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (22 µmol, 76%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-9.7 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 7.50-
7.48 (m, 2H), 7.38-7.36 (m, 3H), 6.60 (dt, J = 7.2, 1.4 Hz, 1H), 5.17 (q, J = 12.6, 5.3 Hz, 1H),
3.69 (s, 3H), 3.52 (s, 3H), 2.59-2.39 (m, 2H), 2.00-1.90 (m, 1H), 1.73 (s, 3H), 1.70-1.55 (m,
2H), 1.35-1.15 (m, 4H), 0.87 (t, J = 6.8 Hz, 1H) ppm; 13C-NMR (100 MHz, CDCl3) δ 168.0,
166.2, 135.8, 130.4, 129.4, 128.3, 127.3, 127.2, 75.7, 51.7, 33.3, 32.8, 27.3, 22.3, 13.8,
12.5 ppm; HRMS-ESI [M+Na+] berechnet für C21H27F3O5Na: 439.1708 gefunden: 439.1767.

Experimenteller Teil
- 111 -
α,α-Diphenyl-L-prolinol 213
[115.13]
NH
O
OH
a) SOCl2, MeOHb) PhMgBr, Et2O
[253.33]
NH
OH
PhPh
2 g (17.37 mmol) L-Prolin wird in MeOH (17 mL) aufgenommen und auf -20 °C gekühlt.
Thionylchlorid (2.5 mL, 34 mmol) wird langsam der Lösung zugetropft und auf RT erwärmt.
Die Reaktion wird zusätzlich für 1 h refluxiert. Das Lösungsmittel wird am Rotations-
verdampfer entfernt und der Rückstand in 60 mL Et2O gelöst. Zu dieser Lösung wird
vorsichtig bei 0 °C Phenylmagnesiumbromid (69.48 mmol) in 23 mL Et2O zugetropft. Das
Gemisch wird auf RT erwärmt und über Nacht gerührt. Anschließend wird die Reaktion durch
vorsichtige Zugabe von 1M HCl (20 mL) bei 0 °C gequencht und für 30 min bei RT gerührt.
Die Phasen werden getrennt und die wässrige Phase mit Ammoniak (20%ig) auf pH = 10
eingestellt. Die basische Phase wird mit Methyl-tert-Butylether (3 x 100 mL) extrahiert. Die
vereinigten organischen Phasen werden über MgSO4 getrocknet, filtriert und im
Rotationsverdampfer eingeengt. Nach Reinigung durch Flash-Chromatographie
(MeOH/CH2Cl2 1:20) wird der erwünschte Aminoalkohol (2.59 g, 10.2 mmol, 59%) als
weißer Feststoff erhalten.
[α]20D =
-58.4 (c 1.0, MeOH); Smp.: 76-79 °C; Rf = 0.17 (MeOH/CH2Cl2 1:20); 1H-NMR
(400 MHz, CDCl3) δ 7.59 (d, J = 7.5 Hz, 2H), 7.51 (d, J = 7.5 Hz, 2H), 7.34-7.27 (m, 4H),
7.21-7.15 (m, 2H), 4.27 (t, J = 7.3 Hz, 1H), 3.08-3.02 (m, 1H), 2.99-2.93 (m, 1H), 1.79-
1.55 (m, 4H) ppm; 13C-NMR (100 MHz, CDCl3) δ 148.0, 145.4, 128.8, 128.5, 127.1, 126.9,
126.4, 125.9, 77.2, 65.1, 47.2, 26.8, 25.8 ppm; HRMS-ESI [M+Na+] berechnet für
C17H19O1N1Na: 276.1364, gefunden: 276.1322.
5-(R)-Hydroxy-4-(R)-methyl-6-methyl-2-heptensäuremethylester 66
[186.24]
OMe
OOH
Die Verbindung wurde nach AAV 3 synthetisiert. Es wurden 87 mg (0.46 mmol, 85%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D = +32.6 (c 1.0 CHCl3); Rf = 0.28 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3) δ 6.91
(dd, J = 15.7, 8.0 Hz, 1H), 5.84 (dd, J = 15.7, 1.2 Hz, 1H), 3.71 (s, 3H), 3.24 (t, J = 5.8 Hz,

Experimenteller Teil
- 112 -
1H), 2.49 (ddqm, J = 8.1, 6.8, 1.2 Hz, 1H), 1.70 (dqq, J = 6.8, 6.8, 5.6 Hz, 1H), 1.49 (br s,
1H), 1.07 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 167.0, 152.0, 120.7, 79.2, 51.4, 39.9, 30.9, 19.7, 16.5,
14.0 ppm; HRMS-ESI [M+Na+] berechnet für C10H18O3Na: 209.1148, gefunden: 209.1137.
5-(R)-Hydroxy-4-(R)-methyl-2-nonensäuremethylester 231
[200.27]
OMe
OOH
Die Verbindung wurde nach AAV 3 synthetisiert. Es wurden 84 mg (0.41 mmol, 76%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D = +11.3 (c 1.0 CHCl3); Rf = 0.31 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3) δ 6.94
(dd, J = 15.8, 7.8 Hz, 1H), 5.85 (dd, J = 15.8, 1.2 Hz,1H), 3.72 (s, 3H), 3.55 (br s, 1H),
2.41 (dddq, J = 7.9, 6.6, 2.9, 1.2 Hz, 1H), 1.49-1.24 (m, 9H), 1.53 (br s, 1H), 1.07 (d,
J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 167.0, 151.4, 121.1, 74.5, 51.5, 42.5,
34.1, 28.1, 22.6, 14.1, 13.9 ppm; HRMS-ESI [M+Na+] berechnet für C11H20O3Na: 223.1305,
gefunden: 223.1267.
5-(R)-Hydroxy-4-(R)-,6,6-trimethyl-2-heptensäuremethylester 230
[200.27]
OMe
OOH
Die Verbindung wurde nach AAV 3 synthetisiert. Es wurden 41 mg (0.20 mmol, 37%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D = +28.3 (c 1.0 CHCl3); Rf = 0.28 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3) δ 6.98
(dd, J = 15.8, 8.4 Hz, 1H), 5.79 (dd, J = 15.8, 1.1 Hz, 1H), 3.71 (s, 3H), 3.25 (t, J = 4.0 Hz,
1H), 2.59 (dddq, J = 8.3, 6.9, 4.0, 1.1 Hz, 1H), 1.45 (br s, 1H), 1.10 (d, J = 6.9 Hz, 3H),
0.93 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3) δ 167.2, 154.2, 119.3, 81.0, 51.5, 38.8, 36.1,
26.7, 15.1 ppm; HRMS-ESI [M+Na+] berechnet für C11H20O3Na: 223.1305, gefunden:
223.1353.

Experimenteller Teil
- 113 -
5-Cyclohexyl-5-(R)-hydroxy-4-(R)-methyl-2-pentensäuremethylester 232
[226,31]
OMe
OOH
Die Verbindung wurde nach AAV 3 synthetisiert. Es wurden 104 mg (0.46 mmol, 84%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D = +16.3 (c 1.0 CHCl3); Rf = 0.36 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3) δ 6.94
(dd, J = 15.8, 7.8 Hz, 1H), 5.84 (dd, J = 15.8, 1.2 Hz, 1H), 3.71 (s, 3H), 3.26 (t, J = 5.8 Hz,
1H), 2.53 (dddq, J = 7.9, 6.6, 5.5, 1.2 Hz, 1H), 1.83 (dm, J = 12.7 Hz, 1H), 1.76-1.53 (m, 4H),
1.42 (br s, 1H), 1.41-0.85 (m, 6H), 1.05 (d, J = 6.6 Hz, 3H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 167.1, 152.3, 120.7, 78.4, 51.5, 40.7, 39.1, 29.8, 27.4, 26.3, 26.2, 25.9, 13.4 ppm;
HRMS-ESI [M+Na+] berechnet für C13H22O3Na: 249.1461, gefunden: 249.1503.
5-Benzyl-5-(R)-hydroxy-4-(R)-methyl-2-pentensäuremethylester 233
[220.26]
OMe
OOH
Die Verbindung wurde nach AAV 3 synthetisiert. Es wurden 83 mg (0.38 mmol, 69%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D = +9.8 (c 1.0 CHCl3); Rf = 0.33 (EtOAc/PE 1:3); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.36-7.23 (m, 5H), 6.92 (dd, J = 15.8, 7.4 Hz, 1H), 5.76 (dd, J = 15.8, 1.2 Hz, 1H),
4.66 (d, J = 5.5 Hz, 1H), 3.68 (s, 3H), 2.75-2.61 (m, 1H), 2.04 (br s, 1H), 1.05 (d, J = 6.8 Hz,
3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.9, 150.6, 142.0, 128.3, 127.8, 126.3, 121.4,
77.0, 51.4, 43.7, 13.9 ppm; HRMS-ESI [M+Na+] berechnet für C13H16O3Na: 243.0992,
gefunden: 243.1016.
5-Furyl-5-(S)-hydroxy-4-(R)-methyl-2-pentensäuremethylester 235
[210.22]
OMe
OOH
O
Die Verbindung wurde nach AAV 3 synthetisiert. Es wurden 65 mg (0.31 mmol, 57%) des

Experimenteller Teil
- 114 -
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D = +16.3 (c 1.0 CHCl3); Rf = 0.30 (EtOAc/PE 1:3); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.36 (s, 1H), 6.90 (ddd, J = 14.7, 7.5 Hz, 1H), 6.27 (dd, J = 3.0, 1.7 Hz, 1H),
6.20 (dd, J = 4.0, 0.7 Hz, 1H), 5.87 (dt, J = 15.4, 1.4 Hz, 1H), 4.77 (q, J = 11.3, 6.5 Hz, 1H),
3.65 (s, 3H), 2.70 (t, J = 7.2 Hz, 1H), 1.05 (d, J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 166.6, 155.3, 144.1, 142.3, 123.8, 110.3, 106.5, 66.4, 51.5, 38.2 ppm; HRMS-ESI
[M+Na+] berechnet für C11H14O3Na: 233.0784, gefunden: 233.0706.
7-Benzyl-5-(R)-hydroxy-4-(R)-methyl-6,2-heptensäuremethylester 234
[246.30]
OMe
OOH
Die Verbindung wurde nach AAV 3 synthetisiert. Es wurden 73 mg (0.26 mmol, 54%) des
erwünschten VMAR-Produkts als farbloses Öl erhalten.
[α]20D = +8.3 (c 1.0 CHCl3); Rf = 0.31 (EtOAc/PE 1:3); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.36-7.22 (m, 5H), 6.99 (dt, J = 15.7, 7.5 Hz, 1H), 6.60 (d, J = 15.7 Hz, 1H),
6.21 (dd, J = 16.0, 6.8 Hz, 1H), 5.93 (dt, J = 15.7, 1.4 Hz, 1H), 4.42 (q, J = 12.6, 6.5 Hz, 1H),
3.71 (s, 3H), 2.52 (t, J = 6.8 Hz, 1H), 1.05 (d, J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 166.7, 144.5, 136.2, 131.2, 130.8, 128.6, 127.9, 126.5, 123.8, 71.5, 51.5, 40.1 ppm;
HRMS-ESI [M+Na+] berechnet für C15H18O3Na: 269.1148, gefunden: 269.1098.
4-(R)-,6-Dimethyl-5-(R)-(Mosher-Ester (S))-2-heptensäuremethylester 66-(S)-MTPA
[402,40]
OMe
OO
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 7 mg (17.4 µmol, 60%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +18.1 (c 0.7 CHCl3); Rf = 0.34 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.54-7.51 (m, 2H), 7.38-7.36 (m, 3H), 6.77 (ddd, J = 15.5, 7.4 Hz, 1H), 5.79 (dd,

Experimenteller Teil
- 115 -
J = 15.6, 1.3 Hz, 1H), 5.97 (t, J = 6.8 Hz, 1H), 3.70 (s, 3H), 3.49 (s, 3H), 2.69-2.64 (m, 1H),
1.94-1.85 (m, 1H), 0.94 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 7.2 Hz, 3H), 0.84 (d, J = 6.8 Hz, 3H)
ppm; 13C-NMR (100 MHz, CDCl3) δ 167.0, 152.0, 120.7, 79.2, 51.4, 39.9, 30.9, 19.7, 16.5,
14.0 ppm; HRMS-ESI [M+Na+] berechnet für C20H25O5F3Na: 425.1552, gefunden: 425.1560.
4-(R)-,6-Dimethyl-5-(R)-(Mosher-Ester (R))-2-heptensäuremethylester 66-(R)-MTPA
[402.40]
OMe
OO(R)
O
F3C OMePh
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 7 mg (17.4 µmol, 60%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +25.3 (c 0.7 CHCl3); Rf = 0.34 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.54-7.51 (m, 2H), 7.38-7.36 (m, 3H), 6.77 (ddd, J = 15.5, 7.4 Hz, 1H), 5.82 (dd,
J = 15.6, 1.3 Hz, 1H), 5.97 (t, J = 6.8 Hz, 1H), 3.71 (s, 3H), 3.49 (s, 3H), 2.65-2.57 (m, 1H),
1.94-1.85 (m, 1H), 0.94 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 7.2 Hz, 3H), 0.84 (d, J = 6.8 Hz, 3H)
ppm; 13C-NMR (100 MHz, CDCl3) δ 167.0, 152.0, 120.7, 79.2, 51.4, 39.9, 30.9, 19.7, 16.5,
14.0 ppm; HRMS-ESI [M+Na+] berechnet für C20H25O5F3Na: 425.1552, gefunden: 425.1560.
4-(R)-,6-Dimethyl-5-(R)-(Mosher-Ester (S))-2-nonensäuremethylester 231-(S)-MTPA
[416,43]
OMe
OO(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 8 mg (18.7 µmol, 67%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +27.6 (c 0.8 CHCl3); Rf = 0.32 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.54-7.51 (m, 2H), 7.38-7.36 (m, 3H), 6.77 (ddd, J = 15.5, 7.4 Hz, 1H), 5.70 (dd,
J = 15.6, 1.3 Hz, 1H), 5.07 (t, J = 6.8 Hz, 1H), 3.69 (s, 3H), 3.51 (s, 3H), 2.69-2.64 (m, 1H),
1.94-1.85 (m, 1H), 0.94 (d, J = 6.8, 3H), 0.88 (d, J = 7.2 Hz, 3H), 0.84 (d, J = 6.8, 3H) ppm;

Experimenteller Teil
- 116 -
13C-NMR (100 MHz, CDCl3) δ 166.3, 166.2, 143.1, 129.6, 128.3, 127.4, 127.3, 124.3, 75.5,
51.6, 36.4, 33.1, 26.8, 22.3, 13.8 ppm; HRMS-ESI [M+Na+] berechnet für C21H27O5F3Na:
439.1703, gefunden: 439.1684.
4-(R)-,6,6’-Trimethyl-5-(R)-(Mosher-Ester (S))-2-heptensäuremethylester 230-(S)-MTPA
[416.43]
OMe
OO(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (20.1 µmol, 63%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +21.2 (c 0.8 CHCl3); Rf = 0.31 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.54-7.51 (m, 2H), 7.38-7.36 (m, 3H), 6.91 (ddd, J = 15.4, 7.4 Hz, 1H), 5.72 (dd,
J = 15.4, 1.3 Hz, 1H), 4.87 (t, J = 4.4 Hz, 1H), 3.70 (s, 3H), 3.49 (s, 3H), 2.79-2.71 (m, 1H),
0.95 (d, J = 6.8 Hz, 3H), 0.90 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3) δ 167.2, 154.2,
119.3, 81.0, 51.5, 38.8, 36.1, 26.7, 15.1 ppm; HRMS-ESI [M+Na+] berechnet für
C21H27O5F3Na: 439.1703, gefunden: 439.1800.
5-Cyclohexyl-4-(R)-mehtyl-5-(R)-(Mosher-Ester(S))-2-pentensäuremethylester
232-(S)-MTPA
[442.47]
OMe
OO(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (20.3 µmol, 75%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +17.2 (c 0.8 CHCl3); Rf = 0.30 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.54-7.51 (m, 2H), 7.38-7.36 (m, 3H), 6.80 (ddd, J = 15.4, 7.4 Hz, 1H), 5.78 (dd,
J = 15.4, 1.3 Hz, 1H), 4.98 (t, J = 5.1 Hz, 1H), 3.70 (s, 3H), 3.49 (s, 3H), 2.74-2.65 (m, 1H),
1.70-1.54 (m, 6H), 1.70-1.54 (m, 6H), 1.25-0.98 (m, 4H), 0.94 (d, J = 6.8 Hz, 3H) ppm;

Experimenteller Teil
- 117 -
13C-NMR (100 MHz, CDCl3) δ 167.1, 152.3, 120.7, 78.4, 51.5, 40.7, 39.1, 29.8, 27.4, 26.3,
26.2, 25.9, 13.4 ppm; HRMS-ESI [M+Na+] berechnet für C23H29O5F3Na: 465.1859, gefunden:
439.1903.
5-Benzyl-5-(R)-(Mosher-Ester (S))-4-(R)-methyl-2-pentensäuremethylester 233-(S)-MTPA
[436.42]
OMe
OO(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 8 mg (18.3 µmol, 68%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +21.2 (c 0.8 CHCl3); Rf = 0.31 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.38-7.25 (m, 10H), 6.66 (ddd, J = 15.4, 7.4 Hz, 1H), 5.86 (d, J = 7.2 Hz, 1H),
5.62 (dd, J = 15.4, 1.3 Hz, 1H), 3.70 (s, 3H), 3.49 (s, 3H), 2.91-2.83 (m, 1H), 1.01 (d,
J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.9, 166.8, 147.8, 136.7, 129.6,
128.7, 128.5, 128.3, 127.4, 80.5, 51.5, 41.6, 15.0 ppm; HRMS-ESI [M+Na+] berechnet für
C23H23O5F3Na: 459.4102, gefunden: 459.4082
5-Furyl-5-(S)-(Mosher-Ester (S))-4-(R)-methyl-2-pentensäuremethylester 235-(S)-MTPA
[426.38]
OMe
OO(S)
O
F3C PhOMe
O
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 8 mg (18.7 µmol, 69%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +21.2 (c 0.8 CHCl3); Rf = 0.31 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.54-7.51 (m, 2H), 7.38-7.36 (m, 3H), 7.35 (s, 1H), 6.90 (ddd, J = 14.7, 7.5 Hz,
1H), 6.27 (dd, J = 3.0, 1.7 Hz, 1H), 6.20 (dd, J = 4.0, 0.7 Hz, 1H), 5.87 (dt, J = 15.4, 1.4 Hz,
1H), 4.77 (q, J = 11.3, 6.5 Hz, 1H), 3.65 (s, 3H), 3.49 (s, 3H), 2.70 (t, J = 7.2 Hz, 1H),
1.05 (d, J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.9, 166.8, 147.8, 136.7,
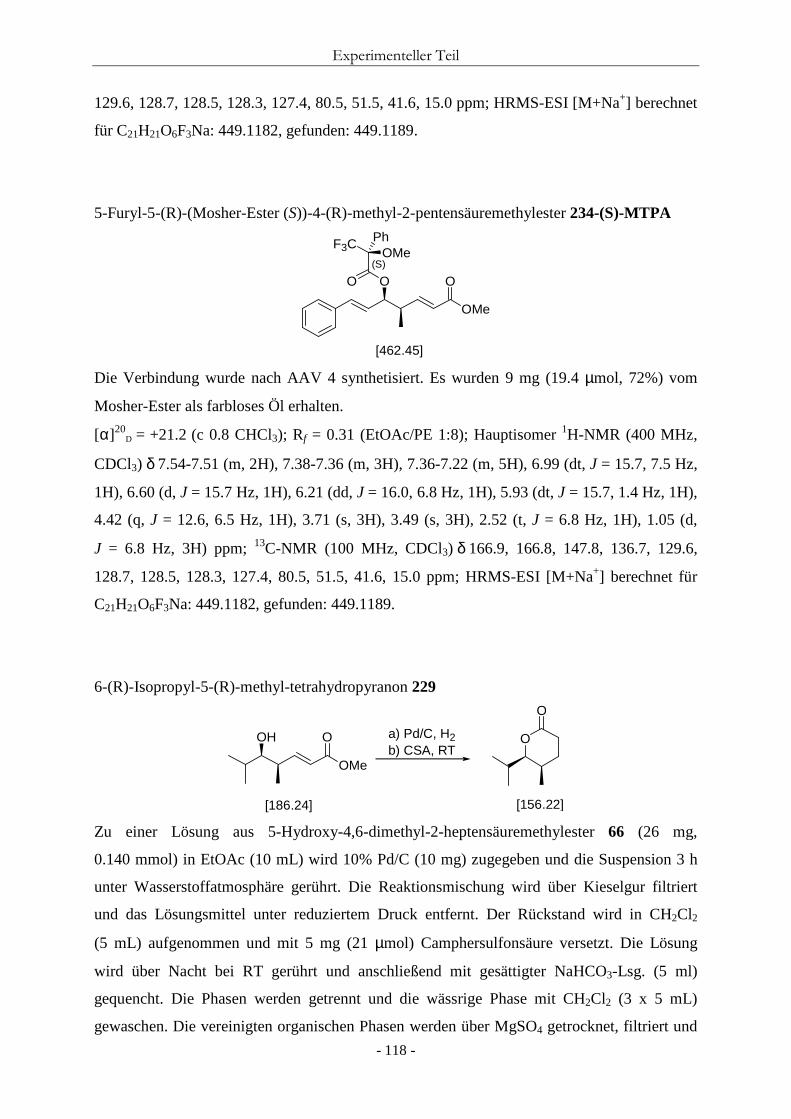
Experimenteller Teil
- 118 -
129.6, 128.7, 128.5, 128.3, 127.4, 80.5, 51.5, 41.6, 15.0 ppm; HRMS-ESI [M+Na+] berechnet
für C21H21O6F3Na: 449.1182, gefunden: 449.1189.
5-Furyl-5-(R)-(Mosher-Ester (S))-4-(R)-methyl-2-pentensäuremethylester 234-(S)-MTPA
[462.45]
OMe
OO(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (19.4 µmol, 72%) vom
Mosher-Ester als farbloses Öl erhalten.
[α]20D = +21.2 (c 0.8 CHCl3); Rf = 0.31 (EtOAc/PE 1:8); Hauptisomer 1H-NMR (400 MHz,
CDCl3) δ 7.54-7.51 (m, 2H), 7.38-7.36 (m, 3H), 7.36-7.22 (m, 5H), 6.99 (dt, J = 15.7, 7.5 Hz,
1H), 6.60 (d, J = 15.7 Hz, 1H), 6.21 (dd, J = 16.0, 6.8 Hz, 1H), 5.93 (dt, J = 15.7, 1.4 Hz, 1H),
4.42 (q, J = 12.6, 6.5 Hz, 1H), 3.71 (s, 3H), 3.49 (s, 3H), 2.52 (t, J = 6.8 Hz, 1H), 1.05 (d,
J = 6.8 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.9, 166.8, 147.8, 136.7, 129.6,
128.7, 128.5, 128.3, 127.4, 80.5, 51.5, 41.6, 15.0 ppm; HRMS-ESI [M+Na+] berechnet für
C21H21O6F3Na: 449.1182, gefunden: 449.1189.
6-(R)-Isopropyl-5-(R)-methyl-tetrahydropyranon 229
[156.22]
O
O
a) Pd/C, H2b) CSA, RT
OMe
OOH
[186.24]
Zu einer Lösung aus 5-Hydroxy-4,6-dimethyl-2-heptensäuremethylester 66 (26 mg,
0.140 mmol) in EtOAc (10 mL) wird 10% Pd/C (10 mg) zugegeben und die Suspension 3 h
unter Wasserstoffatmosphäre gerührt. Die Reaktionsmischung wird über Kieselgur filtriert
und das Lösungsmittel unter reduziertem Druck entfernt. Der Rückstand wird in CH2Cl2
(5 mL) aufgenommen und mit 5 mg (21 µmol) Camphersulfonsäure versetzt. Die Lösung
wird über Nacht bei RT gerührt und anschließend mit gesättigter NaHCO3-Lsg. (5 ml)
gequencht. Die Phasen werden getrennt und die wässrige Phase mit CH2Cl2 (3 x 5 mL)
gewaschen. Die vereinigten organischen Phasen werden über MgSO4 getrocknet, filtriert und

Experimenteller Teil
- 119 -
das Lösungsmittel am Rotationsverdampfer entfernt. Nach Aufreinigung durch Flash-
Chromatographie (PE/EtOAc 4:1) wird das erwünschte Produkt (2.89, 8.17 mmol, 84%) als
farbloses Öl erhalten.
[α]20D = +68.2 (c 1.0 CHCl3); Rf = 0.38 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3)
δ 3.77 (dd, J = 9.8, 2.4 Hz, 1H), 2.48 (dd, J = 8.6, 6.3 Hz, 2H), 2.17-2.11 (m, 1H),
2.10-1.97 (m, 1H), 1.87-1.81 (m, 1H), 1.67-1.63 (m, 1 H), 1.04 (d, J = 6.6 Hz, 3H), 0.92 (d,
J = 7.1 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 172.0, 88.3,
29.7, 26.8, 26.6, 26.1, 19.7, 18.0 ppm.
6.3 Versuchsbeschreibungen zu Kapitel 4
5-Cyano-3-methyl-2,4-pentensäureethylester 250
O
[142.15]
OEtO
NaH, THF, RT
(EtO)2P
O
CN
O
[165.18]
OEtNC
Zu einer Suspension von NaH (80%ig in Paraffinöl, 610 mg, 20.5 mmol) in THF (40 mL)
wird bei 0 °C langsam Diethylcyanomethylphosphonat (3.35 g, 18.7 mmol) zugetropft. Nach
1 h wird vorsichtig Ethyl-3-methyl-4-Oxocrotonat (2.4 g, 16.8 mmol) zugegeben und das
Gemisch auf RT erwärmt und für 1 h gerührt. Bei 0 °C wird vorsichtig 1 M HCl (2 mL) und
gesättigte NH4Cl-Lsg. addiert. Die Phasen werden getrennt und die wässrige Phase mit
Methyl-tert-Butylether (3 x 20 mL) extrahiert. Die vereinigten organischen Phasen werden
über MgSO4 getrocknet, filtriert und im Rotationsverdampfer eingeengt. Nach Reinigung
durch Flash-Chromatographie (EtOAc/PE 1:3) wird der erwünschte Ester (2.2 g, 13.1 mmol,
88%) als weißer Feststoff erhalten.
Rf = 0.43 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3) δ 7.00 (d, J = 16.4 Hz, 1H), 5.97 (s,
1H), 5.67 (d, J = 16.4 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.22 (s, 3H), 1.27 (t, J = 7.1 Hz, 3H)
ppm; 13C-NMR (100 MHz, CDCl3) δ 165.6, 152.9, 147.3, 126.6, 117.3, 101.6, 60.6, 14.2,
12.8 ppm.

Experimenteller Teil
- 120 -
5-Cyano-3-methyl-2,4-pentenaldehyd 251
O
[165.18]
OEtNC
O
[121.13]
NC
a) DiBAl-H, CH2Cl2, -78 °Cb) MnO2, CH2Cl2
Der Ester 250 (1 g, 6 mmol) wird in 6 mL CH2Cl2 aufgenommen und auf -78 °C abgekühlt.
Diisobutylaluminiumhydrid (1 M in CH2Cl2, 15 mL, 15 mmol) wird über 25 min zugetropft.
Die Reaktion wird 30 min bei -78 °C gerührt. Durch Zugabe von Methanol und gesättigter
Rochelle-Salz-Lsg. wird die Reaktion gequencht und auf RT erwärmt. Die wässrige Phase
wird mit Wasser verdünnt und mit CH2Cl2 (3 x 10 mL) extrahiert. Die vereinigten
organischen Phasen werden mit gesättigter NaCl-Lsg. gewaschen, über MgSO4 getrocknet,
filtriert und das Lösungsmittel unter vermindertem Druck entfernt. Nach Reinigung durch
Flash-Chromatographie (EtOAc/PE 2:1) wird der Allylalkohol als farbloses Öl erhalten.
Dieser wird in 6 mL Et2O aufgenommen und mit MnO2 (5 g, 60 mmol) versetzt. Die
Suspension wird über Nacht bei RT gerührt und anschließend über Kieselgur filtriert und das
Filtrat am Rotationsverdampfer eingeengt. Der erwünschte Aldehyd (574 mg, 4.74 mmol,
79%) wird als gelber Feststoff erhalten.
Rf = 0.54 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3) δ 10.10 (d, J = 7.5 Hz, 1H), 7.10 (d,
J = 16.4 Hz, 1H), 6.08 (d, J = 7.5 Hz, 1H), 5.97 (s, 1H), 5.80 (d, J = 16.4 Hz, 1H), 2.24 (s,
3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 165.6, 152.9, 147.3, 126.6, 117.3, 101.6, 60.6, 14.2,
12.8 ppm.
9-Cyano-7-methyl-5-(S)-hydroxy-2,6,8-nonensäuremethylester 252
[221.25]
NC OMe
OOH
Die Verbindung wurde nach AAV 2 synthetisiert. Es wurden 85 mg (0.38 mmol, 70%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
-15.7 (c 1.0, CHCl3); Rf = 0.12 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ
6.98 (d, J = 16.0 Hz, 1H), 6.91 (dt, J = 15.4, 7.5 Hz, 1H), 5.91 (d, J = 15.7 Hz, 1H), 5.78 (d,
J = 8.5 Hz, 1H), 5.32 (d, J = 16.4 Hz, 1H), 4.63 (q, J = 7.2 Hz, 1H), 3.70 (s, 3H),
2.53-2.38 (m, 2H), 2.13 (br s, 1H), 1.78 (s, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.5,
153.9, 143.5, 141.7, 133.6, 124.2, 118.1, 96.4, 67.1, 51.6, 39.7, 12.1 ppm; HRMS-ESI
[M+Na+] berechnet für C12H15O3N1Na: 244.0950, gefunden: 244.0812.

Experimenteller Teil
- 121 -
p-Methoxybenzyltrichloracetimidat 265
[138.16]
NaH, Et2O0 °C - RT
[282.55]
MeOOH
NCl3C
MeOO
NH
ClCl
Cl
Natriumhydrid (60 %ige Suspension in Mineralöl, 1.0 g, 25.5 mmol) wird in Et2O (70 mL)
gelöst. Hierzu wird eine Lösung aus 4-Methoxybenzylalkohol (22 mL, 19.9 g, 175.4 mmol) in
Et2O (70 mL) zugetropft und bei RT für 1 h gerührt. Die Lösung wird auf 0 °C gekühlt und
mit 25 g Trichloracetonitril (175.4 mmol) in Et2O (70 mL) versetzt. Das Gemisch wird nach
2 h auf RT aufgewärmt und weitere 16 h gerührt. Anschließend wird Methanol (2 mL) zu der
Lösung gegeben, das Lösungsmittel unter vermindertem Druck entfernt und der flüssige
Rückstand mit PE und MeOH (je 1 mL) versetzt, worauf ein Feststoff ausfällt, der über eine
kurze Kieselgelsäule abfiltriert wird. Das Acetimidat (36.33 g, 128.6 mmol, 73 %) wird als
leicht gelbes Öl erhalten. 1H-NMR (400 MHz, CDCl3) δ 8.36 (bs, 1H), 7.37 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz,
2H), 5.27 (s, 2H), 3.82 (s, 3H) ppm.
2-Butin-dicobalthexacarbonyl-aldehyd 260
[354.00]
a) Co2(CO)8, CH2Cl2b) Amberlyst 15, Aceton
[142.19]
OEt
OEtO
Co(CO)3(OC)3Co
Zu einer Lösung von Dicobaltoctacarbonyl (2.4 g, 7.04 mmol) in CH2Cl2 (140 mL) wird bei
RT über 30 min 2-Butindiethylacetal (1.11 mL, 7.04 mmol) zugetropft und die Reaktion über
Nacht gerührt. Anschließend wird das Gemisch über eine kurze Aluminiumoxid Säule
filtriert. Das Filtrat wird unter vermindertem Druck eingeengt und der Rückstand in
Aceton (25 mL) aufgenommen. Der Lösung werden zusätzlich 0.5 mL Wasser und 800 mg
Amberlyst 15 addiert und die Suspension 3 h bei RT gerührt. Die Lösung wird filtriert und
das Lösungsmittel unter vermindertem Druck entfernt. Nach säulenchromatographischer
Reinigung (MTBE/PE 1:40) wird der Aldehyd (2.04 g, 5.77 mmol, 82 %) als dunkel rotes Öl
erhalten.
Der Aldehyd wurde ohne weitere Analytik eingesetzt.

Experimenteller Teil
- 122 -
5-(S)-Hydroxy-6-in-2-octensäuremethylester 259
[354.00]
O
Co(CO)3(OC)3Co
OTBS
OMe
[214.37]
a) N-Ts-D-Tryptophan, BCl2Ph, i-PrOH, EtCN, -78 °C
b) CAN, Aceton, -78 °C
OH O
OMe
[168.18]
Eine Suspension von 1.51 g (4.28 mmol) N-Ts-D-Tryptophan in 28 mL CH2Cl2 wird mit
0.55 mL (4.28 mmol) Dichlorphenylboran versetzt und für 1 h gerührt. Das Lösungsmittel
wird unter Stickstoffatmosphäre entfernt. Der Rückstand wird in EtCN gelöst und auf -78 °C
gekühlt. Eine Mischung aus 2-Butin-dicobalthexacarbonyl-aldehyd 260 (1.5 g, 4.28 mmol),
Ketenacetal 35 (1.12 g, 5.56 mmol) und absolutem Isopropanol (0.33 mL, 4.28 mmol) in
4 mL EtCN wird über 15 min addiert. Die Reaktionsmischung wird 1 h bei -78 °C gerührt und
anschließend mit 5 M NaHCO3-Lsg. gequencht. Die Phasen werden getrennt und die wässrige
Phase mit Methyl-tert-Butylether (3 x 5 mL) extrahiert. Die vereinigten organischen Phasen
werden über MgSO4 getrocknet, filtriert und eingeengt. Der Rückstand wird über eine kurze
Kieselgelsäule filtriert und das Filtrat eingeengt. Das zurückbleibende Öl wird in Aceton
(300 mL) gelöst und auf -78 °C abgekühlt. Anschließend wird der Lösung langsam
Cerammoniumnitrat (11.73 g, 21.4 mmol) in 50 mL Aceton zugetropft. Nach 1 h wird die
Reaktion durch Zugabe von gesättigter NaCl-Lsg. (200 mL) gequencht. Die Mischung wird
auf RT erwärmt und die Phasen getrennt. Die wässrige Phase wird mit Methyl-tert-Butylether
(3 x 100 mL) extrahiert. Die vereinigten organischen Phasen werden nochmals mit gesättigter
NaHCO3-Lsg. (100 mL) und gesättigter NaCl-Lsg. (100 mL) gewaschen und über MgSO4
getrocknet, filtriert und eingeengt. Nach Aufreinigung durch Flash-Chromatographie
(EtOAc/PE 1:3) wird das erwünschte Produkt (511 mg, 3.03 mmol, 71%) als farbloses Öl
erhalten.
[α]20D = -24.9 (c 1.0 CHCl3); Rf = 0.27 (EtOAc/PE 1:3); 1H-NMR (400 MHz, CDCl3) δ 6.99
(dt, J = 15.7, 7.5 Hz, 1H), 5.93 (dt, J = 15.7, 1.3 Hz, 1H), 4.45 (br s, 1H), 3.71 (s, 3H),
2.55 (t, J = 7.2 Hz, 2H), 1.97 (br s, 1H), 1.82 (d, J = 2.0 Hz, 3H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 166.6, 143.8, 124.0, 82.7, 79.1, 61.2, 51.5, 3.6 ppm; HRMS-ESI [M+MeCN+Na+]
berechnet für C11H15O3N1Na: 232.0950, gefunden: 232.0886.

Experimenteller Teil
- 123 -
5-(S)-(Mosher-ester(S))-6-in-2-octensäuremethylester 259-(S)-MTPA
OMe
OO
[384.34]
(S)
O
F3C PhOMe
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 9 mg (22 µmol, 76%) des
erwünschten Produkts als farbloses Öl erhalten
[α]20D = -11.9 (c 0.7 CHCl3); Rf = 0.31 (EtOAc/PE 1:6); 1H-NMR (400 MHz, CDCl3) δ 7.51-
7.49 (m, 2H), 7.37-7.35 (m, 3H), 6.79 (dt, J = 15.4, 7.2 Hz, 1H), 5.80 (dt, J = 15.7, 1.2 Hz,
1H), 5.65-5.60 (m, 1H), 3.70 (s, 3H), 3.56 (s, 3H), 2.63 (dt, J = 7.2, 1.2 Hz, 2H), 1.84 (d,
J = 2.0 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.2, 165.9, 141.5, 129.6, 128.4,
127.3, 127.2, 124.7, 84.4, 74.4, 64.7, 55.5, 51.5, 3.6 ppm; HRMS-ESI [M+Na+] berechnet für
C19H19O5F3Na: 407.0182, gefunden: 407.0123.
5-(S)-(Mosher-ester(R))-6-in-2-octensäuremethylester 259-(R)-MTPA
OMe
OO
[384.34]
(R)
O
F3C OMePh
Die Verbindung wurde nach AAV 4 synthetisiert. Es wurden 8 mg (21 µmol, 71%) des
erwünschten Produkts als farbloses Öl erhalten
[α]20D = -38.9 (c 0.7 CHCl3); Rf = 0.31 (EtOAc/PE 1:6); 1H-NMR (400 MHz, CDCl3) δ 7.50-
7.48 (m, 2H), 7.39-7.36 (m, 3H), 6.88 (dt, J = 15.7, 7.2 Hz, 1H), 5.90 (dt, J = 15.7, 1.2 Hz,
1H), 5.58-5.55 (m, 1H), 3.71 (s, 3H), 3.50 (s, 3H), 2.70 (dt, J = 7.2, 1.2 Hz, 2H), 1.81 (d,
J = 2.0 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.2, 165.4, 141.8, 129.6, 128.4,
127.5, 127.3, 124.7, 84.3, 74.3, 65.1, 51.5, 3.5 ppm; HRMS-ESI [M+Na+] berechnet für
C19H19O5F3Na: 407.0182, gefunden: 407.0175.

Experimenteller Teil
- 124 -
5-(S)-(OPMB)-6-in-2-octensäuremethylester 266
OH O
OMe
[168.18]
OPMB O
OMe
[288.33]
BF3.Et2O, CH2Cl2, -78 °C
MeO
CCl3
NH
Der Ester 259 (500 mg, 2.97 mmol) wird in CH2Cl2 aufgenommen (11 ml), auf
-78 °C gekühlt und mit PMB-Trichloracetimidat (870 mg, 3.26 mmol) versetzt. Anschließend
wird dem Gemisch langsam BF3·Et2O (0.2 mL, 1.6 mmol) zugetropft. Nach 2 h wird
Triethylamin (3 ml) addiert und 5 min später zusätzlich noch gesättigte wässrige NaHCO3-
Lsg. (15 ml) zugegeben und die Mischung auf RT erwärmt. Die Phasen werden getrennt und
die wässrige Phase mit CH2Cl2 extrahiert (2 x 40 ml). Die vereinigten organischen Phasen
werden über MgSO4 getrocknet, filtriert und das Lösungsmittel am Rotationsverdampfer
entfernt. Nach säulenchromatographischer Reinigung (PE/EtOAc 6:1) erhält man das Produkt
(710 mg, 2.46 mmol, 83%) als gelbes Öl.
[α]20D = -81.2 (c 1.0 CHCl3); Rf = 0.31 (EtOAc/PE 1:6); 1H-NMR (400 MHz, CDCl3) δ 7.25
(d, J = 8.87 Hz, 2H), 6.97 (dt, J = 15.7, 7.2 Hz, 1H), 6.85 (d, J = 8.87 Hz, 2H), 5.88 (dt,
J = 15.7, 1.7 Hz, 1H), 4.68 (d, J = 11.3 Hz, 1H), 4.40 (d, J = 11.3 Hz, 1H), 4.07-4.13 (m, 1H),
3.78 (s, 3H), 3.70 (s, 3H), 2.56 (tt, J = 7.2, 1.7 Hz, 2H), 1.86 (d, J = 2.0 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 166.7, 159.2, 144.4, 129.7, 129.6, 123.3, 113.8, 83.0, 70.0,
66.9, 55.3, 51.4, 38.8, 3.6 ppm; HRMS-ESI [M+Na+] berechnet für C17H20O4Na: 311.1259,
gefunden: 311.1286.
5-(S)-(OPMB)-6-in-2-octen-1-ol 267
OPMB O
OMe
[288.33]
DibAl-H, CH2Cl2, -78 °COPMB OH
[260.32]
Der Ester 266 (500 mg, 1.73 mmol) wird in 17 mL CH2Cl2 aufgenommen und auf -78 °C
abgekühlt. Diisobutylaluminiumhydrid (1 M in CH2Cl2, 5.19 mL, 5.19 mmol) wird über
25 min zugetropft. Die Mischung wird 30 min bei -78 °C gerührt. Durch Zugabe von
Methanol und gesättigter Rochelle-Salz-Lsg. wird die Reaktion gequencht und auf RT
erwärmt. Die wässrige Phase wird mit Wasser verdünnt und mit CH2Cl2 (3 x 20 mL)
extrahiert. Die vereinigten organischen Phasen werden mit gesättigter NaCl-Lsg. gewaschen,

Experimenteller Teil
- 125 -
über MgSO4 getrocknet, filtriert und das Lösungsmittel unter vermindertem Druck entfernt.
Nach säulenchromatographischer (EtOAc/PE 2:3) Reinigung wird der Alkohol (414 mg,
1.59 mmol, 92 %) als farbloses Öl erhalten.
[α]20D = -63.9 (c 1.0 CHCl3); Rf = 0.28 (EtOAc/PE 2:3); 1H-NMR (400 MHz, CDCl3) δ 7.25
(d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 5.72 (q, J = 7.2, 4.8 Hz, 2H), 4.68 (d,
J = 11.6 Hz, 1H), 4.40 (d, J = 11.3 Hz, 1H), 4.07 (br s, 1H), 4.03 (tq, J = 6.1, 2.0 Hz, 1H),
2.44 (t, J = 6.1 Hz, 2H), 1.86 (d, J = 2.0 Hz, 3H) ppm; 13C-NMR (100 MHz, CDCl3) δ 159.2,
131.9, 130.0, 129.6, 128.0, 113.7, 82.4, 77.7, 69.9, 68.0, 63.6, 55.3, 38.8, 3.6 ppm; HRMS-
ESI [M+Na+] berechnet für C16H20O3Na: 283.1310, gefunden: 283.1292.
5-(S)-(OPMB)-2,3-(R,R)-oxiran-6-octin-1-ol 268
OPMB OH
[260.32]
OPMB OH
[276.32]
Ti(Oi-Pr)4, DET-(-)t-BuOOH, CH2Cl2, -20 °C O
Zu einer Suspension von Molsieb (100 mg, 4Å) in CH2Cl2 (7.6 ml) werden bei -20 °C
nacheinander D-(-)-Diethyltartrat (0.13 ml, 0.76 mmol), Ti(Oi-Pr)4 (0.17 ml, 0.57 mmol) und
tert-Butylhydroperoxid (0.28 ml, 1.56 mmol, 5.5 M in Decan) addiert. Nach 1 h wird der
Allylalkohol 267 (200 mg, 0.76 mmol) in CH2Cl2 (2 ml) über 5 min zugetropft. Die
Reaktionsmischung wird über Nacht bei -20 °C gerührt. Anschließend durch Zugabe von
Wasser (2 ml) gequencht, auf 0°C erwärmt und nach 30 min mit wässriger 30% NaOH
(60 ml, mit NaCl gesättigt) versetzt. Es wird für weitere 40 min gerührt und mit CH2Cl2
(5 ml) verdünnt. Die Phasen werden getrennt und die wässrige Phase mit CH2Cl2 extrahiert
(3 x 20 ml). Die vereinigten organischen Phasen werden mit MgSO4 getrocknet, filtriert und
im Rotationsverdampfer eingeengt. Nach Reinigung durch Flash-Chromatographie
(EtOAc/PE 2:3) wird das Epoxid (172 mg, 0.62 mmol, 82%) als farbloses Öl erhalten.
[α]20D = -41.2 (c 1.0 CHCl3); Rf = 0.15 (EtOAc/PE 2:3); 1H-NMR (400 MHz, CDCl3) δ 7.30
(d, J = 8.9 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 4.68 (d, J = 11.6 Hz, 1H), 4.40 (d, J = 11.6 Hz,
1H), 4.17 (tq, J = 6.5, 2.4 Hz, 1H), 3.85 (d, J = 13.3 Hz, 1H), 3.78 (s, 3H), 3.59 (d,
J = 12.3 Hz, 1H), 3.15 (td, J = 5.8, 2.4 Hz, 1H), 2.94 (dt, J = 13.3 Hz, 1H), 1.96-2.02 (m, 1H),
1.85-1.93 (m, 1H), 1.88 (d, J = 2.0 Hz, 3H), 1.63 (br s, 1H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 159.2, 131.9, 129.9, 129.6, 113.8, 83.0, 70.0, 65.9, 61.6, 58.2, 55.3, 52.8, 38.2,
3.6 ppm; HRMS-ESI [M+Na+] berechnet für C16H20O4Na: 299.1259, gefunden: 299.1287.

Experimenteller Teil
- 126 -
5-(S)-(OPMB)-3-(S)-hydroxy-6-octin-1-ol 269
OPMB OH
[276.32]
OPMBO OH
[278.34]
Red-Al, CH2Cl2, -5 °C OH
Das Epoxid 268 (150 mg, 0.54 mmol) in CH2Cl2 (6 mL) wird auf -15 °C abgekühlt.
Anschließend wird über 20 min Red-Al (3.5 M in Toluol, 0.31 mL, 1.08 mmol) zugetropft
und die Reaktionslösung auf -5 °C erwärmt. Die Lösung wird über Nacht bei -5 °C gerührt.
Die Reaktion wird durch Zugabe von gesättigter Rochelle-Salz-Lsg. (5 mL) gequencht und
für weitere 30 min bei RT gerührt. Die Phasen werden getrennt und die wässrige Phase mit
CH2Cl2 extrahiert (3 x 20 ml). Die vereinigten organischen Phasen werden mit MgSO4
getrocknet, filtriert und im Rotationsverdampfer eingeengt. Nach Reinigung durch Flash-
Chromatographie (EtOAc/PE 2:1) wird das erwünschte Diol (138 mg, 0.49 mmol, 92%) als
farbloses Öl erhalten.
[α]20D = -34.7 (c 1.0 CHCl3); Rf = 0.24 (EtOAc/PE 2:1); 1H-NMR (400 MHz, CDCl3) δ 7.25
(d, J = 8.2 Hz, 2H), 6.85 (d, J = 8.9 Hz, 2H), 4.73 (d, J = 10.9 Hz, 1H), 4.40 (d, J = 11.3 Hz,
1H), 4.27-4.22 (m, 1H), 4.07-4.01 (m, 1H), 3.80-3.75 (m, 5H), 2.70 (br s, 1H), 2.03-1.94 (m,
1H), 1.87 (d, J = 2.0 Hz, 3H), 1.84-1.75 (m, 1H), 1.68-1.62 (m, 2H) ppm; 13C-NMR
(100 MHz, CDCl3) δ 159.4, 129.8, 129.4, 113.9, 82.8, 70.3, 68.3, 61.3, 55.3, 43.1, 38.5,
3.6 ppm; HRMS-ESI [M+Na+] berechnet für C16H22O4Na: 301.1416, gefunden: 301.1414.
5,3-(S,S)-(PMP-dioxan)-6-octin-1-ol 270
PMBO OH
[278.34]
OH O OH
[276.32]
ODDQ, CH2Cl2, 0 °C
PMP
Das Diol 269 (100 mg, 0.36 mmol) wird in CH2Cl2 (1.2 mL) gelöst und auf 0 °C gekühlt.
Anschließend wird in Portionen 139 mg DDQ (0.61 mmol) zugegeben. Die Reaktion wird für
2 h bei 0 °C gerührt und durch Zugabe von gesättigten NaHCO3-Lsg. (1 mL) gequencht. Die
Phasen werden getrennt und die wässrige Phase mit CH2Cl2 extrahiert (3 x 2 ml). Die
vereinigten organischen Phasen werden mit MgSO4 getrocknet, filtriert und im
Rotationsverdampfer eingeengt. Nach Reinigung durch Flash-Chromatographie (EtOAc/PE
3:2) wird das erwünschte Acetal (66 mg, 0.24 mmol, 67%) als gelblicher Feststoff erhalten.

Experimenteller Teil
- 127 -
[α]20D = -27.0 (c 0.9 CHCl3); Rf = 0.24 (EtOAc/PE 3:2); 1H-NMR (400 MHz, CDCl3) δ 7.37
(d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 5.45 (s, 1H), 4.56 (dt, J = 11.3, 2.4 Hz, 1H),
4.06-3.99 (m, 1H), 3.87-3.75 (m, 2H), 3.76 (s, 3H), 1.92-1.84 (m, 1H), 1.83 (d, J = 2.0 Hz,
3H), 1.79-1.74 (m, 2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 159.9, 130.4, 127.6, 113.5,
101.0, 82.0, 75.4, 67.4, 59.9, 55.2, 37.7, 37.6, 3.6 ppm; HRMS-ESI [M+Na+] berechnet für
C16H20O4Na: 299.1259, gefunden: 299.1224.
7-Iodo-5,3-(S,S)-(PMP-dioxan)-6-octen-1-ol 247
O OH
[276.32]
O
PMPa) Bu3SnH, Pd(PPh3)2Cl2, Benzolb) I2, CH2Cl2, 0 °C O OH
[404.24]
O
PMP
I
Das Alkin 270 (50 mg, 0.18 mmol) wird in Benzol (18 mL) aufgenommen und mit
Pd(PPh3)2Cl2 (6 mg, 9 µmol) versetzt. Anschließend wird bei RT langsam 0.25 mL HSnBu3
(1.1 mmol) zugetropft. Nach 20 min wird das Lösungsmittel am Rotationsverdampfer
entfernt. Die Reinigung des Rohprodukts erfolgt durch Flash-Chromatographie (PE/EtOAc
2:1). Das farblose Öl wird in 2 mL CH2Cl2 aufgenommen und auf 0 °C abgekühlt. Über
10 min wird eine Lösung aus Iod (92 mg, 0.36 mmol) in CH2Cl2 (1 mL) zugetropft und für
weitere 10 min gerührt. Anschließend wird ein Gemisch aus gesättigter NaHCO3-Lsg. und
gesättigter Natriumsulfit-Lsg. addiert. Die Phasen werden getrennt und die wässrige Phase mit
CH2Cl2 extrahiert (3 x 5 ml). Die vereinigten organischen Phasen werden mit MgSO4
getrocknet, filtriert und im Rotationsverdampfer eingeengt. Nach Reinigung durch Flash-
Chromatographie (EtOAc/PE 1:2) wird das erwünschte Vinyliodid (56 mg, 0.13 mmol, 77%)
als farbloses Öl erhalten.
[α]20D = -15.3 (c 1.0 CHCl3); Rf = 0.24 (EtOAc/PE 3:2); 1H-NMR (400 MHz, CDCl3) δ 7.37
(d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.9 Hz, 2H), 5.45 (s, 1H), 4.56 (dt, J = 11.3, 2.4 Hz, 1H),
4.06-3.99 (m, 1H), 3.87-3.75 (m, 2H), 3.76 (s, 3H), 1.92-1.84 (m, 1H), 1.83 (d, J = 2.0 Hz,
3H), 1.79-1.74 (m, 2H) ppm; 13C-NMR (100 MHz, CDCl3) δ 159.9, 130.4, 127.6, 113.5,
101.0, 82.0, 75.4, 67.4, 59.9, 55.2, 37.7, 37.6, 3.6 ppm; HRMS-ESI [M+Na+] berechnet für
C16H21O4Na: 427.0382, gefunden: 427.0399.

Experimenteller Teil
- 128 -
5-(R)-(OTBS)-4-(R)-,6-dimethyl-2-heptensäuremethylester 271
[186.12]
OMe
OOH TBSOTf, 2,6-LutidinCH2Cl2
[300.21]
OMe
OOTBS
Die Verbindung wurde nach AAV 5 synthetisiert. Es wurden 357 mg (1.19 mmol, 94%) des
erwünschten Produkts als farbloses Öl erhalten.
[α]20D =
+30.1 (c 1.0, CHCl3); Rf = 0.35 (PE/EtOAc 8:1); 1H-NMR (400 MHz, CDCl3) δ 6.97
(dd, J = 15.7, 8.0 Hz, 1H), 5.75 (dd, J = 15.7, 1.2 Hz, 1H), 3.71 (s, 3H), 3.35 (t, J = 4.8 Hz,
1H), 2.49 (q, J = 6.8 Hz, 1H), 1.74-1.65 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 0.89 (s, 9H),
0.86 (d, J = 7.2 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H), 0.01 (d, J = 5.5 Hz, 6H) ppm; 13C-NMR
(100 MHz, CDCl3) δ 167.2, 153.3, 119.7, 80.0, 51.4, 40.9, 31.9, 26.1, 20.3, 17.5, 15.1,
-3.7, -3.8 ppm; HRMS-ESI [M+Na+] berechnet für C16H32O3Si1Na: 323.2018, gefunden:
323.2005.
5-(R)-(OTBS)-4-(R)-,6-dimethyl-2-heptensäureallylamid 246
[300.21]
OMe
OOTBSAlMe3, CH2Cl2
[325.24]
NH
OOTBSH2N
Allylamin (65 µL, 0.86 mmol) wird in CH2Cl2 (1 mL) aufgenommen und abgekühlt (0 °C).
Langsam wird Trimethylaluminium (2M in Hexan, 0.43 mL, 0.86 mmol) zugetropft und das
Gemisch 20 min bei RT gerührt. Anschließend wird der Ester 271 (130 mg, 0.43 mmol) in
CH2Cl2 (0.5 mL) zugegeben und die Reaktion über Nacht bei RT gerührt. Durch vorsichtige
Zugabe von 1M HCl (2 mL) bei 0 °C wird die Reaktion gequencht. Die Phasen werden
getrennt und die wässrige Phase mit CH2Cl2 extrahiert (3 x 5 ml). Die vereinigten organischen
Phasen werden mit MgSO4 getrocknet, filtriert und im Rotationsverdampfer eingeengt. Nach
Reinigung durch Flash-Chromatographie (EtOAc/PE 1:3) wird das erwünschte Amid
(100 mg, 0.31 mmol, 72%) als farbloses Öl erhalten.
[α]20D =
+20.3 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.82
(dd, J = 15.4, 8.2 Hz, 1H), 5.85 (dq, J = 16.0, 5.8 Hz, 1H), 5.72 (dd, J = 15.4, 1.0 Hz, 1H),
5.45 (br s, 1H), 5.18 (dd, J = 17.4, 1.7 Hz, 1H), 5.15 (dd, J = 10.2, 1.4 Hz, 1H), 3.94 (br t,
J = 5.8 Hz, 2H), 3.35 (t, J = 5.8 Hz, 1H), 2.45 (q, J = 6.8 Hz, 1H), 1.74-1.65 (m, 1H), 1.02 (d,
J = 6.8 Hz, 3H), 0.89 (s, 9H), 0.86 (d, J = 7.2 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H), 0.02 (s,

Experimenteller Teil
- 129 -
6H) ppm; 13C-NMR (100 MHz, CDCl3) δ 165.7, 148.7, 134.3, 122.2, 116.5, 80.1, 80.1,
41.9, 40.8, 31.9, 26.1, 20.4, 17.3, 15.6, -3.6, -3.8 ppm; HRMS-ESI [M+Na+] berechnet für
C18H35O2N1Si1Na: 348.2335, gefunden: 348.2268.
5-(R)-(OTBS)-4-(R)-,6-dimethyl-2-heptensäurepropargylamid 272
[300.21]
OMe
OOTBSAlMe3, CH2Cl2
[323.22]
NH
OOTBSNH2
Propargylamin (42 µL, 0.66 mmol) wird in CH2Cl2 (1 mL) aufgenommen und abgekühlt
(0 °C). Langsam wird Trimethylaluminium (2M in Hexan, 0.33 mL, 0.66 mmol) zugetropft
und das Gemisch 20 min bei RT gerührt. Anschließend wird der Ester 271 (100 mg,
0.33 mmol) in CH2Cl2 (0.5 mL) zugegeben und die Reaktion über Nacht bei RT gerührt.
Durch vorsichtige Zugabe von 1M HCl (2 mL) bei 0 °C wird die Reaktion gequencht. Die
Phasen werden getrennt und die wässrige Phase mit CH2Cl2 extrahiert (3 x 5 ml). Die
vereinigten organischen Phasen werden über MgSO4 getrocknet, filtriert und im
Rotationsverdampfer eingeengt. Nach Reinigung durch Flash-Chromatographie (EtOAc/PE
1:3) wird das erwünschte Amid (93 mg, 0.29 mmol, 88%) als farbloses Öl erhalten.
[α]20D =
+33.1 (c 1.0, CHCl3); Rf = 0.34 (PE/EtOAc 3:1); 1H-NMR (400 MHz, CDCl3) δ 6.84
(dd, J = 15.4, 7.8 Hz, 1H), 5.72 (dd, J = 15.7, 1.0 Hz, 1H), 5.79 (br s, 1H), 5.73 (dd, J = 15.7,
1.0 Hz, 1H), 4.09 (dd, J = 5.5, 2.7 Hz, 1H), 3.33 (t, J = 5.8 Hz, 1H), 2.45 (q, J = 6.8 Hz, 1H),
2.20 (t, J = 2.4 Hz, 3H), 1.73-1.65 (m, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.88 (s, 9H), 0.85 (d,
J = 7.1 Hz, 3H), 0.81 (d, J = 6.8 Hz, 3H), 0.00 (d, J = 1.7 Hz, 6H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 165.5, 149.3, 121.6, 80.0, 71.5, 40.8, 31.9, 29.2, 26.1, 20.4, 18.4, 17.2, 15.6,
-3.6, -3.8 ppm; HRMS-ESI [M+Na+] berechnet für C18H33O2N1Si1Na: 346.2178, gefunden:
346.2150.

Experimenteller Teil
- 130 -
5-(R)-(OTBS)-4-(R)-,6-dimethyl-2-heptensäure-(6,8-(S,S)-(PMP-dioxan)-4-methyl-2,4-
decen-10-ol)-amid 243
OTBS
NH
O
O
OH
O
PMP
I
OTBS
NH
O
O O
PMP
Pd(OAc)2, AgOAc
DMF, RTOH
[325.24]
[601.37]
[404.04]
Zu einer Lösung vom Allylamid 246 (9 mg, 27.7 µmol) in DMF (0.2 mL) wird unter
Lichtausschluss nacheinander Pd(OAc)2 (1 mg, 4 µmol), AgOAc (3 mg, 18 µmol) zugegeben.
Anschließend wird das Iodid 247 (8 mg, 20.6 µmol) in DMF (0.2 mL) addiert und die
Reaktion über Nacht bei RT gerührt. Das Gemisch wird direkt auf Kieselgel aufgetragen und
durch Säulenchromatographie aufgereinigt. Nach der Reinigung wird das erwünschte Produkt
als farbloses Öl erhalten (4 mg, 6.6 µmol, 32%).
[α]20D =
+11.6 (c 0.7, CHCl3); Rf = 0.18 (PE/EtOAc 1:1); 1H-NMR (400 MHz, CDCl3) δ 7.37
(d, J = 8.7 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 6.82 (dd, J = 15.3, 7.3 Hz, 1H), 6.15 (d,
J = 15.7 Hz, 1H), 6.69 (dd, J = 15.3, 1.1 Hz, 1H), 6.68 (dt, J = 15.7, 6.3 Hz, 1H), 5.56 (s, 1H),
5.49 (d, J = 7.9 Hz, 1H), 5.43 (t, J = 5.6 Hz, 1H), 4.56 (dt, J = 11.3, 2.4 Hz, 1H), 4.69 (t,
J = 8.0 Hz, 1H), 4.15-4.09 (m, 1H), 4.04-3.92 (m, 2H), 3.87-3.80 (m, 2H), 3.77 (s, 3H),
3.34 (q, J = 5.9, 4.2 Hz, 1H), 2.46 (q, J = 13.8, 7.6 Hz, 1H), 1.92-1.84 (m, 1H), 1.80 (d,
J = 1.0 Hz, 3H), 1.72-1.66 (m, 2H), 1.43-1.37 (m, 2H), 1.01 (d, J = 6.8 Hz, 3H), 0.89 (s, 9H),
0.86 (d, J = 6.9 Hz, 3H), 0.81 (d, J = 6.8 Hz, 3H), 0.02 (s, 6H) ppm; 13C-NMR (100 MHz,
CDCl3) δ 167.8, 165.7, 159.9, 148.8, 136.1, 135.2, 132.4, 130.8, 128.8, 127.4, 125.4, 122.1,
113.6, 100.7, 80.1, 73.9, 68.1, 55.3, 41.3, 40.8, 38.7, 37.9, 36.5, 31.9, 30.3, 20.4, 18.4, -3.6,
-3.8 ppm; HRMS-ESI [M+H+] berechnet für C34H56O6N1Si1: 602.3877, gefunden: 602.3881.

Literaturverzeichnis
- 131 -
7. LITERATURVERZEICHNIS
[1] J. Rohr, Angew. Chem. 2000, 112, 2967; Angew. Chem., Int. Ed. 2000, 39, 2847.
[2] B. Fugmann, S. Lang-Fugmann, W. Steglich, Römpp, Naturstoffe, 10. Auflage, Georg Thieme Verlag, Stuttgart. New York, 1997, 380.
[3] A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721.
[4] U. Klar, B. Buchmann, W. Schwede, W. Skuballa, J. Hoffmann, R. B. Lichtner, Angew. Chem. 2006, 118, 8110; Angew. Chem., Int. Ed. 2006, 45, 7942.
[5] J. Staunton, K. Weissmann, Nat. Prod. Rep. 2001, 18, 380.
[6] C. A. Wurtz, J. Prakt. Chemie 1872, 5, 457.
[7] A. P. Borodin, Ber.6 1873, 6, 982.
[8] H. E. Zimmermann, M. D. Traxler, J. Am. Chem. Soc. 1957, 79, 1920.
[9] a) I. Paterson, M. M. Mansuri, Tetrahedron 1985, 41, 3569; b) S. Masamune, G. S. Bates, J. W. Corcoran, Angew. Chem. 1977, 89, 602; Angew. Chem., Int. Ed. Engl. 1977, 16, 585.
[10] R. B. Woodward in Perspectives in Organic Chemistry (Hrsg.: A. Todd), Wiley-Interscience, New York, 1956, 160.
[11] R. B. Woodward, E. Logush, K. P. Nambiar, K. Sakan, D. E. Ward, B. W. Au-Yeung, P. Balaram, L. J. Browne, P. J. Card, C. H. Chen, R. B. Chenevert, A. Fliri, K. Frobel, H.-J. Gais, D. G. Garratt, K. Hayakawa, K.W. Heggie, D. P. Hesson, D. Hoppe, I. Hoppe, J. A. Hyatt, D. Ikeda, P. A. Jacobi, K. S. Kim, Y. Kobuke, K. Kojima, K. Krowicki, V. J. Lee, T. Leutert, S. Malchenko, J. Martens, R. S. Matthews, B. S. Ong, J. B. Press, T. V. Rajan Babu, G. Rousseau, H. M. Sauter, M. Suzuki, K. Tatsuta, L. M. Tolbert, E. A. Truesdale, I. Uchida, Y. Ueda, T. Uyehara, A. T. Vasella, W. C. Vladuchick, P. A. Wade, R. M. Williams, H. N.-C. Wong, J. Am. Chem. Soc. 1981, 103, 3210.
[12] a) B. Schetter, R. Mahrwald, Angew. Chem. 2006, 118, 7668; Angew. Chem., Int. Ed. 2006, 45, 7506; b) L. M. Geary, P. G. Hultin Tetrahedron: Asymmetry 2009, 20, 131.
[13] a) D. A. Evans, J. R. Gage, Org. Synth. 1990, 68, 83; b) D. A. Evans, L. R. McGee, J. Am. Chem. Soc. 1981, 103, 2876; c) D. A. Evans, J. Bartroli, T. L. Shih, J. Am. Chem. Soc. 1981, 103, 2127.
[14] D. A. Evans, Pure Appl. Chem. 1981, 53, 1109.
[15] C. H. Heathcock, M. A. Walker, J. Org. Chem. 1991, 56, 5747.
[16] I. Paterson, A. Lister, C. K. McClure, Tetrahedron Lett. 1986, 27, 4787.
[17] a) T. Mukaiyama, K. Narasaka, K. Banno, Chem. Lett. 1973, 1012; b) K. Saigo, M. Osaki, T. Mukaiyama, Chem. Lett. 1975, 989; c) Mukaiyama, T. Org. React. 1982, 28, 203.
[18] R. Mahrwald, Chem. Rev. 1999, 99, 1095.
[19] Y. Yamamoto, H. Yatagati, Y. Naruta, K. Maruyama, J. Am. Chem. Soc. 1980, 102, 7107.
[20] C. Gennari, A. Bernardi, L. Colombo, C. Scolastico, J. Am. Chem. Soc. 1985, 107, 5812.
[21] M. T. Reetz, A. E. Vougioukas, Tetrahedron Lett. 1987, 28, 793.
[22] a) T. Bach, Angew. Chem. 1994, 106, 433; Angew. Chem., Int. Ed. Engl. 1994, 33, 417; b) T. K. Hollis, B. Bosnich, J. Am. Chem. Soc. 1995, 117, 4570; c) H. Gröger, E. M. Vogl, M. Shibasaki, Chem. Eur. J. 1998, 4, 1137.
[23] G. R. Keck, D. Krishnamurthy, J. Am. Chem. Soc. 1995, 117, 2363.
[24] R. C. Fuson, Chem. Rev. 1935, 16, 1.
[25] J. L. Herrmann, G. R. Kieczykowski, R. H. Schlessinger, Tetrahedron Lett. 1973, 14, 2433.
[26] M. W. Rathke, D. Sullivan, Tetrahedron Lett. 1972, 13, 4249.
[27] I. Fleming, Frontier Orbitals and Organic Chemical Reactions, Wiley-Interscience, New York, 1996, 45.

Literaturverzeichnis
- 132 -
[28] a) S. E. Denmark, J. R., Jr. Heemstra, G. L. Beutner, Angew. Chem. 2005, 117, 4760; Angew. Chem., Int. Ed. 2005, 44, 4682; b) M. Kalesse, Top. Curr. Chem. 2005, 244, 43; c) A. Soriente, M. De Rosa, R. Villano, A. Scettri, Curr. Org. Chem. 2004, 8, 993; d) G. Casiraghi, F. Zanardi, G. Appendino, G. Rassu, Chem. Rev. 2000, 100, 1929.
[29] a) C. J. Brennan, J.-M. Campagne, Tetrahedron Lett. 2001, 42, 5195; b) G. Bluet, J.-M. Campagne, Tetrahedron Lett. 1999, 40, 5507; c) G. Bluet, J.-M. Campagne, J. Org. Chem. 2001, 66, 4293; d) G. Bluet, J.-M. Campagne, Synlett 2000, 221; e) G. Bluet, B. Bazán-Tejeda, J.-M. Campagne, Org. Lett. 2001, 3, 3807; f) B. Bazán-Tejeda, G. Bluet, G. Broustal, J.-M. Campagne, Chem. Eur. J. 2006, 12, 8358.
[30] B. L. Pagenkopf, J. Krüger, A. Stojanovic, E. M. Carreira, Angew. Chem. 1998, 110, 3312; Angew. Chem., Int. Ed. 1998, 37, 3124.
[31] I. Paterson, J. D. Smith, R. A. Ward, Tetrahedron 1995, 5, 9413.
[32] a) M. Christmann, U. Bhatt, M. Quitschalle, E. Claus, M. Kalesse, Angew. Chem. 2000, 112, 4535; Angew. Chem., Int. Ed. 2000, 39, 4364; b) J. Hassfeld, M. Kalesse, Tetrahedron Lett. 2002, 43, 5093; c) F. P. Liesener, U. Jannsen, M. Kalesse, Synthesis 2006, 2590; d) F. P. Liesener, M. Kalesse, Synlett 2005, 2236; e) J. Hassfeld, M. Kalesse, Synthesis 2002, 2007; f) T. Brodmann, M. Lorenz, R. Schäckel, S. Simsek, M. Kalesse, Synlett 2009, 174.
[33] a) M. Christmann, M. Kalesse, Tetrahedron Lett. 2001, 42, 1269; b) J. Hassfeld, M. Christmann, M. Kalesse, Org. Lett. 2001, 3, 3561; c) N. Rahn, M. Kalesse, Synlett 2005, 863.
[34] E. M. Carreira, A. S. Roberti, Tetrahedron Lett. 1994, 35, 4323.
[35] a) S. E. Denmark, T. Wynn, G. L. Beutner, J. Am. Chem. Soc. 2002, 124, 13405; b) S. E. Denmark, J. R., Jr. Heemstra, Org. Lett. 2003, 5, 2303; c) S. E. Denmark, J. R., Jr. Heemstra, J. Org. Chem. 2007, 72, 5668; d) S. E. Denmark, G. L. Beutner, J. Am. Chem. Soc. 2003, 125, 7800.
[36] V. Gutmann, The Donor-Acceptor Approach to Molecular Interactions; Plenum Press: New York, 1978, 1.
[37] a) R. Villano, M. R. Acocella, A. Massa, L. Palombi, A. Scettri, Tetrahedron: Asymmetry 2006, 17, 3332; b) S. E. Denmark, S. Fujimori, J. Am. Chem. Soc. 2005, 127, 8971.
[38] a) S.-i. Shirokawa, M. Kamiyama, T. Nakamura, M. Okada, A. Nakazaki, S. Hosokawa, S. Kobayashi, J. Am. Chem. Soc. 2004, 126, 13604; b) T. Nakamura, S.-i. Shirokawa, S. Hosokawa, A. Nakazaki, S. Kobayashi, Org. Lett. 2006, 8, 677; c) S.-i. Shirokawa, M. Shinoyama, I. Ooi, A. Hosokawa, A. Nakazaki, S. Kobayashi, Org. Lett. 2007, 9, 849; d) M. Shinoyama, S.-i. Shirokawa, A. Nakazaki, S. Kobayashi, Org. Lett. 2009, 11, 1277.
[39] a) M. E. Botha, R. G. F. Giles, S. C. Yorke, J. Chem. Soc. Perkin Trans. I 1991, 85; b) D. Cameron, R. M. Heisey, Aust. J. Chem. 2000, 53, 109; c) D. W. Cameron, M. G. Looney, J. A. Patterman, Tetrahedron Lett. 1995, 36, 7555; d) P. Galatsis, J. J. Manwell, S. D. Millan, Tetrahedron Lett. 1996, 37, 5261.
[40] D. J. Morrison, W. E. Piers, M. Parvez, Synlett 2004, 2429.
[41] H. Wittig, W. Herwig, Chem. Ber. 1955, 88, 962.
[42] M. Cereghetti, R. Schmid, P. Schönholzer, A. Rageot, Tetrahedron Lett. 1996, 37, 5343.
[43] M. Reetz, M. Willhun, C. Psiorz, R. Goddard, Chem. Comm. 1999, 12, 1105.
[44] R. Noyori, I. Tomino, Y. Tanimoto, J. Am. Chem. Soc. 1979, 101, 3129.
[45] Y. Chen, S. Yekta, A. K. Yudin, Chem. Rev. 2003, 103, 3155.
[46] H. Kitajima, K. Ito, Y. Aoki, T. Katsuki, Bull. Chem. Soc. Jpn. 1997, 70, 207.
[47] T. R. Kelly, A. Whiting, N. S. Chandrakumar, J. Am. Chem. Soc. 1986, 108, 3510.
[48] H. Ishitani, Y. Yamashita, H. Shimizu, S. Kobayashi, J. Am. Chem. Soc. 2000, 122, 5403.
[49] M. Sato, S. Sunami, Y. Sugita, C. Kaneko, Heterocycles 1995, 41, 1435.
[50] J. Haßfeld, Diplomarbeit, Leibniz Universität Hannover, 2001.

Literaturverzeichnis
- 133 -
[51] a) H. J. C. Yeh, S. K. Balani, H. Yagi. R. M. E. Greene, N. D. Sharma, D. R. Boyd, D. M. Jerina, J. Org. Chem. 1986, 51, 5439; b) L. M. Sweeting, D. C. Crans, G. M. Whitesides, J. Org. Chem. 1987, 52, 2273.
[52] L. V. Heumann, G. E. Keck, Org. Lett. 2007, 27, 4275.
[53] E. M. Carreira, R. A. Singer, W. Lee, J. Am. Chem. Soc. 1994, 116, 8837.
[54] R. A. Singer, E. M. Carreira, J. Am. Chem. Soc. 1995, 117, 12360.
[55] a) Y. Kim, R. A. Singer, E. M. Carreira, Angew. Chem. 1998, 110, 1321; Angew. Chem., Int. Ed. 1998, 37, 1261; b) S. Anne, W. Yong, M. Vandewalle, Synlett 1999, 1435.
[56] a) A. K. Ghosh, P. Mathivanan, J. Capiello, Tetrahedron: Asymmetry 1988, 9, 1; b) G. Desimoni, G. Faita, K. A. Jorgensen, Chem. Rev. 2006, 106, 3561.
[57] R. Annunziata, M. Benaglia, M. Cinquini, F. Cozzi, A. Puglisi, Eur. J. Org. Chem. 2003, 1428.
[58] S. Crosignani, G. Desimoni, G. Faita, P. Righetti, Tetrahedron 1998, 54, 15721.
[59] D. A. Evans, D. W. C. MacMillan, K. R. Campos, J. Am. Chem. Soc. 1997, 119, 10859.
[60] a) D. A. Evans, M. C. Kozlowski, J. A. Murry, C. S. Burgey, K. R. Campos, B. T. Connell, R. J. Staples, J. Am. Chem. Soc. 1999, 121, 669; b) D. A. Evans, J. A. Murry, M. C. Kozlowski, J. Am. Chem. Soc. 1996, 118, 5814.
[61] D. A. Evans, D. M. Fitch, T. E. Smith, V. J. Cee, J. Am. Chem. Soc. 2000, 122, 10033.
[62] E. J. Corey, F. Xu, M. C. Nee, J. Am. Chem. Soc. 1997, 119, 12414.
[63] M. Horikawa, J. Busch-Petersen, E. J. Corey, Tetrahedron Lett. 1999, 40, 3843.
[64] G. Bluet, J. M. Campagne, J. Org. Chem. 2001, 66, 4293.
[65] G. Islas-Gonzales, M. Bois-Choussy, J. Zhu, Org. Biomol. Chem. 2003, 1, 30.
[66] J. Seyden-Penne, Chiral Auxiliaries and Ligands in Asymmetric Synthesis, Wiley, New York, 1995.
[67] D. Sartor, J. Saffrich, G. Helmchen, Synlett 1990, 197.
[68] M. Takasu, H. Yamamoto, Synlett 1990, 194.
[69] S.-i. Kiyooka, Y. Kaneko, M. Komura, H. Matsuo, M. Nakano, J. Org. Chem. 1991, 56, 2276.
[70] S.-i. Kiyooka, M. A. Hena, T. Yabukami, K. Murai, F. Goto, Tetrahedron Lett. 2000, 41, 7511.
[71] R. Fujiyama, K. Goh, S.-i. Kiyooka, Tetrahedron Lett. 2005, 46, 1211.
[72] E. J. Corey, C. L. Cywin, T. D. Roper, Tetrahedron Lett. 1992, 33, 6907.
[73] K. Ishihara, S. Kondo, H. Yamamoto, J. Org. Chem. 2000, 65, 9125.
[74] E. W. McChesney, W. K. Swann, J. Am. Chem. Soc. 1937, 59, 1116.
[75] T. Harada, H. Iwai, H. Takatsuki, K. Fujita, M. Kubo, A. Oku, Org. Lett. 2001, 3, 2101.
[76] X. Wang, S. Adachi, H. Iwai, H. Takatsuki, K. Fujita, M. Kubo, A. Oku, T. Harada, J. Org. Chem. 2003, 68, 10046.
[77] Y. Su, Masterarbeit, Leibniz Universität Hannover, 2007.
[78] M. Horzella, Diplomarbeit, Leibniz Universität Hannover, 2006.
[79] a) J. A. Dale, D. L. Dull, H. S. Mosher, J. Org. Chem. 1969, 34, 2543; b) I. Ohtani, T. Kusumi, Y. Kashman, H. Kakisawa, J. Am. Chem. Soc. 1991, 113, 4092.
[80] S. Simsek, M. Horzella, M. Kalesse, Org. Lett. 2007, 9, 5637.
[81] A. B. III. Smith, J.A. Jurica, S. P. Walsh, Org. Lett. 2008, 10, 5625.
[82] a) K. Furuta,Y. Miwa, K. Iwauaga, H. Yamamoto, J. Am. Chem. Soc. 1988, 110, 6254; b) K. Ishihara, T. Maruyama, M. Mouri, Q. Gao, K. Furuta, H. Yamamoto, Bull. Chem. Soc. Jpn. 1993, 66, 3483.
[83] M. Sato, S. Sunami, Y. Sugita, C. Kaneko, Chem. Pharm. Bull. 1994, 42, 839.
[84] E. J. Corey, Angew. Chem. 2002, 114, 1724; Angew. Chem., Int. Ed. 2002, 41, 1650.
[85] R. K. Jr. Boeckman, J. E. Pero, D. J. Boehmler, J. Am. Chem. Soc. 2006, 128, 11032.

Literaturverzeichnis
- 134 -
[86] D. H. Ryu, E. J. Corey, J. Am. Chem. Soc. 2003, 125, 6388.
[87] K. Futatsugi, H. Yamamoto, Angew. Chem. 2005, 117, 1508; Angew. Chem., Int. Ed. 2005, 44, 1484.
[88] F. Kazmierczak, P. Helquist, J. Org. Chem. 1989, 54, 3988.
[89] M. T. Crimmins, R. S. Al-awar, I. M. Vallin, W. G. Hollis, R. O'Mahony, J. G. Lever, D. M. Bankaitis- Davis, J. Am. Chem. Soc. 1996, 118, 7513.
[90] a) M. G. Brazhnikova, M. K. Kudinova, N. P. Potapova, T. M. Filippova, E. Borowski, Y. Zelinski, J. Golik, Bioorgan. Khim. 1976, 2, 149; b) J. W. Chamberlin, S. Chen, J. Antibiot. 1977, 30, 197.
[91] a) T. A. Mukhtar, G. D. Wright, Chem. Rev. 2005, 105, 529; b) C. Cocito, A. Kaji, Biochimie 1971, 53, 763; c) C. Cocito, M. Giambattista, Mol. Gen. Genet. 1978, 166, 53; d) H. L. Ennis, Biochemistry 1971, 10, 1265; e) M. B. Purvis, D. G. I. Kingston, N. Fujii, H. G. Floss, J. Chem. Soc., Chem. Commun. 1987, 302; f) R. Parfait, C. Cocito, Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 5496.
[92] M. S. Butler, Nat. Prod. Rep. 2008, 25, 47.
[93] a) G. A. Pankuch, L. M. Kelly, G. Lin, A. Bryskier, C. Couturier, M. R. Jacobs, P. C. Appelbaum, Antimicrob. Agents Chemother. 2003, 47, 3270; b) S. Mabe, W. S. Champney, Curr. Microbiol. 2005, 51, 363; c) M. Dupuis, R. Leclercq, Antimicrob. Agents Chemother. 2006, 50, 237.
[94] a) A. I Meyers, J. P. Lawson, D. G. Walker, R. J. Linderman, J. Org. Chem. 1986, 51, 5111; b) P. Helquist, M. Bergdahl, R. Hett, A. R. Gangloff, M. Demillequand, M. Cottard, M. M. Mader, T. Friebe, J. Iqbal, Y. Wu, B. Akermark, T. Rein, N. Kann, Pure Appl. Chem. 1994, 66, 2063; c) R. H. Schlessinger, E. J. Iwanowicz, J. P. Springer, Tetrahedron Lett. 1988, 29, 1489; d) R. D. Wood, B. Ganem, Tetrahedron Lett. 1983, 24, 4391; e) L. Liu, R. S. Tanke, M. J. Miller, J. Org. Chem. 1986, 51, 5332; f) N. Adje, P. Breuilles, D. Uguen, Tetrahedron Lett. 1993, 34, 4631; g) M. S. Mortensen, J. M. Osbourn, G. A. O'Doherty, Org. Lett. 2007, 9, 3105.
[95] F. Tavares, J. P. Lawson, A. I. Meyers, J. Am. Chem. Soc. 1996, 118, 3303.
[96] R. H. Schlessinger, Y.-J. Li, J. Am. Chem. Soc. 1996, 118, 3301.
[97] A. K. Ghosh, W. Liu, J. Org. Chem. 1997, 62, 7908.
[98] C. J. Brennan, J.-M. Campagne, Tetrahedron Lett. 2001, 42, 5195.
[99] H. Zhang, K. A. Lerro, S.-i. Takekuma, D.-J. Baek, C. Moquin-Pattey, M. F. Boehm, K. Nakanishi J. Am. Chem. Soc. 1994, 116. 6823.
[100] D. A. Evans, J. A. Gauchet-Prunet, J. Org. Chem. 1993, 58, 2446.
[101] K. M. Nicholas, R. Petit, J. Organomet. Chem. 1972, 44, C21-C24; b) B. J. Teobald, Tetrahedron 2002, 58, 4133.
[102] P. Ganesh, K. M. Nicholas, J. Org. Chem. 1993, 58, 5587.
[103] a) M. M. Claffrey, C. J. Hayes, C. H. Heathcock, J. Org. Chem. 1999, 64, 8267; b) J. E. Audia, L. Boisvert, A. D. Patten, A. Villalobos, S. J. Danishefsky, J. Org. Chem. 1989, 54, 3738.
[104] J. A. Marshall, S. Xie, J. Org. Chem. 1995, 60, 7230.
[105] A. B. Smith, K. P. Minbiole, P. R. Verhoest, M. Schelhaas, J. Am. Chem. Soc. 2001, 123, 10942.
[106] A. Basha, M. Lipton, S. M. Weinreb, Tetrahedron Lett. 1977, 48, 4171.
[107] J. K. Stille, Angew. Chem. 1986, 98, 504; Angew. Chem., Int. Ed. Engl. 1986, 25, 508.
[108] L. Capella, A. Degl’Innocenti, A. Mordini, G. Reginato, A. Ricci, G. Seconi, Synthesis 1991, 1201.
[109] a) N. Miyaura, A. Suzuki, J. Chem. Soc., Chem Comm. 1979, 866; b) N. Miyaura, A. Suzuki, Chem. Rev. 1995, 95, 2457. c) A. Suzuki, J. Organomet. Chem. 1999, 576, 147.
[110] S. A. Frank, W. R. Roush, J. Org. Chem. 2002, 67, 4316.
[111] a) R. F. Heck, J. P. Nolley, J. Org. Chem. 1972, 37, 2320; b) T. Mizoroki, K. Mori, A. Ozaki, Bull. Chem. Soc. Jpn. 1971, 44, 581.
[112] T. Jeffery, J. Chem. Soc., Chem. Comm. 1984, 1287.
[113] T. Jeffery, J. Chem. Soc., Chem. Comm. 1991, 324.
[114] M. Kalesse, M. Quitschalle, C. P. Khandavalli, A. Saeed, Org. Lett. 2001, 3, 3107.

Spektrenanhang
1.00
00
0.99
10
1.05
62
3.41
22
3.33
95
11.3
22
6.10
82
Inte
gral
7.24
00
6.15
406.
1497
5.10
295.
0884
5.08
59
4.52
964.
5032
3.58
10
1.66
571.
6614
1.64
861.
6444
0.92
86
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
158.
4574
124.
2314
116.
4827
75.2
205
54.7
466
25.6
704
18.0
742
13.1
700
-4.2
555
(ppm)
-100102030405060708090100110120130140150160170
OMe
OTBS
OMe
OTBS

Spektrenanhang
1.00
00
1.04
34
1.13
79
3.73
19
3.75
73
12.3
44
7.46
43
Inte
gral
7.24
00
6.15
236.
1139
5.32
475.
3230
5.30
775.
2863
5.28
465.
2684
4.43
414.
4076
4.38
294.
3582
3.51
70
1.68
87
0.93
54
0.14
80
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
157.
1268
126.
0978
125.
6843
119.
6313
118.
7549
86.7
275
79.2
513
54.7
020
25.7
116
25.5
725
18.4
107
-4.2
619
(ppm)
-100102030405060708090100110120130140150160
OMe
OTBS
OMe
OTBS

Spektrenanhang
1.00
00
1.24
88
1.27
04
1.09
19
3.50
65
11.8
18
6.95
39
Inte
gral
6.52
856.
4858
4.84
534.
8077
4.80
26
4.59
794.
5928
4.47
084.
4452
3.55
11
0.93
29
0.15
82
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
158.
7318
132.
3878
106.
6992
80.2
389
54.7
599
25.6
495
-4.2
688
(ppm)
-100102030405060708090100110120130140150160
OMe
OTBS
OMe
OTBS
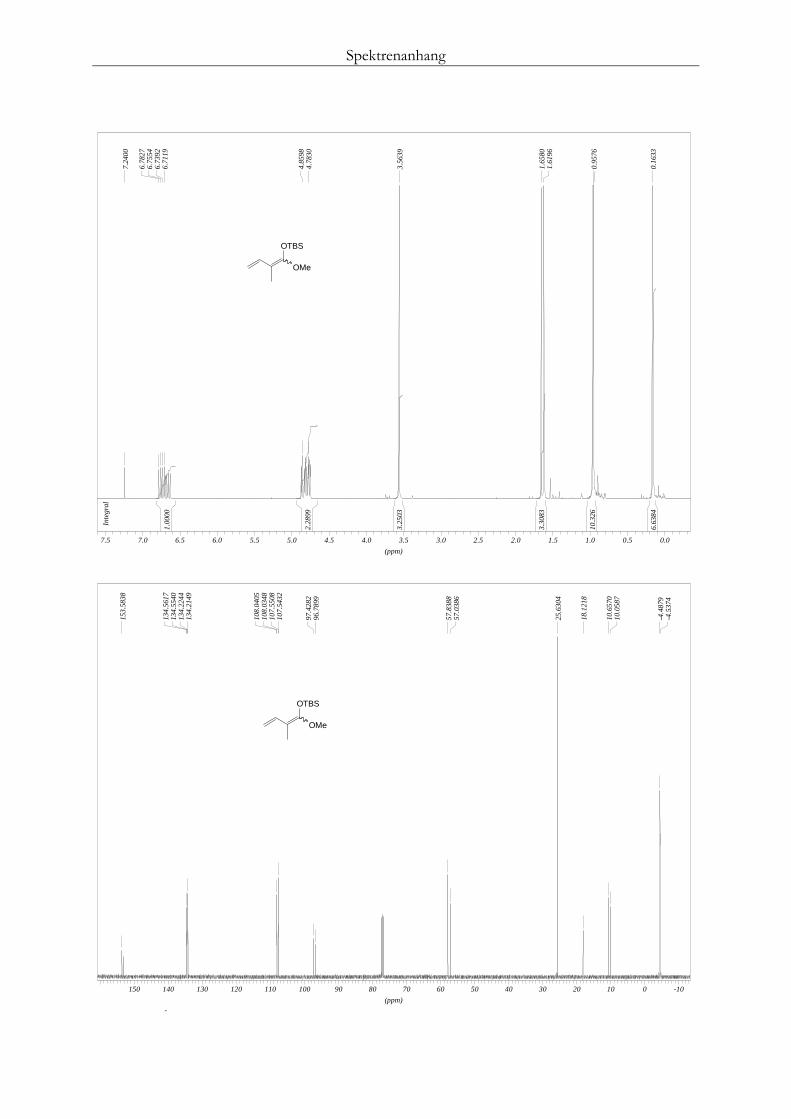
Spektrenanhang
1.00
00
2.28
99
3.25
03
3.30
83
10.3
26
6.63
84
Inte
gral
7.24
00
6.78
276.
7554
6.73
926.
7119
4.85
984.
7830
3.56
39
1.65
801.
6196
0.95
76
0.16
33
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
153.
5838
134.
5617
134.
5540
134.
2244
134.
2149
108.
0405
108.
0348
107.
5508
107.
5432
97.4
282
96.7
899
57.8
388
57.0
386
25.6
304
18.1
218
10.6
570
10.0
587
-4.4
879
-4.5
374
(ppm)
-100102030405060708090100110120130140150
OMe
OTBS
OMe
OTBS

Spektrenanhang
0.92
01
1.00
00
1.07
50
1.06
35
3.13
08
9.90
11
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
153.
5888
134.
5590
134.
2199
108.
0455
107.
5558
97.4
332
96.7
949
57.8
438
57.0
436
25.6
354
10.6
620
10.0
618
-4.4
829
-4.5
344
(ppm)
-100102030405060708090100110120130140150160
H
OTMS
H
OTMS

Spektrenanhang
2.94
15
1.01
00
4.94
11
1.00
00
1.04
06
1.02
74
3.12
74
Inte
gral
7.39
407.
2532
7.04
16
4.04
29
3.17
273.
0055
2.98
08
2.30
00
(ppm)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.5
175.
2326
144.
1560
138.
6288
137.
9906
130.
1733
128.
4529
127.
7022
124.
7834
122.
2037
119.
6906
119.
0676
112.
2392
110.
2634
57.9
490
49.0
000
29.9
799
21.4
710
(ppm)
0102030405060708090100110120130140150160170180
HN
TsHN CO2H
HN
TsHN CO2H

Spektrenanhang
1.00
00
0.97
91
3.25
71
1.08
83
1.12
831.
1486
1.14
69
6.65
16
Inte
gral
(ppm)
1.01.52.02.53.03.54.04.55.05.56.06.57.07.5
(ppm)
0102030405060708090100110120130140150160170
OH
OMe
O
OH
OMe
O

Spektrenanhang
1.00
00
0.96
56
3.13
69
1.08
57
2.18
70
1.06
62
9.27
847.
3094
6.19
94
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
-100102030405060708090100110120130140150160
OTBS
OMe
O
OTBS
OMe
O

Spektrenanhang
1.00
00
0.99
38
3.26
77
1.07
30
1.13
45
1.13
49
9.52
16
Inte
gral
(ppm)
1.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
102030405060708090100110120130140150160170
OH
OMe
O
OH
OMe
O

Spektrenanhang
1.00
00
0.98
46
3.17
77
1.04
79
1.09
46
1.12
12
20.3
50
6.37
67
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
-100102030405060708090100110120130140150160
OTBS
OMe
O
OTBS
OMe
O

Spektrenanhang
1.00
00
0.97
98
0.78
803.
2623
2.25
09
6.89
23
3.28
69
Inte
gral
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
102030405060708090100110120130140150160170
OH
OMe
O
OH
OMe
O

Spektrenanhang
2.11
623.
2454
1.00
00
0.96
87
0.92
30
2.96
18
3.19
39
1.98
41
2.79
61
3.87
67
0.87
882.
6662
Inte
gral
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
(ppm)
102030405060708090100110120130140150160170
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O

Spektrenanhang
2.12
523.
1967
1.00
00
0.97
28
1.06
58
3.09
67
3.19
53
2.11
36
1.99
45
5.16
61
3.43
19
Inte
gral
(ppm)
1.01.52.02.53.03.54.04.55.05.56.06.57.07.5
(ppm)
102030405060708090100110120130140150160
O(R)-MTPA
OMe
O
O(R)-MTPA
OMe
O

Spektrenanhang
1.00
00
0.96
70
3.15
20
1.05
76
1.10
24
1.13
07
3.33
57
2.42
17
1.12
95
5.65
96
Inte
gral
(ppm)
1.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
0102030405060708090100110120130140150160170
OH
OMe
O
OH
OMe
O

Spektrenanhang
2.28
80
3.32
86
1.00
00
0.98
46
1.03
55
3.12
50
3.33
23
2.05
51
5.08
26
5.58
87
Inte
gral
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
(ppm)
102030405060708090100110120130140150160
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O

Spektrenanhang
0.93
95
1.00
00
0.99
55
0.96
07
0.99
59
1.04
95
3.31
96
2.29
42
Inte
gral
(ppm)
2.42.83.23.64.04.44.85.25.66.06.46.87.2
(ppm)
30405060708090100110120130140150160170
OH
OMe
O
O
OH
OMe
O
O

Spektrenanhang
6.61
99
1.00
00
0.98
430.
9401
1.05
79
0.95
91
3.42
74
3.17
27
2.37
48
Inte
gral
(ppm)
2.83.23.64.04.44.85.25.66.06.46.87.27.6
(ppm)
30405060708090100110120130140150160
O(R)-MTPA
OMe
O
O
O(R)-MTPA
OMe
O
O

Spektrenanhang
5.23
31
1.00
00
0.99
29
1.08
83
3.28
82
2.30
05
Inte
gral
(ppm)
2.42.83.23.64.04.44.85.25.66.06.46.87.27.6
(ppm)
2030405060708090100110120130140150160
OH
OMe
O
OH
OMe
O

Spektrenanhang
10.5
51
1.00
00
0.99
89
0.97
40
3.31
91
2.98
47
1.12
33
1.08
10
Inte
gral
(ppm)
2.42.83.23.64.04.44.85.25.66.06.46.87.27.6
(ppm)
2030405060708090100110120130140150160170
O(R)-MTPA
OMe
O
O(R)-MTPA
OMe
O

Spektrenanhang
1.00
00
1.02
39
1.05
08
1.10
633.
1761
1.09
99
1.10
151.
0569
1.08
75
3.09
179.
6463
6.32
17
Inte
gral
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
-100102030405060708090100110120130140150160170
OH
OMe
O
TBSO
OH
OMe
O
TBSO

Spektrenanhang
2.34
673.
5071
1.00
00
0.97
41
1.00
94
2.90
38
3.20
682.
0605
2.06
83
0.99
08
8.68
853.
3688
5.80
29
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
(ppm)
-100102030405060708090100110120130140150160170
O(S)-MTPA
OMe
O
TBSO
O(S)-MTPA
OMe
O
TBSO
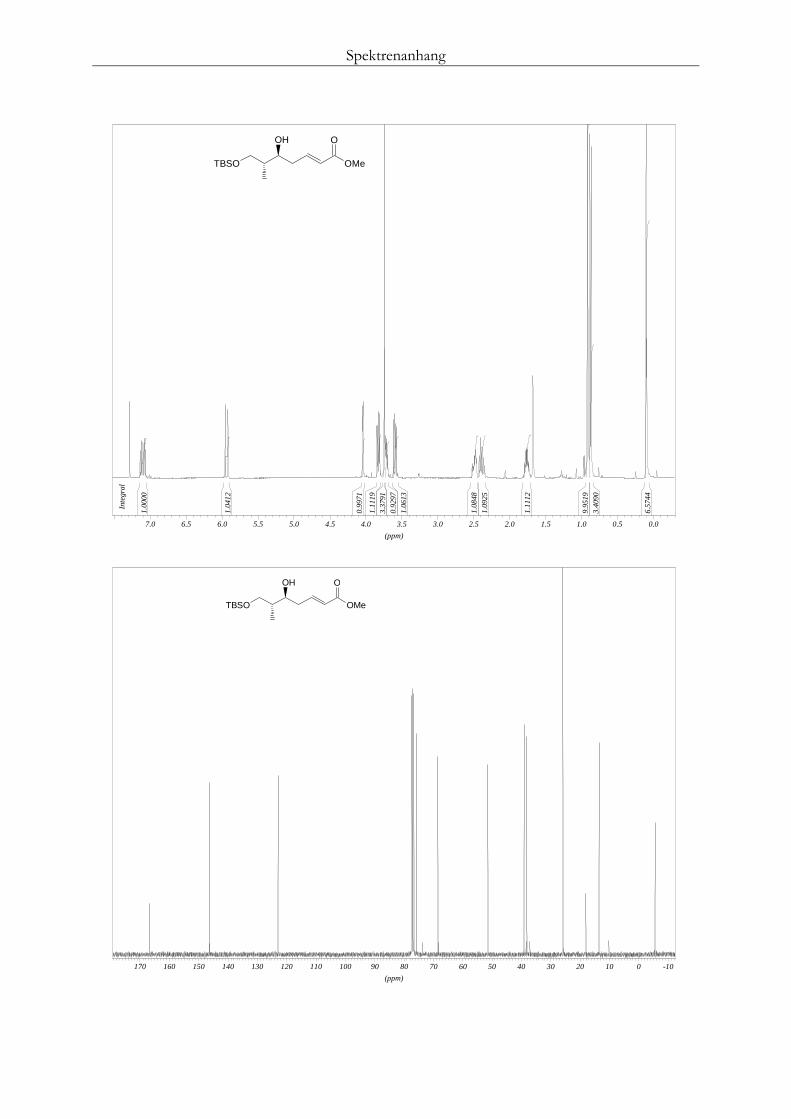
Spektrenanhang
1.00
00
1.04
12
0.99
71
1.11
193.
3791
0.92
971.
0613
1.08
481.
0925
1.11
12
9.95
193.
4090
6.57
44
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
-100102030405060708090100110120130140150160170
OH
OMe
O
TBSO
OH
OMe
O
TBSO

Spektrenanhang
2.15
213.
2815
1.00
00
0.98
43
1.01
95
3.14
92
2.10
612.
7627
1.10
561.
0370
1.20
25
3.00
1910
.041
6.17
32
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
(ppm)
-100102030405060708090100110120130140150160170
O(S)-MTPA
OMe
O
TBSO
O(S)-MTPA
OMe
O
TBSO

Spektrenanhang
1.00
00
0.97
86
0.99
683.
0407
1.03
48
2.08
91
2.91
61
9.45
40
6.11
13
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
(ppm)
0102030405060708090100110120130140150160170
OH
OMe
O
OTBS
OH
OMe
O
OTBS
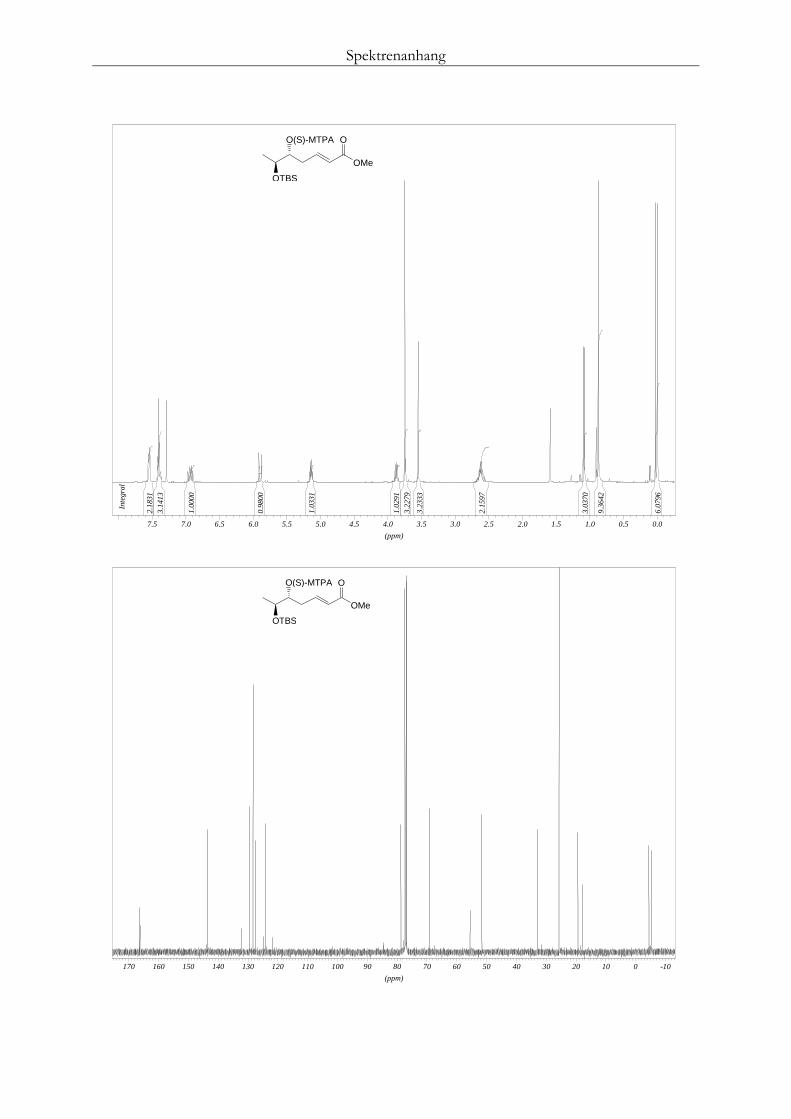
Spektrenanhang
2.18
313.
1413
1.00
00
0.98
00
1.03
31
1.02
913.
2279
3.23
33
2.15
97
3.03
70
9.36
42
6.07
96
Inte
gral
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
(ppm)
-100102030405060708090100110120130140150160170
O(S)-MTPA
OMe
O
OTBS
O(S)-MTPA
OMe
O
OTBS

Spektrenanhang
1.00
00
3.10
92
1.03
20
2.18
25
3.13
35
1.46
93
6.60
39
Inte
gral
7.24
00
6.83
22
3.71
583.
5119
3.49
31
2.32
00
1.84
491.
8431
1.74
761.
7007
1.68
621.
6836
1.63
16
0.93
80
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
168.
4372
138.
9285
129.
4708
75.8
835
51.7
534
33.3
753
18.7
867
17.2
301
12.6
518
(ppm)
0102030405060708090100110120130140150160170
OMe
OOH
OMe
OOH

Spektrenanhang
3.91
57
0.99
43
1.00
00
1.00
35
3.02
31
2.99
23
3.00
02
Inte
gral
7.31
317.
2611
7.25
00
6.79
446.
7910
4.76
404.
7597
3.66
86
2.63
292.
6141
2.59
452.
5800
2.56
38
1.74
57(ppm)
-0.50.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
168.
3876
143.
6917
137.
8692
128.
4420
127.
6875
125.
6451
73.2
581
51.6
944
38.3
842
12.4
937
(ppm)
0102030405060708090100110120130140150160170
OMe
OOH
OMe
OOH

Spektrenanhang
1.00
00
3.03
35
0.99
42
2.05
14
1.24
52
7.06
71
1.54
22
1.09
91
5.95
85
Inte
gral
6.81
86
3.69
193.
4777
3.47
093.
4658
3.45
98
2.31
832.
2987
2.12
551.
8150
1.74
421.
7322
1.72
371.
7152
1.64
52
1.33
901.
3321
1.32
441.
3168
1.21
181.
2025
1.19
310.
9721
0.96
36
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
168.
4372
139.
1819
129.
2555
75.1
957
51.6
734
43.2
712
33.5
487
29.1
399
27.8
138
26.3
506
26.1
277
25.9
696
12.5
794
(ppm)
-100102030405060708090100110120130140150160170
OMe
OOH
OMe
OOH

Spektrenanhang
1.00
00
4.09
98
2.11
50
3.25
40
2.11
68
4.18
42
3.33
58
Inte
gral
7.24
00
6.80
666.
8032
3.70
81
2.32
43
1.82
95
1.46
691.
4584
1.45
411.
2912
0.87
74
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
168.
4676
138.
3912
129.
5737
71.0
651
51.7
477
36.8
600
36.6
638
27.7
986
22.6
125
13.9
988
12.6
404
(ppm)
0102030405060708090100110120130140150160170180
OMe
OOH
OMe
OOH

Spektrenanhang
2.34
93
3.50
60
1.00
00
1.09
86
3.36
86
3.33
01
1.13
89
1.04
99
1.15
66
3.00
91
6.32
97
Inte
gral
7.49
177.
4720
7.36
37
6.60
95
5.05
695.
0381
3.69
443.
4965
3.49
39
2.48
382.
4199
2.01
97
1.73
74
0.94
140.
9371
0.92
430.
9201
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
167.
9628
166.
2880
136.
2097
129.
5127
128.
3220
127.
4189
80.1
494
60.3
652
55.3
334
55.3
182
51.7
591
31.0
585
29.9
782
18.4
000
17.3
845
12.4
594
(ppm)
0102030405060708090100110120130140150160170
OMe
OO(S)-MTPA
OMe
OO(S)-MTPA

Spektrenanhang
10.1
63
1.00
00
1.01
97
3.10
55
3.15
59
1.03
30
1.01
78
3.29
53
7.31
347.
3031
7.28
787.
2400
6.59
25
6.07
296.
0601
6.05
256.
0388
3.69
53
3.41
033.
4078
2.83
112.
8114
2.65
96
1.65
80
(ppm)
1.62.02.42.83.23.64.04.44.85.25.66.06.46.87.2
167.
9113
165.
8727
138.
3798
138.
2674
135.
6191
135.
1733
128.
7202
128.
2934
126.
7635
51.7
992
35.2
710
12.4
346
(ppm)
0102030405060708090100110120130140150160170
OMe
OO(S)-MTPA
OMe
OO(S)-MTPA

Spektrenanhang
2.37
34
3.26
11
1.00
00
1.12
68
3.39
08
3.39
79
2.28
77
5.83
50
3.05
12
7.72
50
Inte
gral
7.47
207.
3646
6.60
10
5.04
495.
0270
3.69
613.
4965
3.49
48
2.47
702.
4582
1.72
80
1.55
91
1.23
571.
2144
1.20
671.
0233
1.00
200.
9934
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
167.
9970
166.
2937
136.
3812
129.
5032
128.
3200
127.
4512
127.
4379
79.7
074
55.3
734
55.3
582
51.7
648
40.8
134
30.0
278
28.8
484
27.8
691
26.1
772
25.9
429
25.8
495
12.4
708
(ppm)
0102030405060708090100110120130140150160170
OMe
OO(S)-MTPA
OMe
OO(S)-MTPA

Spektrenanhang
2.53
52
3.66
05
1.00
00
1.22
97
3.69
29
3.81
95
2.41
71
2.33
00
2.56
06
3.89
93
3.05
95
Inte
gral
7.49
767.
4780
7.37
397.
3637
7.34
497.
2400
6.59
76
5.17
03
3.69
44
3.51
53
2.46
932.
4446
1.72
97
1.29
631.
2886
0.86
55
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
167.
9704
166.
2042
136.
0497
135.
7963
129.
4975
128.
3105
127.
2721
75.9
388
55.3
848
55.3
696
51.7
477
33.3
296
27.3
509
22.3
439
13.8
559
12.4
765
(ppm)
0102030405060708090100110120130140150160170
OMe
OO(S)-MTPA
OMe
OO(S)-MTPA

Spektrenanhang
1.87
71
1.91
60
3.92
87
1.86
46
1.00
00
0.94
51
0.96
97
4.34
17
Inte
gral
7.47
637.
2989
7.27
927.
2451
7.24
007.
1726
7.16
927.
1658
7.15
477.
1470
4.26
174.
2421
4.22
34
3.02
563.
0128
3.00
942.
9966
2.96
242.
9437
2.92
492.
9206
1.72
971.
7246
1.71
521.
7066
1.70
241.
6026
1.58
291.
5642
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
148.
1740
145.
4076
128.
2336
127.
9726
126.
4598
126.
3512
125.
8634
125.
5357
64.4
855
46.7
704
26.2
889
25.5
173
(ppm)
0102030405060708090100110120130140150
NH
Ph
OH
Ph
NH
Ph
OH
Ph

Spektrenanhang
167.
0520
151.
9891
120.
6819
79.1
358
51.4
810
39.9
446
30.9
232
19.7
222
16.4
966
13.9
722
(ppm)
0102030405060708090100110120130140150160170180
OMe
OOH
1.00
00
1.01
22
3.13
98
1.04
28
1.05
00
1.12
54
1.00
58
3.27
33
6.48
35
6.91
506.
8953
5.85
88
3.70
64
3.24
743.
2329
2.49
412.
4787
1.69
13
1.45
92
1.08
221.
0651
0.90
130.
8970
0.88
51
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.0
OMe
OOH
OMe
OOH
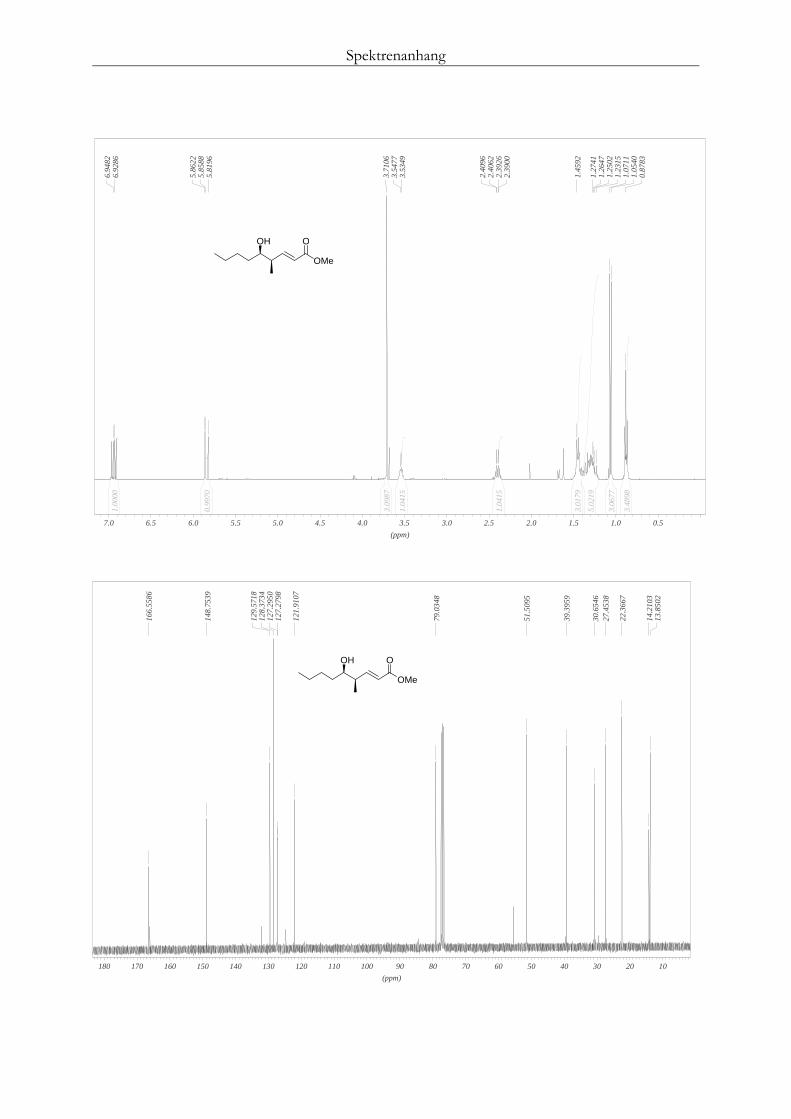
Spektrenanhang
1.00
00
0.99
70
3.09
87
1.04
15
1.04
15
3.01
79
5.02
19
3.06
77
3.48
98
6.94
826.
9286
5.86
225.
8588
5.81
96
3.71
063.
5477
3.53
49
2.40
962.
4062
2.39
262.
3900
1.45
92
1.27
411.
2647
1.25
021.
2315
1.07
111.
0540
0.87
83
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.0
166.
5586
148.
7539
129.
5718
128.
3734
127.
2950
127.
2798
121.
9107
79.0
348
51.5
095
39.3
959
30.6
546
27.4
538
22.3
667
14.2
103
13.8
502
(ppm)
102030405060708090100110120130140150160170180
OMe
OOH
OMe
OOH

Spektrenanhang
1.00
00
1.00
72
3.11
39
1.04
05
1.05
72
1.02
82
3.37
00
9.24
73
6.99
426.
9572
5.81
925.
8136
5.74
025.
7345
3.70
05
3.24
03
2.60
012.
5939
2.57
132.
5650
2.55
87
1.57
811.
5499
1.10
471.
0709
0.91
66
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.0
167.
0520
151.
9891
120.
6819
79.1
358
51.4
810
39.9
446
30.9
232
19.7
222
16.4
966
13.9
722
(ppm)
0102030405060708090100110120130140150160170180
OMe
OOH
OMe
OOH
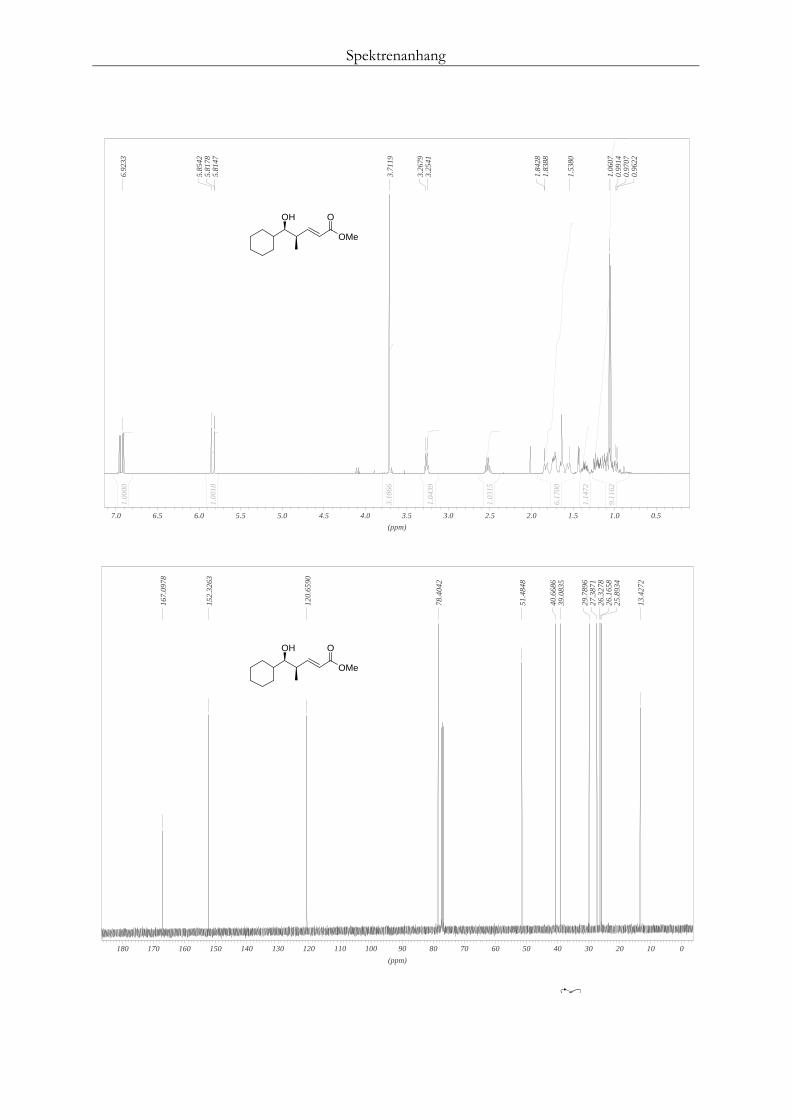
Spektrenanhang
1.00
00
1.00
18
3.18
66
1.04
39
1.03
15
6.17
00
1.14
72
9.11
62
6.92
33
5.85
425.
8178
5.81
47
3.71
19
3.26
793.
2541
1.84
281.
8388
1.53
80
1.06
070.
9914
0.97
070.
9622
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.0
167.
0978
152.
3263
120.
6590
78.4
042
51.4
848
40.6
686
39.0
835
29.7
896
27.3
871
26.3
278
26.1
658
25.8
934
13.4
272
(ppm)
0102030405060708090100110120130140150160170180
OMe
OOH
OMe
OOH

Spektrenanhang
7.34
02
0.34
60
1.00
00
0.33
81
1.00
31
1.05
75
0.35
70
1.07
86
3.11
12
1.38
71
0.33
52
0.98
20
3.19
46
1.14
27
Inte
gral
6.90
81
5.77
265.
7692
5.73
25
4.64
994.
6431
3.66
54
2.70
392.
6860
1.05
061.
0335
(ppm)
1.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
9282
150.
6382
142.
0169
128.
3944
128.
2667
127.
6970
126.
5596
126.
3138
121.
2649
76.8
819
51.4
276
43.6
180
13.9
150
(ppm)
0102030405060708090100110120130140150160170
OMe
OOH
OMe
OOH

Spektrenanhang
1.28
48
0.19
91
1.00
00
1.35
45
1.06
01
0.19
70
1.03
24
1.02
81
0.17
84
3.92
05
1.31
90
1.35
32
3.32
88
Inte
gral
7.35
187.
3492
7.24
00
6.89
116.
8715
6.29
826.
2299
5.85
205.
8127
4.62
69
3.69
27
2.86
522.
8481
2.00
781.
9941
1.11
631.
0992
(ppm)
1.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
8558
154.
5059
149.
7142
142.
1084
121.
6535
110.
1991
107.
2726
70.9
184
51.4
943
41.7
298
14.7
304
(ppm)
102030405060708090100110120130140150160170
OMe
OOH
O
OMe
OOH
O
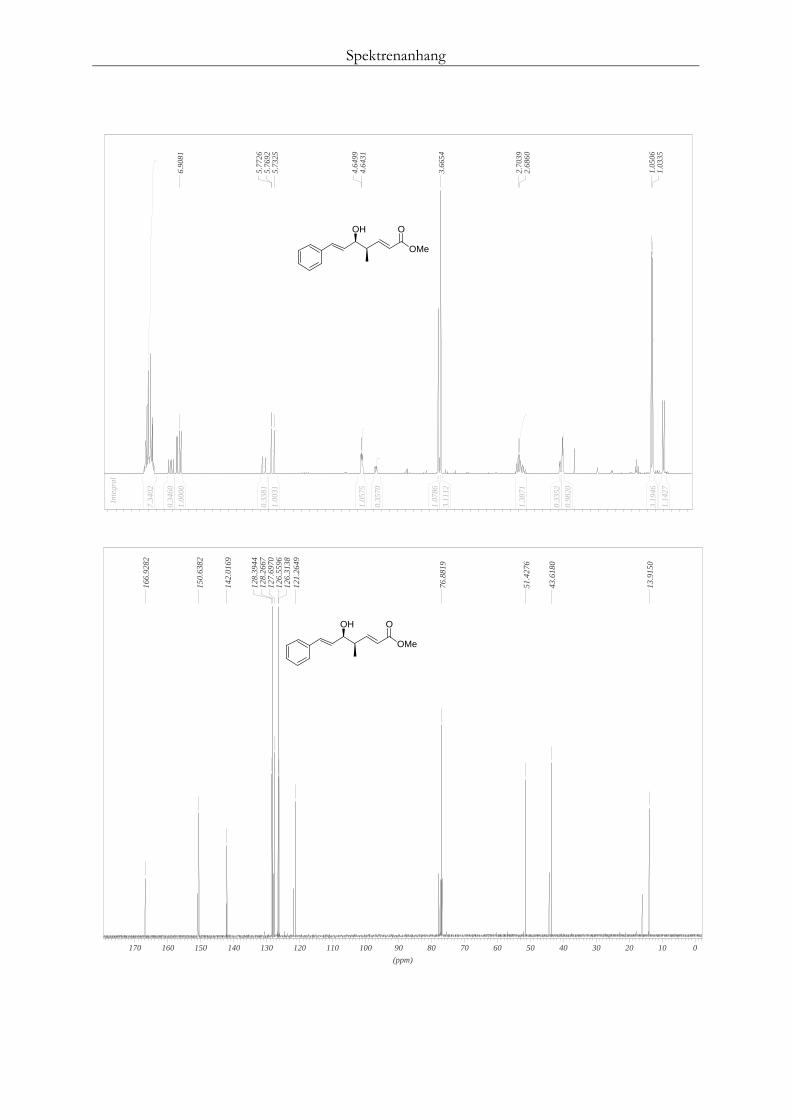
Spektrenanhang
7.34
02
0.34
60
1.00
00
0.33
81
1.00
31
1.05
75
0.35
70
1.07
86
3.11
12
1.38
71
0.33
52
0.98
20
3.19
46
1.14
27
Inte
gral
6.90
81
5.77
265.
7692
5.73
25
4.64
994.
6431
3.66
54
2.70
392.
6860
1.05
061.
0335
(ppm)
1.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
9282
150.
6382
142.
0169
128.
3944
128.
2667
127.
6970
126.
5596
126.
3138
121.
2649
76.8
819
51.4
276
43.6
180
13.9
150
(ppm)
0102030405060708090100110120130140150160170
OMe
OOH
OMe
OOH

Spektrenanhang
1.00
00
1.97
67
1.01
45
1.03
63
1.01
91
1.04
75
0.57
64
3.21
27
6.54
30
Inte
gral
3.79
173.
7857
3.76
693.
7601
2.49
582.
4804
2.03
512.
0146
1.84
831.
8235
1.66
741.
6597
1.06
60
0.93
540.
9175
0.88
170.
8646
(ppm)
0.40.81.21.62.02.42.83.23.64.04.44.85.2
167.
0799
152.
0169
120.
7097
79.1
636
51.5
088
39.9
725
30.9
511
19.7
501
16.5
245
14.0
000
(ppm)
0102030405060708090100110120130140150160170
O
O
O
O

Spektrenanhang
2.08
86
3.09
58
1.00
00
0.97
14
1.02
57
3.08
59
3.13
27
1.03
98
1.07
63
2.94
056.
5009
Inte
gral
7.53
267.
5232
7.38
25
6.81
866.
7981
6.77
936.
7588
5.81
445.
8119
4.98
954.
9767
4.97
244.
9596
3.70
04
3.49
913.
4974
2.67
152.
6545
1.91
311.
9003
1.89
60
0.95
590.
9388
0.89
450.
8766
0.85
44
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
5814
149.
4513
129.
6137
128.
3924
127.
6970
127.
6837
127.
6684
121.
6135
82.9
768
51.5
800
38.5
004
30.0
659
19.4
822
16.5
519
14.6
276
(ppm)
0102030405060708090100110120130140150160170180
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O

Spektrenanhang
2.07
72
3.01
50
1.00
00
0.99
00
1.03
01
3.06
36
3.06
47
1.02
51
1.02
38
3.12
53
6.41
76
Inte
gral
7.52
667.
5173
7.38
67
6.80
836.
7887
5.84
175.
8392
4.98
864.
9860
3.71
403.
7064
3.49
31
2.68
01
1.89
771.
8841
0.99
770.
8749
0.83
13
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
5814
149.
4513
129.
6137
128.
3924
127.
6970
127.
6837
127.
6684
121.
6135
82.9
768
51.5
800
38.5
004
30.0
659
19.4
822
16.5
519
14.6
276
(ppm)
0102030405060708090100110120130140150160170180
O(R)-MTPA
OMe
O
O(R)-MTPA
OMe
O

Spektrenanhang
2.02
722.
9941
0.92
42
0.88
70
1.00
00
3.00
73
3.02
55
0.99
66
2.21
98
4.58
75
2.90
54
3.22
00
Inte
gral
7.51
217.
5028
7.38
33
6.74
696.
7281
5.72
495.
7214
5.08
765.
0705
3.69
443.
5212
3.51
87
2.62
122.
6033
1.59
321.
5753
1.25
531.
2332
1.00
540.
9883
0.84
76
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
166.
5586
148.
7539
129.
5718
128.
3734
121.
9107
79.0
348
55.4
534
55.4
382
51.5
095
39.3
959
30.6
546
27.4
538
22.3
667
14.2
103
13.8
502
(ppm)
102030405060708090100110120130140150160170180
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O
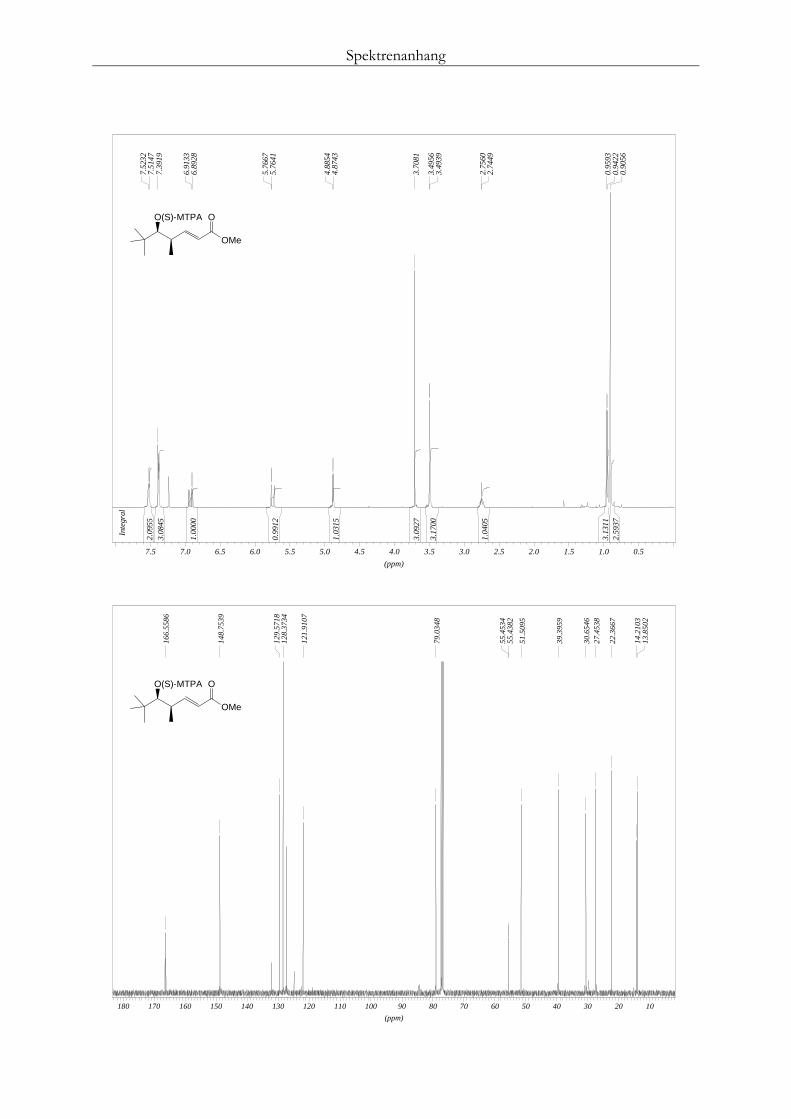
Spektrenanhang
2.09
553.
0845
1.00
00
0.99
12
1.03
15
3.09
27
3.17
00
1.04
05
3.13
112.
5937
Inte
gral
7.52
327.
5147
7.39
19
6.91
336.
8928
5.76
675.
7641
4.88
544.
8743
3.70
81
3.49
563.
4939
2.75
602.
7449
0.95
930.
9422
0.90
56
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
5586
148.
7539
129.
5718
128.
3734
121.
9107
79.0
348
55.4
534
55.4
382
51.5
095
39.3
959
30.6
546
27.4
538
22.3
667
14.2
103
13.8
502
(ppm)
102030405060708090100110120130140150160170180
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O

Spektrenanhang
2.15
91
3.23
45
1.00
00
0.98
40
1.01
06
3.00
75
3.04
20
1.01
75
6.40
51
4.36
65
3.71
50
Inte
gral
7.53
527.
5266
7.38
67
6.78
966.
7699
5.80
165.
7991
4.97
75
3.70
72
3.49
74
2.70
652.
6894
1.60
51
1.23
571.
1700
0.95
850.
9414
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
166.
3280
143.
6421
129.
5508
128.
3258
127.
3636
127.
3484
124.
1132
79.2
158
51.5
534
40.5
105
33.5
411
28.3
969
27.6
081
26.1
010
25.8
438
25.7
657
(ppm)
102030405060708090100110120130140150160170180190
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O

Spektrenanhang
16.0
41
0.40
69
1.00
00
1.09
41
3.26
48
1.57
37
3.20
111.
3298
Inte
gral
7.32
287.
3091
6.66
336.
6437
5.87
765.
8596
5.64
895.
6455
5.60
975.
6063
3.65
43
3.41
63
2.88
402.
8669
1.01
650.
9994
0.88
940.
8723
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
166.
4290
147.
7861
136.
7032
129.
5870
128.
7297
128.
5029
128.
3391
127.
3407
122.
3833
80.5
038
51.5
057
41.6
136
14.9
819
(ppm)
102030405060708090100110120130140150160170
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O
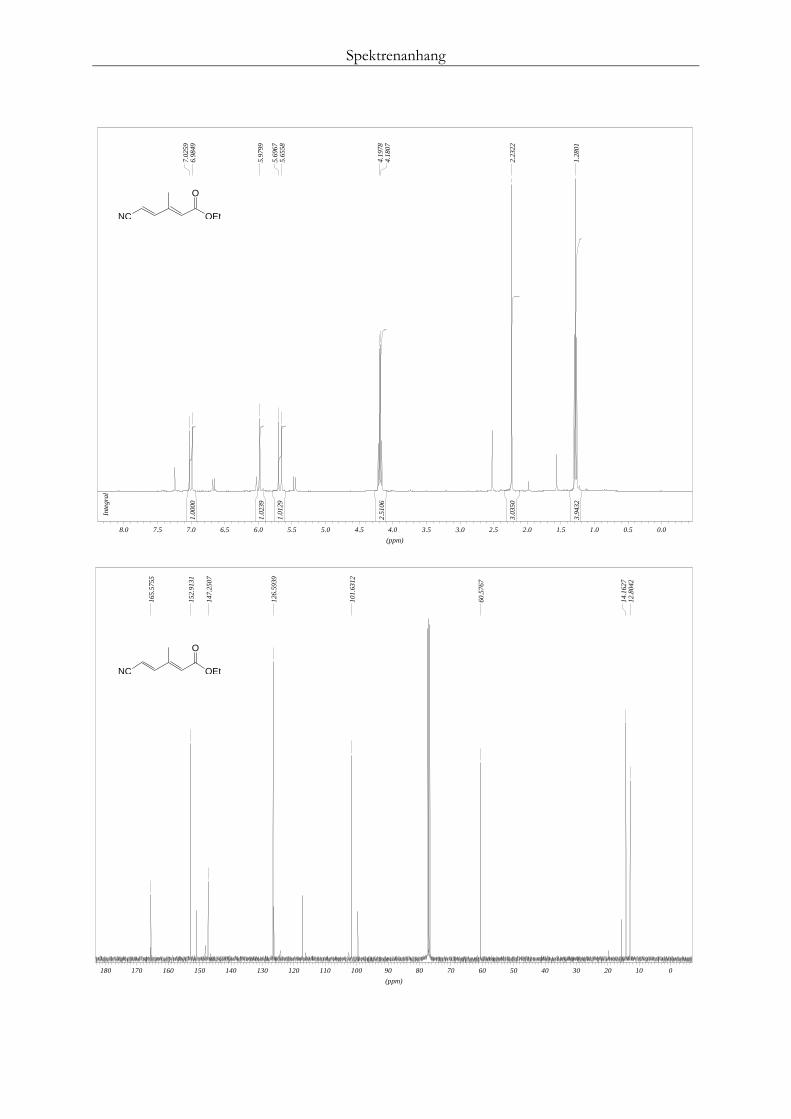
Spektrenanhang
1.00
00
1.02
39
1.01
29
2.51
06
3.03
50
3.94
32
Inte
gral
7.02
596.
9849
5.97
99
5.69
675.
6558
4.19
784.
1807
2.23
22
1.28
01
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0
165.
5755
152.
9131
147.
2507
126.
5939
101.
6312
60.5
767
14.1
627
12.8
042
(ppm)
0102030405060708090100110120130140150160170180
NC OEt
O
NC OEt
O
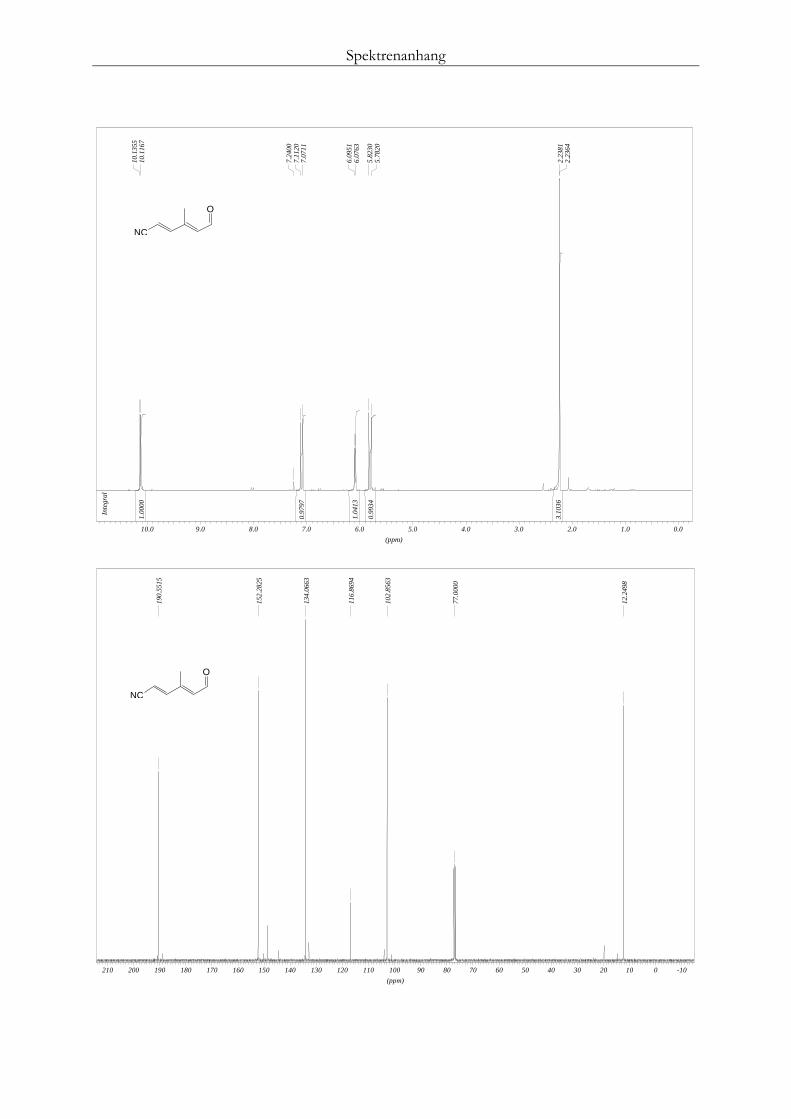
Spektrenanhang
1.00
00
0.97
97
1.04
13
0.99
34
3.10
36
Inte
gral
10.1
355
10.1
167
7.24
007.
1120
7.07
11
6.09
516.
0763
5.82
305.
7820
2.23
812.
2364
(ppm)
0.01.02.03.04.05.06.07.08.09.010.0
190.
5515
152.
2825
134.
0663
116.
8694
102.
8563
77.0
000
12.2
498
(ppm)
-100102030405060708090100110120130140150160170180190200210
NC
O
NC
O

Spektrenanhang
1.12
16
1.02
93
1.00
37
1.17
48
1.13
23
1.00
00
3.36
68
2.22
82
0.94
67
3.09
88
Inte
gral
7.00
036.
9602
6.90
816.
8689
5.91
775.
8784
5.79
915.
7777
5.34
785.
3068
4.62
354.
6073
3.70
12
2.46
932.
4506
2.43
27
2.12
38
1.77
661.
7740
(ppm)
1.62.02.42.83.23.64.04.44.85.25.66.06.46.87.2
166.
5109
153.
8962
143.
5335
141.
7350
133.
6071
124.
1513
118.
0583
96.4
013
67.0
622
51.5
743
39.6
588
12.0
917
(ppm)
0102030405060708090100110120130140150160170180
NC
OH
OMe
O
NC
OH
OMe
O

Spektrenanhang
1.00
00
0.99
50
1.05
84
3.22
02
2.18
10
0.99
91
3.20
97
Inte
gral
7.24
00
6.99
946.
9602
5.94
505.
9057
4.44
35
3.71
40
2.55
21
1.96
771.
9592
1.82
441.
8193
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
6405
143.
7564
124.
0123
82.2
795
79.0
882
77.0
000
61.1
711
51.5
210
40.6
305
3.52
37
(ppm)
0102030405060708090100110120130140150160170
OH
OMe
O
OH
OMe
O
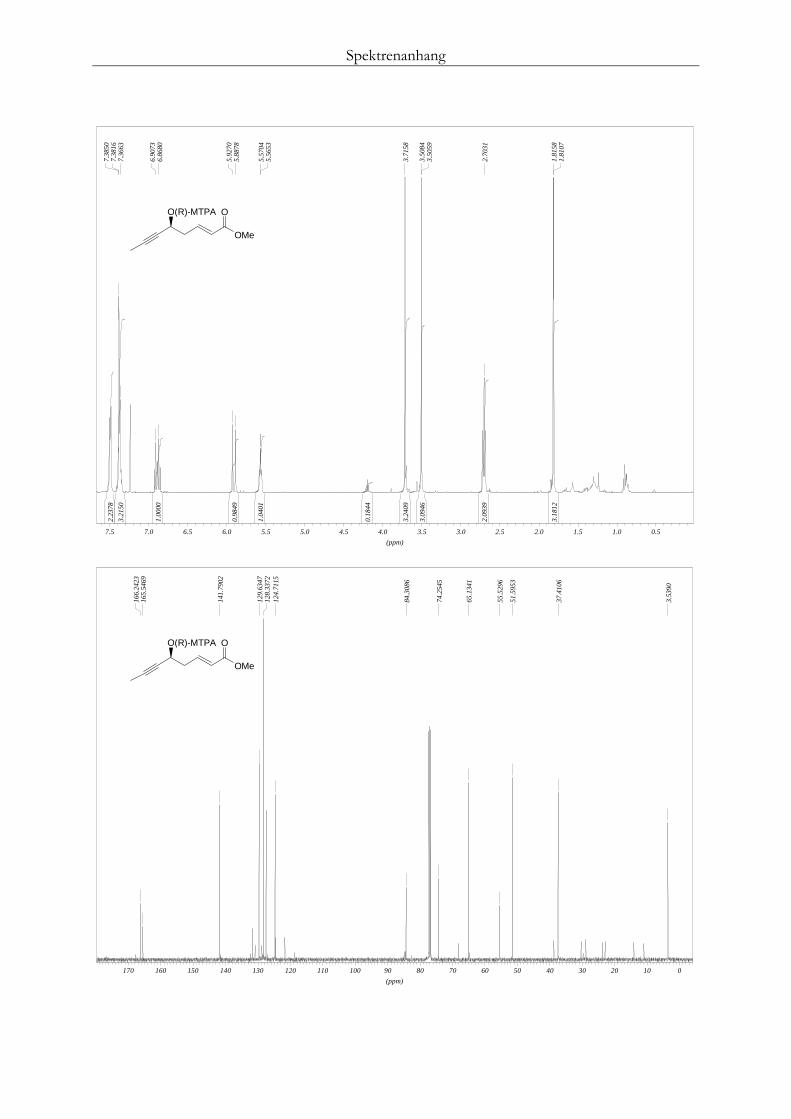
Spektrenanhang
2.23
78
3.21
50
1.00
00
0.98
49
1.04
01
0.18
44
3.24
09
3.09
46
2.09
39
3.18
12
7.38
507.
3816
7.36
63
6.90
736.
8680
5.92
705.
8878
5.57
045.
5653
3.71
58
3.50
843.
5059
2.70
31
1.81
581.
8107
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
2423
165.
5469
141.
7902
129.
6347
128.
3372
124.
7115
84.3
086
74.2
545
65.1
341
55.5
296
51.5
953
37.4
106
3.53
90
(ppm)
0102030405060708090100110120130140150160170
O(R)-MTPA
OMe
O
O(R)-MTPA
OMe
O

Spektrenanhang
2.13
45
3.16
25
1.00
00
1.00
21
1.05
29
3.23
24
3.09
60
2.12
11
3.07
66
Inte
gral
7.51
397.
4942
7.37
657.
3586
7.24
00
6.79
21
5.82
045.
7812
5.61
145.
6063
3.70
12
3.56
22
2.63
40
1.84
661.
8414
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
166.
2004
141.
4797
129.
6004
128.
3696
127.
2550
127.
2436
124.
6867
84.4
419
74.4
222
64.6
958
55.5
163
55.5
030
51.5
343
37.4
335
3.57
51
(ppm)
0102030405060708090100110120130140150160170
O(S)-MTPA
OMe
O
O(S)-MTPA
OMe
O
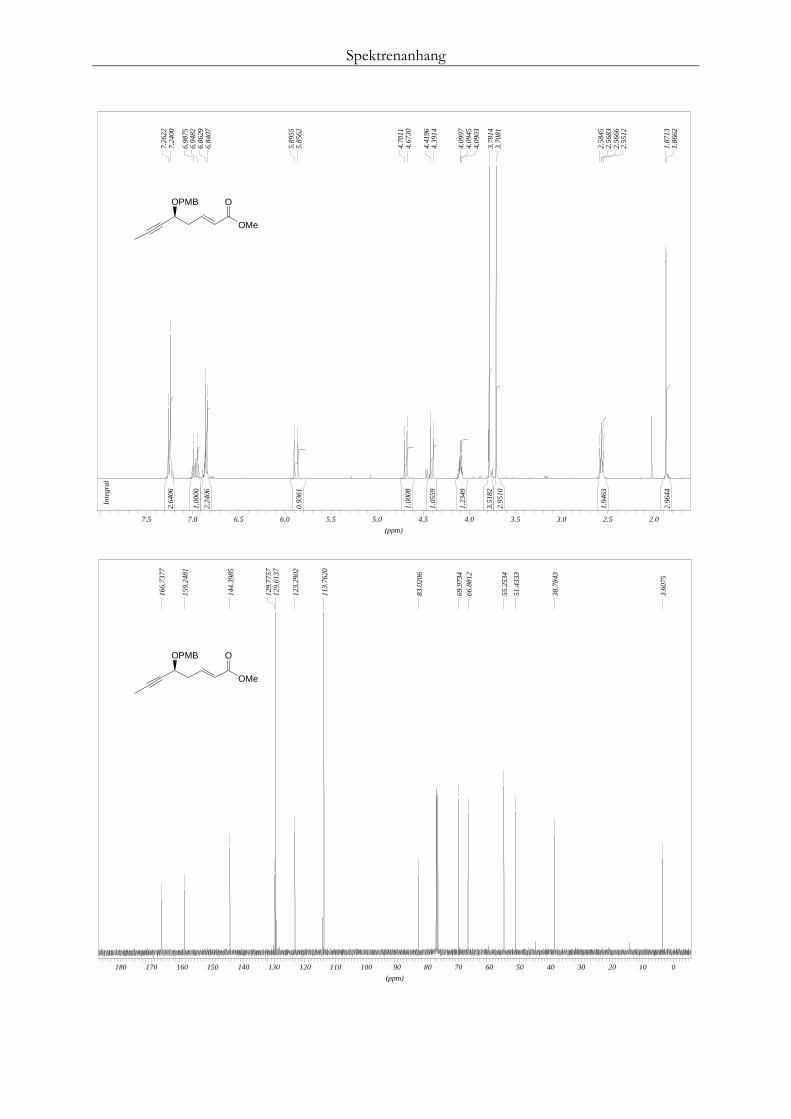
Spektrenanhang
2.64
06
1.00
00
2.24
06
0.93
61
1.00
08
1.05
59
1.23
49
3.51
82
2.95
10
1.94
63
2.96
44
Inte
gral
7.26
227.
2400
6.98
756.
9482
6.86
296.
8407
5.89
555.
8562
4.70
114.
6730
4.41
964.
3914
4.09
974.
0945
4.09
03
3.78
143.
7081
2.58
452.
5683
2.56
662.
5512
1.87
131.
8662
(ppm)
2.02.53.03.54.04.55.05.56.06.57.07.5
166.
7377
159.
2481
144.
3985
129.
7757
129.
6137
123.
2902
113.
7620
83.0
206
69.9
734
66.8
812
55.2
534
51.4
333
38.7
843
3.60
75
(ppm)
0102030405060708090100110120130140150160170180
OPMB
OMe
O
OPMB
OMe
O

Spektrenanhang
2.51
76
2.01
11
1.81
95
1.00
00
1.01
21
2.10
78
0.75
79
3.10
01
1.96
06
3.10
32
Inte
gral
7.26
907.
2477
7.24
00
6.86
126.
8399
5.73
005.
7274
5.72
23
4.69
604.
6670
4.42
304.
3948
4.09
034.
0783
4.02
204.
0169
3.78
06
2.43
612.
4335
1.87
221.
8670
(ppm)
1.62.02.42.83.23.64.04.44.85.25.66.06.46.87.27.6
159.
1909
131.
9267
130.
0538
129.
6004
127.
9657
113.
7162
82.3
995
69.8
572
68.0
224
63.6
213
55.2
591
38.8
015
3.61
90
(ppm)
0102030405060708090100110120130140150160
OPMB OH
OPMB OH

Spektrenanhang
2.00
91
1.82
25
0.95
89
0.97
42
1.00
00
1.11
67
2.95
06
1.00
76
0.98
26
0.97
89
1.87
70
3.04
85
1.34
04
Inte
gral
7.26
227.
2400
6.86
126.
8390
4.71
134.
6823
4.41
454.
3855
4.17
734.
1739
4.16
88
3.86
933.
7772
3.60
57
2.94
202.
9368
2.93
09
1.98
651.
9711
1.95
581.
8815
1.87
64
(ppm)
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
159.
2614
129.
8690
129.
6156
113.
7848
82.9
863
69.9
715
65.8
809
61.6
074
55.2
667
52.8
261
38.1
708
3.62
47
(ppm)
0102030405060708090100110120130140150160170
OPMB OHO
OPMB OHO

Spektrenanhang
2.46
25
1.91
81
1.00
00
0.98
30
0.98
92
1.03
57
5.48
90
0.90
81
2.62
33
1.29
60
2.56
73
Inte
gral
7.26
057.
2400
6.87
156.
8493
4.74
974.
7224
4.42
304.
3948
4.26
094.
2558
4.24
984.
2447
4.23
874.
2336
3.78
40
2.00
611.
9694
1.87
641.
8713
1.81
161.
8056
1.80
051.
7945
1.65
97
(ppm)
1.52.02.53.03.54.04.55.05.56.06.57.07.5
159.
3967
129.
7547
113.
9030
82.8
301
71.2
080
70.2
725
68.3
139
61.3
521
55.2
610
43.0
731
38.4
833
3.57
71
(ppm)
0102030405060708090100110120130140150160
PMBO OHOH
PMBO OHOH

Spektrenanhang
2.40
88
2.27
24
1.00
00
1.07
56
1.10
64
2.20
84
3.99
40
2.34
98
3.66
87
2.21
54
Inte
gral
7.39
017.
3688
7.24
00
6.84
936.
8271
5.45
01
4.56
89
4.02
804.
0186
3.76
18
1.86
451.
8295
1.82
441.
7758
1.74
25
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
159.
9911
130.
4253
113.
5333
100.
9739
82.0
242
77.1
715
75.4
415
67.3
880
59.8
736
55.2
400
37.7
174
3.62
85
(ppm)
0102030405060708090100110120130140150160170180
O OHO
PMP
O OHO
PMP

Spektrenanhang
1.00
00
1.00
22
3.11
43
1.03
02
1.03
70
1.21
15
3.25
22
9.21
34
4.35
45
3.31
16
6.80
34
Inte
gral
7.24
00
6.96
026.
9406
5.78
805.
7846
5.74
875.
7453
3.70
64
3.35
40
2.49
492.
4924
2.47
792.
4753
1.70
411.
6930
1.03
101.
0139
0.88
760.
8766
0.85
860.
8305
0.81
34
0.02
170.
0081
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
167.
2159
153.
2827
119.
6568
80.0
179
51.3
933
40.9
639
31.9
444
26.0
915
20.3
414
18.3
981
17.5
026
15.0
753
-3.7
468
-3.8
535
(ppm)
0102030405060708090100110120130140150160170
OTBS O
OMe
OTBS O
OMe

Spektrenanhang
1.00
00
0.90
39
1.07
10
2.09
62
1.03
10
1.02
16
0.84
54
1.03
94
3.18
97
8.75
87
3.53
26
3.21
92
6.32
76
Inte
gral
7.24
00
6.84
596.
8271
6.80
75
5.79
145.
7402
5.73
685.
7018
4.09
20
2.45
482.
4395
2.20
15
1.69
391.
6836
1.01
220.
9951
0.87
400.
8595
0.84
160.
8134
0.79
64
0.00
890.
0047
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
165.
5316
149.
3674
121.
5888
80.0
389
79.5
492
71.5
338
40.8
210
31.9
140
29.1
647
26.0
991
20.3
814
18.3
942
17.2
339
15.5
764
-3.6
553
-3.8
344
(ppm)
-100102030405060708090100110120130140150160170
OTBS O
NH
OTBS O
NH
Spektrenanhang
1.00
00
0.93
30
1.04
80
0.90
97
0.58
79
1.41
09
1.99
41
1.04
62
1.05
53
1.03
33
3.15
95
15.1
50
6.09
61
Inte
gral
7.24
007.
2387
6.80
276.
7625
5.86
345.
7524
5.74
685.
2768
5.27
565.
2213
5.21
355.
1458
5.13
865.
1358
5.12
795.
0956
5.08
87
3.34
693.
3391
3.31
84
2.47
412.
4691
2.44
062.
4355
1.71
731.
6966
1.68
311.
6624
1.03
070.
9969
0.88
37
0.01
68
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5
165.
5316
149.
3674
121.
5888
80.0
389
79.5
492
71.5
338
40.8
210
31.9
140
29.1
647
26.0
991
20.3
814
18.3
942
17.2
339
15.5
764
-3.6
553
-3.8
344
(ppm)
-100102030405060708090100110120130140150160170
OTBS O
NH
OTBS O
NH

Spektrenanhang
1.
7914
1.90
33
0.79
81
1.00
00
1.91
69
0.87
92
0.92
08
0.93
91
0.99
93
1.05
96
2.18
72
1.66
68
2.66
99
0.97
28
1.10
16
1.22
22
3.70
22
2.43
55
1.79
80
2.95
84
8.53
58
4.13
54
3.58
42
5.26
71
7.24
006.
8568
6.83
946.
8240
6.80
936.
7934
6.17
076.
1393
5.70
685.
7046
5.67
625.
6740
5.56
225.
5051
5.48
945.
4278
4.68
54
4.12
414.
1173
3.99
563.
9648
3.82
713.
8179
3.34
013.
3368
2.46
062.
4468
1.87
991.
7989
1.79
691.
7072
1.69
891.
6854
1.39
971.
3865
1.01
891.
0053
0.88
390.
8673
0.85
350.
8236
0.81
01
0.01
85
(ppm)
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0
167.
7609
165.
6845
159.
9318
148.
7659
136.
0754
135.
1781
132.
4189
131.
0552
130.
8845
128.
7862
127.
3907
125.
3600
122.
1363
113.
6124
100.
7155
80.1
027
73.9
390
68.1
367
55.2
934
41.3
503
40.7
806
38.6
963
37.9
598
36.5
524
31.9
133
30.3
312
29.6
881
28.9
040
26.1
368
23.7
150
22.9
746
20.4
278
18.4
328
17.2
914
15.5
961
14.0
498
13.1
585
10.9
471
-3.6
034
-3.7
919
(ppm)
0102030405060708090100110120130140150160170
OTBS O
NH
OHO O
PMP
OTBS O
NH
OHO O
PMP

Lebenslauf
Persönliche Daten
Name Serkan Simsek
Geburtstag 27.06.1981
Geburtsort Hannover
Familienstand ledig
Schulbildung
08/1987 – 06/2000 Schulausbildung
06/2000 Abitur, Hannover
Studium
11/2000 – 09/2005 Chemiestudium an der Leibniz Universität Hannover
01/2003 Vordiplom im Fach Chemie
09/2005 Diplomprüfung im Fach Chemie
09/2005 – 03/2006 Diplomarbeit an der Leibniz Universität Hannover unter der Leitung von Junior Prof. Dr. M. M. K. Boysen Thema: ”Kohlenhydrate als Auxiliare und Synthesebausteine”
05/2006 – 09/2009 Dissertation an der Leibniz Universität Hannover unter der Leitung von Prof. Dr. M. Kalesse Thema: ”Studien zur asymmetrischen Vinylogen Mukaiyama-Aldolreaktion”
Publikationen
1) ”Oxazaborolidinone promoted vinylogous Aldol Reactions”, Simsek, S.; Horzella, M.; Kalesse, M. Org. Lett. 2007, 9, 5637.
2) “Highly Stereoselective Aldol Reactions in the Total Syntheses of Complex Natural Products”, Brodmann, T.; Lorenz, M.; Schäckel, R.; Simsek, S.; Kalesse, M. Synlett 2009, 174.
3) “ Enantioselective Synthesis of Polyketide Segments through vinylogous Mukaiyama Aldol Reactions”, Simsek, S.; Kalesse, M.; Tetrahedron Lett. 2009, 50, 3485.



















![Olaf Kühl - download.e-bookshelf.de · HSAB hard and soft acids and bases i- ipso I-Effekt isomerer Effekt [Kat] Katalysator, Katalyse L- linkszeigend am untersten asymmetrischen](https://img.pdfslide.org/doc/110x75/5e1a8a229c84f6243d7d64a5/olaf-khl-downloade-hsab-hard-and-soft-acids-and-bases-i-ipso-i-effekt-isomerer.jpg)









