Embed Size (px)
Citation preview

Substrate binding in quinoprotein ethanoldehydrogenase from Pseudomonas aeruginosastudied by electron-nuclear double resonanceChristopher W. M. Kay*, Bina Mennenga†, Helmut Gorisch†, and Robert Bittl*‡
*Institut fur Experimentalphysik, Fachbereich Physik, Freie Universitat, 14195 Berlin, Germany; and †Fachgebiet Technische Biochemie,Institut fur Biotechnologie, Technische Universitat, 13353 Berlin, Germany
Edited by Nicholas J. Turro, Columbia University, New York, NY, and approved February 14, 2006 (received for review November 7, 2005)
Binding of methanol to the quinoprotein ethanol dehydrogenasefrom Pseudomonas aeruginosa has been studied by pulsedelectron-nuclear double resonance at 9 GHz. Shifts in the hyperfinecouplings of the pyrroloquinoline quinone radical provide directevidence for a change in the environment of the cofactor whensubstrate is present. By performing experiments with deuteriatedmethanol, we confirmed that methanol was the cause of the effect.Density functional theory calculations show that these shifts canbe understood if a water molecule, which is often found in x-raystructures of the active site of quinoprotein alcohol dehydroge-nases, is displaced by the substrate. The difference between thebinding of water and methanol is that the water molecule forms ahydrogen bond to O5 of pyrroloquinoline quinone, which themethanol, by virtue of its methyl group, does not. The resultssupport the proposal that aspartate rather than glutamate is thecatalytically active base for a hydride transfer mechanism in qui-noprotein alcohol dehydrogenases.
pyrroloquinoline quinone � alcohol dehydrogenase � density functionaltheory � methanol � electron paramagnetic resonance
Pyrroloquinoline quinone (PQQ, Fig. 1) is one of severalquinone derivatives that function as essential cofactors in a
class of enzymes known as quinoproteins (1–3). X-ray crystal-lographic studies have allowed great progress to be made in theunderstanding of the structure and function of the PQQ-dependent enzymes (for reviews see refs. 4–6). There is specialinterest in the properties of the PQQ cofactor because itspossible role as a vitamin in mammals is currently under debate(7–10).
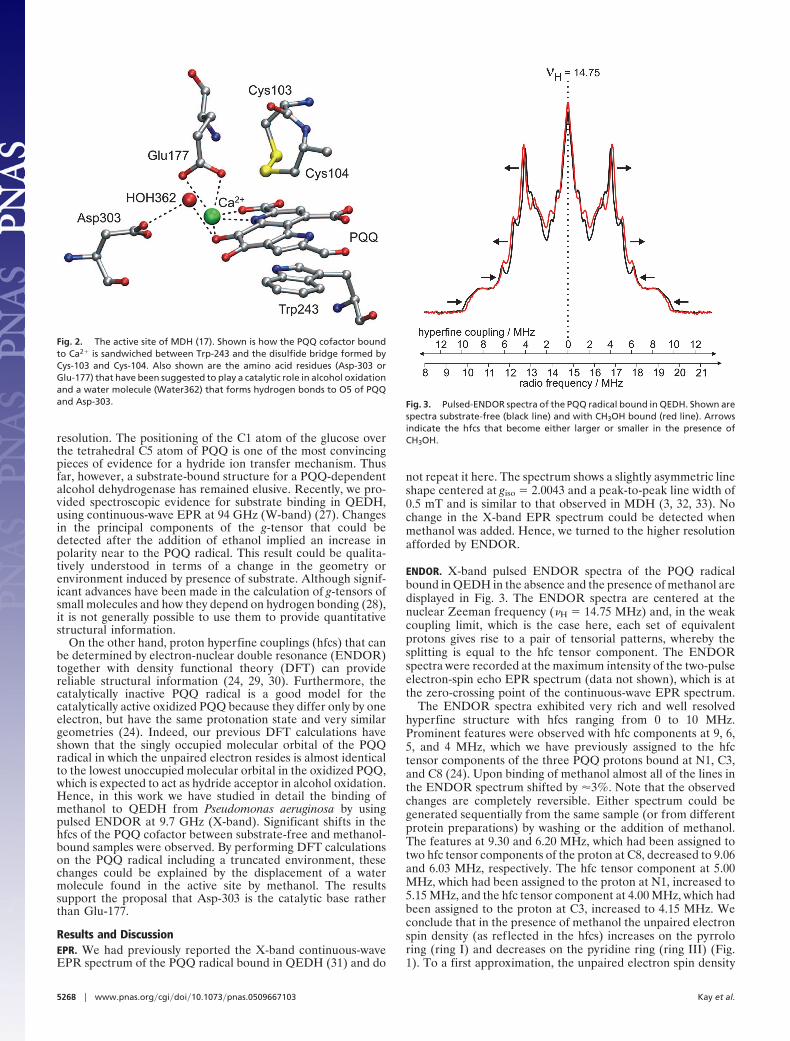
The common structural feature of methanol dehydrogenase(MDH) is an �2�2 tetrameric structure (11–17), in contrast toquinoprotein ethanol dehydrogenase (QEDH), which is a ho-modimeric protein (�2) (18, 19). In both enzymes, the � subunitis a superbarrel composed of eight radially arranged �-sheets.The PQQ cofactor forms a complex with a Ca2� ion buried in theinterior of the superbarrel and is sandwiched between the indolering of a tryptophan and an unusual eight-membered disulfidering structure formed from adjacent cysteines (see Fig. 2).
Two alternatives have been proposed for the reaction mech-anism in quinoprotein dehydrogenases (20). Initially, an addi-tion�elimination mechanism was favored, a suggestion that isnow considered unlikely (21), because a hydride transfer mech-anism is preferred (22) (see Fig. 1). The PQQ is initially oxidized,and after substrate binding, reaction is initiated by amino acidbase-catalyzed proton abstraction of the hydroxyl proton of thealcohol. The characteristics of an Asp-303–Glu mutant protein(numbering scheme of MDH from Methylobacterium extorquens)were considered consistent with Asp-303 acting as the catalyt-ically active base in MDH (16). Recently, however, this widelyaccepted view has been challenged with the results of a molec-ular dynamics study presented by Reddy and Bruice (23), whosuggested that Glu-177 is the catalytic base. In either case,nucleophilic addition of the hydride from the substrate to the C5
position of PQQ is then expected to occur. Subsequently, thePQQ enolizes to form the quinol. Reoxidation proceeds in twosequential one-electron transfer steps to cytochrome via thePQQ radical, which is deprotonated, formally PQQ4�• (24, 25).
Oubrie and coworkers (26) have presented a structure ofPQQ-dependent soluble glucose dehydrogenase in which acomplex of reduced PQQ and glucose was resolved at 1.9-Å
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ENDOR, electron-nuclear double resonance; DFT, density functional theory;QEDH, quinoprotein ethanol dehydrogenase; hfc, hyperfine coupling; MDH, methanoldehydrogenase; PQQ, pyrroloquinoline quinone.
‡To whom correspondence should be addressed. E-mail: [email protected].
© 2006 by The National Academy of Sciences of the USA
Fig. 1. Putative reaction scheme for methanol oxidation by quinoproteinalcohol dehydrogenases. The scheme is based on a hydride transfer mecha-nism with a participation of a nearby amino acid, either Asp or Glu. TheInternational Union of Pure and Applied Chemistry numbering scheme for thecofactor PQQ is also shown. The three carboxylic acid groups of PQQ areconsidered to be deprotonated, hence formally the oxidized state has thestructure PQQ3� (25, 42). The radical form is deprotonated at O4 and O5 andhence has the structure PQQ4�• (24, 25).
www.pnas.org�cgi�doi�10.1073�pnas.0509667103 PNAS � April 4, 2006 � vol. 103 � no. 14 � 5267–5272
CHEM
ISTR
YBI
OPH
YSIC
S
resolution. The positioning of the C1 atom of the glucose overthe tetrahedral C5 atom of PQQ is one of the most convincingpieces of evidence for a hydride ion transfer mechanism. Thusfar, however, a substrate-bound structure for a PQQ-dependentalcohol dehydrogenase has remained elusive. Recently, we pro-vided spectroscopic evidence for substrate binding in QEDH,using continuous-wave EPR at 94 GHz (W-band) (27). Changesin the principal components of the g-tensor that could bedetected after the addition of ethanol implied an increase inpolarity near to the PQQ radical. This result could be qualita-tively understood in terms of a change in the geometry orenvironment induced by presence of substrate. Although signif-icant advances have been made in the calculation of g-tensors ofsmall molecules and how they depend on hydrogen bonding (28),it is not generally possible to use them to provide quantitativestructural information.
On the other hand, proton hyperfine couplings (hfcs) that canbe determined by electron-nuclear double resonance (ENDOR)together with density functional theory (DFT) can providereliable structural information (24, 29, 30). Furthermore, thecatalytically inactive PQQ radical is a good model for thecatalytically active oxidized PQQ because they differ only by oneelectron, but have the same protonation state and very similargeometries (24). Indeed, our previous DFT calculations haveshown that the singly occupied molecular orbital of the PQQradical in which the unpaired electron resides is almost identicalto the lowest unoccupied molecular orbital in the oxidized PQQ,which is expected to act as hydride acceptor in alcohol oxidation.Hence, in this work we have studied in detail the binding ofmethanol to QEDH from Pseudomonas aeruginosa by usingpulsed ENDOR at 9.7 GHz (X-band). Significant shifts in thehfcs of the PQQ cofactor between substrate-free and methanol-bound samples were observed. By performing DFT calculationson the PQQ radical including a truncated environment, thesechanges could be explained by the displacement of a watermolecule found in the active site by methanol. The resultssupport the proposal that Asp-303 is the catalytic base ratherthan Glu-177.
Results and DiscussionEPR. We had previously reported the X-band continuous-waveEPR spectrum of the PQQ radical bound in QEDH (31) and do
not repeat it here. The spectrum shows a slightly asymmetric lineshape centered at giso � 2.0043 and a peak-to-peak line width of0.5 mT and is similar to that observed in MDH (3, 32, 33). Nochange in the X-band EPR spectrum could be detected whenmethanol was added. Hence, we turned to the higher resolutionafforded by ENDOR.
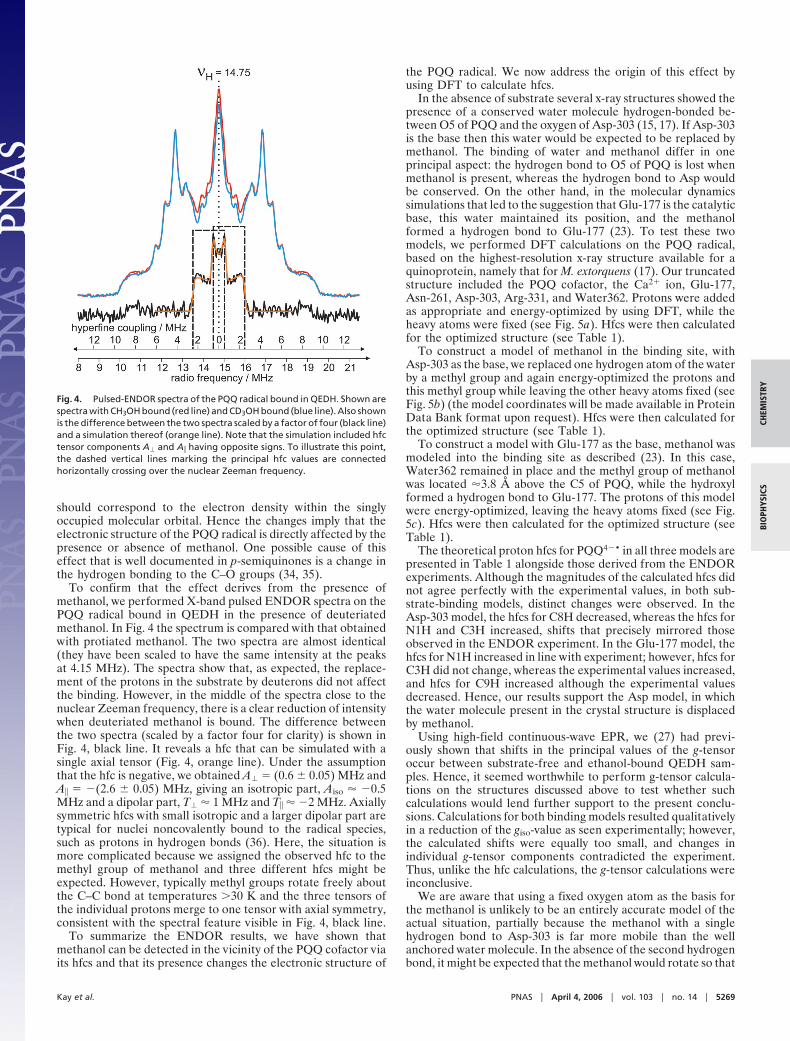
ENDOR. X-band pulsed ENDOR spectra of the PQQ radicalbound in QEDH in the absence and the presence of methanol aredisplayed in Fig. 3. The ENDOR spectra are centered at thenuclear Zeeman frequency (�H � 14.75 MHz) and, in the weakcoupling limit, which is the case here, each set of equivalentprotons gives rise to a pair of tensorial patterns, whereby thesplitting is equal to the hfc tensor component. The ENDORspectra were recorded at the maximum intensity of the two-pulseelectron-spin echo EPR spectrum (data not shown), which is atthe zero-crossing point of the continuous-wave EPR spectrum.
The ENDOR spectra exhibited very rich and well resolvedhyperfine structure with hfcs ranging from 0 to 10 MHz.Prominent features were observed with hfc components at 9, 6,5, and 4 MHz, which we have previously assigned to the hfctensor components of the three PQQ protons bound at N1, C3,and C8 (24). Upon binding of methanol almost all of the lines inthe ENDOR spectrum shifted by �3%. Note that the observedchanges are completely reversible. Either spectrum could begenerated sequentially from the same sample (or from differentprotein preparations) by washing or the addition of methanol.The features at 9.30 and 6.20 MHz, which had been assigned totwo hfc tensor components of the proton at C8, decreased to 9.06and 6.03 MHz, respectively. The hfc tensor component at 5.00MHz, which had been assigned to the proton at N1, increased to5.15 MHz, and the hfc tensor component at 4.00 MHz, which hadbeen assigned to the proton at C3, increased to 4.15 MHz. Weconclude that in the presence of methanol the unpaired electronspin density (as reflected in the hfcs) increases on the pyrroloring (ring I) and decreases on the pyridine ring (ring III) (Fig.1). To a first approximation, the unpaired electron spin density
Fig. 2. The active site of MDH (17). Shown is how the PQQ cofactor boundto Ca2� is sandwiched between Trp-243 and the disulfide bridge formed byCys-103 and Cys-104. Also shown are the amino acid residues (Asp-303 orGlu-177) that have been suggested to play a catalytic role in alcohol oxidationand a water molecule (Water362) that forms hydrogen bonds to O5 of PQQand Asp-303. Fig. 3. Pulsed-ENDOR spectra of the PQQ radical bound in QEDH. Shown are
spectra substrate-free (black line) and with CH3OH bound (red line). Arrowsindicate the hfcs that become either larger or smaller in the presence ofCH3OH.
5268 � www.pnas.org�cgi�doi�10.1073�pnas.0509667103 Kay et al.
should correspond to the electron density within the singlyoccupied molecular orbital. Hence the changes imply that theelectronic structure of the PQQ radical is directly affected by thepresence or absence of methanol. One possible cause of thiseffect that is well documented in p-semiquinones is a change inthe hydrogen bonding to the C–O groups (34, 35).
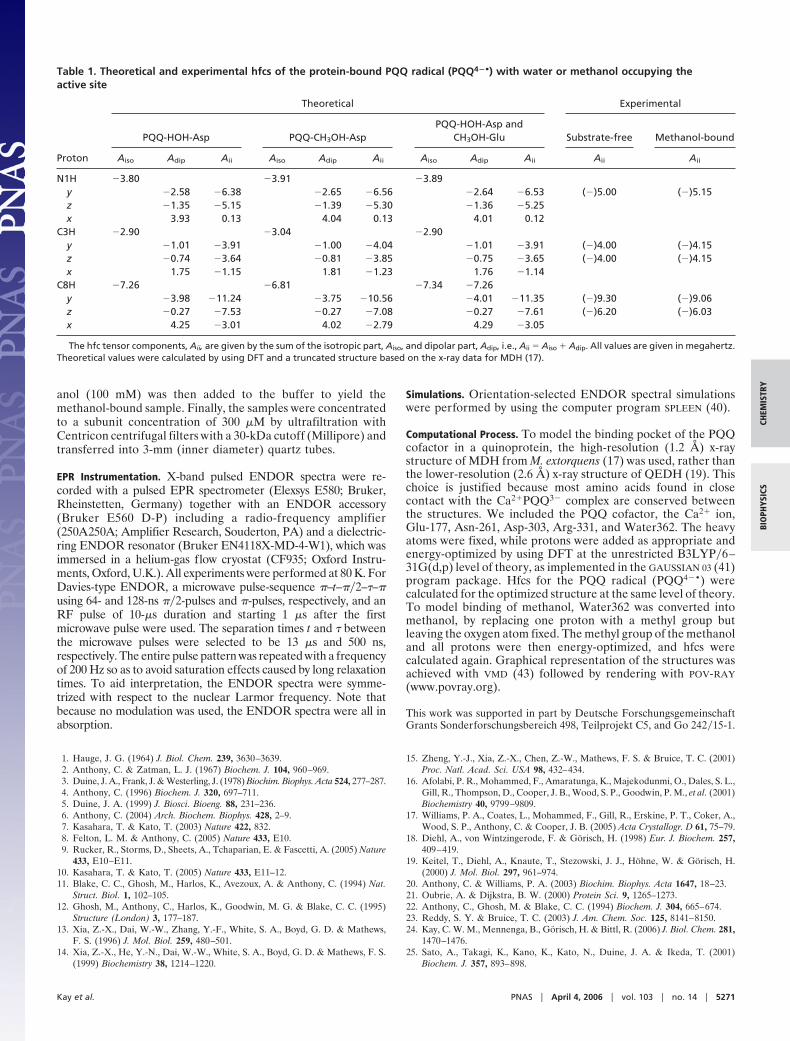
To confirm that the effect derives from the presence ofmethanol, we performed X-band pulsed ENDOR spectra on thePQQ radical bound in QEDH in the presence of deuteriatedmethanol. In Fig. 4 the spectrum is compared with that obtainedwith protiated methanol. The two spectra are almost identical(they have been scaled to have the same intensity at the peaksat 4.15 MHz). The spectra show that, as expected, the replace-ment of the protons in the substrate by deuterons did not affectthe binding. However, in the middle of the spectra close to thenuclear Zeeman frequency, there is a clear reduction of intensitywhen deuteriated methanol is bound. The difference betweenthe two spectra (scaled by a factor four for clarity) is shown inFig. 4, black line. It reveals a hfc that can be simulated with asingle axial tensor (Fig. 4, orange line). Under the assumptionthat the hfc is negative, we obtained A� � (0.6 � 0.05) MHz andA� � �(2.6 � 0.05) MHz, giving an isotropic part, Aiso � �0.5MHz and a dipolar part, T� � 1 MHz and T� � �2 MHz. Axiallysymmetric hfcs with small isotropic and a larger dipolar part aretypical for nuclei noncovalently bound to the radical species,such as protons in hydrogen bonds (36). Here, the situation ismore complicated because we assigned the observed hfc to themethyl group of methanol and three different hfcs might beexpected. However, typically methyl groups rotate freely aboutthe C–C bond at temperatures �30 K and the three tensors ofthe individual protons merge to one tensor with axial symmetry,consistent with the spectral feature visible in Fig. 4, black line.
To summarize the ENDOR results, we have shown thatmethanol can be detected in the vicinity of the PQQ cofactor viaits hfcs and that its presence changes the electronic structure of
the PQQ radical. We now address the origin of this effect byusing DFT to calculate hfcs.
In the absence of substrate several x-ray structures showed thepresence of a conserved water molecule hydrogen-bonded be-tween O5 of PQQ and the oxygen of Asp-303 (15, 17). If Asp-303is the base then this water would be expected to be replaced bymethanol. The binding of water and methanol differ in oneprincipal aspect: the hydrogen bond to O5 of PQQ is lost whenmethanol is present, whereas the hydrogen bond to Asp wouldbe conserved. On the other hand, in the molecular dynamicssimulations that led to the suggestion that Glu-177 is the catalyticbase, this water maintained its position, and the methanolformed a hydrogen bond to Glu-177 (23). To test these twomodels, we performed DFT calculations on the PQQ radical,based on the highest-resolution x-ray structure available for aquinoprotein, namely that for M. extorquens (17). Our truncatedstructure included the PQQ cofactor, the Ca2� ion, Glu-177,Asn-261, Asp-303, Arg-331, and Water362. Protons were addedas appropriate and energy-optimized by using DFT, while theheavy atoms were fixed (see Fig. 5a). Hfcs were then calculatedfor the optimized structure (see Table 1).
To construct a model of methanol in the binding site, withAsp-303 as the base, we replaced one hydrogen atom of the waterby a methyl group and again energy-optimized the protons andthis methyl group while leaving the other heavy atoms fixed (seeFig. 5b) (the model coordinates will be made available in ProteinData Bank format upon request). Hfcs were then calculated forthe optimized structure (see Table 1).
To construct a model with Glu-177 as the base, methanol wasmodeled into the binding site as described (23). In this case,Water362 remained in place and the methyl group of methanolwas located �3.8 Å above the C5 of PQQ, while the hydroxylformed a hydrogen bond to Glu-177. The protons of this modelwere energy-optimized, leaving the heavy atoms fixed (see Fig.5c). Hfcs were then calculated for the optimized structure (seeTable 1).
The theoretical proton hfcs for PQQ4�• in all three models arepresented in Table 1 alongside those derived from the ENDORexperiments. Although the magnitudes of the calculated hfcs didnot agree perfectly with the experimental values, in both sub-strate-binding models, distinct changes were observed. In theAsp-303 model, the hfcs for C8H decreased, whereas the hfcs forN1H and C3H increased, shifts that precisely mirrored thoseobserved in the ENDOR experiment. In the Glu-177 model, thehfcs for N1H increased in line with experiment; however, hfcs forC3H did not change, whereas the experimental values increased,and hfcs for C9H increased although the experimental valuesdecreased. Hence, our results support the Asp model, in whichthe water molecule present in the crystal structure is displacedby methanol.
Using high-field continuous-wave EPR, we (27) had previ-ously shown that shifts in the principal values of the g-tensoroccur between substrate-free and ethanol-bound QEDH sam-ples. Hence, it seemed worthwhile to perform g-tensor calcula-tions on the structures discussed above to test whether suchcalculations would lend further support to the present conclu-sions. Calculations for both binding models resulted qualitativelyin a reduction of the giso-value as seen experimentally; however,the calculated shifts were equally too small, and changes inindividual g-tensor components contradicted the experiment.Thus, unlike the hfc calculations, the g-tensor calculations wereinconclusive.
We are aware that using a fixed oxygen atom as the basis forthe methanol is unlikely to be an entirely accurate model of theactual situation, partially because the methanol with a singlehydrogen bond to Asp-303 is far more mobile than the wellanchored water molecule. In the absence of the second hydrogenbond, it might be expected that the methanol would rotate so that
Fig. 4. Pulsed-ENDOR spectra of the PQQ radical bound in QEDH. Shown arespectra with CH3OH bound (red line) and CD3OH bound (blue line). Also shownis the difference between the two spectra scaled by a factor of four (black line)and a simulation thereof (orange line). Note that the simulation included hfctensor components A� and A� having opposite signs. To illustrate this point,the dashed vertical lines marking the principal hfc values are connectedhorizontally crossing over the nuclear Zeeman frequency.
Kay et al. PNAS � April 4, 2006 � vol. 103 � no. 14 � 5269
CHEM
ISTR
YBI
OPH
YSIC
S

its oxygen could form a coordinate bond with the Ca2�. Such aninteraction would not only increase the polarization of themethanol, thus assisting in the abstraction of the hydroxy protonby Asp, but would also bring the methyl group closer to C5, readyfor hydride transfer.
Direct evidence for the distance between the C5 of PQQ and
the methyl group can, in principle, be obtained from ourENDOR data. The dipolar part of the hfc directly depends onthe distance between a nucleus and the unpaired electron spin,according to:
veN��� ��0
4�hgegN�B�N
r3 �3 cos2� � 1� ,
where ge and gN are the electron and nuclear -factors, �0 is thevacuum permeability, �B and �N are the Bohr and nuclearmagnetons, respectively, h is Planck’s constant, � is the anglebetween the principal axes and r the distance between them, and is the electron spin density that causes the interaction. For arather localized electron, for example, a paramagnetic ion or anitroxide spin-label, can be set to unity. In the PQQ radical,however, the unpaired electron is delocalized over the �-system.The good agreement between the experimental and theoreticalproton hfcs gives us confidence that the predicted electronicdistribution is largely correct and we can use the Muliken spindensities for the heavy atoms given by the DFT calculations. ForC5, � 0.45. Hence, almost 50% of the electron spin density islocalized at this position. The situation here is slightly morecomplicated because we assigned the hfc to the three protons ofa freely rotating methyl group. Nevertheless, if applied withcaution, this approach is still instructive. For example, taking � �1 MHz from our experiment and setting � 0.45, we obtain anaverage r � 3.3 Å, and a closest approach of 2.6 Å, which is closeto the distances obtained from molecular modeling studies ofhydride transfer to C5 of PQQ (37).
To conclude, we used pulsed ENDOR to examine the bindingof methanol to QEDH. Although with a Km value of 94 mMmethanol is not the primary substrate (Km � 0.014 mM forethanol) (38), we still observed significant changes in the hfcs ofthe PQQ radical. We confirmed that the changes arise frommethanol by modifications to the spectra when using deuteriatedmethanol. This approach allowed us to give an estimate of thedistance between the methyl group of methanol and the PQQcofactor. Using DFT, it could be shown that the changes areconsistent with the role of Asp as the catalytically active base, butcontradict the proposal that Glu is the base. The latter proposalwas solely based on in silico molecular dynamics (MD) simula-tions (23). That our experiments yielded a different conclusionindicates that although MD simulations are in many cases veryuseful they ought to be used with caution when no experimentaldata are available. We have shown that ENDOR and DFT area powerful combination that can be used to obtain high-qualitystructural information, and in this case we have provided directevidence for the location of a primary alcohol bound in the activesite of a quinoprotein. The results support the proposal that ahydride transfer mechanism, which is accepted for solubleglucose dehydrogenase, is also active in quinoprotein alcoholdehydrogenases.
Materials and MethodsEnzyme Isolation. The apo-form of QEDH enzyme was heterolo-gously overexpressed in Escherichia coli and purified to homo-geneity with Tris�HCl buffer, in which the enzyme was active andappeared to be completely stable, as described for holo-QEDH(31, 38).
Reconstitution of the Holo-Form of QEDH. The formation of anenzymatically active holo-enzyme was achieved as described(39). In a previous study, we (27) had observed that afterreconstitution or isolation of the native enzyme substrate wasoften bound. Therefore, after reconstitution the preparationswere washed by repeated dilution�concentration cycles in freshlyprepared alcohol-free buffer (100 mM Tris�HCl, pH 8�100 mMNaCl�10 mM CaCl2) to obtain the substrate-free sample. Meth-
Fig. 5. Models of the active site of quinoprotein alcohol dehydrogenasesshowing the PQQ cofactor, the Ca2� ion, Glu-177, Asp-303, Asn-261, Arg-331,and Water362. Heavy atoms were taken from the x-ray structure of MDH (17),and protons were added and energy-optimized by using DFT methods. (a)Substrate-free. Water362 forms hydrogen bonds with Asp-303 and O5 of PQQ.(b) Asp model. Methanol was modeled into the active site by replacing one ofthe protons of Water362 by a methyl group. Methanol maintains a hydrogenbond with Asp-303, but there is no direct interaction with PQQ. (c) Glu model.Methanol was modeled into the active site according to ref. 23 and hydrogen-bonded to Glu-177, while Water362 remained in the position given in the x-raystructure. Hfcs for the PQQ radical were calculated for the three structures andare presented in Table 1.
5270 � www.pnas.org�cgi�doi�10.1073�pnas.0509667103 Kay et al.
anol (100 mM) was then added to the buffer to yield themethanol-bound sample. Finally, the samples were concentratedto a subunit concentration of 300 �M by ultrafiltration withCentricon centrifugal filters with a 30-kDa cutoff (Millipore) andtransferred into 3-mm (inner diameter) quartz tubes.
EPR Instrumentation. X-band pulsed ENDOR spectra were re-corded with a pulsed EPR spectrometer (Elexsys E580; Bruker,Rheinstetten, Germany) together with an ENDOR accessory(Bruker E560 D-P) including a radio-frequency amplifier(250A250A; Amplifier Research, Souderton, PA) and a dielectric-ring ENDOR resonator (Bruker EN4118X-MD-4-W1), which wasimmersed in a helium-gas flow cryostat (CF935; Oxford Instru-ments, Oxford, U.K.). All experiments were performed at 80 K. ForDavies-type ENDOR, a microwave pulse-sequence �–t–��2–�–�using 64- and 128-ns ��2-pulses and �-pulses, respectively, and anRF pulse of 10-�s duration and starting 1 �s after the firstmicrowave pulse were used. The separation times t and � betweenthe microwave pulses were selected to be 13 �s and 500 ns,respectively. The entire pulse pattern was repeated with a frequencyof 200 Hz so as to avoid saturation effects caused by long relaxationtimes. To aid interpretation, the ENDOR spectra were symme-trized with respect to the nuclear Larmor frequency. Note thatbecause no modulation was used, the ENDOR spectra were all inabsorption.
Simulations. Orientation-selected ENDOR spectral simulationswere performed by using the computer program SPLEEN (40).
Computational Process. To model the binding pocket of the PQQcofactor in a quinoprotein, the high-resolution (1.2 Å) x-raystructure of MDH from M. extorquens (17) was used, rather thanthe lower-resolution (2.6 Å) x-ray structure of QEDH (19). Thischoice is justified because most amino acids found in closecontact with the Ca2�PQQ3� complex are conserved betweenthe structures. We included the PQQ cofactor, the Ca2� ion,Glu-177, Asn-261, Asp-303, Arg-331, and Water362. The heavyatoms were fixed, while protons were added as appropriate andenergy-optimized by using DFT at the unrestricted B3LYP�6–31G(d,p) level of theory, as implemented in the GAUSSIAN 03 (41)program package. Hfcs for the PQQ radical (PQQ4�•) werecalculated for the optimized structure at the same level of theory.To model binding of methanol, Water362 was converted intomethanol, by replacing one proton with a methyl group butleaving the oxygen atom fixed. The methyl group of the methanoland all protons were then energy-optimized, and hfcs werecalculated again. Graphical representation of the structures wasachieved with VMD (43) followed by rendering with POV-RAY(www.povray.org).
This work was supported in part by Deutsche ForschungsgemeinschaftGrants Sonderforschungsbereich 498, Teilprojekt C5, and Go 242�15-1.
1. Hauge, J. G. (1964) J. Biol. Chem. 239, 3630–3639.2. Anthony, C. & Zatman, L. J. (1967) Biochem. J. 104, 960–969.3. Duine, J. A., Frank, J. & Westerling, J. (1978) Biochim. Biophys. Acta 524, 277–287.4. Anthony, C. (1996) Biochem. J. 320, 697–711.5. Duine, J. A. (1999) J. Biosci. Bioeng. 88, 231–236.6. Anthony, C. (2004) Arch. Biochem. Biophys. 428, 2–9.7. Kasahara, T. & Kato, T. (2003) Nature 422, 832.8. Felton, L. M. & Anthony, C. (2005) Nature 433, E10.9. Rucker, R., Storms, D., Sheets, A., Tchaparian, E. & Fascetti, A. (2005) Nature
433, E10–E11.10. Kasahara, T. & Kato, T. (2005) Nature 433, E11–12.11. Blake, C. C., Ghosh, M., Harlos, K., Avezoux, A. & Anthony, C. (1994) Nat.
Struct. Biol. 1, 102–105.12. Ghosh, M., Anthony, C., Harlos, K., Goodwin, M. G. & Blake, C. C. (1995)
Structure (London) 3, 177–187.13. Xia, Z.-X., Dai, W.-W., Zhang, Y.-F., White, S. A., Boyd, G. D. & Mathews,
F. S. (1996) J. Mol. Biol. 259, 480–501.14. Xia, Z.-X., He, Y.-N., Dai, W.-W., White, S. A., Boyd, G. D. & Mathews, F. S.
(1999) Biochemistry 38, 1214–1220.
15. Zheng, Y.-J., Xia, Z.-X., Chen, Z.-W., Mathews, F. S. & Bruice, T. C. (2001)Proc. Natl. Acad. Sci. USA 98, 432–434.
16. Afolabi, P. R., Mohammed, F., Amaratunga, K., Majekodunmi, O., Dales, S. L.,Gill, R., Thompson, D., Cooper, J. B., Wood, S. P., Goodwin, P. M., et al. (2001)Biochemistry 40, 9799–9809.
17. Williams, P. A., Coates, L., Mohammed, F., Gill, R., Erskine, P. T., Coker, A.,Wood, S. P., Anthony, C. & Cooper, J. B. (2005) Acta Crystallogr. D 61, 75–79.
18. Diehl, A., von Wintzingerode, F. & Gorisch, H. (1998) Eur. J. Biochem. 257,409–419.
19. Keitel, T., Diehl, A., Knaute, T., Stezowski, J. J., Hohne, W. & Gorisch, H.(2000) J. Mol. Biol. 297, 961–974.
20. Anthony, C. & Williams, P. A. (2003) Biochim. Biophys. Acta 1647, 18–23.21. Oubrie, A. & Dijkstra, B. W. (2000) Protein Sci. 9, 1265–1273.22. Anthony, C., Ghosh, M. & Blake, C. C. (1994) Biochem. J. 304, 665–674.23. Reddy, S. Y. & Bruice, T. C. (2003) J. Am. Chem. Soc. 125, 8141–8150.24. Kay, C. W. M., Mennenga, B., Gorisch, H. & Bittl, R. (2006) J. Biol. Chem. 281,
1470–1476.25. Sato, A., Takagi, K., Kano, K., Kato, N., Duine, J. A. & Ikeda, T. (2001)
Biochem. J. 357, 893–898.
Table 1. Theoretical and experimental hfcs of the protein-bound PQQ radical (PQQ4�•) with water or methanol occupying theactive site
Proton
Theoretical Experimental
PQQ-HOH-Asp PQQ-CH3OH-AspPQQ-HOH-Asp and
CH3OH-Glu Substrate-free Methanol-bound
Aiso Adip Aii Aiso Adip Aii Aiso Adip Aii Aii Aii
N1H �3.80 �3.91 �3.89y �2.58 �6.38 �2.65 �6.56 �2.64 �6.53 (�)5.00 (�)5.15z �1.35 �5.15 �1.39 �5.30 �1.36 �5.25x 3.93 0.13 4.04 0.13 4.01 0.12
C3H �2.90 �3.04 �2.90y �1.01 �3.91 �1.00 �4.04 �1.01 �3.91 (�)4.00 (�)4.15z �0.74 �3.64 �0.81 �3.85 �0.75 �3.65 (�)4.00 (�)4.15x 1.75 �1.15 1.81 �1.23 1.76 �1.14
C8H �7.26 �6.81 �7.34 �7.26y �3.98 �11.24 �3.75 �10.56 �4.01 �11.35 (�)9.30 (�)9.06z �0.27 �7.53 �0.27 �7.08 �0.27 �7.61 (�)6.20 (�)6.03x 4.25 �3.01 4.02 �2.79 4.29 �3.05
The hfc tensor components, Aii, are given by the sum of the isotropic part, Aiso, and dipolar part, Adip, i.e., Aii � Aiso � Adip. All values are given in megahertz.Theoretical values were calculated by using DFT and a truncated structure based on the x-ray data for MDH (17).
Kay et al. PNAS � April 4, 2006 � vol. 103 � no. 14 � 5271
CHEM
ISTR
YBI
OPH
YSIC
S

26. Oubrie, A., Rozeboom, H. J., Kalk, K. H., Olsthoorn, A. J., Duine, J. A. &Dijkstra, B. W. (1999) EMBO J. 18, 5187–5194.
27. Kay, C. W. M., Mennenga, B., Gorisch, H. & Bittl, R. (2005) J. Am. Chem. Soc.127, 7974–7975.
28. Kaupp, M., Remenyi, C., Vaara, J., Malkina, O. L. & Malkin, V. G. (2002)J. Am. Chem. Soc. 124, 2709–2722.
29. Medina, M., Vrielink, A. & Cammack, R. (1994) Eur. J. Biochem. 222, 941–947.30. Weber, S., Richter, G., Schleicher, E., Bacher, A., Mobius, K. & Kay, C. W. M.
(2001) Biophys. J. 81, 1195–1204.31. Kay, C. W. M., Mennenga, B., Gorisch, H. & Bittl, R. (2004) FEBS Lett. 564,
69–72.32. de Beer, R. D., van Ormondt, D., van Ast, M. A., Banen, R., Duine, J. A. &
Frank, J. (1979) J. Chem. Phys. 70, 4491–4495.33. Westerling, J., Frank, J. & Duine, J. A. (1979) Biochem. Biophys. Res. Commun.
87, 719–724.34. Sinnecker, S., Reijerse, E., Neese, F. & Lubitz, W. (2004) J. Am. Chem. Soc.
126, 3280–3290.
35. Flores, M., Isaacson, R. A., Calvo, R., Feher, G. & Lubitz, W. (2003) Chem.Phys. 294, 401–413.
36. MacMillan, F., Lendzian, F., Renger, G. & Lubitz, W. (1995) Biochemistry 34,8144–8156.
37. Jongejan, A., Jongejan, J. A. & Hagen, W. R. (2001) J. Comput. Chem. 22,1732–1749.
38. Rupp, M. & Gorisch, H. (1988) Biol. Chem. Hoppe- Seyler 369, 431–439.39. Mutzel, A. & Gorisch, H. (1991) Agri. Biol. Chem. 55, 1721–1726.40. Gessner, C., Stein, M., Albracht, S. P. & Lubitz, W. (1999) J. Biol. Inorg. Chem.
4, 379–389.41. Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A.,
Cheeseman, J. R., Montgomery, J. J. A., Vreven, T., Kudin, K. N., Burant, J. C.,et al. (2003) GAUSSIAN (Gaussian, Pittsburgh PA).
42. Kano, K., Mori, K., Uno, B., Kubota, T., Ikeda, T. & Senda, M. (1990)Bioelectrochem. Bioenerg. 24, 193–201.
43. Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14,33–38.
5272 � www.pnas.org�cgi�doi�10.1073�pnas.0509667103 Kay et al.





























