Embed Size (px)
Citation preview

Indian Journal of Chemistry
Vol. 53B, December 2014, pp 1584-1595
Synthesis and antitumor activities of novel diacerein
α-aminophosphonates conjugates
Qin Jian-Mei
a, Li Jian-Fei
a, Ye Man-Yi
a, Huang Ri-Zheng
a, Xu Qing
b, Pan Ying-Ming
a,
Wang Heng-Shan*a & Yao Gui-Yang
a
aState Key Laboratory Cultivation Base for Chemistry and Molecular Engineering of Medicinal Resources,
School of Chemistry & Pharmaceutical Science of Guangxi Normal University, Guilin 541004, P. R. China
bCollege of Pharmacy, Guilin Medical University, Guilin 541004, P. R. China
E-mail: [email protected]
Received 24 December 2013; accepted (revised) 7 October 2014
Several diacerein α-aminophosphonates conjugates 4a-k have been synthesized, the structures of compounds have been
characterized by IR, 1H NMR, 13C NMR, 31P NMR, ESI-MS spectra and elementary analysis. In vitro cytotoxicity against
HepG-2, CNE, Spca-2 and Hct-116 cells are evaluated and employing standard MTT assay in comparing with commercial
anticancer drug 5-fluorouracil (5-FU). Some compounds exhibit moderate to high levels of antitumor activity. Especially,
compound 4i exhibit the strongest cytotoxicity against Hct-116 cells with IC50 9.83 µM. All the synthesized compounds
exhibit low cytotoxicity against HUVEC cells. The mechanism of compound 4i has been preliminarily investigated by
Hoechst 33258 staining, JC-1 mitochondrial membrane potential staining and flow cytometry, which indicate that the
compound 4i induced apoptosis in Hct-116 cancer cells. Cell cycle analysis show that compound 4i mainly arrested Hct-116
cells in G1 stage. The effects of 4i on the activation of caspases expression indicate that 4i might induce apoptosis via the
membrane death receptor pathways. In addition, the binding properties of a model analog 4i to DNA have been investigated
by methods (UV-Vis, fluorescence, CD spectroscopy) in comparison with that of diacerein. Results indicate that 4i show
moderate ability to interact ct-DNA.
Keywords: Diacerein, α-aminophosphonic acids, cytotoxicity, cell cycle, DNA binding
Finding new tricks for old drugs is an efficient route
for public sector drug discovery1. Many studies have
discovered many new applications of existing drugs,
such as aspirin2, metformin
3.
Diacerein, clinically used as in anti-inflammatory,
analgesic and antipyretic agent, developed
specifically for the treatment of goarthrosis and
coxarthrosis4-6
. Moreover, diacerein has also been
reported to display another biological activity,
prevention of vascular diseases and treatment of
insulin resistance7. However, the antitumor activity of
diacerein has rarely been studied. Recently, in our
laboratory, we found that diacerein had anti-
proliferative activity against some tumor cells, but
with weak cytotoxicity against normal cells,
motivating us to seek higher activity of diacerein
derivatives. In our precious work, we found that the
introduction of aminophosphonate group to pharmacy
core is able to increase the antitumor activity and
many aminophosphonates derivatives have exhibited
potent inhibition activity against human tumors8-15
.
Based on the concept of hybrid molecules16,17
with a
dual mode of action, a series of diacerein α-amino-
phosphonates conjugates were designed in our study
to find new antitumor agent.
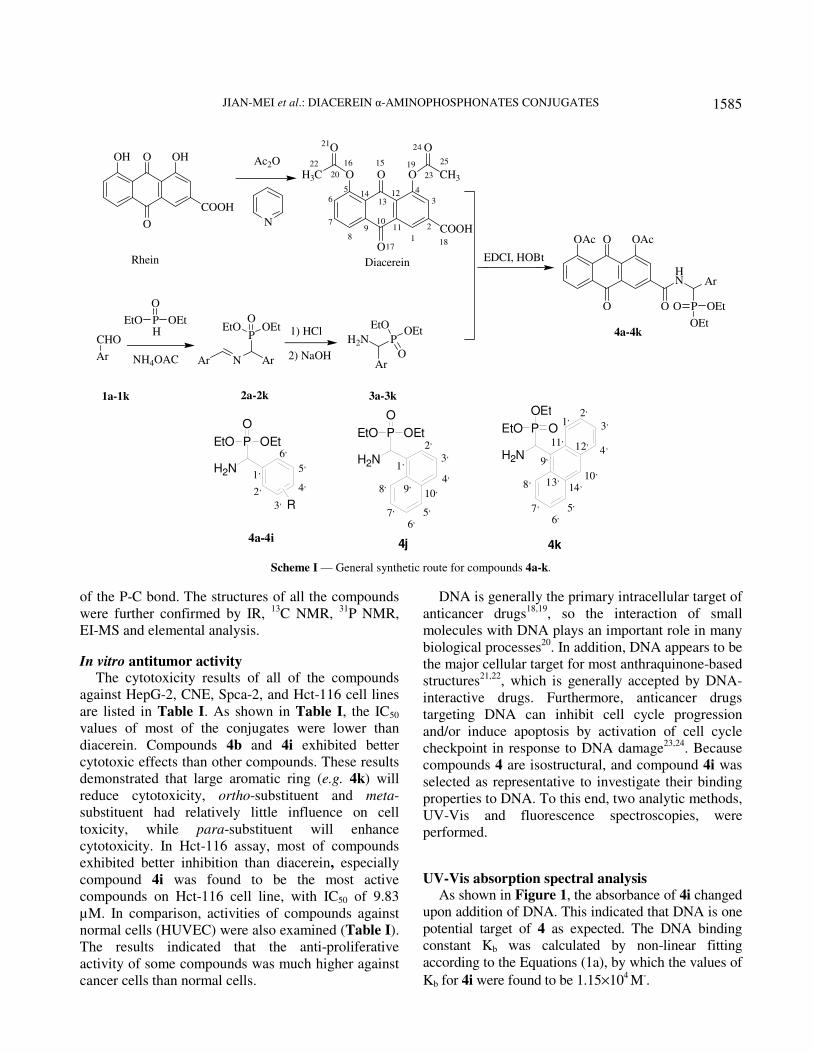
Herein, we report (a) the synthesis of diacerein
α-aminophosphonates conjugates; (b) DNA binding of
potent conjugate 4i; (c) in vitro antitumor activity and
cell selectivity of synthesized conjugates; and (d) the
mechanism of how the novel conjugate 4i killed
Hct-116 cells Scheme I.
Results and Discussion
Chemistry All compounds were obtained as yellowish solids
after column chromatography. Their structures were
fully characterized by 1H NMR. For example, the
corresponding 1H NMR spectrum showed the two
methyleneoxy (OCH2CH3) groups attached with
phosphorus appears as three multiplets at δ 3.28-4.35.
The chemical shifts of the three methyl (OCH2CH3)
hydrogens were different due to the low rate of
environmental exchange caused by the slow rotation
JIAN-MEI et al.: DIACEREIN α-AMINOPHOSPHONATES CONJUGATES
1585
of the P-C bond. The structures of all the compounds
were further confirmed by IR, 13
C NMR, 31
P NMR,
EI-MS and elemental analysis.
In vitro antitumor activity The cytotoxicity results of all of the compounds
against HepG-2, CNE, Spca-2, and Hct-116 cell lines
are listed in Table I. As shown in Table I, the IC50
values of most of the conjugates were lower than
diacerein. Compounds 4b and 4i exhibited better
cytotoxic effects than other compounds. These results
demonstrated that large aromatic ring (e.g. 4k) will
reduce cytotoxicity, ortho-substituent and meta-
substituent had relatively little influence on cell
toxicity, while para-substituent will enhance
cytotoxicity. In Hct-116 assay, most of compounds
exhibited better inhibition than diacerein, especially
compound 4i was found to be the most active
compounds on Hct-116 cell line, with IC50 of 9.83
µM. In comparison, activities of compounds against
normal cells (HUVEC) were also examined (Table I).
The results indicated that the anti-proliferative
activity of some compounds was much higher against
cancer cells than normal cells.
DNA is generally the primary intracellular target of
anticancer drugs18,19
, so the interaction of small
molecules with DNA plays an important role in many
biological processes20
. In addition, DNA appears to be
the major cellular target for most anthraquinone-based
structures21,22
, which is generally accepted by DNA-
interactive drugs. Furthermore, anticancer drugs
targeting DNA can inhibit cell cycle progression
and/or induce apoptosis by activation of cell cycle
checkpoint in response to DNA damage23,24
. Because
compounds 4 are isostructural, and compound 4i was
selected as representative to investigate their binding
properties to DNA. To this end, two analytic methods,
UV-Vis and fluorescence spectroscopies, were
performed.
UV-Vis absorption spectral analysis
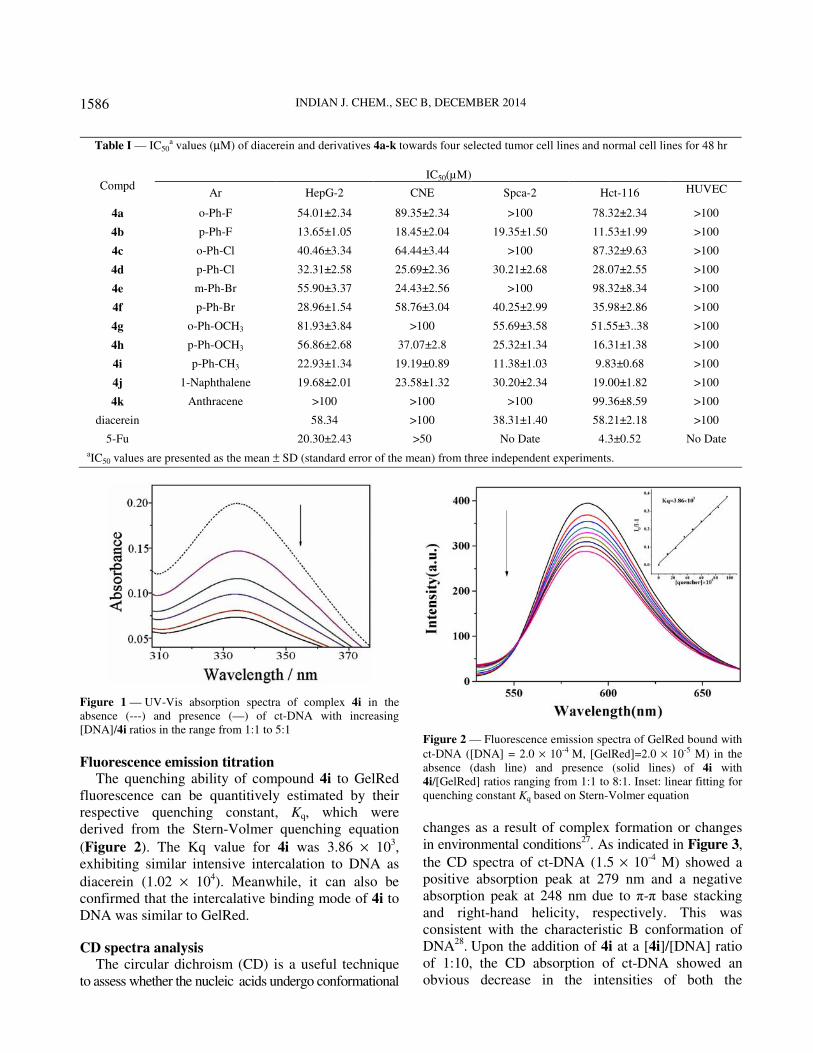
As shown in Figure 1, the absorbance of 4i changed
upon addition of DNA. This indicated that DNA is one
potential target of 4 as expected. The DNA binding
constant Kb was calculated by non-linear fitting
according to the Equations (1a), by which the values of
Kb for 4i were found to be 1.15×104 M
-.
OH OH
COOH
O
O
Rhein Diacerein
Ac2O
N
Ar
CHO
PH
NH4OAC Ar N Ar
PEtO OEt
O1) HCl
2) NaOH
EDCI, HOBt
OAc OAcO
O O
HN Ar
PO OEt
OEt
1a-1k 2a-2k 3a-3k
4a-4k
Ar
H2N P
O
OEtEtO
O O
COOH
O
O
H3C
O
CH3
O
1
2
3
45
6
7
89
1011
1213
14
15
1718
20
22
23
24
2516 19
21
O
OEtEtO
P
H2N
O
OEtEtO
1,
2,
3,
4,
5,
6,
R
P
H2N
O
OEtEtO
1,
4,
5,
6,
2,
3,
7,
8, 9,10,
4a-4i4j
P
H2N
OEtO1,
5,
6,7,
8,
9,
10,
4k
OEt 2,
3,
4,11,12,
13,
14,
Scheme I — General synthetic route for compounds 4a-k.
INDIAN J. CHEM., SEC B, DECEMBER 2014
1586
Figure 1 — UV-Vis absorption spectra of complex 4i in the
absence (---) and presence (—) of ct-DNA with increasing
[DNA]/4i ratios in the range from 1:1 to 5:1
Fluorescence emission titration The quenching ability of compound 4i to GelRed
fluorescence can be quantitively estimated by their
respective quenching constant, Kq, which were
derived from the Stern-Volmer quenching equation
(Figure 2). The Kq value for 4i was 3.86 × 103,
exhibiting similar intensive intercalation to DNA as
diacerein (1.02 × 104). Meanwhile, it can also be
confirmed that the intercalative binding mode of 4i to
DNA was similar to GelRed.
CD spectra analysis The circular dichroism (CD) is a useful technique
to assess whether the nucleic acids undergo conformational
Figure 2 — Fluorescence emission spectra of GelRed bound with
ct-DNA ([DNA] = 2.0 × 10-4 M, [GelRed]=2.0 × 10-5 M) in the
absence (dash line) and presence (solid lines) of 4i with
4i/[GelRed] ratios ranging from 1:1 to 8:1. Inset: linear fitting for
quenching constant Kq based on Stern-Volmer equation
changes as a result of complex formation or changes
in environmental conditions27
. As indicated in Figure 3,
the CD spectra of ct-DNA (1.5 × 10-4
M) showed a
positive absorption peak at 279 nm and a negative
absorption peak at 248 nm due to π-π base stacking
and right-hand helicity, respectively. This was
consistent with the characteristic B conformation of
DNA28
. Upon the addition of 4i at a [4i]/[DNA] ratio
of 1:10, the CD absorption of ct-DNA showed an
obvious decrease in the intensities of both the
Table I — IC50a values (µM) of diacerein and derivatives 4a-k towards four selected tumor cell lines and normal cell lines for 48 hr
IC50(µM)
Compd Ar HepG-2 CNE Spca-2 Hct-116 HUVEC
4a o-Ph-F 54.01±2.34 89.35±2.34 >100 78.32±2.34 >100
4b p-Ph-F 13.65±1.05 18.45±2.04 19.35±1.50 11.53±1.99 >100
4c o-Ph-Cl 40.46±3.34 64.44±3.44 >100 87.32±9.63 >100
4d p-Ph-Cl 32.31±2.58 25.69±2.36 30.21±2.68 28.07±2.55 >100
4e m-Ph-Br 55.90±3.37 24.43±2.56 >100 98.32±8.34 >100
4f p-Ph-Br 28.96±1.54 58.76±3.04 40.25±2.99 35.98±2.86 >100
4g o-Ph-OCH3 81.93±3.84 >100 55.69±3.58 51.55±3..38 >100
4h p-Ph-OCH3 56.86±2.68 37.07±2.8 25.32±1.34 16.31±1.38 >100
4i p-Ph-CH3 22.93±1.34 19.19±0.89 11.38±1.03 9.83±0.68 >100
4j 1-Naphthalene 19.68±2.01 23.58±1.32 30.20±2.34 19.00±1.82 >100
4k Anthracene >100 >100 >100 99.36±8.59 >100
diacerein 58.34 >100 38.31±1.40 58.21±2.18 >100
5-Fu 20.30±2.43 >50 No Date 4.3±0.52 No Date
aIC50 values are presented as the mean ± SD (standard error of the mean) from three independent experiments.
JIAN-MEI et al.: DIACEREIN α-AMINOPHOSPHONATES CONJUGATES
1587
negative and positive absorption bands. The decrease
in the intensities of both positive and negative bands
can usually be observed in the intercalative binding of
small molecules to DNA.
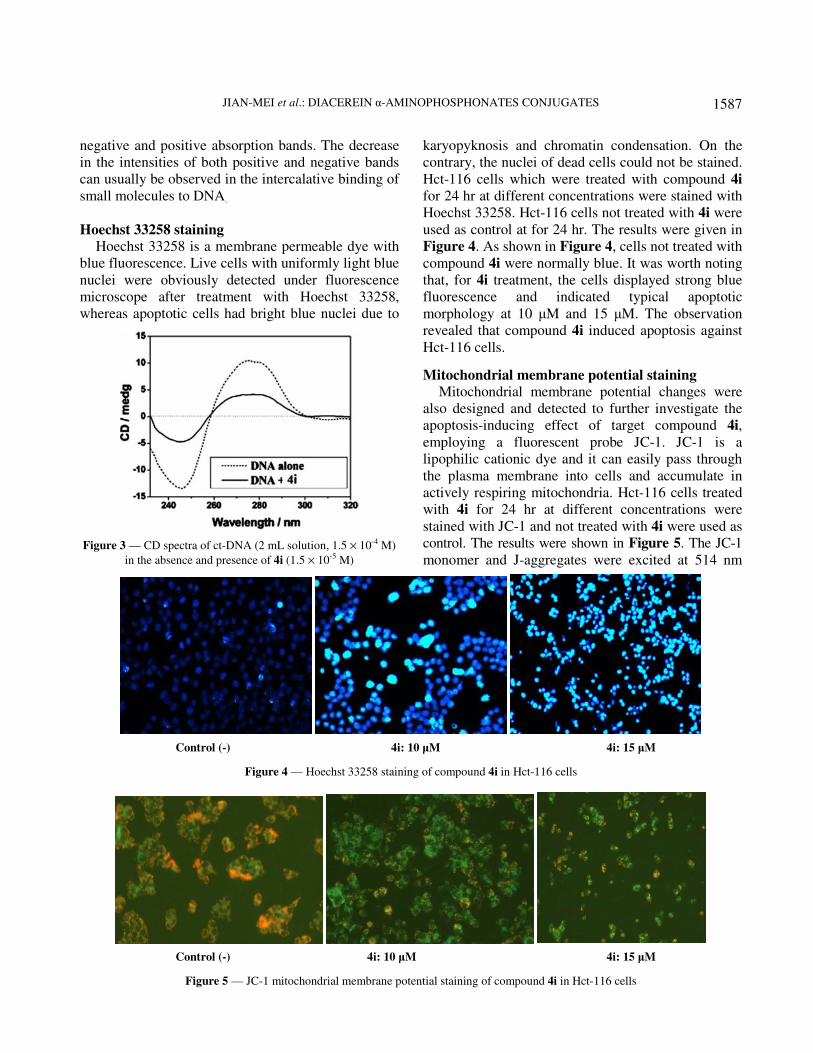
Hoechst 33258 staining
Hoechst 33258 is a membrane permeable dye with
blue fluorescence. Live cells with uniformly light blue
nuclei were obviously detected under fluorescence
microscope after treatment with Hoechst 33258,
whereas apoptotic cells had bright blue nuclei due to
Figure 3 — CD spectra of ct-DNA (2 mL solution, 1.5 × 10-4 M)
in the absence and presence of 4i (1.5 × 10-5 M)
karyopyknosis and chromatin condensation. On the
contrary, the nuclei of dead cells could not be stained.
Hct-116 cells which were treated with compound 4i
for 24 hr at different concentrations were stained with
Hoechst 33258. Hct-116 cells not treated with 4i were
used as control at for 24 hr. The results were given in
Figure 4. As shown in Figure 4, cells not treated with
compound 4i were normally blue. It was worth noting
that, for 4i treatment, the cells displayed strong blue
fluorescence and indicated typical apoptotic
morphology at 10 µM and 15 µM. The observation
revealed that compound 4i induced apoptosis against
Hct-116 cells.
Mitochondrial membrane potential staining
Mitochondrial membrane potential changes were
also designed and detected to further investigate the
apoptosis-inducing effect of target compound 4i,
employing a fluorescent probe JC-1. JC-1 is a
lipophilic cationic dye and it can easily pass through
the plasma membrane into cells and accumulate in
actively respiring mitochondria. Hct-116 cells treated
with 4i for 24 hr at different concentrations were
stained with JC-1 and not treated with 4i were used as
control. The results were shown in Figure 5. The JC-1
monomer and J-aggregates were excited at 514 nm
Control (-) 4i: 10 µM 4i: 15 µM
Figure 4 — Hoechst 33258 staining of compound 4i in Hct-116 cells
Control (-) 4i: 10 µM 4i: 15 µM
Figure 5 — JC-1 mitochondrial membrane potential staining of compound 4i in Hct-116 cells
INDIAN J. CHEM., SEC B, DECEMBER 2014
1588
and 585 nm, respectively, and light emissions were
collected at 515-545 nm (green) and 570-600 nm
(red). For fluorescence microscopy, the results
displayed that cells not treated with the compounds
were normally red (in the web version), while for 4i
treatment, cells exhibited strong green fluorescence
and indicated typical apoptotic morphology.
Therefore, it could be concluded that compound 4i
induced apoptosis against Hct-116 cell line. The
results were identical with that of previous experiment
of Hoechst 33258 staining.
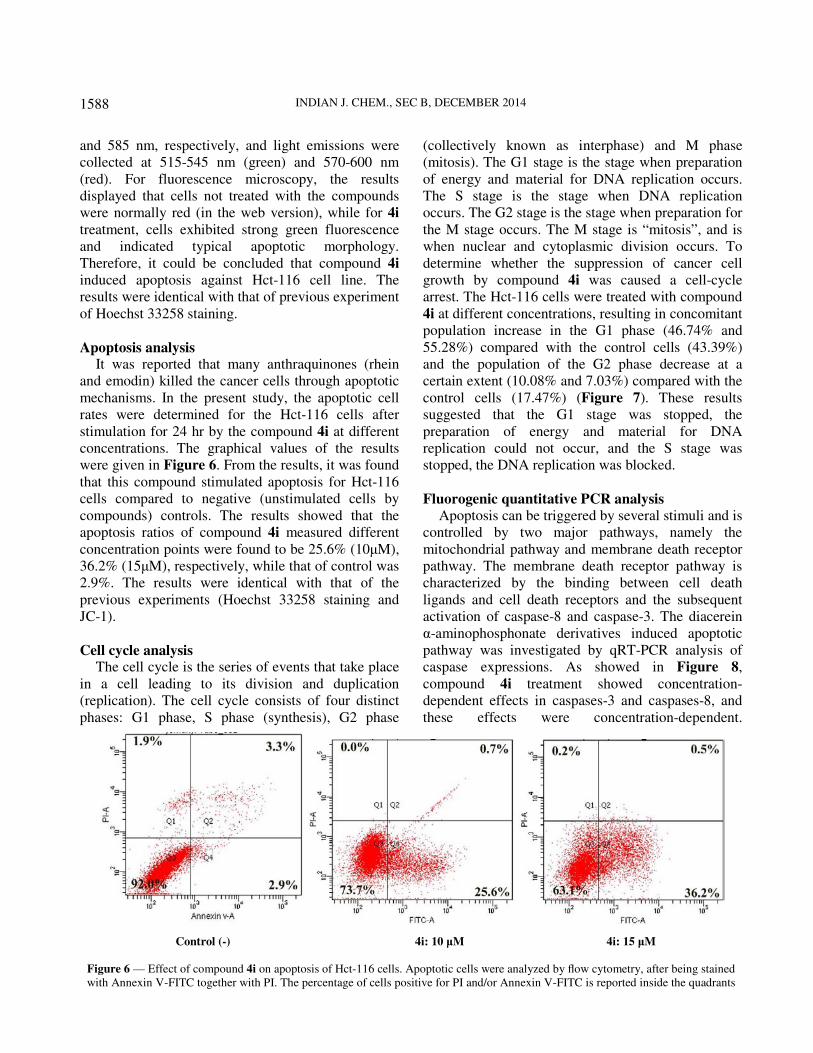
Apoptosis analysis It was reported that many anthraquinones (rhein
and emodin) killed the cancer cells through apoptotic
mechanisms. In the present study, the apoptotic cell
rates were determined for the Hct-116 cells after
stimulation for 24 hr by the compound 4i at different
concentrations. The graphical values of the results
were given in Figure 6. From the results, it was found
that this compound stimulated apoptosis for Hct-116
cells compared to negative (unstimulated cells by
compounds) controls. The results showed that the
apoptosis ratios of compound 4i measured different
concentration points were found to be 25.6% (10µM),
36.2% (15µM), respectively, while that of control was
2.9%. The results were identical with that of the
previous experiments (Hoechst 33258 staining and
JC-1).
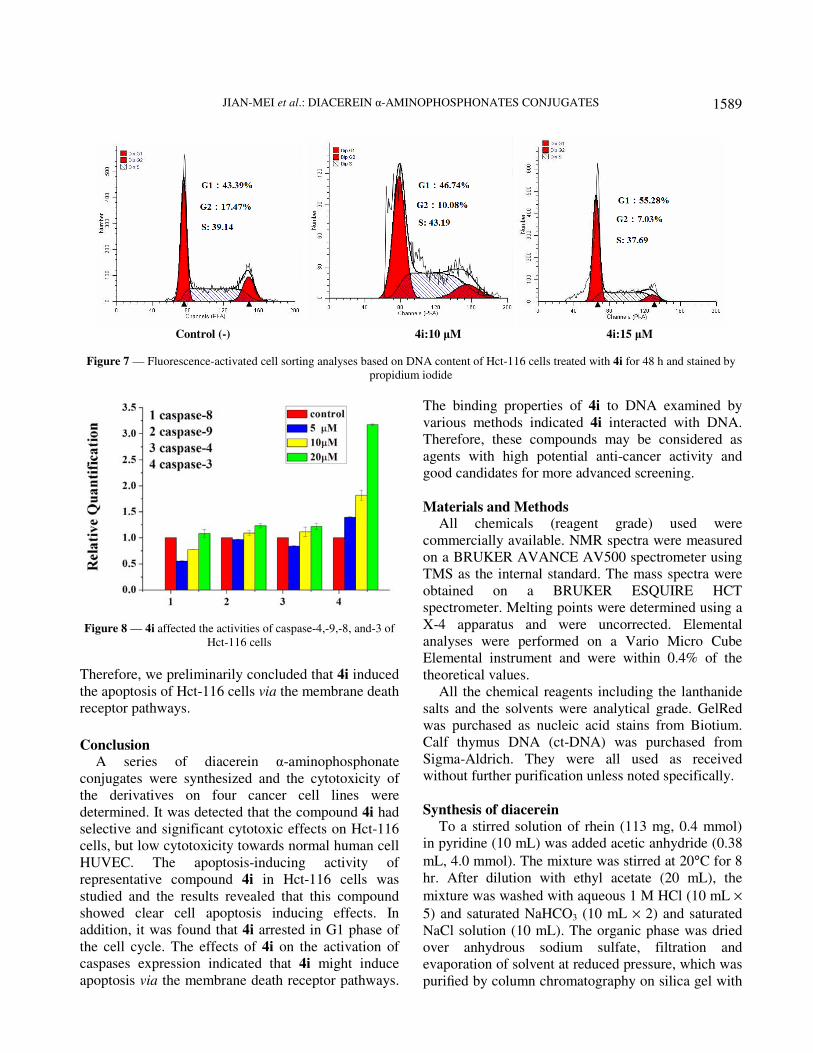
Cell cycle analysis The cell cycle is the series of events that take place
in a cell leading to its division and duplication
(replication). The cell cycle consists of four distinct
phases: G1 phase, S phase (synthesis), G2 phase
(collectively known as interphase) and M phase
(mitosis). The G1 stage is the stage when preparation
of energy and material for DNA replication occurs.
The S stage is the stage when DNA replication
occurs. The G2 stage is the stage when preparation for
the M stage occurs. The M stage is “mitosis”, and is
when nuclear and cytoplasmic division occurs. To
determine whether the suppression of cancer cell
growth by compound 4i was caused a cell-cycle
arrest. The Hct-116 cells were treated with compound
4i at different concentrations, resulting in concomitant
population increase in the G1 phase (46.74% and
55.28%) compared with the control cells (43.39%)
and the population of the G2 phase decrease at a
certain extent (10.08% and 7.03%) compared with the
control cells (17.47%) (Figure 7). These results
suggested that the G1 stage was stopped, the
preparation of energy and material for DNA
replication could not occur, and the S stage was
stopped, the DNA replication was blocked.
Fluorogenic quantitative PCR analysis
Apoptosis can be triggered by several stimuli and is
controlled by two major pathways, namely the
mitochondrial pathway and membrane death receptor
pathway. The membrane death receptor pathway is
characterized by the binding between cell death
ligands and cell death receptors and the subsequent
activation of caspase-8 and caspase-3. The diacerein
α-aminophosphonate derivatives induced apoptotic
pathway was investigated by qRT-PCR analysis of
caspase expressions. As showed in Figure 8,
compound 4i treatment showed concentration-
dependent effects in caspases-3 and caspases-8, and
these effects were concentration-dependent.
Control (-) 4i: 10 µM 4i: 15 µM
Figure 6 — Effect of compound 4i on apoptosis of Hct-116 cells. Apoptotic cells were analyzed by flow cytometry, after being stained
with Annexin V-FITC together with PI. The percentage of cells positive for PI and/or Annexin V-FITC is reported inside the quadrants
JIAN-MEI et al.: DIACEREIN α-AMINOPHOSPHONATES CONJUGATES
1589
Figure 8 — 4i affected the activities of caspase-4,-9,-8, and-3 of
Hct-116 cells
Therefore, we preliminarily concluded that 4i induced
the apoptosis of Hct-116 cells via the membrane death
receptor pathways.
Conclusion
A series of diacerein α-aminophosphonate
conjugates were synthesized and the cytotoxicity of
the derivatives on four cancer cell lines were
determined. It was detected that the compound 4i had
selective and significant cytotoxic effects on Hct-116
cells, but low cytotoxicity towards normal human cell
HUVEC. The apoptosis-inducing activity of
representative compound 4i in Hct-116 cells was
studied and the results revealed that this compound
showed clear cell apoptosis inducing effects. In
addition, it was found that 4i arrested in G1 phase of
the cell cycle. The effects of 4i on the activation of
caspases expression indicated that 4i might induce
apoptosis via the membrane death receptor pathways.
The binding properties of 4i to DNA examined by
various methods indicated 4i interacted with DNA.
Therefore, these compounds may be considered as
agents with high potential anti-cancer activity and
good candidates for more advanced screening.
Materials and Methods All chemicals (reagent grade) used were
commercially available. NMR spectra were measured
on a BRUKER AVANCE AV500 spectrometer using
TMS as the internal standard. The mass spectra were
obtained on a BRUKER ESQUIRE HCT
spectrometer. Melting points were determined using a
X-4 apparatus and were uncorrected. Elemental
analyses were performed on a Vario Micro Cube
Elemental instrument and were within 0.4% of the
theoretical values.
All the chemical reagents including the lanthanide
salts and the solvents were analytical grade. GelRed
was purchased as nucleic acid stains from Biotium.
Calf thymus DNA (ct-DNA) was purchased from
Sigma-Aldrich. They were all used as received
without further purification unless noted specifically.
Synthesis of diacerein
To a stirred solution of rhein (113 mg, 0.4 mmol)
in pyridine (10 mL) was added acetic anhydride (0.38
mL, 4.0 mmol). The mixture was stirred at 20°C for 8
hr. After dilution with ethyl acetate (20 mL), the
mixture was washed with aqueous 1 M HCl (10 mL ×
5) and saturated NaHCO3 (10 mL × 2) and saturated
NaCl solution (10 mL). The organic phase was dried
over anhydrous sodium sulfate, filtration and
evaporation of solvent at reduced pressure, which was
purified by column chromatography on silica gel with
Control (-) 4i:10 µM 4i:15 µM
Figure 7 — Fluorescence-activated cell sorting analyses based on DNA content of Hct-116 cells treated with 4i for 48 h and stained by
propidium iodide

INDIAN J. CHEM., SEC B, DECEMBER 2014
1590
petroleum ether/ethyl acetate (4:1, V:V) to give the
pale yellow solids. Yield 88.5%, yellow solid. m.p.
217~219°C; IR (KBr): 2980 (OH), 1770, 1765 (C=O
stretch in ester), 1701, 1680 cm-1
(C=O streching
vibrations in α,β-unsaturated ketones); 1H NMR (500
MHz, DMSO-d6): δ 2.38 (s, 3H, H-22), 2.39 (s, 3H,
H-25) 7.62 (d, J = 6.0 Hz, 1H, H-6), 7. 93 (t, J = 6.0
Hz, 1H, H-7), 8.01 (d, J = 3.0 Hz, 1H, H-3), 8.12 (d, J
= 6.0 Hz, 1H, H-8), 8.52 ( d, J = 3.0 Hz, 1H, H-1); 13
C
NMR (125 MHz, DMSO-d6): δ 21.0
(C-25), 21.1 (C-22), 123.8 (C-1), 125.3 (C-8), 126.5
(C-14), 129.4 (C-12), 129.7 (C-7), 130.4 (C-6), 134.3
(C-9), 134.8 (C-11), 135.8 (C-3), 136.7 (C-2), 149.8
(C-5), 150.0 (C-4), 165.3 (C-18), 169.3 (C-20), 169.4
(C-23), 180.5 (C-13), 181.1 (C-10); ESI-MS: m/z
369.56 (M + H)+.
Synthesis of α-aminophosphonate
A suspension of aromatic aldehydes (10 mmol) and
ammonium acetate (11 mmol) was stirred for 6 hr at
reflux. The reaction mixture was filtered to give the
white precipitate 2, Diethyl phosphite (5 mmol) was
added to 2 and the resulting solution was stirred for
6 hr at 80°C. After cooling to RT, hydrochloric acid
(4 mmol) was added to the reaction mixture at 0°C,
which was stirred for 2 hr to give the precipitate 3.
The precipitate was filtered and washed with ether
(3 × 15 mL), and then it was added to sodium
hydroxide solution (10%) in water phase to pH 8 and
stirred for 30 min at RT. After extraction with ethyl
acetate (3 × 25 mL), the organic phase was dried over
anhydrous sodium sulfate, the solvent was removed
by evaporation under reduced pressure, gave the oils
of 3, which was purified by column chromatography
on silica gel with petroleum ether/ethyl acetate
(1:2, V:V).
General synthesis of target compounds 4a-k
Diacerin (1 mmol) and HOBt (0.2 mmol) in DMSO
(15 mL) were treated with EDCI (1.2 mmol) and
stirred for 15 min at 0°C. Then 3 was added dropwise
with constant stirring at 0°C, the mixture was
stirred for another 10 hr at RT, then washed
with brine, the organic phase was dried with
anhydrous sodium sulfate, filtered and dried at under
vacuum. The residue was purified by column
chromatography on silica gel with petroleum
ether/ethyl acetate (3:1, V:V), 4a-k were obtained as a
pale yellowish solid.
Cytotoxicity of diacerein derivatives Cell Lines. The following in vitro human cancer
cell lines were used: HepG 2 (Human epidermoid
larynx carcinoma), Hct-116 (Human colorectal cells),
CNE (human nasopharyngeal carcinoma cells), Spca-
2 (human lung adenocarcinoma cell line), HUVEC
(Human Umbilical Vein Endothelial Cells). The cell
lines (HepG 2, Hct-116, CNE, Spca-2) were
purchased from the Cell Resource Center of Shanghai
Institutes for Biological Sciences, the Academy of
Sciences of China.
Cell Culture. HepG 2, Hct-116, CNE and HUVEC
cells were cultured in Dulbecco Modified Eagle
Medium (DMEM; HyClone, USA), containing
4.0 mM L-Glutamine and 4500 mg/L Glucose,
supplemented with 10% (v/v) foetalbovine serum
(FBS; HyClone, USA). Spca-2 was cultured in
RPMI-1640 medium (HyClone, USA), containing
2.05Mm L-Glutamine without Calcium nitrate,
supplemented with 10% (v/v) foetalbovine serum
(FBS; HyClone, USA). The cell culturemedia was
supplemented with penicillin/streptomycin at
100 Units/mL as adherent monolayers. Cell cultures
were kept in a humidified incubator with 5% CO2 at
37°C. Stock solutions were prepared in dimethyl
sulfoxide (DMSO) and further dilutions were made
with fresh culture medium. The concentration of
DMSO in the final culture medium was 1%, which
had no effect on the cell viability.
MTT assay. Chemosensitivity was assessed using
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium
bromide (MTT) assay. Briefly, exponentially growing
HepG 2 cells (2000-3000 cells/well), Hct-116
(3000-4000 cells/well), CNE (2000-3000 cells/well),
HUVEC (2000-3000 cells/well) and Spca-2
(2000-3000 cells/well) were seeded into 96-well
plates and treated with indicated concentrations of
samples for 48hr, and then 10 mL of MTT
(10mg/mL) was added. After incubation for 4 hr at
37°C, the purple formazan crystals (i.e. a reduced
form of MTT) generated from viable cells were
dissolved by adding 100 µL DMSO in each well. The
plates were swirled gently for 10 min to dissolve the
precipitate, and quantified by measuring the optical
density (OD) of the plates at a wavelength of 490 nm
on plate reader (TECAN infinite M1000). Each
concentration was repeated in three wells and the
same experimental conditions were provided for all
compounds and MTT analysis was repeated three
times for each cell line.

JIAN-MEI et al.: DIACEREIN α-AMINOPHOSPHONATES CONJUGATES
1591
Hoechst 333258 staining Cells grown on a sterile cover slip in six-well tissue
culture plates were treated with compounds for a
certain range of time. The culture medium containing
compounds was removed, and the cells were fixed in
4% paraformaldehyde for 10 min. After being washed
twice with PBS, the cells were stained with 0.5 mL of
Hoechst 33258 (Beyotime) for 5 min and then again
washed twice with PBS. The stained nuclei were
observed under a Nikon ECLIPSETE2000-S
fluorescence microscope using 350 nm excitation and
460 nm emissions.
Mitochondrial membrane potential staining JC-1 probe was employed to measure
mitochondrial depolarization in Hct-116 cells. Briefly,
Cells cultured in six-well plates after indicated
treatments were incubated with an equal volume of
JC-1 staining solution (5 µg/mL) at 37°C for 20 min
and rinsed twice with PBS. Mitochondrial membrane
potentials were monitored by determining the relative
amounts of dual emissions from mitochondrial JC-1
monomers or aggregates using a Nikon
ECLIPSETE2000-S fluorescent microscope.
Mitochondrial depolarization is indicated by an
increase in the green/red fluorescence intensity ratio.
Apoptosis analysis
Apoptosis was discriminated with the annexin
V-FITC/propidium iodide test. Cells were seeded at
2 × 106/well in 10% FBS-DMEM into 6-well plates,
and treated with compounds for 24 hr. The cells were
washed twice with cold Phosphate Buffered Saline
(PBS) and then resuspend cells in 1 × Binding Buffer
(0.1 M Hepes/NaOH (pH 7.4), 1.4 M NaCl, 25 mM
CaCl2)) at a concentration of 1 × 106 cells/mL.
Transfer 100 µL of the solution (1 × 105
cells) to a
5 mL culture tube, and add 5 µL of FITC Annexin V
(BD, Pharmingen) and 5 µL propidium iodide (PI) to
each tube. Gently vortex the cells and incubate for
30 min at RT (25°C) in the dark. Add 200 µL PBS to
each tube. Analysis was performed with the system
software (CellQuest; BD Biosciences). Lower left
quadrant, viable cells (annexin V-/PI-); lower right
quadrant, early apoptotic cells (annexin V+/PI-);
upper right quadrant, late apoptotic cells
(annexin V+/PI+); upper left quadrant, necrotic cells
(annexin V-/PI+). The percentage of cells positive for
PI and/or Annexin V-FITC was reported inside the
quadrants.
Cell cycle analysis The cells lines were treated with indicated
concentrations of compound 4i. After incubation for
48 hr, cells were washed twice with ice-cold PBS,
fixed and permeabilized with ice-cold 70% ethanol at
-20°C overnight. The cells were treated with
100 µg/mL RNase A at 37°C for 30 min after washed
with ice-cold PBS, and finally stained with 1 mg/mL
propidium iodide (PI) in the dark at 4°C for 30 min.
Analysis was performed with the system
software(CellQuest; BD Biosciences).
Real-Time PCR of Caspase-3,-4,-8 and-9
Total RNA was extracted from the Hct-116 cells
after treatment with 5 µM, 10 µM, 20 µM diacerein
α-aminophosphonate derivatives for 24 hr using the
Qiagen RNeasy Mini Kit as described previously.
RNA samples were reverse-transcribed for 30 min at
42°C with the High Capacity cDNA Reverse
Transcription Kit (Applied Biosystems). The SYBR®
Green PCR Master Mix (ABI, USA) and specific
primer pairs were used for selected genes, and the
primer pair for actin was used as the reference gene.
qPCRwas performed according to the following
conditions: 2 min at 50°C, 10 min at 95°C, and 40
cycles of 15 s at 95°C and 1 min at 60°C using 0.5 µL
of complementary (c)DNA, 2 × SYBR Green PCR
Master Mix, and 500 nM of the forward and reverse
primers: CASP3-F81, GAGTGCTCGCAGCTCATACCT;
CASP3-R, CCTCACGGCCTGGGATTT; CASP4-
F91, GAAACTCCAAGGGCCAAAGC; CASP4-R,
TCCATTTTCAATTGCCAGGAA; CASP8-F119,
CTTTATGATATTGGGGAACAAC; CASP8-R,
TGGAATAACATCAAGGCATC; CASP9-F247,
CATTGGTTCTGGAGGATTTG; CASP9-R,
CATTTTCTTGGCAGTCAGGT. The threshold cycle
number (Ct) was calculated with ABI software.
Relative transcript quantities were calculated using
the △Ct method with actin as the reference gene
ampli △fied from the same samples. Ct is the
difference in the threshold cycles of messenger
(m)RNA for selected genes relative to those of actin
mRNA. The real-time RT-PCR was performed in
triplicate for each experimental group.
Spectroscopic studies on DNA interaction
The 2 × 10-3
M ct-DNA stock solution was stored
at 4°C for no more than 5 days before use. The
synthesized conjugate (4b and 4i) was prepared as
2 × 10-3
M DMSO stock solutions for DNA binding

INDIAN J. CHEM., SEC B, DECEMBER 2014
1592
studies. The final working solutions of the complexes
for DNA binding studies were diluted by TBS and the
DMSO content was less than10%. For UV-vis
absorption experiments, the working solution of the
complexes was constantly kept at 25 µM. The ct-
DNA stock solution was increasingly added until a
saturation state was achieved. After each addition, the
solution was allowed to incubate for 5 min before the
absorption spectra were recorded. Kb as the
equilibrium DNA binding constant and s as the
binding site size were determined by non-linear fitting
according to the following equation27,28
:
Ct[DNA]/(εa−εf)=Ct[DNA]/(εb−εf)+1/Kb(εb−εf) (1a)
where Ct [DNA] is the DNA concentration in
nucleotides, εa is the molar extinction coefficient of
the compound bound with DNA, εf is the extinction
coefficient of the free compound, and εb is the
extinction coefficient of the compound fully bound to
DNA.
A solution containing 2 × 10-4
M DNA and 2 × 10-5
M GelRed ([DNA]/[GelRed] = 10:1) was prepared for
GelRed-DNA competitive binding studies. Fluo-
rescence emission spectra were recorded under slit
width as 10 nm/10 nm for Ex/Em, respectively. The
quenching constant for comparing the efficiency of
fluorescence quenching, i.e., Kq, of each compound
was obtained by the linear fit of plotting I0/I versus
[Q], according to the classic Stern–Volmer equation:
I0/I = 1+Kq×[Q] (Ref 29), where I0 and I are the peak
emission intensity of the GelRed-DNA system in the
absence and presence of each compound as quencher,
and [Q] is the concentration of quencher. In the
fluorescence polarization experiment, each sample
was pre-incubated for 40 min before the fluorescence
polarization was recorded under the condition of 595
nm emitting wavelength with 350 nm exciting
wavelength, and slit width was set as 5 nm/5 nm for
Ex/Em, respectively.
CD absorption spectra of DNA were measured in
TBS at a 100 nm/min scan rate in the wavelength
range from 200 to 500 nm, with 1.5 × 10-4
M DNA in
the absence and presence of each compound of
1.5 × 10-5
M, respectively. The CD signal of TBS was
taken as the background and subtracted from the
spectra. All the spectroscopic experiments were
performed at 25°C.
Statistical analysis All statistical analyses were performed with
SPSS10. Data were analyzed by one-way analysis of
variance (ANOVA). Mean separations were
performed using the least significant difference
method (LSD test). Each experiment had three
replicates and all experiments were run three times
with similar results. Measurements from all the
replicates were combined and treatment effects
analyzed.
Spectral data
8-(Acetyloxy)-3-({[(diethoxyphosphoryl)(2-fluoro-
phenyl)methyl]amino}carbonyl)-9,10-dioxo-9,10-
dihydro-1-anthracenyl acetate, 4a: Yield 80%. Pale
yellow solid. m.p. 162~164°C. IR (KBr): 1770, 1765
(C=O stretch in ester), 1701, 1680 (C=O stretching
vibrations in α,β-unsaturated ketones), 1550 (NH),
1330 cm-1
(C-N); 1H NMR (500 MHz, CDCl3): δ 8.66
(d, J = 1.8 Hz, 1H, Ar-H), 8.43 (dd,
J = 9.4, 4.3 Hz, 1H, Ar-H), 8.20 (dd, J = 7.8, 1.3 Hz,
1H, Ar-H), 7.94 (d, J = 7.8 Hz, 1H, Ar-H), 7.77 (t, J = 7.9
Hz, 1H, Ar-H), 7.57 (ddd, J = 8.7, 5.1, 1.9 Hz, 2H,
Ar-H), 7.42 (d, J = 8.0 Hz, 1H, NH), 7.05 (d, J = 8.5 Hz,
2H, Ar-H), 5.75 (dd, J = 21.4, 9.3 Hz, 1H, P-CH),
4.20-4.11 (m, 2H, OCH2), 3.98-3.84 (m, 2H, OCH2),
2.44 (s, 3H), 2.43 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H,
CH3), 1.12 (t, J = 7.0 Hz, 3H, CH3); 13
C NMR (125 MHz,
CDCl3): δ 181.4 (C-10), 180.3 (C-13), 169.2 (C-23),
169.0 (C-20), 164.5 (C-18), 160.3 (C-2′), 150.3 (C-4),
150.1 (C-5), 139.3 (C-2), 134.8 (C-3), 134.6 (C-11),
134.4 (C-9), 130.4 (C-6), 130.0 (C-7), 129.8 (C-1′),
129.7 (C-12), 128.7 (C-6′), 128.2 (C-4′), 126.5 (C-14),
125.4 (C-8), 124.3 (C-1), 123.8 (C-5′), 115.3 (C-3′),
48.8 (-OCH2), 47.7 (-OCH2), 66.6 (-PCH, d, J = 3.6 Hz),
21.8 (C-22), 20.8 (C-25), 16.3 (-CH3), 16.1 (-CH3); 31
P
NMR(200 MHz, CDCl3): δ 20.97; ESI-MS: m/z
612.18 (M + H)+. Anal Calcd for C30H27FNO10P: C,
58.92; H, 4.45; N, 2.29; Found: C, 58.95; H, 4.43; N,
2.31%.
8-(Acetyloxy)-3-({[(diethoxyphosphoryl)(4-fluoro-
phenyl)methyl]amino}carbonyl)-9,10-dioxo-9,10- dihydro-1-anthracenyl acetate, 4b: Yield 79%. Pale
yellow solid. m.p. 222~224°C. IR (KBr): 1765, 1762
(C=O stretch in ester), 1709, 1679 (C=O stretching
vibrations in α,β-unsaturated ketones), 1538 (NH),
1319 cm-1
(C-N); 1H NMR (500 MHz, CDCl3): δ 1.11
(t, J = 7.1 Hz, 3H, CH3), 1.29 (t, J = 7.1 Hz, 3H, CH3),
2.42 (s, 3H), 2.43 (s, 3H), 4.00–3.83 (m, 2H, OCH2),
4.19–4.10 (m, 2H, OCH2), 5.76 (dd,

JIAN-MEI et al.: DIACEREIN α-AMINOPHOSPHONATES CONJUGATES
1593
J = 21.3, 9.3 Hz, 1H, P-CH), 7.03 (t, J = 8.6 Hz, 2H,
Ar-H), 7.41 (d, J = 8.1, 1.0 Hz, 1H, NH), 7.64-7.49
(m, 2H, Ar-H), 7.76 (t, J = 7.9 Hz, 1H, Ar-H), 7.91 (d,
J = 7.7 Hz, 1H, Ar-H), 8.18 (dd, J = 7.7 Hz, 1H, Ar-H),
8.41 (d, J = 6.1 Hz, 1H, Ar-H), 8.62 (d, J = 1.7 Hz,
1H, Ar-H); 13
C NMR (125 MHz, CDCl3): δ 181.5 (C-10),
180.0 (C-13), 169.2 (C-23), 169.1 (C-20), 164.6 (C-18),
160.3 (C-4′), 150.3 (C-4), 150.1 (C-5), 144.7 (C-1′),
139.3 (C-2), 138.3 (C-1), 134.8 (C-3), 134.7 (C-11),
134.4 (C-9), 130.5 (C-6), 130.3 (C-7), 129.8 (C-12),
128.9 (C-6′), 128.8 (C-2′), 126.1 (C-14), 125.3 (C-8),
115.5 (C-6′), 115.4 (C-5′), 50.2 (-OCH2), 49.5 (-OCH2),
63.1 (-PCH, d, J = 3.4 Hz), 22.0 (C-22), 20.4 (C-25),
16.5 (-CH3), 16.3 (-CH3); 31
P NMR (200 MHz, CDCl3):
δ 20.61 20.59; ESI-MS: m/z 612.20 (M + H)+. Anal
Calcd for C30H27FNO10P: C, 58.92; H, 4.45; N, 2.29.
Found: C, 58.94; H, 4.44; N, 2.32%.
A-(acetyloxy)-3-({[( 2-chlorophenyl)(diethoxyphos-
phoryl)methyl]amino}carbonyl)-9, 10-dioxo-9,10- dihydro-1-anthracenyl acetate, 4c: Yield 80%. Pale
yellow solid. m.p. 227~229°C. IR (KBr): 1780, 1759
(C=O stretch in ester), 1715, 1685 (C=O stretching
vibrations in α,β-unsaturated ketones), 1542 (NH),
1335 cm-1
(C-N); 1H NMR (500 MHz, CDCl3): δ 8.64
(d, J = 7.8 Hz, 1H, Ar-H), 8.23 (dd, J = 7.8, 1.2 Hz,
1H, Ar-H), 7.92 (d, J = 7.8 Hz, 2H, Ar-H), 7.79 (t, J = 7.9
Hz, 1H, Ar-H), 7.71 (d, J = 7.6 Hz, 1H, NH), 7.46-7.39
(m, 2H, Ar-H), 7.32-7.26 (m, 3H, Ar-H), 6.29 (dd,
J = 21.5, 8.9 Hz, 1H, Ar-H), 4.27–4.18 (m, 2H,
OCH2), 3.98-3.83 (m, 2H, OCH2), 2.45 (s, 6H), 1.36
(t, J = 7.1 Hz, 3H, CH3), 1.10 (t, J = 7.1 Hz, 3H,
CH3); 13
C NMR (125 MHz, CDCl3): δ 181.2 (C-10),
180.2 (C-13), 169.2 (C-23), 169.1 (C-20), 164.4 (C-18),
150.2 (C-4), 150.1 (C-5), 142.5 (C-1′), 139.4 (C-2),
134.9 (C-3), 134.6 (C-11), 134.3 (C-9), 132.6 (C-2′),
130.5 (C-6), 129.8 (C-7), 129.5 (C-12), 128.7 (C-3′),
128.4 (C-6′), 128.0 (C-4′), 126.8 (C-5′), 126.5 (C-14),
125.5 (C-8), 124.3 (C-1), 53.7 (-OCH2), 50.8 (-OCH2),
64.1 (-PCH, d, J = 3.0 Hz), 21.9 (C-22), 20.8 (C-25),
16.4 (-CH3), 16.3 (-CH3); 31
P NMR (200 MHz,
CDCl3): δ 21.92; ESI-MS: m/z 628.18 (M + H)+. Anal
Calcd for C30H27ClNO10P: C, 57.38; H, 4.33; N, 2.23.
Found: C, 57.40; H, 4.32; N, 2.22%.
8-(Acetyloxy)-3-({[(4-chlorophenyl) (diethoxyphos-
phoryl)methyl]amino}carbonyl)-9,10-dioxo-9, 10-
dihydro-1-anthracenyl acetate, 4d: Yield 80%. Pale
yellow solid. m.p. 216~218°C. IR (KBr): 1785, 1750
(C=O stretch in ester), 1726, 1673 (C=O stretching
vibrations in α,β-unsaturated ketones), 1520 (NH),
1321 cm-1
(C-N); 1H NMR (500 MHz CDCl3): δ 1.16
(t, J = 7.0 Hz, 3H, CH3), 1.32 (t, J = 7.1 Hz, 3H,
CH3), 2.43 (s, 3H), 2.45 (s, 3H), 3.82-4.21 (m, 4H,
OCH2), 5.76 (dd, J = 22.4, 9.3Hz 1H, PCH), 7.28-8.40
(m, 9H, Ar-H, 1H, NH); 13
C NMR (125 MHz, CDCl3):
δ 181.4 (C-10), 180.2 (C-13), 169.3 (C-23), 169.2 (C-20),
164.3 (C-18), 150.3 (C-4), 150.1 (C-5), 137.0 (C-1′),
139.4 (C-2), 134.9 (C-3), 134.8 (C-11), 134.3 (C-9),
133.5 (C-4′), 130.6 (C-6), 129.7 (C-7), 129.5 (C-12),
129.2 (C-5′), 129.0 (C-3′), 128.6 (C-2′), 128.5 (C-6′),
126.4 (C-14), 125.5 (C-8), 124.4 (C-1), 51.8 (-OCH2),
49.5 (-OCH2), 63.2 (-PCH, d, J = 3.5Hz), 21.9 (C-22),
20.8 (C-25), 16.4 (-CH3), 16.3 (-CH3); 31
P NMR
(200 MHz, CDCl3): δ 21.34; ESI-MS: m/z 628.20 (M +
H)+. Anal Calcd for C30H27ClNO10P: C, 57.38;
H, 4.33; N, 2.23. Found: C, 55.78; H, 4.52; N, 2.17%.
8-(Acetyloxy)-3-({[(2-bromophenyl)(diethoxyphos-
phoryl)methyl]amino}carbonyl)-9,10-dioxo-9,10- dihydro-1-anthracenyl acetate, 4e: Yield 79%. Pale
yellow solid. m.p. 220~222°C. IR (KBr): 1770, 1740
(C=O stretch in ester), 1715, 1670 (C=O stretching
vibrations in α,β-unsaturated ketones), 1520 (NH),
1310 cm-1
(C-N); 1H NMR (500 MHz CDCl3): δ 1.09
(t, J = 7.1 Hz, 3H, CH3), 1.35 (t, J = 7.2 Hz, 3H,
CH3), 2.43 (s, 3H), 2.45 (s, 3H), 3.72-4.26 (m, 4H,
OCH2), 6.31(dd, J = 21.4, 9.0Hz 1H, PCH),7.17-8.22
(m, 9H, Ar-H, 1H, NH); 13
C NMR (125 MHz,
CDCl3): δ 181.3 (C-10), 180.2 (C-13), 169.2 (C-23),
169.1 (C-20), 164.5 (C-18), 150.3 (C-4), 150.1 (C-5),
145.5 (C-1′), 139.3 (C-2), 134.8 (C-3), 134.5 (C-11),
134.2 (C-9), 131.7 (C-3′), 130.4 (C-6), 129.7 (C-7),
129.5 (C-12), 129.6 (C-6′), 128.3 (C-4′), 127.2 (C-5′),
126.4 (C-14), 125.5 (C-8), 124.3 (C-1), 121.7 (C-2′),
63.7 (-PCH, d, J = 3.3 Hz), 50.7 (-OCH2), 48.5 (-OCH2),
21.8 (C-22), 20.7 (C-25), 16.4 (-CH3), 16.3 (-CH3); 31
P NMR (200 MHz, CDCl3): δ 21.33; ESI-MS: m/z
672.17 (M + H)+. Anal Calcd for C30H27BrNO10P:
C, 53.59; H, 4.05; N, 2.08; Found:C, 53.60; H, 4.07;
N, 2.07%.
8-(Acetyloxy)-3-({[(4-bromophenyl)(diethoxyphos-
phoryl)methyl]amino}carbonyl)-9,10-dioxo-9,10-
dihydro-1-anthracenyl acetate, 4f: Yield 80%. Pale
yellow solid. m.p. 212~214°C. IR (KBr):
1760, 1715 (C=O stretch in ester), 1690, 1663 (C=O
stretching vibrations in α,β-unsaturated ketones),
1515 (NH), 1312 cm-1
(C-N); 1H NMR (500 MHz,
CDCl3): δ 8.62 (d, J = 7.6 Hz, 1H), 8.23 (d, J = 7.7
Hz, 1H), 7.91 (d, J = 7.6 Hz, 2H), 7.79 (t, J = 7.8 Hz,
1H), 7.51 (d, J = 8.4 Hz, 2H),7.44 (dd, J = 7.6, 2.9
Hz, 3H), 5.67 (dd, J = 21.4, 9.3 Hz, 1H), 4.19-4.14
(m, 2H), 4.05-3.97 (m, 1H), 3.85-3.77 (m, 1H), 2.45

INDIAN J. CHEM., SEC B, DECEMBER 2014
1594
(s, 3H), 2.45 (s, 3H), 1.32 (t, J = 7.1 Hz, 3H), 1.15 (t,
J = 7.0 Hz, 3H); 13
C NMR (125 MHz, CDCl3): δ 181.5
(C-10), 180.1 (C-13), 169.5 (C-23), 169.2 (C-20), 164.5
(C-18), 150.4 (C-4), 150.2 (C-5), 139.3 (C-2), 137.0
(C-1′), 134.8 (C-3), 134.6 (C-11), 134.1 (C-9), 131.7
(C-3′), 131.6 (C-5′), 130.3 (C-6), 129.8 (C-7), 129.4
(C-12), 129.5 (C-6′), 129.3 (C-2′), 126.4 (C-14),
125.4 (C-8), 124.3 (C-1), 121.3 (C-4′), 63.2 (-PCH, d,
J = 3.5Hz), 54.2 (-OCH2), 52.6 (-OCH2), 21.9 (C-22),
20.7 (C-25), 16.4 (-CH3), 16.3 (-CH3); 31
P NMR (200
MHz, CDCl3): δ 21.25; ESI-MS: m/z 672.19 (M +
H)+. Anal. Calcd for C30H27BrNO10P: C, 53.59; H, 4.05;
N, 2.08. Found: C, 53.61; H, 4.04; N, 2.05%.
8-(Acetyloxy)-3-({[(diethoxyphosphoryl)(2-methoxy-
phenyl)methyl]amino}carbonyl)-9,10-dioxo-9,10-
dihydro-1-anthracenyl acetate, 4g: Yield 82%. Pale
yellow solid. m.p. 218~220°C. IR (KBr): 1770, 1713
(C=O stretch in ester), 1705, 1665 (C=O stretching
vibrations in α,β-unsaturated ketones), 1530 (NH),
1300 cm-1
(C-N); 1H NMR (500 MHz, CDCl3): δ 8.57
(d, J = 7.8 Hz, 1H), 8.22 (dd, J = 7.8, 1.2 Hz, 1H),
8.03 (d, J = 9.1 Hz, 1H), 7.92 (d, J = 7.8 Hz, 1H),
7.77 (t, J = 7.9 Hz, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.42
(dd, J = 8.0, 1.2 Hz, 1H), 7.30 (t, J = 7.9 Hz, 1H),
7.00-6.94 (m, 2H), 6.12 (dd, J = 21.0, 9.5 Hz, 1H),
4.19-4.13 (m, 2H), 4.01-3.94 (m, 1H), 3.97 (s, 3H)
3.83-3.75 (m, 1H), 2.44 (s, 6H), 1.31 (t, J = 7.1 Hz,
3H), 1.10 (t, J = 7.0 Hz, 3H); 13
C NMR (125 MHz,
CDCl3): δ 181.2 (C-10), 180.3 (C-13), 169.3 (C-23),
169.0 (C-20), 164.1 (C-18), 159.6 (C-2′), 150.3 (C-4),
150.1 (C-5), 139.6 (C-2), 134.8 (C-3), 134.5 (C-11),
134.3 (C-9), 130.4 (C-6), 129.7 (C-7), 129.4 (C-12),
128.4 (C-6′), 128.3 (C-1′), 127.3 (C-4′), 126.6 (C-14),
125.5 (C-8), 123.8 (C-1), 114.2 (C-5′), 113.6 (C-3′),
55.4 (-OCH3), 63.3 (-PCH, d, J = 3.1 Hz), 47.1 (-OCH2),
45.5 (-OCH2), 21.0 (C-22), 20.9 (C-25), 16.4 (-CH3),
16.1 (-CH3); 31
P NMR (200 MHz, CDCl3): δ 20.70;
ESI-MS: m/z 624.22 (M + H)+. Anal Calcd for
C31H30NO11P: C, 59.71; H, 4.85; N, 2.25. Found: C,
59.74; H, 4.93; N, 2.28%.
8-(Acetyloxy)-3-({[(diethoxyphosphoryl)(4-methoxy-
phenyl)methyl]amino}carbonyl)-9,10-dioxo-9,10-
dihydro-1-anthracenyl acetate, 4h: Yield 80%. Pale
yellow solid. m.p. 198~201°C. IR (KBr): 1782, 1720
(C=O stretch in ester), 1700, 1668 (C=O stretching
vibrations in α,β-unsaturated ketones), 1525 (NH),
1315 cm-1
(C-N); 1H NMR (500 MHz CDCl3): δ 1.13
(t, J = 6.6 Hz, 3H, CH3), 1.31 (t, J = 6.6 Hz, 3H,
CH3), 3.76 (s, 3H, OCH3), 2.46 (s, 3H), 2.47 (s, 3H),
3.80-4.18 (m, 4H, OCH2), 5.75 (dd, J = 21.7, 7.6Hz
1H, PCH), 6.83-8.26 (m, 9H, Ar-H, 1H, NH); 13
C NMR
(125 MHz, CDCl3): δ 181.2 (C-10), 180.2 (C-13),
169.2 (C-23), 169.0 (C-20), 164.1 (C-18), 160.0 (C-4′),
150.2 (C-4), 150.1 (C-5), 139.6 (C-2), 134.8 (C-3),
134.4 (C-11), 134.3 (C-9), 130.6 (C-6), 129.7 (C-7),
129.4 (C-12), 128.4 (C-2′), 128.4 (C-6′), 127.5 (C-1′),
126.5 (C-14), 125.5 (C-8), 124.3 (C-1), 113.7 (C-5′),
113.5 (C-3′), 63.4 (-PCH, d, J = 3.1 Hz), 55.2 (-OCH3),
51.5 (-OCH2), 49.5 (-OCH2), 21.1 (C-22), 20.9 (C-25),
16.5 (-CH3), 16.3 (-CH3); 31
P NMR (200 MHz, CDCl3):
δ 20.92; ESI-MS: m/z 624.28 (M + H)+. Anal Calcd
for C31H30NO11P: C, 59.71; H, 4.85; N, 2.25. Found:
C, 59.74; H,4.86; N, 2.28.
8-(Acetyloxy)-3-({[(diethoxyphosphoryl)(4-methphe-
nyl)methyl]amino}carbonyl)-9,10-dioxo-9, 10-dihydro-
1-anthracenyl acetate, 4i: Yield 81%. Pale yellow
solid. m.p. 189~191°C. IR (KBr): 1770, 1723 (C=O
stretch in ester), 1712, 1693 (C=O stretching
vibrations in α,β-unsaturated ketones), 1503 (NH),
1312 cm-1
(C-N); 1H NMR (500 MHz, CDCl3): δ 8.65
(d, J = 7.6 Hz, 1H), 8.24 (dd, J = 7.8, 1.2 Hz, 1H),
7.92 (d, J = 7.7 Hz, 1H), 7.87 (dd, J = 9.1, 4.4 Hz,
1H), 7.79 (t, J = 7.9 Hz, 1H), 7.43 (dd, J = 8.0, 1.1
Hz, 3H), 7.18 (d, J = 8.1 Hz, 2H), 5.69 (dd, J = 20.8,
9.3 Hz, 1H), 4.21-4.11 (m, 2H), 3.99-3.93 (m, 1H),
3.75-3.70 (m, 1H), 2.45 (s, 3H), 2.45 (s, 3H), 2.34 (s,
3H),1.32 (t, J = 7.1 Hz, 3H), 1.12 (t, J = 7.1 Hz, 3H); 13
C NMR (125 MHz, CDCl3): δ 181.2 (C-10), 180.2
(C-13), 169.4 (C-23), 169.1 (C-20), 164.0 (C-18), 150.2
(C-4), 150.0 (C-5), 139.5 (C-2), 136.1 (C-1′), 134.8
(C-3), 134.3 (C-11), 134.3 (C-9), 130.6 (C-6), 129.7
(C-7), 129.6 (C-4′), 129.4 (C-12), 128.7 (C-5′), 128.6
(C-3′), 127.8 (C-6′), 127.6 (C-2′), 126.5 (C-14), 125.5
(C-8), 124.3 (C-1), 65.2 (-PCH, d, J = 3.0 Hz), 57.0 (-
OCH2), 55.8 (-OCH2), 21.1 (C-22), 20.9 (C-25), 20.3 (-
CH3), 16.5 (-CH3), 16.3 (-CH3); 31
P NMR (200 MHz,
CDCl3): δ 21.56; ESI-MS: m/z 608.23 (M + H)+. Anal
Calcd for C31H30NO10P: C, 61.28; H, 4.98; N, 2.31.
Found: C, 61.32; H, 5.01; N, 2.34%.
8-(Acetyloxy)-3-({ [ (diethoxyphosphoryl)(1-naph-
thyl)methyl]amino}carbonyl)-9, 10-dioxo-9, 10- dihydro-1-anthracenyl acetate, 4j: Yield 80%. Pale
yellow solid. m.p. 180~182°C. IR (KBr): 1767, 1716
(C=O stretch in ester), 1692, 1680 (C=O stretching
vibrations in α,β-unsaturated ketones), 1516 (NH),
1296 cm-1
(C-N); 1H NMR (500 MHz CDCl3): δ 0.85
(t, J = 7.0 Hz, 3H, CH3), 1.37 (t, J = 7.1 Hz, 3H,
CH3), 2.49 (s, 3H), 2.50 (s, 3H), 3.50-4.30 (m, 4H,
OCH2), 6.60 (dd, J = 22.3, 9.3Hz 1H, PCH), 7.26-8.33
(m, 12H, Ar-H, 1H, NH); 13
C NMR (125 MHz, CDCl3):

JIAN-MEI et al.: DIACEREIN α-AMINOPHOSPHONATES CONJUGATES
1595
δ 181.2 (C-10), 180.3 (C-13), 169.4 (C-23), 169.2 (C-20),
164.5 (C-18), 150.2 (C-4), 150.1 (C-5), 139.4 (C-2),
134.9 (C-3), 134.5 (C-11), 134.2 (C-9), 133.9 (C-1′),
132.4 (C-10′), 131.3 (C-9′), 130.5
(C-6), 129.7 (C-7), 129.5 (C-12), 128.8 (C-5′), 126.7 (C-
3′), 126.5 (C-14), 126.1 (C-2′), 125.9 (C-4′), 125.5 (C-
8), 125.5 (C-7′), 125.3 (C-6′), 123.9 (C-1), 123.3
(C-8′), 47.2 (-OCH2), 45.6 (-OCH2), 63.2 (-PCH, d, J =
3.1 Hz), 21.1 (C-22), 21.0 (C-25), 16.5 (-CH3), 16.2
(-CH3); 31
P NMR (202 MHz, CDCl3): δ 21.33; ESI-MS:
m/z 644.24 (M + H)+. Anal. Calcd for C34H30NO10P:
C, 63.45; H, 4.70; N, 2.18. Found: C, 63.44; H, 4.73;
N, 2.23%.
8-(Acetyloxy)-3-({[9-anthryl(diethoxyphosphoryl)-
methyl]amino}carbonyl)-9,10-dioxo-9,10-dihydro-
1-anthracenyl acetate, 4k: Yield 80%. Pale yellow
solid. m.p. 168~170°C. IR (KBr): 1785, 1726 (C=O
stretch in ester), 1716, 1683 (C=O stretching
vibrations in α,β-unsaturated ketones), 1506 (NH),
1298 cm-1
(C-N); 1H NMR (500 MHz CDCl3): δ 0.63
(t, J = 7.1 Hz, 3H, CH3), 1.42 (t, J = 7.1 Hz, 3H,
CH3), 2.47 (s, 3H), 2.49 (s, 3H), 3.31-4.35 (m, 4H,
OCH2), 7.24-7.28 (m, 2H, Ar-H), 7.31 (dd, J = 25.7,
8.5 Hz 1H, PCH), 7.38-8.79 (m, 12H, Ar-H, 1H, NH); 13
C NMR (125 MHz, CDCl3): δ 181.5 (C-10), 180.2
(C-13), 169.5 (C-23), 169.1 (C-20), 164.4 (C-18),
150.3 (C-4), 150.2 (C-5), 139.3 (C-2), 134.8 (C-3),
134.4 (C-11), 134.1 (C-9), 132.7 (C-9′), 132.2 (C-12′),
132.0 (C-14′), 131.8 (C-13′), 131.6 (C-11′), 130.5 (C-
6), 129.7 (C-7), 129.5 (C-12), 128.6 (C-5′), 128.3 (C-
4′), 126.5 (C-14), 126.4 (C-1′), 126.0 (C-8′), 125.5
(C-8), 125.5 (C-2′), 125.3 (C-7′), 124.9 (C-3′), 124.7
(C-6′), 124.4 (C-10′), 123.9 (C-1), 46.6 (-OCH2), 48.5
(-OCH2), 63.0 (-PCH, d, J = 3.1 Hz), 21.1 (C-22), 21.0
(C-25), 16.5 (-CH3), 16.2 (-CH3); 31
P NMR
(200 MHz, CDCl3): δ 22.34; ESI-MS: m/z 694.25 (M +
H)+. Anal Calcd for C38H32NO10P: C, 65.80; H, 4.65;
N, 2.02. Found: C, 65.83; H, 4.62; N, 2.05%.
Acknowledgements
This study was supported by 973 projects
(No.2011CB512005, 2012CB723501), the National
Natural Science Foundation of China (No. 81260472,
21101035, 21362002), Guangxi Natural Science
Foundation of China (2011GXNSFD018010 and
No.2010GXNSFF013001), Bagui Scholar project and
the Foundation of Ministry of Education Innovation
Team (NO. IRT1225).
References 1 Dong G Q, Wang S Z, Miao Z Y, Yao J Z, Zhang Y Q, Guo
Z Z, Zhang W N & Sheng C Q, J Med Chem, 55, 2012, 7593.
2 Gilroy D W, Prostag Leukotr Ess, 73, 2005, 203.
3 Antonoff M B & D'Cunha J, Semin Thorac Cardiovasc Surg,
22, 2010, 195.
4 Singh S, Jain A, Mishra S K, Singh S & Singh R, Osteoarthr
Cartilage, 20, 2012, S127.
5 Dhaneshwar S, Patel V, Patil D & Meena G, Bioorg Med
Chem Lett, 23, 2013, 55.
6 Bartels E M, Bliddal H, Schøndorff P K, Altman R D, Zhang
W & Christensen R, Osteoarthr Cartilage, 18, 2010, 289.
7 Ramos-Zavala M G, González-Ortiz M, Martínez-Abundis
E, Robles-Cervantes J A, González-López R & Santiago-
Hernández N J, Diabetes Care, 34, 2011, 1591.
8 Huang, X C, Wang M, Pan Y M, Tian X Y, Wang H S &
Zhang Y, Bioorg Med Chem Lett, 23, 2013, 5283.
9 Ye M Y, Yao G Y, Wei J C, Pan Y M, Liao Z X & Wang H
S, Int J Mol Sci, 14, 2013, 9424.
10 Huang, X C, Wang M, Pan Y M, Yao G Y, Wang H S, Tian
X Y, Qin J K & Zhang Y, Eur J Med Chem, 69, 2013, 508.
11 Yao G Y, Ye M Y, Huang R Z, Li Y J, Pan Y M, Xu Q, Liao
Z X & Wang H S, Bioorg Med Chem Lett, 2013, DOI:
10.1016/j.bmcl.2013.12.030.
12 Jin L H, Song B A, Zhang G P, Xu R Q, Zhang S M, Gao X
W, Hu D Y & Yang S, Bioorg Med Chem Lett, 16, 2006,
1537.
13 Naydenova E, Troev K, Topashka-Ancheva M, Hägele G,
Ivanov I & Kril A, Amino Acids, 33, 2007, 695.
14 Naydenova E D, Todorov P T & Troev K D, Amino Acids,
38, 2010, 23.
15 Mucha A, Kafarski P & Berlicki Ł, J Med Chem, 54, 2011,
5955.
16 Hulsman N, Medema J P, Bos C, Jongejan A, Leurs R, Smit
M J, de Esch I J, Richel D & Wijtmans M, J Med Chem, 50,
2007, 2424.
17 Muregi F W & Ishih A, Drug Develop Res, 71, 2010, 20.
18 Metcalfe C & Thomas J A, Chem Soc Rev, 32, 2003, 215.
19 Tan J, Wang B C & Zhu L C, Bioorg Med Chem, 17, 2009,
614.
20 Tang H, Wang X D, Wei Y B, Huang S L, Huang Z S, Tan J
H, An L K, Wu J Y, Chan A S C & Gu L Q, Eur J Med
Chem, 43, 2008, 973.
21 Cashman D J & Kellogg G E, J Med Chem, 47, 2004, 1360.
22 Chaudhuri P, Majumder H K & Bhattacharya S, J Med
Chem, 50, 2007, 2536.
23 Kastan M B & Bartek J, Nature, 432, 2004, 316.
24 Tao Z F & Lin N H, Anticancer Agent Med Chem, 6, 2006,
377.