Embed Size (px)
Citation preview

48 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Unserer Zeit, 2015, 49, 48 – 58
Wasser und Biomoleküle
Ultraschnelle Dynamik vonStrukturen und Schwingungen THOMAS ELSAESSER
DOI: 10.1002/ciuz.201400678www.chiuz.de
Intermolekulare Wasserstoffbrückenbindungen bestimmen physikalische und chemische Eigenschaften desWassers und wässriger Systeme, etwa hydratisierter Biomoleküle. Aufgrund der begrenzten Bindungsstärkeunterliegen wässrige Systeme ultraschnellen strukturellen Fluktuationen im Zeitbereich zwischen ca. 10–14 und10–11 Sekunden. Schwingungsanregungen von Wasserstoffbrücken zerfallen ebenfalls in diesem Zeitbereich.Die Ultrakurzzeit-Infrarotspektroskopie erlaubt eine zeitaufgelöste Beobachtung dieser elementaren Dynamikin Wasser, hydratisierten DNA-Oligomeren und Phospholipiden.
Abb. 1 (a) Schematische Darstellung der Struktur von flüssigem H2O (oben). Die gewinkelten polaren Wassermoleküle (rot: Sauerstoff, grau:Wasserstoff) bilden ein durch intermolekulare Wasserstoffbrücken (gestrichelte Linien) vermitteltes molekulares Netzwerk. Die Struktur desNetzwerks fluktuiert auf einer Zeitskala von 10–12 s. Unten: Tetraedrische Anordnung von Wassermolekülen. Im Mittel bildet jedes Wassermole-kül 4 Wasserstoffbrücken aus. (b) Schema der DNA-Doppelhelixstruktur (oben), bestehend aus wasserstoffverbrückten Basenpaaren und zweiSträngen aus Zucker- und Phosphatgruppen. Unten: Struktur eines Adenin-Thymin-Basenpaars mit eine N-H..O und einer N-H..N Wasserstoff -brücke sowie Zucker- und Phosphatgruppen. (c) Schema einer hydratisierten Phospholipidmembran (oben, aus [6]) und einer inversen Mizelleaus DOPC-Molekülen (unten). Die Mizelle enthält ein Wasser-Nanovolumen. Rechts: Molekulare Struktur des Phospholipids DOPC.

Die überwiegende Zahl biologischer Prozesse läuft inwässriger Umgebung ab, weshalb Wasser eine der we-
sentlichen Grundlagen des Lebens auf der Erde darstellt. Diephysikalischen und chemischen Eigenschaften von Wasserweisen zahlreiche Besonderheiten auf, etwa die Dichte -anomalie oder das komplexe Phasendiagramm [1].
In der flüssigen Phase bilden die gewinkelten, stark po-laren Wassermoleküle ein ausgedehntes Netzwerk, dasdurch intermolekulare Wasserstoffbrücken (H-Brücken) ver-mittelt wird (Abbildung 1a). Im Mittel bildet jedes Wasser-molekül vier Wasserstoffbrücken aus, zwei, bei denen es alsWasserstoffdonor fungiert und zwei, bei denen das elek-tronegative Sauerstoffatom als Akzeptor dient. Die Strukturdieses auf großen Längenskalen ungeordneten Netzwerkesist nicht statisch, sondern auf Grund thermischer Bewe-gungen und des ständigen Brechens und Wiederherstellensder H-Brückenbindungen starken Fluktuationen unterwor-fen. Diese Fluktuationen treten im Ultrakurzzeitbereich vontypisch 1 ps = 10–12 s auf und haben starken Einfluss aufelektrische und optische Eigenschaften von Wasser [2, 3].
Die ultraschnelle strukturelle Dynamik des Wassers undihr Einfluss auf seine molekularen und makroskopischenEigenschaften haben sich in den letzten Jahren zu einemwichtigen Forschungsgegenstand der Physik und der Che-mie entwickelt. Neben Methoden der stationären Struktur-forschung, Kernspinresonanz und Spektroskopie werdenvor allem Verfahren der Ultrakurzzeitphysik eingesetzt, umfluktuierende mikroskopische Strukturen auf ihrer intrinsi-schen Zeitskala zu erfassen und die relevanten molekularenWechselwirkungen quantitativ zu charakterisieren [4]. Die-se experimentellen Arbeiten wurden durch eingehendetheoretische Analysen und molekulardynamische Simula-tionen ergänzt, durch welche für reines Wasser und einfa-che ionische Systeme in wässriger Umgebung ein weitge-hendes Verständnis des molekularen Geschehens erreichtwurde [5]. Ausgehend von diesen Erkenntnissen sind dieWasserhüllen um Biomoleküle, die Hydrathüllen und dieGrenzschichten von Wasser an biologischen MembranenGegenstände aktueller Forschung.
In diesem Artikel werden jüngste Ergebnisse zu Dyna-mik und Eigenschaften von Wasser und hydratisierten Bio-
molekülen im Ultrakurzzeitbereich, d.h. auf der Zeitskalazwischen 10 Femtosekunden (1 fs = 10–15s) und einigen Pi-kosekunden (1 ps =10–12 s), vorgestellt. In den Experimen-ten kamen Methoden der Femtosekunden-Schwingungs-spektroskopie zum Einsatz, bei denen molekulare Oszilla-toren als Sonden einer fluktuierenden Umgebung dienenund eine Erfassung der molekularen Dynamik in ‚Echtzeit‘gestatten [4]. Neben Ergebnissen zu reinem H2O wird vorallem das Verhalten hydratisierter DNA-Oligomere (Abbil-dung 1b) und Phospholipide (Abbildung 1c) diskutiert.
Schwingungsspektroskopie im Femtosekundenbereich – eine Sonde molekularer DynamikSchwingungsspektren wässriger Systeme
Die Schwingungsspektroskopie ist seit dem Beginn der For-schung an Wasserstoffbrücken eine Schlüsselmethode, umlokale molekulare Umgebungen und Wechselwirkungen zuanalysieren. Die Infrarotabsorptionsspektren wasserstoff-verbrückter Moleküle weisen spezifische Charakteristikaauf, die sie von Spektren isolierter Moleküle unterscheiden[7]. So wird die Streckschwingungsfrequenz der OH- oderNH-Donorgruppe in einer Wasserstoffbrücke durch dieWechselwirkung mit dem Akzeptoratom erniedrigt undhängt empfindlich von der H-Brückenstärke und -geome-trie ab. Gleichzeitig führen Veränderungen im Elektronen-
Chem. Unserer Zeit, 2015, 49, 48 – 58 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 49
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
A B B . 2 I N F R A ROT- A B S O R P T I O N S S PE K T R E N
(a) Wasser, (b) Doppelstrang-DNA-Oligomere, die 23 Adenin-Thymin-Basenpaare enthalten, und (c) inverse Phospho lipid-Mizellen, die ein Nanovolumen von Wasser enthalten (16 H2O-Moleküle pro DOPC Molekül). Aufgetragen ist die Infrarotabsorption als Funktion der Wellenzahl ν~ in cm–1
(ν~ = ν/c mit Übergangsfrequenz ν und Vakuumlichtgeschwin-digkeit c). In (a) und (c) sind die breiten Absorptionsbandenzwischen 3000 und 3800 cm–1 auf die OH-Streckschwingungν(OH) des Wassers zurückzuführen. In (b) ist die entsprechen-de breite Bande die überlagerte Absorption von ν(OH) desWassers und der NH-Streckschwingungen ν(NH) der Basen-paare, deren Gesamtstärke vom Wassergehalt der Probe ab-hängt (strichpunktierte Kurve: 2 Wassermoleküle pro Basen-paar, durchgezogene Kurve: mehr als 20 Wassermoleküle proBasenpaar).

system der verbrückten Moleküle zu einer Erhöhung der Absorptionsstärke. Auch die Schwingungen der Wasser-stoffakzeptorgruppen zeigen charakteristische Frequenz-verschiebungen bei der Bildung von H-Brücken. Schließlichtreten neue niederfrequente Schwingungsmoden, die H-Brü -ckenschwingungen auf, die unter anderem den Abstand deräußeren schweren Atome in der H-Brücke verändern.
In Abbildung 2 sind die linearen Infrarotabsorptions-spektren einiger wässriger Systeme zusammengefasst. Auf-getragen ist jeweils die Infrarotabsorption als Funktion derWellenzahl ν̃ in cm–1. Diese Größe hängt mit der Frequenzν des Schwingungsübergangs in folgender Weise zusam-men: ν̃ = ν/c = 1/λ wobei c die Vakuumlichtgeschwindig-keit (in cm/s) und λ die Lichtwellenlänge (in cm) sind.
Das Infrarotspektrum von flüssigem H2O weist zweistarke Absorptionsbanden mit Maxima bei 3400 und 1650 cm–1 auf (Abbildung 2a), die von der intramolekula-ren OH-Streckschwingung ν(OH) und der OH-Biege-schwingung δ(OH) des Wassermoleküls herrühren. Das Ma-ximum der OH-Streckschwingungsbande bei 3400 cm–1
liegt bei einer deutlich niedrigeren Fre-quenz als für isolierte Wassermolekü-le. Diese Rotverschiebung wird durchdie anziehende Wechselwirkung mitdem Sauer stoffatom benachbarterWassermoleküle verursacht (Abbil-dung 1a), welche die Kraftkonstanteder Schwingung erniedrigt.
Die große spektrale Breite der OH-Streckschwingungsbande (volle Halbwertsbreite 270 cm–1)ist zum einen auf eine Verteilung von Schwingungsfre-quenzen in den leicht unterschiedlichen H-Brückengeome-trien innerhalb des molekularen Netzwerks zurückzufüh-ren. Zum anderen führen ultraschnelle Fluktuationen derNetzwerkstruktur zu einem fluktuierenden elektrischenFeld, das von den polaren H2O-Molekülen (Dipolmoment1,85 Debye (D), 1 D = 3,336 × 10–30 C · m) erzeugt wird.Das elektrische Feld induziert Frequenzsprünge der OH-Streckoszillatoren, die sogenannte spektrale Diffusion, undso eine Verbreiterung des zeitlich gemittelten Absorptions-spektrums (Abbildung 2a).
Neben der Absorption der intramolekularen Schwin-gungen treten deutlich schwächere, spektral breite Ab-sorptionsbanden intermolekularer Moden, sogenannte Li-brationen auf, die sich zwischen ca. 400 und 1600 cm–1 er-strecken, so die schwache, nahezu konstante Absorptionunterhalb der Biegeschwingungsbande in Abbildung 2a. Dieschwache Bande um 2140 cm–1 ist eine Kombination nie-derfrequenter Librationen mit der Biegeschwingung.
Biomoleküle in wässriger Umgebung zeigen wesentlichkomplexere Schwingungsspektren als reines Wasser. Ne-ben den Schwingungen des H2O tritt eine Vielzahl von Ab-sorptionsbanden der verschiedenen funktionellen Gruppendes Biomoleküls auf. Dies ist in Abbildung 2b für kurzeDNA-Oligomere [8] gezeigt, die 23 Adenin-Thymin-Basen-paare enthalten. Im Bereich zwischen 2500 und 3600 cm–1
sind breite NH-Streckschwingungsbanden der Adenin-Thy-min-Basenpaare der DNA zu beobachten, die sowohl mitden CH-Streckschwingungsbanden um 3000 cm–1 als auchmit der OH-Streckschwingungsbande der in den Probenenthaltenen Wassermoleküle überlappen. In wasserarmenProben mit ca. 2 Wassermolekülen pro Basenpaar domi-niert zwischen 3000 und 3600 cm–1 die NH-Streckabsorp-tion (strichpunktierte Linie in Abbildung 2b), für vollstän-dig hydratisierte DNA mit mehr als 20 Wassermolekülenpro Basenpaar hingegen die OH-Streckabsorption des Was-sers (durchgezogene Linie).
Ähnlich komplex sind die Infrarotspektren inverserPhospholipidmizellen (Abbildung 2c), die – eingebettet inein unpolares Lösungsmittel – aus einer Phospholipidhülle(hier DOPC, Abbildung 1c) und einem in ihr enthaltenenWasservolumen bestehen [9]. Lichtstreuungsexperimentelassen auf eine nahezu sphärische Form der Mizellen schlie-ßen. Das Infrarotspektrum in Abbildung 2c enthält Beiträ-ge der DOPC-Moleküle, des Wassers und des umgebendenLösungsmittels Benzol, die teilweise spektral überlappen.
An diesen Beispielen wird deutlich,dass eine eindeutige Zuordnung kom-plexer stationärer Schwingungsban-den zu bestimmten molekularen Ein-heiten in der Regel nicht möglich ist.Nichtlineare mehrdimensionale Ver-fahren der Ultrakurzzeit-Infrarotspek-troskopie geben hier wesentlich spe-zifischere Informationen.
UltrakurzzeitmethodenDie zeitaufgelöste Beobachtung der ultraschnellen Dyna-mik wässriger Systeme erfordert experimentelle Methodenmit einer Zeitauflösung besser als 100 fs. Ein grundlegen-des Verfahren ist die Anrege-Abtastmethode, die für Schwin-gungsanregungen in Abbildung 3a schematisch dargestelltist [10].
Ein erster ultrakurzer Infrarotimpuls mit einer Frequenzim Bereich der fundamentalen Schwingungsabsorptions-bande (mittleres Teilbild) regt einen Bruchteil der moleku-laren Oszillatoren vom v = 0 in den v = 1 Zustand des Os-zillators an (blauer Pfeil im linken Teilbild). Wechselwirktdas so präparierte System mit einem zweiten zeitverzöger-ten Abtastimpuls, beobachtet man eine reduzierte Absorp-tion, d.h. ΔA = Atr(tD)-Ast<0, auf dem v = 0→1 Übergang(blaue Kontur im rechten Teilbild) wobei Atr(tD) die mo-mentane Absorption zum Zeitpunkt tD und Ast die stationä-re Absorption vor der Anregung darstellen. Die Abnahmeder Absorption hat zwei Ursachen: Durch den Anregungs-impuls wurden Moleküle aus dem v = 0 Zustand entfernt,d.h. weniger Moleküle tragen zum Absorptionsprozess bei,und angeregte Moleküle im v = 1 Zustand können durchstimulierte Emission eines Lichtquants wieder in den v = 0Zustand zurückkehren. Der zweite Prozess erhöht die Zahlder Lichtquanten im Abtastimpuls und erscheint deshalbals Absorptionsabnahme.
50 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Unserer Zeit, 2015, 49, 48 – 58
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
DIE MOLEKULARE STRUK TUR
VON FLÜSSIGEM WASSER
UNTERLIEGT E X TREM
SCHNELLEN FLUK TUATIONEN
IM FEMTOSEKUNDENBEREICH

Neben diesen Absorptionsänderungen im Bereich derfundamentalen Schwingungsbande tritt eine neue Absorp-tionsbande auf, die durch Übergänge vom v = 1 in den v = 2Zustand des Oszillators verursacht wird (rote Kontur imrechten Teilbild). Diese neue Bande ist aufgrund der An-harmonizität des Oszillators rotverschoben und existiertnur, solange der Zustand v = 1 besetzt ist. Die Stärke dieserBande nimmt mit wachsender Verzögerungszeit tD des Ab-tastimpulses relativ zum Anregungsimpuls ab, da die Oszil-latoren durch Relaxationsprozesse in den Zustand v = 0 zu-rückkehren. Aus Messungen bei verschiedenen tD lässt sichder Besetzungszerfall zeitaufgelöst rekonstruieren und dieLebensdauer des Zustandes v = 1 bestimmen. Verwendetman ein spektral auflösendes Detektionssystem für den Ab-tastimpuls, können außerdem transiente Spektren ΔA(ν) füreine feste Verzögerungszeit tD gemessen werden – wie sche-matisch im rechten Teilbild der Abbildung 3a gezeigt.
Beim Anrege-Abtastverfahren müssen Anrege- und Ab-tastimpuls mit hoher Genauigkeit synchronisiert und ihre
jeweilige Dauer kurz gegen die zu messende Lebensdauersein. Mit modernen Ultrakurzpulslasern und Verfahren deroptischen Frequenzkonversion lassen sich heute Impulsevon weniger als 100 fs Dauer in einem sehr breiten Spek-tralbereich von 500 bis 4000 cm–1 (Wellenlängen von 20 bis2.5 µm) erzeugen, darüber hinaus Impulse im fernen Infra-rot (um 30 cm–1, Wellenlänge 330 µm) mit einer Dauer vonca. 1 ps.
Kopplungen unterschiedlicher Schwingungen und Fre-quenzfluktuationen von Schwingungsübergängen lassensich mit der zweidimensionalen (2D) Infrarotspektroskopiedirekt sichtbar machen (Ref. [11], Schema in Abbildung3b,c). Während in den Infrarotabsorptions- und den Anre-ge-Abtastspektren jeweils eine Frequenzachse relevant ist,wird bei der 2D-Spektroskopie das transiente Schwingungs -verhalten als Funktion zweier Frequenzen, der Anregungs-frequenz ν1 und der Detektionsfrequenz ν3, aufgezeichnet,eng verwandt mit Verfahren der mehrdimensionalen Kern-spinresonanz. Besteht eine stationäre Schwingungsbande
Chem. Unserer Zeit, 2015, 49, 48 – 58 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 51
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
A B B . 3
(a) Anrege-Abtast-Spektroskopie. Die Gleichgewichtsbesetzungdes v = 0 Zustands eines quantenmechanischen Oszillators ver-ursacht Schwingungsabsorption (mittleres Teilbild) resonantzum v = 0→1 Übergang. Ein ultrakurzer Infrarotimpuls regt denOszillator vom v = 0 in den v = 1 Zustand an (blauer Pfeil im lin-ken Teilbild). Dies führt zu einer Absorptionszunahme ΔA > 0 aufdem rotverschobenenen v = 1→2 Übergang und einer reduzier-ten Absorption ΔA < 0 des v = 0→1 Übergangs (rote Pfeile, Diffe-renzspektrum rechts).
(b) Links: Infrarotabsorptionspektrum mit Beiträgen unterschied-licher Schwingungsübergänge. Rechts: Ein nichtlineares zwei -dimensionales (2D) Spektrum weist Signale auf der Diagonalen ν1 = ν3 auf (gleiche Anregungs- und Detektionsfrequenz, farbigeKreise). Gekoppelte Übergänge verursachen Außerdiagonal-Sig-nale (schwarze Kreise), deren Intensität proportional zur Kopp-lungsstärke ist.
(c) Spektrale Diffusion innerhalb der inhomogen verbreitertenOH-Streckschwingungsbande des Wassers. Ein bei νi angeregterOszillator ändert seine Übergangsfrequenz als Funktion der Zeitnach νf. Im 2D-Spektrum führt dies zu einem Übergang von derursprünglichen elliptischen Form entlang ν1 = ν3 für die Warte-zeit T = 0 (Korrelation zwischen ν1 und ν3) zu einer runden Formbei T > 0 (Verlust der Frequenzkorrelation). Als Maß der spektra-len Verformung dienen Center Lines (CL, weiße Linien), die Sig-nalmaxima bei verschiedenen Detektionsfrequenzen ν3 verbin-den. Die Abnahme der Steigung der CL mit T ist ein Maß für denZerfall der Frequenzkorrelation.
(d) Sequenz der Impulse beim 3-Impuls-Photonecho mit der Ko-härenzzeit τ, der Wartezeit T und der (Echt)Zeit t. Eine doppelteFouriertransformation des gemessenen Signals bzgl. τ und t erzeugt die Frequenzachsen ν1 und ν3.

aus zahlreichen Unterkomponenten (Abbildung 3b links),so führt eine Anregung bei einer Frequenz ν1 zu einem Sig-nal bei der identischen Detektionsfrequenz ν1 = ν3 (blauerund roter Kreis im rechten Teilbild), d.h. man erzeugt fürjede Komponente ein Signal auf der Frequenzdiagonalenν1 = ν3 des 2D-Diagramms. Sind die Unterkomponenten aufgekoppelte Oszillatoren zurückzuführen, so erzeugt die An-regung des ersten Oszillators bei ν1 eine Antwort des zwei-ten Oszillators bei dessen (unterschiedlicher) Frequenz ν3
und umgekehrt, d.h. die Signale liegen im 2D-Spektrum au-ßerhalb der Diagonalen (schwarze Kreise). Das Auftretenund die Größe von Außerdiagonalsignalen sind also direk-te Signaturen der Existenz und der Stärke von Schwin-gungskopplungen, die auf diese Weise quantitativ bestimmtwerden können.
Zweidimensionale Infrarotspektroskopie erlaubt auchdie Sichtbarmachung der spektralen Diffusion, d.h. der Fre-quenzsprünge von Oszillatoren, diedurch eine fluktuierende Umgebungverursacht werden. Eine Verteilungvon Schwingungsfrequenzen inner-halb einer Infrarotbande – in Abbil-dung 3c schematisch am Beispiel derOH-Streckbande des H2O gezeigt –wird zum Zeitpunkt T = 0 angeregt. Zudiesem Zeitpunkt trägt jeder Oszilla-tor bei seiner Anregungsfrequenz zum 2D-Spektrum bei,das deshalb entlang der Frequenzdiagonale ν1 = ν3 orien-tiert ist (oberes rechtes Teilbild). Mit wachsender Warte-zeit T treten im Ensemble der Oszillatoren Frequenzsprün-ge auf, d.h. der anfänglich bei ν1 angeregte Oszillator fin-det sich beispielsweise bei νf wieder (linkes Teilbild). DieserEffekt führt über eine Vielzahl von Frequenzsprüngen zu ei-nem totalen Verlust der Korrelation von Anregungs- undDetektionsfrequenz, weshalb das 2D-Spektrum von seinerelongierten Form entlang ν1=ν3 in eine runde homogeneForm übergeht (unteres Teilbild rechts). Letztere enthältkeinerlei Frequenzkorrelationen, das Ensemble hat ‚sein Ge-dächtnis verloren‘. Misst man 2D-Spektren für verschiede-ne Wartezeiten T, so lässt sich dieser Prozess der spektra-len Diffusion zeitaufgelöst verfolgen.
Das in der 2D-Infrarotspektroskopie an einem moleku-laren Ensemble gemessene Signal ist proportional zur fre-quenzabhängigen makroskopischen Polarisation 3. Ordnungim elektrischen Feld der Lichtimpulse [11–13]. Ein 2D-Spek-trum lässt sich sowohl durch geeignet modifizierte Anrege-Abtastmethoden wie auch durch Photonechoverfahren er-zeugen und nachweisen. Die im Folgenden vorgestelltenErgebnisse beruhen auf der Messung von Heterodyn-Pho-tonechos in der Zeitdomäne. Hierbei wechselwirken 3 ul-trakurze Infrarotimpulse sequentiell mit der Probe (Abbil-dung 3d): der erste Impuls erzeugt eine kohärente Polari-sation resonant zum Schwingungsübergang v = 0→1, diedurch Wechselwirkung mit dem zweiten, um die Kohä-renzzeit τ verzögerten Impuls in eine Besetzung des v = 1-Zustandes (Absorption) oder des v = 0-Zustandes (Raman-
Prozess) verwandelt wird. Auf die Wechselwirkung mit demzweiten Impuls folgt die Wartezeit oder Populationszeit T,während der die so ‚markierten‘ Oszillatoren der Wechsel-wirkung mit der Umgebung unterworfen sind und Prozes-se der Populationsrelaxation, z.B. die Entvölkerung desv = 1-Zustandes auftreten. Zum Zeitpunkt T erzeugt der drit-te Impuls erneut eine Polarisation auf dem v = 0→1- oderv = 1→2-Schwingungsübergang, die ein oszillierendes zeit-abhängiges elektrisches Feld (Echtzeit t) in eine ausge-zeichnete, durch die Ausbreitungsrichtung der 3 Impulsebestimmte Raumrichtung abstrahlt. Dieses makroskopischeFeld enthält die Beiträge aller Oszillatoren, die mit den dreiImpulsen wechselgewirkt haben und wird durch eine in-terferometrische Detektionsmethode in Amplitude und (re-lativer) Phase vermessen. Eine Fouriertransformation diesesSignals bezüglich der Kohärenzzeit τ, d.h. des ersten Zeit-intervalls, und der Echtzeit t, des dritten Zeitintervalls, er-
zeugt das 2D-Spektrum mit den Fre-quenzachsen ν1 und ν3 für eine festeWartezeit T. Wiederholt man die Mes-sung für verschiedene T, lässt sich diein Abbildung 3c gezeigte spektrale Dif-fusion erfassen. Die Zeitauflösung derExperimente beträgt typischerweise100 fs.
Ultraschnelle Dynamik wässriger SystemeStrukturelle Fluktuationen und Schwingungs -anregungen in reinem H2O
An reinem H2O wurden umfangreiche experimentelle Ul-trakurzzeitstudien durchgeführt und mit Hilfe theoretischerBerechnungen und moleküldynamischer Simulationen imDetail analysiert [5]. Ein erster Befund sind die extrem kurzen Lebensdauern der OH-Streckschwingung ν(OH) von 200 fs [14] und der OH-Biegeschwingung δ(OH) von170 fs [15]. Wie im Niveauschema der Abbildung 2a ge-zeigt, liegt der Zustand v = 1 der OH-Streckschwingung beinahezu der gleichen Energie wie der Zustand v = 2 der Bie-geschwingung. Diese Zustände sind im anharmonischenSchwingungspotential stark gekoppelt, sie bilden in einerFermiresonanz Mischzustände des Streck- und des Biegeos-zillators aus. Nach Anregung des Übergangs v = 0→1 desOH-Streckoszillators ν(OH) relaxieren diese Mischzuständeüber den OH-Biegeoszillator δ(OH), d.h. ein erster Relaxa-tionsschritt erfolgt zum v = 1-Zustand der Biegeschwingung,ein zweiter von dort in ihren v = 0-Zustand.
Die aus der OH-Streckanregung stammende und bei derRelaxation in zwei Schritten freiwerdende Überschuss-energie wird auf die Eigenrotation der angeregten Wasser-moleküle und niederfrequente gehinderte Rotationen derUmgebung, sogenannte Librationen übertragen [16, 17].Auf einer Zeitskala von 1 bis 2 ps wird die Überschuss-energie im molekularen Netzwerk (Abbildung 1a) umver-teilt und räumlich delokalisiert, es bildet sich ein ‚heißer‘Grundzustand aus, der durch eine leicht erhöhte Schwin-gungstemperatur gekennzeichnet ist. Das aufgeheizte Pro-
52 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Unserer Zeit, 2015, 49, 48 – 58
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
ENTGEGEN VERBREITETEN
VORSTELLUNGEN VON EINEM
‚GEDÄCHTNIS DES WASSERS‘
IST DIESE FLÜSSIGKEIT
HÖCHST VERGESSLICH
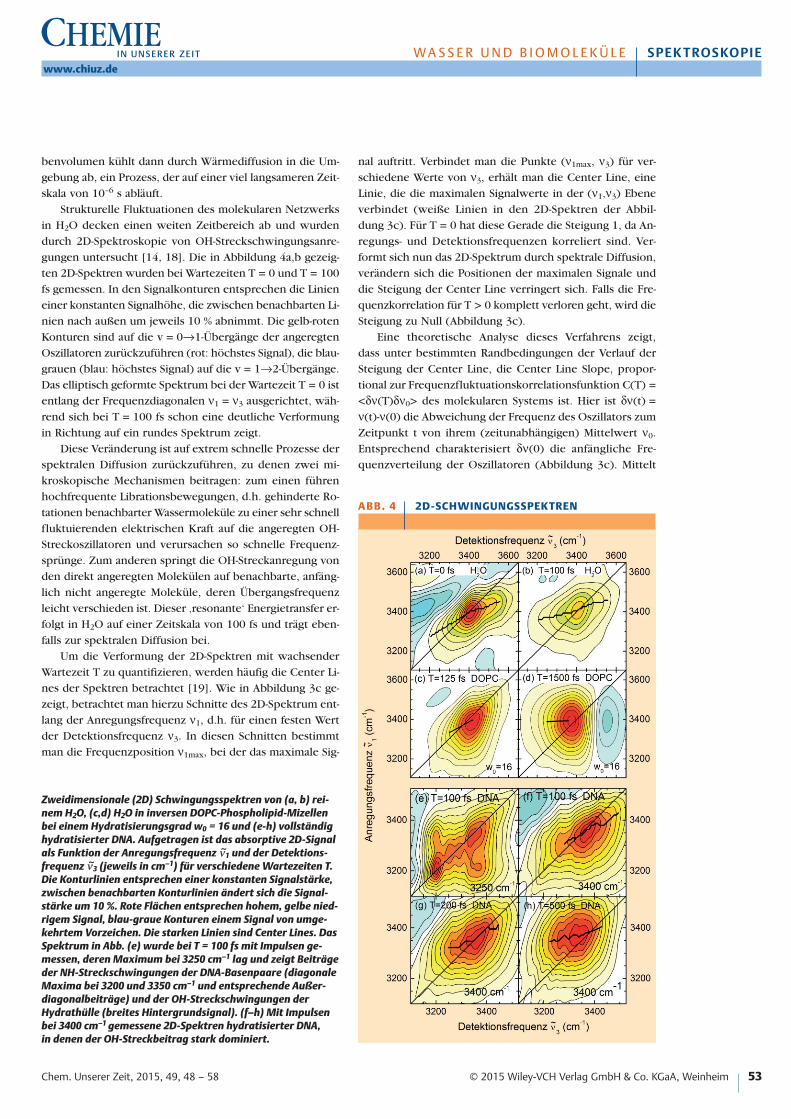
benvolumen kühlt dann durch Wärmediffusion in die Um-gebung ab, ein Prozess, der auf einer viel langsameren Zeit-skala von 10–6 s abläuft.
Strukturelle Fluktuationen des molekularen Netzwerksin H2O decken einen weiten Zeitbereich ab und wurdendurch 2D-Spektroskopie von OH-Streckschwingungsanre-gungen untersucht [14, 18]. Die in Abbildung 4a,b gezeig-ten 2D-Spektren wurden bei Wartezeiten T = 0 und T = 100fs gemessen. In den Signalkonturen entsprechen die Linieneiner konstanten Signalhöhe, die zwischen benachbarten Li-nien nach außen um jeweils 10 % abnimmt. Die gelb-rotenKonturen sind auf die v = 0→1-Übergänge der angeregtenOszillatoren zurückzuführen (rot: höchstes Signal), die blau-grauen (blau: höchstes Signal) auf die v = 1→2-Übergänge.Das elliptisch geformte Spektrum bei der Wartezeit T = 0 istentlang der Frequenzdiagonalen ν1 = ν3 ausgerichtet, wäh-rend sich bei T = 100 fs schon eine deutliche Verformungin Richtung auf ein rundes Spektrum zeigt.
Diese Veränderung ist auf extrem schnelle Prozesse derspektralen Diffusion zurückzuführen, zu denen zwei mi-kroskopische Mechanismen beitragen: zum einen führenhochfrequente Librationsbewegungen, d.h. gehinderte Ro-tationen benachbarter Wassermoleküle zu einer sehr schnellfluktuierenden elektrischen Kraft auf die angeregten OH-Streckoszillatoren und verursachen so schnelle Frequenz-sprünge. Zum anderen springt die OH-Streckanregung vonden direkt angeregten Molekülen auf benachbarte, anfäng-lich nicht angeregte Moleküle, deren Übergangsfrequenzleicht verschieden ist. Dieser ‚resonante‘ Energietransfer er-folgt in H2O auf einer Zeitskala von 100 fs und trägt eben-falls zur spektralen Diffusion bei.
Um die Verformung der 2D-Spektren mit wachsenderWartezeit T zu quantifizieren, werden häufig die Center Li-nes der Spektren betrachtet [19]. Wie in Abbildung 3c ge-zeigt, betrachtet man hierzu Schnitte des 2D-Spektrum ent-lang der Anregungsfrequenz ν1, d.h. für einen festen Wertder Detektionsfrequenz ν3. In diesen Schnitten bestimmtman die Frequenzposition ν1max, bei der das maximale Sig-
nal auftritt. Verbindet man die Punkte (ν1max, ν3) für ver-schiedene Werte von ν3, erhält man die Center Line, eineLinie, die die maximalen Signalwerte in der (ν1,ν3) Ebeneverbindet (weiße Linien in den 2D-Spektren der Abbil-dung 3c). Für T = 0 hat diese Gerade die Steigung 1, da An-regungs- und Detektionsfrequenzen korreliert sind. Ver-formt sich nun das 2D-Spektrum durch spektrale Diffusion,verändern sich die Positionen der maximalen Signale unddie Steigung der Center Line verringert sich. Falls die Fre-quenzkorrelation für T > 0 komplett verloren geht, wird dieSteigung zu Null (Abbildung 3c).
Eine theoretische Analyse dieses Verfahrens zeigt, dass unter bestimmten Randbedingungen der Verlauf derSteigung der Center Line, die Center Line Slope, propor-tional zur Frequenzfluktuationskorrelationsfunktion C(T) =<δν(T)δν0> des molekularen Systems ist. Hier ist δν(t) =ν(t)-ν(0) die Abweichung der Frequenz des Oszillators zumZeitpunkt t von ihrem (zeitunabhängigen) Mittelwert ν0.Entsprechend charakterisiert δν(0) die anfängliche Fre-quenzverteilung der Oszillatoren (Abbildung 3c). Mittelt
Chem. Unserer Zeit, 2015, 49, 48 – 58 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 53
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
A B B . 4 2D-SCHWINGUNGSSPEKTREN
Zweidimensionale (2D) Schwingungsspektren von (a, b) rei-nem H2O, (c,d) H2O in inversen DOPC-Phospholipid-Mizellenbei einem Hydratisierungsgrad w0 = 16 und (e-h) vollständighydratisierter DNA. Aufgetragen ist das absorptive 2D-Signalals Funktion der Anregungsfrequenz ν~1 und der Detektions-frequenz ν~3 (jeweils in cm–1) für verschiedene Wartezeiten T.Die Konturlinien entsprechen einer konstanten Signalstärke,zwischen benachbarten Konturlinien ändert sich die Signal-stärke um 10 %. Rote Flächen entsprechen hohem, gelbe nied-rigem Signal, blau-graue Konturen einem Signal von umge-kehrtem Vorzeichen. Die starken Linien sind Center Lines. DasSpektrum in Abb. (e) wurde bei T = 100 fs mit Impulsen ge-messen, deren Maximum bei 3250 cm–1 lag und zeigt Beiträgeder NH-Streckschwingungen der DNA-Basenpaare (diagonaleMaxima bei 3200 und 3350 cm–1 und entsprechende Außer-diagonalbeiträge) und der OH-Streckschwingungen der Hydrathülle (breites Hintergrundsignal). (f–h) Mit Impulsenbei 3400 cm–1 gemessene 2D-Spektren hydratisierter DNA, in denen der OH-Streckbeitrag stark dominiert.
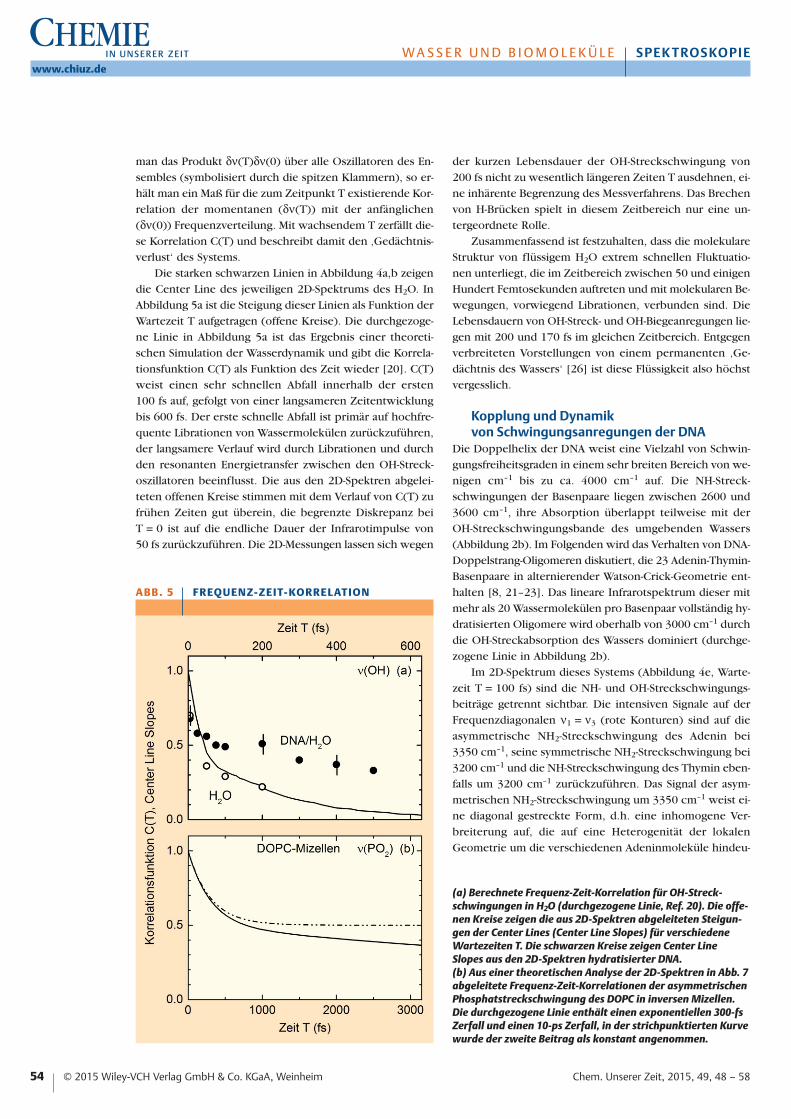
man das Produkt δν(T)δν(0) über alle Oszillatoren des En-sembles (symbolisiert durch die spitzen Klammern), so er-hält man ein Maß für die zum Zeitpunkt T existierende Kor-relation der momentanen (δν(T)) mit der anfänglichen(δν(0)) Frequenzverteilung. Mit wachsendem T zerfällt die-se Korrelation C(T) und beschreibt damit den ‚Gedächtnis-verlust‘ des Systems.
Die starken schwarzen Linien in Abbildung 4a,b zeigendie Center Line des jeweiligen 2D-Spektrums des H2O. InAbbildung 5a ist die Steigung dieser Linien als Funktion derWartezeit T aufgetragen (offene Kreise). Die durchgezoge-ne Linie in Abbildung 5a ist das Ergebnis einer theoreti-schen Simulation der Wasserdynamik und gibt die Korrela-tionsfunktion C(T) als Funktion des Zeit wieder [20]. C(T)weist einen sehr schnellen Abfall innerhalb der ersten 100 fs auf, gefolgt von einer langsameren Zeitentwicklungbis 600 fs. Der erste schnelle Abfall ist primär auf hochfre-quente Librationen von Wassermolekülen zurückzuführen,der langsamere Verlauf wird durch Librationen und durchden resonanten Energietransfer zwischen den OH-Streck-oszillatoren beeinflusst. Die aus den 2D-Spektren abgelei-teten offenen Kreise stimmen mit dem Verlauf von C(T) zufrühen Zeiten gut überein, die begrenzte Diskrepanz bei T = 0 ist auf die endliche Dauer der Infrarotimpulse von 50 fs zurückzuführen. Die 2D-Messungen lassen sich wegen
der kurzen Lebensdauer der OH-Streckschwingung von 200 fs nicht zu wesentlich längeren Zeiten T ausdehnen, ei-ne inhärente Begrenzung des Messverfahrens. Das Brechenvon H-Brücken spielt in diesem Zeitbereich nur eine un-tergeordnete Rolle.
Zusammenfassend ist festzuhalten, dass die molekulareStruktur von flüssigem H2O extrem schnellen Fluktuatio-nen unterliegt, die im Zeitbereich zwischen 50 und einigenHundert Femtosekunden auftreten und mit molekularen Be-wegungen, vorwiegend Librationen, verbunden sind. DieLebensdauern von OH-Streck- und OH-Biegeanregungen lie-gen mit 200 und 170 fs im gleichen Zeitbereich. Entgegenverbreiteten Vorstellungen von einem permanenten ‚Ge-dächtnis des Wassers‘ [26] ist diese Flüssigkeit also höchstvergesslich.
Kopplung und Dynamik von Schwingungsanregungen der DNA
Die Doppelhelix der DNA weist eine Vielzahl von Schwin-gungsfreiheitsgraden in einem sehr breiten Bereich von we-nigen cm–1 bis zu ca. 4000 cm–1 auf. Die NH-Streck-schwingungen der Basenpaare liegen zwischen 2600 und3600 cm–1, ihre Absorption überlappt teilweise mit der OH-Streckschwingungsbande des umgebenden Wassers(Abbildung 2b). Im Folgenden wird das Verhalten von DNA-Doppelstrang-Oligomeren diskutiert, die 23 Adenin-Thymin-Basenpaare in alternierender Watson-Crick-Geometrie ent-halten [8, 21–23]. Das lineare Infrarotspektrum dieser mitmehr als 20 Wassermolekülen pro Basenpaar vollständig hy-dratisierten Oligomere wird oberhalb von 3000 cm–1 durchdie OH-Streckabsorption des Wassers dominiert (durchge-zogene Linie in Abbildung 2b).
Im 2D-Spektrum dieses Systems (Abbildung 4e, Warte-zeit T = 100 fs) sind die NH- und OH-Streckschwingungs-beiträge getrennt sichtbar. Die intensiven Signale auf derFrequenzdiagonalen ν1 = ν3 (rote Konturen) sind auf dieasymmetrische NH2-Streckschwingung des Adenin bei 3350 cm–1, seine symmetrische NH2-Streckschwingung bei3200 cm–1 und die NH-Streckschwingung des Thymin eben-falls um 3200 cm–1 zurückzuführen. Das Signal der asym-metrischen NH2-Streckschwingung um 3350 cm–1 weist ei-ne diagonal gestreckte Form, d.h. eine inhomogene Ver-breiterung auf, die auf eine Heterogenität der lokalenGeometrie um die verschiedenen Adeninmoleküle hindeu-
54 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Unserer Zeit, 2015, 49, 48 – 58
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
A B B . 5 F R EQ U E N Z- Z E I T- KO R R E L AT I O N
(a) Berechnete Frequenz-Zeit-Korrelation für OH-Streck-schwingungen in H2O (durchgezogene Linie, Ref. 20). Die offe-nen Kreise zeigen die aus 2D-Spektren abgeleiteten Steigun-gen der Center Lines (Center Line Slopes) für verschiedeneWartezeiten T. Die schwarzen Kreise zeigen Center Line Slopes aus den 2D-Spektren hydratisierter DNA. (b) Aus einer theoretischen Analyse der 2D-Spektren in Abb. 7abgeleitete Frequenz-Zeit-Korrelationen der asymmetrischenPhosphatstreckschwingung des DOPC in inversen Mizellen.Die durchgezogene Linie enthält einen exponentiellen 300-fsZerfall und einen 10-ps Zerfall, in der strichpunktierten Kurvewurde der zweite Beitrag als konstant angenommen.

tet. Neben den Diagonalsignalen treten außerhalb der Dia-gonalen Crosspeaks bei (ν̃1, ν̃3) = (3350, 3200) cm–1 und(3200, 3350) cm–1 auf. Diese sind auf die Kopplung zwi-schen den unterschiedlichen NH-Streckschwingungen zu-rückzuführen. Eine genauere Analyse zeigt, das die Intensi-tät des Signals bei (3350, 3200) cm–1 als Funktion der War-tezeit T zunimmt, die Anregung der NH2-Streckschwingungbei 3350 cm–1 wird partiell auf die beiden niederfrequen-ten NH-Streckschwingungen bei 3200 cm–1 übertragen. An-sonsten treten im 2D-Spektrum als Funktion der Wartezeitkeine signifikanten Veränderungen der 2D-Signalformen auf,insbesondere keine spektrale Diffusion.
Vergleichende Messungen wurden bei einem sehr nied-rigen Wassergehalt von ca. 2 H2O-Molekülen pro Basenpaardurchgeführt. Das lineare Infrarotabsorptionsspektrum die-ser Proben ist als strichpunktierte Linie in Abbildung 2b ge-zeigt. 2D-Spektren, die hier nicht im einzelnen diskutiertwerden, zeigen NH-Streckschwingungsbeiträge, die mit de-nen in Abbildung 4e nahezu übereinstimmen. Insbesonde-re sind die spektralen Breiten der NH-Streckbeiträge nur geringfügig vomWassergehalt der Proben abhängig.Dies zeigt, dass die Hydrathülle auchder vollständig hydratisierten DNA nurbegrenzten Einfluss auf die Schwin-gungsdynamik der Basenpaare und die2D-Linienformen der NH-Schwingun-gen hat. Letztere sind primär durch dieKopplungen innerhalb des Basenpaa-res und Wasserstoffbrücken zwischen Adenin und Thyminbestimmt.
Die spektral breitbandigen Beiträge im 2D-Spektrum(gelbe Konturen in Abbildung 4e) stammen von OH-Streck-anregungen der Hydrathülle um die DNA-Oligomere. Ihregenaue Form lässt sich wegen der überlagerten NH-Streck-schwingungssignale nicht extrahieren. Deshalb wurden ana-loge Messungen mit Impulsen ausgeführt, deren spektralesMaximum bei 3400 cm–1 mit dem Maximum der OH-Streck-bande zusammenfällt. In den so gemessenen 2D-Spektren(Abbildung 4f-h) dominiert das OH-Strecksignal so stark ge-genüber den NH-Streckbeiträgen, dass die zeitliche Ent-wicklung der spektralen Einhüllenden sichtbar wird. DieEinhüllende geht von einer elliptischen, parallel zur Diago-nalen ν1 = ν3 ausgerichteten Form bei T = 100 fs in eine na-hezu runde Form bei T = 500 fs über, ein klares Kennzei-chen der spektralen Diffusion in der Hydrathülle. Im Ver-gleich zum reinen H2O ist die spektrale Diffusion jedochsignifikant verlangsamt, wie auch der direkte Vergleich mitden Spektren in Abbildung 4a,b zeigt. Letztere erreichen ei-ne runde Form bereits innerhalb der ersten 100 fs. Die Ver-langsamung der spektralen Diffusion wird auch in der zeit-lichen Entwicklung der Center Line Slopes sichtbar, die inAbbildung 5a als ausgefüllte Kreise gezeigt sind. Im Ver-gleich zu H2O ist der anfängliche schnelle Zerfall wesent-lich schwächer ausgeprägt, der Zeitverlauf wird durch ei-nen Zerfall auf einer Zeitskala von ca. 500 fs dominiert.
In der Messung der 2D-Spektren der OH-Streckschwin-gung wird räumlich über alle Wassergeometrien gemittelt.Diese Mittelung schließt Wassermoleküle ein, die direkt mitfunktionellen Gruppen der DNA wechselwirken, d.h. mitden Phosphat- und Zuckergruppen in den DNA-Strängenund mit NH-Gruppen der Basenpaare. Auf Grund der star-ken lokalen Wechselwirkungen – die Wasserstoffbrückenzwischen H2O und Phosphatgruppen sind stärker als H2O-H2O-Brücken – und der sterischen Randbedingungen in denFurchen der DNA-Oberfläche sind strukturelle Fluktuatio-nen der ersten Wasserschicht um die DNA eingeschränkt,was zu einer Verlangsamung der spektralen Diffusion führt.Die äußeren Wasserschichten verhalten sich hingegen ähn-lich reinem H2O und weisen schnelle Fluktuationen auf.Die Wasserkonzentration der DNA-Proben ist mit ca. 7 Mdeutlich geringer als die von reinem H2O (56M). Dies führtzu einer signifikanten Reduktion des resonanten Energie-transfers zwischen OH-Streckoszillatoren benachbarter Was-sermoleküle. Reduzierter Energietransfer ist ebenfalls mit ei-
ner Verlangsamung der spektralen Dif-fusion verbunden.
Diese Mechanismen führen zudem in Abbildung 5a gezeigten ver-langsamten Zerfall der Frequenzkorre-lationsfunktion des DNA/H2O-Systems.Gegenüber dem reinen H2O (offeneKreise und durchgezogene Kurve) istdie Verlangsamung jedoch moderat,die Korrelation nimmt immer noch
deutlich im gezeigten Zeitbereich ab. Damit ist klar, dass inder Literatur geäußerte Spekulationen einer um mehrereGrößenordnung in der Zeit verlangsamten Dynamik der Hy-drathülle, des ‚biologischen Wassers‘, im Vergleich zu rei-nem H2O jeder Grundlage entbehren.
Hydratationsprozesse in PhospholipidenAls Sonden ultraschneller struktureller Dynamik dienten inden bisher vorgestellten Untersuchungen Schwingungsan-regungen von Wassermolekülen. Ein Nachteil dieses Ansat-zes ist die fehlende räumliche Selektivität, d.h. Wassermo-leküle an der Grenzfläche eines Biomoleküls können nichtvon Molekülen im Wasservolumen unterschieden werden.Um die Dynamik an der Grenzfläche genauer zu charakte-risieren, sind molekulare Oszillatoren erforderlich, die sichin definierten Positionen an oder in der Grenzfläche befin-den, ein Konzept, das kürzlich erstmals in hydratisiertenPhospholipidsystemen verwirklicht wurde. Einige Ergeb-nisse werden im Folgenden vorgestellt [9, 24, 25].
Als Sonden der Grenzflächendynamik dienen Schwin-gungsanregungen der Phosphatgruppen, die sich an derOberfläche von Phospholipid-Membranen befinden. Vonden vier Sauerstoffatomen der PO4-Gruppen sind zwei indie Phospholipidstruktur integriert, während die zwei frei-en Sauerstoffatome direkt mit der wässrigen Umgebungwechselwirken. Das Schwingungsspektrum dieser Strukturenthält PO2-Streckschwingungen, und zwar eine symme-
Chem. Unserer Zeit, 2015, 49, 48 – 58 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 55
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
EINE ANGEBLICH STARK
VERLANGSAMTE DYNAMIK
DER HYDRATHÜLLE DES
‚BIOLOGISCHEN WASSERS‘
IM VERGLEICH ZU REINEM H 2O
ENTBEHRT JEDER GRUNDLAGE
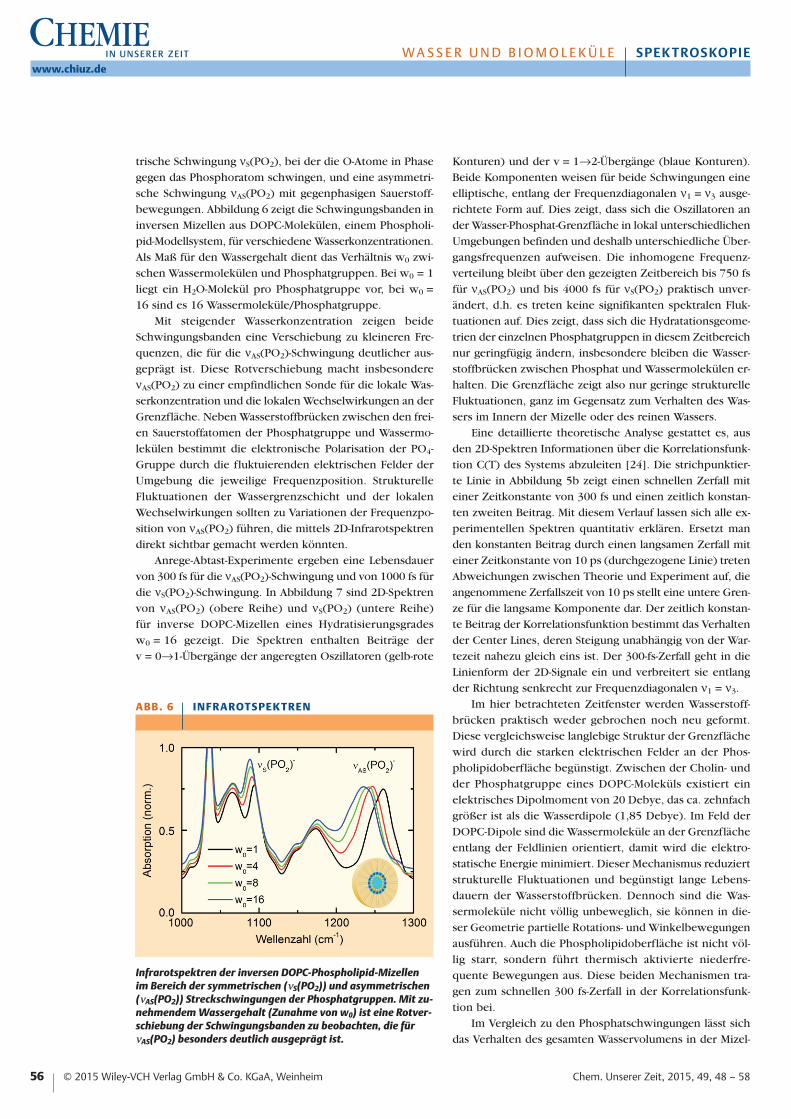
trische Schwingung νS(PO2), bei der die O-Atome in Phasegegen das Phosphoratom schwingen, und eine asymmetri-sche Schwingung νAS(PO2) mit gegenphasigen Sauerstoff-bewegungen. Abbildung 6 zeigt die Schwingungsbanden ininversen Mizellen aus DOPC-Molekülen, einem Phospholi-pid-Modellsystem, für verschiedene Wasserkonzentrationen.Als Maß für den Wassergehalt dient das Verhältnis w0 zwi-schen Wassermolekülen und Phosphatgruppen. Bei w0 = 1liegt ein H2O-Molekül pro Phosphatgruppe vor, bei w0 =16 sind es 16 Wassermoleküle/Phosphatgruppe.
Mit steigender Wasserkonzentration zeigen beideSchwingungsbanden eine Verschiebung zu kleineren Fre-quenzen, die für die νAS(PO2)-Schwingung deutlicher aus-geprägt ist. Diese Rotverschiebung macht insbesondereνAS(PO2) zu einer empfindlichen Sonde für die lokale Was-serkonzentration und die lokalen Wechselwirkungen an derGrenzfläche. Neben Wasserstoffbrücken zwischen den frei-en Sauerstoffatomen der Phosphatgruppe und Wassermo-lekülen bestimmt die elektronische Polarisation der PO4-Gruppe durch die fluktuierenden elektrischen Felder derUmgebung die jeweilige Frequenzposition. StrukturelleFluktuationen der Wassergrenzschicht und der lokalenWechselwirkungen sollten zu Variationen der Frequenzpo-sition von νAS(PO2) führen, die mittels 2D-Infrarotspektrendirekt sichtbar gemacht werden könnten.
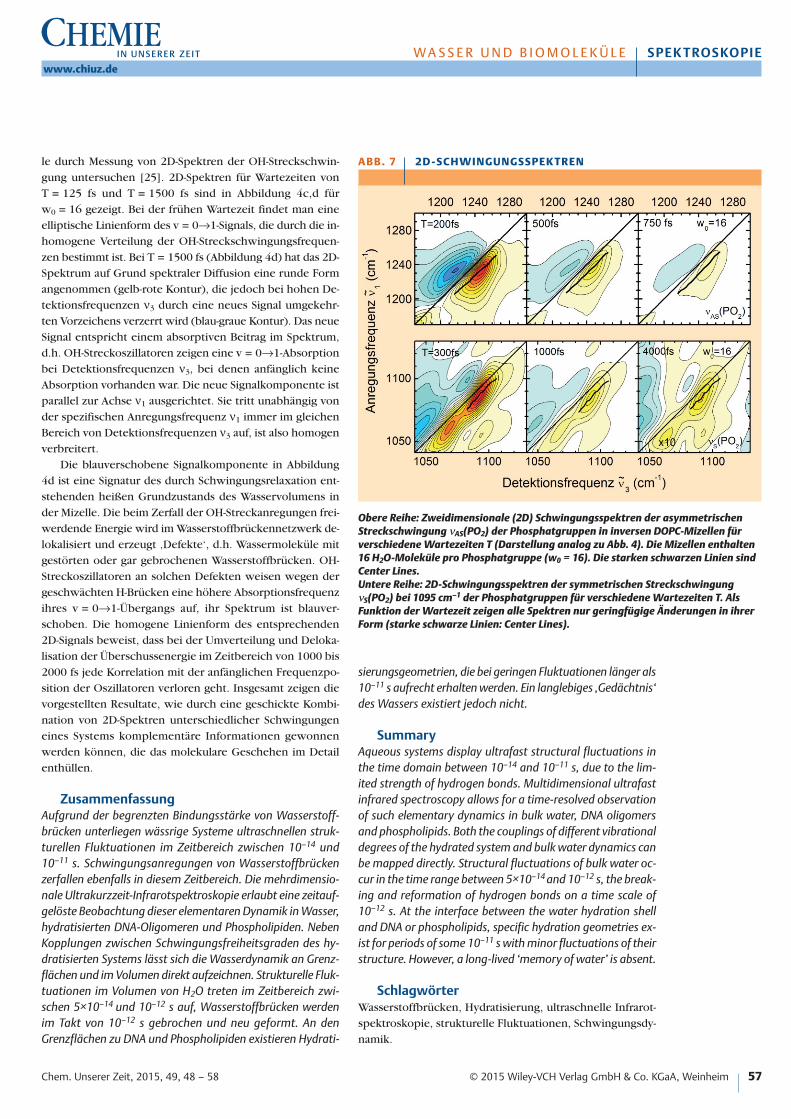
Anrege-Abtast-Experimente ergeben eine Lebensdauervon 300 fs für die νAS(PO2)-Schwingung und von 1000 fs fürdie νS(PO2)-Schwingung. In Abbildung 7 sind 2D-Spektrenvon νAS(PO2) (obere Reihe) und νS(PO2) (untere Reihe) für inverse DOPC-Mizellen eines Hydratisierungsgrades w0 = 16 gezeigt. Die Spektren enthalten Beiträge der v = 0→1-Übergänge der angeregten Oszillatoren (gelb-rote
Konturen) und der v = 1→2-Übergänge (blaue Konturen).Beide Komponenten weisen für beide Schwingungen eineelliptische, entlang der Frequenzdiagonalen ν1 = ν3 ausge-richtete Form auf. Dies zeigt, dass sich die Oszillatoren ander Wasser-Phosphat-Grenzfläche in lokal unterschiedlichenUmgebungen befinden und deshalb unterschiedliche Über-gangsfrequenzen aufweisen. Die inhomogene Frequenz-verteilung bleibt über den gezeigten Zeitbereich bis 750 fsfür νAS(PO2) und bis 4000 fs für νS(PO2) praktisch unver-ändert, d.h. es treten keine signifikanten spektralen Fluk-tuationen auf. Dies zeigt, dass sich die Hydratationsgeome-trien der einzelnen Phosphatgruppen in diesem Zeitbereichnur geringfügig ändern, insbesondere bleiben die Wasser-stoffbrücken zwischen Phosphat und Wassermolekülen er-halten. Die Grenzfläche zeigt also nur geringe strukturelleFluktuationen, ganz im Gegensatz zum Verhalten des Was-sers im Innern der Mizelle oder des reinen Wassers.
Eine detaillierte theoretische Analyse gestattet es, ausden 2D-Spektren Informationen über die Korrelationsfunk-tion C(T) des Systems abzuleiten [24]. Die strichpunktier-te Linie in Abbildung 5b zeigt einen schnellen Zerfall miteiner Zeitkonstante von 300 fs und einen zeitlich konstan-ten zweiten Beitrag. Mit diesem Verlauf lassen sich alle ex-perimentellen Spektren quantitativ erklären. Ersetzt manden konstanten Beitrag durch einen langsamen Zerfall miteiner Zeitkonstante von 10 ps (durchgezogene Linie) tretenAbweichungen zwischen Theorie und Experiment auf, dieangenommene Zerfallszeit von 10 ps stellt eine untere Gren-ze für die langsame Komponente dar. Der zeitlich konstan-te Beitrag der Korrelationsfunktion bestimmt das Verhaltender Center Lines, deren Steigung unabhängig von der War-tezeit nahezu gleich eins ist. Der 300-fs-Zerfall geht in dieLinienform der 2D-Signale ein und verbreitert sie entlangder Richtung senkrecht zur Frequenzdiagonalen ν1 = ν3.
Im hier betrachteten Zeitfenster werden Wasserstoff-brücken praktisch weder gebrochen noch neu geformt.Diese vergleichsweise langlebige Struktur der Grenzflächewird durch die starken elektrischen Felder an der Phos-pholipidoberfläche begünstigt. Zwischen der Cholin- undder Phosphatgruppe eines DOPC-Moleküls existiert einelektrisches Dipolmoment von 20 Debye, das ca. zehnfachgrößer ist als die Wasserdipole (1,85 Debye). Im Feld derDOPC-Dipole sind die Wassermoleküle an der Grenzflächeentlang der Feldlinien orientiert, damit wird die elektro-statische Energie minimiert. Dieser Mechanismus reduziertstrukturelle Fluktuationen und begünstigt lange Lebens-dauern der Wasserstoffbrücken. Dennoch sind die Was-sermoleküle nicht völlig unbeweglich, sie können in die-ser Geometrie partielle Rotations- und Winkelbewegungenausführen. Auch die Phospholipidoberfläche ist nicht völ-lig starr, sondern führt thermisch aktivierte niederfre-quente Bewegungen aus. Diese beiden Mechanismen tra-gen zum schnellen 300 fs-Zerfall in der Korrelationsfunk-tion bei.
Im Vergleich zu den Phosphatschwingungen lässt sichdas Verhalten des gesamten Wasservolumens in der Mizel-
56 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Unserer Zeit, 2015, 49, 48 – 58
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
A B B . 6 I N F R A ROT S PE K T R E N
Infrarotspektren der inversen DOPC-Phospholipid-Mizellenim Bereich der symmetrischen (νS(PO2)) und asymmetrischen(νAS(PO2)) Streckschwingungen der Phosphatgruppen. Mit zu-nehmendem Wassergehalt (Zunahme von w0) ist eine Rotver-schiebung der Schwingungsbanden zu beobachten, die fürνAS(PO2) besonders deutlich ausgeprägt ist.
le durch Messung von 2D-Spektren der OH-Streckschwin-gung untersuchen [25]. 2D-Spektren für Wartezeiten von T = 125 fs und T = 1500 fs sind in Abbildung 4c,d für w0 = 16 gezeigt. Bei der frühen Wartezeit findet man eineelliptische Linienform des v = 0→1-Signals, die durch die in-homogene Verteilung der OH-Streckschwingungsfrequen-zen bestimmt ist. Bei T = 1500 fs (Abbildung 4d) hat das 2D-Spektrum auf Grund spektraler Diffusion eine runde Formangenommen (gelb-rote Kontur), die jedoch bei hohen De-tektionsfrequenzen ν3 durch eine neues Signal umgekehr-ten Vorzeichens verzerrt wird (blau-graue Kontur). Das neueSignal entspricht einem absorptiven Beitrag im Spektrum,d.h. OH-Streckoszillatoren zeigen eine v = 0→1-Absorptionbei Detektionsfrequenzen ν3, bei denen anfänglich keineAbsorption vorhanden war. Die neue Signalkomponente istparallel zur Achse ν1 ausgerichtet. Sie tritt unabhängig vonder spezifischen Anregungsfrequenz ν1 immer im gleichenBereich von Detektionsfrequenzen ν3 auf, ist also homogenverbreitert.
Die blauverschobene Signalkomponente in Abbildung4d ist eine Signatur des durch Schwingungsrelaxation ent-stehenden heißen Grundzustands des Wasservolumens inder Mizelle. Die beim Zerfall der OH-Streckanregungen frei-werdende Energie wird im Wasserstoffbrückennetzwerk de-lokalisiert und erzeugt ‚Defekte‘, d.h. Wassermoleküle mitgestörten oder gar gebrochenen Wasserstoffbrücken. OH-Streckoszillatoren an solchen Defekten weisen wegen dergeschwächten H-Brücken eine höhere Absorptionsfrequenzihres v = 0→1-Übergangs auf, ihr Spektrum ist blauver-schoben. Die homogene Linienform des entsprechenden2D-Signals beweist, dass bei der Umverteilung und Deloka-lisation der Überschussenergie im Zeitbereich von 1000 bis2000 fs jede Korrelation mit der anfänglichen Frequenzpo-sition der Oszillatoren verloren geht. Insgesamt zeigen dievorgestellten Resultate, wie durch eine geschickte Kombi-nation von 2D-Spektren unterschiedlicher Schwingungeneines Systems komplementäre Informationen gewonnenwerden können, die das molekulare Geschehen im Detailenthüllen.
ZusammenfassungAufgrund der begrenzten Bindungsstärke von Wasserstoff-brücken unterliegen wässrige Systeme ultraschnellen struk-turellen Fluktuationen im Zeitbereich zwischen 10–14 und10–11 s. Schwingungsanregungen von Wasserstoffbrückenzerfallen ebenfalls in diesem Zeitbereich. Die mehrdimensio-nale Ultrakurzzeit-Infrarotspektroskopie erlaubt eine zeitauf-gelöste Beobachtung dieser elementaren Dynamik in Wasser,hydratisierten DNA-Oligomeren und Phospholipiden. NebenKopplungen zwischen Schwingungsfreiheitsgraden des hy-dratisierten Systems lässt sich die Wasserdynamik an Grenz-flächen und im Volumen direkt aufzeichnen. Strukturelle Fluk-tuationen im Volumen von H2O treten im Zeitbereich zwi-schen 5×10–14 und 10–12 s auf, Wasserstoffbrücken werdenim Takt von 10–12 s gebrochen und neu geformt. An denGrenzflächen zu DNA und Phospholipiden existieren Hydrati-
sierungsgeometrien, die bei geringen Fluktuationen länger als10–11 s aufrecht erhalten werden. Ein langlebiges ‚Gedächtnis‘des Wassers existiert jedoch nicht.
Summary Aqueous systems display ultrafast structural fluctuations inthe time domain between 10–14 and 10–11 s, due to the lim-ited strength of hydrogen bonds. Multidimensional ultrafastinfrared spectroscopy allows for a time-resolved observationof such elementary dynamics in bulk water, DNA oligomersand phospholipids. Both the couplings of different vibrationaldegrees of the hydrated system and bulk water dynamics canbe mapped directly. Structural fluctuations of bulk water oc-cur in the time range between 5×10–14 and 10–12 s, the break-ing and reformation of hydrogen bonds on a time scale of10–12 s. At the interface between the water hydration shelland DNA or phospholipids, specific hydration geometries ex-ist for periods of some 10–11 s with minor fluctuations of theirstructure. However, a long-lived ‘memory of water’ is absent.
Schlagwörter Wasserstoffbrücken, Hydratisierung, ultraschnelle Infrarot-spektroskopie, strukturelle Fluktuationen, Schwingungsdy-namik.
Chem. Unserer Zeit, 2015, 49, 48 – 58 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 57
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de
A B B . 7 2 D - S C H W I N G U N G S S PE K T R E N
Obere Reihe: Zweidimensionale (2D) Schwingungsspektren der asymmetrischenStreckschwingung νAS(PO2) der Phosphatgruppen in inversen DOPC-Mizellen fürverschiedene Wartezeiten T (Darstellung analog zu Abb. 4). Die Mizellen enthalten16 H2O-Moleküle pro Phosphatgruppe (w0 = 16). Die starken schwarzen Linien sindCenter Lines. Untere Reihe: 2D-Schwingungsspektren der symmetrischen StreckschwingungνS(PO2) bei 1095 cm–1 der Phosphatgruppen für verschiedene Wartezeiten T. AlsFunktion der Wartezeit zeigen alle Spektren nur geringfügige Änderungen in ihrerForm (starke schwarze Linien: Center Lines).

Literatur[1] (a) D. Eisenberg, W. Kauzmann, The Structure and Properties
of Water, Oxford Univ. Press, New York 1969. (b) R. Ludwig, D. Paschek, Wasser: Anomalien und Rätsel, Chem. Unserer Zeit 2005,39, 164–175.
[2] T. Yagasaki, S. Saito, Annu Rev. Phys. Chem. 2013, 64, 55–75.[3] D. Laage, J. T. Hynes, Science 2006, 311, 832–835.[4] M. D. Fayer (Ed.), Ultrafast Infrared Vibrational Spectroscopy,
Taylor&Francis, Boca Raton 2013.[5] H. J. Bakker, J. L. Skinner, Chem. Rev. 2010, 110, 1498–1517.[6] M. Pasenkiewicz-Gierula, Y. Takaoka, H. Miyagawa, K. Kitamura,
A. Kusumi, J. Phys. Chem. A 1997, 101, 3677–3691. [7] P. Schuster, G. Zundel, C. Sandorfy C. (Eds.), The hydrogen bond:
Recent developments in theory and experiments, North Holland,Amsterdam, 1976.
[8] J. R. Dwyer, L. Szyc, E. T. J. Nibbering, T. Elsaesser, J. Phys. Chem. B2008, 112, 11194–11197.
[9] N. E. Levinger, R. Costard, E. T. J. Nibbering, T. Elsaesser, J. Phys.Chem. A 2011, 115, 11952–11959.
[10] E. T. J. Nibbering, T. Elsaesser, Chem. Rev. 2004, 104, 1887–1914.[11] P. Hamm, M. Zanni, Concepts and Methods of 2D Infrared Spectros-
copy, Cambridge University Press, Cambridge 2011.[12] S. Mukamel, Annu. Rev. Phys. Chem. 2000, 51, 691–729.[13] M. C. Asplund, M. T. Zanni, R. M. Hochstrasser, Proc. Natl. Acad. Sci.
USA 2000, 97, 8219–8224.[14] M. L. Cowan, B. D. Bruner, N. Huse, J. R. Dwyer, B. Chugh, E. T. J.
Nibbering, T. Elsaesser, R. J. D. Miller, Nature 2005, 434, 199–202.[15] N. Huse, S. Ashihara, E. T. J. Nibbering, Chem. Phys. Lett. 2005, 404,
389–393.[16] S. Ashihara, N. Huse, A. Espagne, E. T. J. Nibbering, T. Elsaesser,
J. Phys. Chem. A 2007, 111, 743–746.[17] R. Rey, F. Ingrosso, T. Elsaesser, J. T. Hynes, J. Phys. Chem. A 2009,
113, 8949–8962. [18] D. Kraemer, M. L. Cowan, A. Paarmann, N. Huse, E. T. J. Nibbering,
T. Elsaesser, R. J. D. Miller, Proc. Natl. Acad. Sci. USA 2008, 105,437–442.
[19] K. Kwak, S. Park, I. J. Finkelstein, M. D. Fayer, J. Chem. Phys. 2007,127, 124503.
[20] T. l. C. Jansen, B. M. Auer, M. Yang, J. L. Skinner, J. Chem. Phys. 2010,132, 224503.
[21] L. Szyc, M. Yang, E. T. J. Nibbering, T. Elsaesser, Angew. Chem. Int. Ed.2010, 49, 3598–3611.
[22] M. Yang, L. Szyc, T. Elsaesser, J. Phys. Chem. B 2011, 115, 1262–1267.
[23] M. Yang, L. Szyc, T. Elsaesser, J. Phys. Chem. B 2011, 115, 13093–13100.
[24] R. Costard. I. A. Heisler, T. Elsaesser, J. Phys. Chem. Lett. 2014, 5,506–511.
[25] R. Costard. C. Greve, I. A. Heisler, T. Elsaesser, J. Phys. Chem. Lett.2012, 3, 3646–3651.
[26] P. Rademacher, Die Mär vom Wasser mit Gedächtnis, Chem. UnsererZeit 2013, 47, 24–31.
Der AutorThomas Elsaesser, geboren 1957 in Tübingen,studierte Physik an der Universität Heidelberg undder Technischen Universität München, wo er 1986promovierte und sich 1991 habilitierte. Seit 1993 ister Direktor am Max-Born-Institut für NichtlineareOptik und Kurzzeitspektroskopie in Berlin, seit 1994Professor für Experimentalphysik an der Humboldt-Universität. Sein Arbeitsgebiet ist die Erforschungultraschneller Prozesse in kondensierter Materie,wozu er ein breites Spektrum optischer undRöntgenmethoden einsetzt. Er hat mehr als 400 Arbeiten in referierten Zeitschriften publiziertund zahlreiche Auszeichnungen erhalten, darunterden Otto-Klung-Preis für Physik und einen AdvancedResearch Grant des European Research Councils(ERC). Er ist Fellow der American Physical Societyund der Optical Society of America sowie Mitgliedder Berlin-Brandenburgischen Akademie derWissenschaften.
Korrespondenzadresse:Max-Born-Institut für Nichtlineare Optik und Kurzzeitspektroskopie,Max-Born-Str. 2 a, 12489 Berlin, E-Mail: [email protected]
58 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Unserer Zeit, 2015, 49, 48 – 58
W A S S E R U N D B I O M O L E K Ü L E S PE K T ROS KO PI Ewww.chiuz.de





























