Embed Size (px)
Citation preview

A#a ys s aad @ aJ ty Co tro|
Untersuchung von Thallium in Rohstoff- und Umweltproben*
H. Gorbauch 1, H. H. Rump 1, G. Alter 2 und C. H. Schmitt-Henco 2
Institut Fresenius, Im Maisel, D-6204 Taunusstein-4 2 Dyckerhoff Zementwerke AG, D-6200 Wiesbaden, Bundesrepublik Deutschland
Determination of Thallium in Raw Materials and Environmental Samples
Summary. Various authentic samples (fuels, rocks, soils, ores, intermediate products, waste materials, environmental ma- terials) were examined for their thallium content. Based on the compiled analytical results, the problems of sample preparation and analysis are discussed. It became obvious, e.g., that the vaporization analysis at about 1200~ is not in all cases suitable for the preeoncentration of thallium as certain effects might lead to lowered results. Therefore, different decomposition techniques with the subsequent separation procedures are presented. The analytical deter- mination was performed by atomic absorption spectro- photometry and inverse voltammetry, according to concen- tration. Experiences gathered with these methods are reported.
Zusammenfassung. Unterschiedliche authentische Proben wie Brennstoffe, Steine, Erden, Erze, Zwischenprodukte, Abfall- stoffe und Umweltproben wurden auf Thallium untersucht. Ausgehend yon den dokumentarisch zusammengestellten Untersuchungsergebnissen dieser Materialien werden die Probleme der Probenvorbereitung und Analyse diskutiert. Hierbei zeigte sich z. B., dab zur Anreicherung des Thalliums die Verdampfungsanalyse bei ca. 1200~ nicht ffir alle Fglle geeignet ist, da durch bestimmte Effekte Minderbefunde auftreten k6nnen. Es werden daher verschiedene Aufschlu6- verfahren mit den sich anschlieBenden Trennungsmethoden vorgestellt. Als analytische MeBverfahren wurden die AAS und die Inversvoltammetrie eingesetzt. Ober Erfahrungen bei der Anwendung dieser Methoden wird berichtet.
Einf'uhrung
Thallium hat nur eine untergeordnete wirtschaftliche Bedeu- tung. Seine Jahresproduktion wird filteren Angaben zufolge auf ca. 20 t/Jahr geschfitzt, Hervorgerufen durch die im Sommer 1979 bekanntgewordene Umweltbelastung mit Thallium im Bereich von Zementwerken trat das Element jedoch in den Blickpunkt des Interesses. Bis zu diesem Zeitpunkt lagen Angaben tiber Thalliumkonzentrationen im wesentlichen aus dem Bereich der Petrographie und der Lagerstfittenkunde vor. Nach [1] wird die mittlere Konzentra-
* Herrn Prof. Dr. W. Fresenius zum 70. Geburtstag gewidmet Offprint requests to: H. H. Rump
Frese~fius Z Anal Chem (1984) 317:236-240 �9 Springer-Verlag 1984
tion in irdischen Gesteinen der oberen Lithosphfire auf 300 mg/t gesch/itzt. Die Verteilung ist jedoch sehr ungleich- m/il3ig, wobei die Isomorphie des Thalliums mit Kalium und Rubidium (T11,49 •, K 1,33/k, Rb 1,49 A_) eine h6here Kon- zentration in kaliumhaltigen Mineralien erwarten l~6t. Un- tersuchuugen von Standardreferenzgesteinen durch zahlrei- che Institutionen haben gezeigt, dab in Graniten und Grano- dioriten die Konzentrationen im Bereich von 1 mg/kg liegen, in Basalten zwischen 0,05 und 0,25 mg/kg, in einem Kalkstein bei ca. 0,1mg/kg und in Meeres-Sedimenten bei etwa 0,4mg/kg [2-4]. Von Bedeutung ffir die Verbreitung des Thalliums in Gesteinen ist weiterhin, dab es als st/indiger Begleiter der Sulfide yon Eisen, Kupfer und Zink vorkommt. Hier sind Konzentrationen bis zu mehreren Prozent ermittelt worden. Erst seit 1979 wurden verschiedentlich auBer Gestei- hen auch andere Proben wie B6den, Gewfisser und Pflanzen vor allem im Umkreis yon Zement- und Hfittenwerken untersucht [5, 6].
Da die M6glichkeit einer Thalliumemission bei verschie- denen Prozessen gegeben ist und das Element wegen seiner Toxizitfit beispielsweise dutch die US-Environmental-Protec- tion-Agency (EPA) in der Liste der 129 sogenannten ,,Priority Pollutants" sowie in die deutsche technische Anleitung zur Reinhaltung der Luft aufgenommen wurde [7, 8], war es geboten, weitergehende Kenntnisse fiber die Thalliumkon- zentration in solchen Materialien zu erhalten, die bei diesen Prozessen eingesetzt werden. Auch Hinweise auf die Konzen- trationen in potentiellen Energietr/igern, wie Shredder- abffillen, erschienen notwendig.
Von besonderer Bedeutung bei der vorliegenden Untersu- chung war einmal die Entscheidung fiber das zu wfihlende Aufschlul3- bzw. Anreicherungsverfahren, bei sehr inhomo- genen Materialien auch fiber die Probenvorbereitung. Vor allem bei silicatischen Materialien ist auf die Vollst/indigkeit der Thalliumabtrennung bei der Verdampfungsanalyse zu achten. Die weitere Probenvorbereitung, z.B. durch extrak- rive Anreicherung nach Komplexierung, mug sich nach der gewfihlten Bestimmungsmethode richten. Es kamen bei unse- ren Arbeiten die Inversvoltammetrie und die Atomabsorp- tions-Spektralphotometrie mit Flamme und Graphitrohr- kiivette zum Einsatz.
Experimentelles
Probenahme und Probenvorbereitung
Probenahme und Probenvorbereitung richten sich an der grundsgtzlichen Forderung aus, eine ffir das Gesamtmaterial

Ana yt k und
repr/isentative Laborprobe zu erhalten. Dies ist bei homoge- hen oder licht homogenisierbaren Proben wie Flfissigkeiten leicht zu bewerkstelligen. Bei Feststoffen wie Erzen, Sanden, Schlacken, Aschen und St/iuben ergeben sich jedoch ProNe- me durch m6gliche Inhomogenit/iten. Schwierig wird die Probenahme bei sehr inhomogenen Materialien wie M fill oder Shredderschrott. Hinweise finden sich in [9, 10].
Je nach Materialmenge ist die Gr6ge der Rohprobe zu w/ihlen, wobei bei sehr inhomogenen Materialien wie Abf/il- len mit Gesamtrohstoffmengen bis 100 t maximal 0,5 ~o als Rohprobe zu entnehmen sind. Bei gr6Beren Gesamtmengen l~iBt sich der Probenanteil reduzieren. Schfittgfiter wie Erze, Schlacken oder Aschen fallen in der Regel in Haldenform an. Sollten sehr unterschiedliche Korngr613en vorliegen, ist es n6fig, das Material fiber ein Sieb bestimmter Maschenweite abzusieben und die Siebgr6ge mit einer Hammermfihle auf unter 1 cm zu zermahlen. Die Rohprobe oder die so vorberei- tete Rohprobe wird dann fiber den Kegel gemischt. Hierbei rollen die gr6beren Stoffe fiber den Kegelmantel an die Basis und die Feinanteile bleiben an der Kegelspitze liegen. Nach mehrfacher Mischung der Gesamtprobe wird anschlieBend die Kegelspitze abgetragen und im Spiralverfahren ein Kegel- stumpf von geringer H6he hergestellt. Dieser Stumpf wird in 4 Viertel geteilt. Zwei gegenfiberliegende Viertel werden dann zur weiteren Mischung und Verjfingung entnommen, w~h- rend der Rest verworfen wird. In dieser Weise wird welter vorgegangen, bis eine handliche Probenahmemenge erreicht ist. Die weitere Zerkleinerung erfolgt dann mit Schlagkreuz- mfihlen, M6rsermiihlen oder Scheibenschwingmiihlen. Soil- ten sich beim Mischen, Mahlen und Verjfingen schwer zerkleinerbare Anteile ergeben (Metalle, Kunststoffe), so sind diese auszusortieren und zu verwiegen. Sie k6nnen unter Umst~inden nach besonderer Zerkleinerung yon Hand oder durch Entnahme yon Bohrsp~inen pro rata dem Analysen- muster beigegeben werden.
Bei sehr feuchten Feststoffen ist vor dem Mischen und Verjfingen zu trocknen. In einem solchen Fall wird nicht die Rohprobe insgesamt getrocknet, sondern lediglich bei Raum- temperatur konditioniert. Durch Verwiegung vor und nach dieser Konditionierung bei Raumtemperatur kann die soge- nannte grobe Feuchtigkeit ermittelt werden, Dieses so luft- getrocknete Muster lfigt sich ffir die weitere Pr/iparation und Probenvorbereitung verwenden. Will man auf das ursprfing- lich vofliegende Probegut zurfickrechnen, so ist zu beachten, dab das ursprfingliche Probegut zur Ermittlung der groben Feuchtigkeit eingewogen wird und das vorgetrocknete Mate- rial ffir die Analyse. SoUte im restlichen Material gleichfalls die Feuchtigkeit bestimmt werden, so sind die Beziehungen zwischen der groben Feuchtigkeit und der sogenannten hygroskopischen Feuchtigkeit (die aus dem aufbereiteten Material z. B. durch Trocknen bei 105 ~ C ermittelt wird) nicht additiv, sondern sie mfissen verhgltnisgleich umgerechnet werden, z.B. nach der Gleichung
b x l O 0 - a ~o Gesamtwasser = a +
100
a = grobe Feuchtigkeit b = hygroskopische Feuchtigkeit der luftgetrockneten Probe.
Bei der Aufbereitung von sehr inhomogenem Material wird die Rohprobe luftgetrocknet. Anschliel3end wird die grobe Feuchtigkeit berechnet, die entsprechend den oben gemachten Angaben zusammen mit der hygroskopischen Feuchtigkeit auf die Gesamtfeuchtigkeit umgerechnet werden
kann. Diese Vorgehen der Lufttrocknung hat den Vorteil, dab alle anderen SortiermaBnahmen und auch eventuell notwendige Misch- und Verjfingungsvorg~inge kaum noch zu weiteren Feuchtigkeitsverlusten ffihren. Die lufttrockene Rohprobe wird dann yon Hand sortiert und die hierbei erhaltenen Fraktionen werden verwogen. Solten die Fraktio- nen vollst/indig zur Untersuchung gelangen, so werden alle Fraktionen einzeln verwogen und der prozentuale Anteil der Einzelffaktionen berechnet. Die Fraktionen werden nach ihrer Verwiegung wiederum pro rata zum Analysenmuster zusammengesetzt. Ist eine solche Einbeziehung yon aussor- tierten Fraktionen in das Analysenmuster notwendig, so ist es empfehlenswert, diese Fraktionen getrennt aufzuarbeiten und getrennte Analysen durchzuffihren. Die Untersuchungser- gebnisse k6nnen wiederum auf die entsprechende Rohprobe umgerechnet werden. In jedem Fall ist hier auf die Ausgangs- bezugsgr6Be zu achten, wobei Gewiehtsver~inderungen durch Feuchtigkeitsverluste wfihrend der Probenaufbereitung auf- treten k6nnen.
Probenaufschlufl
Bei den meisten Analysenmethoden zur Thalliumbestim- mung ist ein vorheriger Aufschlug sowie eine Anreicherung bzw. Matrixabtrennung erforderlich. Versuche zur Bestim- mung yon Metallen durch Aufgabe der festen Probe in die Graphitrohrkfivette des Atomabsorptionsspektralfotometers wurden nur in wenigen Ausnahmef/illen fortgeffihrt, da die Probleme der Aufgabe homogener Proben sowie der Matrix- st6rungen nur schwer zu beherrschen waren. Wegen dieser zu erwartenden Schwierigkeiten arbeiteten wir grunds~itzlich mit Verfahren zur Abtrennung des Thalliums yon anderen Ele- menten.
Durch h6here Konzentrationen an Matrixelementen (insbesondere in komplex zusammengesetzten Proben) k6nnen sich die Bestimmungsgrenzen deutlich erh6hen. Uber die Ursachen systematischer Fehler ist noch relativ wenig bekannt. Man findet zwar in der Literatur verallgemei- nernde Regeln fiber die Reduktion der Fehler, doch lassen sich derartige Regeln nicht immer anwenden, da sich/ihnliche Spezies der Ionen unter/ihnlichen Untersuchungsbedingun- gen verschieden verhalten k6nnen. Es kommt hinzu, dab keine Referenzstandards zur Verffigung stehen, so dab eine Kompensation der systematischen Fehler durch Eichung nicht m6glich ist. Hier kann teilweise die Aufstockung mit Standardl6sungen weiterhelfen.
Selektive Verdampfung. Dieses von [11] und [12] ausgearbei- tete Verfahren zur Bestimmung flfichtiger Elemente, insbe- sondere Thallium, wurde von uns in solchen F/illen eingesetzt, bei denen Minderbefunde durch unvollstfindige Verdamp- fung des Thalliums nicht auftreten konnten. U'ber der- artige Effekte ist bereits bei [4, 12] berichtet worden. Sie treten vor allem dann auf, wenn die gepulverte Probe zusammenschmilzt und sich so die vorher vorliegende groge Oberfl/iche erheblieh verkleinert. Diese Erscheinungen k6n- nen bei allen Arten von Gesteinen wie Graniten oder Schiefern auftreten. In einigen F/illen ist es notwendig, die Verdampfungstemperatur zu optimieren.
Normalerweise werden bis zu 1 g der zu untersuchenden Substanz in ein Quarzschiffchen eingewogen. Dieses wird mit einem Glasstab ins vordere Drittel eines Quarzverdamp- fungsrohres geschoben. Hinter das Schiffchen wird ein Quarzdiffusionsk6rper gesetzt. Die Verdampfung erfolgt in
237
A allsis @ ali% 8e l ol
A
e
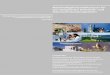
Abb. 1A, B. Thalliumbestimmung mit Inversvoltammetrie (ASV); A Standardl6sung mit a) Blindwert, b) 50 ng T1 c) 100 ng T1; B 100 ng T1 + 100 lal L6sung einer unvollstindig aufgeschlossenen Gummi- probe
Tabelle 1. Thalliumkonzentrationen in Brennstoffen (mg/kg)
Heiz61 S a) < 0,02 b) 0,10
Braunkohle a) 0,005 b) 0,005 c) 1,1
Deutsche Steinkohle a) 0,28 b) 0,31
US-Import-Kohle a) 0,40 b) < 0,005 c) 0,42
Tabelle 2. Schwermetallkonzentrationen in Steinkohlen und schwe- rem Heiz61 (mg/kg)
Stein- S te in - S t e i n S t e i n - Heiz61 S kohle a kohle b kohle c kohle d
Thallium 0,28 0,31 0,40 < 0,0005 < 0,05 Blei 66 85 16 241 0,21 Cadmium 0,4 0,5 0,4 3,3 0,05 Nickel 41 52 20 39 32 Chrom 53 47 41 32 1 Arsen 8,5 8,7 24 2,6 0,2 Beryllium 1,6 1,7 1,8 1,0 < 0,1 Vanadium 56 43 35 < 1 86 Quecksilber 0,68 0,56 3,30 0,50 0,49 Kupfer 45 96 n.b. n.b. 1 Selen 1,7 1,8 4,2 1,0 7 Tellur < 0,2 < 0,2 0,2 < 0,2 < 0,2 Zink 139 116 25 825 1,2
n.b. = nicht bestimmt
der Regel bei 1000~ im Luft- oder Sauerstoffstrom, wobei nur Thallium und einige andere leichtfliichtige Elemente als Oxide flichtig sind und sich in der kihlen Spitze des Quarzrohres abscheiden (Schmelzpunkt von T1203 -- 717 ~ C ;' Sublimation ab ca. 500~
Das Kondensat wird in 2 ml HNO3 konz. gel6st und mit ca. 3 ml destilliertem Wasser nachgespilt. In einem 50 ml- Becherglas wird auf dem Wasserbad zur Trockene einge- dampft. Ftir die praktische Durchf/ihrung wurde ein Rohr- ofen ,,Heraeus ROR" mit einer Nenntemperatur von 1700 ~ C verwendet.
Aufschlufl und Extraktion fiir die Bestimmung mit Inversvol- tammetrie. 0,5 bis 1 g des Probenmaterials (bei h6heren Gehalten kann auch durchaus weniger verwendet werden) werden in einer Abdampfschale von 100 bis 250 ml Inhalt mit 5 ml H2SO 4 konz., 2ml HNO3 fumans (eventuell auch 1 ml H2Oz) zur Oxidation von organischen Substanzen versetzt. Bei h6heren organischen Substanzgehalten muB die Zugabe von HNO3 bzw. H20 2 wiederholt werden. Bei silicatischem Material wird der AufschluB in einer Platinschale zunichst mit HzSO 4 und HNO3 durchgef/ihrt. Nach Zerst6rung der organischen Substanz wird die Probe mit wenigen ml HF versetzt. Die Siuren werden anschlieBend zur Trocknc ab- geraucht. Der Trockenrtickstand wird mit 50ml 1 m HBr- L6sung heiB aufgenommen. Es wird dieser L6sung Bromwas- ser bis zur bleibenden Braunf'irbung zugesetzt und an-
238
schlieBend dreimal mit Diethylether (15m l, 10ml, 10m l) extrahiert. Sollte bei der ersten Extraktion der Extrakt keine Braunfirbung aufweisen, erfolgt nachtrigliche Zugabe von Br-Wasser. Die vereinigten Extrakte werden in einem 50 ml- Becherglas auf dem Wasserbad zur Trockne eingedampft. Der Abdampfrtickstand wird ein- bis zweimal mit einigen Tropfen Salpetersiure fumans oxidiert und wiederum zur Trockne eingeengt.
Extraktion des Aufschlu[3es J~r die Bestimmung mit Atomab- sorptions-Spektrometrie. Der oben erhaltene SiureaufschluB wird mit Ammoniakwasser abgestumpft, wobei darauf zu achten ist, dab kein Niederschlag entsteht. AnschlieBend wird 5 ml gesittigte KMnOg-L6sung zugegeben. Falls dabei Man- gandioxid ausfillt, muB zusitzlich KMnO4-L6sung zugefiigt werden. Die Probe wird anschlieBend mit 10ml (NH4)2 H-Citrat (50~) sowie mit 10ml Titriplex III-L6sung 0,1 m (Merck) versetzt. Mit Ammoniakwasser wird auf pH 9 ein- gestellt. Man gibt 10ml Natrium-diethyldithiocarbaminat (DDTC) 0,1 ~ig hinzu und extrahiert dreimal mit Tetrachlor- methan. Danach gibt man weitere 5 ml DDTC hinzu und extrahiert noch dreimal mit CC14. Die Extrakte werden abgedampft unter Zugabe einiger K6rnchen Natriumnitrat. Die organischen Rickstinde werden im Becherglas mit HNO3 konz. sowie anschlieBend HNO~ fumans nab verascht. Der RLickstand wird in HNO3 i m aufgenommen und auf 5 bzw. 10ml aufgefiillt.
Tabeile3. Thalliumkonzentrationen in Steinen, Erden und Erzen (mg/kg)
Eisenoxid 0,8
Eisenerz a) 0,032 b) 0,012 c) 0,006 d) 0,005 e) 0,045
H/imatiterz 1,9 Purpurerz 0,5 Flul3spat 0,041
Schiefer a) 0,9 b) 0,05 c) 0,15
Kaolin a) 0,005 b) < 0,005
Ton 0,015
Quarzsand Haltern < 0,005
Sand 0,075
Hiittensand 0,18
Anhydrit a) < 0,005 b) 0,02
Gips Lamerden 0,13
Gips Frankenfeld 0,04
Gips Stadtoldendorf 0,04
Dolomit a) 0,055 b) 0,080
Mergel gelb, Am6neburg 0,03 Mergel grtin, Am6neburg 0,10 Mergel schwarz, Am6neburg 0,40 Turonmergel 0,12 L6B, Am6neburg 0,13 Kalkstein, Warstein 0,058 Kalkstein, Medenbach < 0,005 Kalkstein, Steeden < 0,005 Kalkstein, Hohlenfels < 0,005 Kalkstein, Dexheim 0,19 Kalkstein, Nierstein 0,055 Trag 0,39 Turonkalk 0,085 Cenomankalk a) 0,025
b) O,O4 c) 0,025
Tabelle 4. Thalliumkonzentrationen in Vorprodukten und Abfall- stoffen (mg/kg)
Gummi
Flugasche Kohleverbrennung
Siureharz Asphalkohle
Bitumen
REA-Gips
AGM-Abbrand
Flugasche Miillverbrennung
Kohlenschlacke
LKW-Reifen (17 verschiedene Proben)
Brennbare Rfickstinde bei der Aufarbeitung yon Altautos (Shredder)
a) 0,05 b) 0,16
a) 14 b) 3,5 c) 20 d) 10,5
0,035 < 0,05
0,9
a) < 0,005 b) 0,045 c) < 0,005 d) < 0,005 e) 0,64
35,5
2,0
0,16
<0,5
a) 0,08 b) 0,25 c) 0,18
Tabelle 5. Schwermetallkonzentrationen in Kohlenschlacke, Filter- stS.uben der Kohleverbrennung und eines Eluats (1 : 10) aus Filter- staub (mg/kg)
Kohlen- Filter- Filter- Eluat aus schlacke staub a staub b Filterstaub b
Thallium 0,16 10,1 20,0 0,34 Blei 21 812 2170 2,75 Cadmium 0,2 3,7 13 0,98 Nickel 263 724 1270 30 Chrom 418 363 420 0,09 Arsen 16 348 590 0,077 Beryllium 12,4 9,9 29 0,10 Vanadium 85 119 197 0,019 Quecksilber 0,07 0,12 3,5 0,0005 Selen 0,63 7,4 24 < 0,001 Tellur < 0,1 1,5 0,5 < 0,001 Zink 80 1840 500 172 Kupfer 70 560 1180 43
Bestimmungsverfahren
Ober die verschiedenen analytischen Mel3verfabxen liegen zahlreiche Publikationen vor, so fiber ein spektralphotometri- sches Verfahren [13], die AAS mit Flamme und mit Graphit- rohrkfivette [14-17] sowie fiber voltammetrische Methoden [18, 19]. Ober Verfahren zur Steigerung der Empfindlichkeit und Reproduzierbarkeit im Spurenbereich berichten [20].
Parameter ffir die eigenen AAS-Messungen: Ger i t : Varian 775 mit Untergrund-Kompensat ion Wellenl/inge: 276, 8 nm Spalt: 0,5 nm Lampenst rom: 5 mA Standardl6sung: Titrisol (Merck) Erstellung einer Eichkurve durch Herstellen yon L6sun-
gen yon 100 ~tg T1/1 bis 1 mg T1/l. Durchftihrung der Analyse nach dem Standardadditions-Verfahren.
Ffir die inversoltammetrische Bestimmung von Thallium wurde der Rfickstand des entsprechenden Aufschlugverfah- rens im Becherglas mit 20 ml 0,05 M E D T A aufgenommen, mit Stickstoff belfiftet und an einer Quecksilbertropfen- elektrode kathodisch reduziert. Anschliegend wird das so gebildete Amalgam anodisch aufgel6st. Die Stromspan- nungskurve wird mit dem Schreiber aufgezeichnet.
In vielen Matrices k6nnen grenzfl/ichenaktive Stoffe die Analyse st6ren und zwar zum einen, indem sie mechanisch den Zutritt des Depolarisators zur Elektrode hemmen oder indem sie die Elektrodenreduktion wihrend der Messung beeinflussen. Neben einer Abnahme der Spitzenstr6me wird h/iufig eine Verschiebung der Spitzenpotentiale beobachtet.
239

AH II i @u lill Conlvol
Tabelle 6. Thalliumkonzentrationen in ,,Umweltproben" (mg/kg)
Honig (8 Einzelproben)
Heilwisser und Mineralwisser (h6her mineralisiert) (13 Einzelproben)
Stielkorallen, Rotes Meer (Sharma)
Blattkorallen, junge Pflanzen Rotes Meer (Sharma)
Hafer a) Halme, mehrere Proben b) K6rner, mehrere Proben
Pflanzenschutzrnittel Dicuran 500 fltissig < 0,02 Stomp < 0,02 Gesapum 500 flfissig < 0,02 Tribunil 0,50 Nexit stark 0,010 Metasystox R 0,20 Bayleton 100 0,04 Tantizon 0,40 Orthosipholatan < 0,02 Merpelan AZ < 0,02 Unden-flfissig < 0,02
Diingemittel Kalkstickstoff 0,01 Kalkammonsalpeter 26 < 0,005 12-12-17-2 frei Blaukorndiinger 0,015 13-13-21-NPK Diinger mit Bor 0,08 Oskorna-Animalin < 0,005 Kalksalpeter 15 % < 0,005 Ammonsulfatsalpeter + Bor 0,010 Superphosphat + Bor? 0,12 Thomasphosphat 15 % < 0,005 Kali-Magnesia 30% (30% K20 q- 10% MgO) 0,06 Thomaskali 10 x 20 0,005 Kornkali 40 % 0,015 Sehwefelammoniak 0,015 Kainit 0,008
< 0,02
0,003 bis 0,1
< 0,0005
0,005
0,085 bis 0,12 0,005 bis 0,01
Eine Aufspaltung in mehrere Spitzen unter starker Verzer- rung der Stromspannungskurven ist hierbei m6glich. Abbil- dung I gjbt ein Beispiel fiir derartige St6rungen.
U n t e r s u c h u n g s e r g e b n i s s e
Verschiedentlich wurden Ergebnisse der Bestimmung von Thallium mitgeteilt. Ober Konzentrationen in Gewissern, B6den und Pflanzen berichtet [21], wihrend sich [4, 22, 23] mit geologischem Material, [24] mit Zementen und [25] mit Thalliumgehalten in nicht aufbereitetem Erdgas befassen.
In den Tabellen sind die Untersuchungsergebnisse der verschiedensten Materialien zusammengefaBt, wobei Tabel- le 1 die Thalliumkonzentrationen in Brennstoffen wiedergibt.
Einen Oberblick fiber die Konzentrationen weiterer Schwermetalle in verschiedenen Kohlenproben und in einer Heiz61probe gibt Tabelle 2.
In Tabelle 3 sind Untersuchungsergebnisse von Steinen, Erden und Erzen wiedergegeben.
In Tabelle 4 sind die Thalliumkonzentrationen in Vorpro- dukten und Abfallstoffen wiedergegeben.
In Tabelle 5 sind die Untersuchungsergebnisse aufweitere Schwermetalle in Kohlenschlacke, Filterstiuben bei der Koh- leverbrennung und die Ergebnisse einer Elution eines Filter- staubes mit Wasser (entsprechend den Deutschen Einheits- verfahren S 4) wiedergegeben.
In Tabelle 6 sind die Untersuchungsergebnisse in soge- nannten Umweltproben zusammengefagt.
L i t e r a t u r
1. Omelin's Handbuch der anorganischen Chemie, 8. Aufl, Nr 38, 1940
2. Flanagan FJ (1973) Geochim Cosmochim Acta 37:1189 3. Simon FO, Compbell EY, Aruscavage PJ (1977) Res US-Geol
Survey 5: 579 4. Heinrich H (1979) Fresenius Z Anal Chem 294: 345-351 5. LIS-NRW (ed) (1980) Thallium-Report, Bonner Univ Druckerei 6. Schoer J, Nagel U (1980) Naturwiss 67:261-262 7. Keith LH, Telliard WA (1979) Envir Sci Technol 13:416 8. Technische Anleitung zur Reinhaltung der Luft (1982) Hey-
mann, K61n 9. ChemikerausschuB der GDMB (ed) Analyse der Metalle,
2. Aufl, Bd Probenahme. Springer, Berlin 1975 10. Schneider W (1980) Untersuchungsmethoden zur Deponierbar-
keit gewerblicher Abfille. F- u E-Projekt 10302211/05 Umwelt- bundesamt, Berlin
11. Geilmann W (1958) Fresenius Z Anal Chem 160:410-426 12. Geilmann W, Neeb KH (1959) Fresenius Z Anal Chem
165:251-268 13. Sager M, T61g G (1982) Mikrochim Acta 1I:231-246 14. Fuller CW (1976) Anal Chim Acta 81:199-202 15. Berndt H, Messerschmidt J, Alt F, Sommer D (1981) Fresenius Z
Anal Chem 306: 385- 393 16. Keil R (1981) Fresenius Z Anal Chem 309:181 - 185 17. Wachter C, Weisweiler W (1982) Mikrochim. Acta I: 307-315 18. Bonelli JE, Taylor HE, Skogerboe RK (1980) Anal Chim Acta
118:243-256 19. Lukaszewski Z, Pawlak MK, Ciszewski A (1980) Talanta
27:181-185 20. Camman K, Andersson JT (1982) Fresenius Z Anal Chem
310:45-50 21. Landesanstalt ffir Umweltschutz (ed) (1981) Thallium-Studie,
Karlsruhe 22. Langmyhr FJ, Stubergh JR, Thomassen Y, Hanssen JE, Dolezal
J (1974) Anal Chim Acta 71:35-42 23. Hannaker P, Hughes TC (1977) Anal Chem 49:1485-1488 24. Keinhorst H (1980) Staub-Reinh Luft 40:26-29 25. Tunn W (1976) Erd61 und Kohle, Erg-Bd Industrieverlag,
Leinfelden
Eingegangen am 28. Oktober 1983
240