Embed Size (px)
Citation preview

Vorausberechnung des Einflusses starker Elektrolyte -Salzeffekt- auf das
Phasengleichgewichtsverhalten von Lösungsmittelgemischen
Vom Fachbereich Chemie der Universität Oldenburg zur Erlangung des akademischen
Grades eines Doktors der Naturwissenschaften angenommene Dissertation
von
Christian Rose
geb. am 25.05.1966 in Oldenburg
Erstreferent: Prof. Dr. J. Gmehling
Korreferat: P.D. Dr. Axel Brehm
Tag der mündlichen Prüfung: 13.06.2000

Die vorliegende Arbeit entstand in der Zeit von Oktober 1994 bis Mai 2000 am Lehrstuhl für
Technische Chemie der Universität Oldenburg während meiner Tätigkeit als
wissenschaftlicher Mitarbeiter unter der Leitung von Prof. Dr. Jürgen Gmehling.
Mein Dank gilt Herrn Prof. Dr. Jürgen Gmehling für die interessante Themenstellung und die
Bereitstellung der erforderlichen Mittel.
Allen derzeitigen und ehemaligen Mitarbeitern der Arbeitsgruppe sei gedankt für die
freundliche Unterstützung in allen Fragen.
Herrn P. D. Dr. Axel Brehm danke ich für die Übernahme des Korrefferats.
Mein besonderer Dank gilt Herrn Dr. Weidong Yan von der Zheijang Universität, der viel
zum Gelingen dieser Arbeit beigetragen hat.

1
Vorausberechnung des Einflusses starker Elektrolyte –Salzeffekt- auf das Phasengleichgewichtsverhalten von Lösungsmittelgemischen
1. Einleitung 5 1.1 Zielsetzung 10 2. Thermodynamische Grundlagen und Definitionen 2.1 Phasengleichgewicht 11 2.2 Exzeßpotential und Aktivitätskoeffizient 14 2.3 Normierung des Aktivitätskoeffizienten 15 2.4 Chemisches Potential des Ions 17 2.5 Chemisches Potential des Elektrolyten und mittlerer Aktivitätskoeffizient 18 2.6 Definition der Gemischvariablen für starke Elektrolyte 19 2.7 Osmotischer Koeffizient 21 2.8 Gibbs-Duhem-Gleichung 24 3. GE-Ansatz für elektrolythaltige Mischphasen 3.1 Beitrag der ‘long-range’-Wechselwirkungen 27 3.2 Beitrag der ’middle-range’-Wechselwirkungen 30 3.3 Beitrag der ‘short-range’-Wechselwirkungen 33 3.4 Der vollständige Ausdruck für den Aktivitätskoeffizienten 36 3.5 Anzahl der Anpassungsgrößen 37 4. Das Fest-Flüssig-Phasengleichgewicht von Lösungen starker Elektrolyte 4.1 Eutektische Systeme ohne Verbindungsbildung 41 4.2 Systeme mit Verbindungsbildung und kongruentem Schmelzpunkt 43 4.3 Peritektische Systeme 44 4.4 Löslichkeitsprodukt 45 4.5 Temperaturabhängigkeit des Löslichkeitsprodukts 48 5. Anpassung von Modellparametern 5.1 Beschreibung der Datenbasis 52 5.2 Reduzierung der Datenbasis 55 5.3 Parameterbestimmung 57 5.4 Anpassung an binäre, wäßrige Systeme 67 5.4.1 Anpassung systemspezifischer Parameter an Löslichkeitsdaten 67 5.4.2 Simultane Anpassung systemspezifischer Parameter an Löslichkeitsdaten 81 und mittlere Aktivitätskoeffizienten

2
5.4.3 Simultane Anpassung systemspezifischer Parameter an Löslichkeitsdaten, 99 mittlere Aktivitätskoeffizienten, molale osmotische Koeffizienten und Dampfdruckdaten
5.4.4 Beschreibung aller binären, wäßrigen Systeme auf Grundlage 105 allgemein verwendbarer Lösungsmittel-Ion-Parameter 5.5 Binäre nichtwäßrige Systeme 111 5.6 Anpassung an ternäre Systeme 115 6. Messung von Dampf-Flüssig-Gleichgewichten salzhaltiger
Lösungsmittelgemische 6.1 Meßmethoden und Meßprogramm 120 6.2 Gaschromatographische Dampfraumanalyse 121 6.3 Ebulliometrie 125 6.4 Reinheit der eingesetzten Substanzen 128 6.5 Analytische Bestimmung der Phasenzusammensetzung 128 6.6 Kalibrierung 129 6.7 Überprüfung der thermodynamischen Konsistenz 131 6.8 Ergebnisse 136 6.9 Fehlerbetrachtung 141 7. Anhang 7.1 Kalibrationsdaten 144 7.2 Antoine-Konstanten 144 7.3 Experimentelle Ergebnisse 145 7.4 Listen angepaßter Parameter 158 7.5 Liste der verwendeten Standardgrößen 166 7.6 Literaturverzeichnis 168 8. Zusammenfassung 179

3
Verzeichnis der verwendeten Symbole und Abkürzungen Lateinische Symbole: A Debye-Hückel-Konstante a1, a2 Konstanten der Konzentrationsfunktion des zweiten Virialkoeffizienten ai Aktivität der Komponente i ak,l UNIQUAC-Wechselwirkungsparameter b Debye-Hückel-Konstante B zweiter Virialkoeffizient bij binärer Wechselwirkungsparameter c Molarität cij binärer Wechselwirkungsparameter cp molare Wärmekapazität bei konstantem Druck d Dichte D Dielektrizitätskonstante e0 Elementarladung f Fugazität G absoluter Wert der Gibbsschen Enthalpie g molarer Wert der Gibbsschen Enthalpie H absoluter Wert der Enthalpie I Ionenstärke k Boltzmann-Konstante Ksp Löslichkeitsprodukt m Molalität Ms Molgewicht der Lösungsmittelkomponente s n Gesamtzahl der Komponenten eines Systems NA Avogadrokonstante ni Molzahl der Komponente i nion Gesamtanzahl verschiedener Ionen im System nloese Gesamtanzahl verschiedener Lösungsmittel im System P Druck pi Partialdruck der Komponente i Poyi Poynting-Faktor Ps Sättigungsdampfdruck Q Ladung qk relative van der Waalssche Oberfläche der Komponente k r Abstand zweier Teilchen R allgemeine Gaskonstante rk relatives van der Waalssches Volumen der Komponente k S absoluter Wert der Entropie Sm Sättigungsmolalität ui, j Wechselwirkungspotential V Volumen xi Molenbruch der Komponente i in der Flüssigphase xs salzfreier Lösungsmittelbruch yi Molenbruch der Komponente i im Dampf zi Ladungszahl des Ions i

4
Griechische Symbole: α Polarisierbarkeit αij relative Flüchtigkeit ∆ Differenzwert einer Größe φ molaler osmotischer Koeffizient φk Volumenanteil der Komponente k θk Oberflächenanteil der Komponente k ς beliebig zu definierendes Konzentrationsmaß γi Aktivitätskoeffizient der Komponente i ν ν+ −, stöchiometrischer Faktor des Kation bzw. Anions µ Dipolmoment µi chemisches Potential der Komponente i ϕi Fugazitätskoeffizient der Komponente i ϕ rationaler osmotischer Koeffizient Π osmotischer Druck νi Molvolumen der Komponente i Ψk,l Boltzmann-Faktor für die Wechselwirkung der Komponenten k und l Indices tiefgestellt: ber berechnet Diss der Dissoziation Fäll der Fällungsreaktion R der Reaktion Ast im Sättigungszustand solv solvatisiert + des Kations - des Anions Indices hochgestellt: C kombinatorischer Anteil E Exzeßanteil f feste Phase fl flüssige Phase l.r. ‘long-range’ L Flüssigphase m.r. ‘middle-range’ R Restanteil s.r. ‘short-range’ T bei Systemtemperatur T V Dampfphase 0 Standardzustand ∞ Zustand unendlicher Verdünnung ∗ Normierung auf den Zustand unendlicher Verdünnung

5
1. Einleitung
Die Kenntnis des realen Verhaltens elektrolythaltiger Gemische bildet die Grundlage für die
Auslegung und die Simulation einer Reihe industriell genutzter Prozesse. Sei es, daß Salze als
störende Nebenprodukte von Prozeßströmen abgetrennt werden müssen, oder daß sie in neu
entwickelten Prozessen gezielt als Trennhilfsstoffe eingesetzt werden. Beispiele für Prozesse in
denen starke Elektrolyte eine bedeutende Rolle spielen sind u.a. die Entfernung des Schwefels
bei der Rauchgasentschwefelung, die Salzdestillation von Abfallsäuren in der Kaliindustrie,
sowie allgemein die Absorption saurer Gase wie SO2, CO2 und H2S, die bei der Neutralisation
saurer oder alkalischer Prozeßströme anfallen [47]. Die überwiegende Anzahl dieser in-
dustriellen Anwendungen nutzt dabei den Salzeffekt auf das Dampf-Flüssig-Gleichgewicht zur
Überwindung azeotroper Punkte. Prozesse, die die Kenntnis des Fest-Flüssig-Gleichgewichts
voraussetzen finden sich vor allem in der metall- und erdölverarbeitenden Industrie. Für die
Gewinnung geothermaler Energie, müssen Ingenieure die Kristallisations-, die invarianten-
und die eutektischen Punkte hochkonzentrierter Lösungen von Alkali- und Erdalkali-
halogeniden berechnen. Auch bei der Meerwasserentsalzung, der Abwasserreinigung, in der
Galvanotechnik und bei der Gewinnung reiner Salze mit Hilfe fraktionierter Kristallisation hat
die Frage nach der maximalen Löslichkeit von Salzen entscheidende Bedeutung. Extrakti-
onsprozesse, die den Salzeffekt auf das Flüssig-Flüssig-Gleichgewicht zur Beeinflussung von
Mischungslücken und Verteilungskoeffizienten ausnutzen sind in ihrer Effektivität oftmals
durch die Salzlöslichkeit limitiert.
Dies sind einige Anwendungen die sich hinter dem allgemeinen Interesse für ein besseres
Verständnis des realen Verhaltens elektrolythaltiger Mischungen verbergen. Der Wunsch nach
gezieltem Einsatz starker Elektrolyte als Hilfsstoff zur thermischen Trennung komplexer
Fluide ist unzertrennlich an die Entwicklung und den Test thermodynamischer Modelle ge-
knüpft, die die Berechnung möglichst aller Phasengleichgewichte ermöglichen.
Während für Nichtelektrolytsysteme mittlerweile ausgereifte, thermodynamische Modelle für
die Auslegung thermischer Trennprozesse verfügbar sind, wurden für die Berechnung des rea-
len Verhaltens elektrolythaltiger Systeme verhältnismäßig wenig Ansätze entwickelt. Den ge-
meinsamen Ausgangspunkt für die überwiegende Anzahl der bis heute entwickelten Elektro-
lytmodelle bilden noch immer die theoretischen Überlegungen, die Debye und Hückel 1923

6
formulierten [15], und die zumindest für den Bereich starker Verdünnung Gültigkeit besitzen.
Die Zielsetzung der Theorie ist es die Abweichung des Verhaltens starker Elektrolyte von der
klassischen Dissoziationshypothese nach Arrhenius zu erklären [2]. Im Gegensatz zu
Arrhenius gingen Debye und Hückel davon aus, daß der Elektrolyt bis zu relativ hohen Kon-
zentrationen praktisch vollständig dissoziiert vorliegt. Diese Annahme stützt sich nicht allein
auf die besondere elektrische Leitfähigkeit, auch die kollegativen Eigenschaften von Elektro-
lytlösungen weisen in diese Richtung. So sind die beobachteten Dampfdruckerniedrigungen
und osmotischen Drücke größer als sich aus der Anzahl der gelösten Einheiten ergibt, wenn
man keine Dissoziation annimmt. In einer NaCl-Lösung ist z.B. der osmotische Druck etwa
zweimal so hoch, als man nach der Anzahl der gelösten NaCl-Einheiten erwarten würde.
Wenn man jedoch annimmt, daß in der Lösung in Wirklichkeit die Ionen Na + und Cl− -also
doppelt so viele Teilchen- vorliegen, läßt sich dieser Befund problemlos erklären. Da die
Debye-Hückel-Theorie jedoch von einer Reihe Annahmen ausgeht, die mit steigender Kon-
zentration ihre Gültigkeit verlieren, gelten die theoretischen Fragen bis heute nur für den Be-
reich starker Verdünnung als weitgehend geklärt.
Mangels einer einheitlichen Theorie, die das Verhalten derartiger Systeme auch bei hohen
Konzentrationen und Temperaturen zufriedenstellend beschreiben kann, kam es zu einer
Reihe empirischer Erweiterungen und Abwandlungen der Debye-Hückel-Theorie, die sich in
drei großen Gruppen zusammenfassen lassen:
1. Die ersten Versuche den Anstieg des Aktivitätskoeffizienten bei höheren Konzentrationen
zu erklären stützen sich auf die Tatsache, daß der Elektrolyt die Dielektrizitätskonstante des
Lösungsmittels verändert. Dem wird durch die Addition eines konzentrationsabhängigen
Summanden zu dem von Debye und Hückel abgeleiteten Term Rechnung getragen. Solche
Ausdrücke enthalten in der Regel einen oder mehrere anpaßbare Parameter. Modelle dieser
Art wurden unter anderen von Davis [14], Guggenheim [20] und Bromley [7] vorgeschlagen.
Sie alle gehen wie die Debye-Hückel-Theorie davon aus, daß der Grund für das abnorme Ver-
halten der Elektrolytlösungen einzig in elektrostatischen Wechselwirkungen zwischen den
Ionen zu sehen ist.
2. Wesentliche Fortschritte bei der Beschreibung des Verhaltens von Elektrolytlösungen
brachten Modelle, die neben den rein elektrostatischen Wechselwirkungen der Ionen unterein-
ander auch Wechselwirkungen zwischen Ionen und Lösungsmittelmolekülen und den reinen

7
Lösungsmitteln berücksichtigen. Ursache dieser Wechselwirkungen sind Kräfte im Nahord-
nungsbereich der Teilchen, die nur über sehr kurze Distanzen wirken (‘short-range’-Wech-
selwirkungen). Stellvertretend für diese Gruppe von Modellen sei hier der zu Beginn der 70er
Jahre von Pitzer [38, 39, 40, 41, 42] entwickelte Virialansatz genannt. Pitzer erweiterte die
Theorie von Debye und Hückel mit Hilfe statistisch thermodynamischer Methoden und schlug
eine allgemeine Gleichung für die Gibbssche Enthalpie einer Elektrolytlösung vor. Der erste
Summand der Gleichung beschreibt nur die elektrostatischen Wechselwirkungen zwischen
den Ionen, und er ist ebenso wie der von Debye und Hückel abgeleitete Ausdruck neben der
Ionenstärke nur vom Ladungstyp des Elektrolyten abhängig. Der zweite Summand erfaßt
sämtliche binären Wechselwirkungen, die neben den rein elektrostatischen Effekten eine Rolle
spielen. Ein dritter Summand berücksichtigt den Beitrag der bei höheren Konzentrationen
auftretenden ternären Wechselwirkungen.
3. Die dritte Gruppe der Elektrolytmodelle unterscheidet ebenfalls die verschiedenartigen
Beiträge zur Gibbsschen Exzeßenthalpie der Lösung. Zur Beschreibung der Kräfte im Nahord-
nungsbereich wird auf das von Wilson vorgeschlagene Konzept der lokalen Zusammenset-
zung[56] zurückgegriffen. ‘Local-composition’-Modelle werden zur Berechnung der
thermodynamischen Eigenschaften von Nichtelektrolytsystemen erfolgreich eingesetzt. Ein
wesentlicher Vorteil ist dabei die leichte Erweiterung auf Multikomponentensysteme, da nur
binäre Wechselwirkungen betrachtet werden. Ihrer Natur nach berücksichtigen diese Modelle
aber nur Wechselwirkungen, die über sehr kurze Distanzen wirken, da dies die in Nichtelek-
trolytsystemen vorherrschenden Kräfte sind. Die Verhältnisse in Elektrolytlösungen sind in-
folge der zusätzlich auftretenden elektrostatischen Kräfte komplizierter. Das elektrostatische
Wechselwirkungspotential zwischen zwei Ionenladungen nimmt umgekehrt proportional zum
Abstand ab, während für typische ‘short-range’-Wechselwirkungen, wie sie beispielsweise
durch van der Waals Kräfte verursacht werden, eine Abhängigkeit umgekehrt zur 6. Potenz
des Abstandes angenommen werden kann. Die elektrostatischen Effekte zwischen den Ionen
wirken also über sehr viel größere Distanzen (‘long-range’-Wechselwirkungen) und müssen
daher gesondert betrachtet werden. Ein häufig beschrittener Weg zur Modellierung elektro-
lythaltiger Mischphasen besteht in der Annahme, daß ‘short-range’- und ‘long-range’-Effekte
als voneinander unabhängig aufgefaßt, und daher durch unterschiedliche theoretische Ansätze
beschrieben werden können. Das führt folgerichtig zu Modellen, bei denen sich die Gibbssche
Exzeßenthalpie aus zwei Anteilen zusammensetzt: (1) einem ‘long-range’-Anteil zur

8
Beschreibung der elektrostatischen Wechselwirkungen zwischen den Ionen auf der Grundlage
der Theorie von Debye und Hückel, und (2) einem ‘short-range’-Anteil basierend auf einem
‘local-composition’-Modell wie der Wilson-, NRTL- oder UNIQUAC-Gleichung. Zu den
Elektrolytmodellen dieser Gruppe gehören unter andern die Ansätze von Cruz und Renon
[12], Chen [9, 10], Ball und Fürst [3], Sander[52] und Macedo[30].
Neuere Ansätze wie der von Kikic und Fermeglia [28] ergeben sich aus dem Versuch das
Gruppenbeitragskonzept auf Elektrolytsysteme zu übertragen. Lösungsmittelmoleküle werden
dabei formal in Strukturgruppen zerlegt. Neben den Parametern zur Beschreibung der Wech-
selwirkungen zwischen den Strukturgruppen der Lösungsmittelmoleküle müssen zusätzlich
Parameter angepaßt werden, die die ‘short-range’-Effekte zwischen den Strukturgruppen und
den Ionen erfassen. Trotz des großen Potentials, das einem derartigen Gruppenbeitragsmodell
als Vorausberechnungsmethode zukommt, muß festgestellt werden, daß die Abweichungen
zwischen experimentellen und berechneten Werten bei Gruppenbeitragsmodellen durch-
schnittlich dreimal höher liegen als bei den zuvor beschriebenen ‘local-composition’-Model-
len. Häufig erhält man bei der Anwendung von Gruppenbeitragsmodellen nur qualitativ rich-
tige Ergebnisse.
Diese Klassifizierung der heute verfügbaren Elektrolytmodelle darf nicht darüberhinwegtäu-
schen, daß die überwiegende Anzahl der Modelle noch große Schwächen aufweist. So läßt die
Mehrzahl der publizierten Ansätze nicht die Berechnung von Systemen mit mehreren Lö-
sungsmittelkomponenten zu und ist zudem häufig nur für die Berechnung wäßriger Systeme
einsetzbar. Bei den Modellen die geeignet sind auch nichtwäßrige Mischungen aus mehreren
Lösungsmitteln und Salzen zu beschreiben, handelt es sich oft um reine Korrelationsansätze,
die die Beschreibung eines bestimmten Stoffsystems ermöglichen, wobei die Modellparameter
nicht auf andere Systeme übertragbar sind. Ein anderer Teil dieser Methoden gestattet wie-
derum nicht die Berechnung der binären Randsysteme. Nur wenige Ansätze wie die Modelle
von Sander, Macedo und Kikic sind für eine beliebige Anzahl von Elektrolyt- und Lösungs-
mittelkomponenten geeignet. Sie besitzen gleichzeitig ein gewisses Potential als Vorausbe-
rechnungsmethode, wenn die Modellparameter auf der Grundlage einer breiten Datenbasis
bestimmt werden.

9
Li und Polka [29] entwickelten an der Universität Oldenburg ein Modell, das die wesentliche
Konzeption der Verknüpfung eines elektrostatischen Anteils mit einem ‘local-composition’-
Term beibehält. Durch die zusätzliche Berücksichtigung von Ion-Ion- und Ion-Dipol-Wech-
selwirkungen, die weder vom elektrostatischen noch vom ‘local-composition’-Term erfaßt
werden können, überwindet dieser Ansatz jedoch viele Schwächen der älteren Modelle insbe-
sondere bei der Beschreibung binärer Systeme im Bereich hoher Konzentration. Dieser soge-
nannte LIQUAC-Ansatz wurde bereits erfolgreich zur Berechnung und Vorhersage von
Dampf-Flüssig-Gleichgewichten elektrolythaltiger Mischungen getestet.

10
1.1 Zielsetzung
Im ersten Teil der vorliegenden Arbeit soll die Anwendbarkeit des von Li und Polka ent-
wickelten GE-Ansatzes zur Berechnung von Fest-Flüssig-Gleichgewichten überprüft werden.
Dazu war der Aufbau einer Datenbank zur Speicherung und effizienten Nutzung der benötig-
ten Löslichkeitsdaten erforderlich. Ziel ist es den mittleren Aktivitätskoeffizienten des Elektro-
lyten und die Lösungsmittelaktivität über einen großen Temperatur- und Konzentrati-
onsbereich richtig zu beschreiben. Dabei sollen erstmalig Salzlöslichkeiten in die Anpassung
der LIQUAC-Modellparameter mit einfließen. Löslichkeitsdaten lassen sich nur in Verbindung
mit thermodynamischen Standardgrößen als Quelle experimenteller Information nutzen. Ne-
ben der Beschreibung wäßriger Systeme soll geprüft werden, inwieweit sich das Konzept der
Löslichkeitsberechnung auf der Grundlage thermodynamischer Daten unter Berücksichtigung
des realen Verhaltens auf nichtwäßrige Lösungsmittel und deren Mischungen übertragen läßt.
Durch Überarbeitung der Parametermatrix auf der Grundlage der neuen Datenbasis (VLE plus
SLE) sollen einheitliche Parametersätze gefunden werden, die die simultane Beschreibung von
Dampf-Flüssig- und Fest-Flüssig-Gleichgewichten erlauben.
Ziel des zweiten Teils dieser Arbeit ist die Vermessung von Dampf-Flüssig-Gleichgewichten
einer Reihe von Lösungsmittelgemischsystemen, über die noch keine bzw. unvollständige Da-
ten vorliegen. Die Auswahl der Systeme erfolgt dabei im Hinblick auf eine Vervollständigung
der Wechselwirkungsparametermatrix des im ersten Teil angewendeten Berechnungsmodells.
Es sollen Messungen isobarer Siedekurven unter Verwendung einer nach dem dynamischen
Prinzip arbeitenden Gleichgewichtsapparatur mit Kreislaufführung von Dampf- und Flüssig-
phase, und gaschromatographische Dampfraumanalysen mit Hilfe einer Headspace-Apparatur
durchgeführt werden.

11
2. Thermoynamische Grundlagen und Definitionen
2.1 Phasengleichgewicht
Ausgangspunkt für die Beschreibung von Phasengleichgewichten ist das aus dem Gibbsschen
Extremalprinzip (Minimum der Energie, Maximum der Entropie) abgeleitete Isofugazitätskri-
terium, wonach in einem heterogenen System Gleichgewicht vorliegt, wenn neben Druck und
Temperatur auch die Fugazität der einzelnen Komponenten in allen Phasen gleiche Werte auf-
weisen [44, 19, 45]. Für ein zweiphasiges System bedeutet dies also:
(2.1.1)
nKomponenteder Anzahl :nmit 1,2,.....n=i '''
'''
'''
ii ff
PP
TT
===
Die Fugazität ist dabei eine Hilfsgröße, die von Lewis ursprünglich zur Beschreibung des Ver-
haltens realer Gase eingeführt wurde. Sie stellt den mit dem Fugazitätskoeffizienten korrigier-
ten Partialdruck einer Mischphasenkomponente dar:
(2.1.2) Pypf iiiii ϕϕ == mit 1lim0
=→ i
Pϕ
Grundsätzlich läßt sich auch eine flüssige Mischphase mit Hilfe von Fugazitätskoeffizienten
beschreiben. Drückt man den Partialdruck durch das Produkt von Molenbruch und
Gesamtdruck aus, so lautet die Bedingung für den Fall des Gleichgewichts zwischen einer
Dampf- und Flüssigphase:
(2.1.3) y xi iV
i iLϕ ϕ= mit y i : Molenbruch in der Dampfphase
x i : Molenbruch in der Flüssigphase
ϕ iV : Fugazitätskoeffizient im Dampf
ϕ iL : Fugazitätskoeffizient in der Flüssigkeit
(jeweils für Komponente i)

12
Für die Berechnung der Fugazitätskoeffizienten als Funktion von Temperatur, Druck und Zu-
sammensetzung müssen Zustandsgleichungen verwendet werden. Erst in neuerer Zeit ist es
gelungen mit Hilfe geeigneter Mischungsregeln auch bei der Beschreibung polarer Systeme zu
brauchbaren Ergebnissen zu gelangen [21, 22]. Die Verwendung von Zustandsgleichungen für
die Berechnung elektrolythaltiger Mischphasen steckt allerdings noch in den Anfängen. Viel-
versprechende Ansätze wurden beispielsweise von Dahl [13], Simon [53], Hovey [23] und Jin
[27] formuliert. Im allgemeinen geht man hier aber einen anderen Weg. Dazu bedient man sich
des Zusammenhangs zwischen dem chemischen Potential iµ und der Fugazität if :
(2.1.4) 00 ln
i
iii f
fRT+= µµ
Das Potential µ i0 bezieht sich auf einen beliebig wählbaren Standardzustand. 0
if ist die dazu-
gehörige Standardfugazität. Die auf den Standardwert bezogene Fugazität der Komponente i
wird als Aktivität ai bezeichnet und läßt sich über das Produkt von Konzentration und Aktivi-
tätskoeffizient ausdrücken:
(2.1.5) iiii
i af
fγς==
0
ς i ist hier ein beliebiges Konzentrationsmaß. Gl. (2.1.5.) kann zur Beschreibung der Fugazität
in der Flüssigphase herangezogen werden, während die Fugazität in der Dampfphase gemäß
Gl. (2.1.2) über den Fugazitätskoeffizienten ausgedrückt wird. Auf diese Weise erhält man als
Bedingung für das Gleichgewicht zwischen Dampf- und Flüssigphase:
(2.1.6) 0iiiii fxp γϕ =
wobei als Konzentrationsmaß der Molenbruch der Komponente i verwendet wird. Für Lö-
sungsmittelkomponenten ist die Standardfugazität üblicherweise definiert als die Fugazität der
reinen Komponente bei Systemdruck und Systemtemperatur. Sie läßt sich über den Sätti-
gungsdampfdruck der reinen Komponente ausdrücken, wenn man die Abweichung zwischen
System- und Sättigungsdampfdruck durch einen zusätzlichen Korrekturfaktor erfaßt:

13
(2.1.7) isi
sii PoyPf ϕ=0 s
iP :Sättigungsdampfdruck
siϕ :Fugazitätskoeffizient im Sättigungszustand
Poyi : Poynting-Faktor
Der auf diese Weise eingeführte sogenannte Poynting-Faktor kann näherungsweise über das
Molvolumen der reinen Komponenten ν i und der Differenz zwischen System- und Sätti-
gungsdampfdruck berechnet werden:
(2.1.8.) ( )
−≅
RT
PPPoy
sii
i
νexp
ϕ iS ist der Fugazitätskoeffizient für die reine Komponente beim Sättigungsdampfdruck. Aus
Gl. (2.1.7) und (2.1.6) folgt für die Gleichgewichtsbedingung:
(2.1.9) iVi
siS
iiii PoyPxPyϕϕ
γ=
Für Druckdifferenzen zwischen Sättigungsdampfdruck und Systemdruck im Bereich von 100
kPa liegt der Wert des Poynting-Faktors praktisch bei eins, so daß er bei Systemdrucken, die
den Atmosphärendruck nicht wesentlich überschreiten meist vernachlässigt wird. Ebenso
kann das Verhältnis der Fugazitätskoeffizienten für den Sättigungszustand und die Dampf-
phase bei normalen Drucken näherungsweise gleich eins gesetzt werden, vorausgesetzt, es
liegen nur schwach assoziierende Komponenten vor. Gl. (2.1.9) läßt sich somit weiter verein-
fachen:
(2.1.10) siiii PxPy γ=
Gl. (2.1.10) gestattet die Berechnung der Dampfphasenzusammensetzung bei vorgegebener
Zusammensetzung der Flüssigphase. Erforderlich ist dafür die Kenntnis des Sättigungsdampf-
drucks und des Aktivitätskoeffizienten für jede Komponente.

14
2.2 Exzeßpotential und Aktivitätskoeffizient
G.N. Lewis formulierte den Zusammenhang zwischen der Aktivität und dem chemischen Po-
tential einer Komponente i:
(2.2.1) iii aRT ln0 += µµ
Mit der Definition der Aktivität gemäß Gl. (2.1.5) läßt sich das chemische Potential einer
Komponente in einer Mischphase auch schreiben als:
(2.2.2) iiii RTxRT γµµ lnln0 ++=
Andererseits gilt für das chemische Potential einer idealen Mischung:
(2.2.3) iiideali xRT ln0 += µµ
Der letzte Summand in Gl. (2.2.2) beschreibt somit die Abweichung vom idealen Verhalten
und wird als Exzeßpotential bezeichnet:
(2.2.4) iEi RT γµ ln=
Eine Verknüpfung dieser Größe mit der Gibbsschen Enthalpie folgt unmittelbar aus der De-
finition des chemischen Potentials als partielle molare Größe der Gibbsschen Enthalpie:
(2.2.5) ijnPTi
EEi n
G
≠
=,,
∂∂
µ
Demnach läßt sich der Aktivitätskoeffizient durch Ableitung der Gibbsschen Exzeßenthalpie
nach der Molzahl der betreffenden Komponente berechnen. Die Gibbssche Enthalpie stellt da-
bei die Differenz zwischen der Gibbsschen Enthalpie der realen und idealen Mischung dar:
(2.2.6) idealrealE GGG −=

15
Ihr Wert setzt sich additiv aus den Exzeßpotentialen der einzelnen Komponenten zusammen:
(2.2.7) ∑ ∑==i i
iiEii
E nRTnG γµ ln ni ,....,1=
Aufgrund der Definition der Gibbsschen Enthalpie läßt sich eine Zerlegung in Exzeßenthalpie
und -entropie vornehmen:
(2.2.8) EEE TSHG −=
Ein Modellansatz für die Gibbssche Exzeßenthalpie muß demnach beide Einflüsse berück-
sichtigen. Ein mathematischer Ausdruck für die Exzeßenthalpie kann auf der Grundlage der
Vorstellung über die Wechselwirkungskräfte zwischen den Teilchen abgeleitet werden. Für die
Exzeßentropie müssen Vorstellungen über die unterschiedliche Form und Größe der Teilchen
eingehen.
2.3 Normierung des Aktivitätskoeffizienten
Die Beschreibung von Mischphasen mit Hilfe von Aktivitätskoeffizienten erfordert gemäß Gl.
(2.1.5) die Festlegung einer Standardfugazität. Die Standardfugazität ist für die Elektrolyt- und
Lösungsmittelkomponente frei wählbar.
Die für Lösungsmittelkomponenten übliche Standardfugazität ist die reine Komponente bei
Systemdruck und Systemtemperatur. Der Aktivitätskoeffizient ist für diesen Zustand auf den
Wert eins normiert:
(2.3.1) 1lim1
=→ix
iγ
Im Gegensatz dazu verwendet man als Standardzustand für starke Elektrolyte die 1 molale
ideale Lösung. Der Grund dafür liegt darin, daß starke Elektrolyte als reine Komponente unter
den Temperatur und Druckbedingungen der Mischung in einen anderen Aggregatzustand
übergehen. Während Salze in den festen Aggregatzustand übergehen liegen flüchtige Elektro-
lyte wie HCl gasförmig vor, so daß der in Gl. (2.3.1) beschriebene Grenzübergang in beiden
16
Fällen mit einer Phasenumwandlung verbunden ist. Weil starke Elektrolyte im Zustand un-
endlicher Verdünnung nicht molekular sondern als solvatisierte Ionen vorliegen lautet die
Normierungsbedingung:
(2.3.2) 1lim0
=∑ →
∗
jjx
iγ Ionnj ,...,1=
Die Summation ist über alle in der Lösung vorhandene Ionen auszuführen. Der auf diese
Weise normierte Aktivitätskoeffizient wird zwecks Unterscheidung zu der bei Lösungsmittel-
komponenten üblichen Normierung durch einen Stern gekennzeichnet. Die durch Gl. (2.3.1)
eingeführte Normierung wird als ‘symmetrisch’ bezeichnet, während Gl. (2.3.2) die
‘unsymmetrische’ Normierung definiert.
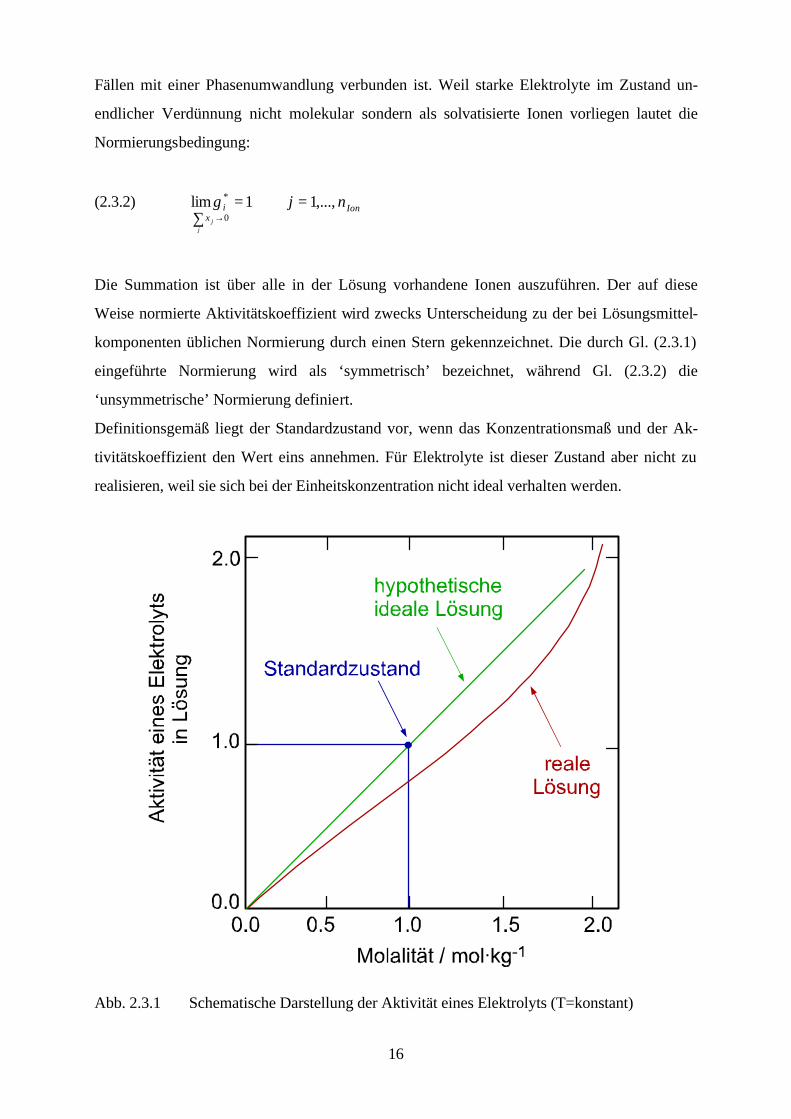
Definitionsgemäß liegt der Standardzustand vor, wenn das Konzentrationsmaß und der Ak-
tivitätskoeffizient den Wert eins annehmen. Für Elektrolyte ist dieser Zustand aber nicht zu
realisieren, weil sie sich bei der Einheitskonzentration nicht ideal verhalten werden.
Abb. 2.3.1 Schematische Darstellung der Aktivität eines Elektrolyts (T=konstant)

17
Deshalb definiert man für sie einen Bezugszustand als den Zustand für den der
Aktivitätskoeffizient mit dem Wert eins normiert ist. Für Lösungsmittelkomponenten ist eine
solche Unterscheidung nicht notwendig, da bei ihnen Standard- und Bezugszustand identisch
sind.
Vergleicht man die Standardzustände der symmetrischen und unsymmetrischen Normierung
miteinander fällt auf, daß es sich im ersten Fall um eine Reinstoffgröße, und damit um einen
real existierenden Standardzustand handelt, während die unendliche Verdünnung ein physika-
lisch nicht realisierbarer Zustand ist, und sie somit einen hypothetischen Standardzustand dar-
stellt. Eine Schwierigkeit, die direkt aus der unsymmetrischen Normierung folgt ist, daß sich in
Mehrkomponenten-Systemen unterschiedliche Standardfugazitäten für die jeweils betrachtete
Komponente ergeben. Rozen [51] wies ferner darauf hin, daß bei unsymmetrischer Nor-
mierung alle Elektrolyte eines Valenztyps im Bereich geringer Konzentration identisch zu sein
scheinen, obwohl die unterschiedlichen Ionen einer Gruppe große Unterschiede in ihrem Hy-
dratationsverhalten zeigen. Aus diesen Gründen wurde verschiedentlich vorgeschlagen bei
nichtflüchtigen Elektrolyten den hypothetischen Zustand der unterkühlten Schmelze, bzw. wie
im Fall der Lösungsmittel, die reine Komponente, also das reine feste Salz, als Bezugszustand
zu wählen.
2.4 Chemisches Potential des Ions
Unter Verwendung des Standardzustandes der unendlich verdünnten Lösung lautet der Aus-
druck für das chemische Potential eines individuellen Ions:
(2.4.1) µ µ ς γi i i iRT= + ∗0 ln( )
Die Werte des Standardpotentials und der Aktivität sind von der Wahl des verwendeten Kon-
zentrationsmaßes ς i abhängig. Während bei Lösungsmittelkomponenten praktisch
ausschließlich der Molenbruch verwendet wird, ist bei Elektrolyten zusätzlich die Molalität m
und die Molarität c gebräuchlich. Entsprechend hat man die Aktivitätskoeffizienten und
Standardpotentiale hinsichtlich der jeweiligen Konzentrationsskala zu unterscheiden:

18
(2.4.2) )ln( )(,0
)(,∗+= xiixii xRT γµµ
(2.4.3) )ln( )(,0
)(,∗+= miimii mRT γµµ
(2.4.4) )ln( )(,0
)(,∗+= ciicii cRT γµµ
Das verwendete Konzentrationsmaß ist hier durch einen entsprechenden Index gekennzeich-
net. Da der Aktivitätskoeffizient in jedem Fall eine dimensionslose Größe ist muß die Kon-
zentration im Fall der Molalität und der Molarität formal auf die Einheitskonzentration bezo-
gen werden. Die auf der Grundlage unterschiedlicher Konzentrationsskalen definierten Aktivi-
tätskoeffizienten lassen sich ineinander umrechnen, da das chemische Potential unabhängig
vom Konzentrationsmaß denselben Wert aufweist. Die Umrechnung zwischen dem Aktivi-
tätskoeffizienten in der Konzentrationsskala des Molenbruchs und der Konzentrationsskala
der Molalität erfolgt nach der Beziehung [50] :
(2.4.5.) ∑+= ∗∗
jjsmixi mM )1()(,)(, γγ Ionnj ,.....,1= mit M s : Masse Lösungsmittel
Bei praktischen Berechnungen hat die Molarität den entscheidenden Nachteil der Volumen-
und damit der Temperaturabhängigkeit. Aus diesem Grunde wird die Molarität seltener als
Konzentrationsmaß verwendet, und die entsprechenden Umrechnungsformeln werden
deshalb hier nicht angegeben.
Im folgenden soll der in der Konzentrationsskala des Molenbruchs definierte Aktivitätskoeffi-
zient entsprechend der Konvention bei den Lösungsmittelkomponenten nicht weiter gekenn-
zeichnet werden. Ein Aktivitätskoeffizient ohne Index bezieht sich also immer auf die Konzen-
trationsskala des Molenbruchs.
2.5 Chemisches Potential des Elektrolyten und mittlerer Aktivitätskoeffizient
Meßtechnisch ist das chemische Potential eines individuellen Ions nicht zugänglich, da eine
Elektrolytlösung zwangsläufig sowohl Kationen als auch Anionen enthält, deren Einflüsse
nicht voneinander getrennt betrachtet werden können. Es ist daher notwendig ein mittleres
chemisches Potential des Elektrolyten einzuführen. Unter der Voraussetzung vollständiger
Dissoziation läßt es sich in einen Beitrag des Kations und einen Beitrag des Anions aufteilen:

19
(2.5.1) −−++± += µνµνµ
+ν und −ν sind die stöchiometrischen Koeffizienten der Dissoziationsreaktion und geben an,
wieviel Mole des betreffenden Ions bei der vollständigen Dissoziation von einem Mol des
Elektrolyten entstehen. Einsetzen des Ausdrucks aus Gl. (2.4.1) für das chemische Potential
des Ions ergibt:
(2.5.2) )ln()ln(00 −+−+−+−+−−++± +++= νννν γγςςµνµνµ RTRT
Die ersten beiden Summanden werden zum Standardpotential des Elektrolyten 0±µ zusam-
mengefaßt. Aus den Argumenten im Logarithmus des dritten und vierten Summanden folgt
die Definition der mittleren Konzentration und des mittleren Aktivitätskoeffizienten:
(2.5.3) −+−+± = ννν ςςς mit −+ += ννν
(2.5.4) −+ ∗−
∗+
∗± = ννν γγγ
Mit diesen Definitionen lautet der Ausdruck für das chemische Potential des Elektrolyten:
(2.5.5) )ln(0±±±± += γςνµµ RT
Für die Umrechnung des mittleren Aktivitätskoeffizienten von der Konzentrationsskala des
Molenbruchs in die Konzentrationsskala der Molalität kann ebenfalls Gl. (2.4.5) verwendet
werden, wenn dort anstelle der individuellen Aktivitätskoeffizienten der Ionen die mittleren
Größen für den Elektrolyten eingesetzt werden.
2.6 Definition der Gemischvariablen für starke Elektrolyte
Bei vollständiger Dissoziation aller Elektrolytkomponenten sind die Molenbrüche der Ionen
und Lösungsmittel folgendermaßen definiert:

20
(2.6.1) ∑ ∑+
=' '
''
s jjs
ss nn
nx sns ...,2,1' = jnj ...,2,1' =
(2.6.2) ∑ ∑+
='
''
's jjs
jj nn
nx
Der Index s bezeichnet eine Lösungsmittelkomponente, der Index j ein Ion. Im Zusammen-
hang mit Elektrolyt-Lösungsmittelgemischsystemen wird häufig der sogenannte salzfreie
Molenbruch verwendet, da er den direkten Vergleich mit dem entsprechenden Nichtelektro-
lytsystem gleicher Lösungsmittelzusammensetzung ermöglicht:
(2.6.3) ∑∑
=='
'
'
'
'
ss
s
ss
ss x
x
n
nx
xs' und xs sind über die Molenbrüche der Ionen miteinander verknüpft:
(2.6.4) )1(' ∑−=j
jss xxx jnj ,...2,1=
Das heute gebräuchlichste Konzentrationsmaß im Zusammenhang mit Elektrolyt-Lösungsmit-
telgemischen ist die Molalität. Sie ist kein relatives Konzentrationsmaß wie der Molenbruch
aber ebenso dichte- und deshalb temperaturunabhängig. Für die Molalität eines Ions in einem
Lösungsmittelgemisch kann allgemein geschrieben werden:
(2.6.5) j
sss
j nMn
m∑
=1000
sns ....,2,1=
Die Molenbrüche der Ionen lassen sich über folgende Beziehung aus den Molalitäten der Io-
nen berechnen:

21
(2.6.6)
∑ ∑−
+
=
'
'
1'
j sssj
jj
Mxm
mx
Da die Abweichung vom idealen Verhalten durch die Ladung der Ionen verursacht wird,
bewirken zweiwertige Ionen eine größere Abweichung als einwertige, dreiwertige eine größere
als zweiwertige usw.. Aus diesem Grund ist es notwendig die ‘Gesamtkonzentration der La-
dungen’ und nicht die Gesamtkonzentration der Ionen einzuführen. Die zu diesem Zweck
eingeführte Ionenstärke ist folgendermaßen definiert:
(2.6.7) ∑=j
jj zmI 2
21
Ionnj ...,2,1=
Die Ladung zj ist die dimensionslose Elementarladung, so daß die Ionenstärke in derselben
Einheit wie die Konzentration ausgedrückt wird. Der Faktor ½ ist in der Definition enthalten,
damit die Ionenstärke für die Lösung eines 1:1 Elektrolyten identisch mit seiner Konzentration
ist. Die durch Gl. (2.6.7) eingeführte Beziehung geht auf G.N. Lewis und M. Randall zurück,
die fanden, daß in verdünnten Lösungen der Logarithmus des Aktivitätskoeffizienten der
Quadratwurzel aus der Ionenstärke direkt proportional ist.
2.7 Osmotischer Koeffizient
Alternativ zum Aktivitätskoeffizienten wird die Abweichung der Lösung vom idealen Verhal-
ten häufig auch durch den osmotischen Koeffizienten angegeben. Er ist definiert als das
Verhältnis des realen osmotischen Drucks der Lösung zum osmotischen Druck einer sich ideal
verhaltenden Lösung gleicher Konzentration:
(2.7.1) ideal
real
ΠΠ
=ϕ
Sind Lösungsmittel und Lösung durch eine semipermeable Membran voneinander getrennt,
die nur das Lösungsmittel hindurchläßt nicht aber den gelösten Stoff, so fließt das Lösungs-

22
mittel solange durch die Membran auf die Seite der Lösung bis das thermodynamische
Gleichgewicht erreicht ist. Der sich dabei aufbauende osmotische Druck kann über die
Isofugazitätsbedingung abgeleitet werden. Ist fs' die Fugazität des reinen Lösungsmittels und
fs'' die Fugazität des Lösungsmittels auf der Seite der Lösung, so gilt:
(2.7.2) ( ) ( )2''
1' PfPf ss =
Die Fugazitäten lassen sich gemäß Gl. (2.1.5) durch das Produkt von Aktivität und Standard-
fugazität beschreiben. Letztere kann über den Sättigungsdampfdruck ausgedrückt werden Gl.
(2.1.7). Berücksichtigt man, daß die Aktivität des reinen Lösungsmittels gleich eins ist, so folgt
unter Verwendung der Näherungsbeziehung Gl.(2.1.8) für den Poynting-Faktor:
(2.7.3) ( ) ( )
−=
−RT
PPPa
RT
PPP
SssS
sSss
SssS
sSs
11 expexpν
ϕν
ϕ
Die Differenz 12 PP − entspricht dem gesuchten osmotischen Druck. Aus Gl. (2.7.3) erhält
man:
(2.7.4) s
sreal aRTPP
νln
12 −=Π=−
Für den Fall idealen Verhaltens kann die Aktivität durch den Molenbruch ersetzt werden:
(2.7.5) s
sideal xRT
νln
−=Π
Aus Gl. (2.7.4) und (2.7.5) ergibt sich unter Verwendung der Definitionsgleichung (2.7.1) ein
einfacher Zusammenhang zwischen Molenbruch und Aktivität des Lösungsmittels:
(2.7.6) ss xa lnln ϕ=

23
Der so definierte sogenannte rationale osmotische Koeffizient stellt also das Verhältnis der
Logarithmen von Aktivität und Molenbruch des Lösungsmittels dar. Unter Annahme voll-
ständiger Dissoziation läßt sich der Molenbruch über die Molalitäten der Ionen ausdrücken:
(2.7.7) ∑∑ +
=+
=
jjs
jjs
ss mMnn
nx
11
Einsetzen in Gl. (2.7.6) ergibt:
(2.7.8)
+
−−= ∑∑ .......
21
ln2
jjs
jjss mMmMa ϕ
Bricht man die Reihe nach dem ersten Glied ab, so erhält man die Definitionsgleichung des
molalen osmotischen Koeffizienten φ :
(2.7.10) ∑−=j
jss mMa ln φ
Letzterer wird in der Praxis häufiger verwendet. Mit abnehmender Konzentration der Lösung
werden die Beiträge der höheren Glieder der Reihenfunktion in Gl. (2.7.9) immer geringer, daß
rationaler und molaler osmotischer Koeffizient mit zunehmender Verdünnung ineinander
übergehen. Der osmotische Koeffizient liefert als Maß für die Abweichung vom idealen Ver-
halten einen signifikanteren Zahlenwert als der Aktivitätskoeffizient. In einer 2-molalen KCl-
Lösung beispielsweise beträgt der Aktivitätskoeffizient des Wassers 1.004, während der mitt-
lere Aktivitätskoeffizient des Elektrolyten mit einem Wert von 0.614 bereits ein stark reales
Verhalten anzeigt. Der Wert des molalen osmotischen Koeffizienten von 0.912 hingegen
weicht viel deutlicher von eins ab als der Wert des Aktivitätskoeffizienten.

24
2.8 Gibbs-Duhem Beziehung
Mit Hilfe des Eulerschen Satzes [57, 33] läßt sich zeigen, daß die Gibbssche Enthalpie einer
Mischung aus den chemischen Potentialen der einzelnen Komponenten zusammengesetzt ist:
(2.8.1) ∑=i
iinG µ
Bildet man aus Gl.(2.8.1) das totale Differential, so ergibt sich:
(2.8.2) ∑ ∑+=i i
iiii dndndG µµ
Andererseits lautet die Fundamentalgleichung der Gibbsschen Enthalpie:
(2.8.3) ∑++−=i
ii dnVdPSdTdG µ
Durch Vergleich von Gl. (2.8.2) mit Gl. (2.8.3) erhält man:
(2.8.4) ∑ =++−i
ii dnVdPSdT 0µ
Diese Gleichung zeigt, daß die chemischen Potentiale der einzelnen Komponenten nicht von-
einander unabhängig sind. Sie ist unter der Bezeichnung Gibbs-Duhem-Gleichung bekannt
und spielt eine wichtige Rolle in der Mischphasenthermodynamik. Für ein binäres System bei
konstantem Druck und konstanter Temperatur folgt als weitere Vereinfachung:
(2.8.5) 2211 µµ dndn −= 0=dP 0=dT
Mit Hilfe dieser Gleichung läßt sich für den Fall einer binären Elektrolytlösung ein Zusam-
menhang zwischen der Aktivität des Lösungsmittels und der Aktivität des Elektrolyten
ableiten. Berücksichtigt man, daß die Standardpotentiale unabhängig von der
Zusammensetzung sind und damit bei der Differentiation wegfallen, ergibt sich durch

25
Einsetzen der Ausdrücke für das chemische Potential des Lösungsmittels Gl. (2.2.2) bzw. des
Elektrolyten Gl.(2.5.5):
(2.8.6) )ln( ln )(∗±−= mss mdmMad γν mit m: Molalität des Elektrolyten
Der Aktivitätskoeffizient des Elektrolyten ist hier in der Konzentrationsskala der Molalität
ausgedrückt. Durch Gl. (2.7.10) läßt sich die Aktivität des Lösungsmittels über den molalen
osmotischen Koeffizienten ausdrücken. Mit:
(2.8.7) ∑ =j
j mm ν
erhält man dann eine Bestimmungsgleichung für den mittleren Aktivitätskoeffizienten des
Elektrolyten bei Kenntnis des molalen osmotischen Koeffizienten:
(2.8.8) ( )mdm
dd m 1ln )( −+=∗± φφγ
Für die Integration dieser Gleichung wird zweckmäßigerweise die unendlich verdünnte Lö-
sung als untere Integrationsgrenze gewählt. Der Aktivitätskoeffizient des Elektrolyten und der
osmotische Koeffizient nehmen dann den Wert eins an:
(2.8.9) ( )∫ −+−=∗±
m
m mdm
0)( 11ln φφγ
Gl. (2.8.9) ermöglicht die Berechnung des mittleren Aktivitätskoeffizienten des Elektrolyten
bei der Molalität m, wenn der Verlauf des osmotischen Koeffizienten im Konzentrationsinter-
vall 0 bis m bekannt ist. Umgekehrt kann natürlich auch der osmotische Koeffizient aus dem
mittleren Aktivitätskoeffizienten abgeleitet werden:
(2.8.10) ∫ ∗±+=
m
mmdm 0
)(ln1
1 γφ

26
Gl. (2.8.9) und (2.8.10) verdeutlichen, daß der Verlauf des osmotischen Koeffizienten in Ab-
hängigkeit von der Konzentration des Elektrolyten mit gegebenem Verlauf des mittleren Ak-
tivitätskoeffizienten festgelegt ist und umgekehrt.

27
3. GE-Ansatz zur Beschreibung elektrolythaltiger Mischphasen
Der in dieser Arbeit verwendete GE-Ansatz zur Beschreibung elektrolythaltiger Mischphasen
wurde 1994 von Li und Polka vorgeschlagen [29]. Er ist für die Berechnung einer beliebigen
Anzahl von Elektrolyt- und Lösungsmitelkomponenten geeignet, und berücksichtigt alle in
einer Elektrolytlösung auftretenden wichtigen Wechselwirkungen. Gemäß der Reichweite der
ihnen zugrundeliegenden intermolekularen Kräfte werden die Wechselwirkungen in drei Grup-
pen zusammengefaßt, so daß sich die Gibbssche Exzeßenthalpie als Summe dreier Anteile er-
gibt:
(3.1) Erangeshort
Erangemiddle
Erangelong
E GGGG −−− ++=
Gemäß Gl. (2.2.5) folgt daraus für die Aktivitätskoeffizienten:
(3.2) rangeshortnrangemiddlenrangelongnn −−− ++= ,,, lnlnlnln γγγγ
Im folgenden soll auf die einzelnen Beiträge eingegangen werden.
3.1 Beitrag der ‘long-range’-Wechselwirkungen
Der ‘long-range’-Term erfaßt die elektrostatischen Effekte zwischen den Ionen. Diese wirken
über den bei weitem größten Abstand und stellen eine Besonderheit der Elektrolytsysteme dar.
Das Wechselwirkungspotential ui,j zweier Ionen nimmt umgekehrt proportional zur ersten Po-
tenz ihres Abstandes ri,j ab.
Der Ausdruck, der diesen elektrostatischen Beitrag zur Gibbsschen Exzeßenthalpie der Lö-
sung erfaßt wurde aus dem von Macedo [30] vorgeschlagenen Ansatz übernommen. Macedo
hatte als erste eine theoretisch abgesicherte Anwendung der Debye-Hückel-Theorie auf den
Fall eines Lösungsmittelgemisches verwirklicht. Sie stützte sich dabei auf eine Arbeit von
Cardoso und O’Connel [8], die zeigen konnten, daß die von Debye und Hückel abgeleiteten
Ausdrücke für die Aktivitätskoeffizienten auch für Elektrolyt-Lösungsmittelgemische gelten,
wenn dort anstelle der Dielektrizitätskonstante des reinen Lösungsmittels der Wert des Lö-
sungsmittelgemisches eingesetzt wird. Dieses Ergebnis ist keinesfalls trivial, da man durch

28
Ableitung der Exzeßfunktion nach den Molzahlen der Komponenten infolge der Abhängigkeit
der Dielektrizitätskonstanten von der Zusammensetzung zu einem ganz anderen Resultat
kommt. Der verwendete Ausdruck für den ‘long-range’-Beitrag zum Aktivitätskoeffizienten
hat für eine Lösungsmittelkomponente s die Form:
3.1.1
+−
+−+= m
m
ms
srls Ib
IbIb
db
dAM1ln(2
1
11
2ln
3..γ
Und für ein Ion j :
3.1.2 m
mjrlj
Ib
IAz
+−=∗
1ln
2..γ
Der Faktor z bezeichnet die Ladungszahl des Ions j, die Debye-Hückel-Konstanten A und b
sind gemäß Gleichung (3.1.3) und (3.1.4) definierte Funktionen der Temperatur und der
Dielektrizitätskonstanten des salzfreien Lösungsmittelsgemisches:
3.1.3 ( )
( ) 2/3
2/130
2
DkT
dNeA sAπ
=
3.1.4
=
DkT
dNeab sA
208π
Gl. (3.1.3) und (3.1.4) gelten für das elektrostatische Einheitensystem. Für die Übertragung in
das SI-System ist es erforderlich die Dielektrizitätskonstante D mit der elektrischen Feldkon-
stante ε0 und dem Faktor 4π zu multiplizieren. Die Berechnung der Dielektrizitätskonstante
des Lösungsmittelgemisches erfolgt unter Verwendung einer von Oster aufgestellten Mi-
schungsregel [36, 37] aus den Dielektrizitätskonstanten der reinen Komponenten. Danach gilt
für ein binäres Gemisch mit den Reinstoffwerten D1 und D2 :
3.1.5 ( )( ) ( )
νν 2'
212
221 1
2121
xDD
DDDD
−−
+−+=

29
Das gesamte molare Volumen ν der Mischung ist wieder näherungsweise additiv aus den
Molvolumina der reinen Komponenten zugänglich:
3.1.6 ∑=n
nnx νν '
Letztere sind über die Reinstoffdichten und Molvolumina gegeben. Gleichung (3.1.5) wurde
von Oster anhand einer Reihe wäßriger Lösungen verschiedener Nichtelektrolytkomponenten
überprüft. Eine zuverlässige Berechnung der Dielektrizitätskonstanten eines Gemisches aus
mehr als zwei Komponenten ist allerdings nicht möglich. Für diesen Fall wird in stark verein-
fachender Weise ein linearer Ansatz vorgeschlagen:
3.1.7 nn
nDxD ∑= '
Die für die Bestimmung der Gemischwerte der Dielektrizitätskonstanten und der Dichte erfor-
derlichen Reinstoffgrößen müssen in Abhängigkeit von der Temperatur bekannt sein. Im Falle
der Dichte wurden dazu die DIPPR-Gleichungen verwendet [17]. Die Dielektrizitätskonstanten
reiner Lösungsmittel sind mit Angabe von Temperaturkoeffizienten in der Datensammlung
von Maryott und Smith aufgeführt [31]. Durch Zusammenfassen der durch Gl. (3.1.3) und
(3.1.4) definierten Debye-Hückel-Konstanten ergeben sich die für Berechnungszwecke besser
handhabbaren Ausdrücke:
3.1.8 ( ) 2/3
2/1510327757.1
DT
dA ⋅=
3.1.9 ( ) 2/1
2/1
359696.6DT
db =

30
3.2 Beitrag der ‘middle-range’-Wechselwirkungen
Sowohl die Wechselwirkungen zwischen Ionen und Dipolen als auch die Wechselwirkungen
zwischen Ionen und induzierten Dipolen nehmen hinsichtlich der Abhängigkeit des Potentials
vom Abstand der wechselwirkenden Teilchen eine Mittelstellung ein. Sie treten nur in Elektro-
lytlösungen auf und werden vom LIQUAC-Modell gesondert im sogenannten ‘middle-range’-
Term berücksichtigt.
Besitzen Lösungsmittelmoleküle einer elektrolythaltigen Mischung ein permanentes Dipolmo-
ment, so kommt es gemäß der folgenden Potentialfunktion zu einer Wechselwirkung der Teil-
ladungen des Dipols mit den Ionenladungen:
3.2.1 2,
cos
ji
jiij Dr
Qu
θµ−=
Hierbei ist Q i die Ladung des Ions, µ j das permanente Dipolmoment des Moleküls, θ der
Winkel, den die Dipolachse mit der Verbindungslinie Ion-Dipol einschließt, und rij der Ab-
stand der Teilchen zwischen denen das Potential u ij besteht. Zusätzlich können Ion-Quadru-
pol-Effekte auftreten, bei denen sich das Potential angenähert proportional zu r n− mit n > 6
verhält.
Für Potentiale, die zwischen Ionen und den von ihnen induzierten Dipolen bestehen, gilt:
3.2.2 4
22
ij
jjiiij r
QQu
αα +−=
dabei sind α i und α j die Polarisierbarkeiten, der auf diese Art miteinander wechselwirkenden
Teilchen.
Eine exakte Bestimmung des Beitrags der Ion-Dipol-Wechselwirkungen zur Gibbsschen Ex-
zeßenthalpie aus den Potentialfunktionen mit Hilfe statistisch mechanischer Methoden ist we-
gen der dabei auftretenden mathematischen Probleme zumindest in analytischer Form nicht
möglich. Hinzu kommt, daß die Wechselwirkungen, bei denen induzierte Momente beteiligt
sind, nicht paarweise additiv sind, d.h. die gesamte Wechselwirkungsenergie des Systems er-

31
gibt sich nicht einfach aus der Summe der Beiträge der einzelnen Teilchenpaare, wie es für
andere Wechselwirkungen der Fall ist. Aus diesem Grunde verwendet das LIQUAC-Modell
einen empirischen Ansatz für die Berechnung des Beitrags der Ion-Dipol-Wechselwirkungen
zur Gibbsschen Exzeßenthalpie der Lösung, der auf dem Virialansatz von Pitzer basiert. Die
Berechtigung für eine solche Vorgehensweise ergibt sich daraus, daß die Virialkoeffizienten
mit den Potentialfunktionen der intermolekularen Kräfte in Zusammenhang gebracht werden
können. Der zweite Summand der Pitzer-Gleichung, der die binären Wechselwirkungen erfaßt,
wurde dahingehend erweitert, daß auch Ion-Molekül und Molekül-Molekül-Paare miteinbezo-
gen werden. Für ein System bestehend aus ns Molen des Lösungsmittels s ),....,2,1( sns =
und n j Molen des Ions j ),...,2,1( jnj = folgt daher:
3.2.3 ( )∑∑∑=
k llkmkl
sss
Erm nnIB
MnRT
G 1.. sns ,.....,2,1=
js nnk += ,.....,2,1 js nnl += ,....,2,1
Die Summationsindices k und l beinhalten sämtliche Komponenten der Lösung und die Virial-
koeffizienten sind symmetrisch ( )B Bkl lk= . Gl. 3.2.3 beinhaltet einen Ion-Ion-, Ion-Molekül-,
und Molekül-Molekül-Beitrag. Im Unterschied zur Pitzer-Gleichung sollen aber nur die Ion-
Dipol-Effekte beschrieben werden. Aus diesem Grund entfällt im LIQUAC-’middle-range’-
Term der Molekül-Molekül-Beitrag und aus Gl. (3.2.3) ergibt sich durch Aufspaltung der
Summation in Ion-Ion und Ion-Molekül-Paare:
3.2.4
+= ∑∑ ∑∑∑ s j c a
acmacjsmjs
sss
Erm nnIBnnIB
MxRT
G)()(
1,,'
..
sns ,....,2,1= jnj ,....,2,1=
cnc ,....,2,1= ana ,....,2,1=
Im zweiten Summenterm wurden sämtliche Kation-Kation- und Anion-Anion-Kombinationen
vernachlässigt. Dies steht im Einklang mit dem von Brönstedt formulierten Prinzip der spezi-

32
fischen Wechselwirkungen [5, 6]. Danach sind die Effekte zwischen gleichartig geladenen
Ionen praktisch rein elektrostatischer Natur, da eine stärkere Annäherung aufgrund der Ab-
stoßungskräfte nicht möglich ist. Die über die rein elektrostatischen Effekte hinausgehenden
spezifischen Wechselwirkungen, die u.a. die Abweichungen von der Debye-Hückel-Theorie
verursachen, sind also im wesentlichen von den Wechselwirkungen zwischen ungleichnamig
geladenen Ionen bestimmt. In Gl. (3.2.4) erfaßt der erste Summand die unter dem Begriff
‘middle-range’-Wechselwirkungen zusammengefaßten Ion-Dipol-Effekte zwischen Lö-
sungsmittelmolekülen und der zweite Summand die entsprechenden Effekte zwischen Katio-
nen und Anionen, die auf induzierte Momente zurückzuführen sind. Die Konzentrationsab-
hängigkeit der Virialkoeffizienten Bs j, und Bc a, ist durch die folgenden Funktionen gegeben:
3.2.5 )exp( 21,,, mmacacac IaIacbB ++=
0.11 −=a 13.02 =a
und
3.2.6 )exp( '2
'1,,, mmjsjsjs IaIacbB ++=
2.1'1 −=a 13.0'
2 =a
b und c sind die für das jeweilige Komponentenpaar charakteristischen Parameter, während
die Konstanten a a a1 2 1, , ' und a 2' vom Stoffsystem unabhängige Größen darstellen.
Aus der Ableitung der Gl. (3.2.3) nach den Molzahlen der Komponenten folgen die Aus-
drücke für die Aktivitätskoeffizienten in der Konzentrationsskala des Molenbruchs. Für die
Lösungsmittelkomponente s erhält man:
3.2.7 [ ]∑ ∑∑∑−+−=
j s jjsmjsmjs
sss
sjmjs
rms mxIBIB
Mx
MmIB
''',','
'''
,.. ')(')(
')(ln γ
[ ]∑∑ +c a
acmacmmacs mmIBIIBM )(')( ,,

33
mit mdI
dBB =' und sns ....,2,1'=
Und für die Aktivitätskoeffizienten von Kation c und Anion a in der unsymmetrischen Nor-
mierung:
3.2.8 ∑∑ ∑∑++=∗
s j aamacjsmjs
sss
crmc mIBmxIB
Mx
z)(')('
'2ln ,,
2..γ
∑∑'
','
2
)('2 c a
acmacc mmIB
z cnc ....,2,1'=
3.2.9 ∑∑ ∑∑++=∗
s j ccmacjsmjs
sss
arma mIBmxIB
Mx
z)(')('
'2ln ,,
2..γ
∑∑c a
acmaca mmIB
z
'','
2
)('2
ana ....,2,1'=
Für die Ableitung des Virialkoeffizienten nach der Ionenstärke B' ergibt sich:
3.2.10 )exp(2
' 2121
,, mm
m
lklk IaIaaI
acB +
+=
3.3 Beitrag der ‘short-range’-Wechselwirkungen
Für die Beschreibung der Molekül-Molekül-, Ion-Molekül-, und Ion-Ion-Wechselwirkungen,
die sich aufgrund der Abhängigkeit des Wechselwirkungspotentials vom intermolekularen Ab-
stand eindeutig zu den ‘short-range’-Effekten zuordnen lassen verwendet das LIQUAC-
Modell die UNIQUAC-Gleichung. Sie basiert auf dem Prinzip der lokalen Zusammensetzung,
berücksichtigt explizit die Temperaturabhängigkeit der Gibbsschen Exzeßenthalpie und stellt
gleichzeitig die Grundlage für die Gruppenbeitragsmethode UNIFAC dar. Die Molekül-Mo-
lekül-Wechselwirkungsparameter wurden zudem bereits für eine Vielzahl binärer Nichtelek-
trolytsysteme bestimmt [18] und können daher direkt übernommen werden. Bei der Anwen-

34
dung der Gleichung auf Elektrolytlösungen werden die Ionen als zusätzliche Komponente
neben den Lösungsmittelmolekülen berücksichtigt, wobei wiederum vollständige Dissoziation
des Elektrolyten vorausgesetzt wird. Die Gibbssche Exzeßenthalpie wird in einen tempera-
turunabhängigen kombinatorischen und in den sogenannten Restanteil zerlegt. Der kombina-
torische Anteil erfaßt die Abweichungen vom idealen Verhalten, die auf die unterschiedliche
Größe und Form der Teilchen zurückzuführen sind (Exzeßentropie), während der Restanteil
die Unterschiede in den binären Wechselwirkungsenergien berücksichtigt (Exzeßenthalpie):
3.3.1 RT
g
RT
g
RT
g ER
Ec
E
+=
3.3.2 ∑ ∑+=k k k
kkk
k
kk
Ec
xxq
zx
xRT
g θφln
2ln js nnk += ....,2,1 z = 10
3.3.3 ∑ ∑
−=
l kk lkll
ER qx
RT
gψθln js nnl += ....,2,1
Die Größen kφ und kθ geben den Volumen- und Oberflächenanteil der betreffenden Kompo-
nente in der Mischung an und sind über das relative van der Waalssche Volumen kr und die
relative van der Waalssche Oberfläche kq definiert1:
3.3.4 ∑
=
lll
kkk rx
rxφ
3.3.5 ∑
=
lll
kkk qx
qxθ
kr stellt ein Maß für die Größe eines Moleküls dar und gibt die Zahl der Plätze an, die es in
einem gedachten dreidimensionalen Gitter mit der Koordinationszahl z besetzt. kq ist ein Maß
für seine Oberfläche und ist über die Anzahl der benachbarten Gitterplätze definiert. Für Mo-
1 Die Formulierung des kombinatorischen Anteils gemäß Gl. (3.3.2) führt zu Schwierigkeiten, wenn eine Komponente mit dem Molenbruch null eingeht (Division durch null). In diesem
Fall ist es zweckmäßig in den Definitionsgleichungen für den Volumen und Oberflächenanteil Gl. (3.3.4) und Gl. (3.3.5) die Molenbrüche im Zähler wegzulassen und die Gleichungen
(3.3.2) und (3.3.3) entsprechend umzuformulieren. Wegen der Übereinstimmung mit den Formulierungen anderer Autoren wurde die obige ältere Schreibweise aber beibehalten.

35
lekülkomponenten lassen sich diese Größen aus Strukturdaten gewinnen [4]. Die relativen van
der Waalsschen Volumina und Oberflächen der Ionen sind prinzipiell aus den Ionenradien
berechenbar, wenn man unter Annahme der Kugelform Volumen und Oberfläche bestimmt
und auf das Volumen bzw. die Oberfläche eines Standardsegments bezieht [1]. Anpas-
sungsversuche unter Verwendung von r- und q-Werten, die aus Kristallradien berechnet wur-
den, waren aber wenig erfolgreich. Ursache sind die zum Teil sehr kleinen Werte, die dabei für
die relativen van der Waalsschen Oberflächen der Kationen resultieren, und im Restanteil der
UNIQUAC-Gleichung zu einer entsprechenden schwachen Gewichtung der Boltzmann-
Faktoren führen. Sander berichtete über ähnliche Schwierigkeiten [52] und schlug vor die r-
und q-Werte der Ionen als Anpassungsgrößen aufzufassen. Dies erfordert aber eine sehr
sorgfältige Bestimmung auf der Grundlage einer breiten Datenbasis, da die Strukturparameter
unabhängig vom Stoffsystem gültig sein müssen. Im Hinblick auf eine Reduzierung der oh-
nehin großen Zahl an Anpassungsgrößen werden die r- und q-Werte der Ionen durchgängig
auf einen Wert von 1.0 festgesetzt. Dieser Wert liegt in der für kleine Moleküle characteristi-
schen Größenordnung.
klψ enthält den binären Wechselwirkungsparameter kla und hat die Bedeutung eines
Boltzmann-Faktors:
3.3.6
−=
T
aklk lψ
Da die Ionen im ‘short-range’-Anteil genauso behandelt werden wie die Lösungsmittelkompo-
nenten, resultiert bei der Ableitung nach den Molzahlen derselbe symmetrisch normierte Aus-
druck für den Aktivitätskoeffizienten:
3.3.7 Rn
Cnn γγγ lnlnln += js nnn += ,....,2,1
3.3.8
−+−−+=
n
n
n
nn
n
n
n
nCn q
zxx θ
φθφφφ
γ 1ln2
1lnln
3.3.9
−
−= ∑ ∑∑
lk
k lk
nllkn
kkn
Rn q
ψθψθ
ψθγ ln1ln

36
Die für Ionen erforderliche Normierung des Aktivitätskoeffizienten auf den Zustand unendli-
cher Verdünnung ergibt sich aus folgendem Zusammenhang:
3.3.10 ∞∗ −= jjj γγγ lnlnln
Der symmetrisch normierte Wert für den Bezugszustand der unendlichen Verdünnung folgt
aus Gl. (3.3.8) und Gl. (3.3.9) durch Nullsetzen der Molenbrüche der Ionen:
3.3.11 ( )jssjjjs
sj
js
sjj
s
j
s
jj q
qr
qr
qr
qrq
zr
r
r
rψψγ −−+
+−−
+−=∞ ln1ln1
2ln1ln
Für den Fall eines Lösungsmittelgemisches stellt s das Referenzlösungsmittel dar und der Be-
zugszustand ist definiert als der Zustand unendlicher Verdünnung der Ionen im Lösungsmittel
s. Eine Normierung auf das jeweils vorliegende Lösungsmittelgemisch ist nicht sinnvoll, da
der Bezugszustand unabhängig von der Zusammensetzung sein muß.
3.4 Der vollständige Ausdruck für den Aktivitätskoeffizienten
Für die Lösungsmittelkomponente s berechnet sich der Aktivitätskoeffizient gemäß:
3.4.1 ...... lnlnlnln rss
rms
rlss γγγγ ++=
Die einzelnen Beiträge ergeben sich dabei aus den Gleichungen (3.1.1), (3.2.7), und (3.3.7).
Der Aktivitätskoeffizient ist in der Skala des Molenbruchs definiert. Der Bezugs- und Stan-
dardzustand ist die reine Lösungsmittelkomponente s unter Systemdruck und Systemtempera-
tur. Für das Ion j folgt in der Konzentrationsskala der Molalität:
3.4.2
+−++= ∑∗∗∗∗
jjs
rsj
rmj
rljmj mM1lnlnlnlnln ......
)(, γγγγ

37
Für die Berechnung des ‘long-range’-Anteils wird Gl. (3.1.2) eingesetzt. Der ‘middle-range’-
Anteil folgt aus Gl. (3.2.8) und (3.2.9), und der ‘short-range’-Anteil aus Gl. (3.3.7) und
(3.3.11). Der zusätzliche Summand ist notwendig, um von der Konzentrationsskala des
Molenbruchs in die Konzentrationsskala der Molalität umzurechnen (siehe Gl. 2.4.5). Der
Bezugszustand ist die unendlich verdünnte Lösung. Der mittlere Aktivitätskoeffizient ergibt
sich dann über die Definitionsgleichung (2.5.4) aus den Aktivitätskoeffizienten von Kation und
Anion:
3.4.3 ∗−
−∗+
+∗± += )()()( lnlnln mmm γ
νν
γνν
γ
3.5 Anzahl der Anpassungsgrößen
Abgesehen vom elektrostatischen Anteil, der neben den Reinstoffgrößen der Lösungsmittel-
komponenten nur von der Ionenstärke und dem Ladungstyp des Elektrolyten abhängt, lassen
sich die vom LIQUAC-Modell benötigten Parameter in drei Gruppen unterteilen:
Die erste Gruppe bilden die UNIQUAC-Parameter akl und alk, die im ‘short-range’-Anteil die
Wechselwirkungen zwischen Lösungsmittelmolekülen beschreiben. Sie wurden der Literatur
[18] entnommen, und sind unabhängig vom LIQUAC-Modell auch für die Beschreibung
elektrolytfreier Lösungsmittelgemische verwendbar.
Die zweite Gruppe bilden die relativen van der Waalsschen Oberflächen und Volumina der
Systemkomponenten. Diese Strukturparameter wurden für Lösungsmittelkomponenten eben-
falls der Literatur [18] entnommen. Für Ionen wurden die r- und q-Werte durchgängig auf den
Wert 1 festgesetzt, und liegen damit in einer Größenordnung die für kleine Moleküle charak-
teristisch ist. Die tatsächlichen relativen van der Waalsschen Volumina der Ionen sind prinzi-
piell aus Ionenradien berechenbar, wenn man unter Annahme der Kugelform Volumen und
Oberfläche bestimmt, und auf das Volumen bzw. die Oberfläche eines Standardsegments be-
zieht [1]. Anpassungsversuche unter Verwendung von r- und q-Werten, die aus Kristallradien
berechnet wurden waren aber wenig erfolgreich. Ursache sind die zum Teil sehr kleinen Werte,

38
die dabei für die van der Waalsschen Oberflächen der Kationen resultieren, und im Restanteil
der UNIQUAC-Gleichung zu einer entsprechend schwachen Gewichtung der Boltzmann-
Faktoren führen. Sander [52] schlug deswegen vor die r- und q-Werte von Ionen als
Anpassungsgrößen aufzufassen. Dies erfordert aber eine sehr genaue Bestimmung auf
Grundlage einer breiten Datenbasis, da diese Strukturparameter unabhängig vom gerade be-
trachteten Stoffsystem universell gültig sein müssen.
Die dritte Gruppe bilden die sogenannten ‘reinen’, für das LIQUAC-Modell spezifischen
Molekül-Ion- und Ion-Ion-Parameter, um deren Bestimmung es im folgenden geht. Zu dieser
Gruppe gehören sowohl die Parameter bkl und ckl, die im ‘middle-range’- Anteil die Konzen-
trationsabhängigkeit der zweiten Virialkoeffizienten beschreiben, als auch die UNIQUAC-
Parameter akl und alk, die im ‘short-range’-Anteil Wechselwirkungen zwischen Lösungsmit-
telmolekülen und Ionen, und zwischen Anionen und Kationen beschreiben. Sie werden itera-
tiv an experimentelle Daten angepaßt.
Für jede Wechselwirkung, an der mindestens ein Ion beteiligt ist benötigt das Modell zwei
Parameter zur Beschreibung der Konzentrationsabhängigkeit des zweiten Virialkoeffizienten
im ‘middle-range’-Term, und zwei weitere, die im UNIQUAC-Anteil die ‘short-range’-Effekte
und die Temperatureffekte berücksichtigen. Gl. (3.5.1) gibt an, wieviele Parameter zur Be-
schreibung eines Systems aus Ns Lösungsmittelkomponenten, Nc Kationen und Na Anionen
nötig sind. NindexNindex bezeichnet die jeweilige Wechselwirkung, und N entspricht der Anzahl
benötigter Parameter:
(3.5.1) ( ) ( )N N N N N N N N Nc s a s c a s s= + + × + −4 1 N s > 1
Die Gültigkeit der Gl. (3.5.1) ist auf Lösungsmittelgemischsysteme beschränkt. Für den einfa-
cheren Fall eines einzigen Lösungsmittels kürzen sich die Virialkoeffizienten der Ion-Molekül-
Wechselwirkung aus dem Ausdruck für den Aktivitätskoeffizienten des Lösungsmittels Gl.
(3.2.8) heraus, so daß nur die Ableitungen nach der Ionenstärke übrigbleiben. Diese enthalten
aber gemäß Gl. (3.2.10) nicht mehr den b-Parameter, der nur in einem Lösungsmittelgemisch
benötigt wird. Im Fall einer einzigen Lösungsmittelkomponente ist Gl. (3.5.1) also so zu
korrigieren, daß jeder Ion-Molekülwechselwirkung nur 3 Parameter zugeordnet werden:
(3.5.2) ( )N N N N N N Nc s a s c a= + × + ×3 4 N s = 1

39
Die Beschreibung des einfachsten Systems bestehend aus einem Lösungsmittel, einem Kation
und einem Anion erfordert demnach 10 Wechselwirkungsparameter. Für ein ternäres System
aus zwei Lösungsmitteln und einem Salz werden bereits 22 Parameter benötigt. Dies scheint
sehr viel, es sollte jedoch erwähnt werden, daß diese Parameter allgemein verwendbar sind,
und nicht nur zur Berechnung des jeweils vorliegenden Stoffsystems dienen.

40
4. Fest-Flüssig-Phasengleichgewichte von Lösungen starker Elektrolyte
Das Phasengleichgewicht fest-flüssig weist gegenüber allen anderen Phasengleichgewichten
die größte Mannigfaltigkeit auf. Der Grund dafür ist, daß neben der flüssigen Phase verschie-
dene feste Phasen auftreten können. Klassifiziert man die Gleichgewichte zwischen festen und
flüssigen Phasen in Zweikomponentensystemen hinsichtlich der Beschaffenheit der festen
Phasen ergeben sich die folgenden drei Gruppen:
A) die festen Phasen treten bei allen Gleichgewichten nur als reine Komponenten auf,
B) die festen Phasen treten nicht nur als reine Komponenten, sondern auch als
Verbindungen der beiden reinen Komponenten in stöchiometrischen Verhältnissen
auf,
C) die Komponenten bilden in der festen Phase homogene Lösungen; ihre gegenseitige
Löslichkeit ist dabei unbegrenzt oder nur auf bestimmte Konzentrationsbereiche
beschränkt.
Diese Klassifizierung ist jedoch nicht ausreichend, weil sie den Unterschied zwischen
Schmelzen und Lösungen nicht hervorhebt.
Lösungen zeichnen sich im Gegensatz zu Schmelzen dadurch aus, daß die Schmelzpunkte der
beiden reinen Komponenten stark unterschiedlich sind. Die Komponente mit dem niedrigeren
Schmelzpunkt bezeichnet man als Lösungsmittel, die andere als den gelösten Stoff. Diese
Unterscheidung ist besonders dann üblich, wenn die eine Komponente bei normaler Tempera-
tur flüssig und die andere fest ist. In diesem Fall bezeichnet man die Kurve, die das Gleich-
gewicht zwischen der flüssigen gesättigten Lösung und der festen Phase des gelösten Stoffes
beschreibt als Löslichkeitskurve. Die Kurve, die das Gleichgewicht zwischen der festen Phase
des Lösungsmittels und der Lösung beschreibt wird als Erstarrungskurve bezeichnet. Bei
Schmelzen ist diese Unterscheidung nicht möglich, weil man beide Komponenten sowohl als
Lösungsmittel als auch als gelösten Stoff betrachten kann. Daher spricht man hier in der Regel
von Erstarrungskurven.
Die Phasendiagramme wäßriger Lösungen starker Elektrolyte zeigen verschiedene Grundfor-
men. Je nachdem ob der Elektrolyt Hydrate ausbildet oder nicht und ob diese Hydrate an
41
ihrem Schmelzpunkt stabil sind oder nicht ergeben sich unterschiedliche
Phasendiagrammtypen:
4.1 Eutektische Systeme ohne Verbindungsbildung
Löslichkeit [mol/kg]
Liquiduslinie
SoliduslinieA
C
I
II
III
IV
B
Daten von: Flöttmann [97], De Coppet [85], Scott und Durham [194], Meusser [140], Eddy [95], Larson [131], Scott und Frazier [195]
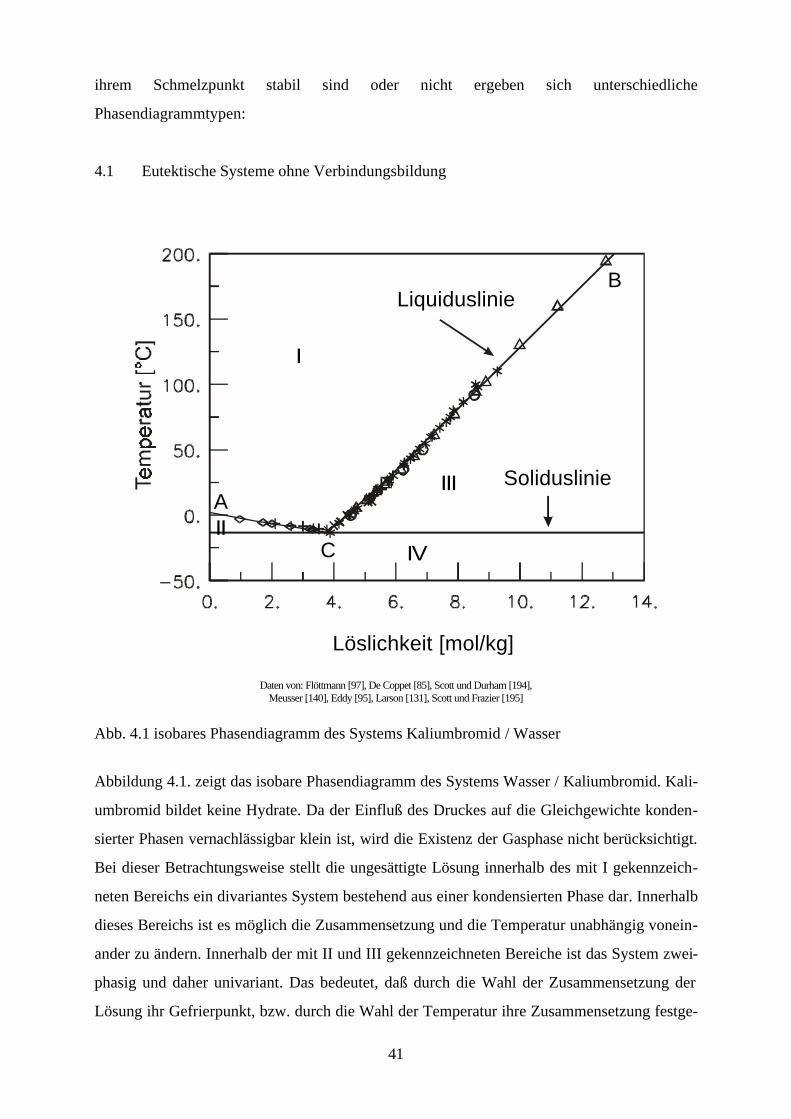
Abb. 4.1 isobares Phasendiagramm des Systems Kaliumbromid / Wasser
Abbildung 4.1. zeigt das isobare Phasendiagramm des Systems Wasser / Kaliumbromid. Kali-
umbromid bildet keine Hydrate. Da der Einfluß des Druckes auf die Gleichgewichte konden-
sierter Phasen vernachlässigbar klein ist, wird die Existenz der Gasphase nicht berücksichtigt.
Bei dieser Betrachtungsweise stellt die ungesättigte Lösung innerhalb des mit I gekennzeich-
neten Bereichs ein divariantes System bestehend aus einer kondensierten Phase dar. Innerhalb
dieses Bereichs ist es möglich die Zusammensetzung und die Temperatur unabhängig vonein-
ander zu ändern. Innerhalb der mit II und III gekennzeichneten Bereiche ist das System zwei-
phasig und daher univariant. Das bedeutet, daß durch die Wahl der Zusammensetzung der
Lösung ihr Gefrierpunkt, bzw. durch die Wahl der Temperatur ihre Zusammensetzung festge-

42
legt ist. Unterhalb der Erstarrungskurve besteht die feste Phase aus reinem Eis, während die
feste Phase unterhalb der Löslichkeitskurve aus reinem Salz besteht. Erstarrungs- und Lös-
lichkeitskurve zusammen bilden die Liquiduskurve, die das Einphasen- von dem Zweiphasen-
gebiet trennt. Die Fläche II entspricht dem univarianten System Eis-Lösung. Dieses System
bleibt univariant, solange die Lösung nicht gesättigt ist. Erniedrigt man die Temperatur der
Lösung, die sich im Gleichgewicht mit dem Eis befindet, so vergrößert sich ihre Konzentration
infolge des Ausfrierens des Eises entlang der Erstarrungskurve, bis bei der dem Punkt C
entsprechenden Temperatur eine gesättigte Lösung entsteht, und neben dem Eis auch festes
Kaliumbromid erscheint. Dieser Punkt wird bei wäßrigen Salzlösungen kryohydratischer
Punkt, bei Lösungen allgemein eutonischer Punkt genannt. Er kennzeichnet die Bedingungen
für die Koexistenz dreier kondensierter Phasen, bei denen das System invariant ist. Aus einer
Lösung mit dieser Konzentration scheiden sich bei weiterem Abkühlen gleichzeitig Eis und
festes Salz ab, und zwar im gleichen Massenverhältnis, in dem sie in der gesättigten Lösung
bei der kryohydratischen Temperatur vorliegen. Dadurch verschwindet die flüssige Phase
vollständig und es bildet sich ein univariantes System von Eis und Salz (Fläche IV). Unterhalb
der Temperatur des kryohydratischen Punktes koexistieren also Eis und Salz bei jeder beliebi-
gen Temperatur und stellen im System zwei selbständige Phasen dar. Der Grund dafür ist, daß
Eis- und Salzkristalle nicht isomorph sind, und es nicht zur Bildung von Mischkristallen
kommen kann. Das Gemisch aus Eis und Salz, dessen Zusammensetzung dem kryohydrati-
schen Punkt entspricht, bezeichnet man als kryohydratische Mischung; es schmilzt bei kon-
stanter Temperatur. Die Trennlinie zwischen dem heterogen zweiphasigen Bereichen (II und
III) und dem homogen zweiphasigen Bereich (IV) bezeichnet man als Soliduslinie.
43
4.2. Systeme mit Verbindungsbildung und kongruentem Schmelzpunkt
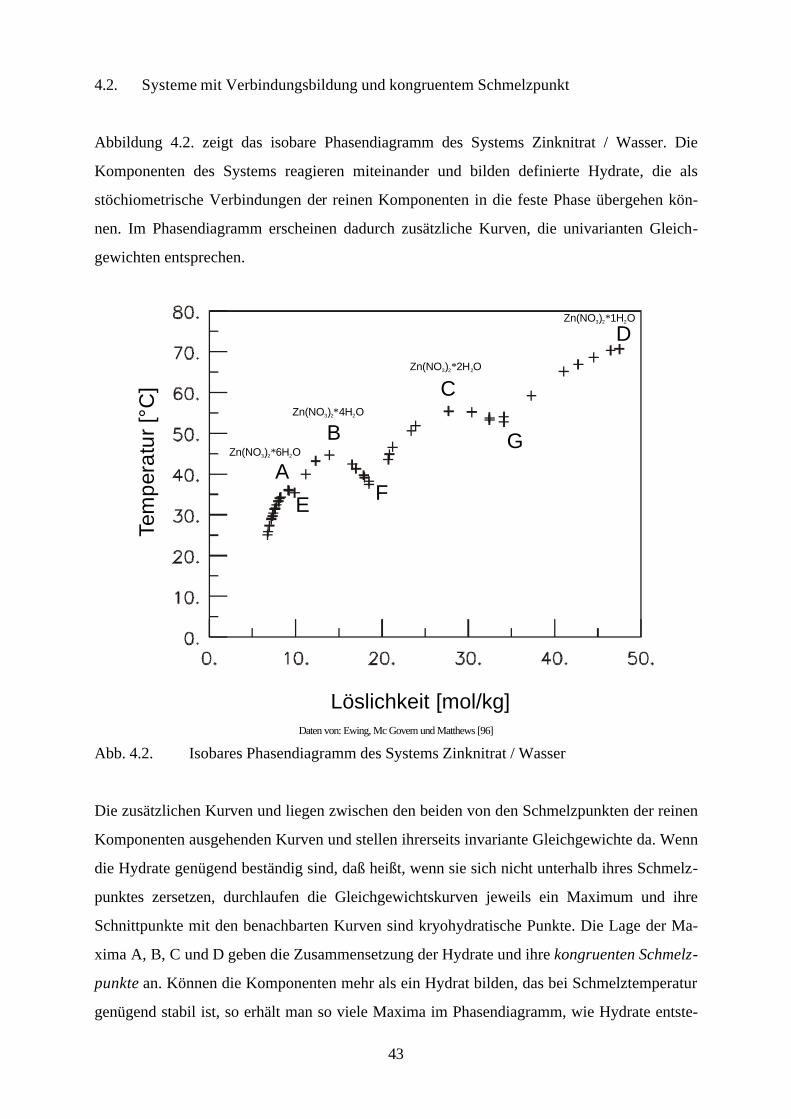
Abbildung 4.2. zeigt das isobare Phasendiagramm des Systems Zinknitrat / Wasser. Die
Komponenten des Systems reagieren miteinander und bilden definierte Hydrate, die als
stöchiometrische Verbindungen der reinen Komponenten in die feste Phase übergehen kön-
nen. Im Phasendiagramm erscheinen dadurch zusätzliche Kurven, die univarianten Gleich-
gewichten entsprechen.
Löslichkeit [mol/kg]
A
B
C
D
EF
GZn(NO ) 6H O3 22∗
Zn(NO ) 1H O3 22∗
Zn(NO ) 2H O3 22∗
Zn(NO ) 4H O3 22∗
Tem
per
atu
r [°
C]
Daten von: Ewing, Mc Govern und Matthews [96]
Abb. 4.2. Isobares Phasendiagramm des Systems Zinknitrat / Wasser
Die zusätzlichen Kurven und liegen zwischen den beiden von den Schmelzpunkten der reinen
Komponenten ausgehenden Kurven und stellen ihrerseits invariante Gleichgewichte da. Wenn
die Hydrate genügend beständig sind, daß heißt, wenn sie sich nicht unterhalb ihres Schmelz-
punktes zersetzen, durchlaufen die Gleichgewichtskurven jeweils ein Maximum und ihre
Schnittpunkte mit den benachbarten Kurven sind kryohydratische Punkte. Die Lage der Ma-
xima A, B, C und D geben die Zusammensetzung der Hydrate und ihre kongruenten Schmelz-
punkte an. Können die Komponenten mehr als ein Hydrat bilden, das bei Schmelztemperatur
genügend stabil ist, so erhält man so viele Maxima im Phasendiagramm, wie Hydrate entste-
44
hen. Die Zahl der kryohydratischen Punkte ist dabei immer um Eins größer als die Zahl der
Maxima. Im Fall des Zinknitrats handelt es sich um das Hexahydrat (A), das Tetrahydrat (B),
das Dihydrat (C), und das Monohydrat (D). Zerlegt man ein solches Phasendiagramm durch
die Ordinaten der Maxima in mehrere Teile so erhält man einfache Diagramme, die man als
selbständig betrachten kann. Viele Salze zwei- und höherwertiger Kationen bilden kristalline,
kongruentschmelzende Hydrate, jedoch ist eine Vorhersage ob und welche Hydrate ein Salz
bildet bisher nicht möglich. Die Berechnung der einzelnen Löslichkeitskurven ist möglich
wenn die thermodynamischen Größen der Hydrate bekannt sind.
4.3. Peritektische Systeme
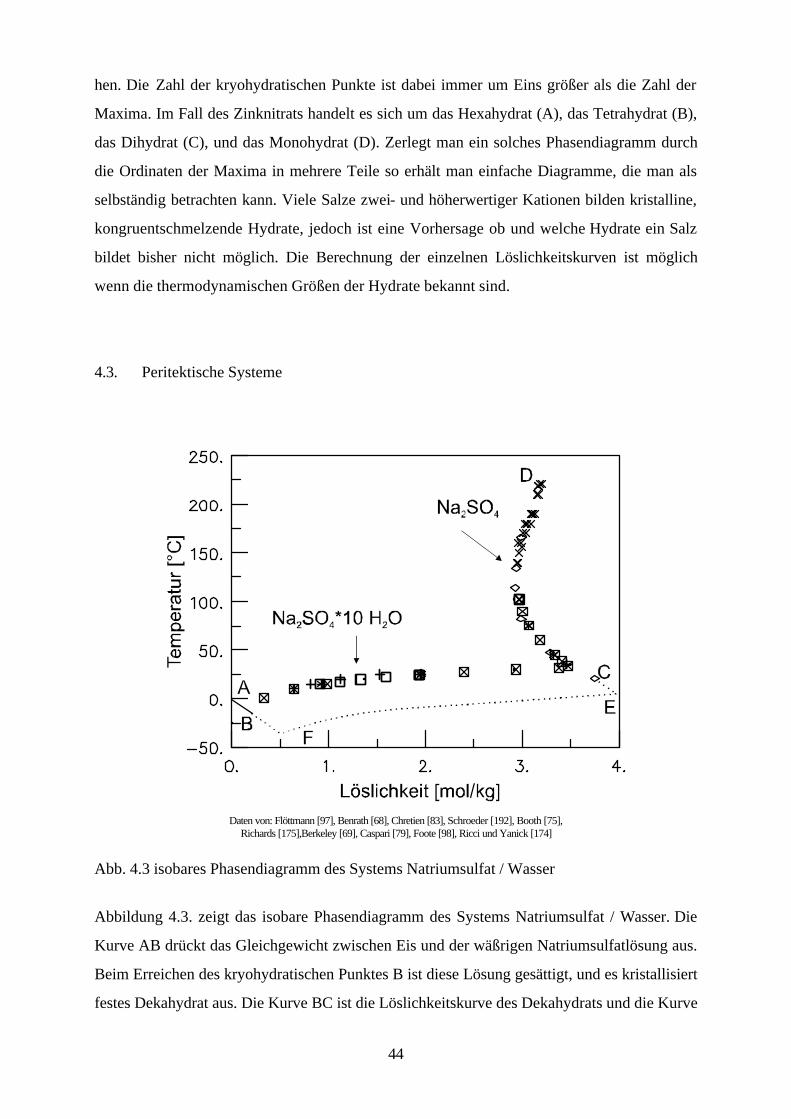
Daten von: Flöttmann [97], Benrath [68], Chretien [83], Schroeder [192], Booth [75],
Richards [175],Berkeley [69], Caspari [79], Foote [98], Ricci und Yanick [174]
Abb. 4.3 isobares Phasendiagramm des Systems Natriumsulfat / Wasser
Abbildung 4.3. zeigt das isobare Phasendiagramm des Systems Natriumsulfat / Wasser. Die
Kurve AB drückt das Gleichgewicht zwischen Eis und der wäßrigen Natriumsulfatlösung aus.
Beim Erreichen des kryohydratischen Punktes B ist diese Lösung gesättigt, und es kristallisiert
festes Dekahydrat aus. Die Kurve BC ist die Löslichkeitskurve des Dekahydrats und die Kurve

45
CD die des Anhydrids. Aus ihrem Verlauf ist ersichtlich, daß die Löslichkeit des Dekahydrats
mit steigender Temperatur zunimmt, während die des Anhydrids sinkt.
Das Dekahydrat wandelt sich bei der peritektischen Temperatur 32.38 °C (Punkt C ) reversibel
in das wasserfreie Salz und in eine gesättigte Lösung um. Zersetzt sich ein aus den Grundkom-
ponenten des Systems gebildetes Hydrat beim Erwärmen bevor es schmilzt, so entsteht ein
neue Komponente und eine neue flüssige Phase. Die flüssige Phase hat dabei eine andere Zu-
sammensetzung als die ursprüngliche Verbindung. Diese Erscheinung ist reversibel, und beim
Abkühlen der entstandenen Schmelze wird wieder die ursprüngliche Verbindung zurückgebil-
det. Bei der Temperatur, bei der diese Zersetzung stattfindet, stellt sich ein Gleichgewicht
zwischen drei kondensierten Phasen ein, und das System ist invariant wie in einem kryohy-
dratischen Punkt. Da aber auch unterhalb dieser Temperatur eine flüssige Phase existieren
kann, bezeichnet man diesen Punkt im Gegensatz zum kryohydratischen Punkt als peritekti-
schen Punkt. Die entsprechende Temperatur wird inkongruenter Schmelzpunkt genannt. Peri-
tektika treten häufig bei Salzhydraten auf, die unterhalb ihres Schmelzpunktes im eigenen
Kristallwasser zerfließen. Zu den bekanntesten Beispielen gehören neben Natriumsulfat auch
Natriumcarbonat und Calciumchlorid.
Kühlt man die gesättigte Natriumsulfatlösung in Anwesenheit des Anhydrids vorsichtig ab, so
kann man sie unterkühlen, daß heißt die Temperatur unter die des peritektischen Punktes C
bringen, ohne daß sich das Dekahydrat ausscheidet. Gelingt es dabei die Temperatur von
24.2°C (Punkt E) zu erreichen, so beginnt die Ausscheidung von kristallinem Heptahydrat, das
sich bei dieser Temperatur in einem metastabilen Gleichgewicht mit dem Anhydrid befindet,
aber unter keinen Bedingungen mit dem Dekahydrat im Gleichgewicht stehen kann. Die
Kurve EF ist die metastabile Löslichkeitskurve des Heptahydrats und der Punkt F dessen me-
tastabiler kryohydratischer Punkt mit dem Eis und der gesättigten Lösung.
Peritektische Punkte, inkongruente Schmelzpunkte und metastabile kryohydratische Punkte
sind ebenso wie die Hydrat- bzw. Aduktbildung nicht voraussagbar.
4.4 Das Löslichkeitsprodukt
Die Mischung eines Salzes mit einem Lösungsmittel stellt je nach Mischungsverhältnis eine
Lösung oder eine Schmelze dar. Der Übergang von hochkonzentrierter Lösung zur Schmelze
ist fließend und letztlich ist es nur eine Definitionsfrage. Das Fest-Flüssig-Phasendiagramm

46
der Lösung setzt sich aus der Erstarrungskurve (vom Schmelzpunkt des reinen Lösungsmittel
bis zum kryohydratischen Punkt) und der Löslichkeitskurve (vom kryohydratischen Punkt bis
zum Schmelzpunkt des Salzes) zusammen. Bei der Berechnung der Erstarrungskurve handelt
es sich um ein reines Phasengleichgewicht zwischen festem und flüssigem Lösungsmittel. Das
Salz ist bis zum kryohydratischen Punkt vollständig gelöst und bedingt entsprechend der
Anzahl solvatisierter Ionen eine Gefrierpunktserniedrigung des Lösungsmittels. Die zu
erfüllende Gleichgewichtsbedingung der Erstarrungskurve ergibt sich demnach allein aus dem
Isofugazitätskriterium. Die Löslichkeitskurve beschreibt dagegen das Gleichgewicht zwischen
festem und gelöstem Salz. Beim Auflösen eines Salzes handelt es sich aber um eine Reaktion,
die mit einer Wärmetönung verbunden ist, und bei der neue Spezies (nämlich solvatisierte
Ionen) entstehen. Die zu erfüllende Gleichgewichtsbedingung der Löslichkeitskurve muß
neben dem Isofugazitätskriterium also noch das chemische Gleichgewicht berücksichtigen.
Phasengleichgewicht herrscht, wenn das chemische Potential der Komponente i in Phase a
genauso groß ist wie in Phase b:
4.4.1 biai ,, µµ =
Die allgemeine Bedingung für chemisches Gleichgewicht lautet:
4.4.2 ∑ = 0ii µν
Kombiniert man diese beiden Gleichungen, dann lautet die Gleichgewichtsbedingung für ein
heterogenes System bestehend aus festem reinem Salz im Gleichgewicht mit seiner gesättigten
Lösung, unter Berücksichtigung der Tatsache, daß das Salz in der flüssigen Phase dissoziiert
und in Form von solvatisierten Ionen vorliegt:
4.4.3 solvsolvf
Salz ,, −−++ += µνµνµ
Drückt man die chemischen Potentiale mit Hilfe der Aktivität aus führt das zu:
4.4.4 ( )νγµνµνµ ±±−−++ ++=+ mRTaRT solvsolvf
Salzf
Salz lnln 0,
0,
,0

47
Die Aktivität des festen reinen Salzes ist definitionsgemäß = 1, wodurch sich Gl. (4.5.4) re-
duziert zu:
4.4.5 ( )νγµνµνµ ±±−−++ ++= mRTsolvsolvf
Salz ln0,
0,
,0
bzw. zu:
4.4.6 ( )( ) ( )νγµνµνµ ±±−−++ =+− mRTsolvsolvf
Salz ln/0,
0,
,0
Ersetzt man die chemischen Potentiale im Standardzustand durch die Gibbsschen Standard-
bildungsenthalpien führt dies zu:
4.4.7 ( )( ) ( )νγνν ±±−−++ =∆+∆−∆ mRTggg solvsolvSalz ln/0,
0,
0
Die Standardglieder auf der linken Seite des Ausdrucks werden zusammengefaßt zu:
4.4.8 ( )0,
0,
00, solvsolvSalzDissK gggg
sp −−++ ∆+∆−∆=∆ νν
Der Index ‘Diss’ kennzeichnet, daß Gl. (4.5.8) für die Dissoziation des Salzes gilt. Betrachtet
man die Rückreaktion, also die Fällung festen Salzes, kehren sich die Vorzeichen um und man
erhält:
4.4.9 ( ) 00,
0,
0, SalzsolvsolvFällK gggg
sp∆−∆+∆=∆− −−++ νν
Das Löslichkeitsprodukt bei Standardbedingungen K sp0 ist je nach betrachteter Reaktion defi-
niert als:
4.4.10
∆=
RT
gK DissspK
sp
0
0 ,exp , oder für die Fällung
∆−=
RT
gK FällspK
sp
0
0 ,exp
beide Ausdrücke sind identisch.
Mit dieser Definition läßt sich Gl. (4.4.6) schreiben als:

48
4.4.11 ( )
∆−== ±± RT
gmK FällspK
Fällsp
0
0,
,expνγ
Gl. (4.4.11) bildet die Grundlage für die Berechnung von Löslichkeitskurven starker Elektro-
lyte. Aus ihr geht hervor, daß sich das Löslichkeitsprodukt entweder direkt aus den Gibbs-
schen Standardbildungsenthalpien berechnen, oder aber, bei Kenntnis der Aktivitätskoeffizi-
enten, aus experimentellen Löslichkeitsdaten gewinnen läßt.
Das Löslichkeitsprodukt ist bei festgelegtem Standardzustand für jeden Elektrolyt eine nur
von der Temperatur abhängige Konstante. Übersteigt das Produkt aus Anionen- und Katio-
nenaktivität den Wert des Löslichkeitsprodukts ist die Lösung übersättigt, und es bildet sich
ein Bodenkörper aus dem reinen festen Salz. Dabei ist es gleich ob die Übersättigung durch
fremdionigen Zusatz oder durch das Salz selbst hervorgerufen wird.
4.5 Temperaturabhängigkeit des Löslichkeitsprodukts
Der Zusammenhang zwischen dem Löslichkeitsprodukt und der Größe 0spKg∆ läßt sich auch
schreiben als:
4.5.1 T
g
RK spK
sp
0
0 1ln
∆−=
Partielle Ableitung nach der Temperatur ergibt bei konstantem Druck:
4.5.2
P
K
P
sp
TT
g
RT
Ksp
∆
−=
∂
∂
∂
∂
0
01ln
und weil
4.5.3 2
0
0
T
h
TT
g
R
P
Ksp
∆−=
∆
∂
∂ (Gibbs-Helmholtz)

49
ergibt sich für die Temperaturabhängigkeit;
4.5.4 2
00ln
RT
h
T
KR
P
sp ∆=
∂
∂
Dieser Ausdruck wird als van’t Hoffsche Reaktionsisobare bezeichnet. Mit ihm ist es möglich
das Löslichkeitsprodukt bei einer beliebigen Temperatur T zu berechnen, wenn das Löslich-
keitsprodukt bei Standardbedingungen K sp0 , und die Standardreaktionsenthalpie 0
Rh∆ bekannt
sind.
Die Enthalpie ist eine Zustandsfunktion. Das bedeutet, daß sie durch die Angabe des Anfangs-
und des Endzustandes des Systems eindeutig bestimmt ist, und nicht davon beeinflußt wird
auf welchem Weg das System vom Anfangs- zum Endzustand gelangt. Es ist daher möglich
die tatsächliche Reaktion aufzuspalten in die Zerlegung der Ausgangsstoffe in die Elemente
und den anschließenden Aufbau der Produkte aus eben diesen Elementen. Heß hat gefunden,
daß sich die Reaktionsenthalpie einer über Zwischenstufen verlaufenden Reaktion additiv aus
den Reaktionsenthalpien der einzelnen Schritte zusammensetzt. Nach dem Heßschen Gesetz
ist die Reaktionsenthalpie gleich der Summe der Bildungsenthalpien der Reaktionsprodukte,
vermindert um die Summe der Bildungsenthalpien der Ausgangsstoffe. Betrachtet man Lös-
lichkeitsgleichgewichte, bei denen das chemische Gleichgewicht nicht eindeutig auf der linken
bzw. rechten Seite der Reaktion liegt, ist die Einteilung in Produkte und Ausgangsstoffe nicht
ohne weiters möglich. In diesen Fällen müssen sowohl die Hin- als auch die Rückreaktion
berücksichtigt werden. Die Standardreaktionsenthalpien beider Reaktionen unterscheiden sich
aber nur in ihrem Vorzeichen und nicht in ihren Absolutbeträgen. Gleichfalls ändert sich die
Definition des Standardlöslichlichkeitsprodukts um das Vorzeichen des Exponenten je nach
betrachteter Reaktion (Dissoziation oder Fällung). Die in Gl. (4.5.9) gegebene Definition des
Löslichkeitsprodukts unter Standardbedingungen gilt für die Fällung des Salzes. Dement-
sprechend wird auch die Standardreaktionsenthalpie als Summe der Standardbildungsenthal-
pien der solvatisierten Ionen vermindert um die Standardbildungsenthalpie des Salzes berech-
net. Der Verlauf des Löslichkeitsproduktes mit der Temperatur ist für die Dissoziations- und
Fällungsreaktion derselbe.
Die Standardreaktionsenthalpie 0Rh∆ ist bei gegebener Temperatur genau wie das Löslich-
keitsprodukt nur vom zugrundeliegenden Standardzustand abhängig. Ihre Temperaturabhän-
gigkeit wird von der Kirchhoffschen Gleichung beschrieben:

50
4.5.5 0,
0
RP
P
R cT
h∆=
∆∂
∂
Zur Umrechnung der Standardreaktionsenthalpie 0Rh∆ bei 0T auf die Reaktionsenthalpie T
Rh∆
bei einer anderen Temperatur T muß die Kirchhoffsche Gleichung integriert werden. Bei
konstantem Druck ist diese Integration durch die Beziehung:
4.5.6 ∫∆+∆=∆ dtchh RPRTR
0,
0
gegeben. Zur numerischen Bestimmung des Integrals muß die Temperaturabhängigkeit der
molaren Wärmekapazitäten bekannt sein. Da molare Wärmekapazitäten solvatisierter Ionen
wenn überhaupt nur bei Standardbedingungen tabelliert sind, und die Temperaturabhängigkeit
nicht bekannt ist, werden die Standardwerte benutzt und angenommen, daß diese temperatu-
runabhängig seien. Die Integration der Gl. (4.5.6) führt dann zu:
4.5.7 ( )00,
0 TTchh RPRTR −∆+∆=∆
Die Kombination von Gl. 4.5.4 mit 4.5.7 führt zu
4.5.8
−
∆+
∆=
2
00,
2
0 1ln
T
TTR
c
RT
h
dT
Kd RPRsp
Die Integration dieses Ausdrucks zwischen der Referenztemperatur 0T und der Systemtempe-
ratur T führt zu
4.5.9
−+
∆+
−
∆−=− 1ln
11lnln
0
0
0,
0
00
TT
T
TR
c
TTR
hKK RPR
spTsp
bzw.:
4.5.10
−+
∆+
−
∆−= 1ln
11lnln
0
0
0,
0
00
TT
T
TR
c
TTR
hKK RPR
spTsp

51
Die Kombination mit Gl. 4.4.10 führt zu
4.5.11
−+
∆+
−
∆−
∆−= 1ln
11ln
0
0
0,
0
0
0
0
TT
T
TR
c
TTR
h
RT
gK RPRKspT
sp
Das Löslichkeitsprodukt bei einer beliebigen Temperatur T ist damit:
4.5.12
−+
∆+
−
∆−
∆−= 1ln
11exp
0
0
0,
0
0
0
0
T
T
T
T
R
c
TTR
h
RT
gK RPRKspT
sp
Die Kombination dieser Gleichung mit Gl.(4.4.11) und der Definitionsgleichung der mittleren
Molalität Gl. (2.5.3) ergibt einen Ausdruck, der es erlaubt bei Kenntnis der mittleren Aktivi-
tätskoeffizienten die Sättigungsmolalität aus dem Löslichkeitsprodukt zu berechnen.
4.5.13 mK
satT sp
T
=±
ν
γ

52
5. Anpassung von Modellparametern
5.1 Beschreibung der Datenbasis
Alle für die Anpassung verfügbaren Daten sind computergerecht in zwei separaten Datenban-
ken gespeichert. Die sogenannte VLE-Datenbank enthält Dampf-Flüssig-Gleichgewichtsdaten,
molale osmotische Koeffizienten von Lösungen, und mittlere Aktivitätskoeffizienten von
Salzen in Lösungen. Die sogenannte SLE-Datenbank enthält Fest-Flüssig-Gleichgewichtsda-
ten elektrolythaltiger Systeme.
Die VLE-Datenbank existierte bereits zu Beginn dieser Arbeit und enthielt gut 1000 Daten-
sätze. Ihr Umfang wurde auf 1855 Datensätze erweitert (Stand Juni 1998). Die Erweiterung
geschah sowohl mit Hilfe eigener Messungen als auch mit Hilfe von publizierten Daten.
Entsprechend der Meßgröße und der innerhalb eines Datensatzes variierten bzw. konstantge-
haltenen Zustandsgrößen ist jedem Datensatz ein sogenannter Isotyp zugeordnet.
ISO Anteil an der VLE- Datenbasis [%]
Meßgröße(n) variierende Zustandsgröße
konstante Zustandsgröße(n)
1 2.04 y,P XS T,C 2 15.36 y,T XS P,C 5 8.03 y XS T,C 6 1.07 y XS P,C 9 0.16 y,P XS,T C 11 2.21 y,P XS,C, T 12 13.04 y,T XS,C P 13 2.47 P XS,C T 15 3.88 y XS,C T 16 0.10 y XS,C P 19 1.13 y XS,C,T P 21 20.48 P C T 22 0.70 T C P 23 17.95 φ C T 24 0.32 φ C P 25 9.27 γ± C T 29 1.72 T C,P
XS=salzfreier Lösungsmittelbruch in der Flüssigphase, y=Molanteil in der Dampfphase, T=Temperatur, P=Druck, C=Elektrolytkonzentration, φ =molaler
osmotischer Koeffizient, γ ± =mittlerer Aktivitätskoeffizient des Elektrolyten
Tabelle 5.2.1 Isotypenverteilung innerhalb der VLE-Datenbank
Isotypen sind Kennzahlen, die Informationen über die jeweilige Meßgröße(n) und die inner-
halb eines Datensatzes variierten bzw. nicht variierten Zustandsgrößen enthalten. Ferner geb-

53
en sie Aufschluß darüber, ob ein System eine oder mehrere Lösungsmittelkomponenten ent-
hält. Die Zuordnung des Isotyps erfolgt immer unter Einhaltung der folgenden Regeln:
• Allen isobaren Datensätzen werden grade, allen isothermen Datensätzen ungrade Isotypen
zugeordnet.
• Allen Datensätze die mehr als eine Lösungsmittekomponente aufweisen wird ein Isotyp
kleiner als 20 zugeordnet.
• Allen Datensätzen in denen die Salzkonzentration konstant ist wird ein Isotyp kleiner als
zehn zugeordnet.
Tabelle 5.2.1 gibt einen Überblick über die 17 verschiedenen Isotypen und ihren jeweiligen
Anteil am Bestand der VLE-Datenbank für Elektrolytsysteme.
Der Isotyp enthält jedoch weder Informationen über die Art noch über die genaue Anzahl der
Systemkomponenten. Wie sich die Datensätze auf binäre, ternäre und quarternäre Systeme
verteilen ist in Abb. 5.2.1 dargestellt. Betrachtet man die Datenbasis hinsichtlich ihrer Vertei-
lung auf bestimmte Komponenten, so wird deutlich, daß es sich bei den binären Systemen
zum weitaus größten Teil um wäßrige Systeme handelt. Bei den ternären Systemen handelt es
sich in der Mehrzahl um Alkohol-Wasser-Systeme. In Abbildung 5.2.2 ist eine genaue Auf-
schlüsselung der Datenbasis auf die Komponenten dargestellt. Sie berücksichtigt nur die
Systeme, deren Komponenten Elemente der LIQUAC-Matrix sind.
Die SLE-Datenbank wurde im Rahmen dieser Arbeit erstellt und enthält 1194 Datensätze
(Stand Juni 1998). Neben Sättigungsmolalitäten starker Elektrolyte in Lösungsmitteln und
Lösungsmittelgemischen, sind auch Dichten von Salzlösungen gespeichert. Allen Fest-Flüs-
sig-Phasengleigewichtsdaten ist durchgängig der Isotyp 32 zugeordnet um sie von den VLE-
Daten zu unterscheiden. Eine weitere Unterteilung in verschiedene Isotypen innerhalb der
SLE-Datenbank wird nicht gemacht. 53 % der SLE-Daten beschreiben binäre Systeme, davon
entfallen wiederum 66 % auf rein wäßrige. Bei den restlichen 47 % handelt es sich um ternäre
Systeme, von denen 26 % aus zwei Lösungsmitteln und einem Salz und 74 % aus zwei Salzen
und einem Lösungsmitteln bestehen. Abbildung 5.2.1 gibt einen Überblick über Zusammen-
setzung der Datenbasis.
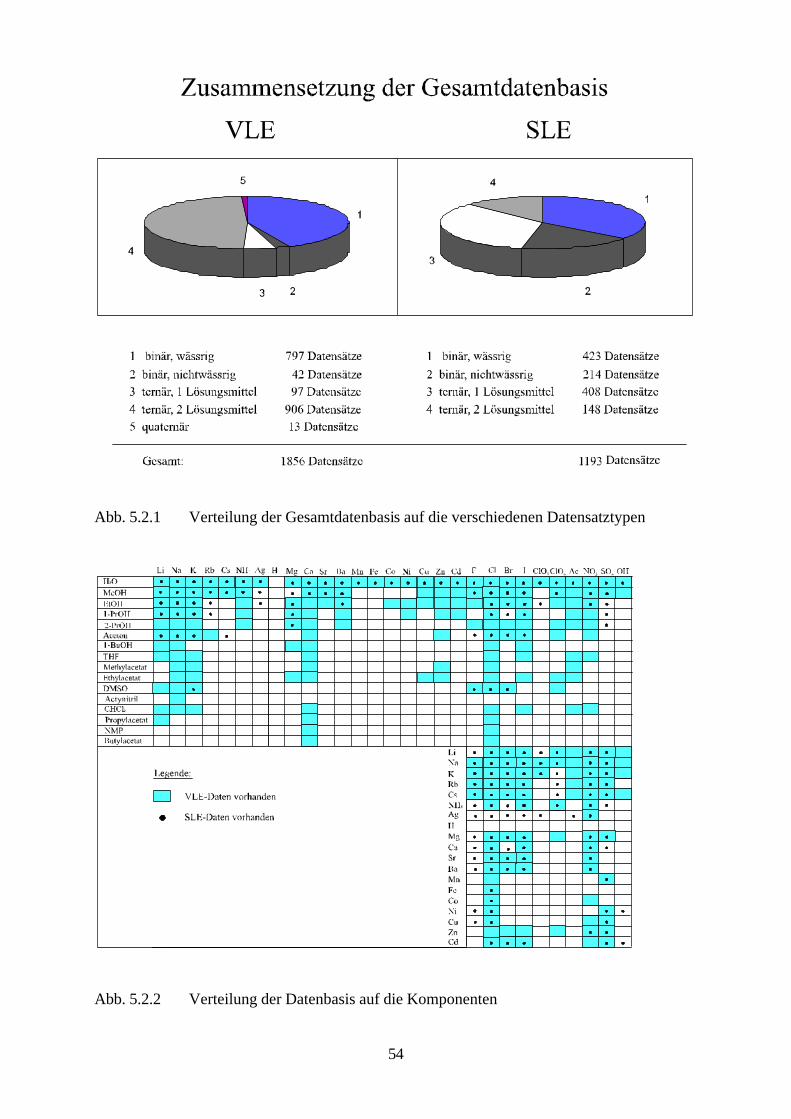
54
Abb. 5.2.1 Verteilung der Gesamtdatenbasis auf die verschiedenen Datensatztypen
Abb. 5.2.2 Verteilung der Datenbasis auf die Komponenten

55
5.2 Reduzierung der Datenbasis
Die für die Anpassung der Wechselwirkungsparameter benutzte Datenbasis sollte so umfang-
reich wie möglich sein. Nichtsdestotrotz können nicht alle Daten zur Anpassung herangezogen
werden. Die Gründe aus denen vorhandene Daten bei der Anpassung nicht berücksichtigt
wurden sind folgende:
Zu geringe Löslichkeit:
Die Löslichkeitsprodukte der Silberhalogenide liegen in der Regel zwischen 0.14 E-10 (AgCl,
T=273.15 K) und 0.31 E-13 (AgI, T= 278.15 K). Der Einfluß solch geringer Konzentrationen
auf das Dampf-Flüssig-Phasengleichgewicht läßt sich mit herkömmlichen Methoden nicht
bestimmen. Dementsprechend existieren keine Dampf-Flüssig-Phasengleichgewichtsdaten
dieser Systeme. Fest-Flüssig-Phasengleichgewichtsdaten können jedoch mit Hilfe von Leitfä-
higkeitsmessungen gewonnen werden. Da sich diese Daten aber auch bei Annahme idealen
Verhaltens gut vorhersagen lassen ( 0 wenn x1 →=±γ ) wurden die Löslichkeitsdaten der
Silberhalogenide bei der Anpassung nicht berücksichtigt.
Unvollständige Dissoziation:
Bereits 1930 fanden Righellato und Davies [49], daß die meisten Elektrolyte unsymmetrischer
Stöchiometrie in wäßriger Lösung nicht ad hoque sondern stufenweise in An- und Kationen
dissoziieren. Die Vernachlässigung dieser Tatsache führt zwangsläufig zu Fehlern bei der
Modellierung. Ein bekanntes Beispiel für dieses Verhalten ist Natriumsulfat. Bei der Model-
lierung einer Natriumsulfatlösung wird in den meisten Fällen von der Reaktion:
Na SO Na SO2 4 422→ ++ −
ausgegangen. Die tatsächliche Reaktion verläuft jedoch in zwei Stufen:
Na SO Na NaSO
NaSO Na SO2 4 4
4 42
→ +
→ +
+ −
− + −
Die Gleichgewichtskonstante für die Dissoziation des Mononatriumsulfat-Anions wird von
Jenkins und Monk [26] mit K=0.19 bei 25°C angegeben. In neueren Arbeiten, wie der von

56
Izatt et al. [24] findet man einen kalorimetrisch bestimmten Wert von K=0.229. Vernachlässigt
man bei der Modellierung die Existenz des Intermediats und führt das reale Verhalten der
Lösung nur auf die Natrium- und Sulfationen zurück erhält man falsche Parameterwerte. Für
die Beschreibung des Dampf-Flüssig-Phasengleichgewichts ist dies irrelevant. Es wird zwar
nicht die tatsächlich vorliegende Lösung sondern nur der Bruttoeffekt des Salzes wiedergege-
ben, trotzdem gelingt eine zufriedenstellende Beschreibung. Die Beschreibung des Löslich-
keitsverhaltens dagegen gelingt nicht, weil man von dem falschen Löslichkeitsprodukt aus-
geht. Da es nicht möglich ist modellimmanent verschiedene Dissoziationsstufen der Elektro-
lyte zu berücksichtigen wurden die Löslichkeitsdaten des Systems Natriumsulfat / Wasser von
der Anpassung ausgeschlossen. Prinzipiell können natürlich alle Salze des Typs A2B bzw. AB2
stufenweise dissoziieren. Eine Vorhersage dieses Verhaltens ist allein aufgrund des Valenztyps
allerdings nicht möglich. Die Frage ob gegebenenfalls Löslichkeitsdaten weiterer Salze von der
Anpassung auszuschließen sind läßt sich demnach nur empirisch anhand der Ergebnisse prü-
fen.
Auf eine andere Variante des gleichen Problems stößt man bei der Modellierung flüchtiger
Elektrolyte. Flüchtige Elektrolyte lösen sich in beträchtlichem Umfang undissoziiert in wäßri-
ger Lösung. Dadurch ist hier der mögliche Übergang in die Dampfphase zu berücksichtigen.
Auch die Gleichgewichtskonstante für diesen Übergang kann modellimmanent nicht berück-
sichtigt werden. Flüchtige Elektrolyte wurden zwar bei der Anpassung berücksichtigt, jedoch
ist klar, daß auch bei ihnen lediglich der Bruttoeffekt auf das Dampf-Flüssig-Gleichgewicht
erfaßt werden kann. Eine Konsequenz dieses Verhaltens ist dabei, daß die Ionenstärke von
Lösungen flüchtiger Elektrolyte falsch berechnet wird.
Fehlerhafte Daten:
In einigen Fällen mußten Datensätze ausgeschlossen werden, die ganz offensichtlich mit gro-
ßen experimentellen Fehlern behaftet waren. Die Beurteilung der Qualität von Phasengleich-
gewichtsdaten elektrolythaltiger Systeme ist besonders schwierig, weil für sie kein thermody-
namisch exakter Konsistenztest existiert. So ist man bei der Auswahl der Datensätze auf rela-
tiv oberflächliche Kriterien wie beispielsweise das Ausmaß der Streuung von Datenpunkten
angewiesen. Dies ist um so schwerwiegender als infolge der größeren Schwierigkeiten bei der
Vermessung von elektrolythaltigen Gemischen die Qualität der Daten häufig unzureichend ist.
Im Vergleich mit der Anzahl an Publikationen über Phasengleichgewichte von Nichtelektro-
lytsystemen stehen insgesamt viel weniger Daten zur Verfügung. Hinsichtlich der Fragestel-

57
lung ob es möglich ist das Fest-Flüssig- und das Dampf-Flüssig-Phasengleichgewicht
elektrolythaltiger Systeme mit einem einheitlichen Parametersatz zu beschreiben sind darüber
hinaus nur die Systeme relevant, für die die beiden folgenden Kriterien erfüllt sind:
• Es müssen Löslichkeitsdaten des Systems vorhanden sein. Dabei ist zu berücksichtigen,
daß jeweils nur ein Ast (die Löslichkeitskurve) des Fest-Flüssig-Phasendiagramms das
Phasengleichgewicht zwischen dem reinen festen Salz und seiner Lösung beschreibt. Daten
bei denen die Lösung mit einer anderen festen Phase außer dem reinen Salz im Gleichge-
wicht steht sind als Löslichkeitsdaten für die Anpassung irrelevant.
• Die thermodynamischen Größen zur Berechnung der Reaktionsenthalpie des Löslichkeits-
produkts müssen verfügbar sein. Von den 19 Kationen und 10 Anionen der LIQUAC-Ma-
trix ist diese Bedingungen für 9 Kat- und 8 Anionen erfüllt.
5.3 Parameterbestimmung
Bei der Parameterbestimmung geht es zum einen um das rein mathematische Verfahren zur
Gewinnung der Parameter, zum anderen geht es um das zugrundeliegende Anpassungskon-
zept, dessen Ziel die Klärung der folgenden Fragen ist:
1. Inwieweit sind die tabellierten Gibbsschen Standardbidungsenthalpien kongruent mit Lös-
lichkeitsdaten und experimentellen mittleren Aktivitätskoeffizienten starker Elektrolyte in
wäßriger Lösung?
2. Inwieweit ist es mit Hilfe des LIQUAC-Modells möglich auf der Grundlage tabellierter
Standardgrößen mittlere Aktivitätskoeffizienten starker Elektrolyte in wäßriger Lösung über
den gesamten Temperatur und Konzentrationsbereich zu beschreiben?
3. Gelingt unter den gegebenen Voraussetzungen die simultane Beschreibung mittlerer Aktivi-
tätskoeffizienten und der Lösungsmittelaktivität?

58
4. Ist das ‘Prinzip des Löslichkeitsprodukts’ auf der Grundlage tabellierter Standardgrößen
wäßriger Systeme für die Beschreibung der Löslichkeit in organischen Lösungsmitteln an-
wendbar?
5. Ist der gewählte Ansatz auch geeignet um das reale Verhalten elektolythaltiger Lösungsmit-
telgemische zu beschreiben?
Im Folgenden soll das Anpassungskonzept erläutert, und die Zielsetzung dieser Arbeit mit
dem von Polka verfolgten Ziel verglichen werden. Dazu ist es wichtig sich einige Besonder-
heiten von Elektrolytmodellen und im besonderen der LIQUAC-Methode vor Augen zu füh-
ren.
Interne Konsistenz:
Grundsätzlich muß innerhalb des LIQUAC-Modells zwischen den Ion-Ion- und den Lö-
sungsmittel-Ion-Parametern unterschieden werden. Während es sich bei den Ersten ihrer
Natur nach um rein salzspezifische Parameter handelt, müssen die Lösungsmittel-Ion-
Parameter allgemein verwendbar sein. Betrachtet man eine wäßrige Natriumchloridlösung, so
müssen die Parameter, die die Wechselwirkung zwischen Natrium und Wasser beschreiben
unabhängig davon sein ob es sich um eine Natriumchlorid oder beispielsweise um eine
Natriumbromidlösung handelt. Ebenso müssen die Chlorid-Wasser-Parameter zur
Beschreibung der Natriumchloridlösung genauso gut für die Beschreibung einer
Kaliumchloridlösung geeignet sein. Die Information darüber, daß sich die
Natriumchloridlösung anders verhält als die Natriumbromidlösung steckt dementsprechend
nur in den Ion-Ion-Parametern.
Die interne Konsistenz der Lösungsmittel-Ion-Parameter wurde dadurch erreicht, daß die Pa-
rameter sequentiell angepaßt wurden. Bei jedem nächsten Anpassungsschritt wurden die im
vorherigen Schritt gewonnenen Parameter genau wie experimentelle Daten behandelt. Interne
Konsistenz begegnet uns auch bei den thermodynamischen Standardgrößen, die im sogenann-
ten ‘Standard Table of Arrangement’ geordnet sind [55], der die Reihenfolge der Evaluierung
dieser Größen festlegt. Prinzipiell ist interne Konsistenz die Grundlage jeder Gruppenbei-
tragsmethode. Eine Konsequenz davon ist, daß es im Nachhinein nicht ohne weiteres möglich

59
ist einen einmal festgelegten Wert oder Parameter zu ändern ohne gleichzeitig alle folgenden
Größen zu beeinflussen. So kann eine vermeintliche Verbesserung eines Wertes zu
unvorhergesehener Verschlechterung an anderer Stelle des in sich geschlossenen Systems füh-
ren. Abbildung (5.3.1) verdeutlicht diesen ‘Dominoeffekt’, den die Veränderung der Lö-
sungsmittel-Ion-Parameter bewirkt beispielhaft:

60
Abb. 5.3.1 Interne Konsistenz der Lösungsmittel-Ion-Parameter

61
Die Datenbasis für die Neuanpassung der Lösungsmittel-Ion-Parameter zur Beschreibung der
Wechselwirkung Li+ / Wasser besteht aus allen binären wäßrigen Systemen in denen diese
Wechselwirkung enthalten ist. Dies sind die wäßrigen Lösungen der Salze Lithium -perchlorat,
-sulfat, -acetat, -hydroxid, -bromid, -chlorid, -nitrat und -jodid. Da sämtliche Parameter zur
Beschreibung dieser (resultierenden) Datenbasis bereits angepaßt wurden, führt die alleinige
Veränderung der Lithium-Wasser-Parameter zwangsläufig zu einer Verschlechterung der
Gesamtbeschreibung. Die einzige Möglichkeit zur Verbesserung besteht darin dem Modell die
Möglichkeit zu geben auch alle anderen Lösungsmittel-Ion-Parameter zur Beschreibung der
betroffenen Systeme zu verändern. In diesem konkreten Fall sind dies die Parameter zur
Beschreibung der Wechselwirkung zwischen Wasser und den Anionen der genannten acht
Lithiumsalze. Die daraus wiederum resultierende Datenbasis ist in Abbildung 5.3.1 doppelt
dargestellt. Auf der linken Seite sind die resultierenden Datenbasen der acht anzupassenden
Wasser-Anion-Parameter dargestellt, auf der rechten Seite sind die gleichen Systeme entspre-
chend der dadurch wiederum betroffenen Kation-Wasser-Parameter geordnet.
Es wird dabei deutlich, daß es innerhalb der bestehenden Parametermatrix nicht möglich ist
nur bestimmte Lösungsmittel-Ion-Parameter zu verändern ohne eine Verschlechterung der
Gesamtbeschreibung der zugrundeliegenden Datenbasis zu verursachen. Auch der Versuch
sämtliche Lösungsmittel-Ion-Parameter gleichzeitig anzupassen kann mit Hilfe des verwende-
ten Optimieralgorithmus nicht gelingen. Das Simplex-Nelder-Mead-Verfahren arbeitet ohne
Gradientenbestimmung. Es verändert nacheinander jeden zu optimierenden Parameter um die
angegebene Schrittweite und berechnet den Wert der Zielfunktion. Da jedoch jede
Veränderung eines einzelnen Lösungsmittel-Ion-Parameters zu einer Erhöhung des
Zielfunktionswertes führt findet der Algorithmus in keinem Fall ein tiefer gelegenes Minimum
der Zielfunktion.
Für die Überarbeitung der Matrix unter Einbeziehung der neu hinzugenommenen Löslich-
keitsdaten bedeutet das, daß nur zwei Wege gangbar sind. Der eine besteht darin die bisheri-
gen Lösungsmittel-Ion-Parameter unverändert zu übernehmen und lediglich die Ion-Ion-Pa-
rameter neu anzupassen, für die keine interne Konsistenz gefordert ist. Dies ist mit dem Nach-
teil verbunden, daß für jedes System eine geringere Anzahl an Anpassungsgrößen zur Verfü-
gung steht. Daß interne Konsistenz durch den Verlust anpaßbarer Größen erkauft wird ist der
Grund dafür, daß eine Allgemeinverwendbarkeit der Parameter immer mit einem Verlust an
Signifikanz einhergeht. Der zweite Weg ist dementsprechend die sequentielle Neuanpassung
aller Parameter ausgehend von den Systemen für die die Datenbasis am umfangreichsten ist.

62
Hierbei stößt man allerdings auf das gleiche Problem. Die in diesem Fall zuerst anzupassenden
Parameter sind die zur Beschreibung der Wechselwirkung Chlorid / Wasser. Da aber von je-
dem in der LIQUAC-Matrix enthaltenen Kation experimentelle Daten über wäßrige Lösungen
seiner Chloride vorhanden sind läuft dieser Versuch der sequentiellen Neuanpassung
wiederum auf die gleichzeitige Anpassung sämtlicher Anpassungsgrößen hinaus. Auch wenn
es in diesem Fall möglich ist das zuvor beschriebene Problem der Simplex-Nelder-Mead-
Methode durch neue Parameterstartwerte zu umgehen, resultiert daraus ein nicht zu
bewältigender Rechenaufwand. Die zugrundeliegende Datenbasis zur Anpassung der Wasser-
Ion-Parameter enthält 674 Datensätze und berücksichtigt 91 verschiedene Systeme. Konkret
bedeutet demnach die gleichzeitige Überarbeitung der Matrix nur für binäre, wäßrige Systeme
die simultane Bestimmung von 451 Parametern an 9532 Datenpunkten. Da dieser
Rechenaufwand mit den verfügbaren Mitteln nicht zu bewältigen ist, kommt diese Möglichkeit
jedoch nicht in Betracht.
Vorgehensweise:
Die zentrale Frage dieser Arbeit ist zunächst die nach der grundsätzlichen Fähigkeit der
LIQUAC-Methode alle verfügbaren Datentypen in einem möglichst großen Temperatur- und
Konzentrationsbereich zu beschreiben. Wie wir gesehen haben schränkt dabei die Notwen-
digkeit der internen Konsistenz aller Lösungsmittel-Ion-Parameter den Bewegungsspielraum
des Modells ein. Um dies zumindest für den Augenblick zu umgehen wurden in einem ersten
Schritt für die Systeme, deren Datenbasis um Löslichkeitsdaten erweitert wurde, alle
Parameter systemspezifisch angepaßt. Auch wenn dieses Vorgehen dem Konzept der
LIQUAC-Methode widerspricht und zu keiner befriedigenden Gesamtbeschreibung führen
kann, bietet es doch die Möglichkeit der bestmöglichen Beschreibung einzelner Systeme.
Auch die Beurteilung thermodynamischer Standardgrößen wird dadurch in begrenztem
Umfang möglich.
Naturgemäß liegen die zuverlässigsten und umfangreichsten Daten über binäre wäßrige Sy-
steme vor. Letztere wurden daher als Ausgangspunkt für die Anpassung der Modellparameter
gewählt.

63
Vor dem Übergang von systemspezifischen zu allgemein verwendbaren Wasser-Ion-Parame-
tern, sollen zwei Dinge geprüft werden. Zunächst soll nur anhand der Systeme, von denen
sowohl experimentelle mittlere Aktivitätskoeffizienten von Salzen in gesättigter Lösungen
(Isotyp 25) als auch Löslichkeitsdaten (Isotyp 32) vorliegen, überprüft werden, inwieweit
beide Datentypen mit den tabellierten Gibbsschen Standardbildungsenthalpien
übereinstimmen.
Anschließend gilt es durch simultane Anpassung an alle binären Datensatztypen zu klären,
inwieweit, das Modell in der Lage ist gleichzeitig den mittleren Aktivitätskoeffizienten des
Elektrolyten und die Lösungsmittelaktivität über den gesamten Temperatur- und Konzentrati-
onsbereich richtig zu beschreiben.
Es ist davon auszugehen, daß sich die Qualität der Beschreibung durch den anschließenden
Übergang zu allgemein verwendbaren Lösungsmittel-Ion-Parametern verschlechtern wird. Ob
diese Verschlechterung in Kauf genommen werden kann oder nicht bleibt anhand der Ergeb-
nisse zu prüfen.
Für die Anpassung der verbleibenden Wechselwirkungsparameter zwischen nichtwäßrigen
Lösungsmittelkomponenten und Ionen werden im Fall ternärer Systeme sowohl die Ion-Ion
als auch die Wasser-Ion-Parameter unverändert übernommen. Aufgrund der sehr geringen
Löslichkeit der meisten Elektrolyte in nichtwäßrigen Lösungsmitteln stellt sich die Datensitua-
tion hier anders dar als bei rein wäßrigen Systemen. Es gibt kaum Daten binärer Systeme aus
einem nichtwäßrigen Lösungsmittel und einem Elektrolyten. Ausnahmen bilden hier nur die
ersten Homologe der Alkohole. Da die Löslichkeit aber schon durch die Zugabe geringer
Mengen Wasser schlagartig zunimmt existieren viele Daten für ternäre Systeme. Für die An-
passung der Wechselwirkungsparameter zwischen nichtwäßrigen Lösungsmittelkomponenten
und Ionen müssen daher ternäre Systeme herangezogen werden.
Trotz der minimalen Datenbasis soll vor der Anpassung der Wechselwirkungsparameter zwi-
schen nichtwäßrigen Lösungsmittelkomponenten und Ionen an ternären Systemen die
Anwendbarkeit des Löslichkeitsprodukts für binäre, nichtwäßrige Systeme überprüft werden.
Die besondere Schwierigkeit liegt darin, daß sämtliche thermodynamischen Daten der solvati-
sierten Ionen nur für das Lösungsmittel Wasser tabelliert sind. Dadurch wird seitens des Lös-
lichkeitsprodukts immer von einer wäßrigen Lösung ausgegangen, was durch das Modell
kompensiert werden muß. Ob und wieweit diese Kompensation gelingt soll wiederum in drei
Schritten geprüft werden. Zunächst werden Lösungsmittel-Ion-Parameter systemspezifisch an
Löslichkeitsdaten angepaßt. Anschließend werden Dampf-Flüssig-Gleichgewichtsdaten in die

64
Anpassung einbezogen und danach der Übergang zu allgemeinverwendbaren Parametern ge-
prüft.
In Tabelle 5.3.1 ist die Vorgehensweise der Anpassung noch einmal schematisch zusammen-
gefaßt. Zu jedem Anpassungsschritt sind von links nach rechts Erläuterungen zur jeweiligen
Datenbasis und der Zielrichtung angegeben.
Tab. 5.3.1 Konzept zur Anpassung der Modellparameter
Mathematisches Verfahren:
Das mathematische Verfahren zur Bestimmung der Modellparameter ist die Minimierung der
folgenden allgemeinen Zielfunktion:
5.3.1 ( ) ( )F a a b c gk l l k k l k l berntnp
, , , , exp, , , min= − =∑∑ Λ Λ Λ2
np : Anzahl Datenpunkte
nt : Anzahl Datensatztypen
Hierbei ist der Gewichtungsfaktor g Λ je nach Datensatztyp definiert. Die Zielgröße Λ ent-
spricht der in Tabelle 5.2.1 angegebenen Meßgröße des jeweiligen Isotyps und wird wie folgt
berechnet:
Anpassungsschritt
Anpassung systemspezifischer Ion-Ionund Wasser-Ion-Parameter
Binäre Löslichkeitsdaten wäßrigerSysteme von denen auch experimen-telle mittlere Aktivitätskoeffizientenim Sättigungszustand verfügbar sind
Datenbasis Ziel / Fragestellung
Beurteilung der Kongruenz mit Gibbs-schen Standardbildungsenthalpien
Binäre Löslichkeitsdaten wäßrigerSysteme von denen auch experimen-telle mittlere Aktivitätskoeffizientenverfügbar sind
Beschreibung des mittleren Aktivitäts-koeffizienten über den gesamten Konzen-trations- und Temperaturbereich
Endgültige Anpassung der Ion-Ion-Parameter, Wasser-Ion-Parameter werden übernommen
Alle binären, wäßrigen Systeme( alle Datensatztypen )
Übergang zu allgemein verwendbarenWasser-Ion-Parametern
Berechnung binärer, nichtwäßrigerSysteme
Alle binären, nichtwäßrigen Systeme Anwendung des Löslichkeitsproduktsfür binäre, nichtwäßrige Systeme
Anpassung der Wechselwirkungspa-rameter zwischen Ionen und nichtwäß-rigenLösungsmitteln
Alle ternären Systeme( 2 Lösungsmit-tel, 1 Salz)
Gleichzeitige Beschreibung von VLEund SLE elektrolythaltiger Lösungs-mittelgemische

65
5.3.2 ( ) PPxy siiiberi /, γ=
5.3.3 ∑=
=nloese
i
siiiber PxP
1
γ
5.3.4 MinTPTPfT berber ≡−= ))()(( )((exp)
5.3.5
⋅⋅−= ∑ s
nion
issber Mxx1
/ln1000 γφ
5.3.6 ν νν γγγ −+ ∗−
∗+
∗± =ber,
Die Zielgröße für den Isotyp 32 ist die Sättigungsmolalität. Sie berechnet sich gemäß Gl.
(4.5.12)
5.3.7 ±= γν /,Tsp
Tbersat Km
Für die Minimierung der Zielfunktion wurde ein Fortran-Programm erstellt, das die von Nelder
und Mead verbesserte Simplexmethode nutzt [34]. Es handelt sich dabei um ein nichtlineares
Regressionsverfahren, das ohne Gradientenbestimmung auskommt und vergleichsweise
schnell arbeitet. Der Zielfunktionswert ist die Summe der prozentualen Abweichungen aller
Datensätze. Diese Summe wird aus den Beträgen der einzelnen Beiträge gebildet. Die
Minimierung der absoluten Abweichung führt aufgrund der unterschiedlich großen Absolut-
werte der jeweiligen Zielgrößen zu keiner guten Gesamtbechreibung.

66
Vergleich der Zielsetzung
Bezüglich der Auswahl der zu berücksichtigenden Systeme und Datensätze unterscheidet sich
diese Arbeit von Polkas Arbeit. Polkas Anliegen bestand darin das LIQUAC-Modell mit kon-
kurrierenden Elektrolytmodellen zu vergleichen. Von daher war er in der Auswahl der unter-
suchten Systeme von vornherein auf diejenigen beschränkt, die sich auch mit den gewählten
Vergleichsmodellen berechnen lassen. Auch der untersuchte Konzentrationsbereich orientiert
sich bei seiner Arbeit nicht ausschließlich an der zur Verfügung stehenden Datenbasis sondern
an der Schnittmenge innerhalb derer alle Modelle sinnvolle Ergebnisse liefern. Innerhalb die-
ses Rahmens geht es ihm darum ein möglichst gutes Gesamtergebnis zu erreichen. So wurde
beispielsweise das System Wasser / Salpetersäure von ihm nur bis zur Konzentration von 5
mol/kg beschrieben obwohl experimentelle Daten bis zu einer Konzentration von 28 mol/kg
zur Verfügung stehen. Ebenso hat er die wäßrigen Lösungen von 24 weiteren Elektrolyten aus
seinen Betrachtungen ausgeschlossen.
Das Ziel dieser Arbeit ist es das Potential der LIQUAC-Methode zu testen. Von daher erklärt
es sich, daß zum einen nur offensichtlich fehlerhafte Daten ausgeschlossen, und zum anderen
von einer besonderen Gewichtung einzelner Datensätze oder einer Beschränkung des Konzen-
trations- und Temperaturbereichs abgesehen wurde. Gerade die Berücksichtigung von Löslich-
keitsdaten stellt besondere Anforderungen an das Modell. Einerseits ist die Berechnung von
Löslichkeitskurven starker Elektrolyte an thermodynamische Daten gekoppelt, die den Verlauf
der mittleren Aktivitätskoeffizienten entlang der Sättigungsgrenze festlegen, andererseits er-
weitert sich durch die Berücksichtigung von Löslichkeitsdaten der zu untersuchende Konzen-
trations- und Temperaturbereich. So liegt die höchste Konzentration die in Polkas Arbeit be-
schrieben wird bei 25.95 mol/kg (Ammoniumnitrat / Wasser), während sie in dieser Arbeit
28.0 mol/kg (Salpetersäure / Wasser) beträgt. Dramatischer stellt sich die Erweiterung des
Temperaturbereichs von 18 bis 150 [°C] auf -18 bis 251 [°C] da.

67
5.4 Anpassung an binäre, wäßrige Systeme
5.4.1 Anpassung systemspezifischer Parameter an Löslichkeitsdaten
Bei Betrachtung der einzelnen Zielfunktionen zeigt sich, daß der mittlere Aktivitätskoeffizient
des Salzes nur in Gl. 5.3.6 und Gl. 5.3.7 eingeht, während für die Beschreibung aller übrigen
Datensatztypen der Aktivitätskoeffizient des Lösungsmittel entscheidend ist. Die
Löslichkeitsdaten verknüpfen gemäß Gl. (5.14) thermodynamische Standardgrößen mit den
experimentellen Aktivitätskoeffizienten der Salze. Die Fehler in den tabellierten
thermodynamischen Standardgrößen wirken sich natürlich direkt auf die Qualität der Berech-
nung aus. So bewirkt ein Fehler der Gibbsschen Standardreaktionsenthalpie von ±0.5 kJ/mol
bei 25°C eine Verfälschung der Gleichgewichtskonstante um ca. 22% [19]. Die Anpassung
von Modellparametern an Löslichkeitsdaten und ein Vergleich der sich ergebenden mittleren
Aktivitätskoeffizienten mit experimentellen Werten stellt einen Schlüssel für die Beurteilung
der thermodynamischen Standardgrößen dar.
In einem ersten Schritt werden deshalb ausschließlich die binären, wäßrigen Systeme
behandelt, von denen sowohl experimentelle Daten über den mittleren Aktivitätskoeffizienten
im Sättigungszustand bei 25°C, als auch Löslichkeitsdaten bei dieser Temperatur vorhanden
sind. Die für die Beschreibung der Systeme notwendigen LIQUAC-Parameter wurden
zunächst nur an Löslichkeitsdaten angepaßt. Die Beschreibung von Löslichkeitsdaten stellt
höhere Anforderungen an das Modell als die Beschreibung herkömmlicher Dampf-Flüssig-
Phasengleichgewichtsdaten. Dies ist zum einen in der Kopplung der Berechnung an
thermodynamische Standardgrößen begründet, gleichzeitig handelt es sich aber um die jeweils
maximale Salzkonzentration. Weiterhin ist auch der Temperaturbereich in dem
Löslichkeitsdaten zur Verfügung stehen in der Regel größer als bei üblichen VLE-Daten-
sätzen. Im Fall einer einzigen Lösungsmittelkomponente kürzen sich die Virialkoeffizienten
der Ion-Molekül-Wechselwirkungen aus dem Ausdruck für den Aktivitätskoeffizienten des
Lösungsmittels Gl.(3.2.7) heraus, so daß nur die Ableitungen nach der Ionenstärke
übrigbleiben. Diese enthalten aber gemäß Gl. (3.2.10) nicht mehr den b-Parameter, der nur in
Lösungsmittelgemischen benötigt wird und deshalb nicht angepaßt wurde. Es resultieren zehn
anpaßbare Größen für jedes der beschriebenen Systeme. Sechs dieser zehn Freiheiten rühren
allein aus der Beschränkung auf systemspezifische Lösungsmittel-Ion-Parameter. Dieses
Vorgehen widerspricht zwar dem Konzept der LIQUAC-Methode, es handelt sich dabei aber

68
nur um einen Zwischenschritt, der durch die genauere Beschreibung auf Grund der
gewonnenen Freiheiten gerechtfertigt ist. Bei exakter Beschreibung der Löslichkeitsdaten
durch das Modell läßt sich die Genauigkeit der thermodynamischen Daten beurteilen, weil das
Maß der Übereinstimmung angepaßter und experimenteller mittlerer Aktivitätskoeffizienten
dann entweder ein Maß für Zuverlässigkeit der zugrundeliegenden thermodynamischen Da-
ten, oder aber, - wenn die thermodynamischen Größen als richtig angenommen werden- ein
Maß für die Kongruenz der Löslichkeitsda ten einerseits und der experimentellen
Aktivitätskoeffizienten andererseits ist.
Die Abbildungen 5.4.1 bis 5.4.7 zeigen die Ergebnisse der Anpassung systemspezifischer
Parameter an Löslichkeitsdaten binärer, wässriger Systeme. Um die Ergebnisse bestmöglich
interpretieren zu können wurden jeweils drei unterschiedliche Arten der Auftragung gewählt
(jeweils Plot a-c). Der oberste Plot einer Abbildung (*a) enthält dabei immer die auf Basis der
gewonnenen Wechselwirkungsparameter berechneten Sättigungsmolalitäten im Vergleich mit
den entsprechenden experimentellen Werten. Das Maß der Übereinstimmung gibt an wie gut
der mittlere Aktivitätskoeffizient im Sättigungszustand im jeweiligen Temperaturintervall
beschrieben wird. Der mittlere Plot (*b) enthält jeweils die absoluten Werte der berechneten
mittleren Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur. Der
jeweils untere Plot (*c) einer Abbildung zeigt eine Vorhersage experimenteller mittlerer
Aktivitätskoeffizienten bei ϑ =25°C als Funktion der Konzentration. Aus dieser Darstellung
wird deutlich in wieweit sich die an Löslichkeitsdaten angepaßten Modellparameter zur
Beschreibung des verdünnten Bereichs eignen.
EineTabelle mit allen verwendeten Standardgrößen findet sich im Anhang (7.5). Die
Standardgrößen dienen der Berechnung des Löslichkeitsprodukts und wurden der Literatur
entnommen [55]. Im Anhang (7.4) sind die angepaßten Parameter gelistet. Das
Abbruchkriterium für die Iteration lag bei 12101 −⋅
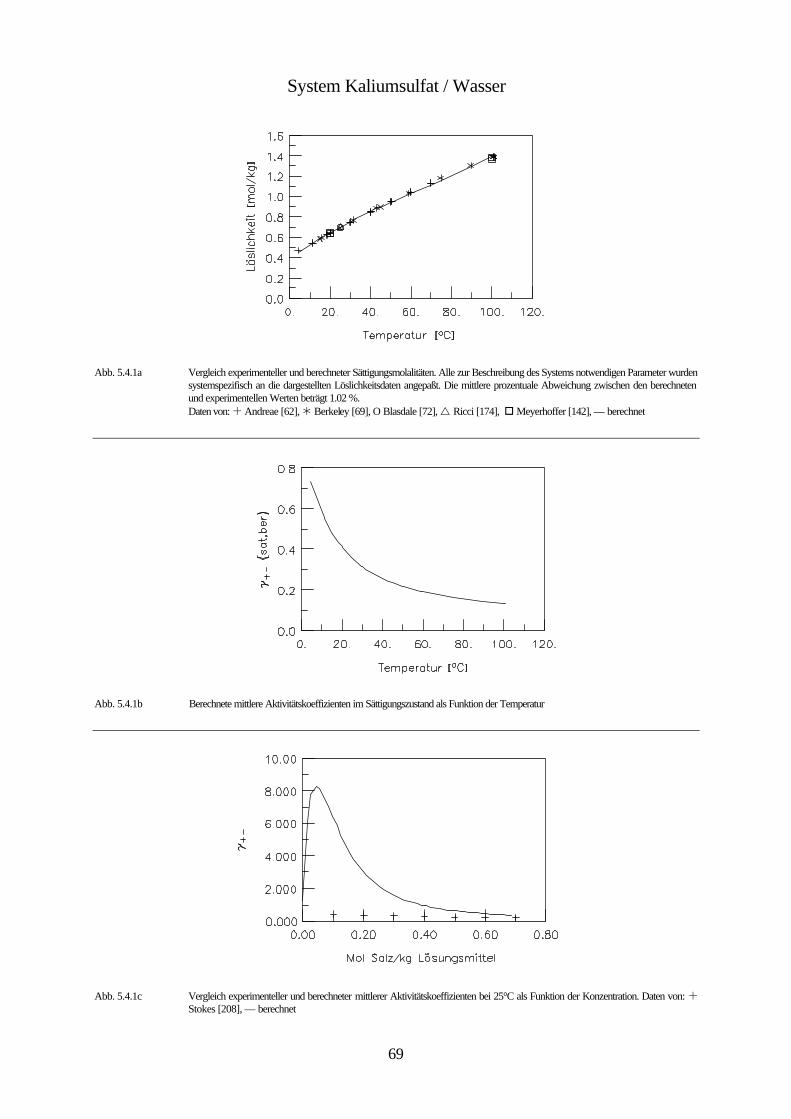
69
System Kaliumsulfat / Wasser
Abb. 5.4.1a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Alle zur Beschreibung des Systems notwendigen Parameter wurden systemspezifisch an die dargestellten Löslichkeitsdaten angepaßt. Die mittlere prozentuale Abweichung zwischen den berechneten und experimentellen Werten beträgt 1.02 %. Daten von: Æ Andreae [62], Ú Berkeley [69], Ο Blasdale [72], r Ricci [174], o Meyerhoffer [142], — berechnet
Abb. 5.4.1b Berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur
Abb. 5.4.1c Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten bei 25°C als Funktion der Konzentration. Daten von: Æ Stokes [208], — berechnet
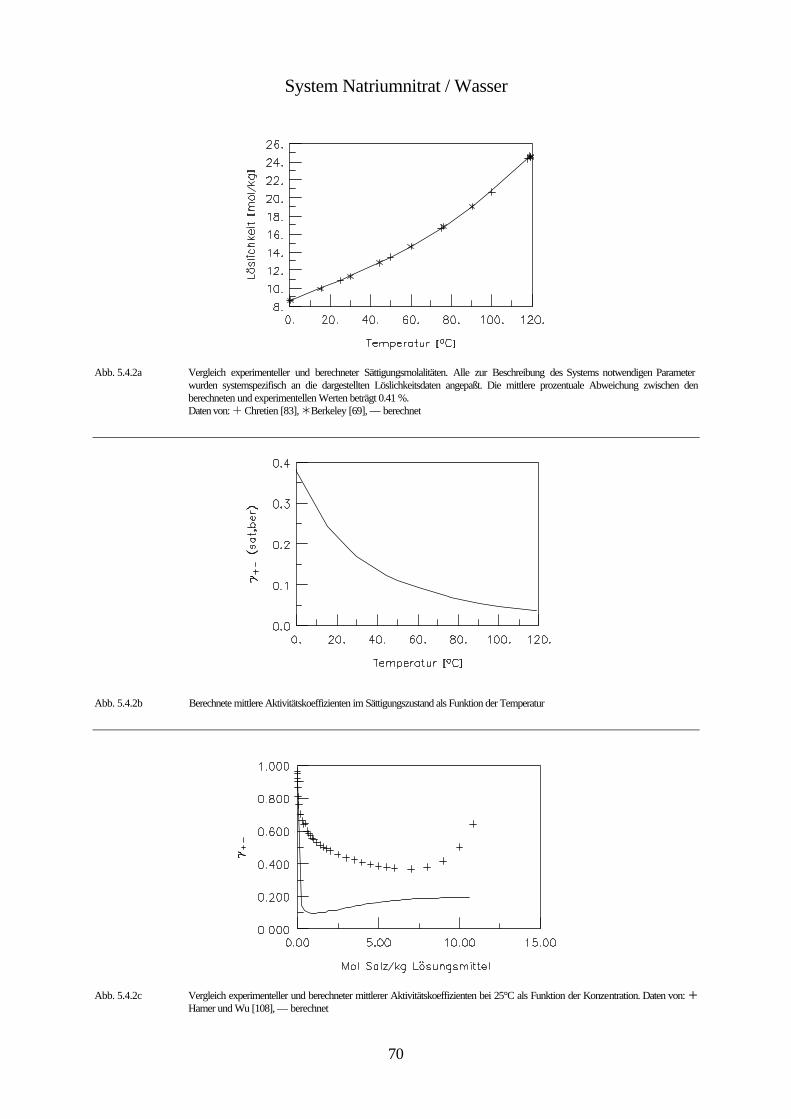
70
System Natriumnitrat / Wasser
Abb. 5.4.2a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Alle zur Beschreibung des Systems notwendigen Parameter wurden systemspezifisch an die dargestellten Löslichkeitsdaten angepaßt. Die mittlere prozentuale Abweichung zwischen den berechneten und experimentellen Werten beträgt 0.41 %. Daten von: Æ Chretien [83], ÚBerkeley [69], — berechnet
Abb. 5.4.2b Berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur
Abb. 5.4.2c Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten bei 25°C als Funktion der Konzentration. Daten von: Ç Hamer und Wu [108], — berechnet
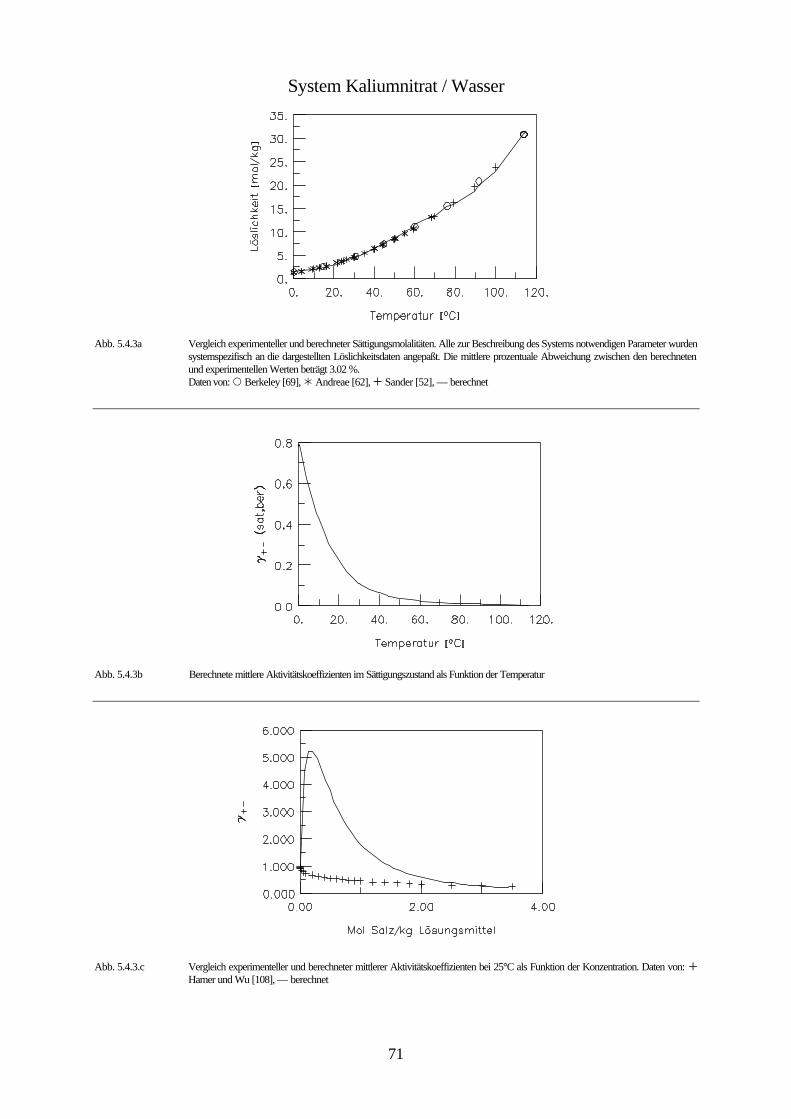
71
System Kaliumnitrat / Wasser
Abb. 5.4.3a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Alle zur Beschreibung des Systems notwendigen Parameter wurden
systemspezifisch an die dargestellten Löslichkeitsdaten angepaßt. Die mittlere prozentuale Abweichung zwischen den berechneten und experimentellen Werten beträgt 3.02 %. Daten von: � Berkeley [69], Ú Andreae [62], Ç Sander [52], — berechnet
Abb. 5.4.3b Berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur
Abb. 5.4.3.c Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten bei 25°C als Funktion der Konzentration. Daten von: Ç Hamer und Wu [108], — berechnet
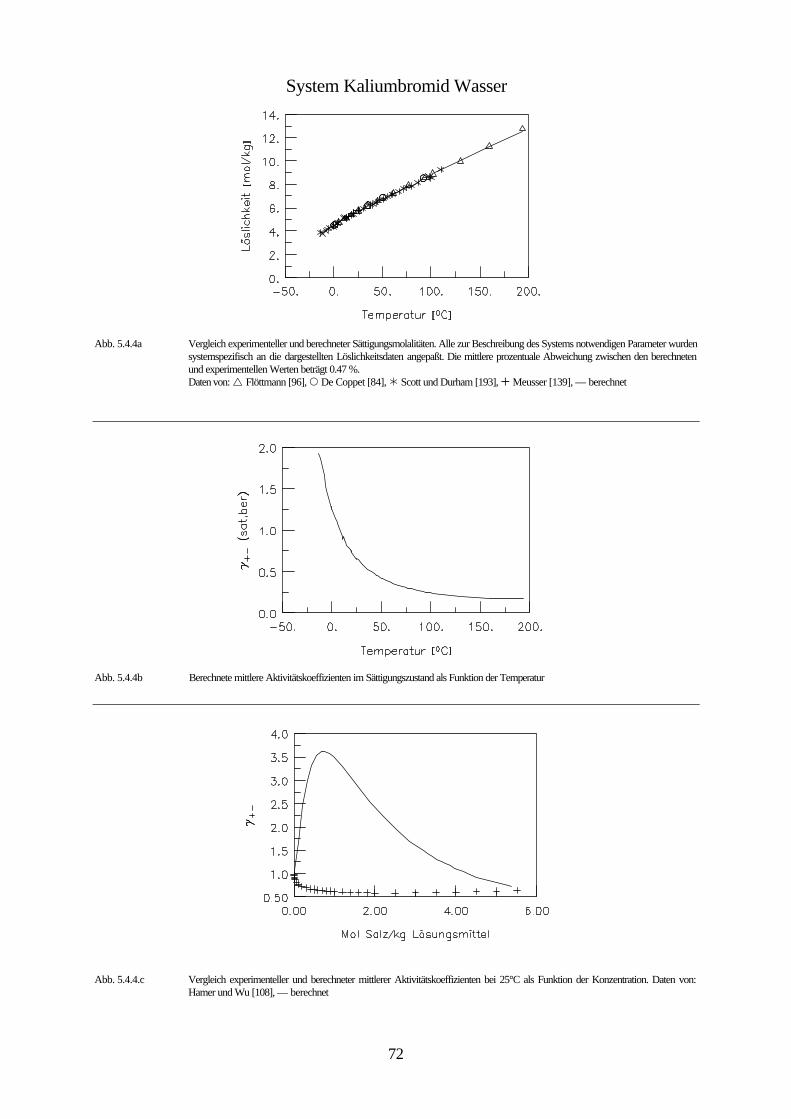
72
System Kaliumbromid Wasser
Abb. 5.4.4a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Alle zur Beschreibung des Systems notwendigen Parameter wurden systemspezifisch an die dargestellten Löslichkeitsdaten angepaßt. Die mittlere prozentuale Abweichung zwischen den berechneten und experimentellen Werten beträgt 0.47 %. Daten von: r Flöttmann [96], � De Coppet [84], Ú Scott und Durham [193], Ç Meusser [139], — berechnet
Abb. 5.4.4b Berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur
Abb. 5.4.4.c Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten bei 25°C als Funktion der Konzentration. Daten von:
Hamer und Wu [108], — berechnet
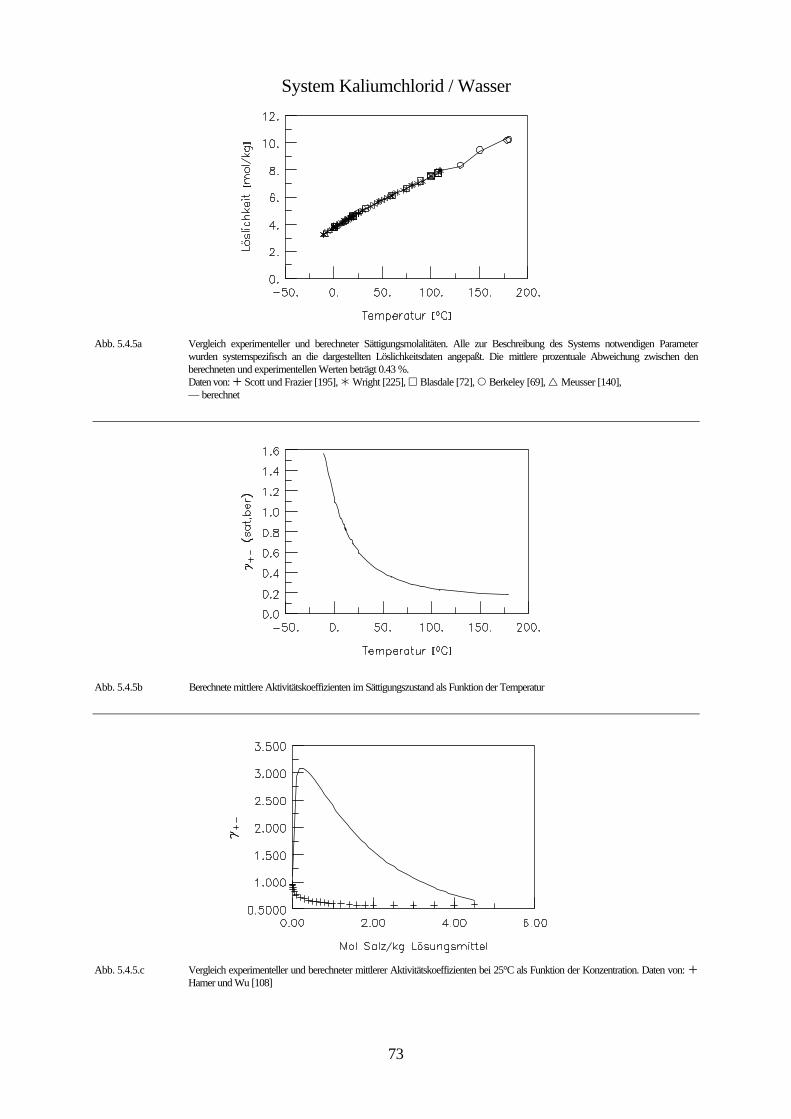
73
System Kaliumchlorid / Wasser
Abb. 5.4.5a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Alle zur Beschreibung des Systems notwendigen Parameter wurden systemspezifisch an die dargestellten Löslichkeitsdaten angepaßt. Die mittlere prozentuale Abweichung zwischen den berechneten und experimentellen Werten beträgt 0.43 %. Daten von: Ç Scott und Frazier [195], Ú Wright [225], £ Blasdale [72], � Berkeley [69], r Meusser [140], — berechnet
Abb. 5.4.5b Berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur
Abb. 5.4.5.c Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten bei 25°C als Funktion der Konzentration. Daten von: Ç
Hamer und Wu [108]
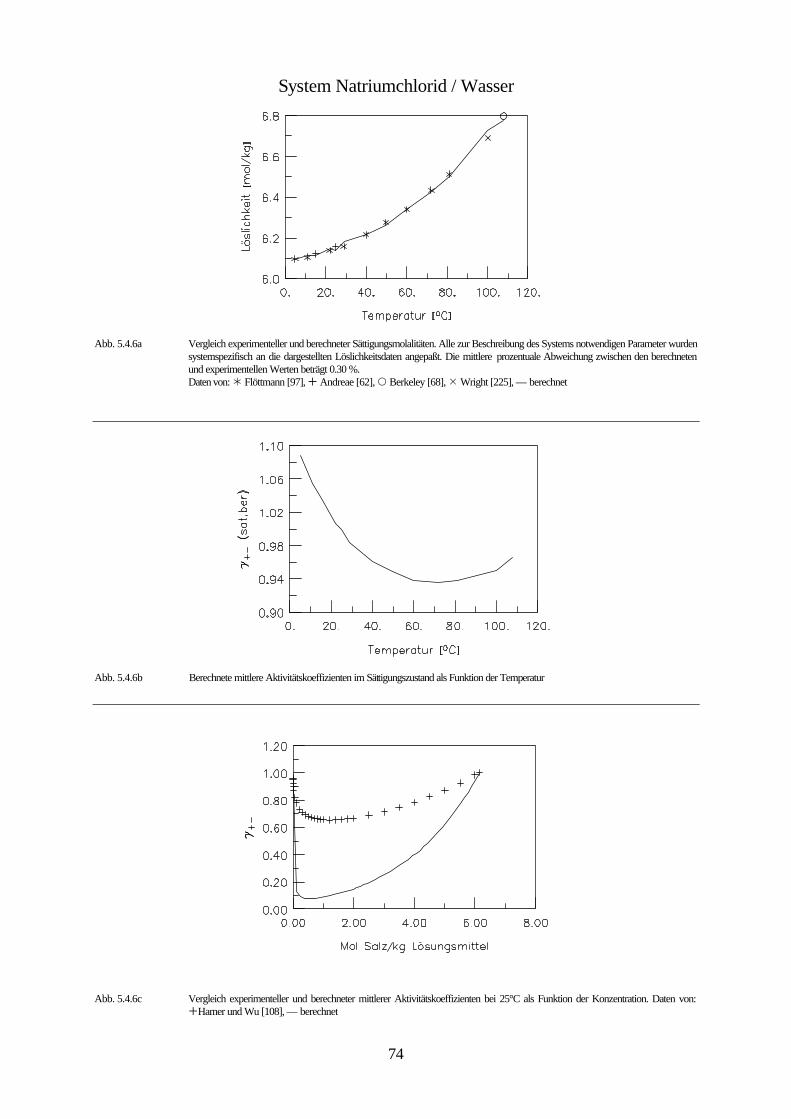
74
System Natriumchlorid / Wasser
Abb. 5.4.6a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Alle zur Beschreibung des Systems notwendigen Parameter wurden systemspezifisch an die dargestellten Löslichkeitsdaten angepaßt. Die mittlere prozentuale Abweichung zwischen den berechneten und experimentellen Werten beträgt 0.30 %. Daten von: Ú Flöttmann [97], Ç Andreae [62], � Berkeley [68], Í Wright [225], — berechnet
Abb. 5.4.6b Berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur
Abb. 5.4.6c Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten bei 25°C als Funktion der Konzentration. Daten von: ÇHamer und Wu [108], — berechnet
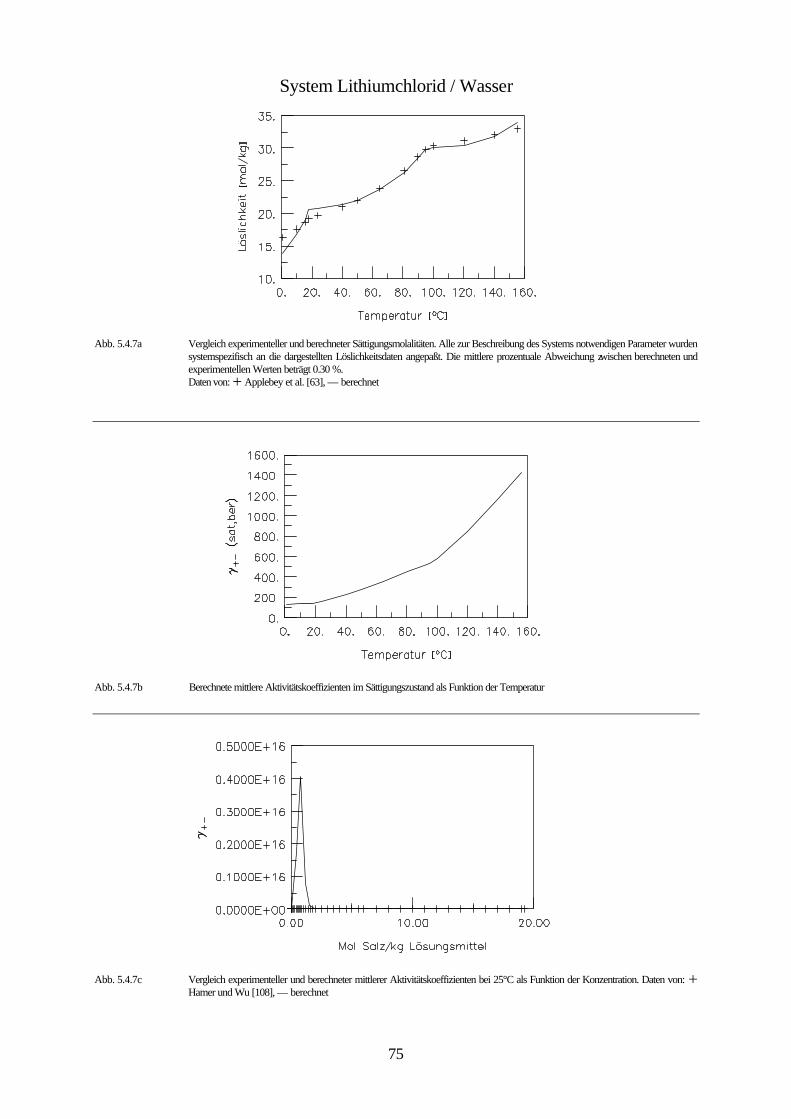
75
System Lithiumchlorid / Wasser
Abb. 5.4.7a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Alle zur Beschreibung des Systems notwendigen Parameter wurden
systemspezifisch an die dargestellten Löslichkeitsdaten angepaßt. Die mittlere prozentuale Abweichung zwischen berechneten und experimentellen Werten beträgt 0.30 %. Daten von: Ç Applebey et al. [63], — berechnet
Abb. 5.4.7b Berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand als Funktion der Temperatur
Abb. 5.4.7c Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten bei 25°C als Funktion der Konzentration. Daten von: Ç Hamer und Wu [108], — berechnet
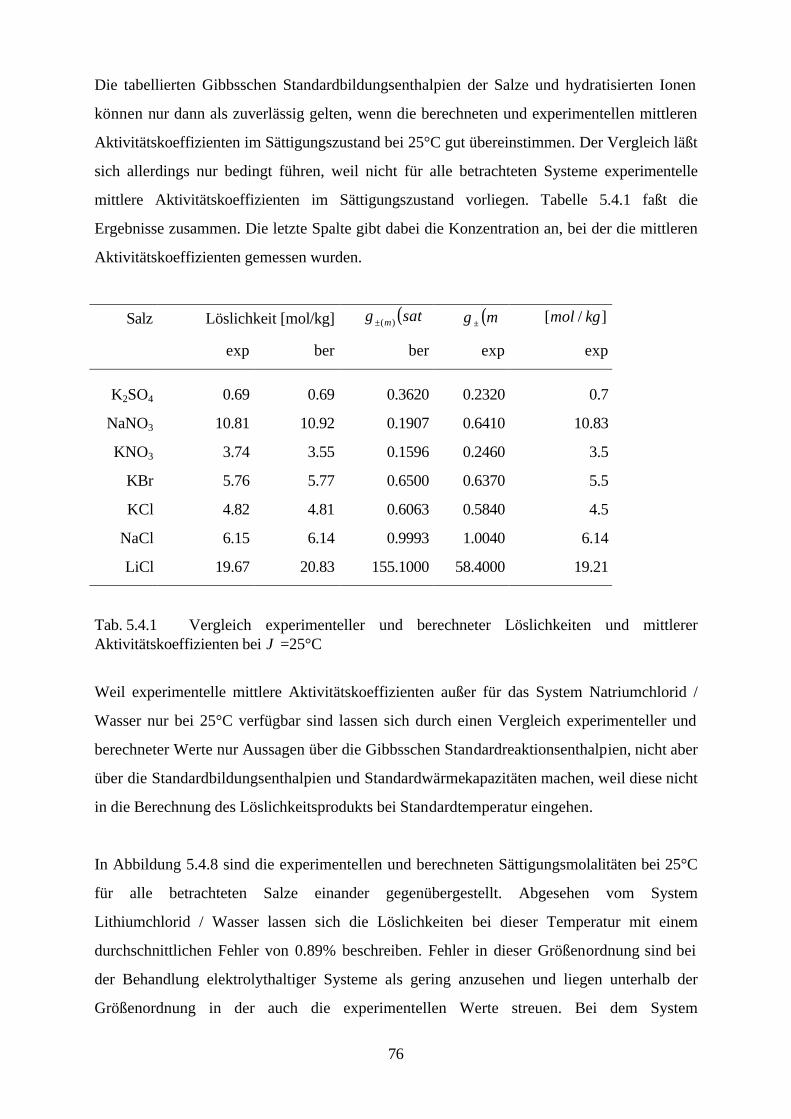
76
Die tabellierten Gibbsschen Standardbildungsenthalpien der Salze und hydratisierten Ionen
können nur dann als zuverlässig gelten, wenn die berechneten und experimentellen mittleren
Aktivitätskoeffizienten im Sättigungszustand bei 25°C gut übereinstimmen. Der Vergleich läßt
sich allerdings nur bedingt führen, weil nicht für alle betrachteten Systeme experimentelle
mittlere Aktivitätskoeffizienten im Sättigungszustand vorliegen. Tabelle 5.4.1 faßt die
Ergebnisse zusammen. Die letzte Spalte gibt dabei die Konzentration an, bei der die mittleren
Aktivitätskoeffizienten gemessen wurden.
Salz Löslichkeit [mol/kg] ( )satm)(±γ ( )m±γ ]/[ kgmol
exp ber ber exp exp
K2SO4 0.69 0.69 0.3620 0.2320 0.7
NaNO3 10.81 10.92 0.1907 0.6410 10.83
KNO3 3.74 3.55 0.1596 0.2460 3.5
KBr 5.76 5.77 0.6500 0.6370 5.5
KCl 4.82 4.81 0.6063 0.5840 4.5
NaCl 6.15 6.14 0.9993 1.0040 6.14
LiCl 19.67 20.83 155.1000 58.4000 19.21
Tab. 5.4.1 Vergleich experimenteller und berechneter Löslichkeiten und mittlerer Aktivitätskoeffizienten bei ϑ =25°C
Weil experimentelle mittlere Aktivitätskoeffizienten außer für das System Natriumchlorid /
Wasser nur bei 25°C verfügbar sind lassen sich durch einen Vergleich experimenteller und
berechneter Werte nur Aussagen über die Gibbsschen Standardreaktionsenthalpien, nicht aber
über die Standardbildungsenthalpien und Standardwärmekapazitäten machen, weil diese nicht
in die Berechnung des Löslichkeitsprodukts bei Standardtemperatur eingehen.
In Abbildung 5.4.8 sind die experimentellen und berechneten Sättigungsmolalitäten bei 25°C
für alle betrachteten Salze einander gegenübergestellt. Abgesehen vom System
Lithiumchlorid / Wasser lassen sich die Löslichkeiten bei dieser Temperatur mit einem
durchschnittlichen Fehler von 0.89% beschreiben. Fehler in dieser Größenordnung sind bei
der Behandlung elektrolythaltiger Systeme als gering anzusehen und liegen unterhalb der
Größenordnung in der auch die experimentellen Werte streuen. Bei dem System
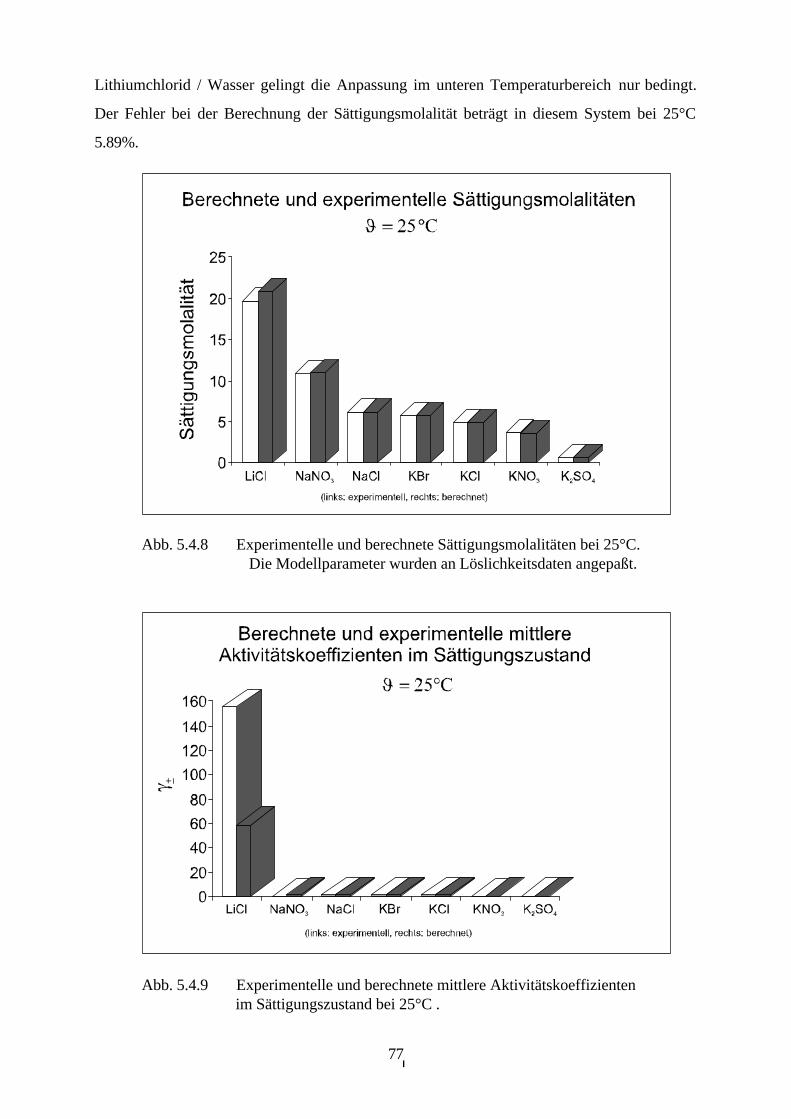
77
Lithiumchlorid / Wasser gelingt die Anpassung im unteren Temperaturbereich nur bedingt.
Der Fehler bei der Berechnung der Sättigungsmolalität beträgt in diesem System bei 25°C
5.89%.
Abb. 5.4.8 Experimentelle und berechnete Sättigungsmolalitäten bei 25°C. Die Modellparameter wurden an Löslichkeitsdaten angepaßt.
Abb. 5.4.9 Experimentelle und berechnete mittlere Aktivitätskoeffizienten im Sättigungszustand bei 25°C .

78
In der Abbildung 5.4.9 sind experimentelle und berechnete mittlere Aktivitätskoeffizienten
einander gegenübergestellt. Die berechneten Werte wurden auf Grundlage der an Löslich-
keitsdaten angepaßten Parameter berechnet. Setzt man die experimentellen Werte als richtig
voraus, dann muß die Abweichung der experimentellen von den berechneten Werten propor-
tional dem Fehler der thermodynamischen Standardgrößen sein. Setzt man dagegen die Stan-
dardgrößen als richtig voraus ist diese Abweichung proportional der Inkongruenz der Gleich-
gewichtsdaten.
Die überdimensionalen Abweichungen im System Lithiumchlorid / Wasser sowohl bei der
Sättigungsmolalität als auch bei den Aktivitätskoeffizienten verzerren allerdings das Bild. Sie
erwecken aufgrund der gewählten Darstellungsweise den Eindruck einer ansonsten guten
Übereinstimmung der Gibbsschen Standardbildungsenthalpien mit den Pha-
sengleichgewichtsdaten bei den anderen Salzen. Diese großen Abweichungen liegen aber
wahrscheinlich nicht in fehlerhaften Standardwerten begründet, sondern darin, daß das ange-
nommene Gleichgewicht zwischen festem Lithiumchlorid und seiner wäßrigen Lösung bei
25°C nicht existiert. Die beiden Knicke in der Löslichkeitskurve des Lithiumchlorids (Abb.
5.4.7a) weisen auf Umwandlungspunkte hin, so daß bei 25°C nicht reines Lithiumchlorid
sondern ein Hydrat als feste Phase mit der Lösung im Gleichgewicht steht. Schließt man
deshalb das Lithiumchlorid aus den Betrachtungen aus ergibt sich folgendes Bild:
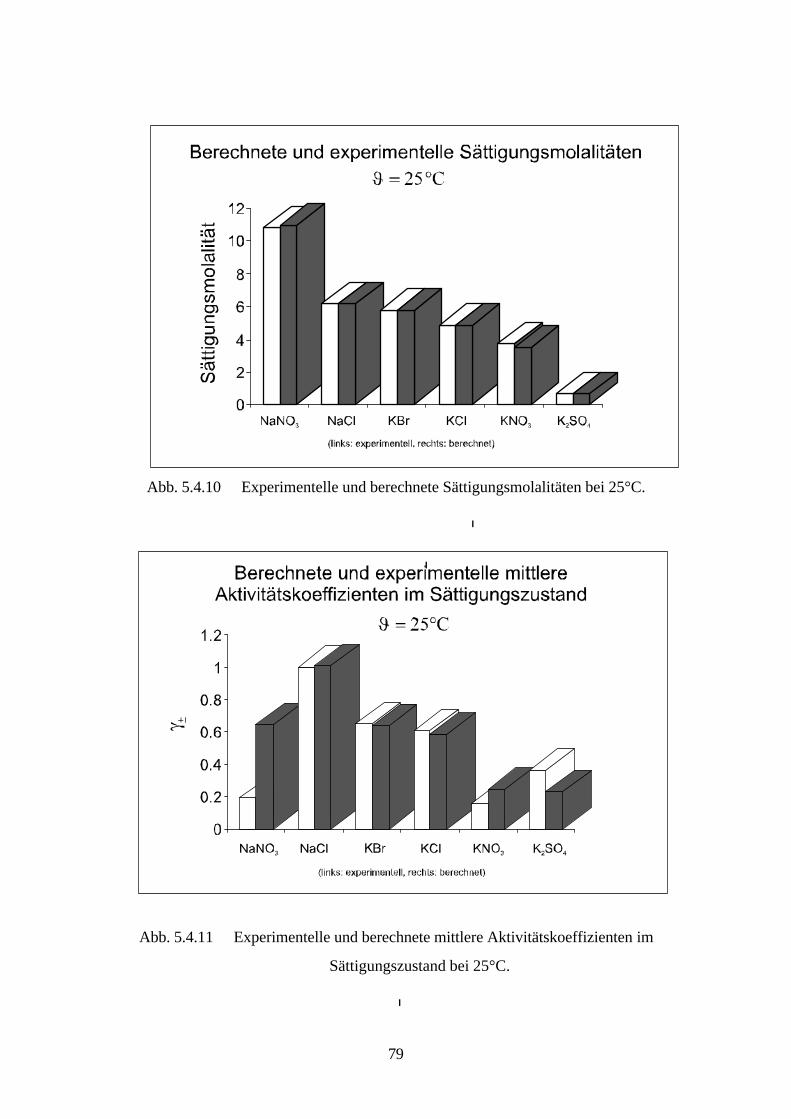
79
Abb. 5.4.10 Experimentelle und berechnete Sättigungsmolalitäten bei 25°C.
Abb. 5.4.11 Experimentelle und berechnete mittlere Aktivitätskoeffizienten im
Sättigungszustand bei 25°C.
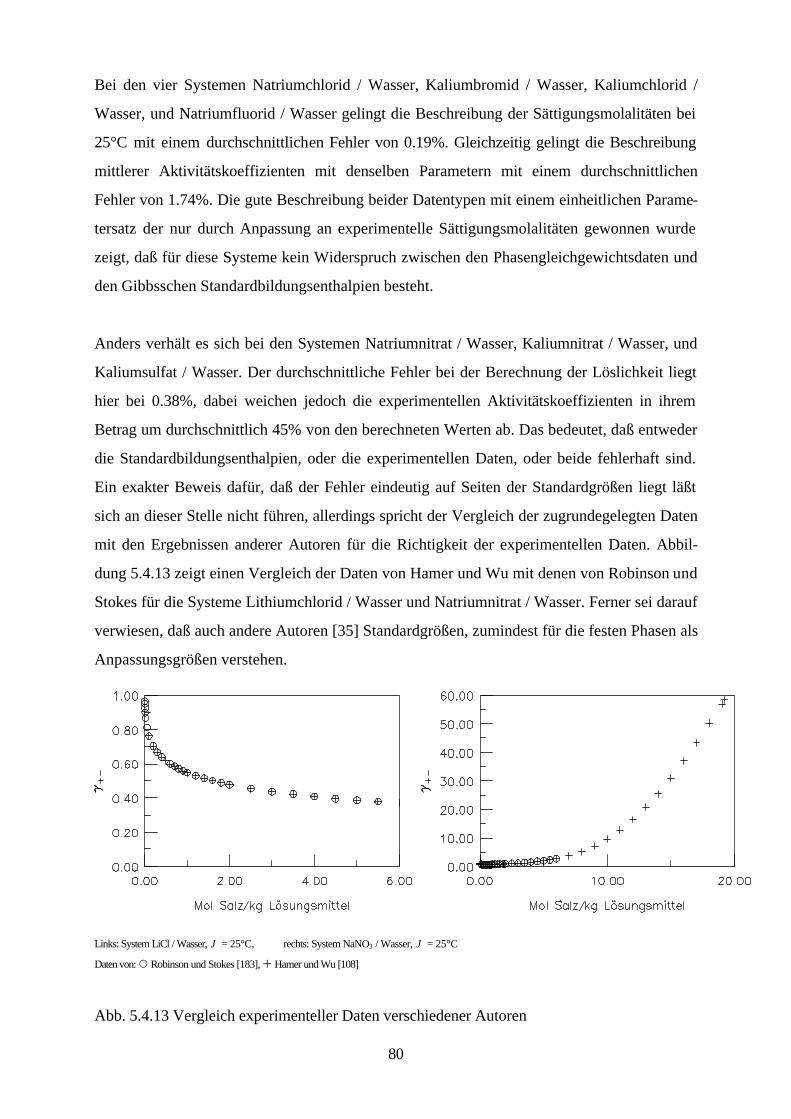
80
Bei den vier Systemen Natriumchlorid / Wasser, Kaliumbromid / Wasser, Kaliumchlorid /
Wasser, und Natriumfluorid / Wasser gelingt die Beschreibung der Sättigungsmolalitäten bei
25°C mit einem durchschnittlichen Fehler von 0.19%. Gleichzeitig gelingt die Beschreibung
mittlerer Aktivitätskoeffizienten mit denselben Parametern mit einem durchschnittlichen
Fehler von 1.74%. Die gute Beschreibung beider Datentypen mit einem einheitlichen Parame-
tersatz der nur durch Anpassung an experimentelle Sättigungsmolalitäten gewonnen wurde
zeigt, daß für diese Systeme kein Widerspruch zwischen den Phasengleichgewichtsdaten und
den Gibbsschen Standardbildungsenthalpien besteht.
Anders verhält es sich bei den Systemen Natriumnitrat / Wasser, Kaliumnitrat / Wasser, und
Kaliumsulfat / Wasser. Der durchschnittliche Fehler bei der Berechnung der Löslichkeit liegt
hier bei 0.38%, dabei weichen jedoch die experimentellen Aktivitätskoeffizienten in ihrem
Betrag um durchschnittlich 45% von den berechneten Werten ab. Das bedeutet, daß entweder
die Standardbildungsenthalpien, oder die experimentellen Daten, oder beide fehlerhaft sind.
Ein exakter Beweis dafür, daß der Fehler eindeutig auf Seiten der Standardgrößen liegt läßt
sich an dieser Stelle nicht führen, allerdings spricht der Vergleich der zugrundegelegten Daten
mit den Ergebnissen anderer Autoren für die Richtigkeit der experimentellen Daten. Abbil-
dung 5.4.13 zeigt einen Vergleich der Daten von Hamer und Wu mit denen von Robinson und
Stokes für die Systeme Lithiumchlorid / Wasser und Natriumnitrat / Wasser. Ferner sei darauf
verwiesen, daß auch andere Autoren [35] Standardgrößen, zumindest für die festen Phasen als
Anpassungsgrößen verstehen.
Links: System LiCl / Wasser, ϑ = 25°C, rechts: System NaNO3 / Wasser, ϑ = 25°C
Daten von: � Robinson und Stokes [183], Ç Hamer und Wu [108]
Abb. 5.4.13 Vergleich experimenteller Daten verschiedener Autoren

81
5.4.2 Simultane Anpassung an mittlere Aktivitätskoeffizienten und Löslichkeitsdaten
Die bisherigen Ergebnisse zeigen, daß die Verwendung der Gibbsschen Standardbildungsent-
halpien zur Berechnung des Löslichkeitsprodukts zumindest bei einigen der untersuchten
Systeme zu Widersprüchen mit den Phasengleichgewichtsdaten führt. Diese Widersprüche
können natürlich auch durch simultane Anpassung nicht aufgehoben werden. Setzt man die
Gibbsschen Standardreaktionsenthalpien als richtig voraus, so ist klar, daß die simultane An-
passung an mitllere Aktivitätskoeffizienten und Sättigungsmolalitäten nur in dem Maße ge-
lingen kann in dem die Daten übereinstimmen. Ist diese Übereinstimmung nur bedingt gege-
ben geht eine gute Beschreibung der Löslichkeitsdaten zu Lasten der Beschreibung der expe-
rimentellen mittleren Aktivitätskoeffizienten und umgekehrt. Da eine höhere oder niedrigere
Gewichtung der verschiedenen Datentypen die Ergebnisse lediglich zur einen oder anderen
Seite verzerrt, aber nichts an der Inkongruenz der Daten ändert, werden auch bei der simulta-
nen Anpassungen beide Datensatztypen gleich stark gewichtet.
Erwartungsgemäß wird bei Anpassung der Modellparameter an Löslichkeitsdaten die Tempe-
raturabhängigkeit der mittleren Aktivitätskoeffizienten entlang der Sättigungsgrenze richtig
beschrieben. Ziel der simultanen Anpassung an mittlere Aktivitätskoeffizienten und Sätti-
gungsmolalitäten ist es gleichzeitig die Konzentrations- und die Temperaturabhängigkeit der
mittleren Aktivitätskoeffizienten richtig vorherzusagen. Das ist notwendig weil die bisherigen
Ergebnisse zeigen, daß trotz guter Beschreibung der Löslichkeitskurve die Konzentrationsab-
hängigkeit der mittleren Aktivitätskoeffizienten bei 25°C für die überwiegende Anzahl der
untersuchten Systeme weder qualitativ noch quantitativ richtig beschrieben werden. Die Ab-
bildungen 5.4.2.1 bis 5.4.2.14 zeigen die Ergebnisse der simultanen Anpassung systemspezifi-
scher Parameter an Löslichkeitsdaten und mittlere Aktivitätskoeffizienten. Die den Darstel-
lungen zugrundeliegenden Parameter sind im Anhang 7.5.2 gelistet
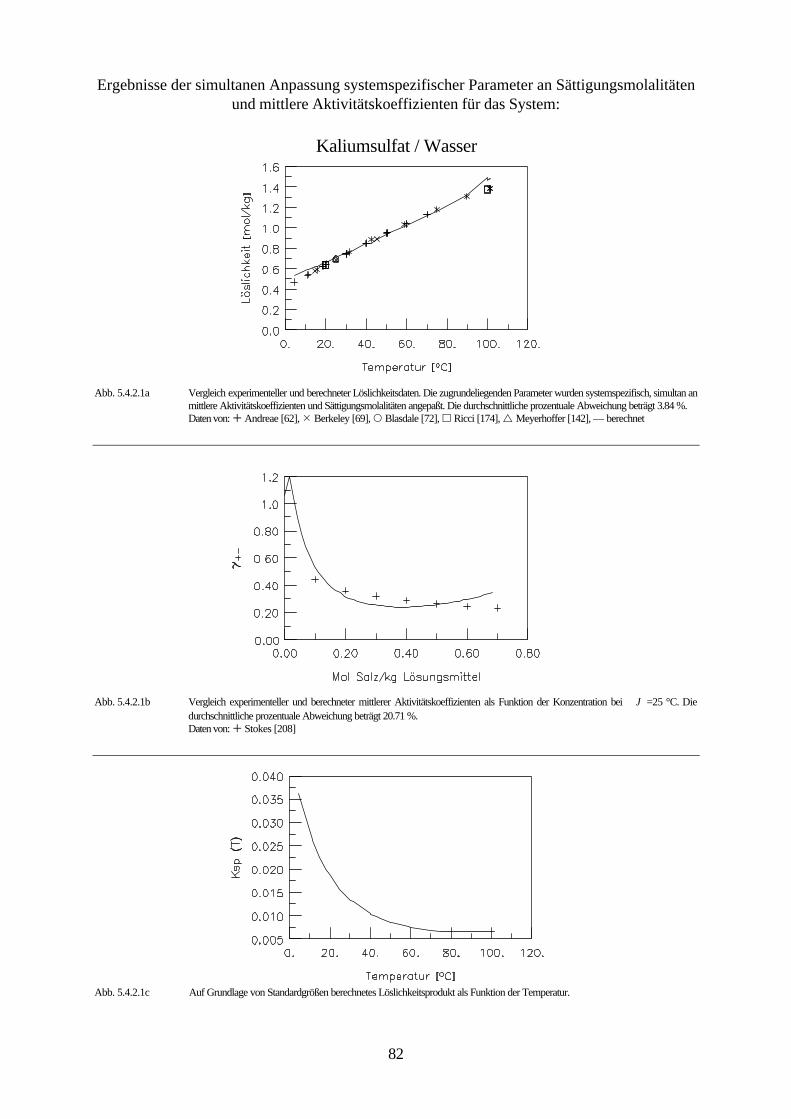
82
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Kaliumsulfat / Wasser
Abb. 5.4.2.1a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 3.84 %. Daten von: Ç Andreae [62], Í Berkeley [69], � Blasdale [72], £ Ricci [174], r Meyerhoffer [142], — berechnet
Abb. 5.4.2.1b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 20.71 %. Daten von: Ç Stokes [208]
Abb. 5.4.2.1c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
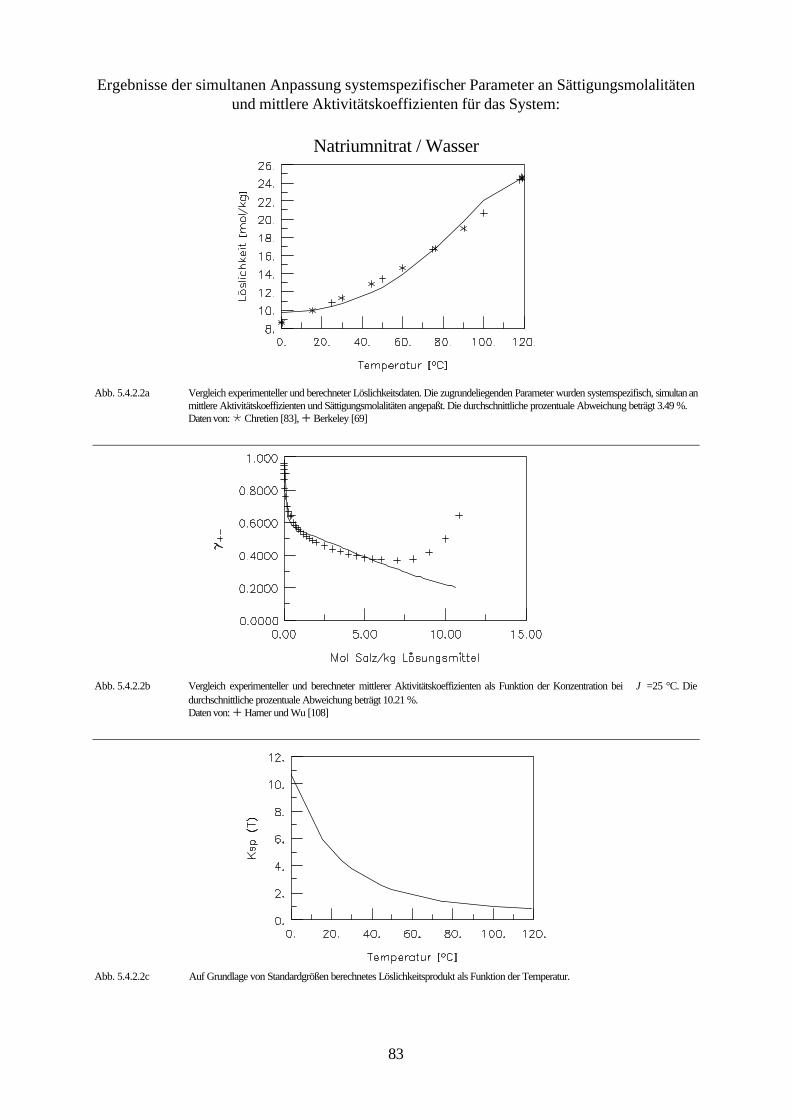
83
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Natriumnitrat / Wasser
Abb. 5.4.2.2a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 3.49 %. Daten von: Ô Chretien [83], Ç Berkeley [69]
Abb. 5.4.2.2b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 10.21 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.2c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
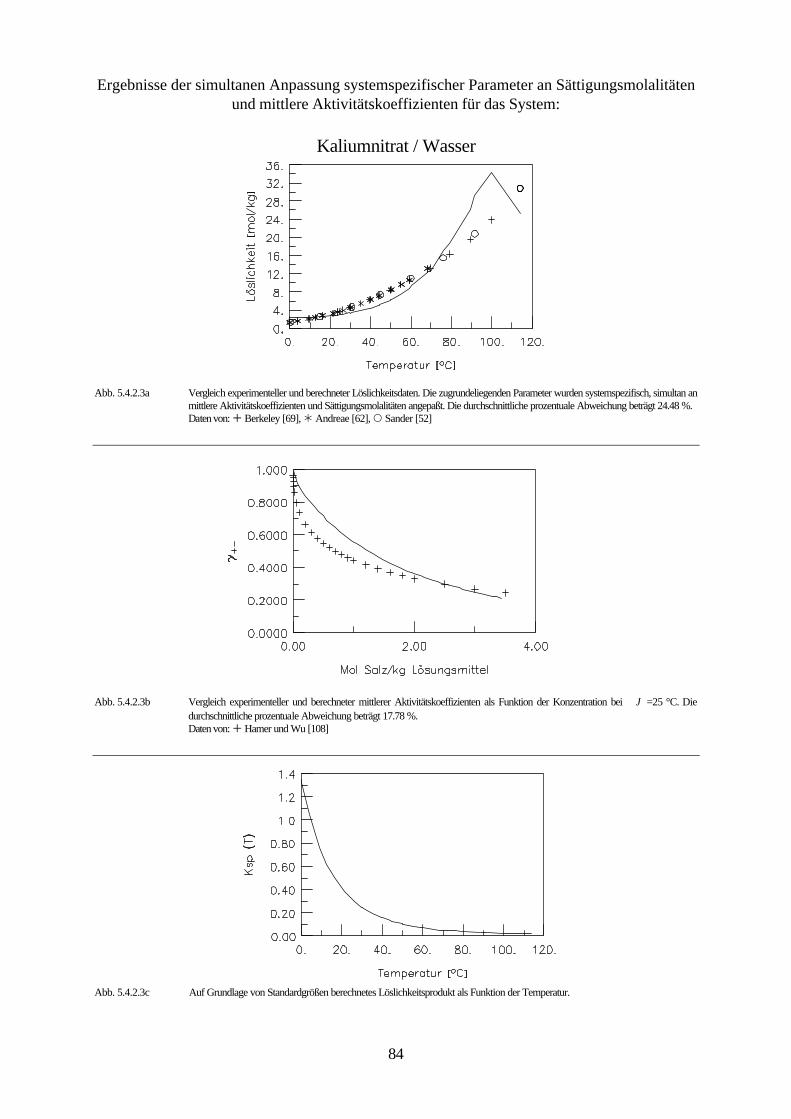
84
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Kaliumnitrat / Wasser
Abb. 5.4.2.3a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 24.48 %. Daten von: Ç Berkeley [69], Ú Andreae [62], � Sander [52]
Abb. 5.4.2.3b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 17.78 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.3c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
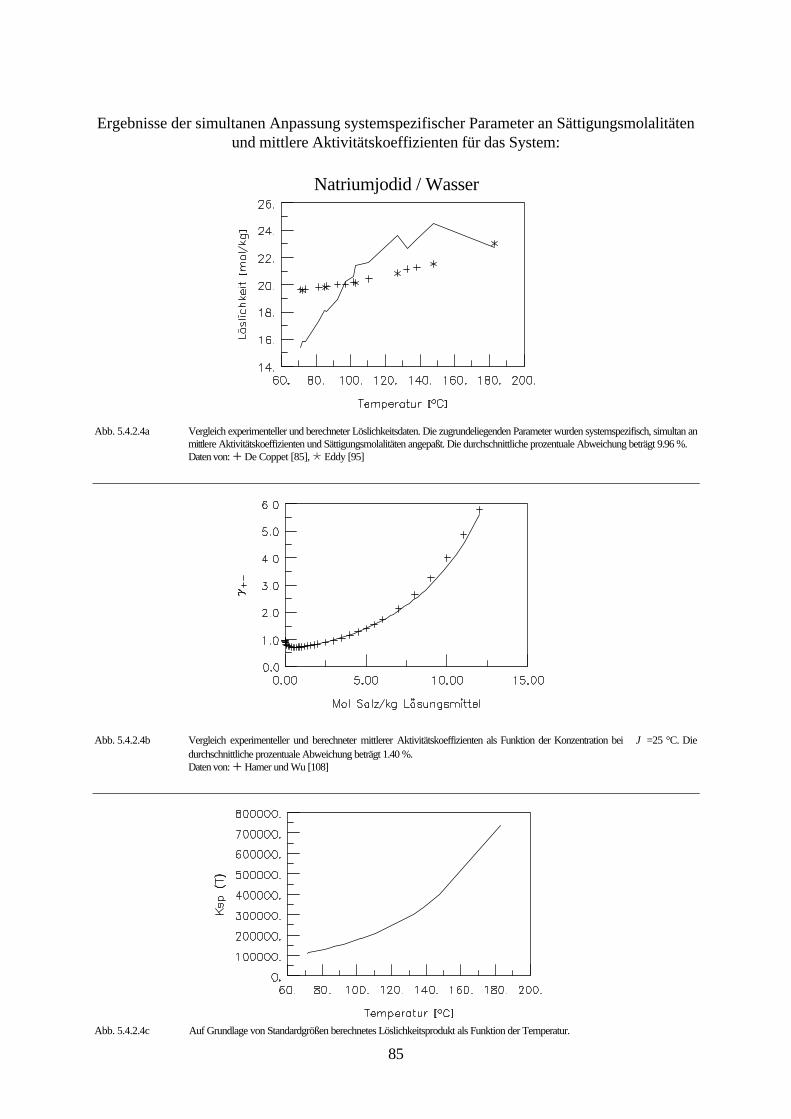
85
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Natriumjodid / Wasser
Abb. 5.4.2.4a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 9.96 %. Daten von: Ç De Coppet [85], Ô Eddy [95]
Abb. 5.4.2.4b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 1.40 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.4c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
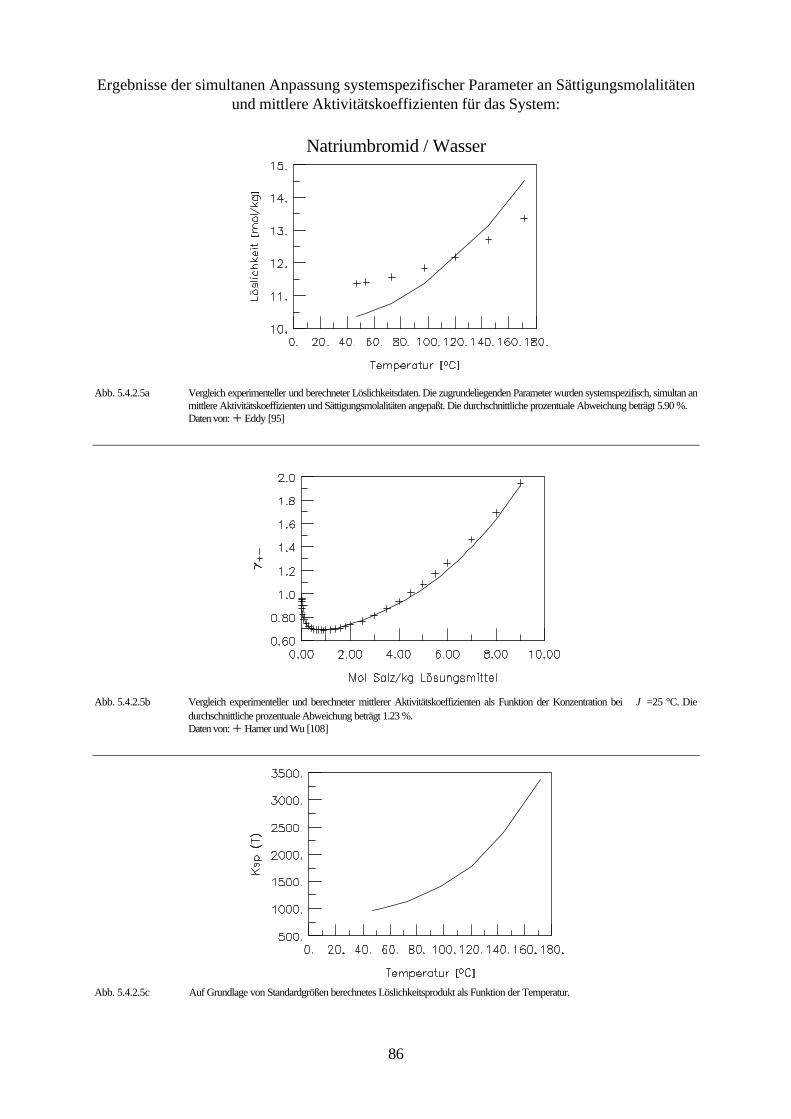
86
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Natriumbromid / Wasser
Abb. 5.4.2.5a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 5.90 %. Daten von: Ç Eddy [95]
Abb. 5.4.2.5b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 1.23 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.5c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
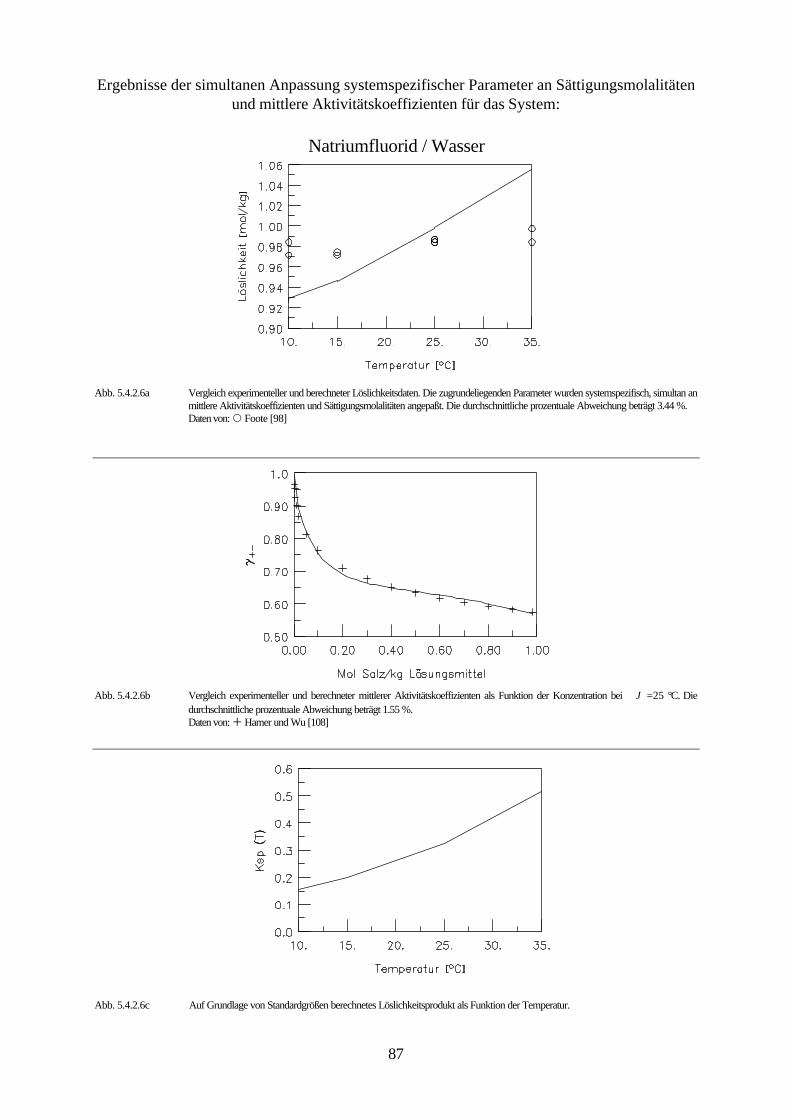
87
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Natriumfluorid / Wasser
Abb. 5.4.2.6a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 3.44 %. Daten von: � Foote [98]
Abb. 5.4.2.6b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die durchschnittliche prozentuale Abweichung beträgt 1.55 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.6c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
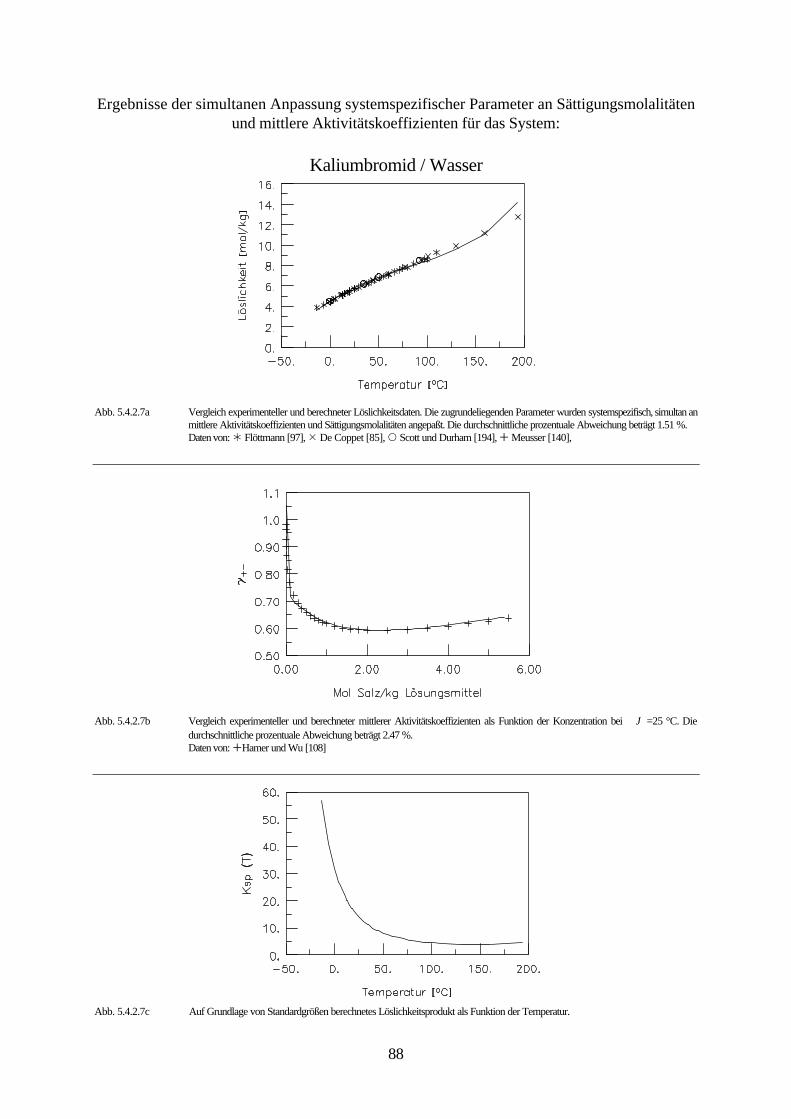
88
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten
und mittlere Aktivitätskoeffizienten für das System:
Kaliumbromid / Wasser
Abb. 5.4.2.7a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 1.51 %. Daten von: Ú Flöttmann [97], Í De Coppet [85], � Scott und Durham [194], Ç Meusser [140],
Abb. 5.4.2.7b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 2.47 %. Daten von: ÇHamer und Wu [108]
Abb. 5.4.2.7c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
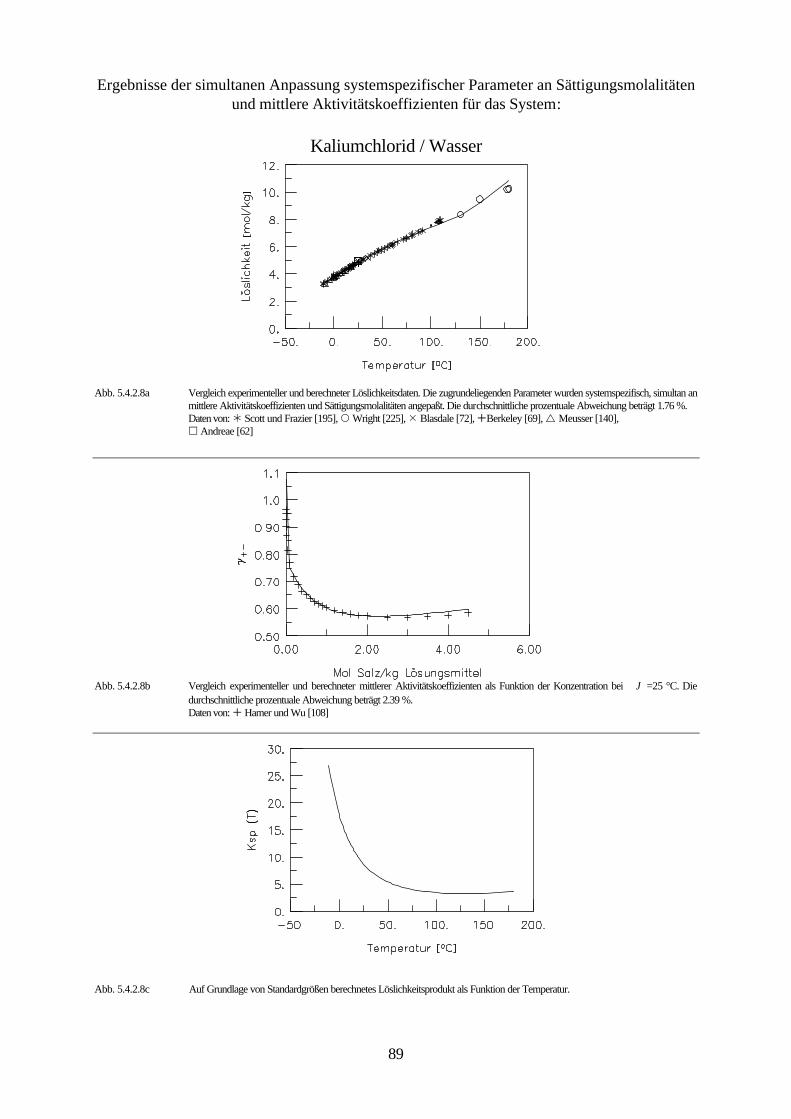
89
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Kaliumchlorid / Wasser
Abb. 5.4.2.8a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 1.76 %. Daten von: Ú Scott und Frazier [195], � Wright [225], Í Blasdale [72], ÇBerkeley [69], r Meusser [140], £ Andreae [62]
Abb. 5.4.2.8b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 2.39 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.8c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
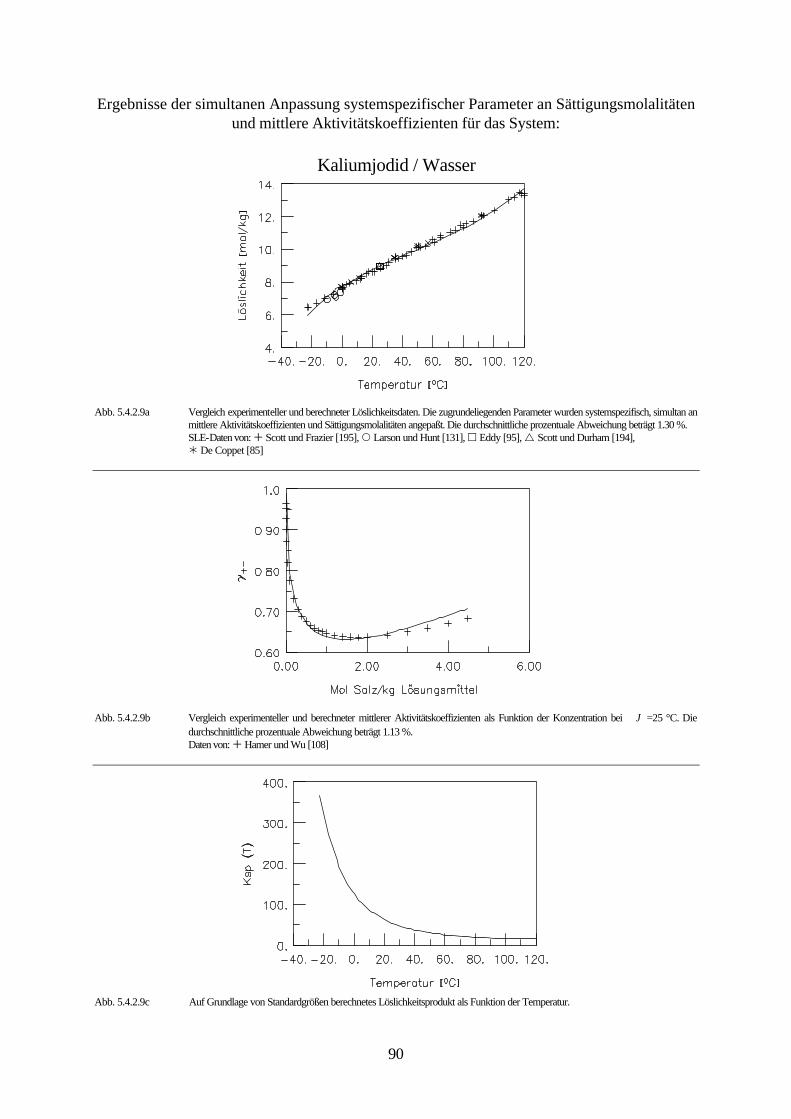
90
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten
und mittlere Aktivitätskoeffizienten für das System:
Kaliumjodid / Wasser
Abb. 5.4.2.9a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 1.30 %. SLE-Daten von: Ç Scott und Frazier [195], � Larson und Hunt [131], £ Eddy [95], r Scott und Durham [194], Ú De Coppet [85]
Abb. 5.4.2.9b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 1.13 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.9c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
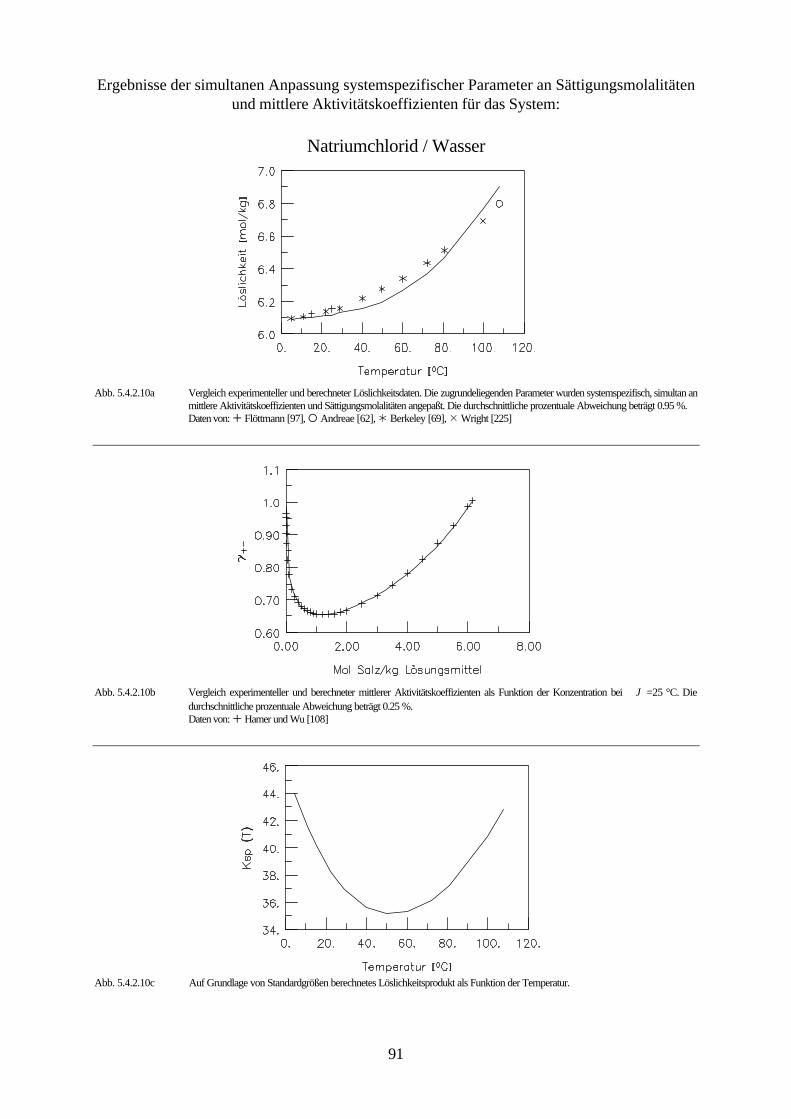
91
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Natriumchlorid / Wasser
Abb. 5.4.2.10a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 0.95 %. Daten von: Ç Flöttmann [97], � Andreae [62], Ú Berkeley [69], Í Wright [225]
Abb. 5.4.2.10b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 0.25 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.10c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
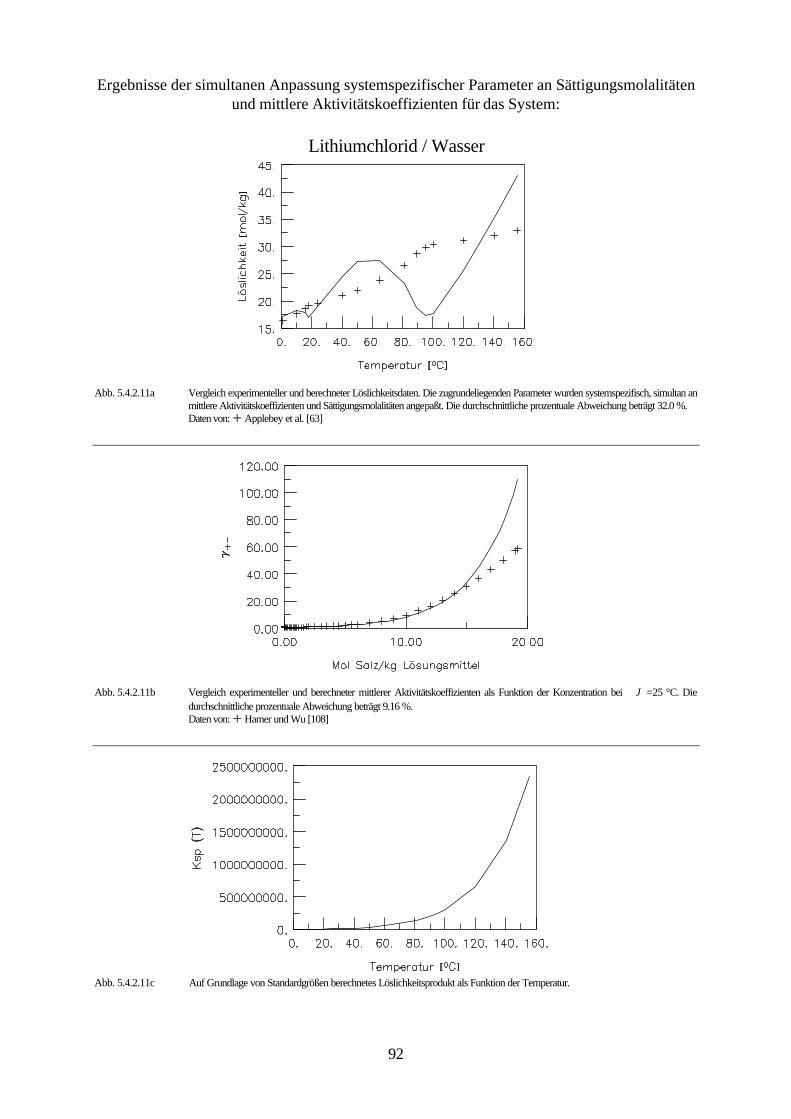
92
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Lithiumchlorid / Wasser
Abb. 5.4.2.11a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 32.0 %. Daten von: Ç Applebey et al. [63]
Abb. 5.4.2.11b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 9.16 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.11c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
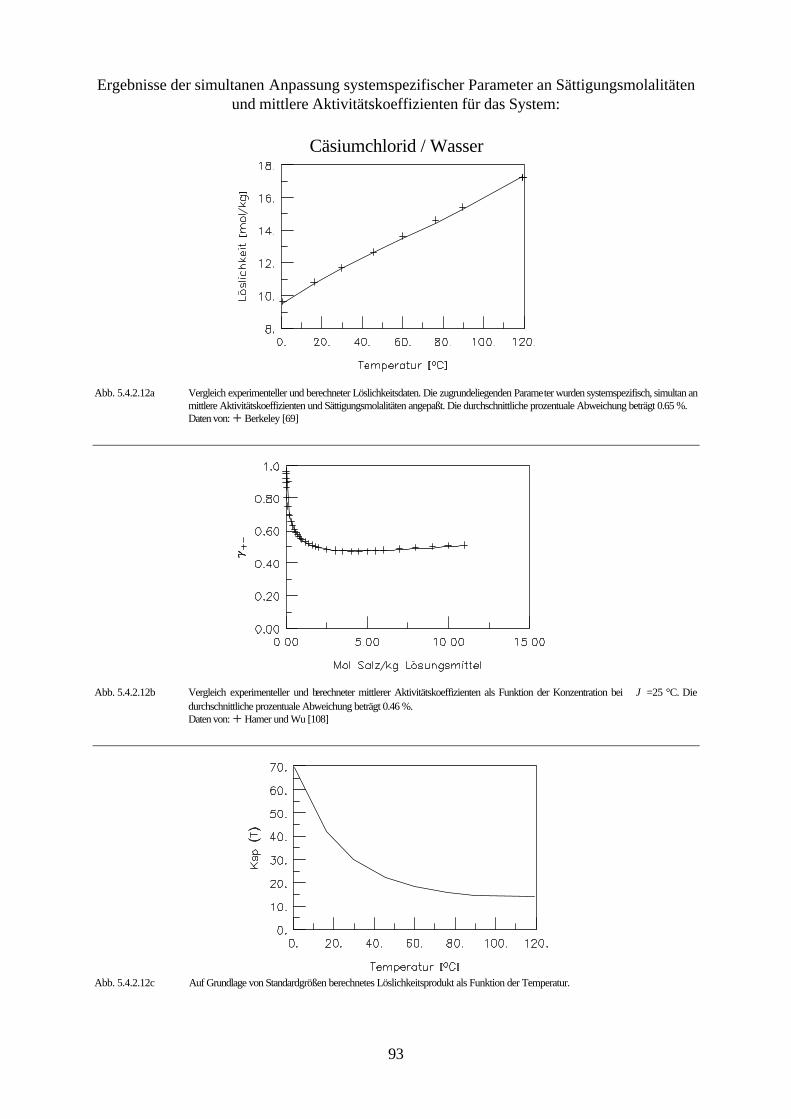
93
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Cäsiumchlorid / Wasser
Abb. 5.4.2.12a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 0.65 %. Daten von: Ç Berkeley [69]
Abb. 5.4.2.12b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 0.46 %. Daten von: Ç Hamer und Wu [108]
Abb. 5.4.2.12c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
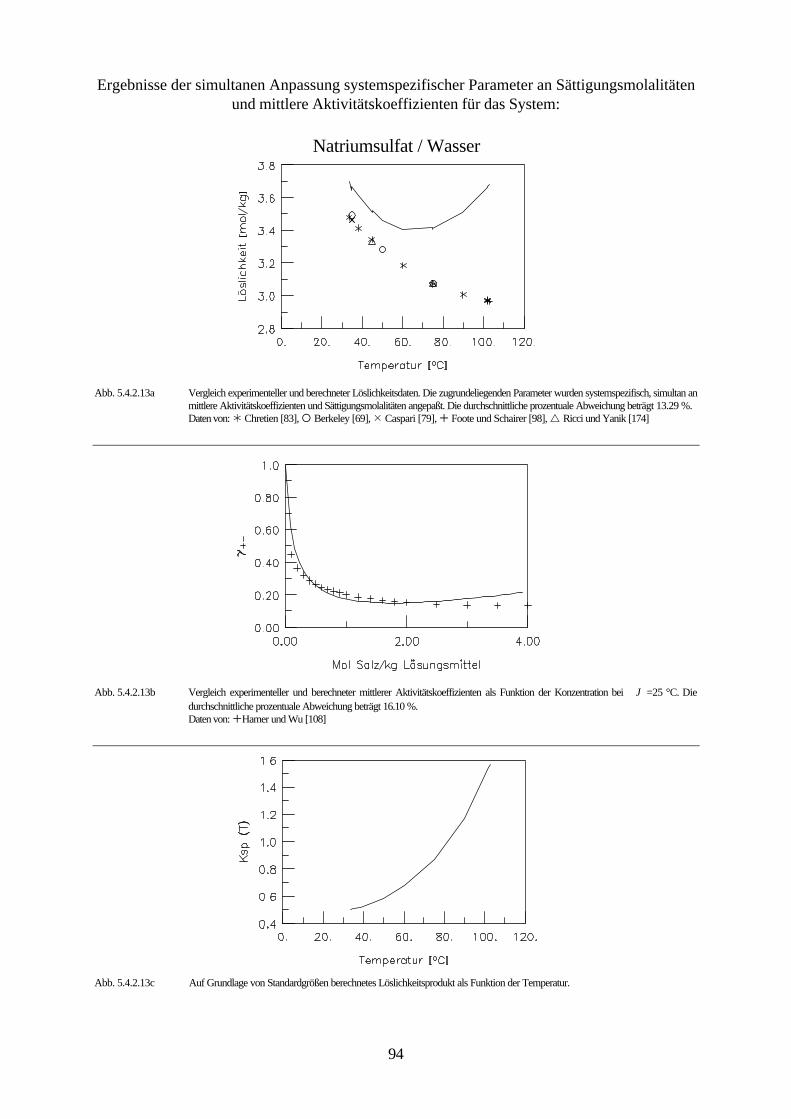
94
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Natriumsulfat / Wasser
Abb. 5.4.2.13a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 13.29 %. Daten von: Ú Chretien [83], � Berkeley [69], Í Caspari [79], Ç Foote und Schairer [98], r Ricci und Yanik [174]
Abb. 5.4.2.13b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 16.10 %. Daten von: ÇHamer und Wu [108]
Abb. 5.4.2.13c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
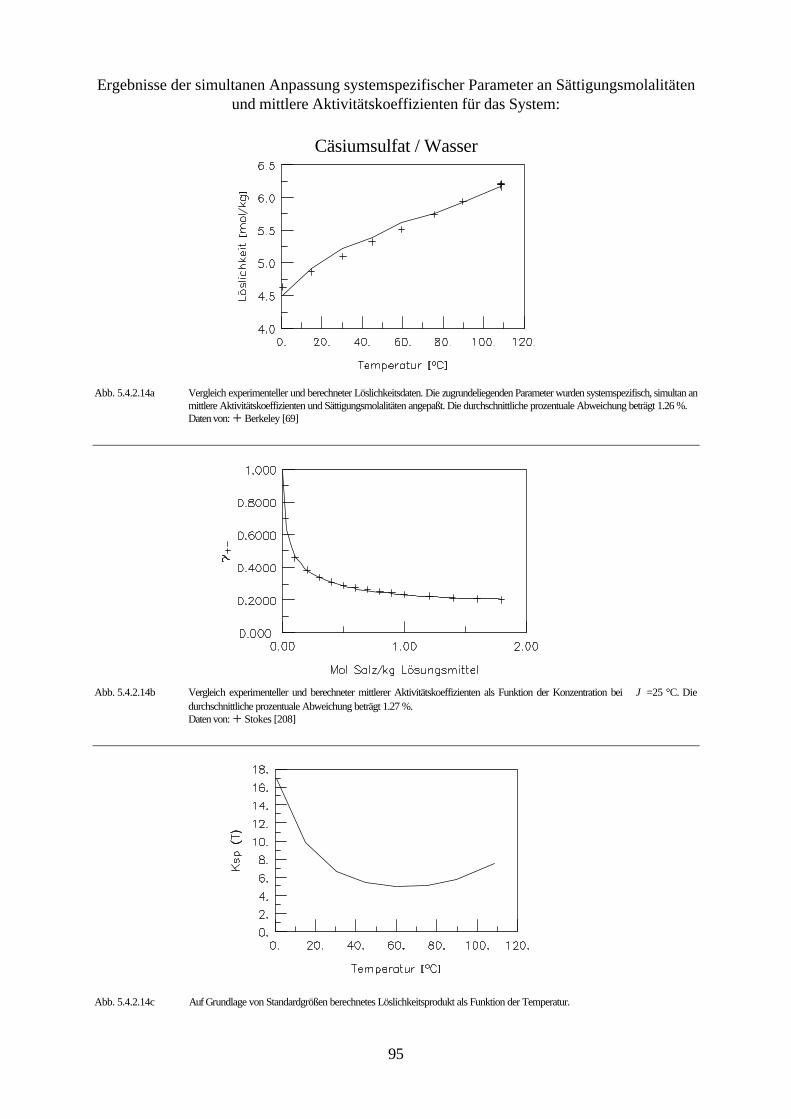
95
Ergebnisse der simultanen Anpassung systemspezifischer Parameter an Sättigungsmolalitäten und mittlere Aktivitätskoeffizienten für das System:
Cäsiumsulfat / Wasser
Abb. 5.4.2.14a Vergleich experimenteller und berechneter Löslichkeitsdaten. Die zugrundeliegenden Parameter wurden systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepaßt. Die durchschnittliche prozentuale Abweichung beträgt 1.26 %. Daten von: Ç Berkeley [69]
Abb. 5.4.2.14b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten als Funktion der Konzentration bei ϑ =25 °C. Die
durchschnittliche prozentuale Abweichung beträgt 1.27 %. Daten von: Ç Stokes [208]
Abb. 5.4.2.14c Auf Grundlage von Standardgrößen berechnetes Löslichkeitsprodukt als Funktion der Temperatur.
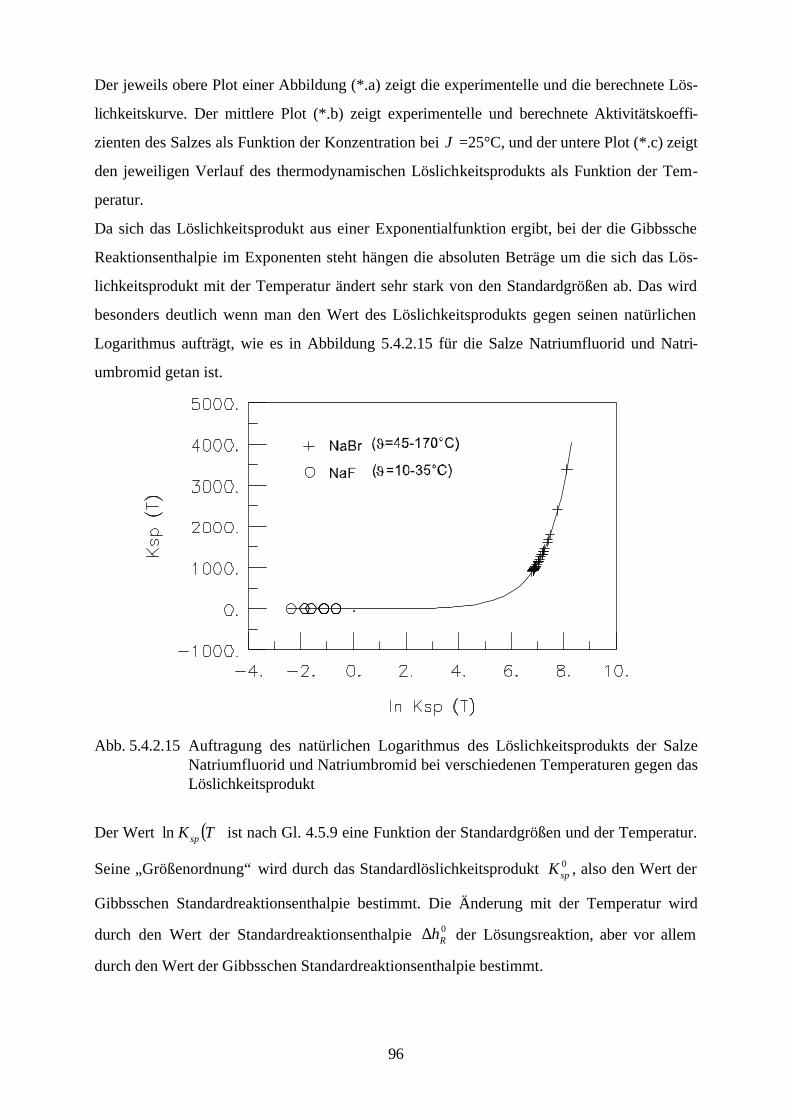
96
Der jeweils obere Plot einer Abbildung (*.a) zeigt die experimentelle und die berechnete Lös-
lichkeitskurve. Der mittlere Plot (*.b) zeigt experimentelle und berechnete Aktivitätskoeffi-
zienten des Salzes als Funktion der Konzentration bei ϑ =25°C, und der untere Plot (*.c) zeigt
den jeweiligen Verlauf des thermodynamischen Löslichkeitsprodukts als Funktion der Tem-
peratur.
Da sich das Löslichkeitsprodukt aus einer Exponentialfunktion ergibt, bei der die Gibbssche
Reaktionsenthalpie im Exponenten steht hängen die absoluten Beträge um die sich das Lös-
lichkeitsprodukt mit der Temperatur ändert sehr stark von den Standardgrößen ab. Das wird
besonders deutlich wenn man den Wert des Löslichkeitsprodukts gegen seinen natürlichen
Logarithmus aufträgt, wie es in Abbildung 5.4.2.15 für die Salze Natriumfluorid und Natri-
umbromid getan ist.
Abb. 5.4.2.15 Auftragung des natürlichen Logarithmus des Löslichkeitsprodukts der Salze Natriumfluorid und Natriumbromid bei verschiedenen Temperaturen gegen das Löslichkeitsprodukt
Der Wert ( )TK spln ist nach Gl. 4.5.9 eine Funktion der Standardgrößen und der Temperatur.
Seine „Größenordnung“ wird durch das Standardlöslichkeitsprodukt 0spK , also den Wert der
Gibbsschen Standardreaktionsenthalpie bestimmt. Die Änderung mit der Temperatur wird
durch den Wert der Standardreaktionsenthalpie 0Rh∆ der Lösungsreaktion, aber vor allem
durch den Wert der Gibbsschen Standardreaktionsenthalpie bestimmt.

97
Ist die Gibbssche Standardreaktionsenthalpie stark negativ wie z.B. beim Natriumbromid (-
16.88 kJ/mol) resultiert daraus ein großer Wert des Löslichkeitsprodukts. Die Temperatur-
abhängigkeit des Löslichkeitsprodukts ist aber trotz des geringen Absolutwertes der
Standardreaktionsenthalpie ( kJ/mol 608.00 −=∆ Rh ) sehr groß. Der Grund dafür liegt in dem
großen Wert des Standardlöslichkeitsprodukts, bzw. der stark negativen Gibbsschen
Standardreaktionsenthalpie. Allgemein lässt sich feststellen, dass die Temperaturabhängigkeit
des Löslichkeitsprodukts umso größer ist, je größer der Betrag der Gibbsschen
Standardreaktionsenthalpie der Lösungsreaktion ist. Der Betrag der
Standardreaktionsenthalpie spielt nur eine untergeordnete Rolle. So beträgt z.B. die Gibbssche
Standardreaktionsenthalpie für die Lösungsreaktion von Natriumfluorid 2.79 kJ/mol. Die
Temperaturabhängigkeit des Löslichkeitsprodukts ist dabei sehr gering, obwohl die
Standardreaktionsenthalpie –35.1 kJ/mol beträgt, und damit 58 mal größer als beim
Natriumbromid. Noch deutlicher wird der große Einfluß der Gibbsschen
Standardreaktionsenthalpie auf die Temperaturabhängigkeit des Löslichkeitsprodukts bei
Lithiumchlorid. Sie beträgt –40.17 kJ/mol, und obwohl die Standardreaktionsenthalpie mit
–37.04 kJ/mol nur geringfügig größer ist als die des Natriumbromids bewirkt ein geringe
Temperaturänderung einen Anstieg des Löslichkeitsprodukts um mehrere Zehnerpotenzen.
Bei einer flach verlaufenden Löslichkeitskurve muß das Modell entsprechend hohe Werte für
den Aktivitätskoeffizienten liefern.
Neben diesen Betrachtungen fällt als weiteres Ergebnis der simultanen Anpassung an Sätti-
gungsmolalitäten und mittlere Aktivitätskoeffizienten auf, daß die Abweichungen im Fall von
2:1 Elektrolyten deutlich größer sind als bei 1:1 Elektrolyten. Vergleicht man die Abweichun-
gen des Systems Natriumchlorid / Wasser mit denen des Systems Natriumsulfat / Wasser so
sind letztere um mehr als den Faktor 10 größer. Der Grund dafür liegt eindeutig darin, daß
sowohl für 2:1, als auch für 1:2 Elektrolyte die Annahme vollständiger Dissoziation nicht zu-
trifft.
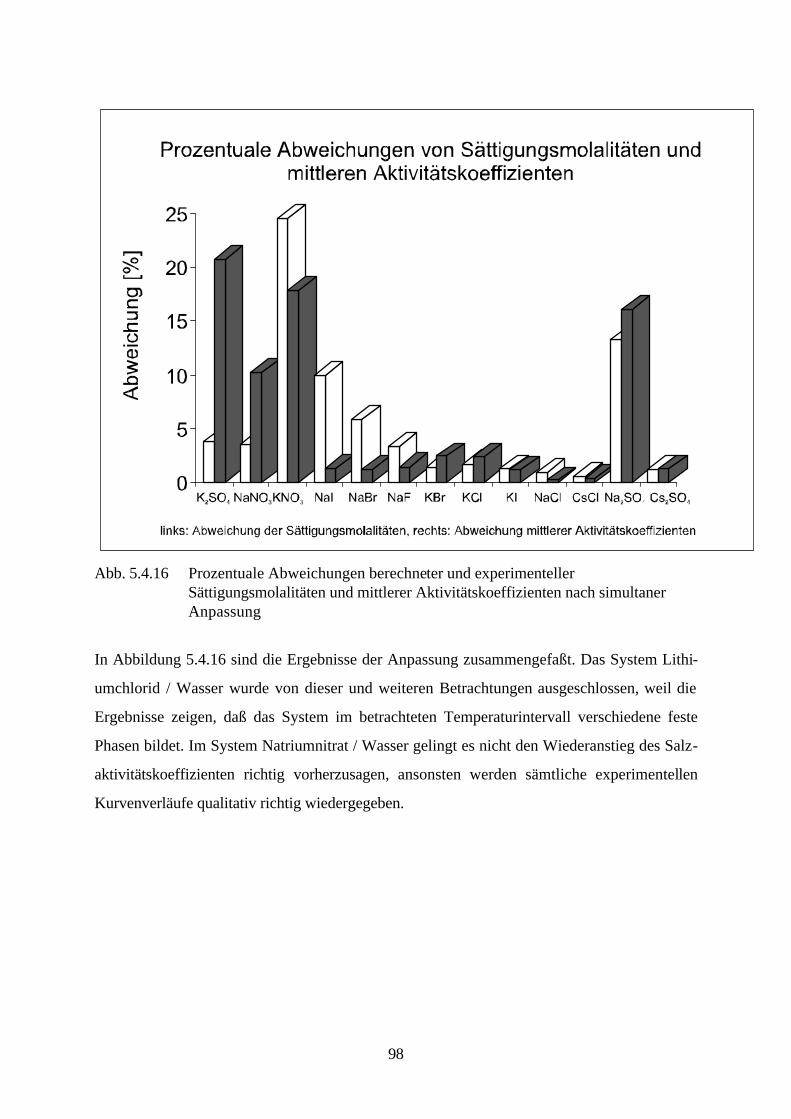
98
Abb. 5.4.16 Prozentuale Abweichungen berechneter und experimenteller Sättigungsmolalitäten und mittlerer Aktivitätskoeffizienten nach simultaner Anpassung
In Abbildung 5.4.16 sind die Ergebnisse der Anpassung zusammengefaßt. Das System Lithi-
umchlorid / Wasser wurde von dieser und weiteren Betrachtungen ausgeschlossen, weil die
Ergebnisse zeigen, daß das System im betrachteten Temperaturintervall verschiedene feste
Phasen bildet. Im System Natriumnitrat / Wasser gelingt es nicht den Wiederanstieg des Salz-
aktivitätskoeffizienten richtig vorherzusagen, ansonsten werden sämtliche experimentellen
Kurvenverläufe qualitativ richtig wiedergegeben.

99
5.4.3 Simultane Anpassung an Löslichkeitsdaten, mittlere Aktivitätskoeffizienten, molale
osmotische Koeffizienten und Dampfdruckdaten
Die bisherigen Anpassungen zeigen in wie weit es möglich ist Löslichkeitsdaten und experi-
mentelle mittlere Aktivitätskoeffizienten simultan zu beschreiben. Die Schwierigkeiten liegen
dabei insbesondere in der Kopplung der in Löslichkeitsdaten enthaltenen experimentellen
Information an Standardgrößen. Die Standardgrößen legen den Verlauf der Salzaktivitäts-
koeffizienten an der Sättigungsgrenze fest. Dabei liegt es in ihrer Natur daß sie mit Fehlern
behaftet sind, genau wie sich jeder experimentelle Wert der Wirklichkeit nur der Meßmethode
entsprechend annähern kann. Aufgrund der vielen experimentellen Daten aus denen die Stan-
dardgrößen gewonnen wurden und der notwendigen internen Konsistenz aller
Standardgrößen stellen sie gewissermaßen den kleinsten gemeinsamen Nenner zur
Beschreibung jeder denkbaren Reaktion dar. Das ist besonders kritisch wenn man bedenkt wie
sensibel sich Fehler in den Standardgrößen auf die Qualität der Berechnung von
Phasengleichgewichten auswirken [19].
Die bisherigen Anpassungen hatten das Ziel den mittleren Aktivitätskoeffizienten als Funktion
von Temperatur und Konzentration richtig zu beschreiben. Dabei wurde nicht berücksichtigt
ob die Lösungsmittelaktivität richtig beschrieben wird. Nach der Gibbs-Duhem-Beziehung
sind die chemischen Potentiale der Komponenten eines Systems aber nicht unabhängig
voneinander. Deshalb soll auf Grundlage der bisherigen Ergebnisse geprüft werden, inwieweit
es möglich ist gleichzeitig die Lösungsmittelaktivität und mittlere Aktivitätskoeffizienten von
Salzen zu beschreiben. Zu diesem Zweck wurde der vorherige Anpassungsschritt auf
Grundlage einer erweiterten Datenbasis wiederholt. Die Modellparameter werden dabei
simultan an Löslichkeitsdaten, mittlere Aktivitätskoeffizienten von Salzen, Dampfdruckdaten
und molale osmotische Koeffizienten angepaßt. Von der Anpassung ausgeschlossen wurden
die Systeme Na2SO4 / Wasser und Cs2SO4 / Wasser weil sie in hohem Maße unvollständig
dissoziieren. Aufgrund der Datenfülle ist es nicht möglich sämtliche Ergebnisse graphisch
darzustellen. Stellvertretend wurden die Systeme Kaliumchlorid / Wasser und Natriumchlorid
/ Wasser ausgewählt und diskutiert. Die Ergebnisse für alle weiteren Systeme sind in Tabelle
5.4.3.1 zusammengefaßt. Angegeben sind die durchschnittlichen, prozentualen
Abweichungen bezogen auf den jeweiligen Datentyp. Neben der Aufschlüsselung auf die
verschiedenen Datensatztypen ist für jedes System der zugrundeliegende Temperatur und
Konzentrationsbereich angegeben. Im Anhang (Kapitel 7.4) sind die zugehörigen Parameter
gelistet.
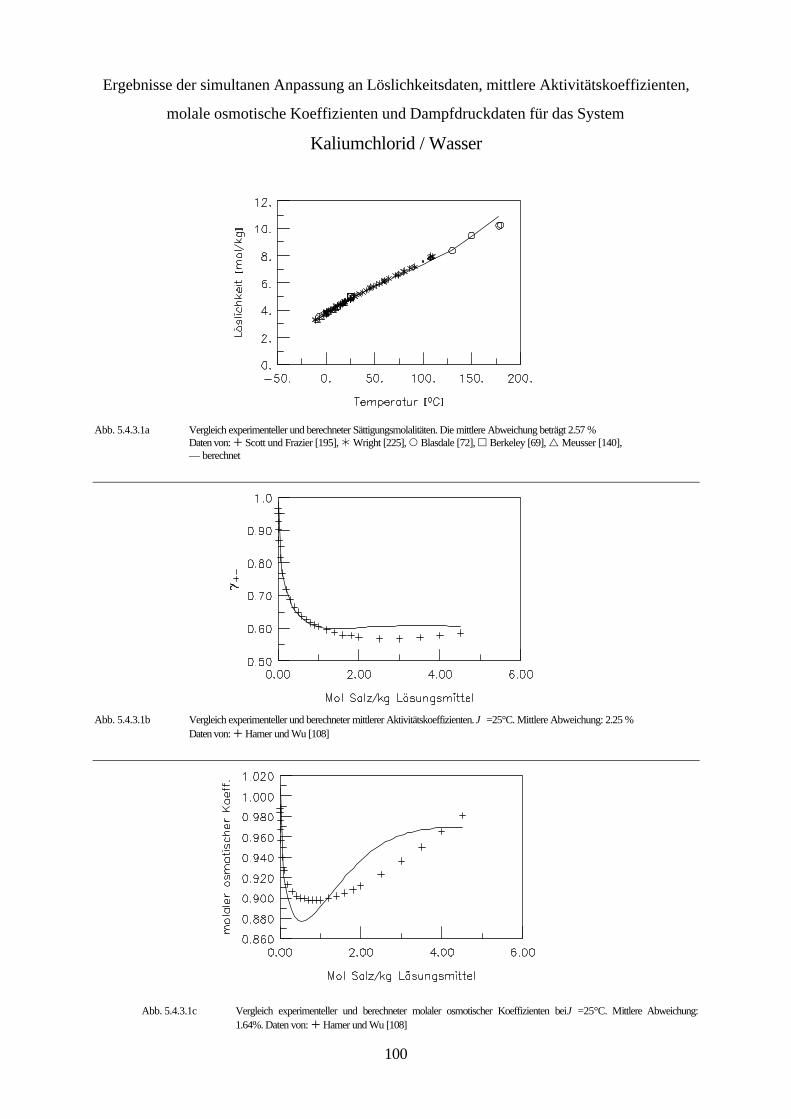
100
Ergebnisse der simultanen Anpassung an Löslichkeitsdaten, mittlere Aktivitätskoeffizienten,
molale osmotische Koeffizienten und Dampfdruckdaten für das System
Kaliumchlorid / Wasser
Abb. 5.4.3.1a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Die mittlere Abweichung beträgt 2.57 % Daten von: Ç Scott und Frazier [195], Ú Wright [225], � Blasdale [72], £ Berkeley [69], r Meusser [140], — berechnet
Abb. 5.4.3.1b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten.ϑ =25°C. Mittlere Abweichung: 2.25 % Daten von: Ç Hamer und Wu [108]
Abb. 5.4.3.1c Vergleich experimenteller und berechneter molaler osmotischer Koeffizienten beiϑ =25°C. Mittlere Abweichung:
1.64%. Daten von: Ç Hamer und Wu [108]
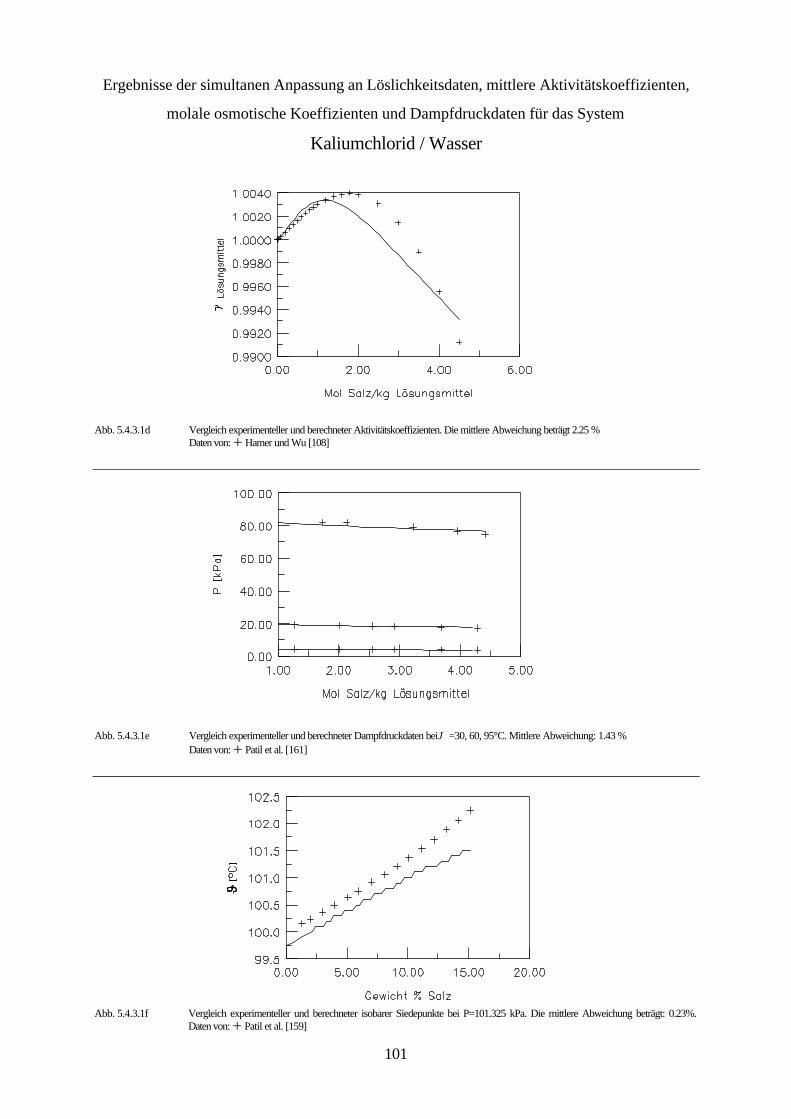
101
Ergebnisse der simultanen Anpassung an Löslichkeitsdaten, mittlere Aktivitätskoeffizienten,
molale osmotische Koeffizienten und Dampfdruckdaten für das System
Kaliumchlorid / Wasser
Abb. 5.4.3.1d Vergleich experimenteller und berechneter Aktivitätskoeffizienten. Die mittlere Abweichung beträgt 2.25 % Daten von: Ç Hamer und Wu [108]
Abb. 5.4.3.1e Vergleich experimenteller und berechneter Dampfdruckdaten beiϑ =30, 60, 95°C. Mittlere Abweichung: 1.43 % Daten von: Ç Patil et al. [161]
Abb. 5.4.3.1f Vergleich experimenteller und berechneter isobarer Siedepunkte bei P=101.325 kPa. Die mittlere Abweichung beträgt: 0.23%. Daten von: Ç Patil et al. [159]
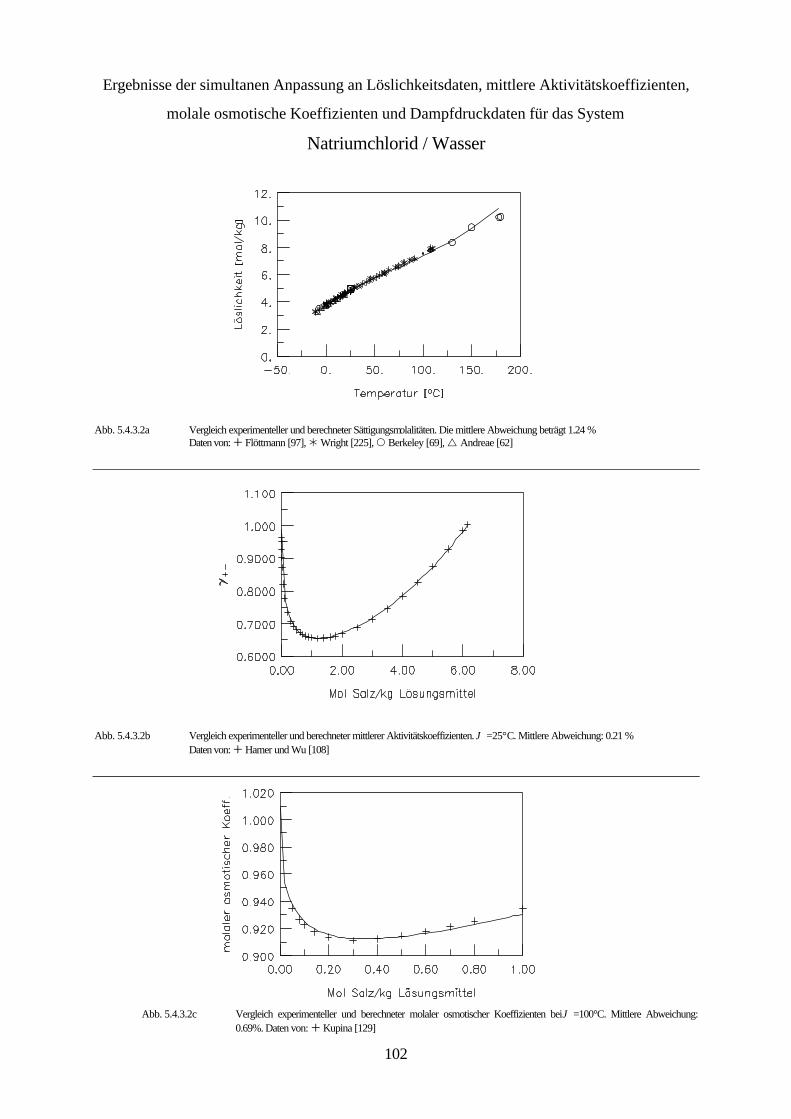
102
Ergebnisse der simultanen Anpassung an Löslichkeitsdaten, mittlere Aktivitätskoeffizienten,
molale osmotische Koeffizienten und Dampfdruckdaten für das System
Natriumchlorid / Wasser
Abb. 5.4.3.2a Vergleich experimenteller und berechneter Sättigungsmolalitäten. Die mittlere Abweichung beträgt 1.24 % Daten von: Ç Flöttmann [97], Ú Wright [225], � Berkeley [69], r Andreae [62]
Abb. 5.4.3.2b Vergleich experimenteller und berechneter mittlerer Aktivitätskoeffizienten.ϑ =25°C. Mittlere Abweichung: 0.21 % Daten von: Ç Hamer und Wu [108]
Abb. 5.4.3.2c Vergleich experimenteller und berechneter molaler osmotischer Koeffizienten beiϑ =100°C. Mittlere Abweichung: 0.69%. Daten von: Ç Kupina [129]
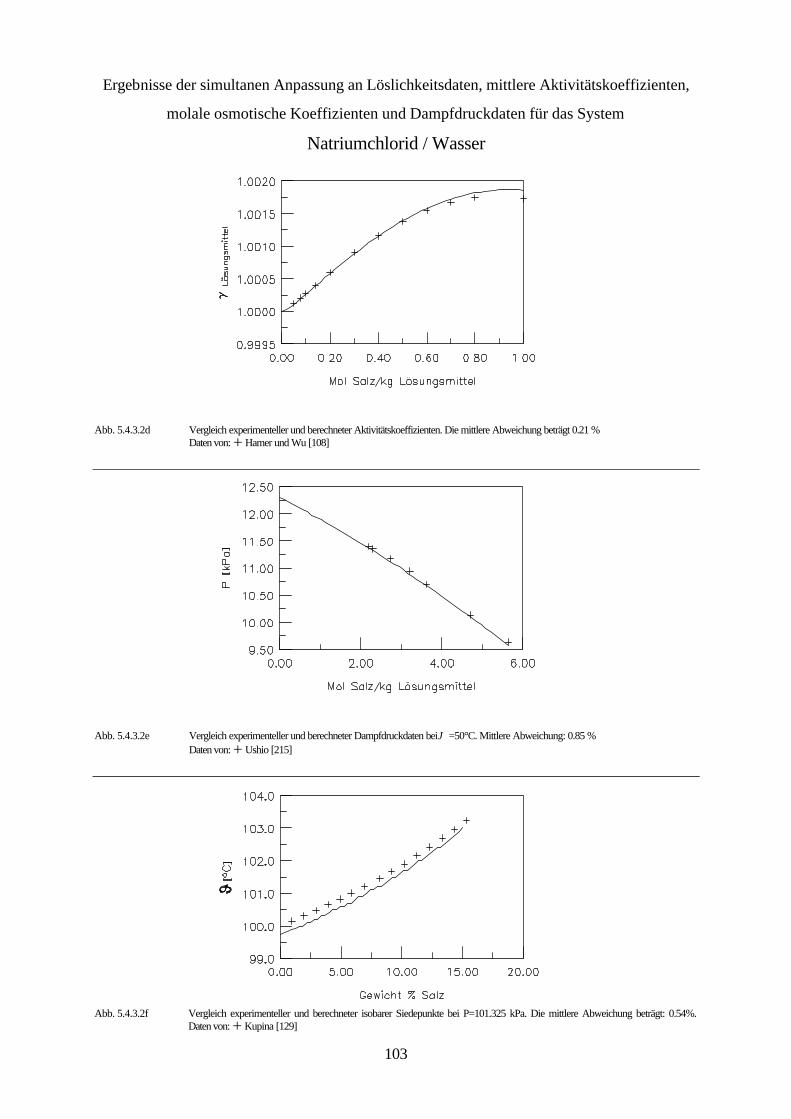
103
Ergebnisse der simultanen Anpassung an Löslichkeitsdaten, mittlere Aktivitätskoeffizienten,
molale osmotische Koeffizienten und Dampfdruckdaten für das System
Natriumchlorid / Wasser
Abb. 5.4.3.2d Vergleich experimenteller und berechneter Aktivitätskoeffizienten. Die mittlere Abweichung beträgt 0.21 % Daten von: Ç Hamer und Wu [108]
Abb. 5.4.3.2e Vergleich experimenteller und berechneter Dampfdruckdaten beiϑ =50°C. Mittlere Abweichung: 0.85 % Daten von: Ç Ushio [215]
Abb. 5.4.3.2f Vergleich experimenteller und berechneter isobarer Siedepunkte bei P=101.325 kPa. Die mittlere Abweichung beträgt: 0.54%. Daten von: Ç Kupina [129]
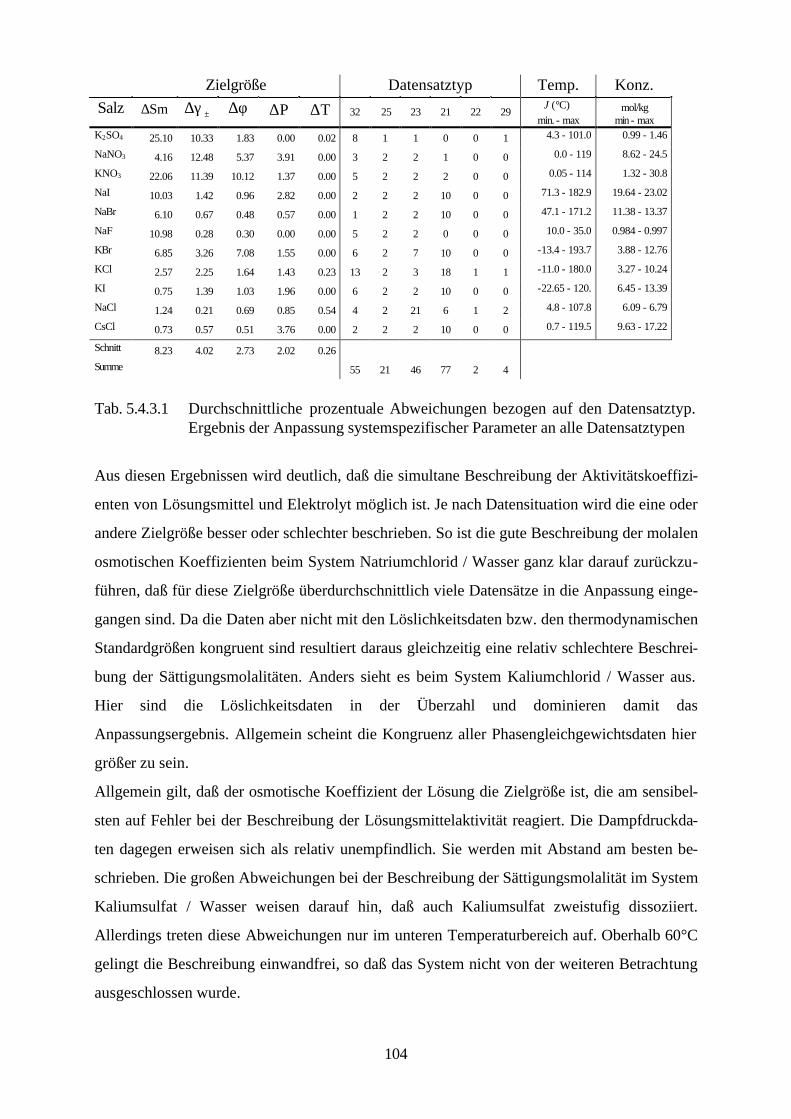
104
Zielgröße Datensatztyp Temp. Konz. Salz ∆Sm ∆γ ± ∆φ ∆P ∆T 32 25 23 21 22 29
C)(°ϑ min. - max
mol/kg min - max
K2SO4 25.10 10.33 1.83 0.00 0.02 8 1 1 0 0 1 4.3 - 101.0 0.99 - 1.46
NaNO3 4.16 12.48 5.37 3.91 0.00 3 2 2 1 0 0 0.0 - 119 8.62 - 24.5
KNO3 22.06 11.39 10.12 1.37 0.00 5 2 2 2 0 0 0.05 - 114 1.32 - 30.8
NaI 10.03 1.42 0.96 2.82 0.00 2 2 2 10 0 0 71.3 - 182.9 19.64 - 23.02
NaBr 6.10 0.67 0.48 0.57 0.00 1 2 2 10 0 0 47.1 - 171.2 11.38 - 13.37
NaF 10.98 0.28 0.30 0.00 0.00 5 2 2 0 0 0 10.0 - 35.0 0.984 - 0.997
KBr 6.85 3.26 7.08 1.55 0.00 6 2 7 10 0 0 -13.4 - 193.7 3.88 - 12.76
KCl 2.57 2.25 1.64 1.43 0.23 13 2 3 18 1 1 -11.0 - 180.0 3.27 - 10.24
KI 0.75 1.39 1.03 1.96 0.00 6 2 2 10 0 0 -22.65 - 120. 6.45 - 13.39
NaCl 1.24 0.21 0.69 0.85 0.54 4 2 21 6 1 2 4.8 - 107.8 6.09 - 6.79
CsCl 0.73 0.57 0.51 3.76 0.00 2 2 2 10 0 0 0.7 - 119.5 9.63 - 17.22
Schnitt 8.23 4.02 2.73 2.02 0.26
Summe 55 21 46 77 2 4
Tab. 5.4.3.1 Durchschnittliche prozentuale Abweichungen bezogen auf den Datensatztyp.
Ergebnis der Anpassung systemspezifischer Parameter an alle Datensatztypen
Aus diesen Ergebnissen wird deutlich, daß die simultane Beschreibung der Aktivitätskoeffizi-
enten von Lösungsmittel und Elektrolyt möglich ist. Je nach Datensituation wird die eine oder
andere Zielgröße besser oder schlechter beschrieben. So ist die gute Beschreibung der molalen
osmotischen Koeffizienten beim System Natriumchlorid / Wasser ganz klar darauf zurückzu-
führen, daß für diese Zielgröße überdurchschnittlich viele Datensätze in die Anpassung einge-
gangen sind. Da die Daten aber nicht mit den Löslichkeitsdaten bzw. den thermodynamischen
Standardgrößen kongruent sind resultiert daraus gleichzeitig eine relativ schlechtere Beschrei-
bung der Sättigungsmolalitäten. Anders sieht es beim System Kaliumchlorid / Wasser aus.
Hier sind die Löslichkeitsdaten in der Überzahl und dominieren damit das
Anpassungsergebnis. Allgemein scheint die Kongruenz aller Phasengleichgewichtsdaten hier
größer zu sein.
Allgemein gilt, daß der osmotische Koeffizient der Lösung die Zielgröße ist, die am sensibel-
sten auf Fehler bei der Beschreibung der Lösungsmittelaktivität reagiert. Die Dampfdruckda-
ten dagegen erweisen sich als relativ unempfindlich. Sie werden mit Abstand am besten be-
schrieben. Die großen Abweichungen bei der Beschreibung der Sättigungsmolalität im System
Kaliumsulfat / Wasser weisen darauf hin, daß auch Kaliumsulfat zweistufig dissoziiert.
Allerdings treten diese Abweichungen nur im unteren Temperaturbereich auf. Oberhalb 60°C
gelingt die Beschreibung einwandfrei, so daß das System nicht von der weiteren Betrachtung
ausgeschlossen wurde.

105
5.4.4 Beschreibung aller binären, wäßrigen Systeme
Die bisherigen Ergebnisse wurden auf Grundlage systemspezifischer Parameter gewonnen.
Sowohl die Ion-Ion-, als auch die Wasser-Ion-Parameter wurden an verschiedene Datensatzty-
pen des jeweils gleichen Systems angepaßt. Es wurden dabei bislang nur Systeme betrachtet,
für die die Datenbasis um Löslichkeitsdaten erweitert wurde. Alle anderen Systeme wurden
aus der Betrachtung ausgeschlossen. Für den so untersuchten Teil der Datenbasis ist klar, daß
der Übergang von systemspezifischen- zu allgemein verwendbaren Wasser-Ion-Parametern
die Qualität der Berechnung nicht verbessern kann. Das Ziel des folgenden
Anpassungsschrittes ist die bestmögliche Beschreibung der gesamten Datenbasis.
Seit der ersten Anpassung von LIQUAC-Parametern durch Li und Polka [29] ist der Datenbe-
stand von damals gut 1000 Datensätzen auf den heutigen Stand von über 3000 Datensätzen
angestiegen. Dementsprechend kann man auch die von Polka [43] angegebenen
Abweichungen als überholt ansehen. In Tabelle 5.4.4.1 sind die Ergebnisse für alle binären
wäßrigen Systeme zusammengefaßt die man mit den von Polka bestimmten Parametern auf
Grundlage der heutigen Datenbasis erhält. Für die Systeme die von Polka nicht berechnet
wurden, sind neue Ion-Ion-Parameter auf Grundlage der vorhandenen Wasser-Ion-Parameter
angepaßt worden. Neben den prozentualen Abweichungen bezogen auf die jeweilige
Zielgröße ist auch die Datentypverteilung und der Temperaturbereich der Daten für jedes
System angegeben. Insgesamt enthält die Datenbasis 674 Datensätze mit 9532 Datenpunkten
und berücksichtigt 91 verschiedene Systeme.
Bei der Neuanpassung wurden die Wasser-Ion-Parameter aus den bereits geschilderten Grün-
den von Polka übernommen, während die Ion-Ion-Parameter auf Grundlage aller binären,
wäßrigen Systeme überarbeitet wurden. Damit stehen für jedes untersuchte System vier
Anpassungsgrößen zur Verfügung. Die Ergebnisse der Neuanpassung sind in Tabelle 5.4.4.2
dargestellt. Die dazu gehörenden Parameter sind in Tabelle 5.4.4.1 (im Anhang) angegeben.
Stellt man die durchschnittlichen prozentualen Abweichungen vor und nach der
Überarbeitung einander gegenüber, ergibt sich das in Abbildung 5.4.4.1 dargestellte
Gesamtergebnis für binäre, wäßrige Systeme.
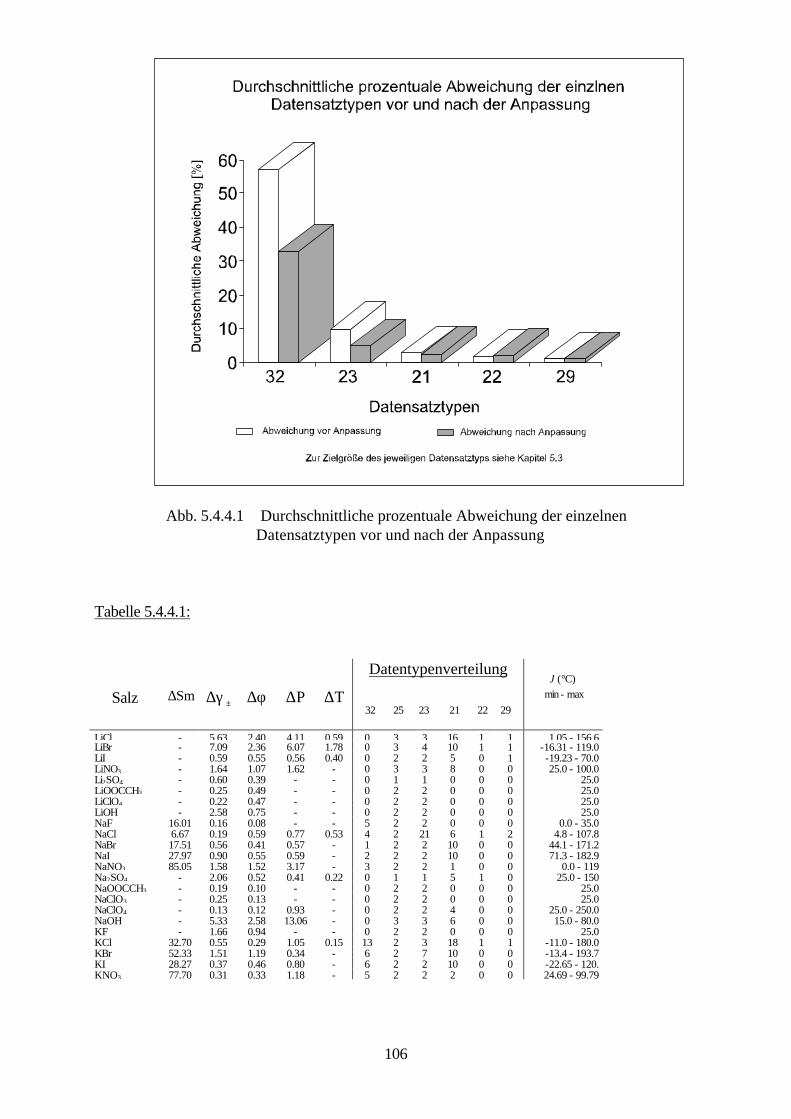
106
Abb. 5.4.4.1 Durchschnittliche prozentuale Abweichung der einzelnen
Datensatztypen vor und nach der Anpassung
Tabelle 5.4.4.1:
Salz
∆Sm
∆γ ±
∆φ
∆P
∆T
Datentypenverteilung
32 25 23 21 22 29
C)(°ϑ
min - max
LiCl - 5.63 2.40 4.11 0.59 0 3 3 16 1 1 1.05 - 156.6 LiBr - 7.09 2.36 6.07 1.78 0 3 4 10 1 1 -16.31 - 119.0 LiI - 0.59 0.55 0.56 0.40 0 2 2 5 0 1 -19.23 - 70.0 LiNO3 - 1.64 1.07 1.62 - 0 3 3 8 0 0 25.0 - 100.0 Li2SO4 - 0.60 0.39 - - 0 1 1 0 0 0 25.0 LiOOCCH3 - 0.25 0.49 - - 0 2 2 0 0 0 25.0 LiClO4 - 0.22 0.47 - - 0 2 2 0 0 0 25.0 LiOH - 2.58 0.75 - - 0 2 2 0 0 0 25.0 NaF 16.01 0.16 0.08 - - 5 2 2 0 0 0 0.0 - 35.0 NaCl 6.67 0.19 0.59 0.77 0.53 4 2 21 6 1 2 4.8 - 107.8 NaBr 17.51 0.56 0.41 0.57 - 1 2 2 10 0 0 44.1 - 171.2 NaI 27.97 0.90 0.55 0.59 - 2 2 2 10 0 0 71.3 - 182.9 NaNO3 85.05 1.58 1.52 3.17 - 3 2 2 1 0 0 0.0 - 119 Na2SO4 - 2.06 0.52 0.41 0.22 0 1 1 5 1 0 25.0 - 150 NaOOCCH3 - 0.19 0.10 - - 0 2 2 0 0 0 25.0 NaClO3 - 0.25 0.13 - - 0 2 2 0 0 0 25.0 NaClO4 - 0.13 0.12 0.93 - 0 2 2 4 0 0 25.0 - 250.0 NaOH - 5.33 2.58 13.06 - 0 3 3 6 0 0 15.0 - 80.0 KF - 1.66 0.94 - - 0 2 2 0 0 0 25.0 KCl 32.70 0.55 0.29 1.05 0.15 13 2 3 18 1 1 -11.0 - 180.0 KBr 52.33 1.51 1.19 0.34 - 6 2 7 10 0 0 -13.4 - 193.7 KI 28.27 0.37 0.46 0.80 - 6 2 2 10 0 0 -22.65 - 120. KNO3 77.70 0.31 0.33 1.18 - 5 2 2 2 0 0 24.69 - 99.79

107
Tabelle 5.4.4.1 (Fortsetzung) ______________________________________________________________________________________________
Salz
∆Sm
∆γ ±
∆φ
∆P
∆T
Datentypenverteilung
32 25 23 21 22 29
C)(°ϑ
min - max
K2SO4 41.50 0.57 0.71 - 0.03 8 1 1 0 0 1 4.32 - 101.1 KOOCCH3 - 0.19 0.26 0.57 - 0 1 2 5 0 0 25.0 - 100.0 KClO3 - 0.27 0.17 - - 0 2 2 0 0 0 25.0 KOH - 2.88 1.01 - - 0 3 3 0 0 0 25.0 RbF - 0.54 0.45 - - 0 1 1 0 0 0 25.0 RbCl - 0.37 0.26 0.77 - 0 2 2 10 0 0 25.0 - 95.0 RbBr - 0.19 0.06 - - 0 2 2 0 0 0 25.0 RbI - 0.18 0.09 - - 0 2 2 0 0 0 25.0 RbNO3 - 0.80 0.92 - - 0 2 1 0 0 0 25.0 Rb2SO4 - 1.02 1.28 - - 0 1 1 0 0 0 25.0 CsF - 0.25 0.22 - - 0 1 1 0 0 0 25.0 CsCl 47.74 0.28 0.23 1.19 - 2 2 2 10 0 0 0.70 - 119.5 CsBr - 0.70 0.66 0.56 - 0 2 2 10 0 0 25.0 - 90.0 CsI - 0.27 0.30 0.22 - 0 2 2 5 0 0 25.0 - 70 CsNO3 - 0.23 0.29 - - 0 2 2 0 0 0 25.0 CsOH - 0.74 0.26 - - 0 2 2 0 0 0 25.0 Cs2SO4 61.28 0.77 0.28 - - 2 1 1 0 0 0 0.70 - 108.6 CsOOCCH3 - 0.21 0.16 - - 0 2 2 0 0 0 25.0 MgCl2 - 2.10 24.46 2.87 0.14 0 1 26 7 1 0 2.0 - 251.0 MgBr2 - 1.15 0.54 - - 0 1 1 0 0 0 25.0 MgI2 - 2.22 1.23 - - 0 1 1 0 0 0 25.0 Mg(NO3)2 - 0.41 0.45 - - 0 1 1 0 0 0 25.0 MgSO4 - - 9.64 - - 0 0 1 0 0 0 25.0 Mg(ClO4)2 - 1.35 0.66 - - 0 1 1 0 0 0 25.0 CaCl2 - 9.46 4.69 2.62 5.96 0 1 4 25 1 2 5.0 - 105.0 CaBr2 - 6.03 2.77 0.81 - 0 1 2 5 0 0 25.0 - 70.0 CaI2 - 2.40 2.20 2.23 - 0 1 1 5 0 0 25.0 - 70.0 Ca(NO3)2 - 1.01 1.06 - - 0 1 1 0 0 0 25.0 SrCl2 - 28.05 11.52 3.83 - 0 1 15 5 0 0 25.0 - 251.0 SrBr2 - 5.36 5.27 0.54 - 0 1 1 5 0 0 25.0 - 70.0 SrI2 - 3.79 4.41 1.90 - 0 1 1 5 0 0 25.0 - 70.0 Sr(NO3)2 - 12.63 5.49 - - 0 1 1 0 0 0 25.0 BaCl2 - 0.25 31.85 0.56 - 0 1 12 5 0 0 25.0 - 251.0 BaBr2 - 0.50 0.51 0.56 - 0 1 1 5 0 0 25.0 - 70.0 BaI2 - 0.30 0.40 - - 0 1 1 0 0 0 25.0 Ba(NO3)2 - 0.45 0.21 - - 0 1 1 0 0 0 25.0 MnCl2 - 1.63 1.36 - - 0 1 1 0 0 0 25.0 MnSO4 - 19.05 - - - 0 1 0 0 0 0 25.0 FeCl2 - 0.27 0.17 - - 0 1 1 0 0 0 25.0 CoCl2 - 3.66 2.88 1.34 0.60 0 1 1 4 3 0 2.0 - 50.0 Co(NO3)2 - 0.81 0.75 - - 0 1 1 0 0 0 25.0 NiCl2 - 2.54 1.97 - - 0 1 2 0 0 0 25.0 NiSO4 - 1.43 4.21 - - 0 1 1 0 0 0 25.0 CuCl2 - 0.50 0.49 - - 0 1 1 0 0 0 25.0 Cu(NO3)2 - 2.40 1.69 - - 0 1 1 0 0 0 25.0 CuSO4 - 1.12 4.54 0.84 - 0 1 1 1 0 0 5.0 - 25.0 ZnCl2 - 6.76 5.69 - - 0 1 1 0 0 0 25.0 ZnBr2 - 19.31 8.13 - - 0 1 1 0 0 0 25.0 ZnI2 - 31.00 14.92 - - 0 1 1 0 0 0 25.0 Zn(NO3)2 - 2.57 3.29 - - 0 1 1 0 0 0 25.0 ZnSO4 - 2.36 3.24 1.70 - 0 1 1 1 0 0 5.0 - 25.0 CdCl2 - 1.38 2.60 - - 0 1 1 0 0 0 25.0 CdBr2 - 1.27 1.68 - - 0 1 1 0 0 0 25.0 CdI2 - 1.60 2.23 - - 0 1 1 0 0 0 25.0 Cd(NO3)2 - 2.57 2.20 - - 0 1 1 0 0 0 25.0 CdSO4 - 1.78 4.69 - - 0 1 1 0 0 0 25.0 AgNO3 - 0.52 0.97 - - 0 3 3 0 0 0 25.0 NH4Cl 181.20 0.23 0.22 0.72 - 1 1 1 4 0 0 -15.36 NH4Br 17.57 - - - - 3 0 0 0 0 0 5.3 - 166.6 NH4ClO4 - 0.20 0.10 - - 0 1 1 0 0 0 25.0 NH4NO3 203.30 0.99 0.79 1.66 - 1 1 1 2 0 0 -16.67 (NH4)2SO4 14.90 - - - - 2 0 0 0 0 0 -18.34 HCl - 11.44 2.94 - - 0 3 3 0 0 0 25.0 HF - 14.27 12.14 - - 0 1 1 0 0 0 25.0 HBr - 2.63 0.91 - - 0 2 2 0 0 0 25.00 HI - 0.88 0.48 - - 0 2 2 0 0 0 25.0 HNO3 - 601.70 23.29 - - 0 2 2 0 0 0 25.0 HClO4 - 2.73 0.98 - - 0 3 3 0 0 0 25.0 Durchschnitt 56.98 9.79 2.76 1.79 1.04 Summe 64 135 216 240 10 9
Tab. 5.4.4.1 Prozentuale Abweichungen und Datentypenverteilung binärer, wäßriger
Systeme auf Grundlage der von Polka [29]bestimmten LIQUAC-Parameter
108
Tabelle 5.4.4.2
Salz
∆Sm
∆γ ±
∆φ
∆P
∆T
Datentypenverteilung
32 25 23 21 22 29
C)(°ϑ
min- max
LiCl - 6.41 2.69 3.78 0.64 0 3 3 16 1 1 1.05 - 156.6 LiBr - 7.20 2.44 5.82 1.78 0 3 4 10 1 1 -16.31 - 119.0 LiI - 0.59 0.54 0.56 0.39 0 2 2 5 0 1 -19.23 - 70.0 LiNO3 - 1.10 0.87 1.63 - 0 3 3 8 0 0 25.0 - 100.0 Li2SO4 - 0.59 0.44 - - 0 1 1 0 0 0 25.0 LiOOCCH3 - 0.20 0.49 - - 0 2 2 0 0 0 25.0 LiClO4 - 0.24 0.47 - - 0 2 2 0 0 0 25.0 LiOH - 2.60 0.71 - - 0 2 2 0 0 0 25.0 NaF 4.54 0.81 0.87 - - 5 2 2 0 0 0 0.0 - 35.0 NaCl 1.56 0.65 0.72 0.81 0.54 4 2 21 6 1 2 4.8 - 107.8 NaBr 7.46 0.83 0.51 0.59 - 1 2 2 10 0 0 44.1 - 171.2 NaI 11.68 1.39 0.70 1.16 - 2 2 2 10 0 0 71.3 - 182.9 NaNO3 21.48 6.52 3.66 1.59 - 3 2 2 1 0 0 0.0 - 119 Na2SO4 - 0.50 0.96 0.41 0.23 0 1 1 5 1 0 25.0 - 150 NaOOCCH3 - 0.11 0.10 - - 0 2 2 0 0 0 25.0 NaClO3 - 0.12 0.11 - - 0 2 2 0 0 0 25.0 NaClO4 - 0.21 0.20 0.85 - 0 2 2 4 0 0 25.0 - 250.0 NaOH - 5.69 2.80 11.39 - 0 3 3 6 0 0 15.0 - 80.0 KF - 1.51 0.87 - - 0 2 2 0 0 0 25.0 KCl 21.17 23.45 7.04 1.22 0.20 13 2 3 18 1 1 -11.0 - 180.0 KBr 31.81 19.24 4.79 0.95 - 6 2 7 10 0 0 -13.4 - 193.7 KI 27.26 6.04 2.06 0.89 - 6 2 2 10 0 0 -22.65 - 120. KNO3 30.30 6.86 4.41 1.32 - 5 2 2 2 0 0 24.69 - 99.79 K2SO4 11.79 42.00 10.10 - 0.03 8 1 1 0 0 1 4.32 - 101.1 KOOCCH3 - 0.13 0.27 0.56 - 0 1 2 5 0 0 25.0 - 100.0 KClO3 - 0.14 0.24 - - 0 2 2 0 0 0 25.0 KOH - 2.60 0.98 - - 0 3 3 0 0 0 25.0 RbF - 0.53 0.45 - - 0 1 1 0 0 0 25.0 RbCl - 0.24 0.19 0.71 - 0 2 2 10 0 0 25.0 - 95.0 RbBr - 0.09 0.06 - - 0 2 2 0 0 0 25.0 RbI - 0.09 0.07 - - 0 2 2 0 0 0 25.0 RbNO3 - 0.17 0.13 - - 0 2 1 0 0 0 25.0 Rb2SO4 - 1.03 1.28 - - 0 1 1 0 0 0 25.0 CsF - 0.25 0.22 - - 0 1 1 0 0 0 25.0 CsCl 31.85 4.84 1.34 1.21 - 2 2 2 10 0 0 0.70 - 119.5 CsBr - 0.35 0.36 0.69 - 0 2 2 10 0 0 25.0 - 90.0 CsI - 0.15 0.17 0.26 - 0 2 2 5 0 0 25.0 - 70 CsNO3 - 0.09 0.12 - - 0 2 2 0 0 0 25.0 CsOH - 0.74 0.26 - - 0 2 2 0 0 0 25.0 Cs2SO4 8.93 10.53 11.52 - - 2 1 1 0 0 0 0.70 - 108.6 CsOOCCH3 - 0.19 0.14 - - 0 2 2 0 0 0 25.0 MgCl2 - 19.66 7.49 9.59 0.28 0 1 26 7 1 0 2.0 - 251.0 MgBr2 - 1.12 0.52 - - 0 1 1 0 0 0 25.0 MgI2 - 1.35 0.72 - - 0 1 1 0 0 0 25.0 Mg(NO3)2 - 0.26 0.48 - - 0 1 1 0 0 0 25.0 MgSO4 - - 0.49 - - 0 0 1 0 0 0 25.0 Mg(ClO4)2 - 1.31 0.64 - - 0 1 1 0 0 0 25.0 CaCl2 - 9.63 4.71 2.57 5.96 0 1 4 25 1 2 5.0 - 105.0 CaBr2 - 5.90 2.63 1.75 - 0 1 2 5 0 0 25.0 - 70.0 CaI2 - 1.43 1.52 2.68 - 0 1 1 5 0 0 25.0 - 70.0 Ca(NO3)2 - 1.04 1.00 - - 0 1 1 0 0 0 25.0 SrCl2 - 26.24 9.62 4.32 - 0 1 15 5 0 0 25.0 - 251.0 SrBr2 - 4.04 5.34 0.54 - 0 1 1 5 0 0 25.0 - 70.0 SrI2 - 3.45 4.82 1.53 - 0 1 1 5 0 0 25.0 - 70.0 Sr(NO3)2 - 5.24 7.57 - - 0 1 1 0 0 0 25.0 BaCl2 - 7.13 9.10 0.90 - 0 1 12 5 0 0 25.0 - 251.0 BaBr2 - 0.59 0.59 0.56 - 0 1 1 5 0 0 25.0 - 70.0 BaI2 - 0.22 0.38 - - 0 1 1 0 0 0 25.0 Ba(NO3)2 - 0.13 0.30 - - 0 1 1 0 0 0 25.0 MnCl2 - 1.61 1.33 - - 0 1 1 0 0 0 25.0 MnSO4 - 18.42 - - - 0 1 0 0 0 0 25.0 FeCl2 - 0.20 0.18 - - 0 1 1 0 0 0 25.0 CoCl2 - 3.44 2.72 1.26 1.13 0 1 1 4 3 0 2.0 - 50.0 Co(NO3)2 - 0.80 0.72 - - 0 1 1 0 0 0 25.0 NiCl2 - 2.85 1.59 - - 0 1 2 0 0 0 25.0 NiSO4 - 1.71 3.71 - - 0 1 1 0 0 0 25.0 CuCl2 - 0.35 0.41 - - 0 1 1 0 0 0 25.0 Cu(NO3)2 - 2.46 1.65 - - 0 1 1 0 0 0 25.0 CuSO4 - 1.78 4.26 0.85 - 0 1 1 1 0 0 5.0 - 25.0 ZnCl2 - 6.80 5.66 - - 0 1 1 0 0 0 25.0 ZnBr2 - 19.10 8.08 - - 0 1 1 0 0 0 25.0 ZnI2 - 30.87 14.86 - - 0 1 1 0 0 0 25.0 Zn(NO3)2 - 2.57 3.28 - - 0 1 1 0 0 0 25.0 ZnSO4 - 2.41 3.12 1.69 - 0 1 1 1 0 0 5.0 - 25.0 CdCl2 - 1.09 2.02 - - 0 1 1 0 0 0 25.0

109
Tabelle 5.4.4.2 (Fortsetzung) ______________________________________________________________________________________________
Salz
∆Sm
∆γ ±
∆φ
∆P
∆T
Datentypenverteilung
32 25 23 21 22 29
C)(°ϑ
min - max
CdBr2 - 1.24 1.63 - - 0 1 1 0 0 0 25.0 CdI2 - 1.72 2.25 - - 0 1 1 0 0 0 25.0 Cd(NO3)2 - 2.54 2.19 - - 0 1 1 0 0 0 25.0 CdSO4 - 2.26 4.12 - - 0 1 1 0 0 0 25.0 AgNO3 - 0.43 0.70 - - 0 3 3 0 0 0 25.0 NH4Cl 137.90 9.18 3.51 1.21 - 1 1 1 4 0 0 -15.36 NH4Br 17.39 - - - - 3 0 0 0 0 0 5.3 - 166.6 NH4ClO4 - 0.11 0.12 - - 0 1 1 0 0 0 25.0 NH4NO3 132.90 10.86 4.59 2.13 - 1 1 1 2 0 0 -16.67 (NH4)2SO4 28.68 - - - - 2 0 0 0 0 0 -18.34 HCl - 7.46 2.39 - - 0 3 3 0 0 0 25.0 HF - 15.70 12.18 - - 0 1 1 0 0 0 25.0 HBr - 1.72 0.70 - - 0 2 2 0 0 0 25.00 HI - 0.72 0.37 - - 0 2 2 0 0 0 25.0 HNO3 - 13.86 6.27 - - 0 2 2 0 0 0 25.0 HClO4 - 2.74 0.98 - - 0 3 3 0 0 0 25.0 Durchschnitt 32.92 4.69 2.50 2.00 1.12 Summe 64 135 216 240 10 9
Tab. 5.4.4.2 Ergebnisse der Anpassung. Datenbasis: binäre wäßrige Systeme
Vergleicht man die durchschnittlichen prozentualen Abweichungen bezogen auf den jeweili-
gen Datensatztyp, so wird deutlich, daß die Löslichkeitsdaten mit Abstand am schlechtesten
beschrieben werden. Sie machen nur 9.5% der Datenbasis aus, verursachen aber 76.5% der
Gesamtabweichung. Der geringe Anteil der Löslichkeitsdaten an der Gesamtdatenbasis trägt
allerdings dazu bei, daß einzelne Datensätze die schlecht beschrieben werden das
Gesamtergebnis stark beeinflussen. So trägt z.B. im Fall des Systems
Ammoniumchlorid/Wasser ein einziger Datenpunkt 137.9% Abweichung zum
Gesamtergebnis bei. Die unzureichende Beschreibung der Löslichkeitsdaten wird nicht durch
das Unvermögen des Modells verursacht. Die vorherigen Anpassungen haben klar gezeigt, daß
das Modell sehr wohl in der Lage ist selbst sehr hohe Sättigungsmolalitäten richtig zu
beschreiben, und dabei gleichzeitig sinnvolle Werte für den Aktivitätskoeffizienten des
Lösungsmittels zu liefern. Die Notwendigkeit der internen Konsistenz der Lösungsmittel-Ion-
Parameter verursacht zwar den Wegfall vieler anpaßbarer Größen, diese Beschränkung kann
aber ebenfalls nicht der Grund für das unzureichende Gesamtergebnis sein. Wenn das Modell
in der Lage ist jeden Datentyp für sich sauber zu beschreiben, dann ist der Grund für ein
unbefriedigendes Gesamtergebnis eindeutig in der Inkongruenz und damit der Qualität der
Daten zu sehen. Die Tatsache, daß die Löslichkeitsdaten mit Abstand am schlechtesten
beschrieben werden weist somit lediglich darauf hin, daß die relativ größten Fehler bei den

110
tabellierten Standardgrößen liegen, die ja ebenfalls wie experimentelle Daten behandelt
wurden. Diesbezüglich bliebe seitens der Thermodynamik höchstens einzuwenden, daß die
Annahme konstanter Wärmekapazitäten insbesondere für Elektrolytsysteme eine grobe
Vereinfachung darstellt. Ist diese Vereinfachung schon für die Beschreibung von Dampf-
Flüssig-Gleichgewichten hinderlich, so wirkt sie sich um so deutlicher bei der Beschreibung
von Löslichkeitsgleichgewichten aus. Allerdings fehlen bislang die experimentellen
Möglichkeiten Wärmekapazitäten solvatisierter Ionen als Funktion der Temperatur zu messen.
Die Annahme konstanter Wärmekapazitäten über einen Temperaturbereich von bis zu 200°C
ist sicher nicht korrekt, allerdings läßt sich das Problem qualitativ schlechter Standardgrößen
nicht auf diesen Umstand allein reduzieren. Aus der Sicht des Modells würde die Möglichkeit
der Berücksichtigung von Dissoziationskonstanten die Qualität der Beschreibung deutlich
steigern. Das gilt sowohl für nicht vollständig dissoziierende Salze als auch für flüchtige
Elektrolyte, bei denen gleichzeitig noch die Gleichgewichtskonstante für den Phasenübergang
Gas-Flüssig zu berücksichtigen ist. Die Berücksichtigung solcher Gleichgewichte ist
theoretisch möglich, wenn sie auch mit einer Reihe numerisch mathematischer Probleme
verbunden ist. Darüber hinaus würde die Berücksichtigung eine große Anzahl weiterer
anpaßbarer Größen bedingen.

111
5.5 Binäre, nichtwäßrige Systeme
Die Beschreibung der Löslichkeit starker Elektrolyte in organischen Lösungsmitteln mit Hilfe
des Löslichkeitsprodukts stellt eine weitere erhöhte Anforderung an das Modell dar. Das
Problem hierbei ist, daß die Standardgrößen nur für das Lösungsmittel Wasser tabelliert sind.
Die Berechnung von Standardgrößen für nichtwässrige Lösungsmittel allein mit Hilfe eines
Born-Haber Kreisprozesses wie er in Abbildung 5.5.1 dargestellt ist kann nicht gelingen. Es
wäre allerdings möglich die gesuchte Solvatationsenergie z.B. mit Hilfe kalorischer Messungen
zu ermitteln, dabei würde man aber nur einen Bruttoenergiebetrag erhalten. Das Problem der
Aufteilung dieses Betrags auf An- und Kationen müsste anschließend durch vergleichende
Messungen der Lösungswärmen verschiedener Salze die jeweils ein gemeinsames Ion
enthalten geschehen. Der zu betreibende Aufwand um konsistente Werte für alle betrachteten
Ionen in verschiedenen Lösungsmitteln zu erhalten würde allerdings den Rahmen dieser
Arbeit weit übersteigen. An dieser Stelle soll lediglich geprüft werden wie die Beschreibung
binärer, nichtwässriger Löslichkeitsdaten gelingt wenn dafür die für wässrige Lösungen
tabellierten Standardgrößen zugrundelegt werden.
Abb. 5.5.1 Born-Haber Kreisprozeß

112
Überträgt man das Konzept des Löslichkeitsprodukts auf organische Lösungsmittel, so muß
das Modell in der Lage sein das zugrundegelegte und -streng genommen- falsche thermody-
namische Löslichkeitsprodukt zu kompensieren. Da die Ion-Ion-Parameter bereits an die
wäßrigen Systeme angepaßt wurden und somit nicht mehr zur Disposition stehen, verbleiben
dafür für jedes untersuchte System nur 6 anpaßbare Größen. Dies sind die vier Lösungsmittel-
Ion-a-Parameter und zwei Lösungsmittel-Ion-c-Parameter. Die b-Parameter kürzen sich wie-
derum aus dem Ausdruck für den Virialkoeffizienten heraus weil es sich um binäre Systeme
handelt. Da die Löslichkeit in organischen Lösungsmitteln unverhältnismäßig viel geringer ist
als in Wasser müssen die Aktivitätskoeffizienten dementsprechend groß werden. Dies sei
durch den Vergleich der Löslichkeit von Natriumchlorid in Wasser und in Methanol demon-
striert:
Die Löslichkeit von Natriumchlorid in Wasser beträgt bei 25°C 6.15 [mol/kg]. Das Löslich-
keitsprodukt beträgt 37.66. Daraus folgt ein Aktivitätskoeffizient von:
5.5.1 γ ± = =37 66615
0 99782 .
..
Die Löslichkeit von Natriumchlorid in Methanol beträgt bei dieser Temperatur nur 0.24
[mol/kg], woraus ein Aktivitätskoeffizient von:
5.5.2 γ ± = =37 660 24
25562 .
..
resultiert. Erschwerend kommt hinzu, daß der mittlere Aktivitätskoeffizient mit zunehmender
Konzentration immer mehr durch den middle-range-Term dominiert wird, in den aber nur die
beiden c-Parameter eingehen. Die a-Parameter verschwinden gewissermaßen im short-range-
Term, der geringeren Einfluß auf den Gesamtwert des mittleren Aktivitätskoeffizienten ausübt.
Die Datenbasis ist auch für binäre nichtwäßrige Systeme umfangreicher geworden. Sie umfaßt
191 Datensätze mit 1521 Datenpunkten. Diese Daten decken 85 verschiedene Systeme ab. Sie
enthält dabei jedoch eine Reihe von Systemen, bei denen die Anzahl verfügbarer Datenpunkte
geringer ist als die Anzahl anpaßbarer Größen. Diese Systeme müssen von der Anpassung
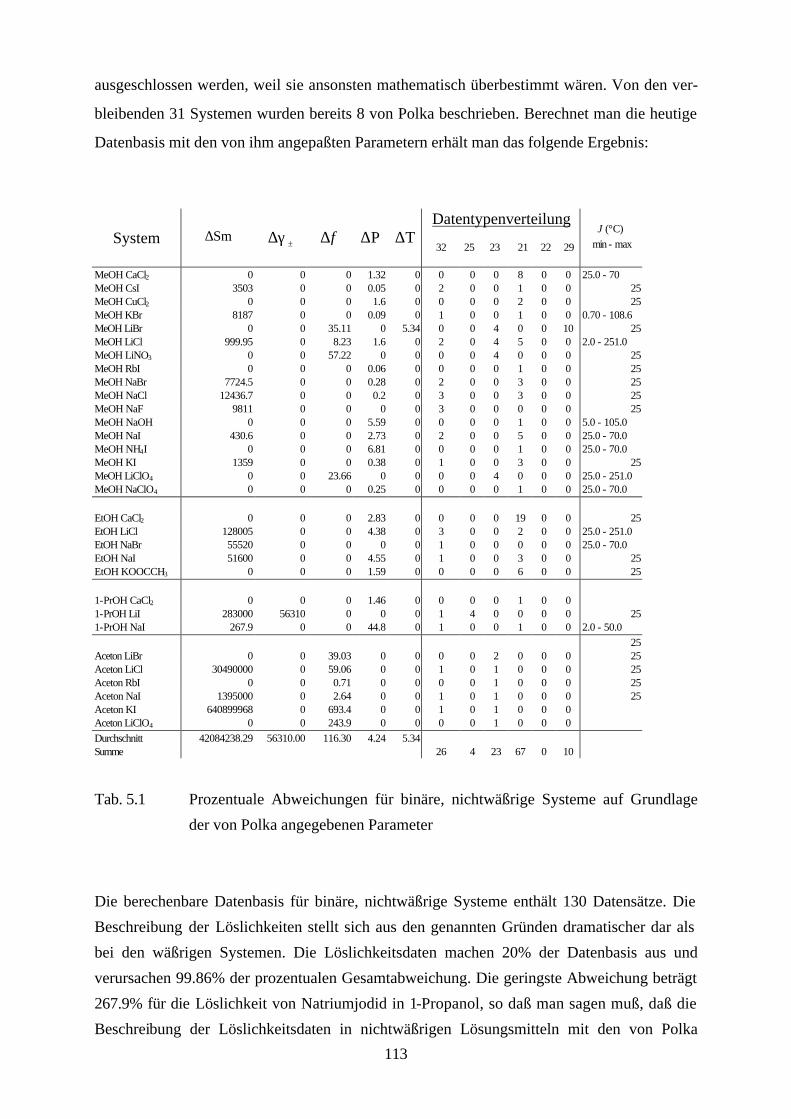
113
ausgeschlossen werden, weil sie ansonsten mathematisch überbestimmt wären. Von den ver-
bleibenden 31 Systemen wurden bereits 8 von Polka beschrieben. Berechnet man die heutige
Datenbasis mit den von ihm angepaßten Parametern erhält man das folgende Ergebnis:
System
∆Sm
∆γ ±
∆φ
∆P
∆T
Datentypenverteilung
32 25 23 21 22 29
C)(°ϑ
min - max
MeOH CaCl2 0 0 0 1.32 0 0 0 0 8 0 0 25.0 - 70 MeOH CsI 3503 0 0 0.05 0 2 0 0 1 0 0 25 MeOH CuCl2 0 0 0 1.6 0 0 0 0 2 0 0 25 MeOH KBr 8187 0 0 0.09 0 1 0 0 1 0 0 0.70 - 108.6 MeOH LiBr 0 0 35.11 0 5.34 0 0 4 0 0 10 25 MeOH LiCl 999.95 0 8.23 1.6 0 2 0 4 5 0 0 2.0 - 251.0 MeOH LiNO3 0 0 57.22 0 0 0 0 4 0 0 0 25 MeOH RbI 0 0 0 0.06 0 0 0 0 1 0 0 25 MeOH NaBr 7724.5 0 0 0.28 0 2 0 0 3 0 0 25 MeOH NaCl 12436.7 0 0 0.2 0 3 0 0 3 0 0 25 MeOH NaF 9811 0 0 0 0 3 0 0 0 0 0 25 MeOH NaOH 0 0 0 5.59 0 0 0 0 1 0 0 5.0 - 105.0 MeOH NaI 430.6 0 0 2.73 0 2 0 0 5 0 0 25.0 - 70.0 MeOH NH4I 0 0 0 6.81 0 0 0 0 1 0 0 25.0 - 70.0 MeOH KI 1359 0 0 0.38 0 1 0 0 3 0 0 25 MeOH LiClO4 0 0 23.66 0 0 0 0 4 0 0 0 25.0 - 251.0 MeOH NaClO4 0 0 0 0.25 0 0 0 0 1 0 0 25.0 - 70.0
EtOH CaCl2 0 0 0 2.83 0 0 0 0 19 0 0 25 EtOH LiCl 128005 0 0 4.38 0 3 0 0 2 0 0 25.0 - 251.0 EtOH NaBr 55520 0 0 0 0 1 0 0 0 0 0 25.0 - 70.0 EtOH NaI 51600 0 0 4.55 0 1 0 0 3 0 0 25 EtOH KOOCCH3 0 0 0 1.59 0 0 0 0 6 0 0 25
1-PrOH CaCl2 0 0 0 1.46 0 0 0 0 1 0 0 1-PrOH LiI 283000 56310 0 0 0 1 4 0 0 0 0 25 1-PrOH NaI 267.9 0 0 44.8 0 1 0 0 1 0 0 2.0 - 50.0
25 Aceton LiBr 0 0 39.03 0 0 0 0 2 0 0 0 25 Aceton LiCl 30490000 0 59.06 0 0 1 0 1 0 0 0 25 Aceton RbI 0 0 0.71 0 0 0 0 1 0 0 0 25 Aceton NaI 1395000 0 2.64 0 0 1 0 1 0 0 0 25 Aceton KI 640899968 0 693.4 0 0 1 0 1 0 0 0 Aceton LiClO4 0 0 243.9 0 0 0 0 1 0 0 0 Durchschnitt 42084238.29 56310.00 116.30 4.24 5.34 Summe 26 4 23 67 0 10
Tab. 5.1 Prozentuale Abweichungen für binäre, nichtwäßrige Systeme auf Grundlage
der von Polka angegebenen Parameter
Die berechenbare Datenbasis für binäre, nichtwäßrige Systeme enthält 130 Datensätze. Die
Beschreibung der Löslichkeiten stellt sich aus den genannten Gründen dramatischer dar als
bei den wäßrigen Systemen. Die Löslichkeitsdaten machen 20% der Datenbasis aus und
verursachen 99.86% der prozentualen Gesamtabweichung. Die geringste Abweichung beträgt
267.9% für die Löslichkeit von Natriumjodid in 1-Propanol, so daß man sagen muß, daß die
Beschreibung der Löslichkeitsdaten in nichtwäßrigen Lösungsmitteln mit den von Polka

114
gefundenen Parametern nicht einmal im Ansatz gelingt. Dies gilt zumindest solange
gleichzeitig binäre, wäßrige Systeme mit den gleichen Löslichkeitsprodukten beschrieben
werden sollen. Die Anpassung aller noch zur Disposition stehenden Freiheiten verbessert
diese Situation nicht wesentlich. Auf der anderen Seite würden die an binäre, nichtwäßrige
Systeme angepaßten Lösungsmittel-Ion-Parameter dann nicht mehr bei der Anpassung an
ternäre Datensätze zur Disposition stehen. Der Anteil ternärer Datensätze an der
Gesamtdatenbasis ist aber fünf mal größer als der Anteil an binären, nichtwäßrigen
Datensätzen. Für eine gute Gesamtbeschreibung ist es demnach sinnvoller auf die Anpassung
der Lösungsmittel-Ion-Parameter an dieser Stelle zu verzichten, um dafür ein besseres
Ergebnis bei den ternären Systemen zu erreichen. Aus diesem Grunde werden auch die
Ergebnisse für die Beschreibung der binären, wäßrigen Systeme im Kapitel 5.6 im Anschluß
an die Ergebnisse für ternäre Systeme vorgestellt. Es bleibt zu betonen, daß es sich nicht um eine Schwäche des Modells handelt. Das Modell ist
sehr wohl in der Lage gleichzeitig verschiedene Typen von Phasengleichgewichtsdaten auch in
nichtwäßrigen Lösungsmitteln zu beschreiben. Dies gelingt selbst wenn die Löslichkeits-
produkte für wäßrige Lösungen den Ausgangspunkt bilden. Was nicht gelingt ist die gleich-
zeitige Beschreibung von Löslichkeiten in Wasser und organischen Lösungsmitteln auf
Grundlage der gleichen thermodynamischen Daten im Rahmen einer 244 Systeme umfassen-
den Datenbasis.

115
5.6 Anpassung an ternäre Systeme
Die Beschreibung elektrolythaltiger Lösungsmittelgemische ist ein Problem, daß alle bisher
besprochenen Schwierigkeiten der Beschreibung elektrolythaltiger Systeme in sich vereint.
Dieser Umstand wird nur dadurch erleichtert, daß im Fall von Lösungsmittelgemischen zwei
zusätzliche anpaßbare Größen in Form der Lösungsmittel-Ion-b-Parameter zur Disposition
stehen. Wegen der begrenzten Löslichkeit der Elektrolyte in unpolaren Lösungsmitteln kom-
men für die Elektrolyt-Lösungsmittelgemische hauptsächlich Alkohol/Wasser-Systeme in
Frage. Insgesamt beinhaltet die berechenbare Datenbasis hier 6221 Datenpunkte in 636 Da-
tensätzen. Diese streuen über 119 verschiedene Systeme, von denen 53 bereits von Polka be-
schrieben wurden. Der Anteil an Alkohol/Wasser-Systemen beträgt 56%. Die Datenbasis ent-
hält 33 verschiedene Elektrolytkomponenten. Der Anteil an Löslichkeitsdaten beträgt 3.14%,
das sind 20 Datensätze. Im Hinblick darauf, daß für viele Systeme nur wenige Datenpunkte
vorliegen, muß die Datenbasis insgesamt als nicht ausreichend bezeichnet werden. Die
vorhandenen Datenlücken zu schließen war deshalb eine wesentliche Zielsetzung des experi-
mentellen Teils der Arbeit, doch kann dies gemessen an dem tatsächlich notwendigen Bedarf
nur ein geringer Beitrag zur Verbesserung der bestehenden Situation sein.
Selbstverständlich müssen auch die nichtwäßrigen Lösungsmittel-Ion-Parameter intern konsi-
stent sein. Durch die Verteilung der relativ geringen Anzahl an Elektrolytkomponenten auf
viele Lösungsmittelgemische stellt sich die Ausgangssituation für die Anpassung jedoch etwas
anders dar als bei den binären, wäßrigen Systemen. Nicht jede Veränderung eines einzelnen
Lösungsmittel-Ion-Parameters führt hier zwangsläufig zu einer Verschlechterung der Ge-
samtabweichung.
Die Berechnungen wurden unter Annahme idealen Verhaltens der Dampfphase und vollstän-
diger Dissoziation des Elektrolyten ausgeführt. Die für die Bestimmung des Sättigungs-
dampfdrucks der Lösungsmittelkomponenten erforderlichen Antoine-Konstanten wurden der
Literatur entnommen [18]. Im Fall isobarer Daten ist eine Iteration erforderlich, die ausgehend
vom experimentell vorgegebenen Druck zu der gesuchten Systemtemperatur führt. Mathema-
tisch läuft dies auf die Berechnung der Nullstelle für die Differenz zwischen berechnetem und
vorgegebenen Druck hinaus. Dafür wurde das Sehnenverfahren angewendet, das lediglich
zwei Startwerte für die Temperatur benötigt und ohne Gradientenbestimmung auskommt. Der
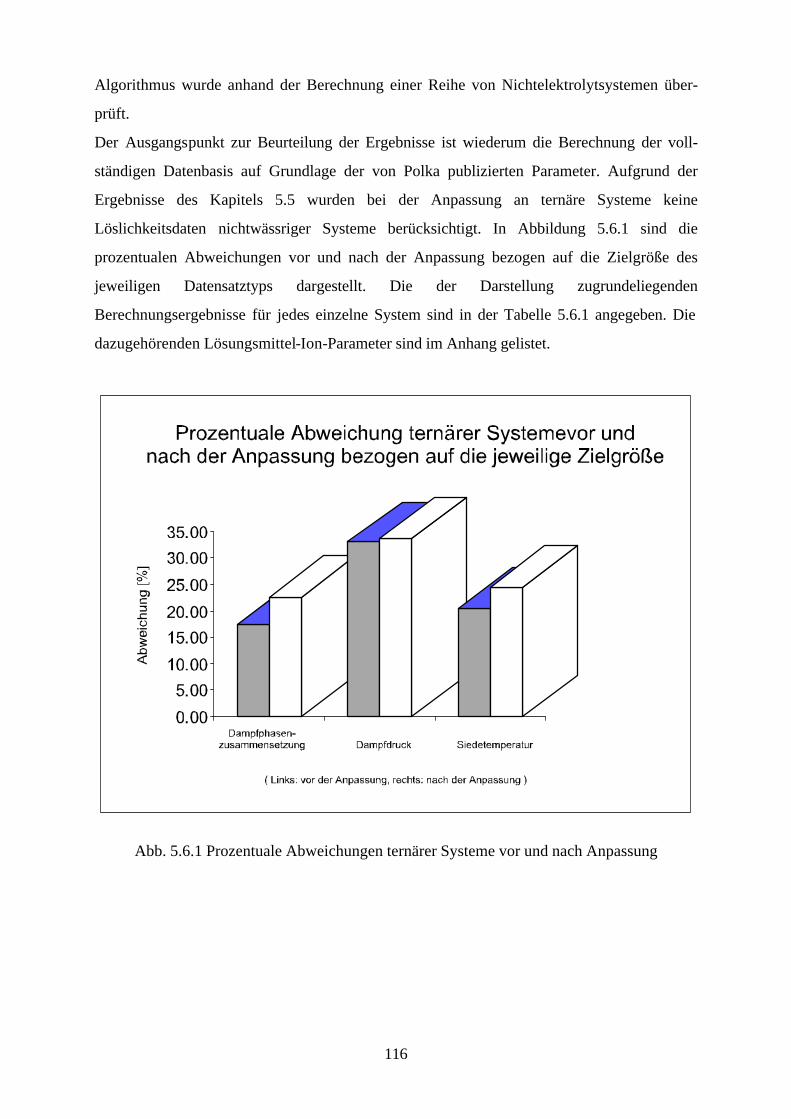
116
Algorithmus wurde anhand der Berechnung einer Reihe von Nichtelektrolytsystemen über-
prüft.
Der Ausgangspunkt zur Beurteilung der Ergebnisse ist wiederum die Berechnung der voll-
ständigen Datenbasis auf Grundlage der von Polka publizierten Parameter. Aufgrund der
Ergebnisse des Kapitels 5.5 wurden bei der Anpassung an ternäre Systeme keine
Löslichkeitsdaten nichtwässriger Systeme berücksichtigt. In Abbildung 5.6.1 sind die
prozentualen Abweichungen vor und nach der Anpassung bezogen auf die Zielgröße des
jeweiligen Datensatztyps dargestellt. Die der Darstellung zugrundeliegenden
Berechnungsergebnisse für jedes einzelne System sind in der Tabelle 5.6.1 angegeben. Die
dazugehörenden Lösungsmittel-Ion-Parameter sind im Anhang gelistet.
Abb. 5.6.1 Prozentuale Abweichungen ternärer Systeme vor und nach Anpassung
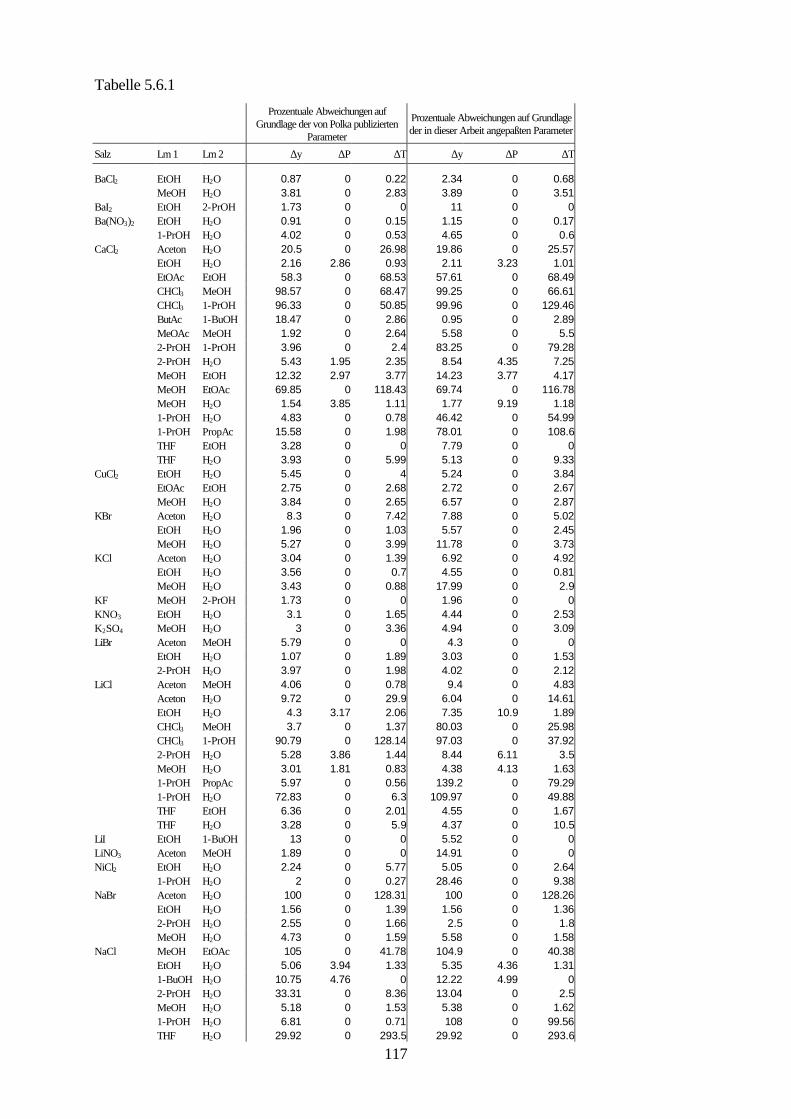
117
Tabelle 5.6.1
Prozentuale Abweichungen auf
Grundlage der von Polka publizierten Parameter
Prozentuale Abweichungen auf Grundlage der in dieser Arbeit angepaßten Parameter
Salz Lm 1 Lm 2 ∆y ∆P ∆T ∆y ∆P ∆T
BaCl2 EtOH H2O 0.87 0 0.22 2.34 0 0.68 MeOH H2O 3.81 0 2.83 3.89 0 3.51 BaI2 EtOH 2-PrOH 1.73 0 0 11 0 0 Ba(NO3)2 EtOH H2O 0.91 0 0.15 1.15 0 0.17 1-PrOH H2O 4.02 0 0.53 4.65 0 0.6 CaCl2 Aceton H2O 20.5 0 26.98 19.86 0 25.57 EtOH H2O 2.16 2.86 0.93 2.11 3.23 1.01 EtOAc EtOH 58.3 0 68.53 57.61 0 68.49 CHCl3 MeOH 98.57 0 68.47 99.25 0 66.61 CHCl3 1-PrOH 96.33 0 50.85 99.96 0 129.46 ButAc 1-BuOH 18.47 0 2.86 0.95 0 2.89 MeOAc MeOH 1.92 0 2.64 5.58 0 5.5 2-PrOH 1-PrOH 3.96 0 2.4 83.25 0 79.28 2-PrOH H2O 5.43 1.95 2.35 8.54 4.35 7.25 MeOH EtOH 12.32 2.97 3.77 14.23 3.77 4.17 MeOH EtOAc 69.85 0 118.43 69.74 0 116.78 MeOH H2O 1.54 3.85 1.11 1.77 9.19 1.18 1-PrOH H2O 4.83 0 0.78 46.42 0 54.99 1-PrOH PropAc 15.58 0 1.98 78.01 0 108.6 THF EtOH 3.28 0 0 7.79 0 0 THF H2O 3.93 0 5.99 5.13 0 9.33 CuCl2 EtOH H2O 5.45 0 4 5.24 0 3.84 EtOAc EtOH 2.75 0 2.68 2.72 0 2.67 MeOH H2O 3.84 0 2.65 6.57 0 2.87 KBr Aceton H2O 8.3 0 7.42 7.88 0 5.02 EtOH H2O 1.96 0 1.03 5.57 0 2.45 MeOH H2O 5.27 0 3.99 11.78 0 3.73 KCl Aceton H2O 3.04 0 1.39 6.92 0 4.92 EtOH H2O 3.56 0 0.7 4.55 0 0.81 MeOH H2O 3.43 0 0.88 17.99 0 2.9 KF MeOH 2-PrOH 1.73 0 0 1.96 0 0 KNO3 EtOH H2O 3.1 0 1.65 4.44 0 2.53 K2SO4 MeOH H2O 3 0 3.36 4.94 0 3.09 LiBr Aceton MeOH 5.79 0 0 4.3 0 0 EtOH H2O 1.07 0 1.89 3.03 0 1.53 2-PrOH H2O 3.97 0 1.98 4.02 0 2.12 LiCl Aceton MeOH 4.06 0 0.78 9.4 0 4.83 Aceton H2O 9.72 0 29.9 6.04 0 14.61 EtOH H2O 4.3 3.17 2.06 7.35 10.9 1.89 CHCl3 MeOH 3.7 0 1.37 80.03 0 25.98 CHCl3 1-PrOH 90.79 0 128.14 97.03 0 37.92 2-PrOH H2O 5.28 3.86 1.44 8.44 6.11 3.5 MeOH H2O 3.01 1.81 0.83 4.38 4.13 1.63 1-PrOH PropAc 5.97 0 0.56 139.2 0 79.29 1-PrOH H2O 72.83 0 6.3 109.97 0 49.88 THF EtOH 6.36 0 2.01 4.55 0 1.67 THF H2O 3.28 0 5.9 4.37 0 10.5 LiI EtOH 1-BuOH 13 0 0 5.52 0 0 LiNO3 Aceton MeOH 1.89 0 0 14.91 0 0 NiCl2 EtOH H2O 2.24 0 5.77 5.05 0 2.64 1-PrOH H2O 2 0 0.27 28.46 0 9.38 NaBr Aceton H2O 100 0 128.31 100 0 128.26 EtOH H2O 1.56 0 1.39 1.56 0 1.36 2-PrOH H2O 2.55 0 1.66 2.5 0 1.8 MeOH H2O 4.73 0 1.59 5.58 0 1.58 NaCl MeOH EtOAc 105 0 41.78 104.9 0 40.38 EtOH H2O 5.06 3.94 1.33 5.35 4.36 1.31 1-BuOH H2O 10.75 4.76 0 12.22 4.99 0 2-PrOH H2O 33.31 0 8.36 13.04 0 2.5 MeOH H2O 5.18 0 1.53 5.38 0 1.62 1-PrOH H2O 6.81 0 0.71 108 0 99.56 THF H2O 29.92 0 293.5 29.92 0 293.6
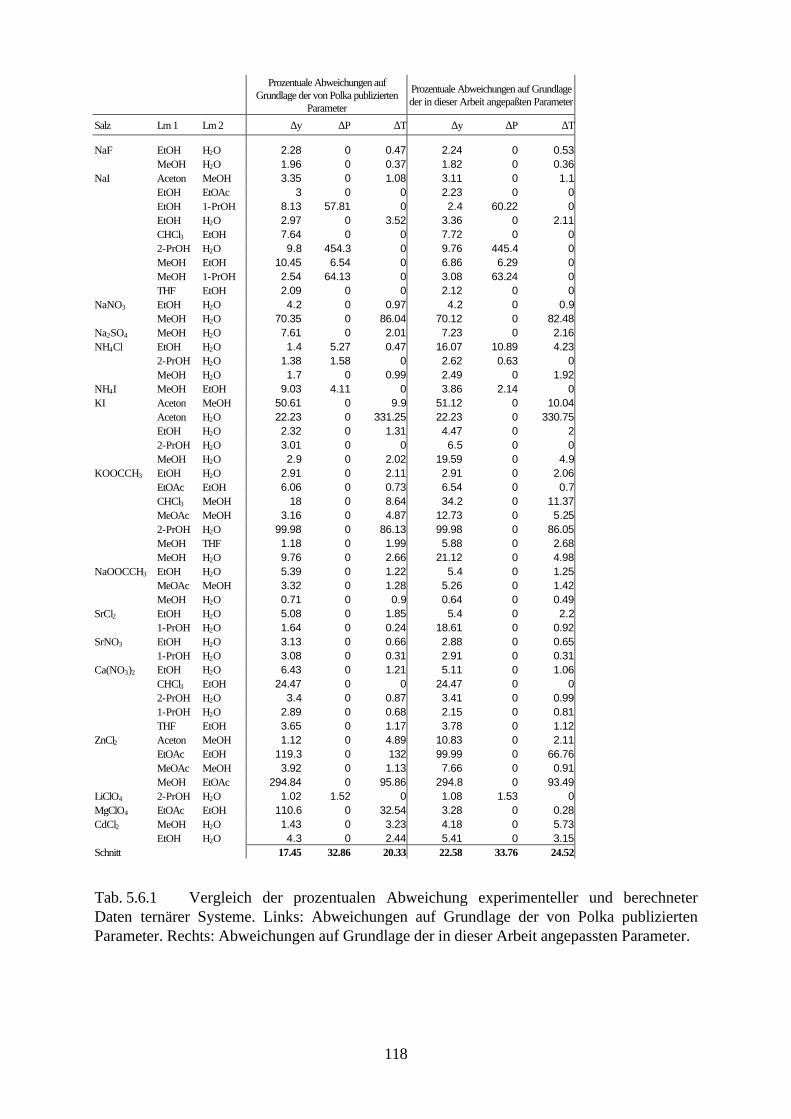
118
Prozentuale Abweichungen auf
Grundlage der von Polka publizierten Parameter
Prozentuale Abweichungen auf Grundlage der in dieser Arbeit angepaßten Parameter
Salz Lm 1 Lm 2 ∆y ∆P ∆T ∆y ∆P ∆T
NaF EtOH H2O 2.28 0 0.47 2.24 0 0.53 MeOH H2O 1.96 0 0.37 1.82 0 0.36 NaI Aceton MeOH 3.35 0 1.08 3.11 0 1.1 EtOH EtOAc 3 0 0 2.23 0 0 EtOH 1-PrOH 8.13 57.81 0 2.4 60.22 0 EtOH H2O 2.97 0 3.52 3.36 0 2.11 CHCl3 EtOH 7.64 0 0 7.72 0 0 2-PrOH H2O 9.8 454.3 0 9.76 445.4 0 MeOH EtOH 10.45 6.54 0 6.86 6.29 0 MeOH 1-PrOH 2.54 64.13 0 3.08 63.24 0 THF EtOH 2.09 0 0 2.12 0 0 NaNO3 EtOH H2O 4.2 0 0.97 4.2 0 0.9 MeOH H2O 70.35 0 86.04 70.12 0 82.48 Na2SO4 MeOH H2O 7.61 0 2.01 7.23 0 2.16 NH4Cl EtOH H2O 1.4 5.27 0.47 16.07 10.89 4.23 2-PrOH H2O 1.38 1.58 0 2.62 0.63 0 MeOH H2O 1.7 0 0.99 2.49 0 1.92 NH4I MeOH EtOH 9.03 4.11 0 3.86 2.14 0 KI Aceton MeOH 50.61 0 9.9 51.12 0 10.04 Aceton H2O 22.23 0 331.25 22.23 0 330.75 EtOH H2O 2.32 0 1.31 4.47 0 2 2-PrOH H2O 3.01 0 0 6.5 0 0 MeOH H2O 2.9 0 2.02 19.59 0 4.9 KOOCCH3 EtOH H2O 2.91 0 2.11 2.91 0 2.06 EtOAc EtOH 6.06 0 0.73 6.54 0 0.7 CHCl3 MeOH 18 0 8.64 34.2 0 11.37 MeOAc MeOH 3.16 0 4.87 12.73 0 5.25 2-PrOH H2O 99.98 0 86.13 99.98 0 86.05 MeOH THF 1.18 0 1.99 5.88 0 2.68 MeOH H2O 9.76 0 2.66 21.12 0 4.98 NaOOCCH3 EtOH H2O 5.39 0 1.22 5.4 0 1.25 MeOAc MeOH 3.32 0 1.28 5.26 0 1.42 MeOH H2O 0.71 0 0.9 0.64 0 0.49 SrCl2 EtOH H2O 5.08 0 1.85 5.4 0 2.2 1-PrOH H2O 1.64 0 0.24 18.61 0 0.92 SrNO3 EtOH H2O 3.13 0 0.66 2.88 0 0.65 1-PrOH H2O 3.08 0 0.31 2.91 0 0.31 Ca(NO3)2 EtOH H2O 6.43 0 1.21 5.11 0 1.06 CHCl3 EtOH 24.47 0 0 24.47 0 0 2-PrOH H2O 3.4 0 0.87 3.41 0 0.99 1-PrOH H2O 2.89 0 0.68 2.15 0 0.81 THF EtOH 3.65 0 1.17 3.78 0 1.12 ZnCl2 Aceton MeOH 1.12 0 4.89 10.83 0 2.11 EtOAc EtOH 119.3 0 132 99.99 0 66.76 MeOAc MeOH 3.92 0 1.13 7.66 0 0.91 MeOH EtOAc 294.84 0 95.86 294.8 0 93.49 LiClO4 2-PrOH H2O 1.02 1.52 0 1.08 1.53 0 MgClO4 EtOAc EtOH 110.6 0 32.54 3.28 0 0.28 CdCl2 MeOH H2O 1.43 0 3.23 4.18 0 5.73 EtOH H2O 4.3 0 2.44 5.41 0 3.15 Schnitt 17.45 32.86 20.33 22.58 33.76 24.52
Tab. 5.6.1 Vergleich der prozentualen Abweichung experimenteller und berechneter Daten ternärer Systeme. Links: Abweichungen auf Grundlage der von Polka publizierten Parameter. Rechts: Abweichungen auf Grundlage der in dieser Arbeit angepassten Parameter.

119
Bei der Beurteilung der durchschnittlich etwas höheren Abweichungen bei den ternären
Systemen muß man berücksichtigen, daß die binären wässrigen Systeme inklusive der
Sättigungsmolalitäten auf Grundlage der neuangepaßten Ion-Ion- und Wasser-Ion-Parametern
um ein vielfaches besser beschrieben werden. Versucht man die Ergebnisse aus Sicht des
Modells zu deuten, haben sie ihre Ursache eindeutig darin, daß der middle-range-Term, den
Gesamtwert des Aktivitätskoeffizienten dominiert. Somit sind es die Ion-Ion-Parameter denen
eine besondere Rolle zukommt. Sind die Ion-Ion-Parameter an wäßrige Systeme angepaßt, so
gelingt die Beschreibung der ternären Systeme allein durch Anpassung der verbleibenden
Lösungsmittel-Ion-Parameter nur bedingt. Je unähnlicher die untersuchten Lösungsmittel in
ihrer Struktur, vor allem aber bezüglich ihrer Dielektrizitätskonstante gegenüber dem
Lösungsmittel Wasser werden, desto geringer wird ihr Vermögen Salze zu lösen, und desto
schlechter gelingt auch die Beschreibung nicht wässriger Systeme. Versucht man die
Ergebnisse aus Sicht der Daten zu interpretieren, so ist klar, daß die Daten um so unsicherer
werden, je geringer die Löslichkeit wird. Derselbe absolute Fehler bedeutet einen ungleich
größeren prozentualen Fehler.

120
6. Messung von Dampf-Flüssig-Gleichgewichten salzhaltiger Lösungsmittelgemische
6.1. Meßmethoden und Meßprogramm
Für die Bestimmung von Dampf-Flüssig-Gleichgewichten salzhaltiger Lösungsmittelgemische
eignen sich sowohl dynamische als auch statische Meßmethoden. Dynamisch meint in diesem
Sinne, daß mindestens eine der beiden Phasen des zu untersuchenden Systems im Kreislauf
geführt wird. Bei statischen Methoden wird das System in eine evakuierte, temperierbare
Meßzelle gegeben und gewartet bis sich das Phasengleichgewicht in der Zelle eingestellt hat.
Die Meßgröße einer statischen Apparatur ist im allgemeinen der sich einstellende Druck, es ist
jedoch auch möglich Proben zu ziehen, und die Zusammensetzung der Dampfphase zu
analysieren. Im Rahmen dieser Arbeit wurden sowohl nach dem statischen, als auch nach dem
dynamischen Prinzip experimentelle Ergebnisse gewonnen. Die dynamischen Messungen
wurden mit Hilfe der Ebulliometrie, die statischen Messungen mit Hilfe gaschromato-
graphischen Dampfraumanalyse durchgeführt.
Die Auswahl der zu vermessenden Systeme erfolgte unter dem Gesichtspunkt fehlender oder
offensichtlich mangelhafter Literaturdaten. Aber auch andere Kriterien wie ausreichende Lös-
lichkeit des Salzes im ausgewählten Lösungsmittelgemisch, und die Vermeidung chemischer
Reaktionen in der Lösung mußten berücksichtigt werden. Ausreichend ist die Löslichkeit,
wenn das gelöste Salz einen meßbaren Effekt auf das Dampf-Flüssig-Gleichgewicht bewirkt.
Diese Grenze wird natürlich durch die Empfindlichkeit der verwendeten Analysegeräte be-
stimmt, dennoch ist klar, daß sich für die Untersuchung schwerlöslicher Salze wie z.B. der
Silberhalogenide Löslichkeits- oder Leitfähigkeitsmessungen besser eignen.
Chemische Reaktionen in der Lösung wie z.B. die Reaktion von Hydroxylionen mit CH-aci-
den Lösungsmitteln, oder die Bildung von Alkoholaten täuschen oft große Löslichkeit vor,
weil das Auflösen der festen Phase äußerlich nicht von reinen Solvatationseffekten zu unter-
scheiden ist. Letztlich aber, ist es widersinnig bei Mischungen in denen die Komponenten
anders als im Sinne reiner Solvatation miteinander reagieren von Lösungen zu sprechen.
Tabelle 6.1. gibt einen Überblick über das Meßprogramm. Im Rahmen dieser Arbeit wurden
150 Datensätze mit der Headspace-Apparatur und weitere 18 mit Hilfe der Ebulliometrie ge-
messen. Beide Meßapparaturen werden im Folgenden beschrieben. Die Tabellen mit den ein-
zelnen experimentellen Ergebnissen finden sich im Anhang. Es wurden jeweils ternäre Sy-

121
steme bestehend aus zwei Lösungsmitteln und einem Salz vermessen. Alle Messungen lagen
im Temperaturbereich zwischen 30°C und 70.6°C, die Salzkonzentration lag jeweils zwischen
0.01[mol/kg] und der Sättigungsgrenze im jeweiligen Lösungsmittelgemisch. Von den 8 un-
tersuchten Lösungsmittelgemischen zeigen die Hälfte der salzfreien Systeme azeotropes Ver-
halten.
Lösungsmittel 1 Lösungsmittel 2 Salz Anzahl Datensätze
2-Propanol Wasser KI 27
Ethanol 1-Butanol LiI 29
Methanol 2-Propanol KF 21
THF Ethanol CaCl2 17
THF Ethanol NaI 21
Chloroform Ethanol NaI 18
Chloroform Ethanol Ca(NO3)2 17
THF Ethanol Ca(NO3)2 10
Aceton Methanol NaI 8
Tab. 6.1 Meßprogramm
6.2. Gaschromatographische Dampfraumanalyse
Die gaschromatographische Dampfraumanalyse (´Headspace-Analyse´) ist eine besondere
Form der statischen Messung, bei der die Gaschromatographie als Analysenmethode dient.
Meßgröße ist die Zusammensetzung der Dampfphase über der elektrolythaltigen flüssigen
Phase. Aufgrund der Arbeitsweise des Probennehmers ist es bei der gaschromatographischen
Dampfraumanalyse nicht möglich den Sättigungsdampfdruck der Mischung zu bestimmen.
Da die Zusammensetzung der flüssigen Phase aus der Einwaage ermittelt werden kann und die
Temperatur frei wählbar ist erhält man isotherme Datensätze des Typs x,y,T.
Die zu vermessenden Proben werden in einem Thermostaten auf der gewünschten Meßtempe-
ratur gehalten, bis sich das Phasengleichgewicht eingestellt hat. Anschließend wird mittels
eines speziellen Probennahmesystems ein Teil der Dampfphase entnommen und in einem
Gaschromatographen analysiert.
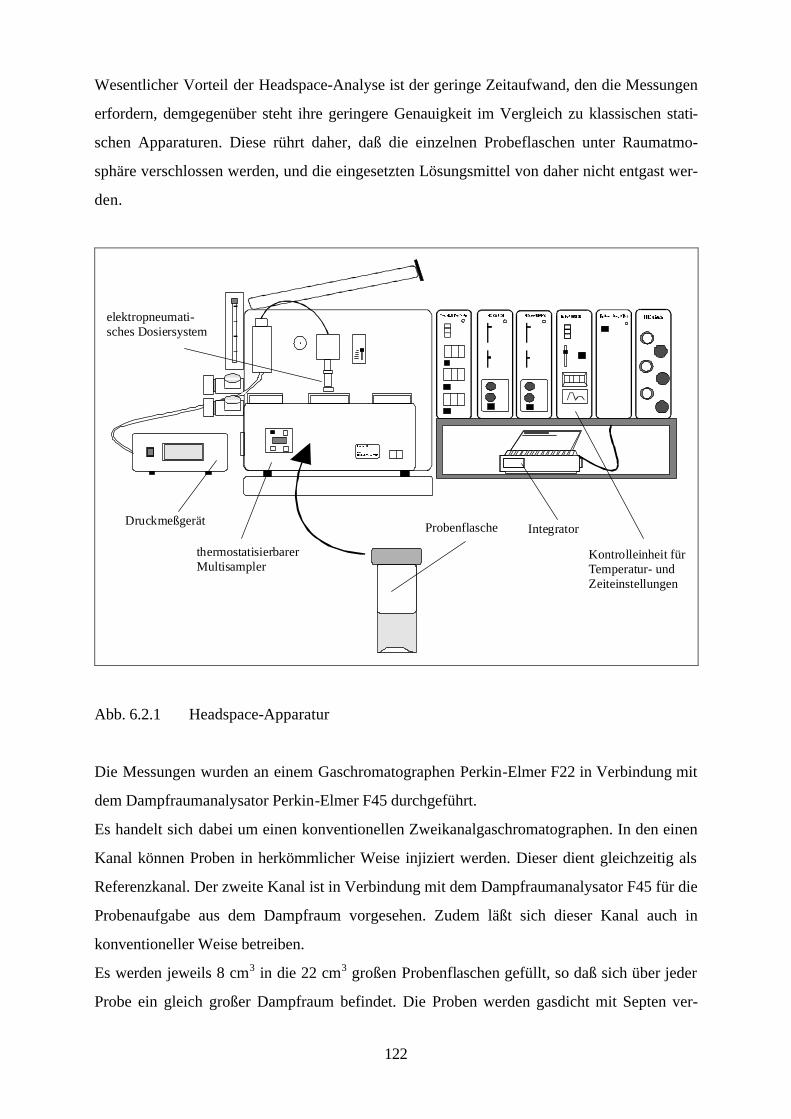
122
Wesentlicher Vorteil der Headspace-Analyse ist der geringe Zeitaufwand, den die Messungen
erfordern, demgegenüber steht ihre geringere Genauigkeit im Vergleich zu klassischen stati-
schen Apparaturen. Diese rührt daher, daß die einzelnen Probeflaschen unter Raumatmo-
sphäre verschlossen werden, und die eingesetzten Lösungsmittel von daher nicht entgast wer-
den.
elektropneumati-sches Dosiersystem
thermostatisierbarerMultisampler
Druckmeßgerät Probenflasche Integrator
Kontrolleinheit fürTemperatur- undZeiteinstellungen
Abb. 6.2.1 Headspace-Apparatur
Die Messungen wurden an einem Gaschromatographen Perkin-Elmer F22 in Verbindung mit
dem Dampfraumanalysator Perkin-Elmer F45 durchgeführt.
Es handelt sich dabei um einen konventionellen Zweikanalgaschromatographen. In den einen
Kanal können Proben in herkömmlicher Weise injiziert werden. Dieser dient gleichzeitig als
Referenzkanal. Der zweite Kanal ist in Verbindung mit dem Dampfraumanalysator F45 für die
Probenaufgabe aus dem Dampfraum vorgesehen. Zudem läßt sich dieser Kanal auch in
konventioneller Weise betreiben.
Es werden jeweils 8 cm3 in die 22 cm3 großen Probenflaschen gefüllt, so daß sich über jeder
Probe ein gleich großer Dampfraum befindet. Die Proben werden gasdicht mit Septen ver-
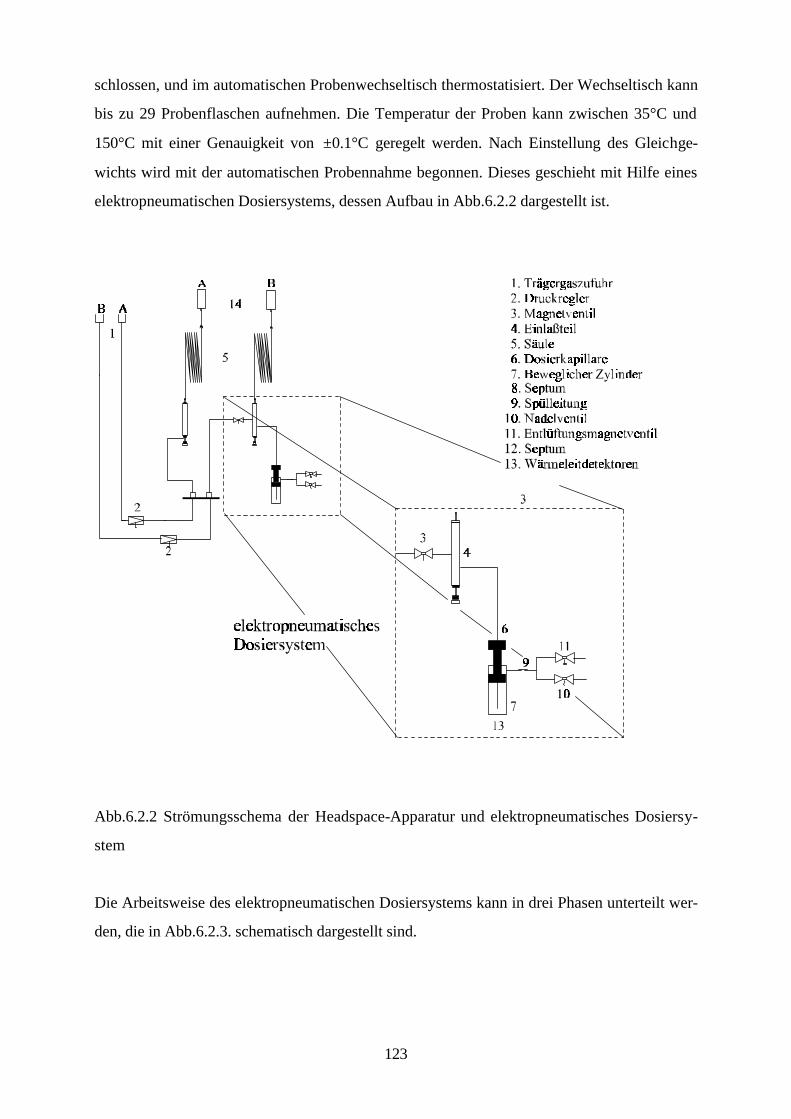
123
schlossen, und im automatischen Probenwechseltisch thermostatisiert. Der Wechseltisch kann
bis zu 29 Probenflaschen aufnehmen. Die Temperatur der Proben kann zwischen 35°C und
150°C mit einer Genauigkeit von ±0.1°C geregelt werden. Nach Einstellung des Gleichge-
wichts wird mit der automatischen Probennahme begonnen. Dieses geschieht mit Hilfe eines
elektropneumatischen Dosiersystems, dessen Aufbau in Abb.6.2.2 dargestellt ist.
Abb.6.2.2 Strömungsschema der Headspace-Apparatur und elektropneumatisches Dosiersy-
stem
Die Arbeitsweise des elektropneumatischen Dosiersystems kann in drei Phasen unterteilt wer-
den, die in Abb.6.2.3. schematisch dargestellt sind.

124
Abb.6.2.3 Schematische Darstellung der Arbeitsphasen der Headspace-Apparatur
In der ersten Phase wird in der jeweiligen Probenflasche ein dem Säulenvordruck entspre-
chender Überdruck aufgebaut. Hierzu wird der gesamte Probenwechseltisch hochgefahren
und die Injektionsnadel durchsticht die Septen. In der zweiten Phase erfolgt die Probennahme.
Der Trägergasstrom wird mittels eines Magnetventils unterbrochen, und der Überdruck in der
Pobenflasche entspannt sich durch die Nadel in die Analysensäule. Die Dauer dieser Entspan-
nung bestimmt die Menge an aufgegebener Probe. Sie wird über Schließzeit des Magnetventils
geregelt, das den Trägergasstrom unterbricht. Um während der Probenahme Kondensati-
onseffekte in der Injektionsnadel zu verhindern wurde die Nadel zum einem versilbert und ist
zum anderen beheizbar. Als dritte Phase erfolgt nun die gaschromatographische Analyse.
Nach Analysenende wird die Injektionsnadel mittels eines starken Trägergasstroms von
eventuellen Resten der Probe freigeblasen. Der Probentisch fährt weiter zur nächsten Probe.

125
6.3 Ebulliometrie
Die Ebulliometrie ist eine der wichtigsten dynamischen Meßmethoden. Die mit ihr zugängli-
chen Meßgrößen sind Siedetemperaturen und Dampfphasenzusammensetzung. Die in dieser
Arbeit verwendeten Ebulliometer sind nach Trampe und Eckert modifizierte Versionen des
Scott-Ebulliometers. Die vollständige Meßanordnung ist in Abb.( 6.3.1 ) dargestellt.
Abb.6.3.1 Aufbau der Ebulliometermeßapparatur
Die Vermessung isobarer Siedekurven erfordert die Kontrolle und Regelung des System-
drucks. Hierzu wird ein Manostat der Firma MKS Baratron Instruments verwendet. Es besteht
aus einem Drucksensor (Modell Baratron 390 HA), einem Druckregler (Baratron 250) zur
Vorgabe des gewünschten Systemdrucks und einem Steuergerät (Baratron 270), das die Ge-
schwindigkeit und das Ausmaß, mit welchen der Druckregler auf Druckschwankungen rea-
giert, vorgibt. An das System ist ein Vakuum angelegt, über das die Druckregulierung auto-
matisch mittels eines Nadelventils erfolgt. Die einzelnen Systemelemente sind durch Rohrlei-

126
tungen miteinander verbunden. Zur Erhöhung der Druckkonstanz sind zusätzlich zwei 5 dm3
Ballasttanks geschaltet. Der Systemdruck, wird so auf ± 0.2 mmHg konstant gehalten.
Die Siedetemperatur wird mit Hilfe eines Quartzthermometers (Modell Hewlett Packard 2804
A) gemessen. Bei Temperaturkalibration mit Hilfe einer Tripelpunktzelle ergab die Messung
der Tripelpunkttemperatur von Wasser einen Wert von ϑ = −0.016 °C und somit eine Abwei-
chung von −0.026 K gegenüber dem tatsächlichen Wert. Das verwendete Quarzthermometer
hat somit eine deutlich höhere absolute Temperaturgenauigkeit als Vergleichsthermometer. Da
es aber wesentlich genauer gelingt, relative als absolute Temperaturen zu bestimmen, wird statt
der absoluten Temperatur die Temperaturdifferenz zwischen beiden Ebulliometern gemessen.
Dabei enthält ein Ebulliometer die zu vermessende Mischung und das andere die salzfreie
Referenzmischung. Durch diese Vorgehensweise wird zudem der Einfluß von
Druckschwankungen minimiert.
Die maximale Auflösung der verwendeten Thermometer beträgt 0.0001°C. Angesichts der
begrenzten Genauigkeit der Druckregelung ist jedoch eine Auflösung von 0.001°C sinnvoll.
Dieses liegt im Bereich der von Swietoslawski angegebenen Genauigkeit der Siedetempera-
turbestimmung mit Hilfe von einfachen Ebulliometern [66].
Wesentliches Problem bei der Siedepunktbestimmung ist eine Überhitzung der flüssigen
Phase, die bis zum Auftreten von Siedeverzügen führen kann, zu vermeiden. Wesentliche
Gründe hierfür sind die hohe Temperatur an der Heizfläche, die Oberflächenspannung und der
hydrostatische Druck in der Flüssigkeit. Die beiden letzten Faktoren verhindern die Bildung
kleiner Dampfblasen an der Heizfläche und begünstigen somit eine Überhitzung der flüssigen
Phase [67].
Diese Effekte lassen sich durch schnelles Rühren teilweise ausgleichen. Dennoch ist der in der
flüssigen Phase bestimmte Siedepunkt nach wie vor zu hoch.

127
Abb.6.3.2 Schematischer Aufbau des Scott-Ebulliometers
Um zu einer exakten Siedepunktbestimmung zu gelangen, wurde im Swietoslawski-Ebullio-
meter ein Cottrell-Rohr verwendet [54]. Zu dessen Funktionsweise sei auf die Literatur
verwiesen [11]. Im Falle des Scott-Ebulliometers ist die Verwendung eines Cottrell-Rohres
nicht möglich. Statt dessen wird der Temperaturfühler so über der Phasengrenzfläche
Flüssigkeit-Dampf positioniert, daß die geringste Abweichung vom tatsächlichen Siedepunkt
zu erwarten ist. Der Vergleich der gemessenen Siedepunkte mit Reinstoffsiedepunkten aus der
Literatur ergab eine gute Übereinstimmung. In dem Referenzebulliometer ist es ausreichend,
das Thermometer mittels eines unten verschlossenen und mit Siliconöl zur besseren
Wärmeübertragung gefüllten Glasrohres einzubringen. Da durch Salzzugabe das Volumen der
flüssigen Phase im zweiten Ebulliometer sich ständig ändert, ist es notwendig die

128
Thermometerposition dem steigenden Flüssigkeitspegel ständig anzupassen. Hierzu wurde im
Rahmen dieser Arbeit das verwendete Quartz-Thermometer modifiziert. Das Thermometer
läßt sich höhenverstellen und ist durch eine Feststellschraube am Temperaturmeßpunkt fixier-
bar (s. Abb.6.3.2).
6.4 Reinheit der eingesetzten Substanzen
Für die Messungen wurden analysenreine Produkte der Firmen Fluka, Scharlau und Sigma-
Aldrich verwendet, außerdem bidestilliertes Wasser. Der zur Stabilisierung dienende Ethanol-
zusatz im Fall des Chloroforms und der noch eventuell vorhandene Restwassergehalt der Lö-
sungsmittel wurde durch Lagerung über Molsieb entfernt. Die eingesetzten Salze wurden im
Vakuumtrockenschrank bei 110°C getrocknet. Eine gaschromatographische Bestimmung
noch vorhandener Verunreinigungen in den Lösungsmitteln ergab einen Reinheitsgrad von >
99.9%, so daß eine weitere Aufbereitung nicht notwendig war. Als mobile Phase bei der
gaschromatographischen Analyse diente entweder Helium oder Stickstoff der Reinheit 5.0.
6.5 Analytische Bestimmung der Phasenzusammensetzung
Die Zusammensetzung der flüssigen Phase geschah an Hand der Einwaage. Die analytische
Bestimmung der Dampfphasenzusammensetzung geschah sowohl bei den Headspace- als
auch bei den ebulliometrischen Messungen gaschromatographisch. Dabei wurden zwei unter-
schiedliche Geräte benutzt. Für Headspace-Messungen wurde ausschließlich ein Gaschroma-
tograph F22 der Firma Perkin-Elmer mit Wärmeleitfähigkeitsdetektor (TCD) verwendet. Die-
ses Gerät arbeitet mit 1,5 m langen, gepackten Stahlsäulen die einen Durchmesser von 2 mm
haben. Die Auswertung des Signals geschah mit Hilfe eines Integrators vom Typ 3390A der
Firma Hewlett Packard.
Die Ebulliometerdaten wurden teilweise mit einem Gaschromatographen vom Typ 6890 der
Firma Hewlett Packard mit Flammenionisationsdetektor (FID) gemessen. Die verwendete
Kapillarsäule vom Typ HP-Innowax ist 30 m lang und hat einen Durchmesser von 0.25 mm.
Das Signal wurde in diesem Fall mit einer Chemstation ausgewertet. Für die unterschiedlichen
Trennprobleme war es erforderlich, die Analysen mit unterschiedlichen

129
Geräteparametern durchzuführen. Folgende Parameter wurden für die vermessenen Systeme
verwendet:
2-Propanol
Wasser
THF
Ethanol
Ethanol
1-Butanol
Chlorform
Ethanol
Aceton
Methanol
Methanol
2-Propanol
Gaschromatograph F 22 F 22 F 22 F 22 HP 6890 F 22
Detektor TCD TCD TCD TCD FID TCD
Säule gepackt gepackt gepackt gepackt kapillar gepackt
stationäre Phase Porapak T® Porapak T® Porapak Q® Porapak T® Polyethylen Porapak T®
mobile Phase He 5.0 He 5.0 He 5.0 He 5.0 N2 He 5.0
Vordruck 1.9 bar 1.975 bar 1.8 bar 1.975 bar 1.14 1.8 bar
Ofentemperatur 145°C 170°C 150°C 170°C 70°C 165°C
Tab.6.5.1 Geräteparameter für gaschromatographische Messungen
6.6 Kalibrierung
Die Größe des von einem Wärmeleitfähigkeitsdetektor erzeugten Signals wird u.a. durch die
spezifische Wärmekapazität der detektierten Substanz bedingt. Das bedeutet, daß der Detektor
nicht für jede Komponente gleich empfindlich ist. Desweiteren besteht kein linearer Zusam-
menhang zwischen der Signalstärke und der absoluten Menge an injizierter Substanz, weshalb
auch Flammenionisationsdetektoren keine strenge Linearität zeigen. Aus diesen beiden Grün-
den ist es nicht möglich die Zusammensetzung der Dampfphase direkt aus den Peakflächen
der Integration abzulesen, und die Anlage muß für jedes untersuchte Lösungsmittelgemisch
kalibriert werden. Erfahrungen haben gezeigt, daß es zwingend notwendig ist die Kalibrierung
und Messung mit derselben Säule durchzuführen, da es nicht möglich ist die exakten physika-
lischen Abmessungen einer GC-Säule zu kopieren. Für die Kalibrierung werden flüssige Pro-
ben definierter Zusammensetzung des salzfreien Lösungsmittelgemisches direkt auf die Säule
gegeben. Wegen der erwähnten nichtlinearen Abhängigkeit der Signalstärke von der absoluten
Menge an eingespritzter Substanz muß das Probevolumen bei der Kalibrierung dem Probevo-
lumen bei der Messung möglichst nah kommen. Für jedes untersuchte System wurde die An-
lage auf mindestens 10 äquidistante Punkte im Molenbruchbereich zwischen 0 und 1 kalibriert.
Jeder Messung wurde dreimal wiederholt und der Mittelwert als wahrer Wert angenommen.
Die Kalibrationskurven wurden erhalten, in dem für jeweils eine der beiden Ge-
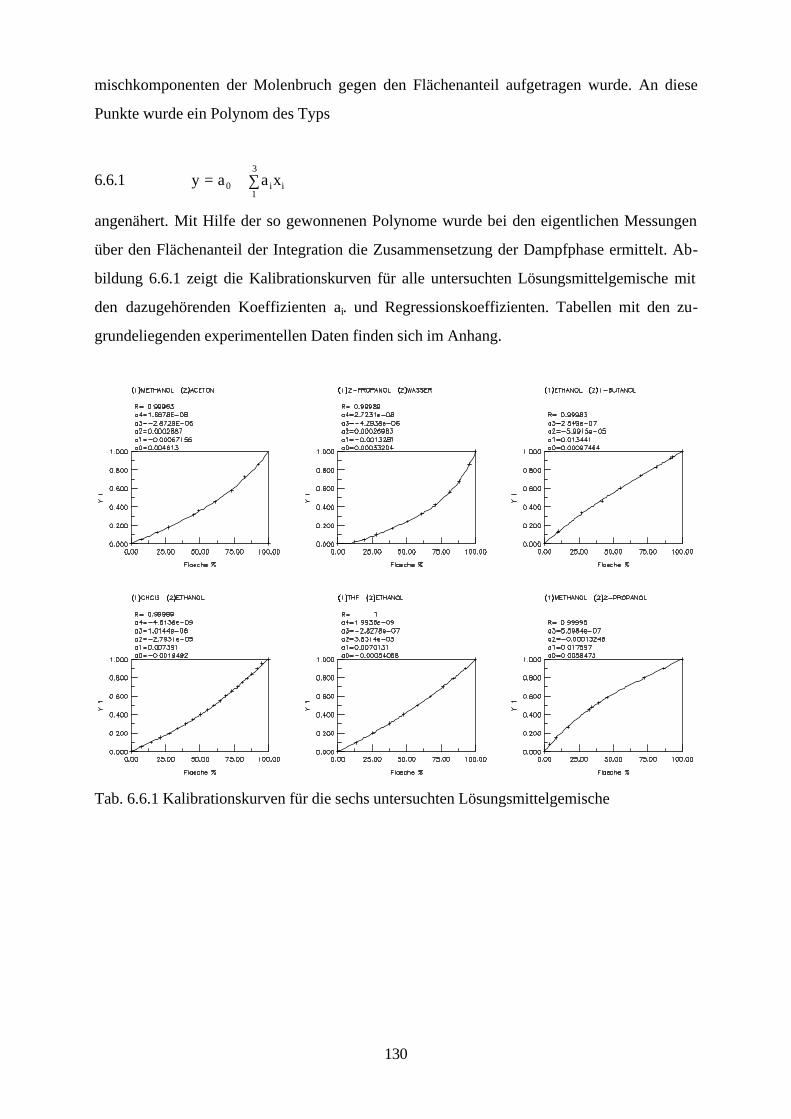
130
mischkomponenten der Molenbruch gegen den Flächenanteil aufgetragen wurde. An diese
Punkte wurde ein Polynom des Typs
6.6.1 y a a xi i= + ∑01
3
angenähert. Mit Hilfe der so gewonnenen Polynome wurde bei den eigentlichen Messungen
über den Flächenanteil der Integration die Zusammensetzung der Dampfphase ermittelt. Ab-
bildung 6.6.1 zeigt die Kalibrationskurven für alle untersuchten Lösungsmittelgemische mit
den dazugehörenden Koeffizienten ai. und Regressionskoeffizienten. Tabellen mit den zu-
grundeliegenden experimentellen Daten finden sich im Anhang.
Tab. 6.6.1 Kalibrationskurven für die sechs untersuchten Lösungsmittelgemische

131
6.7 Überprüfung der Daten auf thermodynamische Konsistenz
Thermodynamische Konsistenz ist eine notwendige aber nicht hinreichende Bedingung für die
Richtigkeit experimenteller Daten. Auf Grundlage der Gibbs-Duhem-Gleichung wurden
Konsistenztests entwickelt, die zur Beurteilung experimenteller Phasengleichgewichtsdaten
binärer Nichtelektrolytsysteme herangezogen werden können. Wie auch bei isopiestrischen
Messungen wird dabei die Tatsache ausgenutzt, daß die chemischen Potentiale der Kompo-
nenten eines Systems nicht unabhängig voneinander sind. Im binären Fall ist es bei Kenntnis
der Aktivität einer Komponente deshalb möglich die Aktivität der anderen zu berechnen. Für
Elektrolytsysteme existiert bis heute kein thermodynamisch exakter Konsistentest.
Ein Grund dafür ist, daß selbst die einfachste Mischung, bestehend aus einem Lösungsmittel
und einem Elektrolyten aufgrund dessen Dissoziation ein mindestens ternäres, gegebenenfalls
sogar polynäres System darstellt. Der von Jaques und Furter [25] vorgeschlagene
Konsistenztest für elektrolythaltige Systeme basiert auf einer pseudobinären Näherung.
Darüber hinaus ist er nur für auf gesättigte Systeme anwendbar.
Die Überprüfung der Konsistenz der im Rahmen dieser Arbeit gemessenen Daten kann sich
daher nur auf die elektrolytfreien Systeme (Kalibrationskurven) erstrecken. Grundlage soll der
von Redlich und Kister [48] vorgeschlagene Flächentest sein. Er beruht auf dem totalen
Differential der Gibbsschen Exzeßenthalpie als Funktion von Temperatur, Druck und Zusam-
mensetzung:
(6.7.1)
Durch verschiedene Substitutionen, den Übergang zu molaren Größen und anschließender
Integration über den gesamten Molenbruch, erhält man für ein binäres System bei der Be-
rücksichtigung von dx1 = -dx2 die folgende Gleichung:
(6.7.2)
dGRT
GRT
PdP
GRT
TdT
GRTn
dnE
E
T n
E
P n
E
i
T P n
i
i i j
=
+
+
∑∂
∂
∂
∂
∂
∂
, , , ,
∂ γγ
∂ ∂gRT
dxh
RTdT
vRT
dPE
x
x
x
x E
x
x E
x
x
= − + ==
=
=
=
=
=
=
=
∫ ∫ ∫ ∫1
1
1
1
1
1
1
1
0
11
21
0
1
20
1
0
1
0ln

132
Für isobare bzw. isotherme Meßreihen entfällt jeweils ein Term. Da sich der Druckeinfluß in
den meisten Fällen vernachlässigen läßt, vereinfacht sich Gl. 5.2. im isothermen Fall zu
(6.7.3)
Im isobaren Fall muß zudem die Exzeßenthalpie hE der Mischung berücksichtigt werden Diese
wird empirisch nach Herington abgeschätzt [74]. Das entsprechende Integral in Gl.5.3. wird
wie folgt angenähert:
(6.7.4)
wobei Tmin die minimale Siedetemperatur und ∆Tmax die maximale Differenz der Siedetempe-
raturen im gesamten Molenbruchbereich ist. Bei zeotropen Systemen entspricht dies der Dif-
ferenz der Siedepunkte der Reinstoffe.
Die Lösung der entsprechenden Gleichungen kann graphisch durch Auftragen des logarith-
mierten Verhältnisses der Aktivitätskoeffizienten gegen den Molenbruch der Komponente 1
erfolgen. Die relative Abweichung der Teilflächen dieses Integrals ober- und unterhalb der
Abzisse ist definiert als:
(6.7.5)
Nach Gl. 6.7.2 muß D gleich Null werden. Als Konsistenzkriterium für den isothermen Fall
soll gelten[75]:
(6.7.6)
Für den isobaren Fall muß Gl. 6.7.4 berücksichtigt werden. Als Konsistenzkriterium soll gel-
ten[75]:
(6.7.7)
lnγγ
1
21
0
1
1
1
0dxx
x
=
=
∫ =
JT
T= 150
∆ max
min
%
DA BA B
=−+
100 %
D ≤ 10 %
D J− ≤ 10%

133
Das Verhältnis der Aktivitätskoeffizienten läßt sich unter Annahme idealen Verhaltens der
Dampfphase und unter Vernachlässigung des Poynting-Faktors und der
Fugazitätskoeffizienten nach
(6.7.8)
und daraus folgend
(6.7.9)
ermitteln. Der Sättigungsdampfdruck wird mit Hilfe der Antoine-Gleichung berechnet:
(6.7.10)
Die jeweils verwendeten Antoine-Konstanten sind im Anhang angegeben. Die Ergebnisse für
die sechs untersuchten Lösungsmittelgemische sind in Abbildung 6.7.1 dargestellt. Die zu-
grundeliegenden Daten sind in Tabelle 6.7.1 zusammengefaßt. Obwohl für die isothermen
Datensätze der gaschromatographischen Dampfraumanalyse keine Daten über den jeweiligen
Systemdruck vorliegen, ist es möglich, auch für diese Daten einen Konsistenztest durchzufüh-
ren, da gemäß Gl. 6.7.9 nur die Sättigungsdampfdrücke der reinen Komponenten für die Be-
rechnung des Aktivitätskoeffizientenverhältnisses erforderlich sind.
γ igesamt i
is
i
P y
P x=
γγ
1
2
2 2 1
1 1 2
=x P yx P y
s
s
log P AB
T Cis
ii
i
= −+
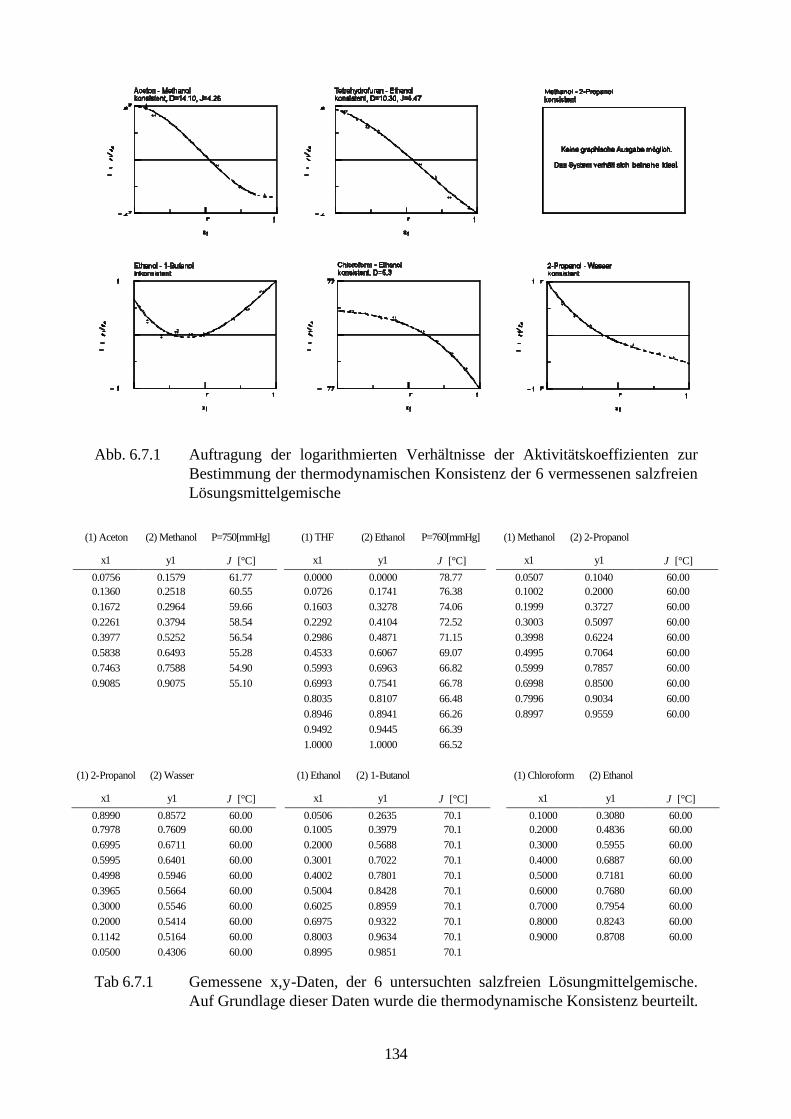
134
Abb. 6.7.1 Auftragung der logarithmierten Verhältnisse der Aktivitätskoeffizienten zur Bestimmung der thermodynamischen Konsistenz der 6 vermessenen salzfreien Lösungsmittelgemische
(1) Aceton (2) Methanol P=750[mmHg] (1) THF (2) Ethanol P=760[mmHg] (1) Methanol (2) 2-Propanol
x1 y1 ϑ [°C] x1 y1 ϑ [°C] x1 y1 ϑ [°C]
0.0756 0.1579 61.77 0.0000 0.0000 78.77 0.0507 0.1040 60.00 0.1360 0.2518 60.55 0.0726 0.1741 76.38 0.1002 0.2000 60.00 0.1672 0.2964 59.66 0.1603 0.3278 74.06 0.1999 0.3727 60.00 0.2261 0.3794 58.54 0.2292 0.4104 72.52 0.3003 0.5097 60.00 0.3977 0.5252 56.54 0.2986 0.4871 71.15 0.3998 0.6224 60.00 0.5838 0.6493 55.28 0.4533 0.6067 69.07 0.4995 0.7064 60.00 0.7463 0.7588 54.90 0.5993 0.6963 66.82 0.5999 0.7857 60.00 0.9085 0.9075 55.10 0.6993 0.7541 66.78 0.6998 0.8500 60.00
0.8035 0.8107 66.48 0.7996 0.9034 60.00 0.8946 0.8941 66.26 0.8997 0.9559 60.00 0.9492 0.9445 66.39 1.0000 1.0000 66.52
(1) 2-Propanol (2) Wasser (1) Ethanol (2) 1-Butanol (1) Chloroform (2) Ethanol
x1 y1 ϑ [°C] x1 y1 ϑ [°C] x1 y1 ϑ [°C]
0.8990 0.8572 60.00 0.0506 0.2635 70.1 0.1000 0.3080 60.00 0.7978 0.7609 60.00 0.1005 0.3979 70.1 0.2000 0.4836 60.00 0.6995 0.6711 60.00 0.2000 0.5688 70.1 0.3000 0.5955 60.00 0.5995 0.6401 60.00 0.3001 0.7022 70.1 0.4000 0.6887 60.00 0.4998 0.5946 60.00 0.4002 0.7801 70.1 0.5000 0.7181 60.00 0.3965 0.5664 60.00 0.5004 0.8428 70.1 0.6000 0.7680 60.00 0.3000 0.5546 60.00 0.6025 0.8959 70.1 0.7000 0.7954 60.00 0.2000 0.5414 60.00 0.6975 0.9322 70.1 0.8000 0.8243 60.00 0.1142 0.5164 60.00 0.8003 0.9634 70.1 0.9000 0.8708 60.00 0.0500 0.4306 60.00 0.8995 0.9851 70.1 Tab 6.7.1 Gemessene x,y-Daten, der 6 untersuchten salzfreien Lösungmittelgemische. Auf Grundlage dieser Daten wurde die thermodynamische Konsistenz beurteilt.
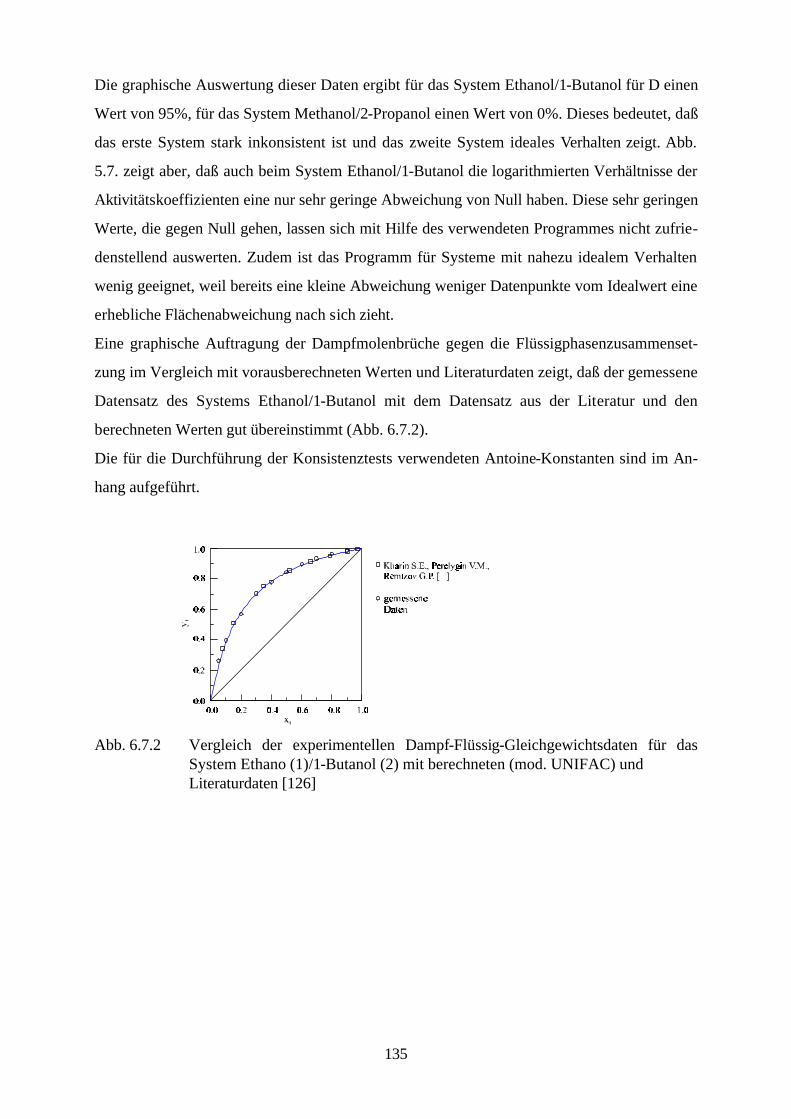
135
Die graphische Auswertung dieser Daten ergibt für das System Ethanol/1-Butanol für D einen
Wert von 95%, für das System Methanol/2-Propanol einen Wert von 0%. Dieses bedeutet, daß
das erste System stark inkonsistent ist und das zweite System ideales Verhalten zeigt. Abb.
5.7. zeigt aber, daß auch beim System Ethanol/1-Butanol die logarithmierten Verhältnisse der
Aktivitätskoeffizienten eine nur sehr geringe Abweichung von Null haben. Diese sehr geringen
Werte, die gegen Null gehen, lassen sich mit Hilfe des verwendeten Programmes nicht zufrie-
denstellend auswerten. Zudem ist das Programm für Systeme mit nahezu idealem Verhalten
wenig geeignet, weil bereits eine kleine Abweichung weniger Datenpunkte vom Idealwert eine
erhebliche Flächenabweichung nach sich zieht.
Eine graphische Auftragung der Dampfmolenbrüche gegen die Flüssigphasenzusammenset-
zung im Vergleich mit vorausberechneten Werten und Literaturdaten zeigt, daß der gemessene
Datensatz des Systems Ethanol/1-Butanol mit dem Datensatz aus der Literatur und den
berechneten Werten gut übereinstimmt (Abb. 6.7.2).
Die für die Durchführung der Konsistenztests verwendeten Antoine-Konstanten sind im An-
hang aufgeführt.
Abb. 6.7.2 Vergleich der experimentellen Dampf-Flüssig-Gleichgewichtsdaten für das System Ethano (1)/1-Butanol (2) mit berechneten (mod. UNIFAC) und Literaturdaten [126]
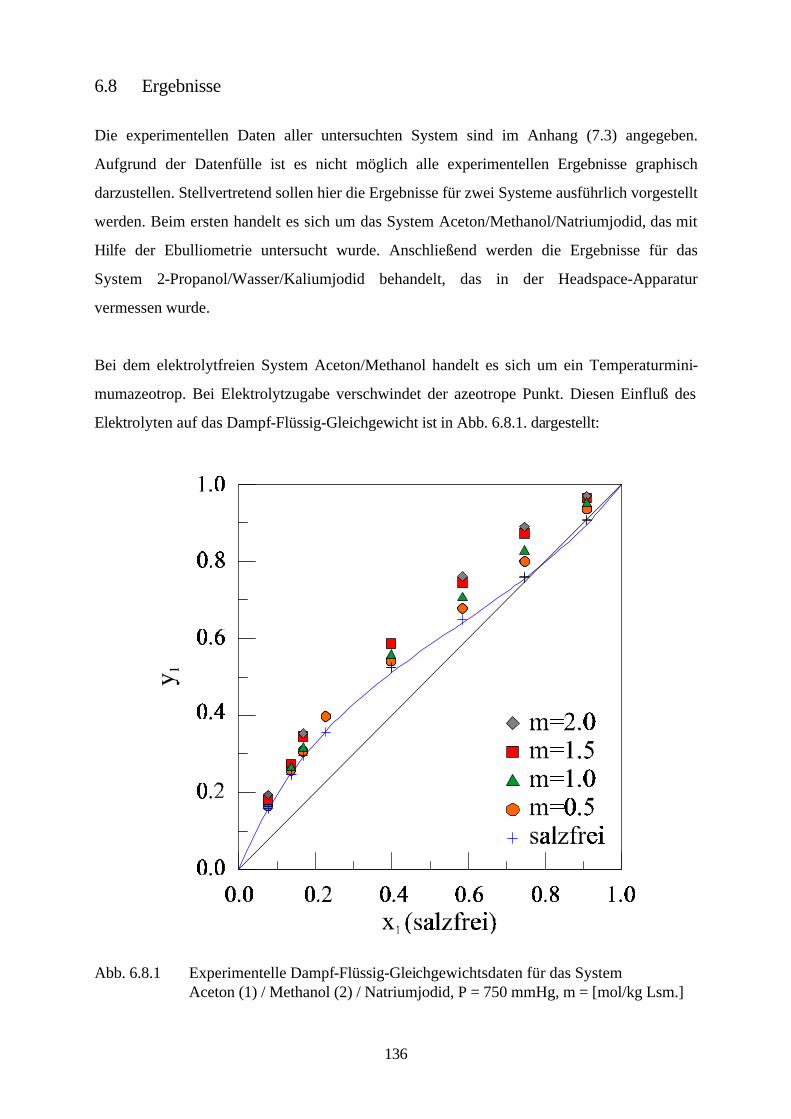
136
6.8 Ergebnisse
Die experimentellen Daten aller untersuchten System sind im Anhang (7.3) angegeben.
Aufgrund der Datenfülle ist es nicht möglich alle experimentellen Ergebnisse graphisch
darzustellen. Stellvertretend sollen hier die Ergebnisse für zwei Systeme ausführlich vorgestellt
werden. Beim ersten handelt es sich um das System Aceton/Methanol/Natriumjodid, das mit
Hilfe der Ebulliometrie untersucht wurde. Anschließend werden die Ergebnisse für das
System 2-Propanol/Wasser/Kaliumjodid behandelt, das in der Headspace-Apparatur
vermessen wurde.
Bei dem elektrolytfreien System Aceton/Methanol handelt es sich um ein Temperaturmini-
mumazeotrop. Bei Elektrolytzugabe verschwindet der azeotrope Punkt. Diesen Einfluß des
Elektrolyten auf das Dampf-Flüssig-Gleichgewicht ist in Abb. 6.8.1. dargestellt:
Abb. 6.8.1 Experimentelle Dampf-Flüssig-Gleichgewichtsdaten für das System Aceton (1) / Methanol (2) / Natriumjodid, P = 750 mmHg, m = [mol/kg Lsm.]

137
Es zeigt sich mit steigender Elektrolytkonzentration eine Anreicherung des Acetons in der
Dampfphase. Es liegt somit ein „Aussalzen“ der weniger polaren Lösungsmittelkomponente
vor. Die Wirkung des Elektrolyten äußert sich auch in der Veränderung des Trennfaktors, der
entsprechend Gl. 6.8.1 definiert ist. Der Trennfaktor wird auch als relative Flüchtigkeit be-
zeichnet.
(6.8.1)
Hat der Trennfaktor einen Wert von αi,j = 1, so liegt ein azeotroper Punkt vor. Die Änderung
des Trennfaktors mit steigender Elektrolytkonzentration ist in Tab. 6.8.1 dargestellt.
x1(s) 0.0756 0.136 0.1651 0.39773 0.5838 0.7463 0.9085
α (m = 0.0) 2.2924 2.1125 2.0981 1.6752 1.3198 1.0692 0.9877
α (m = 0.5) 2.4173 2.1912 2.2057 1.7883 1.501 1.3681 1.4929
α (m = 1.0) 2.493 2.283 2.2813 1.8916 1.7109 1.6164 1.9473
α (m = 1.5) 2.6451 2.4539 2.0253 1.9127 2.0096 2.5072
α (m = 2.0) 2.7069 2.3814 2.6421 2.1483 2.0816 2.3473 2.6562
α (m = 2.5) 2.8578 2.688 2.1876 2.2095 2.5184 3.206
α (m = 3.0) 2.9432 2.7161 2.2615 2.7563 3.1652
α (m = 4.0) 3.0209 2.7932 2.3554 2.5473
α (m = 5.0) 3.0452 2.7908 2.3582
Tab. 6.8.1 Trennfaktoren α1,2 für verschiedene elektrolytfreie Lösungsmittelmolenbrüche
x1 in Abhängigkeit von der Elektrolytkonzentration m [mol/kg Lsm.]
Es zeigt sich vor allem für den salzfreien Molenbruch x1 = 0.7463, der sehr nahe am azeotro-
pen Punkt liegt, daß der Trennfaktor mit steigender Elektrolytkonzentration immer stärker von
1 abweicht und der azeotrope Punkt verschwindet.
In Abb. 6.8.2 ist der Einfluß des Elektrolyten auf die Siedetemperatur der Mischung darge-
stellt. Wie zu erwarten zeigt sich, eine Siedepunkterhöhung mit steigender
Elektrolytkonzentration bis zum Erreichen der Sättigungsgrenze. Nach Erreichen der Sät-
tigungsgrenze ist wie erwartet keine weitere Erhöhung der Siedetemperaturen mehr zu beob-
achten.
α i ji i
j j
y xy x, =
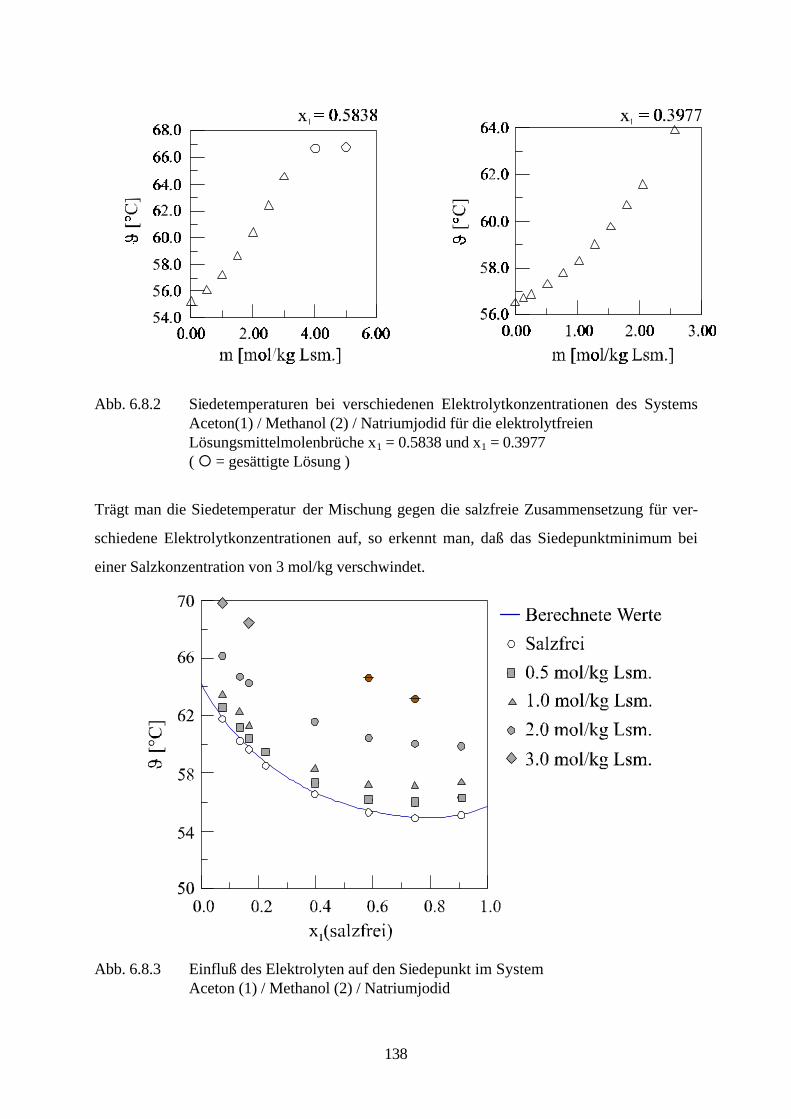
138
Abb. 6.8.2 Siedetemperaturen bei verschiedenen Elektrolytkonzentrationen des Systems Aceton(1) / Methanol (2) / Natriumjodid für die elektrolytfreien Lösungsmittelmolenbrüche x1 = 0.5838 und x1 = 0.3977 ( ¡ = gesättigte Lösung )
Trägt man die Siedetemperatur der Mischung gegen die salzfreie Zusammensetzung für ver-
schiedene Elektrolytkonzentrationen auf, so erkennt man, daß das Siedepunktminimum bei
einer Salzkonzentration von 3 mol/kg verschwindet.
Abb. 6.8.3 Einfluß des Elektrolyten auf den Siedepunkt im System Aceton (1) / Methanol (2) / Natriumjodid
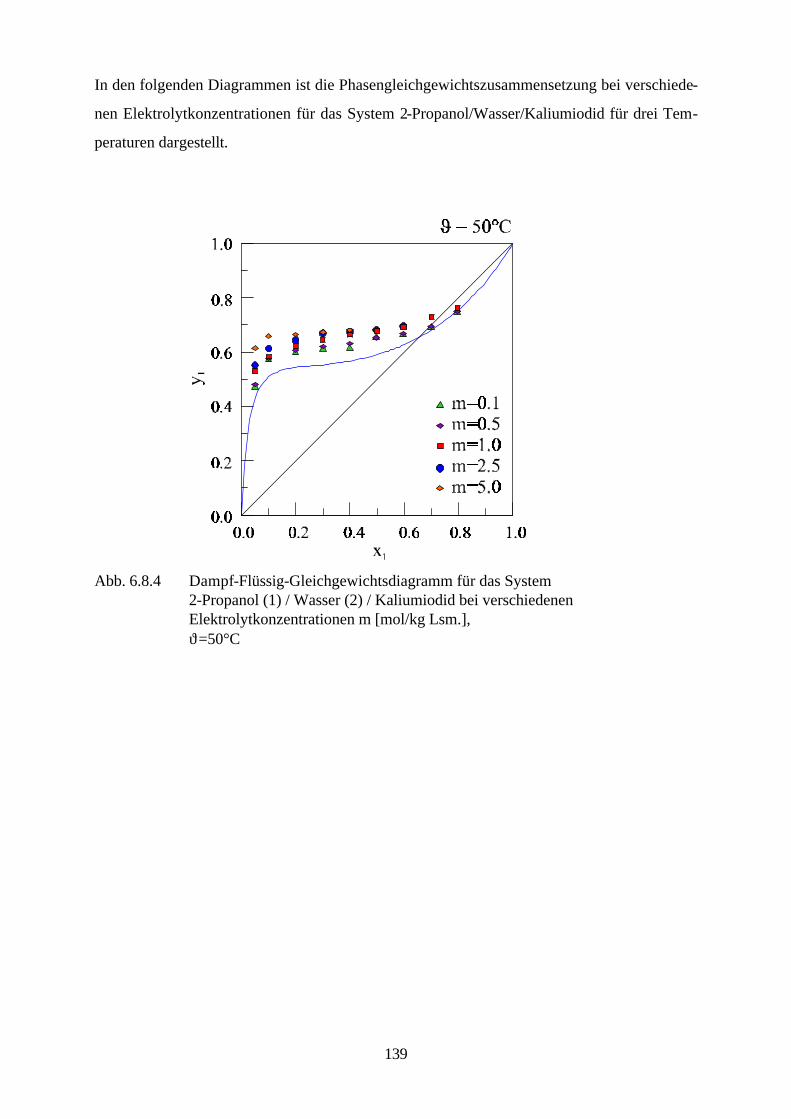
139
In den folgenden Diagrammen ist die Phasengleichgewichtszusammensetzung bei verschiede-
nen Elektrolytkonzentrationen für das System 2-Propanol/Wasser/Kaliumiodid für drei Tem-
peraturen dargestellt.
Abb. 6.8.4 Dampf-Flüssig-Gleichgewichtsdiagramm für das System 2-Propanol (1) / Wasser (2) / Kaliumiodid bei verschiedenen Elektrolytkonzentrationen m [mol/kg Lsm.], ϑ=50°C
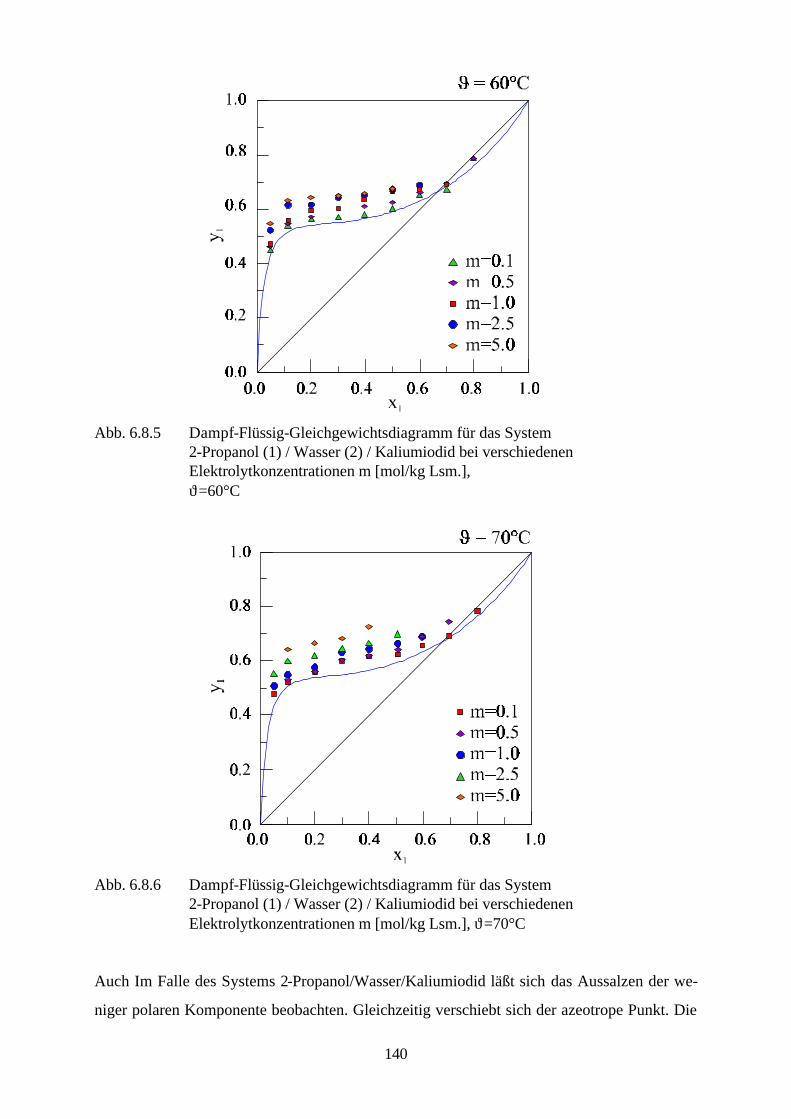
140
Abb. 6.8.5 Dampf-Flüssig-Gleichgewichtsdiagramm für das System 2-Propanol (1) / Wasser (2) / Kaliumiodid bei verschiedenen Elektrolytkonzentrationen m [mol/kg Lsm.], ϑ=60°C
Abb. 6.8.6 Dampf-Flüssig-Gleichgewichtsdiagramm für das System 2-Propanol (1) / Wasser (2) / Kaliumiodid bei verschiedenen Elektrolytkonzentrationen m [mol/kg Lsm.], ϑ=70°C Auch Im Falle des Systems 2-Propanol/Wasser/Kaliumiodid läßt sich das Aussalzen der we-
niger polaren Komponente beobachten. Gleichzeitig verschiebt sich der azeotrope Punkt. Die

141
Zugabe des Elektrolyten hat auch in diesem System deutlichen Einfluß auf die Trennfaktoren.
In Tab. 6.8.2 sind die Trennfaktoren bei verschiedenen Elektrolytkonzentrationen für ϑ = 70°C
zusammengestellt
x1(s) 0.8987 0.799 0.6931 0.5957 0.5053 0.3995 0.2996 0.1999 0.1001 0.05 α (m = 0.01) 0.7137 0.8128 0.9847 1.1884 1.4859 2.0626 2.9549 4.6751 9.5474 15.9186
α (m =
0.025) 0.7275 0.8279 1.0002 1.1924 1.4946 2.1026 3.0645 4.8207 9.4079 15.9497
α (m = 0.05) 0.7749 0.8612 1.073 1.2055 1.5183 2.1411 3.1444 4.8636 9.6811 16.0077 α (m = 0.1) 0.9121 0.9977 1.2903 1.6201 2.3949 3.4905 5.0673 9.7661 17.2818 α (m = 0.5) 1.2897 1.5063 1.735 2.4345 3.5328 5.0817 10.0393 17.5569 α (m = 1.0) 1.5025 1.9272 2.7076 3.9922 5.4088 10.9119 19.4955 α (m = 2.5) 2.2251 2.9216 4.1781 6.4536 13.1796 22.9933 α (m = 5.0) 3.9388 4.9918 7.9138 16.1495
Tab. 6.8.2 Trennfaktoren α1,2 für verschiedene elektrolytfreie Lösungsmittelmolenbrüche x1(s) in Abhängigkeit von der Elektrolytkonzentration m [mol/kg Lsm.] für das System 2-Propanol (1) / Wasser (2) / Kaliumjodid
6.9 Fehlerbetrachtung
Es ist bei den experimentell bestimmten Meßgrößen x, y, T und P zwischen systematischen
und Zufallsfehlern zu unterscheiden, wobei letztere sich nach mehrmaliger Bestimmung der
jeweiligen Meßgröße mittels einer Fehler- und Ausgleichrechnung ermitteln lassen. Im fol-
genden soll eine Fehlerabschätzung durchgeführt werden.
a) Bestimmung der Dampf- und Flüssigphasenzusammensetzung
Zur Bestimmung der Zusammensetzung der Flüssigphase wird auf die eingewogenen Sub-
stanzen zurückgegriffen. Als wesentliche Fehlerquellen sind hierbei die Ungenauigkeit der
Waage (± 0.01g) und teilweises Verdampfen der Lösungsmittel zu nennen. Dieses wird durch
Kühlung der Substanzen und schnelles Einwiegen minimiert. Der Fehler der Flüssigphasenzu-
sammensetzung gegenüber der Einwaage ist bedingt durch das relativ große Füllvolumen und
das geringe Dampfraumvolumen im Ebulliometer und auch in den Probenflaschen der Head-
space-Apparatur gering. Eine rechnerische Bestimmung und damit einer Korrektur dieses
Fehlers ist allerdings nicht möglich, da der Einfluß des Elektrolyten auf das Phasengleichge-

142
wicht und die Exzeßvolumina der Lösungsmittel mit den bisherigen Verfahren nicht erfaßbar
ist, und das Volumen des gelösten Salzes unbekannt ist. Bei der Bestimmung der Dampfphasenzusammensetzung sind als Fehlerquellen vor allem die
Probennahme und die gaschromatographische Analyse zu nennen. Bei der gaschromatogra-
phischen Analyse muß, auch Herstellerangaben zufolge mit einem relativen Fehler von 1
mol% gerechnet werden. Hinzu kommen Fehler durch die Probeninjektion und Probleme bei
der Auswertung der Detektorsignale. Besonders bei den polaren Lösungsmittelkomponenten
(Wasser, Methanol, Ethanol) ist die Reproduzierbarkeit der Flächenanteile schwierig. Somit ist
bei Proben mit einem hohem Anteil der polaren Komponente mit einem größeren Fehler zu
rechnen, der sich durch mehrmalige Analyse und anschließender Mittelwertbildung ausglei-
chen läßt.
b) Bestimmung der Elektrolytkonzentration
Für die Ermittlung der Elektrolytkonzentration wird auf die Einwaage zurückgegriffen. Es
treten auch hier die bereits erwähnten Fehlerquellen auf.
c) Temperaturmessung
Zur Ermittlung der Temperatur des automatischen Probennehmers der Headspace-Apparatur
wird ein Thermometer mit einer Ablesegenauigkeit von maximal 0.1°C verwendet. Die effek-
tive Temperatur in den Probenflaschen läßt sich allerdings nicht ermitteln.
Bei den Ebulliometermessungen treten die bereits erwähnten Probleme der Temperaturmes-
sung auf. Die verwendeten Quarzthermometer zeigen eine deutlich höhere Genauigkeit bei der
Messung von Temperaturdifferenzen als bei der absoluten Temperaturbestimmung, weshalb
eine Referenzmeßzelle verwendet wird. Hinzu kommt die vorherige Eichung mittels einer
Tripelpunktzelle.
d) Druckmessung
Wesentlicher Nachteil der gaschromatographischen Dampfraumanalyse ist, daß es nicht mög-
lich ist, Aussagen über den in den Probenflaschen tatsächlich herrschenden Druck zu machen.

143
Die Druckänderung, die bei der Probennahme in der Probenflasche auftritt, läßt sich entspre-
chend nicht abschätzen.
Dem gegenüber läßt sich bei der Ebulliometerapparatur der Druck genau bestimmen. Die Ab-
lesegenauigkeit beträgt 0.1mmHg. Nach Herstellerangaben beträgt die Standardabweichung
der Druckmessung 0.08% (nach Kalibration mit einer Absolutdruckwaage der Firma Des-
granges&Huot). Der Fehler erhöht sich aber durch die Druckkorrekturen des Manostaten, die
zu periodischen Schwankungen in einer Größenordnung von ±0.2 mmHg liegen.
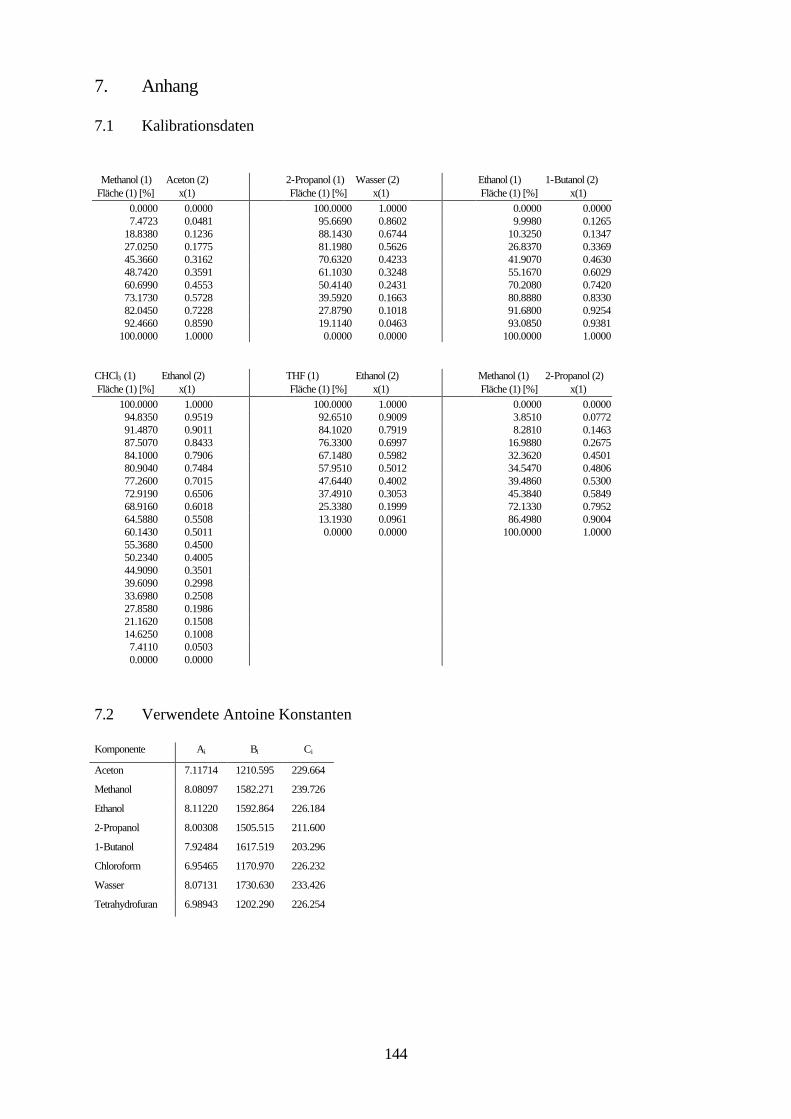
144
7. Anhang 7.1 Kalibrationsdaten
Methanol (1) Aceton (2) 2-Propanol (1) Wasser (2) Ethanol (1) 1-Butanol (2) Fläche (1) [%] x(1) Fläche (1) [%] x(1) Fläche (1) [%] x(1)
0.0000 0.0000 100.0000 1.0000 0.0000 0.0000 7.4723 0.0481 95.6690 0.8602 9.9980 0.1265
18.8380 0.1236 88.1430 0.6744 10.3250 0.1347 27.0250 0.1775 81.1980 0.5626 26.8370 0.3369 45.3660 0.3162 70.6320 0.4233 41.9070 0.4630 48.7420 0.3591 61.1030 0.3248 55.1670 0.6029 60.6990 0.4553 50.4140 0.2431 70.2080 0.7420 73.1730 0.5728 39.5920 0.1663 80.8880 0.8330 82.0450 0.7228 27.8790 0.1018 91.6800 0.9254 92.4660 0.8590 19.1140 0.0463 93.0850 0.9381
100.0000 1.0000 0.0000 0.0000 100.0000 1.0000
CHCl3 (1) Ethanol (2) THF (1) Ethanol (2) Methanol (1) 2-Propanol (2) Fläche (1) [%] x(1) Fläche (1) [%] x(1) Fläche (1) [%] x(1)
100.0000 1.0000 100.0000 1.0000 0.0000 0.0000 94.8350 0.9519 92.6510 0.9009 3.8510 0.0772 91.4870 0.9011 84.1020 0.7919 8.2810 0.1463 87.5070 0.8433 76.3300 0.6997 16.9880 0.2675 84.1000 0.7906 67.1480 0.5982 32.3620 0.4501 80.9040 0.7484 57.9510 0.5012 34.5470 0.4806 77.2600 0.7015 47.6440 0.4002 39.4860 0.5300 72.9190 0.6506 37.4910 0.3053 45.3840 0.5849 68.9160 0.6018 25.3380 0.1999 72.1330 0.7952 64.5880 0.5508 13.1930 0.0961 86.4980 0.9004 60.1430 0.5011 0.0000 0.0000 100.0000 1.0000 55.3680 0.4500 50.2340 0.4005 44.9090 0.3501 39.6090 0.2998 33.6980 0.2508 27.8580 0.1986 21.1620 0.1508 14.6250 0.1008 7.4110 0.0503 0.0000 0.0000
7.2 Verwendete Antoine Konstanten Komponente Ai Bi Ci
Aceton 7.11714 1210.595 229.664
Methanol 8.08097 1582.271 239.726
Ethanol 8.11220 1592.864 226.184
2-Propanol 8.00308 1505.515 211.600
1-Butanol 7.92484 1617.519 203.296
Chloroform 6.95465 1170.970 226.232
Wasser 8.07131 1730.630 233.426
Tetrahydrofuran 6.98943 1202.290 226.254
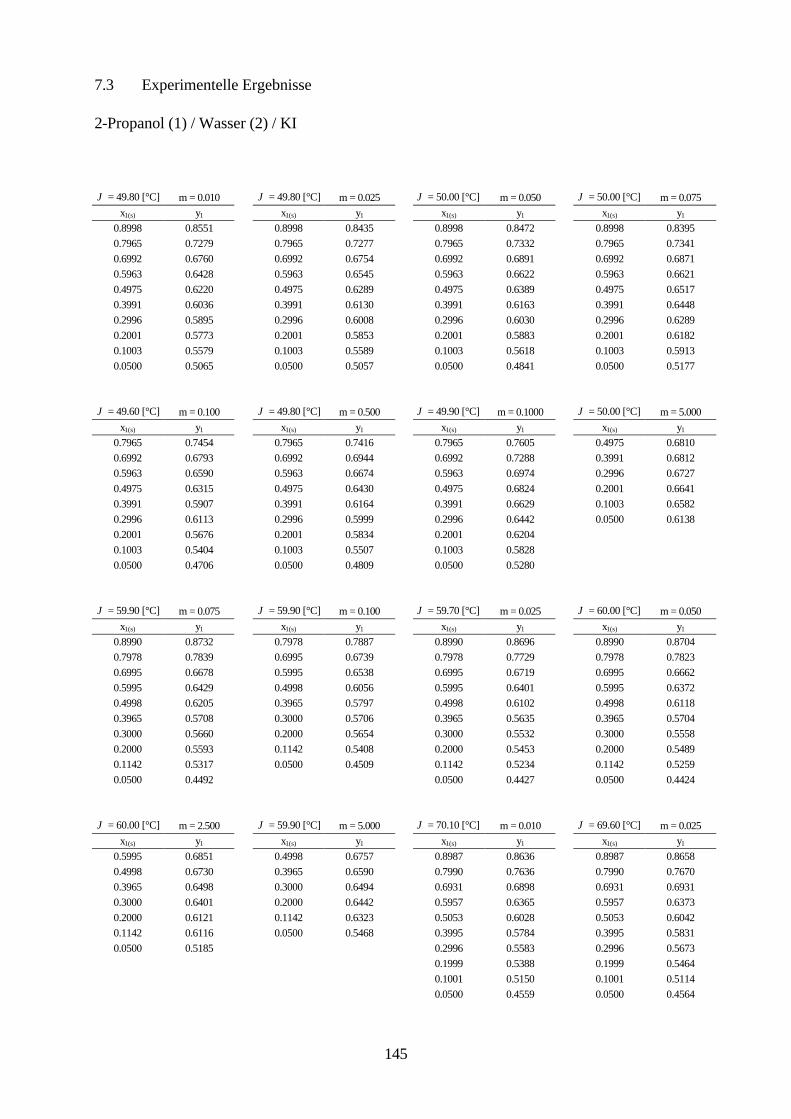
145
7.3 Experimentelle Ergebnisse 2-Propanol (1) / Wasser (2) / KI ϑ = 49.80 [°C] m = 0.010 ϑ = 49.80 [°C] m = 0.025 ϑ = 50.00 [°C] m = 0.050 ϑ = 50.00 [°C] m = 0.075
x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1 0.8998 0.8551 0.8998 0.8435 0.8998 0.8472 0.8998 0.8395 0.7965 0.7279 0.7965 0.7277 0.7965 0.7332 0.7965 0.7341 0.6992 0.6760 0.6992 0.6754 0.6992 0.6891 0.6992 0.6871 0.5963 0.6428 0.5963 0.6545 0.5963 0.6622 0.5963 0.6621 0.4975 0.6220 0.4975 0.6289 0.4975 0.6389 0.4975 0.6517 0.3991 0.6036 0.3991 0.6130 0.3991 0.6163 0.3991 0.6448 0.2996 0.5895 0.2996 0.6008 0.2996 0.6030 0.2996 0.6289 0.2001 0.5773 0.2001 0.5853 0.2001 0.5883 0.2001 0.6182 0.1003 0.5579 0.1003 0.5589 0.1003 0.5618 0.1003 0.5913 0.0500 0.5065 0.0500 0.5057 0.0500 0.4841 0.0500 0.5177
ϑ = 49.60 [°C] m = 0.100 ϑ = 49.80 [°C] m = 0.500 ϑ = 49.90 [°C] m = 0.1000 ϑ = 50.00 [°C] m = 5.000 x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1
0.7965 0.7454 0.7965 0.7416 0.7965 0.7605 0.4975 0.6810 0.6992 0.6793 0.6992 0.6944 0.6992 0.7288 0.3991 0.6812 0.5963 0.6590 0.5963 0.6674 0.5963 0.6974 0.2996 0.6727 0.4975 0.6315 0.4975 0.6430 0.4975 0.6824 0.2001 0.6641 0.3991 0.5907 0.3991 0.6164 0.3991 0.6629 0.1003 0.6582 0.2996 0.6113 0.2996 0.5999 0.2996 0.6442 0.0500 0.6138 0.2001 0.5676 0.2001 0.5834 0.2001 0.6204 0.1003 0.5404 0.1003 0.5507 0.1003 0.5828 0.0500 0.4706 0.0500 0.4809 0.0500 0.5280
ϑ = 59.90 [°C] m = 0.075 ϑ = 59.90 [°C] m = 0.100 ϑ = 59.70 [°C] m = 0.025 ϑ = 60.00 [°C] m = 0.050 x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1
0.8990 0.8732 0.7978 0.7887 0.8990 0.8696 0.8990 0.8704 0.7978 0.7839 0.6995 0.6739 0.7978 0.7729 0.7978 0.7823 0.6995 0.6678 0.5995 0.6538 0.6995 0.6719 0.6995 0.6662 0.5995 0.6429 0.4998 0.6056 0.5995 0.6401 0.5995 0.6372 0.4998 0.6205 0.3965 0.5797 0.4998 0.6102 0.4998 0.6118 0.3965 0.5708 0.3000 0.5706 0.3965 0.5635 0.3965 0.5704 0.3000 0.5660 0.2000 0.5654 0.3000 0.5532 0.3000 0.5558 0.2000 0.5593 0.1142 0.5408 0.2000 0.5453 0.2000 0.5489 0.1142 0.5317 0.0500 0.4509 0.1142 0.5234 0.1142 0.5259 0.0500 0.4492 0.0500 0.4427 0.0500 0.4424
ϑ = 60.00 [°C] m = 2.500 ϑ = 59.90 [°C] m = 5.000 ϑ = 70.10 [°C] m = 0.010 ϑ = 69.60 [°C] m = 0.025 x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1
0.5995 0.6851 0.4998 0.6757 0.8987 0.8636 0.8987 0.8658 0.4998 0.6730 0.3965 0.6590 0.7990 0.7636 0.7990 0.7670 0.3965 0.6498 0.3000 0.6494 0.6931 0.6898 0.6931 0.6931 0.3000 0.6401 0.2000 0.6442 0.5957 0.6365 0.5957 0.6373 0.2000 0.6121 0.1142 0.6323 0.5053 0.6028 0.5053 0.6042 0.1142 0.6116 0.0500 0.5468 0.3995 0.5784 0.3995 0.5831 0.0500 0.5185 0.2996 0.5583 0.2996 0.5673
0.1999 0.5388 0.1999 0.5464 0.1001 0.5150 0.1001 0.5114 0.0500 0.4559 0.0500 0.4564
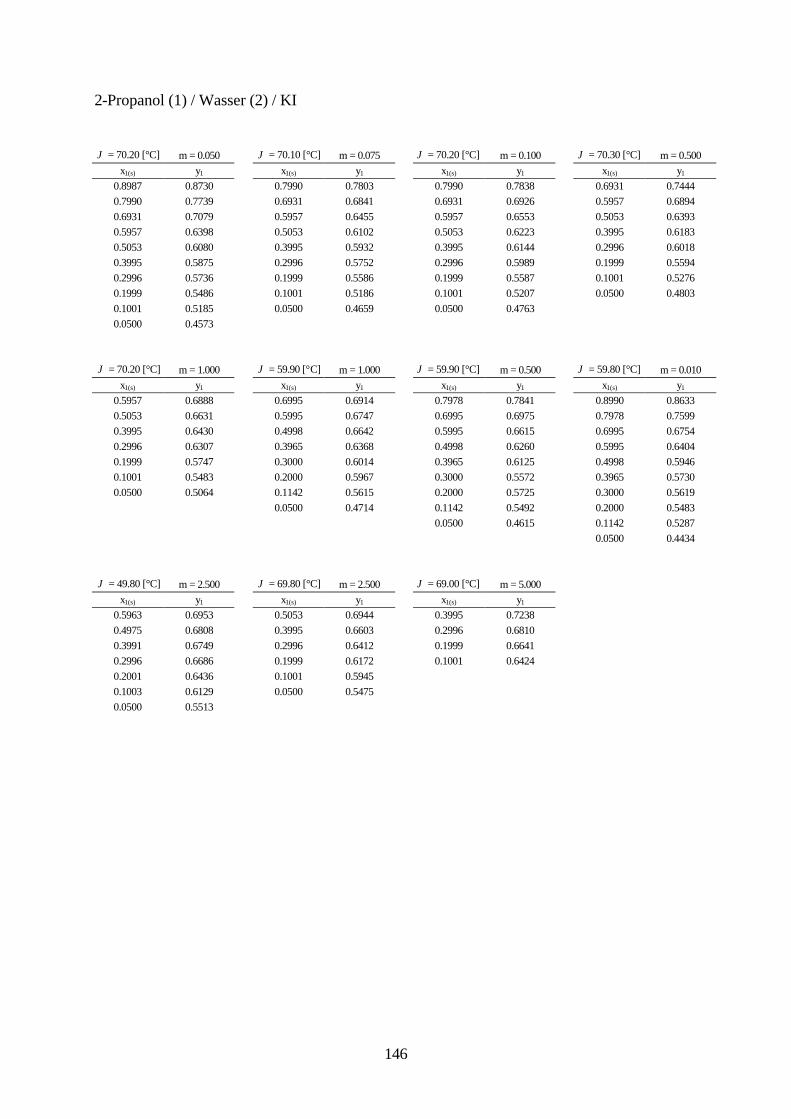
146
2-Propanol (1) / Wasser (2) / KI ϑ = 70.20 [°C] m = 0.050 ϑ = 70.10 [°C] m = 0.075 ϑ = 70.20 [°C] m = 0.100 ϑ = 70.30 [°C] m = 0.500
x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1 0.8987 0.8730 0.7990 0.7803 0.7990 0.7838 0.6931 0.7444 0.7990 0.7739 0.6931 0.6841 0.6931 0.6926 0.5957 0.6894 0.6931 0.7079 0.5957 0.6455 0.5957 0.6553 0.5053 0.6393 0.5957 0.6398 0.5053 0.6102 0.5053 0.6223 0.3995 0.6183 0.5053 0.6080 0.3995 0.5932 0.3995 0.6144 0.2996 0.6018 0.3995 0.5875 0.2996 0.5752 0.2996 0.5989 0.1999 0.5594 0.2996 0.5736 0.1999 0.5586 0.1999 0.5587 0.1001 0.5276 0.1999 0.5486 0.1001 0.5186 0.1001 0.5207 0.0500 0.4803 0.1001 0.5185 0.0500 0.4659 0.0500 0.4763 0.0500 0.4573
ϑ = 70.20 [°C] m = 1.000 ϑ = 59.90 [°C] m = 1.000 ϑ = 59.90 [°C] m = 0.500 ϑ = 59.80 [°C] m = 0.010 x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1
0.5957 0.6888 0.6995 0.6914 0.7978 0.7841 0.8990 0.8633 0.5053 0.6631 0.5995 0.6747 0.6995 0.6975 0.7978 0.7599 0.3995 0.6430 0.4998 0.6642 0.5995 0.6615 0.6995 0.6754 0.2996 0.6307 0.3965 0.6368 0.4998 0.6260 0.5995 0.6404 0.1999 0.5747 0.3000 0.6014 0.3965 0.6125 0.4998 0.5946 0.1001 0.5483 0.2000 0.5967 0.3000 0.5572 0.3965 0.5730 0.0500 0.5064 0.1142 0.5615 0.2000 0.5725 0.3000 0.5619
0.0500 0.4714 0.1142 0.5492 0.2000 0.5483 0.0500 0.4615 0.1142 0.5287 0.0500 0.4434
ϑ = 49.80 [°C] m = 2.500 ϑ = 69.80 [°C] m = 2.500 ϑ = 69.00 [°C] m = 5.000 x1(s) y1 x1(s) y1 x1(s) y1
0.5963 0.6953 0.5053 0.6944 0.3995 0.7238 0.4975 0.6808 0.3995 0.6603 0.2996 0.6810 0.3991 0.6749 0.2996 0.6412 0.1999 0.6641 0.2996 0.6686 0.1999 0.6172 0.1001 0.6424 0.2001 0.6436 0.1001 0.5945 0.1003 0.6129 0.0500 0.5475 0.0500 0.5513
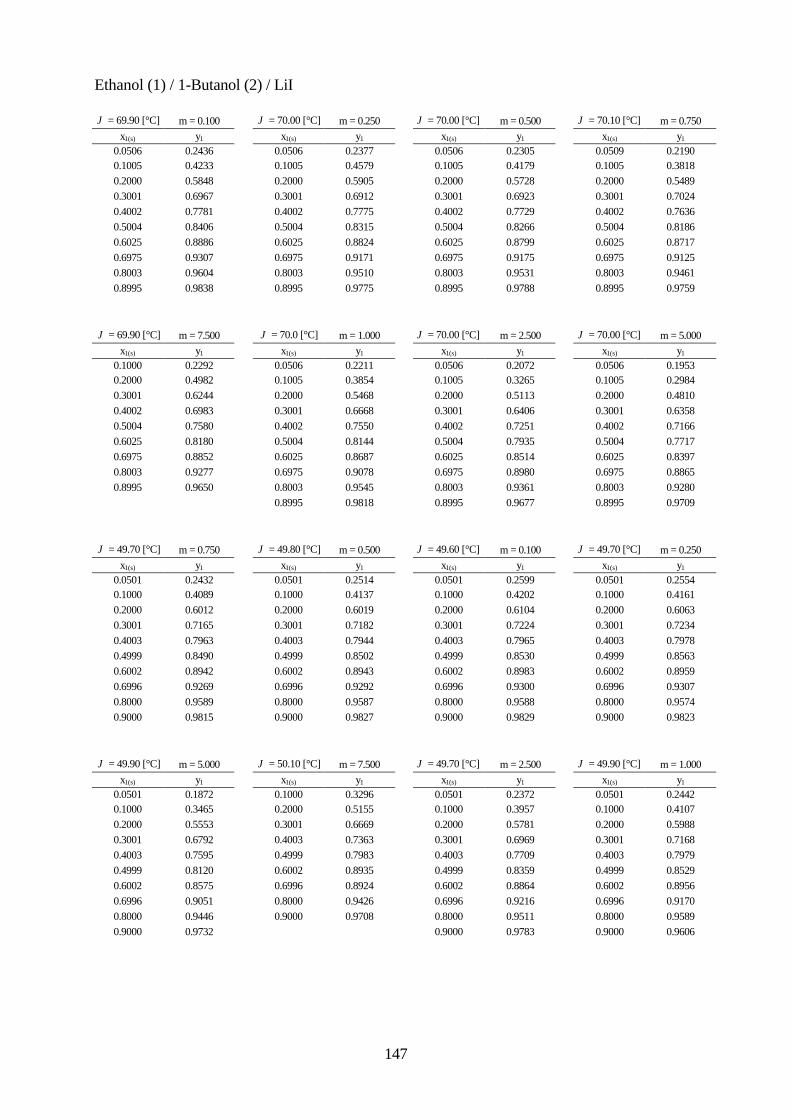
147
Ethanol (1) / 1-Butanol (2) / LiI ϑ = 69.90 [°C] m = 0.100 ϑ = 70.00 [°C] m = 0.250 ϑ = 70.00 [°C] m = 0.500 ϑ = 70.10 [°C] m = 0.750
x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1 0.0506 0.2436 0.0506 0.2377 0.0506 0.2305 0.0509 0.2190 0.1005 0.4233 0.1005 0.4579 0.1005 0.4179 0.1005 0.3818 0.2000 0.5848 0.2000 0.5905 0.2000 0.5728 0.2000 0.5489 0.3001 0.6967 0.3001 0.6912 0.3001 0.6923 0.3001 0.7024 0.4002 0.7781 0.4002 0.7775 0.4002 0.7729 0.4002 0.7636 0.5004 0.8406 0.5004 0.8315 0.5004 0.8266 0.5004 0.8186 0.6025 0.8886 0.6025 0.8824 0.6025 0.8799 0.6025 0.8717 0.6975 0.9307 0.6975 0.9171 0.6975 0.9175 0.6975 0.9125 0.8003 0.9604 0.8003 0.9510 0.8003 0.9531 0.8003 0.9461 0.8995 0.9838 0.8995 0.9775 0.8995 0.9788 0.8995 0.9759
ϑ = 69.90 [°C] m = 7.500 ϑ = 70.0 [°C] m = 1.000 ϑ = 70.00 [°C] m = 2.500 ϑ = 70.00 [°C] m = 5.000 x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1
0.1000 0.2292 0.0506 0.2211 0.0506 0.2072 0.0506 0.1953 0.2000 0.4982 0.1005 0.3854 0.1005 0.3265 0.1005 0.2984 0.3001 0.6244 0.2000 0.5468 0.2000 0.5113 0.2000 0.4810 0.4002 0.6983 0.3001 0.6668 0.3001 0.6406 0.3001 0.6358 0.5004 0.7580 0.4002 0.7550 0.4002 0.7251 0.4002 0.7166 0.6025 0.8180 0.5004 0.8144 0.5004 0.7935 0.5004 0.7717 0.6975 0.8852 0.6025 0.8687 0.6025 0.8514 0.6025 0.8397 0.8003 0.9277 0.6975 0.9078 0.6975 0.8980 0.6975 0.8865 0.8995 0.9650 0.8003 0.9545 0.8003 0.9361 0.8003 0.9280
0.8995 0.9818 0.8995 0.9677 0.8995 0.9709
ϑ = 49.70 [°C] m = 0.750 ϑ = 49.80 [°C] m = 0.500 ϑ = 49.60 [°C] m = 0.100 ϑ = 49.70 [°C] m = 0.250 x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1
0.0501 0.2432 0.0501 0.2514 0.0501 0.2599 0.0501 0.2554 0.1000 0.4089 0.1000 0.4137 0.1000 0.4202 0.1000 0.4161 0.2000 0.6012 0.2000 0.6019 0.2000 0.6104 0.2000 0.6063 0.3001 0.7165 0.3001 0.7182 0.3001 0.7224 0.3001 0.7234 0.4003 0.7963 0.4003 0.7944 0.4003 0.7965 0.4003 0.7978 0.4999 0.8490 0.4999 0.8502 0.4999 0.8530 0.4999 0.8563 0.6002 0.8942 0.6002 0.8943 0.6002 0.8983 0.6002 0.8959 0.6996 0.9269 0.6996 0.9292 0.6996 0.9300 0.6996 0.9307 0.8000 0.9589 0.8000 0.9587 0.8000 0.9588 0.8000 0.9574 0.9000 0.9815 0.9000 0.9827 0.9000 0.9829 0.9000 0.9823
ϑ = 49.90 [°C] m = 5.000 ϑ = 50.10 [°C] m = 7.500 ϑ = 49.70 [°C] m = 2.500 ϑ = 49.90 [°C] m = 1.000 x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1
0.0501 0.1872 0.1000 0.3296 0.0501 0.2372 0.0501 0.2442 0.1000 0.3465 0.2000 0.5155 0.1000 0.3957 0.1000 0.4107 0.2000 0.5553 0.3001 0.6669 0.2000 0.5781 0.2000 0.5988 0.3001 0.6792 0.4003 0.7363 0.3001 0.6969 0.3001 0.7168 0.4003 0.7595 0.4999 0.7983 0.4003 0.7709 0.4003 0.7979 0.4999 0.8120 0.6002 0.8935 0.4999 0.8359 0.4999 0.8529 0.6002 0.8575 0.6996 0.8924 0.6002 0.8864 0.6002 0.8956 0.6996 0.9051 0.8000 0.9426 0.6996 0.9216 0.6996 0.9170 0.8000 0.9446 0.9000 0.9708 0.8000 0.9511 0.8000 0.9589 0.9000 0.9732 0.9000 0.9783 0.9000 0.9606
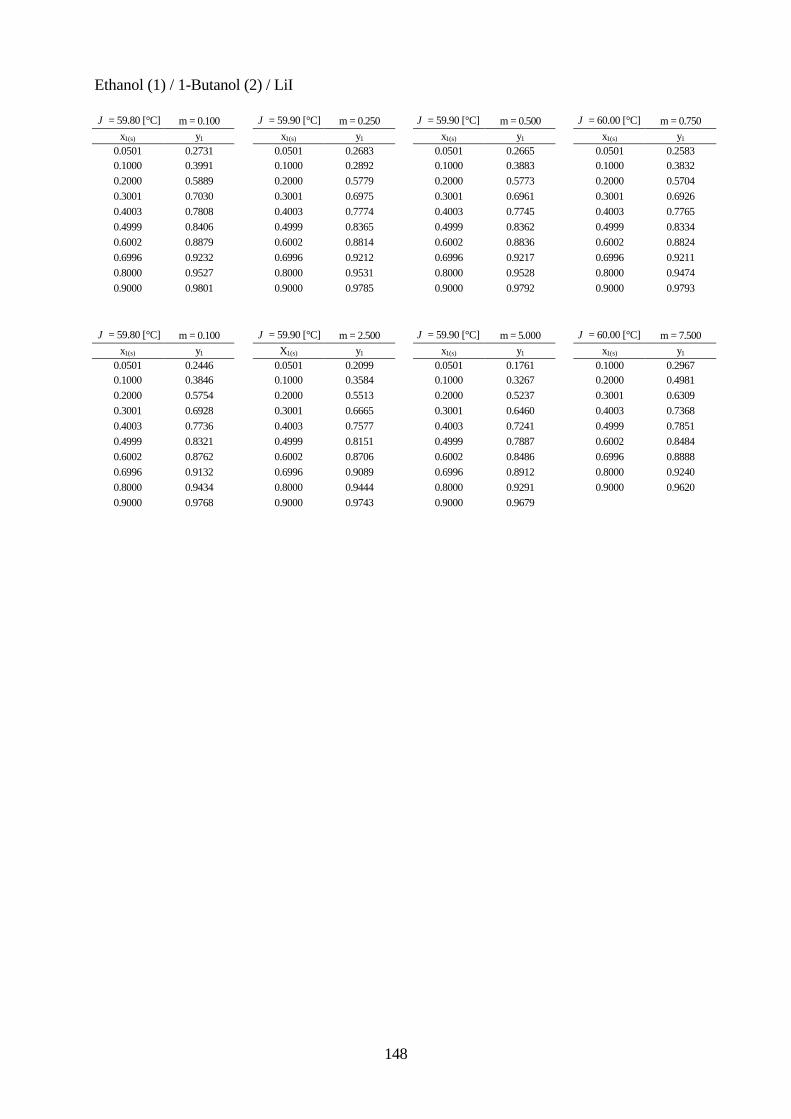
148
Ethanol (1) / 1-Butanol (2) / LiI ϑ = 59.80 [°C] m = 0.100 ϑ = 59.90 [°C] m = 0.250 ϑ = 59.90 [°C] m = 0.500 ϑ = 60.00 [°C] m = 0.750
x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1 0.0501 0.2731 0.0501 0.2683 0.0501 0.2665 0.0501 0.2583 0.1000 0.3991 0.1000 0.2892 0.1000 0.3883 0.1000 0.3832 0.2000 0.5889 0.2000 0.5779 0.2000 0.5773 0.2000 0.5704 0.3001 0.7030 0.3001 0.6975 0.3001 0.6961 0.3001 0.6926 0.4003 0.7808 0.4003 0.7774 0.4003 0.7745 0.4003 0.7765 0.4999 0.8406 0.4999 0.8365 0.4999 0.8362 0.4999 0.8334 0.6002 0.8879 0.6002 0.8814 0.6002 0.8836 0.6002 0.8824 0.6996 0.9232 0.6996 0.9212 0.6996 0.9217 0.6996 0.9211 0.8000 0.9527 0.8000 0.9531 0.8000 0.9528 0.8000 0.9474 0.9000 0.9801 0.9000 0.9785 0.9000 0.9792 0.9000 0.9793
ϑ = 59.80 [°C] m = 0.100 ϑ = 59.90 [°C] m = 2.500 ϑ = 59.90 [°C] m = 5.000 ϑ = 60.00 [°C] m = 7.500 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.0501 0.2446 0.0501 0.2099 0.0501 0.1761 0.1000 0.2967 0.1000 0.3846 0.1000 0.3584 0.1000 0.3267 0.2000 0.4981 0.2000 0.5754 0.2000 0.5513 0.2000 0.5237 0.3001 0.6309 0.3001 0.6928 0.3001 0.6665 0.3001 0.6460 0.4003 0.7368 0.4003 0.7736 0.4003 0.7577 0.4003 0.7241 0.4999 0.7851 0.4999 0.8321 0.4999 0.8151 0.4999 0.7887 0.6002 0.8484 0.6002 0.8762 0.6002 0.8706 0.6002 0.8486 0.6996 0.8888 0.6996 0.9132 0.6996 0.9089 0.6996 0.8912 0.8000 0.9240 0.8000 0.9434 0.8000 0.9444 0.8000 0.9291 0.9000 0.9620 0.9000 0.9768 0.9000 0.9743 0.9000 0.9679
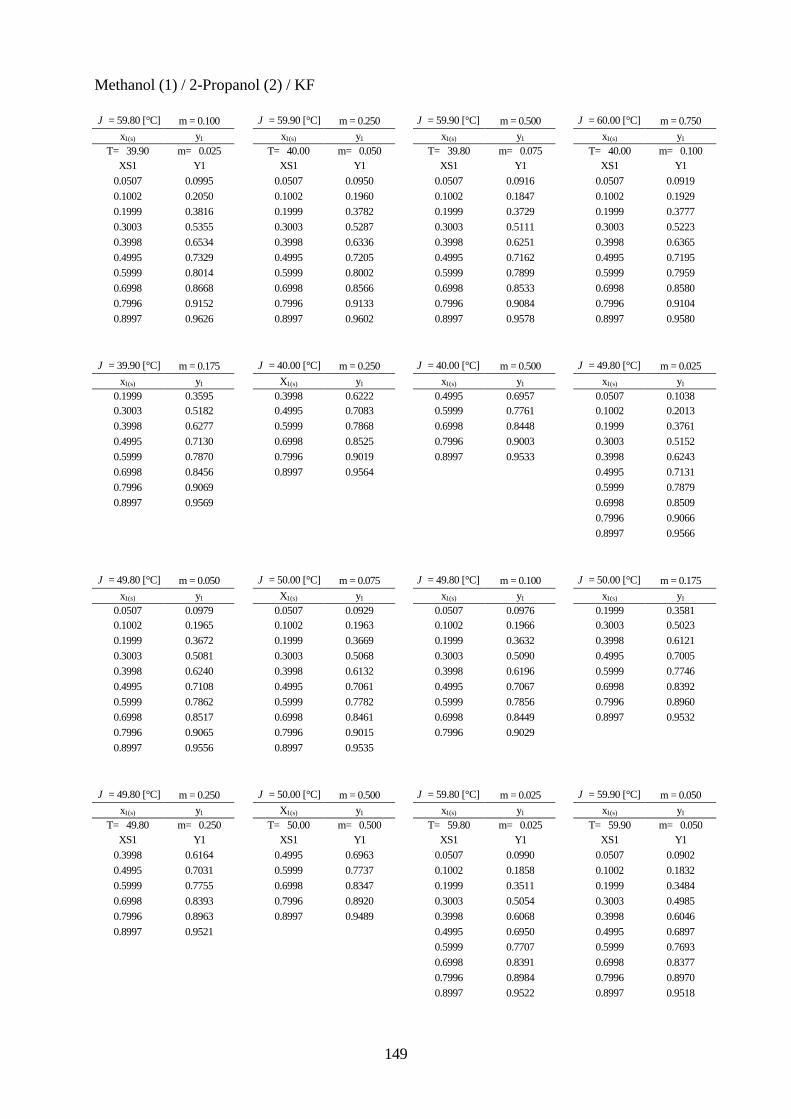
149
Methanol (1) / 2-Propanol (2) / KF ϑ = 59.80 [°C] m = 0.100 ϑ = 59.90 [°C] m = 0.250 ϑ = 59.90 [°C] m = 0.500 ϑ = 60.00 [°C] m = 0.750
x1(s) y1 x1(s) y1 x1(s) y1 x1(s) y1 T= 39.90 m= 0.025 T= 40.00 m= 0.050 T= 39.80 m= 0.075 T= 40.00 m= 0.100
XS1 Y1 XS1 Y1 XS1 Y1 XS1 Y1 0.0507 0.0995 0.0507 0.0950 0.0507 0.0916 0.0507 0.0919 0.1002 0.2050 0.1002 0.1960 0.1002 0.1847 0.1002 0.1929 0.1999 0.3816 0.1999 0.3782 0.1999 0.3729 0.1999 0.3777 0.3003 0.5355 0.3003 0.5287 0.3003 0.5111 0.3003 0.5223 0.3998 0.6534 0.3998 0.6336 0.3998 0.6251 0.3998 0.6365 0.4995 0.7329 0.4995 0.7205 0.4995 0.7162 0.4995 0.7195 0.5999 0.8014 0.5999 0.8002 0.5999 0.7899 0.5999 0.7959 0.6998 0.8668 0.6998 0.8566 0.6998 0.8533 0.6998 0.8580 0.7996 0.9152 0.7996 0.9133 0.7996 0.9084 0.7996 0.9104 0.8997 0.9626 0.8997 0.9602 0.8997 0.9578 0.8997 0.9580
ϑ = 39.90 [°C] m = 0.175 ϑ = 40.00 [°C] m = 0.250 ϑ = 40.00 [°C] m = 0.500 ϑ = 49.80 [°C] m = 0.025 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.1999 0.3595 0.3998 0.6222 0.4995 0.6957 0.0507 0.1038 0.3003 0.5182 0.4995 0.7083 0.5999 0.7761 0.1002 0.2013 0.3998 0.6277 0.5999 0.7868 0.6998 0.8448 0.1999 0.3761 0.4995 0.7130 0.6998 0.8525 0.7996 0.9003 0.3003 0.5152 0.5999 0.7870 0.7996 0.9019 0.8997 0.9533 0.3998 0.6243 0.6998 0.8456 0.8997 0.9564 0.4995 0.7131 0.7996 0.9069 0.5999 0.7879 0.8997 0.9569 0.6998 0.8509
0.7996 0.9066
0.8997 0.9566
ϑ = 49.80 [°C] m = 0.050 ϑ = 50.00 [°C] m = 0.075 ϑ = 49.80 [°C] m = 0.100 ϑ = 50.00 [°C] m = 0.175 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.0507 0.0979 0.0507 0.0929 0.0507 0.0976 0.1999 0.3581 0.1002 0.1965 0.1002 0.1963 0.1002 0.1966 0.3003 0.5023 0.1999 0.3672 0.1999 0.3669 0.1999 0.3632 0.3998 0.6121 0.3003 0.5081 0.3003 0.5068 0.3003 0.5090 0.4995 0.7005 0.3998 0.6240 0.3998 0.6132 0.3998 0.6196 0.5999 0.7746 0.4995 0.7108 0.4995 0.7061 0.4995 0.7067 0.6998 0.8392 0.5999 0.7862 0.5999 0.7782 0.5999 0.7856 0.7996 0.8960 0.6998 0.8517 0.6998 0.8461 0.6998 0.8449 0.8997 0.9532 0.7996 0.9065 0.7996 0.9015 0.7996 0.9029 0.8997 0.9556 0.8997 0.9535
ϑ = 49.80 [°C] m = 0.250 ϑ = 50.00 [°C] m = 0.500 ϑ = 59.80 [°C] m = 0.025 ϑ = 59.90 [°C] m = 0.050 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
T= 49.80 m= 0.250 T= 50.00 m= 0.500 T= 59.80 m= 0.025 T= 59.90 m= 0.050 XS1 Y1 XS1 Y1 XS1 Y1 XS1 Y1
0.3998 0.6164 0.4995 0.6963 0.0507 0.0990 0.0507 0.0902 0.4995 0.7031 0.5999 0.7737 0.1002 0.1858 0.1002 0.1832 0.5999 0.7755 0.6998 0.8347 0.1999 0.3511 0.1999 0.3484 0.6998 0.8393 0.7996 0.8920 0.3003 0.5054 0.3003 0.4985 0.7996 0.8963 0.8997 0.9489 0.3998 0.6068 0.3998 0.6046 0.8997 0.9521 0.4995 0.6950 0.4995 0.6897
0.5999 0.7707 0.5999 0.7693 0.6998 0.8391 0.6998 0.8377 0.7996 0.8984 0.7996 0.8970 0.8997 0.9522 0.8997 0.9518

150
Methanol (1) / 2-Propanol (2) / KF ϑ = 59.60 [°C] m = 0.075 ϑ = 60.00 [°C] m = 0.100 ϑ = 60.00 [°C] m = 0.175 ϑ = 60.00 [°C] m = 0.250
x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1 0.0507 0.0909 0.0507 0.0968 0.1999 0.3567 0.3998 0.5983 0.1002 0.1926 0.1002 0.1924 0.3003 0.4959 0.4995 0.6862 0.1999 0.3558 0.1999 0.3564 0.3998 0.5972 0.5999 0.7629 0.3003 0.4962 0.3003 0.4901 0.4995 0.6857 0.6998 0.8274 0.3998 0.5985 0.3998 0.5962 0.5999 0.7608 0.7996 0.8829 0.4995 0.6915 0.4995 0.6894 0.6998 0.8293 0.8997 0.9472 0.5999 0.7686 0.6998 0.8253 0.7996 0.8869 0.6998 0.8331 0.7996 0.8858 0.8997 0.9485 0.7996 0.8936 0.8997 0.9496 0.8997 0.9309
ϑ = 59.60 [°C] m = 0.500 x1(s) y1
T= 59.90 m= 0.500 XS1 Y1
0.4995 0.6839 0.5999 0.7510 0.6998 0.8230 0.7996 0.8791 0.8997 0.9440
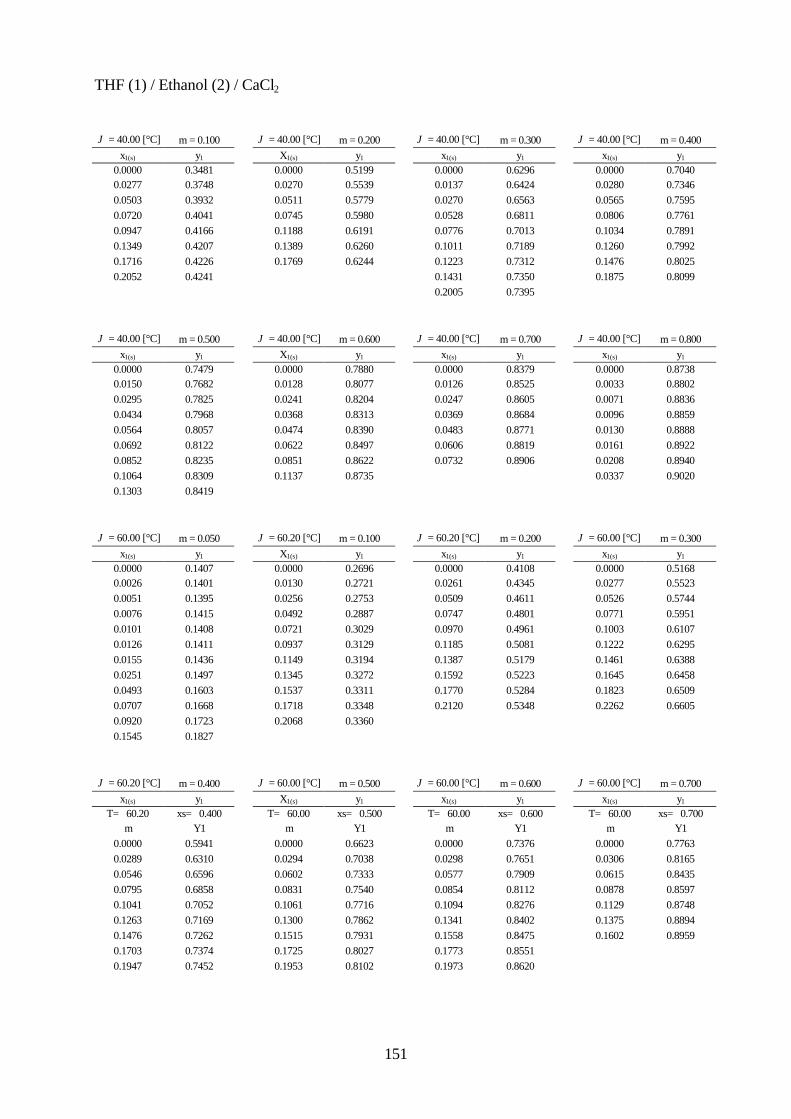
151
THF (1) / Ethanol (2) / CaCl2
ϑ = 40.00 [°C] m = 0.100 ϑ = 40.00 [°C] m = 0.200 ϑ = 40.00 [°C] m = 0.300 ϑ = 40.00 [°C] m = 0.400
x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1 0.0000 0.3481 0.0000 0.5199 0.0000 0.6296 0.0000 0.7040 0.0277 0.3748 0.0270 0.5539 0.0137 0.6424 0.0280 0.7346 0.0503 0.3932 0.0511 0.5779 0.0270 0.6563 0.0565 0.7595 0.0720 0.4041 0.0745 0.5980 0.0528 0.6811 0.0806 0.7761 0.0947 0.4166 0.1188 0.6191 0.0776 0.7013 0.1034 0.7891 0.1349 0.4207 0.1389 0.6260 0.1011 0.7189 0.1260 0.7992 0.1716 0.4226 0.1769 0.6244 0.1223 0.7312 0.1476 0.8025 0.2052 0.4241 0.1431 0.7350 0.1875 0.8099
0.2005 0.7395
ϑ = 40.00 [°C] m = 0.500 ϑ = 40.00 [°C] m = 0.600 ϑ = 40.00 [°C] m = 0.700 ϑ = 40.00 [°C] m = 0.800 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.0000 0.7479 0.0000 0.7880 0.0000 0.8379 0.0000 0.8738 0.0150 0.7682 0.0128 0.8077 0.0126 0.8525 0.0033 0.8802 0.0295 0.7825 0.0241 0.8204 0.0247 0.8605 0.0071 0.8836 0.0434 0.7968 0.0368 0.8313 0.0369 0.8684 0.0096 0.8859 0.0564 0.8057 0.0474 0.8390 0.0483 0.8771 0.0130 0.8888 0.0692 0.8122 0.0622 0.8497 0.0606 0.8819 0.0161 0.8922 0.0852 0.8235 0.0851 0.8622 0.0732 0.8906 0.0208 0.8940 0.1064 0.8309 0.1137 0.8735 0.0337 0.9020 0.1303 0.8419
ϑ = 60.00 [°C] m = 0.050 ϑ = 60.20 [°C] m = 0.100 ϑ = 60.20 [°C] m = 0.200 ϑ = 60.00 [°C] m = 0.300 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.0000 0.1407 0.0000 0.2696 0.0000 0.4108 0.0000 0.5168 0.0026 0.1401 0.0130 0.2721 0.0261 0.4345 0.0277 0.5523 0.0051 0.1395 0.0256 0.2753 0.0509 0.4611 0.0526 0.5744 0.0076 0.1415 0.0492 0.2887 0.0747 0.4801 0.0771 0.5951 0.0101 0.1408 0.0721 0.3029 0.0970 0.4961 0.1003 0.6107 0.0126 0.1411 0.0937 0.3129 0.1185 0.5081 0.1222 0.6295 0.0155 0.1436 0.1149 0.3194 0.1387 0.5179 0.1461 0.6388 0.0251 0.1497 0.1345 0.3272 0.1592 0.5223 0.1645 0.6458 0.0493 0.1603 0.1537 0.3311 0.1770 0.5284 0.1823 0.6509 0.0707 0.1668 0.1718 0.3348 0.2120 0.5348 0.2262 0.6605 0.0920 0.1723 0.2068 0.3360 0.1545 0.1827
ϑ = 60.20 [°C] m = 0.400 ϑ = 60.00 [°C] m = 0.500 ϑ = 60.00 [°C] m = 0.600 ϑ = 60.00 [°C] m = 0.700 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
T= 60.20 xs= 0.400 T= 60.00 xs= 0.500 T= 60.00 xs= 0.600 T= 60.00 xs= 0.700 m Y1 m Y1 m Y1 m Y1
0.0000 0.5941 0.0000 0.6623 0.0000 0.7376 0.0000 0.7763 0.0289 0.6310 0.0294 0.7038 0.0298 0.7651 0.0306 0.8165 0.0546 0.6596 0.0602 0.7333 0.0577 0.7909 0.0615 0.8435 0.0795 0.6858 0.0831 0.7540 0.0854 0.8112 0.0878 0.8597 0.1041 0.7052 0.1061 0.7716 0.1094 0.8276 0.1129 0.8748 0.1263 0.7169 0.1300 0.7862 0.1341 0.8402 0.1375 0.8894 0.1476 0.7262 0.1515 0.7931 0.1558 0.8475 0.1602 0.8959 0.1703 0.7374 0.1725 0.8027 0.1773 0.8551 0.1947 0.7452 0.1953 0.8102 0.1973 0.8620
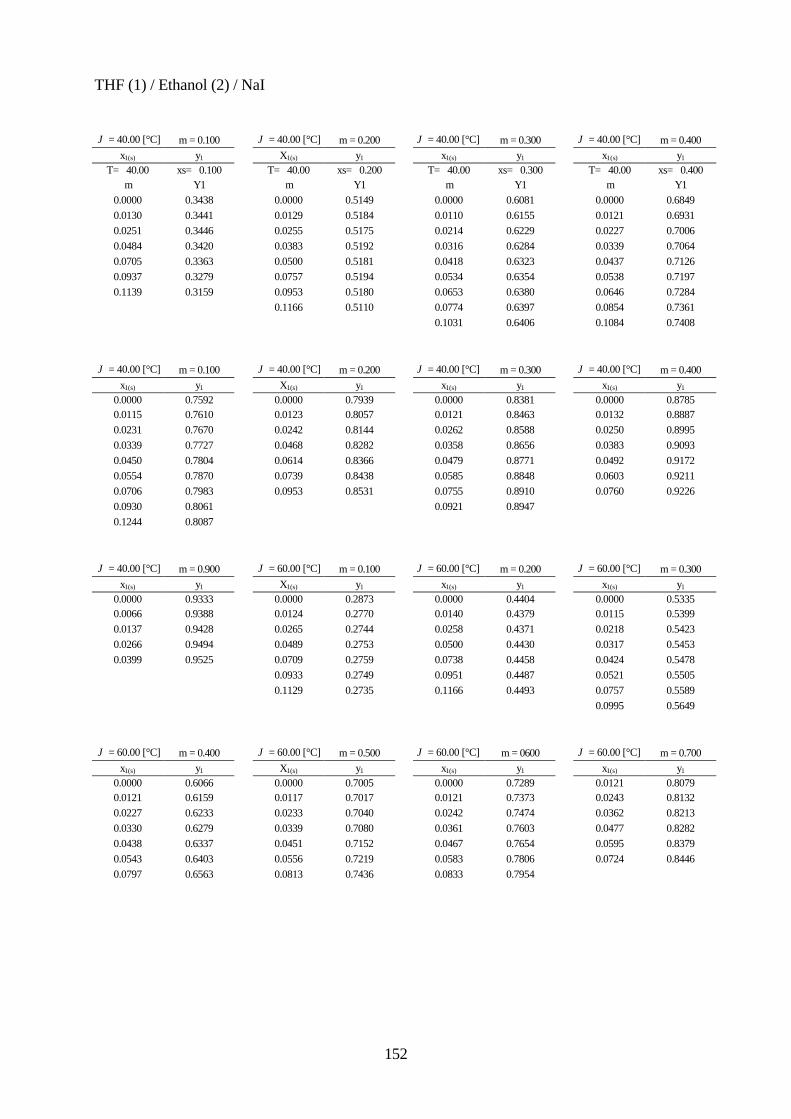
152
THF (1) / Ethanol (2) / NaI ϑ = 40.00 [°C] m = 0.100 ϑ = 40.00 [°C] m = 0.200 ϑ = 40.00 [°C] m = 0.300 ϑ = 40.00 [°C] m = 0.400
x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1 T= 40.00 xs= 0.100 T= 40.00 xs= 0.200 T= 40.00 xs= 0.300 T= 40.00 xs= 0.400
m Y1 m Y1 m Y1 m Y1 0.0000 0.3438 0.0000 0.5149 0.0000 0.6081 0.0000 0.6849 0.0130 0.3441 0.0129 0.5184 0.0110 0.6155 0.0121 0.6931 0.0251 0.3446 0.0255 0.5175 0.0214 0.6229 0.0227 0.7006 0.0484 0.3420 0.0383 0.5192 0.0316 0.6284 0.0339 0.7064 0.0705 0.3363 0.0500 0.5181 0.0418 0.6323 0.0437 0.7126 0.0937 0.3279 0.0757 0.5194 0.0534 0.6354 0.0538 0.7197 0.1139 0.3159 0.0953 0.5180 0.0653 0.6380 0.0646 0.7284
0.1166 0.5110 0.0774 0.6397 0.0854 0.7361 0.1031 0.6406 0.1084 0.7408
ϑ = 40.00 [°C] m = 0.100 ϑ = 40.00 [°C] m = 0.200 ϑ = 40.00 [°C] m = 0.300 ϑ = 40.00 [°C] m = 0.400 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.0000 0.7592 0.0000 0.7939 0.0000 0.8381 0.0000 0.8785 0.0115 0.7610 0.0123 0.8057 0.0121 0.8463 0.0132 0.8887 0.0231 0.7670 0.0242 0.8144 0.0262 0.8588 0.0250 0.8995 0.0339 0.7727 0.0468 0.8282 0.0358 0.8656 0.0383 0.9093 0.0450 0.7804 0.0614 0.8366 0.0479 0.8771 0.0492 0.9172 0.0554 0.7870 0.0739 0.8438 0.0585 0.8848 0.0603 0.9211 0.0706 0.7983 0.0953 0.8531 0.0755 0.8910 0.0760 0.9226 0.0930 0.8061 0.0921 0.8947 0.1244 0.8087
ϑ = 40.00 [°C] m = 0.900 ϑ = 60.00 [°C] m = 0.100 ϑ = 60.00 [°C] m = 0.200 ϑ = 60.00 [°C] m = 0.300 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.0000 0.9333 0.0000 0.2873 0.0000 0.4404 0.0000 0.5335 0.0066 0.9388 0.0124 0.2770 0.0140 0.4379 0.0115 0.5399 0.0137 0.9428 0.0265 0.2744 0.0258 0.4371 0.0218 0.5423 0.0266 0.9494 0.0489 0.2753 0.0500 0.4430 0.0317 0.5453 0.0399 0.9525 0.0709 0.2759 0.0738 0.4458 0.0424 0.5478
0.0933 0.2749 0.0951 0.4487 0.0521 0.5505 0.1129 0.2735 0.1166 0.4493 0.0757 0.5589
0.0995 0.5649
ϑ = 60.00 [°C] m = 0.400 ϑ = 60.00 [°C] m = 0.500 ϑ = 60.00 [°C] m = 0600 ϑ = 60.00 [°C] m = 0.700 x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1
0.0000 0.6066 0.0000 0.7005 0.0000 0.7289 0.0121 0.8079 0.0121 0.6159 0.0117 0.7017 0.0121 0.7373 0.0243 0.8132 0.0227 0.6233 0.0233 0.7040 0.0242 0.7474 0.0362 0.8213 0.0330 0.6279 0.0339 0.7080 0.0361 0.7603 0.0477 0.8282 0.0438 0.6337 0.0451 0.7152 0.0467 0.7654 0.0595 0.8379 0.0543 0.6403 0.0556 0.7219 0.0583 0.7806 0.0724 0.8446 0.0797 0.6563 0.0813 0.7436 0.0833 0.7954
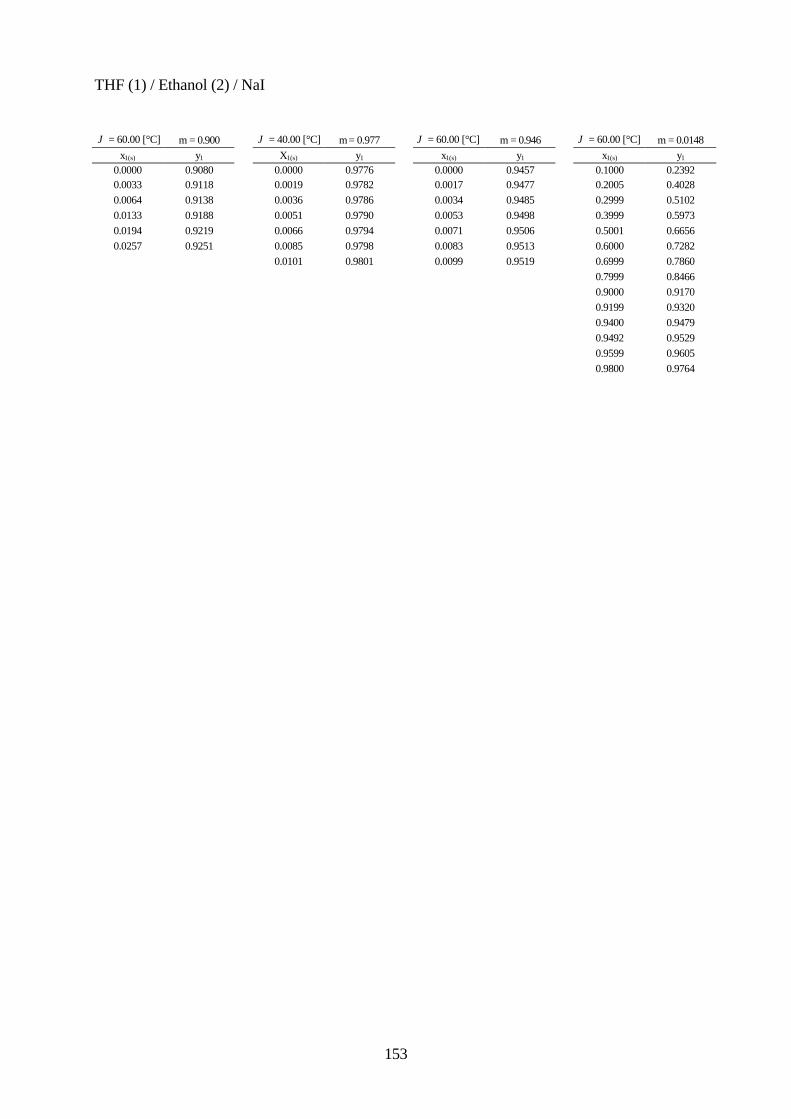
153
THF (1) / Ethanol (2) / NaI ϑ = 60.00 [°C] m = 0.900 ϑ = 40.00 [°C] m = 0.977 ϑ = 60.00 [°C] m = 0.946 ϑ = 60.00 [°C] m = 0.0148
x1(s) y1 X1(s) y1 x1(s) y1 x1(s) y1 0.0000 0.9080 0.0000 0.9776 0.0000 0.9457 0.1000 0.2392 0.0033 0.9118 0.0019 0.9782 0.0017 0.9477 0.2005 0.4028 0.0064 0.9138 0.0036 0.9786 0.0034 0.9485 0.2999 0.5102 0.0133 0.9188 0.0051 0.9790 0.0053 0.9498 0.3999 0.5973 0.0194 0.9219 0.0066 0.9794 0.0071 0.9506 0.5001 0.6656 0.0257 0.9251 0.0085 0.9798 0.0083 0.9513 0.6000 0.7282
0.0101 0.9801 0.0099 0.9519 0.6999 0.7860
0.7999 0.8466
0.9000 0.9170
0.9199 0.9320
0.9400 0.9479
0.9492 0.9529
0.9599 0.9605
0.9800 0.9764
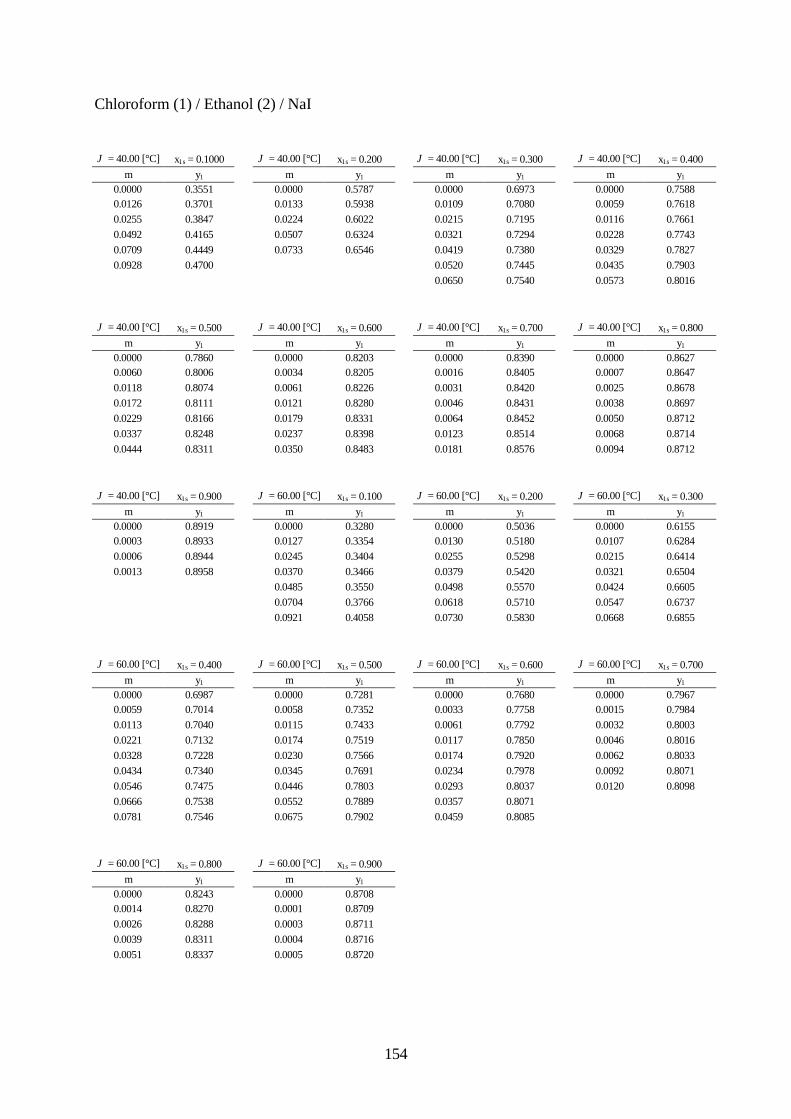
154
Chloroform (1) / Ethanol (2) / NaI ϑ = 40.00 [°C] x1s = 0.1000 ϑ = 40.00 [°C] x1s = 0.200 ϑ = 40.00 [°C] x1s = 0.300 ϑ = 40.00 [°C] x1s = 0.400
m y1 m y1 m y1 m y1 0.0000 0.3551 0.0000 0.5787 0.0000 0.6973 0.0000 0.7588 0.0126 0.3701 0.0133 0.5938 0.0109 0.7080 0.0059 0.7618 0.0255 0.3847 0.0224 0.6022 0.0215 0.7195 0.0116 0.7661 0.0492 0.4165 0.0507 0.6324 0.0321 0.7294 0.0228 0.7743 0.0709 0.4449 0.0733 0.6546 0.0419 0.7380 0.0329 0.7827 0.0928 0.4700 0.0520 0.7445 0.0435 0.7903
0.0650 0.7540 0.0573 0.8016
ϑ = 40.00 [°C] x1s = 0.500 ϑ = 40.00 [°C] x1s = 0.600 ϑ = 40.00 [°C] x1s = 0.700 ϑ = 40.00 [°C] x1s = 0.800 m y1 m y1 m y1 m y1
0.0000 0.7860 0.0000 0.8203 0.0000 0.8390 0.0000 0.8627 0.0060 0.8006 0.0034 0.8205 0.0016 0.8405 0.0007 0.8647 0.0118 0.8074 0.0061 0.8226 0.0031 0.8420 0.0025 0.8678 0.0172 0.8111 0.0121 0.8280 0.0046 0.8431 0.0038 0.8697 0.0229 0.8166 0.0179 0.8331 0.0064 0.8452 0.0050 0.8712 0.0337 0.8248 0.0237 0.8398 0.0123 0.8514 0.0068 0.8714 0.0444 0.8311 0.0350 0.8483 0.0181 0.8576 0.0094 0.8712
ϑ = 40.00 [°C] x1s = 0.900 ϑ = 60.00 [°C] x1s = 0.100 ϑ = 60.00 [°C] x1s = 0.200 ϑ = 60.00 [°C] x1s = 0.300 m y1 m y1 m y1 m y1
0.0000 0.8919 0.0000 0.3280 0.0000 0.5036 0.0000 0.6155 0.0003 0.8933 0.0127 0.3354 0.0130 0.5180 0.0107 0.6284 0.0006 0.8944 0.0245 0.3404 0.0255 0.5298 0.0215 0.6414 0.0013 0.8958 0.0370 0.3466 0.0379 0.5420 0.0321 0.6504
0.0485 0.3550 0.0498 0.5570 0.0424 0.6605 0.0704 0.3766 0.0618 0.5710 0.0547 0.6737 0.0921 0.4058 0.0730 0.5830 0.0668 0.6855
ϑ = 60.00 [°C] x1s = 0.400 ϑ = 60.00 [°C] x1s = 0.500 ϑ = 60.00 [°C] x1s = 0.600 ϑ = 60.00 [°C] x1s = 0.700 m y1 m y1 m y1 m y1
0.0000 0.6987 0.0000 0.7281 0.0000 0.7680 0.0000 0.7967 0.0059 0.7014 0.0058 0.7352 0.0033 0.7758 0.0015 0.7984 0.0113 0.7040 0.0115 0.7433 0.0061 0.7792 0.0032 0.8003 0.0221 0.7132 0.0174 0.7519 0.0117 0.7850 0.0046 0.8016 0.0328 0.7228 0.0230 0.7566 0.0174 0.7920 0.0062 0.8033 0.0434 0.7340 0.0345 0.7691 0.0234 0.7978 0.0092 0.8071 0.0546 0.7475 0.0446 0.7803 0.0293 0.8037 0.0120 0.8098 0.0666 0.7538 0.0552 0.7889 0.0357 0.8071 0.0781 0.7546 0.0675 0.7902 0.0459 0.8085
ϑ = 60.00 [°C] x1s = 0.800 ϑ = 60.00 [°C] x1s = 0.900 m y1 m y1
0.0000 0.8243 0.0000 0.8708 0.0014 0.8270 0.0001 0.8709 0.0026 0.8288 0.0003 0.8711 0.0039 0.8311 0.0004 0.8716 0.0051 0.8337 0.0005 0.8720
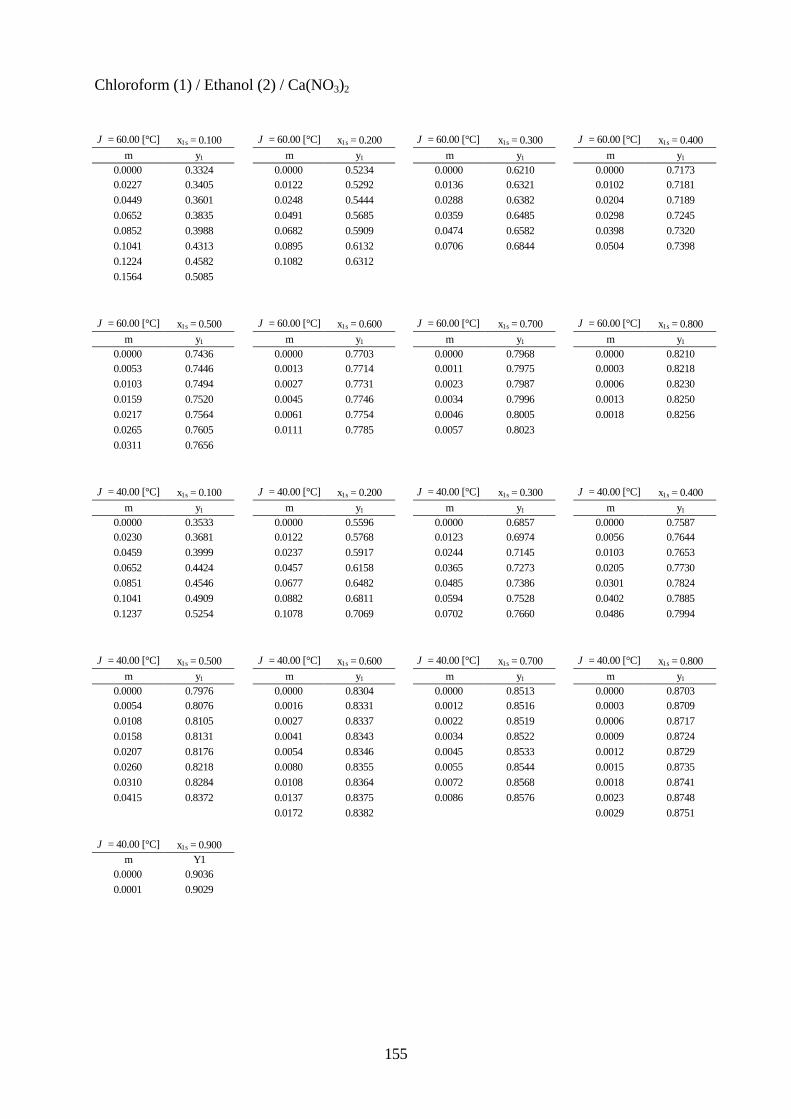
155
Chloroform (1) / Ethanol (2) / Ca(NO3)2 ϑ = 60.00 [°C] x1s = 0.100 ϑ = 60.00 [°C] x1s = 0.200 ϑ = 60.00 [°C] x1s = 0.300 ϑ = 60.00 [°C] x1s = 0.400
m y1 m y1 m y1 m y1 0.0000 0.3324 0.0000 0.5234 0.0000 0.6210 0.0000 0.7173 0.0227 0.3405 0.0122 0.5292 0.0136 0.6321 0.0102 0.7181 0.0449 0.3601 0.0248 0.5444 0.0288 0.6382 0.0204 0.7189 0.0652 0.3835 0.0491 0.5685 0.0359 0.6485 0.0298 0.7245 0.0852 0.3988 0.0682 0.5909 0.0474 0.6582 0.0398 0.7320 0.1041 0.4313 0.0895 0.6132 0.0706 0.6844 0.0504 0.7398 0.1224 0.4582 0.1082 0.6312 0.1564 0.5085
ϑ = 60.00 [°C] x1s = 0.500 ϑ = 60.00 [°C] x1s = 0.600 ϑ = 60.00 [°C] x1s = 0.700 ϑ = 60.00 [°C] x1s = 0.800 m y1 m y1 m y1 m y1
0.0000 0.7436 0.0000 0.7703 0.0000 0.7968 0.0000 0.8210 0.0053 0.7446 0.0013 0.7714 0.0011 0.7975 0.0003 0.8218 0.0103 0.7494 0.0027 0.7731 0.0023 0.7987 0.0006 0.8230 0.0159 0.7520 0.0045 0.7746 0.0034 0.7996 0.0013 0.8250 0.0217 0.7564 0.0061 0.7754 0.0046 0.8005 0.0018 0.8256 0.0265 0.7605 0.0111 0.7785 0.0057 0.8023 0.0311 0.7656
ϑ = 40.00 [°C] x1s = 0.100 ϑ = 40.00 [°C] x1s = 0.200 ϑ = 40.00 [°C] x1s = 0.300 ϑ = 40.00 [°C] x1s = 0.400 m y1 m y1 m y1 m y1
0.0000 0.3533 0.0000 0.5596 0.0000 0.6857 0.0000 0.7587 0.0230 0.3681 0.0122 0.5768 0.0123 0.6974 0.0056 0.7644 0.0459 0.3999 0.0237 0.5917 0.0244 0.7145 0.0103 0.7653 0.0652 0.4424 0.0457 0.6158 0.0365 0.7273 0.0205 0.7730 0.0851 0.4546 0.0677 0.6482 0.0485 0.7386 0.0301 0.7824 0.1041 0.4909 0.0882 0.6811 0.0594 0.7528 0.0402 0.7885 0.1237 0.5254 0.1078 0.7069 0.0702 0.7660 0.0486 0.7994
ϑ = 40.00 [°C] x1s = 0.500 ϑ = 40.00 [°C] x1s = 0.600 ϑ = 40.00 [°C] x1s = 0.700 ϑ = 40.00 [°C] x1s = 0.800 m y1 m y1 m y1 m y1
0.0000 0.7976 0.0000 0.8304 0.0000 0.8513 0.0000 0.8703 0.0054 0.8076 0.0016 0.8331 0.0012 0.8516 0.0003 0.8709 0.0108 0.8105 0.0027 0.8337 0.0022 0.8519 0.0006 0.8717 0.0158 0.8131 0.0041 0.8343 0.0034 0.8522 0.0009 0.8724 0.0207 0.8176 0.0054 0.8346 0.0045 0.8533 0.0012 0.8729 0.0260 0.8218 0.0080 0.8355 0.0055 0.8544 0.0015 0.8735 0.0310 0.8284 0.0108 0.8364 0.0072 0.8568 0.0018 0.8741 0.0415 0.8372 0.0137 0.8375 0.0086 0.8576 0.0023 0.8748
0.0172 0.8382 0.0029 0.8751
ϑ = 40.00 [°C] x1s = 0.900
m Y1 0.0000 0.9036 0.0001 0.9029
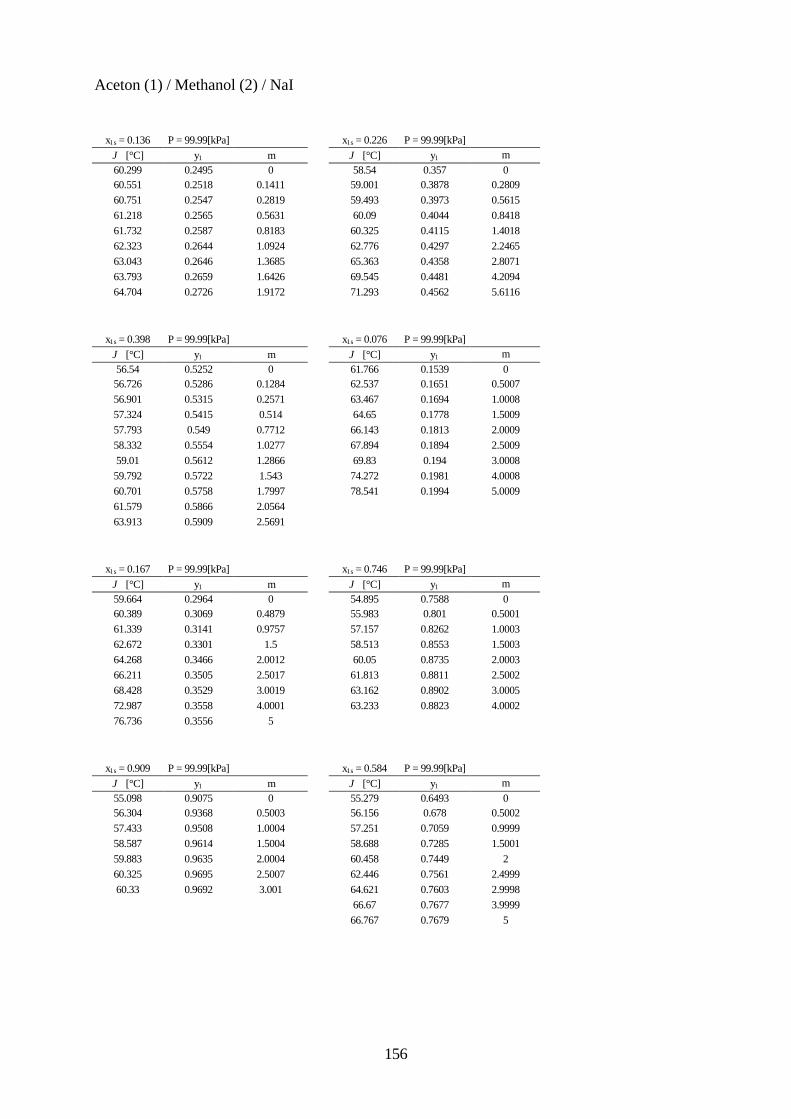
156
Aceton (1) / Methanol (2) / NaI
x1s = 0.136 P = 99.99[kPa] x1s = 0.226 P = 99.99[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 60.299 0.2495 0 58.54 0.357 0 60.551 0.2518 0.1411 59.001 0.3878 0.2809 60.751 0.2547 0.2819 59.493 0.3973 0.5615 61.218 0.2565 0.5631 60.09 0.4044 0.8418 61.732 0.2587 0.8183 60.325 0.4115 1.4018 62.323 0.2644 1.0924 62.776 0.4297 2.2465 63.043 0.2646 1.3685 65.363 0.4358 2.8071 63.793 0.2659 1.6426 69.545 0.4481 4.2094 64.704 0.2726 1.9172 71.293 0.4562 5.6116
x1s = 0.398 P = 99.99[kPa] x1s = 0.076 P = 99.99[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 56.54 0.5252 0 61.766 0.1539 0 56.726 0.5286 0.1284 62.537 0.1651 0.5007 56.901 0.5315 0.2571 63.467 0.1694 1.0008 57.324 0.5415 0.514 64.65 0.1778 1.5009 57.793 0.549 0.7712 66.143 0.1813 2.0009 58.332 0.5554 1.0277 67.894 0.1894 2.5009 59.01 0.5612 1.2866 69.83 0.194 3.0008 59.792 0.5722 1.543 74.272 0.1981 4.0008 60.701 0.5758 1.7997 78.541 0.1994 5.0009 61.579 0.5866 2.0564 63.913 0.5909 2.5691
x1s = 0.167 P = 99.99[kPa] x1s = 0.746 P = 99.99[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 59.664 0.2964 0 54.895 0.7588 0 60.389 0.3069 0.4879 55.983 0.801 0.5001 61.339 0.3141 0.9757 57.157 0.8262 1.0003 62.672 0.3301 1.5 58.513 0.8553 1.5003 64.268 0.3466 2.0012 60.05 0.8735 2.0003 66.211 0.3505 2.5017 61.813 0.8811 2.5002 68.428 0.3529 3.0019 63.162 0.8902 3.0005 72.987 0.3558 4.0001 63.233 0.8823 4.0002 76.736 0.3556 5
x1s = 0.909 P = 99.99[kPa] x1s = 0.584 P = 99.99[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 55.098 0.9075 0 55.279 0.6493 0 56.304 0.9368 0.5003 56.156 0.678 0.5002 57.433 0.9508 1.0004 57.251 0.7059 0.9999 58.587 0.9614 1.5004 58.688 0.7285 1.5001 59.883 0.9635 2.0004 60.458 0.7449 2 60.325 0.9695 2.5007 62.446 0.7561 2.4999 60.33 0.9692 3.001 64.621 0.7603 2.9998
66.67 0.7677 3.9999 66.767 0.7679 5
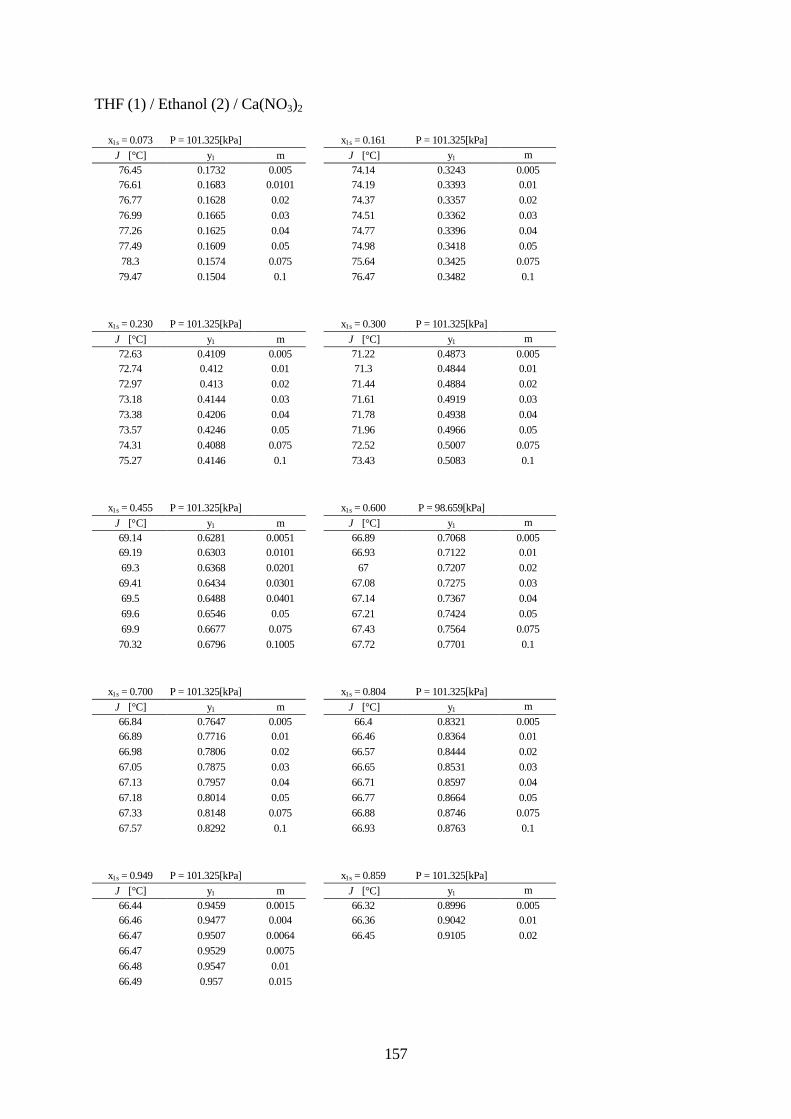
157
THF (1) / Ethanol (2) / Ca(NO3)2
x1s = 0.073 P = 101.325[kPa] x1s = 0.161 P = 101.325[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 76.45 0.1732 0.005 74.14 0.3243 0.005 76.61 0.1683 0.0101 74.19 0.3393 0.01 76.77 0.1628 0.02 74.37 0.3357 0.02 76.99 0.1665 0.03 74.51 0.3362 0.03 77.26 0.1625 0.04 74.77 0.3396 0.04 77.49 0.1609 0.05 74.98 0.3418 0.05 78.3 0.1574 0.075 75.64 0.3425 0.075 79.47 0.1504 0.1 76.47 0.3482 0.1
x1s = 0.230 P = 101.325[kPa] x1s = 0.300 P = 101.325[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 72.63 0.4109 0.005 71.22 0.4873 0.005 72.74 0.412 0.01 71.3 0.4844 0.01 72.97 0.413 0.02 71.44 0.4884 0.02 73.18 0.4144 0.03 71.61 0.4919 0.03 73.38 0.4206 0.04 71.78 0.4938 0.04 73.57 0.4246 0.05 71.96 0.4966 0.05 74.31 0.4088 0.075 72.52 0.5007 0.075 75.27 0.4146 0.1 73.43 0.5083 0.1
x1s = 0.455 P = 101.325[kPa] x1s = 0.600 P = 98.659[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 69.14 0.6281 0.0051 66.89 0.7068 0.005 69.19 0.6303 0.0101 66.93 0.7122 0.01 69.3 0.6368 0.0201 67 0.7207 0.02 69.41 0.6434 0.0301 67.08 0.7275 0.03 69.5 0.6488 0.0401 67.14 0.7367 0.04 69.6 0.6546 0.05 67.21 0.7424 0.05 69.9 0.6677 0.075 67.43 0.7564 0.075 70.32 0.6796 0.1005 67.72 0.7701 0.1
x1s = 0.700 P = 101.325[kPa] x1s = 0.804 P = 101.325[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 66.84 0.7647 0.005 66.4 0.8321 0.005 66.89 0.7716 0.01 66.46 0.8364 0.01 66.98 0.7806 0.02 66.57 0.8444 0.02 67.05 0.7875 0.03 66.65 0.8531 0.03 67.13 0.7957 0.04 66.71 0.8597 0.04 67.18 0.8014 0.05 66.77 0.8664 0.05 67.33 0.8148 0.075 66.88 0.8746 0.075 67.57 0.8292 0.1 66.93 0.8763 0.1
x1s = 0.949 P = 101.325[kPa] x1s = 0.859 P = 101.325[kPa]
ϑ [°C] y1 m ϑ [°C] y1 m 66.44 0.9459 0.0015 66.32 0.8996 0.005 66.46 0.9477 0.004 66.36 0.9042 0.01 66.47 0.9507 0.0064 66.45 0.9105 0.02 66.47 0.9529 0.0075 66.48 0.9547 0.01 66.49 0.957 0.015
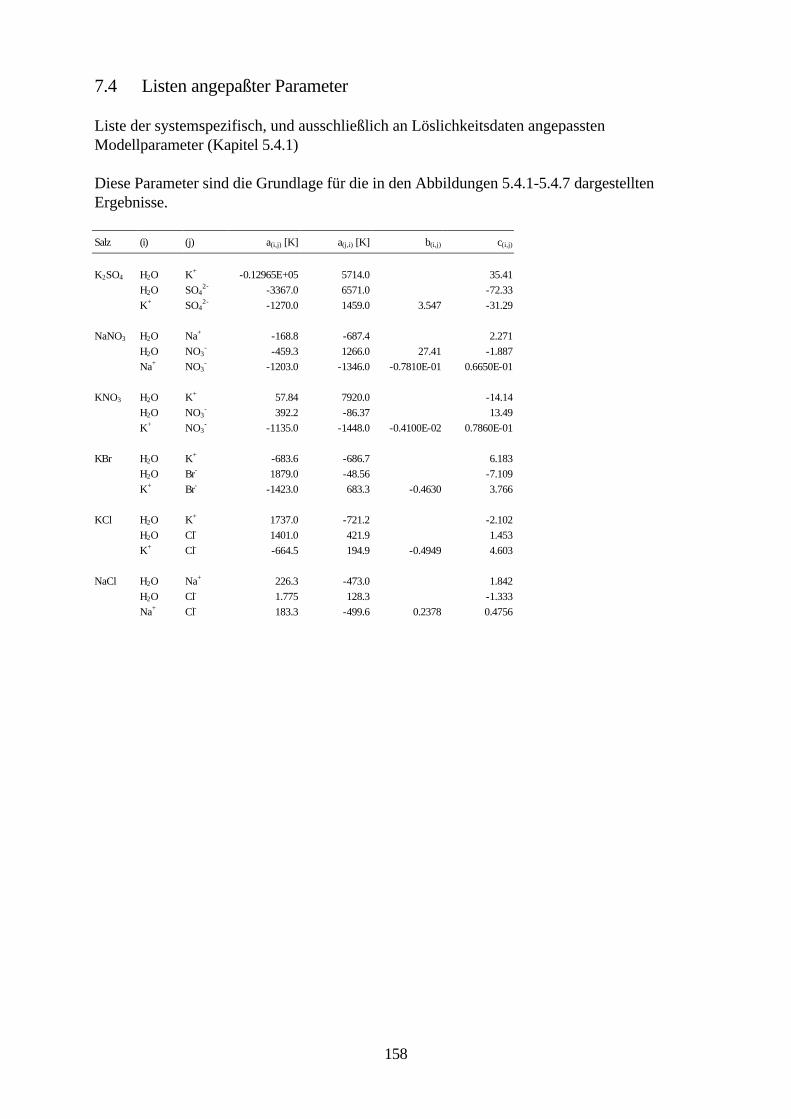
158
7.4 Listen angepaßter Parameter Liste der systemspezifisch, und ausschließlich an Löslichkeitsdaten angepassten Modellparameter (Kapitel 5.4.1) Diese Parameter sind die Grundlage für die in den Abbildungen 5.4.1-5.4.7 dargestellten Ergebnisse. Salz (i) (j) a(i,j) [K] a(j,i) [K] b(i,j) c(i,j)
K2SO4 H2O K+ -0.12965E+05 5714.0 35.41
H2O SO42- -3367.0 6571.0 -72.33
K+ SO42- -1270.0 1459.0 3.547 -31.29
NaNO3 H2O Na+ -168.8
-687.4 2.271
H2O NO3- -459.3 1266.0 27.41 -1.887
Na+ NO3- -1203.0 -1346.0 -0.7810E-01 0.6650E-01
KNO3 H2O K+ 57.84 7920.0 -14.14
H2O NO3- 392.2 -86.37 13.49
K+ NO3- -1135.0 -1448.0 -0.4100E-02 0.7860E-01
KBr H2O K+ -683.6 -686.7 6.183
H2O Br- 1879.0 -48.56 -7.109 K+ Br- -1423.0 683.3 -0.4630 3.766
KCl H2O K+ 1737.0 -721.2 -2.102 H2O Cl- 1401.0 421.9 1.453 K+ Cl- -664.5 194.9 -0.4949 4.603
NaCl H2O Na+ 226.3 -473.0 1.842 H2O Cl- 1.775 128.3 -1.333 Na+ Cl- 183.3 -499.6 0.2378 0.4756
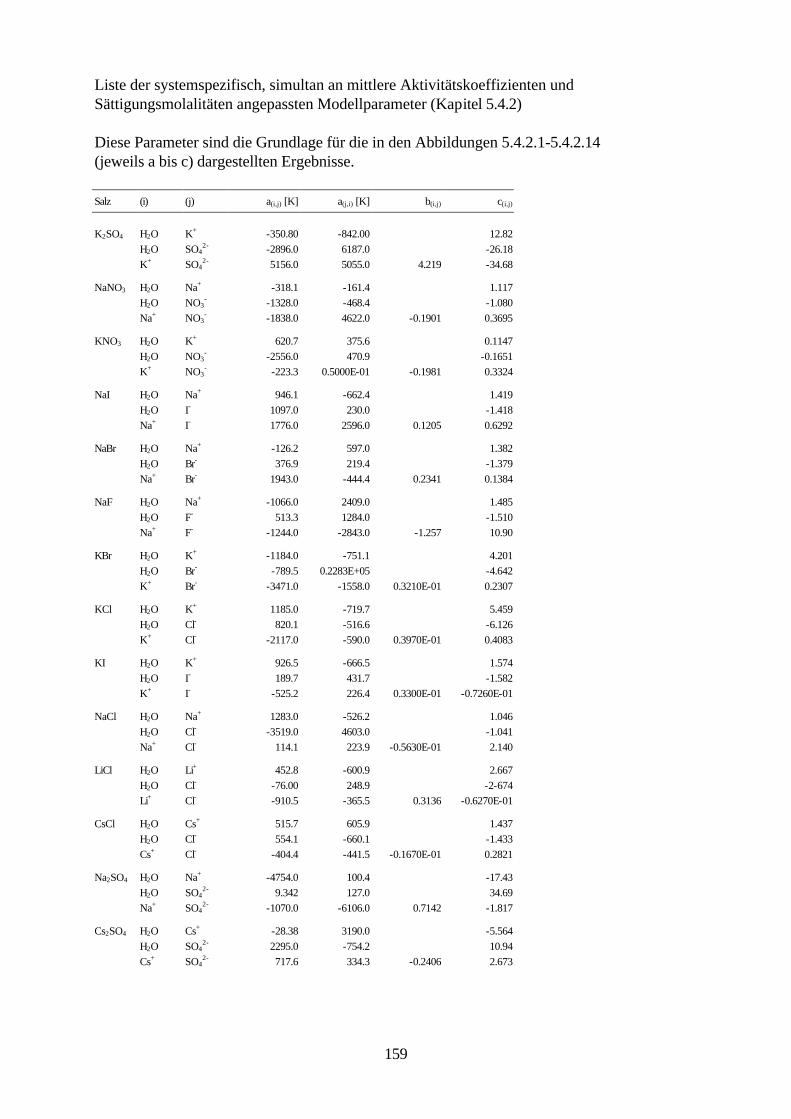
159
Liste der systemspezifisch, simultan an mittlere Aktivitätskoeffizienten und Sättigungsmolalitäten angepassten Modellparameter (Kapitel 5.4.2) Diese Parameter sind die Grundlage für die in den Abbildungen 5.4.2.1-5.4.2.14 (jeweils a bis c) dargestellten Ergebnisse. Salz (i) (j) a(i,j) [K] a(j,i) [K] b(i,j) c(i,j)
K2SO4 H2O K+ -350.80 -842.00 12.82
H2O SO42- -2896.0 6187.0 -26.18
K+ SO42- 5156.0 5055.0 4.219 -34.68
NaNO3 H2O Na+ -318.1 -161.4 1.117
H2O NO3- -1328.0 -468.4 -1.080
Na+ NO3- -1838.0 4622.0 -0.1901 0.3695
KNO3 H2O K+ 620.7 375.6 0.1147
H2O NO3- -2556.0 470.9 -0.1651
K+ NO3- -223.3 0.5000E-01 -0.1981 0.3324
NaI H2O
Na+ 946.1 -662.4 1.419
H2O I- 1097.0 230.0 -1.418 Na+ I- 1776.0 2596.0 0.1205 0.6292
NaBr H2O
Na+ -126.2 597.0 1.382
H2O Br- 376.9 219.4 -1.379 Na+ Br- 1943.0 -444.4 0.2341 0.1384
NaF H2O
Na+ -1066.0 2409.0 1.485
H2O F- 513.3 1284.0 -1.510 Na+ F- -1244.0 -2843.0 -1.257 10.90
KBr H2O K+ -1184.0 -751.1 4.201
H2O Br- -789.5 0.2283E+05 -4.642 K+ Br- -3471.0 -1558.0 0.3210E-01 0.2307
KCl H2O K+ 1185.0 -719.7 5.459 H2O Cl- 820.1 -516.6 -6.126 K+ Cl- -2117.0 -590.0 0.3970E-01 0.4083
KI H2O K+ 926.5 -666.5 1.574 H2O I- 189.7 431.7 -1.582 K+ I- -525.2 226.4 0.3300E-01 -0.7260E-01
NaCl H2O Na+ 1283.0 -526.2 1.046
H2O Cl- -3519.0 4603.0 -1.041 Na+ Cl- 114.1 223.9 -0.5630E-01 2.140
LiCl H2O Li+ 452.8 -600.9 2.667 H2O Cl- -76.00 248.9 -2-674 Li+ Cl- -910.5 -365.5 0.3136 -0.6270E-01
CsCl H2O Cs+ 515.7 605.9 1.437 H2O Cl- 554.1 -660.1 -1.433 Cs+ Cl- -404.4 -441.5 -0.1670E-01 0.2821
Na2SO4 H2O Na+ -4754.0 100.4 -17.43 H2O SO4
2- 9.342 127.0 34.69 Na+ SO4
2- -1070.0 -6106.0 0.7142 -1.817 Cs2SO4 H2O Cs+ -28.38 3190.0 -5.564 H2O SO4
2- 2295.0 -754.2 10.94 Cs+ SO4
2- 717.6 334.3 -0.2406 2.673
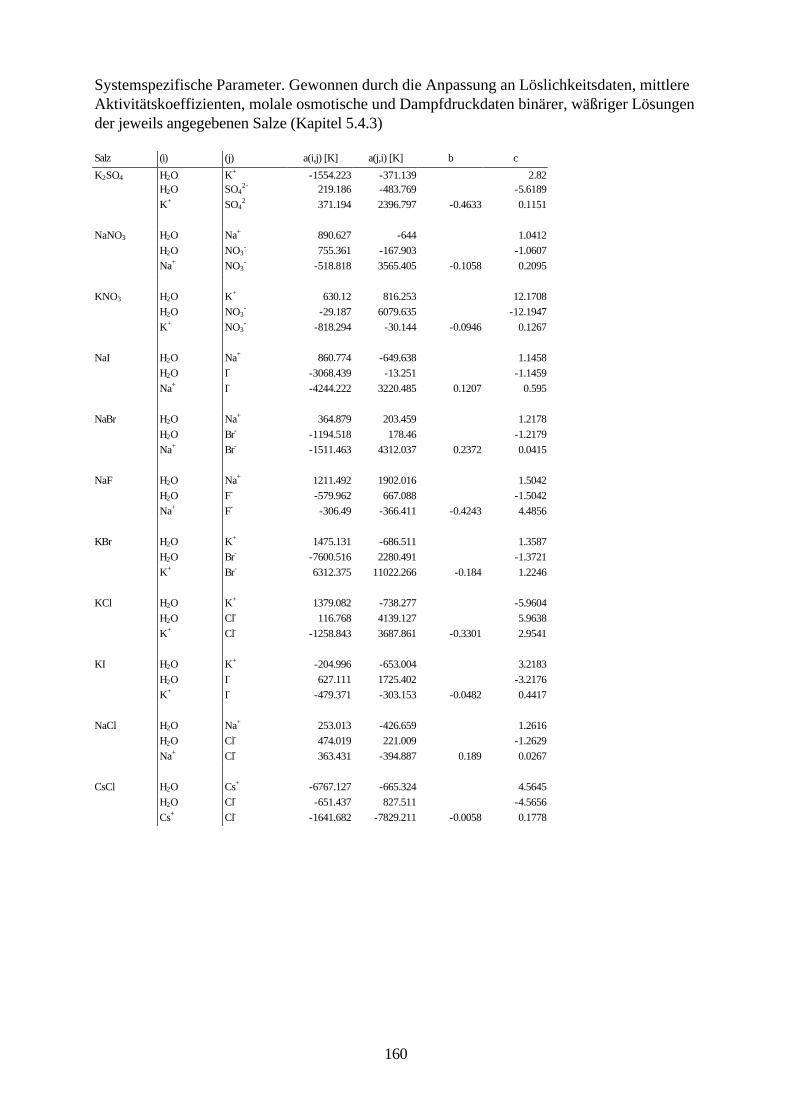
160
Systemspezifische Parameter. Gewonnen durch die Anpassung an Löslichkeitsdaten, mittlere Aktivitätskoeffizienten, molale osmotische und Dampfdruckdaten binärer, wäßriger Lösungen der jeweils angegebenen Salze (Kapitel 5.4.3) Salz (i) (j) a(i,j) [K] a(j,i) [K] b c
K2SO4 H2O K+ -1554.223 -371.139 2.82 H2O SO4
2- 219.186 -483.769 -5.6189 K+ SO4
2 371.194 2396.797 -0.4633 0.1151
NaNO3 H2O Na+ 890.627 -644 1.0412 H2O NO3
- 755.361 -167.903 -1.0607 Na+ NO3
- -518.818 3565.405 -0.1058 0.2095
KNO3 H2O K+ 630.12 816.253 12.1708 H2O NO3
- -29.187 6079.635 -12.1947 K+ NO3
- -818.294 -30.144 -0.0946 0.1267
NaI H2O Na+ 860.774 -649.638 1.1458 H2O I- -3068.439 -13.251 -1.1459 Na+ I- -4244.222 3220.485 0.1207 0.595
NaBr H2O Na+ 364.879 203.459 1.2178 H2O Br- -1194.518 178.46 -1.2179 Na+ Br- -1511.463 4312.037 0.2372 0.0415
NaF H2O Na+ 1211.492 1902.016 1.5042 H2O F- -579.962 667.088 -1.5042 Na+ F- -306.49 -366.411 -0.4243 4.4856
KBr H2O K+ 1475.131 -686.511 1.3587 H2O Br- -7600.516 2280.491 -1.3721 K+ Br- 6312.375 11022.266 -0.184 1.2246
KCl H2O K+ 1379.082 -738.277 -5.9604 H2O Cl- 116.768 4139.127 5.9638 K+ Cl- -1258.843 3687.861 -0.3301 2.9541
KI H2O K+ -204.996 -653.004 3.2183 H2O I- 627.111 1725.402 -3.2176 K+ I- -479.371 -303.153 -0.0482 0.4417
NaCl H2O Na+ 253.013 -426.659 1.2616 H2O Cl- 474.019 221.009 -1.2629 Na+ Cl- 363.431 -394.887 0.189 0.0267
CsCl H2O Cs+ -6767.127 -665.324 4.5645 H2O Cl- -651.437 827.511 -4.5656 Cs+ Cl- -1641.682 -7829.211 -0.0058 0.1778
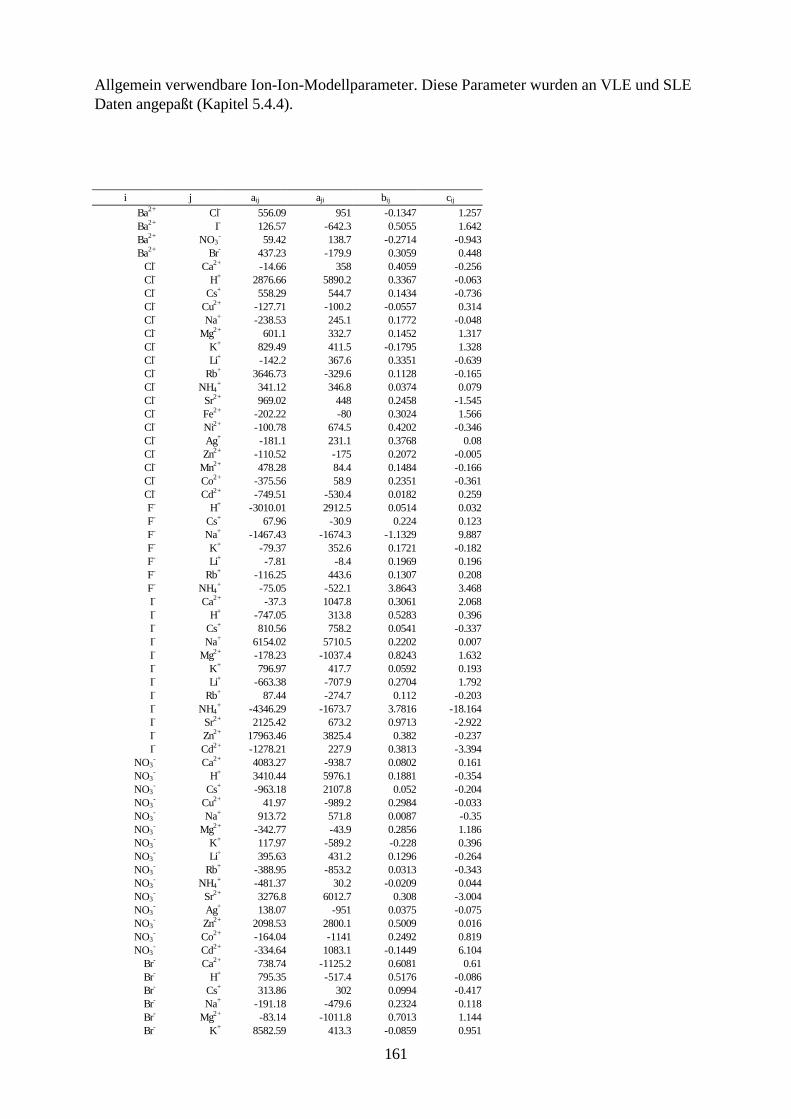
161
Allgemein verwendbare Ion-Ion-Modellparameter. Diese Parameter wurden an VLE und SLE Daten angepaßt (Kapitel 5.4.4).
i j aij aji bij cij Ba2+ Cl- 556.09 951 -0.1347 1.257 Ba2+ I- 126.57 -642.3 0.5055 1.642 Ba2+ NO3
- 59.42 138.7 -0.2714 -0.943 Ba2+ Br- 437.23 -179.9 0.3059 0.448
Cl- Ca2+ -14.66 358 0.4059 -0.256 Cl- H+ 2876.66 5890.2 0.3367 -0.063 Cl- Cs+ 558.29 544.7 0.1434 -0.736 Cl- Cu2+ -127.71 -100.2 -0.0557 0.314 Cl- Na+ -238.53 245.1 0.1772 -0.048 Cl- Mg2+ 601.1 332.7 0.1452 1.317 Cl- K+ 829.49 411.5 -0.1795 1.328 Cl- Li+ -142.2 367.6 0.3351 -0.639 Cl- Rb+ 3646.73 -329.6 0.1128 -0.165 Cl- NH4
+ 341.12 346.8 0.0374 0.079 Cl- Sr2+ 969.02 448 0.2458 -1.545 Cl- Fe2+ -202.22 -80 0.3024 1.566 Cl- Ni2+ -100.78 674.5 0.4202 -0.346 Cl- Ag+ -181.1 231.1 0.3768 0.08 Cl- Zn2+ -110.52 -175 0.2072 -0.005 Cl- Mn2+ 478.28 84.4 0.1484 -0.166 Cl- Co2+ -375.56 58.9 0.2351 -0.361 Cl- Cd2+ -749.51 -530.4 0.0182 0.259 F- H+ -3010.01 2912.5 0.0514 0.032 F- Cs+ 67.96 -30.9 0.224 0.123 F- Na+ -1467.43 -1674.3 -1.1329 9.887 F- K+ -79.37 352.6 0.1721 -0.182 F- Li+ -7.81 -8.4 0.1969 0.196 F- Rb+ -116.25 443.6 0.1307 0.208 F- NH4
+ -75.05 -522.1 3.8643 3.468 I- Ca2+ -37.3 1047.8 0.3061 2.068 I- H+ -747.05 313.8 0.5283 0.396 I- Cs+ 810.56 758.2 0.0541 -0.337 I- Na+ 6154.02 5710.5 0.2202 0.007 I- Mg2+ -178.23 -1037.4 0.8243 1.632 I- K+ 796.97 417.7 0.0592 0.193 I- Li+ -663.38 -707.9 0.2704 1.792 I- Rb+ 87.44 -274.7 0.112 -0.203 I- NH4
+ -4346.29 -1673.7 3.7816 -18.164 I- Sr2+ 2125.42 673.2 0.9713 -2.922 I- Zn2+ 17963.46 3825.4 0.382 -0.237 I- Cd2+ -1278.21 227.9 0.3813 -3.394
NO3- Ca2+ 4083.27 -938.7 0.0802 0.161
NO3- H+ 3410.44 5976.1 0.1881 -0.354
NO3- Cs+ -963.18 2107.8 0.052 -0.204
NO3- Cu2+ 41.97 -989.2 0.2984 -0.033
NO3- Na+ 913.72 571.8 0.0087 -0.35
NO3- Mg2+ -342.77 -43.9 0.2856 1.186
NO3- K+ 117.97 -589.2 -0.228 0.396
NO3- Li+ 395.63 431.2 0.1296 -0.264
NO3- Rb+ -388.95 -853.2 0.0313 -0.343
NO3- NH4
+ -481.37 30.2 -0.0209 0.044 NO3
- Sr2+ 3276.8 6012.7 0.308 -3.004 NO3
- Ag+ 138.07 -951 0.0375 -0.075 NO3
- Zn2+ 2098.53 2800.1 0.5009 0.016 NO3
- Co2+ -164.04 -1141 0.2492 0.819 NO3
- Cd2+ -334.64 1083.1 -0.1449 6.104 Br- Ca2+ 738.74 -1125.2 0.6081 0.61 Br- H+ 795.35 -517.4 0.5176 -0.086 Br- Cs+ 313.86 302 0.0994 -0.417 Br- Na+ -191.18 -479.6 0.2324 0.118 Br- Mg2+ -83.14 -1011.8 0.7013 1.144 Br- K+ 8582.59 413.3 -0.0859 0.951
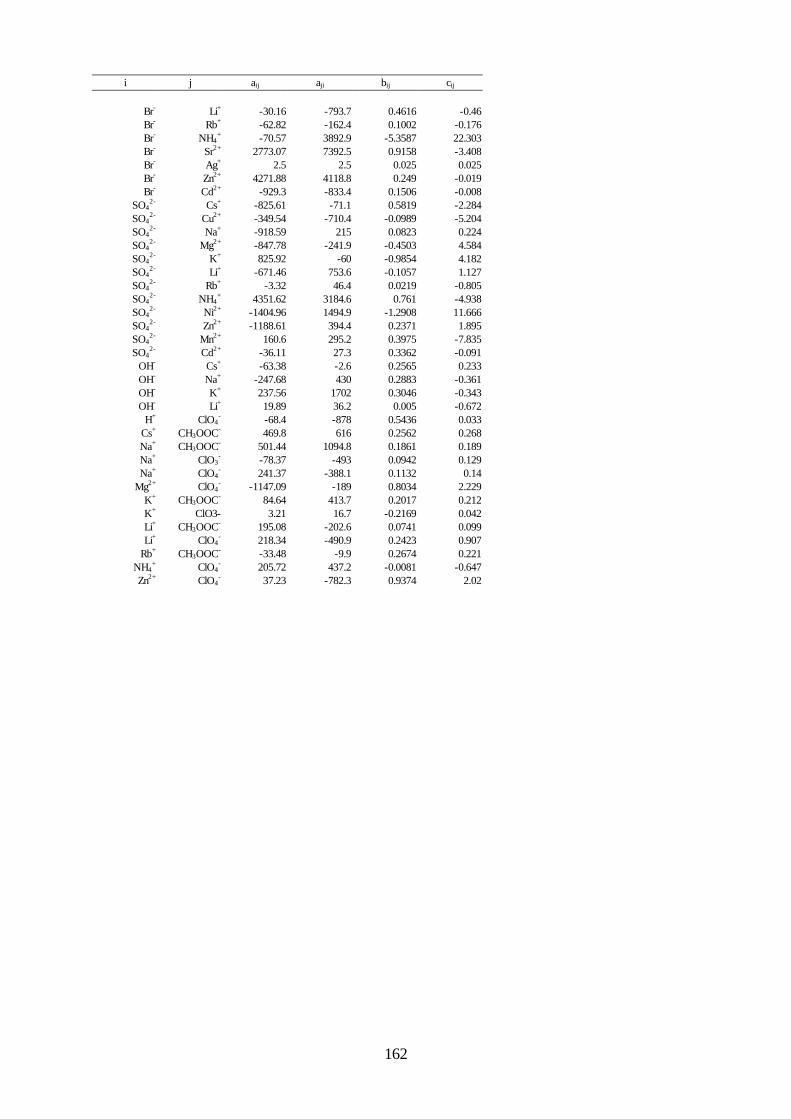
162
i j aij aji bij cij
Br- Li+ -30.16 -793.7 0.4616 -0.46 Br- Rb+ -62.82 -162.4 0.1002 -0.176 Br- NH4
+ -70.57 3892.9 -5.3587 22.303 Br- Sr2+ 2773.07 7392.5 0.9158 -3.408 Br- Ag+ 2.5 2.5 0.025 0.025 Br- Zn2+ 4271.88 4118.8 0.249 -0.019 Br- Cd2+ -929.3 -833.4 0.1506 -0.008
SO42- Cs+ -825.61 -71.1 0.5819 -2.284
SO42- Cu2+ -349.54 -710.4 -0.0989 -5.204
SO42- Na+ -918.59 215 0.0823 0.224
SO42- Mg2+ -847.78 -241.9 -0.4503 4.584
SO42- K+ 825.92 -60 -0.9854 4.182
SO42- Li+ -671.46 753.6 -0.1057 1.127
SO42- Rb+ -3.32 46.4 0.0219 -0.805
SO42- NH4
+ 4351.62 3184.6 0.761 -4.938 SO4
2- Ni2+ -1404.96 1494.9 -1.2908 11.666 SO4
2- Zn2+ -1188.61 394.4 0.2371 1.895 SO4
2- Mn2+ 160.6 295.2 0.3975 -7.835 SO4
2- Cd2+ -36.11 27.3 0.3362 -0.091 OH- Cs+ -63.38 -2.6 0.2565 0.233 OH- Na+ -247.68 430 0.2883 -0.361 OH- K+ 237.56 1702 0.3046 -0.343 OH- Li+ 19.89 36.2 0.005 -0.672
H+ ClO4- -68.4 -878 0.5436 0.033
Cs+ CH3OOC- 469.8 616 0.2562 0.268 Na+ CH3OOC- 501.44 1094.8 0.1861 0.189 Na+ ClO3
- -78.37 -493 0.0942 0.129 Na+ ClO4
- 241.37 -388.1 0.1132 0.14 Mg2+ ClO4
- -1147.09 -189 0.8034 2.229 K+ CH3OOC- 84.64 413.7 0.2017 0.212 K+ ClO3- 3.21 16.7 -0.2169 0.042 Li+ CH3OOC- 195.08 -202.6 0.0741 0.099 Li+ ClO4
- 218.34 -490.9 0.2423 0.907 Rb+ CH3OOC- -33.48 -9.9 0.2674 0.221
NH4+ ClO4
- 205.72 437.2 -0.0081 -0.647 Zn2+ ClO4
- 37.23 -782.3 0.9374 2.02
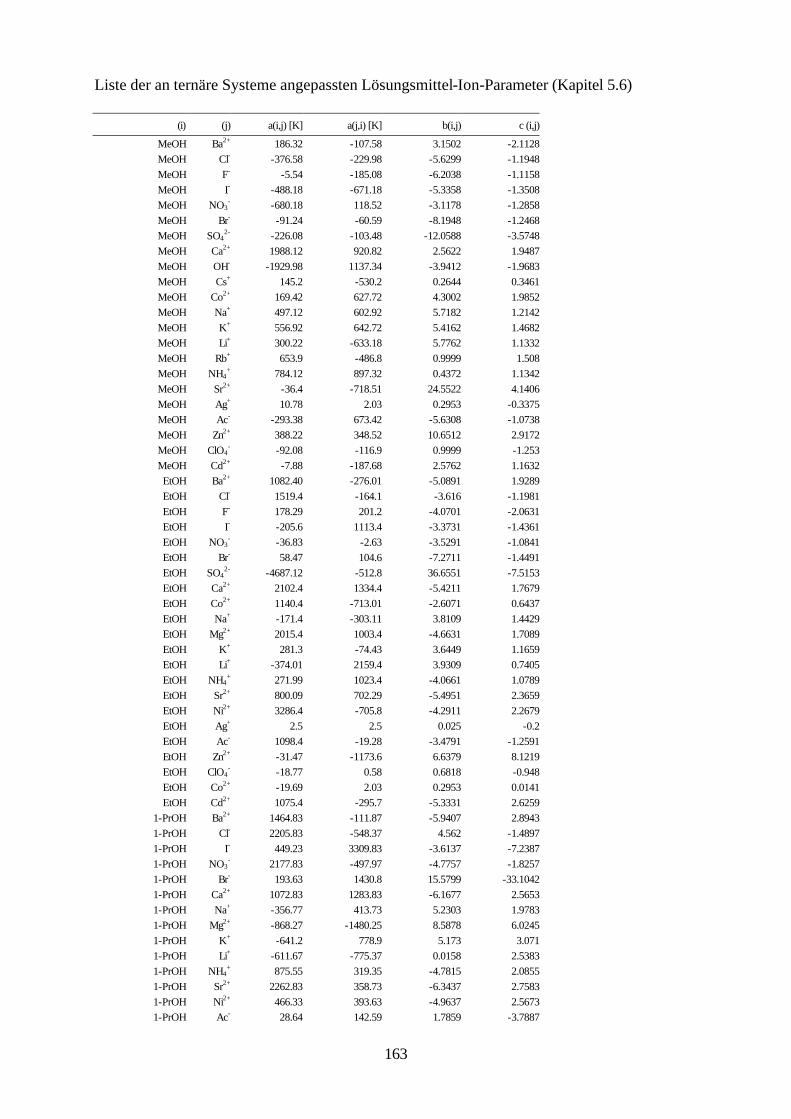
163
Liste der an ternäre Systeme angepassten Lösungsmittel-Ion-Parameter (Kapitel 5.6)
(i) (j) a(i,j) [K] a(j,i) [K] b(i,j) c (i,j)
MeOH Ba2+ 186.32 -107.58 3.1502 -2.1128MeOH Cl- -376.58 -229.98 -5.6299 -1.1948MeOH F- -5.54 -185.08 -6.2038 -1.1158MeOH I- -488.18 -671.18 -5.3358 -1.3508MeOH NO3
- -680.18 118.52 -3.1178 -1.2858MeOH Br- -91.24 -60.59 -8.1948 -1.2468MeOH SO4
2- -226.08 -103.48 -12.0588 -3.5748MeOH Ca2+ 1988.12 920.82 2.5622 1.9487MeOH OH- -1929.98 1137.34 -3.9412 -1.9683MeOH Cs+ 145.2 -530.2 0.2644 0.3461MeOH Co2+ 169.42 627.72 4.3002 1.9852MeOH Na+ 497.12 602.92 5.7182 1.2142MeOH K+ 556.92 642.72 5.4162 1.4682MeOH Li+ 300.22 -633.18 5.7762 1.1332MeOH Rb+ 653.9 -486.8 0.9999 1.508MeOH NH4
+ 784.12 897.32 0.4372 1.1342MeOH Sr2+ -36.4 -718.51 24.5522 4.1406MeOH Ag+ 10.78 2.03 0.2953 -0.3375MeOH Ac- -293.38 673.42 -5.6308 -1.0738MeOH Zn2+ 388.22 348.52 10.6512 2.9172MeOH ClO4
- -92.08 -116.9 0.9999 -1.253MeOH Cd2+ -7.88 -187.68 2.5762 1.1632EtOH Ba2+ 1082.40 -276.01 -5.0891 1.9289EtOH Cl- 1519.4 -164.1 -3.616 -1.1981EtOH F- 178.29 201.2 -4.0701 -2.0631EtOH I- -205.6 1113.4 -3.3731 -1.4361EtOH NO3
- -36.83 -2.63 -3.5291 -1.0841EtOH Br- 58.47 104.6 -7.2711 -1.4491EtOH SO4
2- -4687.12 -512.8 36.6551 -7.5153EtOH Ca2+ 2102.4 1334.4 -5.4211 1.7679EtOH Co2+ 1140.4 -713.01 -2.6071 0.6437EtOH Na+ -171.4 -303.11 3.8109 1.4429EtOH Mg2+ 2015.4 1003.4 -4.6631 1.7089EtOH K+ 281.3 -74.43 3.6449 1.1659EtOH Li+ -374.01 2159.4 3.9309 0.7405EtOH NH4
+ 271.99 1023.4 -4.0661 1.0789EtOH Sr2+ 800.09 702.29 -5.4951 2.3659EtOH Ni2+ 3286.4 -705.8 -4.2911 2.2679EtOH Ag+ 2.5 2.5 0.025 -0.2EtOH Ac- 1098.4 -19.28 -3.4791 -1.2591EtOH Zn2+ -31.47 -1173.6 6.6379 8.1219EtOH ClO4
- -18.77 0.58 0.6818 -0.948EtOH Co2+ -19.69 2.03 0.2953 0.0141EtOH Cd2+ 1075.4 -295.7 -5.3331 2.6259
1-PrOH Ba2+ 1464.83 -111.87 -5.9407 2.89431-PrOH Cl- 2205.83 -548.37 4.562 -1.48971-PrOH I- 449.23 3309.83 -3.6137 -7.23871-PrOH NO3
- 2177.83 -497.97 -4.7757 -1.82571-PrOH Br- 193.63 1430.8 15.5799 -33.10421-PrOH Ca2+ 1072.83 1283.83 -6.1677 2.56531-PrOH Na+ -356.77 413.73 5.2303 1.97831-PrOH Mg2+ -868.27 -1480.25 8.5878 6.02451-PrOH K+ -641.2 778.9 5.173 3.0711-PrOH Li+ -611.67 -775.37 0.0158 2.53831-PrOH NH4
+ 875.55 319.35 -4.7815 2.08551-PrOH Sr2+ 2262.83 358.73 -6.3437 2.75831-PrOH Ni2+ 466.33 393.63 -4.9637 2.56731-PrOH Ac- 28.64 142.59 1.7859 -3.7887

164
Tabelle (Kapitel 5.6) Fortsetzung
(i) (j) a(i,j) [K] a(j,i) [K] b(i,j) c (i,j)
1-PrOH Zn2+ -158.58 51.21 0.1065 2.7651-PrOH Cd2+ 180.6 389.8 -6.845 10.522-PrOH Ba2+ -264.52 1076.58 -36.6242 -63.99422-PrOH Cl- 2204.58 -99.42 -4.8562 -1.50022-PrOH F- 2556.58 333.38 11.4958 27.77582-PrOH I- -254.22 -574.82 10.72 31.96582-PrOH NO3
- 1196.58 -352.22 -4.7212 -1.69422-PrOH Br- 867.88 452.88 -9.7022 -1.62022-PrOH Ca2+ 376.78 46.03 -6.3762 2.72382-PrOH Na+ -326.72 131.78 5.0678 2.11982-PrOH Mg2+ 1086.96 -1266.71 69.1625 -19.10352-PrOH K+ -41.93 -211.72 -10.5442 -30.79422-PrOH Li+ -482.02 516.38 5.2558 1.23282-PrOH NH4
+ 117.58 164.78 -4.8752 1.58282-PrOH Ac- 0.58 0.58 0.0058 0.00582-PrOH ClO4
- 1986.58 -380.62 -4.3762 -0.7961Aceton Cl- 740.73 -332.47 -2.853 -1.1977Aceton F- 5310.91 8399.26 851.8508 -1814.874Aceton I- -72.74 5.92 10.5683 9.1073Aceton NO3
- -248.27 194.83 1.9853 -1.9367Aceton Br- -282.67 536.33 -7.4357 -1.2167Aceton Ca2+ -64.96 86.36 -2.4087 1.2843Aceton Cs+ 1044.51 9457.29 97.6688 -209.638Aceton Na+ 2165.83 790.23 -10.0617 -9.2817Aceton K+ -740.87 153.03 2.8073 1.6573Aceton Li+ 1555.83 1163.83 3.0733 1.2643Aceton Rb+ 366.88 413.21 0.6307 -8.9625Aceton Zn2+ -175.17 66.78 4.5333 3.4133Aceton ClO4
- 0 0 0 01-BuOH Cl- 770.73 846.23 6.8373 -1.39471-BuOH I- 2743.13 3134.13 -6.39 7.62831-BuOH Ca2+ -287.07 203.43 -14.1487 2.37931-BuOH Na+ 842.83 945.83 -6.9897 1.50431-BuOH Mg2+ 1285 839.6 -6.3 2.3241-BuOH Li+ 1936.13 -869.07 7.7427 -7.3797
THF Cl- 206.4 -257.6 0.5731 -2.1THF I- 32.26 365.7 -38.92 -75.58THF NO3
- 1858 508.6 0.5975 -4.233THF Ca2+ 218.1 36.37 -20.42 7.01THF Na+ 508.6 755.7 39.14 77.3THF K+ -2918 389.9 12.46 1.61THF Li+ -355 958 -0.0148 2.838THF Ac- 1000 751.8 -11.63 -3.987
MeAc Cl- 1214.25 637.55 0.7159 -1.2335MeAc Ca2+ 1493.25 -497.15 -21.2075 2.0255MeAc Na+ 504.75 -673.15 0.0653 2.0365MeAc K+ -144.65 -38.13 -0.4997 2.3755MeAc Ac- 239.35 83.08 0.4016 -1.2335MeAc Zn2+ -492.05 456.85 -2.3695 0.8437EtAc Cl- 120.4 397.5 -5.965 -23.52EtAc I- 339.8 410.3 -1.595 -0.923EtAc Ca2+ -397.3 121.6 -0.7972 3.875EtAc Co2+ -3926 -1072 -5.576 45.89EtAc Na+ 333.7 -262.8 2.253 1.944EtAc Mg2+ 338.7 -253.2 -1.043 1.643EtAc K+ 2036 -637 116.4 -13.57EtAc Ac- 2799 -847.8 -115.4 14.42EtAc Zn2+ -337.2 -168.2 0.1109 0.092EtAc ClO4
- 15.72 -0.35 -1.594 -1.612
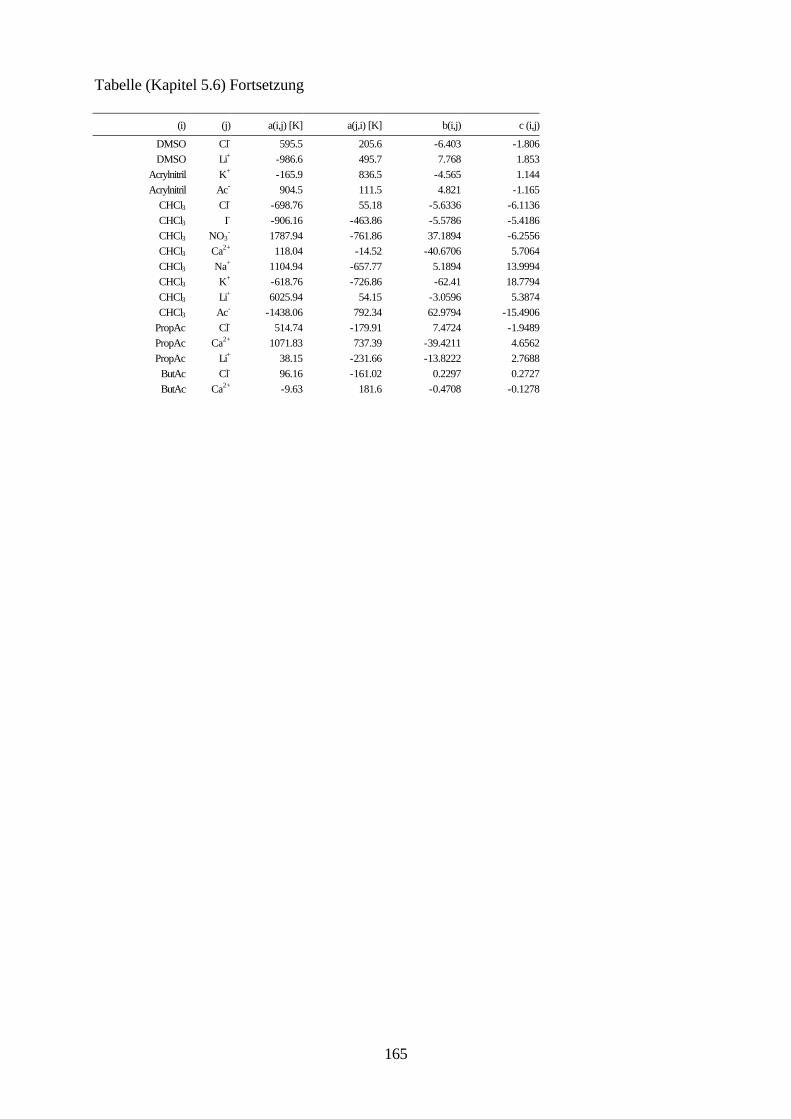
165
Tabelle (Kapitel 5.6) Fortsetzung
(i) (j) a(i,j) [K] a(j,i) [K] b(i,j) c (i,j)
DMSO Cl- 595.5 205.6 -6.403 -1.806DMSO Li+ -986.6 495.7 7.768 1.853
Acrylnitril K+ -165.9 836.5 -4.565 1.144Acrylnitril Ac- 904.5 111.5 4.821 -1.165
CHCl3 Cl- -698.76 55.18 -5.6336 -6.1136CHCl3 I- -906.16 -463.86 -5.5786 -5.4186CHCl3 NO3
- 1787.94 -761.86 37.1894 -6.2556CHCl3 Ca2+ 118.04 -14.52 -40.6706 5.7064CHCl3 Na+ 1104.94 -657.77 5.1894 13.9994CHCl3 K+ -618.76 -726.86 -62.41 18.7794CHCl3 Li+ 6025.94 54.15 -3.0596 5.3874CHCl3 Ac- -1438.06 792.34 62.9794 -15.4906
PropAc Cl- 514.74 -179.91 7.4724 -1.9489PropAc Ca2+ 1071.83 737.39 -39.4211 4.6562PropAc Li+ 38.15 -231.66 -13.8222 2.7688
ButAc Cl- 96.16 -161.02 0.2297 0.2727ButAc Ca2+ -9.63 181.6 -0.4708 -0.1278
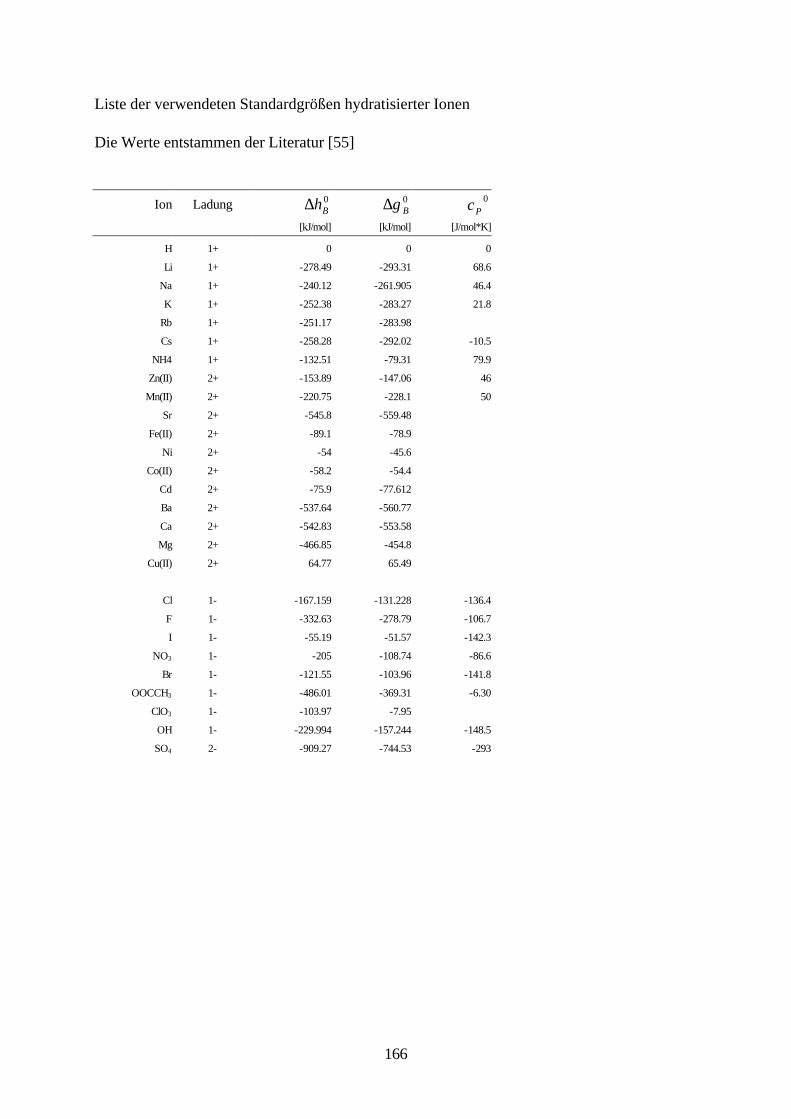
166
Liste der verwendeten Standardgrößen hydratisierter Ionen Die Werte entstammen der Literatur [55]
Ion Ladung 0Bh∆
0Bg∆
0Pc
[kJ/mol] [kJ/mol] [J/mol*K]
H 1+ 0 0 0
Li 1+ -278.49 -293.31 68.6
Na 1+ -240.12 -261.905 46.4
K 1+ -252.38 -283.27 21.8
Rb 1+ -251.17 -283.98
Cs 1+ -258.28 -292.02 -10.5
NH4 1+ -132.51 -79.31 79.9
Zn(II) 2+ -153.89 -147.06 46
Mn(II) 2+ -220.75 -228.1 50
Sr 2+ -545.8 -559.48
Fe(II) 2+ -89.1 -78.9
Ni 2+ -54 -45.6
Co(II) 2+ -58.2 -54.4
Cd 2+ -75.9 -77.612
Ba 2+ -537.64 -560.77
Ca 2+ -542.83 -553.58
Mg 2+ -466.85 -454.8
Cu(II) 2+ 64.77 65.49
Cl 1- -167.159 -131.228 -136.4
F 1- -332.63 -278.79 -106.7
I 1- -55.19 -51.57 -142.3
NO3 1- -205 -108.74 -86.6
Br 1- -121.55 -103.96 -141.8
OOCCH3 1- -486.01 -369.31 -6.30
ClO3 1- -103.97 -7.95
OH 1- -229.994 -157.244 -148.5
SO4 2- -909.27 -744.53 -293
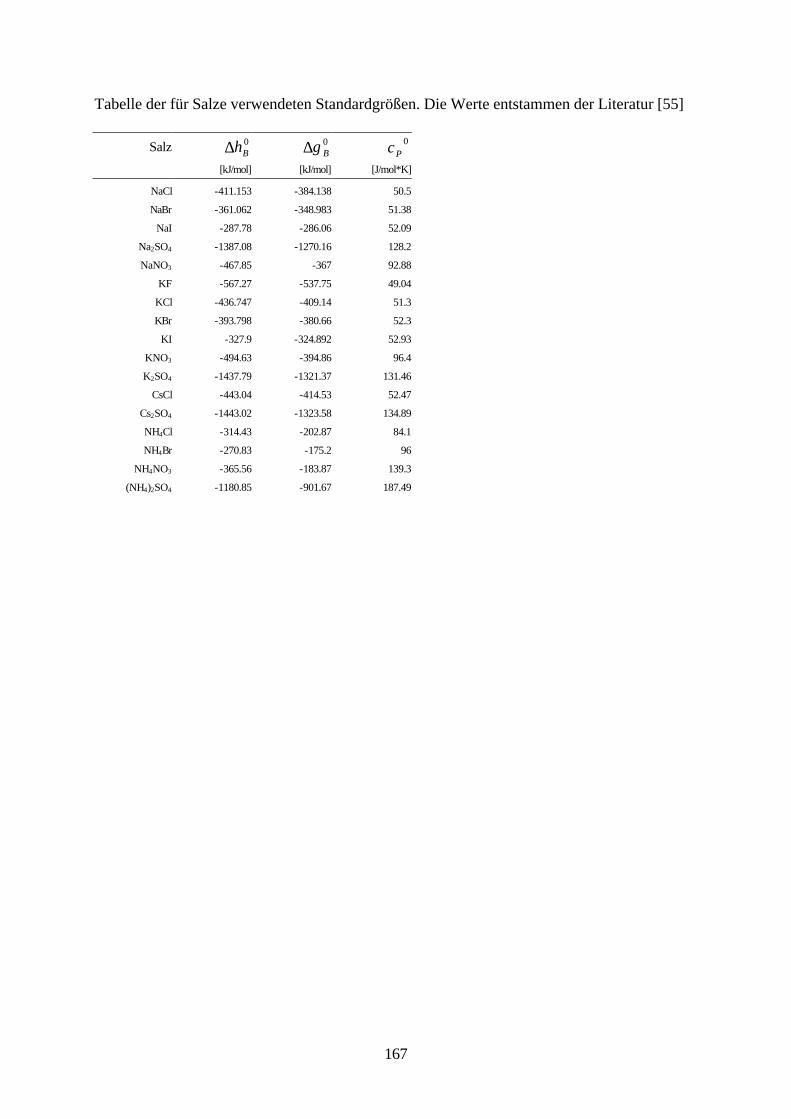
167
Tabelle der für Salze verwendeten Standardgrößen. Die Werte entstammen der Literatur [55]
Salz 0Bh∆
0Bg∆
0Pc
[kJ/mol] [kJ/mol] [J/mol*K]
NaCl -411.153 -384.138 50.5
NaBr -361.062 -348.983 51.38
NaI -287.78 -286.06 52.09
Na2SO4 -1387.08 -1270.16 128.2
NaNO3 -467.85 -367 92.88
KF -567.27 -537.75 49.04
KCl -436.747 -409.14 51.3
KBr -393.798 -380.66 52.3
KI -327.9 -324.892 52.93
KNO3 -494.63 -394.86 96.4
K2SO4 -1437.79 -1321.37 131.46
CsCl -443.04 -414.53 52.47
Cs2SO4 -1443.02 -1323.58 134.89
NH4Cl -314.43 -202.87 84.1
NH4Br -270.83 -175.2 96
NH4NO3 -365.56 -183.87 139.3
(NH4)2SO4 -1180.85 -901.67 187.49

168
7.3 Literaturverzeichnis Quellenangaben im Text (1) Abrams D.S., Prausnitz J.M., AIChE J., 21, 116 (1975) (2) Arrhenius S., Z. Physik. Chem., 1, 631 (1887) (3) Ball F.X., Fürst W., Renon H., AIChE J., 31 (3), 392 (1985) (4) Bondi A., Physical Properties of Molecular Cristals, Liquids and Glasses,
Wiley, New York (1968) (5) Brönstedt J.N., J. Am. Chem. Soc., 44 (5), 65 (1922) (6) Brönstedt J.N., J. Am. Chem. Soc., 45, 2898 (1923) (7) Bromley L.A., AIChE J., 19 (2), 313 (1973) (8) Cardoso M., O’Connell J.P., Fluid Phase Equilib., 33, 315 (1987) (9) Chen C.C., Britt H.I., Boston J.F., Evans L.B., AIChE J., 28 (4), 588 (1982) (10) Chen C.C., AIChE J., 32 (3), 444 (1986) (11) Cottrell F.G., J. Am. Chem. Soc., 41, 721 (1919) (12) Cruz J.L., Renon H., AIChE J., 24 (5), 817 (1978) (13) Dahl S., Macedo E.A., Ind. Eng. Chem. Res., 31, 1195 (1992) (14) Davis C.W., J. Chem. Soc., 2093 (1938) (15) Debye P., Hückel E., Physik Z., 24, 185 (1923) (16) Debye P., Physik. Z., 25, 97 (1924) (17) DIPPR Tables of Physical and Thermodynamic Properties of Pure Compounds,
AIChE, New York (1984) (18) Gmehling J., Onken U., Arlt W., Grenzheuser P., Weidlich U., Rarey J.
Vapor-Liquid Equilibrium Data Collection, DECHEMA Chemistry Data Series, Vol 1, 19, Bände, DECHEMA Frankfurt
(19) Gmehling J., Kolbe B., Thermodynamik, VCH-Verlag, Weinheim (1992) (20) Guggenheim E.A., Turgeon J.C., Trans. Faraday Soc., 51, 747 (1955)

169
(21) Holderbaum T., Gmehling J., Fluid Phase Equilib., 70, 251 (1991) (22) Holderbaum T., Ph. D. Thesis, Dortmund (1991) (23) Hovey J.K., Pitzer K.S., Tanger J.C., J. Phys. Chem., 94, 1175 (1990) (24) Izatt R.M., Eatough J.J., et al., J. Chem. Soc. (A), pp 47-53 (1969) (25) Jaques D., Furter W.F., AIChE J., 18 (2), 343 (1972) (26) Jenkins I.L., Monk C.B., JACS, 72, 2695 (1950) (27) Jin G., Donohue M.D., Ind. Eng. Chem. Res., 27, 1073 (1988) (28) Kikic I., Fermeglia M., Chem. Eng. Sci., 46 (11), 2775 (1991) (29) Li J., Polka H.M., Gmehling J., Fluid Phase Equilib., 94, 89-114 (1994) (30) Macedo E.A., Skovoborg P., Rasmussen P., Chem. Eng. Sci., 45 (4), 875 (1990) (31) Maryott A., Smith E.R.., Tables of Dielectric Constants of Pure Liquids, National
Bureau of Standards, Circular 514, (1951) (32) Mc Millan W.G., Mayer J.E., J. Chem. Phys., 13, 276 (1945) (33) Moore W.J., Hummel D.O., Physikalische Chemie, 3. Aufl., S. 272, Walter de
Gruyter, Berlin (1983) (34) Nelder J.A., Mead R., Comp. J., 7, 308 (1965) (35) Nicolaisen H., Rasmussen P., Sorensen J.M., Correlation and prediction of mineral
solubilities in the reciprocal salt system (Na+, K+) (Cl-, SO42-) - H2O at 0-100°C,
Chem.Eng.Sci., 48 (18), 3149 (1993) (36) Oster G., Water, a Comprehensive Treatise, Bd. 2, Kap. 7, Franks F. (Hrsg.), Plenum
Press, New York (1973) (37) Oster G. J. Am. Chem. Soc., 68, 2036 (1946) (38) Pitzer K.S., J. Phys.Chem., 77 (2), 268 (1973) (39) Pitzer K.S., Mayorga G., J. Phys. Chem., 77 (19), 2300 (1973) (40) Pitzer K.S., Acc. Chem. Res., 10, 371 (1977) (41) Pitzer K.S., Kim J.J., J. Am. Chem. Soc., 96, 5701 (1974) (42) Pitzer K.S., Activity Coefficients in Electrolyte Solutions, Bd. 1 Kap. 7

170
(43) Polka H.M., Experimentelle Bestimmung und Berechnung von Dampf-Flüssig-Gleichgewichten für Systeme mit starken Elektrolyten, Dissertation, Universität Oldenburg (1993)
(44) Prausnitz J.M., Molecular Thermodynamics of Fluid-Phase Equilibria, Prentice-Hall,
New Jersey (1969) (45) Prausnitz J.M., Gmehling J., vt-Hochschulkurs Thermische Verfahrenstechnik-
Phasengleichgewichte, Krausskopf Verlag, Mainz (1980) (46) Pytkowicz R.M. (Hrsg.), CRC Press Inc., Boca Raton, Florida (1979) (47) Ramm W.M., Absorptionsprozesse in der chemischen Technik, Verlag Technik,
Berlin (1952) (48) Redlich O., Kister A.T., Ind.Eng. Chem., 40, 345 (1948) (49) Righellato E.C., Davies C.W., ‘The extent of dissociation of salts in water, Part II. Uni-
bivalent salts’, Trans. Faraday Soc., 26, 592 (1930) (50) Robinson R.A., Stokes R.H., Electrolyte Solutions, 2. Auflage, Butterworth London
(1965) (51) Rozen A.M., Dokl. Akad. Nauk. SSSR 249 (1), 134 (1979) (52) Sander B., Fredenslund Aa., Rasmussen P., Chem. Eng. Sci., 41 (5), 1171 (1986) (53) Simon H.G., Kistenmacher H., Prausnitz J.M., Vortmeyer D., Chem. Eng. Proces., 29,
139 (1991) (54) Swietoslawski W., Ebulliometric Measurements, Kap.1., S.2ff, Reinhold Publishing
Corporation, New York (1945) (55) Wagman D.D., Evans W.H., Parker V.B., Schumm R.H., Halow I., Bailey S.M.,
Churney K.L., Nuttal R.L., The NBS tables of chemical thermodynamic properties, J.Phys.Chem. Ref. Data, 11 (2) (1982)
(56) Wilson G.M., J. Am. Chem. Soc., 86, 127 (1964) (57) Zachmann H.G., Mathematik für Chemiker, Verlag Chemie, Weinheim (1974)

171
Quellenangaben zum verwendeten Datenmaterial (58) Adamcova Z., Svatopluk B., Chem.Prum., 27 (12), 606 (1977) (59) Akerlöf G., Turck H.E., J. Am. Chem. Soc., 57, 1746 (1935) (60) Al-Sahaf T.A., Jabbar N.J., J. Chem. Eng. Data, 38, 522 (1993) (61) Alvarez Gonzales J.R., Labrador Toquero M.D.,..., An. Quim., 75, 670 (1979) (62) Andreae J.L., J. Prakt. Chem., 0, 456 (1884) (63) Applebey M.P., Crawford F.H., Gordon K., J. Chem. Soc, 0, 1665 (1934) (64) Azizov N.D., Akhundov T.S., Zarembo V.I., Zh. Prikl. Khim., 65 (2), 440 (1992) (65) Barthel J., Lauermann G., J. Solution Chem., 15 (10), 869 (1986) (66) Barthel J., Neueder R., Lauermann G., J. Solution Chem., 14 (9), 621 (1985) (67) Bedrossian A.A., Cheh H.Y., AIChE Symp. Ser., 140, Vol.70, 102 (68) Benrath A., Z. Anorg. Allg. Chem., 247, 147 (1941) (69) Berkeley Earl of, Philos. Trans. Roy. Soc. London A, 208, 189 (1904) (70) Bhatnagar O.M., Campbell A.N., Can. J. Chem., 59, 123 (1981) (71) Bixon E., Guerry R., Tassios G., J. Chem. Eng. Data, 24 (1), 10 (1979) (72) Blasdale W.C., J. Am. Chem. Soc., 45, 2935 (1923) (73) Boone J.E., Rousseau R.W., Schoenborn E.M., Advan. Chem. Ser., 155, 36 (1976) (74) Boone J.E., Thesis (75) Booth H.S., Bidwell R.M., J. Am. Chem. Soc., 72, 2567 (1950) (76) Brandani V., Del Re G., Di Giacomo G., Chim. Ind. (milan), 67, 392 (1985) (77) Broul M., Hlavaty K., Linek J., Collect. Czech. Chem. Commun., 34, 3428 (1969) (78) Campbell A.N., Bhatnagar O.N., Can. J. Chem., 57, 2542 (1979) (79) Caspari W.A., J. Chem. Soc., 125, 2381 (1924) (80) Chen F., Zhejiang Daxue Xuebao, 20, 113 (1986) (81) Chen X., Hou Y., J.Chem. Ind. Eng. (China), 1, 114 (1991)

172
(82) Chiavone-Filho O., Rasmussen P., J. Chem. Eng. Data, 38, 367 (1933) (83) Chretien A., Ann. Chim. (Paris), 12, 9 (1929) (84) Cui Zhiyu, Ghao Zhengong, J.Tianjin University 27 (3), 289 (1994) (85) De Coppet, Ann. Chim. (Paris), 30, 411 (1883) (86) Dobroserdov L.L., Bagrov I.V., Zh. Prikl. Khim. (Leningrad), 40, 926 (1967) (87) Dobroserdov L.L., Ilina V.P., Tr. Leningr. Tekhnol. Inst. Pishch. Prom., 14, 143 (1958) (88) Dobroserdov L.L., Ilina V.P., Tr. Leningr. Tekhnol. Inst. Pishch. Prom., 14, 139 (1958) (89) Dobroserdov L.L., Ilina V.P., Tr. Leningr. Tekhnol. Inst. Pishch. Prom., 14, 151 (1958) (90) Dobroserdov L.L., Ilina V.P., Tr. Leningr. Tekhnol. Inst. Pishch. Prom., 14, 147 (1958) (91) Dobroserdov L.L., Tr. Leningr. Tekhnol. Inst. Pishch. Prom., 15, 55 (1958) (92) Dobroserdov L.L., Zh. Prikl. Khim. 32, 2582 (1959) (93) Dobroserdov L.L., Zh. Prikl. Khim., 34, 386 (1961) (94) Duckett L.M., Hollifield J.M., Patterson C.S., J. Chem. Eng. Data, 31, 213 (1986) (95) Eddy R.D., Menzies A.W.C., J. Phys. Chem., 44, 207 (1940) (96) Ewing W.W., Mc Govern J.J., Mathews Jr. G.E., J. Am. Chem. Soc., 55, 4827 (1933) (97) Flöttmann F., Anal. Chem., 73, 1 (1927) (98) Foote H.W., Schairer J.F., J. Am. Chem. Soc., 52, 4202 (1930) (99) Fricke R., Z. Elektrochem. 35 (9), 631 (1929) (100) Froehlich W., J. Am. Chem. Soc, 68, 37 (1929) (101) Furter W.F., Thesis Univ. Of Toronto, Ontario (1958) (102) Galan M.A., Labrador M.D., Alvarez J.R, J. Chem. Eng. Data, 25 , 7 (1980) (103) Galan M.A., Labrador M.D., Alvarez J.R., Advan. Chem. Ser., 155, 85 (1976) (104) Geneva Leopold H., Johnston J., J. Am. Chem. Soc., 49, 1974 (1927) (105) Gibbard H.F., Scatchard G., Rousseau R.A., Creek J.L., J. Chem. Eng. Data 19 (3),
281 (1974) (106) Gu Feiyan, Cui Linlin, Li Xiangjie, ...,International Symposium Beijing, 574 (1994)

173
(107) Hakuta T., Goto T., Ishizaka S., Bull. Soc. Sea Water Sci., Japan, 28, 151 (1974) (108) Hamer W.J., Wu Y.C., J. Phys. Chem. Ref. Data, 1 (4), 1047 (1972) (109) Harrison W.R., Perman E.P., Trans. Faraday Soc., 13, 1 (1927) (110) Hashitani M., Hirata M., Hirose Y., Kagaku Kogaku, 32, 182 (1968) (111) Hashitani M., Hirata M., J. Chem. Eng. Jpn., 1 (2), 116 (1968) (112) Hashitani M., Hirata M., J. Chem. Eng. Jpn., 2 (2), 149 (1969) (113) Hayward A.M., Perman E.P., Trans. Faraday Soc., 27 ,59 (1931) (114) Herrington T.M., Jackson R.J., J. Chem. Soc., Faraday Trans., 63 , 1635 (1973) (115) Hoffmann F., Voigt W., Int. Electron. J. Phys.-Chem. Data 2, 31 (1996) (116) Holmes H.F., Mesmer R.E., J. Chem. Thermodyn. 28, 1325 (1996) (117) Hongo M., Kusunoki M., Kumagae Y., Arai Y., J. Chem. Thermodyn., 24, 649 (1992) (118) Hubert N., Gabes Y., Bourdet J.-B., Schuffenecker L., J. Chem. Eng. Data 40, 891
(1995) (119) Hufschmidt D., Rose C., Unpublished Data (1997) (120) Iliuta M.C., Thyrion F.C., Fluid Phase Equilib. 257, 103 (1995) (121) Iliuta M.C., Thyrion F.C., J. Chem. Eng. Data 41, 402 (1996) (122) Jaques D., Furter W.F., Ind. Eng. Chem. Fundam., 13 (3), 238 (1974) (123) Johnson A.I., Furter W.F., Can. J. Chem. Eng. 38, 78 (1960) (124) Johnson A.I., Furter W.F., Can. J. Technol., 34, 413 (1957) (125) Johnson G.C., Smith R.P., J. Am. Chem. Soc., 63, 1351 (1941) (126) Kharin S.E., Perelygin V.M., Remozov G.P., Izv. Vyssh. Uchebn. Zhaved. Khim.
Khim. Tekhnol, 12, 424 (1969) (127) Kolker A.M., Vatagin V.S., Krestov G.A., Russ. J. Phys. Chem., 59 (6), 843 (1985) (128) Kumagae Y., Mishima K., Hongo M., Kusunoki M., Arai Y., Can. J. Chem. Eng., 70,
1180 (1992) (129) Kupina N.A., Tr. Leningr. Tekhnol. Inst. Im. Lensoveta, 40, 92 (1957)

174
(130) Kupriyanova Z.N.,..., Zh. Prikl. Khim. (Leningrad), 46 (1), 234 (1973) (131) Larson R.G, Hunt H., J. Phys. Chem., 43, 417 (1939) (132) Lee H.C., Miyahara Y., Bull. Chem. Soc. Jpn., 51 (11), 3140 (1978) (133) Li B., Luo Y., Zhu Z., J. Chem. Ind. Eng. (China), 1 (2), 102 (1986) (134) Lin C.L., Lee L.S., Tseng H.C., J. Chem. Eng. Data, 38, 306 (1993) (135) Liu W., Zhu J., Huagong Xuebao, 41 (3), 372 (1990) (136) Maizlish R.S., Tverdovskij I.P., Zh. Fiz. Khim., 27, 1597 (1953) (137) Mato F., Fernandez-Polanco F., Cocero M.J., An. Quim., 83, 114 (1987) (138) Meranda D., Furter W.F., AIChE J., 18 (1), 111 (1972) (139) Meranda D., Furter W.F., AIChE J., 20 (1), 103 (1974) (140) Meusser A., Z. Anorg. Allg. Chem., 44, 79 (1905) (141) Meyer T., Polka H.M., Gmehling J., J. Chem. Eng. Data 36, 340 (1991) (142) Meyerhoffer W., Z. Phys. Chem., 53, 513 (1905) (143) Ming Z., Unpublished Data (1997) (144) Mishima K., Matsubara K., Arai Y., Kagaku Kogaku Ronbunshu, 13, 850 (1987) (145) Mondeja Gonzalez D., Thesis, Technical University Of Lodz (1978) (146) Morrison J.F., Baker J.C., Meredith H.C., Newman .E., J. Chem. Eng. Data, 35, 395
(1990) (147) Natarajan T.S., Srinivasan D., J. Chem. Eng. Data, 25, 218 (1980) (148) Nishi H., Kanai N., Kenkyu Kiyo-Wakayama Kogyo Koto Senmon Gakko, 20, 47
(1985) (149) Nishi H., Nagao E., Kenkyu Kiyo-Wakayama Kogyo Koto Senmon Gakko, 25, 71
(1990) (150) Nishi Y., J. Chem. Eng. Jpn. 8 (3), 187 (1975) (151) Ohe S.,..., Kagaku Kogaku, 34, 325 (1970) (152) Ohe S., Kagaku Kogaku, 34, 1112 (1970) (153) Ohe S., Thesis, Tokyo Metro. Univ. (1971)

175
(154) Ohe S., Yokoyama K., Nakamura S., J. Chem. Eng. Data, 16 (1), 70 (1971) (155) Ohe S., Yokoyama K., Nakamura S., Kogyo Kagaku Zasshi, 72, 313 (1969) (156) Park W.K., Do K.S., Bae H.K., Shim H.S., Rep. Inst. Ind. Technol. Yeung Nam Univ.,
1, 33 (1973) (157) Park W.K., Doh K.S., Park Y.T., Hwahak Konghak, 16 (6), 397 (1978) (158) Patil K. R., Olive F., Coronas A., J. Chem. Eng. Jpn., 27 (5), 680 (1994) (159) Patil K.R., Tripathi A.D., Pathak G., Katti S.S., J. Chem. Eng. Data, 36, 225 (1991) (160) Patil K.R., Chaudhari S.K., Katti S.S., J. Chem. Eng. Data, 37, 136 (1992) (161) Patil K.R., Tripathi A.D., Pathak G., Katti S.S., J. Chem. Eng. Data, 36, 225 (1991) (162) Patil R., Tripathi A.D., Pathak G., Katti S.S., J. Chem. Eng. Data, 35, 166 (1990) (163) Patil R.K., Olive F., Coronas A., J. Chem. Eng. Jpn., 27 (5), 680 (1994) (164) Pena M.P., Vercher E., Martinez-Andreu A., J .Chem. Eng. Data 40, 311 (1995) (165) Pena M.P., Vercher E., Martinez-Andreu A., J. Chem. Eng. Data 41, 1097 (1996) (166) Pinho S.P., Macedo E.A., Fluid Phase Equilib., 116, 209 (1996) (167) Poczopko S., Chmarzynski A., Rocz. Chem., 49 (6), 1127 (1975) (168) Polka H.M., Thesis Oldenburg (1993) (169) Rajendran M., Renganarayanan S., Madhavan P., Srinivasan D., Chem. Eng.
Commun., 74, 179 (1988) (170) Rard J.A., J. Chem. Eng. Data, 37 (4), 433 (1992) (171) Rard J.A., Spedding F.H., J. Chem. Eng. Data, 22, 56 (1977) (172) Renyi S., J. Chem. Eng. (China) 25, 13 (1995) (173) Renz M., Steimle F., Sci. Tech. Froid 1, 83 (1980) (174) Ricci J.E., Yanik N.S., J. Am. Chem. Soc., 59, 491 (1937) (175) Richards T.W., Yngve V., J. Am. Chem. Soc., 40, 164 (1918) (176) Rius Miro A., Alvarez Gonzalez J.R., An. R. Soc. Esp. Fis. Quim., 54 B, 797 (1958) (177) Rius Miro A., Alvarez Gonzalez J.R., Uriante Hulda A.,
An. R. Soc. Esp. Fis. Quim., 56 B, 629 (1960)

176
(178) Rius Miro A., Otero De La Gandara J.L., An. R. Soc. Esp. Fis. Quim., 53 B, 171
(1957) (179) Rius Miro A., Otero De La Gandara J.L., An. R. Soc. Esp. Fis. Quim., 53 B, 185
(1957) (180) Robinson R.A., Mccoach H.J., J. Am. Chem. Soc., 69 , 2244 (1947) (181) Robinson R.A., Stokes R.H,..., Electrolyte Solutions, 478 (1965) (182) Robinson R.A., Stokes R.H.,..., Electrolyte Solutions, 476 (1965) (183) Robinson R.A., Stokes R.H., Trans. Faraday Soc., 45, 612 (1949) (184) Rodebush W.H., J. Am. Chem. Soc., 40, 1204 (1918) (185) Sada E., Morisue T., J. Chem. Eng. Jpn., 8 (3), 196 (1975) (186) Sada E., Morisue T., Miyahara K., J. Chem. Eng. Data, 20 (3), 283 (1975) (187) Sada E., Morisue T., Tsuboi N., Advan. Chem. Ser., 155, 75 (1976) (188) Sada E., Morisue T., Yamaji H., Can. J. Chem. Eng., 53, 350 (1975) (189) Saugier M.T., Noailly M., Cohen-Adad R.,..., J. Therm. Anal., 11, 87 (1977) (190) Schmitt D., Thesis, Karlsruhe 1979 (191) Schreiber E., Schuettau E., Rant D., Schuberth H., Z. Phys. Chem. (Leipzig), 247, 23
(1971) (192) Schroeder W.C., Gabriel A., Partridge E.P., J. Am. Chem. Soc., 57, 1539 (1935) (193) Schuberth H., Z. Phys. Chem. (Leipzig), 255, 165 (1974) (194) Scott A.F., Durham E.J., J. Phys. Chem., 34, 1424 (1930) (195) Scott A.F., Frazier W.R., J. Am. Chem. Soc, 0, 459 (0) (196) Seung-Kyo Oh, Campbell S.W., J. Chem. Eng. Data 40, 504 (1995) (197) Seung-Kyo Oh, J. Chem. Eng. Data 42, 1082 (1997) (198) Shaw R., Butler J.A.V., Proc. Roy. Soc. London, 129 A, 519 (1930) (199) Shouyu W., Shiyou Huagong, 20 (5), 321 (1991) (200) Sidgwick N.V., Ewbank E.K., J. Chem. Soc., 124, 2268 (1925)

177
(201) Simmons J.P., Freimuth H., Russel H., J. Am. Chem. Soc., 58, 1692 (1936) (202) Skabichevskii P.A., Russ. J. Phys. Chem.,43 (10), 1432 (1969) (203) Skabichevskii P.A., Russ. J. Phys. Chem., 44 (5), 739 (1970) (204) Skabichevskii P.A., Russ. J. Phys. Chem., 44 (8), 62 (1970) (205) Smith H.A., Combs R.L., Googin J.M., J. Phys. Chem., 58, 997 (1954) (206) Smith R.P., Hirtle D.S., J. Am. Chem. Soc., 61, 1123 (1939) (207) Smith R.P., J. Am. Chem. Soc., 61, 500 (1939) (208) Stokes R.H., Trans. Farady Soc., 44, 295 (1948) (209) Storonkin A.V., Simanavichus L.E., Vestn. Leningr. Univ., Fiz. Khim., 22 (4), 103
(1957) (210) Sun R., J. Chem. Ind. Eng. (China) 47, 401 (1996). (211) Tatsievskaya G.I., Vitman T.A., Kushner T.M., Serafimov L.A.,
Russ. J. Phys. Chem., 56, 1668 (1982) (212) Tatsievskaya G.I., Vitman T.A., Kushner T.M., Serafimov L.A., Russ. J. Phys. Chem.,
56, 1443 (1982) (213) Tower O.F., Germann A.F.O., J. Am. Chem. Soc., 36, 2449 (1914) (214) Uchizono Y.,..., Kagaku Kogaku Ronbunshu, 9, 485 (1983) (215) Ushio H., Trimm H.H., Patel R.M., Zemany M.D., J. Solution Chem., 10 (1), 39 (1981) (216) Vercher E., Pena M.P., Martinez-Andreu A., J. Chem. Eng. Data 40, 657 (1995) (217) Vercher E., Pena M.P., Martinez-Andreu A., J. Chem. Eng. Data 41, 66-69, (1996) (218) Vercher E., Munoz R., Martinez-Andreu A., J. Chem. Eng. Data, 36, 274 (1991) (219) Vercher E., Pena M.P., Martinez-Andreu A., J. Chem. Eng. Data 41, 748 (1996) (220) Wang S., Shiyou Huagong, 21 (11), 737 (1992) (221) Wang Shouyu, Cai Shuxing, J. Chem. Eng. (China) 22 (5), 36 (1994) (222) Weidong Y., Yijin X., Shijun H., Acta Phys. Chim. Sinica 11 (5), 454 (1995) (223) Wittwer F., Rose C., Unpublished Data (1996) (224) Wittwer F., Thesis Oldenburg (1996)

178
(225) Wright R., J. Chem. Soc., 0, 1334 (1927) (226) Yamamoto H., Terano T., Nishi Y., Tokunaga J., J. Chem. Eng. Data 40, 472 (1995) (227) Yamamoto H., Fukase K., Shibata J., J. Chem. Eng. Data 41, 1066 (1996) (228) Yan W., Rose C., Unpublished Data (1996) (229) Zemp R.J., Francesconi A.Z., J. Chem. Eng. Data, 37, 313 (1992)

179
Zusammenfassung
Mit Hilfe des Salzeffektes lassen sich viele Trennprobleme der industriellen Praxis, wie z.B. die
Verschiebung azeotroper Punkte oder die Beeinflussung von Mischungslücken geschickt
lösen. Grundlage für die optimale Nutzung des Effektes stellen thermodynamische Modelle
dar, mit denen eine zuverlässige Berechnung des Phasengleichgewichts elektrolythaltiger
Mischphasen möglich ist. Das in Oldenburg entwickelte LIQUAC-Modell zur Berechnung der
Gibbsschen Exzeßenthalpie elektrolythaltiger Mischphasen zeichnet sich u.a. dadurch aus, daß
es ähnlich leistungsfähig wie der von Pitzer vorgeschlagene Virialansatz ist, darüber hinaus
aber eine bessere Beschreibung des Konzentrationsbereiches oberhalb 6 molaler Lösungen
ermöglicht. In der vorliegenden Arbeit wurde die Anwendbarkeit des LIQUAC-Modells, durch
simultane Anpassung an Dampf-Flüssig- und Fest-Flüssig-Phasengleichgewichtsdaten erwei-
tert. Das Ziel der Arbeit liegt in der Bestimmung einheitlicher Modellparameter, die die simul-
tane Beschreibung des Dampf-Flüssig- und Fest-Flüssig-Phasengleichgewichtes von
Lösungen starker Elektrolyte in Wasser und nichtwäßrigen Lösungsmittelgemischen
ermöglichen. Entsprechend dieser Zielsetzung erfolgte die Anpassung der Modellparameter
schrittweise unter Berücksichtigung der jeweils zu klärenden Teilaspekte. Die
zugrundeliegende Datenbasis wurde für jeden Anpassungsschritt um die entsprechenden
Datensatztypen erweitert. In einem ersten Schritt galt es die tabellierten Gibbsschen Standard-
bildungsenthalpien für die Berechnung des Löslichkeitsprodukts durch Anpassung systemspe-
zifischer Modellparameter an Löslichkeitsdaten zu prüfen. Anschließend wurde durch simul-
tane Anpassung an Löslichkeitsdaten und experimentelle mittlere Aktivitätskoeffizienten die
gleichzeitige Beschreibbarkeit der Temperatur- und Konzentrationsabhängigkeit der Salzakti-
vitätskoeffizienten überprüft. Die anschließende Erweiterung der Datenbasis um molale
osmotische Koeffizienten und Dampfdruckdaten gab Aufschluß über die Möglichkeit der
gleichzeitigen Beschreibung von Salz- und Lösungsmittelaktivität. Die Ergebnisse zeigen, daß
es mit Hilfe der neugewonnenen Modellparameter gelingt gleichzeitig Dampf-Flüssig- und
Fest-Flüssig-Phasengleichgewichte der wäßrigen Systeme zu beschreiben. Die durch-
schnittlich höheren Abweichungen im Fall der Lösungsmittelgemische rühren in erster Linie
daher, daß die Solvatationsenthalpien der Ionen in organischen Lösungsmitteln nicht bekannt
sind, und daß die Annahme vollständiger Dissoziation der Salze nicht mehr gegeben ist.
Im experimentellen Teil der Arbeit wurden Dampf-Flüssig-Phasengleichgewichte einer Reihe
ternärer Systeme vermessen, über die noch keine oder ungenügende experimentelle Informa-

180
tionen vorliegen. Alle Systeme bestehen aus jeweils zwei Lösungsmitteln und einem Salz, wo-
bei ein Teil der salzfreien Gemische Azeotrope bildet. Es wurden sowohl isobare Siedekurven
mit Hilfe eines nach Trampe und Eckert modifizierten Scott-Ebulliometers, als auch die
Dampfphasenzusammensetzungen verschiedener Isothermen mit Hilfe der Headspace-Gas-
chromatographie bestimmt. Die Meßreihen erstrecken sich über den gesamten Konzentra-
tionsbereich bis hin zur Sättigung. Die Meßergebnisse bestätigen das in den meisten Fällen
beobachtete ‘Aussalzen’ der organischen Lösungsmittelkomponenten im Vergleich zu dem
entsprechenden salzfreien Systemen. Bei der Berechnung der untersuchten Systeme ergaben
sich gute Übereinstimmungen mit den experimentellen Werten.




























