Embed Size (px)
Citation preview

Zur Kenntnis des Pinens 1. ober eine neue partielle Synthese des Pinens aus
einem Pinenderivat vt7n
L. Ruzieka und H. Trebler. (4. x. 'LO )
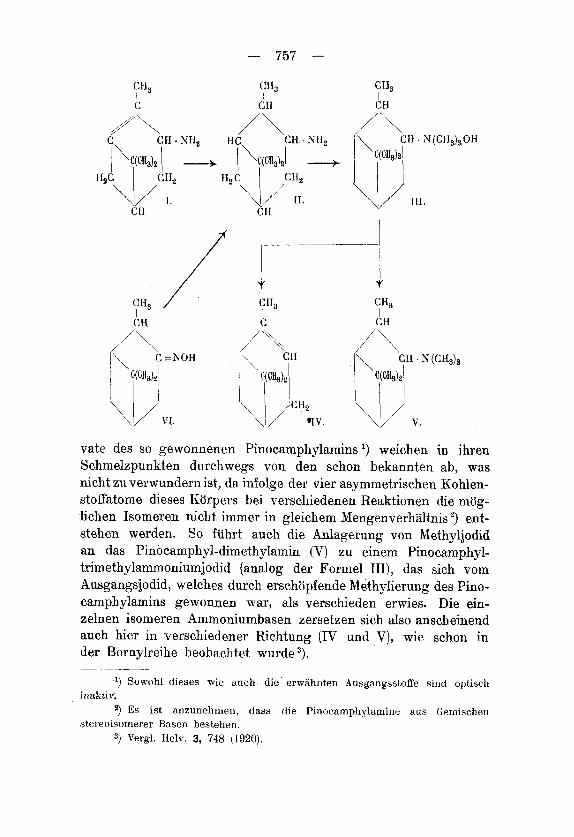
Aus einigen Derivaten des Pinens, die noch das unveriinderte Tetroceanskelett aufweisen, war es moglich, den Kohlenwasserstoff zu regenerieren. Ihreni Mechanismus nach sind nur zwei dieser Methoden derart gestaltet, dass sie als Endreaktioneri fur einen weitergreifenden Aufbau des Pinens in Betracht kommen konnten: die von Tt nZ1ac.h I ) heohachtete Bildung eines Kohlenwasserstoff- gemisches mit 1O'' io n-Pinengehalt bei der Destillation der Nopinol- essigsaure, und hauptsachlich die Zersetzung des Pinocamphyl- xanthogenats von Tschugeef2), die anscheinend zu reinem n-Pinen fuhrt'). Die Gewinnung des Pinens aus seinem Nitrosochlorid ') sowie aus Myrtenol ') sind als Eiidpunkte eines direkten Aufhaus dieses Kohlenwasserstoffs weniger g u t denkbar.
Eine weitere Methode cler ersteren Art schien uns die Zer- setzung des Pinocamphyl-trirnethylammoniumhydroxyds (111) zu sein. Es entsteht dabei in der Tat neben Pinocamphyl-ciimethylainin (V) reines a-Pinen (1V).
Das als Ausgangskorper notige Pinocamphylamin (II), das von l4'nZZach ') durch Reduktion des Pinocaniphonoxinis (VI) und von Tilclen ') aus Nitrosopinen (VII) hergestellt wurde, verschafften wir uns durch katalytische Reduktion des Pinylainins (I). Die Deri-
A . 368, 1 (1909) 'X . 39, 1324 (1908). Yoweit sich aus Clem Hefernt C 1908, 1. 1180 enttiehinc~n Insst.
I.l'ccllnch, A . 253, 251 (1889) Se,itmZer utid Bnvfelt, R. 40, 1368 (1907). A . 313, 367 (1900); s. T s c h u g ~ f C: 1908, 1 1179. Sac. 89, 1560 (1906).
- 757 -
CH, I C
C‘H, CH3 I
CH I
CH
-I Y
CH3 I
CH CH3 CH I /’ !H3 C I
vate des so gewonnenen Pinocamphylamins ’) weichen in ihren Schmelzpunkten durchwegs von den schon bekannten ab, was nicht zu verwundern ist, da infolge der vier asymmetrischen Kohlen- stoffatome dieses Korpers bei verschiedenen Reaktionen die mog- lichen Isomeren nicht immer in gleichem Mengenverhaltnis ’) ent- stehen werden. So fiihrt auch die Anlagerung von Methyljodid an das Pinocamphyl-dimethylamin (V) zu einem Pinocamphyl- trimethylammoniumjodid (analog der Formel III), das sich vom Ausgangsjodid, welches durch erschopfende Methylierung des Pino- camphylamins gewonnen war, als verschieden erwies. Die ein- zelnen isomeren Ammoniumbasen zersetzen sich also anscheinend auch hier in verschiedener Richtung (IV und V), wie schon in der Bornylreihe beobachtet wurde ”). - _____
l) Sowohl tlieses wie such die erwahnten Ausgangsstoffe sind optisch
Es 1st anzunehmen, dass die Pinocamphylamine am Gemischen inaktiv.
stereoisoinerer Basen bestehen. 3, Vergl. Helv. 3, 748 (1920).

768 - -.
Exp e r im e n t e l 1 e r T e i 1.
libcr. die Heystellung des Piiaen-niti.osoclzlorids.
Nach der hiezu meistens beniitzten Vorschrift von Wallncli ') wird zu einem Gemenge von Pinen, Eisessig und AthyInitrit bei tiefer Temperatur rorsichtig konzentrierte Salzsaure zugesetzt und so ein bei 103 O schmelzendes Pinen-nitrosochlorid erhalten. Wir fanden, dass man bei Anwendung alkoholischer Salzsaure ein hijher schmelzendes Produkt erhalten Itann. Durch diesen Ersatz des Wassers wird eine gleichmassigere Durchmischung der Reak- tionsprodukte und ein gelinderer Verlauf der Umsetzung erzielt. Von den in verschiedener Weise variierten Versuchsbedingungen erwiesen sich die folgenden als recht gunstig. Das Gemisch von je 50 gr Pinen (Sdp. ca. 155') uncl Eisessig wird in Kaltemischung zum Erstarren gebracht, rnit 60 gr Athylnitrit versetzt und unter andauernder guter Kuhlung im Laufe von vier Stunden 35 cm3 20proz. alkoholischer Salas'dure zugegeben. Schon waihrend des Salzsaurezusatzes beginnt die Abscheidung des Pinen-nitroso- chlorids, das nach zwijlfstiindigem Stehen im Eisschranke abfiltriert wurde. Das mit etwas Alkohol gewaschene weisse Pulver schmilzt bei 107-108". . Die Ausbeute hangt hekanntlich sehr von der Drehung des Pinens ab.
B e y die Reduktion des ,FTitmvg)inens (UI) zum Pinylamin (I) und Pinocamphon (VTTlj).
CLI, I
C C CH3
I CH
Gearbeitet wurde nach der etwas vereinfachten Vorschrift Wallachs ').
1) A . 253, 251 (1889). 2) A. 268, 198 (1892); 346, 240 (1906).

- 759 -
Zur heissen Losung von 60 gr rollen Nitrosopinensl) in 400 cm3 Eisessig und 100 cm' Wasser wurde allm8hlich unter Erwarmen auf dem Wasserbade 300 gr Zinkstaub zugegeben und noch acht Stunden erhitzt. Das entstandene Pinocamphon wird aus der noch sauren Losung mit Wasserdampf abgeblasen und durch Ausschutteln mit Ather, Trocknen uber Pottasche und Destillieren gereinigt; es siedet bei 90' (12 mm) und die Aus- beute betrug 13 gr. Der essigsaure Ruckstand von der Wasser- dampfdestillation wird noch lieiss vom ungelosten Zinkstaub ab- filtriert, und nach dem Ubersattigen mit konzentrierter Natrdn- lauge ') kann durch nochmalige Wasserdampfdestillation das Pinyl- amin gewonnen werden. Man erhalt so 28 gr vom Sdp. ca. 90' (12 mm).
Redukt ion des Pin,ylanains (I) x u m Pinocamphylanain (Ir). Nach Tilden ') ist diese Reaktion durch Natrium und Alkohol
nur sehr unvollstandig durchfuhrbar. Wir wandten daher die katalytische Reduktion an.
25 gr Pinylamin wurden in 100 cm3 Alkohol mit 2 gr Platin- schwarz nach Willstatter reduziert, wobei rasch eine Mol. Wasser- stoff absorbiert wird. Nach dem Verdampfen des Alkohols wird das erhaltene Pinocamphylamin destilliert ; es geht bei ca. 90 O (12 mm) als farbloses, dunnflussiges 01 uber.
In folgender Tahelle seien die Schmelzpunkte der von Walluch ') und !PiZden ') gewonnenen Derivate des Pinocamphyl- amins mit den unsrigen zusammengestellt. Das Ammoniumjodid war noch unbekannt.
I) Dargestellt nach Wallach, A 245, 352 (1888); 268, 198 (1892). 2) Die Losung kann nach Belieben vorher noch am Dampfbade konzeiitriert
3) Soc. 89, 1560 (1906) 4) A. 313, 367 (1900). 5) Soc. 89, 1560 (1906)
wcrd P ri

760 - -
Das A c e t a t wurde hergestellt durch Erhitzen rnit Essig- sjlureanhydrid am Wasserbade, Fallen rnit Wasser und Umkrystalli- sieren aus Petrolgther.
0,1568 gr Subst. gaben 0,4237 gr CO, und 0,1552 gr I i 2 0
C,,HzlON Ber. C 'i3,77 H 10,850,'o (M. 7355 11,080/0
Das P i k r a t fiillt in konzentrierter alkoholischer Losung aus und wurde aus den gleichen Losungsmitteln umkrystallisiert.
Das C h 1 o r h y d r a t , durch Einleiten von trockenem Salz- sauregas in die atherische Losung des Pinocamphylamins als weisses Pulver gewonnen, beginnt sich bei ca. 300" zu verfarben, um bei ca. 340" unscharf unter Zersetzung zu schmelzen.
Der P i n o c a m p h y 1 a m in - h a r n s t o f f wird durch Erhitzen des Chlorhydrats rnit der berechneten Menge Kaliumcyanat in wassriger Losung erhalten und aus Essigester umkrystallisiert.
0,1355 gr Subst. gaben 17,2 ~1113 N,(180, 724 mm) C,,H20C)S, Ber. N 14,280/0
Gef. ), 14,200/0
P i n o c a 111 p h y 1 - t r i m e t h y 1 a m mo n i urn j o d i d ') (analog Formel. 111) wurde in iiblicher Weise durch erschupfende Methylierung des Amins gewonnen. Nach den1 vollstandigen Ab- destillieren des Alkohols (zuletzt im.Vakuum) wurde der Riick- stand im So.chletapparat rnit Chloroforin extrahiert. Durch Um- krydtallisieren aus Chloroform und Ather wird das Produkt in Form glanzender, schwach gefarbter oder farbloser Schuppen vom Smp. 25.5'' erhalten. Es ist in Wasser, Alkohol und Chloro- form leicht, in Essigester sehr schwer loslich und praktisch un- loslich in Ather und Petrolather.
Das
0,2212 gr Subst. gaben 0,1622 gr Ag.1
C,,H26NJ Ber J 39,300,6 Gel. ,) 39,40 y o
Zersetzung des Pinoc.ccnzl,hlll-t,-iinefh~~ntriinoniiLnahllldi.ox~cls (III,). Das Jodid wurde in wasseriger Losu ng mit iiberschiissigem
Silberoxyd in die Ammoniumbase verwandelt und nach dem Ab- filtrieren das Wasser im Vakuum durch Erwarmen entfernt. Der
~~
l) Die Vcrsuche mit dem Jutlid und der .Immoi~iumbase wurden Zuni Tell voii E. Rohheim aiisgefutirt.

- i61 -
Ruckstand wird dann im absolukn Vakuum ( ' /7 mm) weiter erhitzt, wobei er sich in eine feste, schwach gefarbte Masse venvandelt, die bei 150 O allmahlich, ohne sonst eine aussere Ver- 'kderung zu zeigen, in Form gasformiger Produkte in die auf - 80' gehaltene Vorlage iibergeht. Bei der Verarbeitung von 15 gr Jodid ist die Dauer der Zersetzung mindestens zwei Stunden. Die Zersetzungsprodukte wurden durch Zusatz von verdunnter Essigsaure in einen neutralen und einen basischen Anteil zerlegt. Der erstere wird in wenig Ather aufgenommen und rnit Natrium getrocknet. Die farblose Flussigkeit siedet bei 155-156 O (725 mm, abgekiirztes Normalthermometer), besitzt Pinengeruch und ist ohne Fraktionierung analysenrein.
0,2115 gr Subst. gaben 0,6811 gr CCJ, und 0,2230 gr HzO
C1,H,, (IV) Ber. C 88,23 H l l , ' i 7 O / o Gef. ,, 87,86 ,, 11,80°/o
Das nach der oben angegebenen Vorschrift daraus herge- stellte Pinen-nitrosochlorid schmilzt bei 108 und gibt rnit dem Nitrosochlorid aus Terpentinol keine Depression.
Die essigsaure Losung der basischen Anteile von der Zer- setzung der Ammoniumbase wurde rnit Natronlauge iihersattigt, mit Ather ausgeschiittelt und das erhaltene hasisch riechende 01 mit Methyljodid versetzt, wobei es fast momentan erstarrt. Nach dem Urnkrystallisieren aus Chloroform und Ather schmilzt es hei 300-301 '. Die dem oben beschriebenen Jodid ausserlich ahnliche Substanz besteht aus eineni isoineren Pinocamphyl- trimethyl- ammoniumjodid (analog Forinel 111).
0,0608 gr Subst. gaben 0,0440 gr AgJ
G,,H,,N.I Ber. J 39,300/0 Gef. ,, 39.130/0
Zurich, Cheinisches Institut der Eidg. Techn. Hochschule.





























