
1965 R. HUISCEN und H. BLASCHKE 145
1.3-Dipolare Cycloadditionen, XIXI)
Z U R ADDITION DES ATHOXYCARBONYL-AZENS AN NITRILE
von ROLF HUISCEN und HEINZ BLASCHKE
Eugen MuNer mit besten Wiinschen zum 60. Geburtstag zugeeignet
Aus dem Institut fur Organische Chemie der Universitat Miinchen Eingegangen am 17. Dezember 1964
Bei der Photolyse bzw. Thermolyse des Azidoameisensaureathylesters in Nitrilen werden 5-substituierte 2-Athoxy-1.3.4-oxadiazole erhalten. Eine Mehrzentren- addition des khoxycarbonyl-azens an die CN-Dreifachbindung und mecha-
nistische Alternativen werden diskutiert.
Bei der Thermolyse von Diazoketonen auftretende Ketocnrbene vermogen 1.3-Cyclo- additionen an Nitrile unter Bildung von Oxazolen einzugehenz). Die bei der Photolyse und Thermolyse aromatischer o-Diazo-oxide entstehenden Ketocarbene treten mit Alkinen'), Alkenend), Heteromehrfachbindungen') wie >C=O ' / 'C=S, -C-N und >C=N- zu funf- gliedrigen Ringen zusammen.
LWOWSKI und MATTINGLY5) beobachteten, daR sich das bei der Photolyse des Athyl-azidoformiats (I) bildende Athoxycarbonyl-azen (11) an Cyclohexen zum Aziri- din-Derivat zu addieren vermag. Dies machte eine Prufung der 1.3-Reaktionsweise der carbonylsubstituierten Azene moglich. Wir berichteten schon iiber das Abfangen der Sextett-Zwischenstufe I1 bei der Thermolyse von I in Acetylen-Derivaten, wobei 2-Athoxy-oxazole (111) als Produkte der 1.3-Cycloaddition auftretens).
I
N' jlC.2H5 0
H R R' I11
1) XVIII. Mitteilung: R. HUISGEN und E. AUPDERHAAR, Chem. Ber., im Druck. 2) R. HUISGEN, G. BINSCH und L. GHOSEZ, Chem. Ber. 97, 2628 (1964). 3) R. HUISGEN, G. BINSCH und H. KONIG, Chem. Rer. 97, 2868, 2884 (1964). 4) G. BINSCH, R. HUISGEN und H. KONIG, Chem. Ber. 97, 2893 (1964). 5 ) W. LWOWSKI und T. W. MATTINGLY, Tetrahedron Letters [London] 1962, 227. 6 ) R. HUISGEN und H. BLASCHKE, Tetrahedron Letters [London] 1964, 1409.
Liebigs Ann. Chem. Bd. 686 10
146 R. HUISGEN und H. BLASCHKE Bd. 686
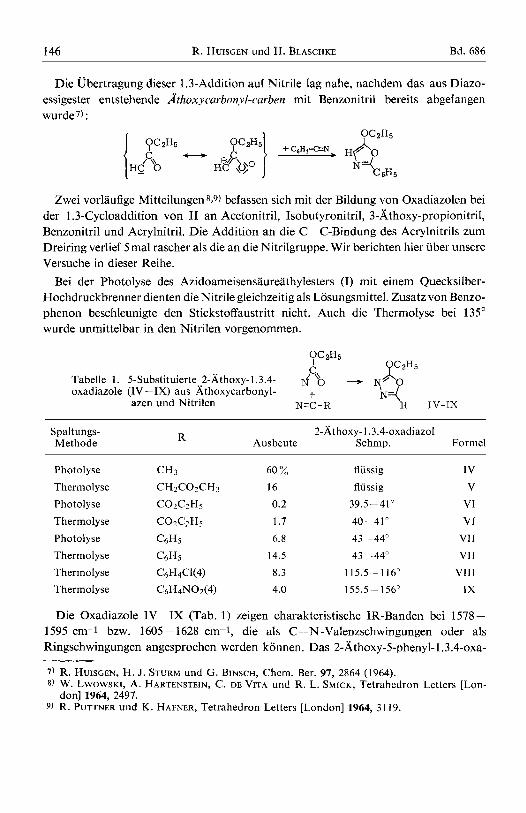
Die Ubertragung dieser 1.3-Addition auf Nitrile lag nahe, nachdem das aus Diazo- essigester entstehende Athoxycarbonyl-curben mit Benzonitril bereits abgefangen wurde7) :
Zwei vorlaufige Mitteilungen 8 9 ) befassen sich mit der Bildung von Oxadiazolen bei der 1.3-Cycloaddition von I1 an Acetonitril, Isobutyronitril, 3-Athoxy-propionitril, Benzonitril und Acrylnitril. Die Addition an die C =C-Bindung des Acrylnitrils zum Dreiring verlief 5mal rascher als die an die Nitrilgruppe. Wir berichten hier iiber unsere Versuche in dieser Reihe.
Bei der Photolyse des Azidoameisensaureathylesters (I) mit einem Quecksilber- Hochdruckbrenner dienten die Nitrile gleichzeitig als Losungsmittel. Zusatz von Benzo- phenon beschleunigte den Stickstoffaustritt nicht. Auch die Thermolyse bei 135" wurde unmittelbar in den Nitrilen vorgenommen.
Tabelle 1 . 5-Substituierte 2-khoxy-1.3.4- rfu\\ 0 - yB0 oxadiazole (IV-IX) aus d;thoxycarbonyl- +
R IV-IX N 4 azen und Nitrilen N-C - R
2-Athoxy-1.3.4-oxadiazol Ausbeute Schmp. Formel R Spaltungs-
Methode
Photolyse Thermolyse Photolyse Thermolyse Photolyse Thermolyse Thermolyse Thermolyse
60 "/, 16 0.2 1.7 6.8
14.5 8.3 4.0
fliissig fliissig
39.5 -41"
40-41" 43 -- 44" 43 - 44"
115.5- 116" 155.5 - 156"
IV V
VI VI
v11 VII
VIII JX
Die Oxadiazole IV-IX (Tab. 1) zeigen charakteristische IR-Banden bei 1578 - 1595 cm-1 bzw. 1605-1628 cm-1, die als C=N-Valenzschwingungen oder als Ringschwingungen angesprochen werden konnen. Das 2-Athoxy-5-phenyl-l.3.4-oxa-
7) R. HUISGEN, H. J. STURM und G. BINSCH, Chem. Ber. 97, 2864 (1964). 8) W. LWOWSKI, A. HARTENSTEIN, C . DE VITA und R. L. SMICK, Tetrahedron Letters [Lon-
9 ) R. PUTTNER und K . HAFNER, Tetrahedron Letters [London] 1964, 3119. don] 1964, 2491.

1965 1.3-Dipolare Cycloadditionen, XIX 147
diazo1 (VII) wurde rnit einem nach BACCHETTI 10) dargestellten Praparat identifiziert. Durch Behandlung des N-[ 1-Chlor-athylidenl- bzw. N-[cl-Chlor-4-substit.-benzyliden]- N’-Bthoxycarbonyl-hydrazins X mit Triathylamin synthetisierten wir Vergleichs-
N-N @ 0 ,OCZH5 - R-C-N-N-C - HCI -+ RXO>OC2H5 R-C: p- :- OZC zH
c1 X XI
praparate von iV, VIII und 1X. Aus dem intermediaren Nitrilimin-N-carbonsaure- ester Xi bildet sich unter Ringschlufi das Oxadiazol. Aus der Analogie in den UV- und IR-Spektren erschlossen wir die Konstitution von V und VI.
Die Uberlegenheit des gesattigten aliphatischen Nitrils (Acetonitrils) ist auffallend (Tab. 1). Bei der Umsetzung rnit Diphenylnitrilimin 11) oder Benzonitriloxydlz), also oktettstabilisierten l.3-Dipolen, entfalteten Benzonitril und Cyanameisensaureester eine weit grofiere dipolarophile Aktivitat als Acetonitril.
Ob die Photolyse oder die Thermolyse von I eine gunstigere Quelle fur khoxycarbonyl- azen bildet, ist noch ungeklart. DaB die Thermolyse von I in Benzonitril mehr 2-Athoxy-5- phenyl-1.3.4-oxadiazol (VII) lieferte, hangt rnit der nachgewiesenen Photolabilitat von VII zusammen.
Die Thermolyse von 1 in Cyanessigsauremethylester lieferte neben 16 % des Oxa- diazols V etwas Oxazolidon-(2) (XII). Zweifellos handelt es sich dabei urn das Resultat einer intramolekularen Einschiebung des Azen-Stickstoffs von I in eine C-H- Bindung des esterartig gebundenen Athyls. Analoge Insertionen wurden auch bei der Thermolyse des Octadecyl-13) und bei der Photolyse des tert.-Butyl-azidoformiats9J4) beobachtet.
Beim Zerfall des Azidoameisensaureathylesters in Cyanameisensuureuthylester trat als Nebenprodukt Nitrilotricarbonsaure-triathylester (XIV) auf (8 % bei der Photolyse, 12% bei der Thermolyse von I). Eine Mitwirkung des Losungsmittels ist nicht anzu- nehmen, da XIV auch bei der Photolyse von I in inerten Solventien erhalten wurdelS.16). Als weiteres Begleitprodukt von VI liel3en sich 2 % Hydrazodicarbonsaurediathylester (XV) bei der Photolyse von I fassen; ein Zusammenhang mit der von anderen Autoren beobachteten 15,16) Bildung des Azodicarbonsaureesters liegt nahe.
10) T. BACCHETTI, Gazz. chim. ital. 91, 866 (1961). 11) R. HUISCEN, R. GRASHEY, M. SEIDEL, G. WALLBILLICH, H. KNUPFER und R. SCHMIDT,
12) R. HUISCEN, W. MACK und E. ANNESER, Tetrahedron Letters [London] 1961, 587. 13) T. J. PROSSER, A. F. MARCANTONIO, C. A. GENCE und D. S. BRESLOW, Tetrahedron
14) R. KREHER und G. H. BOCKHORN, Angew. Chem. 76,681 (1964); Angew. Chem. internat.
15) J. HANCOCK, Tetrahedron Letters [London] 1964, 1585. 16) W. LWOWSKI, T. W. MATTINGLY und T. J. MARICICH, Tetrahedron Letters [London] 1964,
Liebigs Ann. Chem. 653, 105 (1962).
Letters [London] 1964, 2483.
Edit. 3, 589 (1964).
1591. 10*

148 R. HUISCEN und H. BLASCHKE Bd. 686
Als dritten Begleitstoff (bis zu 18 %) isolierten wir Oxazolidon-(2)-carbonsaure-(3)- athylester (XIII). Die Synthese aus XI1 und Chlorameisensaureathylester sicherte die Konstitution. Das Auftreten von XI11 geht vermutlich auf eine khoxycarbonylierung des Oxazolidons-(2) (XII), das oben schon als C -H-Insertionsprodukt von 11 erwahnt wurde, durch Cyanameisensaureathylester zuruck. Ein gesonderter Versuch bestatigte diese Acylierung.
Bezuglich des Mechanismus kann man noch keine Entscheidung treffen. Neben der zum Oxadiazol-Ring fuhrenden Mehrzentrenaddition, also einer 1.3-Dipolaren Cyclo- addition im engeren Sinne 171, ist mit einer primaren Anheftung des Sextett-Stickstoffs am Nitrilstickstoff zu rechnen. Der entstehende Nitrilimin-N-carbonsaureester XI folgt dann der BAccHETTr-Synthese, die ubrigens einen Spezialfall der allgemeinen Cyclisierung von N-Acyl-nitriliniinen zu 1.3.4-Oxadiazolenls) darstellt. Gegen diese von anderen Autoren bevorzugte 8 3 ) Interpretation spricht, daI3 die Basizitat der Nitrile erheblich hinter der des Pyridins zurucksteht, aus dem ein solches Zwitterion erhalten wurde19). Daruber hinaus erscheint uns die Annahme der Zwischenstufe XI unnotig, da bei der nicht minder glalten Bildung von 2-Athoxy-oxazolen (Ill) aus b;thoxycarbonyl-azen und C = C-Bindungens) eine analoge oktettstabilisierte Zwi- schenstufe gar nicht denkbar ist.
F0ZH5
XVI
Ernsthafter Diskussion wert ist dagegen die Moglichkeit, daI3 sich an eine zum 3-substituierten 1 H-Diazirin-carbonsaure-( 1)-athylester (XVI) fiihrende 1.1-Cyclo- addition eine Umlagerung zum fiinfgliedrigen Ring anschlieflt, die iibrigens einige Analogien haben wurde. 1H-Diazirine sind bislang unbekannt.
Ausschalten kann man eine einleitende dipolare Addition des Azidoameisensaureesters an Nitrile unter Bildung von 5-substituierten 1- oder 2-~thoxycarbonyl-tetrazolen. Nur sehr elektronenarme Nitrile wie Perfluoralkylnitrile 20) oder, in geringerem AusmaD, Cyanameisen- saureesterzl) pflegen organische Azide an der C = N-Bindung aufzunehmen. Die Geschwin-
17) R. HUISGEN, Angew. Chem. 75, 742 (1963); Angew. Chern. internat. Edit. 2, 565 (1963). 18) R. HUISGEN, J. SAUER, H. J. STURM und J. H. MARKGRAF, Chem. Eer. 93, 2106 (1960);
19) K. HAFNER, D. ZINSER und K. L. MORITZ, Tetrahedron Letters [London] 1964, 1733. 20) W. R. CARPENTER, J. org. Chemistry 27, 2085 (1962). 21) Unveroffentlichte Versuche von L. MOBIUS, Univ. Miinchen.
R. HUISGEN, Angew. Chem. 72, 359 (1960).

1965 1.3-Dipolare Cycloadditionen, XIX 149
digkeitskonstante fur die Thermolyse von I in Benzonitril liegt in der gleichen GroBenordnung wie die kl-Werte in anderen, nicht mit der Azidgruppe in Wechselwirkung tretenden Solventiens). Mit dem 2-~thoxycarbonyl-tetrazol als Zwischenstufe hatte das Phanomen einer rascheren, induzierten Stickstoff-Entbindung auftreten miissen.
Der DEUTSCHEN FORSCHUNGSGEMEINSCHAFT und dem FONDS DER CHEMISCHEN INDUSTRIE sind wir fur die Forderung des Arbeitsprogramms zu groBem Dank verbunden.
BESCHREIBUNG D E R VERSUCHE Acetonitril
2-Athoxy-5-methyl-l.3.4-oxadiazol (IV). - 5.75 g Azidoameisensaureathylester22) wurden in 200 ccm Acetonitril in geschlossener Quarzapparatur des wassergekiihlten Bestrahlungs- gerats UVM mit Quecksilberbrenner Q 700 (Quarzlampen-Gesellschaft Hanau) belichtet. Nach 5 Stdn. war die Nz-Entwicklung (1.14 Aquivv.) abgeschlossen. Aus der hellgelben Lo- sung wurde iiberschussiges Acetonitril abdestilliert. Bei 81 -83"/14 Torr gingen 3.81 g (60%) IV als blaBgelbes 01 iiber; Riickstand 0.85 g. Nach erneuter Destillation zeigte 1V n"," : 1.4375. - ZR-Spektrum (Film): Kraftige Banden bei 1578 und 1628 cm-1 (vermutlich dem Oxadiazolring zuzuschreiben); C - 0 1035 und 1237 cm-1. ~ UV-Maximum (in Athanol) : 217 mp.
C5HsN20z (128.1) Ber. C 46.87 H 6.29 N 21.87 Gef. 46.74 6.55 21.44 Mo1.-Gew. 144 (osmometr. in Benzol)
Bei Wiederholung des Photolyseversuchs in Gegenwart von 1 .O g Benzophenon war die Nz- Freisetzung (1.06 Aquivv.) erst nach 1 1 Stdn. beendet. Die Aufarbeitung der dunklen Reak- tionslosung erbrachte 3.49 g (55 %) IV, das laut IR-Spektrum von geringerer Reinheit war.
Unabhurigige Synthese von IV: Aus 19.8 g Hydrazincarbonsaureathylester23) und 8.5 g frisch destilliertem Acetaldehyd in 50 ccm Athanol isolierten wir nach 12stdg. Aufbewahren und Einengen 17.4 g (70 %) N-Athyliden-hydrazin-N-carbonsaureathylester in farblosen Spie- Ben. Nach Umlosen aus Athanol Schmp. 98 ~ 100".
C~H10N202 (130.1) Ber. C 46.14 H 7.74 N 21.53 Gef. C 46.42 H 7.78 N 21.39
In die Losung von 3.40g des voranstehenden Hydrazons in 75ccm CHC13 lieBen wir bei -15" die Losung von 2.8 g Chlor in 25 ccm CHC13 unter Riihren einflieBen. Nach 1 Stde. wurde das Solvens entfernt und der X (R=CH3j enthaltende gelbe, viskose Riickstand in 100 ccm trockenem Benzol aufgenommen. Einriihren von 2.65 g Triathylamin fiihrte in 30 Min. zur Abscheidung v m 2.0 g Triathylammoniumchlorid (56 %). Die Vakuum-Destillation des Losungsriickstandes erbrachte 0.339 g I V (10% iiber beide Stufen). Nach Siedepunkt, IR- Spektrum und Brechungsindex mit voranstehendem Praparat identisch.
Cyanessigsauvemethylester 2-Athoxy-5-rnethoxycarbonylmethyl-I .J.I-oxadiazol (V). - In 50 g Cyunessigsauremethyl-
ester, auf 135" (Bad) erwarmt, lieBen wir in 1 Stde. 11.5 g Z unter Riihren eintropfen; nach
22) M. 0. FORSTER und H. E. FIERZ, J . chem. SOC. [London] 93, 81 (1908). 23) 0. DIELS, Ber. dtscb. chem. Ges. 47, 2186 (1914).

150 R. HUISGEN und H. BLASCHKE Bd. 686
3 Stdn. betrug die N2-Entwicklung 98 %. Unter 3 Torr wurde iiberschiissiger Cyanessigester abdestilliert. Auf 3.67 g blaBgelbes 01 mit Sdp. 110--130" (Bad)/O.O5 Torr folgten 0.33 g dunkles 0 1 bei 130-170"/0.005 Torr. Aus der ersten Fraktion schieden sich beim Kiihlen 0.18 g farblose, derbe SpieRe ab, die nach Umkristallisieren aus Athanol bei 88" schmolzen (Lit. 24) 88 -90°). IR-Vergleich und Mischprobe zeigten die Identitat mit Oxazolidon- (2) (XU), dem Produkt der innermolekularen CH-Insertion.
C3H5N02 (87.1) Ber. C 41.38 H 5.79 N 16.09 Gef. C 42.33 H 6.37 N 16.16
Der fliissige Anteil wurde erneut bei 120" (Bad)/O.O5 Torr destilliert: 2.88 g (16%) V ; n5O = 1.4554. - IR-Spektrurn (Film) : Starke Oxadiazol-Banden gleicher Intensitat bei 1585 und 1623 cm-1; Estercarbonyl 1740 cm-I. -- UV-Maxima (in Athanol): 271 und 217 mp. C~H10N204 (186.2) Ber. C 45.16 H 5.41 N 15.05
Gef. 45.07 5.73 14.30 Mo1.-Gew. 188 (osmometr. in Benzol)
Durch Chromatographie an Florisil mit Petrolather/Benzol veranderten sich Brechungsindex und CH-Analyse nicht mehr.
Cyanameisensiiureiithylester
Thermolyse: 11.50 g (100 mMol) I wurden innerhalb 3 Stdn. in 50 ccm frisch destillierten, siedenden Cyanameisensaureathylester unter Riihren eingetragen. Nach 5 Stdn. waren 87 mMol Gas freigesetzt. Der iiberschiissige Cyanameisensaureester wurde iiber eine kleine Kolonne unter Normaldruck, der Rest i. Vak. abdestilliert. Aus 6.72 g hellgelbem 01, das bei 105 - 180"/ 0.1 Torr iiberging und in Ather aufgenommen wurde, schieden sich bei -78" 2.25 g (14%) OxazoIidon-(2)-carbonsaure-(S)-athylester (XIII) vom Schmp. 51 -53" aus; Ather ist zum Umlosen geeignet. - IR-Spektrum (KBr) : Ringcarbony11780 cm-1, Estercarbony11708 cm-1. - NMR-Spektrum (in CDC13, Tetramethylsilan als innerer Standard): C e 3 : C& = 1 : 2. Die Signale der Ringmethylengruppen liegen im gleichen Bereich wie das Quadruplett des Ester- Methylens, namlich um 5.8 T; auch die Koppelungskonstante scheint mit 7.5 Hz gleich zu sein; CH3-Triplett bei 8.67 T.
C6HgN04 (159.1) Ber. C 45.28 H 5.70 N 8.80 Gef. 45.26 6.08 8.60 Mo1.-Gew. 164 (osmometr. in Benzol)
Der Mutterlaugenanteil wurde an 50 g Florisil chromatographiert und mit 600 ccm Petrol- ather/Benzol (19: 1) eluiert. Das Eluat hinterlieR beim Verdampfen 0.926 g (12%) Nitrilo- tricarbonsiiure-triuthylester (XIV) als farbloses, bei 90" (Bad)/0.001 Torr siedendes 01; die Identifizierung erfolgte durch IR-Vergleich mit einem authent. Praparat 2 5 ) .
C9HpjN06 (233.2) Ber. C 46.35 H 6.48 N 6.01 Gef. C 46.68 H 6.58 N 6.79
Weiteres Eluieren der Florisil-Saule mit je 500 ccm Petrolather/Benzol (9: 1) und (1 : I ) erbrachte 1.10 g blal3gelbes 01, das in 3 ccm Ather gelost wurde. Bei -78" kristallisierten 0.308 g (1.7 %) 2-Athoxy-1.3.4-oxadiazol-carbonsaure- (5)-uthylester (VI) in farblosen, bei 35-41" schmelzenden Prismen; Schmp. 40-41" nach Umlosen aus Ather. - UV-Spektrum
24) S. FRANKEL und M. CORNELIUS, Ber. dtsch. chem. Ges. 51, 1662 (1918); vgl. Beilsieins
25) C. F. H. ALLEN und A. BELL, Org. Syntheses, COIL Vol. 111, 415 (1955). Handbuch der organischen Chemie, Bd. IIIjIV, I. Ergiinz.-Werk, S. 8, Anmerkung.

1965 1.3-Dipolare Cycloadditionen, XIX 151
(in Athanol): A,,, = 237 mp, log E = 3.99. - IR-Spektrum (KBr): Starke Oxadiazolbande bei 1591 cm 1; Estercarbonyl 1740 cm-1. C7Hp~N204 (186.2) Ber. C 45.16 H 5.41 N 15.05
Gef. 45.17 5.49 14.85 Mo1.-Gew. 185 (osmometr. in Benzol)
Die Ergebnisse dreier weiterer Versuche finden sich neben denen des voranstehenden in Tabelle 2.
Tabelle 2. Aus Azidoameisensaureathylester mit Cyanameisensaureathylester hervorgegangene Substanzen
Ausbeute XIV xv XI11 Destillation v1 Spaltungs-
methode
Photolyse i. Hochvak. Florisil 0.1 % 4.5 % 2.2% -
- - Photolyse nicht dest. Florisil 0.2 7.5 Thermolyse i. Hochvak. A1203 10 0.3 18% Thermolyse i . Hochvak. Florisil 1.7 12
-
14 -
Synthese des Oxazolidon-(2/-carbonsaure-(3)-athylesters (XIII). - a) 2.18 g (25 mMol) Oxazolidon-(2) 24) wurden mit 0.57 g (25 mg-Atom) Natrium in 50 ccm trockenem Xylol bis zur Auflosung des Metalls gekocht. Alsdann wurden 2.70 g (25 mMol) Chlorameisensaure- iithylester langsam eingeriihrt. Nach weiterem 1 stdg. Kochen wurde das Xylol i. Vak. ab- destilliert. Bei 125" (Bad)/0.001 Torr gingen 2.90 g (73 %) XI11 als farbloses, kristallin erstarren- des 0 1 uber; Schmp. 53" (aus Essigester).
CsHgN04 (159.1) Ber. C 45.28 H 5.70 N 8.80 Gef. C 45.26 H 5.82 N 8.73
b) 0.40 g X V wurden in 10 ccm Cyanameisensaureathylester 4 Stdn. gekocht. Die Hoch- vakuumdestillation ergab ein halbfestes 01, aus dem man beim Anreiben mit Ather 0.37 g (51 %) XI11 erhielt.
Benzonitvil
2-Athoxy-5-phenyl-l.3.4-oxadiazol (Vtl). - a) 13.2 g I , gemischt mit 10 ccm Benzonitril, lieBen wir in 40 ccm Benzonitril, welches auf 130" (Bad) erhitzt wurde, in 2 Stdn. einflieBen. Nach 5 Stdn. hatten sich 95 % Stickstoff entwickelt. Das Benzonitril wurde i. Vak. abgezogen; bei 80-130" (Bad)/0.005 Torr folgten 3.89 g rotes, langsam erstarrendes 01. Bei 130 -160°/ 0.005 Torr gingen weitere 0.45 g dunkles 0 1 uber; Ruckstand 9.52 g. Die erste Fraktion wurde in khan01 aufgenommen und schied bei -78" 1.72 g (7.9%) VII vom Schmp. 42-44" ab. Aus Athanol farblose Blattchen vom Schmp. 43 -44" (Lit.10) 44"). - IR-Spektrum (KBr): Oxadiazol-Schwingungen bei 1580 und 1605 cm-1; C - 0 1020 und 1280 cm-1; aromat. CH-Wagging 690 und 728 cm-1. Mischprobe und IR-Vergleich erwiesen die Identitat mit einem aus N-[u-Chlor-benzylidenl-hydrazin-N'-carbonsaureathylester 10) (X, R = C6H5) bereiteten Praparat.
b) Bei einem zweiten Thermolyseversuch im gleichen Manstab wurde in dem bei 80-133" (Bad)/O.OOl Torr destillierten Rohprodukt (4.82 g) das Oxadiazol VZI durch quantitative

152 R. HUISGEN und 13. BLASCHKE Bd. 686
IR-Analyse ermittelt. Dazu wurde die Extinktion der 3.6-proz. Losung in Tetrachlorathylen bei 728, 1580 und 1605 cm-1 in der 0.02 mm-Fixkiivette gemessen und rnit Eichkurven ver- glichen. Danach enthielt das Rohprodukt 3.16 g (14.5%) VII. Die Messung der Bande bei 1815 cm-1 wies auf 0.96 g (1 1 %) Nitrilotricarbonsuure-triuthylesrer (XIV).
c) Bei der Bestrahlung von 50 mMol Z in 50 ccm Benzonitril rnit der Quecksilberlampe TQ 8 1 in der Quarzapparatur betrug die N2-Entwicklung nach 8 Stdn. 57 %. Die Aufarbeitung, wie oben beschrieben, erbrachte 1.90 g rotes 01; die quantitative IR-Analyse wies auf 0.653 g (6.8 %) VZZ. Aus der ather. Losung wurde bei tiefer Temperatur das bei 38 -40" schmelzende VII in Substanz abgeschieden.
d) In einem zweiten, gleichartigen Photolyseversuch erreichte die Nz-Freisetzung nach 13 Stdn. 77%. In 1.60 g Hochvakuum-Destillat fanden sich laut IR-Analyse nur 0.336 g (3.5%) VIZ. Die Vermutung, daB das Oxadiazol VII nicht photostabil ist, bestatigte sich. 0.292 g VII wurden in 50 ccm Benzonitril 8 Stdn. mit der Lampe TQ 81 bestrahlt. Im Hoch- vakuum-Destillat waren nur noch 0.186 g, also 65 % des eingesetzten VII, vorhanden; 0.1 1 g Destillationsriickstand.
4- Chlor-benzonitril 2-A'thoxy-5-[4-chlor-phenyl]-1.3.4-oxadiazol (VIII). - In 17.9 g geschmolzenes 4-Chlor-
benzonitril wurden bei 135" unter Riihren 5.75 g Zeingetropft, wobei in 3 Stdn. 58 % Stickstoff austraten. Die Aufarbeitung lieferte 1.78 g bei 120- 150" (Bad)/0.001 Torr iibergehendes, bald erstarrendes 0 1 (16% Rohprodukt). Beim Verreiben rnit khan01 bei 0" blieben 0.925 g (8.3 %) VIII ungelost. Die aus Essigester oder Cyclohexan umkristallisierten farblosen, glan- zenden Schuppen schmolzen bei 115.5-116". - ZR-Spektrum (KBr): 1603 (Phenyl); 1584 und 1612 (Oxadiazolbanden); 842 cm-1 (aromat. CH-Wagging).
C ~ O H ~ C I N Z O Z (224.7)
Unabhangige Synthese: 4-Chlor-benzaldehyd-[N-athoxycarbonyll-hydruzon wurde aus den
C ~ O H I I C I N ~ O Z (226.7) Ber. C 52.99 H 4.89 N 12.36 Gef. C 53.35 H 5.01 N 12.13
6.6 g voranstehendes Hydrazon in 120 ccm CHClj lieferten bei -10" mit 2.8 g Chlor 6.5 g N-[a.4-Dichlor-benzyliden~-hydrazin-N-curbons~ure~thylester (X, R = (4)ClC&). 5.82 g Chlo- rierungsprodukt wurden in 100 ccm Benzol rnit 2.50 g Triathylamin bei 0-25" umgesetzt und ergaben 3.50 g (70%) VZZZ; Schmp. 115" (nach mehrfachem Umlosen aus Essigester). Misch- probe und IR-Spektrum dienten zur Identifizierung.
Ber. C 53.46 H 4.04 N 12.47 Gef. C 53.32 H 4.15 N 12.35
Komponenten in khan01 erhalten (80% d. Th.). Schmp. 148" (aus bithanol).
4- Nitro- benzonitril 2-A'thoxy-5-[4-nitro-phenyll-1.3.4-oxadiazol (IX). - Der hohere Schmelzpunkt des Nitrils
veranlaBte uns, die Thermolyse von 54 mMoi Z in 20 g 4-Nitro-benzonitril bei 170" zu unter- suchen, wobei 1.33 Aquiw. Gas austraten. Das iiberschiissige 4-Nitro-benzonitril wurde ab- sublimiert und der dunkle Ruckstand i. Hochvak. destilliert. Bei 160-200"/0.001 Torr gingen 1.44 g kristallin erstarrendes Material iiber. Aus bithanol 0.505 g (4.0%) IX; nach Umlosen aus Essigester Schmp. 155.5-156". - ZR-Spektrum (KBr): Oxadiazolbanden bei 1595 und 1610 (Schulter); 1352 und 1517 (NOz); 732 cm-1 (C-N02).
C10H9N304 (235.2) Ber. C51.06 H3.86 N 17.87 Gef. C51.01 H 3.85 N 18.07

1965 G. W. ROTERMUND und R. KOSTER 153
Unabhangige Synthese: Aus 15.1 g 4-Nitro-benzaldehyd und 11 .O g Hydrazincarbonsaure- athylester in 150 ccm Athanol erhielten wir 20.1 g (85 %) bei 134" schmelzendes Hydrazon (aus khanol) .
C10HllN304 (237.2) Ber. C 50.63 H 4.67 N 17.72 Gef. C 50.96 H 4.92 N 17.98
Wir lieBen 7.1 g Hydrazon in 200 ccm CHC13 bei -8" 1 Stde. rnit 4.0 g Chlor reagieren und engten i. Vak. ein. Der halbkristalline Ruckstand wurde in 200 ccm Benzol rnit 3.40 g Triuthyl- amin behandelt und lieferte 31 % Triathylammoniumchlorid und 58 % Oxadiazol ZX. Nach Mischprobe und IR-Spektrum identisch mit voranstehendem Praparat.
[239/64]
Borverbindungen, IX * ISOMERE DES 9b-BORA-PERHYDROPHENALENS
von GERHARD W. ROTERMUND~) und ROLAND KOSTER
Aus dem Max-Planck-Institut fur Kohlenforschung, Mulheim/Ruhr
Eingegangen am 17. Dezember 1964
Wie bereits vorlaufig mitgeteiltz), konnten wir drei isomere Verbindungen der Bruttoformel C12H21B auf verschiedenen Wegen (Hydroborierung von Cyclo- dodecatrienen ; Pyrolyse von Cyclododecylboranen) rein darstellen bzw. ineinan- der umwandeln. Es handelt sich um das all-cis-9b-Bora-perhydrophenalen (Centrobor I), cis-trans-9b-Bora-perhydrophenalen (Centrobor 11) und 9b-Bora- perhydrobenz[cd]azulen (Centrobor 111). Die physikalischen und chemischen Eigenschaften dieser Verbindungen (Addukte rnit N-Basen, Oxydation, Reaktio-
nen rnit k h y l e n und H-aciden Substanzen) werden beschrieben.
Eine Verbindung der Summenformel ClzHzlB wurde erstmals von WILKE und KoSTER3) aus Triisobutylboran und Cyclododecatrienen-( I .5.9) durch Verdrangung der Isobutylreste erhalten:
B(i-C4H9)3 + C12H18 + C I ~ H ~ ~ B + 3 i-C&
Die Reaktion sollte damals nicht nur das Verhalten von Trialkylboranen gegenuber ungesattigten Kohlenwasserstoffen4) mit mehr als einer C C-Bindufig klaren, sondern
*) VlII. Mitteilung: R. KOSTER, W. LARBIG und G. W. ROTERMUND, Liebigs Ann. Chem.
1) G. W. ROTERMUND, Dissertation Techn. Hochschule Aachen 1962. 2 ) G. W. ROTERMUND und R. KOSTER, Angew. Chem. 74, 329 (1962); Angew. Chem.
3) Gemeinsame Versuche von G. WILKE und R. KOSTER im Max-Planck-Institut fur Kohlen-
4) R. KOSTER, Angew. Chem. 68, 383 (1956); Liebigs Ann. Chem. 618, 31 (1958).
682, 21 (1965).
internat. Edit. 1, 269 (1962).
forschung, Mulheim/Ruhr 1956.
Recommended








![[REGEL-UND STEUERTECHNIK GLOSSAR] - …REGEL-UND STEUERTECHNIK GLOSSAR] Addierglied für digitale Größen summing element for digital quantities Addition addition Ähnlichkeitstransformation](https://img.pdfslide.org/doc/110x75/5b2e9d757f8b9ae16e8c793b/regel-und-steuertechnik-glossar-regel-und-steuertechnik-glossar-addierglied.jpg)








