
Spectrochhics Acta, Vol. 25A, pp. 1589 to 16Q2. Pergamon Press 1969. Printed in Northern Ireland
Die Elektronenanregungsspektren der Dibenzacridine
H.-H. PERKADWUS,* A. KNOP und J. V. KNOP*
Abteilung fiir Molekiilspektroskopie rtm Institut fiir Organische Chemie der Technischen Universitiit Brannschweig
(Received 24 September 1968)
Abstract--The results of SCF-CI calculations have been compared with the experimental data of five dibenzacridines. There proved to be a very good agreement of theory with experiment as to the energy and polarization of the electronic transitions. By measuring the spatial distribu- tion of fluorescence of a stretched foil the direction of the long-wavelength transitions could be absolutely determined and combined with the APF-spectra the transitions could thus be corre- lated.
VON DEN vier isomeren Dibenzanthracenen, dem 1.2,3.4-, 1.2,5.6-, 1.2,6.7- und 1.2,7- %Dibenzanthracen leiten sich sechs isomere Dibenzacridine ab: 1.2,3.4-, 1.2,5.6-, 1.2,6.7-, 1.2,7.8-, 3.4,5.6- und 3.4,6.7-Dibenzacridin. In diesen aromatischen Ver- bindungen sind funf Ringe miteinander kondensiert. Es liegt ein System mit 22 W- Elektronen vor, das im Fall der Dibenzacridine durch den Einbau des Heteroatoms gestijrt ist. Da iiber funfkernige Ringsysteme mit Heteroatome kaum Untersuchun- gen vorliegen, schien es von Interesse, im Zusammenhang mit unseren theoretischen und experimentellen Untersuchungen an Azaaromaten [l-3] eine theoretische Inter- pretation der Elektronenanregungsspektren vorzunehmen und die so erhaltenen Ergebnisse mit experimentellen Daten zu vergleichen. Als theoretische Methode wahlten wir SCF-CI-Modellrechnungen, die sich in letzter Zeit fiir die Behandlung von P-Elektronensystemen sehr gut bewahrt haben [l-5]. Als experimentelle Methoden wurden die Absorptionsspektren, die Fluoreszenz- und Phosphoreszenz- spektren sowie die zugehiirigen Polarisationsgradspektren herangezogen. Letztere gestatten bekanntlich eine relative Zuordnung der Richtung der Elektroneniiber- gange [6]. Im Hinblick auf den Einbau des N-Atoms und die unterschiedliche angu- lare Anellierung ist diese Frage von Interesse.
DURCHF~HRUNQ DER MODELLRECHNUNGEN
Die Modellrechnungen lehnen sich an den bekannten Formalismus einer SCF-Cl- Berechnung an, den wir bei analogen Berechnungen an den Phenanthrolinen [l],
den Dipyridylathylenen [3] und dem Stilben [4] bereits benutzt hatten, und der
* Neue Anschrift: Institut fiir Physikalische Chemie, Universitiit Dtieldorf.
[l] H.-H. PERKAMRPUS, J. V. KNOP, A. KNOP und G. KASSEBEER, 2. Naturfomch. 22a, 1419 (1967).
[2] H.-H. PERKAMPUS, A. KNOP und J. V. KNOP, 2. Nuturforach. %a, 840 (1968). [3] H.-H. PERKAMPTJS, J. V. KNOP und A. KNOP, Ber. Bunaenges. Phys. Chem. 72, 623 (1969). [4] H.-H. PERKAMPUS und J. V. KNOP, Theoret. Chim. Acta 6, 45 (1966). [5] F. D~RR, G. HOHLNEICHER und S. SCHNEIDER, Ber. Bunsengea. Phys. Chem. 70,803 (1966). [B] F. D~RR, Angew. Chem. 78,457 (1966).
1589

1590 H.-H. PERKAMPUS, A. KNOP und J. V. KNOP
kiirzlich von KLESSINQER [7] ausfiihrlich und iibersichtlich dargestellt worden ist. Es eriibrigt sich daher, an dieser Stelle auf die Durchfiihrung der Rechnungen naher einzugehen. Die im folgenden aufgefiihrten Werte der Integrale entsprechen denen, die wir bei den Phenanthrolinen [l] benutzt hatten:
j&o = -2,39 eV; pNN = -2,75 eV
WC = -11,16eV; W, = --14,12 eV
ycc = lo,53 eV; YNN = 11,78 eV.
Die Zweizentrenwechselwirkungsintegrale yrv wurden wieder nach NISHIMOTO und MATA~A [8] angesetzt; speziell fur benachbarte Zentren gilt:
y=v=c: YPV = 5,22 eV
p=N; v = c: Yllv = 5,50 eV
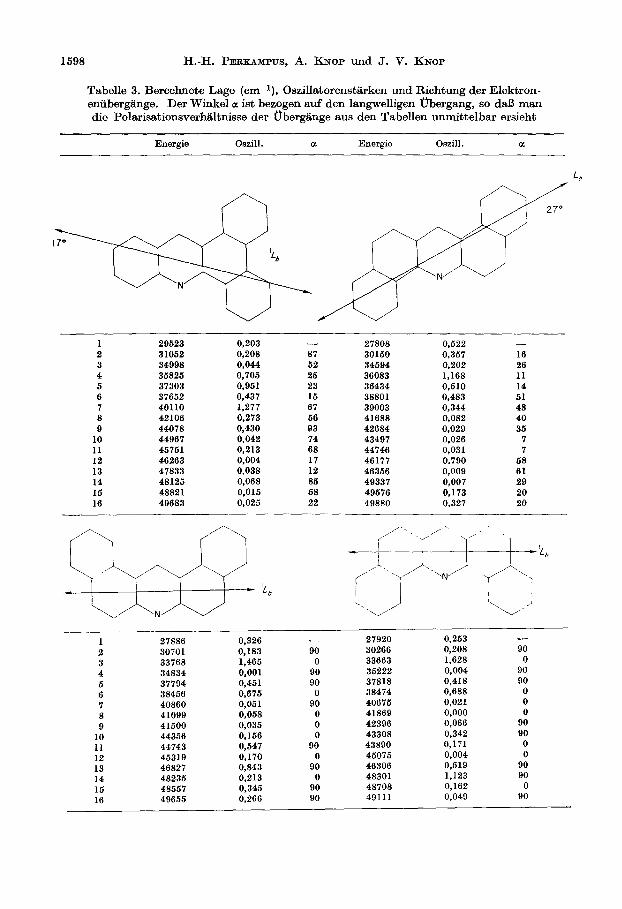
In der Tabelle 3 sind als Ergebnisse die berechneten Energien, Oszillatorenstarken sowie die Winkel a der entsprechenden Ubergangsmomente zum Ubergangsmoment der liingstwelligen Absorptionsbande angefiihrt. Das benutzte Koordinatensystem ist in Abb. 1 am Beispiel des 1.2,5.6-Dibenzacridins dargestellt. Die Modelhech- nungen wurden an der Anlage ICT 1907 des Rechenzentrums der Technischen Universitat Braunschweig durchgefiihrt [9].
Abb. 1. Benutztes Koordinatensystem am Beispiel des 1.2,5.6-Dibenzacridin.
DURCHF~~HRUNO DER MESSUN~EN
Die Elektronenanregungsspektren des 1.2,3.4-, 1.2,5.6-, 1.2,7.8- und 3.4,5.6- Dibenzacridins in Athylalkohol (20°C) sind kiirzlich von Zanker erneut vermessen worden [lo] und sind fiir diese Betrachtungen herangezogen worden. Das Spektrum des 1.2,6.7-Dibenzacridins wurde im Rahmen dieser Untersuchungen neu vermessen. Die berechneten Ubergange sind in allen Spektren als Linien, deren Hijhe der berech- neten Intensitat entspricht, eingetragen. Die Polarisationsgrad-, Fluoreszenz- und Phosphoreszenzspektren sind bei 77°K im Gemisch Athylalkohol/&her (1: 1)
[7] M. KLESSINGER, Forts&r. Chem. Porschg. 9, 354 (1968). [8] K. NISHIMOTO und N. ~MATAOA, 2. Php. Chem. 12, 335 (1957). [9] J. V. KNOP, Rechenprogramm ICT 1967, T.U. Braunschweig.
[lo] V. ZANEER, DMS-UV-Atlas Organischer Verbindungen, Vol. I, Spektren H8/37-44. Verlag Chemie, Butterworth (1966).

Die Elektronenanregungsspektren der Dibenzacridine 1691
gemessen worden. Die dadurch bedingte Rotverschiebung dieser Spektren gegen- iiber den Raumtemperaturspektren betrligt im Mittel fiir die lL,-Bande 160 cm-l, ftir die IL,-Banden 450 cm-l. Siimtliche Angaben der Wellenzahlen in den Tabellen beziehen sich auf die Tieftemperaturspektren.
Die apparative Anordnung ist bereits an anderer Stelle mitgeteilt worden [2]. Die absolute Zuordnung der Richtung der Obergangsmomente erfolgte durch
Untersuchung der r&nnlichen Intensitatsverteilung des Fluoreszenzlichtes einer gestreckten Polyviolfolie bei Anregung mit isotropen Licht (Abb. 8). Auf diese Weise wurde das Obergangsmoment der langstwelligen Absorptionsbande festgelegt, alle anderen konnen auf Grund des APF-Spektrums darauf bezogen werden.
DISKUSSION DER ERCIEBNISSE
1. Bemerkungen zu den Dibenzanthracenen
Fur die Diskussion der theoretischen und experimentellen Ergebnisse an den Dibenzacridinen ist es erforderlich, zuniichst kurz auf die aromatischen Grund- k&per einzugehen.
Von den vier isomeren Dibenzanthracenen sind die Spektren des 1.2,3.4- und 1.2,5.6-Dibenzanthracens von KIESSLINCJ et al. [ll] ausfuhrlich interpretiert worden. Das Spektrum des 1.2,5.6-Dibenzanthracens wurde bereits fruher von STIMLER [12]
diskutiert. Die Spektren des 1.2,6.7- und 1.2,7.8-Dibenzanthracens sind ebenfalls bekannt [13], jedoch bisher nicht ausfuhrlich diskutiert worden. Mit Ausnahme des 1.2,6.7-Dibenzanthracens, d.h. dem 1,2_Benztetracen, ist die langwellige Absorption bei diesen Bkernigen Aromaten der a-Bande nach CLAR [ 131 oder der l_Z&,-Bande nach PLATT [14] zuzuordnen, was insbesondere die Untersuchungen am 1.2,3.4- und 1.2,- 5.6-Dibenzanthracen bestiitigt haben [ 111.
Im Falle des 1.2-Benztetracens bestimmt die lineare Anellierung das spektro- skopische Verhalten. Im Elektronenanregungsspektrum dieser Verbindung sollte deshalb die langwellige Bande der p-Bande bzw. der l&-Bande zugeordnet werden. In Abb. 2 ist das Spektrum dieser Verbindung bei Normaltemperatur dargestellt. Die Lage der berechneten Obergiinge ist in Form von Linien eingetragen. Zwischen den beiden berechneten langwelligen Ubergangen bei 24300 und 314100 cm-l liegt ein weiterer Ubergang bei 27380 cm-l, der jedoch verboten ist. Die Oberglinge bei 24300 und 27380 cm-l stehen fast senkrecht aufeinander (89”, Tabelle 3). Wegen der konstanten Abnahme des Polarisationsgrades in diesem Bereich l&fit sich die Lage des 0-0-Oberganges der schwachen a-oder l.Z&,-Bande such auf Grund des APF- Spektrums nicht festlegen. Das schwache Minimum bei 23200 cm-1 entspricht einer nichttotalsymmetrischen Schwingung (1170 cm-i).
Der Winkel zwischen dem Ubergangsmoment des 1. Elektronenuberganges und des Uberganges bei 31410 cm-l betragt 69”. Der Polarisationsgrad zeigt im APF- Spektrum bei 31500 cm-l einen Minimalwert mit -12 %.Hierbei handelt es sich urn die B- oder lB,-Bande. Die berechneten Richtungen der weiteren Elektronen- ubergange stimmen mit dem APF-Spektrum gut uberein. So ist z.B. der Obergang
[ll] R. KIESSLING, G. HOHLNEICHER und F. D~~RR, 2. Nuturfomch. 22a, 1097 (1967). [12] S. STIMLER, J. Mol. Spectry 16, 84 (1965). [13] E. CLAR, Aromatische Kohlenwasserstoffe. Springer-Verlag (1941) [14] J. R. PLATT, J. Chem. Phys. 17, 484 (1949).
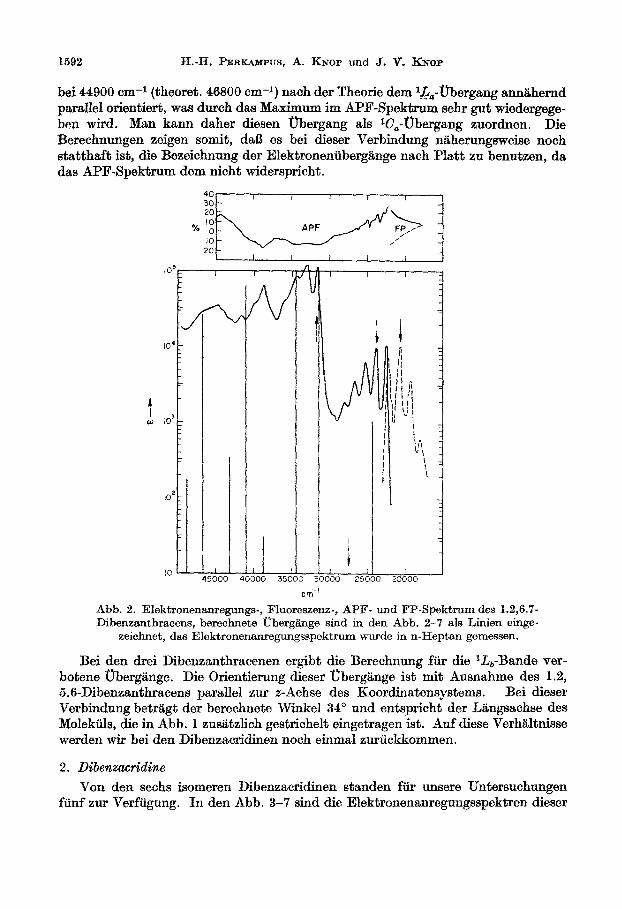
1592 H.-H. PERKANPUS, A. KNOP und 3, V. Kriop
bei 44900 cm-l (theoret. 46800 cm-l) nach der Theorie dem l.j&-Ubergang ann&hernd psrsllef orientiert, was durch das Maximum im APT-Spekt~ sehr gut wiedergege- ben wird, Man kann drther diesen Obergang als lC,-tzbergang zuordnen. Die Berechnungen zeigen somit, dal3 es bei dieser Verbindung nliherungsweise noch st&tth&~ ist, die Bezeichnung der Ele~ronen~berg~nge nach Platt zu benutzen, da das API?-Spektrum dem nicht widerspricht.
P v
- Abb. 2. ~lektr~nen~e~ngs-, Fluoreszenz-, APF- tmd FP-S~~~Irn des 1.2,6.7- Dibenzanthracens, bare&n&e tiberg&nge sind in den Abb. 2-7 als Linien einge-
zeichnet, das Elektronenanregnngsspektrum wurde in n-Heptan gemessen.
Bei den drei Dibenz~nthr~~enen ergibt die Berechnung fur die l.l&-Bande ver- botene Ubergilnge. Die Orientierung dieser UbergBnge ist mit Ausnahme des 1.2, 5.6-Dibenzanthracens parallel znr z-Achse des Koordinatensystems. Bei dieser Verbindung betr> der berechnete Winkel 34” und entspricht der L&ngsachse des Molekiils, die in Abb. 1 zus&tzlioh gestrichelt eingetragen ist. Auf diese VerhLltnisse werden wir bei den Dibenzacridinen noch einmal zuriickkommen.
3. Dibenxacridine Von den sechs isomeren Dibenzacridinen standen fiir unsere Untersuchungen
funf zur Verfiigung. In den Abb. 3-7 sind die Elektronen&nregungsspe~ren dieser

Die Elektronenanregungssppektren der Dibenzacridine 1693
Verbindungen zusammen mit ihren Fluoresxenz-, APF- und FP-Spektren dargestellt . Ala auff&igstes Merkmal ist in den Abb. 3-7 die starke Intensitiitszunahme der fL-Banden zu beobachten. Die Lage dieser Banden bzw. ihrer 0-0-Oberggnge gndert sich bei Einbau des Heteroatoms kaum im Vergleich zu den entsprechenden Dibenzanthracenen. Dies kommt in den berechneten Werten sehr gut zum Ausdruck, wie in Tabelle 1 die Gegeniiberstellung der Dibenzanthracene und Dibenzacridine
IO’ y
IO3 -
IO2 -
IO - I
450( i
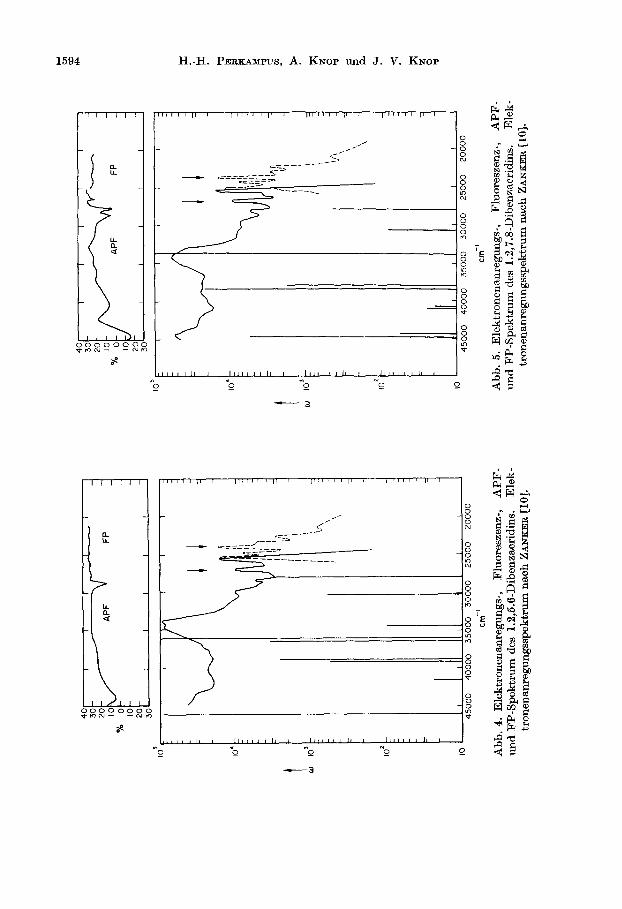
Abb. 3. Elektronenanregungs-, Fluoreszenz-, APF- und FP-Spektrum des 1.2, 3.4-Dibenzacridins. Elektronen~nregungsspektrum nsch ZANKER [lo].
erkennen 1513t. Der Einbau des N-Atoms bewirkt folglich die Aufhebung des uber- gangsverbotes fiir die l_&,-Bande. Die Beeinflussung der Richtung dieses Elektronen- uberganges ist nur gering. Die bevorzugte parallel-Orientierung zur z-Achse bzw. zur Liingsrichtung der Molektile erlaubt es, such hier diese ffberglinge ala I_&,- Ubergsnge zuzuordnen. Durch die starke Intensit&tszunahme der lL,-Bande ist der lL,-vbergang in den Spektren teilweise verdeckt. Das APF-Spektrum erlaubt jedoch, such hier diese ObergiCnge in den Spektren zu erkennen.
Mit Hilfe der APF-Spektren ergibt sich fur die Dibenzacridine die in Tabelle 2
zusammengestellte Zuordnung der I_&- und ll,-Obergiinge. Aus der Gegeniiber- stellung der experimentallen und theoretischen Werte fur die Lage der l&,- und I.&- Banden ersieht man, dag die theoretischen Werte im Mittel urn 2500 bis 3000
H.-H. PERKAMPUS, A. KNOP und J. V. KNOP
.* I’
,_=_--_--- ‘J.
c-2
-- ________-
_:_______
Die Elektronenanmgektren der Dibenzamidine 1596
l I I I I
8 -0
7
I
“0 -
1696 H.-H. PEREAMPUS, A. KNOP und J. V. KNOP
Tabelle 1. Vergleich der theoretischen Werte fur Lage, Oszillatorenstlirke und Richtung des rL,-tfberganges bei den Dibenzanthracenen und Dibenzacridinen
Dibenz
(l-w (cm-‘)
-Anthracen
f (1Lb) Cl&)
(cm-l)
-Acridin
fF-%)
1.2,3.4- 29660 0,000 O0 29520 0,204 --17O 1.2.5.6- 28350 0.000 34O 27810 1.2;7.8-
0.522 27O
3.4,5.6- I 28005 0,000 O0 27890 01326 O0 27920 0,254 O0
1.2,6,7- 27380 0,000 12O 27250 0,245 -7O
Tabelle 2. Lage der 0-0-oberglinge in den lL,-und IL,-Banden, den Fluoreszenz- und Phos- phoreszenzspektren im Vergleich mit den theoretischen Werten fur die Dibenzacridine = DBAc
FlUOreSZCIlZ
‘Lb ‘L, o-o AC.W-Fl. 1’1 -DBAc (cm-‘) (cm-‘) (cm-‘) (cm-‘) (cm-‘) ‘L, - T, T’l’h
exp. 26680 27730 26620 60 18740 8990 098 1.2,3.4
theor. 29520 31050 - - 18780 12270 -
sxp. 25350 28700 25280 70 19710 8990 097 1.2,5.6
theor. 27810 30160 - - 18100 12000
axp. 23980 21090 20610 480 enval-tet - 1.2,6.7 bei 12000
them. 27250 24156 - - 10400 13755 -
exp. 25070 27870 25000 70 19200 8670 235 1.2,7.8 them. 27890 30700 - - 18590 12110 -
3.4J.6 exp.
theor.
25380 27840 26300 SO 19020 8820 195
27920 30265 - - 18065 12210 -
cm-r zu hoch liegen. Dagegen wird die Differenz zwischen den 0-0-Ubergangen der IL,- und lL,-Banden durch die Theorie recht gut wiedergegeben. So liegt z.B. fur das 1.2,3.4-Dibenzacridin diese Differenz bei 1050 cm-l; der theoretische Wert betragt 1530 cm-r. Beim 1.2,6.7-Dibenzacridin liegen die entsprechenden Werte bei 2890 (exp.) und 3090 cm-l (theoret.). Fur die Diskussion der Gesamtspektren sol1 kurz auf die einzelnen Verbindungen eingegangen werden.
1.2,3.4-Dibenzacridin. Aus Abb. 3 in Verbindung mit Tabelle 3 ist zu ersehen, da13 das APF-Spektrum beziiglich der Parallel- und Senkrechtbanden mit der Berech- nung in ffbereinstimmung steht. Der Winkel zwischen dem ‘Lb- und l.L,-ubergang betragt 87”, d.h. da13 diese beiden Obergange praktisch J_ aufeinander stehen. Zu hijheren Anregungsenergien schliefien sich zwischen 35000 und 37000 cm-l drei ubergange an, die fast parallel zum I&,-ubergang orientiert sind und als lBp- fZberglnge zugeordnet werden konnen. Das APF-Spektrum zeigt positive Polarisa- tionsgrade. Bei 40000 cm-l liegt ein annahernd senkrecht polarisierter Ubergang, der mit dem Minimum im APF-Spektrum bei 38500 cm-l gut iibereinstimmt und als rB,-Ubergang zugeordnet werden kann.
Die Elektronenanregungsspektren der Dibenzacridine 1697
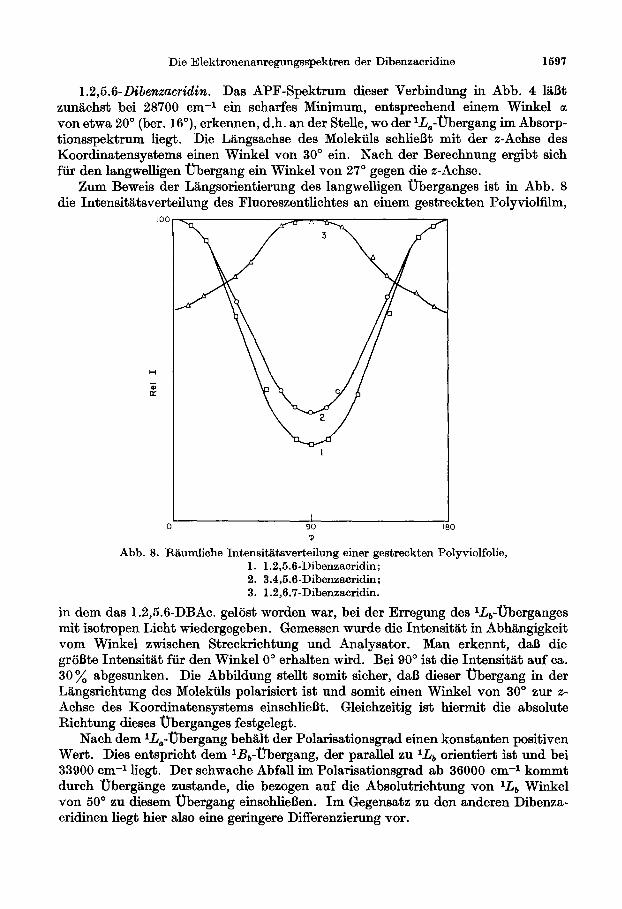
1.2,5.6-Dibenzucridin. Das APF-Spektrum dieser Verbindung in Abb. 4 l&fit zun&chst bei 28700 cm-l ein scharfes Minimum, entsprechend einem Winkel a von etwa 20” (ber. IS’), erkennen, d.h. an der Stelle, wo der I.&-ubergang im Absorp- tionsspektrum liegt. Die Ltingsachse des Molektils schlieBt mit der z-Achse des Koordinatensystems einen Winkel von 30” ein. Nach der Berechnung ergibt sich fur den langwelligen ubergang ein Winkel von 27’ gegen die x-Achse.
Zum Beweis der Llngsorientierung des langwelligen f)berganges ist in Abb. 8 die Intensit5tsverteilung des Fluoreszentlichtes an einem gestreckten Polyviolfilm,
Abb. 8.
IO0
0 90 I 8 ‘p
0
RIiumliche Intensitiitsverteilung einer gestreckten Polyviolfolie, 1. 1.2,5.6-Dibenzacridin; 2. 3.4,&G-Dibenzacridin; 3. 1.2,0.7-Dibenzacridin.
in dem das 1.2,5.6-DBAc. gel&t worden war, bei der Erregung des r.&,-uberganges mit isotropen Licht wiedergegeben. Gemessen wurde die IntensitSit in Abhilngigkeit vom Winkel zwischen Streckrichtung und Analysator. Man erkennt, de6 die grSBte Intensitlit ftir den Winkel 0" erhalten wird. Bei 90’ ist die IntensitLGt auf ca. 30 % abgesunken. Die Abbildung stellt somit sicher, da13 dieser Ubergang in der L&ngsrichtung des Molekiils polarisiert ist und somit einen Winkel von 30” zur z- Achse des Koordinatensystems einschliefit. Gleichzeitig ist hiermit die absolute Richtung dieses Oberganges festgelegt.
Nach dem lL,-ubergang behlllt der Polarisationsgrad einen konstanten positiven Wert. Dies entspricht dem lB,-Vbergang, der parallel zu l_Lb orientiert ist und bei 33900 cm-l liegt. Der schwache Abfall im Polarisationsgrad ab 36000 cm-l kommt durch Obergtinge zustande, die bezogen auf die Absolutrichtung von lLb Winkel von 50” zu diesem Obergang einschliel3en. Im Gegensatz zu den snderen Dibenza- cridinen liegt hier also eine geringere Differenzierung vor.
1598 H.-H. PERKAMPUS, A. KNOP und J. V. KNOP
Tabelle 3. Berechnete Lage (cm-l), Oszillatorenstiirken und Richtung der Elektron- eniibergiinge. Der Winkel a ist bezogen auf den langwelligen tfbergang, so da13 man die Polarisationsverhaltnisse der obergiinge aus den Tabellen unmittelbar ersieht
Energie Oazill. a Energie Oszill. a
1 29623 0,203 - 27808 0,622 - 2 31062 0,208 87 30160 0,367 16 3 34998 0,044 62 34694 0,202 26 4 36826 0,706 26 36083 1,168 11 6 37303 0,961 23 36434 0,610 14 6 37662 0,437 16 38801 0,483 61 7 40110 1,277 67 39003 0,344 48 8 42106 0,273 66 41688 0,082 40 9 44078 0,430 93 42684 0,029 36
10 44967 0,042 74 43497 0.026 7 11 46761 0,213 68 44746 0,031 7 12 46263 0,004 17 46177 0,790 68 13 47833 0,038 12 46366 0,009 61 14 48126 0,068 86 49337 0,007 29 16 48821 0,016 68 49676 0,173 20 16 49683 0,025 22 49880 0,327 20
1 2 3 4 6 6 7 8 0
10 11 12 13 14 16 16
27886 30701 33768 34834 37794 38466 40860 41099 41600 44366 44743 46319 46827 48236 48667
0,326 0,263 - - 27920 0,183 90 30266 0,208 90
1,466 0 33663 1,628 0 0,001 90 36222 0,004 90
0,461 90 37818 0,418 90
0,675 0 38474 0,688 0 0,061 90 40676 0,021 0 0,068 0 41869 0,000 0 0,036 0 42396 0,066 90
0,166 0 43308 0,342 90
0,547 90 43890 0,171 0 0,170 0 46076 0,004 0 0,843 90 46306 0,619 90
0,213 0 48301 1,123 90
0,346 90 48708 0,162 0
0,266 90 49111 0,049 90
Die Elektronenanregungssppektren der Dibenzacridine 1699
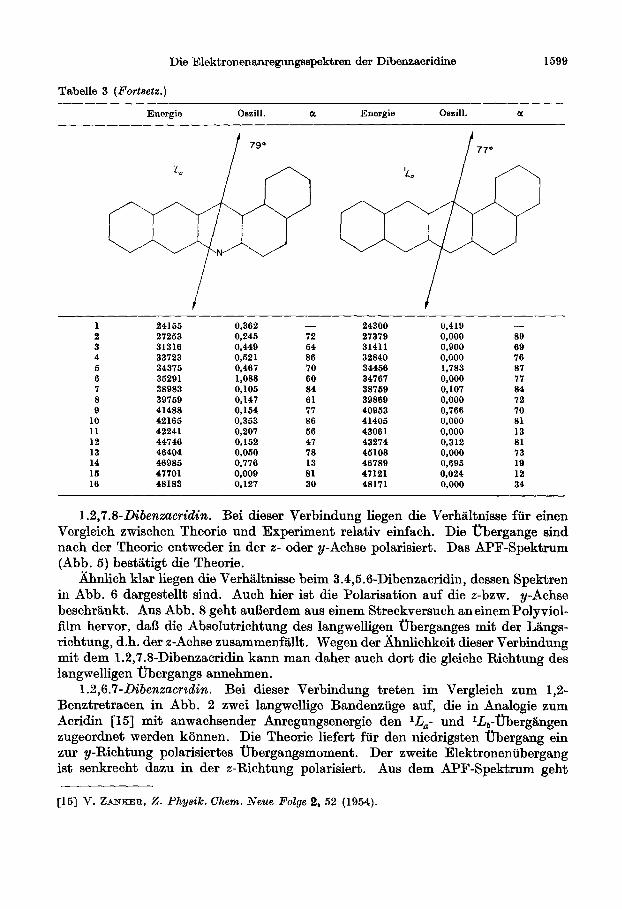
Tab&e 3 (Forteetz.)
Energie Oszill. a Energie Oszill. a
t t
1 24155 0,362 - 24300 0,419 -
2 27253 0,245 72 27379 0,000 89 3 31316 0,449 54 31411 0,900 69 4 33723 0,521 86 32840 0,000 76 5 34375 0,467 70 34456 1,783 87 6 35291 1,088 60 34767 0,000 77 7 38983 0,105 84 38769 0,107 84 8 39759 0,147 61 39869 0,000 72 9 41488 0,154 77 40953 0,766 70
10 42165 0,353 86 41405 0,000 81 11 42241 0,207 56 43061 0,000 13 12 44746 0,152 47 43274 0,312 81 13 46404 0,050 78 45108 0,000 73 14 46985 0,776 13 46789 0,695 19 15 47701 0,009 81 47121 0,024 12 16 48183 0,127 30 48171 0,000 34
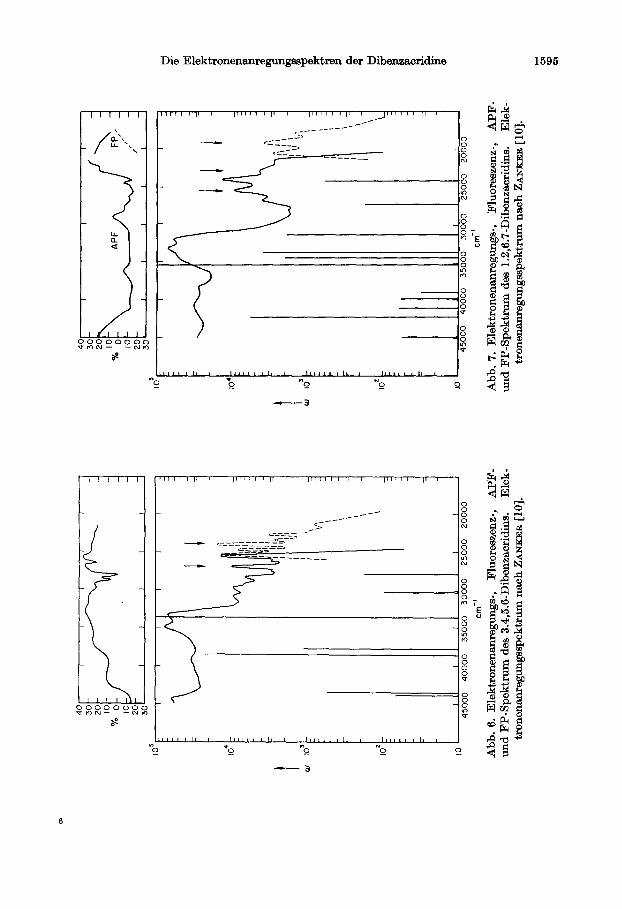
1.2,7.8-Dibenxacridin. Bei dieser Verbindung liegen die Verhaltnisse fti einen Vergleich zwischen Theorie und Experiment relativ einfach. Die Ubergange sind nach der Theorie entweder in der z- oder y-Achse polarisiert. Das APF-Spektrum (Abb. 5) bestatigt die Theorie.
llhnlich klar liegen die Verhaltnisse beim 3.4,5.6-Dibenzacridin, dessen Spektren in Abb. 6 dargestellt sind. Auch hier ist die Polarisation auf die z-bzw. y-Achse beschrankt. Aus Abb. 8 geht augerdem aus einem Streckversuch aneinemPolyviol- film hervor, da13 die Absolutrichtung des langwelligen uberganges mit der Langs- richtung, d.h. der z-Achse zusammen%llt. Wegen der Ahnlichkeit dieser Verbindung mit dem 1.2,7.8-Dibenzacridin kann man daher such dort die gleiche Richtung des langwelligen Ubergangs annehmen.
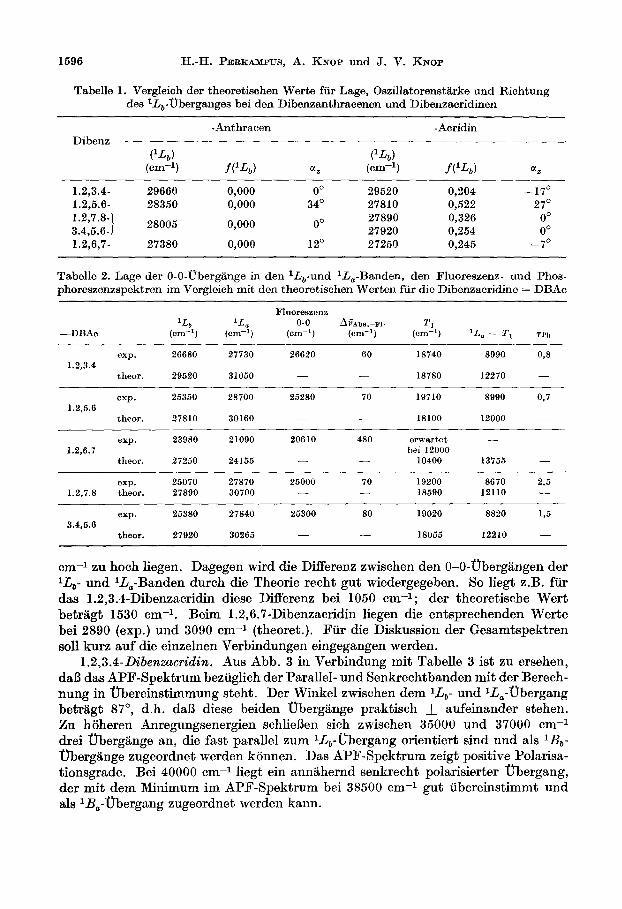
1.2,6.7-Dibenzacrzdin. Bei dieser Verbindung treten im Vergleich zum 1,2- Benztretracen in Abb. 2 zwei langwellige Bandenziige auf, die in Analogie zum Acridin [M] mit anwachsender Anregungsenergie den lL,- und IL,-Obergangen zugeordnet werden konnen. Die Theorie liefert fur den niedrigsten Obergang ein zur y-Richtung polarisiertes Ubergangsmoment . Der zweite Elektroneniibergang ist senkrecht dazu in der z-Richtung polarisiert. Aus dem APF-Spektrum geht
[15] V. ZANKER, 2. Physik. Chem. Neue Folge 2, 52 (1954).

1600 H. H. PERKAMPUS, A. KNOP und J. V. KNOP
zunlichst eindeutig hervor, da13 diese beiden Elektroneniibergiinge senkrecht zuein- ander polarisiert sind. Aus Abb. 8 ist zu ersehen, da13 das Maximum der Fluores- zenzintensitat bei einem Winkel von 90’ erhalten wird. Das bedeutet, da13 die Fluoreszenz einem Ubergang zugehort, der in der kurzen Molekelachse, also der y- Achse unseres Koordinatensystems polarisiert ist und folglich dem IL,-Ubergang zugeordnet werden kann.
Das APF-Spektrum dieser Verbindung zeigt, daD in Bereich bis 45000 cm-l insgesamt mindestens 8 Elektroneniibergange liegen sollten. Das Absorptions- spektrum 18l3t 5 Vbergange eindeutig erkennen. Zwei weitere Ubergange liegen offenbar in der langwelligen bzw. kurzwelligen Flanke des I&-Oberganges bei 33000 cm-l verdeckt. Die Maxima im APF-Spektrum bei 29000 und 36500 cm-l weisen darauf hin, dafi die beiden zugehijrigen Elektroneniibergange gegen die l&,-Band0 gedreht sein miissen. Die theoretisch berechneten Richtungen der fraglichen Obergange stimmen in diesem Bereich mit dem Experiment iiberein, such der starke Anstieg des Polarisationsgrades bei 45000 cm-1 wird durch die Theorie gut wiedergegeben. Der entsprechende, analog zu ‘I;, polarisierte nbergang, liegt bei 46000 cm-i.
Im allgemeinen la& sich zusammenfassend sagen, daB die Lage und Richtung der Obergange fiir die fiinf isomeren Dibenzaoridine durch die Theorie richtig wiedergegeben wird. In der Tabelle 3 sind die berechneten ubergange ftir die besprochenen Dibenzacridine und das 1,2-Benztetracen zusammengestellt.
Die Schwingungsstruktur der Absorptionsspektren der Dibenzacridine ist im Vergleich zu den entsprechenden Dibenzanthracenen wesentlich schwiicher ausge- pragt. Wahrend man beim 1.2,6.7-Dibenzanthracen im Bereich der lL,-Bande neben der vollsymmetrischen Hauptserie mit 1400 cm-l noch weitere 4 Nebenserien mit 250, 480, 740 und 1170 cm-l identiflzieren kann, kann man bei dem 1.2,6.7-Dibenz- acridin nur noch die Hauptserie mit 1350 cm-l erkennen.
Die Schwingungsstruktur der l&,-Bande der iibrigen Dibenzacridine liil3t sich durch folgende Beziehungen wiedergeben :
Hauptserie = q, + n - 1390 cm-l
Sym.
(A,)
I. Nebenserie = Y,, + n * 1390 cm-l + 330 cm-l (A,)
II. Nebenserie = Y,, + n - 1390 cm-l + 670 cm-l (2)
III. Nebenserie = Y,, + n * 1390 cm-l + 1150 cm-l (B,)
Die Zuordnung der Schwingung mit 670 cm-l ist unsicher. Obwohl das APF- und FP-Spektrum in diesem Bereich durchweg ein Minimum aufweisen, ist dieses zu schwach ausgepragt, urn eine Zuordnung als B, zu rechtfertigen. Vermutlich handelt es sich dabei urn die uberlagerung einer total- und einer nichttotalsymmetrischen Schwingung, entsprechend etwa den Linien 644 ( B1,) und 755 (A,,) cm-l im Raman- spektrum des Anthracens [16].
1161 N. ABESBEGOVIC und N. VUKOTIC, J. Chern. phy8. 41, 2575 (1964).
Die Elektronenanregungsspektren der Dibenzaoridine 1601
cm-’
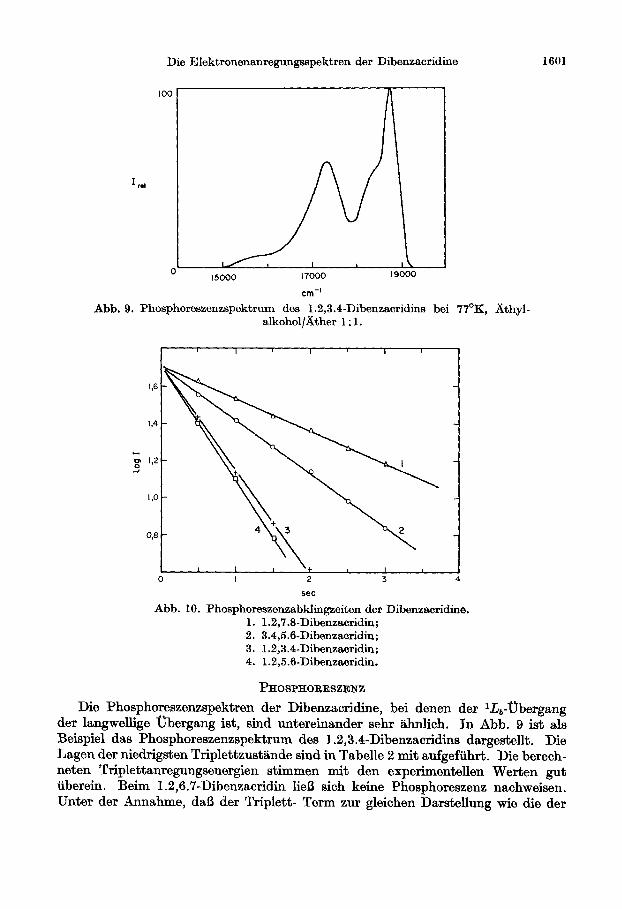
Abb. 9. Phosphoreszenzspektrum des 1.2,3.4-Dibenzacridins bei 77’K, athyl. alkohol/&her 1: 1.
0 I 2
set
3 4
Abb. 10. Phosphoreszenzabklingzeiten der Dibenzacridine.
1. 1.2,7.8-Dibenzacridin; 2. 3.4,5.0-Dibenzacridin; 3. 1.2,3.4-Dibenzacridi; 4. 1.2,5.6-Dibenzacridin.
Die Phosphoreszenzspektren der Dibenzacridine, bei denen der l-Z&-Ubergang der langwellige Ubergang ist, sind untereinander sehr iihnlich. In Abb. 9 ist als Beispiel das Phosphoreszenzspektrum des 1.2,3.4-Dibenzacridins dargestellt. Die Lagen der niedrigsten Triplettzustlinde sind in Tabelle 2 mit aufgefiihrt. Die berech- neten Triplettanregungsenergien stimmen mit den experimentellen Werten gut tiberein. Beim 1.2,6.7-Dibenzacridin lie13 sich keine Phosphoreszenz nachweisen. Unter der Annahme, da13 der Triplett- Term zur gleichen Darstellung wie die der

1602 H.-H. YERKAMPUS, A. KNOP und J. V. KNOP
iibrigen Dibenzacridine gehijrt (vermutlich B2), ist nach dem Singulett-Triplett- Interval1 von durchschnittlich 9000 cm-l der Triplettzustand des 1.2,6.7-DBAc bei 12000 cm-l zu erwarten.
Da sich die Intensit%ten der Fluoreszenz und Phosphoreszenz wie 100: 1 verhalten, war es nicht mbglich, die APPh- und PhP-Spektren zu vermessen. Aus diesem Grunde kann keine eindeutige Zuordnung getroffen werden.
Die Abklingzeiten wurden mit einer bereits friiher beschriebenen Apparatur [17] aufgenommen. Aus Abb. 10 geht hervor, da13 das Abklingen der Phosphoreszenz nach einer e-Funktion ergolgt; die Abklingzeiten sind in Tabelle 2 mit aufgefiihrt.
Die vorstehenden Untersuchungen wurden durch Mittel der Deutschen For- schungsgemeinschaft und des Verbandes der Chemischen Industrie, Fonds der Chemischen Industrie, unterstiizt, wofiir wir an dieser Stelle herzlich danken.
[17] H. R. VOLLBREUHT, Dissertation T. U. Braunschweig (1968).
Recommended

















