Entdeckung neuer Anwendungsgebiete
HDAC-Inhibitoren als Therapiefür neuronale ErkrankungenANDRE FISCHER
Chromatin-Plastizität: Regulierung derUmwelt-Genom-Interaktion
Das menschliche Genom besteht aus 23 Chromosomen-paaren und codiert in jeder somatischen Zelle unseres Kör-pers die gleichen Gene. Jede Zelle transkribiert allerdingsnur einen Bruchteil dieser Gene in mRNA, welche letztlichin Proteine translatiert wird. Gene können demnach aktivoder inaktiv sein und bestimmen so maßgeblich den Phä-notyp einer Zelle. Die Gesamtheit der aktiven Gene (dasTranskriptom) unterscheidet sich also wesentlich zwischenverschiedenen Zelltypen, wie z.B. Leber- oder Nerven-zellen.
Darüber hinaus reagiert ein Organismus auf veränderteUmweltbedingungen mit veränderter Genexpression. Insbe-sondere Nervenzellen zeichnen sich durch eine sehr hohePlastizität aus, d.h. in Antwort auf Umweltreize sind in Ner-venzellen rasche Veränderungen der Genexpression zu beo-bachten. Diese veränderte Genexpression ist essentiell fürLernprozesse und die Abspeicherung von Langzeitgedächt-nisinhalten ist ohne differenzielle Genexpression nicht mög-lich [1].
Jüngste Daten weisen darauf hin, dass eine deregulierte His-ton-Acetylierung in eine Reihe von neurodegenerativen undkognitiven Erkrankungen involviert ist. Histondesacetylase-Inhibitoren gelten daher als aussichtsreiche therapeutischeStrategie für so unterschiedliche neuronale Erkrankungen wieChorea Huntington, Morbus Alzheimer oder Schizophrenie.Dieser Artikel stellt die wichtigsten Ergebnisse dieses neuenForschungsgebiets zusammen.
204 | Pharm. Unserer Zeit | 3/2010 (39) © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
DOI:10.1002/pauz.201000367
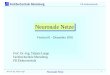
A B B . 1 H I S TO N - AC E T Y L I E R U N G R EG U L I E R T
G E N E X PR E S S I O N
AMe
Me
ARTKQTARKSTGGKAPRKQLATKAARKSAPATGGVKKPH 2 4 9 10 14 18 23 26–28 36
H3Me Me AC
PAC MeAC AC MeMe
PMe
K79
SGRGKGGKGLGKGGAKRHRKVLRDNIQGITKPAIRRLAR 1 3 5 8 12 16 20
PMe AC AC MeAC AC
H4
SGRGKQGGKARAKAKSRSSRAGLQFPVGRVHRLLRKGNY 1 5 9
PAC AC
H2A
PEPAKSAPAPKKGSKKAVTKAQKKDSKKRKRSRKESYSV 5 12 14
PAC AC
H2B
B
H4
H2BH2AH3
H4
H2BH2AH3
H4H
DNA
Aktive Gene
H4
H2BH2AH3
H4
H2BH2AH3
H4H
DNA
Inaktive Gene
HATs HDACs
Phosphorylierung Acetylierung MethylierungP AC Me
ACAC
AC
AC
AC
AC
AC
ACACAC
ACAC
ACAC
AC ACAC AC
AC
AC
ACAC
AC
AC
> A. Die basischen N-terminalen Bereiche der Histone sindzahlreichen posttranslationalen Modifikationen unterwor-fen. Hierbei handelt es sich insbesondere um Lysin-Acetylie-rungen, Lysin- oder Arginin-Methylierungen sowie Serin-Phosphorylierungen. B. Diese Modifikationen beeinflussendie Interaktion des Histonkomplex mit der DNA oder rekru-tieren weitere Proteine und regulieren so die Genexpression. (HDAC: Histondesacetylase; HAT: Histonacetyltransferase)

T H E R A P I E F Ü R N E U R O N A L E E R K R A N K U N G E N | K L I N I K
Daher gibt es strenge Kontrollmechanismen, welche da-rüber entscheiden, wann, wo und wie stark ein Gen expri-miert wird. Neben der Aktivität von Transkriptionsfaktorenhat die dreidimensionale Struktur des Chromatins einen we-sentlichen Einfluss auf die Aktivität von Genen und ist einessentielles Bindeglied zwischen Umwelt und Genom. AlsChromatin bezeichnet man die Gesamtheit von DNA undden daran gebundenen Proteinen. Ist dieses stark konden-siert, nennt man es Heterochromatin, welches mit redu-zierter Genexpression assoziiert ist. Offeneres Chromatinbezeichnet man als Euchromatin, aus dem dann Gene ab-gelesen werden können. Chromatin ist also extrem plastisch.
Die Chromatinplastizität wird vor allem durch Histon-proteine vermittelt. Zusammen mit der DNA bilden dieseals kleinste strukturelle Einheit des Chromatins dieNucleosomen. Ein Nucleosom ist ein oktamerer Komplexaus jeweils zwei der vier kanonischen Histone (H2A, H2B,H3 und H4), um welche sich 147 Basenpaare DNA winden.Zusätzlich gibt es noch Histon 1 (H1), welches als so ge-nannter „Linker“ funktioniert und den Aufbau von Nucleo-somen zu größeren und kompakteren Strukturen reguliert(Abb. 1).
Histone sind hoch konservierte, basische Proteine, dieaus einer globulären und einer flexiblen Domäne bestehen.Aminosäuren in der flexiblen N-terminalen Domäne der His-tone, insbesondere die von Histon H3 (H3) und H4, kön-nen posttranslational modifiziert werden. Die häufigstenModifikationen an Histonen sind die Acetylierung von Ly-sin (K) oder die entsprechende Methylierung. Daneben sindauch Phosphorylierung, Ubiquitinierung, Sumoylierung,ADP-Ribosylierung und Biotinylierung beschrieben worden(Abb. 1) [2].
Bestimmte Modifikationen korrelieren dabei mit aktiverGenexpression, wie z.B. Acetylierung an H3K9 und H3K14oder H4K5, H4K8, H4K12, H4K16, wohingegen andere Mo-difikationen, wie z.B. die H3K27-Methylierung die Gen-expression unterdrücken. Zusätzlich gibt es zumindest fürH3 und H2A verschiedene Proteinvarianten, wodurch sichdie Komplexität der Regulationsmöglichkeiten noch erhöht.Manche Modifikationen sind transient, während andereeher permanent sind und vererbt werden können.
Jede Histonmodifikation wird durch entsprechend ent-gegen gesetzte Aktivitäten von Enzymen reguliert, welchedie Modifikationen anfügen oder abspalten. Histon-Acety-lierung wird durch Histonacetyltransferasen (HAT) kataly-siert, wohingegen Histondesacetylasen (HDAC) diese Ace-tylgruppen wieder abspalten (Abb. 1). Ähnlich regulierenHistonmethyltransferasen und -demethylasen die Methylie-rung von Histonen.
Veränderungen des Chromatins können daher als Signa-turen verstanden werden, die definierte Bereiche des Ge-noms markieren und Gene an- oder abschalten. Da be-stimmte Chromatin-Veränderungen offenbar mit definier-ten Genexpressionsmustern korrelieren, haben Strahl &Allis das Prinzip eines „Epigenetischen Codes“ vorgeschla-gen, der sozusagen dem genetischen Code aufliegt [3].
Chromatinplastizität reguliert kognitiveFunktionen
Neuere Arbeiten an Nagern deuten darauf hin, dass dieChromatin-Plastizität wesentlich zur Gedächtniskonsolidie-rung beiträgt. Lernen und Gedächtnisprozesse können inRatten und Mäusen durch eine Reihe von etablierten Verhal-tenstests untersucht werden.
Die Gedächtniskonsolidierung in diesen Tests erfordertunter anderem Genexpression und De-novo-Proteinsynthe-se im Hippokampus, einer Hirnregion des limbischen Sys-tems, welche auch beim Menschen essentiell für Lernpro-zesse ist. Interessanterweise konnte gezeigt werden, dassdie Gedächtniskonsolidierung mit einer transienten Hoch-regulierung der hippokampalen Histon-Acetylierung korre-liert.
Ratten weisen, nachdem sie einem Lernparadigma aus-gesetzt wurden, eine Stunde später eine transient erhöhteH3-Acetylierung im Hippokampus auf. Diese Befunde konn-ten mittlerweile in Mäusen reproduziert werden. Die tran-sient erhöhte H3-Acetylierung beruht auf der Aktivierungvon synaptischen NMDA-Rezeptoren und der Aktivität desMAP-Kinase-Signalwegs. In Einklang mit diesen Ergebnissenkonnte gezeigt werden, dass genetische Mausmodelle mitveränderter HAT-Aktivität Lerndefizite aufweisen. So führtz.B. eine vermindertet Aktivität des CREB (cAMP responseelement binding factor)-Bindeproteins (CBP) zu reduzier-ter hippokampaler Histon-Acetylierung und deutlichen De-fiziten in der Furchtkonditionierung, dem Wasser-Labyrinth-Test und im Objekt-Erkennungstest [4–7]. Interessanter-weise konnte gezeigt werden, dass diese Lerndefizite durcheine Erhöhung der Histon-Acetylierung ausgeglichen wer-den konnten. Die Histon-Acetylierung wurde dabei durchdie Verabreichung von HDAC-Inhibitoren erreicht. Erstaun-licherweise führte die Gabe von HDAC-Inhibitoren im Ver-gleich zur Placebo-behandelten Gruppe auch in Wildtyp-Mäusen und Ratten zu einer deutlich verbesserten Gedächt-niskonsolidierung.
Zusammenfassend zeigen diese Arbeiten, dass dieHiston-Acetylierung ein wichtiger Mechanismus der Ge-dächtniskonsolidierung ist und dass HDAC-Inhibitorenpotentielle Therapeutika für kognitive Erkrankungen seinkönnten.
HDAC-Inhibitoren als therapeutische Strategiefür neuronale Erkrankungen
Es gibt elf HDAC-Proteine, die aufgrund von Homologien indrei Klassen eingeteilt werden. Die Klasse I HDACs (HDAC1, HDAC 2, HDAC 3 und HDAC 8) sind hauptsächlich imZellkern lokalisiert, wohingegen die Klasse-II-HDACs (HDAC5, HDAC 6, HDAC 7, HDAC 9, HDAC 10) durch zusätzlicheregulatorische Domänen deutlich größer sind und sowohlim Zytoplasma als auch im Zellkern zu finden sind. HDAC11 ist bisher der einzige Vertreter der Klasse-IV-HDACs.HDACs der Klassen I, II und IV benötigen Zink als Cofak-tor. Daneben gibt es noch sieben, als Sirtuine bezeichneteKlasse-III-HDACs, die NAD+ als Cofaktor brauchen.
© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.pharmuz.de 3/2010 (39) | Pharm. Unserer Zeit | 205

Die meisten derzeit zur Verfügung stehenden Substan-zen sind pan-HDAC-Inhibitoren, welche eher unselektiv al-le elf HDAC-Proteine hemmen (Abb. 2). Die HDAC-Inhibi-toren können im Wesentlichen in vier Gruppen eingeteiltwerden:• kurzkettige Fettsäuren wie z.B. Natriumbutyrat (NaB)
oder Valproat;• Hydroxamate wie z.B. Suberoylanilid-Hydroxamsäure
(SAHA, INN: Vorinostat, Zolinza™) oder Trichostatin A(TSA);
• zyklische Tetrapeptide wie Apicidin;• Benzamide wie MS-275 (INN: Entinostat). Diese Inhibitoren zeigen in präklinischen Versuchen deut-liche neuroprotektive und neuroregenerative Eigenschaften(Tab. 1) und einige werden nachfolgend detaillierter vor-gestellt.
Rubinstein-Taybi-SyndromRubinstein-Taybi-Syndrom (RTS) ist eine autosomal-domi-nant vererbte Erkrankung und führt zu mentaler Retardie-rung. Die hauptsächlichen Ursachen für RTS sind Mutatio-nen des CBP-Proteins, was in einer reduzierten HAT-Aktivi-tät und verminderten Histon-Acetylierung resultiert.Heterozygote CBP+/–-Mäuse weisen eine Reihe von charak-teristischen Merkmalen für RTS auf, so z.B. verschlechter-te kognitive Eigenschaften. Interessanterweise konnte durchdie Verabreichung von Vorinostat Defizite in der Histon-Acetylierung ausgeglichen und synaptische Plastizität undLernvermögen in CBP+/–-Mäusen wieder hergestellt werden[8].
Chorea HuntingtonChorea Huntington (CH) ist eine autosomal-dominant ver-erbte neurodegenerative Erkrankung. Auffälligste Symp-tome sind Bewegungsstörungen und kognitive Defizite. Als
Ursache sind Mutationen des Proteins Huntingtin (Htt) an-zusehen, bei denen sich die Basentriplett-Sequenz CAG ab-normal oft wiederholt. Die genaue Wirkungsweise von mu-tiertem Huntingtin ist nicht eindeutig geklärt, aber es führtunter anderem zu einer Deregulation der neuronalen Gen-expression, was vor allem auf eine Inhibierung der CBP-Ak-tivität zurückzuführen ist. Tatsächlich konnten mehrere Stu-dien in Mausmodellen zeigen, dass die Behandlung mit denHDAC-Inhibitoren Vorinostat bzw. NaB neuroprotektivwirkt und kognitive und motorische Fähigkeiten verbes-sert.
CH korreliert auch mit einem Defizit neurotropher Fak-toren wie z.B. BDNF (brain-derived neurotrophic factor).Tatsächlich konnte in Zellkulturexperimenten gezeigt wer-den, dass das Htt-Protein den BDNF-Transport verhindert.Durch HDAC6-Inhibitoren konnte der BDNF-Transport nor-malisiert werden. HDAC6 desacetyliert unter anderem Mi-krotubuli und beeinflusst dadurch den neuronalen Trans-port. Ein deregulierter axonaler Transport ist nicht nur inCH, sondern auch in anderen neurodegenerativen Erkran-kungen wie z.B. Alzheimer zu beobachten. Interessanter-weise zeigten HDAC-Inhibitoren wie Entinostat oder 4-Phe-nylbutyrat, die nicht HDAC6 hemmen, keinen protektivenEffekt, was darauf hinweist, das die Pathologie von CH mitspezifischen Veränderungen der Chromatin-Plastizität undMikrotubuli-Dynamik einhergeht.
Friedreichs AtaxieDie Friedreichs Ataxie (FA) ist eine autosomal-rezessiv ver-erbte neurodegenerative Erkrankung, die auf Mutationendes FRDA-Gens beruht. FRDA codiert für das mitochon-driale Protein Frataxin. Dabei treten in einem Intron desFRDA-Gens abnormal viele GAA-Tripletts auf, welche letzt-lich zu einer deutlich verminderten Expression des FRDA-Gens führen. Zusätzlich weist das FRDA-Gen in Patienten imPromotorbereich eine stark verminderte H3/H4-Acetylie-rung und H3K9-Trimethylierung auf, so dass die damit ein-hergehende Heterochromatinstruktur die Genexpressionzusätzlich verhindert. Herman et al. [9] konnten zeigen,dass im FA-Modell HDAC-Inhibitoren die Histon-Acetylie-rung des FRDA-Locus erhöhen und die FRDA-Expressionnormalisieren. Allerdings hatten herkömmliche pan-HDAC-Inhibitoren wie Vorinostat oder TSA keinen Effekt, wohin-gegen neue HDAC-Inhibitoren wie die Substanz 4b im Pro-motorbereich des FRDA-Gens Histon-Acetylierung und dieGenexpression erhöhten.
Spinale Muskelatrophie Die Spinale Muskelatrophie (SMA) ist eine degenerative Er-krankung der Motoneuronen, bei denen die Expression desSMN1- und SMN2-Gens (SMN: surivial motor neuron 1) re-duziert sind. Dabei bestimmt insbesondere der Gehalt anSMN2 den Grad der Pathogenese. Interessanterweise konn-te gezeigt werden, dass in SMA-Patienten der Promotor desSMN2 Gens hypermethyliert ist und vermutlich überMeCP2-Bindung die Genexpression reprimiert. HDAC-Inhi-
206 | Pharm. Unserer Zeit | 3/2010 (39) www.pharmuz.de © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim
A B B . 2 H DAC- I N H I B I TO R E N Z U R T H E R A PI E N E U RO N A L E R
E R K R A N KU N G E N
Klasse I Klasse IIa
Klasse IIb
Klasse IV
HDAC11
HDAC1
HDAC2
HDAC3
HDAC8
HDAC4
HDAC5
HDAC7
Klasse IIb
HDAC9
HDAC6
HDAC10
MS-275 (Entinostat)
Romidepsin (Istodax®)
NatriumbutyratValproat
4-Phenylbutyrat
Trichostatin ASuberoylanilid-Hydroxamsäure
(Vorinostat, ZolinzaTM)
Tubacin
4b
Das Schema zeigt die Spezifität von HDAC-Inhibitoren, die im Kontext neuronalerKrankheiten experimentell getestet wurden.

T H E R A P I E F Ü R N E U R O N A L E E R K R A N K U N G E N | K L I N I K
bitoren können dazu beitragen, über indirekte Effekte einedurch DNA-Methylierung vermittelte Genrepression aufzu-heben. Tatsächlich konnte gezeigt werden, dass die HDAC-Inhibitoren Vorinostat und Romidepsin die Expression vonSMN2 erhöhen, wohingegen 4-Phenylbutyrat und Valproatkeinen Effekt zeigten. In einer ähnlichen Studie konnte ge-zeigt werden, dass der HDAC-Inhibitor TSA ebenfalls dieSMN2-Expression erhöhen kann
Amyotrophe laterale SkleroseAmyotrophe laterale Sklerose (ALS) ist eine neuro-degenerative Erkrankung, die mit einem progressiven Ver-lust von Motoneuronen einhergeht. Zumindest in 20 % derKrankheitsfälle kann die Pathogenese auf Mutationen imSOD1-Gen zurückgeführt werden. SOD1/G93A-Mäuse sinddaher ein bevorzugtes Tiermodell, um ALS zu untersuchen.Interessanterweise konnte in diesem Modell gezeigt wer-den, dass sich die Behandlung mit 4-Phenylbutyrat (4-PB)deutlich positiv auf die Pathogenese auswirkte und vor al-lem zur Hochregulierung neuroprotektiver Faktoren führ-te. Ähnliche, aber nicht ganz eindeutige Befunde wurdennach Valproat-Behandlung beobachtet.
Morbus ParkinsonMorbus Parkinson (PD) ist durch den selektiven Verlust do-paminerger Neurone in der Substantia Nigra charakterisiert.Als Folge kommt es zu Bewegungsstörungen und kognitivenBeeinträchtigungen. Ein häufig verwendetes Modell für diePathogenese von PD ist die Verabreichung von MPTP (1-Me-
thyl-4-phenyl-1,2,3,6-tetrahydropyridin) oder MPP+ (1-Me-thyl-4-phenyl-pyridinium). In beiden Modellen konnte ge-zeigt werden, dass HDAC-Inhibitoren wie 4-Phenylbutyrat,Valproat, NaB oder TSA neuroprotektiv wirken. Eine zentra-le Hypothese zur Entstehung von PD ist die Deregulationvon α-Synuclein. In vielen PD-Patienten ist α-Synucleinhochreguliert und zeigt eine verstärkte Tendenz, Protein-aggregate zu bilden. Neuere Arbeiten weisen darauf hin,das α-Synuclein auch im Zellkern lokalisiert ist und dort di-rekt HATs bindet, was zur Hypoacetylierung von Histonenführt.
Morbus AlzheimerMorbus Alzheimer (MA) ist die häufigste dementielle neuro-degenerative Erkrankung, an der in Deutschland ca. 2 Mio.Menschen leiden. Hohes Alter ist der wichtigste Risikofak-tor für MA, und, da sich die Zahl der Europäer über 65 Jah-re bis zum Jahr 2025 verdoppeln wird, stellt somit MA ei-ne der größten Herausforderungen für das Gesundheits-system dar. Trotz intensiver Forschung ist es bisher nichtgelungen, wirksame Therapien gegen MA zu finden. Diewichtigsten pathologischen Symptome sind der Verlust vonNervenzellen und bestimmte Ablagerungen im Gehirn, dieextrazellulären amyloiden β-Plaques (Aβ-Plaques) und die in-trazellulären neurofibrillären Bündel (NFB). Die Aβ-Plaquesbestehen aus aggregierten Aβ-Peptiden, welche aus demsehr viel größeren APP-Vorläuferprotein gespalten werden.Genau diese Spaltung ist in den Gehirnen von Alzheimer-Patienten offenbar dereguliert, denn es kommt hier zu ei-
© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.pharmuz.de 3/2010 (39) | Pharm. Unserer Zeit | 207
TA B . 1 E F F E K T VO N H DAC- I N H I B I TO R E N I N PR Ä K L I N I S C H E N V E R S U C H E N
Rubinstein Taybi Syndrom CBP-Knock-out-Maus SAHA Verbesserte neuronale Plastizität,verbessertes Lernvermögen
Chorea Huntington R6/2-Maus, N171-82Q-Maus NaB, 4-Phenylbutyrat, HDACi 4b, Verminderte Sterblichkeit, Neuro-SAHA protektion, verbesserte motorische
Funktionen
Dentatorubral-pallidoluysian Attro-118Q-Maus NaB Neuroprotektion, verbesserte Atrophie motorische Funktionen
Friedreichs Ataxie Lymphozyten von Patienten HDACi 4b Erhöhung der Frataxin-Genexpression
Spinale Muskelatrophie Smn1-Knock-out-Maus NaB, SAHA, TSA, Romidepsin Erhöhte SMN2-Expression; neuro-protektiv, motorische Funktionen
Amyotrophe Laterale Sklerose SOD1/G93A-Maus 4-Phenylbutyrat Neuroprotektion
Morbus Parkinson Primäre Zellkultur NaB, Valproat, TSA Neuroprotektion
Morbus Alzheimer APP-Maus, CK-p25-Maus NaB, Valproat, 4-Phenlybutyrat Verbessertes Lernen, Neuroregenera-tion, reduzierte Amyloid-Pathologie
Schlaganfall Cerebale Ischämie NaB, TSA
Traumatische Hirnverletzung Mausmodell NaB Verbessertes Lernen (in Kombinationmit Verhaltenstherapie)
Schizophrenie Maus, Lymphozyten von Patienten MS-275, Valproat Erhöhte Expression der Reelin- undGAD65-Gene
Angststörungen Mausmodell für Furchtextinktion Valproat, TSA Verbesserte Furchtextinktion
Depression Mausmodell für sozialen Stress MS-275 Anti-depressives Verhalten
Krankheit Modellsystem HDAC-Inhibitoren in Effekt nach Behandlungpräklinischen Tests

ner Akkumulation von Aβ-Peptiden, die 40 bzw. 42 Ami-nosäuren lang sind (Aβ40/42). Bei den NFBs handelt es sichum intrazelluläre Aggregate des Tau-Proteins, einem Ein-weißstoff, der die Funktion von Tubulin reguliert. Für dieEntstehung der NFBs ist eine Hyperphosphorylierung desTau-Proteins entscheidend.
Die Forschung zur Pathogenese der Alzheimer-Erkran-kung sowie zur Entwicklung von therapeutischen Strate-gien widmet sich daher vor allem der Amyloid- und Tau-Pathologie [10].
Zusätzlich deutet eine Vielzahl von Daten darauf hin,dass die Pathogenese von Alzheimer mit der vermindertenExpression von Genen korreliert ist, welche synaptischePlastizität regulieren. Der Grund für diese Deregulation von„Plastizitätsgenen“ ist allerdings weitgehend unverstanden.Mittlerweile konnte in verschiedenen Mausmodellen fürMA gezeigt werden, dass HDAC-Inhibitoren wie NaB, Val-proat oder 4-Phenylbutyrat neuroprotektiv wirken und auchneuroregenerative Eigenschaften aufweisen. So konnte z.B.gezeigt werden, dass die Behandlung mit NaB zur Wieder-herstellung kognitiver Funktionen führt, obwohl bereits biszu 25 % der Vorderhirnneuronen degeneriert waren.
Der Schlaganfall ist eine der häufigsten Todesursachenund wird in den meisten Fällen durch eine cerebrale Ischä-mie ausgelöst. Im Nagermodell führt cerebrale Ischämie zurdeutlich reduzierten Histon-Acetylierung in den betroffe-nen Gebieten. Die Behandlung mit den HDAC-InhibitorenNaB oder TSA vor, aber auch nach dem Schlaganfall wirkteim Tiermodell deutlich neuroprotektiv. Ähnliche Beobach-tungen wurden bzgl. traumatischer Hirnverletzungen ge-macht. Hier konnte im Tiermodell gezeigt werden, dass dieBehandlung mit NaB das Lernvermögen in Nagern, die zu-vor eine Hirnverletzung erfahren hatten, wieder herstellenkonnte.
Neben neurodegenerativen Erkrankungen ist die durchHiston-Acetylierung vermittelte Chromatinplastizität offen-bar auch in die Pathogenese von neuropsychiatrischen Stö-rungen involviert.
SchizophrenieAllein in Europa leiden ca. 4 Mio. Menschen an Schizo-phrenie (1 % der Weltbevölkerung), einer psychischen Stö-rung die meist bei jungen Menschen auftritt und in 80 % derFälle zu lebenslangen Beeinträchtigungen führt. Die Ursa-che der Schizophrenie ist Gegenstand intensiver Forschung,aber eine Reihe von Arbeiten weist darauf hin, dass auchhier eine Deregulation epigenetischer Mechanismen an derPathogenese beteiligt ist.
Tatsächlich wird zur Behandlung der Schizophrenie un-ter anderem Valproat eingesetzt. In behandelten Patientenkonnte zumindest in Lymphozyten eine erhöhte H3/4-Ace-tylierung gemessen werden. Die Analyse in Postmortem-Gewebeproben des präfrontalen Cortex von Schizophre-niepatienten hat man reproduzierbar zeigen können, dassdie Expression der GAD67- und Reelin-Gene in GABAergenNeuronen stark reduziert ist. In Mäusen kann die Injektion
von Valproat die H3-Acetylierung im Bereich der GAD67-und Reelin-Promotoren erhöhen und eine verstärkte Gen-expression induzieren.
Interessanterweise wurde kürzlich gezeigt, dass dermRNA-Gehalt für HDAC1 im präfrontalen Cortex von Pa-tienten, die an Schizophrenie leiden, erhöht ist. Eine intra-peritoneale Applikation des spezifischen HDAC1-InhibitorsMS-275 in Mäuse konnte tatsächlich sehr viel effektiver alsValproat die Expression von GAD67 und Reelin erhöhen.Neuere Befunde zeigen zudem, dass in Schizophrenie-patienten weitere Veränderungen der Chromatinstrukturnachzuweisen sind. Weitere Untersuchungen sind notwen-dig, aber die Verwendung von spezifischen HDAC-Inhibi-toren als Therapiemöglichkeit für Schizophrenie scheint zu-mindest aussichtsreich. Insbesondere auch deshalb, daHDAC-Inhibitoren kognitive Eigenschaften verbessern undStörungen der Kognition einen deutlich prädikativen Cha-rakter haben und meist auf eine Verschlechterung derKrankheit hinweisen.
AngststörungenIn Europa leiden derzeit mehr als 12 Millionen Menschenan Angststörungen. Häufig beinhaltet eine solche Erkran-kung die starke Assoziation eines stressvollen, unangeneh-men Ereignisses mit einer bestimmten Situation (z.B. beipost-traumatischer Belastungsstörung, Phobien). Die Be-handlung von Angststörungen umfasst daher oft eine ko-gnitive Verhaltenstherapie, in welcher der Patient unter kon-trollierten Bedingungen wiederholt dem Angst auslösendenStimulus ausgesetzt wird und die aversive Erinnerung letzt-lich abgeschwächt wird. Interessanterweise deuten kürz-lich erschienene Publikationen darauf hin, dass die HDAC-Inhibitoren TSA oder Valproat die Reduktion von erlernterFurcht erleichtern können.
Depression Depression ist eine affektive Störung und die am häufigstenauftretende psychische Erkrankung. Untersuchungen inTiermodellen, aber auch die Analyse von humanem Post-mortem-Gewebe weisen darauf hin, dass eine deregulierteHiston-Acetylierung in den Basalganglien mit der Ausbil-dung depressiver Symptome korreliert. Im Tiermodell gehtdie Ausbildung depressiven Verhaltens mit veränderter Gen-expression einher. Kürzliche Arbeiten konnten zeigen, dassdurch die Verabreichung von HDAC-Inhibitoren wie MS-275 die pathologische Genexpression normalisiert wird undsich depressives Verhalten signifikant vermindert.
Zusammenfassung Zusammenfassend ist festzustellen, dass HDAC-Inhibitoren ei-ne aussichtsreiche therapeutische Strategie für eine Vielzahlvon neuronalen Erkrankungen darstellen (Tab. 1). Für die Zu-kunft sind vor allem zwei Fragen wichtig. Zunächst wird es es-sentiell sein zu verstehen, ob die deregulierte Histon-Acety-lierung ursächlich in die Pathogenese neuronaler Erkrankun-gen involviert ist. In diesem Fall wäre davon auszugehen, dass
208 | Pharm. Unserer Zeit | 3/2010 (39) www.pharmuz.de © 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim

T H E R A P I E F Ü R N E U R O N A L E E R K R A N K U N G E N | K L I N I K
HDAC-Inhibitoren nicht nur symptomatisch, sondern kausalin den Krankheitsverlauf eingreifen. Außerdem muss die Funk-tion individueller HDACs, HATs und die daraus resultierendenHiston-Modifikationen besser verstanden werden. Zumindestaus Arbeiten in der Hefe ist bekannt, dass einzelne Histon-Mo-difikationen, wie z.B. H4K16, wesentlichen Einfluss auf zellu-läre Prozesse, wie z.B. replikative Lebensdauer, haben kön-nen. Für das Gehirn gibt es hierzu bisher jedoch keine Daten.
Die Tatsache, dass die derzeitigen unselektiven HDAC-Inhi-bitoren ein therapeutisches Potential bei so unterschiedlichenKrankheiten wie Chorea Huntington, Morbus Alzheimer oderSchizophrenie zeigen, deutet darauf hin, dass die Chromatin-plastizität ein zentraler Mechanismus der Pathogenese ist. Dadavon auszugehen ist, dass die mit der Pathogenese einher-gehenden Chromatinveränderungen jeweils spezifisch sind,ist zu vermuten, dass selektive HDAC-Inhibitoren ein noch grö-ßeres therapeutisches Potential haben. Das Ziel muss es letzt-lich sein, spezifische HDACs und andere Histon-modifizieren-de Enzyme als therapeutische Ziele zu identifizieren und se-lektive Inhibitoren zu entwickeln.
Zitierte Literatur[1] Sananbenesi, F., Fischer, A.: The epigenetic bottleneck of neurode-
generative and psychiatric diseases. Biol. Chem. 390 (2009), 1145–1153.
[2] Vaquero, A., Loyola, A., Reinberg, D.: The constantly changing faceof chromatin. Sci. Aging Knowledge Environ. 14 (2003), RE4.
[3] Strahl, B.D., Allis, C.D.: The language of covalent histone modificati-ons. Nature 403 (2000), 41–45.
[4] Fischer, A., et al.: Recovery of learning & memory after neuronal lossis associated with chromatin remodeling. Nature 447 (2007), 178–182.
[5] Fontán-Lozano, A., et al.: Histone deacetylase inhibitors improvelearning consolidation in young and in KA-induced-neurodegenera-tion and SAMP-8-mutant mice. Mol. Cell. Neurosci. 39 (2008), 193–201.
[6] Vecsey, C.G., et al.: Histone deacetylase inhibitors enhance memoryand synaptic plasticity via CREB:CBP-dependent transcriptional acti-vation. J. Neurosci. 27 (2007), 6128–6140.
[7] Chwang, W.B., et al.: ERK/MAPK regulates hippocampal histonephosphorylation following contextual fear conditioning. Learn.Mem. 13 (2006), 322–328.
[8] Alarcon, J.M., et al.: Chromatin acetylation, memory, and LTP areimpaired in CBP+/– mice: a model for the cognitive deficit in Rubin-stein-Taybi syndrome and its amelioration. Neuron 42 (2004), 947–959.
[9] Rai, M., et al.: HDAC inhibitors correct frataxin deficiency in a Fried-reich ataxia mouse model. PLoS ONE 3 (2008). 3(4), e1958.
[10] Haass, C., Selkoe, D.J.: Soluble protein oligomers in neurodegenera-tion: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev.Mol. Cell. Biol. 8 (2007), 112–116.
Der Autor:Dr. Andre Fischer (geb. 1974); 1994–2000 Studiumder Biolgie/Diplomarbeit in Göttingen; 2000–2002Dissertation, Georg-August Universität Göttingen &Max Planck Institute für Experimentelle Medizin;2003 wiss. Assistent, Max Planck Institut für Experi-mentelle Medizin; 2003–2005 wiss. Assistent, Har-vard Medical School, Department of Pathology, USA;2006 wiss. Assistant, Massachussets Institute ofTechnology (MIT), USA; seit 2007 assoziierter Wis-senschaftler, wiss. Assistent, Massachussets Instituteof Technology (MIT), USA; seit 2007 unabhängigerGruppenleiter am European Neuroscience Institute,Göttingen.
Anschrift:Dr. Andre FischerEuropean Neuroscience InstituteGrisebach Str. 537077 Gö[email protected]
© 2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.pharmuz.de 3/2010 (39) | Pharm. Unserer Zeit | 209
EINE AUSWAHL AN BÜCHERN ZUM THEMA „EPIG ENETIK“ |David C. Allis, Thomas Jenuwein, Danny ReinbergEpigeneticsCold Spring Harbor Laboratory Press New York,2007ISBN 978-087969875-165,83 Euro
Dawson ChurchThe Genie in Your Genes: Epigenetic Medicineand the New Biology of IntentionElite Books, 2009ISBN 978-160415011-713,99 Euro
Phillipe JeanteurEpigenetics and Chromatin: Progress in Molecular and Subcellular BiologySpringer, Berlin 2008ISBN 978-354085236-053,45 Euro
Manel Esteller (Hrsg.)Epigenetics in Biology and MedicineCrc Pr Inc, 2008ISBN 978-084937289-6115,99 Euro
Alexander Haslberger, Sabine Greßler (Hrsg.)Epigenetics and Human Health: Linking Hereditary, Environmental and NutritionalAspectsWiley-VCH Verlag, Weinheim 2009ISBN 978-352732427-9119,00 Euro
Wolfgang Sippl, Manfred Jung (Hrsg.)Epigenetic Targets in Drug Discovery (Methodsand Principles in Medicinal Chemistry)Wiley-VCH Verlag, Weinheim 2009ISBN 978-352732355-5129,00 Euro
Herschel S. Zackheim (Hrsg.)Cutaneous T-Cell Lymphoma: MycosisFungoides and Sezary SyndromeInforma Healthcare, 2004ISBN 978-084932101-6292,46 Euro
Michael C. PerryThe Chemotherapy Source BookLippincott Williams & Wilkins, 2007ISBN 978-078177328-776,99 Euro
Recommended