1975 H. Quast und A. GellPri 939 Liebigs Ann. Chem. 1975, 939-945
Heterocyclische Ylide, V1)
Cycloaddition von N-Chinoliniummethylid an N-Methyl- chinoliniumkationen
Helmut Quast") und Andrbs Gellt!ri**'
Institut fur Organische Chemie der Universitat,
D-8700 Wurzburg, Am Hubland
Eingegangen am 25. Mai 1974
Das durch Decarboxylierung von N-(Carboxymelhyl)chinoliniumSetain (7) entstehende Azomethinylid 8 wird durch Cycloaddition an N-Methylchinoliniumkationen abgefangen. Dabei entstehen in ca. 4Oproz. Ausbeuten 14,14a-Dihydro-S-methylbenz[5,6]indolizino[l,2-c]- chinoliniumsalze (10). Durch Verwendung deuterierter Verbindungen wird gezeigt, dalj keine Vertauschung der Ylidfunktion durch Protonenubertragung von der N-Methylgruppe des Chinoliniumkations auf das Ylid 8 stattfindet.
Heterocyclic Ylides, V 1). - Cycloaddition of N-Quinolinium Methylide to N-Methylquinolinium Cations
The azomethinylide 8 formed by decarboxylation of N-(carboxymethy1)quinolinium betaine (7) is trapped by cycloaddition to N-methylquinolinium cations leading to 14,14a-dihydro- 5-methylbenz[S,6]indolizino[l,2-c]quinolinium salts (10) in ca. 40 per cent yields. By using deuterated compounds, it is shown that exchange of the ylide function by proton transfer from the N-methyl group of the quinolinium ion to the ylide 8 can be ruled out.
Den grundlegenden Arbeiten von Krohnke 2) verdankt man die ErschlieBung der heterocyclischen Ylide vom Typ 1.
s tab i l i s ie rende Gruppe 1%
X O Y lb: X, Y = H , Alkyl
*) Korrespondenz bitte an diesen Autor richten. **) Gegenwartige Anschrift: Zentralforschungsinstittit der Ungarischen Akademie der
1) IV. Mitteilung: H. Quast und A . GellPri, Liebigs Ann. Chem. 1975, 929, voranstehend. 2 ) 2a) Zusammenfassungen: F. Krohnke, Angew. Chem. 65, 605 (1953); 75, 181, 317 (1963);
Angew. Chem., Int. Ed. Engl. 2, 225, 380 (1963). - 2b) D . I . Schiitze und F. Kruhnke, Liebigs Ann. Chem. 765, 20, 29 (1972) und fruhere Arbeiten.
Wissenschaften, Budapest 11, Ungarn.
940 H . Quast und A. GellPri 1975
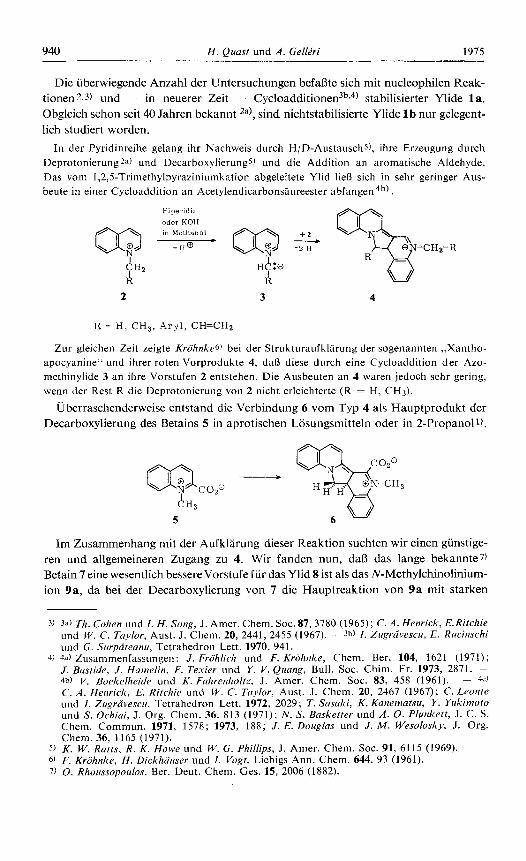
Die uberwiegende Anzahl der Untersuchungen befaljte sich mit nucleophilen Reak- tionen 2.3) und - in neuerer Zeit - Cy~loaddi t ionen~~,~) stabilisierter Ylide la . Obgleich schon seit 40 Jahren bekannt 2a), sind nichtstabilisierte Ylide 1 b nur gelegent- lich studiert worden.
In der Pyridinreihe gelang ihr Nachweis durch H/D-Austauschs), ihre Erzeugung durch Deprotonierung 2" ) und Decarboxylierungs) und die Addition an aromatische Aldehyde. Das vorn 1,2,5-Trimethylpyraziniumkation abgeleitete Ylid lieR sich in sehr geringer Aus- beute in einer Cycloaddition an Acetylendicarbonsaureester a b f a r ~ g e n ~ ~ ) .
Piperidin
oder in Mcthanol KO11 a $ %N-CH2-R
-,,la *
\ / p: *+:G
R 2 3 4
R = H, CH,, A r y l , CH=CH2
Zur gleichen Zeit zeigte Kriihnke6) bei der Strukturaufklarung der sogenannten ,,Xantho- apocyanine" und ihrer roten Vorprodukte 4, dal3 diese durch eine Cycloaddition der Azo- methinylide 3 an ihre Vorstufen 2 entstehen. Die Ausbeuten an 4 waren jedoch sehr gering, wenn der Rest R die Deprotonierung von 2 nicht erleichterte (R = H, CH3).
Uberraschenderweise entstand die Verbindung 6 vom Typ 4 als Hauptprodukt der Decarboxylierung des Betains 5 in aprotischen Losungsmitteln oder in 2-Propanoll).
Im Zusammenhang mit der Aufklarung dieser Reaktion suchten wir einen gunstige- ren und allgemeineren Zugang zu 4. Wir fanden nun, dalj das lange bekannte7) Betain 7 eine wesentlich besserevorstufe fur das Ylid 8 ist als das N-Methylchinohiurn- ion 9a, da bei der Decarboxylierung von 7 die Hauptreaktion von 9a mit starken
3) 3zd Th. Cohen und 1. H . Song, J. Amer. Chem. SOC. 87,3780 (1965); C . A. Henrick, E.Ritchie und W. C . Tuylor, Aust. J . Chem. 20, 2441, 2455 (1967). -- 3h) I . Zugrdvescu, E . Rucinschi und G. Surpdieanu, Tetrahedron Lett. 1970, 941.
J) 4a) Zusammenfassungen: J . Frohlich und F. Krohnke, Chem. Ber. 104, 1621 (1971); J . Basiide, J . Hamelin, F. Tesier und Y . V . Quang, Bull. SOC. Chim. Fr. 1973, 2871. ~
4b) V. Boekelheide und K . Fuhrenholtz, J . Amer. Chem. SOC. 83, 458 (1961). ~ 4c) C. A . Henrick, E. Ritchie und W. C. Taylor, Aust. J. Chem. 20, 2467 (1967); C. Leonie und I . Zugrdvescii. Tetrahedron Lett. 1972, 2029; T . Sasuki, K . Kanemntsu, Y. Yukimoto und S. Ochiai, J. Org. Chem. 36, 813 (1971); N. S . Busketter und A. 0. Plunkett, J. C. S. Chem. Commun. 1971, 1578; 1973, 188; J . E. Douglas und J. M . Wesolosky, J. Org. Chem. 36, 1165 (1971).
5 ) K. W. Ratts, R . K . Howe und W. G . Phillips, J . Amer. Chem. SOC. 91, 61 15 (1969). 6 ) F. Krohnke, H. Dickhuuser und I . Vogi, Liebigs Ann. Chem. 644, 93 (1961). 7 ) 0. Rhoussopoulos, Ber. Deut. Chem. Ges. 15, 2006 (1882).
1975 Heterocyclische Ylide, V 94 1
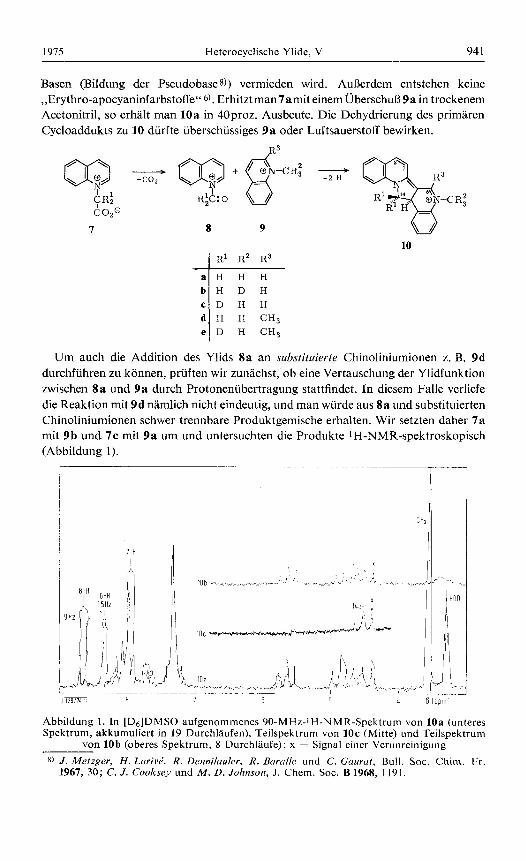
Basen (Bildung der Pseudobases)) vermieden wird. AuRerdem entstehen keine ,,Erythro-apocyaninfarbstoffe" 6). Erhitztman7amit einemUberschuB 9 a in trockenem Acetonitril, so erhalt man 10a in 40proz. Ausbeute. Die Dehydrierung des primaren Cycloaddukts zu 10 diirfte iiberschiissiges 9 a oder Luftsauerstoff bewirken.
R3
7 8 9
10
Um auch die Addition des Ylids 8a an substifuievte Chinoliniumionen z. B. 9d durchfiihren zu konnen, pruften wir zunachst, ob eine Vertauschung der Ylidfunktion zwischen 8a und 9 a durch Protonenubertragung stattfindet. In diesem Falle verliefe die Reaktion mit 9d namlich nicht eindeutig, und man wurde aus 8a und substituierten Chinoliniumionen schwer trennbare Produktgemische erhalten. Wir setzten daher 7 a mit 9b und 7c mit 9 a urn und untersuchten die Produkte 1H-NMR-spektroskopisch (Abbildung 1).
7 H
i 10b x u - x 8 H 6 H
I ~ H Z ' L , ! , 9 HZ
I I I
1 'I loc w I b ~ ~ * w - + + ~ ii -
.. l A - * -Y* yr" - m 8 3 5 L G [ p p m l
von 10b (oberes Spektrum, 8 Durchlaufe); x ~ Signal einer Verunreinigung
Abbildung 1. In [D6]DMSO aufgenommenes 90-MHz-IH-NMR-Spektrum von 10a (unteres Spektrum, akkumuliert in 19 Durchlaufen), Teilspektrum von 1Oc (Mitte) und Teilspektrum
8) J . Metzger, H. LurrvP, R. Denniluuler, R . Burulle und C. Guurut, Bull. SOC Chim. Fr.
1- 1967, 30; C. J. Cooksey und M . D . Johnson, J. Chem. SOC. B 1968, 1191.

942 H. Quast und A . GellPvi 1975
Entsprechend dem eingesetzten Molverhaltnis 7:9 = 1 : 5.37 wurde man bei statisti- scher Verteilung des Deuteriums infolge Protonenubertragung zwischen 8 und 9 im ersten Fall eine gleichzeitige Abnahme der Intensitat des N-Methylsignals und der Signale der CH2-Gruppe um 11 %, im zweiten Fall uni 89% erwarten. Wie sich aus den 1H-NMR-Spektren ablesen IaBt, tritt das jedoch nicht ein. Vielmehr entsteht (innerhalb der Grenzen der MeBgenauigkeit) aus 7a und 9b nur lob, aus 7c und 9a nur 1Oc. Eine Vertauschung der Ylidfunktion kann daher keine wesentliche Rolle spielen.
Die in Abbildung 1 getroffene Zuordnung der IH-NMR-Signale basiert auf dem Vergleich mit den Spektren von Chinoliniumkationen9) sowie von 6 und 10e, denen das Dublett ( J = 1.5 Hz) bei 8 = 8.35 ppm fehlt. Das ABC-System bei 6 = 4-6 ppm ist dern bereits analysiertenl) von 6 auBerordentlich ahnlich. Aus dem Spektrum von 1Oc geht hervor, daB von den drei stark gekoppelten Protonen 14a-H bei hochstem Feld absorbiert.
Wie nach dem Ergebnis der Versuche niit den deutierten Verbindungen zu erwarten, addiert sich das Ylid 8 auch glatt an das 1,2-Dimethylchinoliniumkation 9d zu dem einheitlichen Produkt 10d. Bemerkenswert ist dabei, daB offenbar die Bereitschaft von 8 zur konzertierten oder zweistufigen Cycloaddition seine Basizitat soweit ubertrifft, daB selbst aus der 2-Methylgruppe von 9d kein Proton abgespalten wird, wie das Fehlen von entsprechenden Folgeprodukten (Monomethincyaninen 10)) anzeigt.
Aus den Dihydroverbindungen 10 lassen sich durch photochemische Dehydrierung leicht die entsprechenden Benzindolizinochinoliniumsalze gewinnen 1) .
Die beschriebenen Ergebnisse weisen die Decarboxylierung des Betains 7 als eine besonders ,,saubere" Bildungsweise eines nichtstabilisierten Azomethinylids vom Typ l b aus. Sie durfte auch fur Cycloadditionen anderer, ahnlicher Ylide von Nutzen sein.
Wir danken Frau H . Heinze fur die Aufnahme der IH-NMR-Spektren a n einem 1H-NMR- Spektrometer HFX-90 der Fa. Bruker-Physik AG, das die Stiftung Volkswugenwerk zur Verfugung stellte. Fur groozugige finanzielle Unterstutzung danken wir der Deutschen For- schungsgerneinsch~!~. A. Gelleri dankt besonders der Alexander-von- Humholdt-Stiftung fur ein einjiihriges Forschungsstipendium (I 971/ 1972).
9 ) P. Hurnrn und W. v . Philipshorn, Helv. Chim. Acta 54, 2363 (1971). 10) J . Metzger, H . LurivP, R . Denniluuler, R . Burulle und C. Cuurut, Bull. SOC. Chim. Fr.
11) H. 0. House und C. G . Pitt, J. Org. Chem. 31, 1062 (1966). 12) C. S . Murwel, E. W. Scott und K . L . Amstutz, J. Amer. Chem. Soc. 51, 3638 (1929). 13) A. R . Kutritzky und A . P. Ambler in Physical Methods in Heterocyclic Chemistry
( A . R . Kutritzky), 1 . Aufl., Bd. 2, S. 296, Academic Press, New York-London 1963; A. R . Kutritzky und P . J . Tuylor in Physical Methods in Heterocyclic Chemistry ( A . R . Kur- r i tzky), 1. Aufl., Bd. 4, S. 401, Academic Press, New York-London 1971.
1967, 40, 57.
14) C. F. Wurd, J . Chem. SOC. 121, 1161 (1922).

1975 Heterocyclisch: Ylide, V 943
Experimenteller Teil Allgemeine Vorbemerkungen siehe Lit.')
1-jD31 Methylchinoliniun~perchlorut (9 b). - Das aus p-Toluolsulfonsaure-[D~]methylester.* 1)
und Chinolin erhaltene p-Toluolsulfonatl2) wurde mit waRrjger NaC104-Losung in das Perchlorat ubergefuhrt 1 ) . Im IH-NMR-Spektrum i n Trifluoressigsaure war kein Singulett bei 8 = 4.8 ppml) nachweisbar. - IR (KBr): 2275, 2245 (CD3), 1626, 1607, 1593, 1523 (Chinolinringl3)), 1140 - 1040 cm-1 (C104).
iD3IBromessigsuure. - In Anlehnung an Lit. 14) wurden zu 10.0 g (156 mmol) [DdIEssig- saure (Fa. Merck) und 0.2 g rotem Phosphor bei 95°C unter Ruhren 30.0 g (188 mmol) Brom in 45 min getropft und die Mischung noch 45 min auf 102-105°C erhitzt. Nach Destil- lation i. Vak. erhielt man 20.0 g (90%) vom Sdp. 105-106"Cj18 Torr, Schmp. 49-50°C. -
Tm 1H-NMR-Spektrum (CC14) kein Signal.
I-(Carbo.wymethyl)chinoliniumbromid *). - 10.1 g (78 mmol) Chinolin und 11.4 g (82 mmol) Bromessigsaure wurden in 45 ml trockenem Acetonitril 10 Tage bei 40-45°C geriihrt. Das abgesaugte Rohprodukt (14.0-16.0 g, 67-76%) vom Zen.-P. 189°C wurde aus 150 ml Methanol/Wasser (4: 1) bei tiefer Temperatur umkristallisiert. Man erhielt 11.4-12.2 g (54-58%) farblose Kristalle vom Zen-P . 189-191°C.
C I ~ H I O B ~ N O ~ (268.1) Ber. Br 29.80 N 5.22 Gef. Br 30.01 N 5.23
I - ( Curboxymethy1)chinolinium-tetrafluoroborut. - Aus 13.4 g (50 mmol) des voranstehend beschriebenen Bromids in 60 ml warmem Wasser und 50 ml gesattigter, waI3riger NaBF4- Losung nach Abkuhlen auf 0°C erhielt man 11.4 g (83%) vom Zers.-P. 178--185°C. Um- kristallisieren aus Wasser lieferte 9.9- 10.2 g (72-74 %) farblose Kristalle vom Zers.-P. 190-191°C. - IR (KBr): 1739 (COOH), 1630, 1594, 1522 (Chinolinringl3)), 1080 cm-1 (breit, BF4). - IH-NMR (CF3COOH; 60 MHz): 8 = 6.10 (s; 2H), 8.00-8.70 (m; 5H), 9.20-9.47 ppm (m; 2H).
C ~ ~ H ~ O B F ~ N O ~ (275.0) Ber. N 5.10 Gef. N 5.22
(I-Chino1inio)ucetut-monohydrut (7a' H20) a) 3.48 g (13 mmol) I-(Carboxymethy1)chinoliniumbromid in 20 ml Wasser wurden mit
frisch bereitetem, neutral gewaschenem Silberhydroxid [aus 2.32 g (13.7 mmol) AgN03 und 14.5 ml 1 N KOH] 20 min geruhrt. Die Losung wurde nach Abzentrifugieren des Nieder- schlags bei 40°C i. Vak. bis zur beginnenden Kristallisation eingedampft und mit 20 ml Acetonitril versetzt. Man erhielt 2.1 1-2.20 g (79-83 %) farblose, lichtempfindliche, hygro- skopische Kristalle vom Zen.-P. 172--175°C. Umkristallisieren aus 6 ml Methanol + 10 ml trockenem Acetonitril lieferte bei -20°C 1.86-1.95 g (70-73 %) vom Zen-P . 172 - 176°C (Lit.7) Zers.-P. 168--171°C). - IR(KBr): 1615(breit, COO-~), 1593,1526 cm-1 (Chinolinring13)). - 1H-NMR (D2015); 60 MHz): 8 = 5.67 (s; 2H), 7.77-8.53 (m; 5H), 9.00-9.43 ppm (m; 2 H).
C I ' H ~ N O ~ . H ~ O (205.2) Ber. N 6.85 Gef. N 6.42
Nach Trocknen uber P205 (10-3 Torr, 40"C, 24 h) erhalt man die wasserfreie Verbindung, die sich jedoch rasch unter teilweiser Zersetzung dunkel farbt; Zen.-P. 178°C.
C I I H ~ N O ~ (187.2) Ber. N 7.48 Gef. N 7.30
*) Anmerkung bei der Korrektur (30. Mai 1975): Zur Darstellung des Chlorids s. M. Mottier, Helv. Chim. Acta 58, 337 (1975).
15) In D20 diente Natrium-3-trimethylsilyl-[2,2,3,3-D~]-propionat als interner Standard (8 = 0 ppm): L. Pohl und M . Eckle, Angew. Chem. 81, 395 (1969); Angew. Chem., Int. Ed. Engl. 8, 381 (1969).

944 H . Quast und A. CelCdri 1975
b) 24.5 g (89 mmol) I-(Carboxymethyl)chinolinium-tetrafluoroborat in 200 ml Wasser wurden unter Eiskuhlung rnit 9.1 g (91 mmol) KHC03 versetzt. Der pH-Wert wurde mit ca. 32proz. waoriger HBF4 (d 2 1.22) auf 4- 4.5 eingestellt, die Losung 15 h auf 0°C gekiihlt und nach Filtration i. Vak. bei 40°C bis auf ca. 30 ml eingedampft. Nach Zugabe von 200 ml Methanol wurde erneut filtriert, i. Vak. eingedampft und der erhaltene Sirup rnit 40 ml Acetonitril behandelt. Man gewann 13.7 g (75 %) farblose Kristalle, die in 50 ml warmem Methanol gelost und rnit 120 ml trockenem Acetonitril ausgefallt wurden. Ausb. 11.5 g (63 %) vom Zen.-P. 172--175°C.
Il-Chinolinio)-/D2jncetat (7c). ~ Es wird analog 7a aus dem Bromid rnit Silberhydroxid hergestellt; Zen.-P. 175--178°C. - IR (KBr): 2255, 2150 (CDz), 1615 (breit, COO-), 1592, 1522 cm-1 (Chinolinringl3)). ~ Im 1H-NMR-Spektrum (D20) ist kein CHz-Signal bei S =
5.67 ppm nachweisbar.
CllH7DZN02 (189.2) Ber. N 7.40 Gef. N 7.30
l4,I4a-Dih,vdro-5-methylbenz~5,6Jindoli~i11o~l,2-cJchiuolini1~mperchlorat (10a). - I .27 g (6.2 mmol) 7a .Hz0 und 4.87 g (20 mmol) 9a l ) wurden unter Nz in 30 ml trockenem Aceto- nitril 35 min unter Ruhren und RiickfluR erhitzt. Nach 15stdg. Stehen an der Luft bei Raum- temperatur erhielt man 63 I mg (27 %) rote Kristalle vom Schmp. oberhalb 300°C und nach Einengen der Mutterlauge i. Vak. auf die Halfte und Chromatographie [300 g neutrales A1203, Aktiv-Stufe 111; Eluieren rnit Chloroform/Athanol (2: l)] weitere 321 mg (13 %). Umkristallisieren aus Dimethylformamid/Wasser (6:4) lieferte 803 mg (34%). - IR- und UV- Spektren waren identisch rnit denen von authent. 10aI). - IH-NMR ([D6]DMSO, 19 Durchlaufe; s. Abb. 1): 8 - 3.53 (s; 3H), 4.38-5.00, 5.50-5.80 (ABC-m), 7.16-8.20 (m), 7.96, 8.63 (AB, J = 9Hz), 8.35 ppm (d, J = 1.5 Hz; 1 H).
C20H17CIN204 (384.8) Ber. C 62.42 H 4.45 C1 9.21 N 7.28 Gef. C 62.47 H 4.36 C1 9.34 N 7.28
14,14a-Dihydro-S-[D~]methyC-beriz[ 5,6]indolizinoi I,2-~Jchinoliniumperchlorat (10 b). - Es wird analog 10a aus 883 mg (,4.3 mmol) 7a .H20 und 5.69 g (23.1 mmol) 9 b hergestellt. Ausb. 520 mg (31 %)16), laut UV-Spektrum identisch rnit 10a. - 1H-NMR ([D6]DMSO, 8 Durchlaufe; s. Abb. 1): Bis auf das Fehlen des NCH3-Signals bei 6 = 3.53 ppm identisch mit dem von 10a.
14,14a-,'D~/Dihydro-5-methylhenz~5,6~indo1izino[l,2-cJchinoliniuniperchlornt (1Oc). - Es wird analog 10b aus 7 c und 9a l ) hergestellt. Ausb. 490 mg (29%)16), laut UV-Spektrum identisch rnit 10a. -~ IH-NMR ([D6]DMSO; s. Abb. 1) : Anstelle des ABC-Spektrums von 1Oa verbreitertes Singulett bei 8 = 4.52 ppm.
14,I4~i-Dihydro-5,6-dirneihyIbenz[5,6]indolizino~l,2-cJchinoliniurnperchlorat (10d). - Es wird analog 10a aus 790 mg (3.85 mmol) 7a .H20 und 5.67 g (22 mmol) 9d17) in 35 nil trockenem Acetonitril hergestellt. Ausb. 595 mg (39 % ) l h ) , die aus Dimethylformamid/Wasser (7:3) 510mg (33%) rote Tafeln vom Schmp. oberhalb 300°C ergaben. - UV (Methanol): A,,, (log E) = 481 (4.35), 349.5 (3.86), 313 (4.03), 264 (3.93), 233 nm (4.38). - 1H-NMR ([D6]DMSO): 8 = 2.60 (s; 3H), 3.54 (s; 3H), 4.24--4.96, 5.39- 5.70 (ABC-m), 7.1 1-8.20 (m), 8.59 ppm (d, J = 9.5 Hz).
CzjH19CIN204 (398.8) Ber. CI 8.90 N 7.02 Gef. CI 8.96 N 7.00
16) Rohprodukt; die Mutterlauge wurde nicht chromatographisch aufgearbeitet. 17) W. Konig, J. Prakt. Chem. [2] 85, 514 (1912).

1975 Heterocyclische Ylide, V 945
14,14ci-~D~~Dihydra-5,6-dimethylbenrj5,6]indolizino[l,2-c~chinoliniumperchlorat (IOej. ~
Es wird analog 10d aus 570 mg (2.75 mmol) 7c.H20 hergestellt. Ausb. 380 mg (34%) rote Tafeln, laut UV-Spektrum identisch mit 10d. - 1H-NMR ([D6]DMSO): Anstelle des ABC- Multipletts von 10d verbreitertes Singulett bei 8 = 4.39 ppm.
5,6-Din~e~hylbenz~5,6]indolizino~l,2-c]chinoliniumperchlorn1. ~- Durch Photooxidationl) von 172 mg (0.43 mmol) 10d erhielt man 138 mg (81 %) gelbe Kristalle vom Schmp. oberhalb 300°C (aus Dimethylformamid/Wasser). - UV (Methanol): A,,, (log E ) = 436 (4.25), 348.5 (3.71), 330 (Schulter, 3.93), 309 (4.27), 296 (4.24), 287 (Schulter, 4.21), 265 (Schulter, 4.28), 245 (4.57), 232 nm (Schulter, 4.42).
C ~ I H ~ ~ C I N ~ O ~ (396.8) Ber. CI 8.93 N 7.08 Gef. C1 8.96 N 7.19 [129/74]
Recommended


![Konkave N-heterocyclische Carbene (NHC) als nucleophile ... · Das Konzept der konkaven Reagenzien wurde 1987 von Ulrich Lüning vorgestellt. [14] Hierbei handelt es sich um meist](https://img.pdfslide.org/doc/110x75/5d551ecc88c9938f688bbb7e/konkave-n-heterocyclische-carbene-nhc-als-nucleophile-das-konzept-der.jpg)



![[2 + 3] Cycloaddition Reactions of Azido Metal Complexes ...zfn.mpdl.mpg.de/data/Reihe_B/27/ZNB-1972-27b-0745.pdf · CYCLOADDITION REACTIONS OF AZIDO METAL COMPLEXES 747 tures (TCH,](https://img.pdfslide.org/doc/110x75/60648a2df6edbd732a617206/2-3-cycloaddition-reactions-of-azido-metal-complexes-zfnmpdlmpgdedatareiheb27znb-1972-27b-0745pdf.jpg)





![Photoschaltbare Resorc[4]arene - uni-bielefeld.de...Photoschaltbare Resorc[4]arene Photochrome Supramolekulare Wirtsysteme durch intramolekulare [4+4] Cycloaddition von Anthracen Dissertation](https://img.pdfslide.org/doc/110x75/6080a426c1c70419872280eb/photoschaltbare-resorc4arene-uni-photoschaltbare-resorc4arene-photochrome.jpg)