Synthese des A´-Rings von Labyrinthopeptin A2
und eines Referenzstandards
zum Nachweis der ungewöhnlichen Aminosäure Labionin
vorgelegt von
Diplom-Chemiker
Maik Henkel
aus Luckenwalde
Von der Fakultät II - Mathematik und Naturwissenschaften
der Technischen Universität Berlin
zur Erlangung des akademischen Grades
Doktor der Naturwissenschaften
Dr. rer. nat.
genehmigte Dissertation
Promotionsausschuss:
Vorsitzender: Prof. Dr. rer. nat. Martin Lerch
Berichter/Gutachter: Prof. Dr. rer. nat. Roderich Süßmuth
Berichter/Gutachter: Prof. Dr. rer. nat. Michael Bienert (FMP Berlin)
Tag der wissenschaftlichen Aussprache: 25. April 2011
Berlin 2011
D 83

Die vorliegende Arbeit wurde unter der Leitung von
Herrn Prof. Dr. Roderich Süssmuth
in der Zeit von November 2006 bis Januar 2011 am Institut für Chemie der Fakultät II der
Technischen Universität Berlin angefertigt.

Danksagung
Mein besonderer Dank gilt Herrn Prof. Dr. Roderich Süssmuth für die Überlassung des Themas,
hervorragende Arbeitsbedingungen und die ausgezeichnete Betreuung während der
Durchführung dieser Arbeit.
Herrn Prof. Dr. Michael Bienert danke ich herzlich für die bereitwillige Übernahme der zweiten
Berichterstattung.
Herrn Prof. Dr. Martin Lerch danke ich für die Übernahme des Prüfungsvorsitzes.
Herrn Dr. Mark Brönstrup, Herrn Dr. Luigi Toti und Herrn Dr. Joachim Wink (Sanofi-
Aventis,Frankfurt, Deutschland) danke ich für die Bereitstellung der Labyrinthopeptine.
Herrn Dipl. Chem. Alexander Pesic danke ich für die Hilfe bei der Herstellung der
Labyrinthopeptinderivate, die Durchführung der GC-Analytik mit dem Referenzstandardsubstanz
zum Labioninnachweis und die gute Zusammenarbeit bei diesem Projekt.
Herrn Dipl. Chem. Julian Kretz danke ich für die Durchführung der GC-Analytik der
γ-Methylenglutaminsäurederivate und möchte ihm viel Erfolg bei der Weiterführung dieses
Projekts wünschen.
Herrn Dipl. Chem. Paul Ensle danke ich für die vielen interessanten Gespräche und die
Hilfestellung bei der Benutzung des Peptidsynthesizers.
Frau Dr. Maria Schlangen und Frau Christine Klose danke ich für die Durchführung der
zahlreichen MS-Messungen.
Frau Dr. Elisabeth Irran danke ich für die Kristallstrukturbestimmung.
Für zahlreiche konstruktive Gespräche, das gute Arbeitsklima und Hilfestellungen möchte ich
mich bei meinen Kollegen aus der Arbeitsgruppe Süssmuth bedanken, namentlich: Dr. Srinivas
Banala, Todor Baramov, Dr. Diane Butz, Dr. Brian Davis, Alexander Denisiuk, Paul Ensle,

Dr. Sven C. Feifel, Dr. Elvira Gottardi, Anne Hänchen, Soliman Alsayd Helali, Manuela
Hügelland, Bahar Kalyon, Dr. Simone Keller, Joanna Krawczyk, Bartlomieij Krawczyk, Ginka
Kazakova, Julian Kretz, Benjamin Landmann, Khai Ming Lie, Marius Löhken, Diana Matthes,
Eva Mösker, Dr. Agnes Mühlenweg, Jane Müller, Wolfgang Müller, Jonny Nachtigall, Florian
Oldach, Alexander Pesic, Vanessa Peters, Dr. Stefan Pohle, Marta Ramirez Hernandez, Saskia
Rausch, Dr. Pierre-Loic Saaidi, Georg Sambeth, Nicole Sattler, Dr. Heiko Schadt, Caroline
Schloßer, Dr. Timo Schmiederer, Dr. Kathrin Schneider, Vivien Schubert, Hanna von
Suchodoletz, Kamil Stelmaszyk, Dr. Suada Turkanović, Stefanie Uhlmann, Kati Winter,
Dr. Falko Wolter und David Wagner.
Für ihre stetige Unterstützung und ihr sehr großes Verständnis möchte ich mich bei meiner
Lebenspartnerin Ina bedanken. Mein Dank gilt zudem meinen Eltern, die mir das Studium der
Chemie ermöglicht haben.

Inhaltsverzeichnis
Inhaltsverzeichnis 1. Einleitung .............................................................................................................................. 1
1.1 Conotoxine ....................................................................................................................... 2 1.2 Lantibiotika ...................................................................................................................... 3
1.2.1 Biologische Aktivität der Lantibiotika...................................................................... 4 1.2.2 Biosynthese der Lanthionine und Methyllanthionine ............................................... 6 1.2.3 Klassifizierung von Lantibiotika............................................................................. 10
1.3 Die Labyrinthopeptine.................................................................................................... 13 1.3.1 Struktur von Labyrinthopeptin A2 .......................................................................... 14 1.3.2 Biosynthese von Labyrinthopeptin A2.................................................................... 16 1.3.3 Weitere Labionin-haltige Peptide............................................................................ 18 1.3.4 Biologische Aktivität der Labyrinthopeptine.......................................................... 20
1.4 Totalsynthesen von Lantibiotika .................................................................................... 22 1.4.1 Totalsynthese von Nisin .......................................................................................... 22 1.4.2 Totalsynthese von Bis(desmethyl)-Lacticin 3147 A2 und Lactocin S.................... 23
1.5 Mechanistische Details wichtiger Schlüsselreaktionen.................................................. 24 1.5.1 Stereoselektive Alkylierung von Glycin ................................................................. 25 1.5.2 Synthese von α,α-disubstitutierten Aminosäuren aus zyklischen Sulfiten............. 27 1.5.3. Synthese von α,α–disubstituierten Aminosäuren durch chelatverbrückte Claisen-Umlagerung...................................................................................................................... 29
2. Aufgabenstellung................................................................................................................ 33 3. Ergebnisse und Diskussion ................................................................................................ 35
3.1 Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure .............................................. 35 3.1.1. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure über ein Oxazolidin ..... 36 3.1.2. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure durch eine chelatverbrückte Claisen-Umlagerung............................................................................. 40 3.1.3. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus Pyrazolderivaten.... 41 3.1.4 Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus γ-Methylenglutamin-säure ................................................................................................................................. 42
3.2 Studien zur Synthese von Labionin................................................................................ 46 3.3 Synthese des A´-Rings von Labyrinthopeptin A2.......................................................... 48 3.4 Synthese eines analytischen Referenzstandards zum Labioninnachweis ...................... 57 3.5 Derivatisierung der Naturstoffe Labyrinthopeptin A1 und A2 ...................................... 61
4. Zusammenfassung und Ausblick ...................................................................................... 65 5. Experimenteller Teil .......................................................................................................... 69
5.1. Allgemeine Informationen ............................................................................................ 69 5.2 Synthesevorschriften ...................................................................................................... 73
5.2.1. Vesuch der Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus Oxazolidinen .................................................................................................................... 73 5.2.2. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus Pyrazolderivaten.... 82 5.2.3. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus γ-Methylenglutaminsäure ................................................................................................. 85 5.2.4. Studien zur Synthese von Labionin........................................................................ 98 5.2.5. Synthese des Dipeptides des A´-Ring, des Tripeptids des B´-Rings und des Modellpeptids................................................................................................................. 101 5.2.6. Synthese des A´-Rings von Labyrinthopeptin A2................................................ 112 5.2.7. Synthese eines analytischen Refenzenzstandards zum Labioninnachweis .......... 123 5.2.8. Derivatisierung der Labyrinthopeptine ................................................................ 134
5.3. Analytische Daten ....................................................................................................... 139 5.3.1 NMR-Spektren ausgewählter Verbindungen ........................................................ 139

Inhaltsverzeichnis
5.3.2. MS/MS-Spektrum von Peptid 117 ....................................................................... 170 5.3.3. LC-MS-Analytik der Peptide 122, 123 und 124 .................................................. 171 5.3.4. Röngenstrukturdaten von (2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-(hydroxymethyl)pentandioat .......................................................................... 174
6. Anhang .............................................................................................................................. 180 6.1. Literaturverzeichnis..................................................................................................... 180 6.2. Abkürzungsverzeichnis ............................................................................................... 185

Einleitung
1
1. Einleitung
Peptide beeinflussen eine Vielzahl wichtiger physiologischer und biochemischer
Lebensfunktionen. Bei Wirbeltieren kennt man ca. 100 Peptide mit Funktionen im zentralen
und peripheren Nervensystem, wo sie z.B. als Neurotransmitter und Neuromodulatoren eine
bedeutende Rolle spielen. Darüber hinaus sind sie an der Regulation einer Vielzahl
biochemischer Prozesse, wie beispielsweise Stoffwechsel, Schmerzgeschehen, Reproduktion
und Immunabwehr beteiligt.
Historisch betrachtet, ist die chemische Peptidsynthese eine klassische Methode, die sich in
den letzten Jahrzehnten entscheidend weiter entwickelt hat, obwohl schon um die
Jahrhundertwende des vergangenen Jahrhunderts durch Theodor Curtius und Emil Fischer der
Grundstein dafür gelegt wurde. Bei vielen oft nur in kleinen Mengen isolierten Peptiden
konnte erst mittels chemischer Synthese der endgültige Strukturbeweis erbracht werden[1]
Auch die umfangreichen Studien zur Erforschung der Beziehung zwischen Struktur und
biologischer Aktivität am Beispiel einer Vielzahl sequenzvariierter Analoga, die oft
nichtproteinogene Bausteine und Strukturelemente enthalten, belegen die Bedeutung der
chemischen Peptidsynthese, die nicht zuletzt durch die Realisierung des Merrifieldschen
Konzepts der Festphasen-Peptidsynthese den entscheidenden Aufschwung erhalten hat. Die
Gewinnung von Polypeptiden und Proteinen durch DNA-Rekombination bedeutete einen
weiteren methodischen Fortschritt. Mit der gentechnischen Herstellung von pharmazeutisch
relevanten Proteinen lässt sich das Konzept der Therapie endogener Proteinwirkstoffe
verifizieren, deren wichtigste Indikationsgebiete Herz-Kreislauf-, Tumor- und
Autoimmunerkrankungen sowie Infektionen sind. Mit Sicherheit wird durch diese
Entwicklung die klassische Peptidsynthese nicht in Frage gestellt. Kleine Peptide, wie der
Peptidsüßstoff Aspartam mit dem Bedarf von 5000 Jahrestonnen, und Peptide mittlerer
Kettenlänge, insbesondere mit nichtproteinogenen Aminosäuren oder mit 13C-markierten
Aminosäuren für spezielle NMR-Untersuchungen, werden Objekte der klassischen Synthese
bleiben.
Für die Lösung vielschichtiger biologischer Zielstellungen nimmt der Bedarf an synthetischen
Peptiden ständig zu. Die neuen Aufgabenstellungen erlauben keine isolierte Position einer
allein syntheseorientierten Peptidchemie, vielmehr müssen Synthese, Analytik, Isolierung und
Strukturaufklärung integrativer Teil einer interdisziplinären Zusammenarbeit sein[2]. Die
Natur bringt eine große Vielfalt von bioaktiven Peptiden mit interessanten Strukturen hervor,
auf die im Folgenden genauer eingegangen wird.

Einleitung
2
1.1 Conotoxine
Conotoxine sind eine Gruppe von Toxinen die aus dem Gift der Kegelschneckengattung
Conus isoliert wurden[3, 4]. Dabei handelt es sich um ribosomal synthetisierte Peptide, die aus
15 bis 30 Aminosäuren bestehen und einen hohen Cysteinanteil aufweisen. Diese Cysteine
bilden je nach Art des Conotoxins zwei bis vier Disulfidbrücken aus, die dadurch maßgeblich
zur Struktur und zur Wirkung dieser Peptide beitragen. Eine Kegelschnecke verfügt je nach
Art über eine Vielzahl von Conotoxinen. Diese werden bei der Jagd in ein Beutetier injiziert
und blockieren mit hoher Selektivität spannungsabhängige Ionenkanäle, was zur Lähmung der
Beute führt[5]. Aufgrund dieser Eigenschaften sind Conotoxine als Analgetika von großem
Interesse. Ein Conotoxin, das als Medikament genutzt wird, ist Ziconotid®. Dabei handelt es
sich um die synthetische Form von ω-Conotoxin MVIIA, das aus Conus magus isoliert
wurde[6]. Dieses aus 25 Aminosäuren bestehende Peptid (Abb. 1) ist ein Nicht-Opioid-
Analgetikum das selektiv die spannungsabhängigen Calcium-Ionenkanäle blockiert, die im
gesamten zentralen und peripheren Nervensystem zu finden sind und somit die Freisetzung
von Neurotransmittern und die Signalweitergabe verhindert[7].
A) B)A) B)
Abbildung 1: Struktur von ω-Conotoxin MVIIA als Kugelmodell (A) und 3D-Modell (B) des Peptidrückgrats.
Die Stabilität von ω-Conotoxin MVIIA beruht auf seiner Struktur, in der zwei
Disulfidbrücken das Peptidrückgrat verbinden und einen eingebetteten Ring bilden, der von
der dritten Disulfidbrücke durchstoßen wird (Abb. 1B). Dieses Strukturmotiv verleiht den
Conotoxinen ihre hohe konformationelle Stabilität im Vergleich zu Peptiden ohne
Disulfidbrücken[8].

Einleitung
3
1.2 Lantibiotika
Eine weitere Gruppe bioaktiver schwefelhaltiger Peptide sind Lantibiotika. Das
charakteristische Strukturelement dieser Peptide ist die ungewöhnliche Aminosäure
Lanthionin (Lan) bzw. β-Methyllanthionin (MeLan)[9]. Dabei handelt es sich formal um
Alanin bzw. 2-Aminobutylsäure im Fall von MeLan, die an ihren β-Kohlenstoffatomen über
eine Thioetherverbrückung mit einem weiteren Alanin verbunden sind (Abb. 2).
(S) SHN
O (R)NH
O
(S) (S) SHN
O (R)NH
O
meso-Lanthionin(Lan)
(2S,3S,6R)-3-Methylanthionin(MeLan)
Abbildung 2: Struktur von meso-Lanthionin und Methyllanthionin, die charakteristischen Strukturelemente der Lantibiotika.
Die Aminosäure Lanthionin wurde 1941 bei der Behandlung von Wolle (latein.: lana =
Wolle) mit Natriumcarbonat zum ersten Mal beobachtet.[10] Die Namensgebung der
Lantibiotika leitet sich aus der Bezeichnung lanthioninhaltiger antibiotischer Peptide ab[11].
Zur Klasse der Lantibiotika gehören stark posttranslational modifizierte Peptide mit einer
Kettenlänge von 19 bis 38 Aminosäuren, wobei der Modifizierungsgrad in Lactocin S[12] mit
24% am geringsten und im Lantibiotikum 10789[13] mit 58 % am stärksten ist. Neben dem
charakteristischen Strukturelement Lanthionin, können noch weitere Modifikationen auftreten
(Abb. 3). So kann z.B. ein C-terminales Cystein S-Aminovinyl-D-cystein oder
S-Aminovinyl-3-Methyl-D-cystein bilden, während ein N-terminales Didehydroalanin oder
Didehydrobutyrin zu 2-Oxopropionyl- bzw. 2-Oxobutyrylresten hydrolysiert wird[14].
Aufgrund der zahlreichen posttranslationalen Modifikationen zeigen Lantibiotika eine erhöhte
Stabilität gegenüber Proteaseaktivität[15], oxidativen und sauren Bedingungen[16]. Diese
Eigenschaft von lanthioninhaltigen Peptiden wird auch eingesetzt um die Stabilität bioaktiver
Peptide mit einer Disulfidbrücke durch den Austausch von Cystin gegen Lanthionin zu
erhöhen[17-19].

Einleitung
4
SHN
ONH
O
SHN
ONH
O
HN
O HN
O
NHHN
ONH
O
4 SHN
ONH
SHN
ONH
OOO
O
HOO OH
N
OHN
HOOH
OH2N
O
OHN
NH
Cl
HO
ON
HO
O
OH
meso-Lanthionin(Lan)
(2S,3S,6R)-3-Methylanthionin(MeLan)
2,3-Didehydroalanin(Dha)
2,3-Didehydrobutyrin(Dhb)
(2S,9R)-Lysinoalanin AviCys AviMeCys
2-Oxobutyryl 2-Oxopropionyl 2-Hydroxypropionyl D-Alanin
erythro-3-Hydroxy-L-Aspartat
allo-Isoleucin Chlortryptophan Monohydroxyprolin Dihydroxyprolin
Abbildung 3: Bisher entdeckte Modifikationen in Lantibiotika.
1.2.1 Biologische Aktivität der Lantibiotika
Das neuerliche Interesse an Lantiobiotika hat seinen Ursprung im alarmierenden Anstieg der
Vancomycinresistenzen in Klinikisolaten[20]. Vancomycin, das Reserveantibiotikum zur
Behandlung von Infektionen mit Methicillin-resistenten Staphylococcus aureus (MRSA)-
Stämmen, hat durch vermehrte Resistenzen in seiner Wirkung und Bedeutung nachgelassen,
sodass es von enormer Bedeutung ist, neue Antibiotika zur Behandlung von Infektionen mit
multiresistenten Erregern zu finden. Tab. 1 fasst pharmazeutisch interessante Lantibiotika und
deren möglichen medizinischen Einsatz zusammen.
Das größte Potential der Lantibiotika liegt in der Behandlung von Infektionen mit
multiresistenten S. aureus-Stämmen. Lantibiotika wie Mersacidin und Actagardin zeigen eine
beachtliche Aktivität gegen multiresistente S. aureus Stämme, einschließlich der MRSA-
Stämme[21]. Bisher wurden Infektionen mit diesen Stämmen vorrangig mit Vancomycin
behandelt, was zu einem vermehrten Auftreten von Resistenzen gegen dieses Antibiotikum
Einleitung
5
führte. Diese Vancomycin-resistenten Stämme bleiben aber sensitiv gegenüber Mersacidin[22],
weshalb auf einen anderen Wirkmechanismus als beim Vancomycin geschlossen werden
kann.
Tabelle 1: Lantibiotika und ihre potentielle medizinische Verwendung[21].
Lantibiotikum Produzent Inhibitor-Aktivität
mit kommerziellem
Interesse
Potentielle medizinische
Anwendung
Nisin A Lactococcus lactis Gram-positive und
-negative Bakterien
inkl. Helicobacter
pylori
Mundhygiene, Behandlung
von MRSA- und Entero-
kokkeninfektionen, Mastitis,
Behandlung von Enterokolitis
Lacticin 3147 Lactococcus lactis Gram-positive
Bakterien
Mundhygiene, Behandlung
von MRSA- und
Enterokokkeninfektionen;
Mastitis, Akne
Mutacin 1140 Streptococcus
mutans
Streptococcus mutans Vorbeugung vor Zahnkaries
Mersacidin/
Actagardin
Bacillus subsp./
Actinoplanes subsp.
Staphylokokken
inklusive
methicillinresistenter
Stämme
Behandlung von MRSA- und
Streptokokkeninfektionen
Duramycin Streptomyces subsp.
und Streptoverti-
cillium subsp.
Inhibitor der
Phospholipase A2
Entzündungen
Zystische Fibrose
Cinnamycin Streptomyces
cinnamoneus
Inhibitor der
Phospholipase A2,
ACE und des Herpes
simplex Virus
Entzündungen,
Blutdruckregulation,
viraler Infektionen,
Ancovenin Streptomyces subsp. Inhibitor der ACE Blutdruckregulation

Einleitung
6
Durch Untersuchungen am Nisin und an anderen strukturell verwandten Lantibiotika, zu
denen auch Mersacidin gehört, konnte Lipid II der bakteriellen Zellwand als target
identifiziert werden[23] (Abb. 4). Die Bindung von Nisin an Lipid II führt zu einer
Porenbildung in der Zellmembran. Diese Poren haben eine Lebensdauer von wenigen bis zu
mehreren hundert Millisekunden und eine Größe von 0,2 bis 2 nm[24]. Die Porenbildung wird
durch die H-Brücken-Wechselwirkung des sog. Pyrophosphatkäfigs (Abb. 4A), der aus den
beiden N-terminalen Thioetherbrücken des Nisins besteht, mit dem Pyrophosphat von Lipid II
eingeleitet. Nun kommt es durch eine Konformationsänderung zur Einlagerung des
C-terminalen Bereichs von Nisin in die Lipiddoppelschicht. Durch die Ausbildung eines
Komplexes aus acht Nisin- und vier Lipid II-Molekülen wird eine Pore gebildet und die
strukturelle Integrität der Zellwand zerstört (Abb. 4B)[25].
A) B)
Abbildung 4: A) Wechselwirkung des Pyrophosphatkäfigs des Nisins mit dem Pyrophosphat von Lipid II. B) Einlagerung des Nisins in die Lipiddoppelschicht und Ausbildung der Pore durch einen Komplex aus acht Nisin- und vier Lipid II-Molekülen[25]. Neben der Wirkung auf Lipid II werden noch weitere Aktivitäten von Lantibiotika gefunden,
so inhibiert Nisin z.B. die Transglykosylierung und Transpeptidierung bei der
Zellwandbiosynthese.[25] Die Lantibiotika Duramycin und Cinnamycin zeigen wiederum
einen ganz anderen Wirkmechanismus, da sie als Inhibitor der Phospholipase A2 wirken,
indem sie dessen natürliches Substrat Phosphatidylethanolamin binden[26].
1.2.2 Biosynthese der Lanthionine und Methyllanthionine
Für die Einteilung von Peptiden in die Klasse der Lantibiotika ist die Anwesenheit von
mindestes einer Lanthionin- bzw. Methyllanthioninverbrückung notwendig[11]. Alle
Lantibiotikavorläufer-Peptide (sog. Präpropeptide) enthalten eine C-terminale Strukturregion

Einleitung
7
(Propeptid), in der die posttranslationalen Modifikationen auftreten, und eine relativ lange
N-terminale Leaderregion, die aus 23 bis 59 Aminosäuren besteht und während der
Biosynthese nicht modifiziert wird. Beide Teile des Präpropeptids enthalten die Aminosäuren
Serin und Threonin, während die Aminosäure Cystein nur im Propeptid gefunden wird[24].
Durch den Sequenzvergleich einer Vielzahl von Leaderregionen, hat sich gezeigt, dass zwei
verschieden konservierte Sequenzmotive auftreten. Zum einen ein FNLD-Motiv zwischen den
Positionen -20 und -15 und einem Prolin an der Position -2 vor der Spaltstelle, zum anderen
das konservierte Strukturmotiv GG oder GA und mehrere Asparaginsäuren und
Glutaminsäuren [27] (Abb. 5).
Abbildung 5: Sequenzvergleich der Leaderpeptide von verschiedenen Lantibiotika und die daraus resultierende Einordnung in zwei Gruppen[28].
Die Peptide mit dem FNLD-Motiv benutzen für die Einführung des Lanthionin- bzw.
Methyllanthioninmotivs die Enzymklassen LanB und LanC. Die selektive Dehydratisierung
von Serin- und Threoninresten im Strukturpeptid der Lantibiotika erfolgt durch die LanB-
Enzyme. Diese Enzyme zeigen keine Homologie mit anderen bekannten Proteinen, auch die
Homologie innerhalb der Gruppe der LanB-Enzyme ist mit ca. 30% sehr gering. Ein
möglicher Grund für die geringe Übereinstimmung ist der starke Unterschied zwischen den
Präpropeptiden. So zeigen LanB-Enzyme von strukturell sehr ähnlichen Peptiden wie z.B.
EriB und SpaB eine Homologie von 83%[29]. Die Aktivität und Wirkung von LanB-Enzymen
wurde durch Experimente gezeigt, in denen nach der Inaktivierung von pepC in einem
Pep5-produzierenden Stamm nur noch das dehydratisierte Peptid gefunden wurde[30]. Des
Weiteren konnten nach der Inaktivierung des Gens eriB im Ericingencluster die Peptide
Ericin A und Ericin S nicht produziert werden[29]. Die Aufgabe der Zyklase LanC für die sich
anschließende Michael-Typ-artige Lanthioninsynthese liegt in der Kontrolle der Stereo- und
Regioselektivität und in der Aktivierung des Thiols [31]. Die Analyse der Enzyme NisC und

Einleitung
8
SpaC ergab, dass jedes Enzym eine stöchiometrische Menge an Zink enthält[32]. Die
Kristallstruktur des Enzyms NisC zeigt ein Protein mit 14 α-Helices. Sieben dieser Helices
bilden die innere Schicht eines α/α-Fasses, in deren Zentrum sich ein Zinkion befindet[33].
Dieses Ion ist tetraedrisch durch zwei Cysteine, ein Histidin und ein Wassermolekül
koordiniert (Abb. 6). Zur Katalyse der Zyklisierung im neutralen pH-Bereich muss NisC die
Thiolgrupppe des Cysteins zunächst deprotonieren, um die nukleophile Addition an
α,β-ungesättigte Aminosäuren durch Aktivierung als Thiolat zu beschleunigen. Bei der
Zyklisierung bildet sich ein Enolat-Zwischenprodukt. Bei dessen Protonierung kommt es zur
Inversion der Konfiguration am α-Kohlenstoff im Vergleich zur Konfiguration von Serin oder
Threonin. Das Cystein behält seine Konfiguration bei und es bildet sich (2S,6R)-Lanthionin
oder (2S,3S,6R)-β-Methyllanthionin (Abb. 6)[31, 33].
Abbildung 6: Mechanismus der Thioetherbiosynthese durch das Enzym NisC[33]. Die LanC-Enzyme bestimmen nicht nur Regio- und Stereoselektivität der Lanthioninbildung
sondern auch deren N-terminale bzw. C-terminale Orientierung. So erfolgt diese bei Typ-A-
Lantibiotika immer vom N-Terminus zum C-Terminus, während Typ-B-Lantibiotika vom
C-Terminus zum N-Terminus zyklisiert werden. LanC-Enzyme sorgen auch dafür, dass die
Zyklisierung von Lanthionin in der Natur ähnlich schnell verläuft wie die von
Einleitung
9
β-Methyllanthion. In biomimetischen Studien wurden hier Unterschiede um mehrere
Größenordnungen ermittelt[34].
Im Folgenden sollen Peptide, die das Doppelglycinmotiv im Leaderpeptid aufweisen, näher
betrachtet werden. Diese Peptide benötigen das Enzym LanM zur Synthese der
Thioetherbrücke. Der C-Terminus dieses Enzyms weist eine Sequenzhomologie von 20-27%
gegenüber LanC auf. Es besitzt aber keine Homologie zu LanB-Enzymen, sodass der
Ursprung dieses Enzyms nicht in einer Fusion aus LanB und LanC liegen kann[35]. Die
Funktion des LanM-Enzyms LctM als Dehydratase und Zyklase konnte am Lacticin 481
nachgewiesen werden[36], wobei das Enzym LctM ATP und Mg2+ für die posttranslationale
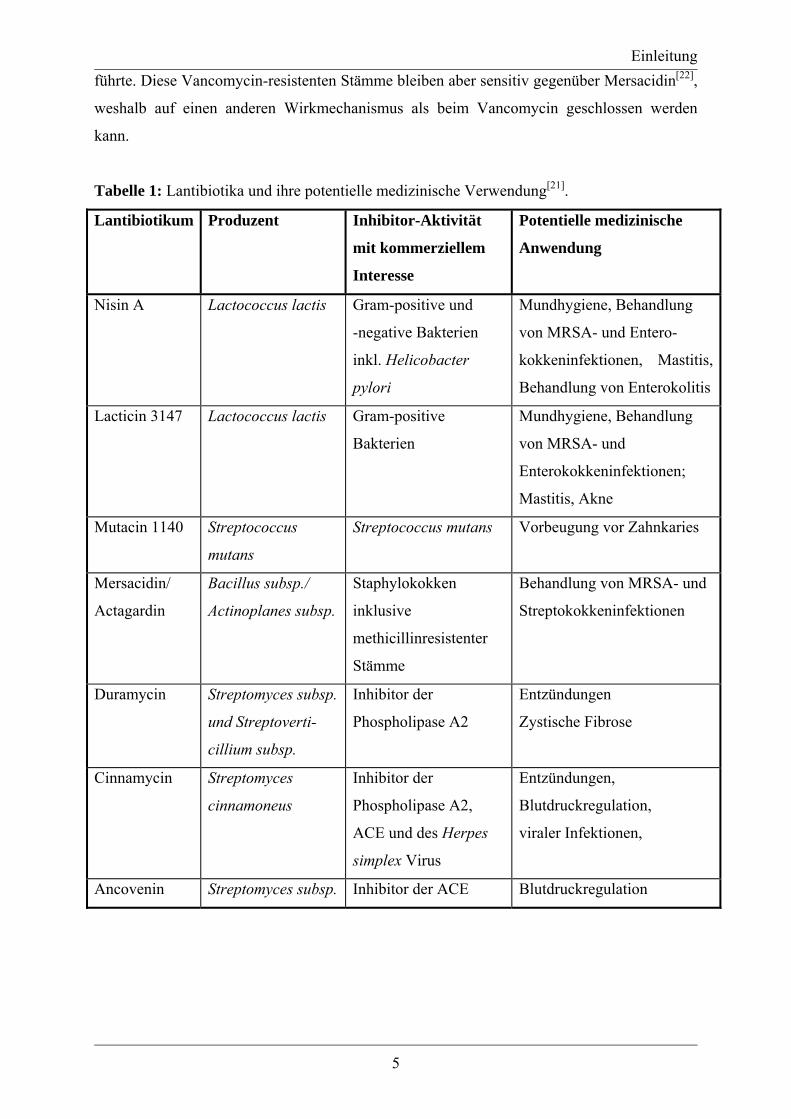
Modifikation benötigt. Die exakte Rolle der Cofaktoren im Reaktionsverlauf war anfangs
unklar, da das Enzym LctM kein ATP-Bindungsmotiv und kein sog. Walkermotiv,
charakteristisch für eine phosphatbindende Schleife, aufweist. Nachfolgende Untersuchungen
zeigten, dass das Magnesiumion an die zwei nichtverbrückenden Sauerstoffatome des β- und
γ-Phosphats des ATPs koordiniert und dadurch das ATP aktiviert. Dem folgt ein
Phosphoryltransfer auf den benachbarten Serin- bzw. Threoninrest und die anschließende
Eliminierung des Phosphatrestes, Deprotonierung des α-Kohlenstoffs des Serin- bzw.
Threoninrestes und Bildung der Doppelbindung (Abb. 7). Zum gegenwärtigen Zeitpunkt ist
noch nicht bekannt, ob die Eliminierung und Deprotonierung des α-Kohlenstoffs konzertiert
ablaufen[37]. Der detaillierte Mechanismus der Zyklisierung mit LanM ist ebenfalls noch nicht
bekannt.
Abbildung 7: Mechanismus der Dehydratisierung eines Serinrestes durch LctM[37].

Einleitung
10
R
HN
OH
O
NH
R
O
HN
O
SHn
Ser/ThrR=H/R=CH3 Cys
R
HN
O
NH
R
O
HN
O
SHn
Dha/DhbR=H/R=CH3 Cys
HN R
SO
O
NHn
DehydratisierungLanB
ZyklisierungLanC
Dehydratisierung/ZyklisierungLanM
Abbildung 8: Möglichkeiten zur Bildung der Thioetherbrücken in Lantibiotika, zum einen mit den Enzymen LanB und LanC, zum anderen direkt mit dem Enzym LanM. Zusammenfassend lässt sich sagen, dass es zwei Wege zur Biosynthese der Lanthioninbrücke
gibt: mit den zwei Enzymen LanB und LanC oder mit einem bifunktionalen Enzym, LanM.
Der grundlegende Mechanismus der Thioethersynthese unterscheidet sich in beiden
Synthesewegen nicht (Abb. 8). Auf welche Art und Weise die Lantibiotika von ihren
bakteriellen Produzenten erzeugt werden, hängt von ihrer genetischen Ausstattung ab.
1.2.3 Klassifizierung von Lantibiotika
Die gegenwärtig bekannten Lantibiotika variieren in ihrer Struktur, Größe und
Wirkungsweise. Allen gemein ist der hohe Grad an posttranslationalen Modifikationen der ca.
ein Viertel aller Aminosäuren in den Peptiden betrifft. Eine Klassifizierungsmöglichkeit
gründet auf deren Topologie und Wirkmechanismus und unterteilt die Lantibiotika in Klasse
A und B [38]. Dabei umfasst Typ A gestreckte, schraubenförmige Peptide, die primär an der
Zellmembran wirken. Der Prototyp dieser Peptide ist das in Abb. 9 gezeigte Nisin. Im
Allgemeinen bestehen diese Peptide aus 20 bis 34 Aminosäuren und besitzen eine positive
Ladung. Der Typ B hingegen umfasst globuläre Lantibiotika, die bei pH 7 keine oder eine
negative Ladung tragen und als Inhibitoren von Enzymen wirken. Bedeutende Vertreter
dieses Typs sind Cinnamycin (Abb. 10) und Mersacidin. Eine zweite Möglichkeit zur
Klassifizierung von Lantibiotika besteht in der Einteilung in drei Klassen, basierend auf ihrem
Biosyntheseweg[39] und auf der Präsenz einer antibiotischen Aktivität[14]. Die
Klasse-I-Lantibiotika werden durch zwei verschiedene Enzyme modifiziert. Das Enzym
LanB, welches die Threonin- und Serinreste dehydratisiert und das Enzym LanC, das die
Lathioninringe formt (siehe 1.2.2). Zur Gruppe der Klasse-I-Lantibiotika gehört z.B. Nisin
und Subtilin (Abb. 9).

Einleitung
11
Nisin A
Subtilin
Nisin A
Subtilin
Abbildung 9: Struktur von Nisin A und Subtilin, zwei Vertreter der Klasse-I-Lantibiotika.
Die Gruppe der Klasse-II-Lantibiotika weist die größte Zahl von Mitgliedern auf und ist
strukturell sehr heterogen, doch benötigen alle Peptide dieser Klasse nur das Enzym LanM für
die Einführung des Lanthioninmotivs. Im Folgenden werden einige Subgruppen von
Klasse-II-Lantibiotika vorgestellt. Lacticin 481 und seine 20 verwandten Lantibiotika[40]
bilden solch eine Subgruppe. Sie alle sind hydrophob und weise keine Ladung bei neutralem
pH auf. Der N-Terminus dieser Peptide ist linear, während der C-Terminus eine globuläre
Struktur bildet, die durch drei überlappende Thioetherbrücken gebildet wird. Diese drei
Thioetherbrücken sind essentiell für die volle Aktivität dieser Peptide[41]. Es wird vermutet,
dass die antibiotische Aktivität dieser Peptide auf einer Porenbildung in der Zellmembran
beruht[40]. Eine weitere Subgruppe beinhaltet Cinnamycin (Abb. 10) und die drei Duramycine
A, B und C. Diese Lantibiotika werden von Streptomyzeten gebildet und weisen eine
globuläre Struktur mit erstaunlich vielen Zyklen auf, inklusive eines Lysinoalanin-Ringes[14].
Sehr interessant ist die Gruppe der Zwei-Komponenten-Lantibiotika, zu denen beispielsweise
Lacticin 3147 und Haloduracin zählen. Jedes Einzelpeptid eines Zwei-Komponenten-
Lantibiotikums zeigt entweder keine oder nur eine schwache antibiotische Aktivität, aber die
Interaktion beider Peptide führt zu einer starken synergetischen antibiotischen Aktivität[42].
Für Lacticin 3147 wird ein Nisin-ähnlicher Wirkmechanismus postuliert, in dem Lacticin
3147 A1 an Lipid II bindet und Lacticin 3147 A2 die Pore in der Zellmembran bildet[43]. In
dieser Gruppe ähneln alle LanA1 Komponenten, mit Ausnahme von Cytolysin, dem

Einleitung
12
Mersacidin und weisen eine eher globuläre Struktur auf, während die LanA2 Komponenten
relativ lang und flexibel sind[42].
Lacticin S
Cynamycin
Lacticin S
Cinnamycin
Lacticin S
Cynamycin
Lacticin S
Cinnamycin
Abbildung 10: Vertreter der Klasse-II-Lantibiotika.
In der Klasse III sind alle lanthioninhaltigen Peptide zusammengefasst, die keine signifikante
antibiotische Aktivität besitzen. Diese Gruppe umfasst bisher die Peptide SapB, SapT und
Labyrinthopeptin A1 bis A3. Letztere werden im Kapitel 1.3 genauer vorgestellt. Die Peptide
dieser Klasse sind 18 bis 21 Aminosäuren lang und weisen eine Funktion als
oberflächenaktive Biomoleküle im Lebenszyklus von Streptomyzeten auf[44]. Aus dieser
Klasse wurden bisher nur die Strukturen von Labyrinthopeptin A2 [45] (Abb. 14) mittels
Röntgenstrukturanalyse, und von SapT [46] (Abb. 11) durch NMR-Spektroskopie, exakt
aufgeklärt.
SapTSapTSapTSapT
Abbildung 11: Struktur von SapT einem Vertreter der Klasse-III-Lantibiotika.
Lacticin 481

Einleitung
13
1.3 Die Labyrinthopeptine
Im Jahre 1988 wurde in der Wüste von Namibia der neuartige Aktinomyzet Actinomadura
namibiensis DSM 6313 im Rahmen eines von der Firma Sanofi-Aventis durchgeführten
Screeningprogramms nach neuen Antibiotikaproduzenten isoliert. Dieser lachsrosafarbende
Stamm formt ein Luftmycel mit spiralförmigen Sporenketten (Abb. 12) [47].
Abbildung 12: Spiralförmige Sporenketten des Luftmycels von Actinomadura namibiensis in licht- und elektronenmikroskopischen Aufnahmen[47].
Aus dem Kulturüberstand von Actinomadura namibiensis wurden drei Peptide mit der
vorläufigen Bezeichnung A1, A2 und A3 isoliert. Die Peptide A1 und A2 konnten einer
Aminosäureanalytik unterzogen werden (Tab. 2).
Tabelle 2: Analytische Daten zu den Peptiden A1-A3[45].
A1 A2 A3 ESI-FTICR-MS 2073,7624 amu
(C92H119N23O25S4) 1922,6930 amu (C85H110N20O24S4)
2188,7946 amu
ASA L-Ala(1), L-Thr(1), L-Asx(1), L-Cys(2), L-Phe(1), L-Glx(1), L-Trp(2), L-Gly(1), L-Val(2), L-Pro(1)
L-Ala(1), L-Thr(1), L-Asx(1), L-Cys(2), L-Phe(1), L-Glx(1), L-Trp(2), L-Gly(1), L-Leu(2)
n.d.
Edman-Abbau Xxx-Asn Xxx-Asp Asp-Xxx-Asn DTT-Reduktion eine Disulfidbrücke eine Disulfidbrücke n.d.
n.d.= nicht detektiert
Die dabei erhaltenen Daten zeigten eine beträchtliche Differenz zur Gesamtmasse des
jeweiligen Moleküls, die mit den bekannten Modifikationen von Peptiden nicht in Einklang
gebracht werden konnte.
Einleitung
14
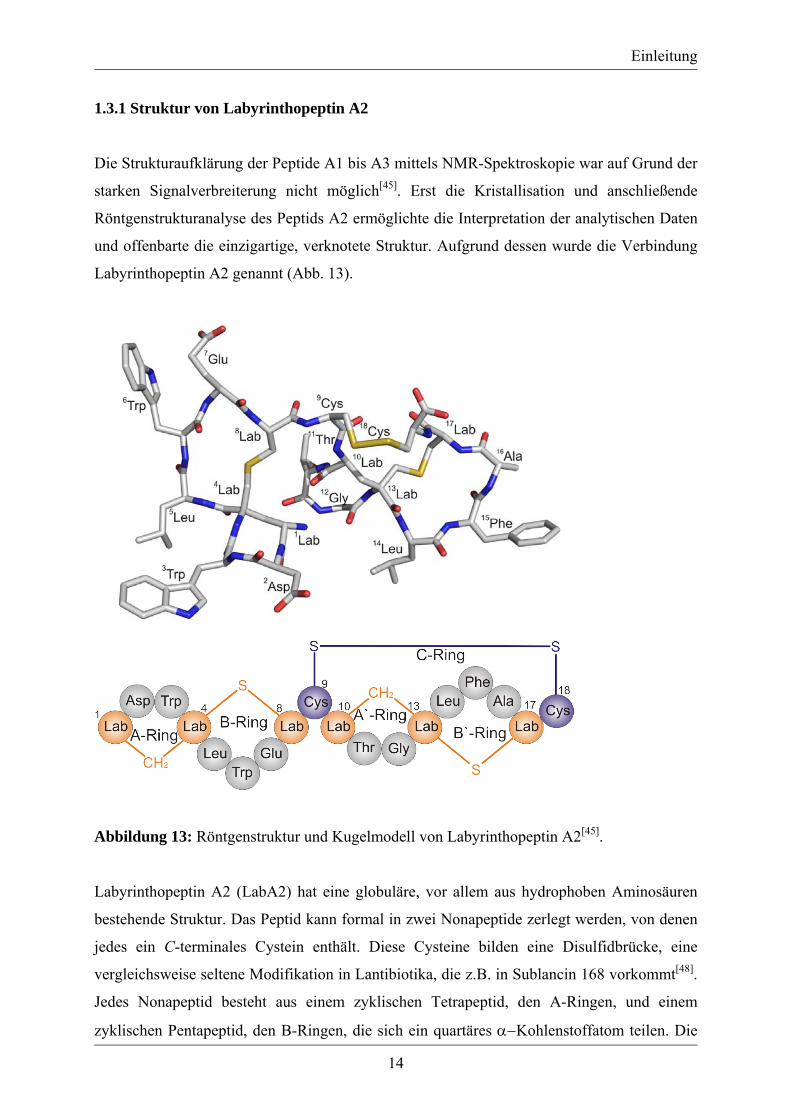
1.3.1 Struktur von Labyrinthopeptin A2
Die Strukturaufklärung der Peptide A1 bis A3 mittels NMR-Spektroskopie war auf Grund der
starken Signalverbreiterung nicht möglich[45]. Erst die Kristallisation und anschließende
Röntgenstrukturanalyse des Peptids A2 ermöglichte die Interpretation der analytischen Daten
und offenbarte die einzigartige, verknotete Struktur. Aufgrund dessen wurde die Verbindung
Labyrinthopeptin A2 genannt (Abb. 13).
Abbildung 13: Röntgenstruktur und Kugelmodell von Labyrinthopeptin A2[45].
Labyrinthopeptin A2 (LabA2) hat eine globuläre, vor allem aus hydrophoben Aminosäuren
bestehende Struktur. Das Peptid kann formal in zwei Nonapeptide zerlegt werden, von denen
jedes ein C-terminales Cystein enthält. Diese Cysteine bilden eine Disulfidbrücke, eine
vergleichsweise seltene Modifikation in Lantibiotika, die z.B. in Sublancin 168 vorkommt[48].
Jedes Nonapeptid besteht aus einem zyklischen Tetrapeptid, den A-Ringen, und einem
zyklischen Pentapeptid, den B-Ringen, die sich ein quartäres α−Kohlenstoffatom teilen. Die
Einleitung
15
A-Ringe von LabA2 werden durch eine Methylenbrücke zwischen den α−Kohlenstoffatomen
von Lab1 und Lab4 sowie zwischen Lab10 und Lab13 gebildet (Abb. 13). Eine Verbrückung
dieser Art war bisher in Peptiden und Proteinen unbekannt. Die quartäre Aminosäure, die
diese Verbrückungen formt, wird Labionin (Abb. 14) genannt[45]. Strukturell verwandte
Verbindungen sind α-Aminoisobuttersäure und Isovalin, die in Peptaibol-Antibiotika aus
Pilzen enthalten sind[49].
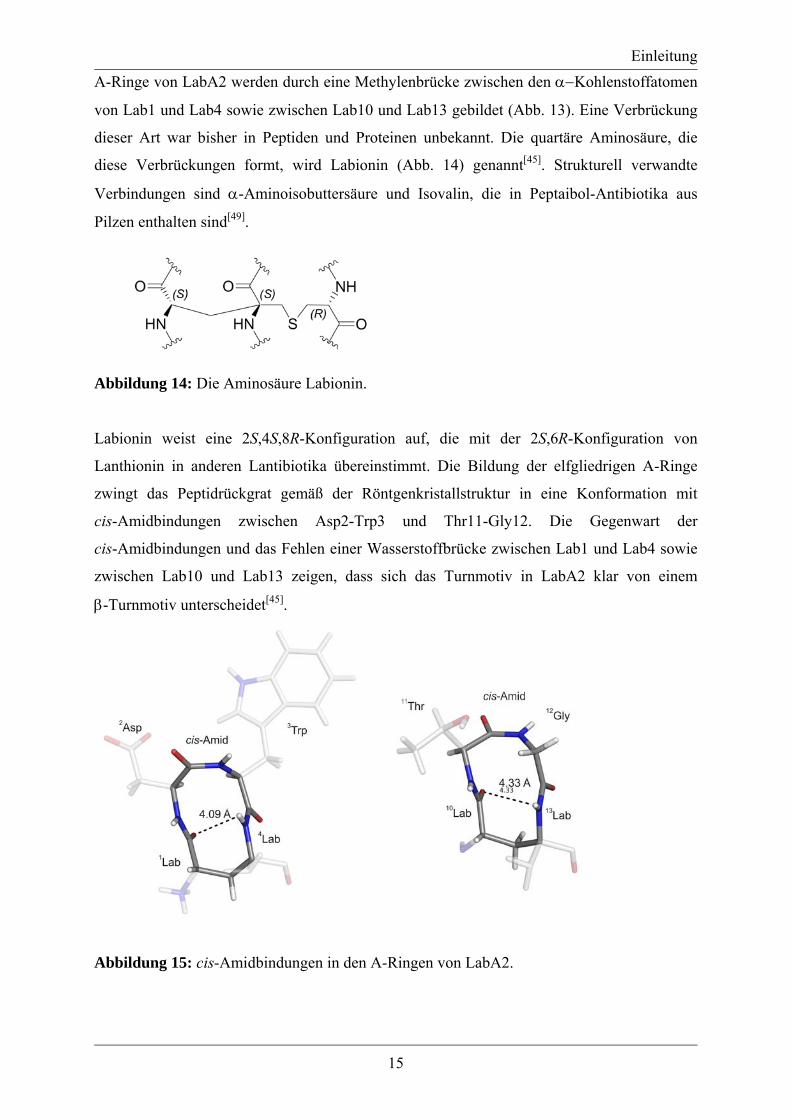
Abbildung 14: Die Aminosäure Labionin.
Labionin weist eine 2S,4S,8R-Konfiguration auf, die mit der 2S,6R-Konfiguration von
Lanthionin in anderen Lantibiotika übereinstimmt. Die Bildung der elfgliedrigen A-Ringe
zwingt das Peptidrückgrat gemäß der Röntgenkristallstruktur in eine Konformation mit
cis-Amidbindungen zwischen Asp2-Trp3 und Thr11-Gly12. Die Gegenwart der
cis-Amidbindungen und das Fehlen einer Wasserstoffbrücke zwischen Lab1 und Lab4 sowie
zwischen Lab10 und Lab13 zeigen, dass sich das Turnmotiv in LabA2 klar von einem
β-Turnmotiv unterscheidet[45].
Abbildung 15: cis-Amidbindungen in den A-Ringen von LabA2.

Einleitung
16
Die B-Ringe im LabA2 werden durch eine Thioetherbrücke gebildet, die ebenfalls vom
Labionin ausgeht. Aufgrund dieser Lanthioninbrücken wird LabA2 als Lantibiotikum
klassifiziert. Zusammenfassend lässt sich sagen, dass es sich bei LabA2 um ein stark
verbrücktes Peptid, mit einer sehr gespannten Struktur handelt. Das herausragende Merkmal
dieses Peptids ist die Aminosäure Labionin (Abb. 14), die im Lab A2 zum ersten Mal
beschrieben wurde[45].
1.3.2 Biosynthese von Labyrinthopeptin A2
Das Biosynthesegencluster von Labyrinthopeptin (Abb. 16) besteht aus fünf Genen einer
6,4 kb großen DNA-Sequenz. Das Strukturgen labA2 von Labyrinthopeptin A2 kodiert 38
Aminosäuren, in denen ein 20 Aminosäuren langes Leaderpeptid enthalten ist.
Abbildung 16: Das Biosynthesegencluster der Labyrinthopeptine[45].
Direkt stromaufwärts von labA2 kodiert das Gen labA1/A3 für das Strukturgen der
Labyrinthopeptine A1 und A3, auf die später noch genauer eingegangen wird. Nun folgen die
Gene labT1 und labT2, die die ATP-abhängigen ABC-Transporter kodieren, welche
vermutlich eine Exportfunktion für die Labyrinthopeptine haben[45]. Das Gen labKC kodiert
das bifunktionale Enzym LabKC, das aus zwei Domänen besteht. Einer konservierten
N-terminalen Domäne mit Merkmalen einer eukaryotischen Serin/Threonin-Kinase, sowie
einer C-terminalen Domäne mit geringer Sequenzhomologie zur LanC-Zyklase. Für eine
erwartete Funktion als LanC-Zyklase fehlen LabKC jedoch wichtige Reste des aktiven
Zentrums, z.B. das Zinkbindemotiv der Nisin-Zyklase. Weiterhin zeigt die
Aminosäuresequenz von LabKC eine sehr große Homologie zum SapB-modifizierenden
Enzym RamC und zu Sequenzen von Genclustern vieler weiterer, bereits vollständig
sequenzierter Actinomyzeten-Stämme[50].
Untersuchungen zur Biosynthese von LabA2 zeigten, dass es sich beim Enzym LabKC um
eine Kinase-Zyklase handelt, die in einem zweistufigen Mechanismus arbeitet. Der erste
Schritt ist die Phosphorylierung der Serine unter Verwendung von GTP als Cosubstrat[50],
anstelle von ATP, wie es zuvor bei der in vitro-Biosynthese von anderen Lantibiotika
beschrieben wurde[36]. Anschließend folgt die Dehydratisierung des phosphorylierten Serin

Einleitung
17
und es entstehen formal zwei α,β-Didehydroalanine. Im zweiten Schritt erfolgt die doppelte
Michael-Addition, die mit dem nucleophilen Angriff eines C-terminalen Cysteins beginnt und
intermediär ein Lanthionin entstehen lässt (Abb. 17). Bei dieser Reaktion wird ein Carbanion
am α-Kohlenstoff des vormaligen α,β-Didehydroalanins stabilisiert. Dieses Anion greift nun
als Kohlenstoffnucleophil das N-terminale α,β-Didehydroalanine an und schließt somit den
Carbazyklus. Ähnliche Funktionen wie die von LabKC werden auch bei den Enzymen zur
Biosynthese weiteren Lantibiotika mit verwandten Biosynthesegenclustern wie z.B.
Streptomyces coelicolor, Streptomyces avermitilis, Streptomyces griseus und
Saccharopolyspora erythrea vermutet[50].
Abbildung 17: Modell zur Labyrinthopeptin A2 Biosynthese, die mit der Phosphorylierung und Dehydratisierung der Serine beginnt, gefolgt von der Zyklisierung der Ringe, der Abspaltung des Leaderpeptids und der Bildung der Disulfidbrücke[50].

Einleitung
18
1.3.3 Weitere Labionin-haltige Peptide
Wie bereits im Kapitel 1.3 angesprochen, wurden im Kulturextrakt von Actinomadura
namibiensis außer LabA2 noch zwei weitere Peptide, LabA1 und LabA3, identifiziert. Bei
längeren Fermentationszeiten konnte man beobachten, dass sich LabA3 in LabA1 umwandelt.
Abbildung 18: Aminosäuresequenz der Präpropeptide von LabA2 und LabA1/A3, identische Aminosäuren im Leaderpeptid sind blau und im Propeptid grau dargestellt[45].
Das in Kapitel 1.3.2 angesprochene Biosynthesegencluster enthält neben dem Strukturgen
labA2, das für Labyrinthopeptin A2 kodiert, noch das Strukturgen labA1/3. Der Vergleich der
Konsensussequenzen dieser Strukturgene zeigt eine große Homologie des Präpropeptides von
LabA1/A3 zum Präpropeptid von LabA2 (Abb. 18). Der Sachverhalt, dass LabA2 und
LabA1/A3 in ungefähr gleichen Mengen bei der Fermentation gebildet werden, spricht für
eine gemeinsame Transkription von labA2 und labA1/A3 desselben Genclusters und deutet
auf ähnliche Prozessierungseffizienzen der Präpropeptide durch LabKC und proteolytische
Enzyme hin. Durch die Kombination der genetischen Informationen, der Strukturdaten von
Lab A2, der massenspektroskopischen Analytik und der Aminosäurenanalytik (Tab. 2)
wurden die Strukturen von LabA1 und LabA3 bisher nur abgeleitet (Abb. 19). Die
Grundstruktur von LabA1 ist mit der von LabA2 identisch, wobei der B´-Ring zu einem
Heptapeptid erweitert wurde und das darin enthaltene Threonin (Abb. 18) zu einem
α,β-Didehydrobutyrin umgewandelt wurde. LabA3 ist mit LabA1 identisch trägt aber
zusätzlich am N-Terminus noch einen Asparaginsäurerest[45] (Abb. 19).
Abbildung 19: Kugelmodell mit der abgeleiteten Struktur von LabA1 und LabA3[45].

Einleitung
19
Die Anordnung der Gene und die Leadersequenz der Strukturgene labA1/A3 und labA2
zeigen signifikante Homologien zu Biosynthesegenclustern aus anderen Aktinomyzeten
(Abb. 20)[45].
Abbildung 20: Biosynthesegencluster von verschiedenen Aktinomyzeten, die eine Homologie zum Biosynthesegencluster der Labyrinthopeptine zeigen[45].
Das trifft besonders auf das ram-Gencluster des Modellorganismus Streptomyces coelicolor
zu. Der Sequenzvergleich mit Genen der Serin/Threonin-Kinase (ramC), des Strukturgens
(ramS) und der ABC-Transporter (ramA, ramB) unterstreichen die evolutionäre Beziehung
beider Gencluster. Das Biosynthesprodukt des ram-Genclusters ist das Lantibiotikum
SapB[51]. Die Kenntnisse der Struktur von SapB sind zum aktuellen Zeitpunkt jedoch nicht
vollständig. Auch die Beteiligung von RamC an der Biosynthese wird bisher nur aufgrund der
LanC-Domäne vermutet[51]. Ein Vergleich der Konsensussequenz zeigt, dass das
SXXS(X)3-5C-Motiv sowohl in den Labyrinthopeptinen als auch im SapB vorhanden ist
(Abb. 20). Dieser Sachverhalt und die starke Homologie zwischen den
Biosynthesegenclustern der Labyrinthopeptine und SapB lassen eine weite hypothetische
Struktur von SapB (Abb. 21) zu[28]. Der Unterschied des publizierten Strukturvorschlags nach
J. Willey et al. liegt darin, dass die zwei Didehydroalaninreste in zwei Labionine eingebunden

Einleitung
20
sind. Die bisher publizierten Daten stimmen für beide Modelle überein, da die beiden
Strukturen bezüglich der Molekülmasse isobar sind[28].
Das Vorhandensein lab-homologer Gencluster (Abb. 20) in Streptomyces avermitilis,
Streptomyces griseus und dem Erythromycin-Produzenten Saccharopolyspora erythrea mit
bislang unbekannten Biosyntheseprodukten lässt auf das Vorhandensein der ungewöhnlichen
Aminosäure Labionin in weiteren Peptiden schließen[45].
xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
Struktur nach Willey et al.
Hypothetische Struktur
Struktur nach Willey et al.
Hypothetische Struktur nach Süßmuth et al.
Struktur nach Willey et al.
Hypothetische Struktur
Struktur nach Willey et al.
Hypothetische Struktur nach Süßmuth et al.
Abbildung 21: Vergleich der publizierten Struktur mit einer alternativen labioninhaltigen Struktur[28] von SapB, beide Strukturen sind im Einklang mit den bisher publizierten Daten[51].
1.3.4 Biologische Aktivität der Labyrinthopeptine
Untersuchungen zur biologischen Aktivität erbrachten eine moderate antivirale Aktivität von
Labyrinthopeptin A2. Beide Peptide verfügen über keine signifikante antibiotische Aktivität,
weshalb sie den Klasse-III-Lantibiotika zugeordnet werden. In weiterführenden
Profilierungsexperimenten wurde Labyrinthopeptin A2 in vivo in einem murinen
Nervenverletzungsmodell[52] gegen neurophatische Schmerzen getestet. Neurophatische
Schmerzen sind eine Dysfunktion des zentralen oder peripheren Nervensystems, die sich
beispielsweise in einer Schmerzüberempfindlichkeit (Allodynie) äußert. In Deutschland
leiden ca. sechs Prozent der Bevölkerung an neurophatischen Schmerzen, das entspricht
knapp fünf Millionen Menschen. Nach einer Operation leiden ca. 20 % der Patienten an
chronischen neurophatischen Schmerzen. Auch sog. Phantomschmerzen, an denen 60 % aller
Patienten nach einer Amputation leiden, gehören zu den neurophatischen Schmerzen. Weitere
Einleitung
21
Ursachen sind beispielsweise Schlaganfall, Multiple Sklerose, Rückenmarksverletzungen,
Stoffwechselkrankheiten wie Diabetes mellitus, virale Infektionen wie Gürtelrose,
Alkoholmissbrauch oder Chemotherapie. In dem erwähnten Nervenverletzungsmodell werden
Mäusen zwei von drei Nervenästen des Hüftnervs (Nervus ischiadicus) durchtrennt und so
neurophatische Schmerzen erzeugt. Nun wird für die Dauer von maximal 20 Sekunden oder
bis zum Zeitpunkt, an dem die Maus ihre Hinterpfote wegzieht ein steigender Druck auf die
Hinterpfote der Maus ausgeübt. Mit diesem Modell kann die Allodynie der Maus in
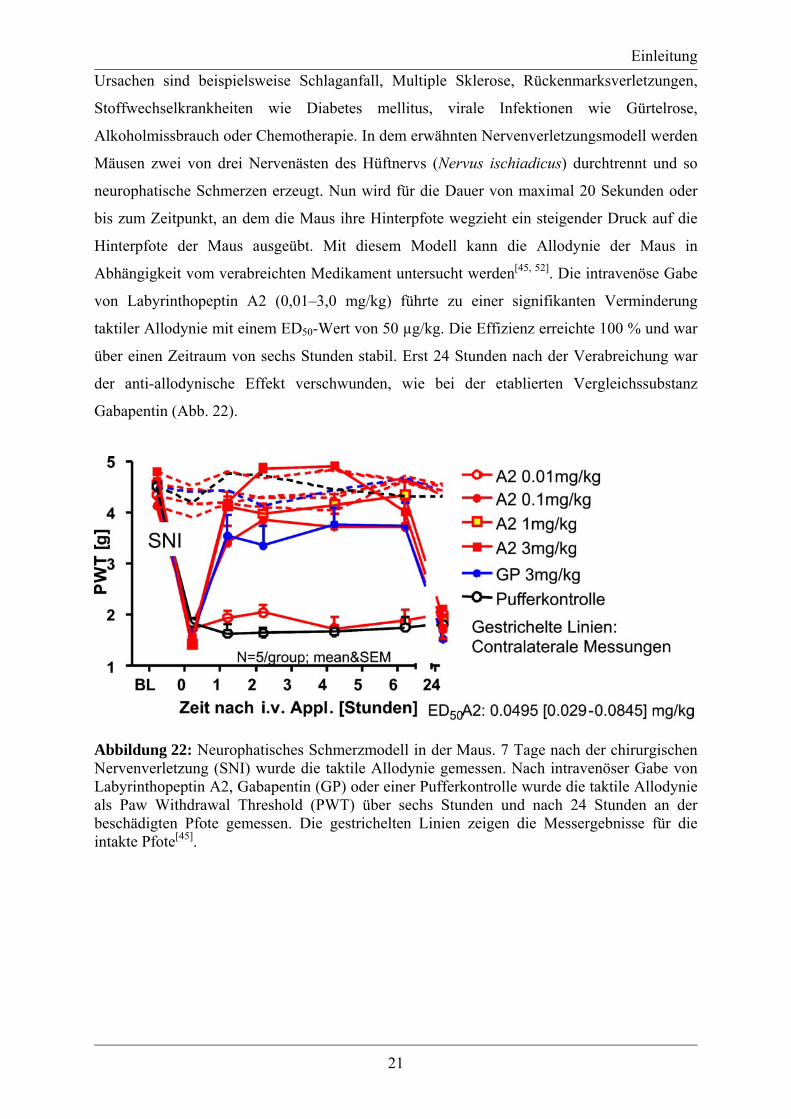
Abhängigkeit vom verabreichten Medikament untersucht werden[45, 52]. Die intravenöse Gabe
von Labyrinthopeptin A2 (0,01–3,0 mg/kg) führte zu einer signifikanten Verminderung
taktiler Allodynie mit einem ED50-Wert von 50 µg/kg. Die Effizienz erreichte 100 % und war
über einen Zeitraum von sechs Stunden stabil. Erst 24 Stunden nach der Verabreichung war
der anti-allodynische Effekt verschwunden, wie bei der etablierten Vergleichssubstanz
Gabapentin (Abb. 22).
Abbildung 22: Neurophatisches Schmerzmodell in der Maus. 7 Tage nach der chirurgischen Nervenverletzung (SNI) wurde die taktile Allodynie gemessen. Nach intravenöser Gabe von Labyrinthopeptin A2, Gabapentin (GP) oder einer Pufferkontrolle wurde die taktile Allodynie als Paw Withdrawal Threshold (PWT) über sechs Stunden und nach 24 Stunden an der beschädigten Pfote gemessen. Die gestrichelten Linien zeigen die Messergebnisse für die intakte Pfote[45].

Einleitung
22
1.4 Totalsynthesen von Lantibiotika
1.4.1 Totalsynthese von Nisin
Die erste Synthese eines Lantibiotikums wurde im Jahre 1988 von der Arbeitsgruppe Shiba
aus Osaka/Japan publiziert[53]. Hierbei handelt es sich um die Totalsynthese von Nisin, dem
zu diesem Zeitpunkt am besten untersuchten Lantibiotikum. Die Totalsynthese erfolgte durch
die Kondensation der in Abb. 23 gekennzeichneten Fragmente in Lösung und unter
Verwendung von säure- und hydrogenolyselabilen Schutzgruppen.
AB
C
DEA
BC
DE
Abbildung 23: Prinzip der Fragmentkondensation zur Totalsynthese von Nisin[53].
Alle Fragmente bis auf das C-terminale Fragment enthalten mindestens einen Ring, der durch
eine Thioetherbrücke gebildet wird. Dieser Thioether wurde durch Desulfurierung aus
Disulfidbrücken gebildet. Dazu wurde für jeden Ring ein lineares Vorläuferpeptid
synthetisiert. Die Positionen des zu bildenden Lanthionins wurden jeweils durch Cysteine
ersetzt, im Fall von β-Methyllanthionin wurden β-Methyl-D-Cystein und Cystein verwendet.
Anschließend wurde eine Disulfidbrücke generiert und diese nun durch Desulfurierung in eine
Lanthioninbrücke umgewandelt[54] (Abb. 24).

Einleitung
23
Abbildung 24: Prinzip der Synthese des B-Ringfragments von Nisin.
1.4.2 Totalsynthese von Bis(desmethyl)-Lacticin 3147 A2 und Lactocin S
Da sich durch die Desulfurierung von Cystin[55] Lanthionin synthetisieren lässt, aber die
Stereoselektivität dieser Reaktion nicht immer gewährleistet ist[56], wurden andere Methoden
für die Lanthioninsynthese entwickelt. Hierzu zählen z.B. die Ringöffnung von
Serinlactonen[57] und Aziridinen[58] und die nukleophile Substitution von Brom-[59] und
Iodalaninen[60] mit geschützten Cysteinderivaten. Im Jahr 2005 folgte die Synthese eines
C-Ring-Analogons von Nisin an der festen Phase[61]. Aufbauend auf diese Experimente
entwickelte die Arbeitsgruppe Vederas eine Methode zur Lantibiotikasynthese an der festen
Phase. Im Jahr 2008 wurde die Synthese von Bis(desmethyl)-Lacticin 3147 A2 mit dieser
Methode veröffentlicht[62]. Dabei wurde erst ein vollgeschütztes Lanthionin ausgehend von
Fmoc-Cys-OtBu und Alloc-BrAla-OAllyl synthetisiert und anschließend auf
2-Chlor-Tritylharz geladen. Nun folgte die SPPS und Zyklisierung des ersten Ringes an der
festen Phase. Für die folgenden Ringe wurde der gleiche Ablauf wiederholt und schließlich
nach Abspaltung vom Harz ein N-terminales didehydroaminosäurenreiches Peptidfragment
gekuppelt (Abb. 25). Ein Jahr später wurde ebenfalls von der Arbeitsgruppe Vederas die
Totalsynthese des Naturstoffs Lactocin S analog zur Synthese von Bis(desmethyl)-Lacticin
3147 A2 veröffentlicht[63]. Da es sich bei Bis(desmethyl)-Lacticin 3147 A2 um ein Derivat
von Lacticin 3147 A2 handelt, bei dem die β-Methyllanthionine durch Lanthionin ersetzt
wurden, um die Synthese zu vereinfachen, wurden bis zum gegenwärtigen Zeitpunkt nur zwei
in der Natur auftretende Lantibiotika synthetisiert: Nisin im Jahr 1988 und Lactocin S im Jahr
2009.

Einleitung
24
Abbildung 25: Schema der Bis(desmethyl)-Lacticin 3147 A2-Synthese an fester Phase[63].
1.5 Mechanistische Details wichtiger Schlüsselreaktionen
α-Aminosäuren gehören zu den bedeutendsten Bausteinen von Naturstoffen und besitzen
zudem in der industriellen Produktion von z.B. Arzneistoffen und Tierfuttermitteln große
Bedeutung. Doch auch im Labormaßstab besteht ein Bedarf an enantiomerenreinen
Aminosäuren, z.B. zur Synthese von Naturstoffen oder deren Derivaten.
Die technische Produktion von Aminosäuren erfolgt einerseits durch die Extraktion von
Proteinhydrolysaten, die durch Fermentation mit Hilfe von Mikroorganismen erzeugt werden.
Aus diesen Hydrolysaten können dann die einzelnen Aminosäuren durch Extraktion,
fraktionelle Kristallisation und Ionenaustauschchromatographie isoliert werden. Eine weitere
biotechnologische Produktionsmethode ist die so genannte Fermentationsmethode. In diesem
Fall werden spezielle Mikroorganismen verwendet, bei denen durch die Verwendung
günstiger Nährmedien und Kulturbedingungen bestimmte Aminosäuren angereichert werden.
Ein Beispiel hierfür ist die Produktion von Glutaminsäure durch Brevibacterium flavum aus
Rübenmelasse. Diese Methode ist aber auf einige Aminosäuren beschränkt und liefert jene
auch nur in begrenzten Konzentrationen. Durch die Verwendung von gentechnisch
veränderten Mikroorganismen lässt sich diese Beschränkung jedoch überwinden, da hier
durch die Ausschaltung von Rückkopplungsmechanismen, die die Bildung von Aminosäuren
regulieren, neue Hochproduzenten für bestimmte Aminosäuren erzeugt werden[64].
Doch lässt sich nicht jede beliebige Aminosäure durch biotechnologische Verfahren
herstellen. Zur großtechnischen chemischen Synthese werden klassische Methoden wie z.B.
die Strecker-Synthese[65] verwendet, bei der die Aminosäuren razemisch synthetisiert werden.

Einleitung
25
Durch die Überführung des Razemats in diastereomere Ester, N-Acylverbindungen oder Salze
und anschließender Kristallisation oder Umsetzung mit Acylasen können die D- und
L-Aminosäuren voneinander getrennt werden[66, 67].
Für die Synthese von Aminosäuren im Labormaßstab werden aber andere Methoden benutzt,
bei der die gewünschte Aminosäure stereoselektiv erzeugt wird. Als Beispiel soll hier die
Aminosäuresynthese nach Schöllkopf über einen Bislactimether[68]genannte werden, welche
sich der chiralen Induktion durch sterische Wechselwirkungen mit dem chiralen Auxilliar,
bedient. Dieser Effekt, aber über eine andere chirale Hilfsgruppe, wird auch bei der
Aminosäuresynthese nach Evans benutzt[69].
1.5.1 Stereoselektive Alkylierung von Glycin
Die in der vorliegenden Arbeit benutzt Methode zur stereoselektiven Alkylierung von Glycin
wurde 1978 in der Arbeitgruppe Yamada[70] entwickelt.
Abbildung 26: Stereoselektive Alkylierung unter Verwendung von (1S,2S,5S)-Hydroxypinanon[70].
Hierzu wird das C-terminal geschützte Glycin 1 mit (1S,2S,5S)-2-Hydroxypinanon 5 in eine
Schiffsche Base 2 überführt. Diese chirale Verbindung wird nun mit LDA deprotoniert (3)
und stereoselektiv 4 alkyliert. Anschließend wird durch saure Hydrolyse die gewünschte
Aminosäure 6 freigesetzt. Wurden jedoch zur Alkylierung Verbindungen (R-X) eingesetzt,
die funktionelle Gruppen im Rest enthielten, wie z.B. Carbonsäuren, Carbonsäureester,
Carbonylverbindungen oder Amide, wird eine Verminderung der Stereoselektivität
festgestellt. Untersuchungen zu diesem Sachverhalt zeigten zudem einen starken Einfluss des
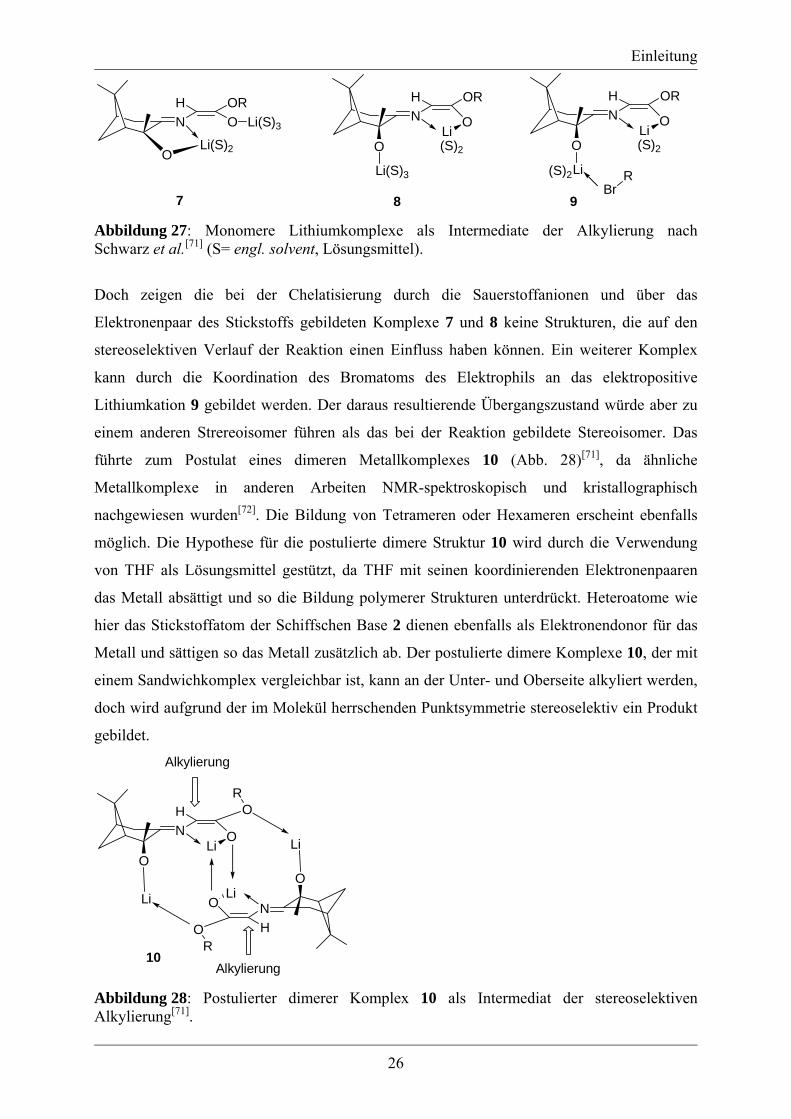
verwendeten Metallsalzes bei der Reaktion[71]. Bei der Reaktion der Schiffschen Base 3 mit
LDA können die monomeren Metallkomplexe 7, 8 oder 9 gebildet werden[71] (Abb. 27).
H2N COOtBu
1
H3O+
O
OH
N COOtBu
OH
2N
OH
3
LDA
RX
5
OtBu
OLi
N CO2tBu
ROH
H2N CO2tBu
R
4 6
Einleitung
26
N
OLi(S)2
H ORO N
O
H OR
OLi(S)3
Li(S)3
(S)2Li
7 8
N
O
H OR
O
Li
(S)2
Li
(S)2 RBr
9 Abbildung 27: Monomere Lithiumkomplexe als Intermediate der Alkylierung nach Schwarz et al.[71] (S= engl. solvent, Lösungsmittel).
Doch zeigen die bei der Chelatisierung durch die Sauerstoffanionen und über das
Elektronenpaar des Stickstoffs gebildeten Komplexe 7 und 8 keine Strukturen, die auf den
stereoselektiven Verlauf der Reaktion einen Einfluss haben können. Ein weiterer Komplex
kann durch die Koordination des Bromatoms des Elektrophils an das elektropositive
Lithiumkation 9 gebildet werden. Der daraus resultierende Übergangszustand würde aber zu
einem anderen Strereoisomer führen als das bei der Reaktion gebildete Stereoisomer. Das
führte zum Postulat eines dimeren Metallkomplexes 10 (Abb. 28)[71], da ähnliche
Metallkomplexe in anderen Arbeiten NMR-spektroskopisch und kristallographisch
nachgewiesen wurden[72]. Die Bildung von Tetrameren oder Hexameren erscheint ebenfalls
möglich. Die Hypothese für die postulierte dimere Struktur 10 wird durch die Verwendung
von THF als Lösungsmittel gestützt, da THF mit seinen koordinierenden Elektronenpaaren
das Metall absättigt und so die Bildung polymerer Strukturen unterdrückt. Heteroatome wie
hier das Stickstoffatom der Schiffschen Base 2 dienen ebenfalls als Elektronendonor für das
Metall und sättigen so das Metall zusätzlich ab. Der postulierte dimere Komplexe 10, der mit
einem Sandwichkomplex vergleichbar ist, kann an der Unter- und Oberseite alkyliert werden,
doch wird aufgrund der im Molekül herrschenden Punktsymmetrie stereoselektiv ein Produkt
gebildet.
N
O
H O
OLi
R
N
O
HO
OLi
R
Li
Li
Alkylierung
Alkylierung10
Abbildung 28: Postulierter dimerer Komplex 10 als Intermediat der stereoselektiven Alkylierung[71].

Einleitung
27
Die Selektivität der Reaktion wird durch tiefe Temperaturen erhöht, was auf eine
Verringerung der intermolekularen Austauschrate zwischen den Metallkomplexen
zurückzuführen ist. Durch die Zugabe von Magnesiumsalzen wird die Selektivität der
Reaktion erhöht[71]. Das ist auf die Fähigkeit der Magnesiumionen zurückzuführen, stärker zu
koordinieren und somit den dimeren Komplex zu stabilisieren. Bis jetzt konnte der Lithium-
Magnesium-Austausch aber noch nicht nachgewiesen werden. So könnte die Stabilisierung
des Komplexes auch über eine zusätzliche Koordination des Magnesiums zustande kommen.
Wie bereits erwähnt, vermindern Elektrophile, die ein Sauerstoff- oder Stickstoffatom
besitzen die Selektivität der Reaktion. Das ist auf eine zusätzliche Koordination des
Alkylierungsreagenzes an das Lithiumionen zurückzuführen, die die Ausbildung eines
dimeren Komplexes behindert. Auch in diesem Fall führt die Zugabe von Magnesiumsalzen
zu einer Erhöhung der Selektivität [71].
Darüber hinaus hat die Natur der eingesetzten Schiffschen Base einen Einfluss auf die
Selektivität und auf die Reaktivität. So wird durch raumerfüllende C-terminale Schutzgruppen
die Stereoselektivität erhöht bzw. erst ermöglicht, da bei der Verwendung von Methylestern
zur Synthese von α-monosubstituierten Aminosäuren ein razemisches Gemisch entsteht[73].
Bei der Verwendung von Alanin als Ausgangsverbindung zur Synthese von
α,α-disubstituierten Aminosäuren wurde der gleiche Einfluss in Abhängigkeit vom
verwendeten Ester beobachtet. Doch führte hier der Einsatz des Methylesters zu 69 % de.
Sperrige Esterreste erhöhen in diesem Fall die Selektivität, jedoch auf Kosten der Ausbeute,
die stark zurückgeht[74]. So beträgt die Ausbeute bei einem tBu-Ester nur noch 7 %[73]. Es
zeigt sich jedoch, dass α-Aminosäuremethylester mit sperrigen Seitenketten, wie
beispielsweise Valin sowohl mit guten Ausbeuten (69 %) als auch mit hoher Selektivität ( 98
% de) mit Methylbromid alkyliert werden können[73].
1.5.2 Synthese von α,α-disubstituierten Aminosäuren aus zyklischen Sulfiten
α,α-disubstituierten Aminosäuren sind wichtige Bausteine in synthetischen Peptiden, da
deren in vivo-Stabilität im Vergleich zu α-monosubstituierten Aminosäuren erhöht ist und
deren erhöhte konformative Stabilität, aufgrund der eingeschränkten Rotation zu einer
Erhöhung der biologischen Aktivität führt[75]. Aufgrund dieser Eigenschaften sind sie
bedeutende Substrate in der biologischen Chemie und der Medizinalchemie. Die Synthese
solcher Aminosäuren in enantiomerenreiner Form ist ein herausforderndes Ziel[76]. In den
meisten Fällen werden hier Aminosäuren[73] oder Aminosäurederivate[77, 78] alkyliert und so
direkt der gewünschte Alkylrest eingeführt. In der Literatur sind jedoch auch wenige

Einleitung
28
Methoden beschrieben, bei denen eine Aminogruppe am α-Kohlenstoffatom erst am Ende der
Synthese eingeführt wird. Das kann über eine Umlagerung[79] oder über die Reaktion mit
Elektrophilen[80] erfolgen.
Die α-Aminogruppe kann aber auch ausgehend von einem Alkohol synthetisiert werden. Dies
kann durch eine Mitsunobu-Reaktion mit einer quartären α-Hydroxysäure[76] erfolgen, bei der
aber die explosive Stickstoffwasserstoffsäure verwendet werden muss oder durch
Verwendung zyklischer Sulfate und Sulfite[81-83]. Als Ausgangsverbindung für eine solche
Synthese dient eine α,β-ungesättigte Carbonsäure, deren Doppelbindung mit der
asymmetrischen Dihydroxylierung nach Sharpless[84] funktionalisiert wird. Das resultierende
Diol wird nun mit Thionylchlorid in ein zyklisches Sulfid umgewandelt, das mit einem Azid
unter Inversion der Konfiguration geöffnet wird[81] (Abb. 29).
CO2Me
HO
HOCO2Me
O
O
SO
CO2Me
N3
HOCO2Me
NH2
HOSOCl2 NaN3 Pd/C; H2
11 12
CO2Me
O
O
SO13bO
NaN3CO2Me
N3
NaO3SO H2SO4CO2Me
N3
HO
Pd/C; H2
14 15
16 17
NaIO4 RuCl3
Abbildung 29: Synthese von α,α-disubstituierten Aminosäuren aus zyklischen Sulfiten und Sulfaten[81].
Die Oxidation des Sulfits 12 zum Sulfat (13b) führt zu einer geringfügigen Verbesserung der
Regioselektivät zugunsten eines Angriffs am α-Kohlenstoffatom, eine Ringöffnung am
β-Kohlenstoffatom kann nur als Nebenprodukt in geringen Mengen beobachtet werden.
Mechanistische Untersuchungen bei denen die Carbonylfunktionalitäten variiert wurden
(Abb. 30), zeigten einen starken Einfluss von Estern bzw. Säureamiden auf die
Regioselektivität. So wurde bei der Verwendung der Säureamide 13a und 13c die
Regioselektivität umgekehrt und der Ring am β-Kohlenstoffatom durch das Azid geöffnet[85].
O
O
SO13bO
O
O
SO13cO
O
O
SO13aO
ON
OMe
Me
O OOMe N
Abbildung 30: Variation der Carbonylfunktionalitäten zur Untersuchung des Reaktionsmechanismus[85].

Einleitung
29
Da die Ester 13b und 13c eine ähnliche Elektronenverteilung aufweisen, zeigte sich, dass die
Elektronenverteilung im Gegensatz zur Ringöffnung von Epoxiden[86] hier keinen Einfluss auf
die Regioselektivität hat[85]. Berechnungen der relativen Energie der Übergangszustände
(Abb. 31) zeigen ein Minimum für 13a im Übergangszustand 13_af und für 13b im
Übergangszustand[85].
Abbildung 31: Berechnete Übergangszustände mit Bindungslängen für den Angriff des Azids am Cβ (f) und Cα (h) an 13a und 13b jeweils syn (s) und anti (a) relativ zur α-Methylgruppe [85].
Diese Übergangszustände führen zu der im Experiment beobachteten Regioselektivität. Die
Bevorzugung des Angriffs am α-Kohlenstoffatom in 13b ist auf einen Lösungsmitteleffekt
zurückzuführen, der zu kleinen Unterschieden im Ladungstransfer in den verschiedenen
Übergangszuständen führt.
1.5.3. Synthese von α,α–disubstituierten Aminosäuren durch chelatverbrückte Claisen-
Umlagerung
Der Isoleucin Antagonist Cyclopentylglycin 18[87] und das antibiotische Furanomycin 19[88]
(Abb. 32) sind natürlich vorkommende γ,δ-ungesättigte Aminosäuren mit interessanten
Bioaktivitäten. Aber nicht nur das macht γ,δ-ungesättigte Aminosäuren zu einem Ziel der
Synthesechemie, sie stellen auch interessante Zwischenstufen zur Synthese von
funktionalisierten Aminosäuren dar[89].
Einleitung
30
H2N CO2H
O
H2N CO2H
18 19 Abbildung 32: Die natürlich vorkommenden γ,δ-ungesättigten Aminosäuren Cyclopentyl-glycin 18 und Furanomycin 19.
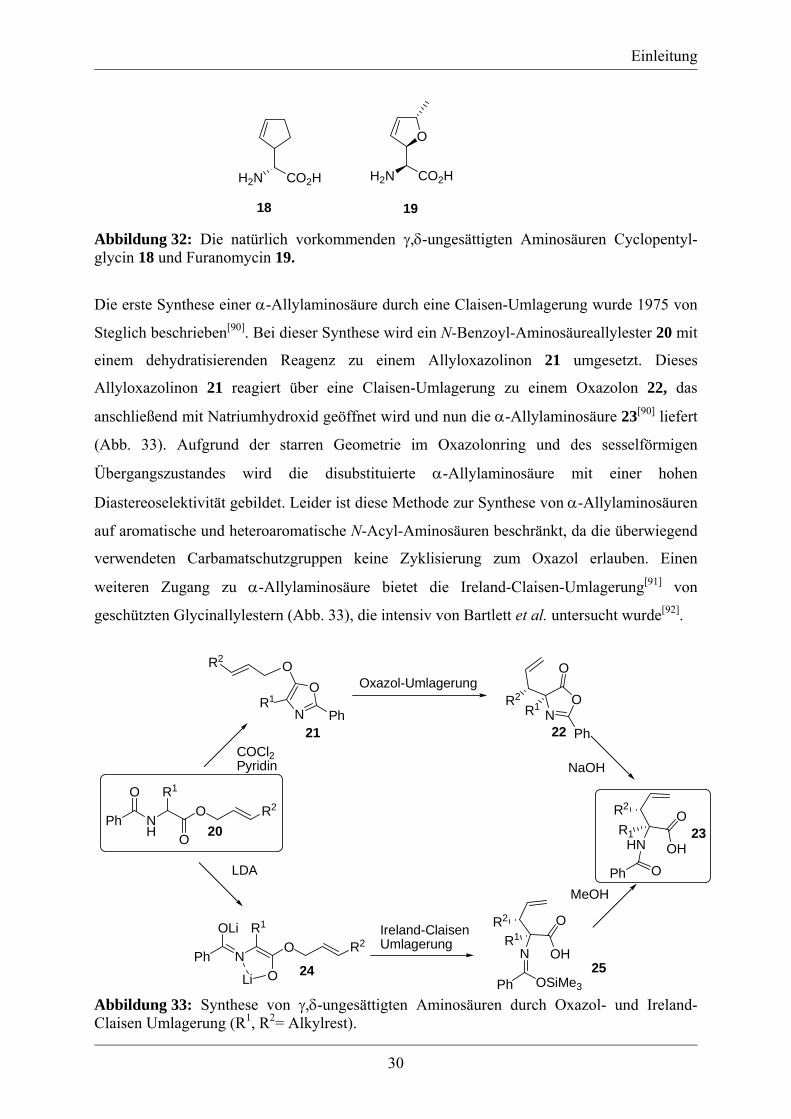
Die erste Synthese einer α-Allylaminosäure durch eine Claisen-Umlagerung wurde 1975 von
Steglich beschrieben[90]. Bei dieser Synthese wird ein N-Benzoyl-Aminosäureallylester 20 mit
einem dehydratisierenden Reagenz zu einem Allyloxazolinon 21 umgesetzt. Dieses
Allyloxazolinon 21 reagiert über eine Claisen-Umlagerung zu einem Oxazolon 22, das
anschließend mit Natriumhydroxid geöffnet wird und nun die α-Allylaminosäure 23[90] liefert
(Abb. 33). Aufgrund der starren Geometrie im Oxazolonring und des sesselförmigen
Übergangszustandes wird die disubstituierte α-Allylaminosäure mit einer hohen
Diastereoselektivität gebildet. Leider ist diese Methode zur Synthese von α-Allylaminosäuren
auf aromatische und heteroaromatische N-Acyl-Aminosäuren beschränkt, da die überwiegend
verwendeten Carbamatschutzgruppen keine Zyklisierung zum Oxazol erlauben. Einen
weiteren Zugang zu α-Allylaminosäure bietet die Ireland-Claisen-Umlagerung[91] von
geschützten Glycinallylestern (Abb. 33), die intensiv von Bartlett et al. untersucht wurde[92].
Ph NH
OO R1
O
R2
N
O
Ph
O
R1
R2
NO
Ph
O
R2R1
HN OH
OR2
R1
OPh
N OH
OR2
R1
Ph OSiMe3
Ph NO
OLi R1
O
R2
Li
COCl2Pyridin NaOH
MeOH
LDA
Oxazol-Umlagerung
Ireland-ClaisenUmlagerung
20
21 22
23
24 25
Abbildung 33: Synthese von γ,δ-ungesättigten Aminosäuren durch Oxazol- und Ireland-Claisen Umlagerung (R1, R2= Alkylrest).

Einleitung
31
Diese Methode toleriert eine Vielzahl an Schutzgruppen und kann zur Synthese von α-mono-
und α,α-disubstituierten Aminosäuren verwendet werden. Die Diastereoselektivität der
Reaktion ist sehr stark abhängig von Parametern wie der verwendeten Base, dem
Lösungsmittel, der N-terminalen Schutzgruppe und dem Substitutionsmuster des
Allylesterrestes. Daher ist diese Methode nicht als allgemeine Methode zur Synthese von
γ,δ-ungesättigten Aminosäuren geeignet[93].
Aus diesem Grund wurde die Claisen-Umlagerung von N-terminal geschützten
Glycinallylestern in der Arbeitsgruppe Kazmaier weiterentwickelt[94]. Hierzu wurden
Glycinesterenolate chelatisiert, da diese Chelate gegenüber den nichtchelatisierten
Aminosäuren wesentlich stabiler sind und Nebenreaktionen wie Eliminierungen unterdrückt
werden können. Die chelatisierten Enolate besitzen eine relativ starre Geometrie und zeigen
dadurch bei der Umlagerung einen hohen Grad an Diastereoselektivität[95]. Für die
Chelatisierung kann eine Vielzahl von Metallsalzen verwendet werden, so z.B. ZnCl2, MgCl2,
EtAlCl2, SnCl2 und CoCl2, wobei die besten Resultate in der Regel bei der Verwendung von
ZnCl2 erzielt wurden. Die Metallchelate zeichnen sich gegenüber den Silylketenacetalen 25
durch eine höhere Reaktivität und Selektivität aus. Die treibende Kraft der Reaktion ist die
Bildung des hochenergetischen Esterenolats 27, der dann zum stabilen Carboxylat 28
umlagert (Abb. 34).
CbzHNO
OCbzN
O
OZn
CbzHN CO2H
LDA
ZnCl2
26 27 28
O
OZn
NZ
O
NZO
Zn HO O O O
Sesselkonformation27
Wannenkonformation27
Abbildung 35: Chelatverbrückte Claisen-Umlagerung von N-terminal geschützten Glycinallylesterderivaten und der Übergangszustand in der Sessel- und Wannenkonformation mit THF an den weiteren Koordinationsstellen des Metallkations.

Einleitung
32
Diese chelatverbrückte Umlagerung liefert eine hohe Diastereoselektivität unabhängig von
der verwendet Schutzgruppe und des Substitutionsmusters am Allylester[96]. So können auch
zyklische Allylester in der Reaktion verwendet werden[97]. Auch sterisch anspruchsvollere
Substrate können für die Umlagerung verwendet werden, so ist die Synthese von quartären
β-Kohlenstoffzentren aus Glycinderivaten [93] sowie die Synthese von α,α-disubstituierten
Aminosäuren aus α-alkylierten Aminosäureallylestern möglich[98]. Es ist mit dieser Reaktion
sogar möglich, quartäre Kohlenstoffzentren in der α- und β-Position zu generieren[99]. Die
Reaktion ist nicht auf aliphatische und aromatische Aminosäurereste beschränkt, auch
Tryptophan, Lysin und Methionin lassen sich in der Reaktion verwenden[93]. Die
Einschränkungen dieser Reaktion, die im Rahmen der vorliegenden Arbeit ermittelt wurden,
werden im Kapitel 3.1.2. diskutiert.

Aufgabenstellung
33
2. Aufgabenstellung
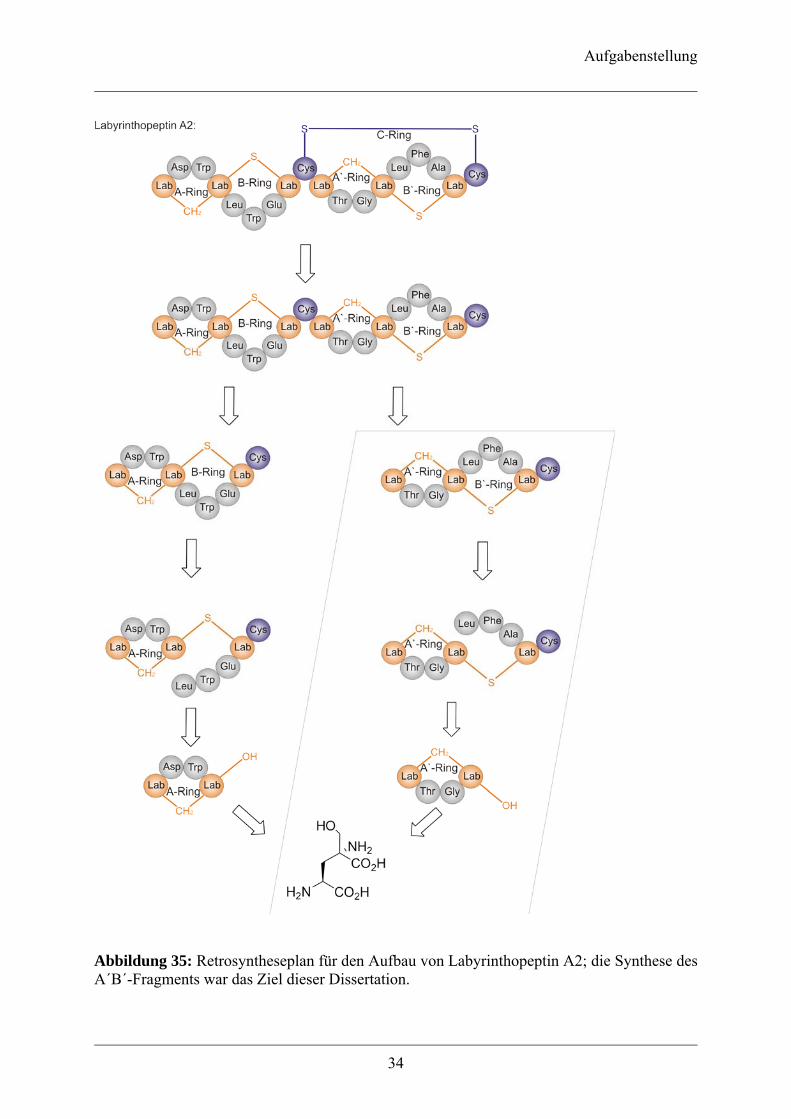
Der Naturstoff Labyrinthopeptin A2 (Abb. 13) stellt durch seine einzigartige Struktur ein
interessantes Ziel der Synthese dar, da in diesem Peptid eine bisher unbekannte Modifikation
auftritt, die Triaminotrisäure Labionin (Abb. 14), deren Existenz auch in weiteren Peptiden
vermutet wird (Kapitel 1.3.3.). Aus diesem Grund ist die Synthese von Labionin und dem
ersten bekannten labioninhaltigen Peptid Labyrinthopeptin A2 von großem akademischem
Interesse. Labionin verleiht dem Labyrinthopeptin A2 eine starre Struktur, die wiederum eine
interessante biologische Aktivität zeigt (Kapitel 1.3.4). Will man nun die Aktivität dieser
Verbindung verstehen und sie gegebenenfalls verbessern, ist deren Synthese unumgänglich.
Denn nur durch die Totalsynthese ist ein Zugang zu einer großen Anzahl von
Labyrinthopeptin-Derivaten möglich, von denen bereits jetzt absehbar ist, dass es keinen
biochemischen Zugang geben wird.
Dazu ist zunächst die Synthese der Aminosäure Labionin notwendig, da sie das zentrale
Strukturelement im Labyrinthopeptin A2 darstellt. Bis zum gegenwärtigen Zeitpunkt ist keine
Synthese einer derart komplexen Aminosäure in der Literatur beschrieben, da Labionin eine
Vielzahl von Aminosäuremodifikationen in sich vereinigt. So etwa ein quartäres
α-Kohlenstoffatom, ein Lanthioninmotiv, drei Amino- und drei Carboxygruppen, die für die
nachfolgende Labyrinthopeptin-Synthese mit den geeigneten, orthogonal abspaltbaren
Schutzgruppen versehen sein müssen. Nach der Synthese des quartären Aminosäurebausteins
soll die Aufbau des A´-Ringes erfolgen. Die A-Ringe bestehen aus 11 Atomen und beinhalten
drei Peptidbindungen von denen jeweils eine als cis-Amidbindung in der Kristallstruktur
vorliegt, was auf den gespannten Charakter der Ringe hinweist. In der Literatur sind bisher
nur zwei Synthesen von derartigen zyklischen Peptiden beschrieben, jedoch ist in diesen
Zyklen keine α,α-disubstituierte Aminosäure enthalten[100, 101]. Anschließend soll die
Synthese des B´-Ringes erfolgen. Dann soll das A´B´-Fragment mit dem AB-Fragment
gekuppelt werden. Das AB-Fragment sollte von Dipl. Chem. Georg Sambeth als Bestandteil
seiner Dissertation synthetisiert werden, und so nach Ausbildung einer Disulfidbrücke
(C-Ring) die Totalsynthese von Labyrinthopeptin A2 vollendet werden (Abb. 35).
Des Weiteren sollten entweder aus dem synthetischen Labionin oder über eine separate
Syntheseroute eine Referenzsubstanz zum Nachweis der Aminosäure Labionin in anderen
Peptiden erzeugt werden und ausgehend vom Naturstoff Labyrinthopeptin A1 und A2
Derivate zur weitergehenden Testierung auf biologische Aktivität generiert werden.
Aufgabenstellung
34
Abbildung 35: Retrosyntheseplan für den Aufbau von Labyrinthopeptin A2; die Synthese des A´B´-Fragments war das Ziel dieser Dissertation.

Ergebnisse und Diskussion
35
3. Ergebnisse und Diskussion
3.1 Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure
Die Schlüsselverbindung zur Totalsynthese von Labyrinthopeptin A2 ist die Aminosäure
Labionin (Abb. 36), da sie die A- und B-Ringe ausbildet und somit die zentrale
Struktureinheit im Labyrinthopeptin bildet. Auf Grund von retrosynthetischen Betrachtungen
wurde das Labionin in zwei Segmente zerlegt: Ein Diaminoglutarsäure-Derivat 29, das ein
quartäres α-Kohlenstoffatom trägt, und ein Cystein (Abb. 36). Die Verknüpfung des
Diaminoglutarsäure-Derivats mit dem Cystein soll zu einem späteren Zeitpunkt über eine
nukleophile Substitution erfolgen. Zu diesem Zweck wurde das Diaminoglutarsäure-Derivat
29 mit einer Alkoholfunktion am β-Kohlenstoff versehen, da sich diese funktionelle Gruppe
in eine gute Abgangsgruppe überführen lässt und im Gegensatz zu einem Thioalkohol eine
hohe Stabilität und Toleranz aufweist.
NHR NHRS
OR
O
ONHR
ORO
OR
NHR NHROH
OR
O
O ORHS
NHR
ORO29
+
Abbildung 36: Retrosynthetische Zerlegung des Labionins in ein Diaminoglutarsäure-Derivat und Cystein.
Die Synthese des Diaminoglutarsäure-Derivats 29 erwies sich als äußerst schwierig. In den
folgenden Kapiteln werden die verschiedenen Synthesewege zur Synthese von 29 vorgestellt.
In Anlehnung an in der Literatur beschriebene Synthesen für quartäre Aminosäuren[77, 98, 102]
wurden nun die ersten Versuche zur Synthese von 29 durchgeführt, bei denen erst das
quartäre Kohlenstoffzentrum aufgebaut wurde und dann die Synthese der Amino- und der
Carboxygruppe in der Seitenkette erfolgen sollte.
Ergebnisse und Diskussion
36
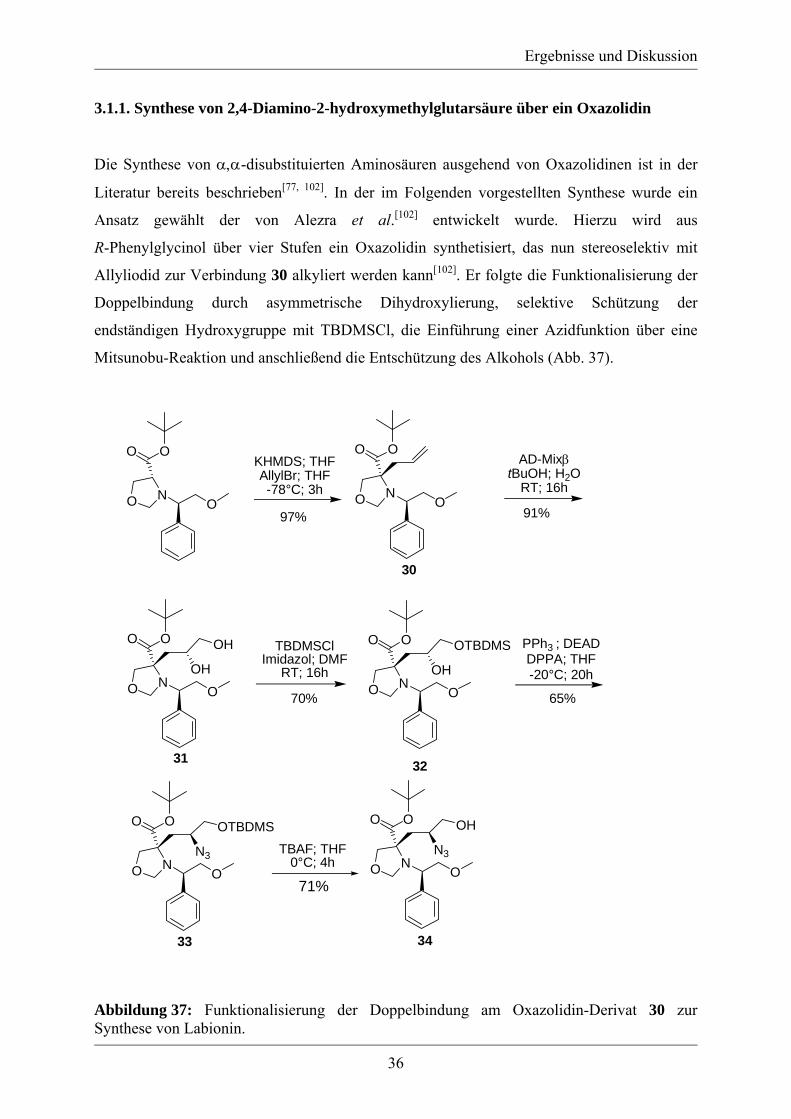
3.1.1. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure über ein Oxazolidin
Die Synthese von α,α-disubstituierten Aminosäuren ausgehend von Oxazolidinen ist in der
Literatur bereits beschrieben[77, 102]. In der im Folgenden vorgestellten Synthese wurde ein
Ansatz gewählt der von Alezra et al.[102] entwickelt wurde. Hierzu wird aus
R-Phenylglycinol über vier Stufen ein Oxazolidin synthetisiert, das nun stereoselektiv mit
Allyliodid zur Verbindung 30 alkyliert werden kann[102]. Er folgte die Funktionalisierung der
Doppelbindung durch asymmetrische Dihydroxylierung, selektive Schützung der
endständigen Hydroxygruppe mit TBDMSCl, die Einführung einer Azidfunktion über eine
Mitsunobu-Reaktion und anschließend die Entschützung des Alkohols (Abb. 37).
sdjflkjsdfjasfjaskfjkasfjkasjs
ONO
O O
N3
OH
ONO
O O
N3
OTBDMS
ONO
O O
OH
OTBDMS
ONO
O O
OH
OH
ONO
O OAD-Mixβ
tBuOH; H2ORT; 16h
91%
TBDMSCl Imidazol; DMF
RT; 16h
70%
PPh3 ; DEADDPPA; THF-20°C; 20h
65%
TBAF; THF 0°C; 4h
71%
30
31 32
33 34
ONO
O O
97%
KHMDS; THF AllylBr; THF -78°C; 3h
Abbildung 37: Funktionalisierung der Doppelbindung am Oxazolidin-Derivat 30 zur Synthese von Labionin.

Ergebnisse und Diskussion
37
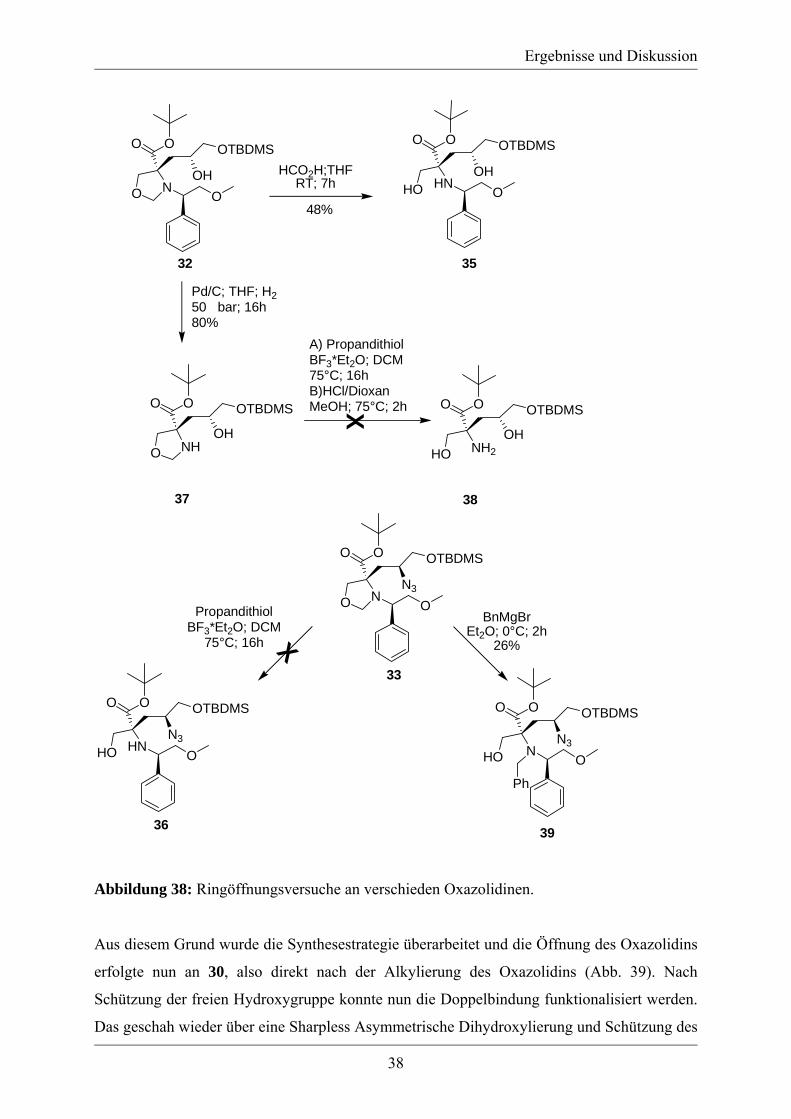
Im Folgenden wurden zeitgleich Versuche zur Oxidation des Alkohols 34 zur entsprechenden
Säure und Versuche zur Öffnung des Oxazolidins durchgeführt. Für die Oxidation des
Alkohols 34 zur Carbonsäure wurden Testversuche im LC-MS-Maßstab durchgeführt, die
jedoch keine Quantifizierung erlaubte. Hierbei wurden die Swern-Oxidation[103], bei der aber
keine Produktbildung beobachtet werden konnte, und die TEMPO-Oxidation[104] untersucht.
Unter den Reaktionsbedingungen der TEMPO-Oxidation konnte die korrespondierende
Carbonsäure von 34 erzeugt werden. Eine Optimierung und Quantifizierung der
Oxidationsreaktion wurde nicht durchgeführt, da die Öffnung des Oxazolidinringes sich als
äußerst schwierig erwies. Die Ringöffnung von Oxazolidinen ist im Allgemeinen unter sauren
Bedingungen möglich[102, 105, 106], jedoch verlief die Öffnung von 32 unter diesen Bedingungen
in schlechten Ausbeuten (48%)(Abb. 38). Aus diesem Grund wurde das Oxazolidin 33 mit
Propandithiol und BF3*Et2O versetzt (Abb. 38). Unter diesen Bedingungen wurden
Ringöffnungen von ähnlichen Substraten beschrieben[107], doch auch nach einer Reaktionszeit
von 24 h unter Rückfluss wurde keine Reaktion beobachtet. Nun wurde mit 32 eine
Hydrogenolyse durchgeführt, um den benzylischen Rest am Amin zu entfernen. Die Reaktion
mit Pd/C und Wasserstoff fand bei Normaldruck nicht statt, ein wiederholter Versuch unter
einem Druck von 50 bar lieferte dann das gewünschte Reaktionsprodukt 37 in einer Ausbeute
von 80%. Die Öffnung des Oxazolidinringes in 37 unter sauren Bedingungen und mit
Propandithiol schlug jedoch fehl (Abb. 38). Da sich die Methylenverbrückung zwischen dem
Stickstoff und dem Sauerstoffatom im Oxazolidin nicht einfach entfernen ließ, musste es in
einen Rest umgewandelt werden, der sich anschließend unter milden Bedingungen aus dem
offenkettigen Molekül entfernen lässt. Eine Möglichkeit bildet im Fall von Oxazolidinen die
Ringöffnung mit Benzylmagnesiumbromid[108]. Die Reaktion von 33 mit
Benzylmagnesiumbromid (Abb. 38) führte zwar zum gewünschten Produkt 39, doch war eine
Ausbeute von nur 26%, bei der kein Edukt mehr zurück gewonnen werden konnte nicht
hinnehmbar, da noch eine Vielzahl weiterer Reaktionsschritte zur Darstellung der quartäre
Aminosäure 29 folgen sollten und diese auch in ausreichenden Mengen für die folgende
Peptidsynthese zur Verfügung stehen musste.
Ergebnisse und Diskussion
38
NHO
O O
OH
OTBDMS
PropandithiolBF3*Et2O; DCM
75°C; 16h
OHNHO
O O OTBDMS
Pd/C; THF; H2 50 bar; 16h80%
ONO
O O OTBDMS
OHNHO
O O OTBDMS
OHOH
N3
HCO2H;THF RT; 7h
48%
NH2HO
O O
OH
OTBDMS
A) PropandithiolBF3*Et2O; DCM75°C; 16hB)HCl/DioxanMeOH; 75°C; 2h
X
X
32 35
36
37 38
ONO
O O OTBDMS
BnMgBrEt2O; 0°C; 2h
26%
ON
O O OTBDMS
N3
N3
33
HO
Ph
39
Abbildung 38: Ringöffnungsversuche an verschieden Oxazolidinen.
Aus diesem Grund wurde die Synthesestrategie überarbeitet und die Öffnung des Oxazolidins
erfolgte nun an 30, also direkt nach der Alkylierung des Oxazolidins (Abb. 39). Nach
Schützung der freien Hydroxygruppe konnte nun die Doppelbindung funktionalisiert werden.
Das geschah wieder über eine Sharpless Asymmetrische Dihydroxylierung und Schützung des
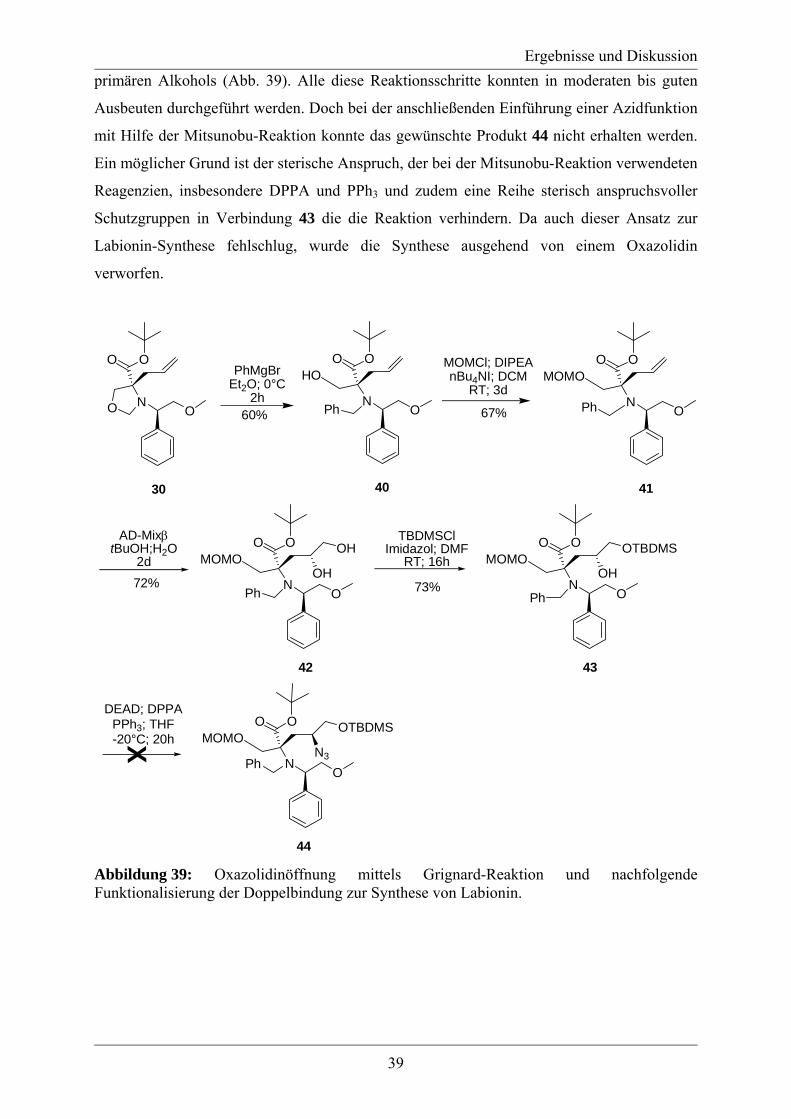
Ergebnisse und Diskussion
39
primären Alkohols (Abb. 39). Alle diese Reaktionsschritte konnten in moderaten bis guten
Ausbeuten durchgeführt werden. Doch bei der anschließenden Einführung einer Azidfunktion
mit Hilfe der Mitsunobu-Reaktion konnte das gewünschte Produkt 44 nicht erhalten werden.
Ein möglicher Grund ist der sterische Anspruch, der bei der Mitsunobu-Reaktion verwendeten
Reagenzien, insbesondere DPPA und PPh3 und zudem eine Reihe sterisch anspruchsvoller
Schutzgruppen in Verbindung 43 die die Reaktion verhindern. Da auch dieser Ansatz zur
Labionin-Synthese fehlschlug, wurde die Synthese ausgehend von einem Oxazolidin
verworfen.
ONO
O O
ON
HOO O
ON
MOMOO O
ON
MOMOO O OH
OHO
N
MOMOO O OTBDMS
OH
ON
MOMOO O OTBDMS
N3
60%
PhMgBr Et2O; 0°C
2h
MOMCl; DIPEA nBu4NI; DCM
RT; 3d
67%
72%
AD-MixβtBuOH;H2O
2d
73%
TBDMSCl Imidazol; DMF
RT; 16h
DEAD; DPPAPPh3; THF-20°C; 20h
30 40 41
42 43
44
X
Ph
Ph Ph
Ph
Ph
Abbildung 39: Oxazolidinöffnung mittels Grignard-Reaktion und nachfolgende Funktionalisierung der Doppelbindung zur Synthese von Labionin.

Ergebnisse und Diskussion
40
3.1.2. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure durch eine
chelatverbrückte Claisen-Umlagerung
Wie bereits im Kapital 1.5.3 ausführlich beschrieben wurde, ist es möglich, aus
α−monoalkylierten Aminosäuren durch eine chelatverbrückte Claisen-Umlagerung α,α-
disubstituierte Aminosäuren zu synthetisieren[93, 98, 99]. Mit Hilfe dieser Umlagerung sollte es
möglich sein, die quartäre Aminosäure 29 aus Serin zu synthetisieren. So könnte theoretisch
der vollgeschütze Serinallylester 46 zu geschütztem α-Allyl-Serin 45 umgelagert werden und
anschließend die Synthese des Aminosäureseitenkette aus der Doppelbindung erfolgen
(Abb. 40).
R1HNOR2
O
R3O
R1HNOR2
O
R3O
CO2R5R4HN
R1HNO
O
R3O
29 45 46
Abbildung 40: Retrosyntheseschema zur Synthese der quartären Aminosäure 29 durch eine chelatverbrückte Claisen-Umlagerung aus einem Serinallylester (R1-6 repräsentieren entsprechende Schutzgruppen).
Zunähst erfolgte die Synthese von Boc-Ser-OAll aus kommerziell verfügbarem Boc-Ser-OH.
Bei der folgenden Umlagerung (Abb. 41) konnte aber keine Produktbildung beobachtet
werden, lediglich das Edukt konnte vollständig zurückgewonnen werden. Des Weiteren
erfolgte eine Testreaktion mit selbst hergestelltem Cbz-Phe-OAll, um die
Reaktionsbedingungen zu überprüfen, da eine Umlagerung mit diesem Aminosäureester in
der Literatur beschrieben wurde[98].
RHNOH
O
X
RHNO
O
X LDAZnCl2; THFX
N-terminale Schutzgruppe R Boc Z Z Z Z Boc Seitenkettensubstituent X OBn OTHP OIPDMS OH OTrt SBn
Abbildung 41: Übersicht über die zu Umlagerungsversuchen verwendeten Aminosäure-derivate.

Ergebnisse und Diskussion
41
Da das Umlagerungsprodukt mit der in der Literatur beschriebenen Ausbeute gebildet wurde,
konnten fehlerhafte Reaktionsbedingungen beim der Umlagerung ausgeschlossen werden. Die
weitere Variation der Schutzgruppen am N-Terminus und in der Seitenkette (Abb. 41) brachte
keinen Erfolg. Die hier verwendeten Aminosäuren tragen im Gegensatz, zu den in der
Literatur verwendeten Aminosäuren[98] ein Heteroatom am β-Kohlenstoff. Daher könnte eine
Fehlkoordination im Übergangszustand, die durch dieses Heteroatom hervorgerufen wird, die
mögliche Ursache für die Unterdrückung der Umlagerung sein.
3.1.3. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus Pyrazolderivaten
Ein weiterer Syntheseansatz beruht auf der Spaltung der Stickstoff-Stickstoff-Bindung in
einem Pyrazolderivat[109-111], die sich leicht über eine [2+3]-dipolare Zykloaddition aus
geeigneten Edukten darstellen lassen. Das Pyrazolderivat 51 wurde in einer Ausbeute von
63% synthetisiert (Abb. 42)[112]. Die Synthese des Pyrazols zeichnet sich durch die geringe
Anzahl von Syntheseschritten und die günstigen Ausgangsverbindungen aus.
CO2tBuHO
CO2tBu
NH
N
EtO2C
CO2tBu
OH
(HCHO)n; DABCOH3PO4; THF; H2O60°C; 16h57%
DABCORT; 16h63% Raney-Ni; H2
MeOH; HCO2HRT; 16h
ClH*H2N CO2Et
H2SO4; NaNO2Et2O; H2ORT; 1min69%
N2 CO2Et
H2NH2N
EtO2C
CO2tBu
OH
Raney-NiH2; MeOH 16h; RT
Boc2O; DMAPTEA; DCMRT; 16h51%
NBoc
N
EtO2C
CO2tBu
OtBu
BocHNH2N
EtO2C
CO2tBu
OtBu
47 48
49 50
51 52
5354
X
H2NCbzHN
EtO2C
CO2tBu
OH
Cbz-Cl; RTNaHCO3H2O; 2dX
162
Abbildung 42: Pyrazolsynthese und Versuche zur anschließenden reduktiven Spaltung der Stickstoff-Stickstoffbindung zur Synthese der quartären Aminosäure 29.

Ergebnisse und Diskussion
42
Jedoch führte die Zykloaddition zu einem razemischen Gemisch von 51. Die anschließende
Spaltung der Stickstoff-Stickstoff-Bindung von 51 verlief quantitativ. Bei dem Versuch, das
Rohprodukt mit einer Schutzgruppe zu versehen (Abb. 42), konnte keine Produktbildung
beobachtet werden. Ein möglicher Grund kann die Bildung eines Lactams, als
Konkurrenzreaktion, unter den basischen Reaktionsbedingungen sein[113]. Auch eine
erfolgreiche Einführung einer Schutzgruppe an 52 hätte im besten Fall zu einer bevorzugten
Schützung des N-Terminus an der γ-Position geführt. Aus diesen Gründen wurde nun 51 mit
einer Boc-Schutzgruppe versehen (Abb. 42). Der elektronenziehende Charakter der
Schutzgruppe oder die dadurch hervorgerufen sterische Hinderung führten dazu, dass nun
keine Spaltung der Stickstoff-Stickstoff-Bindung zu 54 mehr möglich war (Abb. 42).
3.1.4 Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus γ-Methylenglutamin-
säure
In den bisher verwendeten Synthesestrategien erfolgte die Synthese des quartären
α-Kohlenstoffatom immer zu Beginn der Synthese. Nun wurde ein anderer Ansatz verfolgt,
bei dem das quartäre Zentrum am Ende der Synthese aus der Doppelbindung einer
γ-Methylenglutaminsäure erfolgen sollte. Die Synthese der γ-Methylenglutaminsäure erfolgte
durch die stereoselektive Alkylierung[114] eines Glycinesters, deren Mechanismus im Kapitel
1.5.1. ausführlich beschrieben wurde. Als Ausgangssubstanzen für die Alkylierung dienten
2-Brommethylacrylsäuremethylester 57 (Abb. 43) und die Schiffschen Base 62 (Abb. 44).
Der Brommethylacrylsäuremethylester 57 wurde über zwei Stufen aus Acrylsäuremethylester,
Formaldehyd und Bromwasserstoff synthetisiert. Zwar beträgt die Ausbeute über beide Stufen
nur 23%, doch die Ausgangsmaterialien sind extrem günstig und der Aufwand ist sehr gering.
ClH3N CO2tBu CbzHN CO2tBu H2N CO2tBu
Cbz-Clges. NaHCO3-Lösung
RT; 16h
Pd/C; H2 EtOAc; RT; 16h
91% 90%
CO2Me
Br
CO2Me
OH 48%ige HBr; DCMkonz. H2SO4; RT; 16h
68%CO2Me
CH2O; NEt3 H2O; 60°C; 16h
36%55 56 57
58 59 60 Abbildung 43: Synthese der Ausgangssubstanzen für die Darstellung des γ-Methylen-glutaminsäureesters 63.
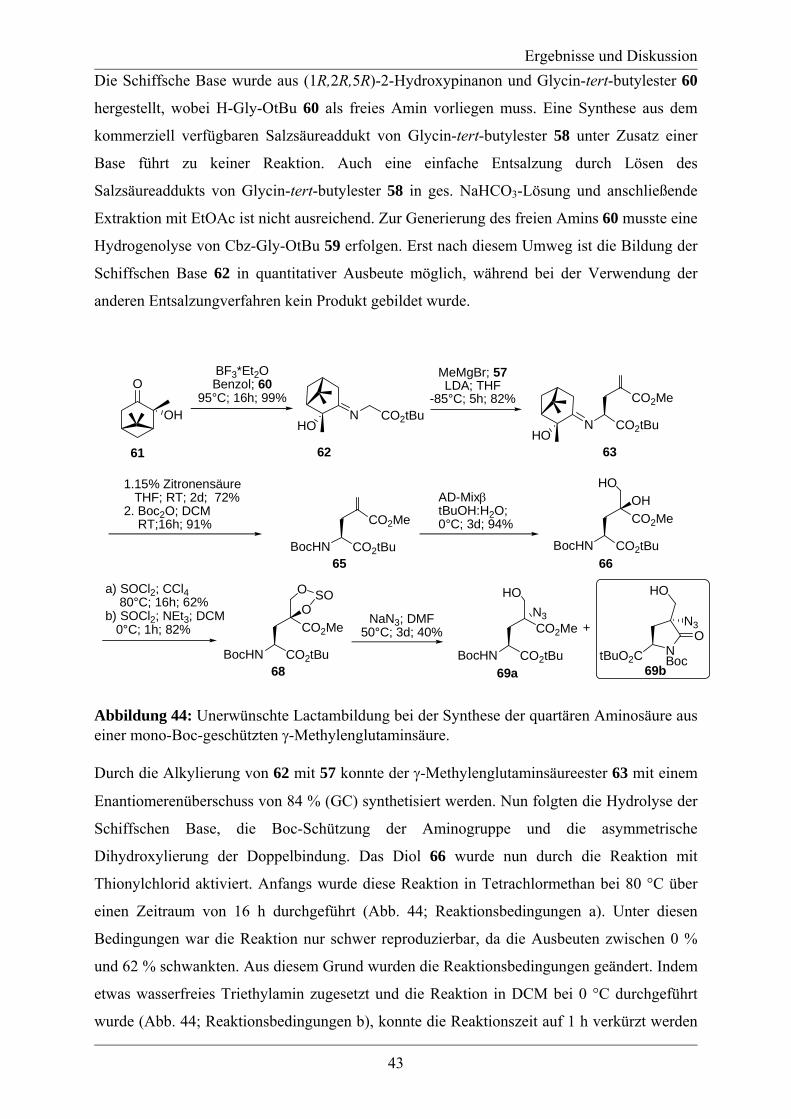
Ergebnisse und Diskussion
43
Die Schiffsche Base wurde aus (1R,2R,5R)-2-Hydroxypinanon und Glycin-tert-butylester 60
hergestellt, wobei H-Gly-OtBu 60 als freies Amin vorliegen muss. Eine Synthese aus dem
kommerziell verfügbaren Salzsäureaddukt von Glycin-tert-butylester 58 unter Zusatz einer
Base führt zu keiner Reaktion. Auch eine einfache Entsalzung durch Lösen des
Salzsäureaddukts von Glycin-tert-butylester 58 in ges. NaHCO3-Lösung und anschließende
Extraktion mit EtOAc ist nicht ausreichend. Zur Generierung des freien Amins 60 musste eine
Hydrogenolyse von Cbz-Gly-OtBu 59 erfolgen. Erst nach diesem Umweg ist die Bildung der
Schiffschen Base 62 in quantitativer Ausbeute möglich, während bei der Verwendung der
anderen Entsalzungverfahren kein Produkt gebildet wurde.
O
OH NHO
CO2tBu
BF3*Et2OBenzol; 60
95°C; 16h; 99%
MeMgBr; 57LDA; THF
-85°C; 5h; 82%
NHO
CO2tBu
CO2Me
1.15% Zitronensäure THF; RT; 2d; 72%2. Boc2O; DCM RT;16h; 91%
BocHN CO2tBu
CO2Me
AD-Mixβ tBuOH:H2O;0°C; 3d; 94%
BocHN CO2tBu
CO2Me
HOOH
BocHN CO2tBu
CO2MeO
O SO
BocHN CO2tBu
CO2Me
HON3
a) SOCl2; CCl4 80°C; 16h; 62%b) SOCl2; NEt3; DCM 0°C; 1h; 82%
NaN3; DMF 50°C; 3d; 40%
62 63
65 66
68 69a
NBoc
O
HO
N3
tBuO2C
61
69b
+
Abbildung 44: Unerwünschte Lactambildung bei der Synthese der quartären Aminosäure aus einer mono-Boc-geschützten γ-Methylenglutaminsäure. Durch die Alkylierung von 62 mit 57 konnte der γ-Methylenglutaminsäureester 63 mit einem
Enantiomerenüberschuss von 84 % (GC) synthetisiert werden. Nun folgten die Hydrolyse der
Schiffschen Base, die Boc-Schützung der Aminogruppe und die asymmetrische
Dihydroxylierung der Doppelbindung. Das Diol 66 wurde nun durch die Reaktion mit
Thionylchlorid aktiviert. Anfangs wurde diese Reaktion in Tetrachlormethan bei 80 °C über
einen Zeitraum von 16 h durchgeführt (Abb. 44; Reaktionsbedingungen a). Unter diesen
Bedingungen war die Reaktion nur schwer reproduzierbar, da die Ausbeuten zwischen 0 %
und 62 % schwankten. Aus diesem Grund wurden die Reaktionsbedingungen geändert. Indem
etwas wasserfreies Triethylamin zugesetzt und die Reaktion in DCM bei 0 °C durchgeführt
wurde (Abb. 44; Reaktionsbedingungen b), konnte die Reaktionszeit auf 1 h verkürzt werden
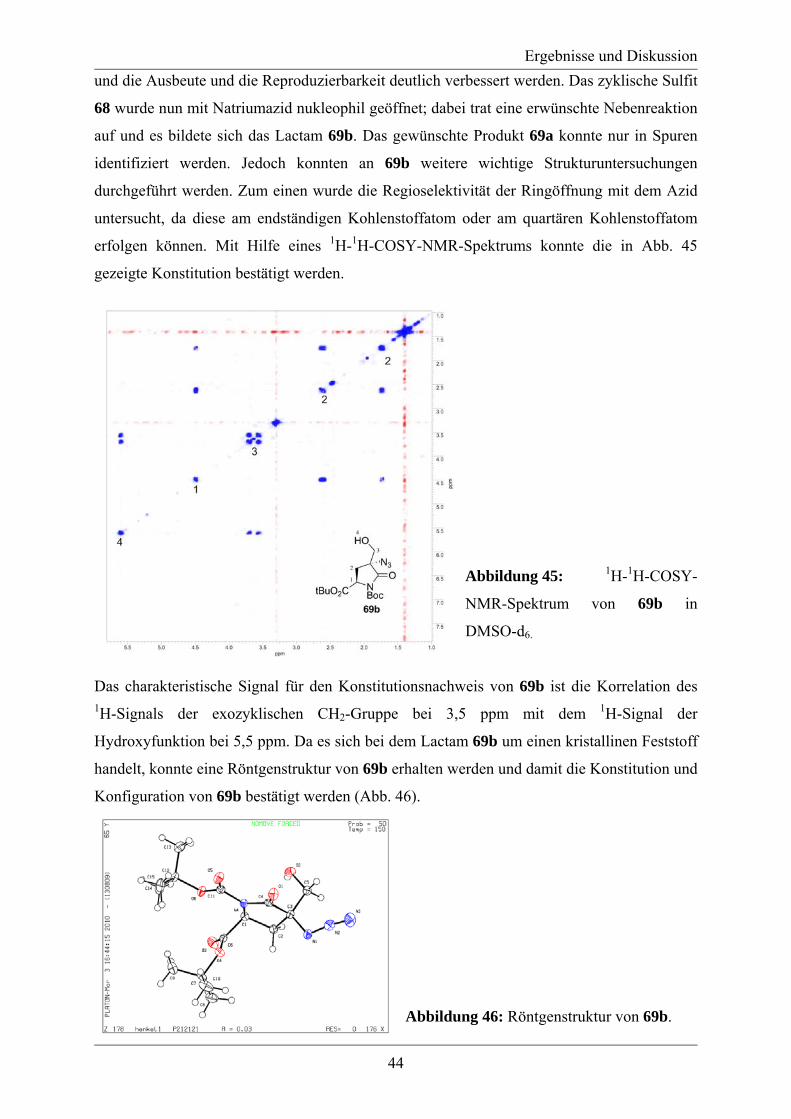
Ergebnisse und Diskussion
44
und die Ausbeute und die Reproduzierbarkeit deutlich verbessert werden. Das zyklische Sulfit
68 wurde nun mit Natriumazid nukleophil geöffnet; dabei trat eine erwünschte Nebenreaktion
auf und es bildete sich das Lactam 69b. Das gewünschte Produkt 69a konnte nur in Spuren
identifiziert werden. Jedoch konnten an 69b weitere wichtige Strukturuntersuchungen
durchgeführt werden. Zum einen wurde die Regioselektivität der Ringöffnung mit dem Azid
untersucht, da diese am endständigen Kohlenstoffatom oder am quartären Kohlenstoffatom
erfolgen können. Mit Hilfe eines 1H-1H-COSY-NMR-Spektrums konnte die in Abb. 45
gezeigte Konstitution bestätigt werden.
Abbildung 45: 1H-1H-COSY-
NMR-Spektrum von 69b in
DMSO-d6.
Das charakteristische Signal für den Konstitutionsnachweis von 69b ist die Korrelation des 1H-Signals der exozyklischen CH2-Gruppe bei 3,5 ppm mit dem 1H-Signal der
Hydroxyfunktion bei 5,5 ppm. Da es sich bei dem Lactam 69b um einen kristallinen Feststoff
handelt, konnte eine Röntgenstruktur von 69b erhalten werden und damit die Konstitution und
Konfiguration von 69b bestätigt werden (Abb. 46).
Abbildung 46: Röntgenstruktur von 69b.

Ergebnisse und Diskussion
45
Die Bildung des unerwünschten Regioisomers konnte bei dieser Reaktion nicht beobachtet
werden. In analoger Weise wurde die in Abb. 44 gezeigte Syntheseroute auch mit einer
Fmoc-Schutzgruppe (Verbindung 144 bis 147; siehe experimenteller Teil) durchgeführt.
Damit sollten die Verbindungen eine UV-Aktivität erhalten um eine schnelle Analyse aller
Reaktionsprodukte mit Hilfe einer chiralen HPLC durchzuführen und die Reaktion damit
besser kontrollieren zu können. Doch zersetzte sich das Fmoc-Derivat (147) von 68 bei der
Ringöffnung mit Natriumazid.
Um die Lactambildung zu unterdrücken musste der sterische Anspruch an der Aminogruppe
von 68 erhöht werden. Zu diesem Zweck wurde eine zweite Boc-Schützung am Carbamat-
Stickstoff von 65 durchgeführt. Wie Abb. 47 zu entnehmen ist, konnte nun die Zyklisierung
unterdrückt werden und die quartäre Aminosäure in geschützter Form mit einer Ausbeute von
12,8 % über 10 Stufen erfolgreich synthetisiert werden. Die Auswertung des 1H-1H-COSY-
Spektrums von 73 in DMSO-d6 zeigt die Korrelation des 1H-Signals der CH2-Gruppe in
β-Position von 73 mit dem 1H-Signal der Hydroxyfunktion, was auf das korrekte Regioisomer
hinweist. Eine Röntgenstruktur von 73 konnte nicht erhalten werden, da es sich bei 73 um ein
Öl handelt, das nicht in einen Feststoff überführt werden konnte. Mit Verbindung 73 konnten
nun weitergehende Untersuchungen zur Zyklisierung der A-Ringe und zur Bildung der
Thioetherbrücke durchgeführt werden.
Boc2N CO2tBu
CO2MeAD-Mixβ tBuOH:H2O0°C; 2d; 99%
Boc2N CO2tBu
CO2Me
HOOH
Boc2N CO2tBu
CO2MeO
O SO
Boc2N CO2tBu
CO2Me
HON3
SOCl2; NEt3DCM; 94%
0°C; 1hNaN3; DMF
50°C; 3d; 40%
70 71
72 73
BocHN CO2tBu
CO2Me
65
Boc2O; DMAPCH3CN; 79%
RT; 16h
Abbildung 47: Synthese der Aminosäure 73 über eine γ-Methylenglutaminsäure unter Verwendung von zwei Boc-Schutzgruppen.

Ergebnisse und Diskussion
46
3.2 Studien zur Synthese von Labionin
Nach der erfolgreichen Synthese von 73 konnte nun die Anknüpfung der Thioetherbrücke
zum Labionin begonnen werden. Der Thioether sollte durch die Reaktion von geschütztem
Cystein mit einem aktivierten Derivat von 73 in Anlehnung an publizierte
Lanthioninsynthesen[59, 60] erfolgen (Abb. 48).
Boc2N CO2tBu
CO2Me
HON3
Boc2N CO2tBu
CO2MeN3
S
CO2All
NHFmoc
Boc2N CO2tBu
CO2Me
XN3
73
Abbildung 48: Retrosyntheseplan zur Synthese von geschütztem Labionin (X =Abgangsgruppe).
Die Hydrogruppe von 73 sollte zunächst unter Verwendung der Appel-Reaktion in ein
Iodatom transformiert werden (Abb. 49), da Beispiele für Appel-Reaktionen in Gegenwart
von Azidfunktionen publiziert wurden[115, 116]. Doch kam es bei der Verwendung von 73 zu
einer Zersetzungsreaktion, sodass weder das Produkt 80 noch das Edukt 73 nach der Reaktion
isoliert werden konnten (Abb. 49). Zur direkten Umwandlung der Hydroxygruppe in ein
Bromatom wurden Reaktionen mit Phosphortribromid [117, 118] und mit
Dibromtriphenylphosphan[119] durchgeführt, bei denen das gewünschte Produkt 79 nicht
detektiert werden konnte (Abb. 49). Aus diesem Grund wurde der Alkohol 73 in den
Methansulfonsäureester 76 umgewandelt, der wiederum in die Verbindung 80 und 79
umgewandelt werden sollte[120, 121]. In beiden Fällen konnte lediglich das Edukt 73 vollständig
zurückgewonnen werden. Ausgehend von 76 wurden nun Versuche unternommen, den
Essigsäurethioester 77 durch Substitution oder durch Verwendung eines
Schwefeltransferreagenzes[122] zu synthetisieren. Doch auch diese Versuche schlugen fehl.
Die Synthese des Trifluormethansulfonsäureesters 78, mit dem eine Reaktion mit Cystein zur
Labionin-Synthese möglich gewesen wäre[123], misslang, da sich 73 unter den verwendeten
Reaktionsbedingungen zersetzte (Abb. 49). In der Literatur findet sich nur ein Beispiel für die
Synthese eines α-Methyllanthionins[124]. Auch beim Labionin handelt es sich um ein
α-Alkyllanthioninderivat[124]. Nach dieser Vorschrift wird α-Methyllanthionin aus einem
geschützten α−Methylserin und einem Cystein synthetisiert, wobei sich die Umwandlung der
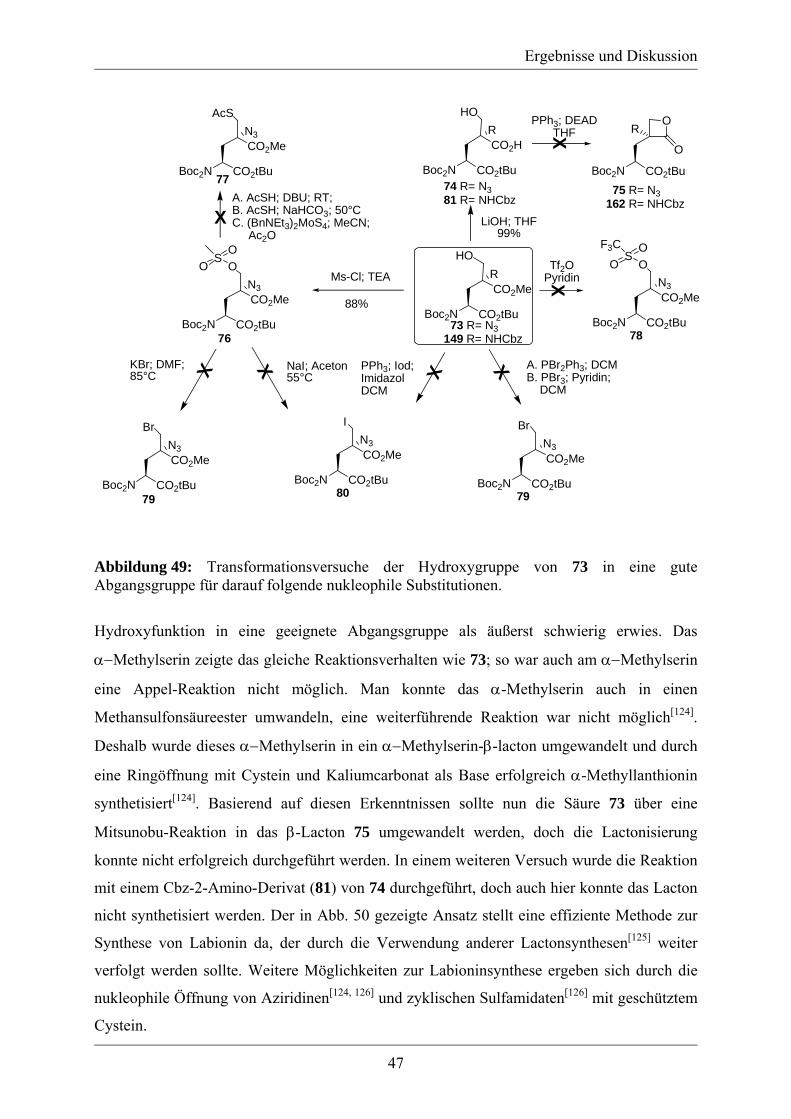
Ergebnisse und Diskussion
47
Boc2N CO2tBu
CO2Me
HOR
Boc2N CO2tBu
CO2Me
BrN3
Boc2N CO2tBu
CO2Me
IN3
A. PBr2Ph3; DCMB. PBr3; Pyridin; DCM
PPh3; Iod;ImidazolDCM
X X
Boc2N CO2tBu
O
O
RPPh3; DEAD
THF
Boc2N CO2tBu
CO2H
HOR
LiOH; THF 99%
Boc2N CO2tBu
CO2Me
ON3
SO
O
Boc2N CO2tBu
CO2Me
BrN3
KBr; DMF; 85°C
NaI; Aceton 55°C
Boc2N CO2tBu
CO2Me
AcSN3
A. AcSH; DBU; RT;B. AcSH; NaHCO3; 50°CC. (BnNEt3)2MoS4; MeCN; Ac2O
X
X X
Boc2N CO2tBu
CO2Me
ON3
SF3C
OO
Tf2O PyridinMs-Cl; TEA
88%
XX
76
77
78
79 7980
73 R= N3149 R= NHCbz
74 R= N3 81 R= NHCbz
75 R= N3162 R= NHCbz
Abbildung 49: Transformationsversuche der Hydroxygruppe von 73 in eine gute Abgangsgruppe für darauf folgende nukleophile Substitutionen.
Hydroxyfunktion in eine geeignete Abgangsgruppe als äußerst schwierig erwies. Das
α−Methylserin zeigte das gleiche Reaktionsverhalten wie 73; so war auch am α−Methylserin
eine Appel-Reaktion nicht möglich. Man konnte das α-Methylserin auch in einen
Methansulfonsäureester umwandeln, eine weiterführende Reaktion war nicht möglich[124].
Deshalb wurde dieses α−Methylserin in ein α−Methylserin-β-lacton umgewandelt und durch
eine Ringöffnung mit Cystein und Kaliumcarbonat als Base erfolgreich α-Methyllanthionin
synthetisiert[124]. Basierend auf diesen Erkenntnissen sollte nun die Säure 73 über eine
Mitsunobu-Reaktion in das β-Lacton 75 umgewandelt werden, doch die Lactonisierung
konnte nicht erfolgreich durchgeführt werden. In einem weiteren Versuch wurde die Reaktion
mit einem Cbz-2-Amino-Derivat (81) von 74 durchgeführt, doch auch hier konnte das Lacton
nicht synthetisiert werden. Der in Abb. 50 gezeigte Ansatz stellt eine effiziente Methode zur
Synthese von Labionin da, der durch die Verwendung anderer Lactonsynthesen[125] weiter
verfolgt werden sollte. Weitere Möglichkeiten zur Labioninsynthese ergeben sich durch die
nukleophile Öffnung von Aziridinen[124, 126] und zyklischen Sulfamidaten[126] mit geschütztem
Cystein.

Ergebnisse und Diskussion
48
Boc2N CO2tBu
N3
Boc2N CO2tBu
CO2HN3
S
CO2All
NHFmoc
Fmoc-Cys-OAllK2CO3; DMF; RT
75
O
O
Abbildung 50: Hypothetische Labioninsynthese durch Öffnung des β-Lactons 75.
3.3 Synthese des A´-Rings von Labyrinthopeptin A2
Die Synthese der Ringe im Labyrinthopeptin A2 (Abb. 13) stellt nach der Synthese des
quartären Aminosäurebausteins 73 eine große Herausforderung bei der Totalsynthese von
Labyrinthopeptin A2 dar. Die Synthese des A´-Rings soll hier vor der Synthese des B´-Rings
erfolgen. Da es sich beim A´-Ring um ein gespanntes Ringsystem handelt, soll dessen
Synthese aus dem offenkettigen Peptid erfolgen.
Der A´-Ring wird durch eine Methylenbrücke zwischen den α-Kohlenstoffatomen der
Aminosäurepositionen Lab10 und Lab13 gebildet, die das Dipeptid Thr-Gly umschließen
(Abb. 51). Der dadurch gebildete Ring besteht aus 11 Atomen und besitzt im Kristall eine
cis-Amidbindung zwischen den Aminosäuren Thr und Gly, was ein Indiz auf eine starke
Ringspannung im A´-Ring ist. Der konformative Einfluss carbazyklischer Ringe in Peptiden
wurde ausführlich in der Arbeitsgruppe Verdine untersucht[127-129], dabei überspannt eine acht
Kohlenstoffatome zählende Brücke ein Penta- bzw. Heptapeptid. Aus diesen Carbozyklen
resultieren stabile α-Helices. Doch im Vergleich zum A´-Ring (Abb. 51), der aus 11 Atomen
besteht, weisen die von Verdine untersuchten zyklischen Peptide, die aus 21 bzw. 27 Atomen
bestehen, eine deutlich geringere Ringspannung auf. Kleinere carbozyklische Peptide wurden
in der Arbeitsgruppe von Vederas bei der Synthese eines Analogons des Lantibiotikums
Lacticin 3147 A2 dargestellt[130]. Dabei handelt es sich um Tetra- bzw. Pentapeptide, die von
einer Brücke aus vier Kohlenstoffatomen überspannt werden. Die resultierenden Ringe
bestehen aus 14 bzw. 17 Atomen und sind damit größer als die A-Ringe im Labyrinthopeptin.
Außerdem bestehen die Brücken im Lacticin 3147 A2 Analogon aus vier
Kohlenstoffatomen[130], während die carbozyklischen Brücken im Labyrinthopeptin nur aus
einem Kohlenstoffatom bestehen, was zu einer deutlich geringeren Spannung in den Ringen
führt.

Ergebnisse und Diskussion
49
Abbildung 51: Der A´- und B´-Ring im Kugelmodell von Labyrinthopeptin A2.
In der Literatur sind lediglich zwei Beispiele für die Synthese von Peptiden dieser Ringgröße
beschrieben[100, 101], bei denen jedoch keine α,α-disubstituierte Aminosäure im Peptid
enthalten ist. Aus diesem Grund wurde die Zyklisierung erst an einem Modellsystem erprobt,
um mögliche Probleme bei der Zyklisierung zu erkennen und ggf. die Notwendigkeit einer
vorgeformten cis-Amidbindung zwischen den Aminosäuren Thr und Gly bei der Zyklisierung
zu untersuchen. In diesem Modellsystem wurde das Labionin durch eine geschützte
Glutaminsäure ersetzt, da die Ringgröße des Modellpeptids mit der des A´-Ringes identisch
ist und sie kommerziell verfügbar ist.
Die cis-Amidbindung im Dipeptid Thr-Gly wurde durch die Verwendung eines
Trimethoxybenzylrest (Tmb) am N-Terminus von Glycin vorgeformt. Die Verwendung von
solchen benzylischen Gruppen zur Formung von cis-Amidbindungen ist in der Literatur
ausführlich beschrieben[131-135]. Eine vorgeformte cis-Amidbindung bringt den N- und
C-Terminus des linearen Peptides in räumliche Nähe und erleichtert dadurch die Zyklisierung
des Peptids[136]. Darüber hinaus wird gegebenenfalls die Bildung von unerwünschten
Oligomeren des Peptides unterdrückt[136]. Die Verwendung von N-alkylierten Aminosäuren ist
besonders dann sinnvoll, wenn in der Aminosäurensequenz des Peptids nur L- oder D-
Aminosäuren und keine Proline enthalten sind[136]. So erweist sich beispielsweise die
Synthese von zyklischen Tripeptiden, die nur aus α-L-Aminosäuren bestehen, ohne die
Verwendung N-alkylierter Aminosäuren als nahezu unmöglich[137-139]. Als ähnlich schwierig
ist die Zyklisierung von Tetrapeptiden, die ebenfalls nur aus α-L-Aminosäuren bestehen, da
ein 12-gliedriger Ring, der nur trans-Amidbindungen enthält, die Zyklisierung behindert[137].
Auch hier kann durch die Verwendung von N-alkylierten Aminosäuren das Peptid in eine für
die Zyklisierung günstige Konformation gebracht werden[140, 141].
Die Synthese von 83 erfolgte durch Peptidkupplung von 82 und Fmoc-Thr(tBu)-OH in
Lösung unter Verwendung von HATU (Abb. 52). Die Reaktionszeit umfasste 4 Tage, um das
Produkt in einer Ausbeute von 60 % zu erhalten. Bei einer Verringerung der Reaktionszeit auf

Ergebnisse und Diskussion
50
einen Tag erzielt man nur eine Ausbeute von 10 %. Die Entschützung des Peptids 83 bzw. 88
am N-Terminus mit DBU unter Verwendung eines Scavenger[142] führte aber zur Bildung des
Diketopiperazins 89 (Abb. 52). Durch die Verwendung einer raumerfüllenden Schutzgruppe
am C-Terminus sollte diese Reaktion unterdrückt werden. Zu diesem Zweck wurde die
Diphenylmethylschutzgruppe (Dpm) verwendet (Verbindung 87 und 88), die zudem durch
Hydrogenolyse abgespalten werden kann. Verbindung 87 wurde über drei Stufen durch
Veresterung und anschließender reduktiven Aminierung ausgehend von
Boc-Gly-OH synthetisiert. Die Verwendung einer säurelabilen Schutzgruppe, wie der tert-
Butyl-Schutzgruppe, hätte zu einem Orthogonalitätsproblem mit der Tmb-Schutzgruppe
geführt. Doch leider bildete sich auch bei der Verwendung der Dpm-Schutzgruppe das
Diketopiperazin 89 in quantitativer Ausbeute (Abb. 52). Die Darstellung des C-terminal
entschützten Dipeptids 84 ausgehend vom 83 war ohne Probleme möglich (Abb. 52), jedoch
ist dieses Peptid für die folgende Synthese des A´-Ringes von Labyrinthopeptin aufgrund der
Schutzgruppenstrategie ungeeignet.
TmbHNOR
O
NOR
O
Tmb
ONHFmoc
tBuO
Fmoc-Ser(tBu)-OH HATU; DCM
82 R= Bn 87 R= Dpm
83 R= Bn 88 R= Dpm
R= Bn 60%R= Dpm 45 %
NH
TmbN
O
O
OtBu
MAH; DBUTHF
98%
89
Pd/C; H2THF
82%
NOH
O
Tmb
ONHFmoc
tBuO
84
Abbildung 52: Synthese des Peptids (Tmb)Thr(tBu)-Gly-OR in verschiedenen Schutzgruppenvarianten mit einer vorgeformten cis-Amidbindung für die Synthese des A´-Ringes von Labyrinthopeptin A2.
Da aufgrund der Diketopiperazinbildung kein Zyklisierungsversuch mit den Tmb-haltigen
Dipeptiden durchgeführt werden konnte, wurden Zyklisierungsversuche ohne die
Tmb-Gruppe durchgeführt und mit drei Kupplungsreagenzien getestet (Abb. 53).
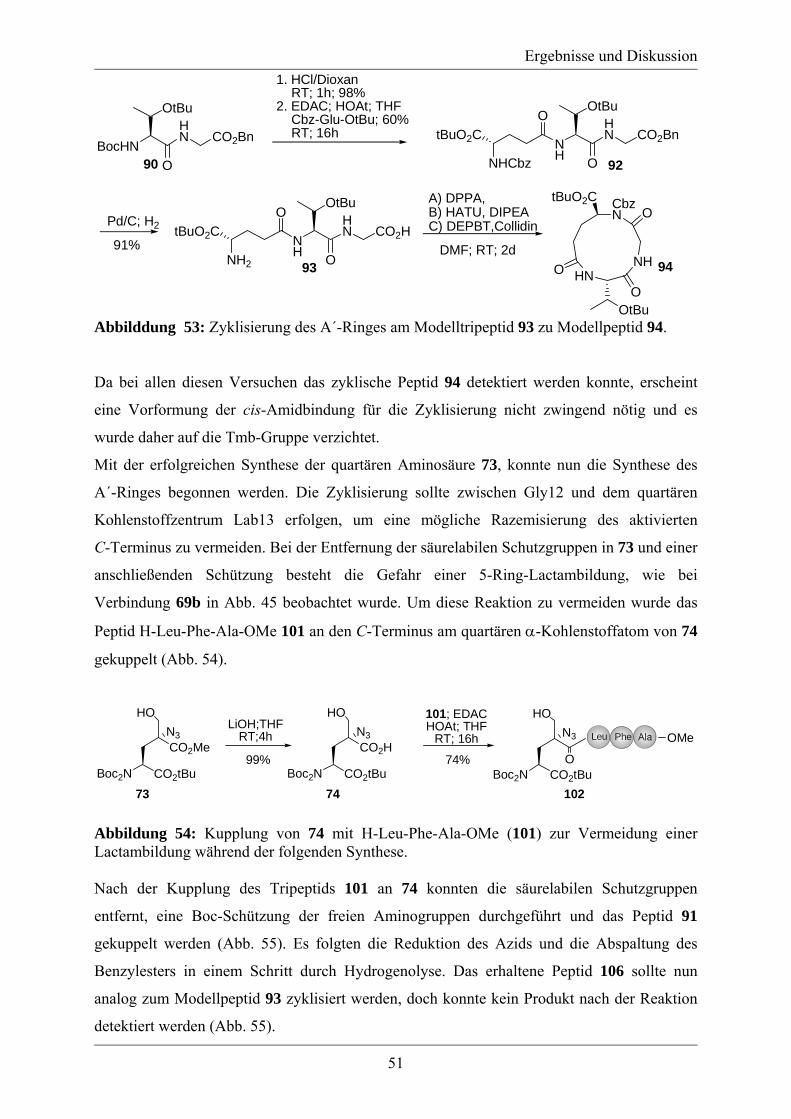
Ergebnisse und Diskussion
51
BocHNO
OtBuHN CO2Bn N
H O
OtBuHN CO2Bn
O
NHCbz
tBuO2C
HNO
OtBu
NHO
CbzN
tBuO2CO
NH O
OtBuHN CO2H
O
NH2
tBuO2C
1. HCl/Dioxan RT; 1h; 98%2. EDAC; HOAt; THF Cbz-Glu-OtBu; 60% RT; 16h
Pd/C; H2
91%
A) DPPA, B) HATU, DIPEAC) DEPBT,Collidin
90 92
93 94DMF; RT; 2d
Abbilddung 53: Zyklisierung des A´-Ringes am Modelltripeptid 93 zu Modellpeptid 94.
Da bei allen diesen Versuchen das zyklische Peptid 94 detektiert werden konnte, erscheint
eine Vorformung der cis-Amidbindung für die Zyklisierung nicht zwingend nötig und es
wurde daher auf die Tmb-Gruppe verzichtet.
Mit der erfolgreichen Synthese der quartären Aminosäure 73, konnte nun die Synthese des
A´-Ringes begonnen werden. Die Zyklisierung sollte zwischen Gly12 und dem quartären
Kohlenstoffzentrum Lab13 erfolgen, um eine mögliche Razemisierung des aktivierten
C-Terminus zu vermeiden. Bei der Entfernung der säurelabilen Schutzgruppen in 73 und einer
anschließenden Schützung besteht die Gefahr einer 5-Ring-Lactambildung, wie bei
Verbindung 69b in Abb. 45 beobachtet wurde. Um diese Reaktion zu vermeiden wurde das
Peptid H-Leu-Phe-Ala-OMe 101 an den C-Terminus am quartären α-Kohlenstoffatom von 74
gekuppelt (Abb. 54).
Boc2N CO2tBu
CO2Me
HON3
73Boc2N CO2tBu
CO2H
HON3
74Boc2N CO2tBu
HON3
102
O
LiOH;THF RT;4h
101; EDACHOAt; THF
RT; 16h Leu Phe OMe
99% 74%
Abbildung 54: Kupplung von 74 mit H-Leu-Phe-Ala-OMe (101) zur Vermeidung einer Lactambildung während der folgenden Synthese. Nach der Kupplung des Tripeptids 101 an 74 konnten die säurelabilen Schutzgruppen
entfernt, eine Boc-Schützung der freien Aminogruppen durchgeführt und das Peptid 91
gekuppelt werden (Abb. 55). Es folgten die Reduktion des Azids und die Abspaltung des
Benzylesters in einem Schritt durch Hydrogenolyse. Das erhaltene Peptid 106 sollte nun
analog zum Modellpeptid 93 zyklisiert werden, doch konnte kein Produkt nach der Reaktion
detektiert werden (Abb. 55).

Ergebnisse und Diskussion
52
Boc2N CO2tBu
HON3
102
O
BocHN
N3
105
O
O
Pd/C; H2MeOH; RT; 2d
TFA; TESRT; 4h
Boc2O; TEA MeCN; RT; 16h
91; EDACHOAt; THFRT; 16h
Thr Gly OBn
Leu Phe OMe
H2N CO2H
HON3
O
Leu Phe OMe
BocHN CO2H
N3
O
Leu Phe OMe Leu Phe OMe
OtBu
BocHN
NH2
106
O
OThr Gly OH
Leu Phe OMe
OtBu
OMeBoc Lab
Thr Gly
CH2 Leu AlaPhe
OH
Lab
OtBu
103
104
107
HOHO
HOA. DPPA; NaHCO3 B. HATU; DIPEAC. DEPBT; Collidin
XDMF; RT; 2d
99%a 95%a
95%a95%a
Abbildung 55: Syntheseversuch des A´-Ringes von Labyrinthopeptin A2 durch Zyklisierung zwischen Gly12 und dem quartären Kohlenstoffzentrum Lab 13( a Umsatz nach LC-MS).
Die Ringspannung im Peptid 107 sollte nicht unbedingt der Grund für die nicht erfolgte
Zyklisierung sein, da im Modellpeptid 93 (Abb. 53) ein Ring gleicher Größe unter den
gleichen Reaktionsbedingungen synthetisiert werden konnte. Der Hauptunterschied zwischen
dem Modellpeptid 93 und dem Peptid 106 ist das quartäre Kohlenstoffzentrum, an dem die
Zyklisierung erfolgt. Aus diesem Grund wurde die Zyklisierungsrichtung umgekehrt. Dazu
war die Synthese eines geeigneten Dipeptids nötig. Um die Orthogonalität der Schutzgruppen
zu gewährleisten, war die Verwendung der α-Azidosäure 97 nötig, mit der dann das Dipeptid
99 synthetisiert werden konnte (Abb. 56).
CuSO4* 5H2O; 95K2CO3; MeOH
RT; 16hN3 CO2H
OBn
N3
OBn
O
HN CO2tBu
Cl-HOBt; TEA; THFEDAC;H-Gly-OtBu
RT; 16h
N3
OBn
O
HN CO2H
73% 44%
TFA; TES; RT; 4h
99%
97
98 99
H2N CO2H
OBn
96
Abb. 56: Synthese des Azidodipeptids 99 zur Synthese des A´-Ringes von Labyrinthopeptin A2.
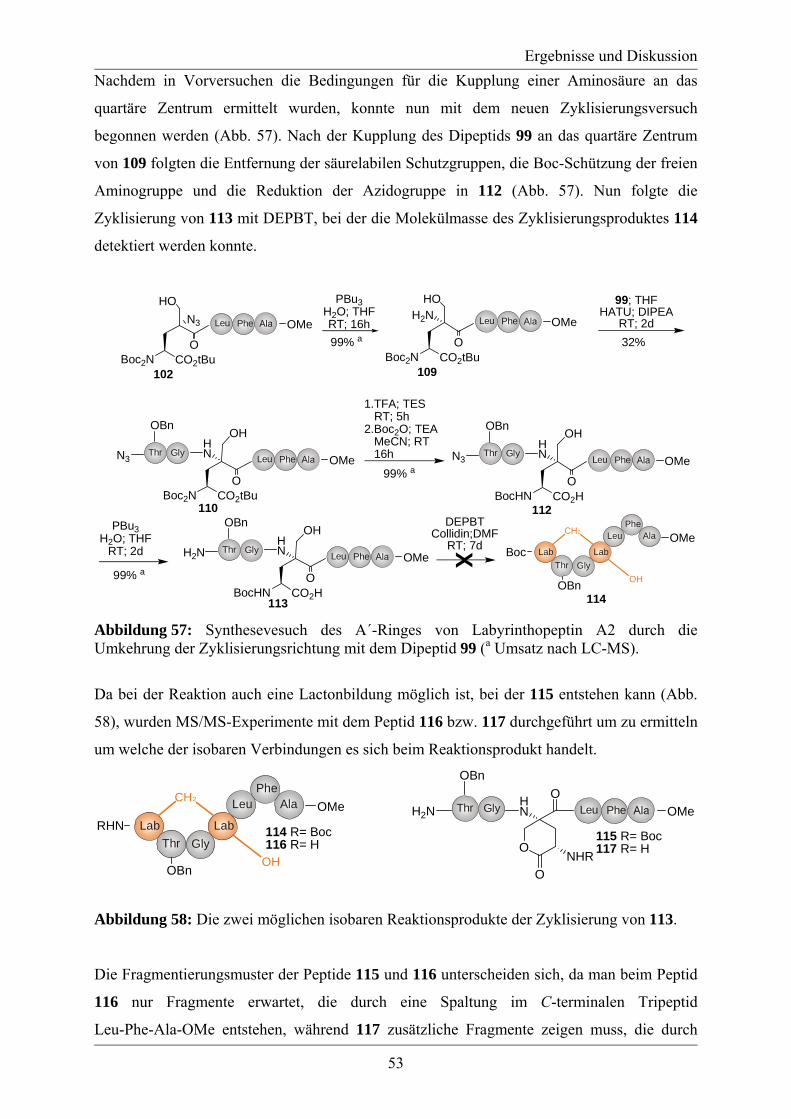
Ergebnisse und Diskussion
53
Nachdem in Vorversuchen die Bedingungen für die Kupplung einer Aminosäure an das
quartäre Zentrum ermittelt wurden, konnte nun mit dem neuen Zyklisierungsversuch
begonnen werden (Abb. 57). Nach der Kupplung des Dipeptids 99 an das quartäre Zentrum
von 109 folgten die Entfernung der säurelabilen Schutzgruppen, die Boc-Schützung der freien
Aminogruppe und die Reduktion der Azidogruppe in 112 (Abb. 57). Nun folgte die
Zyklisierung von 113 mit DEPBT, bei der die Molekülmasse des Zyklisierungsproduktes 114
detektiert werden konnte.
Boc2N CO2tBu
HON3
O
Thr Gly
Leu Phe OMe
Boc2N CO2tBu
HO
O
Leu Phe OMeH2N
Boc2N CO2tBu
OH
O
Leu Phe OMeHNN3
OBn
Thr Gly
BocHN CO2HO
Leu Phe OMeHNN3
OBn
PBu3H2O; THFRT; 16h
99; THFHATU; DIPEA
RT; 2d
1.TFA; TES RT; 5h2.Boc2O; TEA MeCN; RT 16h
PBu3H2O; THF
RT; 2d Thr Gly
BocHN CO2H
OH
O
Leu Phe OMeHNH2N
OBnOMe
Boc LabThr Gly
CH2 Leu AlaPhe
OH
Lab
OBn
DEPBT Collidin;DMF
RT; 7d
OH
102 109
110 112
113 114
X99% a 32%
99% a
99% a
Abbildung 57: Synthesevesuch des A´-Ringes von Labyrinthopeptin A2 durch die Umkehrung der Zyklisierungsrichtung mit dem Dipeptid 99 (a Umsatz nach LC-MS).
Da bei der Reaktion auch eine Lactonbildung möglich ist, bei der 115 entstehen kann (Abb.
58), wurden MS/MS-Experimente mit dem Peptid 116 bzw. 117 durchgeführt um zu ermitteln
um welche der isobaren Verbindungen es sich beim Reaktionsprodukt handelt.
OMeRHN Lab
Thr Gly
CH2 Leu AlaPhe
OH
Lab
OBn
Thr Gly Leu Phe OMeH2N
OBn
114 R= Boc116 R= H
115 R= Boc117 R= HO
HN
O
ONHR
Abbildung 58: Die zwei möglichen isobaren Reaktionsprodukte der Zyklisierung von 113.
Die Fragmentierungsmuster der Peptide 115 und 116 unterscheiden sich, da man beim Peptid
116 nur Fragmente erwartet, die durch eine Spaltung im C-terminalen Tripeptid
Leu-Phe-Ala-OMe entstehen, während 117 zusätzliche Fragmente zeigen muss, die durch

Ergebnisse und Diskussion
54
Spaltung im N-terminalen Dipeptid Thr-Gly entstehen. Ein Vergleich der errechneten
Fragmentmassen aller möglichen Fragmentierungsserien mit dem beobachteten
Fragmentierungsmuster in Abb. 59 zeigt aber deutlich, dass das Produkt der Zyklisierung das
unerwünschte Peptid 115 ist.
Abbildung 59: Schema einer Peptid-fragmentierung im MS/MS-Experiment und Tabelle mit den errechneten Fragmenten. In rot sind die charakteristischen Fragmente für das Peptid 116 abgebildet und in schwarz die gemeinsamen Fragmente beider Peptide 116 und 117. „x“ kennzeichnet ein detektiertes Fragment, während „nd“ ein nicht detektiertes Fragment kennzeichnet.
Aufgrund dieser Ergebnisse wurde die Hydroxygruppe am β-Kohlenstoffatom von 73 mit
einer Schutzgruppe versehen. Die verwendete Schutzgruppe sollte möglichst klein sein, um
eine Abschirmung des quartären Zentrums in 73 zu verhindern. Da eine Schützung als
Allylether synthetisch nicht zugänglich war, wurde der Methylether 118 bzw. 119 vewendet
(Abb. 60).
Boc2N CO2tBu
CO2Me
HON3
73Boc2N CO2tBu
CO2Me
MeON3
118
MeI; Ag2O Me2S; MeCN
RT; 16h
83%Boc2N CO2tBu
CO2H
MeON3
119
LiOH;THF RT; 16h
99%
Abbildung 60: Synthese des Methylethers 118 und anschließende Verseifung des Methylesters.
Bei der Darstellung von 118 aus 73 wurde eine teilweise Epimerisierung am
γ-Kohlenstoffatom festgestellt, da im 1H-NMR eine Signaldopplung in einem Verhältnis von
2:1 beobachtet werden konnte. Eine Trennung der Diastereomere war weder bei Verbindung
118 noch 119 möglich. Daraufhin wurde der Baustein 119 als ein Gemisch aus
nd87,044z1nd682,341c5
nd234,113z2nd535,272c4
nd347,197z3nd422,188c3
nd503,251z4nd266,134c2
nd560,272z5nd209,113c1
nd104,055y1X665,33b5
X251,124y2X518,261b4
nd364,208y3X405,177b3
X520,261y4X249,124b2
X577,283y5nd192,102b1
nd130,050x1X637,335a5
nd277,118x2X490,267a4
nd390,203x3X377,182a3
nd546,256x4X221,129a2
nd603,277x5X164,108a1
nd87,044z1nd682,341c5
nd234,113z2nd535,272c4
nd347,197z3nd422,188c3
nd503,251z4nd266,134c2
nd560,272z5nd209,113c1
nd104,055y1X665,33b5
X251,124y2X518,261b4
nd364,208y3X405,177b3
X520,261y4X249,124b2
X577,283y5nd192,102b1
nd130,050x1X637,335a5
nd277,118x2X490,267a4
nd390,203x3X377,182a3
nd546,256x4X221,129a2
nd603,277x5X164,108a1
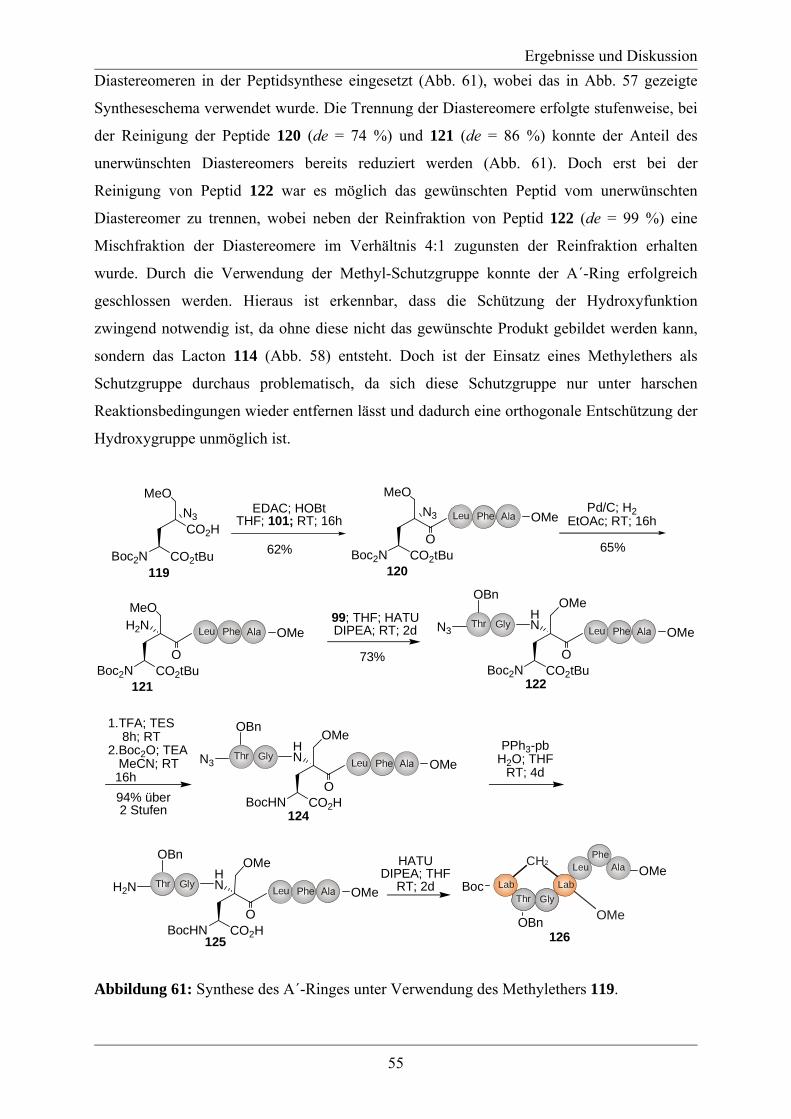
Ergebnisse und Diskussion
55
Diastereomeren in der Peptidsynthese eingesetzt (Abb. 61), wobei das in Abb. 57 gezeigte
Syntheseschema verwendet wurde. Die Trennung der Diastereomere erfolgte stufenweise, bei
der Reinigung der Peptide 120 (de = 74 %) und 121 (de = 86 %) konnte der Anteil des
unerwünschten Diastereomers bereits reduziert werden (Abb. 61). Doch erst bei der
Reinigung von Peptid 122 war es möglich das gewünschten Peptid vom unerwünschten
Diastereomer zu trennen, wobei neben der Reinfraktion von Peptid 122 (de = 99 %) eine
Mischfraktion der Diastereomere im Verhältnis 4:1 zugunsten der Reinfraktion erhalten
wurde. Durch die Verwendung der Methyl-Schutzgruppe konnte der A´-Ring erfolgreich
geschlossen werden. Hieraus ist erkennbar, dass die Schützung der Hydroxyfunktion
zwingend notwendig ist, da ohne diese nicht das gewünschte Produkt gebildet werden kann,
sondern das Lacton 114 (Abb. 58) entsteht. Doch ist der Einsatz eines Methylethers als
Schutzgruppe durchaus problematisch, da sich diese Schutzgruppe nur unter harschen
Reaktionsbedingungen wieder entfernen lässt und dadurch eine orthogonale Entschützung der
Hydroxygruppe unmöglich ist.
EDAC; HOBtTHF; 101; RT; 16h
Boc2N CO2tBu
MeON3
O
Thr Gly
Leu Phe OMe
Boc2N CO2tBu
MeO
O
Leu Phe OMeH2N
Boc2N CO2tBu
OMe
O
Leu Phe OMeHNN3
OBn
Pd/C; H2EtOAc; RT; 16h
99; THF; HATUDIPEA; RT; 2d
1.TFA; TES 8h; RT2.Boc2O; TEA MeCN; RT 16h
PPh3-pbH2O; THF
RT; 4d
Thr Gly
BocHN CO2H
OMe
O
Leu Phe OMeHNH2N
OBnOMe
Boc
OBn
HATU DIPEA; THF
RT; 2d
120
121 122
125 126
LabThr Gly
CH2 Leu AlaPhe
OMe
LabThr Gly
Boc2N CO2tBu
CO2H
MeON3
119
Thr Gly
BocHN CO2H
OMe
O
Leu Phe OMeHNN3
OBn
124
62% 65%
73%
94% über 2 Stufen
Abbildung 61: Synthese des A´-Ringes unter Verwendung des Methylethers 119.

Ergebnisse und Diskussion
56
Durch den Einsatz von geschütztem Labionin (Abb. 50) in der Peptidsynthese könnte dieses
Problem umgangen werden, doch momentan ist, wie bereits in Kapitel 3.2 dargestellt, keine
Labioninsynthese etabliert. Die hier gezeigte erfolgreiche Synthese des A´-Rings bietet jedoch
die Voraussetzung für die Totalsynthese des Labyrinthopeptins. Der A-Ring von
Labyrinthopeptin A2 enthält aber im Vergleich zum A´-Ring raumerfüllendere Aminosäuren
unter anderem die Aminosäure Tryptophan, die leicht Nebenreaktionen wie eine Alkylierung
und Oxidation des Indolrings eingeht[2]. Aus diesen Gründen ist der A´-Ring bzw. das A´B´-
Fragment leichter synthetisch handhabbar als der A-Ring bzw. das AB-Fragment.

Ergebnisse und Diskussion
57
3.4 Synthese eines analytischen Referenzstandards zum Labioninnachweis
Die Aminosäure Labionin (Abb. 14) wurde bisher nur im Labyrinthopeptin A2 durch
Röntgenstrukturanalyse nachgewiesen. Sie wird jedoch auch in anderen Typ III-Lantibiotika
vermutet (s. Kapitel 1.3.3). Eine Methode zur Labioninbestimmung in Peptiden ist der direkte
Nachweis von Labionin im Totalhydrolysat (Abb. 62) des Peptids mittels
Massenspektrometrie. Ein direkter Nachweis von Labionin durch LC-MS konnte bisher nicht
erbracht werden. Dafür gibt es mehrere mögliche Ursachen, zum einen kann die Aminosäure
Labionin durch seine jeweils drei Amino- und Carboxygruppen mehrere Ladungen tragen,
wodurch LC-MS-Messungen negativ beeinflusst werden können, da es aufgrund der
genannten funktionellen Gruppen als einfach, zweifach und dreifach geladenes Ion vorliegen
kann. Das Wert des dreifach geladenen Ions von Labionin im Positivionenmodus beträgt
99,5 m/z, die Detektion solch kleiner und hochgeladenen Massen im LC-MS erweist sich als
schwierig. Zum anderen kann das Schwefelatom der Lanthioninbrücke oxidiert vorliegen,
wodurch die Intensität der Messung ebenfalls negativ beeinflusst wird. Der direkte Nachweis
von Labionin aus dem Hydrolysat des Peptids kann aber auch mit GC-MS erfolgen. Der
Vorteil der GC-MS gegenüber der LC-MS besteht in der höheren Empfindlichkeit und der
Verminderung von Mehrfachladungen. Für die GC-MS-Analyse müssen die Aminosäuren des
Hydrolysats am N-Terminus trifluoracetyliert und am C-Terminus verestert werden. Für den
exakten Nachweis ist jedoch die Verwendung eines Referenzstandards notwendig. Mit der
GC-MS lässt sich auch die Stereochemie von Aminosäuren bestimmen. Dazu muss jedoch
eine chirale Trennsäule benutzt werden und Referenzaminosäuren mit bekannter
Stereochemie vorhanden sein. Nach einer Optimierung der Trennmethode ist es möglich die
Stereoisomere der entsprechenden Aminosäuren voneinander zu trennen und somit sicher
deren Stereochemie zu bestimmen. Da bisher die Synthese von Labionin nicht verwirklicht
werden konnte und Labionin somit nicht als Referenzstandard verwendet werden kann,
erschien es notwendig die Aminosäure Labionin im Peptid durch eine Abbaureaktion in eine
synthetisch leichter zugängliche und besser analysierbare Verbindung zu überführen. Eine
geeignete Abbaureaktion stellt die Desulfurierung des labioninhaltigen Peptids dar (Abb.
62)[42, 143]. Durch diese Reaktion wird Labionin in 2,4-Diamino-2-methylglutarsäure
umgewandelt, die das Ziel der Synthese des Referenzstandards ist.
Ergebnisse und Diskussion
58
Abbildung 62: Hydrolyse eines labioninhaltigen Peptids und Hydrolyse eines labioninhaltigen Peptids nach einer Desulfurierung, mit den jeweils dabei resultierenden Aminosäuren des Hydrolysats (Xxx kennzeichnet beliebige Aminosäuren; R entspricht variablen Aminosäurenseitenketten). Ausgangsmaterial für die Synthese des Refenzstandards ist Cbz-α-Allylalanin 129 das durch
eine chelatverbrückte Claisen-Umlagerung aus Cbz-Ala-OAll 128 (Abb. 63) synthetisiert
wurde. Nun wurde durch eine Sharpless Asymmetrische Dihydroxylierung das Diol 131
synthetisiert und im folgenden Schritt wurde die endständige Hydroxygruppe mit TBDMS
geschützt. Nun konnten die Diastereomere, die aus dem razemischen Cbz-AllylAla-OtBu
(130) entstanden, voneinander getrennt werden (Abb. 63). Eine Analyse der TBDMS-Ether
(+/-132a & b; Abb. 64) auf der chiralen HPLC zeigte, dass die asymmetrische
Dihydroxylierung mit AD-Mixβ in dieser Reaktion zu einem razemischen Gemisch führte, da
zwei Peaks mit nahezu gleicher Intensität beobachtet wurden (Abb. 63).
Abbildung 63: Chromatogramm des TBDMS-Ethers +/- 132a auf der chiralen HPLC, die zwei Signale zeigen das die Verbindung als eine Mischungen aus Enantiomeren vorliegt.
Retentionszeit (min)
Abs
orbt
ion
(AU
)

Ergebnisse und Diskussion
59
Bei der Verwendung vom AD-Mixα für die Dihydroxylierung zeigten die entsprechenden
TBDMS-Ether das gleiche Verhalten auf der chiralen HPLC. Die mangelnde
Stereoselektivität der Sharpless Dihydroxylierung an ähnlichen Substraten ist in der Literatur
beschrieben[144, 145], doch ist die Ursache der geringen Stereoselektivtät bisher unbekannt[146].
Da, wie gesagt, die Diastereomerenpaare voneinander getrennt werden konnten, wurde diese
als Enantiomerenpaar weiter in der Synthese verwendet (Abb. 64). Die sekundäre
Hydroxygruppe wurde in ein Azid umgewandelt, der TBDMS-Ether gespalten und der
entstehende Alkohol zur Säure oxidiert. Nun wurde die tert-Butyl-Schutzgruppe entfernt und
eine Hydrogenolyse durchgeführt (Abb. 64). Bei der Hydrogenolyse der Verbindungen
+/-137a bzw. +/-137b traten eine Reihe von Nebenreaktionen auf. Die Zuordnung der
Stereochemie war durch den Vergleich der Retentionszeiten der synthetischen Verbindungen
+/-137a bzw. +/-137b, mit der Retentionszeit von (2R/4S)-2,4-Diamino-2-methylglutarsäure
möglich, welches aus dem Totalhydrolysat des desulfurierten Labyrinthopeptin A2 gewonnen
werden konnte. Eine Reinigung konnte bisher nur durch präparative DC und anschließende
Extraktion des Kieselgels mit Methanol erfolgen, wobei große Mengen des Kieselgels
mitextrahiert wurden. Eine Trennung über eine C18-Säule zeigte keine Wirkung, da das
Produkt wie auch die Nebenprodukte sehr polare Verbindungen sind, die keine
Wechselwirkung mit C18-Trennmaterial eingehen. Theoretisch sollte die Reinigung dieser
Substanzen mit einer HILIC-Säule möglich sein, da dieses Säulenmaterial speziell für die
Trennung sehr polarer Substanzen wie Aminosäuren entwickelt wurde[147-149]. Eine
enantiomerenreine Synthese des Standards ist möglich, wenn man von enantiomerenreinem
Cbz-α-Allylalanin ausgeht und die in Abb. 74 gezeigte Syntheseroute verwendet. Die
Dihydroxylierung verläuft zwar nicht stereoselektiv, doch lassen sich die Diastereomere gut
als TBDMS-Ether von einander trennen. Die Verbindungen +/-137a und. +/-137b werden
zurzeit innerhalb der Arbeitsgruppe zum Labioninnachweis verwendet.
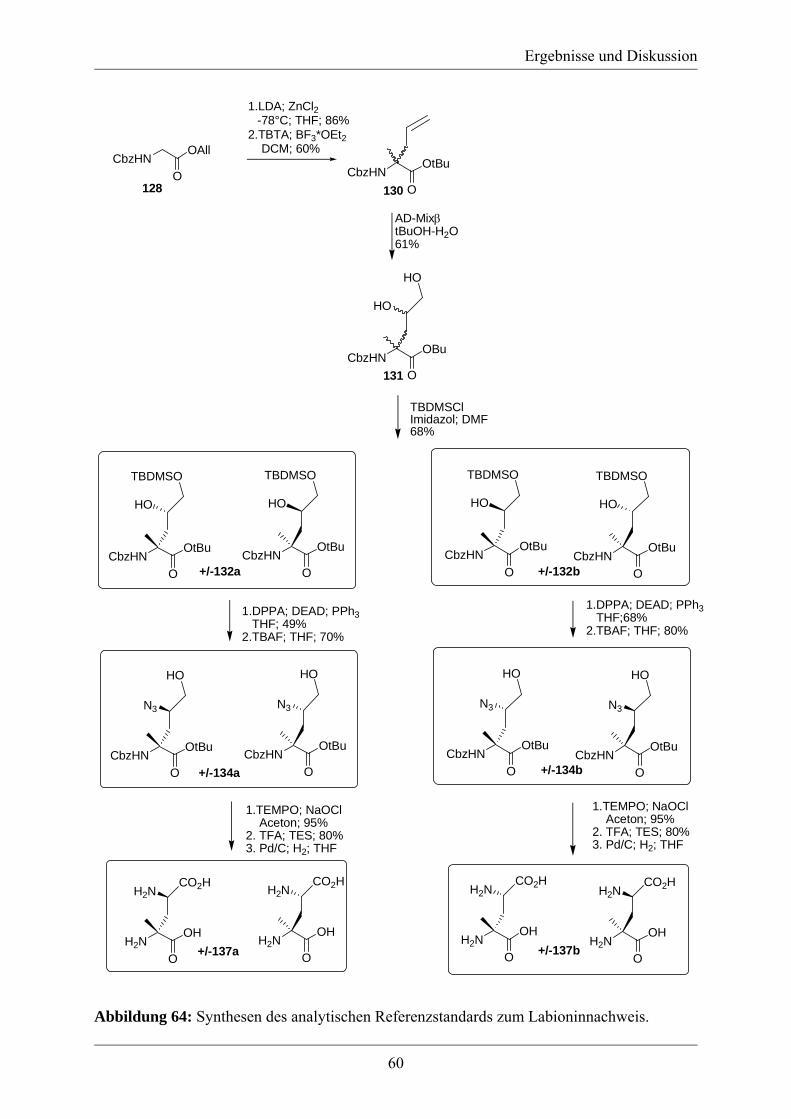
Ergebnisse und Diskussion
60
CbzHNOtBu
O
CbzHNOBu
O
HO
CbzHNOtBu
O
TBDMSO
HO
HO
TBDMSClImidazol; DMF68%
CbzHNOtBu
O
TBDMSO
HO
1.DPPA; DEAD; PPh3 THF; 49%2.TBAF; THF; 70%
1.TEMPO; NaOCl Aceton; 95%2. TFA; TES; 80%3. Pd/C; H2; THF
CbzHNOtBu
O
TBDMSO
HO
CbzHNOtBu
O
TBDMSO
HO
1.DPPA; DEAD; PPh3 THF;68%2.TBAF; THF; 80%
AD-MixβtBuOH-H2O61%
CbzHNOtBu
O
HO
CbzHNOtBu
O
HO
CbzHNOtBu
O
HO
CbzHNOtBu
O
HO
N3N3 N3N3
H2NOH
O
CO2H
H2NOH
O
CO2H
H2NOH
O
CO2H
H2NOH
O
CO2HH2NH2N H2NH2N
1.TEMPO; NaOCl Aceton; 95%2. TFA; TES; 80%3. Pd/C; H2; THF
131
130
+/-132a +/-132b
+/-134a +/-134b
+/-137a +/-137b
CbzHNOAll
O
1.LDA; ZnCl2 -78°C; THF; 86%2.TBTA; BF3*OEt2 DCM; 60%
128
Abbildung 64: Synthesen des analytischen Referenzstandards zum Labioninnachweis.

Ergebnisse und Diskussion
61
3.5 Derivatisierung der Naturstoffe Labyrinthopeptin A1 und A2
Durch die Derivatisierung von biologisch aktiven Naturstoffen und die dadurch im Molekül
hervorgerufenen Änderungen der Struktur, der Ladung oder der Löslichkeit besteht eine
Möglichkeit die Aktivität des betreffenden Naturstoffs zu erhöhen, seine Wirkung besser zu
verstehen und für die Aktivität notwendige Strukturen des Naturstoffs zu erkennen. Im
Rahmen dieser Arbeit wurde Derivatisierungen ausgehend von Labyrinthopeptin A1 und A2
vorgenommen. Diese umfassen eine selektive Desulfurierung der B-Ringe von
Labyrinthopeptin A1 und A2 und führen so zu einer Strukturänderung in den Peptiden (Abb.
65). Diese Reaktion ist ebenfalls der Schlüsselschritt zum Labioninnachweis mit dem im
vorhergehenden Kapitel beschriebenen Aminosäurestandard. Die Desulfurierung kann durch
die Reaktion mit NiCl2 und NaBH4 erfolgen[42, 143]. Dabei ist es von Vorteil nur kleine
Mengen, in diesem Fall 1 mg Substanz, je Reaktionsansatz zu verwenden, da eine
Vergrößerung des Ansatzes die Bildung von nicht charakterisierten Nebenprodukten
begünstigt. Im Fall von Labyrinthopeptin A2 bietet sich eine Desulfurierung mit Raney-
Nickel an, da die Reaktion mit diesem Reagenz hier zu weniger Nebenprodukten führt,
während die Reaktion von Labyrinthopeptin A1 mit Raney-Nickel nicht zum gewünschten
Reaktionsprodukt führte.
Abbildung 65: Derivatisierung von Labyrinthopeptin A2 durch Desulfurierung und NTCB-Spaltung. Durch eine Spaltung mit dem Reagenz NTCB[150] (der Mechanismus dieser Reaktion ist in
Abb. 66 gezeigt) konnte nach einer Öffnung der Disulfidbrücke das AB- und das A´B´-
Fragment von Labyrinthopeptin A2 erzeugt werden. Durch die NTCB-Spaltung von
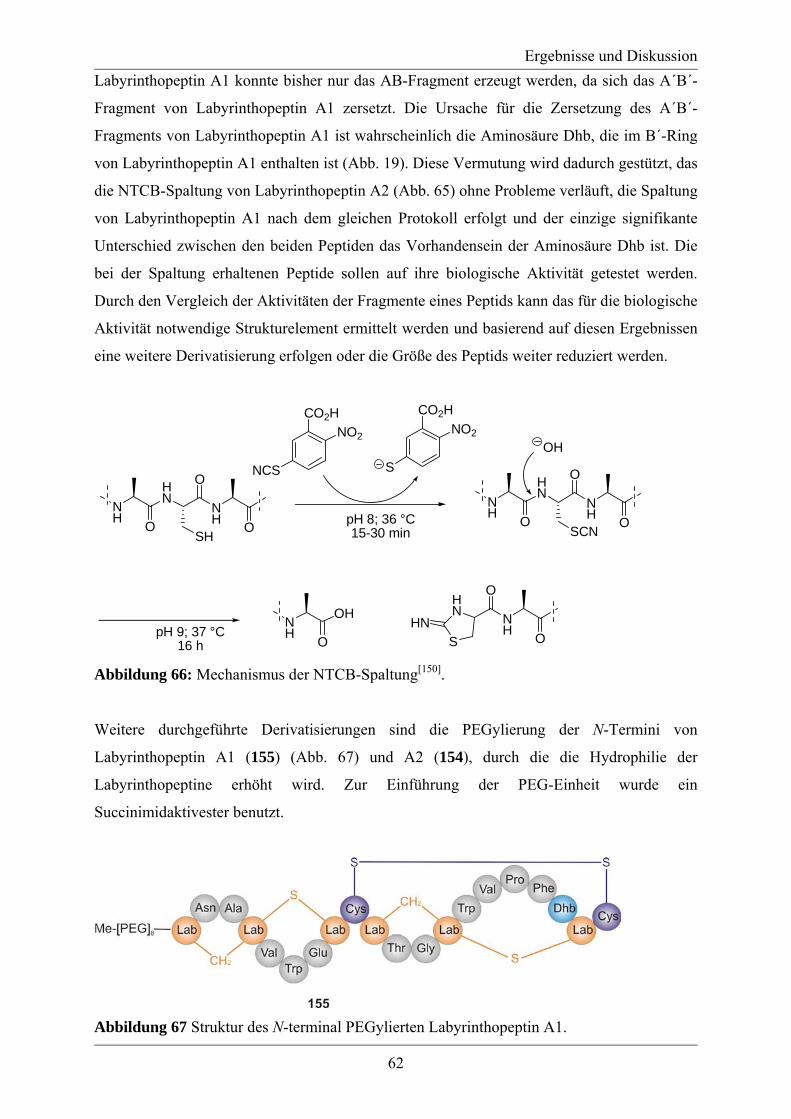
Ergebnisse und Diskussion
62
Labyrinthopeptin A1 konnte bisher nur das AB-Fragment erzeugt werden, da sich das A´B´-
Fragment von Labyrinthopeptin A1 zersetzt. Die Ursache für die Zersetzung des A´B´-
Fragments von Labyrinthopeptin A1 ist wahrscheinlich die Aminosäure Dhb, die im B´-Ring
von Labyrinthopeptin A1 enthalten ist (Abb. 19). Diese Vermutung wird dadurch gestützt, das
die NTCB-Spaltung von Labyrinthopeptin A2 (Abb. 65) ohne Probleme verläuft, die Spaltung
von Labyrinthopeptin A1 nach dem gleichen Protokoll erfolgt und der einzige signifikante
Unterschied zwischen den beiden Peptiden das Vorhandensein der Aminosäure Dhb ist. Die
bei der Spaltung erhaltenen Peptide sollen auf ihre biologische Aktivität getestet werden.
Durch den Vergleich der Aktivitäten der Fragmente eines Peptids kann das für die biologische
Aktivität notwendige Strukturelement ermittelt werden und basierend auf diesen Ergebnissen
eine weitere Derivatisierung erfolgen oder die Größe des Peptids weiter reduziert werden.
O
HN
NH
O
OSH
NH
NCS
CO2HNO2
S
CO2HNO2
O
HN
NH
O
OSCN
NH
OH
O
OHNH
NH
O
O
HN
SHN
pH 8; 36 °C15-30 min
pH 9; 37 °C16 h
Abbildung 66: Mechanismus der NTCB-Spaltung[150].
Weitere durchgeführte Derivatisierungen sind die PEGylierung der N-Termini von
Labyrinthopeptin A1 (155) (Abb. 67) und A2 (154), durch die die Hydrophilie der
Labyrinthopeptine erhöht wird. Zur Einführung der PEG-Einheit wurde ein
Succinimidaktivester benutzt.
Abbildung 67 Struktur des N-terminal PEGylierten Labyrinthopeptin A1.

Ergebnisse und Diskussion
63
Abbildung 68: Struktur des Dimers von Labyrinthopeptin A2. Die Synthese von Wirkstoffdimeren ist eine weit verbreitete Methode in der
Medizinalchemie[151-153]. Für die Synthese eines Labyrinthopeptindimers (Abb. 68) war dabei
die Synthese des Linkers 140 notwendig (Abb. 69). Bedingt durch die positiven Erfahrungen
der Verwendung von Succinimidaktivestern bei der PEGylierung der Labyrinthopeptine A1
und A2, sollte der Linker ebenfalls als Succinimidaktivester (141) eingesetzt werden. Doch
das gewünschte Produkt 141 konnte nicht isoliert werden und somit wurde die Synthese eines
Labyrinthopeptindimers verworfen.
O
O TFA; DCMRT; 4h
OO O
O6
NN
O
O
O
O
A. HOSu; EDAC; DCM DIPEA; RT; 16hB. HOSu; C2O2Cl2 Pyridin; THF; DCM DMF; RT; 2h
Br OO
OO
O6
O
HOO
OOH
O6
O
Hexaethylenglycol; KOHToluol; Bu4NHSO4; RT; 1h
44% 99%139
140 141
138
O
O
X
Abbildung 69: Synthese eines Linkers zur Erzeugung von Labyrinthopeptindimeren.
Weitere Derivatisierungsversuche wurden an der Doppelbindung des Dhbs im
Labyrinthopeptin A1 vorgenommen (Abb. 70). Dabei sollte die Doppelbindung analog zu der
in der Literatur beschriebenen Hydrierungen von Dha bzw. Dhb in anderen Peptiden [154, 155]
hydriert werden (Abb. 70) oder durch den Angriff eines Schwefelnukleophils die
Doppelbindung beseitigt werden[156]. Doch alle diese Versuche scheiterten. Ein möglicher
Grund könnte die Abschirmung der Doppelbindung durch die umgebenden Aminosäuren sein.
Des Weiteren wurde ein Versuch durchgeführt, in dem die Disulfidbrücke von
Labyrinthopeptin A2 in eine Lanthioninbrücke (Abb. 70) durch Monodesulfurierung in
alkalischer Umgebung[56, 157] umgewandelt werden sollte. Dabei wurde das gewünschte
Produkt (163) aber nur in Spuren gebildet und eine Isolierung ist auf Grund des geringen
Unterschieds in der Retentionszeit auf der HPLC nicht möglich. Durch die Verwendung von
Phosphiden[54] in dieser Reaktion kann die Ausbeute vielleicht erhöht werden.

Ergebnisse und Diskussion
64
Abbildung 70: Struktur des C-Ring-Lanthionin Analoga von Labyrinthopeptin A2 (163) und des Hydrierungsprodukt von Labyrinthopeptin A1 (164), die synthetisch nicht darstellbar waren.

Zusammenfassung und Ausblick
65
4. Zusammenfassung und Ausblick
Labyrinthopeptin A2 wurde 1988 im Rahmen eines von der Firma Sanofi-Aventis
durchgeführten Screeningprogramms aus dem Aktinomyzeten Actinomadura namibiensis DM
6313 isoliert[47]. Die Strukturaufklärung dieses Naturstoffs gelang erst im Rahmen einer
Kooperation der Arbeitsgruppen Süßmuth (TU-Berlin) und Sheldrick (Universität
Göttingen)[45]. Labyrinthopeptin A2 weist neben einer Disulfidbrücke und zwei
typischerweise in Lantibiotika vorkommenden Lanthioninbrücken eine strukturelle
Besonderheit auf, die als Labionin bezeichnet wurde. Diese Aminosäure weist eine
Methylenverbrückung zu einem Glycinrest auf, ausgehend von einem α,α-disubstituierten
Kohlenstoffatom, das wiederum die bereits erwähnte Lanthioninbrücke bildet. Diese Brücken
bilden, zusätzlich zur Disulfidbrücke, vier Ringe im Labyrinthopeptin A2, da die Aminosäure
Labionin zweimal im Naturstoff auftritt. Die durch die Methylenbrücken gebildeten A-Ringe
und durch die Thioetherbrücken gebildeten B-Ringe werden N-terminal als Ring A und B
bezeichnet, während sie C-terminal Ring A´ und B´ genannt und von einer Disulfidbrücke
überspannt werden. Durch die Kenntnis der Struktur konnte der Biosynthesegencluster[45] von
Labyrinthopeptin A2 und später auch dessen Biosynthese[50] in unserem Arbeitskreis
aufgeklärt werden.
Von einer anderen Klasse von Peptiden, den Conotoxinen, ist eine analgetische Wirkung
bekannt. Aus diesem Grund wurde Labyrinthopeptin durch die Firma Sanofi-Aventis auf
diese Eigenschaft getestet, wobei sich zeigte dass es zur Behandlung neurophatischer
Schmerzen geeignet ist. Derzeit wird Labyrinthopeptin in der präklinischen Phase als
Analgetikum durch Sanofi-Aventis untersucht.
Aufgrund dieser interessanten Eigenschaft von Labyrinthopeptin A2 und seiner
außerordentlichen Struktur sind die Synthese von Labyrinthopeptin und die Erzeugung von
Derivaten von großem Interesse. Im Rahmen dieser Arbeit wurden umfangreiche Studien zur
Synthese von Vorläufern des Labyrinthopeptins durchgeführt (Abb. 71). So konnte die
geschützte quartäre Aminosäure (2R,4S)-5-tButyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-
(hydroxymethyl)pentandioat (73), die Ausgangsverbindung zur Synthese des zentrale
Strukturmotivs im Labyrinthopeptin A2, Labionin, nach mehrmaliger Änderung der
Synthesestrategie, ausgehend von Glycin über 10 Stufen in einer Ausbeute von 12,8 %
synthetisieren werden (Abb. 71). Dabei handelt es sich um die erste Synthese einer derart
komplexen Aminosäure. Ausgehend von dieser Verbindung konnte nun die Synthese des A´-
Rings von Labyrinthopeptin A2 begonnen werden. Da es sich bei den A-Ringen des

Zusammenfassung und Ausblick
66
Labyrinthopeptin A2 um gespannte Ringe handelt, die eine cis-Amidbindung beinhalten[45],
wurde die Zyklisierung vorher an einem Modellpeptid untersucht, um die Notwendigkeit
eines Einsatzes einer N-alkylierten Aminosäure zu überprüfen. Nach einer Korrektur der
Zyklisierungsrichtung und der zusätzlichen Schützung der freien Hydroxygruppe von 73 war
nun die erfolgreiche Synthese des A´-Ringes möglich (Abb. 71). Des Weiteren konnte die
quartäre Aminosäure 73 unabhängig von dieser Arbeit innerhalb des Arbeitskreises zur
erfolgreichen Synthese des A-Ringes von Labyrinthopeptin A2 genutzt werden. Aufbauend
auf die in dieser Arbeit vorgestellte Synthese von 73 und die folgende Synthese des A´-
Ringes kann nun die Totalsynthese von Labyrinthopeptin A2 und weiterer möglicher
labioninhaltiger Peptide begonnen werden.
Für die Synthese der Aminosäure Labionin wurden im Rahmen dieser Arbeit zahlreiche
Versuche ausgehend von 73 durchgeführt. Diese Versuche blieben ohne Erfolg, da die
Transformation der Hydroxygruppe von 73 in eine geeignete Abgangsgruppe nicht möglich
war. Dieses Problem soll in Zukunft durch die Transformation von 73 in ein
β-Lacton[124], ein Aziridin[124, 126] oder in ein Sulfamidat[124, 126] überwunden werden.
Da sich die Synthese der Aminosäure Labionin als schwierig erwies, aber eine
Standardsubstanz zum Nachweis von Labionin in anderen Peptiden synthetisiert werden
sollte, wurde das Desulfurierungsprodukt von Labionin, das der 2,4-Diamino-2-
methylglutarsäure entspricht, synthetisiert. Die Desulfurierung von Peptiden ist eine gängige
Methode zur Sequenzierung von Lantibiotika und wurde verwendet, um das Labionin im
Peptid zu 2,4-Diamino-2-methylglutarsäure abzubauen und nach Hydrolyse des Peptids
nachzuweisen. Zur sicheren Bestimmung von Labionin bzw. dessen Abbauprodukt der
2,4-Diamino-2-methylglutarsäure ist die Verwendung einer Referenzsubstanz notwendig. Die
Synthese des Standards erfolgte über eine chelatverbrückte Claisen-Umlagerung[98] ausgehend
von Cbz-Gly-OAll. Das bei dieser Reaktion gebildete razemische Cbz-α-Allylalanin wurde
nach einer Schützung des C-Terminus nach Sharpless Asymmetrischer Dihydroxylierung in
das entsprechende Diol transformiert. Die Asymmetrische Dihydroxylierung zeigte bei
diesem Substrat jedoch keine Stereoselektivität, sodass nach der Reaktion vier Stereoisomere
gebildet wurden, bei denen sich die Diastereomere auf der folgenden Stufe trennen ließen. Die
Enantiomerenpaare wurden in weiterfolgenden Reaktionen eingesetzt und die jeweilige +/-
2,4-Diamino-2-methylglutarsäure 137a/b konnte erfolgreich synthetisiert werde (Abb. 71).
Diese Standardsubstanz wird zurzeit innerhalb der Arbeitsgruppe zum Nachweis von
Labionin in anderen Peptiden verwendet.
Des Weiteren wurde eine Vielzahl von Modifikationen an den Naturstoffen Labyrinthopeptin
A1 und A2 vorgenommen (Abb. 71). So konnten die B-Ringe von Labyrinthopeptin A1 und

Zusammenfassung und Ausblick
67
A2 desulfuriert werden (Abb. 71) und somit eine Änderung der Konformation vorgenommen
werden. Ferner konnten beide Peptide N-terminal PEGylierten werden (Abb. 71) und somit
die Polarität dieser Moleküle verändert werden. Außerdem war eine Spaltung der Peptide an
den Cycteinen mit dem Reagenz NTCB möglich (Abb. 71), wodurch die Fragmente AB und
A´B´ von Labyrinthopeptin A2 synthetisiert wurden. Bei der Spaltung von Labyrinthopeptin
A1 konnte nur des AB-Fragment isoliert werden, da sich das A´B´-Fragment während der
Reaktion zersetzte. Durch diese Spaltung sollen die jeweiligen Fragmente in künftigen
Studien auf ihre biologische Aktivität getestet werden, um das für die analgetische Wirkung
relevante Strukturmotiv im Labyrinthopeptin zu bestimmen.

Zusammenfassung und Ausblick
68
Abb
ildun
g 71
: Sc
hem
atis
che
Dar
stel
lung
der
Erg
ebni
sse
dies
er D
isse
rtatio
n. I
n de
r A
bbild
ung
oben
link
s is
t die
Syn
thes
e de
r qu
artä
ren
Am
inos
äure
73
au
s de
r Sc
hiff
sche
n B
ase
62
darg
este
llt.
Die
se
Ver
bind
ung
bild
ete
die
Gru
ndla
ge
für
die
Synt
hese
de
s A
´-R
ings
von
Lab
yrin
thop
eptin
A2.
Obe
n re
chts
sin
d di
e sy
nthe
tisie
rten
anal
ytis
chen
Ref
eren
zsta
ndar
ds d
arge
stel
lt, d
ie z
um N
achw
eis
der
Am
inos
äure
Lab
ioni
n in
and
eren
Kla
sse-
III-L
antib
iotik
a ve
rwen
det w
erde
n. U
nten
in d
er A
bbild
ung
sind
die
Der
ivat
e vo
n La
byrin
thop
eptin
A
1 un
d A
2 ab
gebi
ldet
, die
im R
ahm
en d
iese
r Arb
eit d
arge
stel
lt w
urde
n un
d fü
r Stru
ktur
-Akt
ivitä
ts-S
tudi
en v
erw
ende
t wer
den
solle
n.

Experimenteller Teil
69
5. Experimenteller Teil
5.1. Allgemeine Informationen
Arbeitstechniken:
Arbeiten mit luft- und wasserempfindlichen Verbindungen wurden unter Schutzgas
durchgeführt (Argon bzw. Stickstoff). Hierzu wurde das Reaktionsgefäß durch Erhitzen mit
der Heizpistole von Wasser befreit und unter Schutzgas gesetzt. Feststoffe wurden dem
Reaktionsgefäß im Gegenstrom zugegeben, Lösungsmittel (abs.) und flüssige Reaktanden mit
Spritzen durch ein Septum.
Chemikalien:
Die Chemikalien wurden von den Firmen Karl Roth GmbH und Co. Kg (Karlsruhe), (Sigma-
Aldrich (Taufkirchen), Iris Biotech GmbH (Marktredwitz), Orpegen (Heidelberg), ABCR
(Karlsruhe), Alfa Aesar (Karlsruhe), Merck (Darmstadt) und Acros (Geel, Belgien) bezogen
und ohne weitere Aufreinigung eingesetzt.
Lösungsmittel wurden von der Firma Acros (Geel, Belgien) bezogen und wasserfrei, wie
gekauft verwendet.
Als deuterierte Lösungsmittel für die NMR-Spektroskopie wurden Chloroform-d1 (99.8 %),
Methanol-d4 (99,8%), Wasser-d2 und Dimethylsulfoxid-d6 der Firma DEUTERO GmbH
(Kastellaun) verwendet.
Chromatographie:
Dünnschichtchromatogramme (DC) wurden mit kieselgelbeschichteten Aluminiumfolien der
Firma Merck (Darmstadt) (TLC Kieselgel 60, F254) aufgenommen. Verbindungen wurden
entweder mit UV-Licht einer Wellenlänge von λ = 254 nm oder durch Eintauchen in
Permanganat-Reagenz (3 g KMnO4, 20 g K2CO3, 300 ml dest. Wasser, 5 ml NaOH-Lsg.
(5 %)) mit anschließendem Erhitzen unter Zuhilfenahme einer Heizpistole sichtbar gemacht.
Die Säulenchromatographie wurde mit Kieselgel der Firma Acros (Geel, Belgien)
(0.04-0.064 mm 60) durchgeführt. Eluiert wurde unter erhöhtem Druck (N2) mit
unterschiedlichen Laufmittelgemischen.

Experimenteller Teil
70
Analysenmethoden: 1H-NMR-Spektren wurden mit dem Gerät AM 400 (400 MHz) der Firma Bruker (Karlsruhe)
aufgenommen. Als Lösungsmittel dienten Deuterochloroform (CDCl3), deuteriertes Methanol
(MeOD-d4), deuteriertes Wasser-d2 und deuteriertes DMSO (DMSO-d6). Die chemischen
Verschiebungen sind in δ-Werten (ppm) angegeben. Als Referenz wurde das Restsignal des
undeuterierten Lösungsmittels verwendet. Kopplungskonstanten J sind in Hertz (Hz)
angegeben. Signalmultiziplitäten sind wie folgt gekennzeichnet: (s) Singulett, (d) Duplett,
(dd) Duplett von Duplett, (t) Triplett, (dt) Duplett von Triplett, (td) Triplett von Duplett, (q)
Quartett, (dq) Duplett von Quartett, (m) Multiplett. Die Spektren wurden, sofern nicht anders
angegeben bei 298 K aufgenommen. Zur Auswertung der Spektren wurde das Programm
ACDLabs 5 (Toronto, Kanada) verwendet. 13C-NMR-Spektren wurden mit dem Gerät AM 400 (100 MHz) der Firma Bruker (Karlsruhe) 1H-Breitband entkoppelt aufgenommen. Als Lösungsmittel dienten Deuterochloroform
(CDCl3), deuteriertes Methanol (MeOD-d4), deuteriertes Wasser-d2 und deuteriertes DMSO
(DMSO-d6). Die chemischen Verschiebungen sind in δ-Werten (ppm) angegeben. Als
Referenz wurde das Restsignal des undeuterierten Lösungsmittels verwendet. Die
Zuordnungen der Signale wurden durch DEPT-135 und APT-Experimente ermittelt. Die
Spektren wurden, sofern nicht anders angegeben bei 298 K aufgenommen. Zur Auswertung
der Spektren wurde das Programm ACDLabs 5 verwendet.
MS-Spektren (auch Hochauflösung) wurden auf einem Finnigan MAT 95S aufgenommen.
Die Ionisierung erfolgte durch Elektronenstoß (EI) mit einem Ionisierungspotential von
70 eV.
HPLC-MS-Kopplungen wurden mit dem ESI-Massenspektrometer (electro spray ionisation)
QTrap 2000 ESI-Quadrupol-MS (Niederauflösung) der Firma Applied Biosystems (Forster
City, USA) in Kopplung mit einem Agilent 1200 Series HPLC-System der Firma Agilent
Technologies (Waldbronn) unter Verwendung einer RP-C18-Säule der Fima Phenomenex
(Aschaffenburg) vorgenommen (Lösungsmittel A: Wasser / 0,1 % HCOOH, Lösungsmittel B:
Acetonitril / 0,1 % HCOOH, Flussrate: 1,5 ml/min). Alternativ wurde eine Orbitrap LTX
(Hochauflösung) der Firma Thermo Scientific (Waltham, USA) in Kopplung mit einem
Agilent 1200 Series HPLC-System der Firma Agilent Technologies (Waldbronn) unter
Verwendung einer Hypersil-100 C18-Säule der Firma Thermo Scientific (Waltham, USA)
verwendet (Lösungsmittel A: Wasser / 0,025 % HCOOH, Lösungsmittel B: Methanol / 0,025
% HCOOH, Flussrate: 1,3 ml/min). Zur Auswertung der Spektren wurden die Programme
Xcalibur und Analyst verwendet.

Experimenteller Teil
71
GC-MS-Messungen wurden an einem 5975C der Firma Agilent Technologies (Waldbronn)
aufgenommen. Es wurde in einem Scanbereich von 50-800 amu, einer Detektionstemperatur
von 150 °C und einer Quellentemperatur von 300 °C gearbeitet. Die Ionisierungsenergie
betrug 91 eV. Als Trägergas wurde Helium verwendet. Als Säule wurde eine Agilent 190913-
433 der Firma Agilent Technologies (Waldbronn) eingesetzt (30 m, ID 0,25 mm, Phase: 5%
Phenyl methyl siloxan).
Chirale GC-MS-Messungen wurden an einem GC800 Top Voyager der Firma ThermoQuest
(Rodano, Italien) aufgenommen. Es wurde in einem Scanbereich von 45-465 amu, einer
Detektionstemperatur von 210 °C und einer Quellentemperatur von 220 °C gearbeitet. Die
Injektionstemperatur betrug 250 °C. Die Detektorspannung betrug 350 V mit einer Energie
von 70 eV. Als Trägergas wurde Helium verwendet. Als chirale Säule wurde eine FSLipodex
E der Firma Macherey-Nagel GmbH & Co. KG (Düren) eingesetzt (25 m, ID 0,25 mm, Phase:
Octakis-(2,6-di-O-pentyl-3-O-butyryl)-γ-cyclodextrin).
Drehwerte wurden mit einem Polarimeter P-2000 Series der Firma Jasco Labor- und
Datentechnik GmbH (Groß-Umstadt) aufgenommen. Als Lösungsmittel dienten
Dichlormethan (DCM) und Ethylacetat (EtOAc). Die Drehwerte wurden bei 298 K und den
jeweils angegeben Konzentrationen aufgenommen.
Flash-Chromatographie wurde mit CombiFlash Rf der Firma Teledyne ISCO (Lincoln, USA)
durchgeführt. Als Lösungsmittel wurden Ethylacetat (EtOAc) und Hexan (Hex) verwendet.
Als Säulenmaterial wurden RediSep Rf-Kartuschen der Firma Teledyne ISCO (Lincoln, USA)
verwendet.
Präparative RP-HPLC-Trennungen wurden auf einer Agilent HP 1100 HPLC-Anlage mit UV-
Detektor (Beobachtungswellenlänge: λ = 280 nm bzw. 210 nm) der Firma Agilent
Technologies (Waldbronn) unter Verwendung der RP-C18-Säule Grom-Sil 300 ODS-5 ST
der Firma Grom (Rottenburg-Hailfingen) durchgeführt. Als Lösungsmittelsysteme wurden
Wasser (Lösungsmittel A) und Acetonitril (Lösungsmittel B) bei einer Flussrate von
15 ml/min verwendet. Die jeweiligen Gradienten sind an entsprechender Stelle angegeben
(Auswertungssoftware: Agilent ChemStation).
Die HPLC-Analytik wurde mit einer analytischen 1100-HPLC-Anlage der Firma Agilent
Technologies (Waldbronn) mit DAD-Detektor (Beobachtungswellenlänge: λ = 280 nm bzw.
210 nm) und einer Luna RP-C18-Säule der Firma Phenomenex (Aschaffenburg) durchgeführt
(Lösungsmittel A: Wasser; Lösungsmittel Methanol; Flussrate: 1,5 ml/min; Gradient: 0 min 5
% B → 10 min 100 % B → 12 min 100 % B → 12.1 min 5 % B → 15 min 5 % B;
Auswertungssoftware: Agilent ChemStation).

Experimenteller Teil
72
Die chirale HPLC-Analytik wurde an einem Merck-Hitachi LaChrom System der Firma
Hitachi (Tokyo, Japan) unter Verwendung einer CHIRALPAK AD-H- oder OD-H-Säule der
Firma Chiral Technologies Europe (Illkirch, Frankreich) mittels UV-Detektion durchgeführt
(Beobachtungswellenlänge: λ = 210 nm). Als isokratisches Lösungsmittelsystem diente iso-
PrOH und n-Hexan. ee-Werte [%] wurden über die Verhältnisse der Peakintegrale errechnet
(Auswertungssoftware: VWR HPLC System Manager).
Röntgenstrukturanalysen wurden mit einem Oxford-Diffraction Xcalibur Diffraktometer,
ausgerüstet mit einem CCD area detector Sapphire S und einem Graphit Monochromator
unter Nutzung der Mo-Kα Strahlung (λ = 0.71073 Å) bei 150 K aufgenommen.
Allgemeine Synthesevorschriften für GC-Derivatisierungen
Entfernung der Boc-Schutzgruppen:
Die Aminosäuren werden mit 200 µl einer HCl-Lösung (0,1 M) aufgenommen und für 30 min
auf 110 °C erhitzt. Die Lösungsmittel werden bei 110 °C im Stickstoffstrom entfernt.
Derivatisierung des C-Terminus:
Das Hydrochlorid-Addukt der Aminosäure wird mit einer HCl-Lösung in Ethanol (2 M)
versetzt, die aus Acetylchlorid und Ethanol (abs.) im Verhältnis 1:4 erzeugt wurde.
Derivatisierung des N-Terminus:
Die N-terminal entschützten Aminosäuren werden in 100 µl abs. Dichlormethan gelöst und
mit 50 ml Trifluoressigsäureanhydrid versetzt. Es wird für 10 min auf 110 °C erhitzt.
Nachdem die Proben auf 20 °C gebracht wurden, wird das Lösungsmittel im Stickstoffstrom
entfernt und der Rückstand in 100 ml Toluol (abs.) aufgenommen.

Experimenteller Teil
73
5.2 Synthesevorschriften
5.2.1. Versuch der Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus Oxazolidinen Synthese von (4R)-4-((R)-2,3-dihydroxypropyl)-3-(R)-2-methoxy-1-
phenylethyl)oxazolidin-4-carbonsäure-tert-butylester (31)
Der (R)-4-Allyl-3-((R)-2-methoxy-1-phenylethyl)oxazolidin-4-
carbon-säure-tert-butylester 30[102] (0,5 g; 1,44 mmol) wird in
tBuOH (4,4 ml) und Wasser (4,4 ml) gelöst und bei RT mit
AD-Mixβ (2 g) versetzt. Nachdem 16 h bei RT stark gerührt
wurde, wird die Reaktion durch Zugabe von Na2SO3 (2 g)
beendet und das Reaktionsgemisch mit EtOAc (3x10 ml) extrahiert. Die organische Phase
wird mit Wasser (6 ml) und ges. NaCl-Lösung (6 ml) gewaschen und der Rückstand
chromatographisch gereinigt (Hex: EtOAc; 1:1). Man erhält 0,48 g eines viskosen Öls
(Ausbeute 91%).
Rf 0,21 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 1,5 (s, 9H); 1,88 (dd, J1= 15,31 Hz, J2= 1,88 Hz, 1H); 2,09 (dd, J1= 15,38
Hz, J2= 11,08 Hz, 1H); 3,24 (s, 3H); 3,50 (dd, J1= 9,87 Hz, J2= 4,37 Hz, 1H); 3,53 (m, 1H);
3,69 (dd, J1= 11,15 Hz, J2= 3,56 Hz, 1H); 3,77 (dd, J1= 9,87 Hz, J2= 7,32 Hz, 1H); 4,08 (d,
J= 9,00Hz, 1H); 4,13 (m, 1H); 4,21 (dd, J1= 5,84 Hz, J2= 3,02 Hz, 2H); 4,28 (d, J= 2,82 Hz,
1H); 4,46 (dt, J1= 7,46 Hz, J2= 3,79 Hz, 1H); 7,30 (m, 5H) 13C-NMR (CDCl3): 28,10; 35,46; 58,56; 61,45; 67,30; 67,91; 68,42; 75,01; 77,41; 82,24;
86,56; 127,67; 128,11; 128,28; 128,35; 128,99; 139,59; 171,79
MS (ESI): 382,2227 [MH+]; 404,2042 [MNa+]
[α]D = -14,3 (c = 11,35 mg/ml, T = 22,62 °C, CHCl3)
ONO
O O
OH
OH

Experimenteller Teil
74
Synthese von (4R)-4-((2R)-3-(tert-Butyl-dimethylsilyloxy)-2-hydroxypropyl)-3-((R)-2-
methoxy-1-phenylethyl)oxazolidin-4-carbonsäure-tert-butylester (32)
Das Diol 31 (0,48 g; 1,36 mmol) wird in DMF (6 ml) gelöst und mit
Imidazol (0,13 g; 1,9 mmol) und TBDMSCl (0,2 g; 1,5 mmol)
versetzt. Nun wird 16 h bei RT gerührt und die Reaktion durch
Zugabe von Wasser beendet. Das Reaktionsgemisch wird mit
EtOAc (20 ml) verdünnt und mit ges. NaCl-Lösung (5x 15 ml)
gewaschen. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Anschließend wird das Rohprodukt chromatographisch
gereinigt (Hex: EtOAc; 5:1). Man erhält 0,43 g des gewünschten Produkts (Ausbeute 70%).
Rf 0,47 (Hex: EtOAc; 3:1) 1H-NMR (CDCl3): 0,08 (d, J= 1,34; 6H); 0,9 (s, 9H); 1,49 (s, 9H); 1,86 (dd, J1= 15,11 Hz;
J2= 10,01 Hz; 1H); 2,15 (dd, J1= 15,18 Hz; J2= 1,75 Hz; 1H); 3,24 (s, 3H); 3,52 (m, 2H);
3,70 (dd, J1= 9,81 Hz; J2= 4,97; 1H); 3,77 (dd, J1= 9,81 Hz; J2= 6,98 Hz; 1H); 3,99 (m, 1H);
4,10 (d, J= 8,87 Hz; 1H); 4,18 (d, J= 8,90; 1H); 4,22 (d, J= 2,69; 1H); 4,26 (d, J= 2,69; 1H);
4,43 (dd, J1= 7,12 Hz; J2= 4,70 Hz; 1H); 7,29 (m, 5H) 13C-NMR (CDCl3): -5,29; 25,98; 28,08; 37,05; 58,52; 61,29; 67,50; 67,98; 68,83; 75,09;
77,32; 81,90; 86,33; 127,74; 128,04; 128,23; 128,80; 139,89; 171,98
MS (ESI): 496,3093 [MH+]; 518,2911 [MNa+]
Synthese von (4R)-4-((2S)-2-Azido-3-(tert-butyl-dimethylsilyloxy)-propyl)-3-((R)-2-
methoxy-1-phenylethyl)oxazolidin-4-carbonsäure-tert-butylester (33)
Der TBDMS-Ether 32 (0,5 g; 1,04 mmol) und PPh3 (0,69 g;
2,63 mmol) werden in THF (8 ml) gelöst und bei -20 °C mit DEAD
(1,08 ml; 6,86 mmol) und DPPA (0,55 ml; 2,55 mmol) versetzt.
Nachdem 20 h bei -20°C gerührt wurde, wird das Lösungsmittel im
Vakuum entfernt und der Rückstand chromatographisch gereinigt
(14:1). Man erhält 0,48 g eines Öls (Ausbeute 91%).
Rf 0,18 (Hex: EtOAc; 9:1)
ONO
O O
OH
OTBDMS
ONO
O O OTBDMS
N3

Experimenteller Teil
75
1H-NMR (CDCl3): 0,13 (d, J= 1,34, 6H); 0,95 (s, 9H); 1,46 (s, 9H); 1,84 (dd, J1= 15,18 Hz,
J2= 2,42 Hz, 1H); 2,15 (dd, J1= 15,3 Hz, J2= 8,87 Hz, 1H); 3,20 (s, 3H); 3,41 (dd,
J1= 10,01 Hz, J2= 4,63 Hz, 1H); 3,47 (dd, J1= 10,00 Hz, J2= 6,90, 1H); 3,73 (dd, J1= 10,21
Hz, J2= 7,66 Hz, 1H); 3,83 (dd, J1= 10,10 Hz, J2= 4,30 Hz, 1H); 4,02 (m, 1H); 4,09 (d, J=
8,60 Hz, 1H); 4,18 (d, J= 8,90, 1H); 4,34 (dd, J1= 6,92 Hz, J2= 4,62 Hz, 1H); 4,26 (d, J= 2,69
Hz, 1H); 4,38 (d, J= 10,75 Hz, 1H); 7,33 (m, 5H) 13C-NMR (CDCl3): 25,86; 28,00; 35,75; 58,56; 60,19; 60,81; 66,69; 67,51; 75,86; 76,48;
81,63; 86,11; 126,11; 127,67; 127,80; 128,46; 130,04; 140,83; 172,52
LC-MS (ESI): 521,3 [MH+]
Synthese von (4R)-4-((2S)-2-Azido-3-hydroxy-propyl)-3-((R)-2-methoxy-1-
phenylethyl)oxazolidin-4-carbonsäure-tert-butylester (34)
Das Azid 34 (270 mg; 0,53 mmol) wird in THF (6,5 ml) gelöst und bei
0°C mit TBAF-Lösung (0,85 ml; 1,1 eq.) versetzt. Nachdem 4 h bei 0°C
gerührt wurde, wird die Reaktion durch Zugabe von ges. NH4Cl-Lösung
beendet und das Reaktionsgemisch mit EtOAc (3x) extrahiert. Die
organische Phase wird getrocknet und das Lösungsmittel im Vakuum
entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:EtOAc;
5:1 auf 3:1). Man erhält 147 mg eines Öls (Ausbeute 71%).
Rf 0,25 (Hex: EtOAc; 3:1) 1H-NMR (CDCl3): 1,46 (s, 9H); 2,02 (m, 2H); 3,20 (s, 3H); 3,44 (dd, J1= 10,34 Hz, J2= 6,31
Hz, 1H); 3,50 (dd, J1= 10,40 Hz, J2= 3,90 Hz, 1H); 3,78 (dd, J1= 11,28 Hz, J2= 5,24 Hz, 1H);
3,83 (dd, J1= 11,30 Hz, J2= 6,24 Hz, 1H); 4,04 (m, 1H); 4,09 (d, J= 8,73, 1H); 4,21 (dd, J1=
5,84 Hz, J2= 3,02 Hz, 2H); 4,28 (d, J= 2,82, 1H); 4,33 (dd, J1= 6,18 Hz, J2= 3,90 Hz, 1H);
4,37 (m, 1H); 7,31 (m, 5H) 13C-NMR (CDCl3): 28,05; 34,96; 58,74; 60,37; 61,07; 65,94; 66,54; 75,48; 76,85; 82,09;
86,15; 127,85; 128,02; 128,55; 140,25; 171,46
MS (ESI): 407,2294 [MH+]; 429,2110 [MNa+]
[α]D = -53,3 (c = 10,65 mg/ml, T = 22,23 °C, CHCl3)
ONO
O O OH
N3

Experimenteller Teil
76
Synthese von (2R,4S)-2-((R)-2-Methoxy-1-phenylethylamino)-5-(tbutyl-
dimethylsilyloxy)-4-hydroxy-2-(hydroxymethyl)pentansäure-tert-butylester (35)
Der TBDMS-Ether 32 (100 mg; 0,202 mmol) wird in THF (1 ml)
und Wasser (2,5 ml) gelöst. Nun wird Ameisensäure(99-100 %)
zugegeben bis pH 2 erreicht wird. Das Reaktionsgemisch wird 7 h
bei RT gerührt. Anschließend wird mit ges. K2CO3-Lösung
neutralisiert und mit EtOAc extrahiert. Die organische Phase wird
getrocknet und das Lösungsmittel im Vakuum entfernt. Der
Rückstand wird chromatographisch gereinigt (Hex:EtOAc; 5:1 auf 1:1). Man erhält 47 mg des
gewünschten Produkts (Ausbeute 48%).
Rf 0,35 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 0,02 (s, 6H); 0,84 (s, 9H); 1,47 (m, 11H); 3,18 (m, 3H); 3,36 (s, 3H); 3,64
(m, 3H); 3,82 (dd, J1= 9,6 Hz, J2= 6,11 Hz, 2H), 7,30 (m, 5H) 13C-NMR (CDCl3): -5,38; 25,90; 27,92; 29,71; 58,84; 64,08; 67,21; 68,26; 77,76; 127,21;
127,71; 128,63; 135,03
LC-MS (ESI): 484,3 [MH+]
Synthese von (4R)-4-((2R)-3-(tert-Butyl-dimethylsilyloxy)-2-hydroxypropyl)oxazolidin-4-
carbonsäure-tert-butylester (37)
Der TBDMS-Ether 37 (1 g; 2,02 mmol) wird in THF (55 ml) gelöst
und mit Pd/C (0,1 g) versetzt. Nun wird das Reaktionsgemisch bei
RT unter H2-Atmosphäre (50 bar) 16 h gerührt. Anschließend wird
der Katalysator abfiltriert und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird
chromatographisch gereinigt (Hex: EtOAc; 1:1). Man erhält 0,66 g (Ausbeute 80%) des
gewünschten Produktes.
Rf 0,41 (Hex: EtOAc; 1:1) 1H-NMR (MeOD-d4): 0,07 (s, 6H); 0,9 (s, 9H); 1,45 (d, J= 13,37 Hz, 1H); 1,49 (s, 9H); 1,86
(d, J= 13,30 Hz, 1H); 3,52 (m, 3H); 3,65 (m, 2H); 4,37 (d, J= 10,07 Hz, 1H); 4,50 (d,
J= 10,21 Hz, 1H)
OHNHO
O O
OH
OTBDMS
NHO
O O
OH
OTBDMS

Experimenteller Teil
77
MgBr
13C-NMR (MeOD-d4): -5,15; 19,19; 26,39; 28,31; 33,28; 63,31; 67,67; 68,74; 75,98; 77,36;
82,92; 173,38
LC-MS (ESI): 362,3 [MH+]
Versuch zur Synthese von (2R,4R)-2-Amino-5-(tert-butyl-dimethylsilyloxy)-4-hydroxy-2-
(hydroxymethyl)pentansäure-tert-butylester (38)
Das Oxazolidin 37 wird in MeOH (10 ml) gelöst und 2 h bei 75 °C
gerührt. Nun wird das Lösungsmittel im Vakuum entfernt. Es fand
keine Ringöffnung statt.
Das Oxazolidin 37 (41 mg; 0,166 mmol) wird in DCM (0,5 ml) gelöst und mit 1,3-
Propandithiol (0,167 ml; 1,66 mmol) und Bortrifluoridetherat (0,063 ml; 0,498 mmol)
versetzt. Die Reaktionslösung wird 16 h bei 75 °C gerührt. Nun wird das Lösungsmittel im
Vakuum entfernt. Es fand keine Ringöffnung statt.
Das Oxazolidin 37 (24 mg; 0,1 mmol) wird in THF (0,5 ml) gelöst, Natriumborhydrid
(38 mg; 1 mmol) suspendiert in THF (0,5 ml) zugegeben und das Reaktionsgemisch 16 h bei
RT gerührt. Anschließend wird die Reaktion durch Zugabe von 5% HCl beendet. Das
gewünschte Reaktionsprodukt konnte nur in Spuren gefunden werden, das Hauptprodukt ist
die N-Methyl-Aminosäure.
Das Oxazolidin 37 wird in MeOH (20 ml) gelöst und mit HCl/Dioxan (4M; 0,25 ml) versetzt.
Nun wird das Reaktionsgemisch 2 h bei 70 °C refluxiert. Das gewünschte Reaktionsprodukt
konnte nicht isoliert werden. Es fand lediglich eine vollständige TBDMS-Abspaltung (0,21 g)
statt.
Synthese von Benzylmagnesiumbromid (142)
In wasserfreiem Diethylether (5 ml) werden Mg-Späne (1,21 g; 0,05 mol) suspendiert.
Nun wird Brombenzol (0,26 ml) zugeben bis eine braune Lösung entsteht,
anschließend wird weiteres Brombenzol (5 ml) gelöst in wasserfreiem Diethylether (15 ml)
langsam zutropft. Die Reaktionslösung wird 1 h bei 35 °C gerührt, anschließend wird die
Lösung mit Diethylether (96 ml) verdünnt und direkt weiter verwendet.
NH2HO
O O OTBDMS
OH

Experimenteller Teil
78
Synthese von (2R,4S)-2-((R)-Benzyl-2-methoxy-1-phenylethylamino)-4-azido-5-tert-
butyl-dimethylsilyloxy)-2-(hydroxymethyl)pentansäure-tert-butylester (39)
Das Azid 33 (137 mg; 0,26 mmol) wird in wasserfreiem
Diethylether (10 ml) gelöst und bei 0°C langsam mit einer
Benzylmagnesiumbromidlösung (3 M; 0,26 ml; 3 eq.) versetzt.
Nachdem die Reaktionslösung 2 h bei 0 °C gerührt wurde, wird die
Reaktion durch Zugabe von ges. NH4Cl-Lösung beendet und das
Reaktionsgemisch mit DCM (3x) extrahiert. Das Rohprodukt wird chromatographisch
gereinigt (Hex: EtOAc; 14:1 auf 9:1). Man erhält 40 mg eines gelben Öls (Ausbeute 26%).
Rf 0,27 (Hex: EtOAc; 14:1) 1H-NMR (CDCl3): 0,10 (s, 6H); 0,92 (s, 9H); 1,41 (s, 4H); 1,96 (dd, J1= 13,43 Hz,
J2= 8,72 Hz, 1H); 2,08 (m, 1H); 3,22 (s; 3H); 3,52 (d, J= 15,85 Hz, 1H); 3,66 (m, 2H); 3,81
(m, 2H); 3,91 (m, 2H); 4,32 (dd, J1= 10,28 Hz, J2= 1,54 Hz, 1H); 4,32 (dd, J1= 10,21 Hz, J2=
4,84 Hz, 1H); 7,25 (m, 10H) 13C-NMR (CDCl3): -5,52; 18,15; 25,74; 27,86; 27,86; 33,71; 49,99; 57,69; 58,36; 60,24;
62,88; 67,45; 69,85; 73,94; 82,09; 126,96; 127,43; 127,98; 128,03; 128,24; 129,32; 137,80;
142,26; 172,40
LC-MS (ESI): 599,3 [MH+]
Synthese von (2R)-2-((R)-Benzyl-2-methoxy-1-phenylethylamino)-2-
(hydroxymethyl)pent-4-ensäure-tert-butylester (40)
Das Allyl-Oxazolidin 30 (0,41 g; 1,18 mmol) wird in wasserfreiem
Diethylether gelöst und bei 0 °C mit einer Benzylmagnesiumbromid-
Lösung (3,5 M; 1 ml; 3 eq.) versetzt und bei 0°C gerührt. Nach 2 h wird
die Reaktion durch Zugabe von ges. NH4Cl-Lösung beendet und das
Reaktionsgemisch mit DCM (3x) extrahiert. Die organische Phase wird
getrocknet und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird
chromatographisch gereinigt (Hex: EtOAc; 14:1 auf 9:1). Man erhält 286 mg eines gelben Öls
(Ausbeute 57%).
Rf 0,40 (Hex: EtOAc; 5:1)
ON
O O
Ph
HO
OTBDMS
N3
ON
O O
Ph
HO

Experimenteller Teil
79
1H-NMR (CDCl3): 1,38 (s, 9H); 2,65 (dd, J1= 12,09 Hz, J2= 7,25 Hz, 1H); 2,82 (dd,
J1= 12,09 Hz, J2= 7,25 Hz, 1H); 3,24 (s, 3H); 3,30 (m, 2H); 3,63 (d, J= 15,85 Hz, 1H); 3,78
(d, J= 16,12 Hz, 1H); 3,81 (d, J= 6,85 Hz, 1H); 4,45 (ddd, J1= 12,36 Hz, J2= 6,58 Hz,
J3= 6,31 Hz, 1H); 4,59 (dd, J1= 9,94 Hz, J2= 4,97 Hz, 1H); 5,13 (dd, J1= 10,07 Hz,
J2= 2,01 Hz, 1H); 5,22 (dd, J1=16,52 Hz, J2= 1,48 Hz, 1H); 5,76 (m, 1H); 7,27 (m, 10H) 13C-NMR (CDCl3): 28,13; 37,90; 50,67; 58,02; 58,34; 62,61; 71,74; 73,87; 81,78; 119,28;
126,92; 127,35; 128,00; 128,15; 128,19; 129,18; 132,27; 142,20; 172,51
MS (ESI): 426,2643 [MH+]; 448,2458 [MNa+]
Synthese von (2R)-2-((R)-Benzyl-2-methoxy-1-phenylethylamino)-2-
((methoxymethoxy)methyl)pent-4-ensäure-tert-butylester (41)
40 (270 mg; 0,63 mmol) wird in DCM (4 ml) gelöst und bei 0°C mit
Tetrabutylammoniumiodid (24 mg), DIPEA (1,1 ml; 6,3 mmol) und
MOM-Cl (0,25 ml; 3,15 mmol) versetzt. Nun wird die
Reaktionslösung auf RT erwärmt und unter Lichtausschluss gerührt.
Nach 3 d wird das Reaktionsgemisch mit ges. NaHCO3-Lösung
(5 ml) und Diethylether (10 ml) versetzt. Die Phasen werden getrennt
und die wässrige Phase mit Diethylether (3x 10 ml) extrahiert. Nun wird die vereinigte
organische Phase mit 1% HCl-Lösung und ges. NaCl-Lösung gewaschen. Das Lösungsmittel
wird im Vakuum entfernt und der Rückstand chromatographisch gereinigt (Hex: EtOAc; 9:1).
Man erhält 170 mg eines gelben Öls (Ausbeute 67 %).
Rf 0,64 (Hex: EtOAc; 5:1) 1H-NMR (CDCl3): 1,41 (s, 9H); 2,65 (dd, J1= 13,84 Hz, J2= 7,52 Hz, 1H); 2,81 (dd,
J1= 13,45 Hz, J2= 7,46 Hz, 1H); 3,15 (s, 3H); 3,23 (s, 3H); 3,34 (d, J= 9,67 Hz, 1H); 3,50 (m,
1H); 3,63 (d, J= 15,85 Hz, 1H); 3,70 (s, 2H); 4,10 (d, J= 17,46 Hz, 1H); 4,33 (d,
J1= 17,50 Hz, 1H); 4,43 (m, 2H); 5,09 (m, 2H); 5,82 (m, 1H); 7,23 (m, 10H) 13C-NMR (CDCl3): 28,01; 37,55; 48,74; 55,48; 61,48; 69,24; 70,04; 74,52; 81,40; 96,91;
118,53; 126,10; 127,01; 127,08; 127,86; 127,93; 129,31; 133,73; 140,27; 143,98; 172,52
MS (ESI): 470,2903 [MH+]; 492,2720 [MNa+]
ON
O O
Ph
MOMO

Experimenteller Teil
80
Synthese von (2R,4R)-2-((R)-Benzyl-2-methoxy-1-phenylethylamino)-2-
((methoxymethoxy)methyl)-4,5-dihydroxy-pentansäure-tert-butylester (42)
Der MOM-Ether 41 (0,17 g; 0,36 mmol) wird in einem Gemisch aus
tBuOH und Wasser (je 4,4 ml) gelöst und mit AD-Mix β (0,5 g)
versetzt. Nun wird kräftig gerührt und nach 24 h nochmals AD-Mixβ
(0,5 g) zugegeben. Nach weiteren 24 h wird die Reaktion durch
Zugabe von Na2SO3 beendet. Die Reaktionslösung wird mit Wasser
verdünnt und mit EtOAc (3x) extrahiert. Die organische Phase wird
mit ges. NaCl-Lösung gewaschen und getrocknet. Anschließend wird
das Lösungsmittel im Vakuum entfernt und das Rohprodukt chromatographisch gereinigt
(Hex: EtOAc; 3:1 auf 1:1). Man erhält 125 mg eines gelben Öls (Ausbeute 72%).
Rf 0,12 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 1,13 (s, 9H); 2,38 (m, 2H); 3,20 (s, 3H); 3,23 (s, 3H); 3,35 (m, 3H); 3,47
(m, J= 8,73 Hz, 3H); 3,85 (d, J= 9,95 Hz, 1H); 3,93 (d, J= 9,94 Hz, 1H); 4,38 (m, 4H); 4,78
(dd, J1= 9,47 Hz, J2= 4,90 Hz, 1H); 7,23 (m, 10H) 13C-NMR (CDCl3): 27,69; 34,60; 48,36; 55,78; 61,19; 67,27; 67,75; 69,51; 71,68; 74,13;
81,39; 97,12; 126,57; 126,97; 127,22; 127,73; 128,06; 128,37; 129,32; 138,37; 143,31;
172,38
MS (ESI): 504,2959 [MH+]; 526,2774 [MNa+]
Synthese von (2R,4R)-2-((R)-Benzyl-2-methoxy-1-phenylethylamino)-5-(tert-butyl-
dimethylsilyloxy)-4-hydroxy-2-(hydroxymethyl)pentansäure-tert-butylester (43)
Das Diol 42 (115 mg; 0,23 mmol) wird in DMF (1,1 ml) gelöst
und mit Imidazol (23 mg; 35 mmol) und TBDMSCl (38 mg;
25 mmol) versetzt. Nachdem die Lösung 16 h bei RT gerührt
wurde, wird die Reaktion durch Zugabe von Wasser beendet.
Das Reaktionsgemisch wird mit EtOAc (20 ml) verdünnt und
mit ges. NaCl-Lösung (5x) gewaschen. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:
EtOAc; 5:1). Man erhält 92 mg eines gelben Öls (Ausbeute 73%).
ON
O O
Ph
MOMO
OTBDMS
OH
ON
O O
Ph
MOMOOH
OH

Experimenteller Teil
81
1H-NMR (CDCl3): 0,00 (d, J= 3,76 Hz, 1H); 0,04 (d, J= 5,24 Hz, 1H); 0,87 (s, 5H); 0,88 (s,
4H); 1,29 (s, 6H); 1,43 (s, 4H); 1,95 (dd, J1= 14,20 Hz, J2= 1,75 Hz, 0,3H); 2,06 (dd,
J1= 15,50 Hz, J2= 10,41 Hz, 0,3H); 2,15 (dd, J1= 15,45 Hz, J2= 2,82 Hz, 0,6H); 2,22 (dd,
J1= 15,15 Hz, J2= 7,66 Hz, 0,6H); 3,17 (s, 1,8H); 3,19 (s, 1,2H); 3,22 (s, 1,8H); 3,26 (s,
1,2H); 3,34 (dd, J1= 9,87 Hz, J2= 7,05 Hz, 1H); 3,49 (m, 1H); 3,58 (m, 1H); 3,67 (m, 1H);
4,25 (dd, J1= 16,95 Hz, J2= 14,71 Hz, 1H); 4,38 (m, 2H); 4,47 (dd, J1=16,55 Hz, J2= 6,39 Hz,
1H); 4,64 (t, J= 5,24 Hz, 1H); 7,25 (m, 10H) 13C-NMR (CDCl3): -5,39; -5,29; 25,90; 25,97; 27,85; 27,99; 35,67; 48,77; 48,92; 55,69;
58,40; 58,55; 60,98; 61,47; 67,50; 68,06; 68,14; 68,69; 70,17; 70,26; 71,09; 73,88; 74,38;
81,42; 97,11; 126,16; 126,29; 127,04; 127,18; 127,32; 127,36; 127,89; 128,04; 128,08;
128,12; 129,38; 129,47; 172,61
Versuch zur Synthese von (2R,4S)-2-((R)-Benzyl-2-methoxy-1-phenylethylamino)-4-
Azido-5-(tbutyl-dimethylsilyloxy)-2-(hydroxymethyl)pentansäure-tert-butylester (44)
Der MOM-Ether 43 (90 mg; 0,146 mmol) und
Triphenylphosphin (96 mg; 0,366 mmol) werden in THF
(1,3 ml) gelöst und bei -20°C mit DEAD (169 µl; 1,077 mmol)
und DPPA (78 µl; 0,362 mmol) versetzt. Nachdem 20 h bei
-20 °C gerührt wurde, wird das Lösungsmittel im Vakuum
entfernt. Das gewünschte Produkt konnte nicht isoliert werden.
ON
O O
Ph
MOMO
OTBDMS
N3

Experimenteller Teil
82
5.2.2. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus Pyrazolderivaten
Synthese von 2-Hydroxymethylacrylsäure-tert-butylester (49)[158]
Paraformaldehyd (2 g; 0,066 mol) wird in einem THF-Wasser-Gemisch (je
4,5 ml) suspendiert und mit 1 N Phosphorsäure (0,24 ml) versetzt. Nun wird 2 h bei 80°C
gerührt. Nachdem die Reaktionslösung auf 40°C abgekühlt wurde, wird die Reaktionslösung
mit DABCO (0,74 g; 0,1 eq.) und Acrylsäure-tbutylester (1,9 ml; 0,2 eq.) versetzt. Nachdem
16 h bei 60°C gerührt wurde, wird die Reaktion durch Zugabe von ges. NaCl-Lösung beendet
und mit Diethylether extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel
im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex: EtOAc; 9:1 auf
3:1). Man erhält 0,44 g eines farblose Öls (Ausbeute 21%).
Rf 0,4 (Hex: EtOAc; 3:1) 1H-NMR (CDCl3): 1,48 (s, 9H); 4,19 (t, J= 1,54 Hz, 2H); 5,81 (q, J=1,73 Hz, 1H); 6,19
(q, J=1,34 Hz, 1H) 13C-NMR (CDCl3): 28,05; 68,97; 80,92; 124,47; 138,62
MS(ESI): 159,1012 [MH+] ; 181,0829 [MNa+]
Synthese von Diazoessigsäureethylester (50)[159]
N2 CO2Et
H-Gly-OEt*HCl (8 g; 47,88 mol) wird in Wasser (10 ml) gelöst und mit
Diethylether (10 ml) versetzt. Nun wird eine NaNO2-Lösung (5,8 g in 14 ml Wasser) gefolgt
von 4 N Schwefelsäure (0,57 ml) zugesetzt. Das Reaktionsgemisch wird 1 min im
Scheidetrichter geschüttelt und die gelbe Etherphase in einen Kolben mit Na2CO3-Lösung
überführt. Nun wird die wässrige Phase nochmals mit 4 N Schwefelsäure (0,57 ml) versetzt
und die Phasen wieder getrennt. Dieser Vorgang wird so lange wiederholt bis keine Färbung
der organischen Phase mehr auftritt. Die organische Phase wird mit 1 N NaHCO3-Lösung
gewaschen bis keine Färbung der wässrigen Phase mehr beobachtet wird. Nun wird die
organische Phase getrocknet und das Lösungsmittel im Vakuum bei 20°C entfernt. Man erhält
4,5 g (Ausbeute 69%) einer gelben Flüssigkeit.
Rf 0,62 (Hex: EtOAc; 3:1) 1H-NMR (CDCl3): 1,86 (t, J= 7,12 Hz, 3H); 4,20 (q, J= 7,12 Hz, 2H); 4,70 (br, 1H)
CO2tBuHO

Experimenteller Teil
83
13C-NMR (CDCl3): 14,39; 46,06; 60,81; 166,77
MS(EI): 114,0947 [M+]
Synthese von rac-5-tert-Butyl-3-ethyl-4,5-dihydro-5-(hydroxymethyl)-1H-pyrazol-3,5-
dicarboxylat (51)
In einen Reaktionskolben werden Diazoessigsäureethylester (50)
(0,114 g; 1 mmol), 2-Hydroxymethylacrylsäure-tbutylester (49) (0,156 g;
1 mmol) und DABCO (0,011 g; 1 mmol) gegeben und 16 h bei RT
gerührt. Nun wird das Reaktionsgemisch chromatographisch gereinigt (Hex: EtOAc; 3:1 auf
1:1). Man erhält 203 mg (Ausbeute 75%) eines gelben Öls.
Rf 0,32 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 1,33 (t, J= 7,12 Hz, 3H); 1,46 (s, 9H); 3,03 (d, J= 18,00 Hz, 1H); 3,29 (d,
J= 18,00 Hz, 1H); 3,68 (d, J= 11,15 Hz, 1H); 3,79 (d, J= 11,15 Hz, 1H); 4,29 (q, J= 7,12 Hz,
2H); 6,79 (s, 1H) 13C-NMR (CDCl3): 14,19; 27,91; 37,44; 61,39; 65,32; 74,11; 83,56; 143,61; 161,91; 171,31
MS(ESI): 273,1441[MH+]; 295,1261 [MNa+]
Synthese von rac-2,4-Diamino-1-tert-butyl-5-ethyl -2-(hydroxymethyl)-1,5-dicarboxylat
(52)
Das Pyrazol 51 (10 mg; 0,037 mmol) wird in Methanol (0,8 ml) gelöst
und mit Essigsäure (0,1 ml) versetzt. Nun wird Raney-Nickel zugesetzt
und 16 h unter Wasserstoffatmosphäre (50 bar) gerührt. Die Reaktion
wurde mittels LC-MS untersucht und die gewünschte Produktmasse gefunden, das Produkt
konnte aber nicht isoliert werden.
MS(ESI): 277,1732 [MH+]
NH
N
EtO2C
CO2tBu
OH
H2NH2N
EtO2C
CO2tBu
OH

Experimenteller Teil
84
Synthese von rac-N-Boc-5-tert-Butyl-3-ethyl-4,5-dihydro-5-(tert-butyloxymethyl)-
pyrazol-3,5-dicarboxylat (53)
Das Pyrazolderivat 51 (30 mg; 0,11 mmol) wird in DCM (0,7 ml) gelöst
und mit TEA (3 µl; 0,14 eq.), Boc2O (50 µl; 2 eq.) und DMAP (13 mg;
1 eq.) versetzt. Nun wird 16 h bei RT gerührt. Anschließend wird das Lösungsmittel im
Vakuum entfernt und der Rückstand chromatographisch gereinigt (Hex: EtOAc; 7:1 auf 5:1).
Man erhält 24 mg (Ausbeute 51%) eines gelben Öls.
Rf 0,61 (Hex: EtOAc; 2:1)
H-NMR (CDCl3): 1,35 (t, J= 7,12 Hz, 3H); 1,44 (s, 9H); 1,45 (s, 9H); 1,53 (s, 9H); 3,28 (d,
J= 18,40 Hz, 1H); 3,41 (d, J= 18,60 Hz, 1H); 4,31 (m, 3H); 4,85 (d, J= 11,82 Hz, 1H) 13C-NMR (CDCl3): 14,12; 27,65; 27,83; 28,05; 61,86; 66,06; 70,18; 82,67; 83,39; 143,28;
153,06; 167,89
Versuch zur Synthese von rac-2-Boc-2,4-Diamino-1-tert-butyl-5-ethyl -2-
(hydroxymethyl)-1,5-dicarboxylat (54)
Das Pyrazol 53 (10 mg; 0,023 mmol) wird in Methanol (0,8 ml) gelöst.
Nun wird Raney-Nickel zugesetzt und 16 h unter
Wasserstoffatmosphäre (50 bar) gerührt. Das Produkt konnte nicht identifiziert werden.
NBoc
N
EtO2C
CO2tBu
OtBu
BocHNH2N
EtO2C
CO2tBu
OtBu

Experimenteller Teil
85
5.2.3. Synthese von 2,4-Diamino-2-hydroxymethylglutarsäure aus
γ-Methylenglutaminsäure
Synthese von 2-Hydroxymethylacrylsäuremethylester (56)[160]
Acrylsäuremethylester (90 ml; 990 mmol), Formaldehyd (30%; 37,2 ml; 495
mmol), Triethylamin (83,55 ml, 600 mmol) und Wasser (15 ml) werden in einem Kolben
vorgelegt und bei 60°C gerührt. Nach 16 h wird die Reaktion durch die Zugabe von eiskaltem
Wasser beendet und das Reaktionsgemisch im Vakuum eingeengt. Nun wird mit Ethylacetat
(3x150 ml) extrahiert. Die organische Phase wird mit ges. NaCl-Lösung und Wasser
gewaschen, anschließend getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält
20,6 g (Ausbeute 35,9 %) eines farblosen Öls.
Rf 0,38 (Hex: EtOAc; 2:1) 1H-NMR (CDCl3): 3,79 (s, 3H); 4,33 (s, 2H); 5,84 (q, J=1,34 Hz, 1H); 6,26 (td, J1= 1,01 Hz,
J2=0,81 Hz, 1H) 13C-NMR (CDCl3): 51,88; 62,53; 125,80; 139, 36
MS(CID): 117,0533 [M+]
Synthese von 2-Brommethylacrylsäuremethylester (57)[160]
2-Hydroxymethylacrylsäuremethylester (56) (10 g; 86,15 mmol) wird in DCM
(120 ml) gelöst und bei 0°C langsam mit einer 48%igen
Bromwasserstofflösung (35,2 ml) versetzt. Nach Beendigung der Zugabe wird weitere 5 min
gerührt und anschließend konz. Schwefelsäure (32,2 ml) ebenfalls bei 0°C langsam
zugetropft. Nachdem 1 h bei 0°C gerührt wurde, wird das Kühlbad entfernt und 12 h bei RT
gerührt. Nun wird die Reaktion durch Zugabe von eiskaltem Wasser beendet und die
Reaktionslösung mit DCM (4x500 ml) extrahiert. Die organische Phase wird mit Wasser und
ges. NaCl-Lösung gewaschen, anschließend getrocknet und das Lösungsmittel im Vakuum
entfernt. Der Rückstand wird chromatographisch gereinigt (Hex: EtOAc; 14:1). Man erhält
eine klare, farblose Flüssigkeit (11,4 g; Ausbeute 74 %).
Rf 0,79 (Hex: EtOAc; 2:1)
CO2Me
OH
CO2Me
Br

Experimenteller Teil
86
1H-NMR (CDCl3): 3,79 (s, 3H); 4,16 (d, J=0,81 Hz, 2H); 5,84 (d, J=0,81 Hz, 1H); 6,31 (d,
J= 0,54 Hz, 1H) 13C-NMR (CDCl3): 29,18; 52,22; 129,14; 137, 30;165,26
Synthese von Cbz-Glycin-tert-butylester (59)
H-Gly-OtBu*HCl (5 g; 29,9 mmol) wird in einer ges.
NaHCO3-Lösung (770 ml) gelöst und mit Cbz-Cl (9 ml)
versetzt. Nun wird 16 h bei RT gerührt und anschließend mit Ethylacetat (4x100 ml)
extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt.
Der Rückstand wird chromatographisch gereinigt (Hex: EtOAc; 5:1). Man erhält 7,23 g
(Ausbeute 91 %) einer farblosen Flüssigkeit.
Rf 0,5 (Hex: EtOAc; 3:1) 1H-NMR (CDCl3): 1,45 (s, 9H); 3,85 (d, J=5,24 Hz; 2H); 5,11 (s, 2H); 5,27 (br; 1H); 7,32 (m,
5H) 13C-NMR (CDCl3): 28,04; 43,44; 66,96; 82,22; 136,36, 156,25
MS (ESI): 266,1384 [MH+]; 288,1203 [MNa+]
Synthese von Glycin-tert-butylester (60)
Cbz-Gly-OtBu 59 (6,5 g; 24,5 mmol) wird in Ethylacetat (70 ml) gelöst
und mit Pd/C (10%; 500 mg) versetzt. Nun wird mit Stickstoff gespült und der
Reaktionskolben mit Wasserstoff gefüllt. Nachdem 16 h bei RT gerührt wurde, wird die
Reaktionslösung filtriert und das Lösungsmittel im Vakuum entfernt. Man erhält 3,2 g eines
farblosen Öls in quantitativer Ausbeute.
Rf 0,51 (CHCl3: MeOH; 9:1) 1H-NMR (CDCl3): 1,44 (s, 9H); 3,29 (s, 2H) 13C-NMR (CDCl3): 28,08; 44,70; 81,00; 173,54
MS (ESI): 132,1019 [MH+]; 154,0838 [MNa+]
NH
O
O
O
O
H2NO
O

Experimenteller Teil
87
Synthese von 2-((E)-((1R,2R,5R)-2-hydroxy-2,6,6-trimethylbicyclo[3.1.1]heptan-3-
yliden)amino) essigsäure-tert-butylester (62)[114]
H-Gly-OtBu (60) (2,7 g; 20,6 mmol) wird in Benzol (40 ml) gelöst
und mit (1R,2R,5R)-2-Hydroxypinanon (1,85 g; 11 mmol) und
Bortrifluoridetherat (0,063 ml) versetzt. Nun wird 20 h unter Wasserabscheidung refluxiert
(95°C). Das Lösungsmittel wird im Vakuum entfernt und der Rückstand chromatographisch
gereinigt (Hex: EtOAc; 5:1 auf 3:1). Man erhält 3,09 g (Ausbeute 99 %) eines leicht gelben
Öls.
Rf 0,22 (Hex: EtOAc; 3:1) 1H-NMR (CDCl3): 0,84 (s, 3H); 1,30 (s, 3H); 1,46 (s, 9H); 1,49 (s, 3H); 1,54 (d, J= 10,61 Hz,
1H); 2,00 (ddd, J1= 8,76 Hz, J2= 5,88 Hz, J3= 2,96 Hz, 1H); 2,05 (t, J= 5,91 Hz, 1H); 2,31
(m, 1H); 2,45 (dd, J1= 6,58 Hz, J2= 2,96 Hz, 1H); 4,40 (q, J= 1,07 Hz, 1H) 13C-NMR (CDCl3): 22,90; 27,31; 28,09; 28,12; 38,26; 38,55; 50,25; 53,42; 76,50; 81,32;
169,33; 179,67
MS (ESI): 282,2053 [MH+]; 304,1873 [MNa+]
[α]D = 0,4405 ° (c = 13,6 mg/ml, T = 22,6 °C, EtOAc)[161]
Synthese von (S)-1-tert-Butyl-5-methyl-2-((E)-((1R,2R,5R)-2-hydroxy-2,6,6-
trimethylbicyclo[3.1.1]heptan-3-yliden)amino)-4-methylenpentandioat (63)
Diisopropylamin (0,6 ml; 4,27 mmol) wird in THF (18 ml) gelöst
und bei -30°C mit 2,2 M nButhyllithium-Lösung (1,48 ml) versetzt.
Nachdem 1 h bei -30°C gerührt wurde, wird auf -85°C
herabgekühlt. Währenddessen wird die Schiff´sche Base 62 (0,92 g;
3,26 mmol) gelöst in THF (12 ml) bei 0°C mit einer Methylmagnesiumbromid-Lösung (3 M;
1,62 ml) versetzt und bei 0°C gerührt bis keine Gasentwicklung mehr beobachtet werden
kann. Nun wird diese Lösung zur LDA-Lösung gegeben. Nachdem 1 h bei -85°C gerührt
wurde, wird 2-Brommethylacrylsäuremethylester (57) (0,72 ml) zugegeben und weitere 4 h
bei -85°C gerührt. Über einen Zeitraum von 12 h wird die Lösung auf -30°C erwärmt. Durch
Zugabe von ges. NH4Cl-Lösung wird die Reaktion beendet und mit Ethylacetat (4x10 ml)
extrahiert. Das Lösungsmittel wird im Vakuum entfernt und der Rückstand
chromatographisch gereinigt (Hex: EtOAc; 7:1 auf 3:1). Man erhält 1,02 g (Ausbeute 82 %)
eines gelben Öls.
NHO
CO2tBu
NHO
CO2tBu
CO2Me

Experimenteller Teil
88
Rf 0,26 (Hex: EtOAc; 5:1)
ee = 98% (GC-MS) 1H-NMR (CDCl3): 0,77 (s, 3H); 0,94 (dd, J1= 13,70 Hz, J2= 6,45 Hz, 1H); 1,29 (s, 3H); 1,41
(s, 3H); 1,43 (s, 9H); 1,52 (d, J= 10,61 Hz, 1H); 1,98 (ddd, J1= 8,50 Hz, J2= 5,68 Hz,
J3= 2,62 Hz, 1H); 2,03 (t, J= 5,91 Hz, 1H); 2,29 (m, 1H); 2,39 (dt, J1= 17,93 Hz, J2= 2,18 Hz,
1H); 2,59 (dd, J1= 17,87 Hz, J2= 2,96 Hz, 1H); 2,71 (dd, J1= 13,57 Hz, J2= 9,00 Hz, 1H);
3,00 (dd, J1= 13,16 Hz, J2= 5,37 Hz, 1H); 3,72 (s, 3H); 4,40 (dd, J1= 9,00 Hz, J2= 4,97 Hz,
1H); 5,63 (d, J= 0,81 Hz, 1H); 6,16 (d, J= 1,48 Hz, 1H) 13C-NMR (CDCl3): 22,70; 27,30; 28,04; 28,36; 33,31; 35,70; 38,22; 38,32; 50,06; 51,84;
61,22; 76,53; 81,48; 128,49; 136,43
MS(ESI) :380,2435 [MH+]; 402,2252 [MNa+]
[α]D = 104,6 (c = 7,6 mg/ml; T = 23,8 °C; EtOAc)[161]
Synthese von (S)-1-tert-Butyl-5-methyl-2-amino-4-methylenpentandioat (64)
63 (0,36 g; 1,293 mmol) wird in THF (2,2 ml) gelöst und mit 15%iger
Zitronensäure (1,78 ml) versetzt. Nun wird 2 d bei RT gerührt,
anschließend mit Wasser (10 ml) verdünnt und mit Diethylether (3x10 ml) gewaschen. Nun
wird mit ges. NaHCO3-Lösung auf pH 7-8 eingestellt und mit Diethylether (5x 10 ml)
extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt.
Man erhält 155 mg (72 %) eines farblosen Öls.
Rf 0,16 (Hex: EtOAc; 1:1)
ee = 98 % (GC-MS) 1H-NMR (CDCl3): 1,43 (s, 9H); 2,48 (ddd, J1= 13,90 Hz, J2= 8,26 Hz, J3= 1,07 Hz, 1H); 2,70
(ddd, J1= 13,90 Hz, J2= 6,11 Hz, J3= 1,07 Hz, 1H); 3,56 (dd, J1= 8,26 Hz, J2= 6,11 Hz, 1H);
3,75 (s, 3H); 5,63 (q, J= 1,21 Hz, 1H); 6,23 (d, J= 1,48 Hz, 1H) 13C-NMR (CDCl3): 28,02; 37,97; 51,93; 53,96; 81,32; 127,64; 136,85
MS (ESI): 230,1379 [MH+]; 272,1122 [MNa+]
[α]D = 6,3 (c = 21,9 mg/ml; T = 23,1 °C; EtOAc)[161]
H2N CO2tBu
CO2Me

Experimenteller Teil
89
Synthese von (S)-1-tert-Butyl-5-methyl-2-(Boc-amino)-4-methylenpentandioat (65)
1-tert-Butyl-5-methyl-2-amino-4-methylenpentandioat (64) (155 mg;
0,676 mmol) wird in DCM (4,7 ml) gelöst und mit Boc2O (0,16 ml)
versetzt. Nun wird 16 h bei RT gerührt, anschließend wird das
Lösungsmittel im Vakuum entfernt und der Rückstand chromatographisch gereinigt (Hex;
EtOAc; 9:1). Man erhält 202 mg (Ausbeute 91 %) eines farblosen Öls.
Rf 0,55 (Hex: EtOAc; 3:1)
ee = 72 % (GC-MS) 1H-NMR (CDCl3): 1,41 (s, 9H); 1,43 (s, 9H); 2,62 (dd, J1= 13,90 Hz, J2= 8,40 Hz, 1H); 2,75
(dd, J1= 14,10 Hz, J2= 5,64 Hz, 1H); 3,76 (s, 3H); 4,35 (q, J= 6,45 Hz, 1H); 5,10 (d,
J= 8,46 Hz, 1H) ; 5,63 (d, J= 1,07 Hz, 1H); 6,23 (s, 1H) 13C-NMR (CDCl3): 27,98; 28,28; 35,23; 52,04; 53,37; 79,59; 82,09; 128,12; 136,06; 155,18;
167,17; 171,06
MS(ESI) :330,1901 [MH+]; 352,1717 [MNa+]
[α]D = 3,4 (c = 9,5 mg/ml; T = 23,2 °C; EtOAc)[161]
Synthese von (2S,4S)-5-tert-Butyl-1-methyl-4-(Boc-amino)-2-hydroxy-2-
(hydroxymethyl)pentandioat (66)
Die Boc-geschütze Aminosäure 65 (200 mg; 0,607 mmol) wird einem
tBuOH–Wassergemisch (je 1 ml) gelöst und bei 0° C mit AD-Mixβ
(0,84 g) gelöst in einem tBuOH–Wasser Gemisch (je 2 ml) versetzt.
Nachdem 3 d bei 0°C gerührt wurde, wird die Reaktion durch Zugabe von Na2SO3 beendet
und das Reaktionsgemisch mit Ethylacetat (4x10 ml) extrahiert. Die organische Phase wird
getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält 207 mg (Ausbeute 94 %)
eines farblosen Feststoffs als ein Gemisch aus Diastereomeren.
Rf 0,25 (Hex: EtOAc; 1:1)
de = 80 % (NMR) 1H-NMR (CDCl3):1,44 (s, 9H); 1,45 (s, 9H); 2,09 (m, 2H); 3,59 (dd, J1= 20,08 Hz,
J2= 11,35 Hz, 1H); 3,71 (m, 1H); 3,77 (s, 3H); 4,45 (m, 1H); 4,98 (d, J= 8,87 Hz, 1H) 13C-NMR (CDCl3): 27,91; 28,31; 37,71; 49,88; 52,89; 68,59; 76,42; 80,62; 82,53; 155,99;
171,45; 174;63
BocHN CO2tBu
CO2Me
BocHN CO2tBu
CO2Me
HOOH

Experimenteller Teil
90
MS(ESI) :364,1970 [MH+]; 386,1788 [MNa+]
[α]D =-3,3 (c = 10,34 mg/ml; T = 24,1 °C; DCM)
Synthese von (2S,4S)-5-tert-Butyl-1-methyl-λ4-2-[1,3,2]dioxathiolan-[4]-carbacyclo-4-
(Boc-amino)-pentandioat (68)
Das Diol 67 (40 mg; 0,11 mmol) wird in Tetrachlormethan (1 ml)
gelöst und mit frisch destilliertem Thionylchlorid (0,012 ml;
0,165 mmol) versetzt. Nun wird 16 h bei 80°C gerührt. Die
Reaktionslösung wird mit Triethylamin neutralisiert und das Lösungsmittel im Vakuum
entfernt. Der Rückstand wird chromatographisch gereinigt (Hex: EtOAc; 3:1). Man erhält
28 mg (Ausbeute 62 %) eines braunen Öls.
Das Diol 67 (120 mg; 0,33 mmol) wird in DCM (1 ml) gelöst und bei 0°C mit NEt3 (0,1 ml)
versetzt. Nun wird frisch destilliertes Thionylchlorid (0,036 ml; 0,5 mmol) zugegeben und bei
0°C gerührt. Nach 45 min wird die Reaktion durch Zugabe von Wasser beendet und das
Reaktionsgemisch mit DCM extrahiert. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:
EtOAc; 5:1). Man erhält 111 mg (Ausbeute 82%) eines braunen Öls.
Rf 0,27 (Hex: EtOAc; 3:1)
MS (ESI): 410,1471 [MH+]; 432,1289 [MNa+]
Synthese von (2S,4R)-1-tert-Butyl-2-tert-butyl-4-azido-4-(hydroxymethyl)-5-
oxopyrrolidin-1,2-dicarboxylat (69b)
Das zyklische Sulfit 68 (291 mg; 0,713 mmol) wird in DMF (3,6 ml)
gelöst und mit Natriumazid (69 mg; 1,07 mmol) versetzt. Die
Reaktionslösung wird 3 d bei 50°C gerührt. Nun wird mit Ethylacetat
verdünnt und mit ges. NaCl-Lösung gewaschen. Das Lösungsmittel
wird im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:EtOAc; 5:1
auf 2:1). Man erhält 100 mg (Ausbeute 40 %) des Lactams als einen kristallinen farblosen
Feststoff.
Rf 0,55 (Hex: EtOAc; 1:1)
BocHN CO2tBu
CO2MeO
O SO
NBoc
OtBuO2C
N3
HO

Experimenteller Teil
91
de = 98 % (NMR) 1H-NMR (DMSO-d6): 1,42 (s, 9H); 1,43 (s, 9H); 1,76 (dd, J1= 14,17 Hz, J2= 3,56 Hz, 1H);
2,63 (dd, J1= 14,17 Hz, J2= 9,47 Hz, 1H); 3,58 (dd, J1= 11,01 Hz, J2= 5,64 Hz, 1H); 3,72 (dd,
J1= 11,15 Hz, J2= 5,85 Hz, 1H); 4,50 (dd, J1= 9,40 Hz, J2= 3,49 Hz, 1H); 5,62 (t, J= 5,64 Hz,
1H); 13C-NMR (DMSO-d6): 27,41; 29,64; 56,08; 62,42; 67,71; 81,88; 83,09, 169,40; 169,54
MS(ESI) :357,1765 [MH+]; 379,1580 [MNa+]
[α]D =-17,6 (c = 3,93 mg/ml; T = 23,5 °C; DCM)
Synthese von (S)-1-tert-Butyl-5-methyl-2-(Fmoc-amino)-4-methylenpentandioat (144)
1-tert-Butyl-5-methyl-2-amino-4-methylenpentandioat 64 (100 mg,
0,45 mmol) wird in DCM (2 ml) gelöst und mit FmocOSu (182 mg;
0,54 mmol) versetzt. Nun wird 16 h bei RT gerührt, anschließend wird das Lösungsmittel im
Vakuum entfernt und der Rückstand chromatographisch gereinigt (Hex:EtOAc; 5:1 auf 3:1).
Man erhält 158 mg (Ausbeute 78 %) eines farblosen Öls.
Rf 0,35 (Hex: EtOAc; 3:1) 1H-NMR (CDCl3): 1,45 (s, 9H); 2,68 (dd, J1= 14,04 Hz, J2= 8,26 Hz, 1H); 2,84 (dd,
J1= 14,10 Hz, J2= 5,51 Hz, 1H); 3,77 (s, 3H); 4,13 (t, J= 7,12 Hz, 1H); 4,35 (dd, J1= 7,12 Hz,
J2= 2,28 Hz, 2H); 4,45 (td, J1= 8,26 Hz, J2= 5,78 Hz, 1H); 5,50 (d, J= 8,19 Hz, 1H); 5,65 (s,
1H); 6,25 (d, J= 1,07 Hz, 1H); 7,30 (t, J= 7,46 Hz, 2H); 7,52 (t, J= 7,52 Hz, 2H); 7,58 (dd,
J1= 7,32 Hz, J2= 3,43 Hz, 2H); 7,75 (d, J= 7,52 Hz, 2H) 13C-NMR (CDCl3): 28,01; 35,7; 47,14; 52,17; 53,37; 66,99; 82,49; 119,97; 125,17; 127,06;
127,69; 128,57; 135,82; 141,29; 143,87; 143;95; 155,76; 167,28; 170,76
MS (ESI): 452,2065 [M+H]; 474,1884 [M+Na]
[α]D = 2,4 (c = 4,08 mg/ml; T = 24,0 °C; DCM)
Synthese von (2S,4S)-5-tert-Butyl-1-methyl-4-(Fmoc-amino)-2-hydroxy-2-
(hydroxymethyl)pentandioat (145)
Die Fmoc-geschütze Aminosäure 144 (529 mg; 1,17 mmol) wird in
einem tBuOH–Wassergemisch (je 2 ml) suspendiert und bei 0°C mit
AD-Mixβ (1,62 g) gelöst in einem tBuOH–Wassergemisch (je 4 ml)
versetzt. Nachdem 2 d bei 0 °C gerührt wurde, wird die Reaktion durch Zugabe von
FmocHN CO2tBu
CO2Me
FmocHN CO2tBu
CO2Me
HOOH

Experimenteller Teil
92
Na2SO3 (1,74 g) beendet und das Reaktionsgemisch mit Ethylacetat (4x 25 ml) extrahiert. Die
organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält
350 mg (Ausbeute 62 %) eines weißen Feststoffs.
Rf 0,25 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 1,45 (s, 9H); 2,13 (m, 2H); 3,59 (s, 3H); 3,69 (m, 1H); 4,21 (t, J= 6,65 Hz,
1H); 4,43 (d, J= 6,58 Hz, 2H); 4,53 (m, 1H); 5,19 (d, J= 9,94 Hz, 1H); 7,30 (t, J= 6,78 Hz,
2H); 7,39 (td, J1= 7,35 Hz, J2= 3,43 Hz, 2H); 7,59 (dd, J1= 7,12 Hz, J2= 2,96 Hz, 2H); 7,75
(d, J= 7,52 Hz, 2H) 13C-NMR (CDCl3): 27,91; 37,61; 47,00; 50,37; 52,92; 67,32; 68,50; 76,38; 82,82; 119,99;
124,98; 127,05; 127,69; 127,73; 141,31; 143,66; 143;72; 170,09; 174,67
MS (ESI): 486,2126 [MH+]; 508,1944 [MNa+]
[α]D = 0,3 (c = 12,00 mg/ml; T = 24,1 °C; DCM)
Synthese von (2S,4S)-5-tert-Butyl-1-methyl-λ4-2-[1,3,2]dioxathiolan-[4]-carbacyclo-4-
(Fmoc-amino)-pentandioat (146)
Das Diol 145 (53 mg; 0,11 mmol) wird in Tetrachlormethan (1 ml)
gelöst und mit frisch destilliertem Thionylchlorid (0,012 ml;
0,165 mmol) versetzt. Nun wird 16 h bei 80 °C gerührt. Die
Reaktionslösung wird mit Triethylamin neutralisiert und das Lösungsmittel im Vakuum
entfernt. Das gewünschte Produkt konnte nicht isoliert werden.
Das Diol 145 (42 mg; 0,09 mmol) wird in DCM (0,4 ml) gelöst und bei 0°C mit NEt3
(0,027 ml) versetzt. Nun wird frisch destilliertes Thionylchlorid (0,01 ml) zugegeben und bei
0°C gerührt. Nach 45 min wird die Reaktion durch Zugabe von Wasser beendet und das
Reaktionsgemisch mit DCM extrahiert. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt
(Hex:EtOAc; 3:1). Man erhält 47 mg (Ausbeute 98 %) eines braunen Öls.
Rf 0,17 (Hex: EtOAc; 3:1)
FmocHN CO2tBu
CO2MeO
O SO

Experimenteller Teil
93
Versuch zur Synthese von (2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(Fmoc-amino)-2-
(hydroxymethyl)pentandioat (147)
Das zyklische Sulfit 146 (50 mg; 0,092 mmol) wird in DMF (0,5 ml)
gelöst und mit Natriumazid (9,5 mg; 0,146 mmol) versetzt. Die
Reaktionslösung wird 2 d bei 50 °C gerührt. Nun wird mit Ethylacetat
verdünnt und mit ges. NaCl-Lösung gewaschen. Das Lösungsmittel wird im Vakuum entfernt.
Das gewünschte Produkt konnte nicht isoliert werden.
Synthese von (S)-1-tert-Butyl-5-methyl-2-(bis-Boc-amino)-4-methylenpentandioat (70)
Die Boc-geschützte Aminosäure 65 (194 mg; 0,589 mmol) wird in
Acetonitril (3,2 ml) gelöst und mit DMAP (72 mg; 0,589 mmol) und
Boc2O (0,67 ml; 2,94 mmol) versetzt. Nun wird 16 h bei RT gerührt und das Lösungsmittel
anschließend entfernt. Der Rückstand wird in EtOAc (10 ml) gelöst und mit ges. NH4Cl-
Lösung, Wasser und ges. NaCl-Lösung gewaschen. Die organische Phase wird getrocknet und
das Rohprodukt chromatographisch gereinigt (Hex: EtOAc; 14:1). Man erhält 189 mg
(Ausbeute 79%) eines farblosen Öls.
Rf 0,42 (Hex: EtOAc; 5:1)
ee = 68 % (GC-MS) 1H-NMR (CDCl3): 1,44 (s, 9H); 1,46 (s, 18H); 2,83 (dd, J1= 13,90 Hz, J2= 11,08 Hz, 1H);
3,13 (dd, J1= 13,70 Hz, J2= 3,76 Hz, 1H); 3,74 (s; 3H); 5,05 (dd, J1= 10,88 Hz, J2= 4,43 Hz,
1H); 5,53 (s, 1H); 6,18 (d, J= 1,34 Hz, 1H) 13C-NMR (CDCl3): 27,94; 32,38; 51,84; 57,48; 81,45; 82,72; 128,35; 136,77; 152,20; 166,84;
169,13
MS(ESI): 430,2431 [MH+]; 452,2246 [MNa+]
[α]D = -23,1 (c = 11,3 mg/ml; T = 23,6 °C; EtOAc)[161]
Synthese von (2S,4S)-5-tert-Butyl-1-methyl-4-(bis-Boc-amino)-2-hydroxy-2-
(hydroxymethyl)pentandioat (71)
70 (2,54 g; 5,91 mmol) wird einem tBuOH–Wassergemisch (27 ml:7
ml) gelöst und bei 0°C mit AD-Mixβ (8,33 g) gelöst in Wasser (40 ml)
FmocHN CO2tBu
CO2Me
HON3
Boc2N CO2tBu
CO2Me
Boc2N CO2tBu
CO2Me
HOOH

Experimenteller Teil
94
versetzt. Nachdem 1 d bei 0°C gerührt wurde, wird weiterer
AD-Mix-β (1 g) zugegeben. Nach einem weiteren Tag wird die Reaktion durch Zugabe von
Na2SO3 (10 g) beendet und das Reaktionsgemisch mit Ethylacetat (4x100 ml) extrahiert. Die
organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt. Das
Rohprodukt wird chromatographisch gereinigt (Hex: EtOAc; 5:1 auf 1:1) Man erhält das Diol
als ein Gemisch aus Diastereomeren in quantitativer Ausbeute.
Rf 0,38 (Hex: EtOAc; 1:1)
de = 73 % (NMR) 1H-NMR (CDCl3): 1,42 (s, 9H); 1,50 (s, 18H); 2,35 (dd, J1= 15,04 Hz, J2= 4,03 Hz, 1H); 2,41
(m, 1H); 3,60 (d, J= 11,15 Hz, 1H), 3,74 (s; 3H); 3,74 (d, J= 10,88 Hz, 1H); 5,06 (dd,
J1= 9,94 Hz, J2= 3,90 Hz, 1H) 13C-NMR (CDCl3): 27,88; 28,00; 33,99; 53,24; 54,62; 68,78; 76,42; 81,62; 83,26; 152,06;
169,41; 174,75
MS(ESI): 464,2475 [MH+]; 486,2292 [MNa+]
[α]D =12,4 (c = 10.0 mg/ml; T = 22.8 °C; EtOAc)[161]
Synthese von (2S,4S)-5-tert-Butyl-1-methyl-λ4-2-[1,3,2]dioxathiolan-[4]-carbacyclo-4-
(bis-Boc-amino)-pentandioat (72)
Das Diol 71 (1,3 g; 2,8 mmol) wird in DCM (13 ml) gelöst und bei 0°C
mit NEt3 (0,86 ml) versetzt. Nun wird frisch destilliertes Thionylchlorid
(0,31 ml; 4,32 mmol) zugegeben und bei 0°C gerührt. Nach 45 min wird
die Reaktion durch Zugabe von Wasser beendet und das Reaktionsgemisch mit DCM
extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt.
Der Rückstand wird chromatographisch gereinigt (Hex:EtOAc; 5:1). Man erhält 1,354 g
(Ausbeute 94 %) eines braunen Öls.
Rf 0,46 (Hex: EtOAc; 3:1)
Boc2N CO2tBu
CO2MeO
O SO

Experimenteller Teil
95
Synthese von (2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-
(hydroxymethyl)pentandioat (73)
Das zyklische Sulfit 72 (1,292 g; 2,535 mmol) wird in DMF (13 ml)
gelöst und mit Natriumazid (250 mg; 3,845 mmol) versetzt. Die
Reaktionslösung wird 3 d bei 50°C gerührt. Nun wird mit Ethylacetat
verdünnt und mit ges. NaCl-Lösung gewaschen. Die organische Phase
wird getrocknet und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird
chromatographisch gereinigt (Hex: EtOAc; 5:1 auf 3:1). Man erhält 254 mg (Ausbeute 40 %)
eines farblosen Öls als ein Gemisch aus Diastereomeren.
Rf 0,26 (Hex: EtOAc; 3:1)
de = 78 % (NMR) 1H-NMR (DMSO-d6): 1,35 (s, 9H); 1,44 (s, 18H); 1,97 (dd, J1= 15,11 Hz, J2= 8,66 Hz, 1H);
2,39 (dd, J1= 15,04 Hz, J2= 3,63 Hz, 1H); 3,67 (m, 1H); 3,71 (s; 3H); 3,79 (dd, J1= 11,20 Hz,
J2= 5,10 Hz, 1H); 4,78 (dd, J1= 8,60 Hz, J2= 3,90 Hz, 1H); 5,58 (t, J= 5,37 Hz, 1H) 13C-NMR (DMSO-d6): 27,37; 27,50; 32,52; 52,75; 54,58; 66,92; 69,66; 81,02; 81,35; 151,56;
168,42; 170,25
MS(ESI): 489,2556 [MH+]; 511,2365 [MNa+]
[α]D =-6,3 (c = 8,13 mg/ml; T = 23,47 °C; DCM)
Synthese von (2R,4S)-4-(tert-Butoxycarbonyl)-4-(bis-Boc-amino-2-azido-2-
(hydroxymethyl)butansäure (74)
Das Azid 73 (25 mg; 0,0512 mmol) wird in THF (0,5 ml) gelöst und mit
Lithiumhydroxid (1,77 mg; 0,074 mmol; 1,5 eq) gelöst in Wasser
(0,5 ml) versetzt. Nun wird 4 h bei RT gerührt. Anschließend wird das
Reaktionsgemisch mit Wasser verdünnt und mit EtOAc extrahiert (3x).
Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält
24 mg (Ausbeute 99 %) eines farblosen Öls.
Rf 0,25 ( CHCl3: MeOH; 9:1) 1H-NMR (DMSO-d6): 1,35 (s, 9H); 1,43 (s, 18H); 1,95 (dd, J1= 15,18 Hz, J2= 9,00 Hz, 1H);
2,37 (dd, J1= 15,18 Hz, J2= 3,09 Hz, 1H); 3,62 (d, J= 11,01 Hz, 1H); 3,75 (d, J= 11,01 Hz,
1H); 4,81 (dd, J1= 9,00 Hz, J2= 3,09 Hz, 1H)
Boc2N CO2tBu
CO2Me
HON3
Boc2N CO2tBu
CO2H
HON3

Experimenteller Teil
96
13C-NMR (DMSO-d6): 27,43; 27,55; 32,46; 55,00; 67,08; 69,57; 80,99; 82,23; 151,55;
168,63; 171,46
MS(ESI): 473,2239 [MH-]
[α]D = -14,8 (c = 9,74 mg/ml; T = 23,53 °C; EtOAc)
Synthese von (2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-
(methoxymethyl)pentandioat (118)
Das Azid (73) (150 mg; 0,307 mmol) wird in Acetonitril (400 µl) gelöst
und mit Silberoxid (142 mg; 0,614 mmol), Methyliodid (382 µl; 6,14
mmol) und einer katalytischen Menge von Methylsulfid versetzt.
Nachdem das Reaktionsgemisch 16 h bei RT unter Lichtausschluss
gerührt wurde, wird das Silberoxid abfiltriert, das Lösungsmittel im Vakuum entfernt und der
Rückstand chromatographisch gereinigt (Hex: EtOAc; 9:1). Man erhält 127 mg (Ausbeute
83%) das gewünschte Produkt als ein Gemisch aus Diastereomeren.
Rf 0,54 (Hex: EtOAc; 3:1)
de = 30 % (NMR) 1H-NMR (CDCl3): 1,41 (s, 9H); 1,50 (s, 18H); 2,13 (dd, J1= 15,04 Hz, J2= 8,46 Hz, 0,71H);
2,21 (dd, J1= 14,98 Hz, J2= 2,89 Hz,, 0,35H); 2,39 (dd, J1= 15,00 Hz, J2=10 ,27 Hz, 0,35H);
2,59 (dd, J1= 15,04 Hz, J2= 3,90 Hz,, 0,66H); 3,55 (s, 3H); 3,60 (dd, J1= 14,44 Hz,
J2= 9,34 Hz, 1H); 3,76 (s, 0,92H); 3,79 (s, 2,13H); 3,79 (dd, J1= 30,75 Hz, J2= 9,40 Hz, 1H);
4,95 (dd, J1= 8,53 Hz, J2= 3,96 Hz, 0,64H); 5,07 (dd, J1= 10,34 Hz, J2= 2,82 Hz, 1H) 13C-NMR (CDCl3): 27,85; 27,92; 28,02; 33,31; 33,04; 52,99; 53,13; 54,96; 59,47; 67,91;
68,45; 79,16; 81,59; 81,67; 82,94; 83,02; 151,64; 152,12; 169,08; 169,25; 170,50; 170,70
MS(ESI): 525,2542 [MNa+]
Synthese von (2R,4S)-4-(tert-Butoxycarbonyl)-4-(bis-Boc-amino-2-azido-2-
(methoxymethyl)butansäure (119)
Der Methylether 118 (127 mg; 0,253 mmol) wird in THF (2,5 ml) gelöst
und mit Lithiumhydroxid (9 mg; 0,38 mmol) gelöst in Wasser (2,5 ml)
versetzt. Nun wird 16 h bei RT gerührt und weiteres Lithiumhydroxid
(6 mg; 0,253 mmol) zugesetzt. Nachdem weitere 16 h bei RT gerührt
wurde, wird die Reaktionslösung angesäuert und mit EtOAc extrahiert. Die vereinigte
organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält das
gewünschte Produkt als ein Gemisch aus Diastereomeren in quantitativer Ausbeute.
Boc2N CO2tBu
CO2Me
MeO
N3
Boc2N CO2tBu
CO2H
MeO
N3

Experimenteller Teil
97
Rf 0,3 (CHCl3: MeOH; 9:1)
de = 37 % (NMR) 1H-NMR (CDCl3): 1,41 (s, 9H); 1,49 (s, 5H); 1,50 (s, 13H); 2,23 (m, 1H); 2,42 (dd,
J1= 14,84 Hz, J2= 10,68 Hz, 0,31H); 2,64 (dd, J1= 15,18 Hz, J2= 3,49 Hz, 0,67H); 3,37 (s,
2H); 3,38 (s, 0,89H); 3,64 (dd, J1= 14,91 Hz, J2= 9,40 Hz,, 1H); 3,81 (m, 1H); 4,99 (dd,
J1= 8,93 Hz, J2= 3,43 Hz, 0,67H); 5,14 (dd, J1= 10,48 Hz, J2= 2,82 Hz, 0,31H) 13C-NMR (CDCl3): 27,86 27,88; 28,00; 33,16; 52,02; 52,09; 59,45; 68,22; 81,65; 81,83;
83,23; 83,65; 152,13; 168,95; 170,30
MS(ESI): 487,2403 [MH-]

Experimenteller Teil
98
5.2.4. Studien zur Synthese von Labionin
Synthese von (2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-
(Methylsulfonyloxymethyl)pentandioat (76)
Das Azid (73) (50 mg; 0,102 mmol) wird in DCM (3 ml) gelöst und bei
0°C mit TEA (43 µl; 0,31 mmol; 3 eq.) und Mesylchlorid (10 µl;
0,13 mmol) versetzt. Nun wird 5 h bei RT gerührt. Anschließend mit
EtOAc verdünnt und mit Wasser und ges. NaCl-Lösung gewaschen. Die organische Phase
wird getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält 48 mg (Ausbeute
83 %) eines farbloses Öl als ein Gemisch aus Diastereomeren
Rf 0,26 (Hex: EtOAc; 3:1)
de = 57 % (NMR) 1H-NMR (DMSO-d6): 1,35 (s, 9H); 1,44 (s, 18H); 2,13 (dd, J1= 15,18 Hz, J2= 8,06 Hz, 1H);
2,54 (dd, J1= 15,11 Hz, J2= 4,10 Hz, 1H); 3,23 (s, 3H); 3,76 (s, 3H); 4,48 (d, J= 10,75 Hz,
1H); 4,57 (d, J= 10,60 Hz, 1H); 4,84 (dd, J1= 7,99 Hz, J2= 4,10 Hz, 1H) 13C-NMR (DMSO-d6): 27,39; 27,50; 32,98; 36,69; 53,52; 54,10; 67,16; 72,10; 81,14; 82,78;
151,49; 168,15; 168,43
MS(ESI): 567,2334 [MH+]; 589,2148[MNa+]
[α]D = -6,3 (c = 10,00 mg/ml; T = 23,5 °C; DCM)
Versuch zur Synthese von (2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-
(brommethyl)pentandioat (79)
Der Sulfonsäureester (76) (42 mg; 0,074 mmol) wird in DMF (2 ml)
gelöst und mit Kaliumbromid (88 mg; 0,74mmol; 10 eq) versetzt. Nun
wird 16 h bei 70 °C gerührt. Anschließend wird das Reaktionsgemisch
mit EtOAc verdünnt und mit ges. NaCl-Lösung gewaschen (5x). Die organische Phase wird
getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhielt nur das Edukt der
Reaktion.
Boc2N CO2tBu
CO2Me
BrN3
Boc2N CO2tBu
CO2Me
MsO
N3

Experimenteller Teil
99
Versuch zur Synthese von 2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-
(acetylmercaptomethyl)pentandioat (77)
Der Sulfonsäureester (76) (40 mg; 0,072 mmol) wird in DMF (0,75 ml)
gelöst und mit Kaliumcarbonat (22 mg; 0,16 mmol) versetzt. Nun wird
16 h bei 50 °C gerührt. Anschließend wird das Reaktionsgemisch mit
EtOAc verdünnt und mit ges. NaCl-Lösung gewaschen (5x). Die organische Phase wird
getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält nur das Edukt der
Reaktion.
Synthese von (2R,4S)-5-tert-Butyl-1-methyl-2-amino-4-(bis-Boc-amino)-2-
(hydroxymethyl)pentandioat (148)
Das Azid (73) (50 mg; 0,102 mmol) wird in THF (3 ml) gelöst und mit
Pd/C (20 mg) versetzt. Nun wird die Reaktionslösung 16 h bei RT unter
H2-Atmosphäre (1 bar) gerührt. Anschließend wird der Katalysator
abfiltriert und das Lösungsmittel im Vakuum entfernt. Man erhält 45 mg (Ausbeute 95 %)
eines leicht gelben Öls. Das Rohprodukt wird ohne weitere Reinigung zur Synthese von 149
direkt weiterverwendet.
Rf 0,2 (Hex: EtOAc; 1:1)
MS(ESI): 463,2632 [MH+]
Synthese von (2R,4S)-5-tert-Butyl-1-methyl-2-(Cbz-amino)-4-(bis-Boc-amino)-2-
(hydroxymethyl)pentandioat (149)
Das freie Amin 148 (45 mg; 0,097 mmol) wird in THF (0,25 ml) und
Acetonitril (0,4 ml) gelöst und anschließend mit TEA (15 µl;
0,107 mmol) und Cbz-OSu (27 mg; 0,108 mol) versetzt. Nachdem die
Reaktionslösung 4 d bei RT gerührt wurde, wird das Lösungsmittel im Vakuum entfernt und
der Rückstand in EtOAc aufgenommen. Nun wird mit ges. NaHCO3-Lösung gewaschen. Die
Phasen werden getrennt, die organische Phase wird getrocknet und das Lösungsmittel im
Vakuum entfernt. Das Rohprodukt wird chromatographisch gereinigt (Hex: EtOAc; 9:1 auf
3:1). Man erhält 44 mg (Ausbeute 76%) eines gelben Öls.
Boc2N CO2tBu
CO2Me
HONH2
Boc2N CO2tBu
CO2Me
HONHCbz
Boc2N CO2tBu
CO2Me
AcSN3

Experimenteller Teil
100
Rf 0,57 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 1,41 (s, 9H); 1,48 (s, 18H); 2,37 (dd, J1= 14,91 Hz, J2= 5,10 Hz, 1H); 2,79
(dd, J1= 15,25 Hz, J2= 5,31 Hz, 1H); 3,75 (s, 3H); 3,85 (m, 1H); 4,16 (d, J= 13,57 Hz, 1H);
4,85 (t, J= 5,17 Hz, 1H); 4,99 (d, J= 12,36 Hz, 1H); 5,13 (m, 1H); 6,14 (s, 1H); 7,34 (m, 5H) 13C-NMR (CDCl3): 27,78; 28,01; 33,99; 52,98; 54,89; 64,95; 66,81; 82,05; 83,35; 128,02;
128,44; 129,90; 152,15; 155,45; 169,88; 172,39
MS(ESI): 619,2840 [MNa+]
[α]D = -13,8 (c = 12 mg/ml; T = 24,46 °C; CHCl3)
Synthese von (2R,4S)-4-(tert-Butoxycarbonyl)-4-(bis-Boc-amino-2-(Cbz-amino)-2-
(hydroxymethyl)butansäure (81)
Die Aminosäure 149 (41 mg; 0,069 mmol) wird in THF (0,7 ml) gelöst
und mit Wasser (0,7 ml) versetzt. Anschließend wird die Suspension mit
Lithiumhydroxid (3,2 mg; 0,138 mmol) versetzt und 16 h bei RT
gerührt. Nun wird die Reaktionslösung angesäuert und mit EtOAc extrahiert. Die vereinigte
organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält
30 mg (Ausbeute 75 %) des Produktes.
Rf 0,27 ( CHCl3: MeOH; 9:2)
MS(ESI): 583,2869 [MH+]; 605,2682[MNa+]
Boc2N CO2tBu
CO2H
HONHCbz

Experimenteller Teil
101
5.2.5. Synthese des Dipeptids des A´-Ring, des Tripeptids des B´-Rings und des
Modellpeptids
Synthese von Fmoc-Thr(tBu)-(Tmb)Gly-OBn (83)
Fmoc-Thr(tBu)-OH (805 mg; 2,027 mmol) und Tmb-Gly-OBn *HCl
(500 mg; 1,36 mmol) werden in DCM (50 ml) gelöst und bei 0°C
mit HATU (770 mg; 2,03 mmol) und N-Ethylmorpholin (0,26 ml;
2,05 mmol) versetzt. Nach 15 min wird das Kältebad entfernt. Das
Reaktionsgemisch wird nun 5 d bei RT gerührt und anschließend mit
Wasser verdünnt und mit DCM (3x) extrahiert. Die organische
Phase wird mit ges. NaHCO3-Lösung gewaschen und getrocknet, dass Lösungsmittel wird im
Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:EtOAc; 5:1 auf
3:1). Man erhält 600 mg (Ausbeute 60%) eines farblosen Feststoffs.
Rf 0,26 (Hex:EtOAc; 3:1) 1H-NMR (CDCl3): 1,15 (d, J= 6,54 Hz, 3H); 1,25 (s, 9H); 3,66 (s, 6H); 3,79 (s, 3H); 3,83 (d,
J=17,19 Hz, 1H); 3,97 (m, 1H); 4,25 (t, J= 7,25 Hz, 1H); 4,33 (d, J=17,06 Hz, 1H); 4,40 (m,
1H); 4,44 (d, J=15,72 Hz, 1H), 5,00 (d, J= 12,49 Hz, 1H); 5,05 (d, J= 14,91 Hz, 1H); 5,11 (d,
J= 12,40 Hz, 1H); 5,38 (dd; J1= 7,86 Hz, J2= 3,83 Hz, 1H); 6,03 (s, 2H); 7,31 (m, 7H); 7,40
(t, J= 7,39 Hz, 2H); 6,72 (t, J= 6,72 Hz, 2H); 7,77 (d, J= 7,39 Hz, 2H) 13C-NMR(CDCl3): 18,52; 28,13; 41,25; 46,11; 47,29; 55,36; 55,85; 66,40; 66,76; 69,27;
74,56; 90,11; 104,05; 119,94; 125,21; 125,30; 127,07; 127,07; 127,67; 128,14; 128,45;
135,79; 141,31; 143,97; 155,64; 159,83; 161,52; 169,17; 170,33;
MS (ESI): 725,3449 [MH+]
Synthese von Fmoc-Thr(tBu)-(Tmb)Gly-OH (84)
Das Dipeptid 83 (600 mg, 0,92 mmol) wird in THF (4 ml) gelöst und
mit 5% Pd/C (130 mg) versetzt. Nun wird der Reaktionskolben mit
Stickstoff gespült und anschließend mit Wasserstoff befüllt. Nun wird
2 d bei RT und Normaldruck gerührt. Der Katalysator wird nach der
Reaktion abfiltriert und das Lösungsmittel im Vakuum entfernt. Das
Rohprodukt wird chromatographisch gereinigt (CHCl3: MeOH; 9:0,2 auf 9:1). Man erhält
420 mg (Ausbeute 82%) eines weißen Feststoffs.
NOBn
OO
NHFmoctBuO
OMe
MeO OMe
NOH
OO
NHFmoctBuO
OMe
MeO OMe

Experimenteller Teil
102
Rf 0,46 (CHCl3: MeOH; 9:1) 1H-NMR (CDCl3): 1,13 (d, J= 6,31 Hz, 3H); 1,29 (s, 9H); 3,74 (s, 6H); 3,80 (s, 3H); 4,24 (t,
J= 6,92 Hz, 1H); 4,43 (m, 4H); 4,95 (d, J= 14,51 Hz, 1H); 5,37 (dd; J1= 7,59 Hz,
J2= 4,23 Hz, 1H); 5,93 (d, J= 7,66 Hz, 1H); 6,08 (s, 2H); 7,39 (t, J= 7,39 Hz, 2H); 7,40 (t,
J= 7,39 Hz, 2H); 7,61 (t, J= 7,52 Hz, 2H); 7,77 (d, J= 7,52 Hz, 2H) 13C-NMR(CDCl3): 14,33; 28,22; 42,10; 46,86; 47,39; 55,49; 55,64; 55,75; 60,55; 66,92;
69,06; 75,52; 90,36; 103,58; 120,14; 125,21; 125,31; 127,19; 127,84; 141,45; 143,94; 144,01;
155,76; 159,84; 161,95; 171,14; 171,91
MS 635,2966 [MH+]; 657,2782 [MNa+]
Synthese von Boc-Glycindiphenylmethylester (85)
Boc-Gly-OH (4 g; 22,83 mmol) werden in DMF (130 ml) gelöst
und mit Kaliumcarbonat (3,2 g; 23,15 mmol) versetzt. Nun wird
Bromdiphenylmethan (5,64 g; 22,83 mmol) zugegeben und 16 h
bei RT gerührt. Anschließend wird das Reaktionsgemisch mit EtOAc (200 ml) verdünnt und
mit ges. NaCl-Lösung (4x 200 ml) gewaschen. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:
EtOAc; 9:1). Man erhält 2,24 g (Ausbeute 29%) eines farblosen Öls.
Rf 0,53 (Hex: EtOAc; 5:1) 1H-NMR (CDCl3): 1,44 (s, 9H); 4,02 (d, J= 5,24 Hz; 2H), 4,99 (s, 1H); 6,93 (s, 1H); 7,29 (m,
10H); 13C-NMR (CDCl3): 28,29; 42,72; 77,78; 80,02; 127,11; 128,10; 128,55; 139,59; 155,61;
169,48
MS (ESI): 342,1697 [MH+]; 364,1509 [MNa+]
Synthese von Glycindiphenylmethylester (86)
Boc-Gly-ODpm (85) (200 mg; 0,586 mmol) wird in Dioxan (1,4 ml)
gelöst und bei 0°C mit HCl in Dioxan (4 M; 11 ml) versetzt. Nun
wird 1 h bei RT gerührt anschließend das Lösungsmittel im Vakuum
BocHNO
O
H2NO
O

Experimenteller Teil
103
entfernt. Der Rückstand wird mit ges. NaHCO3-Lösung versetzt und mit EtOAc (2x 50 ml)
extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt.
Man erhält ein farbloses Öl in quantitativer Ausbeute.
Rf 0,77 (CHCl3:MeOH; 9:2) 1H-NMR (MeOH-d4): 3,76 (s, 1H); 3,99 (s, 2H); 6,98 (s,1H); 7,33 (m, 10H) 13C-NMR (MeOH-d4): 41,26; 80,45; 128,13; 129,35; 129,66; 140,76; 167,88
MS (ESI): 242,1146 [MH+]
Synthese von N-2,4,6-Trimethoxybenzylglycindiphenylmethylester (87)
H-Gly-ODpm (86) (141 mg; 0,585 mmol) wird in DCM
(18 ml) gelöst und mit Natriumborhydridtriacetat (172
mg; 0,811 mmol) und 2,4,6-Trimethoxybenzaldehyd
(161 mg; 0,82 mmol) versetzt. Nachdem das Reaktionsgemisch 2 h bei RT gerührt wurde,
wird die Reaktion durch Zugabe von Wasser (15 ml) beendet und mit EtOAc (3x 50 ml)
extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt.
Der Rückstand wird chromatographisch gereinigt (CHCl3: MeOH; 9:0,5). Man erhält 146 mg
(Ausbeute 53%) eines gelben Öls.
Rf 0,5 (CHCl3: MeOH; 9:1) 1H-NMR (CDCl3): 3,5 (s, 2H); 3,71 (s, 6H); 3,78 (s, 3H); 3,87 (s, 2H); 6,06 (s, 2H), 6,88 (s,
1H); 7,29 (m, 10H) 13C-NMR (CDCl3): 41,26; 49,07; 55,27; 55,55; 77,16; 90,34; 127,14; 127,90; 128,43; 139,94;
159,52; 160,84; 170,99
MS (ESI): 422,1949 [MH+]
Synthese von Fmoc-Thr(tBu)-(Tmb)Gly-ODpm (88)
Fmoc-Thr(tBu)-OH (830 mg; 2,088 mmol) und 87 (592 mg;
1,40 mmol) werden in DCM (100 ml) gelöst und bei 0°C mit
HATU (793 mg; 2,087 mmol) und N-Ethylmorpholin
(0,27 ml; 2,133 mmol) versetzt. Nach 15 min wird das
Kältebad entfernt. Das Reaktionsgemisch wird 16 h bei RT
gerührt und anschließend mit Wasser verdünnt und mit DCM (3x) extrahiert. Die organische
HN
O
OOMe
OMe
MeO
NO
OO
NHFmoctBuO
OMe
MeO OMe

Experimenteller Teil
104
Phase wird mit ges. NaHCO3-Lösung gewaschen und getrocknet, dass Lösungsmittel wird im
Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:EtOAc; 2:1). Man
erhält 508 mg (Ausbeute 45 %) eines farblosen Öls.
Rf 0,375 (Hex:EtOAc; 2:1) 1H-NMR (CDCl3): 1,12 (d; J= 6,45 Hz, 3H); 1,26 (s, 9H); 3,55 (s, 6H); 3,77 (s, 3H); 3,83 (d,
J= 17,06 Hz, 1H); 3,96 (ddd; J1= 6,45 Hz, J2= 5,91 Hz, J3= 3,36 Hz, 1H); 4,24 (t, J= 7,12 Hz,
3H); 4,43 (m, 4H); 5,10 (d, J= 14,78 Hz; 1H); 5,41 (dd, J1= 7,79 Hz, J2= 3,76 Hz, 1H); 5,99
(m, 3H), 6,82 (s, 1H); 7,26 (m, 14H); 7,38 (t, J= 7,46 Hz, 2H); 7,62 (t, J= 6,51 Hz, 2H); 7,75
(d, J= 7,52 Hz, 2H) 13C-NMR(CDCl3): 18,41; 28,10; 41,26; 46,27; 47,32; 55,23; 55,80; 66,76; 69,29; 74,62;
77,19; 90,18; 104,10; 119,92; 125,19; 125,27; 127,05; 127,19; 127,27; 127,65; 127,75;
128,31; 128,34; 140,00; 141,32; 144,01; 155,59; 159,86; 161,56; 168,43; 170,99
MS (ESI): 801,3732 [MH+]; 823,3541 [MNa+]
Synthese von cyclo [Thr(tBu)-(Tmb)Gly)] (89)
Das N-(2-Mercaptoethyl)-aminomethyl-Harz (0,6 g; 1,4 mmol/g)
wird in eine mit einer Fritte versehene Spritze gegeben und mit
Fmoc-Thr(tBu)-(Tmb)Gly-ODpm (88) (60 mg, 0,075 mmol)
gelöst in THF (2ml) versetzt. Nun wird DBU (20 µl 0,13 mmol)
zugegeben und 1 h bei RT geschüttelt. Anschließend wird das
Harz abfiltriert, das Lösungsmittel im Vakuum entfernt und das Rohprodukt
chromatographisch gereinigt (DCM:MeOH 9:1 auf 9:2). Man erhält 300 mg eines farblosen
Öls in quantitativer Ausbeute.
Rf 0,63 (CHCl3: MeOH; 9:2) 1H-NMR (DMSO-d6): 1,06 (s, 9H); 1,09 (d, J= 6,31Hz, 3H); 3,15 (d, J= 3,15Hz, 3H); 3,53
(dd, J1= 3,43 Hz, J2= 2,08 Hz, 1H); 3,64 (d, J= 16,79Hz, 1H); 3,75 (s, 6H); 3,79 (s, 3H); 4,02
(dd, J1= 6,51 Hz, J2= 2,08 Hz, 1H); 4,07 (d, J= 13,57Hz, 1H); 4,72 (d, J= 13,57Hz, 1H); 6,26
(s, 2H) 13C-NMR (DMSO-d6): 20,13; 28,22; 37,04; 47,08; 55,24; 55,72; 61,45; 68,92; 73,33; 90,76;
101,92; 159,62; 161,26; 161,26; 165,84; 166,69
MS (ESI): 395,2163 [MH+]; 417,1980 [MNa+]
NH
N
O
O
OtBu
OMe
OMe
MeO

Experimenteller Teil
105
Synthese von Boc-Thr(tBu)-Gly-OBn (90)
H-Gly-OBn*pTos (1,27 g; 3,76 mmol) und Boc-Thr(tBu)-OH
(1 g; 3,63 mmol) werden in THF (63 ml) gelöst und bei 0°C mit
HOAt (1,44 g; 10,57 mmol) versetzt. Nach 5 min wurde EDAC (2,02 g; 10,55 mmol)
zugegeben und 5 h bei RT gerührt. Nun wird das Reaktionsgemisch mit Ethylacetat (250 ml)
verdünnt und mit 5%iger HCl-Lösung (3x60 ml), ges. NaHCO3-Lösung (2x60 ml), Wasser
(2x60 ml) und ges. Natriumchloridlösung (2x60 ml) gewaschen. Die organische Phase wird
getrocknet und das Lösungsmittel im Vakuum entfernt. Der Rückstand wird
chromatographisch gereinigt (Hex: EtOAc; 5:1 auf 2:1) Man erhält 0,52 g (Ausbeute 44%)
eines weißen Feststoffs.
Rf 0,42 (Hex:EtOAc; 3:1) 1H-NMR (DMSO-d6): 1,02 (d, J= 6,04 Hz, 3H); 1,08 (s, 9H); 1,38 (s, 9H); 3,92 (m, 4H); 5,1
(s, 2H); 6,01 (d, J= 8,60 Hz, 1H), 7,33 (m, 5H) 13C-NMR(CDCl3): 17,25; 28,26; 28,38; 41,53; 58,41; 66,90; 67,15; 75,34; 79,67; 128,44;
128,56; 128,66, 135,15; 155,60; 169,44; 170,25
MS (ESI): 423,2475 [M+H]; 445,2291 [M+Na]
Synthese von H-Thr(tBu)-Gly-OBn (91)
Boc-Thr(tBu)-Gly-OBn (92) (0,503 g; 1,19 mmol) wird in Dioxan
(1,5 ml) gelöst und bei 0 °C mit HCl in Dioxan (4 M; 11 ml) versetzt.
Nun wird 1 h bei RT gerührt anschließend das Lösungsmittel im
Vakuum entfernt. Der Rückstand wird mit ges. NaHCO3-Lösung versetzt und mit EtOAc (2x
100 ml) extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum
entfernt. Man erhält ein farbloses Öl in quantitativer Ausbeute (0,382 g).
Rf 0,51 (CHCl3: MeOH; 9:1) 1H-NMR (DMSO-d6): 1,06 (d, J= 6,31 Hz, 3H); 1,08 (s, 9H); 3,02 (d, J= 4,16 Hz, 1H); 3,83
(dd, J1= 6,11 Hz, J2= 4,23 Hz, 1H); 3,92 (dd, J1= 5,51 Hz, J2= 2,55 Hz, 2H); 5,12 (s, 2H);
7,34 (m, 5H) 13C-NMR (DMSO-d6): 20,12; 28,29; 40,67; 59,92; 65,75; 68,10; 73,07; 127,88; 127,99;
128,34; 135,84; 169,65; 173,38
BocHNO
OtBuHN CO2Bn
H2NO
OtBuHN CO2Bn

Experimenteller Teil
106
MS (ESI): 323,2969 [MH+]; 354,1788 [MNa+]
Synthese von Cbz-γ-Glu-Thr(tBu)-Gly-OBn (92)
H-Thr(tBu)-Gly-OBn (91) (0,203 g;
0,63 mmol) und Cbz-Glu(OH)-OtBu
(0,276 g; 0,819 mmol) werden in THF (11 ml) gelöst und bei 0 °C mit HOAt (0,3 g; 2,2
mmol) versetzt. Nach 5 min wird EDAC (0,36 g; 1,88 mmol) zugegeben und 16 h bei RT
gerührt. Nun wird das Reaktionsgemisch mit Ethylacetat (50 ml) verdünnt und mit 5%iger
HCl-Lösung (3x10 ml), ges. NaHCO3-Lösung (2x10 ml), Wasser (2x10 ml) und ges.
Natriumchloridlösung (2x10 ml) gewaschen. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:
EtOAc; 5:1 auf 2:1) Man erhält 240 mg (Ausbeute 60%) eines leicht gelben Öls.
Rf 0,27 (Hex:EtOAc; 3:1) 1H-NMR (DMSO-d6): 0,99 (d, J= 6,18 Hz, 3H); 1,10 (s, 9H); 1,38 (s, 9H); 1,73 (m, 1H); 1,91
(td, J1= 13,43 Hz, J2= 7,79 Hz, 1H); 2,27 (t, J= 7,66 Hz, 2H); 3,86 (m, 2H); 3,92 (d,
J= 5,78 Hz, 2H); 4,29 (dd, J1= 8,73 Hz, J2= 4,16 Hz, 1H); 5,24 (d, J= 5,28 Hz, 2H); 5,11 (s,
2H); 7,34 (m, 10H); 7,61 (d, J= 7,66 Hz, 1H); 7,66 (d, J= 8,73 Hz, 1H); 8,14 (t, J= 5,57 Hz,
1H) 13C-NMR (DMSO-d6): 19,28; 27,70; 28,16; 29,06; 31,59; 40,93; 54,23; 57,45; 65,48; 65,91;
67,33; 73,73; 80,61; 127,84; 127,90; 128,02; 128,14; 128,41; 128,49; 135,93; 137,06; 156,14
169,60; 170,31; 171,33; 173,48
MS (ESI): 642,3384 [MH+]; 664,3196 [MNa+]
Synthese von H-γ-Glu-Thr(tBu)-Gly-OBn (93)
Das Peptid Cbz-γ-Glu-Thr(tBu)-Gly-OBn (92)
(205 mg; 0,32 mmol) wird in Methanol (10 ml)
gelöst und Pd/C (20 mg) versetzt. Nun wird 16 h unter H2 (1 bar) gerührt. Anschließend wird
der Katalysator abfiltriert und das Lösungsmittel im Vakuum entfernt. Man erhält 121 mg
eines farblosen Feststoffs (Ausbeute 91%).
Rf 0,625 (n-Butanol: EtOAc: AcOH: H2O; 2:1:1:1)
NH O
OtBuHN CO2Bn
O
NHCbz
tBuO2C
NH O
OtBuHN CO2H
O
NH2
tBuO2C

Experimenteller Teil
107
1H-NMR (DMSO-d6): 0,99 (d, J= 6,31 Hz, 3H); 1,12 (s, 9H); 1,41 (s, 9H); 1,73 (m, 1H); 1,91
(td, J1= 13,43 Hz, J2= 7,79 Hz, 1H); 2,27 (t, J= 7,66 Hz, 2H); 3,86 (m, 2H); 3,92 (d, J= 5,78
Hz, 2H); 4,29 (dd, J1= 8,73 Hz, J2= 4,16 Hz, 1H); 7,61 (d, J= 7,66 Hz, 1H); 7,66 (d, J= 8,73
Hz, 1H); 8,14 (m, 2H) 13C-NMR (DMSO-d6): 19,39; 27,72; 28,18; 28,41; 30,75; 41,98; 53,32; 55,47; 73,72; 81,04;
171,67; 172,21; 172,77; 174,13
MS (ESI): 418,2538 [MH+]
Synthese von Cyclo[γ-Glu-Thr(tBu)-Gly] (94)
Tabelle 3: Reagenzien zur Zyklisierung von 93
HNO
OtBu
NHO
HN
tBuO2CO
H-γ-Glu-Thr(tBu)-Gly-OH (93) (10 mg; 24 µmol) wird in DMF (0,35 ml) gelöst und mit dem
Kupplungsreagenz und der Base (siehe Tabelle; Mix 1-3) versetzt nun wird 2 d bei RT
geschüttelt. Das gewünschte Produkt konnte mit allen genannten Reaktionsbedingungen
massenspektrometrisch nachgewiesen werden.
MS (ESI): 400,2267 [MH+]; 422,2267 [MNa+]
Synthese von Imidazol-1-Sulforyl-Azid *Hydrochlorid (95)[162]
NaN3 (0,63 g; 9,7 mmol) wird in Acetonitril (10 ml) suspendiert und
bei 0 °C mit Sulfurylchlorid (0,8 ml; 9,9 mmol) versetzt. Nachdem die
Reaktionslösung 20 h bei RT gerührt wurde, wird Imidazol (1,3 g; 19,1 mmol) bei 0 °C
zugegeben und 3 h bei RT gerührt. Nun wird mit EtOAc (20 ml) verdünnt und mit Wasser
(3x20 ml) und ges. NaHCO3-Lösung (2x20 ml) gewaschen. Die organische Phase wird
getrocknet und bei 0 °C mit HCl/ EtOH (hergestellt aus 1 ml Acetylchlorid und 3,75 ml
EtOH) versetzt. Das kristalline Produkt (1,36 g) wird abfiltriert (Ausbeute 67%).
Reagenz Menge
DPPA 15,5 µl 3 eq Mix 1
NaHCO3 10,1 mg 5 eq
HATU 9,5 mg 1 eq Mix 2
Collidin 3,5 µl 3 eq
DEPBT 14,4 mg 1 eq Mix 3
N-Ethylmorpholin 9,5 µl 2 eq
SO
ON3N
N *HCl

Experimenteller Teil
108
1H-NMR (D2O): 7,62 (m, 1H); 8,03 (m, 1H); 9,32 (m, 1H) 13C-NMR (D2O): 120,32; 124, 68; 138,11
MS (ESI): 147,0078 [MH+]
Synthese von (2S,3R)-2-Azido-3-(benzyloxy)butansäure (97)
H-Thr(Bn)-OH (0,8 g; 3,83 mmol), CuSO4*5H2O (20 mg; 0,08 mmol) und
K2CO3 (1,24 g; 9 mmol) werden in Methanol (20 ml) gelöst. Nun wird
Imidazol-1-Sulforyl-Azid *HCl (95) (0,96 g; 4,6 mmol) zugegeben und 16 h
bei RT gerührt. Anschließend wird das Lösungsmittel im Vakuum entfernt und der Rückstand
in Wasser (15 ml) aufgenommen, mit konz. Salzsäure angesäuert und mit EtOAc (3x20ml)
extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt.
Das Rohprodukt wird chromatographisch gereinigt (Hex: EtOAc; 3:1 auf 1:1). Man erhält
0,66 g eines farblosen Öls (Ausbeute 73%).
Rf 0,67 (CHCl3: MeOH; 9:1)
ee = 99 % (LC-MS; Marfey-Derivatisierung) 1H-NMR (DMSO-d6): 1,26 (d, J= 6,31 Hz, 3H); 3,91 (d, J= 2,96 Hz, 1H); 4,14 (ddd,
J1= 12,49 Hz, J2= 6,18 Hz, J3= 2,94 Hz, 1H) ; 4,39 (d, J= 11,95 Hz, 1H); 4,60 (d, J= 12,09
Hz, 1H); 7,28 (m, 5H) 13C-NMR (DMSO-d6): 16,55; 65,33; 70,25; 75,54; 127,03; 127,35; 128,16; 138;33; 170,45
MS (ESI): 234,0880 [MH-]
[α]D = 11,3 (c = 9,33 mg/ml, T = 23,48 °C, EtOAc)
Synthese von 2-((2S,3R)-2-Azido-3-(benzyloxy)butanazido)essigsäure-tert-butylester (98)
97 (0,483 g; 2,05 mmol) und H-Gly-OtBu*HCl (1,02 g; 6,09 mmol)
werden in THF (36 ml) gelöst und bei 0 °C mit TEA (0,28 ml; 2,05
mmol) und Cl-HOBt (1,2 g; 7,08 mmol) versetzt. Nachdem 15 min bei 0 °C gerührt wurde,
wird EDAC (1,15 g; 6,014 mmol) zugeben. Nun wird das Kühlbad entfernt und 16 h bei RT
gerührt. Anschließend wird das Reaktionsgemisch mit Ethylacetat (130 ml) verdünnt und mit
5%iger HCl-Lösung (2x30 ml), ges. NaHCO3-Lösung (2x30 ml), Wasser (2x30 ml) und ges.
Natriumchloridlösung (3x10 ml) gewaschen. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:
N3
OBn
O
HN CO2tBu
N3
OBn
O
OH

Experimenteller Teil
109
EtOAc; 5:1 auf 2:1). Man erhält 0,71 g eines leicht gelben viskosen Öls (Ausbeute
quantitativ).
Rf 0,44 (Hex:EtOAc; 3:1) 1H-NMR (CDCl3): 1,33 (d, J= 6,31 Hz, 3H); 1,49 (s, 9H); 3,84 (dd, J1= 18,20 Hz,
J2= 4,90 Hz, 1H); 4,00 (m, 3H); 4,51 (d, J= 11,69 Hz, 1H); 4,61 (d, J= 11,70 Hz, 1H); 6,97 (t,
J= 4,16 Hz, 1H); 7,29 (m, 5H) 13C-NMR (CDCl3): 16,49; 27,95; 41,90; 67,83; 70,25; 71,66; 75,53; 82,37; 127,73; 128,33;
137,63; 167;85; 168,85
MS (ESI): 349,1864 [MH+]; 371,1679 [MNa+]
Synthese von 2-((2S,3R)-2-Azido-3-(benzyloxy)butanazido)essigsäure (99)
Das Peptid 98 (0,19 g; 0,574 mmol) wird bei 0 °C mit TFA (4 ml) und
TES (0,2 ml; 1,25 mmol) versetzt. Nun wird 4 h bei RT gerührt und
anschließend die TFA im Vakuum entfernt. Man erhält das gewünschte Peptid in quantitativer
Ausbeute.
Rf 0,36 (CHCl3: MeOH; 9:1) 1H-NMR (DMSO-d6): 1,21 (d, J= 6,31 Hz, 3H); 3,81 (m, 3H); 3,93 (dt, J1= 12,29 Hz, J2=
6,21, 1H); 4,48 (d, J= 11,82 Hz, 1H); 4,58 (d, J= 11,9 Hz, 1H); 7,29 (m, 5H); 8,58 (t, J= 5,84
Hz, 1H) 13C-NMR (DMSO-d6): 16,69; 40,74; 65,81; 70,44; 75,15; 127,43; 127,54; 128,19; 138;38;
168,37; 170,85
MS (ESI): 293,1245 [MH+]; 315,1061 [MNa+]
Synthese von Boc-Leu-Phe-OH (150)
Boc-Leu-OSu (1,24 g; 3,77 mmol) wird in THF (55 ml) gelöst und
mit H-Phe-OH (0,62 g; 3,75 mmol) und NaHCO3 (0,32 g;
3,8 mmol) gelöst in Wasser (55 ml) versetzt. Nachdem 16 h bei RT
gerührt wurde, wird das Reaktionsgemisch mit einer 5%igen
NaOH-Lösung auf pH 10 eingestellt und mit Ethylacetat (2x 50 ml) gewaschen. Nun wird mit
5%iger HCl-Lösung auf pH 5 angesäuert und mit Ethylacetat (3x 50 ml) extrahiert. Die
N3
OBn
O
HN CO2H
BocHNO
HN
OH
O

Experimenteller Teil
110
organische Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält
1,34 g (Ausbeute 95%) eines weißen Feststoffs.
Rf 0,36 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 0,88 (t, J= 7,05 Hz, 6H); 1,43 (s, 9H); 1,57 (m, 2H); 2,99 (dd,
J1= 13,97 Hz, J2= 6,31 Hz, 1H); 3,18 (d, J= 13,16 Hz, 1H); 4,19 (m, 1H); 4,31 (d,
J= 5,37 Hz, 1H); 5,06 (d, J= 6,85 Hz, 1H); 7,19 (m, 5H) 13C-NMR (CDCl3): 24,61; 28,28; 37,37; 41,05; 52,89; 53,21; 53,17; 80,53; 127,01; 128,44;
129,46; 135,91; 155,23; 172,52
MS (ESI): 379,22 [MH+]; 401,20 [MNa+]
Synthese von Boc-Leu-Phe-Ala-OMe (100)
Das Dipeptid (150) (0,806 g; 2,13 mmol) wird in THF
(37 ml) gelöst und bei 0°C mit HOAt (1 g; 7,34 mmol)
und H-Ala-OMe*HCl (0,88 g; 6,32 mmol) versetzt. Nach
5 min wird EDAC (1,2 g; 6,27 mmol) zugegeben und 16
h bei RT gerührt. Nun wird das Reaktionsgemisch mit Ethylacetat (130 ml) verdünnt und mit
5%iger HCl-Lösung (2x30 ml), ges. NaHCO3-Lösung (2x30 ml), Wasser (2x30 ml) und ges.
Natriumchloridlösung (2x60 ml) gewaschen. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Man erhält 2,14 g (Ausbeute 89%) eines weißen
Feststoffs.
Rf 0,35 (Hex: EtOAc; 1:1) 1H-NMR (CDCl3): 0,79 (dd, J1= 15,78 Hz, J2= 6,51 Hz, 6H); 1,19 (m, 1H); 1,27 (d, J= 7,25
Hz, 3H); 1,35 (s, 1H); 1,45 (m, 1H); 2,77 (dd, J1= 13,84 Hz, J2= 9,27 Hz, 1H); 2,99 (dd,
J1= 13,90 Hz, J2= 4,50 Hz, 1H); 3,61 (s, 3H); 3,86 (m, 1H); 4,26 (dd, J1= 14,37 Hz,
J2= 7,19 Hz, 1H); 3,86 (m, 1H); 4,56 (dd, J1= 8,16 Hz, J2= 4,37 Hz, 1H); 6,84 (d, J= 8,46 Hz,
1H); 7,18 (m, 5H); 7,69 (d, J= 8,46 Hz, 1H); 8,42 (d, J= 6,98 Hz, 1H) 13C-NMR (CDCl3): 16,94; 21,63; 22,90; 24,23; 28,23; 37,84; 40,99; 47,65; 51,94; 53,01;
53,17; 78,18; 126,26; 127,98; 129,38; 137,52; 155,23; 170,94; 172,14; 172,85
MS (ESI): 464,2740 [MH+]; 486,2556 [MNa+]
[α]D = -20,6 (c = 11,49 mg/ml, T = 22,98 °C, CHCl3)
BocHNO
HN
NH
OOMe
O

Experimenteller Teil
111
Synthese von H-Leu-Phe-Ala-OMe (101)
Das Tripeptid 100 (200 mg, 0,432 mmol) wird in
Dioxan (1 ml) gelöst und bei 0 °C mit HCl in Dioxan
(4 M, 8 ml) versetzt. Das Kältebad wird entfernt und die
Reaktionsmischung 1 h bei RT gerührt. Nun wird mit
EtOAc (40 ml) verdünnt und mit 5%iger HCl-Lösung
(2x 60 ml), ges. NaHCO3-Lösung (2x 60 ml), Wasser
(2x 60 ml) und ges. NaCl-Lösung (2x 60 ml) gewaschen. Die organische Phase wird
getrocknet und das Lösungsmittel im Vakuum entfernt. Man erhält einen weißen Feststoff in
quantitativer Ausbeute (156 mg).
Rf 0,45 (CHCl3: MeOH; 9:1) 1H-NMR (DMSO-d6): 0,79 (dd, J1= 15,78 Hz, J2= 6,65 Hz, 6H); 1,04 (ddd, J1= 13,50 Hz,
J2= 9,00 Hz, J3= 5,71 Hz, 1H); 1,22 (m, 1H); 1,28 (d, J= 7,25 Hz, 3H); 1,58 (m, 1H); 2,78
(dd, J1= 13,84 Hz, J2= 9,00 Hz, 1H); 3,02 (dd, J1= 13,84 Hz, J2= 4,70 Hz, 1H); 3,09 (dd;
J1= 9,07 Hz, J2= 5,17 Hz, 1H); 3,62 (s, 3H), 4,28 (dt; J1= 14,41 Hz, J2= 7,24 Hz, 1H); 4,58
(m, 1H); 7,19 (m, 5H); 7,97 (d, J= 7,66 Hz, 1H); 8,45 (d, J= 7,12 Hz, 1H) 13C-NMR (DMSO-d6): 16,83; 21,65; 23,13; 23,89; 37,87; 43,92; 47,49; 51,81; 52,54; 53,03;
126,14; 127,85; 129,28; 137,44; 170,95; 172,79; 175,10
MS (ESI): 364,2246 [MH+]
[α]D = -29,32 (c = 10,1 mg/ml, T = 22,99 °C, CHCl3)
H2NO
HN
NH
OOMe
O

Experimenteller Teil
112
5.2.6. Synthese des A´-Rings von Labyrinthopeptin A2
Synthese von Peptid 102
Die Aminosäure 74 (100 mg; 0,21 mmol) und das
Peptid H-Leu-Phe-Ala-OMe*HCl (93,3 mg;
0,23 mmol) werden in THF (4 ml) gelöst und bei 0 °C
mit HOAt versetzt. Nach 15 min wird das Kältebad
entfernt, EDAC (116 mg; 0,607 mmol) zugegeben
und bei RT gerührt. Nach 16 h wird mit EtOAc (100 ml) verdünnt und mit 5%iger HCl-
Lösung (3x 30 ml), ges. NaHCO3-Lösung (3x 30 ml), Wasser (3x 30 ml) und ges. NaCl-
Lösung (3x 30 ml) gewaschen. Die organische Phase wird getrocknet und das Lösungsmittel
im Vakuum entfernt. Man erhält 128 mg (Ausbeute 74 %) eines Öls.
Rf 0,65 (Hex: EtOAc; 1:1)
de = 83 % (NMR) 1H-NMR (CDCl3): 0,88 (dd, J1= 6,31 Hz, J2= 3,09 Hz, 6H); 1,34 (d, J= 7,25 Hz, 3H); 1,40 (d,
J= 4,41 Hz, 1H); 1,43 (s, 9H); 1,46 (s, 18H); 1,48 (d, J= 2,15 Hz, 1H); 1,55 (m, 3H); 2,09
(dd, J1= 14,78 Hz, J2= 7,93 Hz, 1H); 2,50 (dd, J1= 14,64 Hz, J2= 3,49 Hz, 1H); 2,94 (dd,
J1= 14,10 Hz, J2= 8,87 Hz, 1H); 3,29 (dd, J1= 13,90 Hz, J2= 5,57 Hz, 1H); 3,69 (s, 3H); 4,18
(dt, J1= 8,66 Hz, J2= 5,54 Hz, 1H); 4,27 (d, J= 11,55 Hz, 1H); 4,39 (d, J= 11,40 Hz, 1H);
4,44 (m, 1H); 4,66 (td, J1= 8,50 Hz, J2= 5,71 Hz, 1H); 5,12 (d, J= 6,72 Hz, 1H); 6,37 (d,
J= 8,33 Hz, 1H); 6,65 (d, J= 6,18 Hz, 1H); 7,05 (d, J= 3, Hz, 1H); 7,22 (m, 5H) 13C-NMR (CDCl3): 17,76; 21,91; 22,80; 24,60; 27,66; 27,86; 28,24; 35,10; 37,34; 39,93;
48,33; 52,29; 54,15; 67,91; 69,58; 77,20; 82,98; 83, 89; 126,79; 128,55; 129,18; 152,39;
168,93; 170,21; 170,29; 170,77; 172,65
MS(ESI): 820,4435 [MH+]; 842,4236 [MNa+]
NH
HN
ONH
O
CO2Me
ON3
HO
NBoc2
CO2tBu
Experimenteller Teil
113
Synthese von Peptid 103
Das Peptid 102 (120 mg; 0,146 mmol) wird in TFA
(0,5 ml) gelöst und mit Triethylsilan (273 µl;
1,71 mmol) versetzt. Nun wird 4 h bei RT gerührt und
anschließend TFA im Vakuum entfernt. Das
Rohprodukt wird direkt weiter verwendet.
MS(ESI): 564,2763 [MH+]; 586,2579 [MNa+]
Synthese von Peptid 104
Das Peptid 103 (82 mg; 0,146 mmol) wird in
Acetonitril (0,5 ml) gelöst und mit TEA (88 µl; 0,63
mmol) und Boc2O (34 µl; 0,160 mmol) versetzt. Nun
wird 16 h bei RT gerührt. Anschließend wird das
Lösungsmittel im Vakuum entfernt. Man 95 mg des
gewünschten Produkts in einer Ausbeute von 98 %.
MS(ESI): 664,3291 [MH+]; 686,3103 [MNa+]
Synthese von Peptid 105
104 (14 mg; 0,021 mol) und H-Thr(tBu)-Gly-OBn
(9,14 mg; 0,028 mmol) werden in THF (1 ml)
gelöst und bei 0 °C mit HOBt (10 mg;
0,073 mmol) versetzt. Nach 15 min wird das
Kältebad entfernt und EDAC (12 mg; 0,063 mmol)
zugegeben. Nun wird 16 h bei RT gerührt.
Anschließend wird das Reaktionsgemisch mit
EtOAc verdünnt und mit 5%iger HCl-Lösung (3x),
ges. NaHCO3-Lösung (3x), Wasser (3x) und ges.
NaCl-Lösung (3x) gewaschen. Die organische
Phase wird getrocknet und das Lösungsmittel im Vakuum entfernt.
MS(ESI): 968,8099 [MH+]; 990,4912 [MNa+]
NH
HN
ONH
O
CO2Me
ON3
HO
NH2
CO2H
NH
HN
ONH
O
CO2Me
ON3
HO
NHBoc
CO2H
NH
HN
ONH
O
CO2MeO
N3
HO
NHBoc
ONH
tBuO OHN
OBnO
Experimenteller Teil
114
Synthese von Peptid 106
Das Peptid 105 (20 mg; 0,021 mmol) wird in
Methanol (1 ml) gelöst und mit Pd/C (4 mg) versetzt.
Nun wird 2 d unter Wasserstoffatmosphäre (1 bar)
gerührt. Anschließend wird der Katalysator abfiltriert
und das Lösungsmittel im Vakuum entfernt. Man
erhält das gewünschte Produkt in einer Ausbeute 95 %
( 17 mg).
LC-MS: 852,5 [MH+]
Versuch zur Synthese von Peptid 107
Tabelle 4: Reagenzien zur Zyklisierung von 106
Das Peptid 106 (5,26 µmol) wird in DMF (1 ml) gelöst und mit dem Kupplungsreagenz und
der Base (Mix 1-3; Tab. 4) versetzt nun wird 2 d bei RT geschüttelt. Das gewünschte Produkt
konnte nicht identifiziert werden.
Reagenz Menge
DPPA 0,4 µl 3 eq Mix 1
NaHCO3 2,4 mg 5 eq
HATU 0,3 mg 3 eq Mix 2
Collidin 0,1 µl 5 eq
DEPBT 0,4 mg 3 eq Mix 3
N-Ethylmorpholin 0,1 µl 5 eq
NH
HN
ONH
O
CO2MeO
H2NHO
NHBoc
ONH
tBuO OHN
OHO
HNNH
OHN
O
CO2Me
OHNHO
ONHtBuO
O
HN
O
NHBoc

Experimenteller Teil
115
Synthese von Peptid 109
Das Peptid 102 (48,5 mg; 0,059 mmol) wird in THF
(3,2 ml) gelöst und unter Argon mit PBu3 (64 µl;
0,25 mmol) und Wasser (32 µl) versetzt. Nun wird 1
6 h bei RT unter Argon gerührt. Anschließend wird
das überschüssige PBu3 im Luftstrom oxidiert und das Lösungsmittel im Vakuum entfernt.
Das erhaltene Produkt wird direkt weiterverwendet.
Rf 0,26 (Hex: EtOAc; 1:1)
MS(ESI): 794,4513 [MH+]; 816,4318 [MNa+]
Synthese von Peptid 108
Das Peptid 109 (6 mg, 0,007
mmol) wird THF (0,4 ml) gelöst
und mit HATU (4,2 mg;
0,01 mmol), DIPEA (3 µl; 17,64
µmol) und Cbz-Gly-OH (2,3 mg;
0,01 mmol) versetzt. Das Reaktionsgemisch wird 16 h bei RT gerührt. Nun wird das
Reaktionsgemisch mit 5% HCl (1 ml) versetzt und 15 min bei RT gerührt, anschließend wird
durch Zugabe von ges. K2CO3-Lösung pH 10 eingestellt und ebenfalls 15 min bei RT gerührt.
Das gewünschte Produkt wurde nur massenspektrometrisch nachgewiesen.
MS(ESI): 985,5146 [MH+]; 1007,4954 [MNa+]
Synthese von Peptid 110
Das Peptid 109 (48,5 mg; 0,06 mmol)
wird in THF (3,5 ml) gelöst und bei
0 °C mit HATU (36 mg; 0,095 mmol),
DIPEA (36 µl; 0,221 mmol) und 99
(26,3 mg; 0,09 mmol) zugegeben. Das Reaktionsgemisch wird 2 d bei RT gerührt.
Anschließend wird das Lösungsmittel im Vakuum entfernt und der Rückstand mittels
NH
HN
ONH
O
CO2Me
OH2NHO
NBoc2
CO2tBu
NH
HN
ONH
O
CO2Me
OHN
HO
NBoc2
CO2tBuOCbzHN
NH
HN
ONH
O
CO2Me
OHN
HO
NBoc2
CO2tBuONH
ON3
OBn

Experimenteller Teil
116
Säulenchromatographie (CHCl3: MeOH; 9:0,5) grob gereinigt. Es wurden 20 mg des
gewünschte Peptid erhalten ( Ausbeute 32 %).
Rf 0,47 (CHCl3: MeOH; 9:1)
MS(ESI): 1068,5607 [MH+]; 1090,5416 [MNa+]
Synthese von Peptid 111
Das Peptid 110 (20 mg,
19 µmol) wird bei 0 °C mit TFA (200
µl) und TES (76 µl) versetzt. Nun wird
5 h bei RT gerührt. Anschließend wird
das Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird ohne weitere Reinigung direkt
weiter verwendet.
Rf 0,37 (CHCl3: MeOH; 9:2)
MS(ESI): 812,3930 [MH+]; 834,3741 [MNa+]
Synthese von Peptid 112
Das Peptid 111 (15 mg; 19 µmol) wird
in Acetonitril (0,3 ml) gelöst und mit
TEA (11 µl; 79 µmol) und Boc2O
(5 µl; 24 µmol) versetzt. Nun wird 16 h
bei RT gerührt. Anschließend wird das
Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird ohne weitere Reinigung direkt
weiter verwendet.
Rf 0,37 (CHCl3: MeOH; 9:1)
NH
HN
ONH
O
CO2Me
OHN
HO
NH2
CO2HONH
ON3
OBn
NH
HN
ONH
O
CO2Me
OHN
HO
NHBoc
CO2HONH
ON3
OBn
Experimenteller Teil
117
Synthese von Peptid 113
Das Peptid 112 (19 µmol) wird in
THF (0,4 ml) gelöst und mit H2O (10
µl) und PBu3 (16 µl; 64 µmol)
versetzt. Nun wird 2 d bei RT gerührt.
Anschließend wird das Lösungsmittel
im Vakuum entfernt. Das Rohprodukt wird ohne weitere Reinigung direkt weiterverwendet.
MS(ESI): 886,4589 [MH+]
Versuch zur Synthese von Peptid 114
Peptid 113 (19 µmol) wird in DMF (2 ml)
gelöst und mit DEPBT (28 mg;
93,5 µmol) und 4-Ethylmorpholin (6 µl;
54 µmol) versetzt. Nun wird 7 d bei RT
gerührt. Anschließend wird das
Reaktionsgemisch mit TFA (0,5 ml)
versetzt, 0,5 h bei RT gerührt und das Lösungsmittel nun im Vakuum entfernt. Das
gewünschte Produkt konnte nicht identifiziert werden.
HNNH
OHN
O
CO2Me
OHNHO
ONHBnO
O
HN
O
NH2
NH
HN
ONH
O
CO2Me
OHN
HO
NHBoc
CO2HONH
OH2N
OBn

Experimenteller Teil
118
Synthese von Peptid 120
Das Tripeptid H-Leu-Phe-Ala-OMe 101 (152 mg;
0,419 mmol) und die Aminosäure 119 (127 mg;
0,253 mmol) werden in THF (5 ml) gelöst und bei
0 °C mit HOBt (170 mg; 1,26 mmol) versetzt.
Nachdem 15 min bei RT gerührt wurde, wird EDAC
(150 mg; 0,785 mmol) zugesetzt, das Kältebad entfernt und 16 h bei RT gerührt.
Anschließend wird das Reaktionsgemisch mit EtOAc (150 ml) verdünnt und mit 5% HCl (2x
30 ml), ges. NaHCO3-Lösung (2x 30 ml), Wasser (30 ml) und ges. NaCl-Lösung (30 ml)
gewaschen. Die organische Phase wird getrocknet und das Lösungsmittel im Vakuum
entfernt. Der Rückstand wird chromatographisch gereinigt (Hex: EtOAc; 2:1). Man erhält
130 mg das Tetrapeptids als ein Gemisch aus Diastereomeren (Ausbeute 62 %).
Rf 0,39 (Hex: EtOAc; 1:1)
de = 74 % (NMR) 1H-NMR (CDCl3): 0,80 (dd, J1= 10,61 Hz, J2= 6,45 Hz, 6H); 1,27 (d, J= 7,25 Hz, 3H); 1,34
(s, 9H); 1,38 (m, 1H); 1,43 (s, 18H); 1,49 (m, 1H); 2,36 (dd, J1= 15,51 Hz, J2= 9,87 Hz, 1H);
2,56 (dd, J1= 15,50 Hz, J2= 2,08 Hz, 1H); 2,75 (dd, J1= 14,04 Hz, J2= 9,60 Hz, 1H); 3,01 (dd,
J1= 13,97 Hz, J2= 4,57 Hz, 1H); 3,22 (s, 3H); 3,52 (d, J= 10,07 Hz, 1H); 3,59 (s, 3H); 3,62 (d,
J= 10,00 Hz, 1H); 4,22 (m, 2H); 4,53 (td, J1= 8,93 Hz, J2= 4,75 Hz, 1H); 4,69 (dd,
J1= 9,81 Hz, J2= 2,15 Hz, 1H); 7,22 (m, 5H); 7,59 (d, J= 8,33 Hz, 1H); 7,96 (d, J= 8,46 Hz,
1H); 8,44 (d, J= 6,98 Hz, 1H) 13C-NMR (CDCl3): 16,89; 21,76; 23,01; 24,02; 27,47; 27,64; 31,75; 37,66; 41,04; 47,67;
51,83; 51,94; 53,27; 55,27; 58,85; 69,21; 76,58; 81,19; 82,47; 126,28; 128,04; 129,27;
137,62; 152,10; 167,69; 168,44; 170,84; 171,17; 172,91
MS(ESI): 834,4606 [MH+]; 856,4415 [MNa+]
NH
HN
ONH
O
CO2Me
ON3
O
NBoc2
CO2tBu

Experimenteller Teil
119
Synthese von Peptid 121
Das Tetrapeptid 120 (40 mg; 0,48 mmol) wird in
EtOAc (10 ml) gelöst und mit Pd/C (20 mg)
versetzt. Nun wird 16 h unter
Wasserstoffatmosphäre (1 bar) gerührt.
Anschließend wird der Katalysator abfiltriert und
der Rückstand chromatographisch gereinigt
(Hex:EtOAc; 1:1). Man erhält 26 mg des entsprechenden Produktes als ein Gemisch aus
Diastereomeren (Ausbeute 65%).
Rf 0,45 (CHCl3: MeOH; 9:1)
de = 86 % (NMR) 1H-NMR (DMSO-d6): 0,80 (dd, J1= 10,81 Hz, J2= 6,51 Hz, 6H); 1,26 (d, J= 7,25 Hz, 3H);
1,33 (s, 9H); 1,43 (s, 18H); 1,84 (dd, J1= 14,91 Hz, J2= 7,89 Hz, 1H); 2,30 (dd, J1= 15,38 Hz,
J2= 3,02 Hz, 1H); 2,74 (dd, J1= 14,04 Hz, J2= 9,60 Hz, 1H); 3,06 (dd, J1= 14,31 Hz,
J2= 4,63 Hz, 1H); 3,14 (s, 3H); 3,18 (d, J= 5,10 Hz, 1H); 3,42 (d, J= 8,73 Hz, 1H); 3,59 (s,
3H); 4,23 (m, 2H); 4,53 (td, J1= 8,93 Hz, J2= 4,84 Hz, 1H); 4,82 (dd, J1= 7,52 Hz,
J2= 3,09 Hz, 1H); 7,18 (m, 6H); 7,87 (m, 2H); 8,44 (d, J= 6,98 Hz, 1H) 13C-NMR (DMSO-d6): 16,88; 21,88; 23,10; 24,13; 27,55; 27,66; 35,43; 37,43; 41,30; 47,70;
51,24; 51,91; 53,31; 54,98; 58,72; 60,07; 77,35; 80,68; 82,33; 126,28; 128,07; 129,21;
137,70; 152,01; 157,83; 158,14; 168,43; 170,81; 170,81; 171,56; 172,87
MS(ESI): 808,4697 [MH+]; 830,4501 [MNa+]
Synthese von Peptid 122
Die Peptide 121 (71 mg;
0,088 mmol) und 99 (74 mg;
0,253 mmol) werden in THF (5 ml)
gelöst und bei 0°C mit DIPEA
(100 µl; 0,58 mmol) und HATU
(100 mg; 0,263 mmol) versetzt.
Nachdem 2 d bei RT gerührt wurde, wird das Lösungsmittel im Vakuum entfernt. Der
Rückstand wird in EtOAc aufgenommen und mit 1% HCl-Lösung, ges. NaHCO3-Lösung und
ges. NaCl-Lösung gewaschen. Das Lösungsmittel wird im Vakuum entfernt und der
Rückstand chromatographisch gereinigt (CHCl3:MeOH; 50:1). Wobei das unerwünschte
NH
HN
ONH
O
CO2Me
OH2N
O
NBoc2
CO2tBu
NH
HN
ONH
O
CO2Me
OHN
O
NBoc2
CO2tBuONH
ON3
OBn

Experimenteller Teil
120
Diastereomer in einer Mischfraktion mit dem Produkt entfernt werden konnte. Man erhält 73
mg (Ausbeute 73%) des gewünschten Hexapeptids.
Rf 0,38 (CHCl3: MeOH; 9:1)
de = 99 % (NMR) 1H-NMR (CDCl3): 0,79 (dd, J1= 13,43 Hz, J2= 6,31 Hz, 6H); 1,32 (m, 1H) 1,39 (d, J= 6,45
Hz, 3H); 1,46 (s, 9H); 1,48 (s, 18H); 1,57 (m , 2H); 2,36 (dd, J1= 15,04 Hz, J2= 1,61 Hz, 1H);
2,68 (dd, J1= 15,04 Hz, J2= 7,93 Hz, 1H); 2,94 (dd, J1= 14,24 Hz, J2= 11,55 Hz, 1H); 3,30 (s,
3H); 3,50 (dd, J1= 14,31 Hz, J2= 3,83 Hz, 1H); 3,70 (s, 3H); 3,74 (d, J= 9,54 Hz, 1H); 3,82
(dd, J1= 16,93 Hz, J2= 5,91 Hz, 1H); 3,87 (dd, J1= 16,85 Hz, J2= 4,38 Hz, 1H); 3,04 (d,
J= 3,90 Hz, 1H); 4,16 (ddd, J1= 12,63 Hz, J2= 6,25 Hz, J3= 3,96 Hz; 1H); 4,49 (d, J= 11,69
Hz, 1H); 4,53 (d, J= 7,32 Hz, 1H); 4,59 (d, J= 11,50 Hz, 1H); 4,72 (ddd, J1= 11,72 Hz,
J2= 8,43 Hz, J3= 3,90 Hz; 1H); 5,44 (dd, J1= 7,66 Hz, J2= 1,61 Hz, 1H); 7,05 (t, J= 7,32 Hz,
1H); 7,14 (m, 3H); 7,29 (m, 10H); 7,41 (d, J= 8,73 Hz, 1H) 13C-NMR (CDCl3): 16,91; 17,64; 21,04; 23,02; 24,80; 27,77; 28,04; 36,28; 39,48; 44,72;
48,34; 52,30; 53,53; 54,37; 54,78; 59,10; 60,74; 67,98; 71,83; 72,48; 75,69; 83,03; 83,36;
126,24; 127,67; 127,94; 128,17; 128,50; 129,20; 137,52; 138,40; 152,30; 169,12; 169,90;
171,33; 172,17; 172,91; 173,62
MS(ESI): 1082,5784 [MH+]; 1104,5583 [MNa+]
Synthese von Peptid 123
Das Peptid 122 (52 mg;
0,048 mmol) wird bei 0 °C in TFA
(510 µl) gelöst und mit TES
(196 µl) versetzt. Nun wird 8 h bei
RT gerührt. Anschließend wird das
Peptid mit eiskaltem Hexan ausgefällt. Man erhält 40 mg des gewünschten Peptids in
quantitativer Ausbeute.
Rf 0,50 (CHCl3: MeOH; 9:2)
MS(ESI): 826,4637 [MH+]; 848,4436 [MNa+]
NH
HN
ONH
O
CO2Me
OHN
O
NH2
CO2HONH
ON3
OBn

Experimenteller Teil
121
Synthese von Peptid 124
Das Peptid 123 (0,048 mmol) wird
in Acetonitril (0,76 ml) gelöst und
mit TEA (28µl; 0,202 mmol) und
Boc2O (13 µl; 0,072 mmol)
versetzt. Nun wird 16 h bei RT
gerührt, anschließend wird das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt
(CHCl3: MeOH; 9:0,5 auf 9:1). Man erhält 42 mg des gewünschten Produkt (Ausbeute 95%).
Rf 0,5 (CHCl3: MeOH; 9:1)
de = 99 % (NMR) 1H-NMR (DMSO-d6): 0,71 (d, J= 6,58 Hz, 3H); 0,77 (d, J= 6,45 Hz, 3H); 1,19 (d, J= 5,91
Hz, 3H); 1,21 (m, 1H); 1,30 (d, J= 7,25 Hz, 3H); 1,36 (s, 9H); 1,51 (m, 1H); 2,03 (dd,
J1= 14,78 Hz, J2= 6,72 Hz, 1H); 2,24 (m, 1H); 2,78 (dd, J1= 13,77 Hz, J2= 10,68 Hz, 1H);
3,12 (dd, J1= 13,77 Hz, J2= 3,83 Hz, 1H); 3,20 (s, 3H); 3,59 (s, 3H); 3,74 (m, 3H); 3,85 (d,
J= 5,10 Hz, 1H); 3,93 (m, 4H); 4,23 (dt, J1= 14,10 Hz, J2= 7,05 Hz, 1H); 4,42 (td,
J1= 9,44 Hz, J2= 4,63 Hz, 1H); 4,45 (d, J= 11,96 Hz, 1H); 4,57 (d, J= 11,80 Hz, 1H); 7,15
(m, 6H); 7,28 (m, 6H); 7,68 (d, J= 6,31 Hz, 1H); 7,73 (d, J= 7,39 Hz, 1H); 7,82 (d, J= 5,37
Hz, 1H); 8,56 (d, J= 7,39 Hz, 1H) 13C-NMR (DMSO-d6): 16,70; 16,81; 21,20; 23,05; 23,89; 28,30; 37,05; 43,21; 47,89; 51,89;
51,89; 52,59; 53,75; 58,56; 61,07; 66,07; 70,48; 72,78; 75,18; 77,88; 126,13; 127,47; 128,03;
128,23; 129,18; 138,18; 138,44; 154,95; 168,86; 169,22; 170,91; 171,67; 172,68
MS(ESI): 926,4876 [MH+]; 948,4423 [MNa+]
Synthese von Peptid 125
Das Peptid 124 (43 mg; 0,043 mmol)
wird in THF (1 ml) gelöst und mit
polymergebundenen PPh3
(0,1 g; 0,086 mmol) und Wasser (30
µl) versetzt. Nun wird das Reaktionsgemisch 4 d bei RT geschüttelt. Anschließend wird das
Lösungsmittel im Vakuum entfernt und das Rohprodukt direkt weiter verwendet.
NH
HN
ONH
O
CO2Me
OHN
O
NHBoc
CO2HONH
ON3
OBn
NH
HN
ONH
O
CO2Me
OHN
O
NHBoc
CO2HONH
ONH2
OBn

Experimenteller Teil
122
Rf 0,25 (CHCl3: MeOH; 9:2)
MS(ESI): 900,4651 [MH+]; 922,4452 [MNa+]
Synthese von Peptid 126
Das Peptid 125 (13 mg; 0,0145 mmol)
wird in THF (9,5 ml) gelöst und bei 0°C
mit DIPEA (13,2 µl; 0,08 mmol) und
HATU (29 mg; 0,076 mmol) versetz.
Nun wird 2 d bei RT gerührt.
Anschließend wird das Lösungsmittel
im Vakuum entfernt. Das gewünschte Produkt konnte bisher nur massenspektrometrisch
charakterisiert werden.
MS(ESI): 882,4607 [MH+]
HNNH
OHN
O
CO2Me
OHN
O
ONHBnO
O
HN
O
NHBoc

Experimenteller Teil
123
5.2.7. Synthese eines analytischen Refenzenzstandards zum Labioninnachweis
Synthese von L-Cbz-Ala-OH (127)
Alanin (7 g; 78,57 mmol) wird in ges. Natriumhydrogencarbonat-Lösung
(320 ml) gelöst und mit Cbz-Cl (16,8 ml; 117,7 mmol) versetzt. Nachdem
das Reaktionsgemisch 16 h bei RT stark gerührt wurde, wird mit Diethylether (3x 250 ml)
gewaschen. Die wässrige Phase wird mit 10%iger HCl-Lösung angesäuert und mit EtOAc (5x
250 ml) extrahiert. Die organische Phase wird mit Natriumsulfat getrocknet und das
Lösungsmittel im Vakuum entfernt. Man erhält 17,19 g (Ausbeute 98 %) eines weißen
Feststoffes.
Rf 0,47 (CHCl3:MeOH; 9:2) 1H-NMR (DMSO-d6): 1,22 (d, J=7,39 Hz, 3H); 3,99 (dt, J1=14,74 Hz, J2=7,34 Hz, 1H); 5,01
(s, 2H); 7,33 (m, 5H) 13C-NMR (DMSO-d6): 17,14; 49,29; 65,42; 127,83; 128,42 ;137,11; 155,92; 174,47
MS (EI): 223,0847 [M+]
Synthese von L-Cbz-Ala-OAll (128)
Cbz-Alanin (5 g; 22,4 mmol) wird in DMF (400 ml) gelöst und mit
Cäsiumcarbonat (15 g; 46,03 mmol) und Allylbromid (2,14 ml;
24,7 mmol) versetzt. Nachdem 16 h bei RT gerührt wurde, wird mit EtOAc (300 ml) verdünnt
und mit ges. NaCl-Lösung (5x 300 ml) gewaschen. Die organische Phase wird mit Na2SO4
getrocknet und das Lösungsmittel im Vakuum entfernt. Das überschüssige Allylbromid wird
am Hochvakuum entfernt. Man erhält 5,77 g (Ausbeute 98 %) eines leicht gelben Öls.
Rf 0,45 (Hex:EtOAc; 3:1) 1H-NMR (DMSO-d6): 1,42 (d, J=7,39 Hz, 3H); 4,41 (dt, J1=14,67 Hz, J2=7,32 Hz, 1H); 4,57
(d, J=4,30 Hz, 2H), 5,02 (s, 2H); 5,19 (d, J=10,86 Hz, 2H); 5,29 (d, J=17,33 Hz, 2H); 5,86
(ddd, J1=22,23 Hz, J2=10,61 Hz, J3=5,44 Hz, 1H); 7,33 (m, 5H); 7,73 (d. J=6,98 Hz, 1H) 13C-NMR (CDCl3): 18,68; 49,64; 65,92; 66,89; 118,72; 128,08; 128,14; 128,50; 131,50;
136,23; 155,54; 174,64
MS (EI): 263,1165 [M+]
CbzHNOH
O
CbzHNO
O

Experimenteller Teil
124
Synthese von rac-Cbz-α-Allyl-Ala-OH (129)
Diisopropylamin (3,5 ml; 25 mmol) wird in THF (23 ml) gelöst und bei
-30 °C mit nButyllithium (1,6 M; 15,7 ml) versetzt. Nun wird 1 h bei
-30 °C gerührt und anschließend auf -78°C herabgekühlt. Der
Reaktionslösung wird nun Cbz-Ala-OAll (3 g, 11,4 mmol) gelöst in THF (17 ml) zugetropft,
gefolgt von in THF (17 ml) gelöstem Zinkchlorid (1,8 g, 13,2 mmol). Nach 4 h wird die
Kühlung abgeschaltet und die Reaktionslösung über 12 h auf RT erwärmt. Nun wird mit
EtOAc (100 ml) verdünnt und mit 5%iger HCl-Lösung gewaschen. Die organische Phase
wird mit Natriumsulfat getrocknet und das Lösungsmittel im Vakuum entfernt. Der
Rückstand wird chromatographisch gereinigt (Hex:EtOAc; 3:1 auf CHCl3:MeOH; 9:2;). Man
erhält 2,4 g (Ausbeute 86 %) eines dunkelbraunen Öls.
Rf 0,54 (CHCl3:MeOH; 9:1) 1H-NMR (MeOD-d4): 1.44 (s, 3H); 2,59(dd, J1= 13,57 Hz, J2= 8,06 Hz, 1H); 2,68 (dd,
J1= 13,40, Hz, J2= 7,23, Hz, 1H); 5,06 (m, 4H); 5,72 (m, 1H); 7,29(m, 6H) 13C-NMR (MeOD-d4): 23,23; 41,94; 59,92; 67,27; 119,35; 128,73; 128,92; 129,41; 133,86;
138,28; 157,14; 177,27
MS (EI): 263,1157 [M] +
Synthese von rac-Cbz-α-Allyl-Alanin-tert-butylester (130)
Cbz-α-Allyl-Alanin (129) (3,9 g, 14,82 mmol) wird in tBuOH (150 ml)
gelöst und mit DMAP (1,1 g; 2,08 mmol) und Boc2O (5,87 ml, 28,31
mmol) versetzt. Nachdem 2 d bei 25 °C gerührt wurde, wird das
Lösungsmittel im Vakuum entfernt und der Rückstand chromatographisch (Hex:EtOAc; 6:1
auf 3:1) gereinigt. Man erhält 1,4 g (Ausbeute 30 %) eines farblosen Öls.
Cbz-α-Allyl-Alanin (129) (2,21 g; 8,4 mmol) wird in DCM (14 ml) gelöst und mit tert-Butyl-
Acetimidat (3,66 g; 16,75 mmol) gelöst in Hexan (18 ml) versetzt. Nun wird
Bortrifluoridetherat (0,169 ml; 1,37 mmol) zugegeben und 16 h bei RT gerührt. Anschließend
wird das Reaktionsgemisch filtriert, das Lösungsmittel im Vakuum entfernt und der
Rückstand chromatographisch gereinigt (Pen: EtOAc; 5:1). Man erhält 1,61 g eines farblosen
Öls (Ausbeute 60 %).
CbzHNOH
O
CbzHNOtBu
O

Experimenteller Teil
125
Rf 0,66 (Hex:EtOAc; 3:1)
ee = 5 % (chirale HPLC; OD-H) 1H-NMR (DMSO-d6): 1,24 (s, 3H);1,31 (s, 9H); 2,37(dd, J1= 13,70 Hz, J2= 7,66 Hz, 1H);
2,53 (m, 1H); 5,02 (m, 4H); 7,31(m, 6H); 7,51 (s, 1H) 13C-NMR (CDCl3): 23,28; 27,83; 41,00; 59,57; 66,18; 82,00; 119,04; 127,93; 127,96; 128,43;
136,65; 154,52; 172, 73
MS (EI): 319,1788 [M+]
Synthese von rac-Cbz-2-amino-4,5-dihydroxy-2-methyl-pentansäure-tert-butylester
(131)
Der tert-Butylester 130 (1,78 g; 5,57 mmol) wird in einem tBuOH-
Wassergemisch (1:1; 26 ml) gelöst und mit AD-Mix-β (7,84 g) versetzt,
anschließend wird 16 h bei RT stark gerührt. Die Reaktion wird durch
Zugabe von Natriumsulfit (8,6g; 68,28 mmol) beendet und mit EtOAc
extrahiert (3x10 ml). Die organische Phase wird über Natriumsulfat getrocknet und das
Lösungsmittel im Vakuum entfernt. Man erhält 1,2 g eines farblosen Öls (Ausbeute 61 %).
Rf 0,22 (Hex:EtOAc; 1:1)
de = 22 % (chirale HPLC; OD-H) 1H-NMR (CDCl3): 1,42 (s, 3,6H); 1,44 (s, 5,2H); 1,54 (s, 1,8H); 1,58 (s, 1,3H); 1,84 (d,
J= 14,24 Hz, 0,56H); 1,98 (dd, J1= 14,37 Hz, J2= 10,34 Hz, 1H); 2,08 (m, 0,74H); 2,22 (d,
J= 14,37 Hz, 0,72H); 3,39 (dd, J1= 10,95 Hz, J2= 7,46 Hz, 1H); 3,52 (m, 1 H); 3,79 ( m, 1 H);
5,045 (m, 2H); 6,16 (s, 0,35H); 6,20 (s; 0,53H); 7,31 (m, 5H) 13C-NMR (CDCl3): 24,06; 27,77; 39,22; 40,53; 58,37; 59,51; 66,37; 66,83; 66,91; 68,77;
69,40; 82,32; 82,39; 128,02; 128,07; 128,10; 128,50; 136,57; 154,68; 173,19; 173,70
MS (ESI): 354,1914 [MH+]
CbzHNOtBu
O
HO
HO

Experimenteller Teil
126
Allgemeine Synthesevorschrift zur Darstellung von Cbz-2-Amino-5-tert-butylsilylether-
4-hydroxy-2-methyl-pentansäure-tert-butylester (+/-132a/b)
Das Diol 131 (1 äq.) wird in DMF (6,25 ml/mmol) gelöst und mit TBDMSCl (1,05 äq.) und
Imidazol (1,1 äq.) versetzt. Nachdem das Reaktionsgemisch 5 h bei RT gerührt wurde, wird
die Reaktion durch Zugabe von Wasser beendet und das Reaktionsgemisch mit EtOAc
(45 ml/mmol) verdünnt. Die organische Phase wird mit ges. NaCl-Lösung gewaschen (4x),
über Natriumsulfat getrocknet und das Lösungsmittel im Vakuum entfernt. Der Rückstand
wird chromatographisch gereinigt (Hex:EtOAc; 9:1) und die bei der Reaktion gebildeten
Diastereomere konnten getrennt werden.
(2S,4S)/ (2R,4R)-Cbz-2-Amino-5-tert-butylsilyloxy-4-hydroxy-2-methyl-pentansäure-tert-
butylester (+/-132a)
CbzHNOtBu
O
TBDMSO
HO
CbzHN
OtBu
O
TBDMSO
HO
Ausbeute: 41 % (921 mg)
Rf 0,57 (Hex:EtOAc; 5:1)
de = 98 % (chirale HPLC; OD-H) 1H-NMR (CDCl3): 0,04 (d, J= 0,94 Hz, 6H); 0,88 (s, 9H); 1,44 (s, 9H); 1,55 (s, 3H); 1,97 (dd,
J1= 14,24 Hz, J2= 10,34 Hz, 1H); 2,14 (d, J= 14,80 Hz, 1H); 2,31 (d, J= 3,90 Hz, 1H); 3,36
(dd, J1= 9,87 Hz, J2= 7,05 Hz, 1H); 3,56 (dd, J1= 10,21 Hz, J2= 2,96 Hz, 1H); 3,73 (ddd,
J1= 9,74 Hz, J2= 6,78 Hz, J3= 2,96 Hz, 1H); 5,07 (dd, J1= 16,25 Hz, J2= 12,48 Hz, 2H);
6,32(s,1H); 7,29 (m, 5H) 13C-NMR (CDCl3): -5,46; -5,38; 18,23; 23,66; 25,84; 27;75; 39,17; 58,36; 66,07; 67,02;
68,40; 81,79; 86,60; 127,84; 127,42; 136,77; 154,53; 172,52
MS (ESI): 468,2767 [MH+]

Experimenteller Teil
127
(2R,4S)/ (2S,4R)-Cbz-2-Amino-5-tert-butylsilyloxy-4-hydroxy-2-methyl-pentansäure-tert-
butylester (+/-132b)
CbzHNOtBu
O
TBDMSO
HO
CbzHN
OtBu
O
TBDMSO
HO
Ausbeute: 27 % (606 mg)
Rf 0,45 (Hex:EtOAc; 5:1)
de = 98 % (chirale HPLC; OD-H) 1H-NMR (CDCl3): 0,04 (d, J= 0,94 Hz, 6H); 0,88 (s, 9H); 1,42 (s, 9H); 1,61 (s, 3H); 1,82 (dd,
J1= 14,78 Hz, J2= 2,15 Hz, 1H); 1,88 (dd, J1= 14,50 Hz, J2= 9,34 Hz, 1H); 2,66 (s, 1H); 3,35
(dd, J1= 9,81 Hz, J2= 3,76 Hz, 1H); 3,77 (dd, J1= 14,78 Hz, J2= 2,15 Hz, 1H); 5,07 (s, 2H);
6,40 (s, 1H); 7,32 (m, 5H) 13C-NMR (CDCl3): -5,38; 18,27; 24,73; 25,85; 27,88; 40,68; 59,66; 66,25; 67,28; 69,30;
81,71; 86,60; 127,91; 128,07; 128,42, 136,81; 155,52; 172,96
MS (ESI): 468,2767 [MH] +
Allgemeine Synthesevorschrift zur Darstellung von Cbz-2-Amino-4-azido-5-tert-
butylsilyloxy-2-methyl-pentansäure-tert-butylester (+/-133a/b)
Der TBDMS-Ether (+/-132a/b) (1 äq.) wird in wasserfreiem THF (10 ml/mmol) gelöst und
mit Triphenylphosphin (2,5 äq.) versetzt. Die Reaktionslösung wird auf -78 °C herabgekühlt
und langsam mit DEAD (7,5 äq.) und DPPA (2,5 äq.) versetzt. Nachdem 16 h bei -78 °C
gerührt wurde, wird das Lösungsmittel im Vakuum entfernt und der Rückstand
chromatographisch gereinigt (Hex: EtOAc; 14:1).
(2S,4S)/ (2R,4R)-Cbz-4-2-Amino-4-azido-5-tert-butylsilyloxy-2-methyl-pentansäure-tert-
butylester (+/-134a)
CbzHNOtBu
O
TBDMSO
N3
CbzHN
OtBu
O
TBDMSO
N3

Experimenteller Teil
128
Ausbeute: 49 % (156 mg)
Rf 0,44 (Hex: EtOAc; 5:1)
de = 97 % (chirale HPLC; OD-H) 1H-NMR (CDCl3): 0,06 (d, J= 1,48 Hz, 6H); 0,90 (s, 9H); 1,44 (s, 9H); 1,56 (s, 3H); 1,93 (dd,
J1= 14,84 Hz, J2= 3,02 Hz, 1H); 2,05 (m, 1H); 3,39 (m, 1H); 3,58 (dd, J1= 10,2 Hz,
J2= 7,39 Hz, 1H); 3,64 (dd, J1= 10,2 Hz, J2= 4,43 Hz, 1H); 5,08 (dd,J1= 21,9 Hz, J2= 12,2 Hz,
2H); 5,79 (s, 1H); 7,33 (m, 5H) 13C-NMR (CDCl3): -5,57; -5,54; 18,20; 24,28; 25,76; 27,81; 36,83; 56,51; 59,14; 60,62;
66,42; 66,95; 82,32; 128,02; 128,05; 128,46, 136,59; 154,92; 172,58
MS (ESI): 493,2830 [MH+]; 515,2646 [MNa+]
(2R,4S)/ (2S,4R)-Cbz-2-Amino-4-azido-5-tert-butylsilyloxy-2-methyl-pentansäure-tert-
butylester (+/-133b)
CbzHNOtBu
O
TBDMSO
N3
CbzHN
OtBu
O
TBDMSO
N3
Ausbeute: 68 % (188 mg)
Rf 0,55 (Hex: EtOAc; 5:1)
de = 92 % (chirale HPLC; OD-H) 1H-NMR (CDCl3): 0,06 (d, J= 1,88 Hz, 6H); 0,90 (s, 9H); 1,54 (s, 9H); 1,57 (s, 3H); 1,87 (dd,
J1= 14,37 Hz, J2= 10,61 Hz, 1H); 2,36 (d, J= 14,10 Hz, 1H); 3,36 (m, 1H); 3,54 (dd,
J1= 10,34 Hz, J2= 7,66 Hz, 1H); 3,69(d, J= 8,19 Hz, 1H); 5,07 (s, 2H); 6,14 (s, 1H); 7,32 (m,
5H) 13C-NMR (CDCl3): -5,58; -5,54; 18,20; 24,40; 25,78; 27,65; 36,63; 58,31; 60,28; 66,27;
66,76; 82,66; 127,86; 128,05; 128,51, 136,61; 154,30; 172,61
MS (ESI): 493,2823 [MH+]; 515,2639 [MNa+]

Experimenteller Teil
129
Allgemeine Synthesevorschrift zur Darstellung von Cbz-2-Amino-4-azido-5-hydroxy-2-
methyl-pentansäure-tbutylester (+/-134a/b)
Das Azid (133a/b) (1 äq.) wird in THF (7,4 ml/mmol) gelöst und bei 0 °C mit einer TBAF-
Lösung (1M in THF; 1,2 äq.) versetzt. Nachdem 4 h bei 0 °C gerührt wurde, wird die
Reaktion durch Zugabe von ges. NH4Cl-Lösung beendet und das Reaktionsgemisch mit
EtOAc (3x) extrahiert. Die organische Phase wird getrocknet und das Lösungsmittel im
Vakuum entfernt. Das Rohprodukt wird chromatographisch gereinigt (Hex:EtOAc; 3:1).
(2S,4S)/ (2R,4R)-Cbz-2-Amino-4-azido-5-hydroxy-2-methyl-pentansäure-tert-butylester
(+/-134a)
CbzHNOtBu
O
HO
N3
CbzHN
OtBu
O
HO
N3
Ausbeute: 70 % (128 mg)
Rf 0,55 (Hex:EtOAc; 1:1)
de = 98 % (chirale HPLC; OD-H) 1H-NMR (CDCl3): 1,45 (s, 9H); 1,56 (s, 3H); 2,06 (dd, J1= 14,84 Hz, J2= 3,96 Hz, 1H); 2,27
(dd, J1= 14,71 Hz, J2= 7,72 Hz, 1H); 3,43 (m, 1H); 3,54 (dd, J1= 11,30 Hz, J2= 7,11 Hz, 1H);
3,62 (dd, J1= 11,15 Hz, J2= 4,23 Hz, 1H); 5,08 (dd, J1= 21,85 Hz, J2= 12,28 Hz, 2H); 5,78 (s,
1H); 7,33 (m, 5H) 13C-NMR (CDCl3): 24,60; 27,78; 36,47; 58,98; 61,13; 65,49; 66,56; 82,80; 128,10; 128,49;
136,45; 154,89; 172,63
MS (ESI): 379,1965 [MH+]; 401,1778 [MNa+]
(2R,4S)/ (2S,4R)-Cbz-2-Amino-4-azido-5-hydroxy-2-methyl-pentansäure-tert-butylester
(+/-134b)
CbzHNOtBu
O
HO
N3
CbzHN
OtBu
O
HO
N3

Experimenteller Teil
130
Ausbeute: 80 % (154 mg)
Rf 0,66 (Hex:EtOAc; 1:1)
de = 98 % (chirale HPLC; OD-H) 1H-NMR (CDCl3): 1,48 (s, 9H); 1,53 (s, 3H); 1,93 (dd, J1= 14,31 Hz, J2= 10,41 Hz, 1H); 2,47
(d, J= 15,04 Hz, 1H); 3,38 (ddd, J1= 12,69 Hz, J2= 7,39 Hz, J3= 3,39 Hz, 1H); 3,54 (m, 1H);
3,67 (d, J= 11,69 Hz, 1H); 5,05 (s, 2H); 6,16 (s, 1H); 7,33 (m, 5H) 13C-NMR (CDCl3): 24,46; 27,61; 36,66; 58,30; 60,92; 65,73; 66,41; 82,89; 127,96; 128,10;
128,52; 136,45; 154,41; 172,79
MS (ESI): 379,1970 [MH+]; 401,1786 [MNa+]
Allgemeine Synthesevorschrift zur Darstellung von Cbz-4-Amino-2-azido-4-(tert-
butyloxycarbonyl)-pentansäure (+/-135a/b)
Der Azidoalkohol (134a/b) (1äq.) wird in Aceton (5 ml/mmol) gelöst und bei 0 °C mit einer
5%igen NaHCO3-Lösung (2,5 ml/mmol) und KBr (0,1 äq.) versetzt. Nun wird der
Reaktionslösung TEMPO (1,1 äq.) zugegeben, gefolgt von NaOCl (2,5 ml/ mmol). Nachdem
1 h bei 0 °C gerührt wurde, wird die Reaktion durch Zugabe von 5%iger HCl beendet und das
Reaktionsgemisch mit EtOAc (3x) extrahiert. Die organische Phase wird getrocknet und das
Lösungsmittel im Vakuum entfernt. Das Rohprodukt wird ohne weitere Reinigung weiter
verwendet.
(4S,2S)/(4R,2R)-Cbz-4-Amino-2-azido-4-(tert-butyloxycarbonyl)-pentansäure (+/-135a)
CbzHNOtBu
O
CO2HN3
CbzHN
OtBu
O
CO2HN3
Ausbeute: 95 % (135 mg)
Rf 0,34 (CHCl3:MeOH; 9:1)
MS (ESI): 393,1765 [MH+]; 415,1581 [M+Na+]

Experimenteller Teil
131
(4R,2S)/(4S,2R)-Cbz-4-Amino-2-azido-4-(tert-butyloxycarbonyl)-pentansäure (+/-135b)
CbzHNOtBu
O
CO2HN3
CbzHN
OtBu
O
CO2HN3
Ausbeute: 91 % (146 mg)
Rf 0,53 (CHCl3:MeOH; 9:1)
MS (ESI): 393,2083 [MH+]
Allgemeine Synthesevorschrift zur Darstellung von Cbz-2-Amino-4-azido-2-methyl-
pentandisäure (+/-136a/b)
Die Aminosäure (+/-135a/b) (1 äq.) wird in TFA (7,7 ml/mmol) gelöst, mit TES (5 äq.)
versetzt und 5 h bei RT gerührt. Anschließend wird die TFA im Vakuum entfernt. Der
Rückstand wird in EtOAc aufgenommen und mit K2CO3-Lösung extrahiert. Nun wird die
wässrige Phase angesäuert und mit EtOAc extrahiert. Die organische Phase wird getrocknet
und das Lösungsmittel im Vakuum entfernt.
(2S,4S)/ (2R,4R)-Cbz-2-Amino-4-azido-2-methyl-pentandisäure (+/-136a)
CbzHNOH
O
CO2HN3
CbzHN
OH
O
CO2HN3
Ausbeute: 80 % (78 mg)
Rf 0,42 (EtOAc:n-BuOH:AcOH:H2O; 2:1:1:1) 1H-NMR (DMSO-d6): 1,37 (s, 3H); 2,26 (dd, J1= 14,91 Hz, J2= 9,54 Hz, 1H); 2,53 (m, 1H);
3,90 (dd, J1= 9,20 Hz, J2= 2,62 Hz, 1H); 5,00 (s, 2H); 7,31 (m, 5H); 7,65 (s, 1H) 13C-NMR (DMSO-d6): 22,89; 36,42; 57,12; 57,15; 65,19; 127,59; 127,77; 128,32; 136,96;
154,88; 171,79; 174,74
MS (ESI): 335,0979 [M-]

Experimenteller Teil
132
(2R,4S)/ (2S,4R)-Cbz-2-Amino-4-azido-2-methyl-pentandisäure (+/-136b)
CbzHNOH
O
CO2HN3
CbzHN
OH
O
CO2HN3
Ausbeute: 67 % (112 mg)
Rf 0,53 (EtOAc:n-BuOH:AcOH:H2O; 2:1:1:1) 1H-NMR (DMSO-d6): 1,36 (s, 3H); 2,26 (dd, J1= 14,91 Hz, J2= 9,54 Hz, 1H); 2,35 (dd,
J1= 14,95 Hz, J2= 2,82 Hz, 1H); 3,91(dd, J1= 9,40 Hz, J2= 2,96 Hz, 1H); 5,00 (dd,
J1= 16,55 Hz, J2= 12,76 Hz, 2H); 7,31 (m, 5H), 7,39 (s, 1H) 13C-NMR (DMSO-d6): 22,87; 36,00; 56,85; 57,88; 65,19; 127,56; 127,71; 128,25; 136,92;
154,72; 171,81; 174,81
MS (ESI): 335,0978 [M-]
Allgemeine Synthesevorschrift zur Darstellung von 2,4-Diamino-2-methyl-
pentandisäure (+/-137a/b)
Die Dicarbonsäure +/-136a/b wird in THF (34 ml/ mmol) gelöst und mit einer Spatelspitze
Pd/C versetzt. Nun wird 16 h unter Wasserstoffatmosphäre bei RT gerührt. Das Rohprodukt
wird direkt für die GC-Analytik derivatisiert (siehe allgemeine Synthesevorschrift für
GC-Derivatisierung).
(2S,4S)/ (2R,4R)-2,4-Diamino-2-methyl-pentandisäure (+/-137a)
H2NOH
O
CO2HNH2
H2NOH
O
CO2HH2N
GC-MS-Analytik nach der entsprechenden Derivatisierung (siehe allgemeine
Synthesevorschrift)
tr = 20,9 min (GC-MS: 70 °C (2 min isothermal), 300 °C, 5 °C/min (1 min isothermal))
GC-MS (PSI): 57 (100); 114 (40); 69 (32); 351 (24); 73 (16); 101 (16); 140 (10); 154 (9); 109
(9); 87 (9); 379 (8); 238 (8); 166 (7); 424 (5); 311 (5)
Experimenteller Teil
133
(2R,4S)/ (2S,4R)-2,4-Diamino-2-methyl-pentandisäure (+/-137a)
H2NOH
O
CO2HH2N
H2NOH
O
CO2HH2N
GC-MS-Analytik nach der entsprechenden Derivatisierung (siehe allgemeine
Synthesevorschrift)
tr = 20,6 min (GC-MS: 70 °C (2 min isothermal), 300 °C, 5 °C/min (1 min isothermal))
GC-MS (PSI): 114 (100); 57 (80); 351 (64); 109 (30); 69 (30); 101 (28); 140 (24); 154 (24);
238 (24); 87 (23); 166 (18); 73 (16); 424 (16); 379 (16); 331 (6); 311 (5); 198 (5)

Experimenteller Teil
134
5.2.8. Derivatisierung der Labyrinthopeptine
Synthese von 3,6,9,12,15,18,21-Heptaoxatricosan-1,23-disäure-tert-butylester (139)
Eine 50%ige KOH-Lösung (11 ml),
Toluol
(11 ml) und Hexaethylenglycol (0,89 ml; 3,54 mmol) werden in einem Kolben vorgelegt. Bei
0 °C werden Tetrabutylammoniumhydrogensulfat (2,65 g; 7,8 mmol) und Bromessigsäure-
tert-butylester
(2,1 ml; 14 mmol) zugegeben. Nach ca. 10 min wird das Kältebad entfernt und 40 min bei RT
gerührt. Das Reaktionsgemisch wird mit 70 ml Wasser verdünnt, die Phasen werden getrennt
und die organische Phase mit ges. NH4Cl-Lösung, ges. NaHCO3-Lösung und ges. NaCl-
Lösung gewaschen. Anschließend wird die organische Phase getrocknet und das
Lösungsmittel im Vakuum entfernt. Der Rückstand wird chromatographisch gereinigt (Hex:
EtOAc; 1:1 auf 0:1). Man erhält 0,8 g (Ausbeute 44 %) eines farblosen Öls.
Rf 0,51 (CHCl3:MeOH; 9:1) 1H-NMR (CDCl3): 1,46 (s, 9H); 3,63 (d, J=4,03Hz, 16H); 3,69 (m, 8H); 4,00 (s, 4H) 13C-NMR (CDCl3): 28,10; 69,04; 70,56; 70,71; 81,50; 169,66
MS (ESI): 511,3118 [MH+]; 533,2925 [MNa+]
Synthese von 3,6,9,12,15,18,21-Heptaoxatricosan-1,23-disäure (140)
Der Diester 139 (800 mg; 1,57 mmol) wird in
Dichlormethan (10 ml) gelöst und mit Trifluoressigsäure
(10 ml) versetzt. Nachdem 4 h bei RT gerührt wurde, wird das Lösungsmittel im Vakuum
entfernt. Man erhält das gewünschte Produkt in quantitativer Ausbeute.
Rf 0,43 (CHCl3:MeOH; 9:2) 1H-NMR (CDCl3): 3,63 (m, 20H); 3,71 (m, 4H); 4,14 (s, 4H), 9,12 (br.s., 2H) 13C-NMR (CDCl3): 68,86; 70,21; 70,27; 70,33; 70,39; 70,47; 71,21; 173,12
MS (ESI): 397,1699 [MH-]
OO O
OO
O
6
OO OH
OHO
O
6

Experimenteller Teil
135
Synthese von 3,6,9,12,15,18,21-Heptaoxatricosan-1,23-disäure-succinimidester (141)
Die Disäure (140) (100 mg; 0,251 mmol) und
HOSu (66 mg; 2,3 eq) werden in DCM (3,5 ml)
gelöst und bei 0 °C mit EDAC (110 mg; 2,3 eq)
versetzt. Nach 20 min wird das Kältebad entfernt und weiter 16 h bei RT gerührt. Nun wird
das Reaktionsgemisch mit EtOAc (20 ml) verdünnt und mit 5% HCl-Lösung (2x10 ml) und
ges. NaHCO3-Lösung (3x10 ml) gewaschen. Anschließend wird das Lösungsmittel im
Vakuum entfernt. Das gewünschte Produkt konnte nicht isoliert werden.
Die Säure (140) (60 mg; 0,15 mmol) wird in DCM (1,5 ml) gelöst und bei 0 °C mit
Oxalylchlorid (77 µl; 0,9 mmol) und einem Tropfen DMF versetzt. Noch 15 min wird das
Kältebad entfernt und 1,5 h bei RT gerührt. Anschließend wird das Lösungsmittel im Vakuum
entfernt. Der Rückstand wird in DCM (1ml) gelöst und mit Pyridin (27 µl; 0,33 mmol) und
HOSu (38 mg; 0,33 mmol) gelöst in THF (0,5 ml) bei -10 °C versetzt. Nun wird das
Reaktionsgemisch 2 h bei RT gerührt. Das gewünschte Produkt konnte nicht isoliert werden.
OO O
O6
NN
O
O
O
O
O
O

Experimenteller Teil
136
Pegylierung von Lab A1/2 (155/154)
In Wasser (10 ml) wird Na3PO4 (71 mg) gelöst und durch Zugabe von 5%iger HCl auf eine
pH-Wert von 7-8 eingestellt. Nun wird Labyrinthopeptin A1 bzw. A2 (10 mg) in dem Puffer
gelöst und mit einer 250 mM Me-PEG8-OSu Lösung (50 µl) versetzt. Das Reaktionsgemisch
wird 6 h bei RT geschüttelt und anschließend gefriergetrocknet. Das Rohprodukt wird mittels
präparativer HPLC (MeCN 45 % auf 51% in 12 min) gereinigt.
Peg-Lab A1 (155): 2 mg (Ausbeute 16 %)
MS (ESI): 1235,5084 [M2+2H]; 2471,0029 [M+H]
tr=4,24 min (MeCN 45 % auf 51% in 12 min)
Peg-Lab A2 (154): 5 mg (Ausbeute 42%)
MS (ESI): 1159,9723 [M2+2H]; 2318,9324 [M+H]
tr=4,27 min (MeCN 45 % auf 51% in 12 min)
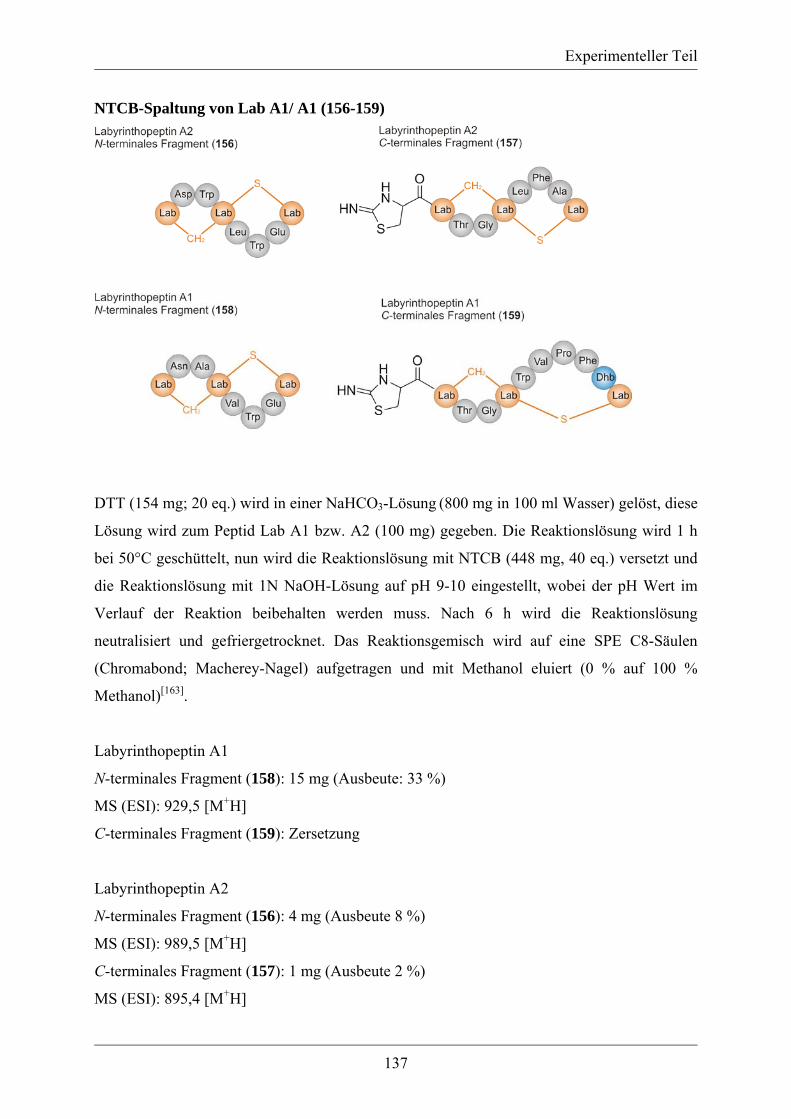
Experimenteller Teil
137
NTCB-Spaltung von Lab A1/ A1 (156-159)
DTT (154 mg; 20 eq.) wird in einer NaHCO3-Lösung (800 mg in 100 ml Wasser) gelöst, diese
Lösung wird zum Peptid Lab A1 bzw. A2 (100 mg) gegeben. Die Reaktionslösung wird 1 h
bei 50°C geschüttelt, nun wird die Reaktionslösung mit NTCB (448 mg, 40 eq.) versetzt und
die Reaktionslösung mit 1N NaOH-Lösung auf pH 9-10 eingestellt, wobei der pH Wert im
Verlauf der Reaktion beibehalten werden muss. Nach 6 h wird die Reaktionslösung
neutralisiert und gefriergetrocknet. Das Reaktionsgemisch wird auf eine SPE C8-Säulen
(Chromabond; Macherey-Nagel) aufgetragen und mit Methanol eluiert (0 % auf 100 %
Methanol)[163].
Labyrinthopeptin A1
N-terminales Fragment (158): 15 mg (Ausbeute: 33 %)
MS (ESI): 929,5 [M+H]
C-terminales Fragment (159): Zersetzung
Labyrinthopeptin A2
N-terminales Fragment (156): 4 mg (Ausbeute 8 %)
MS (ESI): 989,5 [M+H]
C-terminales Fragment (157): 1 mg (Ausbeute 2 %)
MS (ESI): 895,4 [M+H]

Experimenteller Teil
138
Desulfurierung von Lab A1 /A2 (160/161)
Das Peptid Lab A1 bzw. A2 (1 mg) wird einer MeOH-Wasser-Lösung (1,3 ml; 0,64 ml)
suspendiert und mit NiCl2*6H2O (4,74 mg) versetzt. Nun wird NaBH4 (2,5 mg) zugesetzt und
3 h bei 50°C geschüttelt. Bei der Reaktion bildet sich ein schwarzer Niederschlag, der nach
der Reaktion mit TFA (30 µl) gefällt wird. Nach der Zentrifugation wird der Überstand aus
dem Reaktionsgefäß entfernt und die Lösung gefriergetrocknet. Das Rohprodukt wird auf eine
SPE C8-Säule (Chromabond ;Macherey-Nagel) aufgetragen, nun mit einer 50mmol
EDTA-Lösung gewaschen und anschließend mit Methanol eluiert (0 % auf 100 %
Methanol)[163].
Das Peptid Lab A1 /A2 (1 mg) wird in Wasser (0,5 ml) gelöst und mit einer Raney-Nickel
Suspension (0,5 ml; 50 % in Wasser) versetzt. Nun wird 16 h bei 60°C geschüttelt. Das
Rohprodukt wird auf eine SPE C8-Säule (Chromabond ;Macherey-Nagel) aufgetragen, nun
mit einer 50 mmol EDTA-Lösung gewaschen und anschließend mit Methanol eluiert
(0 % auf 100 % Methanol)[163].
Desulf-Lab A1(161): 0,44 mg (Ausbeute 44 %)
MS (ESI): 1008,4424 [M2+2H]; 2015,8732 [M+H]
Desulf-Lab A2 (160): 0,28 mg (Ausbeute 28 %)
MS (ESI): 932,3937 [M2+2H]; 1863,7787 [M+H]
Experimenteller Teil
139
5.3. Analytische Daten 5.3.1 NMR-Spektren ausgewählter Verbindungen rac-Cbz-α-Allylalanin-tert-butylester (130)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
8.394.73 3.991.00 1.00
DMSO-d6
7.51
7.35
7.34
7.32
7.31
7.30 7.
307.
29 5.70
5.70 5.
68 5.68
5.66
5.66 5.
645.
625.
075.
015.
004.
97
2.56 2.54 2.
52 2.37
2.35
2.33
2.31
1.31
1.24
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20-0.15
-0.10
-0.05
0.00
0.05
0.10
0.15
0.20
Chloroform-d
172.73
154.52
136.65
132.
4112
8.43
127.
93
119.04
82.00
66.18
59.57
41.00
27.8
3
23.2
8
Experimenteller Teil
140
rac-Cbz-2-amino-4,5-dihydroxy-2-methyl-pentansäure-tert-butylester (131)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
9.685.03 2.03 1.05 1.05 1.000.92 0.72
Chloroform-d7.
347.
337.
327.
307.
297.
29 6.20
6.16
5.07 5.04
5.01
3.80
3.79
3.79 3.78
3.56 3.53
3.53 3.41
3.39
3.39
3.37
2.24
2.20
2.11
2.08 2.
021.
991.
981.
951.
581.
541.
441.
42
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
0.00
0.05
0.10
0.15
0.20
0.25
0.30
Chloroform-d
173.
6717
3.16
154.
65
136.
53
128.
4712
8.04
127.
99
82.2
9
69.3
7 68.7
3
66.7
966
.34
59.4
758
.33
40.4
939
.19
27.7
324
.03
Experimenteller Teil
141
(2S, 4S)/ (2R, 4R)-Cbz-2-Amino-5-tert-butylsilyloxy-4-hydroxy-2-methyl-pentansäure-
tert-butylester (+/-132a)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
9.579.20 6.114.91 1.94 1.021.01 0.99 0.890.88
Chloroform-d
7.34
7.33
7.30
7.29
7.29 7.28 6.32
5.09 5.
065.
055.
02
3.74
3.73
3.57
3.57 3.55
3.54
3.38
3.37
3.36
3.34 2.
322.
31
2.12 2.
001.
971.
961.
551.
44
0.88
0.04
150 100 50 0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
Chloroform-d
173.
52
154.
53
136.
77
128.
4212
7.84
86.6
081
.79
68.4
067
.02
66.0
7 58.3
6
39.1
7
27.7
525
.84
23.6
6
18.2
3
-5.4
6
Experimenteller Teil
142
(2R, 4S)/ (2S, 4R)-Cbz-2-Amino-5-tert-butylsilyloxy-4-hydroxy-2-methyl-pentansäure-
tert-butylester (+/-132b)
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
9.609.26 6.004.77 1.80 1.000.98 0.970.68 0.67
Chloroform-d
7.37 7.
367.
347.
337.
31 7.31
7.29
7.28
6.40
5.07
3.80 3.
78
3.49
3.48 3.47
3.46 3.
383.
363.
353.
332.
661.
92 1.89
1.87 1.
841.
84
1.61
1.42
0.88
0.04
150 100 50 0
-0.4
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
Chloroform-d
172.90
155.48
136.80
128.
3512
8.01
127.
84
81.62
69.2
5
67.2766.19 59.61
40.70
27.8
325
.80
24.6
8
18.21
-5.4
4
Experimenteller Teil
143
(2S,4S)/ (2R,4R)-Cbz-4-2-Amino-4-azido-5-tert-butylsilyloxy-2-methyl-pentansäure-tert-
butylester (+/-134a)
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
9.00 8.586.95 5.522.18 0.99 0.930.75
Chloroform-d
7.40 7.38 7.
367.
357.
357.
347.
337.
327.
317.
31
5.79 5.13
5.10
5.07
5.04 3.63
3.62
3.61
3.59
3.56
3.41
3.40
3.39
3.38
2.08
2.07 2.05
2.02 1.
951.
921.
561.
44
0.90
0.07 0.
06
150 100 50 0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
Chloroform-d
172.
57
154.
89
136.
54
128.
4512
8.05
82.3
1
66.9
566
.39 60
.58
59.1
1
36.7
2
27.7
825
.75
24.2
5
18.1
9 -5.5
5
Experimenteller Teil
144
(2R,4S)/ (2S,4R)-Cbz-2-Amino-4-azido-5-tert-butylsilyloxy-2-methyl-pentansäure-tert-
butylester (+/-133b)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5 0.0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
9.148.88 6.004.98 2.04 1.03 1.010.940.89 0.88
Chloroform-d
7.35
7.34
7.32
7.30
7.29 6.14
5.07
3.70 3.68
3.58 3.56
3.53
3.38
3.37 3.36
3.36
3.34
2.38
2.34 1.
911.
881.
871.
541.
48
0.90
0.06
150 100 50 0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
Chloroform-d
172.
81
154.
30
136.
61
128.
5112
7.86
82.6
6
66.7
666
.27 60
.28
58.3
1
36.6
3
27.6
525
.78
24.4
0
18.2
0 -5.5
8
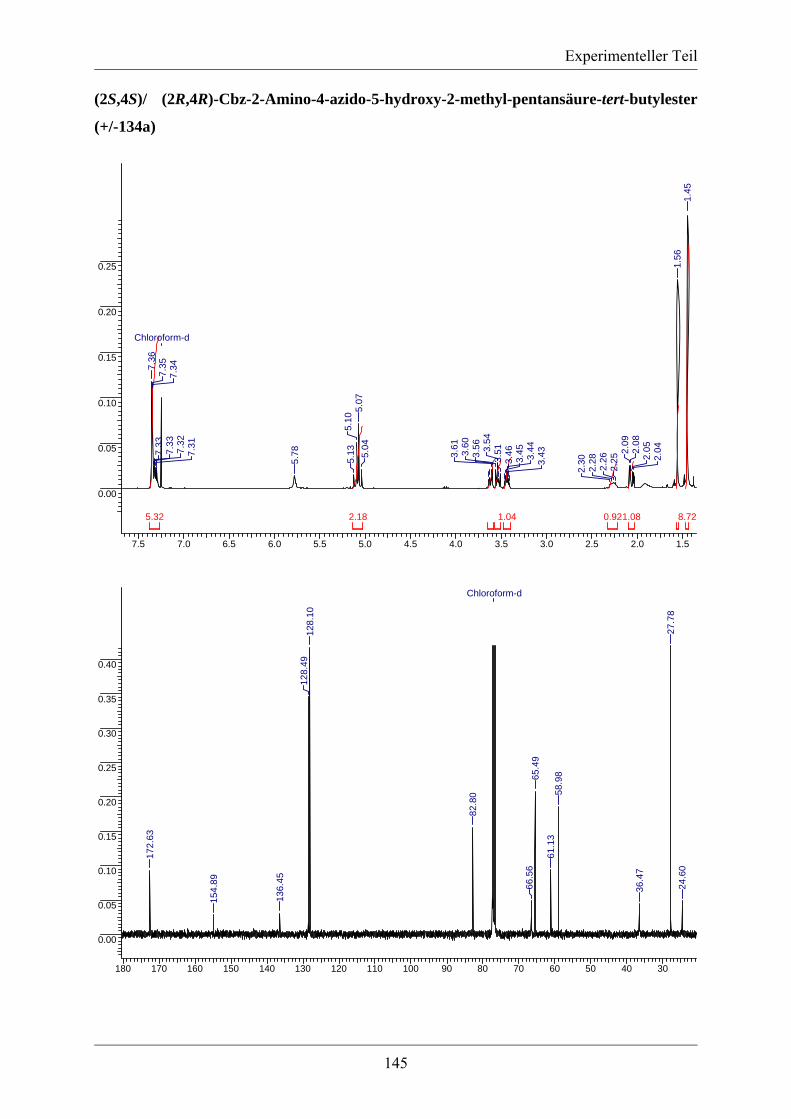
Experimenteller Teil
145
(2S,4S)/ (2R,4R)-Cbz-2-Amino-4-azido-5-hydroxy-2-methyl-pentansäure-tert-butylester
(+/-134a)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
8.725.32 2.18 1.081.04 0.92
Chloroform-d
7.36
7.35
7.34
7.33
7.33
7.32
7.31
5.78
5.13
5.10
5.07
5.04
3.61 3.60
3.56 3.
543.
513.
463.
45 3.44
3.43
2.30
2.28
2.26
2.25
2.09
2.08
2.05
2.04
1.56
1.45
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
Chloroform-d
172.
63
154.
89
136.
45
128.
4912
8.10
82.8
0
66.5
665
.49
61.1
358
.98
36.4
7
27.7
824
.60
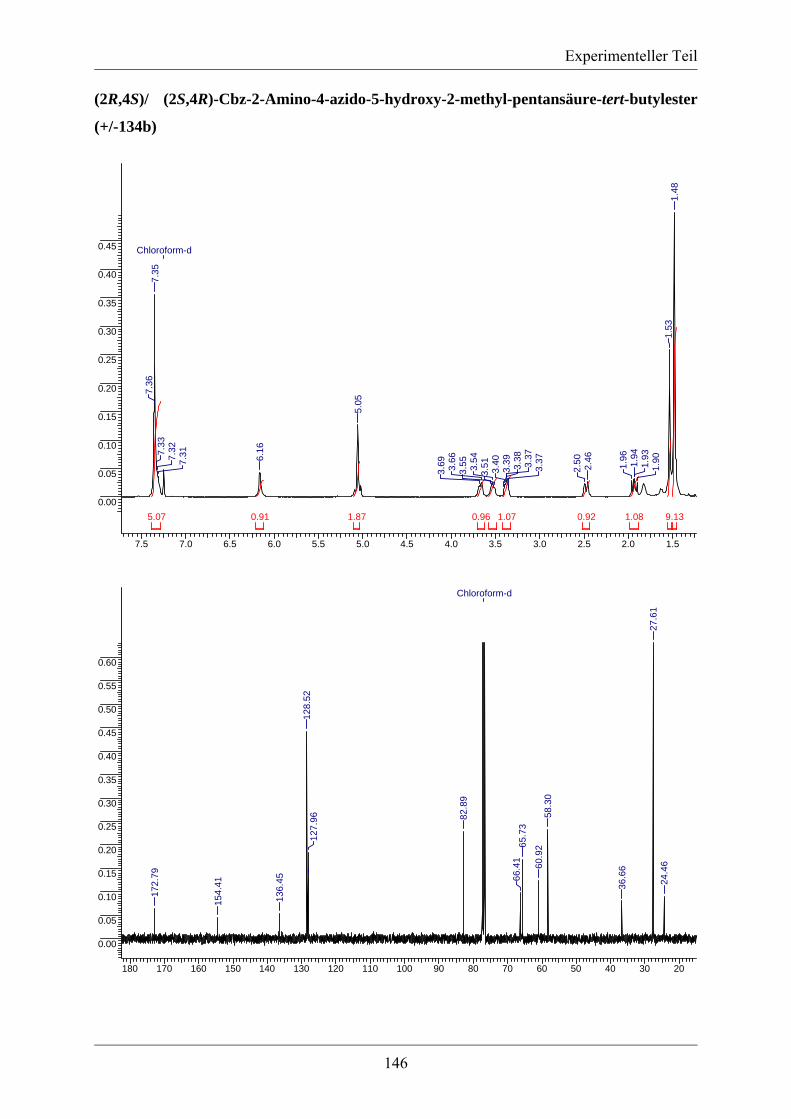
Experimenteller Teil
146
(2R,4S)/ (2S,4R)-Cbz-2-Amino-4-azido-5-hydroxy-2-methyl-pentansäure-tert-butylester
(+/-134b)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
9.135.07 1.87 1.081.070.96 0.920.91
Chloroform-d
7.36
7.35
7.33
7.32
7.31 6.16
5.05
3.69 3.66
3.55 3.54
3.51 3.40
3.39 3.38 3.37
3.37
2.50 2.46
1.96 1.94
1.93
1.90
1.53
1.48
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
Chloroform-d
172.
79
154.
41
136.
45
128.
5212
7.96 82
.89
66.4
165
.73
60.9
258
.30
36.6
6
27.6
124
.46
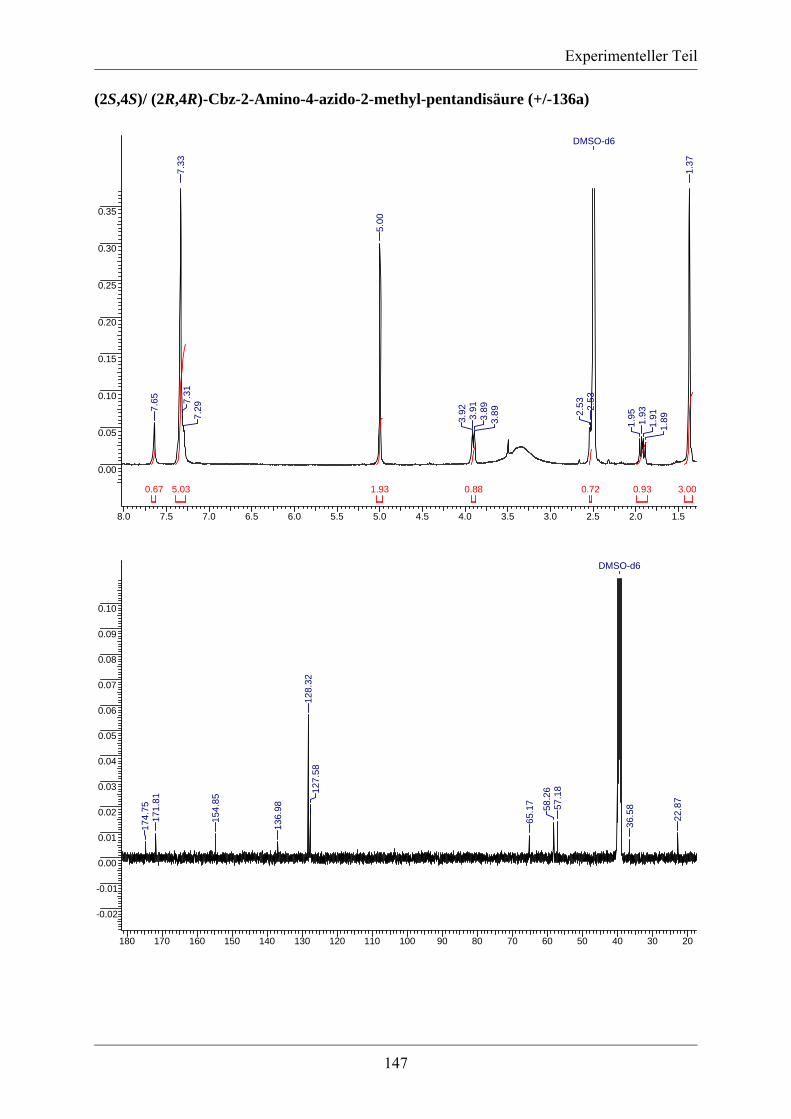
Experimenteller Teil
147
(2S,4S)/ (2R,4R)-Cbz-2-Amino-4-azido-2-methyl-pentandisäure (+/-136a)
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
5.03 3.001.93 0.930.88 0.720.67
DMSO-d67.
65
7.33
7.31
7.29
5.00
3.92 3.91
3.89
3.89 2.
53 2.53
1.95 1.93
1.91
1.89
1.37
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
-0.02
-0.01
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
DMSO-d6
174.
7517
1.81
154.
85
136.
98
128.
3212
7.58
65.1
7 58.2
657
.18
36.5
8
22.8
7
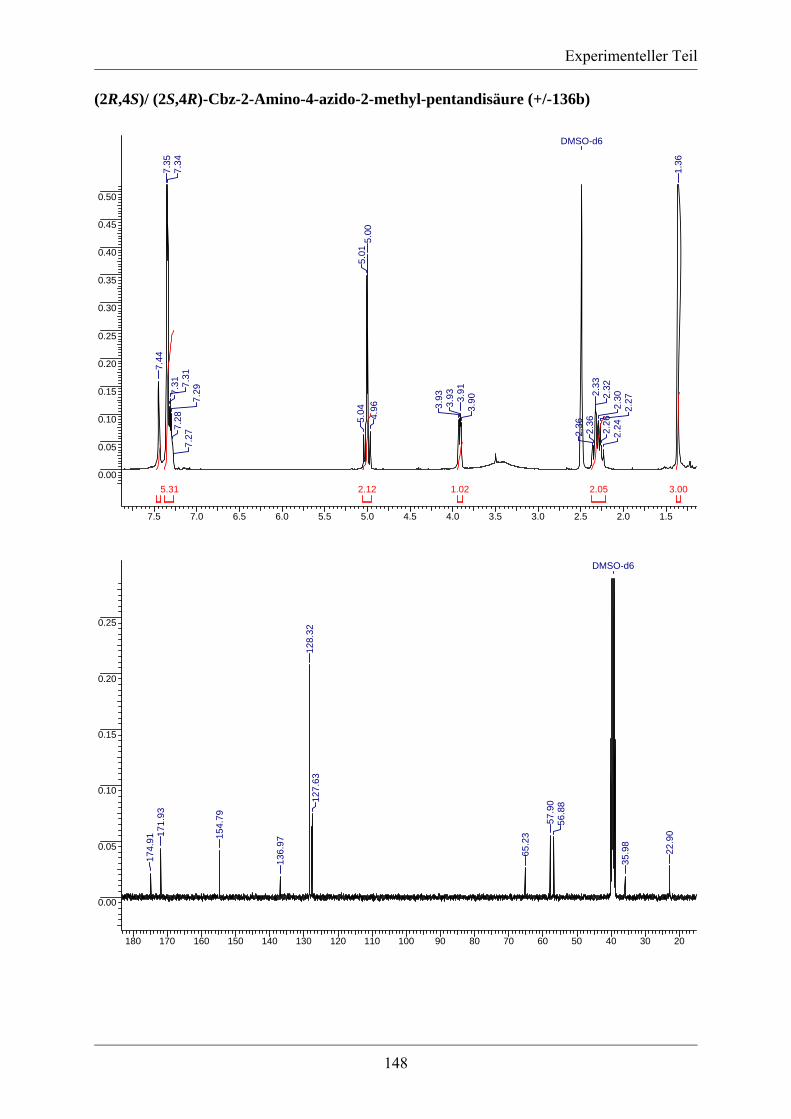
Experimenteller Teil
148
(2R,4S)/ (2S,4R)-Cbz-2-Amino-4-azido-2-methyl-pentandisäure (+/-136b)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
5.31 3.002.12 2.051.02
DMSO-d6
7.44
7.35
7.34
7.31 7.
317.
29
7.28
7.27
5.04
5.01
5.00
4.96 3.
933.
93 3.91
3.90
2.36 2.36
2.33
2.32
2.30
2.27
2.26
2.24
1.36
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
0.00
0.05
0.10
0.15
0.20
0.25
DMSO-d6
174.
9117
1.93
154.
79
136.
97
128.
3212
7.63
65.2
3
57.9
056
.88
35.9
8
22.9
0
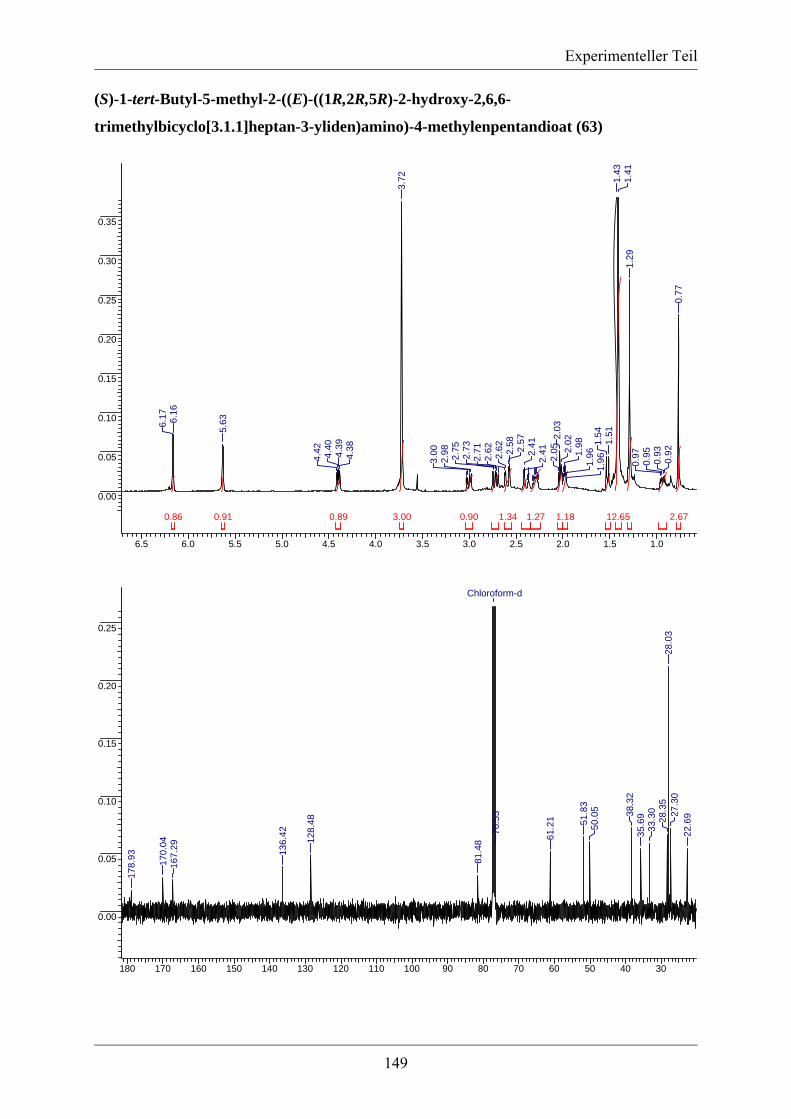
Experimenteller Teil
149
(S)-1-tert-Butyl-5-methyl-2-((E)-((1R,2R,5R)-2-hydroxy-2,6,6-
trimethylbicyclo[3.1.1]heptan-3-yliden)amino)-4-methylenpentandioat (63)
6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
12.653.00 2.671.34 1.27 1.180.91 0.900.890.86
6.17 6.16
5.63
4.42 4.40
4.39
4.38
3.72
3.00
2.98 2.75
2.73
2.71
2.62 2.62 2.58 2.57
2.41
2.41
2.05
2.03
2.02
1.98
1.96
1.96
1.54
1.51
1.43
1.41
1.29
0.97
0.95
0.93
0.92
0.77
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
0.00
0.05
0.10
0.15
0.20
0.25
Chloroform-d
178.
93 170.
0416
7.29 13
6.42
128.
48
81.4
8
76.5
3
61.2
1 51.8
350
.05 38
.32
35.6
933
.30
28.3
528
.03
27.3
022
.69
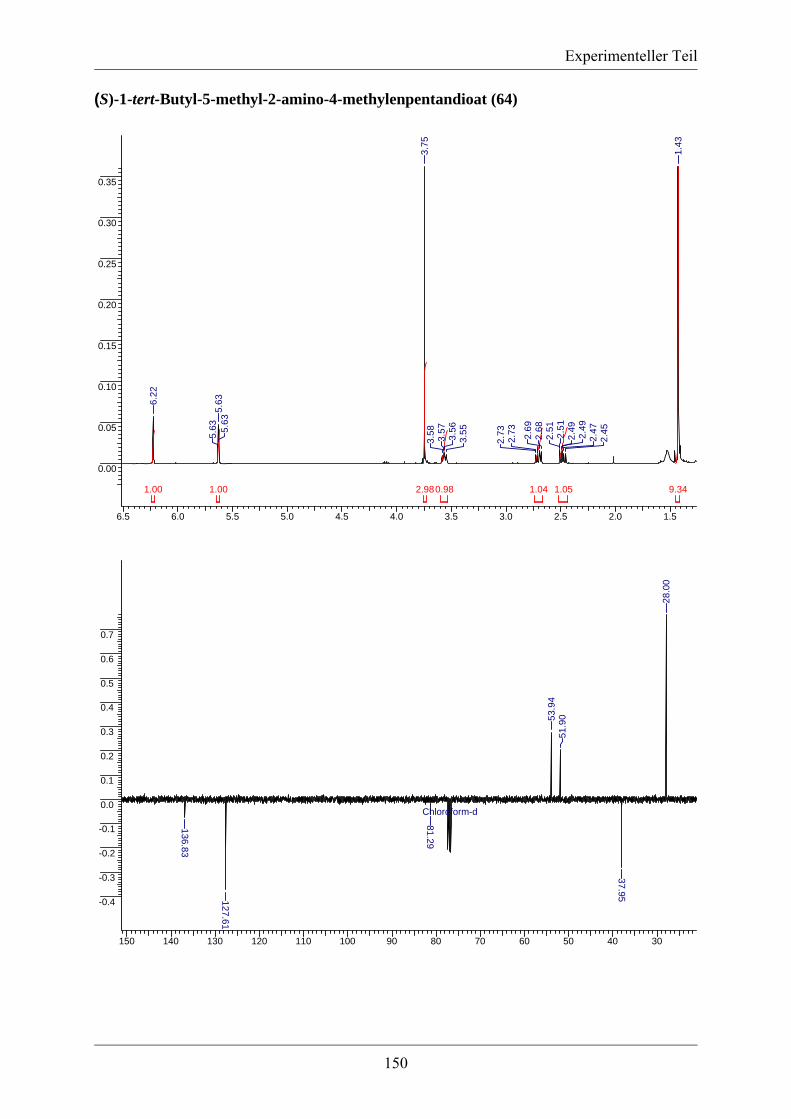
Experimenteller Teil
150
(S)-1-tert-Butyl-5-methyl-2-amino-4-methylenpentandioat (64)
6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
9.342.98 1.051.041.00 1.00 0.98
6.22
5.63
5.63
5.63
3.75
3.58 3.57
3.56
3.55
2.73
2.73 2.69
2.68
2.51
2.51
2.49
2.49
2.47
2.45
1.43
150 140 130 120 110 100 90 80 70 60 50 40 30
-0.4
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Chloroform-d
136.83
127.61
81.29
53.9
451
.90
37.95
28.0
0
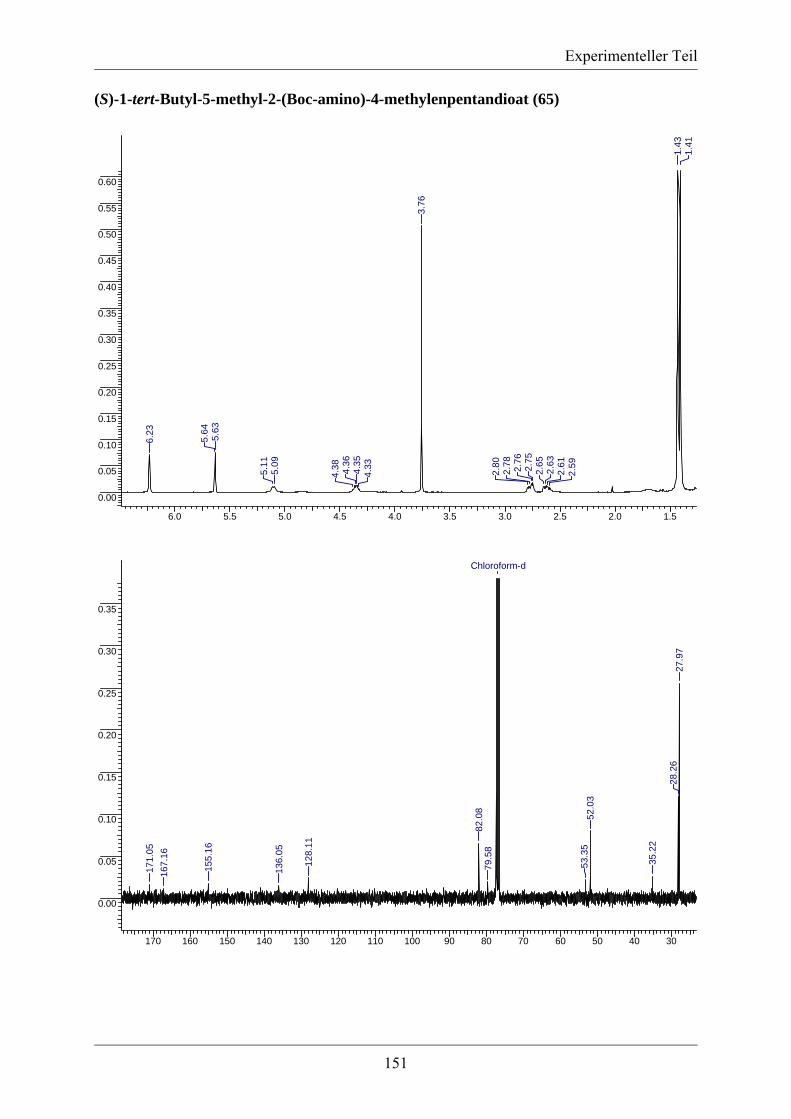
Experimenteller Teil
151
(S)-1-tert-Butyl-5-methyl-2-(Boc-amino)-4-methylenpentandioat (65)
6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
6.23
5.64
5.63
5.11
5.09
4.38 4.36
4.35
4.33
3.76
2.80
2.78 2.76
2.75
2.65
2.63
2.61
2.59
1.43
1.41
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
Chloroform-d
171.
0516
7.16
155.
16
136.
05
128.
11
82.0
879
.58
53.3
552
.03
35.2
2
28.2
627
.97
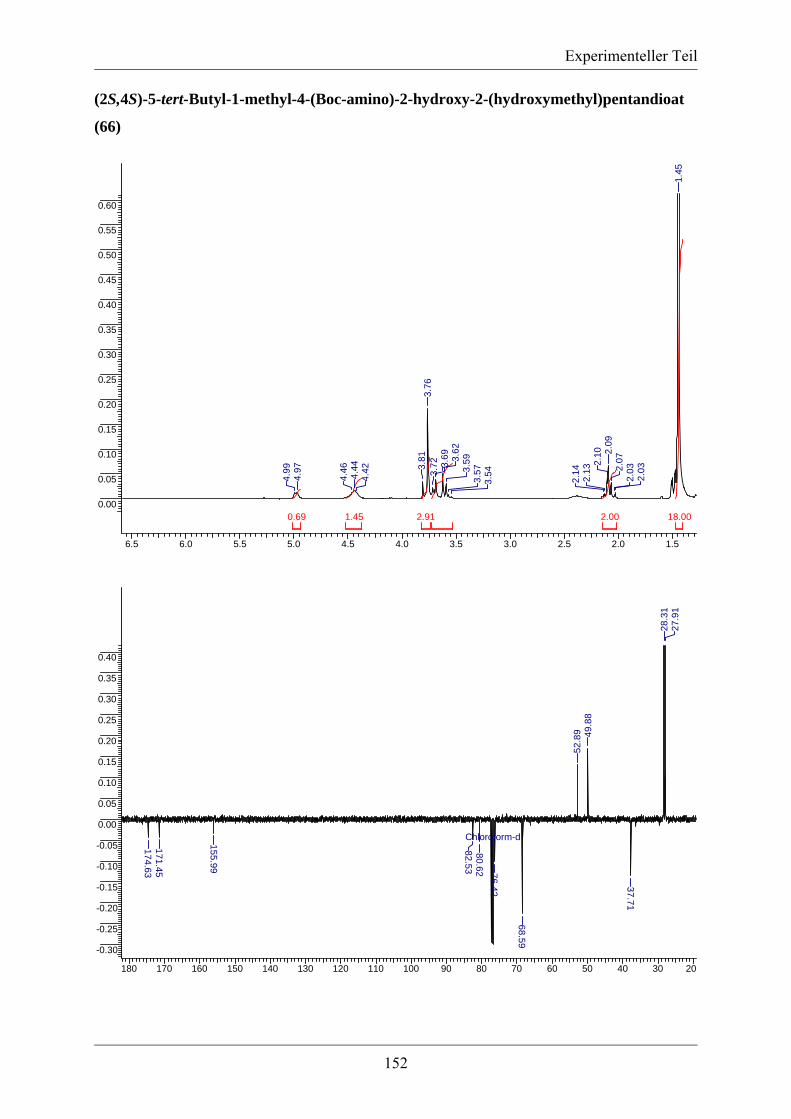
Experimenteller Teil
152
(2S,4S)-5-tert-Butyl-1-methyl-4-(Boc-amino)-2-hydroxy-2-(hydroxymethyl)pentandioat
(66)
6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
18.002.91 2.001.450.69
4.99
4.97
4.46 4.44
4.42 3.
813.
763.
72 3.69 3.
623.
593.
573.
54
2.14
2.13
2.10 2.
092.
072.
032.
03
1.45
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
-0.30
-0.25
-0.20
-0.15
-0.10
-0.05
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
Chloroform-d
174.63171.45
155.99
82.5380.62 76.42
68.59
52.8
9 49.8
8
37.71
28.3
127
.91
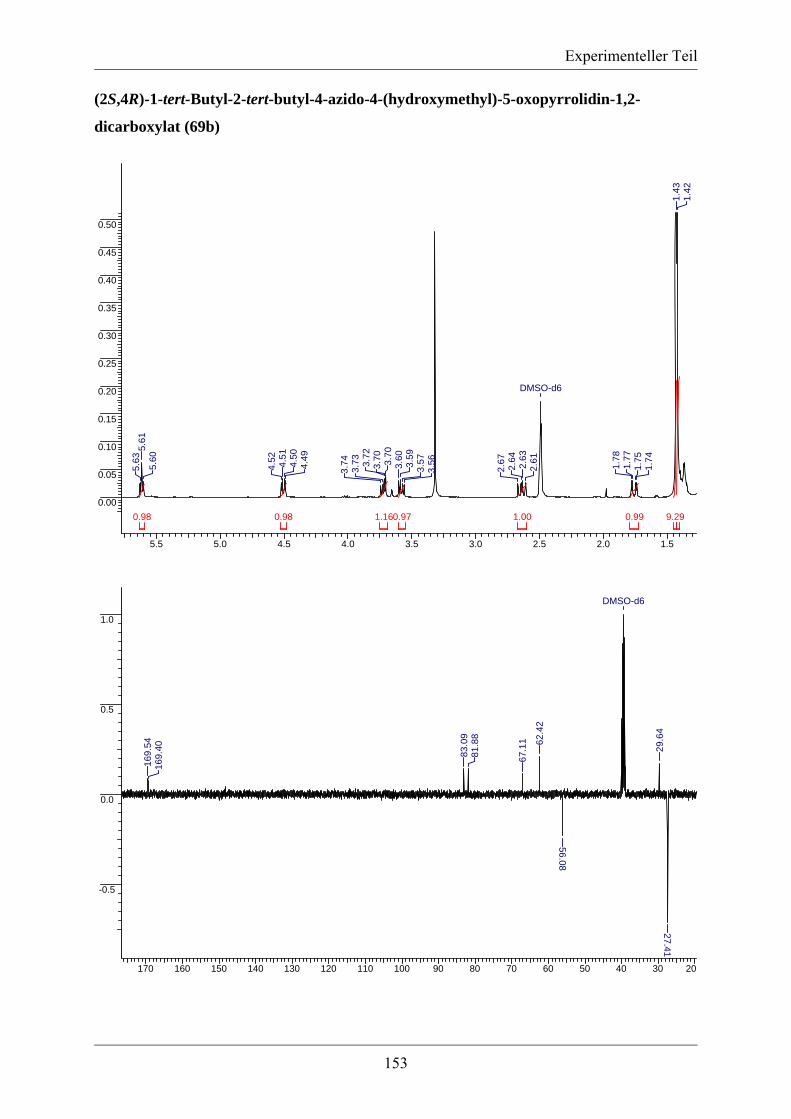
Experimenteller Teil
153
(2S,4R)-1-tert-Butyl-2-tert-butyl-4-azido-4-(hydroxymethyl)-5-oxopyrrolidin-1,2-
dicarboxylat (69b)
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
9.291.16 1.00 0.990.980.98 0.97
DMSO-d6
5.63
5.61
5.60
4.52 4.51
4.50
4.49
3.74
3.73 3.
723.
70 3.70
3.60
3.59
3.57
3.56
2.67 2.64 2.63
2.61 1.78
1.77
1.75
1.74
1.43
1.42
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
-0.5
0.0
0.5
1.0
DMSO-d6
169.
5416
9.40 83.0
981
.88
67.1
1 62.4
2
56.08
29.6
4
27.41
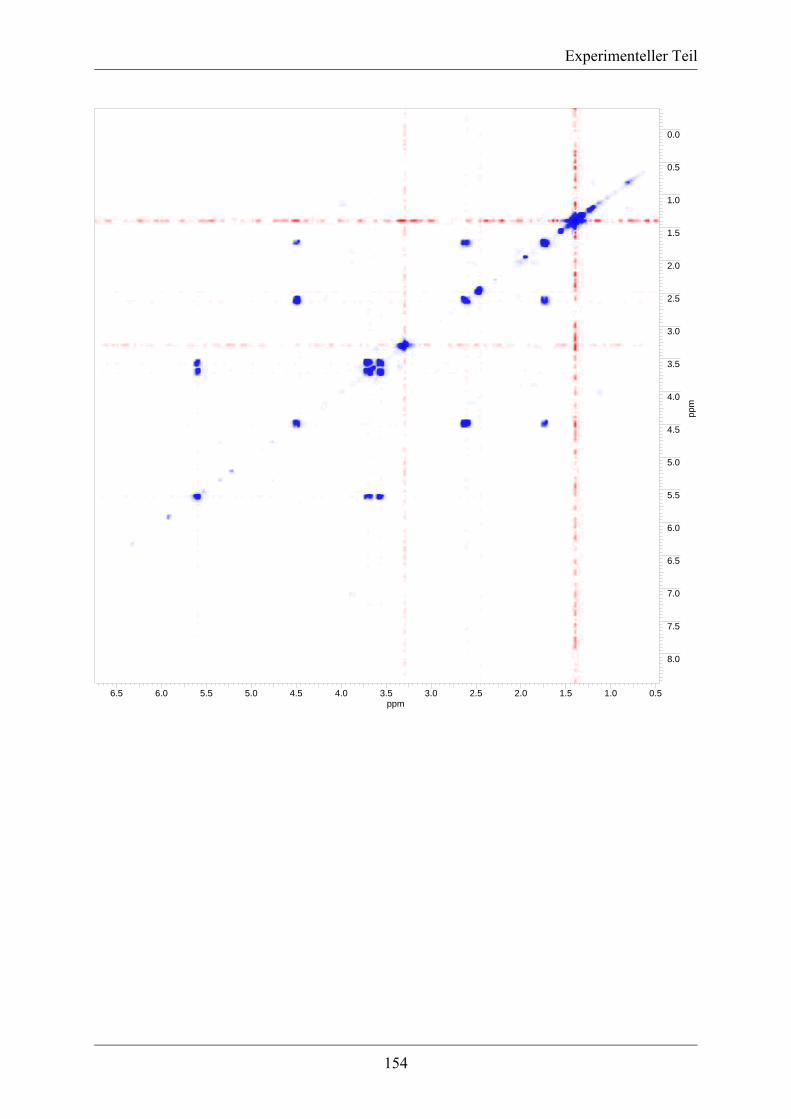
Experimenteller Teil
154
6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5ppm
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5
8.0
ppm
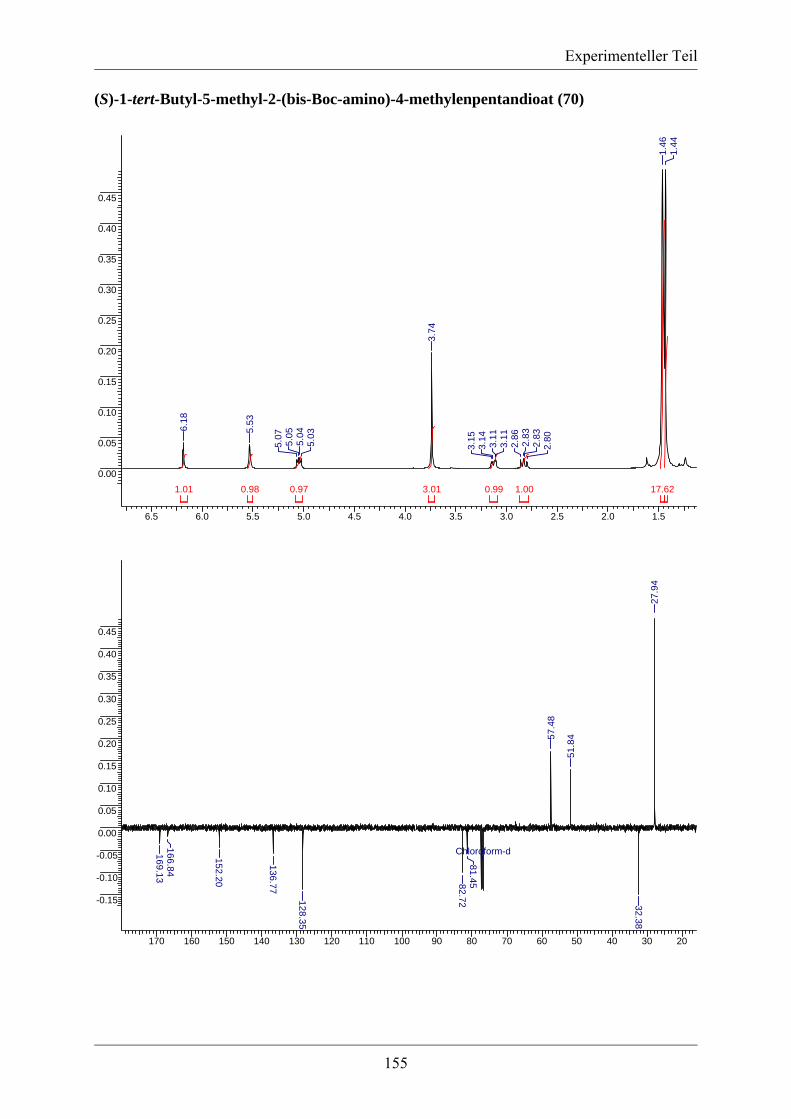
Experimenteller Teil
155
(S)-1-tert-Butyl-5-methyl-2-(bis-Boc-amino)-4-methylenpentandioat (70)
6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
17.623.011.01 1.000.990.98 0.97
6.18
5.53
5.07
5.05
5.04
5.03
3.74
3.15
3.14 3.11
3.11
2.86
2.83
2.83
2.80
1.46
1.44
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
-0.15
-0.10
-0.05
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
Chloroform-d169.13166.84
152.20
136.77
128.35
82.7281.45
57.4
8
51.8
4
32.38
27.9
4
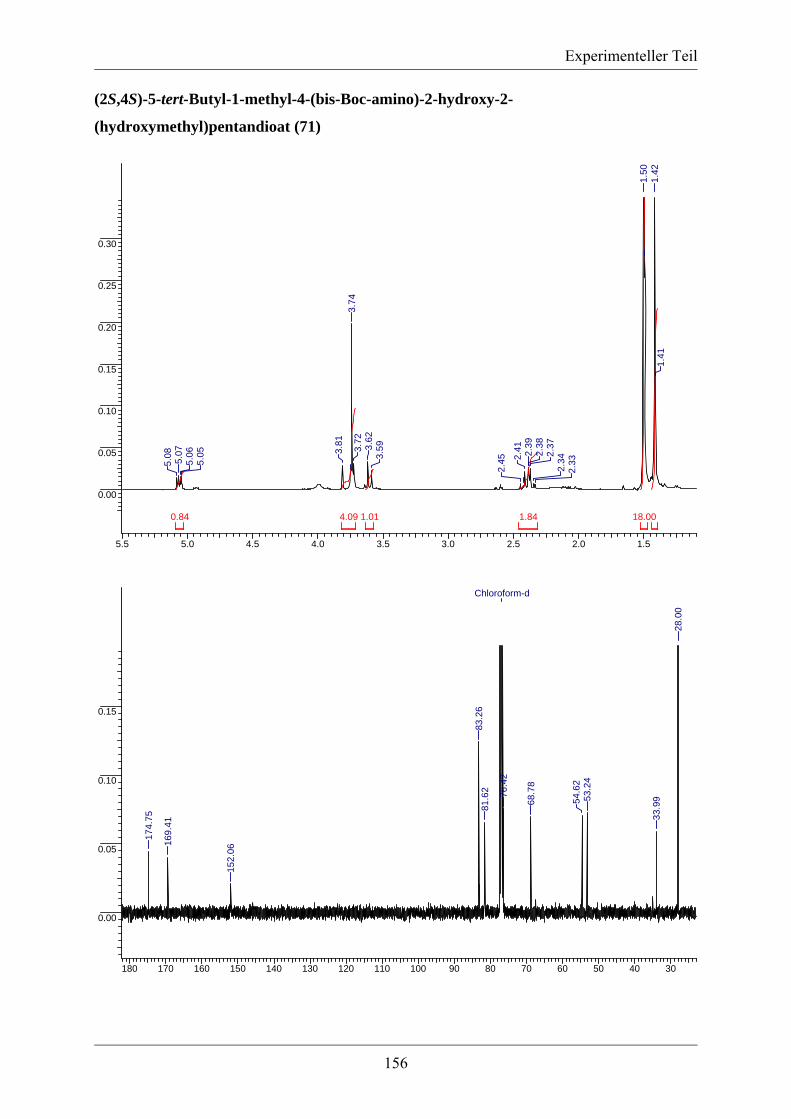
Experimenteller Teil
156
(2S,4S)-5-tert-Butyl-1-methyl-4-(bis-Boc-amino)-2-hydroxy-2-
(hydroxymethyl)pentandioat (71)
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
18.004.09 1.841.010.84
5.08 5.07
5.06
5.05 3.
813.
743.
72 3.62
3.59
2.45 2.
41 2.39
2.38
2.37
2.34
2.33
1.50
1.42
1.41
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
0.00
0.05
0.10
0.15
Chloroform-d
174.
75
169.
41
152.
06
83.2
681
.62 76
.42
68.7
8
54.6
253
.24
33.9
9
28.0
0
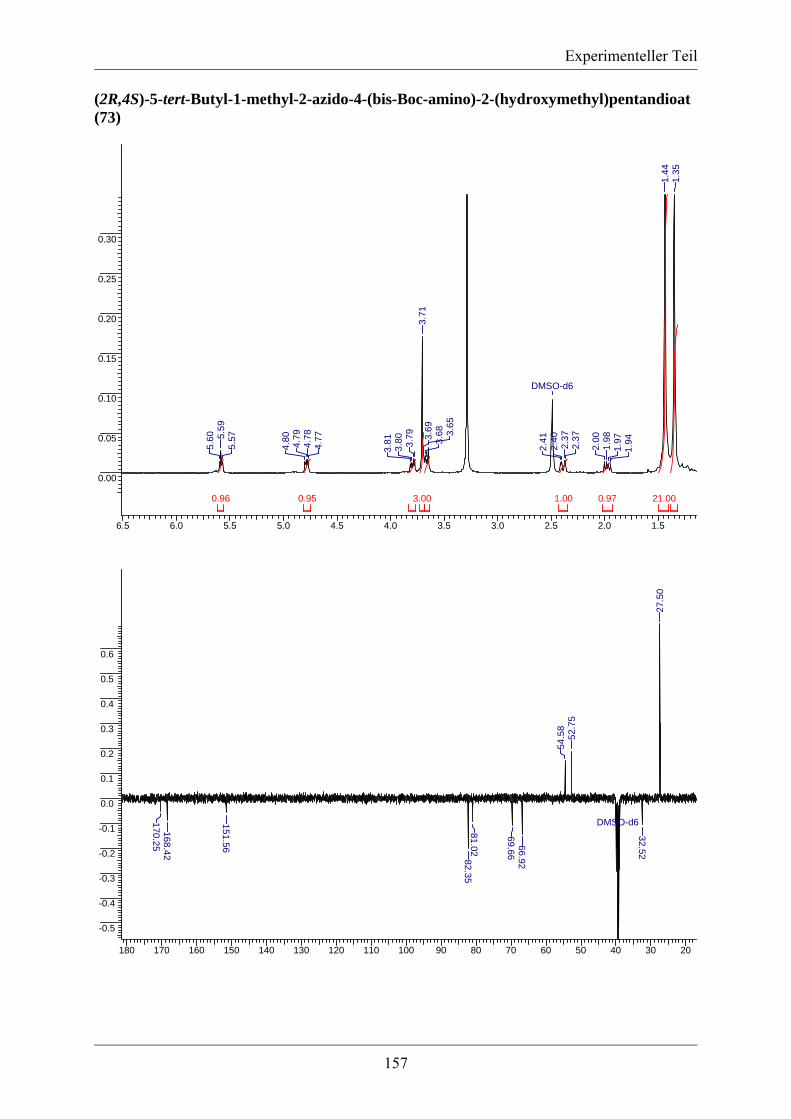
Experimenteller Teil
157
(2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-(hydroxymethyl)pentandioat (73)
6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
21.003.00 1.00 0.970.96 0.95
DMSO-d6
5.60 5.
595.
57
4.80 4.79
4.78
4.77
3.81
3.80 3.79
3.71
3.69
3.68 3.
65
2.41
2.40
2.37
2.37
2.00
1.98
1.97
1.94
1.44
1.35
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
-0.5
-0.4
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
DMSO-d6170.25168.42
151.56
82.3581.02
69.6666.92
54.5
852
.75
32.52
27.5
0
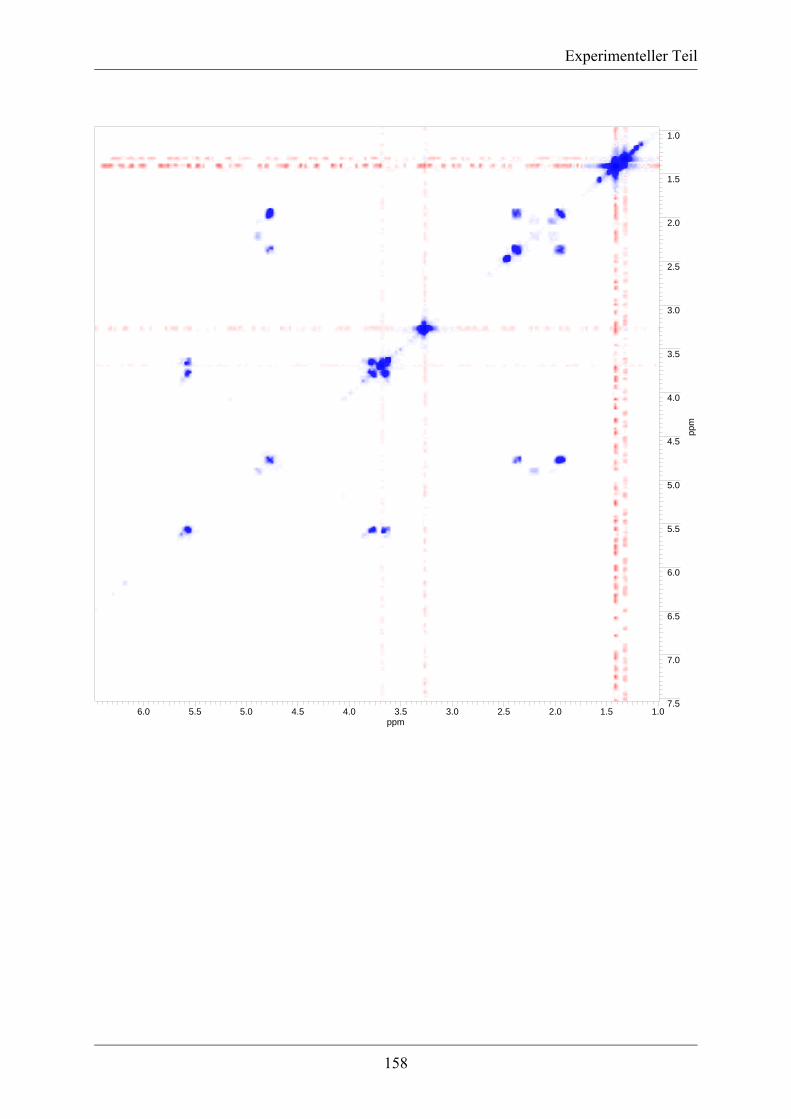
Experimenteller Teil
158
6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0ppm
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
6.5
7.0
7.5pp
m
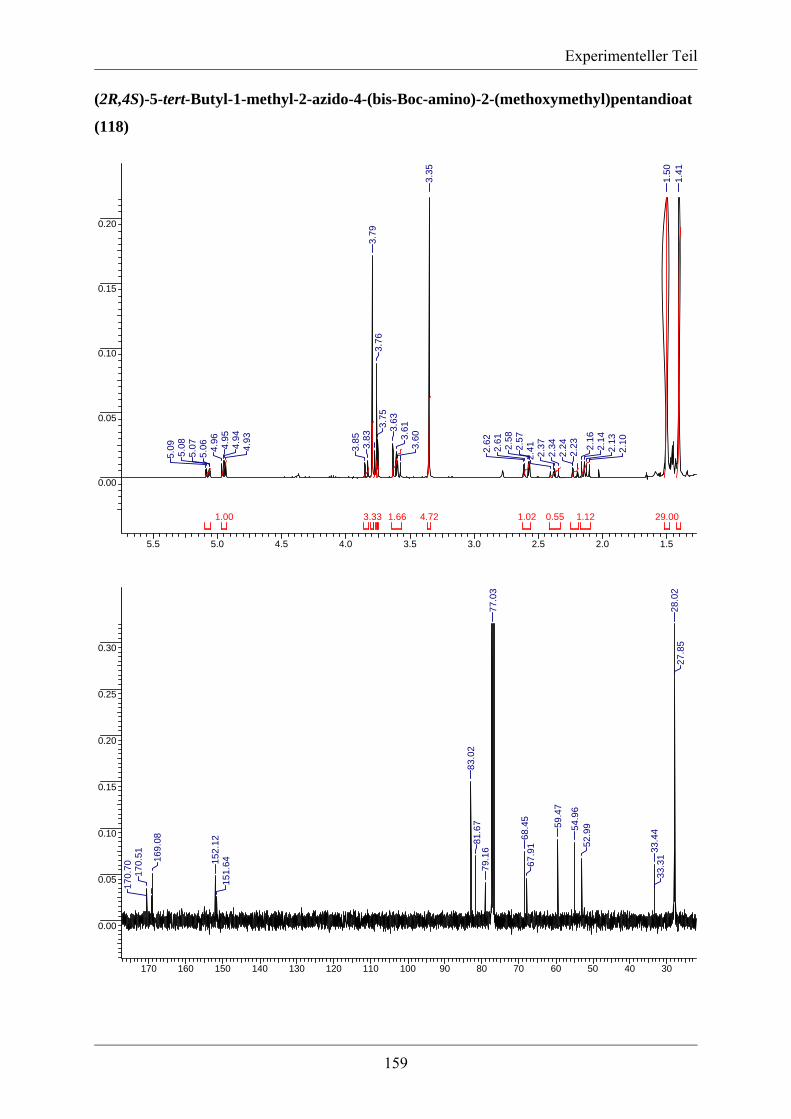
Experimenteller Teil
159
(2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-(methoxymethyl)pentandioat
(118)
5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
29.004.723.33 1.66 1.121.021.00 0.55
5.09 5.08
5.07
5.06 4.
96 4.95
4.94
4.93
3.85 3.83
3.79
3.76
3.75
3.63
3.61
3.60
3.35
2.62
2.61 2.58
2.57
2.41 2.37
2.34
2.24
2.23 2.
162.
142.
132.
10
1.50
1.41
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
0.00
0.05
0.10
0.15
0.20
0.25
0.30
170.
70 170.
51 169.
08
152.
1215
1.64
83.0
281
.67
79.1
677
.03
68.4
567
.91
59.4
7
54.9
652
.99
33.4
433
.31
28.0
227
.85
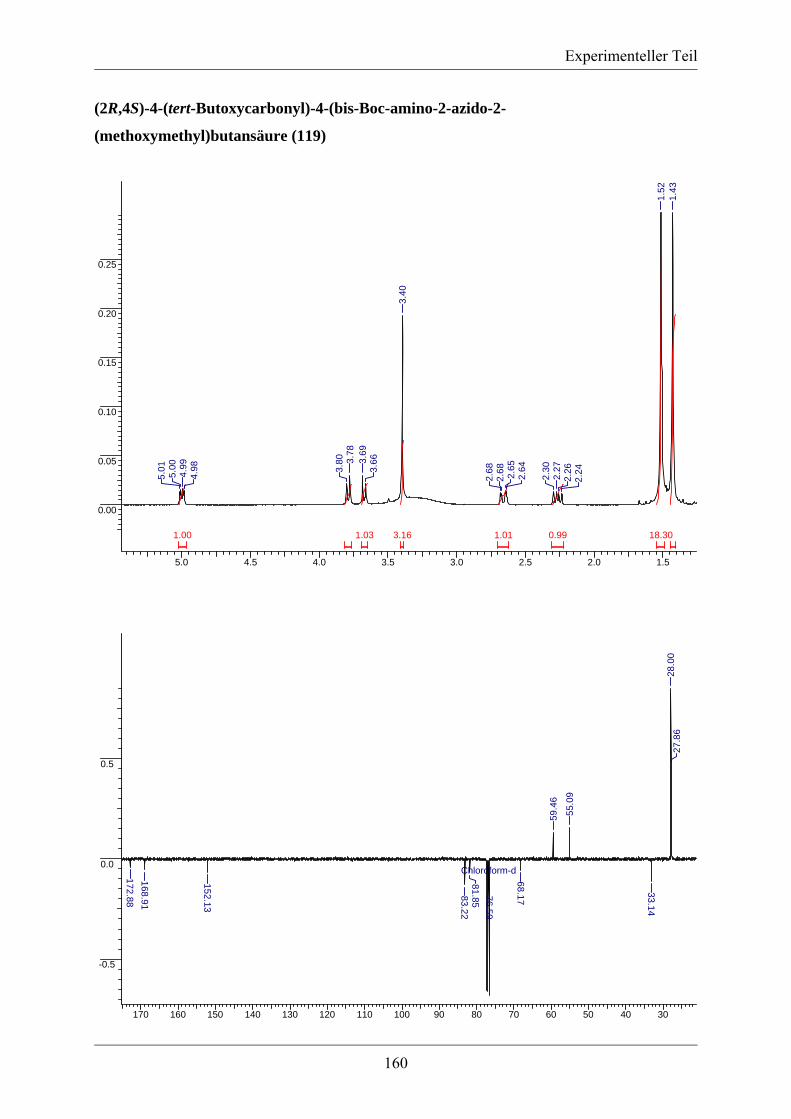
Experimenteller Teil
160
(2R,4S)-4-(tert-Butoxycarbonyl)-4-(bis-Boc-amino-2-azido-2-
(methoxymethyl)butansäure (119)
5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
18.303.161.03 1.011.00 0.99
5.01 5.00
4.99
4.98 3.
80 3.78
3.69
3.66
3.40
2.68
2.68 2.65
2.64
2.30
2.27
2.26
2.24
1.52
1.43
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30
-0.5
0.0
0.5
Chloroform-d172.88168.91
152.13
83.2281.85
76.59
68.17
59.4
6
55.0
9
33.14
28.0
027
.86
Experimenteller Teil
161
Boc-Leu-Phe-Ala-OMe (100)
9 8 7 6 5 4 3 2 1 0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
8.06 6.855.19 3.031.01 1.000.91 0.780.77
DMSO-d6
8.43
8.42 7.70
7.68
7.23
7.22
7.19 7.
187.
177.
166.
856.
83
4.59 4.58 4.57
4.56
4.55 4.
28 4.26
4.25
3.87
3.61
3.02
3.01 2.
992.
97 2.80 2.78
2.77
2.74
1.35
1.28
1.26
0.82
0.80
0.78
0.76
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
-0.5
0.0
0.5
1.0
DMSO-d6
172.
7117
2.01
170.
81
155.
10
137.
39
129.24127.85
126.12
78.0
5
53.0452.87
51.8047.51
40.8
637
.71
28.1024.0922.77
16.81
Experimenteller Teil
162
H-Leu-Phe-Ala-OMe (101)
8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
5.995.15 3.01 2.891.101.00 1.000.99 0.920.90 0.70
DMSO-d6
8.46
8.44
7.98
7.96 7.
257.
247.
22 7.21
7.19
7.18 7.
167.
14 4.57 4.
30 4.28
4.27
4.25
3.62
3.10
3.09
3.08
3.07
3.01
3.00
2.81
2.79
2.76
1.60
1.58
1.58
1.56
1.29
1.28
1.23
1.21
0.82
0.80 0.78
0.76
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10
-0.5
0.0
0.5
1.0
DMSO-d6
175.10172.79
170.95
137.44
129.
2812
7.85
126.
14
53.0
352
.54
51.8
147
.49
43.92
37.87
23.8
923
.13
16.8
3
Experimenteller Teil
163
(2S,3R)-2-Azido-3-(benzyloxy)butansäure (97)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
4.96 4.491.331.011.00 0.99
DMSO-d67.
347.
327.
307.
287.
277.
267.
25
7.24
4.62 4.
594.
414.
384.
164.
15 4.13
4.13
3.92
3.91
1.26
1.25
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
0.00
0.05
0.10
0.15
0.20
0.25
0.30
DMSO-d6
170.
45
138.
33 128.
16
127.
03
75.5
4
70.2
5
65.3
3
16.5
5
Experimenteller Teil
164
2-((2S,3R)-2-Azido-3-(benzyloxy)butanamido)essigsäure-tert-butylester (98)
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
8.934.96 3.011.010.88
Chloroform-d
7.32 7.
317.
317.
29 7.28
7.28
7.26
6.97
6.96
4.62
4.59
4.52
4.49
4.05
4.02 3.99
3.98 3.
953.
943.
873.
863.
82
1.46
1.34
1.32
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
Chloroform-d
168.
3216
7.85
137.
63
128.
3312
7.73
82.3
7
75.5
371
.66
67.8
3
41.9
0
27.9
5
16.4
9
Experimenteller Teil
165
2-((2S,3R)-2-Azido-3-(benzyloxy)butanamido)essigsäure (99)
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
0.0
0.1
0.2
0.3
0.4
0.5
0.6
5.13 3.52 3.001.130.94
DMSO-d6
8.60
8.58
8.57
7.33
7.32
7.28
7.27
7.26
7.25
4.60
4.57
4.50
4.47
3.96
3.95
3.93 3.80
3.78
3.74
1.22
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
0.00
0.05
0.10
0.15
0.20
0.25
0.30
DMSO-d6
170.
8516
8.37
138.
38
128.
1912
7.54
75.1
570
.44
65.8
1
40.7
4 16.6
9
Experimenteller Teil
166
Peptid 120
8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
0.45
0.50
0.55
0.60
20.047.49 7.023.883.85 2.131.15 1.021.01 1.00
7.29 7.
277.
267.
257.
20 7.19
7.17
6.70
6.69
6.65
6.63
4.82
4.81
4.79
4.79
4.74
4.73 4.45 4.43
4.42 3.77 3.75
3.71
3.70
3.54
3.38
3.36
3.34
3.33
2.81
2.79
2.78
2.75
2.74
2.38
2.36
2.34
1.51
1.41
0.85
0.83
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10
-0.02
-0.01
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
DMSO-d6
172.
8217
1.09
170.
7616
8.36
167.
61
151.
98 137.
53
129.
1812
7.96
126.
20
82.3
981
.10
76.5
0 69.1
2
58.7
7
53.1
351
.86
51.7
4 47.5
9
40.9
637
.58
31.6
627
.56
27.3
923
.94 22
.93
21.6
8 16.8
1
Experimenteller Teil
167
Peptid 121
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
0.00
0.05
0.10
0.15
0.20
0.25
17.236.18 6.103.10 2.542.08 1.431.20 1.081.00 0.820.780.77
DMSO-d6
8.31
8.30 7.88
7.86
7.24
7.22
7.19
7.18
7.17
7.16 4.84
4.83
4.82
4.82
4.55 4.53
4.52 4.
26 4.24
4.22
4.21
3.59
3.41
3.19
3.14
3.04
3.02
2.78
2.75
2.74
2.28
1.85
1.84
1.82
1.43
1.34
1.28
1.26
0.83 0.81 0.80
0.78
150 100 50 0
-0.02
-0.01
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
DMSO-d6
174.
24 172.
7817
1.47
170.
7216
9.34
158.
0515
7.74
151.
92
137.
60
129.
12 127.
9812
6.19
82.2
380
.59
77.2
6 59.9
858
.63
54.8
9 51.8
251
.14 47.6
141
.21
37.3
435
.34
27.5
627
.45
24.0
4 23.0
121
.79
16.8
0
Experimenteller Teil
168
Peptid 122
7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0
0.00
0.05
0.10
0.15
0.20
0.25
0.30
18.2911.03 6.002.85 2.762.521.04 0.850.84 0.820.67
Chloroform-d
7.32 7.
307.
287.
257.
157.
13 7.13
7.07 7.
05
5.45 5.45
5.43
5.43
4.72
4.71
4.70 4.
61 4.58
4.50
4.47
4.09 4.
054.
043.
753.
703.
51 3.48
3.30
2.99
2.98 2.97
2.95
2.95
2.92
2.68
2.66 2.11
2.11
2.07
2.07
1.48
1.45
1.43
1.39 1.38
0.81
0.79
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
0.00
0.05
0.10
0.15
0.20
0.25
0.30
0.35
0.40
Chloroform-d
173.
5917
3.29
172.
8717
2.14
171.
3016
9.87
169.
08
152.
26
138.
3613
7.49
129.
17 128.
46
127.
6412
6.20
83.3
282
.99
75.6
672
.44
71.7
967
.94
60.7
059
.07
54.7
554
.33
53.5
052
.27
48.3
044
.68
39.4
436
.45
28.0
027
.74
22.9
921
.01
17.6
016
.88
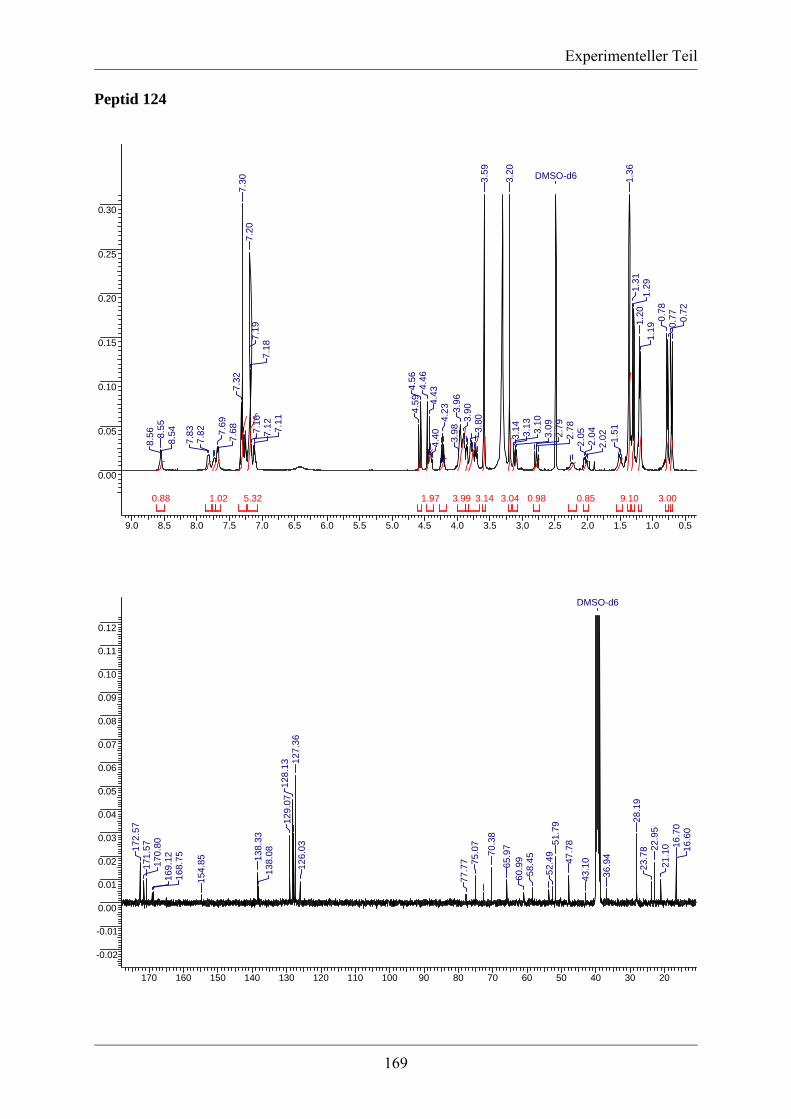
Experimenteller Teil
169
Peptid 124
9.0 8.5 8.0 7.5 7.0 6.5 6.0 5.5 5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5 1.0 0.5
0.00
0.05
0.10
0.15
0.20
0.25
0.30
9.105.32 3.99 3.14 3.04 3.001.971.02 0.980.88 0.85
DMSO-d6
8.56 8.
558.
54
7.83
7.82 7.
697.
687.
327.
307.
207.
197.
187.
167.
12 7.11
4.59
4.56
4.46
4.43
4.40
4.23
3.98
3.96
3.90
3.80
3.59
3.20
3.14 3.13 3.10
3.09
2.79
2.78
2.05
2.04
2.02 1.
511.
361.
311.
291.
201.
190.
780.
77 0.72
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20
-0.02
-0.01
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0.09
0.10
0.11
0.12
DMSO-d6
172.
5717
1.57
170.
8016
9.12
168.
75
154.
85 138.
3313
8.08
129.
0712
8.13 12
7.36
126.
03
77.7
7 75.0
7
70.3
865
.97
60.9
958
.45
52.4
951
.79
47.7
8
43.1
0
36.9
4
28.1
923
.78 22
.95
21.1
0 16.7
016
.60

Experimenteller Teil
170
5.3.2. MS/MS-Spektrum von Peptid 117
Lab-pep-1-9_HC
D768_S
pritze #12-69R
T:0,70-4,32A
V:58
NL:4,96E
3T:FTM
S + c E
SI Full m
s2 768,40@hcd30,00 [50,00-775,00]
100200
300400
500600
700m
/z
0 5 10 15 20 25 30 35 40 45 50 55 60 65 70 75 80 85 90 95
100
Relative Abundance
490,2665
251,1392
164,1072
120,0809
299,1720
518,2611
768,3950
221,1288327,1671
691,1157465,9588
377,1818577,2953
637,3419723,5968
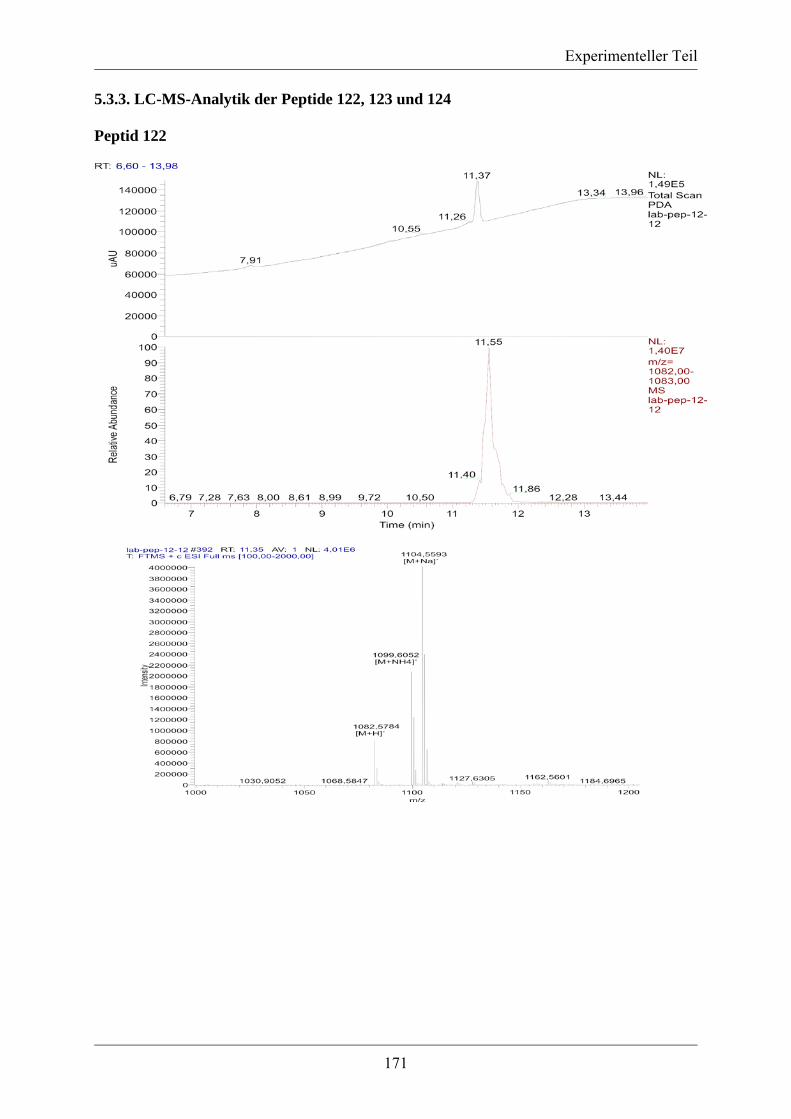
Experimenteller Teil
171
5.3.3. LC-MS-Analytik der Peptide 122, 123 und 124 Peptid 122
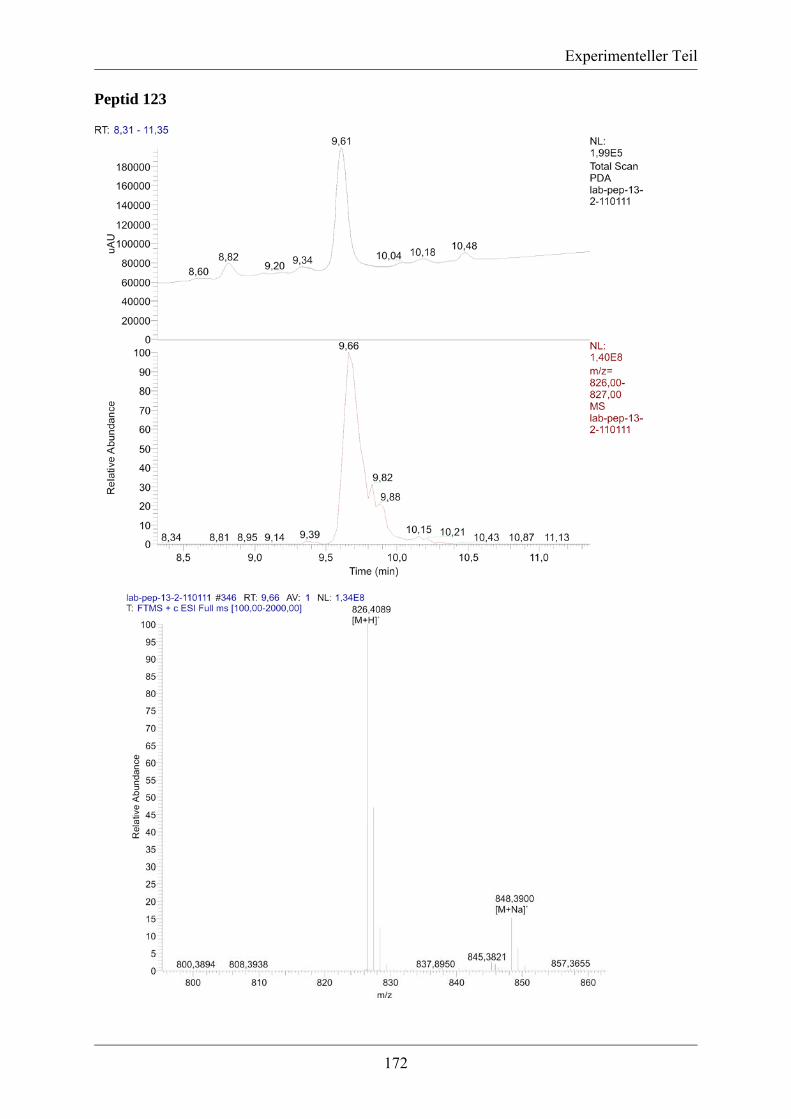
Experimenteller Teil
172
Peptid 123
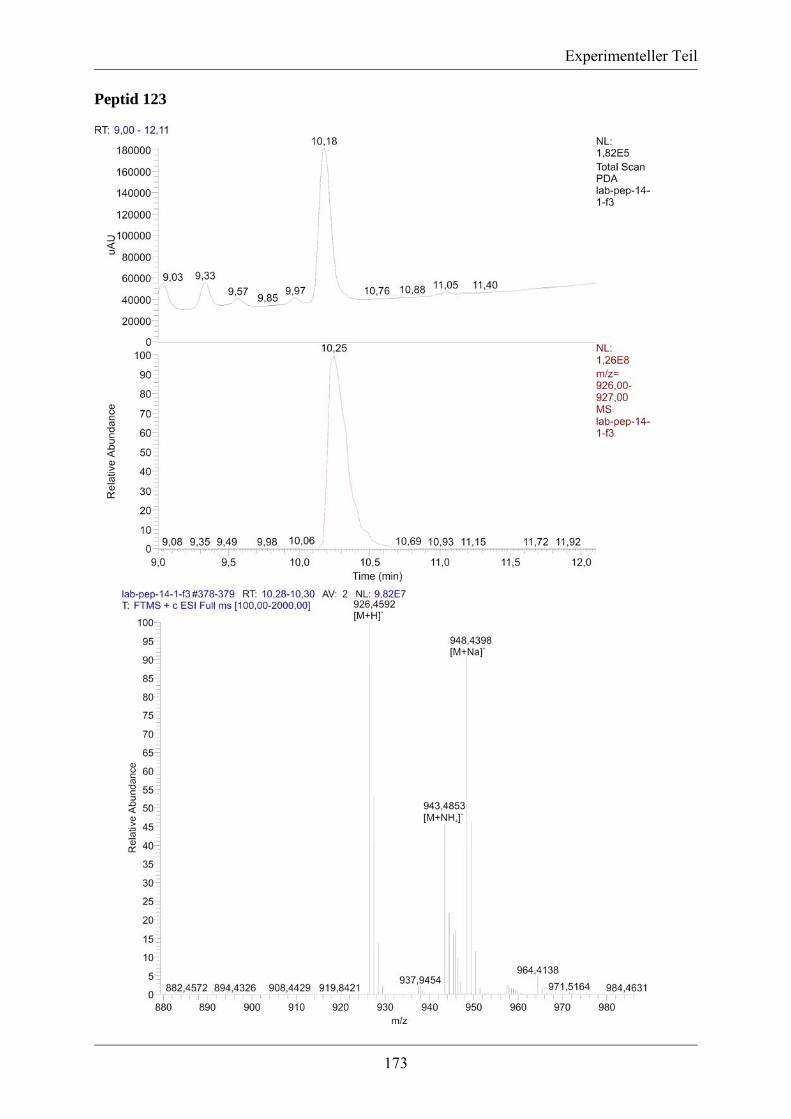
Experimenteller Teil
173
Peptid 123

Experimenteller Teil
174
5.3.4. Röntgenstrukturdaten von (2R,4S)-5-tert-Butyl-1-methyl-2-azido-4-(bis-Boc-amino)-2-(hydroxymethyl)pentandioat
Tabelle 5: Kristalldaten und Strukturerläuterung
Empirische Formel C15 H24 N4 O6 Molekulargewicht 356.38 Temperatur 150(2) K Wellenlänge 0.71073 Å Kristallsystem Orthorhombisch Raumgruppe P212121 (Nr. 19) Dimensionen der a = 6.21698(14) Å α = 90°. Elementarzelle b = 16.9747(5) Å β = 90°. c = 16.9770(4) Å γ = 90°. Volumen 1791.60(8) Å3 Z 4 Dichte (ber.) 1.321 Mg/m3 Absorptionskoeffizient 0.103 mm-1 F(000) 760 Kristallgröße 0.45 x 0.29 x 0.27 mm3 Θ-Bereich für die Datensammlung 3.39 to 25.00°. Bereich der Indizes -7<=h<=7, -19<=k<=20, -16<=l<=20 Anzahl der gemessenen Reflexe 7026 Anzahl der unabhängigen Reflexe 2829 [R(int) = 0.0217] Vollständigkeit von Θ= 25.00° 99.7 % Absorptionskorrektur Semi-empirisch Max. und min. Transmission 0.9727 und 0.9552 Finale R-Werte [I>2σ(I)] R1 = 0.0296, wR2 = 0.0635 R-Werte (alle Daten) R1 = 0.0335, wR2 = 0.0646
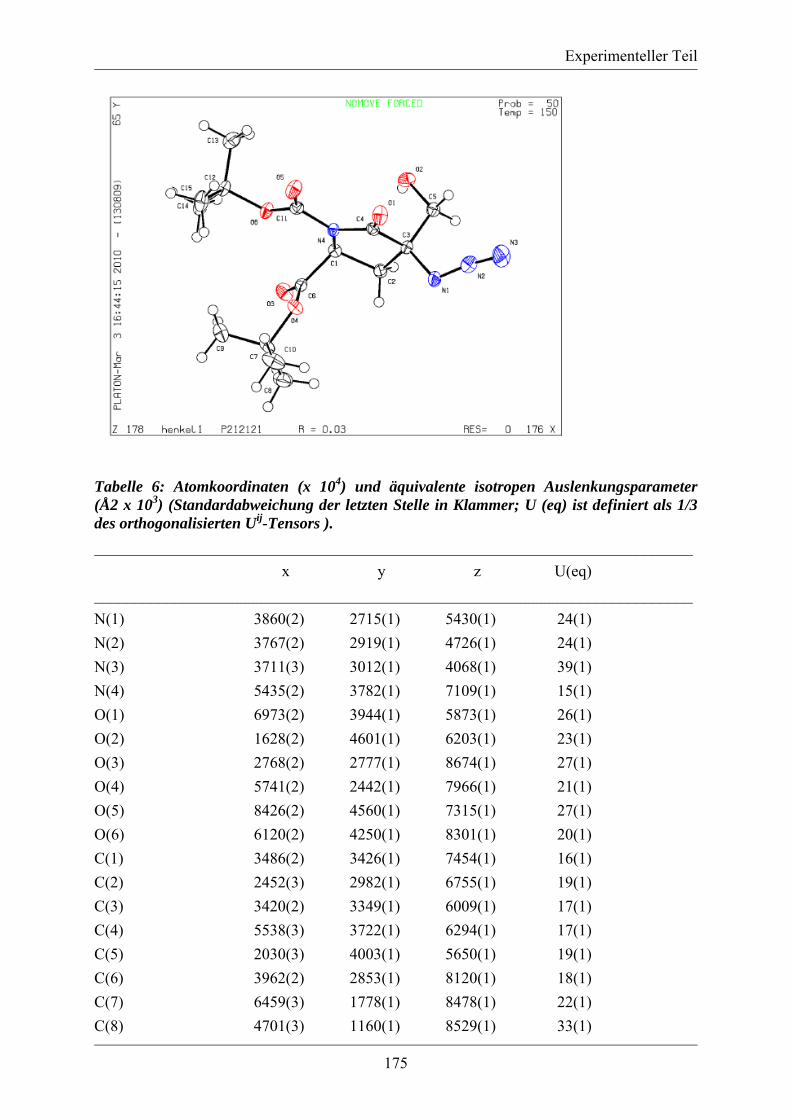
Experimenteller Teil
175
Tabelle 6: Atomkoordinaten (x 104) und äquivalente isotropen Auslenkungsparameter (Å2 x 103) (Standardabweichung der letzten Stelle in Klammer; U (eq) ist definiert als 1/3 des orthogonalisierten Uij-Tensors ). ___________________________________________________________________________ x y z U(eq) ___________________________________________________________________________ N(1) 3860(2) 2715(1) 5430(1) 24(1) N(2) 3767(2) 2919(1) 4726(1) 24(1) N(3) 3711(3) 3012(1) 4068(1) 39(1) N(4) 5435(2) 3782(1) 7109(1) 15(1) O(1) 6973(2) 3944(1) 5873(1) 26(1) O(2) 1628(2) 4601(1) 6203(1) 23(1) O(3) 2768(2) 2777(1) 8674(1) 27(1) O(4) 5741(2) 2442(1) 7966(1) 21(1) O(5) 8426(2) 4560(1) 7315(1) 27(1) O(6) 6120(2) 4250(1) 8301(1) 20(1) C(1) 3486(2) 3426(1) 7454(1) 16(1) C(2) 2452(3) 2982(1) 6755(1) 19(1) C(3) 3420(2) 3349(1) 6009(1) 17(1) C(4) 5538(3) 3722(1) 6294(1) 17(1) C(5) 2030(3) 4003(1) 5650(1) 19(1) C(6) 3962(2) 2853(1) 8120(1) 18(1) C(7) 6459(3) 1778(1) 8478(1) 22(1) C(8) 4701(3) 1160(1) 8529(1) 33(1)
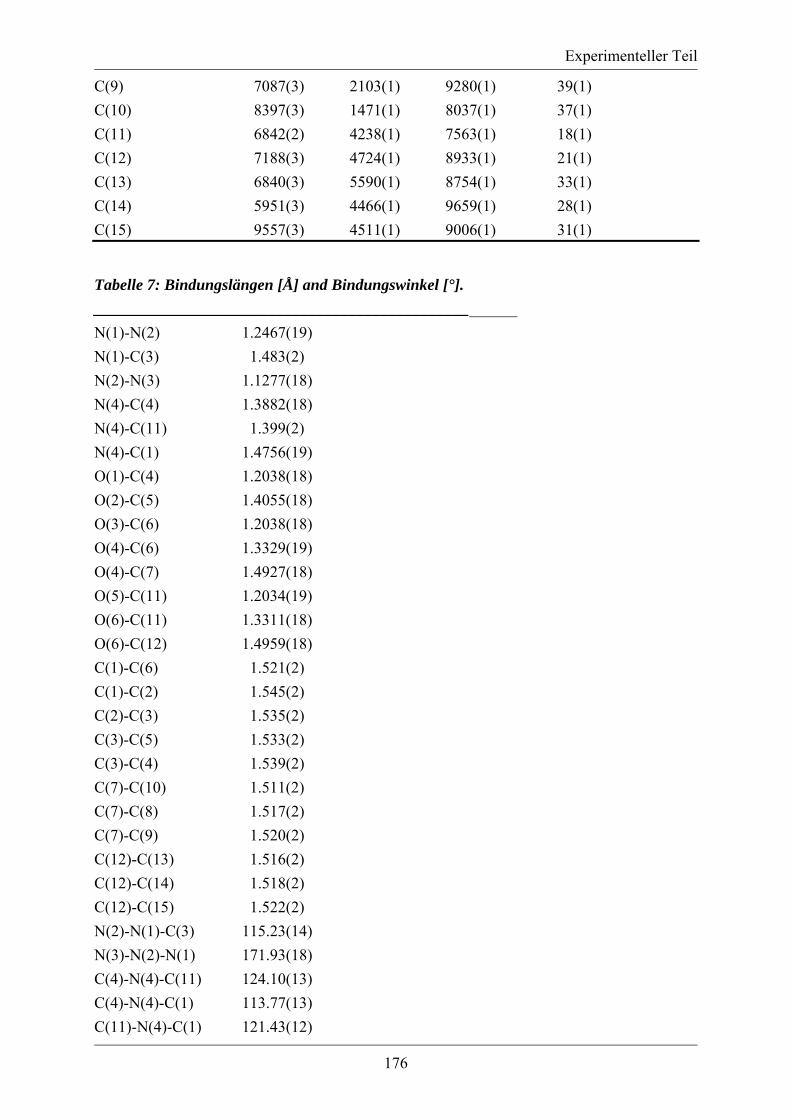
Experimenteller Teil
176
C(9) 7087(3) 2103(1) 9280(1) 39(1) C(10) 8397(3) 1471(1) 8037(1) 37(1) C(11) 6842(2) 4238(1) 7563(1) 18(1) C(12) 7188(3) 4724(1) 8933(1) 21(1) C(13) 6840(3) 5590(1) 8754(1) 33(1) C(14) 5951(3) 4466(1) 9659(1) 28(1) C(15) 9557(3) 4511(1) 9006(1) 31(1)
Tabelle 7: Bindungslängen [Å] and Bindungswinkel [°]. _____________________________________________________ N(1)-N(2) 1.2467(19) N(1)-C(3) 1.483(2) N(2)-N(3) 1.1277(18) N(4)-C(4) 1.3882(18) N(4)-C(11) 1.399(2) N(4)-C(1) 1.4756(19) O(1)-C(4) 1.2038(18) O(2)-C(5) 1.4055(18) O(3)-C(6) 1.2038(18) O(4)-C(6) 1.3329(19) O(4)-C(7) 1.4927(18) O(5)-C(11) 1.2034(19) O(6)-C(11) 1.3311(18) O(6)-C(12) 1.4959(18) C(1)-C(6) 1.521(2) C(1)-C(2) 1.545(2) C(2)-C(3) 1.535(2) C(3)-C(5) 1.533(2) C(3)-C(4) 1.539(2) C(7)-C(10) 1.511(2) C(7)-C(8) 1.517(2) C(7)-C(9) 1.520(2) C(12)-C(13) 1.516(2) C(12)-C(14) 1.518(2) C(12)-C(15) 1.522(2) N(2)-N(1)-C(3) 115.23(14) N(3)-N(2)-N(1) 171.93(18) C(4)-N(4)-C(11) 124.10(13) C(4)-N(4)-C(1) 113.77(13) C(11)-N(4)-C(1) 121.43(12)

Experimenteller Teil
177
C(6)-O(4)-C(7) 121.96(12) C(11)-O(6)-C(12) 122.31(12) N(4)-C(1)-C(6) 113.42(12) N(4)-C(1)-C(2) 103.70(11) C(6)-C(1)-C(2) 109.92(12) C(3)-C(2)-C(1) 105.81(12) N(1)-C(3)-C(5) 111.44(12) N(1)-C(3)-C(2) 108.91(12) C(5)-C(3)-C(2) 113.66(13) N(1)-C(3)-C(4) 110.44(12) C(5)-C(3)-C(4) 108.08(13) C(2)-C(3)-C(4) 104.06(12) O(1)-C(4)-N(4) 127.08(15) O(1)-C(4)-C(3) 125.15(13) N(4)-C(4)-C(3) 107.71(13) O(2)-C(5)-C(3) 110.91(12) O(3)-C(6)-O(4) 127.48(14) O(3)-C(6)-C(1) 121.93(14) O(4)-C(6)-C(1) 110.47(12) O(4)-C(7)-C(10) 102.17(13) O(4)-C(7)-C(8) 109.85(13) C(10)-C(7)-C(8) 111.40(14) O(4)-C(7)-C(9) 108.87(13) C(10)-C(7)-C(9) 111.35(15) C(8)-C(7)-C(9) 112.66(15) O(5)-C(11)-O(6) 126.70(15) O(5)-C(11)-N(4) 124.81(14) O(6)-C(11)-N(4) 108.49(13) O(6)-C(12)-C(13) 108.34(12) O(6)-C(12)-C(14) 101.70(12) C(13)-C(12)-C(14) 111.70(14) O(6)-C(12)-C(15) 111.13(13) C(13)-C(12)-C(15) 112.61(15) C(14)-C(12)-C(15) 110.82(14) _____________________________________________________________
Experimenteller Teil
178
Tabelle 8: Anisotrope Auslenkungsparameter (Å2x 103). ___________________________________________________________________________ U11 U22 U33 U23 U13 U12 ___________________________________________________________________________ N(1) 30(1) 24(1) 18(1) -4(1) -4(1) 4(1) N(2) 19(1) 27(1) 25(1) -6(1) 5(1) 2(1) N(3) 48(1) 49(1) 20(1) -7(1) 7(1) 10(1) N(4) 15(1) 18(1) 14(1) 0(1) 1(1) -1(1) O(1) 20(1) 38(1) 19(1) 0(1) 5(1) -5(1) O(2) 24(1) 20(1) 26(1) -2(1) 6(1) 2(1) O(3) 28(1) 32(1) 21(1) 5(1) 8(1) 4(1) O(4) 19(1) 23(1) 21(1) 6(1) 2(1) 4(1) O(5) 24(1) 34(1) 24(1) -5(1) 5(1) -12(1) O(6) 20(1) 26(1) 16(1) -5(1) 2(1) -5(1) C(1) 13(1) 19(1) 17(1) 2(1) 1(1) 0(1) C(2) 20(1) 19(1) 19(1) 2(1) -2(1) -3(1) C(3) 19(1) 17(1) 15(1) -2(1) -1(1) 0(1) C(4) 18(1) 17(1) 17(1) 2(1) -1(1) 3(1) C(5) 19(1) 21(1) 18(1) -1(1) 1(1) -1(1) C(6) 18(1) 18(1) 17(1) -3(1) -3(1) -1(1) C(7) 23(1) 20(1) 23(1) 9(1) -2(1) 1(1) C(8) 33(1) 24(1) 43(1) 9(1) -4(1) -3(1) C(9) 49(1) 38(1) 30(1) 4(1) -16(1) 5(1) C(10) 30(1) 32(1) 48(1) 15(1) 7(1) 8(1) C(11) 17(1) 17(1) 20(1) -1(1) 0(1) 2(1) C(12) 22(1) 23(1) 17(1) -7(1) -2(1) -3(1) C(13) 42(1) 26(1) 31(1) -6(1) -1(1) -1(1) C(14) 29(1) 34(1) 21(1) -8(1) 1(1) -1(1) C(15) 23(1) 44(1) 28(1) -6(1) -6(1) 1(1)
Experimenteller Teil
179
Tabelle 9: Wasserstoffkoordinaten ( x 104) und isotrope Auslenkungsparameter (Å2x 103). ___________________________________________________________________________ x y z U(eq) ___________________________________________________________________________ H(2) 579 4472 6487 35 H(1) 2493 3849 7643 20 H(2A) 2791 2413 6782 23 H(2B) 870 3048 6761 23 H(5A) 2775 4230 5187 23 H(5B) 647 3777 5468 23 H(8A) 4194 1032 7998 50 H(8B) 3502 1366 8842 50 H(8C) 5272 684 8780 50 H(9A) 8165 2519 9213 58 H(9B) 7689 1679 9604 58 H(9C) 5812 2321 9540 58 H(10A) 7959 1299 7510 55 H(10B) 9017 1025 8325 55 H(10C) 9473 1891 7991 55 H(13A) 5309 5686 8655 49 H(13B) 7676 5737 8287 49 H(13C) 7311 5907 9205 49 H(14A) 4414 4568 9581 42 H(14B) 6462 4763 10118 42 H(14C) 6178 3902 9748 42 H(15A) 9722 3938 8980 47 H(15B) 10111 4703 9512 47 H(15C) 10364 4756 8575 47 _________________________________________________________________________

Anhang
180
6. Anhang
6.1. Literaturverzeichnis
[1] T. Wieland, M. Hasan, P. Pfaender, Justus Liebigs Ann. Chem. 1968, 717, 205-214. [2] H.-D. Jakubke, Spektrum, Akad. Verl., Heidelberg; Berlin; Oxford, 1996. [3] R. Halai, D. J. Craik, Nat. Prod. Rep. 2009, 26, 526-536. [4] J. Ekberg, D. J. Craik, D. J. Adams, Int. J. Biochem. Cell Biol. 2008, 40, 2363-2368. [5] H. Terlau, B. M. Olivera, Physiol Rev 2004, 84, 41-68. [6] M. McIntosh, L. J. Cruz, M. W. Hunkapiller, W. R. Gray, B. M. Olivera, Arch.
Biochem. Biophys. 1982, 218, 329-334. [7] J. A. Williams, M. Day, J. E. Heavner, Expert Opin. Pharmacother. 2008, 9, 1575-
1583. [8] D. J. Craik, D. J. Adams, ACS Chemical Biology 2007, 2, 457-468. [9] R. W. Jack, G. Jung, Curr Opin Chem Biol 2000, 4, 310-317. [10] M. J. Horn, D. B. Jones, S. J. Ringel, J. Biol. Chem. 1941, 138, 141-149. [11] N. Schnell, K.-D. Entian, U. Schneider, F. Götz, H. Zähner, R. Kellner, G. Jung,
Nature 1988, 333, 276-278. [12] C. I. Mortvedt, J. Nissen-Meyer, K. Sletten, I. F. Nes, Appl. Environ. Microbiol. 1991,
57, 1829-1834. [13] F. Castiglione, L. Cavaletti, D. Losi, A. Lazzarini, L. Carrano, M. Feroggio, I.
Ciciliato, E. Corti, G. Candiani, F. Marinelli, E. Selva, Biochemistry 2007, 46, 5884-5895.
[14] J. M. Willey, W. A. van der Donk, Annu. Rev. Microbiol. 2007, 61, 477-501. [15] G. Bierbaum, C. Szekat, M. Josten, C. Heidrich, C. Kempter, G. Jung, H. Sahl, Appl.
Environ. Microbiol. 1996, 62, 385-392. [16] H. G. Sahl, R. W. Jack, G. Bierbaum, Eur. J. Biochem. 1995, 230, 827-853. [17] Y. Rew, S. Malkmus, C. Svensson, T. L. Yaksh, N. N. Chung, P. W. Schiller, J. A.
Cassel, R. N. DeHaven, M. Goodman, J. Med. Chem. 2002, 45, 3746-3754. [18] H. Li, X. Jiang, M. Goodman, J. Pept. Sci. 2001, 7, 82-91. [19] H. Li, X. Jiang, S. B. Howell, M. Goodman, J. Pept. Sci. 2000, 6, 26-35. [20] B. Perichon, P. Courvalin, Antimicrob. Agents Chemother. 2009, 53, 4580-4587. [21] P. D. Cotter, C. Hill, R. P. Ross, Curr. Protein Pept. Sci. 2005, 6, 61-75. [22] H. Brötz, G. Bierbaum, P. E. Reynolds, H. G. Sahl, Eur. J. Biochem. 1997, 246, 193-
199. [23] S.-T. D. Hsu, E. Breukink, E. Tischenko, M. A. G. Lutters, B. de Kruijff, R. Kaptein,
A. M. J. J. Bonvin, N. A. J. van Nuland, Nat. Struct. Mol. Biol. 2004, 11, 963-967. [24] C. Chatterjee, M. Paul, L. Xie, W. A. van der Donk, Chem. Rev. 2005, 105, 633-684. [25] E. Breukink, B. de Kruijff, Nat. Rev. Drug Discov. 2006, 5, 321-323. [26] G. Machaidze, J. Seelig, Biochemistry 2003, 42, 12570-12576. [27] I. F. Nes, J. R. Tagg, Antonie Leeuwenhoek 1996, 69, 89-97. [28] T. Schmiederer, PhD thesis, Technische Universität Berlin (Berlin), 2008. [29] T. Stein, S. Borchert, B. Conrad, J. Feesche, B. Hofemeister, J. Hofemeister, K.-D.
Entian, J. Bacteriol. 2002, 184, 1703-1711. [30] C. Meyer, G. Bierbaum, C. Heidrich, M. Reis, J. Süling, M. I. Iglesias-Wind, C.
Kempter, E. Molitor, H. G. Sahl, Eur. J. Biochem. 1995, 232, 478-489. [31] G. Jung, Angew. Chem., Int. Ed. 2006, 45, 5919-5921. [32] N. M. Okeley, M. Paul, J. P. Stasser, N. Blackburn, W. A. van der Donk, Biochemistry
2003, 42, 13613-13624.

Anhang
181
[33] B. Li, J. P. J. Yu, J. S. Brunzelle, G. N. Moll, W. A. van der Donk, S. K. Nair, Science 2006, 311, 1464-1467.
[34] Y. Zhu, M. D. Gieselman, H. Zhou, O. Averin, W. A. v. d. Donk, Org. Biomol. Chem. 2003, 1, 3304-3315.
[35] R. J. Siezen, O. P. Kuipers, W. M. Vos, Antonie Leeuwenhoek 1996, 69, 171-184. [36] L. Xie, L. M. Miller, C. Chatterjee, O. Averin, N. L. Kelleher, W. A. van der Donk,
Science 2004, 303, 679-681. [37] Y. O. You, W. A. van der Donk, Biochemistry 2007, 46, 5991-6000. [38] G. Jung, Angew. Chem., Int. Ed. 1991, 30, 1051-1068. [39] U. Pag, H. G. Sahl, Curr. Pharm. Des. 2002, 8, 815-833. [40] A. Dufour, T. Hindré, D. Haras, J. P. Le Pennec, FEMS Microbiol. Rev. 2007, 31,
134-167. [41] P. Chen, J. Novak, M. Kirk, S. Barnes, F. X. Qi, P. W. Caufield, Appl. Environ.
Microbiol. 1998, 64, 2335-2340. [42] N. I. Martin, T. Sprules, M. R. Carpenter, P. D. Cotter, C. Hill, R. P. Ross, J. C.
Vederas, Biochemistry 2004, 43, 3049-3056. [43] I. Wiedemann, T. Bottiger, R. R. Bonelli, A. Wiese, S. O. Hagge, T. Gutsmann, U.
Seydel, L. Deegan, C. Hill, P. Ross, H. G. Sahl, Mol. Microbiol. 2006, 61, 285-296. [44] R. D. Tillotson, H. A. B. Wosten, M. Richter, J. M. Willey, Mol. Microbiol. 1998, 30,
595-602. [45] K. Meindl, T. Schmiederer, K. Schneider, A. Reicke, D. Butz, S. Keller, H. Gühring,
L. Vértesy, J. Wink, H. Hoffmann, M. Brönstrup, G. Sheldrick, R. Süssmuth, Angew. Chem., Int. Ed. 2010, 49, 1151-1154.
[46] S. Kodani, M. A. Lodato, M. C. Durrant, F. Picart, J. M. Willey, Mol. Microbiol. 2005, 58, 1368-1380.
[47] J. Wink, R. M. Kroppenstedt, G. Seibert, E. Stackebrandt, Int. J. Syst. Evol. Microbiol. 2003, 53, 721-724.
[48] R. Dorenbos, T. Stein, J. Kabel, C. Bruand, A. Bolhuis, S. Bron, W. J. Quax, J. M. van Dijl, J. Biol. Chem. 2002, 277, 16682-16688.
[49] C. Kubicek, M. Komoń-Zelazowska, E. Sándor, I. Druzhinina, Chem. Biodiversity 2007, 4, 1068-1082.
[50] W. Müller, T. Schmiederer, P. Ensle, R. Süssmuth, Angew. Chem., Int. Ed. 2010, 49, 2436-2440.
[51] S. Kodani, M. E. Hudson, M. C. Durrant, M. J. Buttner, J. R. Nodwell, J. M. Willey, Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 11448-11453.
[52] I. Decosterd, C. J. Woolf, Pain 2000, 87, 149-158. [53] K. Fukase, M. Kitazawa, A. Sano, K. Shimbo, H. Fujita, S. Horimoto, T. Wakamiya,
T. Shiba, Tetrahedron Lett. 1988, 29, 795-798. [54] T. Wakamiya, K. Shimbo, A. Sano, K. Fukase, T. Shiba, Bull. Chem. Soc. Jpn. 1983,
56, 2044-2049. [55] D. N. Harpp, J. G. Gleason, J. Org. Chem. 1971, 36, 73-80. [56] A. K. Galande, J. O. Trent, A. F. Spatola, Peptide Science 2003, 71, 534-551. [57] H. Shao, S. H. H. Wang, C.-W. Lee, G. Oesapay, M. Goodman, J. Org. Chem. 1995,
60, 2956-2957. [58] Y. Hata, M. Watanabe, Tetrahedron 1987, 43, 3881-3888. [59] X. Zhu, Richard R. Schmidt, Eur. J. Org. Chem. 2003, 2003, 4069-4072. [60] V. Swali, M. Matteucci, R. Elliot, M. Bradley, Tetrahedron 2002, 58, 9101-9109. [61] S. Bregant, A. B. Tabor, J. Org. Chem. 2005, 70, 2430-2438. [62] V. R. Pattabiraman, S. M. K. McKinnie, J. C. Vederas, Angew. Chem. 2008, 120,
9614-9617.

Anhang
182
[63] A. C. Ross, H. Liu, V. R. Pattabiraman, J. C. Vederas, J. Am. Chem. Soc. 2009, 132, 462-463.
[64] Y. Izumi, I. Chibata, T. Itoh, Angew. Chem. 1978, 90, 187-194. [65] A. Strecker, Justus Liebigs Ann. Chem. 1850, 75, 27-45. [66] S. Yamada, C. Hongo, M. Yamamoto, I. Chibata, Agric. Biol. Chem. 1976, 40, 1425-
1427. [67] S. Yamada, M. Yamamoto, I. Chibata, J. Org. Chem. 1973, 38, 4408-4412. [68] U. Schöllkopf, T. Tiller, J. Bardenhagen, Tetrahedron 1988, 44, 5293-5305. [69] D. A. Evans, Aldrichim. Acta 1982, 15, 23-32. [70] T. Oguri, N. Kawai, T. Shioiri, S. Yamada, Chem. Pharm. Bull. 1978, 26, 803-808. [71] A. Solladie-Cavallo, M. C. Simon-Wermeister, J. Schwarz, Organometallics 1993, 12,
3743-3747. [72] L. M. Jackman, B. C. Lange, J. Am. Chem. Soc. 1981, 103, 4494-4499. [73] M. Tabcheh, A. El Achqar, L. Pappalardo, M.-L. Roumestant, P. Viallefont,
Tetrahedron 1991, 47, 4611-4618. [74] Klaus W. Laue, S. Kröger, E. Wegelius, G. Haufe, Eur. J. Org. Chem. 2000, 2000,
3737-3743. [75] D. C. Horwell, G. Ratcliffe, E. Roberts, Bioorg. Med. Chem. Lett. 1991, 1, 169-172. [76] J. E. Green, D. M. Bender, S. Jackson, M. J. O`Donnell, J. R. McCarthy, Org. Lett.
2009, 11, 807-810. [77] D. Seebach, J. D. Aebi, M. Gander-Coquoz, R. Naef, Helv. Chim. Acta 1987, 70,
1194-1216. [78] C. Cativiela, M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry 1998, 9, 3517-3599. [79] H.-J. Cristau, X. Marat, J.-P. Vors, J.-L. Pirat, Tetrahedron Lett. 2003, 44, 3179-3181. [80] J. A. Smulik, E. Vedejs, Org. Lett. 2003, 5, 4187-4190. [81] A. Avenoza, C. Cativiela, F. Corzana, J. M. Peregrina, D. Sucunza, M. M. Zurbano,
Tetrahedron: Asymmetry 2001, 12, 949-957. [82] A. Avenoza, J. M. Peregrina, E. San MartIn, Tetrahedron Lett. 2003, 44, 6413-6416. [83] H. Shao, J. K. Rueter, M. Goodman, J. Org. Chem. 1998, 63, 5240-5244. [84] H. C. Kolb, M. S. VanNieuwenhze, K. B. Sharpless, Chem. Rev. 1994, 94, 2483-2547. [85] A. Avenoza, J. H. Busto, F. Corzana, J. I. Garcia, J. M. Peregrina, J. Org. Chem. 2003,
68, 4506-4513. [86] S. Saito, N. Takahashi, T. Ishikawa, T. Moriwake, Tetrahedron Lett. 1991, 32, 667-
670. [87] U. Cramer, A. G. Rehfeldt, F. Spener, Biochemistry 1980, 19, 3074-3080. [88] K. Katagiri, K. Tori, Y. Kimura, T. Yoshida, T. Nagasaki, H. Minato, J. Med. Chem.
1967, 10, 1149-1154. [89] E. C. Roos, H. H. Mooiweer, H. Hiemstra, W. N. Speckamp, B. Kaptein, W. H. J.
Boesten, J. Kamphuis, J. Org. Chem. 1992, 57, 6769-6778. [90] B. Kübel, G. Höfle, W. Steglich, Angew. Chem. 1975, 87, 64-66. [91] R. E. Ireland, R. H. Mueller, J. Am. Chem. Soc. 1972, 94, 5897-5898. [92] P. A. Bartlett, J. F. Barstow, J. Org. Chem. 1982, 47, 3933-3941. [93] U. Kazmaier, Liebigs Ann. Chem. 1997, 1997, 285-295. [94] U. Kazmaier, Angew. Chem. 1994, 106, 1046-1047. [95] U. Kazmaier, R. Grandel, Synlett 1995, 1995, 945,946. [96] U. Kazmaier*, Synlett 1995, 1138-1140. [97] U. Kazmaier, Tetrahedron 1994, 50, 12895-12902. [98] U. Kazmaier, S. Maier, Tetrahedron 1996, 52, 941-954. [99] U. Kazmaier, Tetrahedron Lett. 1996, 37, 5351-5354. [100] R. W. Driver, H. N. Hoang, G. Abbenante, D. P. Fairlie, Org. Lett. 2009, 11, 3092-
3095.

Anhang
183
[101] N.-H. Nam, G. Ye, G. Sun, K. Parang, J. Med. Chem. 2004, 47, 3131-3141. [102] V. Alezra, M. Bonin, A. Chiaroni, L. Micouin, C. Riche, H.-P. Husson, Tetrahedron
Lett. 2000, 41, 1737-1740. [103] A. J. Mancuso, D. Swern, Synthesis 1981, 1981, 165-185. [104] T. Vogler, A. Studer, Synthesis 2008, 2008, 1979-1993. [105] M. Falorni, S. Conti, G. Giacomelli, S. Cossu, F. Soccolini, Tetrahedron: Asymmetry
1995, 6, 287-294. [106] J. Sélambarom, S. Monge, F. Carré, J. P. Roque, A. A. Pavia, Tetrahedron 2002, 58,
9559-9566. [107] A. Ncube, S. B. Park, J. M. Chong, J. Org. Chem. 2002, 67, 3625-3636. [108] H. Takahashi, K. Takahashi, K. Higashiyama, H. Onishi, Chem. Pharm. Bull. 1986,
34, 479-485. [109] A. Alexakis, N. Lensen, J. P. Tranchier, P. Mangeney, J. Org. Chem. 1992, 57, 4563-
4565. [110] Y. Yamashita, S. Kobayashi, J. Am. Chem. Soc. 2004, 126, 11279-11282. [111] A. Alexakis, N. Lensen, P. Mangeney, Synlett 1991, 625,626. [112] P. R. Krishna, E. R. Sekhar, F. Mongin, Tetrahedron Lett. 2008, 49, 6768-6772. [113] K.-D. Gundermann, W. Cohnen, H.-U. Alles, Chem. Ber. 1964, 97, 647-653. [114] J. Wehbe, V. Rolland, A. Fruchier, M.-L. Roumestant, J. Martinez, Tetrahedron:
Asymmetry 2004, 15, 851-858. [115] G. Lu, K. Burgess, Bioorg. Med. Chem. Lett. 2006, 16, 3902-3905. [116] B. P. Mowery, V. Prasad, C. S. Kenesky, A. R. Angeles, L. L. Taylor, J.-J. Feng, W.-
L. Chen, A. Lin, F.-C. Cheng, A. B. Smith, R. Hirschmann, Org. Lett. 2006, 8, 4397-4400.
[117] T. Gagnon, D. Fillion, M.-R. Lefebvre, E. Escher, J. Recept. Signal Transduction 2006, 26, 435-451.
[118] B.-X. Zhao, M. Schaudt, S. Blechert, Chin. J. Chem. 2006, 24, 1080-1085. [119] D. J. St-Cyr, A. G. Jamieson, W. D. Lubell, Org. Lett. 2010, 12, 1652-1655. [120] W. Pilgrim, P. V. Murphy, Org. Lett. 2009, 11, 939-942. [121] M. A. Lago, J. Samanen, J. D. Elliott, J. Org. Chem. 1992, 57, 3493-3496. [122] R. B. Nasir Baig, V. Sai Sudhir, C. Srinivasan, Synlett 2008, 2684-2688. [123] R. Rajan, T. Mathew, R. Buffa, F. Bornancin, M. Cavallari, P. Nussbaumer, G. De
Libero, A. Vasella, Helv. Chim. Acta 2009, 92, 918-927. [124] N. D. Smith, M. Goodman, Org. Lett. 2003, 5, 1035-1037. [125] N. D. Smith, A. M. Wohlrab, M. Goodman, Org. Lett. 2005, 7, 255-258. [126] L. T. Boulton, H. T. Stock, J. Rarhy, D. C. Horwell, J. Chem. Soc. [Perkin 1] 1999,
1421-1429. [127] P. S. Kutchukian, J. S. Yang, G. L. Verdine, E. I. Shakhnovich, J. Am. Chem. Soc.
2009, 131, 4622-4627. [128] Y.-W. Kim, G. L. Verdine, Bioorg. Med. Chem. Lett. 2009, 19, 2533-2536. [129] C. E. Schafmeister, J. Po, G. L. Verdine, J. Am. Chem. Soc. 2000, 122, 5891-5892. [130] V. R. Pattabiraman, J. L. Stymiest, D. J. Derksen, N. I. Martin, J. C. Vederas, Org.
Lett. 2007, 9, 699-702. [131] T. Johnson, M. Quibell, R. C. Sheppard, J. Pept. Sci. 1995, 1, 11-25. [132] T. Johnson, L. C. Packman, C. B. Hyde, D. Owen, M. Quibell, J. Chem. Soc. [Perkin
1] 1996, 719. [133] M. Mergler, F. Dick, B. Sa, C. Stähelin, T. Vorherr, J. Pept. Sci. 2003, 9, 518-526. [134] S. Zahariev, C. Guarnaccia, F. Zanuttin, A. Pintar, G. Esposito, G. Maravi, B. Krust,
A. G. Hovanessian, S. Pongor, J. Pept. Sci. 2005, 11, 17-28. [135] K. Wahlström, O. Planstedt, A. Undén, Tetrahedron Lett. 2008, 49, 3921-3924.

Anhang
184
[136] O. Demmer, A. O. Frank, H. Kessler, in Peptide and Protein Design for Biopharmaceutical Applications (Ed.: K. J. Jensen), Wiley, 2009, pp. 133 - 176.
[137] J. S. Davies, J. Pept. Sci. 2003, 9, 471-501. [138] T. Rückle, P. de Lavallaz, M. Keller, P. Dumy, M. Mutter, Tetrahedron 1999, 55,
11281-11288. [139] H. Kessler, R. Schuck, R. Siegmeier, J. W. Bats, H. Fuess, H. Förster, Liebigs Ann.
Chem. 1983, 1983, 231-247. [140] W. D. F. Meutermans, G. T. Bourne, S. W. Golding, D. A. Horton, M. R. Campitelli,
D. Craik, M. Scanlon, M. L. Smythe, Org. Lett. 2003, 5, 2711-2714. [141] M. El Haddadi, F. Cavelier, E. Vives, A. Azmani, J. Verducci, J. Martinez, J. Pept.
Sci. 2000, 6, 560-570. [142] J. E. Sheppeck, H. Kar, H. Hong, Tetrahedron Lett. 2000, 41, 5329-5333. [143] K. E. Kawulka, T. Sprules, C. M. Diaper, R. M. Whittal, R. T. McKay, P. Mercier, P.
Zuber, J. C. Vederas, Biochemistry 2004, 43, 3385-3395. [144] R. Sateesh Chandra Kumar, G. Venkateswar Reddy, G. Shankaraiah, K. Suresh Babu,
J. Madhusudana Rao, Tetrahedron Lett. 2010, 51, 1114-1116. [145] J. Jeon, J. H. Lee, J.-W. Kim, Y. G. Kim, Tetrahedron: Asymmetry 2007, 18, 2448-
2453. [146] J. K. Cha, N.-S. Kim, Chem. Rev. 1995, 95, 1761-1795. [147] P. Boersema, S. Mohammed, A. Heck, Anal. Bioanal. Chem. 2008, 391, 151-159. [148] T. Yoshida, J. Biochem. Biophys. Methods 2004, 60, 265-280. [149] H. Hayen, Nachr. Chem. 2010, 58, 461-465. [150] H.-Y. Tang, D. W. Speicher, Anal. Biochem. 2004, 334, 48-61. [151] J. Mack, K. Falk, O. Rötzschke, T. Walk, J. L. Strominger, G. Jung, J. Pept. Sci. 2001,
7, 338-300. [152] V. Wittmann, S. Takayama, K. W. Gong, G. Weitz-Schmidt, C.-H. Wong, J. Org.
Chem. 1998, 63, 5137-5143. [153] K. A. McDonnell, S. C. Low, T. Hoehn, R. Donnelly, H. Palmieri, C. Fraley, P.
Sakorafas, A. R. Mezo, J. Med. Chem. 2010, 53, 1587-1596. [154] M. Taniguchi, K. Suzumura, K. Nagai, T. Kawasaki, J. Takasaki, M. Sekiguchi, Y.
Moritani, T. Saito, K. Hayashi, S. Fujita, S. Tsukamoto, K. Suzuki, Bioorg. Med. Chem. 2004, 12, 3125-3133.
[155] M. Takasaki, K. Harada, J. Chem. Soc., Chem. Commun. 1987, 571-573. [156] E. Gross, J. L. Morell, J. Am. Chem. Soc. 1967, 89, 2791-2792. [157] A. Galande, A. Spatola, Lett. Pept. Sci. 2001, 8, 247-251. [158] D. Basavaiah, M. Krishnamacharyulu, A. J. Rao, Synth. Commun. 2000, 30, 2061 -
2069. [159] C. Grundmann, G. Ottmann, Justus Liebigs Ann. Chem. 1953, 582, 163-177. [160] A. Kendhale, R. Gonnade, P. R. Rajamohanan, G. J. Sanjayan, Chem. Commun. 2006,
2756-2758. [161] J. Kretz, Diplomarbeit Technische Universität (Berlin), 2010. [162] E. D. Goddard-Borger, R. V. Stick, Org. Lett. 2007, 9, 3797-3800. [163] A. Pesic, pers. Mitteilung TU Berlin, Berlin, 2009.

Anhang
185
6.2. Abkürzungsverzeichnis
Abu Aminobuttersäure
Ac Acetyl
ACE Angiotensin converting enzyme
AcSH Thioessigsäure
AD asymmetrische Dihydroxylierung
Ala Alanin
All Allyl
äq Äquivalent
Arg Arginin
Asn Asparagin
Asp Asparaginsäure
ATP Adenosintriphosphat
AviCys Aminovinylcystein
AviMeCys Aminovinyl-3-Methylcystein
Bn Benzyl
Boc tert-Butoxycarbonyl
Bu Butyl
Cbz Benzyloxycarbonyl
COSY Correlation spectroscopy; Korrelationspektroskopie
Cys Cystein
d Duplett
DABCO 1,4-Diazabicyclo[2.2.2]octan
DNA Deoxyribonucleic acid; Desoxyribonukleinsäure
DBU 1,8-Diazabicyclo[5.4.0]undec-7-en
DC Dünnschichtchromatographie
DCM Dichlormethan
dd Duplett von Duplett
de diastereomeric excess; Diastereomerenüberschuss
DEAD Diazendicarbonsäurediethylester
DEPBT 3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-on
Dha Didehydroalanin

Anhang
186
Dhb Didehydrobutyrin
DIPEA N,N-Diisopropyl-N-ethylamin
DMAP 4-Dimethylaminopyridin
DMF N,N-Dimethylformamid
DMSO Dimethylsulfoxid
Dpm Diphenylmethyl
DPPA Diphenylphosphorylazid
dt Duplett von Triplett
DTT Dithiothreitol (1,4-Dimercapto-2,3-butandiol)
EDAC
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid
Hydrochlorid
ee enantiomeric excess; Enatiomerenüberschuss
EI Elektronenstoßionisation
ESI Elektronensprayionisation
Et Ethyl
EtOAc Ethylacetat
Fmoc Fluorenylmethoxycarbonyl
GC Gaschromatographie
GC-MS Gaschromatographie-Massenspektrometrie
Gln Glutamin
Glu Glutaminsäure
Gly Glycin
GTP Guanosintriphosphat
HATU O-(7-Azabenzotriazol-1-yl)-N,N,N´,N´-
tetramethyluroniumhexafluorophosphat
Hex Hexan
HILIC Hydrophile Interaktionschromatographie
His Histidin
HOAt 1-Hydroxy-7-azabenzotriazol
HOBt N-Hydroxybenzotriazol
HOSu N-Hydroxysuccinimid
HPLC high-performance liquid chromatography;
Hochleistungs-Flüssigkeitschromatographie
Ile Isoleucin

Anhang
187
IPDMS Isopropyldimethylsilyl
J Kopplungskonstante (NMR)
kb Kilobasen
KHMDS Kaliumhexamethyldisilizan
Lab Labyrinthopeptin
Lan Lanthionin
LC-MS liquid chromatography mass spectrometry
Flüssigkeitschromatograhie-Massenspektometrie
LDA Lithiumdiisopropylamid
Leu Leucin
Lys Lysin
m Multiplett
MAH N-(2-Mercaptoethyl)-aminomethyl-Harz
Me Methyl
MeCys 3-Methylcystein
MeLan β-Methyllanthionin
MeOH Methanol
Met Methionin
MOM Methoxymethyl
MOMCl Methoxymethylchlorid
MRSA Methicilin-resistenter Staphylococus aureus
MS Massenspektrometrie
Ms Methylsulfonyl
MsCl Methylsulfonylchlorid
NMR nuclear magnetic resonance; Kernspinresonanz
NTCB 2-Nitro-5-thiocyanatobenzoesäure
pb polymer bond; Polymer gebunden
PEG Polyethylenglykol
Pen Pentan
Ph Phenyl
Phe Phenylalanin
ppm parts per millon
Pro Prolin
PrOH Propanol

Anhang
188
pTos para-Toluolsulfonsäure
PWT Paw Withdrawal Threhold
q Quartett
Rf Retentionsfaktor
RT Raumtemperatur
s Singulett
Ser Serin
SNI spared nerve injury; neurophatisches Schmerzmodell
SPPS solid phase peptid synthesis; Festphasenpeptidsynthese
Su Succinimidyl
t Triplett
TBAF Tetrabutylammoniumfluorid
TBDMS tert-Butyldimethylsilyl
TBDMSCl tert-Butyldimethylsilylchlorid
tBu tert-Butyl
TBTA tert-Butyl-Acetimidat
td Triplett von Duptett
TEA Triethylamin
TEMPO 2,2,6,6-Tetramethyl-piperidin-1-oxyl
TES Triethylsilan
TFA Trifluoressigsäure
THF Tetrahydrofuran
THP Tetrahydropyranyl
Thr Threonin
Tmb Trimethoxybenzyl
Trp Tryptophan
Tyr Tyrosin
UV Ultraviolett
Val Valin
Recommended