Transduktion hämatopoetischer Stammzellen von Neuweltaffen
mit Foamyviren
Melanie Wurm
2007


Bundesforschungsanstalt für Landwirtschaft (FAL) Braunschweig
Institut für Tierzucht (Mariensee)
Heinrich Heine Universität Düsseldorf
Klinik für Kinder -Onkologie, -Hämatologie und Klinische Immunologie
Transduktion hämatopoetischer Stammzellen von Neuweltaffen mit Foamyviren
Inaugural-Dissertation zur Erlangung des Grades einer
Doktorin der Veterinärmedizin
- Doctor medicinae veterinariae - (Dr. med. vet.)
Vorgelegt von
Melanie Wurm aus Bonn
Hannover 2007

Wissenschaftliche Betreuung: Univ.-Prof. Dr. H. Niemann Univ.-Prof. Dr. H. Hanenberg 1. Gutachter: Univ.-Prof. Dr. H. Niemann 2. Gutachter: Univ.-Prof. Dr. H.Y. Naim Tag der mündlichen Prüfung: 15.05.2007

Meinem Freund Heiko


Inhaltsverzeichnis
1. EINLEITUNG 1
2. LITERATUR 3
2.1 HÄMATOPOETISCHE STAMMZELLEN ALS ZIELZELLEN SOMATISCHER GENTHERAPIE 3 2.2 ZYTOKINE 7 2.3 BIOLOGIE VON RETROVIREN 8 2.3.1 ORTHORETROVIREN 11 2.3.1.1 Replikationszyklus von Orthoretroviren 11 2.3.1.2 Orthoretrovirale Vektoren 12 2.3.1.3 Pseudotypisierung von Orthoretroviren 14 2.3.2 SPUMARETROVIREN 14 2.3.2.1 Merkmale der Spumaretroviren 14 2.3.2.2 Replikationszyklus von Foamyviren 16 2.3.2.3 Foamyvirale Vektoren 17 2.3.2.4 Verpacken von Foamyviren 19 2.4 SELEKTION TRANSGENER ZELLEN IN VITRO UND IN VIVO 20 2.5 INTERAKTIONEN ZWISCHEN VIRUSPARTIKELN UND ZIELZELLEN 23 2.6 NEUWELTAFFEN ALS MODELL FÜR GENTRANSFER 24

3. MATERIAL UND METHODEN 26
3.1 VERWENDETE MATERIALIEN 26 3.1.1 PLASMIDE 26 3.1.2 ZELLEN 31 3.1.3 TIERE 32 3.2 METHODEN 33 3.2.1 MOLEKULARBIOLOGISCHE TECHNIKEN 33 3.2.1.1 Transformation von Bakterien und Isolierung von Plasmid DNA 33 3.2.1.2 Klonierung des Plasmids MH71.MGMTp140kIEG 34 3.2.2 ZELLKULTURTECHNIKEN 35 3.2.2.1 Kultivierung eukaryotischer Zelllinien 35 3.2.2.2 Kryokonservierung eukaryotischer Zellen 36 3.2.2.3 Mykoplasmentest 36 3.2.2.4 Gewinnung primärer Zellen 37 3.2.3 VIRUSPRODUKTION 38 3.2.3.1 Transiente Erzeugung virushaltiger Überstände 38 3.2.3.2 Konzentration von virushaltigen Überständen 40 3.2.4 TITRATION 41 3.2.5 TRANSDUKTION VON HÄMATOPOETISCHEN ZELLLINIEN 42 3.2.5.1 Transduktion von humanen CD34+ Zellen 43 3.2.5.2 Transduktion von Marmoset CD34+ Zellen 44 3.2.6 PROGENITOR-ASSAY 45 3.2.7 VIRUSBINDUNG AN FIBRONEKTIN 46 3.2.8 1,3-BIS-(2-CHLORETHYL)-1-NITROSOUREA (BCNU) 47 3.2.8.1 BCNU Selektion 47 3.2.8.2 BG - BCNU Selektion 48 3.2.8.3 BG - BCNU Selektion von CD34+ Marmoset Zellen 49 3.2.9 KNOCHENMARKENTNAHME BEI MARMOSETS 50 3.2.10 STATISTIK 51

4. ERGEBNISSE 52
4.1 HERSTELLUNG VON FOAMYVIREN 52 4.1.1 OPTIMIERUNG DES TRANSFEKTIONSPROTOKOLLS 52 4.1.2 VERGLEICH VON FOAMYVIRALEN 2,- 3- UND 4-PLASMIDSYSTEMEN 54 4.1.3 FOAMYVIRALE VERPACKUNGSPLASMIDE 55 4.1.4 EINFLUSS DES MGMTP140K TRANSGENS AUF DIE GFP EXPRESSION 57 4.1.5 KONZENTRIERUNG RETROVIRALER VIRUSÜBERSTÄNDE 58 4.1.6 ADHÄSION REKOMBINANTER RETROVIREN AN MOLEKÜLE DER FIBRONEKTIN-MATRIX 59 4.2 GENTRANSFER IN HUMANE CD34+ ZELLEN AUS NABELSCHNURBLUT 63 4.2.1 ANREICHERUNG HUMANER CD34+ ZELLEN 63 4.2.2 RETROVIRALER GENTRANSFER IN HUMANEN CD34+ ZELLEN 63 4.2.3 EINFLUSS DES TRANSGENS MGMTP140K AUF DIE GENTRANSFERRATE 66 4.2.4 TRANSDUKTIONSEFFIZIENZ VERSCHIEDENER FOAMYVIRALER KONSTRUKTE 70 4.3 GENTRANSFER IN CD 34+ MARMOSET ZELLEN 76 4.3.1 GEWINNUNG UND AUFREINIGUNG VON CD34+ MARMOSET ZELLEN 76 4.3.2 RETROVIRALER GENTRANSFER IN CD34+ MARMOSET ZELLEN 77 4.4 MGMTP140K ALS TRANSGEN 84 4.4.1 TRANSDUKTION HÄMATOPOETISCHER ZELLLINIEN 84 4.4.2 SELEKTION VON TRANSDUZIERTEN ZELLLINIEN MITTELS BCNU IN VITRO 88 4.4.3 MEHRMALIGE BEHANDLUNG VON TRANSGENEN HL60 ZELLEN MIT BCNU 91 4.4.4 SELEKTION TRANSGENER HL60 ZELLEN MIT BG UND BCNU IN VITRO 94 4.4.5 SELEKTION TRANSGENER CD34+ MARMOSET ZELLEN MIT BG UND BCNU 95 4.4.6 SELEKTION TRANSGENER CD34+ MARMOSET PROGENITOREN MIT BG UND BCNU 100

5. DISKUSSION 104
5.1 OPTIMIERTE HERSTELLUNG VON FOAMYVIREN, LENTIVIREN UND GAMMARETROVIREN 105 5.2 GENTRANSFER IN HUMANE CD34+ ZELLEN AUS NABELSCHNURBLUT 111 5.3 GENTRANSFER IN CD34+ MARMOSET ZELLEN 113 5.4 MGMTP140K ALS TRANSGEN 115 5.5 SCHLUSSFOLGERUNG 119
6. ZUSAMMENFASSUNG 120
7. SUMMARY 122
8. LITERATURVERZEICHNIS 124

Verzeichnis der Abkürzungen
ADA Adenosindeaminase
BCNU 1, 3-bis-(2-chloroethyl)-1-Nitrosourea
BG O6-Benzylguanin
bGH pA bovine growth hormon polyA site
BSA bovines Serumalbumin
bzw. beziehungsweise
°C Grad Celsius
CAS cis acting sequence
CD cluster of differentition
cDNA complementary DNA
CMV Cytomegalyvirus
CFU colony forming units
ΔU3 U3-Promotor/Enhancer mit Deletion
DMEM Dulbecco’s Modified Eagle’s Medium
DMSO Dimethylsulphoxid
DNA desoxyribonucleic acid
E. coli Escherichia coli
EM140 Verpackungsplasmid pczHFVenvEM140
EMCV Encephalomyelocarditisvirus
Env envelope, retrovirales Hüllprotein
et al. et alteres (und andere)
FACS fluorescence activated cell sorting
FFV Feline Foamy Virus
FKS Fötales Kälberserum
Flt3-L fetal liver tyrosine kinase 3 ligand
FN Fibronektin
FV Foamy Virus
g Gravitationskonstante
Gag group specific antigen

GaLV Gibbonape Leukemia Virus
G-CSF granulocyte-colony stimulating factor
GFP grün fluoreszierendes Protein
GVHD graft versus host disease
HFV Human Foamy Virus
HIV Human Immundeficiency Virus
HLA Humanes Leukozyten-Antigen
IL Interleukin
IMDM Iscove’s Modified Dulbecco’s Medium
IP Interner Promotor
IRES internal ribosome entry site
kb Kilobasen
KMT Knochenmarktransplantation
l Liter
LB-Medium Luria-Bertani-Medium
LTRs long terminal repeats
µ mikro
M Mol
m milli
MACS magnet activated cell sorting
MD9.MGMT Vektorplasmid MD9.MGMTp140kIEG
MGMT O6-methylguanin-DNA-Methyltransferase
MH71.MGMT Vektorplasmid MH71.MGMTp140kIEG
min Minute
MLV Murine Leukemia Virus
mRNA messenger RNA
ORFs open reading frame
Ori origin of replikation
PBS phosphat buffered saline solution
PE Phycoerythrin
PEI Polyethylenimin

PFV Prototype Foamyvirus
PI Propidiumiodine
ψ, Psi Verpackungssignal
resp. respektive
RNA ribonucleic acid
rpm rounds per minute
RPMI Roswell Park Memorial Institute
RT Raumtemperatur
Sav Streptavidin
SCF stem cell factor
SCID severe combined immune deficiency
sec Sekunde
SFFV Spleen Focus Forming Virus
SFV Simian Foamy Virus
SIN self-inactivating
SM04 Verpackungsplasmid pciSM04.seq
SV40 Simian Virus 40
TCD tissue culture dish
TE transduzierende Einheiten
TPO Thrombopoetin
VSV Vesicular Stomatitis Virus
VSV-G Glykoprotein des VSV
X-linked X-chromosomal
z.B. zum Beispiel


Einleitung
1. Einleitung
Hämatopoetische Stammzellen sind auf Grund ihrer guten Zugänglichkeit die am
besten untersuchten Stammzellen des Menschen. Sie sind die einzigen Stammzellen,
die bereits seit Jahrzehnten im Rahmen von Transplantationen klinisch eingesetzt
werden (THOMAS et al. 1957). Seit mehr als 20 Jahren ist der Ersatz eines defekten
oder fehlenden Gens in hämatopoetischen Stammzellen bei monogenen
Erkrankungen ein erklärtes Ziel der somatischen Gentherapie gewesen (WILLIAMS
et al. 1984). Obwohl der Transfer von Genen in hämatopoetische Stammzellen von
Mäusen schon frühzeitig gelang (DICK et al. 1985), konnte in Großtiermodellen erst
in den letzten 10 Jahren eine ausreichende Gentransfereffizienz erzielt werden.
Neben Studien an Hunden (NEFF et al. 2005) wurden auch verschiedene
Affenspezies wie Paviane (Papio cynocephalus anubis) (KIEM et al. 1997) und
Marmosets (Callithrix jacchus) (HIBINO et al. 1999) zur Prüfung gentherapeutischer
Ansätze erfolgreich eingesetzt.
Beim Menschen wurden bis zum März 2004 insgesamt 619 klinische
Gentherapiestudien beim National Institute of Health der U.S.A. angemeldet.
Weltweit wurden über 3.000 Patienten in klinischen Studien zur Gentherapie
eingeschlossen (vgl. www.asgt.org/history.shtml). Diese Phase I-Gentherapiestudien
testeten aber in erster Linie die Sicherheit und die Nebenwirkungen der Applikation
genetisch veränderter Zellen beim Menschen und waren hinsichtlich der erzielten
klinischen Ergebnisse enttäuschend. Bei allen klinischen Stammzellgentherapie-
studien wurden gammaretrovirale Vektoren als Transfersystem für die Übertragung
der therapeutischen Gene eingesetzt. Die erste Gentherapiestudie, bei der eine
Heilung von 9 von 10 mit genetisch modifizierten autologen Stammzellen
behandelten Patienten erzielt werden konnte, wurde in den letzten Jahren in Paris
bei Jungen mit der x-chromosomalen Form des schweren kombinierten
Immundefektes (X-linked SCID) durchgeführt (CAVAZZANA-CALVO et al. 2002;
HACEIN-BEY-ABINA et al. 2002). Leider zeigte diese erste erfolgreiche Studie aber
auch sehr eindrucksvoll, dass die bei Wildtyp-Gammaretroviren beobachte
Aktivierung von Onkogenen auch bei Nutzung dieser einfach Orthoretroviren als
1

Einleitung
replikationsdefiziente Vektoren zu beobachten ist. Drei der behandelten Patienten
entwickelten ein Malignom der T-Zellen, wobei bei mindestens zwei Patienten eine
pathologische Aktivierung des Proto-Onkogens LMO-2 durch den integrierten Vektor
nachgewiesen wurde (HACEIN-BEY-ABINA et al. 2003a; CHECK 2005). Diese
sogenannte Insertionsmutagenese von gammaretroviralen Vektoren wurde in den
letzten Jahren auch bei Mäusen (LI et al. 2002) und in jüngerer Vergangenheit bei
einem Rhesusaffen nachgewiesen (SEGGEWISS et al. 2006).
Wildtyp-Foamyviren sind die einzigen Retroviren, mit denen bisher keine Erkrankung
trotz chronisch-persistierenden Infektionen in ihren natürlichen tierischen Wirten und
in akzidentell infizierten Menschen assoziiert werden konnte. Replikationsdefekte
foamyvirale Vektoren wurden erfolgreich eingesetzt, um Resistenz- und Markergene
in hämatopoetische Stammzellen von Mäusen oder auch Menschen einzubringen
(VASSILOPOULOS et al. 2001; LEURS et al. 2003; POLLOK et al. 2005). Eine
kürzlich publizierte Studie an Hunden konnte zeigen, dass foamyvirale
Vektorsysteme in der Lage sind, hämatopoetische Stamm- und Vorläuferzellen eines
Großtiers effizient zu transduzieren (KIEM et al. 2007).
Generell wird die Übertragbarkeit von Ergebnissen aus Tierversuchen in der Regel
als eher gegeben akzeptiert, wenn das Tiermodel dem Menschen phylogenetisch
nahe steht. Daher befasst sich die vorliegende Untersuchung mit der Fragestellung,
welche Voraussetzungen erfüllt sein müssen, damit foamyvirale Vektoren dazu
genutzt werden können, um hämatopoetische Stammzellen der Primatenart Callitrix
jacchus auch Marmosets genannt, effizient zu transduzieren. In dieser Arbeit wurden
deshalb in vitro ähnliche aufgebaute foamy-, lenti- und oncoretrovirale
Vektorsysteme hinsichtlich ihrer Effizienz, ihres prinzipiellen Aufbaus und
verschiedene Transgene intensiv geprüft.
2

Literatur
2. Literatur
2.1 Hämatopoetische Stammzellen als Zielzellen somatischer Gentherapie
Die Hämatopoese ist ein hierarchisch aufgebautes flüssiges Organsystem, bei dem
sich aus einer pluripotenten Stammzelle alle Blutzellen entwickeln können. Aus
dieser Stammzelle entstehen pluripotente Vorläuferzellen, die in myeloische und
lymphatische Vorläuferzellen unterteilt werden. Aus den myeloischen pluripotenten
Vorläuferzellen entwickeln sich Erythrozyten, Neutrophile, Basophile und Eosinophile
Granulozyten, Thrombozyten, Makrophagen und Mastzellen. Aus den lymphatischen
pluripotenten Vorläuferzellen entstehen die lymphozytären Zellen: T-Zellen, B-Zellen
und natürliche Killerzellen. Der Ort der Entwicklung der Blutzellen ist das
Knochenmark. Bei einem gesunden Individuum gelangen nur die reifen Zellen der
Hämatopoese in das periphere Blut. Einzelne Erkrankungen des blutbildenden
Systems können durch die Transplantation von pluripotenten
„gesunden“ Stammzellen geheilt werden.
Hämatopoetische Stammzellen sind durch die Fähigkeit charakterisiert, nach
Transplantation einen myeloablativ behandelten Organismus langfristig zu
repopulieren und alle hämatopoetischen Zellen sowohl der myeloischen, als auch der
lymphatischen Zellreihe zu bilden (MORRISON et al. 1995). Hämatopoetische
Stamm- und Vorläuferzellen exprimieren auf ihrer Oberfläche neben anderen
Antigenen auch das CD34-Antigen (KRAUSE et al. 1996). Die Anzahl an CD34+
Zellen liegt im peripherem Blut bei unter < 0,1 % und im Knochenmark bei 0,5 bis
2 % aller mononukleären Zellen. Allerdings sind „echte“ hämatopoetische
Stammzellen im Sinne von Pluripotenz und Rekonstitutionspotential unter den CD34+
Zellen trotzdem sehr selten, da CD34 auch auf anderen Zellen exprimiert wird (HAAS
u. MUREA 1995; WANG et al. 1997). Stammzellen lassen sich phänotypisch nicht
von anderen CD34+ Zellen unterscheiden, so dass der sichere Nachweis nur durch
Funktionsprüfungen erfolgen kann.
Über klonogene in vitro Assays kann die Detektion von Progenitorzellen der
Erythropoese und der Granulopoese erfolgen. Diese Form der Detektion wird als
3

Literatur
Progenitor-Assay bezeichnet (METCALF 1977). Doch der in vitro Nachweis von
klonogenen Zellen lässt keine Aussage zu, wie viele Stammzellen tatsächlich im
Transplantat vorhanden sind und nach einer Transplantation wieder im menschlichen
Patienten anwachsen (ORKIN u. MOTULSKY 1995). Um die Zahl der
repopulierenden Zellen besser verifizieren zu können, wurde das
Xenotransplantationsmodell der nonobese diabetic / severe combined
immunodeficiency (NOD/SCID) Mäuse etabliert (LAROCHELLE et al. 1996; VAN
DER LOO et al. 1998).
Die Transpantation von Knochenmark als Quelle für Stammzellen schon seit 1957
klinisch eingesetzt, um eine hämatopoetische Aplasie nach Chemo- und/ oder
Radiotherapien zu behandeln (THOMAS et al. 1957). Daneben findet die
Knochenmarktransplantation (KMT) Anwendung bei der Behandlung von
angeborenen Erkrankungen des blutbildenden Systems (TO et al. 1997; THOMAS
1999). Initial wurden die Stammzelltransplantate aus dem Knochenmark gewonnen,
mittlerweile gibt es eine Alternative, bei der die Stammzellen aus dem peripheren
Blut mittels Zellseparation (Leukopherese) gewonnen werden.
Durch die Gabe von Granulocyte-colony stimulating factor (G-CSF) werden
hämatopoetische Stammzellen aus dem Knochenmark ins periphere Blut mobilisiert.
Da durch dieses Verfahren in der Regel mehr Stammzellen bei der Entnahme aus
dem Spender gewonnen werden, ist nach einer Transplantation dieser Zellen
schneller mit einer Rekonstitution der Hämatopoese des Empfängers zu rechnen
(HAAS et al. 1995). Für eine KMT werden mittlerweile die meisten Stammzellen
mittels Leukopherese gewonnen. Von 1990 bis 1994 stieg der Anteil der KMT mit
durch Leukopherese gewonnenen Stammzellen von 15 % auf 75 % an (TO et al.
1997).
Der Mindestbedarf an hämatopoetischen Stammzellen für eine erfolgreiche
Rekonstruktion der Hämatopoese liegt für einen Menschen bei 2 x 106 CD34+ Zellen
pro Kilogramm Körpergewicht (BURMESTER u. PEZZUTTO 1998).
Bei Transplantationen unterscheidet man zwischen autologen Transplantationen, bei
der eigenes Organmaterial retransplantiert wird und allogenen Transplantationen, bei
denen Organmaterial eines Dritten übertragen wird. Autologe Transplantationen von
4

Literatur
kryokonservierten hämatopoetischen Stammzellen werden beispielsweise bei
therapieinduzierten Panzytopenien, wie nach Chemotherapie oder Bestrahlung,
durchgeführt. Allogene Transplantationen mit Transplantat eines gesunden Spenders
sind unter anderem Teil der Behandlung von Leukämien, angeborenen Immun- oder
Stoffwechselerkrankungen sowie bei der Behandlung des multiplen Myelom.
Für eine allogene Stammzelltransplantation steht bislang nur bei einem Teil der
Patienten ein humanes Leukozytenantigen (human leukozyt antigen, HLA)–
identischer Spender zur Verfügung. In der Regel wird zunächst im Familienkreis
nach einem kompatiblen Spender gesucht, denn für Geschwister besteht eine
Wahrscheinlichkeit von 1:4, dass alle HLA-Allele übereinstimmen (BURMESTER et
al. 1998). Da Familien in der westlichen Welt im Durchschnitt weniger als zwei Kinder
haben, findet man selten einen HLA-kompatiblen Spender in der Familie des
Patienten. Daneben besteht die Möglichkeit, geeignete Knochenmarkspender in
entsprechenden Datenbanken zu finden; weltweit sind etwa 10 Millionen Menschen
in solchen Datenbanken registriert. Von diesen Menschen ist etwa ein Fünftel in
Deutschland registriert (HO 2006). Eine allogene Transplantation geht jedoch mit
einem hohen Therapie-Assoziierten Morbiditäts- und Mortalitätsrisiko einher. An
erster Stelle ist hier die Transplantat-gegen-Wirt-Reaktion (graft versus host disease
- GVHD) zu nennen (THOMAS 1999).
Eine alternative Therapiemöglichkeit zur Behandlung monogener Erkrankungen der
Hämatopoese bietet die Transplantation autologer genetisch korrigierter
Stammzellen. Stammzellen werden nach der Entnahme in vitro mit einem Vektor
behandelt, der das funktionelle Gen in das zelluläre Genom einbringt. Dieses
übernimmt dann die Aufgaben des körpereigenen, defekten Gens (WILLIAMS et al.
1984). Für monogene Erkrankungen, wie zum Beispiel dem schweren kombinierten
Immundefekt (severe combined immunodeficiency - SCID), dem
Adenosindeaminase-Mangel (ADA), der Fanconi-Anämie, der chronischen
Granulomatose, wird eine solche Therapie in Betracht gezogen (KOHN 1996;
WILLIAMS et al. 1997; DIRKSEN et al. 1999). In Zukunft könnten auch
Resistenzgene in Stammzellen eingebracht werden, die sie vor einer Behandlung mit
Chemotherapeutika schützen. So können schwere Nebenwirkungen, wie sie bei der
5

Literatur
chemotherapeutischen Behandlung bei onkologischen Erkrankungen auftreten,
vermindert werden (MAZE et al. 1997).
Das therapeutische Potenzial von gentechnisch veränderten, hämatopoetischen
Zellen konnte erstmals bei 19 Patienten gezeigt werden (CAVAZZANA-CALVO et al.
2000; AIUTI et al. 2002; GASPAR et al. 2004). Im Rahmen dieser Studie wurden, 14
Patienten mit X-linked-SCID und 5 Patienten mit ADA-SCID transplantiert. Obwohl
bei 18 von 19 behandelten Patienten die Grunderkrankung erfolgreich behandelt
werden konnte, ist heute bekannt, dass dieser Erfolg mit schwerwiegenden
Nebeneffekten verbunden war. Mindestens drei Kinder entwickelten eine der
Leukämie ähnliche Funktionsstörung. Bei zwei Patienten konnte gezeigt werden,
dass diese Funktionsstörung mit einer monoklonalen Vermehrung genetisch
modifizierter T-Zellen einherging (ASGT 2003; HACEIN-BEY-ABINA et al. 2003a;
KOHN et al. 2003).
6

Literatur
2.2 Zytokine
Zytokine übermitteln Informationen zwischen verschiedenen Zellen und regulieren so
Wachstum und Differenzierung. Zytokine sind auch notwendig, um Stammzellen zu
Expandierung anzuregen. Derzeit werden in vivo verschiedene Zytokine in
unterschiedlichen Kombinationen eingesetzt. Die wichtigsten Zytokine zum
Überleben von Stamm- und Progenitorzellen in vitro sind Stammzellfaktor (stem cell
factor - SCF) und Thrombopoietin (TPO) (BROUDY 1997; EATON u. DE SAUVAGE
1997; KIMURA et al. 1998). Darüber hinaus werden auch G-CSF, Interleukin (IL)-3
und IL-6 sowie der fetal liver tyrosine kinase 3 ligand (Flt3-L) eingesetzt (LUSKEY et
al. 1992; CROOKS u. KOHN 1993; KURRE et al. 2001). Hierbei scheint Flt3-L in der
Lage zu sein, die Differenzierung von CD34+ Zellen in vitro zu verhindern und so die
Repopulationskapazität bei einer Transplantation zu erhalten (DAO et al. 1997). Es
gibt allerdings auch Hinweise darauf, dass IL-3 in einer hohen Dosierung zu einer
Differenzierung von Stammzellen führt, so dass der klinische Einsatz fraglich ist
(YONEMURA et al. 1996). Kombinationen aus Flt3-L, TPO, IL-6 und SCF haben sich
im Rahmen von in vitro Gentherapieexperimenten (HANENBERG et al. 1997) und in
Untersuchungen an Hunden (GOERNER et al. 1999), Primaten und Menschen
(ABONOUR et al. 2000; AIUTI et al. 2002; CAVAZZANA-CALVO u. FISCHER 2004;
GASPAR et al. 2004) als besonders effizient erwiesen.
7

Literatur
2.3 Biologie von Retroviren
Unabhängig von der Taxonomie werden Retroviren aufgrund der Struktur ihres pro-
viralen Genoms in einfache und komplexe Retroviren unterteilt. Einfache Retroviren
enthalten in der Regel die drei Gene gag, pol und env und darüber hinaus identisch
aufgebaute long terminal repeats (LTRs) an beiden Enden des viralen Genoms
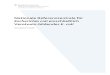
(Abbildung 1).
-gag kodiert die strukturellen Komponenten des Virus: Matrix-, Kapsid- und
Nukleokapsidproteine
-pol kodiert die Enzyme des Virus: Protease, Integrase und reverse
Transkriptase
-env kodiert für die viralen Hüllproteine
-LTRs Long terminal repeats: befinden sich am Anfang und am Ende des
Genoms; sie enthalten Steuersequenzen zur Genexpression und für die
Integration
-ψ Verpackungssignall: ψ (Psi) wird benötigt, damit das Genom der
Retroviren, die virale mRNA, mit in die infektiösen Partikel gelangt (über
Interaktion mit gag)
Komplexe Retroviren z.B. HIV-1, enthalten zusätzlich regulatorische Gene wie tat, ref,
vif, nef, vpu und vpr (MODROW et al. 2002).
8

Literatur
MLV
SFV
gagpol
env
ψ nef
vifvpu
vpr
tatrev5`LTR 3`LTR
HIV U3 U3
12
gag
5`LTR
ψ
gag polenv
5`LTR 3`LTR
ψ
U3
U3U3
pol3`LTR
envIP
tasOrf-2
ψU3
bet
1 432 765 1098 11 kBp
MLV
SFV
gaggagpolpol
env
ψ nefnef
vifvpu
vpr
tatrev5`LTR 3`LTR
HIV U3 U3
12
gag
5`LTR
ψ
gag polenv
5`LTR 3`LTR
ψ
U3
U3U3
pol3`LTR
envIP
tasOrf-2
ψU3
bet
1 432 765 1098 11 kBp
Abbildung 1: Genomaufbau verschiedener Retroviren Die proviralen Genome des Murinen Leukämie Virus (MLV), des Humanen Immundefizienz Virus
(HIV) und des Simian Foamy Virus (SFV) sind hier schematisch dargestellt. Sie werden zu beiden
Seiten von den „long terminal repeats“ (LTRs) begrenzt. Die einzelnen Gene gag, pol, env, vif, vpr,
vpu, nef, tas und Orf-2 sind durch Rechtecke dargestellt. Die Proteine, tat, rev und bet, die von
gespleißter mRNA translatiert werden, sind durch gestrichelte Linien miteinander verbunden. Offene
Leserahmen (ORFs) sind auf gleicher Höhe abgebildet. ORFs verschiedener Leseraster sind auf
unterschiedlichen Ebenen dargestellt. Das SFV hat neben den mit U3 bezeichneten Promotoren im
3`Bereich des env Gens einen Internen Promotor (IP). Mit ψ sind die Verpackungssignale bezeichnet,
die bei FV das diskontinuierliche Verpackungssignal beinhalten (verändert nach MODROW et al.
2002).
Eine charakteristische Besonderheit der Retroviren ist die reverse Transkriptase, die
es ihnen ermöglicht ihr RNA-Genom in DNA umzuschreiben und anschließend in das
Wirtsgenom zu integrieren. Durch diese Integration wird das Provirus ein Teil des
Genoms der Zielzelle und bei der Zellteilung an beide Tochterzellen weitergegeben.
Die Tatsache, dass die genetischen Informationen von Retroviren im Wirtsgenom
stabil integriert werden, macht diese Viren zu idealen Vehikeln für den Transfer von
Genen in Stammzellen.
9

Literatur
Ursprünglich wurden die Viren nach ihrem Erscheinungsbild in Typ A, B, C und D
Retroviren eingeteilt; heute werden sie aufgrund ihrer genetischen
Verwandtschaftsverhältnisse in sieben Genera unterteilt (Tabelle 1). Die Viren der
ersten fünf Genera besitzen onkogenes Potential und werden als Onkoretroviren
bezeichnet.
Tabelle 1: Taxonomie der Virusfamilie Retroviridae
Subfamilie Genus Beispiel Genom
Orthoretroviridae Alpharetroviren Avian Leukosis Virus
Rous Sarcoma Virus
einfach
Betaretroviren Mouse Mammary Tumor Virus
Mason Pfizer Monkey Virus
einfach
Gammaretroviren Murine Leukemia Virus (MLV) einfach
Deltaretroviren Bovine Leukemia Virus komplex
Epsilonretroviren Walleye Dermal Sarcoma Virus komplex
Lentiviren Human Immundeficiency Virus (HIV) komplex
Spumaretroviren Foamyviren Simian Foamy Virus (SFV)
Primate / Prototyp Foamy Virus (PFV)
komplex
10

Literatur
2.3.1 Orthoretroviren
2.3.1.1 Replikationszyklus von Orthoretroviren
Die spezifischen Hüllproteine auf der Oberfläche von Viren, auch als envelope
bezeichnet, binden initial an spezifische Oberflächenmoleküle der Zielzelle. Ohne die
spezifische Bindung können Retroviren Zellen nicht infizieren; beispielsweise kann
HIV Zellen nur über das Oberflächenmolekül CD4 Zellen infizieren. Da nur
Makrophagen, Lymphozyten und Gliazellen CD4 exprimieren, kann HIV nur diese
Zellen, nicht aber andere Zellen des humanen Organismus infizieren.
Der Eintritt in die Zelle ist nicht bei allen Retroviren gleich. Der Viruspartikel wird
entweder von der Zelle durch Endozytose aufgenommen oder die Virushülle
fusioniert mit der Zellmembran und das Kapsid wird freigesetzt. Bei den
Orthoretroviren setzt anschließend die reverse Transkription ein, wobei die virale
RNA durch die reverse Transkription in DNA umgeschrieben wird. Der benötigte
Primer für die reverse Transkription wird im Viruspartikel mitgeführt. Nachdem es zur
Bindung des Primers an die Primerbindungsstelle gekommen ist, erfolgt die
Synthese des DNA-Minusstranges. Vom Polypurintrakt aus findet dann die Synthese
des DNA-Plusstranges statt. Die neu gebildete lineare doppelsträngige DNA wird an
beiden Enden von LTRs abgeschlossen, die am 5’ und 3’ Ende in derselben
Orientierung vorliegen. Sie bestehen aus der U3 Region, die Promotor- und
Enhancer-Elemente enthält und so die Genexpression der viralen Gene kontrolliert,
der R-Region (R = redundant) und der U5 Region anschließt, welche die
Insertionssequenzen zur Integration in das Wirtsgenom enthält. Der
Präintegrationskomplex wird bei Lentiviren aktiv in den Zellkern transportiert.
Dagegen kann der Präintegrationskomplex von Gammaretroviren die Kernmembran
nicht überwinden und gelangt daher nur während der Zellteilung, wenn die
Kernmembran aufgelöst ist, zur DNA der Wirtszelle. Die virale Integrase katalysiert
die Insertion in das Genom der Zelle. Als Teil des Genoms dieser Zelle nutzt das
integrierte Virus (Provirus) jetzt die Transkriptions- und Translationsmachinerie der
11

Literatur
Zelle, um sich zu vermehren. Dabei werden Promotoren in der U3 Region des
Provirus durch zelleigene Transkriptionsfaktoren aktiviert; bei den komplexen
Retroviren ist eine Aktivierung über eigene Transaktivatoren essentiell. Der
Transaktivator des HIV ist tat, das durch basale Aktivität von einer gespleißten RNA
gebildet wird. Ist eine ausreichende Menge tat in der Zelle vorhanden, wird der 5’ U3
Promotor stark aktiviert und die anderen viralen Gene und das komplette
ungespleißte Genom wird abgelesen. Im 3`Bereich des Genoms ist das PolyA-Signal,
dass das Ende der mRNA für die Zelle definiert. Das 5`Ende wird mit einem 5`Cap-
Protein versehen; dieses wird benötigt, damit die mRNA an das Ribosom binden
kann. Beide Prozesse entsprechen denen, die auch jede andere mRNA der Zelle
durchläuft. Nachdem die mRNA in das Zytoplasma transportiert wurde, beginnt an
den Ribosomen die Translation. In komplexen Prozessen werden die Viruspartikel
zusammengefügt.
Eine zentrale Rolle im Vermehrungszyklus spielen die strukturellen Proteine des gag
Gens. Sie sind in der Lage „Pseudopartikel“ zu generieren (IORDANSKII et al. 1997).
„Pseudopartikel“ sind nicht infektiöse Partikel, die aus einer Hülle und inkorporierten
Proteinen bestehen, denen jedoch das Virusgenom fehlt. Die Knospung des Virus
oder der subviralen „Pseudopartikel“ findet an den zellulären Membranen statt, in
denen die env Proteine als Membran- oder als Transmembranproteine eingelassen
sind.
2.3.1.2 Orthoretrovirale Vektoren
Für den Einsatz als Transfervektor werden bis heute einfache Gammaretroviren
verwendet, denen alle Gene (gag, pol und env) entfernt wurde. An Stelle viraler
Gene wird ein therapeutisches Gen oder ein Markergen als Transgen in den Vektor
ligiert. Einige essentielle Elemente bleiben im Transfervektor: das Verpackungssignal
ψ ist notwendig, damit die virale mRNA in den neu gebildeten viralen Partikel
inkorporiert wird; der Vektor enthält die LTRs, die für die Integration in das Genom
12

Literatur
der Zielzelle nötig sind. Dazu gehören Primerbindungsstelle und der Polypurintrakt
als Startpunkt der reverse Transkription. Die Sequenzen, die für die viralen
Strukturproteine, Enzyme und das Hüllprotein kodieren, sind auf andere
Expressionsplasmide verteilt. Die transkribierte RNA von gag, pol und env werden
nicht in die viralen Partikel inkorporiert, da ihnen das Verpackungssignal ψ fehlt. Aus
diesem Grund kann die „Gensequenzen“ für gag, pol und env auch nicht in das
Genom der Zielzelle gelangen.
Die Plasmide, die nur strukturelle Komponenten für den Viruspartikel bereitstellen,
werden Helferplasmide genannt. Durch gleichzeitige Transfektion mit den
Transfervektorplasmiden und Helferplasmiden lassen sich effizient rekombinante,
replikationsdefekte Retroviren generieren (SONEOKA et al. 1995). Die Effektivität
der Virusproduktion kann gesteigert werden, indem die U3-Region des 5`LTRs im
Vektorplasmid durch die Promotorsequenz eines sehr starken Promotors wie z.B. der
intermediate early promotor des humanen Cytomegalyvirus (CMV-Promotor) ersetzt
wird. Die Helferplasmide können ebenfalls von einem CMV-Promotor aus abgelesen
werden. Auf diese Weise lassen sich hochtitrige Virusüberstände generieren (PEAR
et al. 1993; FINER et al. 1994; NAVIAUX et al. 1996). Der CMV-Promotor kann durch
die zusätzliche Gabe von Natriumbutyrat während der Virusproduktion aktiviert
werden (RADSAK et al. 1989; TANAKA et al. 1991). Der Virustiter eines so
erzeugten Überstandes kann 8- bis 12-mal höher sein, als ohne Natriumbutyrat
(LEURS et al. 2003).
Heterologe Promotoren können als interne Promotoren im Vektor vor das Transgen
gesetzt werden. Hierbei wird die Besonderheit der Retroviren, dass die 3’ U3 Region
bei der Integration auf den 5’ LTR übertragen wird, ausgenutzt. Eine Deletion im U3-
Promotor/Enhancer (ΔU3) des 3`LTRs wird bei der reverse Transkription auf den
5`LTR übertragen. Der provirale 5`LTR ist nun inaktiv und das Transgen wird vom
internen Promotor aus abgelesen; das 5’ gelegen virale Genom kann nicht mehr
abgelesen werden.
13

Literatur
2.3.1.3 Pseudotypisierung von Orthoretroviren
Pseudotypisierung beschreibt den Vorgang der Verpackung eines viralen Kapsids in
eine Virushülle (env), die nicht der natürlichen Hülle des Virus entspricht. Einige
Retroviren, wie z.B. Lenti- und Gammaretroviren, lassen sich pseudotypisieren.
Durch die Pseudotypisierung kann der Tropismus des retroviralen Vektors geändert
werden: so kann z.B. das G-Protein des Vesicular Stomatitis Virus (VSV-G) zur
Pseudotypisierung des Murine Leukemia Virus (EMI et al. 1991) und von Lentiviren
(YEE et al. 1994; BARTZ u. VODICKA 1997) verwendet werden. Die
Pseudotypisierung mit VSV-G führt zu einem sehr breiten Wirtsspektrum der
Retroviren (YEE et al. 1994). Die retroviralen Vektoren, die mit VSV-G verpackt
werden, erzeugen hohe Virustiter. Darüber hinaus sind die Viren sehr stabil und es
besteht die Möglichkeit, die Konzentration durch Ultrazentrifugation noch weiter zu
erhöhen (BURNS et al. 1993).
Gammaretroviren können nur sehr ineffektiv mit dem Wildtyp env des HFV
pseudotypisiert werden (LINDEMANN et al. 1997). Erst durch Ersetzen der
Transmembrandomäne des Foamyvirus mit der des amphotropen Hüllproteins
können MLV und HIV mit Hüllproteinen des Feline Foamy Virus (FFV) oder SFV
pseudotypisiert werden (LEURS 2003).
2.3.2 Spumaretroviren
2.3.2.1 Merkmale der Spumaretroviren
Foamyviren (FV) sind mit ca. 13 kb die größten, bisher bekannten Retroviren. Sie
werden aufgrund ihres Aufbaus mit gag, pol und env sowie den zusätzlichen
Leserahmen tas und bet den komplexen Retroviren zugeordnet (RETHWILM 2003).
Einige biologischen Eigenschaften teilen sie sich auch mit den Hepatitisviren, so
dass sie innerhalb der Retroviren eine eigene Subfamilie bilden (LINIAL 1999).
14

Literatur
Die ersten Foamyviren wurden 1971 aus den Zellen eines kenianischen Patienten
mit Nasopharynx-Karzinom isoliert (ACHONG et al. 1971). Aufgrund seines
Ursprungs wurde dieses Virus als Human Foamy Virus (HFV) bezeichnet. Später
konnte gezeigt werden, dass das HFV ein Derivat von einem Schimpansen FV ist
(HERCHENRÖDER et al. 1994); dieses FV wurde daraufhin als Prototyp FV (PFV)
bezeichnet. Zahlreiche Säugetiere stellen für Foamyviren einen natürlichen Wirt da.
Sie wurden in verschiedenen Affenarten wie Schimpansen, Gorillas und Orang-Utans,
sowie in Pferden, Kühen und Katzen nachgewiesen (MEIERING u. LINIAL 2001).
Der Mensch ist kein natürlicher Wirt von FV, allerdings können FV der verschiedenen
Spezies durch Biss auf den Menschen übertragen werden.
Im natürlichen Wirt führt eine Foamyvirus-Infektion zu einer lebenslangen
chronischen Persistenz des Virus in der Rachenschleimhaut der Tiere. Trotz der
Persistenz konnte bisher noch keine Erkrankung mit der Infektion assoziiert werden
(WEISS 1988). Menschen, die durch Bissverletzungen verschiedener Affen mit FV
infiziert worden sind, werden in den USA vom Center of Disease Control in Atlanta
erfasst und beobachtet (SCHWEIZER et al. 1997; CALLAHAN et al. 1999). In den
vergangenen 30 Jahren führten die Infektionen zu keiner nachweisbaren Erkrankung
(LINIAL 2000). Von diesen infizierte Menschen können FV nicht über Blut, Urin,
Speichel oder Samenflüssigkeit auf andere Menschen übertragen werden (WINKLER
et al. 2000; HENEINE et al. 2003).
Die Tatsache, das FV keine Pathogenität bei Mensch und Tieren aufweisen, nicht
von Mensch zu Mensch übertragbar sind und einen breiten Wirts- und
Gewebetropismus haben, machen sie möglicherweise zu idealen Vektoren für die
Gentherapie. In der menschlichen Population kommen Antikörper gegen das Virus
praktisch nicht vor.
15

Literatur
2.3.2.2 Replikationszyklus von Foamyviren
Spumaretroviren weisen einige Unterschiede zu Orthoretroviren auf.
Spumaretroviren enthalten im Gegensatz zu anderen Retroviren im Viruspartikel
doppelsträngige DNA (YU et al. 1999). Die reverse Transkription des viralen RNA
Genoms findet bei Foamyviren schon statt, bevor das Virus die Zelle verlässt. In
experimentellen Untersuchungen, bei denen die Zellen mit 3´-Azido-3`-
deoxythymidin, ein Hemmstoff der reverse Transkription, behandelt worden waren,
konnte die Infektiösität der foamyviralen Viruspartikel kaum beeinflusst werden.
Erfolgte die Behandlung der Zellen erst nach der Infektion mit den Foamyviren,
wurden deutlich weniger neue Viruspartikel gebildet und die produzierten Viren
waren weniger infektiös (MOEBES et al. 1997; YU et al. 1999). Durch den Umstand,
dass schon in der infizierten Zelle das virale Genom als DNA-Strang vorliegt, kann es
zur Reintegration des viralen Genoms, das die Zelle infiziert hat, kommen
(HEINKELEIN et al. 2000a). Die Foamyviren verhalten sich ähnlich wie
Retrotransposons; bis zu 10 % der Viren verlassen die Zelle nicht, sondern
reintegrieren wieder in das Genom (RETHWILM 2003). Es konnten bis zu 20
Integrationen pro Genom in infizierten Zellen gefunden werden (MEIERING et al.
2000).
Ein besonderes Charakteristikum der Foamyviren ist, dass sie nicht nur einen
Promotor in der U3 Region des 3`LTRs, sondern noch einen zusätzlichen internen
Promotor besitzen. Dieser interne Promotor ist im 3`Bereich des env Gens lokalisiert
(LÖCHELT et al. 1993). Beide Promotoren werden vom Transaktivator tas reguliert.
Der foamyvirale LTR ist in der Abwesenheit von tas transkriptionell stumm
(BAUNACH et al. 1993; YU u. LINIAL 1993). Der interne Promotor hat eine
schwache basale Aktivität (YANG et al. 1997) und bildet so in der Frühphase des
Replikationszyklus das Protein tas. Das tas Protein des FV bindet spezifisch an die
U3 Region des FV LTRs. Die Bindung von tas führt zu einer starken Steigerung der
Genexpression (LOCHELT 2003). Das tas ist nicht in der Lage andere Promotoren,
wie z.B. den von HIV, zu aktivieren (OMOTO et al. 2004).
16

Literatur
2.3.2.3 Foamyvirale Vektoren
Replikationskompetente foamyvirale Vektoren wurden zuerst von SCHMID und
RETHWILM (1995) entwickelt. Aufgrund der Größe des Genoms von 13 kb, haben
Foamyviren eine potentiell hohe Kapazität, große heterologe Gensequenzen zu
verpacken. Die Vektoren können eine Vielzahl von Säugerzellen transduzieren und
werden darüber hinaus von menschlichem Serum nicht inaktiviert (RUSSELL u.
MILLER 1996).
Die Vektoren haben Deletionen in der U3-Region und in der 3`Region des viralen
Genoms, die zum Teil in Kultur spontan entstanden sind. Mit diesen Vektoren konnte
eine kleine Transgenkassette in Zellen übertragen werden (SCHMIDT et al. 1995).
Die ersten replikationsinkompetenten foamyviralen Vektoren wurden dann 1996
etabliert (RUSSELL et al. 1996). Durch die Deletion des env Gens wurde der interne
Promotor entfernt und eine Replikation des Virus somit unterbunden. Die
Vektorherstellung erfolgte mit einem Helferplasmid, das für env kodiert.
Durch den Austausch des 5`LTR gegen einen CMV/LTR Hybridpromotor erfolgte die
Vektorherstellung tas unabhängig (FISCHER et al. 1998; HEINKELEIN et al. 1998;
TROBRIDGE u. RUSSELL 1998; WU u. MERGIA 1999). Auf getrennten Konstrukten
wurden gag/pol und env ebenfalls von einem CMV-Promotor aus abgelesen. Die
Expressionskassette hatte einen internen Promotor und in den Vektoren wurden
virale Sequenzen, soweit möglich, entfernt (HEINKELEIN et al. 1998; HEINKELEIN
et al. 2002a). Die foamyviralen Vektoren, die in dieser Arbeit verwendet wurden,
basieren auf den Vektoren MH5 und MH12, die in einer Veröffentlichung von
HEINKELEIN (1998) beschrieben sind.
Hämatopoetische Stammzellen haben nur eine geringe Teilungsaktivität und
befinden sich zum überwiegenden Teil in der G0-Phase (YU 1996). Mit VSV-G
pseudotypisierte Lentiviren können terminal differenzierte, teilungsinaktive Zellen,
wie z. B. Neurone, effektiv transduzieren (NALDINI et al. 1996). Die so
pseudotypisierten Viren können auch hämatopoetische Stammzellen effektiv
transduzieren (VON LAER et al. 2000; LEURS et al. 2003).
17

Literatur
Gammaretrovirale Vektoren sind dagegen auf eine Zellteilung angewiesen, damit der
virale Präintegrationskomplex in das Genom integrieren kann. Der
Präintegrationskomplex kann die intakte Kernmembran, die nur während der
Teilungsphase aufgelöst wird, nicht passieren (ROE et al. 1993; TROBRIDGE u.
RUSSELL 2004). Auch Foamyviren können nur in die DNA von Zellen integrieren,
wenn sich die Zelle teilt. Es wurde gezeigt, dass FV Zellen mit einer geringen
Teilungsaktivität effizienter transduzieren können als Gammaretroviren (RUSSELL et
al. 1996; MERGIA et al. 2001). Der foamyvirale Präintegrationskomplex bleibt in der
Zelle stabil und kann noch 14 Tage nach der Transfektion bei einer Zellteilung in das
Genom integrieren (TROBRIDGE et al. 2004).
Betrachtet man Integrationsstellen von foamyviralen Vektoren im Genom
transduzierter humaner Zellen, so unterscheiden sich diese deutlich von denen lenti-
und gammaretroviraler Vektoren. HIV-Vektoren integrieren besonders häufig in den
Chromosomen 16, 17, 19 und 22, wohingegen MLV in das Chromosom 17 integriert
(SCHRODER et al. 2002; WU et al. 2003; NOWROUZI et al. 2006). Foamyvirale
Vektoren haben keine besondere Präferenz für bestimmte Chromosomen
(NOWROUZI et al. 2006; TROBRIDGE et al. 2006). Darüber hinaus integrieren HIV-
Vektoren besonders häufig in aktive Gene, wie zum Beispiel in Onkogene; MLV
bevorzugt zur Integration in das Wirtsgenom Bereiche in der Nähe von
Transkriptionsstartpunkten. FV integrieren dagegen häufig in „CpG-Inseln“. Hierbei
handelt es sich um Regionen, die in der Nähe von Promotoren liegen oder es sind
nicht kodierende Genomabschnitte, wie sie an den Zentromeren vorkommen
(NOWROUZI et al. 2006; TROBRIDGE et al. 2006).
18

Literatur
2.3.2.4 Verpacken von Foamyviren
Foamyviren besitzen im Gegensatz zu anderen Retroviren ein diskontinuierliches
Verpackungssignal (ψ), das bei Foamyviren auch cis acting sequence
(CAVAZZANA-CALVO et al.) I und II genannt wird (HEINKELEIN et al. 2002a). CAS I
und II werden benötigt, damit das virale Genom in den Viruspartikel verpackt werden
kann. (HEINKELEIN et al. 1998; HEINKELEIN et al. 2000b). CAS I erstreckt sich
vom 5`LTR bis in das gag Gen. CAS II ist im 3`Bereich des pol Gens codiert
(Abbildung 2). Fehlt einer der beiden Regionen auf einer mRNA-Sequenz, kann
diese mRNA nicht verpackt werden. Das pol Protein kann bei FV allein in den
foamyviralen Partikel inkorporiert werden, unabhängig davon ob gag Proteine
anwesend sind (ENSSLE et al. 1996). Prägenomische RNA ist notwendig, damit die
pol Proteine in den Viruspartikel aufgenommen werden können (HEINKELEIN et al.
2002b).
Im Unterschied zu den anderen Retroviren können Foamyviren nur dann Partikel
bilden, wenn neben den Strukturgenen gag und pol auch das foamyvirale env
vorhanden ist (LINDEMANN u. GOEPFERT 2003). Da die Gegenwart des
foamyviralen env Proteins eine Voraussetzung für die Partikelbildung ist (RETHWILM
2003), lassen sich FV nicht mit heterologen Hüllproteinen oder dem VSV-
Glykoprotein pseudotypisieren (PIETSCHMANN et al. 1999).
19

Literatur
2.4 Selektion transgener Zellen in vitro und in vivo Bei manchen Erkrankungen, die gentherapeutisch behandelt werden, bietet das
Transgen den Zellen einen selektiven Überlebensvorteil. Der Einfluss einer in vivo
Selektion wird insbesondere durch die Gentherapie-Studien bei Patienten mit X-
linked-SCID und chronischer Granulomatose deutlich (CAVAZZANA-CALVO et al.
2000; HACEIN-BEY-ABINA et al. 2002).
Ein selektiver Wachstumsvorteil für genkorrigierte Zellen kann entscheidend für
einen klinisch-therapeutischen Erfolg sein. Für die Mehrzahl von Erkrankungen, die
mittels Gentherapie behandelbar sein könnten, existiert jedoch kein solcher
selektiver Wachstumsvorteil aufgrund der alleinigen Expression des therapeutischen
Transgens.
In vivo Selektion von gentechnisch veränderten Zellen durch einen artifiziell
erzeugten Selektionsvorteil der genkorrigierten Zellen kann in Fällen entscheidend
sein, in denen das Transgen selbst den Zellen keinen Selektionsvorteil bietet. Dies
ist beispielsweise bei der chronischen Granulomatose oder der Thalassaemie der
Fall (PFEIFER u. VERMA 2001). Mit Hilfe eines Selektionsvorteils kann die Anzahl
der transgenen Zellen im Organismus auf eine Zahl gesteigert werden, die
Bedingung für eine phänotypische Heilung des Patienten ist.
Das aussichtsreichste Chemotherapieresistenzgen für eine in vivo Selektion von
hämatopoetischen Stammzellen ist das Protein O6-Methylguanin-DNA-
Methyltransferase (MGMT). MGMT behebt DNA Schäden, die durch alkylierende
oder durch chlorethylierende Agenzien verursacht werden. Die bekanntesten
alkylierenden Substanzen sind Nitrosamine. 1,3-bis-(2-chloroethyl)-1-Nitrosourea
(BCNU), dessen Wirkstoffname Carmustin ist, gehört zu den chlorethylierenden
Substanzen. Einige Chemotherapeutika, die zur Behandlung von onkologischen
Erkrankungen eingesetzt werden, gehören zur Gruppe der alkylierenden
Substanzen; Beisiele hierfür sind Temozolomid und Streptozotozin.
Die aufgeführten Substanzen schädigen die DNA, indem sie Basen spezifisch
modifizieren. Dabei wird unter anderem die Alkylgruppe an der O6 Position des
Guanin methyliert, was wiederum zur Quervernetzungen der DNA führen kann
20

Literatur
(PEGG et al. 1995). Daneben treten noch andere DNA Schäden auf, die jedoch von
MGMT nicht korrigiert werden können; MGMT repariert nur Methylierungen an der O6
Position von Guanin. Die Alkylgruppe wird dabei vom Guanin zu einem Cysteinrest in
das katalytisches Zentrum des Proteins transferiert (BRENT u. REMACK 1988).
Benzylguanin (BG) ist ein Guanin-Analogon. BG kann an das katalytische Zentrum
von MGMT binden und es dadurch irreversibel inaktiviert (DOLAN et al. 1991).
Das Expressionslevel von MGMT ist nicht in allen menschlichen Geweben gleich
hoch. Die Leber hat eine hohe MGMT-Aktivität, die myeloischen Vorläuferzellen
kommen dagegen nur auf 6,6% der Aktivität von Hepatozyten (GERSON et al. 1985).
Auch in anderen Untersuchungen wurde gezeigt, dass hämatopoetische
Vorläuferzellen nur ein geringes MGMT Level haben und dadurch gegenüber einer
Behandlung mit BCNU empfindlich sind (GERSON et al. 1996).
Einige Tumore überexprimieren MGMT und sind so gegenüber alkylierenden
Chemotherapeutika resistent (PREUSS et al. 1996). Benzylguanin wird in einigen
Studien dazu verwendet, die Empfindlichkeit der Tumorzellen gegenüber
Chemotherapeutika zu erhöhen, indem durch BG die Aktivität des Wildtyp MGMT
inhibiert wird (Dolan et al. 1991; Kreklau et al. 1999).
In einer Phase I Studie bei Patienten mit Gliom oder Glioblastom, die nicht
empfindlich gegenüber alkylierenden Substanzen waren, konnte bei einer
Behandlung mit BG und BCNU eine Regression des Tumors festgestellt werden. Die
Behandlungsmöglichkeiten sind jedoch durch die gleichzeitige Myelosuppression
limitiert (QUINN et al. 2002).
Im Vergleich zum Wildtyp MGMT haben MGMT Mutanten, wie P140A und G156A,
eine 40- resp. 240-fach höhere Resistenz gegenüber BG. Diese kommt aufgrund
einer sterischen Hinderung zustande, die durch die Mutation entstanden ist und die
die Bindung von BG verhindert (XU-WELLIVER et al. 1998). Beide Mutanten können
in vitro hämatopoetische Zellen (REESE et al. 1996; MAZE et al. 1999) und im Maus
Modell Knochenmark vor den toxischen Schäden einer kombinierten Behandlung mit
BG und BCNU schützen (DAVIS et al. 1997). Auch die Mutante P140K weist diese
schützende Eigenschaft gegenüber der gleichzeitigen Gabe von BG und BCNU
21

Literatur
sowohl in vitro (DAVIS et al. 1999) als auch im Maus Modell in vivo auf (RAGG et al.
2000). Die Resistenz von P140K gegenüber BG ist etwa 40- bis 1000-fach höher als
die des Wildtyp MGMT (DAVIS et al. 1999).
Zur Expression von MGMT Mutanten wurden bisher ausschließlich gammaretrovirale
(CHINNASAMY et al. 2004) und lentivirale Vektoren (RAGG et al. 2000) eingesetzt.
In neueren in vitro Untersuchungen wurde die Effektivität von lentiviralen- und
gammaretroviralen Vektoren zur Expression von MGMT Mutanten verglichen
(SCHAMBACH et al. 2006). MGMTp140k wurde schon mittels gammaretroviraler
Vektoren erfolgreich in hämatopoetische Stammzellen von Hunden eingebracht, die
dann in vivo selektioniert werden konnten (NEFF et al. 2003).
In einem ersten Experiment mit FV als Gentransfervektor für MGMT wurde der
Vektor MD9.MGMTp140kIEG in einem 3-Plasmid-System verwendet; die Effizienz
wurde mit der eines gammaretroviralen Vektors in murinen CD34+ Zellen verglichen
(POLLOK et al. 2005). Die transduzierten Zellen wurden Mäusen transplantiert. Im
peripheren Blut dieser Tiere wurde anschließend die Anzahl transgener Zellen
gemessen, die bei dem gammaretroviralen Vektor deutlich höher war als bei dem
foamyviralen Vektor. Die transgenen Zellen ließen sich in vivo mit einer kombinierten
Behandlung mit BG und BCNU erfolgreich selektionieren.
22

Literatur
2.5 Interaktionen zwischen Viruspartikeln und Zielzellen
Hämatopoetische Stammzellen stellen ein wichtiges Ziel für genetische
Modifikationen mit retroviralen Vektoren dar. Die erzielten Gentransferraten lagen
jedoch nicht in Bereichen, die für eine Therapie zufrieden stellend sein könnten. Erst
mit Hilfe von Fragmenten der Extrazellulärmatrix des Knochenmarks konnten die
Gentransferraten gesteigert werden.
Fibronektin (FN) kommt in der Mikroumgebung des Knochenmarks vor und ist für die
Interaktion von Zellen untereinander und zwischen Zellen und Matrix von Bedeutung
(YODER u. WILLIAMS 1995). Die Transduktionsrate von humanen
hämatopoetischen Progenitorzellen konnte signifikant gesteigert werden, indem die
Transduktion der retroviralen Vektoren auf Fibronektinfragmenten aus dem
peripheren Blut durchgeführt wurde (MORITZ et al. 1994).
Um den Mechanismus des durch Fibronektin verbesserten Gentransfers beschreiben
zu können, wurden funktionelle Domänen von Fibronektin hinsichtlich ihres
Einflusses auf die Transduktionsrate untersucht. Das Fragment CH-296 hat zwei
Bindungsdomänen für Zellen (CBD und CS1) und eine weitere für Heparin. An die
Heparinbindungsdomäne binden Retroviren, die mit amphotropen Hüllproteinen
pseudotypisiert sind, wahrscheinlich über Proteoglykane an die Oberfläche. Viren
und Zellen werden so in eine räumliche Nähe gebracht und die Gentransfer wird
effektiv gesteigert (HANENBERG et al. 1996).
Gibt man Virusüberstand auf ein mit Fibronektin beschichtetes Zellkulturgefäß und
entfernt anschließend wieder den Überstand, so bleibt ein Teil der Viren am
Fibronektin haften. Gibt man wiederholt Virusüberstand auf das Fibronektin und
nimmt ihn wieder ab, so führt dieses zu einem Anstieg der Zahl gebundener
Viruspartikel am Fibronektin und zu eineme signifikanten Anstig der Gentransferrate
(HANENBERG et al. 1997). Auf diese Weise konnte FN für den Gentransfer in
hämatopoetische Zellen vor allem in Studien an Tieren etabliert werden (VON LAER
et al. 2000; NEFF et al. 2003).
23

Literatur
2.6 Neuweltaffen als Modell für Gentransfer
Viele präklinische Studien wurden initial in Mausmodellen durchgeführt. Mäuse
werden für Studien zur somatische Gentherapie schon seit über 20 Jahren
eingesetzt (DICK et al. 1985). In einigen Versuchen konnte Mäusen mit hoher
Effizienz transduzierte hämatopoetische Stammzellen transplantiert werden
(WILLIAMS et al. 1984; DICK et al. 1985; EGLITIS et al. 1985; WILLIAMS 1990).
Allerdings zeigten folgende Studien am Menschen, dass die Gentherapie unter
gleichen Bedingungen nicht zufrieden stellend verlief (CRYSTAL 1995; ORKIN et al.
1995). Unterschiede in der Hämatopoese beider Spezies sind mutmaßlich für diese
Diskrepanz der Versuchsergebnisse mitverantwortlich. In Mäuse kann mit nur fünf
aktiven Stammzellklonen die Hämatopoese aufrecht erhalten werden (SPANGRUDE
et al. 1988; SPANGRUDE et al. 1995), höhere Tiere wie Rhesusaffen haben
hingegen eine polyklonale Hämatopoese, d.h. sie sind auf wesentlich mehr
verschiede Klone angewiesen, um ihre Hämatopoese aufrecht zu erhalten
(SCHMIDT et al. 2002).
Um die Besonderheiten des Menschen in solchen Studie besser zu berücksichtigen,
wurden Tiere ausgewählt, zu denen ein engeres Verwandtschaftsverhältnis besteht.
Für Gentransferstudien der Hämatopoese wurden bereits verschiedene Affenspezies
wie Paviane (Papio cynocephalus anubis) (KIEM et al. 1997), Rhesusaffen (Macaca
mulatta) (DUNBAR et al. 1996), Javaneraffen (Macaca fascicularis) (HANAZONO et
al. 2002) und Marmosets (Callithrix jacchus) (HIBINO et al. 1999) eingesetzt. Sowohl
in Pavianen, als auch in Makaken konnten Foamyviren nachgewiesen werden; in
Marmosets gelang dieser Nachweis nicht (RETHWILM et al. 2005).
Marmosets, die zur Familie der Krallenaffen gehören, haben darüber hinaus weitere
Vorteile, die sie für Gentransferstudien der Hämatopoese besonders interessant
machen. Mit einem Gewicht von 300 bis 500 g und einer Körpergröße zwischen 18
und 30 cm sind diese Affen im Vergleich zu anderen Affenarten sehr klein, lassen
sich leicht halten und sind unkompliziert im Umgang. Die Lebenserwartung von
Marmosets beträgt etwa 10 Jahre in freier Wildbahn und bis zu 20 Jahre in
menschlicher Obhut, was für eine längerfristige Beobachtung der Hämatopoese nach
24

Literatur
einer Transplantation von Vorteil ist. In anderen Tiermodellen, wie z.B. Mäusen,
wurde erst spät erkannt, dass gammaretrovirale Vektoren Malignome induzieren
können (LI et al. 2002). Diese späte Erkenntnis hing wahrscheinlich mit der geringen
Lebenserwartung dieser Tiere zusammen. Mittlerweile wurde auch in einem
Affenmodel nachgewiesen, dass gammaretrovirale Vektoren Malignome induzieren
können (SEGGEWISS et al. 2006).
Hämatopoetische Zellen von Marmosets sind seit Anfang der 1980er Jahre
immunologisch charakterisiert (CRAWFORD et al. 1981). Eine Vielzahl humaner
Zytokine und Antikörper reagieren mit den hämatopoetischen Zellen von Marmosets
(HIBINO et al. 1999). Anti-humane monoklonale Antikörper lassen sich zur
Phänotypisierung von Leukozytensubpopulationen einsetzen (BROK et al. 2001). Für
die Analyse und Anreicherung CD34+ Zellen von Marmosets ist ein monoklonaler
Antikörper „MA24“ etabliert worden (IZAWA et al. 2004). Die Eignung von Marmosets
für hämatopoetische Gentherapiestudien ist erstmals an Vergleichsuntersuchungen
gezeigt worden, bei denen Knochenmarkzellen von Marmosets und humane
Vorläuferzellen mit retroviralen Vektoren transduziert wurden (HIBINO et al. 2001).
25

Material und Methoden
3. Material und Methoden
3.1 Verwendete Materialien
3.1.1 Plasmide Vektorplasmide
Die in der vorliegenden Untersuchung genutzten retroviralen Plasmiden gehören,
anhand ihres Aufbaus charakterisiert, zu den sogenannten self-inaktivating (SIN)
Vektoren, bei denen der Promotor- und Enhancer-Bereich im 3’ LTR deletiert ist.
Dadurch wird dieser defekte U3- Bereich des LTRs bei der Integration in das Genom
der Zielzelle auf den U3- Bereich der 5’ LTRs geschrieben; in der Folge sind beide
LTRs des Provirus transkriptionell stumm. Für die transiente Virusproduktion tragen
alle Plasmide statt des 5’ LTR den starken intermediate early Promotor des Human
Cytomegalyvirus. Zusätzlich tragen alle Vektoren einen Internen Promotor von dem
aus, der das aus der Qualle Aequorea victoria stammende und für das humane
coding optimierte grün fluoreszierende Protein (GFP) als Markergen für den
Gentransfer exprimiert wurde. Dieser Promotor stammt aus dem Spleen Focus
Forming Virus (SFFV), das zu der Familie der Murine Leukemia Viruses gehört. Die
beiden foamyviralen Vektoren MH71 und MD9 tragen außerdem eine
Expressionskassette, mit der cDNA für die p140k- Mutante des humanen MGMT-
Gens und des GFP, die durch eine IRES-site verbunden sind (Abbildung 2).
Die Markervektoren zur Überprüfung des Gentransfers basieren auf dem HIV, dem
MLV resp. dem PFV. Die Vektoren pCAMSΔU3E (MLV-Vektor) und MH71 (SFV-
Vektor), MD9 (SFV-Vektor) wurden von Herrn Dr. Lindemann und Prof. Rethwilm,
Institut für Virologie, Technischen Universität Dresden, zur Verfügung gestellt.
26

Material und Methoden
ΔU3
polgagCMVRU5
GFPU3
gag*CMV GFPU3envRRE
env
cPPT
RU5* *
ΔU3
MLV-Vektor
pCAMSΔU3E
HIV-Vektor
pCL1
SFFV
polgagCMV
RU5ΔU3GFPU3
ψ ψ
SFFV
SFFV
ψ
ψ
PFV-Vektor
MH71
pol*CMVRU5
ΔU3GFPU3gag*CMV
ψ ψ
SFFVPFV-Vektor
MD9
polgagCMV
RU5
ψ ψΔU3GFPU3 IRESMGMT
SFFVPFV-Vektor
MH71.MGMTp140kIEG
PFV-Vektor
MD9.MGMTp140kIEG pol*CMVRU5
ΔU3U3gag*CMV
ψ ψGFPIRESMGMT
SFFV
ΔU3
polgagCMVRU5
GFPU3
gag*CMV GFPU3envRRE
env
cPPT
RU5* *
ΔU3
MLV-Vektor
pCAMSΔU3E
HIV-Vektor
pCL1
SFFV
polgag
polgagCMV
RU5ΔU3GFPU3
ψ ψ
SFFV
SFFV
ψ
ψ
PFV-Vektor
MH71
pol*CMVCMVRU5
ΔU3GFPU3 ΔU3GFPU3gag*CMV
ψ ψ
SFFVPFV-Vektor
MD9
polgag
polgagCMV
RU5
ψ ψΔU3GFPU3 IRESMGMT
SFFV
ΔU3GFPU3 IRESMGMT
SFFVPFV-Vektor
MH71.MGMTp140kIEG
PFV-Vektor
MD9.MGMTp140kIEG pol*CMVCMVRU5
ΔU3U3gag*CMV
ψ ψGFPIRESMGMT GFPIRESMGMT
SFFV
Abbildung 2: Darstellung der verschiedenen Vektorplasmide Schematische Darstellung der verschiedenen Vektorplasmide. Gag und pol bezeichen kodierende
Sequenzbereiche des viralen Genoms. Bei gag* und pol* sind Teile der viralen Gene deletiert worden,
die nicht für das Verpackungssignal (ψ) kodieren. Der mit Env* bezeichnete Sequenzbereiche des
Hüllproteins wurde zum Teile deletiert. Der CMV-Promotor ersetzt die U3- Region des 5’ LTRs. Dann
folgt die redundanten Region (R), der sich die U5- Region des HIV, MLV oder PFV anschließt. SFFV
U3 bezeichnet den U3 Promotor des SFFV, der hier als internen Promotor für das Gen oder die
Genkassette dient. Das Gen, das durch den SSFV-Promotor angetrieben wird, ist das grün
fluoreszierende Protein (GFP). Bei den beiden unteren Vektoren besteht die Expressionskassette aus
dem MGMTp140k (MGMT) Transgen, der internal ribosome entry site (IRES) und dem GFP. In dem 3’
LTR, der auf das Gen bzw. die Genkassette folgt, sind Teile des ΔU3 der Promotor- und Enhancer-
Sequenz deletiert.
27

Material und Methoden
Der foamyviralen Vektor MD9.MGMTp140IEG (MD9.MGMT) wurde aus dem Vektor
MD9 und dem MGMTp140K Gen generiert und stand wie der Vektor pCL1 (HIV-
Vektor) aus Vorarbeiten zur Verfügung. Der Vektor MH71.MGMTp140kIEG
(MH71.MGMT), der von dem Vektor MH71 abgeleitet ist, wurde in Eigenarbeit
kloniert.
Helferplasmide Für die Produktion rekombinanter replikationsdefekter Retroviren wurde ein Teil der
viralen Gene getrennt vom Vektor zur Generierung des Virus zur Verfügung gestellt.
Diese nicht integrierenden Expressionsplasmide werden als Helferplasmide
bezeichnet. Die viralen Gene der verwendeten Helferplasmide stehen unter Kontrolle
des CMV-Promotors. Das Helferplasmid des HIV-Vektors, CD/NL-BH (MOCHIZUKI
et al. 1998) trägt kodierende Sequenzen für gag und pol, sowie für die Gene vif, vpr,
vpu, rev und tat und wurden von Herrn Dr. Reiser, Louisiana State University School
of Medicine, New Orleans, Louisiana 70112, USA zur Verfügung gestellt.
Bei der Herstellung der foamyviralen Virusüberstände wurden verschiedene
Helferplasmide für die Expression von foamyviralem gag und pol verwendet. Das
Plasmid cmvgp1 exprimiert gag und pol als zwei einzelne gespleißte RNAs
(Abbildung 3). Die anderen Helferplasmide kodieren entweder für gag (pcziGag und
p6iGag) oder für pol (pcziPol und p6iPol) und wurden immer so kombiniert, dass die
gag und pol Proteine gemeinsam in einer Zelle zur Verfügung standen. Sowohl die
pczi als auch die p6i Konstrukte haben in ihrem Backbone die bovine growth hormon
polyA site (bGH pA) (Abbildung 4). Diese DNA Sequenz führt dazu, dass das virale
Genprodukt in der Produzentenzelle polyadenyliert wird und verhindert so, dass
weitere Teile des Backbone abgelesen werden. Zudem tragen beide Vektoren eine
Ampicillinresistenzgen, ein Col El für multicopy Plasmide und den SV40ori, der im
pcziPol Helferplasmid im SV40 Promotor lokalisiert ist. In Zellen, die das large-tumor-
antigen des Simian Virus 40 (SV40) tragen, werden Plasmide mit einem SV40ori
verstärkt exprimiert. Origin of Replikation (ori) stellen DNA Sequenzen da, die als
28

Material und Methoden
Startpunkte der DNA Replikation für unterschiedliche Organismen dienen können.
Der pczi Backbone enthält zusätzlich noch ein Zeocinresistenzgen, einen ori für
Bakteriophagen f1 und ein poly Adenylierungssignal SV40pA (alle Teile des pcDNA3
Plasmids von Invitrogen).
In Abbildung 3 ist der virale Teil der Vektoren schematisch dargestellt (Backbone
nicht gezeigt). Die foamyviralen Helferplasmide wurden vom Herrn Dr. Lindemann
zur Verfügung gestellt.
CMVCMV polgag
pA+RU5
ψ ψ
pA+CMVCMV gag
RU5
ψ
pA+CMVCMVRU5
polψ
PFV-Helferplasmid
cmvgp1
PFV-Helferplasmid
pcziGag oder
p6iGag
PFV-Helferplasmid
pcziPol oder
p6iPol
CMVCMV
polgag
pA+RU5
ψvif
vpr
vpu
tat
envdeletion
revHIV-Helferplasmid
CD/NL-BH
CMVCMV polgag
pA+RU5CMVCMVCMV pol
gagpol
gagpA+RU5
ψ ψ
pA+pA+CMVCMV gag
RU5
ψ
pA+CMVCMVCMVRU5
polψ
PFV-Helferplasmid
cmvgp1
PFV-Helferplasmid
pcziGag oder
p6iGag
PFV-Helferplasmid
pcziPol oder
p6iPol
CMVCMVCMV
polpolgag
pA+pA+RU5
ψvif
vprvif
vpr
vpu
tat
envdeletion
envdeletion
revHIV-Helferplasmid
CD/NL-BH
Abbildung 3: Darstellung des viralen Teils der Helferplasmide Schematische Darstellung des viralen Teils der verwendeten Helferplasmide. Die Expression der
viralen Strukturgene gag und pol, sowie der lentiviralen Gene vif, vpr, vpu, tat, rev und dem deletierten
env des lentiviralen Helferplasmids werden vom CMV-Promotor angetrieben. An den CMV-Promotor
schließt sich die redundante Region (R) und die U5 Region mit den Insertionssequenzen zur
Integration in die Produzentenzelle an. Die viralen Gene werden von dem Polyadenylierungssignal
(pA+) abgeschlossen. Psi (ψ) bezeichnet das Verpackungssignal.
29

Material und Methoden
Abbildung 4: Foamyvirale Helferplasmide pcziPol und p6iPol mit Backbone abgebildet Vom Promotor des Human Cytomegalievirus (CMV-Promotor) aus werden Exon 1 und 2, in deren
Mitte ein Intron liegt, direkt gefolgt vom pol Gen abgelesen. Durch das Spleißereignis wird die mRNA
stabiler und kann besser aus dem Zellkern transportiert werden. Beide Plasmide kodieren für das pol
und ein weitestgehend deletiertes env (Δenv) Gen des Prototyp Foamy Virus, hier bezeichnet als
Human Foamy Virus (HFV). Die viralen Gensequenzen werden von einem bovine growth hormon
polyA site (bGH pA) abgeschlossen. Im Backbone tragen beide Plasmide eine Ampicillinresistenzgen
(Amp), einen Col EL für high copy Plasmide und eine Origin of Replikation des Simian Virus 40
(SV40ori). Das Plasmid pcziPol hat im Backbone zusätzlich noch ein SV40pA Signal, ein
Zeocinresistenzgen und einen ori für Bakteriophagen f1. Durch den größeren Backbone ist das
pcziPol Plasmid mit 10185 Basenpaaren (bp) um 1954 bp größer als das p6iPol Plasmid.
Hüllplasmide / env Plasmide Bei allen verwendeten Hüllplasmiden wird das env Gen durch den CMV-Promotor
kontrolliert. Das Expressionskonstrukt pczVSV-G (VSV-G) kodiert für das
Glykoprotein (G) des Vesikular Stomatitis Virus (VSV). Das pALFGaLVTM (GaLV)
kodiert für die cDNA eines chimären env Gens, das aus der Oberflächendomäne des
Gibbonaffen Leukämie Virus sowie der Transmembran- und der zytoplasmatischen
Domäne des amphotropen MLV Hüllproteins zusammengesetzt ist (STITZ et al.
2000). Dieses env Protein kann von der HIV Protease prozessiert werden; eine
30

Material und Methoden
Vorraussetzung für die erfolgreiche Pseudotypisierung. Das Plasmid wurde von Prof.
Dr. Cichutek, Paul Ehrlich Institut, Langen, zur Verfügung gestellt. Als Hüllplasmide
für die foamyviralen Vektoren sind die Plasmide pczHFVenvEM140 (EM140) und
pciSM04 (SM04) von Herrn Dr. Lindemann verwendet worden. Die Hüllproteine
stammen aus verschiedenen Foamyviren. Das EM140 hat seinen Ursprung im PFV,
das SM04 Hüllprotein stammt aus einem SFV. In beiden Hüllproteinen wurden alle
fünf Ubiquitinierungsstellen in der leader-Region ladungsneutral von Lysin zu Arginin
punktmutiert (STANKE et al. 2005). Dadurch konnte das budding des foamyviralen
Hüllproteins deutlich verbessert werden.
3.1.2 Zellen Adhärent wachsende Zelllinien HEK293T Zellen, die mit dem Adenovirus Type 5 immortalisiert wurden, sind von der
humanen embryonalen Nierenzellenlinie HEK293 abgeleitet. Zusätzlich exprimieren
diese Zellen stabil das large-tumor-antigen des Simian Virus 40 (SV40), wodurch
Plasmide mit einem SV40ori besser abgelesen werden können (DUBRIDGE et al.
1987). HT1080 Zellen sind humane Fibrosarkomzellen, die von der Deutschen
Sammlung für Mikroorganismen und Zellkultur GmbH (DSMZ, D-38124
Braunschweig) bezogen wurden.
Hämatopoetische Zelllinien Die Zelllinien HEL, HL60 und K562 sind humanen Ursprungs. HEL Zellen stammen
von einem Patienten mit einer Erythroleukämie, die K562 Zellen von einem Patienten
mit einer chronischen myeloischen Leukämie und die HL60 Zellen von einem
Patienten mit Blastenschub bei einer akuten promyelozytären Leukämie. Die
hämatopoetischen Zelllinien wurden alle von der DSMZ bezogen.
31

Material und Methoden
Primäre Zellen Humanes Nabelschnurblut für die Gewinnung von CD34+ Zellen wurde von Prof.
Kögler, Institut für Transplantationsdiagnostik und Zelltherapeutika des
Universitätsklinikum Düsseldorf, zur Verfügung gestellt. Das Material stammt aus
Nabelschnurblutspenden an die Nabelschnurblutbank, die jedoch aufgrund einer
unzureichenden Zellzahl nicht zur Transplantationszwecken kryokonserviert, sondern
zu Forschungszwecken freigegeben wurden.
Marmoset Knochenmark zur Gewinnung von CD34+ Zellen wurde Tieren entnommen,
die in der Tierversuchsanlage des Universitätsklinikums Düsseldorf gehalten werden.
Für die Knochenmarkentnahme wurden die Tiere narkotisiert.
Zellkulturmedien und Zusätze Die adhärenten Zelllinien wurden in Dulbecco`s Modified Eagle`s Medium (Gibco®
DMEM) mit 4,5 g/l Glukose kultiviert. Die Kultivierung der hämatopoetischen
Zelllinien erfolgt in Roswell Park Memorial Institute (RPMI). Beide Medien wurden
von Invitrogen bezogen. Zur Kultivierung der primären Zellen wurde Iscove`s
Modified Dulbecco`s Medium (IMDM; Sigma-Aldrich Chemie GmbH, D-82024
Taufkirchen) eingesetzt.
Alle Medien wurden vor ihrer Verwendung mit 10 % fötalem Kälberserum (FKS, PAN
Biotech GmbH, D-94501 Aidenbach), 2 mM Glutamin (Gibco® Glutamin, Invitrogen), 100 U/ml Penicillin und 100 µg/ml Streptomycin (Gibco® Penicillin resp. Streptomycin,
Invitrogen) angereichert.
3.1.3 Tiere Marmosets wurden für die oben genannten Versuchszwecke in der zentralen
Tierversuchsanlage der Universitätsklinik Düsseldorf gehalten. Die
Tierversuchsanlage betreibt seit mehr als 10 Jahren eine eigene Marmoset Zucht.
Alle verwendeten Tiere sind in Düsseldorf geboren und aufgewachsen. Die Tiere
32

Material und Methoden
werden dort paarweise gehalten und nur während experimenteller Studien von ihren
Artgenossen getrennt.
Das Versuchsvorhaben „Gentransfer in hämatopoetische Stammzellen von
Marmosets“ ist durch die Bezirksregierung Düsseldorf genehmigt (Aktenzeichen
50.05-230-43/04).
3.2 Methoden
3.2.1 Molekularbiologische Techniken
3.2.1.1 Transformation von Bakterien und Isolierung von Plasmid DNA
Für die Vermehrung der Plasmide wurden verschiedene, Rekombinase-negative
Escherichia (E.) coli Stämme verwendet. Die Plasmide MH71,
MH71.MGMTp140kIEG und pczHFVenvEM140 wurden im E. coli Stamm JM109
(Stratagene, La Jolla, CA 92037, USA) vermehrt. Alle anderen Plasmide wurden in E.
coli One-Shot® TOP10 (Invitrogen) vermehrt. Dazu wurden jeweils 100 µl (d.h. eine
Verpackungseinheit) frisch aufgetaute, transformationskompetente E. coli mit 0,1 µg
Plasmid-DNA für 20 Minuten (min) auf Eis inkubiert. Anschließen wurden die
Reaktionsansätze für 45 Sekunden (sec) in einem Thermoblock auf eine Temperatur
von 42 °C erwärmt um dann wieder für 5 min auf Eis abgekühlt zu werden. Zu den
Reaktionsansätzen wurden 500 µl Luria-Bertani-Medium (LB-Medium; Sigma-
Aldrich) pipettiert. Die Kulturen wurden bei 37 °C für 45 min und 180 Umdrehungen
pro Minute (rpm) auf einem Thermomixer inkubiert. Zur Isolierung einzelner Kolonien
wurden Ausstriche auf Agarplatten (LB-Agar, Sigma-Aldrich) angelegt, denen 100
µg/ml Ampicillin zugesetzt wurde (Ampicillin; Sigma Aldrich). Die Platten wurden über
Nacht bei 37 °C inkubiert. Am folgenden Morgen wurden 5 ml LB-Medium, das
ebenfalls mit 100 µg/ml Ampicillin angereichert war, mit einer Bakterienkolonie
inokuliert und über Tag bei 37 °C und 180 rpm inkubiert. Am gleichen Abend wurden
33

Material und Methoden
5 ml Kulturmedium in 250 ml frisches LB- Medium überführt und über Nacht bei
37 °C und 180 rpm inkubiert. Die Übernachtkultur (Maxi) wurde für 10 min bei 4000 g
zentrifugiert; der Überstand wurde verworfen. Die Aufreinigung der Plasmid DNA für
die Transfektion wurde mit dem PureLink™ HiPure Plasmid Kit (Invitrogen) nach
Anweisung des Herstellers durchgeführt. Die, in Tris-EDTA-Puffer (im Kit enthalten)
aufgenommene Plasmid-DNA wurde spektralphotometrisch mit einem Photometer
(Bio Photometer; Eppendorf AG, D-22339 Hamburg) quantifiziert. Die verschiedenen
Plasmid-Präparationen wurden mittels eines analytischen Restriktionsverdaus (alle in
dieser Arbeit verwendeten Enzyme stammten von New England Biolaps, D-65926
Frankfurt) und anschließender Gelelektrophorese (Biozym LE Agarose, Biozym, D-
31833 Hessisch Oldendorf) kontrolliert.
3.2.1.2 Klonierung des Plasmids MH71.MGMTp140kIEG
Aus dem Plasmid MD9.MGMTp140kIEG wurde das MGMT-Gen mit der
Punktmutation p140k mit Hilfe der Enzyme Asc I und Rsr II herausgeschnitten und in
den Vektor MH71 ligiert. Insgesamt 2 µg des Vektors MH71 und 3 µg des Vektors
MD9.MGMTp140kIEG, aus dem das Insert MGMT herausgeschnitten wurde, sind mit
den Enzymen AscI und RsrII zwei Stunden bei 37 °C verdaut worden. Der Vektor
(MH71) wurde nach dem Verdau eine Stunde bei 37 °C mit einer alkalischen
Phosphatase (CIP, New England Biolabs) dephosphoryliert. In einer anschließenden
Gelelektrophorese mit einem 0,4 %-igen Agarosegel (Biozym LE Agarose) wurden
die einzelnen DNA-Fragmente aufgetrennt. Die spezifische DNA für den Vektor
(MH71) und das Insert (MGMT) wurden aus dem Gel aufgereinigt. Die Extraktion der
DNA aus dem Gel erfolgte mit dem QIAquick® Gel Extraction Kit (Qiagen GmbH, D-
40724 Hilden) nach Anweisung des Herstellers. Im Anschluss an die Extraktion
wurde die DNA Konzentration photometrisch bestimmt. Vektor und Insert wurden im
Verhältnis 1:1 gemischt und anschließend unter Verwendung einer T4-Ligase und
eines Ligase-Puffers (beides New England Biolabs) für 10 min bei Raumtemperatur
ligiert. Die Hälfte des Reaktionsansatzes wurde in frisch aufgetaute E. coli One-Shot®
34

Material und Methoden
TOP 10 transformiert. Nach der Transformation wurden 500 µl LB-Medium zu dem
Ansatz pipettiert und die Kultur bei 37 °C und 180 rpm für 45 min inkubiert. Ein
Ausstrich zur Isolierung einzelner Klone wurde auf LB-Ampicillin-Platten angelegt.
Die Platten wurden über Nacht bei 37 °C inkubiert. Am nächsten Tag wurden 3 ml
LB-Medium, dem ebenfalls Ampicillin zugesetzt war, mit jeweils einer
Bakterienkolonie inokuliert und über Nacht inkubiert. Die Plasmid DNA wurde aus
den Bakterien mittels dem Fast Plasmid Mini Kit (Eppendorf) nach Anweisung des
Herstellers aufgereinigt. Die gewonnene Plasmid DNA wurde mit dem Enzym BSRG
I verdaut, um die Richtigkeit des neuen Plasmids zu überprüfen. Das neue Plasmid
MH71.MGMTp140kIEG wurde zur Vermehrung in den E. coli Stamm JM109
transformiert.
3.2.2 Zellkulturtechniken
3.2.2.1 Kultivierung eukaryotischer Zelllinien
Alle Zelllinien wurden in einem Inkubator bei 37 °C in feuchtigkeitsgesättigter
Atmosphäre mit 5 % CO2 in Zellkulturgefäßen (Corning® Costar®, Corning B.V. Life
Sciences, NE-1119 Schipol-Rilk) kultiviert. Abhängig von der Proliferationsrate
wurden die adhärenten Zellen zwei bis dreimal pro Woche passagiert. Dazu wurde
das Medium entfernt, die Zellen mit Ca2+/Mg2+ -freier, Phosphat gepufferter Saline
(PBS, Dulbecco`s (1x), PAA Laboratories GmbH, D- 35091 Cölbe) gewaschen und
für 5 min mit einer Trypsin / EDTA-Lösung (0,05 % Trypsin / 0,02 % EDTA-Lösung in
Dulbecco`s PBS, PAA) im Brutschrank inkubiert. Die abgelösten Zellen wurden in
Medium resuspendiert und ein Fünftel der Zellen wurde in neue Zellkulturgefäße
überführt. Um die hämatopoetischen Zellen zu passagieren, wurden diese
gleichmäßig im Medium verteilt und 8/10 bis 9/10 des Zellmedium-Überstandes
verworfen. Das Volumen des verworfenen Mediums wurde durch frisches Medium
ersetzt. Die Zellzahlbestimmung erfolgte in einer Neubauer-Zählkammer. Die Zellen
wurden für diesen Vorgang mit Trypanblau (Sigma-Aldrich) gefärbt.
35

Material und Methoden
3.2.2.2 Kryokonservierung eukaryotischer Zellen
Zur langfristigen Aufbewahrung wurden die Zellen in flüssigem Stickstoff tiefgefroren.
Die Suspensionszellen und die adhärenten Zellen, die mit Trypsin abgelöst und in
Medium resuspendiert worden sind, wurden dafür bei 400 g, 5 min und
Raumtemperatur (RT) zentrifugiert. Der Überstand wurde entfernt und die Zellen (1 x
10 6 bis 3 x 106) wurden in Medium resuspendiert, dem 6% Dimethylsulphoxid
(DMSO, Sigma-Aldrich) zugesetzt war. Anschließend wurde die Zellsuspension in
Kryoröhrchen (Corning) überführt. Die Röhrchen wurden in Kryocontainern (Nalgene,
Nalge Nunc International, Rochester, NY 14625, USA) platziert, deren
Zwischenboden mit 2-Propanol (Merck KGaA, D-64271 Darmstadt) befüllt war. Diese
Container wurden bei -80 °C eingefroren. Am darauf folgenden Tag wurde der
Container mit Inhalt in den Stickstofftank überführt.
Zur Rekultivierung wurden die Röhrchen aus dem Stickstofftank entnommen und in
einem Wasserbad bei 37 °C aufgetaut. Die Zellen wurden dafür bei 400 g, 5 min und
RT zentrifugiert und das DMSO-haltige Medium anschließend verworfen. Die Zellen
wurden in frisches Medium aufgenommen und in eine Zellkultur-Flasche oder Platte
überführt.
3.2.2.3 Mykoplasmentest
Alle Zelllinien wurden vor der Verwendung auf eine mögliche Kontamination mit
Mykoplasmen untersucht. Diese Untersuchung wurde alle drei bis vier Monate
wiederholt. Der eingesetzte Mykoplasmen-ELISA (Mycoplasma Detection Kit, Roche
Diagnostics GmbH, D-82372 Penzberg) detektiert Kontaminationen mit folgenden
Mycoplasma species: Mycoplasma (M.) arginini, M. hyorhinis, M. orale und
Archeoplasma laidlawii. Die Nachweisgrenze wird vom Hersteller mit 106–107 colony
forming units (CFU)/ml für M. arginini und M. hyorhinis, mit 105-106 CFU/ml für M.
orale und mit 104-105 CFU/ml für Archeoplasma laidlawii angegeben.
36

Material und Methoden
3.2.2.4 Gewinnung primärer Zellen
Humanes Nabelschnurblut wurde mit 22 g/l Natriumcitrat, 28 g/l Glukose und 8 g/l
Zitronensäure (KAPLAN et al.) versetzt zur Verfügung gestellt. In den Proben waren
zwischen 50 und 70 ml Nabelschnurblut inklusive 10-20 ml ACD enthalten. Das Blut
wurde im Verhältnis 1:1 mit PBS verdünnt. Ficoll-Paque™ Plus (Amersham
Biosciences AB, SE-75184 Uppsala) wurde im Verhältnis 1:1 mit dem verdünnten
Nabelschnurblut überschichtet und mit dem Phasengemisch anschließend eine
Dichtegradientenzentrifugation (400 g, 25 min, RT) durchgeführt. Der „weiße
Ring“ aus mononukleären Zellen wurde abgenommen und die Erythrozyten mit
Ammoniumchlorid-Lösung (155 mM NH4Cl, 10 mM KHCO3, 0,5 mM EDTA, sterilisiert,
Zentralapotheke des Universitätsklinikum Düsseldorf) lysiert. Die Zellen wurden
abschließend noch zweimal mit PBS gewaschen. Aus diesem Zellgemisch wurden
die CD34+ Zellen mittels magnetischer Zellseparation (MACS®; Miltenyi Biotec GmbH,
D-51429 Bergisch Gladbach) sortiert. Zunächst wurde das Oberflächenantigen CD34,
mit CD34 Antikörpern, die an Phycoerythrin (PE) gekoppelt sind, markiert (CD34-PE,
Klon 8G12, BD Biosciences, Tullastasse 8-12, D-69126 Heidelberg), indem die
Zellen mit dem Antikörper 12 min bei Dunkelheit inkubiert wurden. Die
überschüssigen Antikörper wurden entfernt, indem die Zellen einmal mit PBS
gewaschen wurden. In einem zweiten Schritt wurde ein Antikörper gegen PE, der an
ein paramagnetisches Eisenkügelchen (micro beads) gekoppelt ist (Anti-PE Micro
Beads, Miltenyi), nach den Angaben des Herstellers auf die Zellen gebracht. Die mit
dem Kügelchen-Antikörper-Konjugat markierten Zellen ließen sich in einem starken
Magnetfeld anreichern. Die Zellen wurden über eine Metallmatrix-Säule (MACS®
Separation Columns, 25MS Columns, Miltenyi) gegeben, die zuvor in einen starken
Permanentmagneten eingespannt worden war. Die Aufreinigung der Zellen mit der
Metallmatrix-Säule wurde nach der Anleitung des Herstellers durchgeführt. Um die
Reinheit der CD34+ Zellen zu erhöhen, wurde die Positivfraktion ein weiteres Mal
über eine zweite MACS-Säule selektioniert. Die gewonnenen CD34+ Zellen wurden
entweder sofort verwendet oder kryokonserviert.
37

Material und Methoden
Das gewonnene Knochenmark von Marmosets wurde zunächst über einen cell
strainer 70 µm (BD Falkon™, Tullastasse 8-12, D-69126 Heidelberg) gegeben, der
anschließend intensiv mit PBS gespült wurde. Zur Isolierung der mononukleären
Zellen wurde mit dem Knochenmark eine Dichtegradientenzentrifugation
durchgeführt. Aus dieser Fraktion wurden die CD34+ Zellen mittels magnetischer
Zellseparation sortiert. Die Markierung der CD34+ Zellen erfolgte mit einem
biotinylierten CD34 Antikörper MA-24 (IZAWA et al. 2004), der von Prof. Dr. Tani,
Medical Institute of Bioregulation, Kyushu University, Fukuoka, Japan, zur Verfügung
gestellt worden war. In einem zweiten Schritt wurden microbeads, an die Streptavidin
gekoppelt war (Streptavidin Micro Beads, Miltenyi), nach Angaben des Herstellers
auf die Zellen gebracht. Die nun markierten Zellen wurden, wie bereits beschrieben,
über eine Metallmatrix-Säule aufgereinigt. Die Aufreinigung der CD34+ Zellen von
Marmosets erfolgte ebenfalls über zwei Säulen. Zur Bestimmung der Reinheit der
isolierten Zellen im Durchflusszytometer wurden diese mit Antikörperkonjugat SAv-
PE (BD Biosciences) entsprechend den Angaben des Herstellers gefärbt.
3.2.3 Virusproduktion
3.2.3.1 Transiente Erzeugung virushaltiger Überstände
Bei der transienten Virusproduktion wurden alle, für die Viruspartikelbildung
benötigten Gene durch Kotransfektion mit mehreren Plasmiden in eine Zelle
eingebracht. Für alle Versuche wurden HEK293T Zellen zur Virusherstellung
verwendet. Es wurden zwei unterschiedliche Protokolle zur Transfektion von
HEK293T Zellen eingesetzt, die beide von Herrn Dr. Lindemann etabliert wurden und
Polyethylenimin (PEI, hochmolekular, Sigma-Aldrich) verwenden. PEI ist ein
kationisches Polymer, das Nukleinsäure bindet und darüber hinaus kondensierende
Eigenschaften besitzt. DNA-Polykationen-Komplexe werden effizient von
verschiedenen Zellarten aufgenommen und Polykationen selbst wirken einer
intrazellulären Degradierung der DNA entgegen.
38

Material und Methoden
Alle adhärent wachsenden Zellen wurden auf Zellkuturplatten, die für
Gewebekulturen vorbehandelt waren (tissue culture dish), kultiviert. Die Platten
wurden darüber hinaus mit 0,1 % Gelatinelösung (1 g Gelatine auf 1 l doppelt
desiliertes Wasser; Gelatine vom Schwein, Typ A, Sigma-Aldrich) beschichtet. Die
Gelatinelösung wurde für mindestens 30 min und maximal 3 Tage auf der
Zellkulturplatte belassen und im Brutschrank inkubiert. Bevor die Zellen ausgesät
wurden, ist die Gelatinelösung von der Platte abgenommen worden.
.Am ersten Tag der Transfektion wurden 6 x 106 HEK293T Zellen in 10 ml Medium
auf einer 10 cm Zellkulturschale ausgesät.
Am zweiten Tag wurde nach dem zuerst angewandten PEI-Protokoll (Protokoll „ohne
Serum“) vor der Transfektion das Medium, der am Vortag ausgesäten Zellen,
gewechselt. Das Medium vom Vortag wurde verworfen und durch 5 ml DMEM ohne
Serum ersetzt. Das Transfektionsgemisch, das für die beiden verwendeten
Protokolle identisch war, wurde in zwei Kryoröhrchen angesetzt. Im ersten Röhrchen
wurde die PEI-Lösung aus 45 µl PEI (1 mg/ml) und 955 µl DMEM ohne Zusätze
hergestellt und anschließend gut gemischt. Im zweiten Röhrchen wurde die DNA-
Lösung mit insgesamt 15 µg Gesamtmenge DNA (d.h. bei einem 3-Plasmidsystem 5
µg von jedem Plasmid, bei einem 2 Plasmidsystem 7,5 µg von jedem Plasmid) und
985 µl DMEM ohne Zusätze angesetzt. Die Protokolle wurden für die 4-Plasmid
Systeme leicht modifiziert, indem 7 µg DNA von jedem Plasmid eingesetzt wurden
(28 µg total DNA) und die eingesetzte PEI Menge (1 mg/ml) von 45 µl auf 84 µl
erhöht wurde. Anschließend wurde die PEI-Lösung zügig zur DNA-Lösung hinzu
pipettiert und gut gemischt. Das PEI-DNA-Gemisch wurde bei RT für 15 bis 30 min
inkubiert. Bei dem Protokoll „ohne Serum“ wurde wiederum das Medium von den
Zellen entfernt und durch die PEI-DNA-Transfektionslösung ersetzt, der zuvor noch 4
ml DMEM ohne Serum zugesetzt worden war. Nach fünf Stunden wurde das
Transfektionsgemisch abgenommen und durch DMEM mit Zusätzen ersetzt. Bei dem
später angewendeten, zweiten Protokoll „mit Serum“ wurde das Medium von den
Zellen entfernt und durch 4 ml DMEM inkl. der Zusätze in der üblichen Konzentration
und 15 % FKS ersetzt. In dieses Medium wurde dann das Transfektionsgemisch
pipettiert.
39

Material und Methoden
Am dritten Tag wurden nach dem Protokoll „ohne Serum“ 200 µl einer 500 mM
Natriumbutyratlösung (Buttersäure Natriumsalz, Merck) in PBS gelöst und den Zellen
zu pipettiert. Das Medium mit Natriumbutyrat wurde nach sechs Stunden von den
Zellen abgenommen und durch 5 ml frisches DMEM ersetzt. Nach dem Protokoll „mit
Serum“ wurde das alte Medium vom Vortag entfernt und die Zellen einmal mit
Medium gewaschen, bevor 5 ml frisches DMEM auf die Zellen gegeben wurde. Von
der Natriumbutyratlösung wurden dann 200 µl zu den Zellen pipettiert. Auch hier
wurde nach sechs Stunden das natriumbutyrathaltige Medium von den HEK293T
Zellen abgenommen und durch 5 ml DMEM ersetzt.
Am vierten Tag, frühestens aber 19 Stunden nach dem letzten Mediumwechsel,
wurde der virushaltige Überstand geerntet. Die Virusernte war für beide
Transfektionsprotokolle gleich. Der virushaltige Überstand wurde in einer Spritze
aufgezogen und dann mittels eines Spritzenfilters (0,45 µm, Sartorius AG, D-37075
Göttingen) filtriert. Durch das Filtrieren wurden die Produzentenzellen und deren
Überreste aus dem virushaltigen Überstand entfernt. Das virushaltige Medium wurde
entweder frisch verwendet oder bis zum Gebrauch bei -80 °C gelagert.
3.2.3.2 Konzentration von virushaltigen Überständen
Virushaltige Überstände mehreren Platten des gleichen Virus (i.d.R. von 8 Platten)
wurden in einem 50 ml Polypropylenröhrchen (BD Falkon™, BD Biosciences)
gepoolt. Das Röhrchen wurde bei 4 °C für zwei Stunden und 10.000 rpm zentrifugiert
(Zentrifuge: J2-21; Rotor JA-12; Beckmann-Coulter GmbH, D-47807 Krefeld).
Anschließend wurde der Überstand abgesaugt und das nicht sichtbare Virus-
Sediment wurde in 1 bis 3 ml frischem Medium resuspendiert. Das Medium wurde
entsprechend den Bedürfnissen der Zellen, für die das Virus bestimmt war,
ausgewählt. Die Viruspräparationen wurden zumeist frisch verwendet.
40

Material und Methoden
3.2.4 Titration
Um eine Aussage über die Menge der infektiösen Viruspartikel pro Volumeneinheit
im produzierten Überstand treffen zu können, wurde der Virustiter im frischem,
virushaltigen Überstand ermittelt. Am Vortag der Titration wurden auf einer 6-
Wellplatte 35.000 HT1080-Zellen pro Well ausgesät. Die Zielzellen wurden mit einer
zunehmenden Verdünnung der Überstände von 10-1 bis 10-6 transduziert. Die
Verdünnungen wurden in 1,5 ml DMEM angesetzt und anschließend wurde davon 1
ml auf jedes Well pipettiert. Bei Viren, die mit VSV-G verpackt worden sind, wurden
10 µg/ml Protaminsulfat (Sigma-Aldrich) zum Virusüberstand zugegeben. Frühestens
16 Stunden nach der Transduktion erfolgte ein Mediumwechsel, bei dem das
Transduktionsmedium gegen 2 ml frisches DMEM pro Well getauscht wurde. An Tag
vier nach der Transduktion wurden die HT1080-Zellen abgelöst und in PBS, dem 4 %
Paraformaldehyd (Merck) zugesetzt worden sind, aufgenommen. Der Titer wurde
funktionell über die Expression des GFP Markergens bestimmt. Die Analyse des
Anteils an GFP+ Zellen an der Gesamtpopulation wurde durchflusszytometrisch
bestimmt. Unter der Annahme, dass sich die transduzierten Zellen genauso häufig
teilen wie die nicht transduzierten Zellen, kann mit Hilfe der folgenden Formel
bestimmt werden, wie viele ‚transduzierende Einheiten’ (TE) zum Zeitpunkt der
Transfektion im Überstand vorhanden waren.
41

Material und Methoden
0,01
0,1
1
10
100
-1 -2 -3 -4 -5 -6
Verdünnungsstufen (log10)
GFP
pos
itive
Zel
len
(%)
Abbildung 5: Verdünnungsreihe des Virustiter MH71 EM140
Die Virustiter, die als Ergebnis der einzelnen Versuche dargestellt werden, wurden
als arithmetische Mittelwerte aus jeweils 2 Einzelwerten die im linearen Bereich der
½ logarithmischen Kurve liegen ermittelt (Abbildung 5). In jeder Verdünnungsreihe
wurden 6 Verdünnungstufen angesetzt. In der Abbildung 5 sind die
Verdünnungsstufen 1:10, 1:100, 1:1.000, 1:10.000, 1:100.000 und 1:1.000.000
dargestellt. Vergleiche zwischen Mittelwerten werden nur innerhalb einer
Versuchsreihe durchgeführt, da geringgradige Schwankungen in der Virusproduktion
allein durch die Passage der Produzentenzellen für einen weiteren Versuchsansatz
bedingt sein können.
3.2.5 Transduktion von hämatopoetischen Zelllinien
6-Well Zellkulturplatten (BD Falcon), die nicht für Gewebekulturen vorbehandelt
waren, wurden mit dem rekombinanten Fibronektinfragment CH296 (Takara Bio Inc.,
Otsu, Shiga 520-2193, Japan) beschichtet. Die Platten wurden mit 4 µg CH296 pro
42

Material und Methoden
cm2 entweder für 2 Stunden im Inkubator oder über Nacht im Kühlschrank bei 4 °C
beschichtet. Vor der Verwendung wurde das Fibronektin von der Platte abgenommen
und die Platte einmal mit Medium gewaschen. Es wurden 40 ml Virusüberstand, der
von insgesamt 8 Platten stammte, zentrifugiert, das Sediment in 3 ml Medium
aufgenommen und anschließend auf die Wells verteilt. Die so präparierten Platten
wurden in den Inkubator gestellt. Nach einer halben Stunde wurden 100.000 Zellen
der Zelllinien K562, HL60 und HEL, die in 50 bis 100 µl Medium aufgenommen
wurden, zum Viruszentrifugat pipettiert. Nach etwa 16 Stunden (über Nacht) wurde
frisches Medium auf die Zellen gegeben. Am vierten Tag nach der Transduktion
wurden die Zellen von den Fibronektinplatten genommen und zur weiteren
Kultivierung in Zellkulturflaschen überführt. Ein kleiner Teil der Zellen wurde für die
fluorescence activated cell sorting (FACS)-Analyse in FACS-Röhrchen (BD Falcon)
überführt. Die Zellen wurden einmal mit PBS gewaschen und dann in 500 µl einer
Propidiumjodid- (PI, Sigma-Aldrich) FACS-Lösung (PBS mit 1 % FKS und 1 µg PI/ml)
aufgenommen. Die Transduktionseffizienz wurde ermittelt, indem der Anteil GFP
positiver Zellen mittels FACS-Analyse gemessen wurde.
Ein Teil der Zellen wurde weiter kultiviert und der Anteil GFP positiver Zellen wurde
wöchentlich durchflusszytomerisch ermittelt.
3.2.5.1 Transduktion von humanen CD34+ Zellen
Die Transduktion der humanen CD34+ Zellen wurden auf 6-Wellplatten durchgeführt,
die mit Fibronektin beschichtet worden sind. Zur Transduktion wurden zwei
verschiedene Protokolle angewendet, die sich in der Grundstruktur nur minimal
unterscheiden.
Beim ersten Protokoll wurde das Fibronektin zweimal mit jeweils 1 ml Virusüberstand
vorgeladen. Alle verwendeten Virusüberstände waren zuvor kryokonserviert. Direkt
nach der Aufreinigung aus dem Nabelschnurblut wurden 100.000 CD34+ Zellen zum
Virusüberstand pipettiert. Zusätzlich wurden noch die Zytokine SCF, TPO und G-
CSF (Amgen GmbH, D-80992 München) in einer Konzentration von 100 ng/ml zu
43

Material und Methoden
den Ansätzen hinzugegeben. Frühestens 16 Stunden (über Nacht) nach der
Transduktion wurden die Zellen von der Platte abgenommen und in frisches Medium,
wie oben beschrieben, überführt. Vier Tage nach der Transduktion wurden die Zellen
in FACS-Röhrchen überführt und einmal mit PBS gewaschen. Am Tag 4 nach der
Transduktion wurde mittels eines Durchflusszytometers die Anzahl der
transduzierten Zellen bestimmt.
Im zweiten Protokoll wurde frischer Virusüberstand verwendet. Dazu wurden die
Viruskonstrukte von drei Platten gepoolt und zentrifugiert. Das Sediment wurde in 1
ml Medium aufgenommen. Der Virusüberstand wurde eine halbe Stunde vor Zugabe
der Zellen auf die Wells verteilt und die Platten in den Inkubator gestellt. Auch hier
wurden 100.000 CD34+ Zellen nach der Aufreinigung zum Virusüberstand pipettiert.
In einer Konzentration von 100 ng/ml wurden die Zytokine SCF, TPO und G-CSF
(Amgen) den Ansätzen hinzugegeben. Die Zellen wurden ca. 16 Stunden (über
Nacht) nach der Transduktion von der Platte abgenommen und in frisches Medium,
wie oben beschrieben, überführt. Vier Tage nach der Transduktion wurden die Zellen
in FACS-Röhrchen überführt und einmal mit PBS gewaschen. Der Anteil GFP
positiver Zellen wurde durchflusszytometrisch bestimmt.
3.2.5.2 Transduktion von Marmoset CD34+ Zellen
Die Transduktion der CD34+ Zellen von Marmosets erfolgte auf Fibronektin
beschichteten 6-Well Platten. Bei allen Transduktionen von Marmoset CD34+ Zellen
wurde der Virusüberstand von 8 Platten gepoolt, zentrifugiert und in 1 ml Medium
aufgenommen. Die Virusüberstände wurden eine halbe Stunde vor den Zellen auf
die Platten gegeben und in den Inkubator gestellt. Es wurden bei den verschiedenen
Tieren unterschiedlich viele Zellen zur Transduktion eingesetzt. Bei Tier 0224 wurden
60.000 Zellen pro Well ausgesät, bei Tier 3101 40.000 Zellen und bei Tier 0130
wurden 10.000 Zellen zur Transduktion eingesetzt. In allen drei Versuchen wurden
die Zytokine SCF, TPO (Amgen), IL-6 und Flt3-L (Pepro Tech EC Ltd, 29 Margravine
Road, London, UK) in einer Konzentration von 100 ng/ml eingesetzt. Frühestens 16
44

Material und Methoden
Stunden nach der Transduktion wurden die Zellen von der Platte abgenommen und
in frisches Medium überführt, dem wie oben beschrieben, Zytokine zugesetzt wurden.
Die Zellen der ersten beiden Tiere wurden nach 5 Tagen in FACS-Röhrchen
überführt und mit PBS gewaschen. Anschließend wurde der Anteil GFP positiver
Zellen durchflusszytometrisch bestimmt. Bei Tier 3101 wurden nicht alle Zellen für
die FACS-Analyse verwendet, da ein Teil für weitere Untersuchungen kultiviert wurde.
Diese Zellen wurden nach 9 Tagen auf den Anteil an GPF positiver Zellen untersucht.
Die transduzierten Zellen des dritten Tieres 0130 wurden nach 4 Tagen auf den
Anteil der GFP positiver Zellen untersucht.
3.2.6 Progenitor-Assay
Der Progenitor-Assay erlaubt die Detektion klonaler Zellen innerhalb der Fraktion der
CD34+ Zellen. Die Zellen wurden dazu vereinzelt und in niedriger Konzentration in
einem viskösen Medium ausgesät, das Zellbewegungen weitestgehend verhindert.
Reife hämatopoetische Zellen sterben schon nach kurzer Zeit in der Kultur ab. Nach
14 Tagen erfolgte die Auszählung der Kolonien der klonogenen Vorläuferzellen unter
dem Mikroskop. Die CD34+ Zellen wurden am Tag nach der Transduktion
entnommen. Ausschließlich wurden die Zellen in Medium (Metho Cult®, GF H4434,
Stem Cell Technologies Inc, Vancouver, BC (604) 877-0713) ausgesät, in dem
sowohl Zytokine als auch alle anderen Substanzen, die die Zellen für das Wachstum
brauchen, vorhanden waren. Es wurden jeweils zwei Zellkonzentrationen ausgesät:
250 und 500 Zellen pro ml Metho Cult, (im Versuch mit Tier 0130 wurden 200 und
800 Zellen pro ml ausgesät). Jeder Versuch wurde dreifach angesetzt. Von dem
Metho Cult Medium mit den Zellen wurden jeweils 1 ml in Zellkulturschalen mit einem
Durchmesser von 3,5 cm gegeben, die am Boden ein Rastergitter (Nunc, Nalge
Nunc) aufwiesen. Dieses Raster hilft bei der genauen Quantifizierung der Anzahl der
Kolonien pro Platte. Jeweils 6 dieser kleinen Platten wurden in eine große
Zellkulturschale mit einem Durchmesser von 15 cm gestellt. In der Mitte der Schale
wurde eine kleine Platte, die mit PBS gefüllt wurde und keinen Deckel trug, plaziert.
45

Material und Methoden
Die Schalen wurden 14 Tage in einen separaten Brutschrank, der in den 14 Tagen
nur selten geöffnet wurde, inkubiert. Die anschließende Auszählung der Kolonien
erfolgte mit einem Fluoreszenzmikroskop (Axiovert 25, Carl Zeiss Microlmaging
GmbH, D-37081 Göttingen). Bestimmt wurde der Anteil GFP positiver Kolonien an
der Gesamtzahl der gewachsenen Kolonien. Eine Ansammlung von mindestens 50
myeloische oder 200 erythrozytäre Vorläuferzellen wurden als eine Kolonie
angesprochen.
3.2.7 Virusbindung an Fibronektin
12-Wellplatten, die nicht für Gewebekultur vorbehandelt waren (non tissue culture
dish), wurden entweder mit 4 µg/cm2 Fibronektin oder mit 0,5 ml einer PBS-Lösung
mit 2 % Bovinem Serumalbumin (BSA, Sigma-Aldrichs, gelöst in PBS) beschichtet.
Die Platten wurden über Nacht bei 4° C im Kühlschrank inkubiert. Vor der
Verwendung wurde das Fibronektin resp. BSA von der Platte abgenommen und die
Platten einmal mit Medium gewaschen. Die so beschichteten Platten wurden im
Versuch mit einer, für Gewebekultur konventionell vorbehandelten 12-Wellplatte
(tissue culture dish, TCD) verglichen. Kryokonservierte Virusüberstande, die jeweils
1:10 und 1:100 mit Medium verdünnt worden waren, wurde mit einem Volumen von 1
ml je Well auf die Platte gegeben. Anschließend wurden die Platten für eine halbe
Stunde im Brutschrank inkubiert. Bei einem Teil der Platten wurden jetzt die Zellen
zu dem Virusüberstand hinzubegeben, der andere Teil der Platten wurde weiter
behandelt.
Wurde der Virusüberstand von der Platte abgenommen, so wurde die Platte mit
Medium gewaschen. Ein Teil der Platten wurde dreimal für jeweils 30 min mit
Virusüberstand beladen, bevor die Platten dreimal mit Medium gewaschen wurden.
Das Behandlungsschema der einzelnen Platten ist der Tabelle 2 zu entnehmen.
Nach der Behandlung der Platten wurde in jedes Well 1 x 105 HEL Zellen ausgesät.
Jeder Versuch wurde dreifach angesetzt. Nach 6 Tagen wurde der Anteil an GFP
positiven Zellen mittels Durchflusszytometrie ermittelt.
46

Material und Methoden
Tabelle 2: Behandlungsschema Adhäsion von rekombinanten Retroviren
Zellkulturplatten Behandlung
1 x Virus 1 x Virus + 3 x Waschen
3 x Virus + 3 x Waschen
TCD X -- --
BSA X X --
CH296 X X X
3.2.8 1,3-bis-(2-chlorethyl)-1-Nitrosourea (BCNU)
3.2.8.1 BCNU Selektion
Zur Selektion transduzierter HEL, HL60 und K562 Zellen wurden zwei
unterschiedliche Protokolle verwendet. Beim ersten Protokoll wurden transduzierte
HEL, HL60 und K562, sowie nicht transduzierte Zellen dieser Zelllinien in 12-
Wellplatten ausgesät. Dann wurden 2 x 105 Zellen in 1 ml Medium pro Well pipettiert.
Das Medium enthielt keine Zusätze. Das Zytostatikum Carmustin (BCNU,
Carmubris®, Bristol-Meyers Squibb GmbH & Co. KGaA, D-80637 München) wurde in
Ethanol (als Lösungsmittel Bestandteil von Carmubris) gelöst. Die Zellen wurden mit
Konzentrationen von 0, 20, 80 und 160 µM BCNU/ml Medium behandelt. Die Platten
wurden für drei Stunden im Brutschrank inkubiert. Nach drei Stunden (Halbwertzeit
des BCNU 45 min) wurden zusätzliche 2,5 ml Medium mit Zusätzen in jedes Well
pipettiert. Sechs Tage nach der Selektion wurden die Zellen in FACS-Röhrchen
überführt, einmal mit PBS gewaschen und dann in 500 µl einer PI-FACS-Lösung
aufgenommen. Jeder Versuch wurde dreifach angesetzt. Der Anteil an GFP positiven
und toten Zellen wurde mittels Durchflusszytometrie ermittelt.
Im zweiten Protokoll wurden nicht transduzierte und transduzierte HL60 Zellen
verwendet. Es wurden 2 x 105 Zellen in 1 ml Medium mit Zusätzen pro Well einer 12-
47

Material und Methoden
Wellplatte ausgesät. Das in Ethanol gelöste BCNU wurde an mehreren Tagen
hintereinander auf die Zellen aufgetragen (Tabelle 3). Das gelöste BCNU wurde
zwischen den Behandlungen bei -20°C gelagert.
Tabelle 3: BCNU Behandlung der HL60 Zellen
Behandlung mit BCNU (Konzentration) Gruppe
Tag 1 Tag 2 Tag 3 Tag 4
1 20 µM 20 µM 20 µM 20 µM
2 20 µM 20 µM 20 µM
3 20 µM 20 µM
4 20 µM 40 µM 80 µM
5 20 µM 40 µM
6 20 µM
7 40 µM
8 60 µM
9 80 µM
10 0 µM
Jeder Versuch wurde dreifach angesetzt. Eine Woche nach der ersten BCNU
Behandlung wurden die Zellen in FACS-Röhrchen überführt, einmal mit PBS
gewaschen und dann in 500 µl einer PI-FACS-Lösung aufgenommen. Der Anteil
GFP positiver Zellen wurde mittels FACS-Analyse ermittelt.
3.2.8.2 BG - BCNU Selektion
Es wurden je 2 x 105 transduzierte HL60 Zellen, sowie nicht transduzierte Zellen
dieser Zelllinie in 1 ml Medium auf 12-Wellplatten ausgesät. Das BG (Sigma-Aldrich)
wurde in Polyethyenglycol 400 (Merck) gelöst und ein Teil der BG-Stammlösung mit
48

Material und Methoden
Medium zu einer Konzentration von 40 µM verdünnt. Von dieser Lösung wurde
jeweils 1 ml zu den Zellen pipettiert, so dass die Zellen einer Konzentration von 20
µM pro ml Medium ausgesetzt waren. Die Platten wurden eine Stunde lang im
Brutschrank inkubiert. Anschließend wurden die Zellen mit 0, 10, 20, 40 und 60 µM
BCNU/ml Medium (angesetzt wie unter ‚BCNU Selektion’ beschrieben) behandelt.
Die Kontrollgruppe wurde mit 0, 10, 20, 40 und 60 µM BCNU/ml Medium, aber nicht
mit BG behandelt; einige Zellen erhielten nur BG. Fünf Tage nach der Selektion
wurden die Zellen in FACS-Röhrchen überführt, einmal mit PBS gewaschen und
dann in 500 µl einer PI-FACS-Lösung aufgenommen. Der Anteil an GPF positiven
und toten Zellen wurde mittels Durchflusszytometrie ermittelt.
3.2.8.3 BG - BCNU Selektion von CD34+ Marmoset Zellen
Eingefrorene CD34+ Zellen aus dem Knochenmark des Tieres 0224 wurden
aufgetaut und 5 Stunden in IMDM, ergänzt mit 100 ng SCF/ml, kultiviert.
Anschließend wurden die Zellen gezählt und jeweils 38.000 Zellen in jedes Loch
einer 6-Wellplatte ausgesät, die zuvor mit Fibronektin beschichtet und mit
Virusüberstand beladen worden waren. Zur Transduktion wurden die Zytokine SCF,
TPO, IL-6 und Flt-L in einer Konzentration von 100 ng/ml zugesetzt. Die Zellen
wurden anschließend zwei Tage im Brutschrank kultiviert, so dass zwei Tage nach
der Transduktion die Zellen vom Fibronektin abgenommen, geteilt und in einer
Konzentration von 9.500 Zellen pro Well auf einer 12-Wellplatten ausgesät wurden.
Von jedem eingesetzten Viruskonstrukt und der negativen Kontrolle wurden Zellen in
einem Well nicht behandelt, das andere Well wurde mit 20 µM BCNU behandelt. Der
Ansatz wurde eine Stunde im Brutschrank inkubiert. Zwei weitere Wells wurden mit
20 µM pro ml BG behandelt. Nach einer Stunde Inkubation wurde eins der zwei mit
BG behandelten Wells eines jeden Konstruktes zusätzlich mit 20 µM BCNU
behandelt und eine weitere Stunde im Brutschrank inkubiert. Aus jedem Well wurde
ein Teil der Zellen entnommen und in Progenitor-Assays ausgesät. Es wurden
jeweils zwei Zellkonzentrationen ausgesät: 250 und 500 Zellen/ml Metho Cult. Die
49

Material und Methoden
Progenitor-Assays wurden 14 Tage in einem separaten Brutschrank inkubiert. Die
Auszählung erfolgte mit einem Fluoreszenzmikroskop. Der Anteil GFP positiver
Kolonien an der Gesamtzahl der gewachsenen Kolonien wurde bestimmt und die auf
den 12-Wellplatten verbliebenen Zellen wurden weiter kultiviert und dem Medium
wurden die Zytokine SCF, TPO, IL-6 und Flt-L in einer Konzentration von 100 ng/ml
zugesetzt. Am vierten Tag nach der Behandlung wurden die Zellen in FACS-
Röhrchen überführt, einmal mit PBS gewaschen und in 500 µl einer PI-FACS-Lösung
aufgenommen. Der Anteil an GFP positiven wurde mittels Durchflusszytometer
ermittelt. Jeder Versuch wurde dreifach angesetzt.
3.2.9 Knochenmarkentnahme bei Marmosets
Die Affen wurden durch intramuskuläre Injektion eines Narkosemittels in die kaudale
Oberschenkelmuskulatur narkotisiert. Zur Einleitung der Narkose wurde den Tieren
Ketamin (50 mg/kg Körpergewicht, Ketavet®, 100 mg/ml, Pfizer Pharma GmbH, D-
76139 Karlsruhe) verabreicht. Anschließend wurde die Narkose durch Inhalation von
2-3 % Enfluran (Forene®, Abbott GmbH, D-65205 Wiesbaden) über eine
Inhalationsmaske für Katzen aufrechterhalten. Die Punktionsstelle am Innenschenkel
wurde für einen aseptischen Eingriff vorbereitet. Mit einer
Knochenmarkspunktionskanüle (Illinois Sternal/Illiakal-Aspirationskanüle, 16 G,
Tyco/Healthcare Kendall, Mansfield, MA 02048 USA) wurde im distalen Drittel des
Humerus Knochenmark entnommen. Um eine Gerinnung zu verhindern, wurden
Punktionskanüle und Spritze, mit denen das Knochenmark aspiriert wurde, vor der
Entnahme mit Heparin (Heparin-Natrium 25.000 I.E./5ml, Ratiopharm GmbH, D-
89079 Ulm) gespült. Zum Zeitpunkt der Aspiration waren ca. 0,5 ml Heparin in der
Spritze vorgelegt. Die ersten beiden Tiere, 0224 und 3101, waren Marmosets, an
denen eine Finalversuch durchgeführt wurde. Post mortem wurden die Humeri sowie
die Femora entnommen und am proximalen und distalen Ende eröffnet.
Anschließend wurde die Knochenmarkhöhle mit IMDM gespült. Das Medium mit den
Knochenmarkzellen wurde in einem sterilen 50 ml Röhrchen aufgefangen. Bei dem
50

Material und Methoden
dritten Tier, 0130, wurde nur der rechte Humerus, wie oben beschrieben punktiert.
Nach der Punktion wurde die Punktionsstelle etwa 5 min mit einer Kompresse
abgedeckt und zur Stillung der Blutung Druck auf die Wundstelle ausgeübt. Nach der
Stillung der Blutung wurde die Narkose ausgeleitet.
3.2.10 Statistik
Die Ergebnisse der einzelnen Versuche wurden in einem Datenblatt erfasst
(Microsoft® Office Excel 2003; Microsoft Corporation, Redmond, NC, USA). Die
statistische Auswertung der Daten erfolgte mit dem Statistical Analysis System for
Windows, SAS® (Version 9.1; SAS-Institute, Cary, NC, USA) auf einem PC unter
Windows XP SP2 (Microsoft).
Metrische Daten wurden getrennt für jede Variable (Vektor resp. Behandlung)
deskriptiv ausgewertet (Prozedur UNIVARIATE, SAS®). Es wurden die Anzahl der
Messungen, der arithmetische Mittelwert, die Standardabweichung, die Varianz
sowie Minimum und Maximum bestimmt.
Konnte eine Normalverteilung der Virustiter resp. der logarithmierten Werte
festgestellt werden, wurde die Verteilung der Werte in zwei Klassen mittels t-Test
verglichen (Prozedur TTEST, SAS®). Sollte die Verteilung von Ergebnissen zwischen
mehr als 2 Klassen verglichen werden, wurde eine globale Prüfung mittels F-Test
vorgenommen (Prozedur ANOVA, SAS®). Verteilungen nicht normalverteilter
metrischer Daten wurden mit dem Wilcoxon-Test analysiert (Prozedur NPAR1WAY,
SAS®). Verteilungen wurden als signifikant unterschiedlich betrachtet, wenn ein p-
Wert ≤ 0,05 ermittelt werden konnte. Die Unterschiede sind, sofern nicht eine globale
Prüfung der Daten vorgenommen wurde, durch verschiedene Buchstaben in den
Abbildungen gekennzeichnet.
51

Ergebnisse
4. Ergebnisse
4.1 Herstellung von Foamyviren
4.1.1 Optimierung des Transfektionsprotokolls
Lentivirale, foamyvirale und gammaretrovirale Konstrukte wurden mit einem
foamyviralen Hüllprotein EM140 verpackt. Erst mit der Verpackung in einem
strukturgleichen Hüllprotein (EM140) wurde ein echter Vergleich der einzelnen
Konstrukte möglich. Die bisher verwendeten foamyviralen Hüllproteine, die zuvor zur
Pseudotypisierung von gammaretroviralen und lentiviralen Konstrukten eingesetzt
worden sind, waren ergaben nur niedrige Virustiter. Die Transduktionseffizienz von
rekombinanten Retroviren, die mit Hilfe des VSV-G Glykoproteins pseudotypisiert
wurden, haben als Referenzwert gedient, da VSV-G seit einiger Zeit erfolgreich zur
Verpackung lentiviraler Konstrukte genutzt wird und ausreichend Daten vorlagen. Für
die Generierung der lentiviralen Konstrukte wurde das Helferplasmid CD/NL-BH
verwendet (Abbildung 6).
Die Verwendung verschiedener Konstrukte zur Transfektion von Zellen führt zu
deutlichen Unterschieden im Virustiter. Die arithmetischen Mittelwerte, die mit den
einzelnen Konstrukten erzielt wurden, weisen einen statistisch signifikanten
Unterschied auf (p = 0,0039).
Die Optimierung des Transfektionsprotokolls durch den Zusatz von Serum führt bei
allen Konstrukten zu einem Anstieg der Virustiter, verglichen mit den Werten einer
Transfektion ohne Serum. Der Titeranstieg ist bei den Konstrukten pCL1
pseudotypisiert mit EM140, MH71 verpackt mit dem EM140 und pCAMSΔU3E
pseudotypisiert mit dem EM140 statistisch signifikant (p = 0,0002, p = 0,0045 resp. p
= 0,0002). Der Unterschied zwischen dem Transduktionsprotokoll „mit Serum“ und
„ohne Serum“ für das Konstrukt pCL1, pseudotypisiert mit VSV-G, konnte dagegen
statistisch nicht abgesichert werden.
52

Ergebnisse
0,00E+00
2,00E+07
4,00E+07
6,00E+07
8,00E+07
1,00E+08
1,20E+08
1,40E+08
pCL1 CD/NL-BH VSV-G pCL1 CD/NL-BH EM140 MH71 EM140 pCAMSΔU3E EM140
TE /
ml
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
c
b
a a d e f g
Transfektion „ohne Serum“
Transfektion „mit Serum“
Abbildung 6: Vergleich von verschiedenen Transfektionsprotokollen, in denen die
Transfektion „mit Serum“ und „ohne Serum“ durchgeführt wurde
53

Ergebnisse
4.1.2 Vergleich von foamyviralen 2,- 3- und 4-Plasmidsystemen Das foamyvirale Konstrukt MH71 benötigte kein Helferplasmid, denn es trägt schon
die Gene gag und pol, die zur Generierung des Virus notwendig sind. Das Konstrukt
MD9 enthält nur Teile von gag und pol, die lediglich für das Verpackungssignal
kodieren. Um mit den Vektor MD9 Virusüberstand generieren zu können, müssen die
Gene gag und pol auf Helferplasmiden zur Verfügung gestellt werden. Das
Helferplasmid cmvgp1 kodiert für ein vollständiges gag und pol. Bei den andern
verwendeten Helferplasmiden liegen gag und pol auf zwei voneinander getrennten
Helferplasmiden vor. Die Plasmide pcziGag und p6iGag bzw. pcziPol und p6iPol
unterscheiden sich nur im Aufbau des Backbones, nicht aber im viralen Teil des
Vektors.
Zur Optimierung der foamyviralen Virusproduktion wurden verschiedene
Viruskonstrukte verglichen (Abbildung 7). Das Konstrukt MD9 wurde zusammen mit
den Helferplasmiden cmvgp1, einer Kombination aus p6i Gag mit p6i Pol und pczi
Gag mit pczi Pol generiert.
Durch den Einsatz des Helferplasmids p6iGag mit p6iPol konnte die Titerhöhe,
vergleichend zum Konstrukt mit dem Helferplasmid cmvgp1, um das 4,2 fache
gesteigert werden. Durch Verwendung der Helferplasmide pcziGag mit pcziPol ist
eine Steigerung um das 6,0 fache gegenüber dem drei Plasmidsystem möglich. Die
einzelnen Konstrukte weisen deutlich unterschiedliche Virustiter auf. Der Einfluss der
verwendeten Konstrukte auf den Virustiter ist statistisch signifikant (p = 0,0003).
54

Ergebnisse
0,00E+00
2,00E+06
4,00E+06
6,00E+06
8,00E+06
1,00E+07
1,20E+07
MH71EM140 MD9cmvgp1EM140 MD9p6iGag/p6iPolEM140 MD9pcziGag/pcziPolEM140
TE /
ml
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
cmvgp1
EM140
MD9
p6iGag / p6iPol
EM140
MD9
pcziGag / pcziPol
EM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
cmvgp1
EM140
MD9
p6iGag / p6iPol
EM140
MD9
pcziGag / pcziPol
EM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
cmvgp1
EM140
MD9
p6iGag / p6iPol
EM140
MD9
pcziGag / pcziPol
EM140
Abbildung 7: Gegenüberstellung foamyviraler 2- 3- und 4-Plasmidsysteme
4.1.3 Foamyvirale Verpackungsplasmide Das foamyvirale Hüllprotein EM140 wies bei den Produzentenzellen eine Toxizität
auf, die beim Hüllprotein VSV-G nicht beobachtet werden konnte. Die Zellen
fusionierten miteinander und bildeten mehrkernige Riesenzellen aus. Eine Zelle mit
mehreren Kernen ist nicht mehr in der Lage, gerichtete Proteinsynthese zu betreiben
und damit die einzelnen Elemente des Virus zu bilden. Dadurch fällt der Virustiter
letztlich niedriger aus. Das Hüllprotein SM04 ist, ebenso wie das Hüllprotein EM140,
in der Lage, lentivirale und gammaretrovirale Konstrukte zu pseudotypisieren und
wurde daher als alternatives foamyvirales Hüllprotein getestet. Die Hüllproteine
wurden mit zwei unterschiedlichen foamyviralen Konstrukten verglichen (Abbildung
55

Ergebnisse
8). Zu Generierung des Virus wurden die beiden Helferplasmide pcziGag und pcziPol
für das Konstrukt MD9 eingesetzt.
0,00E+00
1,00E+06
2,00E+06
3,00E+06
4,00E+06
5,00E+06
MH71 EM140 MD9 EM140 MH71 SM04 MD9 SM04
TE /
ml
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
pcziGag / pcziPol
EM140
MH71
--
SM04
MD9
pcziGag / pcziPol
SM04
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
pcziGag / pcziPol
EM140
MH71
--
SM04
MD9
pcziGag / pcziPol
SM04
Abbildung 8: Gegenüberstellung foamyviraler Verpackungsplasmide in einem 2- resp.
4-Plasmidsystem
Betrachtet man das Konstrukt MH71, so ist der Titer mit dem Hüllprotein EM140 um
das 1,9 fache höher als der von Zellen mit SM04. Bei dem Konstrukt MD9 ist der
Unterschied zwischen dem Hüllprotein EM140 und SM04 noch deutlicher; der Titer
des Hüllprotein EM140 ist um das 5,6 fache höher als der von Zellen mit Hüllprotein
SM04. Den einzelnen Konstrukten konnte ein signifikanter Einfluss auf die erzielte
Titerhöhe nachgewiesen werden (p= 0,0017).
56

Ergebnisse
4.1.4 Einfluss des MGMTp140k Transgens auf die GFP Expression Die Vektorplasmide MH71.MGMTp140kIEG und MD9.MGMTp140kIEG exprimieren
unter der Kontrolle des SFFV U3’ Promotors eine Expressionskassette mit
MGMTp140k, dass über eine IRES-site mit GFP verbunden ist. Um zu prüfen, in wie
weit die Expression des Markers GFP durch das 5’ gelegene MGMTp140k
beeinflusst wird, wurden die Virustiter der Konstrukte mit und ohne dem Transgen
MGMTp140k einander gegenübergestellt. Für die Plasmide MD9 und MD9.MGMT
wurden das 4-Plasmidsystem mit den Helferplasmiden pcziGag und pcziPol zur
Generierung des Virus genutzt. Bei MH71 und MH71.MGMT handelt es sich um ein
2-Plasmidsystem, in dem gag und pol schon auf dem Vektorplasmid vorhanden sind.
Als Hüllprotein wurde EM140 eingesetzt.
0,00E+00
1,00E+06
2,00E+06
3,00E+06
4,00E+06
MH71 MD9 MH71.MGMT MD9.MGMT
TE /
ml
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
pcziGag / pcziPol
EM140
MH71.MGMT
--
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
pcziGag / pcziPol
EM140
MH71.MGMT
--
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Abbildung 9: Vergleich der foamyviralen Vektorplasmide MH71 und MD9 mit und
ohne MGMTp140k
57

Ergebnisse
Der Abbildung 9 ist zu entnehmen, dass die Konstrukte ohne MGMTp140k immer
einen höheren Virustiter erzielen, als die mit MGMTp140k. Bei dem Konstrukt MH71
ist der Virustiter mit MGMTp140k um 12 % niedriger, bei dem Konstrukt MD9 ist der
Virustiter mit MGMTp140k um 19 % niedriger. Zwischen allen Konstrukten besteht
ein augenscheinlicher Unterschied in der Höhe der Virustiter. Der Einfluss der
verwendeten Konstrukte auf den Virustiter ist statistisch signifikant (p= 0,0297).
4.1.5 Konzentrierung retroviraler Virusüberstände
Der Virustiter kann erhöht werden, indem das Volumen des virushaltigen
Überstandes reduziert wird, ohne dabei infektiöse Viruspartikel zu verlieren. Die
Viruspartikel werden durch Zentrifugation sedimentiert und der restliche Überstand
abgenommen. Die so sedimentierten Viren können in neues Medium aufgenommen
werden und der Virusüberstand weist im Allgemeinen eine deutlich höhere
Konzentration als vor dieser Prozedur auf.
In Abbildung 10 sind die arithmetischen Mittelwerte aus 4 Werten dargestellt, von
denen je 2 aus einem Versuchsansatz stammen. Für die Plasmide MD9 und
MD9.MGMT wurden die Helferplasmide pcziGag und pcziPol zur Generierung des
Virus eingesetzt. Als Hüllprotein wurde EM140 verwendet. Der Virusüberstand von 8
Zellkulturplatten mit je 10 cm Durchmesser wurde gepoolt, zentrifugiert und das
Viruspellet in 1 ml Medium aufgenommen. Durch die Zentrifugation konnte der
Virustiter je nach Konstrukt um das 24,8 fache (MD9), 26,3 fache (MH71.MGMT),
26,5 fache (MH71) resp. um das 30 fache (MD9.MGMT) gegenüber dem
Ausgangswert gesteigert werden.
58

Ergebnisse
0,00E+00
1,00E+07
2,00E+07
3,00E+07
4,00E+07
5,00E+07
6,00E+07
7,00E+07
8,00E+07
9,00E+07
MH71 MD9 MH71.MGMT MD9.MGMT
TE /
ml
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
pcziGag / pcziPol
EM140
MH71.MGMT
--
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MD9
pcziGag / pcziPol
EM140
MH71.MGMT
--
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Virustiter vor dem Zentrifugieren
Virustiter nach dem Zentrifugieren
Abbildung 10: Konzentration foamyviraler Virusüberstände
4.1.6 Adhäsion rekombinanter Retroviren an Moleküle der Fibronektin-Matrix
Transduktionseffizienz und Adhäsion von rekombinanten Retroviren, die mit
verschiedenen Hüllproteinen pseudotypisiert worden waren, wurden auf
unterschiedlich beschichteten Zellkulturplatten getestet. Die Zellkulturplatten waren
mit CH296 oder BSA beschichtet, oder es wurden TCD verwendet.
59

Ergebnisse
GaLV
0
10
20
30
40
1/10 BSA 1/10 BSA 1/100 CH296 1/10 CH296 1/100
GFP
pos
itive
Zel
len
(%)
Beschichtung
Verdünnung
Behandlung
TCD
1/10 1/100
1xV
BSA CH296
1/10
1xV
-- 3xW
1/100
1xV
-- 3xW
1/10
1xV 1xV 3xV
-- 3xW 3xW
1/100
1xV 1xV 3xV
-- 3xW 3xW
Beschichtung
Verdünnung
Behandlung
TCD
1/10 1/100
1xV
BSA CH296
1/10
1xV
-- 3xW
1/100
1xV
-- 3xW
1/10
1xV 1xV 3xV
-- 3xW 3xW
1/100
1xV 1xV 3xV
-- 3xW 3xW
VSV-G
0
20
40
60
80
100
1/10 BSA 1/10 BSA 1/100 CH296 1/10 CH296 1/100
GFP
pos
itive
Zel
len
(%)
Beschichtung
Verdünnung
Behandlung
TCD
1/10 1/100
1xV
BSA CH296
1/10
1xV
-- 3xW
1/100
1xV
-- 3xW
1/10
1xV 1xV 3xV
-- 3xW 3xW
1/100
1xV 1xV 3xV
-- 3xW 3xW
Beschichtung
Verdünnung
Behandlung
TCD
1/10 1/100
1xV
BSA CH296
1/10
1xV
-- 3xW
1/100
1xV
-- 3xW
1/10
1xV 1xV 3xV
-- 3xW 3xW
1/100
1xV 1xV 3xV
-- 3xW 3xW
60

Ergebnisse
EM140
0
20
40
60
80
1/10 BSA 1/10 BSA 1/100 CH296 1/10 CH296 1/100
GFP
pos
itive
Zel
len
(%)
Beschichtung
Verdünnung
Behandlung
TCD
1/10 1/100
1xV
BSA CH296
1/10
1xV
-- 3xW
1/100
1xV
-- 3xW
1/10
1xV 1xV 3xV
-- 3xW 3xW
1/100
1xV 1xV 3xV
-- 3xW 3xW
Beschichtung
Verdünnung
Behandlung
TCD
1/10 1/100
1xV
BSA CH296
1/10
1xV
-- 3xW
1/100
1xV
-- 3xW
1/10
1xV 1xV 3xV
-- 3xW 3xW
1/100
1xV 1xV 3xV
-- 3xW 3xW
Abbildung 11: Bindung verschiedener Virushüllen an unterschiedlich beschichtete
Zellkulturplatten Zellkulturplatten (tissue culture dish) bzw. unbehandelten Platten, die dann mit 2 % bovinem
Serumalbumin (BSA) oder mit dem Fibronektinfragment CH296 beschichtet worden waren, wurden
mit Virusüberstand beladen. Der Vektor pCL1 mit dem Helferplasmid CD/NL-BH wurde mit den
Hüllproteinen GaLV, VSV-G und EM140 pseudotypisiert. Die Virusüberstände wurden 1:10 oder 1:100
verdünnt, bevor die Überstände zur Transduktion eingesetzt wurden. Die Platten wurden alle
mindestens einmal (1xV) mit Virusüberstand vorgeladen. Ein Teil der mit CH296 behandelten Platten
wurde dreimal Vorgeladen (3xV). Um die Adhäsion der Viruspartikel zu testen, wurde ein Teil der
Platten nach der Beladung mit Virusüberstand dreimal gewaschen (3xW), bevor die Zellen auf die
Platten gegeben wurden.
61

Ergebnisse
Der lentivirale Vektor pCL1 wurde zusammen mit dem Helferplasmid CD/NL-BH zur
Generierung der Viren mit den Hüllproteinen GaLV, VSV-G und EM140 eingesetzt.
Der Virustiter der Virusüberstände wurden auf HT1080 Zellen ermittelt. Das GaLV
Konstrukt hatte einen Titer von 850.000 TE / ml, das VSV-G Konstrukt einen Titer
von 29.000.000 TE / ml und das Konstrukt mit der foamyviralen Virushülle einen Titer
von 62.000.000 TE / ml. Die verwendeten Virustiter sind nicht auf ein gemeinsames
Niveau eingestellt, sondern 1:10 und 1:100 mit Medium verdünnt worden. Daher
kann nur einen relative Aussage zur Transduktionseffizienz der einzelnen
Hüllproteine getroffen werden.
Die Adhäsion der Hüllproteine an die beschichteten Zellkulturplatten wurde bestimmt,
indem der Virusüberstand wieder von der Platte entfernt wurde und diese mehrfach
mit Medium gewaschen wurde. Bei den CH296 beschichteten Platten wurde
zusätzlich dreimal Virusüberstand auf die Platte gegeben und wieder entfernt bevor
die Zellkulturplatte auch mehrfach mit Medium gewaschen wurde.
Alle Hüllproteine binden an das CH296 und auch nachdem die Zellkulturplatten
dreimal mit Medium (3xW) gewaschen worden sind, konnten die Zellen noch
transduziert werden. Bei allen verwendeten Hüllproteinen konnte die
Transduktionseffizienz gesteigert werden, indem CH296 dreimal mit Virusüberstand
beladen (vorgeladen) wurde (V 3xW), bevor die Zellkulturplatten dreimal mit Medium
gewaschen wurden. In der Verdünnung 1:10 konnte auf den CH296 beschichteten
Platten durch das dreimalige Vorladen eine Erhöhung der Transduktionseffizienz um
das 1,7- bis 2,7-fache gegenüber den Platten erreicht werden, die nur einmal mit
Virus beladen wurden. In der Verdünnung 1:100 konnte bei allen Hüllproteinen eine
Erhöhung der Transduktionseffizienz um das 3,5 fache erreicht werden, wenn die
Platten dreimal und nicht nur einmal mit Virusüberstand beladen wurden. Die
unterschiedliche Behandlung der Zellkulturplatten hatte einen signifikanten Einfluss
auf den Anteil GFP positiver Zellen (p=0,0111).
62

Ergebnisse
4.2 Gentransfer in humane CD34+ Zellen aus Nabelschnurblut
4.2.1 Anreicherung humaner CD34+ Zellen Nach Aufreinigung der CD34+ Zellen aus etwa 50 bis 70 ml Nabelschnurblut konnten
zwischen 2 x 108 und 4 x 108 mononukleäre Zellen isoliert werden. Aus diesen
mononukleären Zellen konnten durch eine einmalige Aufreinigung in MACS-Säulen
zwischen 0,5 und 1,6 x 106 CD34+ Zellen isoliert werden. Der prozentuale Anteil der
CD34+ Zellen an den Zellen nach der Aufreinigung lag zwischen 45 und 75 %.
4.2.2 Retroviraler Gentransfer in humanen CD34+ Zellen
Lentivirale, gammaretrovirale und foamyvirale Konstrukte wurden mit einem
foamyviralen Hüllprotein verpackt. Zum Vergleich wurde ein lentivirales Konstrukt mit
dem Hüllprotein VSV-G verpackt. Für die Generierung des lentiviralen Konstrukts
wurde des Helferplasmid CD/NL-BH eingesetzt. Die Virustiter wurden vor und nach
der Zentrifugation gemessen. Der Virusüberstand von 8 Zellkulturplatten mit je 10 cm
Durchmesser wurde gepoolt, zentrifugiert und das Viruspellet in 1 ml Medium
aufgenommen.
Durch die Zentrifugation konnte der Virustiter je nach Konstrukt um das 1,4 fache
(pCL1 EM140), 2,9 fache (MH71 EM140), 5,8 fache (pCAMSΔU3E EM140) resp. um
das 7,3 fache (pCL1 VSV-G) gegenüber dem Ausgangswert gesteigert werden
(Abbildung 12). Die zentrifugierten Virusüberstände wurden bis zur weiteren
Verwendung kryokonserviert.
63

Ergebnisse
0,00E+00
1,50E+07
3,00E+07
4,50E+07
6,00E+07
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
TE /
ml
0,00E+00
1,50E+07
3,00E+07
4,50E+07
6,00E+07
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
TE /
ml
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Virustiter vor dem Zentrifugieren
Virustiter nach dem Zentrifugieren
Abbildung 12: Virustiter, gemessen auf HT1080 Zellen vor und nach dem
Zentrifugieren der Virusüberstände
Die Virusüberstände wurden zur Transduktion von humanen CD34+ Zellen eingesetzt.
Fünf Tage nach der Transduktion wurde die Gentransferrate mittels FACS-Analyse
gemessen.
Der Anteil GFP positiver Zellen, und somit die Gentransferrate, betrug bei den
transduzierten CD34+ Zellen 28 bis 77 % (Abbildung 13). Die Höhe des Virustiters
64

Ergebnisse
vor der Transduktion hat keinen Einfluss auf die Gentransferraten der einzelnen
Konstrukte. Das foamyvirale Konstrukt hatte den niedrigsten Virustiter, wies aber mit
74 % GFP positiver Zellen eine der höchsten Gentransferraten auf. Der Virustiter des
Konstruktes pCL1 EM140 war mehr als doppelt so hoch wie der des MH71 EM140
Konstruktes. Der Unterschied in der Gentransferrate betrug jedoch nur 3 %. Das
gammaretrovirale Konstrukt pCAMSΔU3E, pseudotypisiert mit dem EM140, zeigte
einen ähnlich hohen Titer wie das foamyvirale Konstrukt. Die Gentransferrate lag mit
28 % GFP positiver Zellen jedoch etwa 50 % unter der des foamyviralen Konstruktes.
0
20
40
60
80
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E EM140
GFP
pos
itive
Zel
len
(%)
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Abbildung 13: Gentransferraten humanen CD34+ Zellen nach transduktion mit lenti-,
ammaretro- und foamyviralen Viruskonstrukten g
65

Ergebnisse
4.2.3 Einfluss des Transgens MGMTp140k auf die Gentransferrate Die Transduktionseffizienz verschiedener foamyviraler Konstrukte wurde
vergleichend gegenübergestellt. Um den Einfluss des Transgens zu ermitteln,
wurden die Höhe des Virustiters, der Anteil GFP positiver Zellen nach Transduktion
von humanen CD34+ Zellen und der Anteil GFP-positiver Zellen im Progenitor-Assay
bestimmt.
Zur Generierung der Virusüberstände wurden bei den Konstrukten MD9 und
MD9.MGMT die Helferplasmide pcziGag und pcziPol eingesetzt. Als Hüllprotein
wurde für alle Konstrukte das EM140 eingesetzt. Der Virusüberstand von 8
Zellkulturplatten mit einem Durchmesser von 10 cm wurde gepoolt und zentrifugiert.
Das Viruspellet wurde in 1 ml Medium aufgenommen und frisch zur Transduktion der
humanen CD34+ Zellen eingesetzt.
Gegenüber den Ausgangswerten konnte der Virustiter durch das Zentrifugieren je
nach Konstrukt um das 19,7 fache (MD9.MGMT), 25,7 fache (MD9), 29,8 fache
(MH71.MGMT) resp. um das 30 fache (MH71) gesteigert werden (Abbildung 14).
66

Ergebnisse
0,00E+00
1,00E+07
2,00E+07
3,00E+07
4,00E+07
5,00E+07
6,00E+07
7,00E+07
8,00E+07
9,00E+07
/ m
l
1,00E+08
MH71.MGMT MD9 MD9.MGMTMH71
TE
Vektor MH710,00E+00
1,00E+07
2,00E+07
3,00E+07
4,00E+07
MH71
TE
Helferplasmid
Hüllprotein
--
EM140
--
EM140
pcziGag / pcziPol
EM140
pcziGag / pcziPol
EM140
MH71.MGMT MD9 MD9.MGMT
5,00E+07
6,00E+07
7,00E+07
8,00E+07
9,00E+07
/ m
l
1,00E+08
MH71.MGMT MD9 MD9.MGMT0,00E+00
1,00E+07
2,00E+07
3,00E+07
4,00E+07
MH71
TE
5,00E+07
6,00E+07
7,00E+07
8,00E+07
9,00E+07
/ m
l
1,00E+08
MH71.MGMT MD9 MD9.MGMTVektor MH71
Helferplasmid
Hüllprotein
--
EM140
--
EM140
pcziGag / pcziPol
EM140
pcziGag / pcziPol
EM140
MH71.MGMT MD9 MD9.MGMTVektor MH71
Helferplasmid
Hüllprotein
--
EM140
--
EM140
pcziGag / pcziPol
EM140
pcziGag / pcziPol
EM140
MH71.MGMT MD9 MD9.MGMT
Virustiter vor dem Zentrifugieren
Virustiter nach dem Zentrifugieren
Abbildung 14: Virustiter vor und nach dem Zentrifugieren der Virusüberstände
Die zentrifugierten Virusüberstände wurden zur Transduktion von humanen CD34+
Zellen eingesetzt. Ein Teil der Zellen wurde in Progenitor-Assays ausgesät, an dem
anderen Teil wurde 5 Tage nach der Transduktion die Anteil der GFP positiven
Zellen mittels FACS-Analyse ermittelt.
67

Ergebnisse
68
0
10
20
30
40
50
MH71 MH71.MGMT MD9 MD9.MGMT
GFP
pos
itive
Zel
len
(%)
Vektor
Helferplasmid
Hüllprote
MH71
EM140
MH71.MGMT
EM140
MD9
pcziGag / pcziPol
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
-- --
0
10
20
30
40
50
MH71 MH71.MGMT MD9 MD9.MGMT
GFP
pos
itive
Zel
len
(%)
in
Vektor
Helferplasmid
Hüllprote
MH71
EM140
MH71.MGMT
EM140
MD9
pcziGag / pcziPol
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Vektor
Helferplasmid
Hüllprote
MH71
EM140
MH71.MGMT
EM140
MD9
pcziGag / pcziPol
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
bbildung 15: Gentransferraten foamyviraler Konstrukte auf humanen CD34+ Zellen
ie Gentransferraten lagen, gemessen am Anteil GFP positiver Zellen, in diesem
ukt MH71 mit 66,5 Mio TE / ml mehr als 50 %
ber dem des MD9.MGMT mit 25,6 Mio TE / ml. Die Gentransferrate des Konstruktes
MH71 betrug 44,4 % GFP positiver Zellen, die des Konstruktes MD9.MGMT 25,2 %.
Die Gentransferraten der Konstrukte mit MGMTp140k lagen jeweils leicht unterhalb
der von Konstrukten ohne dieses Transgen.
inin
-- ---- --
A
(5 Tage nach der Transduktion mittels Durchflusszytometrie ermittelt)
D
Versuch zwischen 25,2 und 44,4 % (Abbildung 15) und zeigten damit eine deutliche
Korrelation zur Höhe der Virustiter. Dieser Einfluss wird bei einem direkten Vergleich
der Ergebnisse aus Zellen mit den Konstrukten MH71 und MD9.MGMT besonders
deutlich. Der Virustiter lag beim Konstr
ü

Ergebnisse
In Abbildung 16 sind die Ergebnisse des Progenitor-Assays als arithmetrischer
Mittelwert aus drei Ansätzen (Triplikate) mit je 250 Zellen / ml Methylzellulose
dargestellt. Die Anzahl der gezählten Kolonien wurde jeweils als 100 % betrachtet.
Die so bestimmte Gentransferrate betrug 67 % (MH71 resp. MH71.MGMT) resp.
66 % (MD9). Diese hohe Gentransferrate war unabhängig vom zuvor bestimmten
Virustiter und den im FACS gemessenen Gentransferraten. Das Konstrukt
MD9.MGMT wies mit einer Gentransfereffizienz von 53 % einen geringfügig
niedrigeren Wert auf. Die Gentransferrate in der FACS-Analyse war für das Konstrukt
MH71 um 34 % niedriger als im Progenitor-Assay. Für das Konstrukt MH71.MGMT
lag dieser Wert bei 38 %, für das Konstrukt MD9 bei 49 %. Bei dem Konstrukt
MD9.MGMT wurde in der FACS-Analys eine Gentransferrate von 25 % ermittelt und
die Gentransferrate des Progenitor-Assays lag hier um 52 % über dem Niveau der
FACS-Analyse.
69

Ergebnisse
0
8080
20
40
60
GFP
pos
itive
Kol
onie
n (%
)
20
40
60
GFP
pos
itive
Kol
onie
n (%
)
MH71 MH71.MGMT MD9 MD9.MGMTVektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MH71.MGMT
--
EM140
MD9
pcziGag / pcziPol
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
0MH71 MH71.MGMT MD9 MD9.MGMTVektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MH71.MGMT
--
EM140
MD9
pcziGag / pcziPol
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MH71.MGMT
--
EM140
MD9
pcziGag / pcziPol
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
MH71.MGMT
--
EM140
MD9
pcziGag / pcziPol
EM140
MD9.MGMT
pcziGag / pcziPol
EM140
Abbildung 16: Progenitor-Assay von humanen Zellkolonien, die zuvor mit
foamyviralen Vektoren transduziert wurden
4.2.4 Transduktionseffizienz verschiedener foamyviraler Konstrukte
Das Hüllprotein EM140 zeigte auf den Produzentenzellen eine deutliche Toxizität.
Die Titer der unterschiedlichen foamyviralen Hüllproteine wurden bereits verglichen.
An dieser Stelle soll die Transduktionseffizienz verschiedener foamyviraler
Konstrukte und deren Abkömmlinge, die das Transgen MGMTp140k tragen und mit
den Hüllproteinen EM140 und SM04 verpackt sind, auf den Zielzellen verglichen
werden. Die Höhe des Virustiters wurde vor und nach dem Zentrifugieren ermittelt.
Die Gentransferrate der Konstrukte wurde an der Anzahl CD34+ Zellen in der FACS-
Analyse und dem Anteil GFP positiver Zellen im Progenitor-Assay bestimmt. Zur
70

Ergebnisse
Generierung der Virusüberstände wurde bei den Konstrukten MD9 und MD9.MGMT
die Helferplasmide pcziGag und pcziPol eingesetzt. Alle Konstrukte wurden einmal
mit dem Hüllprotein EM140 und einmal mit dem SM04 Hüllprotein verpackt. Von
jedem Konstrukt wurde der Virusüberstand von 8 Zellkulturplatten mit einem
Durchmesser von 10 cm gepoolt und zentrifugiert. Das Viruspellet wurde in 1 ml
Medium aufgenommen und frisch zur Transduktion von humanen CD34+ Zellen
eingesetzt. Durch das Zentrifugieren konnte das Volumen, in dem sich das Virus
befindet, um 97,5 % reduziert und die Konzentration entsprechend gesteigert werden.
Die Virustiter sind logarithmisch dargestellt.
1,00E-01SM04 SM04 EM140 EM140
1,00E+00
1,00E+01
1,00E+03
1,00E+04
1,00E+05
1,00E+06
1,00E+07
1,00E+08
MH71 SM04 MH71.MGMT MD9 SM04 MD9.MGMT MH71 EM140 MH71.MGMT MD9 EM140 MD9.MGMT
TE /
ml
1,00E+02
1,00E-01SM04 SM04 EM140 EM140
1,00E+00
1,00E+01
1,00E+03
1,00E+04
1,00E+05
1,00E+06
1,00E+07
1,00E+08
MH71 SM04 MH71.MGMT MD9 SM04 MD9.MGMT MH71 EM140 MH71.MGMT MD9 EM140 MD9.MGMT
TE /
ml
1,00E+02
Vektor
l
MH71 MD9
pcziPol
MH71MH71.MGMT MD9.MGMT
pcziPol
MH71.MGMT MD9
pcziPol
MD9
ag /
pcziPol
Vektor
l
MH71 MD9
pcziPol
MH71MH71.MGMT MD9.MGMT
pcziPol
MH71.MGMT MD9
pcziPol
MD9
ag /
pcziPol
Helferp asmid -- pcziGag / ---- pcziGag / -- pcziGag / pcziGHelferp asmid -- pcziGag / ---- pcziGag / -- pcziGag / pcziG
Hüllprotein SM04 SM04 EM140SM04 SM04 EM140 EM140 EM140Hüllprotein SM04 SM04 EM140SM04 SM04 EM140 EM140 EM140
Abbildung 17: Virustiter vor und nach der Zentrifugation der Virusüberstände
logarithmisch dargestellt
Durch die Zentrifugation konnte der Virustiter deutlich erhöht werden. In Abhängigkeit
des verwendeten Hüllproteins war die Konzentration von Virusüberstand
71

Ergebnisse
unterschiedlich erfolgreich. Der Virustiter der Konstrukte, die mit dem Hüllprotein
SM04 verpackt worden waren, konnten um das 9 fache (MD9), 11,7 fache (MH71),
14,7 fache (MH71.MGMT) resp. um das 17,9 fache (MD9.MGMT) gegenüber dem
Ausgangswert gesteigert werden. Virustiter der Konstrukte, die mit dem Hüllprotein
EM140 verpackt worden sind, konnten um das 27,6 fache (MH71), 28,3 fache
(MH71.MGMT), 28,3 fache resp. um das 46,5 fache (MD9.MGMT) gegenüber dem
Ausgangswert erhöht werden (Abbildung 17). Es ist deutlich zu erkennen, dass die
Zentrifugation der mit EM140 verpackten Viren zu einer wesentlich stärkeren
Zunahme des Virustiters führt, als es bei SM04 der Fall ist. Im Mittel erhöht sich der
irustiter bei den mit EM140 verpackten Viren um das 32,8 fache, während sich der
Titer bei den mit SM04 verpackten Viren nur um das 13,3 fache erhöhen ließ.
Zusammenfassend kann festgestellt werden, dass das Vermögen zur Konzentration
des Virustiters durch Zentrifugieren bei dem Hüllprotein EM140 etwa 60 % größer ist,
als bei SM04.
Die Höhe der Virustiter verschiedener Konstrukte unterscheiden sich signifikant
(p=0,0209), so dass ein Einfluss der Konstrukte auf die Titerhöhe angenommen
werden kann.
Die zentrifugierten Virusüberstände wurden zur Transduktion von humanen CD34+
Zellen eingesetzt. Ein Teil der Zellen wurde in Progenitor-Assays ausgesät, an einem
anderen Teil wurde 5 Tage nach der Transduktion der Anteil GFP positiver Zellen
mittels FACS-Analyse ermittelt. Die Ergebnisse einer solchen FACS-Messung sind in Abbildung 18 dargestellt.
V
72

Ergebnisse
0
10
20
30
40
50
60G
FP p
ositi
ve Z
elle
n (%
MH71 SM04 MH71.MGMTSM04
MD9 SM04 MD9.MGMTSM04
MH71 EM140 MH71.MGMTEM140
MD9 EM140 MD9.MGMTEM140
)
Vektor
Helferplasmid
Hüllprotein
MH71
--
SM
MD9
pcziGag /
pcziPol
MH71
--
MH71.MGMT
--
MD9.MGMT
pcziGag /
pcziPol
MH71.MGMT
--
MD9
pcziGag /
pcziPol
MD9.MGMT
pcziGag /
pcziPol
0
10
20
30
40
50
60G
FP p
ositi
ve Z
elle
n (%
04 SM04 EM140SM04 SM04 EM140 EM140 EM140
MH71 SM04 MH71.MGMTSM04
MD9 SM04 MD9.MGMTSM04
MH71 EM140 MH71.MGMTEM140
MD9 EM140 MD9.MGMTEM140
)
0
10
20
30
40
50
60G
FP p
ositi
ve Z
elle
n (%
)
MH71 SM04 MH71.MGMTSM04
MD9 SM04 MD9.MGMTSM04
MH71 EM140 MH71.MGMTEM140
MD9 EM140 MD9.MGMTEM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
SM
MD9
pcziGag /
pcziPol
MH71
--
MH71.MGMT
--
MD9.MGMT
pcziGag /
pcziPol
MH71.MGMT
--
MD9
pcziGag /
pcziPol
MD9.MGMT
pcziGag /
pcziPol
Vektor
Helferplasmid
Hüllprotein
MH71
--
SM
MD9
pcziGag /
pcziPol
MH71
--
MH71.MGMT
--
MD9.MGMT
pcziGag /
pcziPol
MH71.MGMT
--
MD9
pcziGag /
pcziPol
MD9.MGMT
pcziGag /
pcziPol
04 SM04 EM140SM04 SM04 EM140 EM140 EM14004 SM04 EM140SM04 SM04 EM140 EM140 EM140
ie Gentransferrate war, gemessen anhand des Anteils GFP positiver Zellen, in
iesem Versuch für das Hüllprotein SM04 zwischen 1,7 und 21,0 % und für das
Hüllprotein EM140 zwischen 18,8 und 58,7 %. Die Gentransferrate zeigte eine
deutliche Abhängigkeit von der Höhe des Virustiters. Die Konstrukte MH71.MGMT
SM04, MH71.MGMT EM140 und MD9.MGMT EM140 hatten einen höheren Virustiter,
als die Varianten ohne MGMTp140k. Die Gentransferraten der Konstrukte mit
MGMTp140k waren geringgradig niedriger als die der Konstrukte ohne dieses
Transgen.
Abbildung 18: Gentransferrate von foamyviralen Konstrukten mit verschiedenen
Hüllproteinen auf humanen CD34+ Zellen (5 Tage nach Transduktion mittels
Durchflusszytometrie ermittelt)
D
d
73

Ergebnisse
Als Ergebnis des Progenitor-Assays sind die arithmetischen Mittelwerte von drei
Ansätzen (Triplikate) mit 250 Zellen / ml Methylzellulose dargestellt (Abbildung 19).
Die Anzahl der gezählten Kolonien wurde als 100 % betrachtet.
0
20
40
60
80
100
MH71 SM04 MH71.MGMTSM04
MD9 SM04 MD9.MGMTSM04
MH71 EM140 MH71.MGMTEM140
MD9 EM140 MD9.MGMTEM140
GFP
pos
itive
Kol
onie
n (%
)
Vektor
Helferplasmid
Hüllprotein
MH71
--
SM04
MD9
pcziGag /
pcziPol
SM04
MH71
--
EM140
MH71.MGMT
--
SM04
MD9.MGMT
pcziGag /
pcziPol
SM04
MH71.MGMT
--
EM140
MD9
pcziGag /
pcziPol
EM140
MD9.MGMT
pcziGag /
pcziPol
EM140
0
20
40
60
80
100
MH71 SM04 MH71.MGMTSM04
MD9 SM04 MD9.MGMTSM04
MH71 EM140 MH71.MGMTEM140
MD9 EM140 MD9.MGMTEM140
GFP
pos
itive
Kol
onie
n (%
)
0
20
40
60
80
100
MH71 SM04 MH71.MGMTSM04
MD9 SM04 MD9.MGMTSM04
MH71 EM140 MH71.MGMTEM140
MD9 EM140 MD9.MGMTEM140
GFP
pos
itive
Kol
onie
n (%
)
Vektor
Helferplasmid
Hüllprotein
MH71
--
SM04
MD9
pcziGag /
pcziPol
SM04
MH71
--
EM140
MH71.MGMT
--
SM04
MD9.MGMT
pcziGag /
pcziPol
SM04
MH71.MGMT
--
EM140
MD9
pcziGag /
pcziPol
EM140
MD9.MGMT
pcziGag /
pcziPol
EM140
Vektor
Helferplasmid
Hüllprotein
MH71
--
SM04
MD9
pcziGag /
pcziPol
SM04
MH71
--
EM140
MH71.MGMT
--
SM04
MD9.MGMT
pcziGag /
pcziPol
SM04
MH71.MGMT
--
EM140
MD9
pcziGag /
pcziPol
EM140
MD9.MGMT
pcziGag /
pcziPol
EM140
Abbildung 19: Progenitor Assay von humanen Zellkolonien, die mit foamyviralen
ektoren transduziert wurden V
Die Gentransferraten der Konstrukte, die mit SM04 verpackt sind, unterscheiden sich
in den Progenitoren weniger deutlich von den mit EM140 verpackten Konstrukten, als
in der FACS-Analyse. Der Anteil GFP positiver Zellen aus den Ansätzen mit EM140
verpackter Viren zeigt eine Korrelation mit der Höhe der im FACS gemessenen
Gentransferrate. Die Konstrukte mit dem MGMTp140k haben eine niedrigere
Gentransferrate als Konstrukte ohne dieses Transgen. Die Konstrukte mit dem
Hüllprotein SM04 zeigen ein uneinheitliches Bild.
74

Ergebnisse
Die Gentransferraten der Progenitor-Assays lagen auch in diesem Versuch deutlich
über den Messwerten der FACS-Analyse. Für die Konstrukte, die mit dem Hüllprotein
SM04 verpackt wurden, lagen die Werte um 61 (MH71) bis 97 % (MD9.MGMT) über
en im FACS gemessenen Gentransferraten. Für das Hüllprotein EM140 lagen die
Zunahmen zwischen 30 (MH71) und 71 % (MD9.MGMT). Die Konstrukte, die das
Transgen MGMT trugen, zeigten eine höhere Zunahme der Gentransferrate
zwischen der FACS-Analyse und den Progenitor-Assays, als die Konstrukte ohne
MGTM. Diese Zunahme wurde unabhängig vom verwendeten Hüllprotein beobachtet.
d
75

Ergebnisse
4.3 Gentransfer in CD 34+ Marmoset Zellen
4.3.1 Gewinnung und Aufreinigung von CD34+ Marmoset Zellen
Die CD 34+ Zellen der Marmosets wurde aus dem Knochenmark isoliert. Das
Knochenmark wurde durch die Punktion der Femures gewonnen. Zur Kontrolle der
Punktionsstelle wurde der Femur des Tieres 0130 nach der Punktion röntgenologisch
untersucht (Abbildung 20). Von den Tieren 0224 und 3101 konnten in einem
Finalversuch etwa 2 ml Knochenmark entnommen werden; bei Tier 0130 konnten
durch die Punktion etwa 0,5 ml Knochenmark entnommen werden. Nach der
Aufreinigung der mononukleären Zellen aus dem Knochenmark wurden 500.000
Zellen von Tier 0224, 340.000 Zellen von Tier 3101 und 70.000 Zellen von Tier 0130
isoliert. Der Anteil CD34+ Zellen an der aufgereinigten CD34 Fraktion, lag bei 95 %.
Abbildung 20: Punktionsstelle im distalen Drittel des rechten Femurs (Tier 0130)
76

Ergebnisse
4.3.2 Retroviraler Gentransfer in CD34+ Marmoset Zellen
Abbildung 21 sind die Virustiter dargestellt, die zur Transduktion der CD34+ Zellen
rstände wurde
ei den lentiviralen Konstrukten pCL1 das Helferplasmid CD/NL-BH eingesetzt.
r
garithmisch dargestellt.
Gegenüber den Ausgangswerten konnte der Virustiter durch das Zentrifugieren je
nach Konstrukt um das 19,7 fache (MD9.MGMT), 25,7 fache (MD9), 29,8 fache
(MH71.MGMT) resp. um das 30 fache (MH71) erhöht werden. Für Tier 0224 lag die
Steigerung der Viruskonzentration durch das Zentrifugieren z
fachen (pCL1 VSV-G) resp. 22,4 fachen (pCAMSΔ lagen
die Werte zwischen 8,9 (pCL1 EM140) resp. 12,6 (pCL1 VSV-G) und bei Tier 0130
zeigten die Titrationen eine Erhöhung um das 6,6 fache (MH71 EM140) resp. um das
12,3 fache (pCAMSΔU3E EM140) gegenüber dem Ausgangswert.
In
von Marmosets eingesetzt worden sind. Zur Generierung der Virusübe
b
Als Hüllprotein wurde das VSV-G zur Pseudotypisierung des pCL1 eingesetzt.
Zusätzlich wurde das Konstrukt pCL1 mit dem foamyviralen Hüllprotein EM140
pseudotypisiert. Das foamyvirale und das gammaretrovirale Konstrukt wurden mit
dem Hüllprotein EM140 verpackt. Der Virusüberstand von 8 Zellkulturplatten mit
einem Durchmesser von 10 cm wurde gepoolt und zentrifugiert. Das Viruspellet
wurde in 1 ml Medium aufgenommen und frisch zur Transduktion der Marmoset
CD34+ Zellen eingesetzt. Durch das Zentrifugieren wurde das Volumen des
Virusüberstandes um 97,5 % verringert. Die zentrifugierten Virusüberstände, die zur
Transduktion der CD34+ Marmoset Zellen eingesetzt wurden, sind hier für jedes Tie
lo
wischen dem 10,5
U3E EM140), bei Tier 3101
77

Ergebnisse
1,00E+09
1,00E-01
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
TE /
ml
1,00E+07
1,00E+08
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1 MH71 pCAMSΔU3E
CD/NL-BH
EM140
--
EM140
--
EM140
1,00E-01
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
TE /
ml
1,00E+09
1,00E+07
1,00E+08
1,00E+09
1,00E+07
1,00E+08
1,00E-01
1,00E+00
1,00E+01
1,00E+02
1,00E+03
1,00E+04
1,00E+05
1,00E+06
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
TE /
ml
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1 MH71 pCAMSΔU3EVektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1 MH71 pCAMSΔU3E
CD/NL-BH
EM140
--
EM140
--
EM140
CD/NL-BH
EM140
--
EM140
--
EM140
ndet worden sind,
garithmisch dargestellt
Die zentrifugierten Virusüberstände wurden zur Transduktion von CD34+ Marmoset
Zellen eingesetzt. Ein Teil der Zellen wurde in Progenitor-Assays ausgesät, an einem
anderen Teil wurde am 5. Tag, bei Tier 3101 zusätzlich am Tag 9 nach der
Tier 0224
Tier 3101
Tier 0130
Abbildung 21: Virustiter nach dem Zentrifugieren der Virusüberstände, die zur
Transduktion der CD34+ Zellen der einzelnen Versuchstiere verwe
lo
78

Ergebnisse
Transduktion der Anteil der GFP positiven Zellen mittels FACS-Analyse ermittelt. Die
Messung der Zellen des Tieres 0224 ist in Scatter-Plots dargestellt (Abbildung 22).
Abbildung 22: Darstellungen der durchflusszytometrischen Messungen der
transduzierten CD34 Zellen des Tieres 0224. A: Zellen im Vorwärt rts-Scatter und das Gate um die lebenden Zellen, B-F Analyse der Gentransfereffizien Konstrukte 5 Tage nach Transfektion
+
s- und Seitwäder einzelnenz
F E D
C B
R1
A ohne Virus ohne Virus
0% 5%
88% 94% 55%
pCL1 VSV-G
pCL1 EM140 MH71 EM140 pCAMSΔU3E EM140
79

Ergebnisse
80
0
20
40
60
80
100
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GFP
pos
itive
Zel
len
(%)
Tier 0224
0
20
40
P po
sitiv
e
60
Zel
len
(%)
80
100
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GF
Tier 3101
am Tag 5 / 9
0
20
80
100
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GF
40
P po
sitiv
e
60
Zel
len
(%)
Tier 3101
am Tag 5 / 9

Ergebnisse
0
20
40
60
80
100
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GFP
pos
itive
Zel
len
(%)
Tier 0130
0
20
40
60
80
100
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GFP
pos
itive
Zel
len
(%)
Tier 0130
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Abbildung 23: Transduktionseffizienzen von CD34+ Marmoset Zellen
Die Auswertung der FACS-Messung ist für alle Tiere in der Abbildung 23 gezeigt. Bei
Tier 0224 fällt auf, dass das foamyvirale Konstrukt mit 93 % die höchste
Gentransferrate aller verwendeten Konstrukte erzielt, der Titer aber niedriger ist, als
der des pCL1 EM140 und des pCAMSΔU3E EM140. Die Transduktionseffizienz des
foamyviralen Konstrukts ist auch bei Tier 3101 hoch. Hier hat das Konstrukt MH71
EM140 den niedrigsten Virustiter, die Transfektionsrate lag mit 78 % GFP positiver
Zellen aber deutlich über der des pCL1 VSV-G und auch 6 % über der des
Gammaretrovirus.
Vergleicht man die an Tag 5 und an Tag 9 durchflusszytometrisch gemessenen
Werte des Tieres 3101, so kann festgestellt werden, dass der Anteil GFP positiver
Zellen bei allen Konstrukten abnimmt und nur das foamyvirale Konstrukt einen gleich
bleibenden Anteil GFP positiver Zellen aufweist. Das Konstrukt pCL1 EM140 führte
nur zu einer geringen Abnahme des Anteils der GFP positiven Zellen, allerdings war
81

Ergebnisse
der Anteil GFP positiver Zellen beim Konstrukt pCAMSΔU3E EM140 37 % geringer
nd beim Konstrukt pCL1 VSV-G 66 % geringer als der Ausgangswertes. Das
foamyvirale Konstrukt führte bei Tier 0130 zu einem niedrigen Titer und erreicht eine
Transfektionsrate von 23 %, die mit der des pCL1 VSV-G (27 %) vergleichbar ist. Der
Virustiter des Konstruktes pCL1 VSV-G betrug 2 x 107 TE / ml und war damit höher
als der des Konstrukts MH71 EM140.
In den Abbildungen der Progenitor-Assays sind die arithmetischen Mittelwerte von
drei Ansätzen (Triplikate) mit 250 Zellen/ml Methylzellulose, bei Tier 0130 mit 200
Zellen/ml Methylzellulose dargestellt (Abbildung 24). Die Anzahl der insgesamt
gezählten Kolonien wurde gleich 100 % gesetzt.
u
Tier 0224
0
20
40
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GFP
pos
itive
60
100
Kol
onie
n (%
)
80 Tier 0224
0
20
40
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GFP
pos
itive
60
100
Kol
onie
n (%
)
80
82

Ergebnisse
Tier 3101
0
20
pCL1 VSV-G pCL1 EM140 M
GF
40
H71 EM140 pCAMSΔU3E
P po
s
60
80
100
itive
Kol
onie
n (%
)
Tier 3101
0
20
pCL1 VSV-G pCL1 EM140 M
GF
40
H71 EM140 pCAMSΔU3E
P po
s
60
80
100
itive
Kol
onie
n (%
)
Tier 0130
0
20
40
60
80
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GFP
pos
itive
Kol
onie
n (%
)
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Tier 0130
0
20
40
60
80
pCL1 VSV-G pCL1 EM140 MH71 EM140 pCAMSΔU3E
GFP
pos
itive
Kol
onie
n (%
)
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
Vektor
Helferplasmid
Hüllprotein
pCL1
CD/NL-BH
VSV-G
pCL1
CD/NL-BH
EM140
MH71
--
EM140
pCAMSΔU3E
--
EM140
keine lebenden
Zellen
Abbildung 24: Progenitor-Assay der transduzierten CD34+ Marmoset Zellen
Die Transduktionsergebnisse der Progenitor-Assays korrelierten eng mit den
Gentransferraten, die bereits im Durchflusszytometer ermittelt werden konnten. In
dem Ansatz pCL1 EM140 waren sowohl in dem Ansatz mit 200 als auch in dem mit
83

Ergebnisse
84
800 Zellen/ml Methylzellulose keine Kolonien gewachsen. In allen drei Versuchen
wies das lentivirale Konstrukt, verpackt mit dem VSV-G, in den Progenitoren
besonders schlechte Gentransferraten auf. Es konnten nur einzelne Kolonien erkannt
werden, die GFP positiv waren. Der Anteil der GFP positiven Kolonien war bei den
Konstrukten pCL1 und pCAMSΔU3E, pseudotypisiert mit dem EM140, immer kleiner
als bei den im FACS gemessenen Gentransferraten. Die an Tag 9 bei Tier 3101 im
FACS gemessenen Gentransferraten waren bei beiden Konstrukten den Werten des
Progenitor-Assays ähnlicher. Die Werte des foamyviralen Konstrukts liegen dagegen
alle über den im FACS ermittelten Anteilen GFP positiver Zellen.
.4 MGMTp140k als Transgen
4.4.1 Transduktion hämatopoetischer Zelllinien
Zellen der Zelllinien HL60, Hel und K562 wurden mit vier unterschiedlichen
Viruskonstrukten transduziert, um transgene Zelllinien zu generieren. Verwendet
wurden die Konstrukte MH71, MH71.MGMT, MD9 und MD9.MGMT. Die
Viruskonstrukte wurden alle mit dem Hüllprotein EM140 verpackt. Für die Konstrukte
MD9 und MD9.MGMT wurden die beiden Helferplasmide pcziGag und pcziPol zur
Generierung des Virus eingesetzt. Der Virusüberstand von 8 Zellkulturplatten mit je
10 cm Durchmesser wurde gepoolt, zentrifugiert und das Viruspellet in 3 ml Medium
aufgenommen. Durch die Zentrifugation konnte der Virustiter je nach Konstrukt um
as 6 fache (MH71.MGMT), 8,7 fache (MD9), 9,4 fache (MD9.MGMT) resp. um das
Virustiter
gen nach dem Zentrifugieren lagen bei 1,7 x 106 TE / ml (MH71.MGMT), 1,4 x 107
E / ml (MD9.MGMT), 1,8 x 107 TE / ml (MD9) und der höchste lag bei 2,5 x107 TE /
4
d
10 fache (MH71) gegenüber dem Ausgangswert gesteigert werden. Die
la
T
ml (MH71).

Ergebnisse
Nach der Zentrifugation sind die Virustiter der einzelnen Konstrukte unterschiedlich
hoch. Diese Unterschiede ließen sich statistisch absichern (p = 0,0117); daraus kann
ein Einfluss der verwendeten Konstrukte auf die Titerhöhe abgeleitet werden.
0
2
60
80
100
ive
Zelle
n (%
)
0
40
MH
71.M
MD
9.
MH
71.M
MD
9.M
GM
T
MH
71
MD
9
MH
71.M
GM
T
MD
9.M
GM
T
MH
71
MD
9
GM
T
MG
MT
MH
71
MD
9
GM
T
HL60 HEL K562
GFP
pos
it
enhang nachgewiesen werden (p = 0,0373).
Abbildung 25: Transduktion von hämatopoetischen Zelllinien mittels foamyviraler
Vektoren, die das Transgen GFP tragen und Konstrukten, die zusätzlich das Gen
MGMTp140k tragen
Die Größe des Anteils transduzierter Zellen (GFP positive Zellen) war unabhängig
vom verwendeten Konstrukt. Insgesamt konnten je nach Konstrukt und Zelllinie
Gentransferraten zwischen 14 und 93 % erreicht werden.
Dagegen zeigte der Anteil der GFP positiver Zellen und somit auch die
Gentransferrate deutliche Unterschiede in Abhängigkeit zur verwendeten Zelllinie.
Hier konnte ein signifikanter Zusamm
85

Ergebnisse
Die HEL Zellen ließen sich mit den verwendeten Konstrukten besonders effizient
transduzieren (Abbildung 25).
Transgene Zelllinien, die in diesem Versuch generiert wurden, sind im Anschluss zur
Ermittlung der Resistenz gegenüber Chemotherapeutika durch MGMTp140k
eingesetzt worden.
Die transgenen Zelllinien wurden weiter kultiviert und die Gentransferrate wurde
wöchentlich gemessen (Abbildung 26).
Bei den Zelllinien HEL und K562 war der Anteil GFP positiver Zellen bei den
Konstrukten MH71 und MD9 ohne MGMT in der ersten Woche immer höher als bei
den Konstrukten mit MGMT. In der zweiten und dritten Woche nach der Transduktion
nimmt der Anteil GFP positiver Zellen an der Gesamtpopulation bei den Konstrukten
ohne MGMT sogar leicht zu, bei den Konstrukten mit MGMT fällt er ab. Die
Gentransferraten von MH71 und MD9 blieben über Wochen hinweg auf einem gleich
bleibend hohen Niveau. Bei den Konstrukten MH71.MGMT und MD9.MGMT nahm
der Anteil GFP positiver Zellen von Woche zu Woche kontinuierlich ab. Bei den HL60
Zellen war die Abnahme bei den MGMT tragenden Konstrukten nicht so auffällig, der
nteil GFP positiver Zellen ist ehr als Konstant zu bezeichnen.
A
86

Ergebnisse
HL60
60
20
40
GFP
pos
itive
Zellen (%
)
01 2 3 4 5 6 7 8
AAAA BBBB CCCC DDDD
0
20
40
60
80
100
1 2 3 4 5 6 7 8
GFP
pos
itive
Zellen (%
)
aaaa bbbb cccc dddd
HEL
0
20
40
60
1 2 3 4 5 6 7 8
GFP
pos
itive
Zellen (%
)
aaaa bbbb cccc dddd
K562
Vektor
Helferplasmid
Hüllprotein
MH71
--
EM140
Woche
MD9
pcziGag /
pcziPol
EM140
MH71.
MGMT
--
EM140
MD9.
MGMT
pcziGag /
pcziPol
EM140 Abbildung 26: Transgene Zelllinien, deren Gentransferraten über acht Wochen hinweg wöchentlich gemessen wurden
87

Ergebnisse
4.4.2 Selektion von transduzierten Zelllinien mittels BCNU in vitro
Die Resistenz transduzierter Zellen, die das Transgen MGMTp140k mittels der
Vektoren MH71.MGMT oder MD9.MGMT erhalten, wurde mit der Resistenz nicht
transduzierter Zellen verglichen. Die Zellen wurden einmalig mit 0, 20, 80 oder 160
µM BCNU behandelt. Die Messwerte aus drei Versuchen sind als arithmetische
Mittelwerte dargestellt (Abbildung 27). Die Grafik enthält die Prozentwerte der
lebenden und GFP positiven Zellen, gemessen an der Gesamtzahl, der im FACS
registrierten Ereignisse.
HL60
0
20
40
60
80
1 2 3 4 5 6 7 8 9 10 11 12
Zelle
n (%
)
88

Ergebnisse
HEL
0
20
40
60
Zelle
n (
80
1 2 3 4 5 6 7 8 9 10 11 12
%)
K562
0
20
40
60
80
1 2 3 4 5 6 7 8 9 10 11 12
Zelle
n (%
)
Virus
BCNU (μM)
--
--
MH71
MGMT
--
MD9
MGMT
--
--
20
MH71
MGMT
20
MD9
MG
20
G
20
MT
--
80
MH71
MGMT
80
MD9
MGMT
80
--
160
MH71
MGMT
160
MD9
MGMT
160
Virus
BCNU (μM)
--
--
MH71
MGMT
--
MD9
MGMT
--
--
20
MH71
MGMT
20
MD9
M MT
--
80
MH71
MGMT
80
MD9
MGMT
80
--
160
MH71
MGMT
160
MD9
MGMT
160
Anteil lebender Zellen (%)
Anteil GFP positiver Zellen unter den lebenden Zellen (%)
Abbildung 27: Selektion verschiedener transduzierter Zelllinien mittels BCNU
89

Ergebnisse
Die verschiedenen Zelllinien zeigten keine signifikanten Unterschiede in der
Überlebensrate nach Behandlung; somit ließ sich eine ähnlich hohe Sensitivität aller
Zellen gegenüber BCNU feststellen. Dagegen bestand zwischen den verwendeten
Konstrukten einen signifikanter Unterschied in der Überlebensrate der Zellen (p =
0,0050).
Betrachtet man den prozentualen Anteil GFP positiver Zellen an der Population
überlebender Zellen, so lässt sich feststellen, dass die Zelllinien HL60 und K562 mit
den Konstrukten MH71.MGMT und MD9.MGMT jeweils den höchsten prozentualen
Anteil GFP positiver Zellen innerhalb der gleichen Zelllinie aufwiesen. HEL Zellen
zeigten nur nach Verwendung des Konstrukts MD9.MGMT einen signifikant höheren
Anteil GFP positiver Zellen (p = 0,0004). Dem Konstrukt MH71.MGMT kann,
bezogen auf diesen Parameter, kein signifikanter Einfluss nachgewiesen werden.
90

Ergebnisse
4.4.3 Mehrmalige Behandlung von transgenen HL60 Zellen mit BCNU
In diesem Versuch sind exemplarisch HL60 Zellen verwendet worden. Es wurden die
Zahl überlebender Zellen und der Anteil GFP positiver Zellen von transduzierten
ellen mit nicht transduzierten Zellen verglichen. Die Zellen wurden über mehrere
die
ehandlung nicht zu einer Zunahme der GFP positiven Zellen am Anteil der
berlebenden Zellen. Dagegen zeigen die Konstrukte MH71.MGMT und MD9.MGMT
eine signifikante Zunahme der GFP positiven Zellen an dem Anteil Überlebender
Zellen (p = 0,0010 resp. p = 0,0013). Demnach ist die effektive Wirkung des
Transgen MGMTp140K als Resistenzgen gegenüber dem Chemotherapeutikum
BCNU nachgewiesen.
Z
Tage hinweg mit BCNU behandelt.
In der Grafik sind die arithmetische Mittelwerte der drei Wiederholungen dargestellt
(Abbildung 28).
Das verwendete Konstrukt und die Art der Behandlung hatten einen signifikanten
Einfluss auf den Anteil überlebender Zellen (p = 0,0011 resp. p = 0,0028). Bei den
beiden Konstrukte MH71 und MD9, die keine Resistenzgene tragen, führt
B
ü
91

Ergebnisse
ohne Virus
0ohne BCNU
20
80
20mM 40mM 60mM 80mM 20-20mM 20-20-20mM
20-20-20-20mM
20-40mM 20-40-80mM
40
60
Zelle
n (%
)
MH71
0
20
40
60
80
Zelle
n (%
)
ohne BCNU 20mM 40mM 60mM 80mM 20-20mM 20-20-20mM
20-20-20-20mM
20-40mM 20-40-80mM
MD9
0
20
40
60
80
ohne BCNU 20mM 40mM 60mM 80mM 20-20mM 20-20-20mM
20-20-20-20mM
20-40mM 20-40-80mM
Zelle
n (%
)
BCNU (μM) -- 20 40 60 80 20-20 20-20-20 20-20-
20-20
20-40 20-40-80BCNU (μM) -- 20 40 60 80 20-20 20-20-20 20-20-
20-20
20-40 20-40-80
92

Ergebnisse
MH71.MGMT
0
20
40
60
80
ohne BCNU 20mM 40mM 60mM 80mM 20-20mM 20-20-20mM
20-20-20-20mM
20-40mM 20-40-80mM
Zelle
n (%
)
MD9.MGMT
0
20
40
60
80
ohne BCNU 20mM 40mM 60mM 80mM 20-20mM 20-20-20mM
20-20-20-20mM
20-40mM 20-40-80mM
Zelle
n (%
)
BCNU (μM) -- 20 40 60 80 20-20 20-20-20 20-20-
20-20
20-40 20-40-80BCNU (μM) -- 20 40 60 80 20-20 20-20-20 20-20-
20-20
20-40 20-40-80
Anteil lebender Zellen (%)
Anteil GFP positive Zellen unter den lebenden Zellen (%)
Abbildung 28: Selektion transduzierter HL60 Zellen mittels BCNU Behandlung über
mehrere Tage
93

Ergebnisse
4.4.4 Selektion transgener HL60 Zellen mit BG und BCNU in vitro
Der Anteil überlebender Zellen und der prozentuale Anteil GFP positiver Zellen am
Anteil der überlebenden Zellen wurde bei transduzierten und nicht transduzierten
HL60 Zellen verglichen, die vor der BCNU Behandlung mit BG inkubiert wurden.
Sowohl die mit BG vorbehandelten, als auch die nicht vorbehandelten Zellen wurden
einmalig mit 0, 10, 20, 40 oder 60 µM BCNU behandelt.
In der Grafik sind die arithmetischen Mittelwerte der drei Wiederholungen (Triplikate)
eines Versuchs dargestellt (Abbildung 29).
Bei allen verwendeten Konstrukten zeigte sich, das die Art der Behandlung einen
signifikanten Einfluss auf den Anteil überlebender Zellen und den Anteil GFP
positiver Zellen an den überlebenden Zellen hat. In der grafischen Darstellungen der
Versuchsergebnisse wird deutlich, dass die Überlebensrate der Zellen mit dem
Transgen MGMTp140k deutlich höher war, als die von Zellen, die mit anderen
Konstrukten transfiziert wurden. Der Unterschied wird nach einer Behandlung der
Zellen mit BG in den Konzentrationen 20 und 40 µM BCNU noch deutlicher. Der
Anteil GFP positiver Zellen an der Anzahl überlebender Zellen nimmt bei den
MGMTp140k tragenden Konstrukten stärker zu, als bei den Konstrukten ohne
MGMTp140k.
94

Ergebnisse
4.4.5 Selektion transgener CD34 Marmoset Zellen mit BG und BCNU
erung des Virus eingesetzt.
entrifugation lagen die Virustiter für die Konstrukte MH71 und MD9.MGMT bei 1 x 7 7 6
influss des verwendeten Konstrukts (p = 0,0281). Das verwendete
auf die numerische Überlebensrate der
Zellen.
+
Die CD34+ Zellen des Marmoset 0224 wurden mit vier unterschiedlichen
Viruskonstrukten transduziert. Die verwendeten Viruskonstrukte wurden alle mit dem
Hüllproteinen EM140 verpackt. Für die Konstrukte MD9 und MD9.MGMT wurden die
beiden Helferplasmide pcziGag und pcziPol zur Generi
Der Virusüberstand von 8 Zellkulturplatten mit je 10 cm Durchmesser wurde gepoolt,
zentrifugiert und das Viruspellet in 1 ml Medium aufgenommen. Nach der
Z
10 , für MD9 bei 1,5 x 10 und für MH71.MGMT bei 2 x 10 TE pro ml.
Die Transduktionseffizienz war bei dem Konstrukt MD9 mit 79 % transduzierter
Zellen am höchsten. Für die Konstrukte MH71 konnte ein Anteil von 68 %, für
MH71.MGMT von 61 % und für MD9.MGMT ein Anteil von 59 % GFP positiver Zellen
ermittelt werden.
Die unterschiedliche Behandlung der Zellen hatte einen signifikanten Einfluss auf
den Anteil der überlebenden Zellen (p = 0,0011). Der Anteil GFP positiver Zellen resp.
transgenen Zellen am Anteil der überlebenden Zellen stand ebenfalls unter einem
signifikanten E
Konstrukt zeigt hingegen keinen Einfluss
95

Ergebnisse
100
ohne Virus80
0
20
40
60
1 2 3 4 5 6 7 8 9 10
Zelle
n (%
)
MH71
0
100
20
40
1 2 3 4 5 6 7 8 9 10
Ze
60
80
llen
(%)
MD9
0
20
40
60
80
100
1 2 3 4 5 6 7 8 9 10
Zelle
n (%
)
MD9
0
20
40
60
80
0
1 2 3 4 5 6 7 8 9 10
Zelle
n (%
)
10
BG
BCNU (μM)
--
--
+
--
+
10
+
20
+
40
+
60
--
10
--
20
--
40
--
60
BG
BCNU (μM)
--
--
+
--
+
10
+
20
+
40
+
60
--
10
--
20
--
40
--
60
96

Ergebnisse
MH71.MGMT
0
20
40
60
80
100
1 2 3 4 5 6 7 8 9 10
Zelle
n (%
)
MD9.MGMT
0
20
40
60
80
100
1 2 3 4 5 6 7 8 9 10
Zelle
n (%
)
BG
BCNU (μM)
--
--
+
--
+
10
+
20
+
40
+
60
--
10
--
20
--
40
--
60
BG
BCNU (μM)
--
--
+
--
+
10
+
20
+
40
+
60
--
10
--
20
--
40
--
60
Anteil lebender Zellen (%)
Anteil GFP positiver Zellen unter den lebenden Zellen (%)
Abbildung 29: Selektion transduzierter HL60 Zellen mittels BG und BCNU
97

Ergebnisse
0
20
40
60
80
100
ohne BG u. BCNU BG 20mM BCNU BG + 20mM BCNU
Zelle
n (%
) ohne Virus
0
20
40
60
80
100
ohne BG u. BCNU BG 20mM BCNU BG + 20mM BCNU
Zelle
n (%
)
MH71
0
20
40
60
80
100
ohne BG u. BCNU BG 20mM BCNU BG + 20mM BCNU
Zelle
n (%
)
MD9
BG
BCNU (μM)
--
--
+
--
+
20
--
20
BG
BCNU (μM)
--
--
+
--
+
20
--
20
98

Ergebnisse
0
20
40
60
80
100
ohne BG u. BCNU BG 20mM BCNU BG + 20mM BCNU
Zelle
n (%
) MH71.MGMT
0
20
40
60
80
100
ohne BG u. BCNU BG 20mM BCNU BG + 20mM BCNU
Zelle
n (%
)
MD9.MGMT
BG
BCNU (μM)
--
--
+
--
+
20
--
20
Anteil lebender Zellen (%)
Anteil GFP positiver Zellen unter den lebenden Zellen (%)
Abbildung 30: Selektion transduzierter CD34+ Zellen des Tieres 0224 mittels BG und
BCNU
99

Ergebnisse
4.4.6 Selektion transgener CD34+ Marmoset Progenitoren mit BG und BCNU
Für diesen Versuch wurden Zellen aus dem vorangegangenen Experiment „Selektion
von transduzierten CD34+ Marmoset Zellen mit BG und BCNU“ verwendet. Nach
Behandlung mit BG und BCNU wurden transduzierte CD34+ Zellen entnommen und
in Methylzellulose ausgesät. Die Kolonien wurden nach 14 Tagen gezählt und die
Ergebnisse fotografisch dokumentiert (Abbildung 31). In der Abbildung stellten sich
monoklonale Zellkolonien, sofern sie nach erfolgreicher Transduktion das Transgen
GFP tragen, unter Fluoreszenzlicht als grüne Kolonie da. Zellen, die mit den
Konstrukten MH71 und MD9 transduziert und mit BG und BCNU behandelt wurden,
zeigen keine Koloniebildung, sondern nur das Überleben einzelner Zellen. Dagegen
konnte bei Zellen, die mit den Konstrukten MH71.MGMT und MD9.MGMT
transduziert und zusätzlich zu dem GFP Gen das Transgen MGMTp140k tragen, ein
deutliches Koloniewachstum festgestellt werden.
Unbehandelt Behandelt mit BG und 20 µM BCNU Beschreibung
CD34+ Marmoset Zellen MD9
monoklonale Zellkolonien von CD34+ Marmoset
Zellen
MD9.MGMT
Abbildung 31: Progenitor-Assay von Tier 0224 im Fluoreszenzlicht, behandelt mit BG
nd BCNU u
100

Ergebnisse
In Abbildung 32 sind die arithmetischen Mittelwerte der gezählten Kolonien aus den
nsätzen gezeigt, in denen 200 Zellen pro ml Methylzellulose ausgesät wurden. A
In den Versuchen, in denen die Zellen zuvor mit Virusüberständen transduziert
wurden, sind etwa 50 % weniger Kolonien gewachsen, als bei den nicht
transduzierten Zellen. Von den nicht mit MH71 oder MD9 transduzierten, wohl aber
mit BG und 20 µM BCNU behandelten Zellen, sind keine Kolonien gewachsen. In
Ansätzen, in denen die Zellen das Transgen MGMTp140k tragen, wuchsen
vereinzelte Kolonien. Bei dem Konstrukt MD9.MGMT konnten einige Kolonien
nachgewiesen werden, die jedoch nicht fluoreszieren.
101

Ergebnisse
0
10
20
30
unbehandelt BG 20 m
Kol
onie
n
40
M BCNU BG + 20 mM BCNU
50
ohne Virus
0
10
20
30
40
50
unbehandelt BG 20 mM BCNU BG + 20 mM BCNU
Kol
onie
n
MH71
0
10
20
30
40
50
unbehandelt BG 20 mM BCNU BG + 20 mM BCNU
Kol
onie
n
MD9
BG
BCNU (μM)
--
--
+
--
+
20
--
20
BG
BCNU (μM)
--
--
+
--
+
20
--
20
102

Ergebnisse
0
10
20
30
40
50
unbehandelt BG 20 mM BCNU BG + 20 mM BCNU
Kol
onie
nMH71.MGMT
0
10
20
30
40
50
unbehandelt BG 20 mM BCNU BG + 20 mM BCNU
Kol
onie
n
MD9.MGMT
BG
BCNU (μM)
--
--
+
--
+
20
--
20
Kolonien die GFP negativ sind
Anteil GFP positiver Kolonien unter den gewachsenen Kolonien
Abbildung 32: Progenitor-Assays transduzierter CD34+ Zellen des Tieres 0224,
behandelt mit BG und BCNU
103

Diskussion
5. Diskussion
Das Ziel der vorliegenden Arbeit war, foamyvirale Vektoren als Gentransfersystem
für humane und Marmoset CD34+ Zellen zu etablieren und ihre Effizienz mit anderen
Gentransfersystemen zu vergleichen. Als Transgen wurden dabei GFP und die
MGMT p140k-Mutante genutzt, um zum einen den Gentransfer zu optimieren und
zum anderen Selektionsstrategien der transduzierten CD34+ Zellen mit bestimmten
Chemotherapeutika systematisch zu untersuchen. Darüber hinaus sollte geprüft
werden, ob sich Marmosets als Großtiermodel für präklinische Gentherapiestudien
zur Validierung der Sicherheit und Effizienz foamyviralen Vektoren eignen.
Gammaretroviren sind bereits erfolgreich in klinischen Studien eingesetzt worden,
um therapeutische Gene stabil in CD34+ Stamm- und Progenitorzellen einzubringen.
Von 19 Patienten, die an X-linked-SCID oder ADA-SCID erkrankt waren, konnten 18
mit Hilfe von gammaretroviralen Vektoren phänotypisch geheilt werden
(CAVAZZANA-CALVO et al. 2000; AIUTI et al. 2002; GASPAR et al. 2004). Aus der
Gruppe von 10 Patienten mit SCID, deren CD34+ Zellen mit gammaretroviralen
Transfervektoren transduziert worden waren, entwickelten drei Patienten ein
Malignom (HACEIN-BEY-ABINA et al. 2003b). Bei zwei Patienten ist bekannt, dass
die Malignome auf Integrationen der gammaretroviralen Transfervektoren in die
Nähe des LMO2 Proto-Onkogen Promotors zurückzuführen sind (HACEIN-BEY-
ABINA et al. 2003b). Die Integration des retroviralen Vektors in diesem
Genomabschnitt führt zu einer aberranten Transkription und Expression des LMO2
Gens. Nach d en Vektoren zur Transduktion von
CD34+ Zellen wurden in verschiedenen Tieren wie Mäusen (LI et al. 2002) und Affen
(SEGGEWISS et al.
eitdem bekannt geworden ist, dass die Integration der Vektoren in der
ome
erden die Vor- und Nachteile, die integrierende
ektorsysteme mit sich bringen, sehr frequent und kontrovers diskutiert (DROPULIC
005; FERGUSON et al. 2005). Von Gammaretroviren ist schon seit längerem
em Einsatz von gammaretroviral
2006) ebenfalls das Entstehen von Malignome beobachtet.
S
Nachtbarschaft von Proto-Onkogenen ursächlich für die Entstehung der Malign
in diesen Studien war, w
V
2
104

Diskussion
bekannt, dass sie durch Insertationsmutagenese zur Entstehung von Malignomen
hren können. Gammaretroviren integrieren bevorzugt in zelluläre Promotorregionen
etroviren
tegrieren zweimal häufiger in diese CpG-Inseln als Foamyviren (NOWROUZI et al.
fü
(WU et al. 2003), dagegen integrieren Lentiviren besonders häufig in solche
Regionen, die transkriptionell aktiv sind (SCHRODER et al. 2002). Die, in der
vorliegenden Untersuchung eingesetzten Foamyviren zeigen dagegen keine
Präferenzen, in aktive Bereiche des Genoms zu integrieren (NOWROUZI et al. 2006;
TROBRIDGE et al. 2006). Auch die Aktivität der Gene hat keinen Einfluss auf die
Integrationsstellen von Foamyviren (TROBRIDGE et al. 2006). Die Bereiche des
Genoms, die besonders viel CpG Nukleotide (CpG-Inseln) enthalten, sind häufig vor
Genen lokalisiert und sind an deren Regulation beteiligt. Gammar
in
2006; TROBRIDGE et al. 2006). Das Integrationsprofil des Foamyvirus unterscheidet
sich somit zu einem nicht unwesentlichen Teil von dem Integrationsprofil anderer
Retroviren.
Für die Verwendung foamyviraler Vektoren spricht neben dem Integrationsprofil
dieser Viren auch die Tatsache, dass seit ihrer Entdeckung vor über 50 Jahren
(RUSTIGIAN et al. 1955) keine Erkrankung mit ihnen assoziiert werden konnte.
Diese Beobachtung kann auch als Hinweis dafür genommen werden, dass auch die
Wildtyp Foamyviren keine Insertionsmutagenese auslösen. Menschen, die durch
Affenbisse mit Foamyviren infiziert wurden, haben bis heute keine Erkrankung
aufgrund der Infektion erfahren (SCHWEIZER et al. 1997; CALLAHAN et al. 1999).
5.1 Optimierte Herstellung von Foamyviren, Lentiviren und Gammaretroviren
Toxizität von foamyviralen Hüllproteinen Um überhaupt die Effizienz des Gentransfers von Gammaretroviren und Lentiviren
direkt mit der von Foamyviren vergleichen zu können, ist es notwendig, ein
Hüllprotein zu verwenden, das sowohl foamyvirale Vektoren als auch
gammaretrovirale und lentivirale Vektoren effektiv verpacken kann. Diese
Grundvoraussetzung erlaubt den direkten Vergleich verschiedener Vektorsysteme,
105

Diskussion
weil nun alle drei rekombinanten Retroviren denselben zellulären Rezeptor zum
Eintritt in die Zelle nutzen und die Effizienz (bei gleichen Titern) wesentlich von der
Biologie des Wildtyp Virus abhängt. Dieser Vergleich ist legitim, wenngleich noch
nicht bekannt ist, welche zelluläre Membranstruktur den Foamyviren als Rezeptor
dient. Alle bisher getesteten Zellen von Vögeln, Reptilien und Säugetieren ließen sich
mit Foamyviren infizieren; daher ist zu vermuten, dass der Rezeptor ubiquitär auf
allen Zellmembranen exprimiert wird (RUSSELL et al. 1996; HILL et al. 1999;
MERGIA u. HEINKELEIN 2003).
In früheren Untersuchungen war ein effizienter Gentransfer mit gammaretroviralen
und lentiviralen Konstrukten, die mit foamyviralen Hüllproteinen pseudotypisiert
worden waren, nicht möglich (LEURS 2003). Diese Versuche wurden entweder mit
dem Wildtyp Hüllprotein des HFV durchgeführt, oder es wurden chimäre Hüllproteine
verwendet, die aus Teilen des HFV und des MLV Hüllproteins bestanden
(LINDEMANN et al. 1997; LEURS 2003). Durch Punktmutationen an den
),
ie zur Bildung von Riesenzellen und letztlich zum Absterben der Zellen führt. Diese
elltoxische Eigenschaft ist sowohl in vitro als auch in vivo zu beobachten. Humane
CD34+ Zellen, die zuvor mit Foamyviren transfiziert worden waren, wachsen nach
r
roviralen oder lentiviralen Vektoren
Ubiquitinierungsstellen der leader-Region des Hüllproteins wurde eine effiziente
Pseudotypisierung anderer Viren ermöglicht. Die Pseudotypisierung dieser
Hüllproteine ist bei gammaretroviralen und lentiviralen Vektoren so effektiv, dass der
auf HT1080 Zellen gemessenen Virustiter zum Teil deutlich über dem der
foamyviralen Vektoren lag.
Ein Nachteil foamyviraler Hüllproteine in Bezug auf ihrer Eignung in
Transfervektorsystemen ist ihre sehr starke fusiogene Wirkung (STANKE et al. 2005
d
z
eine Transplantation in NOD/SCID Mäuse schlechter an, als Zellen, die mit VSV-G
oder GaLV pseudotypisierten, gammaret
transduziert worden waren (LEURS et al. 2003). Die toxischen Eigenschaften des
Hüllproteins EM140 zeigten sich besonders bei der Transduktion von CD34+ Zellen
des Marmosets 0130. Hier hat der sehr hohe Virustiter des Konstruktes pCL1 in
Verbindung mit CD/NL-BH die Zellen, die zur Transduktion eingesetzt worden waren,
soweit geschädigt, dass im Progenitor-Assay keine Zellkolonien gewachsen sind. In
106

Diskussion
einer Untersuchung an transduzierten CD34+ Marmoset Zellen, die mit BG und
BCNU behandelt wurden, sind im Progenitor-Assay 50 % weniger Kolonien in den
transduzierten Ansätzen gewachsen, als in einem Ansatz ohne Virus. Eine
Zytotoxizität von 50 % kann insbesondere dann als zu hoch angesehen werden,
wenn man berücksichtig, dass für die Transduktion und die Transplantation oft nur
eine begrenzte Anzahl CD34+ Zellen zur Verfügung stehen.
Als alternatives foamyvirales Hüllprotein stand das SM04 zur Verfügung. Dieses
Hüllprotein stammt vom SFV und auch hier wurden die Ubiquitinierungsstellen in der
rs mit der fusiogenen Wirkung des Hüllproteins korreliert
leader-Region des Hüllproteins so punktmutiert, dass dieses Hüllprotein in der Lage
ist, auch Gammaretroviren und Lentiviren effektiv zu pseudotypisieren. Im Rahmen
dieser Untersuchung wurde die Transduktionseffizienz beider Verpackungsplasmide
verglichen. Die Fähigkeit der Hüllproteine EM140 und SM04 humane CD34+ Zellen
zu transduzieren wurde anhand verschiedener Versuche evaluiert. Das SM04
Hüllprotein übte auf die Produzentenzellen eine deutlich geringere fusiogene
Wirkung aus, als das EM140. Allerdings waren die Virustiter der foamyviralen
Konstrukte, die mit dem Hüllprotein SM04 verpackt wurden, niedriger als die
Virustiter der Viruskonstrukte die mit dem Hüllprotein EM140 verpackt wurden. Aus
diesen Ergebnissen lässt sich ableiten, dass die Effektivität der Virusproduktion und
damit die Höhe des Virustite
sind.
Fragen des Vektordesigns Mit dem Ziel, einen möglichst effektiven Vektor etablieren zu können, wurden neben
den Hüllproteinen auch die foamyviralen Vektoren selbst auf zelltoxisch
Eigenschaften untersucht. Es konnte festegestellt werden, dass die Toxizität der
Hüllproteine unabhängig vom eingesetzten foamyviralen Vektor auftrat. Neben dem
Vektor MH71 wurde der foamyvirale Vektor MD9, der keine viralen Gene mehr
enthält, auf seine Eignung für den Gentransfer getestet. Da in diesem Vektor keine
viralen Gene mehr vorhanden sind, wird eine Rekombination des Virus in vivo
erschwert. Die einzelnen Gene, die zur Generierung eines Virus benötigt werden,
107

Diskussion
sind auf mehrere „Helferplasmide“ verteilt; diese müssen für die Virusproduktion der
Produzentenzelle gemeinsam und zeitgleich zur Verfügung gestellt werden.
Die Verteilung einzelner Signale auf unterschiedliche Plasmide führt zu einer
Erhöhung der Sicherheit eines Vektorsystems, sie ist jedoch mit dem Nachteil
behaftet, dass bei der Virusproduktion deutlich geringere Virustiter auf HT1080
Zellen erzielt werden. Der Virustiter des 2-Plasmidsystems MH71 EM140 war
ines Vektors nur Helferplasmide enthalten und somit nicht infektiös sind. Ein
rund, warum die Virustiter der beiden 4-Plasmidsysteme variierten, ist
der Helferplasmide lokalisiert, da sich die Plasmide nur
der vorliegenden Arbeit wurden die Titer durch Zentrifugation der Virusüberstände
deutlich höher, als der Virustiter anderer Konstrukte, die auf einem 3- oder 4-
Plasmidsystem basierten. Auffallend war, dass bei dem 3-Plasmidsystem mit dem
Helferplasmid cmvgp1, das für gag und pol kodiert, auf HT1080 Zellen ein niedrigerer
Virustiter, als bei den 4-Plasmidsystemen, in denen gag und pol auf
unterschiedlichen Plasmiden vorliegen erzielt wurde.
Foamyviren besitzen ein diskontinuierliches Verpackungssignal, das am 5’ Ende des
Genoms im LTR und am Anfang des gag Gens liegt und am 3’ Ende von pol
lokalisiert ist. Die gemeinsame Anwesenheit beider Teile des diskontinuierlichen
Verpackungssignals ist notwendig, damit der Vektor in den Viruspartikel verpackt
werden kann (HEINKELEIN et al. 1998). Prinzipiell besteht die Möglichkeit, dass ein
Helferplasmid, welches wie der hier verwendete Vektor, beide Teile des
diskontinuierlichen Verpackungssignals besitzt, also gag und pol enthält, mit dem
Vektor um die Virushülle konkurriert. In diesem Fall können Partikel entstehen, die
statt e
G
wahrscheinlich im Backbone
dort unterscheiden. Es ist beschrieben, dass Plasmide mit einem SV40ori bei der
Anwesenheit des SV40 large-T-Antigens in der Zelle zu einer vermehrten Ablesung
des Vektors führen (PEAR et al. 1993; SONEOKA et al. 1995). Da aber im Backbone
beider Vektoren ein SV40ori ist, kann dieses nicht der alleinige Grund für die
unterschiedlichen Virustiter sein.
Konzentration des viralen Überstandes Die Höhe des Virustiters hat einen großen Einfluss auf die Transduktionseffizienz. In
108

Diskussion
gesteigert. Eine Volumeneinengung des Virusüberstandes wurde mehrfach
beschrieben (BURNS et al. 1993; YANG et al. 2002; LEURS 2003). Rekombinante
t wurden, mittels Zentrifugation
(LEURS 2003). Letztendlich wird nach dem
entrifugieren auch das „verbrauchte“ Medium der Produzentenzellen gegen frisches
mal auf die Bedürfnisse der Zielzellen
Viren, die mit Hüllproteinen wie dem VSV-G pseudotypisiert sind, können durch
Ultrazentrifugation konzentriert werden (BURNS et al. 1993). Bei Viren, deren
Virushülle nicht so stabil wie VSV-G ist, wurde eine erfolgreiche Zentrifugation mit
Geschwindigkeiten zwischen 10.000 und 20.000 x g erreicht (YANG et al. 2002;
LEURS 2003). Foamyvirale Hüllproteine sind sehr stabil und können mittels
Ultrazentrifugation konzentriert werden, sie lassen sich aber auch mit
Geschwindigkeiten zwischen 10.000 und 20.000 x g konzentrieren (LEURS 2003). In
der vorliegenden Untersuchung konnte gezeigt werden, dass auch Retroviren, die
mit dem Hüllprotein EM140 oder dem SM04 verpack
erfolgreich konzentriert werden können. Bisher ist nicht untersucht, ob die
Viruspartikel miteinander eine Verbindung eingehen und sich so bei niedrigen
Drehgeschwindigkeiten sedimentieren lassen. Grundsätzlich werden zwei
Hypothesen diskutiert, warum es durch Zentrifugation zu einer Titererhöhung
kommen kann. Zum einen kann es durch die Einengung des Volumens, indem sich
die Viruspartikel befinden zu einer Konzentrationserhöhung kommen. Zum anderen
wird vermutet, dass Pseudopartikel sehr leicht sind und deshalb nicht sedimentiert
werden; so kommt es zu einer „Aufreinigung“ der Viren. Da die Pseudopartikel nach
ihrer Entfernung nicht mehr die Rezeptoren der Zielzellen blockieren können, erhöht
sich die Wahrscheinlichkeit, dass die Viruspartikel die Zellen transduzieren; in der
Folge steigt der Virustiter an. Auch die Entfernung des Natriumbutyrat, dass zur
Induktion des CMV-Promotors der Plasmide in den Produzentenzellen genutzt wurde,
ist ein Vorteil, der mit der Zentrifugation einhergeht (RADSAK et al. 1989; TANAKA
et al. 1991). Natriumbutyrat ist für die Zellen toxisch und es ist mit einem Verlust von
Zielzellen und einem geringeren Virustiter zu rechnen, wenn es nicht aus dem
Virusüberstand entfernt wird
Z
Medium ersetzt; dieses kann dann opti
abgestimmt werden.
109

Diskussion
Manipulation der Zielzellen auf CH296 Fibronektin ist bereits zur Transduktion von hämatopoetischen Zellen etabliert und
bietet ähnliche Vorteile im Rahmen der Transduktion von hämatopoetischen
Stammzellen (HANENBERG et al. 1997). Die Transduktionseffizienz von Retroviren,
die mit VSV-G pseudotypisiert worden waren, konnte gesteigert werden, indem die
Transduktion auf CH296 beschichteten Platten durchgeführt wurde (DONAHUE et al.
2001; RELANDER et al. 2001). Die Zellen haften sich an die zwei Zellbindedomänen
CBD und CS1 des Fibronektins an, wodurch die Zellen auf dem CH296
beschichteten Untergrund einzeln liegen. Systematische Untersuchungen zur
Bindung von Viruspartikelen wurden nur mit dem amphotropen Hüllprotein und GaLV
durchgeführt. Hier konnte die direkte Bindung von Hüllproteinen des GaLV und des
amphotropen Hüllproteins an die Heparinbindedomäne gezeigt werden
(HANENBERG et al. 1996). Untersuchungen zu Anheftung foamyviraler Hüllproteine
oder auch VSV-G pseudotypisierter Vektoren an CH296 sind bisher nicht
beschrieben.
Um einen Einfluss des Vektortyps auf die Anheftung an das Fibronektin ausschließen
zu können, wurden rekombinante Lentiviren mit den Hüllproteinen GaLV, VSV-G und
EM140 preudotypisiert. Die Transduktionseffizienz war bei Konstrukten, die mit GaLV
oder EM140 pseudotypisiert waren, auf Platten mit CH296 Fragment deutlich höher
als auf Platten, die mit BSA beschichtet waren oder auf TCDs. Dieser Effekt war bei
dem VSV-G Hüllprotein nicht zu erkennen und steht im Widerspruch zu früheren
Untersuchungen, in denen die Transduktionseffizienz durch eine Beschichtung der
Platten mit CH296 gesteigert werden konnte (DONAHUE et al. 2001; RELANDER et
al. 2001). In diesen Arbeiten wurden humane CD34+ Zellen transduziert, während in
der vorliegenden Untersuchung HL60 Zellen verwendet wurden. Dieser Unterschied
könnte eine Erklärung für die Diskrepanz der Untersuchungsergebnisse sein.
Darüber hinaus sind in allen angeführten Untersuchungen unterschiedliche Vektoren
mit VSV-G pseudotypisiert worden und es ist nicht bekannt, inwieweit der Vektor die
Bindung des Viruspartikels beeinflussen kann.
Nachdem der jeweilige Virusüberstand entfernt worden war und die Platten mehrfach
it Medium gewaschen worden sind, waren immer noch so viele Viren an CH296 m
110

Diskussion
gebunden, dass eine Transfektion von Zellen möglich war. In der mit BSA
sfer in humane CD34 Zellen aus Nabelschnurblut
kt MH71. Tragen beide Konstrukte
beschichteten Kontrolle blieben keine Viren haften und es konnten auch keine Zellen
transduziert werden, die nach Entfernung des Virusüberstandes und Waschen der
Platte mit Medium aufgegeben wurden. Wenn CH296 mehrfach mit Virusüberstand
beschichtet wurde (Vorladen), dieser anschließend wieder entfernt und die Platte
gewaschen wurde, konnte eine deutliche Steigerung der Transduktionsrate
gegenüber der einmaligen Beladung mit Virusüberstand beobachtet werden. Dieser
Versuch zeigt, dass sowohl das VSV-G Hüllprotein wie auch das foamyvirale EM140
an Fibronektin binden können. Zusätzlich ist eine Erhöhung der
Transduktionseffizienz bei dem foamyviralen Hüllprotein alleine durch die
Beschichtung der Platten mit Fibronektin möglich.
5.2 Gentran +
In der eigenen Arbeit konnte gezeigt werden, dass foamyvirale Vektoren trotz ihrer
geringen Virustiter auf HT1080 Zellen CD34+ Zellen effizienter als lenti- und
gammaretrovirale Vektoren transduzieren können. Lentivirale Vektoren, die mit VSV-
G pseudotypisiert worden waren, erreichten auf HT1080 Zellen einen höheren
Virustiter als das foamyvirale Konstrukt. Dennoch war die Gentransferrate der
foamyviralen Konstrukte in humane CD34+ Zellen höher als die Rate der lentiviralen
Konstrukte. Untersuchungen mit den foamyviralen Vektoren MH71, MH71.MGMT,
MD9 und MD9.MGMT haben gezeigt, das die Konstrukte mit dem
Chemotherapeutikumresistenzgen MGMT auf den humanen hämatopoetischen
Stammzellen immer niedrigere Gentransferraten erzielten als die Konstrukte, die nur
GFP als Transgen trugen. Die auf HT1080 gemessenen Virustiter lassen jedoch bei
Foamyviren keine präzise Aussage über die Effizienz der Vektoren bei der
Transduktion von CD34+ Zellen zu. Das Viruskonstrukt MD9 zeigte wiederholt
schlechtere Gentransferraten als das Konstru
zusätzlich zum Transgen GFP das Transgen MGMTp140k, so nimmt die
Transduktionseffizienz, gemessen als Anteil GFP positiver Zellen, weiter ab, auch
111

Diskussion
wenn der Virustiter im Einzelfall über dem der Konstrukte ohne MGMTp140k lag. Auf
mögliche Faktoren (IRES-site), die für dieses Problem verantwortlich sein könnten,
wird an späterer Stelle detailliert eingegangen.
An humanen CD34+ Zellen wurden zwei foamyvirale Hüllproteine getestet. Die mit
dem Hüllprotein SM04 verpackten Konstrukte erzielten grundsätzlich auf HT1080
Zellen einen niedrigeren Virustiter, als die mit dem Hüllprotein EM140 verpackten
Konstrukte. Entsprechend wurde in der FACS-Analyse an Tag 5 nach der
Transduktion bei Konstrukten mit SM04 eine deutlich niedrigere Gentransferrate
gemessen als bei Konstrukten mit EM140. Überraschend war, dass in den
Progenitor-Assays die Gentransferrate der mit dem SM04 verpackten Konstrukte nur
napp unterhalb der mit dem EM140 verpackten Konstrukte lag.
iese Ergebnisse können dahingehend interpretiert werden, dass die beiden
foamyviralen Verpackungsplasmide einen Einfluss auf die Geschwindigkeit haben, in
d n werden kann.
k
D
der as foamyvirale Genom in das Genom der Zielzelle integriere
Welche zellulären Rezeptoren die Hüllproteine zum Eintritt in die Zelle nutzen, und
ob beide Hüllproteine den gleichen Rezeptor nutzen, ist noch unbekannt. Es wäre
möglich, dass die Hüllproteine unterschiedlich schnell mit der Zellmembran der
Zielzelle fusionieren und so das Kapsid zeitlich versetzt in das Zytoplasma gelangt.
Das würde bedeuten, dass das Hüllprotein SM04 das virale Kapsid erst später als
das EM140 freigesetzt wird und dadurch die Transduktionsrate erst nach 14 Tagen
der des EM140 entspricht. Gegen diese Vermutung spricht allerdings, dass, wenn
SM04 den Einbau des viralen Genoms stark verzögert, es Kolonien geben müsste,
bei denen nur ein Teil der Zellen GFP positiv und ein Teil der Zellen GFP negativ ist.
Diese Beobachtung konnte nicht gemacht werden. Daher kann nicht von einem
unterschiedlichen Integrationszeitpunkt des viralen Genoms in das Genom der
Zielzelle ausgegangen werden. Die Ursachen für die unterschiedliche Genexpression
bleiben demnach unklar.
112

Diskussion
5.3 Gentransfer in CD34+ Marmoset Zellen
In der eigenen Arbeit konnte gezeigt werden, dass CD34+ Marmoset Zellen sich, wie
ndere Säugerzellen auch, gut mit Foamy-, Lenti- und Gammaretroviren infizieren
rneut, so lässt sich
Messung 5 Tage nach der Transduktion erzielt werden
önnen.
Es ist bekannt, dass Foamyviren nicht wie Lentiviren aktiv die Kernporen überwinden
können. Sie sind auf den Vorgang der Mitose und die damit verbundene Auflösung
a
lassen. Auch wenn der Virustiter des foamyviralen Konstrukts MH71 auf HT1080
Zellen meist unter denen der lenti- und gammaretroviralen Vektoren, die mit dem
gleichen Hüllprotein pseudotypisiert waren, lagen, entsprach die Gentransferrate, die
5 Tage nach der Transduktion ermittelt wurde, dem Niveau der anderen Viren. In
einem Versuch konnte im Vergleich mit der Gentransferrate des Konstrukts pCL1,
pseudotypisiert mit EM140, das sonst in allen Untersuchungen den höchsten
Virustiter aufwies, sogar eine geringfügig höhere Transferrate gezeigt werden.
Bestimmt man die Gentransferrate 9 Tage nach Transduktion e
feststellen, dass die Gentransferrate des foamyviralen Konstrukts gegenüber dem an
Tag 5 gemessenen Wert leicht ansteigt. Die Gentransferraten der lentiviralen und
gammaretroviralen Konstrukte waren dagegen an Tag 9 nach der Transduktion
niedriger als an Tag 5. Die Gentranferrate des mit EM140 pseudotypisierten
lentiviralen Konstrukts zeigte nur eine geringe Abnahme, stattdessen konnt jedoch
bei dem mit VSV-G pseudotypisierten lentiviralen Konstrukt eine Abnahme von 66 %
und bei dem gammaretroviralen Konstrukt eine Abnahme um 37 % gegenüber der
Messung an Tag 5 nach Transduktion bestimmt werden.
Betrachtet man die Gentransferraten 14 Tage nach der Transduktion im Progenitor-
Assay, so ist deutlich zu erkennen, dass die Gentransferrate des foamyviralen
Konstrukts über die Zeit gleich bleibt oder sogar leicht ansteigt, während die
Gentransferraten der anderen Konstrukte deutlich abfallen (Abbildung 24). Vergleicht
man diese Daten mit denen der Progenitor-Assays humaner Zellen (Abbildung 16),
so ist weiterhin zu erkennen, dass bei der Verwendung foamyviraler Konstrukte im
Progenitor-Assay deutlich höhere Gentransferraten als in der
durchflusszytometrischen
k
113

Diskussion
der Kernmembran angewiesen, um ihr eigenes Genom in das Genom der Wirtszelle
tegrieren zu können (BIENIASZ et al. 1995; PATTON et al. 2004; TROBRIDGE et
gestellt, dass das Konstrukt
kann (CARNEIRO et al. 2006). Andere Untersucher vermuten, dass
hosphatidylserin VSV-G nicht als Rezeptor dient (COIL u. MILLER 2004). Wenn
in
al. 2004). Trotzdem sind Foamyviren in der Lage, auch effizient solche Zellen zu
infizieren, die sich selten teilen (TROBRIDGE et al. 2004).
Die längere Persistenz des Präintegrationskomplexes in infizierten Zellen ist
wahrscheinlich durch die Besonderheit der Foamyviren bedingt, dass sie die reverse
Transkription bereits in der Virus produzierenden Zelle abgeschlossen haben. Der
Viruspartikel enthält deshalb doppelsträngige DNA, die deutlich stabiler ist als RNA.
Man nimmt an, dass der foamyvirale Präintegrationskomplex bis zur Zellteilung im
Zytoplasma der Zelle stabil bleibt und dann im Moment der Zellteilung zum
Kernzentrum gelangt und dort in die DNA der Zelle integriert. Diese Art der
Replikation könnte ebenfalls eine Erklärung dafür sein, dass die Gentransferraten
foamyviraler Vektoren sowohl bei humanen als auch bei den von Marmosets
stammenden CD34+ Zellen mit der Zeit ansteigen. Da die Differenz der
Gentransferraten der FACS-Analyse, verglichen mit den Gentransferraten der
Progenitor-Assays, bei den humanen CD34+ größer ist als bei den Marmoset Zellen,
kann vermutet werden, dass die Teilungsrate der Marmoset Zellen höher ist als die
der humanen CD34+ Zellen.
Im Rahmen der vorliegenden Untersuchungen wurde fest
pCL1 pseudotypisiert mit dem VSV-G Hüllprotein in den CD34+ Marmoset Zellen,
verglichen mit den humanen CD34+ Zellen deutlich schlechtere Gentransferraten
erzielt. Einige Untersucher gehen davon aus, dass der zelluläre Rezeptor für das
VSV-G das Phosphatidylserin, ein Lipid der Zellmembran, die Bindung von VSV-G
an die Zellmembran vermittelt (SCHLEGEL et al. 1983a; SCHLEGEL u. WADE
1983b). Dieses Phosphatidylserin ist in allen Zellmembranen vorhanden, jedoch ist
die Zusammensetzung der Zellmembranen mit Phospholipiden nicht bei allen
Lebewesen gleich, sodass es zu unterschiedlich starken Bindung von VSV-G
kommen
P
Phosphatidylserin nicht der Rezeptor wäre, bestünde dennoch die Möglichkeit, dass
114

Diskussion
die Marmoset Zellen deutlich weniger von dem noch unbekannten Rezeptor für VSV-
G auf ihrer Zelloberfläche exprimieren als die humanen Zellen und deshalb
schlechter transduziert werden können. Eine unterschiedlich starke Bindung des
VSV-G an humane und Marmoset Zellen aufgrund einer unterschiedlichen
t wird. Die Nukleinsäure, die nicht in das Wirtsgenom
tegrieren konnte wird im Zytoplasma abgebaut.
Zusammensetzung der Zellmembranen mit Phospholipiden ist ebenfall ein mögliche
Erklärung für die ungleichen Gentransferraten.
Am Tag 5 nach der Transduktion sind bis zu 25 % der CD34+ Marmoset Zellen, die
mit dem mit VSV-G pseudotypisierten lentiviralen Konstrukt transduziert wurden, in
der FACS-Analyse GFP positiv. Dagegen sind 14 Tage nach der Transduktion im
Progenitor-Assay nur noch maximal 0,7 % der Kolonien GFP positiv. Diese
Diskrepanz lässt die Vermutung zu, dass ein Pseudogentransfer stattgefunden hat.
Ein Pseudogentransfer liegt vor, wenn Pseudopartikel, das sind Partikel die nur GFP
enthalten, in das Zytoplasma der Zielzelle gelangen. Das GFP im Zytoplasma der
Zelle wird im Laufe der Zeit wieder abgebaut und die Zelle wird wieder GFP negativ.
Daneben wäre auch ein Nukleinsäuretransfer denkbar, bei dem wohl eine Infektion
der Zelle, aber keine Integration in das Wirtsgenom stattgefunden hat. Die RNA
würde translatiert und im Anschluss GFP gebildet, das dann im Zytoplasma der Zelle
mit der Zeit wieder abgebau
in
5.4 MGMTp140k als Transgen
Bei Erkrankungen wie X-linked-SCID oder der chronischen Granulomatose bietet das
Transgen den genkorrigierte Zellen einen selektiven Wachstumsvorteil, so das es
durch die steigende Zahl an genkorrigierten Zellen zu einer phänotypischen Heilung
der Grunderkrankung kommt (CAVAZZANA-CALVO et al. 2000; PFEIFER et al.
2001; AIUTI et al. 2002). Jedoch bieten nicht alle therapeutischen Transgene, die in
Zukunft gentherapeutisch eingesetzt werden könnten, den Zellen einen solchen
Selektionsvorteil. Mittels Vektoren, die zwei Transgene gleichzeitig exprimieren
können, ist es möglich, ein therapeutisches Transgen und ein Resistenzgen, die über
115

Diskussion
eine IRES-site miteinander verbunden sind, in eine Zelle einzubringen. Ein
besonders aussichtsreiches Resistenzgen ist das MGMTp140k, welches DNA
Schäden, die durch alkylierende Substanzen wie BCNU hervorgerufen werden,
reparieren kann und so zu einer erhöhten Resistenz gegenüber bestimmten
Chemotherapeutika führt (QUINN 2001). Das MGMTp140k ist in mehreren
Gentherapiestudien an Mäusen (KREKLAU et al. 2003; CAI et al. 2006) und Hunden
wahrscheinlich die Lage des GFP
ens verantwortlich. Das GFP Gen liegt am 3’ Ende des Leserahmens und liegt
omit hinter der IRES-site. Es ist bekannt, dass Gene, die hinter der IRES-site liegen,
meist ineffizienter abgelesen werden. Im Progenitor-Assay der humanen CD34+
hiede auf und die Transduktionseffizienz der
(NEFF et al. 2005) schon erfolgreich zur Selektion der transgenen Zellen eingesetzt
worden. Um auch den mit foamyviralen Vektoren transduzierten Zellen einen
Selektionsvorteil zu verschaffen, wurde das MGMTp140k in foamyvirale Vektoren
kloniert und in diesem Vektor eingehend getestet. Dieses Selektionsprinzip wurde
auch in CD34+ Marmoset Zellen getestet, um später einmal Marmsets als
Gentransfermodel nutzen zu können.
Die Virustiter der foamyviralen Vektoren, die das Transgen MGMT tragen, waren
geringfügig niedriger, als die von Konstrukten ohne dieses zusätzliche Transgen. Die
Transduktionseffizienz in den Versuchen mit humanen CD34+ Zellen, gemessen
mittels FACS-Analyse, war bei Konstrukten mit MGMTp140k immer niedriger, als die
Transduktionseffizienz der Viruskonstrukte ohne MGMTp140k. Für die niedrigeren
Virustiter auf HT1080 Zellen und die schlechten Gentransferraten der MGMTp140k
tragenden Konstrukte in der FACS-Analyse ist
G
s
Zellen heben sich die Untersc
Konstrukte mit und ohne MGMTp140k liegen auf einem Niveau. Auch wenn das Gen
am 3’ Ende des Leserahmens schlechter abgelesen wird, so akkumuliert das
Genprodukt, in dem Fall das GFP, in der Zelle und kann dann nachgewiesen werden.
Dieses Ergebnis lässt vermuten, dass der Gentransfer der Konstrukte, die das
zusätzliche Transgen MGMTp140k enthalten, nicht schlechter ist, als die
Gentransferrate der Konstrukte ohne das zusätzliche Transgen, sondern es nur
keine Möglichkeit gibt das Genprodukt nachzuweisen. Es bestünde die Möglichkeit
das MGMT Protein anzufärben, doch da dieses intrazellulär vorliegt, ist eine Färbung
116

Diskussion
mit einigen Einschränkungen verbunden. Zurzeit sind nur MGMT Antikörper
kommerziell erhältlich, die nicht an ein Fluorochrom gekoppelt sind. Aus diesem
Grund müsste der MGMT-Antikörper mit einem fluorochromkonjugierten Antikörper
gekoppelt werden. Diese Zweitantikörper zeichnen sich jedoch sehr häufig durch
eine geringe Spezifität aus und führen unter Umständen zu falsch positivern
Ergebnissen.
Bei den transduzierten hämatopoetischen Zelllinien liegt die Gentransferrate der
MGMTp140k Konstrukte niedriger, als die Zellen mit nur einem Transgen. Kultiviert
man diese Zellen über einen längeren Zeitraum, so kann man deutlich erkennen,
dass die Anzahl GFP positiver Zellen bei beiden MGMTp140k tragenden Konstrukten
mit der Zeit abnimmt. Eine mögliche Erklärung für diese Beobachtung könnte ein
Abschalten des Transgens durch zelluläre Mechanismen sein. Gegen diese
Annahme spricht jedoch, dass die zwei Kontrollkonstrukte, die nur das Transgen
GFP über den gleichen Promotor exprimieren, über die Zeit hinweg ein gleich
bleibendes Expressionsniveau zeigen. Dies lässt vermuten, dass das MGMT-Protein
in Konzentration, die oberhalb des natürlichen Expessionsniveaus liegen, für die
Zelle toxisch ist. Aus verschiedenen Untersuchungen ist bekannt, dass eine
Überexpression von Resistenzgenen für Chemotherapeutika toxische Effekte haben
kann. So wurde z.B. bei der Überexpression der Methylpuringlykosylase, die
alkylierte Basen in der DNA abbaut, auch eine zelltoxische Wirkung festgestellt
(KAINA et al. 1993). Durch die toxische Wirkung des überexprimierten Genprodukts
kommt es zum Absterben der transgenen Zellen und dadurch zu einer negativen
Selektion.
Behandelt man Zellen, die das Transgen MGMTp140k tragen und transduzierte
Zellen, die nur GFP tragen, mit BCNU, dann überleben mehr Zellen mit MGMTp140k
die Behandlung als solche ohne das Transgen. War der Anteil GFP positiver Zellen
an der Gesamtpopulation der MGMTp140k tragenden Konstrukte vor der BCNU
Behandlung niedrig (< 5%), so konnte der Anteil durch die Behandlung deutlich
gesteigert werden (> 50%). Diese Ergebnisse zeigen deutlich, dass das MGMTp140k
im foamyviralen Kontext in der Lage ist, den Zellen eine Resistenz gegenüber
117

Diskussion
alkylierenden Substanzen wie BCNU zu vermitteln. Durch eine zusätzliche
Behandlung mit BG, welches das endogene MGMT irreversibel blockiert, konnte der
Selektionsdruck auf die Zellen noch zusätzlich erhöht werden und die Zellen mit dem
Transgen MGMTp140k konnten noch besser selektioniert werden.
Doch nicht alle überlebende Zellen waren nach der Behandlungen mit hohen BCNU
Konzentrationen oder der Kombination von BCNU und BG auch GFP positiv. Es gibt
wei Möglichkeiten, warum ein Teil der überlebenden Zellen GFP negativ ist. Zum
ative Möglichkeit zwei einzelne Proteine über eine Expressionskassette zu
z
einen könnte das endogene MGMT in der Zelle durch die Behandlung hochreguliert
worden sein, so dass auch Zellen ohne Transgen die chemotherapeutische
Behandlung überleben konnten. Dass das endogene MGMT hochreguliert wird, ist
nur über einen längeren Behandlungszeitraum zu erwarten und die
Behandlungsintervalle, die in dieser Untersuchung angesetzt worden sind, sind für
eine solche Hochregulationen nicht lang genug. Deswegen ist es wahrscheinlicher,
dass es Zellen gibt, die zwar eine Genkassette und damit auch das MGMTp140k
tragen, jedoch das GFP Gen nicht oder nicht in einem Maße exprimieren, dass es
mittels Durchflusszytometer nachgewiesen werden kann. Dieses Problem ist
wahrscheinlich durch die IRES-site bedingt, die zwischen dem MGMT Gen und dem
GFP liegt. Sie sorgt eigentlich dafür, dass beide Gene hintereinander translatiert
werden (JANG u. WIMMER 1990). Es ist jedoch bekannt, das 3’-liegende
Leserahmen, in diesem Fall das GFP, meist ineffizienter abgelesen wird. Diese
Tatsache würde auch die niedrigeren Titer auf HT1080 Zellen der MGMTp140k
tragenden Konstrukte und die damit verbundenen niedrigeren Gentransferraten auf
primären Zellen (5 Tage nach Transduktion) ausreichend erklären.
Eine altern
exprimieren bietet das 2A-Oligopeptid des Maul- und Klauenseuchevirus (RYAN et al.
1991; DE FELIPE et al. 2006). Es ist nur 19 Aminosäuren lang und verursacht bei
der Elongation des Proteins am Ribosom eine Unterbrechung der Elongation nach
dem 2A-Oligopetpid, wodurch die schon translatierte Peptidsequenz zusammen mit
dem 2A-Oligopeptid freigesetzt wird und so von dem noch zu translatierenden
zweiten Peptid getrennt wird. Auf diese Art lassen sich bis zu drei Proteine getrennt
von zwei 2A-Oligopeptiden stromabwärts eines Promotors exprimieren.
118

Diskussion
5.5 Schlussfolgerung
Die Ergebnisse dieser Arbeit zeigen, dass sich von Foamyviren abgeleitete Vektoren
ausgezeichnet zur Transduktion von hämatopoetischen Stammzellen eignen. Sowohl
humane als auch hämatopoetische Stammzellen von Marmosets lassen sich effektiv
transduzieren und die Resultate sind miteinander vergleichbar. Die Gentransferraten
der foamyviralen Vektoren liegen in dieser Untersuchung über den bisher zur
Transduktion von hämatopoetischen Stammzellen eingesetzten lenti- und
gammaretroviralen Vektorsystemen. Foamyviralen Konstrukte lassen sich auch zur
Expression von mehreren Transgenen einsetzen, wobei bezüglich des Mechanismus
der Koexpression noch weiterer Optimierungsbedarf besteht.
So stellt dieses Vektorsystem eine echte Alternative zu den bisher in
Gentherapiestudien verwendeten lenti- und gammaretroviralen Vektoren da. Es kann
empfohlen werden, Marmosets als Primatenmodell zur Erprobung der Effektivität und
Sicherheit von Foamyviralen Vektorsystemen einzusetzen.
119

Zusammenfassung
6 . Zusammenfassung
Melanie Wurm
„Transduktion hämatopoetischer Stammzellen von Neuweltaffen mit Foamyviren“
Die Transplantation von genetisch korrigierten, CD34+ autologen hämatopoetischen
Stammzellen hat 18 von insgesamt 19 Patienten mit schwerem kombiniertem
Immundefekt (SCID) von ihrer, ansonsten im ersten Lebensjahr tödlich verlaufenden,
hereditären Erkrankung geheilt. Dieser klinische Einsatz von replikationsdefekten
gammaretroviralen Vektoren als Gentransfersystem hat allerdings einige Jahre nach
der Gentherapie bei drei der Kinder zur Entstehung von T-Zell-Malignoms geführt.
Bei zwei Patienten konnte gezeigt werden, dass diese Malignome durch die bei
Wildtyp-Gammaretroviren bekannte Insertionsmutagenese mit Aktivierung von
zellulären Onkogenen induziert worden sind. Aufgrund dieser inzwischen in
verschiedenen Tiermodellen ebenfalls nachgewiesenen „Nebenwirkung“ von
gammaretroviralen Vektoren ist es äußerst wichtig, alternative Vektorsysteme zu
entwickeln, die nicht nur Stammzellen effektiv transduzieren können, sondern auch
ein hohes Maß an Sicherheit bieten. Diese neuen Vektorsysteme müssen vor einem
Einsatz beim Menschen in Großtiermodellen sicherheits- und effizienzgetestet
werden. Foamyviren als komplexe Retroviren könnten ein derartiges alternatives
Gentransfersystem sein, da sie als einzige Mitglieder der Retroviridae auch bei
chronisch-persistierender Infektion mit keinerlei Pathogenität bei Tieren oder auch
Menschen assoziiert werden konnten.
In der vorliegenden Arbeit wurde ein Verfahren optimiert, bei dem autologe CD34+
klonogene Zellen der Primatenspecies common marmosets (Callitrix jacchus) in vitro
genetisch verändert werden, um sie dann anschließend nach milder Myeloreduktion
den Tieren zurückzugeben. Initial wurden dabei verschiedene Parameter wie Einsatz
von Molekülen der extrazellulären Matrix, von verschiedenen foamyviralen
Hüllproteinen und Transfektionsprotokollen hinsichtlich des Einflusses auf den
120

Zusammenfassung
Virustiter und die Gentransfereffizienz mit GFP exprimierenden foamy-, lenti- und
ammaretroviralen Vektoren getestet. Um die nach Reinfusion der Zellen in die Tiere
niedrige Gentransfereffienz in repopulierende Stammzellen
usgleichen zu können, wurde im zweiten Teil der Arbeit ein in vivo Selektionssytem,
NA-Methyltransferase (MGMTp140k) als Transgen in foamyviralen
ektoren und dem Chemotherapeutikum 1,3-bis-(2-chloroethyl)-1-Nitrosourea
n die Grundlagen für den Einsatz von
g
zu erwartende
a
bestehend aus der P140K Punktmutante des Chemotherapeutikumresistenzgen O6-
Methylguanin-D
V
(BCNU), an humanen hämatopoetischen Zelllinien und an CD34+ Zellen von
common Marmosets getestet.
Die Resultate zeigten, dass foamyvirale Vektoren in einem kurzen
Transduktionsprotokoll für 16 Stunden auf dem rekombinanten Fibronektinfragment
CH-296 und ohne Vorstimulation der Zielzellen mehr als 60% der humanen CD34+
Nabelschnurblutzellen und mehr als 80% der CD34+ Marmosetzellen genetisch
verändern können. Diese hohe Gentransfereffizienz lag deutlich über der von
gammaretroviralen Vektoren und war vergleichbar mit der von lentiviralen Vektoren,
wenn letztere mit dem gleichen foamyviralen Hüllprotein pseudotypisiert wurden.
Dagegen war das G-Protein des Vesicular Stomatitis Virus (VSV-G) als Hüllprotein
nicht geeignet, Marmoset CD34+ Zellen mit Lentiviren effektiv zu transduzieren. Mit
MGMTp140k als Transgen in zwei verschiedenen foamyviralen Vektoren war essehr
gut möglich, hämatopoetische Zellen von Menschen und Marmosets in vitro mit
BCNU zu selektionieren, so dass der Anteil von genetisch veränderten Zellen über
die Zeit deutlich erhöht werden konnte.
Mit diesen Untersuchungen konnte
foamyviralen Vektoren als effiziente Gentransfersysteme für die Transduktion von
humanen und Marmoset CD34+ Zellen etabliert werden, so dass zur Zeit die
Anwendung von foamyviralen Vektoren in Marmosets ausgetestet wird. Bei guter
Verträglichkeit und effizienter Transduktion von hämatopoetischen Stammzellen der
Tiere wäre dann der Einsatz in klinischen Phase I-Studie beim Menschen zur
Korrektur genetischer Erkrankungen möglich.
121

Summary
7. Summary
Melanie Wurm
‘Transduction of hematopoietic stem cells from new world monkeys with Foamyviruses’
Transplantation of genetically corrected CD34+ autologous hematopoietic stem cells
has cured 18 out of 19 patients with severe combined immune deficiency (SCID), a
ereditary disorder to which patients generally succumb in their first year of life. This
the present thesis, procedures were optimized to genetically modify autologous
d repopulating stem cells, the second
h
clinical application of replication defective gammaretroviral vectors however induced
T cell malignancies in three children within a few years after gene therapy. For two
patients, it was shown that these malignancies occurred due to insertional
mutagenesis, well known for wild-type gammaretroviruses, leading to activation of
cellular proto-oncogenes. Due to the recent findings that this ‘side effect’ of
gammaretroviral vectors also occurred in animal models, it becomes mandatory to
develop alternative vector systems that are characterized not only by their capability
to transduce stem cells, but also by their high degree of safety. Prior to any clinical
application, these novel vector systems have to be evaluated for safety and efficacy
in a large animal model. Foamyviruses as complex retroviruses might be this
candidate vector system as they are the only members of the Retroviridae that
despite chronic persistent infections are non-pathogenic in animals and also humans.
In
CD34+ cells from common marmosets (Callitrix jacchus) with the ultimate aim to re-
infuse these cells to animals undergoing a mild myeloreductive treatment. Initially,
the influence of different parameters like molecules of the extracellular matrix,
different foamyviral envelopes and several transfection protocols on virus titers and
gene transfer efficiencies was analyzed using GFP expressing foamy-, lenti- and
gammaretroviral vectors. Considering the low transduction rates in animals that can
be expected after reinfusion of the transduce
122

Summary
part of the thesis evaluates an +
in vivo selection system in hematopoeitic cell lines and
armoset CD34 cells. This selection system is based on the P140K point mutant of
guanin-DNA-methyltranferase chemotherapy resistant gene as
ansgene in foamyviral vectors in combination with the chemotherapeutic substance
he results demonstrated high efficacy of foamyviral vectors, processed in only 16
m
the O6-methyl
tr
1,3-bis-(2-chlorethyl)-1-Nitrosourea (BCNU).
T
hours on recombinant fibronectin fragment CH296 coated plates. Genetic
modification was observed in 60 % of human and 80 % of marmoset CD34+ cells,
although cells were not stimulated previously. The gene transfection rate was
obviously higher in foamyviruses than in gammaretroviruses; it was equal to the
results from lentiviruses if these were pseudo typed by a foamyviral envelope.
Contrary, the G-protein derived from the vesicular stomatitis virus (VSV-G) was
insufficient when it was used as an envelope. An increase of a low transfection rate
can be triggered by in vitro selection of hematopoietic cells from humans and
marmosets, respectively. This was traced back to the foamyviral vectors that contain
MGMTp140k and the additional treatment of cells with BCNU.
In this study, foamyviral vectors were well evaluated to be very sufficient in gene
transfer into human and marmoset CD34+ cells. Further applications of gene transfer
into marmosets by foamyviral vectors should be tested. Phase I trials that aim at
gene therapy will be sensible, if the transduction of hematopoietic stem cells is
approved to be safe and efficient.
123

Literaturverzeichnis
8. Literaturverzeichnis
ABONOUR, R., D. A. WILLIAMS, L. EINHORN K. M. HALL, J. CHEN, J. COFFMAN,
C. M. TRAYCOFF, A. BANK, I. KATO, M. WARD, S. D. WILLIAMS, R.
HROMAS, M. J. ROBERTSON, F. O
,
. SMITH, D. WOO, B. MILLS, E. F.
SROUR u. K. CORNETTA (2000):
Efficient retrovirus-mediated transfer of the multidrug resistance 1 gene into
autologous human long-term repopulating hematopoietic stem cells.
Nat. Med. 6, 652-658
ACHONG, B. G., P. W. MANSELL, M. A. EPSTEIN u. P. CLIFFORD (1971):
An unusual virus in cultures from a human nasopharyngeal carcinoma.
J. Natl. Cancer Inst. 46, 299-307
AIUTI, A., S. SLAVIN, M. AKER, F. FICARA, S. DEOLA, A. MORTELLARO, S.
MORECKI, G. ANDOLFI, A. TABUCCHI, F. CARLUCCI, E. MARINELLO, F.
CATTANEO, S. VAI, P. SERVIDA, R. MINIERO, M. G. RONCAROLO u. C.
BORDIGNON (2002):
Correction of ADA-SCID by stem cell gene therapy combined with
nonmyeloablative conditioning.
Science 296, 2410-2413
ASGT (2003):
Report from the ASGT Ad Hoc Committee on Retroviral-mediated Gene
Transfer into Hematopoietic Stem cells, April 2003.
www.asgt.org; Abrufdatum: 02.06.2006
BARTZ, S. R., u. M. A. VODICKA (1997):
Production of high-titer human immunodeficiency virus type 1 pseudotyped
with vesicular stomatitis virus glycoprotein.
Methods 12, 337-342
124

Literaturverzeichnis
BAUNACH, G., B. MAURER, H. HAHN, M. KRANZ u. A. RETHWILM (1993):
Functional analysis of human foamy virus accessory reading frames.
J. Virol. 67, 5411-5418
SZ, P. D., R. A. WEISS u. M. O. MCCLURE (1995):
Cell cycle dependence of foamy retr
BIENIA
ovirus infection.
J. Virol. 69, 7295-7299
BREN CK (1988):
Formation of covalent complexes between human O6-alkylguanine-DNA
ynthetic
T, T. P., u. J. S. REMA
alkyltransferase and BCNU-treated defined length s
oligodeoxynucleotides.
Nucleic Acids Res. 16, 6779-6788
BROK, H. P., R. J. HORNBY, G. D. GRIFFITHS, L. A. SCOTT u. B. A. HART (2001):
An extensive monoclonal antibody panel for the phenotyping of leukocyte
subsets in the common marmoset and the cotton-top tamarin.
Cytometry 45, 294-303
BROU
topoiesis.
Blood 90
DY, V. C. (1997):
Stem cell factor and hema
, 1345-1364
BURM
ESTER, G. R., u. A. PEZZUTTO (1998):
Taschenatlas der Immunologie.
Georg Thieme Verlag, Stuttgart, 360 S.
125

Literaturverzeichnis
BURNS, J. C., T. FRIEDMANN, W. DRIEVER, M. BURRASCANO u. J. K. YEE
rus G glycoprotein pseudotyped retroviral vectors:
concentration to very high titer and efficient gene transfer into mammalian and
(1993):
Vesicular stomatitis vi
nonmammalian cells.
Proc. Natl. Acad. Sci. U S A 90, 8033-8037
AI, S., J. R. HARTWELL, R. J. COOPER, B. E. JULIAR, E. KREKLAU, R.
. POLLOK (2006):
C
ABONOUR, W. S. GOEBEL u. K. E
In vivo effects of myeloablative alkylator therapy on survival and differentiation
of MGMT(P140K)-transduced human G-CSF-mobilized peripheral blood cells.
Mol. Ther. 13, 1016-1026
ALLAHAN, M. E., W. M. SWITZER, A. L. MATTHEWS, B. D. ROBERTS, W. C
HENEINE, T. M. FOLKS u. P. A. SANDSTROM (1999):
Persistent zoonotic infection of a human with simian foamy virus in the
absence of an intact orf-2 accessory gene.
J. Virol. 73, 9619-9624
CARNEIRO, F. A., P. A. LAPIDO-LOUREIRO, S. M. CORDO, F. STAUFFER, G.
. A. JULIANO, L. JULIANO, P. M. BISCH
06):
Probing the interaction between vesicular stomatitis virus and
WEISSMULLER, M. L. BIANCONI, M
u. A. T. DA POIAN (20
phosphatidylserine.
Eur. Biophys. J. 35, 145-154
AVAZZANA-CALVO, M., u. A. FISCHER (2004):
Efficacy of gene therapy for SCID is being confirmed.
Lancet 364
C
, 2155-2156
126

Literaturverzeichnis
CAVAZZANA-CALVO, M., S. HACEIN-BEY, G. DE SAINT BASILE, S. DUPUIS-
GIROD, A. THRASHER, N. WULFFRAAT, R. SORENSEN, J. L. CASANOVA,
F. LE DEIST u. A. FISCHER (2000):
Gene therapy of human severe combined immunodeficiency (SCID)-X1
disease.
Blood 96, 2533a
CHECK, E. (2005):
Gene therapy put on hold as third child develops cancer.
Nature 433, 561
CHINN AIRN, J. NEUENFELDT, J. S. TREISMAN, J. P.
HANSON, JR., G. P. MARGISON u. N. CHINNASAMY (2004):
uanine and 1,3-bis(2-
ASAMY, D., L. J. FAIRB
Lentivirus-mediated expression of mutant MGMTP140K protects human
CD34+ cells against the combined toxicity of O6-benzylg
chloroethyl)-nitrosourea or temozolomide.
Hum. Gene Ther. 15, 758-769
OIL, D. A., u. A. D. MILLER (2004): C
Phosphatidylserine is not the cell surface receptor for vesicular stomatitis virus.
J. Virol. 78, 10920-10926
CRAW
FFBRAND u. H. G. PRENTICE (1981):
n of hemopoietic cells in the common marmoset,
rhesus monkey, and man. In search of a model for human marrow
FORD, D. H., G. JANOSSY, C. M. HETHERINGTON, G. E. FRANCIS, A. J.
EDWARDS, A. V. HO
Immunological characterizatio
transplantation.
Transplantation 31, 245-250
127

Literaturverzeichnis
CROOKS, G. M., u. D. B. KOHN (1993):
Growth factors increase amphotropic retrovirus binding to human CD34+ bone
marrow progenitor cells.
Blood 82, 3290-3297
CRYS :
Transfer of genes to humans: early lessons and obstacles to success.
TAL, R. G. (1995)
Science 270, 404-410
DAO, M. A., C. H. HANNUM, D. B. KOHN u. J. A. NOLTA (1997):
FLT3 ligand preserves the ability of human CD34+ progenitors to sustain long-
term hematopoiesis in immune-deficient mice after ex vivo retroviral-mediated
transduction.
Blood 89, 446-456
, B. M., J. S. REESE, O. N. KOC, K. LEE
DAVIS , J. E. SCHUPP u. S. L. GERSON
Selection for G156A O6-methylguanine DNA methyltransferase gene-
enitors and protection from lethality in mice
(1997):
transduced hematopoietic prog
treated with O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea.
Cancer Res. 57, 5093-5099
therapy.
DAVIS, B. M., J. C. ROTH, L. LIU, M. XU-WELLIVER, A. E. PEGG u. S. L. GERSON
(1999):
Characterization of the P140K, PVP(138-140)MLK, and G156A O6-
methylguanine-DNA methyltransferase mutants: implications for drug
resistance gene
Hum. Gene Ther. 10, 2769-2778
128

Literaturverzeichnis
DE FELIPE, P., G. A. LUKE, L. E. HUGHES, D. GANI, C. HALPIN u. M. D. RYAN
proteins from a self-processing polyprotein.
(2006):
E unum pluribus: multiple
Trends Biotechnol. 24, 68-75
D. HUSZAR, R. A. PHILLIPS u. A. BERNSTEIN (1985):
ng-term
mopoietic system of W/Wv mice.
Cell 42
DICK, J. E., M. C. MAGLI,
Introduction of a selectable gene into primitive stem cells capable of lo
reconstitution of the he
, 71-79
DIRKS
ia and beta c deficiency-associated pulmonary alveolar
hereditary diseases of childhood which are potentially
curable by stem cell gene therapy but require different therapeutic approaches.
EN, U., T. MORITZ, S. BURDACH, M. FLASSHOVE u. H. HANENBERG
(1999):
Fanconi anem
proteinosis as two
Klin. Pädiatr. 211, 329-335
DOLA
lls to
ting agents.
Cancer Res. 51
N, M. E., R. B. MITCHELL, C. MUMMERT, R. C. MOSCHEL u. A. E. PEGG
(1991):
Effect of O6-benzylguanine analogues on sensitivity of human tumor ce
the cytotoxic effects of alkyla
, 3367-3372
DONA E., B. P. SORRENTINO, R. G. HAWLEY, D. S. AN, I. S. CHEN u. R.
us and human lentivirus vectors independent of viral
4+ cells.
Mol. Ther. 3
HUE, R.
P. WERSTO (2001):
Fibronectin fragment CH-296 inhibits apoptosis and enhances ex vivo gene
transfer by murine retrovir
tropism in nonhuman primate CD3
, 359-367
129

Literaturverzeichnis
DROPULIC, B. (2005):
Genetic modification of hematopoietic cells using retroviral and lentiviral
cells. vectors: safety considerations for vector design and delivery into target
Curr. Hematol. Rep. 4, 300-304
P.
ein-Barr virus shuttle
Mol. Cell. Biol. 7
DUBRIDGE, R. B., P. TANG, H. C. HSIA, P. M. LEONG, J. H. MILLER u. M.
CALOS (1987):
Analysis of mutation in human cells by using an Epst
system.
, 379-387
DUNBAR, C. E., N. E. SEIDEL, S. DOREN, S. SELLERS, A. P. CLINE, M. E.
ng factor.
Proc. Natl. Acad. Sci. U S A 93
METZGER, B. A. AGRICOLA, R. E. DONAHUE u. D. M. BODINE (1996):
Improved retroviral gene transfer into murine and Rhesus peripheral blood or
bone marrow repopulating cells primed in vivo with stem cell factor and
granulocyte colony-stimulati
, 11871-11876
EATON, D. L., u. F. J. DE SAUVAGE (1997):
Thrombopoietin: the primary regulator of megakaryocytopoiesis and
thrombopoiesis.
Exp. Hematol. 25, 1-7
EMI, N., T. FRIEDMANN u. J. K. YEE (1991):
Pseudotype formation of murine leukemia virus with the G protein of vesicular
stomatitis virus.
J. Virol. 65, 1202-1207
130

Literaturverzeichnis
ENSSLE, J., I. JORDAN, B. MAUER u. A. RETHWILM (1996):
Foamy virus reverse transcriptase is expressed independently from the Gag
protein.
Proc. Natl. Acad. Sci. U S A 93, 4137-4141
em cell gene therapy: dead or alive?
FERGUSON, C., A. LAROCHELLE u. C. E. DUNBAR (2005):
Hematopoietic st
Trends Biotechnol. 23, 589-597
FINER D. FARSON u. M. R. ROBERTS (1994):
kat: a high-efficiency retroviral transduction system for primary human T
, M. H., T. J. DULL, L. QIN,
lymphocytes.
Blood 83, 43-50
FISCH
ER u. A. RETHWILM (1998):
J. Virol. 72
ER, N., M. HEINKELEIN, D. LINDEMANN, J. ENSSLE, C. BAUM, E.
WERDER, H. ZENTGRAF, J. G. MULL
Foamy virus particle formation.
, 1610-1615
GASP
. SCHMIDT, C. VON KALLE, T. BARINGTON, M. A.
RISTENSEN, A. AL GHONAIUM, H. N. WHITE, J. L.
SMITH, R. J. LEVINSKY, R. R. ALI, C. KINNON u. A. J. THRASHER (2004):
bined immunodeficiency by use of a
AR, H. B., K. L. PARSLEY, S. HOWE, D. KING, K. C. GILMOUR, J. SINCLAIR,
G. BROUNS, M
JAKOBSEN, H. O. CH
Gene therapy of X-linked severe com
pseudotyped gammaretroviral vector.
Lancet 364, 2181-2187
ERSON, S. L., K. MILLER u. N. A. BERGER (1985):
O6 alkylguanine-DNA alkyltransferase activity in human myeloid cells.
J. Clin. Invest. 76
G
, 2106-2114
131

Literaturverzeichnis
GERSON, S. L., W. PHILLIPS, M. KASTAN, L. L. DUMENCO u. C. DONOVAN
D34+ hematopoietic progenitors have low, cytokine-unresponsive
plus BCNU.
(1996):
Human C
O6-alkylguanine-DNA alkyltransferase and are sensitive to O6-benzylguanine
Blood 88, 1649-1655
GOER SWEENEY, G. BURON, R. STORB u. H. P.
KIEM (1999):
nsduction
ells in dogs.
NER, M., B. BRUNO, P. A. MC
The use of granulocyte colony-stimulating factor during retroviral tra
on fibronectin fragment CH-296 enhances gene transfer into hematopoietic
repopulating c
Blood 94, 2287-2292
tion and
od progenitor and stem cells.
HAAS, R., u. S. MUREA (1995):
The role of granulocyte colony-stimulating factor in mobiliza
transplantation of peripheral blo
Cytokines Mol. Ther. 1, 249-270
HACEIN-BEY-ABINA, S., F. LE DEIST, F. CARLIER, C. BOUNEAUD, C. HUE, J. P.
DE VILLARTAY, A. J. THRASHER, N. WULFFRAAT, R. SORENSEN, S.
DUPUIS-GIROD, A. FISCHER, E. G. DAVIES, W. KUIS, L. LEIVA u. M.
CAVAZZANA-CALVO (2002):
Sustained correction of X-linked severe combined immunodeficiency by ex
vivo gene therapy.
N. Engl. J. Med. 346, 1185-1193
132

Literaturverzeichnis
HACEIN-BEY-ABINA, S., C. VON KALLE, M. SCHMIDT, F. LE DEIST, N.
WULFFRAAT, E. MCINTYRE, I. RADFORD, J. L. VILLEVAL, C. C. FRASER,
unodeficiency.
M. CAVAZZANA-CALVO u. A. FISCHER (2003a):
A serious adverse event after successful gene therapy for X-linked severe
combined imm
N. Engl. J. Med. 348, 255-256
P. LEBOULCH, A. LIM, C. S. OSBORNE, R. PAWLIUK, E.
NNET, S. ROMANA, I. RADFORD-WEISS, F.
I, E. DELABESSE, E. MACINTYRE, F. SIGAUX, J.
SOULIER, L. E. LEIVA, M. WISSLER, C. PRINZ, T. H. RABBITTS, F. LE
CAVAZZANA-CALVO (2003b):
HACEIN-BEY-ABINA, S., C. VON KALLE, M. SCHMIDT, M. P. MCCORMACK, N.
WULFFRAAT,
MORILLON, R. SORENSEN, A. FORSTER, P. FRASER, J. I. COHEN, G. DE
SAINT BASILE, I. ALEXANDER, U. WINTERGERST, T. FREBOURG, A.
AURIAS, D. STOPPA-LYO
GROSS, F. VALENS
DEIST, A. FISCHER u. M.
LMO2-associated clonal T cell proliferation in two patients after gene therapy
for SCID-X1.
Science 302, 415-419
HANAZONO, Y., T. NAGASHIMA, M. TAKATOKU, H. SHIBATA, N. AGEYAMA, T.
ASANO, Y. UEDA, C. E. DUNBAR, A. KUME, K. TERAO, M. HASEGAWA u.
K. OZAWA (2002):
In vivo selective expansion of gene-modified hematopoietic cells in a
nonhuman primate model.
Gene Ther. 9, 1055-1064
ANENBERG, H., K. HASHINO, H. KONISHI, R. A. HOCK, I. KATO u. D. A.
WILLIAMS (1997):
Optimization of fibronectin-assisted retroviral gene transfer into human CD34+
hematopoietic cells.
Hum. Gene Ther. 8
H
, 2193-2206
133

Literaturverzeichnis
HANENBERG, H., X. L. XIAO, D. DILLOO, K. HASHINO, I. KATO u. D. A. WILLIAMS
(1996):
Colocalization of retrovirus and target cells on specific fibronectin fragments
increases genetic transduction of mammalian cells.
Nat. Med. 2, 876-882
EINKELEIN, M., M. DRESSLER, G. JARMY, M. RAMMLING, H. IMRICH, J. H
THUROW, D. LINDEMANN u. A. RETHWILM (2002a):
Improved primate foamy virus vectors and packaging constructs.
J. Virol. 76, 3774-3783
ELEIN, M., C. LEURS, M. RAMMLING, K. PETERS, H. HANENBERG u. A.
RETHWILM (2002b):
Pregenomic RNA is required for efficient incorporation of pol polyprotein into
foamy virus capsids.
HEINK
J. Virol. 76, 10069-10073
HEINK HMANN, G. JARMY, M. DRESSLER, H. IMRICH, J.
THUROW, D. LINDEMANN, M. BOCK, A. MOEBES, J. ROY, O.
ELEIN, M., T. PIETSC
HERCHENRODER u. A. RETHWILM (2000a):
Efficient intracellular retrotransposition of an exogenous primate retrovirus
genome.
Embo J. 19, 3436-3445
HEINKELEIN, M., M. SCHMIDT, N. FISCHER, A. MOEBES, D. LINDEMANN, J.
ENSSLE u. A. RETHWILM (1998):
vectors.
Characterization of a cis-acting sequence in the Pol region required to transfer
human foamy virus
J. Virol. 72, 6307-6314
134

Literaturverzeichnis
HEINKELEIN, M., J. THUROW, M. DRESSLER, H. IMRICH, D. NEUMANN-
HAEFELIN, M. O. MCCLURE u. A. RETHWILM (2000b):
n Process Citation].
Complex effects of deletions in the 5' untranslated region of primate foamy
virus on viral gene expression and RNA packaging [I
J. Virol. 74, 3141-3148
HENEINE, W., M. SCHWEIZER, P. SANDSTROM u. T. FOLKS (2003):
Human infection with foamy viruses.
Curr. Top. Microbiol. Immunol. 277, 181-196
ERCHENRÖDER, O., R. RENNE, D. LONCAR, E. K. COBB, K. K. MURTHY, J.
sequencing of simian foamy viruses from chimpanzees
H
SCHNEIDER, A. MERGIA u. P. A. LUCIW (1994):
Isolation, cloning, and
(SFVcpz): high homology to human foamy virus (HFV).
Virology 201, 187-199
IBINO, H., K. TANI, K. IKEBUCHI, M. S. WU, H. SUGIYAMA, Y. NAKAZAKI, T.
rimate model for cytokine and
H
TANABE, S. TAKAHASHI, A. TOJO, S. SUZUKI, Y. TANIOKA, Y. SUGIMOTO,
T. NAKAHATA u. S. ASANO (1999):
The common marmoset as a target preclinical p
gene therapy studies.
Blood 93, 2839-2848
IBINO, H., K. TANI, H. SUGIYAMA, S. SUZUKI, M. S. WU, K. IZAWA, H. HASE, Y.
l and adenoviral vectors.
H
NAKAZAKI, T. TANABE, J. OOI, T. IZEKI, A. TOJO, I. SAITOH, Y. TANIOKA
u. S. ASANO (2001):
Haematopoietic progenitor cells from the common marmoset as targets of
gene transduction by retrovira
Eur. J. Haematol. 66, 272-280
135

Literaturverzeichnis
HILL, C. L., P. D. BIENIASZ u. M. O. MCCLURE (1999):
Properties of human foamy virus relevant to its development as a vector for
gene therapy.
J. Gen. Virol. 80, 2003-2009
O, A. D. (2006):
s., Cologne, Proc., S. 12
RDANSKII, S. N., L. F. LIDEMAN, A. K. CHIKOVA u. R. A. GIBADULIN (1997):
mbinant strains of vaccinia
H
Connections determine life fate of adult stem cells.
in: 1st Cong. Germ. Soc. Stem Cell Re
IO
Interaction of gag polyprotein-precursors p55 and p48 with p160gag-pol during
formation of virus-like particles of HIV-1 with reco
virus.
Vopr. Virusol. 42, 205-208
AWA, K., K. TANI, Y. NAKAZAKI, H. HIBINO, H. SUGIYAMA, A. KAWASAKI, E.
armoset CD34 cells isolated by a novel
IZ
SASAKI, C. NISHIOKA, H. ISHII, Y. SODA, H. YAGITA, Y. TANIOKA, A.
TOJO u. S. ASANO (2004):
Hematopoietic activity of common m
monoclonal antibody MA24.
Exp. Hematol. 32, 843-851
ANG, S. K., u. E. WIMMER (1990):
in.
J
Cap-independent translation of encephalomyocarditis virus RNA: structural
elements of the internal ribosomal entry site and involvement of a cellular 57-
kD RNA-binding prote
Genes Dev. 4, 1560-1572
136

Literaturverzeichnis
KAINA, B., G. FRITZ u. T. COQUERELLE (1993):
Contribution of O6-alkylguanine and N-alkylpurines to the formation of sister
chromatid exchanges, chromosomal aberrations, and gene mutations: new
f genetically engineered mammalian cell lines.
Environ. Mol. Mutagen. 22
insights gained from studies o
, 283-292
KAPLA RAMLEY, L. VINCENT, C.
S, Z. ZHU, D.
HICKLIN, Y. WU, J. L. PORT, N. ALTORKI, E. R. PORT, D. RUGGERO, S. V.
438
N, R. N., R. D. RIBA, S. ZACHAROULIS, A. H. B
COSTA, D. D. MACDONALD, D. K. JIN, K. SHIDO, S. A. KERN
SHMELKOV, K. K. JENSEN, S. RAFII u. D. LYDEN (2005):
VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-
metastatic niche.
Nature , 820-827
IEM, H.-P., S. HEYWARD, A. WINKLER, J. POTTER, J. A. ALLEN, A. D. MILLER u.
eudotyped retroviral vectors in a competitive
K
R. G. ANDREWS (1997):
Gene transfer into marrow repopulating cells: comparison between
amphotropic and GALV ps
repopulation assay in baboons.
Blood 90, 4638-4645
IEM, H. P., J. ALLEN, G. TROBRIDGE, E. OLSON, K. KEYSER, L. PETERSON u. K
D. W. RUSSELL (2007):
Foamy-virus-mediated gene transfer to canine repopulating cells.
Blood 109, 65-70
KIMURA, S., A. W. ROBERTS, D. METCALF u. W. S. ALEXANDER (1998):
Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for
thrombopoietin.
Proc. Natl. Acad. Sci. U S A 95, 1195-1200
137

Literaturverzeichnis
KOHN, D. B. (1996):
Gene therapy for hematopoietic and immune disorders.
Bone Marrow Transplant. 18 Suppl 3, S55-58
KOHN . BODINE, H. P. KIEM, F. CANDOTTI, J.
TISDALE, I. RIVIERE, C. A. BLAU, R. E. RICHARD, B. SORRENTINO, J.
, D. B., M. SADELAIN, C. DUNBAR, D
NOLTA, H. MALECH, M. BRENNER, K. CORNETTA, J. CAVAGNARO, K.
HIGH u. J. GLORIOSO (2003):
American society of gene therapy (ASGT) ad hoc subcommittee on retroviral-
mediated gene transfer to hematopoietic stem cells.
Mol. Ther. 8, 180-187
KRAU LER, C. I. CIVIN u. W. S. MAY (1996):
CD34: structure, biology, and clinical utility.
SE, D. S., M. J. FACK
Blood 87, 1-13
KREK
(6)-methylguanine DNA methyltransferase-
sive treatment of human glioma xenografts with
combination O(6)-benzylguanine and 1,3-bis-(2-chloroethyl)-1-nitrosourea.
LAU, E. L., K. E. POLLOK, B. J. BAILEY, N. LIU, J. R. HARTWELL, D. A.
WILLIAMS u. L. C. ERICKSON (2003):
Hematopoietic expression of O
P140K allows inten
Mol. Cancer Ther. 2, 1321-1329
KURR S u. H. P.
Gene transfer into baboon repopulating cells: A comparison of Flt-3 Ligand
ex vivo
E, P., J. MORRIS, P. A. HORN, M. A. HARKEY, R. G. ANDREW
KIEM (2001):
and megakaryocyte growth and development factor versus IL-3 during
transduction.
Mol. Ther. 3, 920-927
138

Literaturverzeichnis
LAROCHELLE, A., J. VORMOOR, H. HANENBERG, J. C. Y. WANG, M. BHATIA, T.
TO, D. A. WILLIAMS
Identification of primitive human hematopoietic cells capable of repopulating
LAPIDOT, T. MORITZ, B. MURDOCH, X. L. XIAO, I. KA
u. J. E. DICK (1996):
NOD/SCID mouse bone marrow: implications for gene therapy.
Nat. Med. 2, 1329-1337
, C. (2003):
LEURS
issenschaftliche Fakultät Düsseldorf, Heinrich-Heine-
Universität, Diss.
LEURS HEINKELEIN, M. SCHMIDT, M.
INDEMANN, C. VON KALLE, A. RETHWILM, D. A. WILLIAMS
u. H. HANENBERG (2003):
ncy mice repopulating human CD34+
Etablierung foamyviraler Vektoren zur Transduktion humaner
hämatopoetischer Stammzellen.
Mathematisch- Naturw
, C., M. JANSEN, K. E. POLLOK, M.
WISSLER, D. L
Comparison of three retroviral vector systems for transduction of nonobese
diabetic/severe combined immunodeficie
cord blood cells.
Hum. Gene Ther 14, 509-519
LI, Z., R, M. SCHMIDT, C. VON KALLE, J. MEYER,
M. FORSTER, C. STOCKING, A. WAHLERS, O. FRANK, W. OSTERTAG, K.
ia induced by retroviral gene marking.
J. DULLMANN, B. SCHIEDLMEIE
KUHLCKE, H. G. ECKERT, B. FEHSE u. C. BAUM (2002):
Murine leukem
Science 296, 497.
MANN, D., M.
LINDE BOCK, M. SCHWEIZER u. A. RETHWILM (1997):
of murine leukemia virus particles with chimeric human
foamy virus envelope proteins.
J. Virol. 71
Efficient pseudotyping
, 4815-4820
139

Literaturverzeichnis
LINDEMANN, D., u. P. A. GOEPFERT (2003):
The foamy virus envelope glycoproteins.
Curr. Top. Microbiol. Immunol. 277, 111-129
LINIAL
pathogenic?
Trends Microbiol. 8
, M. (2000):
Why aren't foamy viruses
, 284-289
LINIAL
l retroviruses.
, M. L. (1999):
Foamy viruses are unconventiona
J. Virol. 73, 1747-1755
OCHELT, M. (2003): L
Foamy virus transactivation and gene expression.
Curr. Top. Microbiol. Immunol. 277, 27-61
LÖCH
er.
ELT, M., W. MURANYI u. R. M. FLÜGEL (1993):
Human foamy virus genome possesses an internal, Bel-1-dependent and
functional promot
Proc. Natl. Acad. Sci. U S A 90, 7317-7321
LUSKEY, B. D., M. ROSENBLATT, K. ZSEBO u. D. A. WILLIAMS (1992):
Stem cell factor, interleukin-3, and interleukin-6 promote retroviral-mediated
gene transfer into murine hematopoietic stem cells.
Blood 80, 396-402
AZE, R., H. HANENBERG u. D. A. WILLIAMS (1997): M
Establishing chemoresistance in hematopoietic progenitor cells.
Mol. Med. Today 3, 350-358
140

Literaturverzeichnis
MAZE, R., C. KURPAD, A. E. PEGG, L. C. ERICKSON u. D. A. WILLIAMS (1999):
40A, but not P140A/G156A, mutant
sferase protects hematopoietic
cells against O6-benzylguanine sensitization to chloroethylnitrosourea
Retroviral-mediated expression of the P1
form of O6-methylguanine DNA methyltran
treatment.
J. Pharmacol. Exp. Ther. 290, 1467-1474
EIERING, C. D., K. E. COMSTOCK u. M. L. LINIAL (2000):
tions of human foamy virus in persistently infected human
M
Multiple integra
erythroleukemia cells.
J. Virol. 74, 1718-1726
MEIERING, C. D., u. M. L. LINIAL (2001):
and infection. Historical perspective of foamy virus epidemiology
Clin. Microbiol. Rev. 14, 165-176
MERGIA, A., S. CHARI, D. L. KOLSON, M. M. GOODENOW u. T. CICCARONE
an foamy virus vector type-1 (SFV-1) in nondividing cells
Virology 280
(2001):
The efficiency of simi
and in human PBLs.
, 243-252
MERG
l. Immunol. 277
IA, A., u. M. HEINKELEIN (2003):
Foamy virus vectors.
Curr. Top. Microbio , 131-159
ns.
METCALF, D. (1977):
In-vitro cloning techniques for hemopoietic cells: clinical applicatio
Ann. Intern. Med. 87, 483-488
141

Literaturverzeichnis
MOCHIZUKI, H., J. P. SCHWARTZ, K. TANAKA, R. O. BRADY u. J. REISER (1998):
High-titer human immunodeficiency virus type 1-based vector systems for
gene delivery into nondividing cells.
J. Virol. 72, 8873-8883
MODROW, S, u. D. FALKE u. U. TRUYEN (2002):
Molekulare Virologie.
eidelberg, 750 S.
MOEBES, A., J. ENSSLE, P. D. BIENIASZ, M. HEINKELEIN, D. LINDEMANN, M.
u. A. RETHWILM (1997):
Human foamy virus reverse transcription that occurs late in the viral replication
Spektrum der Wissenschaft Verlagsgesellschaft mbH, H
BOCK, M. O. MCCLURE
cycle.
J. Virol. 71, 7305-7311
ORITZ, T., V. P. PATEL u. D. A. WILLIAMS (1994):
oietic cells via retroviral vectors.
M
Bone marrow extracellular matrix molecules improve gene transfer into human
hematop
J. Clin. Invest. 93, 1451-1457
MORRISON, S. J., N. UCHIDA u. I. L. WEISSMAN (1995):
The biology of hematopoietic stem cells.
Annu. Rev. Cell. Dev. Biol. 11, 35-71
NALDI . MULLIGAN, F. H. GAGE, I. M.
VERMA u. D. TRONO (1996):
very and stable transduction of nondividing cells by a lentiviral
NI, L., U. BLÖMER, P. GALLAY, D. ORY, R
In vivo gene deli
vector.
Science 272, 263-267
142

Literaturverzeichnis
NAVIAUX, R. K., E. COSTANZI, M. HAAS u. I. M. VERMA (1996):
The pCL system: rapid production of helper-free, high-titer, recombinant
retroviruses.
J. Virol. 70, 5701-5705
NEFF, T., B. C. BEARD, L. J. PETERSON, P. ANANDAKUMAR, J. THOMPSON u.
l of
drug resistance gene therapy.
H. P. KIEM (2005):
Polyclonal chemoprotection against temozolomide in a large-animal mode
Blood 105, 997-1002
NEFF,
MS, M. SCHMIDT, G. E. GEORGES, C. VON KALLE u. H. P. KIEM
Methylguanine methyltransferase-mediated in vivo selection and
rge-animal model.
T., P. A. HORN, L. J. PETERSON, B. M. THOMASSON, J. THOMPSON, D. A.
WILLIA
(2003):
chemoprotection of allogeneic stem cells in a la
J. Clin. Invest. 112, 1581-1588
NOWR KLANKE, M. HEINKELEIN, M. RAMMLING, T.
DANDEKAR, C. VON KALLE u. A. RETHWILM (2006):
ations into a human cell
OUZI, A., M. DITTRICH, C.
Genome-wide mapping of foamy virus vector integr
line.
J. Gen. Virol. 87, 1339-1347
OMOTO, S., E. A. BRISIBE, H. OKUYAMA u. Y. R. FUJII (2004):
Feline foamy virus Tas protein is a DNA-binding transactivator.
J. Gen. Virol. 85, 2931-2935
143

Literaturverzeichnis
ORKIN, S. H., u. A. G. MOTULSKY (1995):
Report and recommendations of the panel to assess the NIH investment in
research on gene therapy.
lrep.htmlwww.nih.gov/news/pane ; Abrufdatum: 07.12.1995
ce of foamy virus vectors.
PATTON, G. S., O. ERLWEIN u. M. O. MCCLURE (2004):
Cell-cycle dependen
J. Gen. Virol. 85, 2925-2930
PEAR M. L. SCOTT u. D. BALTIMORE (1993):
Production of high-titer helper-free retrovirus by transient infection.
, W. S., G. P. NOLAN,
Proc. Natl. Acad. Sci. USA 90, 8392-8396
PEGG, A. E., M. E. DOLAN u. R. C. MOSCHEL (1995):
Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase.
Prog. Nucleic Acid Res. Mol. Biol. 51, 167-223
FEIFER, A., u. I. M. VERMA (2001): P
Gene therapy: promises and problems.
Annu. Rev. Genomics Hum. Genet. 2, 177-211
PIETSCHMANN, T., M. HEINKELEIN, M. HELDMANN, H. ZENTGRAF, A.
N (1999):
Foamy virus capsids require the cognate envelope protein for particle export.
RETHWILM u. D. LINDEMAN
J. Virol. 73, 2613-2621
POLLO GER, S. GOEBEL, H. HANENBERG u. S. CAI
(2005):
In Vivo Selection of Murine Long-Term Repopulating Cells Transduced with a
Foamy Virus Vector That Expresses MGMTP140K.
in: ASH Ann. Meeting, Proc. 106
K, K. E., A. G. ERNSTBER
, 3061
144

Literaturverzeichnis
PREUSS, I., S. HAAS, U. EICHHORN, I. EBERHAGEN, M. KAUFMANN, T. BECK,
6):
thyltransferase in
human tumor and corresponding normal tissue.
R. H. EIBL, P. DALL, T. BAUKNECHT, J. HENGSTLER, B. M. WITTIG, W.
DIPPOLD u. B. KAINA (199
Activity of the DNA repair protein O6-methylguanine-DNA me
Cancer Detect. Prev. 20, 130-136
QUINN
The DNA-repair gene MGMT and the clinical response of gliomas to alkylating
, D. I. (2001):
agents.
N. Engl. J. Med. 344, 686
UINN, J. A., J. PLUDA, M. E. DOLAN, S. DELANEY, R. KAPLAN, J. N. RICH, A. H.
N, O. M. COLVIN, M. M.
M.
-UHLIG, J. E. HERNDON, 2ND,
D. D. BIGNER u. H. S. FRIEDMAN (2002):
plus O(6)-benzylguanine for patients with
ssive malignant glioma.
Q
FRIEDMAN, D. A. REARDON, J. H. SAMPSO
HAGLUND, A. E. PEGG, R. C. MOSCHEL, R. E. MCLENDON, J.
PROVENZALE, S. GURURANGAN, S. TOURT
Phase II trial of carmustine
nitrosourea-resistant recurrent or progre
J. Clin. Oncol. 20, 2277-2283
RADSAK, K., R. FUHRMANN, R. P. FRANKE, D. SCHNEIDER, A. KOLLERT, K. H.
BRUCHER u. D. DRENCKHAHN (1989):
Arch. Virol. 107
Induction by sodium butyrate of cytomegalovirus replication in human
endothelial cells.
, 151-158
145

Literaturverzeichnis
RAGG, S., M. XU-WELLIVER, J. BAILEY, M. D'SOUZA, R. COOPER, S. CHANDRA,
R. SESHADRI, A. E. PEGG u. D. A. WILLIAMS (2000):
Direct reversal of DNA damage by mutant methyltransferase protein protects
mice against dose-intensified chemotherapy and leads to in vivo selection of
hematopoietic stem cells.
Cancer Res. 60, 5187-5195
REESE, J. S., O. N. KOC, K. M. LEE, L. LIU, J. A. ALLAY, W. P. PHILLIPS, JR. u. S.
l transduction of a mutant methylguanine DNA methyltransferase
lls confers resistance to O6-benzylguanine plus 1,3-
bis(2-chloroethyl)-1-nitrosourea.
L. GERSON (1996):
Retrovira
gene into human CD34 ce
Proc. Natl. Acad. Sci. U S A 93, 14088-14093
DER, T., A. BRUN, R. G. HAWLEY, S. KARLSSON u. J. RICHTER (2001):
Retroviral transduction of human CD34+ cells on fibronectin fragment CH-296
is inhibited by high concentrations of vector
RELAN
containing medium.
J. Gene Med. 3, 207-218
RETH
The replication strategy of foamy viruses.
WILM, A. (2003):
Curr. Top. Microbiol. Immunol. 277, 1-26
ROE,
ne leukemia virus DNA depends on mitosis.
T., T. C. REYNOLDS, G. YU u. P. O. BROWN (1993):
Integration of muri
Embo J. 12, 2099-2108
RUSSELL, D. W., u. A. D. MILLER (1996):
Foamy virus vectors.
J. Virol. 70, 217-222
146

Literaturverzeichnis
RUSTIGIAN, R., P. JOHNSTON u. H. REIHART (1955):
Infection of monkey kidney tissue cultures with virus-like agents.
Proc. Soc. Exp. Biol. Med. 88, 8-16
, M. D., A. M. KING u. G. P
RYAN . THOMAS (1991):
disease virus polyprotein is mediated by residues
located within a 19 amino acid sequence.
Cleavage of foot-and-mouth
J. Gen. Virol. 72 ( Pt 11), 2727-2732
SCHA
l and lentiviral SIN vectors for expression of
ematopoietic cells.
Mol. Ther. 13
MBACH, A., J. BOHNE, S. CHANDRA, E. WILL, G. P. MARGISON, D. A.
WILLIAMS u. C. BAUM (2006):
Equal potency of gammaretrovira
O6-methylguanine-DNA methyltransferase in h
, 391-400
SCHLE
dylserine: is
-binding site?
Cell 32
GEL, R., T. S. TRALKA, M. C. WILLINGHAM u. I. PASTAN (1983a):
Inhibition of VSV binding and infectivity by phosphati
phosphatidylserine a VSV
, 639-646
SCHLE
"receptor".
GEL, R., u. M. WADE (1983b):
Neutralized vesicular stomatitis virus binds to host cells by a different
Biochem. Biophys. Res. Commun. 114, 774-778
SCHMIDT, M., u. A. RETHWILM (1995):
Replicating foamy virus-based vectors directing high level expression of
foreign genes.
Virology 210, 167-178
147

Literaturverzeichnis
SCHMIDT, M., P. ZICKLER, G. HOFFMANN, S. HAAS, M. WISSLER, A. MUESSIG,
. KIEM, C. E.
Polyclonal long-term repopulating stem cell clones in a primate model.
J. F. TISDALE, K. KURAMOTO, R. G. ANDREWS, T. WU, H. P
DUNBAR u. C. VON KALLE (2002):
Blood 100, 2737-2743
SCHR RY, J. R. ECKER u. F. BUSHMAN
HIV-1 integration in the human genome favors active genes and local hotspots.
ODER, A. R., P. SHINN, H. CHEN, C. BER
(2002):
Cell 110, 521-529
SCHW
ted from an accidentally infected human individual.
J. Virol. 71
EIZER, M., V. FALCONE, J. GANGE, R. TUREK u. D. NEUMANN-HAEFELIN
(1997):
Simian foamy virus isola
, 4821-4824
SEGG
. S. YOUNG, 3RD, R. E. DONAHUE,
. W. NIENHUIS u. C. E. DUNBAR (2006):
Acute myeloid leukemia is associated with retroviral gene transfer to
a rhesus macaque.
EWISS, R., S. PITTALUGA, R. L. ADLER, F. J. GUENAGA, C. FERGUSON, I.
H. PILZ, B. RYU, B. P. SORRENTINO, W
C. VON KALLE, A
hematopoietic progenitor cells in
Blood 107, 3865-3867
SONE . GRIFFITHS, G. ROMANO,
S. M. KINGSMAN u. A. J. KINGSMAN (1995):
ion system for the production of high titer
es. 23
OKA, Y., P. M. CANNON, E. E. RAMSDALE, J. C
A transient three-plasmid express
retroviral vectors.
Nucleic Acids R , 628-633
148

Literaturverzeichnis
SPANGRUDE, G. J., D. M. BROOKS u. D. B. TUMAS (1995):
Long-term repopulation of irradiated mice with limiting numbers of purified
hematopoietic stem cells: in vivo expansion of stem cell phenotype but not
function.
Blood 85, 1006-1016
SPANGRUDE, G. J., S. HEIMFELD u. I. L. WEISSMAN (1988):
Purification and characterization of mouse hematopoietic stem cells.
Science 241, 58-62
TANKE, N., A. STANGE, D. LUFTENEGGER, H. ZENTGRAF u. D. LINDEMANN
ation of the prototype foamy virus envelope glycoprotein leader
S
(2005):
Ubiquitin
peptide regulates subviral particle release.
J. Virol. 79, 15074-15083
ns derived from
STITZ, J., C. J. BUCHHOLZ, M. ENGELSTADTER, W. UCKERT, U. BLOEMER, I.
SCHMITT u. K. CICHUTEK (2000):
Lentiviral vectors pseudotyped with envelope glycoprotei
gibbon ape leukemia virus and murine leukemia virus 10A1.
Virology 273, 16-20
ANAKA, J., H. SADANARI, H. SATO u. S. FUKUDA (1991): T
Sodium butyrate-inducible replication of human cytomegalovirus in a human
epithelial cell line.
Virology 185, 271-280
THOM
Bone marrow transplantation: a review.
Semin. Hematol. 36
AS, E. D. (1999):
, 95-103
149

Literaturverzeichnis
THOMAS, E. D., H. L. LOCHTE, W. C. LU u. J. W. FERREBEE (1957):
. Med. 157
Intravenous infusion of bone marrow in patients receiving radiation and
chemotherapy.
N. Engl. J , 491-496
O, L. B., D. N. HAYLOCK, P. J. SIMMONS u. C. A. JUTTNER (1997): T
The biology and clinical uses of blood stem cells.
Blood 89, 2233-2258
ROBRIDGE, G., u. D. W. RUSSELL (2004):
oncovirus and lentivirus vectors.
T
Cell cycle requirements for transduction by foamy virus vectors compared to
those of
J. Virol. 78, 2327-2335
TROB ER, M. A. JACOBS, J. M. ALLEN, H. P. KIEM, R.
KAUL u. D. W. RUSSELL (2006):
RIDGE, G. D., D. G. MILL
Foamy virus vector integration sites in normal human cells.
Proc. Natl. Acad. Sci. U S A 103, 1498-1503
TROB
us vectors.
Hum. Gene Ther. 9
RIDGE, G. D., u. D. W. RUSSELL (1998):
Helper-free foamy vir
, 2517-2525
VAN D
. WILLIAMS (1998):
ere combined immunodeficiency (NOD/SCID) mouse
as a model system to study the engraftment and mobilization of human
tem cells.
ER LOO, J. C. M., H. HANENBERG, R. J. COOPER, F. Y. LUO, E. N.
LAZARIDIS u. D. A
Nonobese diabetic/sev
peripheral blood s
Blood 92, 2556-2570
150

Literaturverzeichnis
VASSILOPOULOS, G., G. TROBRIDGE, N. C. JOSEPHSON u. D. W. RUSSELL
nto murine hematopoietic stem cells with helper-free foamy
Blood 98
(2001):
Gene transfer i
virus vectors.
, 604-609
VON L ROSCHER, B. FEHSE u. C.
Amphotropic and VSV-G-pseudotyped retroviral vectors transduce human
ar efficiency.
AER, D., A. COROVIC, B. VOGT, U. HERWIG, S.
BAUM (2000):
hematopoietic progenitor cells with simil
Bone Marrow Transplant. 25 Suppl 2, S75-79
WANG u. J. E. DICK (1997):
Primitive human hematopoietic cells are enriched in cord blood compared to
ing cell (SRC) assay.
, J. C. Y., M. DOEDENS
adult bone marrow or mobilized peripheral blood as measured by the
quantitative in vivo SCID-repopulat
Blood 89, 3919-3924
EISS, R. A. (1988):
disease.
W
Foamy retroviruses. A virus in search of a
Nature 333, 497-498
ILLIAMS, D. A., I. R. LEMISCHKA, D. G. NATHAN u. R. C. MULLIGAN (1984): W
Introduction of new genetic material into pluripotent haematopoietic stem cells
of the mouse.
Nature 310, 476-480
MS, D. A., H. I. LEVITSKY, M
WILLIA . C. DINAUER u. D. W. RUSSELL (1997):
ematology: human diseases, mouse models and new
approaches.
ASH Educ. Book 12
Genetic therapies in h
, 55-74
151

Literaturverzeichnis
WINKLER, I. G., R. M. FLÜGEL, K. ASIKAINEN u. R. L. FLOWER (2000):
Antibody to human foamy virus not detected in individuals treated with blood
products or in blood donors.
Vox. Sang. 79, 118-119
U, M., u. A. MERGIA (1999): W
Packaging cell lines for simian foamy virus type 1 vectors.
J. Virol. 73, 4498-4501
WU, X
me are favored targets for MLV
integration.
., Y. LI, B. CRISE u. S. M. BURGESS (2003):
Transcription start regions in the human geno
Science 300, 1749-1751
XU-WE
rase mutants highly
by O6-benzylguanine.
Cancer Res. 58
LLIVER, M., S. KANUGULA u. A. E. PEGG (1998):
Isolation of human O6-alkylguanine-DNA alkyltransfe
resistant to inactivation
, 1936-1945
YANG , B. ROESSLER u. K. T.
Highly efficient genetic transduction of primary human synoviocytes with
, J., M. S. FRIEDMAN, H. BIAN, L. J. CROFFORD
MCDONAGH (2002):
concentrated retroviral supernatant.
Arthritis Res. 4, 215-219
YANG, P., M. ZEMBA, M. ABOUD, R. M. FLUGEL u. M. LOCHELT (1997):
Deletion analysis of both the long terminal repeat and the internal promoters of
the human foamy virus.
Virus Genes 15, 17-23
152

Literaturverzeichnis
YEE, J. K., T. FRIEDMANN u. J. C. BURNS (1994):
Generation of high-titer pseudotyped retroviral vectors with very broad host
range.
Methods Cell. Biol. 43, 99-112
S (1995):
YODER, M. C., u. D. A. WILLIAM
Matrix molecule interactions with hematopoietic stem cells.
Exp. Hematol. 23, 961-967
u. M. OGAWA (1996):
ic stem cells.
A 93
YONEMURA, Y., H. KU, F. HIRAYAMA, L. M. SOUZA
Interleukin 3 or interleukin 1 abrogates the reconstituting ability of
hematopoiet
Proc. Natl. Acad. Sci. U S , 4040-4044
YU, J. (1996):
Regulation and reconstitution of human hematopoiesis.
J. Formos Med. Assoc. 95, 281-293
U, S. F., u. M. L. LINIAL (1993):
uantitative assay.
Y
Analysis of the role of the bel and bet open reading frames of human foamy
virus by using a new q
J. Virol. 67, 6618-6624
YU, S. . L. LINIAL (1999):
Evidence that the human foamy virus genome is DNA.
F., M. D. SULLIVAN u. M
J. Virol. 73, 1565-1572
153


Danksagung
Mein Dank gilt Prof. Dr. Helmut Hanenberg für die Überlassung des äußerst
interessanten Themas, die fachliche Unterstützung bei virologischen Fragen, seine
tändige Diskussionsbereitschaft und seine Betreuung bei dieser Arbeit.
Herrn erzeit angebotene,
gute f stets gewährte Diskussionsbereitschaft und die
onstruktive Kritik bei der Anfertigung der Arbeit, trotz der großen Entfernung. Vielen
Mein rof. Dr. Heiner Niemann; Institut für Tierzucht,
Bunde nsee, für die Betreuung der Arbeit
nd Verteidigung an der Stiftung Tierärztliche Hochschule Hannover.
Bei de Rethwilm möchte ich
mich für die gute Zusammenarbeit, die fachlich Unterstützung und vielen gute
atschläge im Umgang mit Foamyviren bedanken.
Ich da
eine angenehme Arbeitsatmosphäre sowie für die ständige Gesprächsbereitschaft
und Unterstützung bei Problemlösungen, insbesondere Herrn Dr. Michael Gombert
r die Einführung in die Geheimnisse von Microsoft-Word.
Gedan und Ingrid Wurm und
meine Bastian für die große Geduld und Aufmunterung bei
er Fertigstellung der Arbeit.
esonderer Dank gilt Heiko Nathues, der nicht nur viel Geduld bei der Erstellung
ieser Arbeit mit mir bewies, sondern mich auch im Vorfeld der Finalisierung dieses
erks stets motivierte, nicht den Kopf in den Sand zu stecken. Danke
s
PD. Dr. Peter A. Horn danke ich ganz besonders für die jed
achliche Unterstützung, die
k
Dank auch für die Unterstützung meiner Auslandsaufenthalte.
verbindlicher Dank gilt P
sforschungsanstalt für Landwirtschaft, Marie
u
n Virologen Herrn Dr. Dirk Lindemann und Prof. Axel
R
nke allen Mitarbeiterinnen und Mitarbeitern des KMT-Labor der Kinderklinik für
fü
kt sei an dieser Stelle auch meinen Eltern, Rainer
n Brüdern Bernhard und
d
B
d
W
Recommended




![Ambulante spezialfachärztliche Versorgung: Neue ... · C18.3 Flexura coli dextra [hepatica] C18.4 Colon transversum C18.5 Flexura coli sinistra [lienalis] C18.6 Colon descendens](https://img.pdfslide.org/doc/110x75/5e204c809b010200564bc8f5/ambulante-spezialfachrztliche-versorgung-neue-c183-flexura-coli-dextra-hepatica.jpg)