Embed Size (px)
Citation preview

13
3 Informationen über das Schichtwachstum beim ILGAR-Sulfid-Prozeß aus strukturellen und optischen Eigenschaften
Bei Einführung eines neuen Herstellungsverfahrens zur Abscheidung dünnerSulfid-Schichten soll als erstes dessen charakteristisches Abscheideverhaltengezeigt werden. Neben der stöchiometrischen und morphologischen Charakteri-sierung und der Bestimmung der Kristallstruktur werden die ILGAR-Dünn-schichten spektrophotometrisch vermessen. Da das Schichtwachstum erheblichenEinfluß auf Komposition, Morphologie und Kristallitgröße hat, werden die hiergewonnenen Erkenntnisse bei der Optimierung des Verfahrens für diverseAnwendungsbereiche umgesetzt.
Zur Auswertung der optischen Eigenschaften der nanokristallinenen ILGAR-Sulfidschichten wurde im Rahmen dieser Arbeit ein Programm zur Simulation derAbsorptionskante entwickelt. Hierdurch ergeben sich wichtige Informationen überdas Wachstum der ILGAR-Schichten auf glattem Substrat.
3.1 Standard-ILGAR-Präpara-tionsbedingungen für nano-kristalline Sulfidschichten
Die float-Glas- oder Quarzglas-Substratewurden für zehn Minuten einer Reinigung mitIsopropanol im Ultraschallbad unterzogen.Alle im anschließenden Abscheideprozeßbenutzen Chemikalien und Lösungsmittelwaren von analytischer Reinheit.
Die nanokristallinen Sulfid-Schichten wurdenim ´dip´-Verfahren hergestellt: im Falle vonCdS wurde das Substrat für 10 s in eine CdCl2-Ausgangssalzlösung (Konzentration 3-80mmol/l) eingetaucht. Das Lösungsmittel warein Wasser/Acetonitril-Gemisch mit einemVolumenverhältnis von 1:4. Das Wasser wurdebenötigt, um eine störende Ausfällung zuverhindern. Da Acetonitril als Haupt-bestandteil des Ausgangsbades gewählt wurde,verdampfte das Lösungsmittel unmittelbarnach Herausziehen der Probe aus dem Bad, sodaß auf ein zusätzliches Trocknen der Probeim Inertgasstrom verzichtet werden konnte.Der anschließende, bewußt kurz gehaltene, 15sekündige Begasungsschritt mit H2S erfolgtebei Raumtemperatur. Der gezielt unvoll-
ständige Reaktionsumsatz gewährleistet dieNanokristallinität der erzeugten Sulfid-schichten (siehe die folgenden Abschnitte). Fürdie meisten der in Kapitel 2 beschriebenenExperimente wurde die Zahl der Prozeßzyklenso gewählt, daß unabhängig von der Aus-gangssalz-Konzentration stets eine etwa 60 nmdicke Sulfid-Schicht hergestellt wurde. EineAusnahme bilden spezielle Schichtdicken-variationen.
Für die ZnS-Deposition wurde eine ZnCl2-Konzentration von 20mmol/l benutzt.Lösungsmittel war hier ein Wasser/THF-Gemisch (1:9 v/v). Die Sulfurisierung wurdebei einer Ofentemperatur von T= 80 °C durch-geführt, mit einer ebenfalls kurz gehaltenenBegasungsdauer von 10 sek.. Eine höhereReaktionstemperatur war beim ZnS notwendig,um einen etwas größeren Reaktionsumsatz unddadurch Proben mit homogenerer Schichtdickezu gewährleisten. Wegen der großen optischenBandlücke (Egap ≥ 3.68 eV) wurden für diespektrophotometrischen Untersuchungen derZnS-Schichten beidseitig polierte Quarzglas-substrate verwendet.

14 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
3.2 Komposition, Kristallstrukturund Morphologie von ILGAR-Metall-Sulfid-Schichten
Nach der Vorstellung der verschiedenenVarianten des ILGAR-Verfahrens im Kapitel 2soll nun auf die Materialeigenschaftensulfidischer ILGAR-Dünnschichten einge-gangen werden. Die Materialeigenschaften vonILGAR-Oxid-Schichten werden in derDiplomarbeit von M. Bär [ 11 ] behandelt.
In diesem Abschnitt soll speziell dasWachstum von nanokristallinen ILGAR-CdS-und ILGAR-ZnS-Dünnfilmen auf glattemGlas-Substrat untersucht werden. Bei der Her-stellung dieser Sulfidschichten wird gemäß denStandard-Präparationsbedingungen (Abschnitt3.1) eine nanokristallines Metallsalz-Schichtdirekt in eine nanokristalline Metallsulfid-Verbindung umgewandelt. Die mit der kleinenPartikelgröße einhergehenden Größen-quantisierungseffekte (Abschnitt 3.3.2.1)sollen mithilfe der Spektrophotometrie zurUntersuchung des Kristallitwachstums heran-gezogen werden.
Zuvor wird auf die chemische Zusammen-setzung, Kristallstruktur und Morphologiederartiger nanokristalliner ILGAR-Schichteneingegangen. In Abbildung 3 sind zweiILGAR-Sulfidschichten abgebildet. Typischer-weise beträgt die Probengröße 25x25 mm2, wiesie die ILGAR-ZnS-Probe (Abbildung 3,rechts) besitzt. Daß ein „up-scaling“ desILGAR-Prozesses auf 50x50 mm2 problemlosdurchgeführt werden kann, zeigt die ebenfallshomogen abgeschiedene ILGAR-CdS-Probe(Abbildung 3, links). Beide Proben besitzeneinen Rand (beim CdS oben, beim ZnS untenrechts im Bild) bis zu dem die Probe in dieAusgangssalzlösung eingetaucht war. Bei dersehr transparenten ZnS-Probe wurde einschwarzer Hintergrund gewählt, um Inhomo-genitäten der Probe besser erkennen zukönnen. So sind bei dieser Probe vereinzeltMaterialanhäufungen sichtbar, die in derAbbildung als kleine weiße Punkte inErscheinung treten. Ursache dieserAnhäufungen sind Staubpartikel, die währendder Abscheidung als Kristallisationskeime aufdem Substrat vorlagen. Am rechten Rand derZnS-Probe ist die Probennummer eingeritzt.
Abbildung 3: Über ILGAR, auf float-Glas abgeschiedene Sulfid-Schichten. Eine 100 nm dicke CdS-Probe auf Millimeterpapier (links) und eine 40 nm dicke ZnS-Probe auf schwarzem Hintergrund(rechts).
Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
15
3.2.1 Komposition und Kristallstruktur
Der Nachweis der in den Proben vorhandenenElemente erfolgte über die Energie-dispersiveRöntgenanalyse (energy-dispersive-x-ray-ana-lysis, EDX); mittels der Röntgenfluoreszenz-analyse (RFA) wurden die Stoffmengenanteileder in den Proben vorhandenen Verbindungenbestimmt. Letzere Informationen sind speziellfür die Auswertung der optischen Daten beimILGAR-ZnS notwendig.
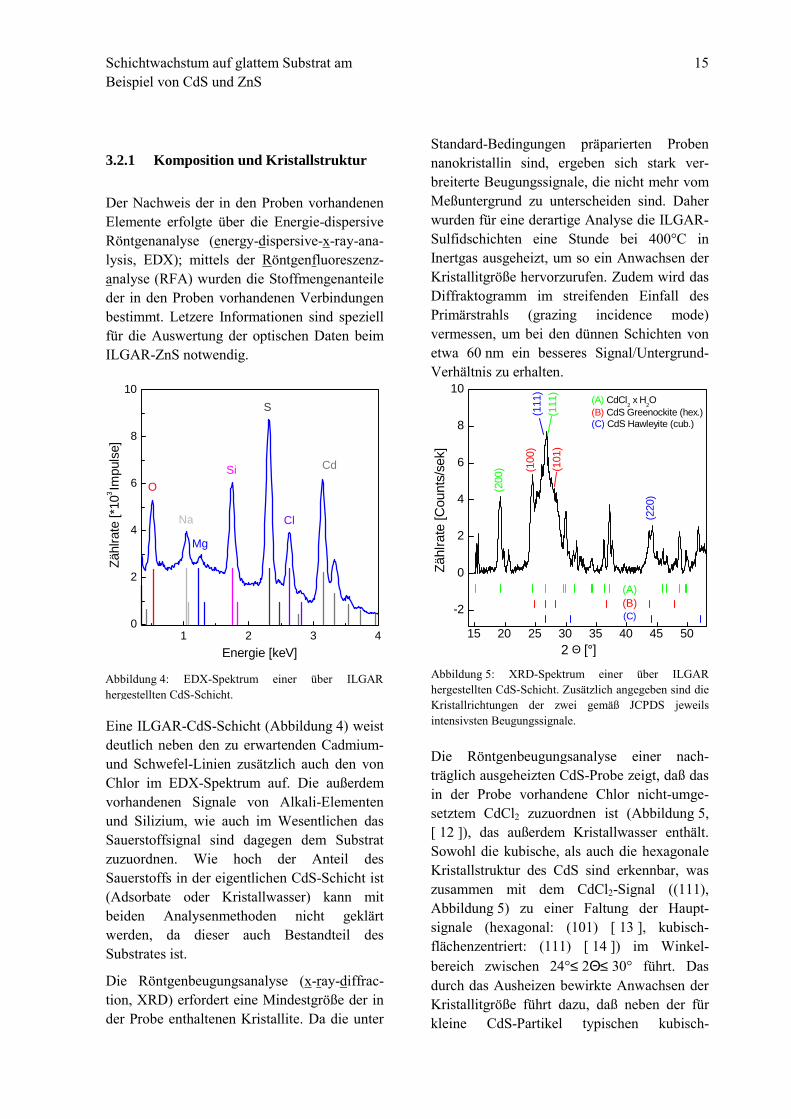
Eine ILGAR-CdS-Schicht (Abbildung 4) weistdeutlich neben den zu erwartenden Cadmium-und Schwefel-Linien zusätzlich auch den vonChlor im EDX-Spektrum auf. Die außerdemvorhandenen Signale von Alkali-Elementenund Silizium, wie auch im Wesentlichen dasSauerstoffsignal sind dagegen dem Substratzuzuordnen. Wie hoch der Anteil desSauerstoffs in der eigentlichen CdS-Schicht ist(Adsorbate oder Kristallwasser) kann mitbeiden Analysenmethoden nicht geklärtwerden, da dieser auch Bestandteil desSubstrates ist.
Die Röntgenbeugungsanalyse (x-ray-diffrac-tion, XRD) erfordert eine Mindestgröße der inder Probe enthaltenen Kristallite. Da die unter
Standard-Bedingungen präparierten Probennanokristallin sind, ergeben sich stark ver-breiterte Beugungssignale, die nicht mehr vomMeßuntergrund zu unterscheiden sind. Daherwurden für eine derartige Analyse die ILGAR-Sulfidschichten eine Stunde bei 400°C inInertgas ausgeheizt, um so ein Anwachsen derKristallitgröße hervorzurufen. Zudem wird dasDiffraktogramm im streifenden Einfall desPrimärstrahls (grazing incidence mode)vermessen, um bei den dünnen Schichten vonetwa 60 nm ein besseres Signal/Untergrund-Verhältnis zu erhalten.
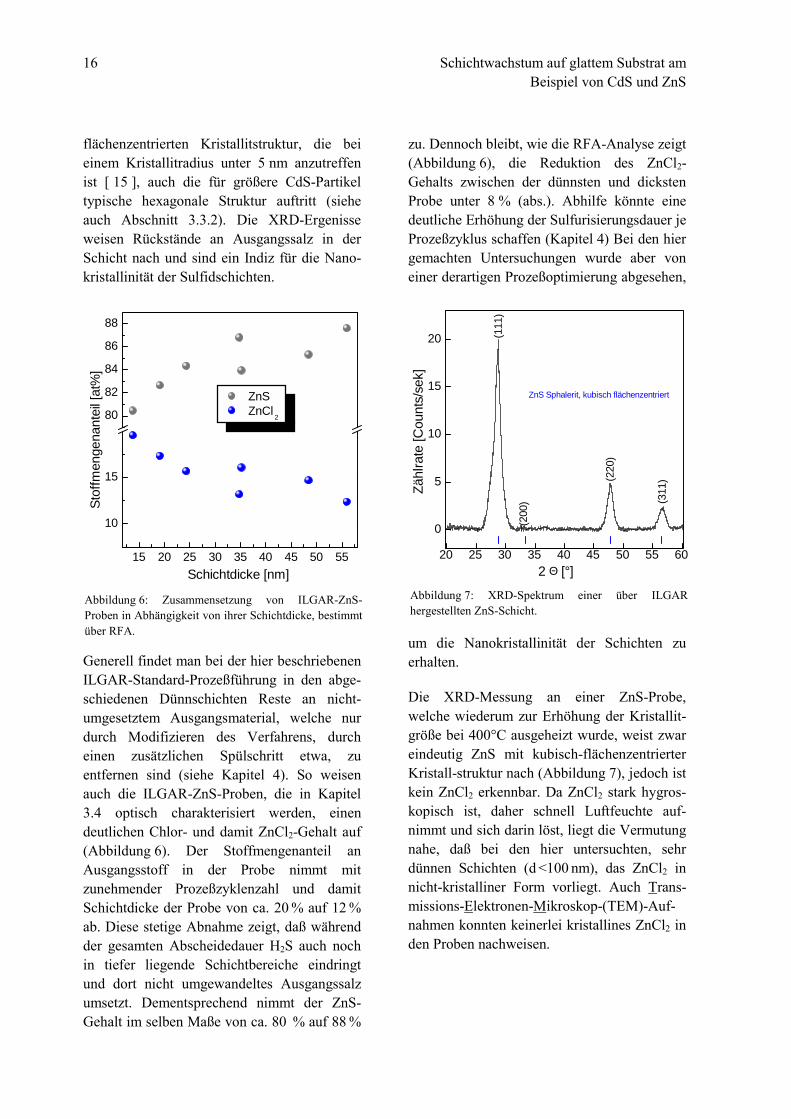
Die Röntgenbeugungsanalyse einer nach-träglich ausgeheizten CdS-Probe zeigt, daß dasin der Probe vorhandene Chlor nicht-umge-setztem CdCl2 zuzuordnen ist (Abbildung 5,[ 12 ]), das außerdem Kristallwasser enthält.Sowohl die kubische, als auch die hexagonaleKristallstruktur des CdS sind erkennbar, waszusammen mit dem CdCl2-Signal ((111),Abbildung 5) zu einer Faltung der Haupt-signale (hexagonal: (101) [ 13 ], kubisch-flächenzentriert: (111) [ 14 ]) im Winkel-bereich zwischen 24°≤ 2Θ≤ 30° führt. Dasdurch das Ausheizen bewirkte Anwachsen derKristallitgröße führt dazu, daß neben der fürkleine CdS-Partikel typischen kubisch-
1 2 3 40
2
4
6
8
10
Cd
Cl
S
Si
Mg
Na
O
Zähl
rate
[*10
3 Im
puls
e]
Energie [keV]
Abbildung 4: EDX-Spektrum einer über ILGARhergestellten CdS-Schicht.
Abbildung 5: XRD-Spektrum einer über ILGARhergestellten CdS-Schicht. Zusätzlich angegeben sind dieKristallrichtungen der zwei gemäß JCPDS jeweilsintensivsten Beugungssignale.
15 20 25 30 35 40 45 50
-2
0
2
4
6
8
10
(111
)
(100
)
(220
)(200
)
(111
)
(101
)
Zähl
rate
[Cou
nts/
sek]
2 Θ [°]
(A)(B)
(A) CdCl2 x H
2O
(B) CdS Greenockite (hex.)(C) CdS Hawleyite (cub.)
(C)
16 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
flächenzentrierten Kristallitstruktur, die beieinem Kristallitradius unter 5 nm anzutreffenist [ 15 ], auch die für größere CdS-Partikeltypische hexagonale Struktur auftritt (sieheauch Abschnitt 3.3.2). Die XRD-Ergenisseweisen Rückstände an Ausgangssalz in derSchicht nach und sind ein Indiz für die Nano-kristallinität der Sulfidschichten.
Generell findet man bei der hier beschriebenenILGAR-Standard-Prozeßführung in den abge-schiedenen Dünnschichten Reste an nicht-umgesetztem Ausgangsmaterial, welche nurdurch Modifizieren des Verfahrens, durcheinen zusätzlichen Spülschritt etwa, zuentfernen sind (siehe Kapitel 4). So weisenauch die ILGAR-ZnS-Proben, die in Kapitel3.4 optisch charakterisiert werden, einendeutlichen Chlor- und damit ZnCl2-Gehalt auf(Abbildung 6). Der Stoffmengenanteil anAusgangsstoff in der Probe nimmt mitzunehmender Prozeßzyklenzahl und damitSchichtdicke der Probe von ca. 20 % auf 12 %ab. Diese stetige Abnahme zeigt, daß währendder gesamten Abscheidedauer H2S auch nochin tiefer liegende Schichtbereiche eindringtund dort nicht umgewandeltes Ausgangssalzumsetzt. Dementsprechend nimmt der ZnS-Gehalt im selben Maße von ca. 80 % auf 88 %
zu. Dennoch bleibt, wie die RFA-Analyse zeigt(Abbildung 6), die Reduktion des ZnCl2-Gehalts zwischen der dünnsten und dickstenProbe unter 8 % (abs.). Abhilfe könnte einedeutliche Erhöhung der Sulfurisierungsdauer jeProzeßzyklus schaffen (Kapitel 4) Bei den hiergemachten Untersuchungen wurde aber voneiner derartigen Prozeßoptimierung abgesehen,
um die Nanokristallinität der Schichten zuerhalten.
Die XRD-Messung an einer ZnS-Probe,welche wiederum zur Erhöhung der Kristallit-größe bei 400°C ausgeheizt wurde, weist zwareindeutig ZnS mit kubisch-flächenzentrierterKristall-struktur nach (Abbildung 7), jedoch istkein ZnCl2 erkennbar. Da ZnCl2 stark hygros-kopisch ist, daher schnell Luftfeuchte auf-nimmt und sich darin löst, liegt die Vermutungnahe, daß bei den hier untersuchten, sehrdünnen Schichten (d <100 nm), das ZnCl2 innicht-kristalliner Form vorliegt. Auch Trans-missions-Elektronen-Mikroskop-(TEM)-Auf-nahmen konnten keinerlei kristallines ZnCl2 inden Proben nachweisen.
15 20 25 30 35 40 45 50 55
10
15
80
82
84
86
88
Stof
fmen
gena
ntei
l [at
%]
Schichtdicke [nm]
ZnS ZnCl
2
Abbildung 6: Zusammensetzung von ILGAR-ZnS-Proben in Abhängigkeit von ihrer Schichtdicke, bestimmtüber RFA.
Abbildung 7: XRD-Spektrum einer über ILGARhergestellten ZnS-Schicht.
20 25 30 35 40 45 50 55 60
0
5
10
15
20
(311
)(220
)
(200
)
(111
)
Zähl
rate
[Cou
nts/
sek]
2 Θ [°]
ZnS Sphalerit, kubisch flächenzentriert

Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
17
3.2.2 Morphologie der ILGAR-Sulfid-Schichten
Das in einem ILGAR-Prozeßzyklus nichtumgesetzte Ausgangsmaterial in der Schichtkann deutlichen Einfluß auf die Schicht-morphologie haben. Wenn sich diese nicht-umgesetzte Ausgangsverbindung beim näch-sten Eintauchen in die Metallsalzlösung wiederauflöst, können einige Metallsulfidkristalliteden Halt in der Schicht verlieren und sichmechanisch herauslösen. Dies tritt um sostärker auf, je mehr Ausgangssalz in derabgeschiedenen Beschichtung vorhanden ist.Neben der Beeinträchtigung der Morphologieführt dies zu einer Verringerung der Wachs-
tumsrate und Kompaktheit der Schicht.
Ein ZnS-Dünnfilm scheint auf glattemSilizium-Substrat geschlossen zu sein(Abbildung 8), doch besitzt die Probe morpho-logische Inhomogenitäten, Materialanhäu-fungen und Gräben, welche die Oberflächen-rauhigkeit stark erhöhen. Eine Ursache fürderartige Stellen, auf denen weniger Zinksulfidzu finden ist, kann das Wiederauflösen nicht-umgesetzten Ausgangsmaterials sein. Ent-netzungseffekte der Ausgangslösung auf einerhydrophob erscheinenden Oberfläche sind nurbei den ersten Prozeßzyklen zu befürchten;danach, wenn bereits eine abgeschiedeneSchicht auf dem Substrat besteht, ist wegen derOberflächenrauhigkeit eine Entnetzung eherunwahrscheinlich. Mit der Integration einesMetallhydroxid erzeugenden Zwischen-schrittes in das ILGAR-Verfahren wird dasWiederauflösen deutlich verringert unddadurch die Morphologie verbessert. Zusätz-lich führt eine solche modifizierte Prozeß-führung zu merklich größeren Kristalliten inder Schicht, was für viele Anwendungsgebiete(Kapitel 4) wünschenswert ist; der beinanokristallinen Schichten vorhandeneGrößenquantisierungseffekt (Q-Effekt, Ab-schnitt 3.3.2.1) tritt aber, wenn überhaupt, hiernur noch sehr schwach ausgeprägt auf.
Abbildung 8: AFM-Aufnahme eines ZnS-Dünnfilms aufSilizium-Substrat (Scanbereich x,y: 3µm; z: 100 nm).
Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
18
3.3 Vereinfachtes Wachstums-modell von ILGAR-CdS-Nanokristalliten
Optische Untersuchungen, wie sie in diesemKapitel gemacht werden, liefern im Fallegroßer Kristallite nur wenig Informationenüber das Kristallitwachstum in den Schichten.Mit dem Verzicht auf einen Hydroxid-zwischenschritt waren die optisch charak-terisierten Proben rauher (Abschnitt 3.2.2),aber dennoch geschlossen und besaßen einemdeutlich ausgeprägten Q-Effekt. Ein störenderEinfluß lokaler, mikroskopischer Inhomo-genitäten auf die gemessenen Transmissions-und Reflexionseigenschaften der Schichtenkann in erster Näherung vernachlässigtwerden, da die Spektralphotometrie über einemakroskopische Probenfläche mittelt.Außerdem wird durch die Messung mit einerIntegrationskugel das bei morphologischenInhomogenitäten auftretende diffus gestreuteLicht im Meßergebnis mitberücksichtigt(Anhang AII).
3.3.1 Abschätzung der Weite deroptischen Bandlücke
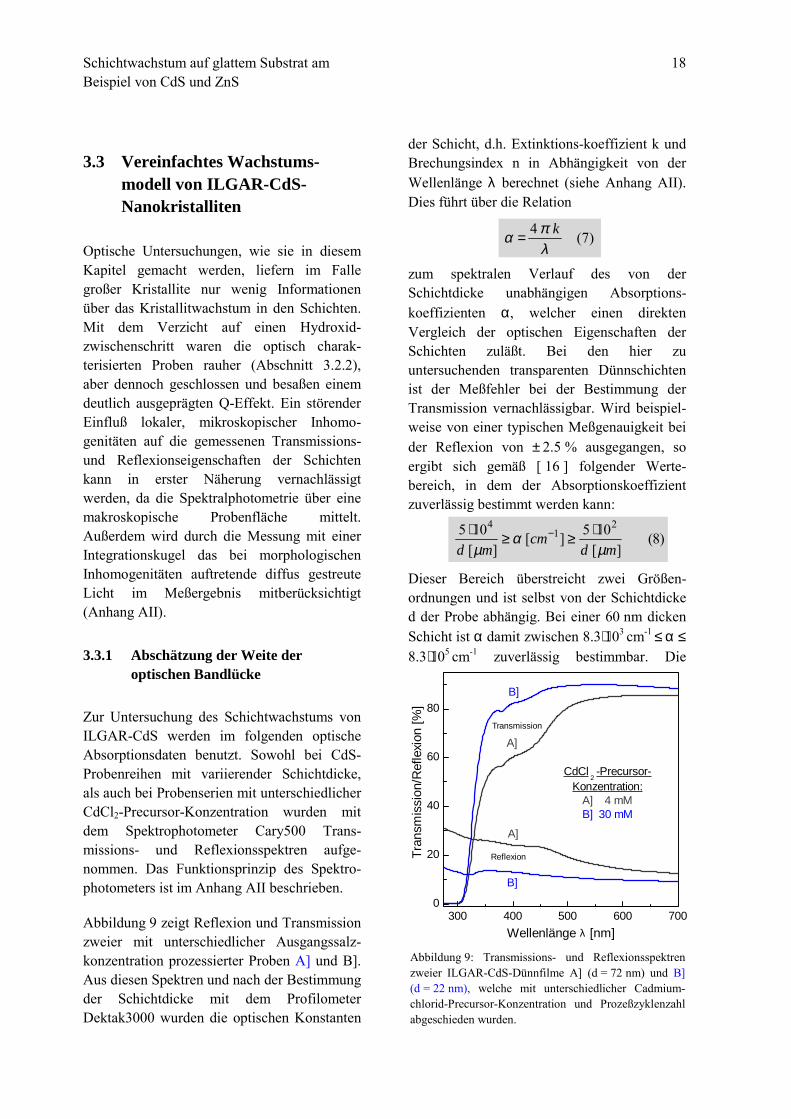
Zur Untersuchung des Schichtwachstums vonILGAR-CdS werden im folgenden optischeAbsorptionsdaten benutzt. Sowohl bei CdS-Probenreihen mit variierender Schichtdicke,als auch bei Probenserien mit unterschiedlicherCdCl2-Precursor-Konzentration wurden mitdem Spektrophotometer Cary500 Trans-missions- und Reflexionsspektren aufge-nommen. Das Funktionsprinzip des Spektro-photometers ist im Anhang AII beschrieben.
Abbildung 9 zeigt Reflexion und Transmissionzweier mit unterschiedlicher Ausgangssalz-konzentration prozessierter Proben A] und B].Aus diesen Spektren und nach der Bestimmungder Schichtdicke mit dem ProfilometerDektak3000 wurden die optischen Konstanten
der Schicht, d.h. Extinktions-koeffizient k undBrechungsindex n in Abhängigkeit von derWellenlänge λ berechnet (siehe Anhang AII).Dies führt über die Relation
zum spektralen Verlauf des von derSchichtdicke unabhängigen Absorptions-koeffizienten α, welcher einen direktenVergleich der optischen Eigenschaften derSchichten zuläßt. Bei den hier zuuntersuchenden transparenten Dünnschichtenist der Meßfehler bei der Bestimmung derTransmission vernachlässigbar. Wird beispiel-weise von einer typischen Meßgenauigkeit beider Reflexion von ± 2.5 % ausgegangen, soergibt sich gemäß [ 16 ] folgender Werte-bereich, in dem der Absorptionskoeffizientzuverlässig bestimmt werden kann:
Dieser Bereich überstreicht zwei Größen-ordnungen und ist selbst von der Schichtdicked der Probe abhängig. Bei einer 60 nm dickenSchicht ist α damit zwischen 8.3⋅103 cm-1
≤ α ≤8.3⋅105 cm-1 zuverlässig bestimmbar. Die
)7(4λπα k=
)8(][
105][][
105 21
4
mdcm
md µα
µ⋅≥≥⋅ −
Abbildung 9: Transmissions- und Reflexionsspektrenzweier ILGAR-CdS-Dünnfilme A] (d = 72 nm) und B](d = 22 nm), welche mit unterschiedlicher Cadmium-chlorid-Precursor-Konzentration und Prozeßzyklenzahlabgeschieden wurden.
300 400 500 600 7000
20
40
60
80
B]
B]
A]
A]
Reflexion
Transmission
CdCl 2
-Precursor-Konzentration:
A] 4 mMB] 30 mM
Tran
smis
sion
/Ref
lexi
on [%
]
Wellenlänge λ [nm]
Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
19
optische Bandlückenenergie Egap ist, wie nochbeschrieben wird, über den Absorptionsverlaufzu berechnen. Es muß daher über die Schicht-dicke der Probe gewährleistet sein, daß das fürdie Ermittlung verwendete α im zuverlässigbestimmbaren Bereich liegt. Dies ist bei allenhier betrachteten CdS-Schichten der Fall.
Nach Auswertung der optischen Spektren derAbbildung 9 ergibt sich nun folgenderspektraler Absorptionsverlauf der Proben(Abbildung 10):
Im Gegensatz zu den Transmissionsspektrenwerden bei den Absorptionsverläufen nun dieUnterschiede zwischen Probe A] und B]deutlich: der Beginn der Absorptionskante beiProbe B] liegt bei einer größeren Anregungs-,d.h. Photonenenergie als bei Probe A], die beieiner weitaus niedrigeren Precursorkonzen-tration von 4 mM hergestellt wurde.
Für die Ermittlung der energetischen Breite deroptischen Bandlücke wird angenommen, daßdas in den CdS-Schichten vorhandene CdCl2
keinerlei Einfluß auf den Absorptionsverlaufim zu betrachtenden Energiebereich zwischen2.5 eV und 3.5 eV hat. Mit einer Interband-Übergangsenergie von 5.94 eV [ 17 ] ist es imrelevanten Absorptionsbereich als transparentanzusehen. Wegen des unvollständigenReaktionsumsatzes sind die in Abbildung 10
gezeigten Absorptionsverläufe die Spektrenvon CdS-CdCl2-Gemischen; die Position derAbsorptionskante ist allerdings nur dem CdSzuzuordnen. Die einzige Folge der An-wesenheit des Ausgangssalzes ist ein zugeringer Absolutbetrag von α im Vergleich zureinen CdS-Schichten, da die eigentliche„Netto-Schichtdicke“ des CdS in der Probegeringer ist als die profilometrisch gemesseneDicke der Schicht.
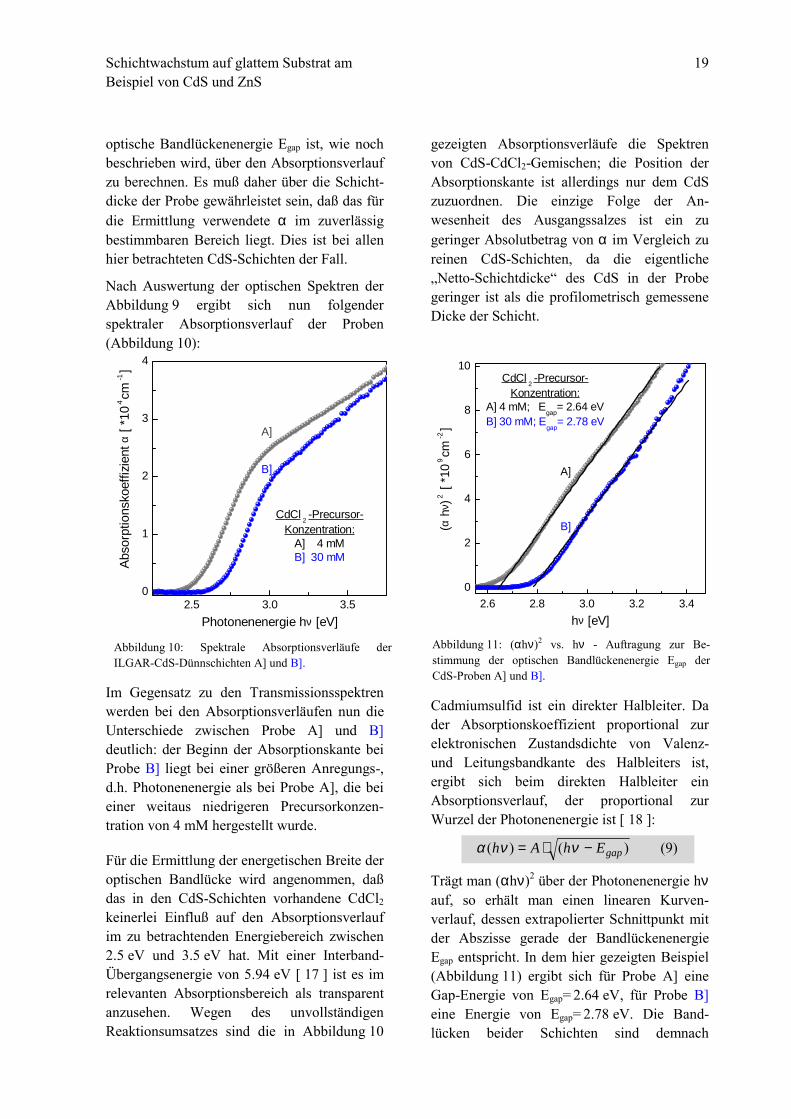
Cadmiumsulfid ist ein direkter Halbleiter. Dader Absorptionskoeffizient proportional zurelektronischen Zustandsdichte von Valenz-und Leitungsbandkante des Halbleiters ist,ergibt sich beim direkten Halbleiter einAbsorptionsverlauf, der proportional zurWurzel der Photonenenergie ist [ 18 ]:
Trägt man (αhν)2 über der Photonenenergie hνauf, so erhält man einen linearen Kurven-verlauf, dessen extrapolierter Schnittpunkt mitder Abszisse gerade der BandlückenenergieEgap entspricht. In dem hier gezeigten Beispiel(Abbildung 11) ergibt sich für Probe A] eineGap-Energie von Egap= 2.64 eV, für Probe B]eine Energie von Egap= 2.78 eV. Die Band-lücken beider Schichten sind demnach
2.6 2.8 3.0 3.2 3.40
2
4
6
8
10
B]
A]
CdCl 2 -Precursor-Konzentration:
A] 4 mM; Egap= 2.64 eVB] 30 mM; E
gap= 2.78 eV
(α hν )
2 [
*10 9
cm -2
]
hν [eV]
Abbildung 11: (αhν)2 vs. hν - Auftragung zur Be-stimmung der optischen Bandlückenenergie Egap derCdS-Proben A] und B].
)9()()( gapEhAh −⋅= ννα
2.5 3.0 3.50
1
2
3
4
B]
A]
CdCl 2
-Precursor-Konzentration:
A] 4 mMB] 30 mMAb
sorp
tions
koef
fizie
nt α
[ *1
0 4 cm
-1 ]
Photonenenergie hν [eV]
Abbildung 10: Spektrale Absorptionsverläufe derILGAR-CdS-Dünnschichten A] und B].

20 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
verschieden, obwohl dieselbe Verbindungvorliegt. Zudem stimmt keine der Band-lückenenergien mit der von Volumen-CdSüberein (Egap(bulk) = 2.48 eV, für die kubische,ebenso wie für die hexagonale Modifikation[ 19 ]). Da bei derart hoch-ohmschen Schichtenein Burstein-Moss-shift [ 18 ] auszuschließenist, muß die Ursache der Blauverschiebung derAbsorptionskante in der geringen Kristallit-größe des CdS zu suchen sein, wo Ladungs-träger lokal eingeschränkt sind.
3.3.2 Größenquantisierungseffekt undKristallitgrößenbestimmung bei denILGAR-CdS-Schichten
Zwei CdS-Proben mit einer Schichtdicke vonetwa 5 nm wurden unter denselben Standard-Prozeßbedingungen wie die in Abschnitt 3.3.1diskutierten Schichten A] und B] fürhochauflösende (High-Resolution)-TEM-Auf-nahmen hergestellt (Abbildung 12).
Hier weist die mit einer CdCl2-Konzentrationvon 30 mM präparierte Schicht ellipsen-förmige Kristallite mit einer Länge der großenAchse von etwa 4 nm auf (Abbildung 12,oben); dagegen besitzt die mit 4 mM CdCl2
prozessierte Probe ungleichmäßig großeKristallite mit einer Achsenlänge zwischen4 nm und 9 nm. Die Kristallitgrößen beiderProben sind von derselben Größenordnung wieder Durchmesser des Bohr´schen Exzitons vonCdS im Grundzustand, dessen Radius (BohrRadius aB) sich über (10) berechnet.
ε ist hierbei die Dielektrizitätskonstante,1/µ= 1/me+1/mh ist die inverse reduzierteeffektive Masse des Elektrons (me) und desLochs (mh), e die Elementarladung und ! diePlanck´sche Konstante. Mit den ent-sprechenden Konstanten für CdS (siehe AV)ergibt sich demnach ein Radius von aB≈ 2 nm.Sind Kristallite im Größenbereich desBohr´schen Exzitons, so befindet sich dasSystem im sogenannten „confinement“-Bereich, d.h. das Material verliert seinetypischen, kristallinen Festkörpereigenschaften[ 20 ], da die Periodizität des Kristallgitters inallen Raumrichtungen eng begrenzt und dahergestört ist.
Zusätzlich in Abbildung 12 (gekennzeichnetdurch ein Quadrat) abgebildet sind die„power“-Spektren einzelner Kristallite. Eshandelt sich um die Fourier-Analyse der durchdas Quadrat begrenzten Fläche, welche deninversen Netzebenenabstand 1/dhkl derbetrachteten (hkl)-Ebene liefert. So weist dergroße Kristallit in Abbildung 12 (unten) einenNetzebenenabstand von d100= 3.6 Å auf. DieserAbstand kann der (100)-Ebene eines CdS-Kristalliten mit hexagonaler Kristallstrukturzugeordnet werden (d100(Lit.) = 3.58 Å, [ 13 ]).Dagegen sind die kleineren Kristallite inAbbildung 12 (oben und unten) mit einemNetzebenenabstand von d111= 3.3 Å der (111)-Ebene eines kubisch-flächenzentrierten CdS-
)10(2
2
eaB µ
ε !=
Abbildung 12: High-Resolution-TEM-Aufnahmen zweierILGAR-CdS-Dünnfilme, welche mit einer Cadmium-chlorid-Konzentration von 30 mM (oben) und 4 mM(unten) abgeschieden wurden. Einzelne Kristallite sindhellgrau umkreist. An den mit einem Quadratumzeichneten Partikeln wurde eine Fourier-Analyse (zurVerdeutlichung als helle Quadrate in der Abbildung)durchgeführt, die zum inversen Netzebenenabstand1/d(hkl) führt.

Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
21
Kristalliten zuzuordnen (d111(Lit.)= 3.36 Å,[ 14 ]). Makrokristallines CdS besitztgewöhnlicherweise die hexagonale Struktur.Bei epitaktisch gewachsenen Schichten istoftmals eine reine kubische Struktur zu finden[ 15 ].
Auch beim ILGAR-Verfahren weisenmakrokristalline CdS-Schichten, welche beiTemperaturen oberhalb T=250°C sulfurisiertwurden, ausschließlich die hexagonaleKristallstruktur auf. Sind jedoch die CdS-Partikel im Radius kleiner als etwa 5 nm, sofindet man bei nanokristallinen CdS-Kolloidenin Lösung ausschließlich die kubischeKristallstruktur vor [ 15 ]. Auch das überILGAR hergestellte, nanokristalline CdSbesitzt laut Röntgenbeugungsanalyse dieseStruktur (Abbildung 5). Eine lokale EDX-Analyse (EDAX) der Kristallite im TEM zeigt,daß diese innerhalb des Nachweisvermögensfrei von Chlorresten sind; das in den CdS-Schichten ebenfalls in den TEM-Probennachgewiesene Chlor, so ergab die EDAX-Messung, befindet sich als nicht-kristallinesMaterial außerhalb der CdS-Kristallite (inAbbildung 12 nicht erkennbar) und muß CdCl2
zugeordnet werden [ 15 ]. ILGAR-CdS-Schichten bestehen somit aus nanokristallinenCdS-Partikeln eingebettet in eine nicht-kristalline Umgebung aus CdCl2.
3.3.2.1 „Größenquantisierungseffekt“ (Q-Size-effect)
Ein qualitativ sehr anschauliches Modell zurquantenmechanischen Beschreibung vonClustern und Nanopartikeln ist das LCAO-Modell (Linear Combination of AtomicOrbitals), welches häufig in der Molekül-theorie angewandt wird. Bei diesem Modellwerden Atomorbitale der im Molekülenthaltenen Atome linear zu bindenden undantibindenden Molekülorbitalen kombiniert,sofern die Orbitale gleiche Symmetrie undähnliche Energie besitzen. Das von Brus [ 21 ]vorgestellte Modell geht hierbei auch bei
II-VI-Verbindungen von sp3-hybridisiertenValenzorbitalen aus (Abbildung 13 a), die dieBasis der Linearkombination bilden. Esergeben sich vollständig besetzte, bindende σ-Molekülorbitale und unbesetzte, antibindendeσ*-Orbitale. Beide Arten von Molekül-orbitalen sind energetisch voneinander getrennt(Abbildung 13 b). Beim Überlagern derAtomorbitale zu Molekülorbitalen muß derenGesamtzahl unverändert bleiben, so daß dieAnzahl an Orbitalen eines Moleküls, Clustersoder Nanoteilchens gleich der Zahl derAtomorbitale aller beteiligten Atome [ 22 ]sein muß. Mit zunehmender Zahl der Atomedieses Verbundes und der damit einher-gehenden zunehmenden Zahl an diskretenEnergieniveaus nimmt der energetischeAbstand zwischen der dem höchsten besetzten„Molekülorbitals“ ( HOMO – HighestOccupied Molecular Orbital) und demniedrigsten, unbesetzten (LUMO – LowestUnoccupied Molecular Orbital) stetig ab,ebenso der energetische Abstand zwischen deneinzelnen Orbitalen (Abbildung 13 c). ImGrenzfall des makros-kopischen, kristallinenFestkörpers liegen die Orbitale energetisch sodicht zusammen, daß sie ein als Energiebandbezeichnetes Quasi-Kontinuum bilden, wassowohl für die bindenden (Valenzband), als
Abbildung 13: Energieníveaus der Nanopartikel c) nachdem LCAO-Modell (a-c) und der Nomenklatur desFestkörpers d).
Zahl der Atome
Ener
gie
Leitu
ngs-
band
Vale
nz-
band
E gap
E exzi
ton
E exzi
ton
Atom-orbital
Molekül-orbital
Cluster-Nanopartikel
Festkörper
sp3
!
!*
a) b) c) d)

22 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
auch für die antibindenden Zustände(Leitungsband) (Abbildung 13 d) zutrifft. Diebeiden Bänder sind durch die Bandlückevoneinander getrennt. Die Nanokristallitestellen somit den Übergang vom Molekül zummakrokristallinen Festkörper dar und weichenin ihren physikalischen Eigenschaften vondenen des makroskopischen Festkörpers ab. Dabei Nanokristalliten noch von diskretenEnergieniveaus auszugehen ist, deren energe-tische Lage von der Kristallitgröße abhängt,bezeichnet man ihr elektrisches und speziell ihroptisches Verhalten als „Größenquanti-sierungseffekt“ (Q-Size-Effect oder auch nurQ-Effect). Die quantitative Berechnung derenergetischen Zustände von Nanopartikeln istin diesem Modell jedoch ziemlich aufwendig,da bei den relevanten Partikelgrößen zwischen101 und 104 Atome in die Rechnungeneingehen (tight-binding-Näherung [ 23 ]).
Ausgehend von einem makroskopischen,kristallinen Halbleiter versucht die Effektive-Massen-Näherung (EMA -Effective-Mass-Ap-proximation) den Q-Size-Effekt zu beschreiben[ 20, 24-26 ].
Durch Lichtabsorption wird ein Elektron vomValenzband in das Leitungsband angeregt; imValenzband bleibt eine positive Ladung, einLoch, zurück. Die beiden Ladungsträgerkönnen sich unabhängig voneinander bewegen,so daß der Kristall elektrisch leitend wird.Elektron und Loch können wegen ihrerCoulomb-Anziehung auch einen schwachgebundenen Zustand einnehmen; manbehandelt beide zusammen dann als Quasi-Teilchen, z.B. als Wannier-Exziton. DiesesExziton ist nur schwach an ein Atom gebundenund über mehrere Atome ausgedehnt. DieBerechnung seiner Energiezustände ähneltstark der des Wasserstoffatoms, sofern dieEnergieflächen für die beiden Ladungenkugelförmig und nicht entartet sind [ 27 ].Wegen der geringen effektiven Massen vonElektron (me" 0.2 m0) und Loch (mh" 0.6 m0)und der großen Dieletrizitätskonstanten ε desHalbleitermaterials (ε" 10 ε0) ist der
Bohr´sche Exzitonenradius groß im Vergleichzur Gitterkonstanten des Festkörpers (a0"5 Å).Dementsprechend ist die Bindungsenergie derExzitonen klein (bei CdS 28 meV, [ 19 ]), sodaß sie bereits unterhalb der Raumtemperaturleicht zu freien Ladungsträgern dissoziierenkönnen. Daher können Exzitonen einesmakroskopischen Halbleiters oft erst bei sehrtiefen Temperaturen nahe dem absolutenNullpunkt eindeutig optisch (z.B. über Photo-lumineszenz) nachgewiesen werden.
Geht man jetzt über zur Lichtabsorption einesnur Nanometer großen Halbleiterteilchens, somuß das erzeugte Elektron-Loch-Paar einenexzitonischen Zustand bilden (ebenfalls einWannier-Exziton); der Aufenthaltsort derLadungsträger ist jetzt räumlich auf diePartikeldimension begrenzt, so daß sie stetsihrer Coulomb-Anziehung ausgesetzt sind.Neben der „erzwungenen“ Coulomb-Wechsel-wirkung führt die geringe Kristallitgröße auchzu einer eingeschränkten Translationsfreiheit,so daß ein Exziton quantenmechanisch alsTeilchen in einem Potentialtopf („Kasten“)beschrieben werden kann [ 28 ]. Das Elektronmit der effektiven Masse me befindet sich im„Leitungsbandkasten“, das Loch mit dereffektiven Masse mh im „Valenzbandkasten“[ 29 ]. Die Energiezustände der Teilchen sinddiskret und gequantelt, weil wegen der nunbestehenden Randbedingungen nur nochbestimmte Wellenfunktionen der Teilchenerlaubt sind. Mit kleinerer Partikelgröße undsomit der Verringerung der Größe desPotentialtopfs reduziert sich die Zahl dermöglichen Energiezustände, d.h. die Zustands-dichte wird kleiner, wogegen sich derenergetische Abstand zwischen zweiZuständen erhöht. Das energetisch niedrigsteNiveau eines jeden Teilchens in seinem„Kasten“ ist der 1s-Zustand. Der niedrigsteerlaubte, elektronische Übergang in kleinenHalbleiterteilchen ist daher ein 1s-1s-Übergang. Die Gesamtenergie E(R) diesesexzitonischen Übergangs setzt sich zusammenaus der Volumen-Exzitonenenergie im Grund-zustand und am absoluten Temperatur-Null-
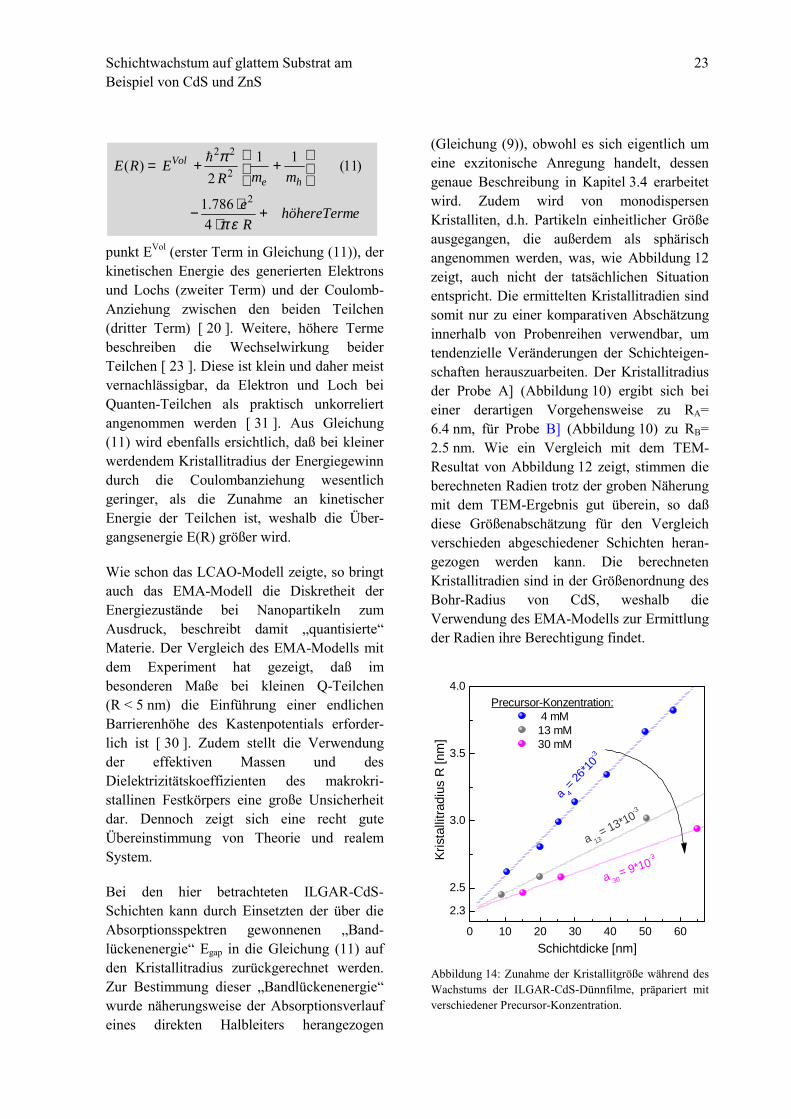
Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
23
punkt EVol (erster Term in Gleichung (11)), derkinetischen Energie des generierten Elektronsund Lochs (zweiter Term) und der Coulomb-Anziehung zwischen den beiden Teilchen(dritter Term) [ 20 ]. Weitere, höhere Termebeschreiben die Wechselwirkung beiderTeilchen [ 23 ]. Diese ist klein und daher meistvernachlässigbar, da Elektron und Loch beiQuanten-Teilchen als praktisch unkorreliertangenommen werden [ 31 ]. Aus Gleichung(11) wird ebenfalls ersichtlich, daß bei kleinerwerdendem Kristallitradius der Energiegewinndurch die Coulombanziehung wesentlichgeringer, als die Zunahme an kinetischerEnergie der Teilchen ist, weshalb die Über-gangsenergie E(R) größer wird.
Wie schon das LCAO-Modell zeigte, so bringtauch das EMA-Modell die Diskretheit derEnergiezustände bei Nanopartikeln zumAusdruck, beschreibt damit „quantisierte“Materie. Der Vergleich des EMA-Modells mitdem Experiment hat gezeigt, daß imbesonderen Maße bei kleinen Q-Teilchen(R < 5 nm) die Einführung einer endlichenBarrierenhöhe des Kastenpotentials erforder-lich ist [ 30 ]. Zudem stellt die Verwendungder effektiven Massen und desDielektrizitätskoeffizienten des makrokri-stallinen Festkörpers eine große Unsicherheitdar. Dennoch zeigt sich eine recht guteÜbereinstimmung von Theorie und realemSystem.
Bei den hier betrachteten ILGAR-CdS-Schichten kann durch Einsetzten der über dieAbsorptionsspektren gewonnenen „Band-lückenenergie“ Egap in die Gleichung (11) aufden Kristallitradius zurückgerechnet werden.Zur Bestimmung dieser „Bandlückenenergie“wurde näherungsweise der Absorptionsverlaufeines direkten Halbleiters herangezogen
(Gleichung (9)), obwohl es sich eigentlich umeine exzitonische Anregung handelt, dessengenaue Beschreibung in Kapitel 3.4 erarbeitetwird. Zudem wird von monodispersenKristalliten, d.h. Partikeln einheitlicher Größeausgegangen, die außerdem als sphärischangenommen werden, was, wie Abbildung 12zeigt, auch nicht der tatsächlichen Situationentspricht. Die ermittelten Kristallitradien sindsomit nur zu einer komparativen Abschätzunginnerhalb von Probenreihen verwendbar, umtendenzielle Veränderungen der Schichteigen-schaften herauszuarbeiten. Der Kristallitradiusder Probe A] (Abbildung 10) ergibt sich beieiner derartigen Vorgehensweise zu RA=6.4 nm, für Probe B] (Abbildung 10) zu RB=2.5 nm. Wie ein Vergleich mit dem TEM-Resultat von Abbildung 12 zeigt, stimmen dieberechneten Radien trotz der groben Näherungmit dem TEM-Ergebnis gut überein, so daßdiese Größenabschätzung für den Vergleichverschieden abgeschiedener Schichten heran-gezogen werden kann. Die berechnetenKristallitradien sind in der Größenordnung desBohr-Radius von CdS, weshalb dieVerwendung des EMA-Modells zur Ermittlungder Radien ihre Berechtigung findet.
ehöhereTermRe
mmRERE
he
Vol
+⋅
⋅−
++=
επ
π
4786.1
)11(112
)(
2
2
22!
Abbildung 14: Zunahme der Kristallitgröße während desWachstums der ILGAR-CdS-Dünnfilme, präpariert mitverschiedener Precursor-Konzentration.
0 10 20 30 40 50 60
2.3
2.5
3.0
3.5
4.0
a 30 = 9*10-3
a 13 = 13*10-3
a 4 = 2
6*10-3
Precursor-Konzentration: 4 mM 13 mM 30 mM
Kris
talli
tradi
us R
[nm
]
Schichtdicke [nm]
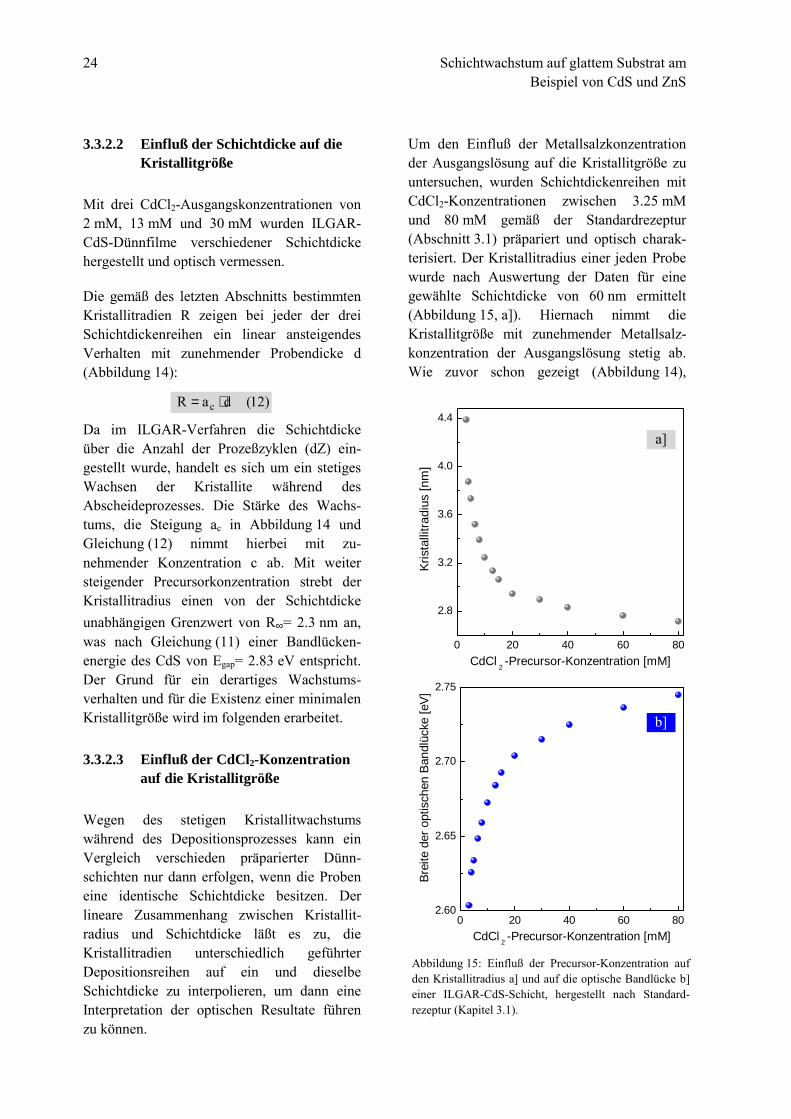
24 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
3.3.2.2 Einfluß der Schichtdicke auf dieKristallitgröße
Mit drei CdCl2-Ausgangskonzentrationen von2 mM, 13 mM und 30 mM wurden ILGAR-CdS-Dünnfilme verschiedener Schichtdickehergestellt und optisch vermessen.
Die gemäß des letzten Abschnitts bestimmtenKristallitradien R zeigen bei jeder der dreiSchichtdickenreihen ein linear ansteigendesVerhalten mit zunehmender Probendicke d(Abbildung 14):
Da im ILGAR-Verfahren die Schichtdickeüber die Anzahl der Prozeßzyklen (dZ) ein-gestellt wurde, handelt es sich um ein stetigesWachsen der Kristallite während desAbscheideprozesses. Die Stärke des Wachs-tums, die Steigung ac in Abbildung 14 undGleichung (12) nimmt hierbei mit zu-nehmender Konzentration c ab. Mit weitersteigender Precursorkonzentration strebt derKristallitradius einen von der Schichtdickeunabhängigen Grenzwert von R∞= 2.3 nm an,was nach Gleichung (11) einer Bandlücken-energie des CdS von Egap= 2.83 eV entspricht.Der Grund für ein derartiges Wachstums-verhalten und für die Existenz einer minimalenKristallitgröße wird im folgenden erarbeitet.
3.3.2.3 Einfluß der CdCl2-Konzentrationauf die Kristallitgröße
Wegen des stetigen Kristallitwachstumswährend des Depositionsprozesses kann einVergleich verschieden präparierter Dünn-schichten nur dann erfolgen, wenn die Probeneine identische Schichtdicke besitzen. Derlineare Zusammenhang zwischen Kristallit-radius und Schichtdicke läßt es zu, dieKristallitradien unterschiedlich geführterDepositionsreihen auf ein und dieselbeSchichtdicke zu interpolieren, um dann eineInterpretation der optischen Resultate führenzu können.
Um den Einfluß der Metallsalzkonzentrationder Ausgangslösung auf die Kristallitgröße zuuntersuchen, wurden Schichtdickenreihen mitCdCl2-Konzentrationen zwischen 3.25 mMund 80 mM gemäß der Standardrezeptur(Abschnitt 3.1) präpariert und optisch charak-terisiert. Der Kristallitradius einer jeden Probewurde nach Auswertung der Daten für einegewählte Schichtdicke von 60 nm ermittelt(Abbildung 15, a]). Hiernach nimmt dieKristallitgröße mit zunehmender Metallsalz-konzentration der Ausgangslösung stetig ab.Wie zuvor schon gezeigt (Abbildung 14),
)12(daR c ⋅=
Abbildung 15: Einfluß der Precursor-Konzentration aufden Kristallitradius a] und auf die optische Bandlücke b]einer ILGAR-CdS-Schicht, hergestellt nach Standard-rezeptur (Kapitel 3.1).
0 20 40 60 802.60
2.65
2.70
2.75
Brei
te d
er o
ptis
chen
Ban
dlüc
ke [e
V]
CdCl 2 -Precursor-Konzentration [mM]
0 20 40 60 80
2.8
3.2
3.6
4.0
4.4Kr
ista
llitra
dius
[nm
]
CdCl 2 -Precursor-Konzentration [mM]
a]
b]
Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
25
bestätigt sich auch hier ein starkes Kristallit-wachstum bei Verwenden niedriger Konzen-trationen, wogegen mit steigender Konzen-tration die Kristallitgröße abnimmt und einenkonstanten Wert, den minimalen kritischenKristallitradius R∞ anstrebt (S. 24). Ent-sprechend tendiert die Breite der optischenBandlücke Egap zu hohen Konzentrationengegen einen Maximalwert (Abbildung 15, b] ).
3.3.2.4 Mechanismus der ILGAR-CdS-Abscheidung
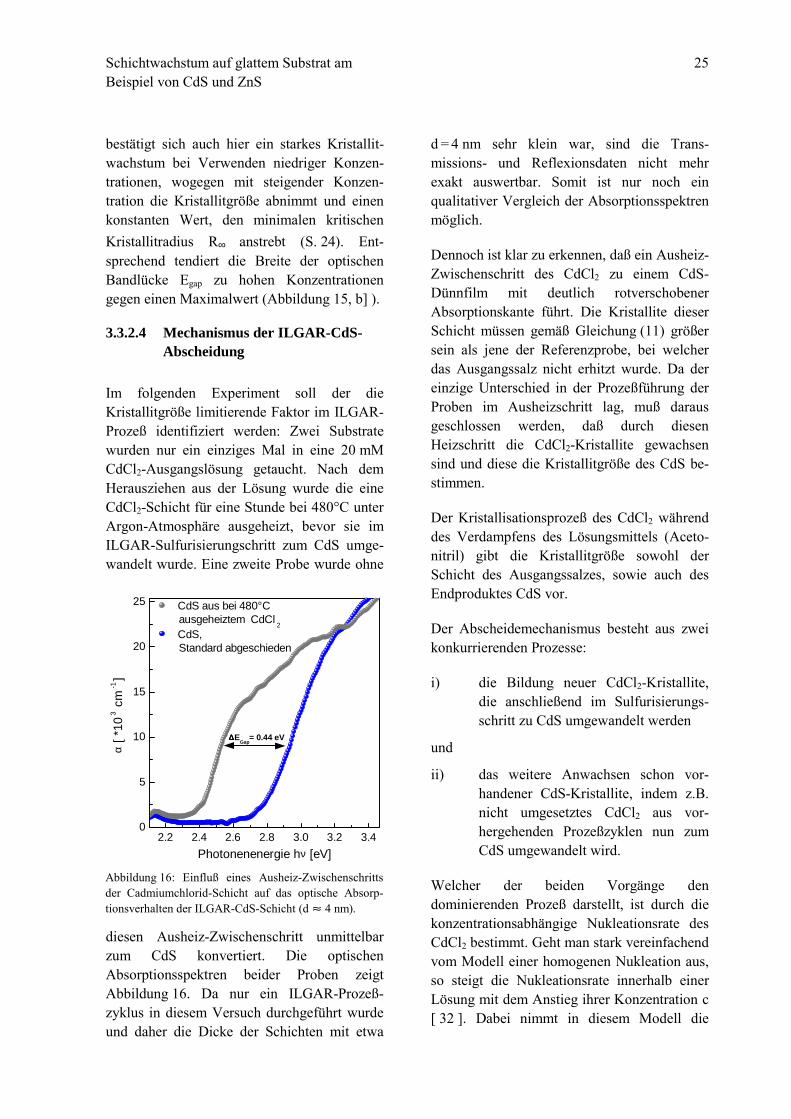
Im folgenden Experiment soll der dieKristallitgröße limitierende Faktor im ILGAR-Prozeß identifiziert werden: Zwei Substratewurden nur ein einziges Mal in eine 20 mMCdCl2-Ausgangslösung getaucht. Nach demHerausziehen aus der Lösung wurde die eineCdCl2-Schicht für eine Stunde bei 480°C unterArgon-Atmosphäre ausgeheizt, bevor sie imILGAR-Sulfurisierungschritt zum CdS umge-wandelt wurde. Eine zweite Probe wurde ohne
diesen Ausheiz-Zwischenschritt unmittelbarzum CdS konvertiert. Die optischenAbsorptionsspektren beider Proben zeigtAbbildung 16. Da nur ein ILGAR-Prozeß-zyklus in diesem Versuch durchgeführt wurdeund daher die Dicke der Schichten mit etwa
d = 4 nm sehr klein war, sind die Trans-missions- und Reflexionsdaten nicht mehrexakt auswertbar. Somit ist nur noch einqualitativer Vergleich der Absorptionsspektrenmöglich.
Dennoch ist klar zu erkennen, daß ein Ausheiz-Zwischenschritt des CdCl2 zu einem CdS-Dünnfilm mit deutlich rotverschobenerAbsorptionskante führt. Die Kristallite dieserSchicht müssen gemäß Gleichung (11) größersein als jene der Referenzprobe, bei welcherdas Ausgangssalz nicht erhitzt wurde. Da dereinzige Unterschied in der Prozeßführung derProben im Ausheizschritt lag, muß darausgeschlossen werden, daß durch diesenHeizschritt die CdCl2-Kristallite gewachsensind und diese die Kristallitgröße des CdS be-stimmen.
Der Kristallisationsprozeß des CdCl2 währenddes Verdampfens des Lösungsmittels (Aceto-nitril) gibt die Kristallitgröße sowohl derSchicht des Ausgangssalzes, sowie auch desEndproduktes CdS vor.
Der Abscheidemechanismus besteht aus zweikonkurrierenden Prozesse:
i) die Bildung neuer CdCl2-Kristallite,die anschließend im Sulfurisierungs-schritt zu CdS umgewandelt werden
und
ii) das weitere Anwachsen schon vor-handener CdS-Kristallite, indem z.B.nicht umgesetztes CdCl2 aus vor-hergehenden Prozeßzyklen nun zumCdS umgewandelt wird.
Welcher der beiden Vorgänge dendominierenden Prozeß darstellt, ist durch diekonzentrationsabhängige Nukleationsrate desCdCl2 bestimmt. Geht man stark vereinfachendvom Modell einer homogenen Nukleation aus,so steigt die Nukleationsrate innerhalb einerLösung mit dem Anstieg ihrer Konzentration c[ 32 ]. Dabei nimmt in diesem Modell die
2.2 2.4 2.6 2.8 3.0 3.2 3.40
5
10
15
20
25
∆∆∆∆EGap
= 0.44 eV
CdS aus bei 480°C ausgeheiztem CdCl 2
CdS, Standard abgeschieden
α [ *
10 3
cm
-1 ]
Photonenenergie hν [eV]
Abbildung 16: Einfluß eines Ausheiz-Zwischenschrittsder Cadmiumchlorid-Schicht auf das optische Absorp-tionsverhalten der ILGAR-CdS-Schicht (d " 4 nm).

26 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
Größe (Radius R) der gebildeten Kristallite ab.Bei hohen Ausgangskonzentrationen werdenalso viele kleine Kristallite gebildet. BeimILGAR-CdS-Prozeß fällt in diesem Fall dasWeiterwachsen der Partikel bezogen auf dieKristallitgrößenverteilung weniger insGewicht. Dies steht im Einklang mit demResultat aus Abbildung 14und 15 a]. Dieumgekehrte Situation liegt bei einer niedrigenKonzentration an Ausgangsstoff vor; dieNukleationsrate ist gering und es bilden sichnur wenige, große Kristallite, die die dieVerteilung vorgeben.
Bei einer sehr hohen CdCl2-Konzentration(c » 100 mM) erlangen die gebildeten Partikeleine kritische, minimale Größe (R∞), welchegerade noch stabil ist. Kleinere Partikel lösensich wieder auf, da sie nur auf diese Art ihrefreie Energie minimieren können [ 32 ]. DasNukleationsverhalten des Precursors erklärt dieEinstellung einer minimalen Kristallitgröße desCdCl2 im Grenzfall hoher Konzentrationenbzw. des CdS nach der Sulfurisierung, wie sieim Abschnitt 3.3.2.2 festgestellt wurde.
Sulfidische ILGAR-Halbleiterschichten inChalkopyrit-Dünnfilmsolarzellen
27
3.4 Beschreibung der Absorptions-kante einer nanokristallinenSchicht
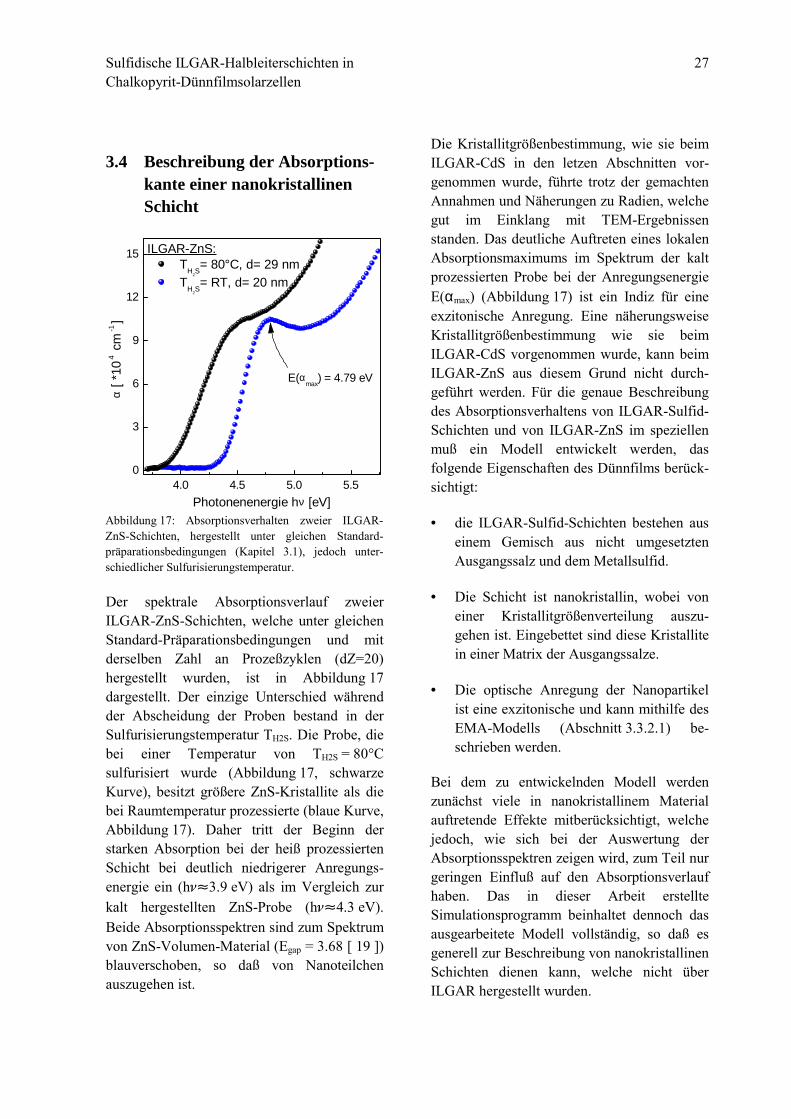
Der spektrale Absorptionsverlauf zweierILGAR-ZnS-Schichten, welche unter gleichenStandard-Präparationsbedingungen und mitderselben Zahl an Prozeßzyklen (dZ=20)hergestellt wurden, ist in Abbildung 17dargestellt. Der einzige Unterschied währendder Abscheidung der Proben bestand in derSulfurisierungstemperatur TH2S. Die Probe, diebei einer Temperatur von TH2S = 80°Csulfurisiert wurde (Abbildung 17, schwarzeKurve), besitzt größere ZnS-Kristallite als diebei Raumtemperatur prozessierte (blaue Kurve,Abbildung 17). Daher tritt der Beginn derstarken Absorption bei der heiß prozessiertenSchicht bei deutlich niedrigerer Anregungs-energie ein (h#"3.9 eV) als im Vergleich zurkalt hergestellten ZnS-Probe (h#"4.3 eV).Beide Absorptionsspektren sind zum Spektrumvon ZnS-Volumen-Material (Egap = 3.68 [ 19 ])blauverschoben, so daß von Nanoteilchenauszugehen ist.
Die Kristallitgrößenbestimmung, wie sie beimILGAR-CdS in den letzen Abschnitten vor-genommen wurde, führte trotz der gemachtenAnnahmen und Näherungen zu Radien, welchegut im Einklang mit TEM-Ergebnissenstanden. Das deutliche Auftreten eines lokalenAbsorptionsmaximums im Spektrum der kaltprozessierten Probe bei der AnregungsenergieE(αmax) (Abbildung 17) ist ein Indiz für eineexzitonische Anregung. Eine näherungsweiseKristallitgrößenbestimmung wie sie beimILGAR-CdS vorgenommen wurde, kann beimILGAR-ZnS aus diesem Grund nicht durch-geführt werden. Für die genaue Beschreibungdes Absorptionsverhaltens von ILGAR-Sulfid-Schichten und von ILGAR-ZnS im speziellenmuß ein Modell entwickelt werden, dasfolgende Eigenschaften des Dünnfilms berück-sichtigt:
• die ILGAR-Sulfid-Schichten bestehen auseinem Gemisch aus nicht umgesetztenAusgangssalz und dem Metallsulfid.
• Die Schicht ist nanokristallin, wobei voneiner Kristallitgrößenverteilung auszu-gehen ist. Eingebettet sind diese Kristallitein einer Matrix der Ausgangssalze.
• Die optische Anregung der Nanopartikelist eine exzitonische und kann mithilfe desEMA-Modells (Abschnitt 3.3.2.1) be-schrieben werden.
Bei dem zu entwickelnden Modell werdenzunächst viele in nanokristallinem Materialauftretende Effekte mitberücksichtigt, welchejedoch, wie sich bei der Auswertung derAbsorptionsspektren zeigen wird, zum Teil nurgeringen Einfluß auf den Absorptionsverlaufhaben. Das in dieser Arbeit erstellteSimulationsprogramm beinhaltet dennoch dasausgearbeitete Modell vollständig, so daß esgenerell zur Beschreibung von nanokristallinenSchichten dienen kann, welche nicht überILGAR hergestellt wurden.
Abbildung 17: Absorptionsverhalten zweier ILGAR-ZnS-Schichten, hergestellt unter gleichen Standard-präparationsbedingungen (Kapitel 3.1), jedoch unter-schiedlicher Sulfurisierungstemperatur.
4.0 4.5 5.0 5.50
3
6
9
12
15
E(αmax) = 4.79 eV
ILGAR-ZnS: TH2S
= 80°C, d= 29 nm TH2S
= RT, d= 20 nm
α [ *
10 4
cm
-1 ]
Photonenenergie hν [eV]

28 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
3.4.1 Lichtabsorption und Interbandüber-gänge bei nanokristallinen Schichten
Es soll zunächst nur ein nanokristallines,monodisperses Quantenteilchen mit bestimm-tem Radius R betrachtet werden. Das folgendeAbsorptionsmodell berücksichtigt zudemausschließlich den exzitonischen (1s-1s)-Übergang (S.22). Weitere Übergänge, wie z.B. der für Kristallite mit Radius BaR ≤ebenfalls erlaubte (1p-1p)-Übergang [ 31 ],sollen in die Rechnung nicht mit einfließen.Das Modell soll einzig die Absorptionskanteder nano-kristallinen Schicht beschreiben.
Ein Nanoteilchen vom Radius R besitzt gemäßdes EMA-Modell nach Gleichung (11) dieexzitonische Anregungsenergie E(R). Imoptischen Absorptionsspektrum des Teilchensist nur bei dieser Anregungsenergie eineAbsorptionslinie zu erkennen (höhereÜbergänge werden nicht betrachtet). Zusätz-lich zu seiner natürlichen Linienbreite könnenAbweichungen von der sphärischenKristallform den Absorptionsverlauf starkverbreitern [ 33 ].
Daneben tritt bei Q-Size-Teilchen imverstärkten Maße eine Linienverbreiterung auf,welche auf einer Phonon-Photon-Wechsel-wirkung beruht. Diese ist im Gegensatz zu denAuswirkungen einer nicht-sphärischen Kri-stallitform nicht über die Partikelpräparationzu beeinflussen. Mit kleiner werdenderKristallitgröße nimmt das Oberflächen-zu-Volumen-Verhältnis der Partikel zu. Ober-flächenmoden treten bei entsprechenderAnregung in Erscheinung und man erhält eindiskretes Vibrationsspektrum, welches demvon Molekülen ähnelt [ 33 ]. Sich ausbreitendePhononmoden an der Oberfläche können fürein Quantenteilchen in einer Matrix nur dannexistieren, wenn diese eine sehr ähnlicheGitterstruktur besitzt wie der betrachteteKristallit selbst [ 33 ]. Diese Bedingung wirdnoch wichtig bei der Interpretation der Ergeb-nisse. Durch Absorption von kurzwelligen
Phononen kann ein exzitonischer Übergang imNanokristallit mit Radius R schon bei deutlichkleineren Anregungsenergien als die obenberechnete E(R) stattfinden oder mittelsEmission von Phononen bei deutlich größeren.Durch die Phonon-Photon-Wechselwirkungbilden sich Absorptionsausläufer zu höherensowie zu niedrigeren Anregungsenergien hinaus.
Unter Berücksichtigung dieser „Phononen“-bedingten Verbreiterung ergibt sich derspektrale Absorptionskoeffizient für einenKristalliten mit Radius R zu [ 34 ]:
Es sind ω! ist die Photonenenergie, ncry derspektrale Brechungsindex des Kristalliten, cdie Lichtgeschwindigkeit, εkri die Dielektri-zitätskonstante des Partikels, V das Volumen,welches das Teilchen einnimmt, und Γ! dieenergetische Verbreiterung aufgrund derPhonon-Photon-Wechselwirkung (auch Pho-non-Photon-Kopplungskonstante genannt). C0,welches selbst eine Funktion derPhotonenenergie ist, ergibt sich zu [ 33 ]:
Die effektive Masse des Elektrons vomentsprechenden Volumenmaterial desKristalliten und die Energie der Spin-Bahn-Aufspaltung ∆, sowie alle anderen hier ver-wendeten Konstanten sind im Falle desILGAR-ZnS dem Anhang AV zu entnehmen.
cvp ist das optische Übergangsmatrixelementund ist hier für den (1s-1s)-Übergangberechnet [ 33 ].
Wie in Abbildung 18 zu erkennen, ist nehmendie Maxima der Absorptionsspektren
)13(][])([
),( 220
Γ+−Γ⋅⋅=
!!
!
ωεωωα
REVC
cnR
krikri
)15(32)(
])([)(3
mit
)14()(
2 222
22
0
∆+∆+⋅⋅=
⋅=
REREREmp
pm
eC
ecv
cve ωπ
!
!
Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
29
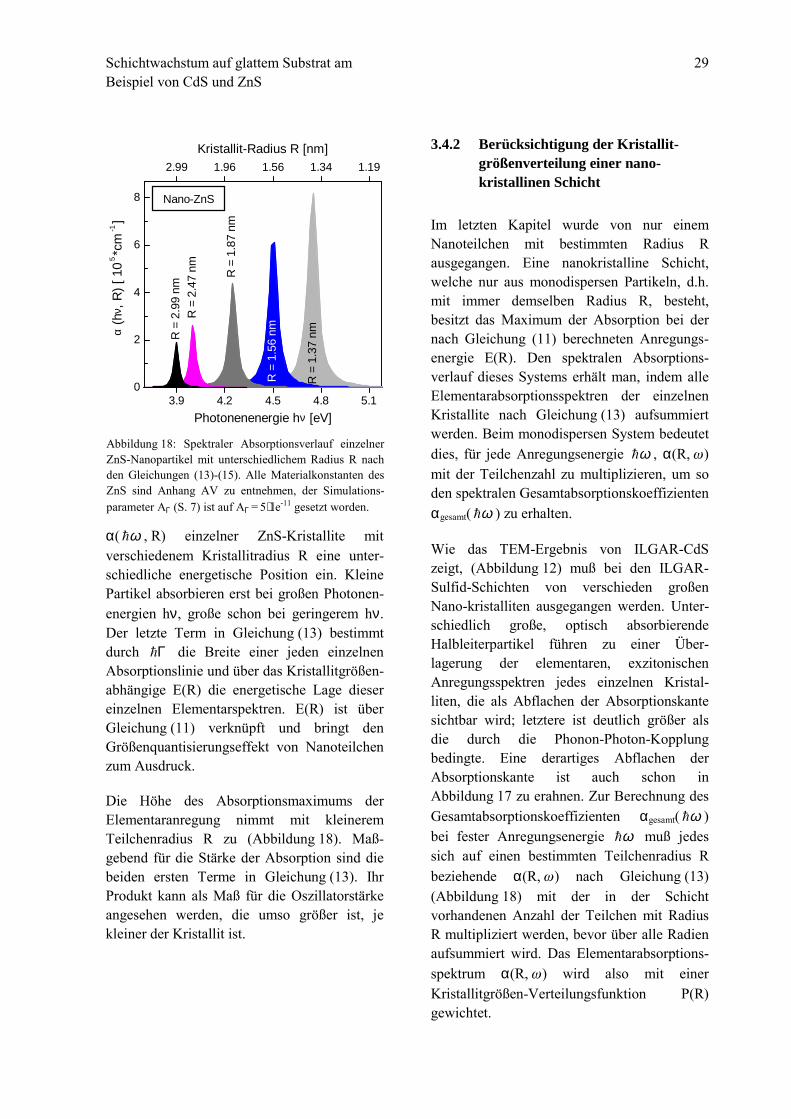
α( ω! , R) einzelner ZnS-Kristallite mitverschiedenem Kristallitradius R eine unter-schiedliche energetische Position ein. KleinePartikel absorbieren erst bei großen Photonen-energien hν, große schon bei geringerem hν.Der letzte Term in Gleichung (13) bestimmtdurch Γ! die Breite einer jeden einzelnenAbsorptionslinie und über das Kristallitgrößen-abhängige E(R) die energetische Lage diesereinzelnen Elementarspektren. E(R) ist überGleichung (11) verknüpft und bringt denGrößenquantisierungseffekt von Nanoteilchenzum Ausdruck.
Die Höhe des Absorptionsmaximums derElementaranregung nimmt mit kleineremTeilchenradius R zu (Abbildung 18). Maß-gebend für die Stärke der Absorption sind diebeiden ersten Terme in Gleichung (13). IhrProdukt kann als Maß für die Oszillatorstärkeangesehen werden, die umso größer ist, jekleiner der Kristallit ist.
3.4.2 Berücksichtigung der Kristallit-größenverteilung einer nano-kristallinen Schicht
Im letzten Kapitel wurde von nur einemNanoteilchen mit bestimmten Radius Rausgegangen. Eine nanokristalline Schicht,welche nur aus monodispersen Partikeln, d.h.mit immer demselben Radius R, besteht,besitzt das Maximum der Absorption bei dernach Gleichung (11) berechneten Anregungs-energie E(R). Den spektralen Absorptions-verlauf dieses Systems erhält man, indem alleElementarabsorptionsspektren der einzelnenKristallite nach Gleichung (13) aufsummiertwerden. Beim monodispersen System bedeutetdies, für jede Anregungsenergie ω! , α(R, $)mit der Teilchenzahl zu multiplizieren, um soden spektralen Gesamtabsorptionskoeffizientenαgesamt( ω! ) zu erhalten.
Wie das TEM-Ergebnis von ILGAR-CdSzeigt, (Abbildung 12) muß bei den ILGAR-Sulfid-Schichten von verschieden großenNano-kristalliten ausgegangen werden. Unter-schiedlich große, optisch absorbierendeHalbleiterpartikel führen zu einer Über-lagerung der elementaren, exzitonischenAnregungsspektren jedes einzelnen Kristal-liten, die als Abflachen der Absorptionskantesichtbar wird; letztere ist deutlich größer alsdie durch die Phonon-Photon-Kopplungbedingte. Eine derartiges Abflachen derAbsorptionskante ist auch schon inAbbildung 17 zu erahnen. Zur Berechnung desGesamtabsorptionskoeffizienten αgesamt( ω! )bei fester Anregungsenergie ω! muß jedessich auf einen bestimmten Teilchenradius Rbeziehende α(R, $) nach Gleichung (13)(Abbildung 18) mit der in der Schichtvorhandenen Anzahl der Teilchen mit RadiusR multipliziert werden, bevor über alle Radienaufsummiert wird. Das Elementarabsorptions-spektrum α(R, $) wird also mit einerKristallitgrößen-Verteilungsfunktion P(R)gewichtet.
3.9 4.2 4.5 4.8 5.10
2
4
6
8
2.99 1.96 1.56 1.34 1.19
Nano-ZnS
R =
1.3
7 nm
R =
1.5
6 nm
R =
1.8
7 nm
R =
2.4
7 nm
R =
2.9
9 nm
Kristallit-Radius R [nm]
α (hν ,
R) [
10 5
*cm
-1 ]
Photonenenergie hν [eV]
Abbildung 18: Spektraler Absorptionsverlauf einzelnerZnS-Nanopartikel mit unterschiedlichem Radius R nachden Gleichungen (13)-(15). Alle Materialkonstanten desZnS sind Anhang AV zu entnehmen, der Simulations-parameter AΓ (S. 7) ist auf AΓ = 5⋅1e-11 gesetzt worden.
30 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
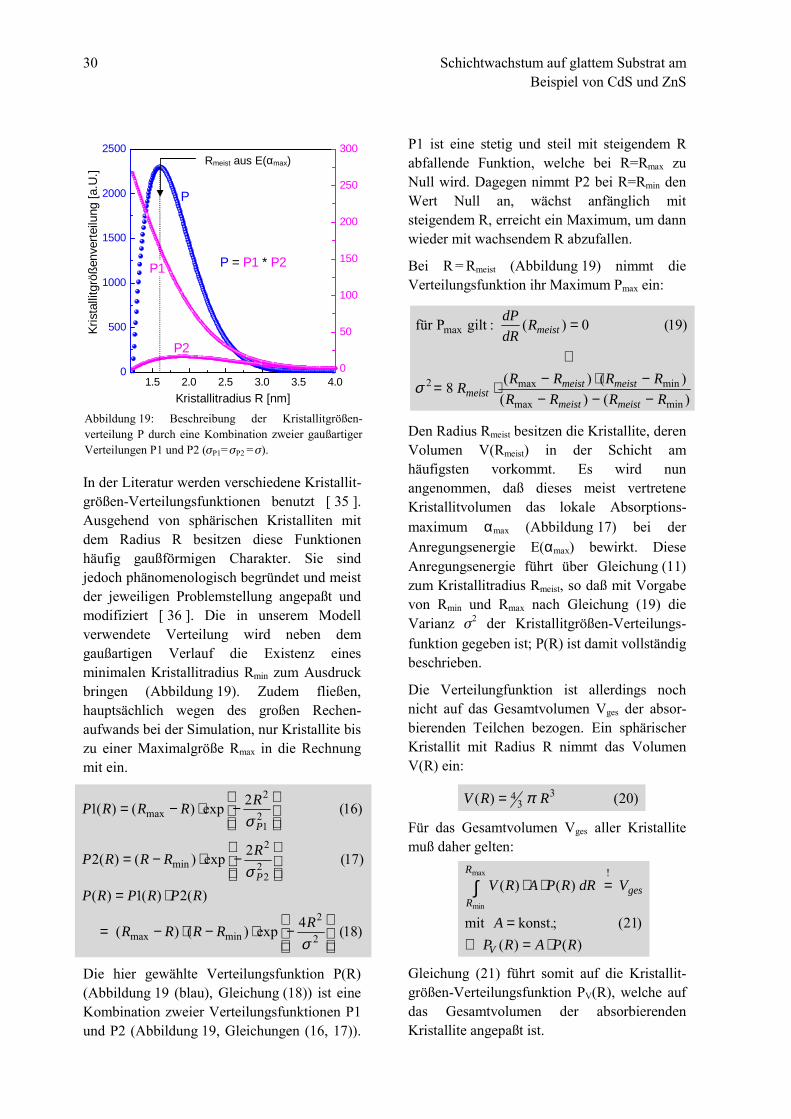
In der Literatur werden verschiedene Kristallit-größen-Verteilungsfunktionen benutzt [ 35 ].Ausgehend von sphärischen Kristalliten mitdem Radius R besitzen diese Funktionenhäufig gaußförmigen Charakter. Sie sindjedoch phänomenologisch begründet und meistder jeweiligen Problemstellung angepaßt undmodifiziert [ 36 ]. Die in unserem Modellverwendete Verteilung wird neben demgaußartigen Verlauf die Existenz einesminimalen Kristallitradius Rmin zum Ausdruckbringen (Abbildung 19). Zudem fließen,hauptsächlich wegen des großen Rechen-aufwands bei der Simulation, nur Kristallite biszu einer Maximalgröße Rmax in die Rechnungmit ein.
Die hier gewählte Verteilungsfunktion P(R)(Abbildung 19 (blau), Gleichung (18)) ist eineKombination zweier Verteilungsfunktionen P1und P2 (Abbildung 19, Gleichungen (16, 17)).
P1 ist eine stetig und steil mit steigendem Rabfallende Funktion, welche bei R=Rmax zuNull wird. Dagegen nimmt P2 bei R=Rmin denWert Null an, wächst anfänglich mitsteigendem R, erreicht ein Maximum, um dannwieder mit wachsendem R abzufallen.
Bei R = Rmeist (Abbildung 19) nimmt dieVerteilungsfunktion ihr Maximum Pmax ein:
Den Radius Rmeist besitzen die Kristallite, derenVolumen V(Rmeist) in der Schicht amhäufigsten vorkommt. Es wird nunangenommen, daß dieses meist vertreteneKristallitvolumen das lokale Absorptions-maximum αmax (Abbildung 17) bei derAnregungsenergie E(αmax) bewirkt. DieseAnregungsenergie führt über Gleichung (11)zum Kristallitradius Rmeist, so daß mit Vorgabevon Rmin und Rmax nach Gleichung (19) dieVarianz !2 der Kristallitgrößen-Verteilungs-funktion gegeben ist; P(R) ist damit vollständigbeschrieben.
Die Verteilungfunktion ist allerdings nochnicht auf das Gesamtvolumen Vges der absor-bierenden Teilchen bezogen. Ein sphärischerKristallit mit Radius R nimmt das VolumenV(R) ein:
Für das Gesamtvolumen Vges aller Kristallitemuß daher gelten:
Gleichung (21) führt somit auf die Kristallit-größen-Verteilungsfunktion PV(R), welche aufdas Gesamtvolumen der absorbierendenKristallite angepaßt ist.
)18(4exp)()(
)(2)(1)(
)17(2exp)()(2
)16(2exp)()(1
2
2
minmax
22
2
min
21
2
max
−⋅−⋅−=
⋅=
−⋅−=
−⋅−=
σ
σ
σ
RRRRR
RPRPRP
RRRRP
RRRRP
P
P
)()()()(8
)19(0)(:giltPfür
minmax
minmax2
max
RRRRRRRRR
RdRdP
meistmeist
meistmeistmeist
meist
−−−−⋅−⋅=
⇓
=
σ
Abbildung 19: Beschreibung der Kristallitgrößen-verteilung P durch eine Kombination zweier gaußartigerVerteilungen P1 und P2 (!P1= !P2 = !).
1.5 2.0 2.5 3.0 3.5 4.00
500
1000
1500
2000
2500
Kris
talli
tgrö
ßenv
erte
ilung
[a.U
.]
Kristallitradius R [nm]
0
50
100
150
200
250
300
P2
P1
P
P = P1 * P2
Rmeist aus E(αmax)
)20()( 33
4 RRV π=
)()()21(;konst.mit
)()(max
min
!
RPARPA
VdRRPARV
V
ges
R
R
⋅=⇒=
=⋅⋅∫

Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
31
Mit PV(R) kann nun eine Gewichtung der füralle Radien R zwischen Rmin und Rmax nachGleichung (13) berechneten, elementarenAbsorptionsspektren erfolgen. Der spektraleGesamtabsorptionskoeffizient αgesamt( ω! ) er-gibt sich dann durch Integration der jetztgewichteten Elementarspektren über Rzwischen Rmin und Rmax (Bildung des Erwar-tungswertes [ 40 ]):
3.5 Anwendung des optischenModells auf nanokristallineILGAR-ZnS-Schichten
Auf Basis des im letzten Kapitel vorgestelltenModells wurde im Laufe dieser Arbeit einSimulationsprogramm zur Beschreibung derAbsorptionskanten von nanokristallinenILGAR-Sulfidschichten ausgearbeitet. Alle imModell nun speziell auf kubisch-flächen-zentriertes ILGAR-ZnS bezogenen Material-konstanten sind in AV zusammengefaßt.
3.5.1 Vorgehensweise bei der Simulation
Im Simulationsprogramm werden Rmin, Rmax
und die Anregungsenergie E(αmax), bei der dieAbsorption ihr lokales Maximum einnimmt(Abbildung 17), vorgegeben. E(αmax) führtüber das EMA-Modell (Gleichung (11)) aufden Kristallitradius Rmeist. Hierdurch ist dieVarianz !2 der Kristallitgrößen-Verteilungs-funktion und die Verteilungsfunktion P(R) fürjeden Radius R zwischen Rmin und Rmax selbstbestimmt.
Die Kristallitgrößen-Verteilungsfunktion P(R)ist noch auf das Gesamtvolumen Vges derabsorbierenden Kristallite anzupassen. ImBereich der Absorptionskante, welche über die
Simulation angefittet werden soll, absorbiertnur das ZnS in den ILGAR-Schichten. Dieoptische Auswertung von Transmissions- undReflexionsdaten des Ausgangssalzes ZnCl2
ergab, daß die Absorption α dieser Verbindungbis zu einer Photonenenergie von h# = 6 eVkleiner α = 110 cm-1 und daher zu vernach-lässigen ist. Das Gesamtvolumen Vges allerabsorbierenden Kristallite entspricht demGesamtvolumen aller in der Schicht auf-tretenden ZnS-Kristallite.
Aus dem über EDX oder RFA bestimmtenStoffmengenanteil an ZnS (Abbildung 6) in derProbe läßt sich gemäß Anhang AI der ZnS-Volumenanteil p der Schicht berechnen. Ist dievom Spektrophotometer beleuchtete Proben-fläche AP (AP=8 mm×6 mm) und die Proben-dicke d bekannt, so ergibt sich das absor-bierende, effektive ZnS-Volumen Vges zu:
Durch Vorgabe des Stoffmengenanteils an ZnSund der Schichtdicke d einer jeden Probeberechnet das Simulationsprogramm den ZnS-Volumenanteil p nach AV und Vges überGleichung (23). Eine Anpassung der Ver-teilungsfunktion P(R) an das Gesamtvolumenergibt dann über Gleichung (21) Pv(R).Nachdem für jeden Partikelradius R zwischenRmin und Rmax nach Gleichung (11) E(R)berechnet wurde, bestimmt das Programmnach Gleichung (13-15) und (22) sodann fürjede Photonenenergie ω! den Gesamt-absorptionskoeffizienten αgesamt( ω! ).
Für den spektralen Verlauf des Brechungs-index nkrist (Gleichung 13) wird der ausTransmissions- und Reflexionsdaten be-stimmte Verlauf des Brechungsindex der be-trachteten ILGAR-ZnS-Schicht verwendet. DiePhonon-Photon-Kopplungskonstante Γ! so-wie Rmin, Rmax und E(αmax) sind Variablen.Diese Werte werden solange variiert, bis dieAbweichung des simulierten αgesamt vomgemessenen αexp im Spektralbereich derAbsorptionskante unter 5 % liegt.
)22()(
)(),(
)(max
min
max
min
∫
∫ ⋅
= R
RV
R
RV
gesamt
dRRP
dRRPR ωα
ωα !
)23(][ 3nmdApV Pges ⋅⋅=
32 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
Das Simulationsprogramm liefert als Ergebnisneben der Kristallitgrößen-VerteilungsfunktionPV(R) die Phonon-Photon-KopplungskonstanteΓ! . Es ermöglicht auf einfache und schnelle
Weise, über die Auswertung optischerTransmissions- und Reflexionsdaten aufInformationen zu Kristallitgrößen in einernanokristallinen Schicht zu schließen. Diesesist bei derart kleinen Partikeln über dieAuswertung von Röntgen-Diffraktogrammennicht möglich, da die Beugungssignale wegenihrer starken Verbreiterung nicht mehr vomUntergrund zu unterscheiden sind. Die TEM-Analyse, mit der die Partikelgröße bestimmtwerden kann, ist mit großem präparativen undinstrumentellem Aufwand verbunden.
3.5.2 Optisches Absorptionsverhalten vonILGAR-ZnS-Schichten
Im folgenden soll eine Schichtdickenvariationvon ILGAR-ZnS betrachtet werden. Es wirduntersucht, in wieweit sich die ZnS-Partikel-größe mit Zunahme der Probendicke d ändert,um somit Aufschluß über das Kristallit-wachstum bei ILGAR-ZnS zu erhalten.
Die Prozeßbedingungen dieser Probenreihewurden in Abschnitt 3.1 angegeben. Damitwurden ILGAR-ZnS-Proben auf Quarzglas mitProzeßzyklenzahlen zwischen dZ = 10 unddZ = 40 hergestellt.
Die chemische Zusammensetzung dieserProben ist in Kapitel 3.2.1 (Abbildung 6)dargelegt. Hieraus ermittelt die Simulation denVolumenanteil p an ZnS in der Probe. Dieserbeläuft sich bei den hier untersuchten Probenauf 71 % (dZ = 10)% p % 79 % (dZ = 40 ).
Neben der Ermittlung der Absorptions-spektren αexp( ω! ) war die Schichtdicke d derProben über die Profilometrie bestimmtworden. Die Röntgenfluoreszenzanalyse(RFA) lieferte zudem die MassenbelegungmRFA der Proben, was bei konstanter Analysen-fläche ein Maß für die Schichtdicke ist:
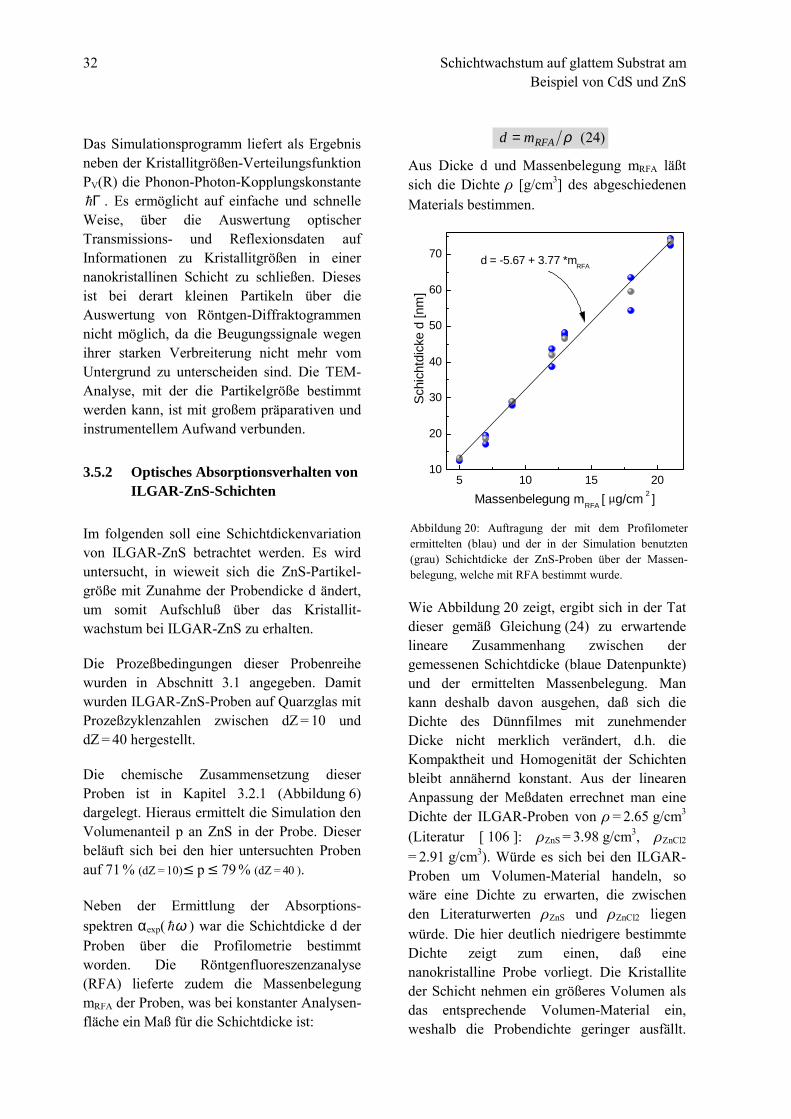
Aus Dicke d und Massenbelegung mRFA läßtsich die Dichte & [g/cm3] des abgeschiedenenMaterials bestimmen.
Wie Abbildung 20 zeigt, ergibt sich in der Tatdieser gemäß Gleichung (24) zu erwartendelineare Zusammenhang zwischen dergemessenen Schichtdicke (blaue Datenpunkte)und der ermittelten Massenbelegung. Mankann deshalb davon ausgehen, daß sich dieDichte des Dünnfilmes mit zunehmenderDicke nicht merklich verändert, d.h. dieKompaktheit und Homogenität der Schichtenbleibt annähernd konstant. Aus der linearenAnpassung der Meßdaten errechnet man eineDichte der ILGAR-Proben von & = 2.65 g/cm3
(Literatur [ 106 ]: &ZnS = 3.98 g/cm3, &ZnCl2
= 2.91 g/cm3). Würde es sich bei den ILGAR-Proben um Volumen-Material handeln, sowäre eine Dichte zu erwarten, die zwischenden Literaturwerten &ZnS und &ZnCl2 liegenwürde. Die hier deutlich niedrigere bestimmteDichte zeigt zum einen, daß einenanokristalline Probe vorliegt. Die Kristalliteder Schicht nehmen ein größeres Volumen alsdas entsprechende Volumen-Material ein,weshalb die Probendichte geringer ausfällt.
5 10 15 2010
20
30
40
50
60
70 d = -5.67 + 3.77 *mRFA
Schi
chtd
icke
d [n
m]
Massenbelegung mRFA [ µg/cm 2 ]
Abbildung 20: Auftragung der mit dem Profilometerermittelten (blau) und der in der Simulation benutzten(grau) Schichtdicke der ZnS-Proben über der Massen-belegung, welche mit RFA bestimmt wurde.
)24(ρRFAmd =
Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
33
ZE(lP
DdwAÜMl
Bereich der mit ±10 % geschätztenFehlerbreite der Schichtdickenmessung.
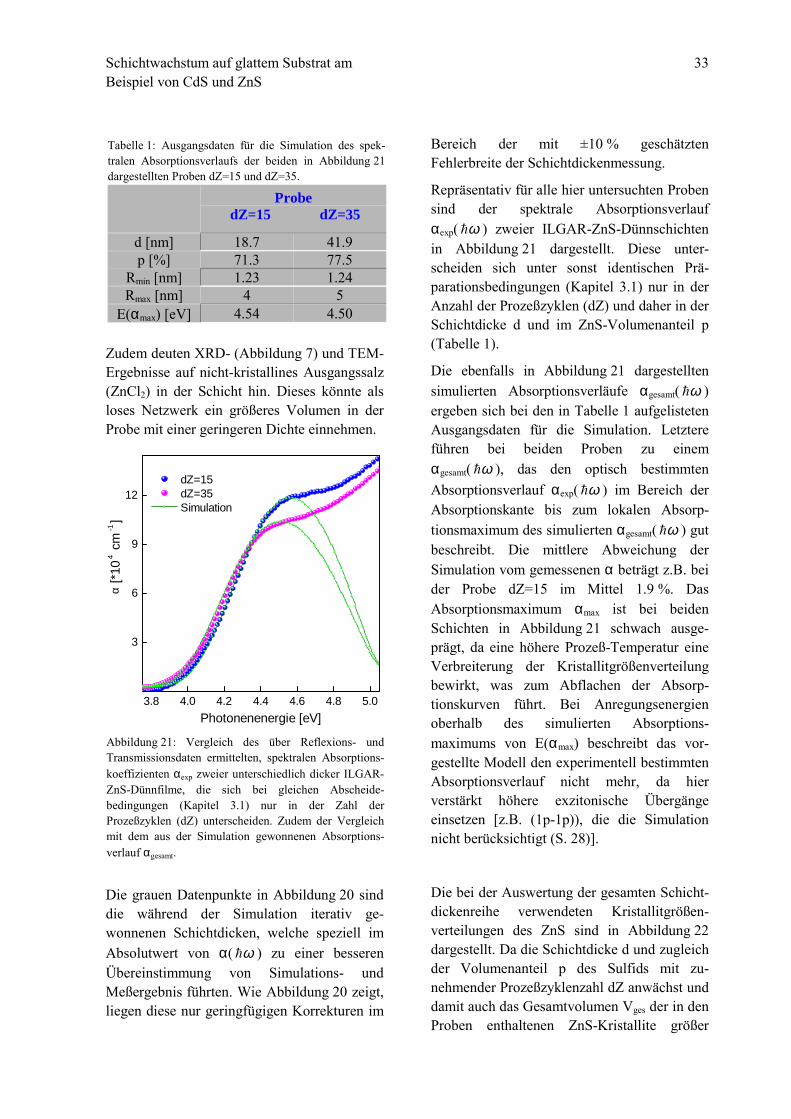
Repräsentativ für alle hier untersuchten Probensind der spektrale Absorptionsverlaufαexp( ω! ) zweier ILGAR-ZnS-Dünnschichtenin Abbildung 21 dargestellt. Diese unter-scheiden sich unter sonst identischen Prä-parationsbedingungen (Kapitel 3.1) nur in derAnzahl der Prozeßzyklen (dZ) und daher in der
ATkZbPmv
Ttd
abelle 1: Ausgangsdaten für die Simulation des spek-ralen Absorptionsverlaufs der beiden in Abbildung 21argestellten Proben dZ=15 und dZ=35.
ProbedZ=15 dZ=35
d [nm] 18.7 41.9p [%] 71.3 77.5
Rmin [nm] 1.23 1.24Rmax [nm] 4 5
E(αmax) [eV] 4.54 4.50
udem deuten XRD- (Abbildung 7) und TEM-rgebnisse auf nicht-kristallines Ausgangssalz
ZnCl2) in der Schicht hin. Dieses könnte alsoses Netzwerk ein größeres Volumen in derrobe mit einer geringeren Dichte einnehmen.
ie grauen Datenpunkte in Abbildung 20 sindie während der Simulation iterativ ge-onnenen Schichtdicken, welche speziell imbsolutwert von α( ω! ) zu einer besserenbereinstimmung von Simulations- undeßergebnis führten. Wie Abbildung 20 zeigt,
iegen diese nur geringfügigen Korrekturen im
Schichtdicke d und im ZnS-Volumenanteil p(Tabelle 1).
Die ebenfalls in Abbildung 21 dargestelltensimulierten Absorptionsverläufe αgesamt( ω! )ergeben sich bei den in Tabelle 1 aufgelistetenAusgangsdaten für die Simulation. Letztereführen bei beiden Proben zu einemαgesamt( ω! ), das den optisch bestimmtenAbsorptionsverlauf αexp( ω! ) im Bereich derAbsorptionskante bis zum lokalen Absorp-tionsmaximum des simulierten αgesamt( ω! ) gutbeschreibt. Die mittlere Abweichung derSimulation vom gemessenen α beträgt z.B. beider Probe dZ=15 im Mittel 1.9 %. DasAbsorptionsmaximum αmax ist bei beidenSchichten in Abbildung 21 schwach ausge-prägt, da eine höhere Prozeß-Temperatur eineVerbreiterung der Kristallitgrößenverteilungbewirkt, was zum Abflachen der Absorp-tionskurven führt. Bei Anregungsenergienoberhalb des simulierten Absorptions-maximums von E(αmax) beschreibt das vor-gestellte Modell den experimentell bestimmtenAbsorptionsverlauf nicht mehr, da hierverstärkt höhere exzitonische Übergängeeinsetzen [z.B. (1p-1p)), die die Simulationnicht berücksichtigt (S. 28)].
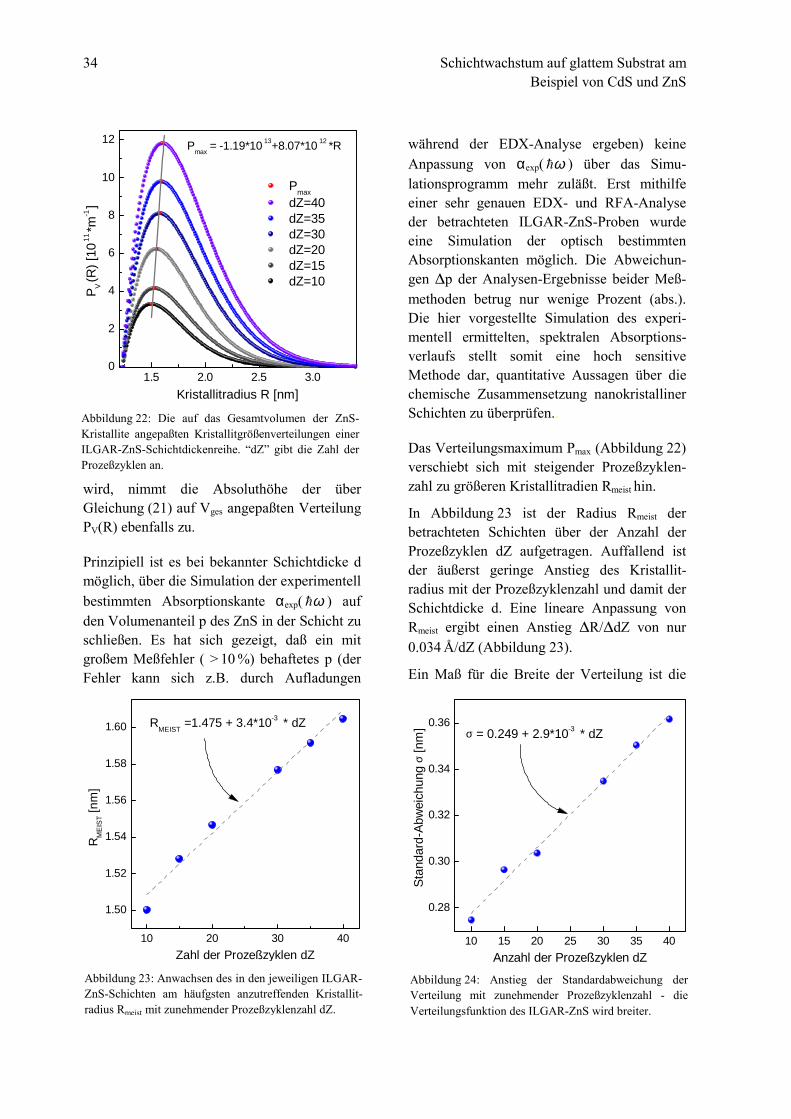
Die bei der Auswertung der gesamten Schicht-dickenreihe verwendeten Kristallitgrößen-verteilungen des ZnS sind in Abbildung 22dargestellt. Da die Schichtdicke d und zugleichder Volumenanteil p des Sulfids mit zu-nehmender Prozeßzyklenzahl dZ anwächst unddamit auch das Gesamtvolumen Vges der in denProben enthaltenen ZnS-Kristallite größer
bbildung 21: Vergleich des über Reflexions- undransmissionsdaten ermittelten, spektralen Absorptions-oeffizienten αexp zweier unterschiedlich dicker ILGAR-nS-Dünnfilme, die sich bei gleichen Abscheide-edingungen (Kapitel 3.1) nur in der Zahl derrozeßzyklen (dZ) unterscheiden. Zudem der Vergleichit dem aus der Simulation gewonnenen Absorptions-
erlauf αgesamt.
3.8 4.0 4.2 4.4 4.6 4.8 5.0
3
6
9
12 dZ=15 dZ=35 Simulation
α [*1
0 4 c
m -1
]
Photonenenergie [eV]
34 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
wird, nimmt die Absoluthöhe der überGleichung (21) auf Vges angepaßten VerteilungPV(R) ebenfalls zu.
Prinzipiell ist es bei bekannter Schichtdicke dmöglich, über die Simulation der experimentellbestimmten Absorptionskante αexp( ω! ) aufden Volumenanteil p des ZnS in der Schicht zuschließen. Es hat sich gezeigt, daß ein mitgroßem Meßfehler ( > 10 %) behaftetes p (derFehler kann sich z.B. durch Aufladungen
während der EDX-Analyse ergeben) keineAnpassung von αexp( ω! ) über das Simu-lationsprogramm mehr zuläßt. Erst mithilfeeiner sehr genauen EDX- und RFA-Analyseder betrachteten ILGAR-ZnS-Proben wurdeeine Simulation der optisch bestimmtenAbsorptionskanten möglich. Die Abweichun-gen 'p der Analysen-Ergebnisse beider Meß-methoden betrug nur wenige Prozent (abs.).Die hier vorgestellte Simulation des experi-mentell ermittelten, spektralen Absorptions-verlaufs stellt somit eine hoch sensitiveMethode dar, quantitative Aussagen über diechemische Zusammensetzung nanokristallinerSchichten zu überprüfen..
Das Verteilungsmaximum Pmax (Abbildung 22)verschiebt sich mit steigender Prozeßzyklen-zahl zu größeren Kristallitradien Rmeist hin.
In Abbildung 23 ist der Radius Rmeist derbetrachteten Schichten über der Anzahl derProzeßzyklen dZ aufgetragen. Auffallend istder äußerst geringe Anstieg des Kristallit-radius mit der Prozeßzyklenzahl und damit derSchichtdicke d. Eine lineare Anpassung vonRmeist ergibt einen Anstieg 'R/'dZ von nur0.034 Å/dZ (Abbildung 23).
Ein Maß für die Breite der Verteilung ist die
Abbildung 23: Anwachsen des in den jeweiligen ILGAR-ZnS-Schichten am häufgsten anzutreffenden Kristallit-radius Rmeist mit zunehmender Prozeßzyklenzahl dZ.
10 20 30 40
1.50
1.52
1.54
1.56
1.58
1.60 RMEIST
=1.475 + 3.4*10-3 * dZ
R ME
IST [
nm]
Zahl der Prozeßzyklen dZ
Abbildung 24: Anstieg der Standardabweichung derVerteilung mit zunehmender Prozeßzyklenzahl - dieVerteilungsfunktion des ILGAR-ZnS wird breiter.
10 15 20 25 30 35 40
0.28
0.30
0.32
0.34
0.36σ = 0.249 + 2.9*10-3 * dZ
Stan
dard
-Abw
eich
ung σ
[nm
]
Anzahl der Prozeßzyklen dZ
Abbildung 22: Die auf das Gesamtvolumen der ZnS-Kristallite angepaßten Kristallitgrößenverteilungen einerILGAR-ZnS-Schichtdickenreihe. “dZ” gibt die Zahl derProzeßzyklen an.
1.5 2.0 2.5 3.00
2
4
6
8
10
12 Pmax = -1.19*10 13+8.07*10 12 *R
Pmax dZ=40 dZ=35 dZ=30 dZ=20 dZ=15 dZ=10
P V (R
) [10
11 *m
-1 ]
Kristallitradius R [nm]

Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
35
Standardabweichung !. Diese ist die Wurzelaus der Varianz !2, des zweiten zentralenMoments der Verteilung. Zwar ist eineeindeutige, statistische Interpretation derzuletzt genannten Größe nur bei der Normal-verteilung möglich [ 40 ], um aber einequalitative Veränderung in der Breite der hierbenutzten Kristallitgrößen-Verteilung PV(R)deutlich zu machen, kann ! herangezogenwerden. In Abschnitt 3.4.2 ergab sich dieVarianz !2 (Gleichung (19)) und damit dieStandard-abweichung ! aus Rmin, Rmax undRmeist. Letzterer wurde nach Gleichung (11)über E(αmax) bestimmt. Das ! der jeweiligenZnS-Schichten, welches in der Simulationbenutzt wurde, steigt linear mit derProzeßzyklenzahl dZ an (Abbildung 24). Einelineare Anpassung dieses Verlaufs ergibt wiebei Rmeist (Abbildung 23) einen äußerstgeringen Anstieg '!/'dZ von 0.029 Å/dZ.
Beide geringen Zuwachsraten, die von Rmeist
und von !, zeigen, daß ein sehr geringesWachstum der Kristallite während desAbscheidevorgangs vorliegt. Dieses ist überdas Kristallisationsverhalten des Ausgangs-salzes ZnCl2 zu erklären. Bei der hierbenutzten hohen Konzentration anAusgangssalz von 20 mM ist davonauszugehen, daß die Nukleationsrate desAusgangsstoffes ZnCl2 groß ist. Beim ILGAR-ZnS-Prozeß fällt in diesem Fall dasWeiterwachsen der Partikel bezogen auf dieKristallitgrößenverteilung weniger ins Gewicht(Abschnitt 3.3.2.4).
Wie bei ILGAR-CdS (Abschnitt 3.3.2.2 bis3.3.2.4) muß von einem großen Einfluß derKonzentration des Ausgangssalzes auf dasWachstumsverhalten der ZnS-Kristallite aus-gegangen werden. Da eine Auftragung der inAbbildung 20 gezeigten Schichtdicke über derjeweiligen Prozeßzyklenzahl dZ der Probeneine konstante Wachstumsrate der Dünnfilmevon 2 nm/dZ ergibt, wachsen Rmeist und !ebenfalls linear mit zunehmender Schichtdicked an. Das Kristallitwachstum der ILGAR-CdS-Schichten zeigte ebenfalls diesen linearen
Zusammenhang (Kapitel 3.3.2.2,Abbildung 14). Diesem wichtigen Ergebnisnach kann davon ausgegangen werden, daß daslineare Wachstumsverhalten eine material-unabhängige, für den ILGAR-Prozeß typischeEigenschaft der Metallsulfidkristallite ist.
Die Tendenz zur Bildung großer Kristallite beiVerwendung niedriger Metallsalzkonzen-trationen und umgekehrt ist bei allen über dasILGAR-Verfahren hergestellten Sulfid-Ver-bindungen zu beobachten. Das Einstellen derKristallitgröße über die Vorgabe der Aus-gangssalzkonzentration und der Anzahl anProzeßzyklen ist gezielt nutzbar, wenn es umAnwendungsbereiche geht, bei denenbesondere Ansprüche an die Kristallitgröße derILGAR-Schichten gestellt werden. DieAbhängigkeit der Kristallitgröße von der Aus-gangssalzkonzentration, so wie in Abschnitt3.3.2.2 für ILGAR-CdS überprüft, wirdinnerhalb dieser Arbeit bei ILGAR-ZnS nichtmehr detailliert untersucht.
Wie das in diesem Kapitel beschriebene Expe-riment gezeigt hat, bewirkt die unter-schiedliche Größe der Kristallite einer nano-kristallinen ILGAR-Schicht ein deutlichesAbflachen des Verlaufs der Absorptionskanteder Probe. Einen zusätzlichen, im Vergleichzum Einfluß der Kristalllitgröße geringerenBeitrag hierzu kommt, wie in Abschnitt 3.4.1erläutert, durch die Phonon-Photon-Kopplungzustande.
Über eine Kristallitgrößenabhängigkeit derPhonon-Photon-Kopplung Γ! gibt es in derLiteratur verschiedene Aussagen: So behauptetC.H. Klein [ 37 ], daß diese keine Ab-hängigkeit von der Kristallitgröße zeige.Andere, z.B. A.P. Alivisatos [ 38 ], gehen voneiner Abhängigkeit von der Kristallitgröße aus,bei der Γ! direkt proportional zu 1/R ist.
Während der Entwicklung des Simulations-programms wurde überprüft, welcher Zu-sammenhang zwischen Γ! und R zu einergenaueren Anpassung an die experimentellbestimmte Absorptionskante αexp( ω! ) führt.
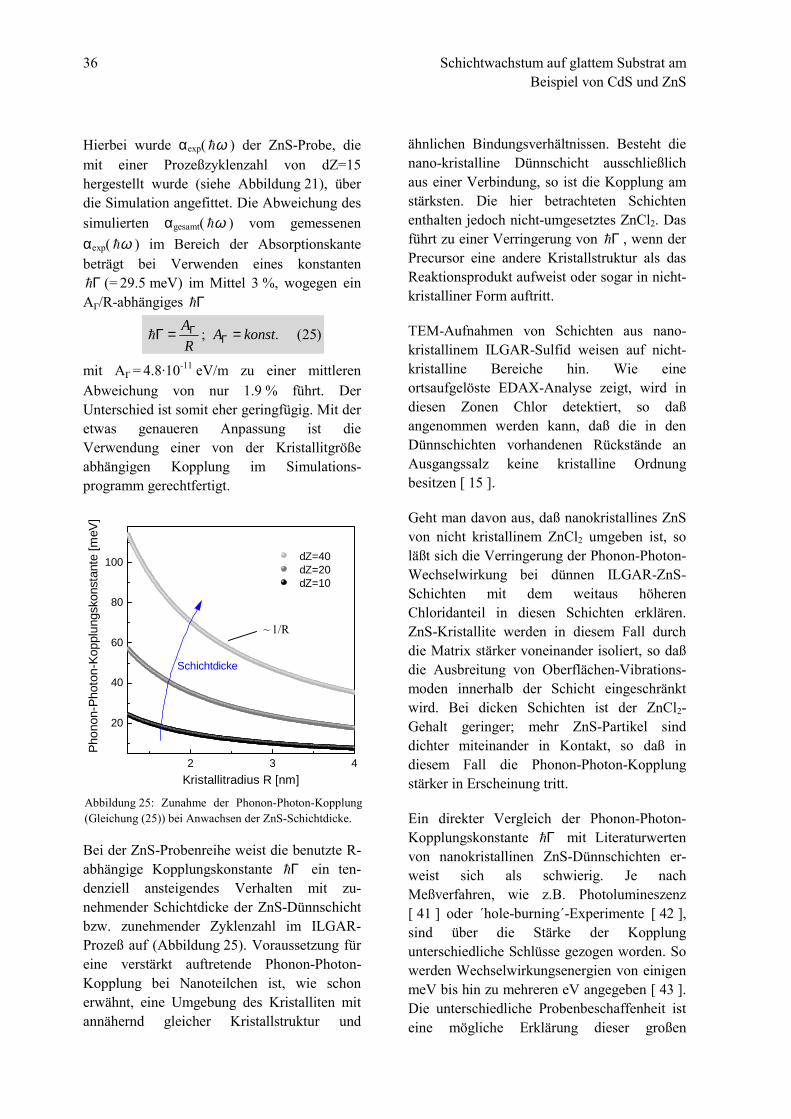
36 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
Hierbei wurde αexp( ω! ) der ZnS-Probe, diemit einer Prozeßzyklenzahl von dZ=15hergestellt wurde (siehe Abbildung 21), überdie Simulation angefittet. Die Abweichung dessimulierten αgesamt( ω! ) vom gemessenenαexp( ω! ) im Bereich der Absorptionskantebeträgt bei Verwenden eines konstantenΓ! (= 29.5 meV) im Mittel 3 %, wogegen ein
A(/R-abhängiges Γ!
mit A( = 4.8·10-11 eV/m zu einer mittlerenAbweichung von nur 1.9 % führt. DerUnterschied ist somit eher geringfügig. Mit deretwas genaueren Anpassung ist dieVerwendung einer von der Kristallitgrößeabhängigen Kopplung im Simulations-programm gerechtfertigt.
Bei der ZnS-Probenreihe weist die benutzte R-abhängige Kopplungskonstante Γ! ein ten-denziell ansteigendes Verhalten mit zu-nehmender Schichtdicke der ZnS-Dünnschichtbzw. zunehmender Zyklenzahl im ILGAR-Prozeß auf (Abbildung 25). Voraussetzung füreine verstärkt auftretende Phonon-Photon-Kopplung bei Nanoteilchen ist, wie schonerwähnt, eine Umgebung des Kristalliten mitannähernd gleicher Kristallstruktur und
ähnlichen Bindungsverhältnissen. Besteht dienano-kristalline Dünnschicht ausschließlichaus einer Verbindung, so ist die Kopplung amstärksten. Die hier betrachteten Schichtenenthalten jedoch nicht-umgesetztes ZnCl2. Dasführt zu einer Verringerung von Γ! , wenn derPrecursor eine andere Kristallstruktur als dasReaktionsprodukt aufweist oder sogar in nicht-kristalliner Form auftritt.
TEM-Aufnahmen von Schichten aus nano-kristallinem ILGAR-Sulfid weisen auf nicht-kristalline Bereiche hin. Wie eineortsaufgelöste EDAX-Analyse zeigt, wird indiesen Zonen Chlor detektiert, so daßangenommen werden kann, daß die in denDünnschichten vorhandenen Rückstände anAusgangssalz keine kristalline Ordnungbesitzen [ 15 ].
Geht man davon aus, daß nanokristallines ZnSvon nicht kristallinem ZnCl2 umgeben ist, soläßt sich die Verringerung der Phonon-Photon-Wechselwirkung bei dünnen ILGAR-ZnS-Schichten mit dem weitaus höherenChloridanteil in diesen Schichten erklären.ZnS-Kristallite werden in diesem Fall durchdie Matrix stärker voneinander isoliert, so daßdie Ausbreitung von Oberflächen-Vibrations-moden innerhalb der Schicht eingeschränktwird. Bei dicken Schichten ist der ZnCl2-Gehalt geringer; mehr ZnS-Partikel sinddichter miteinander in Kontakt, so daß indiesem Fall die Phonon-Photon-Kopplungstärker in Erscheinung tritt.
Ein direkter Vergleich der Phonon-Photon-Kopplungskonstante Γ! mit Literaturwertenvon nanokristallinen ZnS-Dünnschichten er-weist sich als schwierig. Je nachMeßverfahren, wie z.B. Photolumineszenz[ 41 ] oder ´hole-burning´-Experimente [ 42 ],sind über die Stärke der Kopplungunterschiedliche Schlüsse gezogen worden. Sowerden Wechselwirkungsenergien von einigenmeV bis hin zu mehreren eV angegeben [ 43 ].Die unterschiedliche Probenbeschaffenheit isteine mögliche Erklärung dieser großen
2 3 4
20
40
60
80
100
Schichtdicke
dZ=40 dZ=20 dZ=10
Phon
on-P
hoto
n-Ko
pplu
ngsk
onst
ante
[meV
]
Kristallitradius R [nm]
Abbildung 25: Zunahme der Phonon-Photon-Kopplung(Gleichung (25)) bei Anwachsen der ZnS-Schichtdicke.
~ 1/R
)25(.; konstARA ==Γ ΓΓ!

Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS
37
Diskrepanz [ 43 ]. Da das bestimmte Γ! sichauf nanokristallines ZnS mit einer Chlorid-Umgebung bezieht, dieses System in der
Literatur jedoch noch nicht behandelt wurde,kann ein Vergleich mit den Literaturdatennicht angestellt werden.
3.6 Zusammenfassung des Kapitels
Es wurde gezeigt, daß ILGAR-Sulfid-Schichten, die auf glattem Substrat und beiTemperaturen unter 100°C prozessiert sind, eine deutliche Blauverschiebung derAbsorptionskante aufgrund des Q-Size-Effektes aufweisen.
Diese Eigenschaft wurde ausgenutzt, um auf unkomplizierte und rasche Weise dasWachstumsverhalten der ILGAR-Schichten zu untersuchen. Ein Programm zurSimulation des optischen Verlaufs der Absorptionskante αexp( ω! ) wurde ent-wickelt, welches eine Kristallitgrößenverteilung, den Größenquantisierungseffektund die Phonon-Photon-Kopplung berücksichtigt. Anhand der optischenSchichteigenschaften konnte für den zyklischen ILGAR-Prozeß der komplexeWachstumsvorgang von Sulfidschichten identifiziert werden. Er setzt sichzusammen aus dem Anwachsen der Kristallite aus vorangegangenen Zyklen undder Bildung neuer, kleiner Partikel im aktuellen Zyklus. Das Kriterium für dieDominanz einer der beiden Prozesse liefert das Kristallisationsverhalten desAusgangssalzes. Bei einer niedrigen Salzkonzentration, bei der dieNukleationsrate des Salzes gering ist, bilden sich große Kristallite aus, und dasWeiterwachsen der Partikel fällt in diesem Fall bezogen auf dieKristallitgrößenverteilung mehr ins Gewicht. Bei hoher Konzentration gilt dasGegenteil.
Ein in erster Näherung lineares Wachstumsverhalten der Kristallitgröße alsFunktion der Schichtdicke wurde bei ILGAR-ZnS und bei -CdS beobachtet. Diesesspricht für ein für das ILGAR-Verfahren typisches und material-unabhängigesVerhalten. Das die Sulfidpartikel umgebende, nicht umgesetzte Ausgangsmaterialist Voraussetzung für die Ausbildung von Nanoteilchen.
Es konnte nachgewiesen werden, daß der Gehalt an Ausgangssalz in der SchichtEinfluß auf die Phonon-Photon-Kopplung hat. Neben dem Einfluß unterschiedlichgroßer Kristallite auf den spektralen Verlauf der Absorption bewirkt dieseKopplung ein zusätzliches Verbreitern der Absorptionskante. Eine Überprüfungder Kristallitgrößen-Abhängigkeit der Phonon-Photon-Kopplungskonstante Γ!ließ eine gute Anpassung der Simulation an die experimentellen Absorptionsdatenbei Verwenden eines Γ! erkennen, das zum Inversen des Kristallitradiusproportional ist.

38 Schichtwachstum auf glattem Substrat amBeispiel von CdS und ZnS





























