Embed Size (px)
Citation preview

4 Entwicklung eines Targeting-Modulszur HIV-TherapieDer humane T-Zellrezeptor CD4 spielt während einer HIV-Infektion eine Schlüsselrolle.Dieses Protein vermittelt durch seine Wechselwirkung mit dem Oberflächenglyko-protein gp120 des HI-Virus die Assoziation des Virus an die Zellmembran. Nach derInfektion einer Zelle mit dem HI-Virus werden die viruseigenen Oberflächenproteinegp40/gp120 in die Membran eingelagert und auf den Zellen präsentiert (Lever, 1995).Mit der Exposition des gp120 auf der Oberfläche HIV-infizierter Zellen wird dieMöglichkeit eröffnet, spezifisch diese Zellen mit dem natürlichen Rezeptor für HIV, demCD4, zu treffen. Dabei ist die rekombinante Herstellung von CD4 ein wichtiger Aspektzur Entwicklung eines zelltypspezifischen Systems zur HIV-Therapie.
CD4 ist ein aus 435 Aminosäureresten bestehendes Transmembranprotein (Maddonet al., 1985). Es gliedert sich in 4 immunglobulinähnliche extrazelluläre Domänen, einehydrophobe Transmembranhelix und eine kleine cytoplasmatische Domäne. Für dieWechselwirkung mit gp120 ist nur die erste extrazelluläre Domäne (D1) relevant(Perutz, 1992; Kwong et al., 1998).
In dieser Arbeit wurden verschiedene Varianten der Domäne D1 des CD4 (Abb. 4-1)rekombinant hergestellt.
Der Einsatz von D1 kann in HIV-Therapie-Ansätzen die Assoziation des HI-Virus anT-Lymphozyten und Makrophagen verhindern (Traunecker et al., 1988; Hussey et al.,1988; Deen et al., 1988, Fischer et al., 1988). Die bestehenden Verfahren zur rekombi-nanten Herstellung dieses Proteins (Chao et al., 1989; Garlick et al., 1990) ermöglichenfunktionelle Studien, genügen allerdings nicht den Anforderungen, die an dieProduktion eines Therapeutikums gestellt sind. Deshalb sollte im Rahmen dieser Arbeitein Protokoll entwickelt werden, um die Ausbeute und Reinheit eines rekombinant her-gestellten D1 zu optimieren.
Das zu entwickelnde Targeting-Modul zur HIV-Therapie war eine CD4-Variante, wel-che über ein C-terminales Dimerisierungsmotiv an therapeutisch relevante Moleküleassoziiert werden kann.
Grundlage dieses Dimerisierungsmotivs sind kurze polyionische Peptide. Diesebestehen aus einer Abfolge von acht entweder positiv oder negativ geladenen
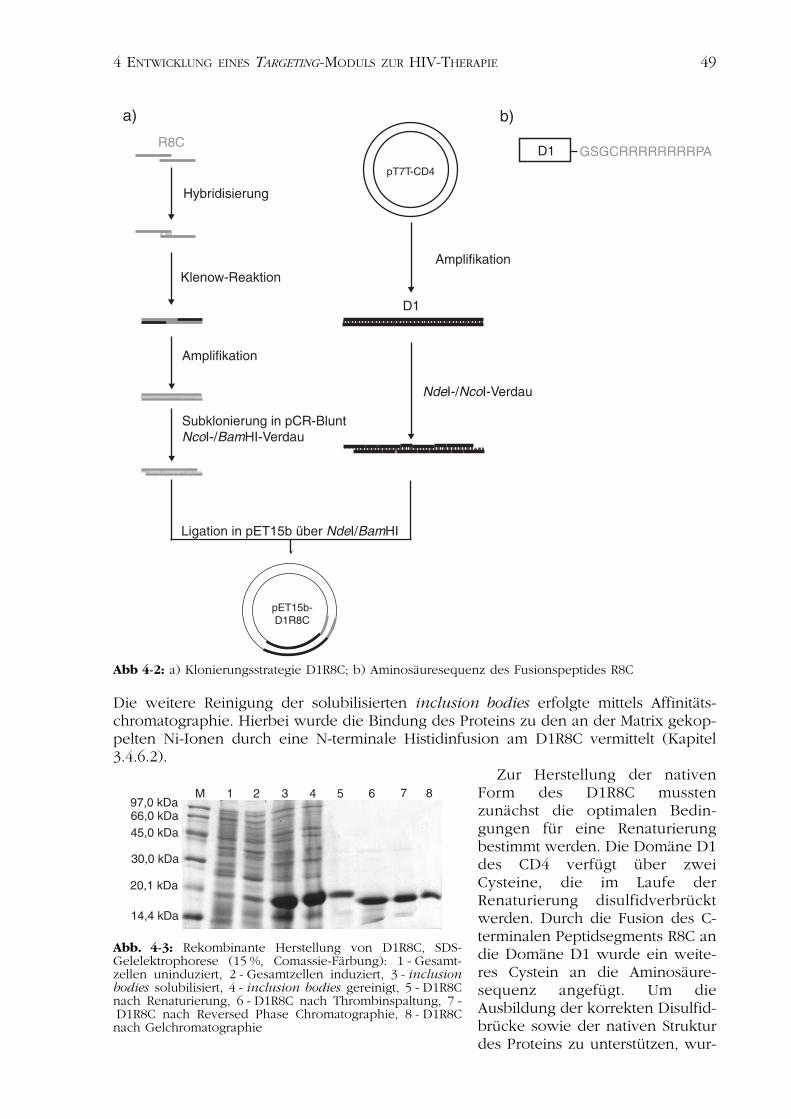
Aminosäureresten und einem zusätzlichenCystein-Rest. Durch Modifizierung vonProteinen mit diesen Peptiden kann, vermit-telt durch ionische Wechselwirkung mitanschließender Disulfidverbrückung, diekovalente Konjugation von zwei Proteinenerreicht werden (Stubenrauch et al., 2001;Richter et al., 2001; Kleinschmidt et al.,2003). Für die Darstellung des Targeting-Moduls wurde die N-terminale extrazelluläreDomäne des CD4 (D1) mit einem Okta-Argininpeptid und einem Cystein N-terminalverlängert. Dieses Konstrukt wird in Folgeals D1R8C bezeichnet (Abb. 4-2b).
Zur Eliminierung HIV-infizierter Zellenerfolgte die Assoziation des CD4-KonstruktsD1R8C an ein Derivat des PseudomonasExotoxin E8C-PE38 (Kleinschmidt et al.,2003). Das erhaltene rezeptorspezifische
Abb. 4-1: Struktur des Konstrukts D1 (Quelle:PDB ID 1GC1)

48
Zytotoxin kann über CD4 HIV-infizierte Zellen binden und durch das angefügte Derivatdes Pseudomonas Exotoxin die Vermehrung der HI-Viren durch Eliminierung der infi-zierten Zellen verhindern.
Der Nachweis der Funktionalität der hergestellten Proteine erfolgte durch Messungder Affinität zum Rezeptor gp120 mit Hilfe biophysikalischer Methoden.Spektroskopische Untersuchungen lieferten Aussagen über die Struktur und Stabilitätder dargestellten Konstrukte.
4.1 Rekombinante Herstellung der CD4-Varianten
4.1.1 Die CD4-Variante D1R8C
4.1.1.1 Herstellung des Plasmids pET15b-D1R8C
Für die Entwicklung des Moduls zur HIV-Therapie war die rekombinante Herstellungeiner CD4-Variante notwendig, welche ein polyionisches Fusionspeptid am C-Terminusbesitzt.
Die Herstellung der für das polyionische Peptid codierenden Sequenz erfolgte überHybridisierung synthetischer Oligonukleotide, die über einen Bereich von 10 bp kom-plementär hybridisierten. Nicht hybridisierte Bereiche wurden durch die DNA-Polymerase Klenow-Fragment synthetisiert. Die Sequenz des polyionischen Fusions-peptids ist im Anhang (Kapitel 8.4) dargestellt.
Die Planung des synthetischen Oligonukleotidpaares erfolgte so, dass am 5'-Endeeine NcoI-Restriktionsschnittstelle entstand, welche die Ligation mit dem durch PCRerhaltenen D1-Fragment ermöglichte. Die am 3'-Ende geplante BamHI-Restriktions-schnittstelle war für die Klonierung in den Expressionsvektor pET15b notwendig.
Durch PCR wurde aus dem Plasmid pT7TCD4 (Conzelmann & Schnell, 1994) die fürD1 codierende Sequenz amplifiziert, hierbei wurden die Restriktionsschnittstellen fürNdeI und NcoI an die Sequenz gefügt (Abb. 4-2).
Die erfolgreiche Klonierung in den Expressionsvektor pET15b bestätigtenSequenzierung und analytischer Restriktionsverdau. Die Klonierung in das PlasmidpET15b ermöglichte die Expression des CD4-Konstrukts mit N-terminaler Histidinfusion.
4.1.1.2 Expression, Renaturierung und Reinigung
Zur Expression des Proteins D1R8C wurde der E.coli-Stamm BL21CodonPlus(DE3)RILmit dem Plasmid pET15b-D1R8C transformiert.
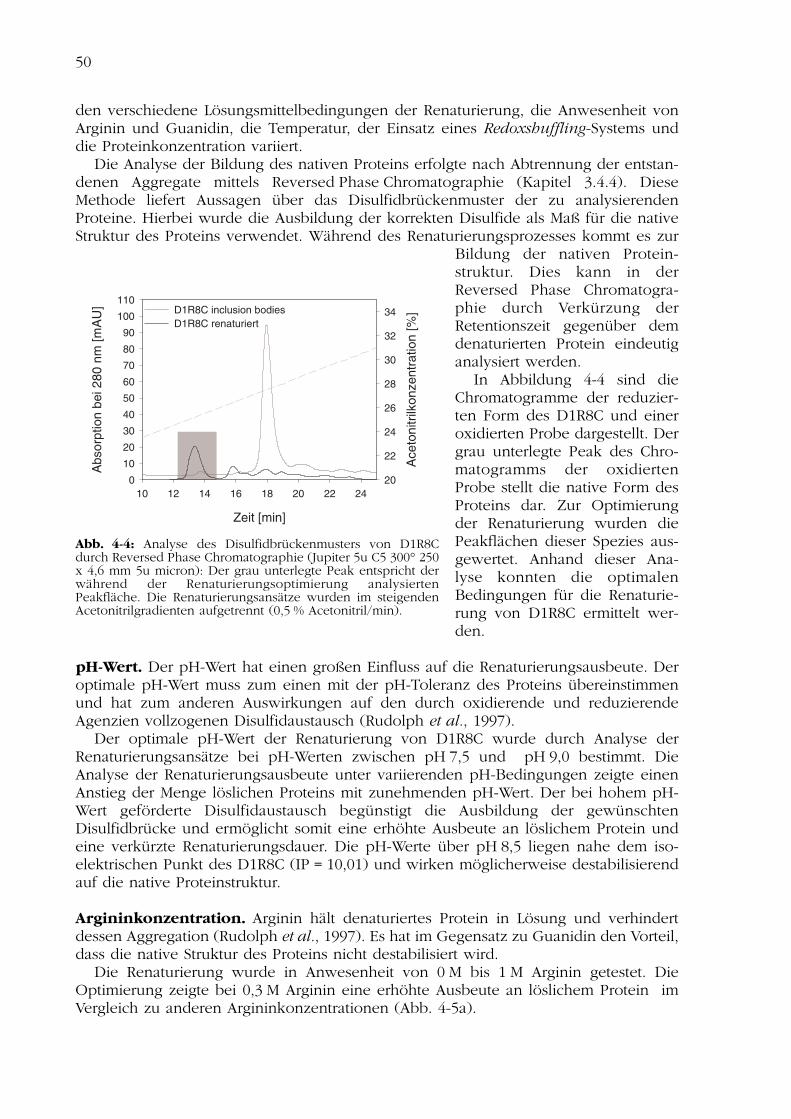
Um ausreichend Zellmaterial zu erhalten, wurde eine 6-l-Fed-batch-Fermentationdurchgeführt (Kapitel 3.3.2). Nach Anzucht des Expressionsstammes in Vollmedium bei37 °C zu einer optischen Dichte OD600 = 54 erfolgte die Expression des Zielproteinsdurch Induktion mit 1 mM IPTG und Inkubation der Zellen bei 37 °C für 3 Stunden. DieHochzelldichtefermentation ergab 755 g Zellmasse. Die Expression der entwickeltenCD4-Variante D1R8C konnte über eine SDS-Gelelektrophorese nachgewiesen werden(Abb. 4-3).
Das Protein D1R8C wurde in Escherichia coli in unlöslicher Form als inclusion bodiesexprimiert. Die inclusion bodies konnten nach Zellaufschluss mittels Hochdruckdis-persion durch ein Standardverfahren isoliert und anschließend in 6 M Guanidin solubi-lisiert werden (Rudolph et al., 1997; Kapitel 3.4.4).
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 49
Die weitere Reinigung der solubilisierten inclusion bodies erfolgte mittels Affinitäts-chromatographie. Hierbei wurde die Bindung des Proteins zu den an der Matrix gekop-pelten Ni-Ionen durch eine N-terminale Histidinfusion am D1R8C vermittelt (Kapitel3.4.6.2).
Zur Herstellung der nativenForm des D1R8C musstenzunächst die optimalen Bedin-gungen für eine Renaturierungbestimmt werden. Die Domäne D1des CD4 verfügt über zweiCysteine, die im Laufe derRenaturierung disulfidverbrücktwerden. Durch die Fusion des C-terminalen Peptidsegments R8C andie Domäne D1 wurde ein weite-res Cystein an die Aminosäure-sequenz angefügt. Um dieAusbildung der korrekten Disulfid-brücke sowie der nativen Strukturdes Proteins zu unterstützen, wur-
GSGCRRRRRRRRPAD1
b)
pT7T-CD4
pET15b-D1R8C
Ligation in pET15b über NdeI/BamHI
Subklonierung in pCR-BluntNcoI-/BamHI-Verdau
Amplifikation
Klenow-Reaktion
Hybridisierung
NdeI-/NcoI-Verdau
Amplifikation
R8C
D1
a)
Abb 4-2: a) Klonierungsstrategie D1R8C; b) Aminosäuresequenz des Fusionspeptides R8C
20,1 kDa
14,4 kDa
7 8
30,0 kDa
45,0 kDa66,0 kDa97,0 kDa
M 1 2 3 4 5 6
Abb. 4-3: Rekombinante Herstellung von D1R8C, SDS-Gelelektrophorese (15 %, Comassie-Färbung): 1 - Gesamt-zellen uninduziert, 2 - Gesamtzellen induziert, 3 - inclusionbodies solubilisiert, 4 - inclusion bodies gereinigt, 5 - D1R8Cnach Renaturierung, 6 - D1R8C nach Thrombinspaltung, 7 -D1R8C nach Reversed Phase Chromatographie, 8 - D1R8Cnach Gelchromatographie
50
den verschiedene Lösungsmittelbedingungen der Renaturierung, die Anwesenheit vonArginin und Guanidin, die Temperatur, der Einsatz eines Redoxshuffling-Systems unddie Proteinkonzentration variiert.
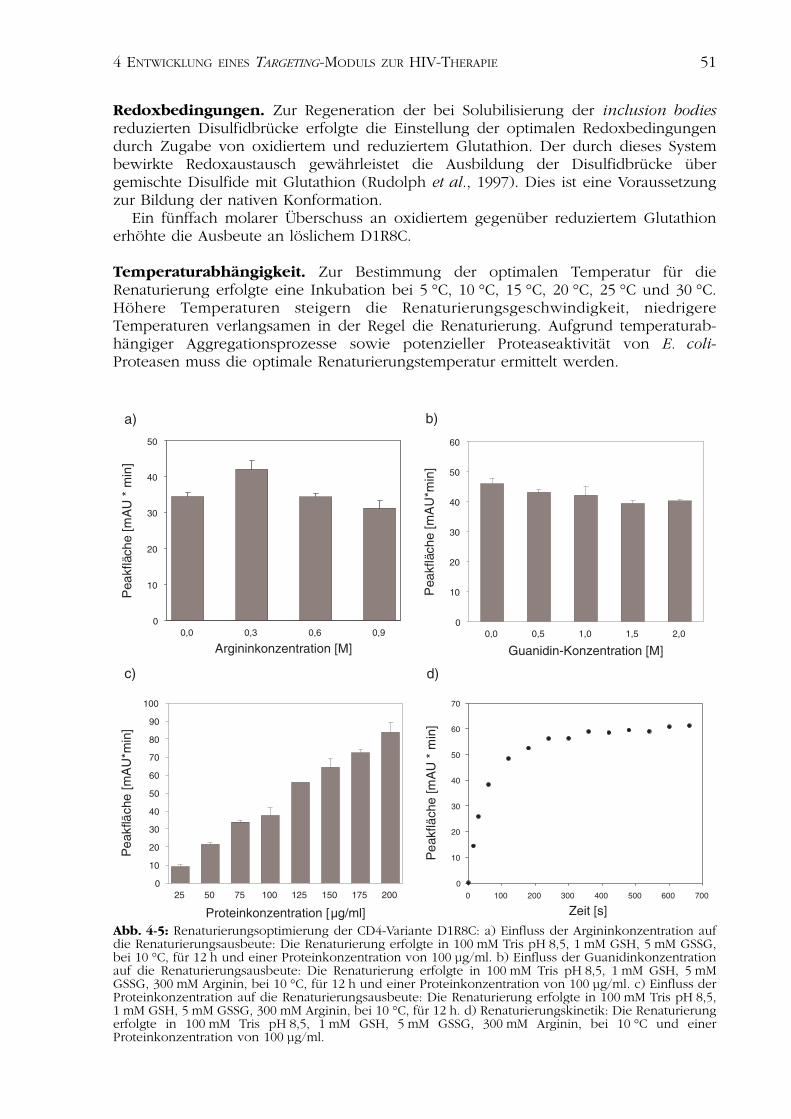
Die Analyse der Bildung des nativen Proteins erfolgte nach Abtrennung der entstan-denen Aggregate mittels Reversed Phase Chromatographie (Kapitel 3.4.4). DieseMethode liefert Aussagen über das Disulfidbrückenmuster der zu analysierendenProteine. Hierbei wurde die Ausbildung der korrekten Disulfide als Maß für die nativeStruktur des Proteins verwendet. Während des Renaturierungsprozesses kommt es zur
Bildung der nativen Protein-struktur. Dies kann in derReversed Phase Chromatogra-phie durch Verkürzung derRetentionszeit gegenüber demdenaturierten Protein eindeutiganalysiert werden.
In Abbildung 4-4 sind dieChromatogramme der reduzier-ten Form des D1R8C und eineroxidierten Probe dargestellt. Dergrau unterlegte Peak des Chro-matogramms der oxidiertenProbe stellt die native Form desProteins dar. Zur Optimierungder Renaturierung wurden diePeakflächen dieser Spezies aus-gewertet. Anhand dieser Ana-lyse konnten die optimalenBedingungen für die Renaturie-rung von D1R8C ermittelt wer-den.
pH-Wert. Der pH-Wert hat einen großen Einfluss auf die Renaturierungsausbeute. Deroptimale pH-Wert muss zum einen mit der pH-Toleranz des Proteins übereinstimmenund hat zum anderen Auswirkungen auf den durch oxidierende und reduzierendeAgenzien vollzogenen Disulfidaustausch (Rudolph et al., 1997).
Der optimale pH-Wert der Renaturierung von D1R8C wurde durch Analyse derRenaturierungsansätze bei pH-Werten zwischen pH 7,5 und pH 9,0 bestimmt. DieAnalyse der Renaturierungsausbeute unter variierenden pH-Bedingungen zeigte einenAnstieg der Menge löslichen Proteins mit zunehmenden pH-Wert. Der bei hohem pH-Wert geförderte Disulfidaustausch begünstigt die Ausbildung der gewünschtenDisulfidbrücke und ermöglicht somit eine erhöhte Ausbeute an löslichem Protein undeine verkürzte Renaturierungsdauer. Die pH-Werte über pH 8,5 liegen nahe dem iso-elektrischen Punkt des D1R8C (IP = 10,01) und wirken möglicherweise destabilisierendauf die native Proteinstruktur.
Argininkonzentration. Arginin hält denaturiertes Protein in Lösung und verhindertdessen Aggregation (Rudolph et al., 1997). Es hat im Gegensatz zu Guanidin den Vorteil,dass die native Struktur des Proteins nicht destabilisiert wird.
Die Renaturierung wurde in Anwesenheit von 0 M bis 1 M Arginin getestet. DieOptimierung zeigte bei 0,3 M Arginin eine erhöhte Ausbeute an löslichem Protein imVergleich zu anderen Argininkonzentrationen (Abb. 4-5a).
Abb. 4-4: Analyse des Disulfidbrückenmusters von D1R8Cdurch Reversed Phase Chromatographie (Jupiter 5u C5 300° 250x 4,6 mm 5u micron): Der grau unterlegte Peak entspricht derwährend der Renaturierungsoptimierung analysiertenPeakfläche. Die Renaturierungsansätze wurden im steigendenAcetonitrilgradienten aufgetrennt (0,5 % Acetonitril/min).
Zeit [min]
10 12 14 16 18 20 22 24
Abs
orpt
ion
bei 2
80 n
m [m
AU
]
0
10
20
30
40
50
60
70
80
90
100
110
Ace
toni
trilk
onze
ntra
tion
[%]
20
22
24
26
28
30
32
34D1R8C inclusion bodiesD1R8C renaturiert
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 51
Redoxbedingungen. Zur Regeneration der bei Solubilisierung der inclusion bodiesreduzierten Disulfidbrücke erfolgte die Einstellung der optimalen Redoxbedingungendurch Zugabe von oxidiertem und reduziertem Glutathion. Der durch dieses Systembewirkte Redoxaustausch gewährleistet die Ausbildung der Disulfidbrücke übergemischte Disulfide mit Glutathion (Rudolph et al., 1997). Dies ist eine Voraussetzungzur Bildung der nativen Konformation.
Ein fünffach molarer Überschuss an oxidiertem gegenüber reduziertem Glutathionerhöhte die Ausbeute an löslichem D1R8C.
Temperaturabhängigkeit. Zur Bestimmung der optimalen Temperatur für dieRenaturierung erfolgte eine Inkubation bei 5 °C, 10 °C, 15 °C, 20 °C, 25 °C und 30 °C.Höhere Temperaturen steigern die Renaturierungsgeschwindigkeit, niedrigereTemperaturen verlangsamen in der Regel die Renaturierung. Aufgrund temperaturab-hängiger Aggregationsprozesse sowie potenzieller Proteaseaktivität von E. coli-Proteasen muss die optimale Renaturierungstemperatur ermittelt werden.
a) b)
c) d)
Argininkonzentration [M]0,0 0,3 0,6 0,9
Pea
kflä
che
[mA
U *
min
]
0
10
20
30
40
50
Guanidin-Konzentration [M]0,0 0,5 1,0 1,5 2,0
Pea
kflä
che
[mA
U*m
in]
0
10
20
30
40
50
60
Proteinkonzentration [µg/ml]
25 50 75 100 125 150 175 200
Pea
kflä
che
[mA
U*m
in]
0
10
20
30
40
50
60
70
80
90
100
Zeit [s]0 100 200 300 400 500 600 700
Pea
kflä
che
[mA
U *
min
]
0
10
20
30
40
50
60
70
Abb. 4-5: Renaturierungsoptimierung der CD4-Variante D1R8C: a) Einfluss der Argininkonzentration aufdie Renaturierungsausbeute: Die Renaturierung erfolgte in 100 mM Tris pH 8,5, 1 mM GSH, 5 mM GSSG,bei 10 °C, für 12 h und einer Proteinkonzentration von 100 µg/ml. b) Einfluss der Guanidinkonzentrationauf die Renaturierungsausbeute: Die Renaturierung erfolgte in 100 mM Tris pH 8,5, 1 mM GSH, 5 mMGSSG, 300 mM Arginin, bei 10 °C, für 12 h und einer Proteinkonzentration von 100 µg/ml. c) Einfluss derProteinkonzentration auf die Renaturierungsausbeute: Die Renaturierung erfolgte in 100 mM Tris pH 8,5,1 mM GSH, 5 mM GSSG, 300 mM Arginin, bei 10 °C, für 12 h. d) Renaturierungskinetik: Die Renaturierungerfolgte in 100 mM Tris pH 8,5, 1 mM GSH, 5 mM GSSG, 300 mM Arginin, bei 10 °C und einerProteinkonzentration von 100 µg/ml.

52
Die Auswertung der Temperaturabhängigkeit zeigte, dass die Inkubation bei 10 °C fürdie Bildung von löslichem D1R8C optimal ist.
Guanidinkonzentration. Die inclusion body-Solubilisate lagen in 4 M Guanidin gelöstvor. Die Verdünnung der Solubilisate im Renaturierungsansatz setzte die Guanidin-Konzentration auf 50 – 100 mM Guanidin herab.
Guanidin destabilisiert die native Struktur von Proteinen. Deshalb konnte davon aus-gegangen werden, dass die Guanidinkonzentration im Faltungsansatz einen negativenEinfluss auf die Rückfaltung hat. Da im präparativen Maßstab eine Pulsrenaturierungdurchgeführt wurde und bei dieser die ständige Zugabe des inclusion body-Solubilisatseine Erhöhung der Guanidin-Konzentration verursachte, war es notwendig, die Bildunglöslicher Proteine bei der Renaturierung in Anwesenheit von 0,5 M bis 2 M Guanidin zubestimmen.
Die Ergebnisse der Renaturierung zeigten, dass eine hohe Guanidin-Konzentrationvon 1,5 M die Renaturierung von D1R8C nur in geringem Maß beeinflusst, so dass inpräparativen Ansätzen eine Pulsrenaturierung möglich ist (Abb. 4-5b).
Proteinkonzentration. Die Konzentration des eingesetzten Proteins hat ebenfallsAuswirkung auf die Ausbeute der Faltung. Hohe Proteinkonzentrationen fördern imLaufe der Renaturierung eine Aggregation der Proteine. Dies führt zu einem Absinkender Ausbeute der Renaturierung mit steigender Proteinkonzentration.
Zur Bestimmung der optimalen Proteinkonzentration im Renaturierungsansatz erfolg-te die Verdünnung des Proteins im Renaturierungsansatz zu folgenden Endkonzen-trationen: 25 µg/ml, 50 µg/ml, 75 µg/ml, 100 µg/ml, 125 µg/ml, 150 µg/ml, 175 µg/mlund 200 µg/ml.
Mit zunehmender Proteinkonzentration erhöhte sich die Ausbeute an löslichemProtein. Auch eine Proteinkonzentration von 200 µg/ml übte keinen negativen Einflussauf die Renaturierungsausbeute von D1R8C aus (Abb. 4-5c).
Kinetik der Renaturierung. Um für eine vorgesehene Pulsrenaturierung abschätzenzu können, wie der zeitliche Abstand zwischen den einzelnen Pulsen zu bemessen ist,wurde die Kinetik der Renaturierung analysiert.
Dazu wurden Aliquots eines Renaturierungsansatzes zu bestimmten Zeitpunkten mit0,05 % TFA angesäuert. Das Absenken des pH-Wertes verhindert den Disulfidaustauschund eine weitere Renaturierung der noch nicht nativ gefalteten Proteine. Die Analyseder Proben mittels Reversed Phase Chromatographie ergab, dass die Renaturierung vonD1R8C innerhalb von 5 Minuten abgeschlossen ist (Abb. 4-5d).
Zusammenfassend konnten für die Renaturierung der CD4-Variante D1R8C folgendeBedingungen ermittelt werden:
Arginin 300 mMGSH 1 mMGSSG 5 mMpH-Wert pH 8,5Temperatur 10 °CProteinkonzentration 200 µg/mlPulsintervall 5 minAnzahl der Pulse 10
Nach der durchgeführten Pulsrenaturierung folgte die Reinigung des Konstruktes überReversed Phase- und Gelchromatographie analog dem im Kapitel 3.4.6.2 beschriebenenProtokoll (Abb. 4-3). Die Ausbeuten der Reinigungsschritte sind in der Tabelle 4-1zusammengefasst.
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 53
Die massenspektrometrische Analyse ergab ein Molekulargewicht des gereinigtenProteins von 13 885,0 Da. Die Abweichung von 305,4 Da zum theoretisch ermitteltenWert (13 579,6 Da) entspricht der Masse eines Glutathions, welches am C-Terminus überdie freie Thiolgruppe des Cysteins an die CD4-Variante D1R8C gekoppelt wurde. DieAbspaltung des Histidinfusionspeptides durch die Serinprotease Thrombin bestätigte dieN-terminale Sequenzierung des Konstrukts D1R8C nach abgeschlossener Reinigung.
Tab. 4-1: Ausbeuten der einzelnen Reinigungsschritte der CD4-Variante D1R8CReinigungsschritt AusbeuteZellen 100,0 gIsolierte inclusion bodies 35,0 gProteingehalt der solubilisierten inclusion bodies 4,8 gProteingehalt der gereinigten inclusion bodies 1,3 gD1R8C nach Reversed Phase Chromatographie 67,6 mgD1R8C nach Rekonstitution 36,4 mgD1R8C nach Gelchromatographie 27,3 mg
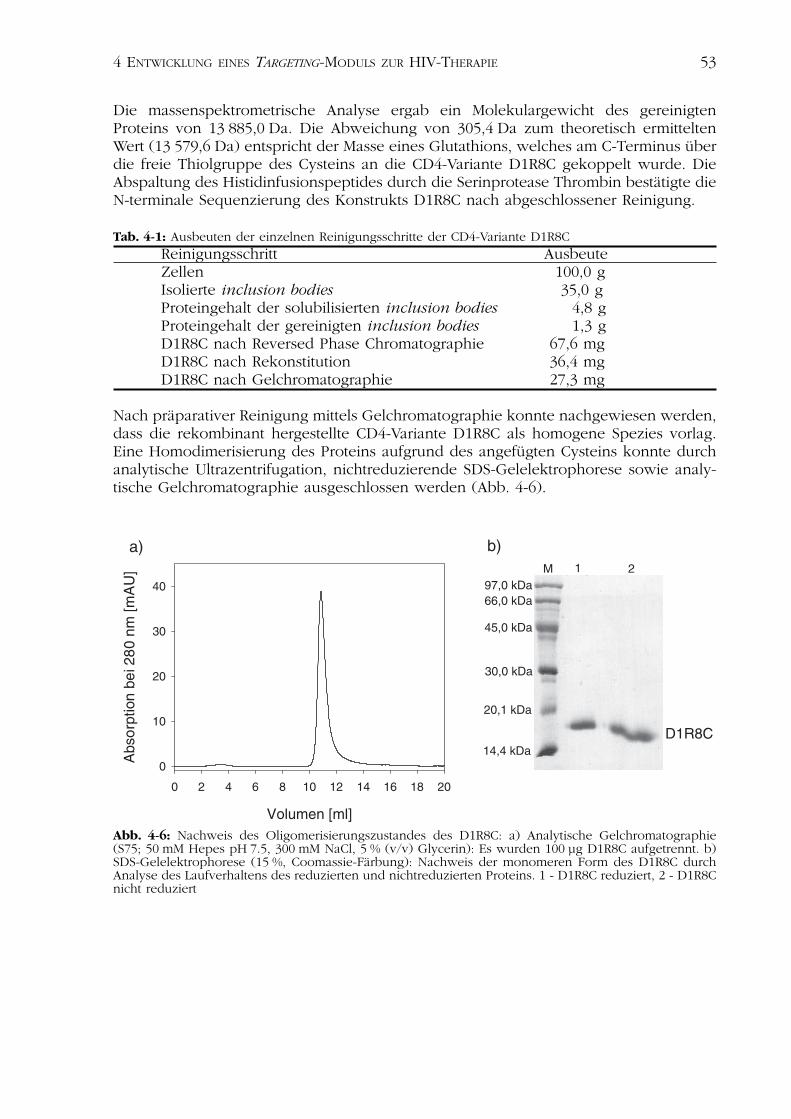
Nach präparativer Reinigung mittels Gelchromatographie konnte nachgewiesen werden,dass die rekombinant hergestellte CD4-Variante D1R8C als homogene Spezies vorlag.Eine Homodimerisierung des Proteins aufgrund des angefügten Cysteins konnte durchanalytische Ultrazentrifugation, nichtreduzierende SDS-Gelelektrophorese sowie analy-tische Gelchromatographie ausgeschlossen werden (Abb. 4-6).
20,1 kDa
14,4 kDa
a) b)
D1R8C
30,0 kDa
45,0 kDa
66,0 kDa97,0 kDa
M 1 2
Volumen [ml]
0 2 4 6 8 10 12 14 16 18 20
Abs
orpt
ion
bei 2
80
nm [m
AU
]
0
10
20
30
40
Abb. 4-6: Nachweis des Oligomerisierungszustandes des D1R8C: a) Analytische Gelchromatographie(S75; 50 mM Hepes pH 7.5, 300 mM NaCl, 5 % (v/v) Glycerin): Es wurden 100 µg D1R8C aufgetrennt. b)SDS-Gelelektrophorese (15 %, Coomassie-Färbung): Nachweis der monomeren Form des D1R8C durchAnalyse des Laufverhaltens des reduzierten und nichtreduzierten Proteins. 1 - D1R8C reduziert, 2 - D1R8Cnicht reduziert

54
4.1.2 Die CD4-Variante D1
4.1.2.1 Herstellung des Plasmids pET15b-D1
Um D1 als Targeting-Modul in Kombination mit anderen biologisch aktivenKomponenten einsetzen zu können, wurde es mit einem polyionischen Fusionspeptidhergestellt. Dieses ermöglicht die spezifische Kopplung an Proteine mit komplementärgeladenen Fusionspeptiden (Richter et al., 2001; Kapitel 1.6). Darüber hinaus kann D1aber auch als isoliertes Protein therapeutisch wirksam sein, da es bei Applikation angp120 des HI-Virus binden kann, die Oberfläche des Virus blockiert und somit eineInfektion der Zelle mit dem Virus verhindert (Kapitel 1.6; Traunecker et al., 1988;Hussey et al., 1988; Deen et al., 1988; Fischer et al., 1988).
Die für D1 codierende Sequenz wurde durch PCR aus dem Plasmid pT7T-CD4(Conzelmann & Schnell, 1994) amplifiziert. Durch diese Reaktion wurden gleichzeitigdie Restriktionsschnittstellen für NdeI und BamHI an das 5'- bzw. 3'-Ende der Sequenzangefügt. Über die eingeführten Schnittstellen erfolgte die Insertion in das Expressions-plasmid pET15b.
Die erfolgreiche Klonierung wurde durch Sequenzierung und analytischenRestriktionsverdau bestätigt.
4.1.2.2 Expression, Renaturierung und Reinigung von D1
Das Protein D1 konnte analog zur beschriebenen CD4-Variante D1R8C im Escherichiacoli Stamm BL21Codon Plus (DE3) RIL im Laufe einer 6 l Fed-batch-Fermentation expri-miert werden (Kapitel 3.3.2). Die Hochzelldichtefermentation ergab 1,115 kg Zell-material.
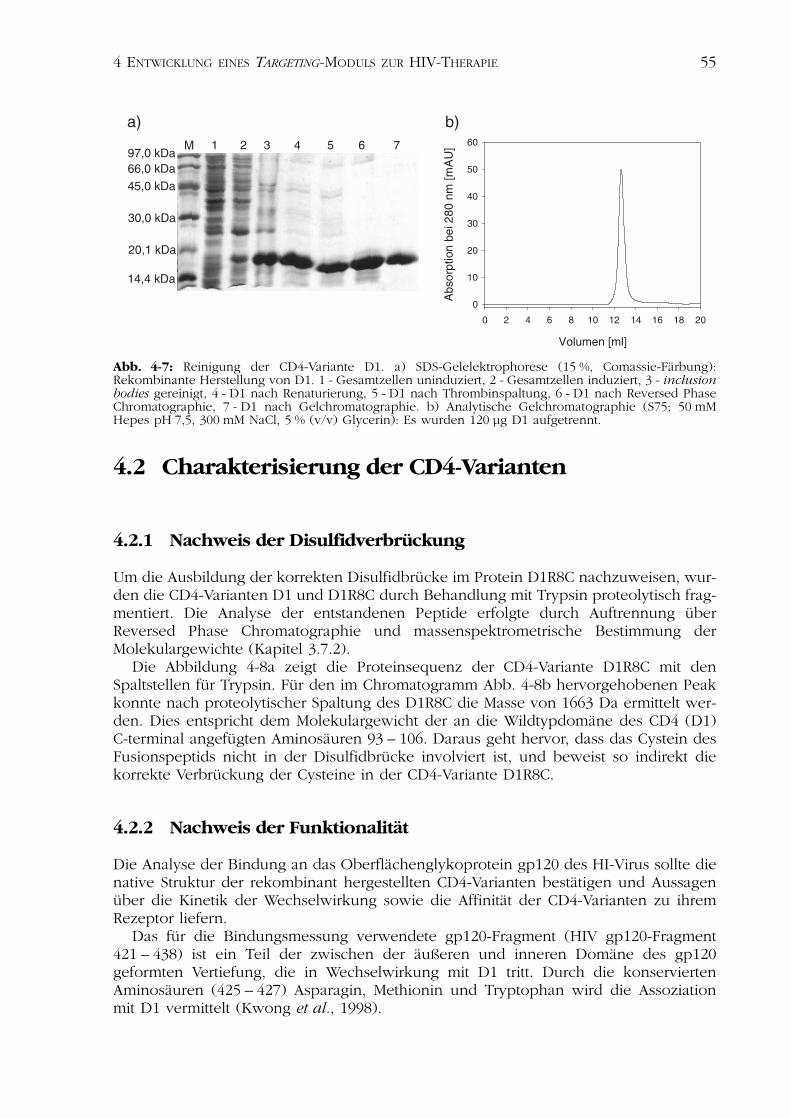
Die Isolierung, Solubilisierung, Reinigung der inclusion bodies sowie die präparativeRenaturierung erfolgten nach der für die Variante D1R8C ermittelten Methode (Kapitel4.1.1.2; Abb. 4-7a).
Die Reinigung des nativen Proteins wurde entsprechend der in Kapitel 3.4.6.2beschriebenen Anleitung durchgeführt. Aus 100 g eingesetztem Zellmaterial konnten119,2 mg reines Protein erhalten werden.
Tab. 4-2: Ausbeuten der einzelnen Reinigungsschritte der CD4-Variante D1Reinigungsschritt AusbeuteZellen 100,0 gIsolierte inclusion bodies 24,4 gProteingehalt der solubilisierten inclusion bodies 2,7 gProteingehalt der gereinigten inclusion bodies 1,8 gD1 nach Reversed Phase Chromatographie 657,2 mgD1 nach Rekonstitution 460,3 mgD1 nach Gelchromatographie 119,2 mg
Die Reinheit des Konstrukts D1 wurde mittels SDS-Gelelektrophorese, analytischerGelchromatographie (Abb. 4-7) und analytischer Reversed Phase Chromatographie(Daten nicht gezeigt) dokumentiert. Die Tabelle 4-2 zeigt die Ausbeuten der einzelnenReinigungsschritte.
Die massenspektrometrische Analyse ergab ein Molekulargewicht des gereinigtenProteins von 11 719,3 Da. Dies entspricht der theoretisch ermittelten Masse des D1.
Die Durchführung eines Ellman-Tests zeigte, dass keine freien Cysteine im Proteinvorlagen (Ellman et al., 1959; Haber, 1972).
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 55
4.2 Charakterisierung der CD4-Varianten
4.2.1 Nachweis der Disulfidverbrückung
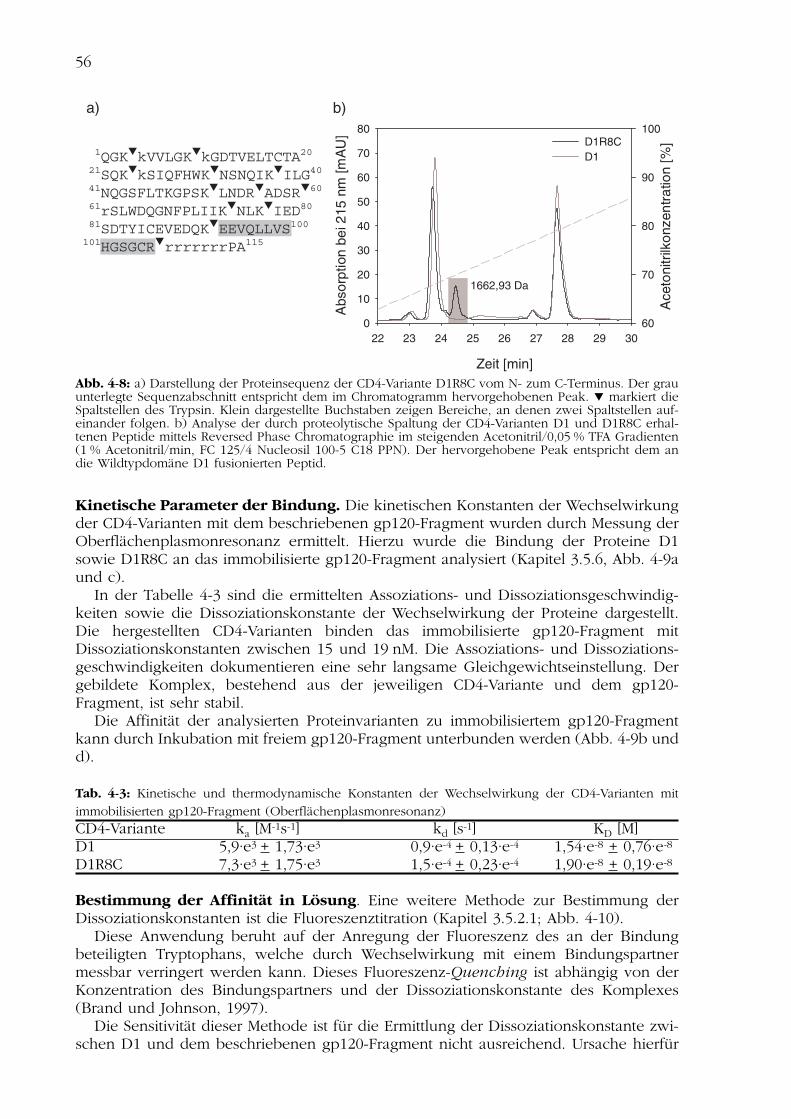
Um die Ausbildung der korrekten Disulfidbrücke im Protein D1R8C nachzuweisen, wur-den die CD4-Varianten D1 und D1R8C durch Behandlung mit Trypsin proteolytisch frag-mentiert. Die Analyse der entstandenen Peptide erfolgte durch Auftrennung überReversed Phase Chromatographie und massenspektrometrische Bestimmung derMolekulargewichte (Kapitel 3.7.2).
Die Abbildung 4-8a zeigt die Proteinsequenz der CD4-Variante D1R8C mit denSpaltstellen für Trypsin. Für den im Chromatogramm Abb. 4-8b hervorgehobenen Peakkonnte nach proteolytischer Spaltung des D1R8C die Masse von 1663 Da ermittelt wer-den. Dies entspricht dem Molekulargewicht der an die Wildtypdomäne des CD4 (D1)C-terminal angefügten Aminosäuren 93 – 106. Daraus geht hervor, dass das Cystein desFusionspeptids nicht in der Disulfidbrücke involviert ist, und beweist so indirekt diekorrekte Verbrückung der Cysteine in der CD4-Variante D1R8C.
4.2.2 Nachweis der Funktionalität
Die Analyse der Bindung an das Oberflächenglykoprotein gp120 des HI-Virus sollte dienative Struktur der rekombinant hergestellten CD4-Varianten bestätigen und Aussagenüber die Kinetik der Wechselwirkung sowie die Affinität der CD4-Varianten zu ihremRezeptor liefern.
Das für die Bindungsmessung verwendete gp120-Fragment (HIV gp120-Fragment421 – 438) ist ein Teil der zwischen der äußeren und inneren Domäne des gp120geformten Vertiefung, die in Wechselwirkung mit D1 tritt. Durch die konserviertenAminosäuren (425 – 427) Asparagin, Methionin und Tryptophan wird die Assoziationmit D1 vermittelt (Kwong et al., 1998).
a) b)
20,1 kDa
14,4 kDa
30,0 kDa
45,0 kDa
66,0 kDa97,0 kDa
M 1 2 3 4 5 6 7
Volumen [ml]
0 2 4 6 8 10 12 14 16 18 20
Abs
orpt
ion
bei 2
80 n
m [m
AU
]
0
10
20
30
40
50
60
Abb. 4-7: Reinigung der CD4-Variante D1. a) SDS-Gelelektrophorese (15 %, Comassie-Färbung):Rekombinante Herstellung von D1. 1 - Gesamtzellen uninduziert, 2 - Gesamtzellen induziert, 3 - inclusionbodies gereinigt, 4 - D1 nach Renaturierung, 5 - D1 nach Thrombinspaltung, 6 - D1 nach Reversed PhaseChromatographie, 7 - D1 nach Gelchromatographie. b) Analytische Gelchromatographie (S75; 50 mMHepes pH 7,5, 300 mM NaCl, 5 % (v/v) Glycerin): Es wurden 120 µg D1 aufgetrennt.
56
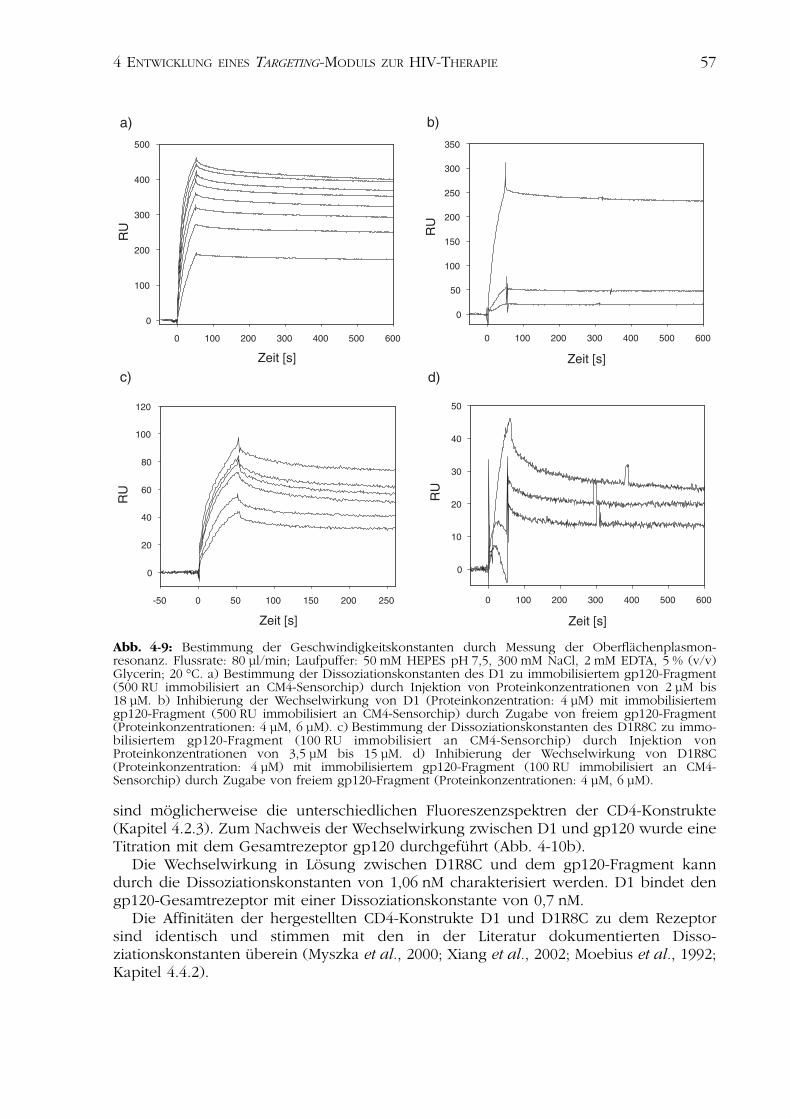
Kinetische Parameter der Bindung. Die kinetischen Konstanten der Wechselwirkungder CD4-Varianten mit dem beschriebenen gp120-Fragment wurden durch Messung derOberflächenplasmonresonanz ermittelt. Hierzu wurde die Bindung der Proteine D1sowie D1R8C an das immobilisierte gp120-Fragment analysiert (Kapitel 3.5.6, Abb. 4-9aund c).
In der Tabelle 4-3 sind die ermittelten Assoziations- und Dissoziationsgeschwindig-keiten sowie die Dissoziationskonstante der Wechselwirkung der Proteine dargestellt.Die hergestellten CD4-Varianten binden das immobilisierte gp120-Fragment mitDissoziationskonstanten zwischen 15 und 19 nM. Die Assoziations- und Dissoziations-geschwindigkeiten dokumentieren eine sehr langsame Gleichgewichtseinstellung. Dergebildete Komplex, bestehend aus der jeweiligen CD4-Variante und dem gp120-Fragment, ist sehr stabil.
Die Affinität der analysierten Proteinvarianten zu immobilisiertem gp120-Fragmentkann durch Inkubation mit freiem gp120-Fragment unterbunden werden (Abb. 4-9b undd).
Tab. 4-3: Kinetische und thermodynamische Konstanten der Wechselwirkung der CD4-Varianten mitimmobilisierten gp120-Fragment (Oberflächenplasmonresonanz)CD4-Variante ka [M-1s-1] kd [s-1] KD [M]D1 5,9·e3 + 1,73·e3 0,9·e-4 + 0,13·e-4 1,54·e-8 + 0,76·e-8
D1R8C 7,3·e3 + 1,75·e3 1,5·e-4 + 0,23·e-4 1,90·e-8 + 0,19·e-8
Bestimmung der Affinität in Lösung. Eine weitere Methode zur Bestimmung derDissoziationskonstanten ist die Fluoreszenztitration (Kapitel 3.5.2.1; Abb. 4-10).
Diese Anwendung beruht auf der Anregung der Fluoreszenz des an der Bindungbeteiligten Tryptophans, welche durch Wechselwirkung mit einem Bindungspartnermessbar verringert werden kann. Dieses Fluoreszenz-Quenching ist abhängig von derKonzentration des Bindungspartners und der Dissoziationskonstante des Komplexes(Brand und Johnson, 1997).
Die Sensitivität dieser Methode ist für die Ermittlung der Dissoziationskonstante zwi-schen D1 und dem beschriebenen gp120-Fragment nicht ausreichend. Ursache hierfür
a) b)
1QGK�kVVLGK�kGDTVELTCTA20 21SQK�kSIQFHWK�NSNQIK�ILG40 41NQGSFLTKGPSK�LNDR�ADSR�60
61rSLWDQGNFPLIIK�NLK�IED80 81SDTYICEVEDQK�EEVQLLVS100101HGSGCR�rrrrrrrPA115
Zeit [min]
22 23 24 25 26 27 28 29 30A
bsor
ptio
n be
i 215
nm
[mA
U]
0
10
20
30
40
50
60
70
80
Ace
toni
trilk
onze
ntra
tion
[%]
60
70
80
90
100D1R8CD1
1662,93 Da
Abb. 4-8: a) Darstellung der Proteinsequenz der CD4-Variante D1R8C vom N- zum C-Terminus. Der grauunterlegte Sequenzabschnitt entspricht dem im Chromatogramm hervorgehobenen Peak. markiert dieSpaltstellen des Trypsin. Klein dargestellte Buchstaben zeigen Bereiche, an denen zwei Spaltstellen auf-einander folgen. b) Analyse der durch proteolytische Spaltung der CD4-Varianten D1 und D1R8C erhal-tenen Peptide mittels Reversed Phase Chromatographie im steigenden Acetonitril/0,05 % TFA Gradienten(1 % Acetonitril/min, FC 125/4 Nucleosil 100-5 C18 PPN). Der hervorgehobene Peak entspricht dem andie Wildtypdomäne D1 fusionierten Peptid.
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 57
sind möglicherweise die unterschiedlichen Fluoreszenzspektren der CD4-Konstrukte(Kapitel 4.2.3). Zum Nachweis der Wechselwirkung zwischen D1 und gp120 wurde eineTitration mit dem Gesamtrezeptor gp120 durchgeführt (Abb. 4-10b).
Die Wechselwirkung in Lösung zwischen D1R8C und dem gp120-Fragment kanndurch die Dissoziationskonstanten von 1,06 nM charakterisiert werden. D1 bindet dengp120-Gesamtrezeptor mit einer Dissoziationskonstante von 0,7 nM.
Die Affinitäten der hergestellten CD4-Konstrukte D1 und D1R8C zu dem Rezeptorsind identisch und stimmen mit den in der Literatur dokumentierten Disso-ziationskonstanten überein (Myszka et al., 2000; Xiang et al., 2002; Moebius et al., 1992;Kapitel 4.4.2).
a) b)
c) d)
Zeit [s]
-50 0 50 100 150 200 250
RU
0
20
40
60
80
100
120
Zeit [s]
0 100 200 300 400 500 600
RU
0
100
200
300
400
500
Zeit [s]
0 100 200 300 400 500 600
RU
0
50
100
150
200
250
300
350
Zeit [s]
0 100 200 300 400 500 600
RU
0
10
20
30
40
50
Abb. 4-9: Bestimmung der Geschwindigkeitskonstanten durch Messung der Oberflächenplasmon-resonanz. Flussrate: 80 µl/min; Laufpuffer: 50 mM HEPES pH 7,5, 300 mM NaCl, 2 mM EDTA, 5 % (v/v)Glycerin; 20 °C. a) Bestimmung der Dissoziationskonstanten des D1 zu immobilisiertem gp120-Fragment(500 RU immobilisiert an CM4-Sensorchip) durch Injektion von Proteinkonzentrationen von 2 µM bis18 µM. b) Inhibierung der Wechselwirkung von D1 (Proteinkonzentration: 4 µM) mit immobilisiertemgp120-Fragment (500 RU immobilisiert an CM4-Sensorchip) durch Zugabe von freiem gp120-Fragment(Proteinkonzentrationen: 4 µM, 6 µM). c) Bestimmung der Dissoziationskonstanten des D1R8C zu immo-bilisiertem gp120-Fragment (100 RU immobilisiert an CM4-Sensorchip) durch Injektion vonProteinkonzentrationen von 3,5 µM bis 15 µM. d) Inhibierung der Wechselwirkung von D1R8C(Proteinkonzentration: 4 µM) mit immobilisiertem gp120-Fragment (100 RU immobilisiert an CM4-Sensorchip) durch Zugabe von freiem gp120-Fragment (Proteinkonzentrationen: 4 µM, 6 µM).
58
4.2.3 Spektroskopische Charakterisierung
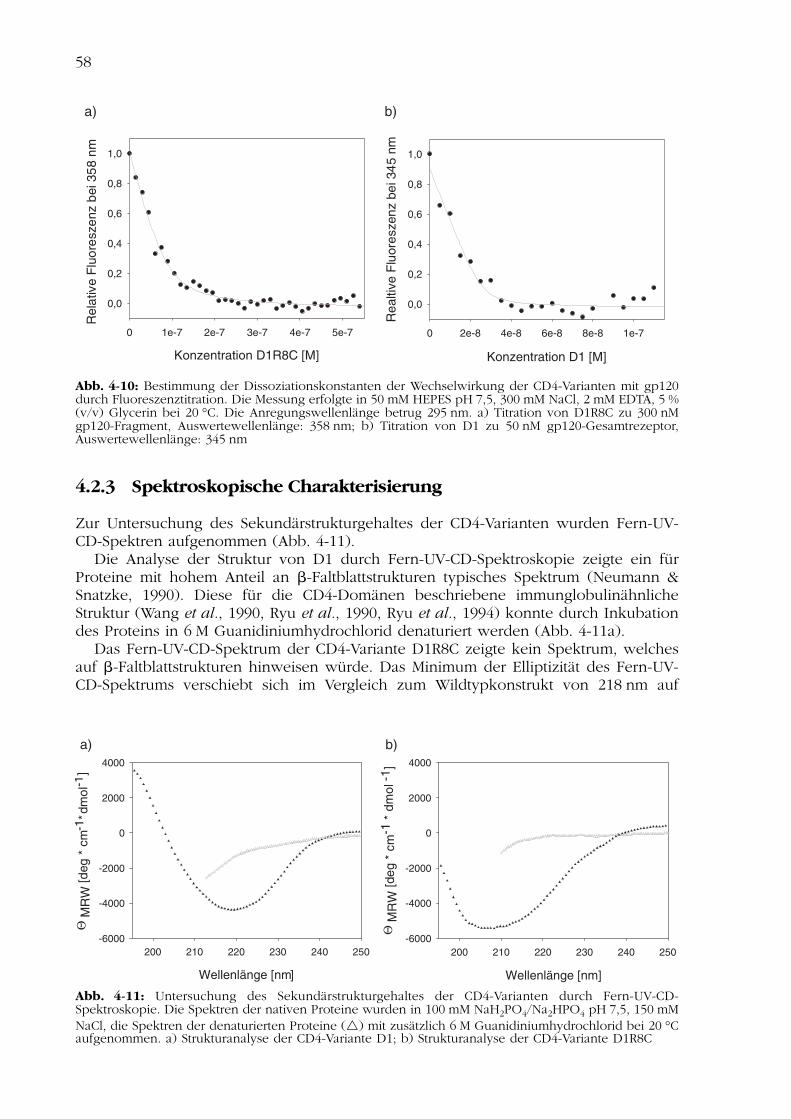
Zur Untersuchung des Sekundärstrukturgehaltes der CD4-Varianten wurden Fern-UV-CD-Spektren aufgenommen (Abb. 4-11).
Die Analyse der Struktur von D1 durch Fern-UV-CD-Spektroskopie zeigte ein fürProteine mit hohem Anteil an �-Faltblattstrukturen typisches Spektrum (Neumann &Snatzke, 1990). Diese für die CD4-Domänen beschriebene immunglobulinähnlicheStruktur (Wang et al., 1990, Ryu et al., 1990, Ryu et al., 1994) konnte durch Inkubationdes Proteins in 6 M Guanidiniumhydrochlorid denaturiert werden (Abb. 4-11a).
Das Fern-UV-CD-Spektrum der CD4-Variante D1R8C zeigte kein Spektrum, welchesauf �-Faltblattstrukturen hinweisen würde. Das Minimum der Elliptizität des Fern-UV-CD-Spektrums verschiebt sich im Vergleich zum Wildtypkonstrukt von 218 nm auf
a) b)
Wellenlänge [nm]
200 210 220 230 240 250
Θ M
RW
[deg
* c
m-1
* d
mol
-1 ]
-6000
-4000
-2000
0
2000
4000
Wellenlänge [nm]
200 210 220 230 240 250
Θ M
RW
[deg
* c
m* d
mol
-1]
-6000
-4000
-2000
0
2000
4000
-1
Abb. 4-11: Untersuchung des Sekundärstrukturgehaltes der CD4-Varianten durch Fern-UV-CD-Spektroskopie. Die Spektren der nativen Proteine wurden in 100 mM NaH2PO4/Na2HPO4 pH 7,5, 150 mMNaCl, die Spektren der denaturierten Proteine ( ) mit zusätzlich 6 M Guanidiniumhydrochlorid bei 20 °Caufgenommen. a) Strukturanalyse der CD4-Variante D1; b) Strukturanalyse der CD4-Variante D1R8C
a) b)
Konzentration D1R8C [M]
0 1e-7 2e-7 3e-7 4e-7 5e-7
Rel
ativ
e F
luor
esze
nz b
ei 3
58
nm
0,0
0,2
0,4
0,6
0,8
1,0
Konzentration D1 [M]
0 2e-8 4e-8 6e-8 8e-8 1e-7
Rea
ltive
Flu
ores
zenz
bei
34
5 nm
0,0
0,2
0,4
0,6
0,8
1,0
Abb. 4-10: Bestimmung der Dissoziationskonstanten der Wechselwirkung der CD4-Varianten mit gp120durch Fluoreszenztitration. Die Messung erfolgte in 50 mM HEPES pH 7,5, 300 mM NaCl, 2 mM EDTA, 5 %(v/v) Glycerin bei 20 °C. Die Anregungswellenlänge betrug 295 nm. a) Titration von D1R8C zu 300 nMgp120-Fragment, Auswertewellenlänge: 358 nm; b) Titration von D1 zu 50 nM gp120-Gesamtrezeptor,Auswertewellenlänge: 345 nm
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 59
208 nm. Dennoch konnte ein hoher Gehalt an Sekundärstruktureinheiten für diesesProtein festgestellt werden, da das Spektrum durch Inkubation von D1R8C in 6 MGuanidiniumhydrochlorid dramatisch verändert wird (Abb. 4-11b).
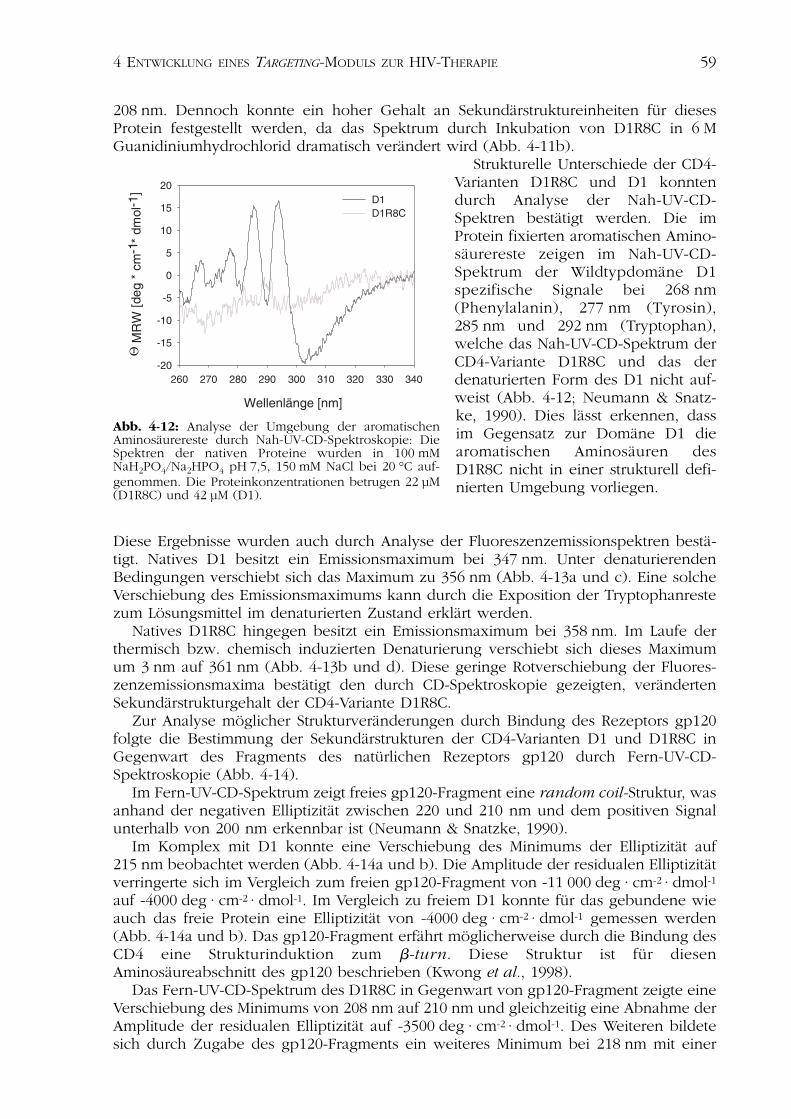
Strukturelle Unterschiede der CD4-Varianten D1R8C und D1 konntendurch Analyse der Nah-UV-CD-Spektren bestätigt werden. Die imProtein fixierten aromatischen Amino-säurereste zeigen im Nah-UV-CD-Spektrum der Wildtypdomäne D1spezifische Signale bei 268 nm(Phenylalanin), 277 nm (Tyrosin),285 nm und 292 nm (Tryptophan),welche das Nah-UV-CD-Spektrum derCD4-Variante D1R8C und das derdenaturierten Form des D1 nicht auf-weist (Abb. 4-12; Neumann & Snatz-ke, 1990). Dies lässt erkennen, dassim Gegensatz zur Domäne D1 diearomatischen Aminosäuren desD1R8C nicht in einer strukturell defi-nierten Umgebung vorliegen.
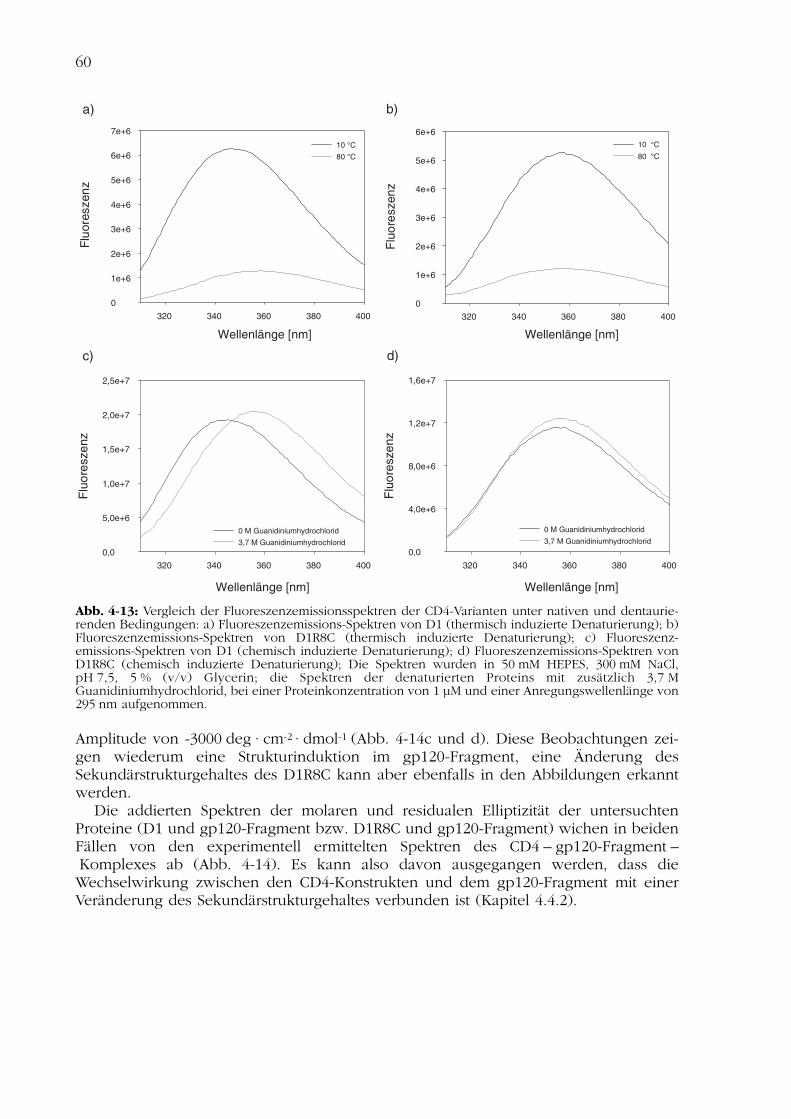
Diese Ergebnisse wurden auch durch Analyse der Fluoreszenzemissionspektren bestä-tigt. Natives D1 besitzt ein Emissionsmaximum bei 347 nm. Unter denaturierendenBedingungen verschiebt sich das Maximum zu 356 nm (Abb. 4-13a und c). Eine solcheVerschiebung des Emissionsmaximums kann durch die Exposition der Tryptophanrestezum Lösungsmittel im denaturierten Zustand erklärt werden.
Natives D1R8C hingegen besitzt ein Emissionsmaximum bei 358 nm. Im Laufe derthermisch bzw. chemisch induzierten Denaturierung verschiebt sich dieses Maximumum 3 nm auf 361 nm (Abb. 4-13b und d). Diese geringe Rotverschiebung der Fluores-zenzemissionsmaxima bestätigt den durch CD-Spektroskopie gezeigten, verändertenSekundärstrukturgehalt der CD4-Variante D1R8C.
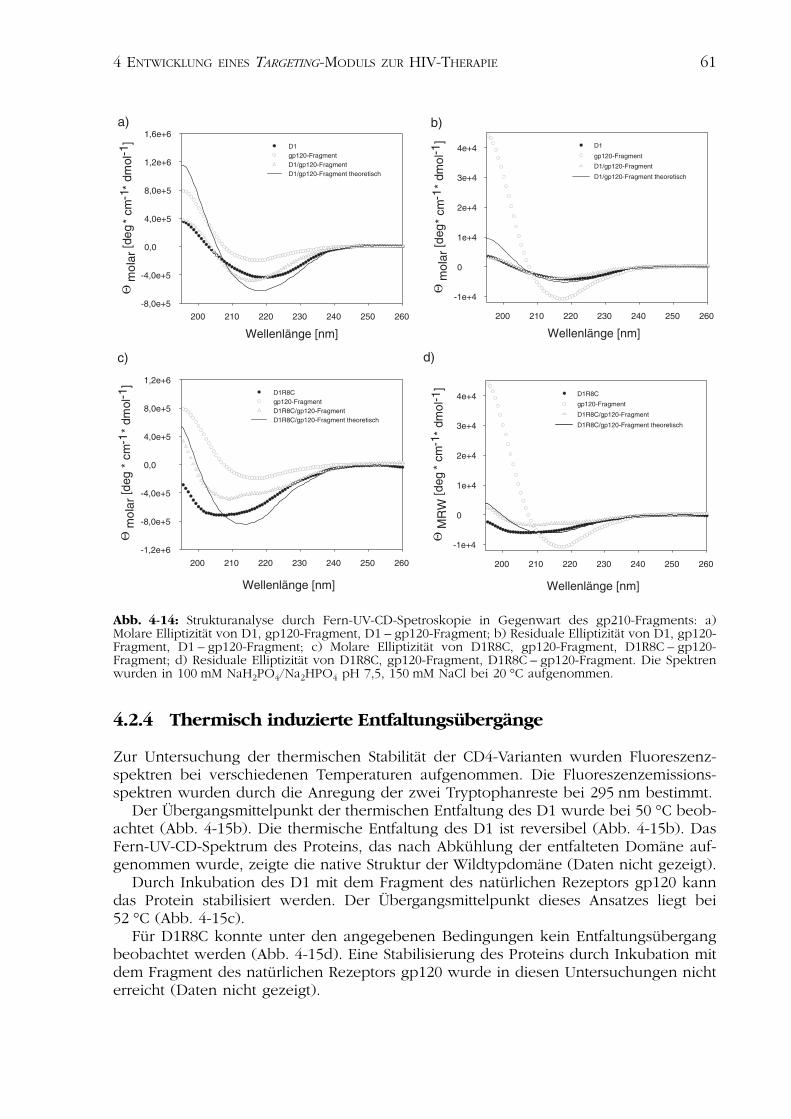
Zur Analyse möglicher Strukturveränderungen durch Bindung des Rezeptors gp120folgte die Bestimmung der Sekundärstrukturen der CD4-Varianten D1 und D1R8C inGegenwart des Fragments des natürlichen Rezeptors gp120 durch Fern-UV-CD-Spektroskopie (Abb. 4-14).
Im Fern-UV-CD-Spektrum zeigt freies gp120-Fragment eine random coil-Struktur, wasanhand der negativen Elliptizität zwischen 220 und 210 nm und dem positiven Signalunterhalb von 200 nm erkennbar ist (Neumann & Snatzke, 1990).
Im Komplex mit D1 konnte eine Verschiebung des Minimums der Elliptizität auf215 nm beobachtet werden (Abb. 4-14a und b). Die Amplitude der residualen Elliptizitätverringerte sich im Vergleich zum freien gp120-Fragment von -11 000 deg · cm-2 · dmol-1auf -4000 deg · cm-2 · dmol-1. Im Vergleich zu freiem D1 konnte für das gebundene wieauch das freie Protein eine Elliptizität von -4000 deg · cm-2 · dmol-1 gemessen werden(Abb. 4-14a und b). Das gp120-Fragment erfährt möglicherweise durch die Bindung desCD4 eine Strukturinduktion zum �-turn. Diese Struktur ist für diesenAminosäureabschnitt des gp120 beschrieben (Kwong et al., 1998).
Das Fern-UV-CD-Spektrum des D1R8C in Gegenwart von gp120-Fragment zeigte eineVerschiebung des Minimums von 208 nm auf 210 nm und gleichzeitig eine Abnahme derAmplitude der residualen Elliptizität auf -3500 deg · cm-2 · dmol-1. Des Weiteren bildetesich durch Zugabe des gp120-Fragments ein weiteres Minimum bei 218 nm mit einer
Wellenlänge [nm]
260 270 280 290 300 310 320 330 340
Θ M
RW
[deg
* c
m*
dmol
-1]
-20
-15
-10
-5
0
5
10
15
20D1D1R8C
-1
Abb. 4-12: Analyse der Umgebung der aromatischenAminosäurereste durch Nah-UV-CD-Spektroskopie: DieSpektren der nativen Proteine wurden in 100 mMNaH2PO4/Na2HPO4 pH 7,5, 150 mM NaCl bei 20 °C auf-genommen. Die Proteinkonzentrationen betrugen 22 µM(D1R8C) und 42 µM (D1).
60
Amplitude von -3000 deg · cm-2 · dmol-1 (Abb. 4-14c und d). Diese Beobachtungen zei-gen wiederum eine Strukturinduktion im gp120-Fragment, eine Änderung desSekundärstrukturgehaltes des D1R8C kann aber ebenfalls in den Abbildungen erkanntwerden.
Die addierten Spektren der molaren und residualen Elliptizität der untersuchtenProteine (D1 und gp120-Fragment bzw. D1R8C und gp120-Fragment) wichen in beidenFällen von den experimentell ermittelten Spektren des CD4 – gp120-Fragment –Komplexes ab (Abb. 4-14). Es kann also davon ausgegangen werden, dass dieWechselwirkung zwischen den CD4-Konstrukten und dem gp120-Fragment mit einerVeränderung des Sekundärstrukturgehaltes verbunden ist (Kapitel 4.4.2).
Wellenlänge [nm]
320 340 360 380 400
Flu
ores
zenz
0
1e+6
2e+6
3e+6
4e+6
5e+6
6e+6
7e+6
10 °C
80 °C
Wellenlänge [nm]
320 340 360 380 400
Flu
ores
zenz
0
1e+6
2e+6
3e+6
4e+6
5e+6
6e+610 °C
80 °C
Wellenlänge [nm]
320 340 360 380 400
Flu
ores
zenz
0,0
5,0e+6
1,0e+7
1,5e+7
2,0e+7
2,5e+7
0 M Guanidiniumhydrochlorid
3,7 M Guanidiniumhydrochlorid
Wellenlänge [nm]
320 340 360 380 400
Flu
ores
zenz
0,0
4,0e+6
8,0e+6
1,2e+7
1,6e+7
0 M Guanidiniumhydrochlorid
3,7 M Guanidiniumhydrochlorid
a) b)
d)c)
Abb. 4-13: Vergleich der Fluoreszenzemissionsspektren der CD4-Varianten unter nativen und dentaurie-renden Bedingungen: a) Fluoreszenzemissions-Spektren von D1 (thermisch induzierte Denaturierung); b)Fluoreszenzemissions-Spektren von D1R8C (thermisch induzierte Denaturierung); c) Fluoreszenz-emissions-Spektren von D1 (chemisch induzierte Denaturierung); d) Fluoreszenzemissions-Spektren vonD1R8C (chemisch induzierte Denaturierung); Die Spektren wurden in 50 mM HEPES, 300 mM NaCl,pH 7,5, 5 % (v/v) Glycerin; die Spektren der denaturierten Proteins mit zusätzlich 3,7 MGuanidiniumhydrochlorid, bei einer Proteinkonzentration von 1 µM und einer Anregungswellenlänge von295 nm aufgenommen.
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 61
4.2.4 Thermisch induzierte Entfaltungsübergänge
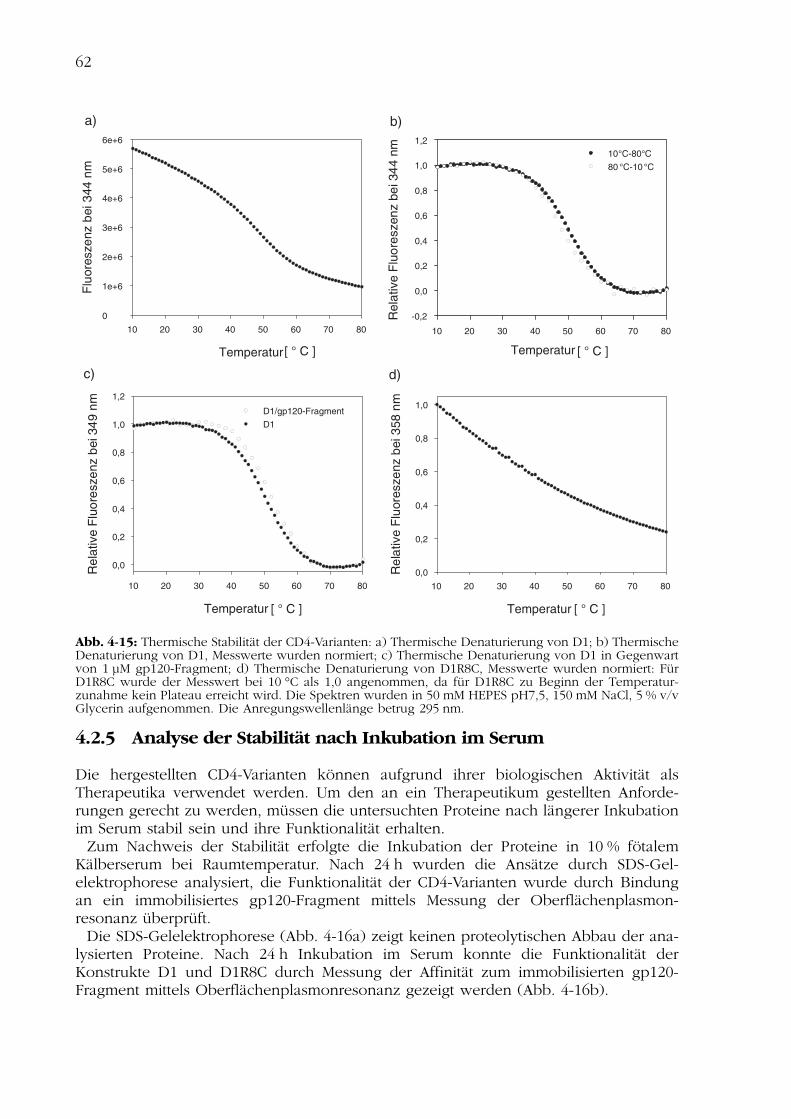
Zur Untersuchung der thermischen Stabilität der CD4-Varianten wurden Fluoreszenz-spektren bei verschiedenen Temperaturen aufgenommen. Die Fluoreszenzemissions-spektren wurden durch die Anregung der zwei Tryptophanreste bei 295 nm bestimmt.
Der Übergangsmittelpunkt der thermischen Entfaltung des D1 wurde bei 50 °C beob-achtet (Abb. 4-15b). Die thermische Entfaltung des D1 ist reversibel (Abb. 4-15b). DasFern-UV-CD-Spektrum des Proteins, das nach Abkühlung der entfalteten Domäne auf-genommen wurde, zeigte die native Struktur der Wildtypdomäne (Daten nicht gezeigt).
Durch Inkubation des D1 mit dem Fragment des natürlichen Rezeptors gp120 kanndas Protein stabilisiert werden. Der Übergangsmittelpunkt dieses Ansatzes liegt bei52 °C (Abb. 4-15c).
Für D1R8C konnte unter den angegebenen Bedingungen kein Entfaltungsübergangbeobachtet werden (Abb. 4-15d). Eine Stabilisierung des Proteins durch Inkubation mitdem Fragment des natürlichen Rezeptors gp120 wurde in diesen Untersuchungen nichterreicht (Daten nicht gezeigt).
Wellenlänge [nm]
200 210 220 230 240 250 260
Θ m
olar
[deg
* dm
ol-1
]
-8,0e+5
-4,0e+5
0,0
4,0e+5
8,0e+5
1,2e+6
1,6e+6D1gp120-FragmentD1/gp120-FragmentD1/gp120-Fragment theoretisch
Wellenlänge [nm]
200 210 220 230 240 250 260
Θ m
olar
[deg
* c
m*
dmol
-1]
-1,2e+6
-8,0e+5
-4,0e+5
0,0
4,0e+5
8,0e+5
1,2e+6D1R8Cgp120-FragmentD1R8C/gp120-FragmentD1R8C/gp120-Fragment theoretisch
Wellenlänge [nm]
200 210 220 230 240 250 260
Θ M
RW
[deg
* dm
ol-1
]
-1e+4
0
1e+4
2e+4
3e+4
4e+4 D1R8C
gp120-Fragment
D1R8C/gp120-Fragment
D1R8C/gp120-Fragment theoretisch
Wellenlänge [nm]
200 210 220 230 240 250 260
Θ m
olar
[deg
* dm
ol-1
]
-1e+4
0
1e+4
2e+4
3e+4
4e+4 D1
gp120-Fragment
D1/gp120-Fragment
D1/gp120-Fragment theoretisch
-1 *
cm
-1
* c
m-1
* c
m-1
a) b)
c) d)
Abb. 4-14: Strukturanalyse durch Fern-UV-CD-Spetroskopie in Gegenwart des gp210-Fragments: a)Molare Elliptizität von D1, gp120-Fragment, D1 – gp120-Fragment; b) Residuale Elliptizität von D1, gp120-Fragment, D1 – gp120-Fragment; c) Molare Elliptizität von D1R8C, gp120-Fragment, D1R8C – gp120-Fragment; d) Residuale Elliptizität von D1R8C, gp120-Fragment, D1R8C – gp120-Fragment. Die Spektrenwurden in 100 mM NaH2PO4/Na2HPO4 pH 7,5, 150 mM NaCl bei 20 °C aufgenommen.
62
4.2.5 Analyse der Stabilität nach Inkubation im Serum
Die hergestellten CD4-Varianten können aufgrund ihrer biologischen Aktivität alsTherapeutika verwendet werden. Um den an ein Therapeutikum gestellten Anforde-rungen gerecht zu werden, müssen die untersuchten Proteine nach längerer Inkubationim Serum stabil sein und ihre Funktionalität erhalten.
Zum Nachweis der Stabilität erfolgte die Inkubation der Proteine in 10 % fötalemKälberserum bei Raumtemperatur. Nach 24 h wurden die Ansätze durch SDS-Gel-elektrophorese analysiert, die Funktionalität der CD4-Varianten wurde durch Bindungan ein immobilisiertes gp120-Fragment mittels Messung der Oberflächenplasmon-resonanz überprüft.
Die SDS-Gelelektrophorese (Abb. 4-16a) zeigt keinen proteolytischen Abbau der ana-lysierten Proteine. Nach 24 h Inkubation im Serum konnte die Funktionalität derKonstrukte D1 und D1R8C durch Messung der Affinität zum immobilisierten gp120-Fragment mittels Oberflächenplasmonresonanz gezeigt werden (Abb. 4-16b).
Temperatur [ ° C ]
10 20 30 40 50 60 70 80
Flu
ores
zenz
bei
344
nm
0
1e+6
2e+6
3e+6
4e+6
5e+6
6e+6
Temperatur
10 20 30 40 50 60 70 80
Rel
ativ
e F
luor
esze
nz b
ei 3
44
nm
-0,2
0,0
0,2
0,4
0,6
0,8
1,0
1,210°C-80°C
80 °C-10°C
Temperatur
10 20 30 40 50 60 70 80
Rel
ativ
e F
luor
esze
nz b
ei 3
49
nm
0,0
0,2
0,4
0,6
0,8
1,0
1,2
D1/gp120-Fragment
D1
Temperatur
10 20 30 40 50 60 70 80
Rel
ativ
e F
luor
esze
nz b
ei 3
58 n
m
0,0
0,2
0,4
0,6
0,8
1,0
a) b)
[ ° C ]
[ ° C ] [ ° C ]
c) d)
Abb. 4-15: Thermische Stabilität der CD4-Varianten: a) Thermische Denaturierung von D1; b) ThermischeDenaturierung von D1, Messwerte wurden normiert; c) Thermische Denaturierung von D1 in Gegenwartvon 1 µM gp120-Fragment; d) Thermische Denaturierung von D1R8C, Messwerte wurden normiert: FürD1R8C wurde der Messwert bei 10 °C als 1,0 angenommen, da für D1R8C zu Beginn der Temperatur-zunahme kein Plateau erreicht wird. Die Spektren wurden in 50 mM HEPES pH7,5, 150 mM NaCl, 5 % v/vGlycerin aufgenommen. Die Anregungswellenlänge betrug 295 nm.
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 63
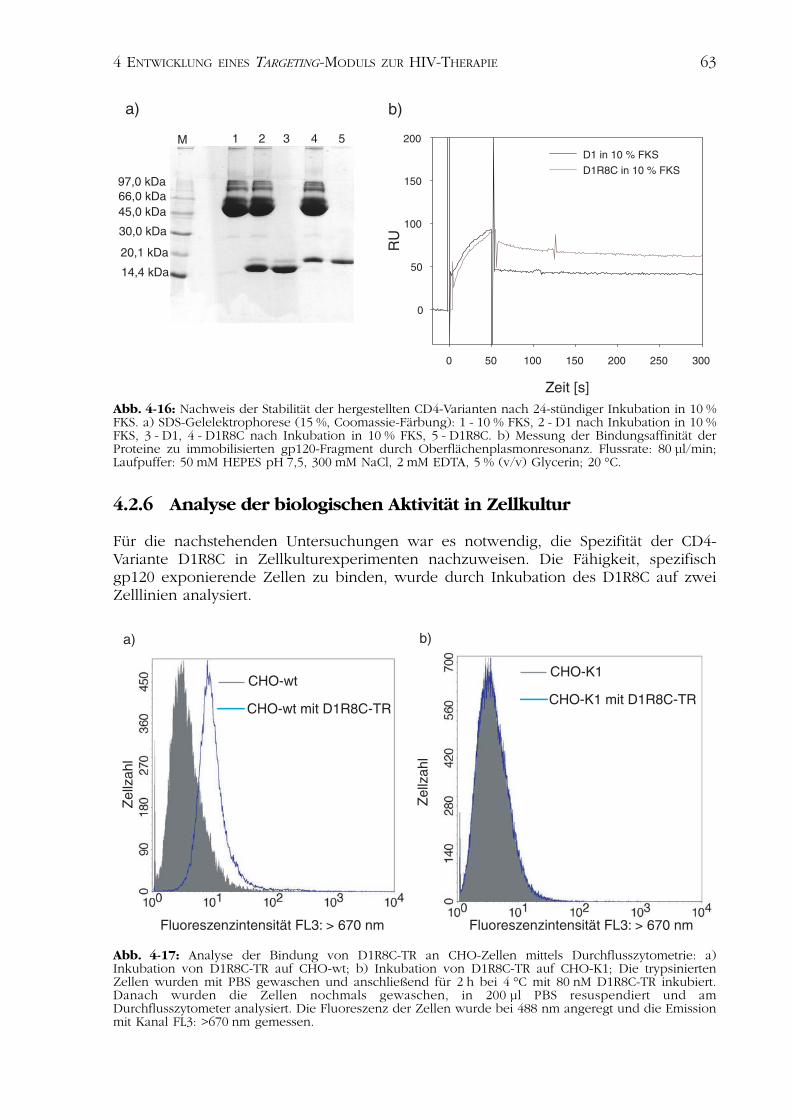
4.2.6 Analyse der biologischen Aktivität in Zellkultur
Für die nachstehenden Untersuchungen war es notwendig, die Spezifität der CD4-Variante D1R8C in Zellkulturexperimenten nachzuweisen. Die Fähigkeit, spezifischgp120 exponierende Zellen zu binden, wurde durch Inkubation des D1R8C auf zweiZelllinien analysiert.
Abb. 4-16: Nachweis der Stabilität der hergestellten CD4-Varianten nach 24-stündiger Inkubation in 10 %FKS. a) SDS-Gelelektrophorese (15 %, Coomassie-Färbung): 1 - 10 % FKS, 2 - D1 nach Inkubation in 10 %FKS, 3 - D1, 4 - D1R8C nach Inkubation in 10 % FKS, 5 - D1R8C. b) Messung der Bindungsaffinität derProteine zu immobilisierten gp120-Fragment durch Oberflächenplasmonresonanz. Flussrate: 80 µl/min;Laufpuffer: 50 mM HEPES pH 7,5, 300 mM NaCl, 2 mM EDTA, 5 % (v/v) Glycerin; 20 °C.
a) b)
Zeit [s]
0 50 100 150 200 250 300
RU
0
50
100
150
200
D1 in 10 % FKS
D1R8C in 10 % FKS
20,1 kDa
14,4 kDa
30,0 kDa
45,0 kDa66,0 kDa97,0 kDa
M 1 2 3 4 5
Fluoreszenzintensität FL3: > 670 nm Fluoreszenzintensität FL3: > 670 nm
Zel
lzah
l
Zel
lzah
l
a) b)
CHO-wt
CHO-wt mit D1R8C-TR
CHO-K1
CHO-K1 mit D1R8C-TR
Abb. 4-17: Analyse der Bindung von D1R8C-TR an CHO-Zellen mittels Durchflusszytometrie: a)Inkubation von D1R8C-TR auf CHO-wt; b) Inkubation von D1R8C-TR auf CHO-K1; Die trypsiniertenZellen wurden mit PBS gewaschen und anschließend für 2 h bei 4 °C mit 80 nM D1R8C-TR inkubiert.Danach wurden die Zellen nochmals gewaschen, in 200 µl PBS resuspendiert und amDurchflusszytometer analysiert. Die Fluoreszenz der Zellen wurde bei 488 nm angeregt und die Emissionmit Kanal FL3: >670 nm gemessen.

64
CHO-wt ist eine CHO-K1-Zelllinie, welche den HIV-1 Glykoproteinrezeptor gp120 aufder Oberfläche exponiert (Weiss & White, 1993). Die Zelllinie CHO-K1 wurde in denfolgenden Experimenten als Kontrollzelllinie verwendet.
Der Fluoreszenzfarbstoff TexasRed Maleimid wurde über das freie Cystein desFusionspeptids kovalent an D1R8C gebunden (Kapitel 3.4.8). Nach Inkubation des mar-kierten Proteins auf den betrachteten Zelllinien CHO-wt und CHO-K1 wurde dieFähigkeit des D1R8C, spezifisch gp120 positive Zellen zu markieren, mittels Durchfluss-zytometrie nachgewiesen (Abb. 4-17; Kapitel 3.6.4).
Diese Methode zeigte nach Inkubation mit dem fluoreszenzmarkierten ProteinD1R8C eine Verschiebung der Population der Zelllinie CHO-wt zu höheren Fluo-reszenzen (Abb. 4-17a). D1R8C ist in der Lage, aufgrund des auf den Zellen exponier-ten Glykoproteins gp120 spezifisch an diese Zelllinie zu binden.
Eine Assoziation des fluoreszenzmarkierten D1R8C an die Zelllinie CHO-K1 konntehingegen nicht beobachtet werden (Abb. 4-17b).
Für die Etablierung des im Anschluss beschriebenen Modellsystems zur HIV-Therapiewurde die CHO-wt-Zelllinie als positive, die CHO-K1-Zelllinie als Kontrollzellliniebetrachtet.
4.3 Eliminierung HIV-infizierter Zellen
Ein Ansatz zur HIV-Therapie ist die Eliminierung der HIV-infizierten Zellen, um dieVermehrung der Viren zu verhindern.
Für ein solches Modell sind die Funktionen von zwei verschiedenen Proteinen rele-vant. Es wurde ein rezeptorspezifisches Zytotoxin entwickelt, welches die zelltypspezi-fische Adressierung und die Funktion zur Eliminierung der betroffenen Zellen gewähr-leisten sollte.
Hierfür wurde das System der Kopplung polyionischer Proteine an ein Derivat desPseudomonas Exotoxin E8C-PE38 (Kleinschmidt et al., 2003) verwendet. DurchKonjugation der CD4-Variante D1R8C an E8C-PE38 sollte ein chimeres Protein gewon-
nen werden, welches spezi-fisch an HIV-infizierte Zellenbindet und die Replikationder HI-Viren verhindert (Abb.4-18; Tsubota et al., 1990).
Die Herstellung und biolo-gische Aktivität bifunktionel-ler Proteine durch Disulfid-verbrückung konnte bereitsdurch Fusion eines tumorspe-zifischen Fv-Antikörpers mitdem Derivat des Pseudomo-nas Exotoxins E8C-PE38gezeigt werden (Kleinschmidtet al., 2003).
4.3.1 Herstellung des rezeptorspezifischen Zytotoxins D1-PE
Optimierung der Kopplung von D1R8C an E8C-PE38. Die in der Literatur angege-benen Bedingungen zur kovalenten Assoziation von Proteinen mittels polyionischerFusionspeptide führten in ersten Kopplungsstudien zu einer vermehrten Aggregationdes Proteins D1R8C, da durch Inkubation in den angegebenen Bedingungen die intra-
Abb. 4-18: Schematische Darstellung von D1R8C und E8C-PE38 alsKopplungsprodukt D1-PE.
IIIII
CEEEEEEEEPSG
CRRRRRRRRPA
G
SG D1Ib
E8C-PE38 D1R8C
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 65
a)
b)
c)
D1R8C
D1R8C
D1R8C
D1-PE
D1-PE
D1-PE
E8C-PE38
E8C-PE38
E8C-PE38
(E8C-PE38)2
(E8C-PE38)2
(E8C-PE38)2
7 81 2 3 4 5 6 109
7 81 2 3 4 5 6M 9
1 2 3 4 5 6M
20,1 kDa
14,4 kDa
30,0 kDa
45,0 kDa
66,0 kDa97,0 kDa
20,1 kDa
14,4 kDa
30,0 kDa
45,0 kDa
66,0 kDa97,0 kDa
Abb. 4-19: Optimierung der Kopplungsbedingungen für die Herstellung des rezeptorspezifischenZytotoxins D1-PE: a) Optimierung der GSH/GSSG-Konzentration: 1 - D1R8C, 2 - E8C-PE38, 3 - 5 µMGSH/20 µM GSSG, 4 - 10 µM GSH/40 µM GSSG, 5 - 50 µM GSH/200 µM GSSG, 6 - 75 µM GSH/300 µMGSSG; 4 µM E8C-PE38 und 8 µM D1R8C wurden 20 h in 50 mM Tris pH 8,0, 2 mM EDTA bei 10 °C inku-biert und anschließend 10 min bei 13 000 rpm zentrifugiert. Die Überstände wurden in einer SDS-Gelelektrophorese (15 %, nichtreduzierend, Comassie-Färbung) analysiert.b) Optimierung der Proteinkonzentration: 1 - 8 µM E8C-PE/4 µM D1R8C 1 h inkubiert, 2 - 8 µM E8C-PE/4 µM D1R8C 1 h inkubiert und zentrifugiert, 3 - 8 µM E8C-PE/4 µM D1R8C 20 h inkubiert, 4 - 8 µME8C-PE/4 µM D1R8C 20 h inkubiert und zentrifugiert, 5 - 4 µM E8C-PE/8 µM D1R8C 1 h inkubiert, 6 - 4 µME8C-PE/8 µM D1R8C 1 h inkubiert und zentrifugiert, 7 - 4 µM E8C-PE/8 µM D1R8C 20 h inkubiert, 8 - 4 µME8C-PE/8 µM D1R8C 20 h inkubiert und zentrifugiert, 9 - D1R8C, 10 - E8C-PE38; Die Proteine wurden in50 mM Tris pH 8,0, 2 mM EDTA, 10 µM GSH, 40 µM GSSG bei 10 °C inkubiert und ein Teil anschließend10 min bei 13 000 rpm zentrifugiert. Die Überstände der Zentrifugation und die unzentrifugierten Ansätzewurden in einer SDS-Gelelektrophorese (15 %, nichtreduzierend, Comassie-Färbung) analysiert.c) Kopplungskinetik: 1 - D1R8C, 2 - E8C-PE38, 3 - 30 min inkubiert, 4 - 1 h inkubiert, 5 - 2 h inkubiert, 6 -4 h inkubiert, 7 - 6 h inkubiert, 8 - 11 h inkubiert, 9 - 20 h inkubiert; 4 µM E8C-PE38 und 8 µM D1R8Cwurden in 50 mM Tris pH 8,0, 2 mM EDTA, 10 µM GSH, 40 µM GSSG bei 10 °C inkubiert und anschlie-ßend 10 min bei 13 000 rpm zentrifugiert. Die Überstände wurden in einer SDS-Gelelektrophorese (15 %,nichtreduzierend, Comassie-Färbung) analysiert.

66
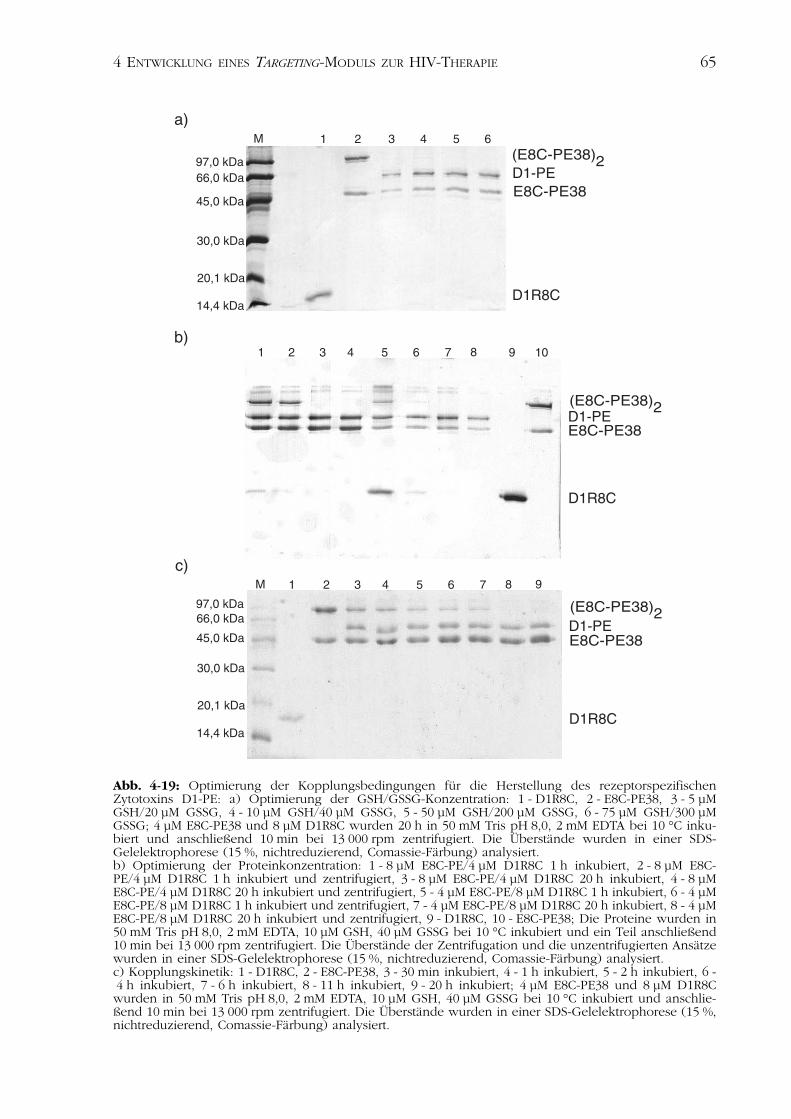
molekulare Disulfidbrücke des D1R8C reduziert und das Protein folglich destabilisiertwurde. Für die kovalente Bindung der CD4-Variante an das Derivat des PseudomonasExotoxin musste ein alternatives Protokoll etabliert werden.
Für die Herstellung des bifunktionellen Proteins D1-PE erfolgte die Inkubation vonE8C-PE38 und D1R8C in verschiedenen molaren Verhältnissen der Proteine und untervariierenden Redoxbedingungen in 50 mM Tris pH 8,0, 2 mM EDTA bei 10 °C. DieDisulfidverbrückung zwischen den Bindungspartnern wurde in einer oxidierenden SDS-Gelelektrophorese analysiert.
Die an die Proteine fusionierten polyionischen Peptide vermitteln die Assoziation derMoleküle. Die kovalente Bindung wird durch Ausbildung einer Disulfidbrücke zwischenzwei Cysteinen erreicht. Um diese kovalente Verbindung zu ermöglichen und gleichzei-tig die intramolekulare Disulfidbrücke der CD4-Variante D1R8C nicht zu zerstören, wares notwendig, die optimalen Redoxbedingungen dieser Kopplungsreaktion zu untersu-chen.
Für die quantitative Umsetzung des D1R8C waren GSH/GSSG-Konzentrationen von10 µM/40 µM ausreichend (Abb. 4-19a). Konzentrationen über von 75 µM GSH und300 µM GSSG förderten die Aggregation der eingesetzten Proteine (Daten nicht gezeigt).
Die Ausbildung der Disulfidbrücke, induziert durch Luftoxidation mit Unterstützungvon 1 µM CuCl2, zeigte keine optimale Konjugation von D1R8C und E8C-PE38 (Datennicht gezeigt).
Für die quantitative Assoziation war die Analyse der molaren Verhältnisse der einge-setzten Bindungspartner notwendig. Eine optimale Ausbeute an Kopplungsprodukt D1-PE konnte bei einem Überschuss an E8C-PE38 beobachtet werden. HöhereKonzentrationen von D1R8C führten zu einer zunehmenden Aggregation und nicht zueiner erhöhten Ausbeute an D1-PE (Abb. 4-19b).
Die Analyse der Kinetik der Kopplung zeigte, dass die Reaktion der Bindung zwi-schen D1R8C und E8C-PE38 nach 30 min beendet ist (Abb. 4-19c). Nach elfstündigerInkubation konnte die vollständige Reduktion der Homodimere (E8C-PE38)2 desDerivats des Pseudomonas Exotoxins beobachtet werden.
Die massenspektrometrische Analyse ergab ein Molekulargewicht des entstandenenProteins D1-PE von 53 885,0 Da. Dies stimmt mit dem theoretisch ermittelten Wert über-ein.
Reinigung des chimeren Proteins D1-PE. Für anschließende Experimente war esnotwendig, das D1-PE in ausreichenden Mengen herzustellen.
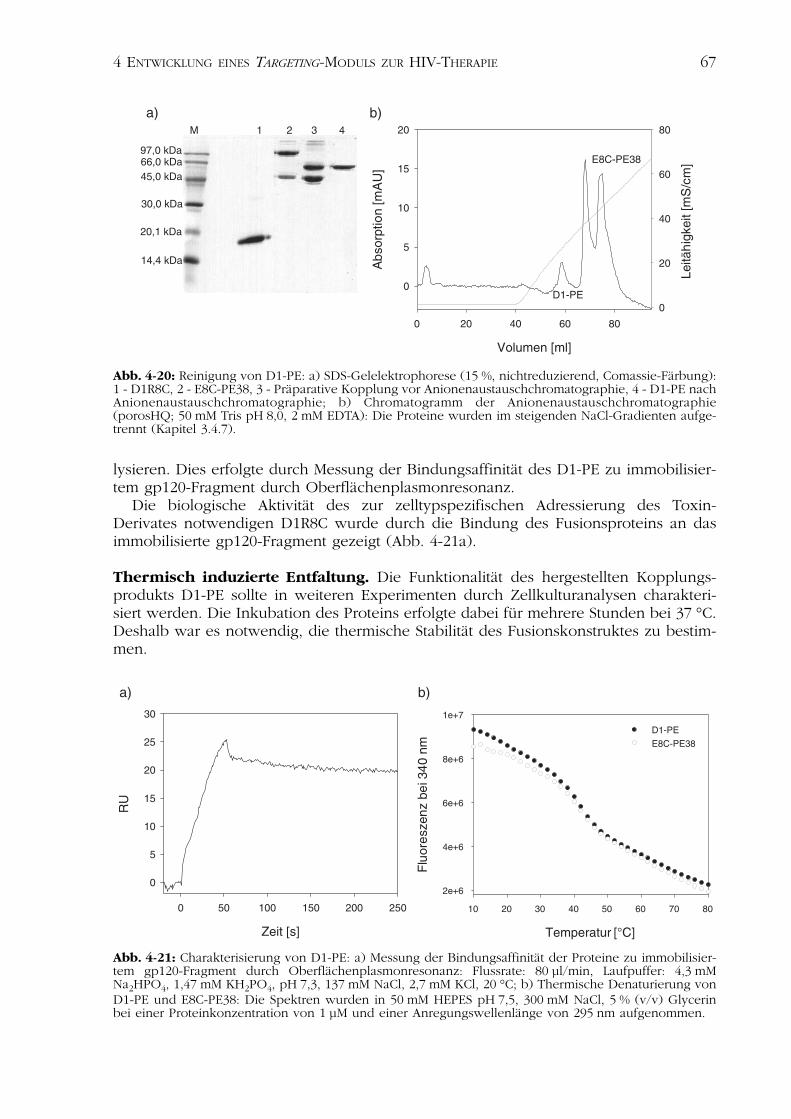
Die präparative Kopplung erfolgte bei einem zweifach molaren Überschuss an E8C-PE38 gegenüber D1R8C (Kapitel 3.4.7). Um unspezifische Effekte beim Nachweis derbiologischen Aktivität des hergestellten Zytotoxins auszuschließen, war es notwendig,nichtgebundenes E8C-PE38 vom entstandenen D1-PE abzutrennen.
Die Reinigung des chimeren Proteins D1-PE erfolgte durch Anionenaustausch-chromatographie (Abb. 4-20). Das Fusionskonstrukt eluierte in einem steigenden NaCl-Gradienten bei 300 mM NaCl und konnte vom nicht gekoppelten Derivat des Pseudo-monas Exotoxins E8C-PE38 abgetrennt werden, welches aufgrund der hohen Ladungs-dichte erst bei 460 nM NaCl vom Anionenaustauschmaterial eluierte (Abb. 4-20b).
4.3.2 Charakterisierung des bifunktionellen Proteins D1-PE
Funktionalität. Für die biologische Aktivität des D1-PE ist die Wechselwirkung vonD1R8C mit gp120 erforderlich.
Um eine zelltypspezifische Adressierung zu gewährleisten, war es notwendig, dieWechselwirkung zwischen dem kovalent gekoppelten D1R8C und dem gp120 zu ana-
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 67
lysieren. Dies erfolgte durch Messung der Bindungsaffinität des D1-PE zu immobilisier-tem gp120-Fragment durch Oberflächenplasmonresonanz.
Die biologische Aktivität des zur zelltypspezifischen Adressierung des Toxin-Derivates notwendigen D1R8C wurde durch die Bindung des Fusionsproteins an dasimmobilisierte gp120-Fragment gezeigt (Abb. 4-21a).
Thermisch induzierte Entfaltung. Die Funktionalität des hergestellten Kopplungs-produkts D1-PE sollte in weiteren Experimenten durch Zellkulturanalysen charakteri-siert werden. Die Inkubation des Proteins erfolgte dabei für mehrere Stunden bei 37 °C.Deshalb war es notwendig, die thermische Stabilität des Fusionskonstruktes zu bestim-men.
a) b)
Volumen [ml]
0 20 40 60 80
Abs
orpt
ion
[mA
U]
0
5
10
15
20
Leitä
hig
keit
[mS
/cm
]
0
20
40
60
80
D1-PE
E8C-PE38
20,1 kDa
14,4 kDa
30,0 kDa
45,0 kDa66,0 kDa97,0 kDa
M 1 2 3 4
Abb. 4-20: Reinigung von D1-PE: a) SDS-Gelelektrophorese (15 %, nichtreduzierend, Comassie-Färbung):1 - D1R8C, 2 - E8C-PE38, 3 - Präparative Kopplung vor Anionenaustauschchromatographie, 4 - D1-PE nachAnionenaustauschchromatographie; b) Chromatogramm der Anionenaustauschchromatographie(porosHQ; 50 mM Tris pH 8,0, 2 mM EDTA): Die Proteine wurden im steigenden NaCl-Gradienten aufge-trennt (Kapitel 3.4.7).
Temperatur [°C]
10 20 30 40 50 60 70 80
Flu
ores
zenz
bei
340
nm
2e+6
4e+6
6e+6
8e+6
1e+7
D1-PE
E8C-PE38
a) b)
Zeit [s]
0 50 100 150 200 250
RU
0
5
10
15
20
25
30
Abb. 4-21: Charakterisierung von D1-PE: a) Messung der Bindungsaffinität der Proteine zu immobilisier-tem gp120-Fragment durch Oberflächenplasmonresonanz: Flussrate: 80 µl/min, Laufpuffer: 4,3 mMNa2HPO4, 1,47 mM KH2PO4, pH 7,3, 137 mM NaCl, 2,7 mM KCl, 20 °C; b) Thermische Denaturierung vonD1-PE und E8C-PE38: Die Spektren wurden in 50 mM HEPES pH 7,5, 300 mM NaCl, 5 % (v/v) Glycerinbei einer Proteinkonzentration von 1 µM und einer Anregungswellenlänge von 295 nm aufgenommen.

68
Es konnte ein Übergangsmittelpunkt von 42 °C ermittelt werden. Dieser stimmt mit demfür das Protein E8C-PE38 ermittelten Wert überein (Abb. 4-21b). D1R8C übt im ProteinD1-PE keinen negativen Einfluss auf die thermische Stabilität des assoziierten Toxinsaus. Durch dieses Experiment konnten jedoch keine Aussagen über die Stabilität vonD1R8C getroffen werden.
Analyse der Stabilität nach Inkubation im Serum. Das hergestellte bifunktionelleProtein D1-PE sollte in weiteren Analysen durch Zellkulturexperimente charakterisiertwerden. Hierzu war es wichtig, die Serumstabilität nachzuweisen.
Analog zu dem in Kapitel 3.7.3 beschriebenen Protokoll erfolgte die Inkubation desFusionskonstruktes in 10 % fötalem Kälberserum. Die Analyse der Stabilität erfolgte mit-tels nichtreduzierender SDS-Gelelektrophorese.
Mit dieser Methode konnte das Fusionskonstrukt nicht direkt nachgewiesen werden,da es vom BSA des Serums überlagert wurde. Daher wurde untersucht, ob dieFusionspartner als isolierte Proteine nach der Inkubation auftraten. Dies war nicht derFall. Somit bleibt die kovalente Bindung zwischen D1R8C und E8C-PE38 im Serumerhalten, die einzelnen Proteine unterliegen keinem proteolytischen Abbau (Kapitel4.2.5; Kleinschmidt, 2004).
4.3.3 Zellkulturstudien
Die Fähigkeit des bifunktionellen Proteins D1-PE, spezifisch HIV-infizierte Zellen zutöten, wurde in Zellkulturexperimenten untersucht.
Hierzu fand eine CHO-K1-Zelllinie Verwendung, welche das Oberflächen-glykoprotein gp120 des HIV auf der Zelloberfläche exponiert (als CHO-wt bezeichnet).Als Negativkontrolle diente eine Zelllinie, die dieses Protein nicht auf der Zelloberflächeträgt (CHO-K1). Vorangegangene Experimente zeigten, dass rekombinant hergestelltesD1R8C in der Lage ist, spezifisch an die gp120-positive Zelllinie CHO-wt zu binden(Kapitel 4.2.6).
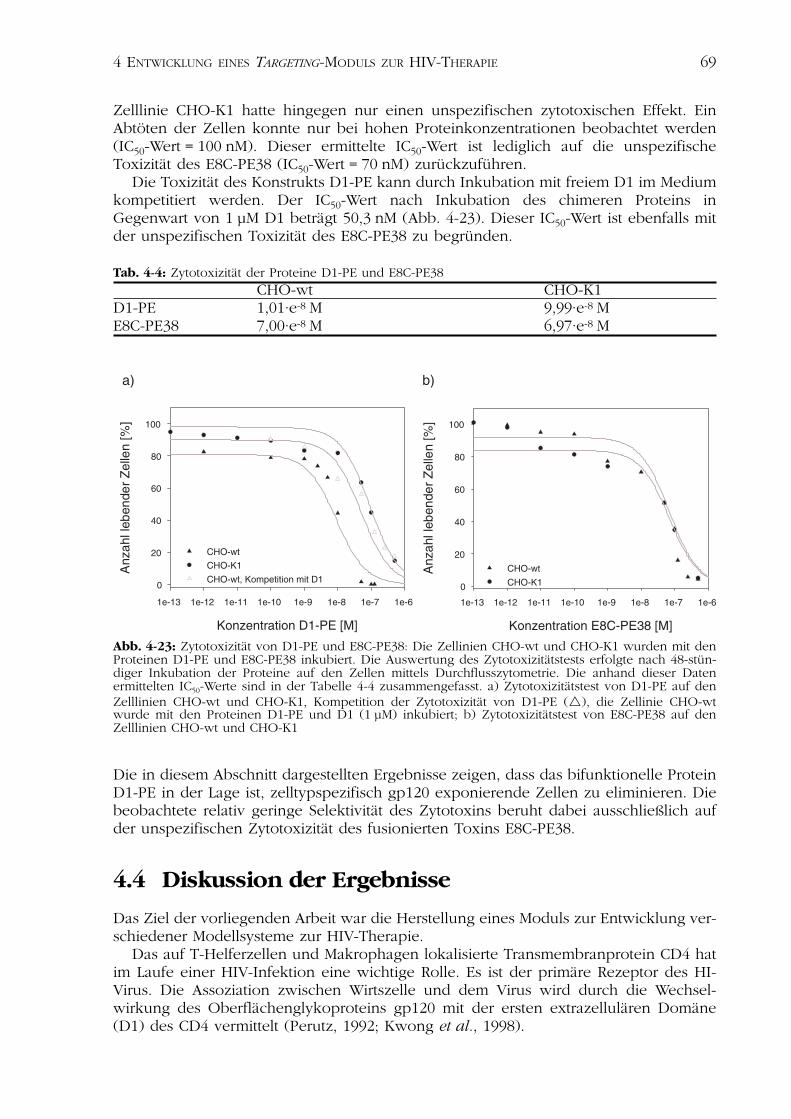
Zum Nachweis der Zytotoxizität wurden die Zellen mit D1-PE in Konzentrationenvon 0,1 pM bis 0,5 µM für 48 h bei 37 °C inkubiert (Kapitel 3.6.4). Die Anzahl der abge-töteten Zellen wurde durch Anfärben der zellulären DNA mit Probidiumjodid detektiert
und mittels Durchflusszytometrie analysiert(Abb. 4-22). Die Auswertung des Zyto-toxizitätstests erfolgte durch Bestimmungdes prozentualen Anteils an lebendenZellen für die jeweilige Protein-konzentration (Abb. 4-23).
Die in der Tabelle 4-4 aufgelisteten IC50-Werte der jeweiligen Proteine wurden ausdem Kurvenverlauf der Auftragung (Abb. 4-23) nach der im Kapitel 3.6.4 beschriebenenGleichung ermittelt.
D1-PE ist in der Lage, sehr effizientgp120 exponierende Zellen zu töten.
Der IC50-Wert der Titration des rezeptor-spezifischen Zytotoxins auf der ZelllinieCHO-wt beträgt 10 nM. Im Gegensatz dazuzeigte das isolierte Toxin E8C-PE38 keinezelltypspezifische Toxizität (IC50-Wert = 69,9 nM). Die Inkubation desProteins D1-PE auf der gp120 negativen
CHO-wtCHO-wt mit D1-PE
Fluoreszenzintensität FL3: > 670 nm
Zel
lzah
l
Abb. 4-22: Histogramm des Zytotoxizitätstestsvon D1-PE auf der Zelllinie CHO-wt: Die Zellenwurden nach dem in Kapitel 3.6.4 beschriebenenProtokoll behandelt und am Durchflusszytometeranalysiert. Die Fluoreszenz des Propidiumjodidswurde bei 488 nm angeregt und die Emission mitKanal FL3: >670 nm gemessen.
4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 69
Zelllinie CHO-K1 hatte hingegen nur einen unspezifischen zytotoxischen Effekt. EinAbtöten der Zellen konnte nur bei hohen Proteinkonzentrationen beobachtet werden(IC50-Wert = 100 nM). Dieser ermittelte IC50-Wert ist lediglich auf die unspezifischeToxizität des E8C-PE38 (IC50-Wert = 70 nM) zurückzuführen.
Die Toxizität des Konstrukts D1-PE kann durch Inkubation mit freiem D1 im Mediumkompetitiert werden. Der IC50-Wert nach Inkubation des chimeren Proteins inGegenwart von 1 µM D1 beträgt 50,3 nM (Abb. 4-23). Dieser IC50-Wert ist ebenfalls mitder unspezifischen Toxizität des E8C-PE38 zu begründen.
Tab. 4-4: Zytotoxizität der Proteine D1-PE und E8C-PE38CHO-wt CHO-K1
D1-PE 1,01·e-8 M 9,99·e-8 ME8C-PE38 7,00·e-8 M 6,97·e-8 M
Die in diesem Abschnitt dargestellten Ergebnisse zeigen, dass das bifunktionelle ProteinD1-PE in der Lage ist, zelltypspezifisch gp120 exponierende Zellen zu eliminieren. Diebeobachtete relativ geringe Selektivität des Zytotoxins beruht dabei ausschließlich aufder unspezifischen Zytotoxizität des fusionierten Toxins E8C-PE38.
4.4 Diskussion der Ergebnisse
Das Ziel der vorliegenden Arbeit war die Herstellung eines Moduls zur Entwicklung ver-schiedener Modellsysteme zur HIV-Therapie.
Das auf T-Helferzellen und Makrophagen lokalisierte Transmembranprotein CD4 hatim Laufe einer HIV-Infektion eine wichtige Rolle. Es ist der primäre Rezeptor des HI-Virus. Die Assoziation zwischen Wirtszelle und dem Virus wird durch die Wechsel-wirkung des Oberflächenglykoproteins gp120 mit der ersten extrazellulären Domäne(D1) des CD4 vermittelt (Perutz, 1992; Kwong et al., 1998).
a) b)
Konzentration D1-PE [M]
1e-13 1e-12 1e-11 1e-10 1e-9 1e-8 1e-7 1e-6
Anz
ahl l
eben
der
Zel
len
[%]
0
20
40
60
80
100
CHO-wt
CHO-K1
CHO-wt, Kompetition mit D1
Konzentration E8C-PE38 [M]
1e-13 1e-12 1e-11 1e-10 1e-9 1e-8 1e-7 1e-6
Anz
ahl l
eben
der
Zel
len
[%]
0
20
40
60
80
100
CHO-wt
CHO-K1
Abb. 4-23: Zytotoxizität von D1-PE und E8C-PE38: Die Zellinien CHO-wt und CHO-K1 wurden mit denProteinen D1-PE und E8C-PE38 inkubiert. Die Auswertung des Zytotoxizitätstests erfolgte nach 48-stün-diger Inkubation der Proteine auf den Zellen mittels Durchflusszytometrie. Die anhand dieser Datenermittelten IC50-Werte sind in der Tabelle 4-4 zusammengefasst. a) Zytotoxizitätstest von D1-PE auf denZelllinien CHO-wt und CHO-K1, Kompetition der Zytotoxizität von D1-PE ( ), die Zellinie CHO-wtwurde mit den Proteinen D1-PE und D1 (1 µM) inkubiert; b) Zytotoxizitätstest von E8C-PE38 auf denZelllinien CHO-wt und CHO-K1

70
Die rekombinante Herstellung des humanen T-Zellrezeptors in ausreichenden Mengenstellt einen wichtigen Aspekt zur Entwicklung eines zelltypspezifischen Systems zurHIV-Therapie dar. Verschiedene Ansätze zeigen die Fähigkeit von rekombinant herge-stelltem CD4, die Infektion von Zellen mit HIV durch Assoziation an dieOberflächenglykoproteine auf dem Virus zu unterbinden (Traunecker et al., 1988;Hussey et al., 1988; Deen et al., 1988; Fischer et al., 1988).
Ein weiterer Aspekt der vorliegenden Arbeit war die Entwicklung eines Targeting-Moduls zum Einsatz in verschiedenen HIV-Therapiemodellen. Diese beruhen auf derAnwendung eines Systems, dessen Bestandteile kovalent miteinander verknüpft werdenkönnen. Die Assoziation dieser Bindungspartner wird durch elektrostatische Wechsel-wirkung vermittelt, die kovalente Bindung erfolgt über Disulfidverbrückung(Stubenrauch et al., 2001; Richter et al., 2001; Kleinschmidt et al., 2003).
Zur Eliminierung HIV-infizierter Zellen erfolgte die Konjugation des CD4-KonstruktsD1R8C an ein Derivat des Pseudomonas Exotoxin E8C-PE38. Das erhaltene rezeptorspe-zifische Zytotoxin kann, vermittelt durch CD4, HIV-infizierte Zellen binden und durchdas angefügte Derivat des Pseudomonas Exotoxin die Vermehrung der HI-Viren durchEliminierung der infizierten Zellen verhindern.
Ähnliche Protokolle zeigen eine Eliminierung gp160-exponierender Zellen durchgenetisch fusionierte rezeptorspezifische Proteine, bestehend aus einem gp41 erkennen-den Protein und einem Toxin (Root et al., 2003).
Die Herstellung eines chimeren Proteins durch kovalente Bindung eines tumorspezi-fischen Fv-Antikörperfragments an das Pseudomonas Exotoxin E8C-PE38 und die zell-typspezifische Eliminierung Tumorantigen präsentierender Zellen durch diesesImmunotoxin zeigten frühere Studien (Kleinschmidt et al., 2003).
4.4.1 Rekombinante Herstellung der CD4-Varianten
Die Darstellung der vorgestellten CD4-Konstrukte erfolgte durch Expression inEscherichia coli.
Vorangegangene Arbeiten zeigen die Expression verschiedener CD4-Varianten, vor-wiegend bestehend aus den zwei N-terminalen Domänen (D1D2) des HIV-Rezeptors,in eukaryontischen Systemen. Dabei kann bis zu 1 mg/l Kultur funktionelles Proteingewonnen werden (Hussey et al., 1988; Traunecker et al., 1988).
Die Überexpression der Proteine in Escherichia coli führt zur Bildung vonAggregaten, den sogenannten inclusion bodies. Die Proteine werden nicht nativ herge-stellt und müssen in weiteren Schritten renaturiert werden.
Die Expression von CD4 in Escherichia coli mit anschließender Sekretion insPeriplasma ist eine Möglichkeit, dieses Protein in Escherichia coli löslich zu exprimie-ren. Diese Studien lieferten 1 bis 2,4 mg natives Protein pro Liter Kultur (Rockenbach etal., 1991; Osburne et al., 1999).
Um die geplanten CD4-Varianten D1 und D1R8C in ausreichenden Mengen zu erhal-ten, erfolgte die Expression in Escherichia coli als inclusion bodies. Das Renaturierungs-protokoll wurde anschließend hinsichtlich der Bildung nativen Proteins verbessert.
Durch definierte Bedingungen, insbesondere den Einsatz eines Redoxshuffling-Systems, bestehend aus oxidiertem und reduzierten Glutathion, sowie den Zusatz desdie Aggregation unterdrückenden Agens Arginin, konnte die Renaturierungsausbeutevon D1 - verglichen mit bestehenden Protokollen - von 20 % auf 37 % gesteigert werden(Tab. 4-5; Chao et al., 1989). Die Renaturierungsdauer wurde im Vergleich zu bestehen-den Protokollen auf 5 min reduziert (Tab. 4-5).
Nach anschließender Reinigung der CD4-Variante D1 konnten aus 100 g Zellen119,2 mg Protein gewonnen werden. Die Analyse nach Reinigung durch analytischeGelchromatographie, SDS-Gelelektrophorese und Reversed Phase Chromatographie

4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 71
zeigte eine Reinheit von bis zu 99 %. Verglichen mit dem bestehenden Protokoll derrekombinanten Herstellung der N-terminalen Domäne des CD4, konnte die Ausbeuteauf das Fünffache gesteigert werden (Tab. 4-5; Chao et al., 1989). Die Reinheit desProteins konnte durch Anwendung von Reversed Phase Chromatographie und Gel-chromatographie noch optimiert werden (Tab. 4-5).
Tab. 4-5: Vergleich der rekombinanten Herstellung verschiedener CD4-KonstrukteRenaturierungsdauer Ausbeute1 Reinheit
D1 5 min 119,20 mg >99 %D18C 5 min 27,30 mg >99 %D1 (Chao et al., 1989) 3 Tage 28,56 mg 85 %1 bezogen auf 100 g Zellen
Die Ausbeute der hergestellten CD4-Variante mit polyionischen Fusionspeptid (D1R8C)sank im Vergleich zur Wildtypdomäne D1 auf 23 % (Tab. 4-5). Dieser erhebliche Unter-schied kann einerseits mit der Einführung eines weiteren Cysteins erklärt werden. DieMöglichkeit der Ausbildung einer Disulfidbrücke erhöht sich im Konstrukt D1R8C aufdrei, während in der Wildtypdomäne D1 nur eine Möglichkeit besteht, eineDisulfidbrücke auszubilden. Im Laufe der Renaturierung des D1R8C können somit meh-rere „falsch verbrückte“ Proteine entstehen, welche durch Aggregation die Konzentra-tion des solubilisierten Proteins mindern und die Ausbeute des nativen Proteins verrin-gern (Kapitel 3.4.4; Abb. 3-2a).
Eine weitere Erklärung ist die Zunahme an positiven Ladungen durch Anfügen despolyionischen Peptids. Der isoelektrische Punkt der CD4-Variante mit C-terminalerFusion steigt um 1,22 pH-Einheiten auf einen IP = 10,01. Diese Zunahme kann eineDestabilisierung des Proteins verursachen, welche die verringerte Ausbeute im Vergleichzum Wildtyp erklärt. Die Renaturierungsausbeute dieser Variante sank auf 5,2 % (Tab.4-1).
Die Reinheit der hergestellten CD4-Variante D1R8C ist mit der Reinheit des Wildtyp-konstrukts vergleichbar (Tab. 4-5). Dies bestätigten analytische Gelchromatographie undSDS-Gelelektrophorese.
Aufgrund des eingeführten C-terminalen Cysteins bestand die Möglichkeit derHomodimerisierung des Konstrukts. Die Kontrolle des Oligomerisierungszustandes desProteins erfolgte durch nichtreduzierende SDS-Gelelektrophorese, analytischeUltrazentrifugation sowie analytische Gelchromatographie. Diese Methoden bestätigtendie monomere Spezies der hergestellten CD4-Variante.
Experimente zur proteolytischen Fragmentierung mit anschließender Identifizierungder entstandenen Fragmente durch Massenspektrometrie zeigten die Ausbildung derkorrekten Disulfidbrücke.
4.4.2 Charakterisierung der CD4-Varianten
Die Funktionalität. Die Kinetik der Bindung der CD4-Varianten zum HIV- Ober-flächenglykoprotein gp120 wurde durch Oberflächenplasmonresonanz bestimmt.
Hierzu wurde ein Teil der Bindungsstelle, die �-Haarnadelschleife, bestehend ausden zwei antiparallelen Strängen �20 – �21 der äußeren Domäne des gp120 (das ent-spricht den Aminosäuren 421 – 438 des gp120; Kwong et al., 1998), als Peptid an einenSensorchip immobilisiert.
Die Kinetik der Wechselwirkung mit den CD4-Varianten wird durch die Assoziations-und Dissoziationsraten beschrieben. Die langsame Assoziation und Dissoziation zeigten,dass die gebildeten Komplexe D1 – gp120-Fragment sowie D1R8C – gp120-Fragment

72
sehr stabil war. Die ermittelten Raten entsprechen den in der Literatur beschriebenenKonstanten.
Die Bindung einer CD4-Variante der zwei N-terminalen Domänen zum gp120-Gesamtrezeptor wurde bei 37 °C mit einer Assoziationsgeschwindigkeit vonka = 6,7·e4 M-1 s-1 sowie einer Dissoziationsgeschwindigkeit von kd = 1,5·e-3 s-1 beschrie-ben (Myszka et al., 2000). Die in dieser Arbeit bestimmten Konstanten beschreibeneinen langsameren Prozess. Dies kann dadurch erklärt werden, dass die Bindungs-messungen bei 20 °C durchgeführt wurden und der Temperaturunterschied eineVeränderung der Bindungskinetik hervorrufen kann.
Die für die Komplexe D1 – gp120-Fragment sowie D1R8C – gp120-Fragment ermittel-ten Dissoziationskonstanten sind mit den in der Literatur ermittelten Konstanten ver-gleichbar (Tab. 4-6; Wu et al., 1996; Myzka et al., 2000). Diese Konstanten beschreibendie Affinität des gp120-Gesamtrezeptors zu einer CD4-Variante, bestehend aus den zweiN-terminalen Domänen des CD4 (D1D2).
Die Affinität des CD4 zu den Aminosäuren 421 – 438 des gp120 ist genauso hoch wiedie Affinität des Proteins zum gesamten Oberflächenrezeptor des HI-Virus (Kapitel 4.2.2;Abb. 4-10).
In einer weiteren Arbeit wurde die Affinität eines Mikroproteins, welches dieBindungsstelle des CD4 zum gp120 darstellt (CD4M33), zum Gesamtrezeptor gp120 ana-lysiert (Martin et al., 2003). Die ermittelte Dissoziationskonstante dieses Konstruktsbeträgt 8,2 nM und entspricht den Werten, welche die Affinität der gebildeten KomplexeD1 – gp120-Fragment sowie D1R8C – gp120-Fragment beschreibt (Tab. 4-6).
Tab. 4-6: Zusammenfassung der Bindungskonstanten der Wechselwirkung CD4 – gp120, ermittelt durchOberflächenplasmonresonanz
T [°C] ka [M-1s-1] kd [s-1] KD [M]D1 20 5,9·e3 0,9·e-4 1,54·e-8
D18C 20 7,3·e3 1,5·e-4 1,90·e-8
D1D2 (Myzka et al., 2000) 37 6,7·e4 1,5·e-3 2,20·e-8
D1D2 (Wu et al., 1996) 25 8,2·e4 1,5·e-3 1,80·e-8
CD4M33 (Martin et al., 2003) 4,5·e4 3,7·e-4 8,20·e-9
D1R8C zeigt in den Bindungstudien vergleichbare Raten und Affinitäten, wie das Wild-typkonstrukt D1 (Tab. 4-6). Die Bindungsstelle hat die native Struktur ausgebildet, dasangefügte polyionische Fusionspeptid beeinflusst die Affinität zum Oberflächen-glykoprotein des HI-Virus nicht.
Tab. 4-7: Zusammenfassung der Affinität der Bindungspartner CD4 und gp120 in LösungT [°C] KD [M]
D1 – gp1201 20 0,70·e-9
D1R8C – gp120 (HIV-1)-Fragment1 20 1,06·e-9
D1D2 – gp120 (Myzka et al., 2000)2 37 5,00·e-9
D1-D4 – gp120 (Xiang et al., 2002)2 37 3,80·e-8
CD4 – gp120 (Moebius et al., 1992)3 37 0,90·e-9
1 Werte, ermittelt durch Fluoreszenztitration2 Werte, ermittelt durch isothermale Titrationskalorimetrie3 Werte, ermittelt durch Immunofluoreszenz
In weiteren Untersuchungen wurde die Affinität der Bindungspartner in Lösung betrach-tet. Die Dissoziationskonstanten von 0,8 bis 1,8 nM sind vergleichbar mit den in derLiteratur durch ITC-Messung ermittelten Werten von 5 nM und 38 nM (Myszka et al.,2000; Xiang et al., 2002). Die durch Immunofluoreszenz ermittelte Dissoziations-konstante zwischen freiem gp120 und einer CD4-exponierenden Maus-Hybridoma-

4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 73
Zelllinie von 0,9 nM ist ebenfalls mit den in dieser Arbeit bestimmten Werten vergleich-bar (Tab. 4-7; Moebius et al., 1992).
Die Struktur und Stabilität der CD4-Konstrukte. Kristallstrukturanalysen zeigen fürVarianten des CD4 eine Faltblattstruktur der Immunglobulin-Superfamilie (Wang et al.,1990; Ryu et al., 1990; Ryu et al., 1994).
Die Analyse des Sekundärstrukturgehaltes des im Rahmen dieser Arbeit hergestelltenCD4-Konstrukts D1 mittels Fern-UV-CD-Spektroskopie zeigte ein für ein überwiegendaus �-Faltblättern aufgebautes Protein typisches Spektrum. Diese Immunglobulin-Faltung spiegelt sich in einem Minimum der negativen residualen Elliptizität bei 218 nmwider. Die Nah-UV-CD-Spektren dieses Proteins zeigen die asymmetrische Umgebungder aromatischen Aminosäurereste und verweisen auf eine kompakte Struktur desProteins. Charakteristisch für eine ausgebildete Struktur des Proteins sind ebenfalls dieFluoreszenzemissionspektren. Im nativen Zustand hat D1 ein Emissionsmaximum bei347 nm. In Gegenwart von Guanidiniumhydrochlorid erfolgten eine Rotverschiebungdes Emissionsmaximums und eine geringe Zunahme der Emissionsintensität. DieseZunahme zeigt einen „Dequenching“-Effekt der Fluoreszenz der aromatischenAminosäurereste im denaturierten Protein. Diese Aminosäurereste sind im nativenProtein in der Nähe eines internen „Quenchers“, z. B. der Disulfidbrücke. Im Verlauf derDenaturierung erfolgt die Auflösung der kompakten Struktur des D1, die aromatischenAminosäuren werden Lösungsmittel-exponiert und unterliegen nicht mehr demQuench-Effekt der Disulfidbrücke, was in der Zunahme der Emissionsintensität zubeobachten ist (Tsunenaga et al., 1987). Der „Dequenching“-Effekt im Verlauf derDenaturierung ist ein weiteres Indiz für eine native Faltung des CD4-Konstrukts D1.
Das Fern-UV-CD-Spektrum der CD4-Variante D1R8C zeigt, dass die Struktur diesesProteins nicht mit der des Wildtypkonstrukts D1 vergleichbar ist. Die CD-Spektren desnativen und denaturierten Zustands des D1R8C unterscheiden sich.
Möglicherweise liegt D1R8C in einem alternativen Faltungszustand, einem moltenglobule vor, der die Funktionalität des Proteins hat (Kapitel 4.2.2), aber dessen Strukturnicht der nativen Struktur entspricht.
Ein ähnlicher Zustand wurde für ein scFV-Fragment eines Antiferritin Antikörpersbeschrieben (Martsev et al., 2000). Dieses Fragment ist im Vergleich zur VL-Domäne desAntikörpers partiell unstrukturiert. Das Fern-UV-CD-Spektrum weist analog zu dem hierbeschriebenen CD4-Konstrukt D1R8C kein typisches Spektrum eines �-Faltblattproteinsauf. Das Minimum der negativen Elliptizität im scFv-Fragment ist im Vergleich zur VL-Domäne um ein Drei- bis Vierfaches höher; das Spektrum unterschiedet sich jedochvom Spektrum des denaturierten scFv-Fragments. Nah-UV-CD-Spektren dieses Proteinszeigen, dass sich die asymmetrische Umgebung der aromatischen Reste im Vergleich zurVL-Domäne verändert hat, was die partielle Entfaltung des Proteins bestätigt.
Für dieses scFv-Fragment wurde wie für weitere Antikörper bzw. Antikörper-fragmente die molten globule Konformation beschrieben (Buchner et al., 1991; Lilie &Buchner, 1995; Martsev et al., 2000; Martsev et al., 2002). Da sich die hier beschriebeneCD4-Variante in den spektroskopischen Untersuchungen ähnlich verhält, kann davonausgegangen werden, dass dieses Protein ebenfalls in einer molten globuleKonformation vorliegt. Im Gegensatz zu der molten globule Konformation eines unter-suchten Maus Antikörpers (MAK33) und dessen Fab-Fragment (Buchner et al., 1991;Lilie & Buchner, 1995) verfügen D1R8C und das scFv-Fragment des AntiferritinAntikörpers (Martsev et al., 2000) über eine biologische Aktivität. Martsev et al. definier-ten den Zustand des scFv-Fragments als den eines funktionellen molten globule (Martsevet al., 2000). Die dargestellten Untersuchungen zeigen, dass D1R8C in einer funktionel-len molten globule Konformation vorliegt.
Die Analyse der Wechselwirkung der CD4-Varianten zeigen, dass beide Konstrukte(D1 und D1R8C) den Rezeptor mit gleicher Affinität binden (Kapitel 4.2.2). Für die

74
Bindung des CD4 an gp120 ist somit nicht die vollständige strukturelle Information desProteins erforderlich.
Die Bindung des gp120-Fragments wird durch die �-Haarnadelschleife der C’- undC’’-Stränge des CD4 vermittelt, welche das Phenylalanin 43 des CD4 in die räumlicheNähe zum �-turn des �20/�21 des gp120 dirigiert (Abb. 1-4; Kwong et al., 1998).Studien der Wechselwirkung des gp120 zu einem CD4-Peptid, welches lediglich aus die-sem Bereich besteht, zeigen, dass dieses Fragment ausreicht, um den Rezeptor mit glei-cher Affinität zu binden wie die vollständige Domäne D1 (Martin et al., 2003; Kapitel4.4.1). Im D1R8C ist die �-Haarnadelschleife der C’- und C’’-Stränge ausgebildet, da dieAffinität zum gp120 vergleichbar ist mit der des Wildtypkonstruktes. EineStrukturinduktion in diesem Bereich durch Bindung des gp120-Fragmentes würde ineiner geringeren Dissoziationskonstante des D1R8C – gp120-Fragment – Komplexeserkennbar sein.
Durch weitere Experimente konnte gezeigt werden, dass durch Bindung des gp120-Fragments eine Strukturänderung sowohl in der CD4-Variante D1R8C als auch imgebundenen gp120-Fragment hervorgerufen werden kann. Fern-UV-CD-Spektren desCD4 – gp120-Fragment – Komplexes weisen differentere CD-Spektren auf als die ausden Einzelspektren addierten.
Frühere Analysen der Fern-UV-CD-Spektroskopie eines Komplexes aus CD4 undgp120 zeigen ebenfalls eine Strukturänderung (Myszka et al., 2000). Diese geht abergrößtenteils vom gp120 aus, da hier der Bereich �2/3 eine erhebliche Umstrukturierungerfährt. Dieser Abschnitt des gp120 liegt im ungebundenen Zustand unstrukturiert vorund bildet während der Bindung des CD4 ein �-Faltblatt aus (Pan et al., 2004).
Da für die vorliegenden Analysen nur ein Teil der Bindungstasche des gp120 zurFern-UV-CD-Spektroskopie verwendet wurde, kann festgestellt werden, dass die beob-achtete Strukturinduktion im Fern-UV-CD-Spektrum des D1 – gp120-Fragment –Komplexes auf eine Strukturänderungen des verwendeten gp120-Fragmentes zurückzu-führen ist. Dieses Fragment liegt im ungebundenen Zustand in einer random coil-Konformation vor, im Komplex mit CD4 bildet sich eine �-Haarnadelschleife �20/21aus, welche teilweise für die Bindung des T-Zellrezeptors verantwortlich ist (Kwong etal., 1998).
Im Komplex des HIV-Fragments mit D1R8C kann eine Strukturinduktion im D1R8Czum �-Faltblatt beobachtet werden. Das Fern-UV-CD-Spektrum des KomplexesD1R8C – gp120-Fragment weist eine Zunahme der Elliptizität zwischen 195 und 200 nmauf, das Minimum der Elliptizität ist von 208 auf 215 nm verschoben. Die Bindung desgp120-Fragments scheint in dem Konstrukt mit polyionischer Fusion eine Struktur zuinduzieren, die der des Wildtypkonstrukts D1 nahe kommt. Die gezeigte Änderung desSekundärstrukturverhaltens im Fern-UV-CD-Spektrum des D1R8C – gp120-Fragment –Komplexes im Vergleich zum ungebundenen Protein bzw. Peptid stellt eine Kombi-nation der Strukturänderungen im Protein D1R8C und im gp120-Fragment dar.
Weitere Untersuchungen zeigten die thermische Stabilität der hergestellten CD4-Varianten. Das Wildtypkonstrukt D1 zeigt einen Übergang bei 50 °C. Die thermischinduzierte Entfaltung konnte bereits ab 40 °C beobachtet werden. Diese Ergebnisse ste-hen im Einklang mit Untersuchungen an einem CD4-Konstrukt, bestehend aus den 4extrazellulären Domänen, welche die thermische Entfaltung ab 40 °C zeigen. Der Über-gangsmittelpunkt liegt bei 59 °C, was eine Stabilisierung durch die Domänen unterein-ander bestätigt (Tendian et al., 1995) und die geringere thermische Stabilität des CD4-Konstruktes D1 erklärt.
D1 kann durch Inkubation mit gp120-Fragment stabilisiert werden. Der Übergangs-mittelpunkt der thermischen Entfaltung des D1 – gp120-Fragment – Komplexes liegt bei52 °C.
D1R8C zeigt in der Fluoreszenz keinen temperaturabhängigen Übergang. Auch diethermische Stabilität des scFv-Fragments mit einer funktionellen molten globule

4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 75
Konformation ist im Gegensatz zu VL-Domäne verringert (Martsev et al., 2000). DieseUntersuchungen bestätigen die Vermutung, dass D1R8C ebenfalls die Konformationeines funktionellen molten globule besitzt.
Durch Kopplung des Proteintoxins E8C-PE38 an D1R8C konnte kein thermisch indu-zierter Entfaltungsübergang des CD4-Konstrukts beobachtet werden. Der dargestellteEntfaltungsübergang zeigt lediglich die thermische Denaturierung des gekoppeltenToxins (Kapitel 4.3.2).
Die Stabilität im Serum ist für beide CD4-Konstrukte vergleichbar und zeigt, dassbeide Proteine als Therapeutikum Verwendung finden können.
Der Nachweis der Funktionalität, die Ausbildung der Immunglobulin-Faltung und diethermische Stabilität des CD4-Konstrukts D1 zeigen, dass die erste extrazelluläreDomäne des CD4 für die biologische Aktivität des CD4 zur Bindung des HIV-Ober-flächenglykoproteins gp120 ausreichend ist. Es konnte im Rahmen dieser Arbeit einProtokoll ermittelt werden, um diese CD4-Variante in ausreichender Quantität undQualität zu erhalten.
4.4.3 Das rezeptorspezifische Zytotoxin D1-PE
Die Komponenten des modular aufgebauten Zytotoxins, D1R8C und E8C-PE38, solltenüber gerichtete Assoziation und kovalente Verknüpfung miteinander verbunden wer-den. Durch die Kombination des D1R8C mit dem Proteintoxin E8C-PE38 ist zum einendie zelltypspezifische Adressierung des Komplexes gesichert und zum anderen wirddurch E8C-PE38 ein biologischer Effekt vermitteln.
Grundlage für dieses System war die durch ionische Assoziation vermittelte kovalen-te Verbrückung polyionischer Peptide (Richter et al., 2001). Die kovalente Konjugationvon durch polyionische Peptide erweiterten Proteinen und die Herstellung chimererProteine stellten bereits frühere Studien dar (Stubenrauch et al., 2001; Kleinschmidt etal., 2003).
Ein Immunotoxin bestehend, aus einem tumorspezifischen Fv-Antikörperfragmentund einem Derivat des Pseudomonas Exotoxin (Kleinschmidt et al., 2003), lieferte dieVorlage für das zu entwickelnde Modellsystem zur HIV-Therapie.
Herstellung des rezeptorspezifischen Zytotoxins. Die Bedingungen für dieAssoziation der Komponenten D1R8C und E8C-PE38 mussten zu Beginn der Studienoptimiert werden. Die bestehenden Protokolle zur Konjugation polyionischer Peptideberuhen auf der durch ein Redoxshuffling-System vermittelten Ausbildung einerDisulfidbrücke.
Die in der Literatur beschriebenen Bedingungen von 1 mM reduzierten und 4 mMoxidiertem Glutathion (Kleinschmidt et al., 2003) führten bei der Herstellung desgeplanten Proteins D1-PE zur vermehrten Aggregation der eingesetzten Proteine. DieseRedoxbedingungen entsprachen denen, die bei der Renaturierung des Proteins D1R8Cverwendet wurden. Durch Anwendung dieser Bedingungen kam es zur Reduktion derintramolekularen Disulfidbrücke des D1R8C, was zu einer Destabilisierung mit anschlie-ßender Aggregation des Proteins führte. Die Konzentrationen der Komponenten desRedoxsystems betrugen für die Konjugation von D1R8C und E8C-PE38 ein Hundertstelder in der Literatur beschriebenen Konzentrationen (Tab. 4-8).
Die Kopplung erfolgte im molaren Unterschuss von D1R8C, da höhereKonzentrationen die Aggregation des Proteins förderten. Die Kopplung war nach30 Minuten beendet (Tab. 4-8). Das eingesetzte D1R8C wurde bei dieser Reaktion voll-ständig an das E8C-PE38 assoziiert. Nicht gekoppeltes E8C-PE38 konnte leicht mittelsIonenaustauschchromatographie vom bifunktionellen Protein abgetrennt werden.

76
Tab. 4-8: Bedingungen der Konjugation von Proteinen, vermittelt durch polyionische FusionspeptideKonz. GSH/GSSG [mM] Assoziationsdauer
D1-PE 0,01/0,04 30 minB3-PE38 (Kleinschmidt et al., 2003) 1,00/4,00 24 hB3-VP1E8C(Stubenrauch et al., 2001) 0,60/2,40 5 hFabE10C–�-GlucR10 (Richter et al., 2001) 0,60/2,40 16 h
Biophysikalische Charakterisierung des D1-PE. Das gereinigte rezeptorspezifischeZytotoxin wurde in verschiedenen Experimenten biophysikalisch charakterisiert.
Die Bindungsfähigkeit des assoziierten D1R8C zum gp120-Fragment wurde durchMessung der Affinität mittels Oberflächenplasmonresonanz gezeigt. Hierbei stellte sichheraus, dass die Bindung des D1R8C durch die Kopplung des Derivats desPseudomonas Exotoxins nicht beeinflusst wird.
Die biologische Aktivität eines fusionierten E8C-PE38 durch Nachweis der Bindungs-fähigkeit von NAD wurde bereits in dem hergestellten Immunotoxin B3-PE38 nachge-wiesen (Kleinschmidt et al., 2003).
Es wurde die thermische Stabilität des Fusionskonstrukts untersucht. Der Übergangs-mittelpunkt der thermischen Denaturierung liegt bei 42 °C und zeigt eine vergleichbareStabilität wie das freie E8C-PE38. Die Assoziation des D1R8C an E8C-PE38 hat keinennegativen Einfluss auf die Stabilität des Konstrukts D1-PE.
Die biophysikalische Charakterisierung zeigte analog zu den Untersuchungen andem Immunotoxin B3-PE38, dass sich die Bindungspartner hinsichtlich ihrer Stabilitätund biologischen Funktion nicht beeinflussen (Kleinschmidt et al., 2003).
Biologische Aktivität des chimeren Proteins D1-PE in Zellkultur. Zur EliminierungHIV-infizierter Zellen durch das beschriebene Protein ist die zelltypspezifischeAdressierung des Systems entscheidend.
D1R8C wurde deshalb auf seine Fähigkeit geprüft, spezifisch an gp120-exponieren-den Zellen zu binden. Die Analyse mittels Durchflusszytometrie ergab, dass fluoreszenz-markiertes D1R8C in der Lage ist, die gp120-exponierende CHO-Zelllinie CHO-wt zumarkieren, während die Kontrollzelllinie CHO-K1 nach der Inkubation mit fluoreszenz-markiertem D1R8C nicht detektiert werden konnte.
Analysen zur Zytotoxizität zeigen, dass das hergestellte chimere Protein D1-PE in derLage ist, zelltypspezifisch gp120-exponierende Zellen zu eliminieren. Der ermittelteIC50-Wert betrug für dieses Konstrukt 10 nM. Die unspezifische Toxizität des D1-PE laglediglich bei 100 nM und ist vergleichbar mit der Toxizität des freien E8C-PE38.
Untersuchungen verschiedener genetisch fusionierter rezeptorspezifischer Zytotoxinelieferten vergleichbare Werte. So konnte für ein Konstrukt, bestehend aus einerHelixstruktur, welche eine starke Affinität zum gp41 aufweist, in Fusion mit einemDerivat des Pseudomonas Exotoxins PE38 ein IC50-Wert für die CHO-Zelllinie CHO-wtvon 1,9 nM gezeigt werden. Die unspezifische Toxizität dieses Konstrukts nachInkubation auf Kontrollzelllinien lag bei über 200 nM (Tab. 4-9; Root et al., 2003).
Weitere Analysen von genetisch hergestellten Fusionen aus CD4-Varianten undToxinderivaten zeigen auf HIV-infizierten Zellen eine Inhibierung der Proteinbio-synthese mit ID50-Werten von 1,5 pM (Tsubota et al., 1990).
Die DNA-Synthese HIV-infizierter Zellen wurde durch ein Konstrukt aus CD4 und derA-Kette des Pflanzentoxins Ricin mit einem ID50-Wert von 0,15 nM inhibiert (Till et al.,1988). Spätere Analysen zeigen, dass dieses Konstrukt keinen Einfluss auf die B- oderT-Zellfunktion besitzt und somit zelltypspezifisch agiert (Till et al., 1990).
Die Eliminierung gp120-exponierender Zellen durch das hier dargestellte bifunktio-nelle Protein kann durch Koinkubation des Wildtypkonstrukts D1 verhindert werden.

4 ENTWICKLUNG EINES TARGETING-MODULS ZUR HIV-THERAPIE 77
Die Toxizität in diesem Ansatz war vergleichbar mit der unspezifischen Toxizität desE8C-PE38.
Dieser Versuch demonstriert, dass D1 in der Lage ist, eine Wechselwirkung zwischenCD4 und gp120 durch Kompetition zu unterbinden. In vergleichbaren Studien wurdedie HIV-Infektion durch Inkubation der Zelllinien mit einer CD4-Variante, bestehend ausden zwei N-terminalen Domänen, inhibiert (Traunecker et al., 1988; Hussey et al., 1988;Deen et al., 1988; Fischer et al., 1988). Die in dieser Arbeit durchgeführten Experimentezur Serumstabilität zeigen, dass das hergestellte Konstrukt D1 in der Lage ist, therapeu-tischen Anforderungen gerecht zu werden, und in ähnlichen Ansätzen Verwendung fin-den kann.
Tab. 4-9: Vergleich der IC50-Werte rezeptorspezifischer ZytotoxineIC50-Wert [M]
CHO-wt CHO-K1D1-PE 1,0·e-8 1,00·e-7
5-Helix–PE38 (Root et al., 2003) 1,9·e-9 2,00·e-7
Die in dieser Arbeit präsentierten Studien beweisen, dass das hergestellte Konstrukt D1-PE gp120-exponiernde Zellen zelltypspezifisch treffen und eliminieren kann. Studien anHIV-infizierten Zellen zeigen allerdings, dass die HIV-Replikation in diesen Zellen nurfür die Dauer der Inkubation des Zytotoxins auf den Zelllinien inhibiert wird. Nachabgeschlossener Behandlung der Zellen mit dem jeweiligen Konstrukt kann in denZellen die Reverse Transkriptase Aktivität des HIV wieder nachgewiesen werden(Tsubota et al., 1990).
Die Kombination aus Hybridmolekül und dem in der HIV-Therapie bereits eingesetz-ten Reverse Transkriptase Inhibitor (AZT) kann jedoch HIV-infizierte Zellen selektiv eli-minieren (Ashorn et al., 1990).
Klinische Studien eines rezeptorspezifischen Zytotoxins, bestehend aus CD4 undeinem Derivat des Pseudomonas Exotoxins, zeigen, dass diese Moleküle noch starkimmunogen sind (Davey et al., 1994). Für einen therapeutischen Einsatz rezeptorspezi-fischer Zytotoxine bedarf es daher noch einiger Optimierungen.
Aufgrund der Verwendung polyionischer Fusionspeptide als Dimerisierungsmodul istes im Gegensatz zu genetisch fusionierten möglich, die zellbindenden und zytotoxi-schen Komponenten separat herzustellen. Vor dem Hintergrund der klinischen Studienist es durch das vorgestellte System möglich, neue zytotoxische Proteine zu finden, dieeine wesentlich geringere Immunogenität aufweisen. Über das vorgestellteDimerisierungsmodul können relativ einfach und schnell verschiedene Toxine an dasKonstrukt D1R8C assoziiert und die Zytotoxizität sowie Immunogenität der Proteinegetestet werden.
Vorteil des hier beschriebenen Systems ist die Möglichkeit des Einsatzes des ProteinsD1R8C in verschiedenen Therapiemodellen. Durch das C-terminale Peptidsegment kanndieses Protein kovalent an andere Moleküle assoziiert werden. So besteht nicht nur dieMöglichkeit, das rekombinant hergestellte CD4-Konstrukt an ein Derivat desPseudomonas Exotoxins zu binden, es ist ebenso denkbar, D1R8C auf der Oberflächeeines virusanalogen Vektorsystems zu immobilisieren und somit einen zelltypspezifi-schen Gentransfer zu erreichen.
Grundlage für dieses System ist das Hüllprotein VP1 des Polyomavirus. VP1 kann invitro unter definierten Bedingungen virusanaloge Partikel bilden. Aufgrund derInsertion einer polyionischen Sequenz in einen oberflächenexponierten Loop des VP1besteht die Möglichkeit, durch ionische Wechselwirkung mit anschließenderDisulfidverbrückung Moleküle auf der Oberfläche der ausgebildeten Kapsidstrukturenkovalent zu binden (Stubenrauch et al., 2001).

78
Durch die Assoziation des D1R8C auf der Oberfläche solcher virusanaloger Partikelkann ein zelltypspezifischer Gentransfer in HIV-infizierte Zellen erreicht werden. DieFähigkeit des D1R8C Zelltypspezifität zu vermitteln wurde im ersten Teil der vorliegen-den Arbeit präsentiert.
Problematisch für einen solchen Gentransfer sind allerdings die niedrigenTransfektionsraten des modularen Vektorsystems. Eine Ursache hierfür kann die wenigeffiziente Nukleinsäureverpackung in die Partikel darstellen. Einen Lösungsansatzbehandeln die folgenden Kapitel.




























