Embed Size (px)
Citation preview

t{. KL]~ENT U. K. O. KNOLLMULLER : Analytische Chemie der Fluorophosphate 193
davon wurden dann na, ch der beschriebenen Arbeitsvorsehrift aufgearbeitet. Der Leerwert enthielt dieselbe Menge Alkohol. Die untersuchten Ester wurden dargest~ellt und bis zur extremen Reinheit im PIochvakuum destilliert.
3. Remdtate
In der Tabelle ist ein Tell der in groBer Anzahl durchgefiihrten Bestimmungen wiedergegeben.
Zusammenfassung
Eine grSi~ere Anzahl yon physiologisch interessanten Estern der Propions~ure, Buttersgure, Crotonsgure, Valeriansgure und Isovalerian- ss mit den Alkoholen Glykolmouomethyl~ther, Athylenglykol, Pro- pylenglykol und Glycerin wurde in der ffir Ester der Essigs~ure bereits beschriebenen Weise mit alk~lischen t tydroxylamin umgesetzt. Die Farbintensit~ten der innerkomplexen Eisen(III)-hydroxamate wurden gemessen. Der molare Extink~ionskoeffizient ist ffir alle untersuchten Ester, unabhgngig yon der Kettenl~nge, einer Verzweigung oder dem Vorhandensein yon Doppelbindungen in der Fetts~urekomponente 1,040, also gleich grol~ wie bei Es~,ern der Essigsgm~e. Ausfiihrliche Beleganalysen werden mitgeteil~.
Literatur
1 GOLDEnlY, he, V., u. P. E. S~OEn~: Analyt. Chemistry 30, 1327 (1958). -- P~Lz, W. : diese Z. 162, 81 (1958). -- ~ P~LZ, W. :Klin. Wschr. 36, 1017 (1958). --
a PILZ, W. :Klin. Wschr. (ira Druck).
Dr. W. Pmz, ~_rztliche Abteilung der Farbe~fubriken Bayer AG., Leverkusen
Aus dem Institut ffir Anorg~nisehe Chemie der Universit&t Miinehen
Beitr~ige zur analytischen Chemie der Fluorophosphate + Von
R. KLEMENT und K. 0. KNOLLMt3LLER
Mit 1 Textabbildung
(Eingegangen am 30. Oktober 1958)
Die durch die Arbeiten yon W. LARGE 3 bekannt gewordenen Fluoro- phosphorsguren und ihre Salze sind in analy~ischer I-Iinsich~ nur wenig untersucht worden. ~Tichtig erscheint es, die Prapara~e auf ihre l~eiuheit prfifen zu kSnnen, weil bei der tterstellung durch thermisehe I~eak~ionen oder durch hydrolytische Vorgiinge in wgl~rigen LSsungen Zersetzungen
* Aus der Diplomarbeit K. O. KNOLL)IffLLEa, Universitat Miinehen 1958. Z. analy~. Chem., Bd. 166 13

194 R . KLEI~ENT u n d K . O. K~OLL~fdLLEI~:
eintreten kSnnen, wobei insbesondere Monophosphat und Fluorid als Verunreinigungen in die Prgparate gelangen k5nnen. Wir haben deshalb sowohl quglitative als anch quantitative Analysenverfahren fiir Fluoro- phosphate, und zwar hier nut fiir Monofiuorophosphat P 0 sF ~- und Difiuorophosphu~ POzF2- im Gemisch mit ~onophosphat ausgearbeitet,
I. Qualitative Trennung yon Monophosphat, Monofluorophosphat und Difluorophosphat
a) Trennung durch Papierelektrophorese Wegen der Forderung, ~uch geringe Mengen des einen oder a~nderen
Bestandteiles nachweisen zu miissen, bietet sich die Papierelektrophorese als brauchbares Hilfsmittel an. Da abet Fluorophosphate sowohl in sauren als anch in alkalischen Medien leicht hydrolytisch ver~ndert werden, kann man nur bei ps 7 mit dem Veronalpuffer nach MICgAELIS arbeiten. Zur Verhinderung allzn groSer Sm'omw~rme wird dessen Ionen- st~rke dutch Verdiinnen mit Wasser im Verh~ltnis 1:1 herabgesetzt. Leider wandern die beiden Fluorophosphat-Anionen gleich schnell, abet dentlich schneller als Monophosphat, n~mlich bei 150 Vol~
PO~ s- : 3,2 cm/h, POsF z- und PO2F2- : 6,5 cm/h.
Nun kann man abet durch spezifische F~llungsreaktionen auf dem Papierstreifen einzelne Ionen an der Auftragstelle festlegen, wghrend das Ion, das yore F~llungsmittel unver~ndert bleibt, im elektrischen ]Peld wandert. So kann Monophosphat sowohl dutch Uranyl- als auch durch Bleiionen quantitativ ausgef~llt werden. Monofiuorophosphat wird nut dutch Bleiionen quantitativ ausgef~llt, w~hrend Uranylionen zun~chst ohne Einwirkung bleiben. Difiuorophosphat wird dutch keines der beiden Kationen niedergeschlagen. Tr~nkt man daher die Auftragstelle zweier Elektrophoresestreifen mit Uranyl- bzw. Bleiacetat in einer Breite yon 1 cm, so t r i t t folgendes ein: Von einem Gemisch der drei Anionen wird auf dem Streifen, der mit Uranylacetat pr~pariert ist, Monophosphat
Ion
1:)O43 -
PO3F ~- PO2F~-
Fixiert mi~ Fixiert mi t UO~(CH~COD~ Pb (0}IsC02)~
J
+ s i + 8
Nicht fixier t
+ + +
+ = Anion wundert -- = Anion wundert nicht S = Sehwanzbildung
unl5slich niedergeschlagen, wghrend Mono- und Difluorophosph~t im elektrischen Felde w~ndern. Tr~gt m~n d~s gleiehe Gemisch guf den Streifen ~uf, der mit Bleiseet~t getr~nkt ist, so werden Monophosph~t und Monofluorophosphat niedergesehl~gen, wghrend Difluorophosph~t

Analytische Chemie der Fluorophosphate 195
wandert. Nun zersetzen sieh aber die Fluorophosphate in L6sung bei Gegenwart soleher Ionen, die mit Fluorid bzw. mit Monophospha$ NiedersehNige oder Komplexe bilden, wie gerade mit Blei- oder Uranyl- ionen, au6erdem mig Zirkonium-, Thorium-, Magnesiumionen (bei An- wesenheit yon Ammoniak) und in untergeordnetem MaBe mit Silber- ionen. Da im Elektrophoresestreifen die Konzentration der zur Fixierung notwendigen Blei- bzw. Uranylionen ziemlieh hoeh ist, wird nieh~ nut das stSrende Ion ausgefiillt, sondern aueh dasjenige, das wandern sollte, teilweise zersetzt. Infolgedessen bildet dieses Ion keinen deutliehen Fleck, sondern es en~wiekelt sieh eine kontinuierliehe Zone, ,,Sehwanz", und gleiehzeitig is~ die Wanderungsweite herabgesetzt. Wie aus der Zusam- menstellung zu entnehmen ist, lassen sieh daher Mono- und Diflnoro- phosphat nieht trennen bzw. unterseheiden. Man muB deshalb zur Iden- tifizierung eines Monophosphat-Fluorophosphatgemisehes folgender- magen verfahren : Zungehst triigt man das Gemiseh auf einen Streifen auf, ohne zu fixieren. Bei der Elektrophorese werden die Fluorophosphate yon Monophosphat gem/~13 den oben angegebenen Wanderungsweiten gut getrennt. Nun pr~ipa- riert man einen Streifen mit Uranylaeetat und trggt das Gemiseh auf. Monophosphat ist jetzt am Startpunkt festge- legt, ein ,,Sehwanz" naeh dem Entwiekeln des Pherogramms zeigt entweder Mono- oder Difluorophosphat oder ein Gemiseh beider an.
AuF/zag- - - s t e / / e +
PO3F - ~ - PO3pZ.poJ2 -
po~ Fz - ++,?-]II~_
L Pore-l: I';I L eo+3- I,,i ii P03 F z - [ ~
PoJ~-/;lll; I
Po+ +- ~ iciiki'erunjs- DTeb~7t
Abb. 1. Papierelek~rophorese yon Gemischen Yon ~ono- phosphat mi t Fillorophosph~ten (naehgezeiohnet). a ohne Yixiertmg, b mi~ lJr~nylaeeta~, c and d mi t J31eiacet, a~ fixiert
Ein drifter Streifen wird an der Auf{ragsgelle mit Bleiaeetat behandelt. Beim Auftragen des unbekannten Gemisehes fallen Monophosphat und Mono- fluorophosphat aus, wghrend ein ,,Sehwanz" nnr aus Difluorophosphat herrtihren kann. Man kann also bei Anwesenhei~ yon Difluorophoslohat nur dieses mit Sieherheit nachweisen (auBer Monophosphat), wenn auf beiden Streifen ein, ,Sehwanz" ents~eht. Dabei wird Monofluorophosphat nicht erfagt. Entsteht ein ,,Sehwanz" auf dem mit Uranylaeetat lorgpa- rierten Streifen, w/~hrend auf dem mit Bleiaeetat behandelten Streifen alle Ionen am Startpunkt bleiben, so liegt neben Monophosphat nur Monofluorophosphat vor (siehe Abb. 1).
13"

196 R. KLE~ENT und K. 0. KNOLL~IOLLEI%:
Monofiuoroohosphat kann also papierelektrophoretisch nich~ ein- deu~ig neben Difluorophosphat bestimmt werden. Seine einwandfreie Identifizierung in Gemischen mit Monophosphat und Difluorophosphat ist darch den nachfolgend beschriebenen Trennungsgang mSglich.
b) Trennung dutch Fi~llungen Man f~llt aus der neutrMen LSsung (zur Vermeidung yon Zersetzungen)
zuerst mit Uranylaeetat das Monophosphat quantitativ aus. Der Nieder- schl~g wird schnell abfiltriert oder abzentrifugiert. Enth~l~ das yon tiber- schiissigem Uranylsalz gelb gefgrbte Filtrat Phosphor und Fluor, was man leicht durch Priifen mit Ammoniummolybdat bzw. mit Zirkonium- Alizarinl6sung feststellen kann, so liegen ia der LSsung entweder Mono- oder Difluorophosphat oder beide gemeinsam vor. Man priift nun in einem kleinen Teil der LSsung mit Benzidinacetat ~uf das Vorhandensein yon Monofluorophosphat, d~s mit diesem l~eagens im Gegensa~z zu Difluorophosphat einen Niederschlag bildet. Bei positivem Ausfall ver- setzt man das Filtrat mit soviel BleiacetatlSsung, bis keine Fgllung yon Bleimonofluorophosphat mehr eintritt. ])as Mare Fil trat yon diesem Niederschlag enth~lt jetzt nur noeh Difiuorophosphut, dessen Anwesen- heir dadurch nachgewiesen wird, dal~ man in dem in zwei Teile geteilten Fil trat auf Phosphat mit Ammoniummolybdat bzw. auf Fluor mit Zirkonium-AlizarinlSsung priift. Da andere Phosphate und ~'luorid, die durch Zerse~zung entstanden sein kSnnten, sehon zuvor ausgef~llt sind, k6nnen die naehgewiesenen Elemente nm�9 yon Difiuorophosphat stammen (siehe das Schema).
F&llungsmittel PO~ s- POaF ~- PO2F 2-
UO~ (CHa C02)2 go2 HP04 POaF 2- PO~ F 2-
Pb(CH3 C02 )2 !, PbPO~ F PO 2 F.-
II. 51achweis yon Fluorid neben Fluorophosphat
Der Nachweis yon Fluorid neben Fluorophosphaten l~13t sieh in esslgsaures LSsung mit Zirkonium-AlizarinlSsung ausffihren, well in diesem Medium die Sanrehydrolyse der Fluorophosphate langsam genug verlguft, um den dureh Fluorid bewirkten (Sbergang der rotvioletten ~grbung des t~eagenses in Gelb sieher beobachten zu kSnnen. Bei An- wesenheit yon Fluorid t r i t t dieser Farbumsehlag sehr schnell ein. Bei Vorliegen eines Fluorophosphates d~uert es aber einige Minuten, bia dureh Hydrolyse genug Fluorid entstanden ist, um den Fa.rbumsehlag zu bewirken. Man fiihrt die Probe folgendermagen aus: In ein Reagens- glas bringt man einige Kristalle Zirkoniumoxychlorid, die man in 1 ml Wasser 16st. Dann ffigt man 5 Tropfen einer 0,2~ LSsung yon N~triumalizarinsulfonat und 5 ml 10~ Essigs~ure hinzn. I-Iierauf erfolgt der Zusatz yon 1 ml ProbelSsung.

Analytische Chemie der Fluorophosphate 197
III. Vollst~indige Analyse yon Fluorophosphaten Die Bestimmung des Phosphors und der Ka.tionen in reinen Fluoro-
phosphaten ist nieht besonders sehwierig, da man heutzutage die bei der Hydrolyse durch verdfinnte Si~ure entstehende Phosphors~ure mit ~Iilfe eines Ionenaustauschers leicht yon den Kationen trennen und dann beide Teile nach bekannten Verfahren bestimmen kann.
Zur Bestimmung des Fluors muB dieses yon der Phosphorsi~ure ab- getrennt werden, wozu am besten ein Destillationsverfahren dient. Wenn man die etwas modifizierte Apparatur nach WILLA~D und WI_wT~a 5 verwende~, kann man in der fiberdestillierten L6sung der Silicofluor- wasserstoffsgure das Fluorid als Bleibromfluorid naeh der passend ver- gnderten 5[ethode yon E~I~LTCK und PIETZKA 1 bestimmen.
IV. Bestimmung yon Monophosphat und Fluorophosphaten nebeneinander
Uns ist es wichtig erschienen, Monophosphat und Mono- bzw. Difluoro- phosphat nebeneinander zu bestimmen, weil jenes in wgl3rigen Systemen immer als t tydrolyseprodukt vorhanden sein kann. Da aueh hier die Forderung besteht, m~r in neutralem Medium mSgliehst bei l~aum- temperatur zu arbeiten, so sind yon vornherein nur wenige .~[Sglichkeiten vorhanden. Als am besten brauehbar zeigt sieh noeh die Fgllung des 5'[onophosphates dutch Silber und seine iudirekte Bestimmung dureh Titration des Silbergehaltes im Niedersehlag oder dutch i~fiektitration des fibersehiissigen Silbers in einem aliquoten Teile des Filtrates. Das Fluorophosphat wird im Filtrat yon diesem Silberphosphat-Niedersehlag bestimmt, naehdem es dureh verdiinnte Salpetersi~ure hydrolysiert ist. Die entstandene Monophosphors~ure ~drd ~deder fiber ihr Silbersalz bestimmt und der erhaltene Wert auf Fluorophosphat bereehnet. ])as welter unten besehriebene Verfahren liefert zwar ffir das 5{onophosphat brauehbare Werte mit einem Fehler yon etwa ~ 0,30/0; aber ffir den J~all des allein untersuehten Monofluorophosphates treten Fehler bis zu --40/0 auf. ])eshalb kann nut eine indirekte ]~estimmung des Fluoro- phosphates neben Monophospha~ angewendet werden, die zu guten Werten fiihrt (siehe Tab. 3, S. 200).
Besehreibung der Versuehe I. Yrennung von Monophosphat, Mono- und Difluo~'ophosphat
dutch Papie~dektrophorese Es wird in der Elektrophoresekammer nach GRaSS~AN~ u. H ~ I ~ 2 auf Papier-
streifen aus Schl. & Sch.-Papier Nr. 2040 a (ausgewasehen) bei einer Spannung yon 150 Volt gearbeitet. Die Strelfen werden mit dem Veronalpuffer naeh l~'[Ie~a~LIS yore pjz-Wert 7 getr/~nkt, dessen Ionenst~rke dureh Verdfinnen mi~ Wasser im Verhfiltnis 1 : 1 herabgesetzt ist.
Zum Nachweis der Anionen nebeneinander tr~gt man die Probel6sung mit ins- gesamt etwa 5 #g P a.uf einen nieht mit einem Fixierungsmittel behandelten Streifen

198 ~ . KLE~CIENT und K. O. K~OLL~i3LL~ :
auf. Zwei andere Streifen werden an der Auftragstelle in einem etwa 1 em breiten, senkreeht zur Lingsr ichtung des Papierstreifens angeordneten Streifen mit 5~ Uranyl- bzw. 5~ BleiaeetatlOsung getr~nkt. Nun ' wird erst die ProbelSsung aufgetragen. Dann l~l]t man die Elektrophorese 1,5 Std lung laufen. Auf dem ersten Streifen erh~lt man nach dem Troeknen, Besprfihen mit perchlorsaurer Ammonium- molybdatlSsung*, Troeknen und Entwiekeln unter UV-Licht 4 zwei blaue Flecke gemiB Abb. 1 a. Auf dem zweiten, rnit Uranylaceta t behandel ten Streifen sieht man naeh der Entwieklung einen lung gezogenen, blauen Fleck (,,Sehwanz"), der auf die Anwesenheit yon 1Vfono- und Difluorophosphat deutet (Abb. i b). Tr i t t der , ,Sehwanz" auch auf dem drit ten, mi t Bleiaeetat behandel ten und entwiekelten Streifen auf, so ist Difluorophosphat sieher vorhanden (Abb. 1 e). Bleibt der , ,Sehwanz" auf diesem dr i t ten Streifen aus, so ist Difluorophosphat sieher abwesend (siehe Abb. 1 d). Monofluorophosphat 1/~gt sieh aber nach diesem Verfahren nicht sieher naehweisen.
I I . Quantitative Bestimmung dee Fluors in Fluorophosphaten
a) Die Destillation des J~luors als Silicofluorwaserstoffsgure. Der seitliche Ansatz eines Claisenkolbens yon 250 ml Inha l t wird so verli~ngert, dug das Ableitungsrohr fiir die Di~mpfe mindestens 10 cm oberhalb der Krf immung ange- setzt wird. In der Kri immung werden 3 - -4 Einstfi lpungen angebracht, die eim ~berspr i t zen des KolbeninhMtes weitgehend verhindern. Der Ansatz wird mi t einem Gummistopfen verschlossen. Durch einen doppelt gebohrten Gummistopfen werden in den Kolben bis auf dessen Boden ein Thermometer und ein Tropftr ichter mit H a h n und Capillarrohr eingefiihrt. Der Kolben s teht in einer Asbestplat te in einem passend eingeschnit tenen Loch. Einige Zent imeter unterhalb seines Bodens befindet sich ein Asbestdrahtnetz . Die Heizung erfolgt durch zwei starke Bunsen- brenner. An das senkrecht nach un ten abgebogene Ablei tungsrohr des Kolbens wird ein gerader Kfihler von 20 cm L~nge angeschlossen. Das Destillat wird durch ein gebogenes Ablauffohr yon etwa 10 cm Lhnge in eine auf einer Heizplatte stehende Porzellanscha.Ie yon 12-- 15 cm Durehmesser geleitet, we es sofort eingedampft wird.
Die Einwaage, die i5 - -25 mg F entha l ten sell, wird mit etwas Wasser in den Kolben gespfilt. Es werden 1 g Qu~rzpulver, eine Spatelspitze Tierkohle und etwa 10 Tropfen 2 n Natronlauge zugegeben. Nun destilliert man zuerst das Wasser bis ~uf einen Rest yon 10 ml ab. Nach dem Abkiihlen versetzt man den KolbeninhMt mit 50 ml Schwefelsiure (100 Teile konz. S iure und 60 Teile Wasser) und verschl iegt den Kolben sofort mit dem Stopfen. Der Tropf~richter wird mi t Wasser gefiillt. Die Quecksilberkugel des Thermometers muB w~hrend der ganzen Bauer der Destilla- t ion in die Fliissigkeit eintauchen. Nun stell t man die Porzellanschale auf die Heiz- platte, fiillt sie mit 5 ml 2 n Natronlauge und etwas Wasser und ffigt einen Tropfen PhenolphthaleinlSsung hinzu, um w i h r end der Destillation zu kontrollieren, dM~ die L6sung d~uernd Mkaliseh bleibt. Der H a h n am Tropftr ichter bleibt gesehlossen, w~hrend man die Appara tur anheizt. Wenn die Tempera tur der Mischung auf 130~ angestiegen ist, h~lt man sie durch ZufluB yon Wasser aus dem Tropftr ichter inner- halb yon 5~ konstant . Bereits w i h r end der Destill~tion wird das Destillat ein- gedampft, ohne es zum Sieden zu erhitzen. Man destilliert etwa 150 ml Fltissigkeit fiber (gemessen am Wa~serverbrauch aus dem Tropftrichter). Bei Bleifluorophospha- t e a empfiehlt sich die Destillation yon 300 ml. Nach Beendigung der Desti l lat ion spfilt man den Kiihler mit Wasser gut aus und dampf t das Destillat auf ein Volumen yon etwa 25 ml ein.
* 1,5 g Ammoniummolybda t werden in 195 nil Wasser gel5st. Dazu werden 5 ml 60~ Perehlors~ure gegeben.
Analytische Chemie der Fluorophosphate 199
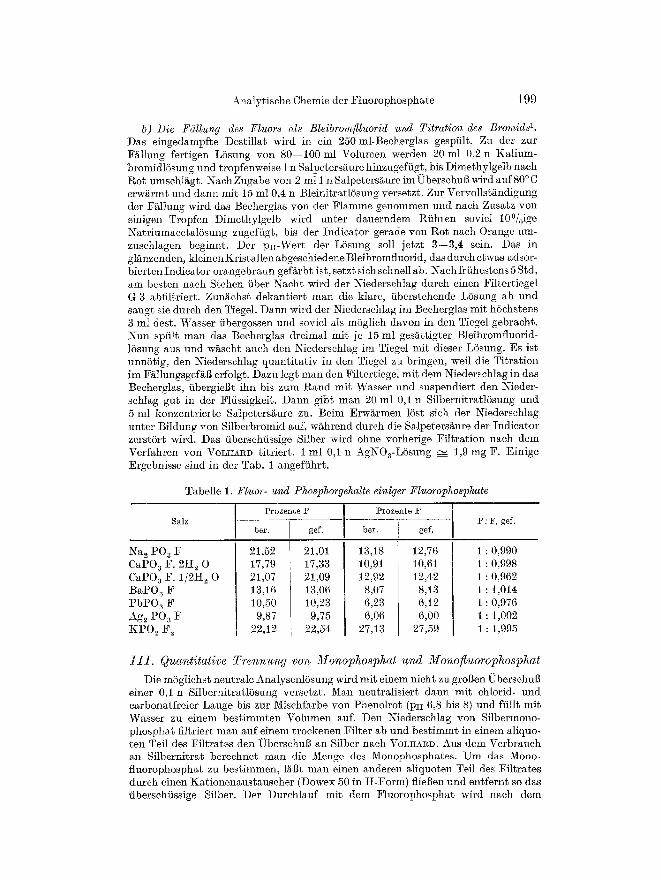
b) Die F~illung des Fluors als Bleibromflluorid und Titration des Bromids 1. Das eingedampfte Destillat wird in ein 250 ml-Beeherglas gespiilt. Zu der zur F~llung fertigen L6sung von 80--100 ml Volumen werden 20 ml 0,2 n Kalium- bromidlSsung End tropfenweise 1 n Salpeters~ure hinzugeffigt, bis Dimethylgelb nach l~ot umsehli~gt. Naeh Zugabe yon 2 ml i n SMpeters~ure im ~bersehuB wird auf 80 ~ C erw~rmt und dann mit 15 ml 0,4 n Bleinitratl6sung versetzt. Zur Vervollst~ndigung der Fi~llung wird das Beeherglas yon der Flamme genommen und nach Zusatz yon einigen Tropfen Dimethylgelb wird unter dauerndem Rfihren soviel 10~ NatriumaeetalSsung zugefiigt, bis der Indicator gerade yon Rot nach Orange mn- zusehlagen beginnt. Der pH-Wert der LSsung soll jetzt 3--3,4 sein. Das in gl~nzenden, kleinen Kristallen abgesehiedene Bleibromfluorid, das dureh etwas adsor- bierten Indicator orangebraun gefs ist, setzt sieh sehnell ab. Nach friihestens 5 Std, am besten naeh Stehen fiber Naeht wird der Niedersehlag durch einen Filtertiegel G 3 abfiltriert. Zun~ehst dekant ier t man die Mare, i iberstehende L6sung ab und saugt sie dureh den Tiegel. Dann wird der Niedersehlag im Becherglas mit hSchstens 3 mI dest. Wasser fibergossen und soviel Ms m6glieh davon in den Tiegel gebracht. Nun spiilt man das Beeherglas dreimal mit je 15 ml ges~ttigter Bleibromfluorid- 16sung aus und wi~scht auch den Niederschlag im Tiegel mit dieser L6sung. Es ist unn5tig, den Niedersehlag quant i ta t iv in den Tiegel zu bringen, weil die Ti t ra t ion im FMlungsgef~B erfolgt. Dazu legt man den Filtertiegel mit dem Niedersehlag in das Becherglas, fibergieBt ihn his zum Rand mit Wasser und suspendiert den Nieder- sehlag gut in der Flfissigkeit. Dann gibt man 20 ml 03 n Silbernitrat]Ssung und 5 ml konzentrierte Salpeters~ure zu. Beim Erwarmen 16st sieh der Niederschlag unter BiIdung von Silberbromid auf, w~hrend dureh die SMpeters~ure der Indicator zerst6rt wird. Das fiberschfissige Silber wird ohne vorherige Fi l t ra t ion naeh dem Verfahren yon ~TOLHARD titriert . 1 ml 0 3 n AgNO3-LSsung --~ 1,9 mg F. Einige Ergebnisse sind in der Tab. 1 angeffihrt.
Tabelle 1. Fluor- and Phosphorgehalte einiger Fluorophosphate
Salz
Na 2 PO 3 F CaPO a F. 2If 2 0
CaPO a F. I/2H 2 0
BaPO 3 F
PbPO 3 F
Ag2 PO~ F KPO2 F2
Prozente P
ber. gef.
21,52 21,01 17,79 17,33 21,07 21,09 13,16 13,06 10,50 10,23
9,87 9,75 22,12 22,54
Prozente
bee i gel.
13,18 12,76 10,91 10,61 12,92 12,42
8,07 8,13 6,23 6,12 6,06 6,00
27,13 27,59
P : F, get".
1 : 0,990 1 : 0,998 1 : 0,962 1 : 1,014 1 : 0,976 1 : 1,002 1 : 1,995
I I I . Quantitative Trennung von Monophosphat und Monofluorophosphat
Die mSglichst neutra]e AnMysenl6sung wird mit einem nicht zu groBen lJberschuB einer 0,1 n Silbernitratl6sung versetzt. Man neutralisiert dann mi t chlorid- und carbonatfreier Lauge bis zur Misehfarbe yon Phenolrot (p~ 6,8 bis 8) und ftillt mi t Wasser zu einem bes t immten Vohmlen auf. Den Niederschlag yon Silbermono- phosphat filtriert man auf einem trockenen Fil ter ab und bes t immt in einem Miquo- t en Teil des Fil trates den IJbersehug an Silber naeh VOLm~nD. Aus dem Verbraueh an Silberni trat bereehnet man die Z~r des Monophosphates. Um das Mono- fluorophosphat zu bestimmen, 1/~l]t man einen anderen Miquoten Teil des Fil trates du tch einen Kat ionenaustauscher (Dowex 50 in H-Form) fliegen und entfernt so das fibersehfissige Silber. Der Durehlauf mit dem Fluorophosphat wird naeh dem
200 KLEMENT und K~OLLMiiLL~: Anatytische Chemie der Fluorophosphate
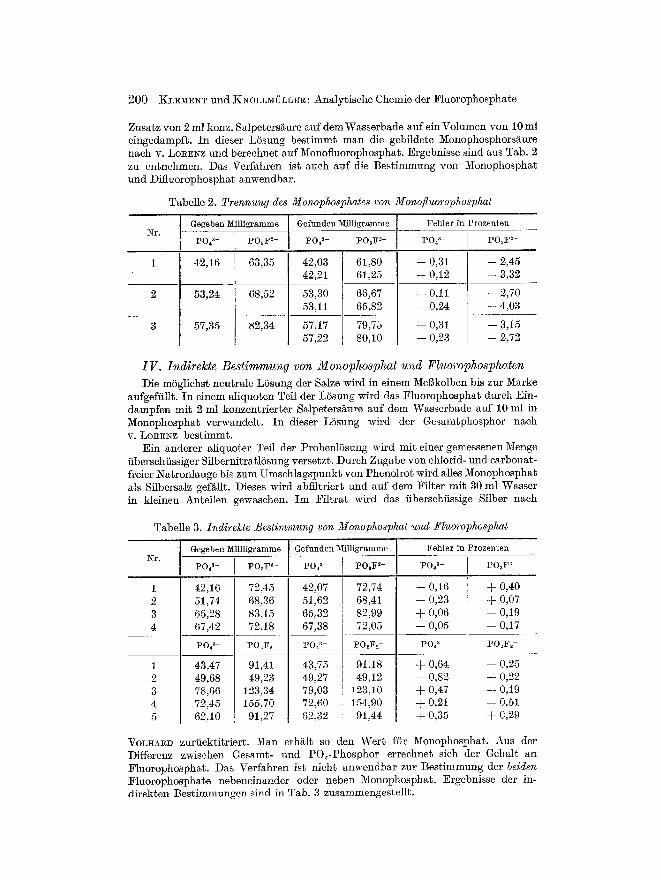
Zusatz yon 2 ml konz. Salpetersi~ure auf demWasserbade auf ein Volumen yon 10 ml eingedampf~. In dieser L5sung bes t immt man die gebildete Monophosphorsi~ure nach v. LOlCE~Z und bereehne$ auf Monofluorophosphat. Ergebnisse sind aus Tab. 2 zu entnehmen. ])as Verfahren ist aueh auf die Best immung yon Monophosphat und ])ifluorophosphat anwendbar.
Tabelle 2. Trennung des Monophosphates yon Monofluorophosphat
~r. Gegeben Minigramme
P04 S- [ PO~ F2-
42,16 63,35
53,24 68,52
57,35
Gefunden ~r i
PO~ S- ] P03 F2-
42,03 61,80 42,21 61,25
53,30 66,67 53,11 65,82
Fehler in Prozenten
p04 a- PO3F 2-
-- 0,31 -- 2,45 ~- 0,12 -- 3,32
-- 0,11 -- 2,70 -- 0,24 -- 4,03
- - 0 , 3 1 - - 3,15 -- 0,23 -- 2,72
IV. Indirekte Bestimmung von Monophosphat und Fluorophosphaten ])ie m5gliehst~ neutrale LSsung der Salze wird in einem MeBkolben bis zur 1V~arke
aufgefiillt. In einem aliquoten Tefl der LSsung wird das Fluorophosphat dutch Ein- dampfen mit 2 ml konzentr ier ter Salpeters/~ure auf dem Wasserbade auf 10 ml in Monophosphat verwandelt. In dieser LSsung wird der Gesamtphosphor nach v. LoREnz best immt.
Ein anderer aliquoter Tell der ProbenlSsung wh'd mit einer gemessenen ~enge fiberschfissiger SflbernitratlSsung versetzt. Durch Zugabe yon chlorid- und carbonat- freier/qatronlauge bis zum Umschlagspunkt yon Phenolrot wird alles Monophosphat als Silbersalz gef~llt. ])ieses wird abfiltriert und auf dem Filter mit 30 ml Wasser in kleinen Antei]en gewasehen. Im Fi l t ra t wird das iiberschfissige Sflber nach
Tabelle 3. Indirekte Bestimmung von Monophosphat und Fluorophosphat
Gegeben 1V[illligramme Gefunden Milligramme Fehler in Prozenten
p04s- P O , F ~-
4 2 , 1 6 72,45 51,74 68,36 65,28 83,15 67,42 72,18
PO, ~- PO2F~
43,47 91,41 49,68 49,23 78,66 123,34 72,45 155,10 62,10 91,27
po43- POsF 2-
4 2 , 0 7 72,74 51,62 68,41 65,32 82,99 67,38 72,05
P043- PO~F2
43,75 91,18 49,27 49,12 79,03 123,10 72,60 154,90 62,32 91,44
p0~3- PO3F 2-
-- 0,16 + 0,40 -- 0,23 ~- 0,07 + 0,06 -- 0,19 -- 0,05 -- 0,17
PO43 PO2F2-
-+- 0,64 -- 0,25 -- 0,82 -- 0,22 ~- 0,47 -- 0,19 + 0,21 -- 0,51 + 0,35 + 0,29
VOLHA~D zuriiektitriert. Man erh~it so den Wert ffir l~ionophospha~. Aus der Differenz zwisehen Gesamt- und PO~-Phosphor errechnet sich der Gehalt an Fluorophosphat. ])as Verfahren ist nicht anwendbar zur Bestimmung der beiden Fluorophosphate nebeneinander oder neben Monophoslohat. Ergebnisse der in- direkten Bestimmungen sind in Tab. 3 zusammengestellt.

Bericht: Spez. analyt. Methoden. 2. Analyse v. Materialien der Industrie usw. 201
Zusammenfassung
Es w e r d e n Vor seh r i f t en m i t g e t e i l t f i ir den q u a l i t a t i v e n N a c h w e i s v o n
M o n o p h o s p h a t , Mono- n n d D i f l u o r o p h o s p h a t n e b e n e i n a n d e r d n r c h
P a p i e r e l e k t r o p h o r e s e u n d d u r e h F ~ l l u n g e n ; fiir den q u a l i t a t i v e n N a e h -
weis y o n F l u o r i d n e b e n F l u o r o p h o s p h a t e n ; fiir die q u a n t i t a t i v e A n a l y s e
y o n F l u o r o p h o s p h a t e n , i n sbesonde re ftir die F l u o r b e s t i m m u n g u n d fi ir
die q u a n t i t a t i v e B e s t i m n m n g y o n M o n o p h o s p h a t u n d F ! u o r o p h o s p h a t e n
n e b e n e i n a n d e r .
I-Ierrn Dr. It . JONAS, Farbenfabriken Bayer, Leverkusen, sei aueh hier bestens gedankt fiir die Versorgung mit reinem Kaliumdifluoroiohosphat. Yiir die materielle Unterst/itzung gebfihrt dem Fends der Chemischen Industrie aufriehtiger Dank.
Literatur 1 EHR.LICI% P., U. G. PIETZI~: diese Z. 133, 84 (1951). - - PIETZE_4, G., n. P. EHtC-
ucH: Angew. Chem. 65, 131 (1953); vgl. diese Z. 141, 149 (1954). -- 2 G~ASS~ANX, W., u. K. HA.W~m: Hoppe-Seylers Z. physiol. Chem. 290, 1 (1952); vgl. diese Z. 140, 293 (1953). - - ~ LARGE, W.: Ber. dtseh, chem. Ges. 60, 965 (1927). -- 4 SA~so~I, B. : Angew. Chem. 65, 423 (1953); vgl. diese Z. 141,464 (1954). -- ~ ~u H. H., U. O. B. ~u Ind. Engng. Chem., anal. Edit. 5, 7 (1933); vgl. diese Z. 96, 213 (1934). -- Vgl. auch Handb. d. analyt. Chemie, herausgegeb, yon W. F~ESEXI(;S u. G. J~ND~R, III . Tell, Band VII a ~, Fluor, bearb, v. g . K L E ~ n ~ , S. 187.
Prof. Dr. 1~. KLEMEI~'T, Mfinchen 22, Kaulbachstral~e 59
Bericht iiber die Fortschritte der analytisehen Chemie
IV. Spezielle analytische Methoden
2. Analyse y o n Materialien der Indnstrie, des Handels
und der Landwirtsehaft
Eine Analysenmethode fiir teehnische Gase~ die aus H2S ~ CS~ und COS bestehen~ wird yon K. E. PEREPELKI~ und JA. Z. Se~oxL'~ 1 angegeben. In einer Reihe yon Waschflaschen wird zuerst der Sehwefelwasserstoff durch Absorption in einer Zink- oder CadmiumacetatlOsung und jodometrisehe Titration bestimmt und dann die Summe CS2 und COS dureh Absorption in alkoholischer Kalilauge. Die ent- standenen Xanthogenate werden ebenfalls jodometrisch titriert, wobei 23{ol Xanthogenat 1 Mol Jod erfordern. In einer zweiten Probe wird dann nach Ent- fernung des Schwefe]wasserstoffs dureh Verbrennung der Gesamtschwefelgehalt bestimmt, indem man das entstandene SO 2 durch 3~ Wasserstoffperoxyd- 15sung oxydiert und die Schwefels~ure aeidimetrisch titriert. Aus den Ti~rations- ergebnissen lassen sieh CS 2 und COS einfach berechnem
1 Zavodskaja Laborat. 23, t414--1417 (1957). W. B 6 ~
Kupfer und Zink in Legierungen bestimmen V. A. C~ADEJEV und A . K . ~D.~XOV ~ dnreh amperometrische Titration. Die Titration ]iii~t sieh in Gegenwart zahlreicher Me~ailionen (3{g 2+, Fe 2§ 3fn 2+, Ni 2+, Co 2+, Zn ~+, Cr a+, A13+, Cd e+





























