Embed Size (px)
Citation preview

Beiträge zur maßanalytisehen Bestimmung der Borsäure. E r s t e 2¢ii~teilu hg1).
Von
Harald Schäfer und Adolf Sieverts.
iEingegangen am 9. Dezember :t940.]
Die nlaßanalytische Bestimmung der ]~orsäure erfolgt wohl stets nach Aktivierung mit Mannit, Invertzucker oder Glycerin durch Titrat ion auf den ]?a-Wert von etwa 8 gegen Indikatoren wie a-Naphtholphthalein oder Phenolphthalein2). Eine große Anzahl von Schwermetallsalzen stört diese übliche Borsäuretitration. Sie verursachen positive Fehler dadurch, daß sie bei der Titrat ion hydrolytisch gespalten werden, und negative Fehler durch Adsorption von Borsäure an den ausfallenden Hydroxydniederschlag. Viele Veröffentlichungen beschäftigen sich damit, die störenden Metalle (z. ]3. A1, Fe, Zn) als Hydroxyde auszufällen. Alle diese Verfahren haben die gleiche Grundlage, nur wird die I~eutralisation der freien und der an die störenden Metalle gebundenen Säure mit ver- schiedenen Mitteln vorgenommen [so werden z. ]3. lqaOI-I, ]~~a2COa, Ba(OH)2, CaCOa, BaCO a Verwendet]. Sie leiden daher auch sämtlich an dem gleichen Fehler, nämlich den Borverlusten infolge Adsorption und Einschluß von Borsäure durch den Hydroxydniederschlag. Diese Bor- mengen lassen sich nur durch mehrfaches Auflösen und Wiederfällen des 1Niederschlags gewinnen. Dadurch bringt man aber wieder größere Mengen der störenden Metalle mit in Lösung, sodaß eine 5Iachfälhmg erforderlich wird [W. Myliusa)] . Verzichtet man auf diese Nachfällung, so kommt man zu dem Urteil von E. T. Wher ry~) , der zu seiner eigenen Metho:le schreibt, daß man zwar die durch die Sesquioxyde mitgerissene Borsäure durch mehrfaches Auflösen und Wiederfällen gewinnen kann, daß aber die Ergebnisse mit jeder Fällung unsicherer werden.
I m folgenden Abschnitt ist eine Auswahl von Veröffentlichungen zusammengestellt, von denen sich die meisten mit der Ausfällung der Schwermetalle als I-Iydroxyde und der darauf folgenden Titrat ion der Borsälire befassen.
1) Nach der gleichnalnigen Dissertation von H a r a l d Schä fe r , Jena 1940 (gekürzt). (D. 27.) -- 3) In der zweiten Mitteilung wird eine Arbeitsweise mit höheren Fructose- oder MannitkonzentrationerL beschrieben werden, die erhebliche Vorteile bietet. -- a) Chem. Ztg. 57, t73, t94 (1933); vgl. diese Ztschrft. 96, ä33, i34 0934). 7- 4) Journ. Americ. Chem. Soc. 30, 1687 (1908); vgl. diese Ztschrft. 55, 346, 599 (1916).
Ztsohrf t . f. anal . Chem. 121, 5. u. 6. H e f t . ~1i

162 I-Iarald Schäfer und Adolf Sieverts:
Einen Überblick über die Verschiedensten Verfahren zur Borbestimm- ung findet man in G m e l i n s Handbuch der anorganischen Chemiel). W. S t r e c k e r und E. K a n n a p p e l 2) empfehlen nach kritischer Nach- prüfung zahlreicher Methoden die Abtrennnng der Borsäure durch Destil- lation als Methylester. W. W. S c o t t 3) sowie t l . L. Po~yne 4) entfernen bei Borbestimmungen in Bormineralien das Aluminium und das Eisen durch Fällung mit N a t r o n l a u g e . Beim Einstellen auf den px-Wert 6 soll der Niederschlag borfrei sein. Nach L. C. P a n 5) wird die Titrat ion von Bot- säure in Nickelbädern nach Ausfällung des Nickels mit N a t r o n l a u g e vorgenommen. An Stelle von Natronlauge verwendet W. M y l i n s 6) B a r i u m h y d r o x y d l ö s u n g zur Entfernung von SiO2, A120» ZnO und C02. Nach seinen Erfahrungen wird die Borsäure nur bei dreimal wieder- holter Fällung vollständig im Fil trat gefunden. Zur Abseheidung noch gelöster l~este von störenden Stoffen werden die vereinigten ]~'iltrate nochmals mit Ba(OH)2 behandelt. Bei Gegenwart von Zink ist außer- dem die Zugabe von Ferrocyanid erforderlich. Die Fällung mit Sodo~- l ö s u n g wird von W. t~. C h o t z i a l o w a 7) zur Abtrennung von Zink, Alu- minium und Eisen verwendet, während ~. P. A l i m a r i n und I . I . t ~ o m m s) zur Fällnng von Kieselsäure und Aluminium A m m o n i u m - c a r b o n a t benutzen. Sie führen bei ihren Analysen einen Korrektur- faktor ein. 3/[. I . iönig und G . S p i t z 9) bestimmen Bor in Gläsern. I m Anschluß an das Aufschließen mit NaKC03 fällen sie die Kieselsäure mit A m m o n i u m e h l o r i d und Z i n k o x y d - A m m o n i a k . Nach dem Ver- kochen des Ammoniaks wird im Fil trat die Borsäure in bekannter Weise titriert. Ähnlich arbeiten P. N i c o l a r d o t und J . Boude t l ° ) . Die Ver- wendung von C a l e i u m e a r b o n a t zur Neutralisation der freien Säure und Ausfätlung der störenden Stoffe wurde zuerst von E. T. W h e r r y und W. I-I. C h a p i n l~) empfohlen. Dieses Verfahren haben I-I. und W. B i l t z in ihr bekanntes Lehrbuch der analytischen Chemie (3. Aufl. t940, S. 392/393) übernommen. Aus W h e r r y s Versuchen folgt, daß zum Teil erst nach fünffaeh wiederholter Auflösung und Fällung der Niedersehläge eine nahezu vollständige Abtrennung der Borsäure gelingt. V i o l e t D i m - b l ë b y 1~) und P. G r i g o r j e l f 1~) verwenden dieses Verfahren mit doppelter
1) 8. Aufl., Nr. 13 (Ber), S. 42--53 (Literatur bis 1~25). - - 2) Diese Ztsehrft. 61, 378 (1922). -- 3) Vgl. diese Ztsehrft. 96, 131 (Œ934). -- ~) Vgl. diese ZtschAt. 101, 58 (1935). -- 5) Metal Clean. Finish. 3, 961 (t931); durch Chem. Zentrbl. 103, I I , 575 (1932)o -- s) Chem. Ztg. 57, 173, t94 (t933); vgl. diese Ztsehrf~. 96, 133, 134 (1934)o -- ~) Vgl. diese Ztsehrft. 118, 277 (1939/40). -- s) Vgl. diese Ztsclirft. 96, 133 (t934). -- 9)Ztsehrf~. f. angew. Chem. 9, 549 (t896); vgl. diese Ztsehrft. 42, t i9 (1903). -- lo) Bull. Soe. chim. de l~"ranee [4J 21, 97 (t9~7). -- 11) Journ. Americ. Chem. S0e. 3O, 1687 (1908); vgl. diese Ztschrft. 55, 346 (t916); 61, 132 (1922). -- 12) Vgl. diese Ztschrft. 96, t3~ (193~). , la) Sprechsaal Keramik, Glas, Email 66, 388 (t933); durch Chem. Zentrbl. 104, I I , 924 (t933).

Beitr&ge zur maßanalytisehen Bestimmung der ]~orsäure I. t63
F~llung zur Borbestimmung in Gläsern, wäl~rend A. Ma t sch ig in und T. K o r s u c h i n a 1) für den gleichen Zweck die Ausfällung störender Stoffe mit B a r i u m e a r b o n a t bei Gegenwart von Bariumehlorid benutzen. H. F u n k und H. W i n t e r 2) untersuchten die Methode der Methy]ester- destillation bei Gegenwart von Aluminium-, Chron» und Eisensalzen und fanden, daß Borsäure nicht quantitativ als Methylester abdestilliert werden kann, wenn größere Mengen dieser Salze vorliegen. Sie ver- suchten daher, die störenden Verbindungen vor der Destillation durch Fallung zu entfernen. Aluminiumfällung mit Ammoniak, Ammonium- earbonat, Ammoniumsulfid und Natriumthiosulfat lieferte immer bor- haltige Niederschl~ge. Borfreie Ausfällung Von Aluminium war möglich in essigsaurer Lösung als Phosphat oder bei Außf~Ilung als tIydroxyd durch Abfangen der Mineralsaure mit Jodid-Jodatiouffer. Das Jodid- Jodatverfahren' war auch zur Abtrennung von Chrom anwendbar. Eisen wurde entweder als Phosphat oder. als Sulfid gefällt. Im Anschluß daran folgte die Destillation des Bors~uremethylesters. Nach E. S chu lek und G. Vas t agh ~) ist dagegen eine Abtrennung störender Stoffe vor der Destillation des Bors~uremethylesters nicht erforderlich. Ganz anders ist schließlich der von F. W. Glaze und A. N. F inn 4) beschrittene Weg. Sie bestimmen Ber in Gl&sern auf folgende Weise: Das Glas wird mit Soda aufgeschlossen und die schwach salzsaure Lösung mit einem Alkohol- Äthergemiseh ausgesehüttelt. Der Verteilungskoeffizient der Borsäure zwischen Äther und Wasser ist unter bestimmten Versuehsbedingungen:
CÄtber - - - - 0 , 4 .
Ctt~o Der Äther wird abgetrennt und darin die Borsäure durch die übliche Titration bestimmt. Ca, Mg, A1, Ma, Fe und As in normalen Mengen stören nicht. Ba, F und viel Fe stören etwas, Zn stört stark.
Als Ursache für die Schwierigkeiten bei der Abtrennung störender Metalle erschien uns, wie schon eingangs ges~gt ist, deren Fällung als amorphe oder gelartige Niedersehläge. Wir versuchten daher die Metalle als krystalline Oxinverblndungen zu entfernen und erzielten so eine glatte Trennung.
Die Fällung der störenden Metalle mit Oxin.
Bei der quantitativen Bestimmung von Metallen mit 0xin a) 10uffert
man die Analysenlösung vor der Fä]]ung und verwendet als l~eagens ent-
weder eine alkoholische Oxinlösung ader eine wäßrige Lösung von 0xin-
i) Vgl. diese Ztschrft. 76, 293 (1929). -- 2) Ztsehrft. f. anorg. Chem. 142, 257 (1925); vgl. diese Ztsehrft. 67, 68 (1925/26). _ z) Diese Ztsehrft. 84, ~67 (1931) und 87, ]65 (1932). -- «) Glass Ind. 17, 156 (1936); durch Chem. Zentrbl. 107, Ii, 2774 (1936). -- 5) i~. Berg, Die analytische Ver- wendung von o-Oxychinolin (Oxin) und seiner Derivate. Die chemische Ana- lyse, Bd. 34, 2. Aufl. Stuttgart 1938.
11"

164 I-[arald Schäfer und Adolf Sieverts:
acetat. I m Hinblick auf die anschließende BorsAuretitration mußte in unserem Falle auf Pufferung verzichtet werden. Ebenso durfte als l~eagens weder Oxinacetat noch alkoholische Oxinlösung verwendet werden. Als l~sagens benutzten wir mit bestem Erfolg eine wäßrige Lösung von Natriumoxinat. Eine Pufferung ist hier nicht erforderlich, da beim FAllen von Metallen mit Oxin-Natrium neutrale Salze entstehen und nicht, wie beim Fällen mit Oxin, freie Shuren.
Herstelhmg der Reagenslösung.
7,5 g o-Oxychinolin werden in 50 ccm n-Natronlauge mit schwacher Erw~rmung fast VollstAndig gelöst. Durch Verdünnen mi t Wasser auf 200 ccm erh~lt man eine 0,25 m-Lösung von Oxin-Natrium. Die ~eagens- lösung ist etwa t0 Tage haltbar. Die nach einigen Tagen entstehende dunkelbraune Fi~rbung ist unschädlich.
Die M e t a l l f ~ l l u n g mit Oxin-Natrium geschieht am zweckmäßigsten folgendermaßen:
Die neutrale oder schwach saure !Vietallsalzlösung (Volumen etwa 50 ccm) wird auf etwa 60 o C erwärmt und mit I~eagenslösung in geringem Überschuß versetzt. Nach dem Umschwenken prüft man durch Zu- t r o p f e n von Bromkresolpurpurlösung auf alkalische 1~eaktion. Is t Oxin-Natrium im Überschuß vorhanden, so fArbt sich der vorher gelbe Indikator tief blau-violett1). Zur Vervollständigung de r F/~llung wird noch 5 Min. erwärmt.
Entiernnng überschüssigen 0xins nach der Fällung.
Die Gegenwart von o-Oxychinolin in einem System wirkt puffernd, da das Oxin sowohl OI-[-ionen abfangt unter Bildung von Oxychinolat als auch H-Ionen unter Salzbildung am Ammoniumstickstoff. I m Inter- esse der TitriersChärfe muß daher Oxin vor der Titrat ion entfernt werden. Der Titrierfehler tr i t t , wie schon Fr . L. H a h n und E. H a r t l e b 2) ge- funden haben, besonders dann in Erscheinung, wenn das Umschlagsgebiet des Indikators bei kleineren pK-Werten liegt als PH--~ 6. Eine weitere Störung erfi~hrt die Titrat ion durch die gelbe Farbe d e s Oxins, die die Erkennung der Indikatorfarbe beeintrAchtigt. Die Entfernung des Oxin- überschusses kann auf verschiedene Weise geschehen. Wir beschreiben hier nur das allgemein anwendbare Verfahren mit Magnesiumchlorid und Tierkohlea).
1) Zusatz von Indikator zur Analysenlösung vor der Fällung ist unzweck- mäßig, weil der Niederschlag die tndikatorfarbe überdeckt und außerdem den Indikator adsorbiert. Das E i n t r o p f e n von Bromkresolpurpur hat sich dagegen bestens bewährt. -- 8) Diese Ztschrft. 71, 225 (1927). -- a) Der Oxinüberschuß und das gcfällte Oxinat können auch durch Schütteln mit Tetrachlorkohlenstoff entfernt werden [vgl. ~Fr. L. H a h n und E. H a r t l e b , diese Ztschrft. 71, 225 (i927)]. Auch kann das überschüssige Oxin durch

Beiträge zm" rnaßanalyt isehen Best immung der Borsäure I. t 6 5
Ungefähr 5 Min. nach erfolgter Oxina t fä l lung g ib t m a n zur Analysen- probe 5 c c m m-Magnes iumchlor id lösung und e rw ä rmt noch 5 Min. Danach f i l t r ie r t l) m a n und wäsch t den Niederschlag mi t Wasse r von l~aum- t e m p e r a t u r aus. Das F i l t r a t wird gegen Methy l ro t s chwach angesäuer t und mit, 0,5 g Tierk0hle2 ) (reinst, t rocken , E. M e r c k ) verse tz t . Nach 5 Min. wird f i l t r ie r t und mi t Wasse r gewaschen. I m farblosen F i l t r a t k a n n d ie Borsäure auf bel iebige Weise b e s t i m m t werden .
Einzelne Anwendungen des Verfahrens. Die A n w e n d u n g des vors tehenden Verfahrens zur A b t r e nnung von
Zink, Blei, Alumin ium, Eisen und Nickel wird in den nächs ten Ab- schn i t t en besprochen3). Die Gegenwar t von Alka l ien und E rda lka l i en in den übl ichen K o n z e n t r a t i o n e n 4) s tör t d ie Borbes t immung n ich t und h a t auch d a n n ke inen Einf luß, wenn andere Metal le als Oxinverb indungen abge t r enn t werden. Zur Bes t immung der Borsäure wurde s te t s wie folgt ve r fahren : Die gegen Methy l ro t angesäuer te Lösung wurde zur Ver- t r e ibung von Kohlensäure aufgekocht 5) und nach erfolgter Abküh lung gegen Methy l ro t neut ra l i s ie r t . Hie rauf wurde gegen a -Naph tho lph tha l e in neu t ra le Inver tzucker lösung a) zugegeben und mi t ca rbona t f re ie r 0 , i n- Lauge gegen a - N a p h t h o l p h t h a l e i n t i t r i e r t .
Borsäure neben Zink. Ti t r i e r t m a n Borsäure neben Zink, so e rhä l t m a n viel zu hohe
Ergebnisse, weil ein Teil der an das Z ink gebundenen Säure mi t - t i t r i e r t wird . Auch is t de r Umschlag des a -Naph tho lph tha l e in s oder Pheno]phtha le ins sehr unscharf . E n t f e r n t m a n aber das Z ink als H y d r - oxyd (vgl. L i t e ra tu rübe r s i ch t , Neu t ra l i sa t ionsmethoden) , so wi rd ein be-
Aufkochen mi t basischem Magnesiumcarbonat gefällt werden. Der Nieder- schlag ist aber nur bei Abwesenheit von ErdMkMien borfrei. Beide Arbeits- weisen sind in der Dissertat ion von H a r a l d S c h ä f e r ausführlich be- schrieben.
1) Sehr schnell geht die F i l t r a t ion vonstat ten, wenn man eine B ü c h n e r - uutsche verwendet, in die man zwei angefeuehtete F i l t e r Nr. 595 von S e h l e i c h e r & S c h ü l l einlegt. Das erste F i l t e r soll etwas kleiner und das zweite F i l t e r etwas größer sein als die Siebplatte der Nutsche. Bei der F i t t ra t ion unter Saugen mit tels der Pumpe erhält man so sofort ein klares F i l t ra t . -- ~) Adsorptionsversuche von Borsaure an Tierkohle zeigten, daß nach dem üblichen Auswaschen der auf dem Fi l te r verbleibenden Kohle die adsorbierte Borsäuremenge vernachlässigt werden kann. -- ~) Es unterl iegt keinem Zweifel, daß das gleiche Verfahren auch zur Abtrennung anderer, hier nicht untersuchter, mi t Oxin fäl lbarer !Vfetalle dienen kann. -- 4) Über die Beeinflussung der Säurestärke der Borsäure durch hochkonzentrierte Lösungen der AlkMi- und ErdMkMisMze wird an anderer Stelle berichte~ werden. -- 5) Beim Verkochen der Kohlensäure ist weder die Verwendung eines l~ückflußkühlers noch das Abpumpen des Kohlendioxyds erforderlich. Vgl. E. S c h u l e k und G. V a s t a g h , diese Ztschrft . 84, 167 (193t). -- «) Die Invertzuckerlösung wurde aus Würfelzucker hergestell t nach der Vor- schrift von W. M y l i u s [Chem. Ztg. 57, t95 (1933)].

166 Harald Schäfer und Adolf Sieverts:
trächtlicher Teil der Bors/~ure vom Niederschlag festgehalten. Beide Er- scheinungen werden durch die folgenden Versuche veranschaulicht:
a) Gegeben: 69,2 mg B20~-f-52mg ZnO [als Zn(NOa) 2 . 6 t i rOl; es wurde ohne Abtrennung des Zinks ti triert . Gefunden: 95 mg ,B203".
b) Gegeben: 69,2 mg B203 + 52mg ZnO ; Zink wurde durch I~eutrali- sation gegen a-~aphtholphthalein als Hydroxyd gef~llt, dieses abfiltriert und ausgewaschen. Im Fil trat wurde die Borsäure titriert. Gefunden: 57,2 mg B203.
I m Gegensatz hierzu läß~ sich das Zink einwandfrei als Oxinat abtrennen. Die Ergebnisse bringt die folgende Tabelle I.
T a b e l l e I. Bors/~urebestimmung nach Abtrennung von Zink als Oxina~.
Gegeben Gegeben Gefunden ~Ynterschied ZnO*) B203 B2Oa B~O3
mg mg mg mg
52 t6,t 78 16,1 78 32,2 78 32,2 78 63,5
*) Gegeben a!s Zn(~03).z. 6 H~O.
t 5,9 --0,2 16,2 + 0,1 32,4 + 0,2 32,2 ~ 0 64,t - - 0,4
B o r s ä u r e n e b e n B l e i .
Auch Bleisalze stören infolge von Hydrolyse [Ausf£l]ung von Pb(OH)2 ] die Titratio11 sehr stark. Die Indikatorumschläge sind ganz unscharf. Eine Borsäurebestimmung in Gegenwart von Blei ist deshalb unmöglich, wie auch der folgende Versuch beweist:
Gegeben: 34,6mg B203 + 2 2 3 m g PbO [als Pb(N0~)2]. Geßunden: 88,5 mg , B2Oa".
Nach Abtrennung des Bleis als Oxinat kann die Borsäure gut be- s t immt werden (Tabelle II) .
T a b e l l e II . Borsäurebestimmung nach Ab$rennung von Blei als Oxinat.
Gegeben Gegeben Gefunden Unterschied 1)bO *) B~Oa B~O3 13203
mg mg mg mg
t t l t t l 334 334 334 334 334
*) Als Pb(NOa) 2.
16,1 32,2 16,1 32,2 32,2 64,5 64,5
16,0 32,8 t5,9 32,2 32,¢ 64,4 64,6
- - 0,1 +0,6 - - 0,2 ± 0 -~- 0,2 - - 0,t + OA
Beiträge zur maßana]ytischer~ Bestimmung der Borsäure I. 167
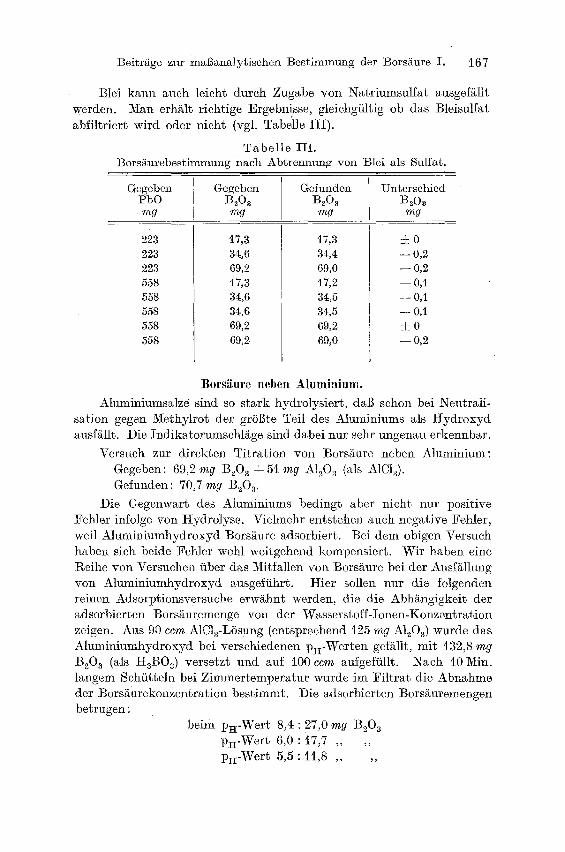
Blei kann auch leicht durch Zugabe von Natriumsulfat ausgefällt werden. Man erhält richtige Ergebnisse, gleichgültig ob das Bleisulfat abfiltriert wird oder nicht (vgl. Tabelle III).
Tabe l l e III. Borsäurebestimmung nach Abtremmng von Blei als Su[fat.
Gegeben Gegeben Gefunden Unterschied PbO B20 a BeO~ B208 mg mg mg mg
223 223 223 558 558 558 558 558
17,3 34,6 69,2 17,3 34,6 34,6 69,2 69,2
17,3 34,4 69,0 t7,2 34,5 34,5 69,2 69,0
±0
--0,2
- - 0,2
--O,i
-- 0,I
- - OA
2c0
--0,2
Borsäure neben Aluminium.
AluminiumsMze sind so stark hydrolysiert, daß schon bei Neutrali- sation gegen Methylrot der größte Teil des Aluminiums als t tydroxyd ausfällt. Die Indikatorumschläge sind dabei nur sehr ungenau erkennbar.
Versuch zur direkten Titrat ion von Borsäure neben Aluminium: Gegeben: 69,2 mg 33202 -~-5t m g AI203 (als AICla). Gefunden: 70,7 mg B~03.
Die Gegenwart des Aluminiums bedingt aber nicht mir positive Fehler infolge von Hydro]yse. Vielmehr entstehen auch negative Fehler, weil Aluminiumhydroxyd Borsäure adsorbiert. Bei dem obigen Versuch haben sich beide Fehler wohl weitgehend kompensiert. Wir haben eine ]~eihe von Versuchen über das Mitfallen von Borsäure bei der Ausfällung von Aluminiumhydroxyd ausgeführt. Hier sollen nur die folgenden reinen Adsorptionsversuche erw/~hnt werden, die die Abhängigkeit der adsorbierten Borsäuremenge von der Wasserstoff-Ionen-Konzentration zeigen. Aus 90 ccm A1Clz-Lösung (entsprechend t25 m g A12Oz) wurde das Aluminiumhydroxyd bei verschiedenen p~t-Werten gefällt, mit t32,8 m g
B20o (als H,BO8) versetzt und auf t00 ccm aufgefüllt. Nach t0 Min. langem Schütteln bei Zimmertemperatur wurde im Filtrat die Abnahme der Borsäurekonzentration bestimmt. Die adsorbierten Borsäuremengen betrugen :
beim prr-Wert 8,4:27,0 mg B203 p~-Wert 6,0 : t7,7 . . . . pa-Wert 5,5 : t1,8 . . . .

168 ]-]:arald Schäfer und Adolf Sleverts:
Man erkennt, daß es nicht möglich sein kann, das Aluminium als Hydroxyd ohne Borverlust abzutrennen, wenn die Adsorption auch in saurer Lösung geringer ist als in alkalischer. Durch Auswaschen des Niederschlages kann die Borsäure nicht quanti tat iv entfernt werden. Auch hier gestat tet die Fällung mit Oxinat eine Äbtrennung des Alu- miniums.
T a b e l l e IV. Borsäurebestin~nung nach Ab~rermung von Aluminium als Oxinat.
Gegeben Gegeben Gefundeß Unterschied A1203") B,20a B203 B203
mg mg mg mg
25 76,5 76,5 76,5 76,5
*) Als AICI 8 . 6 :K20.
32,2 16,1 32,2 64,5 64,5
31,9 16,3 32,5 64,9 64,2
- - 0,3 + 0»2 ~ - 0 , 3
+ 0,4 - - 0,3
Borsäure neben Eisen.
Die Titrat ion von Bors/iure ohne Abtrennung des Eisens ist schon deshalb unmöglich, weil der braune Eisenhydroxydniederschlag das Er- kennen der Indikatorfarbe unmöglich macht.. Dazu kommen die Stör- ungen durch Hydrolyse und Adsorption. Adsorptionsmessungen in Ab- hängigkeit vom p~-Wert, die in gleicher Weise vorgenommen wurden wie beim Aluminium, ergaben, daß die Adsorbierbarkeit von Borsäure an Eisenhydroxyd wie beim Aluminiumhydroxyd mit zunehmender Wasserstoff-Ionen-Konzentration f/~llt. Ein Eisenhydroxydniederschlag (entsprechend 200 mg FeeOa) adsorbierte bei Zimmertemperatur von 132,8 mg B~O 3:
beim p~-Wert 8,6 : i7,9 m g B20 a p~-Wert 6,0 : i0,8 . . . . PH-Vg~ert 5,0 : 6,4 . . . .
Auch hier kann durch Auswaschen nur ein Teil der Bors/~ure ge- wonnen werden. Eine Abtrennung des Eisens als Hydroxyd ist also nicht zu empfehlen1). Gute Ergebnisse liefert wieder das Oxinatver~ahren (Tabelle V, S. 169).
Borsäure neben Nickel.
Diese Trennung hat eine gewisse technische Bedeutung für die Be- st immung von Borsäure in Nickelbi~dern. Der Fehler, der durch die An- wesenheit von Nickelsalzen bei der Borbestimmung entsteht, ist verhgltnis-
1) Bei der Fällung von Eisen als Fe(OH).~ in Gegenwart von Borsäure kann Fe(OH)a (oder Eisenborat ?) kolloid gelöst bleiben.
Beiträge zur maßanalytischen Bestimmung der Bors~ure I.
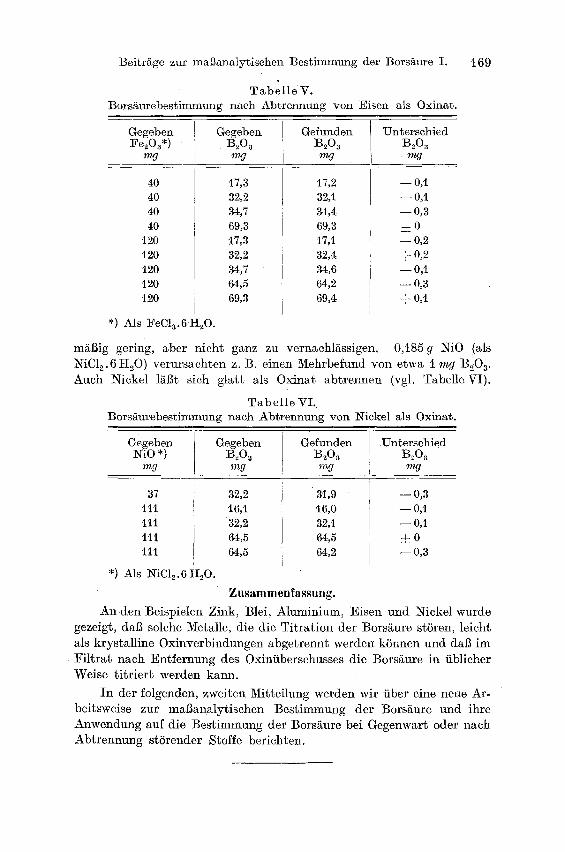
T a b e l l e V. Borsäurebestimmung nach Abtrennung von Eisen als Oxinat.
t69
Gegeben Gegeben Gefunden Unterschied Fe,)Oa*) ' B~O a B 203 B 2 0 a
mg mg mg mg
40 40 40 40
t20 120 120 120 t20
*) Als ~'eCla.6 H,,O.
i7,3 17,2 32,2 32,i 34,7 34,4 69,3 69,3 17,3 17,i 32,2 32,4 34,7 34,6 64,5 64,2 69,3 69,4
mäßig gering, aber nicht ganz zu vernachlässigen,
- - 0 , 1
- - 0,t --0,3 ± 0
- - 0,2 @ 0,2 - - 0 , i
- - 0,3 @ 0,1
0,i85 g NiO .(als NiC12. 6 H~O) verursachten z. ]3. einen Mehrbefund von etwa i mg ]3i0 a. Auch Nickel läßt sich glat t als Oxinat abtrennen (vgl. Tabelle VI).
T a b e l l e ¥ I . ]3orsäm-ebestimmung nach Abtrennung von Nickel als Oxina~.
Gegeben Gegeben Gefunden Unterschied NiO*) B~O a B2Oa B~03
mg mg mg mg
37 111 l i i t l l t l l
*) Als NiCle.6 I-IsO.
32,2 16,1 32,2 64,5 64,5
3t ,9 16,o 32,1 64,5 64,2
- - 0 , 3
- - 0 , 1
- - o , 1
4-0 - - 0,3
Zusammenfassung.
An den ]3eispielen Zink, Blei, Aluminium, Eisen und Nickel wurde gezeigt, daß solche Metalle, die die Titrat ion der ]3orsäure stören, leicht als krystalline 0xinverbindungen abgetrennt werden können und daß im !~'iltrat nach Entfernung des 0xinüberschusses die ]3orsäure in üblicher Weise t i t r ier t werden kann.
In der folgenden, zweiten Mitteilung werden wir über eine neue Ar- beitsweise zur maßanalytisehen Bestimmung der Borsäurë und ihre Anwendung auf die Bestimmung der Borsäure bei Gegenwart oder nach Abtrermung störender Stoffe berichten.

![AUGUSTA VICTORIA BADES - zobodat.at · „von] 2 qm Grundfläche angelegt ist. In dieses Reservoir sind drei ... betrug 0,0729 g, entsprechend Borsäure.... 0,001425 p. ^I. 15. Bestimmung](https://img.pdfslide.org/doc/110x75/5c4d55f793f3c308f758e216/augusta-victoria-bades-von-2-qm-grundflaeche-angelegt-ist-in-dieses-reservoir.jpg)