Embed Size (px)
Citation preview

1
Ratjen & Döring (2003), Lancet, 361:681-689
Zystische Fibrose (Mukoviszidose)
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

2
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)
Gentest:
Screening auf die 36 häufigsten Mutationen (PCR & Hybridisierung)
ca. 90-95% der Mutationen sind damit nachweisbar(starke Schwankungen in verschiedenen Populationen)
wenn negativ: direkte Sequenzierung des gesamten Gens (bis zu 3´000,-CHF)
Zystische Fibrose (Mukoviszidose)
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

3
DNA
Zyklus 1
Zyklus 3
Zyklus 2
Zyklus 4
Zyklus 35
22 =4 Kopien
23 =8 Kopien 16 Kopien 32 Kopien
236 = 68 Milliarden Kopien
DNA zu amplifizieren Schema der PCR
usw.
Häufigkeit:
1:3000 bis 1:6000
Genetik:
- X-chromosomal rezessiv
- Dystrophin-Gen: 2400 kb, 85 Exons, 3685 Aminosäuren
- Deletionen bei 2/3 der Betroffenen
- In-Frame Mutationen: Muskeldystrophie Typ Becker
Muskeldystrophie Typ Duchenne
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

4
Häufigkeit:
1 : 10 000
Klinik:
- Blutungen der Schleimhäute und innere Blutungen
- in 25% der Fälle mildere klinische Formen (bis zu 20% Faktor VIII Aktivität)
Hämophilie A (Faktor VIII-Defizienz)
Inversion of exons 1-22 of the factor VIII gene
Hämophilie A (Faktor VIII-Defizienz)
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

5
Häufigkeit:
1 : 4000 (Männer) – 1 : 6000 (Frauen)
Klinik:
- Entwicklungsverzögerungen (Lernschwierigkeiten bis hin zur schweren geistigen Behinderung)
- Krampfanfälle bei etwa 25% der betroffenen
- Makroorchidie, auffallendes Gesicht
- Verhaltensauffälligkeiten (autistoides Verhalten, ADHS: Aufmerksamkeitsdefizit/Hyperaktivitäts-syndrom)
Fragiles X Syndrom
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

6
CGG expansion in FMR1
normal allele: n = 6-52
premutation: n = 48-200
full mutation: n = 200 bis >1000
test: PCR, Southern-Blot
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

7
Häufigkeit:
1 : 10´000
Klinik:
- häufigste autosomal-dominante neuromuskuläreErkrankung
- erste Symptome im jungen Erwachsenenalter, aber auch kongenitale Formen
- progredienter Verlauf
- Begleitsymptome: Katarakt, geistige Behinderung
Myotone Dystrophie (Curschmann-Steinert)
Genetik:
- autosomal dominanter Vererbungsmodus
- DMPK-Gen, Chromosom 19q13.3
- Triplett-Expansion im 3´-UTR des DMPK-Gens
Myotone Dystrophie (Curschmann-Steinert)
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

8
Stammbaum einer Familie mit myotoner Dystrophie (Curschmann/Steinert)
Häufigkeit:
~ 1 : 30´000
Klinik:
- Erkrankungsmanifestation im 35.-45. Lebensjahr
- Bewegungsstörungen
- Wesensänderung (psychiatrische Auffälligkeiten)
- kognitive Leistungseinbussen
Chorea Huntington (Veitstanz)
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

9
Genetik:
- autosomal dominanter Vererbungsmodus
- IT15-Gen (Huntingtin), Chromosom 4p16.3
- Triplett-Expansion (CAG) im translatierten Bereich
-> Polyglutaminerkrankung
- Antizipation (früheres Erkrankungsalter und Zunahme des Schweregrades in nachfolgenden Generationen)
Chorea Huntington (Veitstanz)
Gentest:
- genetisches Beratungsgespräch
- Problematik der präsymptomatischen/pränatalen molekulargenetischen Testung
- Richtlinien zur Durchführung einer molekulargenetischen Diagnostik
Chorea Huntington (Veitstanz)
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

10
Chorea Huntington (Veitstanz)
Genetische Ursachen der Retinopathia pigmentosa
~ 40 Gene identifiziert
• PhototransduktionRhodopsinPhosphodiesterase,
und -UECNCG1 & 2
ADAR
AR
3q21-245q31.24p16.34p12-cen6q13-21
• Photorezeptor-StrukturproteinePeripherin AD 6p21.1
• Retinol-MetabolismusABCRRLBP1RPE65 AR
ARAR
1p21-1315q261p31
• TranskriptionsfaktorenCRXNRL
ADAD
19q13.314q11.1-12.1
• SpleißfaktorenPRPC8PRP31HPRP3
ADADAD
17p13.319q13.41q21.1
• Unbekannte FunktionCRB1RP2RPGR X
XAR 1q31-32.1
Xp11.4-11.23Xp21.1
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

11
Relevanz für die Diagnostik:
Heterogenität erschwert die molekulare Diagnostik(Nachweis von mehreren Hundert DNA-Varianten in zahlreichen Genen)
-> erfordert neue, hocheffiziente Technologien
Mutationserkennung mittels DNA-Sequenzierung:
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

12
Gendiagnostik bei einem Patienten mitNetzhautablösung
CTG CGG CTG CGA TGC TCA GGGLEU ARG LEU ARG CYS SER GLY
Kontroll-DNAProtein:
Patienten-DNA CTG CGG CTG CGA AGC TCA GGGLEU ARG LEU ARG SER SER GLYProtein:
Mutationsanalyse mit Gen-Chips
DNA Mikroraster (Genchip)enthält > 1000 Genvarianten in Form von kurzen, synthetischen Genabschnitten
CTG CGG CTG CGA TGC TCA GGG
CTG CGG CTG CGA AGC TCA GGG
Kontrolle
Patient
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch
13
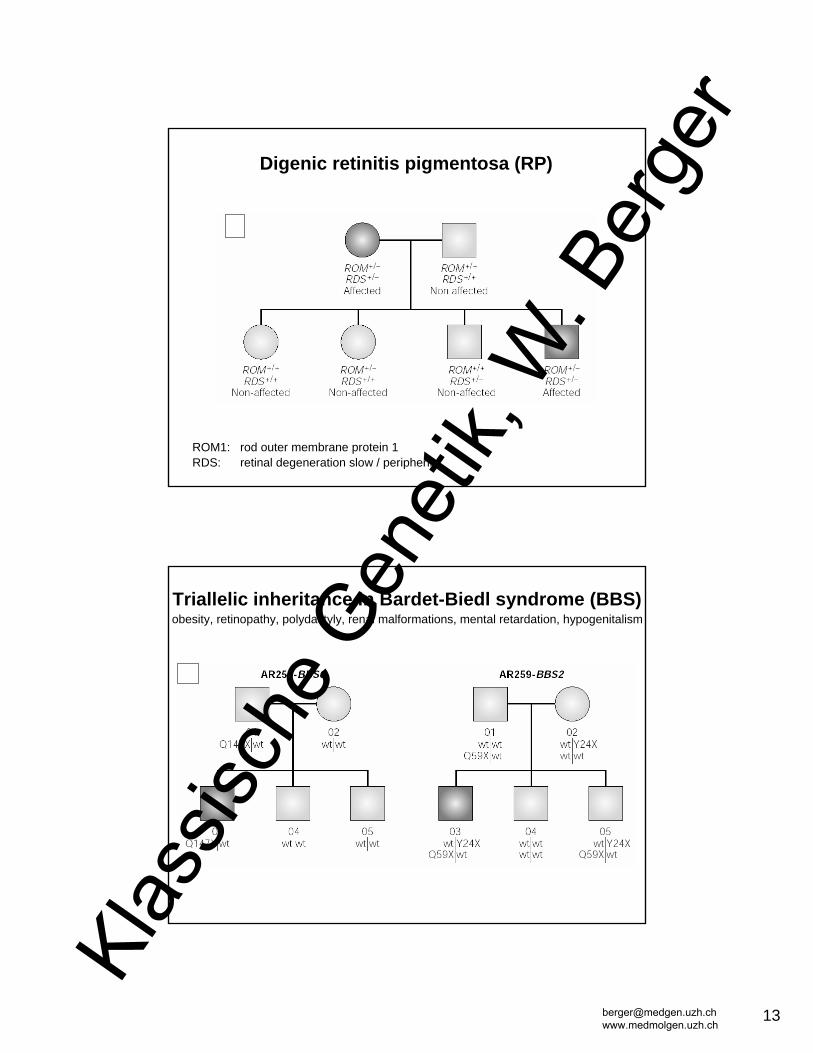
ROM1: rod outer membrane protein 1RDS: retinal degeneration slow / peripherin
Digenic retinitis pigmentosa (RP)
Triallelic inheritance in Bardet-Biedl syndrome (BBS)obesity, retinopathy, polydactyly, renal malformations, mental retardation, hypogenitalism
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

14
1. Entwicklung neuer Hochdurchsatzverfahrenin der Gendiagnostik
2. Zunahme der Diagnostik bei Prädispositionen
3. Aufklärung der Ursachen multifaktoriellerErkrankungen (Herz-Kreislauf, Allergien,Diabetes, Asthma, Krebs, altersbedingteErkrankungen)
4. Verstärkte funktionelle Analyse von Genenund Genvarianten
5. Aufklärung von Krankheitsmechanismen alsGrundlage für die Therapie
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

15
Identifizierung krankheitsassoziierter Mutationen
- grosse Familienstammbäume- umfangreiche Patientenkollektive- strukturelle Chromosomenveränderungen
- Auswahl von Kandidatengenen- Suche nach Mutationen- Untersuchung der Genfunktion
(Biochemie, Zellbiologie, Tiermodelle)
(biochemischer Defekt unbekannt!!!)
1. Kartierung im Genom
2. Identifizierung der Mutation(en)
cen
50-500 kbp
Physical Map
Xp
Xq
> 2000 kbp
Genetic Map
Southern-Blot-Analysis
cont
rol
patients1 2 3 4 5 6
Deletions
Identification of disease-causing mutations
Human X chromosome(160 Mbp, 2000-3000 genes)
1-200 kbp
Gene Map
cont
rol
patients
Mutation-Analysis
SSCP
DNA-Sequencing
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch

16
Retrieval of gene information from databases
rhodopsinsearch term:
http://genome.ucsc.edu/
Deletion of PMP22 Gene
22 23
Nor
mal
ized
flu
ores
cenc
e
Threshold cycle (CT)
Normal Genome
22 22
Nor
mal
ized
fluo
resc
ence
Threshold cycle (CT)
Duplication of PMP22 Gene
2221.5
Nor
mal
ized
fluo
resc
ence
Threshold cycle (CT)
Allele 1 Allele 2Chromosome 17p11.2-12
PMP22Reference Gene
Allele 1 Allele 2Chromosome 17p11.2-12
Allele 1 Allele 2Chromosome 17p11.2-12
Cytogenetic DiseasesDuplications / Deletions (dup/del)
HNPP CMT
Klas
sisch
e Gen
etik,
W. B
erge
r
[email protected] www.medmolgen.uzh.ch















