Embed Size (px)
Citation preview

Bestimmung von Borspuren in Metallen durch Emissionsspektrometrie mit ICP-Anregung nach Abtrennung als Bors iure-Ester*
G. Mezger, E. Grallath, U. Stix und G. Tiilg
Max-Planck-Institut ffir Metallforschung, Institut ffir Werkstoffwissenschaften, Laboratorium ffir Reinststoffe, KatharinenstraBe 17, D-7070 Schw/ihisch Gmfind, Bundesrepublik Deutschland
Determination of Traces of Boron in Metals by Emission Spectrometry with ICP after Separation as Boric Acid Ester
Summary. Procedures for the determination of boron in the ng/g- and low lag/g-range in metals (e.g. Mo, W, Zr, A1, Fe, Co) by emission spectrometry (OES) with inductively coupled hf-plasma (ICP) excitation have been investigated. The procedures involve the suitable dissolution of the samples and the separation of boron either by distillation of boric acid methyl ester with a new circulation distillation apparatus or by liquid extraction of the 2-ethyl-l,3-hexanediol complex. By distillation 0.02 to 20 lag of boron are separated within 15 min from sulphuric or sulphuric-phosphoric acid solutions (< i g of sample) and are concentrated in a few milliliters of NaOH. This separation is applicable to all metals which do not require hydrofluoric acid for dissolution. The ethyl- hexanediol extraction has been optimized for 0.02 to I lag of boron in solutions of Mo, Fe, Zr, and A1. With ICP-OES concentrations of separated boron of >20ng/ml can be determined, corresponding to limits of detection down to 0.02 lag/g, depending on matrix and sample weight.
Zusammenfassung. Zur Bestimmung von Borgehalten im rig/g- und unteren gg/g-Bereich in Metallen (Mo, W, Zr, A1, Fe, Co) wurden Verbundverfahren ausgearbeitet, bei denen Bor nach geeignetem ProbenaufschluB durch Destillation des Bors/iuremethylesters oder durch Ausschfitteln als 2-Ethyl- 1,3-hexandiol-Komplex abgetrennt und emissionsspektro- metrisch mit ICP-Anregung (ICP-OES) bestimmt wird. Mit einer neu entwickelten Kreislaufdestillationsapparatur aus Quarzglas lassen sich Bormengen zwischen 0,02 und 20 gg innerhalb von 15 min aus schwefel- oder schwefel-phosphor- sauren L6sungen von Metallen (Einwaage < I g) quantitativ als Methylester abtrennen und in wenigen Millilitern alkali- scher L6sung konzentrieren. Das Verfahren ist ftir alle Metalle anzuwenden, fiir die man zum L6sen keine HF ben6tigt. Die Ethylhexandiol-Extraktion wurde fiir die Ab- trennung von 0,02 bis 1 lag Bor optimiert, wobei besonders die Matrices Mo, Fe, Zr und A1 untersucht wurden. Die ICP- OES ist ftir die Bestimmung yon Gehalten an abgetrenntem Bor v o n > 20 ng/ml geeignet. Je nach Matrix und H6he der Einwaage k6nnen somit Nachweisgrenzen v o n > 0,02 lag/g erreicht werden.
* Herrn Professor Dr. W. Fresenius zum 70. Geburtstag gewidmet Offprint requests to: E. Grallath
1. Einleitung
Bei der Bestimmung yon Bor in solchen Metallen, die sich gut in fluBs~iurehaltigen S/iuregemischen aufl6sen lassen, haben sich Trenn- und Anreicherungsverfahren bewfihrt, bei denen die Bildung von BF 3 bzw. des BF4-Anions ausgenutzt wird [11].
Diese Verfahren versagen jedoch, wenn bei der destillati- yen Abtrennung des BFa oder beim Ausschiitteln des Tetra- fluoroborat-Anions Matrixbestandteile mitgeschleppt wer- den und die anschliegende Bestimmung st6ren. So gelang es z.B. nicht, geringe Bormengen (< 1 gg) yon molybdfinhalti- gen Matrices so weit abzutrennen, dab die nachfolgende emissionsspektrometrische Bestimmung bei 249,77 nm nicht mehr durch spektrale Interferenzen des Mo beeinfluBt wurde. Auch zur Untersuchung anderer Matrices wfire es yon Vorteil, wenn ein vom Prinzip her verschiedenes Trennverfah- ren zur Kontrolle auf systematische Fehler angewandt wer- den k6nnte.
Bekanntlich 15Bt sich die bereitwillige Bildung yon Estern oder esterartigen Verbindungen der Borsfiure mit Alkoholen oder Polyhydroxyverbindungen ffir die Abtrennung aus sauren L6sungen ausnutzen [16, 33]. Fiir Bormengen in mg- und I~g-Bereich haben sich die Destillation des Bors/iuretri- methylesters nnd die extraktive Abtrennung von Bors~iure- komplexen mit Diolen besonders bewfihrt. Die Ausarbeitung zuverlfissiger Trennverfahren, die in Kombination mit nach- weisstarken Bestimmungsverfahren, wie der Emissionsspek- trometrie mit ICP-Anregung (ICP-OES), die Bestimmung yon Borgehalten in Metallen bis in den ng/g-Bereich erlauben, war Ziel der vorliegenden Arbeit.
2. Trenn- und Bestimmungsverfahren
2.1 Extraktion mit 2-Ethyl-l,3-hexandiol
Borsfiure bildet mit aliphatischen 1,3-Diolen, wie 2-Ethyl-l,3- hexandiol (EHD) oder 2,2-Diethyl-l,3-propandiol farblose (esterartige) Chelate, die im Gegensatz zu den geffirbten Verbindungen mit aromatischen Dihydroxyverbindungen, wie den Naphthalinabk6mmlingen Zephiramin oder Brillant- grfin, nicht ffir die direkte photometrische Bestimmung geeig- net sind [37]. Da die Verteilungen zwischen wfiBrigen L6sun- gen und organischen L6sungsmitteln, insbesondere die des EHD-Komplexes, sehr gfinstig sind [8, 9], kaun EHD gut zur Abtrennung und Anreicherung yon Borspuren benutzt wer-
Fresenius Z Anal Chem (1984) 317: 765-773 Springer Verlag 1984

den. Nach Entfernen des organischen L6sungsmittels kSnnen auf den Rtickstand sowohl photometrische [7, 9, 12] als auch atomemissions- [22] oder -absorptionsspektrometrische Be- stimmungsverfahren angewandt werden. Auch die direkte Bestimmung im organischen Extrakt durch OES [1, 17, 20] oder AAS [13, 27] ist m6glich. Durch Rfickschfitteln des Bors/iure-Diolkomplexes aus der organischen in eine wfiBrige alkalische Phase (NaOH [6, 18], Ca(OH)2/Mannit [22]), l~il3t sich eine zus~itzliche Anreicherung von Bor erzielen.
Der Bors/iure-EHD-Komplex wird in schwefel- oder salzsauren L6sungen im allgemeinen schnell w/ihrend des Schfittelns mit einer L6sung von 5 - 1 0 ~ EHD in Chloro- form [1, 7, 9, 12, 17, 18, 20] oder von 1 0 - 2 0 ~ in Isobutyl- methylketon [5, 13, 19, 27] gebildet. Bei Schfittelzeiten yon 1 bis 5 min kann Bor unter optimalen Bedingungen durch ein- oder mehrmaliges Ausschfitteln praktisch zu 100~ in die organische Phase fiberftihrt werden [1, 7, 12, 17= 19]. Von mSglichen St6relementen werden Ti, Mn, Mound W teilwei- se mitextrahiert [1], was ebenso wie die Mitextraktion von Nb und Ta dutch Zusatz von H202 nach [7] verhindert werden kann. St6rungen durch Fluorid k6nnen durch Zirkoniumzu- satz unterdriickt werden [1, 7, 20].
2.2 Destillation des Borsduremethylesters
Borsfiure und Methanol reagieren in Gegenwart eines wasser- bindenden Kondensationsmittels (z.B. H2SO4, HaPO4, ZnC12) unter Bildung des Bors/iuretrimethylesters [33], der zusammen mit fiberschfissigem Methanol durch Destillation oder Diffusion [16, 33] fiber die Gasphase abgetrennt und in einer alkalischen Vorlagel6sung absorbiert und verseift wet- den kann. Eine einmalige Destillation genfigt nicht zur quantitativen Abtrennung des Bors, die man jedoch dutch wiederholte Zugabe yon Methanol und Destillation erreichen kann [6, 15, 26, 33]; eine zweimalige Destillation soll ausrei- chend sein, um reproduzierbare Ergebnisse zu erhatten [15, 24, 26], was bei eigenen Untersuchungen jedoch nicht bestfi- tigt werden konnte [16]. Bei einem h/iufig angewandten Verfahren, bei dem durch kontinuierliche Zufuhr von Metha- noldampf die Methanolkonzentration in der Reaktions- 16sung aufrechterhalten wird [31, 33], ist das anfallende groBe Destillatvolumen ungfinstig ffir die Bestimmung kleinster Bormengen. Hingegen lassen sich bei einem Kreislaufdestilla- tionsverfahren [2, 10] die abgetrennten Borspuren auf ein kleines, weitgehend konstantes Volumen konzentrieren.
Bei allen Verfahrensvarianten wird das Destillat aus Methanol und Borsfiuremethylester in alkalischen LSsungen (Natronlauge [15, 26], Ca(OH)2-Lsg. [2, 31], methanolische Natriummethylat-Lsg. [10]) aufgefangen, und so der Ester sofort verseift. Beim Eindampfen des Destillates soll die Gegenwart von Alkalien Verluste von Bor verhindern. Die Beobachtungen hierfiber in der Literatur sind jedoch sehr widersprfichlich [37], so dab eine Oberpriifung im speziellen Fall angezeigt ist. Durch Zusatz yon Mannit oder Glycerin wird die Verflfichtigung von Bor beim Eindampfen vermieden [14, 26, 33].
St6rungen dutch die Matrix sind bei der Methylester- destillation nicht zu erwarten. Fluorid liege sich mit Zr, A1 oder Zn maskieren [7, 24, 25], jedoch muB FluBsfiure beim vorange- henden L6sen der Probe vermieden werden, um Borverluste wfihrend der Aufbereitung der Probenl6sungen durch Abrau- chen mit HzSOg/H3PO * zur Entfernung von Wasser (vgl. Kap. 3.3.2) auszuschliel3en. Salpetersfiure lfil3t sich durch Abrauchen vollstfindig entfernen.
766
2.3 Emissionsspektrometrie mit ICP-Anregung
Die Nachweisgrenzen emissionspektrometrischer Verfahren h~ngen auger von den Daten des Spektralapparates (Auf- 15sungsverm6gen, Lichtstfirke, Spektralbereich) stark von der Matrix ab. Die Messung des Bors bis in den ng-Bereich in Gegenwart der Matrix scheitert h/iufig an Querst6rungen. In Einzelf~illen kSnnen zwar durchaus Borgehalte < I gg/g direkt in AufschluB16sungen von Metallen bestimmt werden, z.B. bis 0,4 gg/g Bor in Molybd/in bei Messung im Vakuum- UV (182,528 nm) [23]. F fir noch niedrigere Konzentrationen sowie bei hfiufig wechselnden Matrices ist die Abtrennung des Bors jedoch eine unabdingbare Voraussetzung. Man erreicht dann mit dem ICP Nachweisgrenzen bis in den unteren ng/ml- Bereich.
3. Experimentelles
3.1 Gergite
Laborgerdte aus PE oder PP (Pipetten, MeBkolben, Becher u.a.). Alle mit LSsungen in Berfihrung kommenden Gerfite wurden nach grfindlichem Spfilen mit H20 , HNO 3 oder geeigneten L6sungsmitteln durch Ausdfimpfen mit H20 [30] gereinigt.
QuarzgefdJ3e (100-ml-Bechergl/iser, hohe Form, 25-m1- AufschluBgeffiBe mit Luftkfihler [29], MeBkolben, Vorrats- flaschen) wurden nach Ausspfilen mit UzO mit konz. HC1 ausgekocht, anschlieBend gesptilt und mit HEO mind. 1 h ausged/~mpft.
Reagensgliiser aus PP mit VerschluJ3kappe (H6he 100 ram, i .~ 14mm, Volumen ca. 15 nal) zur Durchffihrung der Diol- extraktion.
Automatische Mikropipetten mit aufsteckbaren Spitzen, Fa. Eppendorf, Hamburg.
SchiittelgeHit: REAX l, Fa. Heidolph-Elektro KG, Kel- heim.
Laborzentrifuge: Typ Labofuge I, Fa. Heraeus-Christ, Osterode.
Platinschalen, 5 cm ~ . Heizplatte mit Thermostatregelung, Corning Hot Plate
PC-35. Wasserbad mit 30ffnungen, elektrisch beheizt (1250 W),
Fa. Memmert, Schwabach. Kreislaufdestillationsapparatur aus Quarzglas (Abb. 1 a),
bestehend aus ReaktionsgeffiB (1), VorlagegeffiB (6) und Kfihler (9), die jeweils miteinander verbunden sind. Das ReaktionsgeffiB (1), Volumen ca. 40ml, ist oben mit einer ()ffnung (NS 10) zum Einffillen yon ProbenlSsung und Rea- gentien versehen und kann durch einen Hahn mit PTFE- Kfiken (4) entleert werden. Ester und Methanoldampf gelan- gen fiber das Verbindungsstfick (5) in die Vorlage mit alkalischer Absorptions15sung. Das Vorlagegef/~B (6), Volu- men ca. 20 ml, ist in seinen Abmessungen so gewfihlt, dab das Destillatvolumen einschlie131ich der zum Sptilen notwen- digen Fltissigkeit unter t0 ml gehalten werden kann. Der im VorlagegeffiB erzeugte Methanoldampf wird fiber Verbin- dungsrohr (8) in den Kfihler (9) geffihrt, dort kondensiert und durch Verbindungsrohr (12) dem Reaktionsgef~13 von unten zugeleitet. Dadurch ist eine gute Durchmischung des Metha- nols mit der spezifisch schwereren Reaktionsl6sung gew/ihr- leistet, was durch das Einleiten von Stickstoff (50-60 Blasen/min) durch das Gaseinleitungsrohr (3) noch gef6rdert wird. Alle Offnungen an Reaktions- bzw. Vorlagegef/il3 sind

El.
1 1 ~ i =
/." ",\
\ )
I i 12
3 cm
J
NI
r ~ , u
'0 0 0 ~0 io 0 Lg.
Abb. 1. a Kreislaufdestillationsapparatur zur Abtrennung von Bor als Borsfiuremethylester; 1 Reaktionsgef/iB (RG), 130mm x 25mm a. ~ ; 2 PTFE-Stopfen (NS 10); 3 Gaseinleitungsrohr aus PTFE, 3mm a. ~ ; 4 Hahnktiken (PTFE); 5 Verbindungsstfick RG/VG; 6 Vorlagegef~il3 (VG), 130 x 18 mm a.~;~ ; 7 PTFE-Stopfen (NS 10); 8 Verbindungsstfick VG/Kfihler, 10 mm a.~ ; 9 Kiihler, 200 mm x 33 mm a. ~ ; 10 NS 14,5, zum Aufsetzen eines Absorptionsrohres mit CaC12 ; 1I Kfihlflfissigkeit Zu- und Ablauf; 12 Verbindungsrohr Kfihler/RG, L/inge 130ram, 3 mm i. ~ . Bemerkung: Verbindungsrohr Ktihler/RG sowie Verbindungssttick RG/VG und Gaseinleitungsohr (3) sollen m6glichst nahe am Hahn enden, b Reaktionsgef'~iB mit Heizmantel (L/ingsschnitt)
mit Stopfen oder Hahnkiiken aus PTFE versehen. Wegen des h6heren WSxmeausdehnungskoeffizienten von PTFE gegen- fiber Quarzglas passen sich diese Teile beim Erw/irmen den Schliffhfilsen gut an und sorgen ffir eine optimale Abdich- tung, die mit den entsprechenden Quarzteilen nicht zu erreichen war.
Heizschnfire von 120 bzw. 60W (Electrothermal Typ HC 1, Fa. Rudolf Brandt G m b H + Co., Wertheim), mit denen Reaktions- und VorlagegeffiB umwickelt sind, dienen zum Heizen der Gef~iBe (Abb. 1 b). Die Heizwicklungen sind zur Isolierung mit Band aus Glasgewebe und Aluminiumfolie ummantelt. Die Regelung der Heizleistung erfolgt durch Proportionalregler mit HeiBleitern als Temperaturffihler, die zwischen der Heizwicklung und dem Isoliermantel ange- bracht sind. Der Kfihler (9) wird mit Wasser-Isopropanol- Gemisch von ca. 4~ aus einem Umw/ilzthermostaten (Typ K 2 RD, MGW Lauda, Lauda) versorgt. Die Kfihlerfff- nung (10) ist mit einem Absorptionsrohr mit CaC12 verschlos- s e n .
ICP-Anordnung: Plasmabrenner aus Quarzglas, betrieben mit Ar/N2; HF-Generator, Modell Linn FS 4, 27,12 MHz, 4,5 kW mit Abstimmeinheit; pneumatischer Zerst/iuber, Typ Meinhard TR 30-B 2, Fa. Kontron, Eching bei Mfinchen, Zerst/iuberkammer nach Scott u. Fassel, Eigenanfertigung aus PTFE.
Spektralapparat: 2-m-Paschen-Runge-Spektrometer, Typ Analymat 2500, Fa. RSV, Hechendorf, mit Elektrometer Typ Digimet.
3.2 Reagentien
Wenn nicht anders vermerkt, wurden Reagentien z.A., Fa. E. Merck, Darmstadt verwendet: HF (48 %ig), HC1 (37 %ig), HNO3 (65%ig) und HzSO 4 (98~ig), durch Destillation unterhalb des Siedepunktes (sub-boiling point distillation) [30] gereinigt; o-H3PO 4 (85 ~ig), Essigs/iure (100 ~ig), H20 2 (30 %ig), Methanol, Methanol getrocknet, max. 0,01% H20, Aceton, Chloroform ohne weitere Reinigung. NaOH-Pl~itz- chen wurden kurz in einer Methanol-H20-Mischung (3 + 1) geschwenkt und nach Abdekantieren der L6sung fiber KOH im Exsiccator getrocknet. H20 wurde durch Bidestillation von deionisiertem H20 in einer Quarzapparatur gereinigt. 2-Ethyl-l,3-hexandiol (EHD)-L6sung (10~ig): 1 ml EHD wurden mit 90 ml Chloroform vermischt und zur Reinigung zweimal mit je 20ml 0,1 M Natronlauge ausgeschtittelt. W~grige L6sungen von: NaOH (0,05tool/I), aus Titrisol- L6sung; NaOH (2tool/I), durch Lfsen von 8g NaOH- P1/itzchen zu 100 ml; Mannit (10 mg/ml), durch L6sen von I g Mannit (z. Borsfiurebestimmung) zu 100ml; HzSO 4 (0,05mol/1), aus gereinigter H2SO 4 hergestellt. Methanol-
767

Schwefels/iure-Mischung (10 + 1): 300 ml Methanol (ge- trocknet) werden im Eisbad gektihlt und vorsichtig unter stetigem Umschwenken mit 30 ml H2SO 4 versetzt; Methanol- Schwefels~iure-Mischung (3+ 1), auf gleiche Weise aus. 300 ml Methanol und 100 ml HzSO 4 bereitet.
Bor-Stamml6sung (100gg/ml): aus Titrisol-L6sung im Quarz-MeBkolben bereitet; sie wurde zum Gebrauch auf 10 gg/ml bzw. auf I gg/ml verdtinnt.
Stickstoff, 99,99 ~ig (Sptilgas ftir die Methylesterdestilla- tion), Stickstoff, 99,996~ig und Argon, 99,997~ig bzw. 99,999 ~ig (Betriebsgase fiir ICP), Fa. Messer Griesheim.
3.3 Verfahrensdurchfiihrung
3.3.1 Probenvorbereitung
Kompakte Proben wurden vor der Analyse durch Beizen mit f~r die jeweilige Matrix geeigneten S~iuremischungen (HF, HNO3, HzSO4) [21] gereinigt. Pulver wurden ohne weitere Vorbereitung eingesetzt.
3.3.2 Aufschl~sse
Soll die Methylesterdestillation durchgeffihrt werden, so ist beim Aufl6sen der Proben die Verwendung yon FluBs/iure zu vermeiden. Beim Abrauchen der Probenl6sungen zur Redu- zierung des Wassergehaltes wird Phosphors/iure zugesetzt, um Borverlusten durch Verfltichtigung vorzubeugen [33].
Fiir eine nachfolgende EHD-Extraktion darf die Sfiure- konzentration in der Probenl6sung nicht zu hoch sein [z. B. H2SO4 (konz.): H20 max. 1:4]. Man sollte daher bestrebt sein, die Proben mit einem Minimum an Sfiure aufzul6sen. Wenn dabei HNO3 verwendet werden muB, so ist nach dem L6sen mit H2SO4/HaPO 4 abzurauchen (vgl. Kap. 4.1). Kleine Mengen an FluBs/iure (_< 0,3 ml) k6nnen dann zuge- setzt werden (z. B. bei Zr), wenn die Temperatur w~ihrend des L6sens niedrig gehalten wird (_< 60 ~ C), Mannit zugegen ist und vor der Extraktion nicht abgeraucht werden muB.
Die AufschluBbedingungen ftir die untersuchten Metalle sind nachfolgend zusammengestellt (A = ffir Destillation, B = ffir EHD-Extraktion). Die Angaben k6nnen nur zur Orientierung dienen, da die optimalen Sfiuremischungen je- weils vonder H6he der Einwaage, der Reinheit des zu 16senden Metalls und vor allem von Form und Zerteilungsgrad der Probe abhiingen.
Molybdiin: A. 1 g Pulver mit 2 ml H20, 3 ml H3PO4, 2ml H/SO4 im 100-ml-Quarzbecherglas versetzen, 1 h i m Eisbad halten, 2 • 0,5 ml HNO3 im Abstand von 0,5 h zusetzen, bis zum Nachlassen der Reaktion kiihlen, dann bis zum vollstfin- digen L6sen erw~irmen, 0,5 h abrauchen, abktihlen lassen, 1 ml H20 + 1 ml H2SO4 zugeben und erneut abrauchen.
Beim L6sen kompakter Stficke wird analog verfahren, jedoch ohne Kiihlung.
B. Wie unter A verfahren, dann in 25-ml-MeBkolben fiberfiih- ren und mit H20 auff[illen.
Wolfram: A. 0 ,2 - 1 g Pulver im 100-ml-Quarzbecherglas mit 1 ml H20, 3 ml H3PO4, 2ml HzSO4 versetzen und tropfen- weise HzO2 zugeben. Wenn n6tig, leicht erw~irmen. Nach dem vollst~indigen L6sen fiberschtissiges H202 durch Erhit- zen zerst6ren und wie bei Mo beschrieben bis auf ein L6sungsvolumen yon 3 - 5 ml abrauchen.
768
B. 1 g Pulver im 25-ml-Quarzme13kolben mit 0,1 ml Mannit- 16sung versetzen und durch sukzessive Zugabe von 6 ml HaO 2 in L6sung bringen. 1 ml H2SO 4 zusetzen und mit H/O aufffillen.
Zirkonium: B. 1 g im 25-ml-QuarzmeBkolben mit 2 ml H20 und 0,1 ml Mannitl6sung versetzen und durch sukzessive Zugabe von insgesamt 4ml HzSO 4 und 0,3ml HF unter leichtem Erwfirmen in L6sung bringen. Mit H20 auf 25 ml aufffillen.
Stahl: A. 0,5 - 1 g im 100-ml-Quarzbecherglas durch sukzessi- ve Zugabe von insgesamt 10 ml einer HC1-HNO3-H/O (1 + 1 + 1)-Mischung aufl6sen. 5 ml H3PO4, 2,5ml HzSO4, 2ml H/O zusetzen, langsam auf 250-270~ erw~irmen und bei dieser Temperatur 1,5h halten (schwerl6sliche Borverbin- dungen! vgl. [28]).
B. 0,1 g analog A in 2 ml HC1-HNO3-Mischung aufl6sen und mit 2 ml H3PO 4 + 2 ml H/SO4 abrauchen. Abkfihlen lassen und nach Zugabe weiterer 2ml H3PO4+2ml H2SO, zur vollstfindigen Entfernung von HNO3 erneut abrauchen. L6sung in 25-ml-MeBkolben fiberftihren und mit H20 aufffil- len.
Cobalt: A. 0,3 g im 100-ml-Quarzbecherglas mit 2ml H20 , 3 ml H3PO4 und 0,2 ml HNO3 versetzen. Nach dem L6sen 3ml H2SO4 + 1 ml H3PO 4 zugeben und wie ftir Stahl be- schrieben weiter verfahren.
Aluminium. 0,5g (A) bzw. bis zu 1,25g (B) im 25-ml- QuarzaufschluBgeFgB mit Luftkiihler mit I ml HzO und 1 ml HC1 versetzen. Unter leichtem Erw~irmen mit Brom gesfittigte HC1 in kleinen Portionen zugeben bis zum vollstfindigen L6sen der Probe. Dabei sollte die Aufschlul316sung immer Brom enthalten, erkennbar an der gelben F~irbung. Ein gelegentlich w/ihrend des Aufschlusses auftretender weiBer Niederschlag geht bei weiterer Zugabe von HC1 wieder in L6sung.
A. L6sung in 100-ml-Quarzbecherglas tiberffihren, 2ml H3PO4 und 2 ml HzSO 4 zuftigen und auf der Heizplatte auf etwa 5 ml einengen.
B. Uberschfissiges Brom verkochen, die L6sung in einen 25-ml-MeBkolben tiberftihren und mit H20 auffiillen.
3.3.3 EHD-Extraktion
4 bis 6ml der sauren Probenl6sung, die 0,02 bis 1 gg Bor enthalten kann, wird in einem PP-Reagensglas mit 2,0ml EHD-L6sung versetzt. Nach VerschlieBen mit einer PE- Kappe wird 5 min geschfittelt und anschlieBend zur Phasen- trennung 1 rain zentrifugiert. Die leichtere, w/iBrige Phase wird abgehoben und verworfen (vgl. [11]). Die verbleibende organische Phase wird durch 5 min Schtitteln mit 6 ml 0,05 M Schwefelsfiure yon mitextrahierten Matrixbestandteilen be- freit. Nach Abtrennung der Waschl6sung wird das Bor durch 5 min Schfitteln mit 0,75 ml 0,05 M Natronlauge aus der organischen Phase rtickgeschiittelt. Nach Zentrifugieren und Trennung der Phasen wird die alkalische L6sung emissions- spektrometrisch gemessen.

3.3.4 Kreislaufdestillation
In das VorlagegeffiB werden 0,10ml 2M Natronlauge und 2ml Methanol gegeben. In das Reaktionsgefil3 wird 1 ml Methanol-Schwefelsiure-Mischung (10 + 1) vorgelegt, die schwefel-phosphorsaure, annfihernd wasserfreie Proben- 16sung (max. 5ml) fiber einen Trichter eingebracht und maximal 15 ml Methanol-Schwefelsfiure-Mischung hinzuge- ffigt, mit denen Probengef/iB und Trichter gespfilt werden. Dabei soll das Volumenverhfiltnis von ProbenlSsung und Methanol-Schwefelsiure-Mischung yon I : 3 nicht fiberschrit- ten werden. Nach VerschlieBen der Stopfen leitet man zunichst mehrere Minuten zur Durchmischung der Reak- tionslSsung einen krfiftigen Stickstoffstrom ein, den man dann auf etwa 1 Blase/s reduziert. Man schaltet nun die Heizung des Reaktionsgef/iBes ein und bei beginnendem Sieden der L6sung auch die Heizung des VorlagegeffiBes. Die Heizleistung der GefiBe wird durch die Regler so vorgegeben, dab beide L6sungen gleichmfiBig und stark sieden, ohne dab sich ihre Volumina wfihrend der Destillationszeit von 15 min wesentlich verfindern. Etwa 3min nach Abschalten der Heizungen 1/il3t man die Absorptionsl6sung durch den Hahn des Geffil3es zur weiteren Aufarbeitung entweder in einen 10-ml-QuarzmeBkolben mit 5-ml-Markierung (Verfahren I) oder in eine Platinschale ab (Verfahren II).
Verfahren I fiir Bormengen > 1 #g. Man sptilt das Vorlage- gefil3 mit Methanol bis der Inhalt des Mel3kolbens die 5-ml- Markierung erreicht hat. Danach werden 2 - 3 ml H20 in das Absorptionsgef/iB gegeben, wobei darauf geachtet wird, dab das Wasser auch in das Innere des Einleitungsrohres ein- dringt. Das Spfilwasser wird dann ebenfalls in den Megkol- ben abgelassen, das Absorptionsgef/il3 mit H/O nachgespiilt und der Mel3kolben auf 10ml aufgeffillt. Auf diese Weise hergestellte L6sungen haben eine Methanolkonzentration von ca. 40 % (V/V) und kSnnen direkt f i r die Messung mit ICP-OES eingesetzt werden (vgl. Kap. 4.2.4).
Verfahren H fiir Bormengen <_ 1 #g. Man spfilt das Vorlage- geffiB grindlich mit Methanol und vereinigt die Spfill6sungen mit der Absorptionsl6sung in der Pt-Schale. Die Verwendung yon H20 zum Spfilen sollte hier unterbleiben. Man ffigt 0,1 ml Mannitl6sung zu und dampft auf dem Wasserbad zur Trock- ne ein (vgl. Kap. 4.2.3). Der Rfickstand wird in 1,00 ml H20 aufgenommen, und die LSsung der emissionsspektrometri- schen Messung zugeffihrt.
Zur Reinigung der Destillationsapparatur spfilt man Reak- tions- und VorlagegefiB grindlich mit H20 und anschliel3end die gesamte Apparatur mit Methanol. F i r gelegentliche Reinigungsdestillationen gibt man 20 ml Methanol-Schwefel- siure-Mischung (3 + 1) in das Reaktionsgef/il3, 1 ml 2M Natronlauge sowie 2 ml Methanol in das VorlagegefS.13 und destilliert 15 min.
3.3.5 Emissionsspektrometrische Bestimmung
Die bei der destillativen Abtrennung oder der EHD-Extrak- tion anfallenden L6sungen, die zwischen 0,02 und 2 ~tg/ml Bor enthalten k6nnen, werden nach der Injektionsmethode [3] in 100-~tl-Portionen mit einer Mikropipette in den Ansaug- trichter des pneumatischen Zerstiubers der ICP-Anordnung eingegeben. Die Signale werden mit einem Schreiber regi- striert und fiber die H6he ausgewertet. Es werden yon jeder Probenl6sung mindestens 4 Einzelmessungen durchgeffihrt.
Parallel dazu werden Eichl6sungen gemessen, deren Borkon- zentrationen denen der Probenl6sungen nahekommen. Die Emissionslinie bei 249,773 nm wird benutzt.
3.3.6 Eichung
Zur Eichung werden der 1 bzw. 10gg/ml enthaltenden BorlSsung entsprechend den Gehalten der Probenl6sungen Bormengen zwischen 0,02 und 20 ~tg entnommen und den parallel zu den Probenaufschlfissen vorbereiteten Siure- mischungen ohne Matrix zugesetzt. Diese EichlSsungen werden dann dem jeweils angewandten Trenn-, Anreiche- rungs- und Bestimmungsverfahren unterworfen. Die Blind- werte des Gesamtverfahrens werden analog ermittelt und von den Eich- bzw. Mel3werten in Abzug gebracht.
Bei Mo, W, Zr und Fe standen Proben zur Verffigung, in denen mit keinem der hier beschriebenen Verfahren Bor nachgewiesen werden konnte. So war es m6glich, auf der Basis ,,borfreier" MatrixlSsungen EichlSsungen herzustellen, die zur f2berpriifung der einzelnen Verfahrensschritte benutzt werden konnten. Da bei der Durchffihrung der EHD- Extraktion mit 25 ml genfigend Aufschlul316sung zur Verffi- gung steht (vgl. Kap. 3.3.2), kann hier gut das Standardaddi- tionsverfahren angewandt werden.
4. Ergebnisse
4.1 Abtrennung des Bors durch Ausschiitteln mit EHD
Von den zum Anfl6sen von Metallproben/iblichen Siuren wird das EHD-Extraktionsverfahren vor allem durch stark oxidierende Stoffe wie HNO3 und K6nigswasser gestSrt, da sie das Reagens zerst6ren. Sie lassen sich durch Abrauchen mit HzSO4/H3PO 4 nach dem Aufschlul3 entfernen. Die Gesamtsiurekonzentration in den ffir die Extraktion vorbe- reiteten Probenl6sungen sollte 20 ~ (V/V) nicht fibersteigen. Aus salzsauren L6sungen liBt sich die Ex[raktion yon Bor ebenfalls durchffihren. Niedrige HF-Konzentrationen (< 12~g/ml) sind tolerierbar, wenn die Matrix fluoridbin- dende Elemente enthilt, wie bei der Analyse yon Zirkonium gezeigt werden konnte. Die Gegenwart yon Mannit, das man beim L6sen von Proben zur Vermeidung von Borverlusten zusetzen kann, beeinfluBt die EHD-Extraktion nicht.
An durch Aufstocken hergestellten ProbenlSsungen wur- de sichergestellt, dab sich das Bor nach einmaligem Schfitteln mit 2ml EHD-L6sung praktisch vollst/indig in der organi- schen Phase befindet. In weiteren Extrakten konnte kein Bor mehr nachgewiesen werden. Tabelle 1 zeigt die Vollstfindig- keit der Extraktion von jeweils 0,5 bzw. 1 p.g Bor, das teils vor dem Aufschlug von 1 g ,,borfreiem" Mo, teils nach dem Aufl6sen der Probe zugegeben wurde. AnschlieBend wurde das Bor durch EHD-Extraktion yon der Molybdinl6sung wie beschrieben abgetrennt und durch ICP-OES gemessen. Als Bezug dienten Borl6sungen entsprechender Konzentration in 0,05 M-Natronlauge.
Die Abtrennung des Bors von mitextrahierbaren Matrix- elementen wurde an Molybdinl6sungen n/iher untersucht, da bei der emissionsspektrometrischen Messung bei 249,773 nm Mo durch zwei Emissionslinien bei 249,780 und 249,758 nm spektrale Interferenzen bewirkt. Der EHD-Extrakt einer Mo- haltigen L6sung wurde nacheinander mehrmals mit je 4 ml 0,05 M HzSO4 gewaschen und der Mo-Gehalt der einzelnen Waschl6sungen durch ICP-OES bei 313,259 nm bestimmt. Es zeigte sich, dab schon durch einmaliges Waschen etwa 90
769
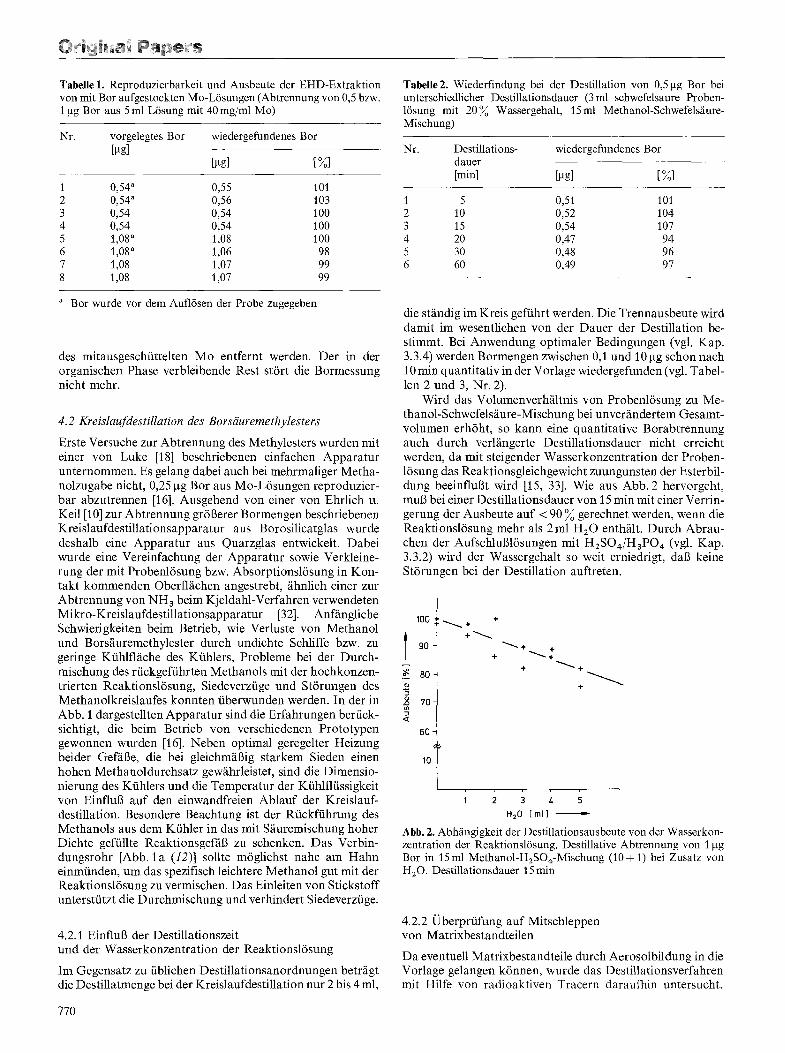
Tabelle 1. Reproduzierbarkeit und Ausbeute der EHD-Extraktion von mit Bor aufgestockten Mo-L6sungen (Abtrennung von 0,5 bzw. 1 ixg Bor aus 5 ml L6sung mit 40 mg/ml Mo)
Nr. vorgelegtes Bor wiedergefundenes Bor [gg]
[gg] [%]
1 0,54" 0,55 101 2 0,54" 0,56 103 3 0,54 0,54 100 4 0,54 0,54 100 5 1,08 a 1,08 100 6 1,08 a 1,06 98 7 1,08 1,07 99 8 1,08 1,07 99
a Bor wurde vor dem Aufl6sen der Probe zugegeben
des mitausgeschtittelten Mo entfernt werden. Der in der organischen Phase verbleibende Rest st6rt die Bormessung nicht mehr.
4.2 Kreislaufdestillation des Borsduremethylesters
Erste Versuche zur Abtrennung des Methylesters wurden mit einer von Luke [18] beschriebenen einfachen Apparatur unternommen. Es gelang dabei auch bei mehrmaliger Metha- nolzugabe nicht, 0,25 lag Bor aus Mo-L6sungen reproduzier- bar abzutrennen [16]. Ausgehend von einer von Ehrlich u. Keil [10] zur Abtrennung gr6Berer Bormengen beschriebenen Kreislaufdestillationsapparatur aus Borosilicatglas wurde deshalb eine Apparatur aus Quarzglas entwickelt. Dabei wurde eine Vereinfachung der Apparatur sowie Verkleine- rung der mit Probenl6sung bzw. Absorptionsl6sung in Kon- takt kommenden Oberfl~ichen angestrebt, ~ihnlich einer zur Abtrennung von NH 3 beim Kjeldahl-Verfahren verwendeten Mikro-Kreislaufdestillationsapparatur [32]. Anffingliche Schwierigkeiten beim Betrieb, wie Verluste von Methanol und Borsfiuremethylester durch undichte Schliffe bzw. zu geringe Kfihlfl/iche des Kiihlers, Probleme bei der Durch- mischung des rfickgefiihrten Methanols mit der hochkonzen- trierten Reaktionsl6sung, Siedeverziige und St6rungen des Methanolkreislaufes konnten tiberwunden werden. In der in Abb. 1 dargestellten Apparatur sind die Erfahrungen bertick- sichtigt, die beim Betrieb yon verschiedenen Prototypen gewonnen wurden [16]. Neben optimal geregelter Heizung beider Geffige, die bei gleichmfiBig starkem Sieden einen hohen Methanoldurchsatz gewS.hrleistet, sind die Dimensio- nierung des Ktihlers und die Temperatur der Ktihlfliissigkeit von Einflul3 auf den einwandfreien Ablauf der Kreislauf- destillation. Besondere Beachtung ist der Rtickfiihrung des Methanols aus dem Ktihler in das mit Sfiuremischung hoher Dichte geffillte ReaktionsgeffiB zu schenken. Das Verbin- dungsrohr [Abb. l a (12)] sollte m6glichst nahe am Hahn einmtinden, um das spezifisch leichtere Methanol gut mit der Reaktionsl6sung zu vermischen. Das Einleiten von Stickstoff untersttitzt die Durchmischung und verhindert Siedeverztige.
4.2.1 Einflug der Destillationszeit und der Wasserkonzentration der Reaktionsl6sung
Im Gegensatz zu fiblichen Destillationsanordnungen betrfigt die Destillatmenge bei der Kreislaufdestillation nur 2 bis 4 ml,
770
Tabelle2. Wiederfindung bei der Destillation von 0,5~tg Bor bei unterschiedlicher Destillationsdauer (3ml schwefelsaure Proben- 16sung mit 20 ~ Wassergehalt, 15 ml Methanol-Schwefels~iure- Mischung)
Nr. Destillations- wiedergefundenes Bor dauer [min] [gg] [%]
1 5 0,51 101 2 10 0,52 104 3 15 0,54 107 4 20 0,47 94 5 30 0,48 96 6 60 0,49 97
die st/indig im Kreis gefiihrt werden. Die Trennausbeute wird damit im wesentlichen yon der Dauer der Destillation be- stimmt. Bei Anwendung optimaler Bedingungen (vgl. Kap. 3.3.4) werden Bormengen zwischen 0,1 und 10 pg schon nach 10 min quantitativ in der Vorlage wiedergefunden (vgl. Tabel- len 2 und 3, Nr. 2).
Wird das Volumenverh/iltnis yon Probenl6sung zu Me- thanol-SchwefelsS.ure-Mischung bei unver/indertem Gesamt- volumen erh6ht, so kann eine quantitative Borabtrennung auch durch verlgngerte Destillationsdauer nicht erreicht werden, da mit steigender Wasserkonzentration der Proben- 16sung das Reaktionsgleichgewicht zuungunsten der Esterbil- dung beeinfluBt wird [15, 33]. Wie aus Abb. 2 hervorgeht, muB bei einer Destillationsdauer von 15 rain mit einer Verrin- gerung der Ausbeute auf < 90 % gerechnet werden, wenn die Reaktionsl6sung mehr als 2 m! H20 enthfilt. Durch Abrau- chen der AufschluB16sungen mit H2SO4/H3PO 4 (vgl. Kap. 3.3.2) wird der Wassergehalt so weit erniedrigt, dab keine St6rungen bei der Destillation auftreten.
100
19~ 80
70-
6 0 -
10-
§ ~ + + ~
~ + + ~ ,
+ ~ +
1 2 ; 4 5 H20 [ ml] ---
Abb. 2. Abhgngigkeit der Destillationsausbeute vonder Wasserkon- zentration der Reaktionsl6sung. Destillative Abtrennung von 1 gg Bor in 15ml Methanol-H2SO4-Mischung (10 + 1) bei Zusatz von H20. Destillationsdauer 15 min
4.2.20berpriifung auf Mitschleppen von Matrixbestandteilen
Da eventuell Matrixbestandteile durch Aerosolbildung in die Vorlage gelangen k6nnen, wurde das Destillationsverfahren mit Hilfe yon radioaktiven Tracern daraufhin untersucht.
@ iIlisalarbeiler
Probenl6sungen, die 50 mg MolybdS_n enthielten, wurden mit 100lal einer im Gleichgewicht befindlichen 99Mo/99mTc- Radionuklidl6sung einer spezifischen AktivitS.t von 5 laCi/ml versetzt und der Destillation unterworfen. In keinem der Destillate konnte anschlieBend durch Messung mit einem 7-Spektrometer mit Ge(Li)-Detektor Aktivitfit von 99M0 oder 99mTc festgestellt werden.
4.2.3 Eindampfen von alkalisch-methanolischen L6sungen
Das bei der Kreislaufdestillation anfallende Destillat kann bei Bormengen _> 1 lag direkt zur Messung verwendet werden (vgl. Kap. 3.3.4). Ffir kleinere Bormengen empfiehlt es sich, das Methanol durch Abdampfen zu entfernen und so eine Konzentrierung der L6sung zu erreichen. Die zum Teil widersprfichlichen Angaben in der Literatur fiber Borverluste beim Eindampfen von Destillaten [33] gaben Anlal3, diesen Verfahrensschritt zu fiberprfifen. ~ 6.
Dazu wurden L6sungen mit 1 bzw. 0,1 lag B, die 0,1 ml | 2 M Natronlauge enthielten, in Platinschalen mit 10 bis 30 ml ~ s- Methanol versetzt und unter folgenden Versuchsbedingun- ~: gen zur Trockne eingedampft: a) im Dampfraum fiber einem ~ ~
i Wasserbad von 90 ~ C, b) aufeiner Heizplatte mit Thermostat- ~ 3- regelung bei einer Plattentemperatur von ca. 120~ Der Eindampfriickstand wurde danach mit 1,00ml HzO auf-
"E 2" genommen und der emissionsspektrometrischen Messung zu- = geffihrt, g I
Tabelle3. Eindampfen alkalisch-methanolischer Borl6sungen unter verschiedenen Bedingungen
Nr. vorgelegte Ein- Bormenge dampf- [gg] bedingung =
wiedergefundenes Bor [~]
Mittel- Standard- n Bestim- wert abw. mungen
1 I a 98,8 2,4 1l 2 1 a b 98,0 1,4 2 3 1 b 90,9 4,5 9 4 0,1 b 85,9 4,3 6 5 0,I b ~ 100,5 1,4 6
a S. Text b Nach vorangegangener Destillation c Nach Zusatz von I mg Mannit eingedampft
Wie aus Tabelle 3 zu entnehmen ist, k6nnen 2- 10 4 Mol NaOH enthaltende methanolische L6sungen in Ubereinstim- mung mit der Literatur [14, 33] ohne Borverluste bis zur Trockne auf der Heizplatte eingedampft werden, wenn Man- nit zugesetzt wurde (Nr. 5). Unter sonst gleichen Eindampf- bedingungen treten wechselnde Borverluste bis ca. 20 % anf, wenn kein Mannit zugegen ist (Nr. 3 und 4). Ffihrt man das Abdampfen des Methanols unter mehr schonenden Bedin- gungen auf dem Dampfbad durch, so bleibt auch ohne Mannitzusatz das Bor praktisch quantitativ im Rfickstand.
Es k6nnen nach dem Eindampfen also auch solche Bestimmungsverfahren angeschlossen werden, bei denen die Gegenwart von Mannit st6rt, wie z.B. das spektralphoto- metrische Curcuminverfahren. Bei allen Versuchen war die Menge des abgedampften Methanols ohne Einflul3 auf das Ergebnis.
4.2.4 Einflul3 der Methanol- und Natronlaugekonzentration des Destillats auf die emissionsspektrometrische Messung
Wird die bei der Destillation anfallende L6sung nicht ein- gedampft, sondern direkt emissionsspektrometrisch gemes- sen, so wirken sich unterschiedliche Methanolkonzentratio- nen des Destillats auf die Empfindlichkeit der Messung aus. Die Intensitfit des Borsignals nimmt mit steigender Methanol- konzentration zu und ist bei einer L6sung mit 90 ~ Methanol am gr613ten. Da jedoch das Untergrundsignal zum Teil noch mehr verstirkt wird, ist die Empfindlichkeitssteigerung nicht auszunutzen. Wie Abb. 3 zeigt, ist das Signal-zu-Untergrund- Verhiltnis bei einer Methanolkonzentration yon ca. 40 ~ am h6chsten. Steigende Natronlaugekonzentrationen bewirken ein Abnehmen des Signal-zu-Untergrund-Verh/iltnisses.
f J
f
i i i
,0 =o ,0 ,-o Q 80 90
M e t h a n o l [ % I -----
100
Abb.3. Einflug der Methanolkonzentration der Probenl6sung auf die emissionsspektrometrische Messung yon 0,1 gg/ml Bor. Natron- laugekonzentration: 1, 2 �9 10 .2 M, 2, 4 �9 10 -2 M
4.2.5 Reproduzierbarkeit und Genauigkeit des Verfahrens mit destillativer Abtrennung
Bormengen yon 0,2 bis 0,4 ~tg wurden aus molybdgmhaltigen schwefel-phosphorsauren Probenl6sungen durch Methyl- esterdestillation nach Kap. 3.3.4, Verfahren II, abgetrennt und emissionsspektrometrisch gemessen. Aus den Ergebnis- sen, die in Tabelle4 zusammengestellt sind, liBt sich eine relative Standardabweichung des Gesamtverfahrens von etwa 5 ~ entnehmen, wenn man alle ermittelten Bormengen auf 1 g Mo-Einwaage bezieht. Auch die Obereinstimmung der ermittelten mit den vorgelegten Bormengen ist befriedigend.
Tabeile 4. Bestimmung yon Bor in Mo-L6sungen nach Abtrennung durch Methylesterdestillation
Nr. vorgelegt B ermittelt
Mo [rag] B [gg] [gg] [gg/g Mo]
1 20 0,20 0,21 10,5 2 20 0,20 0,21 10,5 3 30 0,30 0,30 10,0 4 30 0,30 0,28 9,3 5 40 0,40 0,44 11,0 6 40 0,40 0,43 10,8
771

Or 9 na) P pcr8
Tabelle5. Ergebnisse der Borbestimmungen an verschiedenen Metallproben
Matrix/Probe Einwaage- Abtrennverfahren Borgehalt [gg/g] Bemerkungen bereich [g]
2 s n
Stahl AKP 178-1 (BAM) 1,0 EHD-Extraktion 45,4 1,4 4
0,5 Methylesterdestillation 47,3 1,4 4
Probe 25006 0,1 -0,15 Methylesterdestillation 4,7 0,3 3 Probe 25008 0,1 -0,15 Methylesterdestillation 2,5 0,07 2
Cobalt Cobalt-Pulver 0,2 -0,25 Methylesterdestillation 0,84 0,04 4 Sassoon 0,25 - 0,3 Methylesterdestillation 0,35 0,02 3
Molybd/in Probe 01 1,0 EHD-Extraktion _< 0,5 4
1,0 Methylesterdestillation < 0,05 4
Wolfram Probe 01 1,0 Methylesterdestillation < 0,05 8 Probe 03 0,2 Methylesterdestillation 2,4 0,1 4 Probe 04 0,2 Methylesterdestillation 4,8 0,3 8
1,0 EHD-Extraktion 5,0 0,12 4 Probe 05 0,1 Methylesterdestillation 323 3 4
Zirkonium Zr-Blanco 1,0 EHD-Extraktion < 0,05 4 Zr ,,20 ppm" 0,1 EHD-Extraktion 21,7 0,6 4
Aluminium RM Nr. 25 (BCR)
Zertifiz. Wert: 4l _+ 7 gg/g, Ringanalyse [4]: 44,5--47,2 gg/g
dotierte Probe a dotierte Probe a
dotierte Probe a, andere Labors: 312 bzw. 320gg/g
dotierte Probe, andere Labors: 19,4- 22,7 gg/gb
0,25 - 0,5 Methylesterdestillation 1,23 0,08 8 andere Labors : 1,0 - 1,45 gg/gC
1 , 2 5 EHD-Extraktion 1,23 0,02 8
a Im Rahmen von Ringuntersuchungen des Arbeitsausschusses ,,Sonder- und Refrakt/irmetalle" im ChemikerausschuB der ,,Gesellschaft Deutscher Metallhfitten- und Bergleute" (GDMB) hergestellte Proben
b Bereich der Ergebnisse, die im Rahmen eines Projektes des Referenzbfiro der Europ/iischen Gemeinschaften (BCR), Brfissel, zur Erstellung von Zircaloy-Standards ermittelt wurden (vgl. [11])
c Ergebnisse der Zertifizierungsanalysen zur Erstellung eines Referenzmaterials ,,A199,5 ~" des BCR. Folgende Methoden wurden angewendet: Festk6rpermassenspektrometrie, Methylenblau-Photometrie, Isotopenverdfinnungsanalyse mit Massenspektrometrie, Akti- vierungsanalyse mit Protonen bzw. Deuteronen
Bei Aufschlul3 von 1-g-Proben Molybd~in wurde als Nachweisgrenze des Gesamtverfahrens mit Methylester- destillation und anschlieBender Direktmessung im Destillat (vgl. Kap. 3.4.4, Verfahren I) 0,3 gg/g ermittelt. Bei Durch- f/ihrung der Verfahrensvariante I Imi t Einengen des Destilla- tes ergab sich ftir Molybd/in und Wolfram bei Einwaagen von 1 g eine Nachweisgrenze von 0,05 gg/g. Diese Nachweisgrenze kann bei anderen Metallen noch unterschritten werden, wenn es gelingt, mehr als 1 g Einwaage beim Abrauchen der Aufschlul316sung in L6sung zu halten.
4.3 Analysenergebnisse
Die Ergebnisse einiger analysierter Proben sind in Tabelle 5 angegeben. Mit den zum Vergleich angeft~hrten Ergebnissen, die von anderen Laboratorien im Rahmen von Ringanalysen mit den verschiedensten Methoden (Photometrie mit Methy- lenblau bzw. Curcumin, Emissionsspektrometrie mit Plas- menanregung, Isotopenverdtinnungsanalyse mit Massen- spektrometrie, Festk6rpermassenspektrometrie, Aktivie- rungsanalyse mit Protonen bzw. Deuteronen) erhalten wur-
772
den, zeigt sich gute Ubereinstimmung. Da zur Kontrolle der Richtigkeit keine geeigneten Standardmaterialien zur Verffi- gung stehen, sind solche vergleichenden Untersuchungen eine M6glichkeit zur Priifung auf systematische Fehler. Im Ver- gleich mit dem zertifizierten Wert der einzigen uns zug~ng- lichen Analysenkontrollproben mit analysiertem Borgehalt, AKP 178-1 der BAM, liegen die nach beiden Analysenverfah- ren ermittelten Werte nahe der oberen Grenze des angegebe- nen Streubereichs. Dies ist jedoch in Ubereinstimmung mit den Ergebnissen yon VDEh-Untersuchungen, bei denen photometrische Verfahren angewandt wurden, und von emis- sionsspektrometrischen Messungen mit DC-Plasma [4]. Be- sondere Bedeutung kommt den beschriebenen Analysenver- fahrenjedoch fiir den Konzentrationsbereich _< 1 gg/g zu, wo die Leistungsffihigkeit mit den apparativ und experimentell wesentlich aufwendigeren aktivierungsanalytischen Verfah- ren durchaus vergleichbar ist.
Wit danken Herrn R. Kleinteich ffir die Anfertigung der Destilla- tionsapparatur aus Quarzglas, Fr/iulein A. Reitler ffir ihre Mithilfe bei einigen Untersuchungen und Herrn A. Schiffner fgr die Ausffih- rung der vielf~iltigen Werkstattarbeiten.

Literatur
1. Agazzi EJ (1967) Anal Chem 39:233 2. Annual Book of ASTM Standards (1980) Part 12, Chemical
analysis of metals; Sampling and analysis of metal bearing ores. Philadelphia, E30, pp 114-121
3. Berndt H, Jackwert E (1975) Spectrochim Acta 30B: 169 4. Bosch H, Lohau K, Laqua K (1981) In: Koch KH, Massmann H
(Hrsg) Tagungsbericht der 13. Spektrometertagung in Dfissel- doff. Walter de Gruyter, Berlin New York
5. Choi W-W, Chen KY (1979) J Am Water Works Assoc 71:153 6. Cook EBT, Holan H (1977) Nat Inst Metallurgy, Rep South
Africa, Rep No 1902 7. Donaldson EM (1981) Talanta 28:825 8. Dyrssen D, Uppstr6m L, Zangen M (1969) Anal Chim Acta
46 : 49 9. Dyrssen D, Uppstr6m L, Zangen M (1969) Anal China Acta
46:55 10. Ehrlich P, Keil T (1959) Fresenius Z Anal Chem 165:188 11. Grallath E, Tsch6pel P, K61blin G, Stix U, T61g G (1980)
Fresenius Z Anal Chem 302:40 12. Grotheer EW (1979) Anal Chem 51:2402 13. Horta AMTC, Curtius AJ (1978) Anal Chim Acta 96:207 14. Kuwada K, Motomizu S, T6ei K (1978) Anal Chem 50:1788 15. Luke CM (1955) Anal Chem 27:1150 16. Mezger G (1982) Dissertation, Universitit Stuttgart 17. Patricot M (1977) Analusis 5:236
18. Peterson HP, Zoromski DW (1972) Anal Chem 44:1291 19. Pickett EE, Pau JC-M, Koirtyohann SR (1971) J Assoc Offic
Agric Chem 54: 796 20. Pickett EE, Pau JC-M (1973) J Assoc Offic Agric Chem 56:151 21. Quaglia L, Weber G, David D, Van Audenhove J, Pauwels J
(1976) ITE-Bericht Nr 90, Btiro Eurisotop, Komm Europ Ge- meinsch, Brfissel
22. R6hrl M (1981) SKW Trostberg AG, pers6nl Mitt 23. Scherer V (1983) Osram GmbH Mfinchen, pers6nl Mitt 24. Schulek E, Szakacs O, Szakacs M (1956) Fresenius Z Anal Chem
151:1 25. Siemer DD (1982) Anal Chem 54:1321 26. Spicer GS, Strickland JDH (1958) Anal Chim Acta 18:523 27. Spielholtz GI, Toralballa GC, Willsen JJ (1974) Mikrochim Acta
649 28. Thierig D (1982) Fresenius Z Anal Cbem 310:154 29. T61g G, Lorenz I (1969) Fortschritte der chemischen Forschung,
Bd 11, Heft 4. Springer, Berlin Heidelberg New York, S 541 30. Tsch6pel P, Kotz L, Schulz W, Veber M, T61g G (1980)
Fresenius Z Anal Chem 302:1 31. Werner H (1959) Fresenius Z Anal Chem 168:266 32. Werner W, T61g G (1975) Fresenius Z Anal Chem 276:103 33. Wfinsch G, Umland F (1971) Handbuch der Analytischen
Chemie, Bd IIIa~l : Bor. Springer, Berlin Heidelberg New York
Eingegangen am 20. Juli 1983
773