Embed Size (px)
Citation preview

Tierärztliche Hochschule Hannover
Die Bedeutung des Hypoxie-induzierbaren Faktors 1 in dendritischen Zellen für die Pathophysiologie einer Dextrannatriumsulfat induzierten Colitis
INAUGURAL – DISSERTATION zur Erlangung des Grades einer Doktorin
der Veterinärmedizin - Doctor medicinae veterinariae -
( Dr. med. vet. )
vorgelegt von Katharina Flück
Borken
Hannover 2014

Wissenschaftliche Betreuung: 1. Prof. Dr. Joachim Fandrey Institut für Physiologie Universität Duisburg-Essen Universitätsklinikum Essen 2. Prof. Dr. Gerhard Breves Physiologisches Institut Tierärztliche Hochschule Hannover
1. Gutachter: Prof. Dr. Joachim Fandrey
Prof. Dr. Gerhard Breves
2. Gutachter: PD Dr. Maren von Köckritz-Blickwede
Tag der mündlichen Prüfung: 26.09.2014
Diese Arbeit wurde durch die Deutsche Forschungsgemeinschaft gefördert.

Meiner Familie

Ein Teil dieser Dissertation wurde zur Veröffentlichung vorbereitet.
Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of
regulatory T cells in murine colitis
Katharina Flück1,2, Gerhard Breves2, Joachim Fandrey1, Sandra Winning1
1 Institut für Physiologie, Universität Duisburg-Essen, Essen, Germany;
2 Physiologisches Institut, Tierärztliche Hochschule Hannover, Hannover, Germany

Inhaltsverzeichnis --____________________________________________________________________
Inhaltsverzeichnis
1 Einleitung................................................................................................................ 7
1.1 Immunsystem des Darms .................................................................................7
1.2 Rolle von dendritischen Zellen (DCs) im Immunsystem des Darms .................9
1.2.1 Aktivierung der adaptiven Immunantwort im Darm durch DCs ................ 10
1.2.2 Aufrechterhaltung der intestinalen Homöostase ...................................... 13
1.3 Chronisch entzündliche Darmerkrankungen ................................................... 17
1.4 Murine Modelle für chronisch entzündliche Darmerkrankungen ..................... 20
1.5 Beteiligung von DCs an der Entstehung von chronisch entzündlichen
Darmerkrankungen ........................................................................................ 22
1.6 Der Hypoxie induzierbare Faktor (HIF) ........................................................... 23
1.6.1 Aufbau und Funktion von HIF .................................................................. 23
1.6.2 Sauerstoffabhängige Regulation und Aktivierung von HIF-1 ................... 25
1.6.3 Sauerstoffunabhängige Regulation und Aktivierung von HIF-1 ............... 26
1.6.4 Bedeutung von HIF-1 im Rahmen von Infektionen und Entzündungen ... 28
1.7 Ziel der Dissertation ........................................................................................ 30
2 Material und Methoden ......................................................................................... 31
2.1 Materialien ...................................................................................................... 31
2.1.1 Geräte und Verbrauchsmaterialien .......................................................... 31
2.1.2 Chemikalien ............................................................................................. 33
2.1.3 Puffer und Lösungen ............................................................................... 35
2.1.4 Antikörper ................................................................................................ 37
2.1.5 Zytokine ................................................................................................... 38
2.1.6 Oligonukleotide ........................................................................................ 39
2.1.7 cDNA-Synthese, PCR und Real Time PCR ............................................. 41
2.2 Methoden ........................................................................................................ 42
2.2.1 Tierexperimentelle Methoden .................................................................. 42
2.2.2 Zellkultur .................................................................................................. 47
2.2.3 Molekularbiologische Methoden .............................................................. 48
2.2.4 Histologische Methoden .......................................................................... 60
3 Ergebnisse ........................................................................................................... 64

Inhaltsverzeichnis --____________________________________________________________________
3.1 Zur Publikation vorbereitetes Manuskript ........................................................ 64
3.1.1 Abstract ................................................................................................... 66
3.1.2 Introduction .............................................................................................. 66
3.1.3 Materials and Methods ............................................................................ 68
3.1.4 Results ..................................................................................................... 70
3.1.5 Discussion ............................................................................................... 75
3.1.6 References .............................................................................................. 78
3.1.7 Figures ..................................................................................................... 84
3.1.8 Supplementary Material ........................................................................... 95
3.2 Weitere Präsentationen ................................................................................ 101
3.3 Weitere Ergebnisse ....................................................................................... 102
3.3.1 Anstieg des Disease Activity Index durch DSS induzierte Colitis........... 102
3.3.2 Veränderungen des Colons durch DSS induzierte Colitis ...................... 103
3.3.3 Hochgradige Entzündung des Colons durch DSS Behandlung ............. 104
3.3.4 Verstärkte Alcianblau Färbung in DSS behandelten CD11cCre/HIF-1α+f/+f
Tieren .................................................................................................... 108
4 Diskussion .......................................................................................................... 110
4.1 Auswirkungen einer Colitis auf intestinale DCs ............................................. 110
4.2 Entzündungsparameter HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f DSS behandelter
Tiere .............................................................................................................. 110
4.3 Typ I Interferon Produktion............................................................................ 115
4.4 Aktivierung regulatorischer T-Zellen durch DCs ............................................ 117
4.5 Wechselwirkungen zwischen IECs und DCs................................................. 123
5 Zusammenfassung ............................................................................................. 128
6 Summary ............................................................................................................ 132
7 Literaturverzeichnis ............................................................................................ 135
8 Anhang ............................................................................................................... 162
8.1 Abkürzungsverzeichnis ................................................................................. 162
8.2 Abbildungsverzeichnis .................................................................................. 168
8.3 Tabellenverzeichnis ...................................................................................... 170

Einleitung 7
1 Einleitung
1.1 Immunsystem des Darms
Das Immunsystem ist ein komplexes Netzwerk des Körpers, um Pathogene und
entartete körpereigene Zellen zu erkennen und unschädlich zu machen. Das
Abwehrsystem des Körpers wird dabei in die angeborene und in die adaptive
Immunantwort unterteilt.
Zu den wichtigsten Effektorzellen des angeborenen Immunsystems gehören die
phagozytierenden Zellen, wie Monozyten, Makrophagen und dendritische Zellen
(DC, engl. dendritic cell). Sie können eingedrungene Erreger anhand ihrer
Oberflächenproteine erkennen und vernichten. Diese Zellen stellen zudem ein
Bindeglied zwischen angeborener und adaptiver Immunantwort dar, da sie
aufgenommene Zellen und Partikel in Peptidfragmente zerlegen und durch den
Haupthistokompatibilitätskomplex (MHC) den Zellen der adaptiven Immunantwort
präsentieren können. Diese Zellen werden demnach auch antigenpräsentierende
Zellen (APC, engl. antigen presenting cell) genannt (Medzhitov 2007; Steinman &
Hemmi 2006).
Zu den Zellen der adaptiven Immunantwort gehören unter anderem die B-Zellen,
welche Antikörper oder Immunglobuline (Ig) sezernieren können. Diese können
Antigen-spezifische Epitope erkennen und so gegen extrazelluläre Pathogene und
lösliche Proteine, wie Toxine, schützen (LeBien & Tedder 2008). Eine weitere
Zellpopulation der adaptiven Immunantwort sind die T-Zellen. Diese lassen sich in
mehrere Subgruppen unterteilen und lösen nach Stimulation durch APCs
unterschiedliche Immunantworten aus.
Das mukosale Immunsystem stellt eine große Besonderheit dar, da es mit einer
Oberfläche von ca. 400 m2 wie keine andere Körperoberfläche einer Vielzahl von
Antigenen ausgesetzt ist. Dabei müssen pathogene Faktoren erkannt und eliminiert,
apathogene Substanzen und körpereigene Antigene hingegen toleriert werden. Im
Darm haben sich dafür unterschiedlichste Mechanismen entwickelt. Zum einen bildet
die intestinale Epithelschicht den ersten Schutz gegen eingedrungene Pathogene,
die sogenannte first line of defense. Intestinale Epithelzellen (IEC, engl. intestinal

Einleitung 8
epithelial cell) stehen durch Zell-Zellkontakte, tight junctions, in Verbindung und
verhindern so auch sehr kleinen Molekülen den Durchtritt durch die Darmwand (Shen
& Turner 2006; Madara 1998). Die Becherzellen des Darmepithels produzieren
Mucine und Kleeblatt-Faktoren (TFF, engl. trefoil factor), welche sich als schützende
Schicht auf die IECs legen und so die Anheftung und das Eindringen von
Pathogenen vermindern (Taupin & Podolsky 2003; Shirazi et al. 2000; Ohning 2013).
Außerdem produzieren IECs und Paneth-Zellen antimikrobielle Peptide, wie
Defensine und bakteriolytische Enzyme, welche gegen invasive Erreger schützen
(Müller et al. 2005). Auch die natürliche Darmflora trägt zum Schutz des Darms bei,
da die kommensalen Mikroorganismen die Bindungsstellen für Pathogene besetzen
und so ein Eindringen in den Darm erschweren (Duerkop et al. 2009).
Immunzellen des angeborenen und erworbenen Immunsystems befinden sich verteilt
über den gesamten Darmtrakt in der Lamina propria mucosae oder in organisierten
Strukturen, dem darmassoziierten lymphatischen Gewebe (GALT, engl. gut
associated lymphoid tissue). Dies muss eine Immunreaktion gegen die Darmflora
und Nahrungsbestandteile verhindern, zugleich aber gegen pathogene
Mikroorganismen schützen. Zum GALT gehören die Peyer-Plaques, isolierte
Lymphfollikel, Kryptopatches und die mesenterialen Lymphknoten (Newberry &
Lorenz 2005). Peyer-Plaques befinden sich im gesamten Dünndarm und bestehen
aus mehreren B-Zell-Follikeln mit Keimzentren sowie aus kleineren T-Zell-Regionen.
Sie werden von einen follikelassoziiertem Epithel bedeckt, welches spezialisierte
Epithelzellen, die sogenannten M-Zellen (Mikrofaltenzellen), enthält. Diese besitzen
eine gefaltete Oberfläche und ermöglichen den Eintritt von Antigenen aus dem
Lumen in die Peyer-Plaques. Dort werden die Antigene von APCs aufgenommen,
welche in die T-Zell-Regionen der Peyer-Plaques oder in die mesenterialen
Lymphknoten einwandern, um entsprechende Immunreaktionen auszulösen (Murphy
et al. 2009). In den B-Zell-Follikeln findet zudem ein Klassenwechsel zu IgA statt,
welches in das Darmlumen sekretiert wird und gegen luminale Antigene wirken kann
(Brandtzaeg 2009). Isolierte Lymphfollikel sind kleine Zellaggregate, welche im
gesamten Darmtrakt zu finden sind. Sie bestehen zu einem großen Teil aus B-Zellen,
welche wiederum IgA produzieren können. Wie die Peyer-Plaques sind auch die

Einleitung 9
isolierten Lymphfollikel von M-Zellen bedeckt, welche den Durchtritt von Antigenen
gewähren. Kryptopatches sind im Dünndarm und im Colon an der Basis der
epithelialen Krypten lokalisiert. Sie bestehen unter anderem aus lineage-marker
negativen (lin-)c-kit+ Zellen und DCs. Ihre Aufgabe ist noch nicht vollständig geklärt,
aber es wird vermutet, dass sie an der extrathymischen Differenzierung von T-Zellen
beteiligt sind (Oida et al. 2000). Die mesenterialen Lymphknoten stellen die größten
Lymphknoten des Körpers dar. Sie bestehen aus einem äußerem Cortex (Rinde),
welcher B-Zellen enthält, und einer inneren Medulla (Mark), welche vor allem aus
T-Zellen und DCs besteht. Antigenbeladene APCs aus dem umliegenden intestinalen
Gewebe strömen über afferente Lymphgefäße zu den mesenterialen Lymphknoten,
um dort spezifische Immunreaktionen auszulösen. Naive Lymphozyten treten aus
dem Blut über spezielle postkapilläre Venolen in die Lymphknoten ein.
1.2 Rolle von dendritischen Zellen (DCs) im Immunsystem des Darms
DCs wurden erstmals im Jahre 1973 von Steinman beschrieben (Steinman & Cohn
1973). Sie stellen das Bindeglied zwischen angeborener und erworbener Immunität
dar. Unreife DCs wandern durch die Peripherie und nehmen kontinuierlich
Fremdmaterial auf. Dies geschieht zum einen durch Eintritt von Antigenen durch die
oben beschriebenen M-Zellen, zum anderen sind intestinale DCs in der Lage
Antigene direkt aus dem Darmlumen aufzunehmen. Dafür strecken sie ihre langen
zytoplasmatischen Fortsätze, die Dendriten, durch die Epithelzellen des Darms.
Durch eigene Produktion von tight junction Proteinen können DCs die tight junctions
des Epithels penetrieren ohne die Barriere zu schädigen (Rescigno et al. 2001). DCs
identifizieren sogenannte Pathogen-assoziierte molekulare Muster (PAMP, engl.
pathogen-associated molecular pattern) auf Antigenen durch Mustererkennungs-
Rezeptoren (PRR, engl. pattern-recognition receptor), wie Toll-like Rezeptoren
(TLRs) (Medzhitov & Janeway 2000). Bestandteile von Bakterien, wie z. B.
Lipopolysaccharid (LPS), ein Zellwandmolekül gramnegativer Bakterien, oder auch
virale Bestandteile, wie doppelsträngige DNA, binden an die transmembranären
TLRs und lösen eine Signalkaskade aus, die zu einer spezifischen Immunantwort
führt. DCs können PAMPs nicht nur über TLRs, sondern auch über intrazelluläre

Einleitung 10
Proteine erkennen. Diese Proteine bezeichnet man als NOD1 und NOD2
(Nukleotidbindungs-Oligomerisierungsdomäne). NOD1 bindet γ-Glutamyl-
Diaminopimelinsäure, ein Abbauprodukt von Proteoglykanen gramnegativer
Bakterien. NOD2 bindet Muramyldipeptid, das in den Proteoglykanen aller
Bakterienarten vorkommt (Strober et al. 2006). Durch die Antigenaufnahme wird ein
Reifungsprozess in den DCs ausgelöst. Es kommt zu einer gesteigerten Sekretion
von Zytokinen und Chemokinen. Des Weiteren erhöht sich die Expression der
MHC-Proteine und der kostimulatorischen Moleküle (Banchereau & Steinman 1998).
1.2.1 Aktivierung der adaptiven Immunantwort im Darm durch DCs
Die gereiften DCs wandern nun über die Lymphgefäße in die T-Zellregionen der
mesenterialen Lymphknoten. Dort induzieren sie die Differenzierung und Proliferation
von spezifischen T-Zellen (Banchereau & Steinman 1998). Aufgenommene Antigene
werden im Zytoplasma der DCs in kleine Peptidfragmente gespalten. Diese
Peptidfragmente werden über MHC-Moleküle präsentiert, welche von dem
T-Zellrezeptor (TCR, engl. T-cell receptor) erkannt werden. Für die Zellaktivierung
müssen außerdem die kostimulatorischen Moleküle CD80 und CD86 mit dem CD28
Rezeptor auf der Oberfläche von T-Zellen interagieren. Durch MHC-I-Moleküle
werden CD8+ T-Zellen aktiviert, welche sich überwiegend zu zytotoxischen T-Zellen
ausdifferenzieren (CTL, engl. cytotoxic T-lymphocyte) und z. B. virusbefallene Zellen
direkt abtöten. MHC-II-Moleküle aktivieren CD4+ positive T-Zellen, welche sich in
verschiedene Subgruppen aufteilen lassen (König et al. 1992; Murphy et al. 2009).
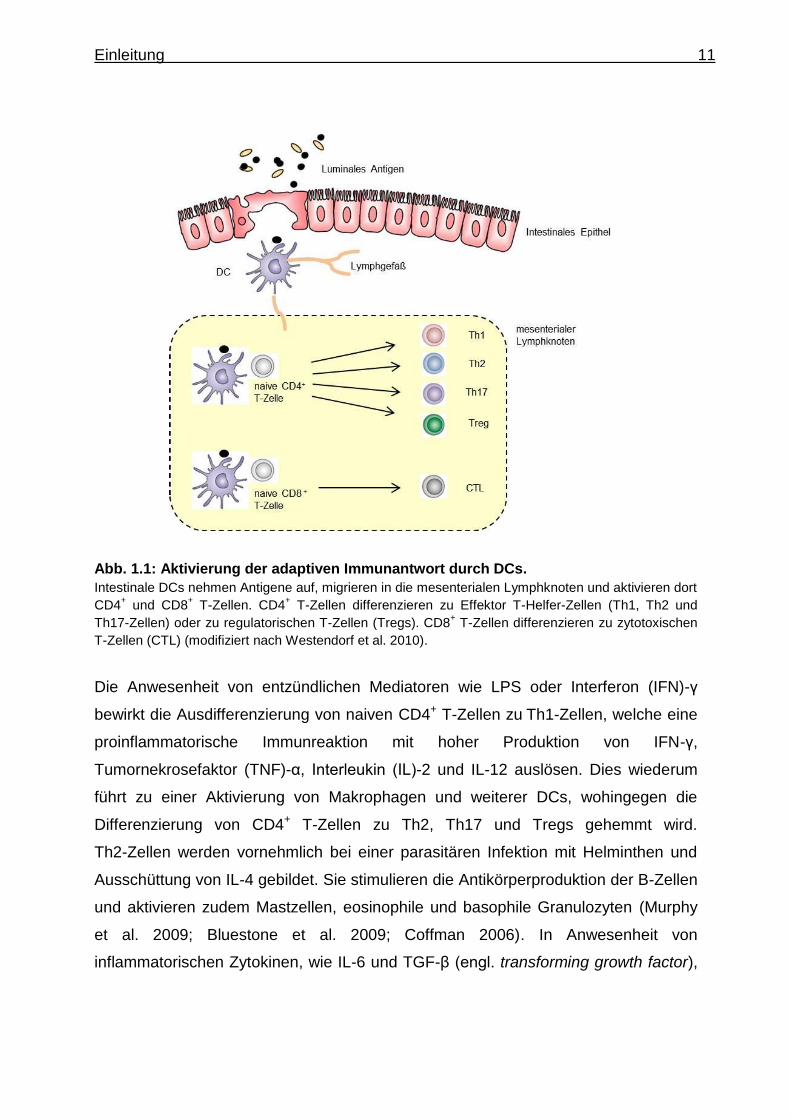
Einleitung 11
Abb. 1.1: Aktivierung der adaptiven Immunantwort durch DCs.
Intestinale DCs nehmen Antigene auf, migrieren in die mesenterialen Lymphknoten und aktivieren dort
CD4+ und CD8
+ T-Zellen. CD4
+ T-Zellen differenzieren zu Effektor T-Helfer-Zellen (Th1, Th2 und
Th17-Zellen) oder zu regulatorischen T-Zellen (Tregs). CD8+ T-Zellen differenzieren zu zytotoxischen
T-Zellen (CTL) (modifiziert nach Westendorf et al. 2010).
Die Anwesenheit von entzündlichen Mediatoren wie LPS oder Interferon (IFN)-γ
bewirkt die Ausdifferenzierung von naiven CD4+ T-Zellen zu Th1-Zellen, welche eine
proinflammatorische Immunreaktion mit hoher Produktion von IFN-γ,
Tumornekrosefaktor (TNF)-α, Interleukin (IL)-2 und IL-12 auslösen. Dies wiederum
führt zu einer Aktivierung von Makrophagen und weiterer DCs, wohingegen die
Differenzierung von CD4+ T-Zellen zu Th2, Th17 und Tregs gehemmt wird.
Th2-Zellen werden vornehmlich bei einer parasitären Infektion mit Helminthen und
Ausschüttung von IL-4 gebildet. Sie stimulieren die Antikörperproduktion der B-Zellen
und aktivieren zudem Mastzellen, eosinophile und basophile Granulozyten (Murphy
et al. 2009; Bluestone et al. 2009; Coffman 2006). In Anwesenheit von
inflammatorischen Zytokinen, wie IL-6 und TGF-β (engl. transforming growth factor),

Einleitung 12
erfolgt die Differenzierung zu Th17-Zellen (Ivanov et al. 2006). Th17-Zellen weisen
eine proinflammatorische Funktion auf und schützen vor mikrobiellen Infektionen und
verstärken die Reaktion neutrophiler Zellen (Murphy et al. 2009). Sie sezernieren
unter anderem IL-6, IL-17, IL-22 und TNF-α.
DCs können Antigene auch B-Zellen präsentieren, welche verschiedene
Immunglobuline sezernieren. Aktivierte B-Zellen stimulieren zudem Th2-Zellen und
regen diese zur Zytokinproduktion an. Dies führt zur weiteren Aktivierung
nahegelegener B-Zellen und somit vermehrter Antikörperproduktion (Murphy et al.
2009).
Die so aktivierten Effektorzellen verlassen die organisierten Strukturen des GALT
oder die mesenterialen Lymphknoten schließlich über die efferenten Lymphgefäße
und gelangen über den Ductus thoracicus und den Blutkreislauf wieder in mukosales
Gewebe (Murphy et al. 2009). Die DCs der Peyer-Plaques und der mesenterialen
Lymphknoten übernehmen hierbei eine entscheidende Aufgabe. Sie vermitteln die
Expression der darmspezifischen Adhäsionsmoleküle α4β7-Integrin und CCR9
(CC Chemokin Rezeptor 9) auf Lymphozyten (Johansson-Lindbom et al. 2003). Das
α4β7-Integrin bindet an das mukosale gefäßspezifische Adressin MAdCAM-1 (engl.
mucosal vascular addressin cell-adhesion molecule 1), welches vor allem auf
intestinalen Endothelzellen vorkommt. CCR9 bindet den Liganden CCL25, welcher
von Epithelzellen des Dünndarms exprimiert wird. Die aktivierten Lymphozyten
gelangen somit direkt zurück in die Lamina propria und das Epithel des Darms, wo
sie gemeinsam mit den Zellen des angeborenen Immunsystems ihre Effektorfunktion
ausüben.
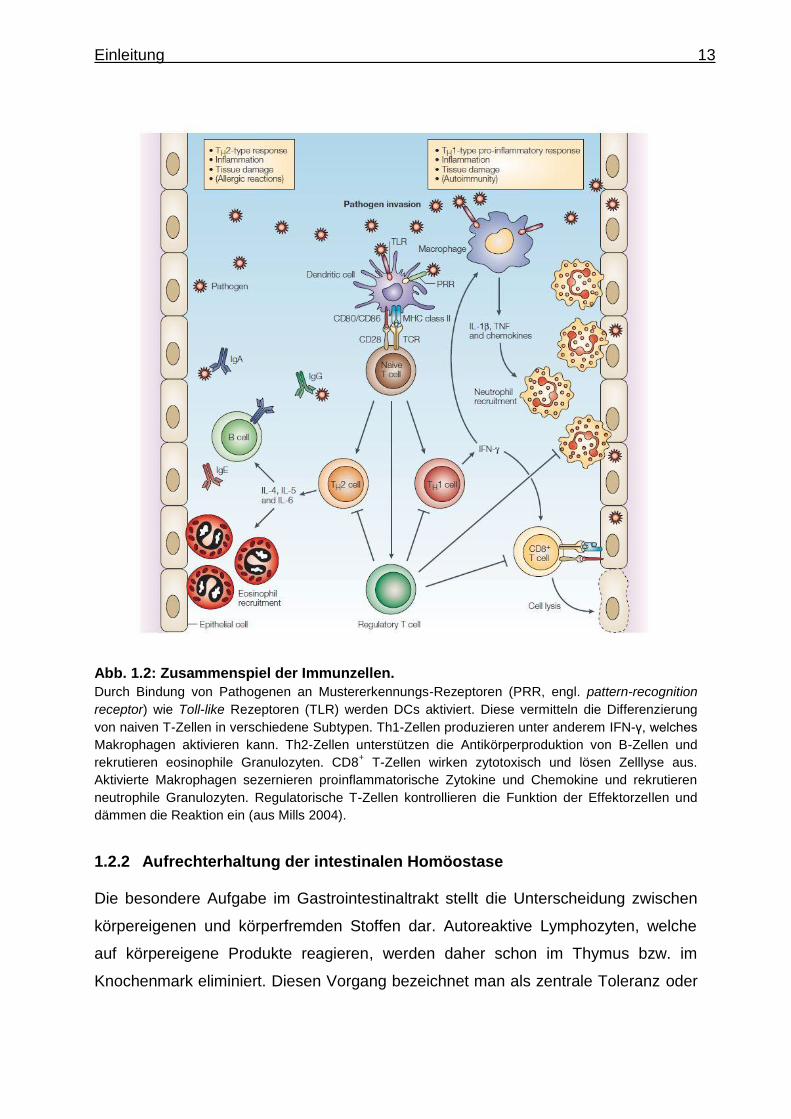
Einleitung 13
Abb. 1.2: Zusammenspiel der Immunzellen.
Durch Bindung von Pathogenen an Mustererkennungs-Rezeptoren (PRR, engl. pattern-recognition
receptor) wie Toll-like Rezeptoren (TLR) werden DCs aktiviert. Diese vermitteln die Differenzierung
von naiven T-Zellen in verschiedene Subtypen. Th1-Zellen produzieren unter anderem IFN-γ, welches
Makrophagen aktivieren kann. Th2-Zellen unterstützen die Antikörperproduktion von B-Zellen und
rekrutieren eosinophile Granulozyten. CD8+ T-Zellen wirken zytotoxisch und lösen Zelllyse aus.
Aktivierte Makrophagen sezernieren proinflammatorische Zytokine und Chemokine und rekrutieren
neutrophile Granulozyten. Regulatorische T-Zellen kontrollieren die Funktion der Effektorzellen und
dämmen die Reaktion ein (aus Mills 2004).
1.2.2 Aufrechterhaltung der intestinalen Homöostase
Die besondere Aufgabe im Gastrointestinaltrakt stellt die Unterscheidung zwischen
körpereigenen und körperfremden Stoffen dar. Autoreaktive Lymphozyten, welche
auf körpereigene Produkte reagieren, werden daher schon im Thymus bzw. im
Knochenmark eliminiert. Diesen Vorgang bezeichnet man als zentrale Toleranz oder

Einleitung 14
negative Selektion. Da viele körpereigene Antigene sich aber der Präsentation im
Thymus oder Knochenmark entziehen können, haben sich verschiedene
Mechanismen der peripheren Toleranz entwickelt. Dabei werden Lymphozyten in
einen anergen Zustand versetzt wodurch eine Immunreaktion ausbleibt. Dies
geschieht z. B. durch das Fehlen entzündungsspezifischer Zytokine oder
kostimulatorischer Moleküle bei der Antigenpräsentation (Murphy et al. 2009; Palmer
2003; Ohashi & DeFranco 2002). Im Darm muss zudem zwischen apathogenen
Nahrungsbestandteilen, der kommensalen Mikroflora und pathogenen Erregern
unterschieden werden. Es werden kontinuierlich Immunantworten ausgelöst, welche
streng kontrolliert werden müssen. Diese Aufgabe übernehmen die regulatorischen
T-Zellen (Tregs). Kennzeichnend für die Identifizierung als Treg ist die Expression
des Transkriptionsfaktors Foxp3 (engl. forkhead box P3) (Hori et al. 2003). Tregs
entwickeln sich zum einen im Thymus und werden daraufhin als natürliche Tregs
(nTregs) bezeichnet. Im Darm kann zum anderen die Bildung von Tregs induziert
werden (iTregs). Diese besondere Aufgabe übernehmen tolerogene DCs.
Tolerogene DCs werden durch IECs induziert. IECs produzieren unter anderem das
Zytokin TSLP (engl. thymic stromal lymphopoietin), welches die Differenzierung von
mukosalen DCs zu tolerogenen DCs fördert. Diese haben die Fähigkeit naive
CD4+ T-Zellen zu Foxp3+ Tregs zu konvertieren (Sun et al. 2007; Annacker et al.
2005). Die Produktion von TGF-ß und IL-10 durch DCs spielt hierbei eine große
Rolle (Coombes et al. 2007; Groux et al. 1999). Des Weiteren ist die Bildung von
Retinsäure (RA, engl. retinoic acid) durch DCs erforderlich (Coombes et al. 2007;
Benson et al. 2007). Retinsäure ist ein Vitamin A Metabolit. Es entsteht durch den
Abbau von Retinol (Vitamin A) durch Alkohol-Dehydrogenasen (ADH) zu Retinal,
welches durch Aldehyd-Dehydrogenasen (Aldh) weiter zu Retinsäure oxidiert wird
(Duester 2000). Es sind mehrere Isoformen der Aldhs beschrieben. Für die DCs in
mesenterialen Lymphknoten spielt Aldh1a2 dabei die größte Rolle (Iwata et al. 2004).
Die durch DCs induzierten Tregs haben große Bedeutung in der Aufrechterhaltung
der intestinalen Homöostase. Es sind verschiedene Suppressionsmechanismen der
Tregs beschrieben. Zum einen inhibieren sie die Proliferation der Effektorzellen
durch Produktion von antiinflammatorischen Zytokinen, wie TGF-β, IL-10 und IL-35.

Einleitung 15
Zum anderen können sie die direkte Zytolyse der Zielzelle, ähnlich wie CTLs, durch
Produktion von Granzymen einleiten. Zielzellen der Tregs sind hierbei vor allem
aktivierte, proinflammatorische T-Zellen. Des Weiteren können sie durch
verschiedene Mechanismen den Metabolismus der Zielzellen stören oder die
Fähigkeit von DCs, naive T-Zellen zu aktivieren, hemmen (Workman et al. 2009).
Abb. 1.3: Suppressionsmechanismen von Tregs.
Durch Sekretion von inhibitorischen Zytokinen, wie TGF-β, IL-10 und IL-35, können Tregs
Effektorzellen hemmen. Die Produktion von Granzymen führt zur Lyse der Zielzelle. Durch Bindung
von IL-2 an den Rezeptor CD25 wird den Effektorzellen IL-2 entzogen, welches sie zur Proliferation
benötigten. Anti-proliferativ wirkendes zyklisches Adenosinmonophosphat (cAMP) kann in Zielzellen
eingeschleust werden oder von Tregs produziertes Adenosin an den Adenosin-Rezeptor 2A auf
Zielzellen binden, so dass die Bildung des proinflammatorischen Zytokins IL-6 unterbleibt. Die
Fähigkeit von DCs T-Zellen zu aktivieren, kann durch Interaktion von LAG-3 mit MHC-II oder von
CTLA-4 mit CD80/CD86 inhibiert werden (aus Workman et al. 2009).
Tregs wirken zudem protektiv im Rahmen einer Colitis. Sie können die Ausprägung
einer Colitis verhindern oder eine bestehende Colitis lindern (Mottet et al. 2003;
Uhlig, Coombes, et al. 2006; Izcue et al. 2006). Es ist außerdem ein positiver
Rückkopplungsmechanismus zwischen Tregs und tolerogenen DCs beschrieben
(Min et al. 2003). Tregs können die Bildung von tolerogenen DCs stimulieren, welche
wiederum weitere Tregs aktivieren.
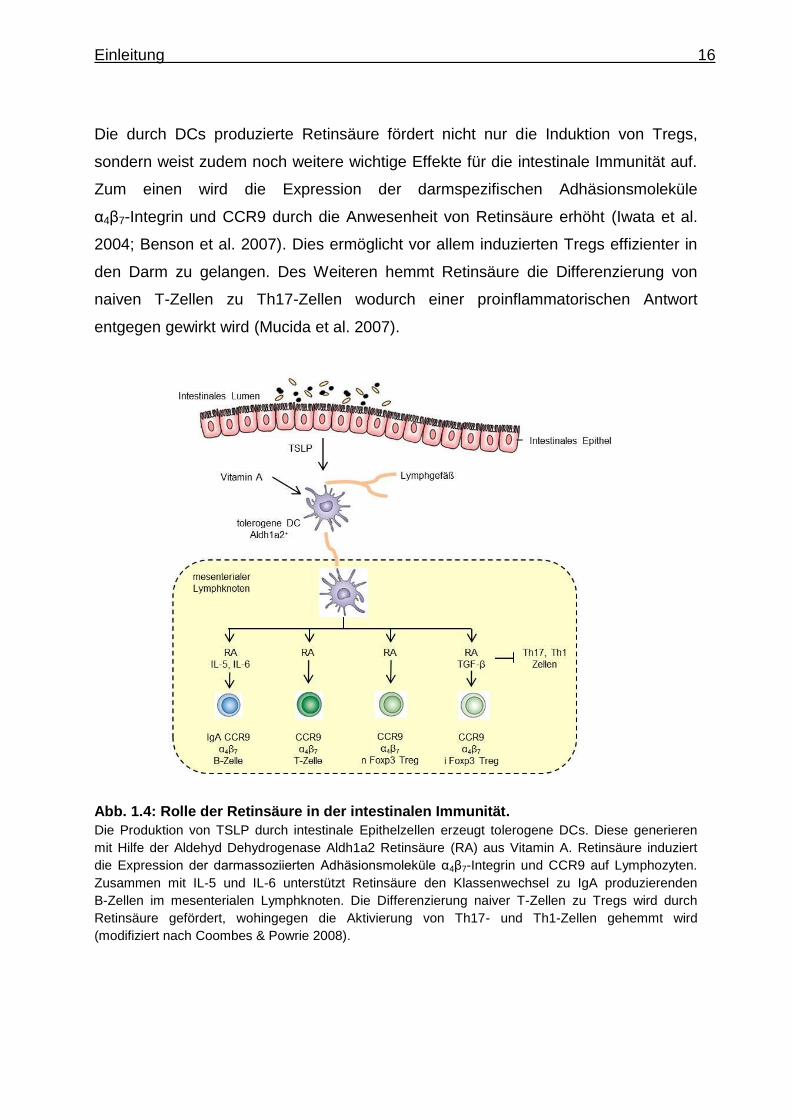
Einleitung 16
Die durch DCs produzierte Retinsäure fördert nicht nur die Induktion von Tregs,
sondern weist zudem noch weitere wichtige Effekte für die intestinale Immunität auf.
Zum einen wird die Expression der darmspezifischen Adhäsionsmoleküle
α4β7-Integrin und CCR9 durch die Anwesenheit von Retinsäure erhöht (Iwata et al.
2004; Benson et al. 2007). Dies ermöglicht vor allem induzierten Tregs effizienter in
den Darm zu gelangen. Des Weiteren hemmt Retinsäure die Differenzierung von
naiven T-Zellen zu Th17-Zellen wodurch einer proinflammatorischen Antwort
entgegen gewirkt wird (Mucida et al. 2007).
Abb. 1.4: Rolle der Retinsäure in der intestinalen Immunität.
Die Produktion von TSLP durch intestinale Epithelzellen erzeugt tolerogene DCs. Diese generieren
mit Hilfe der Aldehyd Dehydrogenase Aldh1a2 Retinsäure (RA) aus Vitamin A. Retinsäure induziert
die Expression der darmassoziierten Adhäsionsmoleküle α4β7-Integrin und CCR9 auf Lymphozyten.
Zusammen mit IL-5 und IL-6 unterstützt Retinsäure den Klassenwechsel zu IgA produzierenden
B-Zellen im mesenterialen Lymphknoten. Die Differenzierung naiver T-Zellen zu Tregs wird durch
Retinsäure gefördert, wohingegen die Aktivierung von Th17- und Th1-Zellen gehemmt wird
(modifiziert nach Coombes & Powrie 2008).

Einleitung 17
1.3 Chronisch entzündliche Darmerkrankungen
Bei chronisch entzündlichen Darmerkrankungen (CED) handelt es sich um
rezidivierende oder kontinuierlich auftretende inflammatorische Erkrankungen des
Gastrointestinaltrakts. CED treten weltweit mit einer erhöhten Häufigkeit in
Nordamerika, Großbritannien und Skandinavien auf. Die Anzahl der Neuerkrankten
(Inzidenz) liegt bei 4-10/100.000 Einwohnern pro Jahr. Die Anzahl der Erkrankten
(Prävalenz) liegt bei 40-100/100.000 Einwohnern (Hendrickson et al. 2002). Meist
manifestiert sich die Krankheit zwischen dem 15. und 30. Lebensjahr (Xavier &
Podolsky 2007). Die Hauptformen sind hierbei der Morbus Crohn (MC) und die
Colitis ulcerosa (CU). MC stellt eine Entzündung dar, welche jeden Abschnitt des
Gastrointestinaltrakts befallen kann. Am häufigsten sind die Ileocaecal-Region und
das terminale Ileum allein betroffen. Histologisch sind granulomatöse Läsionen und
transmurale leukozytäre Infiltrationen zu erkennen. Bei der CU beschränkt sich die
Entzündung auf das Colon, wobei lediglich die Tunica mucosa betroffen ist. Es sind
oberflächliche Ulzerationen und Kryptenabszesse auffindbar. Zudem kann es zu
einem Verlust der Becherzellen kommen (Xavier & Podolsky 2007). Beides sind
multifaktoriell bedingte Erkrankungen mit noch nicht vollständig geklärter Ätiologie.
Es wird davon ausgegangen, dass die Entstehung einer CED durch genetische
Prädisposition begünstigt wird. Die am häufigsten beschriebene genetische Ursache
für MC ist eine Mutation im NOD2/CARD15 Gen (engl. caspase recruitment domain
family member 15) (Strober et al. 2007). Wie bereits oben erwähnt, erkennt NOD2
bakterielle Zellwandpolymere und aktiviert über NF-κB (engl. nuclear factor ‚kappa-
light-chain-enhancer‘ of activated B-cells) weitere zahlreiche proinflammatorische
Gene. Des Weiteren ist NOD2/CARD15 für die Sekretion antimikrobiell wirksamer
α-Defensine verantwortlich (Kucharzik et al. 2006). Mutationen könnten somit zu
einer gestörten Immunreaktion und einer verminderten Fähigkeit des Darmepithels,
Bakterien zu eliminieren, führen (Strober et al. 2007). Für CU sind Mutationen sowohl
im Gen für IFN-γ als auch im MDR1 Gen (engl. multidrug resistance gene)
beschrieben (Abraham & Cho 2009; Annese et al. 2006). Des Weiteren wurden auch
genetische Veränderungen von IL-12 und des IL-23 Rezeptors in Zusammenhang
mit der Entstehung von CED gebracht (Duerr et al. 2006).

Einleitung 18
Einen weiteren Einfluss auf die Entstehung von CED nehmen Umweltfaktoren.
Stress, die Einnahme nichtsteroidaler Antiphlogistika, welche die Epithelbarriere
schädigen, und Rauchen werden als Auslöser für CED diskutiert (Mawdsley &
Rampton 2005; Podolsky 2002). Rauchen wirkt sich dabei positiv auf CU aus, wobei
es MC eher verschlimmert.
Auch die kommensale Mikroflora kann einen Einfluss auf die Entwicklung von
Darmerkrankungen nehmen. Eine Dysregulation zwischen Mikroflora und der
epithelialen Barriere kann zu einer intestinalen Entzündung führen. Es wurde gezeigt,
dass Patienten mit MC und CU zum einen eine erhöhte Permeabilität des
Darmepithels und zum anderen eine veränderte Zusammensetzung der intestinalen
Mikroflora aufweisen (Welcker et al. 2004; Sartor 2008). Des Weiteren hat eine
Therapie mit Antibiotika, wie Metronidazol, bei CED Patienten einen positiven Effekt,
was für eine Beteiligung der Darmflora spricht. Einzelne Erreger, welche die
Erkrankung auslösen, konnten bislang nicht identifiziert werden. Viele Studien
beschäftigen sich mit dem Zusammenhang zwischen MC und dem Erreger
Mycobacterium avium subspecies paratuberculosis (MAP). MAP löst die
Paratuberkulose, eine chronische Enteritis, in Wiederkäuern aus, welche auch
Johne´sche Krankheit genannt wird. Die Paratuberkulose ist eine in Deutschland
meldepflichtige Tierseuche, welche sich in unstillbaren, wässrigen Durchfällen und
starker Abmagerung äußert und zum Tod führen kann (Baumgärtner 2012).
Histologisch ähnelt das Erscheinungsbild dem MC des Menschen. Es ist
gekennzeichnet durch eine granulomatöse Enteritis, die vor allem im Ileum und
Caecum auftritt. Auf Grund dessen wurde eine Beteiligung MAPs an MC diskutiert
(Chiodini et al. 1984). MAP wird über kontaminierte Milch- und Fleischprodukte
übertragen, da es als intrazellulärer Erreger auch starke Erhitzung übersteht (Lund et
al. 2002). Tatsächlich wurde MAP in Patienten mit MC identifiziert (Olsen et al. 2009;
Naser et al. 2004). Allerdings wurde MAP auch zu einem geringeren Anteil in
Patienten mit CU und in gesunden Patienten gefunden (Autschbach et al. 2005;
Davis & Madsen-Bouterse 2012). Es wird somit vermutet, dass MAP nicht als
Auslöser des MC gilt, sondern dass eine persistierende MAP Infektion den Ausbruch
der Krankheit in genetisch prädisponierten Menschen fördert (Sartor 2006).

Einleitung 19
Des Weiteren kann die Entstehung einer CED durch Defekte in der Immunfunktion
begünstigt werden. Patienten mit MC weisen eine überschießende
Th1-Immunreaktion mit erhöhter IFN-γ und IL-12 Produktion auf (Strober et al. 2002;
Sartor 2006). Zusätzlich wurde gezeigt, dass auch Th17-Zellen einen großen
Einfluss auf das Fortschreiten der Erkrankung haben (Elson et al. 2007). Das von
APCs produzierte Zytokin IL-23 stimuliert die Differenzierung von naiven T-Zellen zu
Th17-Zellen. Es beinhaltet die gleiche Untereinheit p40 wie das von Th1-Zellen
sezernierte IL-12. Die Behandlung mit einem IFN-γ Antikörper in Studien zeigte nur
geringen Effekt, wohingegen ein p40 Antikörper, welcher sowohl IL-12 als auch IL-23
blockiert, einen deutlich positiven Effekt bei Patienten mit MC aufwies, was für eine
Beteiligung der Th17-Zellen spricht (Mannon et al. 2004). Bei der CU hingegen wird
davon ausgegangen, dass es sich um eine atypische, Th2 vermittelte Entzündung
handelt. CU Patienten weisen eine erhöhte Produktion von IL-5 und IL-13 auf, nicht
jedoch von IL-4. Es wird davon ausgegangen, dass natürliche Killer T-Zellen durch
APCs, welche einen atypischen MHC-Rezeptor tragen, aktiviert werden. Dadurch
kommt es zu einer gesteigerten Produktion von IL-13, welches das Epithel schädigt
und die Entstehung von Ulzerationen fördert (Fuss et al. 2004). Insgesamt kann
allerdings kein Faktor isoliert betrachtet werden. Meist handelt es sich um ein
Zusammenspiel der Faktoren, welche in Kombination zu schwerwiegenden
entzündlichen Veränderungen im Darm führen können.

Einleitung 20
Abb. 1.5: Auslösende Faktoren einer chronisch entzündlichen Darmerkrankung.
Genetische Prädispositionen und fehlgeleitete Immunreaktionen sind an der Entstehung chronisch
entzündlicher Darmerkrankungen (CED) beteiligt, ebenso wie die kommensale Darmflora und
begünstigende Umweltfaktoren. Meist wirken alle Faktoren synergistisch (modifiziert nach Sartor
2006).
1.4 Murine Modelle für chronisch entzündliche Darmerkrankungen
Zur Untersuchung von CED werden viele verschiedene Tiermodelle heran gezogen
(Hoffmann et al. 2002). Zum einen bildet sich in bestimmten Mausstämmen eine
Spontancolitis. Dabei handelt es sich unter anderem um SAMP1/Yit Mäuse, welche
sich ursprünglich von Seneszenz-Mäusen ableiten (Matsumoto et al. 1998). Des
Weiteren entwickeln C3H/HeJBir Mäusen, welchen der TLR4 fehlt, eine spontane
Colitis (Cong et al. 1998). Außerdem kann eine Colitis in verschiedenen transgenen
Tieren induziert werden. So entwickeln z. B. IL-2 Knock-out oder IL-10 Knock-out
Tiere unter speziellen Bedingungen eine entzündliche Darmerkrankung (Contractor
et al. 1998; Kühn et al. 1993). Unter keimfreien Haltungsbedingungen bleibt eine
Erkrankung aus, wohingegen sich unter SPF-Bedingungen (engl. special pathogen
free) in IL-10 Knock-out Tieren eine milde Entzündung des proximalen Colons und in
IL-2 Tieren eine komplette Colitis entwickelt. Unter konventionellen
Haltungsbedingungen entwickeln IL-10 Knock-out Mäuse eine hochgradige
chronische Enterocolitis (Kühn et al. 1993). Dies spiegelt zudem eine Bedeutung der
mukosalen Mikroflora für die Entwicklung einer Darmerkrankung wider. In

Einleitung 21
immunsupprimierten SCID (engl. severe combined immunodeficiency) oder RAG-
defizienten (engl. recombination activation gene) Mäusen kann durch Transfer von
bestimmten T-Zellpopulationen eine Entzündung ausgelöst werden und die Funktion
der T-Zellen untersucht werden. Der Transfer von CD45RBhigh-T-Lymphozyten
(undifferenzierte T-Lymphozyten) führt hierbei zu einer schweren Colitis, wohingegen
der Transfer von CD45RBlow-T-Lymphozyten (reife T-Lymphozyten) einen protektiven
Effekt aufweist (Hoffmann et al. 2002). Durch diese Modelle erfolgte der Nachweis,
dass sich regulatorische T-Zellen positiv auf eine Colitis auswirken (Izcue et al.
2006). Ein weiteres Colitismodell ist das der chemisch induzierbaren
Darmerkrankungen. Durch rektale Verabreichung von Oxazolon oder TNBS (engl.
trinitrobenzene sulfonic acid) kann eine Colitis ausgelöst werden (Morris et al. 1989;
Boirivant et al. 1998). Des Weiteren entsteht durch die orale Gabe von
Dextrannatriumsulfat (DSS, engl. dextran sodium sulfate) eine schwere Colitis,
welche durch starke leukozytäre Infiltrationen, fokale Kryptenzerstörung, lymphoide
Hyperplasie und epitheliale Ulzeration gekennzeichnet ist (Okayasu et al. 1990;
Cooper et al. 1993). Die Wirkungsweise dieses Modells ist bislang noch nicht
vollständig geklärt. Es wird allerdings meist davon ausgegangen, dass DSS direkt
zytotoxisch auf die Epithelzellen der Colonschleimhaut wirkt und diese somit zerstört.
Daraufhin werden Phagozyten aktiviert, welche eine Immunreaktion mit erhöhter
proinflammatorischer Zytokinproduktion auslösen (Okayasu et al. 1990). Weitere
Studien zeigen jedoch, dass es sich bei der Immunantwort im DSS Modell um eine
Mischform aus einer Th1- und Th2-Antwort handelt (Dieleman et al. 1998). Des
Weiteren konnte zudem gezeigt werden, dass auch in SCID-Mäusen, welche keine
funktionalen T- und B-Lymphozyten besitzen, eine DSS Colitis ausgelöst werden
kann (Dieleman et al. 1994; Axelsson et al. 1996).

Einleitung 22
1.5 Beteiligung von DCs an der Entstehung von chronisch entzündlichen
Darmerkrankungen
Wie bereits erwähnt, haben DCs großen Einfluss auf die Regulation der
Immunantwort. Verschiedene Studien befassen sich mit ihrer Bedeutung bei CED.
Abe et al. zeigten, dass die Ablation von DCs in einem Colitismodell während der
Erkrankung zu einer Verbesserung der Symptome führte. Wurden die DCs jedoch
schon vor Ausbruch der Colitis entfernt, führte dies zu einer Verschlechterung. Die
Behandlung der Mäuse mit einem TLR9-Liganden vor Induktion der Colitis, zeigte
wiederum protektive Wirkung (Abe et al. 2007). DCs spielen hiernach eine zeitlich
abhängige Rolle bei der Entstehung von entzündlichen Darmerkrankungen. Uhlig et
al. beschrieben zudem eine T-Zell unabhängige Entwicklung einer Colitis durch
direkte Aktivierung von DCs durch CD40 Stimulation (Uhlig, McKenzie, et al. 2006).
Des Weiteren besteht ein Zusammenhang zwischen den prädisponierenden Genen
für CED und DCs. Das oben beschriebene NOD2 wird auch auf DCs exprimiert.
Mutationen können zu Dysfunktionen dieser Zellen und somit zu fehlgeleiteten
Immunreaktionen oder schließlich auch zum Ausbruch von CED beitragen (Strober
et al. 2007). In Patienten mit CED konnte eine erhöhte Expression von TLR2 und
TLR4 auf intestinalen DCs festgestellt werden. Patienten mit MC zeigten zudem eine
gesteigerte Expression des Aktivierungsmarkers CD40 auf DCs sowie eine
vermehrte Produktion der proinflammatorischen Zytokine IL-12 und IL-6 durch DCs
(Hart et al. 2005). Des Weiteren konnte nachgewiesen werden, dass DCs im
terminalen Ileum in der Lage sind die Untereinheit p40 der Zytokine IL-12 und IL-23
zu produzieren (Becker et al. 2003). IL-23, welches Th17-Zellen aktivieren kann,
spielt eine bedeutende Rolle in der Pathogenese von CED (Yen et al. 2006; Hue et
al. 2006a; Izcue et al. 2008). Bei Patienten mit MC konnte eine erhöhte Menge an
durch DCs produziertem IL-23 sowie eine verringerte Produktion von IL-10
festgestellt werden (Sakuraba et al. 2009). Des Weiteren sind DCs auch als Quelle
des proinflammatorischen Zytokins TNF-α beschrieben (de Baey et al. 2003).
Baumgart et al. hingegen beschrieben einen Mangel an unreifen DCs bei CED
Patienten (Baumgart et al. 2005). Die Bedeutung von DCs im Rahmen von
Darmentzündungen kann somit als sehr entscheidend angesehen werden.

Einleitung 23
1.6 Der Hypoxie induzierbare Faktor (HIF)
Überschreitet der Bedarf an Sauerstoff das Angebot, so entsteht ein
Sauerstoffmangel, die sogenannte Hypoxie (Fandrey 2007). Geringe
Sauerstoffkonzentrationen sind kennzeichnend für entzündetes Gewebe. Dies wird
zum einen durch mangelhafte Durchblutung bei gleichzeitig erhöhtem
Sauerstoffverbrauch durch rekrutierte Immunzellen bedingt (Karhausen et al. 2005;
Eltzschig & Carmeliet 2011). Im Falle des Darmes liegen noch weitere Ursachen vor.
Die Epithelzellen des Darms nehmen eine besondere Stellung ein. Sie trennen das
fast anoxische Darmlumen von den reich durchbluteten Zellen der Lamina propria
mucosae und sind somit großen Sauerstoffschwankungen ausgesetzt. Im gesunden
Darm weisen die Epithelzellen daher schon eine „physiologische Hypoxie“ auf. Wird
das Epithel nun durch eine Darmentzündung geschädigt, so tritt die Anoxie des
Darmlumens tiefer in das Gewebe ein (Karhausen et al. 2004). Auch die Zellen der
Lamina propria mucosae und der Tela submucosa sind nun hypoxischen
Bedingungen ausgesetzt. Der Transkriptionsfaktor Hypoxie-induzierbarer Faktor
(HIF) nimmt für die Anpassung des Zellstoffwechsels an hypoxische Bedingungen
eine zentrale Rolle ein (Semenza et al. 1997). HIF reguliert die
Sauerstoffhomöostase und induziert die transkriptionelle Genexpression, welche
unter anderem für die Angiogenese, Erythropoiese und anaerobe Glykolyse benötigt
wird.
1.6.1 Aufbau und Funktion von HIF
HIF ist ein heterodimerer Proteinkomplex aus einer sauerstoffabhängig regulierten α-
und einer konstitutiv exprimierten β-Untereinheit, welche auch als ARNT (engl. aryl
hydrocarbon receptor nuclear translocator) bezeichnet wird (Wang et al. 1995). Bei
Mensch und Maus sind bislang drei Isoformen der α-Untereinheit (HIF-1α, -2α, -3α)
sowie drei Isoformen des HIF-β Proteins identifiziert. Die HIF-1α Untereinheit weist
eine extrem kurze Halbwertszeit auf und wird unter normoxischen Bedingungen
kontinuierlich abgebaut und neugebildet. Unter Hypoxie erfolgen die Stabilisierung
des Proteins und die Translokation in den Nukleus. Dort folgt die Dimerisierung mit
der β-Untereinheit. Beide Untereinheiten gehören zur Familie der basischen-Helix-

Einleitung 24
Loop-Helix (bHLH) und PER-ARNT-Sim (PAS) Transkriptionsfaktoren. Die bHLH
Domänen am N-terminalen Ende ermöglichen die Bindung der Untereinheiten an die
Hypoxie-responsiven Elemente (HRE) des jeweiligen Zielgens. Die nachfolgenden
PAS Domänen vermitteln die Dimerisierung zwischen der α- und β-Untereinheit des
HIF-Komplexes. Die Transaktivierungsdomäne (TAD), welche sich im Falle der
HIF-1α Untereinheit in eine N-terminale (NAD) und C-terminale (CAD)
Aktivierungsdomäne aufgliedert, dient der Aktivierung von Zielgenen. Die
sauerstoffabhängigen Abbaudomänen (ODDD, engl. oxygen-dependent degradation
domain) der α-Untereinheit bedingen die posttranslationale, sauerstoffabhängige
Regulation der Aktivität des Transkriptionsfaktors (Wenger et al. 2005; Fandrey et al.
2006).
Abb. 1.6: Struktur des Hypoxie-induzierbaren Faktors HIF-1.
Die basischen Helix-Loop-Helix (bHLH) Domänen dienen der Bindung der Untereinheiten an DNA. Die
PAS Domänen ermöglichen die Dimerisierung der Untereinheiten. Die N- und C-terminalen
sauerstoffabhängigen Abbaudomänen (engl. oxygen-depending degradation domain, NODDD bzw.
CODDD) sowie die transaktivierenden Domänen (NAD bzw. CAD) der α-Untereinheit dienen der
sauerstoffabhängigen Regulation und der Aktivierung von Zielgenen (modifiziert nach Schofield &
Ratcliffe 2004).
Hydroxylierung

Einleitung 25
1.6.2 Sauerstoffabhängige Regulation und Aktivierung von HIF-1
Unter normoxischen Bedingungen vermitteln spezielle Prolylhydroxylasen (PHDs) die
Hydroxylierung von spezifischen Prolinresten innerhalb der ODDDs der
α-Untereinheit. Diese werden von dem von Hippel-Lindau (VHL)
Tumorsuppressorproteinkomplex, welcher zur Familie der E3-Ubiquitin-Ligase
Komplexe gehört, erkannt. Nach vermittelter Ubiquitinierung wird das HIF-1α Protein
daraufhin dem proteasomalen Abbau zugeführt (Ivan et al. 2001). Des Weiteren
hydroxyliert die Asparaginhydroxylase HIF-1-inhibierender Faktor (FIH-1, engl. factor
inhibiting HIF-1) sauerstoffabhängig den Asparaginrest in der C-terminalen
Transaktivierungsdomäne (CAD) der α-Untereinheit. Dies verhindert die Interaktion
der CAD mit dem Koaktivator p300/Creb-bindenden Protein (CPB), wodurch die
transkriptionelle Steuerung von Genen durch den HIF-Komplex inhibiert wird (Mahon
et al. 2001).
Unter hypoxischen Bedingungen kommt es zu einer Hemmung der PHD und der
FIH-1 Aktivität. Eine Veränderung des Eisenhaushaltes der Zelle sowie eine
Inhibition mit NO kann zudem eine Hemmung der PHDs unter Normoxie induzieren
(Zhou et al. 2003; Metzen et al. 2003). Die Hydroxylierung der HIF-1α Untereinheit
unterbleibt, wodurch diese in den Kern translozieren und Koaktivatoren rekrutieren
kann. Die Dimerisierung mit der β-Untereinheit führt schließlich zur transkriptionellen
Aktivität.

Einleitung 26
Abb. 1.7: Sauerstoffabhängige Regulation der HIF-α Aktivität.
Unter normoxischen Bedingungen hydroxylieren die sauerstoffabhängigen Prolylhydroxylasen (PHDs)
und der HIF-1-inhibierende Faktor (engl. factor inhibiting HIF, FIH-1) die Prolin- und Asparaginreste
des HIF-α. Dadurch erfolgen die durch das von Hippel-Lindau Tummorsuppressorprotein vermittelte
Ubiquitinierung und der proteasomale Abbau von HIF-α. Zudem wird die Interaktion der C-terminalen
Transaktivierungsdomäne mit dem Koaktivator p300/CPB verhindert. Unter hypoxischen Bedingungen
werden PHDs und FIH-1 gehemmt. HIF-α wird stabilisiert und transloziert in den Nukleus. Dort
dimerisiert es mit der β-Untereinheit, rekrutiert Koaktivatoren, und bildet somit das transkriptionell
aktive HIF Heterodimer (modifiziert nach Schofield & Ratcliffe 2004).
1.6.3 Sauerstoffunabhängige Regulation und Aktivierung von HIF-1
In verschiedenen Forschungen konnte gezeigt werden, dass eine HIF Aktivierung
nicht nur durch Hypoxie ausgelöst werden kann, sondern auch durch
inflammatorische Stimuli. So induziert LPS durch Aktivierung des
Transkriptionsfaktors NF-κB die HIF-1α mRNA Expression (Frede et al. 2006).
Jantsch et al. konnten zeigen, dass dieser Effekt außerdem von TLR4 und dem
Adaptorprotein MyD88 (engl. myeloid differentiation primary resonse gene 88)
abhängig ist. DCs von MyD88 Knock-out Mäusen zeigten nach LPS Stimulation
keine Induktion von HIF-1α (Jantsch et al. 2011). Weitere Studien beschrieben eine
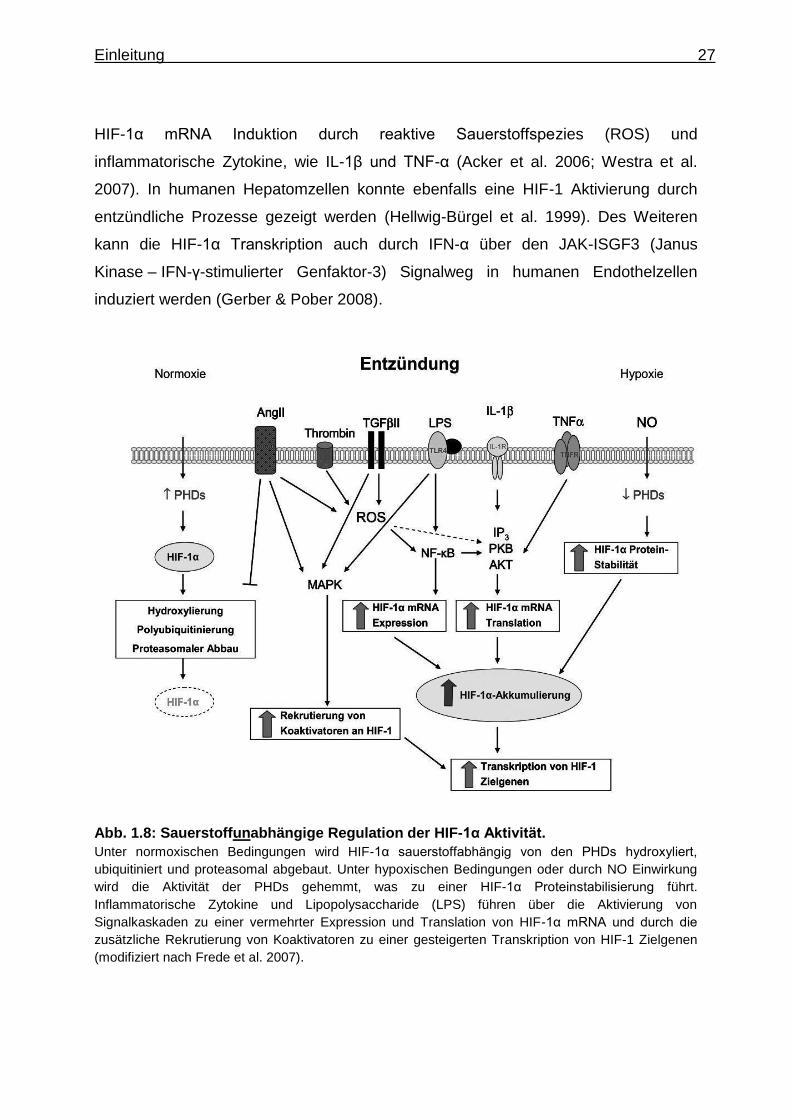
Einleitung 27
HIF-1α mRNA Induktion durch reaktive Sauerstoffspezies (ROS) und
inflammatorische Zytokine, wie IL-1β und TNF-α (Acker et al. 2006; Westra et al.
2007). In humanen Hepatomzellen konnte ebenfalls eine HIF-1 Aktivierung durch
entzündliche Prozesse gezeigt werden (Hellwig-Bürgel et al. 1999). Des Weiteren
kann die HIF-1α Transkription auch durch IFN-α über den JAK-ISGF3 (Janus
Kinase – IFN-γ-stimulierter Genfaktor-3) Signalweg in humanen Endothelzellen
induziert werden (Gerber & Pober 2008).
Abb. 1.8: Sauerstoffunabhängige Regulation der HIF-1α Aktivität.
Unter normoxischen Bedingungen wird HIF-1α sauerstoffabhängig von den PHDs hydroxyliert,
ubiquitiniert und proteasomal abgebaut. Unter hypoxischen Bedingungen oder durch NO Einwirkung
wird die Aktivität der PHDs gehemmt, was zu einer HIF-1α Proteinstabilisierung führt.
Inflammatorische Zytokine und Lipopolysaccharide (LPS) führen über die Aktivierung von
Signalkaskaden zu einer vermehrter Expression und Translation von HIF-1α mRNA und durch die
zusätzliche Rekrutierung von Koaktivatoren zu einer gesteigerten Transkription von HIF-1 Zielgenen
(modifiziert nach Frede et al. 2007).

Einleitung 28
1.6.4 Bedeutung von HIF-1 im Rahmen von Infektionen und Entzündungen
Verschiedene Studien konnten verdeutlichen, dass der Transkriptionsfaktor HIF-1
eine wichtige Rolle im Entzündungsgeschehen spielt. So konnte gezeigt werden,
dass Mäuse mit einem HIF-1α Knock-out in myeloischen Zellen eine deutliche
Beeinträchtigung in ihrer Immunfunktion aufweisen. HIF-1α defiziente Makrophagen
besitzen eine verminderte Fähigkeit zur Phagozytose und sind in ihrer Motilität und
Zellaggregation beeinträchtigt (Cramer et al. 2003). Zudem induziert HIF-1α die
Expression des β2-Integrins, ein Rezeptor, welcher für die Rekrutierung von
Leukozyten eine wichtige Rolle spielt (Kong et al. 2004). Auch Peyssonnaux et al.
konnten zeigen, dass die bakterizide Aktivität und die Produktion von TNF-α in
Makrophagen nach Infektion mit Streptokokken HIF-1α abhängig ist (Peyssonnaux et
al. 2005). Des Weiteren besteht ein Zusammenhang zwischen HIF-1 und der
Induktion von bestimmten TLRs. Kuhlicke et al. konnten eine hypoxische, HIF-1α
abhängige Induktion von TLR2 und TLR9 in verschiedenen Zellen nachweisen
(Kuhlicke et al. 2007).
Weitere Studien beschäftigten sich intensiv mit der Bedeutung von HIF-1 in
intestinalen Epithelzellen im Rahmen entzündlicher Darmveränderungen. Ein Verlust
von HIF-1α in Epithelzellen führte dabei zu einer deutlichen Verschlechterung der
Symptome, wohingegen die Stabilisierung von epithelialem HIF-1 einen protektiven
Effekt aufweist (Karhausen et al. 2004). Die Behandlung mit dem Hydroxylase
Inhibitor Dimethyloxalylglycin (DMOG), welcher den Abbau von HIF-1α verhindert,
führte zudem zu einer reduzierten Apoptoserate der Epithelzellen im DSS
Colitismodell (Cummins et al. 2008). Des Weiteren resultierte eine gesteigerte HIF
Expression durch die genetische Deletion der Prolylhydroxylase 1 (PHD1) in einer
gemilderten Ausprägungsform der Colitis mit reduzierter epithelialer Apoptose und
somit gesteigerter Barrierefunktion (Tambuwala et al. 2010).
Der Einfluss von HIF-1 auf die Funktion von DCs wurde in einigen Studien
beschrieben. Jantsch et al. zeigten, dass Hypoxie die LPS vermittelte Aktivierung von
DCs erhöht. Die Stimulation von murinen DCs mit LPS unter hypoxischen
Bedingungen führte zu einer erhöhten Expression der Aktivierungsmarker CD80 und
CD86, sowie des MHC-II-Oberflächenmoleküls. Ein Knock-down von HIF-1α

Einleitung 29
resultierte hingegen in einer verminderten Expression der Aktivierungsmarker und
einer eingeschränkten Fähigkeit der DCs allogene T-Zellen zu stimulieren (Jantsch et
al. 2008). Diese Ergebnisse stehen im Gegensatz zu Befunden von Mancino et al.,
welche besagen, dass LPS stimulierte DCs unter Hypoxie eine geringere Expression
der Aktivierungsmarker CD40, CD80, CD83 und CD86 aufweisen, wohingegen die
Produktion von TNF-α und IL-1β erhöht ist (Mancino et al. 2008). Auch Bosseto et al.
stellten eine reduzierte Expression von CD80 und CD86 in DCs unter Hypoxie fest.
Die Produktion des proinflammatorischen Zytokins IL-12p70 durch DCs hingegen
war unter Hypoxie erhöht. Eine zusätzliche Infektion der Zellen mit Leishmania führte
wiederum zu einer erhöhten IL-12p70 Produktion unter Normoxie (Bosseto et al.
2010). Des Weiteren konnte ein direkter Zusammenhang zwischen HIF-1α in DCs
und der Produktion von Typ I Interferonen nachgewiesen werden. Ein Knock-out von
HIF-1α in DCs führte zu einer verringerten Produktion von Typ I Interferonen nach
LPS Stimulus, wodurch die Aktivierung von zytotoxischen T-Zellen beeinträchtigt
wurde (Wobben et al. 2013).

Einleitung 30
1.7 Ziel der Dissertation
Dendritische Zellen sind wie alle Immunzellen im entzündeten Gewebe hypoxischen
Bedingungen ausgesetzt und müssen sich diesen anpassen (Eltzschig & Carmeliet
2011). Hierfür wird der Transkriptionsfaktor HIF-1 benötigt, welcher die zelluläre
Adaptation an Hypoxie reguliert. Verschiedene Studien haben die
unterschiedlichsten Effekte von HIF-1 für die Funktion von Immunzellen und speziell
von DCs beschrieben. Im Rahmen einer experimentellen Colitis wurde bislang
allerdings nur die Bedeutung von HIF-1 in intestinalen Epithelzellen untersucht
(Karhausen et al. 2004; Cummins et al. 2008). Hierbei wird HIF-1 eine protektive
Rolle zugeschrieben.
Da DCs von zentraler Bedeutung im Immungeschehen des Darmes sind, sollte in
dieser Arbeit nun der Effekt von dendritischem HIF-1 auf die Ausprägung einer
experimentellen Colitis untersucht werden. Dazu wurde durch orale Gabe von DSS
eine experimentelle Colitis in Kontrollmäusen (HIF-1α+f/+f) und in Mäusen mit einem
Knock-out von HIF-1α in DCs (CD11cCre/HIF-1α+f/+f) induziert. Der Schweregrad der
Entzündung, die Expression von pro- und antiinflammatorischen Zytokinen sowie der
Einfluss von DCs auf weitere Immunzellen wurde untersucht. Da die Antigen-
präsentation von intestinalen DCs vornehmlich in mesenterialen Lymphknoten
stattfindet, wurden diese zusätzlich zum Colon untersucht. Zur Unterstützung dieser
in vivo Untersuchungen wurden zudem DCs aus dem Knochenmark der
beschriebenen Tiere generiert, unter verschiedenen Bedingungen kultiviert und in
ihrer Aktivierbarkeit verglichen.

Material und Methodik
31
2 Material und Methoden
2.1 Materialien
2.1.1 Geräte und Verbrauchsmaterialien
Tabelle 2.1: Geräte
Gerät Hersteller
Absaugpumpe Biotec
Auflichtmikroskop CK 40 Olympus
Brutschrank Hera cell Heraeus Instruments
CO2 Brutschrank Thermo
ELISA-Reader Spectra Count Packard
Spektrophotometer-System Epoch/Take 3 Biotec
Fluoreszenz- und Chemilumineszenzsystem Fusion-FX 7 Peqlab
Fluoreszenzmikroskop Eclipse E1000 Nikon
Gas Mixer Q Ruskinn
Gefrierschrank Liebherr
Heizblock HTM 130 / HTM 130-6 HLC
Hypoxie-Brutschrank BB6620 CUO2 Heraeus Instruments
iCycler iQ5, Multicolor Real-Time PCR Detection System Bio-Rad
Invivo2 400 Hypoxia Workstation Ruskinn
Kamerasystem DS Ri1 Nikon
Kühlschrank Liebherr
Mastercycler Eppendorf
Microm HM 340E Thermo Scientific
Mini Protean 3 Bio-Rad
Mini-Protean® Tetra Cell Bio-Rad
pH-Meter Eutech Instruments
Photometer SmartSpec 3000 Bio-Rad
ScanScope CS2 Aperio

Material und Methodik
32
Schüttler Titramax 101 Heidolph
Sterilbank HERA Safe Heraeus Instruments
Thermocycler Tpersonal / Tprofessional Biometra
UV-Geldokumentationsanlage BioDoc-It UVP
Vortexer Bioproducts
Western Blot-Kammer Mini Trans-Blot© Bio-Rad
Zählkammer nach Neubauer BD
Zentrifugen Eppendorf und Heraeus
Tabelle 2.2: Verbrauchsmaterialien
Bezeichnung Hersteller
8er Kette optisch klar flacher Deckel Sarstedt
Bechergläser Schott
Deckgläser Engelbrecht
Einmalspritzen (2 ml, 5 ml, 10 ml) B. Braun
Erlenmeyerkolben Schott
Gel-blotting-Papiere, GB003 Schleicher & Schuell
hema-screenTM
Immunostics.inc.
Kanülen (G27 ¾") BD
Microtome Blade S35 Feather
96 Multiply® PCR Platte natur Sarstedt
Nitrocellulose-Transfermembran, 0,2 µm Porengröße Schleicher & Schuell
Pinzetten Oehmen
Plastikpipetten (steril, 1 ml, 5 ml, 10 ml, 25 ml) Greiner bio-one
PP-Schraubverschluss Röhrchen (15 ml, 50 ml) Greiner bio-one
Reaktionsgefäß (1,5 ml, 2 ml) Eppendorf
Rotilabo®-Einbettkassetten Roth
Safe-Lock Tubes 0,5 ml Eppendorf

Material und Methodik
33
Scheren Oehmen
SuperFrost® Plus Objektträger R. Langenbrinck
Zellkulturflaschen (T25, T75, T175) Greiner bio-one
Zellkulturplatte steril (6 Vertiefungen) Greiner bio-one
2.1.2 Chemikalien
Tabelle 2.3: Chemikalien
Bezeichnung Hersteller
4,6-Diamidin-2-phenylindol (DAPI) Sigma
Aceton Sigma
Acrylamid/Bisacrylamid (30 %) Bio-Rad
Agarose Invitrogen
Alcian Blue Solution Sigma
Ammoniumchlorid Sigma
Ammoniumpersulfat (APS) Bio-Rad
Bromphenolblau Sigma
Bovines Serum Albumin (BSA) SERVA
Cytoseal XYL Thermo Scientific
Fluorescence Mounting Medium Dako
Dextrannatriumsulfat MP Biomedicals
Diethylpyrocarbonat (DEPC) Sigma
ECL Advance™ Western Blotting Detection Kit GE Healthcare
Eosin Y Lösung Sigma
Essigsäure Fluka
Ethanol Roth
Ethylendiamintetraessigsäure (EDTA) Merck
Ethidiumbromid Sigma
Fetal calf serum (FCS) Biochrom AG

Material und Methodik
34
Glutamin Invitrogen
Glycerin Merck
Glycin Fluka
Guanidinthiocyanat (GTC) Roth
Isopropanol Sigma
Kaliumchlorid AppliChem
Kaliumdihydrogenphosphat Merck
Lipopolysaccharid (LPS) Sigma
Mayers Hämalaunlösung Merck
Mouse IL-6 ELISA MAX™ Deluxe BioLegend
Mouse IL-10 ELISA MAX™ Deluxe BioLegend
Mouse TSLP ELISA MAX™ Deluxe BioLegend
β-Mercaptoethanol Merck
Methanol Sigma
Natriumacetat Sigma
Natriumazid AppliChem
Natriumchlorid Fluka
Natriumdodecylsulfat (SDS) Roth
Natriumhydrogencarbonat Fluka
Natriumdihydrogenphosphat Fluka
Natriumpyruvat Gibco
Normal goat serum (NGS) Sigma
Normal rabbit serum (NRS) Invitrogen
Nukleosidtriphosphate Invitrogen
Paraffin McCormick
Paraformaldehyd (PFA) Sigma
Penicillin Gibco
Phenol AppliChem
Phenol-Chloroform-Isoamylalkohol AppliChem

Material und Methodik
35
Pimonidazole HPI
Ponceau S Sigma
Protease-Inhibitor Roche Diagnostics
RPMI-Medium 1640 Gibco
Salzsäure Fluka
Streptomycin Gibco
Target Retrieval Solution Dako
Tris-aminomethan (Tris-Base) Fluka
Tris-HCl Roth
Trypanblau Merck
Trypsin-EDTA Gibco
Tween 20 Roth
Vector Mouse on Mouse (M.O.M.TM
) Kit Vector Labs
Wasserstoffperoxid (H2O2) Merck
Xylol Roth
2.1.3 Puffer und Lösungen
BmDC-Medium: RPMI 1640
10 % FCS
4 mM Glutamin
1mM Natriumpyruvat
100 U/ml Penicillin
100 µg/ml Streptomycin
0,06 mM β-Mercaptoethanol
Blotpuffer (Western Blot): 25 mM Tris-Base
96 mM Glycin
20 % Methanol

Material und Methodik
36
DEPC-H2O: 500 ml H2O
0,5 ml DEPC
1 h bei RT rühren, autoklavieren
Laufpuffer (Western Blot): 25 mM Tris-Base
192 mM Glycin
3,5 mM SDS
Lower Buffer 1,5 M Tris-Base
(Western Blot): 13,9 mM SDS
pH 8,8
Lysepuffer (Gesamtlysat): 150 mM NaCl
10 mM Tris-Base
1 mM EDTA
0,5 % NP 40
1 % Protease-Inhibitor
PBS: 137 mM NaCl
2,7 mM KCl
10 mM Na2HPO4
2 mM KH2PO4
pH 7,2
PBS-T: 1 x PBS
0,05 % Tween 20

Material und Methodik
37
4 x SDS (Western Blot): 0,4 ml Aqua dest.
1,6 ml 0,5 M Tris pH 6,8
3,2 ml 10 % SDS
0,8 ml β-Mercaptoethanol
0,4 ml 0,5 % Bromphenolblau
10 % Glycerin
TAE (PCR): 40 mM Tris-Base
0,11 % Essigsäure
1 mM EDTA pH 8,0
TBS: 137 mM NaCl
20 mM Tris-HCl
pH 7,6
TBS-T: 1 x TBS
0,1 % Tween 20
Upper Buffer 252 mM Tris-Base
(Western Blot): 6,9 mM SDS
pH 6,8
2.1.4 Antikörper
Tabelle 2.4: Antikörper für Western Blot
Antikörper Typisierung Hersteller
β-Aktin Kaninchen Abcam
HIF-1α Kaninchen Cayman chemical
HRP-konjugiert anti-Kaninchen Ziege Sigma
Material und Methodik
38
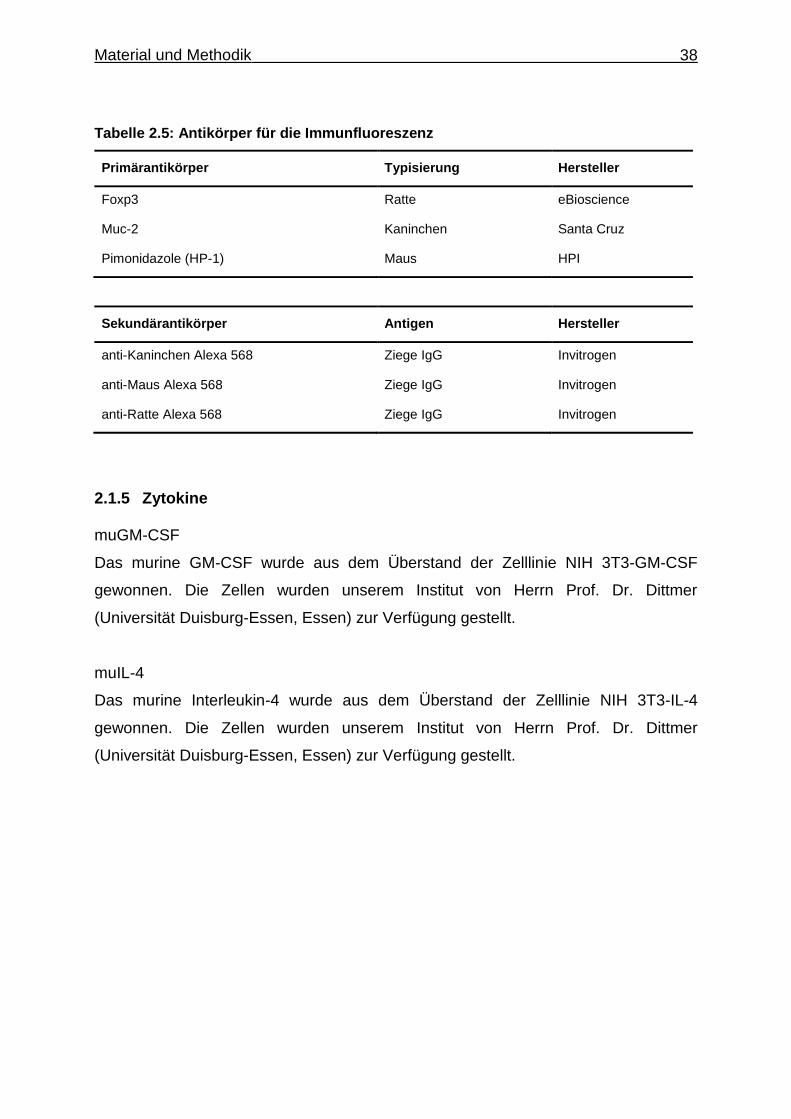
Tabelle 2.5: Antikörper für die Immunfluoreszenz
Primärantikörper Typisierung Hersteller
Foxp3 Ratte eBioscience
Muc-2 Kaninchen Santa Cruz
Pimonidazole (HP-1) Maus HPI
Sekundärantikörper Antigen Hersteller
anti-Kaninchen Alexa 568 Ziege IgG Invitrogen
anti-Maus Alexa 568 Ziege IgG Invitrogen
anti-Ratte Alexa 568 Ziege IgG Invitrogen
2.1.5 Zytokine
muGM-CSF
Das murine GM-CSF wurde aus dem Überstand der Zelllinie NIH 3T3-GM-CSF
gewonnen. Die Zellen wurden unserem Institut von Herrn Prof. Dr. Dittmer
(Universität Duisburg-Essen, Essen) zur Verfügung gestellt.
muIL-4
Das murine Interleukin-4 wurde aus dem Überstand der Zelllinie NIH 3T3-IL-4
gewonnen. Die Zellen wurden unserem Institut von Herrn Prof. Dr. Dittmer
(Universität Duisburg-Essen, Essen) zur Verfügung gestellt.
Material und Methodik
39
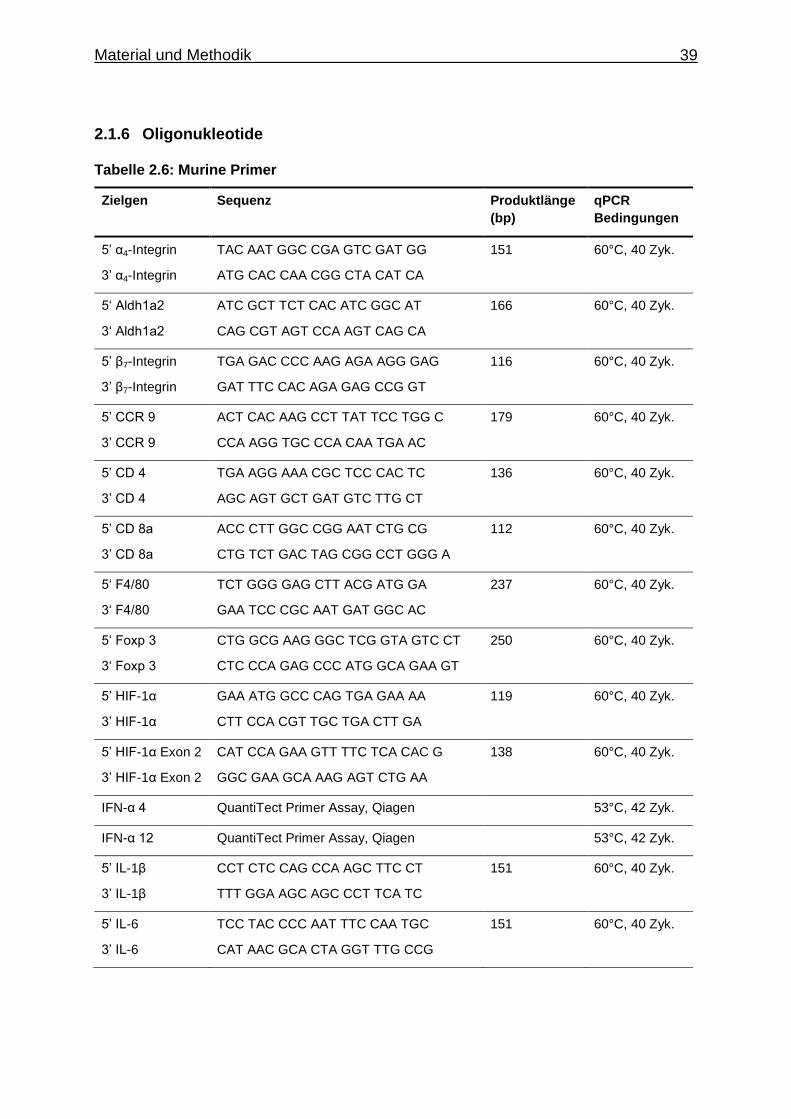
2.1.6 Oligonukleotide
Tabelle 2.6: Murine Primer
Zielgen Sequenz Produktlänge
(bp)
qPCR
Bedingungen
5’ α4-Integrin
3’ α4-Integrin
TAC AAT GGC CGA GTC GAT GG
ATG CAC CAA CGG CTA CAT CA
151 60°C, 40 Zyk.
5‘ Aldh1a2
3‘ Aldh1a2
ATC GCT TCT CAC ATC GGC AT
CAG CGT AGT CCA AGT CAG CA
166 60°C, 40 Zyk.
5’ β7-Integrin
3’ β7-Integrin
TGA GAC CCC AAG AGA AGG GAG
GAT TTC CAC AGA GAG CCG GT
116 60°C, 40 Zyk.
5’ CCR 9
3’ CCR 9
ACT CAC AAG CCT TAT TCC TGG C
CCA AGG TGC CCA CAA TGA AC
179 60°C, 40 Zyk.
5’ CD 4
3’ CD 4
TGA AGG AAA CGC TCC CAC TC
AGC AGT GCT GAT GTC TTG CT
136 60°C, 40 Zyk.
5’ CD 8a
3’ CD 8a
ACC CTT GGC CGG AAT CTG CG
CTG TCT GAC TAG CGG CCT GGG A
112
60°C, 40 Zyk.
5‘ F4/80
3‘ F4/80
TCT GGG GAG CTT ACG ATG GA
GAA TCC CGC AAT GAT GGC AC
237 60°C, 40 Zyk.
5‘ Foxp 3
3‘ Foxp 3
CTG GCG AAG GGC TCG GTA GTC CT
CTC CCA GAG CCC ATG GCA GAA GT
250 60°C, 40 Zyk.
5’ HIF-1α
3’ HIF-1α
GAA ATG GCC CAG TGA GAA AA
CTT CCA CGT TGC TGA CTT GA
119 60°C, 40 Zyk.
5’ HIF-1α Exon 2
3’ HIF-1α Exon 2
CAT CCA GAA GTT TTC TCA CAC G
GGC GAA GCA AAG AGT CTG AA
138 60°C, 40 Zyk.
IFN-α 4 QuantiTect Primer Assay, Qiagen 53°C, 42 Zyk.
IFN-α 12 QuantiTect Primer Assay, Qiagen 53°C, 42 Zyk.
5’ IL-1β
3’ IL-1β
CCT CTC CAG CCA AGC TTC CT
TTT GGA AGC AGC CCT TCA TC
151 60°C, 40 Zyk.
5’ IL-6
3’ IL-6
TCC TAC CCC AAT TTC CAA TGC
CAT AAC GCA CTA GGT TTG CCG
151 60°C, 40 Zyk.
Material und Methodik
40
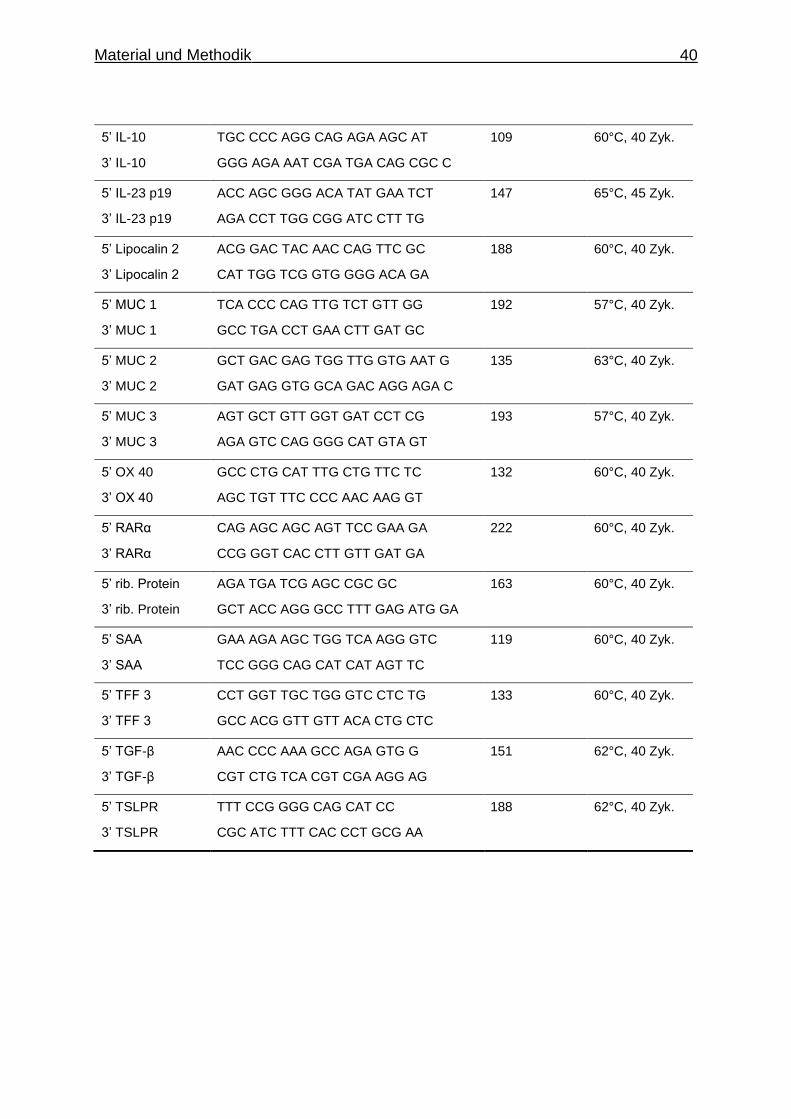
5’ IL-10
3’ IL-10
TGC CCC AGG CAG AGA AGC AT
GGG AGA AAT CGA TGA CAG CGC C
109 60°C, 40 Zyk.
5’ IL-23 p19
3’ IL-23 p19
ACC AGC GGG ACA TAT GAA TCT
AGA CCT TGG CGG ATC CTT TG
147 65°C, 45 Zyk.
5’ Lipocalin 2
3’ Lipocalin 2
ACG GAC TAC AAC CAG TTC GC
CAT TGG TCG GTG GGG ACA GA
188 60°C, 40 Zyk.
5’ MUC 1
3’ MUC 1
TCA CCC CAG TTG TCT GTT GG
GCC TGA CCT GAA CTT GAT GC
192 57°C, 40 Zyk.
5’ MUC 2
3’ MUC 2
GCT GAC GAG TGG TTG GTG AAT G
GAT GAG GTG GCA GAC AGG AGA C
135 63°C, 40 Zyk.
5’ MUC 3
3’ MUC 3
AGT GCT GTT GGT GAT CCT CG
AGA GTC CAG GGG CAT GTA GT
193
57°C, 40 Zyk.
5’ OX 40
3’ OX 40
GCC CTG CAT TTG CTG TTC TC
AGC TGT TTC CCC AAC AAG GT
132 60°C, 40 Zyk.
5’ RARα
3’ RARα
CAG AGC AGC AGT TCC GAA GA
CCG GGT CAC CTT GTT GAT GA
222 60°C, 40 Zyk.
5’ rib. Protein
3’ rib. Protein
AGA TGA TCG AGC CGC GC
GCT ACC AGG GCC TTT GAG ATG GA
163 60°C, 40 Zyk.
5’ SAA
3’ SAA
GAA AGA AGC TGG TCA AGG GTC
TCC GGG CAG CAT CAT AGT TC
119 60°C, 40 Zyk.
5’ TFF 3
3’ TFF 3
CCT GGT TGC TGG GTC CTC TG
GCC ACG GTT GTT ACA CTG CTC
133 60°C, 40 Zyk.
5’ TGF-β
3’ TGF-β
AAC CCC AAA GCC AGA GTG G
CGT CTG TCA CGT CGA AGG AG
151 62°C, 40 Zyk.
5’ TSLPR
3’ TSLPR
TTT CCG GGG CAG CAT CC
CGC ATC TTT CAC CCT GCG AA
188 62°C, 40 Zyk.

Material und Methodik
41
Tabelle 2.7: Murine Genotypisierungsprimer
Zielgen Sequenz Produktlänge
(bp)
PCR
Bedingungen
5‘ HIF-1 α DF
3‘ HIF-1 α DF
GGA GCT ATC TCT CTA GAC C
GCA GTT AAG AGC ACT AGT TG
WT ~ 200
DF ~ 250
57°C, 34 Zyk.
5’ CD 11c Cre
3’ CD 11c Cre
ACT TGG CAG CTG TCT CCA AG
GCG AAC ATC TTC AGG TTC TG
313 63°C, 35 Zyk.
5’ CD 11c Cre
pos. Kontrolle
3’ CD 11c Cre
pos. Kontrolle
CAA ATG TTG CTT GTC TGG TG
GTC AGT CGA GTG CAC AGT TT
206 63°C, 35 Zyk.
2.1.7 cDNA-Synthese, PCR und Real Time PCR
Tabelle 2.8: Reagenzien für cDNA-Synthese, PCR, Real Time PCR, PCR Array
Bezeichnung Hersteller
5x GoTaq® Buffer Invitrogen
100 bp DNA Ladder Invitrogen
GoTaq© DNA Polymerase Promega
M-MLV Reverse Transcriptase Promega
M-MLV RT 5x Reaction Buffer Promega
MESA Green qPCR MastermixPlus for SYBR® ASSAY Eurogentec
NTPs Qiagen
Oligo dT Invitrogen
Primer Invitrogen
RNeasy Mini Kit Qiagen
RT2 First Stand Kit (12) Qiagen
RT2 Profiler PCR Array
Mouse Crohn´s Disease
Qiagen
RT2 SYBR® Green Fluor qPCR Mastermix Qiagen

Material und Methodik
42
2.2 Methoden
2.2.1 Tierexperimentelle Methoden
2.2.1.1 Tierhaltung und –zucht
Alle Mäuse wurden im Zentralen Tierlaboratorium des Universitätsklinikums Essen
unter speziell pathogenfreien (SPF) Bedingungen gehalten. Bis zu fünf Tiere lebten
zusammen in Makrolon©-Filterkäfigen Typ III bei einer Umgebungstemperatur von
20 °C und einem konstanten 12 h-Tag-Nacht-Rhythmus. Die Tiere erhielten
pelletiertes Alleinfuttermittel und Trinkwasser ad libitum.
Die Genehmigung des Tierversuchs wurde vom Landesamt für Natur, Umwelt und
Verbraucherschutz Nordrhein-Westfalen unter dem Aktenzeichen
84-02.04.2011.A098 erteilt.
2.2.1.2 Transgene Mausstämme
Für die vorliegenden Studien wurden transgene Mausstämme mit einem C57BL/6J
Hintergrund verwendet. Es handelte sich bei den verwendeten Tieren um Mäuse mit
einem konditionellen Knock-out von HIF-1α in CD11c+ dendritischen Zellen. Die
genetische Inaktivierung erfolgte mithilfe des Cre/loxP-Systems. In Zielmäusen ist
hierbei das Exon 2 des HIF-1α Gens auf beiden Allelen von loxP-sites flankiert
(HIF-1αf+/f+). Um einen konditionellen Knock-out von HIF-1α zu generieren, wurden
diese homozygot-gefloxten Mäuse mit Mäusen gepaart, bei denen die Sequenz für
die Cre-Rekombinase der Kontrolle des murinen Integrin α X (CD11c) Promoters
unterliegt. Die Expression der Cre-Rekombinase führt zu einem Verlust des Exons 2
des HIF-1α Gens. Die daraus resultierende mRNA führt zur Bildung eines verkürzten,
nicht funktionellen HIF-1α Proteins. Die so generierten Knock-out Mäuse
(CD11cCre/HIF-1αf+/f+) exprimierten das Cre heterozygot in den CD11c+, also
vornehmlich dendritischen Zellen. Geschwistertiere ohne Cre-Rekombinase Aktivität
(HIF-1αf+/f+) dienten als sogenannte Wild-typ Kontrollen.

Material und Methodik
43
Die HIF-1αf+/f+ Mäuse wurden ursprünglich von Prof. Dr. Randall Johnson (University
of California, San Diego) und die CD11cCre Mäuse von Prof. Dr. Boris Reizis
(Columbia University Medical Centre, New York) generiert.
Alle verwendeten Mäuse zeigten unter physiologischen Umständen einen
ungestörten Habitus und züchten normal.
Abb. 2.1: Schematische Darstellung des Cre/loxP-Knockout-Systems.
Durch die Kreuzung von Mäusen mit einem doppelt gefloxten Exon 2 auf dem HIF-1α Gen mit
Mäusen, die eine Expression von Cre unter der Kontrolle des Integrin α X Promoters aufweisen,
entstehen Mäuse mit einem um das Exon 2 verkürzten HIF-1α Gen und funktionslosem Protein in
CD11c+ Zellen.
2.2.1.3 Genotypisierung
Die Mäuse wurden im Alter von etwa vier Wochen abgesetzt und durch das
Ausstanzen eines oder mehrerer Ohrlöcher markiert. Das Ohrgewebe wurde mit
300 µl 50 mM NaOH versetzt und mindestens für eine Stunde bei 100 °C unter
leichten Schüttelbewegungen erhitzt. Anschließend wurden 30 µl 1 M Tris-HCl zu
den Proben gegeben, gevortext und die Gewebereste abzentrifugiert. Es wurden
2,5 µl freigesetzte genomische DNA und 22,5 µl des Mastermixes für die
Genotypisierungs-PCR eingesetzt.
loxP – Maus (HIF-1α+f/+f
)
HIF-1α Gen
Ex.1 Ex.2 Ex.3 Ex.4
Cre-loxP – Maus (CD11cCre/HIF-1α+f/+f
)
Ex.1 Ex.3 Ex.4
CD11cCre - Maus
Integrin α X Gen
Promoter x

Material und Methodik
44
Tabelle 2.9: Mastermix für die Genotypisierung
Volumen (µl)
PCR-Puffer (5x) 5
dNTP 2
je Primer (20 pmol/µl) 0,5
H2O 14,5
Taq-Polymerase 0,05
Tabelle 2.10: Exemplarisches PCR-Programm für die Genotypisierung
Die Annealing-Temperatur und die Zyklenzahl wurden der zu amplifizierenden DNA entsprechend
angepasst.
Temperatur (°C) Dauer (min) Zyklen
Initiale Denaturierung 96 3 1
Denaturierung 96 1
Annealing 60 1 x 30
Elongation 72 3
Finale Elongation 72 10 1
2.2.1.4 DSS Modell
Zur Induktion einer Colitis wurde Dextrannatriumsulfat (DSS, engl. dextran sodium
sulfate) in einer 3%igen Konzentration verwendet (Okayasu et al. 1990; Vowinkel et
al. 2004). Es handelt sich bei DSS um ein sulfatiertes Polysaccharid, welches die
Tiere peroral für 7 Tage via Trinkwasser ad libitum erhielten und aufgrund dessen an
einer Colitis erkrankten. Durch die Gabe von 3 % DSS über 7 Tage wird laut
Vowinkel et al. bei Mäusen des C57BL/6J Stammes die erforderliche Schwellendosis
von 30 mg DSS pro Gramm Körpergewicht vollständig erreicht, um zuverlässig eine
Colitis zu induzieren (Vowinkel et al. 2004). Geringe Trinkmengenunterschiede im
Verlauf des Versuches sind somit vernachlässigbar. Die Tiere waren zu Beginn des

Material und Methodik
45
Versuches mindestens zehn Wochen alt. Wild-typ Tiere (HIF-1αf+/f+) und Knock-out
Tiere (CD11cCre/HIF-1αf+/f+) wurden getrennt voneinander jeweils zu zweit zwei
Tage vor Gabe des DSS in einem Käfig zusammengesetzt. Es wurde ausschließlich
DSS der Firma MP Biomedicals aus ein und derselben Charge verwendet, damit
Schwankungen des Schwefelgehaltes ausgeschlossen werden konnten. Des
Weiteren betrug das Molekulargewicht des DSS hierdurch konstant 36000 – 50000
Dalton, was sehr entscheidend ist, da dieses großen Einfluss auf die Ausprägung der
Colitis hat (Kitajima et al. 2000). Das DSS wurde in unbehandeltem Leitungswasser
gelöst und an Tag 3 des Versuches erneuert. Das Körpergewicht der Tiere wurde
täglich kontrolliert und die prozentuale Veränderung des Gewichtes errechnet. Die
klinische Einstufung der Erkrankung erfolgte mithilfe eines Disease Activity Index
(DAI) (Cooper et al. 1993; Williams et al. 2001; Cummins et al. 2008). Der DAI setzt
sich aus den jeweiligen Punktzahlen für den prozentualen Verlust an Körpergewicht,
die Beschaffenheit des Stuhles und das Ergebnis eines Tests auf okkultes Blut im
Stuhl zusammen. Es handelt sich hierbei um einen Haemoccult-Test, bei welchem
durch Kontakt des Stuhles mit einer Indikatorlösung okkultes Blut durch einen
Farbumschlag detektiert werden kann. Die Einzelpunktzahlen wurden addiert,
woraus ein maximaler Score von 12 resultierte. Die genaue Zusammensetzung des
DAI ist in Tabelle 2.11 dargestellt. Der DAI wurde täglich für jedes einzelne Tier
bestimmt.

Material und Methodik
46
Tabelle 2.11: Disease Activity Index
Score Beschreibung
Gewichtsverlust 0 0 %
1 1-5 %
2 6-10 %
3 11-20 %
4 > 20 %
Stuhlbeschaffenheit 0 geformt
2 breiig
4 flüssig
Blut im Stuhl 0 Hema-Screen negativ
2 Hema-Screen positiv
4 sichtbare Blutung
2.2.1.5 Pimonidazol-Injektion
Zur immunhistochemischen Darstellung von hypoxischem Gewebe wurde den
Mäusen eine Stunde vor Versuchsende 60 mg Pimonidazol pro kg, gelöst in 100 µl
0,9 % NaCl, intraperitoneal injiziert. Pimonidazol (Hydroxyprobe™-1) stellt ein
substituiertes 2-Nitroimidazol dar, welches unter normoxischen Bedingungen leicht
oxidativ metabolisiert und als N-Oxid, Sulfat und Gluconat eliminiert wird. Unter
hypoxischen Bedingungen (pO2 < 10 mm Hg) wird es reduktiv aktiviert und bildet
kovalente Bindungen mit Sulfhydrylgruppen umgebender Protein, Peptide und
Aminosäuren (Arteel et al. 1998; Gross et al. 1995). Die entstanden Produkte können
in Gewebeschnitten anschließend durch spezifische Antikörper detektiert werden, so
dass das hypoxische Gewebe sichtbar gemacht wird (Samoszuk et al. 2004).

Material und Methodik
47
2.2.1.6 Organentnahme
Zur Organentnahme wurden die Tiere an Tag 7 des DSS Versuches durch
Genickbruch getötet und in Rückenlage fixiert. Sowohl die Haut als auch das
Peritoneum wurde durch Längsschnitt durchtrennt. Als erstes wurden die
mesenterialen Lymphknoten entfernt und für die RNA-Isolation bis zur weiteren
Verwendung erst in flüssigem Stickstoff und dann in einem -80 °C Gefrierschrank
gelagert. Das Colon wurde in seiner gesamten Länge freipräpariert, am Übergang
zum Caecum und Rektum durchtrennt und entnommen. Anschließend wurde es
vorsichtig mit PBS gespült, gewogen und die Länge gemessen. 0,5 cm des distalen
Colonendes wurden für die RNA-Isolation verwendet und bis zur Homogenisierung
des Gewebes zunächst in flüssigem Stickstoff und dann in einem -80 °C
Gefrierschrank gelagert. Die nächsten 1 cm des distalen Colons wurden für
histologische Arbeiten in 4 % PFA fixiert. Von einigen Tieren wurde auch das
restliche Colon für die Erstellung sogenannter Swiss Rolls in 4 % PFA fixiert (s.
Abschnitt 2.2.4.1). Für die Durchführung des ELISA wurde von den dafür
verwendeten Tieren die komplette distale Hälfte des Colons zunächst in flüssigem
Stickstoff und anschließend bis zur weiteren Verarbeitung in einem -80 °C
Gefrierschrank gelagert. Für die Gewinnung des Knochenmarks wurden die
Hinterbeine der Tiere präpariert. Dazu wurde die Haut entlang des Hinterbeins
eröffnet und von der darunter liegenden Muskulatur abgelöst. Die Knochen wurden
vollständig von Muskeln und Sehnen befreit und nach dem Durchtrennen des
Fußgelenkes und des Beckens entnommen. Ober- und Unterschenkelknochen
wurden daraufhin in einer 6-Well-Zellkulturplatte mit PBS auf Eis gelegt.
2.2.2 Zellkultur
2.2.2.1 Dendritische Zellen aus dem Knochenmark
Das Knochenmark aus Femur und Tibia von Wild-typ und Knock-out Tieren wurde
mit ca. 5 ml PBS durch die Epiphysenöffnung ausgespült. Das so gewonnene
Knochenmark wurde gesammelt und resuspendiert, um eine Einzelzellsuspension zu
erhalten. Zur Bestimmung der Zellzahl wurden 90 µl der Suspension mit 0,4 %

Material und Methodik
48
Trypanblau 1:10 verdünnt und in einer Neubauerkammer gezählt. Trypanblau färbt
tote Zellen an. Für die Zellzahlbestimmung wurden nur lebende Zellen gezählt. Der
Rest der Zellsuspension wurde für 5 min bei 4 °C und 1500 rpm zentrifugiert und das
Zellpellet danach mit Medium auf 1x107 Zellen pro ml eingestellt. Zur Generierung
von aus dem Knochenmark gewonnenen dendritischen Zellen (engl. bone marrow-
derived dendritic cells, BmDC) wurde von dieser Zellsuspension 100 µl pro
Vertiefung in eine 6-Well-Zellkulturplatte gegeben. Dazu wurde je 1 ml
BmDC-Medium mit 5 ng/ml GM-CSF und 1 ng/ml IL-4 gegeben, so dass eine
Konzentration von 1x106 Zellen pro ml vorlag. Die Zellen wurden in einem
Brutschrank bei 37 °C und 5 % CO2 in wasserdampfgesättigter Umgebung kultiviert.
Bei einem Teil der Zellen erfolgte die Kultivierung in einer Hypoxiekammer mit
3 % O2. Alle zwei Tage wurde 1 ml Medium durch frisches Medium mit Zytokinen
ersetzt. Für die RNA- und Proteinisolierung wurden die BmDCs nach acht Tagen in
den Versuch genommen (Sallusto & Lanzavecchia 1994). Hierbei wurde ein Teil der
Zellen unstimuliert belassen. Ein weiterer Teil wurde mit dem TLR4-Agonisten
Lipopolysaccharid (LPS) in einer Konzentration von 1 µg/ml stimuliert. Beide Ansätze
wurden für den Proteinnachweis vier Stunden und für die RNA-Isolation sechs
Stunden jeweils unter Normoxie und Hypoxie (3 % O2) inkubiert.
2.2.3 Molekularbiologische Methoden
2.2.3.1 RNA Isolierung
Die Isolation der RNA aus den BmDCs erfolgte mithilfe der Phenol-Chloroform-
Extraktionsmethode nach Chomczynski & Sacchi (1987). Dazu wurde das Medium
der BmDCs zu Versuchsende komplett aus den 6-Well-Zellkulturplatten entfernt,
700 µl 4 M GTC in jede Vertiefung gegeben und die Platten über Nacht bei -20 °C
eingefroren. Nach dem Auftauen wurde der Inhalt in ein 2 ml
Eppendorfreaktionsgefäß überführt und 70 µl 2 M Natriumacetat hinzugegeben.
Anschließend wurden die Proben mit 500 µl Phenol und 350 µl
Phenol:Chloroform:Isoamylalkohol versetzt. Die Proben wurden geschüttelt,
gevortext, 60 min auf Eis inkubiert und daraufhin für 30 min bei 4 °C und 13200 rpm
zentrifugiert. Durch dieses Vorgehen erhält man eine Trennung der Probe in drei

Material und Methodik
49
Phasen: die oberste, wässrige Phase enthält die RNA, die Interphase die DNA und
die untere, organische Phase die Proteine. Die obere Phase wurde in ein neues
1,5 ml Eppendorfreaktionsgefäß überführt, 600 µl Isopropanol zugegeben und die
Probe über Nacht bei -20 °C eingefroren. Am nächsten Tag wurden die Proben direkt
für 30 min bei 4 °C und 13200 rpm zentrifugiert. Der Überstand wurde verworfen, die
RNA-Pellets bei Raumtemperatur 30 – 60 min getrocknet und 300 µl GTC und 500 µl
Isopropanol zugegeben. Nach erneuter Inkubation bei -20 °C über Nacht wurden die
Proben wieder für 30 min bei 4 °C und 13200 rpm zentrifugiert und das Pellet nach
Verwerfen des Überstandes mit 500 µl 75%igem Ethanol gewaschen. Daraufhin
wurden die Proben ein letztes Mal bei 4 °C und 13200 rpm für 30 min zentrifugiert,
der Überstand abgegossen, die Pellets getrocknet und anschließend je nach Größe
in 10 – 20 µl DEPC-H2O gelöst und für 10 min bei 60 °C erhitzt. Der RNA Gehalt
wurde photometrisch bei einer Wellenlänge von 260 nm gemessen. Die Reinheit der
Proben wurde über das Verhältnis der gemessenen Absorption bei 260 und 280 nm
bestimmt. Es wurden nur Proben verwendet bei denen ein Quotient zwischen 1,7
und 2,0 vorlag. Die RNA wurde anschließend bei -20 °C eingefroren.
Die Isolation der RNA aus Gewebeproben erfolgte mithilfe des Qiagen RNeasy Mini
Kits. Die RNA wird hierbei an eine Silicagel-Membran gebunden, Kontaminationen
entfernt und nachfolgend die gewaschene RNA von der Membran eluiert. Dazu
wurden die Proben mit je 600 µl RLT-Puffer, zu welchem 1 % frisches
β-Mercaptoethanol gegeben wurde, homogenisiert und über Nacht bei -20 °C
eingefroren. Am nächsten Tag wurden 600 µl 70%iges Ethanol zu den Proben
gegeben und durch Auf- und Abpipettieren gemischt. 700 µl je Probe wurden auf ein
RNeasy-Röhrchen gegeben und für 15 s bei 12000 rpm und Raumtemperatur
zentrifugiert. Der Durchfluss wurde verworfen. Nun wurde der Rest der Proben auf
die Röhrchen gegeben und ebenso zentrifugiert. Der Durchfluss wurde wieder
verworfen. Anschließend wurden je 700 µl RW1 Puffer aus dem RNeasy Mini Kit auf
die Röhrchen pipettiert und für 15 s bei 12000 rpm und Raumtemperatur
zentrifugiert. Der Durchfluss wurde verworfen. Als nächstes wurden 500 µl RPE
Puffer auf die Röhrchen gegeben und unter den gleichen Bedingungen zentrifugiert.

Material und Methodik
50
Der Durchfluss wurde wieder verworfen. Weitere 500 µl RPE Puffer wurden auf je ein
Röhrchen gegeben. Die Proben wurden nun für 2 min bei 12000 rpm und
Raumtemperatur zentrifugiert. Abschließend wurden je 30 µl RNase freies Wasser
zum Eluieren der Membran auf die Röhrchen gegeben und für 1 min bei 13200 rpm
und Raumtemperatur zentrifugiert. Der Durchfluss wurde noch einmal auf die
Membran pipettiert und unter gleichen Umständen zentrifugiert. Der Gehalt der
isolierten RNA wurde wie oben beschrieben photometrisch bestimmt und die RNA
anschließend bei -20 °C gelagert.
2.2.3.2 Reverse Transkription
Für die cDNA-Synthese der BmDCs wurde 1 µg RNA mit 2,5 µl Oligo-dT Primern und
DEPC-H2O auf insgesamt 12 µl gebracht. Für die Gewebeproben wurden 3 µg RNA
eingesetzt und diese mit 5 µl Oligo-dT Primern und DEPC-H2O auf 24 µl aufgefüllt.
Die Proben wurden 10 min bei 68 °C erhitzt und anschließend 5 min auf Eis gekühlt.
Zu den Proben der BmDCs wurde daraufhin 13 µl Mastermix, zu den Gewebeproben
26 µl Mastermix gegeben. Die Umschreibung der RNA erfolgte im Thermocycler bei
45 °C für 90 min und 52 °C für 30 min. Die Denaturierung erfolgte bei 95 °C für
15 min. Die cDNA wurde anschließend bei 4 °C gelagert.
Tabelle 2.12: Mastermix für die cDNA Synthese
Volumen (µl)
M-MLV Buffer (5x) 5
dNTP 5
DEPC-H2O 2,5
M-MLV Reverse Transkriptase 0,5

Material und Methodik
51
2.2.3.3 PCR und quantitative Real Time PCR
cDNA kann mittels der Polymerase-Kettenreaktion (PCR, engl. polymerase chain
reaction) amplifiziert werden. Der Mastermix für die PCR wird wie in Tabelle 2.13
hergestellt, in ein PCR-Reaktionsgefäß überführt und 0,5 µl cDNA hinzugegeben.
Tabelle 2.14 zeigt ein exemplarisches PCR-Programm, bei welchem die
Annealing-Temperatur und die Zyklenzahl abhängig von den verwendeten Primern
angepasst wurden. Die amplifizierte Menge cDNA wurde nun zusammen mit einem
Größenstandard auf ein 2%iges Agarosegel mit 0,07 % Ethidiumbromid aufgetragen,
mittels Gelelektrophorese aufgetrennt und durch UV-Licht in einer
Geldokumentationskammer dargestellt.
Tabelle 2.13: Mastermix für PCR
Volumen (µl)
PCR-Puffer (5x) 5
dNTP 2
je Primer (20 pmol/µl) 0,5
H2O 17
Taq-Polymerase 0,05
Tabelle 2.14: Exemplarisches PCR-Programm
Die Annealing-Temperatur und die Zyklenzahl wurden der zu amplifizierenden DNA entsprechend
angepasst.
Temperatur (°C) Dauer (min) Zyklen
Initiale Denaturierung 96 3 1
Denaturierung 96 1
Annealing 60 1,5 x 30
Elongation 72 1
Finale Elongation 72 10 1

Material und Methodik
52
Die Quantifizierung der cDNA erfolgte mittels Echtzeit-PCR (Real Time PCR). Hierzu
wurden Reaktionsansätze von 50 µl je Probe erstellt und diese, sowie ein Leerwert,
als Doppelbestimmung (2 x 20 µl) in 96-Well-Platten pipettiert. Die PCR-Reaktion mit
gleichzeitiger Fluoreszenz-Messung wurde als Zweistufen-PCR mithilfe des iCycler
Detektionssystems von Bio-Rad durchgeführt. Die Auswertung erfolgte mittels der
ΔΔCT Methode (engl. Cycle Threshold). Dabei werden die ermittelten CT-Werte der
einzelnen Proben auf die CT-Werte des homogen exprimierten Gens ribosomales
Protein normalisiert und die Unterschiede der Genexpression der zu vergleichenden
Proben als n-fache Änderung berechnet (Winer et al. 1999).
Tabelle 2.15: Mastermix für Real Time PCR
Volumen (µl)
MESA Green qPCR MastermixPlus 25
H2O 22
je Primer (20 pmol/µl) 1
cDNA 1
Tabelle 2.16: Exemplarisches Real Time PCR-Programm
Die Annealing-Temperatur und die Zyklenzahl wurden der zu amplifizierenden DNA entsprechend
angepasst.
Temperatur (°C) Dauer (min) Zyklen
Initiale Denaturierung 95 10 1
Denaturierung 95 0,25
Annealing/
Elongation
60 1,5 x 40

Material und Methodik
53
2.2.3.4 PCR Array
Mit Hilfe eines PCR Arrays ist die Quantifizierung sehr vieler mRNAs in einer Probe
gleichzeitig möglich. Im verwendeten Array wurde die Expression von mRNAs
untersucht, welche im Zusammenhang mit Morbus Crohn stehen (RT2 Profiler PCR
Array Mouse Crohn´s Disease, PAMM-169ZA, Qiagen). Es handelte sich um eine
96-Well-Platte auf welcher insgesamt 84 mRNAs untersucht werden konnten.
Zusätzlich waren fünf Referenzgene, eine Kontrolle auf genomische DNA, drei
reverse Transkriptase Kontrollen und drei positive PCR Kontrollen auf der Platte
vorhanden.
Tabelle 2.17: Zusammenstellung der im Array untersuchten Gene
Abkürzung Beschreibung des Genes
Abcb1a ATP-binding cassette, sub-family B (MDR/TAP), member 1A
Adh1 Alcohol dehydrogenase 1 (class I)
Aldob Aldolase B, fructose-bisphosphate
Atg16l1 Autophagy-related 16-like 1 (yeast)
C3 Complement component 3
Casp1 Caspase 1
Ccl11 Chemokine (C-C motif) ligand 11
Ccl12 Chemokine (C-C motif) ligand 12
Ccl20 Chemokine (C-C motif) ligand 20
Ccl25 Chemokine (C-C motif) ligand 25
Ccl5 Chemokine (C-C motif) ligand 5
Ccr1 Chemokine (C-C motif) receptor 1
Ccr2 Chemokine (C-C motif) receptor 2
Ccr5 Chemokine (C-C motif) receptor 5
Ccr9 Chemokine (C-C motif) receptor 9
Cd55 CD55 antigen
Chi3l1 Chitinase 3-like 1

Material und Methodik
54
Cldn8 Claudin 8
Col1a2 Collagen, type I, alpha 2
Cr2 Complement receptor 2
Csta Cystatin A
Cx3cl1 Chemokine (C-X3-C motif) ligand 1
Cx3cr1 Chemokine (C-X3-C) receptor 1
Cxcl1 Chemokine (C-X-C motif) ligand 1
Cxcl10 Chemokine (C-X-C motif) ligand 10
Cxcl11 Chemokine (C-X-C motif) ligand 11
Cxcl12 Chemokine (C-X-C motif) ligand 12
Cxcl2 Chemokine (C-X-C motif) ligand 2
Cxcl3 Chemokine (C-X-C motif) ligand 3
Cxcl9 Chemokine (C-X-C motif) ligand 9
Cxcr1 Chemokine (C-X-C motif) receptor 1
Cxcr3 Chemokine (C-X-C motif) receptor 3
Edn3 Endothelin 3
Egr3 Early growth response 3
Fpr1 Formyl peptide receptor 1
Gcg Glucagon
H2-Aa Histocompatibility 2, class II antigen A, alpha
H2-Eb1 Histocompatibility 2, class II antigen E beta
Hsp90b1 Heat shock protein 90, beta (Grp94), member 1
Hspa5 Heat shock protein 5
Ifng Interferon gamma
Il13 Interleukin 13
Il17a Interleukin 17A
Il1b Interleukin 1 beta
Il1rn Interleukin 1 receptor antagonist
Il23a Interleukin 23, alpha subunit p19

Material und Methodik
55
Il2ra Interleukin 2 receptor, alpha chain
Il5 Interleukin 5
Il6 Interleukin 6
Irf5 Interferon regulatory factor 5
Isg15 ISG15 ubiquitin-like modifier
Itgb2 Integrin beta 2
Lcn2 Lipocalin 2
Ltb Lymphotoxin B
Lyz1 Lysozyme 1
Mmp10 Matrix metallopeptidase 10
Mmp12 Matrix metallopeptidase 12
Mmp1a Matrix metallopeptidase 1a (interstitial collagenase)
Mmp3 Matrix metallopeptidase 3
Mmp7 Matrix metallopeptidase 7
Muc1 Mucin 1, transmembrane
Nod2 Nucleotide-binding oligomerization domain containing 2
Nos2 Nitric oxide synthase 2, inducible
Nr3c2 Nuclear receptor subfamily 3, group C, member 2
Pck1 Phosphoenolpyruvate carboxykinase 1, cytosolic
Pecam1 Platelet/endothelial cell adhesion molecule 1
Reg1 Regenerating islet-derived 1
Reg2 Regenerating islet-derived 2
S100a8 S100 calcium binding protein A8 (calgranulin A)
S100a9 S100 calcium binding protein A9 (calgranulin B)
Saa3 Serum amyloid A 3
Sele Selectin, endothelial cell
Selenbp1 Selenium binding protein 1
Sell Selectin, lymphocyte
Sod2 Superoxide dismutase 2, mitochondrial

Material und Methodik
56
Stat1 Signal transducer and activator of transcription 1
Stat3 Signal transducer and activator of transcription 3
Tdo2 Tryptophan 2,3-dioxygenase
Tff1 Trefoil factor 1
Timp1 Tissue inhibitor of metalloproteinase 1
Tnf Tumor necrosis factor
Tyk2 Tyrosine kinase 2
Ubd Ubiquitin D
Vwf Von Willebrand factor homolog
Actb Actin, beta
B2m Beta-2 microglobulin
Gapdh Glyceraldehyde-3-phosphate dehydrogenase
Gusb Glucuronidase, beta
Hsp90ab1 Heat shock protein 90 alpha (cytosolic), class B member 1
MGDC Mouse Genomic DNA Contamination
RTC Reverse Transcription Control
RTC Reverse Transcription Control
RTC Reverse Transcription Control
PPC Positive PCR Control
PPC Positive PCR Control
PPC Positive PCR Control
Für den PCR-Array wurden Colonproben von DSS behandelten Wild-typ und
Knock-out Tieren benutzt. Dazu wurde die RNA aus Colongewebe mit Hilfe des
Qiagen RNeasy Mini Kits, wie in Abschnitt 2.2.3.1 beschrieben, isoliert. Nach
photometrischer Bestimmung des RNA Gehaltes wurden 500 ng RNA für die
Umschreibung in cDNA mittels des RT2 First Strand Kits eingesetzt. Dazu wurde
zunächst ein Mix aus RNA, RNase freiem Wasser und dem Puffer GE des Kits
erstellt. Dieser Puffer sollte genomische DNA eliminieren. Dieser Mix wurde 5 min bei

Material und Methodik
57
42 °C inkubiert und direkt danach für 1 min auf Eis gelagert. Als nächstes wurde der
in Tabelle 2.18 beschriebene Reverse Transkriptase Mix erstellt. 10 µl des ersten Mix
wurden nun zu 10 µl des Reverse Transkriptase Mix gegeben und vermischt. Dieser
Ansatz wurde 15 min bei 42 °C und danach 5 min bei 95 °C inkubiert. Anschließend
wurden 91 µl RNase freies Wasser dazu gegeben.
Tabelle 2.18: Reverse Transkriptase Mix für PCR Array
Volumen (µl)
5x Puffer BC3 4
Kontrolle P2 1
RE3 Reverse Transkriptase Mix 2
RNase freies Wasser 3
Von der erstellten cDNA wurden nun 102 µl für den PCR Ansatz genutzt. Es wurden
1350 µl 2x RT2 SYBR Green Mastermix und 1248 µl RNase freies Wasser
hinzugeben. 25 µl dieses Ansatzes wurden jeweils in eine Vertiefung der
96-Well-Platte des PCR Arrays gegeben. Das PCR Programm wurde im iCycler von
BioRad durchgeführt.
Tabelle 2.19: Real Time PCR Programm für PCR Array
Temperatur (°C) Dauer (min) Zyklen
Initiale Denaturierung 95 10 1
Denaturierung 95 0,25
Annealing/
Elongation
60 1 x 40

Material und Methodik
58
Die Auswertung erfolgte nach der ΔΔCT Methode mittels einer kostenfrei zur
Verfügung gestellten auf Excel basierenden Analyse Software von SABiociences™.
Gene mit einem CT-Wert von ≥ 35 für die Expression der Amplifikate wurden in der
Analyse ausgeschlossen. Für die Auswertung wurden Gene ausgewählt, die einen
deutlichen Unterschied in der Expression zwischen Wild-typ und Knock-out Tieren
zeigten.
2.2.3.5 Proteinisolierung
Zu Versuchsende wurden die ausdifferenzierten BmDCs in 6-Well-Zellkulturplatten
auf Eis gestellt und das Medium abgesaugt. In jede Vertiefung wurden 65 µl
Lysepuffer pipettiert und die Zellen mit Zellschabern vom Boden der Platten
abgeschabt. Die Zellsuspension wurde in Eppendorfreaktionsgefäße überführt und
20 min auf Eis inkubiert. Anschließend wurde die Suspension für 5 min und 10000
rpm bei 4 °C zentrifugiert. Der Überstand wurde in neue Reaktionsgefäße gegeben.
Für die Bestimmung der Proteinkonzentration wurden 5 µl des Proteinlysates
verwendet.
2.2.3.6 Proteinbestimmung nach Lowry
Der Proteingehalt der Proben wurde nach der Methode von Lowry bestimmt (Lowry
et al. 1951). Dafür wurden je 5 µl eines BSA-Standards (25-0,1 mg/ml BSA) und 5 µl
der Proben in eine 96-Well-Zellkulturplatte pipettiert, mit 10 µl Reagenz A und 80 µl
Reagenz B versetzt und für 10 min bei Raumtemperatur inkubiert. Anschließend
wurde die Extinktion bei 700 nm im ELISA-Reader gemessen und die
Proteinkonzentration anhand der Standardreihe berechnet. Alle Messungen wurden
als Doppelbestimmungen durchgeführt.
2.2.3.7 Sodiumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-Page)
Durch die SDS-Polyacrylamid-Gelelektrophorese nach der Methode von Laemmli
(1970) erfolgte die Auftrennung der Proteine unter denaturierenden Bedingungen. Es
wurden dabei 5%ige Sammelgele und 7,5%ige Trenngele verwendet. Von den

Material und Methodik
59
Lysaten wurden 40 – 60 µg Probe im Verhältnis 1:4 mit 4xSDS-Probenpuffer versetzt
und für 5 min bei 95 °C denaturiert. Anschließend wurden sie auf das Gel
aufgetragen und bei 120 V für 90 min aufgetrennt.
Tabelle 2.20: Zusammensetzung der Trenn- und Sammelgele
Sammelgel (5 %) Trenngel (7,5 %)
A. dest. 2,92 ml 5 ml
Bisacrylamid 30 % 0,83 ml 2,5 ml
4x upper buffer pH 6,8 1,25 ml -
4x lower buffer pH 8,8 - 2,5 ml
2.2.3.8 Western Blot
Der Proteintransfer aus Polyacrylmaid-Gelen auf Nitrocellulosemembranen erfolgte
elektrophoretisch nach der Methode von Towbin (Towbin et al. 1979) bei 110 V für
90 min. Anschließend wurden die Membranen kurz in Ponceau S Lösung getaucht,
um die Effizienz des Transfers zu überprüfen und danach mit TBS-T gewaschen. Die
Blockierung der unspezifischen Bindungen erfolgte mit 5 % Magermilch in TBS-T für
eine Stunde. Die Membranen wurden nun über Nacht mit den primären Antikörpern
bei 4 °C inkubiert. Am nächsten Tag wurden die Membranen dreimal für 5 min mit
TBS-T gewaschen und danach für mindestens eine Stunde mit dem sekundären
Antikörper inkubiert. Die Detektion von Antigenen erfolgte indirekt über die Reaktion
der an den Zweitantikörper gekoppelten Meerrettichperoxidase durch das
ECL-Detektionssystem. Dafür wurden die Membranen dreimal für 5 min mit TBS-T
gewaschen und danach für 5 min mit 600 µl des ECL-Reagenzes inkubiert. Das
Chemilumineszenz-Signal wurde im Fusion FX7-Detektionssystem aufgenommen.
2.2.3.9 Kompetitiver Enzymgekoppelter Immunabsorptionsassay (ELISA)
Der ELISA wurde mit dem jeweiligen Mouse ELISA MAX™ Deluxe Kit von
BioLegend und Proben von Gesamtcolon durchgeführt. Dazu wurde das Colon in

Material und Methodik
60
400 µl PBS homogenisiert, 1 min bei 13200 rpm zentrifugiert und der Überstand in
ein neues Eppendorfreaktionsgefäß gegeben. Davon wurden 5 µl für die
Proteinbestimmung verwendet (s. Abschnitt 2.2.3.6). Es wurden jeweils 200 µg der
Proben eingesetzt und mit Assay Diluent A auf 100 µl aufgefüllt. Zur Durchführung
des ELISA wurde die beigefügte 96-Well-Platte zunächst mit 100 µl Capture Antibody
Lösung über Nacht bei 4 °C inkubiert. Am nächsten Tag wurde viermal mit 300 µl
PBS-T gewaschen. Anschließend wurde die Platte mit 200 µl Assay Diluent A für
1 Stunde bei Raumtemperatur inkubiert. Nach 4 Waschschritten mit 300 µl PBS-T
wurden je 100 µl der Standardreihe (500, 250, 125, 62,5, 31,3, 15,6, 7,8 pg/ml), ein
Leerwert und die Proben als Doppelbestimmung aufgetragen. Die Inkubation erfolgte
für 2 Stunden bei Raumtemperatur. Danach wurde viermal mit 300 µl PBS-T
gewaschen und die Platte mit 100 µl Detection Antibody für 1 Stunde bei
Raumtemperatur inkubiert. Nach 4 weiteren Waschschritten erfolgte die Inkubation
mit 100 µl Avidin-HRP für 30 min bei Raumtemperatur. Danach wurde fünfmal für je
1 min gewaschen und die Platte daraufhin mit 100 µl TMB Lösung im Dunklen
inkubiert. Nach 20 min wurden 100 µl Stopp Lösung hinzugegeben und die
Absorption photometrisch bei einer Wellenlänge von 450 nm und 570 nm gemessen.
Der Leerwert und die Werte der Messung bei 570 nm wurden von den Werten der
Messung bei 450 nm abgezogen und die Konzentration der Proben anhand der
Standardreihe berechnet.
2.2.4 Histologische Methoden
2.2.4.1 Fixierung und Paraffineinbettung von Organen
Die Fixierung der Gewebeproben erfolgte in 4 % Paraformaldehyd für 24 Stunden.
Danach wurden die Proben mit PBS gewaschen und in 70%igem Ethanol gelagert.
Im Institut für Anatomie des Universitätsklinikums Essen erfolgte das Entwässern der
Proben in einer aufsteigenden Alkoholreihe, das Ersetzen des Alkohols durch ein
Intermedium und das Paraffinieren. Gewebeproben des distalen Colons wurden
anschließend im Querschnitt in Paraffin eingebettet. Das restliche Colon wurde als
so genannte Swiss Roll eingerollt, wobei das distale Ende nach innen gelegt wurde

Material und Methodik
61
und in Paraffin eingebettet. Die Blöcke wurden mit dem Rotationsmikroskop in 4 µm
dicke Schnitte geschnitten und auf einen SuperFrost® Plus Objektträger aufgezogen.
2.2.4.2 Hämatoxylin/Eosin-Färbung
Vor der eigentlichen Färbung mussten die Schnitte zunächst deparaffiniert und
rehydriert werden. Für die Deparaffinierung wurden die Schnitte zweimal für 10 min
in Xylol inkubiert. Für die Rehydrierung wurden die Schnitte weiterhin in einer
absteigenden Alkoholreihe beginnend mit Isopropanol, über 90 % Ethanol und 70 %
Ethanol, bis hin zu 50 % Ethanol für jeweils 5 min inkubiert. Anschließend wurden die
Schnitte kurz in vollentsalztes (VE) Wasser getaucht und dann für 1 min in
Hämatoxylin gefärbt. Hämatoxylin färbt alle basophilen Strukturen, wie Zellkerne,
blau an. Die Schnitte wurden nun unter fließendem Leitungswasser gewaschen bis
sich das Wasser entfärbte und ein pH-Wechsel eintrat. Daraufhin wurden die
Schnitte kurz in VE Wasser getaucht, um dann für 40 s in Eosin gefärbt zu werden.
Eosin färbt azidophile Strukturen, wie Zytoplasmaproteine, Mitochondrien oder das
glatte endoplasmatische Retikulum, rot an. Anschließend wurden die Schnitte kurz in
VE Wasser getaucht und dann in einer aufsteigenden Alkoholreihe, beginnend mit
50 % Ethanol, über 70 % Ethanol und 90 % Ethanol, gefolgt von Isopropanol und
zweimal Xylol für jeweils 5 min inkubiert. Abschließend wurden die Schnitte mit dem
Eindeckmedium Cytoseal eingedeckelt und getrocknet. Zur Darstellung von
großflächigen Übersichtsaufnahmen wurde der ScanScope CS2 von Aperio in der
Pathologie des Universitätsklinikums Essen verwendet und diese Bilder mit der
Aperio ImageScope Software betrachtet.
2.2.4.3 Auswertung der HE-Färbung
Die Querschnitte des distalen Colons wurden von zwei verblindeten Untersuchern
betrachtet und die entzündlichen Darmveränderungen anhand dem in Tabelle 2.21
beschriebenen histologischen Score (Cooper et al. 1993; Williams et al. 2001;
Tambuwala et al. 2010) beurteilt.
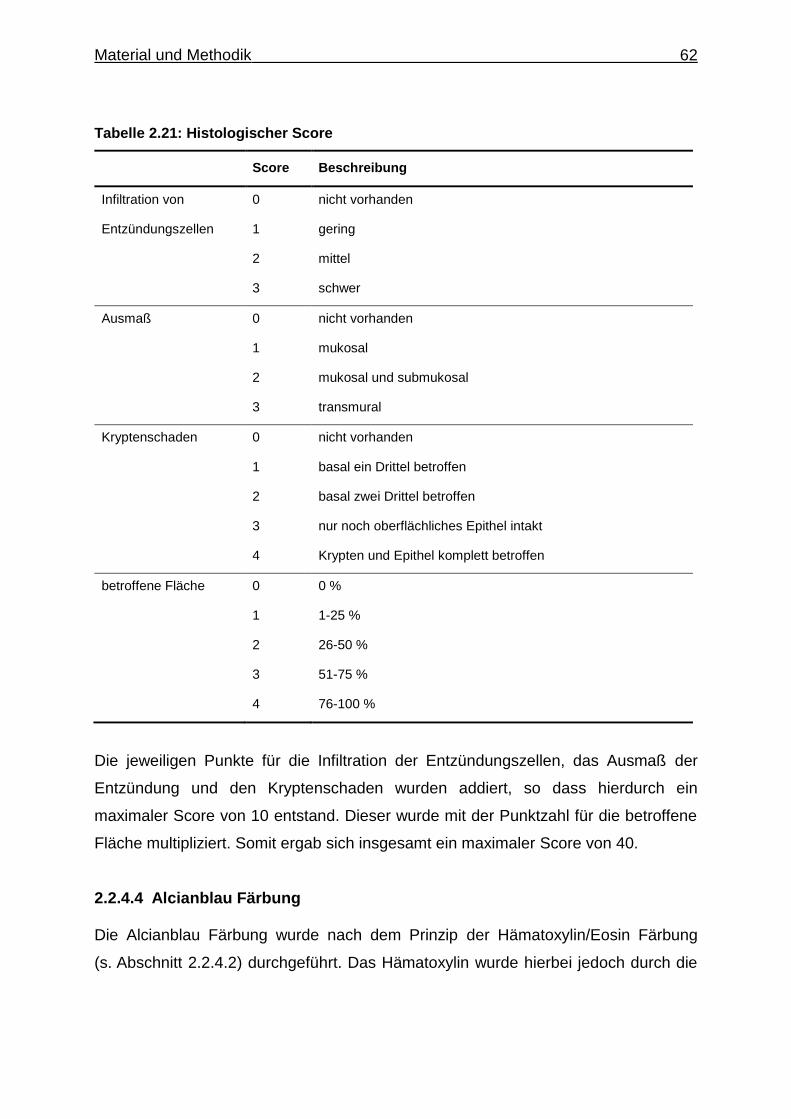
Material und Methodik
62
Tabelle 2.21: Histologischer Score
Score Beschreibung
Infiltration von 0 nicht vorhanden
Entzündungszellen 1 gering
2 mittel
3 schwer
Ausmaß 0 nicht vorhanden
1 mukosal
2 mukosal und submukosal
3 transmural
Kryptenschaden 0 nicht vorhanden
1 basal ein Drittel betroffen
2 basal zwei Drittel betroffen
3 nur noch oberflächliches Epithel intakt
4 Krypten und Epithel komplett betroffen
betroffene Fläche 0 0 %
1 1-25 %
2 26-50 %
3 51-75 %
4 76-100 %
Die jeweiligen Punkte für die Infiltration der Entzündungszellen, das Ausmaß der
Entzündung und den Kryptenschaden wurden addiert, so dass hierdurch ein
maximaler Score von 10 entstand. Dieser wurde mit der Punktzahl für die betroffene
Fläche multipliziert. Somit ergab sich insgesamt ein maximaler Score von 40.
2.2.4.4 Alcianblau Färbung
Die Alcianblau Färbung wurde nach dem Prinzip der Hämatoxylin/Eosin Färbung
(s. Abschnitt 2.2.4.2) durchgeführt. Das Hämatoxylin wurde hierbei jedoch durch die

Material und Methodik
63
Alcianblau Lösung ersetzt. Diese färbt saure Mucopolysaccharide blau an. Die
Schnitte wurden 10 min in der Lösung gefärbt. Anschließend erfolgte die gleiche
Behandlung der Schnitte wie in der HE-Färbung.
2.2.4.5 Immunfluoreszenz
Vor der eigentlichen Färbung mussten die Schnitte auch hierfür zunächst
deparaffiniert und rehydriert werden. Zum Deparaffinieren wurden die Schnitte
dreimal für je 5 min in Xylol inkubiert. Daraufhin wurden die Schnitte in einer
absteigenden Alkoholreihe, beginnend mit zweimal 100 % Ethanol, über 96 %
Ethanol, 90 % Ethanol und 80 % Ethanol, bis zu 70 % Ethanol für jeweils 3 min
inkubiert. Anschließend wurden die Schnitte in VE Wasser gegeben, in welchem eine
Lagerung bei 4 °C bis zu einer Woche möglich war. Für die Antigendemaskierung
wurden die Schnitte 15 min lang in einer 10%igen Demaskierungslösung von Dako in
einer Plastikküvette in einem Schnellkochtopf erhitzt. Danach kühlten sie mindestens
20 min ab. Als nächstes wurden die Schnitte zweimal für 2 min in PBS gewaschen.
Der darauf folgende Proteinblock erfolgt je nach Antikörper in 5 % NGS oder in 5 %
NRS für 1 h bei Raumtemperatur. Anschließend wurden die Schnitte für mindestens
1 h oder über Nacht bei 4 °C mit dem primären Antikörper inkubiert. Die Detektion
des gebundenen Primärantikörpers erfolgte nach zweimaligem Waschen in PBS für
jeweils 2 min mithilfe eines Fluoreszenz-gekoppelten Sekundärantikörpers. Nach
dreimaligem Waschen in PBS für jeweils 2 min erfolgte hiernach die Färbung der
Zellkerne mittels 4,6-Diamidino-2-Phenylindol (DAPI) für 1 min bei Raumtemperatur.
Die Schnitte wurden anschließend noch zweimal für je 5 min in PBS und einmal für
3 min in VE Wasser gewaschen. Für den Nachweis von monoklonalen
Maus-Antikörpern in Mausgewebe wurde das Mouse on Mouse (M.O.M.)-Kit
(Vector Labs) nach Herstellerangaben verwendet. Die Schnitte wurden anschließend
mit dem Dako-Fluorescent Mounting Medium eingedeckelt und bei 4 °C gelagert. Zur
Darstellung der Schnitte wurde ein Nikon Eclipse E1000 Mikroskop mit
entsprechendem Kamerasystem (Nikon DS-Ri1) und eine NIS-Elements F.3.0
Imaging Software benutzt.

Ergebnisse
64
3 Ergebnisse
3.1 Zur Publikation vorbereitetes Manuskript
Das folgende Manuskript wurde zur Veröffentlichung vorbereitet.
Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of
regulatory T cells in murine colitis
Short Title: Dendritic HIF-1α in colitis
Authors:
Katharina Flück1,2, Gerhard Breves2, Joachim Fandrey1, Sandra Winning1
1 Institut für Physiologie, Universität Duisburg-Essen, Essen, Germany;
2 Physiologisches Institut, Tierärztliche Hochschule Hannover, Hannover, Germany

Ergebnisse
65
Abbreviations: Aldh, aldehyde dehydrogenase; BmDC, bone marrow–derived
dendritic cell; DC, dendritic cell; DSS, dextran sodium sulfate; Foxp3, forkhead box
P3; HIF, hypoxia inducible factor; IEC, intestinal epithelial cell; IFN, interferon; IL,
interleukin; MUC, Mucin; RAR, retinoic acid receptor; TFF, trefoil factor; Treg,
regulatory T cell; TGF-β, transforming growth factor β; TSLPR, thymic stromal
lymphopoietin receptor
Correspondence: Joachim Fandrey, Institut für Physiologie, Universität Duisburg-
Essen, Hufelandstr. 55, 45122 Essen, Germany
E-mail: [email protected]
Disclosures: The authors have no conflicts to disclose.
Author Contributions:
Katharina Flück: acquisition of data, analysis and interpretation of data, drafting of
the manuscript
Gerhard Breves: critical revision of the manuscript, study supervision
Joachim Fandrey: critical revision of the manuscript, study supervision
Sandra Winning: critical revision of the manuscript, study supervision
Acknowledgment: We would like to thank Dr. Flo Witte for critical reading of the
manuscript and Agnes Neugebauer for technical support.
HIF-1α+f/+f mice were purchased from The Jackson Laboratory (Bar Harbor, Maine,
USA). CD11cCre mice were originally obtained from Boris Reizis (Columbia
University Medical Center, New York, New York, USA).

Ergebnisse
66
3.1.1 Abstract
Background & Aims: Dendritic cells (DCs) are believed to serve as a bridge between
innate and adaptive immunity; they help to maintain intestinal homeostasis.
Inflammatory bowel disease (IBD) is associated with dysregulation of the mucosal
immune response and hypoxic inflammation; in IBD gene expression by DCs is
regulated by the transcription factor hypoxia inducible factor (HIF)-1. Recent studies
have described only a protective role for epithelial HIF-1 in mouse models of IBD.
Methods: We investigated the role of HIF-1 in DC function in a dextran sodium
sulfate (DSS)-induced model of murine colitis. Wild-type and dendritic cell–specific
HIF-1α knock-out mice were treated with 3% DSS for 7 days.
Results: Knock-out of HIF-1α in DCs led to a significantly larger loss of body weight
in mice with DSS-induced colitis than in control mice. Knock-out mice exhibited
severer intestinal inflammation with increased levels of proinflammatory cytokines
and enhanced production of mucin. Induction of regulatory T cells (Tregs) was
impaired, and the number of forkhead box P3 (Foxp3) Tregs was diminished by
dendritic HIF-1α knock-out.
Conclusion: Our findings demonstrate that in DCs HIF-1α is necessary for the
induction of sufficient numbers of Tregs to control intestinal inflammation.
Keywords: HIF-1α; dendritic cells; DSS; regulatory T cells
3.1.2 Introduction
The intestinal immune system must protect against infection by initiating
defensive responses to pathogens while also maintaining tolerance to self-antigens,
food, and commensal microflora.1 Mucosal dendritic cells (DCs) are fundamental in
balancing this process. Immature DCs are present in peripheral tissues and are
activated by taking up antigens in the intestine, after which they mature and migrate
to the draining lymph nodes.2 Several subsets of DCs are found in the intestine.3
Depending on their cell-surface receptor expression and their cytokine profile, DCs
can induce a proinflammatory Th1 or Th17 immune response or a humoral Th2

Ergebnisse
67
immune response. A specialized function of intestinal DCs is the induction of
regulatory T cells (Tregs).4
Nearly every inflamed tissue is characterized by low oxygen availability
(hypoxia).5 With respect to inflammatory bowel disease (IBD), this feature is likely to
be of special importance because the lumen of the gut is almost anoxic. Inflammatory
breakdown of the intestinal barrier allows anoxia to spread to formerly normoxic
tissue, which must adapt to this condition. The hypoxia inducible factor (HIF)-1
complex is a key transcription factor for cellular adaption to low oxygen tension.6
HIF-1 is a heterodimer formed by two subunits: HIF-1α and HIF-1β. Under normoxic
conditions, hydroxylation of the HIF-1α subunit by specific prolyl hydroxylases
(PHDs) leads to ubiquitin-proteasome–dependent degradation. Under hypoxic
conditions, however, PHD activity is inhibited; thus, HIF-1α can translocate into the
nucleus, dimerize with HIF-1β, and bind to hypoxia-responsive elements of HIF-1
target genes.
Several studies have investigated the function of HIF-1α in intestinal epithelial
cells. Karhausen et al.7 showed that loss of epithelial HIF-1α led to severer intestinal
inflammation, whereas an increase in epithelial HIF protein was protective. Reduction
of epithelial cell apoptosis in murine colitis by inhibition of HIF-1 degradation by the
hydroxylase inhibitor dimethyloxalylglycine supports the importance of HIF-1 in the
intestinal epithelium.8 Frede et al. found that HIF-1 regulates inflammation and
showed that lipopolysaccharide (LPS) and inflammatory cytokines lead to HIF-1α
accumulation.9 Very recent studies also demonstrated that HIF-1α plays a crucial role
in regulatory T cells and DCs. Clambey et al. reported a connection between HIF-1α
levels and the number of Tregs, as well as the expression of Foxp3.10 In vitro studies
by Wobben et al. showed that HIF-1α protein in DCs is needed for adequate
production of interferon type I and for activation of cytotoxic T cells.11
Our study was designed to elucidate the effect of transcription factor HIF-1 on
DCs in an experimental model of colitis by using mice with inactive HIF-1α protein in
CD11c-expressing cells. We treated HIF-1α+f/+f (wild-type) mice and
CD11cCre/HIF-1α+f/+f (knock-out) mice with dextran sodium sulfate (DSS) to induce
colitis and then examined the severity of inflammation.

Ergebnisse
68
3.1.3 Materials and Methods
Mice
C57Bl/6J mice with two loxP sites flanking exon 2 of the hif-1α gene
(HIF-1α+f/+f) were crossed with mice in which the integrin α X (CD11c) promoter
drives Cre recombinase expression (CD11cCre/HIF-1α+f/+f) to achieve a CD11c
cell-specific knock-out of HIF-1α. In these mice, exon 2 encodes for the DNA binding
site of translated HIF-1α protein. Knock-out mice exhibit dramatically reduced
expression of functionally active HIF-1α protein in CD11c-expressing cells and also
exhibit a loss of function of the HIF-1 complex. Sibling mice negative for Cre
recombinase (HIF-1α+f/+f) served as controls. All animals demonstrated physiological
habitus and bred regularly. They were kept and treated in accordance with the
German law for animal welfare and with institutional regulations for animal handling.
Experimental procedures were approved by the Landesamt für Umwelt-, Natur- und
Verbraucherschutz Nordrhein-Westfalen (LANUV NRW; file reference
84-02.04.2011.A098).
DSS colitis model
Colitis was induced by treatment with DSS (MP Biomedicals, Santa Ana,
USA).12 Female mice 10 to 12 weeks old were given 3% DSS dissolved in drinking
water for 7 days. The body weight of each mouse was recorded daily. On day 7, mice
were killed by cervical dislocation, and mesenteric lymph nodes and colonic tissues
were dissected.
Primary murine bone marrow–derived dendritic cells (BmDC)
Dendritic cells were isolated from HIF-1α+f/+f and CD11cCre/HIF-1α+f/+f mice as
previously described.11 Cells were cultivated under normoxic (O2/CO2/N2, 21:5:74
vol%) and hypoxic (O2/CO2/N2, 3:5;92 vol%) conditions. On culture day 8, DCs were
incubated for 6 h with 1 µg/ml LPS (serotype 0111:B4, Sigma, Deisenhofen,
Germany); total RNA was then extracted.

Ergebnisse
69
Western blot analysis
Cells were lysed, and Western blot analysis was performed as previously
described.11 HIF-1α was detected with a rabbit polyclonal anti–HIF-1α antibody
(Cayman Chemical, Michigan, USA), and β-actin was used as a loading control
(Abcam, Cambridge, UK).
RNA preparation and RT-PCR
RNA was isolated from BmDCs by the guanidinium thiocyanate-phenol-
chloroform extraction method.13 Total RNA was isolated from lymph nodes and colon
with the RNeasy Mini Kit (Qiagen, Hilden, Germany). Primer sequences used for
quantitative PCR are listed in supplemental Table S1 (Invitrogen, Darmstadt,
Germany). Quantitative detection of IFN-α subtypes was performed with the
QuantiTect Primer Assay (Qiagen, Hilden, Germany). Real-time PCR was performed
with SYBR green fluorescent dye (Eurogentec, Verviers, Belgium) on the iQ5
Real-Time PCR detection system (Bio-Rad, München, Germany). Amounts of cDNA
were normalized to ribosomal protein, and expression was calculated as induction
relative to respective controls with the Δct method.14
Colon cytokine analysis
Levels of IL-6, IL-10, and thymic stromal lymphopoietin were detected in colon
homogenates according to the manufacturer’s instructions (ELISA MAX Deluxe,
BioLegend, San Diego, USA).
Immunostaining
Tissue hypoxia was detected with the Vector M.O.M. Immunodetection Kit
(Vector Laboratories, Inc., Burlingame, USA) and the Hypoxyprobe-1 Kit
(Hypoxyprobe, Inc., Burlington, USA), according to the manufacturer’s instructions.
Foxp3 was detected with a rat anti-mouse Foxp3 antibody (eBioscience, Inc., San
Diego, USA). MUC2 was detected with a rabbit anti-mouse MUC2 antibody (Santa
Cruz Biotechnology, Inc., Santa Cruz, USA). Alexa Flour 568 second antibodies were
used (Invitrogen, Darmstadt, Germany).

Ergebnisse
70
Statistical analysis
Data were compared by one-way analysis of variance with GraphPad Prism
6 software (GraphPad Software, Inc., La Jolla, USA); the Tukey-Kramer post hoc test
was used to determine the statistical significance of the differences between the
groups. Values are expressed as means ± SEM for n individual experiments.
P values less than 0.05 were considered significant.
3.1.4 Results
DSS-induced colitis leads to profound extension of hypoxia
Colitis was induced by treating HIF-1α+f/+f and CD11cCre/HIF-1α+f/+f mice with
3% DSS in the drinking water for 7 days. Control mice were given drinking water with
3% PBS. Initially, we investigated the extent of hypoxia in the damaged colon by
using 2-nitroimidazole dye. As found in previous studies,5,7 DSS-treated mice
exhibited profound extension of hypoxia into the submucosa (Figure 1); this finding
supports the notion that activation of HIF-1 is likely to occur and to influence gene
expression of mucosal and submucosal cells. To control knock-out efficiency, we
used hypoxia and LPS stimulation to induce HIF-1α protein in bone marrow–derived
dendritic cells (BmDCs) from HIF-1α+f/+f and CD11cCre/HIF-1α+f/+f mice. Immunoblot
assays showed that the HIF-1α protein in DCs from CD11cCre/HIF-1α+f/+f mice is
smaller than that in HIF-1α+f/+f mice because exon 2 is deleted (Figure S1A). The
absence of the DNA binding domain of the transcription factor causes the protein to
be inactive. To quantify knock-out efficiency, we performed mRNA analysis with
specific HIF-1α oligonucleotides binding to HIF-1α cDNA in exon 2. DCs from
CD11cCre/HIF-1α+f/+f mice showed a 90% loss of wild-type HIF-1α (Figure S1B).
Knock-out of HIF-1α in DCs leads to severer inflammation in DSS-induced
colitis
We next studied the phenotype of CD11cCre/HIF-1α+f/+f mice in a DSS colitis
model. Control mice maintained their weight. Both DSS-treated groups experienced
progressive weight loss within 3 days. However, CD11cCre/HIF-1α+f/+f mice lost more
weight than did HIF-1α+f/+f mice; the difference in weight loss was statistically

Ergebnisse
71
significant at days 6 and 7 (P<.01-.001; Figure 2A). Characteristically, colon weight
increased and colon length decreased because of DSS-induced inflammation;
however, the differences between the two groups were not statistically significant
(data not shown).
To investigate the impact of the dendritic HIF-1α knock-out on mucosal
inflammation, we looked for changes in mRNA expression by the colon after DSS
exposure. Levels of IL-6 and IL-23 were significantly higher in DSS-treated
CD11cCre/HIF-1α+f/+f mice than in HIF-1α+f/+f mice (P<.05) (Figure 2B-C).
Furthermore, DSS-treated knock-out mice exhibited significantly higher levels of
lipocalin2, a small secreted protein crucial for regulating the immune response15
(P<.01) (Figure 2D). The number of macrophages was also higher in DSS-treated
CD11cCre/HIF-1α+f/+f mice than in HIF-1α+f/+f mice, as shown by significantly higher
expression of F4/80, a transmembrane protein on the cell surface of macrophages
(P<.01) (Figure 2E). The DSS-induced increase in IL-6 expression in dendritic HIF-1α
knock-out mice was confirmed at the protein level (Figure 2F). In DSS-treated
CD11cCre/HIF-1α+f/+f mice we also detected significantly higher levels of mRNA
expression of the inflammatory cytokine IL-1β and serum amyloid A as an acute
phase protein; the levels of this protein correlate with the degree of intestinal
inflammation.16 In these mice we also found higher levels of HIF-1α, which is known
to accumulate during inflammation9 (Figure S2A-C). Taken together, these findings
demonstrate that DSS-induced inflammation is severer in dendritic HIF-1α knock-out
mice.
In vivo induction of type I interferons is HIF-1α dependent
Previous studies have demonstrated that the secretion of type I interferons by
DCs is HIF-1 dependent in vitro.11 To investigate this finding in vivo, we analyzed
mesenteric lymph nodes from HIF-1α+f/+f and CD11cCre/HIF-1α+f/+f control and
DSS-treated mice for mRNA expression of interferon (IFN)-α4 and IFN-α12. The
induction of IFN-α4 (Figure 3A) and IFN-α12 (Figure 3B) by DSS was significantly
higher in HIF-1α+f/+f mice; this finding confirms the HIF-1α dependency of dendritic
type I interferon production in vivo.

Ergebnisse
72
Induction of IL-10 and TSLPR by LPS in BmDCs and induction of TSLPR in
DSS-induced colitis are HIF-1α dependent
IL-10 is an antiinflammatory cytokine secreted by DCs; it influences the
phenotypes of DCs and T cells.17 To determine whether this antiinflammatory
response is affected by HIF-1α knock-out, we isolated BmDCs from HIF-1α+f/+f and
CD11cCre/HIF-1α+f/+f mice, cultivated them for 8 days under normoxic conditions,
and then stimulated them with 1 µg LPS for 6 h. The induction of IL-10 by LPS was
significantly higher in HIF-1α+f/+f mice than in CD11cCre/HIF-1α+f/+f mice (P<.0001)
(Figure 4A). To better imitate the in vivo situation of mucosal DCs, we cultivated
BmDCs under hypoxic conditions (3% O2) for 8 days. Again, IL-10 induction by LPS
was HIF-1α dependent (Figure 4B). In vivo, mRNA expression of IL-10 in colon tissue
showed a slight but not statistically significant increase in DSS-treated HIF-1α+f/+f and
CD11cCre/HIF-1α+f/+f mice. However, there was no difference between the wild-type
group and the knock-out group (Figure 4D). DSS colitis had no effect on the levels of
IL-10 protein in colon tissues (Figure S3).
Because mucosal DCs are always influenced by intestinal epithelial cells
(IECs), we looked for expression of thymic stromal lymphopoietin receptor (TSLPR)
by DCs. The ligand TSLP is released by IECs and induces the production of
noninflammatory DCs.18 BmDCs exhibited expression of TSLPR in vitro.
Hypoxia-cultivated BmDCs from HIF-1α+f/+f mice exhibited a significantly larger
increase of TSLPR production by LPS stimulation than did BmDCs from
CD11cCre/HIF-1α+f/+f mice (P<.01) (Figure 4C). In vivo, we also found that the
induction of TSLPR in colon tissue from DSS-treated HIF-1α+f/+f mice was
significantly higher than that in colon tissue from DSS-treated CD11cCre/HIF-1α+f/+f
mice (P<.01) (Figure 4E). In DSS colitis, protein levels of TSLP showed a slight but
not statistically significant decrease, probably because of the destruction of a large
number of IECs (Figure 4F). However, there were no significant differences in the
production of TSLP by IECs in the wild-type group and the knock-out group. Taken
together, these findings indicate that HIF-1α significantly influences IL-10 production
and TSLPR expression by DCs.

Ergebnisse
73
Induction of Treg-activating and gut-homing molecules in mesenteric lymph
nodes depends on HIF-1α expression in DCs in DSS-induced colitis
Mucosal DCs migrate into the mesenteric lymph nodes, where they present
antigens to T cells. DCs can induce a proinflammatory Th1 and Th17 response, but
certain mucosal DCs can also promote the differentiation of Tregs to limit
inflammation.19 To determine whether the dendritic HIF-1α knock-out affects the
induction of Tregs, we isolated mesenteric lymph nodes and measured mRNA
expression of Treg-activating molecules. The levels of IL-10 and transforming growth
factor (TGF)-β were significantly higher in DSS-treated HIF-1α+f/+f mice than in
CD11cCre/HIF-1α+f/+f mice (P<.05) (Figure 5A-B).
The induction of Tregs is also dependent on retinoic acid (RA).20 The activity
of aldehyde dehydrogenase (Aldh)1a2, which is necessary for catalyzing retinal to
RA, was significantly higher in DSS-treated HIF-1α+f/+f mice than in DSS-treated
CD11cCre/HIF-1α+f/+f mice (P<.05) (Figure 5C). Furthermore, the levels of RA
receptor α (RARα), a ligand-inducible transcription factor21 expressed by Tregs, were
significantly higher in DSS-treated HIF-1α+f/+f mice than in DSS-treated
CD11cCre/HIF-1α+f/+f mice (P<.0001) (Figure 5D).
The gut-homing T-cell markers CC chemokine receptor (CCR)9 and
α4β7-integrin are dependent on activation by DCs and RA.22 Therefore, we
investigated the effect of dendritic HIF-1α knock-out on these markers in our DSS
colitis model. DSS treatment significantly increased the induction of α4 integrin in
lymph nodes from wild-type and knock-out mice (P<.01) (Figure S4). In contrast, the
expression of CCR9 and β7 integrin was significantly higher in DSS-treated HIF-1α+f/+f
mice than in CD11cCre/HIF-1α+f/+f mice (P<.001-.0001) (Figure 5E-F). Taken
together, these findings indicate that the loss of dendritic HIF-1α leads to impaired
induction of Tregs in mesenteric lymph nodes and to diminished expression of T-cell
gut-homing markers.

Ergebnisse
74
Increased numbers of T cells and Tregs in mesenteric lymph nodes are
dependent on HIF-1α expression in DCs in DSS-induced colitis
To determine whether the dendritic HIF-1α knock-out influences the number of
T cells and in particular of Tregs, we measured mRNA expression of CD4 and CD8a
by mesenteric lymph nodes. We found a significant increase in DSS-mediated
inflammation only in HIF-1α+f/+f mice, whereas the mRNA expression of CD4 and
CD8a did not change in CD11cCre/HIF-1α+f/+f mice (P<.01-.001) (Figure 6A-B).
We next examined the expression of Foxp3, a key transcriptional regulator of
Tregs.23 DSS-treated HIF-1α+f/+f mice exhibited a significantly larger induction of
Foxp3 mRNA than did CD11cCre/HIF-1α+f/+f mice (P<.05) (Figure 6C). Levels of
OX40, reported to be a surface molecule of Tregs,24 were also higher in DSS-treated
HIF-1α+f/+f mice than in CD11cCre/HIF-1α+f/+f mice (P<.05) (Figure 6D). Enhanced
numbers of Foxp3-positive Tregs were confirmed by immunofluorescence.
DSS-treated HIF-1α+f/+f mice exhibited more Foxp3-positive cells (red) in mesenteric
lymph nodes than did knock-out mice (Figure 6E). These findings indicate that
knock-out of dendritic HIF-1α in DSS colitis leads to lower numbers of T cells and
Tregs in mesenteric lymph nodes.
DC-specific knock-out of HIF-1α stimulates mucosal epithelium to increase the
production of mucins in DSS-induced colitis
Because DCs are in close contact with epithelial cells,25 we examined the
influence of the DC-specific HIF-1α knock-out on IECs in a model of DSS colitis.
Goblet cells produce mucins and trefoil factors (TFF), thus forming a protective
mucus layer that coats the gastrointestinal tract.26 We found that the expression of
MUC1, MUC2, and MUC3 by colon mRNA is significantly higher in DSS-treated
CD11cCre/HIF-1α+f/+f mice than in DSS-treated HIF-1α+f/+f mice (P<.05-.01)
(Figure 7A-C). Immunofluorescence studies of MUC2 confirmed these results
(Figure 7D). CD11cCre/HIF-1α+f/+f mice exhibited a more intense production of MUC2
in DSS colitis than did HIF-1α+f/+f mice. Levels of TFF3 mRNA were significantly
higher than control levels only in DSS-treated knock-out mice; there were no
significant differences between DSS-treated wild-type and knock-out mice

Ergebnisse
75
(Figure S5B). In total, these findings show that the loss of HIF-1α by DCs affects the
crosstalk between DCs and IECs in a way that stimulates IECs to produce more
mucins.
3.1.5 Discussion
Dendritic cells play a crucial role in regulating the immune response and are
involved in the pathogenesis of IBD.27 We and others5,7 have reported that mucosal
DCs in inflamed tissue are exposed to hypoxia, and this finding caused us to focus
on the role of dendritic HIF-1 in a murine model of colitis. Recent studies have
demonstrated that epithelial HIF-1 exerts a protective effect in intestinal
inflammation.7,8 In the current report we show for the first time that dendritic HIF-1α
also exerts a pivotal influence on the control of colitis. The absence of dendritic
HIF-1α leads to severe intestinal inflammation with increased levels of inflammatory
cytokines and impaired induction of regulatory T cells.
Inflammatory cytokines such as IL-6 and IL-23 are strongly involved in the
pathogenesis of IBD.28,29 IL-6 can inhibit Treg-mediated suppression in a murine
model of colitis.30 It has been shown that IL-23 promotes inflammation by IL-6 and
IL-17 and drives a Th17 immune response.29 A recent study demonstrated that
lamina propria DCs constitutively express IL-2331; this finding reveals an additional
influence of DCs on IBD. In the present study we found higher levels of IL-6 and
IL-23 in DSS-treated CD11cCre/HIF-1α+f/+f mice than in HIF-1α+f/+f mice. In addition,
we looked for lipocalin2, a known regulator of the gut´s immune response.15
Lipocalin2 is a member of the lipocalin protein family and is apically secreted by
epithelial cells. Chassaing et al. measured fecal lipocalin2 levels with ELISA and
reported that these levels are up-regulated in murine models of colitis.32 After DSS
treatment, our knock-out mice exhibited elevated levels of lipocalin2, a finding
indicating severer inflammation (Figure 2D). Therefore, in future experiments fecal
measurements of lipocalin2 may prove useful in monitoring the progression of
intestinal inflammation.
We further aimed to analyze the influence of dendritic HIF-1α on the
production of type I IFN in a murine colitis model. Wobben et al. found that dendritic

Ergebnisse
76
IFN-α production is HIF-1α dependent.11 We confirmed these findings in vivo,
because our knock-out mice exhibited less expression of IFN-α4 and IFN-α12 mRNA
after DSS treatment. Type I IFNs are known to exert protective effects in intestinal
inflammation by preventing epithelial barrier dysfunction and inhibiting the
inflammatory response of activated macrophages.33,34 This finding is in line with our
finding that HIF-1α+f/+f mice exhibit fewer infiltrated macrophages than do
CD11cCre/HIF-1α+f/+f mice in DSS colitis (Figure 2E).
Tregs can limit inappropriate inflammatory responses and are protective in
intestinal inflammation.35 A subset of intestinal DCs can induce Tregs to control
inflammation. This is why we looked for the impact of dendritic HIF-1α on Treg
induction. Tregs are defined by the expression of Foxp3.23 We found fewer
Foxp3-positive cells in our knock-out mice, and this reduced number of cells may
lead to severer inflammation. The induction of Tregs is further dependent on the
release of retinoic acid (RA) and TGF-β by DCs.22 RA is converted from retinal by
aldehyde dehydrogenases (Aldhs). Aldh1a2 is found primarily in DCs in the
mesenteric lymph nodes.20 Moreover, RA can inhibit the IL-6–mediated induction of
proinflammatory Th17 cells.36 DSS treatment did not induce higher levels of TGF-β,
Aldh1a2, and retinoic acid receptor α (RARα) in our DSS-treated knock-out mice
(Figure 5B-D). Consistent with the findings of Fernández-Martínez et al., who
reported crosstalk between HIF-1α and RARβ,37 we have now shown a connection
between dendritic HIF-1α and RARα expression. Transcription of RARα is directly
induced by RA.21 Together with the decrease in Aldh1a2 expression, this finding
leads to the conclusion that HIF-1α–deficient dendritic cells secrete less RA and
consequently induce fewer Tregs.
Treg induction also relies on DC-secreted IL-10.38 Spontaneous colitis
develops in IL-10 knock-out mice after a few weeks.39 Several studies have
demonstrated that HIF-1α influences IL-10 production. Cai et al. showed that HIF-1α
activates IL-10 production in myocytes and is required for remote preconditioning of
the heart.40 HIF-1α–dependent IL-10 secretion is also required for lung interstitial
macrophages to prevent airway allergy.41 In the current study we found for the first
time that dendritic loss of HIF-1α directly leads to diminished levels of IL-10 in vitro

Ergebnisse
77
(Figure 4A-B) and in the lymph nodes in vivo (Figure 5A), with a severe impact on
intestinal inflammation. The fact that we could not detect significant changes in IL-10
mRNA expression in the colon (Figure 4D) may be due to the small numbers of DCs
in colon tissue.
Intestinal DCs can induce the specific gut-homing markers α4β7 integrin and
CCR9 on Tregs.42 RA enhances the expression of α4β7 integrin and CCR9.20 We
found that the expression of β7 integrin and CCR9 was significantly higher in
DSS-treated HIF-1α+f/+f mice than in knock-out mice (Figure 4E-F), a finding
indicating that dendritic HIF-1α is needed for adequate Treg homing.
Rimoldi et al recently reported that intestinal epithelial cells (IECs) and DCs
interact.18 IECs release TSLP, which conditions mucosal DCs to noninflammatory
tolerogenic DCs. Jang et al. demonstrated that the expression of TSLP in
keratinocytes is HIF-1α dependent.43 We were unable to detect differences in the
expression of TSLP in the colon because the IECs in our model express active
HIF-1α. However, expression of the TSLP receptor (TSLPR) was induced by LPS
only in BmDCs from HIF-1α+f/+f mice (Figure 4C). Correspondingly, in vivo only
HIF-1α+f/+f mice exhibited increased levels of TSLPR in DSS colitis (Figure 4E).
Furthermore, we detected an additional interaction between IECs and DCs.
We found that knock-out of dendritic HIF-1α leads to increased production of
MUC1 - 3. Mucins are produced primarily by goblet cells and play a crucial role in
mucosal protection and repair.26 The expression of mucins in intestinal inflammation
depends on the severity and degree of colitis. Hoebler et al. found that the
expression of MUC1 and MUC3 was higher in DSS-induced colitis, whereas the
expression of MUC2 did not change.44 In contrast, Dharmani et al. found that the
expression of MUC2 and MUC3 did not decrease in a DSS model.45 Clinical studies
also found diverging findings in patients with IBD.26,46,47 Van der Sluis et al. reported
that MUC2 knock-out mice develop spontaneous colitis and are more prone to DSS
colitis.47 Furthermore, recent studies demonstrated an interaction between IL-6 and
the production of mucin. Yokoigawa et al. found that mucins secreted from colon
cancer cells induce the production of IL-6.48 In contrast, Li et al. showed that IL-6
modulates mucin expression of colorectal cancer cells by up-regulating MUC1 and

Ergebnisse
78
down-regulating MUC2.49 Other studies have demonstrated IL-6–mediated induction
of MUC2 and MUC3 in colon carcinoma cells.50,51 In our DSS-treated knock-out mice,
higher levels of IL-6 were associated with higher levels of mucins. This association
may result from a compensatory protective mechanism in the intestinal epithelium,
because mucins are also involved in mucosal repair.52,53
Although new therapeutic strategies for IBD focus primarily on T cells, it is
noteworthy that the phenotype of DCs can also affect intestinal inflammation. In the
present study we demonstrated that loss of dendritic HIF-1α leads to severer
intestinal inflammation with impaired induction of Tregs and enhanced production of
mucins. Targeting intestinal dendritic cells may therefore be an additional therapeutic
option in the treatment of IBD.
3.1.6 References
1. Mowat AM. Anatomical basis of tolerance and immunity to intestinal antigens.
Nat. Rev. Immunol. 2003;3:331–341.
2. Banchereau J, Steinman RM. Dendritic cells and the control of immunity.
Nature 1998;392:245–252.
3. Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat.
Rev. Immunol. 2008;8:435–446.
4. Sun CM, Hall JA, Blank RB, et al. Small intestine lamina propria dendritic cells
promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med.
2007;204:1775–1785.
5. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N. Engl. J. Med.
2011;364:656–665.
6. Semenza GL, Agani F, Booth G, et al. Structural and functional analysis of
hypoxia-inducible factor 1. Kidney Int. 1997;51:553–555.

Ergebnisse
79
7. Karhausen J, Furuta GT, Tomaszewski JE, et al. Epithelial hypoxia-inducible
factor-1 is protective in murine experimental colitis. J. Clin. Invest.
2004;114:1098–1106.
8. Cummins EP, Seeballuck F, Keely SJ, et al. The hydroxylase inhibitor
dimethyloxalylglycine is protective in a murine model of colitis.
Gastroenterology 2008;134:156–165.
9. Frede S, Berchner-Pfannschmidt U, Fandrey J. Regulation of hypoxia-inducible
factors during inflammation. Methods Enzymol. 2007;435:405–419.
10. Clambey ET, McNamee EN, Westrich JA, et al. Hypoxia-inducible factor-1
alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and
function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. U.
S. A. 2012;109:E2784–2793.
11. Wobben R, Hüsecken Y, Lodewick C, et al. Role of hypoxia inducible factor-1α
for interferon synthesis in mouse dendritic cells. Biol. Chem. 2013;394:495–
505.
12. Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of
reliable experimental acute and chronic ulcerative colitis in mice.
Gastroenterology 1990;98:694–702.
13. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid
guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem.
1987;162:156–159.
14. Winer J, Jung CK, Shackel I, et al. Development and validation of real-time
quantitative reverse transcriptase-polymerase chain reaction for monitoring
gene expression in cardiac myocytes in vitro. Anal. Biochem. 1999;270:41–49.

Ergebnisse
80
15. Berger T, Togawa A, Duncan GS, et al. Lipocalin 2-deficient mice exhibit
increased sensitivity to Escherichia coli infection but not to ischemia-
reperfusion injury. Proc. Natl. Acad. Sci. U. S. A. 2006;103:1834–1839.
16. de Villiers WJ, Varilek GW, de Beer FC, et al. Increased serum amyloid a
levels reflect colitis severity and precede amyloid formation in IL-2 knockout
mice. Cytokine 2000;12:1337–1347.
17. Westendorf AM, Fleissner D, Hansen W, et al. T cells, dendritic cells and
epithelial cells in intestinal homeostasis. Int. J. Med. Microbiol. 2010;300:11–
18.
18. Rimoldi M, Chieppa M, Salucci V, et al. Intestinal immune homeostasis is
regulated by the crosstalk between epithelial cells and dendritic cells. Nat.
Immunol. 2005;6:507–514.
19. Rescigno M, Di Sabatino A. Dendritic cells in intestinal homeostasis and
disease. J. Clin. Invest. 2009;119:2441–2450.
20. Iwata M, Hirakiyama A, Eshima Y, et al. Retinoic acid imprints gut-homing
specificity on T cells. Immunity 2004;21:527–538.
21. Blomhoff R, Blomhoff HK. Overview of retinoid metabolism and function. J.
Neurobiol. 2006;66:606–630.
22. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, et al. A functionally
specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T
cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med.
2007;204:1757–1764.
23. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by
the transcription factor Foxp3. Science 2003;299:1057–1061.

Ergebnisse
81
24. Mittrücker HW, Kaufmann SH. Mini-review: regulatory T cells and infection:
suppression revisited. Eur. J. Immunol. 2004;34:306–12.
25. Rescigno M, Urbano M, Valzasina B, et al. Dendritic cells express tight junction
proteins and penetrate gut epithelial monolayers to sample bacteria. Nat.
Immunol. 2001;2:361–367.
26. Shirazi T, Longman RJ, Corfield AP, et al. Mucins and inflammatory bowel
disease. Postgrad. Med. J. 2000;76:473–478.
27. Niess JH. Role of mucosal dendritic cells in inflammatory bowel disease. World
J. Gastroenterol. 2008;14:5138-5148.
28. Yamamoto M, Yoshizaki K, Kishimoto T, et al. IL-6 is required for the
development of Th1 cell-mediated murine colitis. J. Immunol. 2000;164:4878–
4882.
29. Yen D, Cheung J, Scheerens H, et al. IL-23 is essential for T cell-mediated
colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest.
2006;116:1310–1316.
30. Dominitzki S, Fantini MC, Neufert C, et al. Cutting Edge: trans-signaling via the
soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J.
Immunol. 2007;179:2041–2045.
31. Becker C, Wirtz S, Blessing M, et al. Constitutive p40 promoter activation and
IL-23 production in the terminal ileum mediated by dendritic cells. J. Clin.
Invest. 2003;112:693–706.
32. Chassaing B, Srinivasan G, Delgado MA, et al. Fecal lipocalin 2, a sensitive
and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS
One 2012;7:e44328.

Ergebnisse
82
33. Katakura K, Lee J, Rachmilewitz D, et al. Toll-like receptor 9-induced type I IFN
protects mice from experimental colitis. J. Clin. Invest. 2005;115:695–702.
34. Sainathan SK, Bishnupuri KS, Aden K, et al. Toll-like receptor-7 ligand
Imiquimod induces type I interferon and antimicrobial peptides to ameliorate
dextran sodium sulfate-induced acute colitis. Inflamm. Bowel Dis.
2012;18:955–967.
35. Izcue A, Coombes JL, Powrie F. Regulatory T cells suppress systemic and
mucosal immune activation to control intestinal inflammation. Immunol. Rev.
2006;212:256–271.
36. Mucida D, Park Y, Kim G, et al. Reciprocal TH17 and regulatory T cell
differentiation mediated by retinoic acid. Science 2007;317:256–260.
37. Fernández-Martínez AB, Jiménez MIA, Hernández IS, et al. Mutual regulation
of hypoxic and retinoic acid related signalling in tubular proximal cells. Int. J.
Biochem. Cell Biol. 2011;43:1198–1207.
38. Groux H, Cottrez F, Rouleau M, et al. A transgenic model to analyze the
immunoregulatory role of IL-10 secreted by antigen-presenting cells. J.
Immunol. 1999;162:1723–1729.
39. Kühn R, Löhler J, Rennick D, et al. Interleukin-10-deficient mice develop
chronic enterocolitis. Cell 1993;75:263–274.
40. Cai Z, Luo W, Zhan H, et al. Hypoxia-inducible factor 1 is required for remote
ischemic preconditioning of the heart. Proc. Natl. Acad. Sci. U. S. A.
2013;110:17462–17467.
41. Toussaint M, Fievez L, Drion PV, et al. Myeloid hypoxia-inducible factor 1α
prevents airway allergy in mice through macrophage-mediated
immunoregulation. Mucosal Immunol. 2013;6:485–497.

Ergebnisse
83
42. Johansson-Lindbom B, Svensson M, Pabst O, et al. Functional specialization
of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing.
J. Exp. Med. 2005;202:1063–1073.
43. Jang Y, Jeong SH, Park YH, et al. UVB induces HIF-1α-dependent TSLP
expression via the JNK and ERK pathways. J. Invest. Dermatol.
2013;133:2601–2608.
44. Hoebler C, Gaudier E, De Coppet P, et al. MUC genes are differently
expressed during onset and maintenance of inflammation in dextran sodium
sulfate-treated mice. Dig. Dis. Sci. 2006;51:381–389.
45. Dharmani P, Leung P, Chadee K. Tumor necrosis factor-α and Muc2 mucin
play major roles in disease onset and progression in dextran sodium sulphate-
induced colitis. PLoS One 2011;6:e25058.
46. Dorofeyev AE, Vasilenko I V, Rassokhina OA, et al. Mucosal barrier in
ulcerative colitis and Crohn’s disease. Gastroenterol. Res. Pract.
2013;2013:431231.
47. Van der Sluis M, De Koning BA, De Bruijn AC, et al. Muc2-deficient mice
spontaneously develop colitis, indicating that MUC2 is critical for colonic
protection. Gastroenterology 2006;131:117–129.
48. Yokoigawa N, Takeuchi N, Toda M, et al. Enhanced production of interleukin 6
in peripheral blood monocytes stimulated with mucins secreted into the
bloodstream. Clin. Cancer Res. 2005;11:6127–6132.
49. Li YY, Hsieh LL, Tang RP, et al. Macrophage-derived interleukin-6 up-
regulates MUC1, but down-regulates MUC2 expression in the human colon
cancer HT-29 cell line. Cell. Immunol. 2009;256:19–26.

Ergebnisse
84
50. Enss ML, Cornberg M, Wagner S, et al. Proinflammatory cytokines trigger MUC
gene expression and mucin release in the intestinal cancer cell line LS180.
Inflamm. Res. 2000;49:162–169.
51. Shekels LL, Ho SB. Characterization of the mouse Muc3 membrane bound
intestinal mucin 5’ coding and promoter regions: regulation by inflammatory
cytokines. Biochim. Biophys. Acta 2003;1627:90–100.
52. Wallace JL, Vong L, Dharmani P, et al. Muc-2-deficient mice display a sex-
specific, COX-2-related impairment of gastric mucosal repair. Am. J. Pathol.
2011;178:1126–1133.
53. Ho SB, Dvorak LA, Moor RE, et al. Cysteine-rich domains of muc3 intestinal
mucin promote cell migration, inhibit apoptosis, and accelerate wound healing.
Gastroenterology 2006;131:1501–1517.
3.1.7 Figures
Fig. 3.1: DSS colitis leads to profound extension of hypoxia.
Sections (4 µm) of colon tissue from HIF-1α+f/+f
mice were stained with hydroxyprobe (red) and DAPI
(blue). Stained tissue from control mice showed hypoxic epithelium. DSS treatment led to profound
extension of hypoxia into the submucosal regions. Magnification, 200 x; scale bar, 100 µm.
C DSS
HIF-1α+f/+f
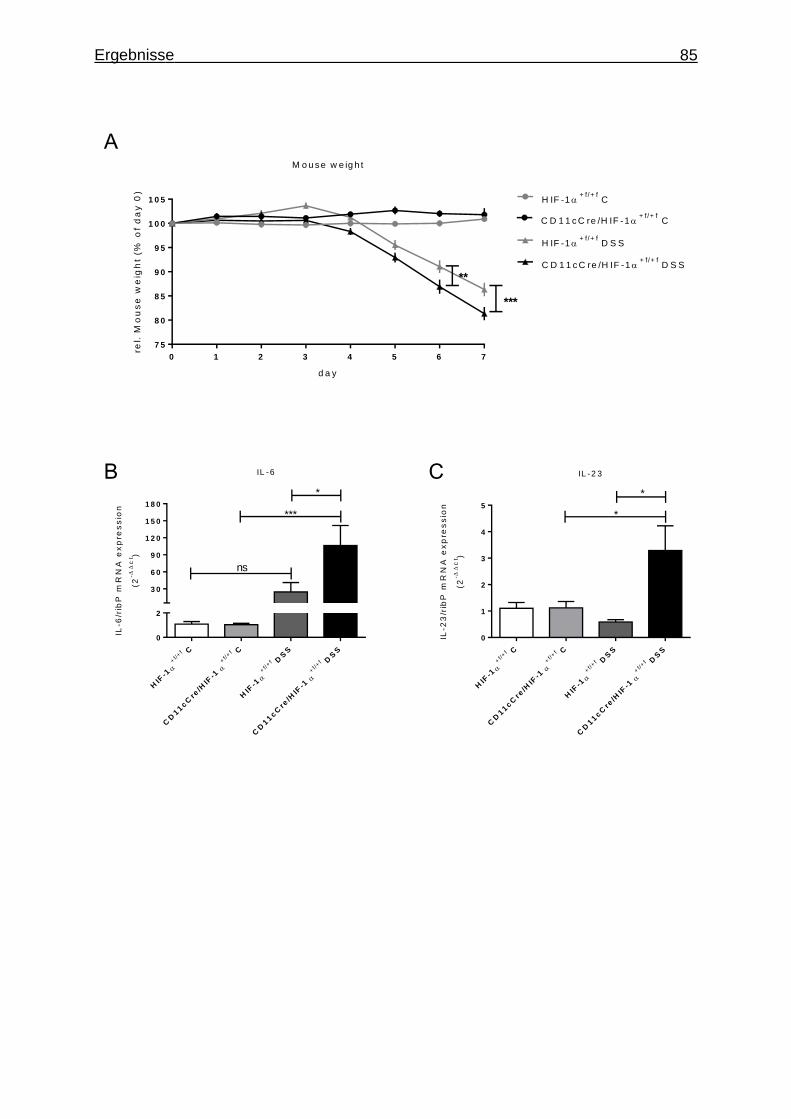
Ergebnisse
85
A
M o u s e w e ig h t
0 1 2 3 4 5 6 7
7 5
8 0
8 5
9 0
9 5
1 0 0
1 0 5
d a y
rel.
Mo
us
e w
eig
ht
(% o
f d
ay
0)
H IF -1+ f/+ f
D S S
C D 1 1 c C re /H IF -1+ f/+ f
C
H IF -1+ f/+ f
C
C D 1 1 c C re /H IF -1+ f/+ f
D S S
**
***
IL -6
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
3 0
6 0
9 0
1 2 0
1 5 0
1 8 0
IL-6
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
***
ns
*
IL -2 3
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4
5
IL-2
3/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
*
*
B C
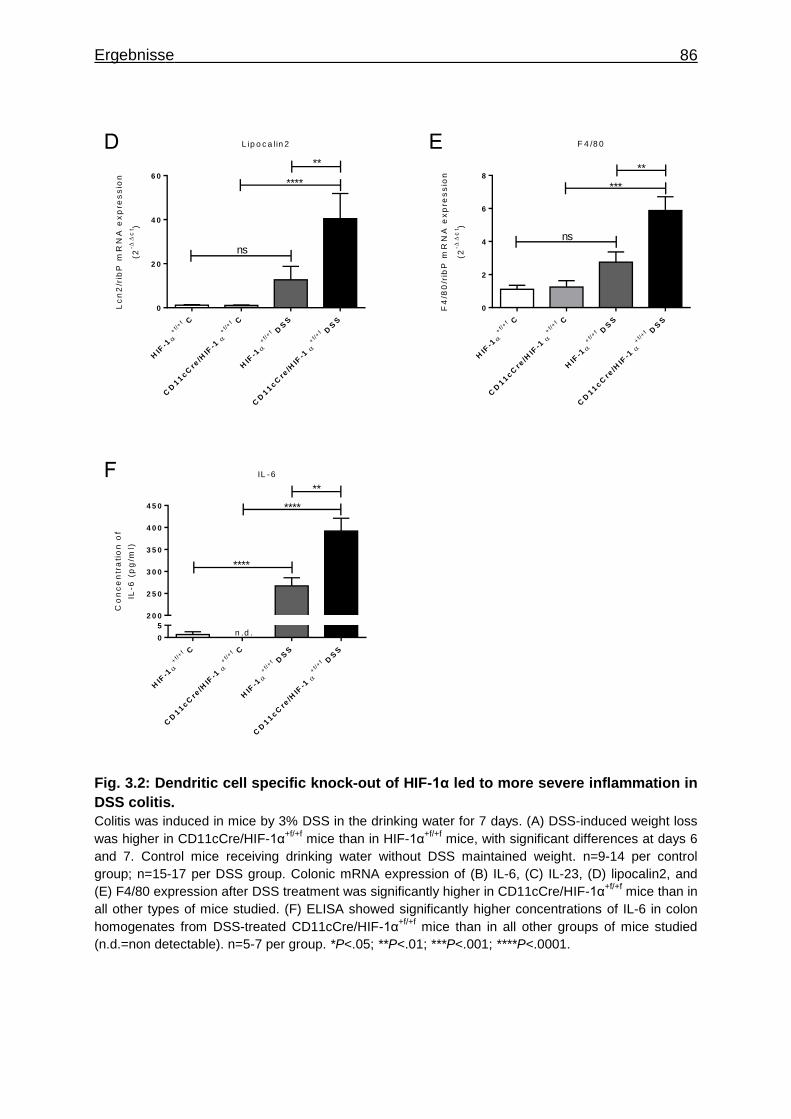
Ergebnisse
86
Fig. 3.2: Dendritic cell specific knock-out of HIF-1α led to more severe inflammation in
DSS colitis.
Colitis was induced in mice by 3% DSS in the drinking water for 7 days. (A) DSS-induced weight loss
was higher in CD11cCre/HIF-1α+f/+f
mice than in HIF-1α+f/+f
mice, with significant differences at days 6
and 7. Control mice receiving drinking water without DSS maintained weight. n=9-14 per control
group; n=15-17 per DSS group. Colonic mRNA expression of (B) IL-6, (C) IL-23, (D) lipocalin2, and
(E) F4/80 expression after DSS treatment was significantly higher in CD11cCre/HIF-1α+f/+f
mice than in
all other types of mice studied. (F) ELISA showed significantly higher concentrations of IL-6 in colon
homogenates from DSS-treated CD11cCre/HIF-1α+f/+f
mice than in all other groups of mice studied
(n.d.=non detectable). n=5-7 per group. *P<.05; **P<.01; ***P<.001; ****P<.0001.
E F 4 /8 0
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
8
F4
/80
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
***
ns
**
L ip o c a lin 2
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2 0
4 0
6 0
Lc
n2
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
****
ns
**
D
IL -6
Co
nc
en
tra
tio
n o
f
IL-6
(p
g/m
l)
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
5
2 0 0
2 5 0
3 0 0
3 5 0
4 0 0
4 5 0
**
****
****
n .d .
F
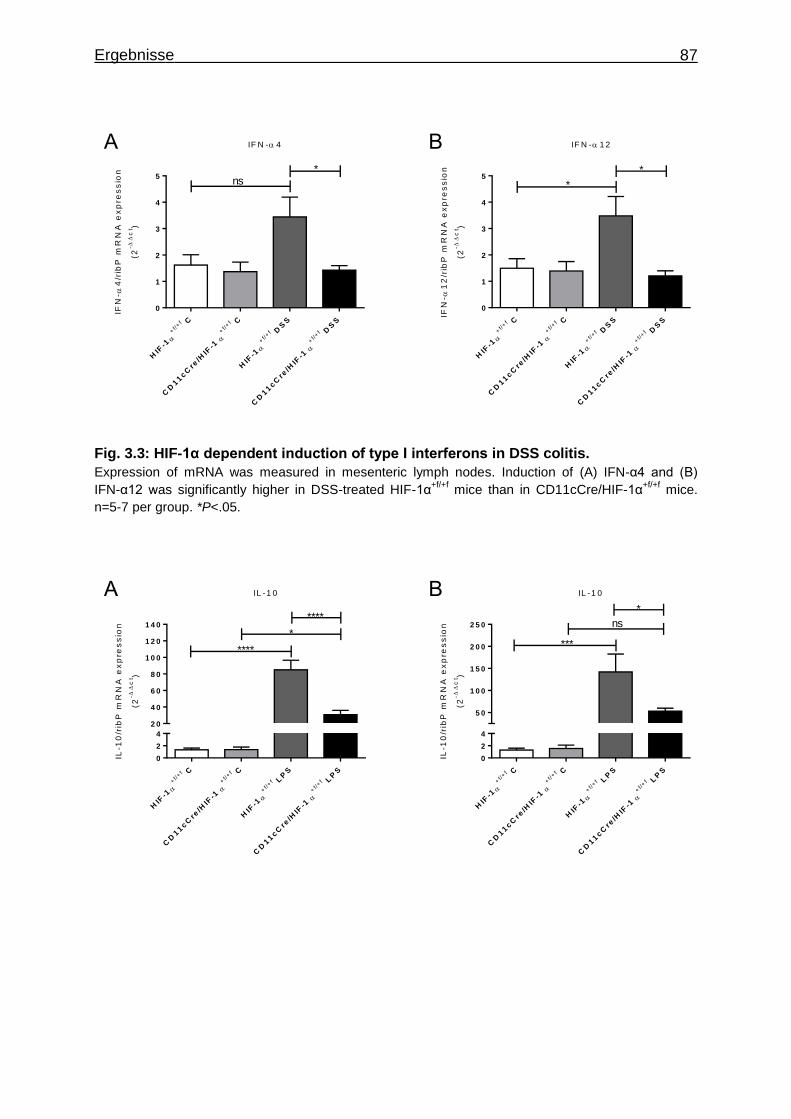
Ergebnisse
87
Fig. 3.3: HIF-1α dependent induction of type I interferons in DSS colitis.
Expression of mRNA was measured in mesenteric lymph nodes. Induction of (A) IFN-α4 and (B)
IFN-α12 was significantly higher in DSS-treated HIF-1α+f/+f
mice than in CD11cCre/HIF-1α+f/+f
mice.
n=5-7 per group. *P<.05.
IF N - 4
IFN
- 4
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4
5ns
*
IF N - 1 2
IFN
- 1
2/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4
5
*
*
B A
IL -1 0
IL-1
0/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f L
PS
CD
11cC
re/H
IF-1+f/+f L
PS
0
2
4
2 0
4 0
6 0
8 0
1 0 0
1 2 0
1 4 0
****
*
****
IL -1 0
IL-1
0/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f L
PS
CD
11cC
re/H
IF-1+f/+f L
PS
0
2
4
5 0
1 0 0
1 5 0
2 0 0
2 5 0
***
ns
*
A B
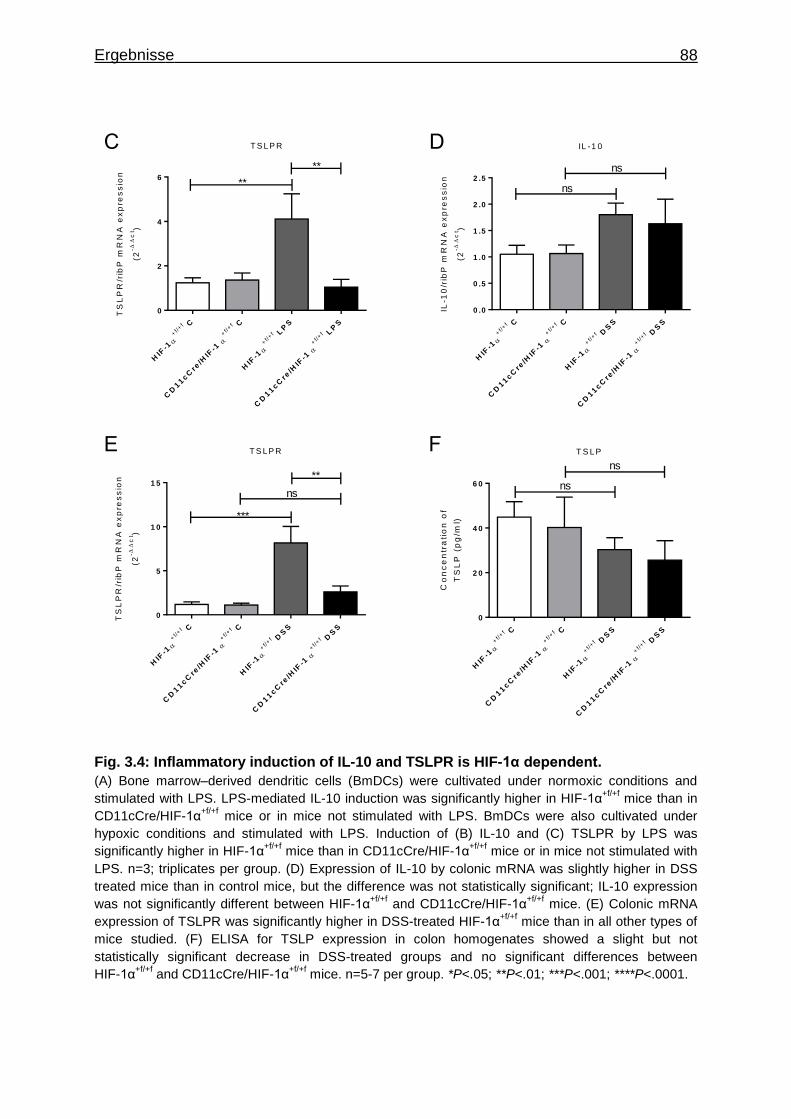
Ergebnisse
88
Fig. 3.4: Inflammatory induction of IL-10 and TSLPR is HIF-1α dependent.
(A) Bone marrow–derived dendritic cells (BmDCs) were cultivated under normoxic conditions and
stimulated with LPS. LPS-mediated IL-10 induction was significantly higher in HIF-1α+f/+f
mice than in
CD11cCre/HIF-1α+f/+f
mice or in mice not stimulated with LPS. BmDCs were also cultivated under
hypoxic conditions and stimulated with LPS. Induction of (B) IL-10 and (C) TSLPR by LPS was
significantly higher in HIF-1α+f/+f
mice than in CD11cCre/HIF-1α+f/+f
mice or in mice not stimulated with
LPS. n=3; triplicates per group. (D) Expression of IL-10 by colonic mRNA was slightly higher in DSS
treated mice than in control mice, but the difference was not statistically significant; IL-10 expression
was not significantly different between HIF-1α+f/+f
and CD11cCre/HIF-1α+f/+f
mice. (E) Colonic mRNA
expression of TSLPR was significantly higher in DSS-treated HIF-1α+f/+f
mice than in all other types of
mice studied. (F) ELISA for TSLP expression in colon homogenates showed a slight but not
statistically significant decrease in DSS-treated groups and no significant differences between
HIF-1α+f/+f
and CD11cCre/HIF-1α+f/+f
mice. n=5-7 per group. *P<.05; **P<.01; ***P<.001; ****P<.0001.
IL -1 0
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0 .0
0 .5
1 .0
1 .5
2 .0
2 .5
IL-1
0/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
ns
ns
D T S LP R
TS
LP
R/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f L
PS
CD
11cC
re/H
IF-1+f/+f L
PS
0
2
4
6**
**
C
T S LP
Co
nc
en
tra
tio
n o
f
TS
LP
(p
g/m
l)
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2 0
4 0
6 0 ns
ns
T S LP R
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
5
1 0
1 5
TS
LP
R/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
ns
***
**
F E
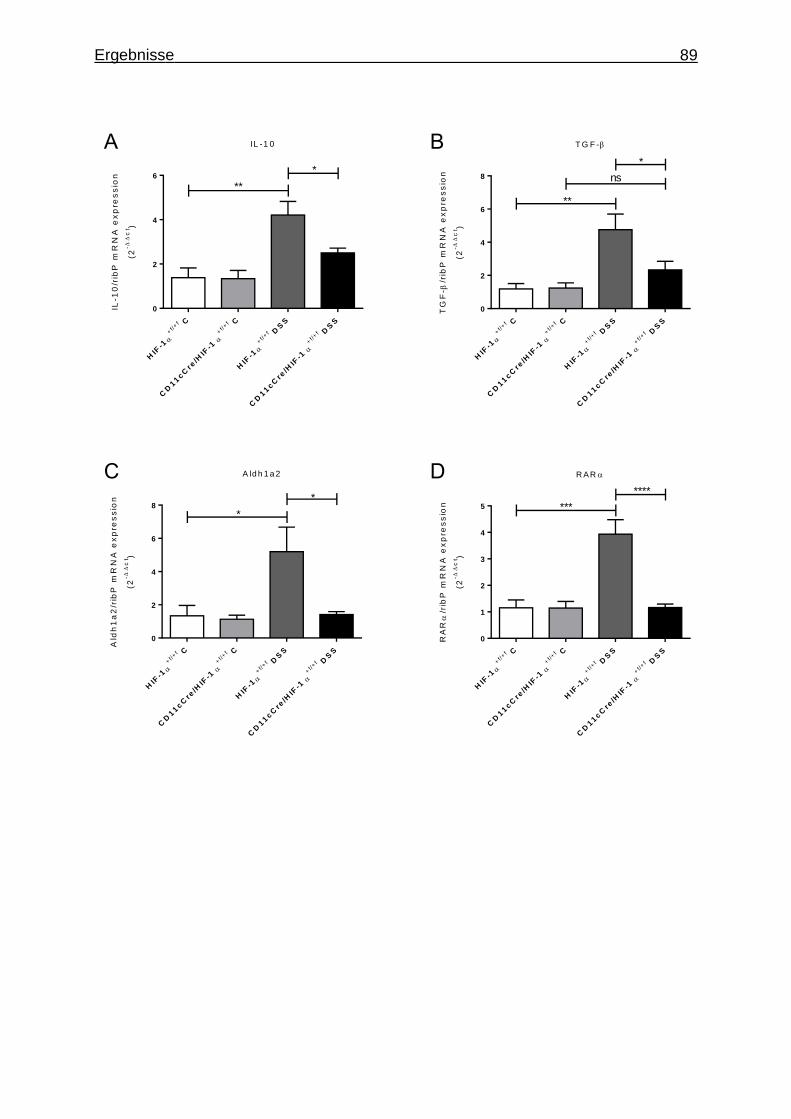
Ergebnisse
89
IL -1 0
IL-1
0/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
**
*
A T G F -
TG
F-
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
8
**
*
ns
B
A ld h 1 a 2
Ald
h1
a2
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
8
*
*
R AR
RA
R
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4
5 ***
****
C D
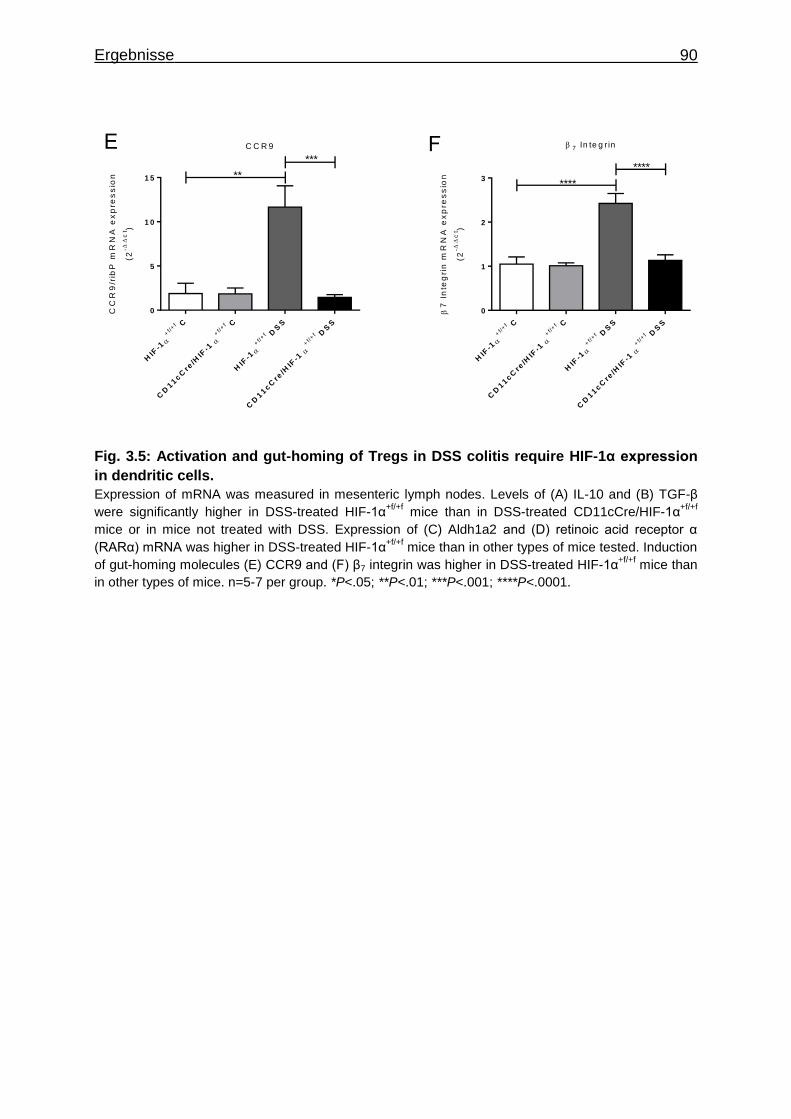
Ergebnisse
90
Fig. 3.5: Activation and gut-homing of Tregs in DSS colitis require HIF-1α expression
in dendritic cells.
Expression of mRNA was measured in mesenteric lymph nodes. Levels of (A) IL-10 and (B) TGF-β
were significantly higher in DSS-treated HIF-1α+f/+f
mice than in DSS-treated CD11cCre/HIF-1α+f/+f
mice or in mice not treated with DSS. Expression of (C) Aldh1a2 and (D) retinoic acid receptor α
(RARα) mRNA was higher in DSS-treated HIF-1α+f/+f
mice than in other types of mice tested. Induction
of gut-homing molecules (E) CCR9 and (F) β7 integrin was higher in DSS-treated HIF-1α+f/+f
mice than
in other types of mice. n=5-7 per group. *P<.05; **P<.01; ***P<.001; ****P<.0001.
7 In te g r in
7
In
teg
rin
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3****
****
C C R 9
CC
R9
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
5
1 0
1 5 **
***F E
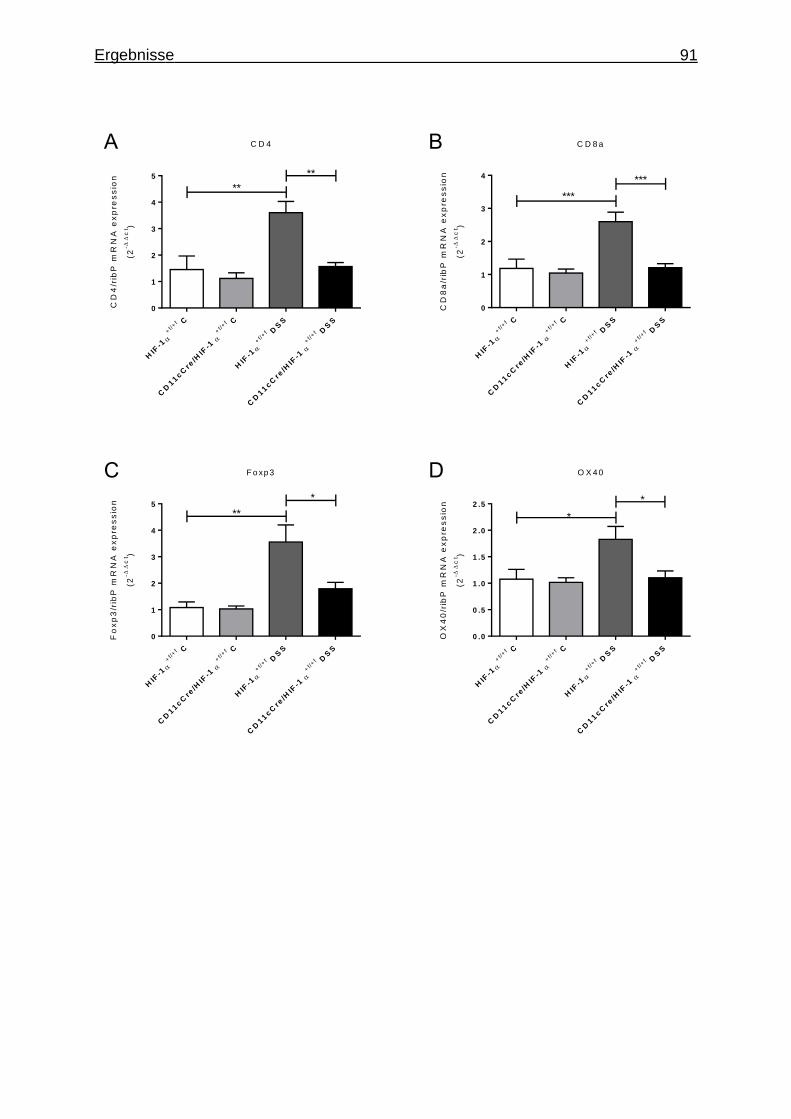
Ergebnisse
91
C D 4
CD
4/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4
5 **
**
C D 8 a
CD
8a
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4 ***
***
A B
F o xp3
Fo
xp
3/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4
5
**
*
O X 4 0
OX
40
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0 .0
0 .5
1 .0
1 .5
2 .0
2 .5
*
*
C D
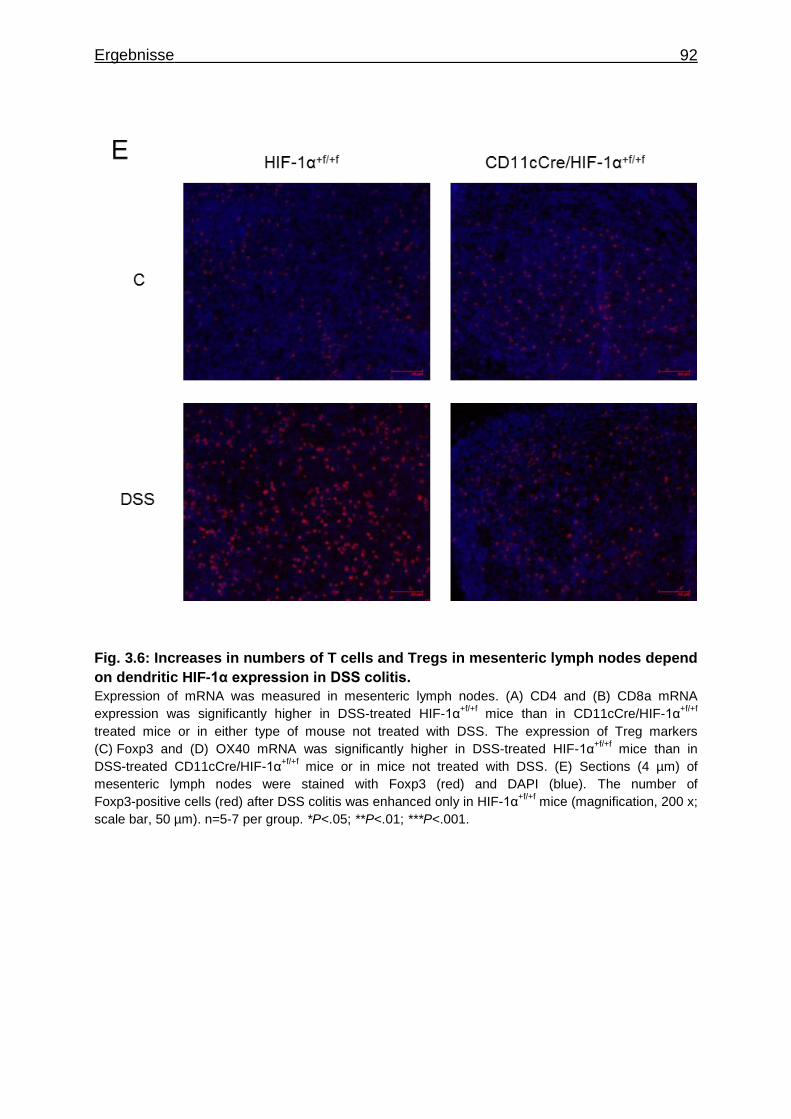
Ergebnisse
92
Fig. 3.6: Increases in numbers of T cells and Tregs in mesenteric lymph nodes depend
on dendritic HIF-1α expression in DSS colitis.
Expression of mRNA was measured in mesenteric lymph nodes. (A) CD4 and (B) CD8a mRNA
expression was significantly higher in DSS-treated HIF-1α+f/+f
mice than in CD11cCre/HIF-1α+f/+f
treated mice or in either type of mouse not treated with DSS. The expression of Treg markers
(C) Foxp3 and (D) OX40 mRNA was significantly higher in DSS-treated HIF-1α+f/+f
mice than in
DSS-treated CD11cCre/HIF-1α+f/+f
mice or in mice not treated with DSS. (E) Sections (4 µm) of
mesenteric lymph nodes were stained with Foxp3 (red) and DAPI (blue). The number of
Foxp3-positive cells (red) after DSS colitis was enhanced only in HIF-1α+f/+f
mice (magnification, 200 x;
scale bar, 50 µm). n=5-7 per group. *P<.05; **P<.01; ***P<.001.
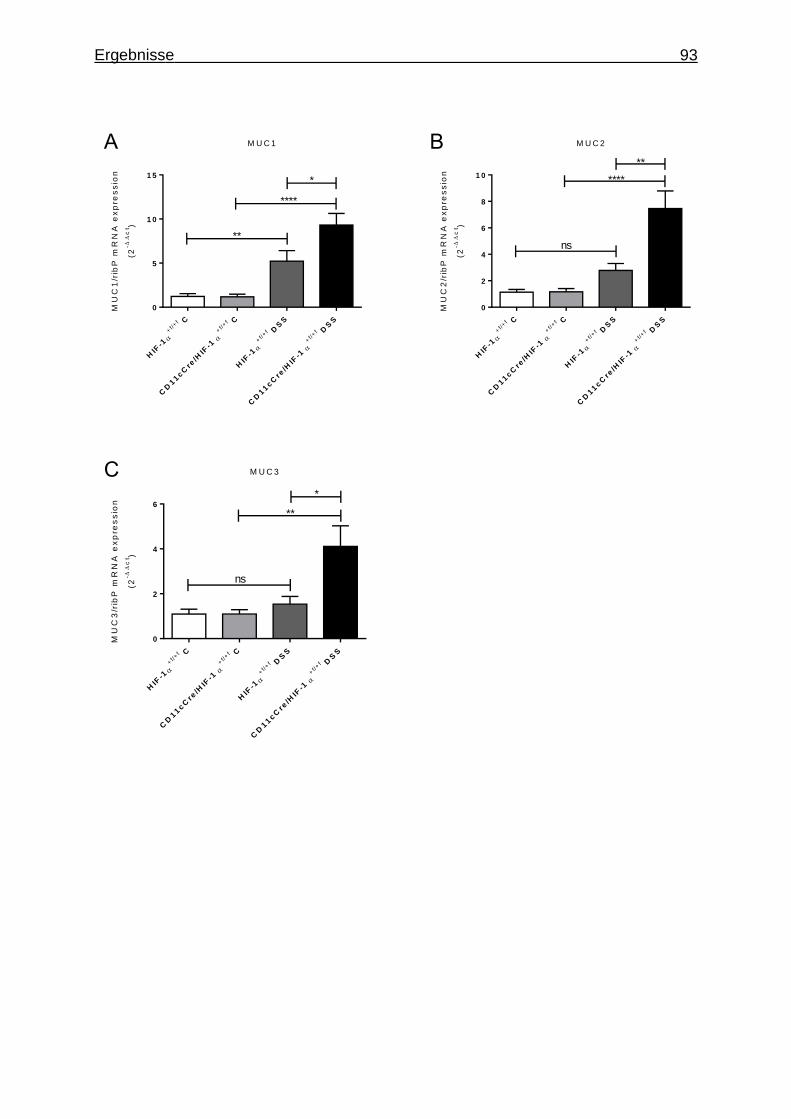
Ergebnisse
93
M U C 1
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
5
1 0
1 5
MU
C1
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
****
**
*
M U C 2
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
8
1 0
MU
C2
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
****
ns
**
B A
M U C 3
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
MU
C3
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
**
ns
*
C
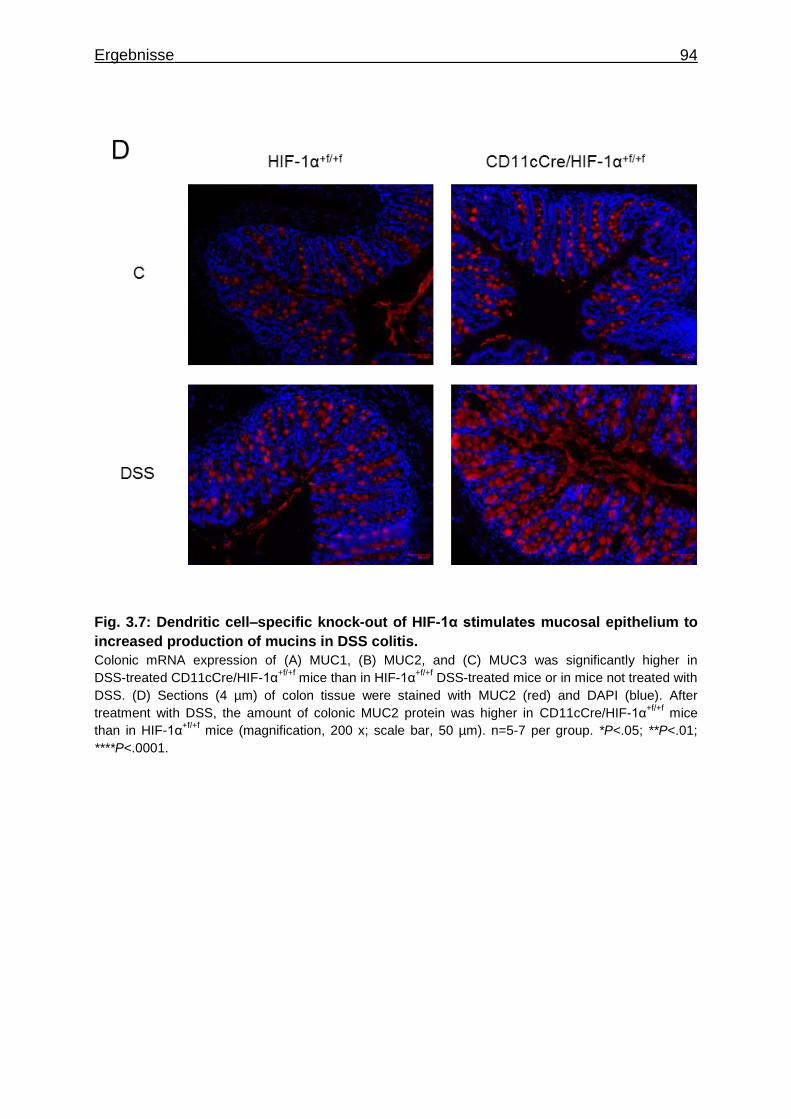
Ergebnisse
94
Fig. 3.7: Dendritic cell–specific knock-out of HIF-1α stimulates mucosal epithelium to
increased production of mucins in DSS colitis.
Colonic mRNA expression of (A) MUC1, (B) MUC2, and (C) MUC3 was significantly higher in
DSS-treated CD11cCre/HIF-1α+f/+f
mice than in HIF-1α+f/+f
DSS-treated mice or in mice not treated with
DSS. (D) Sections (4 µm) of colon tissue were stained with MUC2 (red) and DAPI (blue). After
treatment with DSS, the amount of colonic MUC2 protein was higher in CD11cCre/HIF-1α+f/+f
mice
than in HIF-1α+f/+f
mice (magnification, 200 x; scale bar, 50 µm). n=5-7 per group. *P<.05; **P<.01;
****P<.0001.
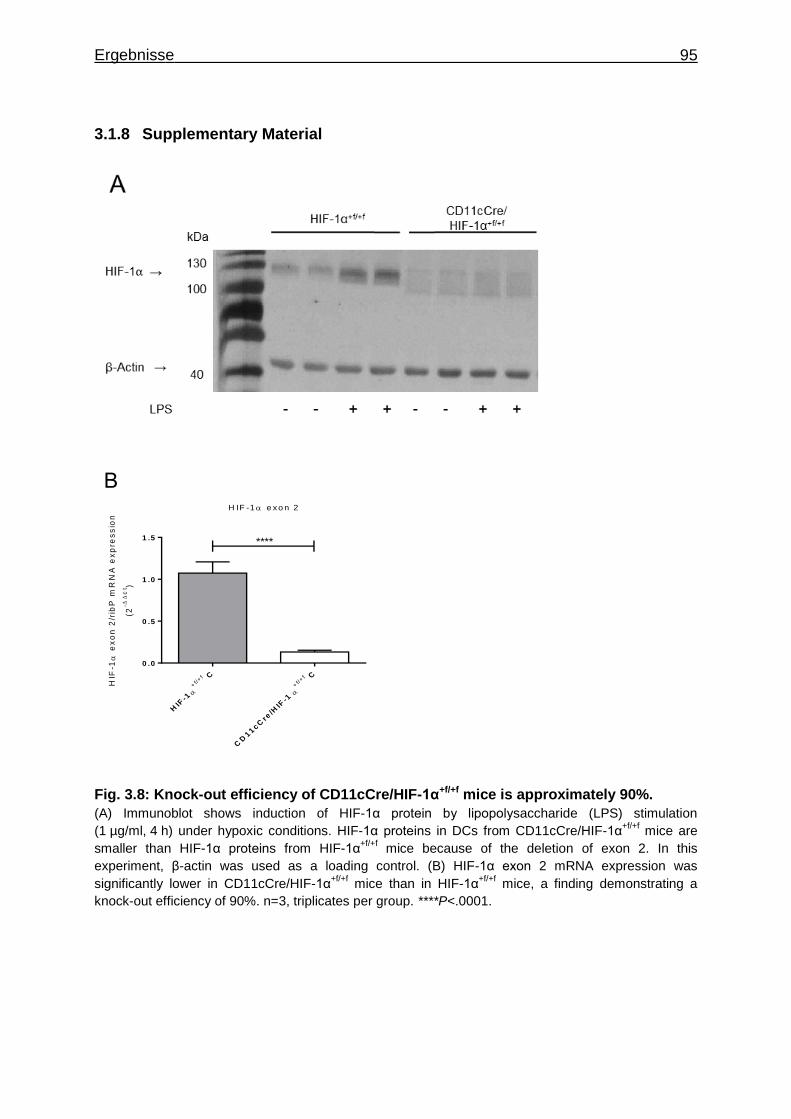
Ergebnisse
95
3.1.8 Supplementary Material
Fig. 3.8: Knock-out efficiency of CD11cCre/HIF-1α+f/+f mice is approximately 90%.
(A) Immunoblot shows induction of HIF-1α protein by lipopolysaccharide (LPS) stimulation
(1 µg/ml, 4 h) under hypoxic conditions. HIF-1α proteins in DCs from CD11cCre/HIF-1α+f/+f
mice are
smaller than HIF-1α proteins from HIF-1α+f/+f
mice because of the deletion of exon 2. In this
experiment, β-actin was used as a loading control. (B) HIF-1α exon 2 mRNA expression was
significantly lower in CD11cCre/HIF-1α+f/+f
mice than in HIF-1α+f/+f
mice, a finding demonstrating a
knock-out efficiency of 90%. n=3, triplicates per group. ****P<.0001.
H IF -1 e x o n 2
HIF
-1
ex
on
2/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
HIF
-1+
f/+
f C
CD
11cC
re/H
IF-1
+
f/+
f C
0 .0
0 .5
1 .0
1 .5 ****
B
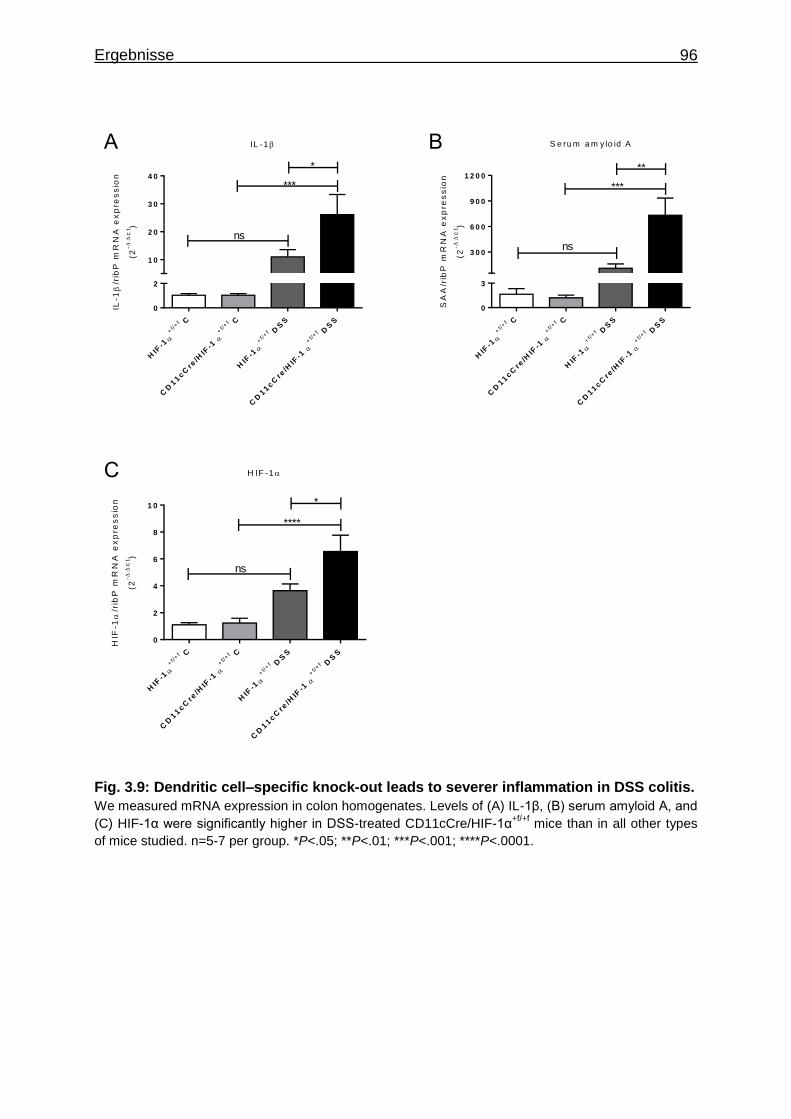
Ergebnisse
96
Fig. 3.9: Dendritic cell–specific knock-out leads to severer inflammation in DSS colitis.
We measured mRNA expression in colon homogenates. Levels of (A) IL-1β, (B) serum amyloid A, and
(C) HIF-1α were significantly higher in DSS-treated CD11cCre/HIF-1α+f/+f
mice than in all other types
of mice studied. n=5-7 per group. *P<.05; **P<.01; ***P<.001; ****P<.0001.
IL -1
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
1 0
2 0
3 0
4 0
IL-1
/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
***
ns
*
A S e ru m a m y lo id A
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
3
3 0 0
6 0 0
9 0 0
1 2 0 0
SA
A/r
ibP
mR
NA
ex
pre
ss
ion
(2-
c
t )
***
ns
**
B
H IF -1
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
8
1 0
HIF
-1
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
****
ns
*
C
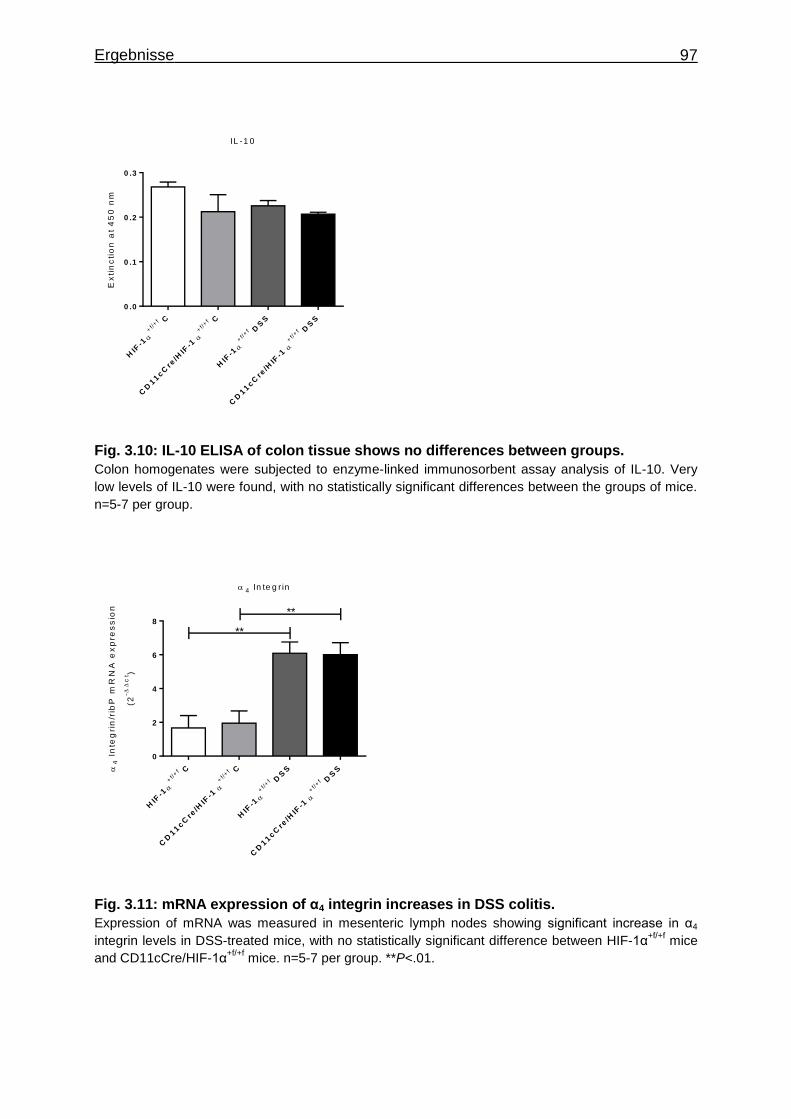
Ergebnisse
97
Fig. 3.10: IL-10 ELISA of colon tissue shows no differences between groups.
Colon homogenates were subjected to enzyme-linked immunosorbent assay analysis of IL-10. Very
low levels of IL-10 were found, with no statistically significant differences between the groups of mice.
n=5-7 per group.
Fig. 3.11: mRNA expression of α4 integrin increases in DSS colitis.
Expression of mRNA was measured in mesenteric lymph nodes showing significant increase in α4
integrin levels in DSS-treated mice, with no statistically significant difference between HIF-1α+f/+f
mice
and CD11cCre/HIF-1α+f/+f
mice. n=5-7 per group. **P<.01.
IL -1 0
Ex
tin
cti
on
at
45
0 n
m
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0 .0
0 .1
0 .2
0 .3
4 In te g r in
4
In
teg
rin
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
2
4
6
8
**
**
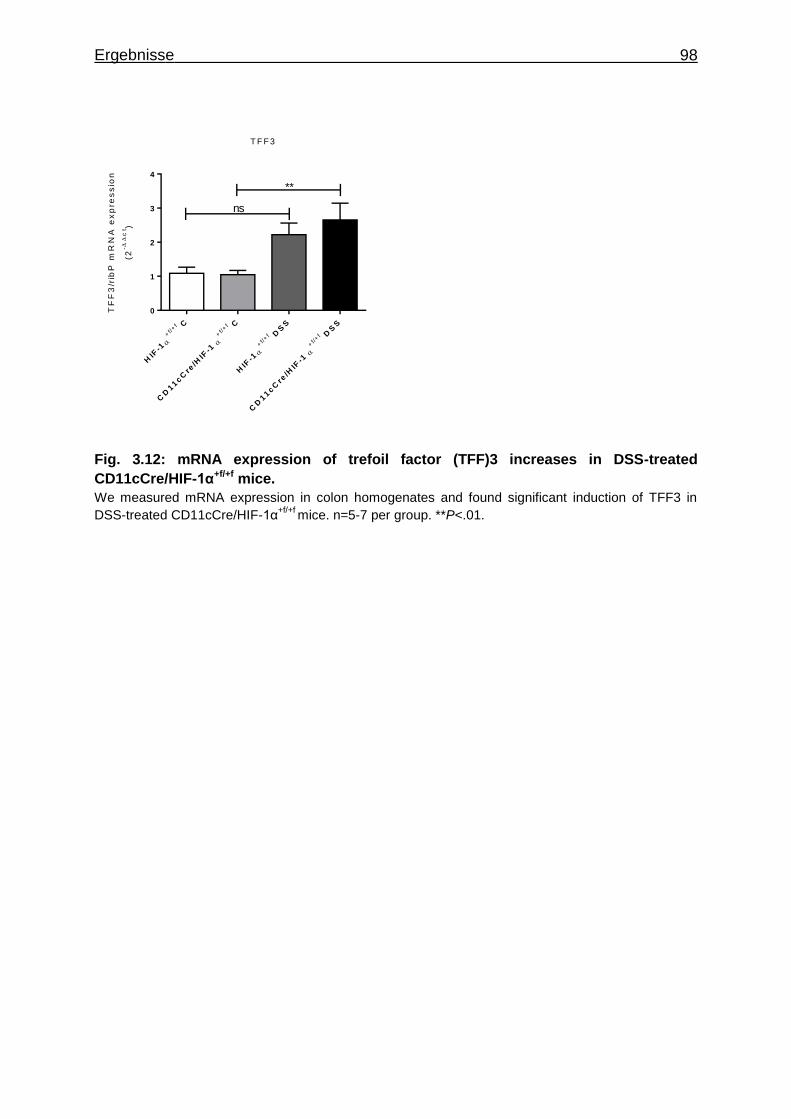
Ergebnisse
98
Fig. 3.12: mRNA expression of trefoil factor (TFF)3 increases in DSS-treated
CD11cCre/HIF-1α+f/+f mice.
We measured mRNA expression in colon homogenates and found significant induction of TFF3 in
DSS-treated CD11cCre/HIF-1α+f/+f
mice. n=5-7 per group. **P<.01.
T F F 3
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1
2
3
4
TF
F3
/rib
P m
RN
A e
xp
res
sio
n
(2-
c
t )
**
ns
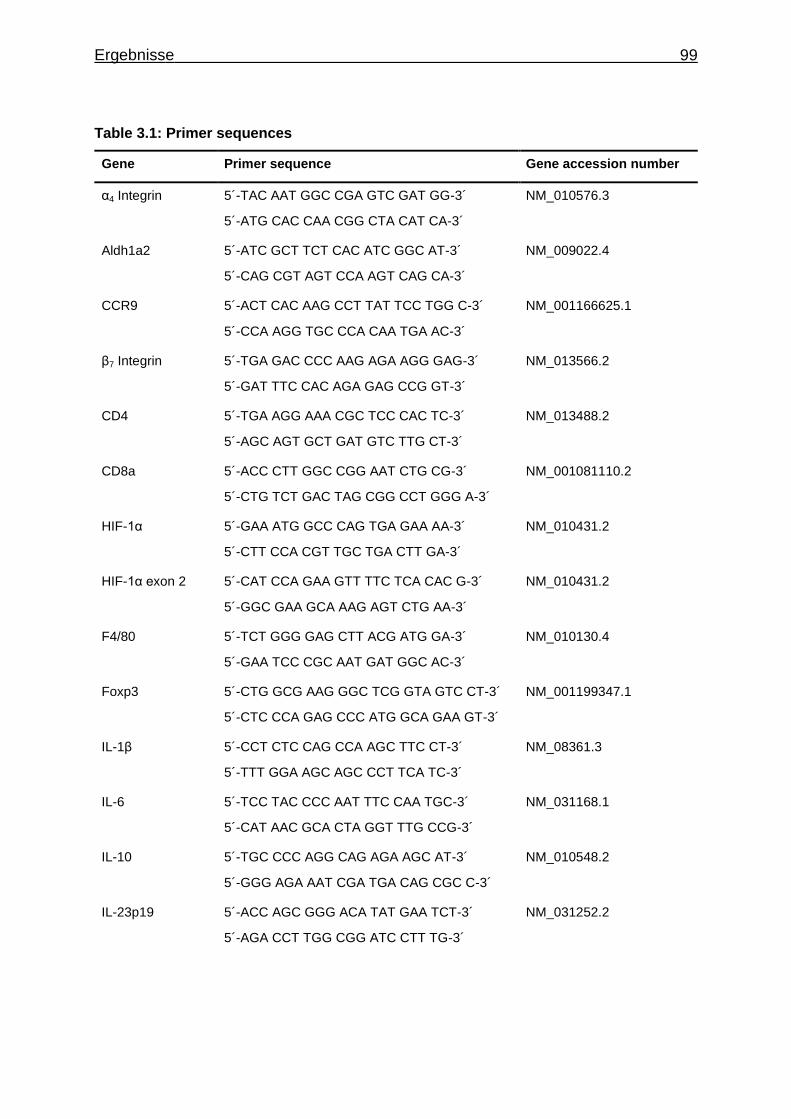
Ergebnisse
99
Table 3.1: Primer sequences
Gene Primer sequence Gene accession number
α4 Integrin 5´-TAC AAT GGC CGA GTC GAT GG-3´
5´-ATG CAC CAA CGG CTA CAT CA-3´
NM_010576.3
Aldh1a2 5´-ATC GCT TCT CAC ATC GGC AT-3´
5´-CAG CGT AGT CCA AGT CAG CA-3´
NM_009022.4
CCR9 5´-ACT CAC AAG CCT TAT TCC TGG C-3´
5´-CCA AGG TGC CCA CAA TGA AC-3´
NM_001166625.1
β7 Integrin 5´-TGA GAC CCC AAG AGA AGG GAG-3´
5´-GAT TTC CAC AGA GAG CCG GT-3´
NM_013566.2
CD4 5´-TGA AGG AAA CGC TCC CAC TC-3´
5´-AGC AGT GCT GAT GTC TTG CT-3´
NM_013488.2
CD8a 5´-ACC CTT GGC CGG AAT CTG CG-3´
5´-CTG TCT GAC TAG CGG CCT GGG A-3´
NM_001081110.2
HIF-1α 5´-GAA ATG GCC CAG TGA GAA AA-3´
5´-CTT CCA CGT TGC TGA CTT GA-3´
NM_010431.2
HIF-1α exon 2 5´-CAT CCA GAA GTT TTC TCA CAC G-3´
5´-GGC GAA GCA AAG AGT CTG AA-3´
NM_010431.2
F4/80 5´-TCT GGG GAG CTT ACG ATG GA-3´
5´-GAA TCC CGC AAT GAT GGC AC-3´
NM_010130.4
Foxp3 5´-CTG GCG AAG GGC TCG GTA GTC CT-3´
5´-CTC CCA GAG CCC ATG GCA GAA GT-3´
NM_001199347.1
IL-1β 5´-CCT CTC CAG CCA AGC TTC CT-3´
5´-TTT GGA AGC AGC CCT TCA TC-3´
NM_08361.3
IL-6 5´-TCC TAC CCC AAT TTC CAA TGC-3´
5´-CAT AAC GCA CTA GGT TTG CCG-3´
NM_031168.1
IL-10 5´-TGC CCC AGG CAG AGA AGC AT-3´
5´-GGG AGA AAT CGA TGA CAG CGC C-3´
NM_010548.2
IL-23p19 5´-ACC AGC GGG ACA TAT GAA TCT-3´
5´-AGA CCT TGG CGG ATC CTT TG-3´
NM_031252.2
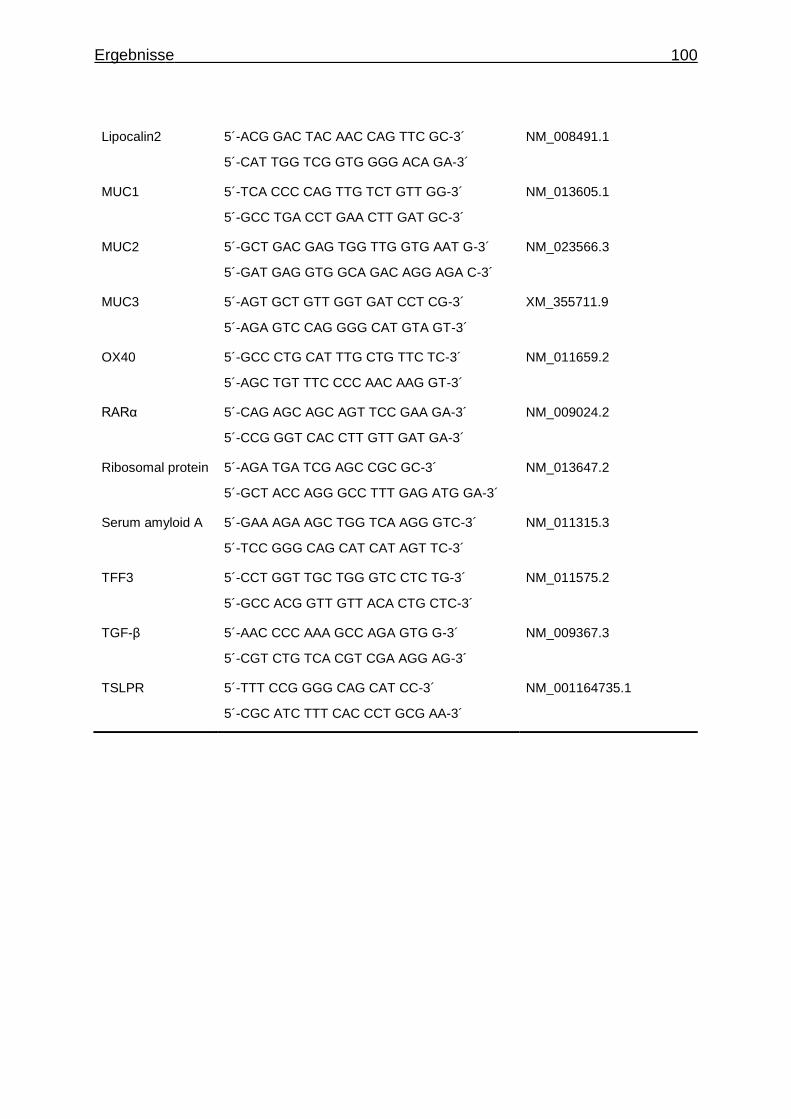
Ergebnisse
100
Lipocalin2 5´-ACG GAC TAC AAC CAG TTC GC-3´
5´-CAT TGG TCG GTG GGG ACA GA-3´
NM_008491.1
MUC1 5´-TCA CCC CAG TTG TCT GTT GG-3´
5´-GCC TGA CCT GAA CTT GAT GC-3´
NM_013605.1
MUC2 5´-GCT GAC GAG TGG TTG GTG AAT G-3´
5´-GAT GAG GTG GCA GAC AGG AGA C-3´
NM_023566.3
MUC3 5´-AGT GCT GTT GGT GAT CCT CG-3´
5´-AGA GTC CAG GGG CAT GTA GT-3´
XM_355711.9
OX40 5´-GCC CTG CAT TTG CTG TTC TC-3´
5´-AGC TGT TTC CCC AAC AAG GT-3´
NM_011659.2
RARα 5´-CAG AGC AGC AGT TCC GAA GA-3´
5´-CCG GGT CAC CTT GTT GAT GA-3´
NM_009024.2
Ribosomal protein 5´-AGA TGA TCG AGC CGC GC-3´
5´-GCT ACC AGG GCC TTT GAG ATG GA-3´
NM_013647.2
Serum amyloid A 5´-GAA AGA AGC TGG TCA AGG GTC-3´
5´-TCC GGG CAG CAT CAT AGT TC-3´
NM_011315.3
TFF3 5´-CCT GGT TGC TGG GTC CTC TG-3´
5´-GCC ACG GTT GTT ACA CTG CTC-3´
NM_011575.2
TGF-β 5´-AAC CCC AAA GCC AGA GTG G-3´
5´-CGT CTG TCA CGT CGA AGG AG-3´
NM_009367.3
TSLPR 5´-TTT CCG GGG CAG CAT CC-3´
5´-CGC ATC TTT CAC CCT GCG AA-3´
NM_001164735.1

Ergebnisse
101
3.2 Weitere Präsentationen
Vortrag
Flück K., (2012). Die Bedeutung der Hypoxie-induzierbaren Faktoren bei chronisch
entzündlichen Darmerkrankungen. 18. Workshop Zell- und Gewebeschädigung:
Mechanismen, Protektion und Therapie, Xanten.
Poster
1. Flück K., Winning S. (2011). Die Bedeutung der Hypoxie-induzierbaren
Faktoren bei chronisch entzündlichen Darmerkrankungen. 10. Forschungstag
der Medizinischen Fakultät, Essen, Abstractband S. 27.
2. Flück K., Fandrey J., Winning S. (2012). The role of hypoxia-inducible factors
in inflammatory bowel disease. 91st Annual Meeting, Deutsche Physiologische
Gesellschaft, Dresden, Abstractband S. 197.
3. Flück K., Breves G., Fandrey J., Winning S. (2012). The role of hypoxia-
inducible factors in inflammatory bowel disease. EPO Meeting Luebeck,
Abstractband S. 29.
4. Flück K., Fandrey J., Winning S. (2012). The role of hypoxia-inducible factors
in inflammatory bowel disease. 11. Forschungstag der Medizinischen Fakultät,
Essen, Abstractband S. 42.
5. Flück K., Fandrey J., Winning S. (2013). The role of hypoxia-inducible factors
in inflammatory bowel disease. 92st Annual Meeting, Deutsche Physiologische
Gesellschaft, Heidelberg, Abstractband S.167.
6. Flück K., Fandrey J., Breves G., Winning S. (2013). Bedeutung des
Hypoxie-induzierbaren Faktors in dendritischen Zellen im Rahmen einer DSS
induzierten akuten Colitis in vivo. 12. Forschungstag der Medizinischen
Fakultät, Essen, Abstractband S. 30.
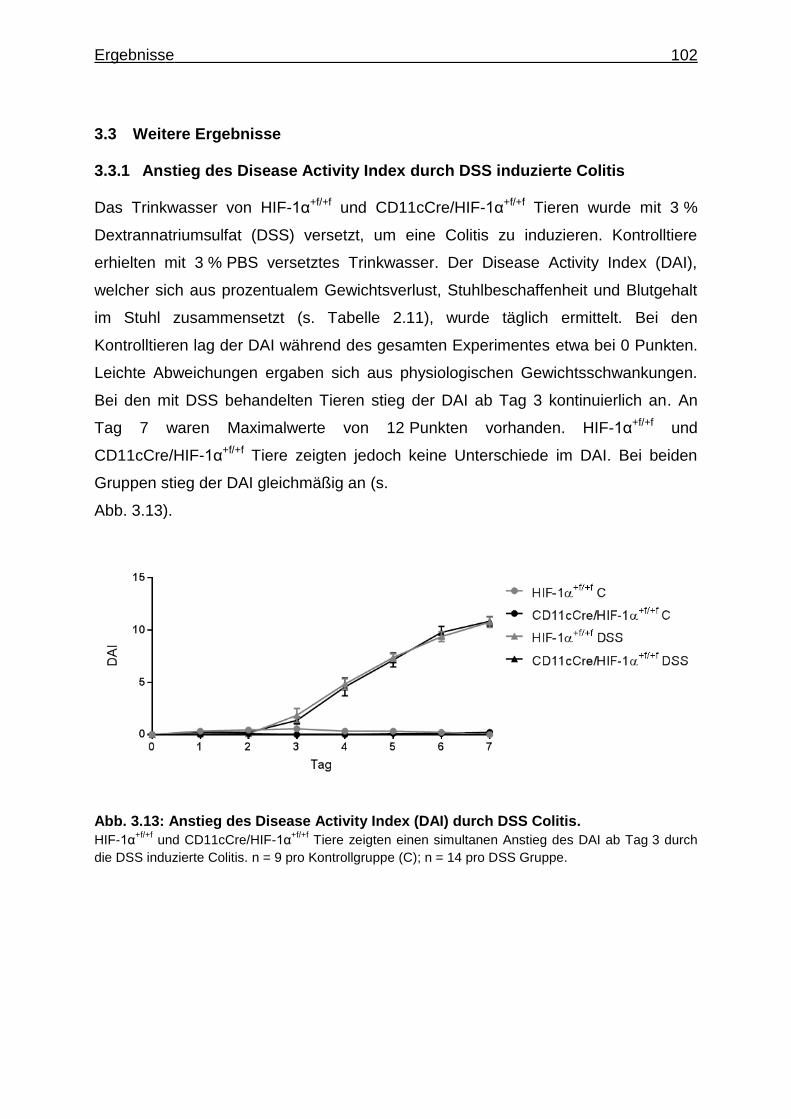
Ergebnisse
102
3.3 Weitere Ergebnisse
3.3.1 Anstieg des Disease Activity Index durch DSS induzierte Colitis
Das Trinkwasser von HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f Tieren wurde mit 3 %
Dextrannatriumsulfat (DSS) versetzt, um eine Colitis zu induzieren. Kontrolltiere
erhielten mit 3 % PBS versetztes Trinkwasser. Der Disease Activity Index (DAI),
welcher sich aus prozentualem Gewichtsverlust, Stuhlbeschaffenheit und Blutgehalt
im Stuhl zusammensetzt (s. Tabelle 2.11), wurde täglich ermittelt. Bei den
Kontrolltieren lag der DAI während des gesamten Experimentes etwa bei 0 Punkten.
Leichte Abweichungen ergaben sich aus physiologischen Gewichtsschwankungen.
Bei den mit DSS behandelten Tieren stieg der DAI ab Tag 3 kontinuierlich an. An
Tag 7 waren Maximalwerte von 12 Punkten vorhanden. HIF-1α+f/+f und
CD11cCre/HIF-1α+f/+f Tiere zeigten jedoch keine Unterschiede im DAI. Bei beiden
Gruppen stieg der DAI gleichmäßig an (s.
Abb. 3.13).
Abb. 3.13: Anstieg des Disease Activity Index (DAI) durch DSS Colitis.
HIF-1α+f/+f
und CD11cCre/HIF-1α+f/+f
Tiere zeigten einen simultanen Anstieg des DAI ab Tag 3 durch
die DSS induzierte Colitis. n = 9 pro Kontrollgruppe (C); n = 14 pro DSS Gruppe.
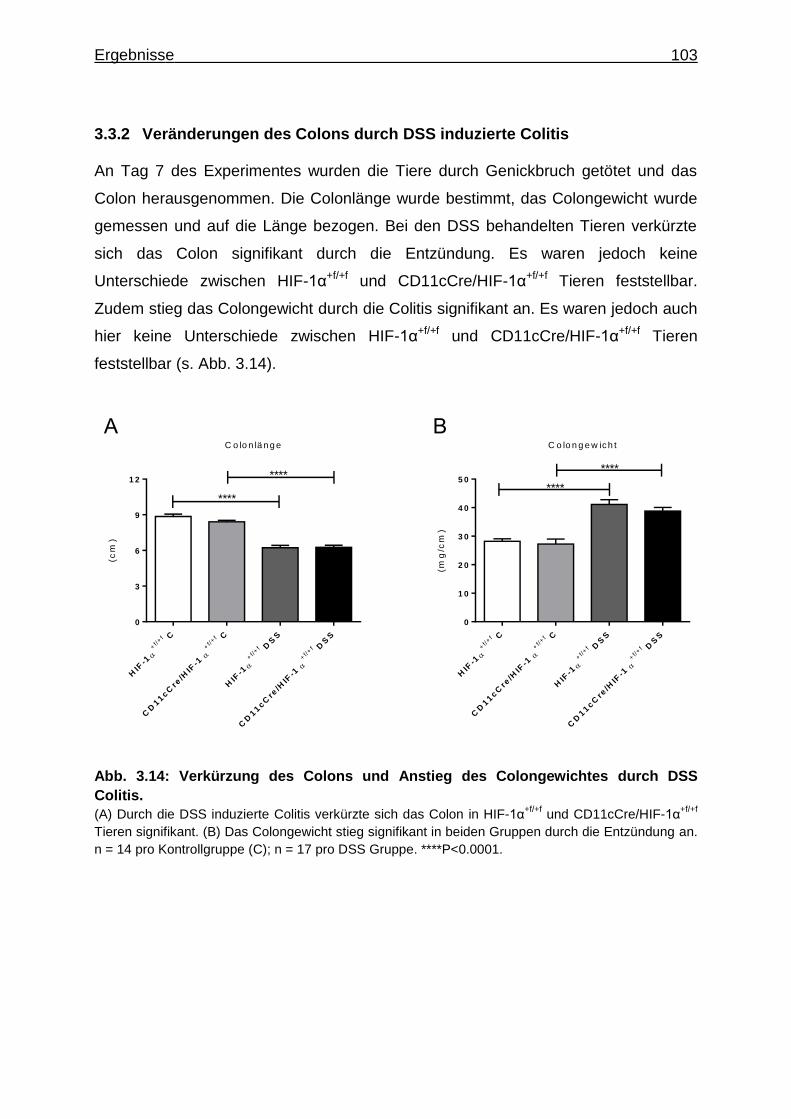
Ergebnisse
103
3.3.2 Veränderungen des Colons durch DSS induzierte Colitis
An Tag 7 des Experimentes wurden die Tiere durch Genickbruch getötet und das
Colon herausgenommen. Die Colonlänge wurde bestimmt, das Colongewicht wurde
gemessen und auf die Länge bezogen. Bei den DSS behandelten Tieren verkürzte
sich das Colon signifikant durch die Entzündung. Es waren jedoch keine
Unterschiede zwischen HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f Tieren feststellbar.
Zudem stieg das Colongewicht durch die Colitis signifikant an. Es waren jedoch auch
hier keine Unterschiede zwischen HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f Tieren
feststellbar (s. Abb. 3.14).
Abb. 3.14: Verkürzung des Colons und Anstieg des Colongewichtes durch DSS
Colitis.
(A) Durch die DSS induzierte Colitis verkürzte sich das Colon in HIF-1α+f/+f
und CD11cCre/HIF-1α+f/+f
Tieren signifikant. (B) Das Colongewicht stieg signifikant in beiden Gruppen durch die Entzündung an.
n = 14 pro Kontrollgruppe (C); n = 17 pro DSS Gruppe. ****P<0.0001.
C o lo n g e w ic h t
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
1 0
2 0
3 0
4 0
5 0
(mg
/cm
)
****
****
C o lo n lä n g e
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
3
6
9
1 2
(cm
)
****
****
A B

Ergebnisse
104
3.3.3 Hochgradige Entzündung des Colons durch DSS Behandlung
Das distale Colon wurde im Querschnitt in Paraffin eingebettet und in 4 µm dicke
Schnitte geschnitten, welche zur Beurteilung der Entzündung mit H&E gefärbt
wurden. Die Kontrolltiere wiesen einen physiologischen Aufbau des Colons auf. Das
Epithel der Tunica mucosa war intakt und die Darmkrypten waren zu erkennen. In
der Lamina propria mucosae waren nur vereinzelte Leukozyten zu finden. HIF-1α+f/+f
und CD11cCre/HIF-1α+f/+f Kontrolltiere zeigten keine Unterschiede im Aufbau des
Colons. Durch die Gabe von 3 % DSS für 7 Tage entwickelte sich eine hochgradige
Entzündung im Colon (s. Abb. 3.15). Es kam zu einer massiven Infiltration von
Leukozyten in die Lamina propria mucosae mit Zerstörung des Epithels und der
Krypten. Die Tela submucosa wies eine Ödematisierung mit deutlicher
Leukozyteninfiltration auf. Auch die Tunica muscularis war verdickt und mit einzelnen
Leukozyten durchsetzt. Bei der Betrachtung der Schnitte war kein Unterschied in
dem Ausmaß der Entzündung zwischen HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f
DSS behandelten Tieren feststellbar. Die Colonquerschnitte wurden daraufhin von
zwei verblindeten Untersuchern nach einem vorgegebenen Score (s. Tabelle 2.21)
ausgewertet und die Ergebnisse in Abb. 3.16 dargestellt. Die Kontrolltiere erhielten
für die jeweiligen Parameter jeweils 0 Punkte, da keinerlei Entzündungszeichen
vorhanden waren. Bei den DSS behandelten Tieren lag ein durchschnittlicher Score
von ca. 14 Punkten vor, was eine deutliche Entzündung beschreibt. Zwischen
HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f DSS behandelten Tieren war jedoch auch
mithilfe der Auswertung des histologischen Score kein Unterschied im
Entzündungsgrad erkennbar.
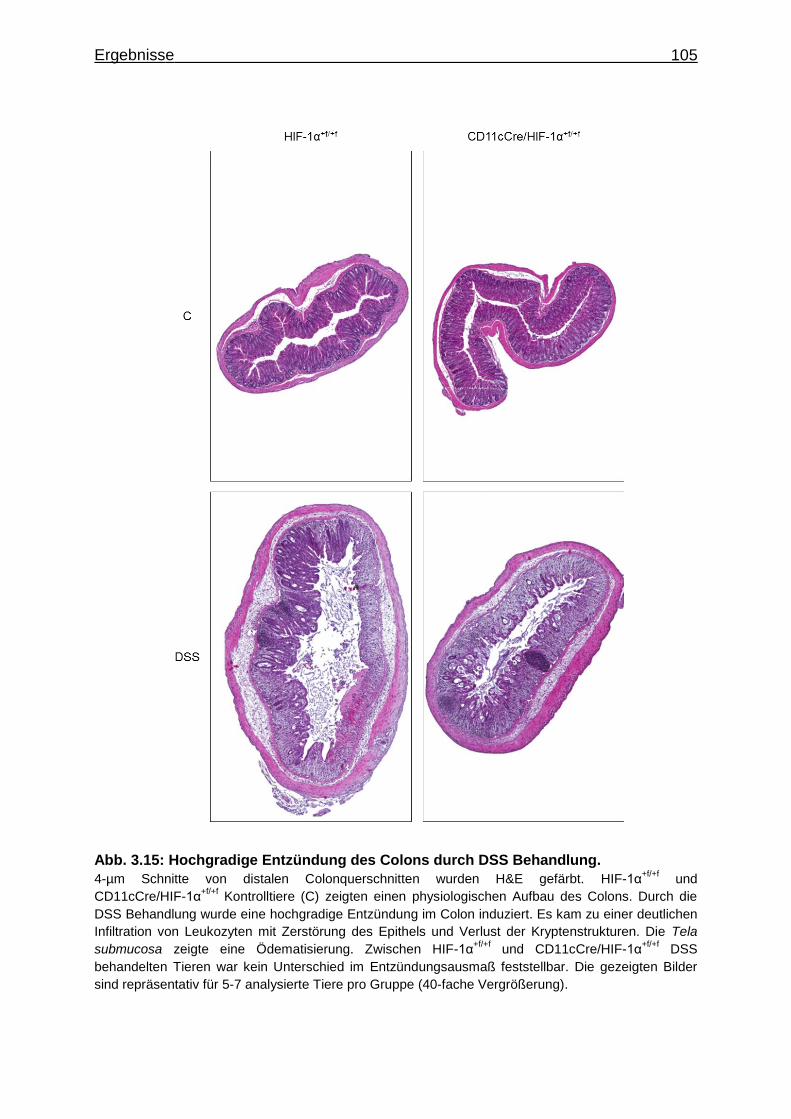
Ergebnisse
105
Abb. 3.15: Hochgradige Entzündung des Colons durch DSS Behandlung.
4-µm Schnitte von distalen Colonquerschnitten wurden H&E gefärbt. HIF-1α+f/+f
und
CD11cCre/HIF-1α+f/+f
Kontrolltiere (C) zeigten einen physiologischen Aufbau des Colons. Durch die
DSS Behandlung wurde eine hochgradige Entzündung im Colon induziert. Es kam zu einer deutlichen
Infiltration von Leukozyten mit Zerstörung des Epithels und Verlust der Kryptenstrukturen. Die Tela
submucosa zeigte eine Ödematisierung. Zwischen HIF-1α+f/+f
und CD11cCre/HIF-1α+f/+f
DSS
behandelten Tieren war kein Unterschied im Entzündungsausmaß feststellbar. Die gezeigten Bilder
sind repräsentativ für 5-7 analysierte Tiere pro Gruppe (40-fache Vergrößerung).
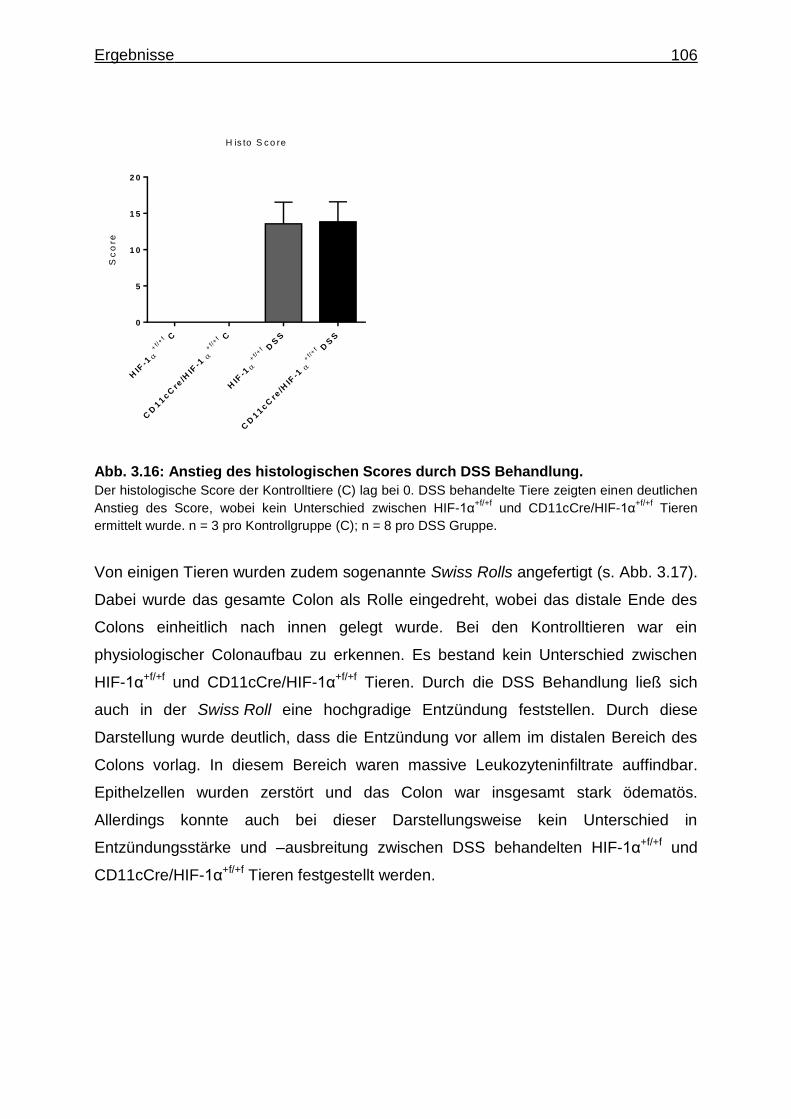
Ergebnisse
106
Abb. 3.16: Anstieg des histologischen Scores durch DSS Behandlung.
Der histologische Score der Kontrolltiere (C) lag bei 0. DSS behandelte Tiere zeigten einen deutlichen
Anstieg des Score, wobei kein Unterschied zwischen HIF-1α+f/+f
und CD11cCre/HIF-1α+f/+f
Tieren
ermittelt wurde. n = 3 pro Kontrollgruppe (C); n = 8 pro DSS Gruppe.
Von einigen Tieren wurden zudem sogenannte Swiss Rolls angefertigt (s. Abb. 3.17).
Dabei wurde das gesamte Colon als Rolle eingedreht, wobei das distale Ende des
Colons einheitlich nach innen gelegt wurde. Bei den Kontrolltieren war ein
physiologischer Colonaufbau zu erkennen. Es bestand kein Unterschied zwischen
HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f Tieren. Durch die DSS Behandlung ließ sich
auch in der Swiss Roll eine hochgradige Entzündung feststellen. Durch diese
Darstellung wurde deutlich, dass die Entzündung vor allem im distalen Bereich des
Colons vorlag. In diesem Bereich waren massive Leukozyteninfiltrate auffindbar.
Epithelzellen wurden zerstört und das Colon war insgesamt stark ödematös.
Allerdings konnte auch bei dieser Darstellungsweise kein Unterschied in
Entzündungsstärke und –ausbreitung zwischen DSS behandelten HIF-1α+f/+f und
CD11cCre/HIF-1α+f/+f Tieren festgestellt werden.
H is to S c o re
HIF
-1+f/+f C
CD
11cC
re/H
IF-1+f/+f C
HIF
-1+f/+f D
SS
CD
11cC
re/H
IF-1+f/+f D
SS
0
5
1 0
1 5
2 0
Sc
ore
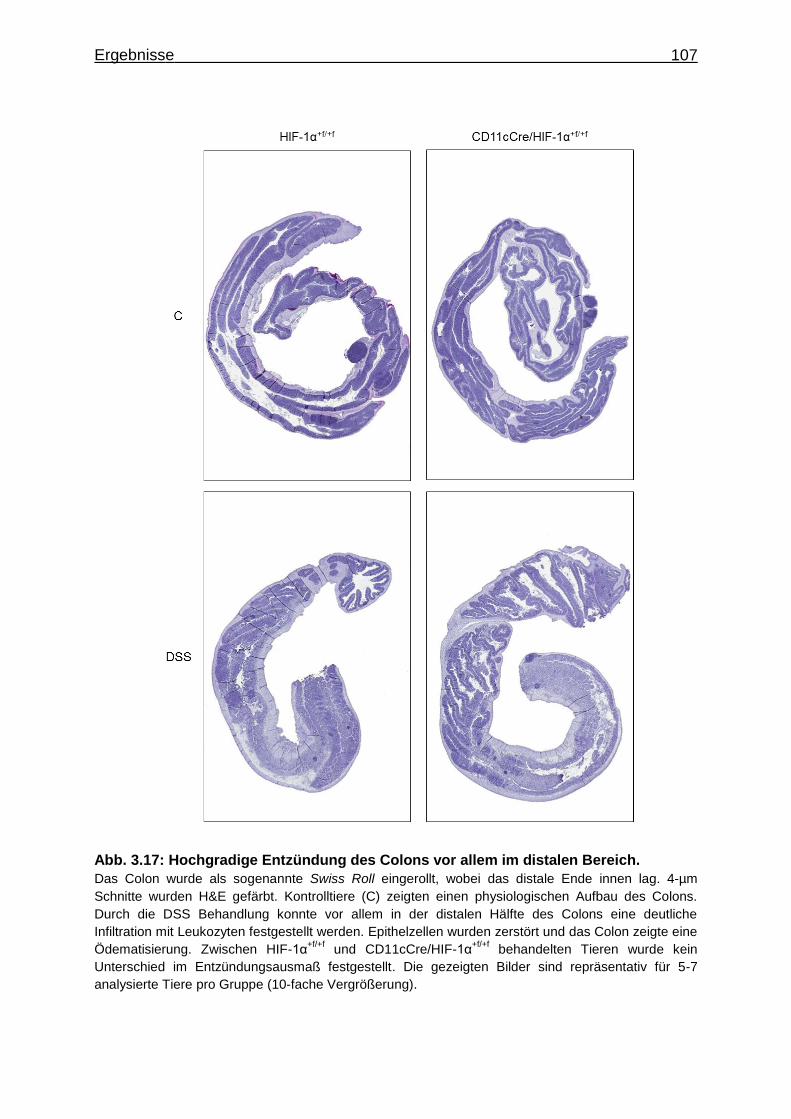
Ergebnisse
107
Abb. 3.17: Hochgradige Entzündung des Colons vor allem im distalen Bereich.
Das Colon wurde als sogenannte Swiss Roll eingerollt, wobei das distale Ende innen lag. 4-µm
Schnitte wurden H&E gefärbt. Kontrolltiere (C) zeigten einen physiologischen Aufbau des Colons.
Durch die DSS Behandlung konnte vor allem in der distalen Hälfte des Colons eine deutliche
Infiltration mit Leukozyten festgestellt werden. Epithelzellen wurden zerstört und das Colon zeigte eine
Ödematisierung. Zwischen HIF-1α+f/+f
und CD11cCre/HIF-1α+f/+f
behandelten Tieren wurde kein
Unterschied im Entzündungsausmaß festgestellt. Die gezeigten Bilder sind repräsentativ für 5-7
analysierte Tiere pro Gruppe (10-fache Vergrößerung).

Ergebnisse
108
3.3.4 Verstärkte Alcianblau Färbung in DSS behandelten CD11cCre/HIF-1α+f/+f
Tieren
4-µm dicke Schnitte von distalen Colonquerschnitten wurden mit Alcianblau gefärbt
(s. Abb. 3.18). Becherzellen im Colon produzieren Mucine, welche aus sauren
Mucopolysacchariden bestehen und durch Alcianblau dunkelblau angefärbt werden.
In den Kontrolltieren wurde ein physiologischer Aufbau des Colons festgestellt.
Alcianblau färbte hierbei vor allem Strukturen in tiefer gelegenen Krypten an. Die
Färbung war sehr regelmäßig. Zwischen HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f
Kontrolltieren war kein Unterschied erkennbar. In den DSS behandelten Tieren
konnte man eine deutliche Änderung des Colonaufbaus erkennen. Die Alcianblau
Färbung war zudem eher vesikulär und zum Lumen orientiert. In den Knock-out
Tieren zeigten sich vermehrt positive Strukturen und die Färbung war deutlich
stärker.
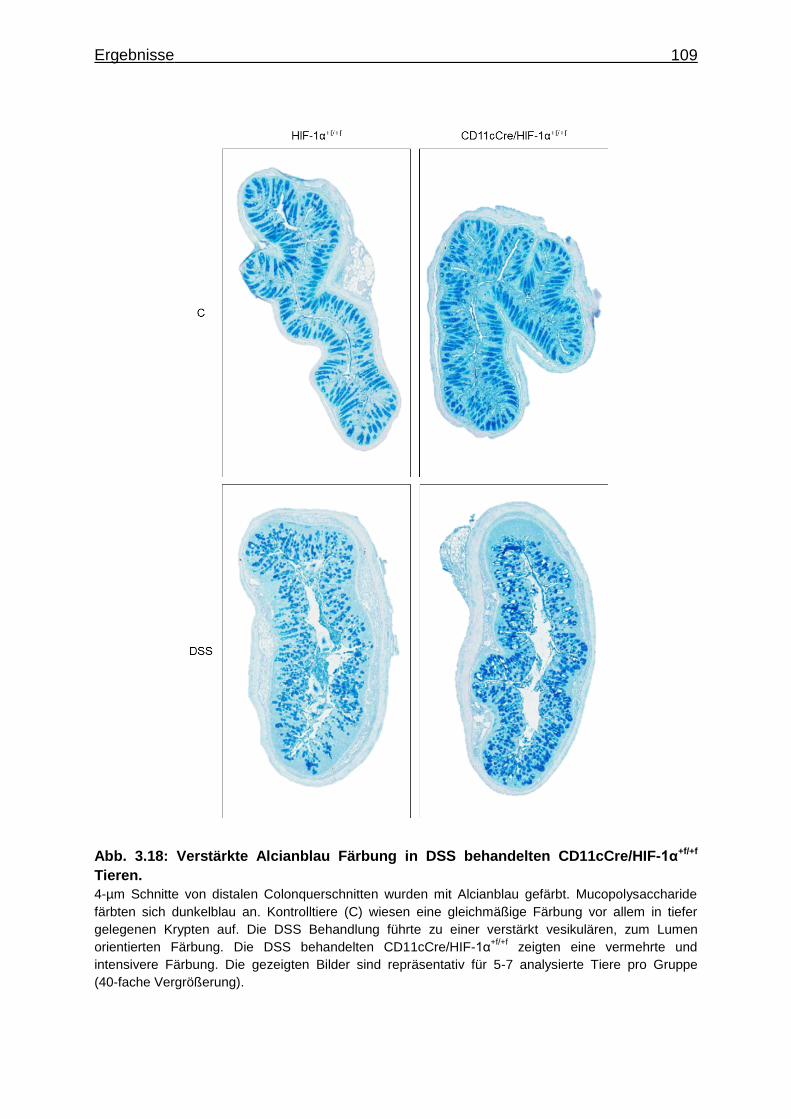
Ergebnisse
109
Abb. 3.18: Verstärkte Alcianblau Färbung in DSS behandelten CD11cCre/HIF-1α+f/+f
Tieren.
4-µm Schnitte von distalen Colonquerschnitten wurden mit Alcianblau gefärbt. Mucopolysaccharide
färbten sich dunkelblau an. Kontrolltiere (C) wiesen eine gleichmäßige Färbung vor allem in tiefer
gelegenen Krypten auf. Die DSS Behandlung führte zu einer verstärkt vesikulären, zum Lumen
orientierten Färbung. Die DSS behandelten CD11cCre/HIF-1α+f/+f
zeigten eine vermehrte und
intensivere Färbung. Die gezeigten Bilder sind repräsentativ für 5-7 analysierte Tiere pro Gruppe
(40-fache Vergrößerung).

Diskussion
110
4 Diskussion
4.1 Auswirkungen einer Colitis auf intestinale DCs
Als Bindeglied zwischen erworbener und angeborener Immunität übernehmen DCs
eine entscheidende Rolle in der intestinalen Immunantwort. Zudem haben sie die
wichtige Aufgabe, die Homöostase des Darms zu regulieren. Mehrere Studien
zeigten, dass entzündliche Prozesse im Darm Veränderungen im Sauerstoffhaushalt
bedingen, welche zu einer sich in das umliegende Gewebe ausbreitenden Hypoxie
führen (Taylor & Colgan 2007; Eltzschig & Carmeliet 2011). Der Transkriptionsfaktor
Hypoxie induzierbarer Faktor (HIF)-1 reguliert die zelluläre Adaptation an diese
sauerstoffarmen Bedingungen (Semenza et al. 1997). In intestinalen Epithelzellen
konnte bislang ein protektiver Effekt von HIF-1 dargestellt werden (Karhausen et al.
2004; Cummins et al. 2008; Tambuwala et al. 2010). Durch die intraperitoneale
Injektion von Pimonidazol, welches in hypoxischer Umgebung aktiviert wird, konnte
in der Immunfluoreszenz gezeigt werden, dass eine Colitis zu einer Ausbreitung der
Hypoxie führt (s. Fig. 3.1 B). Die Immunzellen innerhalb der Tunica mucosa und
Tela submucosa waren deutlich positiv in der Hypoxie Färbung, was auf eine
Aktivierung von HIF-1 schließen lässt. Aus diesem Grund wurde in der vorliegenden
Arbeit die Bedeutung von HIF-1α in DCs im Rahmen einer experimentellen Colitis
untersucht.
4.2 Entzündungsparameter HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f DSS
behandelter Tiere
In Kontrollmäusen (HIF-1α+f/+f) und Knock-out Mäusen ohne funktionelles HIF-1α in
DCs (CD11cCre/ HIF-1α+f/+f) wurde durch siebentägige Gabe von 3 % DSS eine
Colitis induziert. Die Knock-out Mäuse zeigten im Vergleich zu den Kontrollmäusen
einen signifikant größeren Gewichtsverlust an Tag 6 und 7 des Experiments (s. Fig.
3.2 A), wohingegen sich im Disease Activity Index (DAI) (s. Abb. 3.13), in der
Colonlänge und –gewicht (s. Abb. 3.14), sowie in der Histologie (s. Abb. 3.16) keine
Unterschiede zwischen den Gruppen feststellen ließen. Karhausen et al., welche

Diskussion
111
einen protektiven Effekt von epithelialem HIF-1α darstellten, konnten in ihren
Untersuchungen einen Unterschied in der Colonlänge zwischen den untersuchten
Gruppen feststellen. Hierbei handelte es sich jedoch um ein TNBS Colitismodell, bei
welchem TNBS rektal appliziert wurde (Karhausen et al. 2004). Dies könnte eventuell
zu deutlicheren Auswirkungen auf die Colonlänge geführt haben. Auch in weiteren
Studien, die sich mit der Rolle von epithelialem HIF während einer Colitis
beschäftigten, konnten Unterschiede zwischen den betreffenden Gruppen sowohl im
DAI, in der Colonlänge und –gewicht als auch in der Histologie festgestellt werden.
Allerdings wurden in diesen Studien sowohl andere DSS Konzentrationen für
geringere Zeiträume als auch Mäuse mit einem unterschiedlichen genetischen
Hintergrund benutzt (Cummins et al. 2008; Tambuwala et al. 2010). Die Gabe von
3 % DSS über 7 Tage ist für Mäuse mit C57BL/6J Hintergrund ein zuverlässiges
Modell der Colitis Induktion (Vowinkel et al. 2004). Wie man in der Histologie
erkennen kann, lag in den DSS behandelten Tieren eine äußerst hochgradige
Entzündung vor. Dies könnte dazu geführt haben, dass mögliche Unterschiede von
dem Schweregrad der Colitis überdeckt worden sind.
Auf mRNA Ebene ließ sich jedoch ein deutlicher Unterschied zwischen DSS
behandelten HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f Tieren feststellen. Die Expression
der proinflammatorischen Zytokine IL-6 und IL-23 zeigte eine deutliche Erhöhung in
den Knock-out Tieren (s. Fig. 3.2 B-C). Diese Zytokine haben dabei eine große
Bedeutung in murinen Colitismodellen und in der Pathogenese von CED. IL-6 wird
durch viele verschiedene Immunzellen produziert, unter anderem durch DCs,
Makrophagen und Th17-Zellen. In CED Patienten konnten erhöhte Werte von IL-6 in
der intestinalen Mukosa und im Serum sowie eine erhöhte dendritische IL-6
Produktion festgestellt werden (Gross et al. 1992; Hart et al. 2005; Eastaff-Leung et
al. 2010). Des Weiteren konnte durch Gabe eines anti-IL-6-Antikörpers die
Ausprägung einer Colitis in SCID Mäusen unterdrückt werden. Die Behandlung
führte zudem zu verringerten Expressionen von IFN-γ, TNF-α und IL-1β. Dies lässt
darauf schließen, dass auch für die Entwicklung einer proinflammatorischen
Th1-Immunantwort die Wirkung von IL-6 benötigt wird (Yamamoto et al. 2000). Es
konnte außerdem gezeigt werden, dass die Neutralisation des membrangebundenen

Diskussion
112
IL-6 Rezeptors (IL-6R) den Ausbruch einer Colitis in verschiedenen Modellen
verhinderte (Atreya et al. 2000). IL-6 kann zudem an den löslichen IL-6 Rezeptor
binden (sIL-6R, engl. soluble IL-6R), was zur Entstehung des IL-6/sIL-6R-Komplex
führt. Dieser kann über Bindung an das ubiquitär vorkommende Glykoprotein gp130
proinflammatorische Immunantworten auslösen. Dieser Vorgang wird als
IL-6-trans-signaling bezeichnet. Unter anderem wird dadurch die Apoptose
mukosaler T-Zellen inhibiert und die Aufrechterhaltung einer chronischen
Darmerkrankung ermöglicht. Die Neutralisation des sIL-6R in Patienten mit MC führte
zu einer deutlichen Unterdrückung der Aktivität im Rahmen einer Colitis und zu einer
gesteigerten Apoptose von T-Zellen (Atreya et al. 2000). IL-6-trans-signaling führte
außerdem zu einer erhöhten Expression des TGF-β Inhibitors SMAD7. Durch
Überexpression von SMAD7 in transgenen Mäusen waren diese nicht mehr in der
Lage Foxp3+ Tregs zu induzieren und eine Colitis durch die Aktivierung von Tregs zu
unterdrücken (Dominitzki et al. 2007). Fujimoto et al. bewiesen, dass die
Überproduktion von IL-6 die Induktion von Tregs hemmte (Fujimoto et al. 2011). Die
deutlich erhöhte IL-6 Produktion in den DSS behandelten Knock-out Tieren (s. Fig.
3.2 B) lässt somit auf eine verstärkt proinflammatorische Immunantwort schließen,
welche zudem direkt in der Lage sein könnte, regulatorische Antworten zu
unterdrücken.
Viele Studien beschäftigten sich des Weiteren mit der Bedeutung des
proinflammatorischen Zytokins IL-23 in entzündlichen Darmerkrankungen. IL-23
besteht aus den Untereinheiten IL-23p19 und IL-12p40, welches die gemeinsame
Untereinheit mit IL-12 darstellt (Oppmann et al. 2000). IL-23 sowie IL-12 werden von
APCs, vornehmlich DCs, gebildet. IL-12 fördert dabei in Anwesenheit von IFN-γ die
Differenzierung naiver T-Zellen zu Th1-Zellen, wohingegen IL-23 in Verbindung mit
TGF-β und IL-6 die Differenzierung zu Th17-Zellen einleitet (Kikly et al. 2006). Es
konnte gezeigt werden, dass IL-23 in die Pathogenese von Autoimmunerkrankungen,
wie der experimentellen autoimmunen Enzephalomyelitis (EAE) und der
Kollagen-induzierten Arthritis (CIA, engl. collagen-induced arthritis), involviert ist. Die
Produktion von IL-23 steht zudem in engem Zusammenhang mit der Entwicklung
entzündlicher Darmerkrankungen. In CED Patienten konnten sowohl erhöhte IL-23

Diskussion
113
Konzentrationen in intestinalen Geweben als auch eine erhöhte IL-23 Produktion
durch mukosale DCs festgestellt werden (Stallmach et al. 2004; Schmidt et al. 2005;
Sakuraba et al. 2009). In RAG-/- Mäusen, welche auf Grund einer Defizienz reifer
B- und T-Lymphozyten nach T-Zell Transfer eine Colitis entwickeln, führte die
Deletion von IL-23p19 oder IL-12p40 zur Unterdrückung der Colitis. Mäuse mit einer
zusätzlichen Deletion von IL-12p35 hingegen entwickelten eine deutliche Colitis (Hue
et al. 2006b). Die Deletionen von IL-23p19 und IL-12p40 führten zu einer Inhibierung
der IL-23 Produktion, wohingegen durch die Deletion von IL-12p35 die
IL-12 Produktion gehemmt wurde. Es wird daher vermutet, dass IL-23 als
entscheidendes Zytokin für eine intestinale Erkrankung gilt. Studien von Elson et al.
bestätigten diese Vermutung, indem sie zeigten, dass die Neutralisation von
IL-23p19 durch einen monoklonalen Antikörper den Ausbruch einer Colitis verhindern
sowie eine bestehende Colitis abschwächen konnte (Elson et al. 2007). Yen et al.
zeigten des Weiteren, dass die Gabe von IL-23 Protein zu einem deutlich früheren
Ausbruch der Colitis in RAG-/- Mäusen sowie zu einer erhöhten Produktion von IL-6
und IL-17 führte. Zudem konnte eine erhöhte Rekrutierung von Makrophagen und
Granulozyten festgestellt werden (Yen et al. 2006). Izcue et al. konnten eine weitere
Wirkung von IL-23 darstellen. Sie zeigten, dass IL-23 die Funktion regulatorischer
T-Zellen im Darm behindern und dass die Aufhebung immunsupprimierender und
regulatorischer Antworten die Entstehung intestinaler Entzündungen begünstigen
kann (Izcue et al. 2008). Diese Daten stehen im Einklang mit den erhobenen
Ergebnissen dieser Arbeit. Die DSS behandelten Knock-out Tiere zeigten eine
stärkere Entzündung mit einer erhöhten IL-23 Produktion (s. Fig. 3.2 C), einer
erhöhten Anzahl an Makrophagen (s. Fig. 3.2 E) und einer eingeschränkten
regulatorischen T-Zellantwort (s. Fig. 3.6 C-E). Es kann vermutet werden, dass die
HIF-1α Defizienz in DCs vornehmlich zu einer eingeschränkten Induktion
regulatorischer T-Zellen führte. Dies resultierte in einer verstärkten Entzündung mit
erhöhter Produktion proinflammatorischer Zytokine, welche wiederum die Funktion
der Tregs unterdrücken konnten.
Auf der anderen Seite muss jedoch erwähnt werden, dass die durch IL-23 ausgelöste
Th17-Antwort auch positiven Einfluss auf eine Darmentzündung nehmen kann.

Diskussion
114
Sugimoto et al. zeigten, dass das von Th17-Zellen produzierte Zytokin IL-22 zur
Verbesserung einer intestinalen Entzündung führte (Sugimoto et al. 2008). IL-22
aktiviert die Mucinproduktion der IECs und fördert die Restitution geschädigter Mucin
produzierender Becherzellen. Durch Gabe eines IL-22 bindenden Antikörpers wurde
die Wiederherstellung der Becherzellen in der Erholungsphase eines DSS
Colitismodells gehemmt. In den behandelten Knock-out Mäusen dieser Arbeit konnte
eine deutlich erhöhte Mucinproduktion festgestellt werden (s. Fig. 3.7 A-D). Es ist
also möglich, dass der Knock-out von HIF-1α in DCs über ausgelöste
proinflammatorische Th17-Antworten Effekte auf das intestinale Epithel aufwies,
welche kompensatorische Schutzmechanismen und die Restitution des geschädigten
Epithels einleiteten.
Ein weiterer Parameter für die verstärkte Erkrankung der Knock-out Tiere war die
erhöhte Expression von Lipocalin 2 (Lcn2) (s. Fig. 3.2 D). Lcn2 ist ein Mitglied der
Lipocalin Protein Familie und wird unter anderem von neutrophilen Granulozyten und
IECs exprimiert (Cowland & Borregaard 1997; Raffatellu et al. 2009). Es stellt einen
wichtigen Regulator der Immunantwort dar (Berger et al. 2006). Chassing et al.
maßen den fäkalen Lcn2 Gehalt in einem DSS Colitismodell mittels ELISA und
konnten einen kontinuierlichen Anstieg während der Entwicklung der Erkrankung
nachweisen (Chassaing et al. 2012). Dies stellt eine relativ kostengünstige und vor
allem nicht-invasive Methode dar, um den Verlauf der Colitis zu dokumentieren und
könnte in nächsten Versuchen verwendet werden.
Es zeigte sich des Weiteren eine signifikant erhöhte IL-1β Expression in den DSS
behandelten Knock-out Tieren (s. Fig. 3.9 A). Makrophagen stellen die
Hauptproduzenten von IL-1β dar, welches unter anderem Lymphozyten aktiviert und
die Freisetzung von Akute-Phase-Proteinen (APPs) induziert (Murphy et al. 2009).
Serum Amyloid A (SAA) stellt ein wichtiges APP dar, welches zudem mit dem
Schweregrad einer Colitis in Mensch und Maus korreliert (Chambers et al. 1987; de
Villiers et al. 2000). Auch für SAA wiesen die Knock-out Mäuse höhere Werte durch
die DSS Colitis auf (s. Fig. 3.9 B). Verschiedene Studien zeigten zudem, dass CED
Patienten deutlich erhöhte IL-1β Werte aufwiesen (Stallmach et al. 2004; Eastaff-
Leung et al. 2010). Des Weiteren konnte gezeigt werden, dass IL-1β Einfluss auf das

Diskussion
115
intestinale Epithel hat. Zum einen kann es bei einer Shigellen Infektion die tight
junctions der Epithelzellen so stark lockern, dass weitere Mikroorganismen aus dem
Darmlumen in das Gewebe eindringen können und die Infektion sich ausbreitet
(Sansonetti 2004). Zum anderen kann es IECs zur vermehrten Produktion von IL-6
anregen (McGee et al. 1993). Eine erhöhte IL-6 Expression konnte auch in den DSS
behandelten Knock-out Mäusen festgestellt werden (s. Fig. 3.2 B). Es erscheint also
möglich, dass die einzelnen Zytokine sich untereinander beeinflussen und zur
vermehrten Produktion anregen. Da DCs eine entscheidende Bedeutung im
Zusammenspiel der intestinalen Immunität aufweisen, führte der Verlust von
dendritischem HIF-1α somit zu weitgreifenden Veränderungen im
Zytokinexpressionsmuster der lokalen Immunzellen. Dies wiederum könnte das
Ausbilden immunregulatorischer Tregs unterdrückt haben, was sich in einer
verschlechterten Kontrolle der Darminfektion äußerte.
4.3 Typ I Interferon Produktion
DCs stellen die Hauptproduzenten der Typ I Interferone dar (Abe et al. 2007). In vitro
Studien von Wobben et al. zeigten eine HIF-1α abhängige dendritische Produktion
von Typ I Interferonen (Wobben et al. 2013). Die Synthese von IFN-α4, IFN-α12 und
IFN-β durch LPS Stimulation war in isolierten BmDCs von LysMcre/HIF-1α+f/+f
Mäusen, welche einen Verlust von HIF-1α in myeloischen Zellen aufwiesen,
signifikant geringer im Vergleich zu Kontrollmäusen. Dies hatte entscheidende
Auswirkungen auf die Aktivierung zytotoxischer T-Zellen. Durch Kokulturversuche
konnte gezeigt werden, dass DCs mit einem Mangel an HIF-1α Protein deutlich
weniger CD278 mRNA in T-Zellen induzierten, wodurch ein Hinweis auf eine
reduzierte T-Zellaktivierung bestand. Aus diesem Grund wurde in der vorliegenden
Arbeit die Typ I Interferon Produktion untersucht. Ergebnisse von Wobben et al.
konnten dabei zum Teil mit in vivo Daten bestätigt werden, da die Kontrolltiere in den
mesenterialen Lymphknoten nach DSS Behandlung eine signifikant höhere
Expression von IFN-α4 und IFN-α12 aufwiesen als die DSS behandelten Knock-out
Tiere. Typ I Interferone, wozu IFN-α und IFN-β zählen, unterscheiden sich stark von
IFN-γ, welches vornehmlich von Makrophagen, Th1-Zellen, CTLs und NK-Zellen

Diskussion
116
sezerniert wird und gegen intrazelluläre Pathogene wirkt (Murphy et al. 2009). Die
ursprünglich beschriebene Aufgabe der Typ I Interferone war die Abwehr von
Virusinfektionen. Neuere Studien beschrieben jedoch auch weitreichendere
Funktionen und zeigten zudem einen Zusammenhang zwischen Typ I Interferonen
und der Kontrolle einer intestinalen Entzündung. Sainathan et al. untersuchten die
Effekte von Imiquimod auf die Auswirkungen einer DSS Colitis. Imiquimod ist ein
Virostatikum, welches über TLR7 Bindung die Produktion von IFN-α, TNF-α, IL-6 und
IL-12 steigert. Es wird unter anderem gegen Genitalwarzen, aktinische Keratose und
oberflächliche Basalzell-Karzinome eingesetzt (Gupta et al. 2004). Sainathan et al.
zeigten nun, dass sowohl die orale als auch die topische Behandlung mit Imiquimod
zu einer erhöhten Typ I Interferon Produktion im Colon und dadurch zu einer
deutlichen Verbesserung der Symptome der DSS Colitis führte (Sainathan et al.
2012). Katakura et al. konnten des Weiteren nachweisen, dass die Induktion der
Typ I Interferone über TLR9 eine protektive Wirkung in einer experimentellen Colitis
aufwies (Katakura et al. 2005). TLR9 Agonisten, wie nicht-methylierte
Cytosin-Phosphatidyl-Guanin Inseln bakterieller DNA (CpGs), verbesserten eine
DSS induzierte Colitis in RAG defizienten Mäusen, jedoch nicht in SCID Mäusen. Die
Ursache hierfür lag in einer reduzierten IFN-α/β Produktion auf Grund einer
verringerten Anzahl an DCs in den SCID Mäusen. Des Weiteren wurde gezeigt, dass
die positiven Effekte durch die Neutralisation von IFN-α/β mit speziellen Antikörpern
aufgehoben werden konnten. Weitere Untersuchungen konnten nachweisen, dass
die protektive Wirkung von IFN-α/β in einer DSS Colitis zum Teil durch die
Unterdrückung der proinflammatorischen Aktivität der Makrophagen bedingt war.
Zudem wurde vermutet, dass Typ I Interferone Einfluss auf das intestinale Epithel
haben, indem sie die Apoptose der IECs verhindern und somit die epitheliale
Barrierefunktion aufrecht erhalten (Katakura et al. 2005). Auch Abe et al. bewiesen
einen protektiven Einfluss von Typ I Interferonen mit direktem Bezug auf DCs (Abe et
al. 2007). Eine Deletion von DCs während einer DSS induzierten Colitis führte zu
einer Abmilderung der Symptome, wohingegen die Abwesenheit von DCs vor
Induktion der Colitis zu einer deutlichen Verschlechterung führte. Wurden die Mäuse
jedoch vor der Induktion einer DSS Colitis mit TLR9 Agonisten, wie CpGs, behandelt,

Diskussion
117
zeigte dies eine protektive Wirkung. Die Autoren führten diesen positiven Effekt auf
den Einfluss von Typ I Interferonen zurück, welche die Aktivierung
proinflammatorischer Makrophagen unterdrücken konnten. Diese Befunde stehen im
Einklang mit den Ergebnissen dieser Arbeit. Der Verlust von dendritischem HIF-1α
resultierte in einer verringerten Produktion an Typ I Interferonen (s. Fig. 3.3 A-B).
Dies wiederum beeinträchtigte die Unterdrückung der proinflammatorischen
Makrophagenantwort, was sich in der erhöhten Anzahl an F4/80 positiven Zellen
(s. Fig. 3.2 E) und der gesteigerten Produktion proinflammatorischer Zytokine in den
Knock-out Tieren wiederspiegelte (s. Fig. 3.2 B-D).
4.4 Aktivierung regulatorischer T-Zellen durch DCs
Bestimmte intestinale DCs besitzen die Fähigkeit, die Differenzierung CD4+ T-Zellen
zu Foxp3+ regulatorischen T-Zellen zu induzieren (Annacker et al. 2005; Coombes et
al. 2007). Tregs übernehmen eine entscheidende Rolle in der intestinalen Immunität.
Sie können die Symptome einer bestehenden Colitis abmildern oder sogar den
Ausbruch einer Colitis verhindern (Mottet et al. 2003; Uhlig, Coombes, et al. 2006;
Izcue et al. 2006). In CED Patienten konnte zudem eine reduzierte Treg-Anzahl und
eine Zunahme der Th17-Zellen nachgewiesen werden (Eastaff-Leung et al. 2010).
Aus diesem Grund wurden die Auswirkungen des HIF-1α Verlustes in DCs auf die
Fähigkeit der Treg Induktion untersucht.
Die Treg Induktion durch DCs wird von verschiedenen Mediatoren beeinflusst. Dazu
gehören TGF-β, Retinsäure und IL-10. TGF-β wird von äußerst vielen Zellen in einer
inaktiven Vorläuferform sezerniert und liegt in großen Mengen in der intestinalen
Mukosa vor. Für die Ausübung der biologischen Effekte muss TGF-β allerdings noch
aktiviert werden. Es konnte gezeigt werden, dass dies durch die Expression von
ανβ8-Integrinen auf DCs erfolgt (Travis et al. 2007). Der Verlust von ανβ8-Integrinen
auf DCs führte zu einer eingeschränkten Fähigkeit, Foxp3+ Tregs zu induzieren und
zur Entwicklung von Autoimmunerkrankungen und entzündlichen
Darmveränderungen (Travis et al. 2007; Lacy-Hulbert et al. 2007). Coombes et al.
konnten des Weiteren nachweisen, dass CD103+ intestinale DCs sowohl große
Mengen an LTBP3 (engl. latent TGF-β binding protein 3), welches für die Sekretion

Diskussion
118
und Erkennung von inaktivem / latentem TGF-β entscheidend ist, als auch an PLAT
(engl. tissue plasminogen activator) aufweisen, was für die Aktivierung von latentem
TGF-β benötigt wird (Coombes et al. 2007). Sie konnten zudem zeigen, dass die
Differenzierung von CD4+ T-Zellen zu Foxp3+ Tregs durch das von CD103+ DCs
produzierte TGF-β gesteuert wird. Die Inhibierung von TGF-β durch einen
blockierenden Antikörper führte zu einer vollständigen Hemmung der Foxp3
Induktion, wohingegen die Zugabe von exogenem TGF-β zu einer enormen
Steigerung der Foxp3 Induktion führte. Shull et al. zeigten schon vor einigen Jahren,
dass ein vollständiger Verlust von TGF-β1 zur Entwicklung einer Spontancolitis und
im weiteren Verlauf zu Multiorganversagen führte (Shull et al. 1992).
Verschiedene Studien untersuchten den Zusammenhang zwischen HIF-1 und
TGF-β. Bereits 1991 konnten Falanga et al. nachweisen, dass die Synthese von
TGF-β1 in humanen dermalen Fibroblasten durch Hypoxie gesteigert wird (Falanga
et al. 1991). HIF-1α vermittelte zudem in colorektalen Karzinomzellen die durch
TGF-β induzierte Glutathionperoxidase 1 Expression, welche den H2O2 vermittelten
Zelltod hemmt (Huang et al. 2012). In renalen Epithelzellen wird die TGF-β induzierte
Kollagenexpression von HIF-1 gesteuert (Basu et al. 2011). Clambey et al.
untersuchten die Verbindung zwischen Hypoxie und Tregs und konnten auch hierbei
feststellen, dass Hypoxie die relative Menge an Foxp3+ Tregs durch einen TGF-β
abhängigen Mechanismus erhöht (Clambey et al. 2012). Hung et al. konnten
schließlich in neuesten Studien mithilfe eines ChIP Assay den Nachweis erbringen,
dass TGF-β1 ein direktes Zielgen von HIF-1 darstellt (Hung et al. 2013). Intestinale
Makrophagen waren zudem unter dem Einfluss von TGF-β nicht mehr in der Lage,
proinflammatorische Zytokine zu sezernieren (Smythies et al. 2005). Diese
Ergebnisse untermauern insgesamt die Befunde der vorliegenden Arbeit, welche
zeigen, dass der Verlust von HIF-1α in DCs große Auswirkungen auf die TGF-β
Produktion, die Treg Differenzierung und letztendlich auch auf den Schweregrad
einer intestinalen Entzündung hatte.
TGF-β kann allerdings zusammen mit IL-6 auch die Differenzierung von naiven
T-Zellen zu proinflammatorischen Th17-Zellen steuern (Mangan et al. 2006;
Veldhoen et al. 2006; Bettelli et al. 2006). Dies wird jedoch durch die Anwesenheit

Diskussion
119
von Retinsäure inhibiert (Mucida et al. 2007). Viele Studien bewiesen zudem, dass
Retinsäure die Differenzierung naiver T-Zellen zu Foxp3+ Tregs fördert (Sun et al.
2007; Mucida et al. 2007; Coombes et al. 2007; Kang et al. 2007). Retinsäure
entsteht durch den Abbau von Vitamin A (Retinol) zu Retinal, welches durch Aldehyd
Dehydrogenasen weiter zu Retinsäure oxidiert wird (Duester 2000). Es wurde
gezeigt, dass DCs aus mesenterialen Lymphknoten bzw. CD103+ DCs diesen
Prozess durch die Aldehyd Dehydrogenase Aldh1a2 steuern (Iwata et al. 2004;
Coombes et al. 2007). Coombes et al. zeigten, dass die Zufuhr von exogener
Retinsäure in Zusammenhang mit TGF-β zu einer deutlichen Steigerung der
Foxp3+ Treg Induktion führte. Die Inhibierung der Retinsäurerezeptoren (RARs, engl.
retinoic acid receptors) unterdrückte diese Induktion komplett (Coombes et al. 2007).
Balmer et al. beschrieben eine weitere Verbindung zwischen TGF-β und Retinsäure.
Sie zeigten, dass Retinsäure direkt die Expression der Isoformen 1 und 2 des TGF-β
Rezeptors induziert (Balmer 2002). Die Zugabe von Retinsäure führt zudem zu einer
Reduktion der IFN-γ produzierenden T-Zellen (Cantorna et al. 1994). Annacker et al.
belegten diese Befunde, indem sie zeigten, dass mit CD103+ DCs kultivierte T-Zellen
weniger IFN-γ produzierten (Annacker et al. 2005). IFN-γ induziert die Expression
von SMAD7, welches wiederum die Funktion von TGF-β inhibiert. Es besteht also
auch hier ein Zusammenspiel der einzelnen Faktoren, da die durch Retinsäure
reduzierte IFN-γ Produktion in einem Anstieg von TGF-β und folglich in einer
erhöhten Foxp3 Expression resultieren könnte (Coombes & Powrie 2008).
Retinsäure induziert des Weiteren direkt die Transkription der RARs (Blomhoff &
Blomhoff 2006). Fernández-Martínez und Mitarbeiter bewiesen, dass die Regulation
des RARβ von HIF-1α abhängig ist (Fernández-Martínez et al. 2011). In dieser Arbeit
zeigten die HIF-1α+f/+f Kontrolltiere durch die DSS Behandlung einen deutlichen
Anstieg in der Aldh1a2 und RARα Expression, wohingegen der Anstieg in den
CD11cCre/HIF-1α+f/+f Knock-out Tieren unterblieb (s. Fig. 3.5 C-D). Es kann also
vermutet werden, dass der Verlust von dendritischen HIF-1α zu einer
Beeinträchtigung der Retinsäureproduktion und damit eingeschränkter Regulation
durch Tregs im Rahmen einer experimentellen Colitis führte. In weiterführenden

Diskussion
120
Versuchen könnte dies durch die direkte Bestimmung des Retinsäuregehalts z. B.
durch HPLC Verfahren untersucht und gegebenenfalls bestätigt werden.
Intestinale DCs besitzen des Weiteren die Fähigkeit, die darmspezifischen
Adhäsionsmoleküle α4β7-Integrin und CCR9 auf Lymphozyten zu induzieren
(Johansson-Lindbom et al. 2005). Iwata et al. konnten darstellen, dass Retinsäure
die Expression dieser Moleküle deutlich steigert (Iwata et al. 2004). Die Kontrolltiere
wiesen im Vergleich zu den Knock-out Tieren eine signifikant erhöhte Expression des
β7-Integrins und CCR9 durch die DSS Behandlung auf (s. Fig. 3.5 E-F). Es kann
daher angenommen werden, dass dendritisches HIF-1α für die Expression dieser
Moleküle von entscheidender Bedeutung ist und zu einer verbesserten Rekrutierung
der Lymphozyten führt. Zwar werden auch andere Effektorzellen als Tregs rekrutiert,
wie z. B. Th1-Zellen oder CTLs, aber da in den Kontrolltieren eine sehr große Menge
an Foxp3+ Tregs festgestellt werden konnte, ist davon auszugehen, dass diese
Zellen äußerst stark beeinflusst werden und so die Fähigkeit besitzen, die
Entzündung einzudämmen.
Ein weiteres wichtiges Zytokin für die intestinale Immunität stellt IL-10 dar. IL-10
Knock-out Mäuse entwickeln unter SPF Haltungsbedingungen eine milde
Entzündung des proximalen Colons, wohingegen sie unter konventionellen
Bedingungen eine schwere Enterocolitis mit Gewichtsreduktion und Anämie
entwickeln (Kühn et al. 1993). Dies spricht für eine Beteiligung der intestinalen
Mikroflora am Krankheitsgeschehen. In den erkrankten Tieren wurden von Berg et al.
erhöhte Werte von IFN-γ, IL-1α, TNF-α, IL-6 und Stickstoffmonoxid detektiert. Zudem
waren äußerst viele Zellen positiv in der F4/80 Färbung (Berg et al. 1996). Sie
vermuteten daher, dass der Verlust von IL-10 zu einer unkontrollierten Th1-Antwort
mit überschießender Zytokinproduktion durch Makrophagen führte. Die Gabe eines
Antikörpers gegen IFN-γ führte zu einer deutlichen Verbesserung der Symptome in
jungen Tieren. Durch Behandlung von frisch abgesetzten Mäusen mit IL-10 konnte
der Ausbruch der Colitis komplett gehemmt werden (Berg et al. 1996). Weitere
Studien zeigten, dass die Deletion von STAT3, dem Transkriptionsfaktor an welchen
IL-10 bindet, in myeloischen Zellen zu einer erhöhten Produktion von IL-12p40 und
IL-23p40 und dadurch zu einer chronischen Enterocolitis führte (Takeda et al. 1999;

Diskussion
121
Kobayashi et al. 2003). IL-10 wird von verschiedenen Zellen produziert. Das von
Tregs sezernierte IL-10 hat entscheidende Bedeutung für die Regulation und
Suppression von proinflammatorischen Antworten (Mittrücker & Kaufmann 2004;
Workman et al. 2009). Für die Induktion der Tregs ist die IL-10 Produktion durch
tolerogene DCs neben der schon beschriebenen TGF-β und RA Produktion
ausschlaggebend (Groux et al. 1999; Levings et al. 2005; Roncarolo et al. 2006).
Mäuse mit einer Deletion des IL-10 Rezeptors in Foxp3+ Tregs entwickelten im Alter
von 18-20 Wochen eine hochgradige Colitis. Weitere Untersuchungen zeigten, dass
IL-10 Rezeptor defiziente Tregs nicht in der Lage waren, proinflammatorische
Th17-Antworten zu unterdrücken. (Chaudhry et al. 2011). Auch im Rahmen von CED
spielt IL-10 eine Rolle. Isolierte DCs von MC Patienten wiesen eine reduzierte
Produktion von IL-10 auf (Sakuraba et al. 2009). De Baey et al. beschrieben eine
neue Vorläuferform von humanen DCs, genannt M-DC8, welche auf einen LPS
Stimulus 10-mal mehr TNF-α produzierten als normale DCs und im Gegensatz dazu
eine signifikant reduzierte IL-10 Produktion aufwiesen (de Baey et al. 2003). In MC
Patienten fanden sie eine sehr große Anzahl an M-DC8+ Zellen in der entzündeten
Tunica mucosa des Ileums. In der vorliegenden Arbeit wurden zum einen BmDCs
von HIF-1α+f/+f und CD11cCre/HIF-1α+f/+f Tieren isoliert, unter normoxischen
Bedingungen kultiviert und mit LPS stimuliert. Die Kontrolltiere zeigten hierbei im
Vergleich zu den Knock-out Tieren einen signifikanten Anstieg von IL-10 durch LPS
Stimulation (s. Fig. 3.4 A). Zur besseren Imitation einer in vivo Situation wurde der
Versuch mit BmDCs, welche unter hypoxischen Bedingungen kultiviert wurden,
wiederholt. Auch hier zeigten die Kontrolltiere einen signifikanten Anstieg von IL-10
durch LPS Stimulation (s. Fig. 3.4 B). In den DSS behandelten Tieren konnten diese
Befunde in mesenterialen Lymphknoten bestätigt werden. Hier wiesen ebenfalls die
HIF-1α+f/+f Tiere eine höhere Produktion von IL-10 auf (s. Fig. 3.5 A). Es kann somit
angenommen werden, dass HIF-1α für die IL-10 Produktion in DCs von
entscheidender Bedeutung ist und dass ein Verlust von dendritischem HIF-1α auch
durch diesen Mechanismus zu einer Beeinträchtigung der Induktion regulatorischer
T-Zellen führte.

Diskussion
122
Bereits mehrere Studien untersuchten den Zusammenhang zwischen HIF-1 und
IL-10. Rama et al. berichteten, dass durch Hypoxie stimulierte DCs eine erhöhte
Proteinproduktion von IL-10 aufweisen (Rama et al. 2008). Cai et al. bewiesen
zudem, dass HIF-1 die IL-10 Produktion in Myozyten aktiviert und unerlässlich für die
ischämische Präkonditionierung des Herzens ist (Cai et al. 2013). Des Weiteren
konnte gezeigt werden, dass die HIF-1 abhängige IL-10 Produktion in interstitiellen
Makrophagen der Lunge eine Überempfindlichkeitsreaktion in den Atemwegen
unterdrückt (Toussaint et al. 2013). Diese Ergebnisse verdeutlichen insgesamt die
HIF-1 Abhängigkeit der IL-10 Produktion.
Verschiedene Studien untersuchten außerdem den Effekt von HIF-1 in T-Zellen.
Ben-Shoshan et al. zeigten im Jahr 2008, dass hypoxisch stimulierte Jurkat T-Zellen,
humane mononukleäre Zellen sowie murine Milzzellen eine erhöhte Foxp3
Expression aufwiesen. Es konnte bewiesen werden, dass dieser Effekt direkt HIF-1α
abhängig war, da die Expression durch HIF-1α Gen-Silencing mittels siRNA
(engl. short interfering RNA) gehemmt werden konnte. Die hypoxisch stimulierten
Foxp3+ Zellen konnten zudem die Proliferation von T-Effektorzellen effektiver
unterdrücken als normoxische Tregs. Des Weiteren konnte auch in vivo durch eine
Überexpression von HIF-1α in murinen Milzzellen eine erhöhte Menge an Foxp3+
Tregs festgestellt werden (Ben-Shoshan et al. 2008). Higashiyama et al. konnten
zum einen nachweisen, dass infiltrierte T-Zellen in der Tunica mucosa von CED
Patienten eine deutliche HIF-1 Aktivierung aufwiesen (Higashiyama et al. 2012). Zum
anderen untersuchten sie den Knock-out von HIF-1α in T-Zellen im Rahmen einer
DSS Colitis. Hierbei konnten sie feststellen, dass die Knock-out Tiere eine im
Vergleich zu den Kontrolltieren schwerere Entzündung mit erhöhter Th1- und
Th17-Antwort entwickelten. Isolierte Milzzellen der Kontrolltiere zeigten zudem
in vitro eine deutliche Induktion der Foxp3 Expression durch hypoxische Stimulation.
In Knock-out Tieren blieb die Foxp3 Induktion aus, stattdessen konnte sowohl eine
erhöhte Menge an Th17-Zellen als auch eine gesteigerte IL-17 Expression
nachgewiesen werden (Higashiyama et al. 2012). Clambey et al. bestätigten diese
Befunde in eigenen Untersuchungen (Clambey et al. 2012). Sie konnten beweisen,
dass Foxp3 ein direktes HIF-1 Zielgen darstellt und HIF-1 die Quantität und Funktion

Diskussion
123
der Foxp3+ Tregs beeinflusst. Zudem konnten sie in einem T-Zell Transfer
Colitismodell zeigen, dass der Verlust von HIF-1α in Tregs zu einer Beeinträchtigung
der inhibitorischen Fähigkeiten der Tregs und zu einer schwereren Entzündung führt
(Clambey et al. 2012). Diese Studien beschreiben allesamt einen protektiven Effekt
von HIF-1α in T-Zellen im Rahmen einer Colitis. In der vorliegenden Arbeit wurde
nun zum ersten Mal der Einfluss von dendritischem HIF-1α auf eine Colitis
untersucht, wobei auch hier ein bedeutender Effekt dargestellt werden konnte. Die
insgesamt größte Auswirkung des Verlustes von HIF-1α in DCs bestand in der
eingeschränkten Fähigkeit, regulatorische T-Zellen zu induzieren, welche eine
Entzündung eindämmen können. Es konnte somit gezeigt werden, dass auch
dendritisches HIF-1α einen protektiven Einfluss auf eine experimentelle Colitis
ausübt.
4.5 Wechselwirkungen zwischen IECs und DCs
Mehrere Studien haben Interaktionen zwischen IECs und DCs nachgewiesen. Butler
et al. konnten zeigen, dass die Kokultur von DCs mit Caco-2 IECs zu einer
reduzierten Expression von MHC-II, CD80 und CD86 in DCs sowie zu einer
eingeschränkten T-Zellstimulation durch DCs führte (Butler et al. 2006). Des
Weiteren zeigten die kokultivierten DCs eine geringere Aktivierbarkeit durch TLRs
und eine erhöhte TGF-β Produktion. Insgesamt vermuteten die Autoren, dass die
Anwesenheit von IECs zur Ausbildung von tolerogenen DCs führte. Rimoldi et al.
zeigten, dass es die Produktion von TSLP durch IECs war, welche die Entwicklung
tolerogener DCs förderte (Rimoldi et al. 2005). Tolerogene DCs sind für die
intestinale Homöostase von großer Bedeutung. Sie unterdrücken unter anderem
proinflammatorische Th1-Antworten (Rimoldi et al. 2005). Es konnte zudem gezeigt
werden, dass die TSLP Produktion durch Hassall-Körperchen im Thymus DCs dazu
befähigt, CD4+CD25- Thymozyten in Foxp3+CD4+CD25+ Tregs zu differenzieren
(Watanabe et al. 2005). Es könnte daher möglich sein, dass intestinale DCs unter
anderem auch durch den Einfluss von TSLP die Fähigkeit erlangen, Foxp3+ Tregs zu
induzieren (Coombes & Powrie 2008). Studien von Zaph et al. zeigten, dass der
Knock-out von IKKβ in IECs zu einer eingeschränkten TSLP Produktion führte. Dies

Diskussion
124
resultierte in einer verminderten Expression des TSLP Rezeptors (TSLPR) auf DCs.
Diese DCs produzierten erhöhte Mengen der proinflammatorischen Zytokine IL-12
und IL-23 und waren unfähig, eine Trichuris muris Infektion zu unterdrücken (Zaph et
al. 2007). Ein Zusammenhang zwischen TSLP und HIF konnte kürzlich von Jang et
al. nachgewiesen werden (Jang et al. 2013). Sie wiesen eine HIF-1α abhängige
TSLP Expression in Keratinozyten auf. In der vorliegenden Arbeit konnte im Colon
mittels ELISA kein Unterschied zwischen den beiden Mausgruppen in der TSLP
Produktion festgestellt werden, da die IECs der verwendeten Mausstämme
funktionell aktives HIF-1α aufwiesen. Allerdings konnte in isolierten, hypoxisch
kultivierten BmDCs von HIF-1α+f/+f Mäusen eine im Vergleich zu den Knock-out
Tieren signifikant höhere Induktion des TSLPR durch LPS Stimulation nachgewiesen
werden (s. Fig. 3.4 C). Auch in den DSS behandelten Kontrolltieren waren im Colon
höhere Expressionen des TSLPR als in den Knock-out Tieren zu finden
(s. Fig. 3.4 E). Es kann daher angenommen werden, dass der Knock-out von HIF-1α
in DCs die Fähigkeit der TSLPR Expression nachteilig beeinflusst und somit auch die
Ausprägung eines tolerogenen DC-Phänotyps.
Ein weiteres Zusammenspiel der IECs und DCs konnte in dieser Arbeit
nachgewiesen werden, da der Knock-out von dendritischem HIF-1α Auswirkungen
auf die Mucinproduktion des Epithels zeigte. In den CD11cCre/HIF-1α+f/+f Tieren
führte die DSS Colitis zu einer signifikant erhöhten Expression von MUC1, MUC2
und MUC3 im Vergleich zu den Kontrolltieren. Becherzellen produzieren Mucine,
welche sich als schützende Schleimschicht über die Epithelzellen legen. MUC1 bis 4
sind dabei die wichtigsten Mucine im Colon (Corfield et al. 2000). MUC1, MUC3 und
MUC4 stellen membrangebundene Mucine dar, wohingegen MUC2 von den IECs in
das Darmlumen sezerniert wird (Shirazi et al. 2000). Die Mucinexpression während
einer gastrointestinalen Erkrankung wird sehr unterschiedlich beschrieben und ist
meist abhängig von der Schwere und dem Ausmaß der Entzündung. Hoebler et al.
untersuchten die Mucinproduktion von Balb/c Mäusen, in welchen durch die orale
Gabe von 1 % DSS für 5 Tage eine akute Colitis induziert wurde (Hoebler et al.
2006). Die Expression von MUC2 zeigte hierbei keinerlei Veränderungen,
wohingegen die MUC1 und MUC3 Expressionen im Caecum und Colon durch die

Diskussion
125
Colitis deutlich anstiegen. Im Gegensatz dazu, zeigten Dharmani et al. in einem DSS
Modell bei Ratten, dass die MUC2 und MUC3 mRNA Expressionen durch die Colitis
abnahmen. Die Behandlung mit einem anti-TNF-α-Antikörper führte dabei wiederum
zu einem Anstieg der Mucinproduktion (Dharmani et al. 2011). Verschiedene
Untersuchungen mit MUC2 Knock-out Mäusen verdeutlichen eine protektive
Funktion von MUC2. Die Infektion mit Citrobacter rodentium in MUC2 Knock-out
Mäusen führte zu einer hochgradigen Erkrankung mit einer Mortalität von bis zu
90 % (Bergstrom et al. 2010). Van der Sluis et al. zeigten zudem, dass der Verlust
von MUC2 zur Entwicklung einer spontanen Colitis im Alter von fünf Wochen führte
(Van der Sluis et al. 2006). Mit zunehmendem Alter verschlimmerte sich die
Entzündung, bis schließlich ab einem Alter von sechs Monaten Adenokarzinome im
Dünndarm und Rektum auftraten. Dadurch wurde vermutet, dass MUC2 auch eine
suppressive Rolle bei intestinalen Krebserkrankungen aufweist (Van der Sluis et al.
2006). Im DSS Colitismodell zeigten die homozygoten MUC2 Knock-out Tiere eine
deutlich höhere Krankheitsanfälligkeit als die heterozygoten Knock-out Tiere und die
Kontrolltiere, wodurch die protektive Wirkung von MUC2 dargelegt wurde (Van der
Sluis et al. 2006). Klinische Studien, welche die Mucinexpression in Patienten mit MC
und CU untersuchten, beschrieben sehr unterschiedliche Ergebnisse. Sowohl in MC
als auch in CU Patienten wurden gleichbleibende, gesteigerte oder auch reduzierte
Expressionen von MUC1 bis 3 nachgewiesen (Shirazi et al. 2000; Hoebler et al.
2006; Louis et al. 2006; Van der Sluis et al. 2006; Dorofeyev et al. 2013). Es besteht
zudem ein Zusammenhang zwischen einzelnen Mucinen und HIF. In renalen
Karzinomzellen konnte eine HIF-1 abhängige MUC1 Induktion nachgewiesen werden
(Aubert et al. 2009). Louis et al. beschrieben eine HIF-1 abhängige Induktion von
MUC3 in T84 Darmepithelzellen (Louis et al. 2006). In Bronchialepithelzellen konnte
zudem eine HIF-1 Abhängigkeit von MUC5AC festgestellt werden (Polosukhin et al.
2011; Zhou et al. 2012). Ein weiteres interessantes Zusammenspiel findet sich
zwischen Mucinen und dem proinflammatorischen Zytokin IL-6. Yokoigawa et al.
zeigten, dass Mucine von Colonkarzinomzellen der Linie LS180 die Produktion von
IL-6 in Monozyten induzierten, was eine mögliche Ursache für die erhöhten IL-6
Serumspiegel von Patienten mit Krebserkrankungen des Colons darstellen könnte

Diskussion
126
(Yokoigawa et al. 2005). Li et al. beschrieben hingegen, dass von Makrophagen
produziertes IL-6 die Produktion der Mucine beeinflusste (Li et al. 2009). In
Colonkarzinomzellen der Linie HAT-29 erhöhte sich durch Zugabe von IL-6 die
MUC1 Expression, wohingegen sich die MUC2 Expression reduzierte. Andere
Studien wiederum zeigten einen Anstieg von MUC2 und MUC3 durch IL-6 (Enss et
al. 2000; Shekels & Ho 2003). In den DSS behandelten HIF-1α Knock-out Tieren
dieser Arbeit konnte eine signifikant erhöhte Expression der Mucine 1 bis 3 entdeckt
werden (s. Fig. 3.7 A-D). Die IL-6 Expression in diesen Tieren zeigte ebenfalls eine
signifikante Erhöhung (s. Fig. 3.2 B). Ob nun die IL-6 Produktion die Mucinproduktion
beeinflusste oder umgekehrt, konnte nicht geklärt werden. Es wird jedoch vermutet,
dass alle Befunde in einem übergeordneten Zusammenhang stehen. Der Verlust von
dendritischen HIF-1α führte in den Knock-out Tieren zu einer schwereren
Entzündung im DSS Colitismodell als in den Kontrolltieren. Die Hauptursache hierfür
lag vermutlich in der eingeschränkten Fähigkeit der Knock-out DCs regulatorische
T-Zellen zu induzieren, welche die Colitis kontrollieren und einschränken könnten.
Dies resultierte in einer insgesamt gesteigerten proinflammatorischen
Zytokinproduktion. Es könnte möglich sein, dass die epitheliale Mucinproduktion in
den Knock-out Tieren als kompensatorischer Schutzmechanismus auf die verstärkte
Entzündung gesteigert wurde, da Mucine, wie oben erwähnt, eine protektive Funktion
aufweisen. Des Weiteren sind sie aber auch in Reparaturvorgänge involviert.
Wallace et al. zeigten, dass MUC2 defiziente Mäuse eine schwere Beeinträchtigung
in der Restitution der gastrischen Mukosa aufwiesen (Wallace et al. 2011).
Untersuchungen von Ho et al. bewiesen ebenfalls eine Beteiligung von intestinalem
MUC3 an der Wundheilung (Ho et al. 2006). Dies könnte eine weitere Erklärung für
die verstärkte Mucinexpression der Knock-out Tiere darstellen, indem diese
versuchten durch die erhöhte Mucinproduktion dem intestinalen Schaden entgegen
zu wirken.
Zwar entwickelten auch die Kontrolltiere durch die DSS Gabe eine Colitis, jedoch in
einer milderen Ausprägung. Sie zeigten einen signifikant niedrigeren
Gewichtsverlust, eine reduzierte proinflammatorische Immunantwort sowie eine
deutlich effektivere Induktion regulatorischer T-Zellen. Dies lässt darauf schließen,

Diskussion
127
dass HIF-1α in DCs einen positiven Effekt im Rahmen einer DSS induzierten Colitis
aufweist. Wenngleich die Therapiestrategien bei chronisch entzündlichen
Darmerkrankungen überwiegend T-Zellen fokussieren, konnte durch diese Arbeit
gezeigt werden, dass auch der Phänotyp von dendritischen Zellen Auswirkungen auf
eine intestinale Entzündung zeigt. Dies könnte eine neue Therapiemöglichkeit für
chronisch entzündliche Darmerkrankungen darstellen.

Zusammenfassung
128
5 Zusammenfassung
Katharina Flück
Die Bedeutung des Hypoxie-induzierbaren Faktors 1 in dendritischen Zellen für
die Pathophysiologie einer Dextrannatriumsulfat induzierten Colitis
Dendritische Zellen (DCs) vermitteln zwischen angeborener und erworbener
Immunität und sind von entscheidender Bedeutung für die intestinale Homöostase.
Für die Pathogenese chronisch entzündlicher Darmerkrankungen (CED) spielen sie
eine große Rolle. In solch entzündeten Geweben sind DCs niedrigen
Sauerstoffpartialdrücken ausgesetzt. Der Transkriptionsfaktor Hypoxie induzierbarer
Faktor (HIF)-1 reguliert die zelluläre Adaptation an eine sauerstoffarme Umgebung.
Um den Einfluss der regulierenden Untereinheit HIF-1α in DCs auf die Ausprägung
einer experimentellen Colitis zu untersuchen, wurde in Kontroll- (HIF-1α+f/+f) und
Knock-out Mäusen, welche einen Verlust von HIF-1α in DCs aufwiesen
(CD11cCre/HIF-1α+f/+f), durch siebentägige Gabe von 3 % Dextrannatriumsulfat
(DSS, engl. dextran sodium sulfate) eine Colitis induziert. Die Knock-out Tiere
zeigten einen signifikant größeren Gewichtsverlust an Tag 6 und 7 des
Experimentes. Zudem war die mRNA Expression proinflammatorischer Zytokine, wie
Interleukin (IL)-6 und IL-23, in den Knock-out Tieren signifikant erhöht. Kontrolltiere
mit funktionellem dendritischen HIF-1α wiesen hingegen eine deutlich erhöhte
Interferon (IFN)-α Produktion sowie einen tolerogenen DC-Phänotyp (Expression von
TSLP Rezeptor) durch die DSS induzierte Colitis auf. Des Weiteren konnte eine
signifikant erhöhte Anzahl an Foxp3+ regulatorischen T-Zellen (Tregs) in den
erkrankten Kontrolltieren festgestellt werden. Tregs können von bestimmten
intestinalen DCs durch Produktion von IL-10, TGF-β (engl. transforming growth
factor-β) und Retinsäure induziert werden. Expressionen von IL-10 und TGF-β waren
in DSS behandelten HIF-1α+f/+f Tieren signifikant erhöht im Vergleich zu DSS
behandelten CD11cCre/HIF-1α+f/+f Tieren. Retinsäure entsteht in DCs durch den
Abbau von Vitamin A, wozu unter anderem die Aldehyd Dehydrogenase Aldh1a2

Zusammenfassung
129
benötigt wird. Die Expression von Aldh1a2 in Kontrolltieren war im Vergleich zu den
Knock-out Tieren durch die Colitis signifikant erhöht. Zudem konnte in den
Kontrolltieren ein deutlicher Anstieg des Retinsäurerezeptors α (RARα) festgestellt
werden. Retinsäure beeinflusst die Expression der darmspezifischen
Adhäsionsmoleküle α4β7-Integrin und CCR9 (CC Chemokin Rezeptor 9). In den DSS
behandelten Kontrolltieren konnte ein signifikanter Anstieg für das β7-Integrin und
CCR9 nachgewiesen werden. In den Knock-out Tieren zeigte der Verlust von HIF-1α
in DCs Auswirkungen auf das Epithel. Durch die DSS induzierte Colitis konnte in
diesen Tieren eine deutlich gesteigerte Mucinproduktion festgestellt werden. Diese
lässt sich am wahrscheinlichsten als kompensatorischer Schutzversuch durch
geschädigte intestinale Epithelzellen interpretieren.
Insgesamt konnte durch diese Arbeit verdeutlicht werden, dass dendritisches HIF-1α
einen positiven Einfluss auf eine experimentelle Colitis hat, da die Aktivierung
regulatorischer T-Zellen und die Eindämmung der Colitis von funktionellem HIF-1α in
DCs abhängig war.
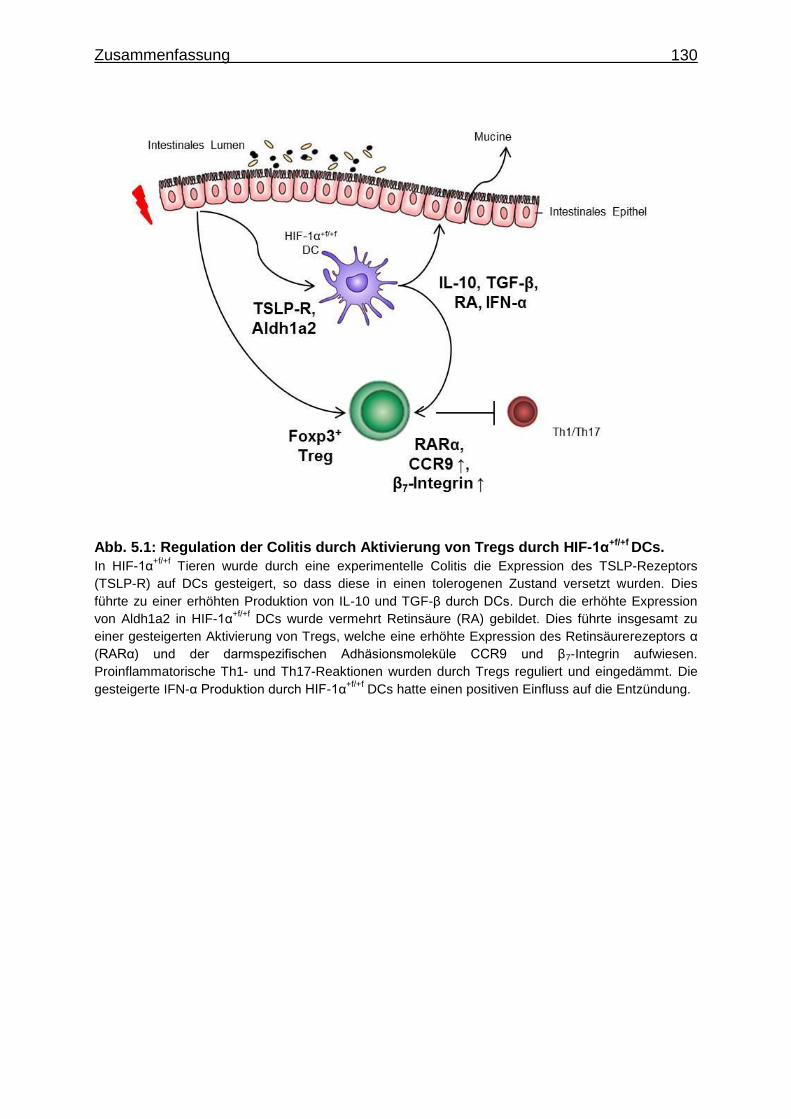
Zusammenfassung
130
Abb. 5.1: Regulation der Colitis durch Aktivierung von Tregs durch HIF-1α+f/+f DCs.
In HIF-1α+f/+f
Tieren wurde durch eine experimentelle Colitis die Expression des TSLP-Rezeptors
(TSLP-R) auf DCs gesteigert, so dass diese in einen tolerogenen Zustand versetzt wurden. Dies
führte zu einer erhöhten Produktion von IL-10 und TGF-β durch DCs. Durch die erhöhte Expression
von Aldh1a2 in HIF-1α+f/+f
DCs wurde vermehrt Retinsäure (RA) gebildet. Dies führte insgesamt zu
einer gesteigerten Aktivierung von Tregs, welche eine erhöhte Expression des Retinsäurerezeptors α
(RARα) und der darmspezifischen Adhäsionsmoleküle CCR9 und β7-Integrin aufwiesen.
Proinflammatorische Th1- und Th17-Reaktionen wurden durch Tregs reguliert und eingedämmt. Die
gesteigerte IFN-α Produktion durch HIF-1α+f/+f
DCs hatte einen positiven Einfluss auf die Entzündung.
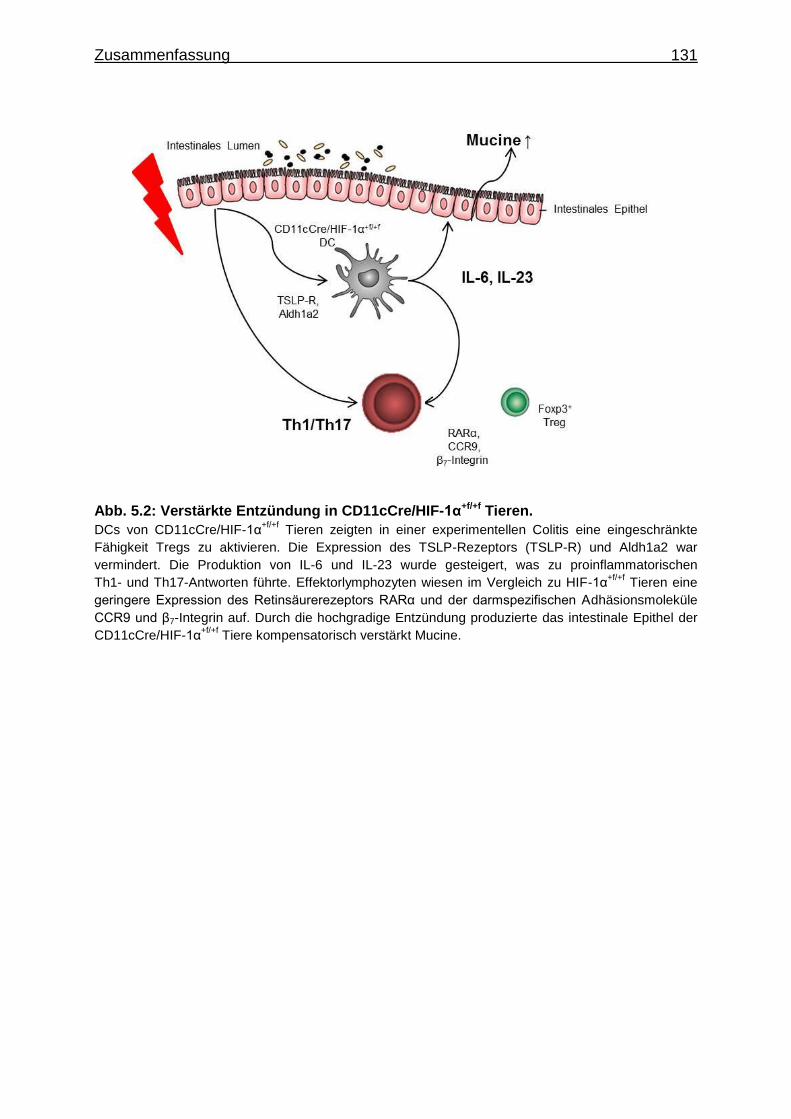
Zusammenfassung
131
Abb. 5.2: Verstärkte Entzündung in CD11cCre/HIF-1α+f/+f Tieren.
DCs von CD11cCre/HIF-1α+f/+f
Tieren zeigten in einer experimentellen Colitis eine eingeschränkte
Fähigkeit Tregs zu aktivieren. Die Expression des TSLP-Rezeptors (TSLP-R) und Aldh1a2 war
vermindert. Die Produktion von IL-6 und IL-23 wurde gesteigert, was zu proinflammatorischen
Th1- und Th17-Antworten führte. Effektorlymphozyten wiesen im Vergleich zu HIF-1α+f/+f
Tieren eine
geringere Expression des Retinsäurerezeptors RARα und der darmspezifischen Adhäsionsmoleküle
CCR9 und β7-Integrin auf. Durch die hochgradige Entzündung produzierte das intestinale Epithel der
CD11cCre/HIF-1α+f/+f
Tiere kompensatorisch verstärkt Mucine.

Summary
132
6 Summary
Katharina Flück
The role of hypoxia-inducible factor 1 in dendritic cells in dextran sodium
sulfate-induced colitis
Dendritic cells (DCs) play a crucial role in connecting innate and adaptive immunity
and in maintaining intestinal homeostasis. They are highly involved in the
pathogenesis of inflammatory bowel disease (IBD). IBD is associated with hypoxic
inflammation where gene expression in DCs is regulated by the transcriptions factor
hypoxia inducible factor (HIF)-1. To investigate how dendritic HIF-1α in DCs
influences the development of an experimental colitis, control mice (HIF-1α+f/+f) and
knock-out mice, which had a deficiency of dendritic HIF-1α (CD11cCre/HIF-1α+f/+f),
were treated with 3 % dextran sodium sulfate (DSS) for 7 days to induce colitis.
Knock-out mice showed significantly higher weight loss at day 6 and 7 of the
experiment. mRNA expression of proinflammatory cytokines as interleukin (IL)-6 and
IL-23 was significantly increased in knock-out mice. Control mice with functional
dendritic HIF-1α showed significantly enhanced interferon (IFN)-α production and a
tolerogenic phenotype of DCs (expression of TSLP receptor) due to DSS-induced
colitis as well as increased amounts of Foxp3+ regulatory T cells (Tregs). Tregs are
induced through production of IL-10, TGF-β (transforming growth factor) and retinoic
acid (RA) by DCs. Expressions of IL-10 and TGF-β were significantly enhanced in
DSS-treated HIF-1α+f/+f mice compared to DSS treated CD11cCre/HIF-1α+f/+f mice.
RA is generated through degradation of vitamin A catalyzed by the aldehyde
dehydrogenase Aldh1a2. Expression of Aldh1a2 in control mice was significantly
increased through colitis compared to knock-out mice as well as expression of the
retinoic acid receptor α (RARα). RA also has influence on expression of the
gut-homing molecules α4β7-integrin and CCR9 (CC chemokine receptor 9).
DSS-treated control mice showed significantly increased levels of β7-integrin and
CCR9 compared to DSS-treated knock-out mice. In addition, the loss of dendritic
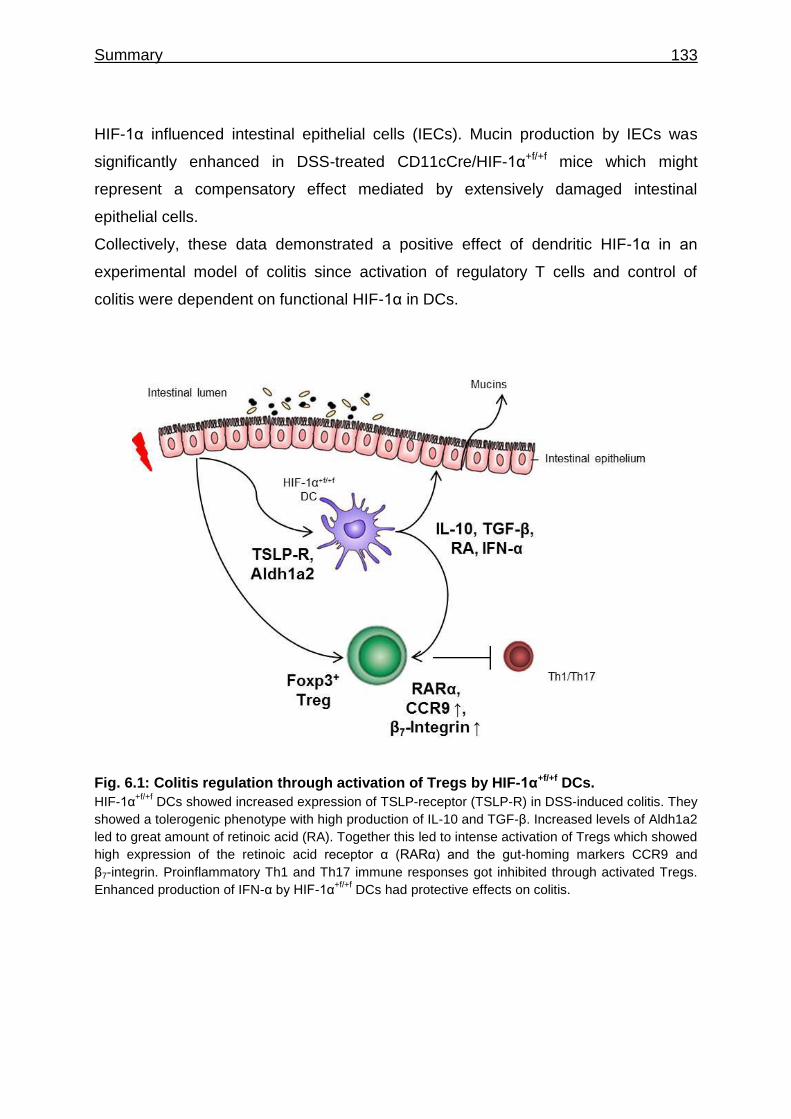
Summary
133
HIF-1α influenced intestinal epithelial cells (IECs). Mucin production by IECs was
significantly enhanced in DSS-treated CD11cCre/HIF-1α+f/+f mice which might
represent a compensatory effect mediated by extensively damaged intestinal
epithelial cells.
Collectively, these data demonstrated a positive effect of dendritic HIF-1α in an
experimental model of colitis since activation of regulatory T cells and control of
colitis were dependent on functional HIF-1α in DCs.
Fig. 6.1: Colitis regulation through activation of Tregs by HIF-1α+f/+f DCs.
HIF-1α+f/+f
DCs showed increased expression of TSLP-receptor (TSLP-R) in DSS-induced colitis. They
showed a tolerogenic phenotype with high production of IL-10 and TGF-β. Increased levels of Aldh1a2
led to great amount of retinoic acid (RA). Together this led to intense activation of Tregs which showed
high expression of the retinoic acid receptor α (RARα) and the gut-homing markers CCR9 and
β7-integrin. Proinflammatory Th1 and Th17 immune responses got inhibited through activated Tregs.
Enhanced production of IFN-α by HIF-1α+f/+f
DCs had protective effects on colitis.
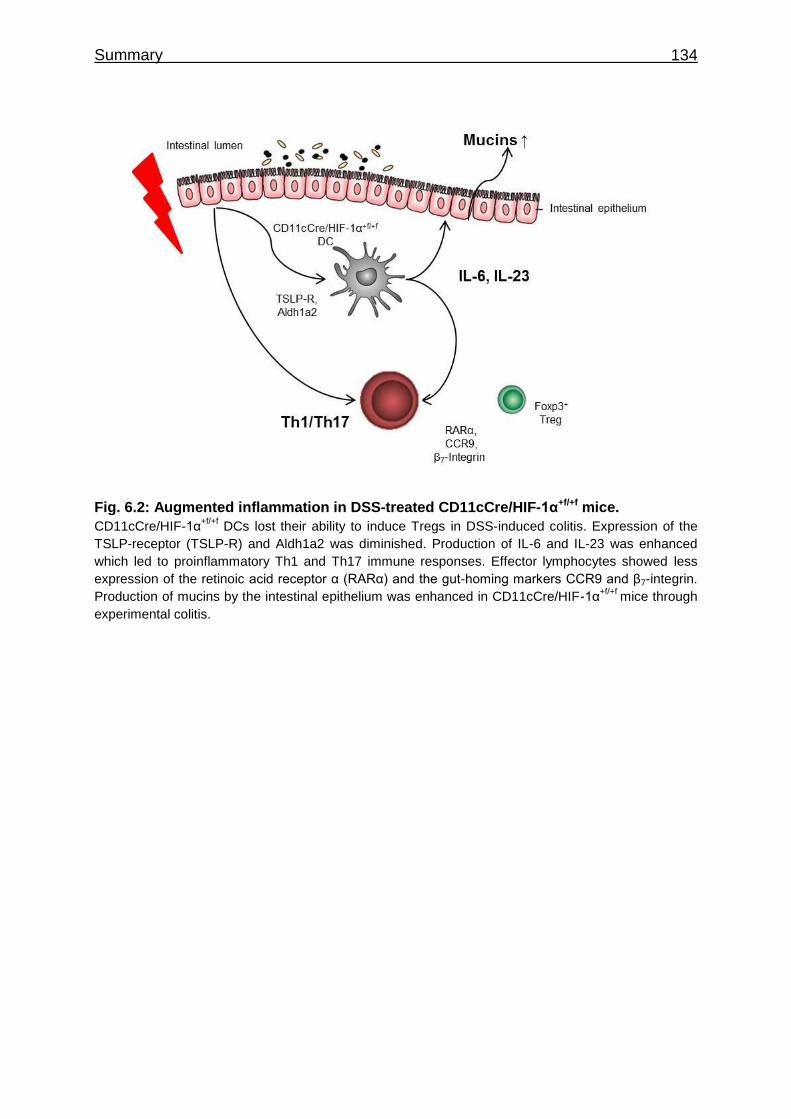
Summary
134
Fig. 6.2: Augmented inflammation in DSS-treated CD11cCre/HIF-1α+f/+f mice.
CD11cCre/HIF-1α+f/+f
DCs lost their ability to induce Tregs in DSS-induced colitis. Expression of the
TSLP-receptor (TSLP-R) and Aldh1a2 was diminished. Production of IL-6 and IL-23 was enhanced
which led to proinflammatory Th1 and Th17 immune responses. Effector lymphocytes showed less
expression of the retinoic acid receptor α (RARα) and the gut-homing markers CCR9 and β7-integrin.
Production of mucins by the intestinal epithelium was enhanced in CD11cCre/HIF-1α+f/+f
mice through
experimental colitis.

Literaturverzeichnis
135
7 Literaturverzeichnis
Abe, K., Nguyen, K.P., Fine, S.D., Mo, J.-H., Shen, C., Shenouda, S., Corr, M.,
Jung, S. et al. (2007). Conventional dendritic cells regulate the outcome of
colonic inflammation independently of T cells. Proceedings of the National
Academy of Sciences of the United States of America, 104(43), pp.17022–7.
Abraham, C. & Cho, J.H. (2009). Inflammatory bowel disease. The New England
journal of medicine, 361(21), pp.2066–78.
Acker, T., Fandrey, J., & Acker, H. (2006). The good, the bad and the ugly in
oxygen-sensing: ROS, cytochromes and prolyl-hydroxylases. Cardiovascular
research, 71(2), pp.195–207.
Annacker, O., Coombes, J.L., Malmstrom, V., Uhlig, H.H., Bourne, T.,
Johansson-Lindbom, B., Agace, W.W., Parker, C.M. et al. (2005). Essential
role for CD103 in the T cell-mediated regulation of experimental colitis. The
Journal of experimental medicine, 202(8), pp.1051–61.
Annese, V., Valvano, M.-R., Palmieri, O., Latiano, A., Bossa, F., & Andriulli, A.
(2006). Multidrug resistance 1 gene in inflammatory bowel disease: a meta-
analysis. World journal of gastroenterology : WJG, 12(23), pp.3636–44.
Arteel, G.E., Thurman, R.G., & Raleigh, J.A. (1998). Reductive metabolism of the
hypoxia marker pimonidazole is regulated by oxygen tension independent of the
pyridine nucleotide redox state. European journal of biochemistry / FEBS,
253(3), pp.743–50.
Atreya, R., Mudter, J., Finotto, S., Müllberg, J., Jostock, T., Wirtz, S., Schütz, M.,
Bartsch, B. et al. (2000). Blockade of interleukin 6 trans signaling suppresses
T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in
crohn disease and experimental colitis in vivo. Nature medicine, 6(5), pp.583–8.

Literaturverzeichnis
136
Aubert, S., Fauquette, V., Hémon, B., Lepoivre, R., Briez, N., Bernard, D., Van
Seuningen, I., Leroy, X. et al. (2009). MUC1, a new hypoxia inducible factor
target gene, is an actor in clear renal cell carcinoma tumor progression. Cancer
research, 69(14), pp.5707–15.
Autschbach, F., Eisold, S., Hinz, U., Zinser, S., Linnebacher, M., Giese, T.,
Löffler, T., Büchler, M.W. et al. (2005). High prevalence of Mycobacterium
avium subspecies paratuberculosis IS900 DNA in gut tissues from individuals
with Crohn’s disease. Gut, 54(7), pp.944–9.
Axelsson, L.G., Landström, E., Goldschmidt, T.J., Grönberg, A., & Bylund-
Fellenius, A.C. (1996). Dextran sulfate sodium (DSS) induced experimental
colitis in immunodeficient mice: effects in CD4(+) -cell depleted, athymic and NK-
cell depleted SCID mice. Inflammation research : official journal of the European
Histamine Research Society … [et al.], 45(4), pp.181–91.
De Baey, A., Mende, I., Baretton, G., Greiner, A., Hartl, W.H., Baeuerle, P., &
Diepolder, H.M. (2003). A subset of human dendritic cells in the T cell area of
mucosa-associated lymphoid tissue with a high potential to produce TNF-alpha.
Journal of immunology (Baltimore, Md. : 1950), 170(10), pp.5089–94.
Balmer, J.E. (2002). Gene expression regulation by retinoic acid. The Journal of
Lipid Research, 43(11), pp.1773–1808.
Banchereau, J. & Steinman, R.M. (1998). Dendritic cells and the control of
immunity. Nature, 392(6673), pp.245–52.
Basu, R.K., Hubchak, S., Hayashida, T., Runyan, C.E., Schumacker, P.T., &
Schnaper, H.W. (2011). Interdependence of HIF-1α and TGF-β/Smad3
signaling in normoxic and hypoxic renal epithelial cell collagen expression.
American journal of physiology. Renal physiology, 300(4), pp.F898–905.
Baumgart, D.C., Metzke, D., Schmitz, J., Scheffold, A., Sturm, A., Wiedenmann,
B., & Dignass, U. (2005). Patients with active inflammatory bowel disease lack

Literaturverzeichnis
137
immature peripheral blood plasmacytoid and myeloid dendritic cells. Gut, 54(2),
pp.228–36.
Baumgärtner, W. (2012). Pathohistologie für die Tiermedizin (2. ed.). Enke Verlag.
Becker, C., Wirtz, S., Blessing, M., Pirhonen, J., Strand, D., Bechthold, O., Frick,
J., Galle, P.R. et al. (2003). Constitutive p40 promoter activation and IL-23
production in the terminal ileum mediated by dendritic cells. The Journal of
clinical investigation, 112(5), pp.693–706.
Ben-Shoshan, J., Maysel-Auslender, S., Mor, A., Keren, G., & George, J. (2008).
Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-
inducible factor-1alpha. European journal of immunology, 38(9), pp.2412–8.
Benson, M.J., Pino-Lagos, K., Rosemblatt, M., & Noelle, R.J. (2007). All-trans
retinoic acid mediates enhanced T reg cell growth, differentiation, and gut
homing in the face of high levels of co-stimulation. The Journal of experimental
medicine, 204(8), pp.1765–74.
Berg, D.J., Davidson, N., Kühn, R., Müller, W., Menon, S., Holland, G.,
Thompson-Snipes, L., Leach, M.W. et al. (1996). Enterocolitis and colon
cancer in interleukin-10-deficient mice are associated with aberrant cytokine
production and CD4(+) TH1-like responses. The Journal of clinical investigation,
98(4), pp.1010–20.
Berger, T., Togawa, A., Duncan, G.S., Elia, A.J., You-Ten, A., Wakeham, A.,
Fong, H.E.H., Cheung, C.C. et al. (2006). Lipocalin 2-deficient mice exhibit
increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion
injury. Proceedings of the National Academy of Sciences of the United States of
America, 103(6), pp.1834–9.
Bergstrom, K.S.B., Kissoon-Singh, V., Gibson, D.L., Ma, C., Montero, M., Sham,
H.P., Ryz, N., Huang, T. et al. (2010). Muc2 protects against lethal infectious

Literaturverzeichnis
138
colitis by disassociating pathogenic and commensal bacteria from the colonic
mucosa. PLoS pathogens, 6(5), p.e1000902.
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T.B., Oukka, M., Weiner, H.L., &
Kuchroo, V.K. (2006). Reciprocal developmental pathways for the generation of
pathogenic effector TH17 and regulatory T cells. Nature, 441(7090), pp.235–8.
Blomhoff, R. & Blomhoff, H.K. (2006). Overview of retinoid metabolism and
function. Journal of neurobiology, 66(7), pp.606–30.
Bluestone, J., Mackay, C.R., O’Shea, J.J., & Stockinger, B. (2009). The functional
plasticity of T cell subsets. Nature reviews. Immunology, 9(11), pp.811–6.
Boirivant, M., Fuss, I.J., Chu, A., & Strober, W. (1998). Oxazolone colitis: A murine
model of T helper cell type 2 colitis treatable with antibodies to interleukin 4. The
Journal of experimental medicine, 188(10), pp.1929–39.
Bosseto, M.C., Palma, P.V.B., Covas, D.T., & Giorgio, S. (2010). Hypoxia
modulates phenotype, inflammatory response, and leishmanial infection of
human dendritic cells. APMIS : acta pathologica, microbiologica, et
immunologica Scandinavica, 118(2), pp.108–14.
Brandtzaeg, P. (2009). Mucosal immunity: induction, dissemination, and effector
functions. Scandinavian journal of immunology, 70(6), pp.505–15.
Butler, M., Ng, C.-Y., van Heel, D., Lombardi, G., Lechler, R., Playford, R.J., &
Ghosh, S. (2006). Modulation of dendritic cell phenotype and function in an in
vitro model of the intestinal epithelium. European journal of immunology, 36(4),
pp.864–74.
Cai, Z., Luo, W., Zhan, H., & Semenza, G.L. (2013). Hypoxia-inducible factor 1 is
required for remote ischemic preconditioning of the heart. Proceedings of the
National Academy of Sciences of the United States of America, 110(43),
pp.17462–7.

Literaturverzeichnis
139
Cantorna, M.T., Nashold, F.E., & Hayes, C.E. (1994). In vitamin A deficiency
multiple mechanisms establish a regulatory T helper cell imbalance with excess
Th1 and insufficient Th2 function. Journal of immunology (Baltimore, Md. : 1950),
152(4), pp.1515–22.
Chambers, R.E., Stross, P., Barry, R.E., & Whicher, J.T. (1987). Serum amyloid A
protein compared with C-reactive protein, alpha 1-antichymotrypsin and alpha 1-
acid glycoprotein as a monitor of inflammatory bowel disease. European journal
of clinical investigation, 17(5), pp.460–7.
Chassaing, B., Srinivasan, G., Delgado, M.A., Young, A.N., Gewirtz, A.T., &
Vijay-Kumar, M. (2012). Fecal lipocalin 2, a sensitive and broadly dynamic non-
invasive biomarker for intestinal inflammation. PloS one, 7(9), p.e44328.
Chaudhry, A., Samstein, R.M., Treuting, P., Liang, Y., Pils, M.C., Heinrich, J.-M.,
Jack, R.S., Wunderlich, F.T. et al. (2011). Interleukin-10 signaling in regulatory
T cells is required for suppression of Th17 cell-mediated inflammation. Immunity,
34(4), pp.566–78.
Chiodini, R.J., Van Kruiningen, H.J., Thayer, W.R., Merkal, R.S., & Coutu, J.A.
(1984). Possible role of mycobacteria in inflammatory bowel disease. I. An
unclassified Mycobacterium species isolated from patients with Crohn’s disease.
Digestive diseases and sciences, 29(12), pp.1073–9.
Chomczynski, P. & Sacchi, N. (1987). Single-step method of RNA isolation by acid
guanidinium thiocyanate-phenol-chloroform extraction. Analytical biochemistry,
162(1), pp.156–9.
Clambey, E.T., McNamee, E.N., Westrich, J.A., Glover, L.E., Campbell, E.L.,
Jedlicka, P., de Zoeten, E.F., Cambier, J.C. et al. (2012). Hypoxia-inducible
factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance
and function during inflammatory hypoxia of the mucosa. Proceedings of the

Literaturverzeichnis
140
National Academy of Sciences of the United States of America, 109(41),
pp.E2784–93.
Coffman, R.L. (2006). Origins of the T(H)1-T(H)2 model: a personal perspective.
Nature immunology, 7(6), pp.539–41.
Cong, Y., Brandwein, S.L., McCabe, R.P., Lazenby, A., Birkenmeier, E.H.,
Sundberg, J.P., & Elson, C.O. (1998). CD4+ T cells reactive to enteric bacterial
antigens in spontaneously colitic C3H/HeJBir mice: increased T helper cell type
1 response and ability to transfer disease. The Journal of experimental medicine,
187(6), pp.855–64.
Contractor, N. V, Bassiri, H., Reya, T., Park, Y., Baumgart, D.C., Wasik, M. a,
Emerson, S.G., & Carding, S.R. (1998). Lymphoid hyperplasia, autoimmunity,
and compromised intestinal intraepithelial lymphocyte development in colitis-free
gnotobiotic IL-2-deficient mice. Journal of immunology (Baltimore, Md. : 1950),
160(1), pp.385–94.
Coombes, J.L. & Powrie, F. (2008). Dendritic cells in intestinal immune regulation.
Nature reviews. Immunology, 8(6), pp.435–46.
Coombes, J.L., Siddiqui, K.R.R., Arancibia-Cárcamo, C. V, Hall, J., Sun, C.-M.,
Belkaid, Y., & Powrie, F. (2007). A functionally specialized population of
mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and
retinoic acid-dependent mechanism. The Journal of experimental medicine,
204(8), pp.1757–64.
Cooper, H.S., Murthy, S.N., Shah, R.S., & Sedergran, D.J. (1993).
Clinicopathologic study of dextran sulfate sodium experimental murine colitis.
Laboratory investigation; a journal of technical methods and pathology, 69(2),
pp.238–49.
Corfield, P., Myerscough, N., Longman, R., Sylvester, P., Arul, S., & Pignatelli,
M. (2000). Mucins and mucosal protection in the gastrointestinal tract: new

Literaturverzeichnis
141
prospects for mucins in the pathology of gastrointestinal disease. Gut, 47(4),
pp.589–94.
Cowland, J.B. & Borregaard, N. (1997). Molecular characterization and pattern of
tissue expression of the gene for neutrophil gelatinase-associated lipocalin from
humans. Genomics, 45(1), pp.17–23.
Cramer, T., Yamanishi, Y., Clausen, B.E., Förster, I., Pawlinski, R., Mackman, N.,
Haase, V.H., Jaenisch, R. et al. (2003). HIF-1alpha is essential for myeloid cell-
mediated inflammation. Cell, 112(5), pp.645–57.
Cummins, E.P., Seeballuck, F., Keely, S.J., Mangan, N.E., Callanan, J.J., Fallon,
P.G., & Taylor, C.T. (2008). The hydroxylase inhibitor dimethyloxalylglycine is
protective in a murine model of colitis. Gastroenterology, 134(1), pp.156–65.
Davis, W.C. & Madsen-Bouterse, S. (2012). Crohn’s disease and Mycobacterium
avium subsp. paratuberculosis: the need for a study is long overdue. Veterinary
immunology and immunopathology, 145(1-2), pp.1–6.
Dharmani, P., Leung, P., & Chadee, K. (2011). Tumor necrosis factor-α and Muc2
mucin play major roles in disease onset and progression in dextran sodium
sulphate-induced colitis. PloS one, 6(9), p.e25058.
Dieleman, L., Palmen, M.J., Akol, H., Bloemena, E., Peña, S., Meuwissen, S.G., &
Van Rees, E.P. (1998). Chronic experimental colitis induced by dextran sulphate
sodium (DSS) is characterized by Th1 and Th2 cytokines. Clinical and
experimental immunology, 114(3), pp.385–91.
Dieleman, L.A., Ridwan, B.U., Tennyson, G.S., Beagley, K.W., Bucy, R.P., &
Elson, C.O. (1994). Dextran sulfate sodium-induced colitis occurs in severe
combined immunodeficient mice. Gastroenterology, 107(6), pp.1643–52.
Dominitzki, S., Fantini, M.C., Neufert, C., Nikolaev, A., Galle, P.R., Scheller, J.,
Monteleone, G., Rose-John, S. et al. (2007). Cutting Edge: trans-signaling via

Literaturverzeichnis
142
the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells.
The Journal of Immunology, 179(4), pp.2041–2045.
Dorofeyev, A.E., Vasilenko, I. V, Rassokhina, O.A., & Kondratiuk, R.B. (2013).
Mucosal barrier in ulcerative colitis and Crohn’s disease. Gastroenterology
research and practice, 2013(Cd), p.431231.
Duerkop, B., Vaishnava, S., & Hooper, L. V (2009). Immune responses to the
microbiota at the intestinal mucosal surface. Immunity, 31(3), pp.368–76.
Duerr, R.H., Taylor, K.D., Brant, S.R., Rioux, J.D., Silverberg, M.S., Daly, M.J.,
Steinhart, H., Abraham, C. et al. (2006). A genome-wide association study
identifies IL23R as an inflammatory bowel disease gene. Science (New York,
N.Y.), 314(5804), pp.1461–3.
Duester, G. (2000). Families of retinoid dehydrogenases regulating vitamin A
function: production of visual pigment and retinoic acid. European journal of
biochemistry / FEBS, 267(14), pp.4315–24.
Eastaff-Leung, N., Mabarrack, N., Barbour, A., Cummins, A., & Barry, S. (2010).
Foxp3+ regulatory T cells, Th17 effector cells, and cytokine environment in
inflammatory bowel disease. Journal of clinical immunology, 30(1), pp.80–9.
Elson, C.O., Cong, Y., Weaver, C.T., Schoeb, T.R., McClanahan, T.K., Fick, R.B.,
& Kastelein, R.A. (2007). Monoclonal anti-interleukin 23 reverses active colitis
in a T cell-mediated model in mice. Gastroenterology, 132(7), pp.2359–70.
Eltzschig, H.K. & Carmeliet, P. (2011). Hypoxia and inflammation. The New
England journal of medicine, 364(7), pp.656–65.
Enss, M.L., Cornberg, M., Wagner, S., Gebert, A., Henrichs, M., Eisenblätter, R.,
Beil, W., Kownatzki, R. et al. (2000). Proinflammatory cytokines trigger MUC
gene expression and mucin release in the intestinal cancer cell line LS180.

Literaturverzeichnis
143
Inflammation research : official journal of the European Histamine Research
Society … [et al.], 49(4), pp.162–9.
Falanga, V., Qian, S.W., Danielpour, D., Katz, M.H., Roberts, A.B., & Sporn, M.B.
(1991). Hypoxia upregulates the synthesis of TGF-beta 1 by human dermal
fibroblasts. The Journal of investigative dermatology, 97(4), pp.634–7.
Fandrey, J. (2007). Regulation der Sauerstoffhomöostase durch Hypoxie-
induzierbaren Faktor-1. BIOspektrum, 1(1-2007), pp.26–28.
Fandrey, J., Gorr, T.A., & Gassmann, M. (2006). Regulating cellular oxygen
sensing by hydroxylation. Cardiovascular research, 71(4), pp.642–51.
Fernández-Martínez, A.B., Jiménez, M.I.A., Hernández, I.S., García-Bermejo,
M.L., Manzano, V.M., Fraile, E.A., & de Lucio-Cazaña, F.J. (2011). Mutual
regulation of hypoxic and retinoic acid related signalling in tubular proximal cells.
The international journal of biochemistry & cell biology, 43(8), pp.1198–207.
Frede, S., Berchner-Pfannschmidt, U., & Fandrey, J. (2007). Regulation of
hypoxia-inducible factors during inflammation. Methods in enzymology, 435,
pp.405–19.
Frede, S., Stockmann, C., Freitag, P., & Fandrey, J. (2006). Bacterial
lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42
MAPK and NF-kappaB. The Biochemical journal, 396(3), pp.517–27.
Fujimoto, M., Nakano, M., Terabe, F., Kawahata, H., Ohkawara, T., Han, Y.,
Ripley, B., Serada, S. et al. (2011). The influence of excessive IL-6 production
in vivo on the development and function of Foxp3+ regulatory T cells. Journal of
immunology (Baltimore, Md. : 1950), 186(1), pp.32–40.
Fuss, I.J., Heller, F., Boirivant, M., Leon, F., Yoshida, M., Fichtner-Feigl, S.,
Yang, Z., Exley, M. et al. (2004). Nonclassical CD1d-restricted NK T cells that

Literaturverzeichnis
144
produce IL-13 characterize an atypical Th2 response in ulcerative colitis. The
Journal of clinical investigation, 113(10), pp.1490–7.
Gerber, S.A. & Pober, J.S. (2008). IFN-alpha induces transcription of hypoxia-
inducible factor-1alpha to inhibit proliferation of human endothelial cells. Journal
of immunology (Baltimore, Md. : 1950), 181(2), pp.1052–62.
Gross, M.W., Karbach, U., Groebe, K., Franko, A.J., & Mueller-Klieser, W. (1995).
Calibration of misonidazole labeling by simultaneous measurement of oxygen
tension and labeling density in multicellular spheroids. International journal of
cancer. Journal international du cancer, 61(4), pp.567–73.
Gross, V., Andus, T., Caesar, I., Roth, M., & Schölmerich, J. (1992). Evidence for
continuous stimulation of interleukin-6 production in Crohn’s disease.
Gastroenterology, 102(2), pp.514–9.
Groux, H., Cottrez, F., Rouleau, M., Mauze, S., Antonenko, S., Hurst, S., McNeil,
T., Bigler, M. et al. (1999). A transgenic model to analyze the immunoregulatory
role of IL-10 secreted by antigen-presenting cells. Journal of immunology
(Baltimore, Md. : 1950), 162(3), pp.1723–9.
Gupta, A.K., Cherman, A.M., & Tyring, S.K. (2004). Viral and nonviral uses of
imiquimod: a review. Journal of cutaneous medicine and surgery, 8(5), pp.338–
52.
Hart, A.L., Al-Hassi, H.O., Rigby, R.J., Bell, S.J., Emmanuel, A. V., Knight, S.C.,
Kamm, M.A., & Stagg, A.J. (2005). Characteristics of intestinal dendritic cells in
inflammatory bowel diseases. Gastroenterology, 129(1), pp.50–65.
Hellwig-Bürgel, T., Rutkowski, K., Metzen, E., Fandrey, J., & Jelkmann, W.
(1999). Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding
of hypoxia-inducible factor-1. Blood, 94(5), pp.1561–7.

Literaturverzeichnis
145
Hendrickson, B.A., Gokhale, R., & Cho, J.H. (2002). Clinical aspects and
pathophysiology of inflammatory bowel disease. Clinical microbiology reviews,
15(1), pp.79–94.
Higashiyama, M., Hokari, R., Hozumi, H., Kurihara, C., Ueda, T., Watanabe, C.,
Tomita, K., Nakamura, M. et al. (2012). HIF-1 in T cells ameliorated dextran
sodium sulfate-induced murine colitis. Journal of leukocyte biology, 91(6),
pp.901–9.
Ho, S.B., Dvorak, L.A., Moor, R.E., Jacobson, A.C., Frey, M.R., Corredor, J.,
Polk, D.B., & Shekels, L.L. (2006). Cysteine-rich domains of muc3 intestinal
mucin promote cell migration, inhibit apoptosis, and accelerate wound healing.
Gastroenterology, 131(5), pp.1501–17.
Hoebler, C., Gaudier, E., De Coppet, P., Rival, M., & Cherbut, C. (2006). MUC
genes are differently expressed during onset and maintenance of inflammation
in dextran sodium sulfate-treated mice. Digestive diseases and sciences, 51(2),
pp.381–9.
Hoffmann, J.C., Pawlowski, N.N., Kühl, A., Höhne, W., & Zeitz, M. (2002). Animal
Models of Inflammatory Bowel Disease: An Overview. Pathobiology, 70(3),
pp.121–130.
Hori, S., Nomura, T., & Sakaguchi, S. (2003). Control of regulatory T cell
development by the transcription factor Foxp3. Science (New York, N.Y.),
299(5609), pp.1057–61.
Huang, Y., Fang, W., Wang, Y., Yang, W., & Xiong, B. (2012). Transforming growth
factor-β1 induces glutathione peroxidase-1 and protects from H2O2-induced cell
death in colon cancer cells via the Smad2/ERK1/2/HIF-1α pathway. International
journal of molecular medicine, 29(5), pp.906–12.
Hue, S., Ahern, P., Buonocore, S., Kullberg, M.C., Cua, D.J., McKenzie, B.S.,
Powrie, F., & Maloy, K.J. (2006a). Interleukin-23 drives innate and T cell-

Literaturverzeichnis
146
mediated intestinal inflammation. The Journal of experimental medicine,
203(11), pp.2473–83.
Hue, S., Ahern, P., Buonocore, S., Kullberg, M.C., Cua, D.J., McKenzie, B.S.,
Powrie, F., & Maloy, K.J. (2006b). Interleukin-23 drives innate and T cell-
mediated intestinal inflammation. The Journal of experimental medicine,
203(11), pp.2473–83.
Hung, S.-P., Yang, M.-H., Tseng, K.-F., & Lee, O.K. (2013). Hypoxia-induced
secretion of TGF-β1 in mesenchymal stem cell promotes breast cancer cell
progression. Cell transplantation, 22(10), pp.1869–82.
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara,
J.M. et al. (2001). HIFalpha targeted for VHL-mediated destruction by proline
hydroxylation: implications for O2 sensing. Science (New York, N.Y.), 292(5516),
pp.464–8.
Ivanov, I.I., McKenzie, B.S., Zhou, L., Tadokoro, C.E., Lepelley, A., Lafaille, J.J.,
Cua, D.J., & Littman, D.R. (2006). The orphan nuclear receptor RORgammat
directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell,
126(6), pp.1121–33.
Iwata, M., Hirakiyama, A., Eshima, Y., Kagechika, H., Kato, C., & Song, S.-Y.
(2004). Retinoic acid imprints gut-homing specificity on T cells. Immunity, 21(4),
pp.527–38.
Izcue, A., Coombes, J.L., & Powrie, F. (2006). Regulatory T cells suppress
systemic and mucosal immune activation to control intestinal inflammation.
Immunological reviews, 212, pp.256–71.
Izcue, A., Hue, S., Buonocore, S., Arancibia-Cárcamo, C. V, Ahern, P.P.,
Iwakura, Y., Maloy, K.J., & Powrie, F. (2008). Interleukin-23 restrains
regulatory T cell activity to drive T cell-dependent colitis. Immunity, 28(4),
pp.559–70.

Literaturverzeichnis
147
Jang, Y., Jeong, S.H., Park, Y.H., Bae, H.C., Lee, H., Ryu, W.I., Park, G.H., & Son,
S.W. (2013). UVB induces HIF-1α-dependent TSLP expression via the JNK and
ERK pathways. Journal of Investigative Dermatology, 133(11), pp.2601–2608.
Jantsch, J., Chakravortty, D., Turza, N., Prechtel, A.T., Buchholz, B., Gerlach,
R.G., Volke, M., Gläsner, J. et al. (2008). Hypoxia and hypoxia-inducible factor-
1 alpha modulate lipopolysaccharide-induced dendritic cell activation and
function. Journal of immunology (Baltimore, Md. : 1950), 180(7), pp.4697–705.
Jantsch, J., Wiese, M., Schödel, J., Castiglione, K., Gläsner, J., Kolbe, S., Mole,
D., Schleicher, U. et al. (2011). Toll-like receptor activation and hypoxia use
distinct signaling pathways to stabilize hypoxia-inducible factor 1α (HIF1A) and
result in differential HIF1A-dependent gene expression. Journal of leukocyte
biology, 90(3), pp.551–62.
Johansson-Lindbom, B., Svensson, M., Pabst, O., Palmqvist, C., Marquez, G.,
Förster, R., & Agace, W.W. (2005). Functional specialization of gut CD103+
dendritic cells in the regulation of tissue-selective T cell homing. The Journal of
experimental medicine, 202(8), pp.1063–73.
Johansson-Lindbom, B., Svensson, M., Wurbel, M.-A., Malissen, B., Márquez,
G., & Agace, W. (2003). Selective generation of gut tropic T cells in gut-
associated lymphoid tissue (GALT): requirement for GALT dendritic cells and
adjuvant. The Journal of experimental medicine, 198(6), pp.963–9.
Kang, S.G., Lim, H.W., Andrisani, O.M., Broxmeyer, H.E., & Kim, C.H. (2007).
Vitamin A metabolites induce gut-homing FoxP3+ regulatory T cells. Journal of
immunology (Baltimore, Md. : 1950), 179(6), pp.3724–33.
Karhausen, J., Furuta, G.T., Tomaszewski, J.E., Johnson, R.S., Colgan, S.P., &
Haase, V.H. (2004). Epithelial hypoxia-inducible factor-1 is protective in murine
experimental colitis. The Journal of clinical investigation, 114(8), pp.1098–106.

Literaturverzeichnis
148
Karhausen, J., Haase, V.H., & Colgan, S.P. (2005). Inflammatory hypoxia: role of
hypoxia-inducible factor. Cell cycle (Georgetown, Tex.), 4(2), pp.256–8.
Katakura, K., Lee, J., Rachmilewitz, D., Li, G., Eckmann, L., & Raz, E. (2005).
Toll-like receptor 9-induced type I IFN protects mice from experimental colitis.
The Journal of clinical investigation, 115(3), pp.695–702.
Kikly, K., Liu, L., Na, S., & Sedgwick, J.D. (2006). The IL-23/Th(17) axis:
therapeutic targets for autoimmune inflammation. Current opinion in
immunology, 18(6), pp.670–5.
Kitajima, S., Takuma, S., & Morimoto, M. (2000). Histological analysis of murine
colitis induced by dextran sulfate sodium of different molecular weights.
Experimental animals / Japanese Association for Laboratory Animal Science,
49(1), pp.9–15.
Kobayashi, M., Kweon, M., Kuwata, H., Schreiber, R.D., Kiyono, H., Takeda, K.,
& Akira, S. (2003). Toll-like receptor-dependent production of IL-12p40 causes
chronic enterocolitis in myeloid cell-specific Stat3-deficient mice. The Journal of
clinical investigation, 111(9), pp.1297–308.
Kong, T., Eltzschig, H.K., Karhausen, J., Colgan, S.P., & Shelley, C.S. (2004).
Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of
beta2 integrin gene expression. Proceedings of the National Academy of
Sciences of the United States of America, 101(28), pp.10440–5.
König, R., Huang, L.Y., & Germain, R.N. (1992). MHC class II interaction with CD4
mediated by a region analogous to the MHC class I binding site for CD8. Nature,
356(6372), pp.796–8.
Kucharzik, T., Maaser, C., Lügering, A., Kagnoff, M., Mayer, L., Targan, S., &
Domschke, W. (2006). Recent understanding of IBD pathogenesis: implications
for future therapies. Inflammatory bowel diseases, 12(11), pp.1068–83.

Literaturverzeichnis
149
Kuhlicke, J., Frick, J.S., Morote-Garcia, J.C., Rosenberger, P., & Eltzschig, H.K.
(2007). Hypoxia inducible factor (HIF)-1 coordinates induction of Toll-like
receptors TLR2 and TLR6 during hypoxia. PloS one, 2(12), p.e1364.
Kühn, R., Löhler, J., Rennick, D., Rajewsky, K., & Müller, W. (1993). Interleukin-
10-deficient mice develop chronic enterocolitis. Cell, 75(2), pp.263–74.
Lacy-Hulbert, A., Smith, A.M., Tissire, H., Barry, M., Crowley, D., Bronson, R.T.,
Roes, J.T., Savill, J.S. et al. (2007). Ulcerative colitis and autoimmunity induced
by loss of myeloid alphav integrins. Proceedings of the National Academy of
Sciences of the United States of America, 104(40), pp.15823–8.
Laemmli, U.K. (1970). Cleavage of structural proteins during the assembly of the
head of bacteriophage T4. Nature, 227(5259), pp.680–5.
LeBien, T.W. & Tedder, T.F. (2008). B lymphocytes: how they develop and function.
Blood, 112(5), pp.1570–80.
Levings, M.K., Gregori, S., Tresoldi, E., Cazzaniga, S., Bonini, C., & Roncarolo,
M.G. (2005). Differentiation of Tr1 cells by immature dendritic cells requires IL-10
but not CD25+CD4+ Tr cells. Blood, 105(3), pp.1162–9.
Li, Y.Y., Hsieh, L.L., Tang, R.P., Liao, S.K., & Yeh, K.Y. (2009). Macrophage-
derived interleukin-6 up-regulates MUC1, but down-regulates MUC2 expression
in the human colon cancer HAT-29 cell line. Cellular immunology, 256(1-2),
pp.19–26.
Louis, N.A., Hamilton, K.E., Canny, G., Shekels, L.L., Ho, S.B., & Colgan, S.P.
(2006). Selective induction of mucin-3 by hypoxia in intestinal epithelia. Journal
of cellular biochemistry, 99(6), pp.1616–27.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., & Randall, R.J. (1951). Protein
measurement with the Folin phenol reagent. The Journal of biological chemistry,
193(1), pp.265–75.

Literaturverzeichnis
150
Lund, B.M., Gould, G.W., & Rampling, A.M. (2002). Pasteurization of milk and the
heat resistance of Mycobacterium avium subsp. paratuberculosis: a critical
review of the data. International journal of food microbiology, 77(1-2), pp.135–
45.
Madara, J.L. (1998). Regulation of the movement of solutes across tight junctions.
Annual review of physiology, 60, pp.143–59.
Mahon, P.C., Hirota, K., & Semenza, G.L. (2001). FIH-1: a novel protein that
interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional
activity. Genes & development, 15(20), pp.2675–86.
Mancino, A., Schioppa, T., Larghi, P., Pasqualini, F., Nebuloni, M., Chen, I.-H.,
Sozzani, S., Austyn, J.M. et al. (2008). Divergent effects of hypoxia on dendritic
cell functions. Blood, 112(9), pp.3723–34.
Mangan, P.R., Harrington, L.E., O’Quinn, D.B., Helms, W.S., Bullard, D.C., Elson,
C.O., Hatton, R.D., Wahl, S.M. et al. (2006). Transforming growth factor-beta
induces development of the T(H)17 lineage. Nature, 441(7090), pp.231–4.
Mannon, P.J., Fuss, I.J., Mayer, L., Elson, C.O., Sandborn, W.J., Present, D.,
Dolin, B., Goodman, N. et al. (2004). Anti-interleukin-12 antibody for active
Crohn’s disease. The New England journal of medicine, 351(20), pp.2069–79.
Matsumoto, S., Okabe, Y., Setoyama, H., Takayama, K., Ohtsuka, J., Funahashi,
H., Imaoka, A., Okada, Y. et al. (1998). Inflammatory bowel disease-like
enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut,
43(1), pp.71–8.
Mawdsley, J.E. & Rampton, D.S. (2005). Psychological stress in IBD: new insights
into pathogenic and therapeutic implications. Gut, 54(10), pp.1481–91.
McGee, D.W., Beagley, K.W., Aicher, W.K., & McGhee, J.R. (1993). Transforming
growth factor-beta and IL-1 beta act in synergy to enhance IL-6 secretion by the

Literaturverzeichnis
151
intestinal epithelial cell line, IEC-6. Journal of immunology (Baltimore, Md. :
1950), 151(2), pp.970–8.
Medzhitov, R. (2007). Recognition of microorganisms and activation of the immune
response. Nature, 449(7164), pp.819–26.
Medzhitov, R. & Janeway, C. (2000). The Toll receptor family and microbial
recognition. Trends in microbiology, 8(10), pp.452–6.
Metzen, E., Zhou, J., Jelkmann, W., Fandrey, J., & Brüne, B. (2003). Nitric oxide
impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases.
Molecular biology of the cell, 14(8), pp.3470–81.
Mills, K.H.G. (2004). Regulatory T cells: friend or foe in immunity to infection? Nature
reviews. Immunology, 4(11), pp.841–55.
Min, W.-P., Zhou, D., Ichim, T.E., Strejan, G.H., Xia, X., Yang, J., Huang, X.,
Garcia, B. et al. (2003). Inhibitory Feedback Loop Between Tolerogenic
Dendritic Cells and Regulatory T Cells in Transplant Tolerance. The Journal of
Immunology, 170(3), pp.1304–1312.
Mittrücker, H.W. & Kaufmann, S.H.E. (2004). Mini-review: regulatory T cells and
infection: suppression revisited. European journal of immunology, 34(2), pp.306–
12.
Morris, G.P., Beck, P.L., Herridge, M.S., Depew, W.T., Szewczuk, M.R., &
Wallace, J.L. (1989). Hapten-induced model of chronic inflammation and
ulceration in the rat colon. Gastroenterology, 96(3), pp.795–803.
Mottet, C., Uhlig, H.H., & Powrie, F. (2003). Cutting edge: cure of colitis by
CD4+CD25+ regulatory T cells. Journal of immunology (Baltimore, Md. : 1950),
170(8), pp.3939–43.

Literaturverzeichnis
152
Mucida, D., Park, Y., Kim, G., Turovskaya, O., Scott, I., Kronenberg, M., &
Cheroutre, H. (2007). Reciprocal TH17 and regulatory T cell differentiation
mediated by retinoic acid. Science (New York, N.Y.), 317(5835), pp.256–60.
Müller, C., Autenrieth, I.B., & Peschel, A. (2005). Innate defenses of the intestinal
epithelial barrier. Cellular and molecular life sciences : CMLS, 62(12), pp.1297–
307.
Murphy, K., Travers, P., & Walport, M. (2009). Janeway Immunologie (7. ed.).
Spektrum Akademischer Verlag Heidelberg.
Naser, S., Ghobrial, G., Romero, C., & Valentine, J.F. (2004). Culture of
Mycobacterium avium subspecies paratuberculosis from the blood of patients
with Crohn’s disease. Lancet, 364(9439), pp.1039–44.
Newberry, R.D. & Lorenz, R.G. (2005). Organizing a mucosal defense.
Immunological reviews, 206, pp.6–21.
Ohashi, P.S. & DeFranco, A.L. (2002). Making and breaking tolerance. Current
opinion in immunology, 14(6), pp.744–59.
Ohning, G. V (2013). Intestinal mucins: the first line of defense. Journal of clinical
gastroenterology, 47(2), pp.104–5.
Oida, T., Suzuki, K., Nanno, M., Kanamori, Y., Saito, H., Kubota, E., Kato, S.,
Itoh, M. et al. (2000). Role of gut cryptopatches in early extrathymic maturation
of intestinal intraepithelial T cells. Journal of immunology (Baltimore, Md. : 1950),
164(7), pp.3616–26.
Okayasu, I., Hatakeyama, S., Yamada, M., Ohkusa, T., Inagaki, Y., & Nakaya, R.
(1990). A novel method in the induction of reliable experimental acute and
chronic ulcerative colitis in mice. Gastroenterology, 98(3), pp.694–702.

Literaturverzeichnis
153
Olsen, I., Tollefsen, S., Aagaard, C., Reitan, L.J., Bannantine, J.P., Andersen, P.,
Sollid, L.M., & Lundin, K.E. (2009). Isolation of Mycobacterium avium
subspecies paratuberculosis reactive CD4 T cells from intestinal biopsies of
Crohn’s disease patients. PloS one, 4(5), p.e5641.
Oppmann, B., Lesley, R., Blom, B., Timans, J.C., Xu, Y., Hunte, B., Vega, F., Yu,
N. et al. (2000). Novel p19 protein engages IL-12p40 to form a cytokine, IL-23,
with biological activities similar as well as distinct from IL-12. Immunity, 13(5),
pp.715–25.
Palmer, E. (2003). Negative selection--clearing out the bad apples from the T-cell
repertoire. Nature reviews. Immunology, 3(5), pp.383–91.
Peyssonnaux, C., Datta, V., Cramer, T., Doedens, A., Theodorakis, E.A., Gallo,
R.L., Hurtado-Ziola, N., Nizet, V. et al. (2005). HIF-1alpha expression regulates
the bactericidal capacity of phagocytes. The Journal of clinical investigation,
115(7), pp.1806–15.
Podolsky, D.K. (2002). Inflammatory bowel disease. The New England journal of
medicine, 347(6), pp.417–29.
Polosukhin, V.V., Cates, J.M., Lawson, W.E., Milstone, A.P., Matafonov, A.G.,
Massion, P.P., Lee, J.W., Randell, S.H. et al. (2011). Hypoxia-inducible factor-
1 signalling promotes goblet cell hyperplasia in airway epithelium. The Journal of
pathology, 224(2), pp.203–11.
Raffatellu, M., George, M.D., Akiyama, Y., Hornsby, M.J., Nuccio, S.-P., Paixao,
T.A., Butler, B.P., Chu, H. et al. (2009). Lipocalin-2 resistance confers an
advantage to Salmonella enterica serotype Typhimurium for growth and survival
in the inflamed intestine. Cell host & microbe, 5(5), pp.476–86.
Rama, I., Bruene, B., Torras, J., Koehl, R., Cruzado, J.M., Bestard, O.,
Franquesa, M., Lloberas, N. et al. (2008). Hypoxia stimulus: An adaptive

Literaturverzeichnis
154
immune response during dendritic cell maturation. Kidney international, 73(7),
pp.816–25.
Rescigno, M., Urbano, M., Valzasina, B., Francolini, M., Rotta, G., Bonasio, R.,
Granucci, F., Kraehenbuhl, J.P. et al. (2001). Dendritic cells express tight
junction proteins and penetrate gut epithelial monolayers to sample bacteria.
Nature immunology, 2(4), pp.361–7.
Rimoldi, M., Chieppa, M., Salucci, V., Avogadri, F., Sonzogni, A., Sampietro,
G.M., Nespoli, A., Viale, G. et al. (2005). Intestinal immune homeostasis is
regulated by the crosstalk between epithelial cells and dendritic cells. Nature
immunology, 6(5), pp.507–14.
Roncarolo, M.G., Gregori, S., Battaglia, M., Bacchetta, R., Fleischhauer, K., &
Levings, M.K. (2006). Interleukin-10-secreting type 1 regulatory T cells in
rodents and humans. Immunological reviews, 212, pp.28–50.
Sainathan, S.K., Bishnupuri, K.S., Aden, K., Luo, Q., Houchen, C.W., Anant, S.,
& Dieckgraefe, B.K. (2012). Toll-like receptor-7 ligand Imiquimod induces type I
interferon and antimicrobial peptides to ameliorate dextran sodium sulfate-
induced acute colitis. Inflammatory bowel diseases, 18(5), pp.955–67.
Sakuraba, A., Sato, T., Kamada, N., Kitazume, M., Sugita, A., & Hibi, T. (2009).
Th1/Th17 immune response is induced by mesenteric lymph node dendritic cells
in Crohn’s disease. Gastroenterology, 137(5), pp.1736–45.
Sallusto, F. & Lanzavecchia, A. (1994). Efficient presentation of soluble antigen by
cultured human dendritic cells is maintained by granulocyte/macrophage colony-
stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor
alpha. The Journal of experimental medicine, 179(4), pp.1109–18.
Samoszuk, M.K., Walter, J., & Mechetner, E. (2004). Improved
immunohistochemical method for detecting hypoxia gradients in mouse tissues

Literaturverzeichnis
155
and tumors. The journal of histochemistry and cytochemistry : official journal of
the Histochemistry Society, 52(6), pp.837–9.
Sansonetti, P.J. (2004). War and peace at mucosal surfaces. Nature reviews.
Immunology, 4(12), pp.953–64.
Sartor, R.B. (2006). Mechanisms of disease: pathogenesis of Crohn’s disease and
ulcerative colitis. Nature clinical practice. Gastroenterology & hepatology, 3(7),
pp.390–407.
Sartor, R.B. (2008). Microbial influences in inflammatory bowel diseases.
Gastroenterology, 134(2), pp.577–94.
Schmidt, C., Giese, T., Ludwig, B., Mueller-Molaian, I., Marth, T., Zeuzem, S.,
Meuer, S.C., & Stallmach, A. (2005). Expression of interleukin-12-related
cytokine transcripts in inflammatory bowel disease: elevated interleukin-23p19
and interleukin-27p28 in Crohn’s disease but not in ulcerative colitis.
Inflammatory bowel diseases, 11(1), pp.16–23.
Schofield, C.J. & Ratcliffe, P.J. (2004). Oxygen sensing by HIF hydroxylases.
Nature reviews. Molecular cell biology, 5(5), pp.343–54.
Semenza, G.L., Agani, F., Booth, G., Forsythe, J., Iyer, N., Jiang, B.H., Leung, S.,
Roe, R. et al. (1997). Structural and functional analysis of hypoxia-inducible
factor 1. Kidney international, 51(2), pp.553–5.
Shekels, L.L. & Ho, S.B. (2003). Characterization of the mouse Muc3 membrane
bound intestinal mucin 5’ coding and promoter regions: regulation by
inflammatory cytokines. Biochimica et biophysica acta, 1627(2-3), pp.90–100.
Shen, L. & Turner, J.R. (2006). Role of epithelial cells in initiation and propagation of
intestinal inflammation. Eliminating the static: tight junction dynamics exposed.
American journal of physiology. Gastrointestinal and liver physiology, 290(4),
pp.G577–82.

Literaturverzeichnis
156
Shirazi, T., Longman, R.J., Corfield, A.P., & Probert, C.S. (2000). Mucins and
inflammatory bowel disease. Postgraduate medical journal, 76(898), pp.473–8.
Shull, M.M., Ormsby, I., Kier, A.B., Pawlowski, S., Diebold, R.J., Yin, M., Allen,
R., Sidman, C. et al. (1992). Targeted disruption of the mouse transforming
growth factor-beta 1 gene results in multifocal inflammatory disease. Nature,
359(6397), pp.693–9.
Van der Sluis, M., De Koning, B.A., De Bruijn, A.C.J.M., Velcich, A., Meijerink,
J.P.P., Van Goudoever, J.B., Büller, H.A., Dekker, J. et al. (2006). Muc2-
deficient mice spontaneously develop colitis, indicating that MUC2 is critical for
colonic protection. Gastroenterology, 131(1), pp.117–29.
Smythies, L.E., Sellers, M., Clements, R.H., Mosteller-Barnum, M., Meng, G.,
Benjamin, W.H., Orenstein, J.M., & Smith, P.D. (2005). Human intestinal
macrophages display profound inflammatory anergy despite avid phagocytic and
bacteriocidal activity. The Journal of clinical investigation, 115(1), pp.66–75.
Stallmach, A., Giese, T., Schmidt, C., Ludwig, B., Mueller-Molaian, I., & Meuer,
S.C. (2004). Cytokine/chemokine transcript profiles reflect mucosal inflammation
in Crohn’s disease. International journal of colorectal disease, 19(4), pp.308–15.
Steinman, R.M. & Cohn, Z.A. (1973). Identification of a novel cell type in peripheral
lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. The
Journal of experimental medicine, 137(5), pp.1142–62.
Steinman, R.M. & Hemmi, H. (2006). Dendritic cells: translating innate to adaptive
immunity. Current topics in microbiology and immunology, 311, pp.17–58.
Strober, W., Fuss, I., & Mannon, P. (2007). The fundamental basis of inflammatory
bowel disease. The Journal of clinical investigation, 117(3), pp.514–21.
Strober, W., Fuss, I.J., & Blumberg, R.S. (2002). The immunology of mucosal
models of inflammation. Annual review of immunology, 20, pp.495–549.

Literaturverzeichnis
157
Strober, W., Murray, P.J., Kitani, A., & Watanabe, T. (2006). Signalling pathways
and molecular interactions of NOD1 and NOD2. Nature reviews. Immunology,
6(1), pp.9–20.
Sugimoto, K., Ogawa, A., Mizoguchi, E., Shimomura, Y., Andoh, A., Bhan, A.K.,
Blumberg, R.S., Xavier, R.J. et al. (2008). IL-22 ameliorates intestinal
inflammation in a mouse model of ulcerative colitis. The Journal of clinical
investigation, 118(2), pp.534–44.
Sun, C.-M., Hall, J.A., Blank, R.B., Bouladoux, N., Oukka, M., Mora, J.R., &
Belkaid, Y. (2007). Small intestine lamina propria dendritic cells promote de
novo generation of Foxp3 T reg cells via retinoic acid. The Journal of
experimental medicine, 204(8), pp.1775–85.
Takeda, K., Clausen, B.E., Kaisho, T., Tsujimura, T., Terada, N., Förster, I., &
Akira, S. (1999). Enhanced Th1 activity and development of chronic enterocolitis
in mice devoid of Stat3 in macrophages and neutrophils. Immunity, 10(1), pp.39–
49.
Tambuwala, M.M., Cummins, E.P., Lenihan, C.R., Kiss, J., Stauch, M., Scholz,
C.C., Fraisl, P., Lasitschka, F. et al. (2010). Loss of prolyl hydroxylase-1
protects against colitis through reduced epithelial cell apoptosis and increased
barrier function. Gastroenterology, 139(6), pp.2093–101.
Taupin, D. & Podolsky, D.K. (2003). Trefoil factors: initiators of mucosal healing.
Nature reviews. Molecular cell biology, 4(9), pp.721–32.
Taylor, C.T. & Colgan, S.P. (2007). Hypoxia and gastrointestinal disease. Journal of
molecular medicine (Berlin, Germany), 85(12), pp.1295–300.
Toussaint, M., Fievez, L., Drion, P., Cataldo, D., Bureau, F., Lekeux, P., &
Desmet, C.J. (2013). Myeloid hypoxia-inducible factor 1α prevents airway
allergy in mice through macrophage-mediated immunoregulation. Mucosal
immunology, 6(3), pp.485–97.

Literaturverzeichnis
158
Towbin, H., Staehelin, T., & Gordon, J. (1979). Electrophoretic transfer of proteins
from polyacrylamide gels to nitrocellulose sheets: procedure and some
applications. Proceedings of the National Academy of Sciences of the United
States of America, 76(9), pp.4350–4.
Travis, M., Reizis, B., Melton, A.C., Masteller, E., Tang, Q., Proctor, J.M., Wang,
Y., Bernstein, X. et al. (2007). Loss of integrin alpha(v)beta8 on dendritic cells
causes autoimmunity and colitis in mice. Nature, 449(7160), pp.361–5.
Uhlig, H.H., Coombes, J., Mottet, C., Izcue, A., Thompson, C., Fanger, A.,
Tannapfel, A., Fontenot, J.D. et al. (2006). Characterization of
Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of
colitis. Journal of immunology (Baltimore, Md. : 1950), 177(9), pp.5852–60.
Uhlig, H.H., McKenzie, B.S., Hue, S., Thompson, C., Joyce-Shaikh, B.,
Stepankova, R., Robinson, N., Buonocore, S. et al. (2006). Differential activity
of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity,
25(2), pp.309–18.
Veldhoen, M., Hocking, R.J., Atkins, C.J., Locksley, R.M., & Stockinger, B.
(2006). TGFbeta in the context of an inflammatory cytokine milieu supports de
novo differentiation of IL-17-producing T cells. Immunity, 24(2), pp.179–89.
De Villiers, W.J., Varilek, G.W., de Beer, F.C., Guo, J.T., & Kindy, M.S. (2000).
Increased serum amyloid a levels reflect colitis severity and precede amyloid
formation in IL-2 knockout mice. Cytokine, 12(9), pp.1337–47.
Vowinkel, T., Kalogeris, T.J., Mori, M., Krieglstein, C.F., & Granger, D.N. (2004).
Impact of dextran sulfate sodium load on the severity of inflammation in
experimental colitis. Digestive diseases and sciences, 49(4), pp.556–64.
Wallace, J.L., Vong, L., Dharmani, P., Srivastava, V., & Chadee, K. (2011). Muc-2-
deficient mice display a sex-specific, COX-2-related impairment of gastric
mucosal repair. The American journal of pathology, 178(3), pp.1126–33.

Literaturverzeichnis
159
Wang, G.L., Jiang, B.H., Rue, E., & Semenza, G.L. (1995). Hypoxia-inducible factor
1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension.
Proceedings of the National Academy of Sciences of the United States of
America, 92(12), pp.5510–4.
Watanabe, N., Wang, Y.-H., Lee, H.K., Ito, T., Wang, Y.-H., Cao, W., & Liu, Y.-J.
(2005). Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+
regulatory T cells in human thymus. Nature, 436(7054), pp.1181–5.
Welcker, K., Martin, A., Kölle, P., Siebeck, M., & Gross, M. (2004). Increased
intestinal permeability in patients with inflammatory bowel disease. European
journal of medical research, 9(10), pp.456–60.
Wenger, R.H., Stiehl, D.P., & Camenisch, G. (2005). Integration of oxygen
signaling at the consensus HRE. Science’s STKE : signal transduction
knowledge environment, 2005(306), p.re12.
Westendorf, A.M., Fleissner, D., Hansen, W., & Buer, J. (2010). T cells, dendritic
cells and epithelial cells in intestinal homeostasis. International journal of
medical microbiology : IJMM, 300(1), pp.11–8.
Westra, J., Brouwer, E., Bos, R., Posthumus, M.D., Doornbos-Van Der Meer, B.,
Kallenberg, C.G.M., & Limburg, P.C. (2007). Regulation of Cytokine-Induced
HIF-1 Expression in Rheumatoid Synovial Fibroblasts. Annals of the New York
Academy of Sciences, 1108(1), pp.340–348.
Williams, K.L., Fuller, C.R., Dieleman, L., DaCosta, C.M., Haldeman, K.M.,
Sartor, R.B., & Lund, P.K. (2001). Enhanced survival and mucosal repair after
dextran sodium sulfate-induced colitis in transgenic mice that overexpress
growth hormone. Gastroenterology, 120(4), pp.925–37.
Winer, J., Jung, C.K., Shackel, I., & Williams, P.M. (1999). Development and
validation of real-time quantitative reverse transcriptase-polymerase chain

Literaturverzeichnis
160
reaction for monitoring gene expression in cardiac myocytes in vitro. Analytical
biochemistry, 270(1), pp.41–9.
Wobben, R., Hüsecken, Y., Lodewick, C., Gibbert, K., Fandrey, J., & Winning, S.
(2013). Role of hypoxia inducible factor-1α for interferon synthesis in mouse
dendritic cells. Biological chemistry, 394(4), pp.495–505.
Workman, C.J., Szymczak-Workman, A.L., Collison, L.W., Pillai, M.R., & Vignali,
D. (2009). The development and function of regulatory T cells. Cellular and
molecular life sciences : CMLS, 66(16), pp.2603–22.
Xavier, R.J. & Podolsky, D.K. (2007). Unravelling the pathogenesis of inflammatory
bowel disease. Nature, 448(7152), pp.427–34.
Yamamoto, M., Yoshizaki, K., Kishimoto, T., & Ito, H. (2000). IL-6 is required for
the development of Th1 cell-mediated murine colitis. Journal of immunology
(Baltimore, Md. : 1950), 164(9), pp.4878–82.
Yen, D., Cheung, J., Scheerens, H., Poulet, F., McClanahan, T., McKenzie, B.,
Kleinschek, M.A., Owyang, A. et al. (2006). IL-23 is essential for T cell-
mediated colitis and promotes inflammation via IL-17 and IL-6. The Journal of
clinical investigation, 116(5), pp.1310–6.
Yokoigawa, N., Takeuchi, N., Toda, M., Inoue, M., Kaibori, M., Yanagida, H.,
Tanaka, H., Ogura, T. et al. (2005). Enhanced production of interleukin 6 in
peripheral blood monocytes stimulated with mucins secreted into the
bloodstream. Clinical cancer research : an official journal of the American
Association for Cancer Research, 11(17), pp.6127–32.
Zaph, C., Troy, A.E., Taylor, B.C., Berman-Booty, L.D., Guild, K.J., Du, Y., Yost,
E., Gruber, A.D. et al. (2007). Epithelial-cell-intrinsic IKK-beta expression
regulates intestinal immune homeostasis. Nature, 446(7135), pp.552–6.

Literaturverzeichnis
161
Zhou, J., Fandrey, J., Schümann, J., Tiegs, G., & Brüne, B. (2003). NO and TNF-
alpha released from activated macrophages stabilize HIF-1alpha in resting
tubular LLC-PK1 cells. American journal of physiology. Cell physiology, 284(2),
pp.C439–46.
Zhou, X., Tu, J., Li, Q., Kolosov, V.P., & Perelman, J.M. (2012). Hypoxia induces
mucin expression and secretion in human bronchial epithelial cells. Translational
research : the journal of laboratory and clinical medicine, 160(6), pp.419–27.

Anhang
162
8 Anhang
8.1 Abkürzungsverzeichnis
Abkürzung Bedeutung
µ Mikro
A. dest. destilliertes Wasser (aqua destillata)
Abb. Abbildung
ADH Alkohol Dehydrogenase
ALDH Aldehyd Dehydrogenase
APC Antigen präsentierende Zelle (antigen presenting cell)
APP Akute-Phase-Protein
APS Ammoniumpersulfat
ARNT Arylhydrocarbon Rezeptor Nuklear Translokator
bHLH Basisches Helix-Loop-Helix
BmDC aus dem Knochenmark generierte Dendritische Zellen
(bone marrow-derived dendritic cells)
bp base pair
BSA bovines Serumalbumin
bzw. beziehungsweise
C Celsius
ca. circa
CAD C-terminale Transaktivierungsdomäne
cAMP zyklisches Adenosinmonophosphat
CBP CREB-bindendes Protein
CCR CC Chemokin Rezeptor
CD Nomenklatur für Zelloberflächenmoleküle (cluster of differentiation)
cDC klassische dendritische Zellen
cDNA komplementäre Desoxyribonukleinsäure
CED Chronisch entzündliche Darmerkrankungen

Anhang
163
ChIP Chromatin-Immunpräzipitation
CO2 Kohlendioxid
Ct cycle threshold
CTL zytotoxische T-Zelle (cytotoxic T-lymphocyte)
CU Colitis ulcerosa
C57BL/6 C57/Black 6
DAPI 4′,6-Diamidin-2-phenylindol
DC Dendritische Zelle (dendritic cell)
DEPC Diethylpyrocarbonat
DF doppelt gefloxt (double-floxed)
DMOG Dimethyloxallylglycin
DNA Desoxyribonucleinsäure
dsDNA doppelsträngige DNA
DSS Dextrannatriumsulfat (dextran sodium sulfate)
ECL Enhanced chemiluminescence
EDTA Ethylendiamintetraessigsäure
ELISA Enzyme-linked immunosorbent assay
engl. englisch
FCS Fötales Kälberserum
Fig. Figure
FIH-1 HIF-inhibierender Faktor-1 (factor inhibiting HIF 1)
Foxp3 Forkhead-Box-Protein P3
g Gramm
GALT Darmassoziierte lymphatische Gewebe (gut associated lymphoid tissue)
GM-CSF Granulozyten-Makrophagen Kolonie-stimulierender Faktor
(granulocyte macrophage colony-stimulating factor)
GTC Guanidinthiocyanat
h Stunde(n)
H&E Hämatoxylin und Eosin

Anhang
164
HIF Hypoxie induzierbarer Faktor
HIF-1αf+/f+
homozygote Mäuse mit einem beidseitig von loxP-sites flankiertem
Exon 2 des HIF-1α Gens
H2O Wasser
H2O2 Wasserstoffperoxid
HOX Hypoxie
HPLC Hochleistungsflüssigkeitschromatographie
(high performance liquid chromatography)
HRE Hypoxie-responsives Element
IBD inflammatory bowel disease
IEC Intestinale Epithelzelle (intestinal epithelial cell)
i.p. intraperitoneal
IFN Interferon
IL Interleukin
KCL Kaliumchlorid
KH2PO4 Kaliumdihydrogenphosphat
KO Knock-out
l Liter
LPS Lipopolysaccharid
LysM Lysozym M
M Molar
m milli
MAdCAM-1 mukosales gefäßspezifisches Adressin 1
(mucosal vascular addressin cell-adhesion molecule 1)
MAP Mycobacterium avium subspezies paratuberculosis
MC Morbus Crohn
MHC Haupthistokompatibilitätskomplex (major histocompatibility complex)
min Minute(n)
mRNA messenger RNA

Anhang
165
MUC Mucin
n nano
n Anzahl
NaCL Natriumchlorid
NAD N-terminale Transaktivierungsdomäne
Na2HPO4 Dinatriumhydrogenphosphat
NaOAc Natriumacetat
NaOH Natriumhydroxid
NF-B nuclear factor ‚kappa-light-chain-enhancer‘ of activated B-cells
NGS normal goat serum
NK-Zellen Natürliche Killerzellen
NOX Normoxie
NRS normal rabbit serum
NTP Nukleosidtriphosphat
O2 Sauerstoff
ODDD Sauerstoffabhängige Degradationsdomäne (oxygen-dependent degradation
domain)
Oligo-dT Oligomer aus Desoxynukleosidtriphosphaten
PAMP Pathogen-assoziierte molekulare Muster
(pathogen-associated molecular patterns)
PAS Per-ARNT-Sim
PBS Phosphat-gepufferte Kochsalzlösung (phosphate buffered saline)
PCR Polymerase Kettenreaktion (polymerase chain reaction)
PFA Paraformaldehyd
pH -log [H+]
PHD Prolylhydroxylase
PRR Mustererkennungs-Rezeptor (pattern-recognition receptor)
pVHL von Hippel-Lindau Tumorsuppressorprotein
qPCR quantitative Echtzeit-PCR

Anhang
166
RA Retinsäure (retinoic acid)
RAG recombination activation gene
RARα Retinsäure Rezeptor α (retinoic acid receptor α)
rib.Protein ribosomales Protein
RNA Ribonukleinsäure
rpm Umdrehungen pro Minute (revolutions per minute)
RPMI Roswell Park Memorial Institute
RT Raumtemperatur
RT(-PCR) Reverse Transkriptase (-PCR)
s Sekunde(n)
s. siehe
S. Seite
SAA Serum Amyloid A
SCID severe combined immunodeficiency
SDS Natriumdodecylsulfat (sodium dodecyl sulfate)
SEM Standardfehler (standard error of mean)
SPF speziell pathogenfrei (special pathogen free)
TAD Transaktivierungsdomäne
TAE Tris-acetate-EDTA
TBS Tris-gepufferte Kochsalzlösung
TCR T-Zellrezeptor (T-cell receptor)
TFF Kleeblatt-Faktor (trefoil factor)
TGF Transformierender Wachstumsfaktor (transforming growth factor)
TLR Toll-like Rezeptor
TNBS trinitrobenzene sulfonic acid
TNF Tumornekrosefaktor
Treg Regulatorische T-Zelle
TRIS Tris-(hydroxymethyl)-aminomethan
TSLP Thymisches stromales Lymphopoietin

Anhang
167
TSLPR TSLP-Rezeptor
TWEEN Polyoxyethylen(20)-sorbitan-monolaurat
U Units
VE vollentsalzt
vHL von Hippel-Lindau Protein
Vol. Volumen
WT Wild-typ
z. B. zum Beispiel
Zyk. Zyklen

Anhang
168
8.2 Abbildungsverzeichnis
Abb. 1.1: Aktivierung der adaptiven Immunantwort durch DCs. ............................... 11
Abb. 1.2: Zusammenspiel der Immunzellen.............................................................. 13
Abb. 1.3: Suppressionsmechanismen von Tregs. .................................................... 15
Abb. 1.4: Rolle der Retinsäure in der intestinalen Immunität. ................................... 16
Abb. 1.5: Auslösende Faktoren einer chronisch entzündlichen Darmerkrankung..... 20
Abb. 1.6: Struktur des Hypoxie-induzierbaren Faktors HIF-1. .................................. 24
Abb. 1.7: Sauerstoffabhängige Regulation der HIF-α Aktivität. ................................ 26
Abb. 1.8: Sauerstoffunabhängige Regulation der HIF-1α Aktivität. .......................... 27
Abb. 2.1: Schematische Darstellung des Cre/loxP-Knockout-Systems. ................... 43
Fig. 3.1: DSS colitis leads to profound extension of hypoxia. ................................... 84
Fig. 3.2: Dendritic cell specific knock-out of HIF-1α led to more severe inflammation
in DSS colitis. .................................................................................................... 86
Fig. 3.3: HIF-1α dependent induction of type I interferons in DSS colitis. ................. 87
Fig. 3.4: Inflammatory induction of IL-10 and TSLPR is HIF-1α dependent. ............. 88
Fig. 3.5: Activation and gut-homing of Tregs in DSS colitis require HIF-1α expression
in dendritic cells. ................................................................................................ 90
Fig. 3.6: Increases in numbers of T cells and Tregs in mesenteric lymph nodes
depend on dendritic HIF-1α expression in DSS colitis. ...................................... 92
Fig. 3.7: Dendritic cell–specific knock-out of HIF-1α stimulates mucosal epithelium to
increased production of mucins in DSS colitis. .................................................. 94
Fig. 3.8: Knock-out efficiency of CD11cCre/HIF-1α+f/+f mice is approximately 90%. . 95
Fig. 3.9: Dendritic cell–specific knock-out leads to severer inflammation in DSS
colitis. ................................................................................................................ 96
Fig. 3.10: IL-10 ELISA of colon tissue shows no differences between groups.......... 97
Fig. 3.11: mRNA expression of α4 integrin increases in DSS colitis. ........................ 97
Fig. 3.12: mRNA expression of trefoil factor (TFF)3 increases in DSS-treated
CD11cCre/HIF-1α+f/+f mice. ............................................................................... 98
Abb. 3.13: Anstieg des Disease Activity Index (DAI) durch DSS Colitis. ................ 102
Abb. 3.14: Verkürzung des Colons und Anstieg des Colongewichtes durch DSS
Colitis. .............................................................................................................. 103

Anhang
169
Abb. 3.15: Hochgradige Entzündung des Colons durch DSS Behandlung. ............ 105
Abb. 3.16: Anstieg des histologischen Scores durch DSS Behandlung. ................ 106
Abb. 3.17: Hochgradige Entzündung des Colons vor allem im distalen Bereich. ... 107
Abb. 3.18: Verstärkte Alcianblau Färbung in DSS behandelten CD11cCre/HIF-1α+f/+f
Tieren. ............................................................................................................. 109
Abb. 5.1: Regulation der Colitis durch Aktivierung von Tregs durch HIF-1α+f/+f DCs.
........................................................................................................................ 130
Abb. 5.2: Verstärkte Entzündung in CD11cCre/HIF-1α+f/+f Tieren. ......................... 131
Fig. 6.1: Colitis regulation through activation of Tregs by HIF-1α+f/+f DCs. ............. 133
Fig. 6.2: Augmented inflammation in DSS-treated CD11cCre/HIF-1α+f/+f mice. ...... 134

Anhang
170
8.3 Tabellenverzeichnis
Tabelle 2.1: Geräte ................................................................................................... 31
Tabelle 2.2: Verbrauchsmaterialien .......................................................................... 32
Tabelle 2.3: Chemikalien .......................................................................................... 33
Tabelle 2.4: Antikörper für Western Blot ................................................................... 37
Tabelle 2.5: Antikörper für die Immunfluoreszenz .................................................... 38
Tabelle 2.6: Murine Primer ....................................................................................... 39
Tabelle 2.7: Murine Genotypisierungsprimer ............................................................ 41
Tabelle 2.8: Reagenzien für cDNA-Synthese, PCR, Real Time PCR, PCR Array und
Sequenzierung .................................................................................................. 41
Tabelle 2.9: Mastermix für die Genotypisierung ....................................................... 44
Tabelle 2.10: Exemplarisches PCR-Programm für die Genotypisierung .................. 44
Tabelle 2.11: Disease Activity Index ......................................................................... 46
Tabelle 2.12: Mastermix für die cDNA Synthese ...................................................... 50
Tabelle 2.13: Mastermix für PCR .............................................................................. 51
Tabelle 2.14: Exemplarisches PCR-Programm ........................................................ 51
Tabelle 2.15: Mastermix für Real Time PCR ............................................................ 52
Tabelle 2.16: Exemplarisches Real Time PCR-Programm ....................................... 52
Tabelle 2.17: Zusammenstellung der im Array untersuchten Gene .......................... 53
Tabelle 2.18: Reverse Transkriptase Mix für PCR Array .......................................... 57
Tabelle 2.19: Real Time PCR Programm für PCR Array .......................................... 57
Tabelle 2.20: Zusammensetzung der Trenn- und Sammelgele ................................ 59
Tabelle 2.21: Histologischer Score ........................................................................... 62
Table 3.1: Primer sequences .................................................................................... 99



















![CME ZertifizierteFortbildung - kielstein.com · CME mit tubulärer Resorption und Exkretion [8], aber auch durch die extreme Vulnerabilität der ständig an der Grenze zur Hypoxie](https://img.pdfslide.org/doc/110x75/5d538c0688c993337f8b9fb8/cme-zertifiziertefortbildung-cme-mit-tubulaerer-resorption-und-exkretion-8.jpg)









